WO2007007910A1 - ヘテロ環置換ベンズイミダゾール誘導体 - Google Patents
ヘテロ環置換ベンズイミダゾール誘導体 Download PDFInfo
- Publication number
- WO2007007910A1 WO2007007910A1 PCT/JP2006/314307 JP2006314307W WO2007007910A1 WO 2007007910 A1 WO2007007910 A1 WO 2007007910A1 JP 2006314307 W JP2006314307 W JP 2006314307W WO 2007007910 A1 WO2007007910 A1 WO 2007007910A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- pyridine
- alkyl
- ethylsulfonyl
- compound
- phenoxy
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
Definitions
- the present invention relates to a darcokinase activator containing a heterocyclic substituted benzimidazole derivative as an active ingredient. Further, the present invention relates to a novel heterocyclic substituted benzimidazole derivative.
- Glucokinase (ATP: D— hexo se 6— pho s pho tr an sf e. Raze, EC 2. 7. 1. 1) is one of four mammalian hexokinases ( Hexokinase IV). Hexokinase is the first stage enzyme in glycolysis and catalyzes the reaction from glucose to glucose 6-phosphate. Darcokinase is mainly expressed in liver and knee beta cells, and plays an important role in glucose metabolism throughout the body by controlling the rate-limiting step of glucose metabolism in these cells. Dalcokinase from the liver and i visceral cells has different N-terminal 15 amino acid sequences due to splicing differences, but the enzymatic properties are the same.
- darcokinase works as a glucose sensor in humans and plays an important role in glucose homeostasis.
- blood glucose control using the dalcokinase sensor system is considered to be possible in many patients with type I diabetes.
- the darcokinase activator is expected to be effective as a therapeutic agent for patients with type I diabetes because it can be expected to promote insulin secretion in the kidney cells and enhance glucose uptake and glucose release in the liver.
- pancreatic beta cell type dalcokinase has been applied to the brain of the rat, especially the feeding center.
- VHM's darucose concentration sensing system is presumed to have a dalcokinase-mediated mechanism similar to that of insulin secretion by visceral cells. Therefore, in addition to liver and knee pancreatic cells, VHM dalcokinase activity can be corrected not only for blood glucose correction but also for obesity, which is a problem in many type II diabetic patients. There is sex.
- a compound having a darcokinase activating effect is used as a therapeutic agent and / or a preventive agent for diabetes, or chronic complication of diabetes such as retinopathy, nephropathy, neurosis, ischemic heart disease, arteriosclerosis Treatment and treatment And / or as a preventive agent, and as a therapeutic and / or prophylactic agent for obesity.
- the compound represented by the above formula is common to the compound according to the present invention in that it has one pyrrole group and pyridine, but the basic skeleton of the above compound is 2,3-d] pyrimidine skeleton, which is different from the basic skeleton according to the present invention. Furthermore, the target diseases possessed by the above formula are subarachnoid hemorrhage and subsequent ischemic stroke, and are different from the target diseases of the compound according to the present invention.
- An object of the present invention is to provide a therapeutic and / or preventive agent for diabetes that binds to dalcokinase and increases the activity of darcokinase, and activates dalcokinase to It is to provide an anti-obesity agent that acts by stimulating.
- the present inventors have intensively studied to develop a novel anti-diabetic drug having a new medicinal effect and an effect higher than that of the existing anti-diabetic drug by an action different from that of the above-described existing drug.
- the inventors have found that the compound represented by (I) has a darcokinase activating action, and have completed the present invention. That is, the present invention
- X 4 represents a carbon atom or a nitrogen atom
- Ring A has the formula (II)
- X represents a carbon atom or a nitrogen atom
- He t has at least one of an oxygen atom or a sulfur atom in the ring, and is selected from the group consisting of a nitrogen atom, a sulfur atom or an oxygen atom in addition to the oxygen atom or the sulfur atom.
- a 5- or 6-membered aliphatic hetero ring optionally having 1 or 2 tera atoms in the ring (the 5- or 6-membered aliphatic hetero ring is — — 6 alkyl, 10 — Ci- 6 alkyl (the Ci- 6 alkyl and mono-O-C- 6 alkyl are halo , Optionally substituted with lower alkoxy), or the same or different with oxo or thixo, optionally substituted with 1 to 3),
- X 5 is 10—, 1 S—, 1 S (O) 1, 1 S (O) 2 —, 1 S (O) 2 N—, 1 C ( ⁇ ) ⁇ 1 or 1 NS ( ⁇ ) 2 — Indicate
- R 1 is Ariru, - CI- e alkyl or - C 3 - 7 or a cycloalkyl, or a nitrogen atom, 1 to 3 hetero atoms in the ring to be selected from the group consisting of sulfur atoms and oxygen atoms A 5- or 6-membered heteroaryl or a group in which the heteroaryl and phenyl or pyridyl are condensed (the R 1 may be substituted with 1 to 4 R 4, which are the same or different);
- R 2 is each independently formyl, 1 OH, 1 Ci- 6 alkyl, —CH 3 — a F a , 1 OCH 3 — a F a , amino, cyan, halogen or one (CH 2 ) n one OH Indicate
- Each R 3 is independently — — 6 alkyl, — (CH 2 > 6 — OH, 1 C ( ⁇ ) — OC — e alkyl, 1 (CH 2 ) 1 e— OC ⁇ — e alkyl, — (CH 2 ) n— NH 2 , Ciano, — C (O) 1-6 alkyl, halogen, —C 2 — 6 alkenyl, — OC — 6 alkyl, — COOH or almost 1 OH, R 4 represents each independently, - 6 alkyl (which alkyl may be the same or different, hydro alkoxy of 1 to 3, halogen, - OC (O) one - 6 alkyl (the alkyl may be substituted with 1 to 3 halogen ) Or may be substituted with monoalkyl),
- — 6 alkyl may be substituted with halogen or N (R 51 ) R 52 ),
- Heterocyclic (heterocycle one CI- 6 alkyl ( ⁇ _ ⁇ C - 6 alkyl, halogen or - may 6 be substituted with alkyl Le) - O-C),
- Phenyl which may be substituted with halogen, ——6 alkyl, —O ——- 6 alkyl Good),
- Halogen CN, formyl, COOH, amino, oxo, hydroxy, hydroxyamidino or nitrite
- R 51 and R 52 each independently represent a hydrogen atom, 16 alkyl, or a nitrogen atom, a 4- to 7-membered heterocycle formed by R 51 and R 52 together,
- R 5 '3 is a hydrogen atom or - indicates a 6 alkyl
- R 54 represents one 6 alkyl, or
- a 4- to 7-membered nitrogen-containing aliphatic heterocycle formed by combining alkyl of R 53 and R 54 with —N—C ( ⁇ ) — or
- a 4- to 7-membered nitrogen-containing aliphatic heterocycle formed by combining R 53 and R 54 alkyl with —N—C ( ⁇ ) —O— (the aliphatic heterocycle is substituted with oxo). And the aliphatic heterocyclic ring may have 1 or 2 double bonds in the ring)
- n an integer from 0 to 2
- q represents an integer of 0 to 2.
- R 1 represents aryl or a heteroatom selected from the group consisting of a nitrogen atom, a sulfur atom and a silicon atom;
- the compound or a pharmaceutically acceptable salt thereof according to the above (1) which is a group condensed with phenyl or pyridyl (the R 1 may be substituted with the same or different 1 to 4 R 4 ).
- a ring may be substituted with the same or different 1 to 3 R 3 , thiazolyl, imidazolyl, isothiazolyl, thiadiazolyl, oxadiazolyl, ⁇ diazolyl, oxazolyl, isoxazolyl, pyrazinyl, pyridyl, pyridazinyl, pyrazolyl or pyrimidinyl
- R 1 represents aryl or a 5- or 6-membered heteroaryl having 1 to 3 heteroatoms in the ring selected from the group consisting of a nitrogen atom, a sulfur atom and an oxygen atom Or a compound obtained by condensing the heteroaryl with phenyl or pyridyl (the R 1 may be substituted with the same or different 1 to 4 R 4 ), or a pharmaceutically acceptable compound thereof Acceptable salt,.
- Het has at least one oxygen atom in the ring, and in addition to the oxygen atom, 1 or a heteroatom selected from the group consisting of a nitrogen atom, a sulfur atom or an oxygen atom 2 may have, hetero ring to aliphatic 5- or 6-membered aliphatic hetero ring (the 5- or 6-membered, - alkyl, one O-C - 6 alkyl (said - C _ 6 alkyl and —O—C i — 6 alkyl may be substituted with halogen, lower alkoxy), or may be substituted with oxo or thixo), or a pharmaceutically acceptable salt thereof.
- Salt :
- Het force It has at least one sulfur atom in the ring, and in addition to the sulfur atom, 1 or 2 heteroatoms selected from the group consisting of nitrogen atoms, sulfur atoms or oxygen atoms
- a 5- or 6-membered aliphatic heterocyclic ring (the 5- or 6-membered aliphatic heterocyclic ring is a 1 C 1-6 alkyl, — ⁇ —C 1-16 alkyl (the — C 1-16 alkyl and —O—C 1-6 alkyl may be substituted with a halogen atom, —O—C 1-6 alkyl), optionally substituted with oxo or thixo) (3) the described compound or a pharmaceutically acceptable salt thereof,
- (6) to X 4 are all carbon atoms, the compound according to the above (3) or a pharmaceutically acceptable salt thereof,
- Equation (I) is transformed into Equation (1— 1)
- R 11 represents phenyl, or a 5- or 6-membered nitrogen-containing heteroaryl having 1 to 4 heteroatoms in the ring selected from the group consisting of a nitrogen atom, a sulfur atom and an oxygen atom. (The R 11 may be the same or different, and may be substituted with 1 to 3 R 4 );
- X 51 represents —0—, 1 S—, 1 S (O) — or — S (O) 2 —, and the other symbols are the same as defined above.
- Acceptable salt
- composition comprising the following (A)-(C) used for the treatment, prevention and prevention of type 2 diabetes mellitus.
- a darcokinase activator comprising the compound according to any one of (1) to (9) or a pharmaceutically acceptable salt thereof as an active ingredient
- the “aryl” preferably means a hydrocarbon aromatic ring having 6 to 14 carbon atoms, and examples thereof include phenyl, naphthyl, biphenyl, anthryl, etc. Among these, phenyl, naphthyl or biphenyl Is preferred, and phenyl is more preferred.
- ( ⁇ —6 alkyl” means straight or branched alkyl having 1 to 6 carbon atoms, such as methyl, ethyl, propyl, isopropyl, butyl, isoptyl, sec-butyl, tert-butyl, pentyl , Isamyl, neopentyl, isopentyl, 1,1-dimethylpropyl, 1-methylpropyl, 2-methylbutyl, 1,2-dimethylpropyl, hexyl, isohexyl, 1-methylpentyl, 2-methylpentyl, 3-methyl Pentyl, 1,1-dimethylbutyl, 1,2-dimethylbutyl, 2,2-dimethylbutyl, 1,3-dimethylbutyl, 2,3-dimethylbutyl, 3,3-dimethylbutyl, 1-ethylbutyl, 2-ethylbutyl, 1, 2, 2_trimethylpropyl, 1-ethyl-2
- C 2 - 6 alkenyl is meant straight chain or alkenyl of 2 to '6 carbon atoms which has a branched, for example, Ariru, 2 one propenyl, 1 Buarticulu, 2-butenyl, 2-methyl-2 —Butenyl, 1-pentenyl and the like.
- C 3 _ 7 cycloalkyl specifically, for example, cyclopropyl,. Cyclobutyl, Shikurobe pentyl, cyclohexyl, cycloheptyl, and the like cyclohexylene.
- Halogen means fluorine, chlorine, bromine X means iodine. .
- Examples of “one (CH 2 ) i- 6 — OH” include hydroxymethylene, hydroxyethylene, and the like.
- —O- 6 alkyl examples include methoxy, ethoxy, propoxy, tert-butoxy and the like.
- Examples of “— (CH 2 ) i- 6 —OC i- 6 alkyl” include methoxymethyl, methoxyethyl, propyloxymethyl, isopropyloxymethyl and the like.
- Examples of “mono-C 6 alkyl” include acetyl, ethylcarbonyl, isopropyl group, propylcarbonyl, and the like.
- Examples of “one C (O) OC i- 6 alkyl” include methoxycarbonyl, ethoxycarbonyl, tert-butoxycarbonyl and the like.
- One (CH 2 ) 1 — 6 -NH 2 J includes, for example, aminomethyl, aminoethyl, aminopropyl and the like.
- Examples of “one NH—C t-e alkyl” include methylamino, ethylamino, propylamino, 2-methylpetituamino and the like.
- “—N-dialkyl)” means a group in which the same or different —6 alkyl as defined above ”and N are bonded, for example, dimethylamino, ethylpropylamino, 2-methylpetituol 1-methylamino and the like.
- the same or different alkyl force in “—N-di--6alkyl)” may be combined with S nitrogen atom to form a ring. Specific examples of the ring include, for example, piperi Gin., Pyrrolidine and the like.
- One CH 3 — a F a means a group in which 1 to 3 hydrogen atoms in methyl are substituted with fluorine atoms, and examples thereof include trifluoromethyl, difluoromethyl, and fluoromethyl.
- One OCH 3 — a F a means a group in which “—CH 3 _ a F a ” as defined above and an oxygen atom are bonded, such as trifluoromethoxy, difluoromethoxy, or fluoromethoxy. Is mentioned.
- a represents an integer of 1 to 3.
- X 4 represent a carbon atom or a nitrogen atom, and among these, all of X to X 4 are carbon atoms, or any one or 2 of to X 4 is a nitrogen atom. More preferably, all of X 4 are carbon atoms.
- Ring A has the formula (II) A 5- to 6-membered heteroaryl having 1 to 3 heteroatoms in the ring, selected from the group consisting of a nitrogen atom, a sulfur atom and an oxygen atom represented by It means a group in which teloaryl and phenyl ring or pyridine ring are condensed. .
- X means a carbon atom or a nitrogen atom.
- Examples of the A ring include thiazolyl, imidazolyl, isothiazolyl, thiadiazolyl, oxadiazolyl, tri-T zolyl, oxazolyl, isoxazolyl, pyrazinyl, pyridyl, pyridazinyl, pyrazolyl or pyrimidinyl, and among these, thiazolyl, thiadialyl , Pyrazinyl, pyridyl, pyridazinyl, triazolyl or pyrazolyl are preferred, pyridyl, pyrazinyl, thiazolyl, thiazolyl, isoxazolyl or pyrazolyl are more preferred, and pyridyl or pyrazinyl is more preferred.
- the A ring may be the same or different and may have 1 or 2 substituents represented by R 3 .
- Each R 3 independently represents — ( 6 alkyl, — (CH 2 ) 6 — OH, 1 C (0) 1 OC ⁇ 6 alkyl, — (CH 2 ) 6 — OCi— 6 alkyl, 1 ( CH 2 ) n— NH 2 , cyan, — C (O) — C alkyl, norogen, —C 2 — 6 alkenyl, — Od— 6 alkyl, mono-COOH or almost — OH.
- R 3 “——6 alkyl” represented by R 3 means the same group as “one Ci-alkyl” defined above. Indicated R 3 "one (CH 2)
- One C (O) —OCi- 6 alkyl represented by R 3 means the same group as “One C (O) —OCi- 6 alkyl” defined above.
- Halogen represented by R 3 means the same group as “halogen” defined above.
- —OCi- 6 alkyl represented by R 3 means the same group as “——6 alkyl” defined above.
- Het has at least one of an oxygen atom or a sulfur atom in the ring, and is selected from the group consisting of a nitrogen atom, a sulfur atom and an oxygen atom in addition to the oxygen atom or the sulfur atom.
- He t is selected from the group consisting of a nitrogen atom, a sulfur atom or an oxygen atom in addition to the oxygen atom or the sulfur atom, having at least one of either an oxygen atom or a sulfur atom.
- a 5- or 6-membered aliphatic heterocycle having one heteroatom in the ring is preferred.
- He t is _C ⁇ 6 alkyl, —O—Ci- 6 alkyl (wherein 6 alkyl and 1 O— 16 alkyl may be substituted with halogen, — ⁇ —Ci-e alkyl) , Thixo or thixo may be the same or different and may be substituted by 1 to 3; '
- halogen of the substituent means the same group as the “halogen” defined above.
- the -alkyl and 1-O- 6 alkyl are halogen such as fluorine, chlorine and bromine or lower alkoxy such as methoxy, ethoxy and isopropoxy, which may be the same or different and may be substituted by 1 to 3 Good. )
- He t may have include, for example, methyl, ethyl, oxo, hydroxy, alkoxy, fluorine, etc. among the substituents described above, and He t represents these substituents. 1 to 3 may be the same or different.
- X 5 is one O—, — S—, — S (O) one, one S (0) 2 —,-S (O) 2 N—, — C ( ⁇ ) — or one NS ( ⁇ ) 2 — Means. '
- X 5 is preferably —O—, 1 S—, 1 S (O) — or 1 S (O) 2 —, and more preferably 1 O—.
- R 1 is Ariru, -C i-6 alkyl or - C 3 - 7 or a cycloalkyl, or a nitrogen atom, 1 heteroatom in the ring to be selected from the group consisting of sulfur and oxygen atoms ⁇ 3 "A 5- or 6-membered heteroaryl, a group in which the heteroaryl and phenyl or pyridyl are condensed, or a bicyclic ring having 9 to 10 members having 2 or 3 nitrogen atoms in the ring. Means group.
- the “aryl” represented by R 1 means the same group as the “aryl” defined above, specifically, for example, phenyl, naphthyl or biphenyl is preferable, and phenyl is more preferable.
- One C i- 6 alkyl” represented by R 1 means the same group as “—C i- 6 alkyl” defined above, specifically, for example, methyl, ethyl, propyl, isopropyl, etc. Can be mentioned.
- R 1 represents "- C 3 - 7 cycloalkyl” refers to "one C 3 - 7 cycloalkyl" defined above means the same groups as, specifically, for example, cyclopropyl, cyclobutyl, cyclopentyl, Examples include cyclohexyl, lu, and cycloheptyl.
- R 1 represented by “a 5- or 6-membered heteroaryl having 1 to 3 heteroatoms selected from the group consisting of a nitrogen atom, a sulfur atom, and an oxygen atom” include pyridyl, Pyrazinyl and pyrimidinyl are preferred, and pyridyl or pyrazinyl is more preferred.
- R 1 among these, phenyl, pyridyl, pyrazinyl, pyrimidinyl and the like are preferable. More preferred are phenyl and pyridyl.
- R 1 may be substituted with 1 to 4 R 4 s that are the same or different, and may preferably be substituted with 1 to 2 R 4 that are the same or different.
- R 4 is primary alkyl (the alkyl may be the same or different, 1 to 3 hydroxy, C androgenic one OC ( ⁇ ) - 6 alkyl (also said alkyl substituted by 1 to 3 halogen Or may be substituted with one OC- 6 alkyl), —C 3 —7 cycloalkyl,
- One O—Ci- 6 alkyl (the d- 6 alkyl may be substituted with halogen or ⁇ ⁇ (R 51 ) R 52 );
- ( 6 alkyl is halogen, amino, CN, hydroxy, ⁇ _ ⁇ 1 Ci- 6 alkyl, — CH 3 — a F a , — OC (O) 1 Ci- 6 alkyl.
- -N ((: 6 alkyl) C ( ⁇ ) O— Ci— 6 alkyl, —NH—C ( ⁇ ) O— C — 6 alkyl, phenyl, 1 N (R 51 ) R 52 , — N HC ( ⁇ ) 1 alkyl, 1 N (Ci- 6 alkyl) 1 C (0) 1 — 6 alkyl or — NH—
- Heterocyclic heterocycle one CI- C6 alkyl (said - 6 alkyl may be substituted by halogen or - ⁇ one alkyl Le)
- Phenyl (which may be substituted with halogen, —6 alkyl, —O—Ci-6 alkyl),
- neurogen Means neurogen, CN, formyl, COH, amino, oxo, 'hydroxy, hydroxyamidino or nitrogen.
- the “octalogen” represented by R 4 means the same group as defined above.
- “—Alkyl” represented by R 4 means a linear or branched alkyl having 1 to 6 carbon atoms, such as methyl, edyl, propyl, isopropyl, butyl, isobutyl, sec-butyl, tert-butyl, Pentyl, isoamyl, neopentyl, isopentyl, 1,1-dimethylpropyl, 1-methylbutyl, 2-methylbutyl, 1,2-dimethylpropyl, hexyl, isohexyl, 1-methylpentyl, 2-methylpentyl, 3-methylpentyl 1,1-dimethylbutyl, 1,2-dimethylbutyl, 2,2-dimethylbutyl, 1,3-dimethylbutyl, 2,3-dimethylbutyl, 3,3-dimethylbutyl, 1-ethylbutyl, 2-ethylbutyl, .1, 2 , 2-trimethylpropyl,
- the “one Ci- 6 alkyl” is 1 to 3 hydroxy, neurogen, —OC ( ⁇ ) — — 6 alkyl (the alkyl may be substituted with 1 to 3 halogen), or —O—. — It may be substituted with 6 alkyls.
- halogen of the substituent examples include the same groups as the halogen defined above.
- Examples of the —OC ( ⁇ ) —Ci- 6 alkyl of the substituent include methylcarbonyloxy, ethylcarbonyloxy, isopropylcarbonyloxy and the like.
- One OC (O) —Ci- 6 alkyl of the substituent may be substituted with 1 to 3 halogen atoms as defined above.
- Examples of one O—C 6 alkyl of the substituent include methoxy, ethoxy, propoxy, isopropoxy and the like.
- R 4 represents "- S ( ⁇ ) 0 - 2 - (- 6 alkyl.”
- The, - S (O) one 2 - and the definition of one - means 6 ⁇ alkyl and is attached group, e.g. 1 S-ethyl, 1 S-methyl, 1 S-isopropyl, 1 S-propyl, 1 S (O) 2 -methyl, 1 S ( ⁇ ) 2 -ethyl and the like.
- — (: 6 alkyl) in the “—S CO) ._ 2 —alkyl” may be substituted with hydroxy.
- R 4 represents a "one C 3 _ 8 cycloalkyl", defined the same group.
- R 4 represents "one C 2 - 6 alkenyl" as is defined the same group.
- R 4 shows + “C (O) N (R 51 ) R 52 ” means a substituted or unsubstituted force rubamoyl group, or N, R 51 and R 52 together. It means a group in which a 4- to 7-membered aliphatic heterocyclic ring formed by bonding with carbonyl.
- substituted or unsubstituted substitution force rubermoyl examples include, for example, force rubamoyl, methylcarbamoyl, ethylcarbamoyl, and isopropyl force rubamoi.
- force rubamoyl methylcarbamoyl, ethylcarbamoyl, and isopropyl force rubamoi.
- C (O) N (R 51 ) R 52 represented by R 4, the 4-to-7-membered aliphatic formed by N, R 51 and R 52 together Examples thereof include azetidinyl, pyrrolidinyl, piperidino, piperazinyl, morpholino and the like. Therefore, C (O) N (R 51 ) R 52 includes: azetidine 1 1 force lponyl, pyrrolidine 1 1 force lponyl, piperidine 1 1 force lponyl, piperazine 1 1 force carbonyl, morpholine— 1-Carbonyl and the like can be mentioned.
- Examples of “—O——6 alkyl” represented by R 4 include the same groups as “—O— ( 6 alkyl”) as defined above.
- the —O—Ci- 6 alkyl may be substituted with halogen or N (R 51 ) R 52 .
- Examples of “—C (O) —— 6 alkyl” represented by R 4 include the same groups as “one C ( ⁇ ) —Ci- 6 alkyl” defined above.
- the “halogen” of the substituent include the same groups as the halogen defined above.
- —N— (C — 6 alkyl) 1 C (O) 0— d— 6 alkyl means — N— (Ci 16 alkyl) — and the above — C (O) ⁇ —C 6 means an alkyl-bonded group, specifically, for example, -N (Me) — C ( ⁇ ) ⁇ 1 tert-butyl.
- —NH—C ( ⁇ ) O— 6 alkyl of the substituent means a group in which —NH— and the —C (O) OC ⁇ e alkyl are bonded, specifically, for example, NH-C (O) O-methyl, -NH-C (0) O-ethyl, NH-C (O) O-isopropyl-NH-C (O) monopropyl, and the like.
- substituent “—N (R 51 ) R 52 ” include the same groups as the above-mentioned “one N (R 51 ) R 52 J.
- the substituent“ one NH—C (O) —Ci “E-alkyl” means —NH—C ( ⁇ ) as defined above.
- Means ⁇ _ 6 alkyl bonding specifically, for example, - NH-C (O) Mechiru, - NH- C (0) - Echiru one NH- C (O) one isopropyl, - NH—C (O) —propyl and the like can be mentioned.
- one C (0) ⁇ - 6 alkyl" and one N-- the alkyl bonding - 6 Al kill one C ( ⁇ ) first and of the definition Specifically, for example, 1 N (methyl)-C (O) -methyl,-N (methyl)-C ( ⁇ ) 1 edil,-N (ethyl) 1 C (O)-isopropyl, 1 N (methyl) 1 C (O) 1 isopropyl, 1 N (isopropyl) 1 C (O) methyl and the like.
- One 2 - as one 6 alkyl means an NH- and the one S (O) ⁇ -2- C -e alkyl bonding, specifically, for example, -NH-S (0 ) 2 - methyl, - NH- S ( ⁇ ) 2 - Echiru, - NH- S ( ⁇ ) 2 -. isopropyl, and the like.
- _ C (O) —C ⁇ 6 alkyl which may have the above-mentioned substituents on the ⁇ _ 6 alkyl specifically includes, for example, fluoromethylcarbonyl, 2, 2, 2-trifluoroethylcarbonyl, cyanomethylcarbonyl, hydroxymethylcarbonyl, 2-hydroxyethylcarbonyl, methoxymethylcarbonyl, aminomethylcarbonyl, N-methylaminocarbonyl, 2-phenylethylcarbonyl, etc. .
- R 4 represents "- C (S) - Ji 6 alkyl" and one C (S) - and the definition "- - 6 alkyl” and is meant bonded groups, specifically, for example, -C (S) -methyl,-C (S) -ethyl,-C (S) monoisopropyl, mono C (S) monopropyl and the like.
- R 4 represents “one (CH 2 ). One four one N (R 53 ) — C ( ⁇ ) — R 54 ”, wherein R 53 represents a hydrogen atom or —C ⁇ 6 alkyl, and R 54 represents —C ⁇ — A force that means 6 alkyl or “— (CH 2 ) 0-4 -N (R 53 ) -C (0) -R 54 J N (R 53 ) One C (O) One In R 54 , a 4- to 7-membered nitrogen-containing aliphatic heterocycle formed by the combination of one N—C (0)-and alkyl of R 53 and R 54 (the heterocycle is substituted with oxo) It may also have 1 or 2 double bonds in the ring.
- R 53 is a hydrogen atom or one - a 6 alkyl
- R 54 is, - CI_ when a 6 alkyl "- (CH 2) 0 - 4 - N (R 53) -C ( ⁇ ) Single R 54 Specifically, for example, -CH 2 -NH- C (O) — Methyl, —CH 2 — NH— C ( ⁇ ) — Ethyl, —CH 2 — NH— C (O) — Isopropyl, —CH 2 —NH—C (O) — Propyl, — CH 2 — N (methyl) -C ( ⁇ ) monomethyl, — CH 2 — N (ethyl) — C ( ⁇ ) -methyl, — NH—C (O) -methyl, one NH—C (O) -ethyl, one NH—C (O) monoisopropyl, — NH—C ( ⁇ ) monopropyl, mono N (methyl) mono C
- R 55 is a hydrogen atom or a - where R 4 represents means flicking 6 ⁇ alkyl, R 56 is a -Ci- 6 alkyl Meaning force, or “One N (R 5 ' 5 ) — C (O) — O — R 56j — N (R 55) — c (O) — 0—
- one N— C (O) 1— means a 4- to 7-membered nitrogen-containing aliphatic heterocycle formed by combining R— and alkyl of R 55 and R 5 6 together.
- R 55 is a hydrogen atom or —Ci- 6 alkyl
- R 56 is 1 C— 6 alkyl
- “—N (R 55 ) 1 C ( ⁇ ) ⁇ 0-R 5 e J Specifically, for example, 1 NH—C ( ⁇ ) 1 O-methyl, —NH—C (0) 1 O—ethyl, 1 NH—C ( ⁇ ) 1 O—isopropyl, —NH—C ( O) 1 O —propyl, 1 N (methyl) —C ( ⁇ ) 1 O—methyl, 1 N (ethyl) 1 C (O) — ⁇ —methyl, and the like.
- One C (O) — aryl represented by R 4 means a group in which force sulfonyl and aryl as defined above are bonded, and specific examples include benzoyl, naphthylcarbonyl and the like. It is done.
- aryl in the “one C (O) -aryl” is a halogen atom as defined above, and is substituted by 1 to 3 substituents. It may be.
- R 4 represents “one C ( ⁇ ) monoaromatic elemental ring”, which is a force luponyl and 5- or 6-membered monocyclic aromatic heterocycle or 9- or 10-membered bicyclic aromatic as defined above. It means a group bonded to a heterocyclic ring.
- the “one C (O) monoaromatic heterocycle” represented by R 4 means a group in which a strong ruponyl and a 4- to 7-membered monocyclic aliphatic heterocycle as defined above are bonded, specifically, Specifically, for example, 1 C (O) -azetidinyl, 1 C (O) —pyrrolidinyl, 1 C (O) -piperidino, 1 C ( ⁇ ) 1 piperidinyl, 1 C ( ⁇ ) -azepanyl, — C ( 0) -piperazinyl, 1 C (0) -morpholino,-C (0) -thiomorpholino, 1 C (O) 1 homopiperazinyl, 1 C (O) 1 imidazolidinyl, 1 C (O) -virazolidinyl, and the like.
- the “heterocycle” represented by R 4 includes the same basic strength as the A ring.
- heterocycle, -C Bok 6 - alkyl, halogen or one O-CI- 6 - 1 to 3 may be substituted with alkyl.
- Examples of the substituent —Ci-e-alkyl, halogen, and mono-O-Ci- 6 -alkyl include the same groups as defined above.
- halogen represented by R 4
- examples of the “halogen” represented by R 4 include the same groups as the “halogen” defined above.
- Phenyl represented by R 4 may be substituted with no, rogen, —Ci- 6 alkyl or —O-alkyl.
- R 1 is a R 4 'has 2 or 3 as a substituent, becomes the same or different two R 4 gar ⁇ , they may form a 4 to 6 membered ring, specifically
- R 2 is each independently formyl, —OH, — — 6 alkyl, — CH 3 — a F a , — OCH 3, a F a , amino, cyan, halogen or mono (CH 2 ) 6 — ⁇ H Means.
- the R 2 includes hydroxy, formyl, —CH 3 —a F a (preferably trifluoromethyl), mono-O CH 3 —a F a , halogen, d- 16 alkyl, amino, CN, — (CH 2 ) H is preferred, hydro Carboxymethyl, formyl, One CH 3 - a F a (preferably triflate Ruo b methyl) one OCH 3 - a F a (preferably triflate Ruo b methoxy), Amino, Halogen, single d-6 alkyl, CN or one (CH 2 ) j— 4 OH is more preferred, hydroxy, formyl, amino, halogen (preferably fluoro and black), —C 6 alkyl or mono (CH 2 ) Further preferred.
- Examples of the compound represented by the formula (I) include:
- the compound (1-1) according to the present invention can be produced, for example, by the following method.
- R 5 represents a hydrogen atom or a hydroxy, a lower alkyl group which may be substituted by alkoxy, etc., etc., r is represents 1 or 2, and other symbols as defined above]
- This step is a method for producing the compound (1-1) according to the present invention by reacting the compound (1) with the compound (2) in the presence of an acid.
- Examples of the acid used in this step include P-toluenesulfonic acid, sulfuric acid, ytterpium triflate, camphorsulfonic acid or a hydrate thereof. .
- the amount of the acid used is generally 0.01 to 10 equivalents, preferably 0.1 to 3 equivalents, relative to 1 equivalent of compound (1).
- the amount of compound (2) used in this step is usually 1 to 100 equivalents, preferably 1 to 5 equivalents, relative to 1 equivalent of compound (1).
- Examples of the compound (2) include ethylene glycol, 1,3-propanediol, 1,4-butanediol, 2- (hydroxymethyl) -1,3-propanediol, 2- (hydroxymethyl) -Butanediol and the like. .
- the reaction solvent is not particularly limited as long as it is not used or does not interfere with the reaction.
- toluene, black mouth form, dimethylformamide and the like can be used. Among these, toluene and black mouth form are used. preferable.
- the reaction temperature is usually 0 to 150 ° C., preferably room temperature to 120 ° C.
- the reaction time is usually 5 minutes to 48 hours, preferably 15 minutes to 12 hours.
- the compound (1-1) thus obtained can be isolated and purified by known separation and purification means such as concentration, concentration under reduced pressure, crystallization, solvent extraction, reprecipitation, chromatography and the like.
- the compound (1-2) according to the present invention can be produced, for example, by the following method.
- R 1G represents a lower alkyl group which may be substituted with hydroxy, alkoxy or the like, r 2 represents 1 or 2, and other symbols are the same as above]
- This step is a method for producing the compound (I-2) according to the present invention by reacting the compound (1) with the compound (3) in the presence of an acid. .
- Examples of the acid used in this step include p-toluenesulfonic acid, sulfuric acid, ytterbium triflate, camphorsulfonic acid, and hydrates thereof.
- the amount of the acid used is generally 0.01 to 10 equivalents, preferably 0.1 to 3 equivalents, relative to 1 equivalent of compound (1).
- the amount of compound (2) used in this step is usually 0.1 to 100 equivalents, preferably 1 to 5 equivalents, relative to 1 equivalent of compound (1).
- Examples of compound (3) include N-acetylethylamine.
- the reaction solvent is not particularly limited as long as it is not used or does not interfere with the reaction.
- Toluene, black mouth form, dimethylformamide and the like can be mentioned. Of these, toluene and black mouth form are preferable.
- the reaction temperature is generally 0 ° C. to 150 ° C., preferably room temperature to 120 ° C. '
- the reaction time is usually 5 minutes to 48 hours, preferably 15 minutes to 12 hours.
- the thus obtained compound (I-12) can be isolated and purified by a known separation and purification means, for example, concentration, concentration under reduced pressure, crystallization, solvent extraction, reprecipitation, chromatography and the like.
- the compound (1-3) according to the present invention can be produced, for example, by the following method.
- This step is a method for producing the compound (4) by reacting the compound (1) with the compound (4-1) and magnesium.
- the compound (4-1) used in this step for example, 4-monobromo-1-butene, 5-bu-mole 1-pentene and the like can be mentioned.
- the amount of the compound (4-1) used is usually 0.5 to 20 equivalents per 1 equivalent of the compound (1).
- the amount of magnesium used is usually 0.5 to 30 equivalents relative to 1 equivalent of compound (1).
- the reaction solvent is not particularly limited as long as it does not interfere with the reaction, and examples thereof include tetrahydrofuran, ether, dichloromethane, chloroform, toluene, etc. Among these, tetrahydrofuran is preferable.
- the reaction time is usually 5 minutes to 12 hours, preferably 5 minutes to 1 hour.
- the reaction temperature is usually ⁇ 78 to 50 ° C., preferably 0 ° C. to room temperature.
- the compound (4) thus obtained can be isolated and purified by known separation and purification means, for example, concentration, concentration under reduced pressure, crystallization, solvent extraction, reprecipitation, chromatography, etc. It can be attached to the next process.
- This step is a method for producing (5) according to the present invention by reacting the compound (4) with sodium periodate and osmium tetroxide.
- the amount of sodium periodate used in this step is usually 0 with respect to 1 equivalent of compound (4). 5 to 20 equivalents, preferably 1 to 5 equivalents.
- the amount of osmium tetroxide used in this step is usually 00 1 to 3 equivalents relative to 1 equivalent of compound (1). '
- the reaction solvent is not particularly limited as long as it does not interfere with the reaction, and examples thereof include tetrahydrofuran, acetonitrile, acetone and the like. Among these, tetrahydrofuran is preferred.
- the reaction temperature is usually from 78 to 50 ° C, preferably from 0 ° to room temperature.
- the reaction time is usually 5 minutes to 24 hours, preferably 30 minutes to 6 hours.
- the thus obtained compound (5) can be isolated or purified by known separation and purification means, for example, concentration, concentration under reduced pressure, crystallization, solvent extraction, reprecipitation, chromatography, etc. It can be attached to the next process.
- This step is a method for producing the compound (6) by reducing the compound (5).
- Examples of the reducing agent used in this step include NaBH 4 , Zn (BH 3 CN) 2 , NaB (O Ac) 3 H, NaBH 3 CN, and the like.
- the amount of the reducing agent used is usually 0.5 to 10 equivalents per 1 equivalent of the compound (6).
- the reaction solvent is not particularly limited as long as it does not interfere with the reaction, and examples thereof include methanol, ethanol, water, tetrahydrofuran, etc. Among these, methanol is preferable.
- the reaction temperature is usually 0 to 60 ° C., preferably 0 to room temperature.
- the reaction time is usually 30 minutes to 24 hours, preferably 1 to 12 hours.
- the compound (6) thus obtained can be isolated or purified by known separation and purification means such as concentration, concentration under reduced pressure, crystallization, solvent extraction, reprecipitation, chromatography, etc., or without isolation and purification. It can be attached to the next process.
- This step is a method for producing the compound (I 13) according to the present invention by subjecting the compound (6) to a cyclization reaction in the presence of an acid. ⁇
- Examples of the acid used in this step include P-toluenesulfonic acid, sulfuric acid, ytterium, riflatate, camphor sulfonic acid, and hydrates thereof.
- the amount of the acid to be used is generally 0.01 to 10 equivalents, preferably 0.1 to 3 equivalents, relative to 1 equivalent of compound (6).
- the reaction solvent is not particularly limited as long as it is not used or does not interfere with the reaction, and examples thereof include toluene, black mouth form, dimethylformamide, etc. Among these, toluene and black mouth form are preferable. .
- the reaction time is usually 5 minutes to 48 hours, preferably 15 minutes to 12 hours.
- the reaction temperature is usually 0 to 180 ° C., preferably room temperature to 120 ° C.
- the compound (1-3) thus obtained can be isolated and purified by known separation and purification means, for example, concentration, concentration under reduced pressure, crystallization, solvent extraction, reprecipitation, chromatography and the like.
- this step can also be produced by introducing a leaving group into a hydroxy group and then adding a base to the reaction solution and cyclization.
- the reagents used for introducing the leaving group are methanesulfonyl chloride, p-toluenesulfonyl chloride, sulfochloride chloride. And methanesulfonyl chloride is preferred.
- the amount of the reagent to be used is usually 5 to 20 equivalents, preferably 0.5 to 10 equivalents, relative to 1 equivalent of compound (6). '
- a base may be used for the reaction, and examples include triethylamine, pyridine, N, N-dimethylaminopyridine and the like, and triethylamine is preferable.
- the amount of the reagent to be used is generally 0.5 to 20 equivalents, preferably 0.5 to 10 equivalents, relative to 1 equivalent of compound (6).
- the reaction solvent is not particularly limited as long as it does not interfere with the reaction, and examples thereof include tetrahydrofuran, ethyl acetate, dioxane, black mouth form, etc. Among these, ethyl acetate is preferable.
- the reaction time is usually 5 minutes to 48 hours, preferably 15 minutes to 12 hours.
- the reaction temperature is usually 120 to 100 ° C., preferably 0 to 40 ° C.
- Examples of the base used for the cyclization include N, N-dimethylaminopyridine, 1,8-diazabicyclo [5.4.0] undecar 7-en, sodium hydride, potassium carbonate and the like.
- the base group may be directly added to the reaction solution into which the leaving group has been introduced, but it is preferable to add the base group to the solution of the crude product after the post treatment.
- the compound (1-3) thus obtained can be isolated and purified by known separation and purification means such as concentration, concentration under reduced pressure, crystallization, solvent extraction, reprecipitation, chromatography and the like.
- the compound (I 14) according to the present invention can be produced, for example, by the following method.
- This step is a method for producing the compound (9-1) by reacting the compound (8) with 2-methylallylmagnesium chloride.
- the amount of the compound 2-methylallylmagnesium chloride used in this step is usually 0.5 to 10 equivalents, preferably 1 to 5 equivalents, relative to 1 equivalent of the compound (8).
- the reaction solvent is not particularly limited as long as it does not interfere with the reaction, and examples thereof include tetrahydrofuran, ether, dichloromethane, chloroform, toluene, etc. Among these, tetrahydrofuran is preferred.
- the reaction time is usually 1 minute to 48 hours, preferably 5 minutes to 1 hour.
- the reaction temperature is usually from 78 to 50 ° C., preferably from 0 to room temperature.
- the compound (9-1) thus obtained can be isolated or purified by a known separation and purification means, for example, concentration, concentration under reduced pressure, crystallization, solvent extraction, reprecipitation, chromatography, etc. It can be applied to the next process. .
- This step consists of the step (step 8-1) of performing a mouth-mouthed positioning using the compound (9-1) and the subsequent step of carrying out a cyclization reaction (step 8-2). This is a method for producing 1-4). (Process 8-1)
- borane used in the hydroporation in this step examples include a porane-tetrahydro complex, a porane-dimethylsulfide complex, and 9-1-BBN, and a porane-tetrahydro complex is preferred. .
- the amount of borane to be used is usually 0.5 to 50 equivalents, preferably 2 to 10 equivalents, relative to 1 equivalent of the compound (9-1).
- the reaction solvent is not particularly limited as long as it does not interfere with the reaction, and examples thereof include tetrahydrofuran, ether, dichloromethane, chloroform, toluene, and the like. Among these, tetrahydrofuran is preferred.
- the reaction time is usually 1 minute to 48 hours, preferably 30 minutes to 3 hours.
- the reaction temperature is usually from 78 to 50 ° C., preferably from 0 to room temperature.
- the reaction solution is treated with sodium hydroxide and hydrogen peroxide.
- the compound (9-2) thus obtained can be isolated or purified by known separation and purification means, for example, concentration, concentration under reduced pressure, crystallization, solvent extraction, reprecipitation, chromatography, etc. It can be applied to the next process.
- This step is a method for producing the compound (I-4) according to the present invention by subjecting the compound (91-2) obtained in the above step 8-1 to a cyclization reaction in the presence of an acid.
- the cyclization reaction can be carried out by the same method as in Step 6 above, a method according to this, or a combination of these with conventional methods.
- the compound (1-4) thus obtained is isolated or purified by known separation and purification means, for example, concentration, concentration under reduced pressure, solvent extraction, crystallization, reprecipitation, chromatography, etc. can do.
- the compound (1-5) according to the present invention can be produced, for example, by the following method.
- This step is a method for producing the compound (12) by reacting the compound (11) with N, O-dimethylhydroxylamine monohydrochloride.
- the reaction in this step is a so-called amide bond forming reaction and is performed using the carboxylic acid represented by the compound (1 1) or a reactive derivative thereof and N, 0-dimethylhydroxylamine monohydrochloride.
- the compound (1 1) or a reactive derivative thereof used is usually 0.1 to 100 equivalents, preferably “0.1 to 3 equivalents”.
- Examples of the “reactive derivative” of the compound (1 1) include mixed acid anhydrides, active esters, active amides and the like, and examples thereof include the method described in International Publication WO 98/0 5 61 Can be obtained by
- the carboxylic acid represented by the compound (1 1) when used, for example, force sulfonyldiimidazole, N, N′-dicyclohexylcarpositimide, 1-ethyl-3- (3-dimethylaminopropyl) It is preferable to carry out the reaction in the presence of a condensing agent such as carpositimide, diphenylphosphoryl azide, dipyridyl disulfide-triphenylphosphine, and preferably sulfonyldiimidazole.
- a condensing agent such as carpositimide, diphenylphosphoryl azide, dipyridyl disulfide-triphenylphosphine, and preferably sulfonyldiimidazole.
- the amount of the condensing agent to be used is not strictly limited, but is usually 0.1 to 100 equivalents, preferably 0.1 to 10 equivalents, relative to compound (1 1).
- the reaction is usually carried out in an inert solvent.
- the inert solvent include tetrahydrofuran, N, N-dimethylformamide, 1,4-dioxane, benzene, toluene, methylene chloride, black mouth form, and tetrachloride. Carbon, 1,2-dichloroethane, pyridine and the like, or a mixture of these solvents.
- the reaction temperature is usually 0 ° C. to the reflux temperature of the reaction solvent, preferably room temperature to the reflux temperature of the reaction solvent.
- the reaction time is usually from 0.1 hours to 72 hours, preferably from 0.5 hours to 24 hours.
- the above reaction can be carried out in the presence of a base and a condensation aid in order to facilitate the reaction.
- a base examples include 4-dimethylaminopyridine, triedylamine and the like.
- the amount of the base to be used is generally 0.1 to 100 equivalents, preferably 0.1 to 1 equivalent, relative to 1 mol of the carboxylic acid represented by compound (11) or a reactive derivative thereof.
- condensation aid examples include N-hydroxybenzotriazole hydrate, N-hydroxysuccinimide and the like. ..
- the amount of the condensation aid used is usually 1 to 100 equivalents, preferably 1 to 5 equivalents, per 1 mol of the carboxylic acid represented by the compound (11) or a reactive derivative thereof.
- the thus obtained (12) can be isolated or purified by known separation and purification means, for example, concentration, concentration under reduced pressure, solvent extraction, crystallization, reprecipitation, chromatography, etc. It can be attached to the next process.
- This step is a method for producing the compound (1 3) by reducing the nitro group of the compound (1 2).
- Examples of the reducing agent used in this step include tin chloride, iron (I 1), Raney nickel, palladium, palladium hydroxide, and the like.
- the amount of the reducing agent used is usually from 0.01 to 30 equivalents, preferably from 0.1 to 10 equivalents, relative to 1 equivalent of the compound (1 2).
- the reaction solvent is not particularly limited as long as it does not interfere with the reaction, and examples thereof include methanol, ethanol, N-methylpyrrolidinone, dimethylformamide, tetrahydrofuran, and acetic acid. Of these, N-methylpyrrolidinone and methanol are preferred. However, acetic acid is preferred when iron (II) is used.
- the reaction temperature is usually 0 to 150 ° C., preferably room temperature to 100 ° C. '
- the reaction time is usually 1 minute to 24 hours, preferably 5 minutes to 12 hours.
- the compound (13) thus obtained can be isolated and purified by known separation and purification means, for example, concentration, concentration under reduced pressure, crystallization, solvent extraction, reprecipitation, chromatography, etc., or without isolation and purification. It can be attached to the next process.
- This step is a method for producing the compound (14) by reacting the compound (13) with the compound (e). .
- This reaction is a so-called amide bond forming reaction and is performed using the carboxylic acid represented by the compound (e) or a reactive derivative thereof.
- Examples of the compound (e) used include pyridine-2-carboxylic acid, pyrazine-1-carbonic acid, pyrimidine-4 mononuclear rubonic acid, pyrimidine-2-carboxylic acid, thiazole-2-carboxylic acid, 3 -Carboxylic acid, 5 -Methyl-isoxazol-3 -Carboxylic acid, 1 -Methyl- 1H -Imidazo 1-ru 4 -Carboxylic acid, Imidazo 1 -2 2-carboxylic acid, imidazole 1-carboxylic acid, [1, 2, 4] triazol 1-carboxylic acid, [1, 2, 4] triazole 3-carboxylic acid, [1, 2, 3 ] Triazol 4 Four strength rubonic acid, 3-methyl- [1, 2, 4] thiadiazol-5-carboxylic acid, [1, 2, 5] thiadiazole-3-carboxylic acid, [1, 2, 3 ] Oxadiazol-3-carboxylic acid, pyr
- the amount of compound (e) or a reactive derivative thereof used is usually 0.1 to 100 equivalents, preferably 0.1 to 20 equivalents, more preferably 0.1 to 3 equivalents to 1 equivalent of compound (13). Is equivalent.
- the reactive derivative of the compound (e) include mixed acid anhydrides, active esters, active amides, and the like. These can be obtained, for example, by the method described in W098 / 05641. be able to.
- the carboxylic acid represented by the compound (e) when used, for example, force sulfonyl diimidazole, N, N′-dicyclohexyl carpositimide, 1-ethyl-3- (3-dimethylaminopropyl) It is preferable to carry out the reaction in the presence of a condensing agent such as carbopositimide, diphenylphosphoryl azide, dipyridyl disulfide-triphenylphosphine, and preferably strong carbonyl imidazole.
- a condensing agent such as carbopositimide, diphenylphosphoryl azide, dipyridyl disulfide-triphenylphosphine, and preferably strong carbonyl imidazole.
- the amount of the condensing agent to be used is not strictly limited, but is usually 0.1 to 100 equivalents, preferably 0.1 to 10 equivalents, relative to compound (e).
- the reaction is usually carried out in an inert solvent.
- the inert solvent include tetrahydrofuran, N, N-dimethylformamide, 1,4-dioxane, benzene, toluene, methylene chloride, black mouth form, and tetrachloride. Examples thereof include carbon, 1,2-dichloroethane, pyridine and the like, or a mixture of these solvents.
- the reaction temperature is usually 0 ° C. to the reflux temperature of the reaction solvent, preferably room temperature to the reflux temperature of the reaction solvent.
- the reaction time is usually 0.1 hour to 72 hours, preferably 0.5 hour to 24 hours.
- the above reaction can be carried out in the presence of a base and a condensation aid in order to facilitate the reaction. Examples of the base include .4-dimethylaminopyridine, triedylamine and the like.
- the amount of the base to be used is generally 1 to 100 equivalents, preferably 0.1 to 1 equivalent, relative to i mol of the carboxylic acid represented by the compound (e) or a reactive derivative thereof.
- condensation aid examples include N-hydroxybenzotriazole hydrate, N-hydroxysuccinimide and the like.
- the amount of the condensation aid used is usually 1 to 100 equivalents, preferably 1 to 5 equivalents, per 1 mol of the carboxylic acid represented by the compound (e) or a reactive derivative thereof.
- the amino group or imino group when an amino group or imino group that does not participate in the reaction is present in the reactant, the amino group or imino group is appropriately protected after protecting with an amino group or a protecting group for the imino group. It is preferable to remove the protecting group later. .
- (14) obtained in this way can be isolated or purified by known separation and purification means, for example, concentration, concentration under reduced pressure, solvent extraction, crystallization, reprecipitation, chromatography, etc. It can be attached to the next process.
- This step is a method for producing a compound (1 4 ⁇ 1) by reacting the compound (1 4) with 3-butenylmagnesium promide.
- the amount of compound 3-butenylmagnesium promide used in this step is usually 0.5 to 10 equivalents, preferably 1 to 5 equivalents, relative to 1 equivalent of compound (14).
- the reaction solvent is not particularly limited as long as it does not interfere with the reaction.
- examples thereof include tetrahydrofuran, ether, dichloromethane, chloroform, toluene, and the like. Among these, tetrahydrofuran is preferred.
- the reaction time is usually 1 minute to 48 hours, preferably 5 minutes to 1 hour.
- the reaction temperature is usually ⁇ 78 to 50 ° C., preferably 0 ° C. to room temperature.
- the compound (14 1 1) obtained in this way can be isolated and purified by known separation and purification means, for example, concentration, concentration under reduced pressure, crystallization, solvent extraction, reprecipitation, chromatography, etc. It can be subjected to the next step without purification.
- This step is a method for producing a compound (1 4-2) by reacting the olefin body obtained in the step (1 2-1) with sodium periodate and osmium tetroxide.
- the amount of sodium periodate used in this step is usually 0.5 to 20 equivalents, preferably 1 to 5 equivalents, relative to 1 equivalent of the olefin body.
- the amount of osmium tetroxide used in this step is usually from 0.01 to 3 equivalents per equivalent of olefin.
- the reaction solvent is not particularly limited as long as it does not hinder the reaction, and examples thereof include tetrahydrofuran, acetonitrile, acetone and the like. Of these, tetrahydrofuran is preferable.
- the reaction temperature is usually ⁇ 78 to 50 ° C., preferably 0 ° C. to room temperature.
- reaction time is usually 5 minutes to 24 hours, preferably 30 minutes to 6 hours.
- (14-12) thus obtained can be isolated or purified by known separation and purification means such as concentration, concentration under reduced pressure, crystallization, solvent extraction, reprecipitation, chromatography, etc. or without isolation and purification. It can be attached to the next process. ' ⁇
- This step is a method for producing the compound (14-3) by oxidizing the compound (14-2) obtained in the previous step (12-2).
- the amount of sodium chlorite used in this step is usually 0.5 to 20 equivalents, preferably 1 to 5 equivalents, relative to 1 equivalent of compound (14-12).
- the amount of 2-methyl-2-butene used in this step is usually 0.5 to 10 equivalents relative to 1 equivalent of compound (14-2).
- the amount of monosodium dihydrogen phosphate used in this step is usually 0.5 to 10 equivalents, preferably 1 to 3 equivalents, relative to 1 equivalent of compound (14-2).
- the reaction solvent is not particularly limited as long as it does not interfere with the reaction, and examples thereof include tetrahydrofuran, acetonitrile, acetone, t-butyl alcohol, water and the like, and these can be used in combination.
- the reaction temperature is usually from 78 to 50 ° C, preferably from 0 ° to room temperature.
- the reaction time is usually 5 minutes to 24 hours, preferably 1 hour to 12 hours.
- the compound (14-3) thus obtained is isolated or purified by a known separation and purification means such as concentration, concentration under reduced pressure, crystallization, solvent extraction, reprecipitation, chromatography, etc. It can be attached to the next process.
- This step is a method in which the ketone group of the compound (14-3) is reduced and converted to the compound (15).
- Examples of the reduction of the ketone include a method using a reducing agent such as sodium borohydride, lithium aluminum hydride, lithium borohydride, disobutyl aluminum hydride, and sodium borohydride is preferable. .
- the amount of the reducing agent used in this step is usually 1 to 10 equivalents, preferably 1 to 5 equivalents, relative to 1 equivalent of the compound (14-13).
- the reaction solvent is not particularly limited as long as it does not interfere with the reaction, and examples thereof include methanol, ethanol, water and the like, and methanol is preferable.
- the reaction temperature is usually from 78 to 50 ° C, preferably from 0 ° to room temperature.
- the reaction time is usually 5 minutes to 24 hours, preferably 5 minutes to 1 hour.
- the compound (15) thus obtained can be isolated or purified by a known separation and purification means, for example, concentration, concentration under reduced pressure, crystallization, solvent extraction, reprecipitation, chromatography, etc. It can be attached to the next process. .
- This step is a method of converting the compound (15) obtained in the step (13-1) into a compound (16) by cyclization.
- the reaction in this step is performed using an acid catalyst.
- Examples of the acid catalyst used in this step include p-toluenesulfonic acid, sulfuric acid, and Um triflates, strong camphor sulfonic acids or their hydrates.
- the amount of the acid used is usually from 0 to 10 equivalents, preferably from 0.1 to 3 equivalents, relative to 1 equivalent of the compound (1 5). '
- the reaction solvent is not particularly limited as long as it is not used or does not interfere with the reaction, and examples thereof include toluene, black mouth form, dimethylformamide, etc. Among these, toluene and black mouth form are preferable. .
- the reaction time is usually 5 minutes to 48 hours, preferably 15 minutes to 12 hours.
- the reaction temperature is usually 0 to 180 ° C., preferably room temperature to 120 ° C.
- the compound (16) thus obtained can be isolated and purified by known separation and purification means such as concentration, concentration under reduced pressure, crystallization, solvent extraction, reprecipitation, chromatography, etc.
- This step is a method for producing the compound (17) by reacting the compound (16) with fuming nitric acid.
- the amount of fuming nitric acid used in this step is usually 5 to 50 equivalents, preferably 1 to 10 equivalents, relative to 1 equivalent of the compound (1 6).
- reaction solvent for example, 'Kuroguchi form, trifluoroacetic acid, sulfuric acid, hydrochloric acid, etc. may be used.
- the reaction time is usually 1 minute to 24 hours, preferably 5 minutes to 3 hours.
- the reaction temperature is usually 0 to 100 ° C., preferably room temperature to 50 ° C.
- the compound (17) thus obtained can be isolated or purified by known separation and purification means such as concentration, concentration under reduced pressure, crystallization, solvent extraction, reprecipitation, chromatography, etc. or without isolation and purification. It can be attached to the next process.
- This step is a method for producing a compound (1 8) by reacting the compound U 7) with Ar—Z—X 8 in the presence of a base.
- the amount of the base used in this step is usually 0.5 to 20 equivalents, preferably 1 to 5 equivalents, relative to 1 equivalent of the compound (1 7).
- Examples of the base include potassium carbonate, sodium carbonate, cesium carbonate, triethylamine, cesium fluoride and the like.
- the reaction solvent is not particularly limited as long as it does not interfere with the reaction.
- Examples thereof include N-methylpyrrolidinone, dimethylformamide, tetrahydrofuran, and acetonitrile. Among these, N-methylpyrrolidinone and dimethylformamide are preferable. .
- the reaction time is usually 1 minute to 12 hours, preferably 5 minutes to 3 hours.
- the reaction temperature is usually room temperature to 1550 ° C., preferably room temperature to 100 ° C.
- the compound (18) thus obtained can be isolated or purified by known separation and purification means such as concentration, concentration under reduced pressure, crystallization, solvent extraction, reprecipitation, chromatography, etc. It can be attached to the next process.
- This step is a method for producing the compound (I-6) according to the present invention by reducing and further cyclizing the compound (18).
- Examples of the reducing agent used in this step include tin chloride (I 1), iron (I I); Raney nickel, palladium hydroxide and the like.
- the amount of the reducing agent to be used is usually 0.01 to 20 equivalents, preferably 0.1 to 10 equivalents, relative to 1 equivalent of compound (18).
- the reaction solvent is not particularly limited as long as it does not interfere with the reaction.
- examples thereof include methanol, ethanol, N-methylpyrrolidinone, dimethylformamide, tetrahydrofuran, and acetic acid.
- methanol Is preferred.
- acetic acid is preferred when iron (I I) is used.
- the reaction temperature is usually 0 to 150 ° C., preferably room temperature to 100 ° C.
- the reaction time is usually 1 minute to 24 hours, preferably 5 minutes to 12 hours.
- the compound (I-16) thus obtained is isolated or purified by a known separation and purification means, for example, concentration, concentration under reduced pressure, crystallization, solvent extraction, reprecipitation, chromatography, etc.
- a known separation and purification means for example, concentration, concentration under reduced pressure, crystallization, solvent extraction, reprecipitation, chromatography, etc.
- Can -The compound (I-16) according to the present invention can be produced, for example, by the following method.
- R 6 represents a hydrogen atom or alkyl, and other symbols are the same as above]
- This step is a method for producing the compound (20) by reacting the compound (1) with trimethylsilylnitrile in the presence of zinc iodide or the like.
- the amount of zinc iodide used in this step is usually 0.01 to 10 equivalents, preferably 0.1 to 1 equivalent, relative to 1 equivalent of compound (1).
- the amount of trimethylsilyl nitrile used in this step is usually 1 to 100 equivalents, preferably 1 to 10 equivalents, relative to 1 equivalent of compound (1).
- the reaction solvent is not particularly limited as long as it is not used or does not interfere with the reaction.
- black mouth form dimethylformamide, and toluene may be used.
- the reaction temperature is usually 0 to 100 ° C., preferably 0 ° C. to room temperature.
- the reaction time is usually 10 minutes to 12 hours, preferably 1 to 12 hours.
- the compound (20) thus obtained can be isolated and purified by known separation and purification means such as concentration, vacuum concentration, crystallization, solvent extraction, reprecipitation, chromatography, etc. It can be attached to the next process.
- This step is a method for producing a compound (2 1) by reacting the compound (20) with 10% hydrochloric acid monomethanol or the like.
- the 10% hydrochloric acid monomethanol used in this step is used as a solvent.
- the reaction temperature is usually 0 to 80 degrees, preferably 0 to 50 degrees.
- the reaction time is usually 10 minutes to 3 hours, preferably 10 minutes to 1 hour.
- the thus obtained compound (21) can be isolated or purified by known separation and purification means, for example, concentration, concentration under reduced pressure, crystallization, solvent extraction, reprecipitation, chromatography, or the like. It can be attached to the next process.
- This step is a method for producing a compound (22) by reacting the compound (21) with a compound R 6 NH 2 in the presence of a base.
- Examples of the compound R 6 NH 2 include methylamine.
- the amount of compound R 6 NH 2 used in this step is generally 0.5 to 30 equivalents, preferably 1 to 10 equivalents, relative to 1 equivalent of compound (21).
- the reaction solvent is not particularly limited as long as it does not interfere with the reaction, and examples thereof include methanol, tetrahydrofuran, black mouth form, etc. Among these, methanol is preferable.
- the reaction time is usually 10 minutes to 12 hours, preferably 10 minutes to 3 hours.
- the reaction temperature is usually 0 to 60 ° C., preferably room temperature to 50 ° C.
- the compound (22) thus obtained can be isolated and purified by known separation and purification means, for example, concentration, concentration under reduced pressure, crystallization, solvent extraction, reprecipitation, chromatography, etc., or without isolation and purification. It can be attached to the next process.
- the compound (22) is reacted with forceful midazolol in the presence of a base, and then a strong base is added to cyclize the compound according to the present invention ( 1-6).
- Bases used in the reaction of the compound (22) with force carbonyl imidazole include triethylamine, N, N-dimethylaminopyridine, 1,8-diazapicyclo [5.4.0] undecaker 7 , Carbonated power and so on.
- the amount of the base is usually 0.5 to 20 equivalents, preferably 1 to 10 equivalents, relative to 1 equivalent of the compound (22).
- the reaction solvent is not particularly limited as long as it does not interfere with the reaction, and examples thereof include dimethylformamide, black mouth form, and tetrahydrofuran. Of these, dimethylformamide is preferred. '
- the reaction time is usually 10 minutes to 24 hours, preferably 1 to 5 hours. .
- the reaction temperature is usually 0 to 100 ° C., preferably 0 to 60 ° C.
- examples of the strong base used after reacting the compound (2 2) with forceful imidazolidone include potassium tert-butoxide.
- the amount of the strong base is usually 1 to 20 equivalents, preferably 1 to 10 equivalents, relative to 1 equivalent of the compound (2 2).
- the reaction temperature is usually 0 to 120 ° C., preferably room temperature to 100 ° C.
- the reaction time is usually 10 minutes to 2.4 hours, preferably 10 minutes to 6 hours.
- the compound (I 16) thus obtained can be isolated and purified by known separation and purification means, for example, concentration, concentration under reduced pressure, crystallization, solvent extraction, reprecipitation, kumatography, etc. Can be purified.
- the compound (1-7) according to the present invention can be produced, for example, by the following method.
- This step is a method for producing a compound (2 4) by subjecting the compound (2 1) to a reduction reaction.
- Examples of the reducing agent used in this step include lithium aluminum hydride or disobutylaluminum hydride.
- the amount of the reducing agent used is usually 0.5 to 10 equivalents, preferably 1 to 5 equivalents, relative to 1 equivalent of the compound (2 1).
- the reaction solvent is not particularly limited as long as it does not interfere with the reaction.
- toluene, chloroform, Rum, tetrahydrofuran, etc. among which tetrahydrofuran is preferred.
- the reaction temperature is usually ⁇ 20 to 80 ° C., preferably 0 to 30 ° C.
- the reaction time is usually 1 minute to 6 hours, preferably 5 minutes to 1 hour.
- the compound (2 4) thus obtained can be isolated or purified by known separation and purification means such as concentration, concentration under reduced pressure, crystallization, solvent extraction, reprecipitation, chromatography, etc. or without isolation and purification. It can be attached to the next process.
- This step is a method for producing the compound (1-7) according to the present invention by reacting the compound (2 4).
- the amount of force sulfonyl diimidazole used in this step is usually 0.5 to 10 equivalents, preferably 1 to 5 equivalents, relative to 1 equivalent of compound (2 4).
- the reaction solvent is not particularly limited as long as it does not hinder the reaction, and examples thereof include dimethylformamide, tetrahydrofuran, black mouth form, etc. Among these, dimethylformamide is preferable.
- the reaction temperature is usually 0 to 1550 ° C., preferably room temperature to 100 ° C.
- the reaction time is usually 5 to 24 hours, preferably 1 to 12 hours.
- the compound (1-7) thus obtained can be isolated and purified by a known separation and purification means, for example, concentration, concentration under reduced pressure, crystallization, solvent extraction, reprecipitation, chromatography and the like.
- the compound (1-8) according to the present invention can be produced, for example, by the following method.
- a leaving group is introduced into the compound (8), and then the compound into which the leaving group is introduced and a succinic acid salt.
- This is a method for producing a compound (2 5) by reacting with lithium.
- the reaction in this step is performed by reacting compound (8) with methanesulfonyl chloride, etc. in the presence of a base, converting a hydroxyl group into a leaving group, and then reacting the compound having the leaving group with sodium cyanide. This can be done by reacting.
- Examples of the base used in this step include triethylamine, pyridine, N, N-dimethylaminopyridine and the like.
- the amount of the base is usually 5 to 50 equivalents, preferably 1 to 1 equivalent to 1 equivalent of the compound (2 5).
- the amount of sodium cyanide used in this step is usually 0.5 to 10 equivalents, preferably 1 to 5 equivalents, relative to 1 equivalent of compound (2 5).
- the reaction solvent is not particularly limited as long as it does not interfere with the reaction, and examples thereof include acetonitrile, acetone, dimethylformamide, dimethylsulfide and the like, and among these, dimethylformamide is preferable.
- the reaction temperature is usually 0 to 100 ° C., preferably 0 to 50 ° C.
- the reaction time is usually 5 minutes to 12 hours, preferably 5 minutes to 6 hours.
- the compound (2 5) thus obtained is isolated or purified by known separation and purification means such as concentration, concentration under reduced pressure, crystallization, solvent extraction, reprecipitation, chromatography, etc. It can be applied to the next process without any problems.
- This step is a method for producing a compound (2 6) by hydrolyzing the nitrile group of the compound (2 5) obtained in the step 25.
- This step is performed in the presence of sodium hydroxide or the like.
- the amount of 5N sodium hydroxide to be used is usually 1 to 100 equivalents, preferably 1 to 10 equivalents, relative to 1 equivalent of the compound (2 5).
- the reaction solvent is not particularly limited as long as it does not interfere with the reaction, and examples thereof include methanol, tetrahydrofuran, water, or a mixed solvent thereof, and a mixed solvent of methanol or tetrahydrofuran and water is preferable. .
- the reaction temperature is usually 0 to 1550 ° C., preferably room temperature to 100 ° C.
- the reaction time is usually 1 to 48 hours, preferably 1 to 24 hours.
- the compound (2 6) thus obtained can be isolated or purified by known separation and purification means such as concentration, concentration under reduced pressure, crystallization, solvent extraction, reprecipitation, chromatography, etc. or without isolation and purification. It can be attached to the next process.
- This step is a method for converting a force Rupokishiru group of step 2.6 the compound obtained in (2 6) to C i _ 6 alkyl ester such as methyl ester (2 7).
- a methyl ester can be obtained by reacting the compound (2 6) with trimethylsilyldiazomethane.
- the amount of trimethylsilyldiazomethane to be used is usually 5 to 20 equivalents, preferably 1 to 10 equivalents, relative to 1 equivalent of the compound (2 6).
- the reaction solvent is not particularly limited as long as it does not hinder the reaction, and examples thereof include methanol, tetrahydrofuran, black mouth form, etc. Among these, methanol is preferable.
- the reaction temperature is usually 0 to 100 ° C., preferably 0 ° C. to room temperature.
- the reaction time is usually 5 minutes to 24 hours, preferably 5 minutes to 2 hours.
- the alkyl ester is converted by a known method, a method according to this, or a combination thereof with a conventional method. Can be manufactured.
- the compound (26) thus obtained can be isolated and purified by known separation and purification means such as concentration, concentration under reduced pressure, crystallization, solvent extraction, reprecipitation, chromatography, etc., or without isolation and purification. It can be attached to the next process.
- This step is a method for producing a compound (28) by reacting the compound (27) obtained in the step 27 with allylic bromide in the presence of a base.
- Examples of the base used in this step include lithium diisopropylamide, sodium hydride, potassium t-butoxide and the like.
- the amount of the base used is usually 0.5 to 10 equivalents, preferably 1 to 5 equivalents, relative to 1 equivalent of the compound (27).
- the amount of odoraryl used in this step is usually 0.5 to 10 equivalents, preferably 1 to 5 equivalents, relative to 1 equivalent of compound (27).
- the reaction solvent is not particularly limited as long as it does not interfere with the reaction.
- Examples thereof include dimethylformamide and tetrahydrofuran. Among these, dimethylformamide is preferable.
- the reaction temperature is usually from 78 to 60 ° C., preferably from 120 ° to room temperature.
- the reaction time is usually 5 minutes to 12 hours, preferably 30 minutes to 6 hours.
- the compound (28) thus obtained can be isolated or purified by known separation and purification means, for example, concentration, concentration under reduced pressure, crystallization, solvent extraction; reprecipitation, chromatography, etc. It can be attached to the next process.
- This step can be carried out by the same method as in step 12-2 and step 13-1, a method according to this, or a combination of these and conventional methods.
- the thus obtained compound (1-8) can be isolated and purified by known separation and purification means, for example, concentration, concentration under reduced pressure, crystallization, solvent extraction, reprecipitation, chromatography and the like.
- This step is a method for producing a compound (6-1) or (6-2) by introducing a leaving group into the compound (6) and then reacting with potassium O-ethyldithiocarbonate. It is.
- the compound (6) is reacted with methanesulfonyl chloride, etc. in the presence of a base to convert the hydroxy group into a leaving group, and then the compound having the leaving group and the strength of 0-ethyldithio This can be done by reacting with a ponate.
- Examples of the base used in this step include triethylamine, pyridine, N, N-dimethylaminopyridine and the like.
- the amount of the base is usually 5 to 50 equivalents, preferably 1 to 10 equivalents, relative to 1 equivalent of the compound (6).
- the amount of potassium O-ethyldithioate used in this step is compound (6) Is usually 0.5 to 10 equivalents, preferably 1 to 5 equivalents.
- the reaction solvent is not particularly limited as long as it does not interfere with the reaction, and examples thereof include acetonitrile, acetone, dimethylformamide, dimethylsulfide and the like, and among these, acetone is preferable.
- the reaction temperature is usually 0 to 100 ° C., preferably 0 to 80 ° C.
- the reaction time is usually 5 minutes to 12 hours, preferably 5 minutes to 6 hours.
- the compound (6-1) or (6-2) thus obtained can be isolated and purified by known separation and purification means, for example, concentration, concentration under reduced pressure, crystallization, solvent extraction, reprecipitation, chromatography, etc. Alternatively, it can be used in the next step without isolation and purification.
- Examples of the base used in this step include 7K sodium oxide, sodium methoxide, potassium carbonate and the like.
- the amount of the base is usually 5 to 50 equivalents, preferably 1 to 10 equivalents, relative to 1 equivalent of the compound (6-1) or (6-2).
- the reaction solvent is not particularly limited as long as it does not hinder the reaction, and examples thereof include methanol and water.
- the reaction temperature is usually 0 to 100 ° C., preferably 0 to 80 ° C.
- the reaction time is usually 5 minutes to 12 hours, preferably 5 minutes to 6 hours.
- the cyclization reaction can be carried out by the same method as in Step 6 above, a method according to this, or a combination thereof with a conventional method.
- the compound (I-9) or (I-10) thus obtained can be isolated and purified by known separation and purification means such as concentration, concentration under reduced pressure, crystallization, solvent extraction, reprecipitation, chromatography, etc. Alternatively, it can be subjected to the next step without isolation and purification. '
- a compound represented by the same symbols as above can be obtained by acidifying the compound (1-9).
- the oxidizing agent used include OXONE.
- the amount of the oxidizing agent used is usually 1 to 10 equivalents, preferably 0.3 to 3 equivalents, relative to the equivalent of compound (1-9).
- the reaction solvent is not particularly limited as long as it does not interfere with the reaction.
- examples thereof include methanol, tetrahydrofuran, chloroform, water, etc.
- a mixed solvent of methanol and water is preferable. .
- the reaction time is usually 10 minutes to 24 hours, preferably 30 minutes to 6 hours.
- the reaction temperature is usually from 20 to 60 ° C., preferably from 0 ° to room temperature.
- the compound thus obtained (I 9 1 1) or (I-9 1 2) can be obtained by known separation and purification means, for example, concentration, concentration under reduced pressure, crystallization, solvent extraction, reprecipitation, chromatography, etc. Can be isolated and purified.
- the compound (1-11) according to the present invention can be produced, for example, by the following method.
- This step is a method for producing the compound (21-1) by hydrolyzing the compound (21).
- This step is performed in the presence of 7K sodium oxide or the like.
- the amount of 5N sodium hydroxide to be used is usually 1 to 100 equivalents, preferably 1 to 10 equivalents, relative to 1 equivalent of compound (25).
- the reaction solvent is not particularly limited as long as it does not hinder the reaction.
- methanol, tetra Hydrofuran, water, or a mixed solvent thereof may be mentioned, and a mixed solvent of methanol or tetrahydrofuran and water is preferable.
- the reaction temperature is usually 0 to 1550 ° C., preferably room temperature to 100 ° C. '
- the reaction time is usually 1 to 48 hours, preferably 1 to 24 hours. .
- the compound (2 1-1) thus obtained can be isolated or purified by known separation and purification means, for example, concentration, concentration under reduced pressure, crystallization, solvent extraction, reprecipitation, chromatography, etc. It can be attached to the next process.
- This step is a method for producing the compound (1-11) according to the present invention by reacting the compound (21-1 with 2,2-dimethoxypropane).
- the amount of 2,2-dimethoxypropane used in this step is usually 0.5 to 100 equivalents, preferably 1 to 10 equivalents, relative to 1 equivalent of the compound (2 1-1).
- the reaction solvent is not particularly limited as long as it is not used or does not interfere with the reaction.
- examples thereof include acetonitrile and tetrahydrofuran, and among these, acetone is preferable.
- the reaction temperature is usually 0 to 1550 ° C., preferably room temperature to 100 ° C.
- the reaction time is usually 10 minutes to 12 hours, preferably 30 minutes to 6 hours.
- the compound (1-111) thus obtained can be isolated and purified by known separation and purification means such as concentration, concentration under reduced pressure, crystallization, solvent extraction, reprecipitation, chromatography and the like.
- the compound (1-1 2) according to the present invention can be produced, for example, by the following method.
- This step is a method for producing a compound (2 9) by reducing the compound (1-8).
- the reducing agent used in this step include lithium aluminum hydride, diisobutylaluminum hydride, sodium borohydride, and the like, and these can be used as necessary.
- the amount of the reducing agent used is usually 5 to 10 equivalents relative to 1 equivalent of the compound (I-8).
- the reaction solvent is not particularly limited as long as it does not interfere with the reaction, and examples thereof include ether, chloroform, methanol, ethanol, water, tetrahydrofuran, etc. Among these, tetrahydride Mouth furan and methanol are preferred.
- the reaction temperature is usually 0 to 60 ° C., preferably 0 to room temperature.
- the reaction time is usually 30 minutes to 24 hours, preferably 1 to 12 hours.
- the compound (29) thus obtained can be isolated or purified by known separation and purification means such as concentration, concentration under reduced pressure, crystallization, solvent extraction, reprecipitation, chromatography, etc. It can be attached to the next process.
- This step is a method for producing the compound (I 1 1 2) according to the present invention by subjecting the compound (2 9) obtained in the above step 35 to a ring reaction in the presence of an acid.
- the cyclization reaction can be carried out by the same method as in Step 6 above, a method according to this, or a combination thereof with a conventional method. .
- the compound (1-12-2) thus obtained can be isolated and purified by known separation and purification means, for example, concentration, concentration under reduced pressure, solvent extraction, crystallization, reprecipitation, chromatographic chromatography, etc. It can be separated and purified.
- R represents a lower alkyl group
- Rpr represents a protecting group in the imidazole ring
- 1 ⁇ and L 2 represent a leaving group, and other symbols are the same as above.
- This step is a method for producing compound (c) by reacting compound (a) with compound (b) in the presence of an acid catalyst.
- step 4 compound (i) is produced by reaction with compound (h) A r—ZH. Any of these may be used, and examples thereof include a fluorine atom, a chlorine atom and a bromine atom, and among these, a fluorine atom is preferred.
- Examples of the acid catalyst used in this step include sulfuric acid, P-toluenesulfonic acid, methanesulfonic acid, hydrochloric acid, and thionyl chloride. .
- the amount of the acid catalyst used is usually from 0.1 to 10 equivalents, preferably from 0.1 to 1 equivalent, based on 1 equivalent of the compound (a).
- Examples of the compound (a) used include 2_fluoro-4-nitrobenzoic acid, 2-fluoro-5-nitrobenzoic acid, 5-fluoro-2-nitrobenzoic acid, and 3-fluoro-5-nitrobenzoic acid. Can be mentioned.
- the lower alkyl group represented by R is the same group as the lower alkyl group defined above.
- the compound (b) is also used as a reaction solvent, and examples thereof include methanol and ethanol.
- the amount of the compound (b) used is usually the amount of solvent relative to 1 equivalent of compound (a).
- the reaction temperature is usually room temperature to the reflux temperature of the reaction solvent, preferably 60 to the reflux temperature of the reaction solvent.
- the reaction time is usually 1 to 120 hours, preferably 24 to 72 hours.
- reaction solvent used in this step examples include methanol, ethanol, toluene, tetrahydrofuran, dimethylformamide and the like.
- the compound (c) thus obtained can be isolated and purified by known separation and purification means, for example, concentration, concentration under reduced pressure, crystallization, solvent extraction, reprecipitation, chromatography, etc., or isolation and purification. It can be attached to the following.
- This step is a method for producing a compound (d) by reducing the nitro group of the compound (c) obtained in the step A.
- the reduction reaction used in this step include catalytic reduction using hydrogen, formic acid, ammonium formate or hydrazine hydrate, etc. and palladium, white gold or nickel catalyst, hydrochloric acid or salt, etc.
- Examples include a reduction method using ⁇ ammonium and iron, and a reduction method using methanol and tin chloride.
- the amount of the reducing agent used in this step varies depending on the type of compound and solvent used, but is usually 1 to 50 equivalents, preferably 2 to 20 equivalents, relative to 1 equivalent of the compound (c).
- the reaction temperature is usually from 1 to 10 ° C., preferably from 0 to 50 ° C.
- the reaction time is usually 1 to 20 hours, preferably 1 to 5 hours.
- the reaction solvent to be used is not particularly limited as long as it does not interfere with the reaction, and examples thereof include methanol, N, N-dimethylformamide, ethyl acetate, tetrahydrofuran and the like and mixed solvents thereof.
- the compound (d) thus obtained can be isolated and purified by known separation and purification means, for example, concentration, concentration under reduced pressure, crystallization, solvent extraction, reprecipitation, chromatography, etc., or isolation and purification. It can be attached to the following.
- This step is a method for producing the compound (f) by reacting the compound (d) obtained in the step B with the compound (e).
- the amide bond forming reaction in this step is performed using a carboxylic acid represented by compound (5) or a reactive derivative thereof.
- Examples of the compound (e) used include, for example, pyridine-2-strong rubonic acid, pyrazine-2-strong rubonic acid, pyrimidine-1-4-carboxylic acid, pyrimidine 1-2-strong rubonic acid, thiazo-1-rurubonic acid, Ixoxazole—3—strong rubonic acid, 5-methyl-isoxazo-luo 3-carboxylic acid, 1-methyl—1H mono-imidazole 4--carboxylic acid, imidazo-luo 2-strong rubonic acid, 1-methyl- 1H—imidazole —2—Strengthen rubonic acid, imidazole 1-strength rubonic acid, [1, 2, 4] Triazol — 1-strength rubonic acid, [1, 2, 4] Triazol-lu , 3] Triazol—4 strong rubonic acid, 3-methyl- [1, 2, 4] thiadiazole-5—strong rubonic acid, [1, 2, 5] thiadiazolulu 3 —carboxylic acid,
- the amount of compound (e) or a reactive derivative thereof used is usually 1 to 100 equivalents, preferably 0.1 to 20 equivalents, more preferably 0.1 to 3 equivalents, relative to 1 equivalent of compound (d). is there.
- Examples of the reactive derivative of the compound (e) include mixed acid anhydrides, active esters, active amides, and the like, which are obtained by, for example, the method described in WO 98/05641. be able to.
- the carboxylic acid represented by the compound (e) when used, for example, force sulfonyldiimidazole, N, N′-dicyclohexyl carpositimide, 1-ethyl-3- (3-dimethylaminopropyl) carposimide It is preferable to carry out the reaction in the presence of a condensing agent such as diphenylphosphoryl azide, dipyridyl disulfide-triphenylphosphine, etc., preferably force sulfonyldiimidazole.
- a condensing agent such as diphenylphosphoryl azide, dipyridyl disulfide-triphenylphosphine, etc.
- the amount of the condensing agent to be used is not strictly limited, but is usually 0.1 to 100 equivalents, preferably 0.1 to 10 equivalents, relative to compound (e).
- the reaction is usually carried out in an inert solvent.
- the inert solvent include tetrahydrofuran, N, N-dimethylformamide, 1,4-dioxan, benzene, toluene, salt methylene chloride, , Carbon tetrachloride, 1,2-dichloroethane, pyridine and the like, or a mixture of these solvents.
- the reaction temperature is usually 0 ° C. to the reflux temperature of the reaction solvent, preferably room temperature to the reflux temperature of the reaction solvent.
- the reaction time is usually 0.1 hour to 72 hours, preferably 0.5 hour to 24 hours.
- the above reaction can be carried out in the presence of a base and a condensation aid in order to facilitate the reaction.
- a base examples include 4-dimethylaminopyridine, triethylamine and the like.
- the amount of the base to be used is generally 1 to 100 equivalents, preferably 0.1 to 1 equivalent, per 1 mol of the carboxylic acid represented by compound (e) or a reactive derivative thereof.
- condensation aid examples include N-hydroxybenzotriazole hydrate, N-hydroxysuccinimide and the like.
- the amount of the condensation aid used is usually 1 to 100 equivalents, preferably 1 to 5 equivalents, per 1 mol of the carboxylic acid represented by the compound (e) or a reactive derivative thereof.
- This step is a method for producing a compound (g) by reacting the compound (f) obtained in the step C with fuming nitric acid.
- the amount of fuming nitric acid used in this step is usually 1 to 100 equivalents, preferably 2 to 20 equivalents, relative to 1 equivalent of the compound (f).
- the reaction temperature is usually 0 to 100 ° C., preferably 10 to 50 ° C.
- the reaction time is usually 0.1 to 48 hours, preferably 0.5 to 12 hours.
- the compound (g) can also be produced by reacting the compound (f) with potassium nitrate in the presence of an acid.
- the amount of potassium nitrate used is usually 1 to 100 equivalents, preferably 1 to 5 equivalents, relative to 1 equivalent of the compound (f). '
- Examples of the acid used include trifluoroacetic acid, hydrochloric acid, sulfuric acid, and nitric acid.
- the amount of the acid used is generally 1 equivalent to the amount of solvent, preferably 1 to 100 equivalents, relative to 1 equivalent of compound (f).
- the reaction temperature is usually from 0 ° C to the reflux temperature of the solvent, preferably from room temperature to 10 ° C.
- the reaction time is usually 0.1 to 72 hours, preferably 0.5 hours to 12 hours.
- the reaction solvent may be any as long as it does not interfere with the reaction, and examples thereof include chloroform and dichloromethane. '
- the compound (g) thus obtained can be isolated and purified by known separation and purification means such as concentration, concentration under reduced pressure, crystallization, solvent extraction, reprecipitation, chromatography, etc., or isolation and purification. It can be attached to the following.
- This step is a method for producing compound (i) by reacting compound (g) obtained in step D with compound (h) in the presence of a base.
- the amount of the compound (h) used is usually 1 to 20 equivalents, preferably 0.5 to 5 equivalents, relative to 1 equivalent of the compound (g).
- Examples of the compound (h) used include 4-methanesulfonylphenol, 4-ethanesulfonylphenol, 3-chloro-4 methanesulfonylphenol, 6-methanesulfonylpyridine, 1-ol, and 6-ethanesulfonyl-pyridine.
- 4 1 (5-Methyl-1, 2, 4-Oxadiazol-3-yl) Phenols.
- the amount of the base used is usually 0.1 to 20 equivalents, preferably 0.5 to 5 equivalents, relative to 1 equivalent of the compound (g).
- the reaction in this step in the reaction of the compound (g) with the compound (h) Any material can be used as long as it can produce the product (i), such as sodium hydride, cesium carbonate, sodium carbonate, potassium carbonate, potassium phosphate, potassium acetate, potassium tert-petitate, And triethylamine. Among these, potassium carbonate, cesium carbonate and the like are preferable.
- compound (h) is a primary or secondary amine
- the reaction in this step may be carried out without using a base.
- the reaction temperature is usually from 0 to the reflux temperature of the reaction solvent, preferably from room temperature to the reflux temperature of the reaction solvent.
- the reaction time is usually 1 to 72 hours, preferably 0.5 to 5 hours.
- the reaction solvent includes an inert solvent, and is not particularly limited as long as the reaction is not hindered. Specifically, for example, pyridine, toluene, tetrahydrofuran, 1,4-dioxane, N, N-dimethylformamide, N , N-dimethylacetamide, dimethyl sulfoxide, 1_methyl_2-pyrrolidinone and the like. .
- the compound (i) thus obtained is isolated and purified by known separation and purification means, for example, concentration, concentration under reduced pressure, crystallization, solvent-extraction, reprecipitation, chromatography, etc., or isolation and purification. It can be attached to the following without any problems.
- This step is a method for producing the compound (j) by reducing the nitro group of the compound (i) obtained in the step E and simultaneously dehydrocyclizing under an acid catalyst.
- reaction conditions in this step can be carried out by the same method as in step B above, a method analogous thereto, or a combination of these with conventional methods.
- the compound (j) thus obtained can be isolated and purified by known separation and purification means, for example, concentration, concentration under reduced pressure, crystallization, solvent extraction, reprecipitation, chromatography, etc., or isolation and purification. It can be attached to the following.
- This step is a method for producing compound (j-1) by reacting compound (j) obtained in step F with compound (k) in the presence of a base.
- the reaction in this step is a method described in the literature, which is a method for introducing a protecting group into an aromatic amino group (for example, Protective Groups in Organic Synthesis (Pr o tecti V e Groupsin Organic Synthesis;) TW G reen, 2nd edition, John Wiley & Sons, 1991, etc.), a method according to it, or a combination of these with conventional methods.
- a protecting group for example, Protective Groups in Organic Synthesis (Pr o tecti V e Groupsin Organic Synthesis;) TW G reen, 2nd edition, John Wiley & Sons, 1991, etc.
- Examples of L 2 in the compound (k) include a halogen atom and the like, and among these, a chlorine atom or a bromine atom is preferable.
- Examples of the compound (k) used include 2- (trimethylsilyl) ethoxymethyl chloride (SEMC 1), methoxymethyl chloride (MOMC 1) and the like.
- the amount of compound (k) to be used is generally 1 to 10 equivalents, preferably 1 to 3 equivalents, relative to 1 equivalent of compound (j).
- Examples of the base used include sodium hydride and the like.
- the amount of the base used is usually 1 to 10 equivalents, preferably 1 to 3 equivalents.
- the reaction temperature is usually from 1 to 50 ° C, preferably from 0 ° C to room temperature.
- the reaction time is usually from 0.1 to 12 hours, preferably from 0.1 to 3 hours.
- the reaction solvent may be any as long as it does not interfere with the reaction, and examples thereof include N, N-dimethylformamide, tetrahydrofuran, and methylene chloride. '
- the compound (j-1) thus obtained can be isolated and purified by known separation and purification means, for example, concentration, concentration under reduced pressure, crystallization, solvent extraction, reprecipitation, kumatography, etc. It can be used in the next step without isolation and purification.
- This step is a method for producing the compound (8-1) by reducing the ester group of the compound (j-1) obtained in the step G.
- Examples of the reducing agent used in this step include lithium aluminum hydride (LiAlH 4 ), lithium borohydride, sodium borohydride and the like.
- the ester body of the compound (j-1) is hydrolyzed to give a carboxylic acid, and then the method described in the literature (for example, SYNLET T, 1995, Vol. 8, pages 839-840, etc. ), A method according to this, or a combination of these with a conventional method, compound (8-1) can be produced. '
- the amount of the reducing agent used is usually 1 to 20 equivalents, preferably 1 to 3 equivalents, relative to 1 equivalent of the compound (j 1).
- the reaction temperature is generally 0 to 80 ° C, preferably 0 ° C to room temperature.
- the reaction time is usually 1 to 24 hours, preferably 0.1 to 3 hours.
- the reaction solvent to be used is not particularly limited as long as the reaction is not hindered.
- methanol, N, N-dimethylformamide, ethyl acetate, tetrahydrofuran and the like and a mixed solvent thereof can be used.
- the compound (8-1) thus obtained can be isolated and purified by known separation and purification means, for example, concentration, concentration under reduced pressure, crystallization, solvent extraction, reprecipitation, chromatography, etc. It can be used in the next step without purification.
- This step is a method for producing the compound (1-1) by oxidizing the hydroxy group of the compound (8-1) obtained in the step H.
- reaction in this step is a method described in the literature (for example, Journal of the American Chemical Society, 1967, 89, 5505-5507). Or a combination of these and conventional methods.
- the compound (1-1) thus obtained is isolated and purified by a known separation and purification means, for example, concentration, concentration under reduced pressure, crystallization, solvent extraction, reprecipitation, chromatography, etc. It can be used in the next step without purification.
- a known separation and purification means for example, concentration, concentration under reduced pressure, crystallization, solvent extraction, reprecipitation, chromatography, etc. It can be used in the next step without purification.
- This step is a protecting group Rpr possessed by the compound (1-1) obtained in the above step I.
- This is a method for producing compound (1) by removing.
- the removal of the protecting group can be carried out by a method described in the literature (for example, Protective Group Organic Synthesis, TW G reen, 2nd edition, John W i 1 ey & Sons, 1 9 9 1, etc.), a method based on it, or a combination of these and conventional methods, for example
- the protecting group is a SEM (trimethylsilylethoxymethyl) group
- the SEM group can be removed by reacting the compound (8-1) with triuro acetic acid.
- the compound (1) thus obtained can be isolated and purified by known separation and purification means, for example, concentration, concentration under reduced pressure, crystallization, solvent extraction, reprecipitation, chromatography and the like.
- This step is a method for producing the compound (8) by removing the protecting group R PI “° of the compound (8-1).
- the reaction in this step can be performed by a method similar to the method for removing the protecting group R p of the compound (1-1), a method analogous thereto, or a combination thereof with a conventional method.
- the compound (8) thus obtained can be isolated or purified by known separation and purification means, for example, concentration, concentration under reduced pressure, crystallization, solvent extraction, reprecipitation, chromatography, etc. It can be attached to the next process.
- the protecting group Rp TM possessed by the compound (1-1) is necessary. Can be removed accordingly.
- the protecting group Rpr possessed by the compound (8-1) can be removed if necessary.
- the amino group or imino group is appropriately reacted after protecting with an amino group or imino group protecting group, and the protecting group is removed after the reaction. It is preferable to remove.
- the heterocyclic-substituted benzimidazole derivative provided by the present invention can exist as a pharmaceutically acceptable salt, which is included in the compound (I) according to the present invention.
- the compounds of the above formulas (1-1) to (I 1 1 2) have a basic group derived from, for example, an amino group, a pyridyl group or the like in the molecule,
- the compound can be converted to the corresponding pharmaceutically acceptable salt by treating with an acid.
- the acid addition salt examples include hydrohalides such as hydrochloride, hydrofluoride, hydrobromide, hydroiodide; nitrate, perchlorate, sulfate, phosphate, Inorganic acid salts such as carbonates; lower alkyl sulfonates such as methane sulfonate, trifluoromethane sulfonate, and ethane sulfonate; aryl sulfonic acids such as benzene sulfonate and p-toluene sulfonate Salts: Organic acid salts such as fumarate, succinate, citrate, tartrate, oxalate and maleate; and acid addition salts which are organic acids such as amino acids such as glutamate and aspartate Can be mentioned.
- hydrohalides such as hydrochloride, hydrofluoride, hydrobromide, hydroiodide
- nitrate, perchlorate, sulfate, phosphate Inorganic
- the compound of the present invention when it has an acidic group in the group, for example, when it has a strong lpoxyl group, etc., it is also applicable by treating the compound with a base. It can be converted to a pharmaceutically acceptable salt.
- the base addition salt and Examples thereof include alkali metal salts such as sodium and potassium, alkaline earth metal salts such as calcium and magnesium, salts with organic salt groups such as ammonium salt, guanidine, triethylamine, and dicyclohexylamine.
- the compounds of the present invention may exist as any free solvate or solvate of the free compound or salt thereof.
- the compound of formula (I) according to the present invention comprises a compound of formula (I) and a carrier substance. Can be used in combination.
- the dosage for prophylaxis or treatment of a compound of formula (I) according to the present invention will, of course, vary depending on the nature of the condition being treated, the particular compound selected and the route of administration.
- the daily dosage is from about 0.001 mg / kg to about 10 Omg / kg body weight, preferably about 0.0 / kg body weight, in single or multiple doses. lmg to about 5 Omg, more preferably about 0.1 lmg strength, et al. 1 Omg. It may be necessary to use dosages that exceed these limits.
- An example of a suitable oral dose is at least about 0.0 lmg to at most 2.0 g for a single or 2-4 doses per day.
- the dosage range is from about 1. Omg to about 20 Omg, once or twice daily. More preferably, the dosage range is about 1 Omg to 10 Omg once a day.
- a typical dosage range is from about 0.001 mg to about 10 Omg (preferably 0. About 1 Omg), more preferably about 0.1 mg to 10 mg of compound of formula (I) per kg body weight per day.
- composition comprises a compound of formula (I) and a pharmaceutically acceptable carrier.
- composition is the result of combining two or more components, combined or aggregated, directly or indirectly, resulting from the dissociation of one or more components, or It includes not only the results of other types of action or interaction between components, but also the active and inactive components (pharmaceutically acceptable excipients) that make up the carrier.
- compositions comprising a compound strength S of the formula (I) in an amount effective in combination with a pharmaceutically acceptable carrier for effective treatment, prevention or delaying the onset of type 2 diabetes are preferred.
- Any suitable route of administration may be employed for administering an effective amount of a compound of the present invention to mammals, particularly humans.
- mammals particularly humans.
- oral, rectal, topical, intravenous, eye, lung, and nose can be used.
- the dosage form include tablets, troches, powders, suspensions, solutions, capsules, creams, aerosols, etc. Oral tablets are preferred.
- any ordinary pharmaceutical medium can be used. Examples thereof include water, glycol, oil, alcohol, flavoring agents, storage, and the like.
- a liquid composition for oral use for example, suspensions, elixirs and solutions can be mentioned, and carriers include, for example, starch, sugar, microcrystalline cellulose. , Diluents, granulating agents, lubricants, binders, disintegrants, etc., and when preparing oral solid compositions, for example, powders, capsules, tablets, etc. Oral solid compositions are preferred.
- Tablets and capsules are the most advantageous oral dosage forms because of their low dosage. If necessary, the tablets Coating can be done with standard aqueous or non-aqueous techniques.
- compounds according to 3 ⁇ 4 (I) are, for example, US Patent Nos. 3, 845, 770, 3, 916, 899, 3, 536, 809, 3, 598, 123, 3, Administration is also possible by means of controlled release and Z or delivery devices as described in 630, 200 and 4,008,719.
- compositions according to the present invention suitable for oral administration is preliminarily determined as powder or granule, or as water-soluble liquid, water-insoluble liquid, oil-in-water emulsion or water-in-oil emulsion. Mention may be made of a force-pelling agent, a force-chipping agent or a tablet containing a certain amount of active ingredient.
- Such compositions can be prepared using any pharmacological method, but all methods also include a method of combining the active ingredient with a carrier comprising one or more necessary ingredients. .
- a composition is prepared by uniformly and thoroughly mixing the active ingredient with a liquid carrier or a well-separated solid carrier or both and then, if necessary, shaping the product into a suitable form.
- a tablet is prepared by compression and molding, optionally with one or more accessory ingredients. Compressed tablets are mixed with binders, lubricants, inert excipients, surfactants or dispersants as appropriate, using suitable machines, and the active ingredients are formed into powders and granules. Prepared by free compression.
- Molded tablets are made by molding in a suitable machine a mixture of the powdered wet compound and an inert liquid diluent.
- each tablet contains about 1 mg to 1 g of active ingredient and each cachet or capsule contains about 1 mg to 50 Omg of active ingredient.
- the compounds of formula (I) can be used in combination with other drugs used in the treatment / prevention / delay of the onset of type 2 diabetes as well as diseases or symptoms associated with type 2 diabetes.
- the other drug can be administered simultaneously or separately with the compound of formula (I) by a commonly used route or dose.
- the pharmaceutical composition according to the invention also contains one or more other active ingredients in addition to the compound of formula (I).
- active ingredients used in combination with a compound of formula (I) may be administered separately or in the same pharmaceutical composition, but are not limited to the following.
- PPAR agonist eg, troglitazone, pioglitazone, nosiglitazone
- ⁇ -Dalcosidease inhibitor eg, poglipose, miglitol, calcalpose
- Insulin secretagogues e.g., acetohexamide, carptamide, chlorpropamide, darivomulide, gliclazide, dalimelpyride, glipizide, glyxidine, glyoxepide, glyburide, glyhexamide, glipinamide, fenbutamide, tolamide, tolamide, tolamide, tolamide, tolamide, tolamide, tolamide Nate glinide, repaglinide), and
- Insulin secretagogues e.g., acetohexamide, carptamide, chlorpropamide, darivomulide, gliclazide, dalimelpyride, glipizide, glyxidine, glyoxepide, glyburide, glyhexamide, glipinamide, fenbutamide, tolamide, tolamide, tolamide, tolamide, tolamide, tolamide, tolamide
- the weight ratio of the compound of formula (I) to the second active ingredient varies within wide limits and, in addition, depends on the effective amount of each active ingredient.
- the weight ratio of compound of formula (I) to P PAR agonist is generally about 1 000: 1 to 1 : 1000, preferably about 200: 1 to 1: 200.
- Combinations of the compound with other active ingredients are within the aforementioned ranges, but in each case, an effective amount of each active ingredient should be used.
- the excellent darcokinase activating action of the compound represented by the formula (I) can be measured by a method described in the literature (for example, Diabetes, Vol. 45, pp. 1671-1677). Page, 1996, etc.) or a similar method.
- Dalcokinase activity occurs when the reporter enzyme glucose-6-phosphate dehydrogenase produces phosphodarconolactone from glucose-6-phosphate rather than directly measuring glucose mono6-phosphate.
- Th io Determine the degree of activation of darcokinase by measuring the amount of NADH. .
- the r e comb i n nt human l ve r GK used in this assay was expressed as FLAG f u s i on p ro t e i in E. c o 1 i and purified by ANT I FLAG M2 AFF I N I TY GEL (S i gma).
- Atsey was performed using a flat bottom 96-we 1 1 p 1 ate at 30 ° C.
- 1 n 1 of DMSQ was added.
- Enzyme miture FLAG—GK, 20 U / m 1 G6 PDH
- the OD value in DMSO control was taken as 100%, and the ⁇ D value at each concentration of the evaluation compound was measured. From the OD value at each concentration, Emax (%) and g C 50 ⁇ ) were calculated and used as an index of the GK activation ability of the compound.
- the compound according to the present invention has an excellent ability to activate GK using Em ax and EC 50 as indices.
- a granule is prepared in the same manner as in Formulation Example 2, and then 3 parts of calcium stearate is added to 96 parts of this granule and compression-molded to prepare a tablet having a diameter of 10 mm. .
- Si 1 icage 160F 2 45 (Me rck) was used as a plate, and a UV detector was used as a detection method.
- Wakogel TM C-300 (Wako Pure Chemicals) is used as the silica gel for the column, and LC-SORB TM SP- B- ⁇ as the silica gel for the reverse phase column.
- n-P r n-propyl group
- the title compound was obtained as a colorless solid by using 2- (hydroxymethyl) -1,4-butanediol in the same manner as in Example 1, a method analogous thereto, or a combination of these and conventional methods. It was.
- the title compound was obtained as a colorless solid by using N-acetylethanolamine in the same manner as in Example 1, a method analogous thereto, or a combination thereof with a conventional method.
- Step 1 (4- (Ethylsulfonyl) phenoxy) —2—Pyridine 1 — 2-yl — 1 H-Benzimidazo 1-Lu 5 _yl) Butane 1 1, 4 obtained in (Step 1) —To a solution of 8 lmg of diol in 4 ml of black mouth form, p-toluenesulfonic acid monohydrate 2 Omg was added, and the reaction solution was stirred overnight with heating under reflux.
- reaction mixture was purified by reversed-phase medium pressure liquid chromatography (ODS—AS-360 I CC (manufactured by YMC) mobile phase: water-acetonitrile—0.1% trifluoroacetic acid) to obtain The obtained fraction was diluted with ethyl acetate, washed successively with saturated aqueous sodium hydrogen carbonate and saturated brine, and dried over anhydrous sodium sulfate. The solvent was distilled off under reduced pressure to obtain the title compound as a white solid.
- ODS reversed-phase medium pressure liquid chromatography
- Example 7 5- (tetrahydrofuran_2_yl) -6- (4- (ethylsulfonyl) phenoxy) 1 2-pyridine-2-yl-1-H-benzimidazole 36 mg obtained in Example 7 was optically analyzed.
- Optical resolution with a column for separation (CHI RALPAK AD-H 2 c X 2 5 c mL (manufactured by Daicel Chemical Industries, Ltd.), mobile phase: hexane noenol 11, flow rate: 1 Oml / min), Enantiomer A (retention time: 14.4 min) and Enantiomer B (retention time: 16.3 min) were obtained as white solids, respectively.
- Example 9 Using 5- (6— (4- (ethylsulfonyl) phenoxy) 1 2-pyrazine 1 2-yl lu 1H-benzimidazole 5-yl) tetrahydrofuran-2-ol obtained in Example 9 Then, the title compound was obtained as a colorless solid by the same method as in Example 7, a method analogous thereto, or a combination thereof with a conventional method.
- Example 12 5- (6 — ((6- (Ethylsulfonyl) pyridine-3-yl) oxy) 1 2-pyridine-1 2-yl 1H-benzimidazole 1-yl) tetrahydrofuran obtained in Example 12
- the title compound was obtained by using 1-2-allenantiomer A in the same manner as in Example 7 (Step 1), a method analogous thereto, or a combination thereof with a conventional method.
- reaction solution was diluted with ethyl acetate, washed successively with saturated aqueous sodium hydrogen carbonate and saturated brine, and dried over anhydrous magnesium sulfate.
- the solvent was distilled off under reduced pressure, and 1,8-diazabicyclo [5.4.0] undeca-7-ene (0.045 ml) was added to a 2 ml 1-mL solution of the residue obtained. Stir for hours.
- Example 6 Using 5-carbaldehyde 6- ((6- (methylsulfonyl) pyridine-3-yl) oxy) —2—pyridine-2-ylru 1H-benzimidazole obtained in Reference Example 13 The title compound was obtained as a colorless solid by the same method as in Example 7, a method analogous thereto, or a combination of these and a conventional method.
- N- (3-Fluoro-41-((Methoxy (methyl) amino) -force lonyl) phenyl) Pyridine-1-2 Synthesis of lpoxamide 10 g of pyridine of 4-nitro-2-fluorobenzoic acid obtained by the method described in Biogarganic & Medicinal Chemistry Letters, 15 (2), 337-343; 200 '5
- To the 80 ml solution add 5.3 g of N, O-dimethylhydroxylamine 'monohydrochloride and 12 g of 1- (3-dimethylaminopropyl) —3-ethylcarbapimide / monohydrochloride The solution was stirred at room temperature overnight.
- the solvent was distilled off under reduced pressure, water was added to the obtained residue, and the deposited precipitate was collected by filtration to obtain a crude product.
- the solvent was distilled off under reduced pressure.
- Step 1 N— (3-Fluoro-4-penter 4-enoylphenyl) N- (3-Fluoro-4-(((Methyl (methyl) amino) carbonyl) phosphine) obtained in the synthesis of pyridine 1-strength lupoxamide (Step 1)
- the title compound was obtained by using the same method as in Example 6 (Step 1), a method analogous thereto, or a combination thereof with a conventional method, using benzyl) pyridine-2-carboxamide.
- reaction solution was diluted with ethyl acetate, washed successively with saturated aqueous sodium hydrogen carbonate and saturated brine, and dried over anhydrous magnesium sulfate.
- the solvent was distilled off under reduced pressure to obtain the title compound.
- the solvent was distilled off under reduced pressure, and the resulting residue was purified by reversed-phase medium pressure liquid chromatography (ODS—AS-36 O-CC (manufactured by YMC) mobile phase: water-acetonitrile—0.1% trifluoroacetic acid).
- ODS reversed-phase medium pressure liquid chromatography
- the obtained fraction was diluted with ethyl acetate, washed successively with saturated aqueous sodium hydrogen carbonate and saturated brine, and dried over anhydrous sodium sulfate.
- the solvent was distilled off under reduced pressure to obtain the title compound as a white solid.
- Example 20 Using 4-ethylsulfonylphenol, the title compound was obtained as a yellow foamy substance by the same method as in Example 20 (Step 6), a method analogous thereto or a combination thereof with a conventional method.
- the purified fraction was diluted with ethyl acetate, washed successively with saturated aqueous sodium hydrogen carbonate and saturated brine, and dried over anhydrous sodium sulfate. The solvent was removed in vacuo to give the title compound as a yellow foam.
- 6-(6— (5-Methyl- (1, 2, 4) oxaziazol luo 3-yl) monopyridin-3-ol was used in the same manner as in Example 20 (Step 6).
- the title compound was obtained as a yellow foam by combining these methods or these and conventional methods.
- Example 7 1- (6— (4- (Ethylsulfonyl) phenoxy) —2—pyridin—2-yl— 1H-benzimidazo 5-L) butane
- 0.052 ml of triethylamine and 0.032 ml of methanesulfonyl chloride were sequentially added, and the reaction solution was stirred at room temperature for 5 minutes.
- the reaction solution was diluted with ethyl acetate, washed successively with saturated aqueous sodium hydrogen carbonate and saturated brine, and dried over anhydrous sodium sulfate.
- the solvent was distilled off under reduced pressure, and 7 Omg of potassium O-ethyldithioate was added to a 3 ml solution of acetone obtained, and the reaction solution was stirred at 50 ° C. for 15 minutes.
- the reaction mixture was diluted with ethyl acetate, washed successively with water and saturated brine, and dried over anhydrous sodium sulfate.
- 0.03 ml of 25% sodium methoxide solution was added, and the reaction solution was stirred at room temperature for 20 minutes, and then 15 mg of p-toluenesulfonic acid monohydrate was added, The solvent was removed under reduced pressure.
- the obtained residue was dissolved in 1 ml of toluene, and the reaction solution was stirred at 70 ° C. overnight. After the solvent was distilled off under reduced pressure, the reaction mixture was purified by reverse-phase medium pressure liquid chromatography (ODS—AS—360-CC (manufactured by YMC) mobile phase: water-acetonitrile 0.1% trifluoroacetic acid). The obtained fractions were each diluted with ethyl acetate, washed successively with saturated aqueous sodium hydrogen carbonate and saturated brine, and dried over anhydrous sodium sulfate.
- ODS—AS—360-CC reverse-phase medium pressure liquid chromatography
- Example 29-1 Of the compounds obtained in Example 29 above, 5- (tetrahydro-2-enyl) -6- (4- (ethylsulfonyl) phenoxy) 1 2-pyridine-2-yloo 1.H-benzimidazole
- the method of manufacturing is referred to as Example 29-1. .
- the reaction mixture was purified by reversed-phase medium-pressure liquid chromatography (ODS—AS-360-CC (manufactured by YMC) mobile phase: water-acetonitrile—0.1% trifluoroacetic acid), and the resulting fractions were each acetic acid.
- ODS reversed-phase medium-pressure liquid chromatography
- the mixture was diluted with ethyl acetate, washed successively with saturated aqueous sodium hydrogen carbonate and saturated Japanese brine, and dried over anhydrous sodium sulfate.
- reaction solution was stirred at 70 ° C. for 5 minutes.
- the reaction mixture was purified by reversed-phase medium-pressure liquid chromatography [ODS-AS-360-CC (manufactured by YMC) mobile phase: water-acetonitrile-0.1% trifluoroacetic acid], and the resulting fraction was converted into ethyl acetate.
- the mixture was diluted with saturated aqueous sodium hydrogen carbonate and saturated brine, and dried over anhydrous sodium sulfate. The solvent was distilled off under reduced pressure to obtain the title compound as a yellow solid.
Abstract
Description





























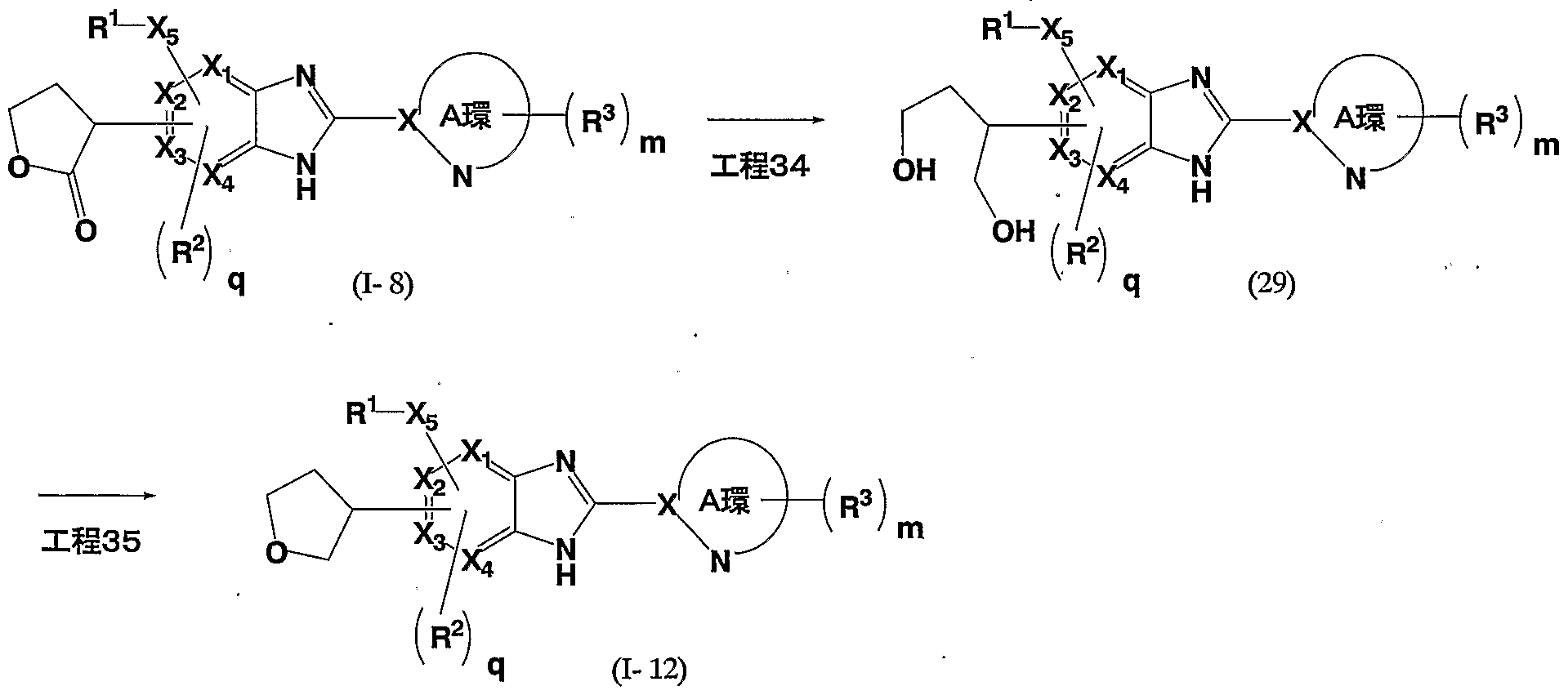






Claims


Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2006267338A AU2006267338B2 (en) | 2005-07-13 | 2006-07-12 | Heterocycle-substituted benzimidazole derivative |
| CA2614544A CA2614544C (en) | 2005-07-13 | 2006-07-12 | Heterocycle-substituted benzimidazole derivative |
| JP2007524727A JP5281287B2 (ja) | 2005-07-13 | 2006-07-12 | ヘテロ環置換ベンズイミダゾール誘導体 |
| US11/988,592 US7994331B2 (en) | 2005-07-13 | 2006-07-12 | Heterocycle-substituted benzimidazole derivative |
| EP06781274.3A EP1905769B1 (en) | 2005-07-13 | 2006-07-12 | Heterocycle-substituted benzimidazole derivative |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2005-204438 | 2005-07-13 | ||
| JP2005204438 | 2005-07-13 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2007007910A1 true WO2007007910A1 (ja) | 2007-01-18 |
Family
ID=37637273
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2006/314307 WO2007007910A1 (ja) | 2005-07-13 | 2006-07-12 | ヘテロ環置換ベンズイミダゾール誘導体 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US7994331B2 (ja) |
| EP (1) | EP1905769B1 (ja) |
| JP (1) | JP5281287B2 (ja) |
| AU (1) | AU2006267338B2 (ja) |
| CA (1) | CA2614544C (ja) |
| WO (1) | WO2007007910A1 (ja) |
Cited By (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007128761A2 (de) | 2006-05-04 | 2007-11-15 | Boehringer Ingelheim International Gmbh | Verwendungen von dpp iv inhibitoren |
| WO2008017381A1 (de) | 2006-08-08 | 2008-02-14 | Sanofi-Aventis | Arylaminoaryl-alkyl-substituierte imidazolidin-2,4-dione, verfahren zu ihrer herstellung, diese verbindungen enthaltende arzneimittel und ihre verwendung |
| WO2008156174A1 (ja) | 2007-06-21 | 2008-12-24 | Taisho Pharmaceutical Co., Ltd. | ピラジンアミド化合物 |
| WO2009021740A2 (de) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituierte tetrahydronaphthaline, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2009082152A2 (en) | 2007-12-20 | 2009-07-02 | Lg Life Sciences Ltd. | Glucokinase activators and pharmaceutical compositions containing the same as an active ingredient |
| WO2010003624A2 (en) | 2008-07-09 | 2010-01-14 | Sanofi-Aventis | Heterocyclic compounds, processes for their preparation, medicaments comprising these compounds, and the use thereof |
| WO2010024110A1 (en) * | 2008-08-29 | 2010-03-04 | Banyu Pharmaceutical Co.,Ltd. | Oxotetrahydrofuran-2-yl-benzimidazole derivative |
| WO2010068601A1 (en) | 2008-12-08 | 2010-06-17 | Sanofi-Aventis | A crystalline heteroaromatic fluoroglycoside hydrate, processes for making, methods of use and pharmaceutical compositions thereof |
| WO2011023754A1 (en) | 2009-08-26 | 2011-03-03 | Sanofi-Aventis | Novel crystalline heteroaromatic fluoroglycoside hydrates, pharmaceuticals comprising these compounds and their use |
| WO2011107494A1 (de) | 2010-03-03 | 2011-09-09 | Sanofi | Neue aromatische glykosidderivate, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2011157827A1 (de) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-on-derivate als inhibitoren von lipasen und phospholipasen |
| WO2011161030A1 (de) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclisch substituierte methoxyphenylderivate mit oxogruppe, verfahren zu ihrer herstellung und ihre verwendung als gpr40 rezeptor modulatoren |
| WO2012004269A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | ( 2 -aryloxy -acetylamino) - phenyl - propionsäurederivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012004270A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclisch substituierte 1,3-propandioxidderivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012010413A1 (de) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylen-substituierte hydroxy-phenyl-hexinsäuren, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012120052A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Mit carbozyklen oder heterozyklen substituierte oxathiazinderivate, verfahren zu deren herstellung, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2012120054A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Di- und trisubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120055A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Di- und trisubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120053A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Verzweigte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120056A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| CN101607923B (zh) * | 2009-07-21 | 2012-12-19 | 焦宁 | 芳基腈类化合物或其衍生物及其合成方法和应用 |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| US8822503B2 (en) | 2009-12-04 | 2014-09-02 | Taisho Pharmaceutical Co., Ltd | 2-pyridone compounds |
| JP2018535263A (ja) * | 2015-11-17 | 2018-11-29 | ダウ アグロサイエンシィズ エルエルシー | 4−((6−(2−(2,4−ジフルオロフェニル)−1,1−ジフルオロ−2−ヒドロキシ−3−(1h−1,2,4−トリアゾール−1−イル)プロピル)ピリジン−3−イル)オキシ)ベンゾニトリル及び調製方法 |
| JP2018535262A (ja) * | 2015-11-17 | 2018-11-29 | ダウ アグロサイエンシィズ エルエルシー | 4−((6−(2−(2,4−ジフルオロフェニル)−1,1−ジフルオロ−2−オキソエチル)ピリジン−3−イル)オキシ)ベンゾニトリル及び製造方法 |
| JP2018538366A (ja) * | 2015-11-17 | 2018-12-27 | ダウ アグロサイエンシィズ エルエルシー | 4−((6−(2−(2,4−ジフルオロフェニル)−1,1−ジフルオロ−2−ヒドロキシ−3−(1h−1,2,4−トリアゾール−1−イル)プロピル)ピリジン−3−イル)オキシ)ベンゾニトリル及び調製方法 |
| US11878968B2 (en) | 2021-07-09 | 2024-01-23 | Plexium, Inc. | Aryl compounds and pharmaceutical compositions that modulate IKZF2 |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP7339881B2 (ja) | 2019-12-26 | 2023-09-06 | 日置電機株式会社 | 部分放電検出装置および部分放電検出方法 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004067629A (ja) * | 2002-08-09 | 2004-03-04 | Yamanouchi Pharmaceut Co Ltd | ミトコンドリア機能活性化剤及び新規なベンゾイミダゾール誘導体 |
| JP2004517087A (ja) * | 2000-12-06 | 2004-06-10 | エフ.ホフマン−ラ ロシュ アーゲー | 縮合複素環式芳香族グルコキナーゼアクチベーター |
| WO2005063738A1 (ja) * | 2003-12-29 | 2005-07-14 | Banyu Pharmaceutical Co.,Ltd | 新規2-へテロアリール置換ベンズイミダゾール誘導体 |
Family Cites Families (30)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6187778B1 (en) * | 1997-08-05 | 2001-02-13 | Pfizer Inc. | 4-aminopyrrole (3, 2-D) pyrimidines as neuropeptide Y receptor antagonists |
| GB9718712D0 (en) * | 1997-09-03 | 1997-11-12 | Merck Sharp & Dohme | Theraputic Agents |
| GB9820113D0 (en) * | 1998-09-15 | 1998-11-11 | Merck Sharp & Dohme | Therapeutic agents |
| JP3869568B2 (ja) * | 1998-11-30 | 2007-01-17 | 本田技研工業株式会社 | 燃料電池用電極 |
| GB9905010D0 (en) * | 1999-03-04 | 1999-04-28 | Merck Sharp & Dohme | Therapeutic agents |
| DE19934432A1 (de) * | 1999-07-22 | 2001-02-01 | Merck Patent Gmbh | Indolderivate |
| JP3712393B2 (ja) | 2000-10-20 | 2005-11-02 | エーザイ株式会社 | 含窒素芳香環誘導体 |
| UA76130C2 (en) | 2000-11-20 | 2006-07-17 | Merck Patent Gmbh | Use of compounds combining properties of selective inhibitors of serotonin re-uptake and agonists of 5-ht1a receptor for treatment of irritable bowel syndrome |
| US6433188B1 (en) * | 2000-12-06 | 2002-08-13 | Wendy Lea Corbett | Fused heteroaromatic glucokinase activators |
| US7064215B2 (en) * | 2001-07-03 | 2006-06-20 | Chiron Corporation | Indazole benzimidazole compounds |
| WO2003080585A1 (fr) * | 2002-03-26 | 2003-10-02 | Banyu Pharmaceutical Co., Ltd. | Derive aminobenzamide |
| DE10217006A1 (de) | 2002-04-16 | 2003-11-06 | Merck Patent Gmbh | Substituierte Indole |
| JP4335136B2 (ja) | 2002-08-09 | 2009-09-30 | メルク エンド カムパニー インコーポレーテッド | チロシンキナーゼ阻害薬 |
| JP4520309B2 (ja) | 2002-09-20 | 2010-08-04 | メルク・シャープ・エンド・ドーム・コーポレイション | 選択的糖質コルチコイド受容体調節剤としてのオクタヒドロ−2−H−ナフト[1,2−f]インドール−4−カルボキサミド誘導体 |
| DE10254596A1 (de) * | 2002-11-22 | 2004-06-03 | Merck Patent Gmbh | Indolderivate |
| US7563815B2 (en) * | 2003-05-05 | 2009-07-21 | Merck & Co., Inc. | Benzamidazoles, compositions containing such compounds and methods of use |
| AU2004238240A1 (en) | 2003-05-09 | 2004-11-25 | Merck & Co., Inc. | Benzimidazoles, compositions containing such compounds and methods of use |
| DE10326940A1 (de) * | 2003-06-16 | 2005-01-05 | Merck Patent Gmbh | Indol-Derivate |
| DE10326939A1 (de) | 2003-06-16 | 2005-01-05 | Merck Patent Gmbh | Indol-Derivate |
| JP2007513128A (ja) | 2003-12-03 | 2007-05-24 | メルク エンド カムパニー インコーポレーテッド | アルツハイマー病および関連状態の治療に有用な1−アルキル−3−チオ置換インドール−2−アルキン酸 |
| EP1699453A4 (en) | 2003-12-19 | 2009-07-01 | Merck & Co Inc | CYCLIC GUANIDINES, COMPOSITIONS AND METHOD OF ADMINISTRATION CONTAINING SUCH COMPOUNDS |
| US7625933B2 (en) | 2004-07-02 | 2009-12-01 | Merck & Co., Inc. | Indoles having anti-diabetic activity |
| CN101094847B (zh) * | 2004-11-02 | 2011-06-15 | Msdk.K.公司 | 芳氧基取代的苯并咪唑衍生物 |
| FR2880889B1 (fr) | 2005-01-14 | 2007-04-06 | Merck Sante Soc Par Actions Si | Derives de l'acide 1h-indole-3-carboxylique, procedes pour leur preparation, compositions pharmaceutiques les contenant et applications en therapeutique |
| JP5094394B2 (ja) * | 2005-04-20 | 2012-12-12 | 武田薬品工業株式会社 | 縮合複素環化合物 |
| US20110059941A1 (en) * | 2005-05-24 | 2011-03-10 | Peter William Rodney Caulkett | 2-phenyl substituted imidazol [4,5b] pyridine/pyrazine and purine derivatives as glucokinase modulators |
| JP2009504652A (ja) | 2005-08-11 | 2009-02-05 | メルク エンド カムパニー インコーポレーテッド | 非ヌクレオシド逆転写酵素阻害剤 |
| EP1915372B1 (en) | 2005-08-12 | 2013-11-20 | Merck Canada Inc. | Indole derivatives as crth2 receptor antagonists |
| WO2007061763A2 (en) | 2005-11-22 | 2007-05-31 | Merck & Co., Inc. | Indole orexin receptor antagonists |
| US7968578B2 (en) | 2006-04-24 | 2011-06-28 | Merck Frosst Canada Ltd. | Indole amide derivatives as EP4 receptor antagonists |
-
2006
- 2006-07-12 JP JP2007524727A patent/JP5281287B2/ja not_active Expired - Fee Related
- 2006-07-12 WO PCT/JP2006/314307 patent/WO2007007910A1/ja active Application Filing
- 2006-07-12 AU AU2006267338A patent/AU2006267338B2/en not_active Ceased
- 2006-07-12 US US11/988,592 patent/US7994331B2/en active Active
- 2006-07-12 CA CA2614544A patent/CA2614544C/en not_active Expired - Fee Related
- 2006-07-12 EP EP06781274.3A patent/EP1905769B1/en active Active
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004517087A (ja) * | 2000-12-06 | 2004-06-10 | エフ.ホフマン−ラ ロシュ アーゲー | 縮合複素環式芳香族グルコキナーゼアクチベーター |
| JP2004067629A (ja) * | 2002-08-09 | 2004-03-04 | Yamanouchi Pharmaceut Co Ltd | ミトコンドリア機能活性化剤及び新規なベンゾイミダゾール誘導体 |
| WO2005063738A1 (ja) * | 2003-12-29 | 2005-07-14 | Banyu Pharmaceutical Co.,Ltd | 新規2-へテロアリール置換ベンズイミダゾール誘導体 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP1905769A4 * |
Cited By (31)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007128761A2 (de) | 2006-05-04 | 2007-11-15 | Boehringer Ingelheim International Gmbh | Verwendungen von dpp iv inhibitoren |
| EP2351568A2 (de) | 2006-05-04 | 2011-08-03 | Boehringer Ingelheim International GmbH | Verwendungen von dpp iv Inhibitoren |
| WO2008017381A1 (de) | 2006-08-08 | 2008-02-14 | Sanofi-Aventis | Arylaminoaryl-alkyl-substituierte imidazolidin-2,4-dione, verfahren zu ihrer herstellung, diese verbindungen enthaltende arzneimittel und ihre verwendung |
| WO2008156174A1 (ja) | 2007-06-21 | 2008-12-24 | Taisho Pharmaceutical Co., Ltd. | ピラジンアミド化合物 |
| WO2009021740A2 (de) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituierte tetrahydronaphthaline, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| US8309586B2 (en) | 2007-12-20 | 2012-11-13 | Lg Life Sciences Ltd. | Glucokinase activators and pharmaceutical compositions containing the same as an active ingredient |
| WO2009082152A2 (en) | 2007-12-20 | 2009-07-02 | Lg Life Sciences Ltd. | Glucokinase activators and pharmaceutical compositions containing the same as an active ingredient |
| WO2010003624A2 (en) | 2008-07-09 | 2010-01-14 | Sanofi-Aventis | Heterocyclic compounds, processes for their preparation, medicaments comprising these compounds, and the use thereof |
| WO2010024110A1 (en) * | 2008-08-29 | 2010-03-04 | Banyu Pharmaceutical Co.,Ltd. | Oxotetrahydrofuran-2-yl-benzimidazole derivative |
| US8609699B2 (en) | 2008-08-29 | 2013-12-17 | Msd K.K. | Oxotetrahydrofuran-2-yl-benzimidazole derivative |
| WO2010068601A1 (en) | 2008-12-08 | 2010-06-17 | Sanofi-Aventis | A crystalline heteroaromatic fluoroglycoside hydrate, processes for making, methods of use and pharmaceutical compositions thereof |
| CN101607923B (zh) * | 2009-07-21 | 2012-12-19 | 焦宁 | 芳基腈类化合物或其衍生物及其合成方法和应用 |
| WO2011023754A1 (en) | 2009-08-26 | 2011-03-03 | Sanofi-Aventis | Novel crystalline heteroaromatic fluoroglycoside hydrates, pharmaceuticals comprising these compounds and their use |
| US8822503B2 (en) | 2009-12-04 | 2014-09-02 | Taisho Pharmaceutical Co., Ltd | 2-pyridone compounds |
| WO2011107494A1 (de) | 2010-03-03 | 2011-09-09 | Sanofi | Neue aromatische glykosidderivate, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2011157827A1 (de) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-on-derivate als inhibitoren von lipasen und phospholipasen |
| WO2011161030A1 (de) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclisch substituierte methoxyphenylderivate mit oxogruppe, verfahren zu ihrer herstellung und ihre verwendung als gpr40 rezeptor modulatoren |
| WO2012004269A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | ( 2 -aryloxy -acetylamino) - phenyl - propionsäurederivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012004270A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclisch substituierte 1,3-propandioxidderivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012010413A1 (de) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylen-substituierte hydroxy-phenyl-hexinsäuren, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012120052A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Mit carbozyklen oder heterozyklen substituierte oxathiazinderivate, verfahren zu deren herstellung, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2012120054A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Di- und trisubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120055A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Di- und trisubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120053A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Verzweigte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120056A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| JP2018535263A (ja) * | 2015-11-17 | 2018-11-29 | ダウ アグロサイエンシィズ エルエルシー | 4−((6−(2−(2,4−ジフルオロフェニル)−1,1−ジフルオロ−2−ヒドロキシ−3−(1h−1,2,4−トリアゾール−1−イル)プロピル)ピリジン−3−イル)オキシ)ベンゾニトリル及び調製方法 |
| JP2018535262A (ja) * | 2015-11-17 | 2018-11-29 | ダウ アグロサイエンシィズ エルエルシー | 4−((6−(2−(2,4−ジフルオロフェニル)−1,1−ジフルオロ−2−オキソエチル)ピリジン−3−イル)オキシ)ベンゾニトリル及び製造方法 |
| JP2018538366A (ja) * | 2015-11-17 | 2018-12-27 | ダウ アグロサイエンシィズ エルエルシー | 4−((6−(2−(2,4−ジフルオロフェニル)−1,1−ジフルオロ−2−ヒドロキシ−3−(1h−1,2,4−トリアゾール−1−イル)プロピル)ピリジン−3−イル)オキシ)ベンゾニトリル及び調製方法 |
| US11878968B2 (en) | 2021-07-09 | 2024-01-23 | Plexium, Inc. | Aryl compounds and pharmaceutical compositions that modulate IKZF2 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP1905769A1 (en) | 2008-04-02 |
| CA2614544A1 (en) | 2007-01-18 |
| CA2614544C (en) | 2013-09-10 |
| AU2006267338A1 (en) | 2007-01-18 |
| JP5281287B2 (ja) | 2013-09-04 |
| US20100087360A1 (en) | 2010-04-08 |
| US7994331B2 (en) | 2011-08-09 |
| EP1905769B1 (en) | 2017-03-29 |
| AU2006267338B2 (en) | 2012-08-16 |
| EP1905769A4 (en) | 2010-12-08 |
| JPWO2007007910A1 (ja) | 2009-01-29 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2007007910A1 (ja) | ヘテロ環置換ベンズイミダゾール誘導体 | |
| US11034685B2 (en) | Aminopyridine derivatives as TAM family kinase inhibitors | |
| JP2022527607A (ja) | Glp-1rアゴニスト及びその使用 | |
| AU2016372048B2 (en) | Heteroarylhydroxypyrimidinones as agonists of the APJ receptor | |
| KR102384668B1 (ko) | Apj 효능제로서의 6-히드록시-5-(페닐/헤테로아릴술포닐)피리미딘-4(1h)-온 | |
| CA2623958C (en) | 2-heteroaryl-substituted indole derivative | |
| US9242977B2 (en) | Trk-inhibiting compound | |
| EP4227299A1 (en) | Benzimidazolone glp-1 receptor agonist and use thereof | |
| JP2018535999A (ja) | アペリン受容体アゴニストおよびその使用方法 | |
| CA3001974A1 (en) | 2,4-dihydroxy-nicotinamides as apj agonists | |
| WO2006049304A1 (ja) | アリールオキシ置換ベンズイミダゾール誘導体 | |
| TW201124400A (en) | Conpounds and compositions as modulators of GPR119 activity | |
| US20120165331A1 (en) | Di/tri-aza-spiro-C9-C11alkanes | |
| EP2734516A1 (en) | Arylpyrazole ethers as inhibitors of leukotriene a4 hydrolase | |
| TW202045498A (zh) | 作為法尼醇x受體調節劑之經取代雙環化合物 | |
| KR20150135458A (ko) | 신규한 피리딘 유도체 | |
| WO2012117996A1 (ja) | Gpr119作動薬 | |
| US20120101110A1 (en) | Diaza-spiro[5.5]undecanes | |
| JP2013047188A (ja) | Gpr119作動薬 | |
| JP2007022937A (ja) | ヘテロ環置換ベンズイミダゾール誘導体 | |
| TW202317553A (zh) | Glp-1受體激動劑及其組合物和用途 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2007524727 Country of ref document: JP |
|
| ENP | Entry into the national phase |
Ref document number: 2614544 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 11988592 Country of ref document: US |
|
| REEP | Request for entry into the european phase |
Ref document number: 2006781274 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006781274 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006267338 Country of ref document: AU |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2006267338 Country of ref document: AU Date of ref document: 20060712 Kind code of ref document: A |