CN107532214B - 用于败血症诊断的方法 - Google Patents
用于败血症诊断的方法 Download PDFInfo
- Publication number
- CN107532214B CN107532214B CN201680023601.1A CN201680023601A CN107532214B CN 107532214 B CN107532214 B CN 107532214B CN 201680023601 A CN201680023601 A CN 201680023601A CN 107532214 B CN107532214 B CN 107532214B
- Authority
- CN
- China
- Prior art keywords
- sepsis
- infection
- subject
- polynucleotide
- biomarkers
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images





















































Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6876—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes
- C12Q1/6883—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for diseases caused by alterations of genetic material
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B25/00—ICT specially adapted for hybridisation; ICT specially adapted for gene or protein expression
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B25/00—ICT specially adapted for hybridisation; ICT specially adapted for gene or protein expression
- G16B25/10—Gene or protein expression profiling; Expression-ratio estimation or normalisation
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B25/00—ICT specially adapted for hybridisation; ICT specially adapted for gene or protein expression
- G16B25/20—Polymerase chain reaction [PCR]; Primer or probe design; Probe optimisation
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16H—HEALTHCARE INFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR THE HANDLING OR PROCESSING OF MEDICAL OR HEALTHCARE DATA
- G16H50/00—ICT specially adapted for medical diagnosis, medical simulation or medical data mining; ICT specially adapted for detecting, monitoring or modelling epidemics or pandemics
- G16H50/20—ICT specially adapted for medical diagnosis, medical simulation or medical data mining; ICT specially adapted for detecting, monitoring or modelling epidemics or pandemics for computer-aided diagnosis, e.g. based on medical expert systems
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/106—Pharmacogenomics, i.e. genetic variability in individual responses to drugs and drug metabolism
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/158—Expression markers
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A90/00—Technologies having an indirect contribution to adaptation to climate change
- Y02A90/10—Information and communication technologies [ICT] supporting adaptation to climate change, e.g. for weather forecasting or climate simulation
Abstract
公开用于败血症诊断的方法。具体来说,本发明涉及用于辅助败血症诊断、预后和治疗的生物标志物的用途,并且涉及一组可用于区分败血症与非感染来源的炎症的生物标志物,所述炎症如由创伤性损伤、手术、自体免疫疾病、血栓形成或全身炎症反应综合征(SIRS)引起。
Description
关于联邦赞助的研究或开发的声明
本发明是依据美国国家卫生研究院(National Institutes of Health)授予的合约AI057229、AI109662和LM007033在政府支持下进行的。政府享有本发明的某些权利。
技术领域
本发明大体上涉及用于败血症诊断的方法。具体来说,本发明涉及用于辅助败血症诊断、预后和治疗的生物标志物的用途,并且更确切地说,涉及可用于区分败血症与非感染来源的炎症的生物标志物,所述炎症如由创伤性损伤、手术、自体免疫疾病、血栓形成或全身炎症反应综合征(SIRS)引起。
背景技术
败血症,一种响应于感染的全身炎症综合征,在美国每年杀死大约750,000人(Angus等人(2001)《危重病监护医学(Crit Care Med)》29:1303-1310)。它也是在美国治疗的单个最昂贵病况,每年花费医疗系统超过240亿美元(Lagu等人(2012)《危重病监护医学》40:754-761);Torio和Andrews(2013)《国内住院病人医院费用:付款人最昂贵的病况(National Inpatient Hospital Costs:The Most Expensive Conditions by Payer)》,2011(统计摘要#160,马里兰州罗克维尔的卫生保健研究与质量管理处(Agency forHealthcare Research and Quality,Rockville,MD),2013年8月)。败血症的早期诊断和治疗对降低死亡率至关重要,每小时的延迟增加死亡风险(Gaieski等人(2010)《危重病监护医学》38:1045-1053;Ferrer等人(2014)《危重病监护医学》42:1749-1755)。败血症由除已知或疑似感染源之外还存在全身炎症反应综合征(SIRS)所定义(Dellinger等人(2013)《重症监护医学(Intensive Care Med)》39:165-228)。然而,SIRS不是特异性针对败血症,因为无菌炎症可作为对创伤、手术、血栓形成和其它非感染性伤害的非特异性反应而出现。因此,败血症可能难以在临床上与由非感染源(如组织创伤)引起的全身炎症区分开(Coburn等人《美国医学会杂志(JAMA)》(2012)308:502-511)。在可从标准微生物培养获得结果之前,没有在诊断时用于区分感染患者的‘黄金标准’血液测试。一种最常见的感染生物标志物降钙素原(procalcitonin)的受试者工作特征曲线下总面积为0.78(范围0.66-0.90)(Tang等人(2007)《柳叶刀感染病(Lancet Infect Dis)》7:210-217;Uzzan等人(2006)《危重病监护医学》34:1996-2003;Cheval等人(2000)《重症监护医学》26增刊2:S153-158;Ugarte等人(1999)《危重病监护医学》27:498-504)。若干群体已评估过细胞因子或基因表达阵列是否可精确诊断败血症;然而,由于宿主反应和人类遗传学的高度可变性,所以尚未发现稳固的诊断特征标志(Cobb等人(2009)《外科学年鉴(Ann Surg)》250:531-539;Xiao等人(2011)《实验医学杂志(J Exp Med)》208:2581-2590;Pankla等人(2009)《基因组生物学(Genome Biol)》10:R127;Tang等人(2009)《危重病监护医学》37:882-888;Wong(2012)《危重病监护(Crit Care)》16:204;Johnson等人(2007)《外科学年鉴》245:611-621)。实际上,“寻找‘完美的’败血症标志物已经是现代医学中最难以实现的梦想之一”(Vega等人(2009)《危重病监护医学》37:1806-1807)。
感染和组织创伤都活化许多相同先天性免疫受体家族,如Toll样受体和NOD样受体,并因此,大大活化了重叠的转录途径。因此,区分仅可归因于感染的保守性下游效应已极其困难。最新的研究已显示,存在可能对病原体反应具有特异性的模式识别受体,如c型凝集素、CEACAM和siglec受体家族(Geijtenbeek等人(2009)《自然·免疫学综述(Nat RevImmunol)》9:465-479;Crocker(2007)《自然·免疫学综述》7:255-266;Kuespert等人(2006)《细胞生物学新见(Curr Opin Cell Biol)》18:565-571)。因此,有可能能够区分感染特异性免疫反应与无菌炎症。
正在进行中的对败血症的新疗法和新的预后和诊断生物标志物的发现已在过去的十年间产生几十个基于微阵列的全基因组表达研究,不同地聚焦于诊断、预后、病原体反应和潜在的败血症病理生理学(Johnson等人,见上文;Maslove等人(2014)《分子医学趋势(Trends Mol Med.)》20(4):204-213)。尽管在理解败血症的基因表达方面有极大的收获,但是极少的洞察已转变成临床实践的改进。重要的是,许多这些研究已寄存到公共存储库中,如NIH基因表达大棚车(GEO)和ArrayExpress,并因此目前存在大量关于败血症的公开可用的数据。具体来说,存在若干比较败血症患者与在重大手术、创伤性损伤之后或在非败血症相关ICU收住(血栓形成、呼吸衰竭等)中出现非感染性炎症(如SIRS)的患者的研究。
一个数据集,具体来说,炎症和宿主对损伤的反应项目(the Inflammation andHost Response to Injury Program)(Glue Grant)(Cobb等人(2005)Proc Natl Acad SciUSA 102:4801-4806),已得到数个关于在创伤后和在败血症中时间对基因表达的影响的重要研究结果。Glue Grant的一个部分纵向检查患者在重度创伤性损伤后的基因表达。若干群体已经相对于时间检查过这些数据;值得注意的研究结果是(1)大于80%的已表达基因在创伤性损伤后显示差异表达(Xiao等人,见上文),(2)不同的基因簇历经显著不同的时间段恢复(Seok等人(2013)《美国国家科学院院刊(Proc Natl Acad Sci USA)》110:3507-3512),(3)不同的炎症情形,如创伤、烧伤和内因性中毒,展现类似的基因表达变化(Seok等人,见上文),以及(4)创伤后基因表达谱与那些健康患者不同的程度和其随时间推移基因表达恢复的程度与临床结果相关(Desai等人(2011)《公共科学图书馆·医学(PLoS Med)》8:e1001093;Warren等人(2009)《分子医学(Mol Med)》15:220-227)。因此,越来越理解构成创伤恢复基础的变化和其对特异性临床结果的影响的重要性。
仍需要针对败血症的灵敏的特异性诊断测试,其可区分败血症与如由创伤性损伤和SIRS引起的非感染来源的炎症。
发明内容
本发明涉及用于败血症诊断的生物标志物的用途。具体来说,本发明人已发现可用于诊断败血症并区分败血症与非感染来源的全身炎症的生物标志物,所述全身炎症如由创伤性损伤、手术、自体免疫疾病、血栓形成或全身炎症反应综合征(SIRS)引起。这些生物标志物可单独或与一种或多种额外生物标志物或相关临床参数组合用于败血症的预后、诊断或监测治疗。
可用于本发明实践的生物标志物包括包含来自基因或基因的RNA转录物的核苷酸序列的多核苷酸,其包括(但不限于)CEACAM1、ZDHHC19、C9orf95、GNA15、BATF、C3AR1、KIAA1370、TGFBI、MTCH1、RPGRIP1和HLA-DPB1。
在某些实施方案中,将一组生物标志物用于败血症诊断。任何大小的生物标志物组可用于本发明实践。用于诊断败血症的生物标志物组通常包含至少3种生物标志物和多到30种的生物标志物,包括其间任何数目的生物标志物,如3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29或30种生物标志物。在某些实施方案中,本发明包括一种生物标志物组,其包含至少3种、至少4种、或至少5种、或至少6种、或至少7种、或至少8种、或至少9种、或至少10种、或至少11种或更多种生物标志物。虽然较小的生物标志物组通常更经济,但是较大的生物标志物组(即,大于30种生物标志物)具有提供更详细信息的优势并且也可用于本发明实践。
在一个实施方案中,生物标志物组包含多种用于诊断败血症的生物标志物,其中所述多种生物标志物包含一种或多种选自以下组成的组的包含来自基因或基因的RNA转录物的核苷酸序列的多核苷酸:CEACAM1、ZDHHC19、C9orf95、GNA15、BATF、C3AR1、KIAA1370、TGFBI、MTCH1、RPGRIP1和HLA-DPB1。在某些实施方案中,生物标志物组包含至少11种生物标志物。在一个实施方案中,生物标志物组包含CEACAM1多核苷酸、ZDHHC19多核苷酸、C9orf95多核苷酸、GNA15多核苷酸、BATF多核苷酸、C3AR1多核苷酸、KIAA1370多核苷酸、TGFBI多核苷酸、MTCH1多核苷酸、RPGRIP1多核苷酸和HLA-DPB1多核苷酸。
在一个方面,本发明包括一种用于诊断受试者的败血症的方法。所述方法包含a)测量来源于受试者的生物样本中多种生物标志物的水平;和b)结合多种生物标志物的相应参考值范围分析生物标志物的水平,其中与未感染的对照受试者的生物标志物的参考值范围相比,生物样本中一种或多种生物标志物的差异表达指示受试者患有败血症。参考值范围可表示在一个或多个未患败血症的受试者(例如健康受试者或未感染受试者)的一个或多个样本中所发现的一种或多种生物标志物的水平。或者,参考值可表示在一个或多个患有败血症的受试者的一个或多个样本中所发现的一种或多种生物标志物的水平。在某些实施方案中,将生物标志物的水平与未感染或已感染/败血症受试者的时间匹配的参考值范围相比。
在某些实施方案中,本发明包括一种使用本文所述的生物标志物组诊断受试者的败血症的方法。所述方法包含:a)从受试者采集生物样本;b)测量生物样本中生物标志物组的每一种生物标志物;以及c)将每一种生物标志物的测量值与生物标志物的相应参考值范围相比,其中与对照受试者的生物标志物的参考值相比,生物样本中生物标志物组的生物标志物的差异表达指示受试者患有败血症。
在一个实施方案中,本发明包括一种用于诊断受试者的败血症的方法,所述方法包含:a)从所述受试者采集生物样本;b)测量所述生物样本中CEACAM1、ZDHHC19、C9orf95、GNA15、BATF、C3AR1、KIAA1370、TGFBI、MTCH1、RPGRIP1和HLA-DPB1的表达水平;和c)结合所述生物标志物的相应参考值范围分析每一种生物标志物的表达水平,其中与未感染的对照受试者的所述生物标志物的参考值范围相比,所述CEACAM1、ZDHHC19、C9orf95、GNA15、BATF和C3AR1生物标志物的表达水平升高和所述KIAA1370、TGFBI、MTCH1、RPGRIP1和HLA-DPB1生物标志物的表达水平降低指示所述受试者患有败血症。
在另一个实施方案中,本发明包括一种用于诊断受试者的败血症的方法,包含根据下式基于生物标志物的水平确定所述受试者的败血症评分:

其中与未感染的对照受试者的参考值范围相比,所述受试者的败血症评分更高指示所述受试者患有败血症。
如本文所述,本发明的方法可用于区分受试者的败血症诊断与非感染来源的炎症,所述炎症如由创伤性损伤、手术、自体免疫疾病、血栓形成或全身炎症反应综合征(SIRS)引起。
生物样本可包含例如全血、血沉棕黄层(buffy coat)、血浆、血清、外周血单个核细胞(peripheral blood mononucleated cell,PBMC)、杆状核细胞、嗜中性粒细胞、单核细胞(monocyte)或T细胞。
生物标志物多核苷酸(例如编码转录物)可例如通过微阵列分析、聚合酶链式反应(PCR)、逆转录酶聚合酶链式反应(RT-PCR)、Northern印迹或基因表达的系列分析(SAGE)检测。
在另一个方面,本发明包括一种确定疑似患有败血症的受试者的感染Z评分的方法,所述方法包含:a)从所述受试者采集生物样本;b)测量所述生物样本中多种败血症生物标志物的水平;以及c)通过将与所述生物标志物的对照参考值相比过度表达的所有生物标志物的表达水平的几何平均值减去与所述生物标志物的对照参考值相比表达不足的所有生物标志物的表达水平的几何平均值,并将差值乘以与所述生物标志物的对照参考值相比过度表达的生物标志物的数目与表达不足的生物标志物的数目的比率,确定所述生物标志物的感染Z评分。
在某些实施方案中,感染Z评分是由多种生物标志物的表达水平计算所得的,所述生物标志物包含一种或多种含有来自选自以下组成的组的基因或基因的RNA转录物的核苷酸序列的多核苷酸:CEACAM1、ZDHHC19、C9orf95、GNA15、BATF、C3AR1、KIAA1370、TGFBI、MTCH1、RPGRIP1和HLA-DPB1。在一个实施方案中,所述多种生物标志物包含CEACAM1多核苷酸、ZDHHC19多核苷酸、C9orf95多核苷酸、GNA15多核苷酸、BATF多核苷酸、C3AR1多核苷酸、KIAA1370多核苷酸、TGFBI多核苷酸、MTCH1多核苷酸、RPGRIP1多核苷酸和HLA-DPB1多核苷酸。
在另一个方面,本发明包括一种治疗患有败血症的受试者的方法,所述方法包含:a)根据本文所述的方法诊断患有败血症的受试者;和b)如果所述受试者的败血症诊断呈阳性,那么给予所述受试者治疗有效量的广谱抗生素。
在另一个方面,本发明包括一种治疗疑似患有败血症的受试者的方法,所述方法包含:a)根据本文所述的方法接收关于所述受试者的诊断信息;和b)如果患者的败血症诊断呈阳性,那么给予所述受试者治疗有效量的广谱抗生素。
在某些实施方案中,通过一种或多种方法分析受试者数据,所述方法包括(但不限于)多变量线性判别分析(LDA)、受试者工作特征(ROC)分析、主成分分析(PCA)、集合数据挖掘方法、微阵列的细胞特异性显著性分析(csSAM)和多维蛋白质鉴定技术(MUDPIT)分析。
在另一个实施方案中,本发明包括一种使用本文所述的生物标志物组评估治疗受试者的败血症的药剂的效应的方法,所述方法包含:在所述受试者用所述药剂治疗之前和之后,结合每一种生物标志物的相应参考值范围分析来源于所述受试者的样本中所述生物标志物组的每一种生物标志物的测量值。
在另一个实施方案中,本发明包括一种使用本文所述的生物标志物组监测治疗受试者的败血症的疗法的功效的方法,所述方法包含:在所述受试者进行所述疗法之前和之后,结合每一种生物标志物的相应参考值范围分析来源于所述受试者的样本中所述生物标志物组的每一种生物标志物的测量值。
在另一个实施方案中,本发明包括一种用于监测治疗受试者的败血症的疗法的功效的方法,所述方法包含:测量在所述受试者进行所述疗法之前来源于所述受试者的第一样本和在所述受试者进行所述疗法之后来源于所述受试者的第二样本中CEACAM1、ZDHHC19、C9orf95、GNA15、BATF、C3AR1、KIAA1370、TGFBI、MTCH1、RPGRIP1和HLA-DPB1生物标志物的表达水平,其中与所述第一样本中所述生物标志物的表达水平相比,所述第二样本中所述CEACAM1、ZDHHC19、C9orf95、GNA15、BATF和C3AR1生物标志物的表达水平升高和所述KIAA1370、TGFBI、MTCH1、RPGRIP1和HLA-DPB1生物标志物的表达水平降低指示所述受试者恶化,并且与所述第一样本中所述生物标志物的表达水平相比,所述第二样本中所述CEACAM1、ZDHHC19、C9orf95、GNA15、BATF和C3AR1生物标志物的表达水平降低和所述KIAA1370、TGFBI、MTCH1、RPGRIP1和HLA-DPB1生物标志物的表达水平升高指示所述受试者改善。
在另一个方面,本发明包括一种用于诊断受试者的败血症的试剂盒。所述试剂盒可包括用于盛放从疑似患有败血症的人类受试者分离的生物样本的容器、至少一种特异性检测败血症生物标志物的药剂;以及用于使所述药剂与所述生物样本或一部分所述生物样本反应以检测所述生物样本中至少一种败血症生物标志物的存在或量的印刷说明书。所述药剂可封装在单独的容器中。所述试剂盒可另外包含一种或多种对照参考样本和用于进行PCR或微阵列分析以检测如本文所述的生物标志物的试剂。
在某些实施方案中,所述试剂盒包括用于检测包含多种用于诊断败血症的生物标志物的生物标志物组的多核苷酸的药剂,其中一种或多种生物标志物选自以下组成的组:CEACAM1多核苷酸、ZDHHC19多核苷酸、C9orf95多核苷酸、GNA15多核苷酸、BATF多核苷酸、C3AR1多核苷酸、KIAA1370多核苷酸、TGFBI多核苷酸、MTCH1多核苷酸、RPGRIP1多核苷酸和HLA-DPB1多核苷酸。在一个实施方案中,所述试剂盒包括用于检测包含以下的生物标志物组的生物标志物的药剂:CEACAM1多核苷酸、ZDHHC19多核苷酸、C9orf95多核苷酸、GNA15多核苷酸、BATF多核苷酸、C3AR1多核苷酸、KIAA1370多核苷酸、TGFBI多核苷酸、MTCH1多核苷酸、RPGRIP1多核苷酸和HLA-DPB1多核苷酸。此外,所述试剂盒可包括用于检测多于一个生物标志物组(如两个或三个生物标志物组)的药剂,其可单独或以任何组合形式和/或与用于败血症诊断的临床参数组合起来使用。
在某些实施方案中,所述试剂盒包含用于分析多种生物标志物多核苷酸的微阵列。在一个实施方案中,所述试剂盒包含微阵列,其包含与CEACAM1多核苷酸杂交的寡核苷酸、与ZDHHC19多核苷酸杂交的寡核苷酸、与C9orf95多核苷酸杂交的寡核苷酸、与GNA15多核苷酸杂交的寡核苷酸、与BATF多核苷酸杂交的寡核苷酸、与C3AR1多核苷酸杂交的寡核苷酸、与KIAA1370多核苷酸杂交的寡核苷酸、与TGFBI多核苷酸杂交的寡核苷酸、与MTCH1多核苷酸杂交的寡核苷酸、与RPGRIP1多核苷酸杂交的寡核苷酸和与HLA-DPB1多核苷酸杂交的寡核苷酸。
在另一个方面,本发明包括一种分析,其包含:a)测量从疑似患有败血症的受试者采集的生物样本中的至少一种生物标志物;和b)将所述生物样本中的至少一种生物标志物的测量值与对照受试者的所述生物标志物的参考值相比,其中与所述参考值相比,所述生物样本中的至少一种生物标志物的差异表达指示所述受试者患有败血症。生物样本可包含例如全血、血沉棕黄层、血浆、血清、外周血单个核细胞(PBMC)、杆状核细胞、嗜中性粒细胞、单核细胞或T细胞。在一个实施方案中,所述分析另外包含确定所述受试者的感染Z评分。
在一个实施方案中,本发明包括一种分析,其包含:a)在从疑似患有败血症的受试者采集的生物样本中,测量本文所述的生物标志物组的每一种生物标志物;和b)将所述生物样本中所述生物标志物组的每一种生物标志物的测量值与对照受试者的每一种生物标志物的参考值相比,其中与所述参考值相比,所述生物样本中生物标志物的差异表达指示所述受试者患有败血症。生物样本可包含例如全血、血沉棕黄层、血浆、血清、外周血单个核细胞(PBMC)、杆状核细胞、嗜中性粒细胞、单核细胞或T细胞。所述分析可另外包含确定所述受试者的感染Z评分。
在另一些实施方案中,测量至少一种生物标志物包含进行微阵列分析、聚合酶链式反应(PCR)、逆转录酶聚合酶链式反应(RT-PCR)、Northern印迹或基因表达的系列分析(SAGE)。在一个实施方案中,使用包含以下的微阵列进行微阵列分析:与CEACAM1多核苷酸杂交的寡核苷酸、与ZDHHC19多核苷酸杂交的寡核苷酸、与C9orf95多核苷酸杂交的寡核苷酸、与GNA15多核苷酸杂交的寡核苷酸、与BATF多核苷酸杂交的寡核苷酸、与C3AR1多核苷酸杂交的寡核苷酸、与KIAA1370多核苷酸杂交的寡核苷酸、与TGFBI多核苷酸杂交的寡核苷酸、与MTCH1多核苷酸杂交的寡核苷酸、与RPGRIP1多核苷酸杂交的寡核苷酸和与HLA-DPB1多核苷酸杂交的寡核苷酸。
在另一个方面,本发明包括一种诊断系统,其包含用于存储数据的存储组件(即,存储器),其中所述存储组件具有用于确定存储在其中的受试者诊断的指令;用于处理数据的计算机处理器,其中所述计算机处理器被耦合到所述存储组件并且被配置成执行存储在所述存储组件中的指令以便接收患者数据并根据算法分析患者数据;以及用于显示关于患者的诊断信息的显示器组件。所述存储组件可包括用于如本文所述(参见实施例1和2)计算感染Z评分或败血症评分的指令。此外,所述存储组件可另外包括用于进行多变量线性判别分析(LDA)、受试者工作特征(ROC)分析、主成分分析(PCA)、集合数据挖掘方法、微阵列的细胞特异性显著性分析(csSAM)和多维蛋白质鉴定技术(MUDPIT)分析的指令。
在某些实施方案中,本发明包括一种计算机实施的用于诊断疑似患有败血症的患者的方法,计算机执行包含以下的步骤:a)接收所输入的患者数据,包含来自所述患者的生物样本中多种败血症生物标志物的水平值;b)分析多种败血症生物标志物的水平并与所述败血症生物标志物的相应参考值范围相比;c)基于所述败血症生物标志物的水平计算所述患者的感染Z评分或败血症评分;d)基于所述感染Z评分的值计算所述患者患有败血症的可能性;以及e)显示关于所述患者的诊断信息。
在某些实施方案中,所输入的患者数据包含来自所述患者的生物样本中至少11种败血症生物标志物的水平值。举例来说,所输入的患者数据可包含CEACAM1多核苷酸、ZDHHC19多核苷酸、C9orf95多核苷酸、GNA15多核苷酸、BATF多核苷酸、C3AR1多核苷酸、KIAA1370多核苷酸、TGFBI多核苷酸、MTCH1多核苷酸、RPGRIP1多核苷酸和HLA-DPB1多核苷酸的水平值。
在另一个实施方案中,本发明包括计算机实施的用于诊断疑似患有败血症的患者的方法,计算机执行包含以下的步骤:a)接收所输入的患者数据,包含来自所述患者的生物样本中CEACAM1、ZDHHC19、C9orf95、GNA15、BATF、C3AR1、KIAA1370、TGFBI、MTCH1、RPGRIP1和HLA-DPB1生物标志物的表达水平值;b)分析每一种生物标志物的水平并与每一种生物标志物的相应参考值范围相比;c)根据下式基于所述生物标志物的表达水平计算所述患者的败血症评分:

d)基于所述败血症评分的值计算所述患者患有败血症的可能性,其中所述患者的败血症评分与未感染的对照受试者的参考值范围相比更高指示所述患者患有败血症;以及e)显示关于所述患者的诊断信息。
所属领域的技术人员鉴于本文的公开内容将易于想到本发明的这些和其它实施方案。
附图说明
图1A和1B示出了比较无菌性SIRS/创伤患者与败血症患者的经标记的主成分分析(PCA)。图1A示出了无菌性SIRS/创伤和败血症患者在转录组学空间呈现为大部分可分离,仅具有最小的不可分离集合。图1B示出了相同标记的PCA,其中标记经过更新以反映从非感染性SIRS/创伤恢复的患者和患有医院获得性败血症的患者;‘后期’组(在入院后>48小时)更难以分离。N=1094,由15项研究组合而来。
图2A-2D示出了11个基因的集合的大小的效应。示出了关于正基因效应量的随机效应模型估计的森林图,从而在每一个发现群组中将SIRS/创伤/ICU与感染/败血症患者相比较。
图3A-3F示出了发现数据集和嗜中性粒细胞验证数据集中11个基因的集合的结果。图3A示出了将发现数据集中的无菌性SIRS/ICU/创伤患者与败血症患者分离的ROC曲线。图3B示出了将Glue Grant嗜中性白细胞验证数据集中感染的创伤患者与时间匹配的未感染的创伤患者分离的ROC曲线。在从受伤起>1天之后,Glue Grant(图3C)血沉棕黄层发现样本和(图3D)嗜中性白细胞验证样本示出了未感染患者相对于在诊断+/-24小时内的患者的平均感染Z评分。在两种情况下,存在因时间和感染状态所致的显著效应。对于(图3E)血沉棕黄层发现样本和(图3F)嗜中性白细胞验证样本,示出了按照从受伤起的时间的感染Z评分的盒形图:将从未感染的患者与感染前>5天、感染前5至1天、诊断(病例)+/-1天和感染诊断后2至5天的患者相比。对于在入院后的每一个时间点从从未感染到感染+/-1天不断增长的趋势,JT趋势检验是显著的(p<0.01)。
图4A-4D示出了罹患VAP的创伤/ICU患者的无对照数据集。这些数据集并不包括未感染的患者,因此它们与时间匹配的Glue Grant患者一起进行经验贝叶斯共同归一化。灰线示出了(图4A)EMEXP3001、(图4B)GSE6377和(图4C)GSE12838嗜中性白细胞和全血样本的Glue Grant局部加权回归曲线。在所有情况下,仅显示自入院起前8天,并且患者在感染诊断后>1天接受检查。图4D示出了将在诊断+/-1天内的患者(图4A-4C中的深灰色点)与时间匹配的未感染的Glue Grant患者相比较的ROC曲线。关于更多数据集细节,参见表5。
图5A和5B示出了健康患者相对于败血症患者的区别。使用感染Z评分检验满足纳入标准(外周全血或嗜中性粒细胞,在败血症诊断48小时内取样)的八个独立的验证数据集。图5A示出了所有n=446名患者的感染Z评分,其组合在单个小提琴图中,误差条示出了中间四分位数。使用威尔科克森秩和检验(Wilcoxon rank-sum test)计算P值。图5B示出了区分败血症患者与健康对照的8个数据集中每一个的单独ROC曲线。平均ROC AUC=0.98。关于更多数据集细节,参见表6。
图6A和6B示出了细胞类型富集分析。示出的是(图6A)在多群组分析中发现为显著的82个基因的完整集合和(图6B)在前向搜索法后所发现的11个基因的集合(82个基因的子集)的人类免疫细胞类型的规范化富集评分(Z评分,黑点)。图6B还示出了Z评分分布的盒形图。
图7A和7B示出了比较健康患者相对于SIRS/创伤患者相对于败血症患者的经标记的PCA。图7A示出了健康患者、SIRS/创伤患者和败血症患者在转录组学空间呈现为大部分可分离,仅具有最小的不可分离集合。图7B示出了相同标记的PCA,其中标记经过更新以反映从非感染性SIRS/创伤恢复的患者和患有医院获得性败血症的患者;‘后期’组(在入院后>48小时)更难以分离。N=1316,由15项研究组合而来。
图8示出了具有完整血细胞计数和微阵列数据的Glue Grant患者的嗜中性白细胞百分比。所有时间点的嗜中性白细胞中位数百分比在75-85%之间。将在住院期间曾经感染的患者与在住院期间从未感染的患者相比。
图9A-9I示出了发现多群组分析中所包括的数据集的小提琴图,包括GPSSSI特有(图9A)、GSE28750(图9B)、GSE32707(图9C)和GSE40012(图9D)。示出的是在入院时将SIRS/ICU/创伤患者与败血症患者相比较的数据集。误差条示出了中间四分位数。使用威尔科克森秩和检验计算P值。图9E-9I示出了关于Glue Grant血沉棕黄层群组的发现多群组分析中所包括的数据集的小提琴图,将未感染的创伤患者与败血症患者在匹配的时间点进行比较,包括[1,3)(图9E)、[3,6)(图9F)、[6,10)(图9G)、[10,18)(图9H)和[18,24)(图9I)。误差条示出了中间四分位数。使用威尔科克森秩和检验计算P值。
图10A和10B示出了从Glue Grant群组分选的单核细胞中感染Z评分的表现。这些是与图3B、3D和3F中的嗜中性粒细胞验证群组相同的患者。图10A示出了四个取样时间仓中每一个的ROC曲线。图10B示出了按照从受伤起的时间的感染Z评分的盒形图。将从未感染的患者与感染前>5天、感染前5至1天、诊断(病例)+/-1天和感染后2至5天的患者相比。
图11A和11B示出了从Glue Grant群组分选的T细胞中感染Z评分的表现。这些是与图3B、3D和3F中的嗜中性粒细胞验证群组相同的患者。图11A示出了四个取样时间仓中每一个的ROC曲线。图11B示出了按照从受伤起的时间的感染Z评分的盒形图。将从未感染的患者与感染前>5天、感染前5至1天、诊断(病例)+/-1天和感染后2至5天的患者相比。
图12A和12B示出了SIRS标准和感染Z评分的线性模型。图12A示出了SIRS数据和微阵列数据可用的Glue Grant患者的逻辑回归模型。SIRS标准以二元变量来表示。第一模型示出了呈组合形式的SIRS标准;第二模型增添了感染Z评分。显著性代码:p<0.001‘***’;0.01‘**’;0.05‘*’。图12B示出了由图12A中的逻辑回归模型所输出的患者的预测对数感染几率的盒形图。
图13A示出了未时间匹配的数据集的感染Z评分。四个数据集在未匹配的时间点比较了SIRS/ICU/创伤患者与败血症患者。这些数据集检验了嗜中性粒细胞(GSE5772,N=93)、全血(EMTAB1548,N=73)和PBMC(GSE9960,N=30;EMEXP3621,N=10)。关于更多数据集细节,参见表7。图13B-13E示出了未时间匹配的数据集的感染Z评分。示出了未匹配的时间点数据集的小提琴图,包括GSE5772(图13B)、GSE9960(图13C)、EMTAB1548(图13D)和EMEXP3621(图13E)。误差条示出了中间四分位数。使用威尔科克森秩和检验进行检验。
图14A和14B示出了具有急性感染的患者与健康对照以及患有自体免疫疾病的患者的感染Z评分的比较。GSE22098比较了健康对照与具有急性自体免疫炎症或急性感染的患者。感染Z评分示出了感染与健康患者和那些具有自体免疫炎症的患者的良好区别。图14A示出了小提琴图;误差条示出了中间四分位数。使用威尔科克森秩和检验来检验具有自体免疫炎症的患者相对于那些患有败血症的患者。图14B示出了自体免疫患者或健康对照相对于败血性患者的ROC标绘图。
图15示出了整体综合多群组分析的示意图。
图16示出了诊断系统的示意图。
图17示出了临床败血症数据集的系统搜索和选择的纲要。
图18A-18C示出了败血症/急性感染与在入院时未感染炎症的患者的ROC曲线图区别。图18A示出了11个基因的评分。图18B示出了FAIM3:PLAC8比率。图18C示出了SepticyteLab。
图19A-19C示出了患有败血症/急性感染的创伤患者与时间匹配的从未感染的创伤患者的ROC曲线图区别。图19A示出了11个基因的评分。图19B示出了FAIM3:PLAC8比率。图19C示出了Septicyte Lab。
具体实施方式
除非另外指明,否则本发明的实践将采用所属领域技能内的药理学、化学、生物化学、重组DNA技术和免疫学的常规方法。在文献中全面解释了这些技术。参见例如J.R.Brown《败血症:症状、诊断和治疗(Sepsis:Symptoms,Diagnosis and Treatment)》(《21世纪系列的公共卫生(Public Health in the 21st Century Series)》,Nova Science Publishers公司,2013);《败血症和非感染性全身炎症:从生物学到危急监护(Sepsis and Non-infectious Systemic Inflammation:From Biology to Critical Care)》(J.Cavaillon,C.Adrie编,Wiley-Blackwell,2008);《败血症:诊断、管理和健康结果(Sepsis:Diagnosis,Management and Health Outcomes)》(《过敏症和感染性疾病(Allergies and InfectiousDiseases)》,N.Khardori编,Nova Science Pub公司,2014);《实验免疫学手册(Handbookof Experimental Immunology)》,第I-IV卷(D.M.Weir和C.C.Blackwell编,布莱克威尔出版公司(Blackwell Scientific Publications));A.L.Lehninger,《生物化学(Biochemistry)》(Worth Publishers公司,现行版);Sambrook等人,《分子克隆:实验室手册(Molecular Cloning:A Laboratory Manual)》(第3版,2001);《酶学方法(Methods InEnzymology)》(S.Colowick和N.Kaplan编,学术出版公司(Academic Press,Inc.))。
本文中(不论上文或下文)所列举的所有出版物、专利和专利申请都以其全文引用的方式并入在此。
I.定义
在描述本发明时,以下术语将被采用并打算如下文所指示定义。
必须注意,除非内容另外清楚地指示,否则如本说明书和所附权利要求书中所用,不使用数量词修饰时包括多个提及物。因此,举例来说,提及“生物标志物”包括两种或更多种生物标志物的混合物等。
术语“约”,特别是关于给定数量,意欲涵盖加上或减去5%的偏差。
在本发明的上下文中,“生物标志物”是指相比于从对照受试者(例如诊断呈阴性的个人、正常或健康受试者或未感染的受试者)获取的可比样本,在从患有败血症的患者获取的样本中差异表达的生物化合物,如多核苷酸。生物标志物可为可检测到和/或定量的核酸、核酸片段、多核苷酸或寡核苷酸。败血症生物标志物包括包含来自基因或基因的RNA转录物的核苷酸序列的多核苷酸,包括(但不限于)CEACAM1、ZDHHC19、C9orf95、GNA15、BATF、C3AR1、KIAA1370、TGFBI、MTCH1、RPGRIP1和HLA-DPB1。
术语“多肽”和“蛋白质”係指氨基酸残基的多聚物并且不限制最小长度的。因此,肽、寡肽、二聚体、多聚体等都包括在所述定义内。全长蛋白质和其片段均由所述定义涵盖。所述术语还包括多肽的表达后修饰,例如糖基化、乙酰化、磷酸化、羟基化、氧化等。
术语“多核苷酸”、“寡核苷酸”、“核酸”和“核酸分子”在本文中用于包括任何长度的聚合物形式的核苷酸(核糖核苷酸或脱氧核糖核苷酸)。此术语仅指代分子的一级结构。因此,所述术语包括三链、双链和单链DNA以及三链、双链和单链RNA。它还包括修饰,如通过甲基化和/或通过加帽,和未修饰形式的多核苷酸。更具体地说,术语“多核苷酸”、“寡核苷酸”、“核酸”和“核酸分子”包括多脱氧核糖核苷酸(含有2-脱氧-D-核糖)、多核糖核苷酸(含有D-核糖)和任何其它类型的多核苷酸(它是嘌呤或嘧啶碱基的N-或C-糖苷)。不意图在术语“多核苷酸”、“寡核苷酸”、“核酸”和“核酸分子”之间存在长度区别,并且这些术语可互换使用。
短语“差异表达”是指相比于对照受试者或未感染的受试者,从患有例如败血症的患者获取的样本中所存在的生物标志物的数量和/或频率的差异。举例来说,生物标志物可为相比于对照受试者的样本以升高水平或降低水平存在于败血症患者的样本中的多核苷酸。或者,生物标志物可为相比于对照受试者的样本在败血症患者的样本中以较高频率或较低频率检测到的多核苷酸。生物标志物可在数量、频率或这两方面存在差异。
如果一个样本中多核苷酸的量在统计学上显著不同于另一个样本中多核苷酸的量,那么多核苷酸在两个样本之间是差异表达的。举例来说,如果多核苷酸存在量为其在另一个样本中的存在量的至少约120%、至少约130%、至少约150%、至少约180%、至少约200%、至少约300%、至少约500%、至少约700%、至少约900%或至少约1000%,或如果其在一个样本中可检测到并且在另一个样本中无法检测到,那么多核苷酸在两个样本中是差异表达的。
或者或另外,如果在罹患败血症的患者的样本中检测到多核苷酸的频率与在对照样本中相比在统计学上显著更高或更低,那么多核苷酸在两组样本中是差异表达的。举例来说,如果检测到多核苷酸在一组样本中比在另一组样本中至少约120%、至少约130%、至少约150%、至少约180%、至少约200%、至少约300%、至少约500%、至少约700%、至少约900%或至少约1000%更频繁或更不频繁地观察到,那么多核苷酸在两组样本中是差异表达的。
“相似度值”是表示在所比较的两个事物之间的相似程度的数值。举例来说,相似度值可为指示在使用特异性表型相关生物标志物的患者的表达谱与一个或多个对照样本中生物标志物的参考值范围或参考表达谱之间的整体相似度(例如与“败血症”表达谱或“无菌炎症”表达谱的相似度)的数值。在患者样本和对照样本中的生物标志物的水平或参考表达谱之间,相似度值可表示为相似性度量(如相关系数),或可简单地表示为表达水平差异或合计的表达水平差异。
术语“受试者”、“个体”和“患者”在本文中可互换使用并且是指需要诊断、预后、治疗或疗法的任何哺乳动物受试者,尤其人类。其它受试者可包括牛、犬、猫、天竺鼠、兔、大鼠、小鼠、马等等。在一些情况下,本发明的方法应用于实验动物、兽医学应用和动物疾病模型(包括(但不限于)啮齿动物,包括小鼠、大鼠和仓鼠;以及灵长类动物)的开发中。
如本文所用,“生物样本”是指从受试者分离的组织、细胞或流体的样本,包括(但不限于)例如血液、血沉棕黄层、血浆、血清、血细胞(例如外周血单个核细胞(PBMC)、杆状核细胞、嗜中性粒细胞、单核细胞或T细胞)、粪便物、尿液、骨髓、胆汁、脊髓液、淋巴液、皮肤样本、皮肤、呼吸道、肠道和泌尿生殖道的外部分泌物、泪液、唾液、乳汁、器官、活检体以及体外细胞培养物成分的样本,包括(但不限于)由细胞和组织在培养基中生长产生的条件培养基,例如重组细胞和细胞组分。
生物标志物的“测试量”是指所测试的样本中所存在的生物标志物的量。测试量可为绝对量(例如μg/ml)或相对量(例如信号的相对强度)。
生物标志物的“诊断量”是指受试者的样本中与败血症的诊断相符的生物标志物的量。诊断量可为绝对量(例如μg/ml)或相对量(例如信号的相对强度)。
生物标志物的“对照量”可为有待与生物标志物的测试量相比较的任何量或量范围。举例来说,生物标志物的对照量可为无败血症的个人体内生物标志物的量。对照量可为绝对量(例如μg/ml)或相对量(例如信号的相对强度)。
术语“抗体”涵盖多克隆和单克隆抗体制剂,以及包括杂交抗体、改变的抗体、嵌合抗体和人源化抗体的制剂,以及:杂交(嵌合)抗体分子(参见例如Winter等人(1991)《自然(Nature)》349:293-299;和美国专利第4,816,567号);F(ab′)2和F(ab)片段;Fv分子(非共价异二聚体,参见例如Inbar等人(1972)《美国国家科学院院刊》69:2659-2662;和Ehrlich等人(1980)《生物化学》19:4091-4096);单链Fv分子(sFv)(参见例如Huston等人(1988)《美国国家科学院院刊》85:5879-5883);二聚和三聚抗体片段构建体;微型抗体(参见例如Pack等人(1992)《生物化学》31:1579-1584;Cumber等人(1992)《免疫学杂志(J Immunology)》149B:120-126);人源化抗体分子(参见例如Riechmann等人(1988)《自然》332:323-327;Verhoeyan等人(1988)《科学(Science)》239:1534-1536;和1994年9月21日公开的英国专利公开第GB 2,276,169号);以及从这类分子获得的任何功能片段,其中这类片段保留亲本抗体分子的特异性结合特性。
考虑用于本发明的“可检测部分”或“可检测标记”包括(但不限于)放射性同位素、荧光染料,如荧光素、藻红蛋白、Cy-3、Cy-5、别藻蓝蛋白、DAPI、德克萨斯红(Texas Red)、罗丹明(rhodamine)、俄勒冈绿(Oregon green)、荧光黄(Lucifer yellow)等、绿色荧光蛋白(GFP)、红色荧光蛋白(DsRed)、青色荧光蛋白(CFP)、黄色荧光蛋白(YFP)、角海葵橙色荧光蛋白(cOFP)、碱性磷酸酶(AP)、β-内酰胺酶、氯霉素乙酰转移酶(CAT)、腺苷脱氨酶(ADA)、氨基糖苷磷酸转移酶(neor、G418r)二氢叶酸还原酶(DHFR)、潮霉素B磷酸转移酶(HPH)、胸苷激酶(TK)、lacZ(编码β-半乳糖苷酶)和黄嘌呤鸟嘌呤磷酸核糖转移酶(XGPRT)、β-葡糖醛酸酶(gus)、胎盘碱性磷酸酶(PLAP)、分泌型胚胎碱性磷酸酶(SEAP)、或萤火虫或细菌萤光素酶(LUC)。酶标签与其同源底物一起使用。所述术语还包括具有已知荧光强度的颜色编码微球体(参见例如由Luminex(德克萨斯州奥斯汀(Austin,TX))生产的具有xMAP技术的微球体;含有量子点纳米晶体的微球体,例如含有不同比率和组合的量子点颜料(例如由LifeTechnologies(加利福尼亚州卡尔斯巴德(Carlsbad,CA))生产的Qdot纳米晶体;玻璃包覆的金属纳米粒子(参见例如由Nanoplex Technologies公司(加利福尼亚州山景城(Mountain View,CA))生产的SERS纳米标签;条形码材料(参见例如亚微米大小的条纹金属棒,如由Nanoplex Technologies公司生产的纳米条形码)、用有色条形码编码的微粒(参见例如由Vitra Bioscience生产的CellCard,vitrabio.com)和具有数字全息代码图像的玻璃微粒(参见例如由Illumina(加利福尼亚州圣地亚哥(San Diego,CA))生产的CyVera微珠。如同与本发明实践相关联的许多标准程序一样,所属领域的技术人员应了解可使用的额外标记。
如本文所用,“诊断”一般包括关于受试者是否可能罹患给定疾病、病症或功能障碍的判定。所属领域的技术人员通常基于一个或多个诊断指标(即,生物标志物)进行诊断,诊断指标的存在、不存在或量指示疾病、病症或功能障碍存在或不存在。
如本文所用,“预后”一般地是指临床病况或疾病的可能病程和结果的预测。患者的预后通常是通过评估疾病的因素或症状而进行,疾病的因素或症状指示疾病的有利或不利的病程或结果。应理解,术语“预后”未必是指以100%准确度预测病况的病程或结果的能力。实际上,所属领域的技术人员应理解,术语“预后”是指特定病程或结果将发生的概率增加;也就是说,当与未展现给定病况的那些个体相比较时,病程或结果更可能发生于展现所述病况的患者中。
“基本上纯化的”是指核酸分子或蛋白质从其天然环境移出并经分离或分开,且至少约60%、优选地约75%、并且最优选地约90%不含与其天然相关联的其它组分。
II.执行本发明的模式
在详细描述本发明之前,应理解,本发明不限于特定配制品或工艺参数,因此当然可变化。还应理解,本文所用的术语仅出于描述本发明的具体实施方案的目的,并且不打算是限制性的。
尽管在本发明的实践中可使用与本文所述的方法和材料类似或等效的许多方法和材料,但是本文描述了优选的材料和方法。
本发明涉及生物标志物单独或与临床参数组合用于败血症诊断的用途。具体来说,本发明人已发现一组生物标志物,其表达谱可用于诊断败血症并区分败血症与非感染来源的全身炎症,所述全身炎症如由创伤性损伤、手术、自体免疫疾病、血栓形成或全身炎症反应综合征引起(参见实施例1)。
A.生物标志物
可用于本发明实践的生物标志物包括包含来自基因或基因的RNA转录物的核苷酸序列的多核苷酸,其包括(但不限于)CEACAM1、ZDHHC19、C9orf95、GNA15、BATF、C3AR1、KIAA1370、TGFBI、MTCH1、RPGRIP1和HLA-DPB1。这些生物标志物的差异表达与败血症相关联并因此,这些生物标志物的表达谱可用于诊断败血症并区分败血症与非感染性发炎病况,所述非感染性发炎病况如由创伤性损伤、手术、自体免疫疾病、血栓形成或全身炎症反应综合征(SIRS)引起。
因此,在一个方面,本发明提供一种用于诊断受试者的败血症的方法,包含测量来源于疑似患有败血症的受试者的生物样本中多种生物标志物的水平,分析所述生物标志物的水平并与所述生物标志物的相应参考值范围相比较,其中与对照样本中的一种或多种生物标志物相比,所述生物样本中一种或多种生物标志物的差异表达指示所述受试者患有败血症。当分析生物样本中生物标志物的水平时,用于比较的参考值范围可表示在一个或多个未患败血症的受试者的一个或多个样本(即,正常或未感染的对照样本)中所发现的一种或多种生物标志物的水平。或者,参考值可表示在一个或多个患有败血症的受试者的一个或多个样本中所发现的一种或多种生物标志物的水平。在某些实施方案中,将生物标志物的水平与未感染或已感染/败血症受试者的时间匹配的参考值相比。
从有待诊断的受试者获得的生物样本通常是全血、血沉棕黄层、血浆、血清或血细胞(例如外周血单个核细胞(PBMC)、杆状核细胞、嗜中性粒细胞、单核细胞或T细胞),但是可为来自含有所表达的生物标志物的体液、组织或细胞的任何样本。如本文所用,“对照”样本是指未发生病变的生物样本,如体液、组织或细胞。也就是说,对照样本是从正常或未感染的受试者(例如已知未患有败血症的个体)获得。生物样本可通过常规技术从受试者获得。举例来说,可通过静脉穿刺获得血液,并且可根据所属领域中众所周知的方法通过手术技术获得实体组织样本。
在某些实施方案中,将一组生物标志物用于败血症诊断。任何大小的生物标志物组可用于本发明实践。用于诊断败血症的生物标志物组通常包含至少3种生物标志物和多到30种的生物标志物,包括其间任何数目的生物标志物,如3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29或30种生物标志物。在某些实施方案中,本发明包括一种生物标志物组,其包含至少3种、至少4种、或至少5种、或至少6种、或至少7种、或至少8种、或至少9种、或至少10种、或至少11种或更多种生物标志物。虽然较小的生物标志物组通常更经济,但是较大的生物标志物组(即,大于30种生物标志物)具有提供更详细信息的优势并且也可用于本发明实践。
在某些实施方案中,本发明包括一组用于诊断败血症的生物标志物,所述生物标志物包含一种或多种包含来自选自以下组成的组的基因或基因的RNA转录物的核苷酸序列的多核苷酸:CEACAM1、ZDHHC19、C9orf95、GNA15、BATF、C3AR1、KIAA1370、TGFBI、MTCH1、RPGRIP1和HLA-DPB1。在一个实施方案中,所述组的生物标志物包含CEACAM1多核苷酸、ZDHHC19多核苷酸、C9orf95多核苷酸、GNA15多核苷酸、BATF多核苷酸、C3AR1多核苷酸、KIAA1370多核苷酸、TGFBI多核苷酸、MTCH1多核苷酸、RPGRIP1多核苷酸和HLA-DPB1多核苷酸。
在某些实施方案中,将感染Z评分用于败血症诊断。感染Z评分是通过将与生物标志物的对照参考值相比过度表达的所有测量的生物标志物的表达水平的几何平均值减去与生物标志物的对照参考值相比表达不足的所有测量的生物标志物的表达水平的几何平均值,并将差值乘以与生物标志物的对照参考值相比过度表达的生物标志物的数目与表达不足的生物标志物的数目的比率而计算得到。与未感染的对照受试者的参考值范围相比,受试者的感染Z评分更高指示受试者患有败血症(参见实施例1)。
在另一些实施方案中,将败血症评分用于败血症诊断。患者的败血症评分可根据下式基于CEACAM1、ZDHHC19、C9orf95、GNA15、BATF、C3AR1、KIAA1370、TGFBI、MTCH1、RPGRIP1和HLA-DPB1生物标志物的表达水平计算得出:

与未感染的对照受试者的参考值范围相比,受试者的败血症评分更高指示受试者患有败血症(参见实施例2)。
在另一个方面,本发明包括一种分析,其包含:a)在从疑似患有败血症的受试者采集的生物样本中,测量本文所述的生物标志物组的每一种生物标志物;和b)将所述生物样本中所述生物标志物组的每一种生物标志物的测量值与对照受试者的每一种生物标志物的参考值相比,其中与所述参考值相比,所述生物样本中生物标志物的差异表达指示所述受试者患有败血症。在某些实施方案中,所述分析另外包含如本文所述确定感染Z评分。
本文所述的方法可用于判定患有全身炎症的患者是否应针对败血症进行治疗。举例来说,如果如本文所述基于生物标志物表达谱或感染Z评分或败血症评分,患者的败血症诊断呈阳性,那么患者被选择进行败血症治疗。
在一个实施方案中,本发明包括一种治疗患有败血症的受试者的方法,所述方法包含:a)根据本文所述的方法诊断患有败血症的受试者;和b)如果所述受试者的败血症诊断呈阳性,那么向所述受试者给予治疗有效量的广谱抗生素。
在另一个实施方案中,本发明包括一种治疗疑似患有败血症的受试者的方法,所述方法包含:a)根据本文所述的方法接收关于所述受试者的诊断信息;和b)如果患者的败血症诊断呈阳性,那么给予所述受试者治疗有效量的广谱抗生素。
在另一个实施方案中,本发明包括一种用于监测治疗受试者的败血症的疗法的功效的方法,所述方法包含:测量在所述受试者进行所述疗法之前来源于所述受试者的第一样本和在所述受试者进行所述疗法之后来源于所述受试者的第二样本中CEACAM1、ZDHHC19、C9orf95、GNA15、BATF、C3AR1、KIAA1370、TGFBI、MTCH1、RPGRIP1和HLA-DPB1生物标志物的表达水平,其中与所述第一样本中所述生物标志物的表达水平相比,所述第二样本中所述CEACAM1、ZDHHC19、C9orf95、GNA15、BATF和C3AR1生物标志物的表达水平升高和所述KIAA1370、TGFBI、MTCH1、RPGRIP1和HLA-DPB1生物标志物的表达水平降低指示所述受试者恶化,并且与所述第一样本中所述生物标志物的表达水平相比,所述第二样本中所述CEACAM1、ZDHHC19、C9orf95、GNA15、BATF和C3AR1生物标志物的表达水平降低和所述KIAA1370、TGFBI、MTCH1、RPGRIP1和HLA-DPB1生物标志物的表达水平升高指示所述受试者改善。所述方法可另外包含计算所述受试者的败血症评分,其中与所述第一样本的败血症评分相比,所述第二样本的败血症评分更高指示所述受试者恶化,并且与所述第一样本的败血症评分相比,所述第二样本的败血症评分更低指示所述受试者改善。
B.检测和测量生物标志物
应理解,样本中的生物标志物可通过所属领域中已知的任何合适的方法测量。生物标志物的表达水平的测量可为直接或间接的。举例来说,可直接定量RNA或蛋白质的丰度水平。或者,可通过测量cDNA、经扩增的RNA或DNA的丰度水平或通过测量RNA、蛋白质或指示生物标志物的表达水平的其它分子(例如代谢物)的数量或活性间接测定生物标志物的量。用于测量样本中生物标志物的方法具有许多应用。举例来说,可测量一种或多种生物标志物以辅助败血症的诊断、确定适于受试者的治疗、监测受试者对治疗的响应或鉴别调节生物标志物在体内或体外的表达的治疗性化合物。
检测生物标志物多核苷酸
在一个实施方案中,生物标志物的表达水平是通过测量生物标志物的多核苷酸水平来测定。特定生物标志物基因的转录物水平可由生物样本中所存在的mRNA或来源于其的多核苷酸的量来测定。多核苷酸可通过多种方法检测和定量,所述方法包括(但不限于)微阵列分析、聚合酶链式反应(PCR)、逆转录酶聚合酶链式反应(RT-PCR)、Northern印迹和基因表达的系列分析(SAGE)。参见例如Draghici《DNA微阵列的数据分析工具(Data AnalysisTools for DNA Microarrays)》,Chapman and Hall/CRC,2003;Simon等人《DNA微阵列研究的设计和分析(Design and Analysis of DNA Microarray Investigations)》,Springer,2004;《实时PCR:当前技术和应用(Real-Time PCR:Current Technology andApplications)》,Logan,Edwards和Saunders编,Caister Academic Press,2009;Bustin《定量PCR的A-Z(A-Z of Quantitative PCR)》(IUL生物技术,第5期),InternationalUniversity Line,2004;Velculescu等人(1995)《科学》270:484-487;Matsumura等人(2005)《细胞微生物学(Cell.Microbiol.)》7:11-18;《基因表达的系列分析(SAGE):方法和方案(分子生物学方法)(Serial Analysis of Gene Expression(SAGE):Methods andProtocols(Methods in Molecular Biology))》,Humana Press,2008;其以全文引用的方式并入本文中。
在一个实施方案中,将微阵列用于测量生物标志物的水平。微阵列分析的优势在于可同时测量每一个生物标志物的表达,并且微阵列可专门设计成提供特定疾病或病况(例如败血症)的诊断表达谱。
微阵列是通过选择包含多核苷酸序列的探针并随后将这类探针固定于固体支撑物或表面而制备。举例来说,探针可包含DNA序列、RNA序列或DNA和RNA的共聚物序列。探针的多核苷酸序列还可包含DNA和/或RNA类似物或其组合。举例来说,探针的多核苷酸序列可为基因组DNA的完全或部分片段。探针的多核苷酸序列还可为合成的核苷酸序列,如合成寡核苷酸序列。探针序列可以酶促方式在体内、以酶促方式在体外(例如通过PCR)或以非酶促方式在体外合成。
本发明的方法中所用的探针优选地固定于固体支撑物,所述固体支撑物可为多孔或无孔的。举例来说,探针可为多核苷酸序列,其在多核苷酸的3'或5'端共价连接到硝化纤维素或尼龙膜或过滤器。这类杂交探针在所属领域中是众所周知的(参见例如Sambrook等人,《分子克隆:实验室手册》(第3版,2001)。或者,固体支撑物或表面可为玻璃或塑料表面。在一个实施方案中,测量与在表面上固定了一群多核苷酸(如一群DNA或DNA模拟物或替代地,一群RNA或RNA模拟物)的固相组成的探针微阵列的杂交水平。固相可为无孔的或任选地可为多孔材料,如凝胶。
在一个实施方案中,微阵列包含具有有序阵列的结合(例如杂交)位点或各自表示本文所述的一种生物标志物的“探针”的支撑物或表面。优选地,微阵列是可寻址的阵列,并且更优选地是可定位寻址的阵列。更特别地,阵列的每一个探针优选地位于固体支撑物上已知的预定位置,使得每一个探针的标识(即,序列)可由其在阵列中(即,在支撑物或表面上)的位置来确定。每一个探针优选地在单个位点处共价连接到固体支撑物。
微阵列可以多种方式制造,下文描述其中数种。无论它们是如何生产的,微阵列共有一些特征。阵列是可重复生产的,从而允许生产具有给定阵列的多个拷贝并且易于彼此相比。优选地,微阵列是由在结合(例如核酸杂交)条件下稳定的材料制成。微阵列一般较小,例如在1cm2与25cm2之间;然而,还可使用较大阵列,例如在筛选阵列中。优选地,微阵列中的给定结合位点或特有的一组结合位点将特异性结合(例如杂交)到细胞中单个基因的产物(例如来源于其的特异性mRNA或特异性cDNA)。然而,一般来说,其它相关或类似序列将与给定结合位点交叉杂交。
如上文所指出,与特定多核苷酸分子特异性杂交的“探针”含有互补多核苷酸序列。微阵列的探针通常由不超过1,000个核苷酸的核苷酸序列组成。在一些实施方案中,阵列的探针由具有10至1,000个核苷酸的核苷酸序列组成。在一个实施方案中,探针的核苷酸序列在10-200个核苷酸的长度范围内并且是一个生物体物种的基因组序列,因此存在多种不同探针,其序列与这种生物体物种的基因组互补并因此能够杂交,循序平铺覆盖所有或一部分基因组。在另一些实施方案中,探针在10-30个核苷酸的长度范围内、在10-40个核苷酸的长度范围内、在20-50个核苷酸的长度范围内、在40-80个核苷酸的长度范围内、在50-150个核苷酸的长度范围内、在80-120个核苷酸的长度范围内或是60个核苷酸的长度。
探针可包含与一部分生物体基因组对应的DNA或DNA“模拟物”(例如衍生物和类似物)。在另一个实施方案中,微阵列的探针是互补RNA或RNA模拟物。DNA模拟物是由能够与DNA特异性沃森-克里克样杂交(Watson-Crick-like hybridization)或能与RNA特异性杂交的亚单位构成的聚合物。核酸可在碱基部分、糖部分或磷酸酯主链(例如硫代磷酸酯)处经修饰。
DNA可例如通过基因组DNA或克隆序列的聚合酶链式反应(PCR)扩增而获得。PCR引物优选地基于基因组的已知序列而选择,将使得基因组DNA的特异性片段扩增。在所属领域中众所周知的计算机程序可用于设计具有所需特异性和最优扩增特性的引物,如Oligo5.0版(National Biosciences)。微阵列上每一个探针的长度将典型地在10个碱基与50,000个碱基之间,通常在300个碱基与1,000个碱基之间。PCR方法在所属领域中是众所周知的,并且例如描述在Innis等人编,《PCR方案:方法和应用指南(PCR Protocols:A Guide ToMethods And Applications)》,学术出版公司,圣地亚哥,加利福尼亚州(1990);其以全文引用的方式并入本文中。所属领域的技术人员应清楚,受控的机器人系统可用于分离和扩增核酸。
一种用于生成多核苷酸探针的优选的替代方式是通过合成多核苷酸或寡核苷酸的合成,例如使用N-膦酸酯或亚磷酰胺化学物质(Froehler等人,《核酸研究(Nucleic AcidRes.)》14:5399-5407(1986);McBride等人,《四面体通讯(Tetrahedron Lett.)》24:246-248(1983))。合成序列的长度通常在约10与约500个碱基之间,更通常在约20与约100个碱基之间并且最优选地在约40与约70个碱基长度之间。在一些实施方案中,合成核酸包括非天然碱基,如(但决不限于)肌苷。如上文所指出,核酸类似物可作为结合位点用于杂交。合适的核酸类似物的实例是肽核酸(参见例如Egholm等人,《自然》363:566-568(1993);美国专利第5,539,083号)。
探针优选地使用考虑结合能、碱基组成、序列复杂性、交叉杂交结合能和二级结构的算法来选择。参见Friend等人,国际专利公开WO 01/05935,2001年1月25日公开;Hughes等人,《自然·生物技术(Nat.Biotech.)》19:342-7(2001)。
所属领域的技术人员还应了解,阳性对照探针(例如已知与目标多核苷酸分子中的序列互补且可杂交的探针)和阴性对照探针(例如已知不与目标多核苷酸分子中的序列互补且不可杂交的探针)应包括在阵列上。在一个实施方案中,沿着阵列的周边合成阳性对照。在另一个实施方案中,在跨越阵列的对角条纹中合成阳性对照。在再一个实施方案中,紧邻探针的位置合成每一个探针的反向互补序列充当阴性对照。在又一个实施方案中,将来自其它物种生物体的序列用作阴性对照或用作“掺入”对照。
探针连接到固体支撑物或表面,所述固体支撑物或表面可例如由玻璃、塑料(例如聚丙烯、尼龙)、聚丙烯酰胺、硝化纤维素、凝胶或其它多孔或无孔材料制造。一种用于将核酸连接到表面的方法是通过印刷在玻璃板上,如由Schena等人,《科学》270:467-470(1995)大体上所述。此方法尤其可用于制备cDNA的微阵列(另外参见DeRisi等人,《自然·遗传学(Nature Genetics)》14:457-460(1996);Shalon等人,《基因组研究(Genome Res.)》6:639-645(1996);和Schena等人,《美国国家科学院院刊》93:10539-11286(1995);其以全文引用的方式并入本文中)。
第二种用于制造微阵列的方法产生高密度寡核苷酸阵列。已知在表面上使用用于原位合成的光刻技术在所界定的位置产生含有数千个与定义序列互补的寡核苷酸的阵列的技术(参见Fodor等人,1991,《科学》251:767-773;Pease等人,1994,《美国国家科学院院刊》91:5022-5026;Lockhart等人,1996,《自然·生物技术》14:1675;美国专利第5,578,832号;第5,556,752号;和第5,510,270号;其以全文引用的方式并入本文中)或其它用于快速合成和沉积所定义的寡核苷酸的方法(Blanchard等人,《生物传感器与生物电子学(Biosensors&Bioelectronics)》11:687-690;其以全文引用的方式并入本文中)。当使用这些方法时,在表面(如衍生化玻璃载片)上直接合成具有已知序列的寡核苷酸(例如60聚体)。通常,所产生的阵列是冗余的,每个RNA具有数个寡核苷酸分子。
还可使用其它用于制造微阵列的方法,例如通过掩蔽(Maskos和Southern,1992,《核酸研究》20:1679-1684;其以全文引用的方式并入本文中)。原则上,可使用任何类型的阵列,例如尼龙杂交膜上的斑点印迹(参见Sambrook等人,《分子克隆:实验室手册》,第3版,2001)。然而,所属领域的技术人员应认识到,极小阵列将常常是优选的,因为杂交体积将较小。
微阵列也可借助于用于寡核苷酸合成的喷墨印刷装置制造,例如使用由Blanchard在美国专利第6,028,189号中;Blanchard等人,1996,《生物传感器与生物电子学》11:687-690;Blanchard,1998,在《基因工程中的合成DNA阵列(Synthetic DNA Arraysin Genetic Engineering)》中,第20卷,J.K.Setlow编,Plenum Press,纽约在第111-123页(其以全文引用的方式并入本文中)所述的方法和系统。具体地说,这类微阵列中的寡核苷酸探针是通过在高表面张力溶剂(如碳酸丙二酯)的“微液滴”中连续沉积个别核苷酸碱基而例如在玻璃载片上以阵列形式合成。微液滴具有小体积(例如100pL或更小,更优选地50pL或更小)并且在微阵列上(例如通过疏水域)彼此分离形成圆形表面张力孔,其界定阵列要素(即,不同探针)的位置。通过此喷墨方法制造的微阵列通常具有高密度,优选地具有每1cm2至少约2,500个不同探针的密度。多核苷酸探针是在多核苷酸的3'或5'端共价连接到支撑物。
可通过微阵列分析测量的生物标志物多核苷酸可为表达RNA或来源于其的核酸(例如cDNA或来源于并入RNA聚合酶启动子的cDNA的扩增RNA),包括天然存在的核酸分子以及合成核酸分子。在一个实施方案中,目标多核苷酸分子包含RNA,包括(但决不限于)总细胞RNA、聚(A)+信使RNA(mRNA)或其片段、细胞质mRNA或由cDNA转录的RNA(即,cRNA;参见例如Linsley和Schelter,1999年10月4日提交的美国专利申请序号09/411,074或美国专利第5,545,522号、第5,891,636号或第5,716,785号)。用于制备总RNA和聚(A)+RNA的方法在所属领域中是众所周知的,并且一般例如描述在Sambrook等人,《分子克隆:实验室手册》(第3版,2001)中。可使用硫氰酸胍溶解随后CsCl离心(Chirgwin等人,1979,《生物化学》18:5294-5299)、基于硅胶的柱(例如RNeasy(加利福尼亚州巴伦西亚市的凯杰公司(Qiagen,Valencia,Calif.))或StrataPrep(加利福尼亚州拉荷亚的Stratagene公司(Stratagene,La Jolla,Calif.)))或使用苯酚和氯仿从所关注的细胞提取RNA,如Ausubel等人编,1989,《分子生物学现代方法(Current Protocols In Molecular Biology)》,第III卷,GreenPublishing Associates公司,John Wiley&Sons公司,纽约,第13.12.1-13.12.5页)中所述。聚(A)+RNA可例如通过用寡聚dT纤维素选择或替代地,通过寡聚dT引导的总细胞RNA的逆转录而选择。RNA可通过所属领域中已知的方法,例如通过与ZnCl2一起培育而片段化,生成RNA片段。
在一个实施方案中,总RNA、mRNA或来源于其的核酸是从败血症患者获取的样本分离。在特定细胞中弱表达的生物标志物多核苷酸可使用正规化技术富集(Bonaldo等人,1996,《基因组研究》6:791-806)。
如上所述,生物标志物多核苷酸可在一个或多个核苷酸处被可检测地标记。所属领域中已知的任何方法可用于标记目标多核苷酸。优选地,此标记沿着RNA的长度均一地并入标记,并且更优选地,标记是以高度的效率进行。举例来说,可通过寡聚dT引导的逆转录标记多核苷酸。随机引物(例如9聚体)可用于逆转录以便在多核苷酸的全长上均一地并入经标记的核苷酸。或者,随机引物可与PCR方法或基于T7启动子的体外转录方法结合使用以扩增多核苷酸。
可检测标记可为发光标记。举例来说,荧光标记、生物发光标记、化学发光标记和比色标记可用于本发明的实践中。可使用的荧光标记包括(但不限于)荧光素、磷光体、罗丹明或聚甲炔染料衍生物。此外,可使用可商购的荧光标记,包括(但不限于)荧光亚磷酰胺,如FluorePrime(Amersham Pharmacia,皮斯卡塔威,新泽西州(Piscataway,N.J.))、Fluoredite(Miilipore,贝德福,马萨诸塞州(Bedford,Mass.))、FAM(ABI,福斯特市,加利福尼亚州(Foster City,Calif.))和Cy3或Cy5(Amersham Pharmacia,皮斯卡塔威,新泽西州)。或者,可检测标记可为放射性标记的核苷酸。
在一个实施方案中,来自患者样本的生物标志物多核苷酸分子以与参考样本的相应多核苷酸分子有差异的方式进行标记。参考物可包含来自正常生物样本(即,对照样本,例如来自未患败血症的受试者的血液)或来自败血症参照生物样本(例如来自患有败血症的受试者的血液)的多核苷酸分子。
选择核酸杂交和洗涤条件,使得目标多核苷酸分子特异性结合或特异性杂交于阵列的互补多核苷酸序列、优选地其互补DNA所位于的特异性阵列位点。含有双链探针DNA定位于上面的阵列优选地经历变性条件以致使DNA在与目标多核苷酸分子接触之前单链化。含有单链探针DNA(例如合成的寡脱氧核酸)的阵列在与目标多核苷酸分子接触之前可能需要变性,例如以去除由于自身互补序列形成的发夹或二聚体。
最优杂交条件将取决于探针和目标核酸的长度(例如寡聚物对大于200个碱基的多核苷酸)和类型(例如RNA或DNA)。所属领域的技术人员应了解,由于寡核苷酸较短,所以可能必需调节其长度以达到相对均一的解链温度,从而获得令人满意的杂交结果。核酸的特异性(即,严格的)杂交条件的通用参数描述在Sambrook等人,《分子克隆:实验室手册》(第3版,2001)和Ausubel等人,《分子生物学现代方法》,第2卷,Current ProtocolsPublishing,纽约(1994)。Schena等人的用于cDNA微阵列的典型杂交条件是在5×SSC+0.2%SDS中在65℃下杂交四小时,随后在25℃下在低严格性洗涤缓冲液(1×SSC+0.2%SDS)中洗涤,随后在25℃下在较高严格性洗涤缓冲液(0.1×SSC+0.2%SDS)中洗涤10分钟(Schena等人,《美国国家科学院院刊》93:10614(1993))。有用的杂交条件还提供在例如Tijessen,1993,《用核酸探针杂交(Hybridization With Nucleic Acid Probes)》,Elsevier Science Publishers公司;和Kricka,1992,《非同位素DNA探针技术(Nonisotopic Dna Probe Techniques)》,学术出版社,圣地亚哥,加利福尼亚州中。尤其优选的杂交条件包括在处于或接近探针的平均解链温度(例如在51℃内,更优选地在21℃内)的温度下在1M NaCl、50mM MES缓冲液(pH 6.5)、0.5%肌氨酸钠和30%甲酰胺中杂交。
当使用荧光标记的基因产物时,在微阵列的每一个位点处的荧光发射可优选地通过扫描共聚焦激光显微术检测到。在一个实施方案中,针对所用两个荧光团中的每一个使用适当激发光线进行单独扫描。或者,可使用允许在特异性针对两个荧光团的波长下同时进行样本照射的激光器,并且可同时分析来自两个荧光团的发射(参见Shalon等人,1996,“使用双色荧光探针杂交分析复杂DNA样本的DNA微阵列系统(A DNA microarray systemfor analyzing complex DNA samples using two-color fluorescent probehybridization),”《基因组研究》6:639-645,其以全文引用的方式并入用于所有目的)。可使用具有计算机控制的X-Y工作台和显微镜物镜的激光荧光扫描仪扫描阵列。使用多线路混合气体激光器实现两个荧光团的依次激发,并且发射光由波长分开并用两个光电倍增管检测到。荧光激光扫描装置描述在Schena等人,《基因组研究》6:639-645(1996)本文中所列举的其它参考文献中。或者,由Ferguson等人,《自然·生物技术》14:1681-1684(1996)所描述的光纤束可用于同时监测大量位点的mRNA丰度水平。
在一个实施方案中,本发明包括微阵列,其包含与CEACAM1多核苷酸杂交的寡核苷酸、与ZDHHC19多核苷酸杂交的寡核苷酸、与C9orf95多核苷酸杂交的寡核苷酸、与GNA15多核苷酸杂交的寡核苷酸、与BATF多核苷酸杂交的寡核苷酸、与C3AR1多核苷酸杂交的寡核苷酸、与KIAA1370多核苷酸杂交的寡核苷酸、与TGFBI多核苷酸杂交的寡核苷酸、与MTCH1多核苷酸杂交的寡核苷酸、与RPGRIP1多核苷酸杂交的寡核苷酸和与HLA-DPB1多核苷酸杂交的寡核苷酸。
多核苷酸也可通过其它方法分析,包括(但不限于)northern印迹、核酸酶保护分析、RNA指纹分析、聚合酶链式反应、连接酶链式反应、Qβ复制酶、等温扩增方法、链置换扩增、基于转录的扩增系统、核酸酶保护(S1核酸酶或RNA酶保护分析)、SAGE以及国际公开第WO 88/10315号和第WO 89/06700号和国际申请第PCT/US87/00880号和第PCT/US89/01025号中所公开的方法;其以全文引用的方式并入本文中。
根据所属领域的一般技术人员已知的常规Northern杂交技术,标准Northern印迹分析可用于确定RNA转录物大小、鉴别交替剪接的RNA转录物和样本中mRNA的相对量。在Northern印迹中,RNA样本首先在变性条件下通过琼脂糖凝胶电泳按大小分离。随后将RNA转移到膜,交联并与标记探针杂交。可使用非同位素或高放射性比度放射性标记的探针,包括随机引导、切口翻译或PCR生成的DNA探针、体外转录的RNA探针和寡核苷酸。此外,可使用仅具有部分同源性的序列(例如来自不同物种的cDNA或可能含有外显子的基因组DNA片段)作为探针。含有全长单链DNA或DNA序列片段的标记探针(例如放射性标记的cDNA)可为至少20个、至少30个、至少50个或至少100个连续核苷酸的长度。探针可通过所属领域的技术人员已知的许多不同方法中的任一种而被标记。最常用于这些研究的标记是放射性元素、酶、当曝露于紫外线时发荧光的化学物质等。多种荧光材料是已知的并且可用作标记。这些荧光材料包括(但不限于)荧光素、罗丹明、金胺、德克萨斯红、AMCA蓝和荧光黄。特定检测材料是在山羊体内制备并经由异硫氰酸酯与荧光素结合的抗兔抗体。蛋白质也可用放射性元素或酶标记。放射性标记可通过目前可用的计数程序中的任一个检测到。可使用的同位素包括(但不限于)3H、14C、32P、35S、36Cl、35Cr、57Co、58Co、59Fe、90Y、125I、131I和186Re。酶标记同样有用,并且可通过目前采用的比色、分光光度、荧光分光光度、电流测定或气体测定技术中的任一个检测到。酶是通过与如碳化二亚胺、二异氰酸酯、戊二醛等桥连分子反应结合到所选粒子。可采用所属领域的技术人员已知的任何酶。这类酶的实例包括(但不限于)过氧化物酶、β-D-半乳糖苷酶、尿素酶、葡萄糖氧化酶加过氧化物酶和碱性磷酸酶。举例来说,参考美国专利第3,654,090号、第3,850,752号和第4,016,043号的关于替代性标记材料和方法的公开内容。
核酸酶保护分析(包括核糖核酸酶保护分析和S1核酸酶分析)可用于检测和定量特异性mRNA。在核酸酶保护分析中,反义探针(经例如放射性标记或非同位素标记)在溶液中与RNA样本杂交。在杂交后,利用核酸酶降解未杂交的单链探针和RNA。使用丙烯酰胺凝胶分离其余受保护的片段。通常,溶液杂交比基于膜的杂交更有效,并且其与最大20-30μg的印迹交杂相比可容纳至多100μg样本RNA。
核糖核酸酶保护分析,是最常见类型的核酸酶保护分析,需要使用RNA探针。寡核苷酸和其它单链DNA探针仅可用于含有S1核酸酶的分析。单链反义探针必须通常与目标RNA完全同源以防止核酸酶裂解探针:目标杂交。
基因表达系列分析(SAGE)也可用于测定细胞样本中的RNA丰度。参见例如Velculescu等人,1995,《科学》270:484-7;Carulli等人,1998,《细胞生物化学杂志(Journal of Cellular Biochemistry)》增刊30/31:286-96;其以全文引用的方式并入本文中。SAGE分析不需要特殊检测装置,并且是用于同时检测大量转录产物表达的优选分析方法之一。首先,从细胞提取聚A+RNA。接着,使用生物素化寡聚(dT)引物将RNA转化成cDNA,并用四碱基识别限制酶(锚定酶:AE)处理,产生在3'端含有生物素基团的经AE处理的片段。接着,经AE处理的片段与抗生蛋白链菌素一起培育以便结合。将结合的cDNA分成两份,并且每一份随后连接到不同双链寡核苷酸接头(连接子)A或B。这些连接子是由以下构成:(1)具有与通过锚定酶作用形成的突出部分的序列互补的序列的突出单链部分,(2)充当标签酶(TE)的IIS型限制酶(在远离识别位点不超过20bp的预定位置处裂解)的5'核苷酸识别序列,和(3)用于构建PCR特异性引物的具有足够长度的额外序列。使用标签酶裂解连接子连接的cDNA,并且仅保留以短链序列标签形式存在的连接子连接的cDNA序列部分。接着,将来自两种不同类型连接子的短链序列标签池彼此连接,随后使用特异性针对连接子A和B的引物进行PCR扩增。因此,扩增产物是以包含结合到连接子A和B的两种邻近序列标签(双标签)的多种序列的混合物形式获得。用锚定酶处理扩增产物,并且在标准键联反应中将游离双标签部分连接成链。随后克隆扩增产物。测定克隆的核苷酸序列可用于获得恒定长度的连续双标签的读出序列。可随后从克隆的核苷酸序列和关于序列标签的信息鉴别与每一个标签对应的mRNA的存在。
定量逆转录酶PCR(qRT-PCR)也可用于测定生物标志物的表达谱(参见例如美国专利申请公开第2005/0048542A1号;其以全文引用的方式并入本文中)。通过RT-PCR的基因表达谱分析的第一步是使RNA模板逆转录成cDNA,随后使其在PCR反应中进行指数扩增。两种最常用的逆转录酶是禽类成髓细胞瘤病毒逆转录酶(avilo myeloblastosis virusreverse transcriptase,AMV-RT)和莫洛尼鼠白血病病毒逆转录酶(Moloney murineleukemia virus reverse transcriptase,MLV-RT)。逆转录步骤通常使用特异性引物、随机六聚体或寡聚dT引物引导,根据表达谱分析的环境和目标而定。举例来说,提取的RNA可按照制造商说明书使用GeneAmp RNA PCR试剂盒(珀金埃尔默公司(Perkin Elmer),加利福尼亚州,美国)逆转录。所得到的cDNA可随后作为模板用于后续PCR反应。
虽然PCR步骤可使用多种热稳定的DNA依赖性DNA聚合酶,但是其通常采用Taq DNA聚合酶,其具有5'-3'核酸酶活性但不具有3'-5'校正核酸内切酶活性。因此,TAQMAN PCR通常利用Taq或Tth聚合酶的5'-核酸酶活性水解与其目标扩增子结合的杂交探针,但是仍可使用任何具有等效5'核酸酶活性的酶。使用两种寡核苷酸引物生成PCR反应典型的扩增子。将第三寡核苷酸或探针设计成检测定位于两个PCR引物之间的核苷酸序列。探针无法通过Taq DNA聚合酶延伸,并且用报道荧光染料和淬灭荧光染料标记。当两种染料如它们在探针上一样紧密定位在一起时,任何激光诱导的来自报道染料的发射是由淬灭染料淬灭。在扩增反应期间,Taq DNA聚合酶以模板依赖性方式裂解探针。所得探针片段在溶液中解离,并且来自所释放的报道染料的信号没有第二荧光团的淬灭效应。新合成的每一分子释放一分子的报道染料,并且检测未淬灭的报道染料提供用于定量解释数据的基础。
TAQMAN RT-PCR可使用可商购的设备进行,如ABI PRISM 7700序列检测系统(珀金埃尔默应用生物系统公司,福斯特市,加利福尼亚州,美国)或Lightcycler(RocheMolecular Biochemicals,曼海姆,德国(Mannheim,Germany))。在一优选实施方案中,5'核酸酶程序是在如ABI PRISM 7700序列检测系统的实时定量PCR装置上运行。所述系统由热循环仪、激光器、电荷耦合装置(CCD)、照相机和计算机组成。所述系统包括用于运行仪器和用于分析数据的软件。5'-核酸酶分析数据首先表示为Ct或阈值循环。在每一循环期间记录荧光值并且其表示在扩增反应中扩增到那一点的产物量。当荧光信号最先记录为统计学上显著时的点是阈值循环(Ct)。
为了使错误和样本间变化的效应最小,通常使用内标进行RT-PCR。理想内标在不同组织当中以恒定水平表达并且不受实验治疗影响。最经常用于将基因表达图谱正规化的RNA是管家基因甘油醛-3-磷酸脱氢酶(GAPDH)和β-肌动蛋白的mRNA。
更新的RT-PCR技术变化形式是实时定量PCR,其经由双重标记的荧光生成探针(即,TAQMAN探针)测量PCR产物的累积。实时PCR与使用每一目标序列的内部竞争者进行正规化的定量竞争性PCR和使用样本内含有的正规化基因或用于RT-PCR的管家基因的定量比较PCR都相容。关于更多细节,参见例如Held等人,《基因组研究》6:986-994(1996)。
生物标志物数据的分析
为了评估患者是否患有败血症或由非感染源(如创伤性损伤、手术、自体免疫疾病、血栓形成或全身炎症反应综合征(SIRS))引起的全身炎症,可通过多种方法分析生物标志物数据以鉴别生物标志物并确定在测试表达谱和参考表达谱之间所观测到的生物标志物水平差异的统计显著性。在某些实施方案中,患者数据是通过一种或多种方法分析,所述方法包括(但不限于)多变量线性判别分析(LDA)、受试者工作特征(ROC)分析、主成分分析(PCA)、集合数据挖掘方法、微阵列的显著性分析(SAM)、微阵列的细胞特异性显著性分析(csSAM)、密度归一化事件的生成树级数分析(SPADE)和多维蛋白质鉴定技术(MUDPIT)分析。(参见例如Hilbe(2009)《逻辑回归模型(Logistic Regression Models)》,Chapman&Hall/CRC Press;McLachlan(2004)《判别分析和统计模式识别(Discriminant Analysisand Statistical Pattern Recognition)》.Wiley Interscience;Zweig等人(1993)《临床化学(Clin.Chem.)》39:561-577;Pepe(2003)《用于分类和预测的医学检验的统计评估(Thestatistical evaluation of medical tests for classification and prediction)》,纽约,NY:Oxford;Sing等人(2005)《生物信息学(Bioinformatics)》21:3940-3941;Tusher等人(2001)《美国国家科学院院刊》98:5116-5121;Oza(2006)《集合数据挖掘(Ensembledata mining)》,NASA Ames研究中心,莫菲特菲尔德(Moffett Field),加利福尼亚州,美国;English等人(2009)《生物医学信息学杂志(J.Biomed.Inform.)》42(2):287-295;Zhang(2007)《生物信息学》8:230;Shen-Orr等人(2010)《免疫学杂志》184:144-130;Qiu等人(2011)《自然·生物技术》29(10):886-891;Ru等人(2006)《色谱杂志A(J.Chromatogr.A.)》1111(2):166-174,Jolliffe主成分分析(《Springer系列丛书--统计(Springer Series inStatistics)》,第2版,Springer,纽约州,2002),Koren等人(2004)《IEEE可视化与计算机图形学学报(IEEE Trans Vis Comput Graph)》10:459-470;其以全文引用的方式并入本文中。)
C.试剂盒
在又一个方面,本发明提供用于诊断败血症的试剂盒,其中所述试剂盒可用于检测本发明的生物标志物。举例来说,试剂盒可用于检测任一个或多个本文所述的生物标志物,所述生物标志物在败血症患者和健康或未感染的受试者的样本中差异表达。试剂盒可包括一种或多种用于检测生物标志物的药剂、用于盛放从疑似患有败血症的人类受试者分离的生物样本的容器;以及用于使药剂与生物样本或一部分生物样本反应以检测生物样本中至少一种败血症生物标志物的存在或量的印刷说明书。药剂可封装在单独的容器中。试剂盒可另外包含一种或多种对照参考样本和用于进行免疫分析或微阵列分析的试剂。
在某些实施方案中,试剂盒包含用于测量所关注的至少十一种生物标志物的水平的药剂。举例来说,试剂盒可包括用于检测一组生物标志物的药剂,所述生物标志物包含CEACAM1多核苷酸、ZDHHC19多核苷酸、C9orf95多核苷酸、GNA15多核苷酸、BATF多核苷酸、C3AR1多核苷酸、KIAA1370多核苷酸、TGFBI多核苷酸、MTCH1多核苷酸、RPGRIP1多核苷酸和HLA-DPB1多核苷酸。此外,试剂盒可包括用于检测大于一个生物标志物组(如两个或三个生物标志物组)的药剂,其可单独或以任何组合形式和/或与用于败血症诊断的临床参数组合起来使用。
在某些实施方案中,试剂盒包含用于分析多种生物标志物多核苷酸的微阵列。试剂盒中所包括的示例性微阵列包含与CEACAM1多核苷酸杂交的寡核苷酸、与ZDHHC19多核苷酸杂交的寡核苷酸、与C9orf95多核苷酸杂交的寡核苷酸、与GNA15多核苷酸杂交的寡核苷酸、与BATF多核苷酸杂交的寡核苷酸、与C3AR1多核苷酸杂交的寡核苷酸、与KIAA1370多核苷酸杂交的寡核苷酸、与TGFBI多核苷酸杂交的寡核苷酸、与MTCH1多核苷酸杂交的寡核苷酸、与RPGRIP1多核苷酸杂交的寡核苷酸和与HLA-DPB1多核苷酸杂交的寡核苷酸。
试剂盒可包含一个或多个用于容纳试剂盒中所含组合物的容器。组合物可呈液体形式或可冻干。用于组合物的合适容器包括例如瓶子、小瓶、注射器和试管。容器可由多种材料形成,包括玻璃或塑料。试剂盒还可包含药品说明书,其含有诊断败血症方法的书面说明。
本发明的试剂盒具有多种应用。举例来说,试剂盒可用于判定受试者是否患有败血症或由非感染源引起的某一其它发炎病况,所述非感染源如创伤性损伤、手术、自体免疫疾病、血栓形成或全身炎症反应综合征(SIRS)。在另一个实例中,试剂盒可用于判定患者是否应针对败血症进行治疗,例如用广谱抗生素治疗。在另一个实例中,试剂盒可用于监测患有败血症的患者的治疗有效性。在另一个实例中,试剂盒可用于鉴别调节一种或多种生物标志物在体外或体内动物模型中的表达的化合物以判定治疗效应。
D.用于败血症诊断的诊断系统和计算机化方法
在另一个方面,本发明包括一种计算机实施的用于诊断疑似患有败血症的患者的方法。计算机执行包含以下的步骤:接收所输入的患者数据,包含来自患者的生物样本中一种或多种败血症生物标志物的水平值;分析一种或多种败血症生物标志物的水平并与败血症生物标志物的相应参考值范围相比;计算患者的感染Z评分或败血症评分;计算患者患有败血症的可能性;以及显示关于患者的诊断信息。在某些实施方案中,所输入的患者数据包含来自患者的生物样本中多种败血症生物标志物的水平值。在一个实施方案中,所输入的患者数据包含CEACAM1、ZDHHC19、C9orf95、GNA15、BATF、C3AR1、KIAA1370、TGFBI、MTCH1、RPGRIP1和HLA-DPB1多核苷酸的水平值。
在另一个方面,本发明包括一种用于执行计算机实施的方法的诊断系统,如所描述。如图16所示,诊断系统100包括计算机110,其含有处理器130、存储组件(即,存储器)120、显示器组件150和通用计算机中通常所存在的其它组件。存储组件120存储可由处理器130访问的信息,包括可由处理器130执行的指令和可由处理器检索、操纵或存储的数据。
存储组件包括用于确定受试者的诊断的指令。举例来说,存储组件包括用于如本文所述(参见实例1和2)基于生物标志物表达水平计算受试者的感染Z评分或败血症评分的指令。此外,存储组件可另外包含用于进行多变量线性判别分析(LDA)、受试者工作特征(ROC)分析、主成分分析(PCA)、集合数据挖掘方法、微阵列的细胞特异性显著性分析(csSAM)和多维蛋白质鉴定技术(MUDPIT)分析的指令。计算机处理器130被耦合到存储组件120并且被配置成执行存储在存储组件中的指令以便接收患者数据并根据一种或多种算法分析患者数据。显示器组件150显示关于患者的诊断信息。
存储组件120可属于任何能够存储可由处理器访问的信息的类型,如硬盘驱动器、存储卡、ROM、RAM、DVD、CD-ROM、USB快闪驱动器、写入存储器和只读存储器。处理器130可为任何熟知的处理器,如来自因特尔公司(Intel Corporation)的处理器。或者,处理器可为专用控制器,如ASIC。
指令可为有待由处理器直接(如机器代码)或间接(如脚本)执行的任何指令集合。为此,术语“指令”、“步骤”和“程序”在本文中可互换使用。指令可以由处理器直接处理的目标代码形式或以任何其它计算机语言形式存储,所述其它计算机语言包括按需求解译或提前编译的脚本或独立的源代码模块的集合。
数据可根据指令由处理器130检索、存储或修改。举例来说,虽然诊断系统不受任何特定数据结构限制,但是数据可存储在计算机寄存器中、以具有多种不同字段或记录的表格、XML文档或平面文件形式存储在关系型数据库中。数据还可以任何计算机可读格式来格式化,计算机可读格式如(但不限于)二进制值、ASCII或统一码。此外,数据可包含足以鉴别相关信息的任何信息,如编号、描述性文本、专有代码、指针、对存储在其它存储器(包括其它网络位置)中的数据的引用或由计算相关数据的函数所使用的信息。
在某些实施方案中,处理器和存储组件可包含能够或不能存储在同一物理外壳内的多个处理器和存储组件。举例来说,一些指令和数据可存储在可移动CD-ROM上并且其它指令和数据存储在只读计算机芯片内。一些或全部指令和数据可存储在物理上远离处理器但仍可由处理器访问的位置中。类似地,处理器可实际包含能够或不能并行操作的处理器集合。
在一个方面,计算机110是与一个或多个客户端计算机140、170通信的服务器。每一个客户端计算机可与服务器110类似地配置,具有处理器、存储组件和指令。每一个客户端计算机140、170可为专供个人190-191使用的个人计算机,具有个人计算机中通常所见的所有内部组件,如中央处理单元(CPU)、显示器150(例如显示由处理器处理的信息的监测器)、CD-ROM、硬盘驱动器、用户输入装置(例如鼠标、键盘、触摸屏或麦克风)160、扬声器、调制解调器和/或网络接口装置(电话、电缆或其它)以及用于将这些要素彼此连接并允许其彼此(直接或间接)通信的所有组件。此外,根据本文所述的系统和方法的计算机可包含任何能够处理指令并传输去往和来自人类和其它计算机(包括不具有本地存储能力的网络计算机)的数据的装置。
虽然客户端计算机140和170可包含全尺寸个人计算机,但是所述系统和方法的许多方面在与能够经网络(如互联网)与服务器无线交换数据的移动装置结合使用时特别有利。举例来说,客户端计算机170可为启用无线的PDA,如黑莓手机、苹果iPhone或其它具有互联网功能的蜂窝电话。在这些方面中,用户可使用小键盘、键区、触摸屏或任何其它用户输入方式输入信息。计算机可具有天线180用于接收无线信号。
服务器110和客户端计算机140、170能够直接和间接通信,如通过网络200。虽然图16中仅描绘很少的计算机,但应了解,典型系统可包括大量连接的计算机,每一个不同计算机处于网络200的不同节点。网络和介入节点可包含装置和通信协议的各种组合,包括互联网、万维网、内联网、虚拟专用网络、广域网、局域网、蜂窝电话网络、使用一个或多个公司专用的通信协议的专用网络、以太网、WiFi和HTTP。这类通信可通过任何能够传输去往和来自其它计算机的数据的装置推进,如调制解调器(例如拨号或电缆)、网络和无线接口。服务器110可为web服务器。
虽然当如上文所指出传输或接受信息时获得某些优势,但是所述系统和方法的其它方面不限于任何特定的信息传输方式。举例来说,在一些方面,信息可经由如磁盘、磁带、闪存驱动器、DVD或CD-ROM等媒体传送。在其它方面,信息可以非电子格式传输并手动输入到系统中。再有,虽然一些功能被指示为在服务器上进行并且其它功能在客户端上进行,但是所述系统和方法的各个方面可由具有单个处理器的单个计算机实施。
III.实验
以下为执行本发明的特定实施方案的实施例。实施例仅出于说明性目的提供,并且不打算以任何方式限制本发明的范围。
已作出努力以确保关于所使用的数字(例如量、温度等)的准确度,但允许一些实验误差和偏差应为理所当然的。
实施例1
败血症和无菌炎症的基于时程的全面多群组分析揭露稳固的诊断基因集合
概述
我们假设仅时间匹配的比较,如在相同临床时间点比较SIRS/创伤与败血症的那些,将获得稳固诊断败血症的基因。我们对公开可用的败血症基因表达数据进行全面的基于时程的多群组分析以鉴别可稳固区分非感染性炎症(如SIRS、创伤和ICU收住)与因急性感染所致的炎症(如在败血症中)的11个基因的保守性集合。此11个基因的集合在发现群组中具有极好的诊断能力,并且随后在15个独立的群组中验证。
结果
全面搜索和经标记的主成分分析(PCA)可视化
我们从包括总共2,903个微阵列的GEO和ArrayExpress中鉴别了27个符合我们标准的独立的基因表达数据集(Pankla等人(2009)《基因组生物学》10:R127;Tang等人(2009)《危重病监护医学》37:882-888;Cvijanovich等人(2008)《生理基因组学(PhysiolGenomics)》34:127-134;Shanley等人(2007)《分子医学》13:495-508;Wong等人(2007)《生理基因组学》30:146-155;Wong等人(2009)《危重病监护医学》37:1558-1566;Wong等人(2010)《儿科危重病监护医学(Pediatr Crit Care Med)》11:349-355;Wong等人(2011)《危重病监护医学》39:2511-2517;Almansa等人(2014)《危重病监护杂志(J Crit Care)》29:307-309;Bermejo-Martin等人(2010)《危重病监护(Crit Care)》14:R167;Martin-Loeches等人(2012)《重症监护医学(Med Intensiva)》36:257-263;Tamayo等人(2012)《危重病监护杂志》27:616-622;Hu等人(2013)《美国国家科学院院刊》110:12792-12797;Parnell等人(2012)《危重病监护》16:R157;Sutherland等人(2011)《危重病监护》15:R149;Tang等人(2006)《脑血流与代谢杂志(J Cereb Blood Flow Metab)》26:1089-1102;Tang等人(2007)《美国呼吸和重症监护医学杂志(Am J Respir Crit Care Med)》176:676-684;Ahn等人(2013)《公共科学图书馆·综合(PLoS One)》8:e48979;Dolinay等人(2012)《美国呼吸和重症监护医学杂志》185:1225-1234;Berdal等人(2011)《传染病杂志(J Infect)》63:308-316;Berry等人(2010)《自然》466:973-977;Fredriksson等人(2008)《公共科学图书馆·综合》3:e3686;McDunn等人(2008)《公共科学图书馆·综合》3:e1564;Chung等人(2006)《美国外科医师学会杂志(J Am Coll Surg)》203:585-598;Parnell等人(2011)《公共科学图书馆·综合》6:e17186;和Emonts,《鹿特丹伊拉斯姆斯大学博士论文(Ph.D.thesis,ErasmusUniversity Rotterdam)》,(2008);其以全文引用的方式并入本文中)。这27个数据集包含仅22个独立的群组,因为来自儿童SIRS/败血性休克基因组学研究者(Genomics ofPediatric SIRS/Septic Shock Investigators,GPSSSI)的六个数据集组合成含有219名SIRS或败血症患者的单个群组(Cvijanovich等人,见上文;Shanley等人,见上文;Wong等人(2007),见上文;Wong等人(2009),见上文;Wong等人(2010),见上文;Wong等人(2011),见上文)。许多所用样本来自Glue Grant创伤数据集,其具有在创伤性损伤后的至多8个时间点取样的总共333名患者(此处使用1301个样本)。这27个数据集含有从全血、嗜中性粒细胞和PBMC取样的患有社区获得性和医院获得性败血症混杂的儿童和成人、男性和女性群组。
首先,我们试图使用尽可能简单的方法以发现未感染的SIRS/创伤患者和败血症/感染患者是否能够通过基因表达分离。我们因此将比较SIRS/创伤与败血症/感染的所有可用数据集共同归一化在单个矩阵中。经标记的PCA(使用通过10倍交叉验证的Lasso惩罚逻辑回归鉴别的168个基因)显示可在适度重叠下将SIRS/创伤患者与败血症患者分离(图1A)。接着,我们将每一个样本标记为“早期”(在入院48小时内)或“后期”(入院超过48小时)。大部分不可分离的样本是‘后期’样本(图1B)。即使当我们将健康患者作为单独的类别包括在内时,此研究结果仍保持成立(图7)。先前研究已显示,在创伤、烧伤或内毒素血症后的基因表达随时间推移发生非线性变化(Cobb等人,见上文;Xiao等人,见上文;Seok等人,见上文;Desai等人,见上文;和McDunn等人,见上文)。表达在初始伤害后的这一连续变化可解释如果对所有时间点同等对待,那么不能在‘后期’样本中区分未感染的SIRS/创伤与败血症。
因此,我们试图定性了解在受伤后的住院过程中,不同群组当中的基因表达是否类似。我们包括在非败血性事件入院之后随时间推移纵向检查基因表达的所有外周血数据集。我们使用CUR矩阵分解以鉴别彼此大部分正交的100个基因,并使用这些以按受伤后天数确定的类别进行经标记的PCA。令人安心的是,在每一时间点的基因表达组最接近于其所限定的时间点(例如天数[1,2)组前面是天数[0,1)并且后面是天数[2,3))。此外,表达随时间推移的变化解释了数据集中的大部分方差,如由不同天数组在前三个经标记的主成分中每一个的变化所证明。总而言之,我们的分析显示基因表达在创伤/ICU收住后的变化(1)随时间推移以非线性方式继续,并且(2)示出了跨数据集间随时间推移的类似变化。
时间匹配的多群组分析
由于在创伤入院后基因表达的变化解释了数据集中的大量方差,并且由于这些变化非线性地继续,所以直接比较入院时的患者与在感染后数天的同一患者将受因从刺激事件恢复所致的‘正常’表达变化以及因医院获得性感染所致的‘异常’变化混淆。即使并非不可能,理清这些变化也将极其困难。因此,不考虑临床时间的比较将无法获得可稳固地区分感染患者与未感染患者的生物标志物(图1)。因此,我们仅聚焦在还包括时间匹配的未感染群组的感染数据集(以允许时间匹配的直接比较)。我们因此将数据集分成两组:(1)比较因创伤、手术或危重病入院的患者对因败血症入院的患者的数据集(GSE28750(Sutherland等人,见上文)、GSE32707(Dolinay等人,见上文)、GSE40012(Parnell等人,见上文)和GPSSSI特有组合数据集(n=408个样本)(Cvijanovich等人,见上文;Shanley等人,见上文;Wong等人(2007),见上文;Wong等人(2009),见上文;Wong等人(2010),见上文;和Wong等人(2011),见上文)和(2)含有具医院获得性感染的患者和天数匹配的未感染患者的Glue Grant数据集,其中我们仅使用血沉棕黄层样本群组中的患者(表1)。Glue Grant创伤群组从损伤起大致0.5、1、4、7、14、21和28天进行取样;这些群组因此划分成其取样时间仓,形成在给定时间仓诊断有感染的患者可与相同时间仓未感染的患者相比的子组。对于血沉棕黄层样本,五个时间仓之存在至少10名患者,并且这些样本因此被取出用于另外的研究。因此,我们使用总共9个群组比较时间匹配的SIRS/创伤与败血症/感染,包含663个样本(326个SIRS/创伤对照和337个败血症/感染病例;表2示出了多群组分析中的群组。
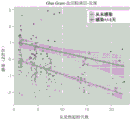
我们随后应用我们先前所描述(Khatri等人(2013)《实验医学杂志(J Exp Med)》210:2205-2221)的多群组基因表达分析框架以留一数据集的方式比较包括所有9个群组的SIRS/创伤与败血症/感染。这一分析的输出结果经历三步阈值处理(所汇集的效应量和Fischer法的错误发现率(FDR)<1%,数据集间异质性p>0.01,并且绝对汇总效应量倍数变化>1.5),得出在SIRS/创伤患者与败血症患者之间跨越所有时间点差异表达的82个基因(所有82个基因的汇总统计资料显示在表8中)。为了获得类别间最佳区分的最简约的重要基因集合,我们执行贪心前向搜索法以鉴别最佳改善跨越所有发现数据集的AUC所得到的82个基因的组合。此处,区分是基于‘感染Z评分’,其将每一个数据集中的每一个样本的基因表达水平(使用正基因与负基因之间几何平均值的差值)组合成标准化评分。这得到最终11个基因的集合(在败血症中6个过度表达并且5个表达不足;表3和图2)。9个发现群组中这11个基因的集合的平均ROC AUC是0.87(范围0.70-0.98;图3A和图9)。
Glue Grant分选细胞群组验证
Glue Grant创伤群组具有两个独立的子群组;一个是血沉棕黄层群组(Affymetrix阵列GPL570上经处理的样本2004-2006),另一个是分选细胞群组,其包括嗜中性粒细胞、单核细胞和T细胞(自定义Glue Grant-人类(GGH)阵列上经处理的样本2008-2011)。这些群组是单独的患者,及时隔离并使用不同技术剖析。虽然纳入标准和登记场所大部分相同,但他们原本是独立的。我们因此在分选细胞Glue Grant群组中验证我们的11个基因标签。此处,我们将分选细胞群组分入与发现血沉棕黄层群组相同的时间仓并分别处理每个时间仓。
从分选细胞子群组,我们预期嗜中性白细胞集合表现最类似于全血样本,因为感染患者和未感染患者在创伤后的嗜中性粒细胞占总白细胞池的75-85%(并且因此大部分基因表达存在于外周血中)(图8)。实际上,在从败血性创伤患者分离时间匹配的未感染的创伤患者时,11个基因的集合表现得极好(4个群组,218个样本;平均AUC 0.83,范围0.73-0.89)(图3B)。出人意料地,在来自这些相同患者的单核细胞和T细胞中11个基因的集合也显示出区分能力(单核细胞AUC范围0.71-0.97,T细胞AUC范围0.69-0.9)(图10A、11A)。由于我们从多群组分析排除了任何分选细胞数据集,所以我们并未预期在这些细胞类型中的诊断能力。令人关注的是,在分选细胞群组中,AUC随着从初始创伤起的时间越长而升高;这可表明因感染所致的炎症更容易区分因为创伤性损伤的‘基因组风暴’开始恢复。
在Glue Grant群组中检验11个基因的集合
正如预期的,在Glue Grant血沉棕黄层群组中,在感染诊断+/-24小时内的患者相比于时间匹配的未感染患者在所有时间点的感染Z评分显著较高;这在嗜中性粒细胞群组中得以验证(重复测量ANOVA p<0.0001;图3C-3D,表9A)。在血沉棕黄层群组中按从受伤起的时间比较感染Z评分显示出随时间推移的显著下降(随时间推移的重复测量ANOVA变化p<0.0001),但是在嗜中性粒细胞验证群组中的效应似乎更小(但仍显著)(随时间推移的重复测量ANOVA变化p<0.05)(图3C-3D,表9A)。群组与从受伤起的时间的相互作用在发现或验证群组中均不显著,表明两组的感染Z评分随时间推移下降可能归因于从创伤性损伤恢复,从而使得炎症减轻(表9A)。
接着,我们分析了感染患者在感染诊断前后的感染Z评分如何变化(在鉴别11个基因的集合中未包括的样本)。我们根据感染诊断的时间将曾在同一医院住院的诊断有感染的患者的样本分成四组(感染前大于5天、感染前5至1天、感染诊断+/-1天内或感染诊断后2至5天,其中除感染诊断+/-1天以外,没有一组包括在用于发现11个基因的集合的多群组分析中)。我们根据从损伤起的天数将这些组进一步划分成仓。在每一个时间仓内,对于发现血沉棕黄层群组和验证嗜中性粒细胞群组,随着它们向感染发展,诊断组的感染Z评分显著增加(Jonckheere趋势(JT)检验p<0.01;图3E-3F)。此外,在所有群组中,当患者可能由于抗生素治疗而开始从感染恢复时,感染诊断后2-5天组的感染Z评分下降。这还可以解释诊断能力随着从初始受伤起的时间增加而增加。我们强调围绕感染诊断时间的感染Z评分的所得‘高峰’验证了感染Z评分与临床感染的相关性,因为>5天前群组、1-5天前群组和2-5后群组都不包括在多群组分析中,但是仍在发现血沉棕黄层群组和验证嗜中性粒细胞群组中显示出假设的趋势。类似结果见于单核细胞和T细胞样本中(与嗜中性粒细胞验证群组相同的患者;图10B、11B)。
令人关注的是,稍后在住院期间感染的患者在入院时的血沉棕黄层样本的感染Z评分显著高于在入院期间从未感染的患者(p<0.01;嗜中性粒细胞验证组p=0.05;图3E-3F)。一种可能性是在受伤严重程度上存在基线差异,并且这可能影响感染Z评分。已知受伤严重的患者对感染更敏感(Osborn等人(2004)《危重病监护医学》32:2234-2240)。为了检验这一假设,我们使用最终医院获得性感染状态的线性回归、受伤严重程度评分和其相互作用以将感染Z评分作为自变量进行预测(表9B)。最终医院获得性感染状态和受伤严重程度评分在预测入院时的感染Z评分方面各自是显著,表明受伤严重程度单独无法解释这些效应。相互作用项是显著的并且在发现血沉棕黄层群组和验证嗜中性粒细胞群组样本中呈阴性,可能表明在入院时感染Z评分越高可指示对稍后感染的敏感性更大。需要其它研究检验这一观测结果。
在Glue Grant中的临床效用
为了检验感染Z评分是否可新增到感染的临床测定,我们在区分有感染和无感染的Glue Grant创伤患者(血沉棕黄层和嗜中性粒细胞群组)时比较了仅使用SIRS标准的逻辑回归与使用SIRS标准加上我们的感染Z评分的逻辑回归。仅使用SIRS标准的逻辑回归模型的AUC是0.64,而SIRS标准加上感染Z评分的总体AUC(在所有时间点使用单个感染系数)是0.81(图12)。连续净重新分类指数(NRI)是多少患者将通过改善疾病标志物正确重新分类的量度;此处,感染Z评分新增到仅SIRS的连续NRI是0.90(95%CI 0.62-1.17),其中大于0.6的连续NRI与预测中的‘强烈’改善相关联(Pencina等人(2012)《医学统计学(StatMed)》31:101-113)。
感染Z评分的独立验证
接着,我们在仅包括最终感染的创伤或ICU患者的三个独立纵向群组中验证我们的评分:GSE6377(McDunn等人,见上文)、GSE12838和EMEXP3001(Martin-Loeches等人,见上文)(表4)。所有三个群组从入院当天追踪患者直到至少感染诊断当天(主要是呼吸机相关肺炎,VAP)。因为三个群组中每一个的所有患者都感染,所以他们不具有时间匹配的未感染对照。为了将验证群组感染病例与未感染的创伤患者相比较,我们使用Glue Grant血沉棕黄层未感染对照。我们使用管家基因将每一个群组内部归一化,并且随后使用经验贝叶斯分批校正与Glue Grant血沉棕黄层患者一起共同归一化。随后,我们在作为变量引用的匹配时间点将验证群组与Glue Grant未感染患者相比较。将创伤/ICU患者与时间匹配的基线相比较是必需的,因为我们早先的研究结果(图3C-3F)显示未感染患者的感染Z评分随时间推移的变化(表9A)。三个独立的纵向创伤/ICU群组显示感染+/-1天内的患者一般可与时间匹配的未感染Glue Grant患者分离,ROC AUC范围介于0.68-0.84(图4)。
我们在入院时使用全血样本比较健康患者与患有细菌或病毒败血症的那些患者的8个额外独立数据集中进一步验证了11个基因的集合(总计N=446:GSE11755(Emonts等人,见上文)、GSE13015(Pankla等人,见上文)、GSE20346(Parnell等人,见上文)、GSE21802(Bermejo-Martin等人,见上文)、GSE25504(Smith等人(2014)《自然通讯(Nat Commun)》5:4649)、GSE27131(Berdal等人,见上文)、GSE33341(Ahn等人,见上文)和GSE40396(Hu等人,见上文),表5)。将所有8个数据集的感染Z评分组合在单个小提琴图中,从而显示出极好的分离(威尔科克森p<1e-63,图5A)。分离健康患者和败血症患者的平均ROC是0.98(范围:0.94-1.0,图5B)。
我们的结果提供了强有力的证据,表明从入院/受伤起随时间推移全血、血沉棕黄层、嗜中性粒细胞和单核细胞的感染Z评分降低。我们也已表明未时间匹配的比较得到不准确的感染分类,尤其对于SIRS/创伤患者的后期获得性感染。因此,将SIRS/创伤患者在入院时的感染Z评分与后期获得性败血症/感染患者的感染Z评分相比较将是诊断能力的不准确量度。然而,因为感染Z评分随时间推移降低的效应是相对单调的,所以入院SIRS/创伤/手术患者与后期获得性败血症/感染的比较将提供感染Z评分的检测ROC AUC下限。换句话说,因为感染Z评分随时间推移降低,所以如果在入院时经测试未患者的感染稍后取样(在与败血症患者匹配的时间),那么他们的感染Z评分将在稍后的时间更低(并且因此可更易于与败血症患者的较高感染Z评分分离)。使用这一推断,我们检查了将SIRS/创伤/手术患者与稍后在住院过程中败血症发病时的相同患者相比或与患有社区获得性和医院获得性败血症的患者的混合群组相比的四个独立数据集。这些数据集研究了全血(EMTAB1548(Almansa等人,见上文))、嗜中性粒细胞(GSE5772(Tang等人(2007),见上文))和PBMC(GSE9960(Tang等人(2009),见上文));EMEXP3621(Vassiliou等人(2013)《危重病监护》17:R199))(表6)。在这四个数据集中的每一个中,感染Z评分将后期获得性感染与入院SIRS或创伤分离,ROCAUC范围介于PBMC 0.48-0.76至全血0.86(图13)。我们强调这些AUC预期由于其时间错配的比较而较低,并且基本上是这些细胞分室中每一个的适当时间匹配的感染Z评分的下限。
最后,我们在确诊后比较健康患者或对急性细菌感染具有自体免疫炎症的那些患者的一个数据集(GSE22098,n=274)中检查了我们的11个基因的集合(Berry等人,见上文;Allantaz等人(2007)《实验医学杂志》204:2131-2144)。精确取样时间无法获得,但是通常证实感染耗费24-72小时,因此预期这些感染样本显示比在诊断时更低的Z评分。另外,感染Z评分仍能够区分健康和自体免疫炎症患者与具有急性感染的那些患者(ROC AUC 0.72;图14)。鉴于研究自体免疫炎症的群组不包括在我们的发现中,因此这提供了感染Z评分针对感染性炎症的特异性的验证。
感染类型对于感染Z评分的效应
为了检查感染Z评分中是否存在任何感染类型特异性差异,我们将感染有革兰氏阳性细菌对革兰氏阴性细菌的患者相比较,以及将感染有病毒感染的患者与感染有细菌感染的患者相比较。未对Glue Grant患者进行分析,因为每一个子群组中时间匹配的感染患者太少。四个数据集具有关于革兰氏阳性感染对革兰氏阴性感染的信息,并且四个数据集具有关于细菌感染对病毒感染的数据;在任一种情况下,基于感染亚型的感染Z评分存在明显趋势的差异(表10)。
基因集合途径评估和转录因子分析
在已验证了11个基因的集合的前提下,我们检查是否存在任何机制可解释为何这些基因共同起作用。我们用Ingenuity Pathway Analysis分析了11个基因的集合,显示数个基因处在IL-6和JUN控制下(图16)。将通过多群组分析鉴别的所有11个基因加载到EncodeQT和PASTAA(针对混杂的实验结果和计算机模拟转录因子预测而选择)。EncodeQT发现6个正基因当中仅一个重要的转录因子(Max),并且负基因中没有(EncodeQT Q值≤0.01,表10A)。PASTAA显示出众所周知的促炎性转录因子的富集,如Nf-KB成员c-Rel、Stat5和干扰素反应因子(IRF)1和10(表10B)。然而,由于这两种用于转录因子分析的方法关于富集的因子集合并不一致,所以无法得出明显的结论。
由于未发现明显的网络驱动器,所以我们接下来研究基因在某些免疫细胞类型中富集是否可解释其与败血症的关系。我们针对人类免疫细胞类型特异性基因表达谱搜索了GEO,并从18个数据集发现了符合我们标准的277个样本。我们使用平均基因表达评分将这些汇总成广泛的免疫细胞类型签名。我们随后使用与感染Z评分相同的方法(正基因与负基因之间的几何平均值的差值)计算标准化富集评分。对于在多群组分析中发现显著富集的82个基因的初始集合以及在前向搜索法后发现的11个基因的集合(进行感染Z评分的基因),我们都这样做了(图6)。发现所有82个重要基因的集合仅在杆状核细胞中高度富集(高于平均值>4个标准差,p<1e-6)。令人关注的是,11个基因的集合在杆状核细胞中显著富集(高于平均值>2个标准差,p=0.015),并且还显示调节性T细胞上调和树突状细胞下调。这表明在无菌SIRS与败血症之间差异基因表达的一个驱动力是存在杆状核细胞;然而,用于诊断的最佳基因集合含有可能并入多个细胞类型立即移位的信息。
论述
在创伤后和在败血症期间基因表达的变化已描述为‘基因组风暴’(Xiao等人(2011)《实验医学杂志》208:2581-2590)。此处我们检查的几十项研究已报道了对响应于SIRS、创伤、手术和败血症出现的基因表达变化的有价值的洞察;然而,先前的单项研究分析无一生产常用临床工具以降低与败血症相关联的发病率和死亡率。我们基于日益增多的对时间依赖性基因表达变化的理解,使用综合多群组分析区分SIRS/创伤中的基因表达与败血症中的基因表达。由此我们发现了针对诊断能力加以优化的11个基因的集合,我们已在15个独立群组中经过验证。在进一步前瞻性临床验证下,这11个基因的集合可辅助临床败血症诊断,继而可对患者监护具有重大影响。
感染性和非感染性炎症可经由激活相同先天性免疫途径(TLR、RLR、NLR等)引起SIRS,因此‘典型’促炎性基因和细胞因子(如TNF和白介素)一般在无菌和感染性炎症中表达(Newton等人(2012)《冷泉港生物学展望(Cold Spring Harb Perspect Biol)》4)。举例来说,一项最新研究显示在无菌炎症(Glue Grant烧伤群组)与四个独立败血症数据集之间基因表达的相关性高,两种病况中高达93%的基因在相同方向上变化(Seok等人(2013)《美国国家科学院院刊》110:3507-3512)。因此,鉴于已知在败血症中表达的细胞因子和趋化因子的‘标准’套件也多半在无菌SIRS中激活,因此搜索在无菌SIRS与败血症之间特异性差异表达的生物标志物的标准假设驱动方法不大可能成功。然而,已显示数个蛋白质家族(如凝集素和CEACAM)仅对病原体相关分子模式具有特异性,因此引起感染特异性先天性免疫信号传导途径的可能性(Geijtenbeek等人(2009)《自然·免疫学综述》9:465-479;Crocker等人(2007)《自然·免疫学综述》7:255-266;Kuespert等人(2006)《细胞生物学新见》18:565-571)。我们因此采取数据驱动的无偏方法跨越多个群组特异性搜索在无菌SIRS/创伤患者与败血症患者之间统计学上均匀差异表达的基因。
我们系统地鉴别了所有公开可用的在SIRS、创伤、危重疾病、急性感染和败血症方面基于微阵列的全基因组表达研究,并贯穿所有数据集分选以鉴别将未感染的SIRS、ICU或创伤患者与具有急性感染或败血症的患者相比的那些数据集。已知受伤后的时间是创伤后基因表达的重要因素(Xiao等人,见上文;Seok等人,见上文;Desai等人,见上文;McDunn等人,见上文)。跨越多个独立群组,我们示出基因表达随时间推移的变化是非线性的,但是遵循类似的轨迹。此外,从创伤正常恢复诱发基因表达随时间推移的巨大变化。因此,同一患者在受伤时或接近受伤时与稍后时间点(如在感染诊断时)的基因表达比较将仅由于恢复过程而产生大量差异表达基因。因此,从由恢复引起的大量变化鉴别因感染所致的基因表达相对较小的变化是非常困难的。因此,我们将我们的发现群组限定于仅在匹配的时间点将SIRS/创伤和败血症/感染患者相比较的那些研究。然而,与创伤或手术不同,败血症不容易界定‘开始’,因为感染显现耗时。我们因此将因败血症入院48小时内或感染诊断+/-24小时内的患者用作病例,因为这些是感染性病征和症状存在的时间,并且临床诊断是必要的。我们使用多群组分析方法(Khatri等人(2013)《实验医学杂志》210:2205-2221;Chen等人(2014)《癌症研究(Cancer Res)》74:2892-2902)以时间匹配的方式比较SIRS/创伤和败血症/感染患者,包括来自9个患者群组的663个样本。我们随后使用前向搜索法优化样本大小加权的ROC AUC,以便选择针对区分能力优化的统计显著基因的简约集合(FDR<1%,绝对汇总效应量>1.5倍)。定义为11个基因的集合的几何平均值的感染Z评分在用于区分SIRS/创伤与败血症/感染患者的发现群组中的平均ROC AUC是0.87。
我们在来自Glue Grant的独立患者组中验证了这个基因集合。在嗜中性粒细胞验证群组中,用于区分败血症与非感染性炎症的平均AUC是0.83,随着从初始受伤起的时间越长而具有朝向更好诊断能力的明显趋势。虽然我们预期全血转录谱主要由嗜中性粒细胞驱动,但是分选细胞中的信号当然将不同于全血。因此,预期使用分选细胞代替全血用于诊断导致区分能力降低。尽管存在这样的限制,但是可在验证群组中同等地进行感染Z评分,尤其从初始受伤起的三天或更多天,当初始创伤性炎症消退并显现医院获得性感染时(Hietbrink等人(2013)《休克(Shock)》40:21-27)。
使用关于Glue Grant中的患者可获得的详尽临床表型数据,我们说明了关于应用感染Z评分的数个重点。首先,感染Z评分显示感染和未感染患者从受伤起随时间推移的下降是类似的。我们还示出,使用Glue Grant中时间可变的未感染基线作为参考阈值允许我们区分三个独立纵向群组中的败血症患者与未感染的创伤患者,ROC AUC范围介于0.68-0.84。因此,对于最大区分能力,如果感染Z评分有待在纵向研究中进行前瞻性测试,那么诊断阈值将需要是从初始受伤起的时间的函数。其次,感染Z评分历经感染前数天增加,在感染诊断时间周围+/-1天内达至峰值并随后降低(可能归因于感染治疗)。此观测结果提升以下可能性:虽然需要进一步研究,但是有可能使用11个基因的集合较早诊断或分层处在罹患败血症风险下的患者。具体来说,我们注意到在感染的临床诊断之前感染Z评分的早期上升不是假阳性而是‘早期阳性’结果。最后,具有较高受伤严重程度评分(ISS)的患者在入院时的感染Z评分较高;初始感染Z评分因此取决于ISS和临床感染的相对时间。然而,在入院24小时内的创伤患者通常不被怀疑具有不明显的感染(开放伤口、腹膜污染等除外),并且因此我们无论如何不会预期感染Z评分具有临床效用。
在Glue Grant血沉棕黄层群组中,对于可获得全部四个SIRS标记的那些患者,SIRS二元参数在区分在感染时的患者与未感染患者方面表现不佳(ROC AUC 0.64)。SIRS标准加上具有整体截止(即,未分解成单独的时间仓)的感染Z评分增加了区分能力(ROC AUC0.81),其中连续NRI是0.9。然而,SIRS仅是用于诊断败血症的数个标准之一。降钙素原是充分研究过的用于区分败血症与SIRS的生物标志物;降钙素原的两个元分析都显示汇总ROCAUC是0.78(范围0.66-0.90)(Tang等人(2007)《柳叶刀感染病》7:210-217;Uzzan等人(2006)《危重病监护医学》34:1996-2003;Cheval等人(2000)《重症监护医学》26增刊2:S153-158;Ugarte等人(1999)《危重病监护医学》27:498-504)。我们的发现群组中的平均AUC是0.87,并且时间匹配的嗜中性粒细胞验证群组的平均AUC是0.83,这两个因此都至少与降钙素原相当。然而,我们强调这些标记中的每一个无需用于分离。没有公开可用的数据集包括在败血症诊断时的降钙素原水平。因此,感染Z评分的任何前瞻性研究应包括传统生物标志物和新的生物标志物,以便针对使用生物标志物组合的较佳诊断表现和头对头比较进行检验。
我们在数个附加的外部数据集中验证了感染Z评分,所述数据集包括罹患VAP/VAT的ICU/创伤患者的三个纵向群组;健康患者与细菌或病毒败血症相比的八个群组;入院SIRS/创伤患者与在混合或稍后时间点使用全血、嗜中性粒细胞、PBMC的患者相比的四个群组;以及患有自体免疫炎症的患者与具有急性感染的患者相比的一个群组。在符合我们纳入标准的每个公开可用的数据集中,感染Z评分具有区分能力。此外,在感染类型信息可用的那些数据集中,感染Z评分不具有关于感染类型(革兰氏阳性对革兰氏阴性和细菌对病毒)的系统化趋势。我们强调,基于Glue Grant数据中基线感染Z评分从受伤起随时间推移降低的研究结果,在任何这些分室中入院SIRS/创伤与稍后时间点的比较将具有比时间匹配研究更糟的诊断能力。因此,四个独立的未时间匹配的群组中感染Z评分的区分能力可为相应血液分室中的真实区分能力的下限。
败血症特异性11个基因的集合中的一些基因此前已与败血症或感染相关联,如CEACAM1、C3AR1、GNA15和HLA-DPB1(Madsen-Bouterse等人(2010)《美国生殖免疫学杂志(AmJ Reprod Immunol)》63:73-92;Wong等人(2012)《危重病监护》16:R213;Kwan等人(2013)公共科学图书馆·综合8:e60501)。基于计算机模拟分析,这些基因的调节控制可富集促炎性因子,如IL-6、JUN、c-Rel、Stat5和IRF 1/10,但是没有单个共有因子解释了网络。可通过细胞类型富集分析较好地解释此处发现的基因集合。我们示出,杆状核细胞和骨髓细胞系高度富集被发现在无菌SIRS与败血症之间显著差异表达的82个基因的全部集合。在杆状核细胞中富集的研究结果特别引人注目,因为先前已显示杆状核细胞有助于区分无菌SIRS与败血症(Cavallazzi等人(2010)《重症监护医学杂志》25:353-357;Drifte等人(2013)《危重病监护医学》41:820-832)。另外,通过自动血液计数器和手动的杆状核细胞计数存在极高变化性(Cornbleet等人(2002)《临床实验室医学(Clin Lab Med)》22:101-136;van der Meer等人(2006)《欧洲血液病学杂志(Eur J Haematol)》76:251-254),并且不存在良好的血清标志物。然而,11个基因的集合可较佳地区分败血症与无菌SIRS,至少部分因为其还包括关于调节性T细胞增加和树突状细胞减少的信息,这两个信息先前已牵涉在败血症中(Saito等人(2008)《东北实验医学杂志(Tohoku J Exp Med)》216:61-68;Venet等人(2008)《白细胞生物学杂志(J Leukoc Biol)》83:523-535;Grimaldi等人(2011)《重症监护医学》37:1438-1446)。11个基因的集合和不同免疫细胞类型之间的关联可有助于解释一些败血症生物学,但是当然这11个基因需要进一步研究。
当前研究到临床使用的潜在转化依据两个因素。首先,11个基因的集合和这些基因的蛋白质产物都应需要以时间匹配的方式进行前瞻性测试。蛋白质分析目前具有比PCR更快的响应时间,尽管PCR技术的许多进步已使得产生结果的时间朝向临床适用性范围减少(Park等人(2011)《生物技术进展(Biotechnol Adv)》29:830-839;Poritz等人(2011)《公共科学图书馆·综合》6,e26047)。其次,我们的结果显示,因从创伤性事件(如受伤或手术)正常恢复所致的基因表达变化意味着必须在任何急性疾病的基因表达研究中适当考虑时间。我们搜索发现了检查SIRS/创伤后时程的数项研究(GSE6377、GSE12838、GSE40012、EMEXP3001)和检查从败血症/感染发病起的时程的数项研究(GSE20346、GSE2713、GSE40012、EMEXP3850)。然而,我们仅发现一项公开可用的随时间推移检查患者群组的微阵列研究(Glue Grant),其中一些群组出现感染并且一些未出现。因此,基于我们的结果,我们推荐未来的败血症诊断研究应设计有感染和未感染的纵向群组,以便能够进行适当的时间匹配的比较(Johnson等人(2007)《外科学年鉴》245:611-621;Maslove等人(2014)《分子医学趋势》20(4):204-213)。
总体而言,我们对SIRS/创伤和败血症公开可用的基因表达数据的全面分析已获得简约的11个基因的集合,其在发现群组和15个独立群组中都具有极好的区分能力。针对这个基因集合优化临床分析以便在临床相关性窗口内得到结果应是可行的。应需要进一步研究,以前瞻性方式证实我们的临床研究结果并研究这些基因上游的分子途径。
方法
研究设计
这项研究的目的是使用综合多群组元分析框架分析多个基因表达数据集,以鉴别可分离具有无菌炎症的患者与具有感染性炎症的患者的基因集合。先前已描述了这一框架(Khatri等人(2013)《实验医学杂志》210:2205-2221;Chen等人(2014)《癌症研究》74:2892-2902)。
搜索
搜索两个公开的基因表达微阵列存储库(NIH GEO、ArrayExpress)有关匹配任何以下搜索术语的所有人类数据集:败血症、SIRS、创伤、休克、手术、感染、肺炎、危重、ICU、发炎、医院内。保留比较健康对照或具有非感染性炎症(SIRS、创伤、手术、自身免疫性)的患者与具有急性感染和/或败血症的患者的数据集用于进一步研究。不包括采用内毒素注射作为SIRS或败血症模型的数据集。
多群组分析
完成比较未感染SIRS/创伤患者对具有感染或败血症的患者的基因表达的多群组元分析。在相同时间点比较SIRS/创伤患者与败血症/感染患者的所有数据集被选择纳入多群组分析中;因此,不包括入院患者与在稍后时间点的败血症患者的比较(参见结果中次模型的调整)。入院数据集限制在来自入院48小时内的患者的样本。Glue Grant创伤数据集被分成从受伤起的天数的时间仓,排除入院后的初始24小时(参见补充方法)。这些时间仓中的每一个作为多群组分析中单独的数据集加以处理,其中时间匹配的从未感染的患者与感染诊断+/-24小时内的患者相比(感染如上文所定义)。因此在这一比较中检查感染诊断后大于24小时的患者。此方法允许检测因感染所致的与因从创伤恢复所致的基因表达随时间推移的‘标准’变化的偏差。
在选择输入数据集后,我们应用两个元分析方法;一个使用Hedge g组合效应量;另一个使用Fisher对数总和法组合p值(参见图15中的示意图)。在n个数据集的情况下,此方法以留一数据集的方式应用n次。设置错误发现率(FDR)阈值(0.01),并在每一轮留一法分析选择在效应量和Fischer对数总和分析中q值低于FDR阈值的基因。随后对基因进行数据集异质性检验,使得在所有输入数据集的异质性检验中,每一个基因的p值需要大于0.01;这去除了在不同数据集中显示显著不同效应的基因。接着,抛弃汇总效应量<1.5倍的所有基因。最后,对根据全部三个以上标准在多群组分析中发现是统计学上显著的所有基因(表8)进行贪心前向搜索模型,其中从最显著的基因开始,将所有其余基因一次一个地添加到基因评分中,并将区分能力增加最大的基因添加到最终的基因清单中。
补充方法
数据集细节
六个公开可用的全血数据集来自儿童SIRS/败血性休克基因组学研究者(GPSSSI)(Cvijanovich等人(2008)《生理基因组学》34:127-134;Shanley等人(2007)《分子医学》13:495-508;Wong等人(2007)《生理基因组学》30:146-155;Wong等人(2009)《危重病监护医学》37:1558-1566;Wong等人(2010)《儿科危重病监护医学》11:349-355;Wong等人(2011)《危重病监护医学》39:2511-2517)。这些数据集含有重叠样本,为此Hector Wong提供了重要的特有患者。随后将那些特有患者一起gcRMA归一化并作为单个数据集(GPSSSI特有)处理。
除了公开可用的数据集以外,我们还使用炎症和宿主对损伤的反应程序(GlueGrant)创伤数据集(Cobb等人,见上文)。Glue Grant数据集由对完整血沉棕黄层或分选细胞(嗜中性粒细胞、单核细胞、T细胞)取样的单独的创伤患者群组组成。纳入标准描述在别处(Desai等人,见上文)。在入院后的以下天数对患者进行取样:0.5、1、4、7、14、21、28天。Glue Grant创伤群组患者如果具有医院内感染(肺炎、泌尿道感染、导管相关血流感染等)、手术感染(排除浅表伤口感染)或经历内脏穿孔手术,那么他们被归类为‘感染’;感染定义可见于gluegrant.org/commonlyreferencedpubs.htm。对于元分析,在感染诊断当天+/-24小时内抽取的样本作为感染病例而被包括。少于20名患者的时间点不包括在多群组分析中。Glue Grant还含有烧伤患者,但是这些由于区分临床相关感染与群集感染烧伤伤口的难度而被排除。Glue Grant的使用是由Glue Grant联盟和斯坦福大学(StanfordUniversity)IRB共同批准(协议29798)。
基因表达归一化
所有Affymetrix数据集以CEL文件形式下载并使用gcRMA(R affy包)再归一化。对来自Agilent芯片的输出和在GenePix扫描器上分析的自定义阵列进行背景校正、阵列内局部加权回归归一化且随后阵列间分位数归一化(R limma包)(G.Smyth,《使用R和Bioconductor的生物信息学和计算生物学解决方案(Bioinformatics and ComputationalBiology Solutions Using R and Bioconductor)》,C.V.Gentleman R,Dudoit S,Irizarry R和Huber W(编),版本(Springer,纽约,2005),第397-420页)。对Illumina数据集进行分位数归一化。使用自定义阵列分析Glue Grant分选细胞数据集(GGH-1、GGH-2);将这些数据集如先前所述归一化并以其处理后状态使用(Xu等人(2011)《美国国家科学院院刊》108:3707-3712)。对于所有基因分析,将针对常见基因的探针平均值设置为基因表达水平。所有探针与基因图谱是在2014年12月14日从GEO最新SOFT文件下载。
经标记的PCA方法
经标记的主成分分析(PCA)方法是在Koren和Carmel(《IEEE可视化与计算机图形学学报》(2004)10:459-470)的章节4.1中在等式6和7中所描述的约束优化的实现方式。此优化计算使数据的不同标记类别中各点间的成对距离达到最大的数据线性变换,同时维持变换数据彼此正交的约束条件。此正交约束条件与PCA所采用的约束条件略微不同,PCA所采用的约束条件要求变换基彼此正交,而不是变换数据本身彼此正交。虽然PCA是投影方案,但是经标记的PCA是归因于此约束条件差异的线性变换的通式。
将原始数据集称为X,其具有m行(数据点)和n列(每一个数据点具有n个要素)。Y是m×1矩阵,具有每一类别的不同清单。换句话说,当且仅当要素i和j是相同标记类别的一部分时,Y(i)等于Y(j)。L是对称m×m矩阵,其(i,j)元素是-1,除非Y(i)等于Y(j)。在这后一情况中,所述元素是0。最后,填写所有对角线元素(其中i等于j),使得行i求和为0。Koren和Carmel在Lemma 3.2中证明转置矩阵(X)*L*X的特征向量提供使数据的不同标记类别中各点间的成对距离达到最大的图谱。然而,这一变换仍是投影方案,这意味着这些特征向量彼此正交。这后一结果限制变换数据的实用性,但是更可归纳。此论文中使用一般线性投影而非寻找求解方程式Av=λBv的向量v,其中A是转置矩阵(X)*L*X,并且B是转置矩阵(X)*X。虽然更加表意,但是这一方法不如经标记的PCA投影方案稳固,因为此通式的解答需要B不是单数。由于B不是单位矩阵,所以不必保持投影中所用的旧的正交约束条件。实际上,此形式的解答需要基相对于原始数据的协方差基彼此正交。
经标记的PCA应用
经标记的PCA方法描述在补充方法中。将含有非感染性SIRS、ICU或创伤患者与败血症患者的比较的所有数据集从探针转化成基因,并随后结合到单个大矩阵中并进行分位数归一化。抛弃所有数据集中不存在的基因。将在任何时间(入院时或医院获得性)患有败血症的患者分组在单个类别中,并应用Lasso惩罚回归以分离无菌SIRS患者与败血症患者(R glmnet包)。基于无菌SIRS对败血症的类别,使用通过惩罚回归选择的基因进行经标记的PCA。随后再标记相同图式以示出哪些样本来自医院获得性(或后期)无菌SIRS或败血症患者。随后将来自惩罚回归的相同基因集合用于经标记的PCA以比较健康、无菌SIRS和败血症患者。随后再标记这一相同图式以示出哪些样本来自医院获得性或后期无菌SIRS或败血症患者。
为了检查时间对于SIRS/创伤和感染中的基因表达的效应,选择包括随时间推移的连续测量结果的所有数据集。仅包括来自Glue Grant数据集的血沉棕黄层阵列,以便不会淹没来自其它数据集的信号。将所选数据集从探针转化成基因,并随后结合到单个大矩阵中并进行分位数归一化。抛弃所有数据集中不存在的基因。为了以无偏方式减少基因集合,使用CUR矩阵分解选择合并的数据集中具有最大正交性的前100个基因(R rCUR包)(Bodor等人(2012)《BMC生物信息学(BMC Bioinformatics)》13:103)。随后,在每一个时间点用作不同类别(在1、2、3、4、5、6、10、20和40天分割)的情况下,进行经标记的PCA。所得PCA以3D图示,按时间点着色,并且使用R rgl包撷取3D空间旋转的短视频。
感染Z评分
将在多群组分析后发现是显著的基因根据其效应是否是正或负而分离(其中‘正’意思指相比于SIRS/创伤的败血症的正效应量,并且‘负’意思指相比于SIRS/创伤的败血症的负效应量)。随后使用单个基因评分测试这些基因集合的类别区分能力。所用基因评分是所有正基因的基因表达水平的几何平均值减去所有负基因的基因表达水平的几何平均值,乘以正基因与负基因计数的比率。这是针对数据集中的每一个样本计算所得的,并且随后将每一个数据集的评分标准化以产生Z评分(‘感染Z评分’)。排除完整数据集中不存在的基因;将个别样本的基因缺失设定成1。为了获得具有负基因表达值的数据集(双通道阵列)的感染Z评分,将完整数据集按数据集中所存在的最小值调整以确保所有值为正(因为负输入的几何平均值产生虚数值)。
检查类别区分能力,比较每一个所检查的数据集中所关注类别的感染Z评分。用小提琴图检查感染Z评分范围,并且由于无法假定感染Z评分范围具有正态分布,所以在25%-75%四分位差下示出并使用威尔科克森秩和检验比较。构建感染Z评分的ROC曲线比较所关注类别(如无菌SIRS与败血症相比较),并连同95%置信区间一起示出总ROC曲线下面积(AUC)。
前向搜索
为了获得区分SIRS/创伤患者与败血症/感染患者的简约基因集合,对在多群组分析中发现是统计学上显著的所有基因进行贪心前向搜索模型,其中从数据集中最显著的基因开始,将所有其余基因一次一个地添加到基因评分中,并将区分能力增加最大的基因添加到最终的基因清单中。此处,区分能力定义为加权ROC AUC,其中检验每一个发现数据集的感染Z评分,并将所得AUC乘以数据集中样本的总数。运行随后使每一步中所有发现数据集的加权AUC的总和达到最大。以此方式,小数据集中极好的类别区分不会胜过极大数据集中类别区分的适度增加。运行在任意界定的阈值停止;我们使用停止阈值一(以使得当运行无法发现将使当前感染Z评分的总发现加权AUC增加大于一时,其将终止)。这最终所得基因集合因此使发现群组中的区分能力达到最大,但没有优化为整体最大。在前向搜索后保留的基因的探针水平数据显示在表8中。
发现群组检查
使用最终基因评分计算每一个发现数据集的感染Z评分。分别分析入院数据集并绘制单独的ROC曲线图。对于医院获得性(Glue Grant)数据集,与分别将不同时间仓归一化相反,将全部群组的感染评分标准化(转化成Z评分)一次以示出随时间推移在相同患者中的变化。随后使用重复测量方差分析分析感染Z评分的显著性。针对在多群组分析中作为单独数据集处理的个别时间仓,绘制ROC曲线。
对于Glue Grant数据集,针对血沉棕黄层和嗜中性白细胞数据集进行两个感染Z评分的时程分析。首先,针对在感染+/-24小时内的患者和未患者感染,使用线性回归随时间推移比较平均感染Z评分。使用重复测量方差分析以使感染组和未感染组彼此相比较并检验变化随时间推移的显著性。接着,针对每一个时间窗构建盒形图,以使得所述时间窗中从未感染的患者的感染Z评分与在感染诊断当天之前>5天、诊断前5-1天或诊断+/-24小时的患者相比。对于每一个时间点(0-1天窗口除外),用Jonckheere趋势(JT)检验测试不同组中感染Z评分的趋势。将在入院时间点([0,1))的感染Z评分作为结果变量在多个线性回归中加以检验,从而检查受伤严重程度评分和时间对感染的贡献效应。
验证
在与发现群组完全分离的数个验证群组中检验最终基因集合。将Glue Grant的分选细胞群组分解成时间仓,并分别计算每一个时间仓的AUC。注意到在受伤后的18天之后在此群组中未捕捉到诊断+/-1天感染,因此未曾显示[18,24)天仓。
验证群组包括随时间推移检查创伤患者的三个数据集(GSE6377、GSE12838和EMEXP3001),其中全部出现感染(主要是呼吸机相关肺炎(VAP))。这些数据集不包括对照,因此它们是与作为基线的Glue Grant未感染患者相比。这三个验证数据集和Glue Grant血沉棕黄层未感染样本首先按四个管家基因(GAPDH、ACTN1、RPL9、KARS)的几何平均值因数线性标度(Vandesompele等人(2002)《基因组生物学》3:RESEARCH0034)。随后将数据集关于重叠基因接合起来,并使用具有参数先验的ComBat经验贝叶斯分批校正工具(R sva包)在数据集之间进行分批校正(Leek等人(2012)《生物信息学》28:882-883)。控制受伤后天数的ComBat校正(以使得天数间的相对差异保持相对不同)。随后计算接合数据集的感染Z评分,并对照未感染Glue Grant群组的局部加权回归曲线绘制验证数据集。随后将验证数据集中在感染诊断+/-24小时内的患者与天数匹配的ComBat共同归一化的未感染Glue Grant血沉棕黄层患者相比,并构建ROC曲线。
将初始搜索中所发现的允许在健康或SIRS/创伤患者与败血症患者之间进行比较的所有其它数据集用于简单的类别区分验证。显示针对全血或嗜中性粒细胞进行的所有数据集。选择在PBMC方面进行的研究用于那些仅检查SIRS/创伤和败血症患者的数据集。排除不包括无菌SIRS组和败血症组的使用PBMC样本的数据集。将所有外周血健康对败血症患者数据集分组成单个小提琴图并共同检验分离(威尔科克森秩和),因为它们全部用于进行相同的比较。分别对每一个单独的数据集绘制ROC曲线以示出每一个数据集内感染Z评分的区分能力。
Glue Grant SIRS评估
为了评估SIRS作为Glue Grant群组中感染的筛选标准的有效性,将所有患者归类为未感染或在感染+/-24小时内,其中感染如上文所定义。患者在感染诊断后>24小时进行检查。根据国际标准指南定义SIRS标准(温度<36C或>38C、呼吸速率>20或PaCO2<32、总WBC<4,000或>12,000并且HR>90)。排除不符任何标准的患者。存储每一名患者每一天的每一个标准作为二元变量。对包括和不包括Z评分的数据进行逻辑回归,并计算两个模型的ROCAUC。随后使用连续净重新分类指数(R PredictABEL包)比较两个模型。
基因集合评估
使用两个在线工具EncodeQT(Auerbach等人(2013)《生物信息学》29:1922-1924)和PASTAA(Roider等人(2009)《生物信息学》25:435-442)针对转录因子结合位点评估最终基因集合。分别评估正基因和负基因,因为它们被假设为在单独的调控下。EncodeQT工具用于转录起始位点上游5000个和下游5000个碱基对。使用PASTAA进行类似分析,检查来自转录起始位点的-200个碱基对区,仅检查那些对于小鼠和人类保守的因子。记录两个分析的前十个显著的转录因子。
细胞类型富集测试
搜索GEO有关相关免疫细胞类型的临床样本的基因表达谱。搜索限制于仅在Affymetrix平台上运行的样本以确保平台效应均质性。所用的所有数据集以RAW格式下载并分别进行gcRMA归一化。对于每一个样本,将定位到相同基因的多个探针的平均值视为基因值。抛弃所有样本中不存在的基因。对于全部对应于相同细胞类型的多个样本,将所述样本的平均值视为最终值,由此形成每一个细胞类型的单个向量。为了获得基因集合在每一个细胞类型向量中的Z评分,获取‘正基因’表达的几何平均值,并减去‘负基因’表达的几何平均值,乘以负基因与正基因的比率(与感染Z评分相同的程序)。随后将所有细胞类型的这些评分标准化,以使得评分表示远离组平均值的标准差的数目。这因此表示给定基因集合在给定细胞类型中相对于其它测试细胞类型的富集程度。
使用符合标准的总共18个GEO数据集:GSE3982(Jeffrey等人(2006)《自然·免疫学》7:274-283)、GSE5099(Martinez等人(2006)《免疫学杂志》177:7303-7311)、GSE8668(Radom-Aizik等人(2008)《应用生理学杂志(J Appl Physiol)》104:236-243)、GSE11292(He等人(2012)《分子系统生物学(Mol Syst Biol)》8:624)、GSE12453(Giefing等人(2013)《公共科学图书馆·综合》8:e84928)、GSE13987(Meyers等人(2009)《免疫学杂志》182:5400-5411)、GSE14879(Eckerle等人(2009)《白血病(Leukemia)》23:2129-2138)、GSE15743(Stegmann等人(2010)《胃肠病学(Gastroenterology)》138:1885-1897)、GSE16020(Vinh等人(2010)《血液(Blood)》115:1519-1529)、GSE16836(Ancuta等人(2009)《BMC基因组学(BMCGenomics)》10:403)、GSE24759(Novershtern等人(2011)《细胞(Cell)》144:296-309)、GSE28490(Allantaz等人(2012)《公共科学图书馆·综合》7:e29979)、GSE28491(Allantaz等人,见上文)、GSE31773(Tsitsiou等人(2012)《过敏及临床免疫学杂志(J Allergy ClinImmunol)》129:95-103)、GSE34515(Frankenberger等人(2012)《欧洲免疫学杂志(Eur JImmunol)》42:957-974)、GSE38043(Huen等人(2013)《国际癌症杂志(Int J Cancer)》133:373-382)、GSE39889(Malcolm等人(2013)《公共科学图书馆·综合》8:e57402)、GSE42519(Rapin等人(2014)《血液》123:894-904)、GSE49910(Mabbott等人(2013)《BMC基因组学》14:632)。
两个基因集合是以此方式测试:在初始多群组分析之后发现两个完整基因集合都是显著的,并且在前向搜索之后发现基因子集最具诊断性。其对应图示出了每一个细胞亚型的Z评分(富集给定基因集合)(黑点)以及Z评分的总体分布的盒形图(任何离群值显示为空心圆)。
统计资料和R
以用于统计学计算的R语言(版本3.0.2)进行所有计算和运算。除非另外规定,否则将p值的显著性水平设置在0.05并且进行双尾分析。
表1.比较SIRS/ICU/创伤与败血症/感染的公开可用的基因表达数据集。CAP,社区获得性肺炎。ARDS,急性呼吸窘迫综合症。


表2.多群组分析中所用的所有数据集。Glue Grant群组标题后的数字指示给定群组中从感染起的天数(例如[1,3)是从受伤起1-3天的患者)。








表11A.EncodeQT的输出结果。使用默认设置分别分析6个正基因和5个负基因。Q值是由Benjamini-Hochberg校正的超几何检验得出。
ENCODEQT-正基因 +/-5000bp

ENCODEQT-负基因 +/-5000bp

表11B.PASTAA的输出结果。使用来自转录起始位点的-200个碱基对,搜索保守性人类/小鼠序列分别分析6个正基因和5个负基因。P值来自超几何检验。
PASTAA-正基因 来自TSS的-200bp,保守性人类/小鼠
| 秩矩阵 | 转录因子 | 相关评分 | P值 |
| 1ZBRK1_01 | N/A | 3.353 | 1.28E-03 |
| 2PAX_Q6 | Pax-1、Pax-2 | 2.967 | 3.70E-03 |
| 3IRF_Q6_01 | Irf-1、Irf-10 | 2.72 | 6.74E-03 |
| 4CREL_01 | C-rel | 2.647 | 7.42E-03 |
| 5GATA4_Q3 | Gata-4 | 2.522 | 1.02E-02 |
| 6PAX4_03 | Pax-4a | 2.522 | 1.02E-02 |
| 7PPAR_DR1_Q2 | Ppar-α、Ppar-β | 2.521 | 1.02E-02 |
| 8STAT5A_04 | Stat5a | 2.503 | 1.02E-02 |
| 9PTF1BETA_Q6 | N/A | 2.372 | 1.43E-02 |
| 10MYB_Q3 | C-myb | 2.371 | 1.43E-02 |
PASTAA-负基因 来自TSS的-200bp,保守性人类/小鼠
| 秩 | 矩阵 | 转录因子 | 相关评分 | P值 |
| 1 | KAISO_01 | N/A | 3.264 | 1.28E-03 |
| 2 | PAX5_01 | Pax-5 | 3.236 | 1.28E-03 |
| 3 | TCF11_01 | Lcr-f1 | 3.066 | 1.78E-03 |
| 4 | STRA13_01 | Stra13 | 2.823 | 4.06E-03 |
| 5 | HNF4ALPHA_Q6 | Hnf-4、Hnf-4α | 2.473 | 9.47E-03 |
| 6 | ARNT_02 | Arnt | 2.346 | 1.31E-02 |
| 7 | USF_Q6 | Usf1、Usf2a | 2.346 | 1.31E-02 |
| 8 | PAX4_01 | Pax-4a | 2.221 | 1.70E-02 |
| 9 | TFIII_Q6 | Tfii-i | 2.22 | 1.70E-02 |
| 10 | AP1_Q6_01 | Fosb、Fra-1 | 2.204 | 1.70E-02 |
参考文献
1.D.C.Angus,W.T.Linde-Zwirble,J.Lidicker,G.Clermont,J.Carcillo,M.R.Pinsky,Epidemiology of severe sepsis in the United States:analysis ofincidence,outcome,and associated costs of care.Crit Care Med 29,1303-1310(2001).
2.T.Lagu,M.B.Rothberg,M.S.Shieh,P.S.Pekow,J.S.Steingrub,P.K.Lindenauer,Hospitalizations,costs,and outcomes of severe sepsis in theUnited States 2003to 2007.Crit Care Med 40,754-761(2012).
3.C.A.Torio,R.A.Andrews.(Agency for Healthcare Research and Quality,Rockville,MD,August 2013).
4.D.F.Gaieski,M.E.Mikkelsen,R.A.Band,J.M.Pines,R.Massone,F.F.Furia,F.S.Shofer,M.Goyal,Impact of time to antibiotics on survival in patients withsevere sepsis or septic shock in whom early goal-directed therapy wasinitiated in the emergency department.Crit Care Med 38,1045-1053(2010).
5.R.Ferrer,I.Martin-Loeches,G.Phillips,T.M.Osborn,S.Townsend,R.P.Dellinger,A.Artigas,C.Schorr,M.M.Levy,Empiric antibiotic treatmentreduces mortality in severe sepsis and septic shock from the first hour:results from a guideline-based performance improvement program*.Crit Care Med42,1749-1755(2014).
6.R.P.Dellinger,M.M.Levy,A.Rhodes,D.Annane,H.Gerlach,S.M.Opal,J.E.Sevransky,C.L.Sprung,I.S.Douglas,R.Jaeschke,T.M.Osborn,M.E.Nunnally,S.R.Townsend,K.Reinhart,R.M.Kleinpell,D.C.Angus,C.S.Deutschman,F.R.Machado,G.D.Rubenfeld,S.Webb,R.J.Beale,J.L.Vincent,R.Moreno,S.S.C.G.C.i.T.P.Subgroup,Surviving Sepsis Campaign:international guidelines for management of severesepsis and septic shock,2012.Intensive Care Med 39,165-228(2013).
7.B.Coburn,A.M.Morris,G.Tomlinson,A.S.Detsky,Does this adult patientwith suspected bacteremia require blood cultures?JAMA 308,502-511(2012).
8.B.M.Tang,G.D.Eslick,J.C.Craig,A.S.McLean,Accuracy of procalcitoninfor sepsis diagnosis in critically ill patients:systematic review and meta-analysis.Lancet Infect Dis 7,210-217(2007).
9.B.Uzzan,R.Cohen,P.Nicolas,M.Cucherat,G.Y.Perret,Procalcitonin as adiagnostic test for sepsis in critically ill adults and after surgery ortrauma:a systematic review and meta-analysis.Crit Care Med 34,1996-2003(2006).
10.C.Cheval,J.F.Timsit,M.Garrouste-Orgeas,M.Assicot,B.De Jonghe,B.Misset,C.Bohuon,J.Carlet,Procalcitonin(PCT)is useful in predicting thebacterial origin of an acute circulatory failure in critically illpatients.Intensive Care Med 26 Suppl 2,S153-158(2000).
11.H.Ugarte,E.Silva,D.Mercan,A.De J.L.Vincent,Procalcitoninused as a marker
J.L.Vincent,Procalcitoninused as a marker
of infection in the intensive care unit.Crit Care Med 27,498-504(1999).
12.J.P.Cobb,E.E.Moore,D.L.Hayden,J.P.Minei,J.Cuschieri,J.Yang,Q.Li,N.Lin,B.H.Brownstein,L.Hennessy,P.H.Mason,W.S.Schierding,D.J.Dixon,R.G.Tompkins,H.S.Warren,D.A.Schoenfeld,R.V.Maier,Validation of theriboleukogram to detect ventilator-associated pneumonia after severeinjury.Ann Surg 250,531-539(2009).
13.W.Xiao,M.N.Mindrinos,J.Seok,J.Cuschieri,A.G.Cuenca,H.Gao,D.L.Hayden,L.Hennessy,E.E.Moore,J.P.Minei,P.E.Bankey,J.L.Johnson,J.Sperry,A.B.Nathens,T.R.Billiar,M.A.West,B.H.Brownstein,P.H.Mason,H.V.Baker,C.C.Finnerty,M.G.Jeschke,M.C.López,M.B.Klein,R.L.Gamelli,N.S.Gibran,B.Arnoldo,W.Xu,Y.Zhang,S.E.Calvano,G.P.McDonald-Smith,D.A.Schoenfeld,J.D.Storey,J.P.Cobb,H.S.Warren,L.L.Moldawer,D.N.Herndon,S.E.Lowry,R.V.Maier,R.W.Davis,R.G.Tompkins,I.a.H.R.t.I.L.-S.C.R.Program,A genomic storm incritically injured humans.J Exp Med 208,2581-2590(2011).
14.R.Pankla,S.Buddhisa,M.Berry,D.M.Blankenship,G.J.Bancroft,J.Banchereau,G.Lertmemongkolchai,D.Chaussabel,Genomic transcriptionalprofiling identifies a candidate blood biomarker signature for the diagnosisof septicemic melioidosis.Genome Biol 10,R127(2009).
15.B.M.Tang,A.S.McLean,I.W.Dawes,S.J.Huang,R.C.Lin,Gene-expressionprofiling of peripheral blood mononuclear cells in sepsis.Crit Care Med 37,882-888(2009).
16.H.R.Wong,Clinical review:sepsis and septic shock--the potential ofgene arrays.Crit Care 16,204(2012).
17.S.B.Johnson,M.Lissauer,G.V.Bochicchio,R.Moore,A.S.Cross,T.M.Scalea,Gene expression profiles differentiate between sterile SIRS andearly sepsis.Ann Surg 245,611-621(2007).
18.V.L.Vega,A marker for posttraumatic-sepsis:searching for the HolyGrail around intensive care units.Crit Care Med 37,1806-1807(2009).
19.T.B.Geijtenbeek,S.I.Gringhuis,Signalling through C-type lectinreceptors:shaping immune responses.Nat Rev Immunol 9,465-479(2009).
20.P.R.Crocker,J.C.Paulson,A.Varki,Siglecs and their roles in theimmune system.Nat Rev Immunol 7,255-266(2007).
21.K.Kuespert,S.Pils,C.R.Hauck,CEACAMs:their role in physiology andpathophysiology.Curr Opin Cell Biol 18,565-571(2006).
22.D.M.Maslove,H.R.Wong,Gene expression profiling in sepsis:timing,tissue,and translational considerations.Trends Mol Med,(2014).
23.J.P.Cobb,M.N.Mindrinos,C.Miller-Graziano,S.E.Calvano,H.V.Baker,W.Xiao,K.Laudanski,B.H.Brownstein,C.M.Elson,D.L.Hayden,D.N.Herndon,S.F.Lowry,R.V.Maier,D.A.Schoenfeld,L.L.Moldawer,R.W.Davis,R.G.Tompkins,P.Bankey,T.Billiar,D.Camp,I.Chaudry,B.Freeman,R.Gamelli,N.Gibran,B.Harbrecht,W.Heagy,D.Heimbach,J.Horton,J.Hunt,J.Lederer,J.Mannick,B.McKinley,J.Minei,E.Moore,F.Moore,R.Munford,A.Nathens,G.O'keefe,G.Purdue,L.Rahme,D.Remick,M.Sailors,M.Shapiro,G.Silver,R.Smith,G.Stephanopoulos,G.Stormo,M.Toner,S.Warren,M.West,S.Wolfe,V.Young,I.a.H.R.t.I.L.-S.C.R.Program,Application of genome-wideexpression analysis to human health and disease.Proc Natl Acad Sci U S A 102,4801-4806(2005).
24.J.Seok,H.S.Warren,A.G.Cuenca,M.N.Mindrinos,H.V.Baker,W.Xu,D.R.Richards,G.P.McDonald-Smith,H.Gao,L.Hennessy,C.C.Finnerty,C.M.López,S.Honari,E.E.Moore,J.P.Minei,J.Cuschieri,P.E.Bankey,J.L.Johnson,J.Sperry,A.B.Nathens,T.R.Billiar,M.A.West,M.G.Jeschke,M.B.Klein,R.L.Gamelli,N.S.Gibran,B.H.Brownstein,C.Miller-Graziano,S.E.Calvano,P.H.Mason,J.P.Cobb,L.G.Rahme,S.F.Lowry,R.V.Maier,L.L.Moldawer,D.N.Herndon,R.W.Davis,W.Xiao,R.G.Tompkins,L.r.S.C.R.P.Inflammation and Host Response to Injury,Genomicresponses in mouse models poorly mimic human inflammatory diseases.Proc NatlAcad Sci U S A 110,3507-3512(2013).
25.K.H.Desai,C.S.Tan,J.T.Leek,R.V.Maier,R.G.Tompkins,J.D.Storey,I.a.t.H.R.t.I.L.-S.C.R.Program,Dissecting inflammatory complications incritically injured patients by within-patient gene expression changes:alongitudinal clinical genomics study.PLoS Med 8,e1001093(2011).
26.H.S.Warren,C.M.Elson,D.L.Hayden,D.A.Schoenfeld,J.P.Cobb,R.V.Maier,L.L.Moldawer,E.E.Moore,B.G.Harbrecht,K.Pelak,J.Cuschieri,D.N.Herndon,M.G.Jeschke,C.C.Finnerty,B.H.Brownstein,L.Hennessy,P.H.Mason,R.G.Tompkins,I.a.H.R.t.I.L.S.C.R.Program,A genomic score prognostic of outcome in traumapatients.Mol Med 15,220-227(2009).
27.N.Cvijanovich,T.P.Shanley,R.Lin,G.L.Allen,N.J.Thomas,P.Checchia,N.Anas,R.J.Freishtat,M.Monaco,K.Odoms,B.Sakthivel,H.R.Wong,G.o.P.S.S.S.Investigators,Validating the genomic signature of pediatricseptic shock.Physiol Genomics 34,127-134(2008).
28.T.P.Shanley,N.Cvijanovich,R.Lin,G.L.Allen,N.J.Thomas,A.Doctor,M.Kalyanaraman,N.M.Tofil,S.Penfil,M.Monaco,K.Odoms,M.Barnes,B.Sakthivel,B.J.Aronow,H.R.Wong,Genome-level longitudinal expression of signalingpathways and gene networks in pediatric septic shock.Mol Med 13,495-508(2007).
29.H.R.Wong,T.P.Shanley,B.Sakthivel,N.Cvijanovich,R.Lin,G.L.Allen,N.J.Thomas,A.Doctor,M.Kalyanaraman,N.M.Tofil,S.Penfil,M.Monaco,M.A.Tagavilla,K.Odoms,K.Dunsmore,M.Barnes,B.J.Aronow,G.o.P.S.S.S.Investigators,Genome-levelexpression profiles in pediatric septic shock indicate a role for alteredzinc homeostasis in poor outcome.Physiol Genomics 30,146-155(2007).
30.H.R.Wong,N.Cvijanovich,G.L.Allen,R.Lin,N.Anas,K.Meyer,R.J.Freishtat,M.Monaco,K.Odoms,B.Sakthivel,T.P.Shanley,G.o.P.S.S.S.Investigators,Genomic expression profiling across the pediatricsystemic inflammatory response syndrome,sepsis,and septic shock spectrum.CritCare Med 37,1558-1566(2009).
31.H.R.Wong,R.J.Freishtat,M.Monaco,K.Odoms,T.P.Shanley,Leukocytesubset-derived genomewide expression profiles in pediatric septicshock.Pediatr Crit Care Med 11,349-355(2010).
32.H.R.Wong,N.Z.Cvijanovich,G.L.Allen,N.J.Thomas,R.J.Freishtat,N.Anas,K.Meyer,P.A.Checchia,R.Lin,T.P.Shanley,M.T.Bigham,D.S.Wheeler,L.A.Doughty,K.Tegtmeyer,S.E.Poynter,J.M.Kaplan,R.S.Chima,E.Stalets,R.K.Basu,B.M.Varisco,F.E.Barr,Validation of a gene expression-based subclassificationstrategy for pediatric septic shock.Crit Care Med 39,2511-2517(2011).
33.R.Almansa,E.Tamayo,M.Heredia,S.Gutierrez,P.Ruiz,E.Alvarez,E.Gomez-Sanchez,D.Andaluz-Ojeda,R. L.Rico,V.Iglesias,J.I.Gomez-Herreras,J.F.Bermejo-Martin,Transcriptomic evidence of impaired immunoglobulin Gproduction in fatal septic shock.J Crit Care 29,307-309(2014).
L.Rico,V.Iglesias,J.I.Gomez-Herreras,J.F.Bermejo-Martin,Transcriptomic evidence of impaired immunoglobulin Gproduction in fatal septic shock.J Crit Care 29,307-309(2014).
34.J.F.Bermejo-Martin,I.Martin-Loeches,J.Rello,A.Antón,R.Almansa,L.Xu,G.Lopez-Campos,T.Pumarola,L.Ran,P.Ramirez,D.Banner,D.C.Ng,L.Socias,A.Loza,D.Andaluz,E.Maravi,M.J.Gómez-Sánchez,M.Gordón,M.C.Gallegos,V.Fernandez,S.Aldunate,C.León,P.Merino,J.Blanco,F.Martin-Sanchez,L.Rico,D.Varillas,V.Iglesias,M.Marcos,F.Gandía,F.Bobillo,B.Nogueira,S.Rojo,S.Resino,C.Castro,R.Ortiz de Lejarazu,D.Kelvin,Host adaptive immunity deficiency insevere pandemic influenza.Crit Care 14,R167(2010).
35.I.Martin-Loeches,E.Papiol,R.Almansa,G.López-Campos,J.F.Bermejo-Martin,J.Rello,Intubated patients developing tracheobronchitis or pneumoniahave distinctive complement system gene expression signatures in the pre-infection period:a pilot study.Med Intensiva 36,257-263(2012).
36.E.Tamayo,A.Fernández,R.Almansa,E.Carrasco,L.Goncalves,M.Heredia,D.AndaluzOjeda,G.March,L.Rico,J.I.Gómez-Herreras,R.O.de Lejarazu,J.F.Bermejo-Martin,Beneficial role of endogenous immunoglobulin subclasses and isotypesin septic shock.J Crit Care 27,616-622(2012).
37.X.Hu,J.Yu,S.D.Crosby,G.A.Storch,Gene expression profiles infebrile children with defined viral and bacterial infection.Proc Natl AcadSci U S A 110,12792-12797(2013).
38.G.P.Parnell,A.S.McLean,D.R.Booth,N.J.Armstrong,M.Nalos,S.J.Huang,J.Manak,W.Tang,O.Y.Tam,S.Chan,B.M.Tang,A distinct influenza infectionsignature in the blood transcriptome of patients with severe community-acquired pneumonia.Crit Care 16,R157(2012).
39.A.Sutherland,M.Thomas,R.A.Brandon,R.B.Brandon,J.Lipman,B.Tang,A.McLean,R.Pascoe,G.Price,T.Nguyen,G.Stone,D.Venter,Development andvalidation of a novel molecular biomarker diagnostic test for the earlydetection of sepsis.Crit Care 15,R149(2011).
40.Y.Tang,H.Xu,X.Du,L.Lit,W.Walker,A.Lu,R.Ran,J.P.Gregg,M.Reilly,A.Pancioli,J.C.Khoury,L.R.Sauerbeck,J.A.Carrozzella,J.Spilker,J.Clark,K.R.Wagner,E.C.Jauch,D.J.Chang,P.Verro,J.P.Broderick,F.R.Sharp,Geneexpression in blood changes rapidly in neutrophils and monocytes afterischemic stroke in humans:a microarray study.J Cereb Blood Flow Metab 26,1089-1102(2006).
41.B.M.Tang,A.S.McLean,I.W.Dawes,S.J.Huang,R.C.Lin,The use of gene-expression profiling to identify candidate genes in human sepsis.Am J RespirCrit Care Med 176,676-684(2007).
42.S.H.Ahn,E.L.Tsalik,D.D.Cyr,Y.Zhang,J.C.van Velkinburgh,R.J.Langley,S.W.Glickman,C.B.Cairns,A.K.Zaas,E.P.Rivers,R.M.Otero,T.Veldman,S.F.Kingsmore,J.Lucas,C.W.Woods,G.S.Ginsburg,V.G.Fowler,Gene expression-basedclassifiers identify Staphylococcus aureus infection in mice and humans.PLoSOne 8,e48979(2013).
43.T.Dolinay,Y.S.Kim,J.Howrylak,G.M.Hunninghake,C.H.An,L.Fredenburgh,A.F.Massaro,A.Rogers,L.Gazourian,K.Nakahira,J.A.Haspel,R.Landazury,S.Eppanapally,J.D.Christie,N.J.Meyer,L.B.Ware,D.C.Christiani,S.W.Ryter,R.M.Baron,A.M.Choi,Inflammasomeregulated cytokines are critical mediators ofacute lung injury.Am J Respir Crit Care Med 185,1225-1234(2012).
44.J.E.Berdal,T.E.Mollnes,T. O.K.Olstad,B.Halvorsen,T.Ueland,J.H.Laake,M.T.Furuseth,A.Maagaard,H.Kjekshus,P.Aukrust,C.M.Jonassen,Excessiveinnate immune response and mutant D222G/N in severe A(H1N1)pandemicinfluenza.J Infect 63,308-316(2011).
O.K.Olstad,B.Halvorsen,T.Ueland,J.H.Laake,M.T.Furuseth,A.Maagaard,H.Kjekshus,P.Aukrust,C.M.Jonassen,Excessiveinnate immune response and mutant D222G/N in severe A(H1N1)pandemicinfluenza.J Infect 63,308-316(2011).
45.M.P.Berry,C.M.Graham,F.W.McNab,Z.Xu,S.A.Bloch,T.Oni,K.A.Wilkinson,R.Banchereau,J.Skinner,R.J.Wilkinson,C.Quinn,D.Blankenship,R.Dhawan,J.J.Cush,A.Mejias,O.Ramilo,O.M.Kon,V.Pascual,J.Banchereau,D.Chaussabel,A.O'Garra,Aninterferon-inducible neutrophil-driven blood transcriptional signature inhuman tuberculosis.Nature 466,973-977(2010).
46.K.Fredriksson,I. P.Keller,N.Petrovic,B.Ahlman,C.Schéele,J.Wernerman,J.A.Timmons,O.Rooyackers,Dysregulation of mitochondrial dynamicsand the muscle transcriptome in ICU patients suffering from sepsis inducedmultiple organ failure.PLoS One 3,e3686(2008).
P.Keller,N.Petrovic,B.Ahlman,C.Schéele,J.Wernerman,J.A.Timmons,O.Rooyackers,Dysregulation of mitochondrial dynamicsand the muscle transcriptome in ICU patients suffering from sepsis inducedmultiple organ failure.PLoS One 3,e3686(2008).
47.J.E.McDunn,K.D.Husain,A.D.Polpitiya,A.Burykin,J.Ruan,Q.Li,W.Schierding,N.Lin,D.Dixon,W.Zhang,C.M.Coopersmith,W.M.Dunne,M.Colonna,B.K.Ghosh,J.P.Cobb,Plasticity of the systemic inflammatory response to acuteinfection during critical illness:development of the riboleukogram.PLoS One3,e1564(2008).
48.T.P.Chung,J.M.Laramie,D.J.Meyer,T.Downey,L.H.Tam,H.Ding,T.G.Buchman,I.Karl,G.D.Stormo,R.S.Hotchkiss,J.P.Cobb,Molecular diagnostics insepsis:from bedside to bench.J Am Coll Surg 203,585-598(2006).
49.G.Parnell,A.McLean,D.Booth,S.Huang,M.Nalos,B.Tang,Aberrant cellcycle and apoptotic changes characterise severe influenza A infection--ameta-analysis of genomic signatures in circulating leukocytes.PLoS One 6,e17186(2011).
50.M.Emonts,Ph.D.thesis,Erasmus University Rotterdam,(2008).
51.P.Khatri,S.Roedder,N.Kimura,K.De Vusser,A.A.Morgan,Y.Gong,M.P.Fischbein,R.C.Robbins,M.Naesens,A.J.Butte,M.M.Sarwal,A common rejectionmodule(CRM)for acute rejection across multiple organs identifies noveltherapeutics for organ transplantation.J Exp Med 210,2205-2221(2013).
52.T.M.Osborn,J.K.Tracy,J.R.Dunne,M.Pasquale,L.M.Napolitano,Epidemiology of sepsis in patients with traumatic injury.Crit Care Med 32,2234-2240(2004).
53.M.J.Pencina,R.B.D'Agostino,O.V.Demler,Novel metrics for evaluatingimprovement in discrimination:net reclassification and integrateddiscrimination improvement for normal variables and nested models.Stat Med31,101-113(2012).
54.C.L.Smith,P.Dickinson,T.Forster,M.Craigon,A.Ross,M.R.Khondoker,R.France,A.Ivens,D.J.Lynn,J.Orme,A.Jackson,P.Lacaze,K.L.Flanagan,B.J.Stenson,P.Ghazal,Identification of a human neonatal immune-metabolic networkassociated with bacterial infection.Nat Commun 5,4649(2014).
55.A.G.Vassiliou,N.A.Maniatis,S.E.Orfanos,Z.Mastora,E.Jahaj,T.Paparountas,A.Armaganidis,C.Roussos,V.Aidinis,A.Kotanidou,Inducedexpression and functional effects of aquaporin-1 in human leukocytes insepsis.Crit Care 17,R199(2013).
56.F.Allantaz,D.Chaussabel,D.Stichweh,L.Bennett,W.Allman,A.Mejias,M.Ardura,W.Chung,E.Smith,C.Wise,K.Palucka,O.Ramilo,M.Punaro,J.Banchereau,V.Pascual,Blood leukocyte microarrays to diagnose systemic onset juvenileidiopathic arthritis and follow the response to IL-1 blockade.J Exp Med 204,2131-2144(2007).
57.K.Newton,V.M.Dixit,Signaling in innate immunity andinflammation.Cold Spring Harb Perspect Biol 4,(2012).
58.R.Chen,P.Khatri,P.K.Mazur,M.Polin,Y.Zheng,D.Vaka,C.D.Hoang,J.Shrager,Y.Xu,S.Vicent,A.J.Butte,E.A.Sweet-Cordero,A meta-analysis of lungcancer gene expression identifies PTK7 as a survival gene in lungadenocarcinoma.Cancer Res 74,2892-2902(2014).
59.F.Hietbrink,L.Koenderman,M.Althuizen,J.Pillay,V.Kamp,L.P.Leenen,Kinetics of the innate immune response after trauma:implications for thedevelopment of late onset sepsis.Shock 40,21-27(2013).
60.S.A.Madsen-Bouterse,R.Romero,A.L.Tarca,J.P.Kusanovic,J.Espinoza,C.J.Kim,J.S.Kim,S.S.Edwin,R.Gomez,S.Draghici,The transcriptome of the fetalinflammatory response syndrome.Am J Reprod Immunol 63,73-92(2010).
61.H.R.Wong,N.Z.Cvijanovich,M.Hall,G.L.Allen,N.J.Thomas,R.J.Freishtat,N.Anas,K.Meyer,P.A.Checchia,R.Lin,M.T.Bigham,A.Sen,J.Nowak,M.Quasney,J.W.Henricksen,A.Chopra,S.Banschbach,E.Beckman,K.Harmon,P.Lahni,T.P.Shanley,Interleukin-27 is a novel candidate diagnostic biomarker forbacterial infection in critically ill children.Crit Care 16,R213(2012).
62.A.Kwan,M.Hubank,A.Rashid,N.Klein,M.J.Peters,Transcriptionalinstability during evolving sepsis may limit biomarker based riskstratification.PLoS One 8,e60501(2013).
63.R.Cavallazzi,C.L.Bennin,A.Hirani,C.Gilbert,P.E.Marik,Is the bandcount useful in the diagnosis of infection?An accuracy study in criticallyill patients.J Intensive Care Med 25,353-357(2010).
64.G.Drifte,I.Dunn-Siegrist,P.Tissières,J.Pugin,Innate immunefunctions of immature neutrophils in patients with sepsis and severe systemicinflammatory response syndrome.Crit Care Med 41,820-832(2013).
65.P.J.Cornbleet,Clinical utility of the band count.Clin Lab Med 22,101-136(2002).
66.W.van der Meer,W.van Gelder,R.de Keijzer,H.Willems,Does the bandcell survive the 21st century?Eur J Haematol 76,251-254(2006).
67.K.Saito,T.Wagatsuma,H.Toyama,Y.Ejima,K.Hoshi,M.Shibusawa,M.Kato,S.Kurosawa,Sepsis is characterized by the increases in percentages ofcirculating CD4+CD25+regulatory T cells and plasma levels of solubleCD25.Tohoku J Exp Med 216,61-68(2008).
68.F.Venet,C.S.Chung,G.Monneret,X.Huang,B.Horner,M.Garber,A.Ayala,Regulatory T cell populations in sepsis and trauma.J Leukoc Biol 83,523-535(2008).
69.D.Grimaldi,S.Louis,F.Pène,G.Sirgo,C.Rousseau,Y.E.Claessens,L.Vimeux,A.Cariou,J.P.Mira,A.Hosmalin,J.D.Chiche,Profound and persistentdecrease of circulating dendritic cells is associated with ICU-acquiredinfection in patients with septic shock.Intensive Care Med 37,1438-1446(2011).
70.S.Park,Y.Zhang,S.Lin,T.H.Wang,S.Yang,Advances in microfluidic PCRfor point-of-care infectious disease diagnostics.Biotechnol Adv 29,830-839(2011).
71.M.A.Poritz,A.J.Blaschke,C.L.Byington,L.Meyers,K.Nilsson,D.E.Jones,S.A.Thatcher,T.Robbins,B.Lingenfelter,E.Amiott,A.Herbener,J.Daly,S.F.Dobrowolski,D.H.Teng,K.M.Ririe,FilmArray,an automated nested multiplexPCR system for multi-pathogen detection:development and application torespiratory tract infection.PLoS One 6,e26047(2011).
72.G.Smyth,in Bioinformatics and Computational Biology SolutionsUsing R and Bioconductor,C.V.Gentleman R,Dudoit S,Irizarry R and Huber W(eds.),Ed.(Springer,New York,2005),pp.pp.397-420.
73.W.Xu,J.Seok,M.N.Mindrinos,A.C.Schweitzer,H.Jiang,J.Wilhelmy,T.A.Clark,K.Kapur,Y.Xing,M.Faham,J.D.Storey,L.L.Moldawer,R.V.Maier,R.G.Tompkins,W.H.Wong,R.W.Davis,W.Xiao,I.a.H.R.t.I.L.-S.C.R.Program,Humantranscriptome array for high-throughput clinical studies.Proc Natl Acad Sci US A 108,3707-3712(2011).
74.Y.Koren,L.Carmel,Robust linear dimensionality reduction.IEEE TransVis Comput Graph 10,459-470(2004).
75.A.Bodor,I.Csabai,M.W.Mahoney,N.Solymosi,rCUR:an R package for CURmatrix decomposition.BMC Bioinformatics 13,103(2012).
76.J.Vandesompele,K.De Preter,F.Pattyn,B.Poppe,N.Van Roy,A.De Paepe,F.Speleman,Accurate normalization of real-time quantitative RT-PCR data bygeometric averaging of multiple internal control genes.Genome Biol 3,RESEARCH0034(2002).
77.J.T.Leek,W.E.Johnson,H.S.Parker,A.E.Jaffe,J.D.Storey,The svapackage for removing batch effects and other unwanted variation in high-throughput experiments.Bioinformatics 28,882-883(2012).
78.R.K.Auerbach,B.Chen,A.J.Butte,Relating genes to function:identifying enriched transcription factors using the ENCODE ChIP-Seqsignificance tool.Bioinformatics 29,1922-1924(2013).
79.H.G.Roider,T.Manke,S.O'Keeffe,M.Vingron,S.A.Haas,PASTAA:identifying transcription factors associated with sets of co-regulatedgenes.Bioinformatics 25,435-442(2009).
80.K.L.Jeffrey,T.Brummer,M.S.Rolph,S.M.Liu,N.A.Callejas,R.J.Grumont,C.Gillieron,F.Mackay,S.Grey,M.Camps,C.Rommel,S.D.Gerondakis,C.R.Mackay,Positive regulation of immune cell function and inflammatory responses byphosphatase PAC-1.Nat Immunol 7,274-283(2006).
81.F.O.Martinez,S.Gordon,M.Locati,A.Mantovani,Transcriptionalprofiling of the human monocyte-to-macrophage differentiation andpolarization:new molecules and patterns of gene expression.J Immunol 177,7303-7311(2006).
82.S.Radom-Aizik,F.Zaldivar,S.Y.Leu,P.Galassetti,D.M.Cooper,Effectsof 30min of aerobic exercise on gene expression in human neutrophils.J ApplPhysiol(1985)104,236-243(2008).
83.F.He,H.Chen,M.Probst-Kepper,R.Geffers,S.Eifes,A.Del Sol,K.Schughart,A.P.Zeng,R.Balling,PLAU inferred from a correlation network iscritical for suppressor function of regulatory T cells.Mol Syst Biol 8,624(2012).
84.M.Giefing,S.Winoto-Morbach,J.Sosna,C. W.Klapper,R.Küppers,S.
W.Klapper,R.Küppers,S. D.Adam,R.Siebert,S.Schütze,Hodgkin-Reed-Sternberg cells in classicalHodgkin lymphoma show alterations of genes encoding the NADPH oxidase complexand impaired reactive oxygen species synthesis capacity.PLoS One 8,e84928(2013).
D.Adam,R.Siebert,S.Schütze,Hodgkin-Reed-Sternberg cells in classicalHodgkin lymphoma show alterations of genes encoding the NADPH oxidase complexand impaired reactive oxygen species synthesis capacity.PLoS One 8,e84928(2013).
85.J.A.Meyers,D.W.Su,A.Lerner,Chronic lymphocytic leukemia and B andT cells differ in their response to cyclic nucleotide phosphodiesteraseinhibitors.J Immunol 182,5400-5411(2009).
86.S.Eckerle,V.Brune,C. E.Tiacci,V.Bohle,C.
E.Tiacci,V.Bohle,C. R.Kodet,M.Paulli,B.Falini,W.Klapper,A.B.Chaubert,K.Willenbrock,D.Metzler,A.
R.Kodet,M.Paulli,B.Falini,W.Klapper,A.B.Chaubert,K.Willenbrock,D.Metzler,A. R.Küppers,M.L.Hansmann,Gene expression profiling of isolated tumour cellsfrom anaplastic large cell lymphomas:insights into its cellular origin,pathogenesis and relation to Hodgkin lymphoma.Leukemia 23,2129-2138(2009).
R.Küppers,M.L.Hansmann,Gene expression profiling of isolated tumour cellsfrom anaplastic large cell lymphomas:insights into its cellular origin,pathogenesis and relation to Hodgkin lymphoma.Leukemia 23,2129-2138(2009).
87.K.A.Stegmann,N.K. H.Veber,S.Ciesek,P.Riese,J.Wiegand,J.Hadem,P.V.Suneetha,J.Jaroszewicz,C.Wang,V.Schlaphoff,P.Fytili,M.Cornberg,M.P.Manns,R.Geffers,T.Pietschmann,C.A.Guzmán,H.G.Ljunggren,H.Wedemeyer,Interferon-alpha induced TRAIL on natural killer cells is associated withcontrol of hepatitis C virus infection.Gastroenterology 138,1885-1897(2010).
H.Veber,S.Ciesek,P.Riese,J.Wiegand,J.Hadem,P.V.Suneetha,J.Jaroszewicz,C.Wang,V.Schlaphoff,P.Fytili,M.Cornberg,M.P.Manns,R.Geffers,T.Pietschmann,C.A.Guzmán,H.G.Ljunggren,H.Wedemeyer,Interferon-alpha induced TRAIL on natural killer cells is associated withcontrol of hepatitis C virus infection.Gastroenterology 138,1885-1897(2010).
88.D.C.Vinh,S.Y.Patel,G.Uzel,V.L.Anderson,A.F.Freeman,K.N.Olivier,C.Spalding,S.Hughes,S.Pittaluga,M.Raffeld,L.R.Sorbara,H.Z.Elloumi,D.B.Kuhns,M.L.Turner,E.W.Cowen,D.Fink,D.Long-Priel,A.P.Hsu,L.Ding,M.L.Paulson,A.R.Whitney,E.P.Sampaio,D.M.Frucht,F.R.DeLeo,S.M.Holland,Autosomal dominantand sporadic monocytopenia with susceptibility to mycobacteria,fungi,papillomaviruses,and myelodysplasia.Blood 115,1519-1529(2010).
89.P.Ancuta,K.Y.Liu,V.Misra,V.S.Wacleche,A.Gosselin,X.Zhou,D.Gabuzda,Transcriptional profiling reveals developmental relationship and distinctbiological functions of CD16+and CD16-monocyte subsets.BMC Genomics 10,403(2009).
90.N.Novershtern,A.Subramanian,L.N.Lawton,R.H.Mak,W.N.Haining,M.E.McConkey,N.Habib,N.Yosef,C.Y.Chang,T.Shay,G.M.Frampton,A.C.Drake,I.Leskov,B.Nilsson,F.Preffer,D.Dombkowski,J.W.Evans,T.Liefeld,J.S.Smutko,J.Chen,N.Friedman,R.A.Young,T.R.Golub,A.Regev,B.L.Ebert,Denselyinterconnected transcriptional circuits control cell states in humanhematopoiesis.Cell 144,296-309(2011).
91.F.Allantaz,D.T.Cheng,T.Bergauer,P.Ravindran,M.F.Rossier,M.Ebeling,L.Badi,B.Reis,H.Bitter,M.D'Asaro,A.Chiappe,S.Sridhar,G.D.Pacheco,M.E.Burczynski,D.Hochstrasser,J.Vonderscher,T.Matthes,Expression profiling ofhuman immune cell subsets identifies miRNA-mRNA regulatory relationshipscorrelated with cell type specific expression.PLoS One 7,e29979(2012).
92.E.Tsitsiou,A.E.Williams,S.A.Moschos,K.Patel,C.Rossios,X.Jiang,O.D.Adams,P.Macedo,R.Booton,D.Gibeon,K.F.Chung,M.A.Lindsay,Transcriptomeanalysis shows activation of circulating CD8+T cells in patients with severeasthma.J Allergy Clin Immunol 129,95-103(2012).
93.M.Frankenberger,T.P.Hofer,A.Marei,F.Dayyani,S.Schewe,C.Strasser,A.Aldraihim,F.Stanzel,R.Lang,R.Hoffmann,O.Prazeres da Costa,T.Buch,L.Ziegler-Heitbrock,Transcript profiling of CD16-positive monocytes reveals a uniquemolecular fingerprint.Eur J Immunol 42,957-974(2012).
94.N.Y.Huen,A.L.Pang,J.A.Tucker,T.L.Lee,M.Vergati,C.Jochems,C.Intrivici,V.Cereda,W.Y.Chan,O.M.Rennert,R.A.Madan,J.L.Gulley,J.Schlom,K.Y.Tsang,Up-regulation of proliferative and migratory genes in regulatory Tcells from patients with metastatic castration-resistant prostate cancer.IntJ Cancer 133,373-382(2013).
95.K.C.Malcolm,E.M.Nichols,S.M.Caceres,J.E.Kret,S.L.Martiniano,S.D.Sagel,E.D.Chan,L.Caverly,G.M.Solomon,P.Reynolds,D.L.Bratton,J.L.Taylor-Cousar,D.P.Nichols,M.T.Saavedra,J.A.Nick,Mycobacterium abscessus induces alimited pattern of neutrophil activation that promotes pathogen survival.PLoSOne 8,e57402(2013).
96.N.Rapin,F.O.Bagger,J.Jendholm,H.Mora-Jensen,A.Krogh,A.Kohlmann,C.Thiede,N.Borregaard,L.Bullinger,O.Winther,K.Theilgaard- B.T.Porse,Comparing cancer vs normal gene expression profiles identifies new diseaseentities and common transcriptional programs in AML patients.Blood 123,894-904(2014).
B.T.Porse,Comparing cancer vs normal gene expression profiles identifies new diseaseentities and common transcriptional programs in AML patients.Blood 123,894-904(2014).
97.N.A.Mabbott,J.K.Baillie,H.Brown,T.C.Freeman,D.A.Hume,An expressionatlas of human primary cells:inference of gene function from coexpressionnetworks.BMC Genomics 14,632(2013).
实施例2
使用公用数据基准测试败血症基因表达诊断
不存在快速可用的黄金标准分子测试可测定具有全身炎症的患者是否可能感染。败血症的漏诊导致延误治疗和死亡率升高,同时不当抗生素增加抗生素抗性并可能引起并发症(Ferrer等人(2014)《危重病监护医学》42(8):1749-1755;McFarland(2008)《未来微生物学(Future Microbiol)》3(5):563-578;Gaieski等人(2010)《危重病监护医学》38(4):1045-1053)。因此,对可分离具有非感染性炎症的患者与患有败血症的患者的新诊断存在迫切的未满足的需要(Cohen等人(2015)《柳叶刀感染病》15(5):581-614)。
难以得到可区分败血症与非感染性炎症的新诊断,因为响应于感染激活的许多细胞途径也响应于组织创伤和非感染性炎症而激活。因此,高通量‘组学’技术,如经由微阵列的基因表达谱分析,是研究败血症的良好方式,因为它们允许同时检验数万个基因。可随后使用统计学技术得出针对诊断优化的新分类器。然而,高通量数据集始终具有比样本更多的变量,并且因此易于出现不可再现的过度拟合结果(Shi等人(2008)《BMC生物信息学》9增刊9:S10;Ioannidis等人(2001)《自然·遗传学(Nat Genet)》29(3):306-309)。此外,为了增加统计功效,在临床上均质的群组中创建各个生物标志物数据集,并且通常使用仅单个类型的微阵列创建。虽然这个设计确实使得检测所研究的各组间的差异的功效更大,但是结果在使用不同实验室技术的不同临床群组中不大可能仍然成立。因此,绝对必需独立验证以估计由高通量研究得出的任何新分类器的普遍适用性。
据我们所知,已使用微阵列数据专门研发出三种基因表达诊断以分离患有败血症的患者与那些具有非感染性炎症的患者。这些是下文称为‘Sepsis MetaScore’(SMS)的11个基因的集合(Sweeney等人(2015)《科学·转化医学(Sci Transl Med)》7(287):287ra271)、FAIM3:PLAC8比率(Scicluna等人(2015)《美国呼吸和重症护理医学杂志(Am JRespir Crit Care Med.)》192(7):826-835)和Septicyte Lab(McHugh等人(2015)《公共科学图书馆·医学》12(12):e1001916)。另外,现存几十个公开可用的检查患有败血症或急性感染的患者的数据集。它们覆盖极广泛范围的临床条件,包括不同年龄组、感染类型、共存病况和对照(非感染性)病况。这个相对未利用的公开资源可因此用于估计跨越巨大数目的患者样本的不同诊断的相对优点和缺点。此处我们使用所有可用的公开基因表达数据研究和直接比较三种败血症基因表达诊断的诊断能力。
方法
我们在2015年12月10日使用以下术语完成了两个公开基因表达存储库(NIH GEO和EBI ArrayExpress)的系统搜索:败血症、SIRS、肺炎、创伤、ICU、感染、急性、休克和手术。我们自动排除了非微阵列、非人类数据。随后,使用相应原稿的摘要进行筛选,我们去除了(1)非临床数据集、(2)未时间匹配的数据集和(3)非全血数据集。随后根据参考群组(与败血症相比)是否是健康对照或未感染的SIRS患者分选其余数据集。示意图显示在图17中。除了来自系统搜索的数据以外,我们包括来自炎症和宿主对损伤的反应(Glue Grant)群组的两个纵向创伤群组,如先前所述(Sweeney等人(2015)《科学·转化医学》7(287):287ra2717)。在不同微阵列上运行的来自相同研究的群组作为独立的群组进行处理。
如果可能的话,将原始资料可用的所有数据集重新归一化。使用gcRMA(在具有完全匹配的探针可用的阵列上)或RMA将Affymetrix阵列重新归一化。Illumina、Agilent、GE和其它市售阵列是经由正规指数背景校正随后分位数归一化而重新归一化。未将自定义阵列重新归一化。所有数据都以log2变换状态使用。使用固定效应模型将探针汇总成数据集内的基因(Ramasamy等人(2008)《公共科学图书馆·医学》5(9):e184)。
进行文献查阅以搜索相比于未感染患者特异性优化败血症诊断的基因表达签名。随后测试所得模型的败血症诊断能力,如通过受试者工作特征曲线下面积(AUC)所测量。从最终结果排除可能无法计算任一个败血症评分(所有上调或所有下调基因缺失)的数据集。对于指定比较(例如非感染性SIRS对在入院时的败血症),计算所有所述类型数据集以及仅非发现数据集的平均值,因为发现数据集经预计显示与独立验证数据集相比估计过高的诊断能力。最后,我们使用成对t检验比较了每一个诊断评分的重叠验证集(例如在GSE74227、E-MEXP-3589和Glue Grant嗜中性粒细胞中比较了11个基因的集合和FAIM3:PLAC8比率诊断败血症的能力,因为这些群组是仅有的验证两个数据集的群组)。
GSE28750(11)(N=21)中的患者样本还用于后期数据集GSE74224(McHugh等人(2015)《公共科学图书馆·医学》12(12):e1001916)(N=105),不过这两个数据集使用不同微阵列类型运行(Affymetrix HG 2.0对Affymetrix Exon 1.0ST)。因此,GSE28750不包括在Septicyte Lab(在GSE74224中发现)的验证计算中,而在计算Sepsis MetaScore的验证平均值时,考虑到发现中存在20%的GSE74224患者的事实,对GSE74224中的AUC进行罚分(罚分AUC*0.8+GSE28750中的实际AUC*0.2)=实际AUC。
为了检验感染类型的混杂,筛选存在(1)革兰氏阳性和革兰氏阴性感染或(2)细菌和病毒感染的每一个数据集。假定如数据的原作者所述的微生物学测定是正确的。协同感染的病例不包括在混杂比较中。为了检验混杂,针对包括所关注的两个类别的数据集计算每一个基因表达评分,并在数据集内经由威尔科克森秩和检验比较类别间的所得评分。
如先前所描述进行元分析。简单来说,使用Hedge g汇总数据集内非感染性SIRS患者与败血症患者之间的差异性基因表达,并接着使用DerSimonian-Laird随机效应模型,随后通过Benjamini-Hochberg校正在数据集之间进行比较。个别基因的森林图在log2空间中示出了每一个数据集内的个别效应以及汇总的‘元效应’。使用R统计学计算语言进行所有分析。显著性检验始终是双尾检验。针对所检查的基因集合重建入院非感染性SIRS对败血症比较的代码和数据可在http://khatrilab.stanford.edu/sepsis获得。上传的数据呈此处所用的重新归一化的形式。Glue Grant数据可供已经Glue Grant联盟批准的研究人员使用;指令在我们的网站上。如果将结果用于原稿中,那么作者申请引用这篇文章和描述原始数据集的文章。
结果
我们进行公开基因表达数据库的系统搜索(图17),并且我们还使用两个独立的Glue Grant创伤群组,分解成从未感染的患者和在败血症诊断+/-24小时内的患者的时间匹配仓,如先前所描述(Sweeney等人(2015)《科学·转化医学》7(287):287ra271)。这产生了总共39个符合标准的数据集,由3241个患者样本构成(Scicluna等人,见上文;McHugh等人,见上文;Dolinay等人(2012)《美国呼吸和重症护理医学杂志》185(11):1225-1234;Parnell等人(2012)《危重病监护》16(4):R157;Wong等人(2007)《生理基因组学》30(2):146-155;Wynn等人(2011)《分子医学》17(11-12):1146-1156;Wong等人(2009)《危重病监护医学》37(5):1558-1566;Shanley等人(2007)《分子医学》13(9-10):495-508;Cvijanovich等人(2008)《生理基因组学》34(1):127-134;Almansa等人(2012)《BMC研究笔记(BMC ResNotes)》5:401;Irwin等人(2012)《BMC医学基因组学(BMC Med Genomics)》5:13;van deWeg等人(2015)《公共科学图书馆·忽视的热带病(PLoS Negl Trop Dis)》9(3):e0003522;Emonts M.《感染性疾病和自体免疫疾病中免疫反应基因的多形现象(Polymorphisms inImmune Response Genes in Infectious Diseases and Autoimmune Diseases)》[博士论文]:鹿特丹伊拉斯姆斯大学;2008;Pankla等人(2009)《基因组生物学》10(11):R127;Zaas等人(2009)《细胞宿主与微生物(Cell Host Microbe)》6(3):207-217;Parnell等人(2011)《公共科学图书馆·综合》6(3):e17186;Bermejo-Martin等人(2010)《危重病监护》14(5):R167;Berry等人(2010)《自然》466(7309):973-977;Smith等人(2014)《自然通讯》5:4649;Berdal等人(2011)《传染病杂志》63(4):308-316;Ahn等人(2013)《公共科学图书馆·综合》8(1):e48979;Mejias等人(2013)《公共科学图书馆·医学》10(11):e1001549;Hu等人(2013)《美国国家科学院院刊》110(31):12792-12797;Herberg等人(2013)《传染病杂志》208(10):1664-1668;Kwissa等人(2014)《细胞宿主与微生物》16(1):115-127;Cazalis等人(2014)《重症监护医学实验(Intensive Care Med Exp)》2(1):20;Suarez等人(2015)《传染病杂志》212(2):213-222;Zhai等人(2015)《公共科学图书馆·病原体(PLoS Pathog)》11(6):e1004869;Conejero等人(2015)《免疫学杂志》195(7):3248-3261;Xiao等人(2011)《实验医学杂志》208(13):2581-2590;和Warren等人(2009)《分子医学》15(7-8):220-227)。
我们的文献综述揭露了经特异性优化以在全血样本中区分非感染性SIRS与败血症的三个基因表达分类器。它们是:我们先前公开的11个基因的集合(SMS)(Sweeney等人,《科学·转化医学》2015);FAIM3:PLAC8比率(Scicluna等人,AJRCCM 2015)和SepticyteLab(McHugh等人,《公共科学图书馆·医学》,2015)。对于每一个样本,根据下式计算11个基因的评分:

FAIM3:PLAC8比率如下计算:PLAC8/FAIM3。Septicyte Lab如下计算:(PLAC8+LAMP1)-(PLA2G7+CEACAM4)。在所有情况下,计算都是在log2变换数据上进行。
三个败血症评分中每一个的稳固性和再现性取决于其组成基因中每一个的稳固并且可再现的表达变化。因此,我们在比较未感染的SIRS/创伤患者与败血症患者的12个全血群组中探索了个别基因在三个检验中的每一个中如何不断地变化。我们关于这些数据集的元分析揭露了3个基因评分中的任一个所包括的16个基因中的每一个(CEACAM4除外)沿所希望的方向变化(FDR<5%)。值得注意的是,CEACAM4,Septicyte Lab中的一个基因,仅在其相应发现群组中显著下调。
接着,我们将数据集分成两种宽泛类型的比较:具有非感染性SIRS或创伤的患者对败血症或急性感染患者(表1);和健康对照对具有败血症或急性感染的患者(表2)。对于这两种类型的比较,我们计算了总平均AUC以及当仅包括独立验证数据集时的AUC;对于三个签名中的每一个,我们排除了其相应发现数据集。
在非感染性SIRS/创伤数据集对败血症数据集(16个群组,1148个样本,表1)中,在比较重叠验证数据集的三个基因表达诊断评分的AUC之间的成对t检验中无显著差异(所有p>0.1,图18和19)。当比较来自所有16个群组(即包括发现群组)的AUC时,SepsisMetaScore AUC显著高于另两个基因评分的AUC(均为p<0.05),FAIM3:PLAC8比率与Septicyte Lab之间无显著差异。然而,这些结果不一定指示Sepsis MetaScore的总体表现较佳,因为Sepsis MetaScore使用这些发现群组中的9个。FAIM3:PLAC8比率显示在GSE32707和GSE40012中的表现降低,但是如先前所论述,它是专门设计用于测试CAP的存在并且可能无法普遍适用于其它形式的非感染性炎症(41,42)。最后,Septicyte Lab在分离儿童SIRS/败血症患者以及有和无感染的住院COPD患者方面的表现显著降低(AUC<0.5)。有可能Septicyte Lab的这一AUC降低归因于与Septicyte Lab的初始发现群组相比临床情况或微阵列类型的差异。
我们接着检查了将健康对照与患有败血症或急性感染的患者相比较的数据集(26个数据集,2417个样本,表2)。这些患者的大部分但不是全部患有败血症;然而,对于败血症诊断测试,预期它应该能够区分大部分感染与健康对照是合理的。此处,Sepsis MetaScore和FAIM3:PLAC8比率都表现得正如所预期,其中平均验证AUC是0.96+/-0.05和0.94+/-0.09(表2)。然而,Septicyte Lab在由1562个样本(全部样本的64%)构成的12个数据集(全部数据集的43%)中的AUC<0.7,使得平均验证AUC=0.71+/-0.20(表2),显著低于SepsisMetaScore和FAIM3:PLAC8比率(都是P<1e-5)。同样,这个表现降低可归因于Septicyte Lab包括CEACAM4,其在元分析中发现是不显著的。有人可能争论不存在对分离健康对照与败血症患者的诊断的临床需要;然而,在区分这两组时的较差表现可指示更深的偏差并且可增加非普遍适用的风险。
理想的败血症诊断不应显示取决于所存在的感染类型的表现变化。为了研究任何诊断是否偏向特定的感染类型,我们搜索贯穿所有包括的数据集以寻找那些比较具有细菌和病毒感染的患者的数据集和那些比较革兰氏阳性和革兰氏阴性感染的数据集。我们随后比较了在检测这些感染类型(相比于健康对照)以及比较评分分布方面的诊断能力。由于评分越高表示感染的可能性越高,所以在一种感染类型中始终较低的评分可指示针对所述类型的诊断表现降低,即使诊断表现的变化不是相比于健康对照检测到的。
存在8个数据集提供关于患者是否具有细菌或病毒感染的信息。一般来说,在所述三个评分中任一个对于细菌和病毒感染的AUC之间存在极少差异(表3);然而,这可能归因于在比较这些感染与健康对照时的小数目和相对高的AUC。然而,尽管存在这些警告,但是Sepsis MetaScore和FAIM3:PLAC8比率都在7/8个测试数据集中显示相比于病毒感染具有细菌感染的患者的平均评分较高,都在2/8个数据集中达到显著性(表5)。相比之下,Septicyte Lab并未在比较细菌和病毒感染时显示出强烈的趋势,在一个数据集中显示与在细菌感染中相比在病毒感染中的平均值显著较高。
存在8个数据集提供关于患者是否具有革兰氏阳性和革兰氏阴性感染的信息。革兰氏阳性和革兰氏阴性感染的比较揭露了Sepsis MetaScore或FAIM3:PLAC8比率的AUC无差异;Septicyte Lab显示一些变异性,但是这可能归因于在革兰氏阳性与革兰氏阴性感染之间在相对于健康对照的诊断表现而非差异方面的高变异性(表4)。Sepsis MetaScore、FAIM3:PLAC8比率和Septicyte lab分别示出了2个、1个和1个数据集,其中在革兰氏阴性患者中的评分显著高于在革兰氏阳性患者中(表6)。
论述
此处,我们在所有可用的时间匹配的全血临床败血症数据集中比较了三个败血症基因表达诊断(Sepsis MetaScore、FAIM3:PLAC8比率和Septicyte Lab)。在比较所有验证非感染性SIRS/创伤和败血症数据集的AUC分布当中不存在显著差异。然而,存在数个FAIM3:PLAC8比率和Septicyte Lab显示AUC<0.7的个别数据集。值得注意的是,当将健康对照与患有败血症或急性感染的患者相比较时,Septicyte Lab还在验证群组中具有显著降低的表现,其中Septicyte lab显示在所有数据集的43%中AUC<0.7。Septicyte Lab首先使用靶向qPCR在来自MARS联盟的大的独立患者群组中进行验证,并且显示总验证AUC是0.88(McHugh等人(2015)《公共科学图书馆·医学》12(12):e1001916);因此,在我们的分析中表现降低可指示在临床条件中的差异、在技术中的差异或这两者。森林图指示GSE74224(Septicyte Lab的发现群组)中CEACAM4的效应量可为离群值,其可导致相对更糟的普遍适用性。
Sepsis MetaScore和FAIM3:PLAC8比率存在相较于病毒感染在细菌感染中朝向较高评分的趋势的一些迹象,而Septicyte Lab显示在病毒感染中的评分较高的一些迹象。全部三个评分存在相较于革兰氏阳性感染在革兰氏阴性感染中朝向较高评分的统计学上不显著的趋势。然而,在它们的个别数据集中并未针对疾病严重程度、年龄、性别或其它临床混杂因素匹配不同病原体类型;因此,这些趋势必须谨慎解释。举例来说,如果细菌感染一般比病毒感染更严重并且革兰氏阴性一般比革兰氏阳性更严重,那么这些评分可通过严重程度指示混杂。在用于此处比较的所有测试的所有群组中,针对混杂因素的进一步测试将因此是必需的。
此前,Sepsis MetaScore在Glue Grant嗜中性粒细胞群组和健康对感染群组的子集中加以验证(Sweeney等人(2015)《科学·转化医学》7(287):287ra271)。在此处测试的额外群组中,Sepsis MetaScore持续显示与先前验证类似的结果。FAIM3:PLAC8比率在后续出版物中在这些数据的一些中加以验证(Sweeney等人(2015)《美国呼吸和重症护理医学杂志》192(10):1260-1261),不过作者指出他们的基因集合最初被设计用于确定入住ICU的疑似患有CAP的患者存在CAP的极狭窄的问题(Scicluna等人(2015)《美国呼吸和重症护理医学杂志》192(10):1261-1262)。Septicyte Lab在群组E-TABM-1548中进行测试(McHugh等人,见上文),但是我们此前已示出由于因从手术恢复所致的基线基因表达谱的预期变化,所述群组不适用于测试败血症诊断(Sweeney等人(2015),见上文)。
公用数据不用于新诊断验证的事实可反映一些研究人员在获取和使用这些数据方面所面临的困难和认知弯路。考虑到将公用数据变形成可易于使用的形式的困难,我们已提供了这些数据的手绘通用储存库以及易于将所关注的分类器应用于数据集的R脚本(khatrilab.stanford.edu/sepsis)。我们建议任何新的用于败血症的基因表达分类器的实践应在这些数据中检验以易于基准测试和在分类器之间比较。我们认识到简单测量AUC无法解释所有可能的临床效用测量。另外,反复在单个临床领域中充分执行的评分即使在不同临床领域失效,仍可具有极大的临床效用,并且应需要谨慎应用。尽管如此,但重要的是阐明任何新的败血症诊断的优点和缺点,以帮助将资源集中在显示最大前景的领域中的其它临床试验。
此处所测试的每一个诊断基因集合都具有优点和缺点。一般来说,对于变成临床上有用的任何败血症诊断,其必须在广泛范围的患者背景中以其最终形式保留良好的诊断能力。此处所用的微阵列群组允许不同基因表达诊断的头对头比较,但是当使用靶向分析(即,无跨越多个微阵列平台的技术变量)时,可显示任何测试低估的诊断表现。因此,在它们转入临床实践之前,应需要任何基因集合的进一步前瞻性验证。另外,考虑到技术准确度的增加,宿主反应的分子谱将可能变成诊断、治疗和可能预防败血症的临床工具集的有价值的部分。
虽然已说明和描述本发明的优选实施例,但是应了解,可在不脱离本发明的精神和范围的情况下进行各种改变。
表1.非感染性SIRS/创伤对败血症/感染数据集。(D):给定评分的发现数据集。*GSE28750是GSE74224的子集,因此作为Septicyte Lab的发现进行计数。**GSE28750是GSE74224的子集并且如方法中所述进行处理。

表2.健康对败血症/急性感染。(D):给定评分的发现数据集


表3.细菌和病毒感染对健康对照的AUC的比较。(D):给定评分的发现数据集。

表4.革兰氏阴性和革兰氏阳性感染对健康对照的AUC的比较。(D):给定评分的发现数据集。





Claims (21)
1.用于测量生物样本中CEACAM1、ZDHHC19、C9orf95、GNA15、BATF、C3AR1、KIAA1370、TGFBI、MTCH1、RPGRIP1和HLA-DPB1的表达水平的试剂在制备用于诊断受试者的败血症的试剂盒中的用途,
其中与未感染的对照受试者的生物标志物的参考值范围相比,所述CEACAM1、ZDHHC19、C9orf95、GNA15、BATF和C3AR1生物标志物的表达水平升高和所述KIAA1370、TGFBI、MTCH1、RPGRIP1和HLA-DPB1生物标志物的表达水平降低指示所述受试者患有败血症。
2.根据权利要求1所述的用途,其中所述生物标志物的表达水平是与感染或未感染的受试者的时间匹配的参考值范围相比。
3.根据权利要求1所述的用途,其中根据下式基于所述生物标志物的表达水平计算所述受试者的败血症评分:

其中与未感染的对照受试者的参考值范围相比,所述受试者的败血症评分更高指示所述受试者患有败血症。
4.根据权利要求1所述的用途,其中区分败血症诊断与非感染性发炎病况。
5.根据权利要求4所述的用途,其中所述非感染性发炎病况选自由以下组成的组:全身炎症反应综合征(SIRS)、自体免疫病症、创伤性受伤和手术。
6.根据权利要求1所述的用途,其中所述受试者是人类。
7.根据权利要求1所述的用途,其中通过微阵列分析、聚合酶链式反应(PCR)、逆转录酶聚合酶链式反应(RT-PCR)、Northern印迹或基因表达的系列分析(SAGE)测量所述生物标志物的表达水平。
8.根据权利要求1所述的用途,其中所述生物样本包含全血、血沉棕黄层、血浆、血清、外周血单个核细胞(PBMC)、杆状核细胞、嗜中性粒细胞、单核细胞或T细胞。
9.用于测量CEACAM1、ZDHHC19、C9orf95、GNA15、BATF、C3AR1、KIAA1370、TGFBI、MTCH1、RPGRIP1和HLA-DPB1的表达水平的试剂在制备用于监测治疗受试者的败血症的疗法的功效的试剂盒中的用途,所述监测包含:测量在所述受试者进行所述疗法之前来源于所述受试者的第一样本和在所述受试者进行所述疗法之后来源于所述受试者的第二样本中CEACAM1、ZDHHC19、C9orf95、GNA15、BATF、C3AR1、KIAA1370、TGFBI、MTCH1、RPGRIP1和HLA-DPB1生物标志物的表达水平,其中与所述第一样本中所述生物标志物的表达水平相比,所述第二样本中所述CEACAM1、ZDHHC19、C9orf95、GNA15、BATF和C3AR1生物标志物的表达水平升高和所述KIAA1370、TGFBI、MTCH1、RPGRIP1和HLA-DPB1生物标志物的表达水平降低指示所述受试者恶化,以及与所述第一样本中所述生物标志物的表达水平相比,所述第二样本中所述CEACAM1、ZDHHC19、C9orf95、GNA15、BATF和C3AR1生物标志物的表达水平降低和所述KIAA1370、TGFBI、MTCH1、RPGRIP1和HLA-DPB1生物标志物的表达水平升高指示所述受试者改善。
10.根据权利要求9所述的用途,其中计算所述受试者的败血症评分,其中与所述第一样本的败血症评分相比,所述第二样本的败血症评分更高指示所述受试者恶化,以及与所述第一样本的败血症评分相比,所述第二样本的败血症评分更低指示所述受试者改善。
11.一种试剂盒,包含用于测量11至30个败血症生物标志物的表达水平的试剂,所述生物标志物包括CEACAM1、ZDHHC19、C9orf95、GNA15、BATF、C3AR1、KIAA1370、TGFBI、MTCH1、RPGRIP1和HLA-DPB1。
12.根据权利要求11所述的试剂盒,另外包含微阵列。
13.根据权利要求12所述的试剂盒,其中所述试剂盒包含微阵列,所述微阵列包含与CEACAM1多核苷酸杂交的寡核苷酸、与ZDHHC19多核苷酸杂交的寡核苷酸、与C9orf95多核苷酸杂交的寡核苷酸、与GNA15多核苷酸杂交的寡核苷酸、与BATF多核苷酸杂交的寡核苷酸、与C3AR1多核苷酸杂交的寡核苷酸、与KIAA1370多核苷酸杂交的寡核苷酸、与TGFBI多核苷酸杂交的寡核苷酸、与MTCH1多核苷酸杂交的寡核苷酸、与RPGRIP1多核苷酸杂交的寡核苷酸和与HLA-DPB1多核苷酸杂交的寡核苷酸。
14.根据权利要求11所述的试剂盒,另外包含呈电子或纸质形式的信息,所述信息包含使每一种生物标志物的检测水平与败血症相关的说明。
15.一种用于执行一种计算机实施的用于诊断疑似患有败血症的患者的方法的诊断系统,所述计算机执行包含以下的步骤:
a)接收所输入的患者数据,包含来自所述患者的生物样本中CEACAM1、ZDHHC19、C9orf95、GNA15、BATF、C3AR1、KIAA1370、TGFBI、MTCH1、RPGRIP1和HLA-DPB1生物标志物的表达水平值;
b)分析每一种生物标志物的水平并与每一种生物标志物的相应参考值范围相比;
c)根据下式基于所述生物标志物的表达水平计算所述患者的败血症评分:

d)基于所述败血症评分的值计算所述患者患有败血症的可能性,其中所述患者的败血症评分与未感染的对照受试者的参考值范围相比更高指示所述患者患有败血症;以及
e)显示关于所述患者的诊断信息,
其中所述诊断系统包含:
a)用于存储数据的存储组件,其中所述存储组件具有用于确定存储在其中的受试者诊断的指令;
b)用于处理数据的计算机处理器,其中所述计算机处理器被耦合到所述存储组件并且被配置成执行存储在所述存储组件中的指令以便接收患者数据并根据一种或多种算法分析患者数据;以及
c)用于显示关于所述患者的诊断信息的显示器组件。
16.根据权利要求15所述的诊断系统,其中所述存储组件另外包含用于进行多变量线性判别分析(LDA)、受试者工作特征(ROC)分析、集合数据挖掘方法、微阵列的细胞特异性显著性分析(csSAM)和多维蛋白质鉴定技术(MUDPIT)分析的指令。
17.一种包含多种体外复合物的组合物,其中所述多种体外复合物包含与11至30个败血症生物标志物的核酸杂交的标记探针,所述生物标志物包括CEACAM1、ZDHHC19、C9orf95、GNA15、BATF、C3AR1、KIAA1370、TGFBI、MTCH1、RPGRIP1和HLA-DPB1,所述标记探针与所述生物标志物基因序列或其互补序列杂交,其中所述核酸是从患有败血症的患者提取,或是从患有败血症的患者提取的核酸的扩增产物。
18.根据权利要求17所述的组合物,其中所述体外复合物是在检测装置中。
19.根据权利要求17所述的组合物,其中所述标记探针包含荧光标记、生物发光标记、化学发光标记、比色标记或同位素标记。
20.能够检测生物样本中的生物标志物CEACAM1、ZDHHC19、C9orf95、GNA15、BATF、C3AR1、KIAA1370、TGFBI、MTCH1、RPGRIP1和HLA-DPB1基因序列的标记探针在制备用于诊断患者的败血症的试剂盒中的用途,
所述标记探针能够与生物标志物CEACAM1、ZDHHC19、C9orf95、GNA15、BATF、C3AR1、KIAA1370、TGFBI、MTCH1、RPGRIP1和HLA-DPB1基因序列或其互补序列杂交,以便产生根据权利要求17所述的组合物,其中
体外复合物的量测定所述生物样本中所述生物标志物核酸的表达水平;以及
所述CEACAM1、ZDHHC19、C9orf95、GNA15、BATF或C3AR1生物标志物核酸的表达水平与对照受试者的所述生物标志物核酸的参考值范围相比增加指示所述患者患有败血症,或所述KIAA1370、TGFBI、MTCH1、RPGRIP1或HLA-DPB1生物标志物核酸的表达水平与对照受试者的所述生物标志物核酸的参考值范围相比降低指示所述患者患有败血症。
21.根据权利要求20所述的用途,其中基于所述生物标志物核酸的表达水平计算所述患者的败血症评分,其中所述患者的败血症评分与所述对照受试者的参考值范围相比更高指示所述患者患有败血症。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201562132293P | 2015-03-12 | 2015-03-12 | |
| US62/132,293 | 2015-03-12 | ||
| PCT/US2016/022233 WO2016145426A1 (en) | 2015-03-12 | 2016-03-12 | Methods for diagnosis of sepsis |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN107532214A CN107532214A (zh) | 2018-01-02 |
| CN107532214B true CN107532214B (zh) | 2021-08-31 |
Family
ID=56878909
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201680023601.1A Active CN107532214B (zh) | 2015-03-12 | 2016-03-12 | 用于败血症诊断的方法 |
Country Status (8)
| Country | Link |
|---|---|
| US (3) | US10533224B2 (zh) |
| EP (1) | EP3268499B1 (zh) |
| JP (3) | JP6995622B2 (zh) |
| CN (1) | CN107532214B (zh) |
| AU (2) | AU2016228508A1 (zh) |
| CA (1) | CA2977422A1 (zh) |
| IL (3) | IL297699A (zh) |
| WO (1) | WO2016145426A1 (zh) |
Families Citing this family (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2017514459A (ja) | 2014-03-14 | 2017-06-08 | イー.ダブリュ. ハンコック,ロバート | 敗血症の診断 |
| GB201616557D0 (en) * | 2016-09-29 | 2016-11-16 | Secretary Of State For Health The | Assay for distinguishing between sepsis and systemic inflammatory response syndrome |
| CA3049582A1 (en) * | 2017-01-08 | 2018-07-12 | The Henry M. Jackson Foundation For The Advancement Of Military Medicine, Inc. | Systems and methods for using supervised learning to predict subject-specific bacteremia outcomes |
| EP3477305A1 (en) | 2017-10-25 | 2019-05-01 | Universität Heidelberg | Delta-like ligand 1 for diagnosing severe infections |
| US10433191B2 (en) * | 2018-01-08 | 2019-10-01 | Cisco Technology, Inc. | Channel management in a virtual access point (VAP) |
| US11538566B2 (en) * | 2018-05-23 | 2022-12-27 | Beckman Coulter, Inc. | Sample analysis with test determination based on identified condition |
| WO2020072823A1 (en) * | 2018-10-04 | 2020-04-09 | Armata Pharmaceuticals, Inc. | Bacteriophage treatment and reduction in inflammatory response |
| WO2020096984A1 (en) * | 2018-11-05 | 2020-05-14 | Institute For Systems Biology | Sepsis biomarker panels and methods of use |
| US11104946B2 (en) | 2019-04-23 | 2021-08-31 | Inflammatix, Inc. | DNA sequences related to diagnosis and treatment of systemic inflammatory response syndrome |
| CN112391460A (zh) * | 2019-08-19 | 2021-02-23 | 四川生美思达生物科技有限公司 | 用于脓毒症的生物标志物组及脓毒症判断方法和试剂盒 |
| CA3198897A1 (en) | 2019-12-13 | 2021-03-19 | Autonomous Medical Devices Inc. | Apparatus and method for point-of-care, rapid, field-deployable diagnostic testing of covid-19, viruses, antibodies and markers |
| KR102281512B1 (ko) * | 2020-03-26 | 2021-07-23 | 경북대학교 산학협력단 | TGFBIp의 아세틸화 수준을 측정하는 제제를 포함하는 염증성 질환 진단용 조성물 |
| CN111563891B (zh) * | 2020-05-09 | 2023-09-26 | 吾征智能技术(北京)有限公司 | 基于颜色认知的疾病预测系统 |
| FR3112211A1 (fr) * | 2020-07-06 | 2022-01-07 | bioMérieux | Procédé pour déterminer le risque de survenue d’une infection associée aux soins chez un patient |
| FR3112207A1 (fr) * | 2020-07-06 | 2022-01-07 | bioMérieux | Procédé pour déterminer le risque de survenue d’une infection associée aux soins chez un patient |
| FR3112209A1 (fr) * | 2020-07-06 | 2022-01-07 | bioMérieux | Procédé pour déterminer le risque de complication chez un patient |
| CN111721941B (zh) * | 2020-07-17 | 2023-08-25 | 南方科技大学 | 一种用于判断脓毒症感染情况的装置及其应用 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103649329A (zh) * | 2010-11-26 | 2014-03-19 | ImmuneXpress有限公司 | 诊断和/或筛选剂及其用途 |
| WO2014201516A2 (en) * | 2013-06-20 | 2014-12-24 | Immunexpress Pty Ltd | Biomarker identification |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE102004009952B4 (de) * | 2004-03-01 | 2011-06-01 | Sirs-Lab Gmbh | Verfahren zur Erkennung von Sepsis |
| SG119251A1 (en) | 2004-08-04 | 2006-02-28 | Agency Science Tech & Res | Methods |
| JP2008538007A (ja) | 2005-04-15 | 2008-10-02 | ベクトン,ディッキンソン アンド カンパニー | 敗血症の診断 |
| DE102005050933A1 (de) | 2005-10-21 | 2007-04-26 | Justus-Liebig-Universität Giessen | Erfindung betreffend Expressionsprofile zur Vorhersage von septischen Zuständen |
| US20150038351A1 (en) | 2012-09-11 | 2015-02-05 | The Regents Of The University Of California | Gene Signatures That Predispose Or Protect Individuals From Low-Dose Radiation Induced Breast Cancer Or Are Associated with Disease-Free Survival |
-
2016
- 2016-03-12 WO PCT/US2016/022233 patent/WO2016145426A1/en active Application Filing
- 2016-03-12 AU AU2016228508A patent/AU2016228508A1/en not_active Abandoned
- 2016-03-12 CA CA2977422A patent/CA2977422A1/en active Pending
- 2016-03-12 IL IL297699A patent/IL297699A/en unknown
- 2016-03-12 CN CN201680023601.1A patent/CN107532214B/zh active Active
- 2016-03-12 EP EP16762690.2A patent/EP3268499B1/en active Active
- 2016-03-12 US US15/526,306 patent/US10533224B2/en active Active
- 2016-03-12 JP JP2017546801A patent/JP6995622B2/ja active Active
-
2017
- 2017-08-30 IL IL254229A patent/IL254229B/en unknown
-
2019
- 2019-11-20 US US16/689,926 patent/US20200131577A1/en not_active Abandoned
-
2021
- 2021-09-30 IL IL286853A patent/IL286853B2/en unknown
- 2021-12-15 JP JP2021203291A patent/JP2022058359A/ja active Pending
-
2022
- 2022-05-18 AU AU2022203348A patent/AU2022203348A1/en active Pending
-
2023
- 2023-03-08 US US18/180,833 patent/US20230227911A1/en active Pending
- 2023-06-30 JP JP2023107866A patent/JP2023138990A/ja active Pending
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103649329A (zh) * | 2010-11-26 | 2014-03-19 | ImmuneXpress有限公司 | 诊断和/或筛选剂及其用途 |
| WO2014201516A2 (en) * | 2013-06-20 | 2014-12-24 | Immunexpress Pty Ltd | Biomarker identification |
Non-Patent Citations (1)
| Title |
|---|
| Identification of candidate genes associated with salivary adenoid cystic carcinomas using combined comparative genomic hybridization and oligonucleotide microarray analyses;Atsushi Kasamatsu,et al;《The International Journal of Biochemistry & Cell Biology》;20051231;1869–1880页 * |
Also Published As
| Publication number | Publication date |
|---|---|
| US20200131577A1 (en) | 2020-04-30 |
| JP2023138990A (ja) | 2023-10-03 |
| EP3268499A4 (en) | 2018-09-05 |
| IL254229A0 (en) | 2017-10-31 |
| CA2977422A1 (en) | 2016-09-15 |
| JP6995622B2 (ja) | 2022-01-14 |
| IL286853B2 (en) | 2023-04-01 |
| EP3268499B1 (en) | 2020-05-13 |
| WO2016145426A1 (en) | 2016-09-15 |
| US20180291449A1 (en) | 2018-10-11 |
| IL254229B (en) | 2021-10-31 |
| IL297699A (en) | 2022-12-01 |
| JP2018514189A (ja) | 2018-06-07 |
| AU2022203348A1 (en) | 2022-06-09 |
| US20230227911A1 (en) | 2023-07-20 |
| CN107532214A (zh) | 2018-01-02 |
| WO2016145426A8 (en) | 2017-04-06 |
| IL286853B (en) | 2022-12-01 |
| US10533224B2 (en) | 2020-01-14 |
| EP3268499A1 (en) | 2018-01-17 |
| JP2022058359A (ja) | 2022-04-12 |
| IL286853A (en) | 2021-10-31 |
| AU2016228508A1 (en) | 2017-09-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN107532214B (zh) | 用于败血症诊断的方法 | |
| US20210348233A1 (en) | Methods for Diagnosis of Tuberculosis | |
| US20110183856A1 (en) | Diagnosis and Prognosis of Infectious Disease Clinical Phenotypes and other Physiologic States Using Host Gene Expression Biomarkers In Blood | |
| US20220251647A1 (en) | Gene expression signatures useful to predict or diagnose sepsis and methods of using the same | |
| JP2019520042A (ja) | 細菌感染及びウイルス感染を診断するための方法 | |
| US20160244834A1 (en) | Sepsis biomarkers and uses thereof | |
| GB2527164A (en) | Apparatus, kits and methods for the prediction of onset of sepsis | |
| CN109451744B (zh) | 用于危重患者的死亡预后的生物标志物 | |
| WO2015179777A2 (en) | Gene expression profiles associated with sub-clinical kidney transplant rejection | |
| AU2021221905A1 (en) | Gene expression profiles associated with sub-clinical kidney transplant rejection | |
| WO2020096796A1 (en) | Method for predicting severe dengue |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant |