WO2010104078A1 - ジアミン誘導体の製造方法 - Google Patents
ジアミン誘導体の製造方法 Download PDFInfo
- Publication number
- WO2010104078A1 WO2010104078A1 PCT/JP2010/053905 JP2010053905W WO2010104078A1 WO 2010104078 A1 WO2010104078 A1 WO 2010104078A1 JP 2010053905 W JP2010053905 W JP 2010053905W WO 2010104078 A1 WO2010104078 A1 WO 2010104078A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- added
- tertiary amine
- represented
- following formula
- Prior art date
Links
- 0 CC(C)(C)OC(*[C@](C1)[C@@](*C(C(*c(cc2)ncc2Cl)=O)=O)CC[C@@]1C(N(*)*)=O)=O Chemical compound CC(C)(C)OC(*[C@](C1)[C@@](*C(C(*c(cc2)ncc2Cl)=O)=O)CC[C@@]1C(N(*)*)=O)=O 0.000 description 4
- LSQWUGYWDZRKNW-UHFFFAOYSA-N CCOC(C(Nc(cc1)ncc1Cl)=O)=O Chemical compound CCOC(C(Nc(cc1)ncc1Cl)=O)=O LSQWUGYWDZRKNW-UHFFFAOYSA-N 0.000 description 2
- HGVDHZBSSITLCT-JLJPHGGASA-N CN(C)C([C@@H](CC[C@@H]1NC(C(Nc(cc2)ncc2Cl)=O)=O)C[C@H]1NC(c1nc(CCN(C)C2)c2[s]1)=O)=O Chemical compound CN(C)C([C@@H](CC[C@@H]1NC(C(Nc(cc2)ncc2Cl)=O)=O)C[C@H]1NC(c1nc(CCN(C)C2)c2[s]1)=O)=O HGVDHZBSSITLCT-JLJPHGGASA-N 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N Cc(cc1)ccc1S(O)(=O)=O Chemical compound Cc(cc1)ccc1S(O)(=O)=O JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
Images


Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/72—Nitrogen atoms
- C07D213/75—Amino or imino radicals, acylated by carboxylic or carbonic acids, or by sulfur or nitrogen analogues thereof, e.g. carbamates
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D513/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00
- C07D513/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00 in which the condensed system contains two hetero rings
- C07D513/04—Ortho-condensed systems
Definitions
- the present invention relates to an optically active diamine derivative which is important for the production of compound (A), a pharmacologically acceptable salt thereof or a hydrate thereof, which is an inhibitor of activated blood coagulation factor X (FXa) Relates to the industrial production method.
- the compound (A) represented by the following formula, a pharmacologically acceptable salt thereof or a hydrate thereof exhibits an inhibitory action of FXa, as disclosed in Patent Documents 1 to 3, It is a compound useful as a prophylactic / therapeutic agent for embolic diseases.
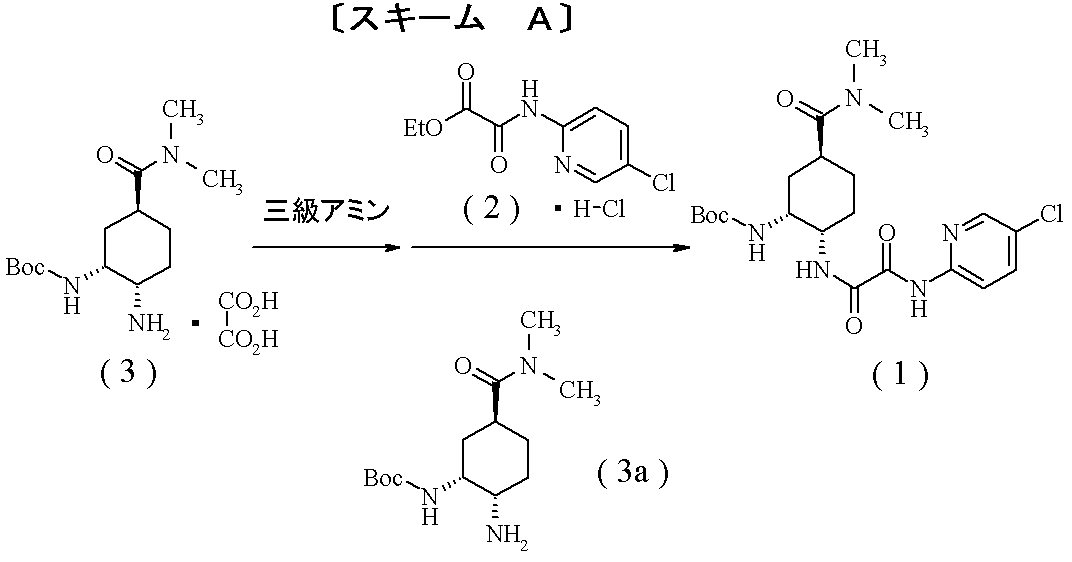
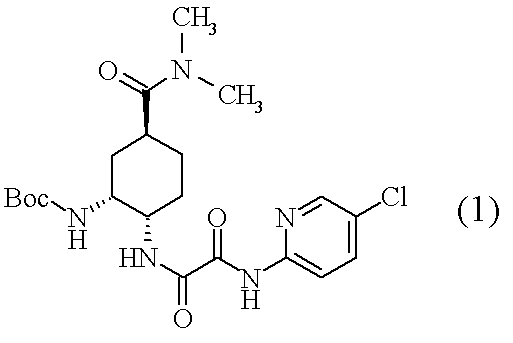
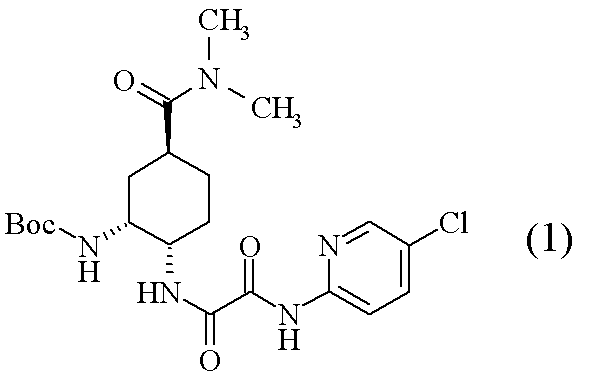
- Compound (1) which is an optically active diamine derivative described in the following scheme is known as a production intermediate of compound (A) which is an FXa inhibitor, a pharmacologically acceptable salt thereof or a hydrate thereof.
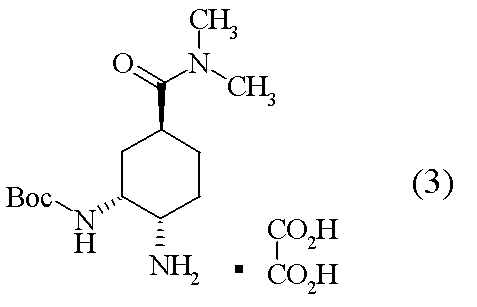
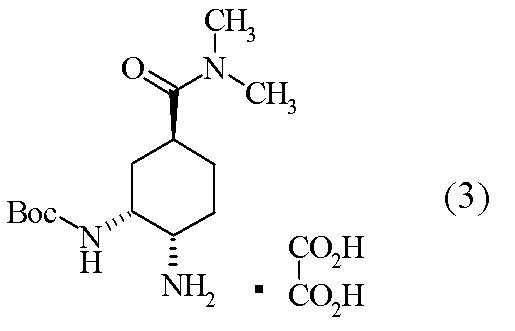
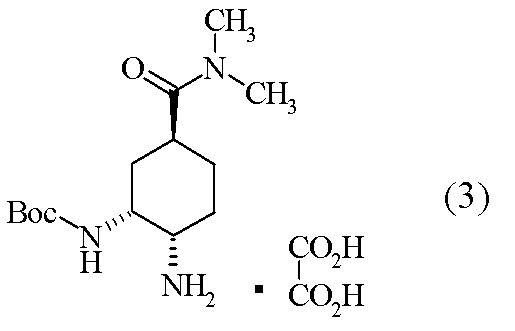
- compound (3) and an excess amount of a tertiary amine are added to neutralize the salt, and then compound (2) is added to produce compound (1). Is disclosed [Scheme A].
- Boc represents a tert-butoxycarbonyl group.
- the inventors of the present invention aim to obtain a new industrial production method of compound (1) by avoiding a significant decrease in yield due to solidification of the reaction system, which is a problem of the conventional production method of compound (1). And intensively studied. As a result of many years of research, compound (2) is first treated with a tertiary amine, followed by addition of compound (3). The order of addition is important; and further, the decomposition of the free form of compound (2) is prevented. Therefore, the present invention has been completed by discovering that a method of adding a tertiary amine in a divided manner is effective.
- the present invention provides the following (1) to (21).
- Boc represents the same as described above.
- a process for producing compound (1) characterized in that (2) The production method according to (1), wherein the C2-C4 nitrile solvent is acetonitrile. (3) The production method according to (1) or (2), wherein the amount of the compound (2) is 1.10 to 1.27 molar equivalent stoichiometrically with respect to the compound (3).
- the tertiary amine is one or two selected from the group consisting of tri (C1-C4 alkyl) amines, 1- (C1-C3 alkyl) piperidine and 4- (C1-C3 alkyl) morpholine
- the production method according to any one of (1) to (3) above.
- [Operation 1] is a compound (2) represented by the following formula in a C2-C4 nitrile solvent:
- Boc represents a tert-butoxycarbonyl group.
- a process for producing a compound (Aa) comprising a step of treating a compound (A) with p-toluenesulfonic acid or a hydrate thereof to obtain a compound (Aa).
- compound (1) which is a production intermediate of compound (A) useful as an inhibitor of FXa, avoids a decrease in reaction yield due to solidification of the reaction system, which is a problem of conventional methods. , 93 to 95% can be produced with high yield. Therefore, the production method of the present invention is useful as a method for producing a compound (A) useful as an FXa inhibitor.
- Compound (A) may be a free form (free base) or a hydrate thereof, and compound (A) may be a pharmacologically acceptable salt or a hydrate of the salt.
- the pharmacologically acceptable salts of compound (A) include hydrochloride, sulfate, hydrobromide, hydroiodide, phosphate, nitrate, benzoate, methanesulfonate, 2-hydroxyethane Sulfonate, p-toluenesulfonate, acetate, propanoate, oxalate, malonate, succinate, glutarate, adipate, tartrate, maleate, fumarate, apple And acid salts and mandelate salts.
- hydrochloride or p-toluenesulfonate is preferable; p-Toluenesulfonate is particularly preferred.
- Compound (1) which is an optically active amide derivative of the present invention can be produced by operating in the order of [Operation 1] and [Operation 2] as shown in the following scheme.
- Boc represents the same as described above.
- Examples of the C2-C4 nitrile solvent in [Operation 1] include acetonitrile, propionitrile, butyronitrile and the like. Of these, acetonitrile is preferred as the reaction solvent.
- tertiary amines composed of the same or different C1-C8 alkyl groups, 1- (C1-C3 alkyl) Pyrrolidine, 1- (C1-C3 alkyl) piperidine, 4- (C1-C3 alkyl) morpholine, 1,8-diazabicyclo [5.4.0] undec-7-ene (DBU), 1,5-diazabicyclo [4 .3.0] non-5-ene (DBN), di (C1-C3 alkyl) aniline, 4-di (C1-C3 alkyl) aminopyridine, and the like.
- alkylamines composed of the same or different C1-C8 alkyl groups, 1- (C1-C3 alkyl) Pyrrolidine, 1- (C1-C3 alkyl) piperidine, 4- (C1-C3 alkyl) morpholine, 1,8-diazabicyclo [5.4.0] undec-7-ene (DBU), 1,5-diazabicyclo
- tertiary amines include trimethylamine, triethylamine, ethyl (diisopropyl) amine, 1-methylpiperidine, 1-ethylpiperidine, 4-methylmorpholine, N, N-dimethylaniline, and N, N-diethylaniline. Is preferred.
- [Operation 1] is an operation for preparing a neutralized compound (2a) represented by the following formula by treating compound (2) with a tertiary amine in a C2-C4 nitrile solvent.
- [Operation 1] More specific preferred embodiments of [Operation 1] include the following [Operation 1 (Method A)] to [Operation 1 (Method C)].
- the amount of the C2-C4 nitrile solvent is preferably in the range of 4 to 6 parts by volume with respect to 1 part by weight of the compound (3).
- the amount of compound (2) used is preferably 1.1 to 1.3 molar equivalents relative to compound (3) stoichiometrically; more preferably about 1.2 molar equivalents.
- the temperature of the reaction solution is preferably 45 ° C. or lower; more preferably in the range of 0 to 35 ° C.
- tertiary amine those described above can be used, but triethylamine is preferable.
- the amount of tertiary amine added is preferably from stoichiometrically 0.87 to 1.08 molar equivalents relative to compound (2); more preferably about 0.98 molar equivalents.
- the amount of tertiary amine added is preferably stoichiometrically 3.2 molar equivalents or more with respect to compound (3); more preferably in the range of 4.9 to 6.5 molar equivalents. In Method C, no additional tertiary amine is added after [Operation 2].
- method A and method B are preferred.
- [Operation 2] in the present invention is an operation of adding the compound (3) represented by the following formula to the mixed solution of [Operation 1].
- Boc represents a tert-butoxycarbonyl group.
- the starting point of addition of compound (3) in Method C is preferably started within 15 minutes after the addition of the tertiary amine; more preferably after the addition of the tertiary amine; addition of the tertiary amine It is more preferable to add the tertiary amine and the compound (3) at the same time when the addition of 1/5 or more of the total amount is completed.
- Method A or Method B When Method A or Method B is employed, a tertiary amine is added to the mixed solution of [Operation 2] with an internal temperature of the mixed solution of 50 to 65 ° C., and then the reaction mixed solution is added to 60 to 80 Stir at °C. After completion of the reaction, the reaction mixture is cooled, water is added, and the crystallized crystals are collected to obtain compound (1).
- the amount of additional tertiary amine added in Method A or Method B is preferably in the range of 3.23 to 4.32 molar equivalents relative to the stoichiometric compound (2) [Addition of tertiary amine Total amount: 4.9 to 6.5 molar equivalents relative to stoichiometric compound (3)].
- the oxalate compound (3) is converted to the free base (free form) compound (3a).
- Boc represents a tert-butoxycarbonyl group.
- the reaction mixture is stirred at 60-80 ° C.
- the stirring time is preferably 6 hours or more; more preferably about 6 to 24 hours.
- the end point of the reaction may be confirmed using a sample of the reaction solution using an instrument such as HPLC.
- the above method A is particularly preferable.
- Specific embodiments of the production of compound (1) using method A are as follows. Compound (2) is added to acetonitrile, 0.87 to 1.08 molar equivalent of triethylamine is added stoichiometrically to compound (2), and then compound (3) is added to the mixture. Subsequently, an additional 3.23 to 4.32 molar equivalents of a tertiary amine stoichiometrically with respect to compound (2) was added to the reaction mixture at 50 to 65 ° C., and the mixture was added at 60 to 80 ° C. for 6 hours. By stirring above, the compound (1) is produced.
- the reaction mixture is cooled, water is added and stirred to complete crystal crystallization.
- the cooling of the reaction mixture is preferably 0 to 60 ° C .; more preferably 0 to 50 ° C.
- the amount of water added is preferably in the range of 6 to 11 parts by volume [6 to 11 (V / W)] with respect to 1 part by weight of the compound (3).
- the crystallized precipitate is completed under cooling, the crystallized crystal is collected by filtration, washed with water and then dried.
- the crystals may be collected by filtration under normal atmospheric pressure or reduced pressure filtration.
- the crystallization temperature is preferably ⁇ 10 to 35 ° C.
- the amount of water is preferably about 5 parts by volume [5 (V / W)] per 1 part by weight of the compound (3).
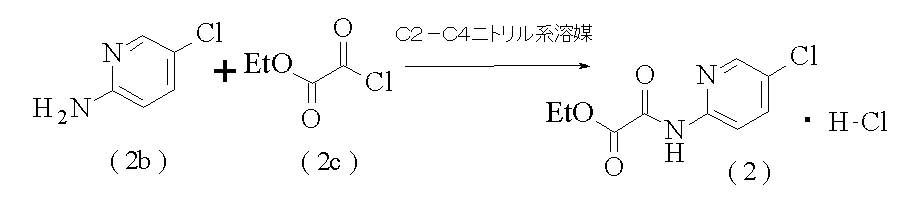
- the compound (2) used in [Operation 1] can be produced as shown below, and specific examples of the production include the method described in Reference Example 2. That is, compound (2) can be produced by adding compound (2c) to commercially available aniline derivative compound (2b) in a C2-C4 nitrile solvent with stirring.
- Examples of the C2-C4 nitrile solvent for this reaction include acetonitrile, propionitrile and butyronitrile, and acetonitrile is preferred.
- the amount of the solvent used is preferably in the range of 10 to 17 parts by volume [10 to 17 (V / W)] with respect to 1 part by weight of the compound (2b).
- the amount of compound (2c) used is preferably in the range of 1.06 to 1.21 molar equivalents relative to compound (2b) stoichiometrically.
- the reaction temperature is preferably in the range of 40 to 80 ° C.
- the reaction time is preferably 1 hour or longer; preferably about 1 to 7 hours.
- the compound (2) thus produced can be isolated by filtration as crystals by crystallization.
- the crystallization temperature is preferably in the range of ⁇ 25 to 35 ° C.
- the crystallized compound (2) can be isolated by filtration. When the compound (2) is used in the following [Operation 1], the filtered compound (2) may be dried under normal pressure or reduced pressure and used as a dry product (dry product); a wet product (wet product) Body).
- Compound (A) useful as an inhibitor of FXa using compound (1) of the present invention and compound (Aa) which is mono-p-toluenesulfonate monohydrate of compound (A)
- a known method disclosed in Patent Document 1 or Patent Document 3 may be used as a method for producing A, and specifically, as shown in the following scheme and Reference Examples 4 and 5 described later.
- Boc represents a tert-butoxycarbonyl group.
- Boc represents the same as described above.
- 28% aqueous ammonia solution (5 ml) was added to (1S, 3S, 6R) -N, N-dimethyl-7-oxabicyclo [4.1.0] heptane-3-carboxamide (1 g) at room temperature. After stirring at 40 ° C. for 5 hours, the solvent was concentrated under reduced pressure to obtain a crude product (1.18 g) of (1S, 3R, 4R) -3-amino-4-hydroxy-N, N-dimethylcyclohexanecarboxamide.
- the obtained crude product was dissolved in water (5 ml), and di-tert-butyl dicarbonate (1.93 g) and 10N aqueous sodium hydroxide solution (1.5 ml) were added at room temperature. After stirring at 40 ° C. for 2 hours, the mixture was extracted 3 times with 4-methyl-2-pentanone (MIBK) (5 ml), and the extraction solvent was distilled off under reduced pressure to give 4-methyl-2-pentanone (MIBK) to the residue. (3 ml) was added and stirred at room temperature. The precipitated crystals were collected by filtration and dried to give the title compound (1.26 g).
- MIBK 4-methyl-2-pentanone
- Ms represents a methanesulfonyl group; Boc represents the same as described above.
- MIBK 4-methyl-2-pentanone
- Boc represents the same as described above.
- DMAC N-dimethylacetamide
- a mixed solvent (300 ml) of n-hexane-ethyl acetate (5: 1) was added to the concentrated residue, and the mixture was stirred at room temperature for 1 hour. Crystals were collected by filtration. To the obtained crystals, a mixed solvent (300 ml) of n-hexane-acetic acid ethyl ester (5: 1) was added, and the operation of stirring and crystal filtration was repeated twice to obtain the title compound (4.6 g).
- Boc represents the same as described above.
- Boc represents the same as described above.
- tert-butyl ⁇ (1R, 2S, 5S) -2-amino-5-[(dimethylamino) carbonyl] cyclohexyl ⁇ carbamate oxalate (1) (100.1 g) in acetonitrile suspension (550 ml) at 60 ° C. Triethylamine (169 ml) was added. At the same temperature, ethyl [5-chloropyridin-2-yl] amino] (oxo) acetate 1 ⁇ hydrochloride 82) (84.2 g) was added, and the mixture was stirred for 6 hours and then at room temperature for 16 hours.
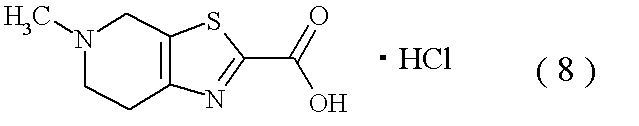
- the reaction mixture was triethylamine (155 ml), 5-methyl-4,5,6,7-tetrahydro [1,3] thiazolo [5,4-c] pyridine-2-carboxylic acid / hydrochloride (8) (52.5 g), 1-hydroxybenzotriazole (33.0 g) and 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide hydrochloride (46.8 g) were added, and the mixture was stirred at room temperature for 16 hours. Triethylamine and water were added, and the mixture was stirred for 1 hour under ice-cooling. The crystals were collected by filtration to give the title compound (103.2 g).
- Boc represents a tert-butoxycarbonyl group.
- 2-Amino-5-chloropyridine (2b) (5.0 g) was added to acetonitrile (60 ml), ethyl oxalyl chloride (2c) (5.9 g) was added at 50 ° C., and the mixture was stirred at the same temperature for 2 hours. .
- the reaction mixture was cooled and the crystals were collected by filtration at 10 ° C. to obtain ethyl [5-chloropyridin-2-yl] amino] (oxo) acetate 1 hydrochloride (2) (9.8 g).
- Test Example 2 From the results of Test Example 1, it was clarified that there is a risk of deterioration in the quality of Compound (1) (Condition 2 of Test Example 1) in the method of adding Compound (3) first, which is a conventional method.
- the compound (2) and triethylamine In order to confirm the influence on the yield and quality of the compound (1) when the addition order of the compound (3), the compound (2) and triethylamine is changed, in addition to the condition 1 of Test Example 1, the following ( The production rates of compound (1) and impurity X in conditions 4) and (condition 5) were measured. The results are shown in Table 2.
- Test Example 3 In Condition 5 (Example 2) of Test Example 2 described above, the influence of time until addition of compound (3) after addition of triethylamine (TEA) was investigated. The results are shown in Table 3.
- Test Example 5 From the results of Test Example 4, since the decomposition of the compound (2a) was promoted by the coexistence of a large excess of triethylamine, the amount of triethylamine added was reduced and the stability of the compound (2a) was evaluated.
- Condition 9 Comparative Example
- Conditions 10 to 12 indicate the production rate of compound (1) and impurity X when triethylamine was added in portions, and the presence or absence of severe smoke during the addition of triethylamine.
- Compound (2) (8.5 g) was added to acetonitrile (50 ml), and triethylamine was added for the first time while stirring at 10 ° C. (amount in Table 5). After completion of the addition, compound (3) (10 g) was added, and then triethylamine was added a second time at 60 ° C. (the total amount of triethylamine is 5.2 molar equivalents of compound (2)).
- the production method of the present invention can be used as a novel industrial production method of a compound (A) useful as an inhibitor of FXa, a pharmacologically acceptable salt thereof, or a hydrate thereof.
Abstract
Description

そこで、上記の問題点を解決し、化合物(A)製造の重要合成中間体である化合物(1)の新規な工業的製造方法を提供することが課題となった。
本発明者らは、化合物(1)の従来の製造方法の問題点である、反応系の固化による収率の大幅低下を回避し、化合物(1)の新規な工業的製造方法の獲得を目指して鋭意検討を行った。
長年にわたる研究の結果、はじめに化合物(2)を三級アミンと処理し、引き続いて化合物(3)を添加する、添加の順番が重要であること;さらに化合物(2)のフリー体の分解を防ぐために三級アミンを分割して添加する方法が有効であること;を見出して本発明を完成した。
(1):下記の式で表される化合物(1)
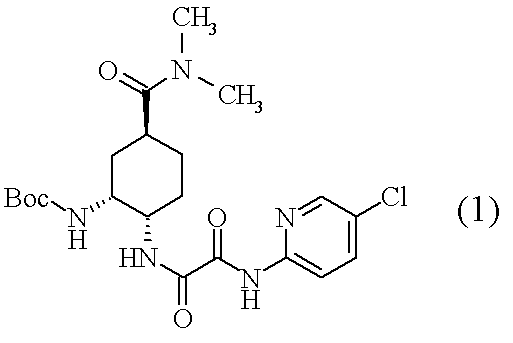
の製造方法であって、
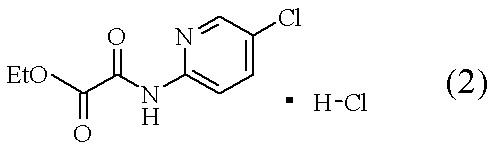
[操作1]:C2-C4ニトリル系溶媒中、下記の式で表される化合物(2)
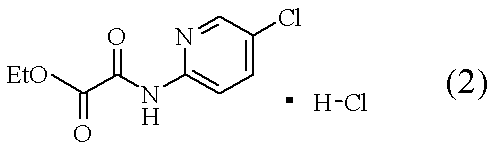
[操作2]:上記の[操作1]の混合液に、下記の式で表される化合物(3)

を添加することを特徴とした、化合物(1)の製造方法。
(2):C2-C4ニトリル系溶媒が、アセトニトリルである(1)に記載の製造方法。
(3):化合物(2)の量が、化学量論的に化合物(3)に対して1.10~1.27モル当量である(1)又は(2)に記載の製造方法。
(4):三級アミンが、トリ(C1-C4アルキル)アミン類、1-(C1-C3アルキル)ピペリジン及び4-(C1-C3アルキル)モルホリンからなる群より選ばれる1種、又は2種以上である(1)~(3)のいずれか1に記載の製造方法。
(5):三級アミンが、トリ(C1-C4アルキル)アミン類から選ばれる1種、又は2種以上である(1)~(3)のいずれか1に記載の製造方法。
(6):三級アミンが、トリエチルアミンである(1)~(3)のいずれか1に記載の製造方法。
(7):[操作1]が、C2-C4ニトリル系溶媒中に、下記の式で表される化合物(2)

(8):[操作1]が、C2-C4ニトリル系溶媒中に三級アミンを加え、次いで下記の式で表される化合物(2)
(9):化合物(2)又は三級アミンの添加が、45℃以下で行うものである(7)又は(8)に記載の製造方法。
(10):化合物(2)又は三級アミンの添加が、0~35℃の範囲で行うものである(7)又は(8)に記載の製造方法。
(11):[操作1]における三級アミンの添加量が、化学量論的に化合物(2)に対して0.87~1.08モル当量である(1)~(10)のいずれか1に記載の製造方法。
(12):[操作2]の操作の後に、さらに三級アミンを追加して添加するものである(1)~(11)のいずれか1に記載の製造方法。
(13):三級アミンを追加して添加する量が、化学量論的に化合物(2)に対して3.02~4.53モル当量の範囲である(12)の製造方法。
(14):50~65℃で三級アミンを追加して添加するものである(12)又は(13)の製造方法。
(15):三級アミンを追加して添加した後、反応混合物を60~80℃で撹拌するものである(12)~(14)ののいずれか1項に記載の製造方法。
(16):撹拌時間が6時間以上である(15)に記載の製造方法。
(17):撹拌時間が6~24時間の範囲である(15)に記載の製造方法。
(18):下記の式で表される化合物(1)の製造方法であって、

[操作1]:アセトニトリル中、下記の式で表される化合物(2)

[操作2]:上記の[操作1]の混合液に、下記の式で表される化合物(3)

を添加した後、50~65℃で化学量論的に化合物(2)に対して3.23~4.32モル当量の三級アミンを追加して添加し、60~80℃で6時間以上撹拌することを特徴とした化合物(1)の製造方法。
(19):下記の式で表される化合物(1)の製造方法であって、

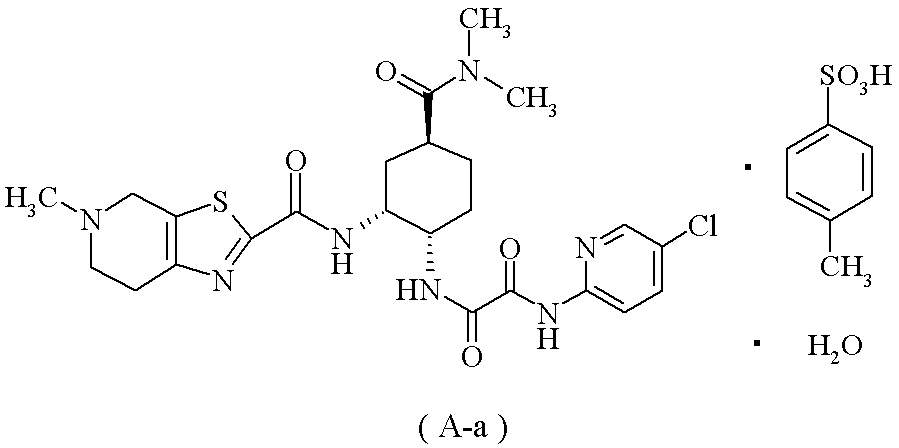
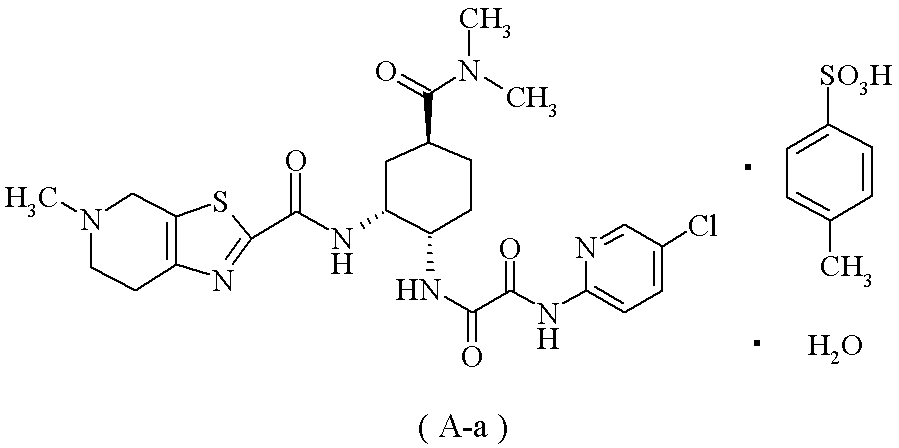
下記の式で表される化合物(A-a)
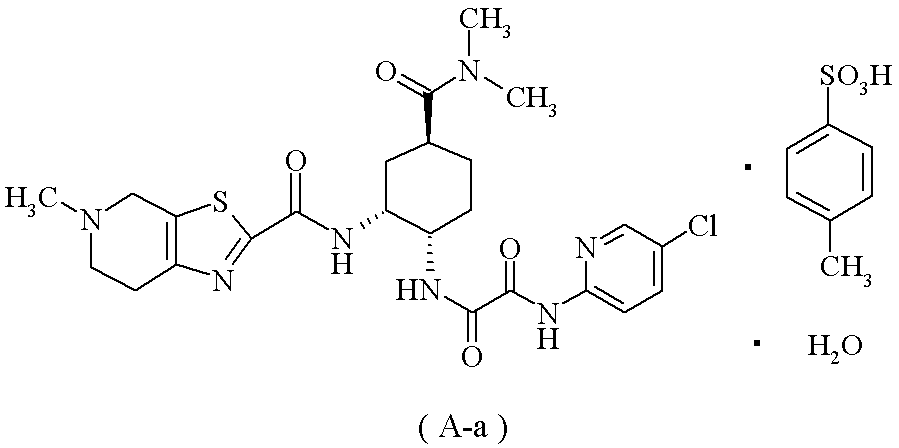
(20):(1)に記載の製造方法により製造した化合物(1)を使用することを特徴とする、下記の式で表される化合物(A-a)の製造方法。

のBoc基を脱保護して下記の式で表される化合物(7)又はその塩を得る工程、

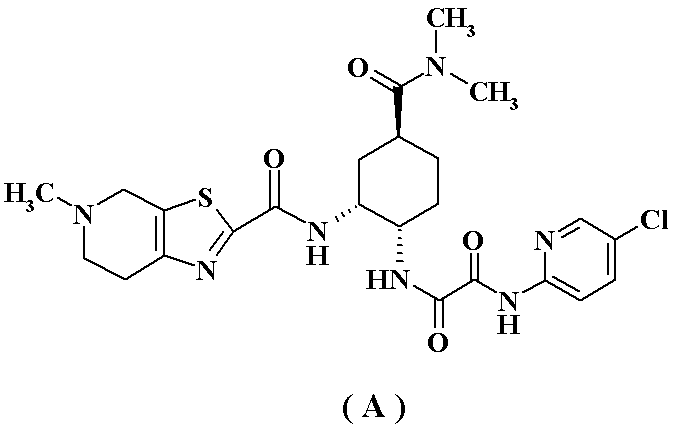
p-トルエンスルホン酸塩が特に好ましい。
N1-(5-クロロピリジン-2-イル)-N2-((1S,2R,4S)-4-[(ジメチルアミノ)カルボニル]-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}シクロヘキシル)エタンジアミド;
N1-(5-クロロピリジン-2-イル)-N2-((1S,2R,4S)-4-[(ジメチルアミノ)カルボニル]-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}シクロヘキシル)エタンジアミド・塩酸塩;
N1-(5-クロロピリジン-2-イル)-N2-((1S,2R,4S)-4-[(ジメチルアミノ)カルボニル]-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}シクロヘキシル)エタンジアミド・モノp-トルエンスルホン酸塩;及び
N1-(5-クロロピリジン-2-イル)-N2-((1S,2R,4S)-4-[(ジメチルアミノ)カルボニル]-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}シクロヘキシル)エタンジアミド・モノp-トルエンスルホン酸塩・1水和物;
が好ましく;N1-(5-クロロピリジン-2-イル)-N2-((1S,2R,4S)-4-[(ジメチルアミノ)カルボニル]-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}シクロヘキシル)エタンジアミド・モノp-トルエンスルホン酸塩・1水和物(A-a)が特に好ましい。


[操作1]:C2-C4ニトリル系溶媒中、下記の式で表される化合物(2)

[操作2]:上記の[操作1]の混合液に、下記の式で表される化合物(3)

を添加して、化合物(1)を製造する。

[操作1(A法)]:「C2-C4ニトリル系溶媒中に化合物(2)を加え、その後45℃以下で三級アミンを添加する。」
C2-C4ニトリル系溶媒の量としては、化合物(3)の1重量部に対し、4~6容量部の範囲が好ましい。化合物(2)の使用量としては、化学量論的に化合物(3)に対して1.1~1.3モル当量が好ましく;1.2モル当量程度がより好ましい。反応液の温度は45℃以下が好ましく;0~35℃の範囲がより好ましい。三級アミンとしては、上記に記載したものを使用することができるが、トリエチルアミンが好ましい。三級アミンの添加量としては、化学量論的に化合物(2)に対して0.87~1.08モル当量が好ましく;0.98モル当量程度がより好ましい。
[操作1(B法)]:「C2-C4ニトリル系溶媒中に三級アミンを加え、その後45℃以下で化合物(2)を添加する。」
C2-C4ニトリル系溶媒の量、化合物(2)の使用量、反応液の温度、使用する三級アミン、三級アミンの添加量、及び三級アミン又は化合物(2)を添加する温度はA法と同様である。
[操作1(C法)]:「C2-C4ニトリル系溶媒中に化合物(2)を加え、その後50~65℃の範囲に昇温して三級アミンを添加する。」
C2-C4ニトリル系溶媒の量、化合物(2)の使用量、及び使用する三級アミンはA法と同様である。三級アミンの添加量としては、化学量論的に化合物(3)に対して3.2モル当量以上が好ましく;4.9~6.5モル当量の範囲がより好ましい。C法では[操作2]の後の三級アミンの追加添加は行わない。
ここで、上記のC法を採用する場合は、三級アミンを加え終わってから、化合物(3)の添加開始までの時間が短いほど、反応溶液の固化による化合物(1)の収量低下を回避できる。C法における化合物(3)の添加開始時点としては、三級アミンの添加後、15分以内に添加を開始するのが好ましく;三級アミンの添加後と同時がより好ましく;三級アミンの添加総量の1/5以上の添加が終了した時点で、三級アミン添加と、化合物(3)の添加を同時に行うのがより好ましい。
反応終了後、反応混合液を冷却し、水を添加し、晶析した結晶を集めて化合物(1)を得る。
A法又はB法における三級アミンを追加して添加する量としては、化学量論的な化合物(2)に対し、3.23~4.32モル当量の範囲が好ましい[三級アミンの添加総量として、化学量論的な化合物(3)に対し、4.9~6.5モル当量]。

三級アミンを追加して添加した後、反応混合液を60~80℃で攪拌する。ここで、攪拌時間は、6時間以上が好ましく;6~24時間程度がより好ましい。また、反応の終点は反応液のサンプルをHPLC等の機器を用いて確認してもよい。
アセトニトリルに化合物(2)を加え、化学量論的に化合物(2)に対して0.87~1.08モル当量のトリエチルアミンを添加した後、混合液に化合物(3)を添加する。次いで、反応混合液に50~65℃で化学量論的に化合物(2)に対して3.23~4.32モル当量の三級アミンを追加して添加し、60~80℃で6時間以上撹拌して、化合物(1)を製造する。


[工程1] tert-ブチル{(1R,2R,5S)-5-[(ジメチルアミノ)カルボニル]-2-ヒドロキシシクロヘキシルカルボニル}カルバマート(4)の合成

(1S,3S,6R)-N,N-ジメチル-7-オクサビシクロ[4.1.0]ヘプタン-3-カルボキサミド(1g)に28%アンモニア水溶液(5ml)を室温下加えた。40℃にて5時間攪拌後、溶媒を減圧濃縮し、(1S,3R,4R)-3-アミノ-4-ヒドロキシ-N,N-ジメチルシクロヘキサンカルボキサミドの粗体(1.18g)を得た。得られた粗体を水(5ml)に溶解後、ジ-tert-ブチルジカルボネート(1.93g)、10N水酸化ナトリウム水溶液(1.5ml)を室温下、加えた。40℃にて、2時間攪拌後、4-メチル-2-ペンタノン(MIBK)(5ml)で3回抽出し、抽出溶媒を減圧留去して、残渣へ4-メチル-2-ペンタノン(MIBK)(3ml)を加え、室温下、攪拌した。析出した結晶を濾取後、乾燥して標題化合物(1.26g)を得た。
1H-NMR(CDCl3)δ:1.44(9H,s),1.48-1.59(2H,m)
,1.77-1.78(2H,m),1.86-1.97(1H,m),2.11-2.
17(1H,m),2.78-2.83(1H,m),2.92(3H,s),3.02
(3H,s),3.53-3.60(1H,m),3.94(1H,br.s),4.5
2-4.68(1H,m).
[工程2] (1R,2R,4S)-2-[(tert-ブトキシカルボニル)アミノ]-4-[(ジメチルアミノ)カルボニル]シクロヘキシルメタンスルホナート(5)の合成

tert-ブチル {(1R,2R,5S)-5-[(ジメチルアミノ)カルボニル]-2-ヒドロキシシクロヘキシルカルバマート(214.59g)の4-メチル-2-ペンタノン(MIBK)溶液(1875ml)に塩化メタンスルホニル(159.07g)を室温にて加えた。室温下、反応液にトリエチルアミン(170.62g)を加え、そのままの温度にて1時間攪拌した。反応液に水を加えた後、有機層を分取した。溶媒を減圧濃縮後、濃縮残渣にMIBK(750ml)を加え、室温下、3時間攪拌した。析出した結晶を濾取、乾燥して標題化合物(242.57g)を得た。
1H-NMR(CDCl3)δ:1.45(9H,s),1.58-1.66(1H,m),1.67-1.76(1H,m),1.84-1.96(2H,m),2.04-2.
15(1H,m),2.17-2.26(1H,m),2.75-2.81(1H,m),2.94(3H,s),3.04(3H,s),3.07(3H,s),4.00-4.08(1H,m),4.69-4.82(2H,m).
[工程3] tert-ブチル{(1R,2R,5S)-2-アジド-5-[(ジメチルアミノ)カルボニル]シクロヘキシル}カルバマート(6)の合成
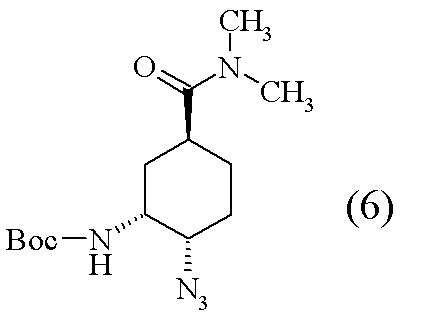
(1R,2R,4S)-2-[(tert-ブトキシカルボニル)アミノ]-4-[(ジメチルアミノ)カルボニル]シクロヘキシルメタンスルホナート(20.0g)のN,N-ジメチルアセトアミド(DMAC)溶液(40ml)に室温にて、アジ化ナトリウム(7.14g)及びドデシルピリジニウム クロリド(7.80g)を加えた。60℃にて、72時間攪拌後、反応液に水、酢酸エチルエステルを加え、有機層を飽和重層水、水にて洗浄後、溶媒を減圧濃縮した。濃縮残渣にn-ヘキサン-酢酸エチルエステル(5:1)の混合溶媒(300ml)を加え、室温下、1時間攪拌した。結晶を濾取した。得られた結晶をn-ヘキサン-酢酸エチルエステル(5:1)の混合溶媒(300ml)を加えて攪拌・結晶濾過の操作を2回繰り返し、標題化合物(4.6g)を得た。
1H-NMR(CDCl3)δ:1.46(9H,s),1.55-1.74(3H,m),1.75-1.82(1H,m),2.02-2.12(2H,m),2.74-2.83(1H,m),2.93(3H,s),3.02(3H,s),3.72-3.78(1H,m),4.07-4.13(1H,m),4.61-4.66(1H,m).
[工程4] tert-ブチル {(1R,2S,5S)-2-アミノ-5-[(ジメチルアミノ)カルボニル]シクロヘキシル}カルバマート オキサレート(3)の合成

(1R,2R,4S)-2-[(tert-ブトキシカルボニル)アミノ]-4-[(ジメチルアミノ)カルボニル]シクロヘキシルメタンスルホナート(20.0g)のトルエン溶液(100ml)に、室温にて、アジ化ナトリウム(7.14g)及びドデシルピリジニウム クロリド(7.80g)を加えた。反応混合液を60℃にて、72時間攪拌後、反応液に水を加え、有機層を飽和重層水、水にて洗浄後、有機層を分離した。有機層にメタノールを加え、7.5%Pd-C及びギ酸アンモニウムを加えて40℃で1時間攪拌した。Pd-Cをろ過除去後、溶媒を減圧濃縮した。残渣に含水アセトニトリル(200ml)及び無水シュウ酸(4.94g)を加え、室温下17時間攪拌し、結晶を濾取した。得られた結晶をアセトニトリル(200ml)に加えて、40℃にて24時間攪拌した。得られた結晶を濾取、乾燥して標題化合物(12.7g)を得た。
1H-NMR(D2O)δ:1.30(9H,s),1.37-1.49(2H,m),1.63(1H,t,J=2.7Hz),1.72-1.83(3H,m),2.77(3H,s)2.80(1H,t,J=12.4Hz),2.96(3H,m),3.32(1H,d,J=12.2Hz),4.10(1H,br).
(参考例2) 2-[(5-クロロピリジン-2-イル)アミノ]-2-オキソアセタート 1・塩酸塩(2)


tert-ブチル {(1R,2S,5S)-2-アミノ-5-[(ジメチルアミノ)カルボニル]シクロヘキシル}カルバマート オキサレート(1)(100.1g)のアセトニトリル懸濁液(550ml)に、60℃にてトリエチルアミン(169ml)を加えた。そのままの温度にて、エチル[5-クロロピリジン-2-イル]アミノ](オキソ)アセタート 1・塩酸塩82)(84.2g)を加え、6時間攪拌後、室温にて16時間攪拌した。反応液に水を加え、10℃にて1時間30分攪拌した。析出した結晶を濾取、乾燥して標題化合物(106.6g,85.4%)を得た。
1H-NMR(CDCl3)δ:1.25-1.55(2H,m),1.45(9H,s),1.60-2.15(5H,m),2.56-2.74(1H,br.s),2.95(3H,s),3.06(3H,s),3.90-4.01(1H,m),4.18-4.27(1H,m),4.70-4.85(0.7H,br),5.70-6.00(0.3H,br.s),7.70(1H,dd,J=8.8,2.4Hz),7.75-8.00(1H,br),8.16(1H,br.d,J=8.8Hz),8.30(1H,d,J=2.4Hz),9.73(1H,s).
(参考例4) N1-(5-クロロピリジン-2-イル)-N2-((1S,2R,4S)-4-[(ジメチルアミノ)カルボニル]-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}シクロヘキシル)エタンジアミド(A)

1H-NMR(CDCl3)δ:1.60-1.98(3H,m),2.00-2.16(3H,m),2.52(3H,s),2.78-2.90(3H,m),2.92-2.98(2H,m),2.95(3H,s),3.06(3H,s),3.69(1H,d,J=15.4Hz),3.75(1H,d,J=15.4Hz),4.07-4.15(1H,m),4.66-4.72(1H,m),7.40(1H,dd,J=8.8,0.6Hz),7.68(1H,dd,J=8.8,2.4Hz),8.03(1H,d,J=7.8Hz),8.16(1H,dd,J=8.8,0.6Hz),8.30(1H,dd,J=2.4,0.6Hz),9.72(1H,s).
MS(ESI)m/z:548(M+H)+.
(参考例5) N1-(5-クロロピリジン-2-イル)-N2-((1S,2R,4S)-4-[(ジメチルアミノ)カルボニル]-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}シクロヘキシル)エタンジアミド モノp-トルエンスルホン酸塩 1水和物(A-a)

1H-NMR(DMSO-d6)δ:1.45-1.54(1H,m),1.66-1.78(3H,m),2.03-2.10(2H,m),2.28(3H,s),2.79(3H,s),2.91-3.02(1H,m),2.93(3H,s),2.99(3H,s),3.13-3.24(2H,m),3.46-3.82(2H,m),3.98-4.04(1H,m),4.43-4.80(3H,m),7.11(2H,d,J=7.8Hz),7.46(2H,d,J=8.2Hz),8.01(2H,d,J=1.8Hz),8.46(1H,t,J=1.8Hz),8.75(1H,d,J=6.9Hz),9.10-9.28(1H,br),10.18(1H,br),10.29(1H,s).
MS(ESI)m/z:548(M+H)+.
元素分析:C24H30ClN7O4S・C7H8O3S・H2O
理論値:C;50.43,H;5.46,N;13.28,Cl;4.80,S;8.69.
実測値:C;50.25,H;5.36,N;13.32,Cl;4.93,S;8.79.
mp(分解):245~248℃.
(実施例1) tert-ブチル (1R,2S,5S)-2-({2-[(5-クロロ-2-ピリジン-2-イル)アミノ]-2-オキソアセチル}アミノ)-5-(ジメチルアミノカルボニル)シクロヘキシルカルバマート(1)

アセトニトリル(60ml)に、2-アミノ-5-クロロピリジン(2b)(5.0g)を添加し、50℃でエチル オキサリル クロリド(2c)(5.9g)を加え、同温度で2時間攪拌した。反応液を冷却して10℃で結晶を濾取し、エチル[5-クロロピリジン-2-イル]アミノ](オキソ)アセタート 1・塩酸塩(2)(9.8g)を得た。
上記の参考例3に記載された、従来法の反応操作を行った。反応系が固化した場合の、化合物(1)の製造収率及び副生物である不純物X*の生成の割合を、下記の(条件1)~(条件3)で評価測定した。その結果を表1に示す。
*副生物である不純物Xは、下記式の構造である。

(条件2):化合物(3)(500mg)をアセトニトリル(2.5ml)に懸濁し、トリエチルアミン(0.7g)を添加後、混合液の温度を55℃で1時間静置して系内を固化させた。1時間後に化合物(2)(0.425g)を添加後、再度反応混合液の温度を75℃に加温して7時間攪拌した。[(条件1)で、反応系を固化させる条件]
(条件3):化合物(3)(500mg)をアセトニトリル(2.5ml)に懸濁し、トリエチルアミン(0.7g)、引き続いて化合物(2)(0.425g)を添加後、混合液の内温を55℃で1時間静置して系内を固化させた。1時間後に、再度反応混合液の温度を75℃に加温して7時間攪拌した。
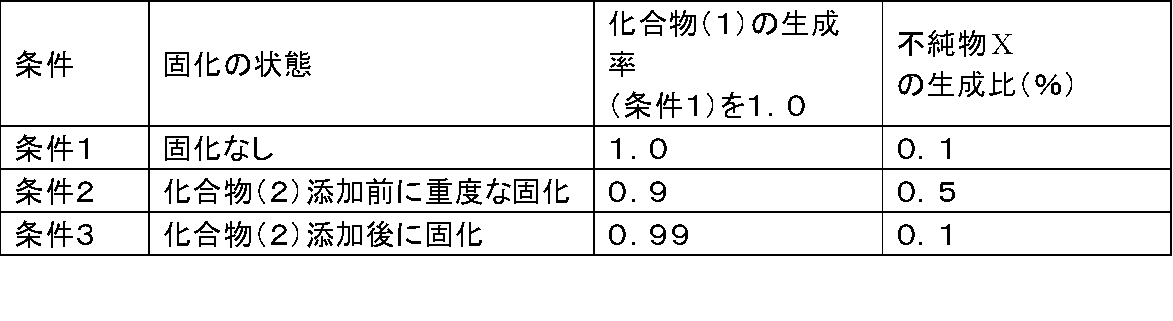
<試験結果>
化合物(2)の添加前に系内が固化した場合(条件2)、攪拌不良に基づく化合物(1)の生成率の低下と不純物Xの生成が増加した。尚、化合物(2)の添加終了後に系内が固化した場合(条件3)は、化合物(1)の収率及び不純物Xの生成率への影響は少なかった。
試験例1の結果から、従来の方法である、化合物(3)を先に添加する方法では化合物(1)の品質低下のリスク(試験例1の条件2)があること明らかになった。化合物(3)、化合物(2)及びトリエチルアミンの添加順番を変更した場合の、化合物(1)の収率、品質への影響を確認する目的で、試験例1の条件1の他に下記の(条件4)、(条件5)における化合物(1)及び不純物Xの生成率を測定した。その結果を表2に示す。
(条件4):化合物(3)(500mg)及び化合物(2)(0.425g)をアセトニトリル(2.5ml)に懸濁し、混合液の内温を60℃でトリエチルアミン(0.7g)を添加後、反応混合液の内温を75℃に加温して7時間攪拌した。
(条件5):化合物(2)(0.425g)をアセトニトリル(2.5ml)に懸濁し、混合液の内温を60℃でトリエチルアミン(0.7g)を添加後、化合物(3)(500mg)を添加し、反応混合液の内温を75℃に加温して7時間攪拌した(実施例2)。

<試験結果>
(条件4)のように、化合物(3)及び化合物(2)を先に混合し、次いでトリエチルアミンを添加した場合は不純物Xの生成比率が大幅に増加した。これは、化合物(3)のBoc基の脱離によるものと考えられた。一方、(条件5)では、トリエチルアミンを添加する際に、トリエチルアミンの蒸気で化合物(3)の一部に非晶質化が観察され、またトリエチルアミンの塩酸塩の生成に伴う発煙が生じ、化合物(3)の添加がしにくい難点があったが、化合物(1)の収率は高く、不純物Xの生成率は低かった(実施例2)。本試験結果から、化合物(2)を先に添加した場合、化合物(1)の収率、品質が良好であることがわかった。
上記の試験例2の条件5(実施例2)において、トリエチルアミン(TEA)の添加後、化合物(3)を添加するまでの時間の影響について精査した。結果を表3に示す。

<試験結果>
トリエチルアミンを添加してから化合物(3)を添加するまでの時間が延長するのに伴って、化合物(1)生成率が低下した。
化合物(2)のフリー体である2-[(5-クロロピリジン-2-イル)アミノ]-2-オキソアセタート(2a)の安定性を試験した。化合物(2)に、トリエチルアミンを化合物(2)に対して4.3モル当量を添加後、45℃、55℃、65℃及び75℃における化合物(2a)の経時的残存率を測定した。結果を図1に示す。
図1に示すように、大過剰のトリエチルアミンの共存下では、温度の上昇にしたがって、化合物(2a)の残存率が低下した。この結果から、化合物(2a)の分解は、過剰量のトリエチルアミンとの共存と温度の影響が考えられた。
試験例4の結果から、大過剰のトリエチルアミンの共存により、化合物(2a)の分解が促進したことから、トリエチルアミン添加量を減らし、化合物(2a)の安定性を評価した。
トリエチルアミンを1.5モル当量添加した場合、化合物(2a)は、1時間当たり0.3%程度の分解にとどまった。このことから、試験例4で見られた化合物(2a)の分解は、トリエチルアミン添加量を減らすことで回避できることがわかった。
トリエチルアミンの添加量により化合物(2a)の分解が回避できることがわかった。つぎに、化合物(2)にトリエチルアミンを1.07~1.15モル当量添加し、さらに化合物(3)を添加した場合に、系内が固化するかどうかを条件6、7(実施例8、9)及び条件8(比較例)で評価した。
アセトニトリル(50ml)に化合物(2)(8.5g)を加え、10℃で攪拌下に表4に記載のトリエチルアミンを添加した。添加終了後、化合物(3)(10g)を添加し、系内の流動性を目視で試験した。

トリエチルアミンが1.10モル当量以下であれば化合物(3)を添加しても固化せず、攪拌に問題がないことがわかった。また、低温でトリエチルアミンを添加すると発煙は軽微であり、トリエチルアミンの蒸気による化合物(3)の非晶質化もなかった。
トリエチルアミンを分割添加した場合の、化合物(1)及び不純物Xの生成率、及びトリエチルアミンの添加時の重度の発煙の有無を条件9(比較例)及び条件10~12(実施例10~13)で試験した。
アセトニトリル(50ml)に化合物(2)(8.5g)を加え、10℃で攪拌下にトリエチルアミンを1回目として添加した(表5の量)。添加終了後、化合物(3)(10g)を添加して、その後60℃でトリエチルアミンを2回目として添加した(トリエチルアミンの総量は対化合物(2)の5.2モル当量)。

トリエチルアミンを分割添加した場合でも、化合物(1)の収率、品質は良好であった。
Claims (21)
- 下記の式で表される化合物(1)

の製造方法であって、
[操作1]:C2-C4ニトリル系溶媒中、下記の式で表される化合物(2)

[操作2]:上記の[操作1]の混合液に、下記の式で表される化合物(3)
を添加することを特徴とした、化合物(1)の製造方法。 - C2-C4ニトリル系溶媒が、アセトニトリルである請求項1に記載の製造方法。
- 化合物(2)の量が、化学量論的に化合物(3)に対して1.10~1.27モル当量である請求項1又は2に記載の製造方法。
- 三級アミンが、トリ(C1-C4アルキル)アミン類、1-(C1-C3アルキル)ピペリジン及び4-(C1-C3アルキル)モルホリンからなる群より選ばれる1種、又は2種以上である請求項1~3のいずれか1項に記載の製造方法。
- 三級アミンが、トリ(C1-C4アルキル)アミン類から選ばれる1種、又は2種以上である請求項1~3のいずれか1項に記載の製造方法。
- 三級アミンが、トリエチルアミンである請求項1~3のいずれか1項に記載の製造方法。
- [操作1]が、C2-C4ニトリル系溶媒中に、下記の式で表される化合物(2)

- [操作1]が、C2-C4ニトリル系溶媒中に三級アミンを加え、次いで下記の式で表される化合物(2)

- 化合物(2)又は三級アミンの添加が、45℃以下で行うものである請求項7又は8に記載の製造方法。
- 化合物(2)又は三級アミンの添加が、0~35℃の範囲で行うものである請求項7又は8に記載の製造方法。
- [操作1]における三級アミンの添加量が、化学量論的に化合物(2)に対して0.87~1.08モル当量である請求項1~10のいずれか1項に記載の製造方法。
- [操作2]の操作の後に、さらに三級アミンを追加して添加するものである請求項1~11のいずれか1項に記載の製造方法。
- 三級アミンを追加して添加する量が、化学量論的に化合物(2)に対して3.02~4.53モル当量の範囲である請求項12の製造方法。
- 50~65℃で三級アミンを追加して添加するものである請求項12又は13の製造方法。
- 三級アミンを追加して添加した後、反応混合物を60~80℃で撹拌するものである請求項12~14のいずれか1項に記載の製造方法。
- 撹拌時間が6時間以上である請求項15に記載の製造方法。
- 撹拌時間が6~24時間の範囲である請求項15に記載の製造方法。
- 下記の式で表される化合物(1)の製造方法であって、

[操作1]:アセトニトリル中、下記の式で表される化合物(2)

[操作2]:上記の[操作1]の混合液に、下記の式で表される化合物(3)
を添加した後、50~65℃で化学量論的に化合物(2)に対して3.23~4.32モル当量の三級アミンを追加して添加し、60~80℃で6時間以上撹拌することを特徴とした化合物(1)の製造方法。 - 下記の式で表される化合物(1)の製造方法であって、
下記の式で表される化合物(A-a)

- 請求項1に記載の製造方法により製造した化合物(1)を使用することを特徴とする、下記の式で表される化合物(A-a)の製造方法。
- 請求項1に記載の製造方法により製造した化合物(1)を使用することを特徴とする、下記の式で表される化合物(A-a)の製造方法であって;

のBoc基を脱保護して、下記の式で表される化合物(7)又はその塩を得る工程、



Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP20100750831 EP2407457B1 (en) | 2009-03-10 | 2010-03-09 | Process for producing diamine derivative |
| CN201080011057.1A CN102348688B (zh) | 2009-03-10 | 2010-03-09 | 用于制备二胺衍生物的方法 |
| JP2011503828A JP5666424B2 (ja) | 2009-03-10 | 2010-03-09 | ジアミン誘導体の製造方法 |
| ES10750831.9T ES2542236T3 (es) | 2009-03-10 | 2010-03-09 | Procedimiento de producción de un derivado de diamina |
| US13/228,928 US8357808B2 (en) | 2009-03-10 | 2011-09-09 | Process for producing diamine derivative |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2009-057031 | 2009-03-10 | ||
| JP2009057031 | 2009-03-10 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US13/228,928 Continuation US8357808B2 (en) | 2009-03-10 | 2011-09-09 | Process for producing diamine derivative |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2010104078A1 true WO2010104078A1 (ja) | 2010-09-16 |
Family
ID=42728367
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2010/053905 WO2010104078A1 (ja) | 2009-03-10 | 2010-03-09 | ジアミン誘導体の製造方法 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US8357808B2 (ja) |
| EP (1) | EP2407457B1 (ja) |
| JP (1) | JP5666424B2 (ja) |
| CN (1) | CN102348688B (ja) |
| ES (1) | ES2542236T3 (ja) |
| WO (1) | WO2010104078A1 (ja) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011115066A1 (ja) * | 2010-03-19 | 2011-09-22 | 第一三共株式会社 | ジアミン誘導体の結晶およびその製造方法 |
| US9809546B2 (en) | 2016-03-14 | 2017-11-07 | Chungwha Chemical Synthesis & Biotech Co., Ltd. | Process for producing diamine derivative |
| JP2019522672A (ja) * | 2016-07-13 | 2019-08-15 | マイラン ラボラトリーズ リミテッドMylan Laboratories Limited | アミン保護(1s,2r,4s)−1,2ーアミノ−n,n−ジメチルシクロヘキサン−4−カルボキサミドの塩 |
| KR20190139463A (ko) | 2018-06-08 | 2019-12-18 | 주식회사 가피바이오 | 에독사반의 제조용 중간체의 제조 방법 및 에독사반의 제조 방법 |
| JP2021514348A (ja) * | 2018-02-14 | 2021-06-10 | モエス、イベリカ、ソシエダッド、リミターダMoehs Iberica S.L. | tert−ブチルN−((1R,2S,5S)−2−((2−((5−クロロピリジン−2−イル)アミノ)−2−オキソアセチル)アミノ)−5−(ジメチルカルバモイル)シクロヘキシル)カルバメートを調製するための方法 |
| WO2024053350A1 (ja) * | 2022-09-09 | 2024-03-14 | 株式会社カネカ | アミノアジド化合物、ジアミン化合物、及びエドキサバンの製造方法 |
Families Citing this family (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DK2140867T4 (da) | 2007-03-29 | 2023-09-25 | Daiichi Sankyo Co Ltd | Farmaceutisk sammensætning |
| EP2407450B1 (en) | 2009-03-13 | 2015-04-22 | Daiichi Sankyo Company, Limited | Method for producing optically active diamine derivative |
| CN103333091A (zh) * | 2011-12-08 | 2013-10-02 | 成都苑东药业有限公司 | 一种抗凝血的二胺衍生物 |
| KR102371784B1 (ko) * | 2014-02-18 | 2022-03-07 | 다이이찌 산쿄 가부시키가이샤 | 활성화 혈액 응고 제 X 인자 (FXa) 의 저해약의 제조 방법 |
| CN104761571B (zh) * | 2015-03-10 | 2017-03-08 | 山东科兴生物制品有限公司 | 一种依度沙班的合成方法 |
| CN106316889B (zh) * | 2015-06-15 | 2020-07-10 | 上海阳帆医药科技有限公司 | 依度沙班中间体的制备方法 |
| CN107033163A (zh) * | 2015-07-16 | 2017-08-11 | 浙江海正药业股份有限公司 | 一种依度沙班对苯甲磺酸盐新晶型及其制备方法和应用 |
| CN105753888B (zh) * | 2016-04-05 | 2017-09-01 | 乐普药业股份有限公司 | 一种游离态依度沙班的制备方法 |
| US10301322B2 (en) | 2016-12-27 | 2019-05-28 | Apotex Inc. | Processes for the preparation of edoxaban and intermediates thereof |
| CN106866452B (zh) * | 2017-03-17 | 2018-10-30 | 浙江天宇药业股份有限公司 | 一种依度沙班中间体的合成方法及中间产物 |
| CN108484641B (zh) * | 2018-06-08 | 2019-04-02 | 北京阳光诺和药物研究有限公司 | 依度沙班对甲苯磺酸盐一水合物的制备方法 |
| US20220251112A1 (en) * | 2019-07-04 | 2022-08-11 | Glenmark Life Sciences Limited | Process for preparation of edoxaban |
| CN111606827B (zh) * | 2020-06-23 | 2022-10-25 | 内蒙古京东药业有限公司 | 一种制备依度沙班手性胺中间体的方法 |
| CN111763169B (zh) * | 2020-06-30 | 2023-06-27 | 浙江苏泊尔制药有限公司 | 一种改良的依度沙班中间体的制备方法 |
| KR102577696B1 (ko) * | 2020-11-16 | 2023-09-14 | 주식회사 보령 | 에독사반 토실산염 또는 그 수화물의 제조방법 |
| CN112940012B (zh) * | 2021-05-17 | 2021-09-03 | 上海翰森生物医药科技有限公司 | 一种依度沙班及其中间体的制备方法 |
| WO2023223346A1 (en) | 2022-05-16 | 2023-11-23 | Mylan Laboratories Limited | An improved process for the preparation of edoxaban intermediate |
| CN116102465A (zh) * | 2023-02-17 | 2023-05-12 | 珠海市海瑞德生物科技有限公司 | 光学活性二胺化合物的制备方法 |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003000657A1 (fr) * | 2001-06-20 | 2003-01-03 | Daiichi Pharmaceutical Co., Ltd. | Derives de diamine |
| WO2003000680A1 (fr) | 2001-06-20 | 2003-01-03 | Daiichi Pharmaceutical Co., Ltd. | Derives de diamine |
| WO2003016302A1 (fr) | 2001-08-09 | 2003-02-27 | Daiichi Pharmaceutical Co., Ltd. | Derives de diamine |
| WO2004058715A1 (ja) | 2002-12-25 | 2004-07-15 | Daiichi Pharmaceutical Co., Ltd. | ジアミン誘導体 |
| WO2007032498A1 (ja) * | 2005-09-16 | 2007-03-22 | Daiichi Sankyo Company, Limited | 光学活性なジアミン誘導体およびその製造方法 |
| WO2007106759A2 (en) | 2006-03-14 | 2007-09-20 | Applied Materials, Inc. | Method to reduce cross talk in a multi column e-beam test system |
Family Cites Families (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5149855A (en) | 1989-12-26 | 1992-09-22 | Mitsubishi Rayon Co., Ltd. | Process for racemizing optically active carboxylic acid esters |
| US5055600A (en) | 1990-04-24 | 1991-10-08 | Rockwell International Corporation | Glycidyl azide polymer (gap) synthesis by molten salt method |
| US5677469A (en) | 1995-05-18 | 1997-10-14 | Sepracor, Inc. | Process for resolving chiral acids with 1-aminoindan-2-ols |
| JPH11180899A (ja) | 1997-12-15 | 1999-07-06 | Mitsui Chem Inc | 活性メチレンアルキル化化合物の製造法 |
| JP3680203B2 (ja) | 1999-06-01 | 2005-08-10 | 東洋化成工業株式会社 | 4−アセチルアミノベンゼンスルホニルアジドの製造方法 |
| JP2001151724A (ja) | 1999-11-19 | 2001-06-05 | Kuraray Co Ltd | 光学活性な2,2,4−トリメチル−3−シクロヘキセンカルボン酸の製造方法 |
| TWI288745B (en) | 2000-04-05 | 2007-10-21 | Daiichi Seiyaku Co | Ethylenediamine derivatives |
| CA2521325C (en) | 2003-04-08 | 2010-09-14 | Novartis Ag | Solid pharmaceutical compositions comprising a s1p receptor agonist and a sugar alcohol |
| WO2005047296A1 (ja) | 2003-11-12 | 2005-05-26 | Daiichi Pharmaceutical Co., Ltd. | チアゾール誘導体の製造法 |
| AR057035A1 (es) | 2005-05-25 | 2007-11-14 | Progenics Pharm Inc | SíNTESIS DE (R)-N-METILNALTREXONA, COMPOSICIONES FARMACÉUTICAS Y USOS |
| JP4227629B2 (ja) | 2006-04-28 | 2009-02-18 | 達實 小野 | 過熱蒸気を応用した塗装用乾燥焼付装置 |
| CA2663981C (en) | 2006-10-09 | 2014-05-27 | Cipla Limited | Process for preparing trityl olmesartan medoxomil and olmesartan medoxomil |
| DK2140867T4 (da) | 2007-03-29 | 2023-09-25 | Daiichi Sankyo Co Ltd | Farmaceutisk sammensætning |
| TW200909437A (en) | 2007-06-21 | 2009-03-01 | Daiichi Sankyo Co Ltd | Process for the preparation of diamine-derivatives |
| MY150973A (en) * | 2008-12-17 | 2014-03-31 | Daiichi Sankyo Co Ltd | Method for producing diamine derivative |
-
2010
- 2010-03-09 ES ES10750831.9T patent/ES2542236T3/es active Active
- 2010-03-09 EP EP20100750831 patent/EP2407457B1/en active Active
- 2010-03-09 JP JP2011503828A patent/JP5666424B2/ja active Active
- 2010-03-09 CN CN201080011057.1A patent/CN102348688B/zh active Active
- 2010-03-09 WO PCT/JP2010/053905 patent/WO2010104078A1/ja active Application Filing
-
2011
- 2011-09-09 US US13/228,928 patent/US8357808B2/en active Active
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003000657A1 (fr) * | 2001-06-20 | 2003-01-03 | Daiichi Pharmaceutical Co., Ltd. | Derives de diamine |
| WO2003000680A1 (fr) | 2001-06-20 | 2003-01-03 | Daiichi Pharmaceutical Co., Ltd. | Derives de diamine |
| WO2003016302A1 (fr) | 2001-08-09 | 2003-02-27 | Daiichi Pharmaceutical Co., Ltd. | Derives de diamine |
| WO2004058715A1 (ja) | 2002-12-25 | 2004-07-15 | Daiichi Pharmaceutical Co., Ltd. | ジアミン誘導体 |
| WO2007032498A1 (ja) * | 2005-09-16 | 2007-03-22 | Daiichi Sankyo Company, Limited | 光学活性なジアミン誘導体およびその製造方法 |
| WO2007106759A2 (en) | 2006-03-14 | 2007-09-20 | Applied Materials, Inc. | Method to reduce cross talk in a multi column e-beam test system |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP2407457A4 * |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011115066A1 (ja) * | 2010-03-19 | 2011-09-22 | 第一三共株式会社 | ジアミン誘導体の結晶およびその製造方法 |
| JP5692873B2 (ja) * | 2010-03-19 | 2015-04-01 | 第一三共株式会社 | ジアミン誘導体の結晶およびその製造方法 |
| US9809546B2 (en) | 2016-03-14 | 2017-11-07 | Chungwha Chemical Synthesis & Biotech Co., Ltd. | Process for producing diamine derivative |
| JP2019522672A (ja) * | 2016-07-13 | 2019-08-15 | マイラン ラボラトリーズ リミテッドMylan Laboratories Limited | アミン保護(1s,2r,4s)−1,2ーアミノ−n,n−ジメチルシクロヘキサン−4−カルボキサミドの塩 |
| JP2021514348A (ja) * | 2018-02-14 | 2021-06-10 | モエス、イベリカ、ソシエダッド、リミターダMoehs Iberica S.L. | tert−ブチルN−((1R,2S,5S)−2−((2−((5−クロロピリジン−2−イル)アミノ)−2−オキソアセチル)アミノ)−5−(ジメチルカルバモイル)シクロヘキシル)カルバメートを調製するための方法 |
| JP7327736B2 (ja) | 2018-02-14 | 2023-08-16 | モエス、イベリカ、ソシエダッド、リミターダ | tert-ブチルN-((1R,2S,5S)-2-((2-((5-クロロピリジン-2-イル)アミノ)-2-オキソアセチル)アミノ)-5-(ジメチルカルバモイル)シクロヘキシル)カルバメートを調製するための方法 |
| KR20190139463A (ko) | 2018-06-08 | 2019-12-18 | 주식회사 가피바이오 | 에독사반의 제조용 중간체의 제조 방법 및 에독사반의 제조 방법 |
| WO2024053350A1 (ja) * | 2022-09-09 | 2024-03-14 | 株式会社カネカ | アミノアジド化合物、ジアミン化合物、及びエドキサバンの製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2407457A1 (en) | 2012-01-18 |
| EP2407457B1 (en) | 2015-04-22 |
| EP2407457A4 (en) | 2012-08-22 |
| US8357808B2 (en) | 2013-01-22 |
| US20120041206A1 (en) | 2012-02-16 |
| CN102348688A (zh) | 2012-02-08 |
| ES2542236T3 (es) | 2015-08-03 |
| JP5666424B2 (ja) | 2015-02-12 |
| JPWO2010104078A1 (ja) | 2012-09-13 |
| CN102348688B (zh) | 2014-07-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5666424B2 (ja) | ジアミン誘導体の製造方法 | |
| JP5780657B2 (ja) | 光学活性ジアミン誘導体の塩の製造方法 | |
| JP5652879B2 (ja) | 光学活性なジアミン誘導体の製造方法 | |
| WO2020210404A1 (en) | Dosage forms and regimens for amino acid compounds | |
| JPWO2014157612A1 (ja) | (1s,4s,5s)−4−ブロモ−6−オキサビシクロ[3.2.1]オクタン−7−オンの製造方法 | |
| JP2014051492A (ja) | ジアミン誘導体の製造法 | |
| JP6832946B2 (ja) | キナーゼ阻害剤およびその中間体の調製方法 | |
| WO2010131663A1 (ja) | オキサミド誘導体 | |
| JP2023116769A (ja) | エドキサバンの製造方法 | |
| US20110172427A1 (en) | Process for preparing certain cinnamide compounds | |
| AU2022265730A1 (en) | Expanded dosage regimens for integrin inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201080011057.1 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10750831 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011503828 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010750831 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1763/MUMNP/2011 Country of ref document: IN |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |