WO2004075815A2 - Diarylcycloalkylderivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel - Google Patents
Diarylcycloalkylderivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel Download PDFInfo
- Publication number
- WO2004075815A2 WO2004075815A2 PCT/EP2004/001584 EP2004001584W WO2004075815A2 WO 2004075815 A2 WO2004075815 A2 WO 2004075815A2 EP 2004001584 W EP2004001584 W EP 2004001584W WO 2004075815 A2 WO2004075815 A2 WO 2004075815A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- ring
- case
- phenyl
- compounds
- Prior art date
Links
- IQJOHJMAEPCYEZ-UHFFFAOYSA-N Cc1c(C(O)=O)c(COC2CC(COCc3c(-c4ccccc4)[o]c(-c4cc(OC)ccc4)n3)CCC2)ccc1 Chemical compound Cc1c(C(O)=O)c(COC2CC(COCc3c(-c4ccccc4)[o]c(-c4cc(OC)ccc4)n3)CCC2)ccc1 IQJOHJMAEPCYEZ-UHFFFAOYSA-N 0.000 description 1
- IHXQLZCNZLKYID-UHFFFAOYSA-N Cc1c(CCl)nc(C(C=C2)=CC3C2=CC=CC3)[o]1 Chemical compound Cc1c(CCl)nc(C(C=C2)=CC3C2=CC=CC3)[o]1 IHXQLZCNZLKYID-UHFFFAOYSA-N 0.000 description 1
- XFLMZIVSUSURBJ-UHFFFAOYSA-N Cc1c(CI)nc(-c2cc(cccc3)c3cc2)[o]1 Chemical compound Cc1c(CI)nc(-c2cc(cccc3)c3cc2)[o]1 XFLMZIVSUSURBJ-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D263/00—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings
- C07D263/02—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings
- C07D263/30—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D263/32—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
Definitions
- Diarylcycloalkyl derivatives processes for their preparation and their use as medicines
- the invention relates to diarylcycloalkyl derivatives and their physiologically tolerable salts and physiologically functional derivatives.
- the invention was based on the object of providing compounds which allow a therapeutically usable modulation of the lipid and / or carbohydrate metabolism and are therefore suitable for the prevention and / or treatment of diseases such as type 2 diabetes and atherosclerosis and their various secondary diseases.
- the compounds are particularly suitable for activating PPARalpha and PPARgamma, the extent of the relative activation being able to vary depending on the compounds.
- the invention therefore relates to compounds of the formula
- Ring A (C 3 -C 8 ) cycloalkanediyl, (C 3 -C 8 ) cycloalkenediyl, it being possible for one or more carbon atoms in the cycloalkanediyl or cycloalkenediyl rings to be replaced by oxygen atoms;
- Phenyl (CC 6 ) alkyl, 0- (C ⁇ -C 6 ) alkyl, 0- (C 1 -C 6 ) alkyl-0- (C 1 -C 3 ) alkyl;
- R5 H, F, Cl, Br, OH, N0 2 , CF 3 , OCF 3 , (d-CeJ-alkyl, 0- (CC 6 ) -alkyl; R3 H, (CC 6 ) alkyl;
- Carbon atoms can be replaced by oxygen atoms
- Preferred compounds of the formula I are those in which
- SCF 3 OCF 2 -CHF2, O-phenyl, 0- (C 1 -C 6 ) alkyl-0- (C 1 -C 3 ) alkyl;
- R5 H, F, Cl, Br, OH, N0 2 , CF 3 , 0CF 3 , (dC ⁇ alkyl, 0- (CC 6 ) alkyl;
- Carbon atoms can be replaced by oxygen atoms.
- Ring B a) phenyl; a or b) 5 - 12-membered heteroaromatic ring, the one to four
- R5 H, F, Cl, Br, OH, N0 2 , CF 3 , OCF 3 , (dC 6 ) alkyl, 0- (dC 6 ) alkyl;
- Ring A, Ring B, R1, R2, R3, R4, R5, X and Y are as defined above.
- R5 means methyl
- SCF 3 OCF 2 -CHF 2 ( O-phenyl, 0- (C 1 -C 6 ) alkyl-0- (C 1 -C 3 ) alkyl;
- the present invention also includes all combinations of the "preferred embodiments" of the invention described herein.
- alkyl radicals in the substituents R1, R2, R3, R4 and R5. can be straight or branched.
- Aryl is understood to mean an aromatic carbocyclic mono- or bicyclic ring system which contains 6 to 10 atoms in the ring or in the rings.
- Heteroaryl is a mono- or bicyclic aromatic ring system with 4 to 11 ring members, in which at least one atom in the ring system is a heteroatom from the series N, O and S.
- the compounds of formula I contain at least two centers of asymmetry and may also contain more.
- the compounds of the formula I can therefore be in the form of their racemates, racemic mixtures, pure enantiomers, diastereomers and diastereomer mixtures.
- the present invention encompasses all of these isomeric forms of the compounds of the formula I. These isomeric forms can, although not described expressly in part, be obtained by known methods.
- Suitable pharmaceutically acceptable acid addition salts of the compounds according to the invention are salts of inorganic acids, such as hydrochloric acid, hydrobromic, phosphoric, metaphosphoric, nitric and sulfuric acid, and organic acids, such as, for example, acetic acid, benzenesulfonic, benzoic, citric, ethanesulfonic, fumaric , Gluconic, glycolic, isethione, lactic, lactobionic, maleic, malic, methanesulfonic, succinic, p-toluenesulfonic and tartaric acid.
- inorganic acids such as hydrochloric acid, hydrobromic, phosphoric, metaphosphoric, nitric and sulfuric acid
- organic acids such as, for example, acetic acid, benzenesulfonic, benzoic, citric, ethanesulfonic, fumaric , Gluconic, glycolic, isethione, lactic, lactobionic,
- Suitable pharmaceutically acceptable basic salts are ammonium salts, alkali metal salts (such as sodium and potassium salts) and alkaline earth salts (such as magnesium and calcium salts) and salts of trometamol (2-amino-2-hydroxymethyl-1, 3-propanediol), diethanolamine, lysine or ethylenediamine ,
- Salts with a non-pharmaceutically acceptable anion are also within the scope of the invention as useful intermediates for the preparation or purification of pharmaceutically acceptable salts and / or for use in non-therapeutic, for example in vitro, applications.
- physiologically functional derivative used here denotes any physiologically compatible derivative of a compound of formula 1 according to the invention, e.g. an ester which, when administered to a mammal, e.g. humans, is able (directly or indirectly) to form a compound of formula I or an active metabolite thereof.
- the physiologically functional derivatives also include prodrugs of the compounds according to the invention, as described, for example, in H. Okada et al., Chem. Pharm. Bull. 1994, 42, 57-61. Such prodrugs can be metabolized in vivo to a compound according to the invention. These prodrugs may or may not work themselves.
- the compounds of the invention can also exist in various polymorphic forms, e.g. as amorphous and crystalline polymorphic forms. All polymorphic forms of the compounds according to the invention belong to the scope of the invention and are a further aspect of the invention.
- This invention further relates to the use of compounds of formula I and their pharmaceutical compositions as PPAR receptor ligands.
- the PPAR receptor ligands according to the invention are suitable as modulators of the activity of the PPAR receptors.
- PPAR Peroxisome Proliferator-Activated Receptors
- PPARdelta which are encoded by different genes
- PPARgamma PPARgammai
- gamma 2 PPARgamma 2
- the different PPAR receptors have a different tissue distribution and modulate different physiological functions.
- the PPAR receptors play a key role in various aspects of regulating a large number of genes, the gene products of which are directly or indirectly linked to the lipid and
- PPARalpha receptors play an important role in the regulation of fatty acid catabolism or lipoprotein metabolism in the liver, while PPARgamma, for example, plays a key role in regulating fat cell differentiation.
- PPAR receptors are also involved in the regulation of many other physiological processes, including those that are not directly related to carbohydrate or lipid metabolism. The activity of the different PPAR receptors can be modulated to different degrees by different fatty acids, fatty acid derivatives and synthetic compounds. Corresponding reviews about functions, physiological effects and
- the present invention relates to compounds of the formula I which are suitable for modulating the activity of PPAR receptors, in particular the activity of PPARalpha and PPARgamma.
- the compounds of the formula I are suitable for the treatment, control and prophylaxis of the indications described below and a number of other associated pharmaceutical applications (see, for example, Joel Berger et al., Annu. Rev. Med. 2002, 53, 409-435; Timothy Wilson et al. J. Med. Chem ._, 2000, Vol. 43, No. 4, 527-550; Steven Kliewer et al., Recent Prog Horm Res.
- Such compounds are particularly suitable for the treatment and / or prevention of
- Diabetes mellitus especially type 2 diabetes including the prevention of the associated complications.
- Dysupidemia and its consequences such as, for example, atherosclerosis, coronary heart disease, cerebrovascular diseases, etc., in particular those (but not limited to), which are characterized by one or more of the following factors:
- Metabolic Syndrome Various other conditions that may be associated with the Metabolic Syndrome include:
- Heart failure such as (but not limited to) on condition after
- - atherosclerosis such as (but not limited to) coronary sclerosis including angina pectoris or heart attack, stroke - vascular restenosis or revision
- Fat line carcinomas such as liposarcomas - solid tumors and neoplasms, such as (but not limited to) carcinomas of the gastrointestinal tract, liver, biliary tract and pancreas, endocrine tumors, carcinomas of the lungs, kidneys and urinary organs, genital tract, prostate carcinomas etc - acute and chronic myeloprolifeative diseases and lymphomas
- Dermatitis e.g. seborrheic dermatitis or light dermatitis
- keratitis and keratoses such as seborrheic keratoses, senile keratoses, actinic keratosis, photo-induced keratoses or keratosis follicularis
- HPV Human papilloma viral infections, such as venereal papillomata, viral warts, e.g. Molluscum contagiosum, leukoplakia
- Papular dermatoses such as Lying planus
- Skin cancer such as Basal cell carcinoma, melanoma or cutaneous T-cell lymphoma
- PCOS Polycystic Ovarian Syndrome
- ARDS - Acute Respiratory Distress Syndrome
- the amount of a compound of formula I required to achieve the desired biological effect depends on a number of factors, e.g. the specific compound chosen, the intended one
- the daily dose is in the range from 0.001 mg to 100 mg (typically from 0.01 mg to 50 mg) per day per kilogram of body weight, for example 0.1-10 mg / kg / day.
- An intravenous dose can range, for example, from 0.001 mg to 1.0 mg / kg, which can suitably be administered as an infusion of 10 ng to 100 ng per kilogram per minute.
- Suitable infusion solutions for these purposes can contain, for example, from 0.1 ng to 10 mg, typically from 1 ng to 10 mg per milliliter.
- Single doses can contain, for example, from 1 mg to 10 g of the active ingredient.
- ampoules for injections can contain, for example, from 1 mg to 100 mg, and orally administrable single-dose formulations, such as tablets or capsules, for example, from 0.05 to 1000 mg, typically from 0.5 to 600 mg.
- the compounds of the formula I themselves can be used as a compound, but they are preferably in the form of a pharmaceutical composition with a compatible carrier.
- the carrier must of course be compatible, in the sense that it is compatible with the other components of the composition and is not harmful to the health of the patient.
- the carrier can be a solid or a liquid or both and is preferably formulated with the compound as a single dose, for example as a tablet, which can contain from 0.05% to 95% by weight of the active ingredient.
- Further pharmaceutically active substances can also be present, including further compounds of the formula I.
- the pharmaceutical compositions according to the invention can be prepared by one of the known pharmaceutical methods which essentially consist in mixing the constituents with pharmacologically acceptable carriers and / or auxiliaries ,
- compositions according to the invention are those which are suitable for oral, rectal, topical, peroral (for example sublingual) and parenteral (for example subcutaneous, intramuscular, intradermal or intravenous) administration, although the most suitable mode of administration in each individual case depends on the type and severity of the to be treated State and on the type of compound used according to formula I is dependent.
- Coated formulations and coated slow-release formulations also fall within the scope of the invention.
- Formulations which are resistant to acid and gastric juice are preferred.
- Suitable enteric coatings include cellulose acetate phthalate, polyvinyl acetate phthalate, hydroxypropylmethyl cellulose phthalai and anionic polymers of methacrylic acid and methyl methacrylate.
- Suitable pharmaceutical preparations for oral administration can be present in separate units, such as, for example, capsules, capsules, lozenges or tablets, each of which contains a certain amount of the compound of the formula I; as powder or granules; as a solution or suspension in an aqueous or non-aqueous liquid; or as an oil-in-water or water-in-oil emulsion.
- these compositions can be prepared by any suitable pharmaceutical method comprising a step in which the active ingredient and the carrier (which can consist of one or more additional ingredients) are brought into contact.
- the compositions are obtained by uniformly and homogeneously mixing the active ingredient with a liquid and / or finely divided solid carrier manufactured, after which the product is molded if necessary.
- a tablet can be produced by compressing or molding a powder or granulate of the compound, optionally with one or more additional components.
- Pressed tablets can be prepared by tabletting the compound in free flowing form, such as a powder or granules, optionally mixed with a binder, lubricant, inert diluent and / or a (several) surface active / dispersing agents in a suitable machine.
- Molded tablets can be made by molding the powdered compound moistened with an inert liquid diluent in a suitable machine.
- compositions suitable for oral (sublingual) administration include lozenges containing a compound of Formula I with a flavor, usually sucrose and acacia or tragacanth, and lozenges containing the compound in an inert base such as gelatin and glycerin or sucrose and gum arabic.
- Suitable pharmaceutical compositions for parenteral administration preferably comprise sterile aqueous preparations of a compound according to formula I, which are preferably isotonic with the blood of the intended recipient. These preparations are preferably administered intravenously, although they can also be administered subcutaneously, intramuscularly or intradermally as an injection. These preparations can preferably be prepared by mixing the compound with water and making the solution obtained sterile and isotonic with the blood. Injectable compositions according to the invention generally contain from 0.1 to 5% by weight of the active compound.
- Suitable pharmaceutical compositions for rectal administration are preferably in the form of single-dose suppositories. These can be prepared by mixing a compound of the formula I with one or more conventional solid carriers, for example cocoa butter, and shaping the resulting mixture.
- Suitable pharmaceutical compositions for topical use on the skin are preferably in the form of an ointment, cream, lotion, paste, spray, aerosol or oil. Vaseline, lanolin, polyethylene glycols, alcohols and combinations of two or more of these substances can be used as carriers.
- the active ingredient is generally present in a concentration of 0.1 to 15% by weight of the composition, for example 0.5 to 2%.
- Suitable pharmaceutical compositions for transdermal applications can be presented as individual patches which are suitable for long-term close contact with the patient's epidermis.
- Such plasters suitably contain the active ingredient in an optionally buffered aqueous solution, dissolved and / or dispersed in an adhesive or dispersed in a polymer.
- a suitable active ingredient concentration is approximately 1% to 35%, preferably approximately 3% to 15%.
- the active ingredient can be released by electrotransport or iontophoresis, as described, for example, in Pharmaceutical Research, 2 (6): 318 (1986).
- the compounds of formula I are distinguished by favorable effects on metabolic disorders. They have a positive influence on fat and sugar metabolism, in particular they lower the triglyceride level and are suitable for the prevention and treatment of type II diabetes and arteriosclerosis and their various complications.
- the compounds according to the invention can be administered alone or in combination with one or more further pharmacologically active substances which, for example, have beneficial effects on metabolic disorders or diseases frequently associated therewith.
- Such drugs are for example
- the active ingredient combination can be administered either by separate administration of the active ingredients to the patient or in the form of combination preparations in which several active ingredients are present in a pharmaceutical preparation.
- Examples include:
- Antidiabetics include all insulins and insulin derivatives such as, for example, Lantus ® (see www.lantus.com) or Apidra ®, and other fast-acting insulins (see US 6,221, 633), GLP-1 receptor modulators as described in WO 01/04146 described , or also such as those that were disclosed in WO 98/08871 by Novo Nordisk AS.
- the orally active hypoglycemic agents preferably include sulphonylureas, biguanides, meglitinides, oxadiazolidinediones, thiazolidinediones, glucosidase inhibitors, glucagon antagonists, oral GLP-1 agonists, DPP-IV inhibitors, potassium channel openers such as those in WO 97/2265 and 62 WO 99/03861, insulin sensitizers, inhibitors of liver enzymes which are involved in stimulating gluconeogenesis and / or glycogenolysis, Modulators of glucose uptake, compounds that alter lipid metabolism that change the lipid composition of the blood, compounds that reduce food intake or intake, PPAR and PXR modulators and active substances that act on the ATP-dependent potassium channel of the beta cells.
- the compounds of the formula I are administered in combination with substances which have an effect on hepatic glucose production, e.g. Glycogen phosphorylase inhibitors (see: WO 01/94300, WO
- the compounds of formula I are used in combination with a sulphonylurea, e.g. Tolbutamide, glibenclamide, glipizide or glimepiride administered.
- a sulphonylurea e.g. Tolbutamide, glibenclamide, glipizide or glimepiride administered.
- the compounds of the formula I are administered in combination with an active ingredient which acts on the ATP-dependent potassium channel of the beta cells, e.g. Tolbutamide, glibenclamide, glipizide, glimepiride or repaglinide.
- an active ingredient which acts on the ATP-dependent potassium channel of the beta cells, e.g. Tolbutamide, glibenclamide, glipizide, glimepiride or repaglinide.
- the compounds of formula I in combination with a biguanide such as e.g. Metformin.
- the compounds of the formula I are used in combination with a thiazolidinedione such as, for example, ciglitazone, pioglitazone, rosiglitazone or those described in WO 97/41097 by Dr. Reddy's Research Foundation disclosed compounds, in particular 5 - [[4 - [(3,4-dihydro-3-methyl-4-oxo-2-quinazolinylmethoxy] phenyl] methyl] -2,4-thiazoIidinedione, in one embodiment the compounds of formula I in combination with a DPPIV inhibitor, such as in W098 / 19998, W099 / 61431, W099 / 67278,
- the compounds of the formula I are used in combination with a PPARgamma agonist, e.g. Rosiglitazone, pioglitazone administered.
- a PPARgamma agonist e.g. Rosiglitazone, pioglitazone administered.
- the compounds of formula I are used in combination with compounds having an inhibitory effect on SGLT-1 and / or 2, e.g. disclosed directly or indirectly in PCT / EP03 / 06841, PCT / EP03 / 13454 and PCT / EP03 / 13455.
- the compounds of the formula I are used in combination with an er-glucosidase inhibitor, e.g. Migl toi or acarbose. In one embodiment, the compounds of formula I are used in combination with more than one of the aforementioned compounds, e.g. in combination with a sulfonylurea and metformin, a sulfonylurea and acarbose, repaglinide and metformin, insulin and a sulfonylurea, insulin and metformin, insulin and troglitazone, insulin and lovastatin, etc. administered.
- an er-glucosidase inhibitor e.g. Migl toi or acarbose.
- the compounds of formula I are used in combination with more than one of the aforementioned compounds, e.g. in combination with a sulfonylurea and metformin, a sulfonylurea and acarb
- the compounds of the formula I are administered in combination with an HMGCoA reductase inhibitor, such as lovastatin, fluvastatin, pravastatin, simvastatin, ivastatin, itavastatin, atorvastatin, rosuvastatin.
- an HMGCoA reductase inhibitor such as lovastatin, fluvastatin, pravastatin, simvastatin, ivastatin, itavastatin, atorvastatin, rosuvastatin.
- the compounds of the formula I are administered in combination with a bile acid absorption inhibitor (see, for example, US 6,245,744, US 6,221,897, US 6,277,831, EP 0683 773, EP 0683 774).
- the compounds of the formula I are administered in combination with a polymeric bile acid adsorber, such as, for example, cholestyramine, colesevelam.
- the compounds of the formula I are used in combination with a cholesterol absorption inhibitor, such as e.g. described in WO 0250027, or ezetimibe, tiqueside, pamaqueside administered.
- a cholesterol absorption inhibitor such as e.g. described in WO 0250027, or ezetimibe, tiqueside, pamaqueside administered.
- the compounds of the formula I are administered in combination with an LDL receptor inducer (see, for example, US 6,342,512).
- the compounds of formula I in combination with bulking agents preferably insoluble bulking agents (see, for example, carob / Caromax ® (Zunft HJ; et al, Carob pulp preparation for treatment of hypercholesterolemia, ADVANCES IN THERAPY (2001 Sep-Oct). 18 (5), 230-6));
- Caromax is a carob-containing product from Nutrinova, Nutrition Specialties & Food Ingredients GmbH, Industriepark availability, 65926 Frankfurt / Main).
- the combination with Caromax ® can be done in one preparation or by separate administration of compounds of formula I and Caromax ® .
- Caromax ® can also be administered in the form of food, such as in baked goods or granola bars.
- the compounds of the formula I are administered in combination with a PPARalpha agonist.
- the compounds of the formula I are used in combination with a mixed PPAR alpha / gamma agonist, e.g. AZ 242 (Tesaglitazar, (S) -3- (4- [2- (4-methanesulfonyloxyphenyl) ethoxy] phenyl) -2-ethoxypropionic acid), BMS 298585 (N - [(4-methoxyphenoxy) carbonyl] -N - [[ 4- [2- (5-methyl-2-phenyl-4-oxazolyl) ethoxy] phenyl] methyl] glycine) or as in WO 99/62872, WO 99/62871, WO 01/40171, WO 01/40169, WO96 / 38428, WO 01/81327, WO 01/21602, WO 03/020269, WO 00/64888 or WO 00/64876.
- AZ 242 Tesaglitazar, (
- the compounds of the formula I in combination with a fibrate such as e.g. Fenofibrate, gemfibrozil, clofibrate, bezafibrate.
- the compounds of the formula I are administered in combination with nicotinic acid or niacin.
- the compounds of formula I in combination with a CETP inhibitor e.g. CP-529, 414 (torcetrapib).
- the compounds of the formula I are administered in combination with an ACAT inhibitor
- the compounds of the formula I in combination with an MTP inhibitor such as e.g. Implitapide administered.
- the compounds of the formula I are administered in combination with an antioxidant.
- the compounds of the formula I are administered in combination with a lipoprotein lipase inhibitor. In one embodiment of the invention, the compounds of the formula I are administered in combination with an ATP citrate lyase inhibitor.
- the compounds of the formula I are administered in combination with a squalene synthetase inhibitor.
- the compounds of the formula I are administered in combination with a lipoprotein (a) antagonist.
- the compounds of the formula I in combination with a lipase inhibitor, such as e.g. Orlistat administered.
- a lipase inhibitor such as e.g. Orlistat administered.
- the further active ingredient is fenfluramine or dexfenfluramine. In another embodiment, the further active ingredient is sibutramine.
- the compounds of the formula I are used in combination with CART modulators (see “Cocaine-amphetamine-regulated transcript influences energy metabolism, anxiety and gastric emptying in mice”)
- NPY antagonists for example naphthalene-1-sulfonic acid ⁇ 4 - [(4-amino-quinazolin-2-ylamino) - methylj-cyclohexylmethyl ⁇ - amide; hydrochloride (CGP 71683A)), MC4 agonists (e.g.
- CRF BP antagonists e.g. Urocortiri
- urocortin agonists e.g. 1- (4-chloro-3-methanesulfonylmethylphenyl) -2- [2- (2,3-dimethyl -1 H-indol-6-yloxy) - ethylaminoj-ethanol; hydrochloride (WO 01/83451)
- MSH melanocyte-stimulating hormone
- CCK-A agonists e.g.
- Leptin agonists as a potential approach to the treatment of obesity. Drugs of the Future (2001), 26 (9), 873-881), DA agonists (bromocriptine, doprexin), lipase / amylase inhibitors ( eg WO 00/40569), PPAR modulators (eg WO 00/78312), RXR modulators or TR - ⁇ - agonists.
- the further active ingredient is leptin.
- the further active ingredient is dexamphetamine, amphetamine,
- Medications with effects on the cardiovascular and blood vessel systems such as ACE inhibitors (e.g. ramipril), medications that affect the cardiovascular and blood vessel systems, such as ACE inhibitors (e.g. ramipril), medications that affect the cardiovascular and blood vessel systems, such as ACE inhibitors (e.g. ramipril), medications that affect the cardiovascular and blood vessel systems, such as ACE inhibitors (e.g. ramipril), medications that affect the following ACE inhibitors (e.g. ramipril), medications that affect the following drugs.
- Angiotensin-renin system work, calcium antagonists, beta-blockers etc.
- the compounds of the formula I are administered in combination with anti-inflammatory drugs. In one embodiment, the compounds of the formula I are administered in combination with medicaments which are used for cancer therapy and cancer prevention.
- HEK human embryo kidney
- pdeltaM-GAL4-Luc-Zeo a luciferase reporter element
- GR-GAL4-humanPPARalpha-LBD PPARalpha fusion protein
- the stable and constitutively expressed fusion protein GR-GAL4-humanPPARalpha-LBD binds in the cell nucleus of the PPARalpha reporter cell line via the GAL4 protein portion to the GAL4-DNA binding motif 5 ' above the luciferase reporter element, which is integrated in the genome of the cell line.
- a PPARalpha ligand Without the addition of a PPARalpha ligand, the expression of the luciferase reporter gene is only low, provided that fatty acid-replicated fetal calf serum (cs-FCS) is used in the test.
- cs-FCS fatty acid-replicated fetal calf serum
- the luciferase formed can be detected on a corresponding substrate using chemiluminescence.
- the PPARalpha reporter cell line was produced in 2 stages: First, the luciferase reporter element was constructed and stably transfected in HEK cells. For this purpose, five binding sites were of the yeast transcription factor GAL4 (in each case 5 '- CGGAGTACTGTCCTCCGAG-3') 5 '-oberjur of a 68 bp-long minimal MMTV promoter (Genbank Accession # V01175) cloned.
- the minimal MMTV promoter section contains a CCAAT box and a TATA element to enable efficient transcription by RNA polymerase II.
- the GAL4-MMTV construct was cloned and sequenced analogously to Sambrook J. et. al.
- the PPARalpha fusion protein (GR-GAL4-humanPPARalpha-LBD) was introduced into the stable cell clone described.
- the cDNA coding for the N-terminal 76 amino acids of the glucocorticoid receptor (Genbank Accession # P04150) was first linked to the cDNA section coding for amino acids 1-147 of the yeast transcription factor GAL4 (Genbank Accession # P04386).
- the cDNA of the ligand binding domain of the human PPARalpha receptor cloned (amino acids S167-Y468; Genbank Accession # S74349).
- the fusion construct thus produced (GR-GAL4-humanPPARalpha-LBD) was cloned into the plasmid pcDNA3 (Invitrogen) in order to enable constitutive expression in it by the cytomegalovirus promoter.
- This plasmid pcDNA3 (Invitrogen) in order to enable constitutive expression in it by the cytomegalovirus promoter.
- the finished PPARalpha reporter cell line which contains a luciferase reporter element and constitutively the PPARalpha fusion protein (GR-GAL4-humanPPARalpha-LBD), was isolated by selection with Zeocin (0.5 mg / ml) and G418 (0.5 mg / ml) )
- the activity of PPARalpha agonists is determined in a 3-day test, which is described below:
- the PPARalpha reporter cell line is up to 80% confluency in DMEM
- the plates are incubated for 24 h in a cell culture incubator at 37 ° C. and 5% CO 2 . day 2
- PPARalpha agonists to be tested are dissolved in a concentration of 10 mM in DMSO. This stock solution is diluted in DMEM medium (# 41965-039, Invitrogen) containing 5% cs-FKS (# SH-30068.03, Hyclone), 2 mM L-glutamine (# 25030-5 024, Invitrogen) and the like described antibiotics (Zeocin, G418, penicillin and streptomycin) is added.
- Test substances are tested in 11 different concentrations in the range from 10 ⁇ M to 100 pM. More potent compounds are found in concentration ranges from
- the medium of the PPARalpha reporter cell line sown on day 1 is completely aspirated and the test substances diluted in medium are immediately added to the cells.
- the substances are diluted and added using a robot (Beckman FX).
- the final volume of the test substances diluted in medium is 100 ⁇ l per well of a 96-well microtiter plate.
- the DMSO concentration in test 5 is below 0.1% v / v in order to avoid cell-toxic effects of the solvent.
- Each plate was coated with a standard PPARalpha agonist, which is also in
- test plates are incubated for 24 h in an incubator at 37 ° C and 5% CO 2 . 0
- the PPARalpha report residues treated with the test substances are removed from the incubator and the medium is suctioned off.
- 50 l of Bright Glo Reagent (Promega) are pipetted into a 96-well microtiter plate 5 per well for lysis of the cells. After a 10 minute incubation in the dark at room temperature, the microtiter plates are measured in the luminescence measuring device (Trilux from Wallac). The measurement time per well of a microtiter plate is 1 sec.
- the PPARalpha-EC50 values for the compounds of Examples 1 to 13 in this assay range from 0.05nM to> 10 ⁇ M.
- a luciferous reporter plasmid pGL3basic-5xGAL4-TK
- pcDNA3-GAL4-humanPPARgammaLBD PPARgamma expression plasmid
- the activated fusion protein GAL4-humanPPARgammaLBD induces the expression of the luciferase reporter gene, which can be detected in the form of a chemiluminescence signal after the addition of a luciferase substrate.
- the two components luciferase reporter plasmid and PPARgamma expression plasmid
- the two components are transiently transfected in HEK cells in the cellular PPARgamma test because the stable and permanent expression of the PPARgamma fusion protein is cell-toxic.
- the luciferase reporter plasmid pGL3basic-5xGAL4-TK is based on the vector pGL3basic from Promega.
- five binding sites of the yeast transcription factor GAL4 (each binding site with the sequence 5 ' -CTCGGAGGACAGTACTCCG-3 ' ), 5 ' above, were cloned into pGL3basic together with a 160 bp long thymidine kinase promoter section (Genbank Accession # AF027128) , 3 ' below the thymidine kinase promoter is the entire luciferase gene from Photinus pyralis (Genbank accession # M 15077), which is already part of the plasmid pGL3basic used.
- the ligand binding domain (LBD) cDNA of the human PPARgamma receptor (amino acids I152-Y475; Accession # g1480099) was then cloned 3 ′ below the GAL4 DNA binding domain.
- Cloning and sequencing of the PPARgamma expression plasmid pcDNA3-GAL4-humanPPARgammaLBD were again carried out analogously to Sambrook J. et. al. (Molecular cloning, Cold Spring Harbor Laboratory Press, 1989).
- the reference plasmid pRL-CMV company Promega
- the plasmid pBluescript-Stragen (+) are used for the cellular PPARgamma test used.
- all four plasmids were prepared with a plasmid preparation kit from Qiagen, which ensures plasmid quality that is as endotoxin-free as possible.
- HEK cells are cultured in DMEM medium (# 41965-039, Invitrogen), to which the following additives have been added: 10% FCS (# 16000-044, Invitrogen), 1% penicillin-streptomycin solution (# 15140 -122, Invitrogen) and 2 mM L-glutamine (# 25030-024, Invitrogen).
- Solution A is first prepared, a transfection mixture which, in addition to DMEM medium, contains all four plasmids already described.
- pro 96-well microtiter plate uses the following amounts to prepare 3 ml of solution A: 2622 ⁇ l antibiotic and serum-free DMEM medium (# 41965-039, Invitrogen), 100 ⁇ l reference plasmid pRL-CMV (1 ng / ⁇ l), 100 ⁇ l luciferase Reporter plasmid pGL3basic- 5xGAL4-TK (10 ng / ⁇ l), 100 ⁇ l PPARgammä expression plasmid pcDNA3-GAL4-humanPPARgammaLBD (100 ng / ⁇ l) and 78 ⁇ l plasmid pBluescript-SK (+) (500 ng / ⁇ l).
- HEK cells from a 175 cm 2 cell culture bottle are washed once with 15 ml PBS (# 14190-094, Invitrogen) and treated with 3 ml trypsin solution (# 25300-054, Invitrogen) for 2 min at 37 ° C.
- the cells are then taken up in 15 ml of DMEM medium (# 41965-039, Invitrogen), which contains 10% FCS (# 16000-044, Invitrogen), 1% penicillin-streptomycin solution (# 15140-122, Invitrogen) and 2 mM L-glutamine (# 25030-024, Invitrogen) is added.
- the suspension After counting the cell suspension in the cell counter, the suspension is diluted to 250,000 cells / ml. For 1 microtiter plate, 15 ml of this cell suspension are mixed with 5 ml of solution C. 200 ⁇ l of the suspension are sown per well of a 96-well microtiter plate with a clear plastic base (# 3610, Corning Costar). The plates are incubated for 24 h in a cell culture incubator at 37 ° C and 5% CO2.
- PPAR agonists to be tested are dissolved in DMSO at a concentration of 10 mM.
- This stock solution is diluted in DMEM medium (# 41965-039, Invitrogen), which is mixed with 2% Ultroser (# 12039-012, Biosepra), 1% penicillin-streptomycin solution (# 15140-122, Invitrogen) and 2 mM L -Glutamine (# 25030-024, Invitrogen) is added.
- Test substances are tested in a total of 11 different concentrations in the range from 10 ⁇ M to 100 pM. More potent compounds are tested in concentration ranges from 1 ⁇ M to 10 pM.
- the medium of the HEK cells transfected and seeded on day 1 is completely aspirated and the test substances diluted in medium immediately to the cells added.
- the substances are diluted and added using a robot (Beckman FX).
- the final volume of the test substances diluted in medium is 100 ⁇ l per well of a 96-well microtiter plate.
- Each plate is loaded with a standard PPARgamma agonist, which is also diluted in 11 different concentrations to demonstrate the operability of the test in each individual plate.
- the test plates are incubated for 48 h in an incubator at 37 ° C and 5% CO 2 .
- Dual-Glo TM reagent Dual-Glo TM Luciferase Assay System
- Dual-Glo TM Stop & Glo reagent Dual-Glo TM Luciferase Assay System; Promega
- the chemiluminescence mediated by the Renilla luciferase is again measured in the measuring device for 1 sec / well.
- the raw data of the luminescence measuring device are transferred to a Microsoft Excel file.
- the quotient Firefly / Renilla-Luciferase activity is determined for each measuring point which is derived from a well of the microtiter plate.
- the dose-response curves and EC50 values of PPAR agonists are calculated from the quotients using the XL.Fit program according to the manufacturer's specifications (IDBS). With the PPAR agonists described in this application, PPARgamma EC50 values in the range from 0.5 nm to> 10 ⁇ M were measured.
- IDBS manufacturer's specifications
- eis 1,3 Cy means: eis substituted cyclohexane-1, 3-diyl with the stereochemistry according to Cahn-Ingold-Prelog, as indicated in the examples.
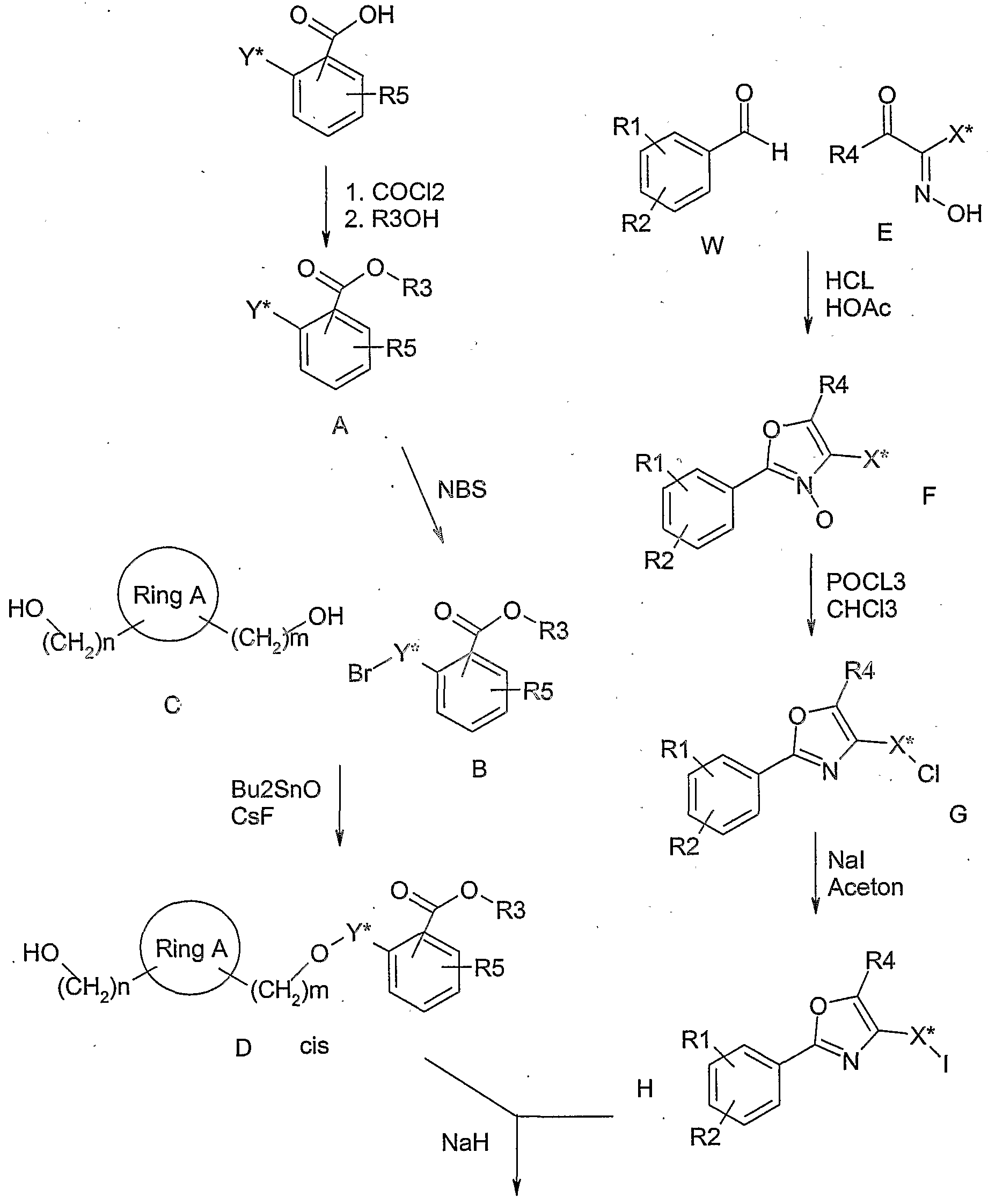
- the compound of the general formula B is reacted with a compound of the general formula C, in which n and m can each represent 0-5, to give a compound of the general formula D, in which R1, R2, R4 m, n and Y have the meanings described above have, component C is first heated with dibutyltin oxide in toluene for several hours on a water separator and then with the addition of dimethylformamide, cesium fluoride and bromide B by stirring for several hours at room temperature. implemented to D.
- the compound of the general formula E is reacted with an aldehyde of the general formula W (for example benzaldehyde, thiophene or furan carbaldehyde) to give a compound of the general formula F, in which R1, R2, R4 and X have the meanings described above, here components E and F are first dissolved in acetic acid and introduced until complete conversion of HCl, giving compounds of general formula F.
- the compound of the general formula F, in which R1, R2, R4 and X have the meanings described above is heated under reflux with POCI3 in chloroform for several hours, giving compounds of the general formula G.
- the compound of the general formula D is reacted with a compound of the general formula H, in which Y has the meaning described above, to give a compound of the general formula J, in which R1, R2, R4, R5, X and Y have the meanings described above.
- D is, for example, in a mixture of dimethylformamide and tetrahydrofuran with a strong base such as Na hydride at room temperature. deprotonated and then alkylated with component H.
- the compound of the general formula J is converted into compounds of the formula M in which R1, R2, R4, R5, X and Y have the meanings described above, for example by heating the ester function with potassium hydroxide in an alcohol (ethanol, tert-butanol) subject to saponification and the
- Other compounds can be obtained according to or by known methods. example 1
- Example 2 Analogously to Example 1, starting from diacetylmonoxime, benzo [1, 3] dioxole-5-carbaldehydes and 2 - ((1 R, 3S) -3-hydroxy-cyclohexyloxymethyl) -6-methyl-benzoic acid methyl ester 2 - [(1 R, 3S) -3- (2-benzo [1, 3] dioxol-5-yl-5-methyl-oxazol-4-ylmethoxy) cyclohexyloxymethyl] -6-methyl-benzoic acid.
- Example 2 Analogously to Example 1, starting from diacetylmonoxime, furan-2-carbaldehydes and 2 - ((1 R, 3S) -3-hydroxy-cyclohexyloxymethyl) -6-methyl-benzoic acid methyl ester 2-methyl-6 - ⁇ (1 R, 3S) -3- [5-methyl-2- (5-methyl-furan-2-yl) -oxazol-4-ylmethoxy] - cyclohexyloxymethyl ⁇ - benzoic acid.
- Example 2 Analogously to Example 1, starting from diacetylmonoxime, 5-methyl-thiophene-2-carbaldehydes and 2 - ((1 R, 3S) -3-hydroxy-cyclohexyloxymethyl) -6-methyl- methyl benzoate 2-methyl-6 - ⁇ (1 R, 3S) -3- [5-methyl-2- (5-methylthiophen-2-yl) - oxazol-4-ylmethoxy] cyclohexyloxymethyl ⁇ - benzoic acid.
- Example 2 Analogously to Example 1, starting from diacetylmonoxime, 4-trifluoromethylsulfanyl-benzaldehydes and 2 - ((1 R, 3S) -3-hydroxy-cyclohexyloxymethyl) - 6-methyl-benzoic acid methyl ester 2 - ⁇ (1 R, 3S) -methyl-6 - ⁇ 3- [5-methyl-2- (4-trifluoromethylsulfanylphenyl) oxazol-4-ylmethoxy] cyclohexyloxymethyl ⁇ benzoic acid was obtained.
- Example 7 Analogously to Example 1, starting from diacetyl monoxime, 3-pentafluoroethyloxybenzaldehyde and 2 - ((1 R, 3S) -3-hydroxy-cyclohexyloxymethyl) -6-methylbenzoic acid methyl ester 2 - ⁇ (1 R, 3S) -methyl-6 - (3- ⁇ 5-methyl-2- [3- (1,1,2,2-tetrafluoroethoxy) phenyl] oxazol-4-ylmethoxy ⁇ cyclohexyloxymethyl) benzoic acid.
- Example 8 Analogously to Example 1, starting from diacetylmonoxime, 4-phenoxybenzaldehyde and 2 - ((1 R, 3S) -3-hydroxycyclohexyloxymethyl) -6-methylbenzoic acid methyl ester 2 - ⁇ (1 R, 3S) -methyl -6- ⁇ 3- [5-methyl-2- (4-phenoxyphenyl) oxazol-4-ylmethoxy] cyclohexyloxymethyl ⁇ benzoic acid was obtained.
- Example 9 Analogously to Example 1, starting from diacetyl monoxime, thiophene-2-carbaldehydes and 2 - ((1 R, 3S) -3-hydroxy-cyclohexyloxymethyl) -6-methyl-benzoic acid methyl ester 2 - ⁇ (1 R, 3S) -methyl-6 - [3- (5-methyl-2-thiophene-2-yl-oxazol-4-ylmethoxy) cyclohexyloxymethyl] benzoic acid was obtained.
- Example 10 Analogously to Example 1, starting from diacetylmonoxime, 3-fluoro-5-trifluoromethyl-benzaldehyde and 2 - ((1 R, 3S) -3-hydroxy-cyclohexyloxymethyl) -6-methyl-benzoic acid methyl ester 2 - ⁇ (1 R, 3S) - ⁇ 3- [2- (3-Fluoro-5-trifluoromethyl-phenyl) -5-methyl-oxazol-4-ylmethoxy] cyclohexyloxymethyl ⁇ -6-methyl-benzoic acid methyl ester was obtained.
- Example 11 Analogously to Example 1, starting from 1-phenyl-1, 2-propanedione-2-oxime, p-toluene aldehyde and 2 - ((1 R, 3S) -3-hydroxy-cyclohexyloxymethyl) -6-methyl-benzoic acid methyl ester was 2-methyl -6 - [(1 R, 3S) -3- (5-phenyl-2-p-tolyl-oxazol-4-ylmethoxy) cyclohexyloxymethyl] - benzoic acid.
- Example 12 Analogously to Example 1, methyl 1 was started from 1-phenyl-1,2-propanedione-2-oxime, m-anisaldehyde and 2 - ((1 R, 3S) -3-hydroxy-cyclohexyloxymethyl) -6-methylbenzoate - ⁇ (1 R, 3S) -3- [2- (3-methoxyphenyl) -5-phenyloxazol-4-ylmethoxy] cyclohexyloxymethyl ⁇ -6-methylbenzoic acid was obtained.
- Example 13 Analogously to Example 1, starting from 2-cyclohexyl-4-iodomethyl-oxazole and 2- ((1 R, 3S) -3-hydroxy-cyclohexyloxymethyl) -6-methyl-benzoic acid methyl ester 2- [(1 R, 3S) -3 - (2-Cyclohexyl-oxazol-4-ylmethoxy) -cyclohexyloxymethyl] -6-methyl-benzoic acid obtained.
Landscapes
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Diabetes (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Obesity (AREA)
- Hematology (AREA)
- Endocrinology (AREA)
- Emergency Medicine (AREA)
- Urology & Nephrology (AREA)
- Vascular Medicine (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Description




























Claims


Priority Applications (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2006501890A JP2006519197A (ja) | 2003-02-27 | 2004-02-19 | ジアリールシクロアルキル誘導体、それらの製造方法、及び医薬としてのそれらの使用 |
| AU2004216520A AU2004216520B2 (en) | 2003-02-27 | 2004-02-19 | Diarylcycloalkyl derivatives, method for their production and their use as medicaments |
| MXPA05008993A MXPA05008993A (es) | 2003-02-27 | 2004-02-19 | Derivados de diarilcicloalquilo, metodo para su produccion y su uso como medicamentos. |
| EP04712502A EP1599454B1 (de) | 2003-02-27 | 2004-02-19 | Diarylcycloalkylderivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| BRPI0407901-9A BRPI0407901A (pt) | 2003-02-27 | 2004-02-19 | derivados de diarilcicloalquila, processos para preparação dos mesmos e uso dos mesmos como medicamentos |
| CA002516573A CA2516573A1 (en) | 2003-02-27 | 2004-02-19 | Diarylcycloalkyl derivatives, method for their production and their use as medicaments |
| IL170313A IL170313A (en) | 2003-02-27 | 2005-08-16 | Diarylcycloalkyl derivatives and pharmaceutical compositions comprising them |
| HR20050740A HRP20050740A2 (en) | 2003-02-27 | 2005-08-26 | Diarylcycloalkyl derivatives, method for their production and their use as medicaments |
| NO20054382A NO20054382L (no) | 2003-02-27 | 2005-09-21 | Diarylsykloalkylderivater, deres fremstilling og deres anvendelse som legemiddel |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE10308353A DE10308353A1 (de) | 2003-02-27 | 2003-02-27 | Diarylcycloalkylderivate, Verfahren zu ihrer Herstellung und ihre Verwendung als Arzneimittel |
| DE10308353.7 | 2003-02-27 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2004075815A2 true WO2004075815A2 (de) | 2004-09-10 |
| WO2004075815A3 WO2004075815A3 (de) | 2004-12-29 |
Family
ID=32920627
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2004/001584 WO2004075815A2 (de) | 2003-02-27 | 2004-02-19 | Diarylcycloalkylderivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
Country Status (21)
| Country | Link |
|---|---|
| US (1) | US7160911B2 (de) |
| EP (1) | EP1599454B1 (de) |
| JP (1) | JP2006519197A (de) |
| KR (1) | KR20050106053A (de) |
| CN (1) | CN100439346C (de) |
| AR (1) | AR043430A1 (de) |
| AU (1) | AU2004216520B2 (de) |
| BR (1) | BRPI0407901A (de) |
| CA (1) | CA2516573A1 (de) |
| CO (1) | CO5690579A2 (de) |
| DE (1) | DE10308353A1 (de) |
| HR (1) | HRP20050740A2 (de) |
| IL (1) | IL170313A (de) |
| MA (1) | MA27741A1 (de) |
| MX (1) | MXPA05008993A (de) |
| NO (1) | NO20054382L (de) |
| PL (1) | PL377741A1 (de) |
| RU (1) | RU2005129999A (de) |
| TW (1) | TW200505880A (de) |
| WO (1) | WO2004075815A2 (de) |
| ZA (1) | ZA200505763B (de) |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006021420A2 (de) * | 2004-08-23 | 2006-03-02 | Sanofi-Aventis Deutschland Gmbh | Verfahren zur herstellung von diarylcycloalkylderivaten |
| WO2006066694A1 (de) * | 2004-12-15 | 2006-06-29 | Sanofi-Aventis Deutschland Gmbh | VERFAHREN ZUR HERSTELLUNG VON OXAZOLEN DURCH KONDENSATION VON AROMATISCHEN ALDEHYDEN MIT α-KETOXIMEN ZU N-OXIDEN UND NACHFOLGENDE REAKTION MIT AKTIVIERTEN SÄUREDERIVATEN |
| WO2007039177A2 (en) | 2005-09-29 | 2007-04-12 | Sanofi-Aventis | Phenyl- and pyridinyl- 1, 2 , 4 - oxadiazolone derivatives, processes for their preparation and their use as pharmaceuticals |
| US7902367B2 (en) | 2004-08-11 | 2011-03-08 | Kyorin Pharmaceutical Co., Ltd. | Cyclic amino benzoic acid derivative |
| US7960384B2 (en) | 2006-03-28 | 2011-06-14 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US8084605B2 (en) | 2006-11-29 | 2011-12-27 | Kelly Ron C | Polymorphs of succinate salt of 2-[6-(3-amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethy]-4-fluor-benzonitrile and methods of use therefor |
| US8093236B2 (en) | 2007-03-13 | 2012-01-10 | Takeda Pharmaceuticals Company Limited | Weekly administration of dipeptidyl peptidase inhibitors |
| US8222411B2 (en) | 2005-09-16 | 2012-07-17 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US8324383B2 (en) | 2006-09-13 | 2012-12-04 | Takeda Pharmaceutical Company Limited | Methods of making polymorphs of benzoate salt of 2-[[6-[(3R)-3-amino-1-piperidinyl]-3,4-dihydro-3-methyl-2,4-dioxo-1(2H)-pyrimidinyl]methyl]-benzonitrile |
| US8906901B2 (en) | 2005-09-14 | 2014-12-09 | Takeda Pharmaceutical Company Limited | Administration of dipeptidyl peptidase inhibitors |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7399777B2 (en) * | 2001-08-31 | 2008-07-15 | Sanofi-Aventis Deutschland Gmbh | Diarylcycloalkyl derivatives, processes for their preparation and their use as pharmceuticals |
| EE05418B1 (et) * | 2001-08-31 | 2011-06-15 | Aventis Pharma Deutschland Gmbh | Diarltskloalklderivaadid, nende kasutamine ravimite valmistamiseks ning neid sisaldav ravim |
| WO2006101108A1 (ja) * | 2005-03-23 | 2006-09-28 | Kyorin Pharmaceutical Co., Ltd. | 新規環状アミノフェニルアルカン酸誘導体 |
| US8153644B2 (en) * | 2007-05-22 | 2012-04-10 | Madrigal Pharmaceuticals, Inc. | Diacylglycerol acyltransferase inhibitors |
| US8115011B2 (en) * | 2007-05-22 | 2012-02-14 | Madrigal Pharmaceuticals, Inc. | Diacylglycerol acyltransferase inhibitors |
| US8211884B2 (en) * | 2008-08-06 | 2012-07-03 | Madrigal Pharmaceuticals, Inc. | Diacylglycerol acyltransferase inhibitors |
| US8324385B2 (en) * | 2008-10-30 | 2012-12-04 | Madrigal Pharmaceuticals, Inc. | Diacylglycerol acyltransferase inhibitors |
| JP2012512252A (ja) * | 2008-12-17 | 2012-05-31 | ブイアイエイ・ファーマシューティカルズ・インコーポレーテッド | ジアシルグリセロールアシルトランスフェラーゼの阻害剤 |
Citations (46)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0683774A1 (de) | 1993-02-15 | 1995-11-29 | The Wellcome Foundation Limited | Hypolipemische verbindungen |
| EP0683773A1 (de) | 1993-02-15 | 1995-11-29 | Wellcome Found | Hypolipidämische, kondensierte 1,4-thiazepine. |
| WO1996038428A1 (fr) | 1995-06-02 | 1996-12-05 | Kyorin Pharmaceutical Co., Ltd. | Derives de n-benzyldioxothiazolidylbenzamide et leur procede de production |
| WO1997026265A1 (en) | 1996-01-17 | 1997-07-24 | Novo Nordisk A/S | Fused 1,2,4-thiadiazine and fused 1,4-thiazine derivatives, their preparation and use |
| WO1997041097A2 (en) | 1996-12-31 | 1997-11-06 | Dr. Reddy's Research Foundation | Novel heterocyclic compounds process for their preparation and pharmaceutical compositions containing them and their use in the treatment of diabetes and related diseases |
| WO1998008871A1 (en) | 1996-08-30 | 1998-03-05 | Novo Nordisk A/S | Glp-1 derivatives |
| WO1998019998A2 (en) | 1996-11-07 | 1998-05-14 | Novartis Ag | N-substituted 2-cyanopyrrolidines |
| WO1999003861A1 (en) | 1997-07-16 | 1999-01-28 | Novo Nordisk A/S | Fused 1,2,4-thiadiazine derivatives, their preparation and use |
| WO1999061431A1 (de) | 1998-05-28 | 1999-12-02 | Probiodrug Gesellschaft für Arzneimittelforschung mbH | Neue effektoren von dipeptidylpeptidase iv |
| WO1999062871A1 (en) | 1998-06-04 | 1999-12-09 | Astrazeneca Ab | New 3-aryl propionic acid derivatives and analogs |
| WO1999062872A1 (en) | 1998-06-04 | 1999-12-09 | Astrazeneca Ab | New 3-aryl-2-hydroxypropionic acid derivative (i) |
| WO1999067279A1 (de) | 1998-06-24 | 1999-12-29 | Probiodrug Gesellschaft für Arzneimittelforschung mbH | Verbindungen von instabilen dp iv-inhibitoren |
| WO1999067278A1 (de) | 1998-06-24 | 1999-12-29 | Probiodrug Gesellschaft für Arzneimittelforschung mbH | Prodrugs von dp iv-inhibitoren |
| WO2000040569A1 (en) | 1999-01-08 | 2000-07-13 | Alizyme Therapeutics Limited | 2-amino-benzoxazinone derivatives for the treatment of obesity |
| WO2000063208A1 (en) | 1999-04-16 | 2000-10-26 | Novo Nordisk A/S | Substituted imidazoles, their preparation and use |
| WO2000064888A1 (en) | 1999-04-28 | 2000-11-02 | Aventis Pharma Deutschland Gmbh | Di-aryl acid derivatives as ppar receptor ligands |
| WO2000064876A1 (en) | 1999-04-28 | 2000-11-02 | Aventis Pharma Deutschland Gmbh | Tri-aryl acid derivatives as ppar receptor ligands |
| WO2000071549A1 (en) | 1999-05-21 | 2000-11-30 | Knoll Gmbh | Thiazoloderivatives and pharmaceutical compositions containing them |
| WO2000078312A1 (en) | 1999-06-18 | 2000-12-28 | Merck & Co., Inc. | Arylthiazolidinedione and aryloxazolidinedione derivatives |
| WO2001004146A2 (en) | 1999-07-09 | 2001-01-18 | Cohesion Technologies, Inc. | Ecarin polypeptides, polynucleotides encoding ecarin, and methods for use thereof |
| WO2001009111A1 (en) | 1999-07-29 | 2001-02-08 | Eli Lilly And Company | Benzofurylpiperazines and benzofurylhomopiperazines: serotonin agonists |
| WO2001021602A1 (en) | 1999-09-22 | 2001-03-29 | Bristol-Myers Squibb Company | Oxa- and thiazole derivatives useful as antidiabetic and antiobesity agents |
| US6221897B1 (en) | 1998-06-10 | 2001-04-24 | Aventis Pharma Deutschland Gmbh | Benzothiepine 1,1-dioxide derivatives, a process for their preparation, pharmaceuticals comprising these compounds, and their use |
| US6221633B1 (en) | 1997-06-20 | 2001-04-24 | Aventis Pharma Deutschland Gmbh | Insulin derivatives having a rapid onset of action |
| WO2001040169A1 (en) | 1999-12-03 | 2001-06-07 | Astrazeneca Ab | Comminuted form of (s)-2-ethoxy -3-[4-(2- {4-methanesulfonyloxyphenyl} ethoxy) phenyl] propanoic acid |
| WO2001040171A1 (en) | 1999-12-03 | 2001-06-07 | Astrazeneca Ab | Crystalline form of (s)-2 ethoxy-3-[4-(2-{4-methanesulfonyloxyphenyl} ethoxy) phenyl] propanoic acid |
| US6245744B1 (en) | 1998-10-02 | 2001-06-12 | Aventis Pharma Deutschland Gmbh | Aryl-substituted propanolamine derivatives, their preparation, pharmaceuticals comprising them, and their use |
| US6277831B1 (en) | 1999-04-09 | 2001-08-21 | Aventis Pharma Deutschland Gmbh | 1,4-benzothiazepine-1,1-dioxide derivatives substituted by sugar residues, process for their preparation, pharmaceuticals comprising these compounds, and their use |
| WO2001072290A2 (en) | 2000-03-31 | 2001-10-04 | Probiodrug Ag | Method for the improvement of islet signaling in diabetes mellitus and for its prevention |
| WO2001081327A1 (fr) | 2000-04-25 | 2001-11-01 | Kyorin Pharmaceutical Co., Ltd. | Nouveau cristal stable de derive thiazolidinedione et son procede de production |
| WO2001083451A1 (fr) | 2000-04-28 | 2001-11-08 | Asahi Kasei Kabushiki Kaisha | Nouveaux composés bicycliques |
| WO2001085695A1 (en) | 2000-05-11 | 2001-11-15 | Bristol-Myers Squibb Co. | Tetrahydroisoquinoline analogs useful as growth hormone secretagogues |
| WO2001091752A1 (en) | 2000-05-30 | 2001-12-06 | Merck & Co., Inc. | Melanocortin receptor agonists |
| WO2001094300A1 (de) | 2000-06-09 | 2001-12-13 | Aventis Pharma Deutschland Gmbh | Acylphenylharnstoffderivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| US6342512B1 (en) | 1999-09-01 | 2002-01-29 | Aventis Pharma Deutschland Gmbh | Sulfonylcarboxamide derivatives, process for their preparation and their use as pharmaceuticals |
| WO2002038541A1 (fr) | 2000-11-10 | 2002-05-16 | Taisho Pharmaceutical Co., Ltd. | Derives de cyanopyrrolidine |
| WO2002050027A1 (de) | 2000-12-21 | 2002-06-27 | Aventis Pharma Deutschland Gmbh | Neue diphenzylazetidinone, verfahren zu deren herstellung, diese verbindungen enthaltende arzneimittel und deren verwendung zur behandlung von lipidstoffwechselstörungen |
| WO2002096864A1 (de) | 2001-05-25 | 2002-12-05 | Aventis Pharma Deutschland Gmbh | Carbonsäureamid substituierte phenylharnstoffderivate, verfahren zu ihrer herstellung als arzneimittel |
| WO2003010418A1 (en) | 2001-07-21 | 2003-02-06 | Edwards Thomas C | Single-degree-of-freedom controlled-clearance univanetm fluid-handling machine |
| WO2003020269A1 (de) | 2001-08-31 | 2003-03-13 | Aventis Pharma Deutschland Gmbh | Diarylcycloalkylderivate, verfahren zu ihrer herstellung und ihre verwendung als ppar-aktivatoren |
| WO2003040174A2 (en) | 2001-11-09 | 2003-05-15 | Probiodrug Ag | Substituted amino ketone compounds |
| WO2003084922A1 (de) | 2002-04-11 | 2003-10-16 | Aventis Pharma Deutschland Gmbh | Acyl-4-carboxy-phenyl-harnstoffderivate, verfahren zu deren herstellung und deren verwendung |
| WO2003084923A1 (de) | 2002-04-11 | 2003-10-16 | Aventis Pharma Deutschland Gmbh | Acyl-3-carboxyphenyl-harnstoffderivate, verfahren zu deren herstellung und deren verwendung |
| WO2004007517A1 (de) | 2002-07-11 | 2004-01-22 | Aventis Pharma Deutschland Gmbh | Neue thiophenglycosidderivate, verfahren zu deren herstellung, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2004052903A1 (de) | 2002-12-12 | 2004-06-24 | Aventis Pharma Deutschland Gmbh | Neue heterocyclische fluorglykosidderivate, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2004052902A1 (de) | 2002-12-12 | 2004-06-24 | Aventis Pharma Deutschland Gmbh | Neue aromatische fluorglycosidderivate, diese verbindungen enthaltende arzneimittel und deren verwendung |
Family Cites Families (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2663336B1 (fr) | 1990-06-18 | 1992-09-04 | Adir | Nouveaux derives peptidiques, leur procede de preparation et les compositions pharmaceutiques qui les contiennent. |
| AU658955B2 (en) * | 1992-01-28 | 1995-05-04 | Sumitomo Chemical Company, Limited | An oxazoline derivative, its production and its use |
| AUPO713297A0 (en) * | 1997-06-02 | 1997-06-26 | Fujisawa Pharmaceutical Co., Ltd. | Oxazole compound |
| CO4970713A1 (es) | 1997-09-19 | 2000-11-07 | Sanofi Synthelabo | Derivados de carboxamidotiazoles, su preparacion, composiciones farmaceuticas que los contienen |
| ATE451346T1 (de) * | 1998-03-10 | 2009-12-15 | Ono Pharmaceutical Co | Carbonsäurederivate und medikamente die diese als aktiven wirkstoff enthalten |
| ID29247A (id) * | 1998-07-01 | 2001-08-16 | Takeda Chemical Industries Ltd | Bahan pengatur fungsi reseptor retinoid |
| US6589969B1 (en) * | 1998-10-16 | 2003-07-08 | Ono Pharmaceutical Co., Ltd. | Carboxylic acid derivatives and drugs containing the same as the active ingredient |
| AU4808300A (en) | 1999-04-30 | 2000-11-17 | Neurogen Corporation | 9h-pyrimido(4,5-b)indole derivatives: crf1 specific ligands |
| WO2001017994A1 (en) * | 1999-09-08 | 2001-03-15 | Glaxo Group Limited | Oxazole ppar antagonists |
| US6414002B1 (en) * | 1999-09-22 | 2002-07-02 | Bristol-Myers Squibb Company | Substituted acid derivatives useful as antidiabetic and antiobesity agents and method |
| EP1182251A1 (de) * | 2000-08-11 | 2002-02-27 | Yissum Research Development Company of the Hebrew University of Jerusalem | Verfahren zum Identifizieren von Verbindungen, die die Ubiquitin vermittelte Proteolyse von IkB inhibieren |
| ATE370130T1 (de) * | 2000-12-25 | 2007-09-15 | Ono Pharmaceutical Co | Dihydronaphthalinderivat verbindungen und arzneimittel, die diese verbindungen als wirkstoff enthalten |
| DE10142734A1 (de) * | 2001-08-31 | 2003-03-27 | Aventis Pharma Gmbh | Diarylcycloalkylderivate, Verfahren zu ihrer Herstellung und ihre Verwendung als Arzneimittel |
| DE10225635C1 (de) | 2002-06-07 | 2003-12-24 | Aventis Pharma Gmbh | N-Benzoylureido-Zimtsäurederivate, Verfahren zu deren Herstellung und deren Verwendung |
| US7094795B2 (en) * | 2003-02-27 | 2006-08-22 | Sanofi-Aventis Deutschland Gmbh | Process for preparing the enantiomeric forms of cis-configured 1,3-cyclohexanediol derivatives |
| DE10308355A1 (de) * | 2003-02-27 | 2004-12-23 | Aventis Pharma Deutschland Gmbh | Aryl-cycloalkyl substituierte Alkansäurederivate, Verfahren zu ihrer Herstellung und ihre Anwendung als Arzneimittel |
| US7148246B2 (en) * | 2003-02-27 | 2006-12-12 | Sanofi-Aventis Deutschland Gmbh | Cycloalkyl derivatives having bioisosteric carboxylic acid groups, processes for their preparation and their use as pharmaceuticals |
| DE10308351A1 (de) * | 2003-02-27 | 2004-11-25 | Aventis Pharma Deutschland Gmbh | 1,3-substituierte Cycloalkylderivate mit sauren, meist heterocyclischen Gruppen, Verfahren zu ihrer Herstellung und ihre Verwendung als Arzneimittel |
-
2003
- 2003-02-27 DE DE10308353A patent/DE10308353A1/de not_active Withdrawn
-
2004
- 2004-02-19 PL PL377741A patent/PL377741A1/pl not_active Application Discontinuation
- 2004-02-19 CN CNB2004800054472A patent/CN100439346C/zh not_active Expired - Fee Related
- 2004-02-19 MX MXPA05008993A patent/MXPA05008993A/es active IP Right Grant
- 2004-02-19 EP EP04712502A patent/EP1599454B1/de not_active Expired - Lifetime
- 2004-02-19 KR KR1020057016020A patent/KR20050106053A/ko not_active Application Discontinuation
- 2004-02-19 AU AU2004216520A patent/AU2004216520B2/en not_active Ceased
- 2004-02-19 BR BRPI0407901-9A patent/BRPI0407901A/pt not_active IP Right Cessation
- 2004-02-19 WO PCT/EP2004/001584 patent/WO2004075815A2/de active Application Filing
- 2004-02-19 RU RU2005129999/04A patent/RU2005129999A/ru not_active Application Discontinuation
- 2004-02-19 JP JP2006501890A patent/JP2006519197A/ja active Pending
- 2004-02-19 CA CA002516573A patent/CA2516573A1/en not_active Abandoned
- 2004-02-25 TW TW093104700A patent/TW200505880A/zh unknown
- 2004-02-27 US US10/789,019 patent/US7160911B2/en not_active Expired - Lifetime
- 2004-02-27 AR ARP040100633A patent/AR043430A1/es not_active Application Discontinuation
-
2005
- 2005-07-19 ZA ZA200505763A patent/ZA200505763B/en unknown
- 2005-08-16 IL IL170313A patent/IL170313A/en not_active IP Right Cessation
- 2005-08-26 MA MA28464A patent/MA27741A1/fr unknown
- 2005-08-26 HR HR20050740A patent/HRP20050740A2/xx not_active Application Discontinuation
- 2005-08-26 CO CO05085505A patent/CO5690579A2/es not_active Application Discontinuation
- 2005-09-21 NO NO20054382A patent/NO20054382L/no unknown
Patent Citations (46)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0683773A1 (de) | 1993-02-15 | 1995-11-29 | Wellcome Found | Hypolipidämische, kondensierte 1,4-thiazepine. |
| EP0683774A1 (de) | 1993-02-15 | 1995-11-29 | The Wellcome Foundation Limited | Hypolipemische verbindungen |
| WO1996038428A1 (fr) | 1995-06-02 | 1996-12-05 | Kyorin Pharmaceutical Co., Ltd. | Derives de n-benzyldioxothiazolidylbenzamide et leur procede de production |
| WO1997026265A1 (en) | 1996-01-17 | 1997-07-24 | Novo Nordisk A/S | Fused 1,2,4-thiadiazine and fused 1,4-thiazine derivatives, their preparation and use |
| WO1998008871A1 (en) | 1996-08-30 | 1998-03-05 | Novo Nordisk A/S | Glp-1 derivatives |
| WO1998019998A2 (en) | 1996-11-07 | 1998-05-14 | Novartis Ag | N-substituted 2-cyanopyrrolidines |
| WO1997041097A2 (en) | 1996-12-31 | 1997-11-06 | Dr. Reddy's Research Foundation | Novel heterocyclic compounds process for their preparation and pharmaceutical compositions containing them and their use in the treatment of diabetes and related diseases |
| US6221633B1 (en) | 1997-06-20 | 2001-04-24 | Aventis Pharma Deutschland Gmbh | Insulin derivatives having a rapid onset of action |
| WO1999003861A1 (en) | 1997-07-16 | 1999-01-28 | Novo Nordisk A/S | Fused 1,2,4-thiadiazine derivatives, their preparation and use |
| WO1999061431A1 (de) | 1998-05-28 | 1999-12-02 | Probiodrug Gesellschaft für Arzneimittelforschung mbH | Neue effektoren von dipeptidylpeptidase iv |
| WO1999062871A1 (en) | 1998-06-04 | 1999-12-09 | Astrazeneca Ab | New 3-aryl propionic acid derivatives and analogs |
| WO1999062872A1 (en) | 1998-06-04 | 1999-12-09 | Astrazeneca Ab | New 3-aryl-2-hydroxypropionic acid derivative (i) |
| US6221897B1 (en) | 1998-06-10 | 2001-04-24 | Aventis Pharma Deutschland Gmbh | Benzothiepine 1,1-dioxide derivatives, a process for their preparation, pharmaceuticals comprising these compounds, and their use |
| WO1999067279A1 (de) | 1998-06-24 | 1999-12-29 | Probiodrug Gesellschaft für Arzneimittelforschung mbH | Verbindungen von instabilen dp iv-inhibitoren |
| WO1999067278A1 (de) | 1998-06-24 | 1999-12-29 | Probiodrug Gesellschaft für Arzneimittelforschung mbH | Prodrugs von dp iv-inhibitoren |
| US6245744B1 (en) | 1998-10-02 | 2001-06-12 | Aventis Pharma Deutschland Gmbh | Aryl-substituted propanolamine derivatives, their preparation, pharmaceuticals comprising them, and their use |
| WO2000040569A1 (en) | 1999-01-08 | 2000-07-13 | Alizyme Therapeutics Limited | 2-amino-benzoxazinone derivatives for the treatment of obesity |
| US6277831B1 (en) | 1999-04-09 | 2001-08-21 | Aventis Pharma Deutschland Gmbh | 1,4-benzothiazepine-1,1-dioxide derivatives substituted by sugar residues, process for their preparation, pharmaceuticals comprising these compounds, and their use |
| WO2000063208A1 (en) | 1999-04-16 | 2000-10-26 | Novo Nordisk A/S | Substituted imidazoles, their preparation and use |
| WO2000064888A1 (en) | 1999-04-28 | 2000-11-02 | Aventis Pharma Deutschland Gmbh | Di-aryl acid derivatives as ppar receptor ligands |
| WO2000064876A1 (en) | 1999-04-28 | 2000-11-02 | Aventis Pharma Deutschland Gmbh | Tri-aryl acid derivatives as ppar receptor ligands |
| WO2000071549A1 (en) | 1999-05-21 | 2000-11-30 | Knoll Gmbh | Thiazoloderivatives and pharmaceutical compositions containing them |
| WO2000078312A1 (en) | 1999-06-18 | 2000-12-28 | Merck & Co., Inc. | Arylthiazolidinedione and aryloxazolidinedione derivatives |
| WO2001004146A2 (en) | 1999-07-09 | 2001-01-18 | Cohesion Technologies, Inc. | Ecarin polypeptides, polynucleotides encoding ecarin, and methods for use thereof |
| WO2001009111A1 (en) | 1999-07-29 | 2001-02-08 | Eli Lilly And Company | Benzofurylpiperazines and benzofurylhomopiperazines: serotonin agonists |
| US6342512B1 (en) | 1999-09-01 | 2002-01-29 | Aventis Pharma Deutschland Gmbh | Sulfonylcarboxamide derivatives, process for their preparation and their use as pharmaceuticals |
| WO2001021602A1 (en) | 1999-09-22 | 2001-03-29 | Bristol-Myers Squibb Company | Oxa- and thiazole derivatives useful as antidiabetic and antiobesity agents |
| WO2001040169A1 (en) | 1999-12-03 | 2001-06-07 | Astrazeneca Ab | Comminuted form of (s)-2-ethoxy -3-[4-(2- {4-methanesulfonyloxyphenyl} ethoxy) phenyl] propanoic acid |
| WO2001040171A1 (en) | 1999-12-03 | 2001-06-07 | Astrazeneca Ab | Crystalline form of (s)-2 ethoxy-3-[4-(2-{4-methanesulfonyloxyphenyl} ethoxy) phenyl] propanoic acid |
| WO2001072290A2 (en) | 2000-03-31 | 2001-10-04 | Probiodrug Ag | Method for the improvement of islet signaling in diabetes mellitus and for its prevention |
| WO2001081327A1 (fr) | 2000-04-25 | 2001-11-01 | Kyorin Pharmaceutical Co., Ltd. | Nouveau cristal stable de derive thiazolidinedione et son procede de production |
| WO2001083451A1 (fr) | 2000-04-28 | 2001-11-08 | Asahi Kasei Kabushiki Kaisha | Nouveaux composés bicycliques |
| WO2001085695A1 (en) | 2000-05-11 | 2001-11-15 | Bristol-Myers Squibb Co. | Tetrahydroisoquinoline analogs useful as growth hormone secretagogues |
| WO2001091752A1 (en) | 2000-05-30 | 2001-12-06 | Merck & Co., Inc. | Melanocortin receptor agonists |
| WO2001094300A1 (de) | 2000-06-09 | 2001-12-13 | Aventis Pharma Deutschland Gmbh | Acylphenylharnstoffderivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2002038541A1 (fr) | 2000-11-10 | 2002-05-16 | Taisho Pharmaceutical Co., Ltd. | Derives de cyanopyrrolidine |
| WO2002050027A1 (de) | 2000-12-21 | 2002-06-27 | Aventis Pharma Deutschland Gmbh | Neue diphenzylazetidinone, verfahren zu deren herstellung, diese verbindungen enthaltende arzneimittel und deren verwendung zur behandlung von lipidstoffwechselstörungen |
| WO2002096864A1 (de) | 2001-05-25 | 2002-12-05 | Aventis Pharma Deutschland Gmbh | Carbonsäureamid substituierte phenylharnstoffderivate, verfahren zu ihrer herstellung als arzneimittel |
| WO2003010418A1 (en) | 2001-07-21 | 2003-02-06 | Edwards Thomas C | Single-degree-of-freedom controlled-clearance univanetm fluid-handling machine |
| WO2003020269A1 (de) | 2001-08-31 | 2003-03-13 | Aventis Pharma Deutschland Gmbh | Diarylcycloalkylderivate, verfahren zu ihrer herstellung und ihre verwendung als ppar-aktivatoren |
| WO2003040174A2 (en) | 2001-11-09 | 2003-05-15 | Probiodrug Ag | Substituted amino ketone compounds |
| WO2003084922A1 (de) | 2002-04-11 | 2003-10-16 | Aventis Pharma Deutschland Gmbh | Acyl-4-carboxy-phenyl-harnstoffderivate, verfahren zu deren herstellung und deren verwendung |
| WO2003084923A1 (de) | 2002-04-11 | 2003-10-16 | Aventis Pharma Deutschland Gmbh | Acyl-3-carboxyphenyl-harnstoffderivate, verfahren zu deren herstellung und deren verwendung |
| WO2004007517A1 (de) | 2002-07-11 | 2004-01-22 | Aventis Pharma Deutschland Gmbh | Neue thiophenglycosidderivate, verfahren zu deren herstellung, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2004052903A1 (de) | 2002-12-12 | 2004-06-24 | Aventis Pharma Deutschland Gmbh | Neue heterocyclische fluorglykosidderivate, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2004052902A1 (de) | 2002-12-12 | 2004-06-24 | Aventis Pharma Deutschland Gmbh | Neue aromatische fluorglycosidderivate, diese verbindungen enthaltende arzneimittel und deren verwendung |
Non-Patent Citations (12)
| Title |
|---|
| "Roten Liste", 2001 |
| "USP Dictionary of USAN and International Drug Names", 2003 |
| FRUCHART J-C, STAELS B, DURIEZ P, METABOLOIS DISEASE AND ARTERIOSCLEROSIS, PHARMATOLOGICAL RESEARCH, vol. 44, no. 5, 2001, pages 345 - 352 |
| JOEL BERGER ET AL., ANNU. REV. MED., vol. 53, 2002, pages 409 - 435 |
| KERSTEN S, DESVERGNE B, WAHLI W: "Roles of PPARs in health and disease", NATURE, vol. 405, 25 May 2000 (2000-05-25), pages 421 - 424, XP002336385, DOI: doi:10.1038/35013000 |
| MOTOJIMA K, CELL STRUCT FUNCT, vol. 18, no. 5, October 1993 (1993-10-01), pages 267 - 277 |
| PHARMACEUTICAL RESEARCH, vol. 2, no. 6, 1986, pages 318 |
| PINEDA I, CHINETTI G, DUVAL C, FRUCHART J. C, STAELS B: "Peroxisome proliferator-activated receptors from transcriptional control to clinical practice", CURR OPIN LIPIDOL, vol. 12, 2001, pages 245 - 254 |
| STEVEN KLIEWER ET AL., RECENT PROG HORM RES., vol. 56, 2001, pages 239 - 263 |
| TIMOTHY WILSON ET AL., J. MED. CHEM., vol. 43, no. 4, 2000, pages 527 - 550 |
| VIDAL-PUIG ET AL., J. CLIN. INVEST., vol. 97, 1996, pages 2553 - 2561 |
| ZUNFT H J EZ AL: "Carob pulp preparation for treatment of hypercholesterolemia", ADVANCE IN THERAPY, vol. 18, no. 5, September 2001 (2001-09-01), pages 230 - 236, XP009029721 |
Cited By (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7902367B2 (en) | 2004-08-11 | 2011-03-08 | Kyorin Pharmaceutical Co., Ltd. | Cyclic amino benzoic acid derivative |
| US7803950B2 (en) | 2004-08-23 | 2010-09-28 | Sanofi-Aventis Deutschland Gmbh | Method for the production of diarylcycloalkyl derivatives |
| AU2005276642B2 (en) * | 2004-08-23 | 2011-12-22 | Sanofi-Aventis Deutschland Gmbh | Method for the production of diarylcycloalkyl derivatives |
| WO2006021420A3 (de) * | 2004-08-23 | 2006-12-21 | Sanofi Aventis Deutschland | Verfahren zur herstellung von diarylcycloalkylderivaten |
| WO2006021420A2 (de) * | 2004-08-23 | 2006-03-02 | Sanofi-Aventis Deutschland Gmbh | Verfahren zur herstellung von diarylcycloalkylderivaten |
| US7825144B2 (en) | 2004-12-15 | 2010-11-02 | Sanofi-Aventis Deutschland Gmbh | Method for the production of oxazoles by condensing aromatic aldehydes with α-ketoximes to N-oxides and then reacting the same with activated acid derivatives |
| US7544809B2 (en) | 2004-12-15 | 2009-06-09 | Sanofi-Aventis Deutschland Gmbh | Method for the preparation of oxazoles by condensing aromatic aldehydes with α-ketoximes to form n-oxides and reacting same with activated acid derivatives |
| WO2006066694A1 (de) * | 2004-12-15 | 2006-06-29 | Sanofi-Aventis Deutschland Gmbh | VERFAHREN ZUR HERSTELLUNG VON OXAZOLEN DURCH KONDENSATION VON AROMATISCHEN ALDEHYDEN MIT α-KETOXIMEN ZU N-OXIDEN UND NACHFOLGENDE REAKTION MIT AKTIVIERTEN SÄUREDERIVATEN |
| US8906901B2 (en) | 2005-09-14 | 2014-12-09 | Takeda Pharmaceutical Company Limited | Administration of dipeptidyl peptidase inhibitors |
| US8222411B2 (en) | 2005-09-16 | 2012-07-17 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| WO2007039177A2 (en) | 2005-09-29 | 2007-04-12 | Sanofi-Aventis | Phenyl- and pyridinyl- 1, 2 , 4 - oxadiazolone derivatives, processes for their preparation and their use as pharmaceuticals |
| US7960384B2 (en) | 2006-03-28 | 2011-06-14 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US8324383B2 (en) | 2006-09-13 | 2012-12-04 | Takeda Pharmaceutical Company Limited | Methods of making polymorphs of benzoate salt of 2-[[6-[(3R)-3-amino-1-piperidinyl]-3,4-dihydro-3-methyl-2,4-dioxo-1(2H)-pyrimidinyl]methyl]-benzonitrile |
| US8084605B2 (en) | 2006-11-29 | 2011-12-27 | Kelly Ron C | Polymorphs of succinate salt of 2-[6-(3-amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethy]-4-fluor-benzonitrile and methods of use therefor |
| US8093236B2 (en) | 2007-03-13 | 2012-01-10 | Takeda Pharmaceuticals Company Limited | Weekly administration of dipeptidyl peptidase inhibitors |
Also Published As
| Publication number | Publication date |
|---|---|
| HRP20050740A2 (en) | 2006-08-31 |
| EP1599454B1 (de) | 2012-12-26 |
| RU2005129999A (ru) | 2006-03-27 |
| TW200505880A (en) | 2005-02-16 |
| EP1599454A2 (de) | 2005-11-30 |
| AU2004216520B2 (en) | 2010-01-07 |
| US20040204462A1 (en) | 2004-10-14 |
| CA2516573A1 (en) | 2004-09-10 |
| AU2004216520A1 (en) | 2004-09-10 |
| ZA200505763B (en) | 2006-05-31 |
| WO2004075815A3 (de) | 2004-12-29 |
| CN100439346C (zh) | 2008-12-03 |
| BRPI0407901A (pt) | 2006-02-14 |
| AR043430A1 (es) | 2005-07-27 |
| US7160911B2 (en) | 2007-01-09 |
| CN1753880A (zh) | 2006-03-29 |
| DE10308353A1 (de) | 2004-12-02 |
| JP2006519197A (ja) | 2006-08-24 |
| NO20054382L (no) | 2005-09-21 |
| CO5690579A2 (es) | 2006-10-31 |
| MA27741A1 (fr) | 2006-02-01 |
| PL377741A1 (pl) | 2006-02-20 |
| IL170313A (en) | 2010-11-30 |
| KR20050106053A (ko) | 2005-11-08 |
| MXPA05008993A (es) | 2005-10-18 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1789403B1 (de) | N-(phenyl-oxazol-4-ylmethoxymethyl)-cyclohexyl-bernsteinsäureamid derivate und verwandte verbindungen als ppar-liganden (peroxisomen-proliferatoren-aktivierte rezeptoren) zur behandlung von hyperlipidämie und diabetes | |
| EP1599454B1 (de) | Diarylcycloalkylderivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel | |
| EP1599453B1 (de) | 3-methyl-2- (3- (2-phenyl-oxazol-4-ylmethoxy)-cyclohexancarbonyl-amino)-buttersäurederivate und verwandte verbindungen als ppar modulatoren zur behandlung von typ 2 diabetes und atherosklerose | |
| EP1599203B1 (de) | 1,3-substituierte cycloalkylderivate mit sauren, meist heterocyclischen gruppen; verfahren zu ihrer herstellung und ihre verwendung als arzneimittel | |
| EP1601643B1 (de) | Arylcycloalkylderivate mit verzweigten seitenketten als ppar rezeptor modulatoren, verfahren zu ihrer herstellung und ihre anwendung als arzneimittel | |
| DE102005029382B3 (de) | Alkoxymethylyl-substituierte Benzoesäurederivate, Verfahren zu ihrer Herstellung und ihre Anwendung als Arzneimittel | |
| EP1599443B1 (de) | Cycloalkyl substituierte alkansaürederivate, verfahren zu ihrer herstellung und ihre anwendung als arzneimittel | |
| EP1789402B1 (de) | 2-{-3-¬2-(phenyl)-oxazol-4-ylmethoxymethyl -cyclohexylmethoxy}-propionsäure derivate als ppar-liganden (peroxisomen-proliferatoren-aktivierte rezeptoren) zur behandlung von hyperlipidämie und diabetes | |
| EP1778655B1 (de) | 2-{-3-'2-(phenyl)-oxazol-4-ylmethoxy!-cyclohexylmethoxy}-propionsäure derivate als ppar-liganden (peroxisomen-proliferatoren-aktivierte rezeptoren) zur behandlung von hyperlipidämie und diabetes | |
| EP1601671B1 (de) | Cycloalkylderivate mit bioisosteren carbonsäure - gruppen, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): BW GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DPEN | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed from 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 1-2005-501328 Country of ref document: PH |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005/05763 Country of ref document: ZA Ref document number: 200505763 Country of ref document: ZA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004712502 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 170313 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2516573 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/2005/008993 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 542028 Country of ref document: NZ Ref document number: P-2005/0653 Country of ref document: YU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 377741 Country of ref document: PL Ref document number: 2006501890 Country of ref document: JP Ref document number: 1020057016020 Country of ref document: KR Ref document number: 2004216520 Country of ref document: AU Ref document number: P20050740A Country of ref document: HR Ref document number: 05085505 Country of ref document: CO Ref document number: 2072/CHENP/2005 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 20048054472 Country of ref document: CN |
|
| ENP | Entry into the national phase |
Ref document number: 2004216520 Country of ref document: AU Date of ref document: 20040219 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004216520 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005129999 Country of ref document: RU |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020057016020 Country of ref document: KR |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004712502 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: PI0407901 Country of ref document: BR |