WO2011010740A1 - 微生物検出法及び微生物検出キット - Google Patents
微生物検出法及び微生物検出キット Download PDFInfo
- Publication number
- WO2011010740A1 WO2011010740A1 PCT/JP2010/062474 JP2010062474W WO2011010740A1 WO 2011010740 A1 WO2011010740 A1 WO 2011010740A1 JP 2010062474 W JP2010062474 W JP 2010062474W WO 2011010740 A1 WO2011010740 A1 WO 2011010740A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- cells
- pcr
- dna
- nucleic acid
- test sample
- Prior art date
Links
Images

















Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6844—Nucleic acid amplification reactions
- C12Q1/6848—Nucleic acid amplification reactions characterised by the means for preventing contamination or increasing the specificity or sensitivity of an amplification reaction
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/02—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving viable microorganisms
- C12Q1/04—Determining presence or kind of microorganism; Use of selective media for testing antibiotics or bacteriocides; Compositions containing a chemical indicator therefor
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N13/00—Treatment of microorganisms or enzymes with electrical or wave energy, e.g. magnetism, sonic waves
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6844—Nucleic acid amplification reactions
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6876—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes
- C12Q1/6888—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for detection or identification of organisms
- C12Q1/689—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for detection or identification of organisms for bacteria
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2561/00—Nucleic acid detection characterised by assay method
- C12Q2561/113—Real time assay
Definitions
- the present invention relates to a method for detecting microorganisms contained in foods and biological samples, microorganisms contained in environments such as industrial water and city water, and a microorganism detection kit. More specifically, the present invention relates to a detection method and a microorganism detection kit that can selectively detect living cells of microorganisms contained in an environment such as foods, biological samples, wiped samples, industrial water, and city water.
- a plate culture method has been used to measure the number of general viable bacteria in foods, biological samples, wiped samples, or the environment.
- the plate culture method takes about 2 days to about a month until results are obtained.
- Patent Document 1 or Patent Document 2 is disclosed as a method for determining the viability of microorganisms such as bacteria using the PCR method.
- the following problems remain in the method for determining the viability of microorganisms such as bacteria using these PCR methods.
- Patent Document 2 discloses a method for discriminating between live cells and dead cells using the fact that the RNA / DNA molar ratio of dead cells is relatively lower than that of live cells. .
- total RNA is extracted, complementary DNA is prepared using reverse transcription reaction, PCR is then performed to calculate its Ct value, and the molar concentration of RNA is determined using a separately prepared calibration curve.
- the region of the chromosomal DNA corresponding to this RNA is amplified by PCR to determine the Ct value, and the molar concentration of chromosomal DNA is calculated from the calibration curve to determine the molar ratio of RNA / DNA.
- RNA derived from dead cells is not stable because it is degraded early with time.
- a food or clinical specimen containing a high concentration of dead cells only 1/10 concentration of living cells can be detected. Therefore, it has been difficult to apply in food hygiene inspections and clinical inspections that require rapid, high sensitivity and accuracy.
- Non-Patent Document 1 A method using ethidium monoazide is also disclosed in Non-Patent Document 1.
- ethidium monoazide is added to the test sample and light is irradiated, DNA is extracted from the sample after irradiation, and a specific region is detected by PCR using the extracted DNA as a template. This is a detection method comprising an amplifying step.
- Non-Patent Document 1 discloses a technique for quantifying the number of living cells semi-quantitatively by combining culturing of microorganisms and a real-time PCR method.
- a method described in Patent Document 4 is disclosed as a method for further clearly distinguishing between living cells and damaged cells of microorganisms.
- This method includes a step of adding a crosslinking agent that crosslinks DNA by light irradiation with a wavelength of 350 nm to 700 nm to a test sample, a step of performing a light irradiation treatment with a wavelength of 350 nm to 700 nm to a test sample to which a crosslinking agent has been added, A step of removing the crosslinking agent contained in the test sample subjected to the light irradiation treatment, a step of adding a culture medium to the test sample from which the cross-linking agent has been removed, and maintaining the temperature, and the irradiated test sample is irradiated again with light having a wavelength of 350 nm to 700 nm.
- a step of adding a cross-linking agent for cross-linking DNA a step of subjecting a test sample to which a cross-linking agent has been added to a light irradiation treatment at a wavelength of 350 nm to 700 nm, extracting DNA from the test sample, and extracting the DNA target
- the method includes a step of amplifying a region by a nucleic acid amplification method and a step of analyzing an amplification product.
- Non-patent Document 2 a method of performing a PCR reaction without extracting DNA from bacteria in a PCR reaction using bacterial DNA as a template has been disclosed (Non-patent Documents 4 and 5).
- Patent Document 5 describes performing random PCR from bacteria in a DNA fingerprinting method, and describes phosphate and dodecyl sulfate as components of a buffer composition for nucleic acid synthesis.
- the above-described method using the topoisomerase inhibitor and / or DNA gyrase inhibitor, or the crosslinking agent is highly effective when the living cells of microorganisms, particularly the living cells of bacteria such as Klebsiella, Citrobacter, Listeria, Salmonella, Although selective detection is possible with sensitivity, a further improved method, in particular, a method for detecting living cells with high sensitivity or high accuracy for Escherichia or Salmonella bacteria has been desired. It is an object of the present invention to provide a new method for selectively detecting living cells of microorganisms contained in foods, biological samples and the like compared to dead cells and damaged cells, and a kit for carrying out the method. To do.
- the present inventors are applicable to various sterilization methods, a method for distinguishing between viability and death of microorganisms suitable for food hygiene inspection with high detection sensitivity, and a method capable of detecting specific pathogens in patients with infectious diseases in hospitals and clinical settings
- the sample was irradiated with light having a wavelength of 350 nm to 700 nm to which an agent that covalently binds to DNA or RNA was added, and the sample was irradiated with light having a wavelength of 350 nm to 700 nm to function as a nucleic acid amplification inhibitor
- the determination can be performed with high sensitivity by adding a suppressive agent, a magnesium salt, and an organic acid salt or phosphate, and amplifying the chromosomal DNA of the microorganism eluted outside the cell by a nucleic acid amplification reaction. As a result, the present invention has been completed.
- the present invention provides a method for detecting a living cell of a microorganism in a test sample by distinguishing it from a dead cell or a damaged cell, and including the following steps. a) adding to the test sample a drug that covalently binds to DNA or RNA by irradiation with light having a wavelength of 350 nm to 700 nm; b) performing a light irradiation treatment at a wavelength of 350 nm to 700 nm on the test sample to which the drug is added; c) A step of amplifying a target region of DNA or RNA of a microorganism contained in a test sample by a nucleic acid amplification method in the presence of a drug that suppresses the function of a nucleic acid amplification inhibitor without extracting nucleic acid from cells.
- the amplification of the target region is performed in a microbial cell.
- the method preferably amplifies the target region in the presence of one or more selected from surfactants, magnesium salts, and organic acid salts or phosphates. It is an aspect.
- the method is preferably performed by repeating the steps a) and b) before the step c).
- the said method makes it the preferable aspect to perform the process of the following e) before the process of said a).
- the said method makes it a preferable aspect that the said enzyme is selected from a proteolytic enzyme, a lipolytic enzyme, and a glycolytic enzyme.
- the said test sample is any one of a foodstuff, a biological sample, drinking water, industrial water, environmental water, drainage, soil, or a wiping sample.
- the microorganism is preferably a bacterium or a virus.
- the bacterium is a gram-negative bacterium.
- the agent that is covalently bonded to DNA or RNA by irradiation with light having a wavelength of 350 nm to 700 nm is ethidium monoazide, ethidium diazide, propidium monoazide.
- Psolaren, 4,5 ′, 8-trimethyl psolaren, and 8-methoxy psolaren are preferred embodiments.
- the agent that suppresses the action of the nucleic acid amplification inhibitor is albumin, dextran, T4 gene 32 protein, acetamide, betaine, dimethyl sulfoxide, formamide, glycerol, polyethylene glycol, soybean trypsin inhibitor, ⁇ 2-macroglobulin.
- Tetramethylammonium chloride, lysozyme, phosphorylase, and lactate dehydrogenase are one or more selected from the preferred embodiments.
- the organic acid salt is preferably selected from acetate, propionate, and citrate.
- the said method makes it a preferable aspect that the said phosphate is a pyrophosphate.
- the target region is preferably a target region of 50 to 5000 bases.
- the said method makes it a preferable aspect that the said target region is a target region corresponding to the gene selected from 5S rRNA gene of the DNA of a test sample, 16S rRNA gene, 23S rRNA gene, and tRNA gene.
- the method has a preferred embodiment in which the nucleic acid amplification method is a PCR method, a LAMP method, an SDA method, an LCR method, a TMA method, a TRC method, an HC method, or a microarray method.
- the said method makes it a preferable aspect to perform the said PCR method by real-time PCR method, and to analyze PCR and an amplification product simultaneously.
- the said method makes it a preferable aspect to perform the analysis of the said amplification product using the standard curve which shows the relationship between the amount of microorganisms produced using the standard sample of microorganisms, and an amplification product.
- the kit of the present invention is a kit for distinguishing and detecting a living cell of a microorganism in a test sample from a dead cell or a damaged cell by a nucleic acid amplification method, which includes the following elements: To do. 1) a drug that covalently binds to DNA or RNA by irradiation with light having a wavelength of 350 nm to 700 nm; 2) a drug that suppresses the action of a nucleic acid amplification inhibitor; and 3) a primer for amplifying a target region of DNA or RNA of a microorganism to be detected by a nucleic acid amplification method.
- the kit preferably includes any one or more selected from surfactants, magnesium salts, organic acid salts or phosphates.
- the kit preferably includes an enzyme having an activity of degrading cells other than microorganisms, protein colloid particles, fat, or carbohydrates present in the test sample.
- the kit has a preferred embodiment in which the nucleic acid amplification method is a PCR method, RT-PCR method, LAMP method, SDA method, LCR method, TMA method, TRC method, HC method, or microarray method.
- the agent that is covalently bonded to DNA or RNA by irradiation with light having a wavelength of 350 nm to 700 nm is ethidium monoazide, ethidium diazide, propidium monoazide.
- Psolaren, 4,5 ′, 8-trimethyl psolaren, and 8-methoxy psolaren are preferred embodiments.
- the agent that suppresses the action of the nucleic acid amplification inhibitor is albumin, dextran, and T4 gene 32 protein, acetamide, betaine, dimethyl sulfoxide, formamide, glycerol, polyethylene glycol, soybean trypsin inhibitor, ⁇ 2-macro.
- a preferred embodiment is one or more selected from globulin, tetramethylammonium chloride, lysozyme, phosphorylase, and lactate dehydrogenase.
- the said kit makes it the preferable aspect that the said organic acid salt is selected from acetate, propionate, and citrate.
- the said kit makes it a preferable aspect that the said phosphate is a pyrophosphate.
- the said kit makes it the preferable aspect that the said enzyme is selected from a proteolytic enzyme, a lipolytic enzyme, and a carbohydrase.
- the electrophoresis photograph of the PCR amplification product by the method of the present invention “Live” indicates a living cell, and “damaged” indicates a damaged cell.
- the electrophoresis photograph which shows the detection result of living microorganisms by the method of this invention. Electrophoresis photograph showing detection results of living microorganism cells according to the prior art. “Live” indicates a living cell, and “damaged” indicates a damaged cell.
- Fluorescence microscopic photograph and stereoscopic microscope photograph of Enterobacter Sakazaki physiological saline suspension after thermal cycle rotation Fluorescence microscopic photograph and stereoscopic microscope photograph of Enterobacter / Sakazaki bacteria saline suspension supernatant after thermal cycle rotation. Fluorescence microscopic photograph and stereoscopic microscope photograph of non-heated Enterobacter Sakazaki pretreatment solution suspension. Fluorescence microscopic photograph and stereoscopic microscope photograph of non-heated Enterobacter / Sakazaki bacteria pretreatment solution suspension supernatant. Fluorescence microscopic photograph and stereoscopic microscope photograph of Enterobacter Sakazaki pretreatment solution suspension after thermal cycle rotation.
- Lanes 2 and 3 PCR reaction supernatant lanes 5 and 6: DNA lanes 7 extracted from the centrifugal pellet after washing twice after PCR reaction, 8: DNA lanes 9 extracted directly from cells, 10: Actually immediately before PCR DNA lanes 13 and 14 extracted from cells used for testing: DNA extracted from cells washed after addition of PCR product: 100-bp DNA ladderB: fix solution BS: non-fixed An electrophoretogram of a suspension of Enterobacter sakazaki after heat treatment in the presence of physiological saline or in the presence of a pretreatment agent and a centrifugal supernatant thereof.
- L 100-bp DNA ladder
- any nucleic acid in general can be used as an object of detection as long as it can be amplified as a result.
- single-stranded DNA, double-stranded DNA, 1 Double-stranded RNA and double-stranded RNA can be exemplified.
- DNA is a detection target, and double-stranded DNA is particularly preferable.
- the method of the present invention is a method for detecting living cells of microorganisms in a test sample by distinguishing them from dead cells or damaged cells, and includes the following steps. a) adding to the test sample a drug that covalently binds to DNA or RNA by irradiation with light having a wavelength of 350 nm to 700 nm; b) performing a light irradiation treatment at a wavelength of 350 nm to 700 nm on the test sample to which the drug is added; c) a step of amplifying a target region of microbial DNA or RNA contained in a test sample by a nucleic acid amplification method in the presence of a drug that suppresses the action of a nucleic acid amplification inhibitor without extracting nucleic acid from cells; And d) analyzing the amplification product.
- test sample is a target for detecting living cells of microorganisms present therein, and the presence is detected by amplification of a specific region of chromosomal DNA or RNA by a nucleic acid amplification method.
- a foodstuff, biological sample, drinking water, industrial water, environmental water, drainage, soil, or a wipe sample etc. are mentioned.
- foods include soft drinks, carbonated drinks, nutrition drinks, fruit juice drinks, lactic acid bacteria drinks and other drinks (including concentrated concentrates and powders for preparation of these drinks); ice creams such as ice cream, ice sherbet and shaved ice; Dairy products such as milk, milk drinks, fermented milk, butter; enteral nutrition foods, liquid foods, milk for childcare, sports drinks; functional foods such as foods for specified health use and health supplements are preferred.
- Biological samples include blood samples, urine samples, spinal fluid samples, synovial fluid samples, pleural fluid samples, sputum samples, stool samples, nasal mucus samples, laryngeal mucus samples, gastric lavage fluid samples, pus juice samples, skin mucosa samples, oral cavity samples
- mucus samples include mucus samples, respiratory mucosa samples, digestive mucosa samples, eye conjunctiva samples, placenta samples, germ cell samples, birth canal samples, breast milk samples, saliva samples, vomit, or blister contents.
- examples of the environmental water include city water, ground water, river water, and rain water.
- the test sample may be a food, biological sample, drinking water, industrial water, environmental water, waste water, soil, or a wipe sample itself as described above, or a diluted or concentrated product thereof.
- pretreatment other than the treatment according to the method of the present invention may be performed. Examples of the pretreatment include heat treatment, filtration, and centrifugation.
- cells other than microorganisms, protein colloid particles, fats and carbohydrates, etc. present in the test sample may be removed or reduced by treatment with an enzyme having an activity of decomposing them.
- Examples of cells other than microorganisms present in the test sample include bovine leukocytes and mammary epithelial cells when the test sample is milk, dairy products, milk or foods made from dairy products.
- the test sample is a biological sample such as a blood sample, urine sample, spinal fluid sample, synovial fluid sample or pleural effusion sample, red blood cells, white blood cells (granulocytes, neutrophils, basophils, monocytes, lymphoid cells) Spheres), and platelets.
- the enzyme is not particularly limited as long as it can decompose the contaminants and does not damage the living cells of the microorganism to be detected.
- a lipolytic enzyme a proteolytic enzyme, and a carbohydrase Enzymes.
- the enzyme one kind of enzyme may be used alone, or two or more kinds of enzymes may be used in combination, but both lipolytic enzyme and proteolytic enzyme, or lipolytic enzyme, proteolytic enzyme It is preferable to use all of saccharide-degrading enzymes.
- lipolytic enzyme examples include lipase and phosphatase
- examples of the proteolytic enzyme include serine protease, cysteine protease, proteinase K, and pronase
- examples of the carbohydrate degrading enzyme include amylase and cellulase.
- a “microorganism” is an object to be detected by the method of the present invention, can be detected by a nucleic acid amplification method, and is a drug that is covalently bound to DNA or RNA by irradiation with light having a wavelength of 350 nm to 700 nm.
- the action is not particularly limited as long as the action on microorganisms is different between live cells, dead cells, and damaged cells, but preferably bacteria, filamentous fungi, yeasts, viruses and the like can be mentioned.
- Bacteria include both gram-positive bacteria and gram-negative bacteria.
- Gram-positive bacteria include Staphylococcus bacteria such as Staphylococcus epidermidis, Streptococcus pneumoniae such as Streptococcus pneumoniae, Listeria monocytogenes Listeria monocytogenes, Bacillus cereus, Bacillus anthracis, Bacillus anthracis, Mycobacterium tuberculosis, Mycobacterium tuberculosis, Mycobacterium Mycobacterium bacteria such as Mycobacterium bovis, Mycobacterium avium, Clostridium botulinum, Clostridium persium Examples include Clostridium bacteria such as fringen (Clostridium perfringens).
- Gram-negative bacteria include Escherichia bacteria such as Escherichia coli, Enterobacter bacteria such as Enterobacter akasakazakii, and Citrobacter bacteria such as Citrobacter koseri.
- the virus include viruses such as influenza viruses having an envelope, and noroviruses, rotaviruses, adenoviruses and the like that do not have an envelope and have only a nucleocapsid.
- EMA does not permeate activated viruses, only permeates only inactivated viruses with nucleocapsids that are severely physically damaged, and EMA can distinguish between activated viruses (Live) and inactivated viruses (Dead). It has been suggested that it is possible. Therefore, it is considered that the present invention can be applied not only to bacteria, filamentous fungi and yeasts but also to viruses.
- a “live cell” is a state (Viable-and-Culturable cell state) that can proliferate when cultured under suitable culture conditions and exhibits the metabolic activity of the microorganism.
- the metabolic activity mentioned here can be exemplified by ATP activity and esterase activity.
- virus particles are also referred to as “cells” for convenience.
- Live cell refers to a state in which a mammalian cell can be infected and propagated with respect to a virus.
- Dead cells are microorganisms that cannot grow even when cultured under suitable culture conditions and do not exhibit metabolic activity (Dead).
- the structure of the cell wall is maintained, the cell wall itself is highly damaged, and a weakly permeable nuclear stain such as propidium iodide penetrates the cell wall.
- virus it means a state in which mammalian cells cannot be infected.
- “Injured cell” (Viable-but-Non Culturable cell) is a cell that has been damaged by human or environmental stress, and therefore proliferates even when cultured under suitable culture conditions. Although it is difficult, the microorganism has a metabolic activity that is reduced compared to living cells, but is significantly more active than dead cells. Regarding virus, it means a state in which, even if a mammalian cell is infected, it cannot grow in the cell.
- live cells”, “dead cells” and “damaged cells” mean live cells, dead cells and damaged cells of microorganisms.
- the unit of the number of living cells, damaged cells, and dead cells is usually expressed by the number of cells (cells) / ml.
- the number of cells is expressed in logarithm, and “a log 10 / ml” represents 10 a / ml.
- the number of living cells can be approximated by the number of colonies formed (cfu / ml (colony forming units / ml)) when cultured on a suitable plate medium under suitable conditions.
- a standard sample of damaged cells can be prepared, for example, by subjecting a living cell suspension to heat treatment, for example, heat treatment in boiling water. In this case, the number of damaged cells is heat treated.
- damaged cells can be prepared in about 50 seconds.
- standard samples of damaged cells can also be prepared by antibiotic treatment, in which case the number of damaged cells is determined by treating the live cell suspension with antibiotics and then removing the antibiotics.
- the preferable conditions on a suitable plate medium can be approximated by the number of colonies formed (cfu / ml).
- the cell number unit is expressed in plaque-forming units (pfu or PFU (plaque-forming units)).
- the method of the present invention is intended for detection of live cells, and the microorganisms distinguished from live cells may be damaged cells or dead cells.
- detection of living cells includes both determination of the presence or absence of living cells in the test sample and determination of the amount of living cells. Further, the amount of living cells is not limited to an absolute amount, and may be an amount relative to a control sample. Further, “detecting a living cell by discriminating it from a dead cell or a damaged cell” means selectively detecting a dead cell or a damaged cell. Note that “discrimination between live cells and dead cells or damaged cells” includes discrimination between live cells and both dead cells and damaged cells.
- the test sample may have an activity of degrading cells other than microorganisms, protein colloid particles, fat, or carbohydrates present in the test sample.
- the process of processing with the enzyme which has may be included.
- the drug intercalates into double-stranded DNA or RNA, and is covalently bonded by light irradiation to crosslink between the molecules.
- the drug is covalently bonded to single-stranded DNA or RNA by light irradiation to inhibit the PCR reaction.
- the drug may be simply referred to as “crosslinking agent”.
- the cross-linking agent has a different action on living cells from damaged cells or dead cells and bovine leukocytes and other somatic cells, leukocytes, platelets, etc., more specifically, damage more than the cell walls of live cells. It is preferable that it is highly permeable to cell walls of cells or dead cells, or somatic cells such as bovine leukocytes, and cell membranes such as leukocytes and platelets.
- cross-linking agent examples include ethidium monoazide, ethidium diazide, psolaren, 4,5 ′, 8-trimethyl psoralen (4,5 ′, 8-trimethyl psolaren), And 8-methoxy psolaren, propidium monoazide and the like.
- a crosslinking agent may be used individually by 1 type, and may use 2 or more types together.
- the treatment conditions with the cross-linking agent can be set as appropriate.
- various concentrations of the cross-linking agent can be added to the suspension of living cells and dead cells or damaged cells of the microorganism to be detected, After leaving the time, the cells can be separated by centrifugation or the like and analyzed by a nucleic acid amplification method to determine conditions that make it easy to distinguish between live cells and dead cells or damaged cells.
- ethidium monoazide has a final concentration of 1 to 100 ⁇ g / ml, 4 to 10 ° C., 5 minutes to 48 hours
- ethidium diazide has a final concentration of 1 to 100 ⁇ g / ml, 4 to 10 5 minutes to 48 hours at 0 ° C., final concentration 1 to 100 ⁇ g / ml for propidium monoazide, 4 to 10 ° C., 5 minutes to 48 hours, final concentration 1 ⁇ 10 ⁇ 5 to 10 ⁇ g / ml for psoralen, 25 to 37 ° C.
- Step b) the test sample to which the cross-linking agent is added is subjected to light irradiation treatment with a wavelength of 350 nm to 700 nm.
- the cross-linking agent is more permeable to the cell walls of dead and damaged cells than the cell walls of living cells. Therefore, it is considered that the cell wall of living cells of microorganisms does not substantially permeate within the action time shown above, and the cell membrane of somatic cells that are damaged cells or dead cells of microorganisms or dead cells permeate.
- the cross-linking agent enters the dead cells of somatic cells, dead cells of microorganisms, and cells of damaged cells, and then hydrogen bonds with chromosomal DNA or RNA and irradiates with light having a wavelength of 350 nm to 700 nm.
- DNA molecules are cross-linked or covalently bonded to RNA.
- chromosomal DNA is distorted, RNA is modified with a cross-linking agent, and finally chromosomal DNA is destroyed (fragmentation / cleavage). ) Or RNA is no longer a template for nucleic acid amplification reaction.
- the light having a wavelength of 350 nm to 700 nm may be light having a wavelength of at least 350 nm to 700 nm, may be single wavelength light, or may be composite light. Further, all the components may be in the range of 350 nm to 700 nm, and may include light having a shorter wavelength than 350 nm and / or light having a longer wavelength of 700 nm or more, but the peak in the intensity distribution is 350 nm. It is preferably in the range of ⁇ 700 nm. It should be noted that it is preferable not to include a component having a short wavelength enough to cleave the chromosomal DNA of a microorganism only by light irradiation.
- chromosomal DNA of damaged or dead cells is preferentially destroyed over live cells
- the target region of chromosomal DNA is amplified by nucleic acid amplification in live cells, whereas chromosomal DNA of damaged and dead cells is amplified.
- the nucleic acid amplification reaction is inhibited, and live cells can be selectively detected compared to damaged cells or dead cells.
- RNA of damaged or dead cells is preferentially modified with a crosslinking agent over living cells
- the target region of RNA is amplified by nucleic acid amplification in live cells, whereas in damaged or dead cells, As a result of modification of the target region of RNA, the nucleic acid amplification reaction is inhibited, and live cells can be selectively detected compared to damaged or dead cells.
- the crosslinking agent is ethidium monoazide
- the test sample to which ethidium monoazide is added is irradiated with light having a wavelength of 350 nm to 700 nm.
- Ethidium monoazide (EMA) is more likely to penetrate damaged and dead cell walls than living cell walls of microorganisms. Therefore, it is considered that EMA does not substantially permeate the cell walls of living cells of microorganisms, and permeates the cell walls of damaged cells of dead cells, cell walls of dead cells, and somatic cells that are dead cells.
- EMA permeates the cell membrane of the cells under sterile water or a hypotonic salt solution.
- EMA enters into dead cells of somatic cells, damaged cells of microorganisms and dead cells, and intercalates with DNA in the nucleus, and then is irradiated with light having a wavelength of 350 nm to 700 nm. Intercalated EMA is converted into nitrene, covalently bonded to DNA in the nucleus, and crosslinks between DNA molecules.
- EMA covalently bound to each base and deoxyribose everywhere in the chromosomal DNA causes large distortion in the chromosomal DNA, and as a result, the chromosomal DNA is destroyed (fragmented).
- double-stranded RNA including partial double-stranded RNA
- EMA entered the dead cells of somatic cells, damaged cells of microorganisms, and dead cells, and intercalated RNA randomly. Thereafter, only EMA intercalated by irradiation with light having a wavelength of 350 nm to 700 nm is converted into nitrene, covalently bonded to RNA, and crosslinked between RNA molecules.
- EMA covalently bonded to each base of RNA causes large distortion in RNA, and as a result, RNA is destroyed (fragmented). Further, for single-stranded DNA or RNA, EMA enters dead cells of somatic cells and damaged cells of microorganisms and cells of dead cells, and EMA is converted to nitrene by light irradiation with a wavelength of 350 nm to 700 nm, Presumed to be covalently linked to DNA or RNA.
- cross-linking agents other than ethidium monoazide are more likely to penetrate damaged and dead cell walls than living cell walls of microorganisms, and emit light with a wavelength of 350 nm to 700 nm (long wavelength ultraviolet light or visible light).
- Any substance that crosslinks DNA or covalently binds to RNA by irradiation and as a result destroys chromosomal DNA or modifies RNA can be used in the present invention.
- the conditions for treatment with EMA can be set as appropriate. For example, various concentrations of EMA are added to a living cell of a microorganism to be detected and a suspension of damaged or dead cells for various times. After placing the cells, irradiate visible light, and if necessary, separate the cells by centrifugation, etc., and analyze them by nucleic acid amplification method to determine the conditions for distinguishing live cells from dead cells and damaged cells. can do. Moreover, the conditions for light irradiation can also be determined by changing the irradiation time and conducting the above experiment.
- the light irradiation condition include a condition in which light having a wavelength of 100 to 750 W is irradiated for 5 minutes to 2 hours from a distance of 10 to 50 cm from the test sample.
- the light irradiation is preferably performed at a low temperature, for example, by cooling the sample with ice.
- the addition of the crosslinking agent in the above step a) and the light irradiation treatment in the step b) may be performed by repeating two cycles or more cycles.
- the concentration of the crosslinking agent is preferably higher in the first step a) than in the second and subsequent steps, and lower in the second and subsequent steps a) than in the first.
- EMA when EMA is applied at a high concentration, for example, 10 ⁇ g / ml or more, the permeability of dead cells to the cell wall or cell membrane increases, but the permeability to living cells also increases (Microbiology and Immunology, 2007, 51, and p). .763-775, Journal of Clinical Microbiology, 2008, 46, p.2305-2313).
- a concentration lower than 10 ⁇ g / ml permeation into living cells can be avoided, but the permeability to dead cells also decreases, and dead cells may be detected by a nucleic acid amplification reaction. Therefore, it is preferable to increase the concentration of the crosslinking agent in the first step a) and decrease the concentration of the crosslinking agent in the second and subsequent steps b).
- the final concentration of ethidium monoazide is 10 to 100 ⁇ g / ml
- the final concentration of ethidium diazide is 10 to 100 ⁇ g / ml
- the final concentration of propidium monoazide is 10 to 100 ⁇ g / ml.
- ml final concentration 2 ⁇ 10 -5 ⁇ 10 [mu] g / ml in psoralen, 4,5 ', 8-trimethyl-flop Solare final concentration 2 ⁇ 10 -5 ⁇ 10 [mu] g / ml in emissions, 8-methoxy-flop Solare final concentration 2 ⁇ 10 in emissions -5 to 10 ⁇ g / ml.
- the first step a it is preferable to shorten the processing time than in the second and subsequent steps b).
- a step of removing unreacted cross-linking agent may be added between step b) of the previous cycle and step a) of the next cycle. Moreover, you may add the process of removing a crosslinking agent between the process b) and the following processes c). Usually, the unreacted crosslinking agent in step a) is almost inactivated in step b). Therefore, the method for removing the cross-linking agent includes a method of centrifuging the test sample, separating the precipitate containing the microorganism and the supernatant containing the cross-linking agent, and removing the supernatant. In this case, after removing the crosslinking agent, it is possible to add a step of washing the microorganisms with a cleaning agent as appropriate.
- Step c) nucleic acid amplification is performed in the presence of a drug that suppresses the action of a nucleic acid amplification inhibitor without extracting nucleic acid from cells in the target region of microbial DNA or RNA contained in the test sample after light irradiation treatment. Amplify by method.
- a nucleic acid amplification reaction is performed by adding an agent that suppresses the action of a nucleic acid amplification inhibitor to a nucleic acid amplification reaction solution containing a test sample.
- a surfactant e.g., sodium EDTA, sodium EDTA, sodium EDTA, sodium EDTA, sodium EDTA, sodium EDTA, sodium EDTA, sodium EDTA, sodium EDTA, sodium EDTA, sodium EDTA, sodium EDTA, sodium EDTA, sodium EDTA, sodium EDTA, sodium EDTA, sodium EDTA, sodium EDTA, sodium EDTA, sodium EDTA, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium
- a nucleic acid amplification inhibitor is a substance that inhibits a nucleic acid amplification reaction or a nucleic acid extension reaction.
- the positive charge inhibitor include calcium ions, polyamines, and heme.
- Examples of the negative charge inhibitor include phenol, phenolic compounds, heparin, and Gram-negative bacterial cell wall outer membrane. Foods and clinical specimens are said to contain many substances that inhibit such nucleic acid amplification reactions.
- Examples of the agent that suppresses the action of the nucleic acid amplification inhibitor as described above include albumin, dextran, T4 gene 32 protein, acetamide, betaine, dimethyl sulfoxide, formamide, glycerol, polyethylene glycol, soybean trypsin inhibitor, ⁇ 2-macroglobulin Examples thereof include one or more selected from phosphorylase and lactate dehydrogenase from tetramethylammonium chloride and lysozyme.
- Examples of the polyethylene glycol include polyethylene glycol 400 and polyethylene glycol 4000.
- Examples of betaine include trimethylglycine and its derivatives.
- glycogen phosphorylase and lactate dehydrogenase examples include glycogen phosphorylase and lactate dehydrogenase derived from rabbit muscle.
- glycogen phosphorylase glycogen phosphorylase b is preferable.
- albumin dextran, T4 gene 32 protein, or lysozyme.
- albumin typified by BSA may reduce nucleic acid amplification inhibition by binding to a nucleic acid amplification inhibitor such as heme (the Abu Al- Soud et al.)
- T4 Gene 32 protein is a single-stranded DNA-binding protein that binds in advance to the single-stranded DNA that is the template in the nucleic acid amplification process, thus preventing the template from being degraded by nucleolytic enzymes and inhibiting the nucleic acid amplification reaction.
- nucleic acid amplification proceeds without being inhibited by binding to a nucleic acid amplification inhibitor similar to BSA (Abu Al-Soud et al.) .
- BSA, T4 Gene 32 protein, and proteinase inhibitor may reduce the proteolytic activity by binding to proteinase and maximize the function of nucleic acid synthase.
- proteolytic enzymes may remain in milk and blood.
- nucleic acid synthase is degraded by the addition of BSA or proteolytic enzyme inhibitors (soybean trypsin inhibitor or ⁇ 2-macrobribulin).
- BSA proteolytic enzyme inhibitors
- Dextran is a polysaccharide generally synthesized by lactic acid bacteria using glucose as a raw material.
- mucin adheres to the intestinal mucosa (Ruas-Madiedo, P., Applied and Environmental Microbiology, 74: 1936-1940, 2008), and dextran is a negative charge inhibitor. It is presumed that there is a sufficient possibility of binding to these inhibitory substances by adsorbing in advance (adsorbed on nucleic acid synthase) or positive charge inhibitory substance (adsorbed on nucleic acid). In addition, it is inferred that lysozyme is adsorbed to a nucleic acid amplification inhibitor thought to be contained in a large amount in milk (Abu Al-Soud et al.).
- albumin T4 gene 32 protein
- dextran a substance represented by albumin
- lysozyme drugs that suppress the action of nucleic acid amplification inhibitors.
- Albumin includes bovine serum albumin, ovalbumin, milk albumin, human serum albumin and the like. Of these, bovine serum albumin is preferred. Albumin may be a purified product and may contain other components such as globulin as long as the effects of the present invention are not impaired. Moreover, a fraction may be sufficient.
- the concentration of albumin in the test sample (nucleic acid amplification reaction solution) is, for example, usually 0.0001 to 1% by mass, preferably 0.01 to 1% by mass, more preferably 0.2 to 0.6% by mass. is there.
- dextran examples include dextran 40 and dextran 500. Of these, dextran 40 is preferred.
- concentration of dextran in the test sample (nucleic acid amplification reaction solution) is, for example, usually 1 to 8%, preferably 1 to 6%, more preferably 1 to 4%.
- the concentration of T4 gene 32 protein (for example, Roche: also called gp32) in the test sample (nucleic acid amplification reaction solution) is usually 0.01 to 1%, preferably 0.01 to 0.1%. Preferably, the content is 0.01 to 0.02%.
- Lysozyme is lysozyme derived from egg white.
- concentration of lysozyme in the test sample (nucleic acid amplification reaction solution is, for example, usually 1 to 20 ⁇ g / ml, preferably 6 to 15 ⁇ g / ml, more preferably 9 to 13 ⁇ g / ml.
- Surfactants include nonionic surfactants such as Triton (registered trademark of Union Carbide), Nonidet (shell), Tween (registered trademark of ICI), Brij (registered trademark of ICI), SDS ( And anionic surfactants such as sodium dodecyl sulfate) and cationic surfactants such as stearyldimethylbenzylammonium chloride.
- Triton include Triton X-100
- Nonidet includes Nonidet P-40
- Tween includes Tween 20, Tween 40, Tween 60, Tween 80
- Brij includes Brij 56.
- the type and concentration of the surfactant in the nucleic acid amplification reaction solution are not particularly limited as long as they promote the penetration of the PCR reagent into the cells of the microorganism and substantially inhibit the nucleic acid amplification reaction.
- SDS for example, it is usually 0.0005 to 0.01%, preferably 0.001 to 0.01%, more preferably 0.001 to 0.005%, more preferably 0.00. 001 to 0.002%.
- Nonidet P-40 is usually 0.001 to 1.5%, preferably 0.002 to 1.2%, more preferably 0.9 to 1.1.
- Tween 20 is usually 0.001 to 1.5%, preferably 0.002 to 1.2%, more preferably 0.9 to 1.1%, and Brij56 is usually 0.00. It is 1 to 1.5%, preferably 0.4 to 1.2%, more preferably 0.7 to 1.1%.
- the enzyme solution used for the nucleic acid amplification reaction contains a surfactant, only the surfactant derived from the enzyme solution may be used, or the same or different surfactant may be added.
- magnesium salts include magnesium chloride, magnesium sulfate, magnesium carbonate and the like.
- concentration of the magnesium salt in the test sample (nucleic acid amplification reaction solution) is, for example, usually 1 to 10 mM, preferably 2 to 6 mM, more preferably 2 to 5 mM.
- organic acid salts include salts of citric acid, tartaric acid, propionic acid, butyric acid, and the like.
- the salt include sodium salt and potassium salt.
- pyrophosphate etc. are mentioned as a phosphate. These may be one kind, or a mixture of two or more kinds.
- the concentration of the organic acid salt or phosphate in the test sample (nucleic acid amplification reaction solution) is, for example, generally 0.1 to 20 mM, preferably 1 to 10 mM, more preferably 1 to 5 mM in a total amount.
- nucleic acid is not extracted from the cells, which is performed before the nucleic acid amplification reaction.
- Extraction of nucleic acids from cells refers to, for example, collecting or purifying nucleic acids by destroying or lysing cells by enzymes or physical means.
- such a process for extracting nucleic acid from cells for example, a process for collecting or purifying nucleic acid by destroying or lysing cells by enzymes or physical means is not performed.
- the target region of DNA or RNA existing in the cell is amplified by a nucleic acid amplification method.
- a nucleic acid amplification method Use microbial cell suspension or suspension of microbial cells treated with proteolytic enzyme, lipolytic enzyme, glycolytic enzyme, etc. as nucleic acid amplification template, and do not extract nucleic acid for template preparation .
- the nucleic acid amplification method preferably includes a step of heat denaturation of the nucleic acid at a high temperature, for example, 90 to 95 ° C., preferably 93 to 95 ° C., more preferably 94 to 95 ° C.
- Amplification of the target region is preferably performed in microbial cells.
- the process is performed in microbial cells. That is, by the high-temperature treatment in the nucleic acid amplification reaction, and in a preferred embodiment, the cell morphology is maintained and the chromosomal DNA remains in the cell by the action of each of the above components, but pinholes or A void is formed, and primers and enzymes necessary for nucleic acid amplification flow into the cell. After an amplification reaction takes place inside the cell, a part of it stays in the cell or flows out of the cell depending on the gene length of the amplification product. Estimated.
- the possibility that a very small part of the chromosomal DNA or RNA flows out of the cell membrane from the pinhole or void in the cell wall or cell wall cannot be denied.
- components necessary for nucleic acid amplification such as primers flow into the cell without substantially destroying or lysing the cell, or a part of the amplification product remains in the cell or the cell.
- the outflow of chromosomal DNA or RNA out of the cell is not included in the “nucleic acid extraction”.
- nucleic acid is collected or purified by a process of extracting nucleic acid from the cell, for example, by destroying or lysing the cell by an enzyme or physical means. Unless the process is performed, this corresponds to “Non-extraction of nucleic acid”. Even if a nucleic acid amplification reaction has occurred outside the cell using chromosomal DNA or RNA eluted from the cell as a template, if the main amplification product is formed in the cell, the nucleic acid amplification reaction can Can be said.
- the amplification product is formed in the microbial cell, it can be evaluated that the nucleic acid amplification reaction has been performed in the microbial cell.
- Nucleic acid amplification methods include PCR methods (White, TJ et al., Trends Genet., 5, 185 (1989)), LAMP method (Loop-Mediated Isothermal Amplification: Principle and application of novel gene amplification method (LAMP method), Nobutomi, Nobuyoshi, Hase Satoshi, BIO INDUSTRY, Vol.18, No.2, 15-23, 2001), SDA method (Strand Displacement Amplification: Edward L. Chan, et al., Arch. Pathol. Lab. Med., 124: 1649-1652, 2000), LCR method (Ligase Chain Reaction: Barany, F., Proc. Natl. Acad.cadSci.
- TMA method Transcription-Mediated -Amplification: Sarrazin C. et al., J. Clin. Microbiol., Vol.39: p.2850-2855 (2001)
- TRC method Transcription-Reverse Transcription-Concerted method : Nakaguchi Y. et al., J Clin. Microbiol., Vol.42: p.4248-4292 (2004)
- HC method Hybrid Capture: Nazarenko I., Kobayashi L. et al., J. Virol. Methods, vol.154: p.76 -81, 2008
- microarray method R ichard P. Spence, et al., J. Clin. Microbiol., Vol.46, No.5, p.1620-1627, 2008.
- the PCR method is particularly preferably used, but is not limited thereto.
- the “target region” refers to a region of chromosomal DNA or RNA that can be amplified by a nucleic acid amplification method using a primer used in the present invention, and can detect a microorganism to be detected. If it does not restrict
- the target region preferably has a sequence specific to the microorganism to be detected. Further, depending on the purpose, it may have a sequence common to a plurality of types of microorganisms. Furthermore, the target area may be single or plural.
- the amount of living cells of the detection target microorganism and the number of living cells of many types of microorganisms can be calculated. Can be measured simultaneously.
- the length of the target region is usually 50 to 5000 bases.
- Primers used for nucleic acid amplification can be appropriately set based on the principles of various nucleic acid amplification methods, and are not particularly limited as long as they can specifically amplify the target region.
- target regions are various specific genes such as 5S rRNA gene, 16S rRNA gene, 23S rRNA gene, tRNA gene, and pathogenic gene.
- One or a part of these genes may be targeted, and a region spanning two or more genes may be targeted.
- a part of the 16S rRNA gene can be amplified by using the primer sets shown in SEQ ID NOs: 1 and 2.
- SEQ ID NOs: 3 and 4 a region spanning part of the 16S ⁇ ⁇ rRNA gene, tRNA gene, and part of the 23S rRNA gene can be amplified.
- the target region includes a pathogenic gene.
- pathogenic genes include Listeria ricin O (hlyA) gene of Listeria, enterotoxin (enterotoxin) gene and invasion (invA) gene of Salmonella, pathogenic E. coli O-157, O-26, O-111, etc.
- Verotoxin gene Enterobacter bacterium outer-membrane-proteinA (ompA) gene (Enterobacter sakazaki) and macromolecular synthesis (MMS) operon (Enterobacter sakazaki), Legionella bacterium macrophage-invasion protein (mip) ) Genes, heat-resistant hemolytic toxin genes of Vibrio parahaemolyticus, heat-resistant hemolytic toxin-like toxin genes, Shiga and intestinal invasive Escherichia coli ipa genes (invasion plasmid antigen gene), invE genes (invasion gene), Staphylococcus aureus enterotoxins Gene, Bacillus cereus cereus De (vomiting toxin) gene and enterotoxin gene, various toxin genes such as Clostridium botulinum and the like.
- ompA Enterobacter bacterium outer-membrane-proteinA
- MMS macromolecular synthesis
- the primers corresponding to the pathogenic gene include, for example, the primer set shown in SEQ ID NOs: 5 and 6 corresponding to the hlyA gene of Listeria, and the primer set corresponding to the SEQ ID NOs: 7 and 8 corresponding to the ompA gene of Enterobacter sakazaki And primer sets for SEQ ID NOs: 9 and 10 corresponding to the MMS operon of Enterobacter sakazaki.
- hemagglutinin (H protein) gene In the case of an influenza virus having an envelope, hemagglutinin (H protein) gene, neuraminidase (N protein) gene, RNA polymerase gene of caliciviridae virus represented by norovirus, gene regions encoding various capsid proteins, etc. Can be mentioned.
- noroviruses rotaviruses and adenoviruses are available as food poisoning viruses.
- the target genes are gene regions encoding RNA polymerase genes and capsid proteins as in the case of noroviruses.
- a primer common to multiple types of microorganisms living cells of multiple types of microorganisms in a test sample can be detected.
- a primer specific to a specific bacterium used, a living cell of a specific bacterial species in a test sample can be detected.
- the conditions of the nucleic acid amplification reaction are specific amplification in accordance with the principle of each nucleic acid amplification method (PCR method, LAMP method, SDA method, LCR method, TMA method, TRC method, HC method, microarray method, etc.) As long as it is not particularly limited, it can be set as appropriate.
- Step d) Analyze amplification products amplified by the nucleic acid amplification method.
- the analysis of the amplification product is performed subsequent to step c) or simultaneously with step c), depending on the nucleic acid amplification method employed in step c).
- step d) can be performed simultaneously with step c).
- the analysis method is not particularly limited as long as the nucleic acid amplification product can be detected or quantified, and examples thereof include electrophoresis.
- real-time PCR Nogva et al., Appl. Environ. Microbiol., Vol. 66, 2000, pp. 4266-4271, Nogva et al., Appl. Environ Microbiol., Vol. 66, 2000, pp. 4029-4036
- the amount and size of the nucleic acid amplification product can be evaluated. Further, according to the real-time PCR method, the PCR amplification product can be quickly quantified.
- the change in fluorescence intensity is generally a noise level and is equal to zero up to 1 to 10 amplification cycles. Therefore, these are regarded as sample blanks with zero amplification products, and their standard deviation SD is calculated.
- a value obtained by multiplying the SD value by 10 is referred to as a threshold value, and the number of PCR cycles that first exceeds the threshold value is referred to as a cycle threshold value (Ct value).
- the presence or absence of an amplification product can also be determined by analyzing the melting temperature (TM) pattern of the amplification product.
- TM melting temperature
- analysis of nucleic acid amplification products can be performed using a standard curve that shows the relationship between the amount of microorganisms prepared using a standard sample of the identified microorganism and the amplification product.
- a standard curve prepared in advance can be used, but it is preferable to use a standard curve prepared by performing each step of the present invention on the standard sample simultaneously with the test sample. If the correlation between the amount of microorganism and the amount of DNA or RNA is examined in advance, DNA or RNA isolated from the microorganism can also be used as a standard sample.
- the kit of the present invention is a kit for distinguishing and detecting live cells of microorganisms in a test sample from dead cells or damaged cells by a nucleic acid amplification method. Including a drug that covalently binds to DNA or RNA upon irradiation with light of a wavelength of, a drug that suppresses the action of a nucleic acid amplification inhibitor, and a primer for amplifying the target region of DNA or RNA of a microorganism to be detected by a nucleic acid amplification method .
- the kit of the present invention can be used for carrying out the method of the present invention.
- any 1 type or multiple types selected from surfactant, magnesium salt, and organic acid salt or phosphate to the kit of this invention.
- an enzyme having an activity of degrading cells other than microorganisms, protein colloid particles, fat, or carbohydrates present in a test sample can be added to the kit of the present invention.
- Enzymes drugs that bind covalently to DNA or RNA, drugs that suppress the action of nucleic acid amplification inhibitors, and surfactants, magnesium salts, and organic acid salts or phosphates, if necessary, all of these components
- a single composition may be included, or a plurality of solutions or compositions containing each component in any combination.
- the nucleic acid amplification reaction is preferably a PCR method, a LAMP method, an SDA method, an LCR method, a TMA method, a TRC method, an HC method, or a microarray method.
- the crosslinking agent and the medium are the same as those described in the method of the present invention.
- the agent that covalently binds to DNA or RNA is ethidium monoazide, ethidium diazide, propidium monoazide, psolaren, It is preferably selected from 4,5 ′, 8-trimethylpsoralen and 8-methoxypsolaren, especially using ethidium monoazide. It is preferable.
- drugs that suppress the action of nucleic acid amplification inhibitors include albumin, dextran, and T4 gene 32 protein, acetamide, betaine, dimethyl sulfoxide, formamide, glycerol, polyethylene glycol, soybean trypsin inhibitor, ⁇ 2-macroglobulin, Any one or more selected from tetramethylammonium chloride, lysozyme, phosphorylase, and lactate dehydrogenase can be exemplified.
- magnesium salt examples include magnesium chloride, magnesium sulfate, and magnesium carbonate.
- organic acid salt examples include salts of citric acid, tartaric acid, propionic acid, butyric acid, and the like.
- salt examples include sodium salt and potassium salt.
- pyrophosphate etc. are mentioned as a phosphate. These may be one kind, or a mixture of two or more kinds.
- the enzyme can decompose non-microorganism cells, protein colloid particles, fats and carbohydrates, etc. present in the test sample, and does not damage the living cells of the target microorganism. If it is, it will not restrict
- the enzyme one kind of enzyme may be used alone, or two or more kinds of enzymes may be used in combination, but both lipolytic enzyme and proteolytic enzyme, or lipolytic enzyme, proteolytic enzyme It is preferable to use all of saccharide-degrading enzymes.
- lipolytic enzyme examples include lipase and phosphatase
- examples of the proteolytic enzyme include serine protease, cysteine protease, proteinase K, and pronase
- examples of the carbohydrate degrading enzyme include amylase and cellulase.
- the kit of the present invention may further contain a diluent, a reaction solution for the reaction of a drug that covalently binds to DNA or RNA, an enzyme and reaction solution for nucleic acid amplification, an instruction describing the method of the present invention, and the like. .
- Example 1 Using enterobacter Sakazaki bacteria, which is representative of coliform bacteria, the conditions for clarifying the distinction between live and dead bacteria were examined.
- Ethidium monoazide (EMA) treatment / light irradiation treatment Ethidium monoazide (EMA: Sigma, St. Louis, MO) was dissolved in 1000 ⁇ g / ml using sterile water, and a 0.20 ⁇ m filter ( The solution was sterilized by filtration using Minisart-plus; Sartorius AG, Gottingen, Germany, and a stock solution was prepared and stored at -20 ° C. protected from light.
- Enterobacter ⁇ Sakazaki live cells and damaged cell suspension milk (1 ml) were subjected to EMA treatment and light irradiation treatment by the following method.
- live cells of Enterobacter sakazaki bacteria and damaged cell suspension milk (1 ml) were subjected to cooling centrifugation at 4 ° C., 15,000 kg ⁇ G for 10 minutes, and after removing the supernatant, 1 ⁇ ml of physiological saline was added. After adding 3 ⁇ l of protease (from Bacillus: Sigma) and treating at 37 ° C.
- PCR amplification In addition to a drug consisting of trisodium citrate dihydrate (TSC; Kanto Chemical) and magnesium chloride hexahydrate (Nacalai Tesque), bovine serum albumin (BSA; Sigma) Dextran (low molecular weight MW 50,000 to 70,000; manufactured by Nacalai Tesque), T4 gene protein 32 (gp32; manufactured by Nippon Gene), sodium lauryl sulfate (SDS; manufactured by Nacalai Tesque), Brij56 (manufactured by Sigma), egg white lysozyme
- a drug containing one or more types was added to 5 ⁇ l of the PCR amplification sample.
- a drug composed of each composition added to a PCR amplification sample may be described as a pretreatment agent. Each composition of the pretreatment agent is shown below.
- Composition 1 2% BSA: 5 ⁇ l 50 mM TSC: 1 ⁇ l 100 mM MgCl 2 : 1.5 ⁇ l 0.05% SDS: 1 ⁇ l
- Composition 2 2% BSA: 5 ⁇ l 50 mM TSC: 1 ⁇ l 100 mM MgCl 2 : 1.5 ⁇ l
- Composition 3 20% dextran: 2.5 ⁇ l 50 mM TSC: 1 ⁇ l 100 mM MgCl 2 : 1.5 ⁇ l
- Composition 4 0.1% gp32: 5 ⁇ l 50 mM TSC: 1 ⁇ l 100 mM MgCl 2 : 1.5 ⁇ l
- Composition 5 2% BSA: 5 ⁇ l 50 mM TSC: 1 ⁇ l 100 mM MgCl 2 : 1.5 ⁇ l 4% Brij56: 12.6 ⁇ l
- Composition 6 2% BSA: 5 ⁇ l 50 mM TSC: 1 ⁇ l 100 mM MgCl 2 : 1.5 ⁇ l 500 ⁇ g / ml egg white lysozyme: 1.0 ⁇ l
- Composition 7 2% BSA: 5 ⁇ l 50 mM TSC: 1 ⁇ l 100 mM MgCl 2 : 1.5 ⁇ l 0.05% SDS: 1 ⁇ l 4% Brij56: 12.6 ⁇ l 500 ⁇ g / ml egg white lysozyme: 1.0 ⁇ l
- Composition 8 2% BSA: 5 ⁇ l 50 mM TSC: 1 ⁇ l 100 mM MgCl 2 : 1.5 ⁇ l 4% Brij56: 12.6 ⁇ l 500 ⁇ g / ml egg white lysozyme: 1.0 ⁇ l
- Composition 9 2% BSA: 5 ⁇ l
- Composition 10 Ingredients only for PCR buffer consisting of the composition of a) to g) described later without including each component of composition 1 to 9
- Primer F 16S rRNA gene detection forward primer 16S_10F (5'-AGTTTGATCCTGGCTC-3 ': SEQ ID NO: 1)
- Primer R 16S rRNA gene detection reverse primer 16S_1500R (5'-GGCTACCTTGTTACGA-3': SEQ ID NO: 2) was used as a PCR primer.
- a PCR buffer comprising the following compositions a) to g)
- the PCR buffer was added to the PCR amplification sample and the pretreatment agent mixture to perform PCR amplification.
- the primer targets long DNA (1491 bp) containing from 16 to 1500 position of 16S rRNA gene.
- Real-time PCR was performed using a real-time PCR apparatus (I cycler iQ, Bio-Rad, Hercules, CA) under the following PCR thermal cycle conditions. 1) 4 °C, 3 minutes (1 cycle) 2) 94 ° C, 30 seconds (1 cycle) 3) 94 ° C, 20 seconds; 55 ° C, 30 seconds; 72 ° C, 1 minute 30 seconds (50 cycles) 4) 95 ° C, 3 minutes (1 cycle)
- Results Table 1 shows the results of real-time PCR.
- the pretreatment agent “Lyso” represents egg white lysozyme.
- b) Damaged cells were prepared by immersing live cells in boiling water for 50 seconds.
- e) Mean Ct value of real-time PCR, expressed as mean ⁇ SD (n 2).
- nd Means that the target gene was not amplified by real-time PCR.
- compositions 1 and composition 2 in Table 1 Based on the results of composition 1 and composition 2 in Table 1, in this system in which PCR is performed directly from bacteria, enterobacter Sakazaki bacteria in physiological saline is identified for viability (live cells and damaged cells are distinguished), and enterobacter in milk The viability of Sakazaki bacteria is clearly identified, and enterobacter Sakazaki bacteria (live cells: EMA untreated) in physiological saline and milk even when evaluated with the Ct value, which is an indicator of the reaction rate of real-time PCR ) And Ct values were not significantly different, and there was no significant difference between EMA treatments of living cells. Thus, it was found that the pretreatment agent may or may not contain the surfactant SDS. The same phenomenon as described above was obtained by comparing the compositions 7 and 8.
- composition 2 and composition 5 life loss is clearly identified in any composition (in physiological saline and milk), but the addition of nonionic surfactant Brij56 is particularly The Ct value of Enterobacter ⁇ Sakazaki bacteria (live cells: EMA-untreated and EMA-treated) in milk was significantly reduced, suggesting that the detection sensitivity of live cells was improved. Furthermore, the comparison of composition 2 and composition 6 clearly shows the viability loss in any composition (in physiological saline and milk), but when lysozyme is added, live cells in milk (EMA untreated) And EMA treatment) were observed to have improved detection sensitivity.
- composition 5 From the comparison of composition 5, 6 and 8, life and loss are clearly identified in any composition (in physiological saline and milk), but as shown in composition 8, Brij56 and egg white lysozyme coexist. As a result, it was found that the detection sensitivity of live cells (EMA-untreated and EMA-treated) was improved.
- Egg white lysozyme acts directly on peptidoglycan of Gram-positive bacteria and hydrolyzes polysaccharides ( ⁇ -1,4 bonds between N-acetylglucosamine and N-acetylmuramic acid), but in the case of Gram-negative bacteria, this polysaccharide
- the egg white lysozyme cannot act because the outer membrane is present outside the peptidoglycan containing the egg white (the side on which the egg white lysozyme acts). From this mechanism of action, egg white lysozyme in composition 8 does not promote the lysis (destruction) of enterobacter Sakazaki, which is a gram-negative bacterium, and is a dead cell of gram-positive bacterium already present in milk.
- composition 8 acts strongly on the cell wall ( ⁇ 5 log 10 cells / ml) in the presence of Brij56, and the surface structure of the Gram-positive bacterial cell wall, which has been considered to be a PCR inhibitory component, has been changed physicochemically. Is no longer functioning.
- the detection sensitivity of live cells by composition 8 is significantly improved, but in live cells suspended in physiological saline, Gram positive as a contaminating component
- compositions 1, 2 and 9 it can be considered that living cells and damaged cells can be distinguished if the action of the nucleic acid amplification reaction inhibitor can be suppressed by BSA without containing magnesium salt or organic acid salt.
- the Ct values of live cells (EMA untreated / EMA treated) and damaged cells (EMA untreated) are delayed by about 3, and in terms of reactivity, magnesium salts and organic acid salts are added. Including compositions 1 and 2 are superior.
- composition 2 it is possible to identify the viability of Enterobacter Sakazaki bacteria suspended in physiological saline using only PCR buffer, but the sensitivity of live cells (EMA untreated and EMA treated) (Ct value) is extremely inferior, and when typical milk is assumed as a normal sample, living cells (EMA-untreated and EMA-treated) and EMA-untreated damaged cells cannot be detected with PCR buffer alone Therefore, it is considered preferable to contain at least an agent that alleviates the influence of a PCR inhibitor typified by albumin, a magnesium salt, and an organic acid salt or phosphate.
- a PCR inhibitor typified by albumin, a magnesium salt, and an organic acid salt or phosphate.
- Example 2 We discriminated between living cells and damaged cells of coliform bacteria and bacteria of the family Enterobacteriaceae.
- Ewingella Americana / JCM4911, and Mollerella wisconsensis (Morrera wisconsensis) / JCM5894 were cultivated at 30 ° C for 16 hours using BHI broth.
- each culture solution is dispensed into 15 ml falcon tubes (Becton Dickinson Labware, NJ), and centrifuged at 4 ° C, 3,000 x G for 10 minutes, and the supernatant is removed Then, 5 ml of physiological saline was added to the precipitate (pellet), and further diluted 10-fold with physiological saline to prepare a living cell suspension of each bacterial species.
- the live cell suspension and damaged cell suspension prepared as described above were used as test samples for the following tests.
- the number of living cells of each coliform group and Enterobacteriaceae in the living cell suspension is counted with a standard agar medium, and at the same time using a spectrophotometer U-2800A (Hitachi, Japan), wavelength 600 Turbidity measurement by nm was performed, and the relationship between the number of living cells and turbidity was confirmed.
- Ethidium monoazide (EMA) treatment / light irradiation treatment Ethidium monoazide (EMA: Sigma, St. Louis, MO) was dissolved in 1000 ⁇ g / ml using sterilized water, and a 0.20 ⁇ m filter ( The solution was sterilized by filtration using Minisart-plus; Sartorius AG, Gottingen, Germany, and stored as a stock solution (EMA solution) at -20 ° C. protected from light. To 1 ml of the test sample (live cell suspension, damaged cell suspension), 10 ⁇ l of EMA solution (1000 ⁇ g / ml) was added and allowed to stand at 4 ° C. for 10 minutes in the dark.
- EMA Ethidium monoazide
- test sample was set on ice at a position 20 cm away from a visible light source (100V PRF 500W Flood eye, Iwasaki Electric Co., Ltd., Tokyo, Japan) and irradiated with visible light for 5 minutes.
- the test sample that has been subjected to EMA treatment and irradiation with visible light is cooled and centrifuged at 4 ° C, 15,000 x G for 10 minutes, the supernatant is removed, and 1 ml of physiological saline is added to the precipitate for washing. The precipitate was recovered by further cooling and centrifugation. After such washing treatment was repeated several times, 10 ⁇ l of sterilized water was added to the precipitate (bacteria) and suspended to prepare a sample for PCR amplification.
- PCR amplification Including bovine serum albumin (BSA; manufactured by Sigma), trisodium citrate dihydrate (TSC; Kanto Chemical), magnesium chloride hexahydrate (manufactured by Nacalai Tesque) 1) to the following A drug having the composition 3) was added to each PCR amplification sample, and 4) a surfactant containing sodium lauryl sulfate (SDS; manufactured by Nacalai Tesque) was added to each 5 ⁇ l PCR amplification sample.
- BSA bovine serum albumin
- TSC trisodium citrate dihydrate
- SDS sodium lauryl sulfate
- the agent having the composition of 1) to 3) and the surfactant of 4) may be combined and described as a pretreatment agent.
- Primer F 16S rRNA gene detection forward primer 16S_10F (SEQ ID NO: 1) and Primer R: 16S rRNA gene detection reverse primer 16S_1500R (SEQ ID NO: 2) were used as PCR primers.
- a PCR buffer consisting of the following compositions a) to g) is used to perform high-sensitivity detection by maximizing the amount of change in the fluorescent substance (first derivative peak) with respect to temperature. The PCR buffer was added to the PCR amplification sample and the pretreatment agent mixture to perform PCR amplification.
- Real-time PCR was performed using a real-time PCR apparatus (I cycler iQ, Bio-Rad, Hercules, CA) under the following PCR thermal cycle conditions. 1) 4 °C, 3 minutes (1 cycle) 2) 94 °C, 30 seconds (1 cycle) 3) 94 ° C, 20 seconds; 55 ° C, 30 seconds; 72 ° C, 90 seconds (50 cycles) 4) 95 ° C, 3 minutes (1 cycle)
- PCR was performed according to the protocol for melting analysis of PCR amplification products (temperature was increased from 60 ° C at 0.1 ° C intervals, held at each temperature for 8 seconds, and repeated 350 times in total, with 95 ° C as the end temperature). The melting temperature of the amplified product was measured.
- PCR amplification was performed in the same manner using a live cell suspension of Enterobacter Sakazaki bacteria (8 log 10 cells (individual cells) / ml). Furthermore, PCR amplification was performed using the PCR buffer as it was without adding a test sample as a blank sample.
- EMA + EMA (light-shielded 10 ⁇ g / ml, 10 minutes, 4 ° C) + visible light irradiation (5 minutes)
- EMA- EMA unprocessed
- PC Enterobacter Sakazaki live cell suspension 5 log of 8 log 10 cells / ml was used.
- NC Negative control using sterile water instead of DNA template M: 100 bp DNA ladder. Damaged bacteria: The living cell suspension was immersed in boiling water for 50 seconds.
- E. coli Escherichia coli DH5 ⁇ (7.91 ⁇ 0.20 log 10 cells / ml)
- S. enteritidis Salmonella enteritidis IIP 604 (8.07 ⁇ 0.02 log 10 cells / ml)
- K. oxytoca Klebsiella oxytoca JCM1665 (8.38 ⁇ 0.08 log 10 cells / ml)
- C. koseri Citrobacter koseri JCM1658 (8.02 ⁇ 0.06 log 10 cells / ml)
- sakazakii Enterobacter sakazakii ATCC 51329 (7.95 ⁇ 0.01 log 10 cells / ml)
- S. fonticola Serratia fonticola JCM1242 (7.47 ⁇ 0.01 log 10 cells / ml)
- B. aquilia Budvicia aquilia JCM3902 (6.98 ⁇ 1.50 log 10 cells / ml)
- E. americana Ewingella americana JCM4911 (7.47 ⁇ 0.43 log 10 cells / ml) H.
- agrestis Buttiauxella agrestis JCM1090 (7.76 ⁇ 0.00 log 10 cells / ml)
- K. ascorbata Kluyvera ascorbata JCM2107 (7.80 ⁇ 0.02 log 10 cells / ml)
- C. davisae Cedecea davisae JCM1685 (7.56 ⁇ 0.10 log 10 cells / ml).
- the Ct value (the number of cycles of the rise of the real-time PCR curve) is 13 to 22 for the EMA-untreated group of living cells, and the Ct value is 16 to 24 for the EMA-treated group of living cells. there were. Further, the EMA-untreated group of damaged cells had Ct values of 15 to 22, and good PCR amplification results were obtained in all cases. However, for the EMA-treated group of damaged cells, the target gene was not amplified in all coliforms and Enterobacteriaceae. Furthermore, as shown in FIG. 1, from the results of electrophoresis, in any coliform group and Enterobacteriaceae, only the EMA-treated group of damaged cells could not detect a band showing a positive PCR amplification product. It was.
- Example 3 We distinguished live and dead cells of coliform bacteria and Enterobacteriaceae inoculated into foods such as milk.
- Test Material and Test Method 1- Bacterial strain and culture method Kluyvera ascorbata / JCM2107, Cedecea davisae / JCM1685, Citrobacter koseri / JCM1658, Klebsiella pneumoniae / NRBC3321, Serratia fonticola / Jella 1243, Yoken aquatilis / NBRC13544, Hafnia alvei / JCM1666, Leclercia adecarboxylata / JCM1667, Pantoea agglomerans / JCM1236, Enterobacter sakazakii / ATCC51329, E.
- the number of living cells of each coliform group and Enterobacteriaceae in the living cell suspension is counted with a standard agar medium, and at the same time using a spectrophotometer U-2800A (Hitachi, Japan), Turbidity measurement was performed at a wavelength of 600 nm, and the relationship between the number of living cells and turbidity was confirmed.
- E. coli group / Enterobacteriaceae / live cell inoculated milk prepared above, and uninoculated milk are centrifuged at 37 ° C., 3,000 ⁇ G for 5 minutes, and present in the fat layer and intermediate layer on the surface of the supernatant The aqueous layer was removed by decantation and the precipitate was collected.
- the collected precipitate (pellet) contains both coliform bacteria / enterobacteriaceae / live cell inoculated milk, and non-inoculated milk, dead cells that have been killed by sterilization presumed to be present in commercially available milk ( Gram-negative bacteria or gram-positive bacteria ( ⁇ 6 log 10 cells) including coliforms are included. Therefore, it was judged that the sediment prepared from coliform bacteria / Enterobacteriaceae / live cell inoculated milk contained dead cells and live cells.
- Ethidium monoazide (EMA) treatment / light irradiation treatment After adding 1 ml of physiological saline to the precipitate after the enzyme treatment and stirring, the EMA solution prepared in the same manner as in Example 2 (1000 ⁇ g / ml) was added, and the mixture was allowed to stand at 4 ° C. for 10 minutes in the dark. Thereafter, in the same manner as in Example 2, irradiation with visible light and washing treatment were performed, and 5 ⁇ l of sterilized water was added to the precipitate to prepare a sample for PCR amplification.
- EMA Ethidium monoazide
- PCR Amplification As in Example 2, a pretreatment agent was added to 5 ⁇ l of a PCR amplification sample.
- Primer F Forward primer for detecting 16S rRNA gene 16S_1234F (5'-CTACAATGGCGCATACAAAGAGAAG-3 ': SEQ ID NO: 3)
- Primer R Reverse primer for detecting 23S rRNA gene 23S_1703R (5'-CCTTCTCCCGAAGTTACGGCACCAT-3': SEQ ID NO: 4) was used as a PCR primer.
- a PCR buffer consisting of the following compositions a) to g) is used to perform high-sensitivity detection by maximizing the amount of change in the fluorescent substance (first derivative peak) with respect to temperature. Then, 41.5 ⁇ l of this PCR buffer was added to the PCR amplification sample and pretreatment mixture to perform PCR amplification.
- the PCR primer contains long DNA (approximately 2450 bp) including positions 1234 to 1258 of the 16S rRNA gene, tRNA gene (76 bp), and positions 1 to 1703 of the 23S rRNA gene and the spacer region (approximately 364 bp). ).
- Real-time PCR was performed using a real-time PCR apparatus (I cycler iQ, Bio-Rad, Hercules, CA) under the following PCR thermal cycle conditions. 1) 95 ° C, 3 minutes (1 cycle) 2) 95 ° C, 30 seconds; 60 ° C, 40 seconds; 68 ° C, 3 minutes (40 cycles) 3) 95 ° C, 3 minutes (1 cycle)
- PCR was performed according to the protocol for melting analysis of PCR amplification products (temperature was increased from 60 ° C at 0.1 ° C intervals, held at each temperature for 8 seconds, and repeated 350 times in total, with 95 ° C as the end temperature). The melting temperature of the amplified product was measured.
- PCR amplification was performed in the same manner using a live cell suspension of Enterobacter Sakazaki (8 log 10 cells / m). Furthermore, PCR amplification was performed using the PCR buffer as it was without adding a test sample as a blank sample.
- Results Table 3 shows the results of real-time PCR. Moreover, the result of electrophoresis of PCR final amplification product is shown in FIG.
- KP K. pneumoniae CK: C. koseri EC: E. coli.
- SE S. enteritidis KA: K. ascorbata CD: C. davisae SF: S. fonticola YR: Y. regensburgei RA: R. aquatilis HA: H. alvei LA: L. adecarboxylata
- PA P. agglomerans ES: E. sakazakii Milk: E.
- Positive control Enterobacter Sakazaki bacteria: 5 ⁇ l of 8 log 10 CFU / ml is used as a PCR template
- Negative Negative control (use 5 ⁇ l of sterile water as PCR template)
- L 100 bp DNA ladder.
- the method of the present invention distinguishing (detecting live cells) the living cells of coliforms (Coliform bacteria) and Enterobacteriaceae inoculated into foods such as milk and dead cells.
- coliforms Cold bacteria
- Enterobacteriaceae Enterobacteriaceae inoculated into foods such as milk and dead cells.
- Control Example 1 Detection of living cells by the conventional method High-concentration Escherichia coli group damaged cells (intestinal tract) by EMA-PCR using 16S rRNA (long DNA) as a template after EMA treatment and DNA purified by DNA extraction (Including bacteriology).
- Test Material and Test Method 1-1) Strains Used and Culture Method The test method was performed based on the method of Japanese Patent No. 4217797 (International Publication No. 2002/052034 pamphlet). Escherichia coli DH5 ⁇ , Salmonella enteritidis IID604, Klebsiella oxytoca JCM1665, and Citrobacter koseri JCM1658 were cultured at 37 ° C. using Brain Heart Infusion (BHI) broth (Eiken, Tokyo). An aliquot of 10 ml was taken from the logarithmic growth phase culture and centrifuged at 4 ° C. for 15 minutes at 8,000 ⁇ G.
- BHI Brain Heart Infusion
- the sample was irradiated with a visible light source (100V PRF 500W Flood eye, Iwasaki Electric Co., Ltd., Tokyo, Japan) on ice for 5 minutes.
- the EMA-treated sample was cooled and centrifuged at 4 ° C., 15,000 ⁇ G for 10 minutes, and after removing the supernatant, the same washing operation was performed with 1 ml of physiological saline.
- PCR buffer represented by the following composition was prepared using Primer F: 16S rRNA gene detection forward primer 16S_10F (SEQ ID NO: 1) and Primer R: 16S rRNA gene detection reverse primer 16S_1500R (SEQ ID NO: 2).
- PCR was performed according to the protocol for melting analysis of PCR amplification products (temperature was increased from 60 ° C at 0.1 ° C intervals, held at each temperature for 8 seconds, and repeated 350 times in total, with 95 ° C as the end temperature). The melting temperature of the amplification product was measured.
- Results Table 4 shows Ct values (number of PCR cycles in which the amplification curve exceeds the boundary value) obtained by performing real-time PCR. Moreover, the result of electrophoresis is shown in FIG.
- the Et. Coli and S. enteritidis living cells did not show a significant change in the Ct value of real-time PCR due to EMA treatment.
- the Ct value shows a high value of about 18 in E. coli and a high value of about 14 in S. enteritidis, compared to untreated by EMA treatment, suppressing PCR amplification.
- PCR showed a positive reaction (Ct values 40 ⁇ 1.4 and 34 ⁇ 1.1).
- FOG. 3 From the identification result of live cells and damaged cells by the PCR final amplification product (FIG. 3), for E. coli DH5 ⁇ and S. enteritidis IID604, a band of the target gene is obtained even in the sample after EMA treatment of the damaged cells, The distinction between live cells and damaged cells could not be fully confirmed.
- Example 4 The extent of lysis (Lysis) of the cells of Enterobacter sakazaki was determined by PCR thermal cycle rotation 50 times in the presence of the pretreatment agent.
- Cells of the test method 10 8 cells / ml of Enterobacter sakazakii ATCC51329 strain (ES), saline, or pretreatment solution shown in Table 5 (hereinafter, to be referred to as "DB (direct buffer)" In each case, and a suspension was prepared (0.25 mL). Each suspension is divided into 25 ⁇ l aliquots and transferred to a 200 ⁇ l PCR tube, followed by a PCR thermal cycle rotation step (50 times) at 95 ° C for 15 seconds, 60 ° C for 20 s, 72 ° C for 30 seconds, and all together again. (Total 0.25 ml), and used as a sample for PCR amplification.
- DB direct buffer
- OmpA_F Forward primer for detecting ompA gene (5′-ggatttaaccgtgaacttttcc-3 ′; SEQ ID NO: 7)
- ompA_R Reverse primer for detecting ompA gene (5′-cgccagcgatgttagaaga-3 ′; SEQ ID NO: 8) .
- PCR buffer composition a) ompA_F (10 pmol / ⁇ l): 2 ⁇ l b) ompA_R (10 pmol / ⁇ l): 2 ⁇ l c) Ex-Taq (5U / ⁇ l: Takara-Bio): 0.25 ⁇ l (Including Tween 20 0.5%, Nonidet P-40 0.5%, glycerol 50%) d) 10 ⁇ Ex-Taq Buffer (Takara-Bio): 2.5 ⁇ l e) dNTP mixture (Takara-Bio): 2 ⁇ l f) 10 ⁇ SYBR Green I (BMA): 4 ⁇ l
- the remaining 247.5 ⁇ l (PCR amplification sample obtained by subtracting 2.5 ⁇ l from 0.25 ml) is cooled and centrifuged (10,000 ⁇ g, 5min, 4 °C), and the supernatant is added to 2.5 ⁇ l.
- 12.25 ⁇ l of solution and 12.75 ⁇ l of PCR buffer were added to perform PCR (Supernatant I).
- physiological saline or 0.25 ⁇ ml of the pretreatment solution shown in Table 5 is added to the pellet after the centrifugation to prepare each suspension, and 2.5 ⁇ l each of the pretreatment solution 12 ⁇ m and PCR buffer 12.75 ⁇ l was added and PCR was performed (suspension suspension II).
- Real-time PCR was performed using a real-time PCR apparatus (I cycler iQ, Bio-Rad, Hercules, CA) under the following PCR thermal cycle conditions. 1) 4 ° C, 3 minutes (1 cycle) 2) 95 ° C, 15 seconds; 60 ° C, 20 seconds; 72 ° C, 30 seconds (50 cycles) 3) 95 ° C, 3 minutes (1 cycle)
- PCR amplification was performed according to the protocol of melting analysis of PCR amplification products (temperature was increased from 60 ° C to 0.1 ° C intervals, held at each temperature for 8 seconds, and repeated a total of 350 times with 95 ° C as the end temperature). The melting temperature of the product was measured.
- the Ct value of Suspension I is 8 or more smaller than the Ct value of Supernatant I, and the maximum percentage of chromosomal DNA eluted in the supernatant is estimated to be around 0.1 to 0.5% (the Ct value of Supernatant I) (The origin of this may be due to the fact that some of the cells of Enterobacter sakazaki bacteria, whose specific gravity has been reduced by thermal cycle rotation, are collected in the supernatant), and the Ct value of suspension I is more than 99% sakazaki Presumed to be DNA in fungal cells. That is, it was suggested that the cells of Enterobacter sakazaki bacteria were not lysed by 99% or more even after 50 PCR thermal cycle rotations in the presence of the pretreatment agent.
- non-heated Ct value was significantly lower than “thermal cycle heating” because the pretreatment agent contained proteins such as egg white lysozyme and bovine serum albumin. Therefore, the hypothesis that it was denatured by 50 PCR thermal cycle rotations was made, and the experiment shown in Table 7 was added. Specific experimental methods are shown below.
- the Ct value of viable Enterobacter sakazaki bacteria in the pretreatment solution subjected to thermal cycle heating is the Ct value when using a pretreatment solution that has not been subjected to physiological saline or thermal cycle rotation. Compared to the non-heated group, the reason is that both the suspension I and the Ct value of the supernatant I of “thermal cycle heating” at least in Table 6 are not considered to be significantly higher values. It was confirmed that it cannot be.
- enterobacter Sakazaki bacteria were subjected to PCR thermal cycle rotation 50 times in the presence of a pretreatment agent, further cooled to room temperature after 4 ° C cooling, then cooled and centrifuged, and the pellet side and supernatant side It is considered that the chromosomal DNA in Enterobacter sakazaki collected in the beginning was not entangled with denatured DNA-binding proteins and denatured enzymes and did not function as a template for PCR.
- Example 5 Whether the Enterobacter Sakazaki cells have been lysed (Lysis) by PCR thermal cycle rotation 50 times in the presence of a pretreatment agent, using a sample before and after PCR thermal cycle rotation, and using a nuclear stain Evaluation was carried out using a flow cytometry method that enables quantification of the number of remaining cells after stereomicroscopic observation and PCR thermal cycle rotation.
- Example 4 Fluorescence microscope observation and stereomicroscope observation Experimental Method Similarly to the method of Example 4, 10 9 cells / ml of Enterobacter sakazaki ATCC51329 strain (ES) cells were suspended in physiological saline or a pretreatment solution shown in Table 5, Each suspension was prepared (0.25 ml). Each suspension is aliquoted into 25 ⁇ l, transferred to a 200 ⁇ l PCR tube, passed through the PCR thermal cycle rotation step (50 times) (95 ° C 15 s, 60 ° C 20 s, 72 ° C 30 s), and again Combined (total 0.25 ml).
- ES Enterobacter sakazaki ATCC51329 strain
- FIGS. 4 to 11 show the observation images of the suspension of the pretreatment agent solution of Sakazaki bacteria and the cooled centrifugal supernatant with a fluorescence microscope, respectively. That is, the experiment was set so that each fluorescence microscope observation image corresponds to the suspension I to the supernatant I in Washing step I in Table 6. In these figures, a stereoscopic microscope image and a superimposed image of the actual microscope image and the fluorescence microscope image are also shown.
- enterobacter sakazaki bacteria cells were also found in the centrifugal supernatant of the physiological suspension of Enterobacter sakazaki bacteria, which correlated with the PCR results in Table 6.
- the cells of Enterobacter sakazaki bacteria mostly maintain the bacterial morphology even after being subjected to PCR thermal cycle rotation 50 times in physiological saline (stereomicroscopic image and fluorescent microscopic image).
- the SYTO9 stained image was also clear, suggesting that chromosomal DNA was retained in the cell. Since there is no cell wall fragment in the stereomicroscopic image of FIG.
- each of the suspension and its supernatant is used for flow cytometry measurement in common with each sample, three 0.25 ml ⁇ 3 were prepared. Specifically, the first was left as it was, the second was cooled and centrifuged (3000 xg, 10 min, 4 ° C), the supernatant was removed, and the precipitate was suspended by adding 0.25 ml of physiological saline. A third sample was subjected to the same cooling and centrifugal treatment, and a supernatant was collected. SYTO9 was added to each of them at a concentration of 1.5 ⁇ l / ml, and stored at 4 ° C. for 15 minutes in the dark, and used as a sample for flow cytometry test.
- the measurement apparatus used a FACS Calibur (BECTON DICKINSON) and an Argon laser of 488 nm, and recognized bacterial cell plots by FSC (forward scattered light measurement) and SSC (side scattered light measurement). If SYTO9 is intercalated into the chromosomal DNA in the cell, green fluorescence can be detected with the FL1 filter with ⁇ max of 530 nm when excited by the same laser, and the FL1 plot was also implemented. In particular, no nuclear stain with propidium iodide (PI) was used, but the red fluorescence from the FL3 filter was also measured for reference. Details of measurement conditions for flow cytometry are shown in Table 8.
- FIG. 12 shows the experimental results of the physiological saline suspension of Enterobacter sakazaki and its supernatant (non-heated, PCR thermal cycle rotation treatment), and the same pretreatment solution suspension (after washing once).
- FIG. 13 shows the experimental results of the supernatant (including re-suspended liquid) and the supernatant (non-heated / PCR thermal cycle rotation treatment).
- Example 6 Real-time PCR measurement using purified chromosomes with the same number of bacterial cells as Enterobacter sakazaki and the amount of chromosomal DNA contained in them
- the overnight enrichment culture solution was washed, and then serially diluted with sterilized water to prepare a 4 ⁇ 10 3 to 4 ⁇ 10 8 cells / ml viable cell suspension of Enterobacter sakazaki bacteria. Thereafter, 2.5 ⁇ l of the suspension was added to 12.25 ⁇ l of the pretreatment solution described in Table 5 (however, sterilized water was changed to 2.7 ⁇ l) according to the method of Example 4, and 12.75 ⁇ l of PCR buffer for detecting ompA gene was added thereto. And the same PCR as in Example 4 was performed. Each PCR tube contained 10 1 to 10 6 cells of bacterial cells of Enterobacter sakazaki.
- the amount of chromosomal DNA obtained from one cell of Enterobacter sakazaki can be regarded as 5 fg (5 ⁇ 10 -15 g)
- the amount of chromosomal DNA contained in each PCR tube is calculated according to this value.
- Purified DNA 2.5 ⁇ l was placed in each PCR tube, and PCR was performed by sequentially adding a pretreatment solution and a PCR buffer in the same manner.
- Results Table 9 shows the degree of DNA purification
- Table 10 shows the results of real-time PCR. From the results in Table 9, since the value of OD 260 / OD 280 was around 2.0, high-purity DNA with less RNA contamination could be prepared from two strains of Enterobacter and Sakazaki. Next, from the results in Table 10, there is no significant difference between the Ct values of the same amount of chromosomal DNA with respect to the number of bacterial cells of each Enterobacter sakazaki, and the purified DNA dissolved in the test tube was 100% PCR. If it functions as a template, it was found that the chromosomal DNA of Enterobacter sakazaki bacteria cells also functions 100% as a PCR template.
- Example 4 the Ct values of suspensions I and II and supernatant I after 50 PCR thermal cycle rotations in the presence of a pretreatment agent for Enterobacter sakazaki in Table 6 were almost 100%.
- the Ct value of supernatant I must be significantly smaller than the Ct value of suspension II, and the supernatant should have a Ct value equivalent to that of suspension I.
- Table 6 shows that I do not support it.
- Example 5 from the quantification result of the bacterial cell number of Enterobacter Sakazaki before and after PCR thermal cycle rotation of FIG. 13 which is a flow cytometry measurement result, there is almost 100% lysis and 10% Is also unlikely to dissolve.
- Example 7 From Examples 4 to 6, the cells of Enterobacter sakazaki are not dissolved in most cases even when PCR reaction (50 times) is performed with a PCR buffer in the presence of a pretreatment agent, and chromosomal DNA is retained in the cells. I understood.
- a TM peak analysis (melting temperature measurement) of the PCR amplification product after the real-time PCR reaction yielded a temperature peak presumed to be an ompA gene product, which was determined to be positive for the real-time PCR reaction.
- a strict sense whether the PCR amplification reaction product is present in the bacterial cell, in the PCR reaction solution, or in both.
- PCR amplification products are mainly dissolved in the PCR reaction solution, but this has not been elucidated yet, and has been further elucidated in the PCR reaction in the presence of the pretreatment agent described in the present application. Absent.
- the PCR reaction in the presence of the pretreatment agent may be carried out in bacterial cells. In the following examples, however, PCR amplification products remain in the bacterial cells. Indicates the possibility of
- fixative A 4% paraformaldehyde
- fixative C Movable Form 10N: 10% formalin Neutral
- Fixation D Movable Form 10NM: 10% formalin Neutral Buffer-Methanol Solution Deodorized; Wako Pure Chemical Industries, Osaka
- Bacterial intracellular chromosomal DNA and cell wall constituent proteins were cross-linked to immobilize the DNA in the cell in advance.
- 500 ⁇ l of physiological saline was used instead of the fixing solution to prepare a sample that was not fixed.
- PCR buffer composition a) 16S_1234F (10 pmol / ⁇ l): 2 ⁇ l b) 23S_1703R (10 pmol / ⁇ l): 2 ⁇ l c) Ex-Taq (5U / ⁇ l: Takara-Bio): 0.25 ⁇ l (Including Tween 20 0.5%, Nonidet P-40 0.5%, glycerol 50%) d) 10 ⁇ Ex-Taq Buffer (Takara-Bio): 2.5 ⁇ l e) dNTP mixture (Takara-Bio): 2 ⁇ l f) 10 ⁇ SYBR Green I (BMA): 4 ⁇ l
- Real-time PCR was performed using a real-time PCR apparatus (I cycler iQ, Bio-Rad, Hercules, CA) under the following PCR thermal cycle conditions. 1) 4 ° C, 3 minutes (1 cycle) 2) 95 ° C, 15 seconds; 60 ° C, 20 seconds; 72 ° C, 3 minutes (30 cycles) 3) 95 ° C, 3 minutes (1 cycle)
- PCR was performed according to the protocol of PCR amplification product melting analysis (increasing the temperature from 60 ° C to 0.1 ° C intervals, holding at each temperature for 8 seconds, repeating it a total of 350 times, with 95 ° C as the end temperature) The melting temperature of the amplification product was measured.
- Example 4 the PCR thermal cycle conditions of Example 4 were changed using the ompA_F (SEQ ID NO: 7) and ompA_R primer (SEQ ID NO: 8) described in Example 4 instead of 16S_1234F and 23S_1703R. Otherwise, in accordance with Example 7 above, electrophoresis of 5 ⁇ l of supernatant after PCR reaction and flow cytometry measurement with SYTO9 on the physiological saline suspension of the pellet from which supernatant was removed after PCR reaction did.
- FIG. 14 shows the results of electrophoresis of the reaction supernatant after PCR (16S-23S rRNA: 2450-bp) after the treatment with the above fixatives, and the Ct value of real-time PCR of the reaction solution is shown immediately below. After the PCR reaction, the supernatant was removed, the pellet was suspended in physiological saline, and the results of flow cytometry measurement using SYTO9 are shown in Table 11. Similarly, FIG. 15 shows an electrophoretogram of the supernatant after the PCR reaction targeting the ompA (469-bp) gene.
- the fixative B may have a function equivalent to that of the non-fixed (S). However, the fixative B strongly cross-links the mammalian cell chromosome and the cell membrane protein. It is reasonable to think that it exhibits a fixed function.
- Example 8 As described above, even if the bacterial chromosome is fixed in the cell and the treatment for preventing the outflow of the chromosome to the external solution is performed in advance, even if PCR thermal cycle rotation is repeated 50 times in the presence of the pretreatment agent, Bacterial cells retain almost 100% bacterial morphology and chromosomes are retained in the cells, but the PCR reaction proceeds and the PCR amplification products are also present in the external solution. It can be concluded that it is mainly carried out in bacterial cells. As shown in Example 8 below, some PCR products were also present in the cells under the above conditions.
- Example 8 In Examples 4 to 7, it has been suggested that there is a high possibility that PCR has been performed in bacterial cells in the presence of a pretreatment agent, that is, in-situ PCR may have occurred.
- In-situ PCR eg, Gerard J. et al., American Journal of Pathology, 139: 847-854, 1991.
- a fixative as shown in Example 7 to obtain chromosomal DNA.
- human HPV genes incorporated into chromosomal DNA in human cells after cross-linking with human cell membrane proteins and treating them with proteolytic enzymes for a short period of time, or treating human immune cells with microwave irradiation. It is a technique for detecting and quantifying various genes.
- a PCR reaction solution is placed on immobilized human immune cells to cause a PCR amplification reaction in human immune cells, and at the same time, even a PCR product of about 500-bp does not flow out of the cell. is there. Since the PCR product does not flow out of the cell, if the PCR reaction is stopped at an early stage of less than 5 to 10 cycles, it is possible to infer not only the detection of the gene in the cell but also the number of incorporation of the gene.
- Example 7 Thereafter, by the same method as in Example 7, it was finally diluted 10-fold to obtain 250 ⁇ l of a physiological saline suspension of Enterobacter sakazaki.
- the estimated concentration of Enterobacter sakazaki in the suspension is approximately 3.5 ⁇ 10 7 cells / ml.
- 2.5 ⁇ l of the sample was used as a PCR amplification sample, added to 12.25 ⁇ l of the pretreatment solution shown in Table 5 (however, sterilized water was changed to 2.7 ⁇ l), and 12.75 ⁇ l of PCR buffer for Gram-negative bacteria detection shown below was added thereto, PCR was performed under the following conditions. During the PCR reaction, 27.5 ⁇ l ⁇ 20 fixing solutions and control samples were prepared.
- the primers used were the 16S rRNA gene detection forward primer 16S_1234F (SEQ ID NO: 3) and the 23S rRNA gene detection reverse primer 23S_1703R (SEQ ID NO: 4) described in Example 3.
- the number of bacteria used in the DNA extraction process is 100 times greater in the pre-PCR sample than in the sample group after the PCR reaction.
- FIG. 15 As shown in Fig. 15, since the PCR amplification product is present in the external solution, the PCR amplification product in the external solution is adsorbed on the bacterial cell wall of Enterobacter sakazaki, and it appears as if the PCR amplification product was inside. There was a possibility of producing a misinterpretation like Therefore, the following experiment was also added.
- PCR buffer composition a) 16S_1234F (10 pmol / ⁇ l): 2 ⁇ l b) 23S_1703R (10 pmol / ⁇ l): 2 ⁇ l c) Ex-Taq (5U / ⁇ l: Takara-Bio): 0.25 ⁇ l (Including Tween 20 0.5%, Nonidet P-40 0.5%, glycerol 50%) d) 10 ⁇ Ex-Taq Buffer (Takara-Bio): 2.5 ⁇ l e) dNTP mixture (Takara-Bio): 2 ⁇ l f) 10 ⁇ SYBR Green I (BMA): 4 ⁇ l
- Real-time PCR was performed using a real-time PCR apparatus (I cycler iQ, Bio-Rad, Hercules, CA) under the following PCR thermal cycle conditions. 1) 4 ° C, 3 minutes (1 cycle) 2) 95 ° C, 15 seconds; 60 ° C, 20 seconds; 72 ° C, 3 minutes (30 cycles) 3) 95 ° C, 3 minutes (1 cycle)
- PCR was performed according to the protocol of PCR amplification product melting analysis (increasing the temperature from 60 ° C to 0.1 ° C intervals, holding at each temperature for 8 seconds, repeating it a total of 350 times, with 95 ° C as the end temperature) The melting temperature of the amplification product was measured.
- FIG. 16 shows the results.
- the cells of Enterobacter sakazaki were fixed using Fixation Solution B, or PCR was not carried out and PCR was performed in the presence of a pretreatment agent (16S-23S: 2450-bp), and the supernatant of the reaction solution was electrophoresed.
- the results are shown in lanes 2 and 3, DNA extracted from the pellets washed twice after the PCR reaction, and electrophoresed in lanes 5 and 6, lanes of fixed and non-fixed Enterobacter Sakazaki bacteria that are the test materials of this experiment.
- Lanes 7 and 8 show the results of direct DNA extraction and electrophoresis from cells
- lanes 9 and 10 show the results of DNA extraction and electrophoresis from fixed and non-fixed Enterobacter and Sakazaki cells immediately before the PCR reaction.
- Lanes 13 and 14 show the results obtained by preparing PCR amplification products, adding them to bacterial cells of Enterobacter sakazaki bacteria, washing them twice, and then extracting and electrophoresis the DNA.
- the PCR gene products in lanes 5 and 6 have been extracted from the cells. Note that even if the bacterial cell has a damaged cell wall, the PCR gene product is free to pass through, and even if the concentration of the product is the same in the external solution and the bacterial body, a comparison of greater at 10 10 times the latter, PCR product quantity often 10 10 times in the external solution, i.e. so that the 1/10 10 amount is dispensed in the cells, lanes 2, 3, From the comparison of the band intensities of 5 and 6, the amount of PCR product remaining in the cells is not considered to be 1/10 10 amount.
- PCR in the method of the present invention may be in-situ PCR. From the results of lanes 7 and 8, chromosomal DNA was detected in both the fixed and non-fixed bacterial cells of Enterobacter sakazaki used as the test material, but lanes 5, 6, 9, and 10 No chromosomal DNA band was obtained. This is probably a problem with the number of Enterobacter sakazaki bacteria cells used for DNA extraction, specifically 0.9 ⁇ 10 6 cells may be insufficient for DNA extraction. In fact, the lanes 7 and 8 of the test material had a high concentration of 0.9 ⁇ 10 8 cells.
- Example 9 Enterobacter Sakazaki cells were boiled in physiological saline or in the presence of a pretreatment agent, and the amount of Sakazaki chromosomal DNA flowing into each supernatant was examined according to the treatment time. .
- FIG. 17 shows how much bacterial chromosomes are eluted in the supernatant when cells of Enterobacter sakazaki are heat-treated with boiling water in physiological saline or in the presence of a pretreatment agent. .
- a small amount of chromosomal DNA is already present while not heated, but this is because the enrichment solution that had been enriched overnight has reached the stationary phase, so some of the dead bacteria have dissolved.
- Chromosomal DNA is thought to have flowed into the external solution and should be ignored during evaluation.
- physiological saline it is assumed that DNA was eluted from bacterial cells of Enterobacter sakazaki by heating. Was not detected.
- the suspension when evaluated in the wells of lanes 10 and 12, the suspension has a band in the well, but the supernatant has no band, and chromosomal DNA does not flow out of the bacterial cells in the presence of the pretreatment agent. It was found that it remained in the fungus body.
- the physiological saline supernatant (lanes 5, 6, and 7) also has a band in the well, but this is due to the centrifugation of some of the Enterobacter Sakazaki bacteria killed by boiling. It is inferred that it was recovered in the supernatant.
- Table 6 and FIG. 12 support this. From the above, it was suggested that although the conditions are strictly different from PCR thermal cycle rotation, it is more difficult for DNA to flow out of bacterial cells by heat treatment in the presence of a pretreatment agent.
- live cells of microorganisms can be distinguished from dead cells or damaged cells and detected with high sensitivity. According to the present invention, it is possible to discriminate between living cells, damaged cells, and dead cells of microorganisms in the environment such as food and biological samples, wiped samples, industrial water, environmental water, wastewater, and the like by simple and rapid nucleic acid amplification methods.
- the method and kit of the present invention can be applied to self-inspection and are excellent in economy.
- hygiene tests of various foods containing damaged or dead cells of E. coli at 5 log 10 cells / ml or more, or rapid diagnosis of pediatric bacteremia in which E. coli circulates in the blood It can also be applied to.
- only live bacteria cells of the coliform group including Enterobacteriaceae from food are highly sensitive (1 CFU / 2.22 ml milk) and compared with the official method (Food Sanitation Law / Ministry Ordinance for Milk). Because it can be detected very quickly (7 hours 30 minutes), it is expected to be used for pre-shipment judgment after production at various food factories represented by milk production factories. Is expected to be high. Furthermore, not only Escherichia coli and Enterobacteriaceae, but also various microorganisms such as pathogenic bacteria, viruses, and other microorganisms, various hygiene tests and clinical trials are possible in order to enable rapid detection and quantification of only low concentrations of living microorganisms. It can be applied to inspection and process management.
Landscapes
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Analytical Chemistry (AREA)
- Genetics & Genomics (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Engineering & Computer Science (AREA)
- Microbiology (AREA)
- Biochemistry (AREA)
- Biotechnology (AREA)
- General Health & Medical Sciences (AREA)
- Molecular Biology (AREA)
- Immunology (AREA)
- Biophysics (AREA)
- Physics & Mathematics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Biomedical Technology (AREA)
- Toxicology (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
Abstract
Description
また、細菌DNAを鋳型とするPCR反応において、細菌からDNAを抽出せずにPCR反応を行う方法が開示されている(非特許文献4、特許文献5)。特許文献5には、DNAフィンガープリンテイング法において細菌からランダムPCRを行うことが記載されており、核酸合成用緩衝剤組成物の成分としてリン酸塩、ドデシル硫酸塩が記載されている。
本発明は、食品や生体試料等に含まれる微生物の生細胞を死細胞や損傷細胞に比べて選択的に検出する新たな方法、及び同方法を実施するためのキットを提供することを課題とする。
a)前記被検試料に、350nm~700nmの波長の光照射によりDNA又はRNAに共有結合する薬剤を添加する工程、
b)前記薬剤を添加した被検試料に、350nm~700nmの波長の光照射処理を行う工程、
c)被検試料に含まれる微生物のDNA又はRNAのターゲット領域を、細胞からの核酸の抽出を行わずに、核酸増幅阻害物質の働きを抑制する薬剤の存在下で核酸増幅法により増幅する工程、及び
d)増幅産物を解析する工程。
本発明は、前記ターゲット領域の増幅が、微生物細胞内で行われることを好ましい態様としている。
また前記方法は、前記c)の工程において、ターゲット領域の増幅を界面活性剤、マグネシウム塩、及び有機酸塩又はリン酸塩から選択されるいずれか一種又は複数種の存在下で行うことを好ましい態様としている。
前記方法は、前記c)の工程の前に、前記a)の工程及びb)の工程を繰り返して行うことを好ましい態様としている。
また前記方法は、前記a)の工程の前に、以下のe)の工程を行うことを好ましい態様としている。
e)被検試料を、被検試料中に存在する微生物以外の細胞、タンパク質コロイド粒子、脂肪、又は糖質を分解する活性を有する酵素で処理する工程。
また前記方法は、前記酵素が、蛋白質分解酵素、脂質分解酵素、及び糖分解酵素から選択されることを好ましい態様としている。
また前記方法は、前記被検試料が、食品、生体試料、飲料水、工業用水、環境用水、排水、土壌、又は拭き取り試料のいずれかであることを好ましい態様としている。
また前記方法は、前記微生物が細菌、又はウイルスであることを好ましい態様としている。
また前記方法は、前記細菌がグラム陰性細菌であることを好ましい態様としている。
また前記方法は、前記350nm~700nmの波長の光照射によりDNA又はRNAに共有結合する薬剤が、エチジウムモノアザイド(ethidium monoazide)、エチジウムジアザイド(ethidium diazide)、プロピジウムモノアザイド(propidium monoazide)、プソラーレン(psolaren)、4,5',8-トリメチルプソラーレン(4,5',8-trimethyl psolaren)、及び8-メトキシプソラーレン(8-methoxy psolaren)から選択されることを好ましい態様としている。
また前記方法は、核酸増幅阻害物質の働きを抑制する薬剤が、アルブミン、デキストラン、T4ジーン32プロテイン、アセトアミド、ベタイン、ジメチルスルフォキシド、ホルムアミド、グリセロール、ポリエチレングリコール、大豆トリプシンインヒビター、α2-マクログロブリン、テトラメチルアンモニウムクロライド、リゾチーム、ホスホリラーゼ、及び乳酸脱水素酵素から選択される1種又は複数種であることを好ましい態様としている。
また前記方法は、前記有機酸塩が、酢酸塩、プロピオン酸塩、及びクエン酸塩から選択されることを好ましい態様としている。
また前記方法は、前記リン酸塩がピロリン酸塩であることを好ましい態様としている。
また前記方法は、前記ターゲット領域が50~5000塩基のターゲット領域であることを好ましい態様としている。
また前記方法は、前記ターゲット領域が、被検試料のDNAの5S rRNA遺伝子、16S rRNA遺伝子、23S rRNA遺伝子、及びtRNA遺伝子から選択される遺伝子に対応するターゲット領域であることを好ましい態様としている。
また前記方法は、前記核酸増幅法が、PCR法、LAMP法、SDA法、LCR法、TMA法、TRC法、HC法、又はマイクロアレイ法であることを好ましい態様としている。
また前記方法は、前記PCR法をリアルタイムPCR法により行い、PCRと増幅産物の解析を同時に行うことを好ましい態様としている。
また前記方法は、前記増幅産物の解析を、微生物の標準試料を用いて作成された微生物量及び増幅産物との関連を示す標準曲線を用いて行うことを好ましい態様としている。
1)350nm~700nmの波長の光照射によりDNA又はRNAに共有結合する薬剤、
2)核酸増幅阻害物質の働きを抑制する薬剤、及び
3)検出対象の微生物のDNA又はRNAのターゲット領域を核酸増幅法により増幅するためのプライマー。
前記キットは、さらに界面活性剤、マグネシウム塩、有機酸塩又はリン酸塩から選択されるいずれか一種又は複数種を含むことを好ましい態様としている。
また前記キットは、被検試料中に存在する微生物以外の細胞、タンパク質コロイド粒子、脂肪、又は糖質を分解する活性を有する酵素を含むことを好ましい態様としている。
また前記キットは、前記核酸増幅法が、PCR法、RT-PCR法、LAMP法、SDA法、LCR法、TMA法、TRC法、HC法、又はマイクロアレイ法であることを好ましい態様としている。
また前記キットは、前記350nm~700nmの波長の光照射によりDNA又はRNAに共有結合する薬剤が、エチジウムモノアザイド(ethidium monoazide)、エチジウムジアザイド(ethidium diazide)、プロピジウムモノアザイド(propidium monoazide)、プソラーレン(psolaren)、4,5',8-トリメチルプソラーレン(4,5',8-trimethyl psolaren)、及び8-メトキシプソラーレン(8-methoxy psolaren)から選択されることを好ましい態様としている。
また前記キットは、核酸増幅阻害物質の働きを抑制する薬剤が、アルブミン、デキストラン、及びT4ジーン32プロテイン、アセトアミド、ベタイン、ジメチルスルフォキシド、ホルムアミド、グリセロール、ポリエチレングリコール、大豆トリプシンインヒビター、α2-マクログロブリン、テトラメチルアンモニウムクロライド、リゾチーム、ホスホリラーゼ、及び乳酸脱水素酵素から選択される1種又は複数種であることを好ましい態様としている。
また前記キットは、前記有機酸塩が、酢酸塩、プロピオン酸塩、及びクエン酸塩から選択されることを好ましい態様としている。
また前記キットは、前記リン酸塩がピロリン酸塩であることを好ましい態様としている。
また前記キットは、前記酵素が、蛋白質分解酵素、脂質分解酵素、及び糖質分解酵素から選択されることを好ましい態様としている。
本発明の方法においては、その検出の対象として、結果的に増幅することが可能であれば、核酸全般のいずれであってもよく、具体的には1本鎖DNA、2本鎖DNA、1本鎖RNA、及び2本鎖RNAを例示することができ、中でもDNAを検出対象とすることが好ましく、2本鎖DNAが特に好ましい。
本発明の方法は、被検試料中の微生物の生細胞を、死細胞又は損傷細胞と識別して検出する方法であって、以下の工程を含む方法である。
a)前記被検試料に、350nm~700nmの波長の光照射によりDNA又はRNAに共有結合する薬剤を添加する工程、
b)前記薬剤を添加した被検試料に、350nm~700nmの波長の光照射処理を行う工程、
c)被検試料に含まれる微生物のDNA又はRNAのターゲット領域を、細胞からの核酸の抽出を行わずに、核酸増幅阻害物質の働きを抑制する薬剤存在下で核酸増幅法により増幅する工程、及び
d)増幅産物を解析する工程。
特に、食品としては、清涼飲料、炭酸飲料、栄養飲料、果汁飲料、乳酸菌飲料等の飲料(これらの飲料の濃縮原液及び調製用粉末を含む);アイスクリーム、アイスシャーベット、かき氷等の氷菓;加工乳、乳飲料、発酵乳、バター等の乳製品;経腸栄養食品、流動食、育児用ミルク、スポーツ飲料;特定保健用食品、健康補助食品等の機能性食品等が好ましい。
さらに、環境用水としては、市水、地下水、河川水、又は雨水等が例示される。
脂質分解酵素としては、リパーゼ、フォスファターゼ等が、タンパク質分解酵素としてはセリンプロテアーゼ、システインプロテアーゼ、プロテイナーゼK、プロナーゼ等が、糖質分解酵素としてはアミラーゼ、セルラーゼ等が挙げられる。
本明細書においては、特記しない限り、「生細胞」、「死細胞」及び「損傷細胞」は、微生物の生細胞、死細胞及び損傷細胞を意味する。
生細胞の細胞数は、好適な平板培地上で好適な条件で培養したときのコロニー形成数(cfu/ml(colony forming units / ml))で近似させることができる。また、損傷細胞の標準試料は、例えば、生細胞懸濁液を加熱処理、例えば沸騰水中で加熱処理することにより調製することができるが、その場合は、損傷細胞の細胞数は、加熱処理する前の生細胞懸濁液のcfu/mlで近似させることができる。尚、損傷細胞を調製するための沸騰水中での加熱時間は、微生物の種類により異なるが、例えば実施例に記載された細菌では、50秒程度で損傷細胞を調製することができる。さらに、損傷細胞の標準試料は、抗生物質処理によっても調製することができるが、その場合は、損傷細胞の細胞数は、生細胞懸濁液を抗生物質で処理した後、抗生物質を除去し、可視光(波長600nm)の透過度、すなわち濁度を測定し、生細胞数濃度が予め判っている生細胞懸濁液の濁度と比較することにより、好適な平板培地上で好適な条件で培養したときのコロニー形成数(cfu/ml)で近似させることができる。
ウイルスでは、細胞数単位は、プラーク形成単位(pfu又はPFU(plaque-forming units))で表される。
被検試料に、350nm~700nmの波長の光照射によりDNA又はRNAに共有結合する薬剤を添加する。すなわち、被検試料中の微生物を、前記薬剤で処理する。
後述するように、前記薬剤は、2本鎖DNA又はRNAにインターカレートし、光照射により共有結合して分子間を架橋する。また、前記薬剤は、1本鎖DNA又はRNAに対しては、光照射により共有結合して、PCR反応を阻害すると推定される。以下、前記薬剤を単に「架橋剤」と記載することがある。
前記架橋剤としては、エチジウムモノアザイド(ethidium monoazide)、エチジウムジアジド(ethidium diazide)、プソラーレン(psolaren)、4,5',8-トリメチルプソラーレン(4,5',8-trimethyl psolaren)、及び8-メトキシプソラーレン(8-methoxy psolaren)、プロピジウムモノアザイド(propidium monoazide)等が挙げられる。架橋剤は、1種類を単独で用いてもよいし、2種又はそれ以上を併用してもよい。
次に、架橋剤を添加した被検試料に350nm~700nmの波長の光照射処理を行う。
上記架橋剤は、生細胞の細胞壁よりも死細胞及び損傷細胞の細胞壁の方が透過しやすい。したがって、前記に示す作用時間内であれば微生物の生細胞の細胞壁は実質的に透過せず、微生物の損傷細胞もしくは死細胞または死細胞になっている体細胞の細胞膜は透過すると考えられる。その結果、架橋剤は、体細胞の死細胞及び微生物の死細胞並びに損傷細胞の細胞内に進入し、続いて、染色体DNA、又はRNAと水素結合し、350nm~700nmの波長の光照射を行うことによりDNAの分子間を架橋し、又はRNAと共有結合し、その結果、染色体DNA内に歪みが生じたり、RNAが架橋剤で修飾され、最終的に、染色体DNAが破壊(断片化・切断)されたり、RNAが核酸増幅反応の鋳型とはならなくなると推定される。
また、生細胞よりも損傷細胞や死細胞のRNAが優先的に架橋剤により修飾を受けると、生細胞ではRNAのターゲット領域が核酸増幅法により増幅されるのに対し、損傷細胞や死細胞ではRNAのターゲット領域が修飾を受ける結果、核酸増幅反応が阻害され、生細胞を損傷細胞や死細胞に比べて選択的に検出することができる。
尚、血液中の白血球、血小板が生細胞の場合、EMAは滅菌水や低張な塩溶液下で前記細胞の細胞膜をより透過する。
特に、DNAについては、EMAが体細胞の死細胞及び微生物の損傷細胞並びに死細胞の細胞内に進入して、核内DNAに無秩序にインターカレートした後、350nm~700nmの波長の光照射によりインターカレートしたEMAがナイトレンに変換され、核内DNAに共有結合し、DNAの分子間を架橋する。そして、染色体DNAの各塩基およびデオキシリボースに対して至るところで共有結合したEMAにより、染色体DNA内に大きな歪みが生じ、その結果、染色体DNAが破壊(断片化)されると推定される。
また、2本鎖RNA(部分的な2本鎖も含む)については、EMAが体細胞の死細胞及び微生物の損傷細胞並びに死細胞の細胞内に進入して、RNAに無秩序にインターカレートした後、350nm~700nmの波長の光照射によりインターカレートしたEMAのみがナイトレンに変換され、RNAに共有結合し、RNAの分子間を架橋する。そして、RNAの各塩基に対して至るところで共有結合したEMAにより、RNA内に大きな歪みが生じ、その結果、RNAが破壊(断片化)されると推定される。
さらに1本鎖DNA又はRNAについては、EMAが体細胞の死細胞及び微生物の損傷細胞並びに死細胞の細胞内に進入して、350nm~700nmの波長の光照射によりEMAがナイトレンに変換されて、DNA又はRNAに共有結合すると推定される。
具体的には例えば、一回目の工程a)では、エチジウムモノアザイドでは5分~1時間、エチジウムジアジドでは5分~1時間、プロピジウムモノアザイドでは5分~1時間、プソラーレンでは5分~1時間、4,5',8-トリメチルプソラーレンでは5分~1時間、8-メトキシプソラーレンでは5分~1時間が挙げられる。また、二回目以降の工程a)では、エチジウムモノアザイドでは6分~48時間、エチジウムジアジドでは6分~48時間、プロピジウムモノアザイドでは6分~48時間、プソラーレンでは6分~48時間、4,5',8-トリメチルプソラーレンでは6分~48時間、8-メトキシプソラーレンでは6分~48時間が挙げられる。
次に、光照射処理後の被検試料に含まれる微生物のDNA又はRNAのターゲット領域を、細胞からの核酸の抽出を行わずに、核酸増幅阻害物質の働きを抑制する薬剤存在下で核酸増幅法により増幅する。
特に、アルブミン、デキストラン、T4ジーン32プロテイン、又はリゾチームを使用することが好ましい。
また、リゾチームは、牛乳中に多数含まれていると考えられる核酸増幅阻害物質と吸着しているものと推察される(前記Abu Al-Soudら)。
核酸増幅反応液中の界面活性剤の種類及び濃度は、微生物の細胞内へのPCR試薬の透過を促進し、核酸増幅反応を実質的に阻害しない限り特に制限されない。具体的には、SDSの場合は、例えば、通常0.0005~0.01%、好ましくは0.001~0.01%、より好ましくは0.001~0.005%、より好ましくは0.001~0.002%である。他の界面活性剤の場合、例えば、Nonidet P-40の場合は、通常、0.001~1.5%、好ましくは0.002~1.2%、より好ましくは0.9~1.1%、Tween 20の場合は、通常、0.001~1.5%、好ましくは0.002~1.2%、より好ましくは0.9~1.1%、Brij56の場合は、通常0.1~1.5%、好ましくは0.4~1.2%、より好ましくは0.7~1.1%である。
核酸増幅反応に用いる酵素溶液に界面活性剤が含まれている場合は、同酵素溶液由来の界面活性剤のみでもよいし、さらに同種又は異なる界面活性剤を追加してもよい。
ターゲット領域の増幅は、好ましくは、微生物細胞内で行われる。本発明においては、実施例に示すように、微生物細胞内で行われている可能性が高い。すなわち、核酸増幅反応における高温処理、及び、好適な態様ではさらに上記各成分の作用によって、細胞の形態は維持され、染色体DNAは細胞内に残されつつも、微生物の細胞膜又は細胞壁にピンホールもしくは空隙が形成され、プライマー及び核酸増幅に必要な酵素等は細胞内に流入し、細胞内で増幅反応が起きた後、増幅産物の遺伝子長によって、一部分が細胞内にとどまる又は細胞外に流出するものと推定される。一方、前記細胞膜又は細胞壁のピンホールもしくは空隙から、染色体DNA又はRNAの極めて一部が細胞外に流出している可能性も否定はできない。
いずれにしても、このように、細胞を実質的に破壊又は溶解せずに、プライマー等の核酸増幅に必要な成分が細胞内に流入すること、増幅産物の一部分が細胞内にとどまること又は細胞外に流出すること、及び、染色体DNA又はRNAが細胞外に流出させることは、「核酸の抽出」には含まれない。また、上記以外の他のメカニズムも否定はできないが、その場合であっても、細胞から核酸を抽出する処理、例えば、酵素や物理的手段によって細胞を破壊又は溶解して、核酸を採取又は精製する処理を行わない限り、「核酸の抽出を行わない」に該当する。
尚、細胞から溶出した染色体DNA又はRNAが鋳型となって、細胞外で核酸増幅反応が生じていたとしても、主たる増幅産物が細胞内で形成されていれば、核酸増幅反応は「微生物細胞内で行われる」といえる。具体的には例えば、増幅産物の80%以上、好ましくは90%以上、より好ましくは99%以上が微生物細胞内で形成されれば、核酸増幅反応が微生物細胞内で行われたと評価できる。
核酸増幅法により増幅した増幅産物を解析する。増幅産物の解析は、工程c)で採用する核酸増幅法に応じて、工程c)に続いて行われるか、又は、工程c)と同時に行われる。例えば、リアルタイムPCRの場合は、工程d)は工程c)と同時に行われ得る。
解析法は、核酸増幅産物の検出又は定量が可能なものであれば特に制限されず、電気泳動法等が例示される。尚、核酸増幅法にPCR法を用いた場合は、リアルタイムPCR法(Nogva et al., Appl. Environ. Microbiol., vol.66, 2000, pp.4266-4271、 Nogva et al., Appl. Environ. Microbiol., vol.66, 2000, pp.4029-4036)を利用することが可能である。
リアルタイムPCR法を採用する場合、一般に増幅サイクル数1~10までは蛍光強度の変化はノイズレベルでありゼロに等しいので、それらを増幅産物ゼロのサンプルブランクと見なし、それらの標準偏差SDを算出し、そのSD値に10を乗じた値をスレッショールド値とし、そのスレッショールド値を最初に上回るPCRサイクル数をサイクルスレッショールド値(Ct値)という。従って、PCR反応溶液に初期のDNA鋳型量が多い程、Ct値は小さな値となり、鋳型DNA量が少ない程、Ct値は大きな値となる。また、鋳型DNA量が同じでも、その鋳型内のPCRのターゲット領域に切断が生じている割合が多くなる程、同領域のPCR反応のCt値は大きな値となる。
上記の各方法は、本発明の方法における諸条件の最適化に際しても使用することができる。
本発明のキットは、核酸増幅法により、被検試料中の微生物の生細胞を、死細胞又は損傷細胞と識別して検出するためのキットであって、350nm~700nmの波長の光照射によりDNA又はRNAに共有結合する薬剤、核酸増幅阻害物質の働きを抑制する薬剤、及び検出対象の微生物のDNA又はRNAのターゲット領域を核酸増幅法により増幅するためのプライマーを含む。本発明のキットは、前記本発明の方法を実施するために用いることができる。
なお、本発明のキットには、界面活性剤、マグネシウム塩、及び有機酸塩又はリン酸塩から選択されるいずれか一種又は複数種を追加することが可能である。
また、本発明のキットには、被検試料中に存在する微生物以外の細胞、タンパク質コロイド粒子、脂肪、又は糖質を分解する活性を有する酵素を追加することが可能である。
また、有機酸塩としては、クエン酸、酒石酸、プロピオン酸、酪酸等の塩が挙げられる。塩の種類としては、ナトリウム塩、カリウム塩等が挙げられる。また、リン酸塩として、ピロリン酸等が挙げられる。これらは、1種でもよく、2種又は3種以上の混合物であってもよい。
脂質分解酵素としては、リパーゼ、フォスファターゼ等が、タンパク質分解酵素としてはセリンプロテアーゼ、システインプロテアーゼ、プロテイナーゼK、プロナーゼ等が、糖質分解酵素としてはアミラーゼ、セルラーゼ等が挙げられる。
大腸菌群(Coliform bacteria)として代表的なエンテロバクター サカザキ菌を使用して、細菌の生細胞と死細胞との識別を明瞭にするための条件の検討を行った。
1-1)使用菌株及び培養方法
Enterobacter sakazakii(エンテロバクター サカザキ)ATCC51329を、ブレイン・ハート・インフュージョン・ブロス(Brain Heart Infusion Broth)(BHIブロス: Eiken Chemical Co., Ltd., Tokyo, Japan)を用いて、37℃、16時間培養した。培養液5mlを、15 ml falcon tube (Becton Dickinson Labware, NJ)に入れ、4℃、3,000×G、10分の冷却遠心処理を行い、上清を除去後、ペレットに5 mlの生理食塩水を加えて、エンテロバクター サカザキ菌のストック・生細胞けん濁液(8.95 ± 0.01 log10 cells/ml, n = 2)を調製した。更に、この生細胞懸濁液を生理食塩水により10倍希釈しエンテロバクター サカザキ菌の生細胞けん濁液(7.95 ± 0.01 log10 cells/ml, n = 2)を調製した。
更に、ストック・損傷細胞けん濁液を市販殺菌牛乳により10倍希釈しエンテロバクター サカザキ菌の損傷細胞けん濁乳(7.95 ± 0.01 log10 cells/ml, n = 2)を調製した。
エチジウムモノアジド(EMA: Sigma, St. Louis, MO)を、滅菌水を用いて1000 μg/mlに溶解し、0.20 μmのフィルター(Minisart-plus; Sartorius AG, Gottingen, Germany)を用いてろ過滅菌を施し、ストックソリューションを調製し-20 ℃にて遮光して保管した。
クエン酸三ナトリウム2水和物(TSC;関東化学)及び塩化マグネシウム6水和物(ナカライテスク社製)からなる薬剤に、更に、ウシ血清アルブミン(BSA;Sigma社製)、デキストラン(低分子M.W. 50,000~70,000;ナカライテスク社製)、T4ジーンプロテイン32(gp32;日本ジーン社製)、ラウリル硫酸ナトリウム(SDS;ナカライテスク社製)、Brij56(Sigma社製)、卵白リゾチーム(和光純薬工業社製)の1又は複数種類を含む薬剤をPCR増幅用試料5 μlに添加した。PCR増幅用試料に添加した各組成からなる薬剤を前処理剤と記載することがある。下記にその前処理剤の各組成を示す。
2% BSA: 5 μl
50 mM TSC: 1 μl
100 mM MgCl2: 1.5 μl
0.05% SDS: 1 μl
2% BSA: 5 μl
50 mM TSC: 1 μl
100 mM MgCl2: 1.5 μl
20% デキストラン: 2.5 μl
50 mM TSC: 1 μl
100 mM MgCl2: 1.5 μl
0.1% gp32: 5 μl
50 mM TSC: 1 μl
100 mM MgCl2: 1.5 μl
2% BSA: 5 μl
50 mM TSC: 1 μl
100 mM MgCl2: 1.5 μl
4% Brij56: 12.6 μl
2% BSA: 5 μl
50 mM TSC: 1 μl
100 mM MgCl2: 1.5 μl
500 μg/ml 卵白リゾチーム: 1.0 μl
2% BSA: 5 μl
50 mM TSC: 1 μl
100 mM MgCl2: 1.5 μl
0.05% SDS: 1 μl
4% Brij56: 12.6 μl
500 μg/ml 卵白リゾチーム: 1.0 μl
2% BSA: 5 μl
50 mM TSC: 1 μl
100 mM MgCl2: 1.5 μl
4% Brij56: 12.6 μl
500 μg/ml 卵白リゾチーム: 1.0 μl
2% BSA: 5 μl
組成1~9までの各成分を含まず後述のa)~g)の組成からなるPCRバッファーのみの成分
前記プライマーは、16S rRNA遺伝子の10~1500位までを含むlong DNA(1491 bp)をターゲットとしている。
b) Primer R (10 pmol/μl): 4 μl
c) Ex-Taq(5U/μl; Takara-Bio): 0.5 μl
(Tween 20 0.5%、Nonidet P-40 0.5%、グリセロール 50%を含む)
d) 10 × Ex-Taq Buffer (Takara-Bio): 5 μl
e) dNTP mixture (Takara-Bio): 4 μl
f) 10 × SYBR Green I (BMA社製) 8 μl
g) 滅菌水: PCR増幅用試料5 μl及び前処理剤と合せて全量55 μlにするための必要量
1) 4 ℃, 3 分 (1 サイクル)
2) 94 ℃, 30 秒 (1 サイクル)
3) 94 ℃, 20 秒; 55 ℃, 30 秒; 72 ℃, 1 分 30 秒(50 サイクル)
4) 95 ℃, 3 分 (1 サイクル)
リアルタイムPCRの結果を表1に示す。
表1中、a)~f)中の記号は以下のとおりである。また、前処理剤の「Lyso」は卵白リゾチームを表す。
a) エンテロバクター サカザキ菌の生細胞数及び損傷細胞数:7.95 ± 0.01 log10 cells/ml(生理食塩水及び市販殺菌牛乳中)。
b) 損傷細胞は生細胞を沸騰水中に50秒浸漬し作製した。
c) EMA未処理を意味する。
d) EMA処理(遮光下10 μg/ml、10 分、4 ℃) + 可視光照射 (5 分)
e) リアルタイムPCRのCt値を意味し、mean ± SD (n = 2)として表示した。
f) nd: リアルタイムPCRで対象遺伝子の増幅が行われなかったことを意味する。

以上から、卵白リゾチームは、Bjii56存在下においてグラム陰性細菌の溶解には関与せず、寧ろPCR夾雑成分として考えられる検体中に予め含まれているグラム陽性細菌の細胞壁を物理化学的に変化させ、PCR阻害成分として機能させなくしたため、結果として対象としているグラム陰性細菌のエンテロバクター サカザキ菌の生細胞の感度を大幅に向上させたと考えられる。そのことは、組成2と組成8の比較からも、生理食塩水中の生細胞の感度(Ct値)には有意な差は見受けられないが、牛乳中(グラム陽性細菌損傷細胞・死細胞を多数含んでいる)の生細胞の感度は組成8が極めて有意に優れている結果が得られていることとも符合する。そして、卵白リゾチームは核酸増幅阻害物質と吸着し、核酸増幅反応を正常に進行させていると考えられる。
組成1、2と9の比較から、マグネシウム塩や有機酸塩を含まなくても、BSAにより核酸増幅反応阻害物質の働きが抑制できれば、生細胞と損傷細胞の識別は可能であると考えられる。なお、組成9については、生細胞(EMA未処理・EMA処理)、及び損傷細胞(EMA未処理)のCt値が約3ほど遅れており、反応性の点で、マグネシウム塩や有機酸塩を含む組成1及び2の方が優れている。
大腸菌群(Coliform bacteria)、及び腸内細菌科の細菌の生細胞と損傷細胞との識別を行った。
1-1)使用菌株及び培養方法
Klebsiella oxytoca(クレブシエラ・オキシトカ)/JCM1665、Citrobacter koseri(シトロバクター・コーセリ)/JCM1658、Enterobacter sakazakii(エンテロバクター・サカザキ)/ATCC51329、Serratia fonticola(セラチア・フォンティコーラ)/JCM1242、Budvicia aquilia(ブドヴィシア・アクイリア)/JCM3902、Rahnella aquatilis(ラーネラ・アクアティリス)/NBRC13544、Hafnia alvei(ハフニア/アルヴェイ)/JCM1666、Leclericia adecarboxylata(レクレリシア・アデカルボキシラタ)/JCM1667、Yokenella regensburgei(ヨケネラ・レゲンスブルゲイ)/JCM2403、Pantoea agglomerans(パントエア・アグロメランス)/JCM1236、Buttiauxella agrestis(ブッティオーキセラ・アグレスティス)/JCM1090、Kluyvera ascorbata(クルイヴェラ・アスコルバータ)/JCM2107、Cedecea davisae(セデセア・ダヴィサエ)/JCM1685、及びE. coli(エシェリヒア・コリ)DH5α、Salmonella enteritidis(サルモネラ・エンテリティディス)/IID604の大腸菌群16属、並びに大腸菌群には属さない腸内細菌科1属について、ブレイン・ハート・インフュージョン・ブロス(Brain Heart Infusion Broth)(BHIブロス:Eiken Chemical Co., Ltd., Tokyo, Japan)を用いて、37℃、16時間培養を行った。
なお、生細胞懸濁液中の各大腸菌群及び腸内細菌科の生細胞数は、標準寒天培地によりカウントし、また、同時に分光光度計U-2800A(Hitachi, Japan)を用いて、波長600 nmによる濁度測定を実施し、生細胞数と濁度の関係を確認した。
エチジウムモノアザイド(EMA:Sigma, St. Louis, MO)を、滅菌水を用いて1000μg/mlに溶解し、0.20μmのフィルター(Minisart-plus; Sartorius AG, Gottingen, Germany)を使用して濾過滅菌し、ストックソリューション(EMA溶液)として、-20℃にて遮光して保管した。
試験試料(生細胞懸濁液、損傷細胞懸濁液)1mlに対し、EMA溶液(1000μg/ml)を10μl添加し、遮光下で4℃、10分間放置した。
EMA処理・可視光照射処理した試験試料について、4℃、15,000×G、10分間の冷却遠心処理を行い、上清を除去した後、沈殿物に1mlの生理食塩水を添加して洗浄処理し、さらに冷却遠心処理して沈殿を回収した。このような洗浄処理を数回繰り返した後に、沈殿物(菌体)に10μlの滅菌水を加えて懸濁し、PCR増幅用試料とした。
ウシ血清アルブミン(BSA;Sigma社製)、クエン酸三ナトリウム2水和物(TSC;関東化学)、塩化マグネシウム6水和物(ナカライテスク社製)を含む以下1)~3)の組成からなる薬剤をPCR増幅用試料に各々添加し、さらに、ラウリル硫酸ナトリウム(SDS;ナカライテスク社製)を含む4)の界面活性剤をPCR増幅用試料5 μlに各々添加した。
なお、以下の記載において、1)~3)の組成からなる薬剤と、4)の界面活性剤を合わせて、前処理剤と記載することがある。
2)50 mM TSC:1μl
3)100 mM MgCl2:1.5μl
4)0.05% SDS:1μl
更に、リアルタイムPCR後の増幅産物の融解解析において、温度に対する蛍光物質の変化量(一次微分ピーク)を最大にして高感度検出を行うために、以下のa)~g)の組成からなるPCRバッファーを調製し、このPCRバッファーを、前記PCR増幅用試料及び前処理剤の混合液に加えてPCR増幅を行った。
b)Primer R(10 pmol/μl):4μl
c)Ex-Taq(5U/μl:Takara-Bio):0.5μl
(Tween 20 0.005%、Nonidet P-40 0.005%、を含む)
d)10×Ex-Taq Buffer(Takara-Bio社製):5μl
e)dNTP mixture(Takara-Bio社製):4μl
f)10×SYBR Green I(BMA社製):8μl
g)滅菌水:16μl
1)4℃、3 分(1 サイクル)
2)94℃、30 秒(1 サイクル)
3)94℃、20 秒;55℃、30 秒;72℃、90 秒(50 サイクル)
4)95℃、3 分(1 サイクル)
陽性コントロールとして、エンテロバクター サカザキ菌の生細胞懸濁液(8 log10 cells(個細胞)/ml)を使用して、同様にPCR増幅を行った。さらに、ブランクサンプルとして、試験試料を加えずに、そのままPCRバッファーを用いてPCR増幅を行った。
0.5×TAEを用いて2%アガロースゲル(2% Seakem GTG agarose:FCM BioProducts, Rockland, Me)を調製した。
10μlのPCR増幅産物を前記アガロースゲルにアプライし、電気泳動を行った。
1μg/mlのエチジウムブロマイド溶液で染色後、そのゲルをデンシトグラフで観察し、その画像をAE-6905H Image Saver HR(Atto Co., Japan)を用いて取り込み保管した。
リアルタイムPCRの結果を表2に示す。また、PCR最終増幅産物の電気泳動の結果を、図1に示す。
EMA + : EMA (遮光下10 μg/ml、10 分、4 ℃) + 可視光照射 (5 分)
EMA - : EMA未処理
PC:エンテロバクター サカザキ菌の生細胞けん濁液 8 log10 cells/mlの5μlを使用した。
NC: DNA鋳型の代わりに滅菌水を用いた陰性コントロール
M: 100 bp DNAラダー。
損傷菌: 生細胞けん濁液を沸騰水に50秒浸漬した。
E. coli: Escherichia coli DH5α (7.91 ± 0.20 log10 cells/ml)
S. enteritidis: Salmonella enteritidis IIP 604 (8.07± 0.02 log10 cells/ml)
K. oxytoca : Klebsiella oxytoca JCM1665 (8.38 ± 0.08 log10 cells/ml)
C. koseri: Citrobacter koseri JCM1658 (8.02 ± 0.06 log10 cells/ml)
E. sakazakii: Enterobacter sakazakii ATCC 51329 (7.95 ± 0.01 log10 cells/ml)
S. fonticola: Serratia fonticola JCM1242 (7.47 ± 0.01 log10 cells/ml)
B. aquilia: Budvicia aquilia JCM3902 (6.98 ± 1.50 log10 cells/ml)
R. aquatilis: Rahnella aquatilis NBRC13544 (7.38 ± 0.14 log10 cells/ml)
E. americana: Ewingella americana JCM4911 (7.47 ± 0.43 log10 cells/ml)
H. alvei: Hafnia alvei JCM1666 (8.04 ± 0.22 log10 cells/ml)
L. adecarboxylata: Leclericia adecarboxylata JCM1667 (7.46 ± 0.20 log10 cells/ml)
M. wisconsensis: Moellerella wisconsensis JCM5894 (7.85 ± 0.34 log10 cells/ml)
Y. regensburgei: Yokenella regensburgei JCM2403 (8.03 ± 0.13 log10 cells/ml)
P. agglomerans: Pantoea agglomerans JCM1236 (7.67 ± 0.78 log10 cells/ml)
B. agrestis: Buttiauxella agrestis JCM1090 (7.76 ± 0.00 log10 cells/ml)
K. ascorbata: Kluyvera ascorbata JCM2107 (7.80 ± 0.02 log10 cells/ml)
C. davisae: Cedecea davisae JCM1685 (7.56 ± 0.10 log10 cells/ml)。

a) 各大腸菌群/腸内細菌科の生細胞数を意味し、各カラムの数値はリアルタイムPCRのCt値を意味する。
b) 沸騰水に50秒浸漬し損傷菌を調製したことを意味する。
c) EMA未処理を意味する。
d) EMA 終濃度10 μg/mlを意味する。
e) Ct値はmean ± SD(n=2)として表示した。
f) 2回PCR測定を行い、2回とも対象遺伝子が増幅されなかったことを意味する。
さらに、図1に示されるとおり、電気泳動の結果から、いずれの大腸菌群及び腸内細菌科においても、損傷細胞のEMA処理群のみが、PCR増幅産物陽性を示すバンドを検出することができなかった。
牛乳等の食品に接種した大腸菌群(Coliform bacteria)及び腸内細菌科の生細胞と死細胞との識別を行った。
1.試験材料及び試験方法
1-1)使用菌株及び培養方法
Kluyvera ascorbata/JCM2107、Cedecea davisae/JCM1685、Citrobacter koseri/JCM1658、Klebsiella pneumoniae(クレブシエラ・ニューモニアエ)/NRBC3321、Serratia fonticola/JCM1242、Yokenella regensburgei/JCM2403、Rahnella aquatilis/NBRC13544、Hafnia alvei/JCM1666、Leclercia adecarboxylata/JCM1667、Pantoea agglomerans/JCM1236、Enterobacter sakazakii/ATCC51329、E. coli DH5α、Salmonella enteritidis/IID604の腸内細菌科1属、大腸菌群12属について、Brain Heart Infusion Broth(BHIブロス:Eiken Chemical Co., Ltd., Tokyo, Japan)を用いて、37℃、16時間培養を行った。
前記により作製した生細胞懸濁液を試験試料に用いて以下の試験に供した。
なお、生細胞懸濁液中の各大腸菌群及び腸内細菌科の細菌の生細胞数は、標準寒天培地によりカウントし、また、同時に分光光度計U-2800A(Hitachi, Japan)を用いて、波長600 nmによる濁度測定を実施し、生細胞数と濁度の関係を確認した。
市販の殺菌牛乳22.2ml(培養法により生細胞は検出されなかった)について、前記で調製した各種大腸菌群又は腸内細菌科の生細胞懸濁液を用いて、9~25 cellsを殺菌牛乳に接種した。
なお、牛乳各22.2mlに対して1種類の大腸菌群又は腸内細菌科の細菌を入れる形態をとった。
更に、サンプルブランクとして生細胞懸濁液を添加しない牛乳22.2 mlを用意した(菌未接種)。
なお、回収した沈殿物(ペレット)には、大腸菌群/腸内細菌科・生細胞接種牛乳、及び菌未接種牛乳ともに、市販の牛乳にすでに存在すると推測される殺菌によって死滅した死細胞群(大腸菌群等を含むグラム陰性細菌又はグラム陽性細菌(≧6 log10 cells))が含まれている。
したがって、大腸菌群/腸内細菌科・生細胞接種牛乳から調製された沈殿物には、死細胞と生細胞が含まれているものと判断した。
前記で調製した大腸菌群/腸内細菌科・生細胞接種牛乳から調製された沈殿物(試験試料)に、予め37℃に保温したBrain Heart Infusion(BHI)ブロスを10ml添加して懸濁し、これに、プロテイナーゼK溶液(シグマ社製:EC. 3. 4. 21. 64、1250 U/ml相当)を生理食塩水により50倍希釈(25 U/ml)した希釈酵素溶液を25μl添加し、37℃で3時間酵素処理を行った。
酵素処理を行った試験試料は、37℃、3,000×G、5分間遠心処理し、上清を除去して再び沈殿物を回収した。
前記酵素処理後の沈殿物に生理食塩水を1ml添加して攪拌した後、実施例2と同様にして調製したEMA溶液(1000μg/ml)を10μl添加し、遮光下で4℃、10分間放置した。
その後、実施例2と同様にして、可視光照射及び洗浄処理を行い、沈殿物に滅菌水5μlを入れてPCR増幅用試料とした。
実施例2と同様に、PCR増幅用試料5 μlに前処理剤を添加した。
PCR増幅には、Primer F:16S rRNA遺伝子検出用フォワードプライマー 16S_1234F(5'-CTACAATGGCGCATACAAAGAGAAG-3':配列番号3)、Primer R:23S rRNA遺伝子 検出用リバースプライマー 23S_1703R(5'-CCTTCTCCCGAAGTTACGGCACCAT-3':配列番号4)をPCRプライマーとして使用した。
更に、リアルタイムPCR後の増幅産物の融解解析において、温度に対する蛍光物質の変化量(一次微分ピーク)を最大にして高感度検出を行うために、以下のa)~g)の組成からなるPCRバッファーを調製し、このPCRバッファー41.5 μlを、前記PCR増幅用試料及び前処理剤の混合液に加えてPCR増幅を行った。
前記PCRプライマーは、16S rRNA遺伝子の1234~1258位、tRNA遺伝子(76 bp)、及び23S rRNA遺伝子の1~1703位までを含み、かつスペーサー領域(約 364 bp)を含むlong DNA(約 2450 bp)をターゲットとしている。
b)Primer R(10 pmol/μl):4μl
c)Ex-Taq(5U/μl:Takara-Bio):0.5μl
(Tween 20 0.5%、Nonidet P-40 0.5%、グリセロール 50%を含む)
d)10×Ex-Taq Buffer(Takara-Bio社製):5μl
e)dNTP mixture(Takara-Bio社製):4μl
f)10×SYBR Green I(BMA社製):8μl
g)滅菌水:16μl
1)95℃、3 分(1 サイクル)
2)95℃、30 秒;60℃、40 秒;68℃、3 分(40 サイクル)
3)95℃、3 分(1 サイクル)
陽性コントロールとして、エンテロバクター サカザキ菌の生細胞懸濁液(8 log10 cells/m)を使用して、同様にPCR増幅を行った。さらに、ブランクサンプルとして、試験試料を加えずに、そのままPCRバッファーを用いてPCR増幅を行った。
0.5×TAEを用いて0.8%アガロースゲル(Seakem GTG agarose:FCM BioProducts, Rockland, Me)を調製した。
5~10μlのPCR増幅産物を前記アガロースゲルにアプライし、電気泳動を行った。
SYBR Gold nucleic acid gel stain(インビトロジェン、Eugene、Oregon、USA)を0.5×TAE により10,000倍希釈した溶液に、電気泳動後のアガロースゲルを15分間浸漬し、染色後、そのゲルをデンシトグラフで観察し、その画像をAE-6905H Image Saver HR (Atto Co., Japan)を用いて取り込み保管した。
リアルタイムPCRを行った結果を表3に示す。また、PCR最終増幅産物の電気泳動の結果を図2に示す。
KP: K. pneumoniae
CK: C. koseri
EC: E. coli.
SE: S. enteritidis
KA: K. ascorbata
CD: C. davisae
SF: S. fonticola
YR: Y. regensburgei
RA: R. aquatilis
HA: H. alvei
LA: L. adecarboxylata
PA: P. agglomerans
ES: E. sakazakii
ミルク: 大腸菌群未接種・市販殺菌牛乳
陽性: 陽性コントロール(エンテロバクター サカザキ菌:8 log10 CFU/mlの5μlをPCRの鋳型として使用)
陰性: 陰性コントロール (滅菌水5μlをPCR鋳型として使用)
L: 100 bp DNA ラダー。

a) 大腸菌群の生菌数を意味し、測定はn = 2~8で行われた。
b) リアルタイムPCRによる増幅産物の融解解析(TMパターン)による全測定回数に対する陽性回数。
c) PCR増幅産物の電気泳動/ゲル染色(SYBR Gold)法による全測定回数に対する陽性回数。
また、PCR増幅産物の融解解析及びPCR増幅産物の電気泳動を行った結果、13菌種全てにおいて生細胞を検出することが可能となった。なお、大腸菌(生細胞)未接種牛乳に関しては、融解解析及び電気泳動いずれのケースにおいてもPCR反応は陰性を呈した。
EMA処理後、DNA抽出により精製したDNAを鋳型とし、16S rRNA (long DNA)をターゲットにしたEMA-PCR法による高濃度大腸菌群損傷細胞(腸内細菌科も含む)の検出を行った。
1-1)使用菌株及び培養方法
本試験の方法は、特許第4217797号(国際公開第2002/052034号パンフレット)の方法に基づいて行った。
Escherichia coli DH5α、Salmonella enteritidis IID604、Klebsiella oxytoca JCM1665、及びCitrobacter koseri JCM1658をブレイン・ハート・インヒュージョン(BHI)ブロス(Eiken、Tokyo)を用いて37℃で培養した。
対数増殖期の培養液から一定分量の10mlを採取し、4℃、15分間、8,000×Gにより冷却遠心処理した。上清を除去後10mlの生理食塩水をペレットに加えて再懸濁させ、同様の洗浄操作を行った後、10mlの生理食塩水を加えて生細胞懸濁液として使用した。生細胞数測定はL寒天平板培地により実施した。
1.5mlマイクロチューブに1mlの生細胞懸濁液を入れ50秒間沸騰水に浸積することにより損傷細胞を調製した(損傷細胞懸濁液)。本処理により得られた損傷細胞は、標準寒天培地でコロニーは形成しなかった。
滅菌水によりEMA(Sigma:St. Louis, Mo, USA)を1,000μg/mlで溶解し、0.20μmのフィルター(Minisart-plus; Sartorius AG, Gottingen, Germany)により無菌ろ過した。
大腸菌E. coli DH5α(7.91 ± 0.20 log10 cells/ml)の生細胞及び損傷細胞の各懸濁液1mlに10μlのEMA溶液(1000μg/ml)を添加し、遮光下4℃、10分間放置した。
その後、氷上にて可視光源(100V PRF 500W Flood eye, Iwasaki Electric Co., Ltd., Tokyo, Japan)から20cm離し、5分間照射した。
EMA処理サンプルを4℃、15,000×G、10分間の冷却遠心処理を行い、上清を除去後に1mlの生理食塩水により同様の洗浄操作を施した。
Salmonella enteritidis IID604(8.47 ± 0.02 log10 cells/ml)、Klebsiella oxytoca JCM1665(8.38 ± 0.08 log10 cells/ml)、及びCitrobacter koseri JCM1658(8.02 ± 0.06 log10 cells/ml)の生細胞及び損傷細胞の各懸濁液についても、大腸菌E. coli DH5αと同様のEMA処理を行った。
EMA処理後の各懸濁液の上清を除去した後、沈殿物(菌体)に10mM Tris-HCl(pH 8.0)を0.5ml添加し、10μlプロテアーゼK溶液(Sigma:1,250 U/ml相当)を添加し、200μlの10% (w/v) SDS溶液を添加して、50℃で、一晩インキュベーションした。
その後、フェノール/クロロホルム抽出、エタノール沈殿法(EP)によりDNA抽出を行った。
抽出精製したDNAに150μlの滅菌水を加え、その濃度をUV 260 nmの吸光度(OD260)により評価した。また、純度をOD260/OD280により評価した。
Primer F:16S rRNA遺伝子検出用フォワードプライマー16S_10F (配列番号1)、Primer R:16S rRNA遺伝子検出用リバースプライマー16S_1500R(配列番号2)を用いて、下記の組成に示されるPCRバッファーを調製した。
b)10×Ex-Taq Buffer (Takara-Bio):5μl
c)dNTP mixture (Takara-Bio):4μl
d)Primer F (10 pmol/μl):4μl
e)Primer R (10 pmol/μl):4μl
f)SYBR Green I(2×)(BMA社製):10μl
g)滅菌水:22.5μl
1)4℃、3 分(1 サイクル)
2)94℃、30 秒(1 サイクル)
3)94℃、20 秒;55℃、30 秒;72℃、90 秒(50 サイクル)
4)95℃、3 分(1 サイクル)
0.5×TAEを用いて2%アガロースゲル(2% Seakem GTG agarose:FCM BioProducts, Rockland, Me)を調製した。
10μlのPCR増幅産物をアガロースゲルにアプライし、電気泳動を行った。
1μg/mlのエチジウムブロマイド溶液で染色後、そのゲルをデンシトグラフで観察し、その画像をAE-6905H Image Saver HR(Atto Co., Japan)を用いて取り込み保管した。
リアルタイムPCRを行って得られたCt値(増幅曲線が境界値を超えるPCRサイクル数)を表4に示す。また、電気泳動の結果を図3に示す。
クレブシェラ菌: K. oxytoca JCM1665 (8.38 ± 0.08 log10 cells/ml)
シトロバクター菌: C. koseri JCM1658 (8.02 ± 0.06 log10 cells/ml)
大腸菌: E. coli DH5α (7.91 ± 0.20 log10 cells/ml)
サルモネラ菌: S. enteritidis IIP 604 (8.47± 0.02 log10 cells/ml)
EMA + : EMA (遮光下10 μg/ml、10 分、4 ℃) + 可視光照射(5 分)。
EMA - : EMA未処理。
NC: DNA鋳型の代わりに滅菌水を用いた陰性コントロール。
M: 100 bp DNAラダー。
損傷菌; 生菌けん濁液を沸騰水に50秒浸漬した。
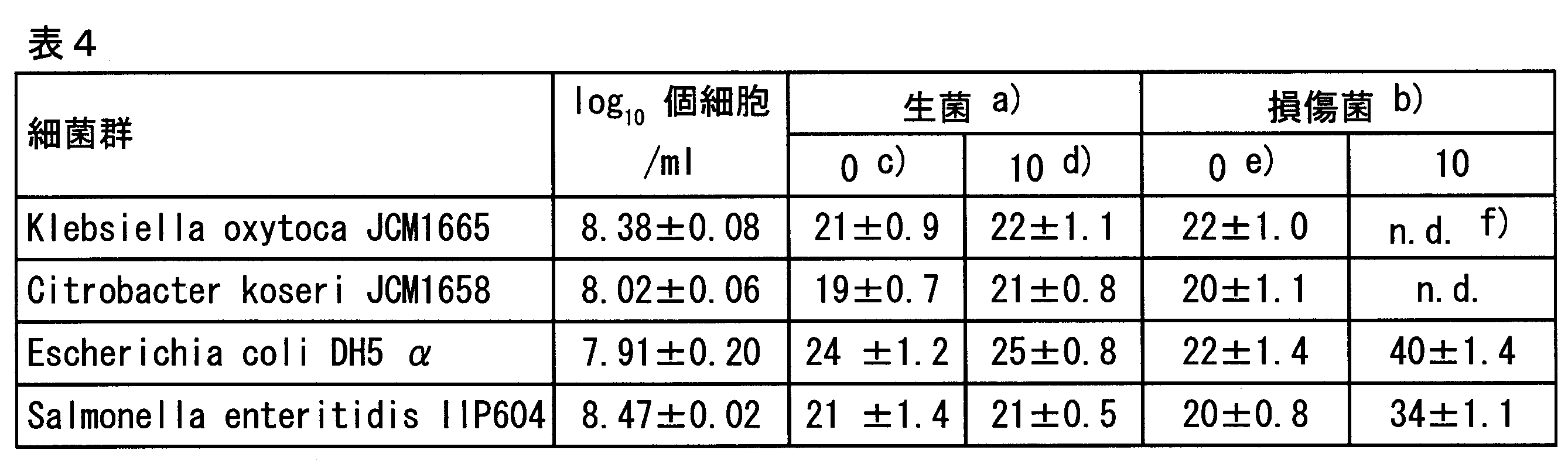
a) クレブシェラ菌、シトロバクター菌、大腸菌、及びサルモネラ菌の生菌を意味し、各カラムの数値はリアルタイムPCRのCt値を意味する。
b) 沸騰水に50秒浸漬し損傷菌を調製したことを意味する。
c) EMA未処理を意味する。
d) EMA 終濃度10 μg/mlを意味する。
e) Ct値はmean ± SD (n = 2)として表示した。
f) n.d. はPCR増幅反応が進行しなかったことを意味し、Ct値が観測できなかったことを意味する。
PCR最終増幅産物による、生細胞及び損傷細胞の識別結果(図3)から、E. coli DH5α、及びS. enteritidis IID604については、損傷細胞のEMA処理後サンプルにおいても対象遺伝子のバンドが得られ、生細胞と損傷細胞の識別は十分に確認することが出来なかった。
エンテロバクター・サカザキ菌の細胞が前処理剤存在下50回のPCRサーマルサイクルローテーションにより、どの程度溶解(Lysis)しているか調べた。
108 cells/mlのエンテロバクター・サカザキATCC51329株(ES)の菌体を、生理食塩水、又は表5に示される前処理剤溶液(以下、「DB (direct buffer)」と記載することがある。)に懸濁させ、それぞれけん濁液を調製(0.25 mL)した。各懸濁液とも、25 μlずつに小分けして200 μl PCRチューブに移し、95℃ 15秒, 60℃ 20 s, 72℃ 30秒のPCRサーマルサイクルローテーション工程(50回)を経た後、再びひとまとめにし(合計0.25 ml)、PCR増幅用試料とした。上記0.25 mlから2.5 μlを採取し、表5記載の前処理剤溶液12.25μl(但し滅菌水を2.7 μlに変更)に加え、そこに以下に示すPCRバッファー12.75 μlを加えてPCR を行った(表6の懸濁液懸濁液Iに相当)。プライマーには、ompA_F: ompA遺伝子検出用フォワードプライマー(5'-ggatttaaccgtgaacttttcc-3';配列番号7)、ompA_R: ompA遺伝子検出用リバースプライマー(5'-cgccagcgatgttagaaga-3';配列番号8)を用いた。
a) ompA_F (10 pmol/μl): 2 μl
b) ompA_R (10 pmol/μl): 2 μl
c) Ex-Taq (5U/μl: Takara-Bio): 0.25 μl
(Tween 20 0.5%、Nonidet P-40 0.5%、グリセロール 50%を含む)
d) 10 × Ex-Taq Buffer (Takara-Bio社製): 2.5 μl
e) dNTP mixture (Takara-Bio社製): 2 μl
f) 10 × SYBR Green I (BMA社製): 4 μl
1) 4 ℃、3分(1サイクル)
2) 95 ℃、 15 秒;60℃ 20 秒;72℃ 30 秒(50サイクル)
3) 95 ℃、3分(1サイクル)

リアルタイムPCR増幅による各Ct値の結果を表6に示す。表中の「非加熱」はPCRサーマルサイクルによるヒート処理(95 ℃、60 ℃、72 ℃によるサーマルサイクルローテーション50回)を施さなかった群であり、サーマクサイクル加熱はそのサーマルサイクルローテーションを50回施した群である。なお、a)で示される懸濁液IVにおけるエンテロバクター・サカザキ菌の生菌数は標準寒天培地平板により測定した結果、107.6 cells/ml であり、また、b)で示される上清Iにおけるエンテロバクター・サカザキ菌の生菌数は同様の方法により、105.7 cells/ml であった。
本試験の基本的性質として、懸濁液IV及び上清Iの生菌数の測定結果から、エンテロバクター・サカザキ菌生理食塩水けん濁液を冷却遠心処理した上清中にも生菌が1%の割合で混入していることが示された。E. sakazakii(生理食塩水中)の非加熱群に関して、懸濁液I又はIIと上清IのCt値の比較において、懸濁液群のCtが5前後小さいのは主として生菌が有意に沈渣(ペレット)に回収されると共に、上清にも極めて少量が回収されているからであると考えられる。すなわち、このような現象は、生菌が特定の割合で沈渣と上清に分配されていることを意味するものである。

エンテロバクター・サカザキ菌の細胞が前処理剤存在下50回のPCRサーマルサイクルローテーションにより溶解(Lysis)しているのかどうかを、PCRサーマルサイクルローテーション前後のサンプルを用い、核染色剤使用による蛍光顕微鏡観察・実体顕微鏡観察、更にはPCRサーマルサイクルローテーション後の残存細胞数の定量を可能にするフローサイトメトリー法を用いて評価した。
1.実験方法
実施例4の方法と同様に、109 cells/mlのエンテロバクター・サカザキATCC51329株(ES)の菌体を、生理食塩水、又は表5に示される前処理剤溶液に懸濁させ、各々けん濁液を調製(0.25 ml)した。各懸濁液とも、25 μlに小分けして200 μl PCRチューブに移し、(95℃ 15 s, 60℃ 20 s, 72℃ 30 s)のPCRサーマルサイクルローテーション工程(50回)を経た後、再び一まとめにした(合計0.25 ml)。それらを半分ずつに分け、一方はそのまま、他方は冷却遠心処理(3000 × g、10 min、4℃)を施し、上清を採取した。それぞれの過程を経た上記0.125 mlにSYTO9を1.5 μl/ mlの割合で添加し、遮光下4 ℃にて15分保持後、各2.5 μlをスライドガラスに載せた後、カバーガラスを掛け、蛍光/実体顕微鏡AxiosKop2 motplus(レンズ: Plan-NEOFLUAR 100×/1.30 oil ∞/0.17; 光源: KublercoDIX ebq 100 isolated; ソフトウェア: AxioVision Rel. 4.6.3; フィルター: FITC and DIC3; 露出時間: FITC 347 ms fixed, DIC3 20 ms fixed; LEJ Leistungs elektronik Jena GmbH, Germany)にセットし、アルゴンレーザー光488 nmを励起光として、530 nmの緑の蛍光が細菌細胞から発せられるかを観察した。
非加熱、又は50回PCRサーマルサイクル工程後のエンテロバクター・サカザキ菌の生理食塩水けん濁液、及びその冷却遠心上清、並びに、非加熱、又は50回PCRサーマルサイクル工程後のエンテロバクター・サカザキ菌の前処理剤溶液けん濁液、及びその冷却遠心上清の蛍光顕微鏡による観察像を、それぞれ図4~11に示す。すなわち各蛍光顕微鏡観察像が表6のWashing step Iの懸濁液I~上清Iに相当するよう実験を設定した。尚、これらの図には、実体顕微鏡像、及び実態顕微鏡像と蛍光顕微鏡像の重ね合わせ像も示した。
1.実験方法
次に、フローサイトメトリーによる実験方法を示す。まず実施例4の方法と同様に、109 cells/mlのエンテロバクター・サカザキATCC51329株(ES)の菌体を、生理食塩水、又は表5に示される前処理剤溶液に懸濁させ、けん濁液を調製(0.25 ml)した。各懸濁液とも、25 μlに小分けして200 μl PCRチューブに移し、(95℃ 15 s, 60℃ 20 s, 72℃ 30 s)のPCRサーマルサイクルローテーション工程(50回)を経た後、再び一まとめにした(合計0.25 ml)。各サンプルに共通してけん濁液及びその上清のそれぞれをフローサイトメトリー測定に使用するので、それぞれ0.25 ml×3本を用意した。具体的には、1本目はそのままとし、2本目は冷却遠心処理(3000 × g、10 min、4℃)後上清を除去し、沈殿に0.25 mlの生理食塩水を加えてけん濁させたもの、3本目は同様の冷却遠心処理を施し、上清を回収したものを調製した。それぞれに対してSYTO9を1.5 μl/mlの濃度で添加し遮光下4 ℃で15分保存し、フローサイトメトリー試験用サンプルとした。

エンテロバクター・サカザキ菌の生理食塩水けん濁液及びその上清(非加熱・PCRサーマルサイクルローテーション処理)の実験結果を図12に、同菌前処理剤溶液けん濁液(1回洗浄後の再けん濁液も含む)及びその上清(非加熱・PCRサーマルサイクルローテーション処理)の実験結果を図13に示す。エンテロバクター・サカザキ菌の生理食塩水けん濁液に関して50回のPCRサーマルサイクルローテーションにより細胞の大部分が溶解(Lysis)していれば細分化されているので、FSC-SSC図の細菌ゲート領域(多角形で囲っている領域が、細菌がプロットされる領域)に入らなくなり、その後のSYTO9による緑の蛍光を意味するFL1(図ではFL1-H)陽性(+)領域(X軸の右半分領域)には何もプロットされなくなるか、もしくはプロットは激減するはずである。しかし図12のエンテロバクター・サカザキ菌の生理食塩水けん濁液の非加熱及びPCRサーマルサイクルローテーションサンプルの結果はそれを支持していない。寧ろ単純数値比較でも、サーマルサイクル後も95%は細菌形態を維持し、且つ染色体DNAも保有していると推察される。元来、フローサイトメトリー測定そのものの複数回数による標準偏差を考慮すれば、その数値差は誤差範囲の可能性が極めて高く、50回のPCRサーマルサイクルローテーションによりエンテロバクター・サカザキ菌の細菌細胞は100%近い割合で形態を維持し、且つ染色体DNAも保有しているとみなせる。
エンテロバクター・サカザキ菌の細菌細胞数と、それに含まれる染色体DNA量と同量の精製染色体を用いたリアルタイムPCR測定
エンテロバクター・サカザキATCC29544及びATCC51329の一夜増菌培養液から、WO2007/094077記載のDNA抽出方法に従い、RNAのコンタミネーションがない精製DNAを得、その260 nm及び280 nmの吸光値(OD260、OD280: DNA溶液50 μg/mlにおいてOD260 = 1.0、セル長1 cm)を測定し、DNA濃度をOD260から算出し、精製DNAの純度をOD260/OD280により評価した。
DNA精製度を表9、リアルタイムPCR結果を表10に示す。表9の結果よりOD260/OD280の値が2.0付近なのでRNAのコンタミネーションの少ない高純度DNAをエンテロバクター・サカザキ菌の2菌株からそれぞれ調製できた。次に、表10の結果から、各エンテロバクター・サカザキ菌の細菌細胞数に対する同量の染色体DNA量のCt値間に有意差はなく、試験管内に溶解していた精製DNAが100% PCRの鋳型として機能しているなら、エンテロバクター・サカザキ菌の細菌細胞の染色体DNAもPCR鋳型として100%機能していることが判明した。


実施例4~6より、前処理剤存在下PCRバッファーによりPCR反応(50回)を行ってもエンテロバクター・サカザキ菌の細胞は大部分溶解せず、細胞内に染色体DNAを保有していることが分かった。その一方で、リアルタイムPCR反応後のPCR増幅産物のTMパターン解析(融解温度測定)により、ompA遺伝子産物と推察される温度ピークが得られリアルタイムPCR反応陽性と判別していた。しかしながら、PCR増幅反応物が細菌細胞内に存在しているのか、PCR反応用液中に存在しているのか、又は双方に存在しているのかは厳密に考えると定かではなく、常識的にはPCR反応用溶液にPCR増幅産物が主として溶解していると考えられるが、これとて解明されているわけではなく、更に本願記載の前処理剤存在下のPCR反応においては、尚更に解明されていない。前記実施例においては、前処理剤存在下のPCR反応が細菌細胞内で行われている可能性があることを示唆されるが、以降の実施例においては細菌細胞内にもPCR増幅産物が残留している可能性を示す。
エンテロバクター・サカザキATCC51329の一夜増菌培養液500 μl(9.3 ×108 cells/ml)を5本用意し、それらを冷却遠心処理(3000×g、10 min、4 ℃)し、上清を除去後、それぞれのペレットに対して一般的な細菌固定液A(4%パラホルムアルデヒド)の他、固定液B(メタノール/酢酸 = 3/1)、固定液C(Mildform 10N: 10%ホルマリン Neutral Buffer Solution Deodorized; 和光純薬工業、大阪)、又は、固定液D(Mildform 10NM: 10% ホルマリン Neutral Buffer-Methanol Solution Deodorized;和光純薬工業、大阪)の500 μlを加え、4 ℃一夜インキュベーションし、細菌細胞内染色体DNAと細胞壁構成蛋白質とをクロスリンクさせDNAを細胞内に予め固定させた。対照として、固定液の代りに生理食塩水500 μlを用いて、固定を行わない試料を作製した。
その後、500 μlの生理食塩水により3回洗浄し、最終的には250 μlの生理食塩水にけん濁させた。洗浄操作によるペレットとしての細菌の回収率は一般に80%と見なされているので、4回遠心していることを考慮すると、推定最終調製液中のエンテロバクター・サカザキ菌濃度は 7.6 × 108 cells/mlである。その250 μl生理食塩水けん濁液を更に10倍希釈した。この希釈液中の推定エンテロバクター・サカザキ菌濃度は7.6 × 107 cells/mlである。その2.5 μlをPCR増幅用試料とし、表5記載の前処理剤溶液 12.25 μl(但し滅菌水を2.7 μlに変更)に加え、そこに以下に示すグラム陰性細菌検出用PCRバッファー12.75 μlを加えた。各固定液で固定されたサンプルについて、それぞれ27.5 μl × 20本準備した。プライマーには、実施例3に記載した16S rRNA遺伝子検出用フォワードプライマー 16S_1234F(配列番号3)、及び、23S rRNA遺伝子 検出用リバースプライマー 23S_1703R(配列番号4)を使用した。
a) 16S_1234F (10 pmol/μl): 2 μl
b) 23S_1703R (10 pmol/μl): 2 μl
c) Ex-Taq (5U/μl: Takara-Bio): 0.25 μl
(Tween 20 0.5%、Nonidet P-40 0.5%、グリセロール 50%を含む)
d) 10 × Ex-Taq Buffer (Takara-Bio社製): 2.5 μl
e) dNTP mixture (Takara-Bio社製): 2 μl
f) 10 × SYBR Green I (BMA社製): 4 μl
1) 4 ℃、3分(1サイクル)
2) 95 ℃、 15 秒;60℃ 20 秒;72℃ 3分(30サイクル)
3) 95 ℃、3分(1サイクル)
更に、固定液A及びBに関しては、16S_1234Fや23S_1703Rの代わりに、実施例4に記載のompA_F(配列番号7)及びompA_Rプライマー(配列番号8)を用いて、実施例4のPCRサーマルサイクル条件を用い、それ以外は本実施例7に従い、PCR反応後の上清5 μlの電気泳動、及び、PCR反応後上清を除去したペレットの生理食塩水けん濁液に関してSYTO9によるフローサイトメトリー測定を実施した。
上記各固定液処理を経たPCR(16S-23S rRNA: 2450-bp)後の反応上清の電気泳動の結果を図14に、その反応溶液のリアルタイムPCRのCt値をその直下に示す。PCR反応後上清を除去し、ペレットを生理食塩水にけん濁し、SYTO9によるフローサイトメトリー測定を行った結果を表11に示す。同様にompA(469-bp)遺伝子をターゲットにしたPCR反応後の上清の電気泳動図を図15に示す。PCR反応後上清を除去し、ペレットを生理食塩水にけん濁し、SYTO9によるフローサイトメトリー測定を行った結果を表12に示す。図14から固定液BのPCR反応液上清には16S-23S遺伝子(2450-bp)産物量が他の固定液より有意に多く、非固定(図中、「S」)と同等のバンド強度であった。細菌の標準的固定法は固定液Aが一般的であり固定液C及び固定液Dも固定液Aに近い成分なので、エンテロバクター・サカザキ菌の細胞内の染色体DNAと細胞壁蛋白質とは強固にクロスリンクを形成していると推察される。図14の結果から固定液Bは非固定(S)と同等の機能しかないのかもしれないが、固定液Bは哺乳動物細胞染色体と細胞膜タンパクを強固にクロスリンクさせるので、細菌においても多少は固定機能を発揮していると考えるのが妥当である。


これは、固定液A、C、及びDの場合、染色体と細胞壁の固定の度合いが高いため、一見、PCR反応工程の熱変性(95 ℃)が良好ではなく染色体DNAが一本鎖になれないため、その後プライマーによる染色体への接着が不良となったものと考えることも可能であるが、図15のompA遺伝子(469-bp)を標的とした同様の実験(30サイクル)結果を考慮すると、固定液AとBを用いた方法で双方ともリアルタイムPCRのCt値及びバンド強度で比較して有意差がないので、その仮説は否定される。
すなわち、図14の固定液A、C、及びDのPCR反応液上清に標的遺伝子増幅産物が少ないのは、PCR反応自体は細菌細胞内に保持された染色体を鋳型として良好に進行しているが、遺伝子産物が図15と比較して5倍程長く(2450-bp)、その結果、菌体内で増幅された遺伝子産物の外部溶液への流出が抑制されたためと考える。この仮説が正しければ、その2450-bpの遺伝子増幅産物はPCR反応後のエンテロバクター・サカザキ菌の細菌細胞からも検出されるはずである。これは、実施例8において実証された。以上のとおり、細菌の染色体を細胞内に固定して外部溶液への染色体の流出を阻止した処理を予め施しておいても、前処理剤存在下ではPCRサーマルサイクルローテーションを50回繰り返しても、細菌細胞は100%近く細菌の形態を保持し、染色体も菌体内に保持されているにも拘わらず、PCR反応は進行しPCR増幅産物は外部溶液にも存在していることから、PCR反応は主として細菌細胞内で行われていると結論できる。尚、下記実施例8で示すように、上記条件下で、PCR産物は一部菌体内にも存在していた。
実施例4~7では、前処理剤存在下PCRが細菌細胞内で行われている可能性が高いこと、すなわちIn-situ PCRが起きていた可能性を示唆してきた。In-situ PCR(例えば、Gerard J. et al., American Journal of Pathology, 139: 847-854, 1991.)は、ヒト免疫細胞を実施例7に示すような固定液で処理し、染色体DNAとヒト細胞膜たんぱく質とをクロスリンクさせ、蛋白質分解酵素で短時間処理し、又は、マイクロ波照射によりヒト免疫細胞の細胞膜処理をした上で、ヒト細胞内で、染色体DNAに組み込まれたHPV遺伝子のような遺伝子を検出・定量する手法である。
同手法は、PCR反応用溶液を、固定化させたヒト免疫細胞の上に載せ、ヒト免疫細胞内でPCR増幅反応を起こさせ、同時に500-bp程度のPCR産物でも細胞外に流出しない手法である。PCR産物が細胞外に流出しないため、PCR反応を5~10サイクル未満の初期で停止すれば、細胞中の遺伝子の検出だけでなく、ある程度それが組み込まれた数まで推察可能な手法である。
そこで、実施例7における固定液Bと非固定(S)に焦点を当て、PCR増幅産物が外部溶液に大量流出している状況でも細菌細胞内に一部PCR増幅産物が残留しているかどうかを検討した。
エンテロバクター・サカザキATCC51329の一夜増菌培養液(4.3 × 108 cells/ml)500 μlを2本用意し、それらを冷却遠心処理(3000 × g、10 min、4 ℃)し、上清を除去後、それぞれのペレットに対して固定液B(メタノール/酢酸 = 3/1)500 μlを加え、4 ℃一夜インキュベーションし、細菌細胞内染色体DNAと細胞壁構成蛋白質とをクロスリンクさせDNAを細胞内に予め固定させた。対照として、固定液 Bの代りに生理食塩水500 μlを用いて、固定を行わない試料を作製した。これ以降は実施例7と同様の方法により、最終的には10倍希釈して250 μlのエンテロバクター・サカザキ菌の生理食塩水けん濁液を得た。懸濁液中の推定エンテロバクター・サカザキ菌濃度はおよそ3.5 × 107 cells/mlである。その2.5 μlをPCR増幅用試料とし、表5記載の前処理剤溶液 12.25 μl(但し滅菌水を2.7 μlに変更)に加え、そこに以下に示すグラム陰性細菌検出用PCRバッファー12.75 μlを加え、下記の条件によりPCRを行った。PCR反応の際には、固定液及び対照サンプル27.5 μl × 20本準備した。プライマーには、実施例3に記載した16S rRNA遺伝子検出用フォワードプライマー 16S_1234F(配列番号3)、及び、23S rRNA遺伝子 検出用リバースプライマー 23S_1703R(配列番号4)を使用した。
DNA抽出工程に供したそれぞれの菌数は、PCR前のサンプルがPCR反応後のサンプル群と比較して100倍多いので、菌数を統一するために、PCRに供する直前のエンテロバクター・サカザキ菌の生理食塩水けん濁液(3.5 × 107 cells/ml)の2.5 μl × 20 (= 1.8 × 106 cells)(固定液B及び対照S)を調製し、PCRを行わず20本一纏めにした後合計3回の遠心処理により洗浄してDNA抽出を行った。
a) 16S_1234F (10 pmol/μl): 2 μl
b) 23S_1703R (10 pmol/μl): 2 μl
c) Ex-Taq (5U/μl: Takara-Bio): 0.25 μl
(Tween 20 0.5%、Nonidet P-40 0.5%、グリセロール 50%を含む)
d) 10 × Ex-Taq Buffer (Takara-Bio社製): 2.5 μl
e) dNTP mixture (Takara-Bio社製): 2 μl
f) 10 × SYBR Green I (BMA社製): 4 μl
1) 4 ℃、3分(1サイクル)
2) 95 ℃、 15 秒;60℃ 20 秒;72℃ 3分(30サイクル)
3) 95 ℃、3分(1サイクル)
図16に結果を示す。固定液Bを用いてエンテロバクター・サカザキ菌の細胞を固定化し、又は固定化せず、前処理剤存在下PCR(16S-23S: 2450-bp)を行い、その反応液上清を電気泳動した結果をレーン2及び3、同PCR反応後の2回洗浄後のペレットからDNA抽出を行い電気泳動した結果をレーン5及び6、本実験の検査材料である固定及び非固定エンテロバクター・サカザキ菌の細胞から直接DNAを抽出し電気泳動した結果をレーン7及び8、PCR反応に供する直前の固定・非固定エンテロバクター・サカザキ菌の細胞からDNAを抽出し電気泳動した結果をレーン9及び10、予めPCR増幅産物を調製しそれをエンテロバクター・サカザキ菌の細菌細胞に添加後2回洗浄し、その後DNAを抽出・電気泳動した結果をレーン13及び14に、それぞれ示す。
エンテロバクター・サカザキ菌の細胞を生理食塩水中、又は、前処理剤存在下、煮沸処理を行いその処理時間に応じて、各上清にサカザキ菌染色体DNAがどの程度流出しているのかを調べた。
エンテロバクター・ザカザキATCC51329の一夜増菌培養液(1.1× 109 cells/ml)を洗浄し、生理食塩水で10倍希釈した後、一端、冷却遠心処理(3000 × g、10 min、4 ℃)によりペレットを回収し、そこへ同量(500 μl)の生理食塩水、又は、表5に示す組成の前処理剤溶液を加え、良くけん濁させた。その後、沸騰水にて0~5分加熱し、加熱後直ぐに冷却した。加熱直後の各けん濁液5 μl、及び、けん濁液の冷却遠心後の上清5 μlを、それぞれ0.8% アガロースゲルにより電気泳動した。
生理食塩水中、又は、前処理剤存在下でエンテロバクター・サカザキ菌の細胞が沸騰水を用いて熱処理された時、上清にどの程度細菌の染色体が溶出しているかを図17に示した。まず、レーン2と9に、非加熱ながら既に僅かな染色体DNAの存在が示唆されるが、これは一夜増菌した増菌液が静止期に到達していたため、死菌の一部が溶解し外部溶液に染色体DNAが流れ出たと考えられ、評価の際は無視するものとする。生理食塩水中では加熱によりエンテロバクター・サカザキ菌の細菌細胞からDNAが溶出していると推察されるが、前処理剤存在下では5分ボイルしてもけん濁液及び上清に染色体DNAのバンドは検出されなかった。しかし、レーン10と12のウェルで評価した場合、けん濁液にはウェルにバンドがあるが、上清にはバンドは存在せず、前処理剤存在下、染色体DNAは細菌細胞から流出せず菌体内に留まっていることが判明した。一方、生理食塩水の上清(レーン5、6、及び7)でもウェル内にバンドが存在するが、これは煮沸により比重が軽くなったエンテロバクター・サカザキ菌の死菌の一部が遠心処理により上清に回収されたものと推察される。表6や図12の結果もそれを支持している。以上により、PCRサーマルサイクルローテーションとは条件が厳密には異なるが、前処理剤存在下の方が、細菌細胞から熱処理によってもDNAが流出困難であることが示唆された。
更には大腸菌群や腸内細菌科に限らず病原細菌を始めとする各種細菌、ウイルス等の微生物に関し、低濃度の生きた微生物のみを迅速に検出・定量可能とするため、各種衛生検査及び臨床検査、工程管理などへも応用が可能である。
Claims (27)
- 被検試料中の微生物の生細胞を、死細胞又は損傷細胞と識別して検出する方法であって、以下の工程を含む方法:
a)前記被検試料に、350nm~700nmの波長の光照射によりDNA又はRNAに共有結合する薬剤を添加する工程、
b)前記薬剤を添加した被検試料に、350nm~700nmの波長の光照射処理を行う工程、
c)被検試料に含まれる微生物のDNA又はRNAのターゲット領域を、細胞からの核酸の抽出を行わずに、核酸増幅阻害物質の働きを抑制する薬剤の存在下で核酸増幅法により増幅する工程、及び
d)増幅産物を解析する工程。 - 前記ターゲット領域の増幅が、微生物細胞内で行われることを特徴とする、請求項1に記載の方法。
- 前記c)の工程において、ターゲット領域の増幅を、界面活性剤、マグネシウム塩、及び有機酸塩又はリン酸塩から選択されるいずれか一種又は複数種の存在下で行うことを特徴とする、請求項1又は2に記載の方法。
- 前記c)の工程の前に、前記a)の工程及びb)の工程を繰り返して行うことを特徴とする請求項1~3のいずれか一項に記載の方法。
- 前記a)の工程の前に、以下のe)の工程を行うことを特徴とする、請求項1~4のいずれか一項に記載の方法。
e)被検試料を、被検試料中に存在する微生物以外の細胞、タンパク質コロイド粒子、脂肪、又は糖質を分解する活性を有する酵素で処理する工程。 - 前記酵素が、蛋白質分解酵素、脂質分解酵素、及び糖分解酵素から選択される請求項5に記載の方法。
- 前記被検試料が、食品、生体試料、飲料水、工業用水、環境用水、排水、土壌、又は拭き取り試料のいずれかである請求項1~6のいずれか一項に記載の方法。
- 前記微生物が細菌、又はウイルスである請求項1~7のいずれか一項に記載の方法。
- 前記細菌がグラム陰性細菌である請求項8に記載の方法。
- 前記350nm~700nmの波長の光照射によりDNA又はRNAに共有結合する薬剤が、エチジウムモノアザイド(ethidium monoazide)、エチジウムジアザイド(ethidium diazide)、プロピジウムモノアザイド(propidium monoazide)、プソラーレン(psolaren)、4,5',8-トリメチルプソラーレン(4,5',8-trimethyl psolaren)、及び8-メトキシプソラーレン(8-methoxy psolaren)から選択される請求項1~9のいずれか一項に記載の方法。
- 核酸増幅阻害物質の働きを抑制する薬剤が、アルブミン、デキストラン、及びT4ジーン32プロテイン、アセトアミド、ベタイン、ジメチルスルフォキシド、ホルムアミド、グリセロール、ポリエチレングリコール、大豆トリプシンインヒビター、α2-マクログロブリン、テトラメチルアンモニウムクロライド、リゾチーム、ホスホリラーゼ、及び乳酸脱水素酵素から選択されるいずれか一種又は複数種である請求項1~10のいずれか一項に記載の方法。
- 前記有機酸塩が、酢酸塩、プロピオン酸塩、及びクエン酸塩から選択される請求項2~11のいずれか一項に記載の方法。
- 前記リン酸塩がピロリン酸塩である請求項2~12のいずれか一項に記載の方法。
- 前記ターゲット領域が50~5000塩基のターゲット領域である請求項1~13のいずれか一項に記載の方法。
- 前記ターゲット領域が、被検試料のDNAの5S rRNA遺伝子、16S rRNA遺伝子、23S rRNA遺伝子、及びtRNA遺伝子から選択される遺伝子に対応するターゲット領域である請求項14に記載の方法。
- 前記核酸増幅法が、PCR法、RT-PCR法、LAMP法、SDA法、LCR法、TMA法、TRC法、HC法、又はマイクロアレイ法である請求項1~15のいずれか一項に記載の方法。
- 前記PCR法をリアルタイムPCR法により行い、PCRと増幅産物の解析を同時に行うことを特徴とする請求項16に記載の方法。
- 前記増幅産物の解析を、微生物の標準試料を用いて作成された微生物量及び増幅産物との関連を示す標準曲線を用いて行うことを特徴とする請求項1~17のいずれか一項に記載の方法。
- 核酸増幅法により、被検試料中の微生物の生細胞を、死細胞又は損傷細胞と識別して検出するためのキットであって、下記の要素を含むキット:
1)350nm~700nmの波長の光照射によりDNA又はRNAに共有結合する薬剤、
2)核酸増幅阻害物質の働きを抑制する薬剤、及び
3)検出対象の微生物のDNA又はRNAのターゲット領域を核酸増幅法により増幅するためのプライマー。 - さらに界面活性剤、マグネシウム塩、及び有機酸塩又はリン酸塩から選択されるいずれか一種又は複数種を含む、請求項19に記載のキット。
- さらに被検試料中に存在する微生物以外の細胞、タンパク質コロイド粒子、脂肪、又は糖質を分解する活性を有する酵素を含む、請求項19又は20に記載のキット。
- 前記核酸増幅法が、PCR法、RT-PCR法、LAMP法、SDA法、LCR法、TMA法、TRC法、HC法、又はDNAマイクロアレイ法である請求項19~21のいずれか一項に記載のキット。
- 前記350nm~700nmの波長の光照射によりDNA又はRNAに共有結合する薬剤が、エチジウムモノアザイド(ethidium monoazide)、エチジウムジアザイド(ethidium diazide)、プロピジウムモノアザイド(propidium monoazide)、プソラーレン(psolaren)、4,5',8-トリメチルプソラーレン(4,5',8-trimethyl psolaren)、及び8-メトキシプソラーレン(8-methoxy psolaren)から選択される請求項19~22のいずれか一項に記載のキット。
- 核酸増幅阻害物質の働きを抑制する薬剤が、アルブミン、デキストラン、及びT4ジーン32プロテイン、アセトアミド、ベタイン、ジメチルスルフォキシド、ホルムアミド、グリセロール、ポリエチレングリコール、大豆トリプシンインヒビター、α2-マクログロブリン、テトラメチルアンモニウムクロライド、リゾチーム、ホスホリラーゼ、及び乳酸脱水素酵素から選択されるいずれか一種又は複数種である請求項19~23のいずれか一項に記載のキット。
- 前記有機酸塩が、酢酸塩、プロピオン酸塩、及びクエン酸塩から選択される請求項20~24のいずれか一項に記載のキット。
- 前記リン酸塩がピロリン酸塩である請求項20~25のいずれか一項に記載のキット。
- 前記酵素が、蛋白質分解酵素、脂質分解酵素、及び糖質分解酵素から選択される請求項21~26のいずれか一項に記載のキット。
Priority Applications (12)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2010275576A AU2010275576B2 (en) | 2009-07-24 | 2010-07-23 | Method and kit for detection of microorganism |
| RU2012106617/10A RU2527897C2 (ru) | 2009-07-24 | 2010-07-23 | Способ и набор для детекции микроорганизмов |
| CN201080033156.XA CN102471768B (zh) | 2009-07-24 | 2010-07-23 | 用于微生物检测的方法及试剂盒 |
| CA2768699A CA2768699C (en) | 2009-07-24 | 2010-07-23 | Method and kit for detection of live microorganisms |
| NZ597138A NZ597138A (en) | 2009-07-24 | 2010-07-23 | Method and kit for detecting microorganisms |
| US13/386,601 US9394572B2 (en) | 2009-07-24 | 2010-07-23 | Method and kit for detection of microorganism |
| SG2012005013A SG177738A1 (en) | 2009-07-24 | 2010-07-23 | Method and kit for detecting microorganisms |
| JP2010529962A JP4825313B2 (ja) | 2009-07-24 | 2010-07-23 | 微生物検出法及び微生物検出キット |
| KR1020127004714A KR101383389B1 (ko) | 2009-07-24 | 2010-07-23 | 미생물 검출법 및 미생물 검출 키트 |
| EP10802360.7A EP2458002B1 (en) | 2009-07-24 | 2010-07-23 | Method and kit for detection of microorganism |
| MX2012001000A MX2012001000A (es) | 2009-07-24 | 2010-07-23 | Metodo y kit para la deteccion de un microorganismo. |
| US15/188,755 US10329604B2 (en) | 2009-07-24 | 2016-06-21 | Method and kit for detection of microorganism |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2009-173566 | 2009-07-24 | ||
| JP2009173566 | 2009-07-24 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US13/386,601 A-371-Of-International US9394572B2 (en) | 2009-07-24 | 2010-07-23 | Method and kit for detection of microorganism |
| US15/188,755 Division US10329604B2 (en) | 2009-07-24 | 2016-06-21 | Method and kit for detection of microorganism |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011010740A1 true WO2011010740A1 (ja) | 2011-01-27 |
Family
ID=43499212
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2010/062474 WO2011010740A1 (ja) | 2009-07-24 | 2010-07-23 | 微生物検出法及び微生物検出キット |
Country Status (12)
| Country | Link |
|---|---|
| US (2) | US9394572B2 (ja) |
| EP (1) | EP2458002B1 (ja) |
| JP (1) | JP4825313B2 (ja) |
| KR (1) | KR101383389B1 (ja) |
| CN (2) | CN103820578B (ja) |
| AU (1) | AU2010275576B2 (ja) |
| CA (1) | CA2768699C (ja) |
| MX (1) | MX2012001000A (ja) |
| NZ (1) | NZ597138A (ja) |
| RU (1) | RU2527897C2 (ja) |
| SG (1) | SG177738A1 (ja) |
| WO (1) | WO2011010740A1 (ja) |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2015139434A (ja) * | 2014-01-30 | 2015-08-03 | 森永乳業株式会社 | 微生物検出法及び微生物検出キット |
| WO2017010001A1 (ja) * | 2015-07-16 | 2017-01-19 | 森永乳業株式会社 | 微生物検出法及び微生物検出キット |
| JP2018068211A (ja) * | 2016-10-28 | 2018-05-10 | 森永乳業株式会社 | 微生物の死細胞及び/又は不活化ウイルスの測定方法 |
| WO2019188552A1 (ja) * | 2018-03-27 | 2019-10-03 | 森永乳業株式会社 | 微生物の細胞及び/又はウイルスの測定方法 |
| WO2020218557A1 (ja) * | 2019-04-26 | 2020-10-29 | bitBiome株式会社 | 単一生物単位の生菌由来核酸の選択的検出、カウント、ゲノム解析 |
| US20210292823A1 (en) * | 2020-03-23 | 2021-09-23 | Feng Zhang | Rapid diagnostics |
| EP4083230A1 (en) * | 2011-07-06 | 2022-11-02 | Quest Diagnostics Investments Incorporated | Direct amplification and detection of viral and bacterial pathogens |
Families Citing this family (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR101609224B1 (ko) * | 2011-01-24 | 2016-04-05 | 다카라 바이오 가부시키가이샤 | 핵산 변형 방법 |
| US10324036B2 (en) | 2012-05-02 | 2019-06-18 | Charles River Laboratories, Inc. | Porous planar cell capture system |
| AU2013256174B2 (en) | 2012-05-02 | 2017-03-02 | Charles River Laboratories, Inc. | Viability staining method |
| JP6482459B2 (ja) | 2012-05-02 | 2019-03-13 | チャールズ リバー ラボラトリーズ, インコーポレイテッド | 細胞サンプル中の生存細胞を検出する方法 |
| KR20150127850A (ko) * | 2012-07-13 | 2015-11-18 | 솔젠트 (주) | 중합효소 연쇄반응을 이용한 분자진단검사용 중합효소 및 보조효소의 dna 오염 제거를 위한 정제 방법 |
| DE102012014981B3 (de) | 2012-07-26 | 2013-07-18 | Sartorius Stedim Biotech Gmbh | Verfahren zur Differenzierung zwischen lebenden und toten Zellen |
| CN102851381A (zh) * | 2012-09-21 | 2013-01-02 | 武汉真福医药科技发展有限公司 | 快速检测单增李斯特菌的lamp试剂盒 |
| CN104278024B (zh) * | 2013-07-05 | 2016-12-28 | 中国人民解放军军事医学科学院微生物流行病研究所 | 用于鉴定人类腺病毒55型的引物组合物以及它们的应用 |
| JP2015188343A (ja) * | 2014-03-27 | 2015-11-02 | セイコーエプソン株式会社 | バイオチップ |
| CN107058517B (zh) * | 2017-03-13 | 2021-05-28 | 新乡医学院第一附属医院 | 一种用于结核分枝杆菌感染检测的试剂盒及检测方法 |
| CN106995805A (zh) * | 2017-03-26 | 2017-08-01 | 海南大学 | 一种溶菌酶标记的工程化噬菌体快速检测微生物 |
| CN107326094A (zh) * | 2017-09-04 | 2017-11-07 | 重庆市畜牧科学院 | 基于环介导等温扩增检测鸡鼻炎型克雷伯氏菌的试剂盒及其检测方法 |
| CN108588188B (zh) * | 2018-04-28 | 2021-11-09 | 天津科技大学 | 一种混合微生物发酵过程中微生物定量检测方法 |
| CN109295174B (zh) * | 2018-10-22 | 2022-05-27 | 益善生物技术股份有限公司 | 一种用于检测真菌感染的试剂组、试剂盒以及检测方法 |
| TR201820388A2 (tr) | 2018-12-25 | 2019-01-21 | Tuerkiye Bilimsel Ve Teknolojik Arastirma Kurumu Tuebitak | Salmonellanin klasi̇k kültür metoduna alternati̇f hizli ve taşinabi̇li̇r mi̇kroakişkan tespi̇t si̇stemi̇ |
| JP7535312B2 (ja) * | 2019-02-28 | 2024-08-16 | デイ ゼロ ダイアグノスティックス, インコーポレイテッド | 核酸増幅用の臨床サンプルを調製する改善された方法 |
| US20220145369A1 (en) * | 2019-03-14 | 2022-05-12 | Merck Patent Gmbh | A novel sampling method for long-term monitoring of microbes |
| CN112063732B (zh) * | 2020-09-17 | 2022-05-31 | 扬州大学 | 一种能够特异性识别阪崎肠杆菌存活细胞的快速定量检测方法及其引物 |
| CN113652358B (zh) * | 2021-09-15 | 2023-09-12 | 合肥工业大学 | 一种基于竹红菌素的光动力处理抑制克罗诺杆菌生长的用途 |
| CN117487896B (zh) * | 2023-12-29 | 2024-06-25 | 清华大学 | 拮抗剂以及含有其的pcr预混液、pcr检测试剂盒和pcr检测方法 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2001008685A (ja) * | 1999-06-25 | 2001-01-16 | Shimadzu Corp | 核酸合成法 |
| WO2009022558A1 (ja) * | 2007-08-16 | 2009-02-19 | Kyushu University | 微生物検出法及び微生物検出キット |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB0008787D0 (en) | 2000-04-10 | 2000-05-31 | Norsk Naeringsmiddelforskning | A method of cell detection |
| WO2002052034A1 (fr) | 2000-12-26 | 2002-07-04 | Joji Oshima | Methodes de bioscopie et d'amplification d'acides nucleiques |
| WO2004104196A1 (ja) | 2003-05-20 | 2004-12-02 | G & G Science Co., Ltd. | 緩衝剤組成物 |
| KR101047750B1 (ko) | 2005-07-21 | 2011-07-07 | 모리나가 뉴교 가부시키가이샤 | 미생물 검출법 및 미생물 검출 키트 |
| KR101010122B1 (ko) | 2006-02-17 | 2011-01-24 | 모리나가 뉴교 가부시키가이샤 | 미생물 검출법 및 미생물 검출 키트 |
-
2010
- 2010-07-23 NZ NZ597138A patent/NZ597138A/xx not_active IP Right Cessation
- 2010-07-23 EP EP10802360.7A patent/EP2458002B1/en active Active
- 2010-07-23 WO PCT/JP2010/062474 patent/WO2011010740A1/ja active Application Filing
- 2010-07-23 MX MX2012001000A patent/MX2012001000A/es not_active Application Discontinuation
- 2010-07-23 CN CN201410067344.4A patent/CN103820578B/zh not_active Expired - Fee Related
- 2010-07-23 SG SG2012005013A patent/SG177738A1/en unknown
- 2010-07-23 AU AU2010275576A patent/AU2010275576B2/en not_active Ceased
- 2010-07-23 CA CA2768699A patent/CA2768699C/en active Active
- 2010-07-23 JP JP2010529962A patent/JP4825313B2/ja active Active
- 2010-07-23 KR KR1020127004714A patent/KR101383389B1/ko active IP Right Grant
- 2010-07-23 US US13/386,601 patent/US9394572B2/en active Active
- 2010-07-23 CN CN201080033156.XA patent/CN102471768B/zh not_active Expired - Fee Related
- 2010-07-23 RU RU2012106617/10A patent/RU2527897C2/ru active
-
2016
- 2016-06-21 US US15/188,755 patent/US10329604B2/en active Active
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2001008685A (ja) * | 1999-06-25 | 2001-01-16 | Shimadzu Corp | 核酸合成法 |
| WO2009022558A1 (ja) * | 2007-08-16 | 2009-02-19 | Kyushu University | 微生物検出法及び微生物検出キット |
Non-Patent Citations (4)
| Title |
|---|
| ABU AI-SOUD W. ET AL.: "Effects of Amplification Facilitators on Diagnostic PCR in the Presence of Blood, Feces, and Meat", J. CLIN. MICROBIOL., vol. 38, no. 12, 2000, pages 4463 - 4470, XP003016479 * |
| KREADER CA ET AL.: "Relief of amplification inhibition in PCR with bovine serum albumin or T4 gene 32 protein", APPL. ENVIRON. MICROBIOL., vol. 62, no. 3, 1996, pages 1102 - 1106, XP002439274 * |
| See also references of EP2458002A4 * |
| TAKASHI FUKUSHIMA ET AL.: "A method to detect only live bacteria during PCR amplification", BIO IND, vol. 25, no. 9, 2008, pages 85 - 93, XP008150360 * |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP4083230A1 (en) * | 2011-07-06 | 2022-11-02 | Quest Diagnostics Investments Incorporated | Direct amplification and detection of viral and bacterial pathogens |
| US11851720B2 (en) | 2011-07-06 | 2023-12-26 | Quest Diagnostics Investments Llc | Direct amplification and detection of viral and bacterial pathogens |
| JP2015139434A (ja) * | 2014-01-30 | 2015-08-03 | 森永乳業株式会社 | 微生物検出法及び微生物検出キット |
| WO2017010001A1 (ja) * | 2015-07-16 | 2017-01-19 | 森永乳業株式会社 | 微生物検出法及び微生物検出キット |
| JP2018068211A (ja) * | 2016-10-28 | 2018-05-10 | 森永乳業株式会社 | 微生物の死細胞及び/又は不活化ウイルスの測定方法 |
| WO2019188552A1 (ja) * | 2018-03-27 | 2019-10-03 | 森永乳業株式会社 | 微生物の細胞及び/又はウイルスの測定方法 |
| WO2020218557A1 (ja) * | 2019-04-26 | 2020-10-29 | bitBiome株式会社 | 単一生物単位の生菌由来核酸の選択的検出、カウント、ゲノム解析 |
| US20210292823A1 (en) * | 2020-03-23 | 2021-09-23 | Feng Zhang | Rapid diagnostics |
| US11639523B2 (en) | 2020-03-23 | 2023-05-02 | The Broad Institute, Inc. | Type V CRISPR-Cas systems and use thereof |
| US11851702B2 (en) * | 2020-03-23 | 2023-12-26 | The Broad Institute, Inc. | Rapid diagnostics |
Also Published As
| Publication number | Publication date |
|---|---|
| KR101383389B1 (ko) | 2014-04-08 |
| EP2458002A4 (en) | 2013-01-30 |
| JPWO2011010740A1 (ja) | 2013-01-07 |
| CN102471768B (zh) | 2014-07-23 |
| CA2768699C (en) | 2017-08-22 |
| CN102471768A (zh) | 2012-05-23 |
| AU2010275576B2 (en) | 2013-02-28 |
| RU2527897C2 (ru) | 2014-09-10 |
| US9394572B2 (en) | 2016-07-19 |
| US20120122082A1 (en) | 2012-05-17 |
| NZ597138A (en) | 2012-12-21 |
| EP2458002B1 (en) | 2015-03-25 |
| US10329604B2 (en) | 2019-06-25 |
| CA2768699A1 (en) | 2011-01-27 |
| EP2458002A1 (en) | 2012-05-30 |
| CN103820578B (zh) | 2015-06-17 |
| RU2012106617A (ru) | 2013-08-27 |
| MX2012001000A (es) | 2012-03-16 |
| JP4825313B2 (ja) | 2011-11-30 |
| AU2010275576A1 (en) | 2012-01-19 |
| CN103820578A (zh) | 2014-05-28 |
| SG177738A1 (en) | 2012-02-28 |
| KR20120048647A (ko) | 2012-05-15 |
| US20160298181A1 (en) | 2016-10-13 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4825313B2 (ja) | 微生物検出法及び微生物検出キット | |
| JP4378537B2 (ja) | 微生物検出法及び微生物検出キット | |
| US9567625B2 (en) | Method for detection of microorganism and kit for detection of microorganism | |
| WO2017010001A1 (ja) | 微生物検出法及び微生物検出キット | |
| JP6139425B2 (ja) | 微生物検出法及び微生物検出キット | |
| JP4217797B2 (ja) | 微生物検出法及び微生物検出キット | |
| JP4217795B2 (ja) | 微生物検出法及び微生物検出キット | |
| JP4217796B2 (ja) | 微生物検出法及び微生物検出キット | |
| NZ564847A (en) | Method for detection of microorganism and kit for detection of microorganism | |
| WO2017009999A1 (ja) | 微生物検出法及び微生物検出キット |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201080033156.X Country of ref document: CN |
|
| ENP | Entry into the national phase |
Ref document number: 2010529962 Country of ref document: JP Kind code of ref document: A |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10802360 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010275576 Country of ref document: AU |
|
| ENP | Entry into the national phase |
Ref document number: 2010275576 Country of ref document: AU Date of ref document: 20100723 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2768699 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12012500144 Country of ref document: PH |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13386601 Country of ref document: US Ref document number: MX/A/2012/001000 Country of ref document: MX |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1201000222 Country of ref document: TH |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010802360 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1571/CHENP/2012 Country of ref document: IN |
|
| ENP | Entry into the national phase |
Ref document number: 20127004714 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012106617 Country of ref document: RU |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112012001657 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 112012001657 Country of ref document: BR Kind code of ref document: A2 Effective date: 20120124 |