TWI703143B - 4-(4-(1-異丙基-7-側氧基-1,4,6,7-四氫螺〔吲唑-5,4’-哌啶〕-1’-羰基)-6-甲氧基吡啶-2-基)苯甲酸之晶型 - Google Patents
4-(4-(1-異丙基-7-側氧基-1,4,6,7-四氫螺〔吲唑-5,4’-哌啶〕-1’-羰基)-6-甲氧基吡啶-2-基)苯甲酸之晶型 Download PDFInfo
- Publication number
- TWI703143B TWI703143B TW107140741A TW107140741A TWI703143B TW I703143 B TWI703143 B TW I703143B TW 107140741 A TW107140741 A TW 107140741A TW 107140741 A TW107140741 A TW 107140741A TW I703143 B TWI703143 B TW I703143B
- Authority
- TW
- Taiwan
- Prior art keywords
- crystalline salt
- scope
- ppm
- patent application
- item
- Prior art date
Links
Images







Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/10—Spiro-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/444—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring heteroatom, e.g. amrinone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C215/00—Compounds containing amino and hydroxy groups bound to the same carbon skeleton
- C07C215/02—Compounds containing amino and hydroxy groups bound to the same carbon skeleton having hydroxy groups and amino groups bound to acyclic carbon atoms of the same carbon skeleton
- C07C215/04—Compounds containing amino and hydroxy groups bound to the same carbon skeleton having hydroxy groups and amino groups bound to acyclic carbon atoms of the same carbon skeleton the carbon skeleton being saturated
- C07C215/06—Compounds containing amino and hydroxy groups bound to the same carbon skeleton having hydroxy groups and amino groups bound to acyclic carbon atoms of the same carbon skeleton the carbon skeleton being saturated and acyclic
- C07C215/10—Compounds containing amino and hydroxy groups bound to the same carbon skeleton having hydroxy groups and amino groups bound to acyclic carbon atoms of the same carbon skeleton the carbon skeleton being saturated and acyclic with one amino group and at least two hydroxy groups bound to the carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/13—Crystalline forms, e.g. polymorphs
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Diabetes (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Gastroenterology & Hepatology (AREA)
- Endocrinology (AREA)
- Emergency Medicine (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
本發明提供4-(4-(1-異丙基-7-側氧基-1,4,6,7-四氫螺[吲唑-5,4’-哌啶]-1’-羰基)-6-甲氧基吡啶-2-基)苯甲酸之tris鹽作為結晶無水或三水合物;以及多晶型、醫藥組成物、劑型,以及其用於治療動物由抑制乙醯輔酶A羧化酶(acetyl-CoA carboxylase,ACC)酵素所調節之疾病(disease)、狀況(condition)或病症(disorder)的用途。
Description
本發明提供4-(4-(1-異丙基-7-側氧基 -1,4,6,7-四氫螺[吲唑-5,4’-哌啶]-1’-羰基)-6-甲氧基吡啶 -2-基)苯甲酸的tris鹽(tris salt);以及晶型、多晶型、醫藥組成物、劑型與其用於治療動物由抑制乙醯輔酶A羧化酶(acetyl-CoA carboxylase,ACC)酵素所調節之疾病(disease)、狀況(condition)或病症(disorder)的用途。
4-(4-(1-異丙基-7-側氧基-1,4,6,7-四氫螺[吲唑-5,4’-哌啶]-1’-羰基)-6-甲氧基吡啶-2-基)苯甲酸(在本文中,以下稱為化合物1)是選擇性的ACC抑制劑且經製備成為US 8,859,577之實例9中的游離酸(free acid),US 8,859,577為國際申請案案號PCT/IB2011/054119的美國國家階段之申請案,這些全部內容為了所有目的以引用方式併入本文。作為游離酸的化合物1具有不合適的物理化學特性。
鹽的形成提供改變藥物的物理化學和所得生物學特性而不修飾其化學結構的手段。鹽形式可對藥物的性質產生顯著影響。選擇合適的鹽形式涉及評估許多因素,包含是否可形成任何鹽。此選擇所包含的其他因素包含可能發現的任何鹽形式的吸濕性、穩定性、溶解性和製程概況(process profile)。
非酒精性脂肪性肝病(nonalcoholic fatty liver disease,NAFLD)是肝臟表現的代謝症候群,是一系列肝臟狀況,包括脂肪變性(steatosis)、非酒精性脂肪性肝炎(non-alcoholic steatohepatitis,NASH)、纖維化、硬化(cirrhosis)和最終的肝細胞癌。NAFLD和NASH被認為是原發性脂肪肝疾病,因為它們佔具有肝臟脂質升高之個體的最大比例。NAFLD/NASH的嚴重程度是基於脂質的存在、發炎細胞浸潤、肝細胞氣球樣變(hepatocyte ballooning)和纖維化程度。雖然並非所有患有脂肪變性的個體都會進展到NASH,但很大一部分都會進展。
越來越清楚的是,肝臟脂質累積引起肝臟胰島素抗性(hepatic insulin resistance)並且有助於第2型糖尿病(type 2 diabetes,T2D)的致病性(pathogenesis)。Savage等人證實ACC1和ACC2皆參與調節肝細胞中的脂肪氧化,而ACC1(大鼠肝臟中的主要同功型(isoform))是脂肪酸合成的唯一調節劑。再者,在它們的模型中,需要組合減少兩種同功型以顯著降低肝臟丙二醯輔酶A(malonyl-CoA)量、增加進食狀態下的脂肪氧化、減少脂質累積、並且改善體內胰島素作用。因此,肝ACC1和ACC2抑制劑可用於治療NAFLD和肝臟胰島素抗性。參閱Savage, D. B., et al., "Reversal of diet-induced hepatic steatosis and hepatic insulin resistance by antisense oligonucleotide inhibitors of acetyl-CoA carboxylases 1 and 2"J. Clin. Invest.
2006;116(3):817-24。亦參閱Oh, W., et al., “Glucose and fat metabolism in adipose tissue of acetyl-CoA carboxylase 2 knockout mice”PNAS
, 102(5)1384-1389(2005)。
因此,需要含有ACC1和/或ACC2抑制劑的口服藥物以治療包含NAFLD、NASH和T2D的疾病。
本發明提供4-(4-(1-異丙基-7-側氧基 -1,4,6,7-四氫螺[吲唑-5,4’-哌啶]-1’-羰基)-6-甲氧基吡啶 -2-基)苯甲酸(在本文中,以下稱為化合物1)的tris鹽(tris salt)之結晶多晶型,其中結晶鹽可為無水或水合物,更具體而言,三水合物;以及多晶型、醫藥組成物、劑型、以及其用於治療動物由抑制乙醯輔酶A羧化酶(acetyl-CoA carboxylase,ACC)所調節之疾病(disease)、狀況(condition)或病症(disorder)的用途。本發明的新穎結晶形具有特別適合作為藥物的性質,包含改善的溶解性和生物可利用性。
化合物1的固體晶形如本文所揭露,其中每一個固體形式可藉由幾種不同的分析參數(單獨或組合)而被獨特地識別,例如但不限於:粉末X射線繞射圖譜峰(peak)或二或更多個峰的組合;固態NMR13
C化學位移或二或多個化學位移的組合;以及Raman峰偏移或二或多個Raman峰偏移的組合。
化合物1含有兩個可離子化的(ionizable)位置: 吡啶環上的氮以及羧酸。本發明提供化合物1(4-(4-(1-異丙基-7-側氧基-1,4,6,7-四氫螺[吲唑-5,4’-哌啶]-1’-羰基)-6-甲氧基吡啶-2-基)苯甲酸)的結晶tris鹽。本發明涉及化合物1的單-tris鹽(mono-tris salt)。單-tris鹽有兩種結晶形式:形式1為無水結晶固體,形式2為三水合物結晶固體。形式3為無定型形式。
吡啶環上的氮以及羧酸。本發明提供化合物1(4-(4-(1-異丙基-7-側氧基-1,4,6,7-四氫螺[吲唑-5,4’-哌啶]-1’-羰基)-6-甲氧基吡啶-2-基)苯甲酸)的結晶tris鹽。本發明涉及化合物1的單-tris鹽(mono-tris salt)。單-tris鹽有兩種結晶形式:形式1為無水結晶固體,形式2為三水合物結晶固體。形式3為無定型形式。
定義
術語「tris」是指2-胺基-2-(羥甲基)丙烷-1,3-二醇(2-amino-2-(hydroxymethyl)propane-1,3-diol),亦被稱為THAM和胺基丁三醇(tromethamine)。化合物1的tris鹽是指使用2-胺基-2-(羥甲基)丙烷-1,3-二醇製造的化合物1的鹽。Tris與化合物1的羧酸部分結合。除非另有說明,否則當提及化合物1的tris鹽時,相對離子(counterion)和化合物1的化學計量比為約1:1。
術語「形式1」是指化合物1的無水結晶2-胺基-2-(羥甲基)丙烷-1,3-二醇(tris)鹽作為單-tris鹽。意指形式1沒有水,但如果材料未完全乾燥,則可能存在殘留溶劑,包含水。
術語「形式2」是指化合物1的三水合物結晶2-胺基-2-(羥甲基)丙烷-1,3-二醇(tris)鹽作為單-tris鹽。
本文所使用的術語「三水合物」是指包含約三個水分子。
術語「約」通常是指在給定值或範圍的10%以內,較佳為5%以內,更佳為1%以內。或者,當由本技藝中普通技術人士考慮時,術語「約」是指在平均值之可接受的標準誤差內。
G或g是克,mg是指毫克。
H或h是小時。
IPA是指異丙醇。
L是公升。
mL是毫升。
MCC是指微晶纖維素。
RH是指相對濕度。
RT或rt是指室溫,其是與環境溫度(約20至25℃)相同。
在所有情況下,1
H核磁共振(NMR)光譜與所提出的結構一致。特徵化學位移(Characteristic chemical shift)(d)以百萬分之一(parts-per-million,ppm)之單位以相對於氘化溶劑中的殘留質子訊號(CHCl3
為7.27ppm;CD2
HOD為3.31ppm)給出,並使用指定主要峰的常規縮寫報告:例如s(單峰);d(雙峰);t(三重峰);q(四重峰);m(多重峰);br(廣的(broad))。
ssNMR是指固態NMR。
PXRD是粉末X射線繞射。
RH是指相對濕度。
術語「實質上相同」當用於描述X射線粉末繞射圖案時是指包含峰在+/-0.2º2Ɵ的標準偏差範圍內的圖案(patterns)。
如本文中所使用,關於特定結晶形式的術語「實質上純的」是指結晶形式包含小於10%,較佳小於5%,較佳小於3%,較佳小於1%重量的任何其他物理形式的化合物1。
由於pKa低且結果差,所以非常少的相對離子(counterion)經評估在化合物1的氮上形成鹽,例如使用硫酸作為相對離子獲得非常吸濕的(hygroscopic)固體。
研究的許多相對離子沒有給出鹽形式,而是分離時的游離酸:組胺酸、離胺酸(含Na和Ca)、L-鳥氨酸(L-ornithine)(含Na)。嘗試使用精胺酸(含Na)導致游離酸,這是因為乾燥時解離。評估許多相對離子的結果如表1所示。藉由將化合物1的儲備溶液(stock solution)加入至相對離子源中(與化合物1以約1:1的比例)而獲得鹽。經計算的相對離子量與化合物1(作為游離酸)的莫耳比為1:1,提供在標示相對離子的欄中的括號中。在括號中提供添加的相對離子的實際量,其中提供不同溶劑中的每種相對離子的結果。製備在給定溶劑中的化合物1(作為游離酸)的儲備溶液,並加入至在給定溶劑中的相對離子,而後加入另外的溶劑以得到總體積,提供在表1中溶劑下方的括號中。觀察每個樣品一週,在此期間將樣品加熱至約50℃約3小時,並攪拌冷卻至室溫。關於甲醇(MeOH)、乙醇(EtOH)和乙酸異丙酯/乙酸乙酯(IPAC/EtOAc)的實驗,觀察樣品總共三週。表1提供在此期間觀察到的內容。對於一些樣品,在第一週後加入2mL庚烷,並在表1中用*表示。關於甲醇與乙腈(acetonitrile,ACN),儲備溶液為15 mg/mL。關於乙醇,儲備溶液為30 mg/mL。關於IPAC/ EtOAc,儲備溶液在50mL IPAC/5mL EtOAc(包含幾滴水)中為13.6mg/mL。總體積(vol.)提供在所檢測之各個列出的溶劑下方的括號中。
決定是否形成固體僅為此評估中的一個因素。雖然使用鈣、鉀與鈉製造鹽時會形成結晶固體,然而結晶固體太吸濕以致於無法持續評估。許多這些鹽沒有獲得吸濕性數據,這是因為它主要是經由可視化而觀察到的。例如,由於鈣與鉀鹽形式是在過濾紙上被收集到,因而固體材料轉變為膠(gum)。分離出化合物1作為膽鹼鹽;發現吸濕性在30%相對濕度下重量變化超過20%。再者,雖然獲得二乙醇胺、二乙胺和哌嗪的鹽的固體,但並未探求這些鹽。參閱例如C. Saal、A. Becker發表之Euro. J. of Pharm Sci 49(2013)614-623;Paullekuhn, G Steffen等人發表之J. Med. Chem. 2007, 50, 6665-6682。
再者,雖然獲得固體,仍使用其他評估(如1
H NMR)以確定是否實際上形成鹽。在一些例子中,獲得的固體是化合物1(作為游離酸)但未與相對離子結合。
在RH高於20%的環境溫度下,形式1和形式2中,形式2是較穩定的形式。溶劑條件影響是否獲得形式1或形式2。確定IPA/水中0.2的臨界水活性提供結晶形式2。當在水存在下使用以下溶劑時提供形式2:5%至15%水/丙酮、4%水/96%乙腈、1%水/99%乙酸丁酯、1%水/99%乙酸異丙酯,1%水/99%乙酸乙酯、2%水/98%二氯乙烯、2%水/98%甲基乙基酮、3至6.0%水/97至94%2-甲基四氫呋喃、和4%水/96%正丙醇。
化合物1亦面臨關於溶解度(solubility)的挑戰。表2提供化合物1(作為游離酸)與各種鹽的溶解度。化合物1(作為游離酸)被證明是製備用於常規口服投與劑量形式(dosage form)之不可接受的形式。雖然化合物1(作為游離酸)是結晶,但熱力學溶解度不一致。鈣鹽在低pH具有極低溶解度。
由於化合物1(作為游離酸)未提供一致的結果,因而探求鹽形式;在沒有限制的情況下,認為游離酸可存在成為多種非化學計量的水合物/溶劑化物。如表2中所示,在各個pH,三個批次的游離酸溶解度不同。
額外比較游離酸和tris鹽的藥代動力學,如表3中所示。化合物1的單-tris鹽(無論它是形式1或形式2)具有類似的藥物動力學。化合物1的單-tris鹽的藥物動力學大幅優於化合物1(作為游離酸)的藥物動力學。雖然用無水形式1製備懸浮液,但在給定的水性介質情況下,也可能存在三水合物形式2。存在懸浮液中的形式未經辨識。
形式1是無水,並且於環境溫度水活性約0.2(20% RH)以下為熱動力學穩定的。形式1的PXRD圖案與圖1所示的圖案實質上相同。形式1在9.6、10.7和11.3具有特徵PXRD峰(以2Ɵ±0.2º2Ɵ表示)。圖1中的PXRD圖案的峰位置和強度如表4中所示。
形式1的Raman光譜與圖2所示的光譜實質上相同。形式1在568、698、989、1218、1511、1561與1615分別±2 cm-1
具有特徵Raman峰偏移(以cm-1
表示)。圖2中的形式1之峰位置(±2 cm-1
)和正規化的強度(normalized intensity)(W=弱,M=中等,S=強)如表5中所列示。
形式1的13
C ssNMR光譜與圖3所示的光譜實質上相同。形式1在22.9、146.2、157.9、161.9以及172.9分別±0.2 ppm具有特徵13
C ssNMR化學位移(以ppm表示)。圖3所示之形式1的13
C化學位移(±0.2 ppm)如表6中所列示。
形式2是三水合物,並且於環境溫度且20% RH,水活性約0.2以上為熱動力學穩定的。形式2的PXRD圖案與圖4所示的圖案實質上相同。形式2在8.4、9.0、10.5、15.0和24.7具有特徵PXRD峰(以2Ɵ±0.2º2Ɵ表示)。圖4中的PXRD圖案的峰位置和強度如表7中所示。
形式2的Raman光譜與圖5所示的光譜實質上相同。形式2在562、692、984、1225、1507、1557以及1610分別±2 cm-1
具有特徵Raman峰偏移(以cm-1
表示)。圖5中的形式2之峰位置(±2 cm-1
)和正規化的強度(normalized intensity)(W=弱,M=中等,S=強)如表8中所列示。
形式2的13
C ssNMR光譜與圖6所示的光譜實質上相同。形式2在19.2、149.5、155.6、163.8以及188.3分別±0.2ppm具有特徵13
C ssNMR化學位移(以ppm表示)。圖6所示之形式2的13
C化學位移(±0.2 ppm)如表9中所列示。
基於本文提供的揭露內容,該技藝中的普通技術人士可理解可藉由不同組合之一些不同的光譜峰或圖案而分別獨特地辨識形式1與形式2。以下描述的是可用於分別識別形式1和形式2的特徵峰值(peak value)的示例性組合,但是這些示例性組合不應被視為限制本文揭露的其他峰值組合。
本發明之一方面是提供4-(4-(1-異丙基-7-側氧基-1,4,6,7-四氫螺[吲唑-5,4’-哌啶]-1’-羰基)-6-甲氧基吡啶-2-基)苯甲酸(化合物1)的結晶2-胺基-2-(羥甲基)丙烷-1,3-二醇鹽。
本發明之另一方面是提供化合物1的結晶鹽,其中4-(4-(1-異丙基-7-側氧基-1,4,6,7-四氫螺[吲唑-5,4’-哌啶]-1’-羰基)-6-甲氧基吡啶-2-基)苯甲酸(化合物1)與鹽的比例為1:1。
本發明之另一方面是提供結晶鹽作為化合物1的無水結晶鹽(形式1)。
本發明之另一方面是提供結晶鹽作為化合物1的三水合物結晶鹽(形式2)。
本發明之另一方面是提供形式1,其中形式1具有PXRD圖案,其包括在繞射角為9.6、10.7以及11.3 2Ɵ分別±0.2º 2Ɵ的峰。
本發明之另一方面是提供形式1,其中形式1具有PXRD圖案,其包括與圖1所示實質上相同的峰(以2Ɵ表示)。
本發明之另一方面是提供形式1,其中形式1具有Raman光譜,其包括在1511、1561以及1615 cm-1
分別±2 cm-1
的峰偏移。
本發明之另一方面是提供形式1,其中形式1具有Raman光譜,其包括在989、1218、1511、1561以及1615 cm-1
分別±2 cm-1
的峰偏移。
本發明之另一方面是提供形式1,其中形式1具有Raman光譜,其包括在568、698、989、1218、1511、1561以及1615 cm-1
分別±2 cm-1
的峰偏移。
本發明之另一方面是提供形式1,其中形式1具有Raman光譜,其包括與圖2所示之實質上相同的峰偏移(以cm-1
表示)。
本發明之另一方面是提供形式1,其中形式1具有13
C ssNMR光譜,其包括在22.9、146.2以及161.9 ppm分別±0.2 ppm的化學位移。
本發明之另一方面是提供形式1,其中形式1具有13
C ssNMR光譜,其包括在22.9、146.2、157.9、161.9以及172.9 ppm分別±0.2 ppm的化學位移。
本發明之另一方面是提供形式1,其中形式1具有13
C ssNMR光譜,其包括與圖3所示之實質上相同的化學位移(以ppm表示)。
本發明之另一方面是提供形式1,其中形式1具有選自由以下所組成之群組的分析參數:包括在1561與1615 cm-1
分別±2 cm-1
的峰偏移之Raman光譜以及包括在22.9、146.2或161.9 ppm分別±0.2 ppm的至少一個化學位移之13
C ssNMR光譜。
本發明之另一方面是提供形式1,其中形式1具有選自由以下所組成之群組的分析參數:包括在1511與1615 cm-1
分別±2 cm-1
的峰偏移之Raman光譜以及包括在22.9、146.2或161.9 ppm分別±0.2 ppm的至少一個化學位移之13
C ssNMR光譜。
本發明之另一方面是提供形式1,其中形式1具有選自由以下所組成之群組的分析參數:包括在1615 cm-1
±2 cm-1
的峰偏移之Raman光譜以及包括在22.9、146.2或161.9 ppm分別±0.2 ppm的至少一個化學位移之13
C ssNMR光譜。
本發明之另一方面是提供形式1,其中形式1具有選自由以下所組成之群組的分析參數:包括在1561 cm-1
±2 cm-1
的峰偏移之Raman光譜以及包括在22.9、146.2或161.9 ppm分別±0.2 ppm的至少一個化學位移之13
C ssNMR光譜。
本發明之另一方面是提供形式1,其中形式1具有選自由以下所組成之群組的分析參數:包括在22.9與161.9 ppm分別±0.2 ppm的化學位移之13
C ssNMR光譜以及包括在1511、1561或1615 cm-1
分別±2 cm-1
的至少一個峰偏移之Raman光譜。
本發明之另一方面是提供形式1,其中形式1具有選自由以下所組成之群組的分析參數:包括在146.2與161.9 ppm分別±0.2 ppm的化學位移之13
C ssNMR光譜以及包括在1511、1561或1615 cm-1
分別±2 cm-1
的至少一個峰偏移之Raman光譜。
本發明之另一方面是提供形式1,其中形式1具有選自由以下所組成之群組的分析參數:包括在繞射角為9.6與10.7 2Ɵ分別±0.2º 2Ɵ的峰之PXRD圖案以及包括在1511、1561或1615 cm-1
分別±2 cm-1
的至少一個峰偏移之Raman光譜。
本發明之另一方面是提供形式1,其中形式1具有選自由以下所組成之群組的分析參數:包括在繞射角為9.6與10.7 2Ɵ分別±0.2º 2Ɵ的峰之PXRD圖案以及包括在22.9、146.2或161.9 ppm分別±0.2 ppm的至少一個化學位移之13
C ssNMR光譜。
本發明之另一方面是提供形式2,其中形式2具有PXRD圖案,其包括在繞射角為8.4、9.0、10.5 2Ɵ分別±0.2º 2Ɵ的峰。
本發明之另一方面是提供形式2,其中形式2具有PXRD圖案,其包括在繞射角為8.4、9.0、10.5、15.0與24.7 2Ɵ分別±0.2º 2Ɵ的峰。
本發明之另一方面是提供形式2,其中形式2具有PXRD圖案,其包括與圖4所示實質上相同的峰(以2Ɵ表示)。
本發明之另一方面是提供形式2,其中形式2具有Raman光譜,其包括在1507、1557以及1610 cm-1
分別±2 cm-1
的峰偏移。
本發明之另一方面是提供形式2,其中形式2具有Raman光譜,其包括在984、1225、1507、1557以及1610 cm-1
分別±2 cm-1
的峰偏移。
本發明之另一方面是提供形式2,其中形式2具有Raman光譜,其包括在562、692、984、1225、1507、1557以及1610 cm-1
分別±2 cm-1
的峰偏移。
本發明之另一方面是提供形式2,其中形式2具有Raman光譜,其包括與圖5所示之實質上相同的峰偏移(以cm-1
表示)。
本發明之另一方面是提供形式2,其中形式2具有13
C ssNMR光譜,其包括在19.2、149.5以及163.8 ppm分別±0.2 ppm的化學位移。
本發明之另一方面是提供形式2,其中形式2具有13
C ssNMR光譜,其包括在19.2、149.5、155.6、163.8以及188.3 ppm分別±0.2 ppm的化學位移。
本發明之另一方面是提供形式2,其中形式2具有13
C ssNMR光譜,其包括與圖6所示之實質上相同的化學位移(以ppm表示)。
本發明之另一方面是提供形式2,其中形式2具有選自由以下所組成之群組的分析參數:
包括在繞射角為8.4與9.0 2Ɵ分別±0.2º 2Ɵ的峰之PXRD圖案;
包括在1557以及1610 cm-1
分別±2 cm-1
的峰偏移之Raman光譜;以及
包括在19.2、149.5或163.8 ppm分別±0.2 ppm的至少一個化學位移之13
C ssNMR光譜。
本發明之另一方面是提供形式2,其中形式2具有選自由以下所組成之群組的分析參數:
包括在繞射角為8.4與10.5 2Ɵ分別±0.2º 2Ɵ的峰之PXRD圖案;
包括在1507以及1610 cm-1
分別±2 cm-1
的峰偏移之Raman光譜;以及
包括在19.2、149.5或163.8 ppm分別±0.2 ppm的至少一個化學位移之13
C ssNMR光譜。
本發明之另一方面是提供形式2,其中形式2具有選自由以下所組成之群組的分析參數:包括在1557 cm-1
±2 cm-1
的峰偏移之Raman光譜;以及包括在19.2、149.5或163.8 ppm分別±0.2 ppm的至少一個化學位移之13
C ssNMR光譜。
本發明之另一方面是提供形式2,其中形式2具有選自由以下所組成之群組的分析參數:包括在1610 cm-1
±2 cm-1
的峰偏移之Raman光譜;以及包括在19.2、149.5或163.8 ppm分別±0.2 ppm的至少一個化學位移之13
C ssNMR光譜。
本發明之另一方面是提供形式2,其中形式2具有選自由以下所組成之群組的分析參數:包括在19.2與149.5分別±0.2 ppm的化學位移之13
C ssNMR光譜以及包括在1507、1557或1610 cm-1
分別±2 cm-1
的至少一個峰偏移之Raman光譜。
本發明之另一方面是提供形式2,其中形式2具有選自由以下所組成之群組的分析參數:包括在149.5與163.8分別±0.2 ppm的化學位移之13
C ssNMR光譜以及包括在1507、1557或1610 cm-1
分別±2 cm-1
的至少一個峰偏移之Raman光譜。
本發明之另一方面是提供形式2,其中形式2具有選自由以下所組成之群組的分析參數:包括在繞射角為8.4與9.0 2Ɵ分別±0.2º 2Ɵ的峰之PXRD圖案;以及包括在1507、1557或1610 cm-1
分別±2 cm-1
的至少一個峰偏移之Raman光譜。
本發明之另一方面是提供形式2,其中形式2具有選自由以下所組成之群組的分析參數:包括在繞射角為8.4與9.0 2Ɵ分別±0.2º 2Ɵ的峰之PXRD圖案;以及包括在19.2、149.5或163.8 ppm分別±0.2 ppm的至少一個化學位移之13
C ssNMR光譜。
本發明亦包含:
化合物1的結晶鹽,包含形式1和/或形式2,如本文所述,用於作為藥物;
包括投與治療有效劑量之化合物1的結晶鹽(包含形式1和/或形式2)至哺乳動物以治療包含NAFLD、NASH與T2D之疾病的方法;以及
化合物1之結晶鹽(包含形式1和/或形式2)之用途,如本文所述,用於製造用於治療包含NAFLD、NASH與T2D之疾病的藥物。
本發明之另一方面是提供包括如本文所述之形式1或形式2之醫藥組成物。在另一方面,本發明提供包括形式1或形式2或本文所述之醫藥組成物中任一者的口服劑量形式。例如,在一實施例中,口服劑量形式為錠劑、藥丸或膠囊。例如,在一實施例中,口服劑量形式為錠劑或膠囊。
本發明之化合物和/或含有所述該化合物之組成物的劑量方案(dosage regimen)是基於多種因素,包含病患的類型、年齡、體重、性別和醫療狀況;狀況的嚴重性;投與途徑;以及所使用之特定化合物的活性。因此,劑量方案可廣泛變化。對於體重約100 kg的正常成人,典型的每日劑量範圍為約0.001 mg至約10 mg /千克體重通常是足夠的,較佳為約0.01 mg/kg至約5.0 mg/kg,更佳為約0.01 mg/kg至約1 mg/kg。然而,根據所治療的個體的年齡和體重,預期的投與途徑、所給予的特定化合物等,可能需要一般劑量範圍的一些變化。對特定患者的劑量範圍和最佳劑量的決定完全在本技藝普通技術人士受益於本案揭露內容的能力範圍內。還應注意本發明化合物可用於持續釋放、控制釋放和延遲釋放配方,這些形式也是本技藝普通技術人士所熟知的。
在另一方面,本發明包括醫藥組成物。此醫藥組成物包括具有醫藥可接受之載體的形式1或形式2。亦可有其它醫藥活性物質存在。如本文所使用,「醫藥可接受之載體」包含生理上相容的任何與所有溶劑、分散介質、塗層、抗細菌和抗真菌劑、等張和吸收延遲劑等。醫藥可接受之載體的實例包含水、鹽水、磷酸鹽緩衝鹽水、右旋糖(dextrose)、甘油、乙醇等中的一種或多種,以及它們的組合,並且在組成物中可包含等張劑,例如糖、氯化鈉、或多元醇(如甘露醇或山梨糖醇)。藥學上可接受之物質,例如潤濕劑或少量輔助物質,例如潤濕或乳化劑、防腐劑或緩衝劑,其增強抗體或抗體部分的儲存壽命(shelf life)或有效性。
固體劑量形式的口服投與可例如以離散單位(discrete unit)存在,例如硬或軟膠囊、藥丸、扁囊劑(cachet)、口含片(lozenge)、或錠劑,每個含有預定量的至少一種本發明之化合物。在另一實施例中,口服投與可為粉末或是顆粒形式。在另一實施例中,口服劑量形式為舌下劑形,例如口含片。在此固體劑量形式中,式I的化合物通常與一或多種佐劑結合。此膠囊或錠劑可含有控制釋放配方。在膠囊、錠劑與藥丸的情況下,劑量形式亦可包括緩衝劑或可用腸溶包衣(enteric coating)製備。
實例
在化合物1的製備中,注意本文所述之一些製備方法可能需要遠程官能性(例如,式I前驅物中的一級胺、二級胺、羧基)的保護。對此保護的需求將根據遠程官能性的本質和製備方法的條件而變化。本領域技術人員容易確定對此種保護的需要。此保護/去保護方法的用途亦在該技藝的技術內。關於保護基團及其用途的一般說明,參閱T.W. Greene, Protective Groups in Organic Synthesis, John Wiley & Sons, New York, 1991。再者,本發明不受限於本文所提供之可變化的特定合成方法。
中間物1:1-異丙基-4,6-二氫螺[吲唑-5,4’-哌啶]-7(1H)-酮,鹽酸鹽。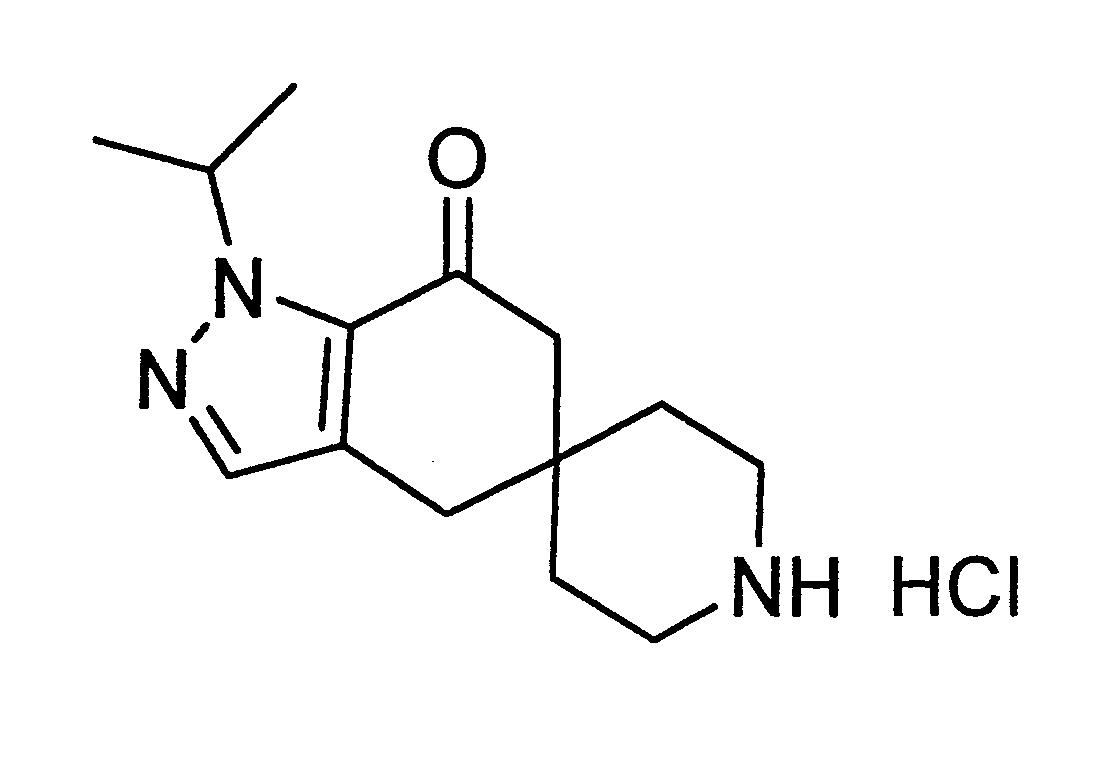
步驟1. 9-側氧基-3-氮雜螺[5.5]十一碳-7-烯-3-羧酸第三丁酯(tert-Butyl 9-oxo-3-azaspiro[5.5]undec-7-ene-3-carboxylate)。
於25-30℃,在乾燥的反應器中,加入4-甲醯基哌啶-1-羧酸第三丁酯(tert-butyl 4-formylpiperidine-1-carboxylate)(108Kg)、環己烷(1080L)和吡咯啶(pyrrolidine)(64.8Kg)。將混合物攪拌5-10分鐘,而後加熱以回流(reflux)12-16 h,同時使用Dean-Stark收集器(trap)收集水。而後,將反應混合物冷卻至50-60℃,在此溫度下施加真空以蒸餾過量的吡咯啶和環己烷。而後,將反應混合物冷卻至25-30℃,加入環己烷(648L),接著加入甲基乙烯基酮(methyl vinyl ketone)(49.63Kg)。將混合物攪拌12-16 h,然後過濾,將濾液置入乾淨且乾燥的反應器中。將溶液冷卻至10-15℃,而後緩慢加入乙酸(54.75Kg)於水(54L)中的溶液,保持溫度低於15℃。在添加結束時,將混合物溫熱至25-30℃並攪拌12-16 h。分離各層,用乙酸乙酯(324 L)萃取水層。將合併的有機層用碳酸氫鈉(32.34 Kg)於水(324L)中的溶液洗滌,而後以硫酸鈉乾燥。用乙酸乙酯(54 L)洗滌固體,並將合併的濾液在低於40℃的減壓下濃縮。將正庚烷(216L)加入反應器中,在減壓和低於40℃下進行蒸餾直至乾燥。將混合物冷卻至25-30℃並將正庚烷(216 L)加入反應器中。在形成固體後,將混合物攪拌1-2 h。而後,過濾固體,用正庚烷(54 L)洗滌,並且在40-50℃下乾燥10-12 h,以產生所欲之材料(90.1 Kg,產率67%)。
步驟2. 10-((二甲基胺基)亞甲基)-9-側氧基-3-氮雜螺[5.5]十一碳-7-烯-3-羧酸(E)-第三丁酯((E)-tert-Butyl 10-((dimethylamino)methylene)-9-oxo-3-azaspiro[5.5] undec-7-ene-3-carboxylate)。
在25-30℃,氮氣氛下,在乾淨且乾燥的反應器中,加入9-側氧基-3-氮雜螺[5.5]十一碳-7-烯-3-羧酸第三丁酯(50 Kg)、N,N-二甲基甲醯胺(500 L)和N,N-二甲基甲醯胺二甲縮醛(N,N-dimethylformamide dimethyl acetal)(135 Kg)。將反應混合物攪拌5-10分鐘,然後加熱至120-130℃達20 h。而後,將混合物冷卻至50-60 ℃,並在低於60℃的高真空下蒸餾溶劑。在低於45℃下加入混合二甲苯(200 L)並在低於60℃的高真空下蒸餾溶劑。用另一批混合二甲苯(200 L)重複此操作。而後,將甲苯(200 L)加入反應器中,並在低於60℃的高真空下蒸餾溶劑。用第二批甲苯(200 L)重複此操作。而後,在低於30℃下加入甲基第三丁基醚(Methyl tert-butyl ether)(100 L)並在低於40℃的高真空下蒸餾溶劑。將混合物冷卻至15-20℃並在低於20℃下加入甲基第三丁基醚(100 L)。將混合物攪拌20-30分鐘,過濾固體,用甲基第三丁基醚(50 L)洗滌,在50-55℃下真空乾燥10 h,以提供所欲之化合物(52.1 Kg,產率87% )。1
H NMR(400 MHz, CDCl3
)δ ppm 7.48(s, 1H), 6.57(d, J=9.97 Hz, 1H), 5.99(d, J=10.16 Hz, 1H), 3.32-3.51(m, 4H), 3.06(s, 6H), 2.72(s, 2H), 1.57-1.66(m, 2H), 1.41-1.53(m, 11H)。
步驟3. 1-異丙基-1,4-二氫螺[吲唑-5,4’-哌啶]-1’-羧酸第三丁酯(tert-Butyl 1-isopropyl-1,4-dihydrospiro [indazole-5,4’-piperidine]-1’-carboxylate)。
在25-30℃,在乾淨且乾燥的反應器中,加入10-((二甲基胺基)亞甲基)-9-側氧基-3-氮雜螺[5.5]十一碳-7-烯-3-羧酸(E)-第三丁酯(80 Kg)、甲苯(704 L)和三甲胺(16 L)。將反應混合物溫熱至70-80℃,並在4-5小時內加入在甲醇中的異丙基聯胺(isopropyl hydrazine)鹽酸鹽溶液(1.25當量,總共141 Kg)。而後,將反應混合物在70-80℃下攪拌8-10 h,然後冷卻至15-25℃。而後,緩慢加入在水(480L)中的檸檬酸(48 Kg)溶液,保持內部溫度低於25℃。加入乙酸乙酯(208 L)並將混合物攪拌10分鐘。分層並將有機層依序用在水(480 L)中的檸檬酸(48 Kg)溶液洗滌,而後僅用水(320 L)洗滌。用乙酸乙酯(320 L)萃取合併的水層。而後,將合併的有機層用硫酸鈉(8 Kg)乾燥,並將溶劑在減壓和低於40℃下蒸發至乾燥。將二氯甲烷(240 L)加入反應器中,並將混合物於25-30℃攪拌直至澄清。在25-30℃下依序加入活性炭(1.84 Kg)、矽酸鎂(1.84Kg)和矽膠(32Kg,100-200目),並將異質混合物攪拌1小時。而後,將漿液在Hyflow床上過濾,藉由混合Hyflow超級細胞(8 Kg)和二氯甲烷(40 L)而製備。用二氯甲烷洗滌(三次120 L)濾餅。將合併的濾液加回到反應器中,並在低於40℃的減壓下蒸發溶劑。而後,加入正庚烷(160 L)並在低於40℃的減壓下蒸餾。將正庚烷(200 L)加入反應器中,並將混合物冷卻至0-5℃。攪拌12-15 h之後,將固體於0℃過濾,用冷的(0-5℃)正庚烷(160 L)洗滌,並於40-50℃真空下乾燥,以提供標題化合物(82.4Kg,75%)。1
H NMR(400 MHz, CDCl3
)δ ppm 7.25(s, 1H), 6.42(dd, J=10.05, 0.49 Hz, 1H)5.84(d, J=9.95 Hz, 1H), 4.42-4.52(m, 1H), 3.36-3.53(m, 4H), 2.62(s, 2H)1.56-1.68(m, 2H)1.45-1.55(m, 17H)。
步驟4. 1-異丙基-4,6-二氫螺[吲唑-5,4’-哌啶]-7(1H)-酮(1-Isopropyl-4,6-dihydrospiro[indazole-5,4’-piperldin]-7(1H)-one),鹽酸鹽。
在25-30℃,在乾淨且乾燥的反應器中,加入1-異丙基-1,4-二氫螺[吲唑-5,4’-哌啶]-1’-羧酸第三丁酯(60 Kg)和甲醇(600 L)。於25-30℃下,在30-40分鐘內分5份加入N-溴琥珀醯亞胺(N-Bromosuccinimide)(32.4 Kg)並繼續攪拌30-60分鐘。緩慢加入硫代硫酸鈉五水合物(5.4 Kg)於水(102 L)中的溶液,保持內部溫度低於30℃。將混合物攪拌20-30分鐘,而後在低於45℃的減壓下蒸發溶劑。將殘餘物冷卻至25-30℃並將2-甲基四氫呋喃(420 L)與水(90 L)一起加入反應器中。將混合物攪拌15-20分鐘,而後分離各層,進一步用2-甲基四氫呋喃(120 L)萃取水層。在25-30℃,用氫氧化鈉(4.8 Kg)在水(120 L)中的溶液處理合併的有機萃取物15-20分鐘。分層,用水(120 L)洗滌有機層,而後用氯化鈉(12 Kg)在水(120 L)中的溶液洗滌,而後用硫酸鈉(6 Kg)乾燥。過濾後,用2-甲基四氫呋喃(30 L)洗滌濾餅,將合併的濾液倒回反應器中。在低於45℃的減壓下將溶劑完全蒸餾,並將殘餘物溶解在四氫呋喃(201 L)中。在25-30℃,將第三丁醇鉀(potassium tert-butoxide)(60.6 Kg)和四氫呋喃(360 L)加入另一個乾淨且乾燥的反應器中。向該混合物中緩慢加入殘餘物在四氫呋喃中的溶液,保持溫度低於30℃。而後將反應混合物升溫至60-65℃並在此溫度下保持1-2 h。完成後,將混合物冷卻至0-10℃,並用鹽酸溶液(1N,196 L)緩慢猝滅(slowly quenched),保持內部溫度低於10℃。將反應混合物溫熱至25-30℃,加入乙酸乙酯(798 L)。攪拌15-20分鐘後,分離各層,進一步用乙酸乙酯(160 L)萃取水層。用水(160 L)洗滌合併的有機層,用硫酸鈉(8 Kg)乾燥、過濾,並用乙酸乙酯(300 L)洗滌濾餅。在低於45℃的減壓下將溶劑完全蒸餾,並在25-30℃,將乙酸乙酯(540 L)加入反應器中,而後加入甲醇(156 L)。將混合物冷卻至0-5℃,此時緩慢加入乙醯氯(acetyl chloride)(79.8 Kg),保持溫度在特定範圍內。而後將混合物溫熱至20-25℃,並在攪拌下於此溫度下保持4-5 h。將所得的漿液過濾,用乙酸乙酯(120 L)洗滌固體,而後在40-45℃乾燥8-10 h,以提供所欲之粗產物(33.5 Kg,65%)。
於25-30℃,在乾淨且乾燥的反應器中將該此粗固體(56.8 Kg)溶解在甲醇(454.4 L)中,進行最終純化步驟。將溶液攪拌30-45分鐘,而後在25-30℃通過0.2微米筒式過濾器進入乾淨且乾燥的反應器中。在低於50℃的減壓下蒸餾甲醇直至殘留約1體積的溶劑。將反應混合物冷卻至25-30℃並經由0.2微米筒式過濾器加入新鮮的乙腈(113.6 L)。在低於50℃的減壓下蒸餾溶劑直至殘留約1體積的溶劑。將反應混合物冷卻至25-30℃,並經由0.2微米筒式過濾器將新鮮乙腈(190 L)加入反應器中。將混合物溫熱至65-70℃並攪拌45分鐘,而後冷卻至25-30℃並攪拌1 h。將所得的漿液過濾,用冷卻的(15℃)乙腈(56.8 L)洗滌濾餅。將固體在減壓下於40-50℃下乾燥8小時,得到中間物1(36.4Kg, 64%)。1
H NMR(400 MHz, CD3
OD)δ ppm 7.43(s, 1H), 5.32-5.42(m, 1H), 3.15-3.25(m, 4H), 2.89(s, 2H), 2.64(s, 2H), 1.69-1.90(m, 4H), 1.37-1.45(m, 6H); ESI [M+H]+
=248。
中間物2:2-(4-(第三丁氧基羰基)苯基)-6-甲氧基異菸鹼酸(2-(4-(tert-Butoxycarbonyl)phenyl)-6-methoxyisonicotinic acid)。
於20-25℃,將2,6-二氯異菸鹼酸(30 Kg)和甲醇(120 L)加入乾淨且乾燥的反應器中。將漿液攪拌5分鐘,而後加熱至65℃(回流(reflux))。而後經由添加漏斗(addition funnel)在至少4小時內緩慢加入甲醇鈉於甲醇中的溶液(30%,87.2 Kg)。用甲醇(15 L)沖洗漏斗,並在65℃下攪拌至少15 h。而後將混合物冷卻至45℃並在減壓下蒸餾直至殘餘體積為~90 L。而後於40-45℃,將碳酸氫鉀(28.2 Kg)和碳酸鉀(21.6 Kg)於水(180 L)中的溶液加入反應器中。用水(21 L)沖洗含有水溶液的反應器,並且將洗液加入反應混合物中。將混合物在低於80℃的減壓下蒸餾直至殘餘體積為~240L,而後冷卻至20-25℃。
在另一個乾淨且乾燥的反應器中加入4-(4,4,5,5-四甲基-1,3,2-二氧代環戊硼烷-2-基)苯甲酸第三丁酯(tert-butyl 4-(4,4,5,5-tetramethyl-1,3,2-dioxoborolan-2-yl)benzoate)(52.3 Kg)和二㗁烷(dioxane)(340 Kg),並於2-25℃攪拌直至完全溶解。而後將前反應器的內容物於40℃加熱以確保完全溶解並轉移到該新反應器中。將反應混合物冷卻至20-25℃,並經由真空/氮氣循環而進行脫氧步驟。將混合物進一步冷卻至0-10℃,並將乙酸鈀(0.65 Kg)加入反應器中,而後在氮氣流下加入三苯基膦(triphenylphosphine)(2.46 Kg)。將混合物溫熱至20-25℃,並經由真空/氮氣循環而進行另一個脫氧步驟。而後將混合物加熱至80℃並在此溫度下保持至少18小時。將混合物冷卻至20-25℃,而後將甲基第三丁基醚(133.2 Kg)和水(30 L)依序加入反應器中。分離各層,用水(110L)稀釋水層,而後用甲基第三丁基醚(110 L)萃取。用檸檬酸(52 Kg)在水(84 L)中的溶液洗滌合併的有機萃取物,且分離各層。進一步用甲基第三丁基醚(88.8 Kg)萃取水層,合併有機層,而後用三分之一的氯化鈉(43 Kg)在水(80 L)中的溶液洗滌三次。在最後一層分離後,經由含有炭匣(charcoal cartridge)的palll過濾器(pall filter)過濾有機層,並用甲基第三丁基醚(11.2 Kg)洗滌濾餅。將濾液在低於50℃的減壓下蒸餾至~90 L,而後在低於50℃下連續與庚烷(120 L)共蒸餾低至~120 L。而後將混合物在1小時內冷卻至20-25℃,而後在此溫度下再攪拌1小時。將漿液過濾,用庚烷(3×18 L)洗滌濾餅三次,而後用乙腈(3×18 L)洗滌三次。將所得的濕固體在真空下乾燥,氮氣於低於45℃流動至少15小時,以提供中間物2(44.6 Kg,87%產率)。1
H NMR(400 MHz, CDCl3
)δ ppm 8.13(s, 2H), 8.09(s, 2H), 7.97(d, J=1.17 Hz, 1H), 7.34(d, J=0.98 Hz, 1H), 4.08(s, 3H), 1.61(s, 9H); ESI [M+H]+
=330。
中間物3:4-(4-(1-異丙基-7-側氧基-1,4,6,7-四氫螺[吲唑-5,4’-哌啶]-1’-羰基)-6-甲氧基吡啶-2-基)苯甲酸第三丁酯(tert-Butyl 4-(4-(1-isopropyl-7-oxo-1,4,6,7-tetrahydrospiro[indazole-5,4’-piperidine]-1’-carbonyl)-6-methoxypyridin-2-yl)benzoate)。
在圓底燒瓶中加入2-(4-(第三丁氧基羰基)苯基)-6-甲氧基異菸鹼酸(中間物2,15.2g,46.2 mmol)和乙酸乙酯(140 mL)。將1,1’-羰基二咪唑(8.98 g,55.4 mmol)加入一個份量(added in one portion)並於室溫攪拌1小時。加入1-異丙基-4,6-二氫螺[吲唑-5,4’-哌啶]-7(1H)-酮鹽酸鹽(中間物1,14.8 g,52.2 mmo),而後加入N,N-二異丙基乙胺(9.1 mL,52.2 mL)並將反應於室溫攪拌18小時。加入2M HCl水溶液(40 mL),而後加入1M硫酸氫鉀(40 mL)和50 mL庚烷。 將所得的混合物於室溫攪拌1小時。 將混合物轉移至分液漏斗中。分離有機相,依序用水(20 mL)、飽和碳酸氫鈉(30 mL)、水(20 mL)、鹽水(20 mL)洗滌,用20 g硫酸鎂和10 g矽膠乾燥、過濾,並且在真空中濃縮。在濃縮結束時,開始形成固體。將殘餘物於80℃在40 mL乙酸乙酯中攪拌並緩慢滴加庚烷(120 mL)。將混合物於80℃攪拌1小時,而後在1小時攪拌下緩慢冷卻至室溫並在室溫下攪拌18小時。經由過濾收集固體,用水和乙酸乙酯-庚烷(1:3)洗滌,並且於50℃真空乾燥18小時,以得到中間物3(19.64 g,76%產率)。
中間物3的替代製備:
於20-25℃,在乾淨且乾燥的反應器中,加入乙腈(219 Kg)和2-(4-(第三丁氧基羰基)苯基)-6-甲氧基異菸鹼酸(中間物2,34.8 Kg)。將混合物攪拌5分鐘,而後分三批連續加入1,1-羰基二咪唑(1,1-carbodiimidazole)(18.9 Kg)。於20-25℃,將漿液進一步攪拌至少1小時,而後將1-異丙基-4,6-二氫吲哚[吲唑-5,4’-哌啶]-7(1H)-酮鹽酸鹽(中間物1,33.0 Kg)加入反應器中,然後經由幫浦加入N,N-二異丙基乙胺(20.5 Kg)。用乙腈(13.7 Kg)洗滌試劑幫浦以及反應器壁,並於20-25℃攪拌至少2小時。完成之後,向混合物中接種4-(4-(1-異丙基-7-側氧基-1,4,6,7-四氫螺[吲唑-5,4’-哌啶]-1’-羰基)-6-甲氧基吡啶-2-基)苯甲酸第三丁酯(中間體3,209g)並且攪拌至少30分鐘。確認開始結晶後,在1小時內加入檸檬酸單水合物(58.5 Kg)於水(257 L)中的溶液。於20-25℃,將所得的漿液進一步攪拌至少2小時,而後過濾,並且用乙腈(68.4 Kg)和水(87 L)的混合物洗滌濾餅。該洗滌液也用於沖洗反應器。將固體在低於55℃的減壓下乾燥,以供應中間物3(43.44 Kg,73%產率)。
化合物1(作為游離酸):4-(4-(1-異丙基-7-側氧基-1,4,6,7-四氫螺[吲唑-5,4’-哌啶]-1’-羰基)-6-甲氧基吡啶-2-基)苯甲酸。
在圓底燒瓶中加入4-(4-(1-異丙基-7-側氧基-1,4,6,7-四氫螺[吲唑-5,4’-哌啶]-1’-羰基)-6-甲氧基吡啶-2-基)苯甲酸第三丁酯(3.7 g,6.6 mmol)和甲苯(25 mL)。攪拌下滴加85%磷酸(3.0 mL),將反應混合物加熱至60℃達4小時。形成無色厚膠。將反應冷卻至室溫並加入水。觀察到白色固體。棄去甲苯有機層,保留水層和固體。加入乙酸乙酯(60 mL)並且加入4N NaOH溶液以將pH調節至~7。分離各層,用乙酸乙酯(50 mL)萃取水層。將合併的乙酸乙酯有機層用硫酸鈉乾燥、過濾、並且真空濃縮,以提供白色固體。將它們於50℃溶解在乙酸乙酯(80 mL)中並且緩慢加入庚烷(90 mL)。除去熱,將混合物冷卻至室溫並攪拌16小時。經由過濾收集所得的固體,用母液沖洗,並且乾燥以提供標題化合物(化合物1游離形式,2.15 g,65%產率),為白色固體。
化合物1(作為游離酸)的替代製備:
於20-25℃,在乾淨乾燥反應器中加入乙腈(130.4 Kg)和4-(4-(1-異丙基-7-側氧基-1,4,6,7-四氫螺[吲唑-5,4’-哌啶]-1’-羰基)-6-甲氧基吡啶-2-基)苯甲酸第三丁酯(中間物3,20.72 Kg)。將混合物攪拌5分鐘,而後在溫和的氮氣吹掃下加入對甲苯磺酸(8.5 Kg)。將反應混合物溫熱至70℃並在此溫度下保持至少6.5小時。完成後,將混合物冷卻至40℃,接種化合物1(104 g),並在至少1小時內緩慢加入水(83 L)。將混合物於40℃進一步攪拌至少4小時,而後在2小時內冷卻至20-25℃。進一步攪拌至少2小時,而後過濾,用乙腈(33 Kg)和水(41 L)的溶液沖洗濾餅。此洗滌也用於沖洗反應器。在低於55℃的減壓下乾燥所得的固體,以提供化合物1(16.5Kg,89%產率)。
形式1(化合物1的無水單-tris)的製備: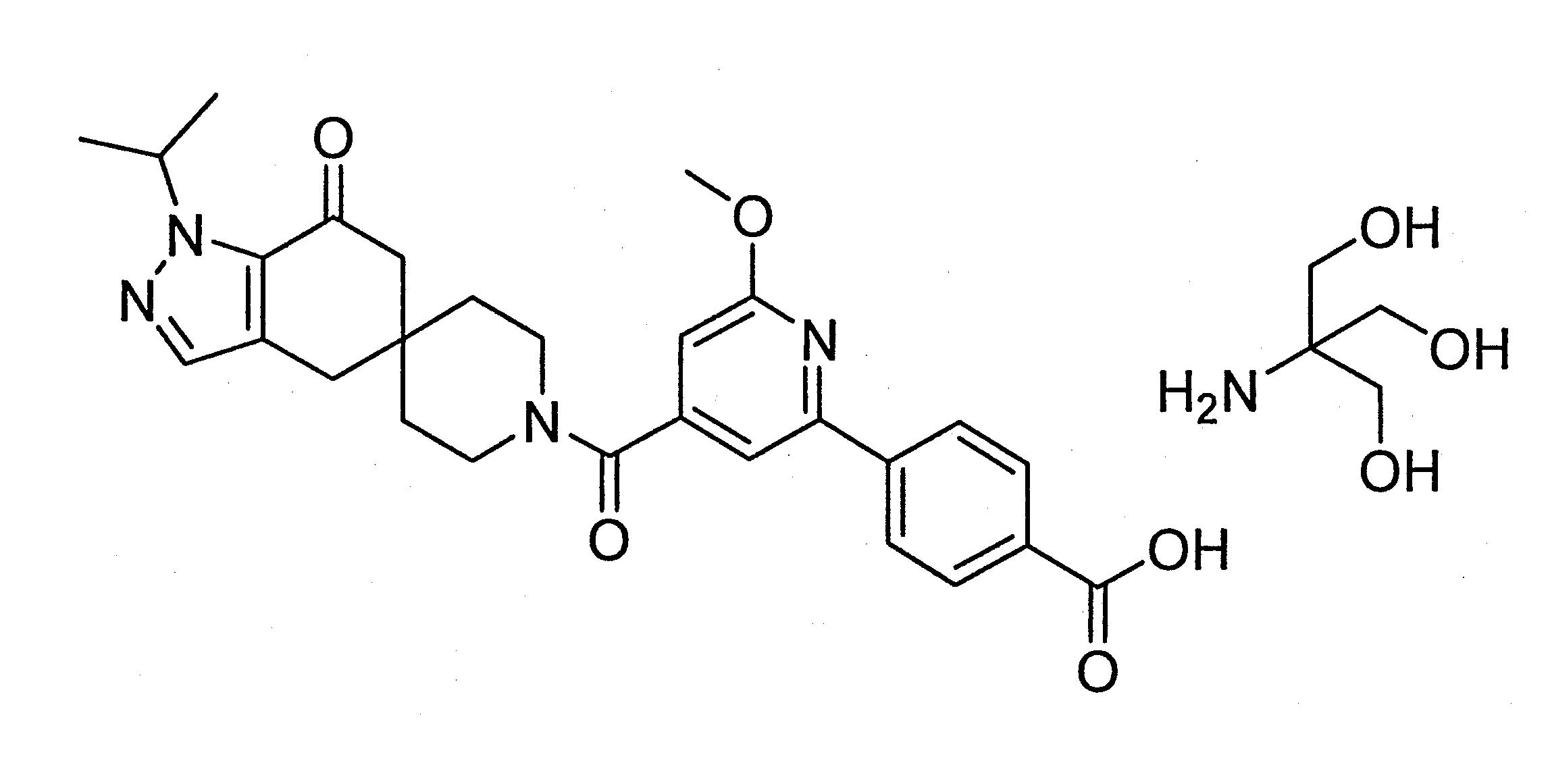
在小瓶中加入4-(4-(1-異丙基-7-側氧基-1,4,6,7-四氫螺[吲唑-5,4’-哌啶]-1’-羰基)-6-甲氧基吡啶-2-苯基)苯甲酸(151 mg,0.300 mmol)和3 mL乙醇。將混合物加熱至80℃達5分鐘以溶解固體,而後冷卻至室溫。加入三(羥甲基)胺基甲烷(Tris(hydroxymethyl)aminomethane) (39 mg,0.32 mmol),並將混合物於室溫攪拌過夜。逐滴加入庚烷(2.25 mL)以產生漿液,將其加熱至50℃以產生澄清溶液。在攪拌下將混合物冷卻至室溫過夜。觀察到白色固體,並將混合物再攪拌3天。將物質過濾並在真空烘箱中於50℃乾燥過夜以產生形式1(151 mg,0.242 mmol,81%產率)。
形式1(化合物1的無水單-tris)的替代製備:
在乾淨且乾燥的反應器中加入乙醇(83 L),而後加入化合物1(9.43 Kg)和tris(2.55 kg),同時將混合物保持在20-25℃的溫度。用乙醇(2 L)沖洗容器壁,並將所得的混合物於65-70℃加熱,在此溫度保持至少30分鐘直至所有固體溶解,而後冷卻至45-50℃。經由10μm在線聚丙烯過濾器(in-line polypropylene filter)進行溫過濾,並用乙醇(9 L)洗滌反應器以及過濾器。經由相同的在線過濾器將正庚烷(24 L)加入到溫溶液中,並於45-50℃,將在乙醇(0.5 L)中的4-(4-異丙基-7-側氧基-1,4,6,7-四氫螺旋體[吲唑-5,4’-哌啶]-1’-羰基)-6-甲氧基吡啶-2-基)苯甲酸無水tris鹽(100 g)接種於混合物。將溫度保持至少2小時,而後在至少2小時內冷卻至20-25℃。攪拌至少5天。 而後過濾漿液,用乙醇(13 L)和正庚烷(6 L)的混合物洗滌濾餅。將固體在低於45℃的減壓下乾燥至少12小時,以提供實例1(11.7 Kg,77%)。
形式2(化合物1的單-tris鹽的三水合物)的製備:
從形式1的轉換獲得形式2。在50 mL EasyMax反應器中,加入形式1(1.7214 g,2.760 mmol)、異丙醇(16.50 mL,215.8 mmol)和水(688 μL,38.190 mmol)。以25℃的反應器夾套溫度,將混合物攪拌(300 rpm)約72小時。而後將反應混合物在15分鐘內溫熱至40℃,並在40℃下保持約24小時,冷卻一次至20℃以移除樣品用於測試。藉由PXRD觀察到形式的混合物;因此,添加額外的水(688 μL,38.190 mmol)。將攪拌速率增加至400 rpm,並且將漿液攪拌6小時,而後冷卻至15℃。將固體在60 mL/40 M過濾器上分離並用96/4異丙醇/水洗滌。藉由PXRD,所得的材料與形式2一致。
形式2(化合物1的單-tris鹽的三水合物)的替代製備:
在乾淨且乾燥的反應器中加入異丙醇(60.4 Kg),並且加入化合物1(16.68 Kg)和tris(4.42 kg),同時將混合物保持在20-25℃的溫度。將混合物攪拌5分鐘,而後加入水(6.7 Kg)並將漿液溫熱至55℃。將現在澄清的溶液經由在線10μm聚丙烯過濾器過濾到預熱的乾淨且乾燥的反應器(50-55℃)中。而後用化合物1的單-tris鹽作為三水合物(167 g)接種溶液。在確認晶種(seed)持續存在後,將混合物在至少2小時內冷卻至15℃,而後在15℃下保持最少16小時。過濾漿液,用冷卻的異丙醇(13.1 Kg)洗滌濾餅。而後將固體在低於25℃的減壓下乾燥,以提供形式2(22.1 Kg,98%產率)。
為了確認形式2中存在三個水分子,在室溫下使用Bruker D8 Venture繞射儀收集數據。參閱圖7。使用Monoclinic類空間群P21
/c(Version 5.1, Bruker AXS, 1997)中的SHELX軟體套件藉由固有相位解出結構。隨後藉由全矩陣最小平方法精進(refine)該結構。使用非等向性位移參數找出並精進所有的非氫原子。
從Fourier差異圖中找到位於氮和氧上的氫原子並且在距離受限的情況下進行精進(refine)。將剩餘的氫原子置於計算的位置並使其搭載於其載體原子上。
最終的R指數為7.2%。Fourier的最終差異顯示沒有丟失或錯位的電子密度。
表10提供關於形式2所收集的數據:
藥物動力學研究:
對於雄性(禁食)食蟹猴進行藥物動力學研究(每種配方n=2)。製備口服藥丸以比較形式1與形式2。此外,為了比較化合物1的tris鹽和化合物1的游離酸,製備懸浮液。分別投與含有2、5或10 mg/kg的口服劑量。藉由注射器經股動靜脈在0、0.25、0.5、1、2、4、7和24小時時間點收集連續血液樣品並轉移至K3
EDTA真空容器中。而後將血液樣品離心,收集血漿並儲存在-20℃或-80℃直至分析。
口服投與的固體錠劑:
藉由混合形式1或形式2與賦形劑的標準混合物而製備口服錠劑,該賦形劑包含:64重量%的微晶纖維素(Avicel PH102)、32重量%的乳糖單水合物(Fast Flo 316)、3重量%的羥乙酸澱粉鈉(sodium starch glycolate) (Explotab®)和1重量%的硬脂酸鎂。將適量的形式1或形式2轉移至研缽中。而後將相應量的賦形劑混合物幾何添加至研缽中,並使用研杵與個別的形式1或形式2充分混合。將混合物轉移到容器中,並在Turbula混合器上混合5分鐘,以提供含有與賦形劑混合的個別形式1或形式2的混合物。對於形式1,該混合物含有19重量%的形式1和81重量%的賦形劑混合物。對於形式2,該混合物含有21重量%的形式2和79重量%的賦形劑混合物。
為了製備錠劑,將每種混合物轉移至配備有0.2362”標準圓形凹形工具的Korsch XP-1單站壓機(single-station press)中。將錠劑壓縮成100 mg錠劑重量,目標錠劑硬度為4至9 kp,壓製速度為每分鐘15錠。
口服投與的懸浮液:
為了製備1L的0.5%(w/v)甲基纖維素(Methocel®
A4M)溶液的代表性批量,將約0.4 L去離子水加熱至80-90℃,而後加入5克的甲基纖維素(Methocel®A4M)並充分混合直至顆粒完全潤濕。而後將混合物從加熱中取出。而後加入冷水(0.6 L),同時在冰浴中持續攪拌,直至所有甲基纖維素顆粒溶解。
從游離酸製備的化合物1之10 mg/mL懸浮液:將化合物1的游離酸(220 mg)轉移到研缽中。使用研杵研磨固體粉末(團塊破碎)。將2 mL的0.5%w/v甲基纖維素(Methocel®
A4M)滴加至粉末。將載體和游離酸充分混合以形成平滑糊狀物。將剩餘的載體以小等分試樣加入,同時混合直至獲得均勻的懸浮液。使用滴管將懸浮液轉移至30 mL玻璃瓶中,並使其體積為22 mL,以達到濃度為10mg/mL的化合物1之游離酸。測得的pH為6.04。將22微升的聚山梨醇酯80(Tween®
80)加入懸浮液中。獲得10mg/mL化合物1的游離酸在0.5%w/v甲基纖維素(Methocel®
A4M),0.1%v/v聚山梨醇酯80(Tween®
80)中的配方。
從化合物1的Tris鹽製備化合物1之2.5 mg/mL懸浮液:將形式1(157 mg)轉移至研缽。使用研杵研磨固體粉末(團塊破碎)。將少量0.5%w/v甲基纖維素(Methocel®
A4M)加入至粉末,形成平滑糊狀物。將剩餘的載體以小等分試樣加入,同時混合直至獲得均勻的懸浮液。將懸浮液轉移至容器中並使最終體積為50 mL。觀察到一些團塊,但攪拌約1小時後得到輕質懸浮液(light suspension)。
分析數據:
吸濕性(Hygroscopicity):
使用由TA Instruments或Surface Measurement Systems製造的動態蒸汽吸附儀器測量吸濕性。將每種形式的樣品暴露於增量RH水平,直至符合在5分鐘內≤0.001%重量變化的重量平衡(視為平穩期(plateau)),或在每個RH水平的最大時間120分鐘。在5分鐘或120分鐘內較短的0.001%重量平衡之後,而後將樣品暴露於下一個RH水平。形式1可能已經失去重量的初始乾燥期存在,未對於形式2使用乾燥。該過程在10%RH開始,RH增加至20%RH,而後在每個間隔後增加10%RH(在5分鐘平衡內≤0.001%或120分鐘,以先發生者為準)。在90%RH,使用相同的平衡標準將RH逆轉回10%RH。測量吸濕性作為在90%RH測量的重量增加百分比的函數。形式1在90%RH/25℃具有約1%的吸濕性。形式2在90%RH/25℃也具有約1%的吸濕性。
表2中提供熱力學溶解度:
將作為游離酸的化合物1或表2中辨識的鹽作為乾粉接收,並且預先稱重至具有0.45 μm聚四氟乙烯(polytetrafluoroethylene,PTFE)膜的Whatman Mini-Uniprep無注射器過濾裝置中。將450微升(450 μL)所欲之培養基加入至過濾器中並且在室溫下攪拌24小時。24小時後,過濾樣品,將濾液注入氮氣檢測器中進行定量。
緩衝液製備:
• pH1.2:稱出1.0g NaCl並轉移至燒杯中。加入約450 mL HPLC等級的水溶解NaCl。用36.6%HCl將溶液滴定至pH 1.2。將溶液轉移至500 mL容量瓶中,用HPLC等級的水加至500 mL。
• pH6.5:用約500mL的50 mM NaPO4
單鹼(monobasic)滴定約250mL的50 mM NaPO4
二元鹼(dibasic)至pH6.5。最終總體積是當向50 mM NaPO4
二元鹼添加50 mM NaPO4
單鹼時溶液達到具有pH 6.5之處的體積。
• pH7.4:用約50 mL的50 mM NaPO4
單鹼(monobasic)滴定約200 mL的50 mM NaPO4
二元鹼(dibasic)至pH7.4。最終總體積是當向50 mM NaPO4
二元鹼添加50 mM NaPO4
單鹼時溶液達到具有pH 7.4之處的體積。
粉末X-射線繞射:
使用配備有Cu輻射源的Bruker AXS D4 Endeavor繞射儀進行粉末X射線繞射分析。發散狹縫設定為0.6 mm,而二次光學系統使用可變狹縫。藉由PSD-Lynx Eye偵側器偵測繞射的輻射。X射線管電壓(X-ray tube voltage)與電流強度(amperage)分別經設定為40 kV與40 mA。在Theta-2Theta測角器中在Cu(k-α平均值)3.0至40.0度2-θ,使用步進尺寸(step size)為0.037度且每步時間為10秒,收集數據。藉由將樣品置於矽低背景樣品架中且在收集期間旋轉以製備樣品。使用Bruker DIFFRAC Plus軟體收集數據,並且藉由EVA繞射加軟體(版本4.2.1)進行分析。在峰搜尋之前未處理PXRD數據檔案。使用EVA軟體中的峰搜尋演算法,用閾值為1所選擇的峰以進行初步峰分配。為確保有效性,手動進行調整;目視檢查自動分配的輸出,並將峰位置調整到峰最大值。通常選擇相對強度≥3%的峰。未解析或與噪聲相符的峰不被選擇。與USP中的PXRD峰位置相關的典型誤差高達+/-0.2°2-θ(USP-941)。
FT-Raman:
使用附接到FT-IR台的Nicolet NXR FT-Raman附件以收集Raman光譜。光譜儀配備有1064 nm Nd:YVO4雷射與液態氮冷卻鍺偵測器。在獲取數據之前,使用聚苯乙烯進行儀器性能和校準驗證。在玻璃NMR管中分析形式1或形式2的樣品,其在光譜收集期間是靜態的。將錠劑樣品收集在錠劑樣品架中,其分析完整錠劑上的一個點。使用0.5W的雷射功率和512次共同添加的掃描以收集光譜。收集範圍是3700-100 cm-1
。使用2cm-1
解析度與Happ-Genzel變跡法(apodization)記錄這些光譜。利用上述Raman方法,與光譜測量相關的可能變化為±2cm-1
。在環境條件(~23℃與30%-60%RH)下,收集樣品(純API與藥物產品)。形式1應與乾燥劑一起儲存,而形式2可在環境條件(15-30℃與環境濕度)下儲存。
在選取峰之前,將強度標度正規化(normalized)為1。使用Thermo Nicolet Omnic 9.7.46軟體,手動識別峰。在峰最大值處選取峰位置,如果每側有斜坡,則僅識別峰;不包含在峰上的肩膀(shoulders on peaks)。對於形式1或形式2,在峰選取期間使用0.004至0.017的絕對閾值,靈敏度為80。使用標準實務(0.5進位,0.4捨去),已將峰位置調整至最接近的整數。在(1-0.75)、(0.74-0.30)、(0.29-0)之間具正規化峰強度的峰分別標示為強、中、以及弱。
固態NMR:
在位於Bruker-BioSpin Avance III 500MHz (1
H頻率)NMR光譜儀中的Bruker-BioSpin CPMAS探針上進行固態NMR(ssNMR)分析。將形式1料裝入用標準驅動蓋密封的4 mm轉子中,並在環境溫度下收集其光譜。將形式2材料裝入4 mm轉子中,用含有O形環的驅動蓋密封該轉子以防止脫水。於25℃收集形式2光譜(藉由PbNO3
的化學位移校準)。裝好的轉子以魔術角度(magic angle)定向並以15.0 kHz旋轉。使用質子解耦的交叉極化魔術角度旋轉(cross-polarization magic angle spinning,CPMAS)實驗收集13
C ssNMR光譜。在獲取光譜期間,使用80-90kHz的相位調節質子去耦場(phase modulated proton decoupling field)。交叉極化接觸時間(cross-polarization contact time)設定為2 ms,並且再循環延遲設定為10秒。調整掃描次數以獲得足夠的訊號與雜訊比率(signal to noise ratio)。在結晶金剛烷的外部標準上使用13
C CPMAS實驗,引用碳化學位移標度,將其高場共振設定為29.5ppm(由純TMS確定)。
使用Bruker-BioSpin TopSpin版本3.5軟體,進行自動峰選取。通常,使用5%相對強度的閾值進行初步峰選擇。目視檢查自動峰選取的輸出,以確保有效性並在必要時進行手動調整。儘管本文報導了特定的13
C固態NMR峰值,但由於儀器、樣品和樣品製備的差異,確實存在這些峰值的範圍。由於峰位置固有的變化,因而這是固態NMR領域的常見實務。對於結晶固體,13
C化學位移x軸值的典型可變性為該值約加或減0.2 ppm。本文所報導的固態NMR峰高度為相對強度。固態NMR強度可依CPMAS實驗參數的實際設定和樣品的熱歷史而改變。
圖1說明在配備有Cu輻射源的Bruker AXS D4 Endeavor繞射儀上進行的形式1的說明性PXRD圖案。
圖2說明使用附接至FT-IR台的Nicolet NXR FT-Raman配件收集的形式1的說明性Raman光譜。
圖3說明位在Bruker-BioSpin Avance III 500 MHz(1
H頻率)NMR光譜儀中的Bruker-BioSpin CPMAS探針上進行的形式1的說明性13
C ssNMR圖案。
圖4說明在配備有Cu輻射源的Bruker AXS D4 Endeavor繞射儀上進行的形式2的說明性PXRD圖案。
圖5說明使用附接至FT-IR台的Nicolet NXR FT-Raman配件收集的形式2的說明性Raman光譜。
圖6說明位在Bruker-BioSpin Avance III 500 MHz(1
H頻率)NMR光譜儀中的Bruker-BioSpin CPMAS探針上進行的形式2的說明性13
C ssNMR圖案。
圖7說明形式2的說明性單晶結構。
Claims (18)
- 一種結晶鹽,其為4-(4-(1-異丙基-7-側氧基-1,4,6,7-四氫螺[吲唑-5,4’-哌啶]-1’-羰基)-6-甲氧基吡啶-2-基)苯甲酸之2-胺基-2-(羥甲基)丙烷-1,3-二醇鹽。
- 如申請專利範圍第1項所述之結晶鹽,其中4-(4-(1-異丙基-7-側氧基-1,4,6,7-四氫螺[吲唑-5,4’-哌啶]-1’-羰基)-6-甲氧基吡啶-2-基)苯甲酸與該鹽的比率為1:1。
- 如申請專利範圍第1或2項所述之結晶鹽,其中該結晶鹽為無水結晶鹽。
- 如申請專利範圍第3項所述之結晶鹽,其中該無水結晶鹽具有PXRD圖案,其包括在9.6、10.7與11.3 2θ分別±0.2° 2θ之繞射角的峰。
- 如申請專利範圍第4項所述之結晶鹽,其中該無水結晶鹽具有Raman光譜,其包括在1511、1561與1615cm-1分別±2cm-1的峰偏移。
- 如申請專利範圍第5項所述之結晶鹽,其中該無水結晶鹽具有13C ssNMR光譜,其包括在22.9、146.2與161.9ppm分別±0.2ppm的化學位移。
- 如申請專利範圍第3項所述之結晶鹽,其中該無水結晶鹽具有選自由以下所組成之群組的分析參數:包括在1511與1615cm-1分別±2cm-1的峰偏移的Raman光譜,以及包括在22.9、146.2或161.9ppm分別±0.2ppm的至少一個化學位移的13C ssNMR光譜。
- 如申請專利範圍第3項所述之結晶鹽,其中該無水結晶鹽為實質上純的。
- 如申請專利範圍第1或2項所述之結晶鹽,其中該結晶鹽為三水合物結晶鹽。
- 如申請專利範圍第9項所述之結晶鹽,其中該三水合物結晶鹽具有PXRD圖案,其包括在8.4、9.0與10.5 2θ分別±0.2° 2θ之繞射角的峰。
- 如申請專利範圍第9項所述之結晶鹽,其中該三水合物結晶鹽具有Raman光譜,其包括在1507、1557與1610cm-1分別±2cm-1的峰偏移。
- 如申請專利範圍第9項所述之結晶鹽,其中該三水合物結晶鹽具有13C ssNMR光譜,其包括在19.2、149.5與163.8ppm分別±0.2ppm的化學位移。
- 如申請專利範圍第9項所述之結晶鹽,其中該三水合物結晶鹽具有選自由以下所組成之群組的分析參數:PXRD圖案,其包括在8.4與9.0 2θ分別±0.2° 2θ之繞射角的峰,Raman光譜,其包括在1557與1610cm-1分別±2cm-1的峰偏移,以及13C ssNMR光譜,其包括在19.2、149.5或163.8ppm分別±0.2ppm的至少一個化學位移。
- 如申請專利範圍第9項所述之結晶鹽,其中該三水合物結晶鹽具有選自由以下所組成之群組的分析參數:PXRD圖案,其包括在8.4與9.0 2θ分別±0.2° 2θ之繞射角的峰,以及Raman光譜,其包括在1507、1557或1610cm-1分別±2cm-1的至少一個峰偏移。
- 如申請專利範圍第9項所述之結晶鹽,其中該三水合物結晶鹽具有選自由以下所組成之群組的分析參數:PXRD圖案,其包括在8.4與9.0 2θ分別±0.2° 2θ之繞射角的峰,以及13C ssNMR光譜,其包括在19.2、149.5或163.8ppm分別±0.2ppm的至少一個化學位移。
- 如申請專利範圍第9項所述之結晶鹽,其中該三水合物結晶鹽為實質上純的。
- 一種醫藥組成物,其包括治療有效量之如申請專利範圍第1至16項中任一項所述之結晶鹽與醫藥上可接受的載體。
- 一種如申請專利範圍第1至16項中任一項所述之結晶鹽的用途,係用於製造一種用來治療哺乳動物選自NAFLD、NASH與T2D之疾病的藥物。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201762589256P | 2017-11-21 | 2017-11-21 | |
| US62/589,256 | 2017-11-21 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| TW201932466A TW201932466A (zh) | 2019-08-16 |
| TWI703143B true TWI703143B (zh) | 2020-09-01 |
Family
ID=64559730
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| TW107140741A TWI703143B (zh) | 2017-11-21 | 2018-11-16 | 4-(4-(1-異丙基-7-側氧基-1,4,6,7-四氫螺〔吲唑-5,4’-哌啶〕-1’-羰基)-6-甲氧基吡啶-2-基)苯甲酸之晶型 |
Country Status (22)
| Country | Link |
|---|---|
| US (1) | US11325909B2 (zh) |
| EP (1) | EP3713942B8 (zh) |
| JP (1) | JP7237082B2 (zh) |
| KR (1) | KR102395451B1 (zh) |
| CN (1) | CN111406056B (zh) |
| AU (1) | AU2018372109B2 (zh) |
| BR (1) | BR112020008413A2 (zh) |
| CA (1) | CA3082867C (zh) |
| DK (1) | DK3713942T3 (zh) |
| ES (1) | ES2942463T3 (zh) |
| FI (1) | FI3713942T3 (zh) |
| HU (1) | HUE061885T2 (zh) |
| IL (1) | IL274820A (zh) |
| MX (1) | MX2020005113A (zh) |
| PL (1) | PL3713942T3 (zh) |
| PT (1) | PT3713942T (zh) |
| RU (1) | RU2020116458A (zh) |
| SG (1) | SG11202003600QA (zh) |
| SI (1) | SI3713942T1 (zh) |
| TW (1) | TWI703143B (zh) |
| WO (1) | WO2019102311A1 (zh) |
| ZA (1) | ZA202002566B (zh) |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11254660B2 (en) | 2018-08-31 | 2022-02-22 | Pfizer Inc. | Combinations for treatment of NASH/NAFLD and related diseases |
| EP3972596A1 (en) * | 2019-05-20 | 2022-03-30 | Pfizer Inc. | Combinations comprising benzodioxol as glp-1r agonists for use in the treatment of nash/nafld and related diseases |
| JP2022058085A (ja) | 2020-02-24 | 2022-04-11 | ファイザー・インク | ジアシルグリセロールアシルトランスフェラーゼ2阻害剤とアセチル-CoAカルボキシラーゼ阻害剤との組合せ |
| JP2021134211A (ja) | 2020-02-24 | 2021-09-13 | ファイザー・インク | Nafld/nashおよび関連疾患の処置のための組合せ |
| CN117940422A (zh) | 2021-08-31 | 2024-04-26 | 辉瑞大药厂 | 2-[(4-{6-[(4-氰基-2-氟芐基)氧基]吡啶-2-基}哌啶-1-基)甲基]-1-[(2s)-氧杂环丁烷-2-基甲基]-1h-苯并咪唑-6-羧酸,1,3-二羟基-2-(羟甲基)丙-2-胺盐的固体形式 |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012042433A1 (en) * | 2010-09-30 | 2012-04-05 | Pfizer Inc. | N1-PYRAZOLOSPIROKETONE ACETYL-CoA CARBOXYLASE INHIBITORS |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7745625B2 (en) | 2004-03-15 | 2010-06-29 | Bristol-Myers Squibb Company | Prodrugs of piperazine and substituted piperidine antiviral agents |
| CH702192A1 (de) | 2009-11-04 | 2011-05-13 | New Dent Ag | Keramisches Implantat. |
| DK2499139T3 (da) | 2009-11-10 | 2014-01-27 | Pfizer | N1-pyrazolospiroketon-acetyl-coa-carboxylasehæmmere |
| NZ609527A (en) | 2010-10-29 | 2014-03-28 | Pfizer | N1/n2-lactam acetyl-coa carboxylase inhibitors |
| CN103492388A (zh) | 2011-04-22 | 2014-01-01 | 辉瑞大药厂 | 用作乙酰辅酶a羧化酶抑制剂类的吡唑并螺酮衍生物 |
-
2018
- 2018-11-14 BR BR112020008413-1A patent/BR112020008413A2/pt unknown
- 2018-11-14 US US16/765,350 patent/US11325909B2/en active Active
- 2018-11-14 EP EP18811658.6A patent/EP3713942B8/en active Active
- 2018-11-14 FI FIEP18811658.6T patent/FI3713942T3/fi active
- 2018-11-14 PL PL18811658.6T patent/PL3713942T3/pl unknown
- 2018-11-14 SI SI201830907T patent/SI3713942T1/sl unknown
- 2018-11-14 DK DK18811658.6T patent/DK3713942T3/da active
- 2018-11-14 KR KR1020207014134A patent/KR102395451B1/ko active IP Right Grant
- 2018-11-14 WO PCT/IB2018/058966 patent/WO2019102311A1/en active Application Filing
- 2018-11-14 MX MX2020005113A patent/MX2020005113A/es unknown
- 2018-11-14 SG SG11202003600QA patent/SG11202003600QA/en unknown
- 2018-11-14 AU AU2018372109A patent/AU2018372109B2/en active Active
- 2018-11-14 PT PT188116586T patent/PT3713942T/pt unknown
- 2018-11-14 ES ES18811658T patent/ES2942463T3/es active Active
- 2018-11-14 JP JP2020545003A patent/JP7237082B2/ja active Active
- 2018-11-14 HU HUE18811658A patent/HUE061885T2/hu unknown
- 2018-11-14 RU RU2020116458A patent/RU2020116458A/ru unknown
- 2018-11-14 CA CA3082867A patent/CA3082867C/en active Active
- 2018-11-14 CN CN201880075399.6A patent/CN111406056B/zh active Active
- 2018-11-16 TW TW107140741A patent/TWI703143B/zh active
-
2020
- 2020-05-08 ZA ZA2020/02566A patent/ZA202002566B/en unknown
- 2020-05-20 IL IL274820A patent/IL274820A/en unknown
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012042433A1 (en) * | 2010-09-30 | 2012-04-05 | Pfizer Inc. | N1-PYRAZOLOSPIROKETONE ACETYL-CoA CARBOXYLASE INHIBITORS |
| US8859577B2 (en) * | 2010-09-30 | 2014-10-14 | Pfizer Inc. | N1-pyrazolospiroketone acetyl-CoA carboxylase inhibitors |
Also Published As
| Publication number | Publication date |
|---|---|
| ES2942463T3 (es) | 2023-06-01 |
| AU2018372109A1 (en) | 2020-05-07 |
| PT3713942T (pt) | 2023-04-17 |
| EP3713942A1 (en) | 2020-09-30 |
| JP2021504442A (ja) | 2021-02-15 |
| RU2020116458A (ru) | 2021-12-23 |
| IL274820A (en) | 2020-07-30 |
| FI3713942T3 (fi) | 2023-05-04 |
| EP3713942B8 (en) | 2023-04-12 |
| US11325909B2 (en) | 2022-05-10 |
| AU2018372109B2 (en) | 2021-07-01 |
| BR112020008413A2 (pt) | 2020-10-06 |
| CN111406056B (zh) | 2023-02-28 |
| US20200354362A1 (en) | 2020-11-12 |
| WO2019102311A1 (en) | 2019-05-31 |
| HUE061885T2 (hu) | 2023-08-28 |
| JP7237082B2 (ja) | 2023-03-10 |
| RU2020116458A3 (zh) | 2021-12-23 |
| CA3082867C (en) | 2022-05-10 |
| CA3082867A1 (en) | 2019-05-31 |
| KR102395451B1 (ko) | 2022-05-09 |
| ZA202002566B (en) | 2023-09-27 |
| EP3713942B1 (en) | 2023-03-08 |
| DK3713942T3 (da) | 2023-04-03 |
| TW201932466A (zh) | 2019-08-16 |
| CN111406056A (zh) | 2020-07-10 |
| KR20200074169A (ko) | 2020-06-24 |
| SI3713942T1 (sl) | 2023-06-30 |
| SG11202003600QA (en) | 2020-06-29 |
| MX2020005113A (es) | 2022-05-11 |
| PL3713942T3 (pl) | 2023-05-29 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| TWI703143B (zh) | 4-(4-(1-異丙基-7-側氧基-1,4,6,7-四氫螺〔吲唑-5,4’-哌啶〕-1’-羰基)-6-甲氧基吡啶-2-基)苯甲酸之晶型 | |
| CN110139860A (zh) | 1,3,5-三嗪衍生物的盐及其晶体、它们的制备方法、药物组合物和用途 | |
| KR102365209B1 (ko) | 세타글립틴의 염 및 그의 제조방법, 약학조성물, 용도 | |
| US20190330161A1 (en) | Crystalline forms of a bromodomain and extraterminal protein inhibitor drug, processes for preparation thereof, and use thereof | |
| CN116249695A (zh) | 结晶PPAR-δ激动剂 | |
| EP2710009B1 (en) | Novel crystalline asenapine hydrochloride salt forms | |
| KR20170033684A (ko) | 리나글립틴 결정형 및 이의 제조방법 | |
| CN116375634A (zh) | 卡瑞斯汀对甲苯磺酸盐的晶型和无定型 | |
| AU2012234325B2 (en) | Benzoic acid salt of Otamixaban | |
| US11149013B2 (en) | Crystal form of urate transporter 1 inhibitor and preparation method and use thereof | |
| CZ20022474A3 (cs) | Ethery dikarboxylátu vápenatého, metody jejich výroby a jejich využití k léčbě vaskulárních chorob a diabetu | |
| JP2022507558A (ja) | チエノピリドン誘導体のカリウム塩一水和物及びその調製方法 | |
| CN112543634A (zh) | Ebna1抑制剂晶体形式及其制备和使用方法 | |
| US11787819B2 (en) | Crystalline salt of a multi-tyrosine kinase inhibitor, method of preparation, and use thereof | |
| JP5589097B2 (ja) | ダサチニブ多結晶体、並びにその調製方法及び薬物組成物 | |
| TW202333694A (zh) | 稠環衍生物的晶型、其製備方法及其應用 | |
| TW202140445A (zh) | 四氮唑類化合物的可藥用鹽及其結晶形式、製備方法和用途 | |
| NZ620339B2 (en) | N-hetero-ring-substituted amide derivative |