NOUVEAUX DERIVES DE CYCLODEXTRINES AMPHIPHILES '
DESCRIPTION
Domaine technique
La présente invention concerne de nouveaux dérivés de cyclodextrines, et leur application, notamment dans les domaines pharmaceutiques, cosmétiques et alimentaires et tout particulièrement le domaine du transfert d'acides nucléiques dans des cellules.
Cette invention concerne encore la préparation de nouveaux dérivés de cyclodextrines et leur application à la production de nouveaux nanosystèmes .
Etat de la technique
Les cyclodextrines, ou cyclomaltooligosaccharides (CDs), sont des oligosaccharides cycliques connus pour leur aptitude à inclure dans leur cavité des molécules diverses, de taille adaptée à celle de la structure hôte. Le caractère généralement apolaire de ces associations conduit généralement à inclure des structures de type hydrophobe. Ceci peut permettre la solubilisation dans l'eau de composés peu ou pas solubles dans l'eau et l'amélioration de leur stabilité. Ces propriétés sont notamment utilisées pour améliorer la biodisponibilité de médicaments.
Les cyclodextrines sont disponibles commercialement en trois tailles différentes, à savoir les alpha-, béta- et gamma-cyclodextrines, qui comportent six, sept et huit résidus d'α-D-glucopyranose. Cette disponibilité impose une limitation importante quant à la taille des molécules hôtes qui peuvent être incluses dans leur cavité. A titre d'exemple, l'α-cyclodextrine, la plus petite, peut complexer des cycles aromatiques tels que les dérivés du benzène, alors que la γ-cyclodextrine, qui présente une cavité plus large,
est capable d'inclure des cycles fusionnés tels que des dérivés de l ' anthracène . Les molécules de taille plus importante, en particulier les macromolécules, ne sont en général pas adaptées à l'inclusion dans les cyclodextrines. De plus, le rapport molaire cyclodextrine : molécule hôte des complexes d'inclusion est en général 1:1 ou supérieur; autrement exprimé, au maximum une molécule est transportée par molécule de cyclodextrine.
La solubilité relativement faible dans l'eau des cyclodextrines, en particulier des cyclodextrines commerciales, et notamment de la plus accessible d'entre- elles sur le plan économique, la béta-cyclodextrine (18 g/L, soit 15 mmol/1, à 25 0C), peut constituer une limite dans leur utilisation en pharmacie. Par ailleurs, les cyclodextrines ne possédant pas de capacité particulière de reconnaissance vis-à-vis de récepteurs biologiques dans l'organisme, ces entités ne peuvent donc pas être utilisées pour l'adressage et la vectorisation de principes actifs.
Afin de remédier à cet état de fait, des cyclodextrines ont été modifiées chimiquement, par exemple les alcools primaires ont été substitués par des groupes monosaccharidiques ou oligosaccharidiques, de façon à améliorer leur solubilité dans l'eau d'une part et, d'autre part, pour incorporer dans leur structure des signaux de reconnaissance cellulaire (demandes internationales PCT WO 95/19994, WO 95/21870 et WO 97/33919).
Cependant, les dérivés de cyclodextrines de l'art antérieur peuvent présenter certaines limitations, notamment vis-à-vis des principes actifs susceptibles d'être transportés, de la capacité de charge de principe actif par unité de masse du dérivé de cyclodextrine, de leur capacité à
« s ' auto-organiser » , de leur capacité à « adresser » une molécule hôte, notamment un principe actif, de leur coût, de leur toxicité, de leur facilité à être synthétisé et/ou de leur solubilité dans certains solvants, en particulier dans l'eau.
II subsiste donc un besoin pour des composés permettant de résoudre en tout ou en partie les problèmes techniques évoqués ci-dessus. En particulier en terme de charge et de type de composé transporté ainsi que pour un procédé de préparation simple à mettre en œuvre, présentant un bon rendement, permettant d'obtenir un produit avec une haute pureté, à un faible coût et/ou qui permette d'obtenir une large gamme de composés.
La présente invention vise encore à proposer de nouveaux dérivés de cyclodextrines qui, tout en comportant une variété de groupements fonctionnels ou éléments de bio-reconnaissance ou de visualisation, sont capables de s ' auto-organiser sous forme de systèmes colloïdaux dispersibles. Description de l'invention
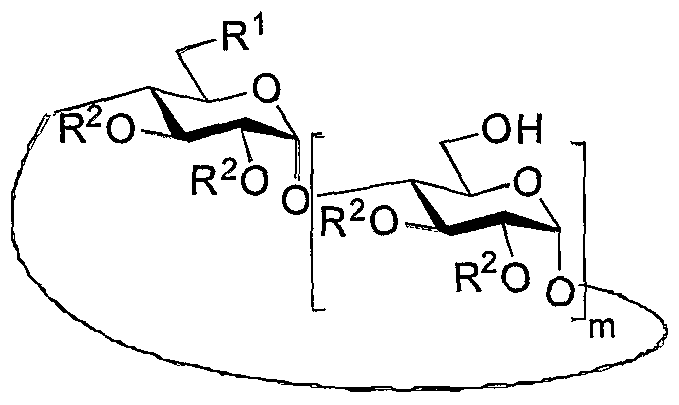
La présente invention concerne une cyclodextrine de Formule (I) suivante :
Formule ( I )
dans laquelle :
- m = 5, 6 où 7
- les radicaux R1, identiques ou différents, représentent :
( 1 ) un groupement OA dans lequel A représente un atome d'hydrogène, un radical alkyle, aryle, ou encore un groupement protecteur, comme un groupement silylé, notamment
tert-butyldiméthylsilyl ou tert-butyldiphénylsilyl, en particulier le groupement OA représente un groupe hydroxyle (OH);
(2) un groupement fonctionnel choisi parmi: • un atome d'halogène ;
• un groupement azoture (N3) ;
• un groupement soufré du type SR3, dans lequel R3 est :
(i) un substituant alkyle ou aryle ou ;
(ii) un élément de bioreconnaissance tel qu'un dérivé d'acide aminé, un peptide, un monosaccharide, un oligosaccharide, un élément de multiplication à plusieurs ramifications, lesquelles ramifications peuvent porter des groupements glucidiques qui peuvent être identiques ou différents, une sonde de visualisation ou de détection fluorescente ou radioactive, ou d'autres groupements fonctionnels ; (iii) CH2- (CH2 )n-B avec n = 1 à 5, B est : - NHX et X est un atome d'hydrogène, un groupement alkyle ou aryle, ou - NZC (=Q)NTW, où Z représente un atome d'hydrogène, un groupe alkyle ou aryle, Q représente un atome d'oxygène ou un atome de soufre et T et W, identiques ou différents, représentent un atome d'hydrogène, un substituant alkyle, aryle ou un élément de reconnaissance cellulaire tel qu'un acide aminé, un peptide, un monosaccharide, un oligosaccharide ou encore un élément de multiplication à plusieurs ramifications portant des groupements glucidiques qui peuvent être identiques ou différents, ou des groupements chargés tels que des groupements ammonium;
• un groupement aminé du type NHR4, dans lequel R4 est : (i) un atome d'hydrogène ; (ii) un substituant alkyle ou aryle ;
(iii) un substituant acyle ou ;
(iv) un substituant du type carbamate, urée ou thiourée, éventuellement substitué par au moins un groupement choisi parmi les groupements alkyle, aryle, des éléments de bioreconnaissance, de visualisation ou de détection, notamment tels que ceux mentionnés ci- dessus pour R3 - les radicaux R2, identiques ou différents, représentent :
(1) un hydrogène H, ou ; (2) un groupement acyle, aryle ou alkyle ; avec au moins un des radicaux R1 différent de OH et au moins un des radicaux R2 différent de H, ainsi que leurs sels et leurs isomères.
Les cyclodextrines représentées précédemment par la formule (I) peuvent concerner à la fois des dérivés de cyclodextrines amphiphiles per(C-6) fonctionnalisés et mono (C- 6)fonctionnalisés. Dans le premier cas, tous les groupements R1 sont identiques, alors que dans le deuxième cas l'un des R1 est différent des autres.
Les cyclodextrines selon l'invention présentent ainsi un caractère amphiphile pouvant leur permettre de présenter une solubilité intéressante dans différents solvants, notamment dans l'eau, et/ou leur permettre de s ' autoorganiser pour former des systèmes formant des suspensions colloïdales.
D'autre part, ces cyclodextrines peuvent être associées à, ou former des complexes avec, des molécules hôtes dans un rapport molécule hôte / cyclodextrine allant de 1/2 à 1000/1, notamment de 1/1 à 100/1, en particulier de 1,1/1 à
20/1, voire de 1,5/1 à 10/1.
On entend par « groupement protecteur » au sens de la présente invention, un groupe permettant d'éviter que la fonction sur laquelle il est présent ne réagisse avec des
réactifs utilisés dans les réactions subies par le composé sur lequel il est présent. En particulier, ce type de groupement est décrit dans les ouvrages « Protective groups in organic synthesis », Wiley, de T.W. Green et P. G.M. Wuts, 3e ed, 1999, ou « Protective groups », Georg Thieme Verlag de P. Kocienski, 3e ed, 2003.
On entend par « alkyle » au sens de la présente invention, un radical carboné, linéaire, ramifié ou cyclique, saturé ou insaturé, notamment comprenant de 1 ou 2 à 12 atomes de carbone.
On entend par « aryle » au sens de la présente invention, un radical comprenant au moins un cycle aromatique, éventuellement substitué, notamment par un alkyle. Ledit aryle peut comprendre de 6 à 20 atomes de carbone. Les radicaux R2, R3 et/ou R4 peuvent être choisis parmi les groupements benzyle, phényle, allyle, méthyle, éthyle, propyle, butyle, pentyle, hexyle ou homologues supérieurs comprenant jusqu'à 12 atomes de carbone, linéaires, ramifiés ou cycliques, saturés ou insaturés, ces groupements pouvant comporter d'autres groupements fonctionnels neutres ou chargés .
On entend par « groupement fonctionnel » au sens de la présente invention, un ensemble mono- ou polyatomique ayant une réactivité caractéristique ou encore un groupement d'atomes ayant une valence propre et caractérisant une fonction.
Les radicaux R2 et/ou R4 peuvent être choisis parmi les groupements acétyle, propionyle, butyroyle, pentanoyle, hexanoyle ou homologues comprenant jusqu'à 22 atomes de carbone, linéaires, ramifiés ou cycliques, saturés ou insaturés, ces groupements pouvant être porteurs d'autres groupements fonctionnels neutres ou chargés .
On entend par « acyle » au sens de la présente invention, un radical carboné comprenant au moins une fonction
carbonyle, de type Ra-C(=O)-, dans lequel Ra représente notamment un radical aryle ou alkyle.
On entend par "élément de bio-reconnaissance" au sens de la présente invention, une structure moléculaire complémentaire d'un récepteur biologique, capable de s'associer spécifiquement à ce dernier, notamment par des liaisons non-covalentes, et en particulier de conduire à une réponse spécifique, par exemple :
- une induction ou inhibition d'un signal de transduction induite après association avec le récepteur,
- une induction et régulation de la biosynthèse d'un enzyme,
- une inhibition de l'activité d'un enzyme par fixation sur son site actif, - une induction d'une réponse immunitaire suite à une affection bactérienne, une infection virale, et/ou
- une inhibition d'un processus inflammatoire par blocage du site actif d'une sélectine.
On entend par "élément de multiplication à plusieurs ramifications", au sens de la présente invention, notamment une chaîne carbonée ramifiée comprenant soit un atome de carbone quaternaire tétrasubstitué comme les dérivés du tris(2-aminométhyl)méthylamine (TRIS) et du pentaérythritol, soit un atome d'azote trisubstitué comme le tris (2- aminoéthyl ) aminé (TREN). Ces éléments de multiplication peuvent encore être incorporés en combinaison avec un deuxième élément de ramification comprenant, notamment, un atome d'azote tertiaire comme les dérivés du tris (2- aminoéthy1 ) aminé (TREN) . On entend par "sonde de visualisation ou de détection", au sens de la présente invention, une structure moléculaire permettant la détection d'un système par une technique physicochimique, telle que la fluorescence ou la radioactivité. Parmi les sondes fluorescentes, on peut
notamment citer les dérivés de la fluorescéine, du dansyle (5-(diméthylamino)-l-naphtalènesulfonyle) ou de la coumarine. Parmi les sondes radioactives, on peut citer les produits marqués par un isotope radioactif. Selon un mode de réalisation, les éléments de reconnaissance cellulaire et/ou les éléments de bioreconnaissance précités sont liés au reste de la molécule directement ou par l'intermédiaire d'un bras espaceur, par exemple un bras espaceur alkyle en C1 à C10 ou hétéroalkyle en C1 à C10, éventuellement substitué. Par exemple, le bras espaceur peut être -CH2CH2-NHC (=S JNHCH2CH2SCH2-, comme illustré dans les exemples 25 et 26.
Selon un mode de réalisation, la cyclodextrine amphiphile répond à la Formule (I) dans laquelle tous les radicaux R1 sont identiques et représentent des atomes d'halogènes, soit Fluor, Chlore, Brome et Iode et notamment choisis parmi l'Iode et le Brome.
En particulier, la cyclodextrine répond à la Formule (I) dans laquelle au moins un groupement R1, voire tous les groupements R1, représentent l'isotope radioactif de l'iode de masse atomique 129. Ce groupement radioactif peut notamment permettre de visualiser les systèmes nanoparticulaires dans lesquels il s'intègre. Ceci peut être particulièrement utile pour des études de transport et de biodistribution de principes actifs in vivo.
En particulier, une cyclodextrine selon l'invention répond à la Formule (III) :
Formule (III) m et R
2 ayant la signification indiquée ci-dessus.
La présence du groupement azoture, dans ces dérivés, peut être intéressante notamment pour promouvoir l'auto- organisation de cyclodextrines amphiphiles dans des systèmes colloïdaux stables du type nanocapsules ou nanosphères .
Lorsque des groupements R1, voire tous les groupements R1, représentent des atomes d'halogènes et que ces derniers sont déplacés par des nucléophiles soufrés, on peut alors obtenir des composés répondant à la Formule (I) dans laquelle R1 représente SR3, ayant la signification indiquée ci- dessus pour la Formule (I).
1
Selon un mode de réalisation particulier, la présente invention concerne un composé répondant à la Formule (IV) :
Formule (IV)
dans laquelle m et R2 ont la signification indiquée ci- dessus, n = l, 2, 3, 4 ou 5 et R représente une fonction aminée telle que :
(i) un groupement aminé du type NHY, Y représentant un atome d'hydrogène ou un substituant alkyle, acyle ou carbamate ; ou (ii) un groupement ammonium quaternaire du type -NY3, Y représentant un substituant alkyle.
En particulier, un composé selon l'invention répond à la Formule (IV) dans laquelle n=2, et R représente le groupement tert-butoxycarbonylamino (NHBoc) ou NH2, le groupement aminé résultant pouvant être protoné.
Dans ces dérivés, la présence d'un groupe espaceur du type co-aminoalcanethiol, et notamment cystéaminyle , peut permettre d'accéder à des dérivés polycationiques amphiphiles.
La présence d'un groupe espaceur du type ω- aminoalcanethiol, et notamment cystéaminyle, peut également permettre d'augmenter la réactivité des groupements aminés, en particulier dans le cas des composés répondant à la formule (IV) avec R = -NHY, Y représentant un atome d'hydrogène, un alkyle ou aryle.
Il est à noter par ailleurs que ce groupe espaceur peut être introduit d'une façon simple en utilisant la cystéamine commerciale ou un ω-aminoalcanethiol homologue comme réactif. Ceci évite notamment l'étape de réduction nécessaire lorsque les groupements aminés sont préparés à partir d'un précurseur de type azoture comme c'est le cas dans les exemples décrits dans le document WO 97/33919.
Ce groupement espaceur préfonctionnalisé peut permettre d'associer la cyclodextrine à un motif hydrophile et de reconnaissance cellulaire tel qu'un dérivé glucidique, ou encore un acide aminé ou un peptide, par des liaisons de type urée, thiourée, amide et thioéther qui sont très stables et donnent lieu à des structures bien définies. La liaison thiourée est créée dans une dernière étape ce qui permet de coupler la cyclodextrine à de nombreux substituants, en particulier des substituants comportant un élément de multiplication à plusieurs ramifications portant divers motifs glucidiques, et/ou des groupements chargés,
et/ou une sonde de visualisation ou de détection fluorescente ou radioactive. Ces thiouréidocystéaminyl-cyclodextrines amphiphiles sont des produits originaux qui montrent une affinité remarquable vis-à-vis des lectines complémentaires.
Les cyclodextrines selon l'invention, notamment de type ureido- et thiouréidocystéaminyl-cyclodextrines, peuvent être représentés par la Formule (V) suivante :
Formule (V) dans laquelle m et R
2 ont la signification indiqué ci-dessus, et
- n représente un nombre entier choisi parmi 1, 2, 3, 4 ou 5,
- Z représente un atome d'hydrogène, un groupe alkyle ou aryle,
- Q représente un atome d'oxygène ou un atome de soufre et
- T et W, identiques ou différents, représentent un atome d'hydrogène, un substituant alkyle, aryle ou un élément de reconnaissance cellulaire tel qu'un acide aminé, un peptide, un monosaccharide, un oligosaccharide ou encore un élément de multiplication à plusieurs ramifications portant des groupements glucidiques qui peuvent être identiques ou différents, notamment portant des substituants, ou des groupements chargés tels que des groupements ammonium, notamment de type -NHY protoné ou -NY3, Y représentant un atome d'hydrogène, un substituant alkyle ou aryle.
Tout particulièrement, lorsque T et/ou W représentent un substituant alkyle, ce substituant est un alkyle de 1 à 12 atomes de carbone linéaire, ramifié ou cyclique.
Lorsque T et/ou W représentent un groupement aryle, celui-ci peut notamment être choisi parmi le phényle, le benzyle, le naphtyle ou des dérivés de ces groupements portant d'autres substituants sur le cycle aromatique. Lorsque T et/ou W représentent un substituant dérivé de monosaccharides, ce dernier peut être un dérivé du glucose, du mannose et du galactose, sous forme α ou β.
Le groupe dérivé du monosaccharide peut être substitué, notamment un ou plusieurs groupes hydroxyle du monosaccharide peuvent être remplacés par des groupes alcoxy de 1 à 16 atomes de carbone, des groupes acyloxy, par exemple le groupe acétoxy, des groupes aminés et amides.
Lorsque T et/ou W représentent des dérivés oligosaccharide, les groupes dérivés dOligosaccharides peuvent être les groupes maltosyle, maltotriosyle, lactosyle, ou encore des tri- ou tétrasaccharides, notamment marqueurs d'affinité cellulaire du type Lewis X ou sialyl Lewis X, ou encore des oligosaccharides dérivés de l'héparine. Ces groupes dérivés d'oligosaccharides peuvent également être substitués par des groupes alcoxy, acyloxy, des groupements aminés, sulfatés ou phosphatés.
Lorsque T et/ou W représentent un groupe comportant un élément de multiplication ramifié, cet élément peut être un groupe dérivé du tris(2-hydroxyméthyl)méthylamine (TRIS), du pentaérythritol ou du tris (2-aminoéthyl) aminé (TREN).
Ces éléments de multiplication ramifiés peuvent comporter dans leurs ramifications des groupes dérivés de mono- ou d'oligosaccharides identiques ou différents. À titre d'exemples, on peut citer les groupes dérivés de mono- ou oligosaccharides cités dans le paragraphe précédent, qui peuvent également comporter des substituants oxygénés ou aminés. Ces groupes glucidiques peuvent être liés à l'élément
de multiplication par une liaison oxygénée, soufrée ou aminée .
Selon un mode particulier de réalisation au moins une des ramifications comporte une sonde, en particulier de type fluorescent ou radioactif, permettant notamment la visualisation ou la détection du système
Un composé particulier selon l'invention répond à la Formule (V) dans laquelle n = 2, Q représente un atome de soufre, Z et T représentent un atome d'hydrogène et W représente le groupement méthyle.
D'autres composés particuliers selon l'invention répondent à la Formule (V) dans laquelle m = 6, n = 2, Q représente un atome de soufre, Z et T représentent un atome d'hydrogène et W représente respectivement, (i) le groupement 2-hydroxyéthyle , (ϋ) le groupement 2-(tert- butoxycarbonylamino)éthyle, (iii) le groupement 2- aminoéthyle, le groupement aminé pouvant être protoné ou (iv) le groupement 2-(α-D-mannopyranosyloxy)éthyle.
D'autres composés particuliers selon l'invention répondent à la Formule (V) dans laquelle m = 6, n ≈ 2, Q représente un atome de soufre, Z et T représentent un atome d'hydrogène et W représente l'élément de ramification 2- [2- azidoéthyl-2 ' - ( tert-butoxycarbonylamino )éthyl ] aminoéthyle ou 2 , 2-bis [ 2- ( tert-butoxycarbonylamino)éthyl ] aminoéthyle .
Un autre composé particulier selon l'invention répond à la Formule (V) dans laquelle m = 6, n = 2, Q représente un atome de soufre, Z représente un atome d'hydrogène, T et W sont identiques et représentent le groupement 2- (tert- butoxycarbonylamino )éthyle .
Un autre composé particulier selon l'invention répond à la Formule (V) dans laquelle m = 6, n = 2, Q représente un atome de soufre, Z représente un atome d'hydrogène, T représente le groupement 2-azidoéthyle et W représente le groupement 2-( teri-butoxycarbonylamino)éthyle .
L'invention concerne également des dérivés répondant à la formule (V) dans laquelle au moins l'un des deux substituants T et W représentent un substituant portant d'autres groupements fonctionnels, en particulier des groupements chargés tels que des groupements ammonium, notamment de type -NHY protoné ou de type -NY3, Y représentant un atome d'hydrogène, un substituant alkyle ou aryle.
En particulier, les cyclodextrines répondent à la Formule (V) dans laquelle tous les radicaux R2 représentent un groupement hexanoyle .
Dans un mode de réalisation particulier de l'invention, les composés répondant à la Formule (IV) dans laquelle Y représente un atome d'hydrogène ou un substituant alkyle ou aryle ainsi que les dérivés répondant à la formule (V) dans laquelle au moins un des substituants T et W portent un groupement de type NHY, Y ayant la même signification ci- dessus mentionnée, peuvent être isolés sous forme de sels d'ammonium ou de base libre.
Dans le cas de sels, le contre-ion peut être un anion monovalent, en particulier un halogénure tel que le chlorure, le bromure ou l'iodure. Aussi bien la base libre que le sel peuvent être utilisés comme précurseurs dans la préparation de thiouréidocystéaminyl-cyclodextrines amphiphiles.
Dans un mode de réalisation préféré, les composés répondent aux formules (I) à (V), dans lesquels tous les
radicaux R2 représentent le groupement hexanoyle et/ou le groupement tétradëcanoyle (myristoyle) et m = 6.
Selon un mode de réalisation, le composé selon l'invention répond à la Formule (VI) suivante :
Formule (VI) dans laquelle
- n ≈ 1, 2, 3, 4 ou 5, et m et R2 ont la signification indiquée ci-dessus, le groupement NCS représente le groupement isothiocyanate .
Des composés particuliers selon l'invention répondent à la Formule (VI) dans laquelle n = 2 , m = 6 et R2 représente respectivement le groupement hexanoyle et le groupement tétradécanoyle (myristoyle).
Selon un autre mode de réalisation, le composé selon l'invention répond à la Formule (VII) suivante :
Formule (VII)
dans laquelle m, R1 et R2 ont la signification indiquée ci-dessus.
La cyclodextrine correspondant à la Formule (VII) présente des groupements hydroxyle sur le carbone en position
6 de chacun des monomères, excepté pour l'un des monomères dont le carbone en C6 porte un substituant Rα différent du groupement hydroxyle.
Des composés particuliers selon l'invention répondent à la Formule (VII) dans laquelle le groupement R1 représente respectivement le groupement 2-(tert- butoxycarbonylamino)éthylthio, le groupement 2- aminoéthylthio, le groupement 2-[iV'-(2-α-D- mannopyranosyloxyéthyl ) thiouréido ] éthylthio et le groupement 2- ( N"- ( 2- (cyclomaltoheptaose-6I-désoxy-6I- yl )éthylthio) thiouréido ) éthylthio .
Des composés particuliers selon l'invention correspondent à la Formule (VII) dans laquelle m = 6 et tous les groupements R2 représentent un groupement hexanoyle.
Tout particulièrement, l'invention concerne des cyclodextrines selon l'invention susceptibles de développer des interactions notamment non-covalentes avec, au moins une molécule hôte, notamment formant ainsi un complexe d'inclusion.
La présente invention concerne également la préparation des composés (cyclodextrines) décrits ci-dessus.
La méthode de synthèse est très flexible en ce qui concerne la nature des groupements R1 et R2. Elle peut notamment permettre d'optimiser les caractéristiques amphiphiles, l'aptitude à l'auto-organisation en milieu aqueux, les propriétés des systèmes colloïdaux résultants
et/ou la capacité de complexer, entre autres, des petites molécules ou des macromolécules.
Le procédé de préparation de cyclodextrine selon l'invention comprend les étapes suivantes consistant à :
(i) introduire au moins un groupement R1 sur au moins un des carbones portant l' hydroxyle primaire ou à protéger au moins une des hydroxyles primaires du composé de départ, notamment d'une cyclodextrine ; (ϋ) introduire au moins un groupement R2 sur au moins un hydroxyle secondaire porté par le carbone en position 3 des monomères formant une cyclodextrine ; et
(iii) récupérer au moins une cyclodextrine selon l'invention, en particulier un composé de Formule (I), obtenue.
Selon une première variante, le procédé peut comprendre une • étape préalable à l'introduction de R2 consistant à protéger au moins un hydroxyle primaire, voire tous les hydroxyles primaires.
Le groupement protecteur introduit initialement à partir d'une cyclodextrine commerciale peut être un groupement du type silyléther, en particulier le tert- butyldiméthylsilyléther, notamment dans le cas des dérivés per(C-β) fonctionnalisés et lorsque les groupements R2 représentent un substituant alkyle.
La protection des hydroxyles primaires peut notamment être effectuée par réaction avec le chlorure de tert- butyldiméthylsilyle et l'imidazole dans un solvant polaire aprotique, de préférence la iV,-V-diméthylformamide.
Les dérivés sélectivement silylés sur les OH primaires peuvent être ensuite alkylés par réaction avec un halogénure d' alkyle, notamment dans un solvant polaire aprotique, comme
la 2V,2V-diméthylformamide, en présence d'une base, en particulier l'hydrure de sodium.
Ultérieurement, les groupements silylés peuvent être hydrolyses, notamment par action d'un acide aqueux, de préférence l'acide acétique ou l'acide trifluoroacétique, ou bien par traitement avec un sel de l'acide fluorhydrique, de préférence le fluorure de tétra-Λ-butylammonium.
Les dérivés déprotégés résultants, présentant des fonctions OH primaires libres et, portant des chaînes alkyle sur les OH secondaires, peuvent être transformés en d'autres dérivés portant des groupements fonctionnels divers sur la face primaire suivant des méthodes de transformation des hydroxyles primaires, en particulier l'halogénation. On peut suivre pour cela les procédés commentés ci-dessous pour la fonctionnalisation des cyclodextrines commerciales sur la face primaire.
Selon une seconde variante, le procédé peut comprendre une étape préalable à l'introduction de R2 consistant à substituer au moins un hydroxyle primaire, voire tous les hydroxyles primaires, par un groupement R1.
En particulier, le procédé peut comprendre une étape d'halogénation des cyclodextrines sur au moins un carbone portant un hydroxyle primaire, voire sur tous les carbones portant un hydroxyle primaire.
En particulier, le procédé selon l'invention comprend une étape de substitution des OH primaires par des groupements halogènes, de préférence l'iode ou le brome, suivie d'une acylation d'au moins un hydroxyle secondaire, voire de tous les hydroxyles secondaires, pour conduire à un composé de Formule (I) dans lequel R2 représente au moins un groupement acyle.
Les dérivés de cyclodextrines persubstitués en position alcool primaire par des groupements halogènes peuvent notamment être préparés à partir de la cyclodextrine commerciale en une seule étape et avec de bons rendements par réaction avec divers réactifs d'halogénation sélective. On peut utiliser pour cela les procédés décrits par J. Defaye et col. dans les documents Supramol. Chem, 2000, 12, pp. 221-
224, Polish J. Chem. 1999, 73, pp. 967-971, Tetrahedron Lett.
1997, 38, pp. 7365-7368, Carbohydr. Res. 1992, 228, pp. 307- 314, et Angew. Chem., Int. Ed. Engl. 1991, 30, pp. 78-80.
Les cyclodextrines per(C-6)halogénée peuvent subir une réaction d'acylation des OH secondaires dans une seconde étape, notamment pour obtenir des composé de Formule (I) dans lesquels au moins un groupement R2 représente un acyle.
On peut suivre le procédé décrit par P. Zhang et al. dans Tetrahedron Lett. 1991, 32, 2769-2770. Cependant, ce procédé peut demander des temps de réaction longs et des températures élevées, conduisant à des rendements modestes, voire faibles, dans le cas de ces dérivés halogènes.
Selon une autre méthode, l'acylation des cyclodextrines per(C-6)halogénées peut être effectuée par réaction avec un anhydride d'acide dans un solvant polaire aprotique, notamment la N, iV-diméthylformamide , en présence d'une base, notamment la JV,iV-diméthylaminopyridine. Dans ces conditions, la réaction peut être complète en 45 min. à température ambiante, avec des rendements en produit pur de l'ordre de 70%.
Dans un mode particulier de l'invention, la préparation de cyclodextrines répondant à la Formule (I) dans laquelle tous les groupements R1 représentent un atome d'halogène, à la Formule (III) et à la Formule (IV) dans laquelle R représente NHY, Y représente un groupement acyle ou carbamate
et le radical R2 représente un groupement acyle, peut être réalisée selon le procédé qui consiste à faire réagir un dérivé de cyclodextrine sélectivement halogène, azidé ou fonctionnalisé avec des groupements NHY en position alcool primaire avec un anhydride d'acide, notamment dans la N,N- diméthylformamide, en présence d'une base, de préférence la N,-V-diméthylaminopyridine .
Les cyclodextrines amphiphiles per(C-6)halogénées répondant à la formule (I) pour laquelle tous les R1 sont identiques peuvent être utilisées comme produits de départ dans la préparation des dérivés incorporant d'autres groupements fonctionnels sur la face primaire. Les groupements halogènes, qui sont de bons groupements partants, peuvent être déplacés par des groupements nucléophiles azotés ou soufrés, tels que l'anion azoture, les aminés ou les thiols. On peut suivre à ce propos les procédés décrits par J. Defaye et col. dans les documents Carbohydr. Res. 1995, 268, 57-61, J. Chem. Soc, Perkin Trans., 2, 1995, pp. 1479-1487 et WO2004087768.
Les composés de formule (III) peuvent être préparés à partir d'un précurseur répondant à la formule (I) dans laquelle les groupements R1 sont tous des atomes d'halogène par réaction avec un anion azoture.
Alternativement, les cyclodextrines de Formule (III) peuvent être obtenues par acylation de la per(6-azido-6- désoxy) cyclodextrine correspondante. On peut suivre à ce propos le procédé décrit par P. Zhang et al. dans Tetrahedron Lett. 1991, 32, 2769-2770 ou bien le nouveau procédé utilisant la iV,-V-diméthylformamide commenté ci-dessus pour 1' acylation des cyclodextrines per(C-6)halogénées.
Ainsi, dans un mode particulier de l'invention, le procédé de préparation de cyclodextrine de Formule (III) consiste à faire réagir un dérivé halogène de cyclodextrine de Formule (I) où R1 est un atome d'halogène avec anion azoture dans la N,iV-diméthylformamide.
Les cyclodextrines de l'invention répondant à la Formule (III) peuvent en outre être utilisées comme produits de départ dans la préparation d'autres dérivés neutres ou chargés grâce à la réactivité du groupement azoture.
Ainsi, une réaction d'addition 1,3-dipolaire catalysée par le cation cuivre (II) avec un alcyne peut permettre d'attacher une variété de substituants au moyen d'un cycle 1,2,3-triazole. On peut suivre pour cela le procédé décrit par Santoyo-Gonzâlez et col. dans Org. Lett. 2003, 5, 1951- 1954.
Par ailleurs, la réduction des groupes azoture peut conduire à la per(C-6 ) aminé correspondante. Cette réduction peut être réalisée par une variété de procédés tels que l'hydrogénation en présence d'un catalyseur hétérogène à base de palladium, platine ou nickel, la réaction avec une phosphine, de préférence la triphényl- ou la tributylphosphine, ou la réaction avec le propanedithiol . On peut suivre pour cela les procédés décrits par J. Defaye et col. dans Carbohydr. Res. 1995, 268, 57-61 ou par L. Jicsinszky et col. dans Comprehensive Supramolecular Chemistry, Vol. 3 (Eds J. Szejtli et T. Osa), Pergamon, Oxford, 1996, pp. 57-198.
Les composés de Formule (IV) peuvent être préparés à partir d'un composé de Formule (I) dans laquelle les radicaux R1 sont identiques et représentent des atomes d'halogène par
réaction avec un nucléophile soufré, notamment de formule HSCH2(CH2)nR.
On peut suivre à ce propos le procédé décrit dans le document WO2004087768. Alternativement, un composé neutre de Formule (IV) (R = NHY, avec Y = acyle ou carbamate) peut être obtenu par acylation du dérivé correspondant où les R2 représentent H, obtenu comme décrit dans le même document cité, par acylation. On peut suivre à ce propos le procédé décrit par P. Zhang et al. dans Tetrahedron Lett. 1991, 32, 2769-2770 ou bien le nouveau procédé utilisant la -V,-V-diméthylformainide commenté ci-dessus pour l' acylation des cyclodextrines per(C- 6)halogénées. Ce procédé ne permet pas d'accéder directement à des dérivés chargés. Cependant, ce type de dérivés peut être obtenu à partir des carbamates correspondants, de préférence le dérivé tert-butoxycarbonyle (formule IV avec R = NHBoc) par hydrolyse acide de la fonction carbamate.
Dans un mode de réalisation particulier, le procédé de préparation de cyclodextrine de Formule (IV), consiste à faire réagir un dérivé de cyclodextrine de Formule (I) dans laquelle les radicaux R1 sont identiques et représentent des atomes d'halogène avec de la cystéamine, un ω-aminothiol , ou un de leur dérivé, notamment dans la i\r,iV-diméthylformamide, en présence d'une base, telle que la triéthylamine ou le carbonate de césium.
Dans un autre mode de réalisation particulier, le procédé de préparation de cyclodextrines répondant à la Formule (IV) dans laquelle R représente un groupement aminé primaire (NH2) consiste à hydrolyser le groupement carbamate dans un précurseur de Formule (IV) dans lequel R représente un groupement NHBoc.
Les composés de Formule (V) (uréido- ou thiouréidocystéaminyl-cyclodextrines) peuvent être préparés par un procédé comprenant les étapes consistant à faire réagir un composé de Formule (IV) où R représente NHY, avec Y représentant un atome d'hydrogène ou un substituant alkyle ou aryle, avec un isocyanate ou isothiocyanate de formule W-NCQ (Q représente un atome d'oxygène ou un atome de soufre, respectivement) dans laquelle W a la signification donnée ci- dessus. Cette réaction peut être effectuée dans un solvant organique tel que la pyridine ou encore dans un mélange d'eau avec un solvant organique miscible tel que l'acétone. On obtient ainsi un composé de Formule (V) (uréido- ou thiouréidocystéaminyl-cyclodextrine) dans laquelle T représente un atome d'hydrogène.
Lorsque Y représente H dans la formule (IV), les thiourées obtenues sont iV^-iV'-disubstituées, alors que lorsque Y représente un substituant alkyle, tel que méthyle, éthyle, propyle ou butyle, les thiourées obtenues sont N,N,Nr- trisubstituées.
Le composé isothiocyanate W-NCS peut être préparé de différentes manières :
- lorsque W est un groupe dérivé d'un monosaccharide ou d'un oligosaccharide, par réaction du thiophosgène sur un aminodésoxyglycose ou un glycoside comportant un groupement aminé dans l'aglycone,. On peut suivre pour cela les procédés rapportés par J. M. Garcia Fernandez et C. Ortiz Mellet dans Adv. Carhohydr. Chem. Biochem. 1999, 55, pp. 35-135. - lorsque W comporte un élément de multiplication ramifié dérivé du tris(2-hydroxyméthyl)méthylamine (TRIS), on peut préparer l' isothiocyanate correspondant par réaction du thiophosgène sur le dérivé aminé portant des substituants glucidiques sur les positions alcool primaire, comme décrit
dans le document Chem. Commun., 2000, pp 1489-1490. Le glycodendron trivalent aminé précurseur peut être obtenu par glycosidation d'un dérivé du TRIS avec la fonction aminé convenablement protégée sous forme de dérivé carbobenzoxy, comme décrit par P. R. Ashton et al. dans J. Org. Chem. 1998, 63, pp 3429-3437.
Lorsque W comporte un élément de multiplication ramifié dérivé du tris ( 2-aminoéthyl) aminé (TREN), on peut préparer l ' isothiocyanate correspondant par réaction du thiophosgène sur un dérivé sélectivement protégé sur deux des groupements aminés primaires, par exemple par le groupement Boc . On peut suivre pour cela le procédé décrit par Benito et al. dans J. Am. Chem. Soc. 2004, 126, 10355-10363. Il est aussi possible d'utiliser un dérivé portant deux groupements protecteurs différents, tel que le groupement Boc et le groupement trifluoroacétyle. Ces types d'éléments de ramification peuvent être préparés par protection sélective partant du TREN commercial ou par réaction du bis (2- aminoéthyl ) aminé , sélectivement protégé sur les groupements aminés primaires par les groupements mentionnés, avec le 2- azidoéthyl-p-toluènesulfonate, suivi de la réduction du groupement azoture puis réaction avec le thiophosgène.
- Lorsque W comporte un élément de multiplication ramifié dérivé du pentaérythritol, les glycodendrons convenablement fonctionnalisés avec un groupement isothiocyanate peuvent être préparés à partir du pentaérythritol commercial par une séquence de réactions qui implique:
(a) une triallylation sélective par traitement avec le bromure d'allyle, ce qui laisse un seul groupe hydroxyle libre ;
(b) l'addition radicalaire d'un 1-thiosucre à la double liaison des groupements allyle.
Cette réaction peut se faire, soit par activation par la lumière ultraviolette, soit en présence d'un initiateur de radicaux libres tel que l'azobis(isobutyronitrile) ou l'acide p-nitroperbenzoique . A titre d'exemple, on peut adapter les conditions réactionnelles décrites par D. A. Fulton et J. F. Stoddart dans Org. Lett. 2000, 2, pp 1113-1116 ou par X. -B. Meng et al. dans Carbohydr. Res. 2002, 337, pp 977-981.
Cette réaction peut permettre l'addition séquentielle des différentes ramifications glucidiques. II est ainsi possible d'accéder à des glycodendrons (polyconjugué qui comporte plusieurs unités glucidiques, identiques ou différentes, liées de façon covalente à un noyau ramifié, le noyau ramifié portant en outre un groupement fonctionnel permettant son greffage sur une autre plateforme) homogènes aussi bien qu'hétérogènes dans lesquels les substituants glucidiques répondent aux structures mentionnées ci-dessus. On peut suivre pour cela le procédé décrit par Gόmez-Garcia et al. dans J. Am. Chem. Soc. 2005, 127, 7970-7971. Cette approche permet également l'incorporation d'un substituant autre qu'un dérivé glucidique à la structure, en particulier une sonde de type fluorescent telle qu'un dérivé de la fluorescéine.
(C) la transformation du groupement alcool primaire restant en groupement isothiocyanate. Cette transformation peut se faire par exemple par transformation du groupe hydroxyle en un bon groupe partant tel que le p- toluènesulfonate ou le trifluorométhanesulfonate, suivi de déplacement nucléophile par l'anion azoture et isothiocyanation de l' azoture résultant par réaction avec la triphénylphosphine et le disulfure de carbone. Pour cette transformation, on peut suivre le procédé décrit dans le document Chem. Commun., 2000, pp 1489-1490.
Les composés répondant à la formule (V) ( thiouréiocystéaminyl-cyclodextrines) avec Q représentant un atome de soufre et Z représentant un atome d'hydrogène peuvent être également préparés à partir d'un précurseur répondant à la Formule (VI), correspondant à la Formule (I) dans laquelle m et R2 ont la signification indiquée ci-dessus et les R1 représentent le groupement -SCH2(CH2JnNCS, avec n = 1, 2, 3, 4 ou 5.
Ces composés de Formule (VI) peuvent être obtenus à partir de l'aminé correspondante de Formule (IV), dans laquelle m et R2 ont la signification indiquée ci-dessus avec n = 1, 2, 3, 4 ou 5 et où R représente NHY, avec Y représente un atome d'hydrogène, par réaction avec un agent d'isothiocyanation, de préférence le thiophosgène.
Le couplage d'un produit de Formule (VI) avec une aminé primaire ou secondaire de formule générale WNHT, ou T et W ont la signification indiquée ci-dessus, conduit à un composé de Formule (V) (thiouréidocystéaminyl-cyclodextrine amphiphile) .
Dans un mode de réalisation particulier, le procédé de préparation de composés de Formule (V) dans laquelle Q représentant un atome de soufre et Z représentant un atome d'hydrogène consiste à faire réagir un précurseur répondant à la Formule (VI) avec une aminé WNHT, W et T ayant la signification ont la signification donnée ci-dessus.
Les composés correspondant à la Formule (VII) dans laquelle l'un des substituants R1 est différent de l'hydroxyle et les autres représentent OH, peuvent être préparés selon le procédé consistant à :
(i) introduire sélectivement un groupement fonctionnel sur une des positions primaires de la cyclodextrine;
(ii) protéger les autres hydroxyles primaires avec un groupement protecteur, en particulier sous forme de silyléther;
(iii) introduire ensuite les substituants sur les hydroxyles primaires; et
(iv) éventuellement hydrolyser les groupements protecteurs .
À partir des 6I-p-tolylsulfonyl cyclodextrines, un procédé particulier de production des composés répondant à la Formule (VII) (préparation décrite dans WO 99/61483), consiste à:
(i) déplacer le groupement p-toluènesulfonate par un nucléophile, notamment halogénure, azoture, dérivé soufré ou dérivé aminé, ;
(ii) protéger des OH primaires restants, notamment sous forme de groupements silyléther, notamment de tert- butyldiméthylsilyléther ;
(iii) acyler ou alkyler des OH secondaires, et ; (iv) hydrolyser ou fluorolyser des groupements silyléther pour régénérer les hydroxyles correspondants.
Les composés de Formule (VII) (dérivés de type cystéaminyl-cyclodextrine amphiphiles) dans laquelle m, et R2 ont la signification indiquée ci-dessus et R1 représente SCH2(CH2JnR, n et R ayant la signification indiquée ci-dessus pour les composés de Formule (IV), peuvent être obtenus à partir d'un dérivé de cyclodextrine monosubstituée en position alcool primaire par un groupement halogène, en particulier les 6I-bromodésoxy ou 6I-désoxyiodo- cyclodextrines. La réaction avec un dérivé de cystéamine protégée sur le groupement aminé, de préférence par le groupement Boc, ou, en général, avec un dérivé d'un ω- aminoalcanethiol iV-protégé, éventuellement iV-alkylé, suivant
la méthodologie décrite ci-dessus pour la préparation des dérivés persubstitués en position alcool primaire, conduit à des monocystéaminyl-cyclodextrines. La séquence de réactions commentée ci-dessus pour accéder aux dérivés de Formule (VII) permet d'obtenir les dérivés amphiphiles de l'invention.
Les dérivés de cyclodextrines monosubstituées en position alcool primaire par des groupements halogènes, utilisés comme produits de départ dans ce procédé, peuvent être préparés à partir du dérivé 6I-C>-p-tolylsulfonylé de la cyclodextrine en une étape et avec un bon rendement par déplacement nucléophile du groupe tosylate par un anion halogénure dans la ^W-diméthylformamide.
De façon générale, les procédés de préparation de cyclodextrines amphiphiles per(C-β) substituées sont aussi applicables à la préparation des dérivés monofonctionnalisés en position primaire, c'est-à-dire substitués au niveau d'un carbone portant un hydroxyle primaire.
Les procédés de préparations de cyclodextrines selon l'invention et décrits ci-dessus ont l'avantage de permettre l'obtention des dérivés souhaités sous forme de produits purs et homogènes en un nombre réduit d'étapes et avec des rendements élevés. En outre, ces procédés permettent d'accéder à des dérivés aussi bien neutres que polychargés, en particulier polycationiques, qui peuvent porter des éléments de bioreconnaissance ou de visualisation.
Les nouveaux dérivés de cyclodextrines selon l'invention, tout en comportant une variété de groupements fonctionnels ou éléments de bio-reconnaissance ou de visualisation, peuvent être capables de s'auto-organiser, notamment ' sous forme de systèmes colloïdaux dispersibles.
En outre, la possibilité d'incorporer dans ces nouveaux dérivés différents groupements sur la face des alcools primaires est intéressante, notamment pour moduler les propriétés d'interaction de la surface extérieure des systèmes colloïdaux nanoparticulaires (nanocapsules ou nanosphères) qu'ils permettent d'obtenir.
Ainsi, selon un autre de ses aspects, l'invention a pour objet des nanostructures comprenant au moins une cyclodextrine de Formule (I) selon l'invention, éventuellement comprenant au moins une molécule hôte.
En particulier, ces nanostructures peuvent incorporer, comprendre, être associées ou former un complexe, avec au moins une molécule hôte. Les nanostructures peuvent tout particulièrement se présenter sous forme de nanosphères, de nanocapsules et ou de nanoparticules .
Les nanostructures, notamment les nanosphères et/ou nanocapsules peuvent comprendre en outre au moins une molécule hôte.
La molécule hôte peut être tout principe actif, notamment une molécule pharmacologiquement active, par exemple des médicaments ou des acides nucléiques.
La molécule hôte peut être une molécule choisie dans le groupe comprenant les nutriments, les oligoéléments, les vitamines , les arômes .
La molécule hôte peut encore être une molécule utilisable en cosmétologie, notamment les molécules citées dans le « International Cosmetic Ingrédient Dictionary and Handbook" de John A. WENNINGER et G.N. McEWEN.
La molécule hôte peut encore être un acide nucléique, notamment sélectionnée dans le groupe comprenant l'ADN, l'ARN, des acides nucléiques modifiés tels que les ribonucléotides ou les désoxyribonucléotides présentant un
groupement sucre ou un groupement carboné modifié, ou bien encore des analogues synthétiques de nucléotides.
Les nanocapsules peuvent enfermer ou contenir une phase organique, notamment telle que le Miglyol 812 (marque déposée) .
Les nanoparticules peuvent comprendre au moins un acide nucléique, notamment sélectionné dans le groupe comprenant l'ADN (linéaire ou plasmidique ) , l'ARN (et notamment les ARN interférants -ARNi- ou encore « silencing » -RNAsi-, les micro-ARN) , des acides nucléiques modifiés, tels que les ribonucléotides ou les désoxyribonucléotides présentant un groupement sucre ou un groupement carboné modifié, ou encore des analogues synthétiques de nucléotides.
En particulier, les cyclodextrines répondant à la Formule (I) dans laquelle R1 représente un • atome d'halogène, présentent une excellente auto-organisation dans des systèmes colloïdaux stables du type nanocapsules ou nanosphères.
Par ailleurs, les composés neutres répondant aux Formules
(I) à (VII) se sont révélés particulièrement intéressants en ce qui concerne leur capacité d'auto-organisation, notamment en nanocapsules ou nanosphères, dans des systèmes colloïdaux stables.
On entend par « nanosphère blanche » ou « nanocapsule blanche » au sens de la présente invention, des nanosphères ou des nanocapsules non chargées en molécule hôte, et notamment en principe actif.
Les nanostructures, en particulier les nanosphères, notamment blanches et/ou homogènes, c'est-à-dire dans lesquelles les cyclodextrines sont toutes de même nature, en particulier présentent toutes la même formule, peuvent être
préparées selon le procédé comprenant les étapes consistant à:
(i) ajouter un solvant organique miscible à l'eauf contenant au moins un composé de Formule (I) à une solution aqueuse, le volume d'eau variant notamment de une à deux fois le volume de solvant organique, sous agitation ;
(ii) puis après nanoprécipitation, c'est-à-dire formation des nanosphères, éliminer le solvant organique.
La suspension aqueuse obtenue, qui peut présenter un aspect opalescent à laiteux (suivant les produits mis en œuvre), peut ensuite être concentrée jusqu'à un volume final souhaité .
Le procédé de préparation de nanosphères présentée ci- dessus peut être appliqué à la préparation de nanosphères hétérogènes. Pour cela, l'étape (i) du procédé de préparation présenté ci-dessus consiste à introduire dans une solution aqueuse au moins deux solutions organiques miscibles à l'eau, contenant des dérivés de cyclodextrine amphiphile différents, notamment de formules chimiques différentes, en proportion variable sous agitation. Le volume de solution aqueuse peut aller de une à deux fois le volume de solvant organique.
Cette méthode de préparation de nanosphères de nature hétérogène permet d'incorporer des cyclodextrines amphiphiles chargées dans la nanosphère même lorsque ces dérivés utilisés seuls ne forment pas de nanosphères.
Les nanostructures, et notamment les nanocapsules, en particulier les nanocapsules blanches, c'est-à-dire des nanocapsules n'incorporant pas de principe actif, peuvent être préparées par un procédé comprenant les étapes consistant à :
(i) préparer une phase acétonique contenant une petite fraction de triglycérides, de préférence une proportion
acétone : triglycérides allant de 1000:1 à 10:1, au moins une cyclodextrine amphiphile, au moins un agent tensioactif lipophile non ionique, et une phase hydrophile contenant de l'eau distillée et au moins un agent tensioactif hydrophile non ionique ;
(ii) introduire la phase organique dans la phase hydrophile sous agitation magnétique ;
(iii) Après formation des nanocapsules (nanoprécipitation) , éliminer le solvant organique, notamment sous pression réduite à +35 0C.
En particulier, dans l'étape (i) on ajoute au moins deux préparations de cyclodextrines, en proportion variable, contenant respectivement des cyclodextrines de Formule (I) de formules chimiques différentes. La suspension aqueuse présentant un aspect laiteux peut ensuite être concentrée jusqu'au volume final souhaité.
Selon un autre de ses aspects, l'invention concerne un procédé de préparation de nanostructures, en particulier de nanosphères et/ou de nanocapsules, comprenant, contenant ou portant au moins une molécule hôte tel que défini ci-dessus, le procédé comprenant les étapes suivantes :
(i) introduire au moins une phase organique contenant un solvant miscible à l'eau, comme l'acétone, un dérivé de cyclodextrine, ou, alternativement, plusieurs préparations comprenant respectivement des cyclodextrines répondant à la Formule (I) de formules chimiques différentes en proportion variable, et le principe actif, dans une phase aqueuse, notamment contenant de l'eau distillée, éventuellement avec un tensioactif, en particulier hydrophile non ionique, sous agitation,
(ii) puis après nanoprécipitation, c'est-à-dire formation des nanocapsules ou de nanosphères, éliminer le solvant organique.
Selon un autre de ses aspects, la présente invention a pour objet des compositions, notamment cosmétique, alimentaire et/ou pharmaceutique, comprenant au moins une cyclodextrine et/ou une nanostructure selon l'invention et une molécule hôte, notamment une molécule pharmacologiquement active .
En particulier l'invention concerne une composition pharmaceutique contenant par dose unitaire de 50 mg à 500 mg de cyclodextrine et/ou de nanostructures selon l'invention et une molécule hôte pharmacologiquement active dans une proportion molaire dérivé de cyclodextrine/molécule hôte qui peut aller de 50:1 à 1:500, notamment de 25:1 à 1:10, en particulier de 20:1 à 1:1, voire de 10:1 à 1,5:1.
Un avantage important de la présente invention, réside dans le fait que l'auto-organisation dans des systèmes colloïdaux nanoparticulaires (nanosphères ou nanocapsules) permet d'augmenter significativement la capacité de charge en principe actif, bien au-delà d'une proportion molaire 1:1. Le principe actif peut se localiser aussi bien dans la cavité de la cyclodextrine qu'en surface ou dans la matrice des nanosphères ou encore dans le cœur lipophile des nanocapsules .
On peut noter que pour les cyclodextrines classiques, non nanoparticulaires, il peut y avoir une limitation importante dans la taille des molécules susceptibles d'être encapsulées, limitation déterminée par la taille de la cavité hydrophobe. La limite se situerait vers 900 Dalton (D) de masse moléculaire. La capacité de prise en charge maximale est de 1:1 en base molaire. Avec les systèmes
nanoparticulaires on va bien au delà. Par exemple, des plasmides de 4000 kD ont été encapsulés.
L'invention concerne encore des dérivés de cyclodextrines capables de former des nanostructures stables de quelques dizaines ou centaines de nanomètres par désolvatation en présence d'un non solvant (nanosphères) , éventuellement en présence d'une phase lipophile
(nanocapsules) . Ces suspensions colloïdales prennent en charge des quantités importantes de molécule hôte, dans une proportion molaire dérivé de cyclodextrine/molécule hôte qui peut aller de 50:1 à 1:500, notamment de 25:1 à 1:10, en particulier de 20:1 à 1:1, voire de 10:1 à 1,5:1, et permettent notamment (i) de modifier, améliorer et contrôler les propriétés pharmacocinétiques de principes pharmacologiquements actifs, (ii) de transporter des composés actifs, et (iii) de faciliter ou améliorer le transfert intracellulaire de composés actifs.
Les cyclodextrines et/ou les nanostructures selon l'invention peuvent être utilisées pour la complexation des acides nucléiques et/ou pour l'introduction d'acides nucléiques dans des cellules .
Les cyclodextrines et/ou les nanostructures selon l'invention peuvent aussi être utilisées pour faciliter et/ou améliorer le transfert intracellulaire de molécule hôte et/ou pour contrôler la libération d'une molécule hôte.
Les cyclodextrines et/ou les nanostructures selon l'invention peuvent aussi être utilisées pour l'introduction d'acides nucléiques dans des cellules, notamment eucaryotes.
Selon un mode de réalisation particulier de la présente invention, les nanoparticules selon l'invention comprennent en outre un acide nucléique.
On entend par acide nucléique l'ADN (linéaire ou plasmidique) , l'ARN (et notamment les ARN interférants -ARNi- ou encore « silencing » -RNAsi-, les micro-ARN) ou les acides nucléiques modifiés, notamment les ribonucléotides ou les désoxyribonucléotides présentant un groupement sucre ou un groupement carboné modifié. Les acides nucléiques peuvent être utilisées sous forme simple brin, double brin ou partiellement double brin.
Les acides nucléiques peuvent également être composés ou contenir des analogues synthétiques de nucléotides, notamment les ribonucléotides présentant un groupement sucre ou groupement carboné modifié. Par exemple, les analogues synthétique de ribonucléotides présentant un groupement sucre modifié présentent un groupement 2 ' -OH remplacé par un groupement sélectionné parmi un atome d'hydrogène, un halogène, un groupement OR/ R7 SH, SR, NH2, NHR, NR2 ou CN, dans lequel R est un groupement alkyle, alcényle ou alcynyle de 1 à 6 carbone et l'halogène est le fluor, le chlore, le brome ou l'iode. Les ribonucléotides présentant un groupement carboné modifié peuvent avoir leur groupement phosphoester lié au ribonucléotide adjacent qui est remplacé par un groupement modifié tel qu'un groupement phosphothioate. Les ribonucléotides peuvent être des ribonucléotides présentant un noyau purine ou pyrimidine modifié. Comme exemples de tels noyaux modifiés, on citera notamment les uridines ou les cytidines modifiées en position 5, telles que la 5- (2- amino)propyl uridine et la 5-bromo uridine, les adénosines et guanosines modifiées en position 8, telle que la 8-bromo guanosine, les nucléotides déazotés, telle que la 7-déaza- adénosine, les nucléotides N- et 0-alkylés, telle que la N6- méthyl adénosine.
Les inventeurs ont notamment constaté en ce qui concerne les cyclodextrines de formule (V) que lorsque le groupement
urée ou thiourée porte des groupements chargés positivement, des effets de synergie dans la complexation des acides nucléiques peuvent se produire, les cyclodextrines étant alors optimisées pour l'introduction (la transfection) de matériel génétique dans des cellules.
Les dérivés de cyclodextrines amphiphiles polycationiques sont particulièrement efficaces pour complexer les acides nucléiques. Ces dérivés s'auto- organisent autour d'un acide nucléique en milieu aqueux, donnant lieu à formation des nanoparticules qui renferment l'acide nucléique et qui sont capables de traverser la membrane cellulaire, comme cela est mis en évidence dans la partie exemples, pour libérer l'acide nucléique qui peut alors exprimer son activité biologique à l'intérieur de la cellule.
Un mode de réalisation particulier de l'invention concerne un procédé in vitro de transfert de molécules hôtes dans des cellules, notamment des cellules eucaryotes consistant à :
(i) mettre en contact le complexe nanostructure/molécule hôte et/ou le complexe cyclodextrine/molécule hôte autre avec des cellules ; (ϋ) laisser en contact les cellules avec le complexe nanostructure/molécule hôte et/ou le complexe cyclodextrine/molécule hôte pendant un temps T de préférence compris entre 4 et 72 heures ;
(iii) retirer le milieu de culture et laver les cellules.
Le temps de contact pourra dépendre des cyclodextrines utilisées pour former les nanostructures, notamment de la présence d'éléments de bio-reconnaissance pouvant se lier
spécifiquement à un récepteur membranaire et faciliter ainsi la pénétration du complexe cyclodextrine/molécule hôte et/ou complexe nanostructure/molécule hôte dans la cellule.
Le temps optimal de contact pourra aisément être déterminé par l'homme du métier en effectuant des expériences à différents temps, par exemple 4, 8, 12, 24, 36, 48, 60 et 72 heures.
L'incorporation d'éléments de bioreconnaissance, par exemple le ou les ligands complémentaires à un récepteur, permet aux dérivés les incorporant d'être reconnus de façon très efficace par les récepteurs membranaires spécifiques.
Notamment, un élément de reconnaissance glucidique sera reconnu spécifiquement par une lectine en fonction des substituants glucidiques incorporés, permettant de cette façon la vectorisation de petites molécules ou macromolécules au niveau de cellules cibles.
L'invention a encore pour objet une composition pharmaceutique comprenant soit (i) un dérivé de cyclodextrine amphiphile de formule (I) ou (ii) un système colloïdal selon l'invention préparé à partir d'une ou plusieurs cyclodextrines amphiphiles de formule (I), (iii) soit un complexe d'un dérivé de cyclodextrine amphiphile de formule (I) ou (iv) un système colloïdal préparé à partir d'une ou plusieurs cyclodextrines amphiphiles de formule (I) et d'une molécule pharmacologiquement active, prëférentiellement avec un véhicule pharmacologiquement acceptable.
L'invention a aussi pour objet une composition pharmaceutique comprenant (i) un dérivé de cyclodextrine amphiphile de formule (I) selon l'invention et/ou une nanostructure selon l'invention et (ii) un acide nucléique, en particulier un fragment d'ADN, de préférence avec un polymère biocompatible tel que le polyéthylèneglycol.
La composition peut comprendre en plus un élément d'adressage, tel qu'un dérivé glucidique ou peptidique ou bien encore un dérivé d'un ligand se liant spécifiquement à un récepteur cellulaire, notamment l'acide folique, ou encore la transferrine. Cet élément d'adressage peut s'intégrer dans la nanoparticule cyclodextrine amphiphile-ADN ou bien peut s'ancrer sur ce système par inclusion d'un motif hydrophobe porté par l'élément de vectorisation, par exemple un substituant dérivé de l ' adamantane , dans la cavité de la cyclodextrine.
Les compositions pharmaceutiques de l'invention, qui peuvent être administrées par voie orale ou parentérale, peuvent être notamment sous forme de solutions, des poudres, des suspensions.
D'autres caractéristiques et avantages de l'invention apparaîtront mieux a la lecture des exemples suivants donnés à titre illustratif et non limitatif.
Exemple 1 : Préparation de l'heptakis(6-désoxy-2,3-di-O- hexanoyl-6-iodo)cyclomaltoheptaose (composé no. 1).
Ce composé répond à la formule I dans laquelle tous les R1 sont identiques, avec m = 6, R1 = I et dans laquelle R2 représente le groupement hexanoyle.
A une solution de l'heptakis(6-deoxy-6- iodo)cyclomaltohepaose (0.5 g, 0.26 mmol) dans la DMF séchée (20 ml), sous argon, a 0 QC, on ajoute la N,N- diméthylaminopyridine (DMAP, 1.35 g, 11.0 mmol, 3 eq) . On ajoute alors l'anhydride hexanoïque (3.4 ml, 14.7 mmol, 4.0 eq) goutte à goutte et le mélange réactionnel est agité
pendant 45 min à température ambiante. La réaction est terminée par addition de MeOH (25 ml). Le mélange est encore agité pendant 1 h, puis versé dans l'eau glacé (50 ml) et extrait par le CH2Cl2 (4 x 50 ml). La phase organique et lavée successivement par l'acide sulfurique dilué (2 N ; 2 x 50 ml) et une solution saturée de NaHCO3 (4 x 50 ml), puis séchée (Na2SO4), concentrée et purifiée par chromatographie sur colonne de gel de silice avec un mélange EtOAc-éther de pétrole 1:12 → 1:10 comme éluant. On obtient ainsi le composé no. 1 (0.58 g, 68%) ayant les caractéristiques suivantes :
.- i?f = 0.40 (EtOAc-éther de pétrole 1: 5)
.- [α]D= +63.8 (c 1.0, CHCl3)
Exemple 2 : Préparation de l'heptakis(6-bromo-6-désoxy-2,3- di-0-hexanoyl)cyclomaltoheptaose (composé no. 2).
Ce composé répond à la formule I dans laquelle tous les R1 sont identiques, avec m = 6, R1 = Br et dans laquelle R2 représente le groupement hexanoyle. Le composé no. 2 est obtenu par acylation de l'heptakis(6-bromo-6-désoxy)cyclomaltoheptaose (0.61 g, 0.39 mmol) dans la DMF séchée (29 ml) avec le DMAP (2.0 g, 16.3 mmol, 3 eq) et l'anhydride hexanoique (5.0 ml, 21.7 mmol, 4.0 eq) , suivant le procédé commenté ci-dessus pour la préparation du composé no. 1. La purification du mélange réactionnel par chromatographie sur colonne de gel de silice avec un mélange EtOAc-éther de pétrole 1:10 1:8 comme éluant conduit au composé no. 2 (715 mg, 63%) ayant les caractéristiques suivantes : .- Rf = 0.60 (EtOAc-éther de pétrole 1:8) .- [α]D= +73.3 (c 0.8, CHCl3)
Exemple 3 : Préparation de l'heptakis(6-azido-6-désoxγ-2,3- di-0-hexanoyl)cycloraaltoheptaose (composé no. 3).
Ce composé répond à la formule III avec m = 6 et dans laquelle R2 représente le groupement hexanoyle.
A une solution de l'heptakis(6-azido-6- désoxy)cyclomaltoheptaose (0.2 g, 0.15 mmol) dans la DMF séchée (20 ml) sous argon, a 0 QC, on ajoute la N,N- diméthylaminopyridine (DMAP ; 783 mg, 6.4 mmol, 3 eq) . On ajoute alors l'anhydride hexanoique (2.0 ml, 8.5 mmol, 4.0 eq) goutte à goutte et le mélange réactionnel est agité pendant 16 h à température ambiante, puis concentré sous pression réduite et dilué avec du CH2Cl2 (20 ml). La solution résultante est lavée par l'acide sulfurique dilué (2 N ; 2 x 10 ml) et concentrée. Le résidu est repris par MeOH (15 ml) et additionné d'une solution aqueuse saturée de NaHCO3 (50 ml). Le mélange est agité a température ambiante pendant I h, puis extrait par CH2Cl2 (3 x 30 ml), la phase organique est séchée (Na2SO4), concentrée, et le résidu purifié par chromatographie sur colonne de gel de silice avec un mélange EtOAc-éther de pétrole 1:15 → 1:9 comme éluant. On obtient ainsi le composé no. 3 (286 g, 71%) ayant les caractéristiques suivantes :
.- R
f ≈ 0.42 (EtOAc-éther de pétrole 1:9) .- [α]
D = +115.0 (c 1.0, CH
2Cl
2)
Exemple 4 : Préparation de l'heptakis(6-désoxy-6-iodo-2,3-di- 0-myristoyl)cyclomaltoheptaose (composé no. 4).
Ce composé répond à la formule I dans laquelle tous les R1 sont identiques, avec m = 6, R1 = I et dans laquelle R2 représente le groupement tétradécanoyle (myristoyle) . A une solution de l'heptakis(6-désoxy-6- iodo)cyclomaltoheptaose (100 mg, 53 μmol) dans la DMF séchée sous argon (5 ml), a 0 se, on ajoute la N,N- diméthylaminopyridine (DMAP, 269 mg, 2.2 mmol, 3 eq) . On ajoute alors l'anhydride tétradécanoique (1.29 g, 2.94 mmol,
4.0 eq) et la suspension résultante est agitée pendant 16 h à température ambiante, puis filtrée. Le solide résultant est lavé par l'eau distillée puis MeOH, puis chauffé à reflux dans un mélange CH2Cl2-MeOH (5:95, 50 mL) pendant 1 h, décanté et purifiée par chromatographie sur colonne de gel de silice avec un gradient éther de pétrole -→ mélange EtOAc- éther de pétrole 1:12 → 1:10 comme éluant. On obtient ainsi le composé no 4 (0.58 g, 68%) ayant les caractéristiques suivantes : .- i?f = 0.49 (EtOAc-éther de pétrole 1:8) .- [α]D= + 49.2 (c 1.0, CH2Cl2)
Exemple 5 : Préparation de l'heptakis(6-bromo-6-désoxy-2,3- di-0-myristoyl)cyclomaltoheptaose (composé no. 5). Ce composé répond à la formule I dans laquelle tous les R1 sont identiques, avec m = 6, R1 = Br et dans laquelle R2 représente le groupement tétradécanoyle (myristoyle) .
Le composé no. 5 est obtenu par acylation de l'heptakis(6-bromo-6-désoxy)cyclomaltoheptaose (100 mg, 63.5 μmol) dans la DMF séchée (5 mL) avec le DMAP (326 mg, 2.67 mmol, 3 eq) et l'anhydride tétradécanoique (1.56 g, 3.55 mmol, 4.0 eq), suivant le procédé décrit ci-dessus pour la préparation du composé no 4. La purification du mélange réactionnel par chromatographie sur colonne de gel de silice avec un gradient éther de pétrole → mélange EtOAc-éther de pétrole 1:12 comme éluant conduit au composé no. 5 (193 mg, 67%) ayant les caractéristiques suivantes :
.- Rf = 0.31 (1:8 EtOAc-éther de pétrole) •- [≈3D = +49.3 (c 1.0, CH2Cl2)
Exemple 6 : Préparation de l'heptakis(6-azido-6-désoxy-2,3- di-O- myristoyl)cyclomaltoheptaose (composé no. 6).
Ce composé répond à la formule III avec m = 6 et dans laquelle R2 représente le groupement tétradécanoyle (myristoyle) .
A une solution de l'heptakis(6-azido-6- désoxy)cyclomaltoheptaose (100 mgf 76.3 μmol) dans la DMF séchée (7 mL), sous argon, a 0 QC, on ajoute la N,N- diméthylaminopyridine (DMAP; 392 mg, 3.2 mmol, 3 eq) . On ajoute alors l'anhydride tétradécanoique (1.87 g, 4.3 mmol, 4.0 eq) et la suspension résultante est agitée pendant 16 h à température ambiante, puis filtrée. Le solide résultant est lavé par l'eau distillée, puis chauffé à reflux dans un mélange CH2Cl2-MeOH (5:95, 25 mL) pendant 1 h, et filtré. On obtient ainsi le composé no. 6 (315 mg, 97%) ayant les caractéristiques suivantes :
Exemple 7 : Préparation de l 'heptakis[ 6- (2-tert- butoxycarbonylaminoethylthio) -6-désoxy-2 ,3-di-O- hexanoyl] cyclomaltoheptaose (composé no. 7).
Ce composé répond à la formule IV avec m = 6, n = 1 et dans laquelle R représente le groupement tert- butoxycarbonylamino (NHBoc) et R2 représente le groupement hexanoyle .
Ce composé est préparé en effectuant les deux étapes suivantes :
a) Préparation de l 'heptakis[6- (2-tert- butoxycarbonylaminoethylthio) -6-désoxy] cyclomaltoheptaose .
A une solution de l'heptakis(6-désoxy-6- iodo)cyclomaltoheptaose (1 g, 0.53 mmol) ou de l'heptakis(6- bromo-6-désoxy) cyclomaltoheptaose (0.84 g, 0.53 mmol) dans la DMF séchée (10 ml), on ajoute le carbonate de césium (1.71 g, 5.25 mmol) et le tert-butyl-2V-(2-mercaptoéthyl)carbamate (5.3 mmol, 1.4 eq) . La suspension résultante est chauffée sous Ar à 70 QC pendant 48 h, puis refroidie à température ambiante,
versée dans l'eau glacée et agitée pendant une nuit. On obtient un solide qui est filtré, lavé d'abord par l'eau distillée, puis par l'éther. Le résidu est purifié par chromatographie sur colonne de gel de silice avec un mélange CH2Cl2-MeOH-H2O 40:10:1 → 30:10:1 comme éluant. On obtient ainsi l ' heptakis [ 6- ( 2-tert-butoxycarbonylaminoethylthio ) -6- désoxyjcyclomaltoheptaose (787 mg, 66%) ayant les caractéristiques suivantes :
. -J?f = 0.60 (40:10:1 CH2Cl2-Me0H-eau) .- [α]D = + 79.7 (c 0.8, MeOH)
b) Préparation du composé no. 7.
A une solution de l 'heptakis [6- (2-tert- butoxycarbonylaminoéthylthio)-6-désoxy]cyclomaltoheptaose (0.2 g, 89 μmol) dans la pyridine séchée (10 ml), sous argon, on ajoute la N,JV-diméthylaminopyridine (DMAP, 456 mg, 3.73 mmol, 3 eq) . On ajoute alors l'anhydride hexanoique (3.4 ml, 14.7 mmol, 4.0 eq) et le mélange réactionnel est agité pendant 5 h à 70 se. La réaction est terminée par addition de MeOH (10 ml) et chauffée encore à 70 se pendant 3 h. Le mélange est alors versé dans l'eau glacé (50 ml) et extrait par CH2Cl2 (4 x 50 ml). La phase organique est lavée successivement par l'acide sulfurique dilué (2 N ; 2 x 50 ml) et une solution saturée de NaHCO3 (4 x 50 ml), puis séchée (Na2SO4), concentrée et purifiée par chromatographie sur colonne de gel de silice avec un mélange EtOAc-éther de pétrole 1:3 comme éluant. On obtient ainsi le composé no. 7 (225 mg, 76%) ayant les caractéristiques suivantes : .- J?f = 0.45 (1:2 EtOAc-éther de pétrole) .- [cc]D = + 84.1 (c 0.9, CHCl3)
Alternativement, le composé no. 7 a été préparé a partir du composé no. 1 (164 mg, 50 μmol) par réaction avec le tert- butyl-2V-(2-mercaptoéthyl)carbamate (82 μl, 0.5 mmol) et le
carbonate de césium (163 mg, 0.5 mmol) dans la DMF séchée (3 mL) suivant le procédé décrit ci-dessus pour la préparation de l ' heptakis [ 6- ( 2-tert-butoxycarbonylaminoéthylthio) -6- désoxy]cyclomaltoheptaose (rendement 88 mg, 49%).
Exemple 8 : Préparation de l 'heptakis [6- (2-aminoéthylthio) -6- désoxy-2,3-di-0-hexanoyl] cyclomaltoheptaose heptachlorhydrate (composé no. 8) .
Ce composé répond à la formule IV avec m = 6, n = 1, R = NH2 et dans laquelle R2 représente le groupement hexanoyle. Ce composé a été isolé sous forme de son sel heptachlorhydrate.
Le composé no. 6 (71 mg, 0.02 mmol) est traité par un mélange d'acide trifluoroacétique (TFA) -eau 1:1 (4 mL) à 40 se pendant 7 h. La solution résultante est concentrée et co- évaporée avec de l'eau distillée. Le résidu est repris par l'acide chlorhydrique dilué (pH 4) et lyophilisé. On obtient ainsi le composé no. 8 (60 mg) ayant les caractéristiques suivantes : .- [α]D= +72.6 (c 1.0, CH3OH)
Exemple 9 : Préparation de l 'heptakis [6-désoxγ-2,3-di-0- hexanoyl-6- (2-isothiocyanatoéthylthio) ] cyclomaltoheptaose (composé no. 9) . Ce composé répond à la formule VI avec m = 6, n = 1 et dans laquelle R2 représente le groupement hexanoyle.
A un mélange hétérogène du composé no. 8 (200 mg, 63 μmol) dans l'acétone (2.4 mL), CaCO3 (176 mg, 1.76 mmol) et l'eau (3.6 mL), est ajouté du CSCl2 (69 μL, 0.88 mmol). La suspension est agitée vigoureusement pendant 2.5 h. On ajoute alors CH2Cl2 (6 mL) et une solution aqueuse saturée de NaHCO3 (6 mL) . La phase organique est décantée, séchée et concentrée. Le résidu est purifié par chromatographie sur colonne de gel de silice avec un mélange EtOAc-éther de
pétrole 1:4 → 1:3 comme éluant. On obtient ainsi le composé no 9 (130 mg, 64%) ayant les caractéristiques suivantes : .- Rf = 0.34 (1:3 EtOAc-éther de pétrole) .- [α]D ≈ +118.8 (c 1.0, CHCl3)
Exemple 10 : Préparation de l 'heptakisf6- (2-tert- butoxycarbonylaminoéthylthio) -6-désoxy-2,3-di-O- myristoyl]cyclomaltoheptaose (composé no. 10).
Ce composé répond à la formule IV avec m = 6, n = 1 et dans laquelle R représente le groupement tert- butoxycarbonylamino (NHBoc) et R2 représente le groupement tétradécanoyle (myristoyle) .
A une solution de l 'heptakis[ 6- (2-tert- butoxycarbonylaminoéthylthio)-6-désoxy]cyclomaltoheptaose (0.2 g, 89 μmol), préparé suivant le procédé décrit dans l'exemple 7, dans la DMF séchée (15 ml), sous argon à 0 se, on ajoute la N,iV-diméthylaminopyridine (DMAP, 456 mg, 3.73 mmol, 3 eq) . On ajoute alors l'anhydride tétradécanoique (2.18 g, 4.97 mmol, 4.0 eq) et la suspension résultante est agitée pendant 5 h à 70 se. La réaction est terminée par addition de MeOH (10 ml) et chauffée encore à température ambiante pendant 48 h, puis concentrée Le résidu est alors repris dans un mélange CH2Cl2-MeOH (5:95, 100 mL) et chauffé à reflux pendant I h, le solide est décanté et purifié par chromatographie sur colonne de gel de silice avec un mélange EtOAc-éther de pétrole 1:2 comme éluant. On obtient ainsi le composé no. 10 (321 mg, 67%) ayant les caractéristiques suivantes :
.- Rf = 0.56 (1: 2 EtOAc-éther de pétrole) .- [α]D= +49.2 (c 1.0, CH2Cl2)
Exemple 11 : Préparation de l'heptakis[6-(2-aminoéthylthio)- 6-désoxy-2 , 3-di-O-myristoyl] cyclomaltoheptaose heptachlorhydrate (composé no. 11).
Ce composé répond à la formule IV avec m = 6, n = l, R = NH2 et dans laquelle R2 représente le groupement tétradécanoyle (myristoyle) . Ce composé a été isolé sous forme de son sel heptachlorhydrate . Le composé no. 10 (112 mg, 21.6 μmol) est traité par un mélange d'acide trifluoroacétique (TFA)-eau 1:1 (4 mL) à 40 se pendant 8 h. La solution résultante est concentrée et co- évaporée avec de l'eau distillée. Le résidu est repris par l'HCl dilué (pH 4) et lyophilisée. On obtient ainsi le composé no. 11 (102 mg) ayant les caractéristiques suivantes :
•- [α]D = +43.3 (c 1.0, CH2Cl2)
Exemple 12 : Préparation de l 'heptakis[6-désoxy-2,3-di-0- myristoyl-6- (2-isothiocyanatoéthylthio) ] cyclomaltoheptaose (composé no. 12).
Ce composé répond à la formule VI avec m = 6, n = 1 et dans laquelle R2 représente le groupement tétradécanoyle (myristoyle) . Un mélange hétérogène du composé no. 11 (102 mg, 21.2 μmol) dans CH2Cl2 (1.6 mL), CaCO3 (59 mg, 0.59 mmol, 4 eq) et l'eau (6.6 mL), est additionné de CSCl2 (23 μL, 0.30 mmol, 2 eq) . La suspension est agitée vigoureusement pendant 16 h. On ajoute alors CH2Cl2 (15 mL) et la phase organique est décantée, lavée par l'eau (6 mL), séchée et concentrée. Le résidu est purifié par chromatographie sur colonne de gel de silice avec un mélange EtOAc-éther de pétrole 1:6 comme éluant. On obtient ainsi le composé no. 12 (54 mg, 54%) ayant les caractéristiques suivantes : .- Rf = 0.38 (1:4 EtOAc-éther de pétrole) .- [α]D= +72.3 (c 0.6, CHCl3)
Exemple 13 : Préparation de l'heptakis[6-désoxy-2,3-di-σ- hexanoyl-6- (2- (N' -
méthylthiouréido)éthylthio) ] cyclomaltoheptaose (composé no. 13).
Ce composé répond à la formule V avec m = 6, n = l, Z = H, Q = S, T = H, dans laquelle W représente le groupement méthyle et R2 représente le groupement hexanoyle.
A une solution du composé no. 8 (85 mg, 27 μmol) dans CH2Cl2 (3 mL), on ajoute Et3N jusqu'à pH 8. On ajoute alors le méthyl isothiocyanate (20.5 mg, 0.28 mmol, 1.5 eq) et le mélange réactionnel est agité à température ambiante pendant 16 h. puis concentré. Le résidu est purifié par chromatographie sur colonne de gel de silice avec un mélange CH2Cl2:Me0H 20:1 comme éluant. On obtient ainsi le composé no. 13 (57 mg, 62%) ayant les caractéristiques suivantes :
.- Rf = 0.41 (20:1 CH2Cl2:MeOH) .- [α]D = +108.1 (c 1.0, CHCl3)
Exemple 14 : Préparation de l'heptakis[6-désoxy-2,3-di-0- hexanoyl-6-(2-(N'-(2- hydroxyéthyl)thiouréido)éthylthio) ] cyclomaltoheptaose (composé no. 14).
Ce composé répond à la formule V avec m ≈ 6, n ≈ l, Z = H, Q = S, T = H, dans laquelle W représente le groupement 2- hydroxyéthyle et R2 représente le groupement hexanoyle.
A une solution du composé no. 9 (112 mg, 35 μmol) dans CH2Cl2 (2 ml), on ajoute une solution d'éthanolamine (0.37 mmol, 22.2 μl, 1.5 eq) dans CH2Cl2 (1 ml), et le mélange est agité à température ambiante pendant 20 h. On ajoute alors l'eau (10 ml) et on extrait par CH2Cl2 (3 x 10 mL) .
L'ensemble des extraits organiques est séché (Na2SO4), concentré et purifié par chromatographie sur colonne de gel de silice avec le mélange CH2Cl2:MeOH 9:1→6:1 comme éluant.
On obtient ainsi le composé no. 14 (82 mg, 64%) ayant les caractéristiques suivantes :
. - Rt = 0 . 34 ( 6 : 1 CH2Cl2-MeOH )
• - [ <x ] D = + 80 . 9 ( c 0 . 97 , MeOH )
Exemple 15 : Préparation de l'heptakis[6-désoxy-6-(2-(iV'-(2- tf-iert-butoxycarbonylaminoéthyl)thiouréido)éthylthio) -2 , 3-di- 0-hexanoyl] cyclomaltoheptaose (composé no. 15).
Ce composé répond à la formule V avec m = 6 , n = 1 , Z = H, Q = S, T = H, dans laquelle W représente le groupement 2- iV-tert-butoxycarbonylaminoéthyle et R2 représente le groupement hexanoyle . A une solution du composé no. 9 (112 mg, 35 μmol) dans CH2Cl2 (2 mL), on ajoute une solution de la mono-N-tert- butoxycarbonyl-éthylenediamine (0.37 mmol, 59 μL, 1.5 eq) dans CH2Cl2 (1 mL), et le mélange est agité à température ambiante pendant 20 h. On ajoute alors l'eau (10 mL) et on extrait par CH2Cl2 (3 x 10 mL) . L'ensemble des extraits organiques est séché (Na2SO4), concentré et purifié par chromatographie sur colonne de gel de silice avec un mélange CH2Cl2-MeOH 20:1 comme éluant. On obtient ainsi le composé no. 15 (129 mg, 85%) ayant les caractéristiques suivantes : .- Rf = 0.27 (20:1 CH2Cl2:Me0H) •- [α]D= +78.4 (c 1.05, CHCl3)
Exemple 16 : Préparation de 1 'heptakis[6-désoxy-6-(2-(-Y'-(2- aminoéthyl)thiouréido)éthylthio)-2 , 3-di-O- hexanoyl] cyclomaltoheptaose heptachlorhydrate (composé no. 16) .
Ce composé répond à la formule V avec m = 6 , n = 1 , Z =
H, T = H, dans laquelle W représente le groupement 2- aminoéthyle et R2 représente le groupement tétradécanoyle (myristoyle) . Ce composé a été isolé sous forme de son sel heptachlorhydrate .
Le composé no. 15 (104 mg, 24 μmol) est traité par un mélange d'acide trifluoroacétique (TFA) -eau 1:1 (2 mL) à 45 se pendant 2 h. La solution résultante est concentrée et co-
évaporée avec de l'eau distillée. Le résidu est repris par l'HCl dilué (pH 4) et lyophilisé. On obtient ainsi le composé no. 16 (90 mg, 96%) ayant les caractéristiques suivantes : •- [α]D = +80.7 (c 1.0, DMSO)
Exemple 17 : Préparation de l'heptakis[6-désoxy-2,3-di-0- hexanoyl-6-(2-(_V'-(2-α-D- mannopyranosyloxyéthyl) thiouréido) éthylthio) ] cyclomaltoheptao se (composé no. 17). Ce composé répond à la formule V avec m. = 6 , n = l, Z = H, Q = S, T = H, dans laquelle W représente le groupement 2- α-D-mannoρyranosyloxyéthyle et R2 représente le groupement hexanoyle .
Une solution du composé no. 8 (85 mg, 27 μmol) dans un mélange eau-acétone 2:1 (3 ml) à pH 8 (NaHCO3) est agitée à température ambiante pendant 20 min. On ajoute alors une solution de 2-isothiocyanatoéthyl-α-D-mannopyranoside (75 mg, 0.28 mmol, 1.5 eq) dans l'eau (1 mL) . Après 2 h on observe l'apparition d'un précipité qui est dissous par addition de CH2Cl2 (0.5 mL) et le mélange réactionnel est agité à température ambiante pendant 16 h. On évapore les solvants organiques et la phase aqueuse est extraite par CH2Cl2 (3 x 5 mL). La phase organique est séchée (Na2SO4), filtrée et concentrée. Le résidu est purifié par chromatographie sur colonne de gel de silice avec un mélange CH2Cl2:MeOH 1:1 → 1:2 comme éluant. On obtient ainsi le composé no. 17 (49 mg, 38%) ayant les caractéristiques suivantes : .- [α]D= +75.1 (c 1.0, CHCl3)
Exemple 18 : Préparation de l'heptakis[6-désoxy-6-(2-(-V'- méthylthiouréido)éthylthio) -2, 3-di-O- myristoyljcyclomaltoheptaose (composé no. 18).
Ce composé répond à la formule V avec m ≈ 6, n = l, Z = H, Q = S, T = H, dans laquelle W représente le groupement
méthyle et R2 représente le groupement tétradécanoyle (myristoyle) .
A une solution du composé no. 11 (65 mg, 14.8 μmol) dans CH2Cl2 (3 mL) , on ajoute Et3N jusqu'à pH 8. On ajoute alors le méthyl isothiocyanate (11.3 mg, 0.16 mmol, 1.5 eq) et le mélange réactionnel est agité à température ambiante pendant 16 h. puis concentré. Le résidu est purifié par chromatographie sur colonne de gel de silice avec un mélange CH2Cl2-MeOH 30:1 comme éluant. On obtient ainsi le composé no. 18 (48 mg, 72%) ayant les caractéristiques suivantes : .- Rf = 0.55 (9:1 CH2Cl2-MeOH) .- [α]D = +60.8 (c 1.0, CHCl3)
Exemple 19 : Préparation de l 'heρtakis[6-désoxy-6-[2-[-V'-[2- bis [2- (iert- butoxycarbonylamino)éthyl] amino] éthyl] thioureido] éthylthio- 2,3-di-0-hexanoyl]cyclomaltoheptaose (composé no. 19).
Ce composé répond à la formule V avec m = 6, n = l, Z = H, Q = S, T = H, dans laquelle W représente l'élément de ramification 2-bis[2-( tert- butoxycarbonylamino)éthyl ]aminoéthyle (voir formule ci- dessous)
et R2 représente le groupement hexanoyle.
Ce composé est préparé en effectuant les deux étapes suivantes:
a) Préparation de la bis [2- (iert- butoxycarbonylamino)éthyl] 2-isothiocyanatoéthylamine (voir formule ci-dessous)).
Une solution de 2-azidoéthyl bis [ 2- ( fcert- butoxycarbonylamino)éthyl] aminé (0.35 g, 0.94 mmol), préparé comme décrit dans le document J. Am. Chem Soc. 2004 , 126, 10355-10363, et la triphénylphosphine (0.27 g, 1.03 mmol, 1.1 eq) dans le dioxane sec (10 mL), sous N2, est additionnée de CS2 (0.57 mL, 9.4 mmol, 10 eq) . La solution est agitée à température ambiante pendant 24 h, puis concentrée et le résidu purifié par chromatographie sur colonne de gel de silice avec un mélange EtOAc-éther de pétrole 30:1 comme éluant. On • obtient ainsi la bis [ 2- ( tert- butoxycarbonylamino)éthyl] 2-isothiocyanatoéthylamine (285 mg, 78%) ayant les caractéristiques suivantes : .- Rf = 0.72 (1:1 EtOAc-ether de pétrole) b) Préparation du composé no. 19.
A une solution du composé no. 8 (50 mg, 13.4 μmol) dans CH2Cl2 (2 mL), on ajoute Et3N jusqu'à pH 8. On agite la solution pendant 10 min et on additionne alors une solution de la bis[2-( iert-butoxycarbonylamino)éthyl] 2- isothiocyanatoéthylamine (51 mg, 0.13 mmol, 1.2 eq) dans CH2Cl2 (3 mL). Le mélange réactionnel est agité à température ambiante pendant 24 h, puis concentré. Le résidu est purifié par chromatographie sur colonne de gel de silice avec le mélange éluant CH2Cl2:Me0H 40:1. On obtient ainsi le composé no. 19 (35 mg, 46%) ayant les caractéristiques suivantes : .- Rf 0.37 (20:1 CH2Cl2-MeOH) • - [α]D = +55.0 (c 1.0, CH2Cl2)
Exemple 20 : Préparation de l'heptakis[6-désoxγ-6-[2-[iV'-[2- bis [2- (tert- butoxycarbonylamino)éthy1] amino] éthy1] thiouréido] éthy1thio- 2,3-di-0-hexanoyl]cyclomaltoheρtaose (composé no. 20).
Ce composé répond a la formule V avec m = 6, n = l, Z =
H, Q = S, T = H, dans laquelle W représente l'élément de ramification 2- [ 2-azidoéthyl-2 ' - ( tert- butoxycarbonylamino)éthyl] aminoéthyle (voir formule ci- dessous)
W=
BocHN^^ N/^N3
et R2 représente le groupement hexanoyle.
Ce composé est préparé en effectuant les étapes suivantes :
a) Préparation de la 2-(tert- butoxycarbonylamino)éthyl 2-aminoéthylamine.
Une solution de triéthylenediamine (10 mL, 93 mmol) dans le dioxane (40 mL), à 0 QC, est additionnée d'une solution de BoC2O (2.75 g, 12.6 mmol) dans le dioxane (40 mL) . Le mélange est agité pendant 4 h à 0 0C, puis on le laisse revenir à la température ambiante. On le concentre sous pression réduite et le résidu est additionné d'eau (20 mL) et extrait par CH2Cl2 (6 x 40 mL). La phase organique est séchée (Na2SO4), filtrée, concentrée et purifiée par chromatographie sur colonne de gel de silice avec un gradient MeOH→MeOH-NH4OH (30% dans l'eau) 100:3 comme éluant. On obtient ainsi la 2- ( tert-butoxycarbonylamino)éthyl 2-aminoéthylamine (2.25 mg, 88%) ayant les caractéristiques suivantes :
. - Rf 0 . 42 ( 20 : 1 MeOH- NH4OH )
b) Préparation de la 2-(tert- butoxycarbonylamino)éthyl 2-(trifluoroacétamido)éthylamine. Une solution de la 2-(tert- butoxycarbonylamino)éthyl 2-aminoéthylamine (2 g, 10 iranol) dans l'acétonitrile (20 mL) est additionnée de trifluoroacétate d'éthyle (4.17 mL, 35 mmol). Le mélange est agité a 90 QC, à reflux, pendant 5 h, puis concentré et le résidu purifié par chromatographie sur colonne de gel de silice avec un mélange EtOAc-EtOH-eau 45:5:3 comme éluant. On obtient ainsi la 2-( tert-butoxycarbonylamino)éthyl-2- (trifluoroacétamido)éthylamine (2.58 mg, 64%) ayant les caractéristiques suivantes : .- Rf 0.65 (30:2:1 MeCN-H2O-NH4OH)
c) Préparation du méthanesuIfonate de 2- azidoéthyle. Une solution de 2-bromoéthanol (5 g, 40 mmol) et d'azoture de sodium (3.12 g, 48 mmol, 1.2 eq) dans l'eau (15 mL) est agité à 60 QC pendant 4 h, puis refroidie à température ambiante et extraite par CH2Cl2 (4 x 20 mL) . La phase organique est séchée (Na2SO4), filtrée et concentrée jusqu'à une huile qui est ensuite reprise dans CH2Cl2 (30 mL) . On ajoute Et3N (1.5 eq, 8.3 mL) puis le chlorure de méthanesulfonyle (5.5 g, 48 mmol, 1.2 eq) goutte à goutte à 0 2C. On laisse revenir à température ambiante et on agite le mélange pendant 4 h. On additionne d'eau (40 mL), on extrait le mélange par CH2Cl2 (3 x 20 mL), puis la phase organique est séchée (Na2SO4), filtrée et concentrée. On obtient ainsi le méthanesulfonate de 2-azidoéthyle (5.31 mg, 80%) ayant les caractéristiques suivantes :
.- données de 1H RMN (300 MHz, CDCl3): δ 4.33 (t, 2 H, 3J" HrH = 5.0 Hz,CiJ2OMs) , 3.58 (t, 2 H, CH2N3), 3.07 (S, 3 H, Me)
.- données de 13C RMN (125.7 MHz, CDCl3): δ 67.7 (CH2OMs), 50.2 (CH2N3), 38.2 (Me).
d) Préparation de la 2-azidoéthyl 2-(ieri- butoxycarbonylamino)éthyl 2- (trifluoroacétamido)éthylamine.
Une solution de 2-( tert-butoxycarbonylamino)éthyl 2- (trifluoroacétamido)éthylamine (1 g, 2.8 mmol) et du méthanesulfonate de 2-azidoéthyle (1.02 g, 4.2 mmol) dans la
DMF (10 mL) est additionnée de Cs2CO3 (1.37 g, 4.2 mmol) et la suspension résultante est agitée à 50 se pendant 24 h, puis concentrée. Le résidu est repris dans CH2Cl2 (40 mL) et lavé par l'eau ( 20 mL). La phase organique est séchée
(Na2SO4), filtrée, concentré et purifiée par chromatographie sur colonne de gel de silice avec un mélange EtOAc-éther de pétrole 1:1 comme éluant. On obtient ainsi la 2-azidoéthyl-2-
( tert-butoxycarbonylamino)éthyl-2- (trifluoroacétamido)éthylamine (531 mg, 80%) ayant les caractéristiques suivantes :
.- Rf 0.61 (1:1 EtOAc-éther de pétrole)
e) Préparation de la 2-aminoéthyl-2-azidoéthyl-2- ( tert-butoxycarbonylamino)éthylamine.
Une solution de la 2-azidoéthyl-2-( tert- butoxycarbonylamino)éthyl-2- (trifluoroacétamido)éthylamine (450 mg, 1.22 mmol) dans un mélange de NH4OH (30%, 4 ml) et MeOH (16 mL) est agitée à 40 se pendant 16 h, puis concentrée. On obtient ainsi la 2-aminoéthyl-2-azidoéthyl 2- ( tert-butoxycarbonylamino)éthylamine, en rendement quantitatif, ayant les caractéristiques suivantes : .- Rf 0.59 (10:1:1 CH3CN-eau-NH4OH)
f) Préparation de la 2-azidoéthyl 2-(fcerfc- butoxycarbonylamino)éthyl-2-isothiocyanatoéthylamine.
Un mélange hétérogène contenant la 2-aminoéthyl-2- azidoéthyl-2-( tert-butoxycarbonylamino)éthylamine (272 mg, 1 mmol) et le CaCO3 (300 mg, 1.5 eq, 1.5 mmol) dans CH2Cl2-eau 1:1 (10 mL) est additionné de Cl2CS (115 μL, 1.5 mmol). Le mélange est agité vigoureusement à température ambiante pendant 1.5 h, puis les deux phases sont séparées, la phase organique est séchée (Na2SO4), filtrée, concentré et purifiée par chromatographie sur colonne de gel de silice avec un mélange EtOAc-éther de pétrole 1:1 comme éluant. On obtient ainsi la 2-azidoéthyl 2-( tert-butoxycarbonylamino)éthyl-2- isothiocyanatoéthylamine (198 mg, 63%) ayant les caractéristiques suivantes : .- Rf 0.72 (1:2 EtOAc-ether de pétrole)
g) Préparation du composé no. 20.
A une solution du composé no. 8 (50 mg, 13.4 μmol) dans CH2Cl2 (2 mL), on ajoute Et3N jusqu'à pH 8. On agite la solution pendant 10 min et on ajoute alors une solution de la 2-azidoéthyl 2-( tert-butoxycarbonylamino)éthyl-2- isothiocyanatoéthylamine (38 mg, 0.13 mmol, 1.2 eq) dans CH2Cl2 (3 mL). Le mélange réactionnel est agité à température ambiante pendant 24 h, puis concentré. Le résidu est purifié par chromatographie sur colonne de gel de silice avec un mélange CH2Cl2:Me0H 40:1 comme éluant. On obtient ainsi le composé no. 20 (55 mg, 80%) ayant les caractéristiques suivantes :
.- Rf 0.37 (20:1 CH2Cl2-MeOH) .- [α]D = +57.4 (c 1.0, CH2Cl2)
Exemple 21 : Préparation de l'heptakis[6-désoxy-6-[2-[.tf',iV'- bis [2- ( tert-butoxycarbonylamino)éthyl]thiouréido]éthylthio] - 2,3-di-0-hexanoyl]cyclomaltoheptaose (composé no. 21).
Ce composé répond à la formule V avec m = 6, n = l, Z = H, Q = S, dans laquelle T et W sont identiques et représentent le groupement 2-( te.rt~butoxycarbonylamino)éthyle et R2 représente le groupement hexanoyle.
A une solution du composé no. 9 (50 mg, 15.5 μmol) dans CH2Cl2 (5 mL), on ajoute Et3N (15 μL, 1 eq, 0.11 mmol) et la bis (2-tert-butoxycarbonylaminoéthyl) aminé (36 mg, 1.1 eq, 0.12 mmol). La solution est agitée pendant 6 h, puis concentrée. Le résidu est purifié par chromatographie sur colonne de gel de silice avec un mélange CH2Cl2:Me0H 40:1 comme éluant. On obtient ainsi le composé no. 21 (76 mg, 92%) ayant les caractéristiques suivantes : . - Rf 0.41 (20:1 CH2Cl2-MeOH) .- [α]D = +57.0 (c 1.0, CH2Cl2)
Exemple 22 : Préparation de l'heptakis[6-désoxy-6-[2-[iV'-(2- azidoéthyl) -Nf-[2- ( tert- butoxycarbonylamino)éthyl]thiouréido] éthylthio] -2,3-di-O- hexanoyl]cyclomaltoheptaose (composé no. 22).
Ce composé répond à la formule V avec m = 6, n ≈ 1 , Z =
H, Q = S, dans laquelle T représente le groupement 2- azidoéthyle, W représente le groupement 2-(tert- butoxycarbonylamino)éthyle et R2 représente le groupement hexanoyle.
Ce composé est préparé en effectuant les étapes suivantes :
a) Préparation de la 2-azidoéthyl 2-N-(tert- butoxycarbonyl)aminoéthyl)aminé.
Une solution de iV-tert-butoxycarbonyléthylenediamine (0.47 g, 2.9 mmol) et du p-toluènesulfonate de 2-azidoéthyle (0.70 g, 1 eq, 2.9 mmol) dans l'acétonitrile (10 mL) est
additionnée de K2CO3 (0.40 g, 2.9 mmol). Le mélange est agité pendant 12 h, puis concentré sous pression réduite et le résidu est additionné d'eau (40 mL) et extrait par CH2Cl2 (2 x 20 mL). La phase organique est séchée (Na2SO4), filtrée, concentrée et purifiée par chromatographie sur colonne de gel de silice avec un mélange CH2Cl2-MeOH 20:1 comme éluant. On obtient ainsi la 2-azidoéthyl 2-N-(tert- butoxycarbonyl)aminoéthyl) aminé (0.43 mg, 65%) ayant les caractéristiques suivantes : .- Rf 0.48 (20:1 CH2Cl2-MeOH)
b) Préparation du composé no. 22.
A une solution du composé no. 9 (50 mg, 15.5 μmol) dans CH2Cl2 (5 mL), on ajoute Et3N (15 μL, 1 eq, 0.11 mmol) et la 2-azidoéthyl-2-iV-Ctert-butoxycarbonyl)aminoéthyl) aminé (27 mg, 1.1 eq, 0.12 mmol). La solution est agitée pendant 3 h, puis concentrée Le résidu est purifié par chromatographie sur colonne de gel de silice avec un mélange CH2Cl2:Me0H 60:1 comme éluant. On obtient ainsi le composé no.22 (48 mg, 92%) ayant les caractéristiques suivantes : .- Rf 0.24 (60:1 CH2Cl2-MeOH) •- [α]D = +47.2 (c 1.0, MeOH)
Exemple 23 : Préparation du 6I-[2-(tert- butoxycarbonylamino)éthylthio]-6I-désoxy-heptakis(2,3-di-0- hexanoyl)cyclomaltoheptaose (composé no. 23).
Ce composé répond à la formule VII avec m = 6 et dans laquelle R1 représente le groupement 2-(tert- butoxycarbonylamino)éthylthio et R2 représente le groupement hexanoyle.
Ce composé est préparé en effectuant les quatre étapes suivantes :
a) Préparation du 6I-[2-(tert- butoxycarbonylami.no)éthylthio] -6I-désoxycyclomaltoheptaose.
A une solution du 6I-déoxy-6I-iodocyclomaltoheptaose (1.42 gf 1.14 mmol) dans la DMF séchée (18 ml), sous Ar, on ajoute le carbonate de césium (0.48 g, 1.48 mmol, 1.3 eg) et le tert-butyl iV-(2-mercaptoéthyl)carbamate (250 μl, 1.48 mmol, 1.3 eq). La suspension résultante est chauffée à 75 se pendant 3 h, puis concentrée. Le résidu solide résultant est lavé successivement par CH2Cl2 et l'acétone et finalement purifié par chromatographie sur colonne de gel de silice avec un mélange CH3CN-eau-NH4OH 6:3:1 comme éluant. On obtient ainsi le 61- [ 2-( tert-butoxycarbonylamino)éthylthio]-6τ- désoxycyclomaltoheptaose (777 mg, 53%) ayant les caractéristiques suivantes : . - Rf = 0.47 (6:3:1 CH3CN-eau-NH4OH) •- [Ct]n = +115.3 (c 1.0, DMSO)
b) Préparation du 6τ-[2- (tert- butoxycarbonylamino)éthylthio]-611~vτi-hexa-O-tert- butyldiméthylsilylcyclomaltoheptaose.
Une solution de 6τ-[2-(tert- butoxycarbonylamino)éthylthio]-6I-désoxycyclomaltoheptaose (378 mg, 0.29 mmol) dans la pyridine (15 mL) est additionnée de TBDMSCl (528 mg, 3.5 mmol, 2 eq) . Le mélange réactionnel est agité pendant 3 jours, puis versé dans l'eau glacée (50 mL) et extrait par CH2Cl2 (4 x 50 ml). La phase organique est lavée successivement par l'acide sulfurique dilué (2 N, 2 x 50 ml), une solution aqueuse saturée de NaHCO3 (4 x 50 ml), séchée (Na2SO4), concentrée et purifié par chromatographie sur colonne de gel de silice avec un mélange CH2Cl2-Me0H-eau 50:10:1 comme éluant. On obtient ainsi le 61- [ 2- ( tert- butoxycarbonylamino)éthylthio] -6II"VII-hexa-0-tert- butyldiméthylsilylcyclomaltoheptaose (494 mg, 86%) ayant les caractéristiques suivantes :
. - Rf = 0 . 38 ( 45 : 5 : 3 AcOEt-EtOH-eau ) • - [ α ] D = +84 . 0 ( c 1 . 0 , CHCl3 )
c) Préparation du 6I-[2-(tert- butoxycarbonylamino)éthylthio] -6I3C"VII-hexa-O-tert- butyldiméthylsilyl-6I-désoxy-heptakis(2, 3-di-O- hexanoyl)cyclomaltoheptaose.
A une solution de 6I-[2-(tert- butoxycarbonylamino)éthylthio]-6II~VII-hexa-O-tert- butyldiméthylsilylcyclomaltoheptaose (0.85 g, 0.43 mmol) dans la DMF séchée (34 ml), sous argon, on ajoute la N,N- diméthylaminopyridine (DMAP, 2.19 g, 17.9 inmol, 3 eq) . On ajoute alors l'anhydride hexanoique (5.54 ml, 23.9 mmol, 4.0 eq) et le mélange réactionnel est agité pendant 4 h à température ambiante. La réaction est terminée par addition de MeOH (70 ml) et on continue l'agitation pendant 24 h. Le mélange est alors concentré, le résidu est repris par le CH2Cl2 (150 ml), lavée successivement par l'acide sulfurique dilué (2 N ; 2 x 50 ml) et une solution saturée de NaHCO3 (4 x 50 ml), puis séchée (Na2SO4), concentrée et purifiée par chromatographie sur colonne de gel de silice avec un mélange EtOAc-éther de pétrole 1:20 -→ 1:9 comme éluant. On obtient ainsi le 61- [ 2- ( tert-butoxycarbonylamino)éthylthio] -β11^11- hexa-O-tert-butyldiméthylsilyl-6I-désoxy-heptakis ( 2 , 3-di-O- hexanoyl)cyclomaltoheptaose (806 mg, 56%) ayant les caractéristiques suivantes :
.- Rf = 0.62 (1:8 EtOAc- éther de pétrole) • - [α]D = +72.1 (c 1.0, CHCl3,
c) Préparation du composé no. 23.
A une solution de la 6I-[2-(tert- butoxycarbonylamino)éthylthio]-6II"VII-hexa-O-tert- butyldiméthylsilyl-6I-désoxy-heρtakis ( 2 , 3-di-O- hexanoyl)cyclomaltoheptaose (78.2 mg, 23.3 μmol) dans le
tétrahydrofurane sec (3 mL) on ajoute le fluorure de tétrabutylammonium (TBAF, solution 1 N dans le THF, 167 μl, 167 μmol, 1.2 eq) . Le mélange réactionnel est agité à température ambiante pendant 16 h, puis concentré et le résidu purifié par chromatographie sur colonne de gel de silice avec un gradient EtOAc → mélange EtOAc-EtOH comme éluant. On obtient ainsi le composé no. 23 (38 mg, 61%) ayant les caractéristiques suivantes :
.- Rf = 0.47 (20:1 EtOAc-EtOH) .- [α]D= +94.4 (c 1.0, CHC13)
Exemple 24 : Préparation du chlorhydrate de 6I-(2- aminoéthylthio) -6I-désoxy-heptakis (2 , 3-di-O- hexanoyl)cyclomaltoheptaose (composé no. 24). Ce composé répond à la formule VII avec m = 6 et dans laquelle R1 représente le groupement 2-aminoéthylthio et R2 représente le groupement hexanoyle. Ce composé est isolé sous forme de son sel heptachlorhydrate .
Le composé no. 23 (71 mg, 0.02 mmol) est traité par un mélange d'acide trifluoroacétique (TFA)-CH2Cl2 1:1 (0.26 mL) à température ambiante pendant 3 h. La solution résultante est concentrée et le résidu est purifié par chromatographie sur colonne de gel de silice avec un gradient EtOAc-EtOH 20:1 → EtOAc-EtOH-eau 45:5:3 comme éluant. Le produit purifié est repris par l'HCl dilué (pH 4) et lyophilisé. On obtient ainsi le composé no. 24 (36 mg, 64%) ayant les caractéristiques suivantes :
.- Rf = 0.19 (20:1 AcOEt-EtOH) •- [α]D = +80.4 (c 1.0, CH2Cl2)
Exemple 25 : Préparation du 6I-désoxy-6I-[2-[ΛT'-(2-α-D- mannopyranosyloxyéthyl) thiouréido] éthylthio] -heptakis (2 , 3-di- 0-hexanoyl)cyclomaltoheptaose (composé no. 25).
Ce composé répond à la formule VII avec m = 6 et dans laquelle R1 représente le groupement 2-[iV'-(2-α-D- mannopyranosyloxyéthyl ) thiouréido]éthylthio (voir formule)
et R2 représente le groupement hexanoyle.
Une solution du composé no. 24 (113 mg, 42 μmol) dans un mélange eau-acétone 2:1 (4 mL) à pH 8 (NaHCO3) est agité pendant 20 min à température ambiante. On ajoute alors une solution de 2-isothiocyanatoéthyl-α-D-mannopyranoside (22 mg, 83 μmol, 2 eq) dans l'eau (0.5 mL). Après 2 h on observe l'apparition d'un précipité qui est dissous par addition de CH2Cl2 (0.5 mL) et le mélange réactionnel est agité à température ambiante pendant 48 h. On évapore les solvants organiques et la phase aqueuse est extraite par CH2Cl2 (3 x 5 mL) . La phase organique est séchée (Na2SO4), filtrée et concentrée. Le résidu est purifié par chromatographie sur colonne de gel de silice avec un gradient EtOAc-EtOH 20:1 → EtOAc-EtOH-H2O 45:5:3 comme éluant. On obtient ainsi le composé no. 25 (46 mg, 39%) ayant les caractéristiques suivantes : .- Rf = 0.52_(45:5:3 EtOAc-EtOH-H2O) .- [α]D ≈ +92.5 (c 1.0, CH2Cl2)
Exemple 26: Préparation du 6I-[2-(W-(2-(cyclomaltoheptaose- 6I-désoxy-6I-yl)éthylthio)thiouréido)éthylthio]-6I-désoxy- heptakis-(2,3-di-0-hexanoyl)cyclomaltoheptaose (composé no. 26).
Ce composé répond à la formule VII avec m = 6, R2 représente le groupement hexanoyle (C5H11CO2) et R1 le groupement ci-dessous:
II est préparé par la suite d'étapes suivantes partant du composé 24 (Exemple 24) et du 6
I-[2-(tert- butoxycarbonylamino)éthylthio]-6
I-désoxycyclomaltoheptaose préparé dans l'exemple 23a. a) 6
I-(2-Aminoéthylthio)-6
I-désoxy-cyclomaltoheptaose trifluoroacétate.
Une solution du 6I-[2-(tert- butoxycarbonylamino)éthylthio] -6I-désoxycyclomaltoheptaose (300 mg, 0.23 mmol) dans un mélange TFA-eau (1:1, 2.7 mL) est agité à température ambiante pendant 2 h, puis concentrée. On obtient ainsi le 6I-(2-aminoéthylthio)-6I-désoxy- cyclomaltoheptaose trifluoroacétate qui est utilisé directement dans la prochaine étape.
b) 6I-Désoxy-6I-(2-isothiocyanatoéthylthio) cyclomaltoheptaose.
A une solution du 61-(2-aminoéthylthio) -β^désoxy- cyclomaltoheptaose trifluoroacétate (270 mg, 0.20 mmol) dans un mélange acétonitrile-eau (1:1, 6 mL), à 0 2C, on ajoute le carbonate de calcium (82 mg, 0,82 mmol) et le thiophosgène (31 μL, 0.41 mmol). La réaction est agité pendant 2 h, puis filtrée, les solvants sont évaporés et le résidu est purifié par chromatographie sur colonne de gel de silice avec un mélange CH3CN-eau 9:1 → 3:1 comme éluant. On obtient ainsi
le 6I-désoxγ-6I- ( 2-isothiocyanatoéthylthio)cyclomaltoheptaose (129 mg, 50%) ayant les caractéristiques suivantes : .- Rt = 0.36 (3:1 CH3CN-eau) •- [α]D ≈ +56.1 (c 1.0, DMSO)
c) β^Désoxy-heptakis (2,3-di-O-hexanoyl) -61-(2- isocyanatoéthylthio)cyclomaltoheptaose.
A une solution du composé 24 (215 mg, 82.6 μmol) dans un mélange dichlorométhane-eau (1:1, 2 πiL), à 0 ec, on ajoute le carbonate de calcium (25 mg, 0,25 mmol) et le thiophosgène
(9.5 μL, 0.12 mmol). La réaction est agitée à température ambiante pendant 4 h. On ajoute alors le dichlorométhane (5 mL) . La phase organique est séparée, lavée par l'eau (3 mL), séchée (Na2SO4) et concentrée. Le résidu est purifié par chromatographie sur colonne de gel de silice avec un mélange
CH2Cl2-MeOH 15:1 → 9:1 comme éluant. On obtient ainsi le 61- désoxy-heptakis ( 2 , 3-di-O-hexanoyl) -61- ( 2- isocyanatoéthylthio)cyclomaltoheptaose (130 mg, 60%) ayant les caractéristiques suivantes: . - Rf = 0.28 (9:1 CH2Cl2-MeOH) .- [α]D ≈ +89.6 (c 0.3, CHCl3)
d) Composé no. 26.
A une solution du composé 24 (121 mg, 46.5 μmol) dans la DMF (2 mL) on ajoute le bicarbonate de sodium (solution saturée dans l'eau) jusqu'à pH 8, et on agite le mélange réactionnel à température ambiante pendant 20 min. On ajoute alors une solution du 6I-désoxy-6I-(2- isothiocyanatoéthylthio)cyclomaltoheptaose (86 mg, 70 μmol) dans la DMF (0.5 mL) et la solution résultante est agité à température ambiante pendant 24 h. Le solvant est évaporé à pression réduite, le résidu est repris dans le dichlorométhane (5 mL), lavé par l'eau (2 mL), la phase organique est séchée (Na2SO4) et concentrée. Le résidu est
purifié par chromatographie sur colonne de gel de silice avec un mélange CH2Cl2-MeOH 10:1 comme éluant, puis par chromatographie de filtration sur gel (Sephadex LH-20, MeOH). On obtient ainsi le composé 26 (80 mg, 45%) ayant les caractéristiques suivantes:
•- [α]D = +91.6 (c 1.0, CHCl3)
Alternativement, le composé 26 (105 mg, 60%) a été préparé par réaction entre le 6I-(2-aminoéthylthio)-6I- désoxy-cyclomaltoheptaose trifluoroacétate (66 mg, 51 μmol) et le 6I-désoxy-heptakis(2,3-di-0-hexanoyl)-6I-(2- isocyanatoéthylthio)cyclomaltoheptaose (121 mg, 46.4 μmol) dans la DMF (3 mL) a pH 8 (NaHCO3) suivant la même procédure.
Exemple 27. Formulation de nanosphères blanches homogènes.
La méthode utilisée pour préparer des nanosphères blanches homogènes consiste à introduire une phase organique d'acétone (ou de tétrahydrofurane ) et d'un dérivé de cyclodextrine amphiphile de l'invention (1 mg-mlf1) dans une phase aqueuse de volume équivalent ou d'un volume double (eau distillée), sous agitation magnétique (500 tr-min"1) à 25 0C. Après nanoprécipitation, le solvant organique est éliminé sous pression réduite à +35 0C. La suspension aqueuse présentant un aspect opalescent à laiteux (suivant les produits mis en œuvre) est ensuite concentrée jusqu'au volume final souhaité, puis filtrée sur filtre 0.8 μm (Millex AA, Millipore, France).
Les suspensions de nanoparticules ont été triplées. La Figure 1 représente une photographie en cryo-transmission des nanosphères observées par microscopie électronique à transmission après cryofracture .
Les suspensions colloïdales sont ensuite caractérisées sur le plan granulométrique (taille moyenne et polydispersité) et, lorsqu'ils incorporent des cyclodextrines amphiphiles
chargées, par leur potentiel zêta. La caractérisation est effectuée en suivant les procédés ci-après : a) Mesure de la taille des nanoparticules.
La diffusion quasi-élastique de la lumière est mise en oeuvre. Un appareil Zetasizer 3000 équipé d'un corrélateur K7132 (Malvern Instruments, RU) a été utilisé. Les conditions de mesure sont les suivantes : angle de détection 90°, température 25 ± 0.10C, viscosité apparente 0.89 mPa.s. Avant de réaliser les mesures de taille, les échantillons sont dilués dans l'eau distillée si nécessaire,. Le diamètre hydrodynamique (Z moyen) et l'indice de polydispersité (IP) des nanoparticules en suspension ont été calculés en intensité par la méthode des cumulants en réalisant 3 mesures d'un même échantillon. La valeur du diamètre hydrodynamique est calculée à partir de la mesure du coefficient de diffusion translationnel des particules animées de mouvement brownien. Un appareil NanoZS (Malvern Instruments) a également été utilisé pour la mesure de taille des nanocapsules . De la même manière, les échantillons sont dilués avant la mesure, si nécessaire dans l'eau additionnée de Montanox 80 (marque déposée).
b) Mesure du potentiel zêta des nanoparticules
Un appareil zétasizer 3000 (laser HeNe 10 mW à 632.8 nm) (Malvern Instruments, Malvern, RU) a été utilisé. Les mesures sont réalisées à une température de 25 ± 0.10C après dilution des échantillons dans une solution de chlorure de sodium (10" 3M) . Les valeurs de potentiel zêta sont obtenues en réalisant 3 mesures d'un même échantillon.
Les études de stabilité physique des particules ont été réalisées sur des périodes s 'étalant de 1 mois1 à 8 mois. Les données correspondantes pour une série de ! cyclodextrines amphiphiles de l'invention sont recueillies dans le Tableau 1
fournissant les caractéristiques granule-métriques (taille moyenne et indice de polydispersité ou IP moyen) , avec évolution dans le temps, de divers nanosphères homogènes issues des composés indiqués dans la colonne de gauche, les numéros de référence des composés en question correspondant à ceux des composés décrits dans les exemples de préparations ci-dessus.
Exemple 28. Formulation de nanosphères blanches mixtes.
La méthode utilisée pour préparer des nanosphères blanches mixtes consiste à introduire une phase organique de deux dérivés de cyclodextrines amphiphiles dans l'acétone, en proportions variables (lmg.mL"1), dans une phase aqueuse de volume équivalent (eau distillée), sous agitation magnétique (500 tr-min"1) à 25 °C. Après nanoprécipitation, le solvant organique est éliminé sous pression réduite à 35 0C. La suspension aqueuse présentant un aspect opalescent est ensuite concentrée jusqu'au volume final souhaité, puis filtrée (0.8 μm, Millex AA, Millipore, France). Les suspensions de nanoparticules sont triplées. Les suspensions colloïdales ont ensuite été caractérisées en terme de taille moyenne et potentiel zêta, suivant les procédés décrits ci- dessous, puis sont stockées à +6°C.
Dans un exemple typique, on utilise comme phase organique une solution des composés no. 2 et no. 8 (1 mg«mL" x), dans des proportions relatives 4:1 et 7:3, dans l'acétone avec comme phase aqueuse l'eau distillée (proportion eau- acétone 1:1 v/v) . On obtient dans le cas d'une proportion 4:1 des composés no. 2 et no. 8, dans une série d'expériences indépendantes, des nanosphères blanches avec une taille moyenne qui varie de 110 à 138 nm avec des IP qui varient de 0.16 à 0.27 et un potentiel zêta qui varie de 43 à 47 mV. Dans le cas d'une proportion 7:3 des composés no. 2 et no. 8, dans une série d'expériences indépendantes, on obtient des
nanosphères blanches avec une taille moyenne qui varie de 88 à 162 nm avec des IP qui varient de 0.14 à 0.2 et un potentiel zêta qui varie de 40 à 45 mV.
Dans un autre exemple typique, on utilise comme phase organique une solution des composés no.25 et 26 ( lmg.mlT1 ) , dans des proportions relatives 1:4, dans l'acétone avec comme phase aqueuse l'eau distillée (proportion eau/ acétone 1 :1 v/v) . On obtient dans le cas d'une proportion 1:4 des composés no. 25 et 26, dans une série d'expériences indépendantes, des nanosphères blanches avec une taille moyenne qui varie de 95 à 120 nm avec des IP de 0.03 à 0.08.
Exemple 29. Formulation de nanocapsules blanches.
La méthode utilisée pour préparer des nanocapsules blanches consiste tout d'abord à préparer une phase acétonique contenant une petite fraction de triglycérides à chaînes caprique/caprylique (Miglyol 812 (marque déposée)), un dérivé de cyclodextrine amphiphile de l'invention, un agent tensioactif lipophile non ionique (Montane 80 (marque déposée)) et une phase hydrophile contenant de l'eau distillée et un agent tensioactif hydrophile non ionique. La phase organique est ensuite introduite dans la phase hydrophile sous agitation magnétique (500 tr^min"1) à 25 0C. Après nanoprécipitation, le solvant organique est éliminé sous pression réduite à 35 0C. La suspension aqueuse présentant un aspect laiteux est ensuite concentrée jusqu'au volume final souhaité. Les lots de suspensions colloïdales ont été triplés. Les nanoparticules ont ensuite été caractérisées en terme de taille moyenne puis ont été stockées à +6°C.
Dans un exemple typique, on utilise comme phase organique l'acétone, le Miglyol 812 (marque déposée) (proportion acétone-Miglyol 812 (marque déposée) 100:1 v/v), le Montane 80 (marque déposée) (4 mg^mL"1) et le composé no.
3 (2 mg'inL"1), et comme phase aqueuse l'eau distillée (proportion eau-acétone 2:1 v/v) contenant le Montanox 80 (marque déposée) (2 mg'inL"1). On obtient dans ce cas là, dans une série d'expériences indépendantes, des nanocapsules blanches avec une taille moyenne qui varie de 203 à 255 nm avec des IP qui varient de 0.04 à 0.24.
Exemple 30. Formulation des nanosphères chargées de principe actif (diazepam) . La méthode utilisée pour préparer des nanosphères chargées de molécule hôte, ici un principe actif (le diazepam) , consiste à introduire une phase organique contenant l'acétone, un dérivé de cyclodextrine amphiphile de l'invention et le diazepam (DZ) dans une phase aqueuse contenant de l'eau distillée avec ou sans tensioactif hydrophile non ionique (par exemple le poloxamer 188), sous agitation magnétique (500 tr^min"1) à 25 0C. Après nanoprécipitation, le- solvant organique est éliminé sous pression réduite à 35 0C. La suspension aqueuse présentant un aspect opalescent est ensuite concentrée jusqu'au volume final souhaité.
Les lots de suspensions colloïdales ont été triplés. Les nanoparticules ont ensuite été caractérisées sur le plan granulométrique . La quantité de diazepam fixée aux nanosphères a été évaluée par différence entre la quantité de diazepam présente dans la suspension colloïdale finale et la quantité de diazepam présente dans le surnageant. Le dosage du diazepam a été réalisé par spectrophotométrie à 285 nm. Le taux d'encapsulation ou d'association du principe actif aux nanoparticules (TEexp) et le rendement ou efficacité d'encapsulation (RE) ont donc été évalués. Les suspensions aqueuses ont été stockées à + 6°C.
Dans un exemple typique, la phase organique est constituée par l'acétone, le composé no. 3 ( 1 mg'inL"1) et le
diazépam (0.5 mg*mL ), et la phase aqueuse est constituée par l'eau distillée (proportion eau-acétone 1:1 v/v) . On obtient dans ce cas là, dans une série d'expériences indépendantes, des nanosphères chargées avec une taille moyenne qui varie de 135 à 175 nm avec des IP qui varient de 0.01 à 0.02, des valeurs de TEexp comprises entre 24 et 28% et des valeurs de RE comprises entre 74 et 89%.
TEth correspond au pourcentage massique de DZ introduit initialement (DZ1) . Il est déterminé de la manière suivante :
100 x DZ1 (mg) / (DZ1 + CD) mg
TEexp correspond au pourcentage de DZ réellement associé aux nanoparticules (DZa)
RE : 10Ox DZa(mg) / DZ1 (mg)
Exemple 31. Formulation de nanocapsules chargées de principe actif (diazépam)
La méthode utilisée pour préparer des nanocapsules chargées de molécule hôte, ici un principe actif (le diazépam), consiste tout d'abord à préparer une phase acétonique contenant une petite fraction de triglycérides à chaînes caprique/caprylique (Miglyol 812 (marque déposée)), un dérivé de cyclodextrine amphiphile de l'invention, un agent tensioactif lipophile non ionique (Montane 80 (marque déposée)), le diazépam (DZ) et une phase hydrophile contenant de l'eau distillée et un agent tensioactif hydrophile non ionique. La phase organique est ensuite introduite dans la phase hydrophile sous agitation magnétique (500 tr.min"1) à 25°C. Après nanoprécipitation, le solvant organique est éliminé sous pression réduite à +350C. La suspension aqueuse présentant un aspect laiteux est ensuite concentrée jusqu'au volume final souhaité. Les lots de suspensions colloïdales ont été triplés. Les nanoparticules ont ensuite été
caractérisées en terme de taille moyenne puis ont été stockées à +60C.
Dans un exemple typique, on utilise comme phase organique, l'acétone, le Miglyol 812 (marque déposée) (proportion acétone — Miglyol 812 (marque déposée) 100 :1 v/v) , le Montane 80 (marque déposée) (4mg.mL"l) et le composé no. 3 (2 mg.mlfl), le diazépam (1 mg.miri) et comme phase aqueuse l'eau distillée (proportion eau-acétone 2 :1 v/v) contenant le Montanox 80 (marque déposée) (4 mg.mlfl). On obtient dans ce cas là, dans une série d'expériences indépendantes, des nanocapsules chargées avec une taille moyenne qui varie de 215 à 225 nm avec des IP qui varient de 0.03 à 0.07 , des valeurs de RE comprises entre 85 et 93%.
Exemple 32. Préparation de complexes avec les composés no. 8 ou 16 et le plasmide pTG11236, caractérisation et capacité de transfection. a) Préparation
Le plasmide pTG11236 (pCMV-SV40-luciferase-SV40pA) , utilisé pour la préparation des complexes d'ADN et pour les tests de transfection est un plasmide de 5739 paires de bases. Les quantités de composés utilisées sont calculées pour obtenir une concentration d'ADN de 0.1 mg^mL"1 (303 μM en équivalent phosphate) et les valeurs désirées des rapports Azote / Phosphate (N/P). Des tests pour des valeurs de N/P =
5, 10, 30 et 50 ont été effectués aussi bien pour le composé no. 8 que pour le composé no. 16. Comme formulation de transfection de référence (contrôle positif), nous avons utilisé les polyplexes formulés avec le même plasmide et le polymère polycationique polyéthylèneimine (PEI ramifié, 25 kDa), et comme contrôle négatif l'ADN nu.
Pour la préparation des complexes, l'ADN est dilué dans l'HEPES (20 mM, pH 7.4) jusqu'à une concentration finale de 303 μM en équivalent phosphate, puis la quantité nécessaire
de composé no. 8 ou no. 16 obtenue à partir d'une solution mère de 20 mg^mL"1 dans le DMSO-eau 1:2 (v/v) est ajoutée afin d'obtenir le rapport N/P désiré. Pour le PEI, on utilise une solution mère 0.1 M dans l'eau. Les mélanges sont vortexés pendant 10 s et utilisés pour la caractérisation et pour les tests de transfection, comme décrit ci-dessous.
b) Caractérisation des complexes ADN-composé no.° 8 ou no. 16 b-1) Mesure de taille et polydispersité des nanoparticules d'ADN.
La diffusion quasi-élastique de la lumière est mise en œuvre comme décrit ci-dessus en utilisant un appareil CoulterN4 MD. Les mesures sont effectuées directement sur les préparations ci-dessus soit à une concentration d'ADN de 0.1 mg'mL"1 (303 μM en équivalent phosphate).
Dans le cas des nanoparticules d'ADN formulées pour un rapport N/P de 10" et 30 avec les composés no. 8 ou no. 16, on obtient, dans une série d'expériences indépendantes, des nanoparticules avec une taille moyenne qui varie de 150 (Déviation Standard — SD - = 100) à 200 (SD = 190) nm. A titre de comparaison, les polyplexes formulés avec le PEI pour un rapport N/P de 10 ont une taille moyenne de 160 nm (SD = 100) .
b-2) Analyse électrophorétique des nanoparticules d'ADN.
10 μl de chaque formulation de complexes préparés comme mentionnés ci-dessus sont dilués une fois dans du tampon de charge (glycérol + bleu de bromophénol + Tris-acétate-EDTA ou TAE). 10 μl de cette solution sont déposés sur un gel à 0,8% d'agarose. Le reste de l'échantillon est dilué une fois avec une solution de décomplexant à 8% de sodium dodécyl sulfate (SDS) dans le TAE, puis 10 μl sont également déposés sur un gel. L'électrophorèse est réalisée pendant 90 min à 50 V. Le
gel est ensuite développé avec le bromure d'éthidium (Sigma) dans le TAE (20 μL d'une solution de bromure d'éthidium de concentration 10 mg^mL"1 dans 200 mL du TAE). L'ADN est ainsi visualisé et photographié avec un transilluminateur UV. Les Figures 2 et 3 sont des photographies des analyses électrophorétiques, réalisées sur gel d'agarose en présence de bromure d'éthidium, des complexes ADN/nanoparticules obtenus avec le composé no. 8 selon des rapport N/P respectivement de 5, 10 , 30 et 50. L'analyse de la Figure 2 montre l'absence totale de bandes correspondant à l'ADN en ce qui concerne les nanoparticules d'ADN issues de préparation ayant un rapport N/P ≥ 10 (couloir 3 à 5) comparativement à l'ADN nu qui est clairement visible couloir 1. Ce résultat indique une complexation totale de l'ADN. Cependant, pour un rapport N/P = 5 (couloir 6), l'ADN reste encore partiellement accessible à l'intercalation par le bromure d'éthidium.
Des résultats très similaires ont été obtenus avec les complexes formulés avec le composé no. 16, celui-ci formant des complexes avec l'ADN dès un rapport N/P 5, l'ADN n'étant plus accessible à l'intercalation du bromure d'éthidium. L'intégrité du plasmide dans chaque échantillon est confirmée par électrophorèse sur gel après décomplexation avec le SDS, comme le montrent la Figure 3 pour un temps d'incubation de 10 min.
Exemple 33. Tests de transfection in vitro des nanoparticules d'ADN.
Des cellules murines d'embryon BNL-CL2 sont cultivées en plaques 96-puits jusqu'à une densité de 2 x 104 cellules/puits dans un milieu de culture DMEM (Dulbelcco's Modified Eagle Médium, Gibco-BRL) contenant 10% de sérum de veau fœtal (SVF; Sigma) à 37 QC dans une atmosphère humide avec une proportion 5% CO2/95% air.
Les complexes entre le composé no. 8, no. 16 ou le polyéthylèneimine (PEI), et le plasmide pTG11236 sont dilués dans 100 μL de DMEM de façon à avoir 0.5 μg d'ADN dans le puits . Le milieu de culture est enlevé et remplacé par 100 μL de solution de complexe nanoparticule/ADN dans le DMEM. Après 4 h et 24 h, 50 et 100 μL de DMEM contenant 30% et 10% de SVF, respectivement, sont additionnés. Après 48 h, la transfection est arrêtée, le milieu de culture est retiré et les cellules sont lavées par le PBS (2 x 100 μL), puis lysées avec 50 μL de tampon de lyse (Promega, Charbonnières, France). Les lysats sont congelés à -32 se jusqu'au dosage de luciférase et des protéines.
Le dosage de la luciférase permet d'évaluer l'efficacité de transfection. Ce dosage est basé sur une réaction de chimioluminescence dont le principe repose sur l ' oxydation par la luciférase de son substrat (la luciférine) avec production concomitante d'un photon. La mesure de l'activité de la luciférase est réalisée avec un appareil Biolumat LB96P WMP100 (BERTHOLD) pendant 10 s après l'injection de 50 μl de réactif contenant la luciférine (kit de dosage Proméga) dans chaque puits contenant 10 μl d'extrait cellulaire. Pour chaque dosage, une gamme étalon de 1 fg/μl à 1 ng/μl est réalisée avec de la R-luciférase (Bio-Rad) ce qui permet de convertir les mesures RLU en femtogrammes (fg) de luciférase par puits. Le dosage des protéines dans chaque puits (cf. ci- après) permet de convertir ces mesures en fg de luciférase par mg de protéine.
Le dosage des protéines s'effectue au moyen du test BCA (Pierce). Le dosage des protéines est réalisé sur 15 μl de lysat cellulaire auxquels sont rajoutés 300 μl de réactif de Pierce (BCA Protein Assay Reagent). Il s'agit d'un dosage colorimétrique qui est réalisé avec un spectrophotomètre UV/visible MRX (DYNEX Technologies). Pour chaque dosage, une
gamme étalon est réalisée avec de la BSA (Bovine Sérum Albumin, BIO-RAD), de 0 à 30 μg par puits. La lecture est faite à 570 nm, après 30 mn d'incubation à 37 0C et 15 min à température ambiante. Grâce à la gamme étalon de BSA, les DO peuvent être exprimées en mg de protéines/puits.
Le rapport des quantités de protéines correspondant aux cellules transfectées et non transfectées (100 % de viabilité cellulaire) permet d'évaluer la viabilité cellulaire des complexes (exprimée en %). Toutes les formulations sont testées au moins 3 fois. Les résultats obtenus ont été traités au moyen du logiciel STATGRAPHICS Plus5.1 (marque déposée). L'analyse de variance Anova est effectuée sur les valeurs des efficacités de transfection (LoglO(fg luciférase / mg protéine)) et sur la viabilité cellulaire. Il est possible d'analyser l'impact (sur la transfection ou la viabilité cellulaire) de la variation d'un ou de plusieurs facteurs en appliquant respectivement une analyse de variance mono- ou multifactorielle. La méthode utilisée pour la comparaison des moyennes est le HSD (honestly significant différence) intervais de Tukey. Cette technique du HSD de Tukey permet de comparer toutes les paires de moyennes et d'y montrer des différences avec un risque alpha.
Deux facteurs, la nature de l'agent de complexation (composés no. 8, no. 16, ou PEI) et le rapport N/P, ont été analysés comme source de variation des pourcentages de viabilité cellulaire (Figure 4) et du logarithme des niveaux de transfection (Figure 5 et 6).
La Figure 4, représente les moyennes et intervalles significatifs à 95 % de Tukey de la viabilité cellulaire après transfection de cellules BNL-CL2 par des complexes formulés avec les composés no. 8, no. 16 ou PEI et l'ADN plasmidique pTG11236, en fonction du rapport N/P allant de 5
à 50, et déterminée pour une concentration en plasmide de 0,5 μg/puits.
L'analyse de la Figure 4 indique d'excellentes viabilités cellulaires après transfection des complexes formulés avec le composé no. 8 ou no. 16.
Pour des rapports N/P > 10, les transfections réalisées avec des complexes formulés avec l'ADN et les composés no. 8 ou no. 16 révèle que la viabilité cellulaire est significativement supérieure à celle obtenue après transfection des cellules, pour les mêmes rapports N/P, par les polyplexes issus des complexes PEI/ADN.
La Figure 5 représente les moyennes et intervalles significatifs à 95 % de Tukey de la transformation logarithmique de l'expression de luciférase (fg de luciférase / mg de protéine) par les cellules BNL-CL2 transfectées avec des complexes formés par les composés no. 8, no. 16 ou PEI et l'ADN plasmidique pTG11236 pour un rapport N/P = 5, 10, 30 et 50, et pour 0,5 μg d'ADN par puits sur des cellules BNL-CL2. La Figure 6 représente les moyennes et intervalles significatifs à 95 % de Tukey de la transformation logarithmique de l'expression de luciférase (fg de luciférase / mg de protéine) par les cellules BNL-CL2 transfectées avec des complexes formulés avec les composés no. 8, no. 16 ou PEI et l'ADN plasmidique pTG11236 pour N/P = 10 et pour 0,5 μg d'ADN par puits.
L'analyse des Figures 5 et 6 indique des niveaux de transfection qui sont, pour des rapports N/P ≥ 10, de l'ordre de 100 fois supérieurs à ceux de l'ADN nu, et environ 100 fois inférieurs à ceux mesurés pour les polyplexes N/P 10 formulés avec le PEI.
Pour les complexes formulés avec le composé no. 16, cette analyse statistique indique des niveaux de transfection qui sont de l'ordre de 10.000 fois supérieurs a ceux de l'ADN nu, et équivalents à ceux mesurés pour les polyplexes N/P 10 formulés avec le PEI qui correspondent à l'optimum de la formulation de référence ( figure 6 ) .