DERIVES DE N-(IMIDAZOLYLBUTYLE) BENZENESULFONAMIDE AYANT UNE ACTIVITE ANTITHROMBOTIQUE
La présente invention a pour objet des dérivés de 5 N- (îmidazolylbutyle) benzènesulfonamide, leur préparation et leur application en thérapeutique.
Les composés de l'invention répondent à la formule (Ii
dans laquelle et R'
x représentent chacun indépendamment l'un de l'autre, 0 soit un atome d'hydrogène, soit un atome d'halogène, soit un groupe (C -C
j) alkyle,
R2 représente soit un groupe pιpérιdm-1-yle éventuellement substitué en position 4 par un ou plusieurs groupes choisis parmi les groupes hydroxy, (C1-C4) alkyle droit ou ramifié, 5 hydroxy (C1-C4) alkyle, (C1-C4) alcoxy (C1-C4) alkyle,
(C1-C ) alcoxy, (C1-C4) alkylthio, nitπle, monofluorométhyle, difluorométhyle, trifluorométhyle, 2-fluoroéthoxy, 2, 2,2-tπfluoroéthoxy, (C3-C6) cycloalkyle, -COOR' et -CONR'R" (R' étant un groupe (C1-C4) alkyle et R" étant un atome 0 d'hydrogène ou un groupe (C1-C4) alkyle) ou par un groupe =CYZ [Y et Z étant choisis indépendamment l'un de l'autre parmi les atomes d'hydrogène et d'halogène et les groupes (C1-C4) alkyle (éventuellement substitué par un ou plusieurs atomes d'halogène), cyano et -COOR', R' étant tel que défini
(r = 1 à 3) ou par
un groupe =NOCH
3 , soit un groupe spiro [ (C
3-C
6) cycloalkane- 1 , 4 ' -pipéridin] -1-yle, soit un groupe 1 , 2 , 3 , 6-tétrahydropy- ridin-1-yle éventuellement substitué en position 4 par un groupe (C
1-C
4) alkyle droit ou ramifié (éventuellement substitué par un ou plusieurs atomes d'halogène) ou
(C3-C6) cycloalkyle, soit un groupe hexahydro-lH-azépin- 1-yle éventuellement substitué en position 4 par un groupe trifluorométhyle ou =CF2, soit un groupe heptahydroazocin-1- yle, soit un groupe octahydro-lH-azonin- 1-yle,
soit un groupe (A-B étant un groupe -CONR" ,
m = l à 2 et p = l à 2 ) , soit un groupe
(Q étant un atome de carbone ou d'azote, D un groupe (C
1-C
4)alkyle ou -CH
2CF
3, et r = 1 à 3), R
3 représente soit un groupe (C^-Cs) alkyle droit ou ramifié, soit un groupe -COR
5 où R
5 est un groupe {C
1 -C
4 ) alkyle droit ou ramifié, -(CH
2)
nOCH
3, -CH
20 (C
2H
40)
nCH
3/ -(CH
2)
nCF
3, -(CH
2)
n0H (n = 1 à 4) , soit un groupe -S0
2R
6, soit un groupe -C0NHR
6, soit un groupe -S0
2N(R
6)
2 où R
6 est un groupe (C
x-C
4) alkyle droit ou ramifié, R
4 représente soit un atome d'hydrogène, soit un atome d'halogène et
A représente soit un groupe phényle éventuellement substitué par un ou plusieurs substituants choisis parmi les atomes d'halogènes et les groupes (C -Cj) alkyle droit ou ramifié, (Cx-C^ alcoxy droit ou ramifié, trifluorométhyle, trifluoro- méthoxy, formyle, -CH2OR10, -CH2OCOR10, -CH2OCONR10R1;L, -COOR10, -CONR10R1:I, nitro, -NR10R1:L, -NHCOR10, et -NH (CH2) qOR10 avec R10 et Rl étant chacun indépendamment l'un de l'autre un atome d'hydrogène ou un groupe (C1-C4) alkyle droit ou ramifié et q compris entre 0 et 6, soit un hétérocycle choisi parmi les groupes pyridinyle, thiényle, furyle, pyrimidyle et thiazolyle les dits groupes pouvant être substitués comme ci-dessus, soit un groupe cyclo (C5-C8) alkyle .
Selon l'invention les composés préférés sont les composés de formule (I) dans laquelle
Rx et R'i représentent chacun indépendamment l'un de l'autre soit un atome d'hydrogène, soit un atome d'halogène, soit un groupe (C1-C4) alkyle,
R2 représente soit un groupe pipéridin- 1-yle éventuellement substitué en position 4 par un ou plusieurs groupes choisis parmi les groupes (Cτ_-C4) alkyle droit ou ramifié, hydroxy (C1-C4) alkyle, (Cx-C4) alcoxy (C-L-Cς) alkyle, (C1-C4) alcoxy, (C1-C4) alkylthio, nitrile, difluorométhyle, trifluorométhyle, 2 , 2 , 2-trifluoroethoxy et (C3-C6) cycloalkyle ou par un groupe =CYZ (Y et Z étant choisis indépendamment l'un de l'autre parmi les atomes d'hydrogène et d'halogène et les groupes (C1-C4) alkyle) , ou par un groupe =NOCH3 , soit un groupe spiro [ (C3-C6) cycloalkane-1 , 4 ' -pipéridin] -1-yle, soit un groupe 1 , 2 , 3 , 6-tétrahydropyridin-l-yle éventuellement substitué en position 4 par un groupe (C1-C4) alkyle droit ou ramifié (éventuellement substitué par un ou plusieurs atomes d'halogène), soit un groupe hexahydro-lH-azépin- 1-yle éventuellement substitué en position 4 par un groupe =CF2 , soit un groupe octahydro-lH-azonin-1-yle, soit un groupe
(A-B étant un groupe -CONR" , m = 1 à 2 et
D \ p = 1 à 2) , soit un groupe N- (Q étant un
(CH2)r
atome d'azote ou de carbone, D un groupe (C1-C ) alkyle ou -CH2CF3, et r = 1 à 3) ,
R3 représente soit un groupe (C^-C^) alkyle droit ou ramifié, soit un groupe -COR5 où R5 est un groupe (Cx-C4) alkyle droit ou ramifié, -CH20 (C2H40) nCH3 , (CH2)n0H, (CH2)nOCH3, soit un groupe -C0NHR6 et
A représente soit un groupe phényle éventuellement substitué par un ou plusieurs substituants choisis parmi les atomes d'halogènes et les groupes (C1-C4) alkyle droit ou ramifié, (C1-C4) alcoxy droit ou ramifié, trifluorométhyle,
trifluorométhoxy et -NR10Rι;L avec R10 et Rλ l étant chacun indépendamment l'un de l'autre un atome d'hydrogène ou un groupe (C1-C4) alkyle droit ou ramifié, soit un groupe pyπdinyle ou thiényle pouvant être substitués comme ci -dessus, soit un groupe cyclo (C5-C8) alkyle .
Parmi ces composés, les composés de choix sont ceux pour lesquels R représente un groupe (CT-C^ alkyle, et R' un atome d'hydrogène, R2 représente un groupe pιpérιdm-1-yle substitué en position 4 par un groupe (C1-C4) alkyle droit ou ramifié ou par un groupe =CF2, R3 représente un groupe -COR5 où R5 est un groupe (C-L-C^ alkyle droit ou ramifié et A représente un groupe thiényle éventuellement substitué comme ci -dessus ou un groupe cyclo (C5-C8) alkyle .
La configuration préférentielle de la partie acide aminé centrale
est [ S]
Les composés de l'invention peuvent exister sous forme de racémates ou d ' énantiomères purs ou de mélange d'énantio- mères . Ils peuvent également exister sous forme d'acide ou de base libres ou de sels d'addition pharmaceutiquement acceptables . Toutes ces formes font partie de l'invention.
Dans les schémas qui suivent, le groupe -CPh3 représente le groupe tπphénylméthyle .
Selon l'invention les composés de formule (la) dans laquelle R3 représente un groupe -COR5 où R5 est un groupe (C1-C4) alkyle droit ou ramifié ou un groupe -(CH2)nCF3 (n égal 1 à 4), peuvent être synthétisés selon le schéma 1. On fait réagir un
Schéma 1
CPh,
CPh,
composé de formule (II) dans laquelle R
τ et R '
λ représentent chacun soit un atome d'hydrogène, soit un groupe (C
1-C
4) alkyle et R
2 est tel que défini précédemment avec un composé de formule (III) dans laquelle A représente soit un groupe phényle éventuellement substitué comme ci -dessus, soit un hétérocycle choisi parmi les groupes pyridmyle, thiényle, furyle, pyπmidyle et thiazolyle, les dits groupes pouvant être substitués comme ci -dessus et R
4 et R
5 sont tels que définis précédemment, dans un solvant aprotique tel que le dichloromethane en présence d'une base comme la triethylamme et on obtient un composé de formule (IV) que l'on traite par un mélange acide acétique : éthanol , acide acétique: eau ou acide acétique : tétrahydrofurane : eau à la température de reflux.
Dans une variante de l'invention illustrée par le schéma 2, pour préparer les composés de formule (la) dans laquelle R5 est un groupe (C1-C4) alkyle droit ou ramifié, -(CH2)nOCH3, -CH20(C2H40)nCH3/ -(CH2)nCF3 ou -(CH2)nOH (n égal 1 à 4), on peut faire réagir un composé de formule (II) avec un composé de formule (V) dans laquelle A représente soit un groupe phényle substitué comme ci -dessus, soit un hétérocycle choisi parmi les groupes pyridmyle, thiényle, furyle, pynmidyle et thiazolyle, les dits groupes pouvant être substitués comme ci-dessus, soit un groupe cyclo (C5-C8) alkyle, et R4 et R5 sont tels que définis précédemment, dans un solvant aprotique tel que le dichloromethane en présence d'une base comme la triethylamme et on obtient un composé de formule (VI) que l'on traite par un mélange acide acétique : éthanol , acide acétique: eau ou acide acétique : tétrahydrofurane : eau à la température de reflux.
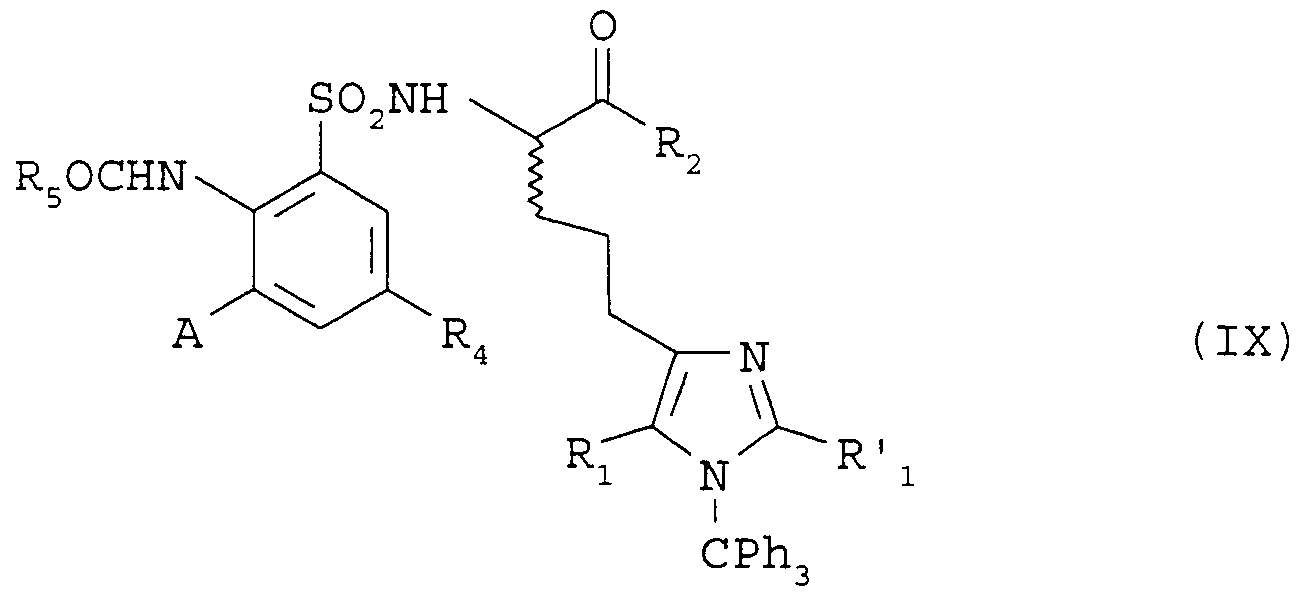
Dans une variante selon 1 ' invention on peut également utiliser le procédé illustré dans le schéma 3. On fait réagir un composé de formule (VII) dans laquelle Rλ et R'i représentent chacun soit un atome d'hydrogène, soit un groupe (C1-C4) alkyle et R2 , R4 et R5 sont tels que définis précédemment avec un composé de formule (VIII) dans laquelle R représente un groupe (Ci-C^ alkyle et A est tel que défini
Schéma 2
(la!
ASn(R) (VIII)
lia)
précédemment, dans un solvant comme le diméthylformamide en présence d'un catalyseur comme le tétrakis (triphénylphosphi- ne) palladium (0) pour former un composé de formule (IX) que l'on chauffe à la température de reflux en milieu acide, par exemple dans un mélange acide acétique : eau.
Si l'on souhaite obtenir un composé de formule (la) dans laquelle R4 est un atome d'hydrogène, alors on peut soumettre le composé de formule (la) correspondant dans laquelle R4 est un atome d'halogène à une hydrogénolyse . Lorsqu'on désire obtenir un composé de formule (la) dans laquelle R± et/ou R' représentent un atome d'halogène, alors on traite le composé de formule (la) correspondant dans laquelle Rλ et/ou R'x représentent un atome d'hydrogène par un agent d ' halogénation comme par exemple le N-bromosuccinimide ou le N- chlorosucci - mmide dans un solvant tel que le diméthyl ormamide .
Alternativement, pour préparer les composés de formule (Ib) dans laquelle R3 représente soit un groupe -C0R5j soit un groupe -S02R6, soit un groupe -C0ΝHR6, soit un groupe -S02N(R6)2, où R5 est un groupe (C1-C4) alkyle droit ou ramifié, -(CH2)nOCH3, -CH20 (C2H40) nCH3; -(CH2)nCF3, -(CH2)n0P, P groupe protecteur (n égal 1 à 4) et R6 est tel que défini précédemment, on utilise le procédé illustré dans le schéma 4.
On fait réagir un composé de formule (II) avec un composé de formule (X) dans laquelle A et R4 sont tels que définis précédemment et on obtient un composé de formule (XI) que l'on fait réagir avec un chlorure d'acide de formule R5C0C1 ou un alkylisocyanate de formule R6NC0 ou un chlorure de sulfonyle de formule R6S02C1 ou un chlorure de sulfamoyle de formule (R6)2NS02C1 et on obtient un composé de formule (XII) que l'on traite par un mélange acide acétique : éthanol , acide acétique: eau ou acide acétique : tétrahydrofurane : eau à la température de reflux.
Lorsqu'on désire obtenir un composé de formule (Ib) dans laquelle R± et/ou R' représentent un atome d'halogène, alors
Schéma 4
(Ib)
on traite le composé de formule (Ib) correspondant dans laquelle R
λ et/ou R '
λ représentent un atome d'hydrogène par un agent d ' halogénation comme par exemple le N-bromosuccini - mide ou le N-chlorosuccinimide dans un solvant tel que le diméthylformamide .
Si l'on souhaite obtenir un composé de formule (Ib) dans laquelle R4 est un atome d'hydrogène, alors on soumet le composé de formule (Ib) correspondant dans laquelle R4 est un atome d'halogène à une hydrogénolyse .
Pour préparer les composés de formule (le) dans laquelle R12 correspond à R3 quand R3 représente un groupe (C -Cs) alkyle droit ou ramifié, on utilise le procédé illustré dans le schéma 5. On fait réagir un composé de formule (II) avec un composé de formule (XIV) dans laquelle R12 représente un groupe (CIL-CS) alkyle droit ou ramifié et A et R4 sont tels que définis précédemment et on obtient un composé de formule (XV) que l'on traite par un mélange acide acétique : éthanol , acide acétique: eau ou acide acétique : tétrahydrofurane : eau à la température de reflux.
Lorsqu'on désire obtenir un composé de formule (le) dans laquelle Rτ et/ou R ' ± représentent un atome d'halogène, alors on traite le composé de formule (le) correspondant dans laquelle Rx et/ou R ' λ représentent un atome d'hydrogène par un agent d' halogénation comme par exemple le N-bromosuccini - mide ou le N-chlorosuccinimide dans un solvant tel que le diméthylformamide .
Si l'on souhaite obtenir un composé de formule (le) dans laquelle R4 est un atome d'hydrogène, alors on soumet le composé de formule (le) correspondant dans laquelle R4 est un atome d ' halogène à une hydrogénolyse .
Schéma 5
Les composés de départ sont disponibles dans le commerce ou décrits dans la littérature ou peuvent être préparés selon des méthodes qui y sont décrites ou qui sont connues de l'homme du métier.
Ainsi les composés de formule (II) sont préparés selon une méthode analogue à celle décrite dans la demande de brevet européen EP 0643047.
Certains composés de formule (III) sont décrits dans la demande de brevet européen EP 0718307.
Les composés de formule (VII) sont préparés selon une méthode analogue à celle décrite dans la demande de brevet européen
EP 0718307.
Les composés de formule (X) et (XIV) sont décrits dans la demande de brevet européen EP 0713865.
La préparation du 5-éthyl-lH-ιmιdazole est réalisée selon la méthode décrite par Horne D. A., (1994), Heterocycles, 39 , n°l, 139.
La préparation de la 4-cyclopropylpyrιdme est décrite par Eisch J. J., (1974), J. Org . Chem. 3_9, n° 21, 5110.
La 4-dιflurométhylènepipéπdine est préparée selon une méthode analogue à celle décrite par Sch idt . et coll.,
(1995), Liebigs Ann., 1319-1326.
La préparation du N- cyclopentylformamide est décrite par Bossio R. et coll., (1993), Synthesis, 8., 783-785.
Les exemples 1 à 11 qui suivent illustrent la préparation de quelques composés de formule (II) ; les exemples 12 à 27 illustrent la préparation de quelques composés de formule (I) selon l'invention.
Les microanalyses et les spectres IR et RMΝ confirment la structure des composés obtenus.
Les numéros des composés exemplifiés renvoient à ceux du tableau donné plus loin qui illustre les structures chimiques et les propriétés physiques de quelques composés selon 1 ' invention.
Les rapports entre parenthèses représentent le rapport (acide :base) .
Exemple 1 chlorhydrate de { S) -5-éthyl-α- [ (4-éthylpιpéπdm-l-yl , carbonyl] -1- (triphénylméthyl) -lH-ιmιdazole-4-butanamme (1:1)
1.1. 5-éthyl-4-ιodo-lH-ιmιdazole
A une solution de 8,6 g (89 mmoles) de 5-éthyl-lH-ιmιdazole dans 600 ml d'une solution aqueuse de soude 2 N sous agitation, on ajoute goutte à goutte à 0 °C en 3 heures 22,7 g (89 mmoles) d'iode en solution dans 800 ml de chloroforme. On poursuit l'agitation pendant 4 heures à cette température puis on évapore le chloroforme sous pression réduite. On refroidit la phase aqueuse à 0 °C, on neutralise à l'aide d'une solution aqueuse d'acide chlorhydπque 12 N et on extrait par 3 fois 1 1 d'acétate d'éthyle. On rassemble les phases organiques, on les lave avec 100 ml d'une solution saturée en chlorure de sodium, on sèche sur sulfate de sodium et on concentre sous pression réduite. On purifie le résidu obtenu par chromatographie sur colonne de gel de silice en éluant par un mélange méthanol : dichloromethane (1,5:98,5). On obtient 10,5 g de produit sous forme de poudre blanche. Rendement = 53 % Point de fusion = 155 °C
1.2. 5-éthyl-4-ιodo-l- f (4-méthylphényl) sulfonyl] -1H- îmidazole
A une solution de 4,8 g (21,6 mmoles) de 5-éthyl -4-ιodo-lH- îmidazole dans 25 ml de diméthylformamide anhydre sous agitation, on ajoute par petites quantités à 0 °C sous azote 0,91 g (22,7 mmoles) d ' hydrure de sodium à 60 % dans l'huile. On poursuit l'agitation pendant 0,5 heure à 0 °C et on ajoute 4,35 g (22,7 mmoles) de chlorure de 4- (méthylphényl) sulfo- nyle. On maintient l'agitation pendant une heure à 0 °C, on laisse remonter la température du mélange à la température ambiante, on poursuit l'agitation pendant une heure puis on concentre sous pression réduite. On reprend le résidu par 400 ml d'acétate d'éthyle et on lave successivement par 100 ml d'une solution aqueuse d'acide chlorhydrique 0,5 N, 100 ml d'eau et 100 ml d'une solution saturée en chlorure de sodium. Finalement on sèche sur sulfate de sodium et on
concentre sous pression réduite.
On obtient 6,1 g de produit sous forme d'un solide blanc après précipitation dans un mélange acétate d ' éthyle :pentane Rendement = 75 % Point de fusion = 95 °C
1.3. (S) -2- [ [ (1, 1-diméthyléthoxy) carbonyl] amino] -5- [5-éthyl- 1- [ (4-méthylphényl) sulfonyl] -lH-imidazol-4-yl] pent-4- ynoate de méthyle 1.3.1. { S) -2- [[ (1 , 1-diméthyléthoxy) carbonyl] amino] pent-4- ynoate de méthyle a) acide (S) -2 -[[( 1 , 1-diméthyléthoxy) carbonyl] amino] pent-4 - ynoïque
Dans un ballon de 250 ml sous atmosphère d'azote, on introduit 11,5 g (77 mmoles) de chlorhydrate d'acide
( S) -2-aminopent-4-ynoïque, 100 ml de dioxane, 50 ml d'e'au et 80 ml de soude 2 N. A cette solution on ajoute 17,9 g (82 mmoles) de tert-butyldicarbonate et on agite pendant 3 heures à la température ambiante. On ajoute 200 ml d'acétate d'éthyle et on acidifie à pH 2 par addition d'une solution d'acide chlorhydrique 2 N. On sépare les phases et on extrait la phase aqueuse par 50 ml d'acétate d'éthyle. On sèche sur sulfate de magnésium et on évapore à sec. On obtient 18,78 g de produit sous forme d'une huile incolore que l'on utilise telle quelle dans l'étape suivante.
b) (S) -2- [ [ (1, 1-diméthyléthoxy) carbonyl] amino] pent-4-ynoate de méthyle
Dans un ballon de 250 ml, sous atmosphère d'azote, on ajoute 13 g (154 mmoles) d' hydrogenocarbonate de sodium à une solution de 18,78 g (77 mmoles) d'acide (S) -2- [ [ (1, 1-diméthylé- thoxy) carbonyl] aminopent-4 -ynoïque dans 150 ml de diméthyl- formamide. On ajoute 20 ml (318 mmoles) d' iodure de méthyle et on agite le mélange pendant 18 heures à la température ambiante. On verse le mélange sur de l'eau et on extrait à l'acétate d'éthyle. On lave la phase organique à l'eau puis on la sèche sur sulfate de magnésium. On évapore à sec. On obtient 15,85 g de produit sous forme d'une huile jaune que l'on utilise telle quelle dans l'étape suivante.
1.3.2. (S) -2- [ [ (1 , 1-dιméthyléthoxy) carbonyl] amino] -5- [5- éthyl-1- [ (4 -méthylphényl ) sulfonyl] -lH-ιmιdazol-4 - yl] pent-4-ynoate de méthyle On chauffe pendant 8 heures à 50 °C sous argon un mélange de 9,87 g (26,3 mmoles) de 5-éthyl-4-ιodo-l- [ (4 -méthylphényl) sulfonyl] -lH-imidazole, 8,94 g (39,4 mmoles) de (S) -2- [ [ (1, 1- diméthyléthoxy) carbonyl] amino] pent-4 -ynoate de méthyle, 0,25 g (1,3 mmoles) de îodure de cuivre, 10,84 ml (105 mmoles) de diéthylamme et 0,92 g (1,3 mmoles) de dichlorobis (triphénylphosphme) palladium dans 26 ml de diméthylformamide . On concentre le milieu réactionnel sous pression réduite, on reprend le résidu obtenu dans 300 ml d'acétate d'éthyle et on lave successivement par 3 fois 100 ml d'eau et 100 ml d'une solution saturée en chlorure de sodium. Finalement on sèche sur sulfate de sodium et on concentre sous pression réduite. On purifie le résidu par chromatographie sur colonne de gel de silice en éluant par un mélange acétate d ' éthyle :hexane (3:7). On obtient 11 g de produit sous forme d'une huile visqueuse. Rendement = 88 %
1.4. (S) -2- [ [ (1, 1-dιméthyléthoxy) carbonyl] amino] -5- [5-éthyl- 1- [ (4 -méthylphényl) sulfonyl] -lH-ιmιdazol-4 -yl] pentanoa- te de méthyle On hydrogène pendant 10 heures à la température ambiante sous une pression de 0,35 MPa (50 psi) un mélange de 13,5 g (28,4 mmoles) de (S) -2- [[ (1 , 1-dιméthyléthoxy) carbonyl] amino] - 5- [5-éthyl-l- [ (4 -méthylphényl) sulfonyl] -lH-ιmιdazol-4-yl] pent -4 -ynoate de méthyle en présence de 1,8 g de palladium sur charbon à 10 % dans 50 ml de méthanol . On filtre le milieu réactionnel sur célite et on concentre le filtrat sous pression réduite. On purifie le résidu par chromatographie sur colonne de gel de silice en éluant par un mélange cyclohexane: acétate d'éthyle (7:3). On obtient 10,5 g de produit sous forme d'une huile visqueuse . Rendement = 77 %
1.5. (S) -2- [ [ (1, 1-diméthyléthoxy) carbonyl] amino] -5- (5-éthyl-
1H- imidazol -4 -yl) pentanoate de méthyle On agite à la température ambiante pendant 4 heures un mélange de 10,4 g (21,6 mmoles) de { S) -2 - [ [ (1 , 1-diméthyléthoxy) carbonyl] amino] -5- [5-éthyl-l- [ (4 -méthylphényl) sulfonyl] - 1H- imidazol -4 -yl] pentanoate de méthyle et 8,79 g (65,2 mmoles) d'hydrate de 1-hydroxybenzotriazole dans 150 ml de méthanol . On concentre le milieu réactionnel sous pression réduite, on reprend le résidu par 100 ml d'éther et on lave avec 450 ml d'une solution aqueuse d'acide ehlorhydrique 0,7 N. On sépare les phases, on ajuste le pH de la phase aqueuse à 8-9 avec une solution d1 hydrogenocarbonate de sodium et on extrait par 2 fois 500 ml d'acétate d'éthyle. On rassemble les phases organiques, on sèche sur sulfate de sodium et on concentre sous pression réduite.
On obtient 8,79 g de composé sous forme d'une huile visqueuse utilisée telle quelle dans l'étape suivante. Rendement = 88 %
1.6. ( S) -2 - [[ (1, 1-diméthyléthoxy) carbonyl] amino] -5- [5-éthyl- 1- (triphénylméthyl) -lH-imidazol-4 -yl] pentanoate de méthyle A une solution de 5,95 g (18,3 mmoles) de (S) -2 - [ [ (1 , 1 - diméthyléthoxy) carbonyl] amino] -5- (5-éthyl-lH- imidazol -4 - yl) pentanoate de méthyle dans 70 ml de dichloromethane, on ajoute successivement à 0 °C 2,9 ml (20,3 mmoles) de triéthylamine et 5,77 g (20,7 mmoles) de chlorure de triphénylméthyle . On laisse la température du mélange revenir à la température ambiante, on poursuit l'agitation pendant 18 heures à cette température puis on concentre sous pression réduite. On reprend le résidu par 300 ml d'acétate d'éthyle et on lave successivement par 200 ml d'une solution aqueuse d'acide ehlorhydrique 0,1 N, 200 ml d'une solution saturée d ' hydrogenocarbonate de sodium et 100 ml d'une solution saturée de chlorure de sodium. On sèche sur sulfate de magnésium et on concentre sous pression réduite. On purifie le résidu obtenu par chromatographie sur colonne de gel de silice en éluant par un mélange dichloromethane : méthanol (99:1) .
On obtient 9,4 g de produit sous forme d'une huile visqueuse. Rendement = 90,6 %
1.7. acide { S) -a- [[ (1, 1-diméthyléthoxy) carbonyl] amino] -5- éthyl-1- (triphénylméthyl) -lH-imidazole-4-pentanoïque A 9,4 g (16,6 mmoles) de (S) -2- [[ (1 , 1-diméthyléthoxy) carbonyl] amino] -5- [5-éthyl-l- (triphénylméthyl) -lH-imidazol-4 -yl] pentanoate de méthyle dans un mélange de 48 ml de méthanol et 16 ml d'eau sous agitation à 0 °C, on ajoute 0,83 g (19,8 mmoles) d ' hydroxyde de lithium monohydraté. On laisse la température du mélange revenir à la température ambiante et on poursuit l'agitation à cette température pendant 24 heures. On évapore sous pression réduite et on acidifie la phase aqueuse à pH 2 à 0 °C avec une solution aqueuse d'acide ehlorhydrique 1 N avant d'extraire par 2 fois 300 ml de dichloromethane. On rassemble les phases organiques, on les lave par 100 ml d'une solution saturée de chlorure de sodium, on sèche sur sulfate de magnésium et on concentre sous pression réduite. On triture le résidu dans l'éther, on filtre et on sèche sous pression réduite.
On obtient 8,87 g de produit sous forme d'une poudre blanche. Rendement = 96,7 % Point de fusion = 141 °C
1.8. { S) - [1- [ (4-éthylpipéridin-l-yl) carbonyl] -4- [5-éthyl-l- (triphénylméthyl) -lH-imidazol-4-yl] butyl] carbamate de 1, 1-diméthyléthyle 1.8.1. chlorhydrate de 4-éthylpipéridine a) 4-éthylpipéridine-l-carboxylate de 1 , 1-diméthyléthyle On hydrogène pendant 4 heures à 50 °C, sous une atmosphère de 0,42 Pa (60 psi), 20 g (190 mmoles) de 4-éthénylpyridine en présence de 2 g d'oxyde de platine (IV), on filtre le milieu réactionnel sur célite et on concentre le filtrat sous pression réduite. On reprend le résidu dans 150 ml d'eau, on ajuste le pH à 8 avec une solution saturée aqueuse de carbonate de sodium et on ajoute goutte à goutte 44 g (190 mmoles) de dicarbonate de bis- (1 , 1-diméthyléthyle) en solution dans 100 ml de tétrahydrofurane. On laisse la température du milieu réactionnel revenir à la température
ambiante et on maintient l'agitation pendant 18 heures à cette température. On évapore sous pression réduite et on extrait la phase aqueuse par 2 fois 300 ml d'acétate d'éthyle. On rassemble les phases organiques et on lave par 100 ml d'une solution saturée de chlorure de sodium. On sèche sur sulfate de sodium et on concentre sous pression réduite. On purifie le résidu ainsi obtenu par chromatographie sur colonne de gel de silice en éluant par un mélange cyclohexane : acétate d'éthyle (9:1). On obtient 13,8 g de produit sous forme d'une huile. Rendement = 34 %
b) chlorhydrate de 4-éthylpιpérιdme
On traite par un courant de gaz ehlorhydrique pendant 1 heure à 0 °C une solution de 13,8 g (64,8 mmoles) de 4-éthylpιpérι- dme-1-carboxylate de 1 , 1-diméthyléthyle dans 200 ml d'éther On laisse revenir la température du mélange à la température ambiante, on poursuit l'agitation à cette température pendant 18 heures et on concentre sous pression réduite. On triture le résidu ainsi obtenu dans l'éther, on filtre et on sèche sous pression réduite.
On obtient 6,62 g de produit sous forme d'une poudre blanche que l'on utilise telle quelle dans l'étape suivante. Rendement = 70 % Point de fusion = 138 °C
1.8.2. (s - [1- [ (4 -éthylpιpérιdm-1-yl) carbonyl] -4- [5-éthyl- 1- (triphénylméthyl) -lH-ιmιdazol-4-yl] butyl] carbamate de 1 , 1-diméthyléthyle A un mélange de 0,6 g (1,08 mmoles) d'acide (S) -α- [ [ (1 , 1- diméthyléthoxy) carbonyl] amino] -5-éthyl-l- (triphénylméthyl) - lH-ιmιdazole-4-pentanoïque et 0,18 g (1,2 mmoles) de chlorhydrate de 4-éthylpιpérιdme dans 8 ml de dichloromethane, on ajoute successivement à 0 °C sous azote et en agitant, 0,37 g (2,9 mmoles) de dusopropyléthylamme et
0,46 g (1,2 mmoles) d ' hexafluorophosphate de [ (benzotriazol- 1-yl) oxy] tris (diméthylammo) phosphonium. On laisse la température du mélange revenir à la température ambiante, on poursuit l'agitation à cette température pendant 18 heures et
on concentre le milieu réactionnel sous pression réduite. On reprend le résidu par 100 ml d'acétate d'éthyle, on lave successivement par 80 ml d'une solution aqueuse d'acide ehlorhydrique 1 N, 50 ml d'une solution saturée d'hydrogé- nocarbonate de sodium, 50 ml d'une solution saturée de chlorure de sodium, on sèche sur sulfate de magnésium et on concentre sous pression réduite. On purifie le résidu par chromatographie sur colonne de gel de silice en éluant par un mélange méthanol : dichloromethane (2:98). On obtient 0,7 g de produit sous forme d'une huile visqueuse. Rendement = 98 %
1.9. chlorhydrate de (S) -5-éthyl- - [ (4 -éthylpipéπdin-1- yl) carbonyl] -1- (triphénylméthyl ) -lH-ιmidazole-4 - butanamine (1:1)
On traite par un courant de gaz ehlorhydrique pendant 15 minutes à 0 °C une solution de 0,7 g (1,07 mmoles) de { S) - [1- [ (4-éthylpipéridin-l-yl) carbonyl] -4- [5-éthyl-l- (triphénylméthyl) -lH-imidazol-4-yl] butyl] carbamate de 1 , 1-diméthyléthyle dans 50 ml de benzène. On laisse revenir la température du mélange à la température ambiante, on poursuit l'agitation à cette température pendant 1,5 heures et on concentre sous pression réduite. On triture le résidu ainsi obtenu dans l'éther, on filtre et on sèche sous pression réduite.
On obtient 0,61 g de produit sous forme d'une poudre blanche que l'on utilise telle quelle dans l'étape suivante. Rendement = 97 %
Exemple 2 chlorhydrate de (S) -5-méthyl-α- [ [4 - (trifluorométhyl) pipéridin- 1-yl] carbonyl] -1- (triphénylméthyl) - lH-ιmιdazole-4 - butanamme (1:1)
2.1. { S) - [4- [5-méthyl-l- (triphénylméthyl) -lH-ιmιdazol-4 -yl] -
1- [ [4- (trifluorométhyl) pipéridin- 1-yl] carbonyl] butyl] carbamate de 1 , 1-diméthyléthyle
2.2.1. acide (S) -α- [[ (1 , 1-dιméthyléthoxy) carbonyl] amino] -5- méthyl-1- (triphénylméthyl) -lH-ιmιdazole-4-pentanoïque
On le prépare selon la méthode décrite à l'exemple 1.7 à partir du (S) -2- [ [ (1 , 1-dιméthyléthoxy) carbonyl] amino] -5- [5- méthyl-1- (triphénylméthyl) -lH-ιmιdazol-4 -yl] pentanoate de méthyle .
2.2.2. { S) - [4- [5-méthyl-l- (triphénylméthyl) -lH-ιmιdazol-4- yl] -1- [ [4- (trifluorométhyl ) pipéridin- 1-yl] carbonyl] butyl] carbamate de 1 , 1-diméthyléthyle
A un mélange de 1,08 g (2 mmoles) d'acide { S) -α- [ [ (1 , 1- diméthyléthoxy) carbonyl] ammo] -5-méthyl-l- (triphénylméthyl) - lH-ιmιdazole-4-pentanoïque, 0,306 g (2 mmoles) de 4- (trifluorométhyl) pipéπdme et 1,04 ml (6 mmoles) de dusopropyléthylamme dans 25 ml de dichloromethane, on ajoute par petites quantités à 0 °C sous argon et en agitant 0,834 g (2,2 mmoles) d' hexafluorophosphate de [ (benzotπazol- 1-yl) oxy] tris (diméthylam o) phosphonium. On laisse la température du mélange revenir à la température ambiante, on poursuit l'agitation à cette température pendant 18 heures et on concentre le milieu réactionnel sous pression réduite. On reprend le résidu par 100 ml d'acétate d'éthyle, on lave successivement par 50 ml d'une solution aqueuse d'acide ehlorhydrique I N, 50 ml d'une solution saturée d' hydrogenocarbonate de sodium, 50 ml d'une solution saturée de chlorure de sodium, on sèche sur sulfate de sodium et on concentre sous pression réduite. On purifie le résidu par chromatographie sur colonne de gel de silice en éluant par un mélange méthanol : dichloromethane (2:98).
On obtient 1,2 g de produit sous forme d'une huile visqueuse. Rendement = 89 %
2.2. chlorhydrate de (S) -5-méthyl-α- [ [4 - (trifluorométhyl ) pipéridin-1-yl] carbonyl] -1- (triphénylméthyl) - 1H- imidazole-4-butanamine (1:1) On traite par un courant de gaz ehlorhydrique pendant 20 minutes à 0 °C une solution de 1,2 g (1,78 mmoles) de { S) - [4- [5-méthyl-l- (triphénylméthyl) -lH-imidazol-4-yl] -1- [ [4- (trifluorométhyl) pipéridin-1-yl] carbonyl] butyl] carbamate de 1, 1-diméthyléthyle dans 100 ml de benzène. On laisse sous agitation à cette température pendant 1 heure et on concentre sous pression réduite. On triture le résidu ainsi obtenu dans l'éther, on filtre et on sèche sous pression réduite. On obtient 1,05 g de produit sous forme d'une poudre blanche que l'on utilise telle quelle dans l'étape suivante. Rendement = 97 % Point de fusion = 78 °C
Exemple 3 chlorhydrate de { S) -α- [ (4 -méthoxypipéridin- l -yl ) carbonyl ] - 5 - méthyl - 1 - ( triphénylméthyl ) - lH-imidazole - 4 -bu anamine ( 1 : 1 )
3.1. chlorhydrate de 4-méthoxypipéridine
3.1.1. 4-hydroxypipéridine-l-carboxylate de 1 , 1-diméthyléthyle A une solution de 5,06 g (50 mmoles) de pipéridin-4-ol dans 50 ml de méthanol, on ajoute goutte à goutte à la température ambiante 12 g (55 mmoles) de dicarbonate de bis- (1 , 1-diméthyléthyle) en solution dans 50 ml de méthanol. On laisse le milieu réactionnel sous agitation à cette température pendant 2 heures et on concentre sous pression réduite. On purifie le résidu ainsi obtenu par chromatographie sur colonne de gel de silice en éluant par un mélange dichloromethane : méthanol (95:5) .
On obtient 9,74 g de produit sous forme d'une huile. Rendement = 97 %
3.1.2. 4-méthoxypipéridine-l-carboxylate de 1 , 1-diméthyléthyle A un mélange de 8 g (39,8 mmoles) de 4-hydroxypipéridine-l- carboxylate de 1 , 1-diméthyléthyle et de 4,95 ml (79,5 mmoles]
de îodométhane en solution dans 40 ml de diméthylformamide, on ajoute par petites portions à 0 °C sous argon et sous agitation 1,59 g (39,8 mmoles) d ' hydrure de sodium à 60 % dans l'huile et on poursuit l'agitation à cette température pendant 2 heures. On verse le milieu réactionnel sur 100 ml d'une solution saturée de chlorure d'ammonium et on extrait par 2 fois 200 ml d'acétate d'éthyle. On rassemble les phases organiques, on lave successivement par 100 ml d'eau et 100 ml d'une solution saturée de chlorure de sodium, on sèche sur sulfate de magnésium et on concentre sous pression réduite. On purifie le résidu par chromatographie sur colonne de gel de silice en éluant par un mélange acétate d ' éthyle : cyclohe- xane (2:8) . On obtient 7,1 g de produit sous forme d'une huile. Rendement = 85 %
3.1.3. chlorhydrate de 4 -méthoxypipéridme
On traite par un courant d'acide ehlorhydrique gazeux pendant
30 minutes à 0 °C une solution de 7 g (32,5 mmoles) de 4 -méthoxypipéridme- 1-carboxylate de 1 , 1-diméthyléthyle dans 100 ml de tétrahydrofurane. On laisse la température du mélange revenir à la température ambiante et on poursuit l'agitation pendant 18 heures à cette température. On concentre sous pression réduite, on triture le résidu dans l'éther, on filtre et on sèche sous pression réduite.
On obtient 4,2 g de produit sous forme d'une poudre blanche que l'on utilise telle quelle dans l'étape suivante. Rendement = 86 % Point de fusion = 132 °C
3.2. {S) - [1- [ (4-méthoxypιpérιdm-l-yl) carbonyl] -4- [5-méthyl- 1- (triphénylméthyl) -lH-ιmιdazol-4-yl] butyl] carbamate de 1, 1-diméthyléthyle A un mélange de 0,918 g (1,7 mmoles) d'acide (S) - - [ [ (1 , 1- diméthyléthoxy) carbonyl] amino] -5-méthyl-l- (triphénylméthyl) - lH-ιmιdazole-4-pentanoιque, 0,281 g (1,87 mmoles) de chlorhydrate de 4 -méthoxypipéridme et 0,63 ml (3,57 mmoles) de diisopropyléthylamme dans 12 ml de dichloromethane, on ajoute par petites quantités à 0 °C sous azote et en agitant
0,71 g (1,87 mmoles) d ' hexafluorophosphate de [ (benzotriazol - 1-yl) oxy] tris (diméthylam o) phosphonium. On laisse la température du mélange revenir à la température ambiante, on poursuit l'agitation à cette température pendant 18 heures et on concentre le milieu réactionnel sous pression réduite. On reprend le résidu par 100 ml d'acétate d'éthyle, on lave successivement par 50 ml d'une solution aqueuse d'acide ehlorhydrique 0,5 N, 50 ml d'une solution saturée d' hydrogenocarbonate de sodium, 50 ml d'une solution saturée de chlorure de sodium, on sèche sur sulfate de sodium et on concentre sous pression réduite. On purifie le résidu par chromatographie sur colonne de gel de silice en éluant par un mélange méthanol : dichloromethane (2:98). On obtient 1,05 g de produit sous forme d'une huile visqueuse.
Rendement = 97 %
3 . 3 . chlorhydrate de ( S) -α- [ ( 4 -méthoxypιpérιdm- l -yl ) carbonyl ] - 5 -méthyl - l - ( triphénylméthyl ) - lH- ιmιdazole - 4 - butanamme ( 1 : 1 )
On traite par un courant de gaz ehlorhydrique pendant 20 minutes à 0 °C une solution de 1,05 g (1,65 mmoles) de { S) - [1- [ (4-méthoxypιpérιdm-l-yl) carbonyl] -4- [5-méthyl-l- (triphénylméthyl) -lH-ιmιdazol-4-yl] butyl] carbamate de 1 , 1-diméthyléthyle dans 50 ml de benzène. On laisse sous agitation à cette température pendant 30 minutes et on concentre sous pression réduite. On triture le résidu ainsi obtenu dans l'éther, on filtre et on sèche sous pression réduite . On obtient 0,93 g de produit sous forme d'une poudre blanche que l'on utilise telle quelle dans l'étape suivante. Rendement = 98 % Point de fusion = 112 °C
Exemple 4 chlorhydrate de ( S) - 5 -méthyl -α - [ ( 4 -méthylènepιpéndm- 1 - yl ) carbonyl ] - 1 - ( triphénylméthyl ) - lH- ιmιdazole - 4 -butanamme ( 1 : 1 )
4.1. chlorhydrate de 4-méthylènepιpérιdιne
4.1.1. 4-méthylènepιpéπdme-l-carboxylate de 1 , 1-dιméthyl- éthyle
A un mélange de 9,81 g (27,5 mmoles) de bromure de méthyltriphénylphosphonium dans 60 ml de tétrahydrofurane anhydre, on ajoute à la température ambiante, sous atmosphère d'azote, 17 ml d'une solution 1,6 M de n-butyllithium dans 1 ' hexane . On laisse le mélange pendant 4 heures sous agitation à la température ambiante et on ajoute rapidement une solution de 5 g (25 mmoles) de 4-oxopιpérιdme-l-carboxy- late de 1 , 1-diméthyléthyle dans 20 ml de tétrahydrofurane anhydre. On chauffe le milieu réactionnel pendant 10 heures à la température de reflux, on le verse sur 400 ml d'une solution saturée de chlorure d'ammonium et on extrait par 2 fois 300 ml d'éther. On rassemble les phases organiques, on les sèche sur sulfate de sodium et on concentre sous pression réduite. On purifie le résidu par chromatographie sur colonne de gel de silice en éluant par un mélange acétate d'éthyle: n-hexane (5 : 95) . On obtient 2,8 g de produit sous forme d'une huile vitreuse. Rendement = 57 %
4 . 1 . 2 . chlorhydrate de 4 -méthylènepιpéπdme
On obtient ce composé à partir du 4-méthylènepιpérιdme-l- carboxylate de 1 , 1-diméthyléthyle selon la méthode décrite dans l'exemple 3.1.3.
4.2. chlorhydrate de { S) -5-méthyl-α- [ (4 -méthylènepipéridm- 1-yl) carbonyl] -1- (triphénylméthyl) -lH-ιmιdazole-4- butanamme (1:1)
On fait réagir le (S) -α- [[ (1, 1-dιméthyléthoxy) carbonyl] ammo] -5-méthyl-l- (triphénylméthyl) -lH-ιmιdazol-4-pentanoιque avec du chlorhydrate de 4-méthylènepιpéπdme selon la méthode décrite dans l'exemple 3.2 et on obtient le
(S) - [4- [5-méthyl-l- (triphénylméthyl) -lH-ιmιdazol-4 -yl] -1- [ (4- méthylènepipéπdm- 1 -yl ) carbonyl ] butyl ] carbamate de 1 , 1-diméthyléthyle, produit amorphe Point de fusion = 85 °C
On traite ce produit par un courant d'acide ehlorhydrique gazeux selon la méthode décrite dans l'exemple 3.3. On obtient 0,74 g de produit. Rendement = 100 % Point de fusion = 148 °C
Exemple 5 chlorhydrate de { S) -α- [ (4 -cyclopropylpιpérιdm- l -yl ) carbonyl ] - 5 -méthyl - l - ( triphénylméthyl ) - lH-ιmιdazole -4 - butanamme ( 1 : 1 )
5 . 1 . chlorhydrate de 4 -cyclopropylpιpérιdme
5 . 1 . 1 . 4 -cyclopropylpιpéπdme
On hydrogène à 50 °C dans un appareil de Parr sous une pression de 0,35 MPa (50 psi) 13 g (109 mmoles) de 4 -cyclopropylpyridme en solution dans 150 ml d'acide acétique en présence de 0,7 g d'oxyde de platine (IV) . On filtre le milieu réactionnel sur célite et on concentre le filtrat sous pression réduite.
On obtient 12,85 g de produit que l'on utilise tel quel dans l'étape suivante.
Rendement = 94 %
5.1.2. 4-cyclopropylpιpéπdme-l-carboxylate de 1,1-dιmé- thyléthyle On solubilise 5 g (40 mmoles) de 4-cyclopropylpιpérιdme dans 40 ml de dichloromethane, on refroidit le mélange à 0 °C et on ajoute goutte à goutte 6,98 g (32 mmoles) de dicarbonate de bis- (1, 1-diméthyléthyle) et 4,85 g (48 mmoles) de triethylamme. On concentre le milieu réactionnel et on purifie le résidu par chromatographie sur colonne de gel de silice en éluant par un mélange dichloromethane : méthanol (99:1) .
On obtient 4 g de produit. Rendement = 44 %
5.1.3. chlorhydrate de 4-cyclopropylpipéridine On traite par un courant de gaz ehlorhydrique pendant 30 minutes à 0 °C une solution agitée de 6,5 g (28,8 mmoles) de 4-cyclopropylpipéridine-l-carboxylate de 1 , 1-diméthylé- thyle dans 100 ml de benzène. On laisse la température du milieu réactionnel revenir à la température ambiante, on maintient l'agitation pendant 4 heures à cette température et on concentre sous pression réduite. On triture le résidu ainsi obtenu dans l'éther, on filtre et on sèche sous pression réduite.
On obtient 4,1 g de produit sous forme d'une poudre blanche que l'on utilise telle quelle dans l'étape suivante. Rendement = 88 % Point de fusion = 186 °C
5.2. ( S) - [1- [ (4-cyclopropylpipéridin-l-yl) carbonyl] -4- [5- méthyl-1- (triphénylméthyl) -lH-imidazol-4-yl] butyl] carbamate de 1 , 1-diméthyléthyle On place sous agitation un mélange de 6 g (11 mmoles) d'acide (S) -α- [ [ (1, 1-diméthyléthoxy) carbonyl] amino] -5-méthyl-l- (triphénylméthyl) -lH-imidazole-4-pentanoïque, 1,79 g (11 mmoles) de chlorhydrate de 4-cyclopropylpipéridine et 9,6 ml (55,5 mmoles) de diisopropyléthylamine dans 100 ml de dichloromethane et on ajoute par petites quantités à 0 °C sous argon et en agitant 4,62 g (12,2 mmoles) d' hexafluorophosphate de [ (benzotriazol-1-yl) oxy] tris (diméthylamino) phosphonium. On laisse la température du mélange revenir à la température ambiante, on poursuit l'agitation à cette température pendant 4 heures et on concentre le milieu réactionnel sous pression réduite. On reprend le résidu par 300 ml d'acétate d'éthyle, on lave successivement par 100 ml d'une solution aqueuse d'acide ehlorhydrique 1 N, 100 ml d'une solution saturée d ' hydrogenocarbonate de sodium et 100 ml d'une solution saturée de chlorure de sodium, on sèche sur sulfate de sodium et on concentre sous pression réduite. On purifie le résidu par chromatographie sur colonne de gel de silice en éluant par un mélange méthanol : dichloromethane (1:99) . On obtient 5 g de produit.
Rendement = 70 %
5.3. chlorhydrate de {S) -α- [ (4-cyclopropylpipéridin-l-yl) carbonyl] -5-méthyl-l- (triphénylméthyl) -lH-imidazole-4- butanamine (1:1)
On traite par un courant de gaz ehlorhydrique pendant 30 minutes à 0 °C une solution agitée de 5 , 3 g (8 mmoles) de ( S) - [1- [ (4-cyclopropylpipéridin-l-yl) carbonyl] -4- [5-méthyl-l- (triphénylméthyl) - lH-imidazol -4 -yl] butyl] carbamate de 1, 1-diméthyléthyle dans 200 ml de benzène. On laisse la température du milieu réactionnel revenir à la température ambiante, on maintient l'agitation pendant 3 heures et on concentre sous pression réduite. On reprend le résidu ainsi obtenu dans deux fois 280 ml de dichloromethane et on sèche sous pression réduite.
On obtient 4,7 g de produit sous forme d'une poudre blanche que l'on utilise telle quelle dans l'étape suivante. Rendement = 100 % Point de fusion = 124 °C
Exemple 6 chlorhydrate de (S) -5-méthyl-α- [ [4- (difluorométhylène) pipéridin- 1-yl] carbonyl] -1- (triphénylméthyl) -lH-imidazole-4 - butanamine (1:1)
6.1. chlorhydrate de 4- (difluorométhylène) pipéridine 6.1.1. 4- (difluorométhylène) pipéridine-1-carboxylate de
1 , 1-diméthyléthyle A 12 ml (120 mmoles) de difluorobromomethane en solution dans 180 ml de triglyme et maintenus sous agitation, on ajoute goutte à goutte à 0 °C, sous atmosphère d'argon, 45,6 ml (252 mmoles) d' hexaméthylphosphorotriamide en solution dans 30 ml de triglyme. On laisse la température du milieu réactionnel revenir à la température ambiante, on maintient l'agitation pendant 30 minutes à cette température et on refroidit à nouveau à 0 °C. On ajoute alors 11,94 g (60 mmoles) de 4-oxopipéridine-l-carboxylate de 1 , 1-diméthyléthyle en solution dans 30 ml de triglyme, on laisse la température du mélange revenir à la température
ambiante et on agite pendant 30 minutes à cette température. On chauffe le milieu réactionnel pendant 2 heures à 80 °C, on le refroidit, on le verse sur 1 litre d'eau et on l'extrait par 3 fois 400 ml de pentane . On lave à l'eau, on sèche sur sulfate de sodium et on évapore. On purifie le résidu par chromatographie sur colonne de gel de silice en éluant par un mélange cyclohexane : acétate d'éthyle (97:3). On obtient 8,5 g de produit. Rendement = 61 %
6.1.2. chlorhydrate de 4- (difluorométhylène) ipéridme On obtient ce composé sous forme de poudre blanche à partir du 4- (difluorométhylène) pιpérιdme-1-carboxylate de 1,1-dιmé- thyléthyle selon la méthode décrite dans l'exemple 3.1.3. Rendement = 100 %
Point de fusion = 196 °C
6.2. chlorhydrate de (S) -5-méthyl-α- [ [4- (difluorométhylène) - p péπdm-1-yl] carbonyl] -1- (triphénylméthyl) -1H- ιmιdazole-4 -butanamine (1:1)
On fait réagir le { S) -α- [[ (1, 1-dιméthyléthoxy) carbonyl] ammo] -5-méthyl-l- (triphénylméthyl) -lH-ιmιdazol-4-pentanoique avec du chlorhydrate de 4- (difluorométhylène) pipéridme selon la méthode décrite dans l'exemple 3.2 et on obtient le (S) - [4- [5-méthyl-l- (triphénylméthyl) -lH-ιmιdazol-4-yl] -1- [ [4-
(difluorométhylène) pιpérιdm-1-yl) carbonyl] butyl] carbamate de
1, 1-diméthyléthyle sous forme d'un solide vitreux.
Rendement = 75 %
Point de fusion = 86 °C On traite ce produit par un courant d'acide ehlorhydrique gazeux selon la méthode décrite dans l'exemple 3.3.
On obtient le produit sous forme de poudre blanche.
Rendement = 99 %
Point de fusion = 117 °C
Exemple 7 chlorhydrate de (S) -5-méthyl-α- [ (4 - (méthylthio) pipéridin- 1- yl] carbonyl] -1- (triphénylméthyl) - lH-imidazole-4 -butanamine (1:1)
7.1. chlorhydrate de 4- (méthylthio) pipéridine
7.1.1. 4- [ (méthylsulfonyl) oxy] pipéridine-1-carboxylate de 1 , 1-diméthyléthyle
A une solution de 13,9 g (69 mmoles) de 4-hydroxypipéridine- 1-carboxylate de 1 , 1-diméthyléthyle et de 5,6 ml (76 mmoles) de triéthylamine dans 80 ml de dichloromethane, on ajoute goutte à goutte, à 0 °C, sous azote, 5 , 6 ml (72 mmoles) de chlorure de méthanesulfonyle . On laisse le milieu réactionnel pendant 6 heures sous agitation à cette température et on le concentre sous pression réduite. On reprend le résidu par 200 ml d'acétate d'éthyle et on lave successivement par 2 fois 100 ml d'une solution aqueuse d'acide ehlorhydrique 1 N, 100 ml d'eau et 100 ml d'une solution saturée en chlorure de sodium. On sèche sur sulfate de sodium et on concentre sous pression réduite. On purifie le résidu par chromatographie sur colonne de gel de silice en éluant par de l'acétate d'éthyle.
On obtient 16,2 g de produit sous forme de cristaux blancs. Rendement = 95 % Point de fusion = 93,9 °C
7.1.2. 4 - (méthylthio) pipéridine-1-carboxylate de 1 , 1-diméthyléthyle
On agite pendant 72 heures à la température ambiante un mélange de 2,47 g (10 mmoles) de 4- [ (méthylsulfonyl) oxy] pipéridine- 1-carboxylate de 1, 1-diméthyléthyle, 0,71 g (10,1 mmoles) de thiométhoxyde de sodium et 0,37 g (1 mmole) de iodure de tétrabutylammonium dans 10 ml de tétrahydrofurane puis on concentre sous pression réduite. On purifie le résidu par chromatographie sur colonne de gel de silice en éluant par un mélange n-hexane : acétate d'éthyle (9:1) . On obtient 1,5 g de produit sous forme d'une huile visqueuse. Rendement = 63 %
7.1.3 chlorhydrate de 4- (méthylthio) pipéridine On obtient ce composé à partir du 4 - (méthylthio) pipéridine- 1- carboxylate de 1 , 1-diméthyléthyle selon la méthode décrite dans 1 ' exemple 3.1.3. Rendement = 100 %
Point de fusion = 156,5 °C
7.2. chlorhydrate de (S) -5-méthyl-α- [ [4 - (méthylthio) pipé- ridin-1-yl] carbonyl] -1- (triphénylméthyl) -lH-imidazole- 4 -butanamine (1:1)
On fait réagir le (S) -α- [[ (1, 1-diméthyléthoxy) carbonyl] amino] -5-méthyl-l- (triphénylméthyl) -lH-imidazol - -pentanoïque avec du chlorhydrate de 4- (méthylthio) pipéridine selon la méthode décrite dans l'exemple 3.2 et on obtient le (S) - [4- [5-méthyl-l- (triphénylméthyl) -lH-imidazol-4 -yl] -1- [ [4-
(méthylthio) pipéridin- 1-yl] carbonyl] butyl] carbamate de
1 , 1-diméthyléthyle sous forme d'une poudre amorphe.
Rendement = 93 %
Point de fusion = 101,2 °C On traite ce produit par un courant d'acide ehlorhydrique gazeux selon la méthode décrite dans l'exemple 3.3.
On obtient le produit sous forme de poudre amorphe.
Rendement = 100 %
Point de fusion = 127,7 °C
Exemple 8 chlorhydrate de (S) -5-méthyl-α- [ (4-méthyl-l , 2 , 3 , 6- tétrahydropyridin- 1-yl) carbonyl] -1- (triphénylméthyl) -1H- imidazole-4 -butanamine (1:1)
8.1. chlorhydrate de 4-méthyl-l , 2 , 3 , 6-tétrahydropyridine 8.1.1. 4 -méthyl-1 , 2 , 3 , 6-tétrahydropyridine-l-carboxylate de
1 , 1-diméthyléthyle A une solution de 4,95 g (25 mmoles) de 4 -oxopipéridine-1- carboxylate de 1 , 1-diméthyléthyle dans 30 ml de tétrahydrofurane anhydre, on ajoute à 0 °C, sous azote, une solution de 18 ml (28,8 mmoles) de méthyllithium dans l'éther et on poursuit l'agitation pendant 2 heures à cette température. On ajoute alors goutte à goutte 3 ml (38 mmoles) de chlorure de
méthanesulfonyle , on poursuit l'agitation pendant 4 heures à 0 °C puis on concentre le milieu réactionnel sous pression réduite. On reprend le résidu par 200 ml d'acétate d'éthyle et on le lave successivement par 2 fois 100 ml d'une solution aqueuse d'acide ehlorhydrique 0,1 N, 100 ml d'eau et 100 ml d'une solution saturée en chlorure de sodium. On sèche sur sulfate de sodium et on concentre sous pression réduite. On reprend le résidu dans 100 ml de toluène et 15 ml de triéthylamine, on chauffe pendant 18 heures à la température de reflux et on concentre sous pression réduite. On purifie le résidu par chromatographie sur colonne de gel de silice en éluant par un mélange n-hexane :éther (95:5) .
On obtient 0,9 g de produit sous forme d'une huile visqueuse. Rendement = 18 %
8.1.2. chlorhydrate de 4-méthyl-l , 2 , 3 , 6-tétrahydropyridine On obtient ce composé à partir du 4-méthyl-l , 2 , 3 , 6- tétrahy- dropyridine-1 -carboxylate de 1 , 1-diméthyléthyle selon la méthode décrite dans l'exemple 3.1.3. Rendement = 100 %
8.2. chlorhydrate de (S) -5-méthyl-α- [ (4-méthyl-l , 2 , 3 , 6- téthrahydropyridin-1-yl) carbonyl] -1- (triphénylméthyl) - lH-imidazole-4 -butanamine (1:1) On fait réagir le (S) -α- [[ (1 , 1-diméthyléthoxy) carbonyl] amino] -5-méthyl-l- (triphénylméthyl) -lH-imidazol-4 -pentanoïque avec du chlorhydrate de 4-méthyl-l, 2 , 3 , 6-tétrahydropyridine selon la méthode décrite dans l'exemple 3.2 et on obtient le (S) - [4- [5-méthyl-l- (triphénylméthyl) -lH-imidazol-4-yl] -1- [ (4- méthyl -1,2,3,6 -1étrahydropyridin-1-yl ) carbonyl] butyl] carbamate de 1 , 1-diméthyléthyle sous forme d'une poudre amorphe .
Rendement = 90 % Point de fusion = 90,7 °C On traite ce produit par un courant d'acide ehlorhydrique gazeux selon la méthode décrite dans l'exemple 3.3. On obtient le produit sous forme de poudre blanche. Rendement = 100 % Point de fusion = 118 °C
Exemple 9 chlorhydrate de 1- [2 -amino-5- [5-méthyl -1- (triphénylméthyl ) - lH-imidazol-4-yl] -1-oxopentyl] -hexahydro-5H-l , 4-diazépin-5- one
9.1. chlorhydrate d' hexahydro-5H-l , 4 -diazépin-5-one
9.1.1. 1- (phénylméthyl) hexahydro-5H-l , 4-diazépin-5-one A une solution de 11 g (58,12 mmoles) de 1-phénylméthylpipé- ridin-3-one dans 60 ml d'acide formique, on ajoute en 10 minutes une solution de 9,86 g (87,18 mmoles) d'acide hydroxylamine-O-sulfonique dans l'acide acétique. On chauffe le milieu réactionnel pendant 4 heures à la température de reflux. On laisse refroidir le mélange et on le verse sur un mélange glace: eau puis on neutralise avec une solution aqueuse de soude à 5 % . On extrait avec du chloroforme, on récupère la phase organique, on la sèche et on évapore à sec. On purifie le résidu par chromatographie sur colonne de gel de silice en éluant par un mélange dichloromethane : méthanol (2:98) . On obtient 6,94 g de produit. Rendement = 58,5 %
9.1.2. chlorhydrate d' hexahydro-5H-l , 4-diazépin-5-one
On met 5,5 g (26,2 mmoles) de 1- (phénylméthyl) hexahydro-lH- 1 , 4-diazépin-5-one en solution dans 100 ml de méthanol, on ajoute 0,7 g de palladium sur charbon à 10 % et on chauffe pendant 3 heures à 45 °C sous une pression de 0,29 MPa (42 psi) . On filtre le milieu réactionnel, on évapore les solvants et on reprend le résidu dans 30 ml d' éthanol. On chauffe, on filtre l'insoluble, on le rince à 1 ' éther et on évapore le solvant .
On obtient 2,44 g de produit sous forme d'une poudre blanchâtre que l'on utilise telle quelle dans l'étape suivante .
9.2. chlorhydrate de 1- [2-ammo-5- [5-méthyl -1 - (triphénylméthyl) -lH-ιmιdazol-4-yl] -1-oxopentyl] hexahydro-5H-l , 4- dιazépm-5-one 9.2.1. ( S) - [4- [5-méthyl-l- (triphénylméthyl) -lH-ιmιdazol-4- yl] -1- [ (5-oxohexahydro-5H-l , 4-dιazépm-l-yl) carbonyl] butyl] carbamate de 1, 1-diméthyléthyle A une solution de 0,6 g (4 mmoles) de chlorhydrate d' hexahydro-5H-l, 4 -dιazépm-5-one dans 40 ml de dichloromethane, on ajoute à 0 °C, 2,15 g (4 mmoles) d'acide (S) -α- [ [ (1, 1-dιméthyléthoxy) carbonyl] ammo] -5-méthyl-l-
(triphénylméthyl) -lH-imidazol -4 -pentanoïque puis successivement 2 , 8 ml (16 mmoles) de N, N-dusopropyléthylamme et 1,5 g (4 mmoles) d' hexafluorophosphate de 0- (benzotπazol -1- yl ) -N, N, N' , N' -tetramethyluronium. On laisse la température du milieu réactionnel revenir à la température ambiante, on poursuit l'agitation pendant une nuit à cette température et on concentre sous vide. On reprend le résidu par 200 ml d'acétate d'éthyle et on lave successivement par 3 fois 30 ml d'une solution aqueuse d'acide ehlorhydrique 1 Ν, 2 fois 20 ml d'une solution saturée en hydrogenocarbonate de sodium puis 20 ml d'une solution saturée de chlorure de sodium. On sèche sur sulfate de magnésium et on concentre sous pression réduite. On purifie le résidu par chromatographie sur colonne de gel de silice en éluant par un gradient dichloromethane: méthanol (98:2 à 97:3).
On obtient 1,87 g de produit sous forme d'une mousse blanchâtre . Rendement = 74 %
9.2.2. chlorhydrate de 1- [2-ammo-5- [5-méthyl-l- (triphénylméthyl) -lH-ιmιdazol-4-yl] -1-oxopentyl] hexahydro-5H- 1, 4-dιazépm-5-one On traite par un courant d'acide ehlorhydrique gazeux pendant 10 secondes à 0 °C, une solution de 1,87 g (2,94 mmoles) de { S) - [4- [5-méthyl-l- (triphénylméthyl) -lH-ιmιdazol-4-yl] -1- [ (5- oxohexahydro-5H-l, 4-dιazépm-l-yl) carbonyl] butyl] carbamate de 1, 1-diméthyléthyle dans 200 ml de toluène. On laisse la température du mélange revenir à la température ambiante puis on concentre sous presion réduite. On dissout le résidu dans
un volume minimal de dichloromethane et on ajoute 200 ml d'éther. On triture le mélange, on filtre et on sèche. On obtient 1,64 g de produit que l'on utilise tel quel dans l'étape suivante. Rendement = 97 %
Exemple 10 chlorhydrate de { S) -α-ammo-N-cyclopentyl -N, 5 -dιméthyl - l - ( triphénylméthyl ) - 1H- îmidazole - 4 -pentanamide
10 . 1 . chlorhydrate de N-méthylcyclopentanamme
10 . 1 . 1 . N-cyclopentylf ormamide
On chauffe pendant 4 heures à la température de reflux un mélange de 10 g (117 mmoles) de cyclopentanamme et de 10,8 ml (140 mmoles) de formiate d'éthyle puis on concentre le milieu réactionnel sous pression réduite. On purifie le résidu par chromatographie sur colonne de gel de silice en éluant par un gradient d'acétate d' éthyle : cyclohexane (1:9 à 6:4) . On obtient 10 g de produit sous forme d'une huile. Rendement = 75 %
10.1.2. cyclopentylméthylcarbamate de 1 , 1-diméthyléthyle A une solution de 4,37 g (38 mmoles) de N- cyclopentyl for- mamide dans 20 ml de tétrahydrofurane anhydre, on ajoute goutte à goutte, à 0 °C sous azote, 50 ml (50 mmoles) d'une solution 1 M d'hydrure de lithium et d' aluminium dans le tétrahydrofurane. On laisse la température du mélange revenir à la température ambiante et on chauffe à la température de reflux pendant 8 heures. On refroidit le milieu réactionnel à 0 °C, on l'acidife à pH 2 avec une solution aqueuse d'acide ehlorhydrique 1 Ν et on ajuste le pH à 8 avec du carbonate de potassium. On ajoute alors goutte à goutte 8,6 g (40 mmoles) de dicarbonate de bis (1 , 1-diméthyléthyle) en solution dans 40 ml de méthanol. On laisse la température du mélange revenir à la température ambiante et on poursuit l'agitation pendant 15 heures à cette température. On extrait le milieu réactionnel par 2 fois 300 ml d'éther et on rassemble les phases organiques. On les lave par 2 fois 200 ml d'une
solution aqueuse d'acide ehlorhydrique 1 N puis par 200 ml d'une solution saturée de chlorure de sodium. On sèche sur sulfate de sodium, on filtre et on concentre sous pression réduite. On purifie le résidu par chromatographie sur colonne de gel de silice en éluant par un mélange cyelohexane : éther (95:5) .
On obtient 2,91 g de produit sous forme d'une huile. Rendement = 38 %
10.1.3. chlorhydrate de N-méthylcyclopentanamme
On traite par un courant d'acide ehlorhydrique gazeux pendant 5 minutes à 0 °C, une solution de 2,9 g (14,5 mmoles) de cyclopentylméthylcarbamate de 1 , 1-diméthyléthyle . On laisse le mélange sous agitation pendant 4 heures à cette tempéra- ture puis on concentre sous pression réduite.
On obtient 1,96 g de produit sous forme d'une poudre blanche hygroscopique .
Rendement = 100 %
Point de fusion = 123-126 °C
10.2. chlorhydrate de ( S) -α-ammo-N-cyclopentyl -N, 5- dιméthyl-1- (triphénylméthyl) -lH-ιmιdazole-4- pentanamide 10.2.1. (S) - [1- [ (cyclopentylméthylammo) carbonyl] -4- [5- méthyl-1- (triphénylméthyl) -lH-ιmιdazol-4-yl] butyl] carbamate de 1 , 1-diméthyléthyle A une solution de 2,57 g (4,76 mmoles) de (S) -α- [ [ (1, 1- diméthyléthoxy) carbonyl] ammo] -5-méthyl-l- (triphénylméthyl) - lH-ιmιdazol-4-pentanoïque dans 15 ml de dichloromethane, on ajoute successivement à 0 °C sous azote, 0,68 g (5 mmoles) de chlorhydrate de N-méthylcyclopentanamme , 2,15 ml (12,3 mmoles) de Ν,Ν-dιιsopropyléthylamme et 1,98 g (5,24 mmoles) d' hexafluorophosphate de O- (benzotπazol-1-yl) -N, N, N' , N' - tetramethyluronium. On laisse la température du milieu réactionnel revenir à la température ambiante, on poursuit l'agitation pendant 15 heures à cette température et on concentre sous vide. On reprend le résidu par 150 ml d'acétate d'éthyle et on lave successivement par 100 ml d'une solution aqueuse d'acide ehlorhydrique 1 Ν, 100 ml d'une
solution saturée d' hydrogenocarbonate de sodium puis 100 ml d'une solution saturée de chlorure de sodium. On sèche sur sulfate de sodium et on concentre sous pression réduite. On purifie le résidu par chromatographie sur colonne de gel de silice en éluant par un gradient acétate d'éthyle: cyelohexane (3:7 à 8:2).
On obtient 2,26 g de produit sous forme d'un solide amorphe. Rendement = 77 % Point de fusion = 86-90 °C
10.2.2. chlorhydrate de { S) -α-ammo-N- cyclopentyl -N, 5- dιméthyl-1- (triphénylméthyl) -lH-ιmιdazole-4-pen- tanamide
On traite par un courant d'acide ehlorhydrique gazeux pendant 5 minutes à 0 °C, une solution de 2 , 2 g (3,5 mmoles) de
(S) - [1- [ (cyclopentylméthylammo) carbonyl] -4- [5-méthyl-l-
(triphénylméthyl) -lH-imidazol -4 -yl] butyl] carbamate de
1 , 1-diméthyléthyle . On laisse le mélange sous agitation pendant 5 heures à cette température puis on concentre sous pression réduite.
On obtient 2 g de produit que l'on utilise tel quel dans l'étape suivante.
Rendement = 100 %
Point de fusion = 138-142 °C
Exemple 11 chlorhydrate de { S) -α-ammo-N, 5 -dιméthyl -N-pyrrolιdm- l - yl-1- (triphénylméthyl) -lH-ιmιdazole-4-pentanamιde
11.1. chlorhydrate de N-méthylpyrrolιdm- 1 -amme
11.1.1. pyrrolιdm-1-ylcarbamate de 1 , 1-diméthyléthyle A une solution de 1 g (8,15 mmoles) de chlorhydrate de pyrrolιdm-1-amme et de 1,62 g (7,4 mmoles) de dicarbonate de bis (1 , 1-diméthyléthyle) dans 8 ml de dichloromethane, on ajoute goutte à goutte 1,13 ml (8,15 mmoles) de triethylamme. On laisse le mélange pendant 15 heures sous agitation et on concentre sous pression réduite. On reprend le résidu par 100 ml d'éther et on lave successivement par 10 ml d'eau et 100 ml d'une solution aqueuse saturée de
chlorure de sodium. On sèche sur sulfate de sodium, on filtre sur silice et on concentre sous pression réduite. On obtient 1 g de produit. Rendement = 67 % Point de fusion = 108 °C
11.1.2. méthylpyrrolιdm-1-ylcarbamate de 1, 1-dιméthyl- éthyle
A une solution de 1,34 g (7 mmoles) de pyrrolιdm-1-ylcarba- mate de 1 , 1-diméthyléthyle et de 1,75 ml (28 mmoles) de îodure de méthyle dans 3 ml de tétrahydrofurane anhydre, on ajoute goutte à goutte à -78 °C sous azote, 7,7 ml (7,7 mmoles) d'une solution de lithium bis (tπméthylsilyl) amide 1 M dans le tétrahydrofurane. On laisse la température du mélange revenir à la température ambiante et on poursuit l'agitation pendant 30 minutes à cette température. On ajoute 150 ml d'éther et on lave successivement par 100 ml d'eau et 100 ml d'une solution saturée en chlorure de sodium. On sèche sur sulfate de sodium, on filtre et on concentre sous pression réduite. On purifie le résidu par chromatographie sur colonne de gel de silice en éluant par un mélange cyelohexane -.acétate d'éthyle (9:1).
On obtient 0,75 g de produit sous forme d'une huile. Rendement = 55 %
11 . 1 . 3 . chlorhydrate de N-méthylpyrrolιdm- 1 -amme
On prépare ce produit à partir de 0,75 g (3,7 mmoles) de méthylpyrrolιdm-1-ylcarbamate de 1 , 1-diméthyléthyle selon la méthode décrite en 10.1.3. On obtient 0,5 g de produit sous forme d'une huile visqueuse. Rendement = 100 %
11.2. chlorhydrate de { S) -α-ammo-N, 5 -dιméthyl -N-pyrrol ι - dm-l-yl-1- (triphénylméthyl) -lH-ιmιdazole-4- pentanamide
11.2.1. {S) - [1- [ (mëthylpyrrolιdm-1-ylammo) carbonyl] -
4- [5-méthyl-l- (triphénylméthyl) -lH-ιmιdazol-4-yl] butyl] carbamate de 1 , 1-diméthyléthyle On prépare le produit selon le mode opératoire décrit en
10.2.1. à partir de 1,8 g (3,3 mmoles) de (S) -α- [ [ (1, 1- diméthyléthoxy) carbonyl] ammo] -5-méthyl-l- (triphénylméthyl) - lH-ιmιdazol-4-pentanoιque et de 0,48 g (3,5 mmoles) de chlorhydrate de N-méthylpyrrolιdιn-1-amιne . On obtient 1,8 g de produit sous forme d'un solide amorphe. Rendement = 88 % Point de fusion = 70-75 °C
11.2.2. chlorhydrate de { S) -α-ammo-N, 5-dιméthyl-N-pyrrolι- dm-l-yl-1- (triphénylméthyl) -lH-ιmιdazole-4- pentanamide On prépare le produit selon le mode opératoire décrit en 10.2.2. à partir de 1,8 g (2,8 mmoles) de (S) - [1- [ (méthylpyr- rolidm-l-ylammo) carbonyl] -4- [5-méthyl-l- (triphénylméthyl) - lH-ιmιdazol-4-yl] butyl] carbamate de 1 , 1-diméthyléthyle .
On obtient 1,65 g de produit sous forme d'un solide amorphe.
Rendement = 100 %
Point de fusion = 130-135 °C
Exemple 12 (composé n° 67) chlorhydrate de { S) -N- [3- [ [ [4- (5-éthyl-lH-ιmιdazol-4-yl) -1- [ (4-éthylpιpérιdm-l-yl) carbonyl] butyl] am o] sulfonyl] [1,1' -biphényl] -2-yl] propanamide (1:1)
12.1. (S) -N- [3- [ [ [1- [ (4 -éthylpipéπdm- 1-yl) carbonyl] -4- [5- éthyl-1- (triphénylméthyl) -lH-ιmιdazol-4-yl] butyl] am o] sulfonyl] [1,1' -biphényl] -2-yl] -N- (1-oxopropyl) propanamide A un mélange de 0,435 g (1,25 mmoles) de chlorure de [bis (1-oxopropyl) ammo] [1 , 1 ' -biphenyle] -3-sulfonyle et de 0,61 g (1,04 mmoles) de chlorhydrate de (S) -5-éthyl-α- [ (4- éthylpipéridm- 1-yl) carbonyl] -1- (triphénylméthyl) -1H- ιmιdazole-4 -butanamine dans 8 ml de dichloromethane, on ajoute goutte à goutte à 0 °C sous azote, 0,48 ml (3,4 mmoles) de triethylamme. On laisse le mélange pendant 4 heures sous agitation et on concentre sous pression réduite. On reprend le résidu par 100 ml d'acétate d'éthyle, on lave successivement par 50 ml d'une solution aqueuse d'acide ehlorhydrique 1 Ν, 50 ml d'une solution saturée
d ' hydrogenocarbonate de sodium et 50 ml d'une solution saturée de chlorure de sodium, on sèche sur sulfate de magnésium et on concentre sous pression réduite. On obtient 0,85 g de produit sous forme d'une huile visqueuse que l'on utilise telle quelle dans l'étape suivante. Rendement = 95 %
12.2. chlorhydrate de ( S) -N- [3- [ [ [4- (5-éthyl-lH-imidazol-4- yl) -1- [ (4-éthylpipéridin-l-yl) carbonyl] butyl] amino] sulfonyl] [1, 1 ' -biphényl] -2-yl] propanamide (1:1)
On chauffe pendant 16 heures à la température de reflux 0,85 g (0,95 mmole) de (S) -N- [3- [ [ [1- [ (4-éthylpipéridin-l- yl) carbonyl] -4- [5-éthyl-l- (triphénylméthyl) -lH-imidazol-4 - yl] butyl] amino] sulfonyl] [1,1' -biphényl] -2-yl] -N- (1-oxopropyl) propanamide en solution dans un mélange de 30 ml d'acide acétique et 10 ml d'eau et on concentre le milieu réactionnel sous pression réduite. On reprend le résidu par 150 ml d'acétate d'éthyle, on lave successivement par 50 ml d'une solution saturée d' hydrogenocarbonate de sodium et par 50 ml d'une solution saturée de chlorure de sodium, on sèche sur sulfate de sodium et on concentre sous pression réduite. On purifie le résidu par chromatographie sur colonne de gel de silice en éluant par un mélange méthanol : dichloromethane (4:96) . On obtient 0,44 g de produit sous forme de base que l'on reprend dans 10 ml d'une solution d'acide ehlorhydrique 0,1 Ν dans 1 ' isopropanol et on concentre sous pression réduite. On purifie le résidu par chromatographie sur colonne RP 18 en éluant par un mélange acétonitrile : eau (3:7) . On obtient après lyophilisation 0,42 g de produit sous forme d'une poudre blanche. Rendement = 69 % Point de fusion = 132 °C [ ]p° = + 112 ° ; (c = 0,2 ; méthanol)
Exemple 13 (composé n° 40) chlorhydrate de (S) -N- [3 - [ [ [4- (5-méthyl-lH-imidazol-4 -yl) - 1- [ [4- (trifluorométhyl) pipéridin-1-yl] carbonyl] butyl] amino] sulfonyl] [1 , 1 ' -biphényl] -2-yl] propanamide (1:1)
13.1. ( S) -N- [3- [ [ [4- [5-méthyl-l- (triphénylméthyl) - 1H- imidazol-4-yl] -1- [ [4- (trifluorométhyl) pipéridin-1- yl] carbonyl] butyl] amino] sulfonyl] [1,1' -biphényl] -2- yl] -N- (1-oxopropyl) propanamide A un mélange de 0,65 g (1,72 mmoles) de chlorure de [bis(l- oxopropyl) amino] [1 , 1 ' -biphenyle] -3 -suifonyle et de 1,05 g (1,72 mmoles) de chlorhydrate de (S) -5-méthyl-α- [ [4 -trifluorométhyl) pipéridin- 1-yl] carbonyl] -1- (triphénylméthyl) - 1H- imidazole-4 -butanamine dans 20 ml de dichloromethane, on ajoute goutte à goutte à 0 °C sous argon, 0,79 ml
(5,7 mmoles) de triéthylamine. On laisse revenir la température du mélange à la température ambiante, on poursuit l'agitation pendant 18 heures à cette température et on concentre sous pression réduite. On reprend le résidu par 100 ml d'acétate d'éthyle, on lave successivement par 50 ml d'une solution aqueuse d'acide ehlorhydrique 1 Ν, 50 ml d'une solution saturée d 'hydrogenocarbonate de sodium et 50 ml d'une solution saturée de chlorure de sodium, on sèche sur sulfate de sodium et on concentre sous pression réduite. On purifie le résidu par chromatographie sur colonne de gel de silice en éluant par un mélange méthanol : dichloromethane (2:98) .
On obtient 1,04 g de produit sous forme d'une huile visqueuse . Rendement = 70 %
13.2. chlorhydrate de (S) -N- [3 - [ [ [4- (5-méthyl-lH-imidazol- 4-yl) -1- [ [4- (trifluorométhyl) pipéridin- 1-yl] carbonyl] butyl] amino] sulfonyl] [1,1' -biphényl] -2-yl] propanamide (1:1)
On chauffe pendant 10 heures à la température de reflux 1,02 g (1,1 mmoles) de (S) -N- [3- [[ [4- [5-méthyl-l- (triphénylméthyl) -lH-imidazol-4-yl] -1- [ [4- (trifluorométhyl) pipéri- din-l-yl] carbonyl] butyl] amino] sulfonyl] [1,1' -biphényl] -2-yl] -
N- (1-oxopropyl) propanamide en solution dans un mélange de 25 ml d'acide acétique et 25 ml d'eau et on concentre le milieu réactionnel sous pression réduite. On reprend le résidu par 100 ml d'acétate d'éthyle, on lave par 50 ml d'une solution saturée d ' hydrogenocarbonate de sodium, on sèche sur sulfate de sodium et on concentre sous pression réduite. On purifie le résidu par chromatographie sur colonne de gel de silice en éluant par un mélange méthanol : dichloromethane (5:95) . On obtient 0,42 g de produit sous forme de base que l'on reprend dans 12 ml d'une solution d'acide ehlorhydrique 0,1 Ν dans 1 ' isopropanol et on concentre sous pression réduite. On purifie le résidu par chromatographie sur colonne RP 18 en éluant par un mélange acétonitrile :eau (3:7) . On obtient après lyophilisation 0,33 g de produit. Rendement = 46 %
Point de fusion = 146-150 [αj2û = + 80 o (c = 0 2 ; méthanol)
Exemple 14 (composé n° 34) chlorhydrate de (S) -N- [2 - [ [ [1- [ (4-méthoxypipéridin-l-yl) carbonyl] -4- (5-méthyl-lH-imidazol -4-yl) butyl] amino] sulfonyl] - 6-thién-2-ylphényl] propanamide (1:1)
14.1. (S) -1- [2- (3-éthyl-l,l-dioxo-5-thién-2-yl-2H-l,2,4- benzothiadiazin-2-yl) -5- [5-méthyl-l- (triphénylméthyl) -lH-imidazol-4-yl] -1-oxopentyl] -4-méthoxypi- péridine 14.1.1. chlorure de 2- [ (1-chloropropylidène) amino] -3 -thién- 2 -ylbenzènesulfonyle A un mélange de 3,8 g (15 mmoles) d'acide 2-amino-3- (thién-2- yl) benzènesulfonique et de 4 ml (49,5 mmoles) de pyridine dans 30 ml de dichloromethane, on ajoute goutte à goutte à
0 °C, 2,86 ml (33 mmoles) de chlorure de propionyle. On laisse le milieu réactionnel sous agitation pendant 5 heures à cette température puis on concentre sous pression réduite. On reprend le résidu par 40 ml de dichloromethane, on ajoute par petites quantités 7,8 g (37,5 mmoles) de pentachlorure de phosphore et on laisse le mélange sous agitation pendant
1 heure à 0 °C puis pendant 2 heures à la température
ambiante. On ajoute 200 ml d'éther au milieu réactionnel, on filtre, on lave le filtrat successivement avec 2 fois 200 ml d'eau glacée et 50 ml d'une solution saturée de chlorure de sodium, on sèche sur sulfate de sodium et on concentre sous pression réduite. On purifie le résidu par chromatographie
® sur Florisil en éluant rapidement par de l' éther.
On obtient 3,18 g de produit après cristallisation dans le pentane .
Rendement = 61 % Point de fusion = 74 °C
14.1.2. (S) -1- [2- (3-éthyl-l, l-dioxo-5-thién-2 -yl-2H-l , 2,4- benzothiadiazin-2-yl) -5- [5-méthyl-l- (triphénylméthyl) -lH-imidazol-4-yl] -1-oxopentyl] -4-méthoxypι - péridine
A un mélange de 0,42 g (1,2 mmoles) de chlorure de 2-[(l- chloropropylidène) amino] -3 -thién-2-ylbenzènesulfonyle et de 0,69 g (1,2 mmoles) de chlorhydrate de (S) -α- [ (4-méthoxypi- péridin-1-yl) carbonyl] -5-méthyl-l- (triphénylméthyl) - 1H- imidazole-4 -butanamine dans 15 ml de dichloromethane, on ajoute goutte à goutte à 0 °C, 0,55 ml (3,96 mmoles) de triéthylamine . On laisse revenir la température du mélange à la température ambiante, on poursuit l'agitation pendant 18 heures à cette température et on concentre sous pression réduite. On reprend le résidu par 100 ml d'acétate d'éthyle, on lave successivement par 50 ml d'une solution aqueuse d'acide ehlorhydrique 1 N, 50 ml d'une solution saturée d' hydrogenocarbonate de sodium et 50 ml d'une solution saturée de chlorure de sodium, on sèche sur sulfate de sodium et on concentre sous pression réduite.
On obtient 1,07 g de produit sous forme d'une huile visqueuse que l'on utilise telle quelle dans l'étape suivante. Rendement = 100 %
14.2. chlorhydrate de ( S) -N- [2 - [ [ [1- [ (4-méthoxypipéridin-l- yl) carbonyl] -4- (5-méthyl-lH-imidazol-4-yl) butyl] amino] sulfonyl] -6-thién-2-ylphényl] propanamide (1:1) On chauffe pendant 6 heures à la température de reflux 1,07 g (1,2 mmoles) de (S) -1- [2- (3-éthyl-l , l-dioxo-5-thién-2-yl-2H-
1,2, 4-benzothιadιazm-2-yl) -5- [5-méthyl-l- (triphénylméthyl) - lH-ιmιdazol-4-yl] - 1-oxopentyl] -4 -méthoxypipéridme en solution dans un mélange de 50 ml d'acide acétique et 50 ml d'eau et on concentre le milieu réactionnel sous pression réduite. On reprend le résidu par 150 ml d'acétate d'éthyle, on lave successivement par 50 ml d'une solution saturée d ' hydrogenocarbonate de sodium et 50 ml d'une solution saturée de chlorure de sodium, on sèche sur sulfate de sodium et on concentre sous pression réduite. On purifie le résidu par chromatographie sur colonne de gel de silice en éluant par un mélange méthanol : dichloromethane (5:95). On obtient 0,43 g de produit sous forme de base que l'on reprend dans 12 ml d'une solution d'acide ehlorhydrique 0,1 N dans 1 ' isopropanol et on concentre sous pression réduite. On purifie le résidu par chromatographie sur colonne RP 18 en éluant par un mélange acétonitrile : eau (3:7) .
On obtient après lyophilisation 0,34 g de produit sous forme d'une poudre blanche.
Rendement = 45 % Point de fusion = 126 °C
[α]20 = + 87 o (c = Qι2 ; méthanol))
Exemple 15 (composé n° 47) chlorhydrate de (S) -N- [2- [ [ [4- (5-méthyl-lH-ιmιdazol-4-yl) -1- [ (4-méthylènepιpéridm-l-yl) carbonyl] butyl] ammo] sulfonyl] -6- thιényl-2-ylphényl] propanamide (1:1)
On prépare ce composé selon la méthode décrite dans l'exemple 14 à partir du chlorhydrate de (S) -5-méthyl-α- [ (4-méthylène- pipéridin- 1-yl) carbonyl] -1- (triphénylméthyl) -lH-ιmιdazole-4- butanamine et du chlorure de 2- [ (1-chloropropylidêne) amino] - 3 -thién-2-ylbenzènesulf onyle . Point de fusion = 115-120 °C
[α] 20 = + 54 o (c = 0,2 ; méthanol)
Exemple 16 (composé n° 45) chlorhydrate de ( S) -N- [2 - [ [ [1- [ (4 -cyclopropylpιpéndm-1-yl ) carbonyl] -4- (5-méthyl- lH-imidazol -4 -yl) butyl] ammo] sulfonyl] - 6 -thiényl -2 -ylphényl ] propanamide (1:1)
16.1. (S) -4-cyclopropyl-l- [2- (3-éthyl-l, 1-dιoxo- 5-thién-2- yl-2H-l, 2,4-benzothιadιazm-2-yl) -5- [5-méthyl -1-
(triphénylméthyl) -lH-ιmιdazol-4-yl] -1-oxopentyl] pipéridme A un mélange de 0,38 g (1,1 mmoles) de chlorure de 2-[(l- chloropropylidêne) ammo] -3-thιén-2 -ylbenzènesulfonyle et de 0,64 g (1,1 mmoles) de chlorhydrate de (S) -α- [ (4-cyclopro- pylpιpéπdm-1-yl) carbonyl] -5-méthyl-l- (triphénylméthyl) -1H- ιmιdazole-4 -butanamine dans 20 ml de dichloromethane, on ajoute goutte à goutte à 0 °C, 0,5 ml (3,63 mmoles) de triethylamme. On laisse revenir la température du mélange à la température ambiante, on poursuit l'agitation pendant 2 heures à cette température et on concentre sous pression réduite. On reprend le résidu par 100 ml d'acétate d'éthyle, on lave successivement par 50 ml d'une solution aqueuse d'acide ehlorhydrique 1 Ν, 50 ml d'une solution saturée d ' hydrogenocarbonate de sodium et 50 ml d'une solution saturée de chlorure de sodium, on sèche sur sulfate de sodium et on concentre sous pression réduite. On obtient 1,1 g de produit sous forme d'une huile visqueuse que l'on utilise telle quelle dans l'étape suivante. Rendement = 100 %
16.2. chlorhydrate de { S) -N- [2- [ [ [1- [ (4-cyclopropylpιpé- rιdm-1-yl) carbonyl] -4- (5-méthyl-lH-ιmιdazol-4-yl) butyl] ammo] sulfonyl] -6-thιényl-2-ylphényl] propanamide (1:1) On chauffe pendant 4 heures à la température de reflux 1,1 g (1,1 mmoles) de { S) -4-cyclopropyl-l- [2- (3-éthyl-l , l-dιoxo-5- thιén-2-yl-2H-l, 2 , 4-benzothιadιazm-2-yl) -5- [5-méthyl-l-
(triphénylméthyl) -lH-ιmιdazol-4-yl] -1-oxopentyl] pipéridme en solution dans un mélange de 25 ml d'acide acétique et 25 ml d'eau et on concentre le milieu réactionnel sous pression réduite. On reprend le résidu par 150 ml d'acétate d'éthyle,
on lave successivement par 50 ml d'une solution saturée d ' hydrogenocarbonate de sodium et 50 ml d'une solution saturée de chlorure de sodium, on sèche sur sulfate de sodium et on concentre sous pression réduite. On purifie le résidu par chromatographie sur colonne de gel de silice en éluant par un mélange méthanol : dichloromethane (2:98 à 8:92). On obtient 0,528 g de produit sous forme de base. Rendement = 80 % On reprend 0,528 g de base dans 10 ml d'une solution d'acide ehlorhydrique 0,1 N dans 1 ' isopropanol et on concentre sous pression réduite. On purifie le résidu par chromatographie sur colonne RP 18 en éluant par un mélange acétonitrile : eau (3:7) . On obtient après lyophilisation 0,27 g de produit sous forme d'une poudre blanche.
Rendement = 39 %
Point de fusion = 108-110 °C [α] 20 = + 98 o (c = Q, 2 ; méthanol)
Exemple 17 (composé n° 20) chlorhydrate de (S) -N- [3 ' - (éthylammo) -3 - [ [ [1- [ (4-éthylpι- pérιdm-1-yl) carbonyl] -4- (5-méthyl-lH-ιmιdazol-4-yl) butyl] ammo] sulfonyl] [1 , 1 ' -biphényl] -2 -yl] propanamide (2:1)
17.1. { S) -1- [2- [7-bromo-3-éthyl-5- (3 -mtrophényl) -1,1- dιoxo-2H-l, 2 , 4-benzothιadιazm-2-yl] -5- [5-méthyl-l- (triphénylméthyl) -lH-ιmιdazol-4-yl] -1-oxopentyl] -4- éthylpipéπdme 17.1.1. chlorure de 5-bromo-2- [ (1-chloropropylιdène) ammo] - 3 ' -nitro [1,1' -biphenyle] -3-sulfonyle a) sel de pyridme de l'acide 5-bromo-3 ' -nιtro-2- [ (1-oxopro- pyl) ammo] [1,1' -biphenyle] -3 -sulfonique A une solution de 3,15 g (8,45 mmoles) d'acide 2-ammo-5- bromo-3 ' -nitro [1 , 1 ' -biphenyle] -3 -sulfonique et 2,4 ml (29,6 mmoles) de pyridme dans 10 ml de dichloromethane on ajoute goutte à goutte à 0 °C sous atmosphère d'azote, 1,62 ml (18,6 mmoles) de chlorure de propionyle. On laisse la température du mélange revenir à la température ambiante et on poursuit l'agitation pendant 18 heures. On concentre le
milieu réactionnel sous pression réduite.
On utilise le résidu tel quel dans l'étape suivante.
b) chlorure de 5-bromo-2- [ (1-chloropropylιdène) amino] -3 ' - nitro [1, 1 ' -biphenyle] -3 -suifonyle
On dissout le résidu obtenu précédemment dans 20 ml de dichloromethane et on ajoute à 0 °C sous atmosphère d'azote, 4,6 g (21,2 mmoles) de pentachlorure de phosphore. On laisse la température du mélange revenir à la température ambiante et on maintient le mélange pendant 5 heures sous agitation à cette température. On concentre le milieu réactionnel sous pression réduite, on reprend le résidu par 150 ml d'éther et on filtre sur fritte. On lave le filtrat par 2 fois 100 ml d'eau puis 100 ml d'une solution saturée de chlorure de sodium, on le sèche sur sulfate de magnésium et on concentre sous pression réduite.
On obtient 3 g de produit sous forme d'une huile vitreuse que l'on utilise telle quelle dans l'étape suivante. Rendement = 77 %
17.1.2. (S) -1- [2- [7-bromo-3-éthyl-5- (3-nιtrophényl) -1,1- dιoxo-2H-l, 2 , 4-benzothιadιazm-2-yl] -5- [5-méthyl-l- (triphénylméthyl) -l.tf-ιmιdazol-4 -yl] -1-oxopentyl] -4- éthylpipérid e A un mélange de 0,53 g (1,15 mmoles) de chlorure de 5-bromo- 2- [ (1-chloropropylιdène) amino] -3 ' -nitro [1 , 1 ' -biphenyle] -3- sulfonyle et de 0,58 g (1,02 mmoles) de chlorhydrate de { S) -α- [ (4-éthylpιpérιdm-l-yl) carbonyl] -5-méthyl-l- ( riphénylméthyl) -lH-ιmιdazole-4 -butanamine dans 5 ml de dichloromethane, on ajoute goutte à goutte à 0 °C, 0,42 ml (3,5 mmoles) de triethylamme. On laisse revenir la température du mélange à la température ambiante, on poursuit l'agitation pendant 18 heures à cette température et on concentre sous pression réduite. On reprend le résidu par 100 ml d'acétate d'éthyle, on lave successivement par 50 ml d'une solution aqueuse d'acide ehlorhydrique 1 N, 50 ml d'une solution saturée d ' hydrogenocarbonate de sodium et 50 ml d'une solution saturée de chlorure de sodium, on sèche sur sulfate de sodium et on concentre sous pression réduite.
On obtient 1 g de produit sous forme d'une huile visqueuse que l'on utilise telle quelle dans l'étape suivante. Rendement = 100 %
17.2. (S) -N- [5-bromo-3- [[ [1- [ (4-éthylpιpéπdm-l-yl) carbonyl] -4- (5-méthyl-lH-ιmιdazol-4-yl) butyl] ammo] sulfonyl] -3 ' -nitro [1, 1 ' -biphényl] -2-yl] propanamide On chauffe pendant 8 heures à la température de reflux 1 g (1 mmole) de (S) -1- [2- [7-bromo-3 -éthyl-5- (3-nιtrophényl) -1 , 1- dιoxo-2H-l, 2,4-benzothιadιazm-2-yl] -5- [5-méthyl-l- (triphénylméthyl) -lH-ιmιdazol-4-yl] -1-oxopentyl] -4-éthylpιpérιdme en solution dans un mélange de 30 ml d'acide acétique et 20 ml d'eau et on concentre le milieu réactionnel sous pression réduite. On reprend le résidu par 100 ml d'acétate d'éthyle, on lave par 50 ml d'une solution saturée d ' hydrogenocarbonate de sodium, on sèche sur sulfate de sodium et on concentre sous pression réduite. On purifie le résidu par chromatographie sur colonne de gel de silice en éluant par un mélange méthanol : dichloromethane (5:95) . On obtient 0,42 g de produit sous forme d'un solide blanc. Rendement = 60 % Point de fusion = 215 °C
17.3. { S) -N- [3 ' -amino-3- [ [ [1- [ (4 -éthylpιpéπdm-1-yl) carbonyl] -4- (5-méthyl-lH-ιmιdazol-4-yl) butyl] ammo] sulfonyl] [1,1' -biphényl] -2-yl] propanamide On hydrogène pendant 10 heures à la température ambiante à 0,35 MPa (50 psi), 0,4 g (0,56 mmole) de (S) -N- [5-bromo-3- [ [ [1- [ (4-éthylpιpéπdm-l-yl) carbonyl] -4- (5-méthyl-lH- ιmιdazol-4-yl) butyl] ammo] sulfonyl] -3 ' -nitro [1 , 1 ' -biphényl] - 2-yl] propanamide dans 20 ml d' éthanol en présence de 0,1 g de palladium sur charbon à 10 %. On filtre le milieu réactionnel sur célite et on concentre le filtrat sous pression réduite. On reprend le résidu par 100 ml d'acétate d'éthyle, on lave par 50 ml d'une solution saturée d ' hydrogenocarbonate de sodium, on sèche sur sulfate de sodium et on concentre sous pression réduite.
On obtient 0,33 g de produit sous forme d'un solide blanc. Rendement = 100 %
Point de fusion = 160 °C
17.4. chlorhydrate de { S) -N- [3 ' - (éthylammo) -3- [ [ [1- [ (4 - éthylpιpérιdm-1-yl) carbonyl] -4- ( 5-méthyl -lH-imida- zol-4-yl)butyl] amino] sulfonyl] [1 , 1 ' -biphényl] -2 -yl] propanamide (2:1) A 0,33 g (0,56 mmole) de (S) -N- [3 ' -ammo-3- [ [ [1- [ (4 -éthylpi- pérιdm-1-yl) carbonyl] -4- (5-méthyl- lH-imidazol-4 -yl) butyl] ammo] sulfonyl] [1 , 1 ' -biphényl] -2-yl] propanamide dans 10 ml d'éthanol, on ajoute 0,043 ml (0,78 mmole) d ' acétaldéhyde et 70 mg de palladium sur charbon à 10 % et on agite le mélange pendant 8 heures à la température ambiante sous une pression de 0,35 MPa (50 psi) . On filtre le milieu réactionnel sur célite, on ajoute au filtrat 4 ml d'une solution d'acide ehlorhydrique 0,1 Ν dans 1 ' isopropanol et on concentre sous pression réduite. On purifie le résidu par chromatographie sur colonne RP 18 en éluant par un mélange acétonitrile : eau (3:7) . On obtient après lyophilisation 0,2 g de produit sous forme de poudre blanche. Rendement = 53 % Point de fusion = 163 °C [α]20 = + 130 o (c = 0,2 ; méthanol)
Exemple 18 (composé n° 29) chlorhydrate de (S) -N- [3 - [ [ [1- [ (4 -éthylpιpérιdm-1-yl) carbonyl] -4- (5-méthyl-lH-ιmιdazol-4-yl) butyl] ammo] sulfonyl] [1,1' -biphényl] -2-yl] -2- (2-méthoxyéthoxy) acétamide (1:1)
18.1. (S) -2 -ammo-N- [1- [ (4-éthylpιpéπdm-l-yl) carbonyl] -4- [5-méthyl-l- (triphénylméthyl) -lH-ιmιdazol-4-yl] butyl] [1,1' -biphényl] -3-sulfonamιde A un mélange de 0,5 g (0,87 mmole) de chlorhydrate de { S) -5-éthyl-α- [ (4-éthylpιpéπdm-l-yl) carbonyl] -1- (triphé- nylméthyl) -lH-ιmιdazole-4-butanamme et de 0,281 g (1,05 mmoles) de chlorure de 2-ammo [1 , 1 ' -biphenyle] -3 -suifonyle en solution dans 10 ml de dichlorométnane, on ajoute goutte à goutte à 0 °C sous atmosphère d'argon, 0,365 ml (2,61 mmoles) de triethylamme. On laisse le mélange sous agitation pendant
1 heure à cette température puis on concentre sous pression réduite. On reprend le résidu dans 50 ml d'acétate d'éthyle, on lave successivement par 20 ml d'une solution d'acide ehlorhydrique 1 N, 20 ml d'une solution saturée d'hydrogé- nocarbonate de sodium et 20 ml d'une solution saturée de chlorure de sodium puis on sèche sur sulfate de magnésium. Finalement on filtre et on concentre à sec. On purifie le résidu par chromatographie sur colonne de gel de silice en éluant par un mélange méthanol : dichloromethane (1:99 puis 3 :97) .
On obtient 0,53 g de produit sous forme d'un solide blanc. Rendement = 79 %
18.2. chlorhydrate de { S) -N- [3- [ [ [1- [ (4-éthylpιpérιdm-l- yl) carbonyl] -4- (5-méthyl-lH-ιmιdazol-4 -yl) butyl] ammo] sulfonyl] [1,1' -biphényl] -2-yl] -2- (2-méthoxy- éthoxy) acétamide (1:1) A 1 g (1,3 mmoles) de (S) -2 -ammo-N- [1- [ (4-éthylpιpérιdm- 1- yl ) carbonyl] -4- [5-méthyl-l- (triphénylméthyl) -lH-ιmιdazol-4 - yl] butyl] [1 , 1 ' -biphényl] -3 -suifonamide en solution dans 10 ml de diméthylacétamide, on ajoute à température ambiante sous argon, 2 g (13 mmoles) de chlorure de 2- (2 -méthoxyéthoxy) acé- tyle. On laisse le milieu réactionnel sous agitation à cette température pendant 0,5 heure puis on le refroidit par un bain de glace. On ajoute 100 ml d'acétate d'éthyle et 100 ml d'une solution aqueuse d'acide ehlorhydrique 1 Ν et on récupère la phase organique. On la lave successivement par 50 ml d'une solution saturée d' hydrogenocarbonate de sodium et 50 ml d'une solution saturée de chlorure de sodium puis on concentre sous vide. On reprend le résidu par un mélange acide acétique : eau: tétrahydrofurane (2:1:1), on chauffe le mélange pendant 3 heures à 80 °C et on concentre sous vide. On reprend le résidu par 100 ml d'acétate d'éthyle, on lave successivement par 50 ml d'une solution d'acide ehlorhydrique 1 Ν, 50 ml d'une solution saturée d ' hydrogenocarbonate de sodium et 50 ml d'une solution saturée de chlorure de sodium puis on sèche sur sulfate de magnésium. Finalement on filtre et on concentre sous vide. On purifie le résidu par chromatographie sur colonne de gel de silice en éluant par un
mélange dichloromethane : méthanol (98:2 puis 90:10).
On obtient 0,54 g de produit sous forme de base.
On prépare le chlorhydrate dans 10 ml d'une solution d'acide ehlorhydrique 0,1 N dans 1 ' isopropanol . On obtient après lyophilisation 0,57 g de produit.
Rendement = 64,6 %
Point de fusion = 98 °C [α]20 _ + 55 t 5 o (c = o,2 ; méthanol)
Exemple 19 (composé n° 30) chlorhydrate de { S) -N- [2-cyclopentyl-6- [ [ [1- [ (4-éthylpιpé- πdm-l-yl) carbonyl] -4- (5-méthyl- lH-imidazol -4 -yl) butyl] ammo] sulfonyl] phényl] -N ' -éthylurée (1:1)
19.1. { S) -2 -ammo-3 - cyclopentyl -N- [1- [ (4-éthylpιpéπdm-l- yl) carbonyl] -4- [5-méthyl-l- (triphénylméthyl) -1H- ιmιdazol-4-yl] butyl] benzènesuifonamide A une solution de 0,70 g (2,7 mmoles) de chlorure de 2-ammo- 3 -cyclopentylbenzènesulfonyle et de 1,54 g (2,7 mmoles) de chlorhydrate de (S) -5-méthyl-α- [ (4-éthylpιpéπdm-l-yl) carbonyl] -1- (triphénylméthyl) -lH-ιmιdazole-4-butanamme dans 6 ml de dichloromethane, on ajoute goutte à goutte à 0 °C, 0,95 ml (6,75 mmoles) de triethylamme. On laisse le milieu réactionnel sous agitation pendant 5 heures à cette température puis on concentre sous pression réduite. On reprend le résidu dans 100 ml d'acétate d'éthyle, on lave successivement par 50 ml d'une solution d'acide ehlorhydrique 1 Ν, 50 ml d'une solution saturée d ' hydrogenocarbonate de sodium et 50 ml d'une solution saturée de chlorure de sodium, on sèche sur sulfate de magnésium et on concentre sous pression réduite. On purifie le résidu par chromatographie sur colonne de gel de silice en éluant par un mélange dichloromethane : méthanol (98:2) . On obtient 1,8 g de produit sous forme d'une huile visqueuse. Rendement = 88 %
19.2. { S) -N- [2-cyclopentyl-6- [ [ [1- [ (4-éthylpιpéndm-l- yl) carbonyl] -4- [5-méthyl-l- (triphénylméthyl) -1H- ιmιdazol-4-yl] butyl] ammo] sulfonyl] phényl] -N ' - ethylurée On chauffe pendant 38 heures à 50 °C une solution de 0,75 g (1 mmole) de { S) - 2 -ammo- 3 -cyclopentyl -N- [1- [ (4-éthylpιpé- πdm-l-yl) carbonyl] -4- [5-méthyl-l- (triphénylméthyl) - 1H- ιmιdazol-4-yl] butyl] benzènesulfonamide et de 0,32 ml (4 mmoles) d'isocyanate d'éthyle dans le diméthylformamide puis on concentre le milieu réactionnel sous pression réduite. On purifie le résidu obtenu par chromatographie sur colonne de gel de silice en éluant par un mélange méthanol : dichloromethane (2:98).
On obtient 0,53 g de produit sous forme d'un solide blanc Rendement = 65 %
Point de fusion = 115 °C
19.3. chlorhydrate de (S) -N- [2 -cyclopentyl -6- [ [ [1- [ (4 - éthylpιpérιdm-1-yl) carbonyl] -4- ( 5-méthyl -1H- îmidazol -4 -yl ) butyl ] ammo] sul fonyl ] phényl ] -N ' - ethylurée (1:1) On agite pendant 40 heures à la température ambiante sous une pression de 0,35 MPa (50 psi) un mélange de 0,53 g (0,64 mmole) de (S) -N- [2-cyclopentyl-6- [ [ [1 - [ (4-éthylpιpé- πdm-l-yl) carbonyl] -4- [5-méthyl-l- (triphénylméthyl) - 1H- ιmιdazol-4-yl] butyl] ammo] suifonyl] phényl] -N' -ethylurée et de 0,2 g de palladium sur charbon à 10 % dans 8 ml d'une solution d'acide ehlorhydrique 0,1 Ν dans 1 ' isopropanol . On filtre le mélange sur célite et on concentre le filtrat sous pression réduite. On purifie le résidu ainsi obtenu sur colonne RP 18 en éluant par un mélange acétonitπle : eau (4:6) On obtient après lyophilisation 0,30 g de produit. Rendement = 77 % Point de fusion = 147 °C [α]20 = + 86 ° (c = 0,2 ; méthanol)
Exemple 20 (composé n° 70) chlorhydrate de { S) -N- [3 - [ [ [4- (5-chloro-lH-imidazol-4 -yl ) - 1- [ (4-éthylpipéridin-l-yl) carbonyl] butyl] amino] sulfonyl] [1,1' -biphényl] -2-yl] propanamide (1:1)
20.1. { S) -N- [3- [ [ [1- [ (4-éthylpipéridin-l-yl) carbonyl] -4- (lH-imidazol-4-yl) butyl] amino] sulfonyl] [1,1' -biphényl] -2-yl] propanamide 20.1.1. chlorure de 2- [ (1 -ehloropropylidène) amino] - [1 , 1 ' - biphenyle] -3 -suifonyle
On le prépare selon la méthode décrite dans l'exemple 14.1.1. à partir de l'acide 2 -amino [1 , 1 ' -biphenyle] -3 -suifonique . On obtient le produit sous forme d'une huile visqueuse que l'on utilise telle quelle dans l'étape suivante.
20.1.2. (S) -N- [3- [ [ [1- [ (4-éthylpipéridin-l-yl] carbonyl] -4- (lH-imidazol-4-yl) butyl] amino] sulfonyl] [1,1' -biphényl] -2-yl] propanamide A un mélange de 1,8 g (5,3 mmoles) de chlorure de 2-[(l- ehloropropylidène) amino] - [1, 1 ' -biphenyle] -3 -suifonyle et de 2,7 g (4,8 mmoles) de chlorhydrate de (S) -α- [ (4 -éthylpipé- ridin-1-yl) carbonyl] -1- (triphénylméthyl) -lH-imidazole-4 - butanamine dans 30 ml de dichloromethane, on ajoute goutte à goutte à 0 °C sous azote, 2,2 ml (15,84 mmoles) de triéthylamine . On laisse revenir la température du mélange à la température ambiante, on poursuit l'agitation pendant 18 heures à cette température et on concentre sous pression réduite. On reprend le résidu par 150 ml d'acétate d'éthyle, on lave successivement par 100 ml d'une solution aqueuse d'acide ehlorhydrique 1 Ν, 50 ml d'une solution saturée d ' hydrogenocarbonate de sodium et 50 ml d'une solution saturée de chlorure de sodium, on sèche sur sulfate de sodium et on concentre sous pression réduite. On reprend le résidu par un mélange de 60 ml d'acide acétique et de 40 ml d'eau et on chauffe le mélange pendant 6 heures à la température de reflux puis on concentre le milieu réactionnel sous pression réduite. On reprend le résidu par 200 ml d'acétate d'éthyle, on lave successivement par 50 ml d'une solution saturée d ' hydrogenocarbonate de sodium et
50 ml d'une solution saturée de chlorure de sodium, on sèche sur sulfate de sodium et on concentre sous pression réduite. On purifie le résidu par chromatographie sur colonne de gel de silice en éluant par un mélange méthanol : dichloromethane (5:95) .
On obtient 2,2 g de produit sous forme d'une huile visqueuse. Rendement = 81 %
20.2. chlorhydrate de (S) -N- [3- [ [ [4- (5-chloro-lH-ιmιdazol- 4-yl) -1- [ (4-éthylpιpérιdm-l-yl) carbonyl] butyl] ammo] sulfonyl] [1 , 1 ' -biphényl] -2-yl] propanamide (1:1) On agite pendant 5 heures à 0 °C une solution de 0,56 g (1 mmole) de (S) -N- [3- [[ [1- [ (4-éthylpιpéπdm-l-yl] carbonyl] - 4- (lH-imidazol-4-yl) butyl] am o] sulfonyl] [1,1' -biphényl] -2- yl] propanamide et de 0,116 g (1,1 mmoles) de N-chloro- suc - cmimide dans 2 ml de diméthylformamide puis on concentre le milieu réactionnel sous pression réduite. On purifie le résidu par chromatographie sur colonne de gel de silice en éluant par un mélange méthanol : dichloromethane (2:98) . On obtient 0,3 g de produit sous forme de base. Rendement = 50 %
On reprend la base dans 15 ml d'une solution d'acide ehlorhydrique 0,1 Ν dans 1 ' isopropanol et on concentre sous pression réduite. On purifie le résidu ainsi obtenu sur colonne RP 18 en éluant par un mélange acétonitrile : eau (6:4) .
On obtient après lyophilisation 0,30 g de produit. Rendement = 94 % Point de fusion = 100 °C [«]D° = + 102 ° (c = 0,2 ; méthanol)
Exemple 21 (composé n° 23) chlorhydrate de (S) -N- [2- [ [ [1- [ ( -éthylpιpérιdm-1-yl ) carbonyl] -4- (5-méthyl-lH-ιmιdazol-4 -yl ) butyl] ammo] sulfonyl] 6 -pyrîdin-2 -ylphényl ] propanamide (2:1)
21.1. { S) -N- [4-bromo-2- [ [ [1- [ (4 -éthylpιpérιdm-1-yl) carbonyl] -4- [5-méthyl-l- (triphénylméthyl) -1H- ιmιdazol-4-yl] butyl] ammo] sulfonyl] -6-ιodophényl] propanamide A une solution de 2,28 g (4 mmoles) de chlorhydrate de { S) -5-méthyl-α- [ (4-éthylpιpérιdm-l-yl) carbonyl] -1- (triphénylméthyl) -lH-ιmιdazole-4 -butanamine dans 25 ml de dichloromethane, on ajoute successivement à 0 °C 2,24 g (4,4 mmoles) de chlorure de 2 - [bis (1-oxopropyl) amino] -5- bromo-3-ιodobenzènesulfonyle puis goutte à goutte 1,84 ml
(13,2 mmoles) de triethylamme. On laisse le mélange pendant 6 heures sous agitation à cette température et on concentre sous pression réduite. On reprend le résidu par 200 ml d'acétate d'éthyle, on lave successivement par 100 ml d'une solution aqueuse d'acide ehlorhydrique 1 Ν, 100 ml d'une solution saturée d ' hydrogenocarbonate de sodium et 100 ml d'une solution saturée de chlorure de sodium, on sèche sur sulfate de sodium et on concentre sous pression réduite. On reprend le résidu dans 150 ml de tétrahydrofurane, on fait passer un courant d'ammoniac gazeux à 0 °C pendant 2 heures et on concentre sous pression réduite. On purifie le résidu par chromatographie sur colonne de gel de silice en éluant par un mélange dichloromethane :méthanol (98:2). On obtient 2,64 g de produit sous forme d'une huile vitreuse. Rendement = 70 %
21.2. (S) -N- [4-bromo-2- [ [ [1- [ (4-éthylpιpérιdm-l-yl) carbonyl] -4- [5-méthyl-l- (triphénylméthyl) - 1H- ιmιdazol-4-yl) butyl] ammo] sulfonyl] -6-pyrιdm-2- ylphényl] propanamide
On chauffe à 80 °C sous argon pendant 5 heures un mélange de 1,9 g (2 mmoles) de { S) -N- [4-bromo-2- [ [ [1- [ (4-éthylpιpéπdm- 1-yl) carbonyl] -4- [5-méthyl-l- (triphénylméthyl) -lH-imidazol- 4 -yl] butyl] ammo] sulfonyl] -6 -îodophényl] propanamide, 0,883 g
(2,4 mmoles ) de 2 - (tributylstannyl) pyridine , 0,1 g (0,17 mmole) de bis (dibenzylidèneacétone) palladium (0), 0,033 g (0,17 mmole) d'iodure de cuivre et 0,98 g (0,34 mmole) de triphénylarsine dans 4 ml de diméthylfor- mamide . On reprend le milieu réactionnel dans 150 ml d'acétate d'éthyle, on lave par deux fois 100 ml d'une solution aqueuse d'ammoniaque à 10 % puis par 50 ml d'une solution saturée de chlorure de sodium. On sèche sur sulfate de sodium et on concentre sous pression réduite. On purifie le résidu par chromatographie sur colonne de gel de silice en éluant par un mélange dichloromethane : méthanol (98:2). On obtient 1,15 g de produit sous forme d'une huile visqueuse . Rendement = 64 %
21.3. (S) -N- [4-bromo-2- [ [ [1- [ (4 -éthylpipéridin-1-yl) carbonyl] -4- ( 5 -méthyl-lH-imidazol -4-yl) butyl] amino] sulfonyl] -6 -pyridin-2 -ylphényl] propanamide On chauffe pendant 1 heure à la température de reflux, 1,14 g (1,26 mmoles) de (S) -N- [4-bromo-2 - [ [ [1- [ (4-éthylpipéridin-l- yl) carbonyl] -4- [5-méthyl-l- (triphénylméthyl) -lH-imidazol -4- yl) butyl] amino] sulfonyl] -6 -pyridin-2 -ylphényl] propanamide dans un mélange de 30 ml d'acide acétique et 15 ml d'eau. On concentre le milieu réactionnel sous pression réduite et on reprend le résidu dans 150 ml d'acétate d'éthyle. On lave successivement par 50 ml d'une solution saturée d' hydrogenocarbonate de sodium et 50 ml d'une solution saturée de chlorure de sodium, on sèche sur sulfate de sodium et on concentre sous pression réduite . On purifie le résidu par chromatographie sur colonne de gel de silice en éluant par un mélange méthanol : dichloromethane (5:95) .
On obtient 0,66 g de produit sous forme d'une huile vitreuse. Rendement = 79,5 %
21.4. chlorhydrate de (S) -N- [2- [ [ [1- [ (4-éthylpιpérιdm-l- yl) carbonyl] -4- (5-méthyl-lH-ιmιdazol-4 -yl) butyl] ammo] sulfonyl] -6-pyrιdm-2-ylphényl] propanamide (2:1) On chauffe pendant 3 heures à la température de reflux un mélange de 0,65 g (0,98 mmole) de (S) -N- [4-bromo-2- [ [ [1- [ (4 - éthylpιpérιdm-1-yl) carbonyl] -4- (5-méthyl-lH-ιmιdazol-4- yl) butyl] ammo] sulfonyl] -6 -pyridin-2 -ylphényl] propanamide, 1,24 g (20 mmoles) de formiate d'ammonium et 0,065 g de palladium sur charbon dans 10 ml de méthanol contenant 0 , 2 ml d'acide acétique puis on concentre le milieu réactionnel sous pression réduite. On reprend le résidu par 100 ml d'acétate d'éthyle, on lave successivement par 50 ml d'une solution saturée d ' hydrogenocarbonate de sodium et 50 ml d'une solution saturée de chlorure de sodium, on sèche sur sulfate de sodium et on concentre sous pression réduite. On purifie le résidu par chromatographie sur colonne de gel de silice en éluant par un mélange méthanol : dichloromethane (5:95). On obtient 0,55 g de base sous forme d'une huile visqueuse. Rendement = 96 %
On reprend la base dans 25 ml d'une solution d'acide ehlorhydrique 0,1 Ν dans 1 ' isopropanol et on concentre sous pression réduite. On purifie le résidu ainsi obtenu sur colonne RP 18 en éluant par un mélange acétonitrile : eau (6:4) .
On obtient 0,44 g de produit. Rendement = 68 % Point de fusion = 138-144 °C [α] 0 = + 121 ° (c = 0,2 ; méthanol)
Exemple 22 (composé n° 22) chlorhydrate de { S) -N- [2 - [ [ [1- [ (4-éthylpipéridin- 1-yl) carbonyl] -4- (5-méthyl-lH-imidazol-4 -yl) butyl] amino] sulfonyl] - 4-fluoro-6 -thién-2 -ylphényl] propanamide (1:1)
22.1. chlorure de 2 - [bis (1-oxopropyl) amino] -5-fluoro-3 - thién-2 -ylbenzènesulfonyle
22.1.1. sel de N, N-diéthyléthanamine de l'acide
2- [bis (1-oxopropyl) amino] -5-fluoro-3-thién-2- ylbenzènesulfonique
On chauffe pendant 24 heures à la température de reflux un mélange de 10,92 g (40 mmoles) d'acide 2 -amino-5-fluoro-3 - thién-2 -ylbenzènesulfonique et de 5,6 ml (40 mmoles) de triethylamine dans 77 ml (600 mmoles) d'anhydride propionique puis on concentre sous pression réduite. On cristallise le résidu dans un mélange acétate d' éthyle : éther . On obtient 16,9 de produit. Rendement = 90 % Point de fusion = 239 °C
22.1.2. chlorure de 2 - [bis (1-oxopropyl) amino] -5-fluoro-3 - thién-2 -ylbenzènesulfonyle
A une solution de 1,60 g (34,1 mmoles) de sel de
N, N-diéthyléthanamine de l'acide 2 - [bis (1-oxopropyl) amino] -5- fluoro-3-thién-2-ylbenzènesulfonique dans 60 ml de dichloromethane, on ajoute à 0 °C sous azote 14,22 g (68,2 mmoles) de pentachlorure de phosphore. On maintient le mélange pendant 5 heures sous agitation à cette température, on laisse la température remonter à la température ambiante et on poursuit l'agitation pendant 1 heure à cette température. On ajoute 200 ml d'éther, on filtre et on concentre sous pression réduite. On reprend le résidu par
800 ml d'éther, on filtre et on concentre sous pression réduite. On purifie le résidu par chromatographie sur colonne ® Florisil en éluant par un mélange éther :pentane (1:9) puis
(1:1) .
On obtient 6,4 g de produit sous forme d'une huile visqueuse.
Rendement = 55 %
22.2. chlorhydrate de { S) -N- [2 - [ [ [1- [ (4 -éthylpιpérιdm-1 - yl) carbonyl] -4- (5-méthyl-lH-ιmιdazol-4 -yl) butyl] amino] sulfonyl] -4 -fluoro-6-thιén-2-ylphényl] propanamide (1:1) A une solution de 9,83 g (17,2 mmoles) de chlorhydrate de ( S) -α- [ (4-éthylpιpérιdm-l-yl) carbonyl] -5-méthyl- 1- (triphénylméthyl) -lH-ιmιdazole-4 -butanamine dans 60 ml de dichloromethane, on ajoute sous azote à 0 °C, 6,16 g (18,1 mmoles) de chlorure de 2- [bis (1-oxopropyl) ammo] -5- fluoro-3-thιén-2-ylbenzènesulfonyle puis 5,5 mmoles de triethylamme. On laisse la température du milieu réactionnel revenir lentement à la température ambiante, on poursuit l'agitation à cette température pendant 15 heures et on concentre sous pression réduite. On reprend le résidu dans 300 ml d'acétate d'éthyle et on lave successivement par 300 ml d'une solution aqueuse d'acide ehlorhydrique 1 Ν, 300 ml d'une solution saturée d' hydrogenocarbonate de sodium puis 300 ml d'une solution saturée en chlorure de sodium. On sèche sur sulfate de sodium, on filtre et on concentre sous pression réduite.
On chauffe le résidu pendant 12 heures dans 225 ml d'un mélange acide acétique: eau (2:1) et on concentre sous pression réduite. On reprend le résidu dans 400 ml de dichloromethane et on lave successivement par 200 ml d'une solution saturée d' hydrogenocarbonate de sodium puis 200 ml d'une solution saturée en chlorure de sodium. On sèche sur sulfate de sodium, on filtre et on concentre sous pression réduite. On purifie le résidu ainsi obtenu par chromatographie sur colonne de gel de silice en éluant par un mélange dichloromethane :méthanol (96:4).
On obtient 7,7 g de produit sous forme de base.
Rendement = 74 %
Point de fusion = 145-150 °C
On prépare le chlorhydrate en ajoutant 45,1 ml d'une solution d'acide ehlorhydrique 0,1 Ν dans 1 ' isopropanol à 2,5 g (4,5 mmoles) de base.
On obtient 2,7 g de produit sous forme de chlorhydrate. Point de fusion = 145 °C [α]2° = + 112 ° (c = 0,2 ; méthanol)
Exemple 23 (composé n° 53) chlorhydrate de { S) -N- [2 -[[ [1- [ [4- (difluorométhylène) pipéridin-1-yl] carbonyl] -4- (5-méthyl-lH-imidazol-4-yl) butyl] amino] sulfonyl] -6-thién-2 -ylphényl] propanamide (1:1)
On prépare ce produit selon le mode opératoire décrit dans l'exemple 19 à partir de 0,81 g (1,36 mmoles) de chlorhydrate de (S) -5-méthyl-α- [ [4- (difluorométhylène) pipéridin- 1-yl] carbonyl] -1- (triphénylméthyl) -lH-imidazole-4 -butanamine et de 0,47 g (1,36 mmoles) de chlorure de 2- [ (1 -ehloropropylidène) amino] -3 -thién-2 -ylbenzènesulfonyle .
On obtient 0,58 g de produit sous forme de poudre blanche. Rendement = 67 % Point de fusion = 144-145 °C [ ]o° = + 107,9 ° (c = 0,2 ; méthanol)
Exemple 24 (composé n° 25) chlorhydrate de (S) -N- [6-cyclopentyl-2- [ [ [1- [ (4 -éthylpipé- ridin-1-yl) carbonyl] -4- (5-méthyl-lH-imidazol-4 -yl) butyl] amino] suifonyl] phényl] propanamide (1:1)
24.1. chlorure de 3-cyclopentyl-2 - (diacétylaminc ) benzène suifonyle 24.1.1. sel de N, N-diéthyléthanamine de l'acide 3-cyclopentyl-2- (diacétylamino) benzènesulfonique
On chauffe pendant 48 heures à la température de reflux 6,5 g (19 mmoles) de sel de N, N-diéthyléthanamine de l'acide 2-amino-3-cyclopentylbenzènesulfonique en solution dans du chlorure d'acétyle puis on concentre le milieu réactionnel sous pression réduite.
On obtient 8,22 g de produit que l'on utilise tel quel dans l'étape suivante.
Rendement = 95 %
Point de fusion = 186 °C
24.1.2. chlorure de 3-cyclopentyl-2 - (diacétylamino) benzènesulfonyle On prépare ce composé selon le mode opératoire décrit dans l'exemple 22.1 à partir de 8,2 g (18,1 mmoles) d'acide
3 -cyclopentyl -2 - (diacétylamino) benzenesulfonique . On obtient 3,81 g de produit sous forme d'une huile visqueuse que l'on utilise telle quelle dans l'étape suivante. Rendement = 57 %
24.2. chlorhydrate de (S) -N- [6-cyclopentyl-2- [ [ [1- [ (4- éthylpιpérιdm-1-yl) carbonyl] -4- ( 5-méthyl - 1H- ιmιdazol-4-yl) butyl] ammo] sulfonyl] phényl] propanamide (1:1) On prépare ce produit selon le mode opératoire décrit dans l'exemple 22.1 à partir de 0,743 g (2 mmoles) de chlorure de 3 -cyclopentyl -2- (diacétylamino) benzenesulfonyle et de 1,14 g (2 mmoles) de { S) -α- [ (4 -éthylpιpéπdm-1-yl) carbonyl] -5- méthyl-1- (triphénylméthyl) - lH-imidazole-4 -butanamine . On obtient 0,73 g de produit. Rendement = 60 % Point de fusion = 124 °C [α] 0 = + 89 o (c = o,2 ; méthanol)
Exemple 25 (composé n° 64) chlorhydrate de (S) -N- [2- [ [ [4- (5-méthyl-lH-ιmιdazol-4-yl) -1- [ (5-oxohexahydro-lH-l , 4-dιazépm-l-yl) carbonyl] butyl] ammo] sulfonyl] -6-thién-2 -ylphényl] propanamide (1:1)
25.1. 1- [2- [[ (2-ammo-3-thιén-2-ylphényl) sulfonyl] ammo] -5- [5-méthyl-l- (triphénylméthyl) -lH-ιmιdazol-4-yl] -1- oxopentyl] hexahydro-5H-l , 4-dιazépm-5-one On place sous argon 0,618 g (2,26 mmoles) de chlorure de 2 -ammo-3 -thιén-2 -ylbenzènesulfonyle dans 80 ml de dichloromethane et on ajoute 1,29 mg (2,26 mmoles) de chlorhydrate de 1- [2-amino-5- [5-méthyl-l- (triphénylméthyl) - lH-ιmιdazol-4-yl] -1-oxopentyl] hexahydro-5H-l , 4-dιazépm-5- one . On refroidit le mélange à 0 °C au bain de glace et on ajoute lentement 0,94 ml (6,74 mmoles) de triethylamme. On laisse la température du milieu réactionnel revenir à la température ambiante et on poursuit l'agitation pendant une nuit à cette température. On ajoute alors 10 ml d'une solution aqueuse d'acide ehlorhydrique 0,5 Ν, on lave la phase organique par une solution aqueuse d'acide
ehlorhydrique 0,5 N puis par une solution saturée de chlorure de sodium et on sèche sur sulfate de magnésium. On purifie le résidu par chromatographie sur colonne de gel de silice en éluant par un mélange dichloromethane : méthanol (97,5:2,5). On obtient 1,33 g de produit. Rendement = 76 %
25.2. (S) -N- [2- [ [ [4- [5-méthyl-l- (triphénylméthyl) - 1H- ιmιdazol-4-yl] -1- [ (5-oxohexahydro- 1H-1 , 4-dιazépm-l- yl) carbonyl] butyl] ammo] sulfonyl] - 6-thién-2 - ylphényl] propanamide On place 1,33 g (1,72 mmoles) de 1- [2 [ [ (2-ammo-3 -thιén-2 - ylphényl) sulfonyl] am o] -5- [5-méthyl-l- (triphénylméthyl) -1H- ιmιdazol-4-yl] -1-oxopentyl] hexahydro-5H-l , 4-dιazépm-5-one dans 1,3 ml de diméthylacétamide et on ajoute 0,3 ml
(3,44 mmoles) de chlorure de propionyle On laisse le mélange pendant 2 heures sous agitation puis on ajoute de l'acétate d'éthyle. On évapore à sec et on reprend le résidu dans du dichloromethane. On lave par 2 fois 20 ml d'une solution aqueuse d'acide ehlorhydrique 1 Ν, on sèche sur sulfate de magnésium et on évapore.
On obtient 1,4 g de produit que l'on utilise tel quel dans l'étape suivante. Rendement = 100 %
25.3. chlorhydrate de (S) -N- [2 - [ [ [4- ( 5-méthyl -lH-imidazol - 4-yl) -1- [ (5-oxohexahydro-lH-l, 4-dιazépm-l-yl) carbonyl] butyl] ammo] sulfonyl] -6 -thién-2 -ylphényl] propanamide (1:1) On place 0,25 g (0,31 mmole) de (S) -N- [2 - [ [ [4- [5-méthyl-l- (triphénylméthyl) -lH- mιdazol-4-yl] -1- [ (5-oxohexahydro-lH- 1 , 4-dιazépm-l-yl) carbonyl] butyl] ammo] sulfonyl] -6-thιén-2- ylphényl] propanamide dans 4 ml de tétrahydrofurane et on ajoute 2 ml d'acide acétique et 2 ml d'eau. On chauffe le mélange pendant une nuit à 50 °C, on évapore le milieu à sec et on reprend le résidu par 100 ml de dichloromethane. On lave avec une solution aqueuse d' hydrogenocarbonate de sodium et on sèche sur sulfate de magnésium. On filtre et on purifie le résidu par chromatographie sur colonne de gel de silice en
éluant par un mélange dichloromethane : méthanol (90:10). On dissout la base dans une solution d'acide ehlorhydrique 0,1 N dans 1 ' isopropanol et on purifie le produit par chromatographie sur colonne de silice RP 18 en éluant par un mélange acétonitrile/eau.
On obtient 0,026 g de produit sous forme de chlorhydrate.
Rendement = 14 %
Point de fusion = 155 °C
[ ]n° = + 80 ° (c = 0,2 ; méthanol)
Exemple 26 (composé n° 65) chlorhydrate de (S) -N-cyclopentyl -N, 5-dιméthyl-α- [ [ [2- [ (1- oxopropyl) ammo] -3-thιén-2-ylphényl] sulfonyl] ammo] - 1H- ιmιdazole-4-pentanamιde (1:1)
26.1. (S) -α- [ [ (2-ammo-3-thιén-2-ylphényl) sulfonyl] ammo] - N- cyclopentyl -N, 5-dιméthyl- 1- (triphénylméthyl) - 1H- ιmιdazole-4-pentanamιde A une solution de 1,95 g (3,5 mmoles) de chlorhydrate de (S) -α-ammo-N-cyclopentyl -Ν, 5-dιméthyl- 1- (triphénylméthyl) - lH-ιmιdazole-4 -pentanamide dans 15 ml de dichloromethane, on ajoute successivement à 0 °C sous azote 1 g (3,66 mmoles) de chlorure de 2-ammo-3-thιén-2-ylbenzène sulfonyle puis 1,12 ml (7,85 mmoles) de triethylamine dans 5 ml de dichloromethane. On laisse la température du milieu réactionnel revenir à la température ambiante et on poursuit l'agitation pendant 15 heures à cette température. On concentre le milieu réactionnel sous pression réduite. On reprend le résidu par 150 ml d'acétate d'éthyle et on lave successivement par 50 ml d'une solution aqueuse d'acide ehlorhydrique 1 Ν, 50 ml d'une solution saturée d' hydrogenocarbonate de sodium puis par 50 ml d'une solution saturée de chlorure de sodium. On sèche sur sulfate de sodium. On purifie le résidu par chromatographie sur colonne de gel de silice en éluant par un mélange dichloromethane: méthanol (97 : 3) .
On obtient 2,35 g de produit sous forme d'un solide amorphe. Rendement = 87 % Point de fusion = 102-107 °C
26.2. chlorhydrate de { S) -N- cyclopentyl -N, 5-diméthyl - α- [ [ [2- [ (1-oxopropyl) amino] -3 -thién-2 -ylphényl] sulfonyl] amino] -lH-imidazole-4 -pentanamide (1:1) A une solution de 2,23 g (2,94 mmoles) de (S) -α- [ [ (2-amino- 3 -thién- 2 -ylphényl) sulfonyl] amino] -N-cyclopentyl -N, 5-diméthyl -1- (triphénylméthyl) -lH-imidazole-4 -pentanamide dans 1,5 ml de diméthylacétamide, on ajoute goutte à goutte à la température ambiante 0,51 ml (5,9 mmoles) de chlorure de propionyle. On laisse le mélange pendant 10 heures sous agitation puis on ajoute 150 ml d'acétate d'éthyle. On lave par 150 ml d'eau puis par 100 ml d'une solution saturée de chlorure de sodium. On sèche sur sulfate de sodium, on filtre et on concentre sous pression réduite. On reprend le résidu par 60 ml d'acétate d'éthyle et 30 ml d'eau puis on chauffe à la température de reflux pendant 2 heures. On ajoute alors 150 ml de dichloromethane et on lave successivement par 100 ml d'une solution saturée d' hydrogenocarbonate de sodium et 100 ml d'une solution saturée de chlorure de sodium. On sèche sur sulfate de sodium, on filtre et on concentre sous pression réduite. On reprend le résidu ainsi obtenu par 40 ml d'une solution d'acide ehlorhydrique 0,1 Ν dans 1 ' isopropanol et on concentre sous pression réduite. On purifie le résidu par chromatographie sur colonne de gel de silice RP 18 en éluant par un mélange acétonitrile : eau (2:8). On obtient 1,15 g de produit.
Rendement = 64 %
Point de fusion = 140-145 °C [α] 0 = + 103 o (c = o,2 ; méthanol)
Exemple 27 (composé n° 66) chlorhydrate de ( S) -N, 5-diméthyl-α- [[ [2- [ (1-oxopropyl) amino] -3 -thién-2 -ylphényl] sulfonyl] amino] -N-pyrrolidin- 1 -yl - lH- imidazole-4 -pentanamide (1:1)
27.1. { S) -α- [[ (2 -amino-3-thién-2-ylphényl) sulfonyl] amino] - N, 5-diméthyl -N-pyrrolidin- l -yl - 1 - (triphénylméthyl) - lH-imidazole-4 -pentanamide On prépare ce composé selon le mode opératoire décrit en 26.1 à partir de 0,8 g (2,94 mmoles) de chlorure de 2-amino-3-
thién-2-ylbenzène sulfonyle et 1,56 g (2,8 mmoles) de chlorhydrate de (S) -α~amino-N, 5 -diméthyl -N-pyrrol idin- l - yl-1- (triphénylméthyl) -lH-imidazole-4 -pentanamide . On obtient 1,84 g de produit sous forme d'un solide amorphe. Rendement = 83 %
Point de fusion = 102-106 °C
27.2. chlorhydrate de (S) -N, 5-diméthyl-α- [ [ [2 - [ (1-oxopro- pyl) amino] -3 -thién-2 -ylphényl] sulfonyl] amino] -N- pyrrolidin-l-yl-lH-imidazole-4-pentanamide (1:1)
On prépare ce composé selon le mode opératoire décrit en 26.2 à partir de 1,8 g (2,37 mmoles) de (S) -α- [ [ (2 -amino-3 -thién-
2 -ylphényl) sulfonyl] amino] -N, 5-diméthyl -N-pyrrol idin-1-yl -1-
(triphénylméthyl) -lH-imidazole-4 -pentanamide . On obtient 1 g de produit.
Rendement = 69 %
Point de fusion = 154-160 °C
[α]2° = 4- 119 ° (c = 0,2 ; méthanol)
Légende du tableau :
dans la colonne "Sel",
"HCl" correspond à un chlorhydrate et le rapport entre parenthèses est le rappport (acide :base) ,
dans la colonne " [α] π "
c = 0 , 2 ; méthanol sauf pour le composé 1 (c = 0, 22 ; méthanol), pour le composé 6 (c = 0,26 ; méthanol), pour le composé 12 (c = 0,25 ; méthanol) et pour les composés 51, 54, et 56 (c = 0,4 ; méthanol)
Tableau
Les composés de l'invention ont fait l'objet d'études pharmacologiques qui ont mis en évidence leurs propriétés antithrombotiques et leur intérêt comme substances à activité thérapeutique .
1. Détermination des constantes d'inhibition (Ki) vis à vis de la thrombine
Sur une microplaque de 96 puits, on dépose dans chaque puits 25 μl d'une solution de composé à tester (on étudie 7 concentrations), 50 μl d'une solution de substrat chromogène (on étudie 2 concentrations ; S2238 Chromogénix™) en solution dans du tampon Tris à pH 7,5 (Tris 50 mM, NaCl 100 mM et BSA 0,1 %) et finalement 25 μl d'une solution de thrombine à 300 U/ml . On suit la libération de 4-nitroaniline à 405 nm à l'aide d'un lecteur de plaques.
On détermine le K^ par la méthode de Dixon.
Les composés de 1 ' invention sont des inhibiteurs de la thrombine et leur K± est compris entre 0,001 et 100 μM.
2. Coagulation du plasma de rat par la thrombine humaine ex -vivo
On traite des rats mâles CD pesant 150 à 200 g avec le composé à tester ou avec le véhicule, par voie i.v., orale ou sous-cutanée. Ensuite on anesthésie les animaux au Nembutal™ (60 mg/kg ; 0,1 ml/kg) , on prélève le sang sur du citrate trisodique à 3,8% (1 vol/9 vol de sang) au niveau du sinus rétro-orbital et on prépare le plasma par centrifugation à 3600 g pendant 15 minutes à la température ambiante. On incube alors à 37 °C, 200 μl de plasma avec 200 μl d'une solution de thrombine humaine, la concentration finale en thrombine humaine étant de 0,75 unités NIH/ml et on note le temps de coagulation. L'effet anticoagulant est exprimé par la dose qui augmente le temps de coagulation de 100 %. Ils inhibent la coagulation du plasma de rat à des doses de 0,01 à 5 mg/kg i.v. Ils sont également actifs par les voies orale et sous-cutanée.
Les composés de l'invention peuvent être utiles dans toutes les indications cliniques liées à la thrombose ou dans celles
où des complications thrombotiques pourraient intervenir.
A cet effet ils peuvent être présentés sous toutes formes appropriées à l'administration orale, parentérale ou intraveineuse, telles que comprimés, dragées, gélules, capsules, suspensions ou solutions buvables ou injectables, etc. en association avec des excipients convenables. Toutes ces formes sont dosées pour permettre une administration de 1 à 1000 mg par jour et par patient, en une ou plusieurs doses.