TWI710552B - 作為神經元組織胺受體-3拮抗劑之化合物及其用途 - Google Patents
作為神經元組織胺受體-3拮抗劑之化合物及其用途 Download PDFInfo
- Publication number
- TWI710552B TWI710552B TW108134419A TW108134419A TWI710552B TW I710552 B TWI710552 B TW I710552B TW 108134419 A TW108134419 A TW 108134419A TW 108134419 A TW108134419 A TW 108134419A TW I710552 B TWI710552 B TW I710552B
- Authority
- TW
- Taiwan
- Prior art keywords
- compound
- pharmaceutically acceptable
- methyl
- concentrated
- stirred
- Prior art date
Links
- 150000001875 compounds Chemical class 0.000 title claims abstract description 246
- NTYJJOPFIAHURM-UHFFFAOYSA-N Histamine Chemical compound NCCC1=CN=CN1 NTYJJOPFIAHURM-UHFFFAOYSA-N 0.000 title abstract description 20
- 239000005557 antagonist Substances 0.000 title abstract description 14
- 229960001340 histamine Drugs 0.000 title abstract description 10
- 230000001537 neural effect Effects 0.000 title description 2
- 239000008194 pharmaceutical composition Substances 0.000 claims abstract description 92
- 150000003839 salts Chemical class 0.000 claims description 60
- -1 carrier Substances 0.000 claims description 54
- QMNUDYFKZYBWQX-UHFFFAOYSA-N 1H-quinazolin-4-one Chemical compound C1=CC=C2C(=O)N=CNC2=C1 QMNUDYFKZYBWQX-UHFFFAOYSA-N 0.000 claims description 51
- 239000003814 drug Substances 0.000 claims description 35
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 31
- 125000000217 alkyl group Chemical group 0.000 claims description 29
- 229940079593 drug Drugs 0.000 claims description 25
- 239000002904 solvent Substances 0.000 claims description 25
- 201000003631 narcolepsy Diseases 0.000 claims description 21
- 208000015114 central nervous system disease Diseases 0.000 claims description 20
- 238000011282 treatment Methods 0.000 claims description 20
- 238000004519 manufacturing process Methods 0.000 claims description 16
- 229910052739 hydrogen Inorganic materials 0.000 claims description 13
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 13
- 239000001257 hydrogen Substances 0.000 claims description 11
- 125000003545 alkoxy group Chemical group 0.000 claims description 9
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 6
- 239000002671 adjuvant Substances 0.000 claims description 6
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 5
- 150000002431 hydrogen Chemical class 0.000 claims 3
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims 2
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 abstract description 37
- 201000010099 disease Diseases 0.000 abstract description 31
- 238000002360 preparation method Methods 0.000 abstract description 25
- 208000035475 disorder Diseases 0.000 abstract description 6
- 230000006870 function Effects 0.000 abstract description 5
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 150
- 238000006243 chemical reaction Methods 0.000 description 92
- 239000000203 mixture Substances 0.000 description 87
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 83
- 239000000243 solution Substances 0.000 description 78
- 238000000034 method Methods 0.000 description 64
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 48
- DFHCZVVWCSOMKA-UHFFFAOYSA-N 2-amino-5-(pentafluoro-lambda6-sulfanyl)benzoic acid Chemical compound Nc1ccc(cc1C(O)=O)S(F)(F)(F)(F)F DFHCZVVWCSOMKA-UHFFFAOYSA-N 0.000 description 47
- 239000007787 solid Substances 0.000 description 46
- 239000003921 oil Substances 0.000 description 45
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 44
- 235000019198 oils Nutrition 0.000 description 44
- 239000012267 brine Substances 0.000 description 43
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 43
- 239000011734 sodium Substances 0.000 description 41
- 238000005481 NMR spectroscopy Methods 0.000 description 40
- 239000012074 organic phase Substances 0.000 description 38
- 239000000725 suspension Substances 0.000 description 38
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 38
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 37
- 239000000706 filtrate Substances 0.000 description 26
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 25
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 24
- 239000004480 active ingredient Substances 0.000 description 24
- 239000000843 powder Substances 0.000 description 24
- 238000002953 preparative HPLC Methods 0.000 description 24
- 238000010992 reflux Methods 0.000 description 24
- 239000000126 substance Substances 0.000 description 24
- 238000000746 purification Methods 0.000 description 23
- 229920006395 saturated elastomer Polymers 0.000 description 21
- 239000000741 silica gel Substances 0.000 description 21
- 229910002027 silica gel Inorganic materials 0.000 description 21
- 238000003786 synthesis reaction Methods 0.000 description 21
- 239000012298 atmosphere Substances 0.000 description 20
- 238000004440 column chromatography Methods 0.000 description 20
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 19
- 239000004698 Polyethylene Substances 0.000 description 19
- 230000015572 biosynthetic process Effects 0.000 description 19
- 239000002552 dosage form Substances 0.000 description 18
- 239000012453 solvate Substances 0.000 description 18
- 239000012065 filter cake Substances 0.000 description 17
- 238000004458 analytical method Methods 0.000 description 16
- 125000003118 aryl group Chemical group 0.000 description 16
- 125000001424 substituent group Chemical group 0.000 description 16
- 238000005516 engineering process Methods 0.000 description 15
- 238000003756 stirring Methods 0.000 description 15
- 102000004384 Histamine H3 receptors Human genes 0.000 description 14
- 108090000981 Histamine H3 receptors Proteins 0.000 description 14
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 14
- XOFLBQFBSOEHOG-UUOKFMHZSA-N γS-GTP Chemical compound C1=2NC(N)=NC(=O)C=2N=CN1[C@@H]1O[C@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=S)[C@@H](O)[C@H]1O XOFLBQFBSOEHOG-UUOKFMHZSA-N 0.000 description 14
- 239000007788 liquid Substances 0.000 description 13
- 208000024891 symptom Diseases 0.000 description 13
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 description 12
- LIBYGKUWXRBMPA-UHFFFAOYSA-N 1-(3-bromopropoxy)-4-nitrobenzene Chemical compound [O-][N+](=O)C1=CC=C(OCCCBr)C=C1 LIBYGKUWXRBMPA-UHFFFAOYSA-N 0.000 description 12
- VIAOUXPCADEEEG-UHFFFAOYSA-N 4-(4-nitrophenoxy)piperidine;hydrochloride Chemical compound Cl.C1=CC([N+](=O)[O-])=CC=C1OC1CCNCC1 VIAOUXPCADEEEG-UHFFFAOYSA-N 0.000 description 12
- WLKUSVNHZXUEFO-UHFFFAOYSA-N 4-fluoro-2-methoxy-1-nitrobenzene Chemical compound COC1=CC(F)=CC=C1[N+]([O-])=O WLKUSVNHZXUEFO-UHFFFAOYSA-N 0.000 description 12
- 125000003341 7 membered heterocyclic group Chemical group 0.000 description 12
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 12
- 150000003840 hydrochlorides Chemical class 0.000 description 12
- WXJMPQQWPDNSNJ-UHFFFAOYSA-N [4-(4-aminophenoxy)piperidin-1-yl]-cyclopentylmethanone Chemical compound NC1=CC=C(OC2CCN(CC2)C(=O)C2CCCC2)C=C1 WXJMPQQWPDNSNJ-UHFFFAOYSA-N 0.000 description 11
- 125000004429 atom Chemical group 0.000 description 11
- 125000004432 carbon atom Chemical group C* 0.000 description 11
- 239000000839 emulsion Substances 0.000 description 11
- 238000009472 formulation Methods 0.000 description 11
- 239000007937 lozenge Substances 0.000 description 11
- CKJNUZNMWOVDFN-UHFFFAOYSA-N methanone Chemical compound O=[CH-] CKJNUZNMWOVDFN-UHFFFAOYSA-N 0.000 description 11
- 239000000047 product Substances 0.000 description 11
- 239000002253 acid Substances 0.000 description 10
- 239000002585 base Substances 0.000 description 10
- 125000002837 carbocyclic group Chemical group 0.000 description 10
- 239000003795 chemical substances by application Substances 0.000 description 10
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 10
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 9
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 9
- 239000002775 capsule Substances 0.000 description 9
- 239000003937 drug carrier Substances 0.000 description 9
- 239000003755 preservative agent Substances 0.000 description 9
- 230000001225 therapeutic effect Effects 0.000 description 9
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 8
- 239000000443 aerosol Substances 0.000 description 8
- 230000003042 antagnostic effect Effects 0.000 description 8
- 239000003153 chemical reaction reagent Substances 0.000 description 8
- 239000000460 chlorine Substances 0.000 description 8
- 235000008504 concentrate Nutrition 0.000 description 8
- 239000012141 concentrate Substances 0.000 description 8
- 229920001577 copolymer Polymers 0.000 description 8
- 230000000694 effects Effects 0.000 description 8
- 230000005764 inhibitory process Effects 0.000 description 8
- 229910052757 nitrogen Inorganic materials 0.000 description 8
- 238000007911 parenteral administration Methods 0.000 description 8
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 8
- 241000124008 Mammalia Species 0.000 description 7
- 239000000872 buffer Substances 0.000 description 7
- 239000000969 carrier Substances 0.000 description 7
- 150000002148 esters Chemical class 0.000 description 7
- 238000004128 high performance liquid chromatography Methods 0.000 description 7
- 239000004615 ingredient Substances 0.000 description 7
- 239000000543 intermediate Substances 0.000 description 7
- 229940125425 inverse agonist Drugs 0.000 description 7
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 7
- 230000000670 limiting effect Effects 0.000 description 7
- 239000000463 material Substances 0.000 description 7
- 239000012528 membrane Substances 0.000 description 7
- 239000002480 mineral oil Substances 0.000 description 7
- 235000010446 mineral oil Nutrition 0.000 description 7
- DNIAPMSPPWPWGF-UHFFFAOYSA-N propylene glycol Substances CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 7
- PWQLFIKTGRINFF-UHFFFAOYSA-N tert-butyl 4-hydroxypiperidine-1-carboxylate Chemical group CC(C)(C)OC(=O)N1CCC(O)CC1 PWQLFIKTGRINFF-UHFFFAOYSA-N 0.000 description 7
- 229940124597 therapeutic agent Drugs 0.000 description 7
- ZNWWCOXXAHRYAH-UHFFFAOYSA-N 1-[3-(3-methoxy-4-nitrophenoxy)propyl]pyrrolidine Chemical compound COC=1C=C(OCCCN2CCCC2)C=CC=1[N+](=O)[O-] ZNWWCOXXAHRYAH-UHFFFAOYSA-N 0.000 description 6
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N Aniline Chemical compound NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 6
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 6
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 6
- 239000005977 Ethylene Substances 0.000 description 6
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 6
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 6
- 239000007891 compressed tablet Substances 0.000 description 6
- 239000012043 crude product Substances 0.000 description 6
- 239000007941 film coated tablet Substances 0.000 description 6
- 239000003112 inhibitor Substances 0.000 description 6
- 239000008297 liquid dosage form Substances 0.000 description 6
- 239000012071 phase Substances 0.000 description 6
- 239000011541 reaction mixture Substances 0.000 description 6
- 239000012896 selective serotonin reuptake inhibitor Substances 0.000 description 6
- 229940124834 selective serotonin reuptake inhibitor Drugs 0.000 description 6
- 239000000375 suspending agent Substances 0.000 description 6
- 238000004809 thin layer chromatography Methods 0.000 description 6
- SFAXHRYTDJJHII-MRVPVSSYSA-N 3-[(2r)-2-methylpyrrolidin-1-yl]propan-1-ol Chemical compound C[C@@H]1CCCN1CCCO SFAXHRYTDJJHII-MRVPVSSYSA-N 0.000 description 5
- SFAXHRYTDJJHII-QMMMGPOBSA-N 3-[(2s)-2-methylpyrrolidin-1-yl]propan-1-ol Chemical compound C[C@H]1CCCN1CCCO SFAXHRYTDJJHII-QMMMGPOBSA-N 0.000 description 5
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 5
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 5
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 5
- 239000000556 agonist Substances 0.000 description 5
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 5
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 5
- 239000003995 emulsifying agent Substances 0.000 description 5
- 239000007924 injection Substances 0.000 description 5
- 238000002347 injection Methods 0.000 description 5
- 239000002207 metabolite Substances 0.000 description 5
- 229920001223 polyethylene glycol Polymers 0.000 description 5
- 230000002829 reductive effect Effects 0.000 description 5
- 238000006467 substitution reaction Methods 0.000 description 5
- AHQMTHUYLMBHRJ-UHFFFAOYSA-N (1-methylpiperidin-4-yl)-[4-(4-nitrophenoxy)piperidin-1-yl]methanone Chemical compound CN1CCC(CC1)C(=O)N1CCC(CC1)OC1=CC=C(C=C1)[N+](=O)[O-] AHQMTHUYLMBHRJ-UHFFFAOYSA-N 0.000 description 4
- FTZHTTRIEAFVCK-GFCCVEGCSA-N (2R)-1-[3-(3-methoxy-4-nitrophenoxy)propyl]-2-methylpyrrolidine Chemical compound COC=1C=C(OCCCN2[C@@H](CCC2)C)C=CC=1[N+](=O)[O-] FTZHTTRIEAFVCK-GFCCVEGCSA-N 0.000 description 4
- GDQGOVBBDFTHCY-GFCCVEGCSA-N (2R)-2-methyl-1-[3-(4-nitrophenoxy)propyl]pyrrolidine Chemical compound C[C@H]1N(CCC1)CCCOC1=CC=C(C=C1)[N+](=O)[O-] GDQGOVBBDFTHCY-GFCCVEGCSA-N 0.000 description 4
- FTZHTTRIEAFVCK-LBPRGKRZSA-N (2S)-1-[3-(3-methoxy-4-nitrophenoxy)propyl]-2-methylpyrrolidine Chemical compound COC=1C=C(OCCCN2[C@H](CCC2)C)C=CC=1[N+](=O)[O-] FTZHTTRIEAFVCK-LBPRGKRZSA-N 0.000 description 4
- GDQGOVBBDFTHCY-LBPRGKRZSA-N (2s)-2-methyl-1-[3-(4-nitrophenoxy)propyl]pyrrolidine Chemical compound C[C@H]1CCCN1CCCOC1=CC=C([N+]([O-])=O)C=C1 GDQGOVBBDFTHCY-LBPRGKRZSA-N 0.000 description 4
- QJOQQGBYJZYHOJ-ZDUSSCGKSA-N (3S)-1-[3-(3-methoxy-4-nitrophenoxy)propyl]-3-methylpiperidine Chemical compound COC=1C=C(OCCCN2C[C@H](CCC2)C)C=CC=1[N+](=O)[O-] QJOQQGBYJZYHOJ-ZDUSSCGKSA-N 0.000 description 4
- CTUPSSVYEBTCNI-ZDUSSCGKSA-N (3s)-3-methyl-1-[3-(4-nitrophenoxy)propyl]piperidine Chemical compound C1[C@@H](C)CCCN1CCCOC1=CC=C([N+]([O-])=O)C=C1 CTUPSSVYEBTCNI-ZDUSSCGKSA-N 0.000 description 4
- XNQIOISZPFVUFG-RXMQYKEDSA-N (R)-alpha-methylhistamine Chemical compound C[C@@H](N)CC1=CN=CN1 XNQIOISZPFVUFG-RXMQYKEDSA-N 0.000 description 4
- KCOPAESEGCGTKM-UHFFFAOYSA-N 1,3-oxazol-4-one Chemical compound O=C1COC=N1 KCOPAESEGCGTKM-UHFFFAOYSA-N 0.000 description 4
- OMAZPJVZNWEHDZ-UHFFFAOYSA-N 1-[3-(2-methoxy-4-nitrophenoxy)propyl]pyrrolidine Chemical compound COC1=CC([N+]([O-])=O)=CC=C1OCCCN1CCCC1 OMAZPJVZNWEHDZ-UHFFFAOYSA-N 0.000 description 4
- LYPWWOZNYSSZJA-UHFFFAOYSA-N 1-[3-(3-methoxy-4-nitrophenoxy)propyl]-3-methylpyrrolidine Chemical compound COC=1C=C(OCCCN2CC(CC2)C)C=CC=1[N+](=O)[O-] LYPWWOZNYSSZJA-UHFFFAOYSA-N 0.000 description 4
- DFNHWMSTHVIRFR-UHFFFAOYSA-N 1-cyclobutylpiperidin-4-ol Chemical compound C1CC(O)CCN1C1CCC1 DFNHWMSTHVIRFR-UHFFFAOYSA-N 0.000 description 4
- MHYLVJJGZMOYTO-UHFFFAOYSA-N 2-amino-5-(3-pyrrolidin-1-ylpropoxy)phenol Chemical compound NC1=C(C=C(C=C1)OCCCN1CCCC1)O MHYLVJJGZMOYTO-UHFFFAOYSA-N 0.000 description 4
- SCVJRXQHFJXZFZ-KVQBGUIXSA-N 2-amino-9-[(2r,4s,5r)-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-3h-purine-6-thione Chemical compound C1=2NC(N)=NC(=S)C=2N=CN1[C@H]1C[C@H](O)[C@@H](CO)O1 SCVJRXQHFJXZFZ-KVQBGUIXSA-N 0.000 description 4
- RPUZDJUPQBDKNK-UHFFFAOYSA-N 2-methoxy-4-(3-pyrrolidin-1-ylpropoxy)aniline Chemical compound COc1cc(OCCCN2CCCC2)ccc1N RPUZDJUPQBDKNK-UHFFFAOYSA-N 0.000 description 4
- OZHSXRILMAFJAO-UHFFFAOYSA-N 2-methoxy-4-[3-(3-methylpyrrolidin-1-yl)propoxy]aniline Chemical compound COC1=C(N)C=CC(=C1)OCCCN1CC(CC1)C OZHSXRILMAFJAO-UHFFFAOYSA-N 0.000 description 4
- ORIWCCBREONBBE-GFCCVEGCSA-N 2-methoxy-4-[3-[(2R)-2-methylpyrrolidin-1-yl]propoxy]aniline Chemical compound COC1=C(N)C=CC(=C1)OCCCN1[C@@H](CCC1)C ORIWCCBREONBBE-GFCCVEGCSA-N 0.000 description 4
- ORIWCCBREONBBE-LBPRGKRZSA-N 2-methoxy-4-[3-[(2S)-2-methylpyrrolidin-1-yl]propoxy]aniline Chemical compound COC1=C(N)C=CC(=C1)OCCCN1[C@H](CCC1)C ORIWCCBREONBBE-LBPRGKRZSA-N 0.000 description 4
- MNNSNZNJZFYETA-ZDUSSCGKSA-N 2-methoxy-4-[3-[(3S)-3-methylpiperidin-1-yl]propoxy]aniline Chemical compound COC1=C(N)C=CC(=C1)OCCCN1C[C@H](CCC1)C MNNSNZNJZFYETA-ZDUSSCGKSA-N 0.000 description 4
- VJRGVFZTMPJJCL-UHFFFAOYSA-N 2-nitro-5-(3-pyrrolidin-1-ylpropoxy)phenol Chemical compound [N+](=O)([O-])C1=C(C=C(C=C1)OCCCN1CCCC1)O VJRGVFZTMPJJCL-UHFFFAOYSA-N 0.000 description 4
- RBPKQHNYKJHOQM-UHFFFAOYSA-N 3-(3-methylpyrrolidin-1-yl)propan-1-ol Chemical compound CC1CCN(CCCO)C1 RBPKQHNYKJHOQM-UHFFFAOYSA-N 0.000 description 4
- PSSJPGZIAICNGX-VIFPVBQESA-N 3-[(3s)-3-methylpiperidin-1-yl]propan-1-ol Chemical compound C[C@H]1CCCN(CCCO)C1 PSSJPGZIAICNGX-VIFPVBQESA-N 0.000 description 4
- JMWNSZLQYVCNFM-UHFFFAOYSA-N 3-methoxy-4-(3-pyrrolidin-1-ylpropoxy)aniline Chemical compound COC1=CC(N)=CC=C1OCCCN1CCCC1 JMWNSZLQYVCNFM-UHFFFAOYSA-N 0.000 description 4
- SIZYEJRMTCSNJO-UHFFFAOYSA-N 3-methyl-1-[3-(4-nitrophenoxy)propyl]pyrrolidine Chemical compound CC1CN(CC1)CCCOC1=CC=C(C=C1)[N+](=O)[O-] SIZYEJRMTCSNJO-UHFFFAOYSA-N 0.000 description 4
- CKZDBHBPTICGST-UHFFFAOYSA-N 4-(1-cyclobutylpiperidin-4-yl)oxyaniline Chemical compound C1=CC(N)=CC=C1OC1CCN(C2CCC2)CC1 CKZDBHBPTICGST-UHFFFAOYSA-N 0.000 description 4
- WXGSXSDPNOYJPS-UHFFFAOYSA-N 4-(1-cyclopropylpiperidin-4-yl)oxyaniline Chemical compound C1=CC(N)=CC=C1OC1CCN(C2CC2)CC1 WXGSXSDPNOYJPS-UHFFFAOYSA-N 0.000 description 4
- YRADBIIYODPUEQ-UHFFFAOYSA-N 4-(3-methoxy-4-nitrophenoxy)piperidine;hydrochloride Chemical compound Cl.C1=C([N+]([O-])=O)C(OC)=CC(OC2CCNCC2)=C1 YRADBIIYODPUEQ-UHFFFAOYSA-N 0.000 description 4
- XSGWSJJKVBKZIG-UHFFFAOYSA-N 4-[3-(3-methylpyrrolidin-1-yl)propoxy]aniline Chemical compound C1C(C)CCN1CCCOC1=CC=C(N)C=C1 XSGWSJJKVBKZIG-UHFFFAOYSA-N 0.000 description 4
- FBHYLOHAZWGJIM-GFCCVEGCSA-N 4-[3-[(2r)-2-methylpyrrolidin-1-yl]propoxy]aniline Chemical compound C[C@@H]1CCCN1CCCOC1=CC=C(N)C=C1 FBHYLOHAZWGJIM-GFCCVEGCSA-N 0.000 description 4
- FBHYLOHAZWGJIM-LBPRGKRZSA-N 4-[3-[(2s)-2-methylpyrrolidin-1-yl]propoxy]aniline Chemical compound C[C@H]1CCCN1CCCOC1=CC=C(N)C=C1 FBHYLOHAZWGJIM-LBPRGKRZSA-N 0.000 description 4
- PCNDYFSRKBIZIT-ZDUSSCGKSA-N 4-[3-[(3s)-3-methylpiperidin-1-yl]propoxy]aniline Chemical compound C1[C@@H](C)CCCN1CCCOC1=CC=C(N)C=C1 PCNDYFSRKBIZIT-ZDUSSCGKSA-N 0.000 description 4
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 4
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 4
- 241001465754 Metazoa Species 0.000 description 4
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 4
- 229910019142 PO4 Inorganic materials 0.000 description 4
- 239000002202 Polyethylene glycol Substances 0.000 description 4
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 4
- 239000003963 antioxidant agent Substances 0.000 description 4
- 235000006708 antioxidants Nutrition 0.000 description 4
- 239000008365 aqueous carrier Substances 0.000 description 4
- 239000007864 aqueous solution Substances 0.000 description 4
- 230000008901 benefit Effects 0.000 description 4
- 229910052799 carbon Inorganic materials 0.000 description 4
- 230000008859 change Effects 0.000 description 4
- 238000004587 chromatography analysis Methods 0.000 description 4
- 239000006071 cream Substances 0.000 description 4
- 125000000753 cycloalkyl group Chemical group 0.000 description 4
- 239000002270 dispersing agent Substances 0.000 description 4
- 235000003599 food sweetener Nutrition 0.000 description 4
- 125000000623 heterocyclic group Chemical group 0.000 description 4
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 4
- 238000001727 in vivo Methods 0.000 description 4
- 150000002500 ions Chemical class 0.000 description 4
- LXCFILQKKLGQFO-UHFFFAOYSA-N methylparaben Chemical compound COC(=O)C1=CC=C(O)C=C1 LXCFILQKKLGQFO-UHFFFAOYSA-N 0.000 description 4
- 238000012986 modification Methods 0.000 description 4
- 230000004048 modification Effects 0.000 description 4
- 235000021317 phosphate Nutrition 0.000 description 4
- 125000004482 piperidin-4-yl group Chemical group N1CCC(CC1)* 0.000 description 4
- 229920000573 polyethylene Polymers 0.000 description 4
- 229920000139 polyethylene terephthalate Polymers 0.000 description 4
- 239000005020 polyethylene terephthalate Substances 0.000 description 4
- 229920000642 polymer Polymers 0.000 description 4
- 230000002265 prevention Effects 0.000 description 4
- 239000000651 prodrug Substances 0.000 description 4
- 229940002612 prodrug Drugs 0.000 description 4
- QELSKZZBTMNZEB-UHFFFAOYSA-N propylparaben Chemical compound CCCOC(=O)C1=CC=C(O)C=C1 QELSKZZBTMNZEB-UHFFFAOYSA-N 0.000 description 4
- 239000008299 semisolid dosage form Substances 0.000 description 4
- 239000007858 starting material Substances 0.000 description 4
- 239000003765 sweetening agent Substances 0.000 description 4
- 239000006188 syrup Substances 0.000 description 4
- 235000020357 syrup Nutrition 0.000 description 4
- UNNMTKGKCWNQNU-UHFFFAOYSA-N tert-butyl 4-(4-nitrophenoxy)piperidine-1-carboxylate Chemical compound C1CN(C(=O)OC(C)(C)C)CCC1OC1=CC=C([N+]([O-])=O)C=C1 UNNMTKGKCWNQNU-UHFFFAOYSA-N 0.000 description 4
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 4
- 239000002562 thickening agent Substances 0.000 description 4
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 4
- 239000000080 wetting agent Substances 0.000 description 4
- DUBLPBOUYRUPIW-UHFFFAOYSA-N 1-cyclobutyl-4-(4-nitrophenoxy)piperidine Chemical compound C1=CC([N+](=O)[O-])=CC=C1OC1CCN(C2CCC2)CC1 DUBLPBOUYRUPIW-UHFFFAOYSA-N 0.000 description 3
- FLMRWMXFSGTJHC-UHFFFAOYSA-N 1-cyclopentyl-4-(4-nitrophenoxy)piperidine Chemical compound C1=CC([N+](=O)[O-])=CC=C1OC1CCN(C2CCCC2)CC1 FLMRWMXFSGTJHC-UHFFFAOYSA-N 0.000 description 3
- VIKJYAYQGBFZGK-UHFFFAOYSA-N 1-cyclopropyl-4-(4-nitrophenoxy)piperidine Chemical compound C1=CC([N+](=O)[O-])=CC=C1OC1CCN(C2CC2)CC1 VIKJYAYQGBFZGK-UHFFFAOYSA-N 0.000 description 3
- RQFUZUMFPRMVDX-UHFFFAOYSA-N 3-Bromo-1-propanol Chemical compound OCCCBr RQFUZUMFPRMVDX-UHFFFAOYSA-N 0.000 description 3
- TWOVRSMFLFYABX-UHFFFAOYSA-N 4-(3-pyrrolidin-1-ylpropoxy)aniline Chemical compound C1=CC(N)=CC=C1OCCCN1CCCC1 TWOVRSMFLFYABX-UHFFFAOYSA-N 0.000 description 3
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 3
- BTJIUGUIPKRLHP-UHFFFAOYSA-N 4-nitrophenol Chemical compound OC1=CC=C([N+]([O-])=O)C=C1 BTJIUGUIPKRLHP-UHFFFAOYSA-N 0.000 description 3
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 description 3
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 3
- 229920000858 Cyclodextrin Polymers 0.000 description 3
- QMMFVYPAHWMCMS-UHFFFAOYSA-N Dimethyl sulfide Chemical compound CSC QMMFVYPAHWMCMS-UHFFFAOYSA-N 0.000 description 3
- 229940121891 Dopamine receptor antagonist Drugs 0.000 description 3
- 108010010803 Gelatin Proteins 0.000 description 3
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 3
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 3
- 229940121991 Serotonin and norepinephrine reuptake inhibitor Drugs 0.000 description 3
- 229920002472 Starch Polymers 0.000 description 3
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical class OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 3
- SJKDGWSQHQSZHT-UHFFFAOYSA-N [3-(dichloromethyl)-4-nitrophenyl]-pentafluoro-$l^{6}-sulfane Chemical compound [O-][N+](=O)C1=CC=C(S(F)(F)(F)(F)F)C=C1C(Cl)Cl SJKDGWSQHQSZHT-UHFFFAOYSA-N 0.000 description 3
- RDMSETAQUASNPN-UHFFFAOYSA-N [4-(4-aminophenoxy)piperidin-1-yl]-(1-methylpiperidin-4-yl)methanone Chemical compound NC1=CC=C(OC2CCN(CC2)C(=O)C2CCN(CC2)C)C=C1 RDMSETAQUASNPN-UHFFFAOYSA-N 0.000 description 3
- WREOTYWODABZMH-DTZQCDIJSA-N [[(2r,3s,4r,5r)-3,4-dihydroxy-5-[2-oxo-4-(2-phenylethoxyamino)pyrimidin-1-yl]oxolan-2-yl]methoxy-hydroxyphosphoryl] phosphono hydrogen phosphate Chemical compound O[C@@H]1[C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)O[C@H]1N(C=C\1)C(=O)NC/1=N\OCCC1=CC=CC=C1 WREOTYWODABZMH-DTZQCDIJSA-N 0.000 description 3
- 150000001241 acetals Chemical class 0.000 description 3
- 239000013543 active substance Substances 0.000 description 3
- 239000004599 antimicrobial Substances 0.000 description 3
- 235000010323 ascorbic acid Nutrition 0.000 description 3
- 229960004853 betadex Drugs 0.000 description 3
- 125000002619 bicyclic group Chemical group 0.000 description 3
- 230000004071 biological effect Effects 0.000 description 3
- 239000002738 chelating agent Substances 0.000 description 3
- 229910052801 chlorine Inorganic materials 0.000 description 3
- 239000000544 cholinesterase inhibitor Substances 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 239000011248 coating agent Substances 0.000 description 3
- 230000001149 cognitive effect Effects 0.000 description 3
- 239000003086 colorant Substances 0.000 description 3
- 229940125758 compound 15 Drugs 0.000 description 3
- 230000008094 contradictory effect Effects 0.000 description 3
- 238000007796 conventional method Methods 0.000 description 3
- 239000013078 crystal Substances 0.000 description 3
- 125000004122 cyclic group Chemical group 0.000 description 3
- UTRMXCLQXJZZNA-UHFFFAOYSA-N cyclohexyl-[4-(4-nitrophenoxy)piperidin-1-yl]methanone Chemical compound C1(CCCCC1)C(=O)N1CCC(CC1)OC1=CC=C(C=C1)[N+](=O)[O-] UTRMXCLQXJZZNA-UHFFFAOYSA-N 0.000 description 3
- YHTGNDWRWGZHSP-UHFFFAOYSA-N cyclopentyl-[4-(4-nitrophenoxy)piperidin-1-yl]methanone Chemical compound C1(CCCC1)C(=O)N1CCC(CC1)OC1=CC=C(C=C1)[N+](=O)[O-] YHTGNDWRWGZHSP-UHFFFAOYSA-N 0.000 description 3
- 238000011161 development Methods 0.000 description 3
- MTHSVFCYNBDYFN-UHFFFAOYSA-N diethylene glycol Chemical compound OCCOCCO MTHSVFCYNBDYFN-UHFFFAOYSA-N 0.000 description 3
- 239000003085 diluting agent Substances 0.000 description 3
- 238000010790 dilution Methods 0.000 description 3
- 239000012895 dilution Substances 0.000 description 3
- 239000003210 dopamine receptor blocking agent Substances 0.000 description 3
- 239000007888 film coating Substances 0.000 description 3
- 238000009501 film coating Methods 0.000 description 3
- 239000000796 flavoring agent Substances 0.000 description 3
- 229910052731 fluorine Inorganic materials 0.000 description 3
- 150000008423 fluorobenzenes Chemical class 0.000 description 3
- 235000013355 food flavoring agent Nutrition 0.000 description 3
- 239000000499 gel Substances 0.000 description 3
- 239000008273 gelatin Substances 0.000 description 3
- 229920000159 gelatin Polymers 0.000 description 3
- 235000019322 gelatine Nutrition 0.000 description 3
- 235000011852 gelatine desserts Nutrition 0.000 description 3
- 235000011187 glycerol Nutrition 0.000 description 3
- 229910052736 halogen Inorganic materials 0.000 description 3
- 150000002367 halogens Chemical class 0.000 description 3
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 3
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 3
- 238000001802 infusion Methods 0.000 description 3
- 230000002401 inhibitory effect Effects 0.000 description 3
- 239000008101 lactose Substances 0.000 description 3
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 3
- 239000006210 lotion Substances 0.000 description 3
- 239000000314 lubricant Substances 0.000 description 3
- 239000011159 matrix material Substances 0.000 description 3
- 230000003340 mental effect Effects 0.000 description 3
- 229940071648 metered dose inhaler Drugs 0.000 description 3
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 3
- 239000000693 micelle Substances 0.000 description 3
- 238000002156 mixing Methods 0.000 description 3
- 125000002950 monocyclic group Chemical group 0.000 description 3
- 230000001095 motoneuron effect Effects 0.000 description 3
- OFBQJSOFQDEBGM-UHFFFAOYSA-N n-pentane Natural products CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 3
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 3
- 239000002674 ointment Substances 0.000 description 3
- 230000003287 optical effect Effects 0.000 description 3
- 210000000056 organ Anatomy 0.000 description 3
- 229910052760 oxygen Inorganic materials 0.000 description 3
- 125000004430 oxygen atom Chemical group O* 0.000 description 3
- 230000036407 pain Effects 0.000 description 3
- 239000002245 particle Substances 0.000 description 3
- 230000000144 pharmacologic effect Effects 0.000 description 3
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 3
- 239000010452 phosphate Substances 0.000 description 3
- 229960003651 pitolisant Drugs 0.000 description 3
- 239000003380 propellant Substances 0.000 description 3
- 125000002572 propoxy group Chemical group [*]OC([H])([H])C(C([H])([H])[H])([H])[H] 0.000 description 3
- 235000010232 propyl p-hydroxybenzoate Nutrition 0.000 description 3
- RONWGALEIBILOG-VMJVVOMYSA-N quinine sulfate Chemical compound [H+].[H+].[O-]S([O-])(=O)=O.C([C@H]([C@H](C1)C=C)C2)C[N@@]1[C@@H]2[C@H](O)C1=CC=NC2=CC=C(OC)C=C21.C([C@H]([C@H](C1)C=C)C2)C[N@@]1[C@@H]2[C@H](O)C1=CC=NC2=CC=C(OC)C=C21 RONWGALEIBILOG-VMJVVOMYSA-N 0.000 description 3
- 239000000376 reactant Substances 0.000 description 3
- 238000011160 research Methods 0.000 description 3
- 239000003775 serotonin noradrenalin reuptake inhibitor Substances 0.000 description 3
- 238000002603 single-photon emission computed tomography Methods 0.000 description 3
- 239000002002 slurry Substances 0.000 description 3
- 239000011780 sodium chloride Substances 0.000 description 3
- 239000003381 stabilizer Substances 0.000 description 3
- 239000008107 starch Substances 0.000 description 3
- 235000019698 starch Nutrition 0.000 description 3
- 238000007910 systemic administration Methods 0.000 description 3
- 239000003826 tablet Substances 0.000 description 3
- WNJGYLABZMLNPD-UHFFFAOYSA-N tert-butyl 4-(3-methoxy-4-nitrophenoxy)piperidine-1-carboxylate Chemical compound C1=C([N+]([O-])=O)C(OC)=CC(OC2CCN(CC2)C(=O)OC(C)(C)C)=C1 WNJGYLABZMLNPD-UHFFFAOYSA-N 0.000 description 3
- 238000012360 testing method Methods 0.000 description 3
- DTQVDTLACAAQTR-UHFFFAOYSA-N trifluoroacetic acid Substances OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 3
- PUPZLCDOIYMWBV-UHFFFAOYSA-N (+/-)-1,3-Butanediol Chemical compound CC(O)CCO PUPZLCDOIYMWBV-UHFFFAOYSA-N 0.000 description 2
- SFLSHLFXELFNJZ-QMMMGPOBSA-N (-)-norepinephrine Chemical compound NC[C@H](O)C1=CC=C(O)C(O)=C1 SFLSHLFXELFNJZ-QMMMGPOBSA-N 0.000 description 2
- RGHPCLZJAFCTIK-YFKPBYRVSA-N (2s)-2-methylpyrrolidine Chemical compound C[C@H]1CCCN1 RGHPCLZJAFCTIK-YFKPBYRVSA-N 0.000 description 2
- DIJAKMCTWBGMNQ-RGMNGODLSA-N (3s)-3-methylpiperidine;hydrochloride Chemical compound Cl.C[C@H]1CCCNC1 DIJAKMCTWBGMNQ-RGMNGODLSA-N 0.000 description 2
- RGHPCLZJAFCTIK-RXMQYKEDSA-N (R)-2-methylpyrrolidine Chemical compound C[C@@H]1CCCN1 RGHPCLZJAFCTIK-RXMQYKEDSA-N 0.000 description 2
- GVJHHUAWPYXKBD-UHFFFAOYSA-N (±)-α-Tocopherol Chemical compound OC1=C(C)C(C)=C2OC(CCCC(C)CCCC(C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-UHFFFAOYSA-N 0.000 description 2
- LVGUZGTVOIAKKC-UHFFFAOYSA-N 1,1,1,2-tetrafluoroethane Chemical compound FCC(F)(F)F LVGUZGTVOIAKKC-UHFFFAOYSA-N 0.000 description 2
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 2
- DDMOUSALMHHKOS-UHFFFAOYSA-N 1,2-dichloro-1,1,2,2-tetrafluoroethane Chemical compound FC(F)(Cl)C(F)(F)Cl DDMOUSALMHHKOS-UHFFFAOYSA-N 0.000 description 2
- AZNKKZHZGDZSIF-UHFFFAOYSA-N 1-fluoro-2-methoxy-4-nitrobenzene Chemical compound COC1=CC([N+]([O-])=O)=CC=C1F AZNKKZHZGDZSIF-UHFFFAOYSA-N 0.000 description 2
- WFQDTOYDVUWQMS-UHFFFAOYSA-N 1-fluoro-4-nitrobenzene Chemical compound [O-][N+](=O)C1=CC=C(F)C=C1 WFQDTOYDVUWQMS-UHFFFAOYSA-N 0.000 description 2
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 2
- WXTMDXOMEHJXQO-UHFFFAOYSA-N 2,5-dihydroxybenzoic acid Chemical compound OC(=O)C1=CC(O)=CC=C1O WXTMDXOMEHJXQO-UHFFFAOYSA-N 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- PYRKKGOKRMZEIT-UHFFFAOYSA-N 2-[6-(2-cyclopropylethoxy)-9-(2-hydroxy-2-methylpropyl)-1h-phenanthro[9,10-d]imidazol-2-yl]-5-fluorobenzene-1,3-dicarbonitrile Chemical compound C1=C2C3=CC(CC(C)(O)C)=CC=C3C=3NC(C=4C(=CC(F)=CC=4C#N)C#N)=NC=3C2=CC=C1OCCC1CC1 PYRKKGOKRMZEIT-UHFFFAOYSA-N 0.000 description 2
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 2
- MJKVTPMWOKAVMS-UHFFFAOYSA-N 3-hydroxy-1-benzopyran-2-one Chemical compound C1=CC=C2OC(=O)C(O)=CC2=C1 MJKVTPMWOKAVMS-UHFFFAOYSA-N 0.000 description 2
- JDJFUMBJQINVKP-UHFFFAOYSA-N 3-methylpyrrolidine;hydrochloride Chemical compound Cl.CC1CCNC1 JDJFUMBJQINVKP-UHFFFAOYSA-N 0.000 description 2
- ZMJQROKRSPSLFH-UHFFFAOYSA-N 3-pyrrolidin-1-ylpropan-1-ol Chemical compound OCCCN1CCCC1 ZMJQROKRSPSLFH-UHFFFAOYSA-N 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 2
- 239000005711 Benzoic acid Substances 0.000 description 2
- 239000004322 Butylated hydroxytoluene Substances 0.000 description 2
- NLZUEZXRPGMBCV-UHFFFAOYSA-N Butylhydroxytoluene Chemical compound CC1=CC(C(C)(C)C)=C(O)C(C(C)(C)C)=C1 NLZUEZXRPGMBCV-UHFFFAOYSA-N 0.000 description 2
- 241000282472 Canis lupus familiaris Species 0.000 description 2
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 2
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 2
- 241000282693 Cercopithecidae Species 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 2
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 2
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 2
- 239000001116 FEMA 4028 Substances 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- 241000282412 Homo Species 0.000 description 2
- QIGBRXMKCJKVMJ-UHFFFAOYSA-N Hydroquinone Chemical compound OC1=CC=C(O)C=C1 QIGBRXMKCJKVMJ-UHFFFAOYSA-N 0.000 description 2
- 238000004566 IR spectroscopy Methods 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 2
- 102000018697 Membrane Proteins Human genes 0.000 description 2
- 108010052285 Membrane Proteins Proteins 0.000 description 2
- 241000699670 Mus sp. Species 0.000 description 2
- UFWIBTONFRDIAS-UHFFFAOYSA-N Naphthalene Chemical compound C1=CC=CC2=CC=CC=C21 UFWIBTONFRDIAS-UHFFFAOYSA-N 0.000 description 2
- 235000019483 Peanut oil Nutrition 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- ZTHYODDOHIVTJV-UHFFFAOYSA-N Propyl gallate Chemical compound CCCOC(=O)C1=CC(O)=C(O)C(O)=C1 ZTHYODDOHIVTJV-UHFFFAOYSA-N 0.000 description 2
- 241000700159 Rattus Species 0.000 description 2
- 239000008156 Ringer's lactate solution Substances 0.000 description 2
- 229930006000 Sucrose Natural products 0.000 description 2
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 2
- XTXRWKRVRITETP-UHFFFAOYSA-N Vinyl acetate Chemical compound CC(=O)OC=C XTXRWKRVRITETP-UHFFFAOYSA-N 0.000 description 2
- 208000036142 Viral infection Diseases 0.000 description 2
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 2
- GAMPNQJDUFQVQO-UHFFFAOYSA-N acetic acid;phthalic acid Chemical compound CC(O)=O.OC(=O)C1=CC=CC=C1C(O)=O GAMPNQJDUFQVQO-UHFFFAOYSA-N 0.000 description 2
- 230000002378 acidificating effect Effects 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- 150000001298 alcohols Chemical class 0.000 description 2
- 150000001335 aliphatic alkanes Chemical group 0.000 description 2
- 150000001448 anilines Chemical class 0.000 description 2
- MWPLVEDNUUSJAV-UHFFFAOYSA-N anthracene Chemical compound C1=CC=CC2=CC3=CC=CC=C3C=C21 MWPLVEDNUUSJAV-UHFFFAOYSA-N 0.000 description 2
- 230000000845 anti-microbial effect Effects 0.000 description 2
- 239000011668 ascorbic acid Substances 0.000 description 2
- 238000011914 asymmetric synthesis Methods 0.000 description 2
- 229960000686 benzalkonium chloride Drugs 0.000 description 2
- 235000010233 benzoic acid Nutrition 0.000 description 2
- CADWTSSKOVRVJC-UHFFFAOYSA-N benzyl(dimethyl)azanium;chloride Chemical compound [Cl-].C[NH+](C)CC1=CC=CC=C1 CADWTSSKOVRVJC-UHFFFAOYSA-N 0.000 description 2
- WHGYBXFWUBPSRW-FOUAGVGXSA-N beta-cyclodextrin Chemical compound OC[C@H]([C@H]([C@@H]([C@H]1O)O)O[C@H]2O[C@@H]([C@@H](O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O3)[C@H](O)[C@H]2O)CO)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@@H]3O[C@@H]1CO WHGYBXFWUBPSRW-FOUAGVGXSA-N 0.000 description 2
- 239000011230 binding agent Substances 0.000 description 2
- 230000033228 biological regulation Effects 0.000 description 2
- 238000009835 boiling Methods 0.000 description 2
- 229910052794 bromium Inorganic materials 0.000 description 2
- 235000010354 butylated hydroxytoluene Nutrition 0.000 description 2
- 229940095259 butylated hydroxytoluene Drugs 0.000 description 2
- 239000001768 carboxy methyl cellulose Substances 0.000 description 2
- 239000004359 castor oil Substances 0.000 description 2
- 235000019438 castor oil Nutrition 0.000 description 2
- 229920002301 cellulose acetate Polymers 0.000 description 2
- OSASVXMJTNOKOY-UHFFFAOYSA-N chlorobutanol Chemical compound CC(C)(O)C(Cl)(Cl)Cl OSASVXMJTNOKOY-UHFFFAOYSA-N 0.000 description 2
- 238000000576 coating method Methods 0.000 description 2
- 239000012230 colorless oil Substances 0.000 description 2
- 239000008139 complexing agent Substances 0.000 description 2
- 229940125773 compound 10 Drugs 0.000 description 2
- 125000004093 cyano group Chemical group *C#N 0.000 description 2
- 125000004966 cyanoalkyl group Chemical group 0.000 description 2
- BGTOWKSIORTVQH-UHFFFAOYSA-N cyclopentanone Chemical compound O=C1CCCC1 BGTOWKSIORTVQH-UHFFFAOYSA-N 0.000 description 2
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 2
- 238000013461 design Methods 0.000 description 2
- 230000006866 deterioration Effects 0.000 description 2
- 229910052805 deuterium Inorganic materials 0.000 description 2
- RWRIWBAIICGTTQ-UHFFFAOYSA-N difluoromethane Chemical compound FCF RWRIWBAIICGTTQ-UHFFFAOYSA-N 0.000 description 2
- 239000004205 dimethyl polysiloxane Substances 0.000 description 2
- USIUVYZYUHIAEV-UHFFFAOYSA-N diphenyl ether Chemical compound C=1C=CC=CC=1OC1=CC=CC=C1 USIUVYZYUHIAEV-UHFFFAOYSA-N 0.000 description 2
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 2
- 239000007884 disintegrant Substances 0.000 description 2
- 238000009826 distribution Methods 0.000 description 2
- VYFYYTLLBUKUHU-UHFFFAOYSA-N dopamine Chemical compound NCCC1=CC=C(O)C(O)=C1 VYFYYTLLBUKUHU-UHFFFAOYSA-N 0.000 description 2
- 238000001647 drug administration Methods 0.000 description 2
- 229920001971 elastomer Polymers 0.000 description 2
- BEFDCLMNVWHSGT-UHFFFAOYSA-N ethenylcyclopentane Chemical compound C=CC1CCCC1 BEFDCLMNVWHSGT-UHFFFAOYSA-N 0.000 description 2
- 230000004907 flux Effects 0.000 description 2
- 238000005187 foaming Methods 0.000 description 2
- 238000001640 fractional crystallisation Methods 0.000 description 2
- 239000007789 gas Substances 0.000 description 2
- 239000003349 gelling agent Substances 0.000 description 2
- 230000014509 gene expression Effects 0.000 description 2
- ZEMPKEQAKRGZGQ-XOQCFJPHSA-N glycerol triricinoleate Natural products CCCCCC[C@@H](O)CC=CCCCCCCCC(=O)OC[C@@H](COC(=O)CCCCCCCC=CC[C@@H](O)CCCCCC)OC(=O)CCCCCCCC=CC[C@H](O)CCCCCC ZEMPKEQAKRGZGQ-XOQCFJPHSA-N 0.000 description 2
- 229940093915 gynecological organic acid Drugs 0.000 description 2
- 125000001072 heteroaryl group Chemical group 0.000 description 2
- 125000005842 heteroatom Chemical group 0.000 description 2
- 150000004677 hydrates Chemical class 0.000 description 2
- 239000000017 hydrogel Substances 0.000 description 2
- 238000000338 in vitro Methods 0.000 description 2
- 230000003993 interaction Effects 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- 229910052740 iodine Inorganic materials 0.000 description 2
- NNPPMTNAJDCUHE-UHFFFAOYSA-N isobutane Chemical compound CC(C)C NNPPMTNAJDCUHE-UHFFFAOYSA-N 0.000 description 2
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 2
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 2
- 239000007951 isotonicity adjuster Substances 0.000 description 2
- ZLVXBBHTMQJRSX-VMGNSXQWSA-N jdtic Chemical compound C1([C@]2(C)CCN(C[C@@H]2C)C[C@H](C(C)C)NC(=O)[C@@H]2NCC3=CC(O)=CC=C3C2)=CC=CC(O)=C1 ZLVXBBHTMQJRSX-VMGNSXQWSA-N 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 239000002502 liposome Substances 0.000 description 2
- 239000003589 local anesthetic agent Substances 0.000 description 2
- 229960005015 local anesthetics Drugs 0.000 description 2
- 230000007774 longterm Effects 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 2
- 238000007726 management method Methods 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- 239000002609 medium Substances 0.000 description 2
- 230000002503 metabolic effect Effects 0.000 description 2
- 230000004060 metabolic process Effects 0.000 description 2
- 239000004292 methyl p-hydroxybenzoate Substances 0.000 description 2
- 229960002216 methylparaben Drugs 0.000 description 2
- 239000004005 microsphere Substances 0.000 description 2
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 150000005181 nitrobenzenes Chemical class 0.000 description 2
- 125000004433 nitrogen atom Chemical group N* 0.000 description 2
- 230000002474 noradrenergic effect Effects 0.000 description 2
- SFLSHLFXELFNJZ-UHFFFAOYSA-N norepinephrine Natural products NCC(O)C1=CC=C(O)C(O)=C1 SFLSHLFXELFNJZ-UHFFFAOYSA-N 0.000 description 2
- 229960002748 norepinephrine Drugs 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 235000005985 organic acids Nutrition 0.000 description 2
- 150000007530 organic bases Chemical class 0.000 description 2
- 150000002894 organic compounds Chemical class 0.000 description 2
- 239000003002 pH adjusting agent Substances 0.000 description 2
- 239000006072 paste Substances 0.000 description 2
- 239000000312 peanut oil Substances 0.000 description 2
- 239000008188 pellet Substances 0.000 description 2
- ZQBAKBUEJOMQEX-UHFFFAOYSA-N phenyl salicylate Chemical compound OC1=CC=CC=C1C(=O)OC1=CC=CC=C1 ZQBAKBUEJOMQEX-UHFFFAOYSA-N 0.000 description 2
- YBYRMVIVWMBXKQ-UHFFFAOYSA-N phenylmethanesulfonyl fluoride Chemical compound FS(=O)(=O)CC1=CC=CC=C1 YBYRMVIVWMBXKQ-UHFFFAOYSA-N 0.000 description 2
- 239000002953 phosphate buffered saline Substances 0.000 description 2
- HDOWRFHMPULYOA-UHFFFAOYSA-N piperidin-4-ol Chemical compound OC1CCNCC1 HDOWRFHMPULYOA-UHFFFAOYSA-N 0.000 description 2
- NNACHAUCXXVJSP-UHFFFAOYSA-N pitolisant Chemical group C1=CC(Cl)=CC=C1CCCOCCCN1CCCCC1 NNACHAUCXXVJSP-UHFFFAOYSA-N 0.000 description 2
- 229920000435 poly(dimethylsiloxane) Polymers 0.000 description 2
- 229920006254 polymer film Polymers 0.000 description 2
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 2
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 2
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 2
- 230000002335 preservative effect Effects 0.000 description 2
- 230000000069 prophylactic effect Effects 0.000 description 2
- 239000004405 propyl p-hydroxybenzoate Substances 0.000 description 2
- 229960003415 propylparaben Drugs 0.000 description 2
- 102000004169 proteins and genes Human genes 0.000 description 2
- 108090000623 proteins and genes Proteins 0.000 description 2
- AOJFQRQNPXYVLM-UHFFFAOYSA-N pyridin-1-ium;chloride Chemical compound [Cl-].C1=CC=[NH+]C=C1 AOJFQRQNPXYVLM-UHFFFAOYSA-N 0.000 description 2
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 2
- JWVCLYRUEFBMGU-UHFFFAOYSA-N quinazoline Chemical compound N1=CN=CC2=CC=CC=C21 JWVCLYRUEFBMGU-UHFFFAOYSA-N 0.000 description 2
- 239000002287 radioligand Substances 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- 239000005060 rubber Substances 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- QZAYGJVTTNCVMB-UHFFFAOYSA-N serotonin Chemical compound C1=C(O)C=C2C(CCN)=CNC2=C1 QZAYGJVTTNCVMB-UHFFFAOYSA-N 0.000 description 2
- ZLGIYFNHBLSMPS-ATJNOEHPSA-N shellac Chemical compound OCCCCCC(O)C(O)CCCCCCCC(O)=O.C1C23[C@H](C(O)=O)CCC2[C@](C)(CO)[C@@H]1C(C(O)=O)=C[C@@H]3O ZLGIYFNHBLSMPS-ATJNOEHPSA-N 0.000 description 2
- 229920002379 silicone rubber Polymers 0.000 description 2
- 239000004945 silicone rubber Substances 0.000 description 2
- 210000003491 skin Anatomy 0.000 description 2
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 2
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 2
- HRZFUMHJMZEROT-UHFFFAOYSA-L sodium disulfite Chemical compound [Na+].[Na+].[O-]S(=O)S([O-])(=O)=O HRZFUMHJMZEROT-UHFFFAOYSA-L 0.000 description 2
- 229940001584 sodium metabisulfite Drugs 0.000 description 2
- 235000010262 sodium metabisulphite Nutrition 0.000 description 2
- 239000007909 solid dosage form Substances 0.000 description 2
- 235000010199 sorbic acid Nutrition 0.000 description 2
- 239000004334 sorbic acid Substances 0.000 description 2
- 229940075582 sorbic acid Drugs 0.000 description 2
- 239000000600 sorbitol Substances 0.000 description 2
- 235000010356 sorbitol Nutrition 0.000 description 2
- 239000007921 spray Substances 0.000 description 2
- 238000010561 standard procedure Methods 0.000 description 2
- 239000011550 stock solution Substances 0.000 description 2
- 238000007920 subcutaneous administration Methods 0.000 description 2
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 2
- 239000005720 sucrose Substances 0.000 description 2
- 238000009495 sugar coating Methods 0.000 description 2
- 229910052717 sulfur Inorganic materials 0.000 description 2
- 239000000829 suppository Substances 0.000 description 2
- 239000004094 surface-active agent Substances 0.000 description 2
- 239000000454 talc Substances 0.000 description 2
- 229910052623 talc Inorganic materials 0.000 description 2
- 235000019640 taste Nutrition 0.000 description 2
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- 210000001519 tissue Anatomy 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- 238000011200 topical administration Methods 0.000 description 2
- 150000003626 triacylglycerols Chemical class 0.000 description 2
- CYRMSUTZVYGINF-UHFFFAOYSA-N trichlorofluoromethane Chemical compound FC(Cl)(Cl)Cl CYRMSUTZVYGINF-UHFFFAOYSA-N 0.000 description 2
- 235000015112 vegetable and seed oil Nutrition 0.000 description 2
- 239000008158 vegetable oil Substances 0.000 description 2
- 239000003981 vehicle Substances 0.000 description 2
- 230000009385 viral infection Effects 0.000 description 2
- 239000001993 wax Substances 0.000 description 2
- LSPHULWDVZXLIL-UHFFFAOYSA-N (+/-)-Camphoric acid Chemical compound CC1(C)C(C(O)=O)CCC1(C)C(O)=O LSPHULWDVZXLIL-UHFFFAOYSA-N 0.000 description 1
- BZMMRNKDONDVIB-UHFFFAOYSA-N (1-ethoxycyclopropyl)oxy-trimethylsilane Chemical compound CCOC1(O[Si](C)(C)C)CC1 BZMMRNKDONDVIB-UHFFFAOYSA-N 0.000 description 1
- ASGMFNBUXDJWJJ-JLCFBVMHSA-N (1R,3R)-3-[[3-bromo-1-[4-(5-methyl-1,3,4-thiadiazol-2-yl)phenyl]pyrazolo[3,4-d]pyrimidin-6-yl]amino]-N,1-dimethylcyclopentane-1-carboxamide Chemical compound BrC1=NN(C2=NC(=NC=C21)N[C@H]1C[C@@](CC1)(C(=O)NC)C)C1=CC=C(C=C1)C=1SC(=NN=1)C ASGMFNBUXDJWJJ-JLCFBVMHSA-N 0.000 description 1
- UAOUIVVJBYDFKD-XKCDOFEDSA-N (1R,9R,10S,11R,12R,15S,18S,21R)-10,11,21-trihydroxy-8,8-dimethyl-14-methylidene-4-(prop-2-enylamino)-20-oxa-5-thia-3-azahexacyclo[9.7.2.112,15.01,9.02,6.012,18]henicosa-2(6),3-dien-13-one Chemical compound C([C@@H]1[C@@H](O)[C@@]23C(C1=C)=O)C[C@H]2[C@]12C(N=C(NCC=C)S4)=C4CC(C)(C)[C@H]1[C@H](O)[C@]3(O)OC2 UAOUIVVJBYDFKD-XKCDOFEDSA-N 0.000 description 1
- GHYOCDFICYLMRF-UTIIJYGPSA-N (2S,3R)-N-[(2S)-3-(cyclopenten-1-yl)-1-[(2R)-2-methyloxiran-2-yl]-1-oxopropan-2-yl]-3-hydroxy-3-(4-methoxyphenyl)-2-[[(2S)-2-[(2-morpholin-4-ylacetyl)amino]propanoyl]amino]propanamide Chemical compound C1(=CCCC1)C[C@@H](C(=O)[C@@]1(OC1)C)NC([C@H]([C@@H](C1=CC=C(C=C1)OC)O)NC([C@H](C)NC(CN1CCOCC1)=O)=O)=O GHYOCDFICYLMRF-UTIIJYGPSA-N 0.000 description 1
- IUSARDYWEPUTPN-OZBXUNDUSA-N (2r)-n-[(2s,3r)-4-[[(4s)-6-(2,2-dimethylpropyl)spiro[3,4-dihydropyrano[2,3-b]pyridine-2,1'-cyclobutane]-4-yl]amino]-3-hydroxy-1-[3-(1,3-thiazol-2-yl)phenyl]butan-2-yl]-2-methoxypropanamide Chemical compound C([C@H](NC(=O)[C@@H](C)OC)[C@H](O)CN[C@@H]1C2=CC(CC(C)(C)C)=CN=C2OC2(CCC2)C1)C(C=1)=CC=CC=1C1=NC=CS1 IUSARDYWEPUTPN-OZBXUNDUSA-N 0.000 description 1
- YJLIKUSWRSEPSM-WGQQHEPDSA-N (2r,3r,4s,5r)-2-[6-amino-8-[(4-phenylphenyl)methylamino]purin-9-yl]-5-(hydroxymethyl)oxolane-3,4-diol Chemical compound C=1C=C(C=2C=CC=CC=2)C=CC=1CNC1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O YJLIKUSWRSEPSM-WGQQHEPDSA-N 0.000 description 1
- FMCAFXHLMUOIGG-JTJHWIPRSA-N (2s)-2-[[(2r)-2-[[(2s)-2-[[(2r)-2-formamido-3-sulfanylpropanoyl]amino]-3-methylbutanoyl]amino]-3-(4-hydroxy-2,5-dimethylphenyl)propanoyl]amino]-4-methylsulfanylbutanoic acid Chemical compound O=CN[C@@H](CS)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C(=O)N[C@@H](CCSC)C(O)=O)CC1=CC(C)=C(O)C=C1C FMCAFXHLMUOIGG-JTJHWIPRSA-N 0.000 description 1
- FMCAFXHLMUOIGG-IWFBPKFRSA-N (2s)-2-[[(2s)-2-[[(2s)-2-[[(2r)-2-formamido-3-sulfanylpropanoyl]amino]-3-methylbutanoyl]amino]-3-(4-hydroxy-2,5-dimethylphenyl)propanoyl]amino]-4-methylsulfanylbutanoic acid Chemical compound O=CN[C@@H](CS)C(=O)N[C@@H](C(C)C)C(=O)N[C@H](C(=O)N[C@@H](CCSC)C(O)=O)CC1=CC(C)=C(O)C=C1C FMCAFXHLMUOIGG-IWFBPKFRSA-N 0.000 description 1
- VIJSPAIQWVPKQZ-BLECARSGSA-N (2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-acetamido-5-(diaminomethylideneamino)pentanoyl]amino]-4-methylpentanoyl]amino]-4,4-dimethylpentanoyl]amino]-4-methylpentanoyl]amino]propanoyl]amino]-5-(diaminomethylideneamino)pentanoic acid Chemical compound NC(=N)NCCC[C@@H](C(O)=O)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(C)(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(C)=O VIJSPAIQWVPKQZ-BLECARSGSA-N 0.000 description 1
- NAWXUBYGYWOOIX-SFHVURJKSA-N (2s)-2-[[4-[2-(2,4-diaminoquinazolin-6-yl)ethyl]benzoyl]amino]-4-methylidenepentanedioic acid Chemical compound C1=CC2=NC(N)=NC(N)=C2C=C1CCC1=CC=C(C(=O)N[C@@H](CC(=C)C(O)=O)C(O)=O)C=C1 NAWXUBYGYWOOIX-SFHVURJKSA-N 0.000 description 1
- STBLNCCBQMHSRC-BATDWUPUSA-N (2s)-n-[(3s,4s)-5-acetyl-7-cyano-4-methyl-1-[(2-methylnaphthalen-1-yl)methyl]-2-oxo-3,4-dihydro-1,5-benzodiazepin-3-yl]-2-(methylamino)propanamide Chemical compound O=C1[C@@H](NC(=O)[C@H](C)NC)[C@H](C)N(C(C)=O)C2=CC(C#N)=CC=C2N1CC1=C(C)C=CC2=CC=CC=C12 STBLNCCBQMHSRC-BATDWUPUSA-N 0.000 description 1
- IWZSHWBGHQBIML-ZGGLMWTQSA-N (3S,8S,10R,13S,14S,17S)-17-isoquinolin-7-yl-N,N,10,13-tetramethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3-amine Chemical compound CN(C)[C@H]1CC[C@]2(C)C3CC[C@@]4(C)[C@@H](CC[C@@H]4c4ccc5ccncc5c4)[C@@H]3CC=C2C1 IWZSHWBGHQBIML-ZGGLMWTQSA-N 0.000 description 1
- UDQTXCHQKHIQMH-KYGLGHNPSA-N (3ar,5s,6s,7r,7ar)-5-(difluoromethyl)-2-(ethylamino)-5,6,7,7a-tetrahydro-3ah-pyrano[3,2-d][1,3]thiazole-6,7-diol Chemical compound S1C(NCC)=N[C@H]2[C@@H]1O[C@H](C(F)F)[C@@H](O)[C@@H]2O UDQTXCHQKHIQMH-KYGLGHNPSA-N 0.000 description 1
- OOKAZRDERJMRCJ-KOUAFAAESA-N (3r)-7-[(1s,2s,4ar,6s,8s)-2,6-dimethyl-8-[(2s)-2-methylbutanoyl]oxy-1,2,4a,5,6,7,8,8a-octahydronaphthalen-1-yl]-3-hydroxy-5-oxoheptanoic acid Chemical compound C1=C[C@H](C)[C@H](CCC(=O)C[C@@H](O)CC(O)=O)C2[C@@H](OC(=O)[C@@H](C)CC)C[C@@H](C)C[C@@H]21 OOKAZRDERJMRCJ-KOUAFAAESA-N 0.000 description 1
- HUWSZNZAROKDRZ-RRLWZMAJSA-N (3r,4r)-3-azaniumyl-5-[[(2s,3r)-1-[(2s)-2,3-dicarboxypyrrolidin-1-yl]-3-methyl-1-oxopentan-2-yl]amino]-5-oxo-4-sulfanylpentane-1-sulfonate Chemical compound OS(=O)(=O)CC[C@@H](N)[C@@H](S)C(=O)N[C@@H]([C@H](C)CC)C(=O)N1CCC(C(O)=O)[C@H]1C(O)=O HUWSZNZAROKDRZ-RRLWZMAJSA-N 0.000 description 1
- MPDDTAJMJCESGV-CTUHWIOQSA-M (3r,5r)-7-[2-(4-fluorophenyl)-5-[methyl-[(1r)-1-phenylethyl]carbamoyl]-4-propan-2-ylpyrazol-3-yl]-3,5-dihydroxyheptanoate Chemical compound C1([C@@H](C)N(C)C(=O)C2=NN(C(CC[C@@H](O)C[C@@H](O)CC([O-])=O)=C2C(C)C)C=2C=CC(F)=CC=2)=CC=CC=C1 MPDDTAJMJCESGV-CTUHWIOQSA-M 0.000 description 1
- VUEGYUOUAAVYAS-JGGQBBKZSA-N (6ar,9s,10ar)-9-(dimethylsulfamoylamino)-7-methyl-6,6a,8,9,10,10a-hexahydro-4h-indolo[4,3-fg]quinoline Chemical compound C1=CC([C@H]2C[C@@H](CN(C)[C@@H]2C2)NS(=O)(=O)N(C)C)=C3C2=CNC3=C1 VUEGYUOUAAVYAS-JGGQBBKZSA-N 0.000 description 1
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 description 1
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 description 1
- GHOKWGTUZJEAQD-ZETCQYMHSA-N (D)-(+)-Pantothenic acid Chemical compound OCC(C)(C)[C@@H](O)C(=O)NCCC(O)=O GHOKWGTUZJEAQD-ZETCQYMHSA-N 0.000 description 1
- PMJHHCWVYXUKFD-SNAWJCMRSA-N (E)-1,3-pentadiene Chemical compound C\C=C\C=C PMJHHCWVYXUKFD-SNAWJCMRSA-N 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- SRKGZXIJDGWVAI-GVAVTCRGSA-M (e,3r)-7-[6-tert-butyl-4-(4-fluorophenyl)-2-propan-2-ylpyridin-3-yl]-3,5-dihydroxyhept-6-enoate Chemical compound CC(C)C1=NC(C(C)(C)C)=CC(C=2C=CC(F)=CC=2)=C1\C=C\C(O)C[C@@H](O)CC([O-])=O SRKGZXIJDGWVAI-GVAVTCRGSA-M 0.000 description 1
- ZYZCALPXKGUGJI-DDVDASKDSA-M (e,3r,5s)-7-[3-(4-fluorophenyl)-2-phenyl-5-propan-2-ylimidazol-4-yl]-3,5-dihydroxyhept-6-enoate Chemical compound C=1C=C(F)C=CC=1N1C(\C=C\[C@@H](O)C[C@@H](O)CC([O-])=O)=C(C(C)C)N=C1C1=CC=CC=C1 ZYZCALPXKGUGJI-DDVDASKDSA-M 0.000 description 1
- DEVSOMFAQLZNKR-RJRFIUFISA-N (z)-3-[3-[3,5-bis(trifluoromethyl)phenyl]-1,2,4-triazol-1-yl]-n'-pyrazin-2-ylprop-2-enehydrazide Chemical compound FC(F)(F)C1=CC(C(F)(F)F)=CC(C2=NN(\C=C/C(=O)NNC=3N=CC=NC=3)C=N2)=C1 DEVSOMFAQLZNKR-RJRFIUFISA-N 0.000 description 1
- ICLYJLBTOGPLMC-KVVVOXFISA-N (z)-octadec-9-enoate;tris(2-hydroxyethyl)azanium Chemical compound OCCN(CCO)CCO.CCCCCCCC\C=C/CCCCCCCC(O)=O ICLYJLBTOGPLMC-KVVVOXFISA-N 0.000 description 1
- NPNPZTNLOVBDOC-UHFFFAOYSA-N 1,1-difluoroethane Chemical compound CC(F)F NPNPZTNLOVBDOC-UHFFFAOYSA-N 0.000 description 1
- 229940051271 1,1-difluoroethane Drugs 0.000 description 1
- VEFLKXRACNJHOV-UHFFFAOYSA-N 1,3-dibromopropane Chemical compound BrCCCBr VEFLKXRACNJHOV-UHFFFAOYSA-N 0.000 description 1
- KKHFRAFPESRGGD-UHFFFAOYSA-N 1,3-dimethyl-7-[3-(n-methylanilino)propyl]purine-2,6-dione Chemical compound C1=NC=2N(C)C(=O)N(C)C(=O)C=2N1CCCN(C)C1=CC=CC=C1 KKHFRAFPESRGGD-UHFFFAOYSA-N 0.000 description 1
- HTSGKJQDMSTCGS-UHFFFAOYSA-N 1,4-bis(4-chlorophenyl)-2-(4-methylphenyl)sulfonylbutane-1,4-dione Chemical compound C1=CC(C)=CC=C1S(=O)(=O)C(C(=O)C=1C=CC(Cl)=CC=1)CC(=O)C1=CC=C(Cl)C=C1 HTSGKJQDMSTCGS-UHFFFAOYSA-N 0.000 description 1
- DNUTZBZXLPWRJG-UHFFFAOYSA-N 1-Piperidine carboxylic acid Chemical compound OC(=O)N1CCCCC1 DNUTZBZXLPWRJG-UHFFFAOYSA-N 0.000 description 1
- KQZLRWGGWXJPOS-NLFPWZOASA-N 1-[(1R)-1-(2,4-dichlorophenyl)ethyl]-6-[(4S,5R)-4-[(2S)-2-(hydroxymethyl)pyrrolidin-1-yl]-5-methylcyclohexen-1-yl]pyrazolo[3,4-b]pyrazine-3-carbonitrile Chemical compound ClC1=C(C=CC(=C1)Cl)[C@@H](C)N1N=C(C=2C1=NC(=CN=2)C1=CC[C@@H]([C@@H](C1)C)N1[C@@H](CCC1)CO)C#N KQZLRWGGWXJPOS-NLFPWZOASA-N 0.000 description 1
- WZZBNLYBHUDSHF-DHLKQENFSA-N 1-[(3s,4s)-4-[8-(2-chloro-4-pyrimidin-2-yloxyphenyl)-7-fluoro-2-methylimidazo[4,5-c]quinolin-1-yl]-3-fluoropiperidin-1-yl]-2-hydroxyethanone Chemical compound CC1=NC2=CN=C3C=C(F)C(C=4C(=CC(OC=5N=CC=CN=5)=CC=4)Cl)=CC3=C2N1[C@H]1CCN(C(=O)CO)C[C@@H]1F WZZBNLYBHUDSHF-DHLKQENFSA-N 0.000 description 1
- QXOGPTXQGKQSJT-UHFFFAOYSA-N 1-amino-4-[4-(3,4-dimethylphenyl)sulfanylanilino]-9,10-dioxoanthracene-2-sulfonic acid Chemical compound Cc1ccc(Sc2ccc(Nc3cc(c(N)c4C(=O)c5ccccc5C(=O)c34)S(O)(=O)=O)cc2)cc1C QXOGPTXQGKQSJT-UHFFFAOYSA-N 0.000 description 1
- DURPTKYDGMDSBL-UHFFFAOYSA-N 1-butoxybutane Chemical compound CCCCOCCCC DURPTKYDGMDSBL-UHFFFAOYSA-N 0.000 description 1
- VFWCMGCRMGJXDK-UHFFFAOYSA-N 1-chlorobutane Chemical compound CCCCCl VFWCMGCRMGJXDK-UHFFFAOYSA-N 0.000 description 1
- VUQPJRPDRDVQMN-UHFFFAOYSA-N 1-chlorooctadecane Chemical compound CCCCCCCCCCCCCCCCCCCl VUQPJRPDRDVQMN-UHFFFAOYSA-N 0.000 description 1
- MIVZCINZDWIBJS-UHFFFAOYSA-N 1-methylpiperidine-4-carbonyl chloride Chemical compound CN1CCC(C(Cl)=O)CC1 MIVZCINZDWIBJS-UHFFFAOYSA-N 0.000 description 1
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 1
- WGFNXGPBPIJYLI-UHFFFAOYSA-N 2,6-difluoro-3-[(3-fluorophenyl)sulfonylamino]-n-(3-methoxy-1h-pyrazolo[3,4-b]pyridin-5-yl)benzamide Chemical compound C1=C2C(OC)=NNC2=NC=C1NC(=O)C(C=1F)=C(F)C=CC=1NS(=O)(=O)C1=CC=CC(F)=C1 WGFNXGPBPIJYLI-UHFFFAOYSA-N 0.000 description 1
- VCUXVXLUOHDHKK-UHFFFAOYSA-N 2-(2-aminopyrimidin-4-yl)-4-(2-chloro-4-methoxyphenyl)-1,3-thiazole-5-carboxamide Chemical compound ClC1=CC(OC)=CC=C1C1=C(C(N)=O)SC(C=2N=C(N)N=CC=2)=N1 VCUXVXLUOHDHKK-UHFFFAOYSA-N 0.000 description 1
- OEPOKWHJYJXUGD-UHFFFAOYSA-N 2-(3-phenylmethoxyphenyl)-1,3-thiazole-4-carbaldehyde Chemical compound O=CC1=CSC(C=2C=C(OCC=3C=CC=CC=3)C=CC=2)=N1 OEPOKWHJYJXUGD-UHFFFAOYSA-N 0.000 description 1
- FMKGJQHNYMWDFJ-CVEARBPZSA-N 2-[[4-(2,2-difluoropropoxy)pyrimidin-5-yl]methylamino]-4-[[(1R,4S)-4-hydroxy-3,3-dimethylcyclohexyl]amino]pyrimidine-5-carbonitrile Chemical compound FC(COC1=NC=NC=C1CNC1=NC=C(C(=N1)N[C@H]1CC([C@H](CC1)O)(C)C)C#N)(C)F FMKGJQHNYMWDFJ-CVEARBPZSA-N 0.000 description 1
- VVCMGAUPZIKYTH-VGHSCWAPSA-N 2-acetyloxybenzoic acid;[(2s,3r)-4-(dimethylamino)-3-methyl-1,2-diphenylbutan-2-yl] propanoate;1,3,7-trimethylpurine-2,6-dione Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O.CN1C(=O)N(C)C(=O)C2=C1N=CN2C.C([C@](OC(=O)CC)([C@H](C)CN(C)C)C=1C=CC=CC=1)C1=CC=CC=C1 VVCMGAUPZIKYTH-VGHSCWAPSA-N 0.000 description 1
- YSUIQYOGTINQIN-UZFYAQMZSA-N 2-amino-9-[(1S,6R,8R,9S,10R,15R,17R,18R)-8-(6-aminopurin-9-yl)-9,18-difluoro-3,12-dihydroxy-3,12-bis(sulfanylidene)-2,4,7,11,13,16-hexaoxa-3lambda5,12lambda5-diphosphatricyclo[13.2.1.06,10]octadecan-17-yl]-1H-purin-6-one Chemical compound NC1=NC2=C(N=CN2[C@@H]2O[C@@H]3COP(S)(=O)O[C@@H]4[C@@H](COP(S)(=O)O[C@@H]2[C@@H]3F)O[C@H]([C@H]4F)N2C=NC3=C2N=CN=C3N)C(=O)N1 YSUIQYOGTINQIN-UZFYAQMZSA-N 0.000 description 1
- TVTJUIAKQFIXCE-HUKYDQBMSA-N 2-amino-9-[(2R,3S,4S,5R)-4-fluoro-3-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-7-prop-2-ynyl-1H-purine-6,8-dione Chemical compound NC=1NC(C=2N(C(N(C=2N=1)[C@@H]1O[C@@H]([C@H]([C@H]1O)F)CO)=O)CC#C)=O TVTJUIAKQFIXCE-HUKYDQBMSA-N 0.000 description 1
- NPRYCHLHHVWLQZ-TURQNECASA-N 2-amino-9-[(2R,3S,4S,5R)-4-fluoro-3-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-7-prop-2-ynylpurin-8-one Chemical compound NC1=NC=C2N(C(N(C2=N1)[C@@H]1O[C@@H]([C@H]([C@H]1O)F)CO)=O)CC#C NPRYCHLHHVWLQZ-TURQNECASA-N 0.000 description 1
- CFWRDBDJAOHXSH-SECBINFHSA-N 2-azaniumylethyl [(2r)-2,3-diacetyloxypropyl] phosphate Chemical compound CC(=O)OC[C@@H](OC(C)=O)COP(O)(=O)OCCN CFWRDBDJAOHXSH-SECBINFHSA-N 0.000 description 1
- LFOIDLOIBZFWDO-UHFFFAOYSA-N 2-methoxy-6-[6-methoxy-4-[(3-phenylmethoxyphenyl)methoxy]-1-benzofuran-2-yl]imidazo[2,1-b][1,3,4]thiadiazole Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(C=1)=CC=CC=1OCC1=CC=CC=C1 LFOIDLOIBZFWDO-UHFFFAOYSA-N 0.000 description 1
- 125000004918 2-methyl-2-pentyl group Chemical group CC(C)(CCC)* 0.000 description 1
- 125000004493 2-methylbut-1-yl group Chemical group CC(C*)CC 0.000 description 1
- QTWJRLJHJPIABL-UHFFFAOYSA-N 2-methylphenol;3-methylphenol;4-methylphenol Chemical compound CC1=CC=C(O)C=C1.CC1=CC=CC(O)=C1.CC1=CC=CC=C1O QTWJRLJHJPIABL-UHFFFAOYSA-N 0.000 description 1
- WMPPDTMATNBGJN-UHFFFAOYSA-N 2-phenylethylbromide Chemical compound BrCCC1=CC=CC=C1 WMPPDTMATNBGJN-UHFFFAOYSA-N 0.000 description 1
- SLRMQYXOBQWXCR-UHFFFAOYSA-N 2154-56-5 Chemical compound [CH2]C1=CC=CC=C1 SLRMQYXOBQWXCR-UHFFFAOYSA-N 0.000 description 1
- ODJQKYXPKWQWNK-UHFFFAOYSA-N 3,3'-Thiobispropanoic acid Chemical compound OC(=O)CCSCCC(O)=O ODJQKYXPKWQWNK-UHFFFAOYSA-N 0.000 description 1
- QBWKPGNFQQJGFY-QLFBSQMISA-N 3-[(1r)-1-[(2r,6s)-2,6-dimethylmorpholin-4-yl]ethyl]-n-[6-methyl-3-(1h-pyrazol-4-yl)imidazo[1,2-a]pyrazin-8-yl]-1,2-thiazol-5-amine Chemical compound N1([C@H](C)C2=NSC(NC=3C4=NC=C(N4C=C(C)N=3)C3=CNN=C3)=C2)C[C@H](C)O[C@H](C)C1 QBWKPGNFQQJGFY-QLFBSQMISA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-M 3-carboxy-2,3-dihydroxypropanoate Chemical compound OC(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-M 0.000 description 1
- 125000004919 3-methyl-2-pentyl group Chemical group CC(C(C)*)CC 0.000 description 1
- ROLMZTIHUMKEAI-UHFFFAOYSA-N 4,5-difluoro-2-hydroxybenzonitrile Chemical compound OC1=CC(F)=C(F)C=C1C#N ROLMZTIHUMKEAI-UHFFFAOYSA-N 0.000 description 1
- HXCWFNAKKROIHH-UHFFFAOYSA-N 4-(4-nitrophenoxy)piperidine-1-carboxylic acid Chemical compound C1CN(C(=O)O)CCC1OC1=CC=C([N+]([O-])=O)C=C1 HXCWFNAKKROIHH-UHFFFAOYSA-N 0.000 description 1
- IYLZZNHDXBTZMO-UHFFFAOYSA-N 4-[2-(ethylamino)-1-hydroxyethyl]benzene-1,2-diol Chemical compound CCNCC(O)C1=CC=C(O)C(O)=C1 IYLZZNHDXBTZMO-UHFFFAOYSA-N 0.000 description 1
- WYFCZWSWFGJODV-MIANJLSGSA-N 4-[[(1s)-2-[(e)-3-[3-chloro-2-fluoro-6-(tetrazol-1-yl)phenyl]prop-2-enoyl]-5-(4-methyl-2-oxopiperazin-1-yl)-3,4-dihydro-1h-isoquinoline-1-carbonyl]amino]benzoic acid Chemical compound O=C1CN(C)CCN1C1=CC=CC2=C1CCN(C(=O)\C=C\C=1C(=CC=C(Cl)C=1F)N1N=NN=C1)[C@@H]2C(=O)NC1=CC=C(C(O)=O)C=C1 WYFCZWSWFGJODV-MIANJLSGSA-N 0.000 description 1
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 1
- 125000004920 4-methyl-2-pentyl group Chemical group CC(CC(C)*)C 0.000 description 1
- XFJBGINZIMNZBW-CRAIPNDOSA-N 5-chloro-2-[4-[(1r,2s)-2-[2-(5-methylsulfonylpyridin-2-yl)oxyethyl]cyclopropyl]piperidin-1-yl]pyrimidine Chemical compound N1=CC(S(=O)(=O)C)=CC=C1OCC[C@H]1[C@@H](C2CCN(CC2)C=2N=CC(Cl)=CN=2)C1 XFJBGINZIMNZBW-CRAIPNDOSA-N 0.000 description 1
- FCSKOFQQCWLGMV-UHFFFAOYSA-N 5-{5-[2-chloro-4-(4,5-dihydro-1,3-oxazol-2-yl)phenoxy]pentyl}-3-methylisoxazole Chemical compound O1N=C(C)C=C1CCCCCOC1=CC=C(C=2OCCN=2)C=C1Cl FCSKOFQQCWLGMV-UHFFFAOYSA-N 0.000 description 1
- RSIWALKZYXPAGW-NSHDSACASA-N 6-(3-fluorophenyl)-3-methyl-7-[(1s)-1-(7h-purin-6-ylamino)ethyl]-[1,3]thiazolo[3,2-a]pyrimidin-5-one Chemical compound C=1([C@@H](NC=2C=3N=CNC=3N=CN=2)C)N=C2SC=C(C)N2C(=O)C=1C1=CC=CC(F)=C1 RSIWALKZYXPAGW-NSHDSACASA-N 0.000 description 1
- HCCNBKFJYUWLEX-UHFFFAOYSA-N 7-(6-methoxypyridin-3-yl)-1-(2-propoxyethyl)-3-(pyrazin-2-ylmethylamino)pyrido[3,4-b]pyrazin-2-one Chemical compound O=C1N(CCOCCC)C2=CC(C=3C=NC(OC)=CC=3)=NC=C2N=C1NCC1=CN=CC=N1 HCCNBKFJYUWLEX-UHFFFAOYSA-N 0.000 description 1
- NIXOWILDQLNWCW-UHFFFAOYSA-M Acrylate Chemical compound [O-]C(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-M 0.000 description 1
- NIXOWILDQLNWCW-UHFFFAOYSA-N Acrylic acid Chemical group OC(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 1
- 241000251468 Actinopterygii Species 0.000 description 1
- 229920001450 Alpha-Cyclodextrin Polymers 0.000 description 1
- 229910000497 Amalgam Inorganic materials 0.000 description 1
- 241001061225 Arcos Species 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- JQUCWIWWWKZNCS-LESHARBVSA-N C(C1=CC=CC=C1)(=O)NC=1SC[C@H]2[C@@](N1)(CO[C@H](C2)C)C=2SC=C(N2)NC(=O)C2=NC=C(C=C2)OC(F)F Chemical compound C(C1=CC=CC=C1)(=O)NC=1SC[C@H]2[C@@](N1)(CO[C@H](C2)C)C=2SC=C(N2)NC(=O)C2=NC=C(C=C2)OC(F)F JQUCWIWWWKZNCS-LESHARBVSA-N 0.000 description 1
- PKMUHQIDVVOXHQ-HXUWFJFHSA-N C[C@H](C1=CC(C2=CC=C(CNC3CCCC3)S2)=CC=C1)NC(C1=C(C)C=CC(NC2CNC2)=C1)=O Chemical compound C[C@H](C1=CC(C2=CC=C(CNC3CCCC3)S2)=CC=C1)NC(C1=C(C)C=CC(NC2CNC2)=C1)=O PKMUHQIDVVOXHQ-HXUWFJFHSA-N 0.000 description 1
- 241000283707 Capra Species 0.000 description 1
- 208000001573 Cataplexy Diseases 0.000 description 1
- 241000700198 Cavia Species 0.000 description 1
- 239000004709 Chlorinated polyethylene Substances 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- 102000008186 Collagen Human genes 0.000 description 1
- 108010035532 Collagen Proteins 0.000 description 1
- 229940127007 Compound 39 Drugs 0.000 description 1
- FKLJPTJMIBLJAV-UHFFFAOYSA-N Compound IV Chemical compound O1N=C(C)C=C1CCCCCCCOC1=CC=C(C=2OCCN=2)C=C1 FKLJPTJMIBLJAV-UHFFFAOYSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- DSLZVSRJTYRBFB-LLEIAEIESA-L D-glucarate(2-) Chemical compound [O-]C(=O)[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O DSLZVSRJTYRBFB-LLEIAEIESA-L 0.000 description 1
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 1
- AEMOLEFTQBMNLQ-AQKNRBDQSA-N D-glucopyranuronic acid Chemical compound OC1O[C@H](C(O)=O)[C@@H](O)[C@H](O)[C@H]1O AEMOLEFTQBMNLQ-AQKNRBDQSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- XBPCUCUWBYBCDP-UHFFFAOYSA-N Dicyclohexylamine Chemical class C1CCCCC1NC1CCCCC1 XBPCUCUWBYBCDP-UHFFFAOYSA-N 0.000 description 1
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- 239000001692 EU approved anti-caking agent Substances 0.000 description 1
- 241000196324 Embryophyta Species 0.000 description 1
- 239000004593 Epoxy Substances 0.000 description 1
- 241000283086 Equidae Species 0.000 description 1
- IMROMDMJAWUWLK-UHFFFAOYSA-N Ethenol Chemical compound OC=C IMROMDMJAWUWLK-UHFFFAOYSA-N 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- 108091006027 G proteins Proteins 0.000 description 1
- 102000013446 GTP Phosphohydrolases Human genes 0.000 description 1
- 102000030782 GTP binding Human genes 0.000 description 1
- 108091000058 GTP-Binding Proteins 0.000 description 1
- 108091006109 GTPases Proteins 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 244000043261 Hevea brasiliensis Species 0.000 description 1
- 241001272567 Hominoidea Species 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- 239000004354 Hydroxyethyl cellulose Substances 0.000 description 1
- 229920000663 Hydroxyethyl cellulose Polymers 0.000 description 1
- 102000004310 Ion Channels Human genes 0.000 description 1
- 150000000994 L-ascorbates Chemical class 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 1
- 239000004166 Lanolin Substances 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- CERQOIWHTDAKMF-UHFFFAOYSA-M Methacrylate Chemical compound CC(=C)C([O-])=O CERQOIWHTDAKMF-UHFFFAOYSA-M 0.000 description 1
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 1
- LVDRREOUMKACNJ-BKMJKUGQSA-N N-[(2R,3S)-2-(4-chlorophenyl)-1-(1,4-dimethyl-2-oxoquinolin-7-yl)-6-oxopiperidin-3-yl]-2-methylpropane-1-sulfonamide Chemical compound CC(C)CS(=O)(=O)N[C@H]1CCC(=O)N([C@@H]1c1ccc(Cl)cc1)c1ccc2c(C)cc(=O)n(C)c2c1 LVDRREOUMKACNJ-BKMJKUGQSA-N 0.000 description 1
- OPFJDXRVMFKJJO-ZHHKINOHSA-N N-{[3-(2-benzamido-4-methyl-1,3-thiazol-5-yl)-pyrazol-5-yl]carbonyl}-G-dR-G-dD-dD-dD-NH2 Chemical compound S1C(C=2NN=C(C=2)C(=O)NCC(=O)N[C@H](CCCN=C(N)N)C(=O)NCC(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC(O)=O)C(N)=O)=C(C)N=C1NC(=O)C1=CC=CC=C1 OPFJDXRVMFKJJO-ZHHKINOHSA-N 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- QOVYHDHLFPKQQG-NDEPHWFRSA-N N[C@@H](CCC(=O)N1CCC(CC1)NC1=C2C=CC=CC2=NC(NCC2=CN(CCCNCCCNC3CCCCC3)N=N2)=N1)C(O)=O Chemical compound N[C@@H](CCC(=O)N1CCC(CC1)NC1=C2C=CC=CC2=NC(NCC2=CN(CCCNCCCNC3CCCCC3)N=N2)=N1)C(O)=O QOVYHDHLFPKQQG-NDEPHWFRSA-N 0.000 description 1
- 208000012902 Nervous system disease Diseases 0.000 description 1
- 208000025966 Neurological disease Diseases 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- 239000004677 Nylon Substances 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 1
- 241000282579 Pan Species 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 1
- 229920012485 Plasticized Polyvinyl chloride Polymers 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- 239000005062 Polybutadiene Substances 0.000 description 1
- 229920002367 Polyisobutene Polymers 0.000 description 1
- 108010039918 Polylysine Proteins 0.000 description 1
- 229920001710 Polyorthoester Polymers 0.000 description 1
- 229920001213 Polysorbate 20 Polymers 0.000 description 1
- 239000004372 Polyvinyl alcohol Substances 0.000 description 1
- HCBIBCJNVBAKAB-UHFFFAOYSA-N Procaine hydrochloride Chemical compound Cl.CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 HCBIBCJNVBAKAB-UHFFFAOYSA-N 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-N Propionic acid Chemical class CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 1
- 241000283984 Rodentia Species 0.000 description 1
- 235000019485 Safflower oil Nutrition 0.000 description 1
- 229920001800 Shellac Polymers 0.000 description 1
- 241000282887 Suidae Species 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 229920002253 Tannate Polymers 0.000 description 1
- 239000003490 Thiodipropionic acid Substances 0.000 description 1
- 241000009298 Trigla lyra Species 0.000 description 1
- 239000007983 Tris buffer Substances 0.000 description 1
- BZHJMEDXRYGGRV-UHFFFAOYSA-N Vinyl chloride Chemical compound ClC=C BZHJMEDXRYGGRV-UHFFFAOYSA-N 0.000 description 1
- 229930003427 Vitamin E Natural products 0.000 description 1
- LJOOWESTVASNOG-UFJKPHDISA-N [(1s,3r,4ar,7s,8s,8as)-3-hydroxy-8-[2-[(4r)-4-hydroxy-6-oxooxan-2-yl]ethyl]-7-methyl-1,2,3,4,4a,7,8,8a-octahydronaphthalen-1-yl] (2s)-2-methylbutanoate Chemical compound C([C@H]1[C@@H](C)C=C[C@H]2C[C@@H](O)C[C@@H]([C@H]12)OC(=O)[C@@H](C)CC)CC1C[C@@H](O)CC(=O)O1 LJOOWESTVASNOG-UFJKPHDISA-N 0.000 description 1
- SPXSEZMVRJLHQG-XMMPIXPASA-N [(2R)-1-[[4-[(3-phenylmethoxyphenoxy)methyl]phenyl]methyl]pyrrolidin-2-yl]methanol Chemical compound C(C1=CC=CC=C1)OC=1C=C(OCC2=CC=C(CN3[C@H](CCC3)CO)C=C2)C=CC=1 SPXSEZMVRJLHQG-XMMPIXPASA-N 0.000 description 1
- IOSLINNLJFQMFF-XMMPIXPASA-N [(2R)-1-[[4-[[3-[(4-fluorophenyl)methylsulfanyl]phenoxy]methyl]phenyl]methyl]pyrrolidin-2-yl]methanol Chemical compound FC1=CC=C(CSC=2C=C(OCC3=CC=C(CN4[C@H](CCC4)CO)C=C3)C=CC=2)C=C1 IOSLINNLJFQMFF-XMMPIXPASA-N 0.000 description 1
- PSLUFJFHTBIXMW-WYEYVKMPSA-N [(3r,4ar,5s,6s,6as,10s,10ar,10bs)-3-ethenyl-10,10b-dihydroxy-3,4a,7,7,10a-pentamethyl-1-oxo-6-(2-pyridin-2-ylethylcarbamoyloxy)-5,6,6a,8,9,10-hexahydro-2h-benzo[f]chromen-5-yl] acetate Chemical compound O([C@@H]1[C@@H]([C@]2(O[C@](C)(CC(=O)[C@]2(O)[C@@]2(C)[C@@H](O)CCC(C)(C)[C@@H]21)C=C)C)OC(=O)C)C(=O)NCCC1=CC=CC=N1 PSLUFJFHTBIXMW-WYEYVKMPSA-N 0.000 description 1
- YTLZIKSTJAMOAK-UHFFFAOYSA-N [4-(4-aminophenoxy)piperidin-1-yl]-cyclohexylmethanone Chemical compound NC1=CC=C(OC2CCN(CC2)C(=O)C2CCCCC2)C=C1 YTLZIKSTJAMOAK-UHFFFAOYSA-N 0.000 description 1
- SMNRFWMNPDABKZ-WVALLCKVSA-N [[(2R,3S,4R,5S)-5-(2,6-dioxo-3H-pyridin-3-yl)-3,4-dihydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl] [[[(2R,3S,4S,5R,6R)-4-fluoro-3,5-dihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-hydroxyphosphoryl]oxy-hydroxyphosphoryl] hydrogen phosphate Chemical compound OC[C@H]1O[C@H](OP(O)(=O)OP(O)(=O)OP(O)(=O)OP(O)(=O)OC[C@H]2O[C@H]([C@H](O)[C@@H]2O)C2C=CC(=O)NC2=O)[C@H](O)[C@@H](F)[C@@H]1O SMNRFWMNPDABKZ-WVALLCKVSA-N 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- DHKHKXVYLBGOIT-UHFFFAOYSA-N acetaldehyde Diethyl Acetal Natural products CCOC(C)OCC DHKHKXVYLBGOIT-UHFFFAOYSA-N 0.000 description 1
- 150000001242 acetic acid derivatives Chemical class 0.000 description 1
- OIPILFWXSMYKGL-UHFFFAOYSA-N acetylcholine Chemical compound CC(=O)OCC[N+](C)(C)C OIPILFWXSMYKGL-UHFFFAOYSA-N 0.000 description 1
- 229960004373 acetylcholine Drugs 0.000 description 1
- 150000008043 acidic salts Chemical class 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 125000002252 acyl group Chemical group 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 239000000853 adhesive Substances 0.000 description 1
- 230000001070 adhesive effect Effects 0.000 description 1
- 239000003463 adsorbent Substances 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 230000002776 aggregation Effects 0.000 description 1
- 238000004220 aggregation Methods 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 150000001447 alkali salts Chemical class 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 125000004183 alkoxy alkyl group Chemical group 0.000 description 1
- 150000001350 alkyl halides Chemical class 0.000 description 1
- 125000002947 alkylene group Chemical group 0.000 description 1
- VWYANPOOORUCFJ-UHFFFAOYSA-N alpha-Fernenol Chemical compound CC1(C)C(O)CCC2(C)C3=CCC4(C)C5CCC(C(C)C)C5(C)CCC4(C)C3CCC21 VWYANPOOORUCFJ-UHFFFAOYSA-N 0.000 description 1
- HFHDHCJBZVLPGP-RWMJIURBSA-N alpha-cyclodextrin Chemical compound OC[C@H]([C@H]([C@@H]([C@H]1O)O)O[C@H]2O[C@@H]([C@@H](O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](CO)[C@H]([C@@H]([C@H]3O)O)O3)[C@H](O)[C@H]2O)CO)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@@H]3O[C@@H]1CO HFHDHCJBZVLPGP-RWMJIURBSA-N 0.000 description 1
- 229940043377 alpha-cyclodextrin Drugs 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- CEGOLXSVJUTHNZ-UHFFFAOYSA-K aluminium tristearate Chemical compound [Al+3].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CEGOLXSVJUTHNZ-UHFFFAOYSA-K 0.000 description 1
- 229940063655 aluminum stearate Drugs 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 229920000469 amphiphilic block copolymer Polymers 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 230000008485 antagonism Effects 0.000 description 1
- 239000000729 antidote Substances 0.000 description 1
- 229940075522 antidotes Drugs 0.000 description 1
- 239000003429 antifungal agent Substances 0.000 description 1
- 229940121375 antifungal agent Drugs 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 239000012736 aqueous medium Substances 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 229940072107 ascorbate Drugs 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- XRWSZZJLZRKHHD-WVWIJVSJSA-N asunaprevir Chemical compound O=C([C@@H]1C[C@H](CN1C(=O)[C@@H](NC(=O)OC(C)(C)C)C(C)(C)C)OC1=NC=C(C2=CC=C(Cl)C=C21)OC)N[C@]1(C(=O)NS(=O)(=O)C2CC2)C[C@H]1C=C XRWSZZJLZRKHHD-WVWIJVSJSA-N 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 230000001580 bacterial effect Effects 0.000 description 1
- 239000011324 bead Substances 0.000 description 1
- 235000013871 bee wax Nutrition 0.000 description 1
- 239000012166 beeswax Substances 0.000 description 1
- 229940092738 beeswax Drugs 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 229940077388 benzenesulfonate Drugs 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical class OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229960001950 benzethonium chloride Drugs 0.000 description 1
- UREZNYTWGJKWBI-UHFFFAOYSA-M benzethonium chloride Chemical compound [Cl-].C1=CC(C(C)(C)CC(C)(C)C)=CC=C1OCCOCC[N+](C)(C)CC1=CC=CC=C1 UREZNYTWGJKWBI-UHFFFAOYSA-M 0.000 description 1
- 150000001558 benzoic acid derivatives Chemical class 0.000 description 1
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 235000011175 beta-cyclodextrine Nutrition 0.000 description 1
- 229920002988 biodegradable polymer Polymers 0.000 description 1
- 239000004621 biodegradable polymer Substances 0.000 description 1
- 230000009141 biological interaction Effects 0.000 description 1
- 239000012472 biological sample Substances 0.000 description 1
- SIPUZPBQZHNSDW-UHFFFAOYSA-N bis(2-methylpropyl)aluminum Chemical compound CC(C)C[Al]CC(C)C SIPUZPBQZHNSDW-UHFFFAOYSA-N 0.000 description 1
- 210000001124 body fluid Anatomy 0.000 description 1
- 239000010839 body fluid Substances 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- 150000001642 boronic acid derivatives Chemical class 0.000 description 1
- 125000001246 bromo group Chemical group Br* 0.000 description 1
- KXVUSQIDCZRUKF-UHFFFAOYSA-N bromocyclobutane Chemical compound BrC1CCC1 KXVUSQIDCZRUKF-UHFFFAOYSA-N 0.000 description 1
- 239000001273 butane Substances 0.000 description 1
- 150000004648 butanoic acid derivatives Chemical class 0.000 description 1
- 229940038926 butyl chloride Drugs 0.000 description 1
- 229920005549 butyl rubber Polymers 0.000 description 1
- 150000003940 butylamines Chemical group 0.000 description 1
- 235000010410 calcium alginate Nutrition 0.000 description 1
- 239000000648 calcium alginate Substances 0.000 description 1
- 229960002681 calcium alginate Drugs 0.000 description 1
- 159000000007 calcium salts Chemical class 0.000 description 1
- OKHHGHGGPDJQHR-YMOPUZKJSA-L calcium;(2s,3s,4s,5s,6r)-6-[(2r,3s,4r,5s,6r)-2-carboxy-6-[(2r,3s,4r,5s,6r)-2-carboxylato-4,5,6-trihydroxyoxan-3-yl]oxy-4,5-dihydroxyoxan-3-yl]oxy-3,4,5-trihydroxyoxane-2-carboxylate Chemical compound [Ca+2].O[C@@H]1[C@H](O)[C@H](O)O[C@@H](C([O-])=O)[C@H]1O[C@H]1[C@@H](O)[C@@H](O)[C@H](O[C@H]2[C@H]([C@@H](O)[C@H](O)[C@H](O2)C([O-])=O)O)[C@H](C(O)=O)O1 OKHHGHGGPDJQHR-YMOPUZKJSA-L 0.000 description 1
- BPKIGYQJPYCAOW-FFJTTWKXSA-I calcium;potassium;disodium;(2s)-2-hydroxypropanoate;dichloride;dihydroxide;hydrate Chemical compound O.[OH-].[OH-].[Na+].[Na+].[Cl-].[Cl-].[K+].[Ca+2].C[C@H](O)C([O-])=O BPKIGYQJPYCAOW-FFJTTWKXSA-I 0.000 description 1
- BMLSTPRTEKLIPM-UHFFFAOYSA-I calcium;potassium;disodium;hydrogen carbonate;dichloride;dihydroxide;hydrate Chemical compound O.[OH-].[OH-].[Na+].[Na+].[Cl-].[Cl-].[K+].[Ca+2].OC([O-])=O BMLSTPRTEKLIPM-UHFFFAOYSA-I 0.000 description 1
- MIOPJNTWMNEORI-UHFFFAOYSA-N camphorsulfonic acid Chemical compound C1CC2(CS(O)(=O)=O)C(=O)CC1C2(C)C MIOPJNTWMNEORI-UHFFFAOYSA-N 0.000 description 1
- 125000004452 carbocyclyl group Chemical group 0.000 description 1
- 150000001722 carbon compounds Chemical class 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 229910002092 carbon dioxide Inorganic materials 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 229940096529 carboxypolymethylene Drugs 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229940081733 cetearyl alcohol Drugs 0.000 description 1
- 229960000541 cetyl alcohol Drugs 0.000 description 1
- 229910052729 chemical element Inorganic materials 0.000 description 1
- 125000003636 chemical group Chemical group 0.000 description 1
- 238000001311 chemical methods and process Methods 0.000 description 1
- 239000007910 chewable tablet Substances 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- 229960004926 chlorobutanol Drugs 0.000 description 1
- YACLQRRMGMJLJV-UHFFFAOYSA-N chloroprene Chemical compound ClC(=C)C=C YACLQRRMGMJLJV-UHFFFAOYSA-N 0.000 description 1
- 229940001468 citrate Drugs 0.000 description 1
- 150000001860 citric acid derivatives Chemical class 0.000 description 1
- 239000003240 coconut oil Substances 0.000 description 1
- 235000019864 coconut oil Nutrition 0.000 description 1
- 229920001436 collagen Polymers 0.000 description 1
- 238000004040 coloring Methods 0.000 description 1
- 239000013066 combination product Substances 0.000 description 1
- 229940127555 combination product Drugs 0.000 description 1
- 238000002648 combination therapy Methods 0.000 description 1
- 229940125797 compound 12 Drugs 0.000 description 1
- 229940125810 compound 20 Drugs 0.000 description 1
- 229940126086 compound 21 Drugs 0.000 description 1
- 229940125961 compound 24 Drugs 0.000 description 1
- 229940125846 compound 25 Drugs 0.000 description 1
- 229940125851 compound 27 Drugs 0.000 description 1
- 229940127204 compound 29 Drugs 0.000 description 1
- 229940125877 compound 31 Drugs 0.000 description 1
- 229940125878 compound 36 Drugs 0.000 description 1
- 229940125807 compound 37 Drugs 0.000 description 1
- 229940127573 compound 38 Drugs 0.000 description 1
- 229940126540 compound 41 Drugs 0.000 description 1
- 229940125936 compound 42 Drugs 0.000 description 1
- 229940127271 compound 49 Drugs 0.000 description 1
- 229940125898 compound 5 Drugs 0.000 description 1
- 229940126545 compound 53 Drugs 0.000 description 1
- 229940127113 compound 57 Drugs 0.000 description 1
- 229940125900 compound 59 Drugs 0.000 description 1
- 229940126179 compound 72 Drugs 0.000 description 1
- 238000007906 compression Methods 0.000 description 1
- 230000006835 compression Effects 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 230000005494 condensation Effects 0.000 description 1
- 238000013267 controlled drug release Methods 0.000 description 1
- 238000013270 controlled release Methods 0.000 description 1
- 239000012050 conventional carrier Substances 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 235000005687 corn oil Nutrition 0.000 description 1
- 239000002285 corn oil Substances 0.000 description 1
- 239000006184 cosolvent Substances 0.000 description 1
- 235000012343 cottonseed oil Nutrition 0.000 description 1
- 239000002385 cottonseed oil Substances 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 229930003836 cresol Natural products 0.000 description 1
- 229920006037 cross link polymer Polymers 0.000 description 1
- 239000002577 cryoprotective agent Substances 0.000 description 1
- 229940097362 cyclodextrins Drugs 0.000 description 1
- RVOJTCZRIKWHDX-UHFFFAOYSA-N cyclohexanecarbonyl chloride Chemical compound ClC(=O)C1CCCCC1 RVOJTCZRIKWHDX-UHFFFAOYSA-N 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- WEPUZBYKXNKSDH-UHFFFAOYSA-N cyclopentanecarbonyl chloride Chemical compound ClC(=O)C1CCCC1 WEPUZBYKXNKSDH-UHFFFAOYSA-N 0.000 description 1
- 238000007405 data analysis Methods 0.000 description 1
- 230000007423 decrease Effects 0.000 description 1
- 125000002704 decyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 239000008121 dextrose Substances 0.000 description 1
- 239000008355 dextrose injection Substances 0.000 description 1
- 150000008050 dialkyl sulfates Chemical class 0.000 description 1
- LMEDOLJKVASKTP-UHFFFAOYSA-N dibutyl sulfate Chemical compound CCCCOS(=O)(=O)OCCCC LMEDOLJKVASKTP-UHFFFAOYSA-N 0.000 description 1
- PXBRQCKWGAHEHS-UHFFFAOYSA-N dichlorodifluoromethane Chemical compound FC(F)(Cl)Cl PXBRQCKWGAHEHS-UHFFFAOYSA-N 0.000 description 1
- 235000019404 dichlorodifluoromethane Nutrition 0.000 description 1
- UMNKXPULIDJLSU-UHFFFAOYSA-N dichlorofluoromethane Chemical compound FC(Cl)Cl UMNKXPULIDJLSU-UHFFFAOYSA-N 0.000 description 1
- 229940099364 dichlorofluoromethane Drugs 0.000 description 1
- 229940087091 dichlorotetrafluoroethane Drugs 0.000 description 1
- 235000005911 diet Nutrition 0.000 description 1
- 230000037213 diet Effects 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- 125000004177 diethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- VAYGXNSJCAHWJZ-UHFFFAOYSA-N dimethyl sulfate Chemical compound COS(=O)(=O)OC VAYGXNSJCAHWJZ-UHFFFAOYSA-N 0.000 description 1
- 150000002016 disaccharides Chemical class 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- 125000003438 dodecyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- JRBPAEWTRLWTQC-UHFFFAOYSA-N dodecylamine Chemical compound CCCCCCCCCCCCN JRBPAEWTRLWTQC-UHFFFAOYSA-N 0.000 description 1
- 229960003638 dopamine Drugs 0.000 description 1
- 239000008298 dragée Substances 0.000 description 1
- 238000009510 drug design Methods 0.000 description 1
- 238000009509 drug development Methods 0.000 description 1
- 238000001962 electrophoresis Methods 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- BJXYHBKEQFQVES-NWDGAFQWSA-N enpatoran Chemical compound N[C@H]1CN(C[C@H](C1)C(F)(F)F)C1=C2C=CC=NC2=C(C=C1)C#N BJXYHBKEQFQVES-NWDGAFQWSA-N 0.000 description 1
- 239000003248 enzyme activator Substances 0.000 description 1
- 210000002615 epidermis Anatomy 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- HQQADJVZYDDRJT-UHFFFAOYSA-N ethene;prop-1-ene Chemical group C=C.CC=C HQQADJVZYDDRJT-UHFFFAOYSA-N 0.000 description 1
- NLFBCYMMUAKCPC-KQQUZDAGSA-N ethyl (e)-3-[3-amino-2-cyano-1-[(e)-3-ethoxy-3-oxoprop-1-enyl]sulfanyl-3-oxoprop-1-enyl]sulfanylprop-2-enoate Chemical compound CCOC(=O)\C=C\SC(=C(C#N)C(N)=O)S\C=C\C(=O)OCC NLFBCYMMUAKCPC-KQQUZDAGSA-N 0.000 description 1
- GWNFQAKCJYEJEW-UHFFFAOYSA-N ethyl 3-[8-[[4-methyl-5-[(3-methyl-4-oxophthalazin-1-yl)methyl]-1,2,4-triazol-3-yl]sulfanyl]octanoylamino]benzoate Chemical compound CCOC(=O)C1=CC(NC(=O)CCCCCCCSC2=NN=C(CC3=NN(C)C(=O)C4=CC=CC=C34)N2C)=CC=C1 GWNFQAKCJYEJEW-UHFFFAOYSA-N 0.000 description 1
- 229960003750 ethyl chloride Drugs 0.000 description 1
- DEFVIWRASFVYLL-UHFFFAOYSA-N ethylene glycol bis(2-aminoethyl)tetraacetic acid Chemical compound OC(=O)CN(CC(O)=O)CCOCCOCCN(CC(O)=O)CC(O)=O DEFVIWRASFVYLL-UHFFFAOYSA-N 0.000 description 1
- 239000005038 ethylene vinyl acetate Substances 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 239000003925 fat Substances 0.000 description 1
- 235000019197 fats Nutrition 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- 230000002349 favourable effect Effects 0.000 description 1
- RWTNPBWLLIMQHL-UHFFFAOYSA-N fexofenadine Chemical group C1=CC(C(C)(C(O)=O)C)=CC=C1C(O)CCCN1CCC(C(O)(C=2C=CC=CC=2)C=2C=CC=CC=2)CC1 RWTNPBWLLIMQHL-UHFFFAOYSA-N 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 239000012467 final product Substances 0.000 description 1
- RFHAOTPXVQNOHP-UHFFFAOYSA-N fluconazole Chemical compound C1=NC=NN1CC(C=1C(=CC(F)=CC=1)F)(O)CN1C=NC=N1 RFHAOTPXVQNOHP-UHFFFAOYSA-N 0.000 description 1
- 229960004884 fluconazole Drugs 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- 239000006260 foam Substances 0.000 description 1
- 238000004108 freeze drying Methods 0.000 description 1
- 238000007710 freezing Methods 0.000 description 1
- 230000008014 freezing Effects 0.000 description 1
- 229940050411 fumarate Drugs 0.000 description 1
- 238000010230 functional analysis Methods 0.000 description 1
- 230000002538 fungal effect Effects 0.000 description 1
- 229960003692 gamma aminobutyric acid Drugs 0.000 description 1
- BTCSSZJGUNDROE-UHFFFAOYSA-N gamma-aminobutyric acid Chemical compound NCCCC(O)=O BTCSSZJGUNDROE-UHFFFAOYSA-N 0.000 description 1
- WIGCFUFOHFEKBI-UHFFFAOYSA-N gamma-tocopherol Natural products CC(C)CCCC(C)CCCC(C)CCCC1CCC2C(C)C(O)C(C)C(C)C2O1 WIGCFUFOHFEKBI-UHFFFAOYSA-N 0.000 description 1
- 210000004211 gastric acid Anatomy 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 229940114119 gentisate Drugs 0.000 description 1
- 229940050410 gluconate Drugs 0.000 description 1
- 229940097042 glucuronate Drugs 0.000 description 1
- 229930195712 glutamate Natural products 0.000 description 1
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 1
- 229960002743 glutamine Drugs 0.000 description 1
- 150000004676 glycans Chemical class 0.000 description 1
- YQEMORVAKMFKLG-UHFFFAOYSA-N glycerine monostearate Natural products CCCCCCCCCCCCCCCCCC(=O)OC(CO)CO YQEMORVAKMFKLG-UHFFFAOYSA-N 0.000 description 1
- SVUQHVRAGMNPLW-UHFFFAOYSA-N glycerol monostearate Natural products CCCCCCCCCCCCCCCCC(=O)OCC(O)CO SVUQHVRAGMNPLW-UHFFFAOYSA-N 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 239000003979 granulating agent Substances 0.000 description 1
- 238000000227 grinding Methods 0.000 description 1
- 159000000011 group IA salts Chemical class 0.000 description 1
- JAXFJECJQZDFJS-XHEPKHHKSA-N gtpl8555 Chemical compound OC(=O)C[C@H](N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N1CCC[C@@H]1C(=O)N[C@H](B1O[C@@]2(C)[C@H]3C[C@H](C3(C)C)C[C@H]2O1)CCC1=CC=C(F)C=C1 JAXFJECJQZDFJS-XHEPKHHKSA-N 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- 150000008282 halocarbons Chemical class 0.000 description 1
- 125000005843 halogen group Chemical group 0.000 description 1
- 239000007902 hard capsule Substances 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- UKACHOXRXFQJFN-UHFFFAOYSA-N heptafluoropropane Chemical compound FC(F)C(F)(F)C(F)(F)F UKACHOXRXFQJFN-UHFFFAOYSA-N 0.000 description 1
- 125000003187 heptyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- BXWNKGSJHAJOGX-UHFFFAOYSA-N hexadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCO BXWNKGSJHAJOGX-UHFFFAOYSA-N 0.000 description 1
- UBHWBODXJBSFLH-UHFFFAOYSA-N hexadecan-1-ol;octadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCO.CCCCCCCCCCCCCCCCCCO UBHWBODXJBSFLH-UHFFFAOYSA-N 0.000 description 1
- 239000005556 hormone Substances 0.000 description 1
- 229940088597 hormone Drugs 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 150000002430 hydrocarbons Chemical class 0.000 description 1
- 239000008173 hydrogenated soybean oil Substances 0.000 description 1
- 239000008172 hydrogenated vegetable oil Substances 0.000 description 1
- 238000005984 hydrogenation reaction Methods 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- 229920001477 hydrophilic polymer Polymers 0.000 description 1
- 229960004337 hydroquinone Drugs 0.000 description 1
- 235000019447 hydroxyethyl cellulose Nutrition 0.000 description 1
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 1
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 1
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 1
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 description 1
- 238000003384 imaging method Methods 0.000 description 1
- 238000002513 implantation Methods 0.000 description 1
- 239000011261 inert gas Substances 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- 229910052500 inorganic mineral Inorganic materials 0.000 description 1
- 230000002452 interceptive effect Effects 0.000 description 1
- 210000000936 intestine Anatomy 0.000 description 1
- 238000001361 intraarterial administration Methods 0.000 description 1
- 238000007917 intracranial administration Methods 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 238000007916 intrasternal administration Methods 0.000 description 1
- 238000007919 intrasynovial administration Methods 0.000 description 1
- 238000007913 intrathecal administration Methods 0.000 description 1
- 238000007915 intraurethral administration Methods 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- 238000007914 intraventricular administration Methods 0.000 description 1
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 1
- 125000002346 iodo group Chemical group I* 0.000 description 1
- 229920000554 ionomer Polymers 0.000 description 1
- 239000001282 iso-butane Substances 0.000 description 1
- TWBYWOBDOCUKOW-UHFFFAOYSA-M isonicotinate Chemical compound [O-]C(=O)C1=CC=NC=C1 TWBYWOBDOCUKOW-UHFFFAOYSA-M 0.000 description 1
- 238000010983 kinetics study Methods 0.000 description 1
- 238000009533 lab test Methods 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 235000019388 lanolin Nutrition 0.000 description 1
- 229940039717 lanolin Drugs 0.000 description 1
- 239000007942 layered tablet Substances 0.000 description 1
- 235000010445 lecithin Nutrition 0.000 description 1
- 239000000787 lecithin Substances 0.000 description 1
- 229940067606 lecithin Drugs 0.000 description 1
- 239000003446 ligand Substances 0.000 description 1
- 229940057995 liquid paraffin Drugs 0.000 description 1
- 229910003002 lithium salt Inorganic materials 0.000 description 1
- 159000000002 lithium salts Chemical class 0.000 description 1
- RENRQMCACQEWFC-UGKGYDQZSA-N lnp023 Chemical compound C1([C@H]2N(CC=3C=4C=CNC=4C(C)=CC=3OC)CC[C@@H](C2)OCC)=CC=C(C(O)=O)C=C1 RENRQMCACQEWFC-UGKGYDQZSA-N 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 239000008176 lyophilized powder Substances 0.000 description 1
- 230000002934 lysing effect Effects 0.000 description 1
- ZLNQQNXFFQJAID-UHFFFAOYSA-L magnesium carbonate Chemical compound [Mg+2].[O-]C([O-])=O ZLNQQNXFFQJAID-UHFFFAOYSA-L 0.000 description 1
- 239000001095 magnesium carbonate Substances 0.000 description 1
- 229910000021 magnesium carbonate Inorganic materials 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 238000004949 mass spectrometry Methods 0.000 description 1
- 238000002483 medication Methods 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 239000001525 mentha piperita l. herb oil Substances 0.000 description 1
- AGJSNMGHAVDLRQ-HUUJSLGLSA-N methyl (2s)-2-[[(2r)-2-[[(2s)-2-[[(2r)-2-amino-3-sulfanylpropanoyl]amino]-3-methylbutanoyl]amino]-3-(4-hydroxy-2,3-dimethylphenyl)propanoyl]amino]-4-methylsulfanylbutanoate Chemical compound SC[C@H](N)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C(=O)N[C@@H](CCSC)C(=O)OC)CC1=CC=C(O)C(C)=C1C AGJSNMGHAVDLRQ-HUUJSLGLSA-N 0.000 description 1
- AGJSNMGHAVDLRQ-IWFBPKFRSA-N methyl (2s)-2-[[(2s)-2-[[(2s)-2-[[(2r)-2-amino-3-sulfanylpropanoyl]amino]-3-methylbutanoyl]amino]-3-(4-hydroxy-2,3-dimethylphenyl)propanoyl]amino]-4-methylsulfanylbutanoate Chemical compound SC[C@H](N)C(=O)N[C@@H](C(C)C)C(=O)N[C@H](C(=O)N[C@@H](CCSC)C(=O)OC)CC1=CC=C(O)C(C)=C1C AGJSNMGHAVDLRQ-IWFBPKFRSA-N 0.000 description 1
- ZBELDPMWYXDLNY-UHFFFAOYSA-N methyl 9-(4-bromo-2-fluoroanilino)-[1,3]thiazolo[5,4-f]quinazoline-2-carboximidate Chemical compound C12=C3SC(C(=N)OC)=NC3=CC=C2N=CN=C1NC1=CC=C(Br)C=C1F ZBELDPMWYXDLNY-UHFFFAOYSA-N 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 229940050176 methyl chloride Drugs 0.000 description 1
- 235000010981 methylcellulose Nutrition 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 244000005700 microbiome Species 0.000 description 1
- 239000011707 mineral Substances 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- PYLWMHQQBFSUBP-UHFFFAOYSA-N monofluorobenzene Chemical class FC1=CC=CC=C1 PYLWMHQQBFSUBP-UHFFFAOYSA-N 0.000 description 1
- 150000002772 monosaccharides Chemical class 0.000 description 1
- IJDNQMDRQITEOD-UHFFFAOYSA-N n-butane Chemical compound CCCC IJDNQMDRQITEOD-UHFFFAOYSA-N 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- PSZYNBSKGUBXEH-UHFFFAOYSA-M naphthalene-1-sulfonate Chemical compound C1=CC=C2C(S(=O)(=O)[O-])=CC=CC2=C1 PSZYNBSKGUBXEH-UHFFFAOYSA-M 0.000 description 1
- 229920003052 natural elastomer Polymers 0.000 description 1
- 229920001194 natural rubber Polymers 0.000 description 1
- IOMMMLWIABWRKL-WUTDNEBXSA-N nazartinib Chemical compound C1N(C(=O)/C=C/CN(C)C)CCCC[C@H]1N1C2=C(Cl)C=CC=C2N=C1NC(=O)C1=CC=NC(C)=C1 IOMMMLWIABWRKL-WUTDNEBXSA-N 0.000 description 1
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 230000000926 neurological effect Effects 0.000 description 1
- 239000002858 neurotransmitter agent Substances 0.000 description 1
- 150000002823 nitrates Chemical class 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- 239000012875 nonionic emulsifier Substances 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 239000000346 nonvolatile oil Substances 0.000 description 1
- 229920001778 nylon Polymers 0.000 description 1
- QYSGYZVSCZSLHT-UHFFFAOYSA-N octafluoropropane Chemical compound FC(F)(F)C(F)(F)C(F)(F)F QYSGYZVSCZSLHT-UHFFFAOYSA-N 0.000 description 1
- 125000002347 octyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 235000019645 odor Nutrition 0.000 description 1
- 229940049964 oleate Drugs 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- PIDFDZJZLOTZTM-KHVQSSSXSA-N ombitasvir Chemical compound COC(=O)N[C@@H](C(C)C)C(=O)N1CCC[C@H]1C(=O)NC1=CC=C([C@H]2N([C@@H](CC2)C=2C=CC(NC(=O)[C@H]3N(CCC3)C(=O)[C@@H](NC(=O)OC)C(C)C)=CC=2)C=2C=CC(=CC=2)C(C)(C)C)C=C1 PIDFDZJZLOTZTM-KHVQSSSXSA-N 0.000 description 1
- 239000003605 opacifier Substances 0.000 description 1
- 238000005457 optimization Methods 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 150000003891 oxalate salts Chemical class 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 239000003346 palm kernel oil Substances 0.000 description 1
- 229940023569 palmate Drugs 0.000 description 1
- 125000001312 palmitoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- WLJNZVDCPSBLRP-UHFFFAOYSA-N pamoic acid Chemical compound C1=CC=C2C(CC=3C4=CC=CC=C4C=C(C=3O)C(=O)O)=C(O)C(C(O)=O)=CC2=C1 WLJNZVDCPSBLRP-UHFFFAOYSA-N 0.000 description 1
- 229940014662 pantothenate Drugs 0.000 description 1
- 235000019161 pantothenic acid Nutrition 0.000 description 1
- 239000011713 pantothenic acid Substances 0.000 description 1
- 229940056211 paraffin Drugs 0.000 description 1
- 239000012188 paraffin wax Substances 0.000 description 1
- LCCNCVORNKJIRZ-UHFFFAOYSA-N parathion Chemical compound CCOP(=S)(OCC)OC1=CC=C([N+]([O-])=O)C=C1 LCCNCVORNKJIRZ-UHFFFAOYSA-N 0.000 description 1
- 239000011236 particulate material Substances 0.000 description 1
- GTLACDSXYULKMZ-UHFFFAOYSA-N pentafluoroethane Chemical compound FC(F)C(F)(F)F GTLACDSXYULKMZ-UHFFFAOYSA-N 0.000 description 1
- 235000019477 peppermint oil Nutrition 0.000 description 1
- 229960004692 perflenapent Drugs 0.000 description 1
- KAVGMUDTWQVPDF-UHFFFAOYSA-N perflubutane Chemical compound FC(F)(F)C(F)(F)C(F)(F)C(F)(F)F KAVGMUDTWQVPDF-UHFFFAOYSA-N 0.000 description 1
- 229950003332 perflubutane Drugs 0.000 description 1
- 229960004065 perflutren Drugs 0.000 description 1
- 230000000737 periodic effect Effects 0.000 description 1
- 210000001428 peripheral nervous system Anatomy 0.000 description 1
- 239000003208 petroleum Substances 0.000 description 1
- 229940124531 pharmaceutical excipient Drugs 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- 230000003285 pharmacodynamic effect Effects 0.000 description 1
- 125000000951 phenoxy group Chemical group [H]C1=C([H])C([H])=C(O*)C([H])=C1[H] 0.000 description 1
- DHFYLDMPSGAGTP-UHFFFAOYSA-N phenoxymethanol Chemical class OCOC1=CC=CC=C1 DHFYLDMPSGAGTP-UHFFFAOYSA-N 0.000 description 1
- 229960000969 phenyl salicylate Drugs 0.000 description 1
- WVDDGKGOMKODPV-ZQBYOMGUSA-N phenyl(114C)methanol Chemical compound O[14CH2]C1=CC=CC=C1 WVDDGKGOMKODPV-ZQBYOMGUSA-N 0.000 description 1
- 235000011007 phosphoric acid Nutrition 0.000 description 1
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 1
- PDDXOPNEMCREGN-UHFFFAOYSA-N phosphoric acid;trioxomolybdenum;hydrate Chemical compound O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.OP(O)(O)=O PDDXOPNEMCREGN-UHFFFAOYSA-N 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 239000011574 phosphorus Substances 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 239000002504 physiological saline solution Substances 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 239000004014 plasticizer Substances 0.000 description 1
- 229920001490 poly(butyl methacrylate) polymer Polymers 0.000 description 1
- 229920000747 poly(lactic acid) Polymers 0.000 description 1
- 229920003229 poly(methyl methacrylate) Polymers 0.000 description 1
- 229920002857 polybutadiene Polymers 0.000 description 1
- 229920001610 polycaprolactone Polymers 0.000 description 1
- 229920002721 polycyanoacrylate Polymers 0.000 description 1
- 229940068886 polyethylene glycol 300 Drugs 0.000 description 1
- 229940068918 polyethylene glycol 400 Drugs 0.000 description 1
- 229940057838 polyethylene glycol 4000 Drugs 0.000 description 1
- 229920001228 polyisocyanate Polymers 0.000 description 1
- 239000005056 polyisocyanate Substances 0.000 description 1
- 239000004626 polylactic acid Substances 0.000 description 1
- 229920000656 polylysine Polymers 0.000 description 1
- 239000004926 polymethyl methacrylate Substances 0.000 description 1
- 229920005862 polyol Polymers 0.000 description 1
- 150000003077 polyols Chemical class 0.000 description 1
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 1
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 1
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 1
- 239000000244 polyoxyethylene sorbitan monooleate Substances 0.000 description 1
- 229920006324 polyoxymethylene Polymers 0.000 description 1
- 229920001282 polysaccharide Polymers 0.000 description 1
- 239000005017 polysaccharide Substances 0.000 description 1
- 229920001296 polysiloxane Polymers 0.000 description 1
- 229920000053 polysorbate 80 Polymers 0.000 description 1
- 239000011118 polyvinyl acetate Substances 0.000 description 1
- 229920002689 polyvinyl acetate Polymers 0.000 description 1
- 229920002451 polyvinyl alcohol Polymers 0.000 description 1
- 229920000915 polyvinyl chloride Polymers 0.000 description 1
- 239000004800 polyvinyl chloride Substances 0.000 description 1
- 230000001242 postsynaptic effect Effects 0.000 description 1
- 159000000001 potassium salts Chemical class 0.000 description 1
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 1
- 229960001309 procaine hydrochloride Drugs 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- ALDITMKAAPLVJK-UHFFFAOYSA-N prop-1-ene;hydrate Chemical group O.CC=C ALDITMKAAPLVJK-UHFFFAOYSA-N 0.000 description 1
- UMSVPCYSAUKCAZ-UHFFFAOYSA-N propane;hydrochloride Chemical compound Cl.CCC UMSVPCYSAUKCAZ-UHFFFAOYSA-N 0.000 description 1
- 238000011321 prophylaxis Methods 0.000 description 1
- 239000000473 propyl gallate Substances 0.000 description 1
- 235000010388 propyl gallate Nutrition 0.000 description 1
- 229940075579 propyl gallate Drugs 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- QQONPFPTGQHPMA-UHFFFAOYSA-N propylene Natural products CC=C QQONPFPTGQHPMA-UHFFFAOYSA-N 0.000 description 1
- RUOJZAUFBMNUDX-UHFFFAOYSA-N propylene carbonate Chemical compound CC1COC(=O)O1 RUOJZAUFBMNUDX-UHFFFAOYSA-N 0.000 description 1
- 125000004805 propylene group Chemical group [H]C([H])([H])C([H])([*:1])C([H])([H])[*:2] 0.000 description 1
- 125000006239 protecting group Chemical group 0.000 description 1
- 239000003223 protective agent Substances 0.000 description 1
- 238000000425 proton nuclear magnetic resonance spectrum Methods 0.000 description 1
- 208000020016 psychiatric disease Diseases 0.000 description 1
- 238000001959 radiotherapy Methods 0.000 description 1
- 239000008175 ready-to-use sterile solution Substances 0.000 description 1
- 102000005962 receptors Human genes 0.000 description 1
- 108020003175 receptors Proteins 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- 238000007363 ring formation reaction Methods 0.000 description 1
- 235000005713 safflower oil Nutrition 0.000 description 1
- 239000003813 safflower oil Substances 0.000 description 1
- YGSDEFSMJLZEOE-UHFFFAOYSA-M salicylate Chemical compound OC1=CC=CC=C1C([O-])=O YGSDEFSMJLZEOE-UHFFFAOYSA-M 0.000 description 1
- 229960001860 salicylate Drugs 0.000 description 1
- 150000003902 salicylic acid esters Chemical class 0.000 description 1
- DCKVNWZUADLDEH-UHFFFAOYSA-N sec-butyl acetate Chemical compound CCC(C)OC(C)=O DCKVNWZUADLDEH-UHFFFAOYSA-N 0.000 description 1
- 229940076279 serotonin Drugs 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 239000004208 shellac Substances 0.000 description 1
- 229940113147 shellac Drugs 0.000 description 1
- 235000013874 shellac Nutrition 0.000 description 1
- 238000010898 silica gel chromatography Methods 0.000 description 1
- 239000008354 sodium chloride injection Substances 0.000 description 1
- 229960003928 sodium oxybate Drugs 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 239000007901 soft capsule Substances 0.000 description 1
- 239000008137 solubility enhancer Substances 0.000 description 1
- 239000002195 soluble material Substances 0.000 description 1
- 239000003549 soybean oil Substances 0.000 description 1
- 235000012424 soybean oil Nutrition 0.000 description 1
- 230000003595 spectral effect Effects 0.000 description 1
- 238000002798 spectrophotometry method Methods 0.000 description 1
- 238000004611 spectroscopical analysis Methods 0.000 description 1
- 238000010186 staining Methods 0.000 description 1
- 239000008174 sterile solution Substances 0.000 description 1
- 239000008223 sterile water Substances 0.000 description 1
- 210000002784 stomach Anatomy 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 238000010254 subcutaneous injection Methods 0.000 description 1
- 239000007929 subcutaneous injection Substances 0.000 description 1
- 150000003890 succinate salts Chemical class 0.000 description 1
- 239000007940 sugar coated tablet Substances 0.000 description 1
- 229940097346 sulfobutylether-beta-cyclodextrin Drugs 0.000 description 1
- 150000003467 sulfuric acid derivatives Chemical class 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 239000013589 supplement Substances 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- 230000005062 synaptic transmission Effects 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- 150000003892 tartrate salts Chemical class 0.000 description 1
- 239000006068 taste-masking agent Substances 0.000 description 1
- 229920001897 terpolymer Polymers 0.000 description 1
- RUPAXCPQAAOIPB-UHFFFAOYSA-N tert-butyl formate Chemical group CC(C)(C)OC=O RUPAXCPQAAOIPB-UHFFFAOYSA-N 0.000 description 1
- ZUHZGEOKBKGPSW-UHFFFAOYSA-N tetraglyme Chemical compound COCCOCCOCCOCCOC ZUHZGEOKBKGPSW-UHFFFAOYSA-N 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- CZDYPVPMEAXLPK-UHFFFAOYSA-N tetramethylsilane Chemical compound C[Si](C)(C)C CZDYPVPMEAXLPK-UHFFFAOYSA-N 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- RTKIYNMVFMVABJ-UHFFFAOYSA-L thimerosal Chemical compound [Na+].CC[Hg]SC1=CC=CC=C1C([O-])=O RTKIYNMVFMVABJ-UHFFFAOYSA-L 0.000 description 1
- 229940033663 thimerosal Drugs 0.000 description 1
- 150000003567 thiocyanates Chemical class 0.000 description 1
- 235000019303 thiodipropionic acid Nutrition 0.000 description 1
- 230000009974 thixotropic effect Effects 0.000 description 1
- 238000004448 titration Methods 0.000 description 1
- 238000003325 tomography Methods 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 239000012049 topical pharmaceutical composition Substances 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 238000000844 transformation Methods 0.000 description 1
- ODLHGICHYURWBS-LKONHMLTSA-N trappsol cyclo Chemical compound CC(O)COC[C@H]([C@H]([C@@H]([C@H]1O)O)O[C@H]2O[C@@H]([C@@H](O[C@H]3O[C@H](COCC(C)O)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](COCC(C)O)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](COCC(C)O)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](COCC(C)O)[C@H]([C@@H]([C@H]3O)O)O3)[C@H](O)[C@H]2O)COCC(O)C)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@@H]3O[C@@H]1COCC(C)O ODLHGICHYURWBS-LKONHMLTSA-N 0.000 description 1
- 229940029284 trichlorofluoromethane Drugs 0.000 description 1
- 229940117013 triethanolamine oleate Drugs 0.000 description 1
- YFNKIDBQEZZDLK-UHFFFAOYSA-N triglyme Chemical compound COCCOCCOCCOC YFNKIDBQEZZDLK-UHFFFAOYSA-N 0.000 description 1
- 235000011178 triphosphate Nutrition 0.000 description 1
- 239000001226 triphosphate Substances 0.000 description 1
- UNXRWKVEANCORM-UHFFFAOYSA-N triphosphoric acid Chemical compound OP(O)(=O)OP(O)(=O)OP(O)(O)=O UNXRWKVEANCORM-UHFFFAOYSA-N 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 229920011532 unplasticized polyvinyl chloride Polymers 0.000 description 1
- 238000012800 visualization Methods 0.000 description 1
- 239000011709 vitamin E Substances 0.000 description 1
- 235000019165 vitamin E Nutrition 0.000 description 1
- 229940046009 vitamin E Drugs 0.000 description 1
- 238000009736 wetting Methods 0.000 description 1
- 229940051225 xyrem Drugs 0.000 description 1
Images



Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/517—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with carbocyclic ring systems, e.g. quinazoline, perimidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/20—Hypnotics; Sedatives
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Veterinary Medicine (AREA)
- Life Sciences & Earth Sciences (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Biomedical Technology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Neurosurgery (AREA)
- Neurology (AREA)
- Psychology (AREA)
- Epidemiology (AREA)
- Anesthesiology (AREA)
- Pulmonology (AREA)
- Psychiatry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
本發明關於作為H3R拮抗劑之式(A)化合物、以及彼等的製備和用途、及進一步關於包含此等化合物之醫藥組成物和彼等作為組織胺3受體(H3R)功能的調節劑之用途。本發明亦關於該等化合物或醫藥組成物在治療或預防某些與人類H3R功能有關的病症和疾病之用途。
Description
本發明關於醫藥技術領域,特別是某些化合物、彼等的製備和用途,以及包含該等化合物之醫藥組成物。例如,本發明關於某些化合物、彼等的製備和對應醫藥組成物,其可能用於製造用於預防、治療、改善患者的某病症或疾病(其包括CNS病症或嗜睡症)的藥物。一般認為本發明之化合物及/或醫藥組成物尤其是藉由調節(例如,拮抗)神經元組織胺受體-3(H3R)而起作用來發揮彼等的治療益處。
突觸後組織胺結合至其H3R上不僅限制組織胺的通量,而且還限制各種其他重要的神經遞質(例如,乙醯膽鹼、多巴胺、麩胺酸鹽、正腎上腺素、血清素、GABA、等等)(例如,T.A. Esbenshade, et. al, Mol. Intervention, v. 6, pp. 77-88 2006)。相比之下,H3R拮抗劑(也稱為逆促效劑)可增加這些神經傳遞通量,以治療各種CNS病症(例如認知、精神、神經運動、疼痛、等等)。(http://molpharm.aspetjournals.org/content/molpharm/90/5 /649.full.pdf讀取於2017年2月26日;對H3R拮抗劑的最新專利申請案的回顧,D. Lazewska et. al., Expert Opin. Therapeutic Patents, v.28, pp. 175- 196 2018以及其中引用的參考文獻)。
過去十年來正在進行的治療嗜睡症之H3R拮抗劑的臨床試驗(例如review-https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5344488/pdf/nss-9-039.pdf讀取於2018年2月26日)導致第一個H3R拮抗劑-匹托利生(pitolisant)的EU銷售批准(品牌名稱=Wakix;http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/002616/human_med_001955.jsp&mid=WC0b01ac058001d124,讀取於2018年2月26日)。而Wakix和羥丁酸鈉(sodium oxybate)(品牌名稱-Xyrem;獲得EU銷售批准:http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human /medicines/000593/human_med_001163.jsp&mid=WC0b01ac058001d124,讀取於2018年2月26日)是目前所銷售之專門治療嗜睡症和猝倒症的藥物(~70%的發作性睡眠病患者受累),初始滴定的患者中有顯著部分(~20-40%)由於不良療效及/或毒性而中斷進一步的藥物給藥(分別https: //www.ncbi.nlm.nih.gov/pmc/articles/PMC5414617/pdf/nss-9-127. pdf和https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5727622/ pdf/ main.pdf皆讀取於2018年2月26日)。因此且像受其他CNS病症影響的患者一樣,發作性睡眠病患者迫切需要更好的藥物以顯著降低這些部分(fraction)。
本申請案所揭示之化合物和醫藥調配物被認為是藉由尤其是拮抗H3R而起作用來發揮彼等的治療益處。
以下僅為本發明之一些態樣的概述,但不限於此。本說明書的所有參考文獻均以彼等之全文藉引用的方式併入本文。當本說明書的揭示內容與引文不同時,以本說明書的揭示內容為準。本發明提供調節拮抗H3R之化合物和醫藥組成物、彼等的製備、及對應醫藥組成物。本發明之化合物及/或醫藥組成物可能用於製造供預防、治療、改善患者的與H3R有關之某病症或疾病,其包括CNS病症或嗜睡症。
具體而言,在一態樣中,本發明關於一種具有式(A)之化合物或其醫藥上可接受的鹽類,
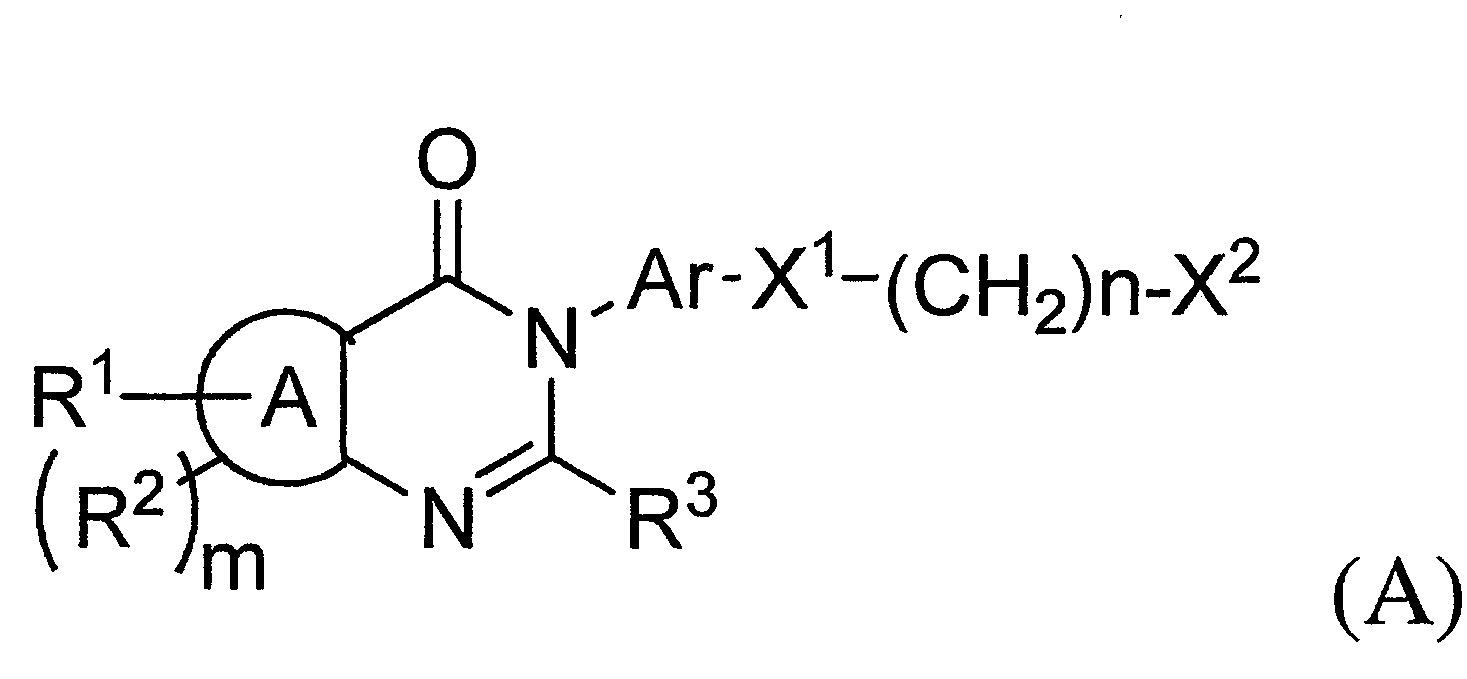 ,
其中,R
1和R
2獨立地為H或SF
5,先決條件為至少R
1和R
2之一者為SF
5;
R
3為C
1-6烷基或C
3-6環烷基;
環A為C
6~8芳基;
Ar為C
5-6芳基,其中該C
5-6芳基係隨意地經1、2、3或4個R
e取代;
R
e為H、鹵素、CN、NO
2、OH、C
1-6烷氧基或C
1-6烷基;
X
1為S或O;
X
2為
,
其中,R
1和R
2獨立地為H或SF
5,先決條件為至少R
1和R
2之一者為SF
5;
R
3為C
1-6烷基或C
3-6環烷基;
環A為C
6~8芳基;
Ar為C
5-6芳基,其中該C
5-6芳基係隨意地經1、2、3或4個R
e取代;
R
e為H、鹵素、CN、NO
2、OH、C
1-6烷氧基或C
1-6烷基;
X
1為S或O;
X
2為
 ,其中R為N或C;
R
3和R
4為C
1-6烷基、或R
3和R
4與彼等所連接的R一起形成C
3-7碳環、3-至7-員雜環,且其中C
3-7碳環、3-至7-員雜環各自係獨立地隨意地經1、2、3、4、5或6個R
f取代;
R
f為C
1-6烷基、C
3-6環烷基、-C(=O)R
a,且其中C
1-6烷基、C
3-6環烷基、-C(=O)R
a各自係獨立地隨意地經C
1-6烷基取代;
R
a為C
1-6烷基、C
3-6環烷基或3-至7-員雜環;
m為從0至3的整數;
n為從0至5的整數。
在一個實施態樣中,R
3為C
1-4烷基或C
3-6環烷基。
在一個實施態樣中,A為苯基。
在一個實施態樣中,Ar為苯基,其中苯基係隨意地經1、2、3或4個R
e取代。
在一個實施態樣中,Ar為下列子式:
,其中R為N或C;
R
3和R
4為C
1-6烷基、或R
3和R
4與彼等所連接的R一起形成C
3-7碳環、3-至7-員雜環,且其中C
3-7碳環、3-至7-員雜環各自係獨立地隨意地經1、2、3、4、5或6個R
f取代;
R
f為C
1-6烷基、C
3-6環烷基、-C(=O)R
a,且其中C
1-6烷基、C
3-6環烷基、-C(=O)R
a各自係獨立地隨意地經C
1-6烷基取代;
R
a為C
1-6烷基、C
3-6環烷基或3-至7-員雜環;
m為從0至3的整數;
n為從0至5的整數。
在一個實施態樣中,R
3為C
1-4烷基或C
3-6環烷基。
在一個實施態樣中,A為苯基。
在一個實施態樣中,Ar為苯基,其中苯基係隨意地經1、2、3或4個R
e取代。
在一個實施態樣中,Ar為下列子式:
 (W-1a),其中w1為0、1、2、3或4。
在一個實施態樣中,R
e為H、F、Cl、Br、I、CN、NO
2、OH、甲基、乙基、異丙基、正丙基、正丁基、三級丁基、甲氧基、乙氧基。
在一個實施態樣中,R
3和R
4為C
1-4烷基。
在一個實施態樣中,R為N,R
3和R
4與彼等所連接的氮原子一起形成3-至7-員雜環,且其中3-至7-員雜環係獨立地隨意地經1、2、3、4或5個R
f取代;
在一個實施態樣中,R為C,R
3和R
4與彼等所連接的碳原子一起形成C
3-7碳環、3-至7-員雜環,且其中C
3-7碳環、3-至7-員雜環各自係獨立地隨意地經1、2、3、4或5個R
f取代。
在一個實施態樣中,R
f為C
1-4烷基、C
3-6環烷基、-C(=O)R
a。
在一個實施態樣中,R
f為-C(=O)R
a、甲基、乙基、異丙基、
(W-1a),其中w1為0、1、2、3或4。
在一個實施態樣中,R
e為H、F、Cl、Br、I、CN、NO
2、OH、甲基、乙基、異丙基、正丙基、正丁基、三級丁基、甲氧基、乙氧基。
在一個實施態樣中,R
3和R
4為C
1-4烷基。
在一個實施態樣中,R為N,R
3和R
4與彼等所連接的氮原子一起形成3-至7-員雜環,且其中3-至7-員雜環係獨立地隨意地經1、2、3、4或5個R
f取代;
在一個實施態樣中,R為C,R
3和R
4與彼等所連接的碳原子一起形成C
3-7碳環、3-至7-員雜環,且其中C
3-7碳環、3-至7-員雜環各自係獨立地隨意地經1、2、3、4或5個R
f取代。
在一個實施態樣中,R
f為C
1-4烷基、C
3-6環烷基、-C(=O)R
a。
在一個實施態樣中,R
f為-C(=O)R
a、甲基、乙基、異丙基、
 。
在一個實施態樣中,R
a為甲基、乙基、
。
在一個實施態樣中,R
a為甲基、乙基、
 或
或
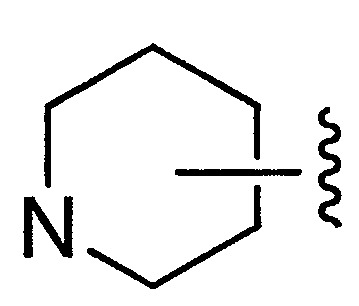 。
在一個實施態樣中,X
2為下列子式中之一者:
。
在一個實施態樣中,X
2為下列子式中之一者:
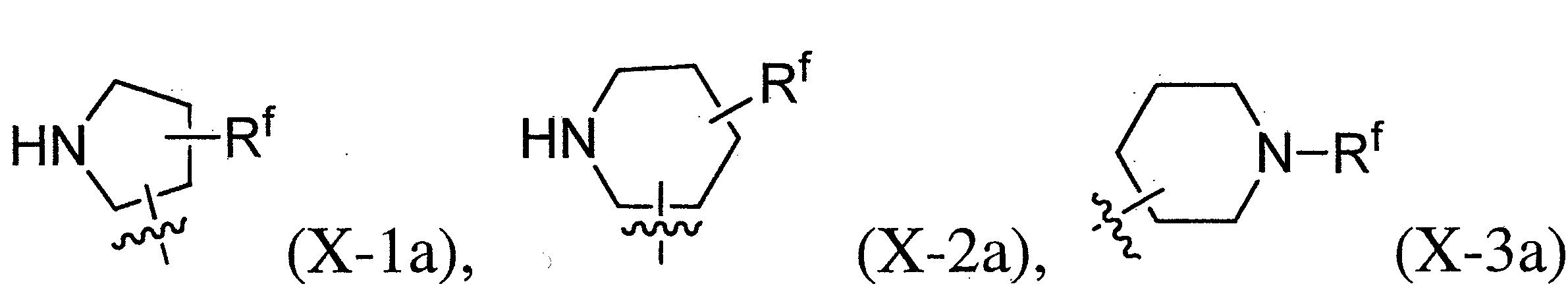 。
在一個實施態樣中,X
2為下列子式中之一者:
。
在一個實施態樣中,X
2為下列子式中之一者:
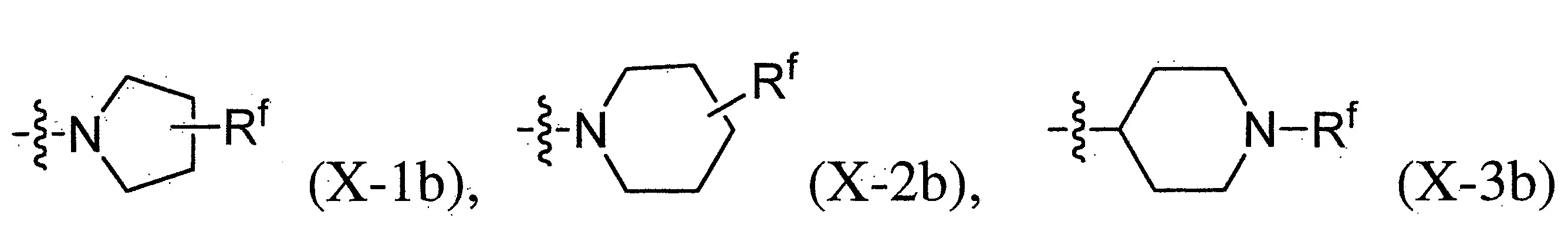 在一個實施態樣中,本文提供一種具有式(B)之化合物,
在一個實施態樣中,本文提供一種具有式(B)之化合物,
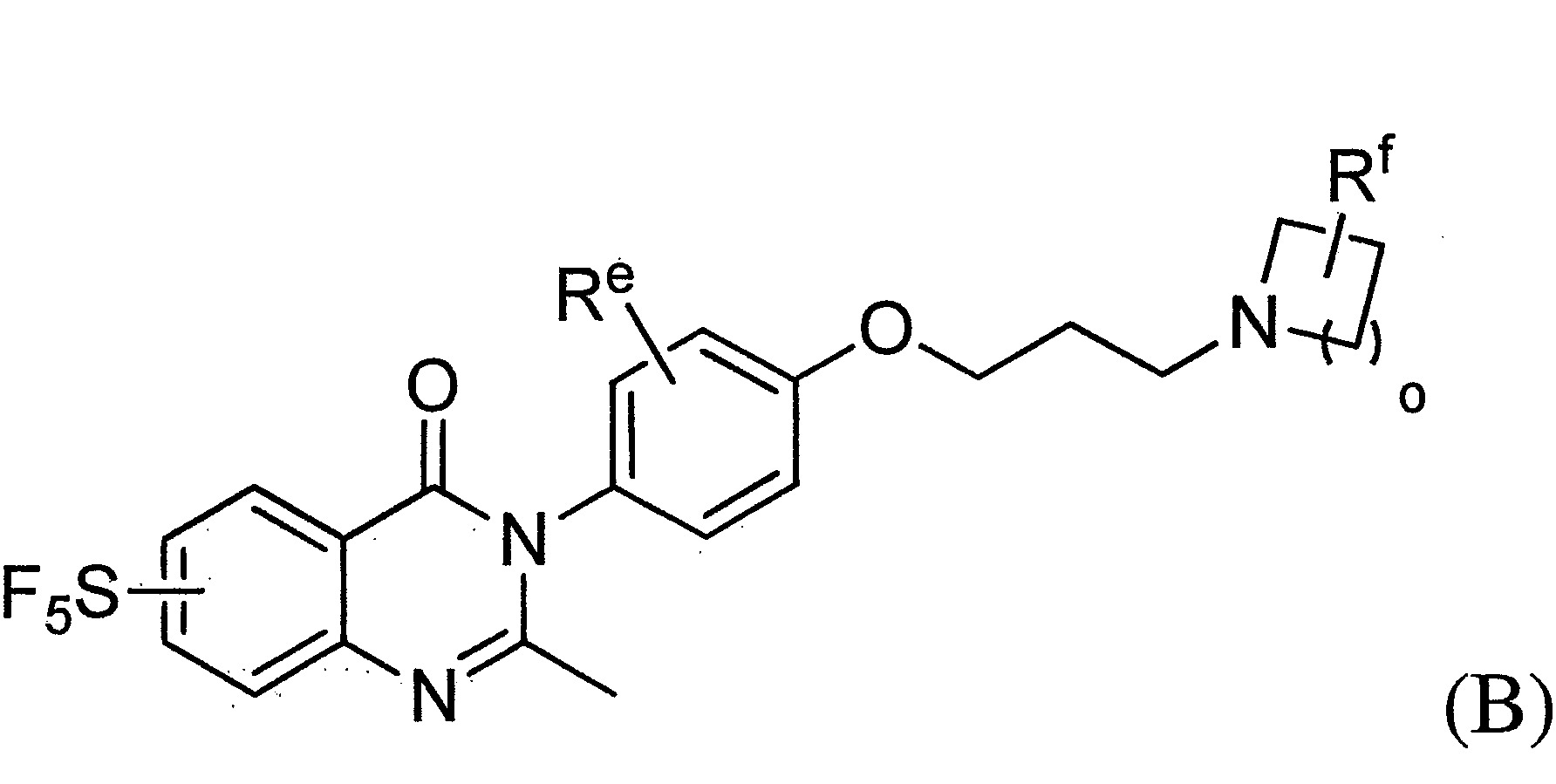 ,
其中o為從0至4的整數;及
R
e、R
f係如本文所定義。
在一個實施態樣中,本文提供一種具有式(C)之化合物,
,
其中o為從0至4的整數;及
R
e、R
f係如本文所定義。
在一個實施態樣中,本文提供一種具有式(C)之化合物,
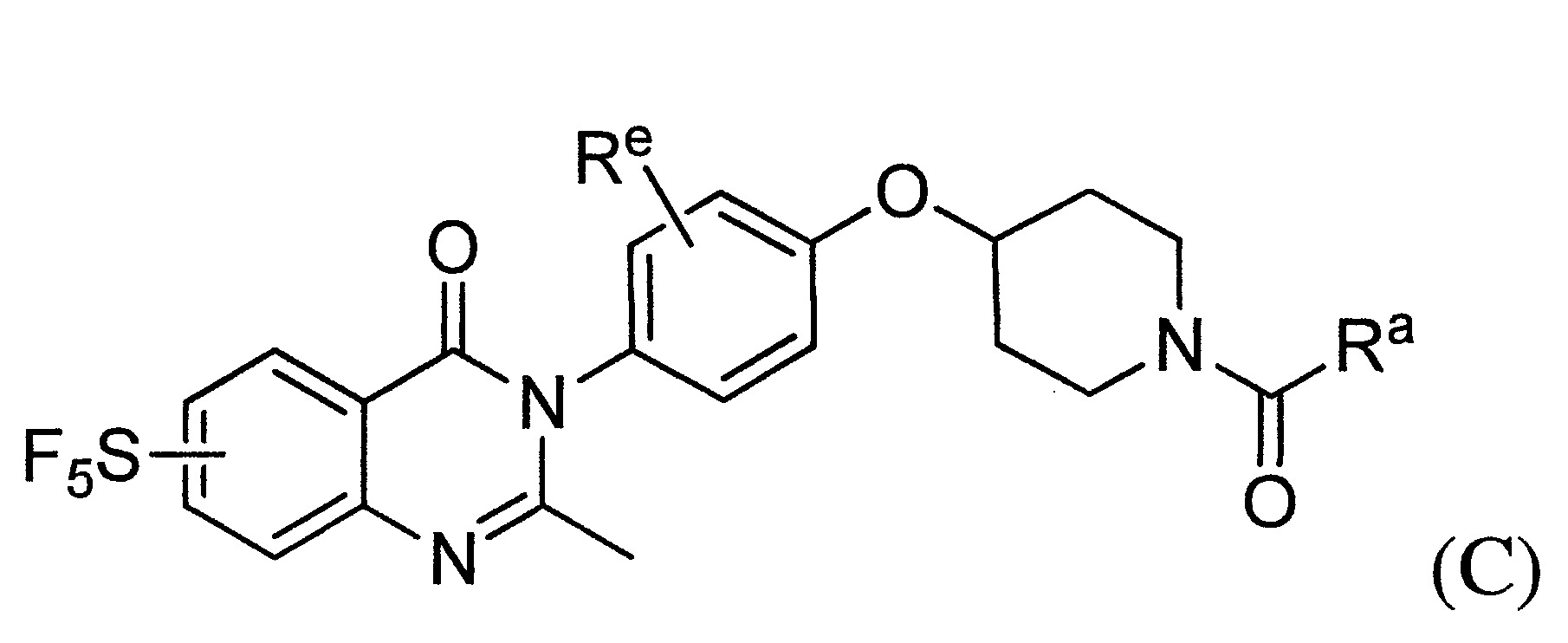 ,
R
e、R
a係如本文所定義。
在一個實施態樣中,本文提供一種具有式(D)之化合物,
,
R
e、R
a係如本文所定義。
在一個實施態樣中,本文提供一種具有式(D)之化合物,
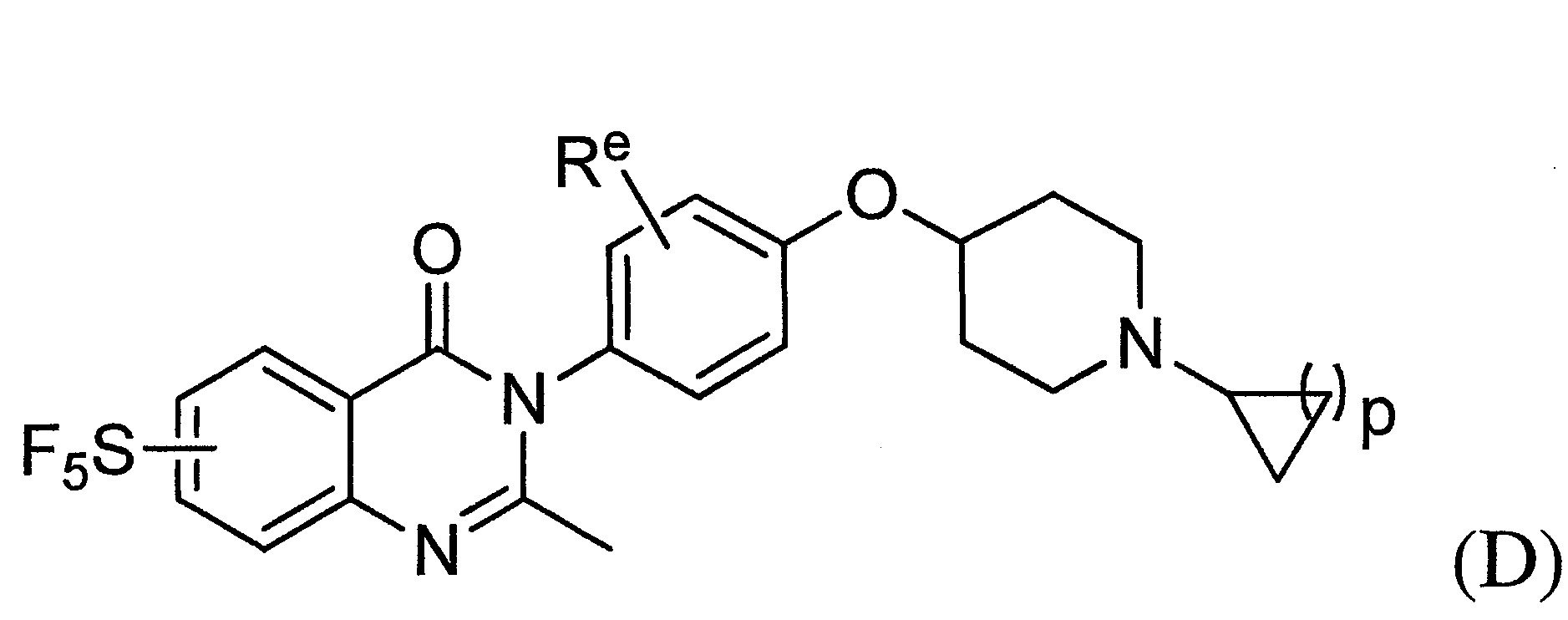 ,
其中p為從1至5的整數;及
R
e係如本文所定義。
在另一態樣中,本文提供一種包含本發明化合物之醫藥組成物。
在一個實施態樣中,醫藥組成物進一步包含醫藥上可接受的載體、佐劑、載劑或其組合。
在一個實施態樣中,醫藥組成物進一步包含一或多種其他治療劑,且其中該其他治療劑用於治療CNS病症或嗜睡症。
在一個實施態樣中,CNS病症為認知、精神、神經運動或疼痛。
在一個實施態樣中,其他治療劑係進一步包含:選自由多巴胺受體拮抗劑、血清素-去甲基腎上腺素再吸收抑制劑(serotonin-norepinephrine reuptake inhibitor)、選擇性血清素再吸收抑制劑、去甲基腎上腺素激導性再吸收抑制劑(noradrenergic reuptake inhibitor)、非選擇性血清素再吸收抑制劑、及乙醯膽鹼酯酶抑制劑所組成群組之活性成分。
在另一態樣中,本文提供化合物或醫藥組成物之用途,其係用於製造供預防、管理、治療或減輕患者中CNS病症或嗜睡症的藥物。
在另一態樣中,本文提供化合物或醫藥組成物之用途,其係用於製造供拮抗H3受體的藥物。
在另一態樣中,本文提供用於預防、管理、治療或減輕患者中由病毒感染引起的CNS病症或嗜睡症之化合物或醫藥組成物。
在另一態樣中,本文提供用於拮抗H3受體之化合物或醫藥組成物。
在另一態樣中,本文提供一種預防、管理、治療或減輕患者中CNS病症或嗜睡症之方法,其包含將治療有效量的該化合物或該醫藥組成物投予至有需要的患者。
在另一態樣中,本文提供一種在患者中拮抗H3受體之方法,其包含將治療有效量的該化合物或該醫藥組成物投予至有需要的患者。
在又另一態樣中,本發明係關於製造式(A)~(D)化合物及其醫藥上可接受的鹽類之方法。
在本發明之化合物、醫藥組成物、和方法的一些實施態樣中,式(A)~(D)化合物為選自彼等以下詳細說明中所描述或例舉的種類之化合物,或為該類化合物之醫藥可接受的鹽。
另一較佳實施態樣,本發明係關於製備醫藥組成物之方法,該醫藥組成物各自包含治療有效量的至少一種式(A)~(D)化合物或式(A)~(D)化合物之醫藥上可接受的鹽類。根據本發明之醫藥組成物可另外包含至少一種醫藥上可接受的賦形劑、載體、佐劑、溶劑、撐體或其組合。
若調配成固定劑量,則該組合產物使用本文所述的(或如熟習該項技術者已知的)劑量範圍內的本發明化合物以及在其劑量範圍內的其他醫藥活性劑或治療。當組合調配物不適合時,本發明之化合物亦可與已知的抗CNS病症或嗜睡症劑依序地投予。在任何組合治療中,本發明不限於投予的順序;式(A)~(D)化合物可在已知抗CNS病症或嗜睡症劑投予之前或之後投予。該等技術係在熟習該項技術者以及主治醫師的技術範圍內。
又另一實施態樣為一種藉由將本發明之醫藥調配物投予至有需要的對象而將本發明化合物投予至該對象(例如,人類)之方法。
又另一實施態樣為一種藉由混合至少一種本發明醫藥上可接受的化合物和隨意地一或多種醫藥上可接受的添加劑或賦形劑來製備本發明醫藥調配物之方法。
為了從本發明所述化合物製備醫藥組成物,惰性醫藥上可接受的載體可為固體或液體。固體形式製劑包括粉劑、錠劑、分散性粒劑、膠囊、珠劑、扁囊劑及栓劑。粉劑及錠劑可由從約5至約95百分比的活性成分組成。適當固體載體為該項技術中已知的,例如,碳酸鎂、硬脂酸鎂、滑石、糖或乳糖。錠劑、粉劑、扁囊劑及膠囊可用作為適合於口服投予的固體劑型。製造各種組成物之醫藥上可接受的載體及方法之實例可見於A. Gennaro(ed.), Remington's Pharmaceutical Sciences, 18
thEdition,(1990), Mack Publishing Co., Easton, Pa。
液體形式製劑包括溶液、懸浮液以及乳液。例如,有用於腸胃外注射的水或水-丙二醇溶液、或添加用於口服溶液、懸浮液以及乳液的甜味劑及不透明劑。液體形式製劑亦可包括用於鼻內投予的溶液。
適合於吸入的氣霧劑製劑可包括溶液及粉末形式的固體,其可與醫藥上可接受的載體(例如惰性壓縮氣體,例如:氮氣)組合。
亦包含固體形式製劑,其意欲在使用前不久轉化為用於口服或腸胃外投予的液體形式製劑。該等液體形式包含溶液、懸浮液以及乳液。
本發明化合物亦可為經皮遞送。經皮組成物可為霜劑、乳劑、氣霧劑及/或乳液之形式,且可包括在該項技術領域中習知用於此目的之基質或儲器類型的經皮貼片。
本發明化合物亦可皮下(subcutaneously)遞送。
較佳地化合物係以口服或靜脈內投予。
較佳地,醫藥製劑為單位劑型。在該形式中,製劑會被分成含有適量的活性成分之適當大小單位劑量,例如達到所需目的之有效量。
製劑的單位劑量中之活性化合物的量可根據特定應用在從約1mg至約1000mg,較佳從約1mg至約500mg,更佳從約1mg至約300mg,又更佳從約1mg至約200mg進行改變或調整。
所使用的實際劑量可取決於患者的需求和待治療病況的嚴重性而改變。用於特定情況的適當劑量方案的測定在該項技術範圍內。為方便起見,可根據需求將每日總劑量分開且在一天內分批投予。本發明化合物及/或其醫藥上可接受的鹽類之投予的量和頻率將根據臨床主治醫師考慮該等諸如患者之年齡、病況和大小以及待治療症狀的嚴重性之因素判斷來調整。用於口服投予之典型推薦的每日劑量方案範圍可在從約1mg/天至約300mg/天,較佳為10mg/天至200mg/天,以單劑至二分劑量形式。
本文所揭示的任何實施態樣可與其他實施態樣組合,只要彼等不相互矛盾,即使實施態樣在本發明不同態樣下描述。此外,在一個實施態樣中的任何技術特徵可應用於其他實施態樣中的對應技術特徵,只要彼等不相互矛盾,即使實施態樣在本發明不同態樣下描述。
前述僅概述本文所揭示的某些態樣,且本質上不意欲為限制的。這些態樣和其他態樣以及實施態樣將更充分地描述於下。
,
其中p為從1至5的整數;及
R
e係如本文所定義。
在另一態樣中,本文提供一種包含本發明化合物之醫藥組成物。
在一個實施態樣中,醫藥組成物進一步包含醫藥上可接受的載體、佐劑、載劑或其組合。
在一個實施態樣中,醫藥組成物進一步包含一或多種其他治療劑,且其中該其他治療劑用於治療CNS病症或嗜睡症。
在一個實施態樣中,CNS病症為認知、精神、神經運動或疼痛。
在一個實施態樣中,其他治療劑係進一步包含:選自由多巴胺受體拮抗劑、血清素-去甲基腎上腺素再吸收抑制劑(serotonin-norepinephrine reuptake inhibitor)、選擇性血清素再吸收抑制劑、去甲基腎上腺素激導性再吸收抑制劑(noradrenergic reuptake inhibitor)、非選擇性血清素再吸收抑制劑、及乙醯膽鹼酯酶抑制劑所組成群組之活性成分。
在另一態樣中,本文提供化合物或醫藥組成物之用途,其係用於製造供預防、管理、治療或減輕患者中CNS病症或嗜睡症的藥物。
在另一態樣中,本文提供化合物或醫藥組成物之用途,其係用於製造供拮抗H3受體的藥物。
在另一態樣中,本文提供用於預防、管理、治療或減輕患者中由病毒感染引起的CNS病症或嗜睡症之化合物或醫藥組成物。
在另一態樣中,本文提供用於拮抗H3受體之化合物或醫藥組成物。
在另一態樣中,本文提供一種預防、管理、治療或減輕患者中CNS病症或嗜睡症之方法,其包含將治療有效量的該化合物或該醫藥組成物投予至有需要的患者。
在另一態樣中,本文提供一種在患者中拮抗H3受體之方法,其包含將治療有效量的該化合物或該醫藥組成物投予至有需要的患者。
在又另一態樣中,本發明係關於製造式(A)~(D)化合物及其醫藥上可接受的鹽類之方法。
在本發明之化合物、醫藥組成物、和方法的一些實施態樣中,式(A)~(D)化合物為選自彼等以下詳細說明中所描述或例舉的種類之化合物,或為該類化合物之醫藥可接受的鹽。
另一較佳實施態樣,本發明係關於製備醫藥組成物之方法,該醫藥組成物各自包含治療有效量的至少一種式(A)~(D)化合物或式(A)~(D)化合物之醫藥上可接受的鹽類。根據本發明之醫藥組成物可另外包含至少一種醫藥上可接受的賦形劑、載體、佐劑、溶劑、撐體或其組合。
若調配成固定劑量,則該組合產物使用本文所述的(或如熟習該項技術者已知的)劑量範圍內的本發明化合物以及在其劑量範圍內的其他醫藥活性劑或治療。當組合調配物不適合時,本發明之化合物亦可與已知的抗CNS病症或嗜睡症劑依序地投予。在任何組合治療中,本發明不限於投予的順序;式(A)~(D)化合物可在已知抗CNS病症或嗜睡症劑投予之前或之後投予。該等技術係在熟習該項技術者以及主治醫師的技術範圍內。
又另一實施態樣為一種藉由將本發明之醫藥調配物投予至有需要的對象而將本發明化合物投予至該對象(例如,人類)之方法。
又另一實施態樣為一種藉由混合至少一種本發明醫藥上可接受的化合物和隨意地一或多種醫藥上可接受的添加劑或賦形劑來製備本發明醫藥調配物之方法。
為了從本發明所述化合物製備醫藥組成物,惰性醫藥上可接受的載體可為固體或液體。固體形式製劑包括粉劑、錠劑、分散性粒劑、膠囊、珠劑、扁囊劑及栓劑。粉劑及錠劑可由從約5至約95百分比的活性成分組成。適當固體載體為該項技術中已知的,例如,碳酸鎂、硬脂酸鎂、滑石、糖或乳糖。錠劑、粉劑、扁囊劑及膠囊可用作為適合於口服投予的固體劑型。製造各種組成物之醫藥上可接受的載體及方法之實例可見於A. Gennaro(ed.), Remington's Pharmaceutical Sciences, 18
thEdition,(1990), Mack Publishing Co., Easton, Pa。
液體形式製劑包括溶液、懸浮液以及乳液。例如,有用於腸胃外注射的水或水-丙二醇溶液、或添加用於口服溶液、懸浮液以及乳液的甜味劑及不透明劑。液體形式製劑亦可包括用於鼻內投予的溶液。
適合於吸入的氣霧劑製劑可包括溶液及粉末形式的固體,其可與醫藥上可接受的載體(例如惰性壓縮氣體,例如:氮氣)組合。
亦包含固體形式製劑,其意欲在使用前不久轉化為用於口服或腸胃外投予的液體形式製劑。該等液體形式包含溶液、懸浮液以及乳液。
本發明化合物亦可為經皮遞送。經皮組成物可為霜劑、乳劑、氣霧劑及/或乳液之形式,且可包括在該項技術領域中習知用於此目的之基質或儲器類型的經皮貼片。
本發明化合物亦可皮下(subcutaneously)遞送。
較佳地化合物係以口服或靜脈內投予。
較佳地,醫藥製劑為單位劑型。在該形式中,製劑會被分成含有適量的活性成分之適當大小單位劑量,例如達到所需目的之有效量。
製劑的單位劑量中之活性化合物的量可根據特定應用在從約1mg至約1000mg,較佳從約1mg至約500mg,更佳從約1mg至約300mg,又更佳從約1mg至約200mg進行改變或調整。
所使用的實際劑量可取決於患者的需求和待治療病況的嚴重性而改變。用於特定情況的適當劑量方案的測定在該項技術範圍內。為方便起見,可根據需求將每日總劑量分開且在一天內分批投予。本發明化合物及/或其醫藥上可接受的鹽類之投予的量和頻率將根據臨床主治醫師考慮該等諸如患者之年齡、病況和大小以及待治療症狀的嚴重性之因素判斷來調整。用於口服投予之典型推薦的每日劑量方案範圍可在從約1mg/天至約300mg/天,較佳為10mg/天至200mg/天,以單劑至二分劑量形式。
本文所揭示的任何實施態樣可與其他實施態樣組合,只要彼等不相互矛盾,即使實施態樣在本發明不同態樣下描述。此外,在一個實施態樣中的任何技術特徵可應用於其他實施態樣中的對應技術特徵,只要彼等不相互矛盾,即使實施態樣在本發明不同態樣下描述。
前述僅概述本文所揭示的某些態樣,且本質上不意欲為限制的。這些態樣和其他態樣以及實施態樣將更充分地描述於下。
詳述說明和特定實施態樣
為求簡潔起見,本發明說明書中所引用之公開文獻的揭示(包括專利案及專利申請案)以其全文引用方式併入本文中。
在本文中大部分化學名稱係使用IUPAC命名法產生。一些化學名稱係以該項技術已知的不同命名法或替代或商業名稱產生。在名稱及結構之間存在衝突的情況下,以結構為準。
定義和一般術語
現在將詳細參考本發明的某些實施態樣,彼等的實例係以所附結構和化學式說明。本發明意欲涵蓋可包括在如申請專利範圍所定義之本發明範圍內的所有替代、修改和等效物。熟習該項技術者將認可許多類似於或等同於本文所述者的彼等方法和材料,這些方法和材料可用於實踐本發明。本發明決不限於本文所述的方法和材料。如果所併入的文獻、專利和類似材料中的一或多者與本申請案不同或相矛盾(包括但不限於定義的術語、術語用法、所述技術或類似者),則以本申請案為準。
應進一步理解,為了清楚起見,在單獨實施態樣的情况下所述之本發明的一些特徵也可在單一實施態樣中以組合形式提供。反之,為了簡潔起見,在單一實施態樣的情况下所述之本發明的各種特徵也可單獨或以任意適合的子組合提供。
除非另有定義,否則本文所使用的技術和科學術語具有與本發明所屬領域的技術人員通常所理解者相同的意義。本文中所提及的所有專利和出版品以其全文引用的方式併入。
在本文中,除非另有指示,否則下列定義應適用。為了本發明之目的,化學元素係根據元素週期表,CAS版本,及化學和物理手冊,第75版,1994來定義。另外,有機化學之一般原理係描述於Michael B. Smith和Jerry March 之"Organic Chemistry", Thomas Sorrell, University Science Books, Sausalito:1999及"March's Advanced Organic Chemistry", John Wiley & Sons, New York:2007中,其全部內容特此以引用方式併入。
如上所述,且遍及本揭示,除非另有指示,否則下列術語應理解為具有下列意義。若缺少定義,則以熟悉該項技術者已知的定義為準。若本文提供的定義與任何引用的出版品中提供之定義不同,則以本文提供的定義為準。
在本文中,術語“包括”、“含有”、和“包含”係以其開放非限制性的意義使用。
在本文中,單數形式“一(a、an)”及“該(the)”包括複數個指示物,除非上下文另有明確的指示。
為提供更簡潔的描述,本文給出的一些數量表述沒有用術語“約”限定。應瞭解,無論術語“約”明確使用與否,本文給出的每個數量意欲指實際所給值,且亦意指根據一般技藝人士合理推斷之該所給值的近似值,包括由於該所給值之實驗及/或測量條件的等效值及近似值。每當產率以百分比給出時,該產率係指給出產率之實體的質量相對於可在特定化學計量條件下獲得之相同實體的最大量。除非不同地指示,否則以百分比給出之濃度係指質量比。
化學定義
在本文中,“烷基”係指具有1至12個碳原子的飽和直鏈-或支鏈-烴基。代表性的烷基包括但不限於甲基、乙基、正丙基、異丙基、2-甲基-1-丙基、2-甲基-2-丙基、2-甲基-1-丁基、3-甲基-1-丁基、2-甲基-3-丁基、2,2-二甲基-1-丙基、2-甲基-1-戊基、3-甲基-1-戊基、4-甲基-1-戊基、2-甲基-2-戊基、3-甲基-2-戊基、4-甲基-2-戊基、2,2-二甲基-1-丁基、3,3-二甲基-1-丁基、2-乙基-1-丁基、丁基、異丁基、三級丁基、正戊基、異戊基、新戊基、正己基、等等,以及較長的烷基,諸如庚基、辛基、等等。
在本說明書的各處,本文所揭示的化合物之取代基係以群組或範圍揭示。特別意欲本發明包括這些群組和範圍的成員之各個和每個個別亞組合。例如,術語“C
1-6烷基”特別意欲個別揭示甲基、乙基、C
3烷基、C
4烷基、C
5烷基、和C
6烷基。
術語“鹵素”或“鹵基”在本發明中可互換使用且係指氟基(F)、氯基(Cl)、溴基(Br)或碘基(I)。
術語“烷氧基”係指經由氧原子鍵結至母分子部分之如先前所定義的烷基。除非另有規定,烷氧基含有1-12個碳原子。在一個實施態樣中,烷氧基含有1-6個碳原子。在其他實施態樣中,烷氧基含有1-4個碳原子。在又其他實施態樣中,烷氧基含有1-3個碳原子。烷氧基可隨意地經本文所揭示的一或多個取代基取代。在本文中,“烷氧基烷基”意指-(伸烷基)-O-(烷基),其中各“烷基”獨立地為上述定義的烷基。
“芳基”意指單-、雙-、或三環芳族基團,其中該基團的所有環均為芳族。對於雙-或三環系統,各個芳族環彼此稠合。示例性芳基包括但不限於苯基、萘以及蒽。
術語“氰烷基”表示如本文所定義的烷基,其中烷基的氫原子經氰基(-CN)置換。氰烷基的烷基部份提供接至分子其餘部份的連接點。
術語“氘”在本文中意指具有一個質子及一個中子的氫之穩定同位素。
在本文中可互換使用的術語“碳環基”和“碳環”係指呈單環、雙環或三環環系統之具有3至12個碳原子的單價或多價環,其為飽和或含有一或多個不飽和度,但是芳族環不能存在於該碳環基中。
術語“羥基”意指-OH基。
在本文中可互換使用的術語“雜環基”和“雜環”係指含有3-12個碳原子的單價或多價單環、雙環或三環,其中該環中的一或多個原子各自獨立地經雜原子置換,該雜原子系如本文所定義,且該環可為飽和或包含一或多個不飽和度,但是芳族環不可存在於該芳族環中。
術語“環烷基”係指呈單環、雙環或三環環系統之具有3至12個環碳原子的單價或多價飽和環。
熟習該項技術者將認知上述所列或說明之雜芳基和環烷基的種類並非詳盡無遺,且也可選擇在這些所定義之術語範圍內的其他種類。
如本文所述,本文所揭示的化合物可隨意地經一或多個取代基取代,或如本發明的特定類別、亞類和種類所舉例說明的。
在本文中,術語“經取代”意指特定基團或部分具有一個或多個適當取代基。在本文中,術語“未經取代”意指特定基團沒有取代基。在本文中,術語“隨意經取代”意指特定基團未經取代或經特定數量的取代基取代。其中術語“經取代”係用以描述結構系統,該取代係意指發生在系統上任何化學價允許的位置。
在本文中,詞句“一或多個取代基”表示可發生在系統上任何化學價允許的位置之一至最大可能數目的取代基。在某一實施態樣中,一或多個取代基意指1、2、3、4、或5個取代基。在另一實施態樣中,一或多個取代基意指1、2、或3個取代基。
本文以不飽和價表示的任何原子假設為具有足夠數目的氫原子以滿足原子的價。
當任何變體(例如,烷基、伸烷基、雜芳基、R
1、R
2、或R
a)在本文所提供之任何化學式或說明中在超過一個位置出現時,每次出現時該變體的定義與其在每次其他出現時的定義無關。
數字範圍,在本文中,意指包括連續的整數。例如,表示為“從0至4”或“0-4”之範圍包括0、1、2、3和4,而表示為"10-20%"之範圍包括10%、11%、12%、13%、14%、15%、16%、17%、18%、19%和20%。類似地,數字範圍亦意欲包括連續小數整數。例如,表示為"1-2%"之範圍將包括1.0%、1.1%、1.2%、1.3%、1.4%、1.5%、1.6%、1.7%、1.8%、1.9%以及2.0%。
當顯示多官能部分時,連接至核心的點以線或連字號(hyphen)表示。例如,“芳氧基-”係指其中氧原子為連接至核心分子的點而芳基則連接至該氧原子的基團。
其他定義
在本文中,術語“對象”包含哺乳動物或非哺乳動物。哺乳動物的實例包括但不限於哺乳動物綱的任何成員:人類;非人類靈長類動物諸如黑猩猩、及其他人猿和猴種;農場動物諸如牛、馬、羊、山羊、豬;家畜諸如兔子、狗和貓;及實驗室動物包括囓齒動物諸如大鼠、小鼠和天竺鼠、等等。非哺乳動物的實例包括但不限於鳥類、魚類等等。在本發明之一個實施態樣中,哺乳動物為人類。
“患者”包括人類及動物二者。
術語“抑制劑”係指阻斷或以其他方式干擾特定生物活性的分子,諸如化合物、藥物、酵素活化劑、或激素。
術語“調節劑”係指增加或減少、或以其他方式影響所給蛋白質、受體及/或離子通道之活性的分子,諸如本發明化合物。
術語“有效量”或“治療有效量”係指藥劑提供所需生物結果的足夠量。該結果可減少及/或緩解疾病或病情的病徵、症狀、或病因,或任何其他所需生物系統的改變。例如,關於治療用途的“有效量”為化合物的量、或包含該化合物之組成物的量,其為提供疾病狀態、症狀或病情之臨床上相關改變所需者。在任何個案中適當“有效”量可由一般技藝人士使用例行實驗檢測確定。因此,詞句“有效量”通常係指活性物質具有治療上所需效果的量。
在本文中,術語“治療(treat或treatment)”包含“預防性”和“治癒性”治療二者。“預防性”治療意指表示疾病、疾病的症狀、或病情的發展之延緩、抑制可能出現的症狀、或減少疾病或症狀的發展或復發之風險。“治癒性”治療包括降低現有疾病、症狀或病況的嚴重性或抑制現有疾病、症狀或病況的惡化。因此,治療包括改善或預防現有疾病症狀的惡化、預防其他症狀的發生、改善或預防症狀的潛在代謝病因、抑制病症或疾病,例如:阻止病症或疾病的發展、緩解病症或疾病、導致病症或疾病的消退、緩解疾病或病症引起的病況、或停止疾病或病症的症狀。
在本文中,術語化合物之"投予(administration或administering)"應理解意指將本發明之化合物、包含本發明化合物或化合物的前藥之醫藥組成物提供給有需要的個體。熟習非限制性技術者係理解可以有效量的本發明化合物治療目前患有神經和精神病症的患者或藉由預防性治療患有該病症的患者。
術語"組成物"在本文中意欲包括一種包含特定含量的特定成分之產物,以及從特定含量的特定成分之組合直接地或間接地產生的任何產物。該術語與醫藥組成物有關,意欲包括一種包含活性成分和組成載體的惰性成分之產物,以及從任何二或更多種成分的組合、複合或聚集、或從諸如引起一或多種成分解離的反應或交互作用之其他類型直接地或間接地產生的任何產物。因此,本發明之醫藥組成物包括藉由混合本發明化合物和醫藥上可接受的載體製造的任何組成物。
其他化學說明
本文所給的任何化學式意欲表示具有以結構式描述之結構的化合物以及一些變體或形式。例如,本文所給的任何化學式之化合物可具有不對稱或掌性(chiral)中心,且因此以不同的立體異構形式存在。通式化合物的所有立體異構物(包括光學異構物、鏡像異構物、和非鏡像異構物),以及其混合物被認為是落在該化學式的範圍內。此外,一些結構可以幾何異構物(即,順式和反式異構物)、互變異構物或阻轉異構物存在。所有該等異構物形式及其混合物在本文中都被認為是本發明的一部分。因此,本文所給的任何化學式意欲表示外消旋物、一或多種鏡像異構物形式、一或多種非鏡像異構物形式、一或多種互變異構物或阻轉異構物形式、及其混合物。
“立體異構物”係指具有相同化學組成但原子或基團在空間上的排列不同之化合物。立體異構物包括鏡像異構物、非鏡像異構物、構形異構物(旋轉異構物)、幾何異構物(順式/反式)、阻轉異構物等等。
“掌性”係指具有鏡像伴體的非重疊性之性質的分子,而術語“非掌性”係指可與其鏡像伴體重疊的分子。
“鏡像異構物”係指化合物之彼此為不重疊的鏡像之二種立體異構物。
“非鏡像異構物”係指具有二或更多個掌性中心且其分子彼此不為鏡像的立體異構物。非鏡像異構物具有不同的物理性質,例如熔點、沸點、光譜性質或生物活性。非鏡像異構物的混合物可在高解析分析程序諸如電泳和層析法諸如HPLC下分離。
本文中所使用的立體化學定義及慣例通常按照S. P. Parker, Ed., McGraw-Hill Dictionary of Chemical Terms(1984) McGraw-Hill Book Company, New York;及Eliel, E.和Wilen, S., "Stereochemistry of Organic Compounds", John Wiley & Sons, Inc., New York, 1994。
許多有機化合物以光學活性形式存在,即,彼等具有旋轉偏振光的平面之能力。在描述光學活性化合物中,字首D和L、或R和S係用於表示該分子相對於其掌性中心之絕對組態。字首d和l或(+)和(-)係用於指定平面偏振光被該化合物旋轉的符號,具有(-)或l意指該化合物為左旋性。具有字首(+)或d的化合物為右旋性。特定立體異構物可稱為鏡像異構物,並且該等立體異構物的混合物稱為鏡像異構物混合物。鏡像異構物的50:50混合物稱為外消旋混合物或外消旋物,其可在化學反應或方法中沒有立體選擇性或立體特異性的情況下發生。
本文所揭示之化合物的任何不對稱原子(例如碳或類似物)可為外消旋或鏡像異構地富含的組態,例如(R)-、(S)-或(R,S)-組態。在一些實施態樣中,各個不對稱原子具有至少50%鏡像異構物過量、至少60%鏡像異構物過量、至少70%鏡像異構物過量、至少80%鏡像異構物過量、至少90%鏡像異構物過量、至少95%鏡像異構物過量、或至少99%鏡像異構物過量之(R)-或(S)-組態。
取決於起始材料和程序的選擇,化合物可依據不對稱碳原子的數目以可能的立體異構物之一者或其混合物(諸如外消旋物和非鏡像異構物混合物)的形式存在。光學活性(R)-和(S)-異構物可使用掌性合成組元(synthon)或掌性試劑來製備,或使用習用的技術解析。若化合物含有雙鍵,則取代基可為E或Z構型。若化合物含有經二取代之環烷基,則相對於相同環烷基架構的另一個取代基,環烷基取代基可具有順式或反式構型。
任何所得立體異構物的混合物可根據組分的物理化學差異而分離成純的或實質上純的幾何異構物、鏡像異構物、非鏡像異構物,例如藉由層析法及/或分級結晶。任何所得終產物或中間物的外消旋物可藉由熟習該項技術者已知的方法(例如藉由其非鏡像異構物鹽之分離)解析成光學鏡像體(antipodes)。外消旋產物亦可藉由掌性層析法(例如,使用掌性吸附劑的高性能液相層析(HPLC))解析。較佳鏡像異構物亦可藉由不對稱合成來製備。參見,例如Jacques等人,Enantiomers, Racemates and Resolutions(Wiley Interscience, New York, 1981);
Principlesof
Asymmetric Synthesis(2nd Ed. Robert E. Gawley, Jeffrey Aubé, Elsevier, Oxford, UK, 2012);Eliel, E.L. Stereochemistry of Carbon Compounds(McGraw-Hill, NY, 1962);Wilen, S.H. Tables of Resolving Agents and Optical Resolutions p. 268(E.L. Eliel, Ed., Univ. of Notre Dame Press, Notre Dame, IN 1972);Chiral Separation Techniques: A Practical Approach(Subramanian, G. Ed., Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2007)。
非鏡像異構物混合物可藉由熟習該項技術者已知的方法(諸如,例如藉由層析法及/或分級結晶)而根據其物理化學差異分離成彼等各個的非鏡像異構物。鏡像異構物可如下分離:藉由與適當的光學活性化合物(例如掌性助劑諸如掌性醇或Mosher氏醯氯)之反應,或形成非鏡像異構物鹽的混合物)而將鏡像異構物混合物轉化成非鏡像異構物混合物,分離非鏡像異構物並將各個非鏡像異構物轉化(例如水解或脫鹽)成相對應純鏡像異構物。鏡像異構物亦可藉由使用掌性HPLC管柱來分離。
本發明化合物可形成醫藥上可接受的鹽類,其亦在本發明的範圍內。“醫藥上可接受的鹽類”係指式A化合物的游離酸或鹼的鹽,其為無毒的、為生理上可耐受的、為與調配的醫藥組成物相容,且除此以外適合於調配及/或投予至對象。除非另有指示,否則本文所提及的化合物應理解為包括所提及的該化合物之醫藥上可接受的鹽類。
化合物鹽類包括與無機酸類及/或有機酸類形成的酸性鹽類,以及與無機鹼類及/或有機鹼類形成的鹼性鹽類。此外,在所給化合物同時含有鹼性部分(諸如但不限於吡啶或咪唑),以及酸性部分(諸如但不限於羧酸)的情況下,熟習該項技術者將理解該化合物可以兩性離子(“內鹽”)存在;該等鹽類包括在本文中的術語“鹽”內。本發明化合物的鹽類可例如藉由化合物與某量之適當酸或鹼(諸如等量)在介質諸如鹽沉澱於其中者或在水性介質中反應,接著凍乾來製備。
示例性鹽類包括但不限於硫酸鹽、檸檬酸鹽、乙酸鹽、草酸鹽、氯化物、溴化物、碘化物、硝酸鹽、硫酸氫鹽、磷酸鹽、酸式磷酸鹽、異菸酸鹽、乳酸鹽、水楊酸鹽、酸式檸檬酸鹽、酒石酸鹽、油酸鹽、鞣酸鹽、泛酸鹽、酒石酸氫鹽、抗壞血酸鹽、琥珀酸鹽、馬來酸鹽、龍膽酸鹽、富馬酸鹽、葡糖酸鹽、葡醣醛酸鹽、葡萄糖二酸鹽、甲酸鹽、苯甲酸鹽、麩胺酸鹽、甲磺酸鹽(“甲磺酸鹽”)、乙磺酸鹽、苯磺酸鹽、對-甲苯磺酸鹽、和巴莫酸鹽(pamoate)(即,1,1'-亞甲基-雙(2-羥基-3-萘甲酸鹽))。醫藥上可接受的鹽類可包括內含另一分子諸如乙酸根離子、琥珀酸根離子或其他相對離子。該相對離子可為使母化合物上的電荷穩定之任何有機或無機部分。此外,醫藥上可接受的鹽類在其結構中可具有大於一個帶電原子。其中多個帶電原子為醫藥上可接受的鹽類的部分之情況可具有多個相對離子。因此,醫藥上可接受的鹽類可具有一或多個帶電原子及/或一或多個相對離子。
示例性的酸加成鹽類包括乙酸鹽類、抗壞血酸鹽類、苯甲酸鹽類、苯磺酸鹽類、硫酸氫鹽類、硼酸鹽類、丁酸鹽類、檸檬酸鹽類、樟腦酸鹽類、樟腦磺酸鹽類、富馬酸鹽類、鹽類酸鹽類、氫溴酸鹽類、氫碘酸鹽類、乳酸鹽類、馬來酸鹽類、甲磺酸鹽類、萘磺酸鹽類、硝酸鹽類、草酸鹽類、磷酸鹽類、丙酸鹽類、水楊酸鹽類、琥珀酸鹽類、硫酸鹽類、酒石酸鹽類、硫氰酸鹽類、甲苯磺酸鹽類等等。
示例性的鹼性鹽類包括銨鹽類、鹼金屬鹽類(諸如鈉鹽類、鋰鹽類和鉀鹽類)、鹼土金屬鹽類(諸如鈣鹽類及鎂鹽類)、與有機鹼類(例如有機胺類,諸如二環己胺類、三級丁基胺類)形成的鹽類、與胺基酸諸如精胺酸、離胺酸等等形成的鹽類。鹼性含氮基團可用諸如低級烷基鹵化物(例如,甲基、乙基、和丁基的氯化物、溴化物和碘化物)、硫酸二烷酯類(例如,硫酸二甲酯、硫酸二乙酯、和硫酸二丁酯)、長鏈鹵化物(例如癸基、月桂基、和硬脂基的氯化物、溴化物和碘化物)、芳烷基鹵化物(例如苯甲基溴化物和苯乙基溴化物)、及其他試劑來進行四級化(quarternized)。
另外,通常認為適合於從醫藥化合物形成醫藥上有用的鹽類之酸類和鹼類係討論於例如P. Stahl等人,Camille G.(eds.) Handbook of Pharmaceutical Salts. Properties, Selection and Use.(2002) Zurich: Wiley-VCH;S. Berge等人,Journal of Pharmaceutical Sciences(1977) 66(1) 1-19;P. Gould, International J. of Pharmaceutics(1986) 33 201-217;Anderson等人,The Practice of Medicinal Chemistry (1996), Academic Press, New York;及The Orange Book(Food & Drug Administration, MD,可向FDA購買)。該些揭示以引用方式併入本文中。
另外,本文所述的任何化合物也意指該類化合物的任何未溶劑合的形式、或水合物、溶劑合物、或多晶型物及其混合物,即使該等形式未明確列出。“溶劑合物”意指本發明化合物與一或多種溶劑分子的物理結合。此物理結合涉及不同程度的離子和共價鍵結,包括氫鍵結。在某些情況下,溶劑合物將能夠分離,例如當一或多種溶劑分子併入結晶固體的晶格中時。“溶劑合物”包含溶液相和可分離的溶劑合物。適當溶劑合物包括與醫藥上可接受的溶劑如諸水、乙醇、等等所形成的溶劑合物。在一些實施態樣中,溶劑為水則溶劑合物為水合物。
一或多種本發明化合物可隨意地轉化為溶劑合物。製備溶劑合物的方法通常是已知的。因此,例如M. Caira等人,J. Pharmaceutical Sci., 93(3), 601-611(2004)描述抗真菌劑氟康唑(fluconazole)在乙酸乙酯中以及從水中製備溶劑合物。E. C. van Tonder 等人,AAPS PharmSciTech., 5(1), article 12(2004);及A. L. Bingham 等人,Chem. Commun., 603-604(2001)描述溶劑合物、半溶劑合物、水合物、等等的類似製備。典型的非限制性方法包括在高於環境溫度下將本發明化合物溶解於適量溶劑(有機溶劑或水或其混合物)中,並以足以形成晶體的速率冷卻溶液,該晶體接著藉由標準方法分離。分析技術(諸如例如紅外線光譜)顯示溶劑(或水)存在於晶體中而呈溶劑合物(或水合物)。
本發明亦關於式(A)化合物之醫藥活性代謝物,及該等代謝物在本發明方法中的用途。“醫藥活性代謝物”意指式(A)化合物或其鹽之體內代謝的藥理活性產物。化合物的活性代謝物可使用該項技術領域已知或可獲得的習用技術來確定。參見,例如,Bertolini等人,
J. Med. Chem. 1997,
40, 2011-2016;Shan等人,
J. Pharm. Sci. 1997,
86(7), 765-767;Bagshawe,
Drug Dev. Res. 1995,
34, 220-230;Bodor,
Adv. Drug Res. 1984,
13, 255-331;Bundgaard, Design of Prodrugs (Elsevier Press, 1985);及Larsen, Design and Application of Prodrugs, Drug Design and Development (Krogsgaard-Larsen等人編輯,Harwood Academic Publishers, 1991)。
本文所給的任何化學式亦意欲表示化合物的未標記形式以及同位素標記形式。同位素標記的化合物除了一或多個原子被具有所選擇的原子量或質量數的原子替換之外,具有以本文所給化學式描述之結構。可併入本發明化合物之同位素的實例包括氫、碳、氮、氧、磷、氟、氯和碘的同位素,諸如分別為
2H、
3H、
11C、
13C、
14C、
15N、
18O、
17O、
31P、
32P、
35S、
18F、
36Cl、和
125I。該等同位素標記的化合物可用於代謝研究(例如用
14C)、反應動力學研究(用例如
2H或
3H)、檢測或成像技術[諸如正子斷層攝影(PET)或單光子發射電腦斷層掃描儀(SPECT)],包括藥物或基質組織分佈分析、或用於患者的放射性治療。特別是,
18F或
11C標記的化合物可能特別適合於PET或SPECT研究。此外,以較重的同位素例如氘(如
2H)取代可提供由更大的代謝穩定性而產生的一些治療優勢,例如活體內半衰期增加或劑量需求減少。同位素標記的本發明化合物通常可藉由進行下述流程或實施例和製備中所揭示的程序藉由容易獲得之同位素標記的試劑取代非同位素標記的試劑來製備。
關於本文所述的化合物之術語“鹽”、溶劑合物”、“多晶型物”、等等的使用意欲同樣適用於本發明化合物之鏡像異構物、立體異構物、旋轉異構物、互變異構物、阻轉異構物、和外消旋物的鹽、溶劑合物和多晶型物形式。
本發明化合物之說明
本發明係關於特定分子及其醫藥上可接受的鹽類或異構物。本發明進一步關於可用於調節H3R的分子及其醫藥上可接受的鹽類、溶劑合物、酯類、或異構物。
本發明關於如本文所述之化合物及其醫藥上可接受的鹽類、溶劑合物、酯類、或異構物,以及包含一或多種如本文所述之化合物及其醫藥上可接受的鹽類或異構物之醫藥組成物。本發明之一態樣為提供用於調節哺乳動物H3R之具有式(A)化合物或其醫藥上可接受的鹽類之化合物、組成物、套組、及解毒劑:
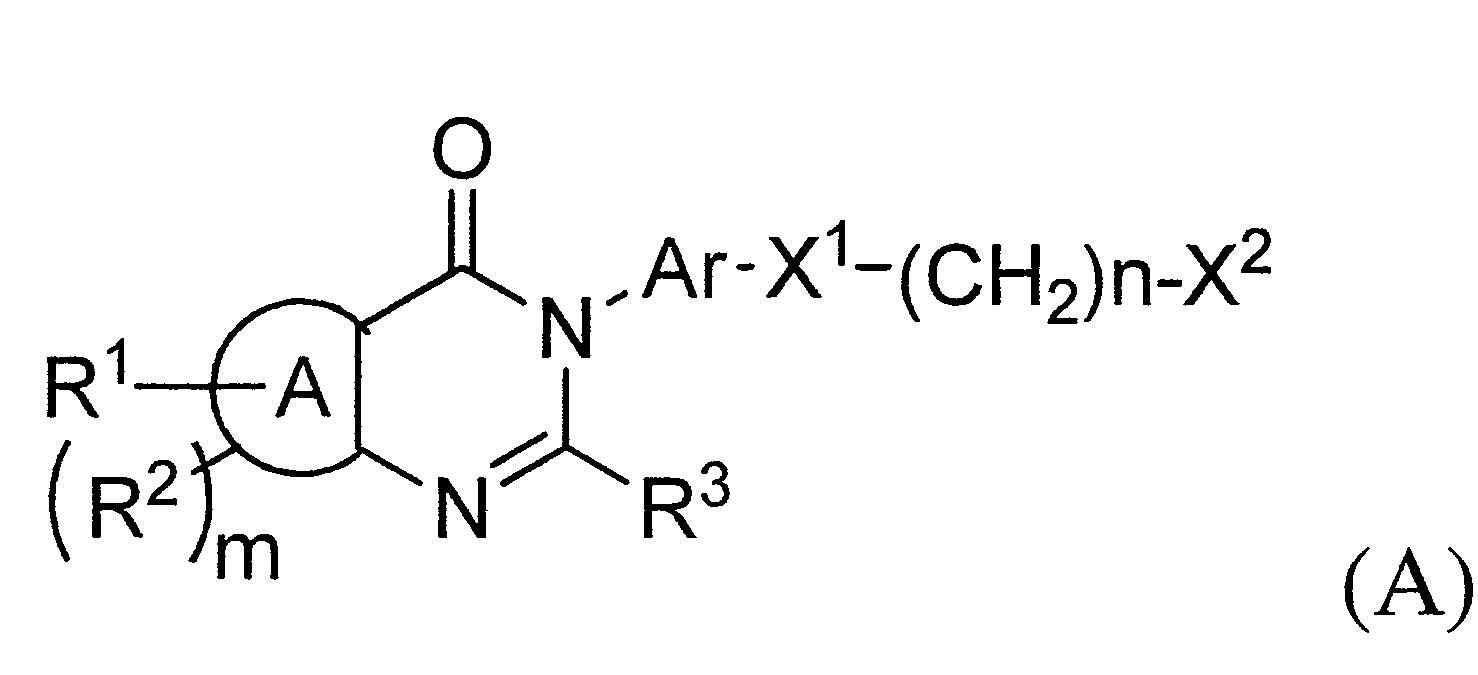 ,
其中,R
1和R
2獨立地為H或SF
5,先決條件為至少R
1和R
2中之一者為SF
5;
R
3為C
1-6烷基或C
3-6環烷基;
環A為C
6~8芳基;Ar為C
5-6芳基,其中C
5-6芳基係隨意地經1、2、3或4個R
e取代;
R
e為H、鹵素、CN、NO
2、OH、C
1-6烷氧基或C
1-6烷基;
X
1為S或O;
X
2為
,
其中,R
1和R
2獨立地為H或SF
5,先決條件為至少R
1和R
2中之一者為SF
5;
R
3為C
1-6烷基或C
3-6環烷基;
環A為C
6~8芳基;Ar為C
5-6芳基,其中C
5-6芳基係隨意地經1、2、3或4個R
e取代;
R
e為H、鹵素、CN、NO
2、OH、C
1-6烷氧基或C
1-6烷基;
X
1為S或O;
X
2為
 ,其中R為N或C;
R
3和R
4為C
1-6烷基,或R
3和R
4與彼等所連接的R一起形成C
3-7碳環、3-至7-員雜環,且其中C
3-7碳環、3-至7-員雜環各自係獨立地隨意地經1、2、3、4、5或6個R
f取代;
R
f為C
1-6烷基、C
3-6環烷基、-C(=O)R
a,且其中C
1-6烷基、C
3-6環烷基、-C(=O)R
a各自係獨立地隨意地經C
1-6烷基取代;
R
a為C
1-6烷基、C
3-6環烷基或3-至7-員雜環;
m為從0至3的整數;
n為從0至5的整數。
在一個實施態樣中,R
3為C1-4烷基或C3-6環烷基。
在一個實施態樣中,A為苯基。
在一個實施態樣中,Ar為苯基,其中苯基係隨意地經1、2、3或4個R
e取代。
在一個實施態樣中,Ar為下列子式:
,其中R為N或C;
R
3和R
4為C
1-6烷基,或R
3和R
4與彼等所連接的R一起形成C
3-7碳環、3-至7-員雜環,且其中C
3-7碳環、3-至7-員雜環各自係獨立地隨意地經1、2、3、4、5或6個R
f取代;
R
f為C
1-6烷基、C
3-6環烷基、-C(=O)R
a,且其中C
1-6烷基、C
3-6環烷基、-C(=O)R
a各自係獨立地隨意地經C
1-6烷基取代;
R
a為C
1-6烷基、C
3-6環烷基或3-至7-員雜環;
m為從0至3的整數;
n為從0至5的整數。
在一個實施態樣中,R
3為C1-4烷基或C3-6環烷基。
在一個實施態樣中,A為苯基。
在一個實施態樣中,Ar為苯基,其中苯基係隨意地經1、2、3或4個R
e取代。
在一個實施態樣中,Ar為下列子式:
 (W-1a),其中w1為0、1、2、3或4。
在一個實施態樣中,R
e為H、F、Cl、Br、I、CN、NO
2、OH、甲基、乙基、異丙基、正丙基、正丁基、三級丁基、甲氧基、乙氧基。
在一個實施態樣中,R
3和R
4為C
1-4烷基。
在一個實施態樣中,R為N,R
3和R
4與彼等所連接的氮原子一起形成3-至7-員雜環,及其中該3-至7-員雜環係獨立地隨意地經1、2、3、4或5個R
f取代;
在一個實施態樣中,R為C,R
3和R
4與彼等所連接的碳原子一起形成C
3-7碳環、3-至7-員雜環,且其中C
3-7碳環、3-至7-員雜環各自係獨立地隨意地經1、2、3、4或5個R
f取代。
在一個實施態樣中,R
f為C
1-4烷基、C
3-6環烷基、-C(=O)R
a。
在一個實施態樣中,R
f為-C(=O)R
a、甲基、乙基、異丙基、
(W-1a),其中w1為0、1、2、3或4。
在一個實施態樣中,R
e為H、F、Cl、Br、I、CN、NO
2、OH、甲基、乙基、異丙基、正丙基、正丁基、三級丁基、甲氧基、乙氧基。
在一個實施態樣中,R
3和R
4為C
1-4烷基。
在一個實施態樣中,R為N,R
3和R
4與彼等所連接的氮原子一起形成3-至7-員雜環,及其中該3-至7-員雜環係獨立地隨意地經1、2、3、4或5個R
f取代;
在一個實施態樣中,R為C,R
3和R
4與彼等所連接的碳原子一起形成C
3-7碳環、3-至7-員雜環,且其中C
3-7碳環、3-至7-員雜環各自係獨立地隨意地經1、2、3、4或5個R
f取代。
在一個實施態樣中,R
f為C
1-4烷基、C
3-6環烷基、-C(=O)R
a。
在一個實施態樣中,R
f為-C(=O)R
a、甲基、乙基、異丙基、
 。
在一個實施態樣中,R
a為甲基、乙基、
。
在一個實施態樣中,R
a為甲基、乙基、
 ,
,
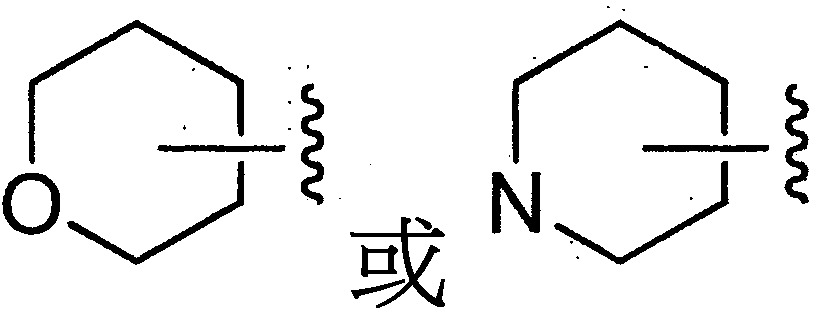 。
在一個實施態樣中,X
2為下列子式中之一者:
。
在一個實施態樣中,X
2為下列子式中之一者:
 。
在一個實施態樣中,X
2為下列子式中之一者:
。
在一個實施態樣中,X
2為下列子式中之一者:
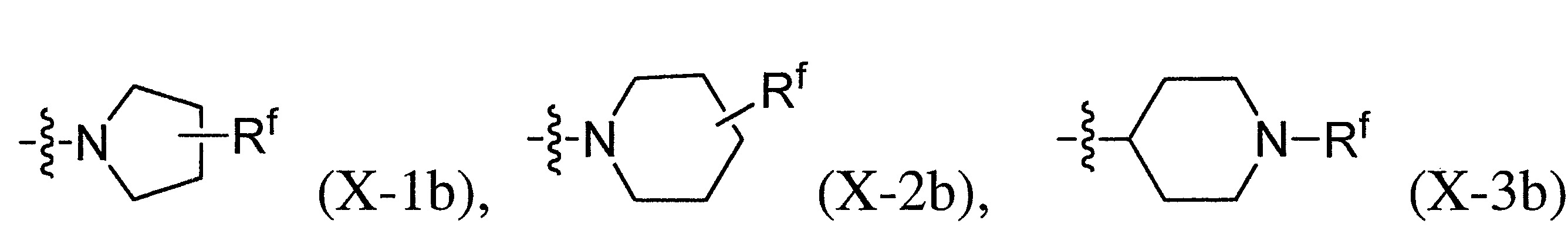 。
在一個實施態樣中,本文提供一種具有式(B)之化合物,
。
在一個實施態樣中,本文提供一種具有式(B)之化合物,
 ,
其中o為從0至4的整數;及
R
e、R
f係如本文所定義。
在一個實施態樣中,本文提供一種具有式(C)之化合物,
,
其中o為從0至4的整數;及
R
e、R
f係如本文所定義。
在一個實施態樣中,本文提供一種具有式(C)之化合物,
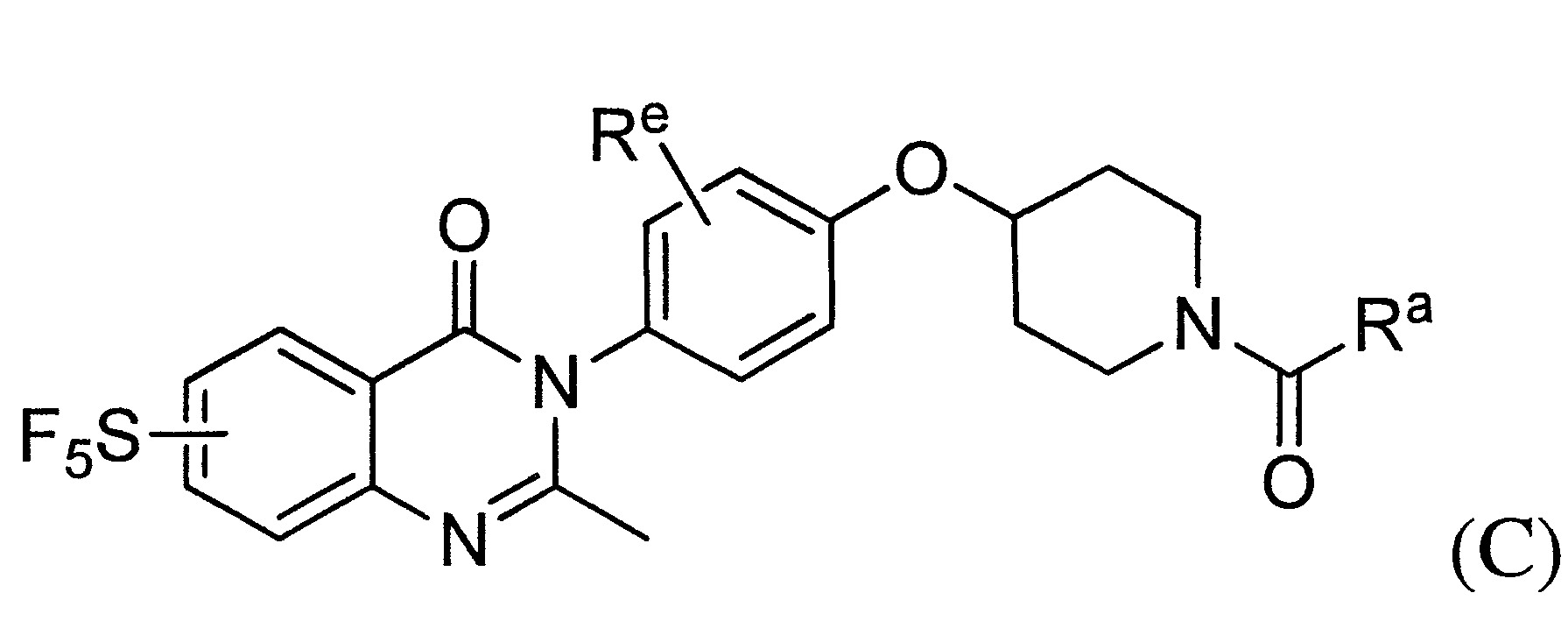 ,
R
e、R
a係如本文所定義。
在一個實施態樣中,本文提供一種具有式(D)之化合物,
,
R
e、R
a係如本文所定義。
在一個實施態樣中,本文提供一種具有式(D)之化合物,
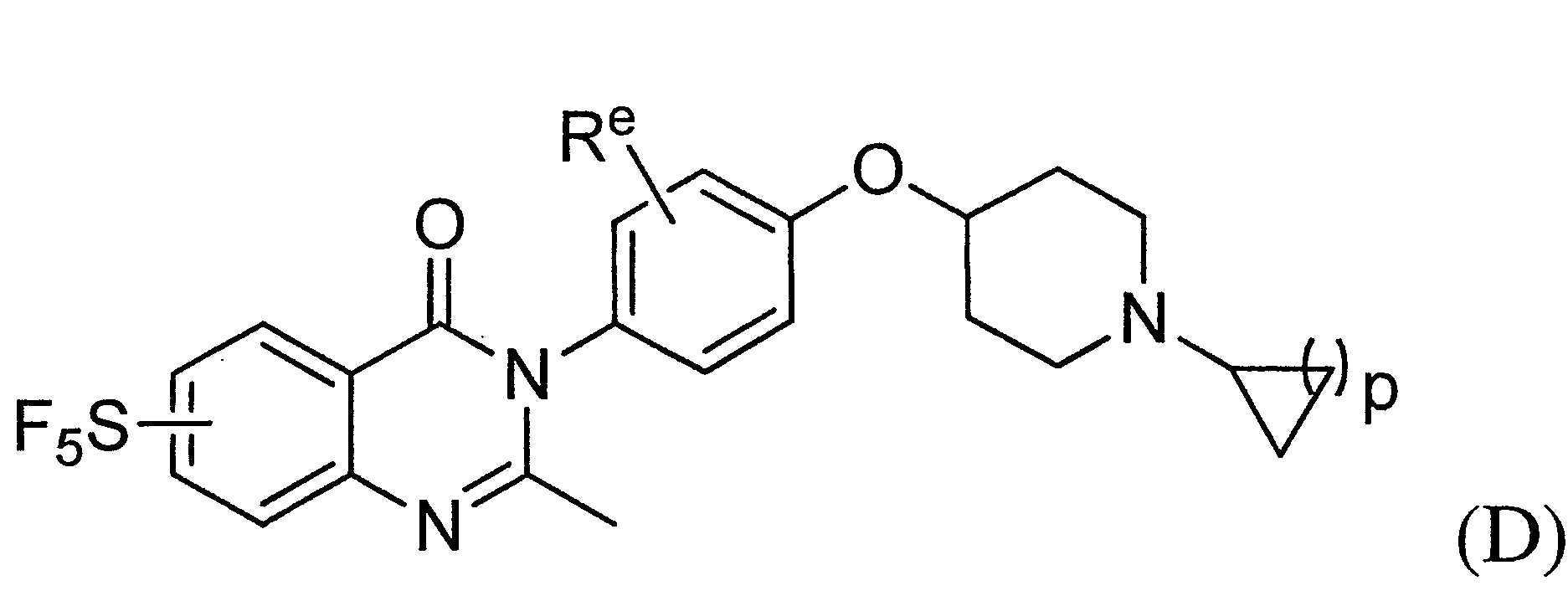 ,
其中p為從1至5的整數;及
R
e係如本文所定義。
在又另一實施態樣中,本文提供一種具有下列結構之一者的化合物或醫藥上可接受的鹽類,
,
其中p為從1至5的整數;及
R
e係如本文所定義。
在又另一實施態樣中,本文提供一種具有下列結構之一者的化合物或醫藥上可接受的鹽類,

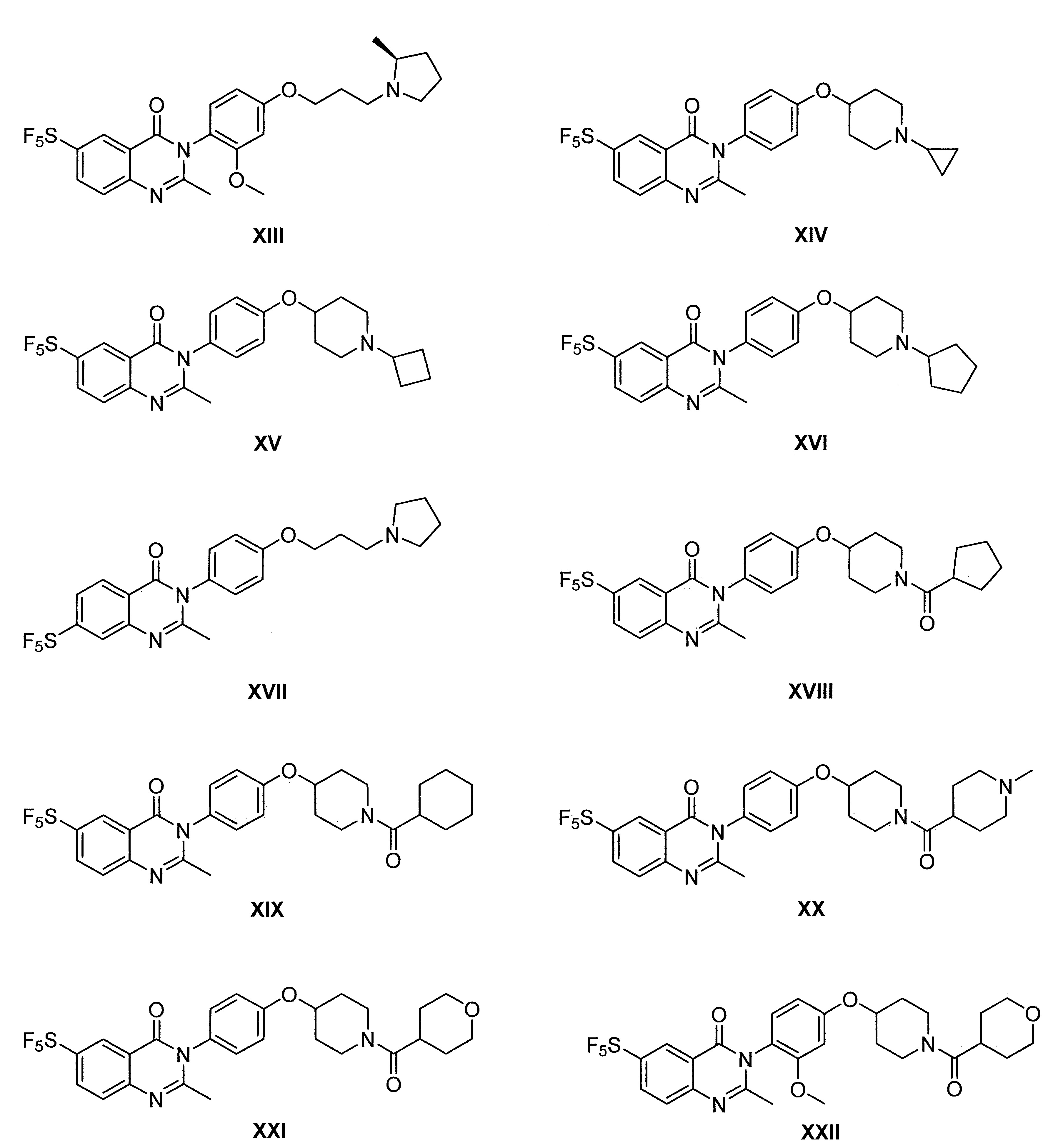 在另一態樣中,本文中提供一種包含本發明化合物之醫藥組成物。
在一個實施態樣中,醫藥組成物進一步包含醫藥上可接受的載體、佐劑、載劑或其組合。
在一個實施態樣中,醫藥組成物進一步包含一或多其他種治療劑,及其中該其他種治療劑係用於治療CNS病症或嗜睡症。
在一個實施態樣中,CNS病症為認知、精神、神經運動或疼痛。
在一個實施態樣中,其他治療劑係進一步包含:選自由多巴胺受體拮抗劑、血清素-去甲基腎上腺素再吸收抑制劑、選擇性血清素再吸收抑制劑、去甲基腎上腺素激導性再吸收抑制劑、非選擇性血清素再吸收抑制劑、及乙醯膽鹼酯酶抑制劑所組成群組之活性成分。
在另一態樣中,本文提供化合物或醫藥組成物之用途,其係用於製造供預防、管理、治療或減輕患者中CNS病症或嗜睡症的藥物。
在另一態樣中,本文提供化合物或醫藥組成物之用途,其係用於製造供拮抗H3受體的藥物。
在另一態樣中,本文提供化合物或醫藥組成物,其係用於預防、管理、治療或減輕患者中由病毒感染引起的CNS病症或嗜睡症的藥物。
在另一態樣中,本文提供用於拮抗H3受體之化合物或醫藥組成物。
在另一態樣中,本文提供一種預防、管理、治療或減輕患者中CNS病症或嗜睡症之方法,其包含將治療有效量的該化合物或該醫藥組成物投予至有需要的患者。
在另一態樣中,本文提供一種在患者中拮抗H3受體之方法,其包含將治療有效量的該化合物或該醫藥組成物投予至有需要的患者。
在又另一態樣中,本發明係關於製造式(A)~(D)之化合物和其醫藥上可接受的鹽類之方法。
本發明化合物的醫藥組成物及製備和投予
本發明提供一種醫藥組成物,其包含本發明化合物,例如,實施例化合物。根據本發明的特定實例,醫藥組成物可進一步包含醫藥上可接受的賦形劑、載體、佐劑、溶劑及其組合。
本發明提供一種治療、預防或改善疾病或病症之方法,其包含投予安全有效量之含有本發明化合物及一或多種治療活性劑的組合。其中,藥物的組合包含一或多個用於治療CNS病症或嗜睡症的其他藥物。
用於治療CNS病症或嗜睡症的其他藥物包括但不限於:進一步包含:選自由多巴胺受體拮抗劑、血清素-去甲基腎上腺素再吸收抑制劑、選擇性血清素再吸收抑制劑、去甲基腎上腺素激導性再吸收抑制劑、非選擇性血清素再吸收抑制劑、及乙醯膽鹼酯酶抑制劑所組成群組之活性成分。
本文揭示之醫藥組成物的化合物之量係指可有效檢測到調節生物學樣品及患者之功能失調H3R的量。活性成分可將提供最佳藥物療效(其不限於所要治療效果、投予途徑和治療期間)的劑量投予至需要該治療的對象。取決於疾病的性質以及嚴重性、患者的體重、患者接著所遵循的特殊飲食、同時用藥及熟習該項技術者將理解的其他因素,劑量將隨患者而異。根據具體應用,在製劑的單位劑量中之活性化合物的量可在從約1mg至約1000mg,較佳從約1mg至約500mg,更佳從約1mg至約300mg,仍更佳從約1mg至約200mg改變或調整。
所使用的實際劑量可取決於患者的需求和治療病況的嚴重性而改變。用於特定情況的適當劑量方案的測定在該項技術範圍內。為方便起見,可根據需求將每日總劑量分開且在一天內分批投予。本發明化合物及/或其醫藥上可接受的鹽類之投予的量和頻率將根據臨床主治醫師考慮該等諸如患者之年齡、病況和大小以及所治療症狀的嚴重性之因素判斷來調整。用於口服投予之典型推薦的每日劑量方案範圍可在從約1mg/天至約300mg/天,較佳為10mg/天至200mg/天,其可以單劑量或多劑量投予。在又另一實施態樣中,每個患者每天約1mg至50mg。
也應理解本發明的一些化合物可以游離形式存在而用於治療,或適當的話,呈其醫藥上可接受的衍生物或前藥的形式。醫藥上可接受的衍生物包括醫藥上可接受的鹽類、酯、該等酯的鹽、或在投予至有需要的患者時,其直接或間接地提供如本文另外描述的化合物或其治療上有效的代謝物或殘餘物之任何其他加合物或衍生物。
本發明之醫藥組成物可以散裝形式製備及包裝,由此散裝形式可提取安全有效量的本文所揭示之式(A)化合物,並接著投予至患者,諸如以粉末或糖漿。通常,將介於0.0001至10mg/kg體重的劑量水平每日投予至患者以獲得功能失調H3R的有效調節。或者,本發明之醫藥組成物可以單位劑型製備及包裝,其中各個物理上分離單位含有安全有效量的本文揭示之式(A)化合物。當製備成單位劑型時,本發明之醫藥組成物通常含有從約0.5mg至1g,或1mg至500mg,或5mg至200mg,或更佳地,25mg至100mg的本發明化合物。
當本發明醫藥組成物除了本發明化合物之外亦含有一或多種其他活性成分時,本發明化合物對第二種活性成分的重量比可改變並且取決於各成分的有效劑量。因此,例如,當本發明化合物與另一個藥劑組合時,本發明化合物與其他藥劑的重量比通常範圍從約1000:1至約1:1000,諸如約200:1至1:200。本發明化合物與其他活性成分的組合通常也在上述範圍內,但在各情況下,應使用組合中各活性成分的有效劑量。
"醫藥上可接受的賦形劑"在本文中意指涉及賦予醫藥組成物形式或一致性的醫藥上可接受的材料、組成物或載劑(vehicle)。當混合時,各賦形劑必須與醫藥組成物的其他成分相容,使得當投予至患者時相互作用會實質上減少本發明化合物的療效並會導致醫藥上不可接受的組成物。另外,各賦形劑當然必須具有足夠高的純度以使其能為醫藥上可接受的。
適當醫藥上可接受的賦形劑將依據所選特定劑量形式而變化。另外,可選擇適當醫藥上可接受的賦形劑用於組成物中的特定功能。例如,可選擇一些具有促進製造均勻劑型之能力的醫藥上可接受的賦形劑。可選擇一些具有促進製造穩定劑型之能力的醫藥上可接受的賦形劑。可選擇一些當投予至患者時具有促進本發明的化合物從一個器官或身體的部位攜帶或傳遞至另一個器官或身體部位之能力的醫藥上可接受的賦形劑。可選擇一些具有提高患者依從性之能力的醫藥上可接受的賦形劑。
適當醫藥上可接受的賦形劑包括下列賦形劑之類型:稀釋劑、填充劑、黏合劑、崩解劑、潤滑劑、助滑劑、製粒劑、塗佈劑、潤濕劑、溶劑、共溶劑、懸浮劑、乳化劑、甜味劑、調味劑、味道掩蓋劑、著色劑、防結塊劑、濕潤劑、螯合劑、塑化劑、增黏劑、抗氧化劑、防腐劑、穩定劑、界面活性劑以及緩衝劑。熟悉該項技術者將理解一些醫藥上可接受的賦形劑可達成一種以上的功能且可依據調配物中存在多少賦形劑以及調配物中存在什麼其他成分而達成替代功能。
技術人員擁有該項技術的知識和技能以使他們能夠選擇適當醫藥上可接受的賦形劑以適當量用於本發明。此外,技術人員可獲得之描述醫藥上可接受的賦形劑且可用於選擇適當醫藥上可接受的賦形劑之辦法。實例包括Remington's Pharmaceutical Sciences(Mack Publishing Company)、The Handbook of Pharmaceutical Additives (Gower Publishing Limited)、及The Handbook of Pharmaceutical Excipients(the American Pharmaceutical Association and the Pharmaceutical Press)。
在Remington:The Science and Practice of Pharmacy, 21st edition, 2005, ed. D.B. Troy, Lippincott Williams & Wilkins, Philadelphia, 和Encyclopedia of Pharmaceutical Technology, eds. J. Swarbrick and J. C. Boylan, 1988-1999, Marcel Dekker, New York中,其各內容以引用方式併入本文中,揭示可用於調配醫藥上可接受的組成物之各種載體及其製備的已知技術。除非任何習知載體介質與本發明化合物不相容,諸如因產生的任何不良生物效應或以有害的方式與醫藥上可接受的組成物的任何其他組分相互作用,否則其使用預期在本發明的範圍內。
使用熟習該項技術者已知的技術及方法來製備本發明之醫藥組成物。該項技術常用的一些方法係描述於Remington's Pharmaceutical Sciences(Mack Publishing Company)中。
因此,本發明之另一態樣係關於一種製備醫藥組成物之方法。該醫藥組成物含有本文所揭示之化合物及醫藥上可接受的賦形劑、載體、佐劑、載劑或其組合,該方法包含混合各種成分。含有本文所揭示之化合物的醫藥組成物可在例如常溫和常壓下製備。
本發明化合物通常將調配成適用於藉由所需的投予途徑投予至患者的劑型。例如,劑型包括適用於(1) 口服投予,諸如錠劑、膠囊、膠囊型錠劑、丸劑、含片、粉劑、糖漿、酏劑、懸浮液、溶液、乳液、小藥囊及扁囊劑;(2) 胃腸外投予,諸如無菌溶液、懸浮液、及用於復原之粉劑;(3) 經皮投予,諸如經皮貼片;(4) 直腸投予,諸如栓劑;(5) 吸入,諸如氣霧劑、溶液及乾粉;以及(6) 局部投予,諸如霜劑、軟膏、洗劑、溶液、糊劑、噴霧劑、泡沫劑及凝膠之劑型。
本文所提供之醫藥組成物可以下列提供:壓縮錠、研製錠、可咀嚼口含錠、速溶錠、多重壓縮錠、或腸溶膜衣錠、糖衣錠或膜衣錠。腸溶膜衣錠為壓縮錠,其以可抵抗胃酸作用但在腸中溶解或崩解之物質塗覆,因此保護活性成分抵抗胃中之酸性環境。腸溶膜衣包括但不限於脂肪酸、脂肪、苯基水楊酸酯、蠟、蟲膠、胺化蟲膠、及乙酸纖維素鄰苯二甲酸。糖衣錠劑為由糖衣包圍之壓縮錠劑,該糖衣可有益於掩蓋不良味道或氣味且防止錠劑氧化。膜衣錠為以水溶性材料之薄層或薄膜覆蓋的壓縮錠。膜衣包括但不限於羥乙基纖維素、羧甲基纖維素鈉、聚乙二醇4000及乙酸纖維素鄰苯二甲酸。膜衣賦予與糖衣相同的一般特性。多重壓縮錠劑為藉由一個以上壓縮循環製成之壓縮錠,其包括分層錠、及壓製膜衣錠或乾壓膜衣錠。
錠劑劑型可自呈粉狀、結晶或粒狀形式之活性成分,單獨或與一或多種本文所述載體或賦形劑(包括黏合劑、崩解劑、控釋聚合物、潤滑劑、稀釋劑及/或著色劑)組合來製備。矯味劑及甜味劑尤其可用於形成咀嚼錠及口含錠。
本文所提供之醫藥組成物可以軟膠囊或硬膠囊來提供,其可自明膠、甲基纖維素、澱粉或藻酸鈣來製造。硬明膠膠囊(亦稱為乾填充膠囊(DFC))由兩部分組成,一部分套在另一部分上,因此完全包覆活性成分。軟彈性膠囊(SEC)為軟球形外殼,諸如明膠外殼,其係藉由添加甘油、山梨醇或類似多元醇來塑化。軟明膠外殼可含有防腐劑以防止微生物生長。適當防腐劑為彼等如本文所述者,包括對羥基苯甲酸甲酯和對羥基苯甲酸丙酯、及山梨酸。可將本文所提供之液體、半固體及固體劑型封裝於膠囊中。適當液體及半固體劑型包括在碳酸丙二酯、植物油或三酸甘油酯中之溶液及懸浮液。含有該等溶液之膠囊可如美國專利第4,328,245號;第4,409,239號和第4,410,545號中所述製備。該等膠囊亦可如熟習該項技術者已知的來塗覆以改良或維持活性成分之溶解。
本文所提供之醫藥組成可以液體及半固體劑型(包括乳液、溶液、懸浮液、酏劑及糖漿)提供。乳液為二相系統,其中一種液體以小球體型式分散遍佈於另一種液體中,其可為水包油或油包水。乳液可包括醫藥上可接受的非水性液體或溶劑、乳化劑、和防腐劑。懸浮液可包括醫藥上可接受的懸浮劑及防腐劑。水性醇溶液可包括醫藥上可接受的縮醛,諸如低碳烷醛之二(低碳烷基)縮醛,例如二乙醇縮乙醛;及具有一或多個羥基之水互溶性溶劑,諸如丙二醇及乙醇。酏劑為透明、甜味化及水醇性溶液。糖漿為糖(例如蔗糖)之濃水溶液,且亦可含有防腐劑。就液體劑型而言,例如,在聚乙二醇中之溶液可以足夠量之醫藥上可接受的液體載體(例如水)稀釋以便於量測投予。
其他可用液體及半固體劑型包括但不限於含有本文所提供之活性成分及下列二烷基化的單-或聚-伸烷基二醇的劑型:1,2-二甲氧基甲烷、二甘醇二甲醚、三甘醇二甲醚、四甘醇二甲醚、聚乙二醇-350-二甲醚、聚乙二醇-550-二甲醚、聚乙二醇-750-二甲醚,其中350、550及750係指聚乙二醇之大約平均分子量。此等調配物可另外包含一或多種抗氧化劑,諸如丁基羥基甲苯(BHT)、丁基羥基甲氧苯(BHA)、沒食子酸丙酯、維生素E、氫醌、羥基香豆素、乙醇胺、卵磷脂、腦磷脂、抗壞血酸、蘋果酸、山梨醇、磷酸、亞硫酸氫鹽、偏亞硫酸氫鈉、硫二丙酸及其酯、和二硫胺甲酸鹽。
在適當的情況下,用於口服投予的劑量單位調配物可微膠囊化。調配物亦可製備以延長或維持釋放,例如藉由將顆粒材料塗覆或嵌入聚合物、蠟、或類似物中。
本文所提供之用於口服投予的醫藥組成物亦可以脂質體、微胞、微球體或奈米系統型式提供。微胞劑型可如美國專利第6,350,458號中所述製備。
本文所提供之醫藥組成物可以非發泡性或發泡性顆粒及粉劑提供,以復原為液體劑型。用於非發泡性顆粒或粉劑之醫藥上可接受的載體及賦形劑可包括稀釋劑、甜味劑及潤濕劑。用於發泡性顆粒或粉劑之醫藥上可接受的載體及賦形劑可包括有機酸及二氧化碳來源。
著色劑及矯味劑可用於所有上述劑型中。
本文所揭示之化合物也可偶合至作為標靶藥物載體之可溶性聚合物。該等聚合物可包括經棕櫚醯基取代之聚乙烯吡咯啶酮、哌喃共聚物、聚羥丙基甲基丙烯醯胺基酚、聚羥乙基天冬醯胺酚或聚環氧乙烷聚離胺酸。該等化合物此外可偶合至適合於達成藥物控釋的可生物降解聚合物類,例如聚乳酸、聚-ε-己內酯、聚羥基丁酸、聚原酸酯、聚縮醛、聚二羥基哌喃、聚氰基丙烯酸酯、及水凝膠的交聯或兩性嵌段共聚物。
本文所提供之醫藥組成物可調配成立即或改良釋放劑型,包括延遲-、持續-、脈衝-、受控-、標靶-及程式化釋放形式。
本文所提供之醫藥組成物可與不損害所需治療作用的其他活性成分或與補充所需作用的物質共同調配。
本文所提供之醫藥組成物可藉由用於局部或全身性投予之注射、輸注或植入來腸胃外投予。胃腸外投予,在本文中,包括靜脈內、動脈內、腹膜內、鞘內、心室內、尿道內、胸骨內、顱內、肌肉內、滑膜內、及皮下投予。
本文所提供之醫藥組成物可調配成適合於胃腸外投予之任何劑型,包括溶液、懸浮液、乳液、微胞、脂質體、微球體、奈米系統、及適用於在注射前於液體中之溶液或懸浮液的固體形式。該等劑型可根據熟習醫藥科學之技術者已知的習用方法來製備(參見Remington:The Science and Practice of Pharmacy,同上)。
意欲用於胃腸外投予之醫藥組成物可包括一或多種醫藥上可接受的載體及賦形劑,包括但不限於水性載劑、水互溶性載劑、非水性載劑、抵抗微生物生長之抗微生物劑或防腐劑、穩定劑、溶解度增強劑、等滲劑、緩衝劑、抗氧化劑、局部麻醉劑、懸浮及分散劑、潤濕或乳化劑、錯合劑、鉗合或螯合劑、冷凍保護劑、凍乾保護劑、增稠劑、pH調節劑、及惰性氣體。
適當水性載劑包括但不限於水、鹽水、生理鹽水或磷酸鹽緩衝鹽水(PBS)、氯化鈉注射液、林格氏注射液、等滲右旋糖注射液、無菌水注射液、右旋糖及乳酸林格氏注射液。非水性載劑包括但不限於植物源不揮發油、蓖麻油、玉米油、棉籽油、橄欖油、花生油、薄荷油、紅花子油、芝麻油、大豆油、氫化植物油、氫化大豆油及椰子油之中鏈三酸甘油酯、棕櫚籽油。水互溶性載劑包括但不限於乙醇、1,3-丁二醇、液體聚乙二醇(例如聚乙二醇300及聚乙二醇400)、丙二醇、甘油、N-甲基-2-吡咯啶酮、N,N-二甲基乙醯胺、及二甲亞碸。
適當抗微生物劑或防腐劑包括但不限於酚、甲酚、汞劑、苯甲醇、氯丁醇、對羥苯甲酸甲酯及對羥苯甲酸丙酯、硫柳汞(thimerosal)、氯化苄烷銨(benzalkonium chloride,例如氯化苄甲乙氧銨(benzethonium chloride))、對羥苯甲酸甲酯及對羥苯甲酸丙酯、及山梨酸。適當等滲劑包括但不限於氯化鈉、甘油及右旋糖。適當緩衝液包括但不限於磷酸鹽及檸檬酸鹽。適當抗氧化劑為彼等本文中所述者,包括亞硫酸氫鹽及偏亞硫酸氫鈉。適當局部麻醉劑包括但不限於普羅卡因鹽酸鹽(procaine hydrochloride)。適當懸浮及分散劑為彼等本文所述者,包括羧甲基纖維素鈉、羥丙基甲基纖維素及聚乙烯基吡咯啶酮。適當乳化劑包括彼等本文所述者,包括聚氧乙烯山梨醇酐單月桂酸酯、聚氧乙烯山梨醇酐單油酸酯80及三乙醇胺油酸酯。適當鉗合或螯合劑包括但不限於EDTA。適當pH調節劑包括但不限於氫氧化鈉、鹽酸、檸檬酸及乳酸。適當錯合劑包括但不限於環糊精,包括α-環糊精、β-環糊精、羥丙基-β-環糊精、磺酸基丁基醚-β-環糊精、及磺酸基丁基醚7-β-環糊精(CAPTISOL
®、CyDex、Lenexa、Kans.)。
本文所提供之醫藥組成物可調配成用於單劑量或多劑量投予。單劑量調配物包裝於安瓿瓶、小瓶或注射器中。多劑量腸胃外調配物必須含有細菌或真菌抑制濃度的抗微生物劑。如該項技術中已知及實施的,所有腸胃外調配物必須無菌。
在一個實施態樣中,醫藥組成物係以備用無菌溶液來提供。在另一實施態樣中,醫藥組成物係以在使用前以無菌載劑復原之無菌乾燥可溶性產物提供,包括凍乾粉及皮下錠。在又另一實施態樣中,醫藥組成物係以備用無菌懸浮液提供。在又另一實施態樣中,醫藥組成物係以在使用前用載劑復原之無菌乾燥不溶性產物提供。在仍另一實施態樣中,醫藥組成物係以備用無菌乳液提供。
醫藥組成物可調配成用於以植入貯庫投予之懸浮液、固體、半固體或觸變性液體。在一個實施態樣中,本文所提供之醫藥組成物係分散於固體內基質中,該內基質被不溶於體液但允許醫藥組成物中之活性成分擴散通過之外聚合物膜所包圍。
適當的內基質包括聚甲基丙烯酸甲酯、聚甲基丙烯酸丁酯、塑化或未塑化聚氯乙烯、塑化耐綸、塑化聚對苯二甲酸乙二酯、天然橡膠、聚異戊二烯、聚異丁烯、聚丁二烯、聚乙烯、乙烯-乙酸乙烯酯共聚物、矽氧橡膠、聚二甲基矽氧烷、矽氧碳酸酯共聚物、親水聚合物(諸如丙烯酸酯及甲基丙烯酸酯之水凝膠)、膠原、交聯聚乙烯醇、及交聯且部分水解之聚乙酸乙烯酯。
適當的外聚合物膜包括聚乙烯、聚丙烯、乙烯/丙烯共聚物、乙烯/丙烯酸乙酯共聚物、乙烯/乙酸乙烯酯共聚物、矽氧橡膠、聚二甲基矽氧烷、氯丁二烯橡膠、氯化聚乙烯、聚氯乙烯、氯乙烯與乙酸乙烯酯之共聚物、二氯亞乙烯、乙烯及丙烯離聚物、聚對苯二甲酸乙二酯、丁基橡膠、環氧氯丙烷橡膠、乙烯/乙烯醇共聚物、乙烯/乙酸乙烯酯/乙烯醇三元共聚物、及乙烯/乙烯氧基乙醇共聚物。
在其他態樣中,本發明之醫藥組成物係製備成適合經由吸入投予至患者的劑型,例如乾粉、氣霧劑、懸浮液、或溶液組成物。在一個實施態樣中,本發明關於一種適合於藉由吸入而以乾粉投予至患者的劑型。在一個實施態樣中,本發明關於一種適合於藉由吸入而以乾粉投予至患者的劑型。藉由吸入遞送至肺部的乾粉組成物通常包含呈細碎粉末的本文所揭示之化合物或其醫藥上可接受的鹽類與一或多種呈細碎粉末之醫藥上可接受的賦形劑一起。特別適合使用於乾粉之醫藥上可接受的賦形劑為熟習該項技術者已知的且包括乳糖、澱粉、甘露糖醇和單醣、雙醣以及多醣。細碎粉末可藉由例如微粉化和研磨來製備。通常,尺寸減小(例如微粉化)的化合物可藉由約1至約10微米的D
50值(例如使用雷射繞射測量)來定義。
氣霧劑可藉由將本文所揭示之化合物或其醫藥上可接受的鹽類懸浮或溶解在液化推進劑中來形成。適當的推進劑包括鹵碳、烴及其他液化氣體。代表性推進劑包括:三氯氟甲烷(推進劑11)、二氯氟甲烷(推進劑12)、二氯四氟乙烷(推進劑114)、四氟乙烷(HFA-134a)、1,1-二氟乙烷(HFA-152a)、二氟甲烷(HFA-32)、五氟乙烷(HFA-12)、七氟丙烷(HFA-227a)、全氟丙烷、全氟丁烷、全氟戊烷、丁烷、異丁烷、和戊烷。包含式(A)化合物或其醫藥上可接受的鹽類之氣霧劑通常可經由計量吸入器(MDI)投予至患者。該等裝置為熟習該項技術者已知的。
該氣霧劑可含有通常與MDI一起使用之額外醫藥上可接受的賦形劑諸如界面活性劑、潤滑劑、共溶劑及其他賦形劑,以改良調配物之物理穩定性、改良閥性能、改良溶解性或改良味道。
適於經皮投予之醫藥組成物可以意欲保持與患者表皮長期密切接觸之獨立貼片存在。例如,活性成分可如Pharmaceutical Research, 3(6), 318(1986)中一般所述藉由離子電滲法自貼片遞送。
適於局部投予之醫藥組成物可調配成軟膏、霜劑、懸浮液、洗劑、粉劑、溶液、糊劑、凝膠、噴霧、氣霧劑或油狀物。軟膏、霜劑及凝膠例如可用水性或油性基質且添加適當增稠劑及/或膠凝劑及/或溶劑來調配。該等基質因此例如可包括水及/或油(諸如液狀石蠟或植物油(諸如花生油或蓖麻油))或溶劑(例如聚乙二醇)。根據基質之性質可使用的增稠劑及膠凝劑包括軟石蠟、硬脂酸鋁、鯨蠟硬脂醇、聚乙二醇、羊毛脂、蜂臘、羧聚乙烯(carboxypolymethylene)和纖維素衍生物、及/或甘油單硬脂酸酯及/或非離子乳化劑。
洗劑可用水性或油性基質調配且通常亦含有一種或多種乳化劑、穩定劑、分散劑、懸浮劑或增稠劑。
供外部施用之粉劑可藉助任何適當粉末基質(例如滑石、乳糖或澱粉)來形成。滴劑可用水性或非水性基質調配且亦包含一或多種分散劑、助溶劑、懸浮劑或防腐劑。
局部製劑可經由每日施用一次或多次至感染區域來投予;可有利地使用封閉敷料覆蓋皮膚區。可經由黏合劑儲存系統達成連續或長期遞送。
本發明之化合物及組成物的用途
本文所揭示之本發明的化合物或醫藥組成物可用於製造供治療、預防、改善或緩和對象的病症或疾病之藥物、以及其他用於調節(例如,拮抗)H3R之藥物,且本發明之化合物具有優越的藥物動力學及藥效學性質、毒副作用較少。
具體而言,本發明組成物之化合物的量可有效且可檢測地調節(例如,拮抗)H3R。本發明之化合物或醫藥組成物可用於預防、治療、或緩解與H3R有關之疾病,其中該等疾病包括CNS病症或嗜睡症。
治療
在一個實施態樣中,本文揭示之治療包含將安全有效劑量之本發明化合物或含有本發明化合物的醫藥組成物投予至有需要的患者。本文揭示的各實例包含治療上述疾病之方法,包含將安全有效量之本發明化合物或含有本發明化合物的醫藥組成物投予至有需要的患者。
在一個實施態樣中,本發明化合物或其醫藥組成物可藉由適當投予途徑來投予,包括全身性投予或局部投予。全身性投予包括口服投予、胃腸外投予、經皮投予及直腸投予。胃腸外投予係指除經腸或經皮外之投予途徑且通常藉由注射或輸注。胃腸外投予包括靜脈內、肌內、和皮下注射或輸注。局部投予包括施用於皮膚以及眼內、陰道內、吸入及鼻內投予。在一個實施態樣中,本發明化合物或其醫藥組成物可口服投予。在另一實施態樣中,本發明化合物或其醫藥組成物可藉由吸入投予。在另一實施態樣中,本發明化合物或其醫藥組成物可鼻內投予。
在一個實施態樣中,本發明化合物或其醫藥組成物可投予一次或根據投予方案(dosing regimen)投予,其中多劑量係在一段時間內以不同時間間隔投予。例如,劑量可每天投予至一、二、三、或四次。在一個實施態樣中,劑量為每天投予一次。在另一實施態樣中,劑量為每天投予兩次。劑量可投予直到達到所需治療效果或無限期地維持所需治療效果。本發明化合物或其醫藥組成物的適當投予方案取決於化合物的藥物動力學性質,諸如其吸收、分佈、及代謝和消除的半衰期,彼等可由技術人員測定。此外,適合於本發明化合物或其醫藥組成物之投予方案(包括在投予該方案期間)取決於待治療的病症、待治療的病症之嚴重性、待治療的患者之年齡及身體狀況、待治療患者的病史、並行治療的性質、所需治療的效果、在技術人員的知識及專業中的類似因素。該等技術人員將進一步理解特定個體患者對投予方案的耐受性或隨著時間的推移,當個體患者需要改變時,適當投予方案可能需要調整。
本發明化合物可與一或多種其他治療劑一起、或之前、或之後投予。本發明化合物可藉由相同或不同投予途徑分開投予,或者與其他藥劑一起在相同的醫藥組成物中一起投予。
本發明之醫藥組成物或組合就約50-70kg個體而言可為約1-1000mg的活性成分,較佳為約1-500mg或約1-250mg或約1-150mg或約0.5-100mg或約1-50mg的活性成分。化合物、醫藥組成物、或其組合的治療有效劑量係取決於對象的種類、體重、年齡和個體狀況、待治療的障礙或疾病或其嚴重性。普通技術的醫師、臨床醫生或獸醫可容易地確定預防,治療或抑制障礙或疾病進展所需的各活性成分之有效量。
以上引用的劑量性質與使用有利的哺乳動物(例如小鼠、大鼠、狗、非人靈長類,諸如猴或其分離的器官、組織及其製劑)之活體外和活體內試驗相關。本發明化合物可以溶液形式(例如較佳水溶液)體外施用,及經由局部、吸入、腸內或腸胃外、有利地靜脈內(例如懸浮液或水溶液)活體內施用。
在一個實施態樣中,本文揭示之化合物的治療有效劑量為每日從約0.1mg至約1,000mg。醫藥組成物應提供約0.1mg至約1,000mg的化合物之劑量。在一特定實施態樣中,製備醫藥劑量單位形式以提供從約1mg至約1,000mg、約10mg至約500mg、約20mg至約200mg、約25mg至約100mg、或約30mg至約60mg的活性成分或每劑量單位形式的必需成分的組合。在一特定實施態樣中,製備醫藥劑量單位形式以提供約1mg、5mg、10mg、20mg、25mg、50mg、100mg、250mg、500mg、1000mg的活性成分。
本發明之較佳實施態樣
一般合成程序
提供下列實施例以使可更充分地理解本發明。然而,應該理解該等實施態樣僅提供實踐本發明之方法,且本發明不限於該等實施態樣。
通常,本文揭示之化合物可藉由本文所述的方法製備,其中取代基係如上述式(A)所定義,除非另有說明。提供下列非限制性流程和實施例以進一步舉例說明本發明。
熟習該項技術之專業人員將理解所述的化學反應可容易地改變而適合於製備本文所揭示之許多其他化合物,及用於製備本文所揭示之化合物的替代方法被視為在本文所揭示的範圍內。彼等具有該項技術者將理解如下列實施例所證明的,可改變用以製造本發明所涵蓋的化合物之起始材料和額外步驟。在一些情況下,某些反應官能性之保護可能是達成一些上述轉變必需的。通常,對於熟習有機合成的技術者將顯而易見保護基團的該需求以及附接和去除該等基團所需的條件。例如,根據本發明之非示例性化合物的合成可藉由熟習該項技術者顯而易見的修改(例如藉由適當地保護干擾基團,藉由利用除了所描述者之外的該技術中已知的其他適當試劑,及/或藉由進行反應條件之習知修改)而成功地進行。或者,已知的反應條件或本發明中所揭示的反應將被認為具有製備本文揭示之其他化合物的適用性。
在下述實施例中,除非另有指示,否則所有的溫度定以攝氏度陳述。從商品供應商諸如Aldrich Chemical Company、Arcos Chemical Company、Alfa Aesar Chemical Company和J&K Chemical Company購買試劑,且除非另有指示,否則使用時都未經過進一步純化。
化合物的製備
本發明化合物(包括其鹽類、酯、水合物、或其溶劑合物)可使用任何已知的有機合成技術來製備,並且可根據許多可能的合成途徑中之任一者來合成。
製備本發明化合物之反應可在由熟習有機合成的技術者容易地選擇之適當溶劑中進行。適當溶劑可實質上不與起始材料(反應物)、中間物或產物在進行反應之溫度(例如可在溶劑之冷凍溫度至溶劑之沸騰溫度的範圍內之溫度)下進行。既定反應可在一種溶劑或一種以上溶劑之混合物中進行。視特定反應步驟而定,適合於特定反應步驟之溶劑可由技術嫻熟的技工選擇。
可根據該項技術中已知的任何適當方法監測反應。例如,產物形成可藉由光譜方法進行監測,諸如核磁共振光譜法(例如
1H或
13C)、紅外光譜法、分光光度測定法(例如UV-可見光)、質譜、或藉由層析方法諸如高效液相層析(HPLC)、液相層析-質譜(LCMS)或薄層層析(TLC)。化合物可由熟習該項技術者以多種方法,包括高效液相層析(HPLC)(“Preparative LC-MS Purification: Improved Compound Specific Method Optimization”Karl F. Blom, Brian Glass, Richard Sparks, Andrew P. Combs J. Combi. Chem. 2004, 6(6), 874-883,其以全文引用的方式併入本文中)及正相矽膠層析進行純化。
本發明化合物可使用下述方法,與合成有機化學技術中已知的合成方法或熟習該項技術者所理解的其變體來合成。較佳的方法包括但不限於下述彼等方法。具體而言,本發明之式(B~D)化合物可藉由按照下列示例性一般合成流程中概述的步驟合成,且合成流程中包括的反應物或反應物的化學基團之縮寫係定義在實施例中。
關於具有式(B)、(C)和(D)之化合物的一般合成流程(1-7)如下所示:
流程1:具有式(B)之化合物的一般合成
在另一態樣中,本文中提供一種包含本發明化合物之醫藥組成物。
在一個實施態樣中,醫藥組成物進一步包含醫藥上可接受的載體、佐劑、載劑或其組合。
在一個實施態樣中,醫藥組成物進一步包含一或多其他種治療劑,及其中該其他種治療劑係用於治療CNS病症或嗜睡症。
在一個實施態樣中,CNS病症為認知、精神、神經運動或疼痛。
在一個實施態樣中,其他治療劑係進一步包含:選自由多巴胺受體拮抗劑、血清素-去甲基腎上腺素再吸收抑制劑、選擇性血清素再吸收抑制劑、去甲基腎上腺素激導性再吸收抑制劑、非選擇性血清素再吸收抑制劑、及乙醯膽鹼酯酶抑制劑所組成群組之活性成分。
在另一態樣中,本文提供化合物或醫藥組成物之用途,其係用於製造供預防、管理、治療或減輕患者中CNS病症或嗜睡症的藥物。
在另一態樣中,本文提供化合物或醫藥組成物之用途,其係用於製造供拮抗H3受體的藥物。
在另一態樣中,本文提供化合物或醫藥組成物,其係用於預防、管理、治療或減輕患者中由病毒感染引起的CNS病症或嗜睡症的藥物。
在另一態樣中,本文提供用於拮抗H3受體之化合物或醫藥組成物。
在另一態樣中,本文提供一種預防、管理、治療或減輕患者中CNS病症或嗜睡症之方法,其包含將治療有效量的該化合物或該醫藥組成物投予至有需要的患者。
在另一態樣中,本文提供一種在患者中拮抗H3受體之方法,其包含將治療有效量的該化合物或該醫藥組成物投予至有需要的患者。
在又另一態樣中,本發明係關於製造式(A)~(D)之化合物和其醫藥上可接受的鹽類之方法。
本發明化合物的醫藥組成物及製備和投予
本發明提供一種醫藥組成物,其包含本發明化合物,例如,實施例化合物。根據本發明的特定實例,醫藥組成物可進一步包含醫藥上可接受的賦形劑、載體、佐劑、溶劑及其組合。
本發明提供一種治療、預防或改善疾病或病症之方法,其包含投予安全有效量之含有本發明化合物及一或多種治療活性劑的組合。其中,藥物的組合包含一或多個用於治療CNS病症或嗜睡症的其他藥物。
用於治療CNS病症或嗜睡症的其他藥物包括但不限於:進一步包含:選自由多巴胺受體拮抗劑、血清素-去甲基腎上腺素再吸收抑制劑、選擇性血清素再吸收抑制劑、去甲基腎上腺素激導性再吸收抑制劑、非選擇性血清素再吸收抑制劑、及乙醯膽鹼酯酶抑制劑所組成群組之活性成分。
本文揭示之醫藥組成物的化合物之量係指可有效檢測到調節生物學樣品及患者之功能失調H3R的量。活性成分可將提供最佳藥物療效(其不限於所要治療效果、投予途徑和治療期間)的劑量投予至需要該治療的對象。取決於疾病的性質以及嚴重性、患者的體重、患者接著所遵循的特殊飲食、同時用藥及熟習該項技術者將理解的其他因素,劑量將隨患者而異。根據具體應用,在製劑的單位劑量中之活性化合物的量可在從約1mg至約1000mg,較佳從約1mg至約500mg,更佳從約1mg至約300mg,仍更佳從約1mg至約200mg改變或調整。
所使用的實際劑量可取決於患者的需求和治療病況的嚴重性而改變。用於特定情況的適當劑量方案的測定在該項技術範圍內。為方便起見,可根據需求將每日總劑量分開且在一天內分批投予。本發明化合物及/或其醫藥上可接受的鹽類之投予的量和頻率將根據臨床主治醫師考慮該等諸如患者之年齡、病況和大小以及所治療症狀的嚴重性之因素判斷來調整。用於口服投予之典型推薦的每日劑量方案範圍可在從約1mg/天至約300mg/天,較佳為10mg/天至200mg/天,其可以單劑量或多劑量投予。在又另一實施態樣中,每個患者每天約1mg至50mg。
也應理解本發明的一些化合物可以游離形式存在而用於治療,或適當的話,呈其醫藥上可接受的衍生物或前藥的形式。醫藥上可接受的衍生物包括醫藥上可接受的鹽類、酯、該等酯的鹽、或在投予至有需要的患者時,其直接或間接地提供如本文另外描述的化合物或其治療上有效的代謝物或殘餘物之任何其他加合物或衍生物。
本發明之醫藥組成物可以散裝形式製備及包裝,由此散裝形式可提取安全有效量的本文所揭示之式(A)化合物,並接著投予至患者,諸如以粉末或糖漿。通常,將介於0.0001至10mg/kg體重的劑量水平每日投予至患者以獲得功能失調H3R的有效調節。或者,本發明之醫藥組成物可以單位劑型製備及包裝,其中各個物理上分離單位含有安全有效量的本文揭示之式(A)化合物。當製備成單位劑型時,本發明之醫藥組成物通常含有從約0.5mg至1g,或1mg至500mg,或5mg至200mg,或更佳地,25mg至100mg的本發明化合物。
當本發明醫藥組成物除了本發明化合物之外亦含有一或多種其他活性成分時,本發明化合物對第二種活性成分的重量比可改變並且取決於各成分的有效劑量。因此,例如,當本發明化合物與另一個藥劑組合時,本發明化合物與其他藥劑的重量比通常範圍從約1000:1至約1:1000,諸如約200:1至1:200。本發明化合物與其他活性成分的組合通常也在上述範圍內,但在各情況下,應使用組合中各活性成分的有效劑量。
"醫藥上可接受的賦形劑"在本文中意指涉及賦予醫藥組成物形式或一致性的醫藥上可接受的材料、組成物或載劑(vehicle)。當混合時,各賦形劑必須與醫藥組成物的其他成分相容,使得當投予至患者時相互作用會實質上減少本發明化合物的療效並會導致醫藥上不可接受的組成物。另外,各賦形劑當然必須具有足夠高的純度以使其能為醫藥上可接受的。
適當醫藥上可接受的賦形劑將依據所選特定劑量形式而變化。另外,可選擇適當醫藥上可接受的賦形劑用於組成物中的特定功能。例如,可選擇一些具有促進製造均勻劑型之能力的醫藥上可接受的賦形劑。可選擇一些具有促進製造穩定劑型之能力的醫藥上可接受的賦形劑。可選擇一些當投予至患者時具有促進本發明的化合物從一個器官或身體的部位攜帶或傳遞至另一個器官或身體部位之能力的醫藥上可接受的賦形劑。可選擇一些具有提高患者依從性之能力的醫藥上可接受的賦形劑。
適當醫藥上可接受的賦形劑包括下列賦形劑之類型:稀釋劑、填充劑、黏合劑、崩解劑、潤滑劑、助滑劑、製粒劑、塗佈劑、潤濕劑、溶劑、共溶劑、懸浮劑、乳化劑、甜味劑、調味劑、味道掩蓋劑、著色劑、防結塊劑、濕潤劑、螯合劑、塑化劑、增黏劑、抗氧化劑、防腐劑、穩定劑、界面活性劑以及緩衝劑。熟悉該項技術者將理解一些醫藥上可接受的賦形劑可達成一種以上的功能且可依據調配物中存在多少賦形劑以及調配物中存在什麼其他成分而達成替代功能。
技術人員擁有該項技術的知識和技能以使他們能夠選擇適當醫藥上可接受的賦形劑以適當量用於本發明。此外,技術人員可獲得之描述醫藥上可接受的賦形劑且可用於選擇適當醫藥上可接受的賦形劑之辦法。實例包括Remington's Pharmaceutical Sciences(Mack Publishing Company)、The Handbook of Pharmaceutical Additives (Gower Publishing Limited)、及The Handbook of Pharmaceutical Excipients(the American Pharmaceutical Association and the Pharmaceutical Press)。
在Remington:The Science and Practice of Pharmacy, 21st edition, 2005, ed. D.B. Troy, Lippincott Williams & Wilkins, Philadelphia, 和Encyclopedia of Pharmaceutical Technology, eds. J. Swarbrick and J. C. Boylan, 1988-1999, Marcel Dekker, New York中,其各內容以引用方式併入本文中,揭示可用於調配醫藥上可接受的組成物之各種載體及其製備的已知技術。除非任何習知載體介質與本發明化合物不相容,諸如因產生的任何不良生物效應或以有害的方式與醫藥上可接受的組成物的任何其他組分相互作用,否則其使用預期在本發明的範圍內。
使用熟習該項技術者已知的技術及方法來製備本發明之醫藥組成物。該項技術常用的一些方法係描述於Remington's Pharmaceutical Sciences(Mack Publishing Company)中。
因此,本發明之另一態樣係關於一種製備醫藥組成物之方法。該醫藥組成物含有本文所揭示之化合物及醫藥上可接受的賦形劑、載體、佐劑、載劑或其組合,該方法包含混合各種成分。含有本文所揭示之化合物的醫藥組成物可在例如常溫和常壓下製備。
本發明化合物通常將調配成適用於藉由所需的投予途徑投予至患者的劑型。例如,劑型包括適用於(1) 口服投予,諸如錠劑、膠囊、膠囊型錠劑、丸劑、含片、粉劑、糖漿、酏劑、懸浮液、溶液、乳液、小藥囊及扁囊劑;(2) 胃腸外投予,諸如無菌溶液、懸浮液、及用於復原之粉劑;(3) 經皮投予,諸如經皮貼片;(4) 直腸投予,諸如栓劑;(5) 吸入,諸如氣霧劑、溶液及乾粉;以及(6) 局部投予,諸如霜劑、軟膏、洗劑、溶液、糊劑、噴霧劑、泡沫劑及凝膠之劑型。
本文所提供之醫藥組成物可以下列提供:壓縮錠、研製錠、可咀嚼口含錠、速溶錠、多重壓縮錠、或腸溶膜衣錠、糖衣錠或膜衣錠。腸溶膜衣錠為壓縮錠,其以可抵抗胃酸作用但在腸中溶解或崩解之物質塗覆,因此保護活性成分抵抗胃中之酸性環境。腸溶膜衣包括但不限於脂肪酸、脂肪、苯基水楊酸酯、蠟、蟲膠、胺化蟲膠、及乙酸纖維素鄰苯二甲酸。糖衣錠劑為由糖衣包圍之壓縮錠劑,該糖衣可有益於掩蓋不良味道或氣味且防止錠劑氧化。膜衣錠為以水溶性材料之薄層或薄膜覆蓋的壓縮錠。膜衣包括但不限於羥乙基纖維素、羧甲基纖維素鈉、聚乙二醇4000及乙酸纖維素鄰苯二甲酸。膜衣賦予與糖衣相同的一般特性。多重壓縮錠劑為藉由一個以上壓縮循環製成之壓縮錠,其包括分層錠、及壓製膜衣錠或乾壓膜衣錠。
錠劑劑型可自呈粉狀、結晶或粒狀形式之活性成分,單獨或與一或多種本文所述載體或賦形劑(包括黏合劑、崩解劑、控釋聚合物、潤滑劑、稀釋劑及/或著色劑)組合來製備。矯味劑及甜味劑尤其可用於形成咀嚼錠及口含錠。
本文所提供之醫藥組成物可以軟膠囊或硬膠囊來提供,其可自明膠、甲基纖維素、澱粉或藻酸鈣來製造。硬明膠膠囊(亦稱為乾填充膠囊(DFC))由兩部分組成,一部分套在另一部分上,因此完全包覆活性成分。軟彈性膠囊(SEC)為軟球形外殼,諸如明膠外殼,其係藉由添加甘油、山梨醇或類似多元醇來塑化。軟明膠外殼可含有防腐劑以防止微生物生長。適當防腐劑為彼等如本文所述者,包括對羥基苯甲酸甲酯和對羥基苯甲酸丙酯、及山梨酸。可將本文所提供之液體、半固體及固體劑型封裝於膠囊中。適當液體及半固體劑型包括在碳酸丙二酯、植物油或三酸甘油酯中之溶液及懸浮液。含有該等溶液之膠囊可如美國專利第4,328,245號;第4,409,239號和第4,410,545號中所述製備。該等膠囊亦可如熟習該項技術者已知的來塗覆以改良或維持活性成分之溶解。
本文所提供之醫藥組成可以液體及半固體劑型(包括乳液、溶液、懸浮液、酏劑及糖漿)提供。乳液為二相系統,其中一種液體以小球體型式分散遍佈於另一種液體中,其可為水包油或油包水。乳液可包括醫藥上可接受的非水性液體或溶劑、乳化劑、和防腐劑。懸浮液可包括醫藥上可接受的懸浮劑及防腐劑。水性醇溶液可包括醫藥上可接受的縮醛,諸如低碳烷醛之二(低碳烷基)縮醛,例如二乙醇縮乙醛;及具有一或多個羥基之水互溶性溶劑,諸如丙二醇及乙醇。酏劑為透明、甜味化及水醇性溶液。糖漿為糖(例如蔗糖)之濃水溶液,且亦可含有防腐劑。就液體劑型而言,例如,在聚乙二醇中之溶液可以足夠量之醫藥上可接受的液體載體(例如水)稀釋以便於量測投予。
其他可用液體及半固體劑型包括但不限於含有本文所提供之活性成分及下列二烷基化的單-或聚-伸烷基二醇的劑型:1,2-二甲氧基甲烷、二甘醇二甲醚、三甘醇二甲醚、四甘醇二甲醚、聚乙二醇-350-二甲醚、聚乙二醇-550-二甲醚、聚乙二醇-750-二甲醚,其中350、550及750係指聚乙二醇之大約平均分子量。此等調配物可另外包含一或多種抗氧化劑,諸如丁基羥基甲苯(BHT)、丁基羥基甲氧苯(BHA)、沒食子酸丙酯、維生素E、氫醌、羥基香豆素、乙醇胺、卵磷脂、腦磷脂、抗壞血酸、蘋果酸、山梨醇、磷酸、亞硫酸氫鹽、偏亞硫酸氫鈉、硫二丙酸及其酯、和二硫胺甲酸鹽。
在適當的情況下,用於口服投予的劑量單位調配物可微膠囊化。調配物亦可製備以延長或維持釋放,例如藉由將顆粒材料塗覆或嵌入聚合物、蠟、或類似物中。
本文所提供之用於口服投予的醫藥組成物亦可以脂質體、微胞、微球體或奈米系統型式提供。微胞劑型可如美國專利第6,350,458號中所述製備。
本文所提供之醫藥組成物可以非發泡性或發泡性顆粒及粉劑提供,以復原為液體劑型。用於非發泡性顆粒或粉劑之醫藥上可接受的載體及賦形劑可包括稀釋劑、甜味劑及潤濕劑。用於發泡性顆粒或粉劑之醫藥上可接受的載體及賦形劑可包括有機酸及二氧化碳來源。
著色劑及矯味劑可用於所有上述劑型中。
本文所揭示之化合物也可偶合至作為標靶藥物載體之可溶性聚合物。該等聚合物可包括經棕櫚醯基取代之聚乙烯吡咯啶酮、哌喃共聚物、聚羥丙基甲基丙烯醯胺基酚、聚羥乙基天冬醯胺酚或聚環氧乙烷聚離胺酸。該等化合物此外可偶合至適合於達成藥物控釋的可生物降解聚合物類,例如聚乳酸、聚-ε-己內酯、聚羥基丁酸、聚原酸酯、聚縮醛、聚二羥基哌喃、聚氰基丙烯酸酯、及水凝膠的交聯或兩性嵌段共聚物。
本文所提供之醫藥組成物可調配成立即或改良釋放劑型,包括延遲-、持續-、脈衝-、受控-、標靶-及程式化釋放形式。
本文所提供之醫藥組成物可與不損害所需治療作用的其他活性成分或與補充所需作用的物質共同調配。
本文所提供之醫藥組成物可藉由用於局部或全身性投予之注射、輸注或植入來腸胃外投予。胃腸外投予,在本文中,包括靜脈內、動脈內、腹膜內、鞘內、心室內、尿道內、胸骨內、顱內、肌肉內、滑膜內、及皮下投予。
本文所提供之醫藥組成物可調配成適合於胃腸外投予之任何劑型,包括溶液、懸浮液、乳液、微胞、脂質體、微球體、奈米系統、及適用於在注射前於液體中之溶液或懸浮液的固體形式。該等劑型可根據熟習醫藥科學之技術者已知的習用方法來製備(參見Remington:The Science and Practice of Pharmacy,同上)。
意欲用於胃腸外投予之醫藥組成物可包括一或多種醫藥上可接受的載體及賦形劑,包括但不限於水性載劑、水互溶性載劑、非水性載劑、抵抗微生物生長之抗微生物劑或防腐劑、穩定劑、溶解度增強劑、等滲劑、緩衝劑、抗氧化劑、局部麻醉劑、懸浮及分散劑、潤濕或乳化劑、錯合劑、鉗合或螯合劑、冷凍保護劑、凍乾保護劑、增稠劑、pH調節劑、及惰性氣體。
適當水性載劑包括但不限於水、鹽水、生理鹽水或磷酸鹽緩衝鹽水(PBS)、氯化鈉注射液、林格氏注射液、等滲右旋糖注射液、無菌水注射液、右旋糖及乳酸林格氏注射液。非水性載劑包括但不限於植物源不揮發油、蓖麻油、玉米油、棉籽油、橄欖油、花生油、薄荷油、紅花子油、芝麻油、大豆油、氫化植物油、氫化大豆油及椰子油之中鏈三酸甘油酯、棕櫚籽油。水互溶性載劑包括但不限於乙醇、1,3-丁二醇、液體聚乙二醇(例如聚乙二醇300及聚乙二醇400)、丙二醇、甘油、N-甲基-2-吡咯啶酮、N,N-二甲基乙醯胺、及二甲亞碸。
適當抗微生物劑或防腐劑包括但不限於酚、甲酚、汞劑、苯甲醇、氯丁醇、對羥苯甲酸甲酯及對羥苯甲酸丙酯、硫柳汞(thimerosal)、氯化苄烷銨(benzalkonium chloride,例如氯化苄甲乙氧銨(benzethonium chloride))、對羥苯甲酸甲酯及對羥苯甲酸丙酯、及山梨酸。適當等滲劑包括但不限於氯化鈉、甘油及右旋糖。適當緩衝液包括但不限於磷酸鹽及檸檬酸鹽。適當抗氧化劑為彼等本文中所述者,包括亞硫酸氫鹽及偏亞硫酸氫鈉。適當局部麻醉劑包括但不限於普羅卡因鹽酸鹽(procaine hydrochloride)。適當懸浮及分散劑為彼等本文所述者,包括羧甲基纖維素鈉、羥丙基甲基纖維素及聚乙烯基吡咯啶酮。適當乳化劑包括彼等本文所述者,包括聚氧乙烯山梨醇酐單月桂酸酯、聚氧乙烯山梨醇酐單油酸酯80及三乙醇胺油酸酯。適當鉗合或螯合劑包括但不限於EDTA。適當pH調節劑包括但不限於氫氧化鈉、鹽酸、檸檬酸及乳酸。適當錯合劑包括但不限於環糊精,包括α-環糊精、β-環糊精、羥丙基-β-環糊精、磺酸基丁基醚-β-環糊精、及磺酸基丁基醚7-β-環糊精(CAPTISOL
®、CyDex、Lenexa、Kans.)。
本文所提供之醫藥組成物可調配成用於單劑量或多劑量投予。單劑量調配物包裝於安瓿瓶、小瓶或注射器中。多劑量腸胃外調配物必須含有細菌或真菌抑制濃度的抗微生物劑。如該項技術中已知及實施的,所有腸胃外調配物必須無菌。
在一個實施態樣中,醫藥組成物係以備用無菌溶液來提供。在另一實施態樣中,醫藥組成物係以在使用前以無菌載劑復原之無菌乾燥可溶性產物提供,包括凍乾粉及皮下錠。在又另一實施態樣中,醫藥組成物係以備用無菌懸浮液提供。在又另一實施態樣中,醫藥組成物係以在使用前用載劑復原之無菌乾燥不溶性產物提供。在仍另一實施態樣中,醫藥組成物係以備用無菌乳液提供。
醫藥組成物可調配成用於以植入貯庫投予之懸浮液、固體、半固體或觸變性液體。在一個實施態樣中,本文所提供之醫藥組成物係分散於固體內基質中,該內基質被不溶於體液但允許醫藥組成物中之活性成分擴散通過之外聚合物膜所包圍。
適當的內基質包括聚甲基丙烯酸甲酯、聚甲基丙烯酸丁酯、塑化或未塑化聚氯乙烯、塑化耐綸、塑化聚對苯二甲酸乙二酯、天然橡膠、聚異戊二烯、聚異丁烯、聚丁二烯、聚乙烯、乙烯-乙酸乙烯酯共聚物、矽氧橡膠、聚二甲基矽氧烷、矽氧碳酸酯共聚物、親水聚合物(諸如丙烯酸酯及甲基丙烯酸酯之水凝膠)、膠原、交聯聚乙烯醇、及交聯且部分水解之聚乙酸乙烯酯。
適當的外聚合物膜包括聚乙烯、聚丙烯、乙烯/丙烯共聚物、乙烯/丙烯酸乙酯共聚物、乙烯/乙酸乙烯酯共聚物、矽氧橡膠、聚二甲基矽氧烷、氯丁二烯橡膠、氯化聚乙烯、聚氯乙烯、氯乙烯與乙酸乙烯酯之共聚物、二氯亞乙烯、乙烯及丙烯離聚物、聚對苯二甲酸乙二酯、丁基橡膠、環氧氯丙烷橡膠、乙烯/乙烯醇共聚物、乙烯/乙酸乙烯酯/乙烯醇三元共聚物、及乙烯/乙烯氧基乙醇共聚物。
在其他態樣中,本發明之醫藥組成物係製備成適合經由吸入投予至患者的劑型,例如乾粉、氣霧劑、懸浮液、或溶液組成物。在一個實施態樣中,本發明關於一種適合於藉由吸入而以乾粉投予至患者的劑型。在一個實施態樣中,本發明關於一種適合於藉由吸入而以乾粉投予至患者的劑型。藉由吸入遞送至肺部的乾粉組成物通常包含呈細碎粉末的本文所揭示之化合物或其醫藥上可接受的鹽類與一或多種呈細碎粉末之醫藥上可接受的賦形劑一起。特別適合使用於乾粉之醫藥上可接受的賦形劑為熟習該項技術者已知的且包括乳糖、澱粉、甘露糖醇和單醣、雙醣以及多醣。細碎粉末可藉由例如微粉化和研磨來製備。通常,尺寸減小(例如微粉化)的化合物可藉由約1至約10微米的D
50值(例如使用雷射繞射測量)來定義。
氣霧劑可藉由將本文所揭示之化合物或其醫藥上可接受的鹽類懸浮或溶解在液化推進劑中來形成。適當的推進劑包括鹵碳、烴及其他液化氣體。代表性推進劑包括:三氯氟甲烷(推進劑11)、二氯氟甲烷(推進劑12)、二氯四氟乙烷(推進劑114)、四氟乙烷(HFA-134a)、1,1-二氟乙烷(HFA-152a)、二氟甲烷(HFA-32)、五氟乙烷(HFA-12)、七氟丙烷(HFA-227a)、全氟丙烷、全氟丁烷、全氟戊烷、丁烷、異丁烷、和戊烷。包含式(A)化合物或其醫藥上可接受的鹽類之氣霧劑通常可經由計量吸入器(MDI)投予至患者。該等裝置為熟習該項技術者已知的。
該氣霧劑可含有通常與MDI一起使用之額外醫藥上可接受的賦形劑諸如界面活性劑、潤滑劑、共溶劑及其他賦形劑,以改良調配物之物理穩定性、改良閥性能、改良溶解性或改良味道。
適於經皮投予之醫藥組成物可以意欲保持與患者表皮長期密切接觸之獨立貼片存在。例如,活性成分可如Pharmaceutical Research, 3(6), 318(1986)中一般所述藉由離子電滲法自貼片遞送。
適於局部投予之醫藥組成物可調配成軟膏、霜劑、懸浮液、洗劑、粉劑、溶液、糊劑、凝膠、噴霧、氣霧劑或油狀物。軟膏、霜劑及凝膠例如可用水性或油性基質且添加適當增稠劑及/或膠凝劑及/或溶劑來調配。該等基質因此例如可包括水及/或油(諸如液狀石蠟或植物油(諸如花生油或蓖麻油))或溶劑(例如聚乙二醇)。根據基質之性質可使用的增稠劑及膠凝劑包括軟石蠟、硬脂酸鋁、鯨蠟硬脂醇、聚乙二醇、羊毛脂、蜂臘、羧聚乙烯(carboxypolymethylene)和纖維素衍生物、及/或甘油單硬脂酸酯及/或非離子乳化劑。
洗劑可用水性或油性基質調配且通常亦含有一種或多種乳化劑、穩定劑、分散劑、懸浮劑或增稠劑。
供外部施用之粉劑可藉助任何適當粉末基質(例如滑石、乳糖或澱粉)來形成。滴劑可用水性或非水性基質調配且亦包含一或多種分散劑、助溶劑、懸浮劑或防腐劑。
局部製劑可經由每日施用一次或多次至感染區域來投予;可有利地使用封閉敷料覆蓋皮膚區。可經由黏合劑儲存系統達成連續或長期遞送。
本發明之化合物及組成物的用途
本文所揭示之本發明的化合物或醫藥組成物可用於製造供治療、預防、改善或緩和對象的病症或疾病之藥物、以及其他用於調節(例如,拮抗)H3R之藥物,且本發明之化合物具有優越的藥物動力學及藥效學性質、毒副作用較少。
具體而言,本發明組成物之化合物的量可有效且可檢測地調節(例如,拮抗)H3R。本發明之化合物或醫藥組成物可用於預防、治療、或緩解與H3R有關之疾病,其中該等疾病包括CNS病症或嗜睡症。
治療
在一個實施態樣中,本文揭示之治療包含將安全有效劑量之本發明化合物或含有本發明化合物的醫藥組成物投予至有需要的患者。本文揭示的各實例包含治療上述疾病之方法,包含將安全有效量之本發明化合物或含有本發明化合物的醫藥組成物投予至有需要的患者。
在一個實施態樣中,本發明化合物或其醫藥組成物可藉由適當投予途徑來投予,包括全身性投予或局部投予。全身性投予包括口服投予、胃腸外投予、經皮投予及直腸投予。胃腸外投予係指除經腸或經皮外之投予途徑且通常藉由注射或輸注。胃腸外投予包括靜脈內、肌內、和皮下注射或輸注。局部投予包括施用於皮膚以及眼內、陰道內、吸入及鼻內投予。在一個實施態樣中,本發明化合物或其醫藥組成物可口服投予。在另一實施態樣中,本發明化合物或其醫藥組成物可藉由吸入投予。在另一實施態樣中,本發明化合物或其醫藥組成物可鼻內投予。
在一個實施態樣中,本發明化合物或其醫藥組成物可投予一次或根據投予方案(dosing regimen)投予,其中多劑量係在一段時間內以不同時間間隔投予。例如,劑量可每天投予至一、二、三、或四次。在一個實施態樣中,劑量為每天投予一次。在另一實施態樣中,劑量為每天投予兩次。劑量可投予直到達到所需治療效果或無限期地維持所需治療效果。本發明化合物或其醫藥組成物的適當投予方案取決於化合物的藥物動力學性質,諸如其吸收、分佈、及代謝和消除的半衰期,彼等可由技術人員測定。此外,適合於本發明化合物或其醫藥組成物之投予方案(包括在投予該方案期間)取決於待治療的病症、待治療的病症之嚴重性、待治療的患者之年齡及身體狀況、待治療患者的病史、並行治療的性質、所需治療的效果、在技術人員的知識及專業中的類似因素。該等技術人員將進一步理解特定個體患者對投予方案的耐受性或隨著時間的推移,當個體患者需要改變時,適當投予方案可能需要調整。
本發明化合物可與一或多種其他治療劑一起、或之前、或之後投予。本發明化合物可藉由相同或不同投予途徑分開投予,或者與其他藥劑一起在相同的醫藥組成物中一起投予。
本發明之醫藥組成物或組合就約50-70kg個體而言可為約1-1000mg的活性成分,較佳為約1-500mg或約1-250mg或約1-150mg或約0.5-100mg或約1-50mg的活性成分。化合物、醫藥組成物、或其組合的治療有效劑量係取決於對象的種類、體重、年齡和個體狀況、待治療的障礙或疾病或其嚴重性。普通技術的醫師、臨床醫生或獸醫可容易地確定預防,治療或抑制障礙或疾病進展所需的各活性成分之有效量。
以上引用的劑量性質與使用有利的哺乳動物(例如小鼠、大鼠、狗、非人靈長類,諸如猴或其分離的器官、組織及其製劑)之活體外和活體內試驗相關。本發明化合物可以溶液形式(例如較佳水溶液)體外施用,及經由局部、吸入、腸內或腸胃外、有利地靜脈內(例如懸浮液或水溶液)活體內施用。
在一個實施態樣中,本文揭示之化合物的治療有效劑量為每日從約0.1mg至約1,000mg。醫藥組成物應提供約0.1mg至約1,000mg的化合物之劑量。在一特定實施態樣中,製備醫藥劑量單位形式以提供從約1mg至約1,000mg、約10mg至約500mg、約20mg至約200mg、約25mg至約100mg、或約30mg至約60mg的活性成分或每劑量單位形式的必需成分的組合。在一特定實施態樣中,製備醫藥劑量單位形式以提供約1mg、5mg、10mg、20mg、25mg、50mg、100mg、250mg、500mg、1000mg的活性成分。
本發明之較佳實施態樣
一般合成程序
提供下列實施例以使可更充分地理解本發明。然而,應該理解該等實施態樣僅提供實踐本發明之方法,且本發明不限於該等實施態樣。
通常,本文揭示之化合物可藉由本文所述的方法製備,其中取代基係如上述式(A)所定義,除非另有說明。提供下列非限制性流程和實施例以進一步舉例說明本發明。
熟習該項技術之專業人員將理解所述的化學反應可容易地改變而適合於製備本文所揭示之許多其他化合物,及用於製備本文所揭示之化合物的替代方法被視為在本文所揭示的範圍內。彼等具有該項技術者將理解如下列實施例所證明的,可改變用以製造本發明所涵蓋的化合物之起始材料和額外步驟。在一些情況下,某些反應官能性之保護可能是達成一些上述轉變必需的。通常,對於熟習有機合成的技術者將顯而易見保護基團的該需求以及附接和去除該等基團所需的條件。例如,根據本發明之非示例性化合物的合成可藉由熟習該項技術者顯而易見的修改(例如藉由適當地保護干擾基團,藉由利用除了所描述者之外的該技術中已知的其他適當試劑,及/或藉由進行反應條件之習知修改)而成功地進行。或者,已知的反應條件或本發明中所揭示的反應將被認為具有製備本文揭示之其他化合物的適用性。
在下述實施例中,除非另有指示,否則所有的溫度定以攝氏度陳述。從商品供應商諸如Aldrich Chemical Company、Arcos Chemical Company、Alfa Aesar Chemical Company和J&K Chemical Company購買試劑,且除非另有指示,否則使用時都未經過進一步純化。
化合物的製備
本發明化合物(包括其鹽類、酯、水合物、或其溶劑合物)可使用任何已知的有機合成技術來製備,並且可根據許多可能的合成途徑中之任一者來合成。
製備本發明化合物之反應可在由熟習有機合成的技術者容易地選擇之適當溶劑中進行。適當溶劑可實質上不與起始材料(反應物)、中間物或產物在進行反應之溫度(例如可在溶劑之冷凍溫度至溶劑之沸騰溫度的範圍內之溫度)下進行。既定反應可在一種溶劑或一種以上溶劑之混合物中進行。視特定反應步驟而定,適合於特定反應步驟之溶劑可由技術嫻熟的技工選擇。
可根據該項技術中已知的任何適當方法監測反應。例如,產物形成可藉由光譜方法進行監測,諸如核磁共振光譜法(例如
1H或
13C)、紅外光譜法、分光光度測定法(例如UV-可見光)、質譜、或藉由層析方法諸如高效液相層析(HPLC)、液相層析-質譜(LCMS)或薄層層析(TLC)。化合物可由熟習該項技術者以多種方法,包括高效液相層析(HPLC)(“Preparative LC-MS Purification: Improved Compound Specific Method Optimization”Karl F. Blom, Brian Glass, Richard Sparks, Andrew P. Combs J. Combi. Chem. 2004, 6(6), 874-883,其以全文引用的方式併入本文中)及正相矽膠層析進行純化。
本發明化合物可使用下述方法,與合成有機化學技術中已知的合成方法或熟習該項技術者所理解的其變體來合成。較佳的方法包括但不限於下述彼等方法。具體而言,本發明之式(B~D)化合物可藉由按照下列示例性一般合成流程中概述的步驟合成,且合成流程中包括的反應物或反應物的化學基團之縮寫係定義在實施例中。
關於具有式(B)、(C)和(D)之化合物的一般合成流程(1-7)如下所示:
流程1:具有式(B)之化合物的一般合成
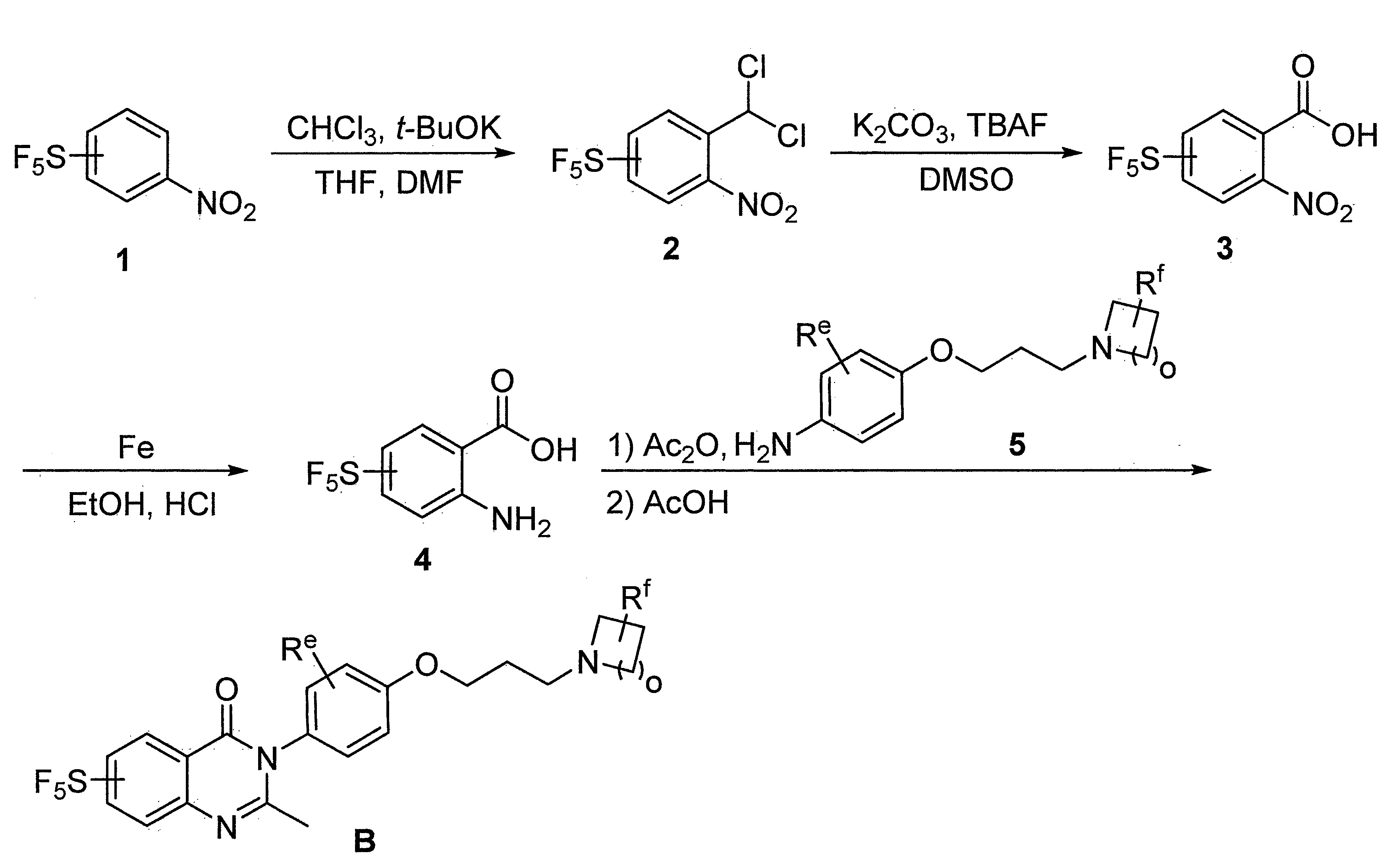 按照流程1,可根據參考文獻(Journal of Organic Chemistry, 2011, 76, 4781-4786;Tetrahedron Letters, 2004, 45, 1071-1074;Organic Letters, 2009, 11, 15, 3230-3233;Journal of Medicinal Chemistry, 2008, 51, 4780-4789;US2005182045A1)中所揭示的相關程序進行關於具有式(B)之化合物的合成,但不限於所揭示的這些程序。將硝基苯衍生物1用氯仿鹼化並接著水解以形成酸3,其以在HCl中之Fe還原而產生苯胺4。因此,酸4與5之縮合,接著環化反應提供化合物(B)。
流程2:中間化合物5的合成
按照流程1,可根據參考文獻(Journal of Organic Chemistry, 2011, 76, 4781-4786;Tetrahedron Letters, 2004, 45, 1071-1074;Organic Letters, 2009, 11, 15, 3230-3233;Journal of Medicinal Chemistry, 2008, 51, 4780-4789;US2005182045A1)中所揭示的相關程序進行關於具有式(B)之化合物的合成,但不限於所揭示的這些程序。將硝基苯衍生物1用氯仿鹼化並接著水解以形成酸3,其以在HCl中之Fe還原而產生苯胺4。因此,酸4與5之縮合,接著環化反應提供化合物(B)。
流程2:中間化合物5的合成
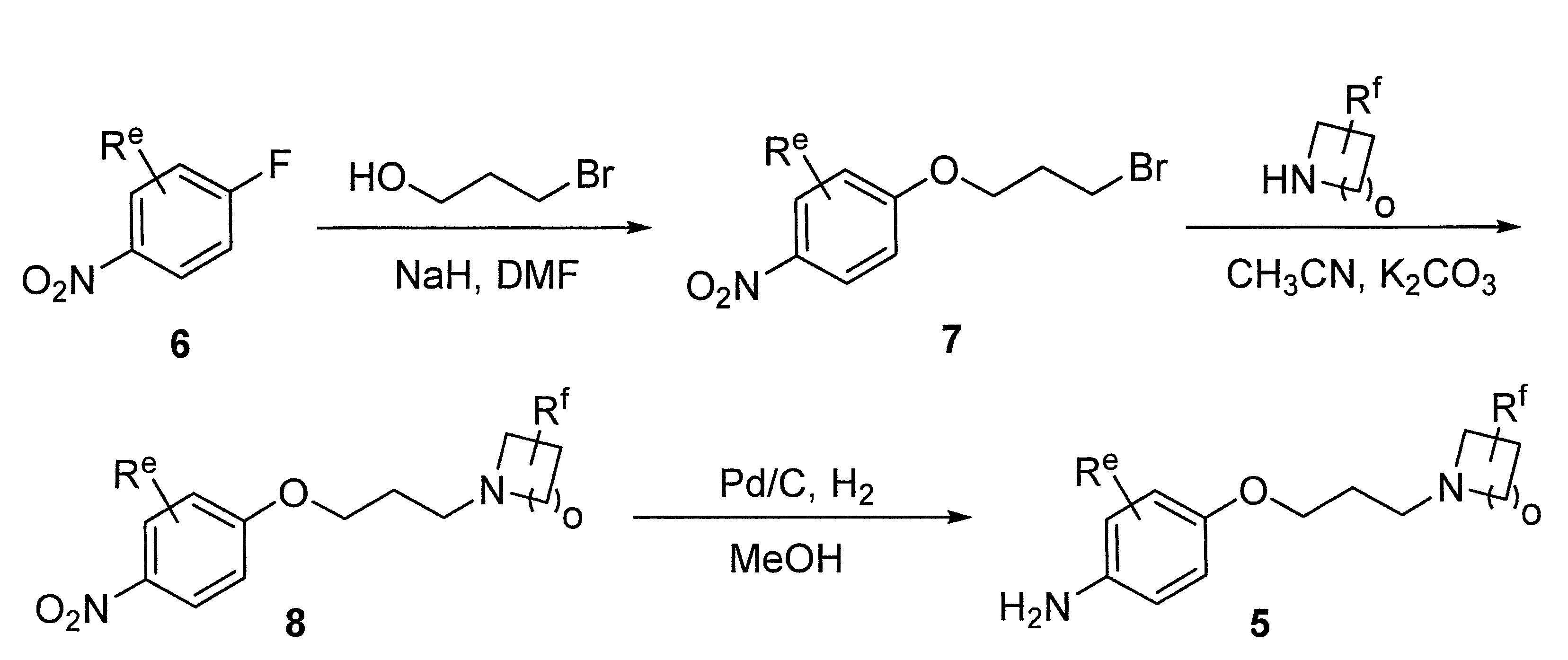 按照流程2,可根據參考文獻(US2005182045A1;WO2017201161A1)中所揭示的相關程序進行關於具有式(5)之化合物的合成,但不限於所揭示的這些程序。將氟苯衍生物6以羥烷基溴化物取代而產生苯基醚7,接著將其胺化為化合物8。因此,在Pd/C存在下的隨後氫化可提供中間物5。
流程3:中間化合物8的替代合成
按照流程2,可根據參考文獻(US2005182045A1;WO2017201161A1)中所揭示的相關程序進行關於具有式(5)之化合物的合成,但不限於所揭示的這些程序。將氟苯衍生物6以羥烷基溴化物取代而產生苯基醚7,接著將其胺化為化合物8。因此,在Pd/C存在下的隨後氫化可提供中間物5。
流程3:中間化合物8的替代合成
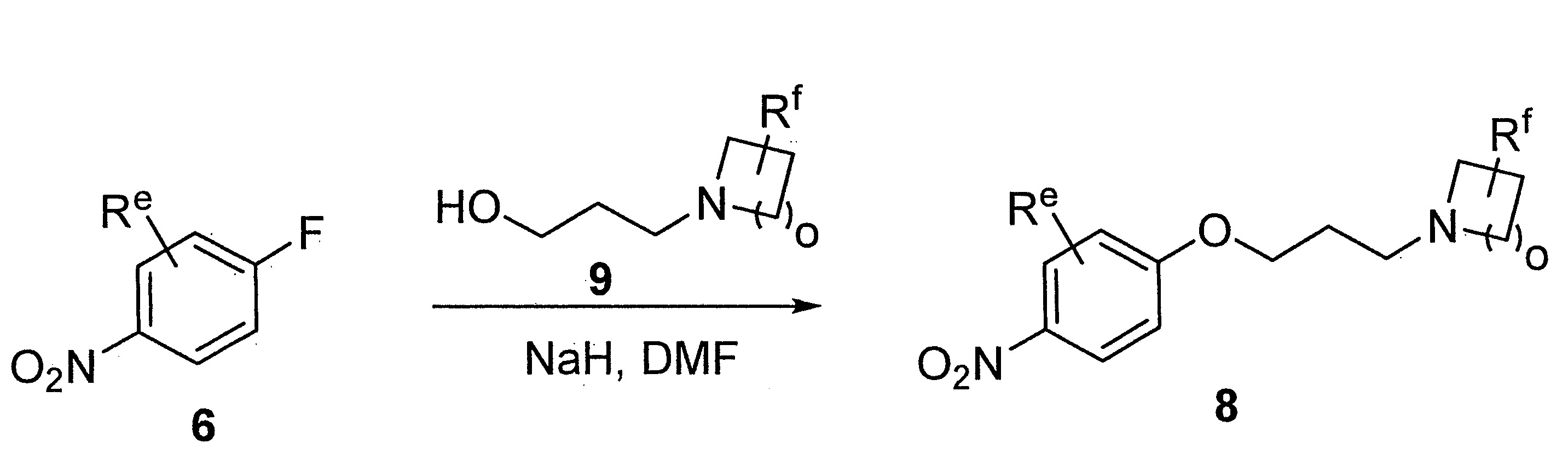 或者,按照流程3,可根據參考文獻(US2005182045A1;WO2017201161A1)中所揭示的相關程序進行關於具有式(8)之化合物的合成,但不限於所揭示的這些程序。將氟苯衍生物6以醇衍生物9取代而形成苯基醚化合物8。
流程4:具有式(C)之化合物的一般合成
或者,按照流程3,可根據參考文獻(US2005182045A1;WO2017201161A1)中所揭示的相關程序進行關於具有式(8)之化合物的合成,但不限於所揭示的這些程序。將氟苯衍生物6以醇衍生物9取代而形成苯基醚化合物8。
流程4:具有式(C)之化合物的一般合成
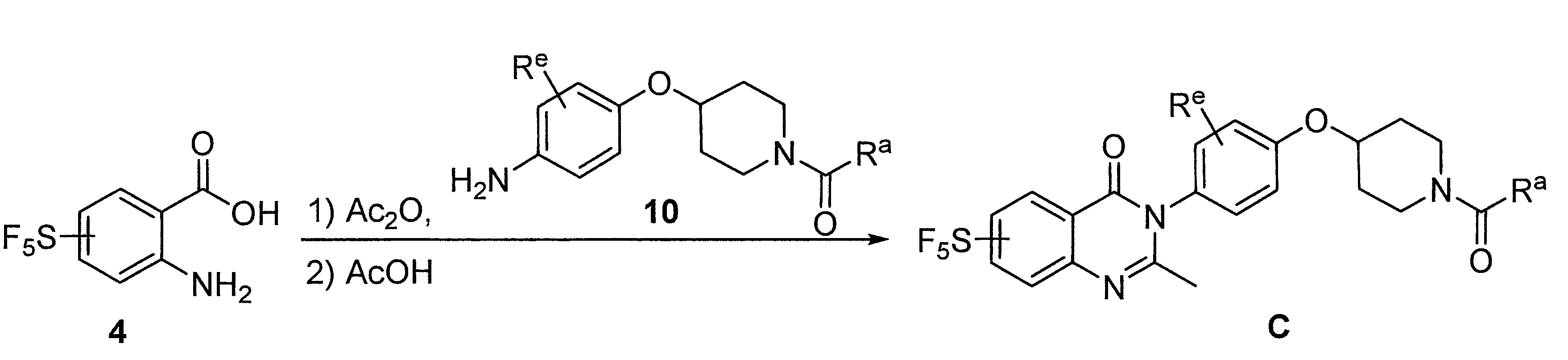 按照流程4,可根據參考文獻(US2005182045A1;WO2017201161A1)中所揭示的相關程序進行關於具有式(C)之化合物的合成,但不限於所揭示的這些程序。將苯胺衍生物4用10環化以提供化合物(C)。
流程5:中間化合物10的合成
按照流程4,可根據參考文獻(US2005182045A1;WO2017201161A1)中所揭示的相關程序進行關於具有式(C)之化合物的合成,但不限於所揭示的這些程序。將苯胺衍生物4用10環化以提供化合物(C)。
流程5:中間化合物10的合成
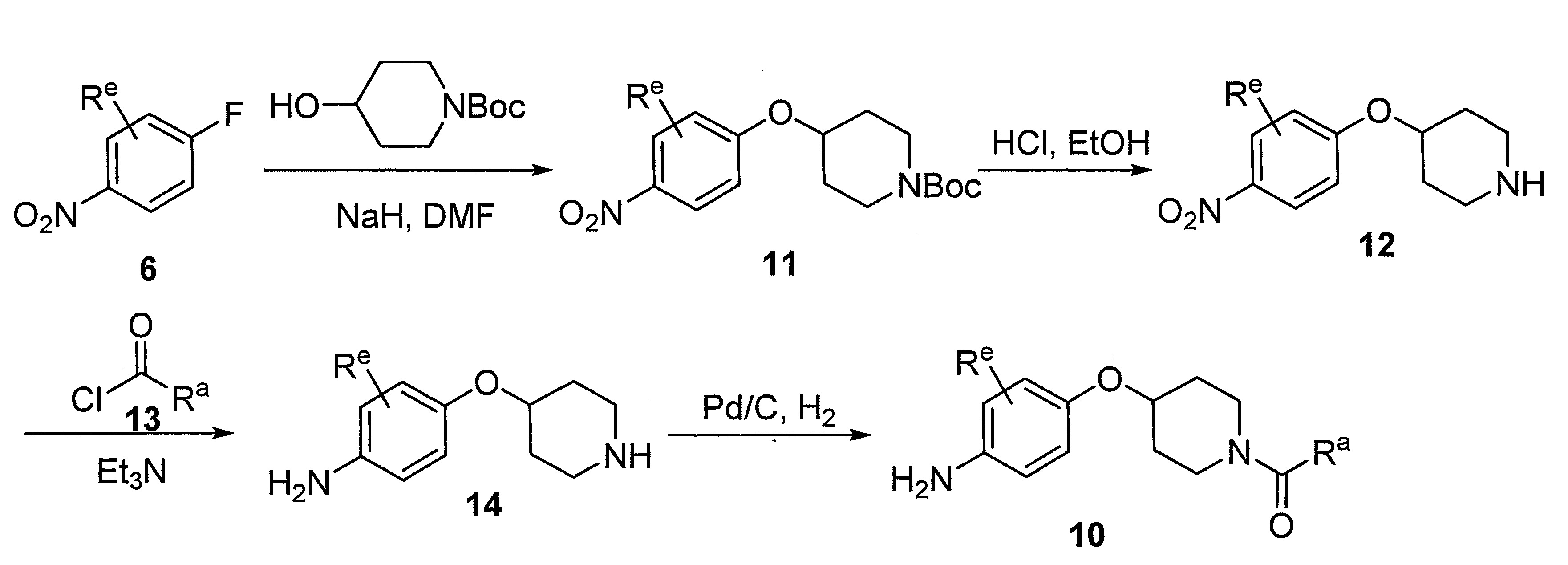 按照流程5,可根據參考文獻(US2005182045A1;Journal of Medicinal Chemistry, 2008, 51, 21, 6889-6901;US2009306375A1)中所揭示的相關程序進行關於具有式(10)之化合物的合成,但不限於所揭示的這些程序。將氟苯衍生物6以
N-Boc-哌啶-4-醇取代而形成硝基苯衍生物11,其進行去保護以產生胺12。因此,將化合物12醯化接著還原以提供化合物10。
流程6:具有式(D)之化合物的一般合成
按照流程5,可根據參考文獻(US2005182045A1;Journal of Medicinal Chemistry, 2008, 51, 21, 6889-6901;US2009306375A1)中所揭示的相關程序進行關於具有式(10)之化合物的合成,但不限於所揭示的這些程序。將氟苯衍生物6以
N-Boc-哌啶-4-醇取代而形成硝基苯衍生物11,其進行去保護以產生胺12。因此,將化合物12醯化接著還原以提供化合物10。
流程6:具有式(D)之化合物的一般合成
 按照流程6,可根據參考文獻(US2005182045A1;WO2017201161A1)中所揭示的相關程序進行關於具有式(D)之化合物的合成,但不限於所揭示的這些程序。將苯胺衍生物4用化合物15環化以提供化合物D。
流程7:中間化合物15的合成
按照流程6,可根據參考文獻(US2005182045A1;WO2017201161A1)中所揭示的相關程序進行關於具有式(D)之化合物的合成,但不限於所揭示的這些程序。將苯胺衍生物4用化合物15環化以提供化合物D。
流程7:中間化合物15的合成
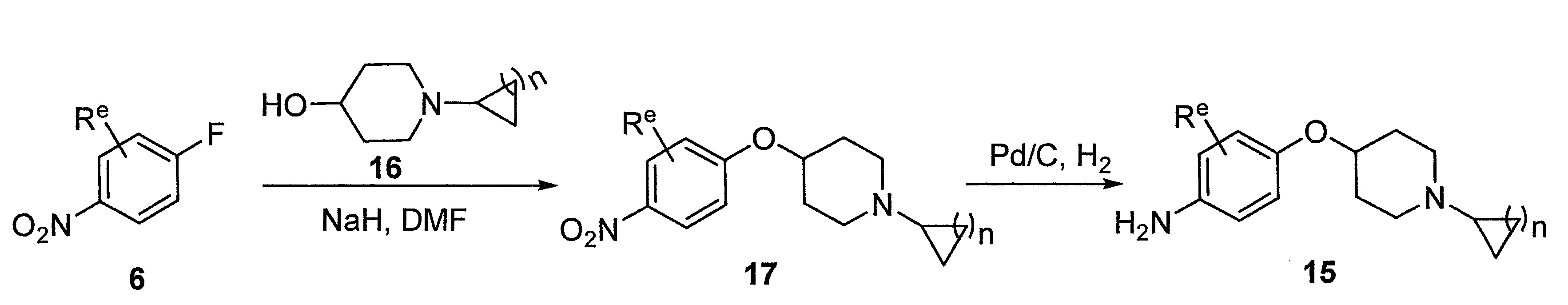 按照流程7,可根據參考文獻(US2005182045A1)中所揭示的相關程序進行關於具有式(15)之化合物的合成,但不限於所揭示的這些程序。將氟苯衍生物6以醇衍生物16取代而形成苯基醚17,將其還原以提供化合物15。
示例性化合物之製備和示性
本揭示所包含的化合物可經由不同的流程製備。經由各種流程之22種示例性化合物的詳細製備方法係描述於下,並列出示性結果。
除非另有說明,否則所有試劑均購自商業供應商而無需進一步純化。必要時使用以標準方法乾燥的溶劑。用於薄層層析法(TLC)的板為預塗在鋁板上的E. Merck矽膠60F254(0.24 nm 厚度),並接著在UV光(365 nm和254 nm)下或透過用在乙醇中的5%的十二鉬磷酸(dodecamolybdophosphoric acid)染色及隨後加熱可視化。使用來自商業供應商的矽膠(200-400篩目)進行管柱層析。在室溫下以Agilent 400-MR NMR光譜儀(1H為400.00MHz)記錄
1H NMR光譜。溶劑信號用作為
1H NMR之參考(CDCl
3,7.26ppm;CD
3OD,3.31ppm;DMSO-d
6,2.50ppm;D
2O,4.79ppm)。下列縮寫係用於解釋多重性:s=單峰,d=雙峰,t=三重峰,q=四重峰,br. s.=寬單峰,dd=雙二重峰,td=三雙重峰,dt=雙三重峰,dq=雙四重峰,m=多重峰。在實驗細節中所使用之其他縮寫如下:δ=距四甲基矽烷的低磁場(downfield)方向的化學位移(ppm),Ar=芳基,Ac=醯基,Boc=三級丁氧基羰基,Bn=苯甲基,DCM=二氯甲烷,DCE=二氯乙烷,DMF=N,N’-二甲基甲醯胺,NMP = N-甲基-2-吡咯啶酮,DIBAL-H=二異丁基氫化鋁,DIPEA=二異丙基乙胺,DMAP=4-(二甲胺基)吡啶,DMSO=二甲亞碸,EA=乙酸乙酯,Et=乙基,Me=甲基,Hz=赫茲,HPLC=高效液相層析,J=偶合常數(NMR),min=分鐘,h=小時,NMR=核磁共振,prep=製備型,PE=石油醚,t-Bu=三級丁基,iPr=異丙基,TBAF=四丁基氟化銨,tert=三級,TFA=三氟乙酸,THF=四氫呋喃,TLC=薄層層析法。
實施例
應指出的是,以下詳述之本發明實施態樣僅為用於解釋本發明的示例,而不應被解釋為限制本發明。沒有特定技術或條件的實施例可以根據該項技術領域之文件中的技術或條件或根據產品說明書實施。沒有製造商的試劑或儀器可透過習知購買獲得。具有該項技術者將理解可改變起始材料且製造本發明所包含之化合物所採用的額外步驟,如下列實施例所證明的。
按照流程7,可根據參考文獻(US2005182045A1)中所揭示的相關程序進行關於具有式(15)之化合物的合成,但不限於所揭示的這些程序。將氟苯衍生物6以醇衍生物16取代而形成苯基醚17,將其還原以提供化合物15。
示例性化合物之製備和示性
本揭示所包含的化合物可經由不同的流程製備。經由各種流程之22種示例性化合物的詳細製備方法係描述於下,並列出示性結果。
除非另有說明,否則所有試劑均購自商業供應商而無需進一步純化。必要時使用以標準方法乾燥的溶劑。用於薄層層析法(TLC)的板為預塗在鋁板上的E. Merck矽膠60F254(0.24 nm 厚度),並接著在UV光(365 nm和254 nm)下或透過用在乙醇中的5%的十二鉬磷酸(dodecamolybdophosphoric acid)染色及隨後加熱可視化。使用來自商業供應商的矽膠(200-400篩目)進行管柱層析。在室溫下以Agilent 400-MR NMR光譜儀(1H為400.00MHz)記錄
1H NMR光譜。溶劑信號用作為
1H NMR之參考(CDCl
3,7.26ppm;CD
3OD,3.31ppm;DMSO-d
6,2.50ppm;D
2O,4.79ppm)。下列縮寫係用於解釋多重性:s=單峰,d=雙峰,t=三重峰,q=四重峰,br. s.=寬單峰,dd=雙二重峰,td=三雙重峰,dt=雙三重峰,dq=雙四重峰,m=多重峰。在實驗細節中所使用之其他縮寫如下:δ=距四甲基矽烷的低磁場(downfield)方向的化學位移(ppm),Ar=芳基,Ac=醯基,Boc=三級丁氧基羰基,Bn=苯甲基,DCM=二氯甲烷,DCE=二氯乙烷,DMF=N,N’-二甲基甲醯胺,NMP = N-甲基-2-吡咯啶酮,DIBAL-H=二異丁基氫化鋁,DIPEA=二異丙基乙胺,DMAP=4-(二甲胺基)吡啶,DMSO=二甲亞碸,EA=乙酸乙酯,Et=乙基,Me=甲基,Hz=赫茲,HPLC=高效液相層析,J=偶合常數(NMR),min=分鐘,h=小時,NMR=核磁共振,prep=製備型,PE=石油醚,t-Bu=三級丁基,iPr=異丙基,TBAF=四丁基氟化銨,tert=三級,TFA=三氟乙酸,THF=四氫呋喃,TLC=薄層層析法。
實施例
應指出的是,以下詳述之本發明實施態樣僅為用於解釋本發明的示例,而不應被解釋為限制本發明。沒有特定技術或條件的實施例可以根據該項技術領域之文件中的技術或條件或根據產品說明書實施。沒有製造商的試劑或儀器可透過習知購買獲得。具有該項技術者將理解可改變起始材料且製造本發明所包含之化合物所採用的額外步驟,如下列實施例所證明的。
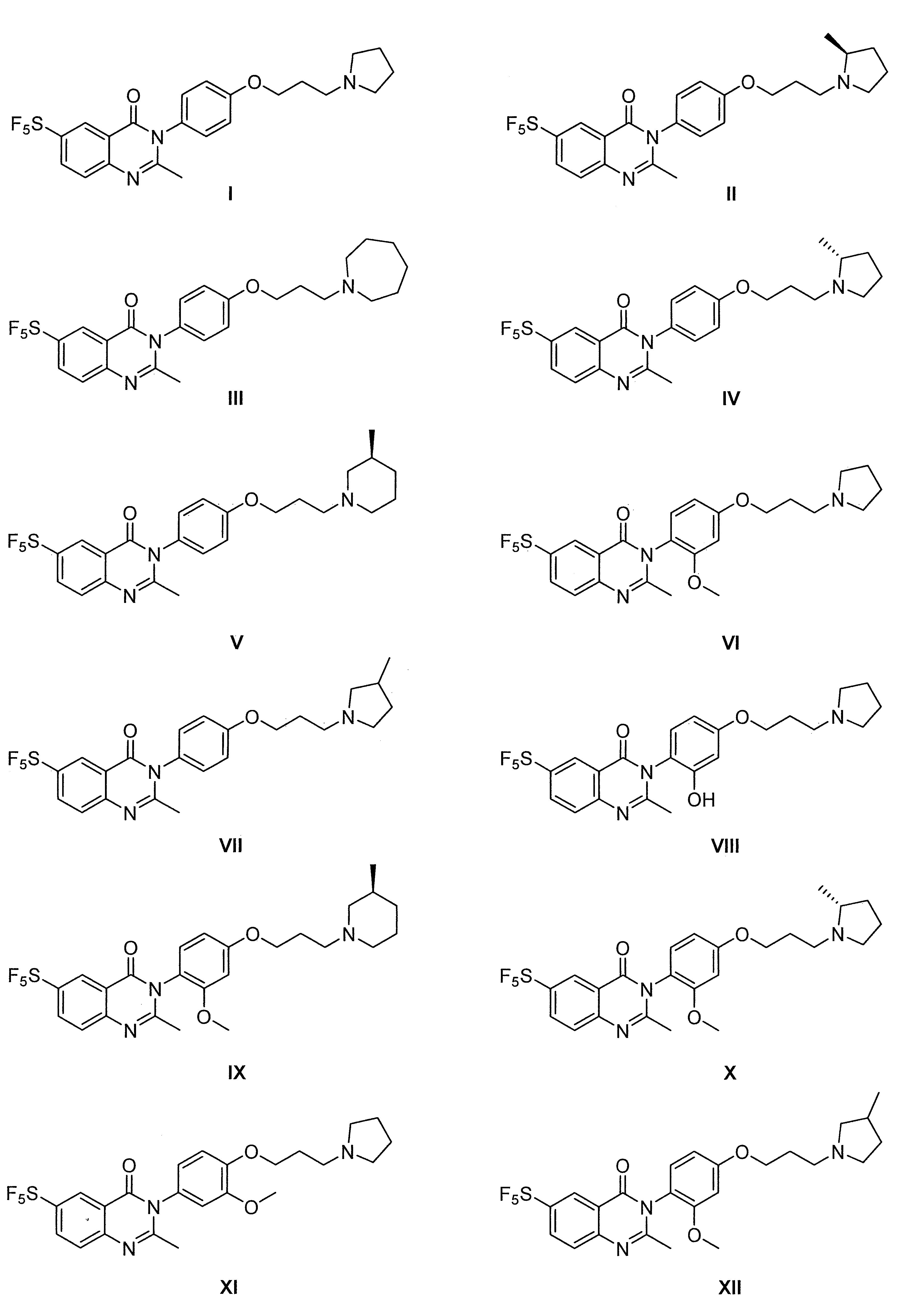
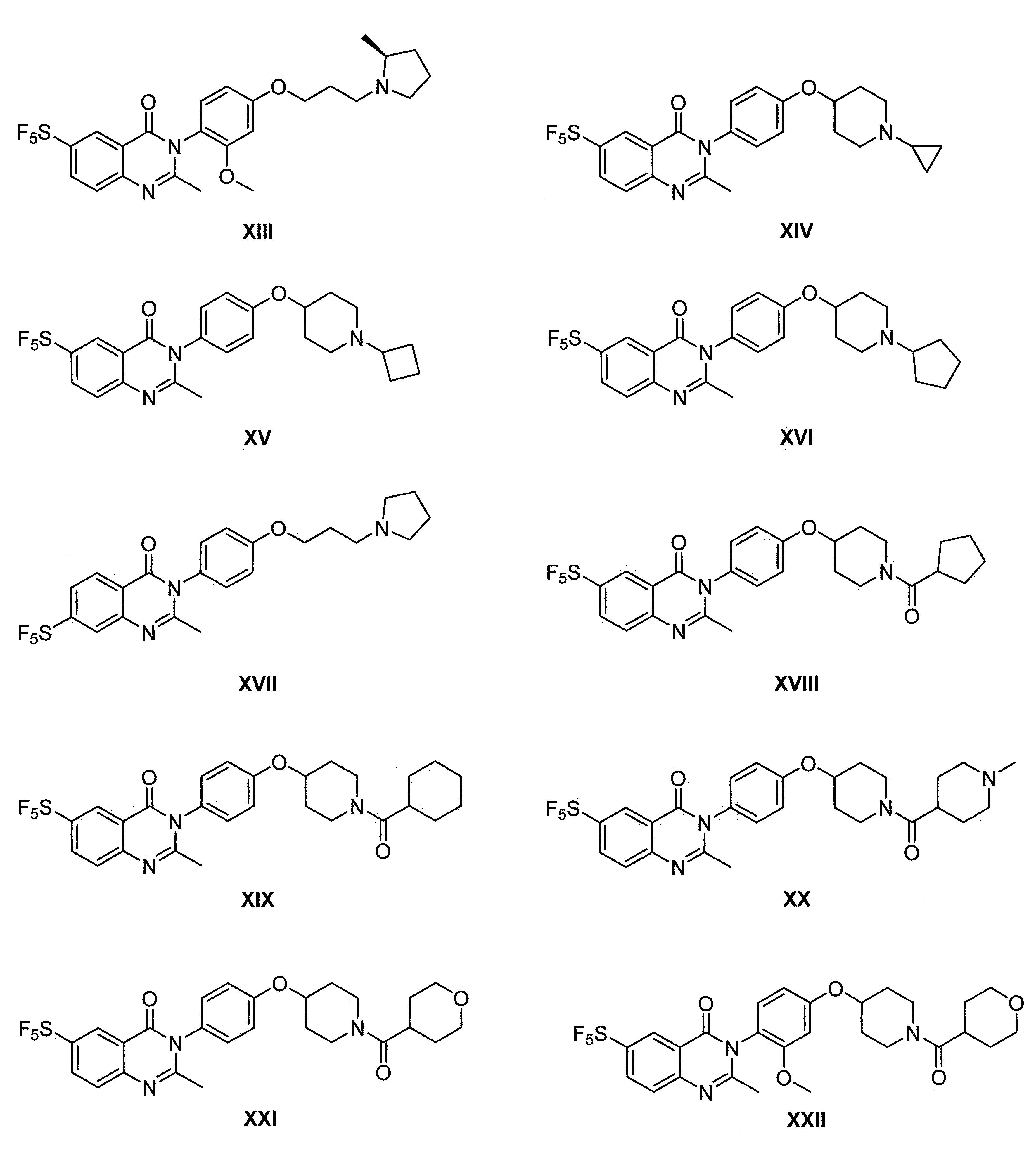 實施例1:2-甲基-3-(4-(3-(吡咯啶-1-基)丙氧基)苯基)-6-(五氟硫基)喹唑啉-4(3H)-酮(I)
實施例1:2-甲基-3-(4-(3-(吡咯啶-1-基)丙氧基)苯基)-6-(五氟硫基)喹唑啉-4(3H)-酮(I)
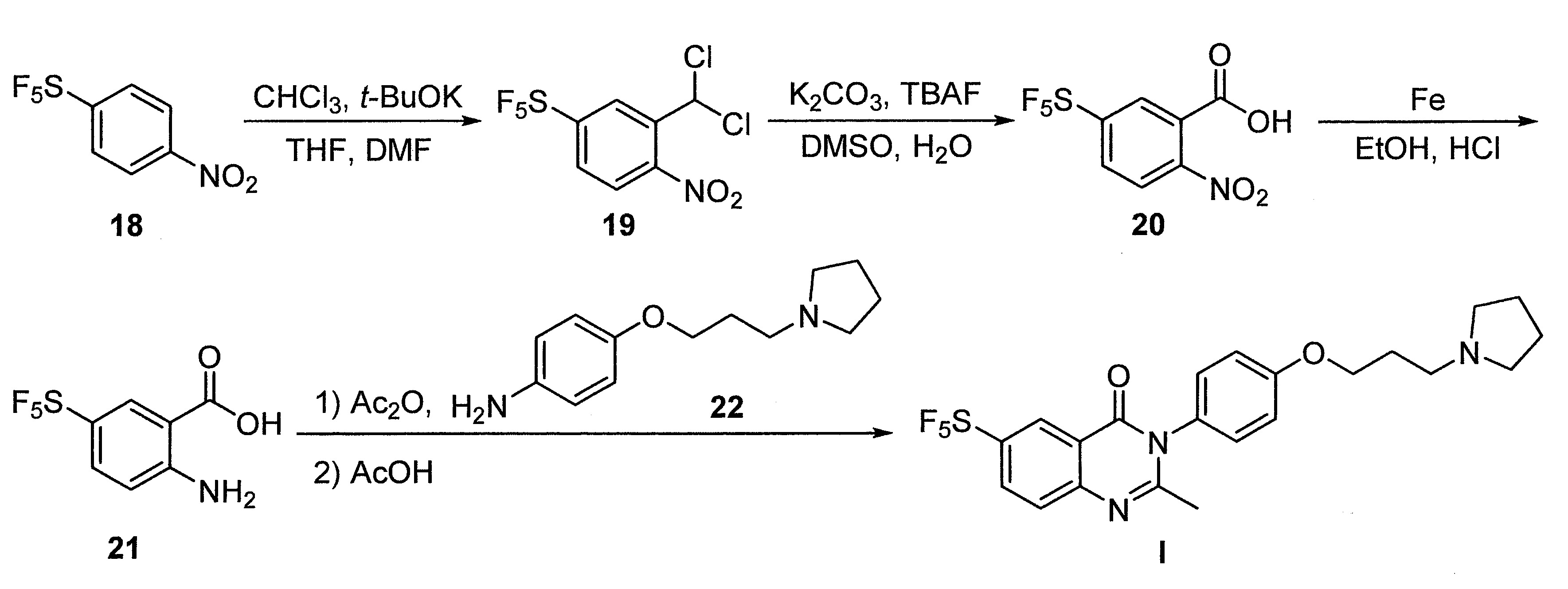 步驟1:2-(二氯甲基)-1-硝基-4-(五氟硫基)苯(19)
在N
2氛圍下於-78℃將1-硝基-4-(五氟硫基)苯(18) (6.0g,24.1mmol)和CHCl
3(3.0g,25.3mmol)在DMF(30mL)中之溶液滴加至t-BuOK(8.1g,72.2mmol)在乾燥DMF (60mL)和THF(60mL)中之溶液。將反應在-78℃下攪拌0.5h及接著用HCl(2 M)水溶液淬滅直到pH=4。將所得混合物用水(200mL)稀釋並用EA(2×100mL)萃取。將合併的有機相用鹽水(100mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由管柱層析法(矽膠,PE/EA=100:1)將殘餘物純化以提供呈黃色油之標題化合物19(6.0g,75%)。
1H NMR(400MHz, CDCl
3) δ=8.56(s, 1H), 8.09(d, J=9.2Hz, 1H), 7.96(dd, J=2.0, 8.8Hz, 1H), 7.52(s, 1H)。
步驟2:2-硝基-5-(五氟硫基)苯甲酸(20)
將K
2CO
3(0.6g,4.5mmol)和TBAF(72.3mg,0.3mmol)加至2-(二氯甲基)-1-硝基-4-(五氟硫基)苯(19)(0.5g,1.5mmol)在DMSO(5mL)中之攪拌溶液。將反應在25℃下攪拌16h並接著用鹽酸水溶液(1M)酸化至pH=4。將所得混合物用EA(2×5mL)萃取。將合併的有機相用鹽水(10mL)洗滌,用無水MgSO
4乾燥,過濾和濃縮。藉由管柱層析法(矽膠,PE/EA=5:1和DCM/MeOH=10:1)將殘餘物純化以提供呈灰白色固體之標題化合物20(0.2g,50%)。
1H NMR(400MHz, CDCl
3)δ=8.31(s, 1H), 8.07(d, J=8.8Hz, 1H), 7.92(d, J=8.8Hz, 1H)。
步驟3:2-胺基-5-(五氟硫基)苯甲酸(21)
將Fe粉(123.0mg,2.2mmol)一次全部加至2-硝基-5-(五氟硫基)苯甲酸(20)(0.2g,0.7mmol)在EtOH(1mL)和濃HCl(0.3mL)中之攪拌懸浮液。將反應在回流下加熱1h並接著濃縮。將殘餘物溶解在水(5mL)中並用EA(2×5mL)萃取。將合併的有機相用鹽水(10mL)洗滌,用無水MgSO
4乾燥,過濾和濃縮以提供呈棕色固體之標題化合物21(190 mg,98%)。
1H NMR(400MHz, CD
3OD) δ=8.20(d, J=2.4Hz, 1H), 7.58(dd, J=2.8, 9.2Hz, 1H), 6.77(d, J=9.2Hz, 1H);
MS(ESI):[M+H]
+=263.7。
步驟4:2-甲基-3-(4-(3-(吡咯啶-1-基)丙氧基)苯基)-6-(五氟硫基)喹唑啉-4(3H)-酮(I)
將2-胺基-5-(五氟硫基)苯甲酸(21)(0.2g,0.8mmol)在Ac
2O(2mL)中之溶液在回流下加熱4h並接著濃縮。將4-(3-(吡咯啶-1-基)丙氧基)苯胺(22)(0.2g,0.9mmol)和AcOH(2mL)加至殘餘物。將反應在80℃下攪拌2h並接著濃縮。藉由製備型HPLC將殘餘物純化以提供呈灰色固體之標題化合物I(120mg,32%)。
1H NMR(400MHz, CD
3OD) δ=8.65(d, J=2.4Hz, 1H), 8.40(dd, J=2.4, 9.0Hz, 1H), 7.91(d, J=8.8Hz, 1H), 7.39(d, J=8.8Hz, 2H), 7.21(d, J=8.8Hz, 2H), 4.23(t, J=5.6Hz, 2H), 3.78-3.69(m, 2H), 3.51-3.42(m, 2H), 3.22-3.09(m, 2H), 2.46(s, 3H), 2.35-2.25(m, 2H), 2.25-2.15(m, 2H), 2.13-2.01(m, 2H);
MS(ESI):[M+H]
+=489.9。
實施例2:(S)-2-甲基-3-(4-(3-(2-甲基吡咯啶-1-基)丙氧基)苯基)-6-(五氟硫基)喹唑啉-4(3H)-酮(II)
步驟1:2-(二氯甲基)-1-硝基-4-(五氟硫基)苯(19)
在N
2氛圍下於-78℃將1-硝基-4-(五氟硫基)苯(18) (6.0g,24.1mmol)和CHCl
3(3.0g,25.3mmol)在DMF(30mL)中之溶液滴加至t-BuOK(8.1g,72.2mmol)在乾燥DMF (60mL)和THF(60mL)中之溶液。將反應在-78℃下攪拌0.5h及接著用HCl(2 M)水溶液淬滅直到pH=4。將所得混合物用水(200mL)稀釋並用EA(2×100mL)萃取。將合併的有機相用鹽水(100mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由管柱層析法(矽膠,PE/EA=100:1)將殘餘物純化以提供呈黃色油之標題化合物19(6.0g,75%)。
1H NMR(400MHz, CDCl
3) δ=8.56(s, 1H), 8.09(d, J=9.2Hz, 1H), 7.96(dd, J=2.0, 8.8Hz, 1H), 7.52(s, 1H)。
步驟2:2-硝基-5-(五氟硫基)苯甲酸(20)
將K
2CO
3(0.6g,4.5mmol)和TBAF(72.3mg,0.3mmol)加至2-(二氯甲基)-1-硝基-4-(五氟硫基)苯(19)(0.5g,1.5mmol)在DMSO(5mL)中之攪拌溶液。將反應在25℃下攪拌16h並接著用鹽酸水溶液(1M)酸化至pH=4。將所得混合物用EA(2×5mL)萃取。將合併的有機相用鹽水(10mL)洗滌,用無水MgSO
4乾燥,過濾和濃縮。藉由管柱層析法(矽膠,PE/EA=5:1和DCM/MeOH=10:1)將殘餘物純化以提供呈灰白色固體之標題化合物20(0.2g,50%)。
1H NMR(400MHz, CDCl
3)δ=8.31(s, 1H), 8.07(d, J=8.8Hz, 1H), 7.92(d, J=8.8Hz, 1H)。
步驟3:2-胺基-5-(五氟硫基)苯甲酸(21)
將Fe粉(123.0mg,2.2mmol)一次全部加至2-硝基-5-(五氟硫基)苯甲酸(20)(0.2g,0.7mmol)在EtOH(1mL)和濃HCl(0.3mL)中之攪拌懸浮液。將反應在回流下加熱1h並接著濃縮。將殘餘物溶解在水(5mL)中並用EA(2×5mL)萃取。將合併的有機相用鹽水(10mL)洗滌,用無水MgSO
4乾燥,過濾和濃縮以提供呈棕色固體之標題化合物21(190 mg,98%)。
1H NMR(400MHz, CD
3OD) δ=8.20(d, J=2.4Hz, 1H), 7.58(dd, J=2.8, 9.2Hz, 1H), 6.77(d, J=9.2Hz, 1H);
MS(ESI):[M+H]
+=263.7。
步驟4:2-甲基-3-(4-(3-(吡咯啶-1-基)丙氧基)苯基)-6-(五氟硫基)喹唑啉-4(3H)-酮(I)
將2-胺基-5-(五氟硫基)苯甲酸(21)(0.2g,0.8mmol)在Ac
2O(2mL)中之溶液在回流下加熱4h並接著濃縮。將4-(3-(吡咯啶-1-基)丙氧基)苯胺(22)(0.2g,0.9mmol)和AcOH(2mL)加至殘餘物。將反應在80℃下攪拌2h並接著濃縮。藉由製備型HPLC將殘餘物純化以提供呈灰色固體之標題化合物I(120mg,32%)。
1H NMR(400MHz, CD
3OD) δ=8.65(d, J=2.4Hz, 1H), 8.40(dd, J=2.4, 9.0Hz, 1H), 7.91(d, J=8.8Hz, 1H), 7.39(d, J=8.8Hz, 2H), 7.21(d, J=8.8Hz, 2H), 4.23(t, J=5.6Hz, 2H), 3.78-3.69(m, 2H), 3.51-3.42(m, 2H), 3.22-3.09(m, 2H), 2.46(s, 3H), 2.35-2.25(m, 2H), 2.25-2.15(m, 2H), 2.13-2.01(m, 2H);
MS(ESI):[M+H]
+=489.9。
實施例2:(S)-2-甲基-3-(4-(3-(2-甲基吡咯啶-1-基)丙氧基)苯基)-6-(五氟硫基)喹唑啉-4(3H)-酮(II)
 步驟1:1-(3-溴丙氧基)-4-硝基苯(24)
在25℃下將1,3-二溴丙烷(5.8g,28.8mmol)和K
2CO
3(6.0g,43.2mmol)加至4-硝基酚(23)(2.0g,14.4mmol)在CH
3CN(30mL)中之攪拌懸浮液。將反應在80℃下攪拌16h並接著濃縮。將殘餘物分溶在EA(20mL)和水(10mL)之間。將有機相用鹽水(10mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由管柱層析法(矽膠,PE/EA=10:1)將殘餘物純化以提供呈灰白色固體之標題化合物24(2.1g,57%)。
步驟2:(S)-2-甲基-1-(3-(4-硝基苯氧基)丙基)吡咯啶(25)
在25℃下將K
2CO
3(276mg,2.0mmol)和(S)-2-甲基吡咯啶(170mg,2.0mmol)加至1-(3-溴丙氧基)-4-硝基苯(24)(260mg,1.0mmol)在CH
3CN(4mL)中之攪拌懸浮液。將反應在80℃下攪拌16h並接著濃縮。將殘餘物分溶在EA(10mL)和水(5mL)之間。將有機相用鹽水(5mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由管柱層析法(矽膠,DCM/MeOH=10:1)將殘餘物純化以提供呈黃色油之標題化合物25(251mg,95%)。
MS(ESI):[M+H
+]=264.9。
步驟3:(S)-4-(3-(2-甲基吡咯啶-1-基)丙氧基)苯胺(26)
在25℃下將Pd/C(25mg,10%)一次全部加至(S)-2-甲基-1-(3-(4-硝基苯氧基)丙基)吡咯啶(25)(251mg,1.0mmol)在MeOH(5mL)中之攪拌懸浮液。將反應在H
2氛圍下於25℃攪拌16h並接著通過Celite
®過濾。用MeOH(5mL)洗滌濾餅。將合併的濾液濃縮以提供呈紅色油之標題化合物26(222mg,100%)。
MS(ESI):[M+H
+]=234.9。
步驟4:(S)-2-甲基-3-(4-(3-(2-甲基吡咯啶-1-基)丙氧基)苯基)-6-(五氟硫基)喹唑啉-4(3H)-酮(II)
將2-胺基-5-(五氟硫基)苯甲酸(21)(50mg,0.2mmol)在Ac
2O(1mL)中之溶液在回流下加熱4h並接著濃縮。在25℃下將(S)-4-(3-(2-甲基吡咯啶-1-基)丙氧基)苯胺(26) (54mg,0.2mmol)和AcOH(1mL)加至殘餘物。將反應在80℃下攪拌1h並接著濃縮。將殘餘物分溶在EA(10mL)與飽和NaHCO
3水溶液(5mL)之間。將有機相用鹽水(5mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由製備型HPLC將殘餘物純化以提供呈棕色固體之標題化合物II(40mg,呈HCl鹽,35%)。
1H NMR(400MHz, DMSO-d
6) δ=10.52(br. s., 1H), 8.42 (d, J=2.4Hz, 1H), 8.33(dd, J=2.6, 9.0Hz, 1H), 7.87(d, J=8.8Hz, 1H), 7.40(d, J=8.8Hz, 2H), 7.13(d, J=8.8Hz, 2H), 4.19-4.13(m, 2H), 3.49-3.37(m, 2H), 3.13-3.05(m, 2H), 2.36-2.08(m, 7H), 1.99-1.90(m, 2H), 1.69-1.61(m, 1H), 1.41(d, J=6.8Hz, 3H);
MS(ESI):[M+H
+]=504.1。
實施例3:3-(4-(3-(氮𠰢-1-基)丙氧基)苯基)-2-甲基-6-(五氟硫基)喹唑啉-4(3H)-酮(III)
步驟1:1-(3-溴丙氧基)-4-硝基苯(24)
在25℃下將1,3-二溴丙烷(5.8g,28.8mmol)和K
2CO
3(6.0g,43.2mmol)加至4-硝基酚(23)(2.0g,14.4mmol)在CH
3CN(30mL)中之攪拌懸浮液。將反應在80℃下攪拌16h並接著濃縮。將殘餘物分溶在EA(20mL)和水(10mL)之間。將有機相用鹽水(10mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由管柱層析法(矽膠,PE/EA=10:1)將殘餘物純化以提供呈灰白色固體之標題化合物24(2.1g,57%)。
步驟2:(S)-2-甲基-1-(3-(4-硝基苯氧基)丙基)吡咯啶(25)
在25℃下將K
2CO
3(276mg,2.0mmol)和(S)-2-甲基吡咯啶(170mg,2.0mmol)加至1-(3-溴丙氧基)-4-硝基苯(24)(260mg,1.0mmol)在CH
3CN(4mL)中之攪拌懸浮液。將反應在80℃下攪拌16h並接著濃縮。將殘餘物分溶在EA(10mL)和水(5mL)之間。將有機相用鹽水(5mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由管柱層析法(矽膠,DCM/MeOH=10:1)將殘餘物純化以提供呈黃色油之標題化合物25(251mg,95%)。
MS(ESI):[M+H
+]=264.9。
步驟3:(S)-4-(3-(2-甲基吡咯啶-1-基)丙氧基)苯胺(26)
在25℃下將Pd/C(25mg,10%)一次全部加至(S)-2-甲基-1-(3-(4-硝基苯氧基)丙基)吡咯啶(25)(251mg,1.0mmol)在MeOH(5mL)中之攪拌懸浮液。將反應在H
2氛圍下於25℃攪拌16h並接著通過Celite
®過濾。用MeOH(5mL)洗滌濾餅。將合併的濾液濃縮以提供呈紅色油之標題化合物26(222mg,100%)。
MS(ESI):[M+H
+]=234.9。
步驟4:(S)-2-甲基-3-(4-(3-(2-甲基吡咯啶-1-基)丙氧基)苯基)-6-(五氟硫基)喹唑啉-4(3H)-酮(II)
將2-胺基-5-(五氟硫基)苯甲酸(21)(50mg,0.2mmol)在Ac
2O(1mL)中之溶液在回流下加熱4h並接著濃縮。在25℃下將(S)-4-(3-(2-甲基吡咯啶-1-基)丙氧基)苯胺(26) (54mg,0.2mmol)和AcOH(1mL)加至殘餘物。將反應在80℃下攪拌1h並接著濃縮。將殘餘物分溶在EA(10mL)與飽和NaHCO
3水溶液(5mL)之間。將有機相用鹽水(5mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由製備型HPLC將殘餘物純化以提供呈棕色固體之標題化合物II(40mg,呈HCl鹽,35%)。
1H NMR(400MHz, DMSO-d
6) δ=10.52(br. s., 1H), 8.42 (d, J=2.4Hz, 1H), 8.33(dd, J=2.6, 9.0Hz, 1H), 7.87(d, J=8.8Hz, 1H), 7.40(d, J=8.8Hz, 2H), 7.13(d, J=8.8Hz, 2H), 4.19-4.13(m, 2H), 3.49-3.37(m, 2H), 3.13-3.05(m, 2H), 2.36-2.08(m, 7H), 1.99-1.90(m, 2H), 1.69-1.61(m, 1H), 1.41(d, J=6.8Hz, 3H);
MS(ESI):[M+H
+]=504.1。
實施例3:3-(4-(3-(氮𠰢-1-基)丙氧基)苯基)-2-甲基-6-(五氟硫基)喹唑啉-4(3H)-酮(III)
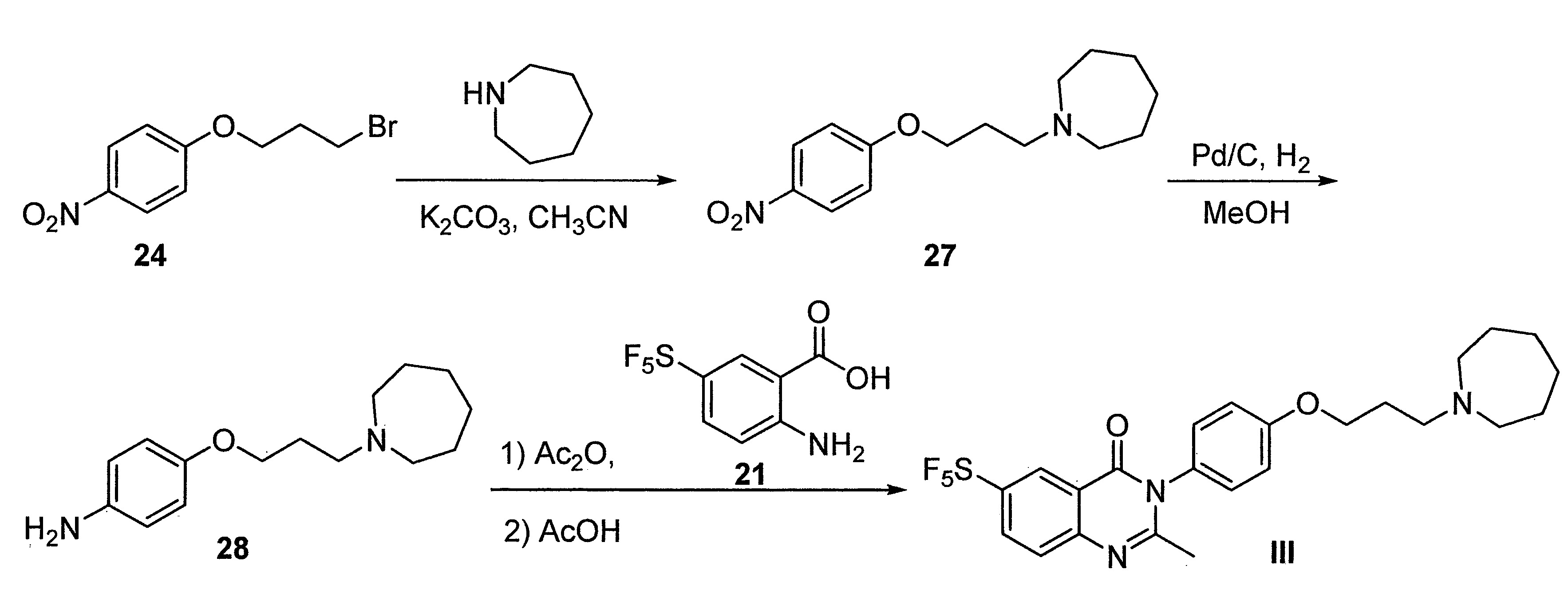 步驟1:1-(3-(4-硝基苯氧基)丙基)氮𠰢(27)
在25℃下將K
2CO
3(276mg,2.0mmol)和氮𠰢(198mg,2.0mmol)加至1-(3-溴丙氧基)-4-硝基苯(24)(260mg,1.0mmol)在CH
3CN(4mL)中之攪拌懸浮液。將反應在80℃下攪拌16h並接著濃縮。將殘餘物分溶在EA(10mL)和水(5mL)之間。將有機相用鹽水(5mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由管柱層析法(矽膠,DCM/MeOH =10:1)將殘餘物純化以提供呈黃色油之標題化合物27(217mg,78%)。
MS(ESI):[M+H
+]=278.9。
步驟2:4-(3-(氮𠰢-1-基)丙氧基)苯胺(28)
在25℃下將Pd/C(20mg,10%)一次全部加至1-(3-(4-硝基苯氧基)丙基)氮𠰢(27)(217mg,0.8mmol)在MeOH(5mL)中之攪拌懸浮液。將反應在H
2氛圍下於25℃攪拌16h。將所得混合物通過Celite
®過濾。用MeOH(5mL)洗滌濾餅。將合併的濾液濃縮以提供呈紅色油之標題化合物28(193mg,100%)。
MS(ESI):[M+H
+]=248.9。
步驟3:3-(4-(3-(氮𠰢-1-基)丙氧基)苯基)-2-甲基-6-(五氟硫基)喹唑啉-4(3H)-酮(III)
將2-胺基-5-(五氟硫基)苯甲酸(21)(50mg,0.2mmol)在Ac
2O(1mL)中之溶液在回流下加熱4h並接著濃縮。在25℃下將4-(3-(氮𠰢-1-基)丙氧基)苯胺(28)(57mg,0.2mmol)和AcOH(1mL)加至殘餘物。將反應在80℃下攪拌1h並接著濃縮。將殘餘物分溶在EA(10mL)與飽和NaHCO
3水溶液(5mL)之間。將有機相用鹽水(5mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由製備型HPLC將殘餘物純化以提供呈棕色固體之標題化合物III(50mg,呈HCl鹽,45%)。
1H NMR(400MHz, DMSO-d
6) δ=10.37(br. s., 1H), 8.42(d, J=2.4Hz, 1H), 8.32(dd, J=2.6, 9.0Hz, 1H), 7.86(d, J=8.8Hz, 1H), 7.40(d, J=8.8Hz, 2H), 7.12(d, J=8.8Hz, 2H), 4.14(t, J=5.8Hz, 2H), 3.42-3.36(m, 2H), 3.27-3.22(m, 2H), 3.18-3.10(m, 2H), 2.28-2.21(m, 2H), 2.18(s, 3H), 1.89-1.77(m, 4H), 1.69-1.55(m, 4H);
MS(ESI):[M+H
+]=518.1。
實施例4:(R)-2-甲基-3-(4-(3-(2-甲基吡咯啶-1-基)丙氧基)苯基)-6-(五氟硫基)喹唑啉-4(3H)-酮(IV)
步驟1:1-(3-(4-硝基苯氧基)丙基)氮𠰢(27)
在25℃下將K
2CO
3(276mg,2.0mmol)和氮𠰢(198mg,2.0mmol)加至1-(3-溴丙氧基)-4-硝基苯(24)(260mg,1.0mmol)在CH
3CN(4mL)中之攪拌懸浮液。將反應在80℃下攪拌16h並接著濃縮。將殘餘物分溶在EA(10mL)和水(5mL)之間。將有機相用鹽水(5mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由管柱層析法(矽膠,DCM/MeOH =10:1)將殘餘物純化以提供呈黃色油之標題化合物27(217mg,78%)。
MS(ESI):[M+H
+]=278.9。
步驟2:4-(3-(氮𠰢-1-基)丙氧基)苯胺(28)
在25℃下將Pd/C(20mg,10%)一次全部加至1-(3-(4-硝基苯氧基)丙基)氮𠰢(27)(217mg,0.8mmol)在MeOH(5mL)中之攪拌懸浮液。將反應在H
2氛圍下於25℃攪拌16h。將所得混合物通過Celite
®過濾。用MeOH(5mL)洗滌濾餅。將合併的濾液濃縮以提供呈紅色油之標題化合物28(193mg,100%)。
MS(ESI):[M+H
+]=248.9。
步驟3:3-(4-(3-(氮𠰢-1-基)丙氧基)苯基)-2-甲基-6-(五氟硫基)喹唑啉-4(3H)-酮(III)
將2-胺基-5-(五氟硫基)苯甲酸(21)(50mg,0.2mmol)在Ac
2O(1mL)中之溶液在回流下加熱4h並接著濃縮。在25℃下將4-(3-(氮𠰢-1-基)丙氧基)苯胺(28)(57mg,0.2mmol)和AcOH(1mL)加至殘餘物。將反應在80℃下攪拌1h並接著濃縮。將殘餘物分溶在EA(10mL)與飽和NaHCO
3水溶液(5mL)之間。將有機相用鹽水(5mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由製備型HPLC將殘餘物純化以提供呈棕色固體之標題化合物III(50mg,呈HCl鹽,45%)。
1H NMR(400MHz, DMSO-d
6) δ=10.37(br. s., 1H), 8.42(d, J=2.4Hz, 1H), 8.32(dd, J=2.6, 9.0Hz, 1H), 7.86(d, J=8.8Hz, 1H), 7.40(d, J=8.8Hz, 2H), 7.12(d, J=8.8Hz, 2H), 4.14(t, J=5.8Hz, 2H), 3.42-3.36(m, 2H), 3.27-3.22(m, 2H), 3.18-3.10(m, 2H), 2.28-2.21(m, 2H), 2.18(s, 3H), 1.89-1.77(m, 4H), 1.69-1.55(m, 4H);
MS(ESI):[M+H
+]=518.1。
實施例4:(R)-2-甲基-3-(4-(3-(2-甲基吡咯啶-1-基)丙氧基)苯基)-6-(五氟硫基)喹唑啉-4(3H)-酮(IV)
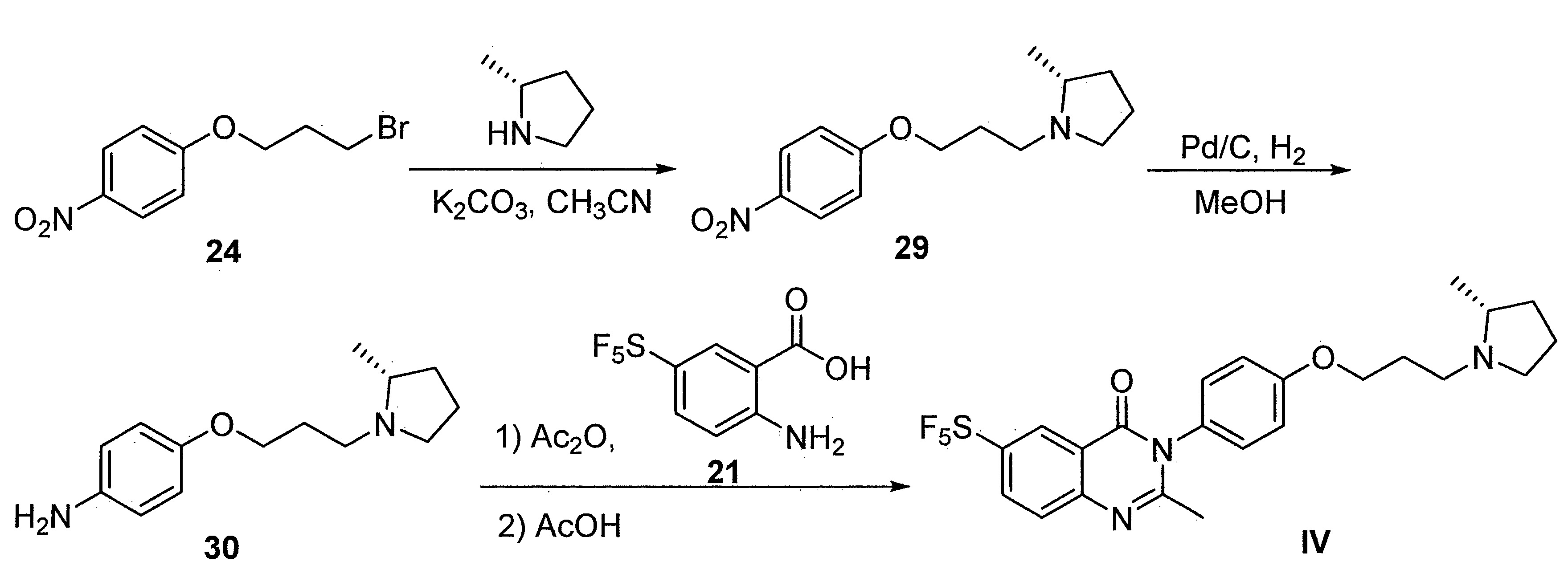 步驟1:(R)-2-甲基-1-(3-(4-硝基苯氧基)丙基)吡咯啶(29)
在25℃下將K
2CO
3(138mg,1.0mmol)和(R)-2-甲基吡咯啶(85mg,1.0mmol)加至1-(3-溴丙氧基)-4-硝基苯(24)(130mg,0.5mmol)在CH
3CN(2mL)中之攪拌懸浮液。將反應在80℃下攪拌16h並接著濃縮。將殘餘物分溶在EA(10mL)和水(5mL)之間。將有機相用鹽水(5mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由管柱層析法(矽膠,DCM/MeOH=10:1)將殘餘物純化以提供呈黃色油之標題化合物29(100mg,76%)。
MS(ESI):[M+H
+]=264.9。
步驟2:(R)-4-(3-(2-甲基吡咯啶-1-基)丙氧基)苯胺(30)
在25℃下將Pd/C(10mg,10%)一次全部加至(R)-2-甲基-1-(3-(4-硝基苯氧基)丙基)吡咯啶(29)(100mg,0.38mmol)在MeOH(3mL)中之攪拌懸浮液。將反應在H
2氛圍下於25℃攪拌16h並接著通過Celite
®過濾。用MeOH(3mL)洗滌濾餅。將合併的濾液濃縮以提供呈棕色油之標題化合物30(89mg,粗製),將其直接用於下一步驟而無需進一步純化。
MS(ESI):[M+H
+]=234.9。
步驟3:(R)-2-甲基-3-(4-(3-(2-甲基吡咯啶-1-基)丙氧基)苯基)-6-(五氟硫基)喹唑啉-4(3H)-酮(IV)
將2-胺基-5-(五氟硫基)苯甲酸(21)(50mg,0.2mmol)在Ac
2O(1mL)中之溶液在回流下加熱4h並接著濃縮。在25℃下將(R)-4-(3-(2-甲基吡咯啶-1-基)丙氧基)苯胺(30)(89mg,0.4mmol)和AcOH(1mL)加至殘餘物。將反應在80℃下攪拌1h。之後,將所得混合物濃縮。將殘餘物分溶在EA(10mL)與飽和水NaHCO
3溶液(5mL)之間。將有機相用鹽水(5mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由製備型HPLC將殘餘物純化以提供呈棕色固體之標題化合物IV(50mg,呈HCl鹽,43%)。
1H NMR(400MHz, DMSO-d
6) δ=10.50(br. s., 1H), 8.42(d, J=2.4Hz, 1H), 8.32(dd, J=2.4, 9.2Hz, 1H), 7.86(d, J=9.2Hz, 1H), 7.40(d, J=8.8Hz, 2H), 7.13(d, J=8.8Hz, 2H), 4.16(br. s., 2H), 3.67-3.60(m, 2H), 3.48-3.36(m, 2H), 3.16-3.00(m, 2H), 2.23-2.16(m, 5H), 2.04-1.87(m, 2H), 1.67-1.62(m, 1H), 1.41(d, J=6.4Hz, 3H);
MS(ESI):[M+H
+]=504.0。
實施例5:(S)-2-甲基-3-(4-(3-(3-甲基哌啶-1-基)丙氧基)苯基)-6-(五氟硫基)喹唑啉-4(3H)-酮(V)
步驟1:(R)-2-甲基-1-(3-(4-硝基苯氧基)丙基)吡咯啶(29)
在25℃下將K
2CO
3(138mg,1.0mmol)和(R)-2-甲基吡咯啶(85mg,1.0mmol)加至1-(3-溴丙氧基)-4-硝基苯(24)(130mg,0.5mmol)在CH
3CN(2mL)中之攪拌懸浮液。將反應在80℃下攪拌16h並接著濃縮。將殘餘物分溶在EA(10mL)和水(5mL)之間。將有機相用鹽水(5mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由管柱層析法(矽膠,DCM/MeOH=10:1)將殘餘物純化以提供呈黃色油之標題化合物29(100mg,76%)。
MS(ESI):[M+H
+]=264.9。
步驟2:(R)-4-(3-(2-甲基吡咯啶-1-基)丙氧基)苯胺(30)
在25℃下將Pd/C(10mg,10%)一次全部加至(R)-2-甲基-1-(3-(4-硝基苯氧基)丙基)吡咯啶(29)(100mg,0.38mmol)在MeOH(3mL)中之攪拌懸浮液。將反應在H
2氛圍下於25℃攪拌16h並接著通過Celite
®過濾。用MeOH(3mL)洗滌濾餅。將合併的濾液濃縮以提供呈棕色油之標題化合物30(89mg,粗製),將其直接用於下一步驟而無需進一步純化。
MS(ESI):[M+H
+]=234.9。
步驟3:(R)-2-甲基-3-(4-(3-(2-甲基吡咯啶-1-基)丙氧基)苯基)-6-(五氟硫基)喹唑啉-4(3H)-酮(IV)
將2-胺基-5-(五氟硫基)苯甲酸(21)(50mg,0.2mmol)在Ac
2O(1mL)中之溶液在回流下加熱4h並接著濃縮。在25℃下將(R)-4-(3-(2-甲基吡咯啶-1-基)丙氧基)苯胺(30)(89mg,0.4mmol)和AcOH(1mL)加至殘餘物。將反應在80℃下攪拌1h。之後,將所得混合物濃縮。將殘餘物分溶在EA(10mL)與飽和水NaHCO
3溶液(5mL)之間。將有機相用鹽水(5mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由製備型HPLC將殘餘物純化以提供呈棕色固體之標題化合物IV(50mg,呈HCl鹽,43%)。
1H NMR(400MHz, DMSO-d
6) δ=10.50(br. s., 1H), 8.42(d, J=2.4Hz, 1H), 8.32(dd, J=2.4, 9.2Hz, 1H), 7.86(d, J=9.2Hz, 1H), 7.40(d, J=8.8Hz, 2H), 7.13(d, J=8.8Hz, 2H), 4.16(br. s., 2H), 3.67-3.60(m, 2H), 3.48-3.36(m, 2H), 3.16-3.00(m, 2H), 2.23-2.16(m, 5H), 2.04-1.87(m, 2H), 1.67-1.62(m, 1H), 1.41(d, J=6.4Hz, 3H);
MS(ESI):[M+H
+]=504.0。
實施例5:(S)-2-甲基-3-(4-(3-(3-甲基哌啶-1-基)丙氧基)苯基)-6-(五氟硫基)喹唑啉-4(3H)-酮(V)
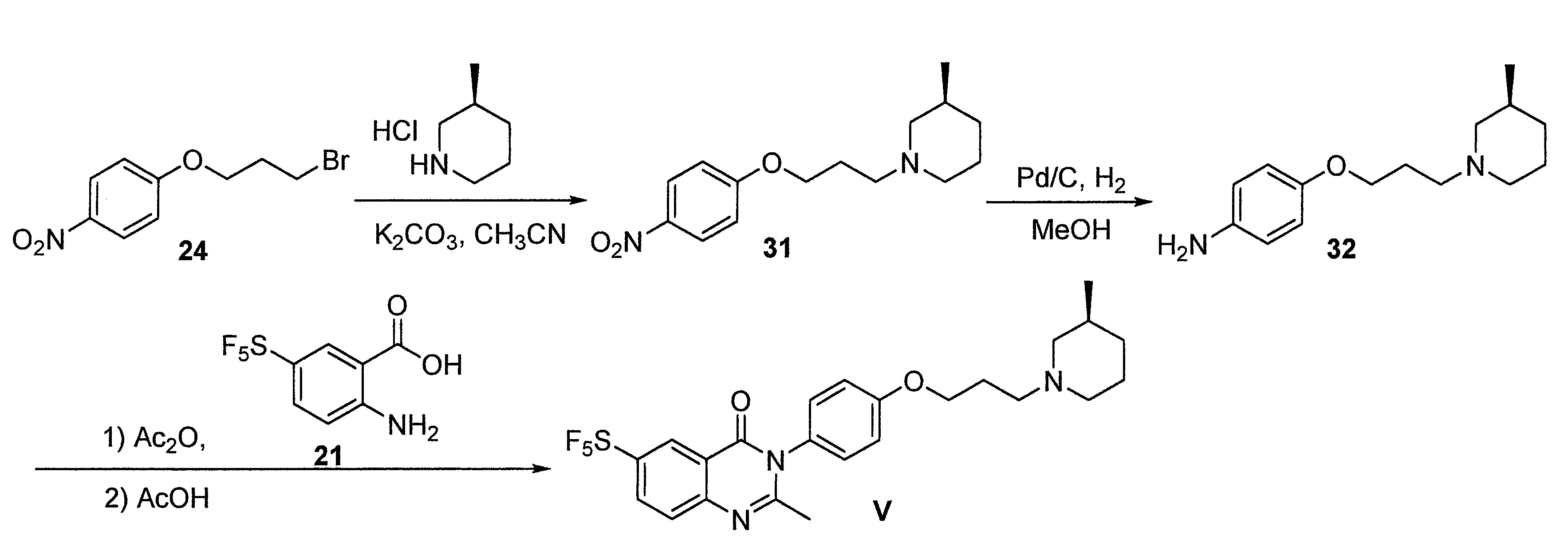 步驟1:(S)-3-甲基-1-(3-(4-硝基苯氧基)丙基)哌啶(31)
在25℃下將K
2CO
3(552mg,4.0mmol)和(S)-3-甲基哌啶鹽酸鹽(270mg,2.0mmol)加至1-(3-溴丙氧基)-4-硝基苯(24)(260mg,1.0mmol)在CH
3CN(6mL)中之攪拌懸浮液。將反應在80℃下攪拌16h並接著濃縮。將殘餘物分溶在EA(20mL)和水(10mL)之間。將有機相用鹽水(10mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由管柱層析法(矽膠,DCM/MeOH=10:1)將殘餘物純化以提供呈黃色油之標題化合物31(248mg,89%)。
MS(ESI):[M+H
+]=278.9。
步驟2:(S)-4-(3-(3-甲基哌啶-1-基)丙氧基)苯胺(32)
在25℃下將碳載鈀(25mg,10%)一次全部加至(S)-3-甲基-1-(3-(4-硝基苯氧基)丙基)哌啶(31)(248mg,0.9mmol)在MeOH(5mL)中之攪拌懸浮液。將反應在H
2氛圍下於25℃攪拌16h並接著通過Celite®過濾。用MeOH(5mL)洗滌濾餅。將合併的濾液濃縮以提供呈紅色油之標題化合物32(221mg,100%)。
MS(ESI):[M+H
+]=248.9。
步驟3:(S)-2-甲基-3-(4-(3-(3-甲基哌啶-1-基)丙氧基)苯基)-6-(五氟硫基)喹唑啉-4(3H)-酮(V)
將2-胺基-5-(五氟硫基)苯甲酸(21)(50mg,0.2mmol)在Ac
2O(1mL)中之溶液在回流下加熱4h並接著濃縮。在25℃下將(S)-4-(3-(3-甲基哌啶-1-基)丙氧基)苯胺(32)(57mg,0.23mmol)和AcOH(1mL)加至殘餘物。將反應在80℃下攪拌1h。之後,將所得混合物濃縮。將殘餘物分溶在EA(10mL)與飽和NaHCO
3水溶液(5mL)之間。將有機相用鹽水(5mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由製備型HPLC將殘餘物純化以提供呈棕色固體之標題化合物V(50mg,呈HCl鹽,45%)。
1H NMR(400MHz, DMSO-d
6) δ=10.36(br. s., 1H), 8.42(d, J=2.4Hz, 1H), 8.32(dd, J=2.6, 9.0Hz, 1H), 7.86(d, J=8.8Hz, 1H), 7.40(d, J=8.8Hz, 2H), 7.12(d, J=8.8Hz, 2H), 4.14(t, J=5.8Hz, 2H), 3.50-3.39(m, 2H), 3.22-3.14(m, 2H), 2.83-2.75(m, 1H), 2.60-2.52(m, 1H), 2.31-2.21(m, 2H), 2.18(s, 3H), 1.98-1.73(m, 5H), 0.90(d, J=6.8Hz, 3H);
MS(ESI):[M+H
+]=518.1。
實施例6:3-(2-甲氧基-4-(3-(吡咯啶-1-基)丙氧基)苯基)-2-甲基-6-(五氟硫基)喹唑啉-4(3H)-酮(VI)
步驟1:(S)-3-甲基-1-(3-(4-硝基苯氧基)丙基)哌啶(31)
在25℃下將K
2CO
3(552mg,4.0mmol)和(S)-3-甲基哌啶鹽酸鹽(270mg,2.0mmol)加至1-(3-溴丙氧基)-4-硝基苯(24)(260mg,1.0mmol)在CH
3CN(6mL)中之攪拌懸浮液。將反應在80℃下攪拌16h並接著濃縮。將殘餘物分溶在EA(20mL)和水(10mL)之間。將有機相用鹽水(10mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由管柱層析法(矽膠,DCM/MeOH=10:1)將殘餘物純化以提供呈黃色油之標題化合物31(248mg,89%)。
MS(ESI):[M+H
+]=278.9。
步驟2:(S)-4-(3-(3-甲基哌啶-1-基)丙氧基)苯胺(32)
在25℃下將碳載鈀(25mg,10%)一次全部加至(S)-3-甲基-1-(3-(4-硝基苯氧基)丙基)哌啶(31)(248mg,0.9mmol)在MeOH(5mL)中之攪拌懸浮液。將反應在H
2氛圍下於25℃攪拌16h並接著通過Celite®過濾。用MeOH(5mL)洗滌濾餅。將合併的濾液濃縮以提供呈紅色油之標題化合物32(221mg,100%)。
MS(ESI):[M+H
+]=248.9。
步驟3:(S)-2-甲基-3-(4-(3-(3-甲基哌啶-1-基)丙氧基)苯基)-6-(五氟硫基)喹唑啉-4(3H)-酮(V)
將2-胺基-5-(五氟硫基)苯甲酸(21)(50mg,0.2mmol)在Ac
2O(1mL)中之溶液在回流下加熱4h並接著濃縮。在25℃下將(S)-4-(3-(3-甲基哌啶-1-基)丙氧基)苯胺(32)(57mg,0.23mmol)和AcOH(1mL)加至殘餘物。將反應在80℃下攪拌1h。之後,將所得混合物濃縮。將殘餘物分溶在EA(10mL)與飽和NaHCO
3水溶液(5mL)之間。將有機相用鹽水(5mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由製備型HPLC將殘餘物純化以提供呈棕色固體之標題化合物V(50mg,呈HCl鹽,45%)。
1H NMR(400MHz, DMSO-d
6) δ=10.36(br. s., 1H), 8.42(d, J=2.4Hz, 1H), 8.32(dd, J=2.6, 9.0Hz, 1H), 7.86(d, J=8.8Hz, 1H), 7.40(d, J=8.8Hz, 2H), 7.12(d, J=8.8Hz, 2H), 4.14(t, J=5.8Hz, 2H), 3.50-3.39(m, 2H), 3.22-3.14(m, 2H), 2.83-2.75(m, 1H), 2.60-2.52(m, 1H), 2.31-2.21(m, 2H), 2.18(s, 3H), 1.98-1.73(m, 5H), 0.90(d, J=6.8Hz, 3H);
MS(ESI):[M+H
+]=518.1。
實施例6:3-(2-甲氧基-4-(3-(吡咯啶-1-基)丙氧基)苯基)-2-甲基-6-(五氟硫基)喹唑啉-4(3H)-酮(VI)
 步驟1:1-(3-(3-甲氧基-4-硝基苯氧基)丙基)吡咯啶(34)
在0℃下將NaH(48mg,1.2mmol,在礦物油中之60%)加至3-(吡咯啶-1-基)丙-1-醇(142mg,1.1mmol)在DMF(3mL)中之攪拌溶液。將反應在0℃下攪拌30min並接著添加4-氟-2-甲氧基-1-硝基苯(33)(171mg,1.0mmol)。將反應混合物在0-25℃下攪拌16h。將所得混合物用EA(10mL)和水(3mL)稀釋。將有機相用鹽水(3mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由管柱層析法(矽膠,PE/EA=1:1和DCM/MeOH=10:1)將殘餘物純化以提供呈黃色油之標題化合物34(200mg,71%)。
MS(ESI):[M+H
+]=280.9。
步驟2:2-甲氧基-4-(3-(吡咯啶-1-基)丙氧基)苯胺(35)
在25℃下將Pd/C(20mg,10%)一次全部加至1-(3-(3-甲氧基-4-硝基苯氧基)丙基)吡咯啶(34)(200mg,0.7mmol)在MeOH(5mL)中之攪拌懸浮液。將反應在H
2氛圍下於25℃攪拌16h並接著通過Celite®過濾。用MeOH(5mL)洗滌濾餅。將合併的濾液濃縮以提供呈紅色油之標題化合物35(178mg,100%)。
MS(ESI):[M+H
+]=250.9。
步驟3:3-(2-甲氧基-4-(3-(吡咯啶-1-基)丙氧基)苯基)-2-甲基-6-(五氟硫基)喹唑啉-4(3H)-酮(VI)
將2-胺基-5-(五氟硫基)苯甲酸(21)(50mg,0.2mmol)在Ac
2O(1mL)中之溶液在回流下加熱4h並接著濃縮。在25℃下將2-甲氧基-4-(3-(吡咯啶-1-基)丙氧基)苯胺(35)(57mg,0.2mmol)和AcOH(1mL)加至殘餘物。將反應在80℃下攪拌1h。之後,將所得混合物濃縮。將殘餘物分溶在EA(10 mL)與飽和NaHCO
3水溶液(5mL)之間。將有機相用鹽水(5mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由製備型HPLC將殘餘物純化以提供呈棕色固體之標題化合物VI(50mg,呈HCl鹽,45%)。
1H NMR(400MHz, DMSO-d
6) δ=10.94(br. s., 1H), 8.41(d, J=2.8Hz, 1H), 8.32(dd, J=2.6, 9.0Hz, 1H), 7.85(d, J=9.2Hz, 1H), 7.34(d, J=8.8Hz, 1H), 6.81(d, J=2.4Hz, 1H), 6.71(dd, J=2.2, 8.6Hz, 1H), 4.18(t, J=5.8Hz, 2H), 3.75(s, 3H), 3.59-3.53(m, 2H), 3.33-3.26(m, 2H), 3.05-2.96(m, 2H), 2.25-2.18(m, 2H), 2.16(s, 3H), 2.04-1.97(m, 2H), 1.94-1.83(m, 2H);
MS(ESI):[M+H
+]=520.0。
實施例7:2-甲基-3-(4-(3-(3-甲基吡咯啶-1-基)丙氧基)苯基)-6-(五氟硫基)喹唑啉-4(3H)-酮(VII)
步驟1:1-(3-(3-甲氧基-4-硝基苯氧基)丙基)吡咯啶(34)
在0℃下將NaH(48mg,1.2mmol,在礦物油中之60%)加至3-(吡咯啶-1-基)丙-1-醇(142mg,1.1mmol)在DMF(3mL)中之攪拌溶液。將反應在0℃下攪拌30min並接著添加4-氟-2-甲氧基-1-硝基苯(33)(171mg,1.0mmol)。將反應混合物在0-25℃下攪拌16h。將所得混合物用EA(10mL)和水(3mL)稀釋。將有機相用鹽水(3mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由管柱層析法(矽膠,PE/EA=1:1和DCM/MeOH=10:1)將殘餘物純化以提供呈黃色油之標題化合物34(200mg,71%)。
MS(ESI):[M+H
+]=280.9。
步驟2:2-甲氧基-4-(3-(吡咯啶-1-基)丙氧基)苯胺(35)
在25℃下將Pd/C(20mg,10%)一次全部加至1-(3-(3-甲氧基-4-硝基苯氧基)丙基)吡咯啶(34)(200mg,0.7mmol)在MeOH(5mL)中之攪拌懸浮液。將反應在H
2氛圍下於25℃攪拌16h並接著通過Celite®過濾。用MeOH(5mL)洗滌濾餅。將合併的濾液濃縮以提供呈紅色油之標題化合物35(178mg,100%)。
MS(ESI):[M+H
+]=250.9。
步驟3:3-(2-甲氧基-4-(3-(吡咯啶-1-基)丙氧基)苯基)-2-甲基-6-(五氟硫基)喹唑啉-4(3H)-酮(VI)
將2-胺基-5-(五氟硫基)苯甲酸(21)(50mg,0.2mmol)在Ac
2O(1mL)中之溶液在回流下加熱4h並接著濃縮。在25℃下將2-甲氧基-4-(3-(吡咯啶-1-基)丙氧基)苯胺(35)(57mg,0.2mmol)和AcOH(1mL)加至殘餘物。將反應在80℃下攪拌1h。之後,將所得混合物濃縮。將殘餘物分溶在EA(10 mL)與飽和NaHCO
3水溶液(5mL)之間。將有機相用鹽水(5mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由製備型HPLC將殘餘物純化以提供呈棕色固體之標題化合物VI(50mg,呈HCl鹽,45%)。
1H NMR(400MHz, DMSO-d
6) δ=10.94(br. s., 1H), 8.41(d, J=2.8Hz, 1H), 8.32(dd, J=2.6, 9.0Hz, 1H), 7.85(d, J=9.2Hz, 1H), 7.34(d, J=8.8Hz, 1H), 6.81(d, J=2.4Hz, 1H), 6.71(dd, J=2.2, 8.6Hz, 1H), 4.18(t, J=5.8Hz, 2H), 3.75(s, 3H), 3.59-3.53(m, 2H), 3.33-3.26(m, 2H), 3.05-2.96(m, 2H), 2.25-2.18(m, 2H), 2.16(s, 3H), 2.04-1.97(m, 2H), 1.94-1.83(m, 2H);
MS(ESI):[M+H
+]=520.0。
實施例7:2-甲基-3-(4-(3-(3-甲基吡咯啶-1-基)丙氧基)苯基)-6-(五氟硫基)喹唑啉-4(3H)-酮(VII)
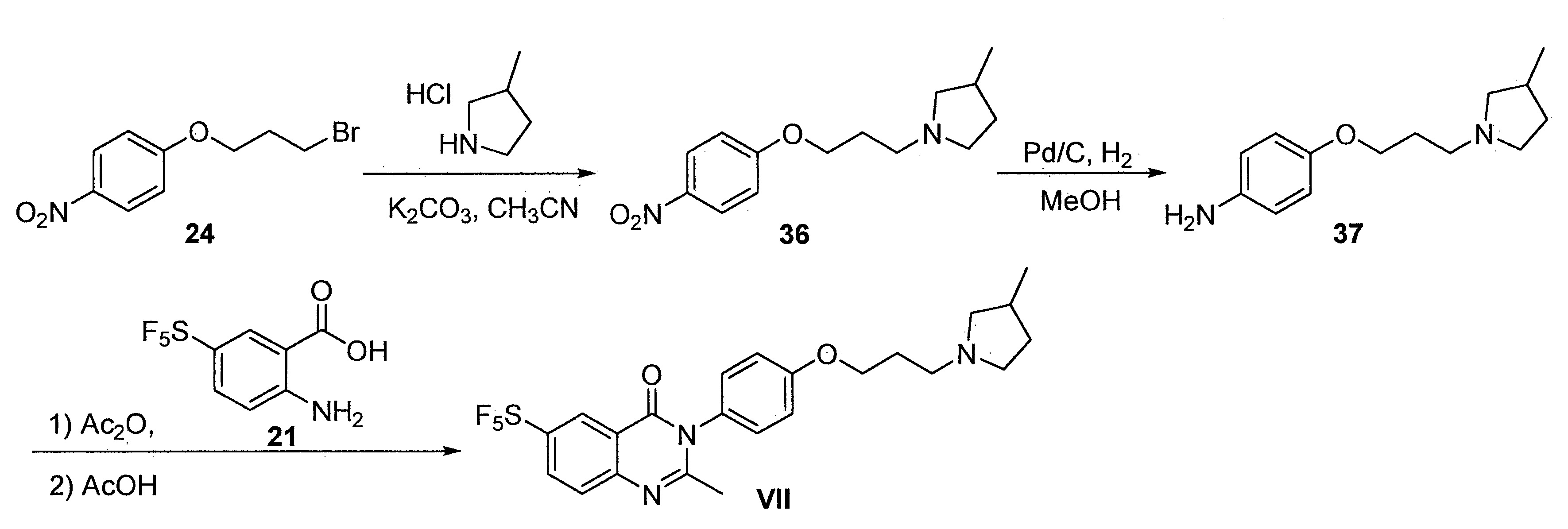 步驟1:3-甲基-1-(3-(4-硝基苯氧基)丙基)吡咯啶(36)
在25℃下將K
2CO
3(207mg,1.5mmol)和3-甲基吡咯啶鹽酸鹽(122mg,1.0mmol)加至1-(3-溴丙氧基)-4-硝基苯(24)(130mg,0.5mmol)在CH
3CN(3mL)中之攪拌懸浮液。將反應在80℃下攪拌16h並接著濃縮。將殘餘物分溶在EA(10mL)和水(5mL)之間。將有機相用鹽水(5mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由管柱層析法(矽膠,DCM/MeOH=10:1)將殘餘物純化以提供呈黃色油之標題化合物36(110mg,83%)。
步驟1:3-甲基-1-(3-(4-硝基苯氧基)丙基)吡咯啶(36)
在25℃下將K
2CO
3(207mg,1.5mmol)和3-甲基吡咯啶鹽酸鹽(122mg,1.0mmol)加至1-(3-溴丙氧基)-4-硝基苯(24)(130mg,0.5mmol)在CH
3CN(3mL)中之攪拌懸浮液。將反應在80℃下攪拌16h並接著濃縮。將殘餘物分溶在EA(10mL)和水(5mL)之間。將有機相用鹽水(5mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由管柱層析法(矽膠,DCM/MeOH=10:1)將殘餘物純化以提供呈黃色油之標題化合物36(110mg,83%)。
MS(ESI):[M+H+]=264.9。
在25℃下將Pd/C(10mg,10%)一次全部加至3-甲基-1-(3-(4-硝基苯氧基)丙基)吡咯啶(36)(110mg,0.4mmol)在MeOH(3mL)中之攪拌懸浮液。將反應在H2氛圍下於25℃攪拌16h並接著通過Celite®過濾。用MeOH(3mL)洗滌濾餅。將合併的濾液濃縮以提供呈棕色油之標題化合物37(98mg,粗製),將其直接用於下一步驟而無需進一步純化。
MS(ESI):[M+H+]=234.9。
將2-胺基-5-(五氟硫基)苯甲酸(21)(50mg,0.19mmol)在Ac2O(1mL)中之溶液在回流下加熱4h並接著濃縮。在25℃下將4-(3-(3-甲基吡咯啶-1-基)丙氧基)苯胺(37)(89mg,0.38mmol)和AcOH(1mL)加至殘餘物。將反應在80℃下攪拌1h。之後,將所得混合物濃縮。將殘餘物分溶在EA(10mL)與飽和NaHCO3水溶液(5mL)之間。將有機相用鹽水(5mL)洗滌,用無水Na2SO4乾燥,過濾和濃縮。藉由製備型HPLC將殘餘物純化以提供呈棕色固體之標題化合物VII(57mg,呈HCl鹽,56%)。
1H NMR(400MHz,DMSO-d6)δ=10.97(br.s.,1H),8.42(d,J=2.4Hz,1H),8.32(dd,J=2.4,9.2Hz,1H),7.86(d,J=9.2Hz,1H),7.40(d,J=8.8Hz,2H),7.12(d,J=9.2Hz,2H),4.15(t,J=6.0Hz,2H),3.67-3.51(m,1.5H),3.33-3.00(m,4H),2.67-2.57(m,0.5H),2.38-2.04(m,7H),1.68-1.46(m,1H),1.07(dd,J=2.6,6.6Hz,3H);MS(ESI):[M+H+]=504.0。

在-78℃下將BBr3(1.9g,7.5mmol)滴加至1-(3-(3-甲氧基-4-硝基苯氧基)丙基)吡咯啶(34)(0.7g,2.5mmol)在DCM(10mL)中之攪拌溶液。將反應攪拌4h並逐漸升溫至0℃。在-10℃下將反應混合物用MeOH淬滅(10mL)並接著用飽和NaHCO3水溶液(20mL)稀釋。將所得混合物用EA(2×20mL)萃取。將合併的有機相用鹽水(30mL)洗滌,用無水Na2SO4乾燥,過濾和濃縮。藉由製備型HPLC將殘餘物純化以提供呈灰白色固體之標題化合物38(90mg,
14%)。
1H NMR(400MHz,CD3OD)δ=8.09(d,J=9.2Hz,1H),6.72-6.58(m,2H),4.21(t,J=5.6Hz,2H),3.79-3.64(m,2H),3.47-3.36(m,2H),3.21-3.06(m,2H),2.34-2.13(m,4H),2.11-1.96(m,2H);MS(ESI):[M+H]+=266.9。
將Pd/C(10mg,10%)加至2-硝基-5-(3-(吡咯啶-1-基)丙氧基)酚(38)(90mg,0.34mmol)在MeOH(1mL)中之攪拌溶液。將反應在H2氛圍下於25℃攪拌16h並接著過濾。將濾液濃縮以提供呈灰白色油之標題化合物39(80mg,粗製),將其用於下一步驟而無需進一步純化。
MS(ESI):[M+H]+=236.9。
將2-胺基-5-(五氟硫基)苯甲酸(21)(50mg,0.2mmol)在Ac2O(1mL)中之溶液在回流下加熱2h並接著濃縮。將2-胺基-5-(3-(吡咯啶-1-基)丙氧基)酚(39)(54mg,0.2mmol)和AcOH(1mL)加至殘餘物。將反應在80℃下攪拌1h並接著濃縮。藉由製備型HPLC將殘餘物純化以提供呈灰白色固體之標題化合物VIII(25mg,26%)。
1H NMR(400MHz,CD3OD)δ=8.66(d,J=2.0Hz,1H),
8.42(d,J=9.2Hz,1H),7.92(d,J=9.2Hz,1H),7.24(d,J=8.4Hz,1H),6.76-6.59(m,2H),4.26-4.10(m,2H),3.83-3.65(m,2H),3.54-3.38(m,2H),3.23-3.06(m,2H),2.52(s,3H),2.38-2.14(m,4H),2.13-1.97(m,3H);MS(ESI):[M+H]+=506.0。

在0℃下將K2CO3(1191mg,8.6mmol)和(S)-3-甲基哌啶鹽酸鹽(587mg,4.3mmol)加至3-溴丙-1-醇(300mg,2.2mmol)在THF(10mL)中之攪拌懸浮液。將反應在0-25℃下攪拌16h。將所得混合物用EA(15mL)稀釋並通過Celite®過濾。將濾餅用EA(10mL)洗滌。將合併的濾液用水(10mL)和鹽水(10mL)洗滌,用無水Na2SO4乾燥,過濾和濃縮以提供呈無色油之標題化合物40(350mg,粗製),將其用於下一步驟而無需進一步純化。
步驟2:(S)-1-(3-(3-甲氧基-4-硝基苯氧基)丙基)-3-甲基哌啶(41)
在0℃下將NaH(97mg,2.4mmol,在礦物油中之60%)加至(S)-3-(3-甲基哌啶-1-基)丙-1-醇(40)(350mg,2.2mmol)在DMF(4mL)中之攪拌溶液。將反應在0℃下攪拌0.5h並接著添加4-氟-2-甲氧基-1-硝基苯(33)(347mg,2.0mmol)。將反應混合物在0-25℃下攪拌16h。將所得混合物用EA(30mL)和水(5mL)稀釋。將有機相用鹽水(5mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由管柱層析法(矽膠,PE/EA=1:1和DCM/MeOH=10:1)將殘餘物純化以提供呈黃色油之標題化合物41(300mg,45%二步驟)。
MS(ESI):[M+H
+]=308.9。
步驟3:(S)-2-甲氧基-4-(3-(3-甲基哌啶-1-基)丙氧基)苯胺(42)
在25℃下將Pd/C(30mg,10%)一次全部加至(S)-1-(3-(3-甲氧基-4-硝基苯氧基)丙基)-3-甲基哌啶(41)(300mg,1.0mmol)在MeOH(4mL)中之攪拌懸浮液。將反應在H
2氛圍下於25℃攪拌16h並接著通過Celite
®過濾。用MeOH(5mL)洗滌濾餅。將合併的濾液濃縮以提供呈黑色油之標題化合物42(270mg,粗製),將其用於下一步驟而無需進一步純化。
MS(ESI):[M+H
+]=278.9。
步驟4:(S)-3-(2-甲氧基-4-(3-(3-甲基哌啶-1-基)丙氧基)苯基)-2-甲基-6-(五氟硫基)喹唑啉-4(3H)-酮(IX)
將2-胺基-5-(五氟硫基)苯甲酸(21)(50mg,0.2mmol)在Ac
2O(1mL)中之溶液在回流下加熱4h並接著濃縮。在25℃下將(S)-2-甲氧基-4-(3-(3-甲基哌啶-1-基)丙氧基)苯胺(42)(64mg,0.23mmol)和AcOH(1mL)加至殘餘物。將反應在80℃下攪拌1h。之後,將所得混合物濃縮。將殘餘物分溶在EA(10mL)與飽和NaHCO
3水溶液(5mL)之間。將有機相用鹽水(5mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由製備型HPLC將殘餘物純化以提供呈灰白色固體之標題化合物IX(40mg,呈HCl鹽,30%)。
1H NMR(400MHz, DMSO-d
6) δ=10.54(br. s., 1H), 8.42(d, J=2.4Hz, 1H), 8.32(dd, J=2.2, 9.0Hz, 1H), 7.85(d, J=9.2Hz, 1H), 7.34(d, J=8.8Hz, 1H), 6.80(d, J=2.0Hz, 1H), 6.71(dd, J=2.0, 8.8Hz, 1H), 4.17(t, J=5.6Hz, 2H), 3.75(s, 3H), 3.49-3.38(m, 2H), 3.23-3.13(m, 2H), 2.88-2.71(m, 1H), 2.59-2.52(m, 1H), 2.32-2.21(m, 2H), 2.16(s, 3H), 2.07-1.95(m, 1H), 1.88-1.73(m, 3H), 1.12-1.03(m, 1H), 0.90(d, J=6.8Hz, 3H);
MS(ESI):[M+H
+]=548.1。
實施例10:(R)-3-(2-甲氧基-4-(3-(2-甲基吡咯啶-1-基)丙氧基)苯基)-2-甲基-6-(五氟硫基)喹唑啉-4(3H)-酮(X)
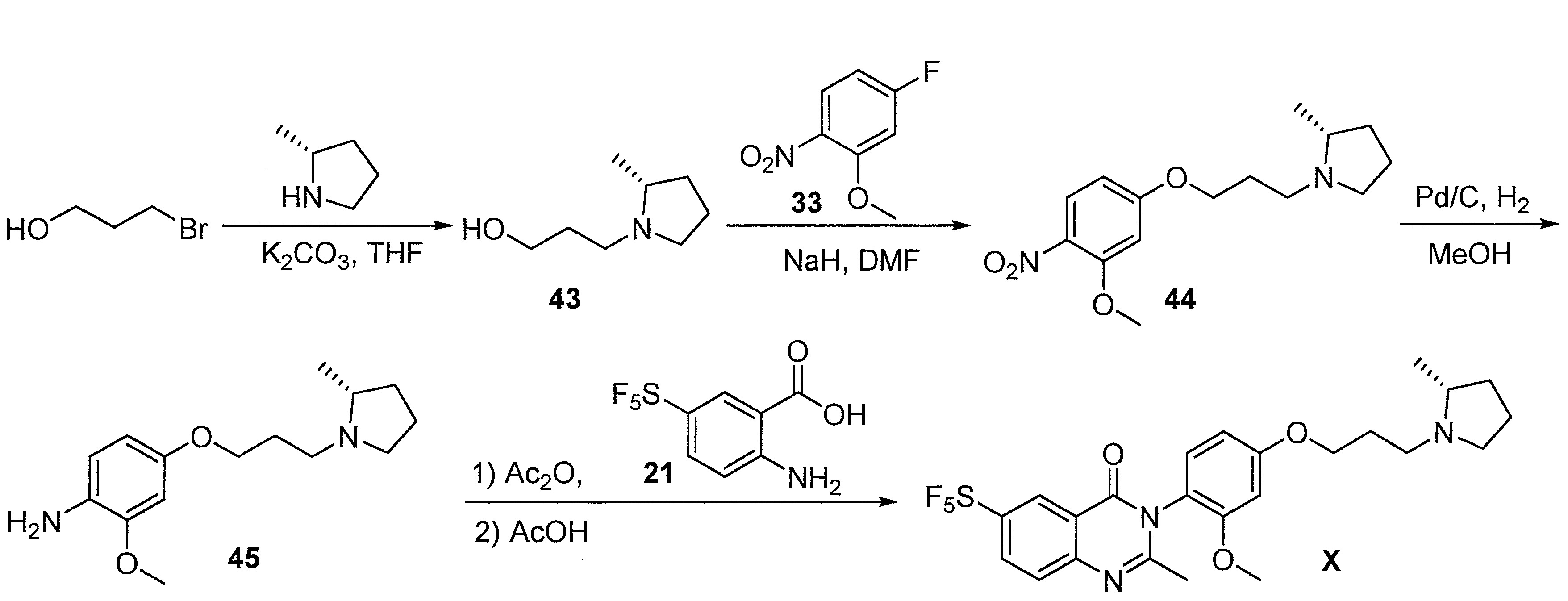 步驟1:(R)-3-(2-甲基吡咯啶-1-基)丙-1-醇(43)
在0℃下將K
2CO
3(381mg,2.8mmol)和(R)-2-甲基吡咯啶(43)(200mg,2.4mmol)加至3-溴丙-1-醇(192mg,1.4mmol)在THF(3mL)中之攪拌懸浮液。將反應在0-25℃下攪拌16h。將所得混合物用EA(5mL)稀釋並通過Celite
®過濾。將濾餅用EA(5mL)洗滌。將合併的濾液用水(5mL)和鹽水(5mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮以提供呈黃色油之標題化合物44(127mg,64%),將其用於下一步驟而無需進一步純化。
步驟2:(R)-1-(3-(3-甲氧基-4-硝基苯氧基)丙基)-2-甲基吡咯啶(44)
在0℃下將NaH(39mg,1.0mmol,在礦物油中之60%)加至(R)-3-(2-甲基吡咯啶-1-基)丙-1-醇(43)(127mg,0.9mmol)在DMF(2mL)中之攪拌溶液。將反應在0℃下攪拌30min並接著添加4-氟-2-甲氧基-1-硝基苯(33)(138mg,0.8mmol)。將反應混合物在0-25℃下攪拌16h。將所得混合物用EA(10mL)和水(2mL)稀釋。將有機相用鹽水(2mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由管柱層析法(矽膠,PE/EA=1:1和DCM/MeOH=10:1)將殘餘物純化以提供呈黃色油之標題化合物44(81mg,34%,二步驟)。
MS(ESI):[M+H
+]=294.9。
步驟3:(R)-2-甲氧基-4-(3-(2-甲基吡咯啶-1-基)丙氧基)苯胺(45)
在25℃下將Pd/C(8mg,10%)一次全部加至(R)-1-(3-(3-甲氧基-4-硝基苯氧基)丙基)-2-甲基吡咯啶(44)(81mg,0.3mmol)在MeOH(1mL)中之攪拌懸浮液。將反應在H
2氛圍下於25℃攪拌16h並接著通過Celite
®過濾。用MeOH(5mL)洗滌濾餅。將合併的濾液濃縮以提供呈紫色油之標題化合物45(73mg,粗製),將其用於下一步驟而無需進一步純化。
MS(ESI):[M+H
+]=264.9。
步驟4:(R)-3-(2-甲氧基-4-(3-(2-甲基吡咯啶-1-基)丙氧基)苯基)-2-甲基-6-(五氟硫基)喹唑啉-4(3H)-酮(X)
將2-胺基-5-(五氟硫基)苯甲酸(21)(50mg,0.2mmol)在Ac
2O(1mL)中之溶液加熱至回流經4h並接著濃縮。在25℃下將(R)-2-甲氧基-4-(3-(2-甲基吡咯啶-1-基)丙氧基)苯胺(45)(61mg,0.2mmol)和AcOH(1mL)加至殘餘物。將反應在80℃下攪拌1h。之後,將所得混合物濃縮。將殘餘物分溶在EA(10mL)與飽和NaHCO
3水溶液(5mL)之間。將水相分離。將有機相用鹽水(5mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由製備型HPLC將殘餘物純化以提供呈棕色固體之標題化合物X(30mg,呈HCl鹽,23%)。
1H NMR(400MHz, DMSO-d
6) δ=10.74(br. s., 1H), 8.41(d, J=2.4Hz, 1H), 8.33(dd, J=2.2, 9.0Hz, 1H), 7.86(d, J=9.2Hz, 1H), 7.34(d, J=8.4Hz, 1H), 6.82(s, 1H), 6.72(dd, J=1.6, 8.8Hz, 1H), 4.19(br. s., 2H), 3.75(s, 3H), 3.65-3.58(m, 1H), 3.50-3.34(m, 2H), 3.13-3.02(m, 2H), 2.31-2.17(m, 3H), 2.17(s, 3H), 2.01-1.90(m, 2H), 1.72-1.60(m, 1H), 1.42(d, J=6.0Hz, 3H);
MS(ESI):[M+H
+]=534.1。
實施例11:3-(3-甲氧基-4-(3-(吡咯啶-1-基)丙氧基)苯基)-2-甲基-6-(五氟硫基)喹唑啉-4(3H)-酮(XI)
步驟1:(R)-3-(2-甲基吡咯啶-1-基)丙-1-醇(43)
在0℃下將K
2CO
3(381mg,2.8mmol)和(R)-2-甲基吡咯啶(43)(200mg,2.4mmol)加至3-溴丙-1-醇(192mg,1.4mmol)在THF(3mL)中之攪拌懸浮液。將反應在0-25℃下攪拌16h。將所得混合物用EA(5mL)稀釋並通過Celite
®過濾。將濾餅用EA(5mL)洗滌。將合併的濾液用水(5mL)和鹽水(5mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮以提供呈黃色油之標題化合物44(127mg,64%),將其用於下一步驟而無需進一步純化。
步驟2:(R)-1-(3-(3-甲氧基-4-硝基苯氧基)丙基)-2-甲基吡咯啶(44)
在0℃下將NaH(39mg,1.0mmol,在礦物油中之60%)加至(R)-3-(2-甲基吡咯啶-1-基)丙-1-醇(43)(127mg,0.9mmol)在DMF(2mL)中之攪拌溶液。將反應在0℃下攪拌30min並接著添加4-氟-2-甲氧基-1-硝基苯(33)(138mg,0.8mmol)。將反應混合物在0-25℃下攪拌16h。將所得混合物用EA(10mL)和水(2mL)稀釋。將有機相用鹽水(2mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由管柱層析法(矽膠,PE/EA=1:1和DCM/MeOH=10:1)將殘餘物純化以提供呈黃色油之標題化合物44(81mg,34%,二步驟)。
MS(ESI):[M+H
+]=294.9。
步驟3:(R)-2-甲氧基-4-(3-(2-甲基吡咯啶-1-基)丙氧基)苯胺(45)
在25℃下將Pd/C(8mg,10%)一次全部加至(R)-1-(3-(3-甲氧基-4-硝基苯氧基)丙基)-2-甲基吡咯啶(44)(81mg,0.3mmol)在MeOH(1mL)中之攪拌懸浮液。將反應在H
2氛圍下於25℃攪拌16h並接著通過Celite
®過濾。用MeOH(5mL)洗滌濾餅。將合併的濾液濃縮以提供呈紫色油之標題化合物45(73mg,粗製),將其用於下一步驟而無需進一步純化。
MS(ESI):[M+H
+]=264.9。
步驟4:(R)-3-(2-甲氧基-4-(3-(2-甲基吡咯啶-1-基)丙氧基)苯基)-2-甲基-6-(五氟硫基)喹唑啉-4(3H)-酮(X)
將2-胺基-5-(五氟硫基)苯甲酸(21)(50mg,0.2mmol)在Ac
2O(1mL)中之溶液加熱至回流經4h並接著濃縮。在25℃下將(R)-2-甲氧基-4-(3-(2-甲基吡咯啶-1-基)丙氧基)苯胺(45)(61mg,0.2mmol)和AcOH(1mL)加至殘餘物。將反應在80℃下攪拌1h。之後,將所得混合物濃縮。將殘餘物分溶在EA(10mL)與飽和NaHCO
3水溶液(5mL)之間。將水相分離。將有機相用鹽水(5mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由製備型HPLC將殘餘物純化以提供呈棕色固體之標題化合物X(30mg,呈HCl鹽,23%)。
1H NMR(400MHz, DMSO-d
6) δ=10.74(br. s., 1H), 8.41(d, J=2.4Hz, 1H), 8.33(dd, J=2.2, 9.0Hz, 1H), 7.86(d, J=9.2Hz, 1H), 7.34(d, J=8.4Hz, 1H), 6.82(s, 1H), 6.72(dd, J=1.6, 8.8Hz, 1H), 4.19(br. s., 2H), 3.75(s, 3H), 3.65-3.58(m, 1H), 3.50-3.34(m, 2H), 3.13-3.02(m, 2H), 2.31-2.17(m, 3H), 2.17(s, 3H), 2.01-1.90(m, 2H), 1.72-1.60(m, 1H), 1.42(d, J=6.0Hz, 3H);
MS(ESI):[M+H
+]=534.1。
實施例11:3-(3-甲氧基-4-(3-(吡咯啶-1-基)丙氧基)苯基)-2-甲基-6-(五氟硫基)喹唑啉-4(3H)-酮(XI)
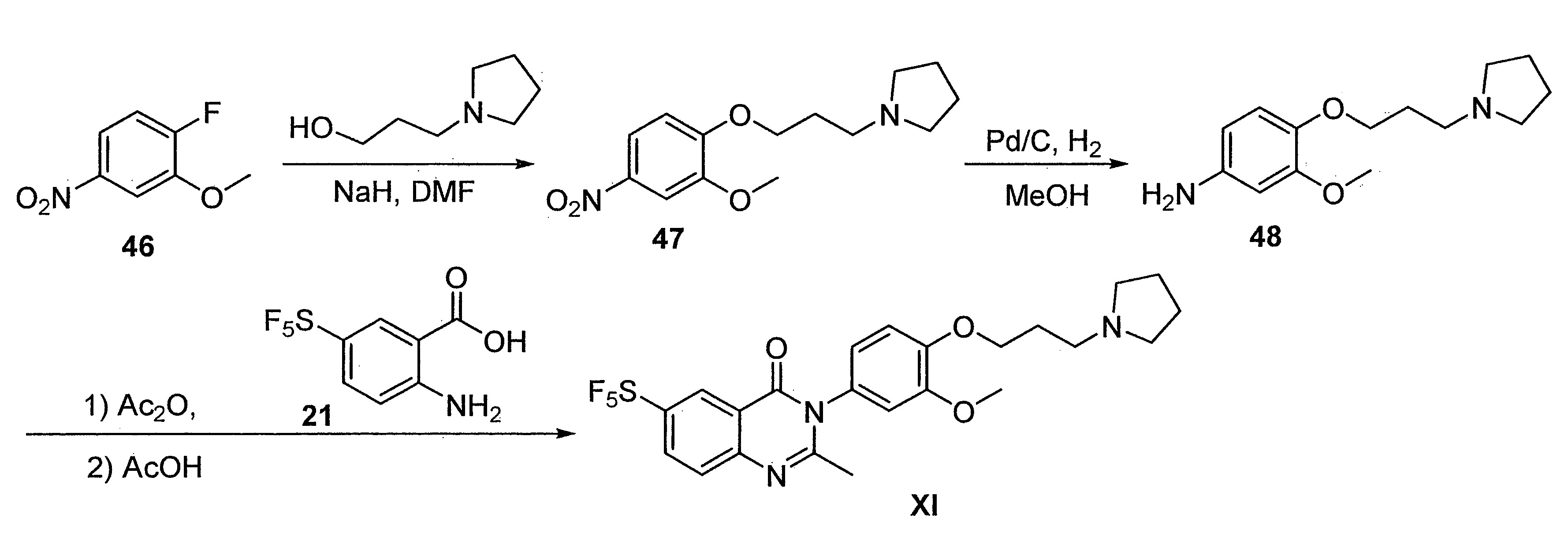 步驟1:1-(3-(2-甲氧基-4-硝基苯氧基)丙基)吡咯啶(47)
在0℃下將NaH(48mg,1.2mmol,在礦物油中之60%)加至3-(吡咯啶-1-基)丙-1-醇(142mg,1.1mmol)在DMF (3mL)中之攪拌溶液。將反應在0℃下攪拌0.5h並接著添加1-氟-2-甲氧基-4-硝基苯(46)(171mg,1.0mmol)。將反應在0-25℃下攪拌16h。將所得混合物用EA(10mL)和水(3mL)稀釋。將有機相用鹽水(3mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由管柱層析法(矽膠,PE/EA=1:1和DCM/MeOH=10:1)將殘餘物純化以提供呈黃色油之標題化合物47(170mg,61%)。
MS(ESI):[M+H
+]=280.9。
步驟2:3-甲氧基-4-(3-(吡咯啶-1-基)丙氧基)苯胺(48)
在25℃下將Pd/C(20mg,10%)一次全部加至1-(3-(2-甲氧基-4-硝基苯氧基)丙基)吡咯啶(47)(170mg,0.6mmol)在MeOH(5mL)中之攪拌懸浮液。將反應在H
2氛圍下於25℃攪拌16h並接著通過Celite
®過濾。用MeOH(5mL)洗滌濾餅。將合併的濾液濃縮以提供呈紅色油之標題化合物48(152mg,100%)。
MS(ESI):[M+H
+]=250.9。
步驟3:3-(3-甲氧基-4-(3-(吡咯啶-1-基)丙氧基)苯基)-2-甲基-6-(五氟硫基)喹唑啉-4(3H)-酮(XI)
將2-胺基-5-(五氟硫基)苯甲酸(21)(50mg,0.2mmol)在Ac
2O(1mL)中之溶液加熱至回流經4h並接著濃縮。在25℃下將3-甲氧基-4-(3-(吡咯啶-1-基)丙氧基)苯胺(48)(57mg,0.2mmol)和AcOH(1mL)加至殘餘物。將反應在80℃下攪拌1h。之後,將所得混合物濃縮。將殘餘物分溶在EA(10 mL)與飽和NaHCO
3水溶液(5mL)之間。將有機相用鹽水(5mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由製備型HPLC將殘餘物純化以提供呈棕色固體之標題化合物XI(50mg,呈HCl鹽,40%)。
1H NMR(400MHz, DMSO-d
6) δ=10.55(br. s., 1H), 8.43(d, J=2.4Hz, 1H), 8.32(dd, J=2.6, 9.0Hz, 1H), 7.86(d, J=9.2Hz, 1H), 7.18-7.11(m, 2H), 7.00(dd, J=2.2, 8.6Hz, 1H), 4.14(t, J=5.8Hz, 2H), 3.76(s, 3H), 3.63-3.58(m, 2H), 3.32-3.26(m, 2H), 3.06-2.98(m, 2H), 2.23(s, 3H), 2.21-2.14(m, 2H), 2.05-1.97(m, 2H), 1.93-1.85(m, 2H);
MS(ESI):[M+H
+]=520.0。
實施例12:3-(2-甲氧基-4-(3-(3-甲基吡咯啶-1-基)丙氧基)苯基)-2-甲基-6-(五氟硫基)喹唑啉-4(3H)-酮(XII)
步驟1:1-(3-(2-甲氧基-4-硝基苯氧基)丙基)吡咯啶(47)
在0℃下將NaH(48mg,1.2mmol,在礦物油中之60%)加至3-(吡咯啶-1-基)丙-1-醇(142mg,1.1mmol)在DMF (3mL)中之攪拌溶液。將反應在0℃下攪拌0.5h並接著添加1-氟-2-甲氧基-4-硝基苯(46)(171mg,1.0mmol)。將反應在0-25℃下攪拌16h。將所得混合物用EA(10mL)和水(3mL)稀釋。將有機相用鹽水(3mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由管柱層析法(矽膠,PE/EA=1:1和DCM/MeOH=10:1)將殘餘物純化以提供呈黃色油之標題化合物47(170mg,61%)。
MS(ESI):[M+H
+]=280.9。
步驟2:3-甲氧基-4-(3-(吡咯啶-1-基)丙氧基)苯胺(48)
在25℃下將Pd/C(20mg,10%)一次全部加至1-(3-(2-甲氧基-4-硝基苯氧基)丙基)吡咯啶(47)(170mg,0.6mmol)在MeOH(5mL)中之攪拌懸浮液。將反應在H
2氛圍下於25℃攪拌16h並接著通過Celite
®過濾。用MeOH(5mL)洗滌濾餅。將合併的濾液濃縮以提供呈紅色油之標題化合物48(152mg,100%)。
MS(ESI):[M+H
+]=250.9。
步驟3:3-(3-甲氧基-4-(3-(吡咯啶-1-基)丙氧基)苯基)-2-甲基-6-(五氟硫基)喹唑啉-4(3H)-酮(XI)
將2-胺基-5-(五氟硫基)苯甲酸(21)(50mg,0.2mmol)在Ac
2O(1mL)中之溶液加熱至回流經4h並接著濃縮。在25℃下將3-甲氧基-4-(3-(吡咯啶-1-基)丙氧基)苯胺(48)(57mg,0.2mmol)和AcOH(1mL)加至殘餘物。將反應在80℃下攪拌1h。之後,將所得混合物濃縮。將殘餘物分溶在EA(10 mL)與飽和NaHCO
3水溶液(5mL)之間。將有機相用鹽水(5mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由製備型HPLC將殘餘物純化以提供呈棕色固體之標題化合物XI(50mg,呈HCl鹽,40%)。
1H NMR(400MHz, DMSO-d
6) δ=10.55(br. s., 1H), 8.43(d, J=2.4Hz, 1H), 8.32(dd, J=2.6, 9.0Hz, 1H), 7.86(d, J=9.2Hz, 1H), 7.18-7.11(m, 2H), 7.00(dd, J=2.2, 8.6Hz, 1H), 4.14(t, J=5.8Hz, 2H), 3.76(s, 3H), 3.63-3.58(m, 2H), 3.32-3.26(m, 2H), 3.06-2.98(m, 2H), 2.23(s, 3H), 2.21-2.14(m, 2H), 2.05-1.97(m, 2H), 1.93-1.85(m, 2H);
MS(ESI):[M+H
+]=520.0。
實施例12:3-(2-甲氧基-4-(3-(3-甲基吡咯啶-1-基)丙氧基)苯基)-2-甲基-6-(五氟硫基)喹唑啉-4(3H)-酮(XII)
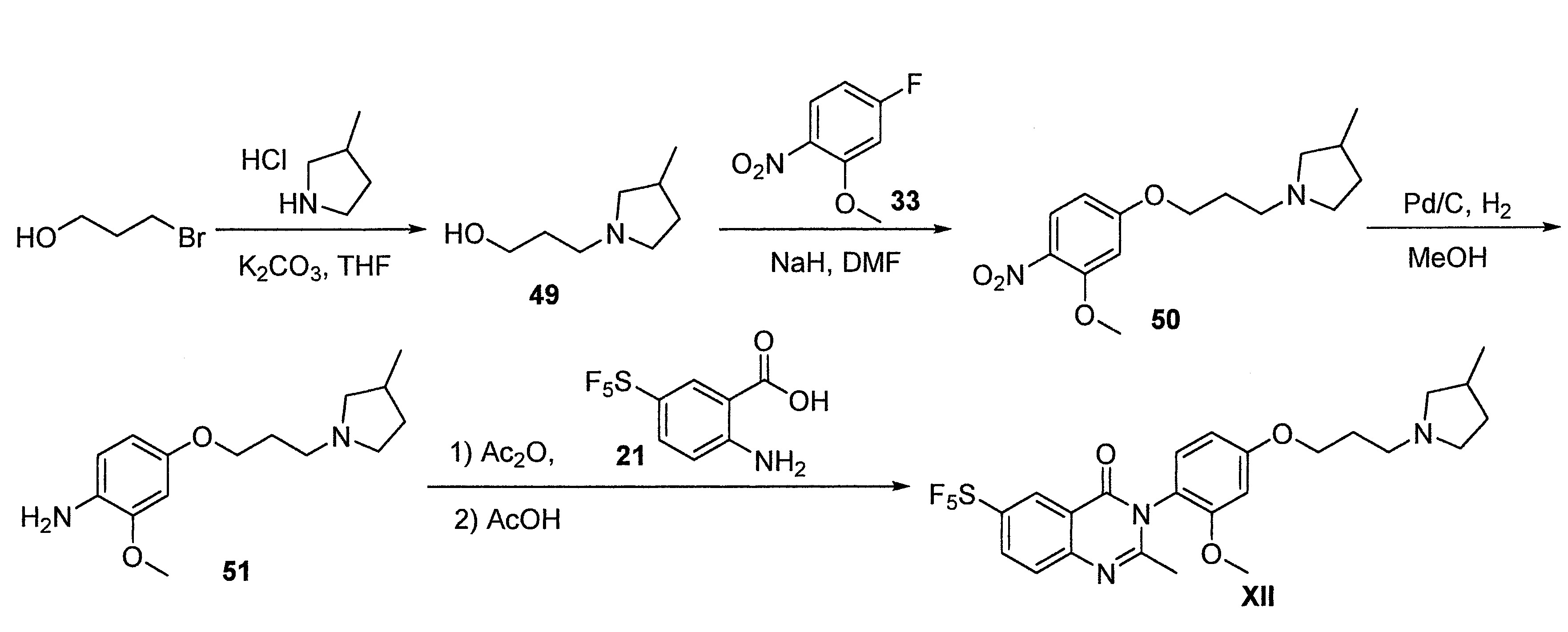 步驟1:3-(3-甲基吡咯啶-1-基)丙-1-醇(49)
在0℃下將K
2CO
3(532mg,3.9mmol)和3-甲基吡咯啶鹽酸鹽(200mg,1.6mmol)加至3-溴丙-1-醇(134mg,1.0mmol)在THF(3mL)中之攪拌懸浮液。將反應在0-25℃下攪拌16h。將所得混合物用EA(5mL)稀釋,通過Celite
®過濾。將濾餅用EA(5mL)洗滌。將合併的濾液用水(5mL)和鹽水(5mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮以提供呈黃色油之標題化合物49(86mg,62%)。
步驟2:1-(3-(3-甲氧基-4-硝基苯氧基)丙基)-3-甲基吡咯啶(50)
在0℃下將NaH(26mg,0.7mmol,在礦物油中之60%)加至3-(3-甲基吡咯啶-1-基)丙-1-醇(49)(86mg,0.6mmol)在DMF(2mL)中之攪拌溶液。將反應在0℃下攪拌30min並接著添加4-氟-2-甲氧基-1-硝基苯(33)(93mg,0.6mmol)。將反應混合物在0-25℃下攪拌16h。將所得混合物用EA(10mL)和水(2mL)稀釋。將有機相用鹽水(2mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由管柱層析法(矽膠,PE/EA=1:1和DCM/MeOH=10:1)將殘餘物純化以提供呈黃色油之標題化合物50(38mg,21%)。
MS(ESI):[M+H
+]=294.9。
步驟3:2-甲氧基-4-(3-(3-甲基吡咯啶-1-基)丙氧基)苯胺(51)
在25℃下將Pd/C(4mg,10%)一次全部加至1-(3-(3-甲氧基-4-硝基苯氧基)丙基)-3-甲基吡咯啶(50)(38mg,0.1mmol)在MeOH(1mL)中之攪拌懸浮液。將反應在H
2氛圍下於25℃攪拌16h並接著通過Celite
®過濾。用MeOH(5mL)洗滌濾餅。將合併的濾液濃縮以提供呈油之標題化合物51(34mg,粗製),將其直接用於下一步驟而無需進一步純化。
MS(ESI):[M+H
+]=264.9。
步驟4:3-(2-甲氧基-4-(3-(3-甲基吡咯啶-1-基)丙氧基)苯基)-2-甲基-6-(五氟硫基)喹唑啉-4(3H)-酮(XII)
將2-胺基-5-(五氟硫基)苯甲酸(21)(25mg,0.1mmol)在Ac
2O(1mL)中之溶液在回流下加熱4h並接著濃縮。在25℃下將2-甲氧基-4-(3-(3-甲基吡咯啶-1-基)丙氧基)苯胺(51)(34mg,0.1mmol)和AcOH(1mL)加至殘餘物。將反應在80℃下攪拌1h。之後,將所得混合物濃縮。將殘餘物分溶在EA(10mL)與飽和NaHCO
3水溶液(5mL)之間。將水相分離。將有機相用鹽水(5mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由製備型HPLC將殘餘物純化以提供呈棕色固體之標題化合物XII(33mg,呈HCl鹽,45%)。
1H NMR(400MHz, DMSO-d
6) δ=11.12(br. s., 1H), 8.42(d, J=2.4Hz, 1H), 8.33(dd, J=2.2, 9.0Hz, 1H), 7.85(d, J=9.2Hz, 1H), 7.34(d, J=8.8Hz, 1H), 6.81(d, J=1.6Hz, 1H), 6.71(dd, J=2.0, 8.8Hz, 1H), 4.17(t, J=5.8Hz, 2H), 3.75(br. s., 3H), 3.66-3.53(m, 2H), 3.33-3.07(m, 4H), 2.66-2.57(m, 0.5H), 2.37-2.29(m, 0.5H), 2.25-2.18(m, 2H), 2.16(s, 3H), 2.15-2.04(m, 1H), 1.67-1.47(m, 1H), 1.07(dd, J=4.6, 6.2Hz, 3H);
MS(ESI):[M+H
+]=534.1。
實施例13:(S)-3-(2-甲氧基-4-(3-(2-甲基吡咯啶-1-基)丙氧基)苯基)-2-甲基-6-(五氟硫基)喹唑啉-4(3H)-酮(XIII)
步驟1:3-(3-甲基吡咯啶-1-基)丙-1-醇(49)
在0℃下將K
2CO
3(532mg,3.9mmol)和3-甲基吡咯啶鹽酸鹽(200mg,1.6mmol)加至3-溴丙-1-醇(134mg,1.0mmol)在THF(3mL)中之攪拌懸浮液。將反應在0-25℃下攪拌16h。將所得混合物用EA(5mL)稀釋,通過Celite
®過濾。將濾餅用EA(5mL)洗滌。將合併的濾液用水(5mL)和鹽水(5mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮以提供呈黃色油之標題化合物49(86mg,62%)。
步驟2:1-(3-(3-甲氧基-4-硝基苯氧基)丙基)-3-甲基吡咯啶(50)
在0℃下將NaH(26mg,0.7mmol,在礦物油中之60%)加至3-(3-甲基吡咯啶-1-基)丙-1-醇(49)(86mg,0.6mmol)在DMF(2mL)中之攪拌溶液。將反應在0℃下攪拌30min並接著添加4-氟-2-甲氧基-1-硝基苯(33)(93mg,0.6mmol)。將反應混合物在0-25℃下攪拌16h。將所得混合物用EA(10mL)和水(2mL)稀釋。將有機相用鹽水(2mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由管柱層析法(矽膠,PE/EA=1:1和DCM/MeOH=10:1)將殘餘物純化以提供呈黃色油之標題化合物50(38mg,21%)。
MS(ESI):[M+H
+]=294.9。
步驟3:2-甲氧基-4-(3-(3-甲基吡咯啶-1-基)丙氧基)苯胺(51)
在25℃下將Pd/C(4mg,10%)一次全部加至1-(3-(3-甲氧基-4-硝基苯氧基)丙基)-3-甲基吡咯啶(50)(38mg,0.1mmol)在MeOH(1mL)中之攪拌懸浮液。將反應在H
2氛圍下於25℃攪拌16h並接著通過Celite
®過濾。用MeOH(5mL)洗滌濾餅。將合併的濾液濃縮以提供呈油之標題化合物51(34mg,粗製),將其直接用於下一步驟而無需進一步純化。
MS(ESI):[M+H
+]=264.9。
步驟4:3-(2-甲氧基-4-(3-(3-甲基吡咯啶-1-基)丙氧基)苯基)-2-甲基-6-(五氟硫基)喹唑啉-4(3H)-酮(XII)
將2-胺基-5-(五氟硫基)苯甲酸(21)(25mg,0.1mmol)在Ac
2O(1mL)中之溶液在回流下加熱4h並接著濃縮。在25℃下將2-甲氧基-4-(3-(3-甲基吡咯啶-1-基)丙氧基)苯胺(51)(34mg,0.1mmol)和AcOH(1mL)加至殘餘物。將反應在80℃下攪拌1h。之後,將所得混合物濃縮。將殘餘物分溶在EA(10mL)與飽和NaHCO
3水溶液(5mL)之間。將水相分離。將有機相用鹽水(5mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由製備型HPLC將殘餘物純化以提供呈棕色固體之標題化合物XII(33mg,呈HCl鹽,45%)。
1H NMR(400MHz, DMSO-d
6) δ=11.12(br. s., 1H), 8.42(d, J=2.4Hz, 1H), 8.33(dd, J=2.2, 9.0Hz, 1H), 7.85(d, J=9.2Hz, 1H), 7.34(d, J=8.8Hz, 1H), 6.81(d, J=1.6Hz, 1H), 6.71(dd, J=2.0, 8.8Hz, 1H), 4.17(t, J=5.8Hz, 2H), 3.75(br. s., 3H), 3.66-3.53(m, 2H), 3.33-3.07(m, 4H), 2.66-2.57(m, 0.5H), 2.37-2.29(m, 0.5H), 2.25-2.18(m, 2H), 2.16(s, 3H), 2.15-2.04(m, 1H), 1.67-1.47(m, 1H), 1.07(dd, J=4.6, 6.2Hz, 3H);
MS(ESI):[M+H
+]=534.1。
實施例13:(S)-3-(2-甲氧基-4-(3-(2-甲基吡咯啶-1-基)丙氧基)苯基)-2-甲基-6-(五氟硫基)喹唑啉-4(3H)-酮(XIII)
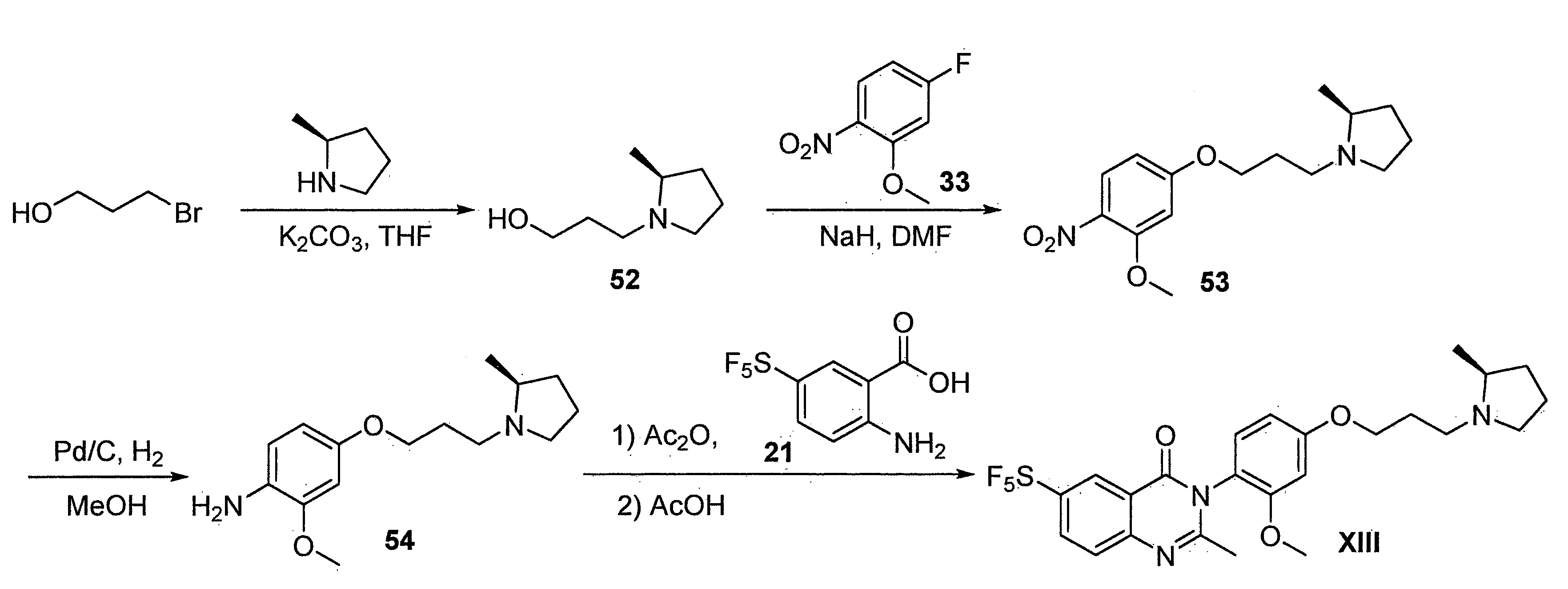 步驟1:(S)-3-(2-甲基吡咯啶-1-基)丙-1-醇(52)
在0℃下將K
2CO
3(417mg,3.0mmol)和(S)-2-甲基吡咯啶(312mg,3.7mmol)加至3-溴丙-1-醇(300mg,2.2mmol)在THF(2mL)中之攪拌懸浮液。將反應在0-25℃下攪拌16h。將所得混合物用EA(10mL)稀釋並通過Celite
®過濾。將濾餅用EA(10mL)洗滌。將合併的濾液用水(5mL)和鹽水(5mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮以提供呈無色油之標題化合物52(363mg,粗製),將其用於下一步驟而無需進一步純化。
步驟2:(S)-1-(3-(3-甲氧基-4-硝基苯氧基)丙基)-2-甲基吡咯啶(53)
在0℃下將NaH(111mg,2.8mmol,在礦物油中之60%)加至(S)-3-(2-甲基吡咯啶-1-基)丙-1-醇(52)(363mg,粗製)在DMF(4mL)中之攪拌溶液。將反應在0℃下攪拌0.5h並接著添加4-氟-2-甲氧基-1-硝基苯(33)(395mg,2.3mmol)。將反應在0-25℃下攪拌16h。將所得混合物用EA(30mL)和水(5mL)稀釋。將有機相用鹽水(5mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由管柱層析法(矽膠,PE/EA=1:1和DCM/MeOH=10:1)將殘餘物純化以提供呈黃色油之標題化合物53(370mg,58%二步驟)。
MS(ESI):[M+H
+]=294.9。
步驟3:(S)-2-甲氧基-4-(3-(2-甲基吡咯啶-1-基)丙氧基)苯胺(54)
在25℃下將Pd/C(40mg,10%)一次全部加至(S)-1-(3-(3-甲氧基-4-硝基苯氧基)丙基)-2-甲基吡咯啶(53) (370mg,1.3mmol)在MeOH(4mL)中之攪拌懸浮液。將反應在H
2氛圍下於25℃攪拌16h並接著通過Celite
®過濾。用MeOH(5mL)洗滌濾餅。將合併的濾液濃縮以提供呈黑色油之標題化合物54(330mg,粗製),將其用於下一步驟而無需進一步純化。
MS(ESI):[M+H
+]=264.9。
步驟4:(S)-3-(2-甲氧基-4-(3-(2-甲基吡咯啶-1-基)丙氧基)苯基)-2-甲基-6-(五氟硫基)喹唑啉-4(3H)-酮(XIII)
將2-胺基-5-(五氟硫基)苯甲酸(21)(50mg,0.2mmol)在Ac
2O(1mL)中之溶液加熱至回流經4h並接著濃縮。在25℃下將(S)-2-甲氧基-4-(3-(2-甲基吡咯啶-1-基)丙氧基)苯胺(54)(101mg,0.4mmol)和AcOH(1mL)加至殘餘物。將反應在80℃下攪拌1h。之後,將所得混合物濃縮。將殘餘物分溶在EA(10mL)與飽和NaHCO
3水溶液(5mL)之間。將有機相用鹽水(5mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由製備型HPLC將殘餘物純化以提供呈棕色固體之標題化合物XIII(24mg,呈HCl鹽,22%)。
1H NMR(400MHz, DMSO-d
6) δ=10.73(br. s., 1H), 8.41(br. s., 1H), 8.33(d, J=8.0Hz, 1H), 7.85(d, J=8.8Hz, 1H), 7.34(d, J=8.8Hz, 1H), 6.82(br. s., 1H), 6.72(d, J=8.0Hz, 1H), 4.19(br. s., 2H), 3.75(br. s., 3H), 3.45-3.40(m, 2H), 3.15-3.02(m, 2H), 2.36-2.07(m, 7H), 1.95(br. s., 2H), 1.73-1.58 (m, 1H), 1.42(d, J=5.6Hz, 3H);
MS(ESI):[M+H
+]=534.1。
實施例14:3-(4-((1-環丙基哌啶-4-基)氧基)苯基)-2-甲基-6-(五氟硫基)喹唑啉-4(3H)-酮(XIV)
步驟1:(S)-3-(2-甲基吡咯啶-1-基)丙-1-醇(52)
在0℃下將K
2CO
3(417mg,3.0mmol)和(S)-2-甲基吡咯啶(312mg,3.7mmol)加至3-溴丙-1-醇(300mg,2.2mmol)在THF(2mL)中之攪拌懸浮液。將反應在0-25℃下攪拌16h。將所得混合物用EA(10mL)稀釋並通過Celite
®過濾。將濾餅用EA(10mL)洗滌。將合併的濾液用水(5mL)和鹽水(5mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮以提供呈無色油之標題化合物52(363mg,粗製),將其用於下一步驟而無需進一步純化。
步驟2:(S)-1-(3-(3-甲氧基-4-硝基苯氧基)丙基)-2-甲基吡咯啶(53)
在0℃下將NaH(111mg,2.8mmol,在礦物油中之60%)加至(S)-3-(2-甲基吡咯啶-1-基)丙-1-醇(52)(363mg,粗製)在DMF(4mL)中之攪拌溶液。將反應在0℃下攪拌0.5h並接著添加4-氟-2-甲氧基-1-硝基苯(33)(395mg,2.3mmol)。將反應在0-25℃下攪拌16h。將所得混合物用EA(30mL)和水(5mL)稀釋。將有機相用鹽水(5mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由管柱層析法(矽膠,PE/EA=1:1和DCM/MeOH=10:1)將殘餘物純化以提供呈黃色油之標題化合物53(370mg,58%二步驟)。
MS(ESI):[M+H
+]=294.9。
步驟3:(S)-2-甲氧基-4-(3-(2-甲基吡咯啶-1-基)丙氧基)苯胺(54)
在25℃下將Pd/C(40mg,10%)一次全部加至(S)-1-(3-(3-甲氧基-4-硝基苯氧基)丙基)-2-甲基吡咯啶(53) (370mg,1.3mmol)在MeOH(4mL)中之攪拌懸浮液。將反應在H
2氛圍下於25℃攪拌16h並接著通過Celite
®過濾。用MeOH(5mL)洗滌濾餅。將合併的濾液濃縮以提供呈黑色油之標題化合物54(330mg,粗製),將其用於下一步驟而無需進一步純化。
MS(ESI):[M+H
+]=264.9。
步驟4:(S)-3-(2-甲氧基-4-(3-(2-甲基吡咯啶-1-基)丙氧基)苯基)-2-甲基-6-(五氟硫基)喹唑啉-4(3H)-酮(XIII)
將2-胺基-5-(五氟硫基)苯甲酸(21)(50mg,0.2mmol)在Ac
2O(1mL)中之溶液加熱至回流經4h並接著濃縮。在25℃下將(S)-2-甲氧基-4-(3-(2-甲基吡咯啶-1-基)丙氧基)苯胺(54)(101mg,0.4mmol)和AcOH(1mL)加至殘餘物。將反應在80℃下攪拌1h。之後,將所得混合物濃縮。將殘餘物分溶在EA(10mL)與飽和NaHCO
3水溶液(5mL)之間。將有機相用鹽水(5mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由製備型HPLC將殘餘物純化以提供呈棕色固體之標題化合物XIII(24mg,呈HCl鹽,22%)。
1H NMR(400MHz, DMSO-d
6) δ=10.73(br. s., 1H), 8.41(br. s., 1H), 8.33(d, J=8.0Hz, 1H), 7.85(d, J=8.8Hz, 1H), 7.34(d, J=8.8Hz, 1H), 6.82(br. s., 1H), 6.72(d, J=8.0Hz, 1H), 4.19(br. s., 2H), 3.75(br. s., 3H), 3.45-3.40(m, 2H), 3.15-3.02(m, 2H), 2.36-2.07(m, 7H), 1.95(br. s., 2H), 1.73-1.58 (m, 1H), 1.42(d, J=5.6Hz, 3H);
MS(ESI):[M+H
+]=534.1。
實施例14:3-(4-((1-環丙基哌啶-4-基)氧基)苯基)-2-甲基-6-(五氟硫基)喹唑啉-4(3H)-酮(XIV)
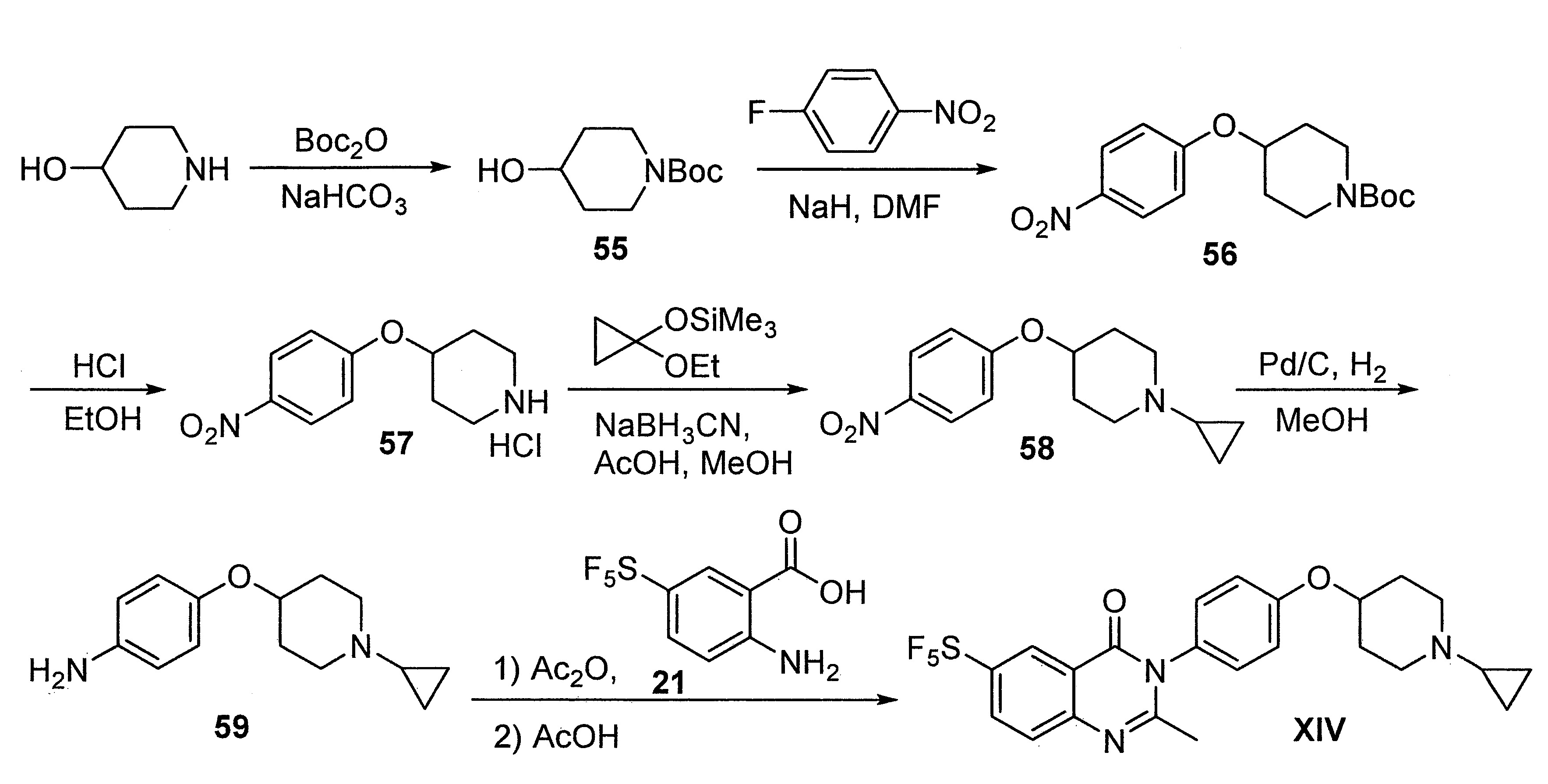 步驟1:4-羥基哌啶-1-甲酸三級丁基酯(55)
將Boc
2O(9.5g,43.6mmol)加至哌啶-4-醇(3.0g,21.8mmol)在DCM(30mL)中之攪拌漿液。將所得混合物在25℃下攪拌1h,並接著添加飽和NaHCO
3(30mL)。將反應在25℃下攪拌16h,及接著分離。將有機相用鹽水(30mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮以提供呈白色固體之標題化合物55(4.0g,91%)。
步驟2:4-(4-硝基苯氧基)哌啶-1-甲酸三級丁基酯(56)
在0℃下將NaH(0.5g,11.9mmol,在礦物油中之60%)加至4-羥基哌啶-1-甲酸三級丁基酯(55)(2.0g,9.9mmol)在DMF(20mL)中之攪拌溶液。將所得漿液在0℃下攪拌30min並接著添加1-氟-4-硝基苯(1.7g,11.9mmol)。將所得混合物在25℃下攪拌16h及接著用水(60mL)淬滅並用EA(2×20mL)萃取。將合併的有機相用鹽水(2×30mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由管柱層析法(矽膠,PE/EA=20:1-10:1)將殘餘物純化以提供呈淺黃色固體之標題化合物56(2.0g,62%)。
1H NMR(400MHz, CDCl
3)δ=8.20(d, J=9.2Hz, 2H), 6.95(d, J=9.2Hz, 2H), 4.65-4.53(m, 1H), 3.78-3.60(m, 2H), 3.46-3.31(m, 2H), 2.05-1.89(m, 2H), 1.88-1.69(m, 2H), 1.47(s, 9H)。
步驟3:4-(4-硝基苯氧基)哌啶鹽酸鹽(57)
將濃HCl水溶液(10mL)加至4-(4-硝基苯氧基)哌啶-1-甲酸三級丁基酯(56)(2.0g,6.2mmol)在EtOH(10mL)中之攪拌溶液。將所得混合物在25℃下攪拌4h並接著濃縮以提供呈黃色固體之標題化合物57(1.6g,99%)。
MS(ESI):[M+H]
+=222.9。
步驟4:1-環丙基-4-(4-硝基苯氧基)哌啶(58)
將NaBH
3CN(0.3g,4.5mmol)加至4-(4-硝基苯氧基)哌啶鹽酸鹽(57)(0.5g,2.3mmol)和(1-乙氧基環丙氧基)三甲矽烷(0.6g,3.4mmol)在AcOH(5mL)和MeOH(5mL)中之攪拌溶液。將反應加熱至65℃,攪拌18h並接著濃縮。將殘餘物溶於EA(20mL)中,並用NaOH水溶液(20mg,1M)洗滌。將有機相用無水MgSO
4乾燥,過濾和濃縮以提供呈灰白色油之標題化合物58(0.6g,粗製)。將粗製產物用於下一步驟而無需進一步純化。
MS(ESI):[M+H]
+=262.9。
步驟5:4-((1-環丙基哌啶-4-基)氧基)苯胺(59)
將Pd/C(30mg,10%)加至1-環丙基-4-(4-硝基苯氧基)哌啶(58)(0.3g,1.2mmol)在MeOH中之攪拌溶液。將反應在H
2下於25℃攪拌16h並接著過濾。將濾液濃縮以提供呈灰白色油之標題化合物59(0.2g,粗製)。將粗製產物用於下一步驟而無需進一步純化。
MS(ESI):[M+H]
+=232.9。
步驟6:3-(4-((1-環丙基哌啶-4-基)氧基)苯基)-2-甲基-6-(五氟硫基)喹唑啉-4(3H)-酮(XIV)
將2-胺基-5-(五氟硫基)苯甲酸(21)(50mg,0.2mmol)在Ac
2O(1mL)中之溶液在回流下加熱2h並接著濃縮。將4-((1-環丙基哌啶-4-基)氧基)苯胺(59)(50mg,0.2mmol)和AcOH(1mL)加至殘餘物。將反應在80℃下攪拌2h並接著濃縮。藉由製備型HPLC將殘餘物純化以提供呈灰白色固體之標題化合物XIV(40mg,42%)。
1H NMR(400MHz, CD
3OD) δ=8.64(s, 1H), 8.46-8.35(m, 1H), 7.98-7.87(m, 1H), 7.51-7.36(m, 2H), 7.35-7.22(m, 2H), 4.87-4.72(m, 1H), 3.77(d, J=12.3Hz, 1H), 3.64-3.54(m, 2H), 3.49-3.37(m, 1H), 3.02-2.88(m, 1H), 2.54-2.40(m, 3H), 2.39-2.10(m, 3H), 2.08-1.87(m, 1H), 1.22-0.92(m, 4H);
MS(ESI):[M+H]
+=502.1。
實施例15:3-(4-((1-環丁基哌啶-4-基)氧基)苯基)-2-甲基-6-(五氟硫基)喹唑啉-4(3H)-酮(XV)
步驟1:4-羥基哌啶-1-甲酸三級丁基酯(55)
將Boc
2O(9.5g,43.6mmol)加至哌啶-4-醇(3.0g,21.8mmol)在DCM(30mL)中之攪拌漿液。將所得混合物在25℃下攪拌1h,並接著添加飽和NaHCO
3(30mL)。將反應在25℃下攪拌16h,及接著分離。將有機相用鹽水(30mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮以提供呈白色固體之標題化合物55(4.0g,91%)。
步驟2:4-(4-硝基苯氧基)哌啶-1-甲酸三級丁基酯(56)
在0℃下將NaH(0.5g,11.9mmol,在礦物油中之60%)加至4-羥基哌啶-1-甲酸三級丁基酯(55)(2.0g,9.9mmol)在DMF(20mL)中之攪拌溶液。將所得漿液在0℃下攪拌30min並接著添加1-氟-4-硝基苯(1.7g,11.9mmol)。將所得混合物在25℃下攪拌16h及接著用水(60mL)淬滅並用EA(2×20mL)萃取。將合併的有機相用鹽水(2×30mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由管柱層析法(矽膠,PE/EA=20:1-10:1)將殘餘物純化以提供呈淺黃色固體之標題化合物56(2.0g,62%)。
1H NMR(400MHz, CDCl
3)δ=8.20(d, J=9.2Hz, 2H), 6.95(d, J=9.2Hz, 2H), 4.65-4.53(m, 1H), 3.78-3.60(m, 2H), 3.46-3.31(m, 2H), 2.05-1.89(m, 2H), 1.88-1.69(m, 2H), 1.47(s, 9H)。
步驟3:4-(4-硝基苯氧基)哌啶鹽酸鹽(57)
將濃HCl水溶液(10mL)加至4-(4-硝基苯氧基)哌啶-1-甲酸三級丁基酯(56)(2.0g,6.2mmol)在EtOH(10mL)中之攪拌溶液。將所得混合物在25℃下攪拌4h並接著濃縮以提供呈黃色固體之標題化合物57(1.6g,99%)。
MS(ESI):[M+H]
+=222.9。
步驟4:1-環丙基-4-(4-硝基苯氧基)哌啶(58)
將NaBH
3CN(0.3g,4.5mmol)加至4-(4-硝基苯氧基)哌啶鹽酸鹽(57)(0.5g,2.3mmol)和(1-乙氧基環丙氧基)三甲矽烷(0.6g,3.4mmol)在AcOH(5mL)和MeOH(5mL)中之攪拌溶液。將反應加熱至65℃,攪拌18h並接著濃縮。將殘餘物溶於EA(20mL)中,並用NaOH水溶液(20mg,1M)洗滌。將有機相用無水MgSO
4乾燥,過濾和濃縮以提供呈灰白色油之標題化合物58(0.6g,粗製)。將粗製產物用於下一步驟而無需進一步純化。
MS(ESI):[M+H]
+=262.9。
步驟5:4-((1-環丙基哌啶-4-基)氧基)苯胺(59)
將Pd/C(30mg,10%)加至1-環丙基-4-(4-硝基苯氧基)哌啶(58)(0.3g,1.2mmol)在MeOH中之攪拌溶液。將反應在H
2下於25℃攪拌16h並接著過濾。將濾液濃縮以提供呈灰白色油之標題化合物59(0.2g,粗製)。將粗製產物用於下一步驟而無需進一步純化。
MS(ESI):[M+H]
+=232.9。
步驟6:3-(4-((1-環丙基哌啶-4-基)氧基)苯基)-2-甲基-6-(五氟硫基)喹唑啉-4(3H)-酮(XIV)
將2-胺基-5-(五氟硫基)苯甲酸(21)(50mg,0.2mmol)在Ac
2O(1mL)中之溶液在回流下加熱2h並接著濃縮。將4-((1-環丙基哌啶-4-基)氧基)苯胺(59)(50mg,0.2mmol)和AcOH(1mL)加至殘餘物。將反應在80℃下攪拌2h並接著濃縮。藉由製備型HPLC將殘餘物純化以提供呈灰白色固體之標題化合物XIV(40mg,42%)。
1H NMR(400MHz, CD
3OD) δ=8.64(s, 1H), 8.46-8.35(m, 1H), 7.98-7.87(m, 1H), 7.51-7.36(m, 2H), 7.35-7.22(m, 2H), 4.87-4.72(m, 1H), 3.77(d, J=12.3Hz, 1H), 3.64-3.54(m, 2H), 3.49-3.37(m, 1H), 3.02-2.88(m, 1H), 2.54-2.40(m, 3H), 2.39-2.10(m, 3H), 2.08-1.87(m, 1H), 1.22-0.92(m, 4H);
MS(ESI):[M+H]
+=502.1。
實施例15:3-(4-((1-環丁基哌啶-4-基)氧基)苯基)-2-甲基-6-(五氟硫基)喹唑啉-4(3H)-酮(XV)
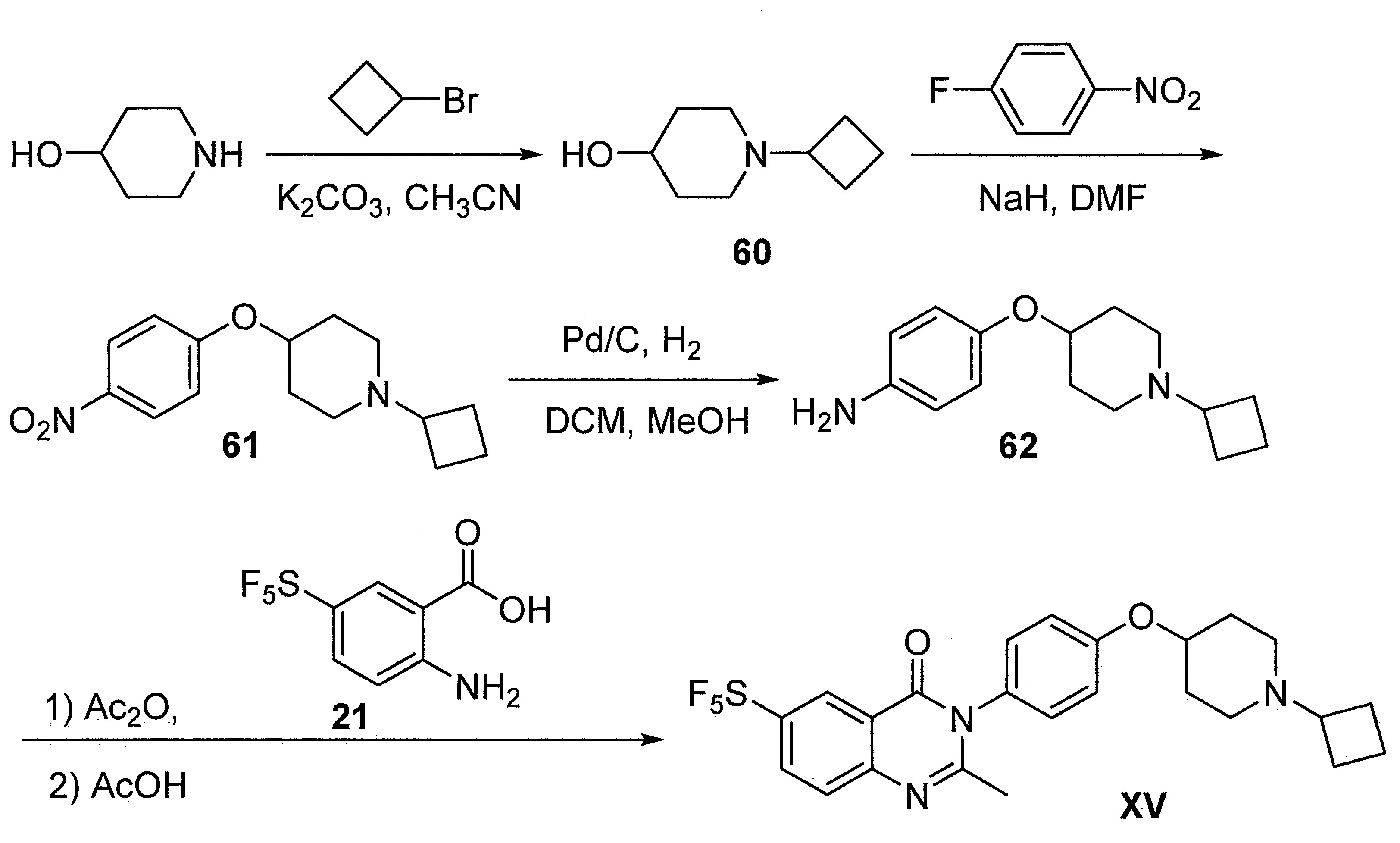 步驟1:1-環丁基哌啶-4-醇(60)
將K
2CO
3(3.4g,24.7mmol)加至哌啶-4-醇(1.0g,9.9mmol)和溴環丁烷(1.6g,11.9mmol)在CH
3CN(20mL)中之攪拌漿液。將反應在回流下加熱48h並接著冷卻至25℃。將所得混合物過濾並將濾餅用CH
3CN(30mL)洗滌。將合併的濾液濃縮以提供呈灰白色油之標題化合物60(1.4g,粗製)。將粗製產物用於下一步驟而無需進一步純化。
1H NMR(400MHz, CDCl
3) δ=3.79-3.61(m, 1H), 3.09(d, J=13.8Hz, 1H), 2.74-2.55(m, 2H), 2.08-1.82(m, 6H), 1.79-1.48(m, 5H), 1.46-1.31(m, 1H)。
步驟2:1-環丁基-4-(4-硝基苯氧基)哌啶(61)
在0℃下將NaH(0.7g,18.0mmol,在礦物油中之60%)加至1-環丁基哌啶-4-醇(60)(1.4g,9.0mmol)在DMF(20mL)中之攪拌溶液。將所得混合物在0℃下攪拌30min並接著添加1-氟-4-硝基苯(1.3g,9.0mmol)。將所得混合物在25℃下攪拌16h及然後用水(60mL)淬滅並用EA萃取(2×20mL)。將合併的有機相用鹽水(2×30mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由管柱層析法(矽膠,DCM/MeOH=100:1-20:1)將殘餘物純化以提供呈黃色固體之標題化合物61(0.8g,32%)。
1H NMR(400MHz ,CDCl
3) δ=8.18(d, J=9.2Hz, 2H), 6.94(d, J=9.2Hz, 2H), 4.46(d, J=3.2Hz, 1H), 2.80-2.69(m, 1H), 2.68-2.51(m, 2H), 2.27-2.13(m, 2H), 2.12-1.96(m, 4H), 1.96-1.78(m, 4H), 1.77-1.56(m, 2H);
MS(ESI):[M+H]
+=276.9。
步驟3:4-((1-環丁基哌啶-4-基)氧基)苯胺(62)
將Pd/C(70mg,10%)加至1-環丁基-4-(4-硝基苯氧基)哌啶(61)(0.7g,2.5mmol)在MeOH(15mL)中之攪拌溶液。將反應在H
2氛圍下於25℃攪拌16h並接著過濾。將濾液濃縮以提供呈棕色固體之標題化合物62(0.6g,96%)。
1H NMR(400MHz, CDCl
3) δ=6.75(d, J=8.8Hz, 2H), 6.62(d, J=8.8Hz, 2H), 4.13(br. s., 1H), 3.43(br. s., 2H), 2.78-2.68(m, 1H), 2.68-2.52(m, 2H), 2.25-2.09(m, 2H), 2.06-1.88(m, 4H), 1.84-1.74(m, 2H), 1.73-1.57(m, 2H);
MS(ESI):[M+H]
+=246.9。
步驟4:3-(4-((1-環丁基哌啶-4-基)氧基)苯基)-2-甲基-6-(五氟硫基)喹唑啉-4(3H)-酮(XV)
將2-胺基-5-(五氟硫基)苯甲酸(21)(0.1g,0.4mmol)在Ac
2O(1mL)中之溶液在回流下加熱2h並接著濃縮。將4-((1-環丁基哌啶-4-基)氧基)苯胺(62)(86mg,0.35mmol)和AcOH(1mL)加至殘餘物。將反應在80℃下攪拌2h並接著濃縮。藉由製備型HPLC將殘餘物純化以提供呈灰白色固體之標題化合物XV(90mg,50%)。
1H NMR(400MHz, CD
3OD) δ=8.65(s, 1H), 8.43(d, J=8.4Hz, 1H), 7.94(d, J=9.2Hz, 1H), 7.48-7.36(m, 2H), 7.35-7.21(m, 2H), 4.82-4.69(m, 1H), 3.83-3.70(m, 1H), 3.60(d, J=12.5Hz, 1H), 3.41(d, J=12.1Hz, 1H), 3.21-3.11(m, 1H), 3.10-2.93(m, 1H), 2.50(s, 3H), 2.45-2.27(m, 5H), 2.27-2.10(m, 2H), 2.06-1.73(m, 3H);
MS(ESI):[M+H]
+=516.1。
實施例16:3-(4-((1-環戊基哌啶-4-基)氧基)苯基)-2-甲基-6-(五氟硫基)喹唑啉-4(3H)-酮(XVI)
步驟1:1-環丁基哌啶-4-醇(60)
將K
2CO
3(3.4g,24.7mmol)加至哌啶-4-醇(1.0g,9.9mmol)和溴環丁烷(1.6g,11.9mmol)在CH
3CN(20mL)中之攪拌漿液。將反應在回流下加熱48h並接著冷卻至25℃。將所得混合物過濾並將濾餅用CH
3CN(30mL)洗滌。將合併的濾液濃縮以提供呈灰白色油之標題化合物60(1.4g,粗製)。將粗製產物用於下一步驟而無需進一步純化。
1H NMR(400MHz, CDCl
3) δ=3.79-3.61(m, 1H), 3.09(d, J=13.8Hz, 1H), 2.74-2.55(m, 2H), 2.08-1.82(m, 6H), 1.79-1.48(m, 5H), 1.46-1.31(m, 1H)。
步驟2:1-環丁基-4-(4-硝基苯氧基)哌啶(61)
在0℃下將NaH(0.7g,18.0mmol,在礦物油中之60%)加至1-環丁基哌啶-4-醇(60)(1.4g,9.0mmol)在DMF(20mL)中之攪拌溶液。將所得混合物在0℃下攪拌30min並接著添加1-氟-4-硝基苯(1.3g,9.0mmol)。將所得混合物在25℃下攪拌16h及然後用水(60mL)淬滅並用EA萃取(2×20mL)。將合併的有機相用鹽水(2×30mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由管柱層析法(矽膠,DCM/MeOH=100:1-20:1)將殘餘物純化以提供呈黃色固體之標題化合物61(0.8g,32%)。
1H NMR(400MHz ,CDCl
3) δ=8.18(d, J=9.2Hz, 2H), 6.94(d, J=9.2Hz, 2H), 4.46(d, J=3.2Hz, 1H), 2.80-2.69(m, 1H), 2.68-2.51(m, 2H), 2.27-2.13(m, 2H), 2.12-1.96(m, 4H), 1.96-1.78(m, 4H), 1.77-1.56(m, 2H);
MS(ESI):[M+H]
+=276.9。
步驟3:4-((1-環丁基哌啶-4-基)氧基)苯胺(62)
將Pd/C(70mg,10%)加至1-環丁基-4-(4-硝基苯氧基)哌啶(61)(0.7g,2.5mmol)在MeOH(15mL)中之攪拌溶液。將反應在H
2氛圍下於25℃攪拌16h並接著過濾。將濾液濃縮以提供呈棕色固體之標題化合物62(0.6g,96%)。
1H NMR(400MHz, CDCl
3) δ=6.75(d, J=8.8Hz, 2H), 6.62(d, J=8.8Hz, 2H), 4.13(br. s., 1H), 3.43(br. s., 2H), 2.78-2.68(m, 1H), 2.68-2.52(m, 2H), 2.25-2.09(m, 2H), 2.06-1.88(m, 4H), 1.84-1.74(m, 2H), 1.73-1.57(m, 2H);
MS(ESI):[M+H]
+=246.9。
步驟4:3-(4-((1-環丁基哌啶-4-基)氧基)苯基)-2-甲基-6-(五氟硫基)喹唑啉-4(3H)-酮(XV)
將2-胺基-5-(五氟硫基)苯甲酸(21)(0.1g,0.4mmol)在Ac
2O(1mL)中之溶液在回流下加熱2h並接著濃縮。將4-((1-環丁基哌啶-4-基)氧基)苯胺(62)(86mg,0.35mmol)和AcOH(1mL)加至殘餘物。將反應在80℃下攪拌2h並接著濃縮。藉由製備型HPLC將殘餘物純化以提供呈灰白色固體之標題化合物XV(90mg,50%)。
1H NMR(400MHz, CD
3OD) δ=8.65(s, 1H), 8.43(d, J=8.4Hz, 1H), 7.94(d, J=9.2Hz, 1H), 7.48-7.36(m, 2H), 7.35-7.21(m, 2H), 4.82-4.69(m, 1H), 3.83-3.70(m, 1H), 3.60(d, J=12.5Hz, 1H), 3.41(d, J=12.1Hz, 1H), 3.21-3.11(m, 1H), 3.10-2.93(m, 1H), 2.50(s, 3H), 2.45-2.27(m, 5H), 2.27-2.10(m, 2H), 2.06-1.73(m, 3H);
MS(ESI):[M+H]
+=516.1。
實施例16:3-(4-((1-環戊基哌啶-4-基)氧基)苯基)-2-甲基-6-(五氟硫基)喹唑啉-4(3H)-酮(XVI)
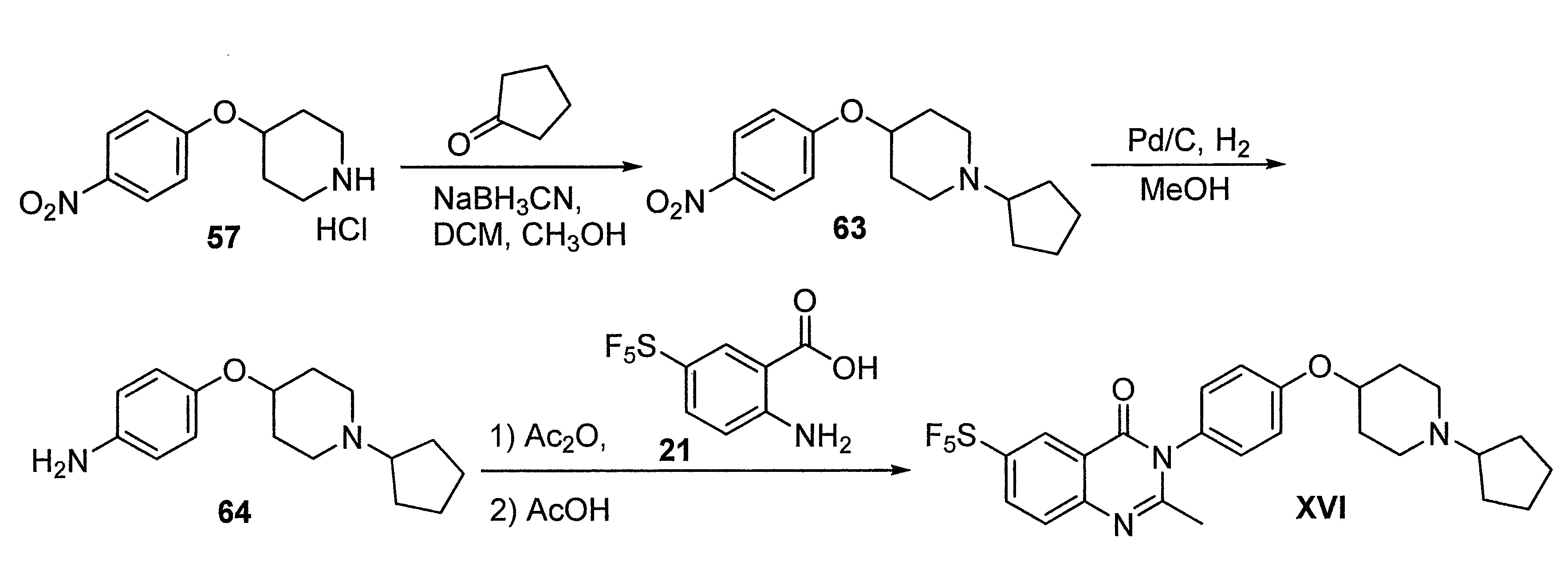 步驟1:1-環戊基-4-(4-硝基苯氧基)哌啶(63)
將一滴AcOH加至4-(4-硝基苯氧基)哌啶鹽酸鹽(57)(0.2g,0.9mmol)和環戊酮(0.1g,1.1mmol)在DCM(2mL)中之攪拌溶液並接著在25℃下攪拌1h。將MeOH(2mL)和NaBH
3CN(68mg,1.1mmol)加至反應。將反應在25℃下攪拌2h並接著用飽和NH
4Cl水溶液(5mL)淬滅。將所得混合物用EA(2×5mL)萃取。將合併的有機相用鹽水(5mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮以提供呈棕色油之標題化合物63(260mg,粗製)。將粗製產物用於下一步驟而無需進一步純化。
MS(ESI):[M+H]
+=290.9。
步驟2:(4-(4-胺基苯氧基)哌啶-1-基)(環戊基)甲酮(64)
將Pd/C(30mg,10%)加至環戊基(4-(4-硝基苯氧基)哌啶-1-基)甲酮(63)(260mg,0.9mmol)在MeOH(3mL)中之攪拌溶液。將反應在H
2氛圍下於25℃攪拌16h並接著過濾。將濾液濃縮以提供呈灰白色油之標題化合物64(230mg,粗製)。將粗製產物用於下一步驟而無需進一步純化。
MS(ESI):[M+H]
+=260.9。
步驟3:3-(4-((1-(環戊烷羰基)哌啶-4-基)氧基)苯基)-2-甲基-6-(五氟硫基)喹唑啉-4(3H)-酮(XVI)
將2-胺基-5-(五氟硫基)苯甲酸(21)(50mg,0.2mmol)在Ac
2O(1mL)中之溶液在回流下加熱2h並接著濃縮。將(4-(4-胺基苯氧基)哌啶-1-基)(環戊基)甲酮(64)(70mg,0.2mmol)和AcOH(1mL)加至殘餘物。將反應在80℃下攪拌2h並接著濃縮。藉由製備型HPLC將殘餘物純化以提供呈灰白色固體之標題化合物XVI(30mg,28%)。
1H NMR(400MHz, CD
3OD) δ=8.60(d, J=2.4Hz, 1H), 8.29(dd, J=2.4, 9.2Hz, 1H), 7.84(d, J=9.2Hz, 1H), 7.42-7.32(m, 2H), 7.30-7.18(m, 2H), 3.79-3.70(m, 1H), 3.69-3.46(m, 3H), 3.42-3.35(m, 2H), 3.24-3.12(m, 1H), 2.53-2.43(m, 1H), 2.33(s, 3H), 2.29-2.04(m, 4H), 2.02-1.82(m, 3H), 1.79-1.68(m, 3H);
MS(ESI):[M+H]
+=530.1。
實施例17:2-甲基-7-(五氟硫基)-3-{4-[3-(吡咯啶-1-基)-丙氧基]苯基}-3,4-二氫喹唑啉-4-酮(XVII)
步驟1:1-環戊基-4-(4-硝基苯氧基)哌啶(63)
將一滴AcOH加至4-(4-硝基苯氧基)哌啶鹽酸鹽(57)(0.2g,0.9mmol)和環戊酮(0.1g,1.1mmol)在DCM(2mL)中之攪拌溶液並接著在25℃下攪拌1h。將MeOH(2mL)和NaBH
3CN(68mg,1.1mmol)加至反應。將反應在25℃下攪拌2h並接著用飽和NH
4Cl水溶液(5mL)淬滅。將所得混合物用EA(2×5mL)萃取。將合併的有機相用鹽水(5mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮以提供呈棕色油之標題化合物63(260mg,粗製)。將粗製產物用於下一步驟而無需進一步純化。
MS(ESI):[M+H]
+=290.9。
步驟2:(4-(4-胺基苯氧基)哌啶-1-基)(環戊基)甲酮(64)
將Pd/C(30mg,10%)加至環戊基(4-(4-硝基苯氧基)哌啶-1-基)甲酮(63)(260mg,0.9mmol)在MeOH(3mL)中之攪拌溶液。將反應在H
2氛圍下於25℃攪拌16h並接著過濾。將濾液濃縮以提供呈灰白色油之標題化合物64(230mg,粗製)。將粗製產物用於下一步驟而無需進一步純化。
MS(ESI):[M+H]
+=260.9。
步驟3:3-(4-((1-(環戊烷羰基)哌啶-4-基)氧基)苯基)-2-甲基-6-(五氟硫基)喹唑啉-4(3H)-酮(XVI)
將2-胺基-5-(五氟硫基)苯甲酸(21)(50mg,0.2mmol)在Ac
2O(1mL)中之溶液在回流下加熱2h並接著濃縮。將(4-(4-胺基苯氧基)哌啶-1-基)(環戊基)甲酮(64)(70mg,0.2mmol)和AcOH(1mL)加至殘餘物。將反應在80℃下攪拌2h並接著濃縮。藉由製備型HPLC將殘餘物純化以提供呈灰白色固體之標題化合物XVI(30mg,28%)。
1H NMR(400MHz, CD
3OD) δ=8.60(d, J=2.4Hz, 1H), 8.29(dd, J=2.4, 9.2Hz, 1H), 7.84(d, J=9.2Hz, 1H), 7.42-7.32(m, 2H), 7.30-7.18(m, 2H), 3.79-3.70(m, 1H), 3.69-3.46(m, 3H), 3.42-3.35(m, 2H), 3.24-3.12(m, 1H), 2.53-2.43(m, 1H), 2.33(s, 3H), 2.29-2.04(m, 4H), 2.02-1.82(m, 3H), 1.79-1.68(m, 3H);
MS(ESI):[M+H]
+=530.1。
實施例17:2-甲基-7-(五氟硫基)-3-{4-[3-(吡咯啶-1-基)-丙氧基]苯基}-3,4-二氫喹唑啉-4-酮(XVII)
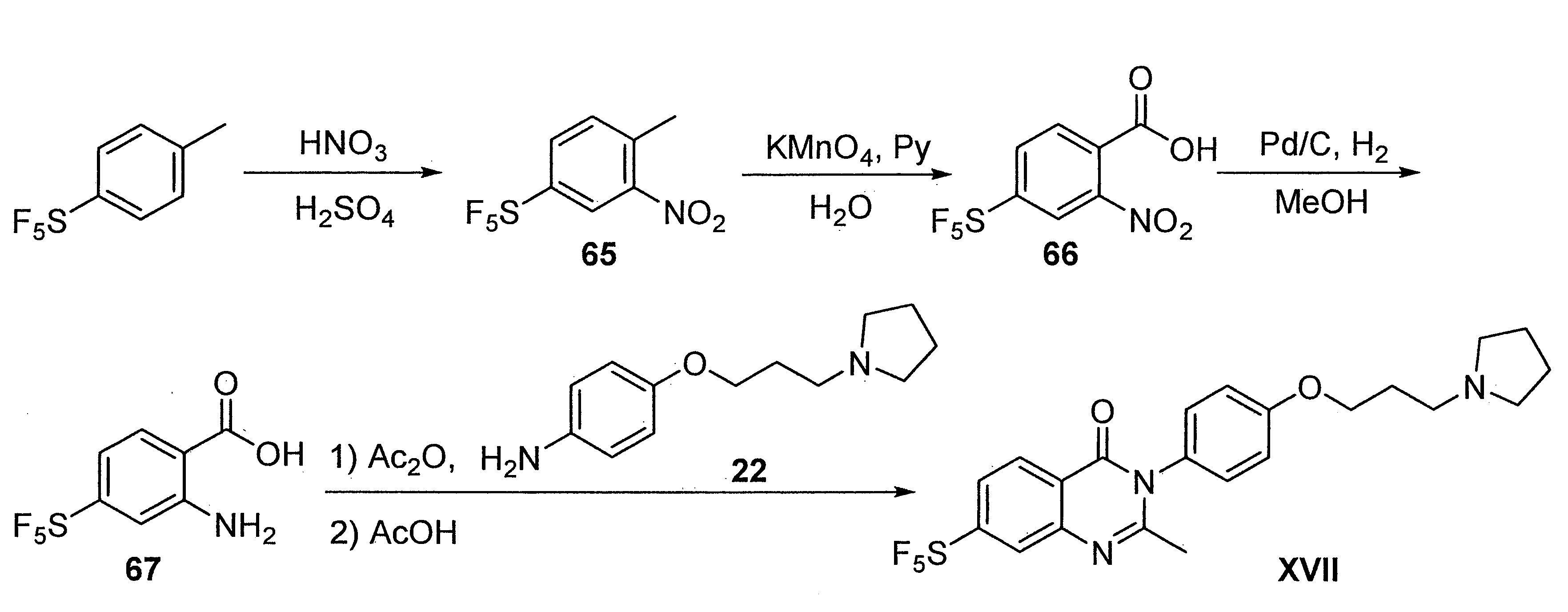 步驟1:1-甲基-2-硝基-4-(五氟硫基)苯(65)
在0℃下將H
2SO
4(2mL)和HNO
3(2mL)的混合物加至1-甲基-4-(五氟硫基)苯(500mg,2.3mmol)在H
2SO
4(2mL)中之攪拌溶液。將反應在25℃下攪拌3h並接著倒進水(20mL)中。將所得混合物用EA(20mL)萃取。將有機相用H
2O(3×20mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由管柱層析法(矽膠,PE和PE/DCM=-2:1)將殘餘物相純化以提供呈淺黃色油之標題化合物65(581mg,96%)。
1H NMR(400 MHz, CDCl
3) δ=8.39(s, 1H), 7.89(d, J=6.0Hz, 1H), 7.49(d, J=8.8Hz, 1H), 2.68(s, 3H)。
步驟2:1-甲基-2-硝基-4-(五氟硫基)苯(66)
將KMnO
4(2.1g,13.3mmol)加至1-甲基-2-硝基-4-(五氟硫基)苯(65)(581mg,2.2mmol)在吡啶(5mL)和H
2O(10mL)中之攪拌溶液。將反應在95℃下攪拌48h並接著添加額外的KMnO
4(2.1g,13.3mmol)。將混合物在95℃下攪拌12h。將所得混合物用HCl水溶液(1M)酸化直到pH=1及用EA(50mL)萃取。將有機相用鹽水(3×50mL)洗滌和濃縮。藉由管柱層析法(矽膠,DCM/MeOH=10:1)將殘餘物純化以提供呈白色固體之標題化合物66(450mg,70%)。
MS(ESI):[M-H
+]=291.9。
步驟3:2-胺基-4-(五氟硫基)苯甲酸(67)
將Pd/C(50mg,10%)加至1-甲基-2-硝基-4-(五氟硫基)苯(66)(450mg,1.54mmol)在MeOH(15mL)中之攪拌溶液。將反應在H
2氛圍下於25℃攪拌10h並接著過濾。將濾液濃縮以提供呈淺黃色固體之標題化合物67(350mg,86%)。
MS(ESI):[M+H
+]=263.8。
步驟4:2-甲基-3-(4-(3-(吡咯啶-1-基)丙氧基)苯基)-7-(五氟硫基)喹唑啉-4(3H)-酮(XVII)
將2-胺基-5-(五氟硫基)苯甲酸(21)(200mg,0.8mmol)在Ac
2O(2mL)中之溶液在回流下加熱2h並接著濃縮。將2-胺基-4-(五氟硫基)苯甲酸(67)(235mg,1.07mmol)和AcOH(2mL)加至殘餘物。將混合物在80℃下攪拌1h並接著濃縮。藉由製備型HPLC將殘餘物純化以提供呈灰白色固體之標題化合物XVII(330mg,63%)。
1H NMR(400 MHz, CD
3OD) δ=8.38(d, J=8.4Hz, 1H), 8.11(s, 1H), 7.99(d, J=8.4Hz, 1H), 7.34(d, J=8.8Hz, 2H), 7.18(d, J=8.4Hz, 2H), 4.22(t, J=5.8Hz, 2H), 3.72-3.69(m, 2H), 3.46(t, J=5.8Hz, 2H), 3.17-3.11(m, 2H), 2.31(s, 3H) , 2.29-2.25(m, 2H), 2.21-2.15(m, 2H), 2.06-2.01(m, 2H);
MS(ESI):[M+H
+]
=490.1。
實施例18:3-(4-((1-(環戊烷羰基)哌啶-4-基)氧基)苯基)-2-甲基-6-(五氟硫基)喹唑啉-4(3H)-酮(XVIII)
步驟1:1-甲基-2-硝基-4-(五氟硫基)苯(65)
在0℃下將H
2SO
4(2mL)和HNO
3(2mL)的混合物加至1-甲基-4-(五氟硫基)苯(500mg,2.3mmol)在H
2SO
4(2mL)中之攪拌溶液。將反應在25℃下攪拌3h並接著倒進水(20mL)中。將所得混合物用EA(20mL)萃取。將有機相用H
2O(3×20mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由管柱層析法(矽膠,PE和PE/DCM=-2:1)將殘餘物相純化以提供呈淺黃色油之標題化合物65(581mg,96%)。
1H NMR(400 MHz, CDCl
3) δ=8.39(s, 1H), 7.89(d, J=6.0Hz, 1H), 7.49(d, J=8.8Hz, 1H), 2.68(s, 3H)。
步驟2:1-甲基-2-硝基-4-(五氟硫基)苯(66)
將KMnO
4(2.1g,13.3mmol)加至1-甲基-2-硝基-4-(五氟硫基)苯(65)(581mg,2.2mmol)在吡啶(5mL)和H
2O(10mL)中之攪拌溶液。將反應在95℃下攪拌48h並接著添加額外的KMnO
4(2.1g,13.3mmol)。將混合物在95℃下攪拌12h。將所得混合物用HCl水溶液(1M)酸化直到pH=1及用EA(50mL)萃取。將有機相用鹽水(3×50mL)洗滌和濃縮。藉由管柱層析法(矽膠,DCM/MeOH=10:1)將殘餘物純化以提供呈白色固體之標題化合物66(450mg,70%)。
MS(ESI):[M-H
+]=291.9。
步驟3:2-胺基-4-(五氟硫基)苯甲酸(67)
將Pd/C(50mg,10%)加至1-甲基-2-硝基-4-(五氟硫基)苯(66)(450mg,1.54mmol)在MeOH(15mL)中之攪拌溶液。將反應在H
2氛圍下於25℃攪拌10h並接著過濾。將濾液濃縮以提供呈淺黃色固體之標題化合物67(350mg,86%)。
MS(ESI):[M+H
+]=263.8。
步驟4:2-甲基-3-(4-(3-(吡咯啶-1-基)丙氧基)苯基)-7-(五氟硫基)喹唑啉-4(3H)-酮(XVII)
將2-胺基-5-(五氟硫基)苯甲酸(21)(200mg,0.8mmol)在Ac
2O(2mL)中之溶液在回流下加熱2h並接著濃縮。將2-胺基-4-(五氟硫基)苯甲酸(67)(235mg,1.07mmol)和AcOH(2mL)加至殘餘物。將混合物在80℃下攪拌1h並接著濃縮。藉由製備型HPLC將殘餘物純化以提供呈灰白色固體之標題化合物XVII(330mg,63%)。
1H NMR(400 MHz, CD
3OD) δ=8.38(d, J=8.4Hz, 1H), 8.11(s, 1H), 7.99(d, J=8.4Hz, 1H), 7.34(d, J=8.8Hz, 2H), 7.18(d, J=8.4Hz, 2H), 4.22(t, J=5.8Hz, 2H), 3.72-3.69(m, 2H), 3.46(t, J=5.8Hz, 2H), 3.17-3.11(m, 2H), 2.31(s, 3H) , 2.29-2.25(m, 2H), 2.21-2.15(m, 2H), 2.06-2.01(m, 2H);
MS(ESI):[M+H
+]
=490.1。
實施例18:3-(4-((1-(環戊烷羰基)哌啶-4-基)氧基)苯基)-2-甲基-6-(五氟硫基)喹唑啉-4(3H)-酮(XVIII)
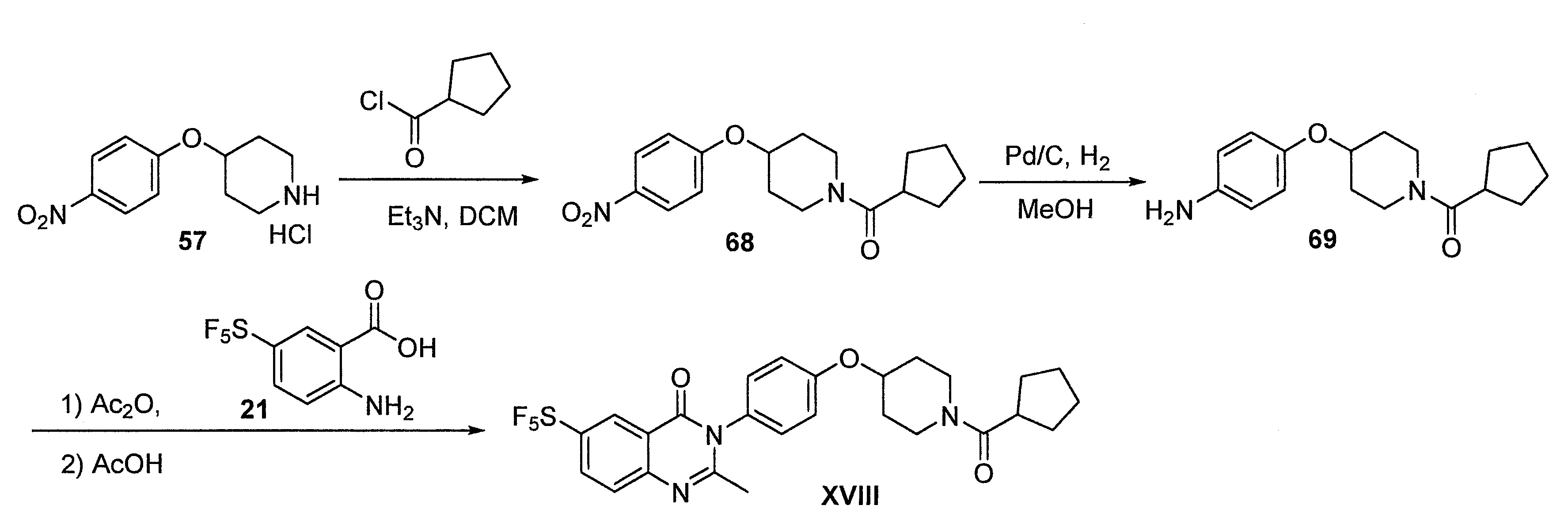 步驟1:環戊基(4-(4-硝基苯氧基)哌啶-1-基)甲酮(68)
在0℃下將Et
3N(117mg,1.2mmol)和環戊烷羰基氯(56mg,0.43mmol)加至4-(4-硝基苯氧基)哌啶鹽酸鹽(57)(100mg,0.39mmol)之攪拌溶液。將反應在25℃下攪拌2h並接著用DCM(4mL)稀釋。將所得混合物用鹽水(5mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮以提供呈棕色油之標題化合物68(120mg,98%),將其用於下一步驟而無需進一步純化。
MS(ESI):[M+H]
+=318.9。
步驟2:(4-(4-胺基苯氧基)哌啶-1-基)(環戊基)甲酮(69)
將Pd/C(10mg,10%)加至環戊基(4-(4-硝基苯氧基)哌啶-1-基)甲酮(68)(120mg,0.38mmol)在MeOH(2mL)中之攪拌溶液。將反應在H
2氛圍下於25℃攪拌16h並接著過濾。將濾液濃縮以提供呈棕色油之標題化合物69(100mg,92%),將其用於下一步驟而無需進一步純化。
MS(ESI):[M+H]
+=288.9。
步驟3:3-(4-((1-(環戊烷羰基)哌啶-4-基)氧基)苯基)-2-甲基-6-(五氟硫基)喹唑啉-4(3H)-酮(XVIII)
將2-胺基-5-(五氟硫基)苯甲酸(21)(50mg,0.2mmol)在Ac
2O(1mL)中之溶液加熱至回流經2h並接著濃縮至乾。將(4-(4-胺基苯氧基)哌啶-1-基)(環戊基)甲酮(69)(70mg,0.2mmol)和AcOH(1mL)加至殘餘物。將反應在80℃下攪拌2h並接著濃縮。藉由製備型HPLC將殘餘物純化以提供呈灰白色固體之標題化合物XVIII(30mg,28%)。
1H NMR(400MHz, CD
3OD) δ=8.58(d, J=2.4Hz, 1H), 8.24(dd, J=2.4, 9.2Hz, 1H), 7.81(d, J=9.2Hz, 1H), 7.31(d, J=8.8Hz, 2H), 7.19(d, J=8.8Hz, 2H), 4.79-4.72(m, 1H), 4.62(br. s., 1H), 3.95-3.82(m, 2H), 3.64-3.53(m, 2H), 3.16-3.06(m, 1H), 2.38-2.21(m, 3H), 2.12-1.97(m, 2H), 1.91-1.57(m, 9H);
MS(ESI):[M+H]
+=558.1。
實施例19:3-(4-((1-(環己烷羰基)哌啶-4-基)氧基)苯基)-2-甲基-6-(五氟硫基)喹唑啉-4(3H)-酮(XIX)
步驟1:環戊基(4-(4-硝基苯氧基)哌啶-1-基)甲酮(68)
在0℃下將Et
3N(117mg,1.2mmol)和環戊烷羰基氯(56mg,0.43mmol)加至4-(4-硝基苯氧基)哌啶鹽酸鹽(57)(100mg,0.39mmol)之攪拌溶液。將反應在25℃下攪拌2h並接著用DCM(4mL)稀釋。將所得混合物用鹽水(5mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮以提供呈棕色油之標題化合物68(120mg,98%),將其用於下一步驟而無需進一步純化。
MS(ESI):[M+H]
+=318.9。
步驟2:(4-(4-胺基苯氧基)哌啶-1-基)(環戊基)甲酮(69)
將Pd/C(10mg,10%)加至環戊基(4-(4-硝基苯氧基)哌啶-1-基)甲酮(68)(120mg,0.38mmol)在MeOH(2mL)中之攪拌溶液。將反應在H
2氛圍下於25℃攪拌16h並接著過濾。將濾液濃縮以提供呈棕色油之標題化合物69(100mg,92%),將其用於下一步驟而無需進一步純化。
MS(ESI):[M+H]
+=288.9。
步驟3:3-(4-((1-(環戊烷羰基)哌啶-4-基)氧基)苯基)-2-甲基-6-(五氟硫基)喹唑啉-4(3H)-酮(XVIII)
將2-胺基-5-(五氟硫基)苯甲酸(21)(50mg,0.2mmol)在Ac
2O(1mL)中之溶液加熱至回流經2h並接著濃縮至乾。將(4-(4-胺基苯氧基)哌啶-1-基)(環戊基)甲酮(69)(70mg,0.2mmol)和AcOH(1mL)加至殘餘物。將反應在80℃下攪拌2h並接著濃縮。藉由製備型HPLC將殘餘物純化以提供呈灰白色固體之標題化合物XVIII(30mg,28%)。
1H NMR(400MHz, CD
3OD) δ=8.58(d, J=2.4Hz, 1H), 8.24(dd, J=2.4, 9.2Hz, 1H), 7.81(d, J=9.2Hz, 1H), 7.31(d, J=8.8Hz, 2H), 7.19(d, J=8.8Hz, 2H), 4.79-4.72(m, 1H), 4.62(br. s., 1H), 3.95-3.82(m, 2H), 3.64-3.53(m, 2H), 3.16-3.06(m, 1H), 2.38-2.21(m, 3H), 2.12-1.97(m, 2H), 1.91-1.57(m, 9H);
MS(ESI):[M+H]
+=558.1。
實施例19:3-(4-((1-(環己烷羰基)哌啶-4-基)氧基)苯基)-2-甲基-6-(五氟硫基)喹唑啉-4(3H)-酮(XIX)
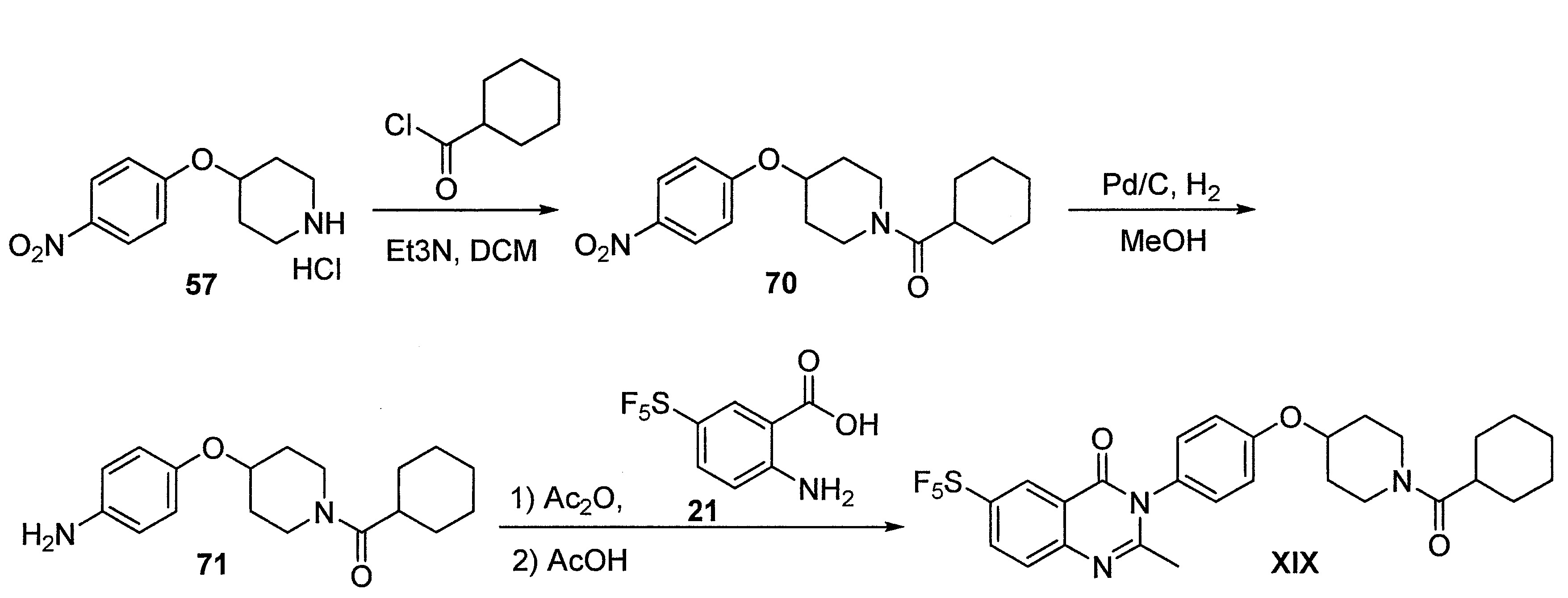 步驟1:環己基(4-(4-硝基苯氧基)哌啶-1-基)甲酮(70)
在25℃下將環己烷羰基氯(170mg,1.2mmol)加至4-(4-硝基苯氧基)哌啶鹽酸鹽(57)(250mg,0.9mmol)和Et
3N(117mg,1.2mmol)在DCM(3mL)中之攪拌溶液。將反應在25℃下攪拌16h並接著用DCM(6mL)稀釋。將反應用鹽水(5mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮以提供呈棕色油之標題化合物70(300mg,93%)。
MS(ESI):[M+H]
+=332.9。
步驟2:(4-(4-胺基苯氧基)哌啶-1-基)(環戊基)甲酮(71)
將Pd/C(30mg,10%)加至環己基(4-(4-硝基苯氧基)哌啶-1-基)甲酮(70)(300mg,0.9mmol)在MeOH(6mL)中之攪拌溶液。將反應在H
2氛圍下於25℃攪拌16h並接著過濾。將濾液濃縮以提供呈棕色油之標題化合物71(250mg,92%)。
1H NMR(400MHz, CD
3OD) δ=6.92(d, J=8.4Hz, 2H), 6.79(d, J=8.8Hz, 2H), 4.42-4.34(m, 1H), 3.62-3.50(m, 2H), 3.48-3.33(m, 2H), 2.36-2.25(m, 1H), 1.98-1.86(m, 4H), 1.57-1.45(m, 4H), 1.30-1.18(m, 6H)。
步驟3:3-(4-((1-(環己烷羰基)哌啶-4-基)氧基)苯基)-2-甲基-6-(五氟硫基)喹唑啉-4(3H)-酮(XIX)
將2-胺基-5-(五氟硫基)苯甲酸(21)(50mg,0.2mmol)在Ac
2O(1mL)中之溶液在回流下加熱2h並接著濃縮。將(4-(4-胺基苯氧基)哌啶-1-基)(環己基)甲酮(71)(70mg,0.2mmol)和AcOH(1mL)加至殘餘物。將反應在80℃下攪拌1h並接著濃縮。藉由製備型HPLC將殘餘物純化以提供呈灰白色固體之標題化合物XIX(20mg,18%)。
1H NMR(400MHz, CD
3OD) δ=8.66(s, 1H), 8.12(d, J=7.6Hz, 1H), 7.91(d, J=8.0Hz, 1H), 7.25-7.13(m, 2H), 7.13-7.00(m, 2H), 4.62(s, 1H), 3.91-3.72(m, 2H), 3.71-3.59(m, 1H), 3.57-3.40(m, 1H), 2.56-2.47(m, 1H), 2.41(s, 3H), 2.06-1.94(m, 2H), 1.93-1.78(m, 4H), 1.78-1.64(m, 3H), 1.62-1.47(m, 2H), 1.38-1.17(m, 3H);
MS(ESI):[M+H]
+=572.1。
實施例20:2-甲基-3-(4-((1-(1-甲基哌啶-4-羰基)哌啶-4-基)氧基)苯基)-6-(五氟硫基)喹唑啉-4(3H)-酮(XX)
步驟1:環己基(4-(4-硝基苯氧基)哌啶-1-基)甲酮(70)
在25℃下將環己烷羰基氯(170mg,1.2mmol)加至4-(4-硝基苯氧基)哌啶鹽酸鹽(57)(250mg,0.9mmol)和Et
3N(117mg,1.2mmol)在DCM(3mL)中之攪拌溶液。將反應在25℃下攪拌16h並接著用DCM(6mL)稀釋。將反應用鹽水(5mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮以提供呈棕色油之標題化合物70(300mg,93%)。
MS(ESI):[M+H]
+=332.9。
步驟2:(4-(4-胺基苯氧基)哌啶-1-基)(環戊基)甲酮(71)
將Pd/C(30mg,10%)加至環己基(4-(4-硝基苯氧基)哌啶-1-基)甲酮(70)(300mg,0.9mmol)在MeOH(6mL)中之攪拌溶液。將反應在H
2氛圍下於25℃攪拌16h並接著過濾。將濾液濃縮以提供呈棕色油之標題化合物71(250mg,92%)。
1H NMR(400MHz, CD
3OD) δ=6.92(d, J=8.4Hz, 2H), 6.79(d, J=8.8Hz, 2H), 4.42-4.34(m, 1H), 3.62-3.50(m, 2H), 3.48-3.33(m, 2H), 2.36-2.25(m, 1H), 1.98-1.86(m, 4H), 1.57-1.45(m, 4H), 1.30-1.18(m, 6H)。
步驟3:3-(4-((1-(環己烷羰基)哌啶-4-基)氧基)苯基)-2-甲基-6-(五氟硫基)喹唑啉-4(3H)-酮(XIX)
將2-胺基-5-(五氟硫基)苯甲酸(21)(50mg,0.2mmol)在Ac
2O(1mL)中之溶液在回流下加熱2h並接著濃縮。將(4-(4-胺基苯氧基)哌啶-1-基)(環己基)甲酮(71)(70mg,0.2mmol)和AcOH(1mL)加至殘餘物。將反應在80℃下攪拌1h並接著濃縮。藉由製備型HPLC將殘餘物純化以提供呈灰白色固體之標題化合物XIX(20mg,18%)。
1H NMR(400MHz, CD
3OD) δ=8.66(s, 1H), 8.12(d, J=7.6Hz, 1H), 7.91(d, J=8.0Hz, 1H), 7.25-7.13(m, 2H), 7.13-7.00(m, 2H), 4.62(s, 1H), 3.91-3.72(m, 2H), 3.71-3.59(m, 1H), 3.57-3.40(m, 1H), 2.56-2.47(m, 1H), 2.41(s, 3H), 2.06-1.94(m, 2H), 1.93-1.78(m, 4H), 1.78-1.64(m, 3H), 1.62-1.47(m, 2H), 1.38-1.17(m, 3H);
MS(ESI):[M+H]
+=572.1。
實施例20:2-甲基-3-(4-((1-(1-甲基哌啶-4-羰基)哌啶-4-基)氧基)苯基)-6-(五氟硫基)喹唑啉-4(3H)-酮(XX)
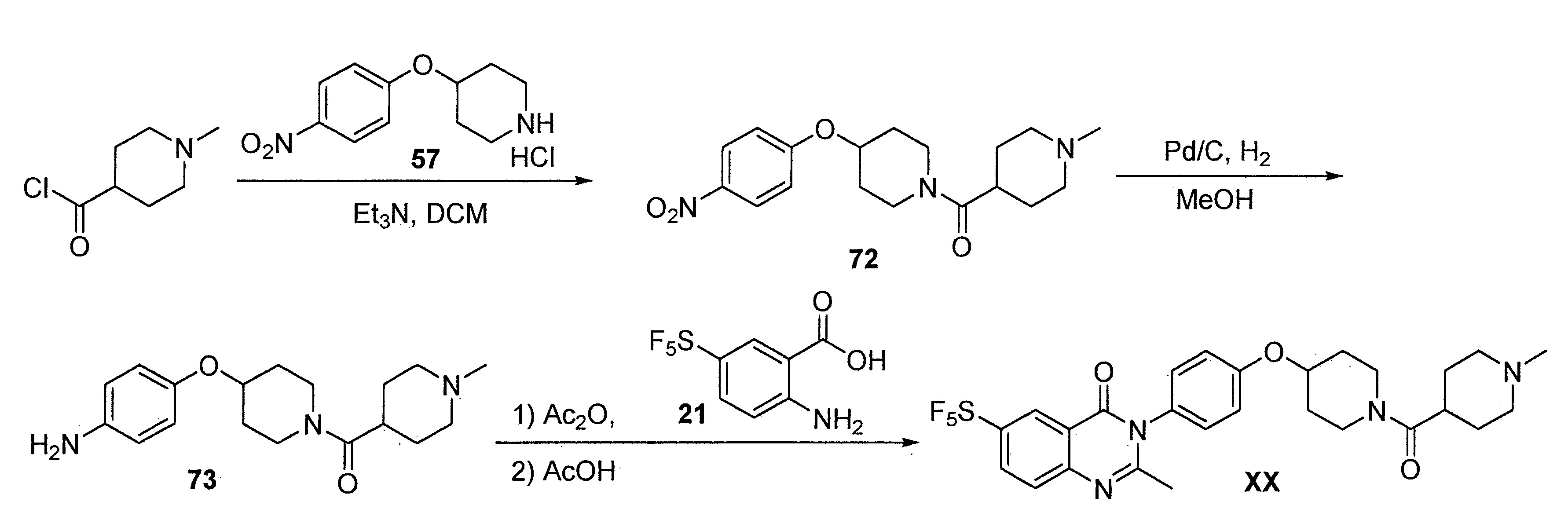 步驟1:(1-甲基哌啶-4-基)(4-(4-硝基苯氧基)哌啶-1-基)甲酮(72)
在0℃下將Et
3N(254mg,2.5mmol)和1-甲基哌啶-4-羰基氯(122mg,0.8mmol)一次全部加至4-(4-硝基苯氧基)哌啶鹽酸鹽(57)(163mg,0.6mmol)在DCM(3mL)中之攪拌懸浮液。將反應在0-25℃下攪拌16h。將混合物分溶在DCM(10mL)和水(5mL)之間。將有機相用鹽水(3mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由製備型HPLC將殘餘物純化以提供呈白色固體之標題化合物72(100mg,46%)。
MS(ESI):[M+H
+]=347.9。
步驟2:(4-(4-胺基苯氧基)哌啶-1-基)(1-甲基哌啶-4-基)甲酮(73)
在25℃下,向(1-甲基哌啶-4-基)(4-(4-硝基苯氧基)哌啶-1-基)甲酮(72)(100mg,0.3mmol)在MeOH(4mL)中之攪拌懸浮液中一次全部添加Pd/C(10mg,10%)。將反應在H
2氛圍下於25℃攪拌16h並接著通過Celite
®過濾。用MeOH(5mL)洗滌濾餅。將合併的濾液濃縮以提供呈油之標題化合物73(91mg,粗製),將其用於下一步驟而無需進一步純化。
MS(ESI):[M+H
+]=317.9。
步驟4:2-甲基-3-(4-((1-(1-甲基哌啶-4-羰基)哌啶-4-基)氧基)苯基)-6-(五氟硫基)喹唑啉-4(3H)-酮(XX)
將2-胺基-5-(五氟硫基)苯甲酸(21)(50mg,0.2mmol)在Ac
2O(1mL)中之溶液在回流下加熱2h並接著濃縮。在25℃下將(4-(4-胺基苯氧基)哌啶-1-基)(1-甲基哌啶-4-基)甲酮(73)(73mg,0.2mmol)和AcOH(1mL)加至殘餘物。將反應在80℃下攪拌1h。之後,將所得混合物濃縮。將殘餘物分溶在EA(10mL)與飽和NaHCO
3水溶液(5mL)之間。將有機相用鹽水(5mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由製備型HPLC將殘餘物純化以提供呈棕色固體之標題化合物XX(37mg,呈HCl鹽,26%)。
1H NMR(400MHz, DMSO-d
6) δ=10.42(br. s., 1H), 8.43 (d, J=2.4Hz, 1H), 8.34(dd, J=2.4, 8.8Hz, 1H), 7.88(d, J=8.4Hz, 1H), 7.38(d, J=8.4Hz, 2H), 7.17(d, J=7.2Hz, 2H), 4.73(br. s., 1H), 4.00-3.88(m, 2H), 3.49-3.34(m, 3H), 3.27-3.12(m, 2H), 2.98-2.90(m, 2H), 2.72-2.66(m, 3H), 2.21(s, 3H), 2.07-1.79(m, 6H), 1.66-1.53(m, 2H);
MS(ESI):[M+H
+]=587.1。
實施例21:2-甲基-3-(4-((1-(四氫-2H-哌喃-4-羰基)哌啶-4-基)氧基)苯基)-6-(五氟硫基)喹唑啉-4(3H)-酮(XXI)
步驟1:(1-甲基哌啶-4-基)(4-(4-硝基苯氧基)哌啶-1-基)甲酮(72)
在0℃下將Et
3N(254mg,2.5mmol)和1-甲基哌啶-4-羰基氯(122mg,0.8mmol)一次全部加至4-(4-硝基苯氧基)哌啶鹽酸鹽(57)(163mg,0.6mmol)在DCM(3mL)中之攪拌懸浮液。將反應在0-25℃下攪拌16h。將混合物分溶在DCM(10mL)和水(5mL)之間。將有機相用鹽水(3mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由製備型HPLC將殘餘物純化以提供呈白色固體之標題化合物72(100mg,46%)。
MS(ESI):[M+H
+]=347.9。
步驟2:(4-(4-胺基苯氧基)哌啶-1-基)(1-甲基哌啶-4-基)甲酮(73)
在25℃下,向(1-甲基哌啶-4-基)(4-(4-硝基苯氧基)哌啶-1-基)甲酮(72)(100mg,0.3mmol)在MeOH(4mL)中之攪拌懸浮液中一次全部添加Pd/C(10mg,10%)。將反應在H
2氛圍下於25℃攪拌16h並接著通過Celite
®過濾。用MeOH(5mL)洗滌濾餅。將合併的濾液濃縮以提供呈油之標題化合物73(91mg,粗製),將其用於下一步驟而無需進一步純化。
MS(ESI):[M+H
+]=317.9。
步驟4:2-甲基-3-(4-((1-(1-甲基哌啶-4-羰基)哌啶-4-基)氧基)苯基)-6-(五氟硫基)喹唑啉-4(3H)-酮(XX)
將2-胺基-5-(五氟硫基)苯甲酸(21)(50mg,0.2mmol)在Ac
2O(1mL)中之溶液在回流下加熱2h並接著濃縮。在25℃下將(4-(4-胺基苯氧基)哌啶-1-基)(1-甲基哌啶-4-基)甲酮(73)(73mg,0.2mmol)和AcOH(1mL)加至殘餘物。將反應在80℃下攪拌1h。之後,將所得混合物濃縮。將殘餘物分溶在EA(10mL)與飽和NaHCO
3水溶液(5mL)之間。將有機相用鹽水(5mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由製備型HPLC將殘餘物純化以提供呈棕色固體之標題化合物XX(37mg,呈HCl鹽,26%)。
1H NMR(400MHz, DMSO-d
6) δ=10.42(br. s., 1H), 8.43 (d, J=2.4Hz, 1H), 8.34(dd, J=2.4, 8.8Hz, 1H), 7.88(d, J=8.4Hz, 1H), 7.38(d, J=8.4Hz, 2H), 7.17(d, J=7.2Hz, 2H), 4.73(br. s., 1H), 4.00-3.88(m, 2H), 3.49-3.34(m, 3H), 3.27-3.12(m, 2H), 2.98-2.90(m, 2H), 2.72-2.66(m, 3H), 2.21(s, 3H), 2.07-1.79(m, 6H), 1.66-1.53(m, 2H);
MS(ESI):[M+H
+]=587.1。
實施例21:2-甲基-3-(4-((1-(四氫-2H-哌喃-4-羰基)哌啶-4-基)氧基)苯基)-6-(五氟硫基)喹唑啉-4(3H)-酮(XXI)
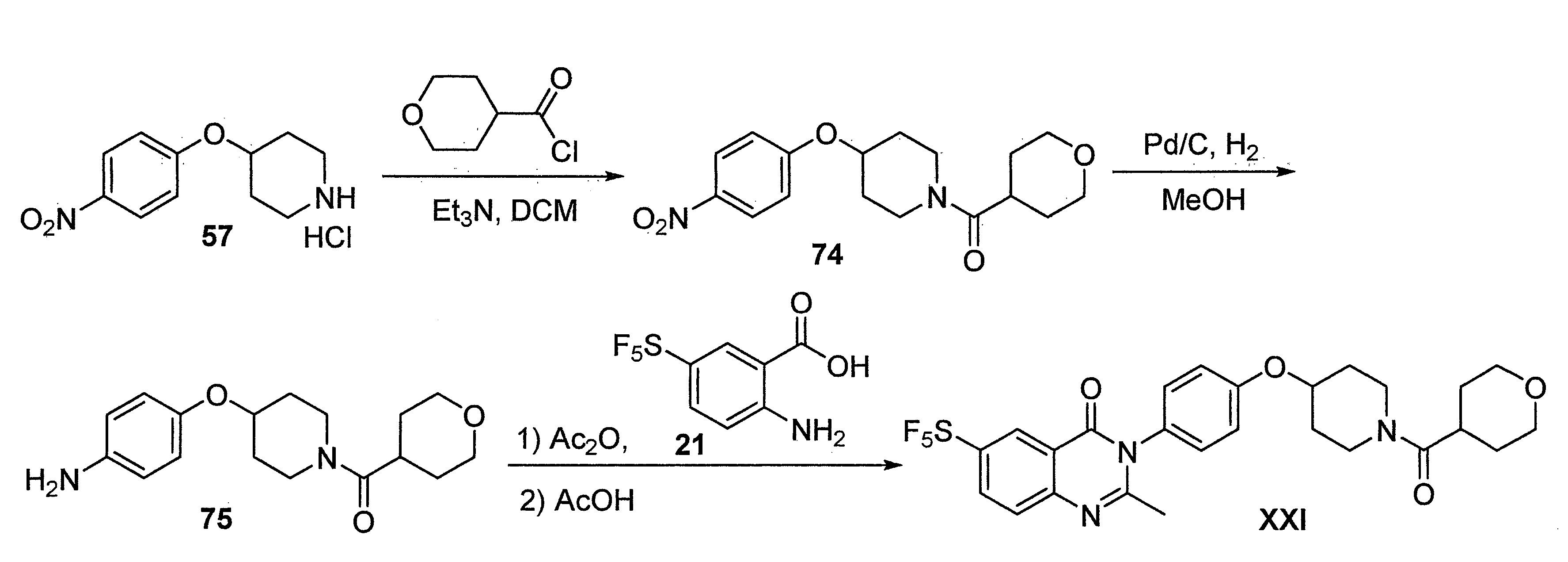 步驟1:(4-(4-硝基苯氧基)哌啶-1-基)(四氫-2H-哌喃-4-基)甲酮(74)
將四氫-2H-哌喃-4-羰基氯(126.0mg,0.8mmol)在0℃下滴加至4-(4-硝基苯氧基)哌啶鹽酸鹽(57)(0.2g,0.8mmol)在Et
3N(235.0mg,2.3mmol)中之溶液。將反應攪拌30min及然後使升溫至25℃,接著用DCM(4mL)稀釋。將所得混合物用(5mL)鹽水洗滌,用無水Na
2SO
4乾燥,過濾和濃縮以提供呈棕色油之標題化合物74(250mg,97%)。
MS(ESI):[M+H]
+=334.9。
步驟2:(4-(4-胺基苯氧基)哌啶-1-基)(四氫-2H-哌喃-4-基)甲酮(75)
將Pd/C(25mg,10%)加至(4-(4-硝基苯氧基)哌啶-1-基)(四氫-2H-哌喃-4-基)甲酮(74)(250mg,0.7mmol)在MeOH中之攪拌溶液。將反應在H
2氛圍下於25℃攪拌16h並接著過濾。將濾液濃縮以提供呈棕色油之標題化合物75(230mg,粗製)。將粗製產物用於下一步驟而無需進一步純化。
MS(ESI):[M+H]
+=305.0。
步驟3:2-甲基-3-(4-((1-(四氫-2H-哌喃-4-羰基)哌啶-4-基)氧基)苯基)-6-(五氟硫基)喹唑啉-4(3H)-酮(XXI)
將2-胺基-5-(五氟硫基)苯甲酸(21)(50mg,0.2mmol)在Ac
2O(1mL)中之溶液在回流下加熱2h並接著濃縮。將(4-(4-胺基苯氧基)哌啶-1-基)(四氫-2H-哌喃-4-基)甲酮(75)(70mg,0.2mmol)和AcOH(1mL)加至殘餘物。將反應在80℃下攪拌2h並接著濃縮。藉由製備型HPLC將殘餘物純化以提供呈灰白色固體之標題化合物XXI(15mg,14%)。
1H NMR(400MHz, CD
3OD) δ=8.59(d, J=2.0Hz, 1H), 8.26(dd, J=2.0, 9.2Hz, 1H), 7.82(d, J=9.2Hz, 1H), 7.32(d, J=8.4Hz, 2H), 7.19(d, J=8.0Hz, 2H), 4.76(d, J=3.2Hz, 1H), 3.97(d, J=9.2Hz, 2H), 3.94-3.79(m, 2H), 3.66-3.56(m, 2H), 3.56-3.46(m, 2H), 3.35(s, 3H), 3.06-2.97(m, 1H), 2.17-1.94(m, 2H), 1.92-1.72(m, 4H), 1.64(d, J=12.8Hz, 2H);
MS(ESI):[M+H]
+=574.2。
實施例22:3-(2-甲氧基-4-((1-(四氫-2H-哌喃-4-羰基)哌啶-4-基)氧基)苯基)-2-甲基-6-(五氟硫基)喹唑啉-4(3H)-酮(XXII)
步驟1:(4-(4-硝基苯氧基)哌啶-1-基)(四氫-2H-哌喃-4-基)甲酮(74)
將四氫-2H-哌喃-4-羰基氯(126.0mg,0.8mmol)在0℃下滴加至4-(4-硝基苯氧基)哌啶鹽酸鹽(57)(0.2g,0.8mmol)在Et
3N(235.0mg,2.3mmol)中之溶液。將反應攪拌30min及然後使升溫至25℃,接著用DCM(4mL)稀釋。將所得混合物用(5mL)鹽水洗滌,用無水Na
2SO
4乾燥,過濾和濃縮以提供呈棕色油之標題化合物74(250mg,97%)。
MS(ESI):[M+H]
+=334.9。
步驟2:(4-(4-胺基苯氧基)哌啶-1-基)(四氫-2H-哌喃-4-基)甲酮(75)
將Pd/C(25mg,10%)加至(4-(4-硝基苯氧基)哌啶-1-基)(四氫-2H-哌喃-4-基)甲酮(74)(250mg,0.7mmol)在MeOH中之攪拌溶液。將反應在H
2氛圍下於25℃攪拌16h並接著過濾。將濾液濃縮以提供呈棕色油之標題化合物75(230mg,粗製)。將粗製產物用於下一步驟而無需進一步純化。
MS(ESI):[M+H]
+=305.0。
步驟3:2-甲基-3-(4-((1-(四氫-2H-哌喃-4-羰基)哌啶-4-基)氧基)苯基)-6-(五氟硫基)喹唑啉-4(3H)-酮(XXI)
將2-胺基-5-(五氟硫基)苯甲酸(21)(50mg,0.2mmol)在Ac
2O(1mL)中之溶液在回流下加熱2h並接著濃縮。將(4-(4-胺基苯氧基)哌啶-1-基)(四氫-2H-哌喃-4-基)甲酮(75)(70mg,0.2mmol)和AcOH(1mL)加至殘餘物。將反應在80℃下攪拌2h並接著濃縮。藉由製備型HPLC將殘餘物純化以提供呈灰白色固體之標題化合物XXI(15mg,14%)。
1H NMR(400MHz, CD
3OD) δ=8.59(d, J=2.0Hz, 1H), 8.26(dd, J=2.0, 9.2Hz, 1H), 7.82(d, J=9.2Hz, 1H), 7.32(d, J=8.4Hz, 2H), 7.19(d, J=8.0Hz, 2H), 4.76(d, J=3.2Hz, 1H), 3.97(d, J=9.2Hz, 2H), 3.94-3.79(m, 2H), 3.66-3.56(m, 2H), 3.56-3.46(m, 2H), 3.35(s, 3H), 3.06-2.97(m, 1H), 2.17-1.94(m, 2H), 1.92-1.72(m, 4H), 1.64(d, J=12.8Hz, 2H);
MS(ESI):[M+H]
+=574.2。
實施例22:3-(2-甲氧基-4-((1-(四氫-2H-哌喃-4-羰基)哌啶-4-基)氧基)苯基)-2-甲基-6-(五氟硫基)喹唑啉-4(3H)-酮(XXII)
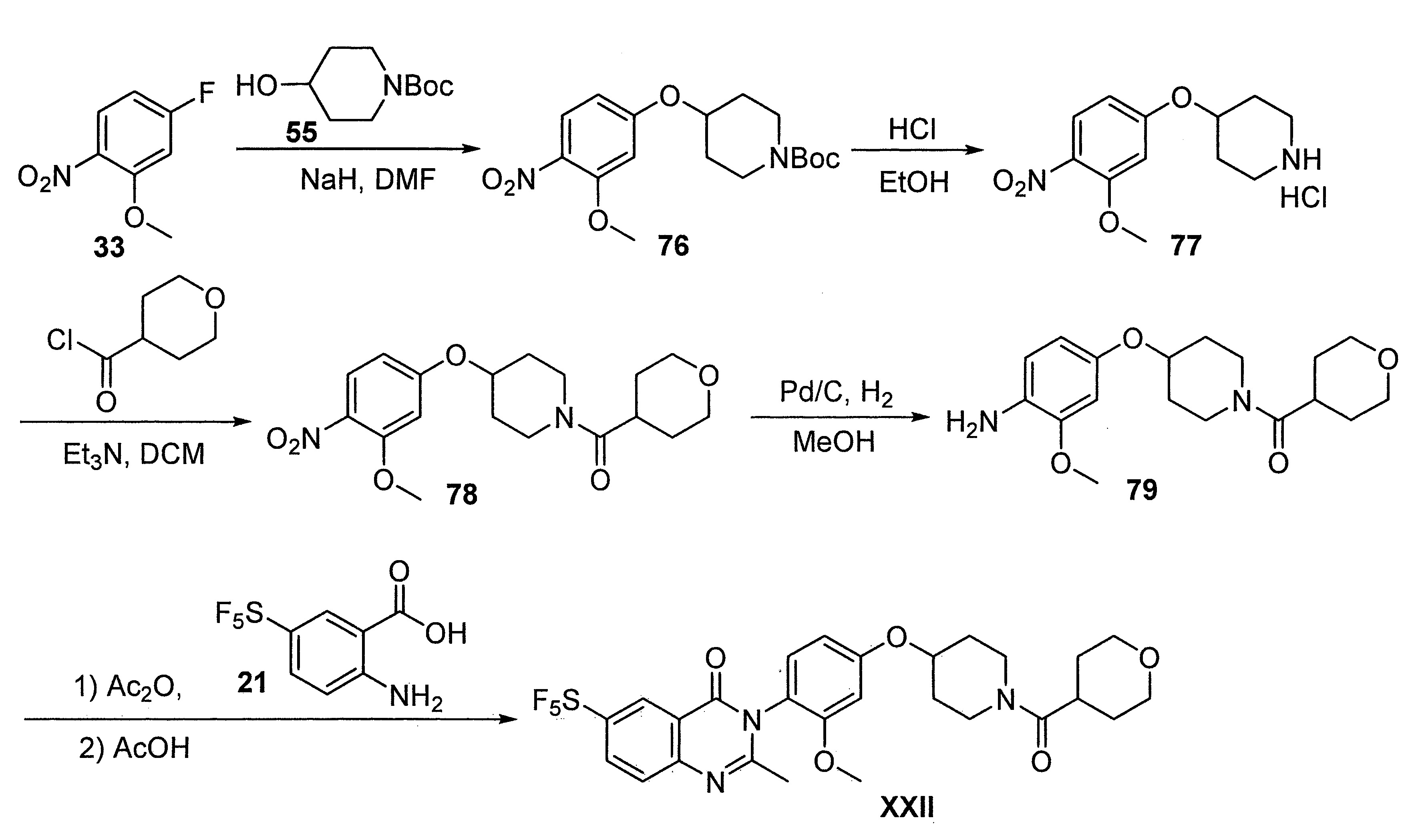 步驟1:4-(3-甲氧基-4-硝基苯氧基)哌啶-1-甲酸三級丁基酯(76)
在0℃下經0.5h將NaH(0.5g,11.7mmol)分批加至4-羥基哌啶-1-甲酸三級丁基酯(55)(1.4g,7.0mmol)在DMF (10mL)中之攪拌溶液,並再接著添加4-氟-2-甲氧基-1-硝基苯(33)(1.0g,5.8mmol)。將反應在25℃下攪拌16h。將反應混合物用飽和NH
4Cl水溶液(20mL)淬滅並用EA(2×20mL)萃取。將合併的有機相用鹽水(2×20mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由管柱層析法(矽膠,PE/EA=100:1-5:1)將殘餘物純化以提供呈黃色固體之標題化合物76(1.7g,83%)。
1H NMR(400MHz, CDCl
3) δ=8.00(d, J=8.8Hz, 1H), 6.54(s, 1H), 6.50(d, J=9.2Hz, 1H), 4.62-4.50(m, 1H), 3.94(s, 3H), 3.74-3.63(m, 2H), 3.44-3.30(m, 2H), 2.00-1.90(m, 2H), 1.84-1.72(m, 2H), 1.47(s, 9H)。
步驟2:4-(3-甲氧基-4-硝基苯氧基)哌啶鹽酸鹽(77)
將濃鹽酸水溶液(10mL)加至4-(3-甲氧基-4-硝基苯氧基)哌啶-1-甲酸三級丁基酯(76)(1.7g,4.8mmol)在EtOH(20mL)中之攪拌溶液。將反應在回流下加熱2h並接著濃縮以提供呈黃色固體之標題化合物77(1.3g,93%)。
1H NMR(400MHz, CD
3OD) δ=7.94(d, J=9.2Hz, 1H), 6.82(d, J=2.0Hz, 1H), 6.71(dd, J=2.0, 9.2Hz, 1H), 4.90-4.86(m, 1H), 3.95(s, 3H), 3.47-3.37(m, 2H), 3.30-3.22(m, 2H), 2.30-2.18(m, 2H), 2.12-2.00(m, 2H);
MS(ESI):[M+CH
3CN+H]
+=293.9。
步驟3:(4-(3-甲氧基-4-硝基苯氧基)哌啶-1-基)(四氫-2H-哌喃-4-基)甲酮(78)
將四氫-2H-哌喃-4-羰基氯(247mg,1.7mmol)在0℃下滴加至4-(3-甲氧基-4-硝基苯氧基)哌啶鹽酸鹽(77)(0.4g,1.4mmol)和Et
3N(0.4g,4.2mmol)在DCM(5mL)中之攪拌溶液。將反應在25℃下攪拌1h且然後用EA(15mL)接著飽和NH
4Cl水溶液(10mL)稀釋。將有機相用鹽水(10mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮以提供呈黃色油之標題化合物78(0.4g,79%)。
MS(ESI):[M+H]
+=364.9。
步驟4:(4-(4-胺基-3-甲氧基苯氧基)哌啶-1-基)(四氫-2H-哌喃-4-基)甲酮(79)
將Pd/C(40mg,10%)加至(4-(3-甲氧基-4-硝基苯氧基)哌啶-1-基)(四氫-2H-哌喃-4-基)甲酮(78)(0.4g,1.1mmol)在MeOH(4mL)中之攪拌溶液。將反應在H
2氛圍下於25℃攪拌16h並接著過濾。將濾液濃縮以提供呈油之標題化合物79(360mg,98%)。
MS(ESI):[M+H]
+=334.9。
步驟5:3-(2-甲氧基-4-((1-(四氫-2H-哌喃-4-羰基)哌啶-4-基)氧基)苯基)-2-甲基-6-(五氟硫基)喹唑啉-4(3H)-酮(XXII)
將2-胺基-5-(五氟硫基)苯甲酸(21)(50mg,0.2mmol)在Ac
2O(1mL)中之溶液在回流下加熱2h並接著濃縮。將(4-(4-胺基-3-甲氧基苯氧基)哌啶-1-基)(四氫-2H-哌喃-4-基)甲酮(79)(76mg,0.2mmol)和AcOH(1mL)加至殘餘物。將反應在80℃下攪拌1h並接著濃縮。藉由製備型HPLC將殘餘物純化以提供呈灰白色固體之標題化合物XXII(20mg,17%)。
1H NMR(400MHz, CD
3OD) δ=8.59(d, J=2.4Hz, 1H), 8.31(dd, J=2.2, 9.0Hz, 1H), 7.84(d, J=8.8Hz, 1H), 7.26(d, J=8.4Hz, 1H), 6.85(s, 1H), 6.79(d, J=8.8Hz, 1H), 4.83-4.73(m, 1H), 4.05-3.93(m, 2H), 3.93-3.84(m, 2H), 3.82(s, 3H), 3.72-3.58(m, 2H), 3.57-3.45(m, 2H), 3.07-2.96(m, 1H), 2.34(s, 3H), 2.13-1.95(m, 2H), 1.94-1.70(m, 4H), 1.69-1.57(m, 2H);
MS(ESI):[M+H]
+=604.1。
實施例23:藥理研究
在實施例中,以具有式I-XXII之化合物詳細描述藥理性質。
組織胺H3受體GTPγS結合親和力之評估
組織胺H3受體介導CNS和末梢神經系統中的組織胺信號。組織胺H3受體直接與Gi/Go偶合,導致隨後的GTPγS結合。雖然GTPγS結合分析係以與放射性配體結合分析相似的方式使用膜製劑進行,但此等為功能分析,且可用於區分促效劑、拮抗劑和逆促效劑活性。使用提供對G蛋白α次單元具有高親和力的放射性配體之[
35S]-鳥苷-5'-O-(3-硫基)三磷酸鹽進行該等分析,該配體對α次單元的固有GTP酶活性具有高度抵抗力,使其保持結合足夠的時間以允許對放射性進行計數。使用具有高組織胺H3受體表現的HEK293/Ga15/hH3R細胞株進行GTPγS結合分析,以確定代表性化合物的H3受體GTPγS結合親和力。逆促效劑和拮抗劑活性的結果係顯示於表1中,而關於逆促進效劑模式和拮抗劑模之代表性化合物的H3R GTPγS結合親和力和%抑制曲線分別顯示於圖1A、1B和2A、2B中。
1. 用於測定組織胺H3受體配體的拮抗劑和逆促效劑藥理參數之[
35S]-GTPγS結合分析:
材料:GTPγS,[35S](PerkinElmer,Cat# NEG030H001MC), DMSO(Amresco, Cat#0231),MicroScint™-20(PerkinElmer, Cat # 6013621),CelLytic(TM)M細胞分解劑(Sigma, Cat # C2978-250ML),(
R)(−)-α-甲基組織胺(Sigma, Cat # H128),GDP(Sigma, Cat # G7127),GF/C 盤(PE, CAT#6005174)。
實驗程序:1) HEK293/Ga15/hH3R的膜製備;2) 標準結合分析。簡而言之,關於膜巴拉松(parathion),將HEK293/Ga15/ hH3R細胞生長至匯合,收穫並將細胞沉澱物(cell pellet)懸浮在TEL緩衝液(50mM Tris-HCl緩衝液,1mM EGTA,0.1mM PMSF)中。均質化和在1,000g下離心10min。將上清液以46,000g離心30min。將膜沉澱物懸浮在具有0.32M蔗糖之50mM Tris(pH 7.0)中。等分試樣為1mg蛋白質/mL。保持冷凍並儲存於-80℃直至使用。藉由溶於DMSO中製成10mM的儲備液來製備所有化合物。使用10mM儲備液作為最高濃度(1μM),以在96孔盤中使用DMSO進行10點、3倍稀釋方案以製造化合物劑量盤。如下進行H3R GTPγS結合分析:在37℃下將膜解凍,在冰上冷卻,添加GDP和膜入分析緩衝液(50mM Tris-HCl,100mM NaCl,5 mM MgCl
2,pH 7.4,和0.2%,BSA)。在冰上停留20min。關於逆促效劑模式:將20μL試驗化合物(從1μM之10點、3倍稀釋)、20uL緩衝液、140μL膜溶液(GDP 10μM,膜蛋白20μg/孔)加至分析盤中,並在室溫下預培養30min。添加20μL[
35S]-GTPγS(最終200 pM)並在室溫下培養60min。關於拮抗劑模式:將20μL促效劑(
R-α-甲基組織胺,最終濃度1μM)、20μL試驗化合物(最終最高濃度1μM,3-倍稀釋,10點)、20μL[
35S]-GTPγS(最終200 pM)、140μL膜溶液(總計200μL,GDP10μM,膜蛋白20μg/孔)加至分析盤中。在室溫下培養60min。在GF/C(無PEI塗布)盤上過濾分析盤以終止分析。將GF/C盤乾燥1小時。添加50μL閃爍液並在MicroBeta上計數。
數據分析:使用下列公式將CPM值計算為%抑制:
關於逆促效劑模式:%抑制=(DMSO對照CPM-化合物CPM)/(DMSO對照CPM-GTP對照CPM)×100。
關於拮抗劑模式:%抑制=(
R-α-甲基-組織胺對照CPM-化合物CPM)/(
R-α-甲基-組織胺對照CPM-DMSO對照CPM)×100。
使用Prism5與log(抑制劑)對反應方程式計算IC
50。
2. 實驗結果:
表1:代表性化合物之HEK293/Ga15/hH3RGTPγS分析中的IC
50之數據總結。
步驟1:4-(3-甲氧基-4-硝基苯氧基)哌啶-1-甲酸三級丁基酯(76)
在0℃下經0.5h將NaH(0.5g,11.7mmol)分批加至4-羥基哌啶-1-甲酸三級丁基酯(55)(1.4g,7.0mmol)在DMF (10mL)中之攪拌溶液,並再接著添加4-氟-2-甲氧基-1-硝基苯(33)(1.0g,5.8mmol)。將反應在25℃下攪拌16h。將反應混合物用飽和NH
4Cl水溶液(20mL)淬滅並用EA(2×20mL)萃取。將合併的有機相用鹽水(2×20mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮。藉由管柱層析法(矽膠,PE/EA=100:1-5:1)將殘餘物純化以提供呈黃色固體之標題化合物76(1.7g,83%)。
1H NMR(400MHz, CDCl
3) δ=8.00(d, J=8.8Hz, 1H), 6.54(s, 1H), 6.50(d, J=9.2Hz, 1H), 4.62-4.50(m, 1H), 3.94(s, 3H), 3.74-3.63(m, 2H), 3.44-3.30(m, 2H), 2.00-1.90(m, 2H), 1.84-1.72(m, 2H), 1.47(s, 9H)。
步驟2:4-(3-甲氧基-4-硝基苯氧基)哌啶鹽酸鹽(77)
將濃鹽酸水溶液(10mL)加至4-(3-甲氧基-4-硝基苯氧基)哌啶-1-甲酸三級丁基酯(76)(1.7g,4.8mmol)在EtOH(20mL)中之攪拌溶液。將反應在回流下加熱2h並接著濃縮以提供呈黃色固體之標題化合物77(1.3g,93%)。
1H NMR(400MHz, CD
3OD) δ=7.94(d, J=9.2Hz, 1H), 6.82(d, J=2.0Hz, 1H), 6.71(dd, J=2.0, 9.2Hz, 1H), 4.90-4.86(m, 1H), 3.95(s, 3H), 3.47-3.37(m, 2H), 3.30-3.22(m, 2H), 2.30-2.18(m, 2H), 2.12-2.00(m, 2H);
MS(ESI):[M+CH
3CN+H]
+=293.9。
步驟3:(4-(3-甲氧基-4-硝基苯氧基)哌啶-1-基)(四氫-2H-哌喃-4-基)甲酮(78)
將四氫-2H-哌喃-4-羰基氯(247mg,1.7mmol)在0℃下滴加至4-(3-甲氧基-4-硝基苯氧基)哌啶鹽酸鹽(77)(0.4g,1.4mmol)和Et
3N(0.4g,4.2mmol)在DCM(5mL)中之攪拌溶液。將反應在25℃下攪拌1h且然後用EA(15mL)接著飽和NH
4Cl水溶液(10mL)稀釋。將有機相用鹽水(10mL)洗滌,用無水Na
2SO
4乾燥,過濾和濃縮以提供呈黃色油之標題化合物78(0.4g,79%)。
MS(ESI):[M+H]
+=364.9。
步驟4:(4-(4-胺基-3-甲氧基苯氧基)哌啶-1-基)(四氫-2H-哌喃-4-基)甲酮(79)
將Pd/C(40mg,10%)加至(4-(3-甲氧基-4-硝基苯氧基)哌啶-1-基)(四氫-2H-哌喃-4-基)甲酮(78)(0.4g,1.1mmol)在MeOH(4mL)中之攪拌溶液。將反應在H
2氛圍下於25℃攪拌16h並接著過濾。將濾液濃縮以提供呈油之標題化合物79(360mg,98%)。
MS(ESI):[M+H]
+=334.9。
步驟5:3-(2-甲氧基-4-((1-(四氫-2H-哌喃-4-羰基)哌啶-4-基)氧基)苯基)-2-甲基-6-(五氟硫基)喹唑啉-4(3H)-酮(XXII)
將2-胺基-5-(五氟硫基)苯甲酸(21)(50mg,0.2mmol)在Ac
2O(1mL)中之溶液在回流下加熱2h並接著濃縮。將(4-(4-胺基-3-甲氧基苯氧基)哌啶-1-基)(四氫-2H-哌喃-4-基)甲酮(79)(76mg,0.2mmol)和AcOH(1mL)加至殘餘物。將反應在80℃下攪拌1h並接著濃縮。藉由製備型HPLC將殘餘物純化以提供呈灰白色固體之標題化合物XXII(20mg,17%)。
1H NMR(400MHz, CD
3OD) δ=8.59(d, J=2.4Hz, 1H), 8.31(dd, J=2.2, 9.0Hz, 1H), 7.84(d, J=8.8Hz, 1H), 7.26(d, J=8.4Hz, 1H), 6.85(s, 1H), 6.79(d, J=8.8Hz, 1H), 4.83-4.73(m, 1H), 4.05-3.93(m, 2H), 3.93-3.84(m, 2H), 3.82(s, 3H), 3.72-3.58(m, 2H), 3.57-3.45(m, 2H), 3.07-2.96(m, 1H), 2.34(s, 3H), 2.13-1.95(m, 2H), 1.94-1.70(m, 4H), 1.69-1.57(m, 2H);
MS(ESI):[M+H]
+=604.1。
實施例23:藥理研究
在實施例中,以具有式I-XXII之化合物詳細描述藥理性質。
組織胺H3受體GTPγS結合親和力之評估
組織胺H3受體介導CNS和末梢神經系統中的組織胺信號。組織胺H3受體直接與Gi/Go偶合,導致隨後的GTPγS結合。雖然GTPγS結合分析係以與放射性配體結合分析相似的方式使用膜製劑進行,但此等為功能分析,且可用於區分促效劑、拮抗劑和逆促效劑活性。使用提供對G蛋白α次單元具有高親和力的放射性配體之[
35S]-鳥苷-5'-O-(3-硫基)三磷酸鹽進行該等分析,該配體對α次單元的固有GTP酶活性具有高度抵抗力,使其保持結合足夠的時間以允許對放射性進行計數。使用具有高組織胺H3受體表現的HEK293/Ga15/hH3R細胞株進行GTPγS結合分析,以確定代表性化合物的H3受體GTPγS結合親和力。逆促效劑和拮抗劑活性的結果係顯示於表1中,而關於逆促進效劑模式和拮抗劑模之代表性化合物的H3R GTPγS結合親和力和%抑制曲線分別顯示於圖1A、1B和2A、2B中。
1. 用於測定組織胺H3受體配體的拮抗劑和逆促效劑藥理參數之[
35S]-GTPγS結合分析:
材料:GTPγS,[35S](PerkinElmer,Cat# NEG030H001MC), DMSO(Amresco, Cat#0231),MicroScint™-20(PerkinElmer, Cat # 6013621),CelLytic(TM)M細胞分解劑(Sigma, Cat # C2978-250ML),(
R)(−)-α-甲基組織胺(Sigma, Cat # H128),GDP(Sigma, Cat # G7127),GF/C 盤(PE, CAT#6005174)。
實驗程序:1) HEK293/Ga15/hH3R的膜製備;2) 標準結合分析。簡而言之,關於膜巴拉松(parathion),將HEK293/Ga15/ hH3R細胞生長至匯合,收穫並將細胞沉澱物(cell pellet)懸浮在TEL緩衝液(50mM Tris-HCl緩衝液,1mM EGTA,0.1mM PMSF)中。均質化和在1,000g下離心10min。將上清液以46,000g離心30min。將膜沉澱物懸浮在具有0.32M蔗糖之50mM Tris(pH 7.0)中。等分試樣為1mg蛋白質/mL。保持冷凍並儲存於-80℃直至使用。藉由溶於DMSO中製成10mM的儲備液來製備所有化合物。使用10mM儲備液作為最高濃度(1μM),以在96孔盤中使用DMSO進行10點、3倍稀釋方案以製造化合物劑量盤。如下進行H3R GTPγS結合分析:在37℃下將膜解凍,在冰上冷卻,添加GDP和膜入分析緩衝液(50mM Tris-HCl,100mM NaCl,5 mM MgCl
2,pH 7.4,和0.2%,BSA)。在冰上停留20min。關於逆促效劑模式:將20μL試驗化合物(從1μM之10點、3倍稀釋)、20uL緩衝液、140μL膜溶液(GDP 10μM,膜蛋白20μg/孔)加至分析盤中,並在室溫下預培養30min。添加20μL[
35S]-GTPγS(最終200 pM)並在室溫下培養60min。關於拮抗劑模式:將20μL促效劑(
R-α-甲基組織胺,最終濃度1μM)、20μL試驗化合物(最終最高濃度1μM,3-倍稀釋,10點)、20μL[
35S]-GTPγS(最終200 pM)、140μL膜溶液(總計200μL,GDP10μM,膜蛋白20μg/孔)加至分析盤中。在室溫下培養60min。在GF/C(無PEI塗布)盤上過濾分析盤以終止分析。將GF/C盤乾燥1小時。添加50μL閃爍液並在MicroBeta上計數。
數據分析:使用下列公式將CPM值計算為%抑制:
關於逆促效劑模式:%抑制=(DMSO對照CPM-化合物CPM)/(DMSO對照CPM-GTP對照CPM)×100。
關於拮抗劑模式:%抑制=(
R-α-甲基-組織胺對照CPM-化合物CPM)/(
R-α-甲基-組織胺對照CPM-DMSO對照CPM)×100。
使用Prism5與log(抑制劑)對反應方程式計算IC
50。
2. 實驗結果:
表1:代表性化合物之HEK293/Ga15/hH3RGTPγS分析中的IC
50之數據總結。
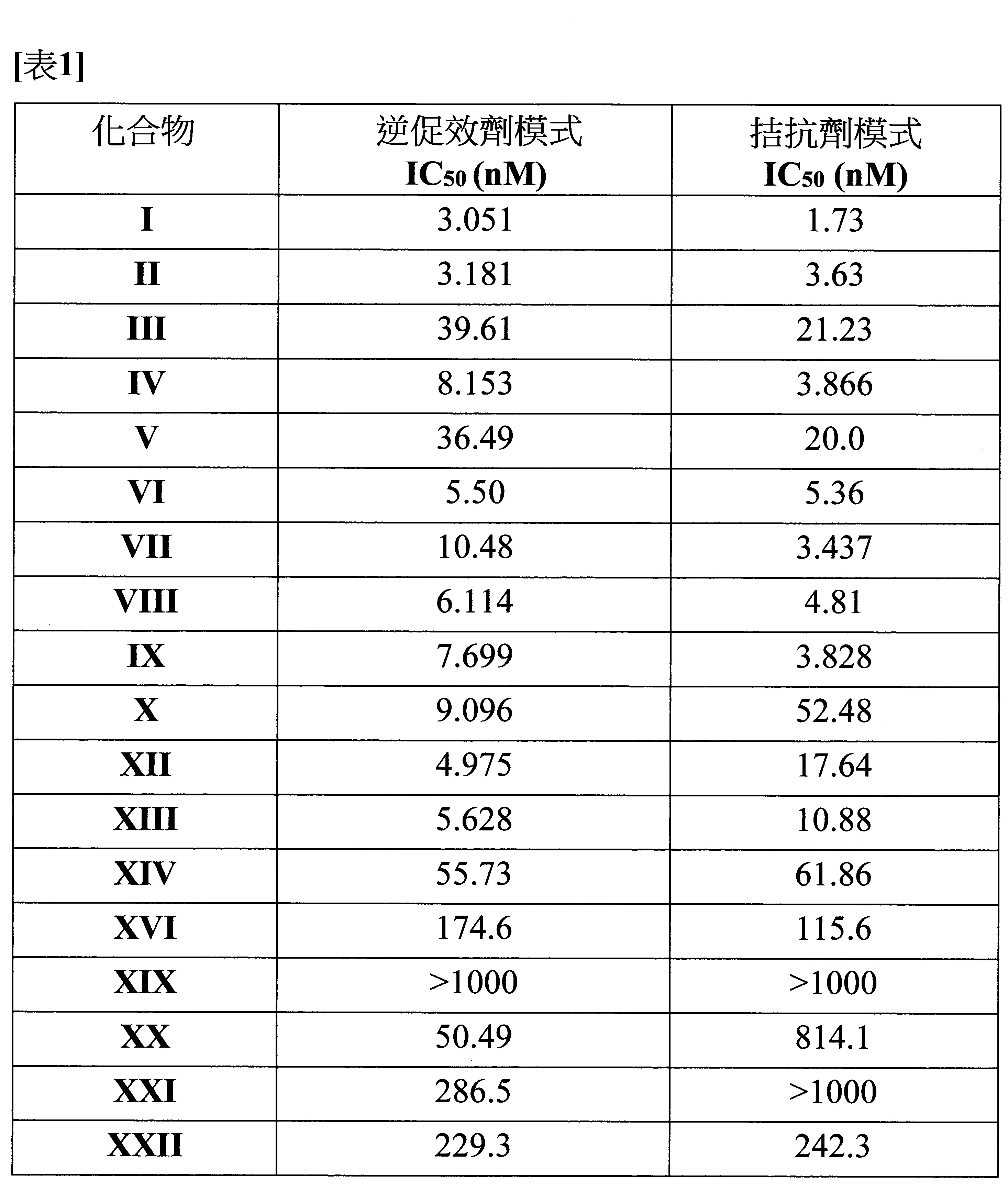 最後,應指出還有實施本發明之其他方法。因此,本發明之實施態樣將描述為實施例,但是本發明不限於所描述的內容,可在本發明的範圍內或在申請專利範圍中所加的等同物進行進一步的修改。
本文引用的所有出版品或專利均以引用的方式併入本發明中。
遍及本說明書提及“一實施態樣”、“一些實施態樣”、“一個實施態樣”、“另一實施例”、“一實例”、“一特定實施例”或“一些實施例”意指關於實施態樣或實施例描述的特定特徵、結構、材料、或特性包括在本揭示的至少一個實施態樣或實施例中。因此,出現在整個說明書中的各處之短語諸如“在一些實施態樣中”、“在一個實施態樣”、“在一實施態樣”、“在另一實施例中”、“在一實施例中”、“在一特定實施例中”或“在一些實施例中”的不一定相同之本揭示的實施態樣或實施例。此外,特定特徵、結構、材料、或特性可在一或多個實施態樣或實施例中以任何適當方式組合。
雖然已經顯示和描述說明性實施態樣,但熟習該項技術者將理解上述實施態樣不可解釋為限制本揭示,且在這些實施態樣可進行變化、替代和修改而不脫離本揭示的精神、原則和範圍。
最後,應指出還有實施本發明之其他方法。因此,本發明之實施態樣將描述為實施例,但是本發明不限於所描述的內容,可在本發明的範圍內或在申請專利範圍中所加的等同物進行進一步的修改。
本文引用的所有出版品或專利均以引用的方式併入本發明中。
遍及本說明書提及“一實施態樣”、“一些實施態樣”、“一個實施態樣”、“另一實施例”、“一實例”、“一特定實施例”或“一些實施例”意指關於實施態樣或實施例描述的特定特徵、結構、材料、或特性包括在本揭示的至少一個實施態樣或實施例中。因此,出現在整個說明書中的各處之短語諸如“在一些實施態樣中”、“在一個實施態樣”、“在一實施態樣”、“在另一實施例中”、“在一實施例中”、“在一特定實施例中”或“在一些實施例中”的不一定相同之本揭示的實施態樣或實施例。此外,特定特徵、結構、材料、或特性可在一或多個實施態樣或實施例中以任何適當方式組合。
雖然已經顯示和描述說明性實施態樣,但熟習該項技術者將理解上述實施態樣不可解釋為限制本揭示,且在這些實施態樣可進行變化、替代和修改而不脫離本揭示的精神、原則和範圍。
本揭示的實施態樣之這些和其他的態樣及優點從下列說明且參考所附流程和圖式將變得顯而易見並且更容易理解,其中:
[圖1] 顯示代表性化合物在拮抗劑模式下之H3受體GTPγS結合親和力,
其中[圖1A]顯示在逆促效劑模式下之組織胺H3 GTPγS結合和%抑制曲線,
[圖1B] 顯示在逆促效劑模式下之組織胺H3 GTPγS結合和%抑制曲線;
[圖2] 顯示代表性化合物在逆促效劑模式下之H3受體GTPγS結合親和力,
其中[圖2A]顯示在拮抗劑模式下之組織胺H3 GTPγS結合和%抑制曲線,
[圖2B] 顯示在拮抗劑模式下之代表性組織胺H3 GTPγS結合和%抑制曲線。

Claims (17)
- 一種具有下列結構之化合物或其醫藥上可接受的鹽類,

- 如請求項1之化合物或其醫藥上可接受的鹽類,其中,R1係選自氫和甲基;及R2係選自氫、羥基、和甲氧基。
- 如請求項1之化合物或其醫藥上可接受的鹽類,其中,R1為氫;及R2係選自氫、羥基、和甲氧基。
- 如請求項1之化合物或其醫藥上可接受的鹽類,其中R1係選自氫和甲基;及R2為氫。
- 如請求項1之化合物或其醫藥上可接受的 鹽類,其中該化合物為2-甲基-3-(4-(3-(吡咯啶-1-基)丙氧基)苯基)-6-(五氟硫基)喹唑啉-4(3H)-酮(I)。
- 如請求項1之化合物或其醫藥上可接受的鹽類,其中該化合物為(S)-2-甲基-3-(4-(3-(2-甲基吡咯啶-1-基)丙氧基)苯基)-6-(五氟硫基)喹唑啉-4(3H)-酮(II)。
- 如請求項1之化合物或其醫藥上可接受的鹽類,其中該化合物為(R)-2-甲基-3-(4-(3-(2-甲基吡咯啶-1-基)丙氧基)苯基)-6-(五氟硫基)喹唑啉-4(3H)-酮(IV)。
- 如請求項1之化合物或其醫藥上可接受的鹽類,其中該化合物為3-(2-甲氧基-4-(3-(吡咯啶-1-基)丙氧基)苯基)-2-甲基-6-(五氟硫基)喹唑啉-4(3H)-酮(VI)。
- 如請求項1之化合物或其醫藥上可接受的鹽類,其中該化合物為3-(2-羥基-4-(3-(吡咯啶-1-基)丙氧基)苯基)-2-甲基-6-(五氟硫基)喹唑啉-4(3H)-酮(VIII)。
- 如請求項1之化合物或其醫藥上可接受的鹽類,其中該化合物為(R)-3-(2-甲氧基-4-(3-(2-甲基吡咯啶-1-基)丙氧基)苯基)-2-甲基-6-(五氟硫基)喹唑啉-4(3H)-酮(X)。
- 如請求項1之化合物或其醫藥上可接受的鹽類,其中該化合物為3-(2-甲氧基-4-(3-(3-甲基吡咯啶-1-基)丙氧基)苯基)-2-甲基-6-(五氟硫基)喹唑啉-4(3H)-酮(XII)。
- 如請求項1之化合物或其醫藥上可接受的鹽類,其中該化合物為(S)-3-(2-甲氧基-4-(3-(2-甲基吡 咯啶-1-基)丙氧基)苯基)-2-甲基-6-(五氟硫基)喹唑啉-4(3H)-酮(XIII)。
- 一種醫藥組成物,其包含治療有效量的如請求項1至12中任一項之化合物或其醫藥上可接受的鹽類和醫藥上可接受的賦形劑、載體、佐劑、溶劑、或其任何組合。
- 一種治療有效量的如請求項1至12中任一項之化合物或其醫藥上可接受的鹽類之用途,其係用於製造供治療患者中樞神經系統疾病的藥物。
- 一種治療有效量的如請求項1至12中任一項之化合物或其醫藥上可接受的鹽類之用途,其係用於製造供治療患者嗜睡症的藥物。
- 一種治療有效量的如請求項13之醫藥組成物之用途,其係用於製造供治療患者中樞神經系統疾病的藥物。
- 一種治療有效量的如請求項13之醫藥組成物之用途,其係用於製造供治療患者嗜睡症的藥物。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| WOPCT/CN2018/109115 | 2018-09-30 | ||
| PCT/CN2018/109115 WO2020062251A1 (en) | 2018-09-30 | 2018-09-30 | Compounds as neuronal histamine receptor-3 antagonists and uses thereof |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| TW202028180A TW202028180A (zh) | 2020-08-01 |
| TWI710552B true TWI710552B (zh) | 2020-11-21 |
Family
ID=69945563
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| TW108134419A TWI710552B (zh) | 2018-09-30 | 2019-09-24 | 作為神經元組織胺受體-3拮抗劑之化合物及其用途 |
Country Status (7)
| Country | Link |
|---|---|
| US (5) | US10618886B1 (zh) |
| EP (1) | EP3856719B1 (zh) |
| JP (1) | JP6996029B2 (zh) |
| CN (2) | CN114042070A (zh) |
| ES (1) | ES2947089T3 (zh) |
| TW (1) | TWI710552B (zh) |
| WO (1) | WO2020062251A1 (zh) |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11400065B2 (en) | 2019-03-01 | 2022-08-02 | Flamel Ireland Limited | Gamma-hydroxybutyrate compositions having improved pharmacokinetics in the fed state |
| US11504347B1 (en) | 2016-07-22 | 2022-11-22 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| US11583510B1 (en) | 2022-02-07 | 2023-02-21 | Flamel Ireland Limited | Methods of administering gamma hydroxybutyrate formulations after a high-fat meal |
| US11602513B1 (en) | 2016-07-22 | 2023-03-14 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| US11602512B1 (en) | 2016-07-22 | 2023-03-14 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| US11779557B1 (en) | 2022-02-07 | 2023-10-10 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| US11839597B2 (en) | 2016-07-22 | 2023-12-12 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| US11986451B1 (en) | 2016-07-22 | 2024-05-21 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| US12186296B1 (en) | 2016-07-22 | 2025-01-07 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| US12478604B1 (en) | 2016-07-22 | 2025-11-25 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN114042070A (zh) * | 2018-09-30 | 2022-02-15 | 凯瑞康宁生物工程(武汉)有限公司 | 作为神经元组胺受体-3拮抗剂的化合物及其用途 |
| WO2022113008A1 (en) | 2020-11-27 | 2022-06-02 | Richter Gedeon Nyrt. | Histamine h3 receptor antagonists/inverse agonists for the treatment of autism spectrum disorder |
| CN115462369B (zh) * | 2022-09-29 | 2023-09-22 | 安徽省立医院(中国科学技术大学附属第一医院) | 一种玻璃化冷冻外泌体保存方法 |
| CN117942344A (zh) * | 2024-01-11 | 2024-04-30 | 良渚实验室 | 作为组胺3型受体拮抗剂的化合物及其应用 |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7521455B2 (en) * | 2004-02-13 | 2009-04-21 | Banyu Pharmaceutical Co. Ltd. | Fused ring 4-oxopyrimidine derivative |
Family Cites Families (40)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6458772B1 (en) | 1909-10-07 | 2002-10-01 | Medivir Ab | Prodrugs |
| DE852392C (de) | 1944-08-30 | 1952-10-13 | Hoechst Ag | Verfahren zur Darstellung von Selencyanverbindungen bzw. Selenazolen von aromatischen Aminen |
| FR2492258A1 (fr) | 1980-10-17 | 1982-04-23 | Pharmindustrie | Nouveau medicament a base d'amino-2 trifluoromethoxy-6 benzothiazole |
| US4410545A (en) | 1981-02-13 | 1983-10-18 | Syntex (U.S.A.) Inc. | Carbonate diester solutions of PGE-type compounds |
| US4328245A (en) | 1981-02-13 | 1982-05-04 | Syntex (U.S.A.) Inc. | Carbonate diester solutions of PGE-type compounds |
| US4409239A (en) | 1982-01-21 | 1983-10-11 | Syntex (U.S.A.) Inc. | Propylene glycol diester solutions of PGE-type compounds |
| FR2662695A1 (fr) | 1990-06-05 | 1991-12-06 | Rhone Poulenc Sante | Amino-2 polyfluoroalcoxy-6 benzoselenazoles, leur preparation et les medicaments les contenant. |
| FR2699077B1 (fr) | 1992-12-16 | 1995-01-13 | Rhone Poulenc Rorer Sa | Application d'anticonvulsivants dans le traitement de lésions neurologiques liées à des traumatismes. |
| BR9714222A (pt) | 1996-12-17 | 2000-04-18 | Du Pont | Método para o controle de doenças de plantas |
| US6350458B1 (en) | 1998-02-10 | 2002-02-26 | Generex Pharmaceuticals Incorporated | Mixed micellar drug deliver system and method of preparation |
| WO1999051613A1 (en) | 1998-04-03 | 1999-10-14 | Medivir Ab | Prodrugs of phosphorous-containing pharmaceuticals |
| US6489350B1 (en) | 1999-09-15 | 2002-12-03 | Elan Pharmaceuticals, Inc. | Methods for treating neuropathic pain using heteroarylmethanesulfonamides |
| GB0002952D0 (en) | 2000-02-09 | 2000-03-29 | Pharma Mar Sa | Process for producing kahalalide F compounds |
| JP2004059452A (ja) | 2002-07-25 | 2004-02-26 | Asahi Glass Co Ltd | ペンタフルオロサルファー置換ベンズイミダゾール化合物およびその製造方法 |
| JPWO2005115993A1 (ja) | 2004-05-31 | 2008-03-27 | 萬有製薬株式会社 | キナゾリン誘導体 |
| MY144903A (en) | 2004-06-17 | 2011-11-30 | Novartis Ag | Pyrrolopyridine derivatives and their use as crth2 antagonists |
| US8193211B2 (en) | 2004-09-30 | 2012-06-05 | Supernus Pharmaceuticals, Inc. | Controlled release compositions of gamma-hydroxybutyrate |
| US7790731B2 (en) | 2005-02-14 | 2010-09-07 | Banyu Pharmaceutical Co. Ltd. | Crystal form of 2-methyl-3-{4-[3-(1-pyrrolidinyl)propoxy]phenyl}-5-trifluoromethyl-4(3H)-quinazolinone |
| EP1900733A4 (en) | 2005-06-07 | 2009-12-30 | Banyu Pharma Co Ltd | PROCESS FOR PREPARING A 4 (3H) -CHINAZOLINONE DERIVATIVE |
| CA2612922A1 (en) | 2005-06-21 | 2006-12-28 | Neurosearch A/S | 2-(phenylamino)benzimidazole derivatives and their use as modulators of small-conductance calcium-activated potassium channels |
| WO2008010223A2 (en) | 2006-07-17 | 2008-01-24 | Ramot At Tel Aviv University Ltd. | Conjugates comprising a psychotropic drug or a gaba agonist and an organic acid and their use in treating pain and other cns disorders |
| ITTO20070665A1 (it) | 2007-09-24 | 2009-03-25 | Rottapharm Spa | Derivati amidinici, tioureici e guanidinici di 2-amminobenzotiazoli e amminobenzotiazine, nuovi agenti farmacologici per il trattamento delle patologie neurodegenerative. |
| WO2009102462A1 (en) | 2008-02-15 | 2009-08-20 | Abbott Laboratories | Thienopyrroles and pyrrolothiazoles as new therapeutic agents |
| EP3067352A3 (en) | 2008-05-07 | 2016-10-19 | Cardioxyl Pharmaceuticals Inc. | Novel nitroso compounds as nitroxyl donors and methods of use thereof |
| EP2421808B1 (en) | 2009-04-23 | 2013-07-31 | Concert Pharmaceuticals Inc. | 4-hydroxybutyric acid analogs |
| KR101632318B1 (ko) | 2009-11-05 | 2016-06-27 | 재단법인 의약바이오컨버젼스연구단 | 벤조헤테로사이클 유도체 또는 이의 약학적으로 허용가능한 염을 유효성분으로 포함하는 암 예방 및 치료용 조성물 |
| KR101976218B1 (ko) | 2011-02-14 | 2019-05-07 | 콘서트 파마슈티컬즈, 인크. | 4-히드록시부티르산 중수소화 유사체 |
| US8802673B2 (en) | 2011-03-24 | 2014-08-12 | Hoffmann-La Roche Inc | Heterocyclic amine derivatives |
| JP6006794B2 (ja) | 2011-07-29 | 2016-10-12 | カリオファーム セラピューティクス,インコーポレイテッド | 核内輸送調節因子およびその使用 |
| SG11201406759YA (en) | 2012-04-26 | 2014-11-27 | Bristol Myers Squibb Co | Imidazothiadiazole derivatives as protease activated receptor 4 (par4) inhibitors for treating platelet aggregation |
| EP2888222A1 (en) | 2012-08-22 | 2015-07-01 | Concert Pharmaceuticals Inc. | Deuterated 4-hydroxybutyric acid analogs |
| WO2014152263A1 (en) | 2013-03-15 | 2014-09-25 | Karyopharm Therapeutics Inc. | Exo olefin-containing nuclear transport modulators and uses thereof |
| WO2014205393A1 (en) | 2013-06-21 | 2014-12-24 | Karyopharm Therapeutics Inc. | Nuclear transport modulators and uses thereof |
| WO2015057884A1 (en) | 2013-10-16 | 2015-04-23 | The Regents Of The University Of California | Methods for treating seizure disorders and pain |
| ITMI20132041A1 (it) | 2013-12-06 | 2015-06-07 | Ct Lab Farm Srl | Derivati dell'acido gamma-idrossibutirrico, loro preparazione e loro uso medico. |
| CA2999367C (en) | 2015-09-23 | 2023-09-26 | Xw Laboratories Inc. | Prodrugs of gamma-hydroxybutyric acid, compositions and uses thereof |
| KR20180100705A (ko) * | 2016-01-29 | 2018-09-11 | 메이지 세이카 파루마 가부시키가이샤 | 신규 화합물 및 이의 약학적으로 허용 가능한 염 |
| KR102444509B1 (ko) | 2016-05-18 | 2022-09-19 | 미라티 테라퓨틱스, 인크. | Kras g12c 억제제 |
| US11602530B2 (en) | 2016-11-28 | 2023-03-14 | Biogen Ma Inc. | CRM1 inhibitors for treating epilepsy |
| CN114042070A (zh) * | 2018-09-30 | 2022-02-15 | 凯瑞康宁生物工程(武汉)有限公司 | 作为神经元组胺受体-3拮抗剂的化合物及其用途 |
-
2018
- 2018-09-30 CN CN202111508899.4A patent/CN114042070A/zh active Pending
- 2018-09-30 JP JP2021517823A patent/JP6996029B2/ja active Active
- 2018-09-30 EP EP18934682.8A patent/EP3856719B1/en active Active
- 2018-09-30 CN CN201880092531.4A patent/CN112004802B/zh active Active
- 2018-09-30 ES ES18934682T patent/ES2947089T3/es active Active
- 2018-09-30 WO PCT/CN2018/109115 patent/WO2020062251A1/en not_active Ceased
-
2019
- 2019-09-24 TW TW108134419A patent/TWI710552B/zh active
- 2019-09-30 US US16/587,344 patent/US10618886B1/en active Active
-
2020
- 2020-02-18 US US16/793,139 patent/US10730853B2/en active Active
- 2020-06-23 US US16/909,779 patent/US10968202B2/en active Active
-
2021
- 2021-03-05 US US17/193,678 patent/US11420955B2/en active Active
-
2022
- 2022-07-18 US US17/867,681 patent/US12172980B2/en active Active
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7521455B2 (en) * | 2004-02-13 | 2009-04-21 | Banyu Pharmaceutical Co. Ltd. | Fused ring 4-oxopyrimidine derivative |
Non-Patent Citations (1)
| Title |
|---|
| George A. Patani et al."Bioisosterism: A Rational Approach in Drug Design." Chemical Reviews. 1996,96,3147-3176. * |
Cited By (36)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US12128021B1 (en) | 2016-07-22 | 2024-10-29 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| US11504347B1 (en) | 2016-07-22 | 2022-11-22 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| US12478604B1 (en) | 2016-07-22 | 2025-11-25 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| US11602513B1 (en) | 2016-07-22 | 2023-03-14 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| US11602512B1 (en) | 2016-07-22 | 2023-03-14 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| US11766418B2 (en) | 2016-07-22 | 2023-09-26 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| US12263150B2 (en) | 2016-07-22 | 2025-04-01 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| US11826335B2 (en) | 2016-07-22 | 2023-11-28 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| US11839597B2 (en) | 2016-07-22 | 2023-12-12 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| US11896572B2 (en) | 2016-07-22 | 2024-02-13 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| US11986451B1 (en) | 2016-07-22 | 2024-05-21 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| US12097175B2 (en) | 2016-07-22 | 2024-09-24 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| US12097176B2 (en) | 2016-07-22 | 2024-09-24 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| US12109186B2 (en) | 2016-07-22 | 2024-10-08 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| US12115144B2 (en) | 2016-07-22 | 2024-10-15 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| US12115143B2 (en) | 2016-07-22 | 2024-10-15 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| US12115145B2 (en) | 2016-07-22 | 2024-10-15 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| US12115142B2 (en) | 2016-07-22 | 2024-10-15 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| US12239625B2 (en) | 2016-07-22 | 2025-03-04 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| US12138239B2 (en) | 2016-07-22 | 2024-11-12 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| US12144793B2 (en) | 2016-07-22 | 2024-11-19 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| US12263151B2 (en) | 2016-07-22 | 2025-04-01 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| US12257223B2 (en) | 2016-07-22 | 2025-03-25 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| US12186298B2 (en) | 2016-07-22 | 2025-01-07 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| US12186296B1 (en) | 2016-07-22 | 2025-01-07 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| US12226388B2 (en) | 2016-07-22 | 2025-02-18 | Flamel Ireland Limited | Modified release gamma- hydroxybutyrate formulations having improved pharmacokinetics |
| US12226389B2 (en) | 2016-07-22 | 2025-02-18 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| US12226377B2 (en) | 2019-03-01 | 2025-02-18 | Flamel Ireland Limited | Gamma-hydroxybutyrate compositions having improved pharmacokinetics in the fed state |
| US11400065B2 (en) | 2019-03-01 | 2022-08-02 | Flamel Ireland Limited | Gamma-hydroxybutyrate compositions having improved pharmacokinetics in the fed state |
| US12167992B2 (en) | 2019-03-01 | 2024-12-17 | Flamel Ireland Limited | Gamma-hydroxybutyrate compositions having improved pharmacokinetics in the fed state |
| US12167991B2 (en) | 2019-03-01 | 2024-12-17 | Flamel Ireland Limited | Gamma-hydroxybutyrate compositions having improved pharmacokinetics in the fed state |
| US12303478B2 (en) | 2019-03-01 | 2025-05-20 | Flamel Ireland Limited | Gamma-hydroxybutyrate compositions having improved pharmacokinetics in the fed state |
| US11779557B1 (en) | 2022-02-07 | 2023-10-10 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| US12295926B1 (en) | 2022-02-07 | 2025-05-13 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| US11583510B1 (en) | 2022-02-07 | 2023-02-21 | Flamel Ireland Limited | Methods of administering gamma hydroxybutyrate formulations after a high-fat meal |
| US12551457B1 (en) | 2022-02-07 | 2026-02-17 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
Also Published As
| Publication number | Publication date |
|---|---|
| CN114042070A (zh) | 2022-02-15 |
| US20220380339A1 (en) | 2022-12-01 |
| EP3856719A1 (en) | 2021-08-04 |
| EP3856719A4 (en) | 2021-09-08 |
| JP2021530556A (ja) | 2021-11-11 |
| JP6996029B2 (ja) | 2022-01-17 |
| US20200369645A1 (en) | 2020-11-26 |
| ES2947089T3 (es) | 2023-08-01 |
| EP3856719B1 (en) | 2023-06-07 |
| US12172980B2 (en) | 2024-12-24 |
| CN112004802B (zh) | 2021-12-28 |
| WO2020062251A1 (en) | 2020-04-02 |
| US20210188808A1 (en) | 2021-06-24 |
| CN112004802A (zh) | 2020-11-27 |
| US10618886B1 (en) | 2020-04-14 |
| US20200102289A1 (en) | 2020-04-02 |
| US11420955B2 (en) | 2022-08-23 |
| EP3856719C0 (en) | 2023-06-07 |
| US20200181112A1 (en) | 2020-06-11 |
| US10730853B2 (en) | 2020-08-04 |
| US10968202B2 (en) | 2021-04-06 |
| TW202028180A (zh) | 2020-08-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| TWI710552B (zh) | 作為神經元組織胺受體-3拮抗劑之化合物及其用途 | |
| US11905247B2 (en) | Compounds as neurokinin-1 receptor antagonists and uses thereof | |
| US12234226B2 (en) | Compounds as nuclear transport modulators and uses thereof | |
| JP7308911B2 (ja) | ニューロンヒスタミン受容体-3アンタゴニストとしての化合物およびその使用 | |
| HK40069211A (zh) | 作为神经元组胺受体-3拮抗剂的化合物及其用途 | |
| HK40035049A (zh) | 作为神经元组胺受体-3拮抗剂的化合物及其用途 | |
| HK40035049B (zh) | 作为神经元组胺受体-3拮抗剂的化合物及其用途 | |
| HK40029219A (zh) | 作为神经激肽-1受体拮抗剂的化合物及其用途 | |
| HK40029219B (zh) | 作为神经激肽-1受体拮抗剂的化合物及其用途 | |
| HK40072528A (zh) | 双环杂芳基衍生物及其制备与用途 |