WO2008026577A1 - Procédé de production d'un dérivé de phénoxypyridine - Google Patents
Procédé de production d'un dérivé de phénoxypyridine Download PDFInfo
- Publication number
- WO2008026577A1 WO2008026577A1 PCT/JP2007/066635 JP2007066635W WO2008026577A1 WO 2008026577 A1 WO2008026577 A1 WO 2008026577A1 JP 2007066635 W JP2007066635 W JP 2007066635W WO 2008026577 A1 WO2008026577 A1 WO 2008026577A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- azetidine
- piperidine
- methyl
- dimethylamino
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/72—Nitrogen atoms
- C07D213/73—Unsubstituted amino or imino radicals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/04—Antineoplastic agents specific for metastasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/72—Nitrogen atoms
- C07D213/75—Amino or imino radicals, acylated by carboxylic or carbonic acids, or by sulfur or nitrogen analogues thereof, e.g. carbamates
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/78—Carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/81—Amides; Imides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
Definitions
- the present invention relates to an antitumor agent having a hepatocyte growth factor receptor (hereinafter abbreviated as “HGFR”) inhibitory action, antitumor action, angiogenesis inhibitory action, cancer metastasis inhibiting action, etc.
- HGFR hepatocyte growth factor receptor
- the present invention relates to a method for producing a phenoxypyridine derivative (hereinafter abbreviated as “the present compound”) useful as a cancer metastasis inhibitor, and a production intermediate in the production method.
- HGFR expressed in these tumor cells is constitutively or stimulated by hepatocyte growth factor (hereinafter abbreviated as “HGF”) to cause tyrosine kinase autophosphorylation in the intracellular region. It is thought to be involved in cancer malignancy (abnormal growth, invasion or increased metastatic potential).
- HGF hepatocyte growth factor
- HGFR is also expressed in vascular endothelial cells, and it has been reported that HGF stimulates HGFR and promotes proliferation and migration of vascular endothelial cells, and is therefore involved in tumor angiogenesis. (Non-patent document 2).
- Non-patent Documents 3 and 4 Furthermore, it has been reported that blocking the NK4 force HGF—HGFR signal, which is an HGF antagonist peptide, suppresses invasion of cancer cells and inhibits tumor angiogenesis.
- a compound having an HGFR inhibitory action is expected to be useful as an antitumor agent, an angiogenesis inhibitor or a cancer metastasis inhibitor.
- Patent Document 1 Pamphlet of International Publication No. 2005/082855
- Non-Patent Document 1 Oncology Reports, 5, 1013-1024 (1998)
- Non-Patent Document 2 Advances in Cancer Research, 67, 257-279 (1995)
- Non-patent literature 3 British Journal of Cancer, 84, 864-873 (2001)
- Non-Patent Document 4 Cancer Sci., 94, 321-327 (2003)
- An object of the present invention is to provide a method for producing a phenoxypyridine derivative having an HGFR inhibitory action, an antitumor action, an angiogenesis inhibitory action, a cancer metastasis inhibiting action, and the like, and a production intermediate in the production method To find.
- the present invention provides the following [1] to [21].
- R 1 has 1) a substituent selected from the following substituent group a! /, May! /, Azetidine 1-yl group, and 2) has a substituent selected from the following substituent group a. ! /, May! /, Pyrrolidine 1-yl group, 3) has a substituent selected from the following substituent group a! /, May! /, Piperidine 1-yl group, 4) having a substituent selected from the following substituent group a! /, May! /, Piperazine 1-yl group, 5) having a substituent selected from the following substituent group a! / But!
- R ub means n-propyl group, n-butyl group, pyrrolidine-3-yl group, piperidine-3-yl group, piperidine-4-yl group or tetrahydropyran-4-yl group .
- Rub may have a substituent selected from the following substituent group b. ) Means a group represented by
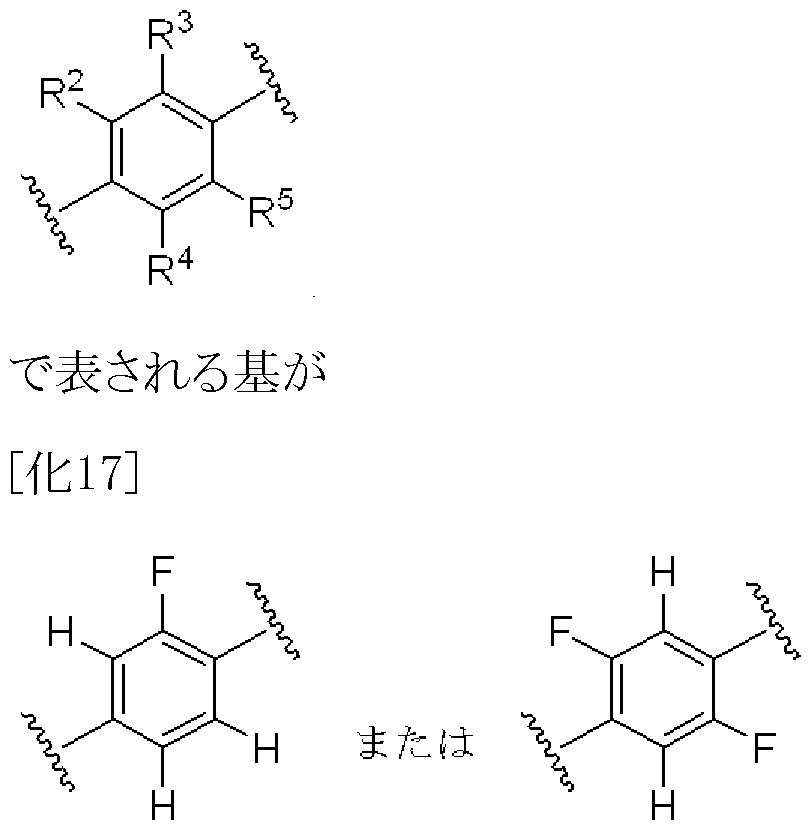
- R 2 , R 4 and R 5 are the same or different and each represents a hydrogen atom or a fluorine atom.
- hydroxyl group consists of a hydroxyl group, a dimethylaminoacetoxy group, a methyl group, an ethyl group, a dimethylamino group, a azetidinyl group, a pyrrolidinyl group, a piperidinyl group, and a piperazinyl group.
- the above groups are a hydroxyl group, a methyl group , May have a dimethylamino group, azetidyl group, pyrrolidinyl group or piperidinyl group.
- R ° means a hydrogen atom or a fluorine atom.
- R 7 represents (1) a halogen atom, (2) a hydroxyl group, (3) a nitro group on the C alkyl group or benzene ring,
- R 2 , R 4 and R 5 have the same meanings as defined in [1].
- R 7 has the same meaning as defined in [2].
- Ar means a phenyl group which may have one or two substituents selected from a halogen atom, a methyl group, a methoxy group, a nitro group, a cyano group and a trifluoromethyl group.
- HN R Ua R llb (wherein R lla and R l lb are defined in claim 1) Means the same.)
- R 1 is 4- [2 (dimethylamino) ethyl] piperazine 1-yl group, 4 pyrrolidine 1-ylpiperidine 1-yl group, 4 [(dimethylamino) methyl] piperidine 1-yl group, 4 Azetidine 1-ylpiperidine 1-yl group, 4 (dimethylamino) -azetidine 1-yl] piperidine 1-yl group, 4 4-methylbiperazine 1-yl) piperidine 1-yl group, 4 1-methylbiperidine 4 ) Piperazine 1-yl group, 4 1 Methylazetidine-3-yl) Piperazine 1-yl group, 4 (dimethyla group, 4 (pyrrolidine-1-ylmethyl) piperidine-1-yl group, (3 S) - (dimethyl Amino) pyrrolidin - 1-I group, (3R) - 3- (Jimechiruamino) pyrrolidine - 1-I group, Azechijin 1
- R 1 has 1) a substituent selected from the following substituent group a! /, May! /, Azetidine 1-yl group, and 2) has a substituent selected from the following substituent group a. ! /, May! /, Pyrrolidine 1-yl group, 3) has a substituent selected from the following substituent group a! /, May! /, Piperidine 1-yl group, 4) having a substituent selected from the following substituent group a! /, May! /, Piperazine 1-yl group, 5) having a substituent selected from the following substituent group a!
- R Ua means a hydrogen atom or a methyl group.
- R Ub means an n-propyl group, an n-butyl group, a pyrrolidine-3-yl group, a piperidine-3-yl group, a piperidine-4-inole group, or a tetrahydropyran-4-yl group.
- R llb may have a substituent selected from the following substituent group b. ) Means a group represented by
- R 2 , R 4 and R 5 are the same or different and each represents a hydrogen atom or a fluorine atom o
- R 71 is a hydrogen atom, C alkyl group or benzene ring on a (1) halogen atom, (2) water
- hydroxyl group consists of a hydroxyl group, a dimethylaminoacetoxy group, a methyl group, an ethyl group, a dimethylamino group, a azetidinyl group, a pyrrolidinyl group, a piperidinyl group, and a piperazinyl group.
- the above groups are a hydroxyl group, a methyl group , May have a dimethylamino group, azetidyl group, pyrrolidinyl group or piperidinyl group.
- R 7 represents (1) a halogen atom, (2) a hydroxyl group, (3) a nitro group, (4) on the C alkyl group or benzene ring.
- Cyan group (5) trifluoromethyl group, (6) C alkyl group, (7) C alkoxy group, (8 ) Amino groups, (9) mono-C alkylamino groups and (10) di-C alkylamino groups
- Means a benzyl group optionally having 1 or 2 substituents selected from Ar means a phenyl group which may have one or two substituents selected from a halogen atom, a methyl group, a methoxy group, a nitro group, a cyano group and a trifluoromethyl group. Or a salt thereof;
- R 2 R 3 R 4 and R 5 have the same meaning as defined in [13].
- R 7 has the same meaning as defined in [14]. Or a salt thereof;
- R 1 is 4- [2 (dimethylamino) ethyl] piperazine 1- yl group, 4 pyrrolidine Group, 4 [(dimethylamino
- the present invention provides a method for producing a phenoxypyridine derivative having an HGFR inhibitory action, an antitumor action, an angiogenesis inhibitory action, a cancer metastasis inhibitory action and the like suitable for industrial mass synthesis.
- the present invention can provide a production intermediate that can be used in the production method.
- FIG. 1 is a diagram showing a powder X-ray diffraction pattern of the crystal obtained in Example 9 (Method 3).
- the structural formula of a compound may represent a certain isomer for convenience.
- the present invention includes all geometric isomers generated in the structure of a compound, optical isomers based on asymmetric carbons, It includes isomers such as stereoisomers and tautomers and mixtures of isomers, and is not limited to the description of the formula for convenience, and may be either one isomer or a mixture. Therefore, the compound of the present invention has an asymmetric carbon atom in the molecule and may have an optically active substance and a racemate. In the present invention, the compound is not limited to one, but includes both.
- any one of crystal forms may be a single crystal form or a mixture of crystal forms.
- the compounds according to the present invention include anhydrides and hydrates.
- the “salt” is not particularly limited as long as it forms a salt with the compound according to the present invention.
- a salt with an inorganic acid a salt with an organic acid, a salt with an inorganic base, an organic base And salts with acidic or basic amino acids.
- the salt with inorganic acid include salts with hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid and the like.
- salts with organic acids include acetic acid, succinic acid, fumaric acid, maleic acid, tartaric acid, succinic acid, lactic acid, stearic acid, benzoic acid, methanesulfonic acid, ethanesulfonic acid, p-toluenesulfonic acid. And salt.
- Preferable examples of the salt with an inorganic base include alkali metal salts such as sodium salt and potassium salt, alkaline earth metal salts such as calcium salt and magnesium salt, aluminum salt and ammonium salt.
- Preferable examples of salts with organic bases include salts with jetylamine, diethanolamine, medamine, N, N dibenzylethylenediamine and the like.
- Preferable examples of the salt with acidic amino acid include salts with aspartic acid, glutamic acid and the like.
- Preferable examples of the salt with basic amino acid include salts with arginine, lysine, ornithine and the like.
- Halogen atom means a fluorine atom, a chlorine atom, a bromine atom or an iodine atom.
- C alkyl group means a linear or branched alkyl having 1 to 6 carbon atoms.
- Specific examples include methyl group, ethyl group, 1 propyl group (n propyl group), 2-propyl group (i propyl group), 2-methyl-1 propyl group (i butyl group), 2-methyl group.
- Examples thereof include a tert-2-propyl group (t-butyl group), a 1-butyl group (n-butyl group), and a 2-butyl group (s-butyl group).
- C alkoxy group means that an oxygen atom is bonded to the terminal of the above-defined “C alkyl group”.
- 1-6 1 alkyl group means a group substituted with a methylamino group, ethylamino 6
- 1-propylamino group (n-propylamino group), 2-propylamino group (i-propylamino group), 2-methyl-1 propylamino group (i-butylamino group), 2-methyl-2-pro
- Examples include a pyramino group (t-butylamino group), a 1-butylamino group (n-butylamino group), and a 2-butylamino group (s-butylamino group).
- Gae C alkylamino group means that two hydrogen atoms in an amino group are identical to each other.
- the "condensing agent" in the above [1] and [6] is 4- (4,6-dimethoxy [1.3.5] triazin-2-yl) -4 -metnylmorpholinium cnionde hydrate 2_cnloro_4, o_dimethoxy_l, ⁇ 5 , 5_tnazine, 2,4,6_trichloro_l, 3,5_triazine, dicyclohexyl carboaiimide (DCC), l_ethyto 3, (3'_dimeth ylamino) carbodiimide HCl salt (EDC or WSC HCl), 0- (lH_benzotiazo ⁇ 1-yl)-N , N, N ', N' ⁇ tetramethy luronium hexafluorophosphate (HBTU), O- (1H-benzotiazoto 1-yl)-A force that means ⁇ , ⁇ , ⁇ ',
- the "base” in the above [3] is a power meaning potassium carbonate, sodium carbonate, pyridine, triethylamine, diisopropylethylamine or the like, preferably potassium carbonate.
- the "salt" of "amamine or a salt thereof” in [3] is not particularly limited as long as it forms a salt with ammine, and examples thereof include salts with hydrochloric acid, acetic acid, trifluoroacetic acid and the like. It is done.
- the “base” in the above [4] means pyridine, triethylamine, diisopropylethylamine, potassium carbonate, sodium carbonate and the like, preferably pyridine.
- the "Hoffman rearrangement agent" in [5] means iodine diacetate, ditrifluoroacetate benzene, sodium hypochlorite, potassium hypobromite, bromine, iodine, etc., preferably With diacetate diacetate and trifluoroacetate odobenzene is there.
- no, rogenating agent means thionyl chloride, oxalyl chloride, phosphorus trichloride, phosphorus oxychloride, phosphorus pentachloride, etc., preferably thionyl chloride.
- the crystals of phenyl) 1 N, 1 (4-fluorophenyl) cyclopropane 1,1-dicarboxamide have a diffraction angle (2 ⁇ ⁇ 0.2 °) 6.3 °, 12. 3 ° in powder X-ray diffraction.
- the value of the diffraction angle is within a range of about ⁇ 0.2 °. It should be understood as including numerical values. Therefore, a crystal in which the diffraction angle in the powder X-ray diffraction is exactly the same within the error range of ⁇ 0.2 ° is included in the present invention.
- R 1 has 1) a substituent selected from the following substituent group a! /, May! /, Azetidine 1-yl group, and 2) has a substituent selected from the following substituent group a. /, May! /, Pyrrolidine 1-yl group, 3) having a substituent selected from the following substituent group a! /, May! /, Piperidine 1-yl group, 4 ) Having a substituent selected from the following substituent group a! /, May! /, Piperazine 1-yl group, 5) having a substituent selected from the following substituent group a!
- R Ua means a hydrogen atom or a methyl group.
- R ub means n-propyl group, n-butyl group, pyrrolidine-3-yl group, piperidine-3-yl group, piperidine-4-yl group or tetrahydropyran-4-yl group .
- R Ub may have a substituent selected from the following substituent group b.
- R 1 Means a group represented by Preferable examples of R 1 include 4 [2 (dimethylamino) ethyl] piperazine 1-yl group, 4 pyrrolidine 1-ylpiperidine 1-yl group, 4 [(dimethylamino) methyl] piperidine 1-yl group, 4 azetidine 1-ylpiperidine 1-yl group, 4 1 [3- (dimethylamino) azetidine 1-yl] piperidine 1-yl group, 4 1- (4-methylbiperazine 1-yl) piperidine 1-yl group, 4 1-methylbiperidine 4-yl) piperazine 1-yl group, 4 (1-methylazetidine-3-ynole) piperazine 1-yl group, 4 (dimethylamino) piperidine 1-yl group, 4 (azetidine 1-ylmethyl) piperidine-1 —Yl group, 4-(pyrrolidine-1-ylmethyl) piperidine-1-yl group, (3S) -3
- R 1 More preferable examples of R 1 include 4- (4-methylbiperazine-1-yl) piperidine-1-yl group, 3-hydroxyazetidine-1-yl group, (3R) -3 —Hydroxypyrrolidine 1-yl group, (3S) 3-hydroxypyrrolidine 1-yl group or methyl (1-methylpiperidine 4-yl) amino group.
- Substituent group a means a group consisting of a hydroxyl group, a dimethylaminoacetoxy group, a methyl group, an ethyl group, a dimethylamino group, an azetidyl group, a pyrrolidinyl group, a piperidinyl group, and a piperazinyl group.
- each group described in the substituent group a may have a hydroxyl group, a methyl group, a dimethylamino group, an azetidyl group, a pyrrolidinyl group, or a piperidinyl group. Les.
- Substituent group b means a group consisting of methyl group, ethyl group, n-propyl group, acetyl group, dimethylamino group, jetylamino group, azetidyl group, pyrrolidinyl group and piperazinyl group.
- each group described in the substituent group b may have a methyl group or a dimethylamino group.
- R 2 , R 4 and R 5 are the same or different and each represents a hydrogen atom or a fluorine atom.
- R 2 , R 4 and R 5 may be any one of 1) when they are all hydrogen atoms, 2) when they are all fluorine atoms, or 3) when they are hydrogen atoms or fluorine atoms, but preferably R 2 , R 3 , R 4 and R 5 are 2 or 3 hydrogen atoms.
- R 6 means a hydrogen atom or a fluorine atom.
- R 6 is a fluorine atom.
- R 7 represents (1) a halogen atom, (2) a hydroxyl group on the C alkyl group or benzene ring, (3)
- 1-6 1-6 means a benzyl group optionally having 1 or 2 substituents selected from a killiamino group.
- R 7 is a benzyl group.
- R 71 is a hydrogen atom, C alkyl group or (1) halogen atom on the benzene ring, (2)
- R 71 include a hydrogen atom or a benzyl group.
- Ar represents one or two substituents selected from a halogen atom, a methyl group, a methoxy group, a nitro group, a cyano group, and a trifluoromethyl group! /, May! /, A phenyl group .
- Ar is a phenyl group.
- This step is a step for producing compound (VIII) by reacting compound (IX) with compound (X) in the presence of a halogenating agent or a condensing agent.
- compound (IX) a compound described in the examples below, a known compound, a commercially available compound, or a compound that can be easily produced by a person skilled in the art from a commercially available compound can be used. .
- compound (X) a compound described in the examples below, a known compound, a commercially available compound, or a compound that can be easily produced from a commercially available compound by a method commonly used by those skilled in the art can be used. .
- the solvent used in this step is not particularly limited as long as it dissolves the starting materials to some extent and does not inhibit the reaction.
- tetrahydrofuran 1,2-dimethoxyethane, tert- Ether solvents such as butyl methyl ether, cyclopentinolemethinore ethere, jetinoree ethereol, diisopropinoreatenore, dibutinolee ethereol, dicyclopentenore ether, aromatic hydrocarbons such as benzene and toluene Solvents, aliphatic hydrocarbon solvents such as heptane and hexane, N, N-dimethylformamide, N-methyl-2-pyrrolidone or a mixed solvent thereof can be used, and tetrahydrofuran is preferred.
- tert- Ether solvents such as butyl methyl ether, cyclopentinolemethinore ethere, jetinoree ethereol, diisopropinoreatenore, dibutinolee ethereol, dicyclopentenore ether,
- the halogenating agent means thionyl chloride, oxalyl chloride, phosphorus trichloride, phosphorus oxychloride, phosphorus pentachloride, etc., preferably thionyl chloride.
- Yarn combinations are 4_ (4,6_dimethoxy [1.3.ri] triazin_2_yl) _4_methylmorpholinium chloride hydrate 2_chloro_4,6_dimethoxy_l, 3,5_tnazine, 2,4,6_trichioro_l, ⁇ 5,5_tnazine, die yclohexyl carbodiimide (DCC), l_y 3, (3'_dimethylamino) carbodiimide HCl salt (EDC or WSC HCl), 0_ (lH_benzotiazo ⁇ l-yl) —N, N, N ', N, — tetramethyluronium hexafl uorophosphate (HBTU), 0— (lH—benzotiazo ⁇ 1—yl) — ⁇ , ⁇ , ⁇ ', ⁇ ' — A force that means tetramethyluronium tetrafluoroborate
- the reaction temperature usually varies depending on the starting material, solvent, and other reagents used in the reaction, and is preferably 0 ° C to 50 ° C (internal temperature in the reaction vessel), more preferably 0 ° C. -30 ° C (internal temperature in the reaction vessel).
- the reaction time usually varies depending on the starting material, solvent, other reagents used in the reaction, and the reaction temperature.
- the reaction solution is stirred at the above reaction temperature for 1 to 48 hours. It is more preferable to stir for 4 to 24 hours.
- Compound (X) can be used in an amount of 1.0 to 3.0 molar equivalents relative to compound (IX), but preferably 1.0 to 1.3 molar equivalents. .
- the halogenating agent can be used at 1.0 to 2.0 molar equivalents relative to compound (IX), but preferably 1.1 molar equivalents.
- the condensing agent is a force S that can be used in a molar amount of 1.0 to 3.0 times that of Compound (IX), preferably 1 .;! To 1.3 times a molar equivalent can be used. .
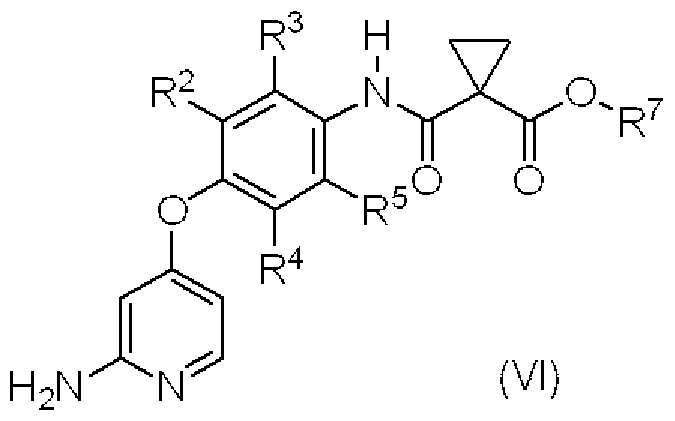
- This step is a step for producing compound (VI) by reacting compound (VIII) with a Hoffman rearrangement agent.
- the solvent used in this step is not particularly limited as long as it dissolves the starting materials to some extent and does not inhibit the reaction.
- N, N dimethylformamide, N, N dimethyla Cetamide, dimethyl sulfoxide, N-methyl 2-pyrrolidone and the like can be used, and N, N-dimethylformamide and N-methyl 2-pyrrolidone are preferred.
- Hoffman rearrangement agent means iodobenzene diacetate, iodobenzene ditrifluoroacetate, sodium hypochlorite, potassium hypobromite, bromine, iodine, etc. is there.
- the reaction temperature usually varies depending on the starting material, solvent, and other reagents used in the reaction, and is preferably 10 ° C to 50 ° C (internal temperature in the reaction vessel), more preferably 20 ° C. ⁇ 30 ° C (internal temperature in the reaction vessel).
- the reaction time usually varies depending on the starting material, solvent, other reagents used in the reaction, and the reaction temperature.
- the reaction solution is stirred at the above reaction temperature;! ⁇ 24 hours It is preferable to stir for 3 to 5 hours.
- the Hoffman rearrangement agent has a force S that can be used at 1.0 to 3.0-fold molar equivalents relative to compound (VIII), preferably 1.0 to; it can.
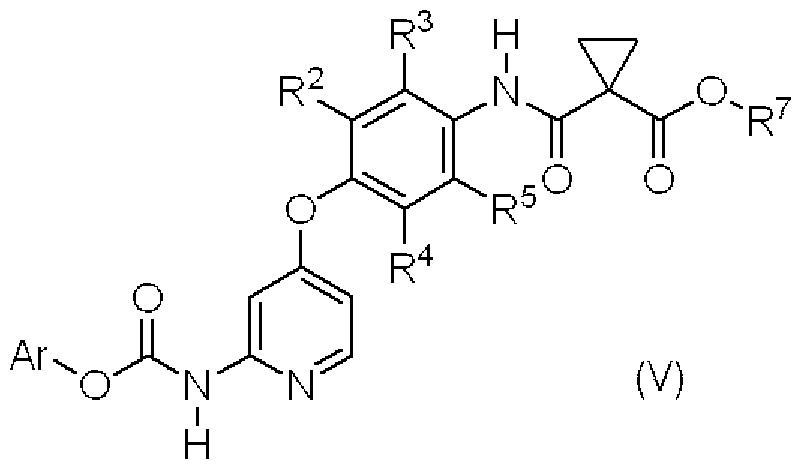
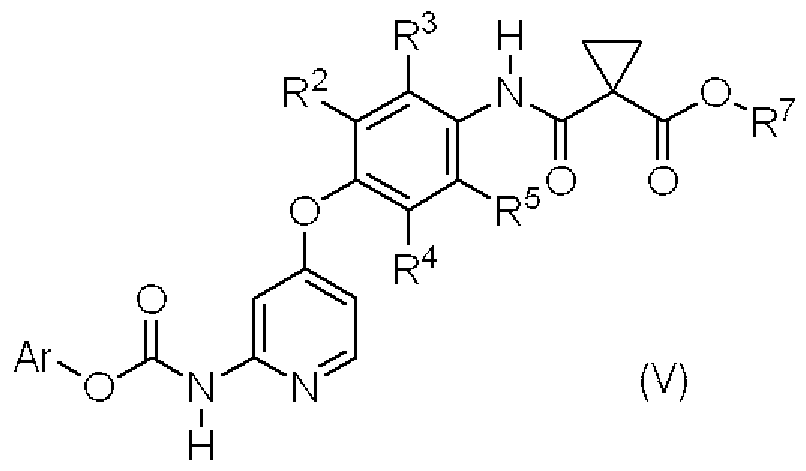
- This step is a step for producing the compound (V) by reacting the compound (VI) with the compound (VII) in the presence of a base.
- the compound (VII) As the compound (VII), a known compound, a commercially available compound, or a compound that can be easily purchased can be used.
- the solvent used in this step is one that dissolves the starting materials to some extent and is reactive. As long as it does not inhibit the reaction, there is no particular limitation, but for example, tetrahydrofuran, 1,2-dimethoxyethane, tert butyl methyl ether, cyclopentinoremethinoreatenore, jetinoreteinole, diisopropinolei
- ether solvents such as tenole, dibutino eletenole, dicyclopentinole ether, aromatic hydrocarbon solvents such as benzene and toluene, fatty hydrocarbon solvents such as heptane and hexane, acetonitrile or a mixed solvent thereof.
- a mixed solvent of tetrahydrofuran and acetonitrile is preferable.
- the base is a force S meaning pyridine, triethylamine, diisopropylethylamine, potassium carbonate, sodium carbonate or the like, and preferably pyridine.
- the reaction temperature usually varies depending on the starting materials, solvent, and other reagents used in the reaction, and is preferably 10 ° C to 50 ° C (internal temperature in the reaction vessel), more preferably 0 ° C. ⁇ 30 ° C (inner temperature in reaction vessel).
- the reaction time usually varies depending on the starting material, solvent, other reagents used in the reaction, and the reaction temperature.
- the reaction solution is stirred at the above reaction temperature;! ⁇ 24 hours It is preferable to stir for 2 to 5 hours.
- Compound (VII) can be used in a molar amount equivalent to 1.0 to 3.0 times that of Compound (VI) S, preferably 1.; it can.
- the base can be used in a force S capable of using a 1.0 to 3.0-fold molar equivalent to the compound (VI), preferably 1.;! To 2.0-fold molar equivalent.
- This step is a step for producing compound (IV) or a salt thereof by reacting compound (V) with an appropriate amine or a salt thereof in the presence or absence of a base.
- an appropriate amine or a salt thereof in the presence or absence of a base.
- amine compounds described in the examples below, known compounds, compounds that can be purchased, or compounds that can be easily produced by commercially available methods by those skilled in the art can be used.
- the solvent used in this step is not particularly limited as long as it dissolves the starting materials to some extent and does not inhibit the reaction.
- N, N dimethylformamide, N methyl 2-pyrrolidone , N, N dimethylacetamide, dimethylsulfoxy N methyl 2-pyrrolidone is preferred.
- the base means potassium carbonate, sodium carbonate, pyridine, triethylamine, diisopropinoethylamine and the like, and potassium carbonate is preferable.
- the reaction temperature usually varies depending on the starting material, solvent, and other reagents used in the reaction, and is preferably 10 ° C to 100 ° C (internal temperature in the reaction vessel), more preferably 20 ° C to 50 ° C (internal temperature in reaction container).
- the reaction time usually varies depending on the starting material, solvent, other reagents used in the reaction, and the reaction temperature.
- the reaction solution is stirred at the above reaction temperature;! ⁇ 24 hours It is more preferable to stir for! ⁇ 4 hours.
- Amine or its salt has a force S that can be used at 1.0 to 3.0 molar equivalents relative to compound (V), preferably 1.; Can be used.
- the base can be used in an amount of 1.0 to 3.0 molar equivalents relative to compound (V). Preferably, 1.;! To 1.3 molar equivalents can be used.
- This step is a step for producing compound (II) or a salt thereof by hydrolysis or catalytic hydrogenation of compound (IV) or a salt thereof.
- Compound (II) or a salt thereof can be produced by hydrolyzing compound (IV) or a salt thereof in the presence of an acid or a base.
- the solvent used in this step is not particularly limited as long as it dissolves the starting materials to some extent and does not inhibit the reaction.
- alcohol solvents such as methanol, ethanol, propanol, and butanol , Tetrahydrofuran, 1,2-Dimetho carten, tert Butinolemethinoreatenore, Cyclopentinoremethinoleatenore, Jetinore Etenore, Diisopropinoreatenore, Dibutinoreethenore, Dicyclopentenoreatenore
- a solvent, water or a mixed solvent thereof can be used, and preferably , A mixed solvent of water and methanol, ethanol or tetrahydrofuran
- the acid means hydrochloric acid or the like.
- the base means sodium hydroxide, potassium hydroxide, potassium carbonate, sodium carbonate and the like.
- the reaction temperature usually varies depending on the starting material, solvent, and other reagents used in the reaction, and is preferably 0 ° C to 80 ° C (internal temperature in the reaction vessel), more preferably 30 ° C. ⁇ 50 ° C (internal temperature in the reaction vessel).
- the reaction time usually varies depending on the starting material, solvent, other reagents used in the reaction, and the reaction temperature.
- the reaction solution is stirred at the above reaction temperature;! ⁇ 24 hours It is preferable to stir for 2 to 5 hours.
- the acid can be used in a force S that can be used at 1.0 to 5.0 molar equivalents relative to compound (IV), and preferably 1.0 to 2.0 molar equivalents.
- the base can be used in an amount of 1.0 to 5.0 molar equivalents relative to compound (IV). Preferably, 1.0 to 2.0 molar equivalents can be used.
- compound (II) or a salt thereof is produced by catalytic hydrogenation of compound (IV) or a salt thereof in the presence of a reduction catalyst in a hydrogen atmosphere.
- the solvent used in this step is not particularly limited as long as it dissolves the starting materials to some extent and does not inhibit the reaction.
- alcohol solvents such as methanol, ethanol, propanol, and butanol are used.
- Ether solvents N, N dimethylformamide N-methyl 2-pyrrolidone, formic acid, water, or a mixed solvent thereof can be used, and preferably a mixed solvent of water, methanol, and tetrahydrofuran, a mixed solvent of water, ethanol, and tetrahydrofuran, or water and ethanol. It is a mixed solvent.
- the reduction catalyst means palladium carbon, palladium hydroxide, platinum oxide, Raney nickel, and the like, and is preferably a radium carbon.
- This process can be carried out in a hydrogen atmosphere of 0.1 IMPa (normal pressure) to 1. OMPa in a hydrogen atmosphere, and preferably in a hydrogen atmosphere of 0.1 MPa to 0.3 MPa.
- this step can be performed without using hydrogen gas.
- the reaction temperature usually varies depending on starting materials, solvents, and other reagents used in the reaction, and is preferably 0 ° C to 50 ° C (internal temperature in the reaction vessel), more preferably 20 ° C. ⁇ 30 ° C (internal temperature in the reaction vessel).
- the reaction time usually varies depending on the starting material, solvent, other reagents used in the reaction, and the reaction temperature.
- the reaction solution is stirred at the above reaction temperature for 1 to 48 hours. 3 to; more preferably, stirring for 18 hours.
- the reduction catalyst is a force capable of using 0.5 to 5 molar equivalents relative to compound (IV). Preferably, 0.5 to 1.5 molar equivalents can be used.
- This step is a step for producing compound (I) by reacting compound (II) or a salt thereof with compound (III) in the presence of a condensing agent, in the presence or absence of a base.
- the compound (III) As the compound (III), a known compound, a commercially available compound, or a compound that can be easily produced from a commercially available compound by a method commonly used by those skilled in the art can be used.
- the solvent used in this step is not particularly limited as long as it dissolves the starting materials to some extent and does not inhibit the reaction.
- tetrahydrofuran, 1,2-dimethoxyethane, tert butyl Ether solvents such as methyl ether, cyclopentinolemethinole ethere, jetinoleo ethere, diisopropinoleo ethere, dibutinoleo ethere, dicyclopentinole ether, alcohols such as ethanol, 1 propanol, 2-propanol Solvent, N, N dimethylformamide, N methyl-2-pyrrolidone, N, N dimethylacetamide, or a mixed solvent thereof, preferably a mixed solvent of tetrahydrofuran and N, N dimethylformamide, Or mixed solution of tetrahydrofuran and 2-propanol It is.
- Ito te gosei 1 J is 4_ (4,6_dimethoxy [1.3.5] triazin_2_yl) _4_methylmorpholinium chloride hydrate 2_chloro_4,6_dimethoxy_l, 3,5_triazine, 2,4,6_trichloro_l, 3,5_triazine, die yclohexyl carbodiimide (DCC), 1 -ethyl-3, (3 '-dimethylamino) carbodiimide HCl salt (EDC or WSC HCl), 0_ ( lH_benzotiazo ⁇ l-yl) —N, N, N ′, N, — tetramethyluronium hexafl uorophosphate (HBTU), 0— (lH—benzotiazo ⁇ 1—yl) — ⁇ , ⁇ , ⁇ ', ⁇ '— tetramethyluronium t etrafluo
- the base means N methylmorpholine, pyridine, triethylamine, diisopropylethylamine, 1-methylimidazole, potassium carbonate, sodium carbonate and the like. Preferred is N methylmorpholine.
- the reaction temperature usually varies depending on the starting material, solvent, and other reagents used in the reaction, and is preferably 10 ° C to 50 ° C (internal temperature in the reaction vessel), more preferably 20 ° C. ⁇ 30 ° C (internal temperature in the reaction vessel).
- the reaction time usually varies depending on the starting material, solvent, other reagents used in the reaction, and the reaction temperature.
- the reaction solution is stirred at the above reaction temperature for 1 to 48 hours. 3 to; more preferably, stirring for 18 hours.
- Compound (III) can be used at 1.0 to 3.0-fold molar equivalents relative to Compound (II), but preferably 1.0 to 2.0-fold molar equivalents.
- the condensing agent can be used in an amount of 1.0 to 3.0 molar equivalents, preferably 1.0 to 2.0 molar equivalents, relative to compound (II).
- the base can be used in an amount of 1.0 to 10-fold molar equivalent relative to compound (II). Preferably, a 2.0 to 4.0-fold molar equivalent can be used.
- the aqueous layer was extracted with ethyl acetate.
- the organic layer was collected, washed successively with 1N aqueous sodium hydroxide solution and saturated brine, and dried over anhydrous sodium sulfate.
- the organic layer after drying was concentrated under reduced pressure to obtain the title compound (8.03 g, quantitative) as colorless crystals.
- the separated organic layer was washed with saturated aqueous sodium hydrogen carbonate solution (100 ml), water (100 ml) and saturated brine (100 ml), and then dried over sodium sulfate.
- the solvent was distilled off under reduced pressure to obtain the title compound (8.19 g, quantitative) as a colorless oil.
- lithium aluminum hydride (405 mg) was suspended in tetrahydrofuran (10 ml) with stirring and cooling in an ice-water bath, and then 1- (azetidine 1 yl) -2- (4 monobenzoic reaction solution was added to the reaction mixture. The mixture was stirred for 3 hours at ° C. After cooling the reaction solution to room temperature, water (0.4 ml), 5N aqueous sodium hydroxide solution (0.4 ml) and water (1.2 ml) were added and stirred for 13 hours. The insoluble liquid was filtered off through celite and washed with ethyl acetate (100 ml). The crude product of the title compound (1 ⁇ 287 mg) was obtained as a pale yellow oil by distillation under reduced pressure.
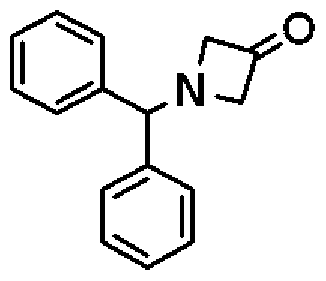
- Azetidine hydrochloride (326 mg) was added to a solution of benzohydrylazetidine 1-3-one (750 mg) in dichloromethane (12 ml), and the mixture was stirred at room temperature. To this was added sodium triacetoxyborohydride (1. Olg), and the mixture was stirred at room temperature for 25 hours. Sodium carbonate (until foaming subsided), water (50 ml), and ethyl acetate (100 ml) were added to the reaction solution. The organic layer was separated. This was washed with saturated brine and dried over anhydrous sodium sulfate. The organic layer after drying was concentrated under reduced pressure.
- Jetyl ether (15 ml) was added thereto to suspend the crystals. The crystals were collected by filtration and washed with jetyl ether. This was air-dried to obtain the title compound (4.20 g, 71.7%) as pale yellow crystals.
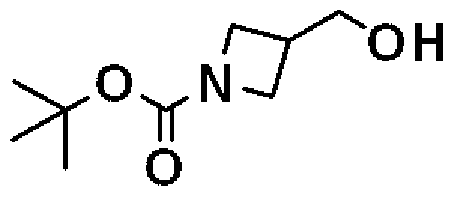
- Methyl azetidine-3-carboxylate hydrochloride crude product (equivalent to 1.93 g equivalent as a pure product) was dissolved in water (26 ml), sodium hydrogen carbonate (3.2 g) with ice bath cooling and stirring, then g t A solution of butyl dicarbonate (2.91 g) in tetrahydrofuran (13 ml) was added, and the mixture was stirred at the same temperature for 0.5 hr. The reaction was stirred at room temperature for 19.5 hours. Tetrahydrofuran in the reaction solution was distilled off and extracted with ethyl acetate. The organic layer was washed with saturated brine (70 ml) and dried over anhydrous sodium sulfate.
- the aqueous layer was extracted with ethyl acetate.
- the organic layers were combined, washed with saturated brine, and dried over anhydrous sodium sulfate.
- the target fraction was concentrated to obtain the title compound (1.80 g, 33.3%) as a colorless oil.
- the raw material fraction was concentrated and recovered (2.10 g, 42.0%).
- the partitioned organic layer was washed successively with 1N aqueous sodium hydroxide solution, water and saturated brine, and dried over anhydrous sodium sulfate.
- Ethyl acetate (2 ml) and tert butyl methyl ether (10 ml) were added to the residue (1 ⁇ 83 g) obtained by distilling off the solvent to precipitate crystals.
- the crystals were collected by filtration and dried by aeration to give the title compound (1.14 g 65%) as pale yellow crystals.
- Benzohydryl-3-azetidinecarboxyl acid (3.12 g) was suspended in tetrahydrofuran (60 ml) and cooled in an ice-ethanol bath under a nitrogen atmosphere. Triethylamine (1.96 ml) was added dropwise, and then a solution of ketyl carbonate (1.34 ml) in tetrahydrofuran (5 ml) was added dropwise over 20 minutes. After dropping, the mixture was stirred at the same temperature for 30 minutes. The reaction mixture was filtered, and the filtrate was washed with tetrahydrofuran (30 ml).
- 1,1-Cyclopropanedicarboxylic acid (5.02 g) was dissolved in tetrahydrofuran (50 ml) under a nitrogen atmosphere, and then triethylamine (5.38 ml) was added dropwise with stirring in an ice-water bath. After stirring at the same temperature for 30 minutes, thionyl chloride (2.82 ml) was added dropwise with cooling in an ice-water bath. After stirring at the same temperature for 30 minutes, a solution of benzyl alcohol (4.39 ml) in tetrahydrofuran (25 ml) was added while cooling in an ice-water bath, and the mixture was gradually warmed to room temperature and stirred overnight.
- 1,1-Cyclopropanedicarboxylic acid 50 g was dissolved in acetonitrile (500 ml) under a nitrogen atmosphere, and then N-methylimidazole (31 ml) was added dropwise with stirring in an ice-water bath. After stirring at the same temperature for 30 minutes, thionyl chloride (29 ml) was added dropwise. After stirring at the same temperature for 30 minutes, a mixed solution of benzyl alcohol (45.7 g) and N-methylimidazole (31 ml) was added while cooling in an ice-water bath, and the mixture was stirred at the same temperature for 6 hours. The reaction solution was adjusted to pH 8 by adding 2N aqueous sodium hydroxide solution (900 ml).
- aqueous solution was added tert-butyl methyl ether (500 ml) and stirred.
- the organic layer and the aqueous layer were separated, and the organic layer was extracted with 5% aqueous sodium hydrogen carbonate solution (200 ml).
- Ethyl acetate (1000 ml) was added and stirred for a while. The organic layer was separated, washed with saturated brine, and dried over anhydrous sodium sulfate.
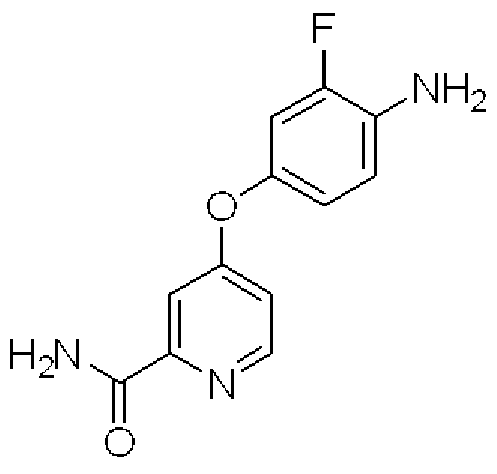
- tert-butoxy potassium (214 g) was dissolved in dimethyl sulfoxide (750 ml) and tetrahydrofuran (250 ml), and this solution was mixed with 4 amino-3 fluorophenol 1/2
- a solution of naphthalene 2,6 disulfonate (242 g) and 4-chlorodipyridine-2-carboxyamide (1 OOg) in dimethyl sulfoxide (1000 ml) was added dropwise with ice-cooling and stirring. After stirring for 30 minutes at room temperature, the mixture was stirred for 2 hours using an oil bath with an external temperature of 90 ° C.
- the reaction mixture was allowed to cool to room temperature, water (3,000 ml) was added, and the mixture was stirred for 2 hr.
- the precipitated solid was collected by filtration and washed with water (500 ml, twice).
- the filtrate was suspended in water (2000 ml), stirred for 30 minutes, collected again by filtration, and washed with water (500 ml, twice).
- the title compound (119 g, 75.3%) was obtained by drying with hot air at 60 ° C.
- the eluate was concentrated under reduced pressure, and tert-butyl methyl ether (3 ml) was added to stimulate the solution. As a result, precipitation was observed. Further, tert butyl methyl ether (40 ml) was added and stirred overnight. The resulting precipitate was collected by filtration, washed with tert butyl methyl ether (3 ml), and air-dried to obtain the title compound (1.61 g) of interest.
- Carboxylic acid benzyl ester (189 mg) was dissolved in ethyl acetate (6 ml), and 4N hydrogen chloride-ethyl acetate solution (0.3 ml) was added. The resulting mixture was concentrated under reduced pressure, and methanol (0.5 ml) and ethyl acetate (4 ml) were added. The precipitate was filtered and absorbed moisture, and was collected with methanol (10 ml). The recovered solution was concentrated again under reduced pressure, and methanol (0.5 ml) and tert butyl methyl ether (4 ml) were added. The precipitate was filtered to obtain the title compound (102 mg).
- Carboxylic acid benzyl ester (800 mg) was dissolved in a mixed solution of tetrahydrofuran (4 ml) and ethanol (4 ml), palladium carbon (400 mg) was added, and the mixture was stirred at room temperature for 4 hours under a hydrogen atmosphere of 0.15 MPa. Water (4 ml) was added to the reaction solution and filtered, and the residue was washed with 50% aqueous ethanol (8 ml) and water (4 ml), and the filtrate was concentrated. Tetrahydrofuran (8 ml) and ethanol (8 ml) were added to the concentrated residue and concentrated.
- Tetrahydrofuran (8 ml)
- ethyl acetate 8 ml
- ethanol 2 ml
- Tetrahydrofuran 8 ml
- ethanol 16 ml
- the crystals were suspended in tetrahydrofuran (16 ml) and stirred at room temperature for 40 minutes. The crystals were filtered and dried to give the title compound (550 mg) as white crystals.
- the filtrate was concentrated by adding 20 ml of ethanol.
- the operation of adding ethanol (10 ml) to the obtained mixture and concentrating was repeated 4 times.
- the mixture was added dropwise to ethyl acetate (40 ml) with stirring while filtering while hot. After stirring at room temperature for 25 hours and 30 minutes, the crystals were filtered and dried while washing with a mixed solution of ethanol (2 ml) and ethyl acetate (2 ml) to obtain the title compound (1.56 g) as a white solid. .
- Ethyl acetate (7.5 ml) was added to the reaction mixture, and 5% aqueous sodium hydrogen carbonate solution (7.5 ml) was added to separate the layers, and the layers were separated.
- a 1N aqueous hydrochloric acid solution (5 ml) was added to the organic layer, and the layers were separated.
- Tetrahydrofuran (7.5 ml) was added to the aqueous layer, 2N aqueous sodium hydroxide solution (3 ml) was added for neutralization, and ethyl acetate (7.5 ml) was added to separate the layers.
- the organic layer was washed with water and concentrated. Add ethyl acetate to the concentrated residue Concentration was repeated 3 times.
- the organic layer was washed successively with 1N aqueous sodium hydroxide solution (15 ml), water (15 ml) and saturated brine (15 ml) and then dried over anhydrous magnesium sulfate.
- the desiccant was filtered and the filtrate was concentrated under reduced pressure.
- the target compound fraction was concentrated under reduced pressure to give the title compound (822.7 mg, 93%) as a white powder.
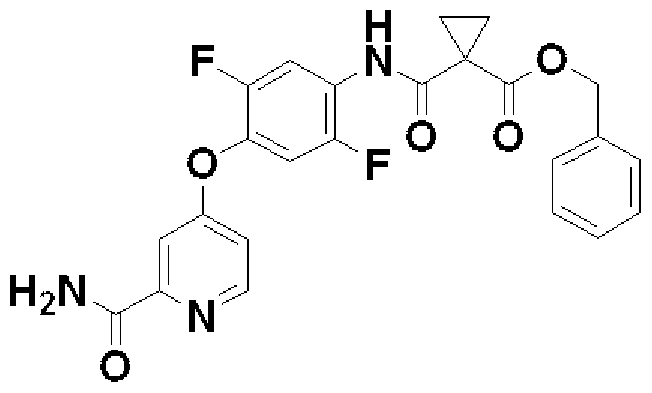
- benzyl 1 [( ⁇ 4 [(2 aminoviridine 4yl) oxy] -2,5-difluorophenyl ⁇ amino) carbonyl] cyclopropanecarboxylate (1.15 g) was dissolved in tetrahydrofuran (12 ml). .
- pyridine (0 ⁇ 424 ml) and phenyl chloroformate (0.657 ml) were added and stirred for 20 minutes.
- saturated aqueous sodium hydrogen carbonate solution 36 ml
- hexane 36 ml
- the obtained solid was washed with hexane, dried by aeration, and then dried with hot air (60 ° C.) for 5 hours. Water (150 ml) was added to this solid and stirred for 2 hours. The solid was collected by filtration, and the resulting solid was washed with water. The solid was dried in warm air (60 ° C.) for 3 days to give the title compound (1.111 7 g, 76%) as a white solid.
- benzyl 1 1 ( ⁇ [4 1 ( ⁇ 2 [(phenoxycarbonyl) amino] pyridine 4 inole ⁇ oxy) 2,5 difluorophenyleno] amino ⁇ carboninole) cyclopropane carboxylate (200 mg)
- 3 hydroxyazetidine hydrochloride 39 ⁇ lmg
- N, N-dimethylformamide (4 ⁇ Oml) were added triethylamine (0.100ml) at room temperature and stirred for 6 hours and 10 minutes.
- 3-hydroxyazetidine hydrochloride (10. Omg) and triethylamine (0.025 ml) were added at room temperature, and the mixture was stirred for 1 hour and 20 minutes.
- Example 15 1 1 “( ⁇ 2, 5 Difluoro-4 1” (2 — ⁇ “(3 Hydroxyzetidine 1 1 Fuenino: Carboninore ⁇
- N Benzinore 1 [( ⁇ 2, 5 Difluoro-4 [(2-— ⁇ [(3 Hydroxyzetidine 1 yl) carbonyl] amino ⁇ pyridine 4 yl) oxy] phenyl ⁇ amino) carbonyl] cyclopropanecarboxylate under nitrogen atmosphere (84 ⁇ 2 mg) was dissolved in tetrahydrofuran-methanol (1: 1) (2 ml). 10% palladium carbon (33.2 mg) was added to make the reaction system a hydrogen atmosphere, and the mixture was stirred at room temperature for 20 hours. After substituting the reaction system with nitrogen, tritylamine (0 ⁇ 0435 ml) was added and stirred for 30 minutes. The catalyst was filtered and washed with methanol. The filtrate was concentrated under reduced pressure to obtain the title compound (75.3 mg, 88%) as a white solid.
- EESSII--MMSS ((mm // zz)) :: 554400 [[M ⁇ —— H ⁇ ]] __ ..
- benzyl 1 1 ( ⁇ [4 1 ( ⁇ 2 — [(phenoxycarbonyl) amino] pyridine 4 inole ⁇ oxy) 2,5 difluorophenyleno] amino ⁇ carbinole) cyclopropane carboxylate (200 mg ) was suspended in N-methylpyrrolidinone (2.0 ml). At room temperature, 1-methyl-4- (methylamino) piperidine (0.104 ml) was added and stirred. Anti A saturated aqueous sodium hydrogen carbonate solution (10 ml) was added to the reaction solution and stirred. Extracted with ethyl acetate (20 ml).
- the organic layer was washed with water (10 ml), saturated aqueous ammonium chloride solution (10 ml) and saturated brine (10 ml), and dried over anhydrous sodium sulfate.
- the desiccant was filtered and the filtrate was concentrated under reduced pressure.
- the target product fraction was concentrated under reduced pressure, and the residue was dried under reduced pressure to give the title compound (2.3 mg, 29%) as a white powder.
- Example 18 1 ( ⁇ “(2.5 Difluoro-4 ⁇ 2—“ 3 Methyl-3- (1-methylbiperi_gin—4-ynole) ureido ⁇ pyridine—4-yl ⁇ _oxy) phenenole ⁇ amino ⁇ Kanorepo Ninore
- the target compound fraction was concentrated under reduced pressure to give the title compound (178.2 mg, 90%) as a white solid.
- the desiccant was filtered and the filtrate was concentrated under reduced pressure.
- the target fraction was concentrated under reduced pressure to obtain the title compound (132.8 mg, 73%) as a white solid.
- the production method of the present invention can be carried out by carrying out the same reaction as in the above examples using the amines described in the above production examples or known amines as one of the starting materials.
- HGFR Hepatocyte growth factor receptor
- VEGFR2 Vascular endothelial growth factor receptor2, jfl tube endothelial growth factor receptor 2
- PDGFR ⁇ Platelet derived growth factor receptor / 3, jfl platelet derived growth factor receptor / 3
- EGFR Extracellular growth factor receptor
- FBS Fetal bovine serum
- PBS Phosphate buffered saline
- EGTA O— Bis (2-aminoethyleneglycol) — N, N, N, N′— Tetraacetic acid, glycol ether diamine tetraacetic acid
- BSA Bovine Serum Albumin
- Hepes N— [2— Hydroxyethylj piperazine— N'— [2— ethanesulionic acid], Hepes (buffer)) ATP (Adenosine 5'-Triphosphate, adenosine 5, triphosphate) EDTA (Ethylenediaminetetraacetic acid)
- the cytoplasmic domain of HGFR (Genbank accession number J02958) is a 1-3 kb DNA fragment beginning with lysine and containing a stop codon, Park et al. (Proc. Natl. Acad. Sci. USA 84 (18), 6379— 6383, 1987)
- This DNA fragment was obtained from human placental cDNA library (purchased from Clontech) by using two kinds of primers (SEQ ID NO: 5,-CCGGCCGGATCCAAAAAGAGAAAGC AAATTAAA—3 'and SEQ ID NO: 5'-TTAATTCTGC AGCTATGATGT CTCCCAGAAGGA-3 ', Invitrogen.
- the product was isolated using the PCR method (TaKaRa EX Taq TM Kit, purchased from TaKaRa). This DNA fragment was cloned into a baculovirus replacement vector (pFastBac TM -HT (purchased from GIBCO BRL)) to obtain a recombinant construct. This was transferred to insect cells (Spodoptera frugip erda9 (Sf9)) and a HGFR recombinant baculovirus solution was prepared (recombinant baculovirus was prepared using the standard text (Bac-to-Bac Baculovirus Expression System (GIBCO BRL)).
- cytoplasmic fragments starting from lysine 791 (VEGFR2, Genbank accession number L04947).
- a cytoplasmic fragment starting from lysine 398 (FGFR1, Genban k acquisition number X52833) or a cytoplasmic fragment starting from lysine 558 (PDGFR ⁇ , Genbank acquisition number M21616), where EGFR is a product of Sigma (product number No. E—2645).
- the precipitated infected cells were suspended in 80 ml of ice-cold PBS and centrifuged at 4 ° C for 5 minutes at lOOO rpm, and the supernatant was removed. Precipitated infected cells were washed with 40 ml of ice-cold Lysis Buffer (50 mM Tris—HCl ( ⁇ 8 ⁇ 5), 5 mM)
- This column is divided into 30 ml Buffer A, 6 ml Buffer B (20 mM Tris-HCl (pH 8.5), 5 mM 2-mercaptoethanol, 1 M KC1, 10% (v / v) glycerol), 6 ml Buffer.
- the cytoplasmic domain of VEGFR2 the cytoplasmic domain of FGFR1 or the cytoplasmic domain of PDGFR / 3 were fused with 6 histidines at the N-terminus. Or His6-PDGFR / 3).
- T purchased from Nippon Sealing Co., Ltd.
- 6 H 1 diluted 15-fold with distilled water
- 10 1) diluted 30-fold His 6 -HGFR (60 1-fold with 0.4% BSA solution) and dimethylsulfo
- the test substance dissolved in xidide (4 1 diluted 100-fold with 0.1% BSA) was added to make the total volume 30 a 1.
- 4 ⁇ M ATP purchased from Sigma
- diluted with distilled water was added to 10–1 caroten, incubated at 30 ° C for 10 minutes, and then 10 1 500 mM EDTA (pH 8.0) (Wako Jun) (Purchased from Yakuhin Kogyo) was added to obtain a kinase reaction solution.
- tyrosine phosphorylated biotin—poly was performed using the Homogenous Time-Resolved Fluorescence (HTRF) method (Analytical Biochemistry, 269, 94—104, 1999). That is, 20 ⁇ 1 of the above kinase reaction solution and 30 ⁇ 1 diluted solution (5 OmM Hepes (pH 7.4), 20 mM MgCl, 4 mM MnCl, 0.5 mM Na VO, 0
- the inhibitory action on VEGFR2, FGFR1 or EGFR tyrosine kinase activity is the same as the above-mentioned inhibitory action on HGFR tyrosine kinase activity using 15ng His6-VEGFR2 and 15ng His6-FGFR1 or 23ng EGFR instead of HGFR. It measured by the method of.
- the inhibitory effect on PDGFR / 3 tyrosine kinase activity was obtained by using 50 ng of His6—PD GFR / 3 to obtain a kinase reaction solution as described above, and then tyrosine phosphorylated biotin—poly (GT) was detected and evaluated.
- Human gastric cancer cells (MKN-45) were suspended in RPMI 1640 medium (purchased from Sigma) containing 1% FBS. Add 0.1 ml / well of the cell suspension (1 X 10 4 cells / ml) to a 96-well plate for cell culture (purchased from NUNC, product number 167008), and add 5% CO incubator.
- a sputum culture was performed in a plate (37 ° C). After incubation, add 0.1 ml of the test substance diluted with RPMI1640 medium containing 1% FBS to each well, and further in a 5% CO incubator (37 ° C).
- Test example 3 ELISA method HGFR itself, phosphorylation action
- Human gastric cancer cells ( ⁇ -45) were suspended in RPMI1640 medium (purchased from Sigma) containing 1% FBS. Add 0.1 ml / well of the cell suspension (1 X 10 5 cells / ml) to a 96-well plate for cell culture (purchased from NUNC, product number 167008), and add 5% CO incubator.
- a sputum culture was performed in a plate (37 ° C). After the culture, the supernatant was removed from each tool, and RPMI1640 medium containing 0.05 ml of 1% FBS was added. Add 0.05 ml of test substance dissolved in dimethyl sulfoxide (diluted with RPMI1640 medium containing 1% FBS) and add 5% CO2.
- the cells were cultured in an incubator (37 ° C) for 1 hour. The supernatant is removed from each well, and each well is washed with PBS 150 1 and then added to a soluble buffer (50 mM Hepes (pH 7.4), 150 mM NaCl, 10% (v / v) glycerol, 1% Triton X—100, 1.5 mM MgCl
- ImM EDTA (pH 8.0), lOOmM NaF, ImM PMSF, 10 g / ml Aproti nin, 50 ⁇ g Z ml Leupeptin, 1 ⁇ g ml Pepstatin A, ImM Na VO) 10
- Each well of the plate prepared in 2. was washed 3 times with 200 1 PBS, 150 1 3% BSA / PBS was added thereto, and incubated at room temperature for 2 hours.
- Each well was washed 3 times with 200 ⁇ l of PBS, 50 1 of the above-described cell extract was added thereto, and incubated at 4 ° C.
- each well was washed 3 times with 250 1 wash solution (0.1% BSA, 20 mM Tris-HCl (pH7.6), 137 mM NaCl, 0.05% Tween-20), and the reaction solution (1% BSA , 20 mM Tris—HCl (pH 7. 6), 137 mM NaCl, 0.05% D.
- Example 9 About the crystal obtained in Example 9 (Method 3), about 5 mg of a sample was pulverized in a mortar, and then placed on an aluminum pan for measurement under the following conditions.
- Goniometer TTR—III horizontal goniometer
- Fig. 1 shows the powder X-ray diffraction pattern of the crystal obtained in Example 9 (Method 3), and Table 4 shows the typical peak and relative intensity of the diffraction angle (2 ⁇ ) of the crystal. . [0120] [Table 4]
- the method for producing a phenoxypyridine derivative according to the present invention includes an antitumor agent for various tumors such as spleen cancer, stomach cancer, colon cancer, breast cancer, prostate cancer, lung cancer, kidney cancer, brain tumor and ovarian cancer. It is possible to provide a phenoxypyridine derivative useful as an angiogenesis inhibitor or a cancer metastasis inhibitor.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Public Health (AREA)
- General Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Oncology (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Pyridine Compounds (AREA)
Description





































































Claims


















Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN2007800195205A CN101454286B (zh) | 2006-08-31 | 2007-08-28 | 苯氧基吡啶衍生物的制备方法 |
| EP07793075.8A EP2058302B1 (en) | 2006-08-31 | 2007-08-28 | Method for producing phenoxypyridine derivative |
| KR1020087029577A KR101432318B1 (ko) | 2006-08-31 | 2007-08-28 | 페녹시피리딘 유도체의 제조 방법 |
| AU2007289787A AU2007289787B2 (en) | 2006-08-31 | 2007-08-28 | Method for producing phenoxypyridine derivative |
| JP2008532065A JP5145231B2 (ja) | 2006-08-31 | 2007-08-28 | フェノキシピリジン誘導体の製造方法 |
| CA2661702A CA2661702C (en) | 2006-08-31 | 2007-08-28 | Method for producing phenoxypyridine derivative |
| IL197002A IL197002A (en) | 2006-08-31 | 2009-02-11 | Method for producing phenoxypyridine derivative |
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US82419206P | 2006-08-31 | 2006-08-31 | |
| US60/824192 | 2006-08-31 | ||
| JP2006-285327 | 2006-10-19 | ||
| JP2006285327 | 2006-10-19 | ||
| US85511706P | 2006-10-30 | 2006-10-30 | |
| US60/855117 | 2006-10-30 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2008026577A1 true WO2008026577A1 (fr) | 2008-03-06 |
Family
ID=39135860
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2007/066635 WO2008026577A1 (fr) | 2006-08-31 | 2007-08-28 | Procédé de production d'un dérivé de phénoxypyridine |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US7790885B2 (ja) |
| EP (1) | EP2058302B1 (ja) |
| JP (1) | JP5145231B2 (ja) |
| CN (1) | CN101454286B (ja) |
| AU (1) | AU2007289787B2 (ja) |
| CA (1) | CA2661702C (ja) |
| IL (1) | IL197002A (ja) |
| WO (1) | WO2008026577A1 (ja) |
Cited By (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009104520A1 (ja) * | 2008-02-18 | 2009-08-27 | エーザイ・アール・アンド・ディー・マネジメント株式会社 | フェノキシピリジン誘導体の製造方法(2) |
| US7998948B2 (en) | 2007-11-30 | 2011-08-16 | Eisai R&D Management Co., Ltd. | Pharmaceutical composition for treating esophageal cancer |
| WO2012133416A1 (ja) | 2011-03-29 | 2012-10-04 | エーザイ・アール・アンド・ディー・マネジメント株式会社 | フェノキシピリジン誘導体の製造方法(3) |
| US8288538B2 (en) | 2005-08-24 | 2012-10-16 | Eisai R&D Management Co., Ltd. | Pyridine derivatives and pyrimidine derivatives (3) |
| JP5190365B2 (ja) * | 2006-08-23 | 2013-04-24 | エーザイ・アール・アンド・ディー・マネジメント株式会社 | フェノキシピリジン誘導体の塩またはその結晶およびそれらの製造方法 |
| US8637672B2 (en) | 2010-04-29 | 2014-01-28 | Deciphera Pharmaceuticals, Llc | Cyclopropyl dicarboxamides and analogs exhibiting anti-cancer and anti-proliferative activities |
| US9012458B2 (en) | 2010-06-25 | 2015-04-21 | Eisai R&D Management Co., Ltd. | Antitumor agent using compounds having kinase inhibitory effect in combination |
| US9334239B2 (en) | 2012-12-21 | 2016-05-10 | Eisai R&D Management Co., Ltd. | Amorphous form of quinoline derivative, and method for producing same |
| US10259791B2 (en) | 2014-08-28 | 2019-04-16 | Eisai R&D Management Co., Ltd. | High-purity quinoline derivative and method for manufacturing same |
| US10517861B2 (en) | 2013-05-14 | 2019-12-31 | Eisai R&D Management Co., Ltd. | Biomarkers for predicting and assessing responsiveness of endometrial cancer subjects to lenvatinib compounds |
| US11090386B2 (en) | 2015-02-25 | 2021-08-17 | Eisai R&D Management Co., Ltd. | Method for suppressing bitterness of quinoline derivative |
| US11369623B2 (en) | 2015-06-16 | 2022-06-28 | Prism Pharma Co., Ltd. | Anticancer combination of a CBP/catenin inhibitor and an immune checkpoint inhibitor |
| US11547705B2 (en) | 2015-03-04 | 2023-01-10 | Merck Sharp & Dohme Llc | Combination of a PD-1 antagonist and a VEGF-R/FGFR/RET tyrosine kinase inhibitor for treating cancer |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7790885B2 (en) | 2006-08-31 | 2010-09-07 | Eisai R&D Management Co., Ltd. | Process for preparing phenoxypyridine derivatives |
| US20090227556A1 (en) * | 2008-01-31 | 2009-09-10 | Eisai R&D Management Co., Ltd. | Receptor tyrosine kinase inhibitors comprising pyridine and pyrimidine derivatives |
| CN102731362B (zh) * | 2012-07-09 | 2014-03-19 | 南京药石药物研发有限公司 | 1-羧酸叔丁酯-3-氟-氮杂环丁烷衍生物的制备方法 |
| EA033834B1 (ru) * | 2014-07-31 | 2019-11-29 | Exelixis Inc | Способ получения кабозантиниба, меченного фтором-18, и его аналогов |
| CN112159351B (zh) * | 2020-09-21 | 2021-12-07 | 广州南鑫药业有限公司 | 一种多靶点抗肿瘤药物的制备方法 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002032872A1 (en) * | 2000-10-20 | 2002-04-25 | Eisai Co., Ltd. | Nitrogenous aromatic ring compounds |
| WO2004076412A2 (en) * | 2003-02-26 | 2004-09-10 | Sugen, Inc. | Aminoheteroaryl compounds as protein kinase inhibitors |
| WO2005082855A1 (ja) | 2004-02-27 | 2005-09-09 | Eisai Co., Ltd. | 新規ピリジン誘導体およびピリミジン誘導体(2) |
| WO2005117867A2 (en) * | 2004-04-23 | 2005-12-15 | Bristol-Myers Squibb Company | Monocyclic heterocycles as kinase inhibitors |
| WO2007023768A1 (ja) * | 2005-08-24 | 2007-03-01 | Eisai R & D Management Co., Ltd. | 新規ピリジン誘導体およびピリミジン誘導体(3) |
Family Cites Families (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AR036042A1 (es) | 2001-05-30 | 2004-08-04 | Sugen Inc | Derivados aralquilsufonil-3-(pirrol-2-ilmetiliden)-2-indolinona, sus composiciones farmaceuticas y metodo para la modulacion de la actividad catalitica de una proteina quinasa |
| DE60233736D1 (de) | 2001-06-22 | 2009-10-29 | Kirin Pharma K K | Chinolinderivat und chinazolinderivat, die die selbstphosphorylierung des hepatocytus-proliferator-rezeptors hemmen, und diese enthaltende medizinische zusammensetzung |
| US6790852B2 (en) | 2002-04-18 | 2004-09-14 | Hoffmann-La Roche Inc. | 2-(2,6-dichlorophenyl)-diarylimidazoles |
| AR037647A1 (es) | 2002-05-29 | 2004-12-01 | Novartis Ag | Derivados de diarilurea utiles para el tratamiento de enfermedades dependientes de la cinasa de proteina |
| SI1603570T1 (sl) * | 2003-02-26 | 2013-03-29 | Sugen, Inc. | Spojine aminoheteroarila kot zaviralci proteinkinaze |
| US20050014753A1 (en) * | 2003-04-04 | 2005-01-20 | Irm Llc | Novel compounds and compositions as protein kinase inhibitors |
| EP1473043A1 (en) | 2003-04-29 | 2004-11-03 | Boehringer Ingelheim Pharma GmbH & Co.KG | Pharmaceutical combination for the treatment of diseases involving cell proliferation, migration or apotosis of myeloma cells, or angiogenesis |
| US7037909B2 (en) | 2003-07-02 | 2006-05-02 | Sugen, Inc. | Tetracyclic compounds as c-Met inhibitors |
| BRPI0412040A (pt) | 2003-07-02 | 2006-09-05 | Sugen Inc | hidrazidas de indolinona como inibidores de c-met |
| CA2530589A1 (en) | 2003-07-02 | 2005-01-20 | Sugen Inc. | Arylmethyl triazolo and imidazopyrazines as c-met inhibitors |
| US7122548B2 (en) | 2003-07-02 | 2006-10-17 | Sugen, Inc. | Triazolotriazine compounds and uses thereof |
| CA2531485C (en) | 2003-07-07 | 2013-03-26 | Merck Patent Gmbh | Malonamide derivatives |
| WO2005016920A1 (en) | 2003-08-15 | 2005-02-24 | Vertex Pharmaceuticals Incorporated | Pyrrole compositions useful as inhibitors of c-met |
| EP2609919A3 (en) | 2003-09-26 | 2014-02-26 | Exelixis, Inc. | c-Met modulators and methods of use |
| US7169781B2 (en) | 2003-10-17 | 2007-01-30 | Hoffmann-La Roche Inc. | Imidazole derivatives and their use as pharmaceutical agents |
| US7173031B2 (en) | 2004-06-28 | 2007-02-06 | Bristol-Myers Squibb Company | Pyrrolotriazine kinase inhibitors |
| WO2006014325A2 (en) | 2004-07-02 | 2006-02-09 | Exelixis, Inc. | C-met modulators and method of use |
| US7652009B2 (en) * | 2004-11-30 | 2010-01-26 | Amgem Inc. | Substituted heterocycles and methods of use |
| NZ568654A (en) | 2005-12-05 | 2012-02-24 | Pfizer Prod Inc | Method of treating abnormal cell growth |
| EP2062886B1 (en) * | 2006-08-23 | 2011-11-30 | Eisai R&D Management Co., Ltd. | Salt of phenoxypyridine derivative or crystal thereof and process for producing the same |
| US7790885B2 (en) | 2006-08-31 | 2010-09-07 | Eisai R&D Management Co., Ltd. | Process for preparing phenoxypyridine derivatives |
| US20120232049A1 (en) | 2007-02-23 | 2012-09-13 | Eisai R&D Management Co., Ltd. | Pyridine or pyrimidine derivative having excellent cell growth inhibition effect and excellent anti-tumor effect on cell strain having amplification of hgfr gene |
| US20090227556A1 (en) * | 2008-01-31 | 2009-09-10 | Eisai R&D Management Co., Ltd. | Receptor tyrosine kinase inhibitors comprising pyridine and pyrimidine derivatives |
-
2007
- 2007-08-27 US US11/892,785 patent/US7790885B2/en not_active Expired - Fee Related
- 2007-08-28 WO PCT/JP2007/066635 patent/WO2008026577A1/ja active Application Filing
- 2007-08-28 AU AU2007289787A patent/AU2007289787B2/en not_active Ceased
- 2007-08-28 CN CN2007800195205A patent/CN101454286B/zh not_active Expired - Fee Related
- 2007-08-28 CA CA2661702A patent/CA2661702C/en not_active Expired - Fee Related
- 2007-08-28 EP EP07793075.8A patent/EP2058302B1/en active Active
- 2007-08-28 JP JP2008532065A patent/JP5145231B2/ja not_active Expired - Fee Related
-
2009
- 2009-02-11 IL IL197002A patent/IL197002A/en not_active IP Right Cessation
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002032872A1 (en) * | 2000-10-20 | 2002-04-25 | Eisai Co., Ltd. | Nitrogenous aromatic ring compounds |
| WO2004076412A2 (en) * | 2003-02-26 | 2004-09-10 | Sugen, Inc. | Aminoheteroaryl compounds as protein kinase inhibitors |
| WO2005082855A1 (ja) | 2004-02-27 | 2005-09-09 | Eisai Co., Ltd. | 新規ピリジン誘導体およびピリミジン誘導体(2) |
| WO2005117867A2 (en) * | 2004-04-23 | 2005-12-15 | Bristol-Myers Squibb Company | Monocyclic heterocycles as kinase inhibitors |
| WO2007023768A1 (ja) * | 2005-08-24 | 2007-03-01 | Eisai R & D Management Co., Ltd. | 新規ピリジン誘導体およびピリミジン誘導体(3) |
Non-Patent Citations (7)
| Title |
|---|
| ADVANCES IN CANCER RESEARCH, vol. 67, 1995, pages 257 - 279 |
| ANALYTICAL BIOCHEMISTRY, vol. 269, 1999, pages 94 - 104 |
| BRITISH JOURNAL OF CANCER, vol. 84, 2001, pages 864 - 873 |
| CANCER SCI., vol. 94, 2003, pages 321 - 327 |
| ONCOLOGY REPORTS, vol. 5, 1998, pages 1013 - 1024 |
| PARK ET AL., PROC. NATL. ACAD. SCI. U.S.A., vol. 84, no. 18, 1987, pages 6379 - 6383 |
| See also references of EP2058302A4 |
Cited By (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8288538B2 (en) | 2005-08-24 | 2012-10-16 | Eisai R&D Management Co., Ltd. | Pyridine derivatives and pyrimidine derivatives (3) |
| JP5190365B2 (ja) * | 2006-08-23 | 2013-04-24 | エーザイ・アール・アンド・ディー・マネジメント株式会社 | フェノキシピリジン誘導体の塩またはその結晶およびそれらの製造方法 |
| US7998948B2 (en) | 2007-11-30 | 2011-08-16 | Eisai R&D Management Co., Ltd. | Pharmaceutical composition for treating esophageal cancer |
| WO2009104520A1 (ja) * | 2008-02-18 | 2009-08-27 | エーザイ・アール・アンド・ディー・マネジメント株式会社 | フェノキシピリジン誘導体の製造方法(2) |
| US8637672B2 (en) | 2010-04-29 | 2014-01-28 | Deciphera Pharmaceuticals, Llc | Cyclopropyl dicarboxamides and analogs exhibiting anti-cancer and anti-proliferative activities |
| US9012458B2 (en) | 2010-06-25 | 2015-04-21 | Eisai R&D Management Co., Ltd. | Antitumor agent using compounds having kinase inhibitory effect in combination |
| RU2590158C2 (ru) * | 2011-03-29 | 2016-07-10 | ЭЙСАЙ АрЭНДДи МЕНЕДЖМЕНТ КО., ЛТД. | Способ получения производного феноксипиридина |
| JPWO2012133416A1 (ja) * | 2011-03-29 | 2014-07-28 | エーザイ・アール・アンド・ディー・マネジメント株式会社 | フェノキシピリジン誘導体の製造方法(3) |
| US8759530B2 (en) | 2011-03-29 | 2014-06-24 | Eisai R&D Management Co., Ltd. | Method for producing phenoxypyridine derivative |
| WO2012133416A1 (ja) | 2011-03-29 | 2012-10-04 | エーザイ・アール・アンド・ディー・マネジメント株式会社 | フェノキシピリジン誘導体の製造方法(3) |
| US9334239B2 (en) | 2012-12-21 | 2016-05-10 | Eisai R&D Management Co., Ltd. | Amorphous form of quinoline derivative, and method for producing same |
| US10517861B2 (en) | 2013-05-14 | 2019-12-31 | Eisai R&D Management Co., Ltd. | Biomarkers for predicting and assessing responsiveness of endometrial cancer subjects to lenvatinib compounds |
| US10407393B2 (en) | 2014-08-28 | 2019-09-10 | Eisai R&D Management Co., Ltd. | High-purity quinoline derivative and method for manufacturing same |
| US10259791B2 (en) | 2014-08-28 | 2019-04-16 | Eisai R&D Management Co., Ltd. | High-purity quinoline derivative and method for manufacturing same |
| US10822307B2 (en) | 2014-08-28 | 2020-11-03 | Eisai R&D Management Co., Ltd. | High-purity quinoline derivative and method for manufacturing same |
| US11186547B2 (en) | 2014-08-28 | 2021-11-30 | Eisai R&D Management Co., Ltd. | High-purity quinoline derivative and method for manufacturing same |
| US11090386B2 (en) | 2015-02-25 | 2021-08-17 | Eisai R&D Management Co., Ltd. | Method for suppressing bitterness of quinoline derivative |
| US11547705B2 (en) | 2015-03-04 | 2023-01-10 | Merck Sharp & Dohme Llc | Combination of a PD-1 antagonist and a VEGF-R/FGFR/RET tyrosine kinase inhibitor for treating cancer |
| US12083112B2 (en) | 2015-03-04 | 2024-09-10 | Eisai R&D Management Co., Ltd. | Combination of a PD-1 antagonist and a VEGFR/FGFR/RET tyrosine kinase inhibitor for treating cancer |
| US11369623B2 (en) | 2015-06-16 | 2022-06-28 | Prism Pharma Co., Ltd. | Anticancer combination of a CBP/catenin inhibitor and an immune checkpoint inhibitor |
Also Published As
| Publication number | Publication date |
|---|---|
| AU2007289787A1 (en) | 2008-03-06 |
| CA2661702A1 (en) | 2008-03-06 |
| EP2058302B1 (en) | 2013-04-17 |
| EP2058302A4 (en) | 2010-10-06 |
| IL197002A0 (en) | 2009-11-18 |
| JPWO2008026577A1 (ja) | 2010-01-21 |
| CN101454286B (zh) | 2011-07-20 |
| US20080214815A1 (en) | 2008-09-04 |
| JP5145231B2 (ja) | 2013-02-13 |
| IL197002A (en) | 2013-02-28 |
| CA2661702C (en) | 2014-05-13 |
| US7790885B2 (en) | 2010-09-07 |
| AU2007289787B2 (en) | 2012-03-29 |
| CN101454286A (zh) | 2009-06-10 |
| EP2058302A1 (en) | 2009-05-13 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2008026577A1 (fr) | Procédé de production d'un dérivé de phénoxypyridine | |
| Jaime-Figueroa et al. | Design, synthesis and biological evaluation of Proteolysis Targeting Chimeras (PROTACs) as a BTK degraders with improved pharmacokinetic properties | |
| TWI585088B (zh) | 作爲激酶抑制劑之咪唑并[1,2-b]嗒衍生物 | |
| AU2007288793B2 (en) | Salt of phenoxypyridine derivative or crystal thereof and process for producing the same | |
| JP4077028B2 (ja) | 新規ピリジン誘導体およびピリミジン誘導体(3) | |
| TWI650322B (zh) | 嘧啶並吡咯類化合物、其製備方法、藥用組合物及其應用 | |
| KR101567763B1 (ko) | 키나제 억제제로서 활성인 치환된 인다졸 유도체 | |
| WO2005082854A1 (ja) | 新規ピリジン誘導体およびピリミジン誘導体(1) | |
| KR102627756B1 (ko) | 브루톤 타이로신 키나제 억제제 | |
| JPWO2008102870A1 (ja) | Hgfr遺伝子増幅細胞株に優れた細胞増殖阻害効果および抗腫瘍効果を示すピリジン誘導体またはピリミジン誘導体 | |
| CA3009669A1 (en) | Bruton's tyrosine kinase inhibitors | |
| US9624218B2 (en) | Pyrido[2,3-d]pyrimidin-4-one compounds as tankyrase inhibitors | |
| CN110753690B (zh) | 吡啶衍生物 | |
| WO2014014314A1 (ko) | 이중 저해 활성을 갖는 헤테로고리 유도체 | |
| JP2022525676A (ja) | キノリン誘導体、それらの調製のための方法及びがんの治療のためのその使用 | |
| Hirayama et al. | Design, synthesis and biological activity of YM-60828 derivatives. Part 2: Potent and orally-bioavailable factor Xa inhibitors based on benzothiadiazine-4-one template | |
| JP6012634B2 (ja) | Nk1アンタゴニスト活性を有するスピロアミン系化合物 | |
| KR101432318B1 (ko) | 페녹시피리딘 유도체의 제조 방법 | |
| BR112018014705B1 (pt) | Compostos inibidores de tirosina quinase de Bruton e composição farmacêutica |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200780019520.5 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07793075 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008532065 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020087029577 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 197002 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007289787 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2661702 Country of ref document: CA |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007793075 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1772/CHENP/2009 Country of ref document: IN |
|
| NENP | Non-entry into the national phase |
Ref country code: RU |