WO2009115478A2 - Verfahren zur in vitro erfassung und unterscheidung von pathophysiologischen zuständen - Google Patents
Verfahren zur in vitro erfassung und unterscheidung von pathophysiologischen zuständen Download PDFInfo
- Publication number
- WO2009115478A2 WO2009115478A2 PCT/EP2009/053042 EP2009053042W WO2009115478A2 WO 2009115478 A2 WO2009115478 A2 WO 2009115478A2 EP 2009053042 W EP2009053042 W EP 2009053042W WO 2009115478 A2 WO2009115478 A2 WO 2009115478A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- sepsis
- gene
- patients
- infection
- patient
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6876—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes
- C12Q1/6883—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for diseases caused by alterations of genetic material
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/106—Pharmacogenomics, i.e. genetic variability in individual responses to drugs and drug metabolism
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/158—Expression markers
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/178—Oligonucleotides characterized by their use miRNA, siRNA or ncRNA
Definitions
- the present invention relates to a method for the in vitro detection and / or differentiation and / or course monitoring of pathophysiological conditions according to claim 1, the use of a plurality of polynucleotides and / or their gene loci and / or their transcripts to form at least one multigene biomarker for the production of a multiplex Assays according to claim 4; the use of at least one polynucleotide and / or their gene loci and / or their transcripts for the production of an assay according to claim 11, as well as a kit for carrying out the method according to claim 14.
- the present invention relates to the use of polynucleotides for detecting gene activities of at least one multigene biomarker, for the preparation of an adjunct for diagnosis in patients with certain pathophysiological conditions, such as sepsis and sepsis-like conditions, having similar features to an "In Vitro Diagnostic Multivariate Index Assay "(IVDMIA).
- IVDMIA Intelligent Multivariate Index Assay
- Sepsis blood poisoning
- blood poisoning is a life-threatening infection that affects the entire organism, is associated with high mortality, is becoming more prevalent and affects people of all ages.
- the mortality rate of severe sepsis has not improved significantly in recent decades, with the last two breakthroughs in innovation since the introduction of blood culture (around 1880) being the introduction of antibiotics over 60 years ago and the onset of intensive care about 50 years ago.
- novel diagnostic agents must be made available.
- Sepsis is caused by infectious agents. As there is currently no specific therapy for sepsis, the success of the treatment largely depends on the successful control of the underlying infection and the quality of the intensive care treatment. Crucial for survival is the early administration of an antibiotic that also successfully combats the causative agent [Kumar et. al., 2006]. However, deficits in sepsis diagnostics delay the initiation of therapy and the choice of a suitable antibiotic. Since the identification of sepsis with the current methods of blood culture only succeed in less than 25% of sepsis cases and the findings in the case of pathogen detection after 2-3 days, the initial choice of antibiotic or antimycotic (anti-fungal substances) must " calculated on suspicion, in 20-30% of cases this choice is not correct.
- the present invention relates to genes and / or fragments thereof and their use for generating multigene biomarkers which are specific to a condition and / or investigation.
- the invention further relates to marker primers derived PCR primers and probes for hybridization or duplication methods.
- Sepsis is still one of the most difficult clinical pictures in modern intensive care, with not only the therapy but also the diagnosis being a challenge for the clinician.
- generalized inflammatory conditions such as SIRS and sepsis are very common in patients in intensive care units and contribute significantly to mortality [Marshai et al., 2003; Alberti et al., 2003].
- the mortality is about 20% in SIRS, about 40% in sepsis and increases in development of multiple organ dysfunction up to 70-80% [Brun-Buisson et al., 1995; LeGalII et al., 1995; Brun-Buisson et al., 2003].
- SIRS and sepsis are of interdisciplinary clinical-medical importance, as it increasingly the treatment successes of the most advanced therapeutic procedures numerous medical specialties (eg traumatology, neurosurgery, heart / lung surgery, visceral surgery, transplantation medicine, hematology / oncology, etc. ), which are all, without exception, at risk of SIRS and sepsis.
- medical specialties eg traumatology, neurosurgery, heart / lung surgery, visceral surgery, transplantation medicine, hematology / oncology, etc.
- This is also reflected in the continuous increase in the frequency of sepsis: between 1979 and 1987, there was an increase of 139%, from 73.6 to 176 cases per 100,000 hospital patients [MMWR Morb Mortal WkIy Rep 1990].
- SIRS systemic inflammatory response syndrome
- SIRS systemic inflammatory response syndrome
- Sepsis severe sepsis
- septic shock a distinction is made between the clinically defined severity levels “systemic inflammatory response syndrome” (SIRS), "sepsis”, “severe sepsis” and “septic shock.”
- SIRS is the systemic response of the inflammatory system to a non-infectious stimulus
- at least two of the following clinical criteria must be met: fever> 38 ° C or hypothermia ⁇ 36 ° C, leukocytosis> 12g / l or leukopenia ⁇ 4g / l or left shift in the differential blood count, heart rate above 90 / min, a tachypnoea> 20 breaths / min or a PaCO 2 (partial pressure of carbon dioxide in the arterial blood) ⁇ 4.3 kPa.
- This Definit ion has a high sensitivity but low specificity. For intensive medical care, it is of little help
- Sepsis is defined as those clinical conditions in which the SIRS criteria are fulfilled and the cause of an infection is proven or at least very probable.
- Infection is defined as a pathological process caused by invasion of pathogens or potentially pathogenic organisms into a normally sterile tissue. If the body fails to limit this infection to the site of origin, the pathogens or their toxins induce inflammation in the organs or tissues of the body remote from the site of infection. Immediate intensive care treatment, the targeted administration of antibiotics and the surgical rehabilitation of the infectious focus are needed to achieve recovery. Severe sepsis is characterized by the additional occurrence of organ dysfunctions.
- Common organ dysfunctions are changes in the state of consciousness, an oliguria, a Lactic acidosis or sepsis-induced hypotension with a systolic blood pressure of less than 90 mmHg or a pressure drop of more than 40 mmHg from baseline. If such hypotension can not be cured by the administration of crystalloids and / or colloids and in addition to the catecholamine requirement of the patient, it is called a septic shock. This is detected in about 20% of all sepsis patients.
- the first group includes score systems such as APACHE, SAPS and SIRS, which can stratify patients based on a variety of physiological indices. While some studies have shown diagnostic potential for the APACHE Il Score, other studies have demonstrated that APACHE II and SAPS II can not differentiate between sepsis and SIRS [Carrigan et al., 2004].
- the second group contains protein markers that are detected from plasma and serum.
- protein markers that are detected from plasma and serum.
- protein markers include, for example, CA125, S100B, copeptin, glycine N-acyltransferase (GNAT), protachykinin and / or its fragments, aldose 1-epimerase (mutarotase), Chp, carbamoyl phosphate synthetase 1, LASP-1 (Brahms Diagnostika GmbH Germany), IL -1 Ra, MCP-1, MPIF-1, TNF-R1, MIG, BLC, HVEM, IL-15, MCP-2, M-CSF, MIP-3b, MMP-9, PARC, ST-2; IL-6, slL-2R, CD141, MMP-9, EGF, ENA-78, EOT, Gro-beta, IL-1b, leptin, MIF, MIP-1a, OSM, protein C, P-selectin, and HCC4 (Molecular St
- PCT procalcitonin
- CRP C-reactive protein
- Procalcitonin is a 16 amino acid peptide that plays a role in inflammatory reactions. This marker has been increasingly used over time as a new infection marker in intensive care units [Sponholz et al., 2006]. This marker is used as an infection marker and serves to determine the severity of sepsis, with the dynamics of the values is more important than the absolute values themselves, for. To differentiate between infectious and non-infectious complication in cardiac surgery patients [Sponholz et al., 2006].
- CRP C-reactive protein
- PCT is considered more suitable as a CRP to differentiate non-infectious versus infectious SIRS as well as bacterial versus viral infection [Simon et al., 2004].
- the third group contains biomarkers or profiles identified at the transcriptome level. These molecular parameters should allow a better correlation of the molecular inflammatory / immunological host response with the severity of sepsis, but also provide information on individual prognosis.
- biomarkers are currently being searched extensively by various scientific groups and commercial organizations, such as changes in blood cytokine concentrations caused by bacterial cell wall components such as lipopolysaccharides [Mathiak et al., 2003], or use of gene expression profiles in a blood sample to determine differences in survivors and non-survivors sepsis patients [Pachot et al., 2006].
- Gene expression profiles or classifiers are for the determination of the severity of sepsis [WO 2004/087949], the distinction between a local or systemic infection [unpublished DE 10 2007 036 678.9], the identification of the source of infection [WO 2007/124820] or gene expression signatures for the distinction between multiple etiologies and pathogen-associated signatures [Ramilo et al., 2007].
- the currently available protein markers due to the insufficient specificity and sensitivity of the consensus criteria according to [Bone et al., 1992] the currently available protein markers, as well as due to the time required to detect the cause of the infection by blood culture, there is an urgent need for new methods that take into account the complexity of the disease.
- Many gene expression studies that either single genes and / or combinations of Genes named as classifiers, as well as numerous descriptions of statistical methods for deriving a score and / or index [WO03084388; US6960439] belong to the state of the art.
- MammaPrint The microarray-based, 70-gene signature called MammaPrint (Aqendia, NL) allows predicting the risk of recurrence and metastasis in women with breast cancer. It is being investigated whether the risk of developing distant metastases in the next few years could be considered high or low and that they would benefit from chemotherapy. The approval of this test by the FDA has led to the development of guidelines for a new class of diagnostic tests known as IVDMIA (in vitro diagnostic multivariate index assay). The MammaPrint signature is measured and calculated on a microarray in the manufacturer's laboratories.
- Oncotype DX multigene assav (Genomic Health, USA) is used to assess the likelihood of breast cancer recurrence in patients and to evaluate the response of patients to chemotherapy. 21 genes are summarized as "Recurrence-Score.” The measurement takes place in the rooms of the company, it is also the TaqMan- PCR technology used.
- the AlloMap gene expression test from XDx (USA) is used to monitor any rejection reactions in patients with heart transplantation, which show approximately 30% of patients within one year. So far, were to Diagnosis several biopsies necessary.
- the test is based on 1 1 quantitative PCR assays (plus 9 controls and references) using the TaqMan technology (Hoffman-La Roche) in the manufacturer's premises. The sample material is blood. Already two months after the transplantation, the results are reliable and predict the absence of rejection reactions for the next 80 days.
- a kit according to claim 14 solves the problem as well.
- the present invention relates to a system comprising the following elements:
- the system provides a solution to the problem of detecting disease states such as For example, the distinction between infectious and non-infectious multi-organ failure but also for other relevant applications and issues in this context.
- the present invention relates to a method for in vitro detection and / or differentiation and / or course monitoring of pathophysiological conditions selected from the group consisting of: SIRS, sepsis and their degrees of severity; sepsis-like states; septic shock; infectious / non-infectious multi-organ failure; Survival probability in sepsis; Focus of an infection; Responder / non-responder for a given therapy; Causes of a pathophysiological condition, in particular classification of an infection according to gram-positive and / or gram-negative bacteria; the method comprising the steps of:
- the at least one reference gene is a housekeeping gene, wherein the housekeeping gene is in particular selected from polynucleotides of the group consisting of SEQ ID NO: 676 to SEQ ID NO: 686 and / or their gene loci and / or their transcripts and / or Fragments of it.
- RNA As polynucleotide sequences, preference is given to using gene loci, sense and / or antisense strands of pre-mRNA and / or mRNA, small RNA, in particular scRNA, snoRNA, microRNA, siRNA, dsRNA, ncRNA or transposable elements.
- the index is preferably determined by statistical techniques such as supervised machine and static learning classification techniques such as e.g. (diagonal, linear, quadratic) discriminant analysis, super vector machines, generalized partial least squares, k-nearest neighbors, random forests, k-nearest neighbor.
- supervised machine and static learning classification techniques such as e.g. (diagonal, linear, quadratic) discriminant analysis, super vector machines, generalized partial least squares, k-nearest neighbors, random forests, k-nearest neighbor.
- the invention further relates to the use of a plurality of polynucleotides selected from the group consisting of SEQ ID NO: 1 to SEQ ID NO: 669 and / or their gene loci and / or their transcripts and / or fragments thereof, for the formation of at least one Multigenbiomarkers for the production of a multiplex Assays as an aid to assessing whether a patient has a pathophysiological condition and / or to determine the severity and / or the course of the pathophysiological condition.
- the multigene biomarker is preferably a combination of a plurality of polynucleotide, in particular gene sequences, based on whose gene activities a classification is carried out by means of an interpretation function and / or an index or score is formed.
- the gene activities are determined by enzymatic methods, in particular amplification methods, preferably polymerase chain reaction (PCR), preferably real-time PCR; and / or by means of hybridization methods, in particular those on microarrays.
- enzymatic methods in particular amplification methods, preferably polymerase chain reaction (PCR), preferably real-time PCR; and / or by means of hybridization methods, in particular those on microarrays.
- Differential expression signals of the polynucleotide sequences contained in the multigene biomarker may be advantageously and unambiguously assigned to a pathophysiological condition, course, and / or therapy monitoring.
- an index is formed from the individual determined gene activities, which after appropriate calibration is a measure of the severity and / or the course of the pathophysiological state, in particular sepsis or the sepsis-like state.
- This index or score can be displayed on an easily interpretable scale to give the treating physician a quick diagnostic tool.
- the gene activity data obtained for the production of software for the description of at least one pathophysiological condition and / or an investigation question and / or used as aids for diagnostic purposes and / or for patient data management systems.
- RNA small RNA, in particular scRNA, snoRNA, microRNA, siRNA, dsRNA, ncRNA or transposable elements, genes and / or gene fragments used which have a sequence homology of at least about 10%, in particular about 20%, preferably about 50%, particularly preferably about 80% to the polynucleotide sequences according to SEQ ID NO: 1 to SEQ ID NO: 669.
- the invention further relates to the use of at least one polynucleotide selected from the group consisting of SEQ ID NO: 1 to SEQ ID NO: 152 and / or their gene loci and / or their transcripts and / or fragments thereof, for the preparation of an assay for Assessing whether a patient has a pathophysiological condition and / or assessing the severity and / or pathophysiological status.
- the pathophysiological condition is advantageously selected from the group consisting of: SIRS, sepsis and their degrees of severity; sepsis-like states; septic shock; infectious / non-infectious multi-organ failure; local / systemic infection; Improvement / worsening of a pathophysiological condition, especially sepsis; Responder / non-responder for a given therapy; Focus of an infection; Causes of a pathophysiological condition, in particular classification according to gram-positive and / or gram-negative.
- the sample nucleic acid is RNA, in particular total RNA or mRNA, or DNA, in particular cDNA.
- the invention further relates to a kit for carrying out the method according to the invention, comprising at least one multigene biomarker which comprises a plurality of polynucleotide sequences which are selected from the pool of SEQ ID NO: 1 to SEQ ID NO.
- the multigene biomarker is specific for a pathophysiological condition of a patient and such conditions which are selected from the group consisting of: SIRS, sepsis and their degrees of severity; sepsis-like states; septic shock; infectious / non-infectious multi-organ failure; Survival probability in sepsis; local / systemic infection; Responder / non-responder for a given therapy; Focus of an infection; Causes of a pathophysiological condition, in particular classification of an infection according to Gram-positive or Gram-negative pathogens.
- the polynucleotide sequences of the kit preferably also include gene loci, sense and / or antisense strands of pre-mRNA and / or mRNA, small RNA, in particular scRNA, snoRNA, microRNA, siRNA, dsRNA, ncRNA or transposable elements.
- the polynucleotide sequences with the SEQ IDs given in Tab. 11 and 16 are preferably used.
- the polynucleotide sequences with the SEQ IDs given in Tab. 20 and 21 are preferably used.
- the present invention makes it possible to assess a potential infectious complication in patients with SIRS or possible sepsis.
- This system includes the choice of patients and the determination of their gene expression signals in an interpretable index that the physician can use as a diagnostic tool.
- This system combines the measured gene expression data from defined sequence groups selected from SEQ ID NO: 1 to SEQ ID NO: 669 and / or their gene loci and / or their transcripts and / or fragments thereof, as well as housekeeping genes.
- those specific genes and / or gene fragments are used which have a sequence homology of at least about 10%, in particular about 20%, preferably about 50%, particularly preferably about 80% have the polynucleotide sequences according to SEQ ID NO: 1 to SEQ ID NO: 669 or to the housekeeping genes.
- Table 32 shows the highly relevant sequence pool, which is important for various clinical questions.
- Tab. 8, 11 and 16 show a preferred choice of sequences which, when integrated into the above system, are essential for the distinction between SIRS and sepsis.
- Applicant has developed a method that uses large sequence pools to detect and / or differentiate states or to answer defined questions of investigation. Examples can be found in the following patents: Distinction between SIRS, sepsis and sepsis-like conditions [WO 2004/087949; WO 2005/0831 15], Development of Criteria for Predicting the Disease Course in Sepsis [WO 05/106020], distinction between non-infectious and infectious causes of multiorgan failure [WO 2006/042581], in vitro classification of gene expression profiles of patients with infectious / non-infectious multiorgan failure [WO 2006/100203], Determination of the Local Causes of a Fever of Unclear Genesis [WO 2007/144105], polynucleotides for the detection of gene activities for the distinction between local and systemic infection [DE 10 2007 036 678.9].
- the invention relates to polynucleotide sequences, to a method and also to kits for generating multigene biomarkers which have features of an "in vitro diagnostic multivariate index assay” (IVDMIA) in one and / or several modules.
- IVDMIA in vitro diagnostic multivariate index assay
- Multi-organ failure is the failure of two or more vital organ systems occurring simultaneously or in rapid succession.
- Multi-organ dysfunction syndrome precedes MOV as initial organ failure [Zeni et al., 1997].
- MODS Multi-organ dysfunction syndrome
- the prognosis of MOV is closely related to the number of involved organ systems. Mortality is 22% in the first 24 hours of an organ failure and 41% after 7 days. When three organ systems fail, mortality increases to 80% on the first day and to 100% on the first day [Knaus et al., 1985].
- MODS and MOV can be both infectiologic and non-infectiological.
- Fever of unclear origin Fever of unknow origin (FUO) is clinically defined as a fever in which the temperature is higher than 38.8 ° C over a period of more than 3 weeks, without a week after that Examination time is a clear diagnosis of the cause.
- FUO Fever of unknow origin
- four classes of FUO have been described: FUO classic, nosocomial, immunodeficient or HIV-related origin [Roth and Basello, 2003].
- FUO has also been described as "a more well-known disease with an unusual appearance than a rare disorder" [Amin and Kauffman, 2003].
- Examination Question A clinically relevant question that is important for the treatment of a patient, for example: prediction of disease progression, therapy monitoring, focus of infection, chances of survival, predisposition, etc.
- a systemic infection is an infection in which the pathogens have spread through the bloodstream throughout the organism.
- SIRS Systemic Inflammatory Response Syndrome, according to Bone [Bone et al., 1992] and Levy [Levy et al., 2003] a generalized, inflammatory, non-infectious condition of a patient.
- Biological fluid Biological fluids within the meaning of the invention are understood to be all body fluids of mammals, including humans.
- a gene is a section of the deoxyribonucleic acid (DNA) that contains the basic information needed to produce a biologically active ribonucleic acid (RNA) as well as regulatory elements that activate or inactivate this production.
- genes are also understood as meaning all derived DNA sequences, partial sequences and synthetic analogs (for example peptido-nucleic acids (PNA)).
- PNA peptido-nucleic acids
- Genlocus is the position of a gene in the genome. If the genome consists of several chromosomes, the position within the chromosome on which the gene is located is meant. Different manifestations or variants of this gene are referred to as alleles, all located at the same site on the chromosome, namely the gene locus.
- the term "gene locus” includes, on the one hand, the pure genetic information for a specific gene product and, on the other hand, all regulatory DNA segments as well as any additional DNA sequences that are in any functional relationship with the gene at the gene locus in the immediate vicinity (1 Kb) but outside the 5 'and / or 3' end of a gene locus
- the gene locus is specified by the accession number and / or RefSeq ID of the major RNA product derived from this locus ,
- Gene activity is the measure of the ability of a gene to be transcribed and / or to form translation products.
- Gene Expression The process of forming a gene product and / or expression of a genotype into a phenotype.
- Multigenbiomarker Combination of several gene sequences whose gene activities form a combined overall result (eg a classification and / or an index) by means of an interpretation function. This result is specific to a condition and / or an investigation question.
- Hybridization Conditions Physical and chemical parameters well known to those skilled in the art that may affect the establishment of a thermodynamic equilibrium of free and bound molecules. In the interest of optimal hybridization conditions, the duration of contact of the probe and sample molecules, cation concentration in the hybridization buffer, temperature, volume, and concentrations and ratios of the hybridizing molecules must be matched.
- Amplification conditions Constant or cyclic reaction conditions that allow the amplification of the starting material in the form of nucleic acids.
- the reaction mixture are the individual components (deoxyribonucleotides) for the resulting nucleic acids, as well as short oligonucleotides, which can attach to complementary regions in the starting material, as well as a nucleic acid synthesis enzyme, called polymerase.
- the cation concentrations, pH, volume and the duration and temperature of the individual reaction steps which are well known to the person skilled in the art are of importance for the course of the amplification.
- Primer is an oligonucleotide which can be used as a starting point for nucleic acid-replicating enzymes, such as, for example, B. the DNA polymerase is used. Primers may consist of both DNA and RNA (primer 3, see, e.g., http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi of MIT).
- a probe is a
- Nucleic acid fragment having a molecular tag (for example fluorescent markers, in particular Scorpion ®, molecular beacons, Minor Groove Binding- probes, TaqMan ® probes, isotopic label, etc.) are provided can be used and for the sequence-specific detection of target DNA and / or target RNA molecules.
- a molecular tag for example fluorescent markers, in particular Scorpion ®, molecular beacons, Minor Groove Binding- probes, TaqMan ® probes, isotopic label, etc.
- PCR is the abbreviation for the term "polymerase chain reaction.”
- the polymerase chain reaction is a method to amplify DNA in vitro outside a living organism using a DNA-dependent DNA polymerase according to the present invention, to amplify short parts - up to about 3,000 base pairs - of a DNA strand of interest, which may be a gene or just a part of a gene or non-coding DNA sequences.
- PCR a number of PCR methods are known in the art, all of which are encompassed by the term "PCR”. This applies in particular to "real-time PCR” (see also the explanations below).
- PCR primers typically, PCR requires two primers to set the starting point of DNA synthesis on each of the two strands of DNA, thereby limiting the range of duplication on both sides.
- primers are well known to those skilled in the art, for example from the website "Primer3", see for example http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi of MIT.
- RNA transcript For the purposes of the present application, a transcript is understood to mean any RNA product which is produced on the basis of a DNA template.
- Small RNAs in general. Representatives of this group are in particular, but not limited to: a) small cytoplasmic RNA (scRNA), which is one of several small RNA molecules in the cytoplasm of a eukaryote. b) snRNA (small nuclear RNA), one of the many small RNA forms found only in the nucleus. Some of the snRNAs play a role in splicing or in other RNA-processing reactions.
- scRNA small cytoplasmic RNA
- snRNA small nuclear RNA
- RNAs 1 small non-protein codinq RNAs 1 which include the so-called small nucleolar RNAs (snoRNAs), microRNAs (miRNAs), short interfering RNAs (siRNAs) and small double-stranded RNAs (dsRNAs), which increase gene expression on many levels, including chromatin architecture, RNA editing, RNA stability, translation, and possibly also transcription and splicing. In general, these RNAs are multiply processed from the introns and exons of longer primary transcripts, including protein-coding transcripts. Although only 1, 2% of the human genome encodes proteins, a large part is nevertheless transcribed.
- ncRNA non-protein-coding RNAs
- snoRNAs Small nucleolar RNAs
- miRNAs have a length of about 60 to 300 nucleotides.
- miRNAs miRNAs
- siRNAs short interfering RNAs
- miRNAs are derived from endogenous short hairpin precursor structures and usually use other loci with similar - but not identical - sequences as the target of translational repression.
- siRNAs arise from longer double-stranded RNAs or long hairpins, often of exogenous origin. They usually target homologous sequences at the same locus or elsewhere in the genome, where they participate in so-called gene silencing, a phenomenon also called RNAi. However, the boundaries between miRNAs and siRNAs are fluid.
- the term "small RNA” may also include so-called transposable elements (TEs) and in particular retroelements, which are also understood for the purposes of the present invention by the term "small RNA”.
- RefSeq ID This name refers to entries in the NCBI database (www.ncbi.nlm.nih.gov). This database provides non-redundant Reference standards for genomic information. This genomic information includes, among others, chromosomes, mRNAs, RNAs, and proteins. Each RefSeq ID represents a single, naturally occurring molecule of an organism. The biological sequences representing a RefSeq are derived from GenBank entries (also NCBI) but are a collection of informational elements. These information elements come from primary research at the DNA, RNA and protein levels.
- accession number represents the entry number of a polynucleotide in the NCBI gene bank known to the person skilled in the art. In this database, both RefSeq IDs and less well characterized and redundant sequences are managed as entries and made available to the public (www.ncbi .nlm.nih.gov / gene bank / index.html).
- PCR polymerase chain reaction
- Suitable PCR primers are, for example, primers having the sequences of SEQ ID NO: 687 to SEQ ID NO: 742. However, it is known to the person skilled in the art that a variety of other primers can be used to practice the present invention.
- PCR is one of the most important methods in molecular biology and molecular medicine. Today it is used in a very broad thematic spectrum, eg. In the detection of viruses or germs, in sequencing, in the detection of kinship, the generation of transcription profiles and the quantification of nucleic acids [Valasek and Repa, 2005; Klein, 2002]. In addition, any sequence sections of the nucleic acid inventory of an organism can be cloned in a simple manner with the aid of the PCR.
- the variety of developed PCR variants allows u. a. a targeted or random change in the DNA sequence and even the synthesis of larger, in this form previously nonexistent sequence sequences.
- RNA can be detected with high sensitivity and RNA can also be qualitatively detected by reverse transcription (RT) [Wong et al., 2005; Bustin 2002].
- RT reverse transcription
- a further development of this method is the real-time PCR, which was first introduced in 1991 and enables not only qualitative statements but also quantification.
- Real-time PCR also called quantitative PCR (qPCR)
- qPCR quantitative PCR
- the detection takes place here already during the amplification.
- the fluorophores Based on fluorescently labeled probes, the fluorophores, amplification can be monitored in real time.
- the fluorescent PCR products and thus the intensity of light-induced fluorescence emission increase. Since the increase in fluorescence and the amount of newly synthesized PCR products over a wide range are proportional to each other, the data obtained from the data obtained Templates are determined. Gel electrophoretic separation of the amplificates is no longer necessary. The results are directly available, resulting in significant time savings. Since the reactions take place in closed vessels and no further pipetting steps are required after the start of the PCR, the risk of contamination is reduced to a minimum.
- the fluorophores used are either nucleic acid-binding fluorescent dyes such as SYBRGreen or sequence-specific fluorescent probes such as Taq-Man probes, LightCycler probes and molecular beacons [Kubista et al., 2006].
- SYBRGreen is a dye whose fluorescence increases strongly as the molecule binds to double-stranded DNA. This cost-effective solution is particularly suitable for the parallel execution of multiple reactions with different primer pairs. Disadvantages lie in the low specificity, since SYBRGreen binds sequences nonspecifically to each double-stranded DNA, and in that no multiplex measurements can be performed.
- each DNA double strand breaks down into its two single strands at a characteristic temperature, the melting temperature. Since the double-stranded DNA of specific PCR products has a higher melting point than nonspecifically resulting primer dimers, a distinction based on the fluorescence decrease with increasing temperature is possible.
- the detection with fluorescence-based probes is highly specific, but also very costly.
- the PCR approach contains, in addition to the PCR primers, a sequence-specific TaqMan hybridization probe which has a quencher and a reporter dye.
- the probe is complementary to a sequence lying between the primers.
- the fluorescence is suppressed by the proximity of the quencher.
- the quencher swallows the fluorescence emission of the excited fluorophore.
- this probe hybridizes to the target sequence, it is hydrolyzed by the Taq polymerase during PCR, the reporter dye is spatially removed from the quencher and emits detectable fluorescence upon excitation.
- the PCR approach contains two fluorescently-labeled PCR primers Probes (donor and acceptor fluorescent dye).
- Probes donor and acceptor fluorescent dye
- a fluorescence signal which can be measured outwardly only arises in the case of directly adjacent hybridization of the two probes with the specific target sequence. In a subsequent melting curve analysis, even the presence and type of single point mutations within the hybridization regions of the probes can be detected.
- Another example is the Molecular Beacons. These oligonucleotides contain mutually complementary sequences at the 5 ' and 3 ' ends, which hybridize in an unbound state and form a hairpin structure. Reporter fluorophore and quencher, located at both ends, are in close proximity. Only when the probe binds to the template, the two dyes are spatially separated, so that after excitation again fluorescence is measurable.
- Scorpion and Sunrise primers form two further modifications for sequence-specific probes [Whitcombe et al. 1999].
- the quantitative determination of a template can be done by absolute or relative quantification.
- absolute quantification the measurement is based on external standards, e.g. Plasmid DNA in different dilutions, instead.
- relative quantification uses so-called housekeeping or reference genes as reference [Huggett et al., 2005]. These reference genes are constantly expressed and thus offer the opportunity to standardize different expression analyzes. The selection of housekeeping genes must be done individually for each experiment.
- housekeeping genes having the sequences of SEQ ID NO: 676 to SEQ ID NO: 686 are preferably used.
- the generated experiment data are evaluated using the device's own software. For the plot, the measured fluorescence intensity is plotted against the number of cycles. The resulting curve is divided into three areas. In the first phase, ie at the beginning of the reaction, the background noise predominates, a signal of the PCR product is not yet detectable. The second phase corresponds to the exponential growth phase. In this segment, the DNA template almost doubles in each reaction step. Critical to the evaluation is the cycle at which detectable fluorescence occurs and the exponential phase of amplification begins. This threshold Cycle (CT) value or Crossing Point provides the basis for calculating the starting amount of existing target DNA. In the case of an absolute quantification, the software determines the crossing points of the different reference dilutions and quantifies the template quantity on the basis of the calculated standard curve. In the last phase, the reaction finally reaches a plateau.
- CT Cycle
- Quantitative PCR is an important tool for gene expression studies in clinical research. With the ability to accurately quantify mRNA, the search for new drugs can be used to analyze the effects of certain factors on cells, to observe the differentiation of progenitor cells into different cell types, or to track gene expression in host cells in response to infection. By comparing wild-type and cancer cells at the RNA level, it is possible to identify genes in cell culture that have a decisive influence on carcinogenesis.
- real-time PCR is used primarily for the qualitative and quantitative detection of viruses and bacteria. In clinical routine, especially in the field of intensive care, the doctor needs a quick and clear finding. On the basis of real-time PCR, tests can be carried out that deliver the result on the same day. This is an enormous advance in the clinical diagnosis of sepsis.
- so-called isothermal amplification methods such as NASBA or SDA or other technical variants can be used for the detection of the target sequence preceding the detection.
- a preferred method for selecting multigene biomarker sequences comprises the following steps:
- Patient selection is based on the extreme group procedure
- control genes are also suitably determined, for example those with the sequences of SEQ ID NO: 670 to SEQ ID NO: 675.
- a preferred embodiment of the present invention is also in a use in which the gene activities are determined by means of a hybridization method, in particular on at least one microarray.
- the advantage of a microarray lies in the higher information density of the biochip compared to the amplification method. So it is e.g. It is easily possible to provide several 100 probes on a microarray in order to examine several questions simultaneously in a single examination.
- the gene activity data obtained by means of the invention can also be used advantageously for electronic further processing, e.g. B. be used for recording in the electronic medical record.
- a further embodiment of the invention consists of the use of recombinantly or synthetically produced, specific nucleic acid sequences, partial sequences, individually or in part, as multigene biomarkers in sepsis assays and / or for evaluating the effect and toxicity in drug screening and / or for the preparation of therapeutics and of substances and mixtures intended as therapeutic agents for the prevention and treatment of SIRS and sepsis.
- the sample is selected from: tissue, body fluids, in particular blood, serum, plasma, urine, saliva or cells or cell components; or a mixture of them.
- samples especially cell samples, be subjected to lytic treatment to release their cell contents.
- polynucleotide sequences of SEQ ID NO: 1 to SEQ ID NO: 669 from blood and blood cells and probes derived therefrom, which can be used for generating multigene biomarkers, are disclosed (see Table 32).
- Tables 1 and 16 exemplify sequence selection for multigene biomarkers for distinguishing infectious / non-infectious states
- Tables 20 and 21 exemplify sequence selection for multigene biomarkers to distinguish Gram-positive and Gram-negative infections.
- Class prediction uses data / samples / patients assigned to existing or defined classes / groups (so-called training data set) to develop an analytical method (classification algorithm) that reflects the differences between the groups. Independent samples (so-called test data set) are used to evaluate the separation quality of the classification rule.
- the procedure can be divided into the following steps:
- Each group is then split to create 2 equivalent subsets, a training record and a test record.
- Profiles for the training record ideally contain data that reflects a maximum difference between the groups.
- DA Discriminant Analysis
- RF Random Forests
- GPLS Generalized Partial Least Squares
- SVM Support Vector Machines
- kNN k-Nearest Neighbors
- DA Discriminant Analysis
- Random Forests The Random Forests classification is based on the combination of decision trees, [Breiman, 2001]. The process of the algorithm is something like this:
- GPLS Generalized Partial Least Squares
- Support Vector Machine The Support Vector Machine classifier is a generalized linear classifier. The input data is mapped into a higher dimensional space and in this space an optimal separating (hyper) plane is constructed. These barriers, linear in higher-dimensional space, transform into nonlinear barriers in the space of input data, [Vapnik, 1999].
- k-nearest neighbors In the k-closest neighbors method, the class affiliation of an observation (of a patient) becomes based on the nearest k neighbors in its vicinity decided. As a rule, the neighborhood is determined by Euclidean distance, and the class membership is then decided by a majority vote [Hastie et al., 2001].
- a method to determine a multigene biomarker should be developed that reflects an infectious complication such as sepsis.
- the biomarker and associated index value also known as “score” form the basis of a so-called “in vitro diagnostic multivariate index assay” [IVDMIA, FDA Guidelines, 2003] for improving the diagnosis of systemic infections.
- the classification rule resulting from the method should allow a differentiation of SIRS and sepsis patients with improved sensitivity and specificity compared to the established biomarker procalcitonin, but is not limited to this question.
- populations are most clearly defined that represent their presence or absence.
- SIRS non-infectious
- a plan for the collection or selection of the associated RNA samples is determined. From the selected samples, gene expression profiles are measured on a suitable platform, preprocessed and subjected to a quality control. Systematic measurement errors are corrected and outliers eliminated.
- Step 3 Classification procedure.
- Various classification methods are tested for their ability to separate with respect to the pathophysiological conditions to be differentiated. For this purpose methods of cross-validation are used. A classification method with the smallest classification error is selected, with the smallest necessary number of genes being co-determined. As a reasonable rule, it has been found that the number of genes should always be smaller than the number of samples in the training data set in order to avoid over-fitting. Finally, the resulting classification rule is defined.
- Patient selection is important when setting up the training data set.
- a sensitivity of approximately 75% in the training data and approximately 65% in the test data set was achieved for the time being.
- this relatively low classification quality could not be explained by the poor optimization of the classifier but by the insufficiently precise selection of sepsis patients.
- sepsis patients after peritonitis were classified much more correctly than sepsis patients after a "VAP" (ventilator-associated pneumonia) .
- VAP ventilation-associated pneumonia
- extreme groups can be useful. Thereafter, in a study, only those patient groups are taken into account that reflect the examined effect as clearly as possible. The selected samples represent an idealized case in which many effects occurring in practice (e.g., frequency of disease) are not taken into account.
- Liu Liu [Liu et al., 2005] suggested forming extreme groups for the training data set of a microarray-based classifier. Using cancer survivor survival as an example, it has been shown that the use of extreme groups (patients who died in a short time vs.
- the sepsis selection was also made using clinical sepsis diagnosis.
- the severity of the disease between the group of sepsis patients and the control group of SIRS patients was not considered. This may be the reason for the low classification quality and its dependence on Be classification algorithm.
- Johnson Johnson et al., 2007
- patients were divided into two groups after trauma, with one infectious complication and no infection.
- the advantage of this study was that patients in the two groups differed little in comorbidity and pretreatment.
- the preselection is not representative for all sepsis patients and the generalization of the sepsis-relevant gene expression pattern revealed here to patients with a different background (to other risk groups) is not self-evident.
- Applicants' patient database included 400 ITS patients who were suspected to be at risk of sepsis over a two-and-a-half-year period and detailed documentation of the patient's clinical history throughout their stay. The RNA samples were collected for approximately 7-14 sepsis-relevant days.
- Step 1 Quality control: Based on the preselection of a patient collective confirmed by the expert's knowledge, the associated gene expression data were subjected to various similarity analyzes in order to rule out atypical hybridization results [Buneß et al., 2005], which generated the final training data matrix.
- 2nd step normalization or preprocessing of the data: Different methods of background correction and normalization were compared. Methods with a variance-stabilizing transformation were best shown [Rocke and Durbin, 2001]. Normalization by Box-Cox [Box and Cox, 1964], with subsequent median and MAD standardization, was the best normalization procedure. Its advantage, namely the normalization of individual profiles (compared to the normalization of the entire data matrix according to, for example, Huber [Huber et al., 2003]), was specifically used in the bootstrap in particular.
- Step 3 Filter: A filter was used to identify the best classifier genes. The filter consisted of the following steps:
- Step 4th step Classification: The best of the selected transcripts were then used for classification.
- the classification step compared different linear and non-linear methods [Hastie et al., 2001]: DLDA, LDA, RF, GPLS, SVM, and kNN.
- Step 5 Internal validation: To assess the quality of the classification, the 10-fold cross-validation was used, with the cross-validation repeated several times (20 or even 1000 times).
- Step 6 Selection of the transcripts: The final selection of the transcripts for the classifier was carried out with the aid of bootstrap.
- FIG. 1 shows the classification error for the linear discriminant analysis (LDA). Since the curve reaches its minimum at about 12 features, the results obtained with this number of genes were further presented.
- Tab. 2 summarized the results of the various classification methods, which were obtained by means of 20 repetitions of a 10-fold cross-validation.
- Table 2 shows that the estimated sensitivity in the range of 95% and the estimated specificity - with the exception of DLDA - are in the range of more than 90%. The most promising are the results using LDA and SVM. In the case of these two classification methods, only a few patients were predominantly wrongly classified, so that a misclassification rate of at most 5% is achieved. Due to the great complexity of the SVM method and the resulting computational effort, which would cause the optimization of an SVM classifier, as well as the better biological interpretability of a classifier based on the LDA, the Applicant decided to use the classifier based on the LDA to develop. The classification rule resulting from the LDA was converted into a score. The score is shown for an exemplary collective of 96 patients in FIG. A value> 10 indicates that infection (ie sepsis) is very likely. A value between +10 and -10 indicates that there is some risk of sepsis. Finally, a value ⁇ -10 indicates that infection is very unlikely.
- the quality of the multigene biomarkers according to the invention was compared with the established biomarkers PCT and CRP, for which the associated ROCs for the training data set were calculated (FIG. 3).
- AUC (PCT) 0.326
- AUC (CRP) 0.656
- AUC (PCT & CRP) 0.940
- AUC (multigene biomarker) 0.997.
- FIG. 4a shows the distribution of score values as a function of the clinical diagnosis.
- the distribution of the PCT and CRP values for the same data set is shown in FIG. 4b. While the index values or scores are consistent with the clinical diagnosis, PCT distribution in particular shows that severe SIRS is more likely to be classified as sepsis and uncomplicated sepsis rather than non-infectious.
- a non-specific distribution is shown by the markers CRP and WBC (leukocyte count).
- the quality of the multigene biomarkers according to the invention and of the method according to the invention was investigated on expression data of other patients of a foreign collection center. Again, clinical and molecular biology ratings were consistent in 90% of cases.
- FIG. 5 shows the score curve in the course of the disease for individual patients.
- the multigene biomarker of the present invention reflects the clinical diagnosis.
- the validation analysis included patient profiles of the applicant's patient database, whose expression profiles were not included in the training dataset were represented. Due to the lack of a golden standard for the diagnosis of sepsis, this independent test dataset was examined in stratified subgroups. Patient profiles were divided and classified according to the severity of the disease (see Fig. 4). In fact, patients with uncomplicated SIRS were almost exclusively classified as non-infectious. Patients with severe SIRS (SIRS with additional multi-organ dysfunction (MOD)) were predominantly identified as noninfectious. Patients with uncomplicated sepsis were predominantly classified as systemically infectious. The infectious complication was most often diagnosed among patients with severe sepsis or septic shock. This finding was confirmed in a group of patients recruited and diagnosed in an independent center ( Figure 6).
- FIG. 2 shows a score (a) and its distribution for the training data record (b);
- FIG. 3 shows the quality of a multigene biomarker compared to established monomolecular biomarkers PCT and CRP or their combination (via
- Fig. 4 shows a distribution of biomarker values as a function of the clinical
- Diagnosis (a) multigene biomarker score, (b) PCT, CRP and WBC; 5 shows a course of the score for three patients (the gray area marks the
- Fig. 6 shows a distribution of the scores for expression data of a foreign
- FIG. 7 is a schematic representation of the microarray design and the three
- qPCR run shows an example of a qPCR run (marker EPC1)
- Fig. 1 1 is a schematic representation of the derived score
- Fig. 12 is an illustration of the differences in expression between the
- Patient Groups Boxplots of markers created from 31 patient samples (19 with sepsis diagnosis, 12 with SIRS); the legend explains the used ones
- Fig. 13 is a box plot of the normalized real-time PCR data for the
- Biomarker candidate CDKN1 C (SEQ ID NO: 104) for distinguishing Gram-positive and Gram-negative infection
- Fig. 14 is a box plot of the normalized real-time PCR data for the biomarker
- FIG. 15 is a box plot of the normalized real-time PCR data for the biomarker candidate METTL7B (SEQ ID NO: 145) to distinguish Gram-positive and Gram-negative infection; and FIG. 16 shows a box plot for the non-coding marker with SEQ ID NO: 207; on the y-axis, the mean Ct value during real-time amplification is shown.
- a method for the determination of multigene biomarkers will be disclosed.
- the classification rule resulting from the procedure should allow a differentiation of SIRS and sepsis patients.
- Another classification rule is to allow the distinction between the focus of infection pneumonia and peritonitis.
- transcripts were identified which, unaffected by patient heterogeneity due to age, comorbidities, and medications, reflect the molecular differences between groups of sepsis patients. Depending on the patient population studied, the number of biomarkers required for successful classification varies.
- biomarker candidates Two examples will illustrate the potential of biomarker candidates:
- FIG. 7 shows a schematic representation of the focused sepsis microarray. Spotted on epoxy-silanized glass slides (Nexterion E-Slides, manufacturer Schott, FRG), each gene-specific oligonucleotide is represented in triplicate. The three identical sub-arrays are hybridized to a patient sample. In addition to the marker-specific oligonucleotides, probes for controls (monitoring of the entire sample preparation and hybridization process) are also represented on the array.
- the marker genes addressed on the array are assigned with high significance to the signaling pathways in the human cell shown in FIG. 8 and the associated functionalities. A high relevance for immunological and inflammatory processes and thus also sepsis is given.
- the Ingenuity Pathway Analysis software (Ingenuity Systems, USA, www.ingenuity.com) was used to clarify the functional context of the identified markers.
- the markers are classified into functional networks, which may then have relevance for physiological and pathological processes.
- the markers are of high significance for immunological and inflammatory processes, which suggests a close connection to sepsis from a functional point of view. This provides biological plausibility, a prerequisite for biomarkers.
- RNA from patient blood was analyzed by reverse transcription (Superscriptll, Invitrogen, USA) in a reaction volume of 30 ⁇ l ??? transcribed into cDNA.
- the primer used was a PolydT primer (18mer). Aminoallyl-dUTP was added to the reaction, thus substituting 80% of the dTTP amount in the mRNA strand with AA-dUTP (Table 4).
- Table 4 Sample pipetting approach for cDNA synthesis. 4 ⁇ g of totaIRNA and 2.5 ⁇ g of OligodT primer were submitted. RNAse-free water was added to 30 ⁇ l total volume.
- the entire samples are placed on each column, which is centrifuged at 1 10OOxg for 10 min. After washing twice with 450 .mu.l RNase-free H 2 O and intervening centrifugation at 1 1000xg for 10 min, the columns are inverted on a new 1.5 ml reaction vessel and centrifuged for 3 min at 15000xg. The eluate obtained now purified single-stranded cDNA with a volume of about 20 - 40 ul, which is reduced in the Speedvac to dryness.
- Fluorescent dyes are used to detect the hybridization signals.
- a fluorescent dye was used by Dyomics (manufacturer: Dyomics GmbH, Jena, FRG).
- DY-647 (Cy5 analogues) are purchased as N-hydroxysuccinimide esters (NHS esters) and used for fluorescent labeling. The chemical coupling of the dyes takes place on the incorporated AA dUTPs.
- the cDNA is dissolved in 10 .mu.l H 2 O and divided with 5 ul each on two tubes. The dissolved samples are incubated at 42 ° C for 5 min. Subsequently, 3 ⁇ l of bicarbonate buffer are added to each sample. The fluorescent dye is dissolved in DMSO (manufacturer SIGMA-Aldrich, FRG). 75 ⁇ g of dye are used per sample. This photosensitive reaction takes place in the dark for 1 h. After this time, the samples are filled with H 2 O to a final volume of 30 ⁇ l. The samples are pipetted together with 80 ⁇ l each of H 2 O and 100 ⁇ l membrane binding solution and purified by Promega Kit (Promega Wizard-SV gel and PCR CleanUP System, PROMEGA, USA) according to the manufacturer's instructions.
- Promega Kit Promega Wizard-SV gel and PCR CleanUP System, PROMEGA, USA
- the columns are centrifuged dry for 1 min at 16000xg and eluted twice with 50 ul H 2 O (1 min, 10000xg). Thereafter, each sample is spiked with 10 ⁇ l of Cot-1 DNA (Invitrogen, USA) and 400 ⁇ l of H 2 O. The concentration of the labeled samples is carried out by means of Microcon YM-30 (10000xg, 10 min centrifugation). The columns are inverted and placed on a new tube and centrifuged at 15,000xg for 3 min. The volume of the cDNA / Cot-1 DNA mixture is adjusted to 32 ⁇ l. The fluorescently labeled cDNA / Cot-1 DNA mixture (32 ⁇ l) is admixed with 58 ⁇ l of hybridization mixture (Table 5).
- the mixture After denaturation at 98 ° C. for 3 minutes, the mixture is pipetted into the hybridization chambers of the TECAN hybridization automaton (HS-400, manufacturer Tecan, Austria).
- the contained formamide lowers the melting temperature of the hybrid and thus allows a good hybridization.
- the addition of 10% SDS improves the wetting of the biomolecules on the glass slide.
- the yeast t-RNA / poly-A mix ensures that there are no non-specific bonds and background noise. As a result, poly (A) binds to the poly (T) end of the labeled cDNA, with the yeast t-RNA blocking all nonspecific sequences.
- Table 5 The hybridization mixture for a sample.
- the arrays are initially washed with hybridization solution and then incubated with the samples. The process is incubated for ten hours at a temperature of 42 ° C in hybridization chambers of the Tecan HS-400 device with constant agitation of the hybridization mixture on the array surface carried out. Finally, the arrays are washed and dried in three automated steps.
- the expression levels of the transcripts studied were compared to the Wilcoxon rank sum test for infection status (infectious vs. non-infectious).
- Table 8 shows the differential gene expression in the patient groups measured on the microarray.
- Table. 8 differential gene expression between patient groups; p-values for analyzes 1 and 2: grayed out are the markers that show a significant difference between the groups for the respective analysis; Analysel (non-infectious vs. infectious cause of multiple organ failure): CPB vs patient. septic patients with a focus on peritonitis or pneumonia; Analysis2 (focus of infection, distinction of focus peritonitis from focus pneumonia): 18 septic patients with focus on peritonitis. 12 patients with the focus on pneumonia
- Example 2 Preparation of a Classifier for the Identification of SIRS and Sepsis Patients by Real-Time-PCR
- the markers for classification were selected from the biomarker pool (see Example 1) and show strong differential gene expression in patient groups with and without diagnosed sepsis.
- Relative quantification of gene expression means a statement on the abundance of the target transcript, for example with respect to a calibrator. This can be a reference value determined from the expression values of constantly expressed genes (so-called reference genes or housekeeping genes). For each organism and tissue, such reference genes are specific and must be carefully selected for each study. Based on the gene expression profiles from the whole blood of the sepsis and control patients, the most stable genes with the lowest variability were selected and used in the quantitative PCR for normalization.
- Table 1 1 is a list of the primers used in real-time PCR and their SeqlDs. For each target sequence several primer combinations are possible, the table represents only one of many possibilities.
- the patients' whole blood was removed from the intensive care unit by the patients using the PAXGene kit according to the manufacturer's instructions (Qiagen) and the RNA was isolated.
- RNA was then removed from the batch by alkaline hydrolysis.
- the reaction mixtures were not purified, but made up to 50 ⁇ l with water.
- the Platinum SYBR Green qPCR SuperMix UDG kit from Invitrogen was used.
- the patient cDNA was diluted 1: 100 with water and 1 ⁇ l of each was used in the PCR.
- One PCR plate (BIORAD) with all 31 patients and no template controls (NTC) was pipetted in 3-fold per marker.
- the iQ TM 5 Multicolor Real-Time PCR Detection System from BIORAD was used with the associated evaluation software. The results of such a qPCR run are shown in FIG. With the aid of the evaluation software, such images were generated for each of the 18 markers and 5 housekeepers, from which the corresponding Ct values could then be derived. The Ct values are automatically calculated by the program in the area of the linear increase of the curves. In the example of EPC1, the Ct values for the sepsis patients were in the range of 25.08 - 27.71 and for the SIRS patients in the range of 28.08 - 35.91.
- the measured expression signals were saved in Excel format and averaged over the 3-fold determinations.
- the marker MON2 with 15 missing values and the patients 6065 and 81 1 1 with 13 or a missing value were excluded from the analysis.
- the training record contained 18 infectious (62%) and 1 1 non-infectious (38%) samples.
- the 3 most stable housekeeper genes were determined from the 5 measured. Subsequently, for each patient, the mean of the 3 selected housekeeper genes was subtracted from the marker genes.
- the linear discriminant analysis [Hastie et al., 2001] was used with a simple cross-validation. The calculation was carried out using the function Ida from the R library MASS. For p markers, the weights of the discriminant function f L D were given by the formula is calculated from the training data, wherein a sample has been omitted sequentially. This sample was classified retrospectively, for which in the previous formula for x, the Ct values of the sample were used. The weights of the discriminant function were calculated so that a positive value of the function means the assignment to the group with an infectious complication and a negative value of the function means the assignment to the group without an infectious complication. The classification procedure was repeated for an increasing number of markers.
- FIG. 10 shows the scores for the 12 marker classification for samples 933 and 790. A schematic representation of the derived score value and the division into 4 areas is shown. If the calculated score is above 6.5, the patient is 95% likely to have sepsis (infectious). If the score is below -6.5, then the probability of not having sepsis for the patient is also 95% (non-infectious).
- Table 13a shows the raw data (Ct values) from the qPCR assays
- Table 13b the weights of the linear discriminant function for increasing number of markers and the associated score values for independent samples 790 and 933 are shown.
- Table 13b Weights of the linear discriminant function for increasing number of markers and the associated score values for independent samples 790 and 933.
- Example 3 Preparation of a Classifier for the Identification of SIRS and Sepsis Patients by Conventional PCR
- the markers for classification were selected from the biomarker pool (see Example 1) and show strong differential gene expression in patient groups with and without diagnosed sepsis.
- Table 15 lists the gene expression gene marker gene products used for classification and their description.
- Table 16 is a list of the primers used in the PCR and the associated Seq IDs. For each target sequence several primer combinations are possible, the table represents only one of many possibilities.
- Table 15 Gene expression gene marker gene products used for classification and their description
- Table 16 List of primers used. For each target sequence several primer combinations are possible, the table represents only one of many possibilities.
- the patients' whole blood was removed from the intensive care unit by the patients using the PAXGene kit according to the manufacturer's instructions (Qiagen) and the RNA was isolated.
- RNA was then removed from the batch by alkaline hydrolysis.
- the reaction mixtures were not purified, but made up to 50 ⁇ l with water.
- the patient cDNA was diluted 1: 500 (or in the case of 4 markers 1:50, SNAPC, EPC1, KIAA0146 and MON2) with water and 1 ⁇ l of each were used in the PCR.
- One PCR plate (96 wells, Nerbe Plus) was pipetted per marker with all 31 patients and no template controls (NTC) in a 3-fold determination.
- a mastermix without template was prepared, this was quilted into 12 ⁇ l aliquots in the PCR plate and to each of the patient cDNA were pipetted (see composition of the PCR reaction mixture).
- a 1, 1-fold SYBR Green solution was prepared.
- 100 ⁇ l of a 100 ⁇ SYBR Green stock solution (prepared from a 10,000 ⁇ SYBR Green stock solution from BMA, BioWhittaker Molecular Applications) were pipetted into 8.9 ml of water and mixed.
- 90 ⁇ l of this solution were added to each PCR batch and this mixture was then transferred to a black plate (96 wells, Greiner). Subsequently, this plate was measured in a fluorescence measuring instrument (TECAN GENios) at 485 nm excitation wavelength / 535 nm emission wavelength.
- the measured expression signals were saved in Excel format, averaged over the 3-fold determinations and the NTC values were subtracted for each marker.
- Patient 6065 with 15 missing values was excluded from the analysis.
- Individual missing values were replaced with the knn algorithm (the function pamr.knnimpute from the R library pamr was used for this).
- the averaged signals were transformed log-2.
- the mean of the 3 housekeeper genes was subtracted from the associated marker genes for each patient. Classification:
- the separability of the training data set was checked by means of simple cross-validation. Subsequently, two independent samples were classified, one from each of the two groups of patients studied (patients 933 and 790). The raw measurement signals were preprocessed in the same way as the training data.
- FIG. 11 The arrangement of the genes and the associated values are summarized in FIG. 11. The differences in expression between the groups are shown: Boxplots of the 15 markers created from 31 patient samples (19 with sepsis diagnosis, 12 with SIRS). By means of the box plots, gene by gene was shown the distribution of the Ct values per group. These Ct values were generated from each patient sample by real-time PCR on the patient's cDNA (Biorad IQ5) and normalized by the Ct values of three reference genes. The x-axis gives the p-value and the Hodge-Lehmann estimator of the Wilcoxon rank-sum test. In the classification, 1 became a sensitivity in the simple cross-validation of 100% and a specificity of 83%, which corresponds to a misclassification of 2 non-infectious samples.
- FIG. 12 shows a schematic representation of the derived score value and the division into 4 ranges. If the calculated score is above 6.5, the patient is 95% likely to have sepsis. If the score is below -6.5, then the probability of not having sepsis for the patient is also 95%. The classification result was projected on this scale. The score of sample 933 took the value of -38.7 and was classified as non-infectious; the score of sample 790 took the value of 9.1 and was classified as infectious.
- Table 18a contains the raw data from the fluorescence measurements with SYBR Green at TECAN GENios.
- Tab. 18b shows the raw data of the independent patient samples as well as the legend for the gene names and their assignment to the SeqlDs.
- Example 4 Exciter Type - Gram Vs. Gram - Differential gene expression in
- biomarkers In genome-wide gene expression analyzes on microarray platforms, biomarkers have been identified that express different levels in septic patients with infections of Gram-negative and Gram-positive bacteria. Starting from this biomarker list with 1 14 markers, it was shown for three markers that these differences in gene expression can be represented by quantitative PCR. For these 3 markers, gene-specific primers were identified and their gene activity determined by quantitative PCR.
- Table 19 List of patients studied. Not highlighted: Patients with gram-negative infection; highlighted in light gray: patients with gram-positive infection.
- RNA Place 50-500ng RNA in a microcentrifuge tube and make up to 1 ⁇ l with nuclease-free water.
- 1 ⁇ l array script 9 ⁇ l of the mix are added to the RNA sample and then incubated for 2 hours at 42 ° C.
- the poly-A overhang at the 3 'end of the mRNA integrates the T7 oligo (dT) nucleotide so that the mRNA, regardless of its sequence, is transcribed into cDNA using ArrayScript. After the 2 hour incubation, the reaction vessel is put back on ice.
- RNA is simultaneously degraded by RNase H.
- the prepared mix is added to the sample and incubated for 14 hours.
- the T7 Enzyme Mix contains T7 RNA polymerase, a highly promoter-specific RNA polymerase that requires a DNA template.
- the T7 oligo (dT) nucleotide used for reverse transcription has a T7 promoter sequence, which is now recognized by the T7 RNA polymerase.
- the in vitro Transcription is thus an amplification and labeling step in one. Subsequent to the incubation, 75 ⁇ l of nuclease-free water are added.
- RNA, primers, enzymes and salt are removed.
- Another purification step after in vitro transcription removes enzymes, salt, and non-embedded nucleotides.
- bead arrays which are arranged on carriers, the bead chips.
- the required buffers, solutions and hybridization chambers are provided by the manufacturer in the form of the bead-chip kit (HumanWG-6 BeadChip Kit, Illumina, www.illumina.com).
- 1, 5 ⁇ g of the respective cRNA sample is made up to 10 ⁇ l with RNase-free water. 20 ⁇ l GEX-HYB solution is added to the sample. 200 ⁇ l of GEX-HCB are loaded into the humidification buffer reservoirs of the hybridization chamber and the bead chips (Human WG-6 BeadChip, Illumina, www.illumina.com) are inserted into the hybridization chamber. 30 ⁇ l of sample is applied to the sample port of the array. The hybridization chamber is closed carefully and the samples are incubated at 58 ° C for 16-20 hours.
- the bead chips are immersed in E1 BC wash solution and washed in high-temp buffer at 55 ° C. This is followed by a rinsing step at room temperature with E1 BC solution, an ethanol washing step and another E1 BC washing step. This is followed by a blocking step with block E1 buffer and a labeling step with block E1 + streptavidin-Cy3, in which the fluorescently labeled streptavidin binds to the biotinylated nucleotides of the cRNA. It will be again washed with E1 BC buffer and then the bead chip is dried by centrifugation (2min at 500 rpm). The Bead Chip can then be scanned in with the Bead Array Reader (Illumina Beadstation 500, www.illumina.com).
- the bead chip is read out fluorometrically.
- the scanner has a resolution of 0.8 ⁇ m and thus the fluorescence is measured at least 9 pixels from each of the 48687 array types placed on an array.
- Each bead type is at least 5 times redundant.
- the Bumina Studio 2.0 program provided by Illumina the fluorescence values of a bead type are averaged and output as an "average signal.”
- the background signal is detected, which is subtracted from each averaged signal.
- the negative controls determine the detection p-value of each individual bead type, which provides information on whether it is a true signal or whether the measured intensity matches the background. For further analysis, only the bead types are used in which at least one of the ten arrays has reached a detection p-value below 0.01.
- the normalization proposed by the data processing program Bead Studio 2.0 (part of the Illumina Beadstation 500) was selected using Cubic Splines. In accordance with the recommendations [MAQC Consortium, 2006], the following correction steps were additionally added.
- the data was further processed with the statistics software (http://www.r.project.org). From all the bead types selected for further analysis, the smallest averaged signal value is determined. This minimum is subtracted from each averaged signal, so that now the smallest averaged signal takes the value 0.
- the constant 16 is added to each averaged signal before logarithmizing to the base 2. After the logarithm receives the smallest average signal is 4. At the same time, the averaged signal is prevented from accepting a negative value.
- the ratio between the expression values is given as a "fold change.” This value indicates the factor by which the transcript was expressed differently in one sample than in the other sample.
- the difference is formed from the average values of the normalized data of both groups, where the FoId Change of gram-positive with respect to gram-negative is given:
- the number 2 is exponentiated with the logarithmic FoId Change to obtain a theoretical change. If the theoretical change takes a value less than one, the change results from the negative reciprocal of the theoretical change. In the opposite case, the FoId Change corresponds to the Theoretical FoId Change:
- a positive change means that the corresponding gene is expressed more strongly in the case of a gram-positive infection than in a gram-negative infection.
- the p-value for the t-test and the Wilcoxon test are also calculated. Assuming that the null hypothesis of the test is correct, the p-value indicates the probability that the measured value will be by chance comes about. If this probability is less than a given bound, it is assumed that the difference is not random.
- Tab. 20 shows the identified biomarkers:
- Table 20 Differential gene expression of transcripts in Gram-positive and Gram-negative sepsis measured on the Illumina gene expression platform
- the three markers and a representative primer pair for quantification by means of real-time PCR are shown in Tab. 21. Furthermore, so-called reference genes are used for relative quantification, which are constantly expressed in the respective tissue. The reference genes used in this experiment are also shown.
- the patients' whole blood in the intensive care unit was approved by the patients using the PAXGene kit according to the manufacturer's instructions (Qiagen). After collection of the whole blood, the total RNA of the samples was isolated using the PAXGene Blood RNA Kit according to the manufacturer's instructions (Qiagen).
- RNA to complementary DNA RNA was transcribed with the reverse transcriptase Superscript II (Invitrogen) in a 20 ⁇ l batch, and the RNA was subsequently removed from the batch by alkaline hydrolysis. The reaction mixtures were then purified by means of microconcolumns.
- the iQ TM 5 Multicolor Real-Time PCR Detection System from BIORAD was used with the associated evaluation software.
- the third root is drawn from the product of the three reference genes.
- the quotient of relative size R and the normalization factor is formed:
- Fig. 13 shows the differential expression of the CDKN1 C gene in septic patients with gram-positive and gram-negative infection.
- the box plot shows the mean normalized Ct values for every 5 patients. These values were determined using real-time PCR on the patient's cDNA.
- Figure 14 shows the differential expression of the CTSL gene in septic patients with gram-positive and gram-negative infection. In the box plot are also the middle normalized Ct values are shown for every 5 patients. These values were determined using real-time PCR on the patient's cDNA.
- Figure 15 shows the differential expression of the gene METTL7B in septic patients with Gram-positive and Gram-negative infection.
- the box plot shows the mean normalized Ct values for every 5 patients. These values were determined using real-time PCR on the patient's cDNA.
- Table 22 shows raw data (Ct values, mean values from triplicates) from the qPCR assays for the marker CDKN1 C (SeqlD 104).
- Table 23 contains normalized raw data from the qPCR assays for the marker CDKN1 C (SeqlD 104).
- Table 24 contains raw data (Ct values, mean values from triplicates) from the qPCR assays for the marker CTSL (SeqlD 149).
- Table 25 shows, according to Vandesompele [Vandesompele et al., 2002], normalized raw data from the qPCR assays for the marker CTSL (SeqlD 149).
- Table 26 contains raw data (Ct values, mean values from triplicates) from the qPCR assays for the marker METTL7B (SeqlD 145).
- Table 27 shows, according to Vandesompele [Vandesompele et al., 2002], normalized raw data from the qPCR assays for the marker METTL7B (SeqlD 145).
- Table 22 Raw data (Ct values, means) from the qPCR assays for the marker CDKN1 C (SeqlD 104). Light gray: patients with gram-negative infection, dark gray: patients with gram-positive infection.
- Table 23 Raw data normalized by Vandesompele from the qPCR assays for marker CDKN 1 C (SeqlD 104). Light gray: patients with gram-negative infection, dark gray: patients with gram-positive infection.
- Table 24 Raw data (Ct values, means) from the qPCR assays for marker CTSL (SeqlD 149). Light gray: patients with gram-negative infection, dark gray: patients with gram-positive infection.
- Table 25 Raw data normalized by Vandesompele from the qPCR assays for marker CTSL (SeqlD 149). Light gray: patients with gram-negative infection, dark gray: patients with gram-positive infection.
- Table 26 Raw data (Ct values, means) from the qPCR assays for the marker METTL7B (SeqlD 145). Light gray: patients with gram-negative infection, dark gray: patients with gram-positive infection.
- Table 27 Raw data normalized by Vandesompele from the qPCR assays for the marker METTL7B (SeqlD 145). Light gray: patients with gram-negative infection, dark gray: patients with gram-positive infection.
- the FoId change of the patients is thus calculated as follows:
- Table 28 shows the medical parameters of the patients included in the analysis, as validated by the clinic.
- Table 28 Medical parameters of the patients included in the analysis. Light gray: patients with gram-negative infection, dark gray: patients with gram-positive infection.
- Example 5 Non-coding RNA - Differential gene expression of a transcript without protein-coding function (so-called non-coding RNA) in SIRS and sepsis patients by means of real-time PCR.
- the marker with SeqlD 207 (Accession No. AA868082) for non-coding RNA is part of the biomarker list above.
- Tab. 30 shows an example of a primer pair for the amplification of the noncoding marker with the SeqlD 207 in the real-time PCR. 10 patients were studied (5 sepsis patients, 5 SIRS patients).
- the patients' whole blood was removed from the intensive care unit by the patients using the PAXGene kit according to the manufacturer's instructions (Qiagen) and the RNA was isolated.
- RNA to complementary DNA RNA to complementary DNA
- cDNA RNA to complementary DNA
- the reverse transcriptase Superscript II Invitrogen
- the gene-specific primers in a 20 ⁇ l assay (10 mM dNTP mix and 2 ⁇ M gene-specific primers ( SeqID 207) and the RNA were then removed from the batch by alkaline hydrolysis, the reaction mixtures were purified with microconcolumns, the eluted cDNA was evaporated in the SpeedVac and then taken up in 50 ⁇ l of water.
- the Platinum SYBR Green qPCR SuperMix UDG kit from Invitrogen was used.
- the patient cDNA was diluted 1: 100 with water and 2 ⁇ l of each were used in the PCR. All batches were pipetted in 3-fold.
- the iQ TM 5 Multicolor Real-Time PCR Detection System from BIORAD was used with the associated evaluation software.
- Figure 16 shows a box plot for the non-coding marker with SeqlD 207 made from 10 patient samples (5 with sepsis diagnosis, 5 with SIRS). The mean Ct value during real-time amplification is shown on the y axis. There is a clear separation between sepsis and SIRS patients.
- Table 32 establishes the relationship between the sequence listing number of each polynucleotide and its publicly available accession number.
- MAQC MicroArray Quality Control
- Valasek MA Repa JJ (2005) The power of real-time PCR.
- WO 2006/100203 Use of geneactivity classifiers for the in vitro classification of gene expression profiles of patients with infectious / non-infectious WO 2004/087949 A method for the detection of acute, generalized inflammatory conditions (SIRS), sepsis, sepsis-like conditions and systemic infections
- SIRS generalized inflammatory conditions
- WO 2006/042581 A method for distinguishing between non-infectious and infectious causes of a multi-organ failure
Landscapes
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Wood Science & Technology (AREA)
- Analytical Chemistry (AREA)
- Zoology (AREA)
- Genetics & Genomics (AREA)
- Engineering & Computer Science (AREA)
- Pathology (AREA)
- Immunology (AREA)
- Microbiology (AREA)
- Molecular Biology (AREA)
- Biotechnology (AREA)
- Biophysics (AREA)
- Physics & Mathematics (AREA)
- Biochemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
Abstract
Description

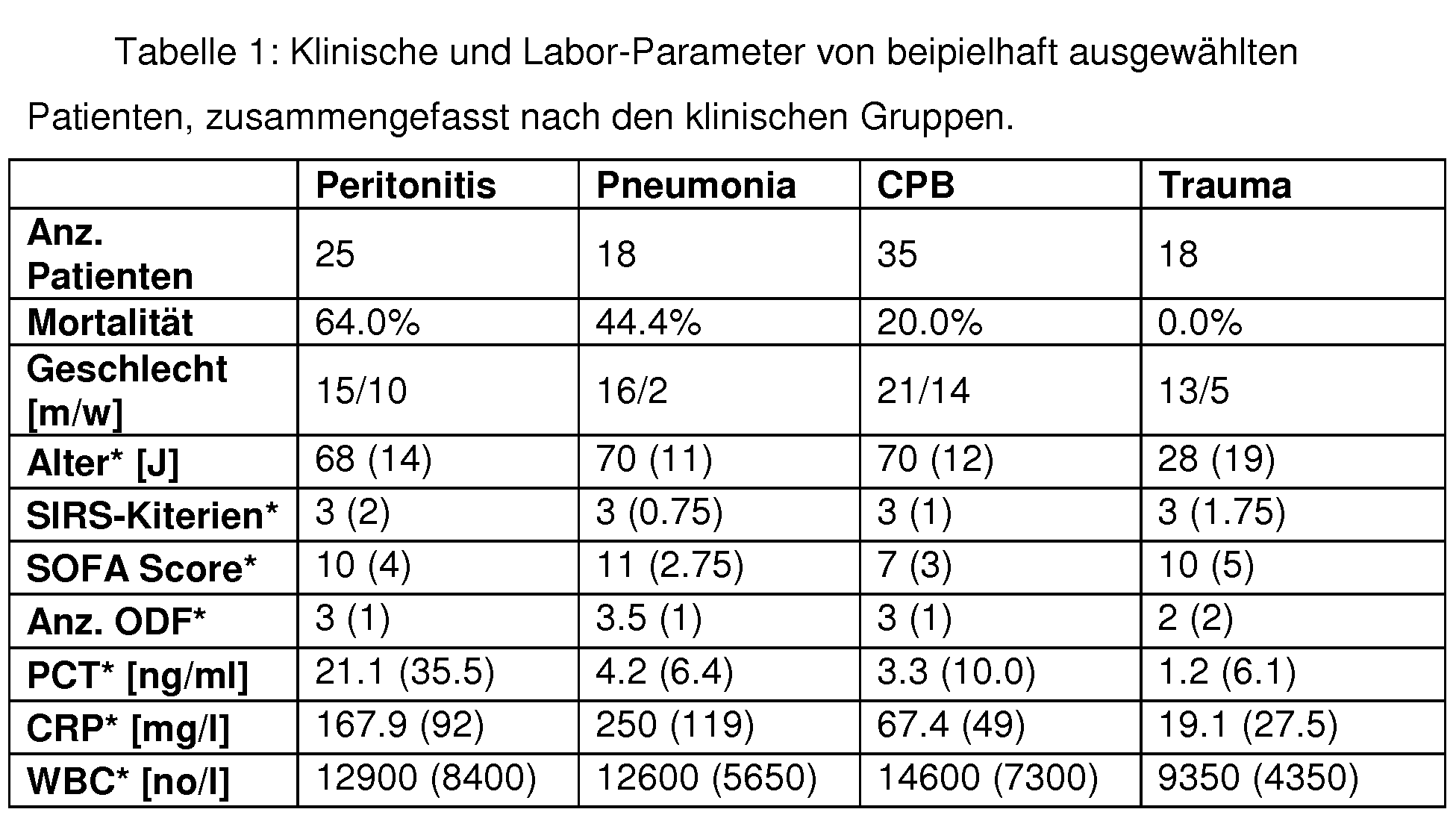



















































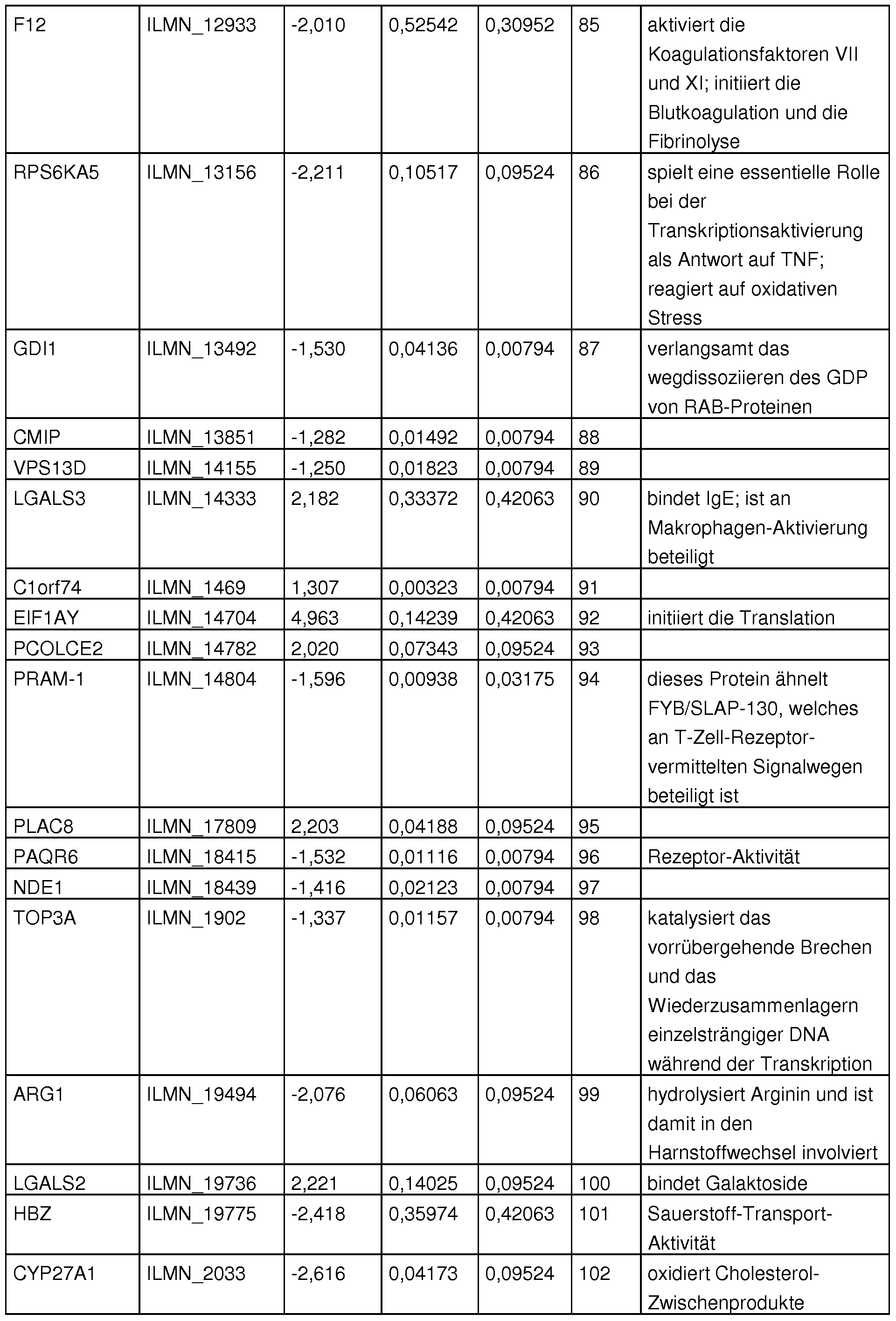

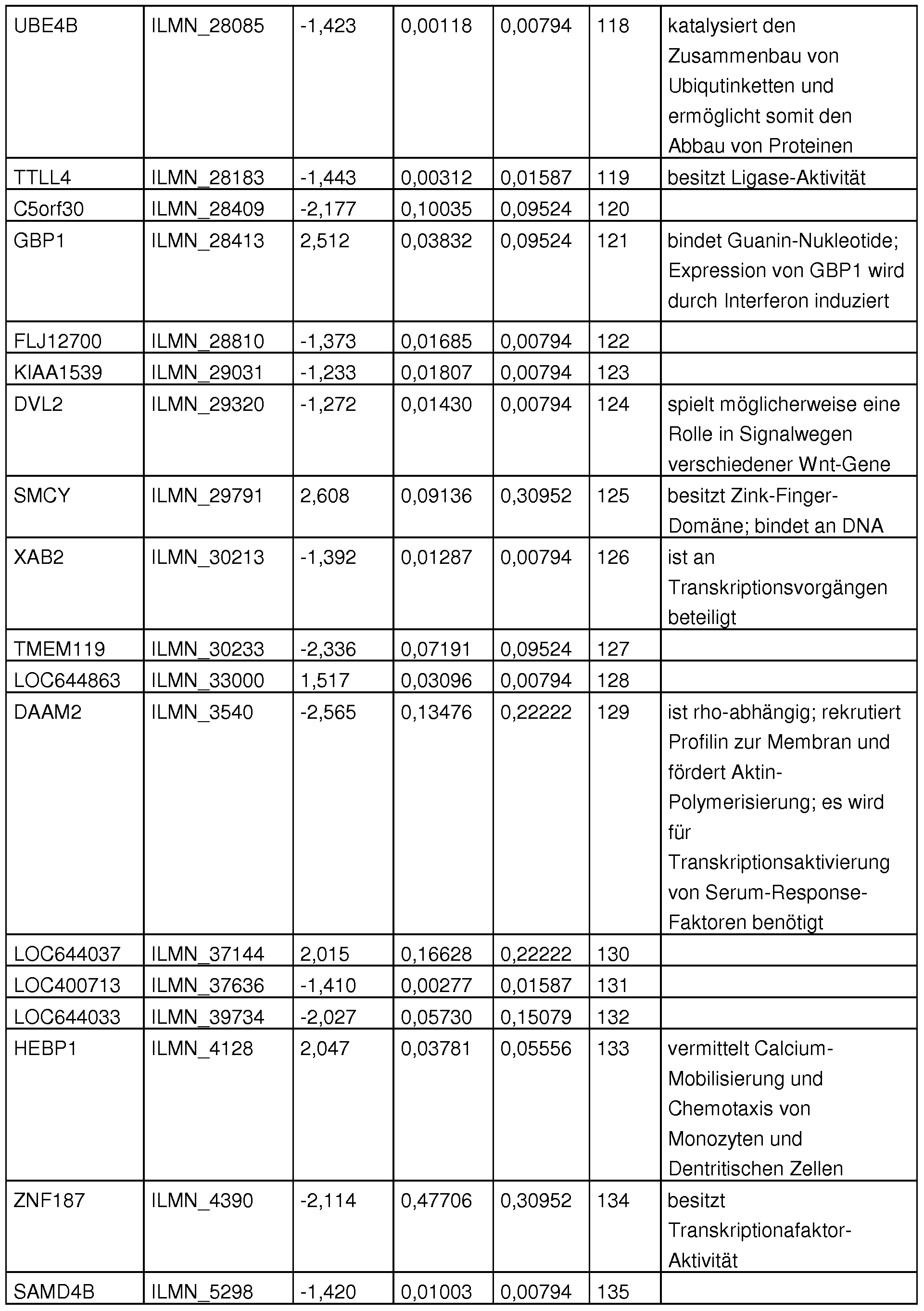

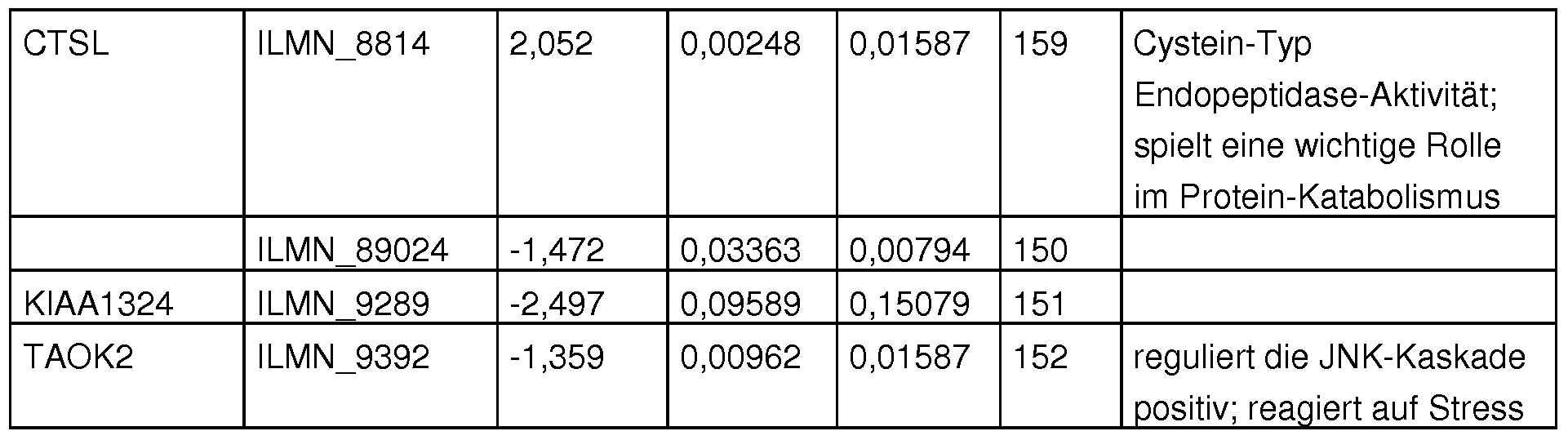











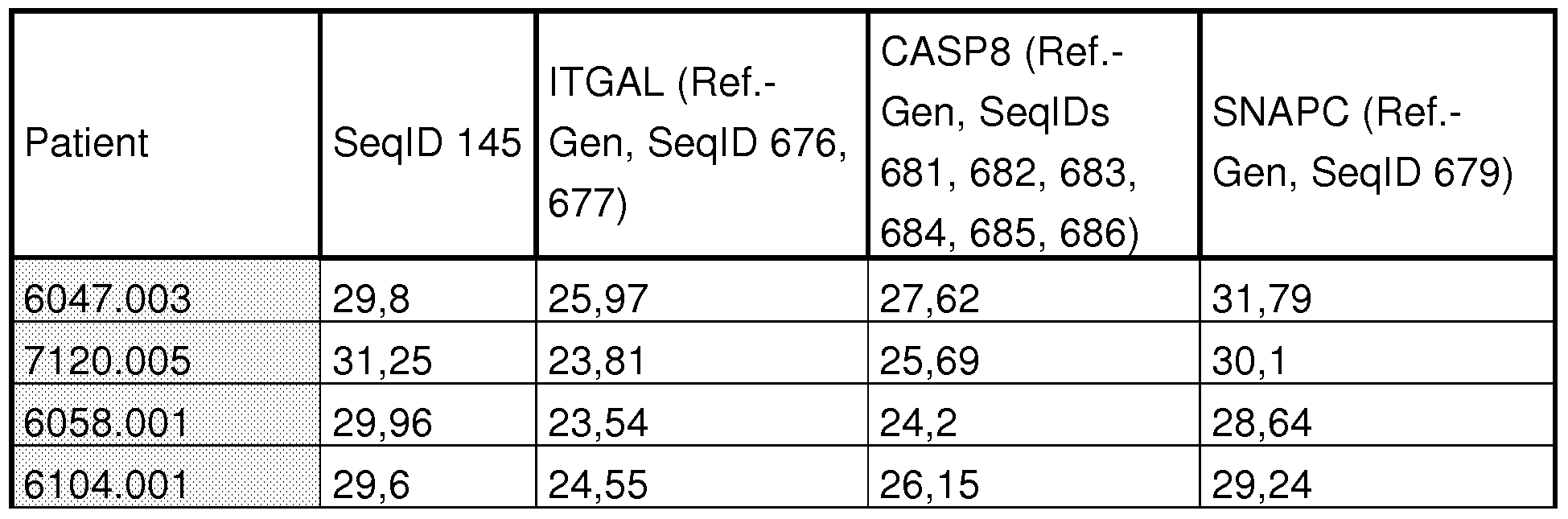









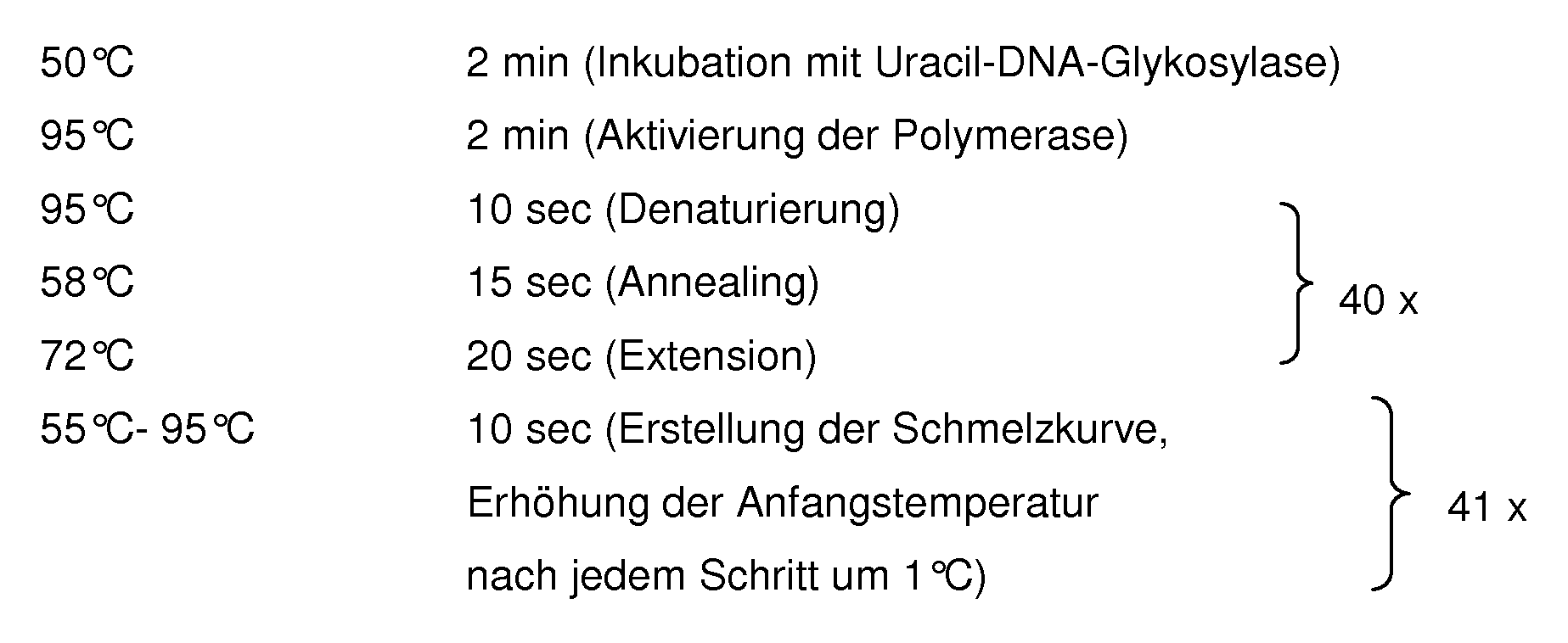







Claims
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011500171A JP2011517401A (ja) | 2008-03-17 | 2009-03-16 | 病態生理学的症状の体外検出及び識別方法 |
| CA2718741A CA2718741A1 (en) | 2008-03-17 | 2009-03-16 | Method for the in vitro detection and differentiation of pathophysiological conditions |
| GB201017326A GB2470707C (en) | 2008-03-17 | 2009-03-16 | Method for the in vitro detection and differentiation of pathophysiological conditions |
| US12/933,169 US8765371B2 (en) | 2008-03-17 | 2009-03-16 | Method for the in vitro detection and differentiation of pathophysiological conditions |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE102008000715.3 | 2008-03-17 | ||
| DE102008000715A DE102008000715B9 (de) | 2008-03-17 | 2008-03-17 | Verfahren zur in vitro Erfasssung und Unterscheidung von pathophysiologischen Zuständen |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2009115478A2 true WO2009115478A2 (de) | 2009-09-24 |
| WO2009115478A3 WO2009115478A3 (de) | 2009-11-12 |
Family
ID=40679552
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2009/053042 WO2009115478A2 (de) | 2008-03-17 | 2009-03-16 | Verfahren zur in vitro erfassung und unterscheidung von pathophysiologischen zuständen |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US8765371B2 (de) |
| JP (1) | JP2011517401A (de) |
| CA (1) | CA2718741A1 (de) |
| DE (1) | DE102008000715B9 (de) |
| GB (1) | GB2470707C (de) |
| WO (1) | WO2009115478A2 (de) |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011035904A1 (en) * | 2009-09-24 | 2011-03-31 | Ludwig-Maximilians-Universität München | A combination of markers for predicting the mortality risk in a polytraumatized patient |
| WO2011036091A1 (de) * | 2009-09-23 | 2011-03-31 | Sirs-Lab Gmbh | Verfahren zur in vitro erfassung und unterscheidung von pathophysiologischen zuständen |
| EP2309001A1 (de) * | 2009-09-23 | 2011-04-13 | SIRS-Lab GmbH | Verfahren zur in vitro Erfassung und Unterscheidung von pathophysiologischen Zuständen |
| WO2011058765A1 (ja) * | 2009-11-13 | 2011-05-19 | 国立大学法人九州大学 | トコトリエノールの抗がん作用標的遺伝子ならびにバイオマーカー及びその使用方法 |
| WO2012170947A2 (en) * | 2011-06-10 | 2012-12-13 | Isis Pharmaceuticals, Inc. | Methods for modulating factor 12 expression |
| EP2643483A1 (de) * | 2010-11-26 | 2013-10-02 | Immunexpress Pty Ltd | Diagnose- und/oder screening-mittel und verwendungen dafür |
| JP2014508525A (ja) * | 2011-03-08 | 2014-04-10 | アナリティック イエナ アーゲー | ヒトゲノムに対応するポリヌクレオチドの初期セットから患者の宿主応答の重症度をインビトロ決定するためのポリヌクレオチドのサブセットを同定するための方法 |
| US9150864B2 (en) | 2010-11-08 | 2015-10-06 | Isis Pharmaceuticals, Inc. | Methods for modulating factor 12 expression |
| FR3044325A1 (fr) * | 2015-12-01 | 2017-06-02 | Biomerieux Sa | Procede d'evaluation du risque de complications chez les patients qui presentent un syndrome de reponse inflammatoire systemique (sirs) |
| US20210068735A1 (en) * | 2016-02-17 | 2021-03-11 | Nuralogix Corporation | System and method for detecting physiological state |
Families Citing this family (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20110076685A1 (en) * | 2009-09-23 | 2011-03-31 | Sirs-Lab Gmbh | Method for in vitro detection and differentiation of pathophysiological conditions |
| FR2988479B1 (fr) * | 2012-03-23 | 2014-12-19 | Hospices Civils Lyon | Procede de determination de la susceptibilite aux infections nosocomiales |
| US10190169B2 (en) | 2013-06-20 | 2019-01-29 | Immunexpress Pty Ltd | Biomarker identification |
| AU2014299322B2 (en) * | 2013-06-28 | 2018-08-09 | Acumen Research Laboratories Pte. Ltd. | Sepsis biomarkers and uses thereof |
| US10260111B1 (en) | 2014-01-20 | 2019-04-16 | Brett Eric Etchebarne | Method of detecting sepsis-related microorganisms and detecting antibiotic-resistant sepsis-related microorganisms in a fluid sample |
| US11047010B2 (en) | 2014-02-06 | 2021-06-29 | Immunexpress Pty Ltd | Biomarker signature method, and apparatus and kits thereof |
| GB201402293D0 (en) | 2014-02-11 | 2014-03-26 | Secr Defence | Biomarker signatures for the prediction of onset of sepsis |
| EP3211989A4 (de) | 2014-10-27 | 2018-09-26 | Pioneer Hi-Bred International, Inc. | Verbesserte molekulare zuchtverfahren |
| US11980147B2 (en) | 2014-12-18 | 2024-05-14 | Pioneer Hi-Bred International Inc. | Molecular breeding methods |
| FR3101423A1 (fr) * | 2019-10-01 | 2021-04-02 | bioMérieux | Procédé pour déterminer la capacité d’un individu à répondre à un stimulus |
| EP4038202A1 (de) * | 2019-10-01 | 2022-08-10 | Biomérieux | Verfahren zum bestimmen der ansprechbarkeit eines individuums auf einen stimulus |
| CN111413497A (zh) * | 2020-04-15 | 2020-07-14 | 上海市东方医院(同济大学附属东方医院) | 组蛋白甲基转移酶ezh2在制备用于诊断脓毒症的生物标记物中的用途 |
Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004087949A2 (de) * | 2003-04-02 | 2004-10-14 | Sirs-Lab Gmbh | Verfahren zur erkennung akuter generalisierter entzündlicher zustände (sirs), sepsis, sepsisähnlichen zuständen und systemischen infektionen |
| DE10315031A1 (de) * | 2003-04-02 | 2004-10-14 | Sirs-Lab Gmbh | Verfahren zur Erkennung von Sepsis und/oder sepsisähnlichen Zuständen |
| DE10336511A1 (de) * | 2003-08-08 | 2005-02-24 | Sirs-Lab Gmbh | Verfahren zur Erkennung von schwerer Sepsis |
| DE10340395A1 (de) * | 2003-09-02 | 2005-03-17 | Sirs-Lab Gmbh | Verfahren zur Erkennung akuter generalisierter entzündlicher Zustände (SIRS) |
| WO2005106020A1 (de) * | 2004-03-30 | 2005-11-10 | Sirs-Lab Gmbh | Verfahren zur vorhersage des individuellen krankheitsverlaufs bei sepsis |
| WO2006042581A1 (de) * | 2004-10-13 | 2006-04-27 | Sirs-Lab Gmbh | Verfahren zur unterscheidung zwischen nichtinfektiösen und infektiösen ursachen eines multiorganversagens |
| WO2006100203A2 (de) * | 2005-03-21 | 2006-09-28 | Sirs-Lab Gmbh | Verwendung von genaktivitäts-klassifikatoren für die in vitro klassifizierung von genexpressionsprofilen von patienten mit infektiösem/nichtinfektiösem multiorganversagen |
| WO2008107114A2 (de) * | 2007-03-02 | 2008-09-12 | Sirs-Lab Gmbh | Kontrollgene zur normalisierung von genexpressionsanalysedaten |
| WO2009018962A1 (de) * | 2007-08-03 | 2009-02-12 | Sirs-Lab Gmbh | Verwendung von polynukleotiden zur erfassung von genaktivitäten für die unterscheidung zwischen lokaler und systemischer infektion |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6960439B2 (en) | 1999-06-28 | 2005-11-01 | Source Precision Medicine, Inc. | Identification, monitoring and treatment of disease and characterization of biological condition using gene expression profiles |
| US7465555B2 (en) | 2002-04-02 | 2008-12-16 | Becton, Dickinson And Company | Early detection of sepsis |
| DE102004009952B4 (de) | 2004-03-01 | 2011-06-01 | Sirs-Lab Gmbh | Verfahren zur Erkennung von Sepsis |
| EP1774044A4 (de) | 2004-06-03 | 2009-06-10 | Univ Johns Hopkins | Verfahren zur überprüfung von zellproliferation oder neoplastischer störungen |
| EP1869463A4 (de) | 2005-04-15 | 2010-05-05 | Becton Dickinson Co | Sepsisdiagnose |
| DE102006019480A1 (de) | 2006-04-26 | 2007-10-31 | Sirs-Lab Gmbh | Verfahren zur in-vitro Überwachung postoperativer Veränderungen nach Lebertransplantation |
| DE102006027842B4 (de) | 2006-06-16 | 2014-07-31 | Analytik Jena Ag | Verfahren zur Feststellung der Infektionsquelle bei Fieber unklarer Genese |
| WO2008095050A1 (en) | 2007-01-30 | 2008-08-07 | Pharmacyclics, Inc. | Methods for determining cancer resistance to histone deacetylase inhibitors |
-
2008
- 2008-03-17 DE DE102008000715A patent/DE102008000715B9/de not_active Expired - Fee Related
-
2009
- 2009-03-16 WO PCT/EP2009/053042 patent/WO2009115478A2/de active Application Filing
- 2009-03-16 US US12/933,169 patent/US8765371B2/en not_active Expired - Fee Related
- 2009-03-16 CA CA2718741A patent/CA2718741A1/en not_active Abandoned
- 2009-03-16 GB GB201017326A patent/GB2470707C/en not_active Expired - Fee Related
- 2009-03-16 JP JP2011500171A patent/JP2011517401A/ja active Pending
Patent Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004087949A2 (de) * | 2003-04-02 | 2004-10-14 | Sirs-Lab Gmbh | Verfahren zur erkennung akuter generalisierter entzündlicher zustände (sirs), sepsis, sepsisähnlichen zuständen und systemischen infektionen |
| DE10315031A1 (de) * | 2003-04-02 | 2004-10-14 | Sirs-Lab Gmbh | Verfahren zur Erkennung von Sepsis und/oder sepsisähnlichen Zuständen |
| DE10336511A1 (de) * | 2003-08-08 | 2005-02-24 | Sirs-Lab Gmbh | Verfahren zur Erkennung von schwerer Sepsis |
| DE10340395A1 (de) * | 2003-09-02 | 2005-03-17 | Sirs-Lab Gmbh | Verfahren zur Erkennung akuter generalisierter entzündlicher Zustände (SIRS) |
| WO2005106020A1 (de) * | 2004-03-30 | 2005-11-10 | Sirs-Lab Gmbh | Verfahren zur vorhersage des individuellen krankheitsverlaufs bei sepsis |
| WO2006042581A1 (de) * | 2004-10-13 | 2006-04-27 | Sirs-Lab Gmbh | Verfahren zur unterscheidung zwischen nichtinfektiösen und infektiösen ursachen eines multiorganversagens |
| WO2006100203A2 (de) * | 2005-03-21 | 2006-09-28 | Sirs-Lab Gmbh | Verwendung von genaktivitäts-klassifikatoren für die in vitro klassifizierung von genexpressionsprofilen von patienten mit infektiösem/nichtinfektiösem multiorganversagen |
| WO2008107114A2 (de) * | 2007-03-02 | 2008-09-12 | Sirs-Lab Gmbh | Kontrollgene zur normalisierung von genexpressionsanalysedaten |
| WO2009018962A1 (de) * | 2007-08-03 | 2009-02-12 | Sirs-Lab Gmbh | Verwendung von polynukleotiden zur erfassung von genaktivitäten für die unterscheidung zwischen lokaler und systemischer infektion |
Non-Patent Citations (2)
| Title |
|---|
| DEIGNER H P: "Vortrag im Rahmen der Konferenz 6th World Congress on Trauma, Shock, Inflammation and Sepsis ? Pathophysiology, Immune Consequences and Therapy, 4. März 2004, München" INTERNET CITATION, [Online] XP002327554 Gefunden im Internet: URL:http://www.sirs-lab.de/content/de/pdf/kompetenzen/deigner.pdf> [gefunden am 2005-05-09] * |
| LANDRÉ, J.: "Towards transcription-based sepsis diagnosis - recent progress." LECTURE AT THE EDUCATIONAL WORKSHOP OF DYNEX LABORATORIES, OCTOBER 24, 2006, PRAGUE, CZECH REPUBLIC., [Online] 2006, XP002535917 PRAGUE, CZECH REPUBLIC. Gefunden im Internet: URL:http://separraytor.com/files/0610_landre_lec_towards.pdf> [gefunden am 2009-07-07] * |
Cited By (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011036091A1 (de) * | 2009-09-23 | 2011-03-31 | Sirs-Lab Gmbh | Verfahren zur in vitro erfassung und unterscheidung von pathophysiologischen zuständen |
| EP2309001A1 (de) * | 2009-09-23 | 2011-04-13 | SIRS-Lab GmbH | Verfahren zur in vitro Erfassung und Unterscheidung von pathophysiologischen Zuständen |
| EP2985352A1 (de) * | 2009-09-23 | 2016-02-17 | Analytik Jena AG | Verfahren zur in vitro erfassung und unterscheidung von pathophysiologischen zuständen |
| WO2011035904A1 (en) * | 2009-09-24 | 2011-03-31 | Ludwig-Maximilians-Universität München | A combination of markers for predicting the mortality risk in a polytraumatized patient |
| WO2011058765A1 (ja) * | 2009-11-13 | 2011-05-19 | 国立大学法人九州大学 | トコトリエノールの抗がん作用標的遺伝子ならびにバイオマーカー及びその使用方法 |
| US9150864B2 (en) | 2010-11-08 | 2015-10-06 | Isis Pharmaceuticals, Inc. | Methods for modulating factor 12 expression |
| EP2643483A1 (de) * | 2010-11-26 | 2013-10-02 | Immunexpress Pty Ltd | Diagnose- und/oder screening-mittel und verwendungen dafür |
| CN103649329A (zh) * | 2010-11-26 | 2014-03-19 | ImmuneXpress有限公司 | 诊断和/或筛选剂及其用途 |
| EP2643483A4 (de) * | 2010-11-26 | 2014-04-30 | Immunexpress Pty Ltd | Diagnose- und/oder screening-mittel und verwendungen dafür |
| JP2014508525A (ja) * | 2011-03-08 | 2014-04-10 | アナリティック イエナ アーゲー | ヒトゲノムに対応するポリヌクレオチドの初期セットから患者の宿主応答の重症度をインビトロ決定するためのポリヌクレオチドのサブセットを同定するための方法 |
| US20140128277A1 (en) * | 2011-03-08 | 2014-05-08 | Analytik Jena Ag | Method for Identifying a Subset of Polynucleotides from an Initial Set of Polynucleotides Corresponding to the Human Genome for the In Vitro Determination of the Severity of the Host Response of a Patient |
| WO2012170947A2 (en) * | 2011-06-10 | 2012-12-13 | Isis Pharmaceuticals, Inc. | Methods for modulating factor 12 expression |
| US9187749B2 (en) | 2011-06-10 | 2015-11-17 | Isis Pharmaceuticals, Inc. | Methods for modulating factor 12 expression |
| WO2012170947A3 (en) * | 2011-06-10 | 2013-02-21 | Isis Pharmaceuticals, Inc. | Methods for modulating factor 12 expression |
| FR3044325A1 (fr) * | 2015-12-01 | 2017-06-02 | Biomerieux Sa | Procede d'evaluation du risque de complications chez les patients qui presentent un syndrome de reponse inflammatoire systemique (sirs) |
| US11339438B2 (en) | 2015-12-01 | 2022-05-24 | Biomerieux | Method for assessing the risk of complications in patients with systemic inflammatory response syndrome (sirs) |
| AU2016361646B2 (en) * | 2015-12-01 | 2023-01-19 | Hospices Civils De Lyon | Method for assessing the risk of complications in patients with systemic inflammatory response syndrome (SIRS) |
| US20210068735A1 (en) * | 2016-02-17 | 2021-03-11 | Nuralogix Corporation | System and method for detecting physiological state |
| US11497423B2 (en) * | 2016-02-17 | 2022-11-15 | Nuralogix Corporation | System and method for detecting physiological state |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2009115478A3 (de) | 2009-11-12 |
| US8765371B2 (en) | 2014-07-01 |
| DE102008000715B9 (de) | 2013-01-17 |
| DE102008000715A1 (de) | 2009-09-24 |
| GB201017326D0 (en) | 2010-11-24 |
| GB2470707B (en) | 2013-03-06 |
| JP2011517401A (ja) | 2011-06-09 |
| GB2470707A (en) | 2010-12-01 |
| CA2718741A1 (en) | 2009-09-24 |
| DE102008000715B4 (de) | 2012-02-02 |
| US20110098195A1 (en) | 2011-04-28 |
| GB2470707C (en) | 2013-03-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DE102008000715B9 (de) | Verfahren zur in vitro Erfasssung und Unterscheidung von pathophysiologischen Zuständen | |
| US20110076685A1 (en) | Method for in vitro detection and differentiation of pathophysiological conditions | |
| DE102011005235B4 (de) | Verfahren zum Identifizieren einer Teilmenge von Polynucleotiden aus einer dem Humangenom entsprechenden Ausgangsmenge von Polynucleotiden zur in vitro Bestimmung eines Schweregrads der Wirtsantwort eines Patienten | |
| KR102082016B1 (ko) | 알츠하이머병을 나타내는 마이크로rna 바이오마커들 | |
| EP3103046B1 (de) | Biomarkersignaturverfahren sowie vorrichtung und kits dafür | |
| CN107532214B (zh) | 用于败血症诊断的方法 | |
| EP1863928B1 (de) | Verwendung von genaktivitäts-klassifikatoren für die in vitro klassifizierung von genexpressionsprofilen von patienten mit infektiösem/nichtinfektiösem multiorganversagen | |
| EP2218795B1 (de) | Verfahren zur Unterscheidung zwischen nichtinfektiösen und infektiösen Ursachen eines Multiorganversagens | |
| CN116218988A (zh) | 用于诊断结核病的方法 | |
| WO2011036091A1 (de) | Verfahren zur in vitro erfassung und unterscheidung von pathophysiologischen zuständen | |
| JP2019530878A (ja) | 重篤患者における死亡率の予後診断に使用するためのバイオマーカー | |
| US10584383B2 (en) | Mitochondrial non-coding RNAs for predicting disease progression in heart failure and myocardial infarction patients | |
| KR102096498B1 (ko) | 대장암 진단 또는 재발 예측을 위한 마이크로RNA-4732-5p 및 이의 용도 | |
| EP2092087B1 (de) | Prognostische marker für die klassifizierung von kolorektalen karzinomen basierend auf expressionsprofilen von biologischen proben | |
| CN114566224B (zh) | 一种用于鉴别或区分不同海拔人群的模型及其应用 | |
| WO2015117205A1 (en) | Biomarker signature method, and apparatus and kits therefor | |
| EP2985352A1 (de) | Verfahren zur in vitro erfassung und unterscheidung von pathophysiologischen zuständen | |
| CN114839369B (zh) | 急性高原反应微生物标志物及其应用 | |
| EP2092086B1 (de) | Prognostische marker für dievorhersage des fünfjährigen progressionsfreien überlebens von patienten mit kolorektalen karzinomen basierend auf expressionsprofilen von biologischen proben | |
| KR20240054437A (ko) | 패혈증 진단 또는 예후 예측용 신규 바이오마커 및 이의 용도 | |
| CN116875685A (zh) | 一种肝硬化合并门静脉系统血栓形成的诊断模型及其应用 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09722590 Country of ref document: EP Kind code of ref document: A2 |
|
| DPE1 | Request for preliminary examination filed after expiration of 19th month from priority date (pct application filed from 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2011500171 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2718741 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 1017326 Country of ref document: GB Kind code of ref document: A Free format text: PCT FILING DATE = 20090316 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1017326.8 Country of ref document: GB |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12933169 Country of ref document: US |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 09722590 Country of ref document: EP Kind code of ref document: A2 |