TW202045480A - 化合物及使用方法 - Google Patents
化合物及使用方法 Download PDFInfo
- Publication number
- TW202045480A TW202045480A TW109106570A TW109106570A TW202045480A TW 202045480 A TW202045480 A TW 202045480A TW 109106570 A TW109106570 A TW 109106570A TW 109106570 A TW109106570 A TW 109106570A TW 202045480 A TW202045480 A TW 202045480A
- Authority
- TW
- Taiwan
- Prior art keywords
- alkyl
- alkenyl
- cycloalkyl
- aryl
- heteroaryl
- Prior art date
Links
Images


Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D217/00—Heterocyclic compounds containing isoquinoline or hydrogenated isoquinoline ring systems
- C07D217/12—Heterocyclic compounds containing isoquinoline or hydrogenated isoquinoline ring systems with radicals, substituted by hetero atoms, attached to carbon atoms of the nitrogen-containing ring
- C07D217/14—Heterocyclic compounds containing isoquinoline or hydrogenated isoquinoline ring systems with radicals, substituted by hetero atoms, attached to carbon atoms of the nitrogen-containing ring other than aralkyl radicals
- C07D217/16—Heterocyclic compounds containing isoquinoline or hydrogenated isoquinoline ring systems with radicals, substituted by hetero atoms, attached to carbon atoms of the nitrogen-containing ring other than aralkyl radicals substituted by oxygen atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/4365—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system having sulfur as a ring hetero atom, e.g. ticlopidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/437—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a five-membered ring having nitrogen as a ring hetero atom, e.g. indolizine, beta-carboline
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/472—Non-condensed isoquinolines, e.g. papaverine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/472—Non-condensed isoquinolines, e.g. papaverine
- A61K31/4725—Non-condensed isoquinolines, e.g. papaverine containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D217/00—Heterocyclic compounds containing isoquinoline or hydrogenated isoquinoline ring systems
- C07D217/02—Heterocyclic compounds containing isoquinoline or hydrogenated isoquinoline ring systems with only hydrogen atoms or radicals containing only carbon and hydrogen atoms, directly attached to carbon atoms of the nitrogen-containing ring; Alkylene-bis-isoquinolines
- C07D217/06—Heterocyclic compounds containing isoquinoline or hydrogenated isoquinoline ring systems with only hydrogen atoms or radicals containing only carbon and hydrogen atoms, directly attached to carbon atoms of the nitrogen-containing ring; Alkylene-bis-isoquinolines with the ring nitrogen atom acylated by carboxylic or carbonic acids, or with sulfur or nitrogen analogues thereof, e.g. carbamates
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D217/00—Heterocyclic compounds containing isoquinoline or hydrogenated isoquinoline ring systems
- C07D217/12—Heterocyclic compounds containing isoquinoline or hydrogenated isoquinoline ring systems with radicals, substituted by hetero atoms, attached to carbon atoms of the nitrogen-containing ring
- C07D217/14—Heterocyclic compounds containing isoquinoline or hydrogenated isoquinoline ring systems with radicals, substituted by hetero atoms, attached to carbon atoms of the nitrogen-containing ring other than aralkyl radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D217/00—Heterocyclic compounds containing isoquinoline or hydrogenated isoquinoline ring systems
- C07D217/22—Heterocyclic compounds containing isoquinoline or hydrogenated isoquinoline ring systems with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to carbon atoms of the nitrogen-containing ring
- C07D217/26—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/10—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D473/00—Heterocyclic compounds containing purine ring systems
- C07D473/02—Heterocyclic compounds containing purine ring systems with oxygen, sulphur, or nitrogen atoms directly attached in positions 2 and 6
- C07D473/04—Heterocyclic compounds containing purine ring systems with oxygen, sulphur, or nitrogen atoms directly attached in positions 2 and 6 two oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D495/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms
- C07D495/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D495/04—Ortho-condensed systems
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Oncology (AREA)
- Hematology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Plural Heterocyclic Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Abstract
本發明係關於具有鐵依賴型細胞死亡誘導活性之化合物、使用該等化合物治療患有癌症之個體之方法、及與第二治療劑之組合治療。
Description
麩胱甘肽過氧化物酶4 (GPX4)可直接還原磷脂氫過氧化物。GPX4消耗會誘導脂質過氧化依賴性細胞死亡。藥物誘導之抗療法狀態中之癌細胞強烈依賴於GPX4之脂質過氧化物酶活性來防止發生鐵依賴型細胞死亡性細胞死亡。研究已展示,親脂性抗氧化劑(例如菲羅他仃(Ferrostatin))可拯救細胞以免受GPX4抑制誘導之鐵依賴型細胞死亡。舉例而言,間質態GPX4敲除細胞可在菲羅他仃存在下存活,然而,在菲羅他仃之供應終止時,該等細胞發生鐵依賴型細胞死亡(例如參見Viswanathan等人,Nature 547:453-7, 2017)。亦已以實驗方式測得,可藉由阻斷鐵依賴型細胞死亡路徑之其他組分來恢復GPX4i (例如脂質ROS清除劑(菲羅他仃、利普羅他仃(Liproxstatin))、脂氧合酶抑制劑、鐵螯合劑及半胱天冬酶(caspase)抑制劑),而細胞凋亡抑制劑則不能恢復。該等發現指示了非細胞凋亡性、鐵依賴性、氧化性細胞死亡(亦即鐵依賴型細胞死亡)。因此,GPX4抑制劑可用於誘導鐵依賴型細胞死亡性癌細胞死亡且由此治療癌症。
本發明係關於具有鐵依賴型細胞死亡誘導活性之化合物及使用該等化合物治療癌症之方法。在某些實施例中,本文提供式I化合物: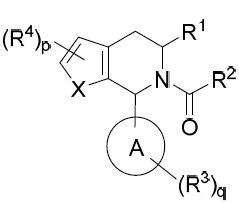 I
或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽,其中:
環A係C4
-C10
環烷基、雜環基、芳基或雜芳基;
X係-O-、-S-、-NR9
-、-CR5
=CR5
-或-CR5
=N-;
p為0、1或2;
q為0、1、2或3;
R1
係C1
-C6
烷基、C2
-C6
烯基、C2
-C6
炔基、C1
-C6
鹵代烷基、C3
-C10
環烷基、-CN、-OR7
、-C(O)OR6
、-C(O)N(R7
)2
、-OC(O)R6
、-S(O)2
R8
、-S(O)2
N(R7
)2
、-S(O)N(R7
)2
、-S(O)R8
、-N(R7
)2
、-NO2
、-C1
-C6
烷基-OR7
或-Si(R15
)3
;
R2
係-C1
-C2
鹵代烷基、-C2
-C3
烯基、-C2
-C3
鹵代烯基、C2
炔基或-CH2
OS(O)2
-苯基,其中C1
-C2
烷基鹵基及-C2
-C3
烯基鹵基視情況經一或兩個-CH3
取代,且C2
炔基及苯基視情況經一個-CH3
取代;
每一R3
獨立地係鹵基、-CN、-OH、-OR8
、-NH2
、-NHR8
、-N(R8
)2
、-S(O)2
R8
、-S(O)R8
、-S(O)2
N(R7
)2
、-S(O)N(R7
)2
、-NO2
、-Si(R12
)3
、-SF5
、-C(O)OR6
、-C(O)N(R7
)2
、-NR12
C(O)R8
、-NR12
C(O)OR8
、-OC(O)N(R7
)2
、-OC(O)R8
、-C(O)R6
、-OC(O)CHR8
N(R12
)2
、C1
-C6
烷基、C2
-C6
烯基、C2
-C6
炔基、C3
-C10
環烷基、雜環基、芳基、雜芳基、-C1
-C6
烷基C3
-C10
環烷基、-C2
-C6
烯基C3
-C10
環烷基、-C1
-C6
烷基雜環基、-C2
-C6
烯基雜環基、-C1
-C6
烷基芳基、-C2
-C6
烯基芳基、C1
-C6
烷基雜芳基或-C2
-C6
烯基雜芳基;其中R3
之每一C1
-C6
烷基、C2
-C6
烯基、C2
-C6
炔基、C3
-C10
環烷基、雜環基、芳基、雜芳基、-C1
-C6
烷基C3
-C10
環烷基、-C2
-C6
烯基C3
-C10
環烷基、-C1
-C6
烷基雜環基、-C2
-C6
烯基雜環基、-C1
-C6
烷基芳基、-C2
-C6
烯基芳基、C1
-C6
烷基雜芳基或-C2
-C6
烯基雜芳基獨立地視情況經一至三個R10
取代;
每一R4
獨立地係鹵基、-CN、-OH、-OR8
、-NH2
、-NHR8
、-N(R8
)2
、-S(O)2
R8
、-S(O)R8
、-S(O)2
N(R7
)2
、-S(O)N(R7
)2
、-NO2
、-Si(R15
)3
、-C(O)OR6
、-C(O)N(R7
)2
、-NR12
C(O)R8
、-OC(O)R8
、-C(O)R6
、-NR12
C(O)OR8
、-OC(O)N(R7
)2
、-OC(O)CHR8
N(R12
)2
、C1
-C6
烷基、C2
-C6
烯基、C2
-C6
炔基、C3
-C10
環烷基、雜環基、芳基、雜芳基、-C1
-C6
烷基C3
-C10
環烷基、-C2
-C6
烯基C3
-C10
環烷基、-C1
-C6
烷基雜環基、-C2
-C6
烯基雜環基、-C1
-C6
烷基芳基、-C2
-C6
烯基芳基、C1
-C6
烷基雜芳基或-C2
-C6
烯基雜芳基;其中R4
之每一C1
-C6
烷基、C2
-C6
烯基、C2
-C6
炔基、C3
-C10
環烷基、雜環基、芳基、雜芳基、-C1
-C6
烷基C3
-C10
環烷基、-C2
-C6
烯基C3
-C10
環烷基、-C1
-C6
烷基雜環基、-C2
-C6
烯基雜環基、-C1
-C6
烷基芳基、-C2
-C6
烯基芳基、C1
-C6
烷基雜芳基或-C2
-C6
烯基雜芳基視情況獨立地視情況經一至三個R10
取代;
每一R5
獨立地係氫、鹵基、-CN、-OH、-OR8
、-NH2
、-NHR8
、-N(R8
)2
、-S(O)2
R8
、-S(O)R8
、-S(O)2
N(R7
)2
、-S(O)N(R7
)2
、-NO2
、-Si(R15
)3
、-C(O)OR6
、-C(O)N(R7
)2
、-NR12
C(O)R8
、-OC(O)R8
、-C(O)R6
、-NR12
C(O)OR8
、-OC(O)N(R7
)2
、-OC(O)CHR8
N(R12
)2
、C1
-C6
烷基、C2
-C6
烯基、C2
-C6
炔基、C3
-C10
環烷基、雜環基、芳基、雜芳基、-C1
-C6
烷基C3
-C10
環烷基、-C2
-C6
烯基C3
-C10
環烷基、-C1
-C6
烷基雜環基、-C2
-C6
烯基雜環基、-C1
-C6
烷基芳基、-C2
-C6
烯基芳基、C1
-C6
烷基雜芳基或-C2
-C6
烯基雜芳基;其中R5
之每一C1
-C6
烷基、C2
-C6
烯基、C2
-C6
炔基、C3
-C10
環烷基、雜環基、芳基、雜芳基、-C1
-C6
烷基C3
-C10
環烷基、-C2
-C6
烯基C3
-C10
環烷基、-C1
-C6
烷基雜環基、-C2
-C6
烯基雜環基、-C1
-C6
烷基芳基、-C2
-C6
烯基芳基、C1
-C6
烷基雜芳基或-C2
-C6
烯基雜芳基視情況獨立地視情況經一至三個R10
取代;
每一R6
獨立地係氫、C1
-C6
烷基、C2
-C6
烯基、C2
-C6
炔基、C3
-C10
環烷基、雜環基、芳基、雜芳基、-C1
-C6
烷基C3
-C10
環烷基、-C2
-C6
烯基C3
-C10
環烷基、-C1
-C6
烷基雜環基、-C2
-C6
烯基雜環基、-C1
-C6
烷基芳基、-C2
-C6
烯基芳基、C1
-C6
烷基雜芳基或-C2
-C6
烯基雜芳基;其中每一R6
獨立地進一步經一至三個R11
取代;
每一R7
獨立地係氫、C1
-C6
烷基、C2
-C6
烯基、C2
-C6
炔基、C3
-C10
環烷基、雜環基、芳基、雜芳基、-C1
-C6
烷基C3
-C6
環烷基、-C2
-C6
烯基C3
-C6
環烷基、-C1
-C6
烷基雜環基、-C2
-C6
烯基雜環基、-C1
-C6
烷基芳基、-C2
-C6
烯基芳基、-C1
-C6
烷基雜芳基、-C2
-C6
烯基雜芳基,或兩個R7
與其所連接之氮原子一起形成4至7員雜環基;其中每一R7
或由此形成之環獨立地進一步經一至三個R11
取代;
每一R8
獨立地係C1
-C6
烷基、C2
-C6
烯基、C2
-C6
炔基、C3
-C10
環烷基、雜環基、芳基、雜芳基、-C1
-C6
烷基C3
-C10
環烷基、-C2
-C6
烯基C3
-C10
環烷基、-C1
-C6
烷基雜環基、-C2
-C6
烯基雜環基、-C1
-C6
烷基芳基、-C2
-C6
烯基芳基、-C1
-C6
烷基雜芳基或-C2
-C6
烯基雜芳基;其中每一R8
獨立地進一步經一至三個R11
取代;
R9
係氫或C1
-C6
烷基;
每一R10
獨立地係鹵基、-CN、-OR12
、-NO2
、-N(R12
)2
、-S(O)R13
、-S(O)2
R13
、-S(O)N(R12
)2
、-S(O)2
N(R12
)2
、-Si(R12
)3
、-C(O)R12
、-C(O)OR12
、-C(O)N(R12
)2
、-NR12
C(O)R12
、-OC(O)R12
、-OC(O)OR12
、-OC(O)N(R12
)2
、-NR12
C(O)OR12
、-OC(O)CHR12
N(R12
)2
、C1
-C6
烷基、C1
-C6
鹵代烷基、C2
-C6
烯基、C2
-C6
炔基、C3
-C10
環烷基、雜環基、芳基或雜芳基,其中R10
之每一C1
-C6
烷基、C1
-C6
鹵代烷基、C2
-C6
烯基、C2
-C6
炔基、C3
-C10
環烷基、雜環基、芳基或雜芳基視情況獨立地經一至三個R11
取代;
每一R11
獨立地係鹵基、-CN、-OR12
、-NO2
、-N(R12
)2
、-S(O)R13
、-S(O)2
R13
、-S(O)N(R12
)2
、-S(O)2
N(R12
)2
、-Si(R12
)3
、-C(O)R12
、-C(O)OR12
、-C(O)N(R12
)2
、-NR12
C(O)R12
、-OC(O)R12
、-OC(O)OR12
、-OC(O)N(R12
)2
、-NR12
C(O)OR12
、-OC(O)CHR12
N(R12
)2
、C1
-C6
烷基、C1
-C6
鹵代烷基、C2
-C6
烯基、C2
-C6
炔基、C3
-C10
環烷基、雜環基、芳基或雜芳基;
每一R12
獨立地係氫、C1
-C6
烷基或C3
-C10
環烷基;
每一R13
獨立地係C1
-C6
烷基或C3
-C10
環烷基;且
每一R15
獨立地係C1
-C6
烷基、C2
-C6
烯基、芳基、雜芳基、-C1
-C6
烷基芳基、-C2
-C6
烯基芳基、-C1
-C6
烷基雜芳基及-C2
-C6
烯基雜芳基。
I
或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽,其中:
環A係C4
-C10
環烷基、雜環基、芳基或雜芳基;
X係-O-、-S-、-NR9
-、-CR5
=CR5
-或-CR5
=N-;
p為0、1或2;
q為0、1、2或3;
R1
係C1
-C6
烷基、C2
-C6
烯基、C2
-C6
炔基、C1
-C6
鹵代烷基、C3
-C10
環烷基、-CN、-OR7
、-C(O)OR6
、-C(O)N(R7
)2
、-OC(O)R6
、-S(O)2
R8
、-S(O)2
N(R7
)2
、-S(O)N(R7
)2
、-S(O)R8
、-N(R7
)2
、-NO2
、-C1
-C6
烷基-OR7
或-Si(R15
)3
;
R2
係-C1
-C2
鹵代烷基、-C2
-C3
烯基、-C2
-C3
鹵代烯基、C2
炔基或-CH2
OS(O)2
-苯基,其中C1
-C2
烷基鹵基及-C2
-C3
烯基鹵基視情況經一或兩個-CH3
取代,且C2
炔基及苯基視情況經一個-CH3
取代;
每一R3
獨立地係鹵基、-CN、-OH、-OR8
、-NH2
、-NHR8
、-N(R8
)2
、-S(O)2
R8
、-S(O)R8
、-S(O)2
N(R7
)2
、-S(O)N(R7
)2
、-NO2
、-Si(R12
)3
、-SF5
、-C(O)OR6
、-C(O)N(R7
)2
、-NR12
C(O)R8
、-NR12
C(O)OR8
、-OC(O)N(R7
)2
、-OC(O)R8
、-C(O)R6
、-OC(O)CHR8
N(R12
)2
、C1
-C6
烷基、C2
-C6
烯基、C2
-C6
炔基、C3
-C10
環烷基、雜環基、芳基、雜芳基、-C1
-C6
烷基C3
-C10
環烷基、-C2
-C6
烯基C3
-C10
環烷基、-C1
-C6
烷基雜環基、-C2
-C6
烯基雜環基、-C1
-C6
烷基芳基、-C2
-C6
烯基芳基、C1
-C6
烷基雜芳基或-C2
-C6
烯基雜芳基;其中R3
之每一C1
-C6
烷基、C2
-C6
烯基、C2
-C6
炔基、C3
-C10
環烷基、雜環基、芳基、雜芳基、-C1
-C6
烷基C3
-C10
環烷基、-C2
-C6
烯基C3
-C10
環烷基、-C1
-C6
烷基雜環基、-C2
-C6
烯基雜環基、-C1
-C6
烷基芳基、-C2
-C6
烯基芳基、C1
-C6
烷基雜芳基或-C2
-C6
烯基雜芳基獨立地視情況經一至三個R10
取代;
每一R4
獨立地係鹵基、-CN、-OH、-OR8
、-NH2
、-NHR8
、-N(R8
)2
、-S(O)2
R8
、-S(O)R8
、-S(O)2
N(R7
)2
、-S(O)N(R7
)2
、-NO2
、-Si(R15
)3
、-C(O)OR6
、-C(O)N(R7
)2
、-NR12
C(O)R8
、-OC(O)R8
、-C(O)R6
、-NR12
C(O)OR8
、-OC(O)N(R7
)2
、-OC(O)CHR8
N(R12
)2
、C1
-C6
烷基、C2
-C6
烯基、C2
-C6
炔基、C3
-C10
環烷基、雜環基、芳基、雜芳基、-C1
-C6
烷基C3
-C10
環烷基、-C2
-C6
烯基C3
-C10
環烷基、-C1
-C6
烷基雜環基、-C2
-C6
烯基雜環基、-C1
-C6
烷基芳基、-C2
-C6
烯基芳基、C1
-C6
烷基雜芳基或-C2
-C6
烯基雜芳基;其中R4
之每一C1
-C6
烷基、C2
-C6
烯基、C2
-C6
炔基、C3
-C10
環烷基、雜環基、芳基、雜芳基、-C1
-C6
烷基C3
-C10
環烷基、-C2
-C6
烯基C3
-C10
環烷基、-C1
-C6
烷基雜環基、-C2
-C6
烯基雜環基、-C1
-C6
烷基芳基、-C2
-C6
烯基芳基、C1
-C6
烷基雜芳基或-C2
-C6
烯基雜芳基視情況獨立地視情況經一至三個R10
取代;
每一R5
獨立地係氫、鹵基、-CN、-OH、-OR8
、-NH2
、-NHR8
、-N(R8
)2
、-S(O)2
R8
、-S(O)R8
、-S(O)2
N(R7
)2
、-S(O)N(R7
)2
、-NO2
、-Si(R15
)3
、-C(O)OR6
、-C(O)N(R7
)2
、-NR12
C(O)R8
、-OC(O)R8
、-C(O)R6
、-NR12
C(O)OR8
、-OC(O)N(R7
)2
、-OC(O)CHR8
N(R12
)2
、C1
-C6
烷基、C2
-C6
烯基、C2
-C6
炔基、C3
-C10
環烷基、雜環基、芳基、雜芳基、-C1
-C6
烷基C3
-C10
環烷基、-C2
-C6
烯基C3
-C10
環烷基、-C1
-C6
烷基雜環基、-C2
-C6
烯基雜環基、-C1
-C6
烷基芳基、-C2
-C6
烯基芳基、C1
-C6
烷基雜芳基或-C2
-C6
烯基雜芳基;其中R5
之每一C1
-C6
烷基、C2
-C6
烯基、C2
-C6
炔基、C3
-C10
環烷基、雜環基、芳基、雜芳基、-C1
-C6
烷基C3
-C10
環烷基、-C2
-C6
烯基C3
-C10
環烷基、-C1
-C6
烷基雜環基、-C2
-C6
烯基雜環基、-C1
-C6
烷基芳基、-C2
-C6
烯基芳基、C1
-C6
烷基雜芳基或-C2
-C6
烯基雜芳基視情況獨立地視情況經一至三個R10
取代;
每一R6
獨立地係氫、C1
-C6
烷基、C2
-C6
烯基、C2
-C6
炔基、C3
-C10
環烷基、雜環基、芳基、雜芳基、-C1
-C6
烷基C3
-C10
環烷基、-C2
-C6
烯基C3
-C10
環烷基、-C1
-C6
烷基雜環基、-C2
-C6
烯基雜環基、-C1
-C6
烷基芳基、-C2
-C6
烯基芳基、C1
-C6
烷基雜芳基或-C2
-C6
烯基雜芳基;其中每一R6
獨立地進一步經一至三個R11
取代;
每一R7
獨立地係氫、C1
-C6
烷基、C2
-C6
烯基、C2
-C6
炔基、C3
-C10
環烷基、雜環基、芳基、雜芳基、-C1
-C6
烷基C3
-C6
環烷基、-C2
-C6
烯基C3
-C6
環烷基、-C1
-C6
烷基雜環基、-C2
-C6
烯基雜環基、-C1
-C6
烷基芳基、-C2
-C6
烯基芳基、-C1
-C6
烷基雜芳基、-C2
-C6
烯基雜芳基,或兩個R7
與其所連接之氮原子一起形成4至7員雜環基;其中每一R7
或由此形成之環獨立地進一步經一至三個R11
取代;
每一R8
獨立地係C1
-C6
烷基、C2
-C6
烯基、C2
-C6
炔基、C3
-C10
環烷基、雜環基、芳基、雜芳基、-C1
-C6
烷基C3
-C10
環烷基、-C2
-C6
烯基C3
-C10
環烷基、-C1
-C6
烷基雜環基、-C2
-C6
烯基雜環基、-C1
-C6
烷基芳基、-C2
-C6
烯基芳基、-C1
-C6
烷基雜芳基或-C2
-C6
烯基雜芳基;其中每一R8
獨立地進一步經一至三個R11
取代;
R9
係氫或C1
-C6
烷基;
每一R10
獨立地係鹵基、-CN、-OR12
、-NO2
、-N(R12
)2
、-S(O)R13
、-S(O)2
R13
、-S(O)N(R12
)2
、-S(O)2
N(R12
)2
、-Si(R12
)3
、-C(O)R12
、-C(O)OR12
、-C(O)N(R12
)2
、-NR12
C(O)R12
、-OC(O)R12
、-OC(O)OR12
、-OC(O)N(R12
)2
、-NR12
C(O)OR12
、-OC(O)CHR12
N(R12
)2
、C1
-C6
烷基、C1
-C6
鹵代烷基、C2
-C6
烯基、C2
-C6
炔基、C3
-C10
環烷基、雜環基、芳基或雜芳基,其中R10
之每一C1
-C6
烷基、C1
-C6
鹵代烷基、C2
-C6
烯基、C2
-C6
炔基、C3
-C10
環烷基、雜環基、芳基或雜芳基視情況獨立地經一至三個R11
取代;
每一R11
獨立地係鹵基、-CN、-OR12
、-NO2
、-N(R12
)2
、-S(O)R13
、-S(O)2
R13
、-S(O)N(R12
)2
、-S(O)2
N(R12
)2
、-Si(R12
)3
、-C(O)R12
、-C(O)OR12
、-C(O)N(R12
)2
、-NR12
C(O)R12
、-OC(O)R12
、-OC(O)OR12
、-OC(O)N(R12
)2
、-NR12
C(O)OR12
、-OC(O)CHR12
N(R12
)2
、C1
-C6
烷基、C1
-C6
鹵代烷基、C2
-C6
烯基、C2
-C6
炔基、C3
-C10
環烷基、雜環基、芳基或雜芳基;
每一R12
獨立地係氫、C1
-C6
烷基或C3
-C10
環烷基;
每一R13
獨立地係C1
-C6
烷基或C3
-C10
環烷基;且
每一R15
獨立地係C1
-C6
烷基、C2
-C6
烯基、芳基、雜芳基、-C1
-C6
烷基芳基、-C2
-C6
烯基芳基、-C1
-C6
烷基雜芳基及-C2
-C6
烯基雜芳基。
在某些實施例中,化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽或包括其之醫藥組合物展現GPX4抑制活性,且在某些實施例中與其他GPX4抑制劑相比展現改變或增強之穩定性(例如代謝穩定性)及/或增強之活性或其他特性。在某些實施例中,本文所闡述之化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽或包括其之醫藥組合物較其他GPX對GPX4具有選擇性。在某些實施例中,該等化合物係用於抑制細胞中之GPX4之方法中,其包括使細胞與有效量之本文所闡述之化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽或包括其之醫藥組合物接觸以抑制細胞中之GPX4。在某些實施例中,細胞係癌細胞。
在某些實施例中,提供誘導細胞中之鐵依賴型細胞死亡之方法,其包括使細胞與有效量之本文所提供之化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽或醫藥組合物接觸。
在某些實施例中,提供治療有需要之患者之癌症之方法,其包括投與有效量之本文所提供之化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽或醫藥組合物。在某些實施例中,提供治療有需要之患者之惡性實體腫瘤之方法,其包括向患者投與有效量之本文所提供之化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽或醫藥組合物。在某些實施例中,惡性實體腫瘤係肉瘤、癌或淋巴瘤。
相關申請案之交叉參考
本申請案係2019年2月27日提出申請之PCT申請案第PCT/US2019/019854號之部分接續申請案、2019年2月27日提出申請之美國專利申請案第16/287,805號之部分接續申請案,且在35 U.S.C. §119(e)下主張2019年8月28日提出申請之美國臨時專利申請案第62/893,092號之權益,該等申請案之內容之全部內容併入本文中。
除非上下文另外明確指示,否則如本說明書及隨附申請專利範圍中所使用,單數形式「一(a、an)」及「該」包含複數個指示物。因此,舉例而言,所提及「蛋白質」包含一種以上蛋白質,且所提及「化合物」係指一種以上化合物。
另外,除非另外陳述,否則所使用之「或」意指「及/或」。類似地,「包括(comprise、comprises、comprising)」、「包含(include、includes及including)」可互換使用且並不意欲加以限制。
另外應理解,在使用術語「包括」闡述各個實施例之情形下,熟習此項技術者應理解,在一些具體情況下,可替代地使用語言「基本上由……組成」或「由……組成」來闡述一實施例。
應理解,前述一般說明(包含圖式)及下列詳細說明二者皆僅為實例性及解釋性且並不限制本發明。本文所用之各部分標題僅出於組織性目的且不應視為限制所闡述標的物。
1. 定義
參照本發明,除非另外具體定義,否則本文之闡述中所使用之技術及科學術語具有熟習此項技術者通常所理解之含義。因此,下列術語意欲具有如下文所闡述之含義。
「鐵依賴型細胞死亡」係指業內理解為涉及生成由鐵調介之反應性氧物質且特徵部分地在於脂質過氧化之細胞死亡形式。
「鐵依賴型細胞死亡誘導劑」或「鐵依賴型細胞死亡活化劑」係指誘導、促進或激活鐵依賴型細胞死亡之藥劑。
「GPX4抑制劑」係指任一抑制麩胱甘肽過氧化物酶4 (GPX4)之活性之藥劑。GPX4抑制劑可為直接或間接抑制劑。GPX4係磷脂氫過氧化物酶,其催化過氧化氫及有機過氧化物之還原,由此保護細胞免於膜脂質過氧化或氧化應力。GPX4在活性位點中具有硒半胱胺酸,該硒半胱胺酸由過氧化物氧化成次硒酸以提供脂質醇。麩胱甘肽用於將次硒酸(-SeOH)還原回硒醇(-SeH)。倘若此催化循環遭到破壞,則經由細胞內鐵調介過程(稱為鐵依賴型細胞死亡)發生細胞死亡。
本文所用之「個體」係指哺乳動物,例如狗、貓、馬或兔。在某些實施例中,個體係非人類靈長類動物,例如猴、黑猩猩或大猩猩。在某些實施例中,個體係人類,有時在本文中稱為患者。
如本文中所使用,疾病、病症或症候群之「治療(treating或treatment)」包含(i)預防個體發生疾病、病症或症候群,亦即使得在可暴露於或易患該疾病、病症或症候群但尚未經歷或顯示該疾病、病症或症候群之症狀之動物中不發生該疾病、病症或症候群之臨床症狀;(ii)抑制疾病、病症或症候群,亦即阻止其發生;及(iii)減輕疾病、病症或症候群,亦即使疾病、病症或症候群消退。如業內已知,可能需要根據全身性遞送與局域性遞送、年齡、體重、一般健康狀況、性別、飲食、投與時間、藥物相互作用及病狀嚴重程度來進行調節,且可使用常規實驗由熟習此項技術者尤其根據本發明中所提供之導則來確定。
「治療有效量」係指在投與動物(例如人類)以治療疾病時足以實現疾病、病症或病狀之該治療之量。在某些實施例中,該治療提供治療益處,例如改善症狀或減緩疾病進展。舉例而言,治療有效量可為足以降低如本文所闡述之疾病或病狀之症狀之量。
「烷基」係指具有1至20個碳原子(C1
-C20
或C1-20
)、1至12個碳原子(C1
-C12
或C1-12
)或1至8個碳原子(C1
-C8
或C1-8
)之直鏈或具支鏈烴基。實例性「烷基」包含(但不限於)甲基、乙基、正丙基、異丙基、正丁基、第二丁基、第三丁基、正戊基及第二戊基以及諸如此類。
「烯基」係指具有2至20個碳原子(C2
-C20
或C2-20
)、2至12個碳原子(C2
-C12
或C2-12
)或2至8個碳原子(C2
-C8
或C2-8
)且具有至少一個雙鍵之直鏈或具支鏈烴基。實例性「烯基」包含(但不限於)乙烯基、乙烯基、烯丙基、異丙烯基、1-丙烯基、2-甲基-1-丙烯基、1-丁烯基、2-丁烯基、3-丁烯基、2-乙基-1-丁烯基、3-甲基-2-丁烯基、1-戊烯基、2-戊烯基、3-戊烯基、4-戊烯基、4-甲基-3-戊烯基、1-己烯基、2-己烯基、3-己烯基、4-己烯基及5-己烯基以及諸如此類。
「炔基」係指具有2至12個碳原子(C2
-C12
或C2-12
)、2至8個碳原子(C2
-C8
或C2-8
)且含有至少一個三鍵之直鏈或具支鏈烴基。實例性「炔基」包含乙炔基、1-丙炔基、2-丙炔基、1-丁炔基、2-丁炔基、3-丁炔基、1-戊炔基、2-戊炔基、3-戊炔基、4-戊炔基、1-己炔基、2-己炔基、3-己炔基、4-己炔基及5-己炔基以及諸如此類。
「伸烷基」、「伸烯基」及「伸炔基」分別係指相應烷基、烯基及炔基之直鏈或具支鏈二價烴基團。在某些實施例中,「烷基」、「烯基」及「炔基」可代表相應「伸烷基」、「伸烯基」及「伸炔基」,例如(舉例而言且不限於)環烷基烷基-、雜環烷基烷基-、芳基烷基-、雜芳基烷基-、環烷基烯基-、雜環烷基烯基-、芳基烯基-、雜芳基烯基-、環烷基炔基-、雜環烷基炔基-、芳基炔基-、雜芳基炔基-及諸如此類,其中環烷基、雜環烷基、芳基及雜芳基作為取代基經由相應伸烷基、伸烯基或炔基進行連結。
提及取代基之「低碳」係指具有一至六個碳原子之基團。
「烷基鹵基」或「鹵代烷基」係指其中一或多個(例如一至三個或一個)氫原子由鹵素(例如Cl、F等)代替之具有1至20個碳原子(C1
-C20
或C1-20
)、1至12個碳原子(C1
-C12
或C1-12
)或1至8個碳原子(C1
-C8
或C1-8
)之直鏈或具支鏈烴基。在某些實施例中,術語「烷基鹵基」係指其中一個氫原子由鹵素(例如Cl、F等)代替之如本文所定義之烷基。在某些實施例中,術語「烷基鹵基」係指烷基氯。
「烯基鹵基」或「鹵代烯基」係指其中一或多個(例如一至三個或一個)氫原子由鹵素(例如Cl、F等)代替之具有2至20個碳原子(C2
-C20
或C2-20
)、2至12個碳原子(C2
-C12
或C2-12
)或2至8個碳原子(C2
-C8
或C2-8
)且具有至少一個雙鍵之直鏈或具支鏈烴基。在某些實施例中,術語「烯基鹵基」係指其中一個氫原子由鹵素(例如Cl、F等)代替之如本文所定義之烯基。在某些實施例中,術語「烯基鹵基」係指烯基氯。
「雜烷基」係指其中1至3個碳原子由雜原子代替之具有1至20個碳原子(C1
-C20
或C1-20
)、1至12個碳原子(C1
-C12
或C1-12
)或1至8個碳原子(C1
-C8
或C1-8
)之直鏈或具支鏈烴基。可代替碳原子之雜原子及/或雜原子基團包含(但不限於) -O-、-S-、-NR40
-、-PH-、-C(O)-、-S(O)-、-S(O)2
-、-S(O)NR40
-、-S(O)2
NR40
-及諸如此類(包含其組合),其中每一R40
獨立地係氫或低碳烷基。
「環烷基」係指任一由碳原子組成之穩定單環或多環系統,其任一環係飽和的。「環烯基」係指任一由碳原子組成且至少一個環部分地不飽和之穩定單環或多環系統。環烷基之實例包含(但不限於)環丙基、環丁基、環戊基、環己基、環庚基、環辛基、雙環烷基及三環烷基(例如金剛烷基)。
「雜環烷基」或「雜環基」係指其中1至3個碳原子由雜原子代替之4至14員、單-或多環(例如雙環)、非芳香族烴環。可代替碳原子之雜原子及/或雜原子基團包含(但不限於) -O-、-S-、-S-O-、-NR40
-、-PH-、-C(O)-、-S(O)-、-S(O)2
-、-S(O)NR40
-、-S(O)2
NR40
-及諸如此類(包含其組合),其中每一R40
獨立地係氫或低碳烷基。實例包含噻唑啶基、噻二唑基、三嗪基、嗎啉基、吡咯啶酮基、吡咯啶基、六氫吡啶基、六氫吡嗪基、2,3-二氫呋喃基、二氫吡喃基、乙內醯脲基、戊內醯胺基、環氧乙烷基、環氧丙烷基、四氫呋喃基、四氫吡喃基、二氫吡啶基、四氫吡啶基、四氫嘧啶基、四氫噻吩基、四氫噻喃基及諸如此類。在某些實施例中,「雜環烷基」或「雜環基」係經取代或未經取代之4至7員單環,其中1至3個碳原子由如上文所闡述之雜原子代替。
在某些實施例中,「雜環烷基」或「雜環基」係4至10或4至9或5至9或5至7或5至6員單-或多環(例如雙環),其中1至3個碳原子由如上文所闡述之雜原子代替。在某些實施例中,在「雜環烷基」或「雜環基」係經取代或未經取代之雙環時,一個環可為芳香族,條件係至少一個環係非芳香族,不論至分子之其他部分之連接點如何(例如二氫吲哚基、異二氫吲哚基及諸如此類)。
「芳基」係指6至14員、單-或雙碳環,其中單環係芳香族且雙環中之至少一個環係芳香族。除非另外陳述,否則只要化合價規則允許,基團之化合價可位於該基團內之任一環之任一原子上。「芳基」之實例包含苯基、萘基、茚基、聯苯、菲基、稠四苯基及諸如此類。
「雜芳基」意指芳香族雜環(包含單環及多環(例如雙環)系統),其中一或兩個環之至少一個碳原子經獨立地選自氮、氧及硫之雜原子代替,或一或兩個環之至少兩個碳原子經獨立地選自氮、氧及硫之雜原子代替。在某些實施例中,雜芳基可為5至6員單環或7至11員雙環系統。「雜芳基」之實例包含吡咯基、吡唑基、咪唑基、吡嗪基、噁唑基、異噁唑基、噻唑基、呋喃基、噻吩基、吡啶基、嘧啶基、苯并噻唑基、嘌呤基、苯并咪唑基、吲哚基、異喹啉基、喹喔啉基、喹啉基及諸如此類。
「橋接雙環」係指具有至少一個橋之飽和或部分不飽和之任一雙環環系統,亦即碳環或雜環。如由IUPAC所定義,「橋」係連接兩個橋頭之一或多個原子或化學鍵之無支鏈鏈,其中「橋頭」係鍵結至三個或更多個骨架原子(排除氫)之環系統的任一骨架原子。在某些實施例中,橋接雙環基團具有5至12個環成員及0至4個獨立地選自氮、氧及硫之雜原子。該等橋接雙環基團包含下文所陳述之彼等基團,其中每一基團於任一可取代碳或氮原子處連接至分子之其餘部分。實例性橋接雙環包含(但不限於): 。
「稠合環」係指兩個或更多個環共用至少一個鍵及兩個原子之環系統。「稠合芳基」及「稠合雜芳基」係指分別具有至少一個與另一環共用至少一個鍵及兩個原子之芳基及雜芳基之環系統。
。
「稠合環」係指兩個或更多個環共用至少一個鍵及兩個原子之環系統。「稠合芳基」及「稠合雜芳基」係指分別具有至少一個與另一環共用至少一個鍵及兩個原子之芳基及雜芳基之環系統。
「鹵素」或「鹵基」係指氟、氯、溴及碘。
「醯基」係指-C(O)R43
,其中R43
係氫或如本文所定義之視情況經取代之烷基、雜烷基、環烷基、環烷基烷基、雜環烷基、雜環烷基烷基、芳基、芳基烷基、雜芳基或雜芳基烷基。實例性醯基包含(但不限於)甲醯基、乙醯基、環己基羰基、環己基甲基羰基、苯甲醯基、苄基羰基及諸如此類。
「烷基氧基」或「烷氧基」係指-OR44
,其中R44
係視情況經取代之烷基。
「芳基氧基」係指-OR45
,其中R45
係視情況經取代之芳基。
「羧基」係指-COO-
或COOM,其中M係H或相對離子(例如陽離子,例如Na+
、Ca2+
、Mg2+
等)。
「胺甲醯基」係指-C(O)NR46
R46
,其中每一R46
獨立地選自H或視情況經取代之烷基、環烷基、環烷基烷基、雜環烷基、雜環烷基烷基、芳基、芳基烷基、雜芳基或雜芳基烷基。
「酯」係指諸如-C(=O)OR47
等基團,其可替代地闡釋為-C(O)OR47
,其中R47
係選自視情況經取代之烷基、環烷基、環烷基烷基、雜環烷基、雜環烷基烷基、芳基、芳基烷基、雜芳基及雜芳基烷基。
「醚」係指基團-烷基-O-烷基,其中術語烷基係如本文所定義。
「硫烷基」係指-SR48
,其中R48
係選自視情況經取代之烷基、環烷基、環烷基烷基、雜環烷基、雜環烷基烷基、芳基、芳基烷基、雜芳基及雜芳基烷基。舉例而言,-SR48
(其中R48
係烷基)係烷基硫烷基。
「磺醯基」係指-S(O)2
-,其可具有各種取代基以形成不同磺醯基(包含磺酸、磺醯胺、磺酸酯及碸)。舉例而言,-S(O)2
R49
(其中R49
係烷基)係指烷基磺醯基。在-S(O)2
R49
之某些實施例在,R49
係選自視情況經取代之烷基、環烷基、環烷基烷基、雜環烷基、雜環烷基烷基、芳基、芳基烷基、雜芳基及雜芳基烷基。
「亞磺醯基」係指-S(O)-,其可具有各種取代基以形成不同亞磺醯基(包含亞磺酸、亞磺醯胺醯胺及亞磺醯基酯)。舉例而言,-S(O)R50
(其中R50
係烷基)係指烷基亞磺醯基。在-S(O)R50
之某些實施例在,R50
係選自視情況經取代之烷基、環烷基、環烷基烷基、雜環烷基、雜環烷基烷基、芳基、芳基烷基、雜芳基及雜芳基烷基。
「矽基」係指Si,其可具有各種取代基,例如-SiR51
R51
R51
,其中每一R51
獨立地選自烷基、環烷基、環烷基烷基、雜環烷基、雜環烷基烷基、芳基、芳基烷基、雜芳基及雜芳基烷基。如本文所定義,存在於矽基中之任一雜環烷基或雜芳基具有1至3個獨立地選自O、N及S之雜原子。
「胺基」或「胺」係指基團-NR52
R52
或-N+
R52
R52
R52
,其中每一R52
獨立地選自氫及視情況經取代之烷基、環烷基、雜環烷基、烷基氧基、芳基、雜芳基、雜芳基烷基、醯基、-C(O)-O-烷基、硫烷基、亞磺醯基、磺醯基及諸如此類。實例性胺基包含(但不限於)二甲基胺基、二乙基胺基、三甲基銨、三乙基銨、甲基磺醯基胺基、呋喃基-氧基-磺酸胺基及諸如此類。
「醯胺」係指諸如-C(=O)NR53
R53
等基團,其中每一R53
獨立地選自H及視情況經取代之烷基、環烷基、環烷基烷基、雜環烷基、雜環烷基烷基、芳基、芳基烷基、雜芳基及雜芳基烷基。
「胺基甲酸酯」係指諸如-O-C(=O)NR53
R53
或-NR53
-C(=O)OR53
等基團,其中每一R53
獨立地選自H及視情況經取代之烷基、環烷基、環烷基烷基、雜環烷基、雜環烷基烷基、芳基、芳基烷基、雜芳基及雜芳基烷基。
「磺醯胺」係指-S(O)2
NR54
R54
,其中每一R54
獨立地選自H及視情況經取代之烷基、雜烷基、雜芳基、雜環、烯基、炔基、芳基烷基、雜芳基烷基、雜環基烷基、伸烷基-C(O)-OR55
或伸烷基-O-C(O)-OR55
,其中R55
係選自H、烷基、雜烷基、環烷基、雜環基、芳基、雜芳基、烯基、炔基、芳基烷基、雜環烷基、雜芳基烷基、胺基及亞磺醯基。
「金剛烷基」係指以下結構式之化合物: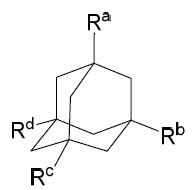 其中可選取代可存在於Ra
、Rb
、Rc
及Rd
中之一或多者上。金剛烷基包含由一或多個取代基(包含烷基、鹵基、-OH、-NH2
及烷氧基)取代之經取代金剛烷基,例如1-或2-金剛烷基。實例性衍生物包含甲基金剛烷、鹵代金剛烷、羥基金剛烷及胺基金剛烷(例如金剛烷胺(amantadine))。
其中可選取代可存在於Ra
、Rb
、Rc
及Rd
中之一或多者上。金剛烷基包含由一或多個取代基(包含烷基、鹵基、-OH、-NH2
及烷氧基)取代之經取代金剛烷基,例如1-或2-金剛烷基。實例性衍生物包含甲基金剛烷、鹵代金剛烷、羥基金剛烷及胺基金剛烷(例如金剛烷胺(amantadine))。
本文所用之「N-保護基團」係指彼等意欲在合成程序期間保護氮原子免於不期望反應之基團。實例性N-保護基團包含(但不限於)醯基(例如乙醯基及第三丁基乙醯基、三甲基乙醯基)、烷氧基羰基(例如甲基氧基羰基及第三丁基氧基羰基(Boc))、芳基氧基羰基(例如苄基氧基羰基(Cbz)及茀基甲氧基羰基(Fmoc))及芳醯基(例如苯甲醯基)。N-保護基團闡述於Greene's Protective Groups in Organic Synthesis,第5版,P. G. M. Wuts編輯,Wiley (2014)中。
「可選」或「視情況」係指所闡述事件或情況可能發生或可能不發生,且該闡述包含該事件或情況發生之情形及該事件或情況不發生之情形。舉例而言,「視情況經取代之烷基」係指可或可不經取代之烷基且該闡述涵蓋經取代烷基及未取代烷基二者。
本文所用之「經取代」意指,基團之一或多個氫原子經通常用於醫藥化學中之取代基原子或基團代替。每一取代基可相同或不同。適宜取代基之實例包含(但不限於)烷基、烯基、炔基、環烷基、芳基、芳基烷基、雜環烷基、雜芳基、-OR56
(例如羥基、烷基氧基(例如甲氧基、乙氧基及丙氧基)、醚、酯、胺基甲酸酯等)、羥基烷基、-C(O)O-烷基、-O-烷基-O-烷基、鹵代烷基、烷基-O-烷基、SR56
(例如-SH、-S-烷基、-S-芳基、-S-雜芳基、芳基烷基-S-等)、S+
R56 2
、S(O)R56
、SO2
R56
、NR56
R57
(例如一級胺(亦即NH2
)、二級胺、三級胺、醯胺、胺基甲酸酯、脲等)、醯肼、鹵基、腈、硝基、硫化物、亞碸、碸、磺醯胺、-SH、羧基、醛、酮基、羧酸、酯、醯胺、亞胺及醯亞胺(例如-C(O)NR56
C(O)R57
) (包含其硒基及硫基衍生物),其中R56
及R57
各自獨立地係烷基、烯基、炔基、雜烷基、環烷基、環烷基烷基、雜環烷基、雜環烷基烷基、芳基、芳基烷基、雜芳基或雜芳基烷基,且其中每一取代基可視情況進一步經取代。在具有芳香族碳環之官能基經取代之實施例中,該等取代之數量通常為小於約10個取代或約1至5個取代且在某些實施例中約1或2個取代。
「醫藥上可接受之鹽」意欲包含使用相對無毒酸或鹼製得之活性化合物之鹽,此取決於在本文所闡述化合物上所發現之特定取代基。在如本文所揭示之化合物含有相對酸性官能基時,可藉由使該等化合物之中性形式與足夠量之純淨或存於適宜惰性溶劑中之期望鹼接觸來獲得鹼加成鹽。醫藥上可接受之鹼加成鹽之實例包含鈉鹽、鉀鹽、鈣鹽、銨鹽、有機胺基鹽或鎂鹽或類似鹽。在如本文所揭示之化合物含有相對鹼性官能基時,可藉由使該等化合物之中性形式與足夠量之純淨或存於適宜惰性溶劑中之期望酸接觸來獲得酸加成鹽。醫藥上可接受之酸加成鹽之實例包含衍生自以下無機酸者:如鹽酸、氫溴酸、硝酸、碳酸、磷酸、部分中和性磷酸、硫酸、部分中和性硫酸、氫碘酸或亞磷酸及諸如此類;以及衍生自以下相對無毒有機酸之鹽:如乙酸、丙酸、異丁酸、馬來酸、丙二酸、苯甲酸、琥珀酸、辛二酸、富馬酸、扁桃酸、鄰苯二甲酸、苯磺酸、對甲苯磺酸、檸檬酸、酒石酸、甲磺酸及諸如此類。亦包含胺基酸之鹽(例如精胺酸鹽及諸如此類)及諸如葡糖醛酸或半乳糖醛酸及諸如此類等有機酸之鹽。本發明之某些特定化合物可含有允許將化合物轉化成鹼或酸加成鹽之鹼性及酸性官能基。適宜鹽之列表可參見Remington's Pharmaceutical Sciences,第17版,Mack Publishing Company, Easton, Pa., (1985)及Journal of Pharmaceutical Science, 66:2 (1977),該等文獻中每一者之全部內容以引用方式併入本文中。
「醫藥上可接受之載劑」或「醫藥上可接受之賦形劑」係指具有以下特徵之賦形劑、載劑或佐劑:其可與至少一種化合物一起投與個體且不會破壞其藥理學活性,其通常安全、無毒且在以足以遞送治療量藥劑之劑量投與時並不在生物上或其他方面不期望。
本文所給出之任一化合物或結構亦意欲代表化合物之未經標記形式以及經同位素標記之形式。該等形式之化合物亦可稱為「同位素富集之類似物」。經同位素標記之化合物具有本文所繪示之結構,只是一或多個原子經具有所選原子質量或質量數之原子代替。可納入所揭示化合物中之同位素之實例包含氫、碳、氮、氧、磷、氟、氯及碘之同位素,例如分別為2
H、3
H、11
C、13
C、14
C、13
N、15
N、15
O、17
O、18
O、31
P、32
P、35
S、18
F、36
Cl、123
I及125
I。本發明之各種經同位素標記之化合物,例如納入放射性同位素(例如3
H及14
C)者)。該等經同位素標記之化合物可用於代謝研究、反應動力學研究、檢測或成像技術(例如正電子發射斷層掃描術(PET)或單光子發射電腦斷層掃描術(SPECT),包含藥物或基質組織分佈分析)或患者之放射性治療。
術語「同位素富集之類似物」包含本文所闡述化合物之「氘化類似物」,其中一或多個氫(例如碳原子上之氫)由氘代替。該等化合物展現增加之抗代謝性且由此可用於增加任一化合物在投與哺乳動物(例如人類)時之半衰期。例如參見Foster, 「Deuterium Isotope Effects in Studies of Drug Metabolism,」 Trends Pharmacol. Sci. 5(12):524-527 (1984)。該等化合物係藉由業內熟知方式來合成,例如藉由採用其中一或多個氫已由氘代替之起始材料。
經氘標記或取代之本發明治療化合物可具有改良之關於分佈、代謝及排泄(ADME)之DMPK (藥物代謝及藥物動力學)性質。使用較重同位素(例如氘)取代可提供某些源於較大代謝穩定性之治療優點,例如活體內半衰期延長、劑量需求降低及/或治療指數改良。18
F、3
H、11
C標記化合物可用於PET或SPECT或其他成像研究。經同位素標記之本發明化合物及其前藥通常可藉由實施在反應圖中或在下文所闡述實例及製備中所揭示之程序藉由使用易於獲得之經同位素標記之試劑取代未經同位素標記的試劑來製備。應理解,此背景中之氘可視為本文所闡述化合物中之取代基。
此一較重同位素(具體而言氘)之濃度可由同位素富集因子定義。在本發明化合物中,任一未特定地指定為特定同位素之原子意欲代表該原子之任一穩定同位素。除非另外陳述,否則在將某一位置特定地指定為「H」或「氫」時,該位置應理解為具有其天然豐度同位素組成之氫。因此,在本發明化合物中,特定地指定為氘(D)之任一原子意欲代表氘。
一些化合物以互變異構體形式存在。互變異構異構體彼此處於平衡中。舉例而言,含有醯胺之化合物可以與醯亞胺酸互變異構體之平衡存在。不論展示哪種互變異構體且不論互變異構體之間之平衡性質如何,熟習此項技術者應理解,該等化合物包括醯胺及醯亞胺酸互變異構體二者。因此,應理解,含有醯胺之化合物包含其醯亞胺酸互變異構體。同樣,應理解,含有醯亞胺酸之化合物包含其醯胺互變異構體。
如本文所揭示之化合物或其醫藥上可接受之鹽包含不對稱中心且可由此產生對映異構體、非對映異構體及其他立體異構體形式,該等形式可根據絕對立體化學定義為(R
)-或(S
)-或對於胺基酸而言定義為(D)-或(L)-。本發明意欲包含所有該等可能之異構體以及其外消旋及光學純形式。光學活性(+)及(-)、(R
)-及(S
)-或(D)-及(L)-異構體可使用對掌性合成子或對掌性試劑來製備,或使用習用技術(例如層析及分段結晶)來拆分。製備/分離個別對映異構體之習用技術包含自適宜光學純前體對掌性合成或使用(例如)對掌性高壓液相層析(HPLC)拆分外消旋物(或鹽或衍生物之外消旋物)。在本文所闡述化合物含有烯烴雙鍵或其他幾何不對稱性中心時,除非另外陳述,否則該等化合物意欲包含E及Z幾何異構體。
「立體異構體」係指由藉由相同鍵鍵結之相同原子構成但具有不可互換之不同三維結構之化合物。本發明涵蓋各種立體異構體及其混合物且包含「對映異構體」,對映異構體係指兩種其分子彼此係非重疊鏡像之立體異構體。
「非對映異構體」係具有至少兩個不對稱原子、但彼此不為鏡像之立體異構體。
如本文所繪示化合物之相對中心係以圖形形式使用「粗鍵」形式(粗體或平行線)來指示且絕對立體化學係使用楔形鍵(粗體或平行線)來繪示。
2. 化合物
在某些實施例中,本文提供式I化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽: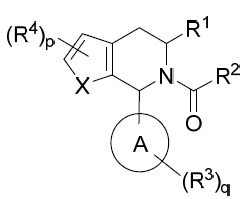 I
其中:
環A係C4
-C10
環烷基、雜環基、芳基或雜芳基;
X係-O-、-S-、-NR9
-、-CR5
=CR5
-或-CR5
=N-;
p為0、1或2;
q為0、1、2或3;
R1
係C1
-C6
烷基、C2
-C6
烯基、C2
-C6
炔基、C1
-C6
鹵代烷基、C3
-C10
環烷基、-CN、-OR7
、-C(O)OR6
、-C(O)N(R7
)2
、-OC(O)R6
、-S(O)2
R8
、-S(O)2
N(R7
)2
、-S(O)N(R7
)2
、-S(O)R8
、-N(R7
)2
、-NO2
、-C1
-C6
烷基-OR7
或-Si(R15
)3
;
R2
係-C1
-C2
鹵代烷基、-C2
-C3
烯基、-C2
-C3
鹵代烯基、C2
炔基或-CH2
OS(O)2
-苯基,其中C1
-C2
鹵代烷基及-C2
-C3
烯基鹵基視情況經一或兩個-CH3
取代,且C2
炔基及苯基視情況經一個-CH3
取代;
每一R3
獨立地係鹵基、-CN、-OH、-OR8
、-NH2
、-NHR8
、-N(R8
)2
、-S(O)2
R8
、-S(O)R8
、-S(O)2
N(R7
)2
、-S(O)N(R7
)2
、-NO2
、-Si(R12
)3
、-SF5
、-C(O)OR6
、-C(O)N(R7
)2
、-NR12
C(O)R8
、-NR12
C(O)OR8
、-OC(O)N(R7
)2
、-OC(O)R8
、-C(O)R6
、-OC(O)CHR8
N(R12
)2
、C1
-C6
烷基、C2
-C6
烯基、C2
-C6
炔基、C3
-C10
環烷基、雜環基、芳基、雜芳基、-C1
-C6
烷基C3
-C10
環烷基、-C2
-C6
烯基C3
-C10
環烷基、-C1
-C6
烷基雜環基、-C2
-C6
烯基雜環基、-C1
-C6
烷基芳基、-C2
-C6
烯基芳基、C1
-C6
烷基雜芳基或-C2
-C6
烯基雜芳基;其中R3
之每一C1
-C6
烷基、C2
-C6
烯基、C2
-C6
炔基、C3
-C10
環烷基、雜環基、芳基、雜芳基、-C1
-C6
烷基C3
-C10
環烷基、-C2
-C6
烯基C3
-C10
環烷基、-C1
-C6
烷基雜環基、-C2
-C6
烯基雜環基、-C1
-C6
烷基芳基、-C2
-C6
烯基芳基、C1
-C6
烷基雜芳基或-C2
-C6
烯基雜芳基獨立地視情況經一至三個R10
取代;
每一R4
獨立地係鹵基、-CN、-OH、-OR8
、-NH2
、-NHR8
、-N(R8
)2
、-S(O)2
R8
、-S(O)R8
、-S(O)2
N(R7
)2
、-S(O)N(R7
)2
、-NO2
、-Si(R15
)3
、-C(O)OR6
、-C(O)N(R7
)2
、-NR12
C(O)R8
、-OC(O)R8
、-C(O)R6
、-NR12
C(O)OR8
、-OC(O)N(R7
)2
、-OC(O)CHR8
N(R12
)2
、C1
-C6
烷基、C2
-C6
烯基、C2
-C6
炔基、C3
-C10
環烷基、雜環基、芳基、雜芳基、-C1
-C6
烷基C3
-C10
環烷基、-C2
-C6
烯基C3
-C10
環烷基、-C1
-C6
烷基雜環基、-C2
-C6
烯基雜環基、-C1
-C6
烷基芳基、-C2
-C6
烯基芳基、C1
-C6
烷基雜芳基或-C2
-C6
烯基雜芳基;其中R4
之每一C1
-C6
烷基、C2
-C6
烯基、C2
-C6
炔基、C3
-C10
環烷基、雜環基、芳基、雜芳基、-C1
-C6
烷基C3
-C10
環烷基、-C2
-C6
烯基C3
-C10
環烷基、-C1
-C6
烷基雜環基、-C2
-C6
烯基雜環基、-C1
-C6
烷基芳基、-C2
-C6
烯基芳基、C1
-C6
烷基雜芳基或-C2
-C6
烯基雜芳基視情況獨立地視情況經一至三個R10
取代;
每一R5
獨立地係氫、鹵基、-CN、-OH、-OR8
、-NH2
、-NHR8
、-N(R8
)2
、-S(O)2
R8
、-S(O)R8
、-S(O)2
N(R7
)2
、-S(O)N(R7
)2
、-NO2
、-Si(R15
)3
、-C(O)OR6
、-C(O)N(R7
)2
、-NR12
C(O)R8
、-OC(O)R8
、-C(O)R6
、-NR12
C(O)OR8
、-OC(O)N(R7
)2
、-OC(O)CHR8
N(R12
)2
、C1
-C6
烷基、C2
-C6
烯基、C2
-C6
炔基、C3
-C10
環烷基、雜環基、芳基、雜芳基、-C1
-C6
烷基C3
-C10
環烷基、-C2
-C6
烯基C3
-C10
環烷基、-C1
-C6
烷基雜環基、-C2
-C6
烯基雜環基、-C1
-C6
烷基芳基、-C2
-C6
烯基芳基、C1
-C6
烷基雜芳基或-C2
-C6
烯基雜芳基;其中R5
之每一C1
-C6
烷基、C2
-C6
烯基、C2
-C6
炔基、C3
-C10
環烷基、雜環基、芳基、雜芳基、-C1
-C6
烷基C3
-C10
環烷基、-C2
-C6
烯基C3
-C10
環烷基、-C1
-C6
烷基雜環基、-C2
-C6
烯基雜環基、-C1
-C6
烷基芳基、-C2
-C6
烯基芳基、-C1
-C6
烷基雜芳基或-C2
-C6
烯基雜芳基視情況獨立地視情況經一至三個R10
取代;
每一R6
獨立地係氫、C1
-C6
烷基、C2
-C6
烯基、C2
-C6
炔基、C3
-C10
環烷基、雜環基、芳基、雜芳基、-C1
-C6
烷基C3
-C10
環烷基、-C2
-C6
烯基C3
-C10
環烷基、-C1
-C6
烷基雜環基、-C2
-C6
烯基雜環基、-C1
-C6
烷基芳基、-C2
-C6
烯基芳基、-C1
-C6
烷基雜芳基或-C2
-C6
烯基雜芳基;其中每一R6
獨立地進一步經一至三個R11
取代;
每一R7
獨立地係氫、C1
-C6
烷基、C2
-C6
烯基、C2
-C6
炔基、C3
-C10
環烷基、雜環基、芳基、雜芳基、-C1
-C6
烷基C3
-C6
環烷基、-C2
-C6
烯基C3
-C6
環烷基、-C1
-C6
烷基雜環基、-C2
-C6
烯基雜環基、-C1
-C6
烷基芳基、-C2
-C6
烯基芳基、-C1
-C6
烷基雜芳基、-C2
-C6
烯基雜芳基,或兩個R7
與其所連接之氮原子一起形成4至7員雜環基;其中每一R7
或由此形成之環獨立地進一步經一至三個R11
取代;
每一R8
獨立地係C1
-C6
烷基、C2
-C6
烯基、C2
-C6
炔基、C3
-C10
環烷基、雜環基、芳基、雜芳基、-C1
-C6
烷基C3
-C10
環烷基、-C2
-C6
烯基C3
-C10
環烷基、-C1
-C6
烷基雜環基、-C2
-C6
烯基雜環基、-C1
-C6
烷基芳基、-C2
-C6
烯基芳基、-C1
-C6
烷基雜芳基或-C2
-C6
烯基雜芳基;其中每一R8
獨立地進一步經一至三個R11
取代;
R9
係氫或C1
-C6
烷基;
每一R10
獨立地係鹵基、-CN、-OR12
、-NO2
、-N(R12
)2
、-S(O)R13
、-S(O)2
R13
、-S(O)N(R12
)2
、-S(O)2
N(R12
)2
、-Si(R12
)3
、-C(O)R12
、-C(O)OR12
、-C(O)N(R12
)2
、-NR12
C(O)R12
、-OC(O)R12
、-OC(O)OR12
、-OC(O)N(R12
)2
、-NR12
C(O)OR12
、-OC(O)CHR12
N(R12
)2
、C1
-C6
烷基、C1
-C6
鹵代烷基、C2
-C6
烯基、C2
-C6
炔基、C3
-C10
環烷基、雜環基、芳基或雜芳基,其中R10
之每一C1
-C6
烷基、C1
-C6
鹵代烷基、C2
-C6
烯基、C2
-C6
炔基、C3
-C10
環烷基、雜環基、芳基或雜芳基視情況獨立地經一至三個R11
取代;
每一R11
獨立地係鹵基、-CN、-OR12
、-NO2
、-N(R12
)2
、-S(O)R13
、-S(O)2
R13
、-S(O)N(R12
)2
、-S(O)2
N(R12
)2
、-Si(R12
)3
、-C(O)R12
、-C(O)OR12
、-C(O)N(R12
)2
、-NR12
C(O)R12
、-OC(O)R12
、-OC(O)OR12
、-OC(O)N(R12
)2
、-NR12
C(O)OR12
、-OC(O)CHR12
N(R12
)2
、C1
-C6
烷基、C1
-C6
鹵代烷基、C2
-C6
烯基、C2
-C6
炔基、C3
-C10
環烷基、雜環基、芳基或雜芳基;
每一R12
獨立地係氫、C1
-C6
烷基或C3
-C10
環烷基;
每一R13
獨立地係C1
-C6
烷基或C3
-C10
環烷基;且
每一R15
獨立地係C1
-C6
烷基、C2
-C6
烯基、芳基、雜芳基、-C1
-C6
烷基芳基、-C2
-C6
烯基芳基、-C1
-C6
烷基雜芳基及-C2
-C6
烯基雜芳基。
I
其中:
環A係C4
-C10
環烷基、雜環基、芳基或雜芳基;
X係-O-、-S-、-NR9
-、-CR5
=CR5
-或-CR5
=N-;
p為0、1或2;
q為0、1、2或3;
R1
係C1
-C6
烷基、C2
-C6
烯基、C2
-C6
炔基、C1
-C6
鹵代烷基、C3
-C10
環烷基、-CN、-OR7
、-C(O)OR6
、-C(O)N(R7
)2
、-OC(O)R6
、-S(O)2
R8
、-S(O)2
N(R7
)2
、-S(O)N(R7
)2
、-S(O)R8
、-N(R7
)2
、-NO2
、-C1
-C6
烷基-OR7
或-Si(R15
)3
;
R2
係-C1
-C2
鹵代烷基、-C2
-C3
烯基、-C2
-C3
鹵代烯基、C2
炔基或-CH2
OS(O)2
-苯基,其中C1
-C2
鹵代烷基及-C2
-C3
烯基鹵基視情況經一或兩個-CH3
取代,且C2
炔基及苯基視情況經一個-CH3
取代;
每一R3
獨立地係鹵基、-CN、-OH、-OR8
、-NH2
、-NHR8
、-N(R8
)2
、-S(O)2
R8
、-S(O)R8
、-S(O)2
N(R7
)2
、-S(O)N(R7
)2
、-NO2
、-Si(R12
)3
、-SF5
、-C(O)OR6
、-C(O)N(R7
)2
、-NR12
C(O)R8
、-NR12
C(O)OR8
、-OC(O)N(R7
)2
、-OC(O)R8
、-C(O)R6
、-OC(O)CHR8
N(R12
)2
、C1
-C6
烷基、C2
-C6
烯基、C2
-C6
炔基、C3
-C10
環烷基、雜環基、芳基、雜芳基、-C1
-C6
烷基C3
-C10
環烷基、-C2
-C6
烯基C3
-C10
環烷基、-C1
-C6
烷基雜環基、-C2
-C6
烯基雜環基、-C1
-C6
烷基芳基、-C2
-C6
烯基芳基、C1
-C6
烷基雜芳基或-C2
-C6
烯基雜芳基;其中R3
之每一C1
-C6
烷基、C2
-C6
烯基、C2
-C6
炔基、C3
-C10
環烷基、雜環基、芳基、雜芳基、-C1
-C6
烷基C3
-C10
環烷基、-C2
-C6
烯基C3
-C10
環烷基、-C1
-C6
烷基雜環基、-C2
-C6
烯基雜環基、-C1
-C6
烷基芳基、-C2
-C6
烯基芳基、C1
-C6
烷基雜芳基或-C2
-C6
烯基雜芳基獨立地視情況經一至三個R10
取代;
每一R4
獨立地係鹵基、-CN、-OH、-OR8
、-NH2
、-NHR8
、-N(R8
)2
、-S(O)2
R8
、-S(O)R8
、-S(O)2
N(R7
)2
、-S(O)N(R7
)2
、-NO2
、-Si(R15
)3
、-C(O)OR6
、-C(O)N(R7
)2
、-NR12
C(O)R8
、-OC(O)R8
、-C(O)R6
、-NR12
C(O)OR8
、-OC(O)N(R7
)2
、-OC(O)CHR8
N(R12
)2
、C1
-C6
烷基、C2
-C6
烯基、C2
-C6
炔基、C3
-C10
環烷基、雜環基、芳基、雜芳基、-C1
-C6
烷基C3
-C10
環烷基、-C2
-C6
烯基C3
-C10
環烷基、-C1
-C6
烷基雜環基、-C2
-C6
烯基雜環基、-C1
-C6
烷基芳基、-C2
-C6
烯基芳基、C1
-C6
烷基雜芳基或-C2
-C6
烯基雜芳基;其中R4
之每一C1
-C6
烷基、C2
-C6
烯基、C2
-C6
炔基、C3
-C10
環烷基、雜環基、芳基、雜芳基、-C1
-C6
烷基C3
-C10
環烷基、-C2
-C6
烯基C3
-C10
環烷基、-C1
-C6
烷基雜環基、-C2
-C6
烯基雜環基、-C1
-C6
烷基芳基、-C2
-C6
烯基芳基、C1
-C6
烷基雜芳基或-C2
-C6
烯基雜芳基視情況獨立地視情況經一至三個R10
取代;
每一R5
獨立地係氫、鹵基、-CN、-OH、-OR8
、-NH2
、-NHR8
、-N(R8
)2
、-S(O)2
R8
、-S(O)R8
、-S(O)2
N(R7
)2
、-S(O)N(R7
)2
、-NO2
、-Si(R15
)3
、-C(O)OR6
、-C(O)N(R7
)2
、-NR12
C(O)R8
、-OC(O)R8
、-C(O)R6
、-NR12
C(O)OR8
、-OC(O)N(R7
)2
、-OC(O)CHR8
N(R12
)2
、C1
-C6
烷基、C2
-C6
烯基、C2
-C6
炔基、C3
-C10
環烷基、雜環基、芳基、雜芳基、-C1
-C6
烷基C3
-C10
環烷基、-C2
-C6
烯基C3
-C10
環烷基、-C1
-C6
烷基雜環基、-C2
-C6
烯基雜環基、-C1
-C6
烷基芳基、-C2
-C6
烯基芳基、C1
-C6
烷基雜芳基或-C2
-C6
烯基雜芳基;其中R5
之每一C1
-C6
烷基、C2
-C6
烯基、C2
-C6
炔基、C3
-C10
環烷基、雜環基、芳基、雜芳基、-C1
-C6
烷基C3
-C10
環烷基、-C2
-C6
烯基C3
-C10
環烷基、-C1
-C6
烷基雜環基、-C2
-C6
烯基雜環基、-C1
-C6
烷基芳基、-C2
-C6
烯基芳基、-C1
-C6
烷基雜芳基或-C2
-C6
烯基雜芳基視情況獨立地視情況經一至三個R10
取代;
每一R6
獨立地係氫、C1
-C6
烷基、C2
-C6
烯基、C2
-C6
炔基、C3
-C10
環烷基、雜環基、芳基、雜芳基、-C1
-C6
烷基C3
-C10
環烷基、-C2
-C6
烯基C3
-C10
環烷基、-C1
-C6
烷基雜環基、-C2
-C6
烯基雜環基、-C1
-C6
烷基芳基、-C2
-C6
烯基芳基、-C1
-C6
烷基雜芳基或-C2
-C6
烯基雜芳基;其中每一R6
獨立地進一步經一至三個R11
取代;
每一R7
獨立地係氫、C1
-C6
烷基、C2
-C6
烯基、C2
-C6
炔基、C3
-C10
環烷基、雜環基、芳基、雜芳基、-C1
-C6
烷基C3
-C6
環烷基、-C2
-C6
烯基C3
-C6
環烷基、-C1
-C6
烷基雜環基、-C2
-C6
烯基雜環基、-C1
-C6
烷基芳基、-C2
-C6
烯基芳基、-C1
-C6
烷基雜芳基、-C2
-C6
烯基雜芳基,或兩個R7
與其所連接之氮原子一起形成4至7員雜環基;其中每一R7
或由此形成之環獨立地進一步經一至三個R11
取代;
每一R8
獨立地係C1
-C6
烷基、C2
-C6
烯基、C2
-C6
炔基、C3
-C10
環烷基、雜環基、芳基、雜芳基、-C1
-C6
烷基C3
-C10
環烷基、-C2
-C6
烯基C3
-C10
環烷基、-C1
-C6
烷基雜環基、-C2
-C6
烯基雜環基、-C1
-C6
烷基芳基、-C2
-C6
烯基芳基、-C1
-C6
烷基雜芳基或-C2
-C6
烯基雜芳基;其中每一R8
獨立地進一步經一至三個R11
取代;
R9
係氫或C1
-C6
烷基;
每一R10
獨立地係鹵基、-CN、-OR12
、-NO2
、-N(R12
)2
、-S(O)R13
、-S(O)2
R13
、-S(O)N(R12
)2
、-S(O)2
N(R12
)2
、-Si(R12
)3
、-C(O)R12
、-C(O)OR12
、-C(O)N(R12
)2
、-NR12
C(O)R12
、-OC(O)R12
、-OC(O)OR12
、-OC(O)N(R12
)2
、-NR12
C(O)OR12
、-OC(O)CHR12
N(R12
)2
、C1
-C6
烷基、C1
-C6
鹵代烷基、C2
-C6
烯基、C2
-C6
炔基、C3
-C10
環烷基、雜環基、芳基或雜芳基,其中R10
之每一C1
-C6
烷基、C1
-C6
鹵代烷基、C2
-C6
烯基、C2
-C6
炔基、C3
-C10
環烷基、雜環基、芳基或雜芳基視情況獨立地經一至三個R11
取代;
每一R11
獨立地係鹵基、-CN、-OR12
、-NO2
、-N(R12
)2
、-S(O)R13
、-S(O)2
R13
、-S(O)N(R12
)2
、-S(O)2
N(R12
)2
、-Si(R12
)3
、-C(O)R12
、-C(O)OR12
、-C(O)N(R12
)2
、-NR12
C(O)R12
、-OC(O)R12
、-OC(O)OR12
、-OC(O)N(R12
)2
、-NR12
C(O)OR12
、-OC(O)CHR12
N(R12
)2
、C1
-C6
烷基、C1
-C6
鹵代烷基、C2
-C6
烯基、C2
-C6
炔基、C3
-C10
環烷基、雜環基、芳基或雜芳基;
每一R12
獨立地係氫、C1
-C6
烷基或C3
-C10
環烷基;
每一R13
獨立地係C1
-C6
烷基或C3
-C10
環烷基;且
每一R15
獨立地係C1
-C6
烷基、C2
-C6
烯基、芳基、雜芳基、-C1
-C6
烷基芳基、-C2
-C6
烯基芳基、-C1
-C6
烷基雜芳基及-C2
-C6
烯基雜芳基。
亦提供式IA化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽: IA
其中環A、X、R1
、R2
、R3
、R4
、p及q中之每一者獨立地如本文所定義。
IA
其中環A、X、R1
、R2
、R3
、R4
、p及q中之每一者獨立地如本文所定義。
亦提供式IB化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽: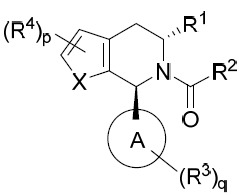 IB
其中環A、X、R1
、R2
、R3
、R4
、p及q中之每一者獨立地如本文所定義。
IB
其中環A、X、R1
、R2
、R3
、R4
、p及q中之每一者獨立地如本文所定義。
亦提供式II化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽: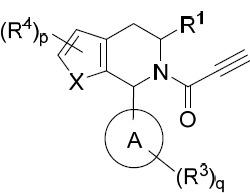 II
其中環A、X、R1
、R3
、R4
、p及q中之每一者獨立地如本文所定義。
II
其中環A、X、R1
、R3
、R4
、p及q中之每一者獨立地如本文所定義。
亦提供式IIA化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽: IIA
其中環A、X、R1
、R3
、R4
、p及q中之每一者獨立地如本文所定義。
IIA
其中環A、X、R1
、R3
、R4
、p及q中之每一者獨立地如本文所定義。
亦提供式IIB化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽: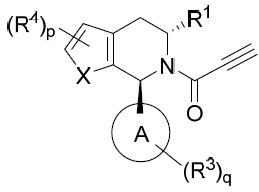 IIB
其中環A、X、R1
、R3
、R4
、p及q中之每一者獨立地如本文所定義。
IIB
其中環A、X、R1
、R3
、R4
、p及q中之每一者獨立地如本文所定義。
亦提供式III化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽: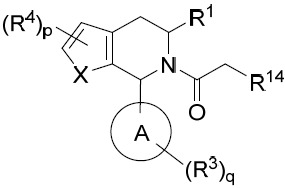 III
其中環A、X、R1
、R3
、R4
、p及q中之每一者獨立地如本文所定義,且R14
係鹵基。
III
其中環A、X、R1
、R3
、R4
、p及q中之每一者獨立地如本文所定義,且R14
係鹵基。
亦提供式IIIA化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽: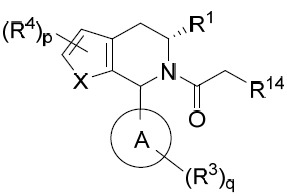 IIIA
其中環A、X、R1
、R3
、R4
、p及q中之每一者獨立地如本文所定義,且R14
係鹵基。
IIIA
其中環A、X、R1
、R3
、R4
、p及q中之每一者獨立地如本文所定義,且R14
係鹵基。
亦提供式IIIB化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽: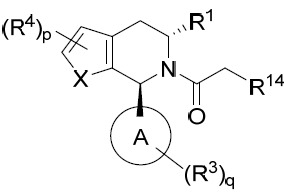 IIIB
其中環A、X、R1
、R3
、R4
、p及q中之每一者獨立地如本文所定義,且R14
係鹵基。
IIIB
其中環A、X、R1
、R3
、R4
、p及q中之每一者獨立地如本文所定義,且R14
係鹵基。
在某些實施例中,
環A係芳基或雜芳基;
X係-O-、-S-、-NH-、-CH=CH-或-CH=N-;
p為0、1或2;
q為1;
R1
係C1
-C6
烷基、-C(O)O-C1
-C6
烷基或-C(O)N(C1
-C6
烷基)2
;
R3
係鹵基、-NHR8
、-S(O)2
N(R7
)2
、-C(O)OR6
、-C(O)N(R7
)2
或雜環基;
每一R4
獨立地係-OR8
;
R6
係C1
-C6
烷基;
每一R7
獨立地係氫、C1
-C6
烷基或C3
-C10
環烷基,其中每一R7
獨立地進一步經一至三個R11
取代;
每一R8
獨立地係C1
-C6
烷基或C3
-C10
環烷基;其中每一R8
獨立地進一步經一至三個R11
取代;且
每一R11
獨立地係-O-C1
-C6
烷基。
在某些實施例中,X係-O-、-S-或-NR9
-。在某些實施例中,X係-O-、-S-或-NH-。在某些實施例中,X係-O-。在某些實施例中,X係-S-。在某些實施例中,X係-NR9
-。在某些實施例中,X係-NH-。
在某些實施例中,X係-CR5
=CR5
-或-CR5
=N-。在某些實施例中,X係-CH=CH-或-CH=N-。在某些實施例中,X係-CR5
=CR5
-。在某些實施例中,X係-CR5
=N-。
在某些實施例中,R5
係R4
。
在某些實施例中,在X係-CH=CH-、p為1或2、每一R4
係甲氧基、環A係苯基且q為1時,則R3
不為金剛烷基胺、氟或-C(O)NH-環丙基。在某些實施例中,在X係-CH=CH-、p為1或2、每一R4
係甲氧基、R1
係甲基、正丁基或-C(O)OCH3
、環A係苯基且q為1時,則R3
不為金剛烷基胺、氟及-C(O)NH-環丙基。在某些實施例中,在X係-CH=CH-、p為1或2、每一R4
係甲氧基、R1
係甲基、正丁基或-C(O)OCH3
、R2
係-CH2
Cl或C2
炔基、環A係苯基且q為1時,則R3
不為金剛烷基胺、氟或-C(O)NH-環丙基。
在某些實施例中,在X係-CR5
=CR5
-、p為1或2、環A係苯基、環己基或呋喃基且q為0或1時,則至少一個R4
不為甲氧基。在某些實施例中,在R1
係-C(O)OCH3
且R2
係-CH2
Cl、環A係苯基、環己基或呋喃基、q為0或1、R3
係-NO2
、Br或-OCH3
且p為1或2時,則至少一個R4
不為甲氧基。在某些實施例中,在R2
係-CH2
Cl、X係-CR5
=CR5
-、p為1或2、環A係苯基、環己基或呋喃基且q為0或1時,則至少一個R4
不為甲氧基。在某些實施例中,在R1
係-C(O)OCH3
、R2
係-CH2
Cl、X係-CR5
=CR5
-、p為1或2、環A係苯基、環己基或呋喃基且q為0或1時,則至少一個R4
不為甲氧基。
在某些實施例中,在R1
係-C(O)OCH3
且R2
係-CH2
Cl時,則X不為-CR5
=CR5
-。在某些實施例中,在X係-CH=CH-、p為1或2、環A係苯基且q為1時,則至少一個R4
不為甲氧基。
在某些實施例中,化合物不為N-環丙基-4-((1S,3S)-6-甲氧基-3-甲基-2-丙炔醯基-1,2,3,4-四氫異喹啉-1-基)苯甲醯胺、4-((1S,3S)-2-(2-氯乙醯基)-6-甲氧基-3-甲基-1,2,3,4-四氫異喹啉-1-基)-N-環丙基苯甲醯胺、1-((1S,3S)-3-丁基-1-(4-氟苯基)-6-甲氧基-3,4-二氫異喹啉-2(1H)-基)-2-氯乙烷-1-酮、4-((1S,3S)-3-丁基-2-(2-氯乙醯基)-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)-N-環丙基苯甲醯胺、4-((1S,3S)-3-丁基-6-甲氧基-2-丙炔醯基-1,2,3,4-四氫異喹啉-1-基)-N-環丙基苯甲醯胺、1-((1S,3S)-1-(4-(金剛烷-1-基胺基)苯基)-3-丁基-6-甲氧基-3,4-二氫異喹啉-2(1H)-基)-2-氯乙烷-1-酮、1-((1S,3S)-1-(4-(金剛烷-1-基胺基)苯基)-3-丁基-6-甲氧基-3,4-二氫異喹啉-2(1H)-基)丙-2-炔-1-酮、(1S,3R)-1-(4-(金剛烷-1-基胺基)苯基)-2-(2-氯乙醯基)-6-甲氧基-1,2,3,4-四氫異喹啉-3-甲酸甲酯、(1S,3R)-1-(4-(金剛烷-1-基胺基)苯基)-6-甲氧基-2-丙炔醯基-1,2,3,4-四氫異喹啉-3-甲酸甲酯、4-((1S,3S)-3-丁基-2-(2-氯乙醯基)-6,7-二甲氧基-1,2,3,4-四氫異喹啉-1-基)-N-環丙基苯甲醯胺或4-((1S,3S)-3-丁基-6,7-二甲氧基-2-丙炔醯基-1,2,3,4-四氫異喹啉-1-基)-N-環丙基苯甲醯胺。
在某些實施例中,在R2
係-C1
-C2
鹵代烷基時,則環A不為苯并[d][1,3]二氧雜環戊烯。
在某些實施例中,R5
係R4
。
亦提供式IV化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽: IV
其中環A、R1
、R3
、R4
、p及q中之每一者獨立地如本文所定義。
IV
其中環A、R1
、R3
、R4
、p及q中之每一者獨立地如本文所定義。
亦提供式IV化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽: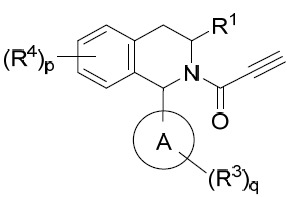 IV
其中:
環A係芳基或雜芳基;
p為0、1或2;
q為1;
R1
係C1
-C6
烷基、-C(O)O-C1
-C6
烷基或-C(O)N(C1
-C6
烷基)2
;
R3
係鹵基、-NHR8
、-S(O)2
N(R7
)2
、-C(O)OR6
、-C(O)N(R7
)2
或雜環基;
每一R4
獨立地係-OR8
;
R6
係C1
-C6
烷基;
每一R7
獨立地係氫、C1
-C6
烷基或C3
-C10
環烷基,其中每一R7
獨立地進一步經一至三個R11
取代;
每一R8
獨立地係C1
-C6
烷基或C3
-C10
環烷基,其中每一R8
獨立地進一步經一至三個R11
取代;且
每一R11
獨立地係-O-C1
-C6
烷基。
IV
其中:
環A係芳基或雜芳基;
p為0、1或2;
q為1;
R1
係C1
-C6
烷基、-C(O)O-C1
-C6
烷基或-C(O)N(C1
-C6
烷基)2
;
R3
係鹵基、-NHR8
、-S(O)2
N(R7
)2
、-C(O)OR6
、-C(O)N(R7
)2
或雜環基;
每一R4
獨立地係-OR8
;
R6
係C1
-C6
烷基;
每一R7
獨立地係氫、C1
-C6
烷基或C3
-C10
環烷基,其中每一R7
獨立地進一步經一至三個R11
取代;
每一R8
獨立地係C1
-C6
烷基或C3
-C10
環烷基,其中每一R8
獨立地進一步經一至三個R11
取代;且
每一R11
獨立地係-O-C1
-C6
烷基。
亦提供式IVA化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽: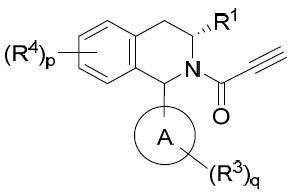 IVA
其中環A、R1
、R3
、R4
、p及q中之每一者獨立地如本文所定義。
IVA
其中環A、R1
、R3
、R4
、p及q中之每一者獨立地如本文所定義。
亦提供式IVB化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽: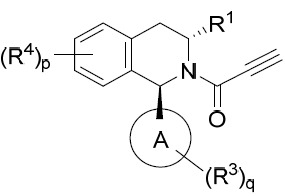 IVB
其中環A、R1
、R3
、R4
、p及q中之每一者獨立地如本文所定義。
IVB
其中環A、R1
、R3
、R4
、p及q中之每一者獨立地如本文所定義。
亦提供式V化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽: V
其中環A、R1
、R3
、R4
、p及q中之每一者獨立地如本文所定義,且R14
係鹵基。
V
其中環A、R1
、R3
、R4
、p及q中之每一者獨立地如本文所定義,且R14
係鹵基。
亦提供式V化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽: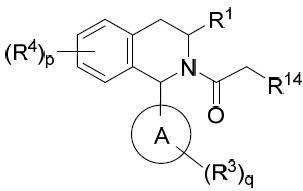 V
其中:
環A係芳基或雜芳基;
p為0、1或2;
q為1;
R1
係C1
-C6
烷基、-C(O)O-C1
-C6
烷基或-C(O)N(C1
-C6
烷基)2
;
R3
係鹵基、-NHR8
、-S(O)2
N(R7
)2
、-C(O)OR6
、-C(O)N(R7
)2
或雜環基;
每一R4
獨立地係-OR8
;
R6
係C1
-C6
烷基;
每一R7
獨立地係氫、C1
-C6
烷基或C3
-C10
環烷基,其中每一R7
獨立地進一步經一至三個R11
取代;
每一R8
獨立地係C1
-C6
烷基或C3
-C10
環烷基;其中每一R8
獨立地進一步經一至三個R11
取代;且
每一R11
獨立地係-O-C1
-C6
烷基。
V
其中:
環A係芳基或雜芳基;
p為0、1或2;
q為1;
R1
係C1
-C6
烷基、-C(O)O-C1
-C6
烷基或-C(O)N(C1
-C6
烷基)2
;
R3
係鹵基、-NHR8
、-S(O)2
N(R7
)2
、-C(O)OR6
、-C(O)N(R7
)2
或雜環基;
每一R4
獨立地係-OR8
;
R6
係C1
-C6
烷基;
每一R7
獨立地係氫、C1
-C6
烷基或C3
-C10
環烷基,其中每一R7
獨立地進一步經一至三個R11
取代;
每一R8
獨立地係C1
-C6
烷基或C3
-C10
環烷基;其中每一R8
獨立地進一步經一至三個R11
取代;且
每一R11
獨立地係-O-C1
-C6
烷基。
亦提供式VA化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽: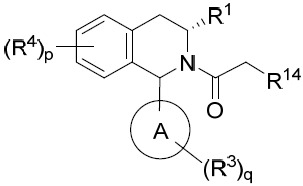 VA
其中環A、R1
、R3
、R4
、p及q中之每一者獨立地如本文所定義,且R14
係鹵基。
VA
其中環A、R1
、R3
、R4
、p及q中之每一者獨立地如本文所定義,且R14
係鹵基。
亦提供式VB化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽: VB
其中環A、R1
、R3
、R4
、p及q中之每一者獨立地如本文所定義,且R14
係鹵基。
VB
其中環A、R1
、R3
、R4
、p及q中之每一者獨立地如本文所定義,且R14
係鹵基。
亦提供式VI化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽: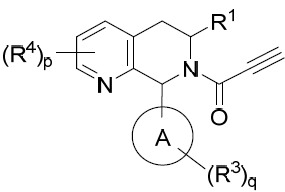 VI
其中環A、R1
、R3
、R4
、p及q中之每一者獨立地如本文所定義。
VI
其中環A、R1
、R3
、R4
、p及q中之每一者獨立地如本文所定義。
亦提供式VI化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽: VI
其中:
環A係芳基或雜芳基;
p為0、1或2;
q為1;
R1
係C1
-C6
烷基、-C(O)O-C1
-C6
烷基或-C(O)N(C1
-C6
烷基)2
;
R3
係鹵基、-NHR8
、-S(O)2
N(R7
)2
、-C(O)OR6
、-C(O)N(R7
)2
或雜環基;
每一R4
獨立地係-OR8
;
R6
係C1
-C6
烷基;
每一R7
獨立地係氫、C1
-C6
烷基或C3
-C10
環烷基,其中每一R7
獨立地進一步經一至三個R11
取代;
每一R8
獨立地係C1
-C6
烷基或C3
-C10
環烷基;其中每一R8
獨立地進一步經一至三個R11
取代;且
每一R11
獨立地係-O-C1
-C6
烷基。
VI
其中:
環A係芳基或雜芳基;
p為0、1或2;
q為1;
R1
係C1
-C6
烷基、-C(O)O-C1
-C6
烷基或-C(O)N(C1
-C6
烷基)2
;
R3
係鹵基、-NHR8
、-S(O)2
N(R7
)2
、-C(O)OR6
、-C(O)N(R7
)2
或雜環基;
每一R4
獨立地係-OR8
;
R6
係C1
-C6
烷基;
每一R7
獨立地係氫、C1
-C6
烷基或C3
-C10
環烷基,其中每一R7
獨立地進一步經一至三個R11
取代;
每一R8
獨立地係C1
-C6
烷基或C3
-C10
環烷基;其中每一R8
獨立地進一步經一至三個R11
取代;且
每一R11
獨立地係-O-C1
-C6
烷基。
亦提供式VIA化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽: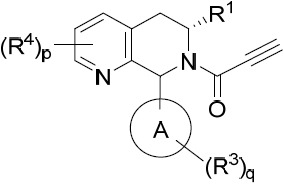 VIA
其中環A、R1
、R3
、R4
、p及q中之每一者獨立地如本文所定義。
VIA
其中環A、R1
、R3
、R4
、p及q中之每一者獨立地如本文所定義。
亦提供式VIB化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽: VIB
其中環A、R1
、R3
、R4
、p及q中之每一者獨立地如本文所定義。
VIB
其中環A、R1
、R3
、R4
、p及q中之每一者獨立地如本文所定義。
亦提供式VII化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽: VII
其中環A、R1
、R3
、R4
、p及q中之每一者獨立地如本文所定義,且R14
係鹵基。
VII
其中環A、R1
、R3
、R4
、p及q中之每一者獨立地如本文所定義,且R14
係鹵基。
亦提供式VII化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽: VII
其中:
環A係芳基或雜芳基;
p為0、1或2;
q為1;
R1
係C1
-C6
烷基、-C(O)O-C1
-C6
烷基或-C(O)N(C1
-C6
烷基)2
;
R3
係鹵基、-NHR8
、-S(O)2
N(R7
)2
、-C(O)OR6
、-C(O)N(R7
)2
或雜環基;
每一R4
獨立地係-OR8
;
R6
係C1
-C6
烷基;
每一R7
獨立地係氫、C1
-C6
烷基或C3
-C10
環烷基,其中每一R7
獨立地進一步經一至三個R11
取代;
每一R8
獨立地係C1
-C6
烷基或C3
-C10
環烷基;其中每一R8
獨立地進一步經一至三個R11
取代;且
每一R11
獨立地係-O-C1
-C6
烷基。
VII
其中:
環A係芳基或雜芳基;
p為0、1或2;
q為1;
R1
係C1
-C6
烷基、-C(O)O-C1
-C6
烷基或-C(O)N(C1
-C6
烷基)2
;
R3
係鹵基、-NHR8
、-S(O)2
N(R7
)2
、-C(O)OR6
、-C(O)N(R7
)2
或雜環基;
每一R4
獨立地係-OR8
;
R6
係C1
-C6
烷基;
每一R7
獨立地係氫、C1
-C6
烷基或C3
-C10
環烷基,其中每一R7
獨立地進一步經一至三個R11
取代;
每一R8
獨立地係C1
-C6
烷基或C3
-C10
環烷基;其中每一R8
獨立地進一步經一至三個R11
取代;且
每一R11
獨立地係-O-C1
-C6
烷基。
亦提供式VIIA化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽: VIIA
其中環A、R1
、R3
、R4
、p及q中之每一者獨立地如本文所定義,且R14
係鹵基。
VIIA
其中環A、R1
、R3
、R4
、p及q中之每一者獨立地如本文所定義,且R14
係鹵基。
亦提供式VIIB化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽: VIIB
其中環A、R1
、R3
、R4
、p及q中之每一者獨立地如本文所定義,且R14
係鹵基。
VIIB
其中環A、R1
、R3
、R4
、p及q中之每一者獨立地如本文所定義,且R14
係鹵基。
亦提供式VIII化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽: VIII
其中環A、R1
、R2
、R3
、R4
、p及q中之每一者獨立地如本文所定義。
VIII
其中環A、R1
、R2
、R3
、R4
、p及q中之每一者獨立地如本文所定義。
在某些實施例中,環A或部分 係:
係: 其中U、V、W、X、Y及Z中之0至3者獨立地係N、S或O,且剩餘變量係CH或CR3
且每一
其中U、V、W、X、Y及Z中之0至3者獨立地係N、S或O,且剩餘變量係CH或CR3
且每一 獨立地代表單鍵或雙鍵,從而符合基於U、V、W、X、Y及Z之化合價需求。
獨立地代表單鍵或雙鍵,從而符合基於U、V、W、X、Y及Z之化合價需求。
在某些實施例中,環A或部分 係:
係: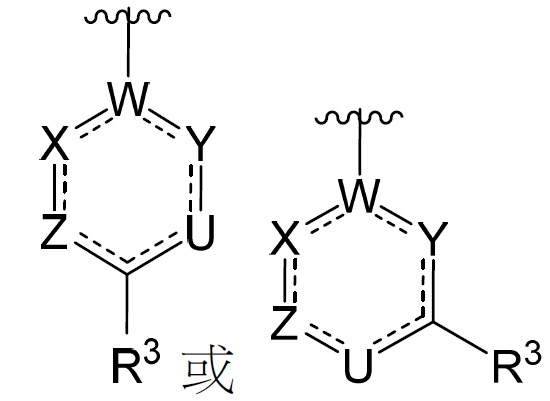 ,其中U、W、X、Y及Z中之1至3者係N、S或O,且剩餘變量係CH或CR3
且代表單鍵或雙鍵,從而符合基於U、W、X、Y及Z之化合價需求。
,其中U、W、X、Y及Z中之1至3者係N、S或O,且剩餘變量係CH或CR3
且代表單鍵或雙鍵,從而符合基於U、W、X、Y及Z之化合價需求。
在某些實施例中,環A係芳基或雜芳基。在某些實施例中,環A係單環芳基或單環雜芳基。在某些實施例中,環A係雜環基。在某些實施例中,環A係4至7員雜環基。在某些實施例中,環A係芳基。在某些實施例中,環A係苯基。在某些實施例中,環A係雜芳基。在某些實施例中,環A係吡啶基。在某些實施例中,環A係吡唑基。在某些實施例中,環A係苯基、吡啶基、六氫吡啶基、六氫吡嗪基或嗎啉基。
在某些實施例中,環A係芳基或雜芳基,其中之每一者由一至三個R3
取代。在某些實施例中,環A係芳基或雜芳基,其中之每一者由一至三個R3
取代,其中至少一個R3
係C3
-C10
環烷基、雜環基、芳基或雜芳基;其中R3
之每一C3
-C10
環烷基、雜環基、芳基及雜芳基視情況經一至三個R10
取代。
在某些實施例中,環A係芳基或雜芳基,其中之每一者由一至三個R3
取代,其中至少一個R3
係C3
-C10
環烷基、雜環基、芳基或雜芳基;且其中R3
之每一C3
-C10
環烷基、雜環基、芳基及雜芳基視情況經一至三個R10
取代;
每一R10
獨立地係-OR12
、-N(R12
)2
、-S(O)2
R13
、-OC(O)CHR12
N(R12
)2
或C1
-C6
烷基,其中R10
之C1
-C6
烷基視情況獨立地經一至三個R11
取代;
每一R11
獨立地係鹵基、-OR12
、-N(R12
)2
、-Si(R12
)3
、-C(O)OR12
、-NR12
C(O)OR12
、-OC(O)CHR12
N(R12
)2
、C1
-C6
烷基或雜環基;
每一R12
獨立地係氫、C1
-C6
烷基或C3
-C10
環烷基;且
每一R13
獨立地係C1
-C6
烷基或C3
-C10
環烷基。
在某些實施例中,環A係雙環[1.1.1]戊烷-1-基、苯基、六氫吡啶基、吡唑基、吡啶基或喹啉基,其中之每一者視情況由一個、兩個或三個R3
取代。在某些實施例中,環A係雙環[1.1.1]戊烷-1-基、苯基、六氫吡啶基、吡唑基、吡啶基或喹啉基,其中之每一者由一個、兩個或三個R3
取代。在某些實施例中,環A係雙環[1.1.1]戊烷-1-基、苯基、六氫吡啶基、吡唑基、吡啶基或喹啉基,其中之每一者由兩個或三個R3
取代。
在某些實施例中,環A係芳基或雜芳基,其中之每一者由兩個或三個R3
取代。在某些實施例中,環A係芳基或雜芳基,其中之每一者由兩個或三個R3
取代;其中至少一個R3
係鹵基。
在某些實施例中,環A係環己基。在某些實施例中,環A係C4
-C10
環烷基。在某些實施例中,環A係C4
-C7
環烷基。在某些實施例中,環A係雙環[1.1.1]戊烷基。在某些實施例中,環A係選自環丁基、環戊基、環己基及環庚基。
在某些實施例中,環A或部分 係:
係: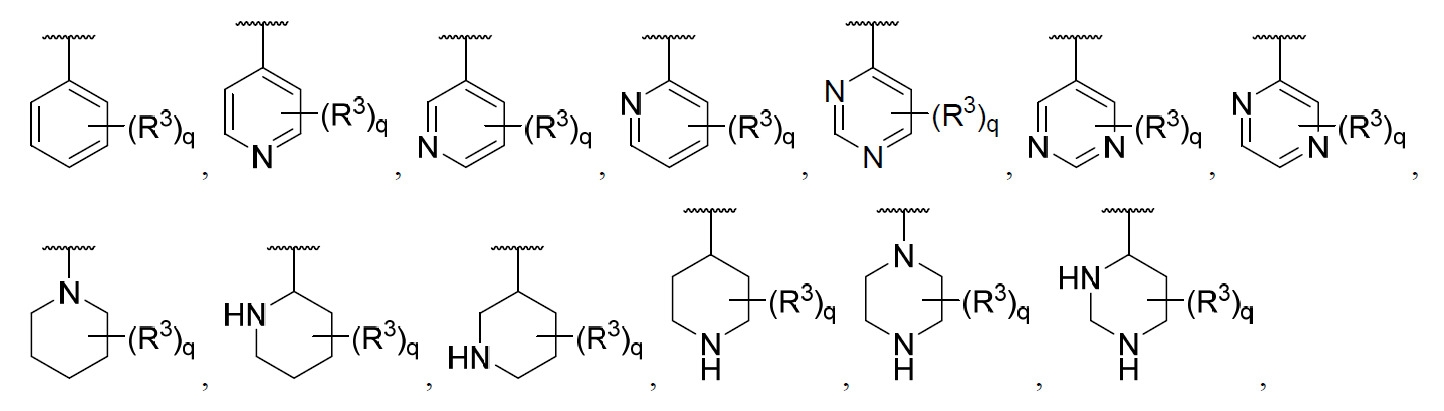
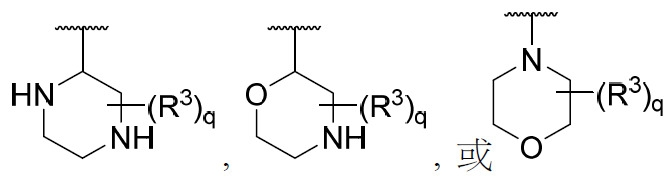 ,其中q及每一R3
獨立地如本文所定義。
,其中q及每一R3
獨立地如本文所定義。
在某些實施例中,環A或部分 係:
係: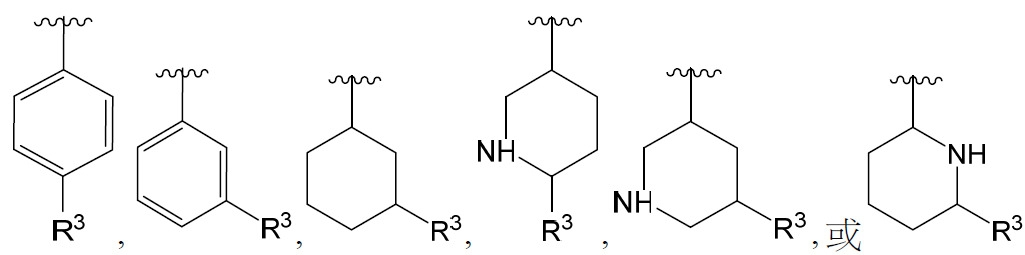 ,其中R3
獨立地如本文所定義。
,其中R3
獨立地如本文所定義。
在某些實施例中,環A係選自以下之橋接雙環:
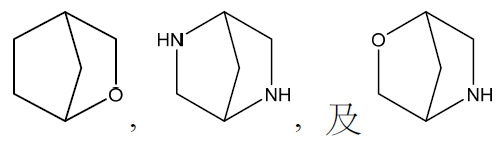 ,其中每一者經一至三個R3
取代。在某些實施例中,環A係選自以下之橋接雙環:
,其中每一者經一至三個R3
取代。在某些實施例中,環A係選自以下之橋接雙環: 其中每一R3
連接至橋接雙環上之碳原子。
其中每一R3
連接至橋接雙環上之碳原子。
在某些實施例中,環A或部分 係:
係: 。
。
亦提供式VIII化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽: VIII
其中R1
、R2
、R3
、R4
、p及q中之每一者獨立地如本文所定義。
VIII
其中R1
、R2
、R3
、R4
、p及q中之每一者獨立地如本文所定義。
亦提供式VIIIA化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽: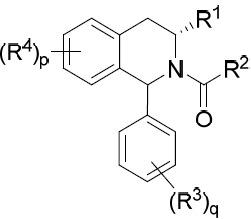 VIIIA
其中R1
、R2
、R3
、R4
、p及q中之每一者獨立地如本文所定義。
VIIIA
其中R1
、R2
、R3
、R4
、p及q中之每一者獨立地如本文所定義。
亦提供式VIIIB化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽: VIIIB
其中R1
、R2
、R3
、R4
、p及q中之每一者獨立地如本文所定義。
VIIIB
其中R1
、R2
、R3
、R4
、p及q中之每一者獨立地如本文所定義。
在某些實施例中,R1
係C1
-C6
烷基、C2
-C6
烯基、C2
-C6
炔基、C1
-C6
鹵代烷基、C3
-C10
環烷基、-CN、-C(O)OR6
、-C(O)N(R7
)2
、-N(R7
)2
、-OR7
或-C1
-C6
烷基-OR7
。
在某些實施例中,R1
係-C(O)OR6
或-C(O)N(R7
)2
。
在某些實施例中,R1
係C1
-C6
烷基。在某些實施例中,在某些實施例中,R1
係C2
-C6
烷基。在某些實施例中,R1
係C3
-C6
烷基。在某些實施例中,R1
係C5
-C6
烷基。在某些實施例中,R1
係C2
-C3
烷基。在某些實施例中,R1
係C4
-C6
烷基。在某些實施例中,R1
係甲基。在某些實施例中,R1
係正丁基。
在某些實施例中,R1
係-CH2
-R16
,其中R16
係C1
-C5
烷基、C2
-C5
烯基、C2
-C5
炔基、C1
-C5
鹵代烷基或-C1
-C5
烷基-OR7
。
在某些實施例中,R1
係C2
-C6
烯基、C2
-C6
炔基、C1
-C6
鹵代烷基、C3
-C10
環烷基、-CN、-OR7
、-C(O)N(R7
)2
、-OC(O)R6
、-S(O)2
R8
、-S(O)2
N(R7
)2
、-S(O)N(R7
)2
、-S(O)R8
、-N(R7
)2
、-NO2
、-C1
-C6
烷基-OR7
或-Si(R15
)3
。
在某些實施例中,R1
不為甲基。在某些實施例中,R1
不為正丁基。在某些實施例中,R1
不為-C(O)OR6
。在某些實施例中,R1
不為-C(O)OCH3
。
亦提供式IX化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽: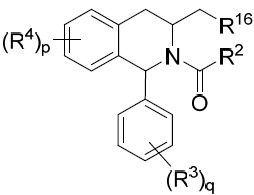 IX
其中R2
、R3
、R4
、R16
、p及q中之每一者獨立地如本文所定義。在某些實施例中,R16
係氫或C2
-C5
烷基。
IX
其中R2
、R3
、R4
、R16
、p及q中之每一者獨立地如本文所定義。在某些實施例中,R16
係氫或C2
-C5
烷基。
亦提供式IXA化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽: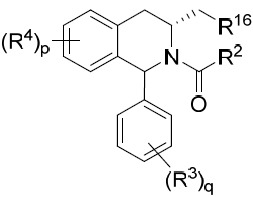 IXA
其中R2
、R3
、R4
、R16
、p及q中之每一者獨立地如本文所定義。在某些實施例中,R16
係氫或C2
-C5
烷基。
IXA
其中R2
、R3
、R4
、R16
、p及q中之每一者獨立地如本文所定義。在某些實施例中,R16
係氫或C2
-C5
烷基。
亦提供式IXB化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽: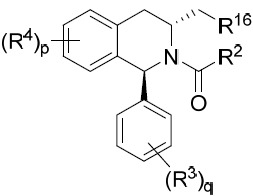 IXB
其中R2
、R3
、R4
、R16
、p及q中之每一者獨立地如本文所定義。在某些實施例中,R16
係氫或C2
-C5
烷基。
IXB
其中R2
、R3
、R4
、R16
、p及q中之每一者獨立地如本文所定義。在某些實施例中,R16
係氫或C2
-C5
烷基。
在某些實施例中,R2
係-C1
-C2
鹵代烷基、-C2
-C3
烯基、-C2
-C3
鹵代烯基、C2
炔基,其中C1
-C2
鹵代烷基及-C2
-C3
烯基鹵基視情況經一或兩個-CH3
取代,且C2
炔基視情況經一個-CH3
取代。在某些實施例中,R2
係-C1
-C2
鹵代烷基。在某些實施例中,R2
係-C2
-C3
烯基。在某些實施例中,R2
係C2
-C3
鹵代烯基。在某些實施例中,R2
係C2
炔基。
在某些實施例中,至少一個R3
係鹵基、-NH2
、-NHR8
、-N(R8
)2
、-S(O)2
R8
、-S(O)R8
、-S(O)2
N(R7
)2
、-S(O)N(R7
)2
、-NO2
、-Si(R12
)3
、-SF5
、-C(O)OR6
、-C(O)N(R7
)2
、-NR12
C(O)R8
、-NR12
C(O)OR8
、-OC(O)R8
、-C(O)R6
或-OC(O)CHR8
N(R12
)2
。
在某些實施例中,至少一個R3
係鹵基。
在某些實施例中,至少一個R3
係NHR8
。在某些實施例中,至少一個R3
係-N(R8
)2
。在某些實施例中,q為2,且一個R3
係鹵基且另一R3
係-N(R8
)2
。在某些實施例中,q為3,且兩個R3
獨立地係鹵基且一個R3
係-N(R8
)2
。
在某些實施例中,至少一個R3
係-C(O)OR6
或-C(O)R6
。
在某些實施例中,至少一個R3
係-S(O)2
N(R7
)2
、-S(O)N(R7
)2
或-C(O)N(R7
)2
。
在某些實施例中,至少一個R3
係-S(O)2
R8
、-S(O)R8
、-NR12
C(O)R8
、-NR12
C(O)OR8
、-OC(O)R8
或-OC(O)CHR8
N(R12
)2
。
在某些實施例中,每一R3
獨立地係鹵基、-CN、-OR8
、-NHR8
、-S(O)2
R8
、-S(O)2
N(R7
)2
、-NO2
、-Si(R12
)3
、-SF5
、-C(O)OR6
、-C(O)N(R7
)2
、-NR12
C(O)R8
、-NR12
C(O)OR8
、-OC(O)R8
、-OC(O)CHR8
N(R12
)2
、C1
-C6
烷基、C3
-C10
環烷基、雜環基、雜芳基或-C1
-C6
烷基雜環基;其中R3
之每一C1
-C6
烷基、C3
-C10
環烷基、雜環基、雜芳基或-C1
-C6
烷基雜環基獨立地視情況經一至三個R10
取代。
在某些實施例中,每一R3
獨立地係鹵基、-CN、-OR8
、-NHR8
、-S(O)2
R8
、-S(O)2
N(R7
)2
、-NO2
、-Si(R12
)3
、-SF5
、-C(O)OR6
、-C(O)N(R7
)2
、-NR12
C(O)R8
、-NR12
C(O)OR8
、-OC(O)R8
、-OC(O)CHR8
N(R12
)2
、C1
-C6
烷基、C3
-C10
環烷基、雜環基、雜芳基或-C1
-C6
烷基雜環基;其中每一C1
-C6
烷基、C3
-C10
環烷基、雜環基、雜芳基或-C1
-C6
烷基雜環基獨立地視情況經一至三個獨立地選自以下之取代基取代:-OR12
、-N(R12
)2
、-S(O)2
R13
、-OC(O)CHR12
N(R12
)2
及視情況經一至三個鹵基、-OR12
、-N(R12
)2
、-Si(R12
)3
、-C(O)OR12
、-NR12
C(O)OR12
、-OC(O)CHR12
N(R12
)2
、C1
-C6
烷基或雜環基取代之C1
-C6
烷基;其中
每一R12
獨立地係氫、C1
-C6
烷基或C3
-C10
環烷基;且
每一R13
獨立地係C1
-C6
烷基或C3
-C10
環烷基。
在某些實施例中,每一R3
獨立地係-NH2
、氟、甲基、吡啶-4-甲醯胺基、吡啶-3-胺基、戊基氧基羰基胺基、N-(3-胺基雙環[1.1.1]戊烷-1-基)胺基、嗎啉-4-基、甲氧基羰基、二甲基胺甲醯基、環丙基胺甲醯基、環己基、環丁基胺甲醯基、環丁基胺基磺醯基、金剛烷基胺基、(金剛烷-1-基胺基)甲基、3-甲基-1,2,4-噁二唑-5-基、2-甲基吡啶-4-甲醯胺基、(雙環[1.1.1]戊烷-1-基胺基)甲基、(金剛烷-1-基)胺甲醯基或(2-甲氧基乙基)胺甲醯基。
在某些實施例中,q為0或1,且R3
係-NH2
、氟、甲基、吡啶-4-甲醯胺基、吡啶-3-胺基、戊基氧基羰基胺基、N-(3-胺基雙環[1.1.1]戊烷-1-基)胺基、嗎啉-4-基、甲氧基羰基、二甲基胺甲醯基、環丙基胺甲醯基、環己基、環丁基胺甲醯基、環丁基胺基磺醯基、金剛烷基胺基、(金剛烷-1-基胺基)甲基、3-甲基-1,2,4-噁二唑-5-基、2-甲基吡啶-4-甲醯胺基、(雙環[1.1.1]戊烷-1-基胺基)甲基、(金剛烷-1-基)胺甲醯基或(2-甲氧基乙基)胺甲醯基。
在某些實施例中,每一R4
獨立地係鹵基、-CN、-OH、-OR8
、-NH2
、-NHR8
、-N(R8
)2
、-S(O)2
R8
、-S(O)R8
、-S(O)2
N(R7
)2
、-S(O)N(R7
)2
、-NO2
、-Si(R15
)3
、-C(O)OR6
、-C(O)N(R7
)2
、-NR12
C(O)R8
、-OC(O)R8
、-C(O)R6
、C1
-C6
烷基、C2
-C6
烯基、C2
-C6
炔基或C3
-C10
環烷基;其中R4
之每一C1
-C6
烷基、C2
-C6
烯基、C2
-C6
炔基或C3
-C10
環烷基獨立地視情況經一至三個R10
取代。
在某些實施例中,每一R4
獨立地係鹵基、-CN、-OR7
、C1
-C6
烷基、C2
-C6
炔基或C3
-C10
環烷基;其中R4
之每一C1
-C6
烷基、C2
-C6
炔基或C3
-C10
環烷基獨立地視情況經一至三個R10
取代。
在某些實施例中,每一R4
獨立地係鹵基、-CN、-OH、C1
-C6
烷基、C2
-C6
炔基或C3
-C10
環烷基。
在某些實施例中,每一R4
獨立地係鹵基、-CN、-OH、-OR8
、C1
-C6
烷基或C2
-C6
炔基;其中R4
之C1
-C6
烷基視情況經一至三個R10
取代。
在某些實施例中,每一R4
獨立地係鹵基、-CN、-OH、-OR8
、C1
-C6
烷基、C2
-C6
炔基;其中R4
之C1
-C6
烷基視情況經一至三個獨立地選自以下之取代基取代:-OR12
、-N(R12
)2
、-S(O)2
R13
、-OC(O)CHR12
N(R12
)2
及視情況經一至三個鹵基、-OR12
、-N(R12
)2
、-Si(R12
)3
、-C(O)OR12
、-NR12
C(O)OR12
、-OC(O)CHR12
N(R12
)2
、C1
-C6
烷基或雜環基取代之C1
-C6
烷基;其中
每一R12
獨立地係氫、C1
-C6
烷基或C3
-C10
環烷基;且
每一R13
獨立地係C1
-C6
烷基或C3
-C10
環烷基。
在某些實施例中,每一R5
獨立地係鹵基、-CN、-OH、-OR8
、-NH2
、-NHR8
、-N(R8
)2
、-S(O)2
R8
、-S(O)R8
、-S(O)2
N(R7
)2
、-S(O)N(R7
)2
、-NO2
、-Si(R15
)3
、-C(O)OR6
、-C(O)N(R7
)2
、-NR12
C(O)R8
、-OC(O)R8
、-C(O)R6
、C1
-C6
烷基、C2
-C6
烯基、C2
-C6
炔基或C3
-C10
環烷基;其中R5
之每一C1
-C6
烷基、C2
-C6
烯基、C2
-C6
炔基或C3
-C10
環烷基獨立地視情況經一至三個R10
取代。
在某些實施例中,每一R5
獨立地係鹵基、-CN、-OR7
、C1
-C6
烷基、C2
-C6
炔基或C3
-C10
環烷基;其中R5
之每一C1
-C6
烷基、C2
-C6
炔基或C3
-C10
環烷基獨立地視情況經一至三個R10
取代。
在某些實施例中,每一R5
獨立地係鹵基、-CN、-OH、C1
-C6
烷基、C2
-C6
炔基或C3
-C10
環烷基。
在某些實施例中,每一R5
獨立地係鹵基、-CN、-OH、-OR8
、C1
-C6
烷基或C2
-C6
炔基;其中R5
之C1
-C6
烷基視情況經一至三個R10
取代。
在某些實施例中,每一R5
獨立地係鹵基、-CN、-OH、-OR8
、C1
-C6
烷基、C2
-C6
炔基;其中R5
之C1
-C6
烷基視情況經一至三個獨立地選自以下之取代基取代:-OR12
、-N(R12
)2
、-S(O)2
R13
、-OC(O)CHR12
N(R12
)2
及視情況經一至三個鹵基、-OR12
、-N(R12
)2
、-Si(R12
)3
、-C(O)OR12
、-NR12
C(O)OR12
、-OC(O)CHR12
N(R12
)2
、C1
-C6
烷基或雜環基取代之C1
-C6
烷基;其中
每一R12
獨立地係氫、C1
-C6
烷基或C3
-C10
環烷基;且
每一R13
獨立地係C1
-C6
烷基或C3
-C10
環烷基。
在某些實施例中,每一R6
獨立地係氫、C1
-C6
烷基、C2
-C6
烯基或-C1
-C6
烷基C3
-C10
環烷基;其中每一R6
獨立地進一步經一至三個R11
取代。
在某些實施例中,每一R6
獨立地係氫、C1
-C6
烷基、C2
-C6
烯基或-C1
-C6
烷基C3
-C10
環烷基;其中每一R6
獨立地進一步經一至三個鹵基、-OR12
、-N(R12
)2
、-Si(R12
)3
、-C(O)OR12
、-NR12
C(O)OR12
、-OC(O)CHR12
N(R12
)2
、C1
-C6
烷基或雜環基取代;其中
每一R12
獨立地係氫、C1
-C6
烷基或C3
-C10
環烷基。
在某些實施例中,每一R7
獨立地係氫、C1
-C6
烷基、C3
-C10
環烷基、雜環基、雜芳基、-C1
-C6
烷基C3
-C6
環烷基、-C1
-C6
烷基雜環基,或兩個R7
與其所連接之氮原子一起形成4至7員雜環基;其中每一R7
或由此形成之環獨立地進一步經一至三個R11
取代。
在某些實施例中,每一R7
獨立地係氫、C1
-C6
烷基、C3
-C10
環烷基、雜環基、雜芳基、-C1
-C6
烷基C3
-C6
環烷基、-C1
-C6
烷基雜環基,或兩個R7
與其所連接之氮原子一起形成4至7員雜環基;其中每一R7
或由此形成之環獨立地進一步經一至三個鹵基、-OR12
、-N(R12
)2
、-Si(R12
)3
、-C(O)OR12
、-NR12
C(O)OR12
、-OC(O)CHR12
N(R12
)2
、C1
-C6
烷基或雜環基取代;其中
每一R12
獨立地係氫、C1
-C6
烷基或C3
-C10
環烷基。
在某些實施例中,每一R8
獨立地係C1
-C6
烷基、C2
-C6
炔基、C3
-C10
環烷基、-C1
-C6
烷基C3
-C10
環烷基或-C1
-C6
烷基芳基;其中每一R8
獨立地進一步經一至三個R11
取代。
在某些實施例中,每一R8
獨立地係C1
-C6
烷基、C2
-C6
炔基、C3
-C10
環烷基、-C1
-C6
烷基C3
-C10
環烷基或-C1
-C6
烷基芳基;其中每一R8
獨立地進一步經一至三個鹵基、-OR12
、-N(R12
)2
、-Si(R12
)3
、-C(O)OR12
、-NR12
C(O)OR12
、-OC(O)CHR12
N(R12
)2
、C1
-C6
烷基或雜環基取代;其中
每一R12
獨立地係氫、C1
-C6
烷基或C3
-C10
環烷基。
在某些實施例中,每一R10
獨立地係-OR12
、-N(R12
)2
、-S(O)2
R13
、-OC(O)CHR12
N(R12
)2
或C1
-C6
烷基,其中R10
之C1
-C6
烷基視情況獨立地經一至三個R11
取代;
每一R11
獨立地係鹵基、-OR12
、-N(R12
)2
、-Si(R12
)3
、-C(O)OR12
、-NR12
C(O)OR12
、-OC(O)CHR12
N(R12
)2
、C1
-C6
烷基或雜環基;
每一R12
獨立地係氫、C1
-C6
烷基或C3
-C10
環烷基;且
每一R13
獨立地係C1
-C6
烷基或C3
-C10
環烷基。
在某些實施例中,每一R15
獨立地係C1
-C6
烷基。
在某些實施例中,p為0。在某些實施例中,p為0或1。在某些實施例中,p為1或2。在某些實施例中,p為1。在某些實施例中,p為2。
在某些實施例中,q為0。在某些實施例中,q為0或1。在某些實施例中,q為1或2。在某些實施例中,q為1。在某些實施例中,q為2。在某些實施例中,q為3。
亦提供選自表1之化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽:表 1 

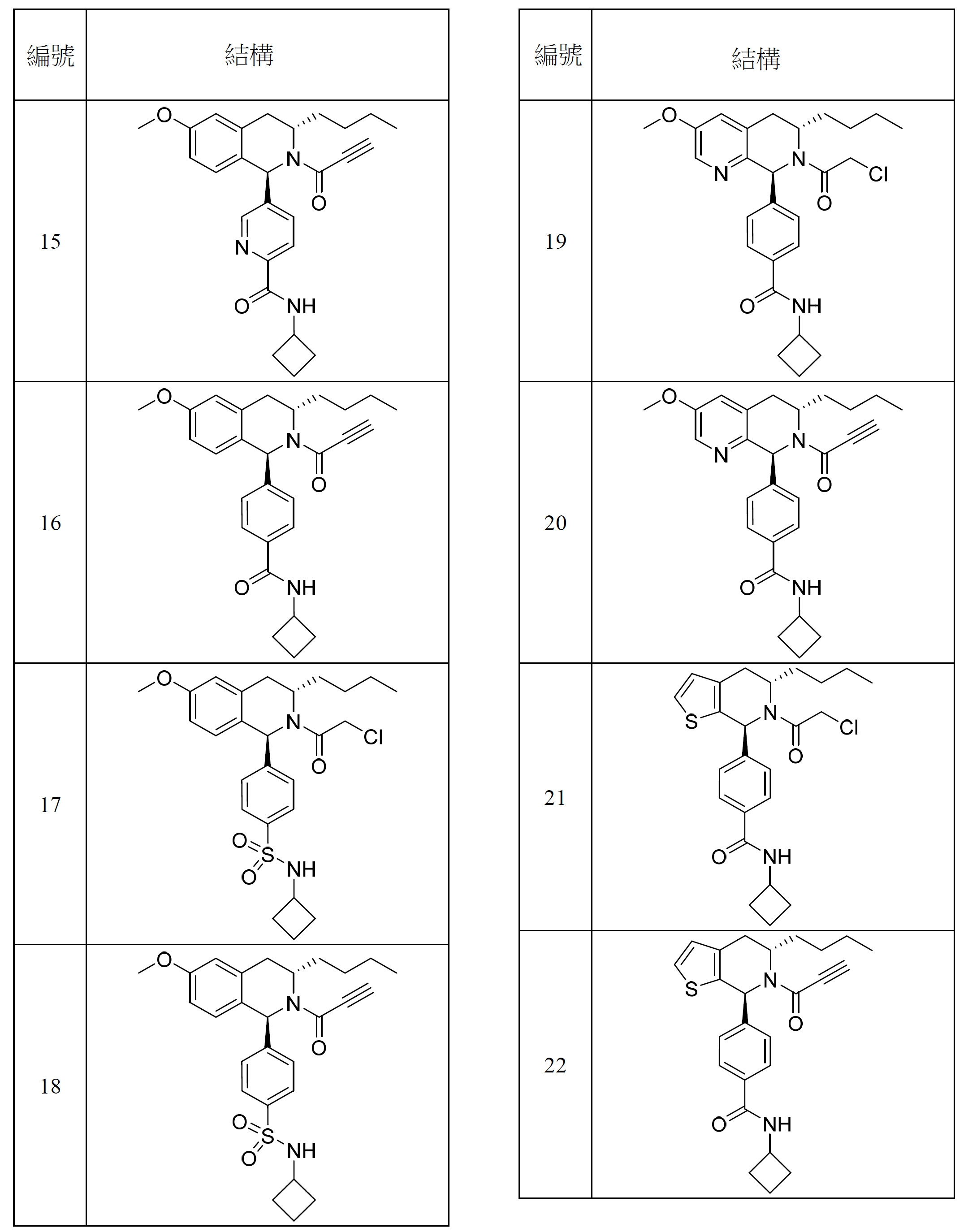
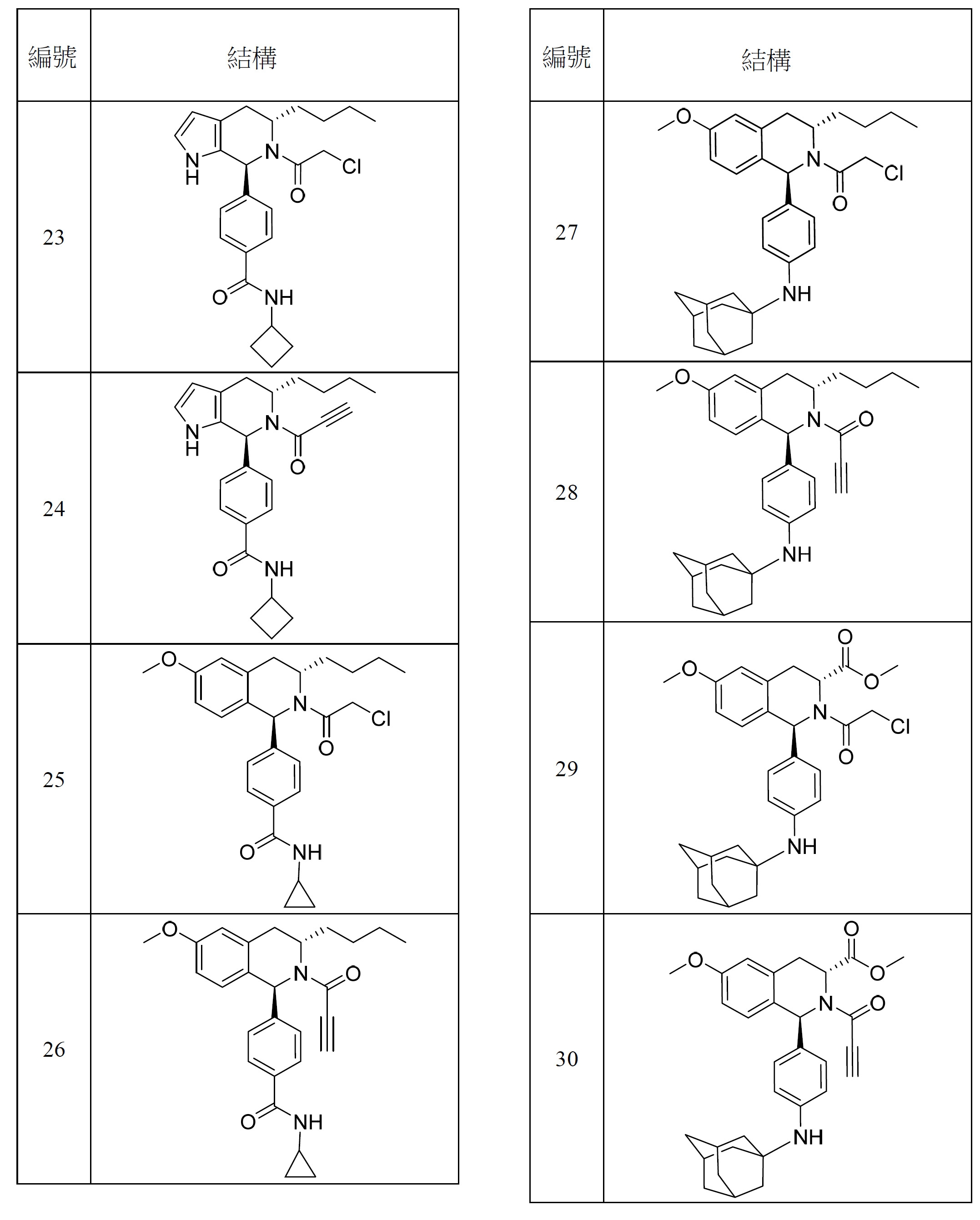
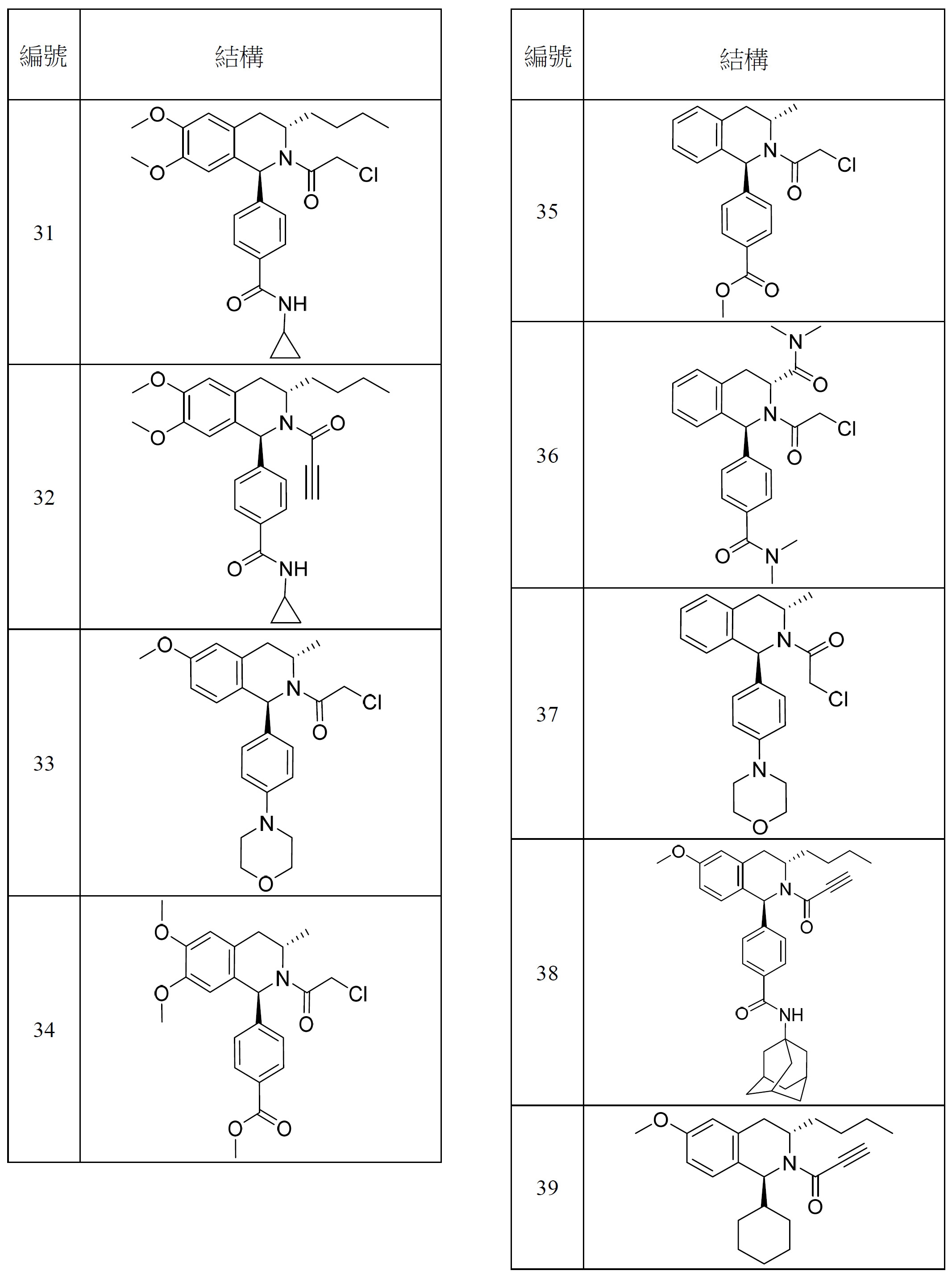
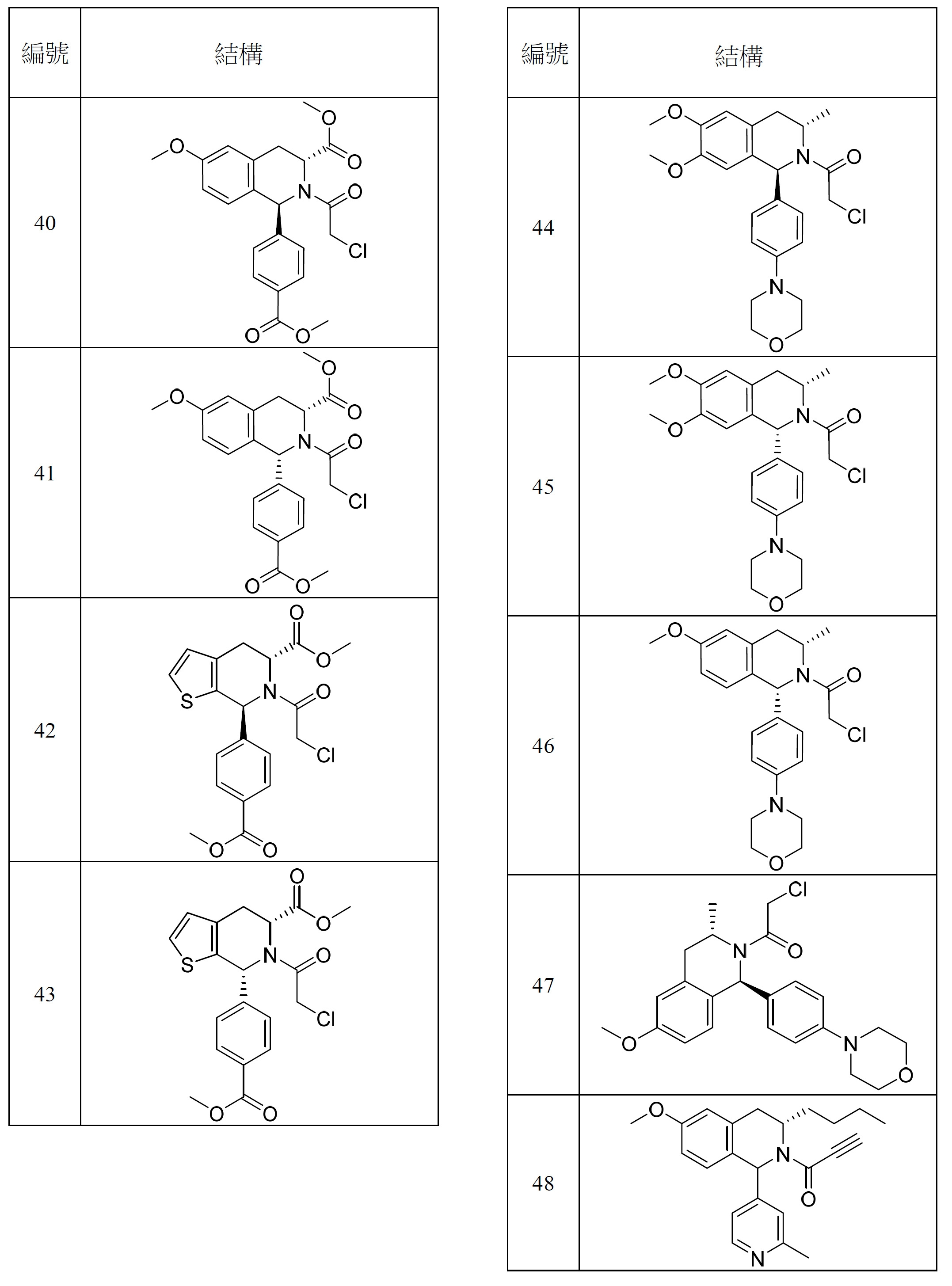
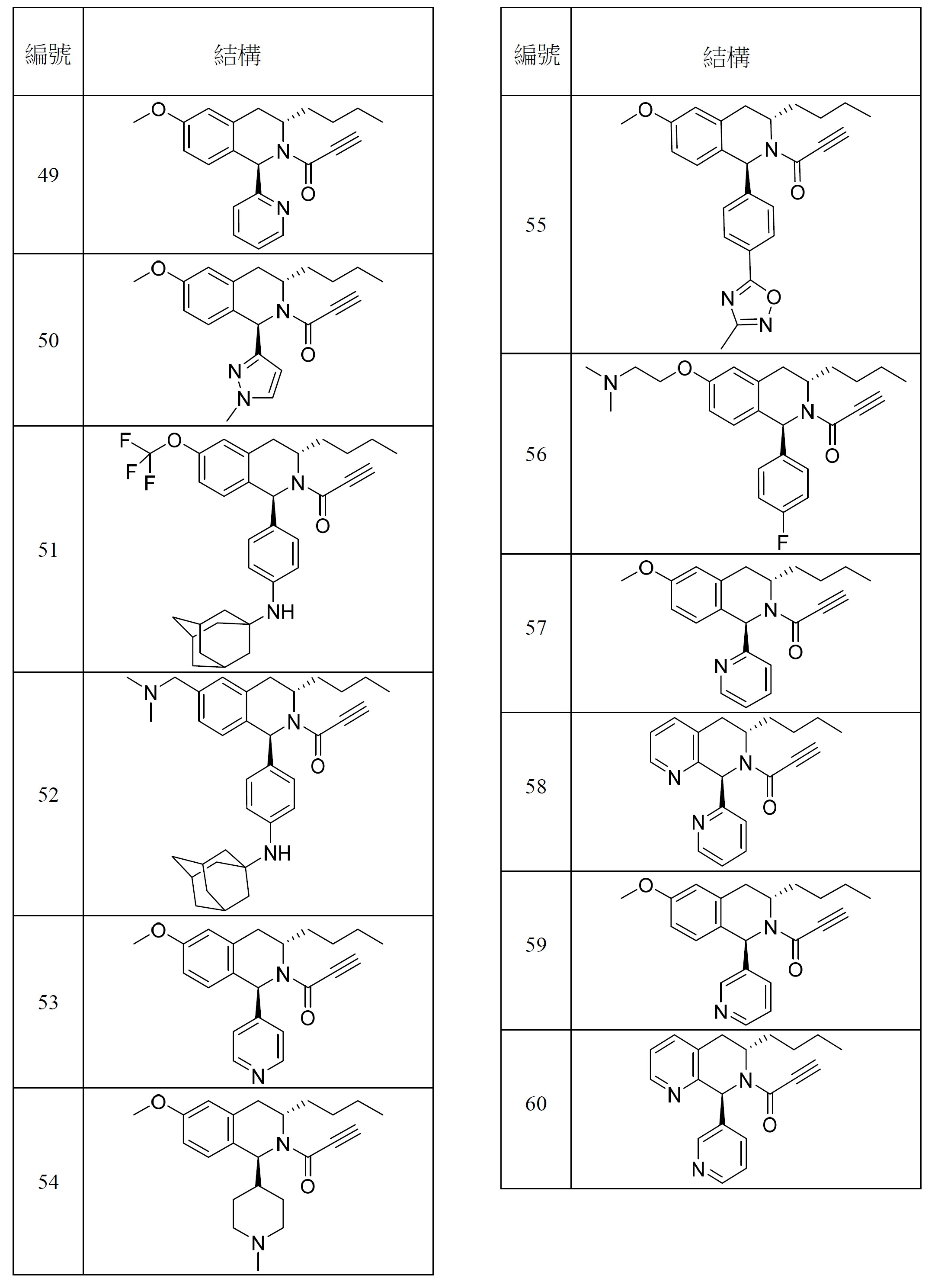
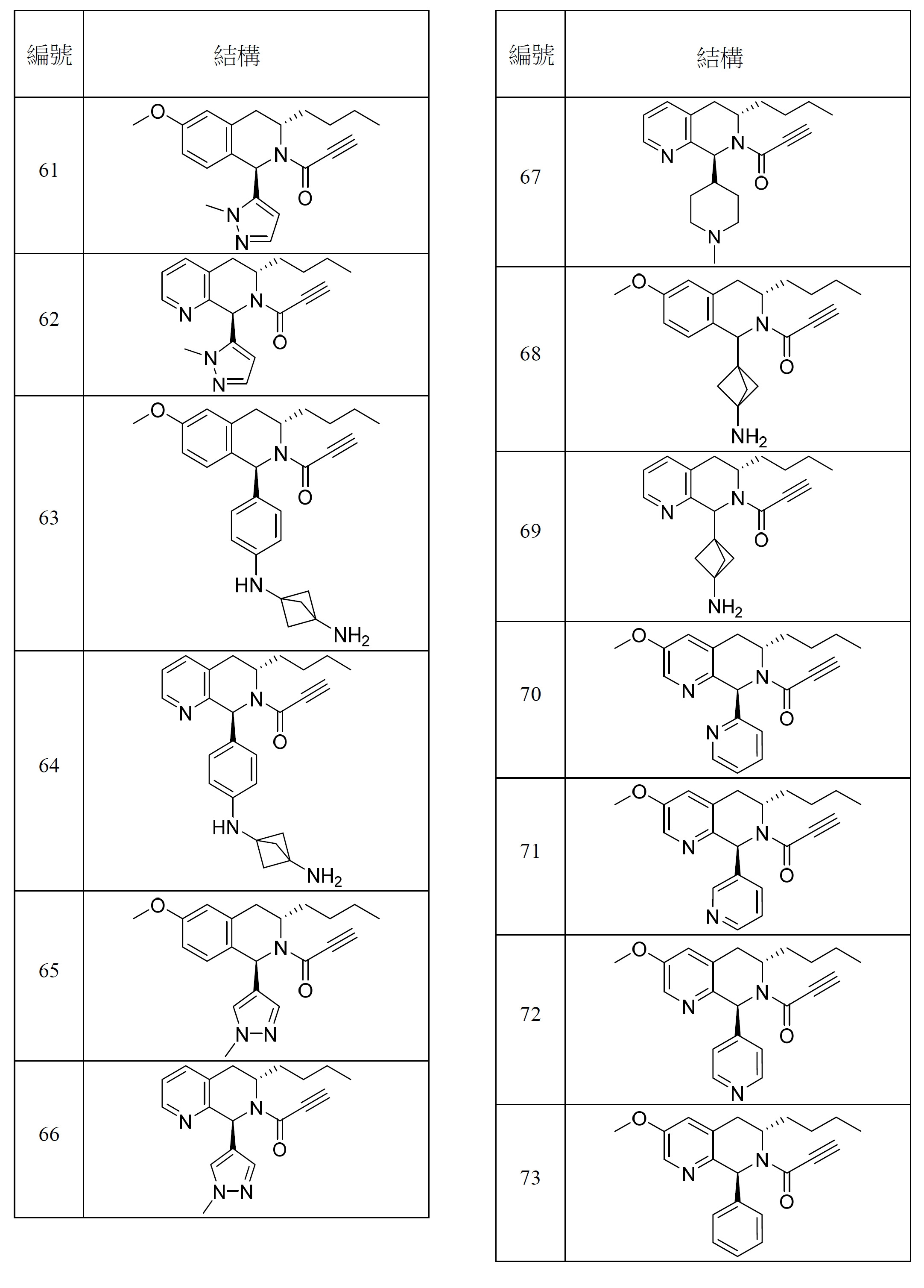

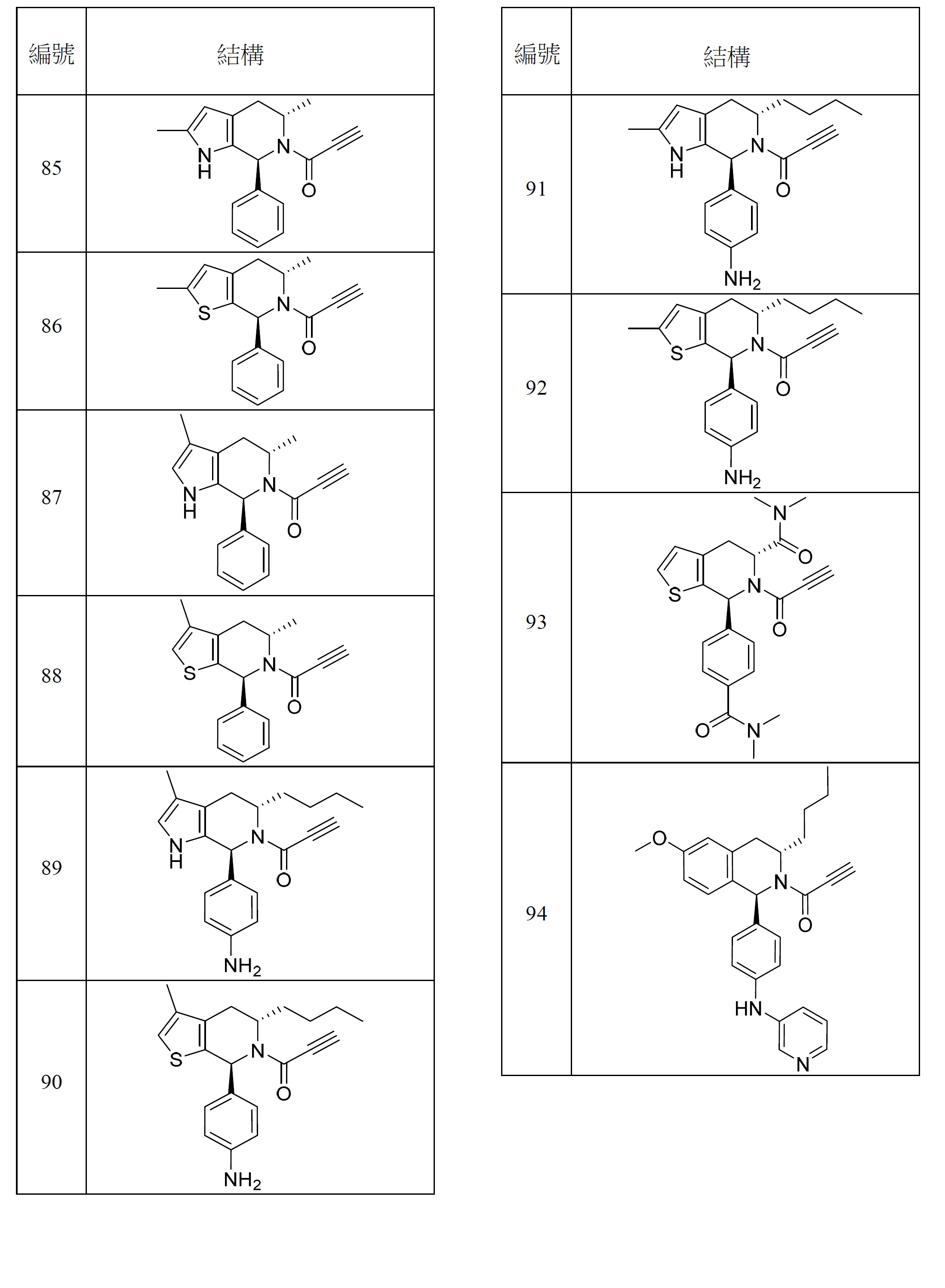
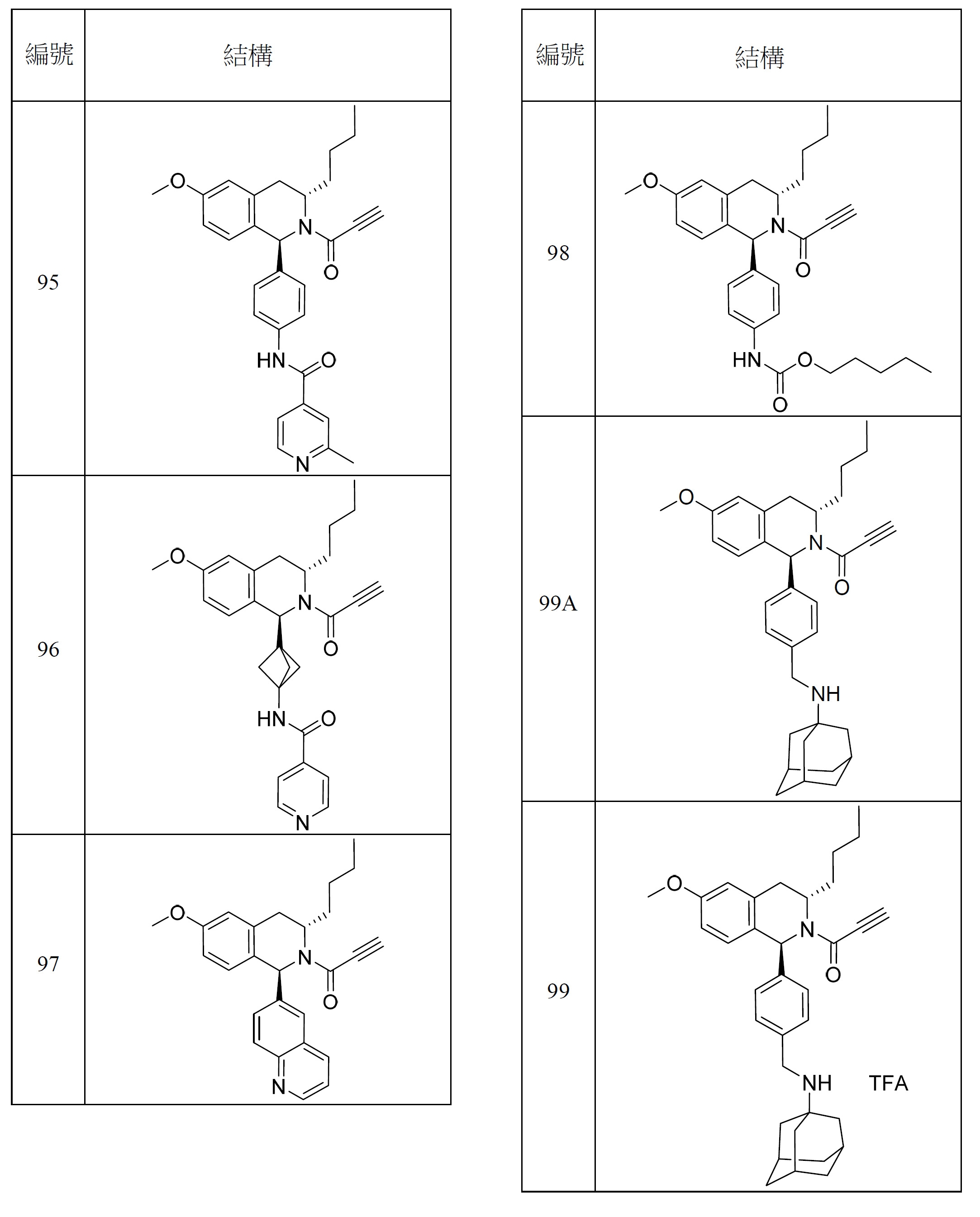
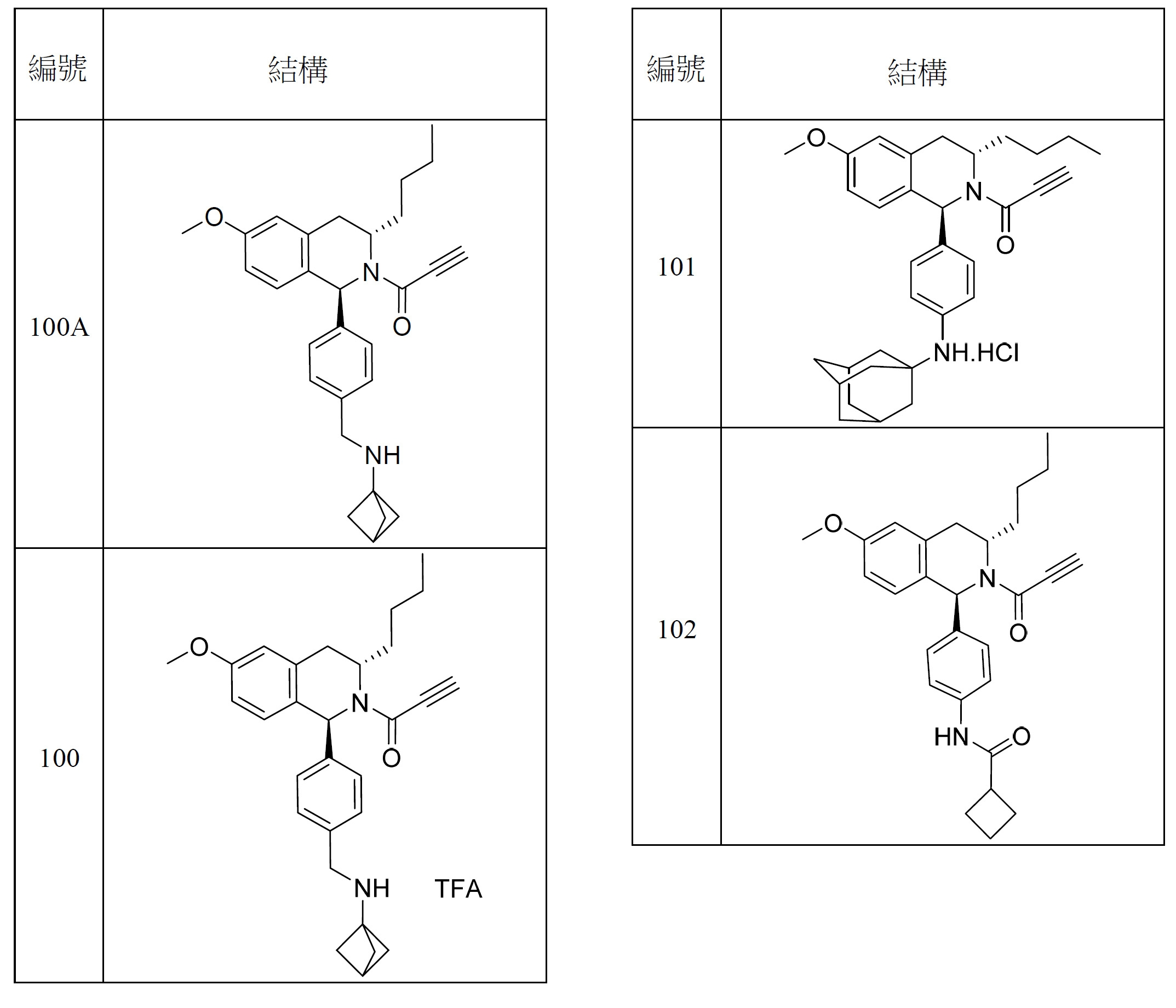
3. 使用方法
在某些實施例中,本文所闡述之化合物係用於治療癌症之方法中。在某些實施例中,治療癌症之方法包括向有需要之個體投與治療有效量之本文所闡述之任一化合物。
在某些實施例中,該等化合物係用於抑制細胞中之GPX4之方法中,該方法包括使細胞與有效量之本文所闡述之化合物或組合物接觸以抑制細胞中之GPX4。在某些實施例中,細胞係癌細胞。在某些實施例中,該方法包括向有需要之患者投與有效量之本文所闡述之化合物或組合物。
在某些實施例中,該等化合物係用於誘導細胞中之鐵依賴型細胞死亡之方法中,該方法包括使細胞與有效量之本文所提供之化合物或組合物接觸。在某些實施例中,該方法包括向有需要之患者投與有效量之本文所闡述之化合物或組合物。
在某些實施例中,提供治療有需要之患者之癌症之方法,其包括投與有效量之本文所提供之化合物或組合物。
在某些實施例中,該等化合物係用於治療有需要之個體之癌症之方法中,該方法包括向患有癌症之個體投與治療有效量之本文所揭示之鐵依賴型細胞死亡誘導化合物。用於使用該等化合物治療之各種癌症包含(但不限於)腎上腺皮質癌、肛門癌、膽管癌、膀胱癌、骨癌、神經膠質瘤、星形細胞瘤、神經母細胞瘤、乳癌、子宮頸癌、結腸癌、子宮內膜癌、食道癌、頭頸癌、腸癌、肝癌、肺癌、口腔癌、卵巢癌、胰臟癌、腎癌、前列腺癌、唾液腺癌、皮膚癌、胃癌、睪丸癌、喉癌、甲狀腺癌、子宮癌、陰道癌、肉瘤及軟組織癌。在某些實施例中,使用該等化合物來治療胰臟癌。
在某些實施例中,癌症係腎細胞癌(RCC)、胰臟癌、肺癌、乳癌或前列腺癌。在某些實施例中,提供治療有需要之患者之腎細胞癌(RCC)之方法,其包括投與有效量之本文所提供之化合物或組合物。在某些實施例中,提供治療有需要之患者之胰臟癌之方法,其包括投與有效量之本文所提供之化合物或組合物。在某些實施例中,提供治療有需要之患者之肺癌之方法,其包括投與有效量之本文所提供之化合物或組合物。在某些實施例中,提供治療有需要之患者之乳癌之方法,其包括投與有效量之本文所提供之化合物或組合物。在某些實施例中,提供治療有需要之患者之前列腺癌之方法,其包括投與有效量之本文所提供之化合物或組合物。
在某些實施例中,提供治療有需要之患者之惡性實體腫瘤之方法,其包括向患者投與有效量之本文所提供之化合物或組合物。在某些實施例中,惡性實體腫瘤係癌。在某些實施例中,惡性實體腫瘤係淋巴瘤。在某些實施例中,惡性實體腫瘤係肉瘤。
在某些實施例中,用於使用該化合物治療之癌症可尤其選自腎上腺皮質癌、肛門癌、膽管癌、膀胱癌、骨癌(例如骨肉瘤)、腦癌(例如神經膠質瘤、星形細胞瘤、神經母細胞瘤等)、乳癌、子宮頸癌、結腸癌、子宮內膜癌、食道癌、頭頸癌、血液學癌症(例如白血病及淋巴瘤)、腸癌(小腸)、肝癌、肺癌(例如支氣管癌、小細胞肺癌、非小細胞肺癌等)、口腔癌、卵巢癌、胰臟癌、腎癌、前列腺癌、唾液腺癌、皮膚癌(例如基底細胞癌、黑色素瘤)、胃癌、睪丸癌、喉癌、甲狀腺癌、子宮癌、陰道癌、肉瘤及軟組織癌。在某些實施例中,癌症係腎細胞癌(RCC)。在某些實施例中,癌症係胰臟癌。在某些實施例中,癌症係肺癌。在某些實施例中,癌症係乳癌。在某些實施例中,癌症係前列腺癌。
在某些實施例中,用於使用該化合物治療之癌症係胰臟癌。在某些實施例中,用於使用該等化合物治療之胰臟癌係胰臟腺癌或轉移性胰臟癌。在某些實施例中,用於使用該等化合物治療之癌症係I期、II期、III期或IV期胰臟腺癌。
在某些實施例中,用於使用該等化合物治療之癌症係肺癌。在某些實施例中,用於使用該等化合物治療之肺癌係小細胞肺癌或非小細胞肺癌。在某些實施例中,用於使用該等化合物治療之非小細胞肺癌係腺癌、鱗狀細胞癌或大細胞癌。在某些實施例中,用於使用該等化合物治療之肺癌係轉移性肺癌。
在某些實施例中,用於使用該等化合物治療之癌症係血液學癌症。在某些實施例中,血液學癌症係選自急性淋巴母細胞性白血病(ALL)、急性骨髓樣白血病(AML)、淋巴瘤(例如何傑金氏淋巴瘤(Hodgkin’s lymphoma)、非何傑金氏淋巴瘤(Non-Hodgkin’s lymphoma)、伯基特氏淋巴瘤(Burkitt's lymphoma))、慢性淋巴球性白血病(CLL)、慢性骨髓性白血病(CML)、毛細胞慢性骨髓性白血病(CML)及多發性骨髓瘤。
在某些實施例中,用於使用該等化合物治療之癌症係選自急性淋巴母細胞性白血病(ALL)、急性骨髓樣白血病(AML)、慢性淋巴球性白血病(CLL)、慢性骨髓性白血病(CML)、毛細胞慢性骨髓性白血病(CML)之白血病及多發性骨髓瘤。
在某些實施例中,用於使用該化合物治療之癌症係選自何傑金氏淋巴瘤、非何傑金氏淋巴瘤及伯基特氏淋巴瘤之淋巴瘤。
在某些實施例中,用於使用該化合物治療之癌症係特徵在於間質特徵或間質表型之癌症。在一些癌症中,間質特徵之獲得與癌症之遷移(例如內滲)及侵襲性有關。間質特徵可尤其包含增強之遷移能力、侵襲性、升高之抗細胞凋亡性及增加之細胞外基質(ECM)組分產生。除該等生理學特性外,間質特徵亦可包含某些生物標記物之表現,該等生物標記物尤其包含E-鈣黏蛋白、N-鈣黏蛋白、整聯蛋白、FSP-1、α-SMA、波形蛋白(vimentin)、β-連環蛋白、膠原I、膠原II、膠原III、膠原IV、纖連蛋白、層黏蛋白5、SNAIL-1、SNAIL-2、Twist-1、Twist-2及Lef-1。在某些實施例中,選擇用於使用本文化合物治療之癌症尤其包含乳癌、肺癌、頭頸癌、前列腺癌及結腸癌。在某些實施例中,間質特徵可為癌症類型所固有或由使用化學療法及/或輻射療法之癌症治療所誘導或選擇用於該治療。
在某些實施例中,用於使用該化合物治療之癌症鑑別為具有或經測定具有活化或致癌RAS活性。在某些實施例中,RAS係K-RAS、H-RAS或N-RAS。在某些實施例中,活化或致癌RAS係活化或致癌RAS突變。
在某些實施例中,選擇用於使用該等化合物治療之癌症經測定具有或鑑別為具有活化或致癌RAS活性。在某些實施例中,活化或致癌RAS活性係活化或致癌RAS突變。在某些實施例中,活化或致癌RAS活性係活化或活化K-RAS活性、尤其活化或致癌K-RAS突變。在某些實施例中,活化或致癌RAS活性係活化或活化N-RAS活性、尤其活化或致癌N-RAS突變。在某些實施例中,活化或致癌RAS活性係活化或活化H-RAS活性、尤其活化或致癌H-RAS突變。
在某些實施例中,該等化合物可用於治療一或多種其他化學治療劑、尤其細胞毒性化學治療劑之難治性癌症;或治療抵抗輻射治療之癌症 。在某些實施例中,該等化合物用於治療已對活化其他細胞死亡路徑(例如細胞凋亡、有絲分裂驟變、壞死、衰老及/或自體吞噬)之化學治療劑產生耐受性之癌症。
在某些實施例中,用於使用該等化合物治療之癌症鑑別為化學療法難治性或抗性。在某些實施例中,癌症對於以下中之一或多者而言係難治性或抗性:烷基化劑、抗癌抗生素劑、抗代謝劑(例如葉酸鹽拮抗劑、嘌呤類似物、嘧啶類似物等)、拓撲異構酶抑制劑、抗微管劑(例如紫杉烷(taxane)、長春花生物鹼)、激素劑(例如芳香酶抑制劑)、植物源藥劑及其合成衍生物、抗血管生成劑、分化誘導劑、細胞生長停滯誘導劑、細胞凋亡誘導劑、細胞毒性劑、影響細胞生物能學(亦即影響細胞ATP含量及調控該等含量之分子/活性)之藥劑、生物劑(例如單株抗體)、激酶抑制劑以及生長因子及其受體之抑制劑。
在某些實施例中,用於使用該等化合物治療之癌症係鑑別為對以下中之一或多者而言係難治性或抗性之癌症:阿法替尼(afatinib)、阿來替布(afuresertib)、阿雷替尼(alectinib)、阿立塞替(alisertib)、阿伏昔地(alvocidib)、安吖啶(amsacrine)、胺萘非特(amonafide)、阿姆替尼(amuvatinib)、阿西替尼(axitinib)、阿紮胞苷(azacitidine)、硫唑嘌呤(azathioprine)、巴非替尼(bafetinib)、巴拉澤替(barasertib)、苯達莫司汀(bendamustine)、博來黴素(bleomycin)、伯舒替尼(bosutinib)、硼替佐米(bortezomib)、白消安(busulfan)、卡博替尼(cabozantinib)、喜樹鹼(camptothecin)、卡奈替尼(canertinib)、卡培他濱(capecitabine)、卡巴他賽(cabazitaxel)、卡鉑(carboplatin)、卡莫司汀(carmustine)、西尼澤替(cenisertib)、塞瑞替尼(ceritinib)、氮芥苯丁酸(chlorambucil)、順鉑(cisplatin)、克拉屈濱(cladribine)、氯法拉濱(clofarabine)、克萊拉尼(crenolanib)、克裡唑蒂尼(crizotinib)、環磷醯胺(cyclophosphamide)、阿糖胞苷(cytarabine)、達拉非尼(dabrafenib)、達卡巴嗪(dacarbazine)、達克替尼(dacomitinib)、更生黴素(dactinomycin)、達努替普(danusertib)、達沙替尼(dasatinib)、柔紅黴素(daunorubicin)、地西他濱(decitabine)、地那西布(dinaciclib)、多西他賽(docetaxel)、多韋替尼(dovitinib)、多柔比星(doxorubicin)、表柔比星(epirubicin)、依吡替尼(epitinib)、甲磺酸埃雷布林(eribulin mesylate)、埃羅替尼(errlotinib)、依諾替康(etirinotecan)、依託泊苷(etoposide)、依維莫司(everolimus)、依西美坦(exemestane)、氟尿苷(floxuridine)、氟達拉濱(fludarabine)、氟尿嘧啶(fluorouracil)、吉非替尼(gefitinib)、吉西他濱(gemcitabine)、羥基脲(hydroxyurea)、依魯替尼(ibrutinib)、埃克替尼(icotinib)、伊達比星(idarubicin)、異環磷醯胺(ifosfamide)、伊馬替尼(imatinib)、伊美司他(imetelstat)、依帕替布(ipatasertib)、伊立替康(irinotecan)、伊沙匹隆(ixabepilone)、拉帕替尼(lapatinib)、來那度胺(lenalidomide)、來他替尼(lestaurtinib)、洛莫司汀(lomustine)、盧西他尼(lucitanib)、馬賽替尼(masitinib)、雙氯乙基甲胺(mechlorethamine)、美法侖(melphalan)、巰基嘌呤(mercaptopurine)、胺甲喋呤(methotrexate)、米哚妥林(midostaurin)、絲裂黴素(mitomycin)、米托蒽醌(mitoxantrone)、木利替尼(mubritinib)、奈拉濱(nelarabine)、奈拉替尼(neratinib)、尼羅替尼(nilotinib)、尼達尼布(nintedanib)、美琥他辛(omacetaxine mepesuccinate)、奧蘭替尼(orantinib)、奧沙利鉑(oxaliplatin)、太平洋紫杉醇(paclitaxel)、帕博西利(palbociclib)、帕利伐米斯(palifosfamide tris)、帕唑帕尼(pazopanib)、培利替尼(pelitinib)、培美曲塞(pemetrexed)、噴司他汀(pentostatin)、普卡黴素(plicamycin)、普納替尼(ponatinib)、波齊替尼(poziotinib)、普拉曲沙(pralatrexate)、丙卡巴肼(procarbazine)、奎紮替尼(quizartinib)、雷替曲塞(raltitrexed)、瑞格菲尼(regorafenib)、魯索替尼(ruxolitinib)、塞利西裡(seliciclib)、索拉菲尼(sorafenib)、鏈脲菌素(streptozocin)、索凡替尼(sulfatinib)、舒尼替尼(sunitinib)、他莫昔芬(tamoxifen)、坦度替尼(tandutinib)、替莫唑胺(temozolomide)、替西羅莫司(temsirolimus)、替尼泊苷(teniposide)、塞來替尼(theliatinib)、硫鳥嘌呤(thioguanine)、噻替派(thiotepa)、托泊替康(topotecan)、烏拉莫司汀(uramustine)、戊柔比星(valrubicin)、凡德他尼(vandetanib)、威羅菲尼(vemurafenib) (Zelborae)、長春新鹼(vincristine)、長春鹼(vinblastine)、長春瑞濱(vinorelbine)及長春地辛(vindesine)。
在某些實施例中,用於使用該化合物治療之癌症鑑別為對一或多種選自以下之化學治療劑係難治性或抗性:環磷醯胺、氮芥苯丁酸、美法侖、雙氯乙基甲胺、異環磷醯胺、白消安、洛莫司汀、鏈脲菌素、替莫唑胺、達卡巴嗪、順鉑、卡鉑、奧沙利鉑、丙卡巴肼、烏拉莫司汀、胺甲喋呤、培美曲塞、氟達拉濱、阿糖胞苷、氟尿嘧啶、氟尿苷、吉西他濱、卡培他濱、長春鹼、長春新鹼、長春瑞濱、依託泊苷、太平洋紫杉醇、多西他賽、多柔比星、柔紅黴素、表柔比星、伊達比星、米托蒽醌、博來黴素、絲裂黴素、羥基脲、托泊替康、伊立替康、安吖啶、替尼泊苷及埃羅替尼。
在某些實施例中,用於使用該等化合物治療之癌症係抵抗離子化輻射療法之癌症。癌症之抗輻射性可為輻射療法所固有或自其產生。在某些實施例中,用於使用該化合物治療之癌症尤其係抗輻射性腎上腺皮質癌、肛門癌、膽管癌、膀胱癌、骨癌(例如骨肉瘤)、腦癌(例如神經膠質瘤、星形細胞瘤、神經母細胞瘤等)、乳癌、子宮頸癌、結腸癌、子宮內膜癌、食道癌、頭頸癌、血液學癌症(例如白血病及淋巴瘤)、腸癌(小腸)、肝癌、肺癌(例如支氣管癌、小細胞肺癌、非小細胞肺癌等)、口腔癌、卵巢癌、胰臟癌、腎癌、前列腺癌、唾液腺癌、皮膚癌(例如基底細胞癌、黑色素瘤)、胃癌、睪丸癌、喉癌、甲狀腺癌、子宮癌或陰道癌。在某些實施例中,癌症係胰臟癌、乳癌、神經膠母細胞瘤、晚期非小細胞肺癌、膀胱癌、肉瘤或軟組織癌。
4. 組合治療
在某些實施例中,組合使用本文所闡述之化合物與一或多種其他(例如第二治療劑)治療性治療來治療癌症。在某些實施例中,該等化合物可作為單一療法來使用,或如下文進一步所提供以與一或多種治療性治療之組合療法形式來使用,尤其與一或多種化學治療劑進行組合。在某些實施例中,組合使用該等化合物與第二治療劑,其中該等化合物係以使癌症或癌細胞對第二治療劑敏化之含量(例如以不引起顯著細胞死亡之化合物含量)來使用。在某些實施例中,該等化合物可與輻射療法組合使用以使細胞對輻射療法敏化或輔助於輻射療法(例如以足以活化細胞死亡路徑之劑量)。
在某些實施例中,使用本文所闡述之化合物及輻射療法之組合來治療患有癌症之個體。在某些實施例中,該方法包括向患有癌症之個體投與治療有效量之本發明化合物,及使用有效量之輻射療法輔助性治療個體。在某些實施例中,在使用輻射治療之前、同時或之後將化合物投與有需要之個體。
在某些實施例中,該方法包括向患有癌症之個體投與有效量之本文所闡述之化合物以使癌症對輻射治療敏化,及投與治療有效量之輻射療法以治療癌症。在某些實施例中,向個體投與有效量之X射線及γ射線。在某些實施例中,向個體投與有效量之粒子輻射,其中粒子輻射係選自電子束、質子束及中子束輻射。在某些實施例中,輻射療法係分次的。
在某些實施例中,向患有癌症之個體投與治療有效量之本文所闡述之化合物或其第一醫藥組合物,且輔助性投與治療有效量之第二化學治療劑及其第二醫藥組合物。
在某些實施例中,第二化學治療劑係選自鉑類藥劑、烷基化劑、抗癌抗生素劑、抗代謝劑(例如葉酸鹽拮抗劑、嘌呤類似物、嘧啶類似物等)、拓撲異構酶I抑制劑、拓撲異構酶II抑制劑、抗微管劑(例如紫杉烷、長春花生物鹼)、激素劑(例如芳香酶抑制劑)、植物源藥劑及其合成衍生物、抗血管生成劑、分化誘導劑、細胞生長停滯誘導劑、細胞凋亡誘導劑、細胞毒性劑、影響細胞生物能學(亦即影響細胞ATP含量及調控該等含量之分子/活性)之藥劑、抗癌生物劑(例如單株抗體)、激酶抑制劑以及生長因子及其受體之抑制劑。
在某些實施例中,第二化學治療劑係血管生成抑制劑,例如(但不限於)以下各項之抑制劑:可溶性VEGFR-1、NRP-1、血管生成素2、TSP-1、TSP-2、血管抑素(angiostatin)及相關分子、內皮抑素、血管抑制因子(vasostatin)、鈣網織蛋白、血小板因子-4、TIMP、CDAI、Meth-1、Meth-2、IFN-α、IFN-β、IFN-γ、CXCL10、IL-4、IL-12、IL-18、凝血酶原(三環結構域-2)、抗凝血酶III片段、泌乳素、VEGI、SPARC、骨橋蛋白、乳腺絲胺酸蛋白酶抑制劑(maspin)、癌抑素
(COL4A2之片段)或多育麴菌素相關蛋白。在某些實施例中,血管生成抑制劑係貝伐珠單抗(bevacizumab) (Avastin)、伊曲康唑(itraconazole)、羧基醯胺基三唑、TNP-470 (煙麴黴素(fumagillin)類似物)、CM101、IFN-α、IL-12、血小板因子-4、舒拉明(suramin)、SU5416、凝血酶敏感蛋白(thrombospondin)、VEGFR拮抗劑、血管抑制類固醇+肝素、軟骨源血管生成抑制因子(CDAI)、基質金屬蛋白酶抑制劑、血管抑素、內皮抑素、2-甲氧基雌二醇、替康蘭(tecogalan)、四硫鉬酸鹽、沙立度胺(thalidomide)、凝血酶敏感蛋白、泌乳素、αVβ3抑制劑、利諾胺(linomide)、雷莫蘆單抗(ramucirumab)、他喹莫德(tasquinimod)、蘭尼單抗(ranibizumab)、索拉菲尼(Nexavar)、舒尼替尼(Sutent)、帕唑帕尼(Votrient)或依維莫司(Afinitor)。
在某些實施例中,第二化學治療劑係細胞週期蛋白依賴性激酶(CDK)抑制劑(例如CDK4/CDK6抑制劑)。實例包含(但不限於)帕博西利(Ibrance)、瑞博西利(Ribociclib) (視情況進一步與來曲唑(letrozole)組合)、阿貝西利(abemaciclib) (LY2835219;Verzenio)、P1446A-05及特拉西利(Trilaciclib) (G1T28)。
在某些實施例中,第二化學治療劑係布魯頓氏酪胺酸激酶(Bruton's tyrosine kinase,BTK)抑制劑,例如(但不限於)依魯替尼(PCI-32765)、阿卡替尼(acalabrutinib)、ONO-4059 (GS-4059)、司培替尼(spebrutinib) (AVL-292, CC-292)、BGB-3111及HM71224。
在某些實施例中,第二化學治療劑係BRAF抑制劑。實例包含(但不限於) BAY43-9006 (索拉菲尼,Nexavar)、PLX-4032 (威羅菲尼)、GDC-0879、PLX-4720、達拉非尼及LGX818。
在某些實施例中,第二化學治療劑係EGFR抑制劑。實例包含(但不限於)吉非替尼、埃羅替尼、阿法替尼、布吉替尼(brigatinib)、埃克替尼、西妥昔單抗(cetuximab)、奧希替尼(osimertinib)、帕尼單抗(panitumumab)、布吉替尼、拉帕替尼、cimaVax-EGF及維爾拉特(veristrat)。
在某些實施例中,第二化學治療劑係人類表皮生長因子受體2 (HER2)抑制劑。實例包含(但不限於)曲妥珠單抗(trastuzumab)、帕妥珠單抗(pertuzumab) (視情況進一步與曲妥珠單抗組合)、馬妥昔單抗(margetuximab)及NeuVax。
在某些實施例中,本文揭示增加個體對免疫治療性或免疫原性化學治療劑之反應性之方法,該方法包括向有需要之個體投與有效量之本文所闡述之化合物及有效量之免疫治療劑及/或免疫原性化學治療劑。在某些實施例中,該方法進一步包含向個體投與脂氧合酶抑制劑。在某些實施例中,個體具有細胞微環境富集基質細胞之腫瘤。在某些實施例中,投與本文所闡述之化合物可殺死腫瘤細胞微環境中之一或多種基質細胞。在某些實施例中,投與有效量之免疫治療劑及/或免疫原性化學治療劑可殺死一或多種腫瘤細胞。本文亦提供包括本文所闡述之化合物及免疫治療劑、脂氧合酶抑制劑或免疫原性化學治療劑之組合。在某些實施例中,免疫治療劑係選自CTLA4、PDL1或PD1抑制劑。在某些實施例中,免疫治療劑可選自CTLA4抑制劑(例如伊匹單抗(ipilimumab))、PD1抑制劑(例如派姆單抗(pembrolizumab)或尼沃魯單抗(nivolumab))或PDL1抑制劑(例如阿替珠單抗(atezolizumab)或德瓦魯單抗(durvalumab))。在某些實施例中,免疫治療劑係派姆單抗。在其他實施例中,免疫原性化學治療劑係選自蒽環、多柔比星、環磷醯胺、太平洋紫杉醇、多西他賽、順鉑、奧沙利鉑或卡鉑之化合物。在某些實施例中,本文提供包括本文所闡述之化合物及脂氧合酶抑制劑之組合。在某些實施例中,脂氧合酶抑制劑係選自PD147176及/或ML351。在某些實施例中,脂氧合酶抑制劑可為15-脂氧合酶抑制劑(例如參見Sadeghian等人,Expert Opinion on Therapeutic Patents, 2015, 26:1, 65-88)。
在某些實施例中,第二化學治療劑係選自烷基化劑,包含(但不限於)阿多來新(adozelesin)、六甲蜜胺(altretamine)、苯達莫司汀、比折來新(bizelesin)、白消安、卡鉑、卡波醌(carboquone)、卡莫氟(carmofur)、卡莫司汀、氮芥苯丁酸、順鉑、環磷醯胺、達卡巴嗪、雌氮芥(estramustine)、依託格魯(etoglucid)、福莫司汀(fotemustine)、合普舒凡(hepsulfam)、異環磷醯胺、英丙舒凡(improsulfan)、伊洛福芬(irofulven)、洛莫司汀、甘露舒凡(mannosulfan)、雙氯乙基甲胺、美法侖、二溴甘露醇(mitobronitol)、奈達鉑(nedaplatin)、尼莫司汀(nimustine)、奧沙利鉑、哌泊舒凡(piposulfan)、潑尼莫司汀(prednimustine)、丙卡巴肼、雷莫司汀(ranimustine)、沙鉑(satraplatin)、司莫司汀(semustine)、鏈脲菌素、替莫唑胺、噻替派、曲奧舒凡(treosulfan)、三亞胺醌(triaziquone)、三乙烯三聚氰胺、四硝酸三鉑、曲磷胺(trofosphamide)及烏拉莫司汀;抗生素,包含(但不限於)阿柔比星(aclarubicin)、胺柔比星(amrubicin)、博來黴素、更生黴素、柔紅黴素、多柔比星、依沙蘆星(elsamitrucin)、表柔比星、伊達比星、美諾立爾(menogaril)、絲裂黴素、新製癌菌素(neocarzinostatin)、噴司他汀、吡柔比星(pirarubicin)、普卡黴素、戊柔比星及佐柔比星(zorubicin);抗代謝物,包含(但不限於)胺喋呤(aminopterin)、阿紮胞苷、硫唑嘌呤、卡培他濱、克拉屈濱、氯法拉濱、阿糖胞苷、地西他濱、氟尿苷、氟達拉濱、5-氟尿嘧啶、吉西他濱、羥基脲、巰基嘌呤、胺甲喋呤、奈拉濱、培美曲塞、雷替曲塞、替加氟-尿嘧啶(tegafur-uracil)、硫鳥嘌呤、甲氧苄啶(trimethoprim)、三甲曲沙(trimetrexate)及阿糖腺苷(vidarabine);免疫療法;抗體療法,包含(但不限於)阿倫單抗(alemtuzumab)、貝伐珠單抗、西妥昔單抗、加利昔單抗(galiximab)、吉妥珠單抗(gemtuzumab)、帕尼單抗、帕妥珠單抗、利妥昔單抗(rituximab)、貝倫妥單抗(brentuximab)、托西莫單抗(tositumomab)、曲妥珠單抗、90 Y替伊莫單抗(90 Y ibritumomab tiuxetan)、伊匹單抗、曲美目單抗(tremelimumab)及抗CTLA-4抗體;激素或激素拮抗劑,包含(但不限於)阿那曲唑(anastrozole)、雄激素、布舍瑞林(buserelin)、己烯雌酚(diethylstilbestrol)、依西美坦、氟他胺(flutamide)、氟維司群(fulvestrant)、戈舍瑞林(goserelin)、艾多昔芬(idoxifene)、來曲唑、柳培林(leuprolide)、甲地孕酮(magestrol)、雷洛昔芬(raloxifene)、他莫昔芬及托瑞米芬(toremifene);紫杉烷,包含(但不限於) DJ-927、多西他賽、TPI 287、拉洛他賽(larotaxel)、奧他賽(ortataxel)、太平洋紫杉醇、DHA-太平洋紫杉醇及替司他賽(tesetaxel);類視色素,包含(但不限於)阿曲諾英(alitretinoin)、貝沙羅汀(bexarotene)、芬維a胺(fenretinide)、異維A酸(isotretinoin)及維A酸(tretinoin);生物鹼,包含(但不限於)秋水仙胺(demecolcine)、高三尖杉酯鹼(homoharringtonine)、長春鹼、長春新鹼、長春地辛、長春氟寧(vinflunine)及長春瑞濱;抗血管生成劑,包含(但不限於) AE-941 (GW786034,新伐司他(Neovastat))、ABT-510、2-甲氧基雌二醇、來那度胺及沙立度胺;拓撲異構酶抑制劑,包含(但不限於)安吖啶、貝洛替康(belotecan)、伊多替卡(edotecarin)、依託泊苷、磷酸依託泊苷(etoposide phosphate)、依沙替康(exatecan)、伊立替康(亦活性代謝物SN-38 (7-乙基-10-羥基-喜樹鹼))、硫蒽酮(lucanthone)、米托蒽醌、匹杉瓊(pixantrone)、盧比替康(rubitecan)、替尼泊苷、托泊替康及9-胺基喜樹鹼;激酶抑制劑,包含(但不限於)阿西替尼(AG 013736)、達沙替尼(BMS 354825)、埃羅替尼、吉非替尼、夫拉平度(flavopiridol)、甲磺酸伊馬替尼(imatinib mesylate)、拉帕替尼、二磷酸莫替沙尼(motesanib diphosphate) (AMG 706)、尼羅替尼(nilotinib) (AMN107)、塞利西裡、索拉菲尼、蘋果酸舒尼替尼(sunitinib malate)、AEE-788、BMS-599626、UCN-01 (7-羥基星狀孢菌素)、威羅菲尼、達拉非尼、司美替尼(selumetinib)、悖論粉碎劑(paradox breaker) (例如PLX8394或PLX7904)、LGX818、BGB-283、培西達替尼(pexidartinib) (PLX3397)及瓦他拉尼(vatalanib);靶向信號轉導抑制劑,包含(但不限於)硼替佐米、格爾德黴素(geldanamycin)及雷帕黴素(rapamycin);生物反應改良劑,包含(但不限於)咪喹莫特(imiquimod)、干擾素-α及介白素-2;及其他化學治療劑,包含(但不限於) 3-AP (3-胺基-2-羧基醛縮胺基硫脲)、阿曲生坦(altrasentan)、胺格魯米特(aminoglutethimide)、阿那格雷(anagrelide)、天門冬醯胺酶、苔蘚蟲素-1(bryostatin-1)、西侖吉肽(cilengitide)、伊利司莫(elesclomol)、甲磺酸埃雷布林(E7389)、伊沙匹隆(ixabepilone)、氯尼達明(lonidamine)、馬索羅酚(masoprocol)、米托胍腙(mitoguanazone)、奧利默森(oblimersen)、舒林酸(sulindac)、睪內酯(testolactone)、噻唑呋啉(tiazofurin)、mTOR抑制劑(例如西羅莫司(sirolimus)、替西羅莫司、依維莫司、地伏莫司(deforolimus)、INK28、AZD8055、PI3K抑制劑(例如BEZ235、GDC-0941、XL147、XL765、BMK120)、細胞週期蛋白依賴性激酶(CDK)抑制劑(例如CDK4抑制劑或CDK6抑制劑,例如帕博西利(PD-0332991)、利伯西利(Ribocyclib) (LEE011)、阿貝西利(LY2835219)、P1446A-05、阿貝西利(LY2835219)、特拉西利(G1T28)等)、AKT抑制劑、Hsp90抑制劑(例如格爾德黴素、根赤殼菌素(radicicol)、坦螺旋黴素(tanespimycin))、法尼基轉移酶抑制劑(例如替吡法尼(tipifarnib))、芳香酶抑制劑(阿那曲唑-來曲唑-依西美坦);MEK抑制劑,包含(但不限於) AS703026、AZD6244 (司美替尼)、AZD8330、BIX 02188、CI-1040 (PD184352)、GSK1120212 (亦稱為曲美替尼(trametinib)或JTP-74057)、克美替尼(cobimetinib)、PD0325901、PD318088、PD98059、RDEA119 (BAY 869766)、TAK-733及U0126-EtOH;酪胺酸激酶抑制劑,包含(但不限於) AEE788、AG-1478 (酪胺酸磷酸化抑制劑(Tyrphostin),AG-1478)、AG-490、阿帕替尼(Apatinib) (YN968D1)、AV-412、AV-951(替肟紮尼(Tivozanib))、阿西替尼、AZD8931、BIBF1120 (瓦格替夫(Vargatef))、BIBW2992 (阿法替尼)、BMS794833、BMS-599626、布立尼布(Brivanib) (BMS-540215)、丙胺酸布立尼布(Brivanib alaninate) (BMS-582664)、西地尼布(Cediranib) (AZD2171)、大黃苷酸(Chrysophanic acid) (大黃酚(Chrysophanol))、克萊拉尼(CP-868569)、CUDC-101、CYC116、多韋替尼二乳酸(Dovitinib Dilactic acid) (TKI258二乳酸)、E7080、埃羅替尼鹽酸鹽(Erlotinib Hydrochloride)(Tarceva,CP-358774、OSI-774、NSC-718781)、弗雷替尼(Foretinib) (GSK1363089、XL880)、吉非替尼(ZD-1839或Iressa)、伊馬替尼(Gleevec)、甲磺酸伊馬替尼、Ki8751、KRN 633、拉帕替尼(Tykerb)、利尼伐尼(Linifanib) (ABT-869)、馬賽替尼(Masivet, AB1010)、MGCD-265、莫替沙尼(Motesanib) (AMG-706)、MP-470、木利替尼(TAK 165)、奈拉替尼(HKI-272)、NVP-BHG712、OSI-420 (去甲基埃羅替尼(Desmethyl Erlotinib)、CP-473420)、OSI-930、帕唑帕尼HCl、PD-153035 HCl、PD173074、培利替尼(EKB-569)、PF299804、普納替尼(AP24534)、PP121、RAF265 (CHIR-265)、Raf265衍生物、瑞格菲尼(BAY 73-4506)、甲苯磺酸索拉菲尼(Sorafenib Tosylate) (Nexavar)、蘋果酸舒尼替尼(Sutent)、替拉替尼(Telatinib) (BAY 57-9352)、TSU-68 (SU6668)、凡德他尼(Zactima)、瓦他拉尼二鹽酸鹽(Vatalanib dihydrochloride) (PTK787)、WZ3146、WZ4002、WZ8040、奎紮替尼、卡博替尼、XL647、EGFR siRNA、FLT4 siRNA、KDR siRNA、抗糖尿病劑(例如二甲雙胍(metformin))、PPAR激動劑(羅格列酮(rosiglitazone)、吡格列酮(pioglitazone)、苯紮貝特(bezafibrate)、環丙貝特(ciprofibrate)、氯貝特(clofibrate)、吉非羅齊(gemfibrozil)、非諾貝特(fenofibrate)、因格列紮(indeglitazar))、DPP4抑制劑(西他列汀(sitagliptin)、維格列汀(vildagliptin)、沙格列汀(saxagliptin)、度格列汀(dutogliptin)、吉格列汀(gemigliptin)、阿格列汀(alogliptin))或EGFR抑制劑(包含(但不限於) AEE-788、AP-26113、BIBW-2992 (Tovok)、CI-1033、GW-572016、Iressa、LY2874455、RO-5323441、Tarceva (埃羅替尼,OSI-774)、CUDC-101及WZ4002)。
在某些實施例中,第二化學治療劑係選自阿法替尼、阿來替布、阿雷替尼、阿立塞替、阿伏昔地、安吖啶、胺萘非特、阿姆替尼、阿西替尼、阿紮胞苷、硫唑嘌呤、巴非替尼、巴拉澤替、苯達莫司汀、博來黴素、伯舒替尼、硼替佐米、白消安、卡博替尼、喜樹鹼、卡奈替尼、卡培他濱、卡巴他賽、卡鉑、卡莫司汀、西尼澤替、塞瑞替尼、氮芥苯丁酸、順鉑、克拉屈濱、氯法拉濱、克萊拉尼、克裡唑蒂尼、環磷醯胺、阿糖胞苷、達拉非尼、達卡巴嗪、達克替尼、更生黴素、達努替普、達沙替尼、柔紅黴素、地西他濱、地那西布、多西他賽、多韋替尼、多柔比星、表柔比星、依吡替尼、甲磺酸埃雷布林、埃羅替尼、依諾替康、依託泊苷、依維莫司、依西美坦、氟尿苷、氟達拉濱、氟尿嘧啶、吉非替尼、吉西他濱、羥基脲、依魯替尼、埃克替尼、伊達比星、艾代拉裡斯(idelalisib)、異環磷醯胺、伊馬替尼、伊美司他、依帕替布、伊立替康、伊沙匹隆、拉帕替尼、來那度胺、來他替尼、洛莫司汀、盧西他尼、馬賽替尼、雙氯乙基甲胺、美法侖、巰基嘌呤、胺甲喋呤、米哚妥林、絲裂黴素、米托蒽醌、木利替尼、奈拉濱、奈拉替尼、尼羅替尼、尼達尼布、美琥他辛、奧拉帕尼(olaparib)、奧蘭替尼、奧沙利鉑、太平洋紫杉醇、帕博西利、帕利伐米斯、帕唑帕尼、培利替尼、培美曲塞、噴司他汀、普卡黴素、普納替尼、波齊替尼、普拉曲沙、丙卡巴肼、奎紮替尼、雷替曲塞、瑞格菲尼、魯索替尼、塞利西裡、索拉菲尼、鏈脲菌素、索凡替尼、舒尼替尼、他莫昔芬、坦度替尼、替莫唑胺、替西羅莫司、替尼泊苷、塞來替尼、硫鳥嘌呤、噻替派、托泊替康、烏拉莫司汀、戊柔比星、凡德他尼、威羅菲尼(Zelboraf)、長春新鹼、長春鹼、長春瑞濱、長春地辛及諸如此類。在某些實施例中,在使用化學治療劑治療之前、同時或之後投與本文之化合物。
在某些實施例中,治療癌症之方法包括投與治療有效量之本文所闡述之化合物及治療有效量之用於治療癌症的生物劑。在某些實施例中,生物劑係選自抗BAFF (例如貝利木單抗(belimumab));抗CCR4 (例如莫加珠單抗(mogamulizumab));抗CD19/CD3 (例如比林莫單抗(blinatumomab));抗CD20 (例如奧妥珠單抗(obinutuzumab)、利妥昔單抗、替伊莫單抗、奧法木單抗(ofatumumab)、托西莫單抗);抗CD22 (例如帕西妥莫單抗(moxetumomab pasudotox));抗CD30 (例如貝倫妥單抗-維多汀(brentuximab vedotin));抗CD33 (例如吉妥珠單抗);抗CD37 (例如奧樂妥珠單抗(otlertuzumab));抗CD38 (例如達雷木單抗(daratumumab));抗CD52 (例如阿倫單抗);抗CD56 (例如羅沃珠單抗-莫登素(lorvotuzumab mertansine));抗CD74 (例如米拉珠單抗(milatuzumab));抗CD105;抗CD248 (TEM1) (例如恩妥昔單抗(ontuxizumab));抗CTLA4 (例如曲美目單抗、伊匹單抗);抗EGFL7 (例如帕圖珠單抗(parsatuzumab));抗EGFR (HER1/ERBB1) (例如帕尼單抗、尼妥珠單抗(nimotuzumab)、奈昔木單抗(necitumumab)、西妥昔單抗、英加妥珠單抗(imgatuzumab)、弗妥昔單抗(futuximab));抗FZD7 (例如凡體妥單抗(vantictumab));抗HER2 (ERBB2/neu) (例如馬妥昔單抗、帕妥珠單抗、阿多-曲妥珠單抗艾坦辛(ado-trastuzumab emtansine)、曲妥珠單抗);抗HER3 (ERBB3);抗HGF(例如利妥木單抗(rilotumumab)、芬克拉妥珠單抗(ficlatuzumab));抗IGF-1R (例如蓋尼塔單抗(ganitumab)、芬妥木單抗(figitumumab)、西妥木單抗(cixutumumab)、達洛珠單抗(dalotuzumab));抗IGF-2R;抗KIR (例如利利單抗(lirilumab)、昂妥珠單抗(onartuzumab));抗MMP9;抗PD-1 (例如尼沃魯單抗、匹利珠單抗(pidilizumab)、蘭布魯珠單抗(lambrolizumab));抗PD-L1 (例如阿替珠單抗);抗PDGFRa (例如雷莫蘆單抗、特維妥單抗(tovetumab));抗PD-L2;抗PIGF (例如ziv-阿柏西普(ziv-aflibercept));抗RANKL (例如地諾單抗(denosumab));抗TNFRSF 9 (CD 137/4-1 BB) (例如烏瑞魯單抗(urelumab));抗TRAIL-RI /DR4、R2/D5 (例如杜拉樂明(dulanermin));抗TRAIL-R1/D4 (例如馬帕木單抗(mapatumumab));抗TRAIL-R2/D5 (例如可那木單抗(conatumumab)、來沙木單抗(lexatumumab)、阿波單抗(apomab));抗VEGFA (例如貝伐珠單抗、ziv-阿柏西普);抗VEGFB (例如ziv-阿柏西普);及抗VEGFR2 (例如雷莫蘆單抗)。
5. 調配物及投與
在某些實施例中,可藉由標準技術使用一或多種生理上可接受之載劑或賦形劑來調配化合物之醫藥組合物。適宜醫藥載劑闡述於本文及Remington: The Science and Practice of Pharmacy,第21版(2005)中。治療性化合物及其生理上可接受之鹽、水合物及溶劑合物可經調配以藉由任一適宜途徑(尤其包含經局部、經鼻、經口、非經腸、經直腸或藉由吸入)來投與。在某些實施例中,可藉由真皮內、真皮下、靜脈內、肌內、鼻內、大腦內、氣管內、動脈內、腹膜腔內、膀胱內、胸膜內、冠狀動脈內或腫瘤內注射使用注射器或其他器件來投與醫藥組合物。亦涵蓋穿皮式投與以及吸入或氣溶膠投與。可經口、經直腸或經陰道投與錠劑、膠囊及溶液。
對於經口投與而言,醫藥組合物可採用(例如)藉由習用方式使用醫藥上可接受之賦形劑製得之錠劑或膠囊形式。可製備包括活性成分以及諸如以下等賦形劑之錠劑及膠囊:(a)稀釋劑或填充劑,例如乳糖、右旋糖、蔗糖、甘露醇、山梨醇、纖維素(例如乙基纖維素、微晶纖維素)、甘胺酸、果膠、聚丙烯酸酯及/或磷酸氫鈣、硫酸鈣;(b)潤滑劑,例如二氧化矽、滑石粉、硬脂酸、其鎂或鈣鹽、金屬硬脂酸鹽、膠質二氧化矽、氫化植物油、玉米澱粉、苯甲酸鈉、乙酸鈉及/或聚乙二醇;(c)黏合劑,例如矽酸鎂鋁、澱粉膏糊、明膠、黃蓍膠、甲基纖維素、羧甲基纖維素鈉、聚乙烯基吡咯啶酮及/或羥丙基甲基纖維素;(d)崩解劑,例如澱粉(包含馬鈴薯澱粉或澱粉鈉)、羥基乙酸鹽、瓊脂、海藻酸或其鈉鹽或泡騰混合物;(e)潤濕劑,例如月桂基硫酸鈉;及/或(f)吸收劑、著色劑、矯味劑及甜味劑。根據習用混合、粒化或塗覆方法來製備組合物。
在某些實施例中,載劑係(例如)用以增強本文化合物之溶解性及/或生物可用性之環糊精。在某些實施例中,用於醫藥組合物中之環糊精可選自α-環糊精、β-環糊精、γ-環糊精、其衍生物及其組合。在某些實施例中,環糊精係選自β-環糊精、γ-環糊精、其衍生物及其組合。
在某些實施例中,該等化合物可與選自羧基烷基環糊精、羥基烷基環糊精、磺基烷基醚環糊精及烷基環糊精之環糊精或其衍生物一起調配。在各個實施例中,環糊精中之烷基係甲基、乙基、丙基、丁基或戊基。
在與本發明化合物一起用於調配物中時,可存在約0.1 w/v至約30% w/v、約0.1 w/v至約20% w/v、約0.5% w/v至約10% w/v或約1% w/v至約5% w/v之環糊精。在某些實施例中,存在約0.1% w/v、約0.2% w/v、約0.5% w/v、約1% w/v、約2% w/v、約3% w/v、約4% w/v、約5% w/v、約6% w/v、約7% w/v、約8% w/v、約9% w/v、約10% w/v、約12% w/v、約14% w/v、約16% w/v、約18% w/v、約20% w/v、約25% w/v或約30% w/v或更多之環糊精。
錠劑可根據業內已知方法進行膜包衣或腸包衣。用於經口投與之液體製劑可採用(例如)溶液、糖漿或懸浮液之形式,或可將其呈遞為乾燥產物以供在使用前使用水或其他適宜媒劑重構。該等液體製劑可藉由習用方式使用醫藥上可接受之載劑及添加劑來製備,例如懸浮劑(例如山梨醇糖漿、纖維素衍生物或氫化食用脂肪);乳化劑(例如卵磷脂或阿拉伯樹膠);非水性媒劑(例如杏仁油、油性酯、乙醇或經分餾植物油);及防腐劑(例如對羥基苯甲酸甲酯或對羥基苯甲酸丙酯或山梨酸)。若適宜,該等製劑亦可含有緩衝鹽、矯味劑、著色劑及/或甜味劑。若期望,則用於經口投與之製劑可適宜地經調配以受控釋放活性化合物。
該等化合物可經調配以(例如)藉由濃注或連續輸注實施非經腸投與。用於注射之調配物可以單位劑型存在,例如存於安瓿或存於多劑量容器中,同時視情況添加有防腐劑。可注射組合物可為水性等滲溶液或懸浮液。在用於非經腸投與之某些實施例中,可使用表面活性劑(例如Cremaphor)或親脂性溶劑(例如三甘油酯或脂質體)來製備該等化合物。該等組合物可經滅菌及/或含有佐劑,例如防腐劑、穩定劑、潤濕劑或乳化劑、促溶劑、調節滲透壓之鹽及/或緩衝劑。或者,化合物可呈粉末形式以便在使用之前使用適宜媒劑(例如無菌無致熱源水)重構。另外,其亦可含有其他治療有效性物質。
對於吸入投與,該化合物可使用適宜推進劑(例如二氯二氟甲烷、三氯氟乙烷、二氯四氟乙烷、二氧化碳或其他適宜氣體),方便地以氣溶膠噴霧呈現形式,自加壓包裝或噴霧器遞送。在加壓氣溶膠之情形下,可藉由提供遞送量測計量的閥門來確定劑量單位。用於吸入器或吹入器中之明膠之膠囊及藥筒可經調配含有化合物與適宜粉末基質(例如乳糖或澱粉)之粉末混合物。
用於穿皮式施用之適宜調配物包含有效量之化合物及載劑。較佳載劑包含可吸收性藥理上可接受之溶劑,以助於穿透個體皮膚。舉例而言,穿皮式器件係呈包含背襯元件之繃帶或貼片形式;含有該化合物(視情況具有載劑)之儲液器,視情況以速率控制屏障,依控制的預定速率長時間遞送化合物至宿主皮膚;及將器件固定至皮膚之構件。亦可使用基質穿皮式調配物。
用於局部施用至(例如)皮膚及眼睛之適宜調配物較佳係業內熟知之水溶液、軟膏、乳霜或凝膠。調配物可含有增溶劑、穩定劑、張力增強劑、緩衝劑及防腐劑。
在某些實施例中,化合物亦可調配為(例如)含有習用栓劑基質(例如可可脂或其他甘油酯)或凝膠形成劑(例如卡波姆(carbomer))之直腸用組合物,例如栓劑或保留灌腸劑。
在某些實施例中,化合物可調配為儲積製劑。該等長效調配物可藉由植入(例如經皮下或肌內)或藉由肌內注射投與。該化合物可利用適宜聚合或疏水性材料(例如作為於可接受油中之乳液)、離子交換樹脂、生物可降解聚合物來調配,或呈難溶衍生物(例如呈難溶鹽)來調配。
若需要,則醫藥組合物可呈含有一或多個含有活性成分之單位劑型之封裝或分配器件來傳遞。包裝可包含(例如)金屬或塑膠箔,例如泡罩包裝。包裝或分配器件可附帶投藥說明書。
6. 有效量及投藥法
在某些實施例中,將化合物之醫藥組合物依用於預防、治療或控制如本文所闡述之病狀或疾病之治療有效劑量投與個體,較佳為人類。將藥組合物以足以誘發個體中之有效治療反應之量投與個體。有效治療反應係至少部分地遏阻或減緩病狀或疾病之症狀或併發症之反應。適於達成此反應之量定義為「治療有效劑量」或「治療有效量」。化合物劑量可尤其考慮溫血動物(哺乳動物)之物種、體重、年齡、所治療病狀、所治療病狀之嚴重程度、投與形式、投與途徑。劑量大小亦可由伴隨在特定個體中投與特定治療化合物出現之任何不良效應的存在、性質及程度決定。
在某些實施例中,本發明化合物或其組合物之適宜劑量為約1 ng/kg至約1000 mg/kg、0.01 mg/kg至900 mg/kg、0.1 mg/kg至800 mg/kg、約1 mg/kg至約700 mg/kg、約2 mg/kg至約500 mg/kg、約3 mg/kg至約400 mg/kg、4 mg/kg至約300 mg/kg或約5 mg/kg至約200 mg/kg。在某些實施例中,化合物之適宜劑量可為約1 mg/kg、5 mg/kg、10 mg/kg、15 mg/kg、20 mg/kg、25 mg/kg、30 mg/kg、35 mg/kg、40 mg/kg、45 mg/kg、50 mg/kg、60 mg/kg、70 mg/kg、80 mg/kg、90 mg/kg、100 mg/kg、125 mg/kg、150 mg/kg、175 mg/kg、200 mg/kg、250 mg/kg、300 mg/kg、400 mg/kg、500 mg/kg、600 mg/kg、700 mg/kg、800 mg/kg、900 mg/kg或1000 mg/kg。在某些實施例中,化合物之劑量可每天投與一次或分成子劑量且以多個劑量投與(例如每天兩次、三次或四次)。
在某些實施例中,該等化合物可與 一或多種第二化合物一起藉由相同途徑或藉由不同投與途徑依序或同時投與。在依序投與時,選擇投與之間之時間以尤其有益於組合治療之治療效能及/或安全性。在某些實施例中,可首先投與本文之化合物且隨後投與第二化合物,或替代地,首先投與第二化合物且隨後投與本發明化合物。舉例而言且並不加以限制,投與之間之時間為約1 hr、約2 hr、約4hr、約6 hr、約12 hr、約16 hr或約20 hr。在某些實施例中,投與之間之時間為約1、約2、約3、約4、約5、約6或約7天或更長。在某些實施例中,投與之間之時間為約1週、2週、3週或4週或更長。在某些實施例中,投與之間之時間為約1個月或2個月或更長。
在同時投與時,該化合物可與第二化合物同時藉由相同或不同途徑分開投與,或以單一組合物藉由相同途徑投與。在某些實施例中,第二化合物之投與量及頻率可使用用於特定化合物之標準劑量及標準投與頻率。例如參見Physicians’ Desk Reference,第70版,PDR Network, 2015;其以引用方式併入本文中。
在本發明化合物與第二化合物組合投與之某些實施例中,以治療有效劑量投與第二化合物之劑量。在某些實施例中,適宜劑量可為約1 ng/kg至約1000 mg/kg、約0.01 mg/kg至約900 mg/kg、約0.1 mg/kg至約800 mg/kg、約1 mg/kg至約700 mg/kg、約2 mg/kg至約500 mg/kg、約3 mg/kg至約400 mg/kg、約4 mg/kg至約300 mg/kg或約5 mg/kg至約200 mg/kg。在某些實施例中,第二化合物之適宜劑量可為約1 mg/kg、5 mg/kg、10 mg/kg、15 mg/kg、20 mg/kg、25 mg/kg、30 mg/kg、35 mg/kg、40 mg/kg、45 mg/kg、50 mg/kg、60 mg/kg、70 mg/kg、80 mg/kg、90 mg/kg、100 mg/kg、125 mg/kg、150 mg/kg、175 mg/kg、200 mg/kg、250 mg/kg、300 mg/kg、400 mg/kg、500 mg/kg、600 mg/kg、700 mg/kg、800 mg/kg、900 mg/kg或1000 mg/kg。在某些實施例中,第二化合物之劑量之導則提供於Physicians’ Desk Reference,第70版,PDR Network (2015)中,該文獻以引用方式併入本文中。
應理解,該等化合物之最佳劑量、毒性及治療效能可端視個別化合物之相對功效而有所變化且可藉由標準醫藥程序在細胞培養物或實驗動物中(例如)藉由測定LD50
(對50%之群體致死之劑量)及ED50
(在50%之群體中治療有效之劑量)來測定。毒性效應與治療效應之間之劑量比率係治療指數且可表示為比率LD50
/ED50
。展現較大治療指數之化合物或其組合較佳。儘管可使用展現毒性副效應之某些藥劑,但應謹慎設計遞送系統以使該等藥劑靶向受影響組織位點以最小化對正常細胞之潛在損害且由此減少副效應。
可使用自(例如)細胞培養分析及動物研究獲得之數據來調配用於人類之劑量範圍。該等小分子化合物之劑量較佳在具有較少毒性或沒有毒性之循環濃度(包含ED50
)範圍內。端視所用劑型及投與途徑而定,劑量可在此範圍內變化。對於本文所揭示方法中所使用之任何化合物而言,可首先自細胞培養分析來估計治療有效劑量。可在動物模型中調配劑量以達成循環血漿濃度範圍(包含IC50
,即達成症狀之半最大抑制之測試化合物之濃度),如在細胞培養物中所測定。該資訊可用於更準確地確定人類可用之劑量。可藉由(例如)高效液相層析(HPLC)量測血漿中之含量。
7. 製備方法
提供下列實例以進一步闡釋本發明方法及用於該等方法中之化合物及組合物。所闡述實例僅係闡釋性且並不意欲以任何方式限制本發明範圍。本申請案中所提及之所有文章及參考文獻(包含專利)之揭示內容之全部內容皆以引用方式併入本文中。
可根據本文所提供之指導併入已知化學反應及相關程序(例如分離及純化)來合成本發明化合物。用於製備本發明化合物之代表性方法及程序闡述於下文及實例中。首字母縮略詞係根據慣例使用之縮寫,其可發現於文獻及科學期刊中。
在某些實施例中,提供製備式I化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或鹽之製程: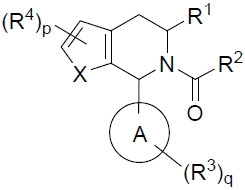 I
I
其中環A、X、R1
、R2
、R3
、R4
、p及q中之每一者獨立地如本文所定義,該製程包括使式1-5化合物與式1-6化合物: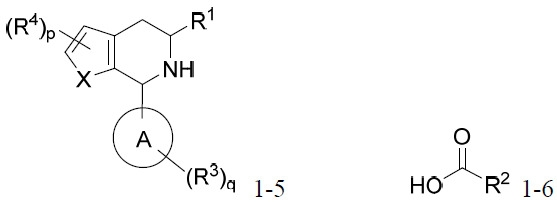 在足以提供式I化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或鹽之反應條件下接觸。
在足以提供式I化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或鹽之反應條件下接觸。
在某些實施例中,提供製備式I-5化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或鹽之製程:
其中環A、X、R1
、R3
、R4
、p及q中之每一者獨立地如本文所定義,該製程包括使式1-3化合物: 在足以提供式1-5化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或鹽之反應條件下環化。
在足以提供式1-5化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或鹽之反應條件下環化。
應理解,可改變起始材料及反應條件,改變反應序列,並採用額外步驟以產生本發明所涵蓋之化合物,如下列實例所顯示。可利用可用於合成所揭示化合物之已知化學反應之一般參考文獻(例如參見Smith及March, March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure,第五版,Wiley Interscience, 2001;或Carey及Sundberg, Advanced Organic Chemistry,部分B. Reaction and Synthesis;第五版,Springer, 2007;或Li, J.J. Name Reactions, A Collection of Detailed Mechanisms and Synthetic Applications;第五版,Springer, 2014)。
應瞭解,除非另外陳述,否則在給出典型或較佳製程條件(亦即反應溫度、施加、反應物莫耳比率、溶劑、壓力等)之情形下,亦可使用其他製程條件。最佳反應條件可隨所使用之特定反應物或溶劑而變化,但該等條件可由熟習此項技術者藉由常規最佳化程序來確定。
另外,可能需要習用保護基團來防止某些官能基發生不期望反應。各種官能基之適宜保護基團以及保護及去保護特定官能基之適宜條件在業內已眾所周知。舉例而言,諸多保護基團闡述於Wuts, P. G. M., Greene, T. W., & Greene, T. W. (2006)。Greene’s protective groups in organic synthesis。Hoboken, N.J., Wiley-Interscience及其中所引用之參考文獻中。
另外,本發明化合物可含有一或多個對掌性中心。因此,若期望,則該等化合物可以純淨立體異構體形式(亦即以個別對映異構體或非對映異構體形式)或以富含立體異構體之混合物形式製備或分離。除非另外指示,否則所有該等立體異構體(及富集混合物)皆包含於本發明範圍內。可使用(例如)業內熟知之光學活性起始材料或立體選擇性試劑來製備純立體異構體(或富集混合物)。或者,可使用(例如)對掌性管柱層析、對掌性拆分劑及諸如此類來分離該等化合物之外消旋混合物。
下列反應之起始材料係通常已知之化合物或可藉由已知程序或其明顯修改形式來製備。舉例而言,許多起始材料可自諸如以下等商業供應商獲得:Aldrich Chemical Co. (Milwaukee, Wisconsin, USA)、Bachem (Torrance, California, USA)、Emka-Chemce或Sigma (St. Louis, Missouri, USA)。其他材料可藉由諸如以下等標準參考文件中所闡述之程序或其明顯修改形式來製備,例如Fieser and Fieser’s Reagents for Organic Synthesis,第1-15卷(John Wiley, and Sons, 1991);Rodd’s Chemistry of Carbon Compounds,第1-5卷及增刊(Elsevier Science Publishers, 1989);organic Reactions,第1-40卷(John Wiley, and Sons, 1991);March’s Advanced Organic Chemistry, (John Wiley, and Sons,第5版,2001);及Larock’s Comprehensive Organic Transformations (VCH Publishers Inc., 1989)。
一般合成
在某些實施例中,本文所揭示之化合物可根據下文所展示之一般反應圖。舉例而言,式I化合物可根據下文在反應圖1中所概述之一般合成來製備,其中適宜試劑可購自商業來源或經由已知方法或自本文所提供實例改編之方法來合成。在反應圖1中,環A、X、R1
、R2
、R3
、R4
、p及q中之每一者獨立地如本文所定義。
反應圖 1 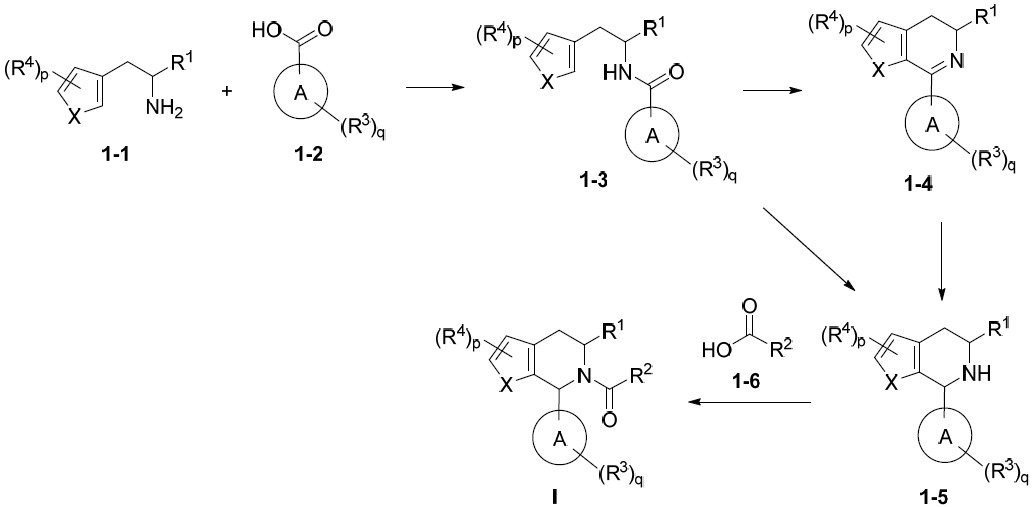 在反應圖1中,可藉由使胺1-1
與酸1-2
在標準醯胺鍵形成反應條件下偶合來提供化合物1-3
。可藉由首先形成化合物1-4
且隨後使用氫化物(例如NaBH4
、LiAlH4
等)還原來環化化合物1-3
以提供化合物1-5
。或者,可在適宜條件(例如非質子溶劑)下在酸觸媒存在下自化合物1-3
直接提供化合物1-5
。然後可藉由使化合物1-5
與化合物1-6
在適於提供式I化合物之反應條件下偶合來提供式I化合物。在每一反應完成時,可回收中間體或最終化合物中之每一者,且視情況藉由習用技術(例如中和、萃取、沈澱、層析、過濾及諸如此類)純化。
用於反應圖1中之適當起始材料及試劑可夠得或藉由熟習此項技術者已知之方法來製備。如反應圖2中所展示,可提供對掌性或對映異構體富集之起始材料以藉由將對掌性或對映異構體富集之胺基醇轉化成氧雜噻唑啶二氧化物2-2
來用於反應圖1之方法中。在反應圖2中,X、R1
、R4
及p獨立地如本文所定義,M係金屬鹵化物(例如MgBr)且PG係保護基團(例如Boc)。
在反應圖1中,可藉由使胺1-1
與酸1-2
在標準醯胺鍵形成反應條件下偶合來提供化合物1-3
。可藉由首先形成化合物1-4
且隨後使用氫化物(例如NaBH4
、LiAlH4
等)還原來環化化合物1-3
以提供化合物1-5
。或者,可在適宜條件(例如非質子溶劑)下在酸觸媒存在下自化合物1-3
直接提供化合物1-5
。然後可藉由使化合物1-5
與化合物1-6
在適於提供式I化合物之反應條件下偶合來提供式I化合物。在每一反應完成時,可回收中間體或最終化合物中之每一者,且視情況藉由習用技術(例如中和、萃取、沈澱、層析、過濾及諸如此類)純化。
用於反應圖1中之適當起始材料及試劑可夠得或藉由熟習此項技術者已知之方法來製備。如反應圖2中所展示,可提供對掌性或對映異構體富集之起始材料以藉由將對掌性或對映異構體富集之胺基醇轉化成氧雜噻唑啶二氧化物2-2
來用於反應圖1之方法中。在反應圖2中,X、R1
、R4
及p獨立地如本文所定義,M係金屬鹵化物(例如MgBr)且PG係保護基團(例如Boc)。
反應圖 2  參照反應圖2,在標準偶合條件下使化合物2-1
偶合至化合物2-2
以產生化合物2-3
。通常在適宜觸媒(例如CuI)存在下使用適宜溶劑/溶劑混合物來實施反應。對化合物2-3
去保護以提供化合物2-4
。在反應完成時,可藉由習用技術(例如中和、萃取、沈澱、層析、過濾及諸如此類)回收每一中間體。
在反應圖1及反應圖2之方法之一些實施例中,起始化合物(例如化合物I-1
及化合物I-2
)上之各種取代基(例如環A、R1
、R2
、R3
等)係如針對式I所定義。然而,亦應瞭解,可使用化學衍生及/或官能基互變來進一步改質反應圖1或反應圖2中之任一化合物以提供各種式I化合物。
可使用上述合成途徑且修改熟習此項技術者可用之化學合成程序來合成其他本發明化合物。實例性合成方法提供於實例中。應理解。闡述實例性化合物之合成之每一程序係說明書之一部分,且由此在本文中納入本發明之實施方式中。
參照反應圖2,在標準偶合條件下使化合物2-1
偶合至化合物2-2
以產生化合物2-3
。通常在適宜觸媒(例如CuI)存在下使用適宜溶劑/溶劑混合物來實施反應。對化合物2-3
去保護以提供化合物2-4
。在反應完成時,可藉由習用技術(例如中和、萃取、沈澱、層析、過濾及諸如此類)回收每一中間體。
在反應圖1及反應圖2之方法之一些實施例中,起始化合物(例如化合物I-1
及化合物I-2
)上之各種取代基(例如環A、R1
、R2
、R3
等)係如針對式I所定義。然而,亦應瞭解,可使用化學衍生及/或官能基互變來進一步改質反應圖1或反應圖2中之任一化合物以提供各種式I化合物。
可使用上述合成途徑且修改熟習此項技術者可用之化學合成程序來合成其他本發明化合物。實例性合成方法提供於實例中。應理解。闡述實例性化合物之合成之每一程序係說明書之一部分,且由此在本文中納入本發明之實施方式中。
合成實例 中間體 1 : 中間體 (S)-1-(3- 甲氧基苯基 ) 己烷 -2- 胺之合成  (S)-2-胺基己烷-1-醇:在0℃下,經1 h時段向(S)-2-胺基己酸(30.0 g, 228.6 mmol, 1當量)於THF (300 mL)中之溶液中添加氫化鋰鋁(1M於THF中,458 mL, 457.3 mmol, 2當量)。將反應混合物升溫至室溫,然後將混合物在70℃及N2氣氛下攪拌14 h。將反應混合物冷卻至室溫,使用二乙醚(50 mL)稀釋反應液,在費希爾後處理(fisher – workup)之後,經由燒結漏斗使用二乙醚過濾反應混合物,在減壓下濃縮濾液以得到產物,粗產物未經進一步純化即用於下一步驟中。1
H NMR (400 MHz, CDCl3) δ ppm 0.89 (s, 3 H), 1.29 - 1.39 (m, 6 H), 2.00 (s, 3 H), 2.81 (s, 1 H), 3.23 - 3.25 (m, 1 H), 3.55 - 3.56 (m, 1 H)。
(S)-(1-羥基己烷-2-基)胺基甲酸第三丁基酯:在0℃下,向(S)-2-胺基己烷-1-醇(24.5 g, 209.06 mmol, 1當量)於DCM (250 mL)在之溶液中逐滴添加TEA (58.76 mL, 418.12 mmol, 2當量),將其攪拌5 min,然後添加二碳酸二-第三丁基酯(57.63 mL, 250.87 mmol, 1.2當量)。在室溫下攪拌14 h之後,使用水(30 mL)稀釋,使用DCM (2×150 mL)萃取。使用水洗滌合併之有機層,然後使用NaHCO3水溶液(約30 mL)洗滌且最後使用鹽水溶液(75 mL)洗滌,藉由Na2S04乾燥,並在真空中濃縮。對殘餘物實施Combiflash矽膠層析(配備有於DCM中之MeOH作為洗脫劑)以得到(S)-(1-羥基己烷-2-基)胺基甲酸第三丁基酯。1
H NMR (400 MHz, CDCl3) δ ppm 0.90 (s, 3 H), 1.25 - 1.33 (m, 6 H), 1.38 - 1.41 (m, 9 H), 3.48 - 3.55 (m, 1 H), 3.62 - 3.68 (m, 2 H), 4.57 (bs,1 H)。
在室溫下,將1H -咪唑(25 g, 368.1 mmol, 4當量)及三乙胺(39 mL, 276.1 mmol, 3當量)溶於無水二氯甲烷(200 mL,商業無水溶劑)中且將混合物冷卻至0℃ (外部溫度,使用冰維持)。然後,經由加料漏斗經約30分鐘時段逐滴緩慢添加亞硫醯氯(7.3 mL, 101.2 mmol, 1.1當量),同時將浴溫度維持在0℃下。然後將反應混合物在0℃下再攪拌10 mins。然後將反應混合物冷卻至-78℃。然後在室溫下經由加料漏斗將在無水二氯甲烷(100 mL,商業無水溶劑)中製得之(S)-(1-羥基己烷-2-基)胺基甲酸第三丁基酯(20 g, 92.03 mmol, 1當量)之溶液逐滴添加至在-78℃下攪拌45 min時段的反應混合物中。將反應混合物在-78℃下再攪拌3小時。然後,去除乾冰-丙酮浴且將反應混合物在室溫下攪拌16 h。在完成反應之後(tlc,於己烷中之10% EA),使用DCM稀釋混合物,使用水(200 mL × 3)及鹽水(200 mL)洗滌。藉由無水Na2SO4乾燥有機相,過濾並在減壓下於旋轉蒸發儀上濃縮以獲得粗產物。藉由矽膠管柱層析使用乙酸乙酯之己烷溶液作為洗脫劑純化粗製物。使用於己烷中之10 - 25% EA洗脫產物以得到(4S)-4-丁基-1,2,3-氧雜噻唑啶-3-甲酸第三丁基酯2-氧化物。1
H NMR (400 MHz, CDCl3) δ ppm 0.91 (t,J
= 6.8 Hz, 3 H), 1.27 - 1.38 (m, 4 H), 1.52 (s, 9 H), 1.67 - 1.73 (m, 1 H), 1.99 - 2.10 (m, 1 H), 3.97 - 4.02 (m, 1 H), 4.70 - 4.78 (m, 2 H)。
(S)-4-丁基-1,2,3-氧雜噻唑啶-3-甲酸第三丁基酯2,2-二氧化物:以20 g × 2批實施此反應。在0℃下,將氯化釕(III) (0.463 g, 2.23 mmol, 0.014當量)添加至(4S)-4-丁基-1,2,3-氧雜噻唑啶-3-甲酸第三丁基酯2-氧化物(42.0 g, 159.48 mmol, 1當量)於乙腈(400 mL)及水(200 mL)中之經攪拌溶液中,隨後逐份添加偏過碘酸鈉(37.43 g, 175.43 mmol, 1.1當量)。將雙相混合物在室溫下攪拌2小時。經由燒結漏斗過濾反應混合物,使用乙酸乙酯洗滌。添加水(250 mL)且將混合物萃取至乙酸乙酯(2 × 150 mL)中。使用水(150 mL)、鹽水(150 mL)洗滌合併之有機物,使用Na2SO4乾燥,過濾並在減壓下濃縮以獲得粗產物,藉由管柱層析使用於己烷中之10%乙酸乙酯作為洗脫劑進行純化以得到(S)-4-丁基-1,2,3-氧雜噻唑啶-3-甲酸第三丁基酯2,2-二氧化物。1
H NMR (400 MHz, CDCl3) δ ppm 0.89 - 0.92 (m, 3 H), 1.24 - 1.37 (m, 4 H), 1.53 (s, 9 H), 1.77 - 1.83 (m, 1 H), 1.88 - 1.89 (m, 1 H), 4.26 - 4.30 (m, 2 H), 4.59 - 4.63 (m, 1 H)。
(S)-(1-(3-甲氧基苯基)己烷-2-基)胺基甲酸第三丁基酯:在-20℃ (鹽及冰混合物浴)下,以15min時間向碘化銅(0.95 g, 5.017 mmol, 0.1當量)於二乙醚(150 mL)中之溶液中逐滴添加(3-甲氧基苯基)溴化鎂(1M於THF中) (98.5 mL, 100.35 mmol, 2當量)。將反應混合物在-20℃ (鹽及冰混合物浴)下攪拌30 min。此後,在-20℃ (鹽及冰混合物浴)下,以25min時間向反應物質中逐滴添加(S)-4-丁基-1,2,3-氧雜噻唑啶-3-甲酸第三丁基酯2,2-二氧化物(14 g, 50.172 mmol, 1當量)於二乙醚(100 mL)中之溶液。將所得混合物在-20℃下攪拌4 h。最後,使用10%檸檬酸水溶液(70 mL)在-20℃ (鹽及冰混合物浴)下終止反應。將混合物升溫至室溫並攪拌10 min。經由矽藻土墊過濾混合物,使用乙酸乙酯充分洗滌。使用水(100 mL)、鹽水(100 mL)洗滌濾液,藉由無水硫酸鈉乾燥,過濾並濃縮以得到粗產物,藉由急速管柱層析使用於正己烷中之15%乙酸乙酯作為洗脫劑進行純化以獲得(S)-(1-(3-甲氧基苯基)己烷-2-基)胺基甲酸第三丁基酯。LCMS (ES) m/z = 208.2 [M+H]+
,未觀察到boc質量。1
H NMR (400 MHz, CDCl3) δ ppm 0.86 - 0.87 (m, 3 H), 1.25 - 1.28 (m, 6 H), 1.40 (s, 9 H), 2.73 (bs, 2 H), 3.79 (s, 3 H), 4.29 (bs, 1 H), 6.71 - 6.76 (m, 3 H), 7.17 -7.21 (m, 1 H),未觀察到醯胺NH。
(S)-1-(3-甲氧基苯基)己烷-2-胺:在0℃下,向(S)-(1-(3-甲氧基苯基)己烷-2-基)胺基甲酸第三丁基酯(12.0 g, 39.033 mmol, 1當量)於二氯甲烷(50 mL)中之溶液中緩慢添加於1,4-二噁烷中之4M HCl (150 mL)。將混合物在室溫下攪拌5 h。藉由TLC監測反應進展,在反應完成之後,在減壓下濃縮反應混合物。藉由NaHCO3
飽和水溶液鹼化所獲得粗製物。使用EtOAc (3 × 200 mL)萃取化合物。使用鹽水(100 mL)洗滌有機層,藉由無水硫酸鈉乾燥,過濾並濃縮以得到(S)-1-(3-甲氧基苯基)己烷-2-胺。LCMS (ES) m/z = 208.1 [M+H]+
(S)-2-胺基己烷-1-醇:在0℃下,經1 h時段向(S)-2-胺基己酸(30.0 g, 228.6 mmol, 1當量)於THF (300 mL)中之溶液中添加氫化鋰鋁(1M於THF中,458 mL, 457.3 mmol, 2當量)。將反應混合物升溫至室溫,然後將混合物在70℃及N2氣氛下攪拌14 h。將反應混合物冷卻至室溫,使用二乙醚(50 mL)稀釋反應液,在費希爾後處理(fisher – workup)之後,經由燒結漏斗使用二乙醚過濾反應混合物,在減壓下濃縮濾液以得到產物,粗產物未經進一步純化即用於下一步驟中。1
H NMR (400 MHz, CDCl3) δ ppm 0.89 (s, 3 H), 1.29 - 1.39 (m, 6 H), 2.00 (s, 3 H), 2.81 (s, 1 H), 3.23 - 3.25 (m, 1 H), 3.55 - 3.56 (m, 1 H)。
(S)-(1-羥基己烷-2-基)胺基甲酸第三丁基酯:在0℃下,向(S)-2-胺基己烷-1-醇(24.5 g, 209.06 mmol, 1當量)於DCM (250 mL)在之溶液中逐滴添加TEA (58.76 mL, 418.12 mmol, 2當量),將其攪拌5 min,然後添加二碳酸二-第三丁基酯(57.63 mL, 250.87 mmol, 1.2當量)。在室溫下攪拌14 h之後,使用水(30 mL)稀釋,使用DCM (2×150 mL)萃取。使用水洗滌合併之有機層,然後使用NaHCO3水溶液(約30 mL)洗滌且最後使用鹽水溶液(75 mL)洗滌,藉由Na2S04乾燥,並在真空中濃縮。對殘餘物實施Combiflash矽膠層析(配備有於DCM中之MeOH作為洗脫劑)以得到(S)-(1-羥基己烷-2-基)胺基甲酸第三丁基酯。1
H NMR (400 MHz, CDCl3) δ ppm 0.90 (s, 3 H), 1.25 - 1.33 (m, 6 H), 1.38 - 1.41 (m, 9 H), 3.48 - 3.55 (m, 1 H), 3.62 - 3.68 (m, 2 H), 4.57 (bs,1 H)。
在室溫下,將1H -咪唑(25 g, 368.1 mmol, 4當量)及三乙胺(39 mL, 276.1 mmol, 3當量)溶於無水二氯甲烷(200 mL,商業無水溶劑)中且將混合物冷卻至0℃ (外部溫度,使用冰維持)。然後,經由加料漏斗經約30分鐘時段逐滴緩慢添加亞硫醯氯(7.3 mL, 101.2 mmol, 1.1當量),同時將浴溫度維持在0℃下。然後將反應混合物在0℃下再攪拌10 mins。然後將反應混合物冷卻至-78℃。然後在室溫下經由加料漏斗將在無水二氯甲烷(100 mL,商業無水溶劑)中製得之(S)-(1-羥基己烷-2-基)胺基甲酸第三丁基酯(20 g, 92.03 mmol, 1當量)之溶液逐滴添加至在-78℃下攪拌45 min時段的反應混合物中。將反應混合物在-78℃下再攪拌3小時。然後,去除乾冰-丙酮浴且將反應混合物在室溫下攪拌16 h。在完成反應之後(tlc,於己烷中之10% EA),使用DCM稀釋混合物,使用水(200 mL × 3)及鹽水(200 mL)洗滌。藉由無水Na2SO4乾燥有機相,過濾並在減壓下於旋轉蒸發儀上濃縮以獲得粗產物。藉由矽膠管柱層析使用乙酸乙酯之己烷溶液作為洗脫劑純化粗製物。使用於己烷中之10 - 25% EA洗脫產物以得到(4S)-4-丁基-1,2,3-氧雜噻唑啶-3-甲酸第三丁基酯2-氧化物。1
H NMR (400 MHz, CDCl3) δ ppm 0.91 (t,J
= 6.8 Hz, 3 H), 1.27 - 1.38 (m, 4 H), 1.52 (s, 9 H), 1.67 - 1.73 (m, 1 H), 1.99 - 2.10 (m, 1 H), 3.97 - 4.02 (m, 1 H), 4.70 - 4.78 (m, 2 H)。
(S)-4-丁基-1,2,3-氧雜噻唑啶-3-甲酸第三丁基酯2,2-二氧化物:以20 g × 2批實施此反應。在0℃下,將氯化釕(III) (0.463 g, 2.23 mmol, 0.014當量)添加至(4S)-4-丁基-1,2,3-氧雜噻唑啶-3-甲酸第三丁基酯2-氧化物(42.0 g, 159.48 mmol, 1當量)於乙腈(400 mL)及水(200 mL)中之經攪拌溶液中,隨後逐份添加偏過碘酸鈉(37.43 g, 175.43 mmol, 1.1當量)。將雙相混合物在室溫下攪拌2小時。經由燒結漏斗過濾反應混合物,使用乙酸乙酯洗滌。添加水(250 mL)且將混合物萃取至乙酸乙酯(2 × 150 mL)中。使用水(150 mL)、鹽水(150 mL)洗滌合併之有機物,使用Na2SO4乾燥,過濾並在減壓下濃縮以獲得粗產物,藉由管柱層析使用於己烷中之10%乙酸乙酯作為洗脫劑進行純化以得到(S)-4-丁基-1,2,3-氧雜噻唑啶-3-甲酸第三丁基酯2,2-二氧化物。1
H NMR (400 MHz, CDCl3) δ ppm 0.89 - 0.92 (m, 3 H), 1.24 - 1.37 (m, 4 H), 1.53 (s, 9 H), 1.77 - 1.83 (m, 1 H), 1.88 - 1.89 (m, 1 H), 4.26 - 4.30 (m, 2 H), 4.59 - 4.63 (m, 1 H)。
(S)-(1-(3-甲氧基苯基)己烷-2-基)胺基甲酸第三丁基酯:在-20℃ (鹽及冰混合物浴)下,以15min時間向碘化銅(0.95 g, 5.017 mmol, 0.1當量)於二乙醚(150 mL)中之溶液中逐滴添加(3-甲氧基苯基)溴化鎂(1M於THF中) (98.5 mL, 100.35 mmol, 2當量)。將反應混合物在-20℃ (鹽及冰混合物浴)下攪拌30 min。此後,在-20℃ (鹽及冰混合物浴)下,以25min時間向反應物質中逐滴添加(S)-4-丁基-1,2,3-氧雜噻唑啶-3-甲酸第三丁基酯2,2-二氧化物(14 g, 50.172 mmol, 1當量)於二乙醚(100 mL)中之溶液。將所得混合物在-20℃下攪拌4 h。最後,使用10%檸檬酸水溶液(70 mL)在-20℃ (鹽及冰混合物浴)下終止反應。將混合物升溫至室溫並攪拌10 min。經由矽藻土墊過濾混合物,使用乙酸乙酯充分洗滌。使用水(100 mL)、鹽水(100 mL)洗滌濾液,藉由無水硫酸鈉乾燥,過濾並濃縮以得到粗產物,藉由急速管柱層析使用於正己烷中之15%乙酸乙酯作為洗脫劑進行純化以獲得(S)-(1-(3-甲氧基苯基)己烷-2-基)胺基甲酸第三丁基酯。LCMS (ES) m/z = 208.2 [M+H]+
,未觀察到boc質量。1
H NMR (400 MHz, CDCl3) δ ppm 0.86 - 0.87 (m, 3 H), 1.25 - 1.28 (m, 6 H), 1.40 (s, 9 H), 2.73 (bs, 2 H), 3.79 (s, 3 H), 4.29 (bs, 1 H), 6.71 - 6.76 (m, 3 H), 7.17 -7.21 (m, 1 H),未觀察到醯胺NH。
(S)-1-(3-甲氧基苯基)己烷-2-胺:在0℃下,向(S)-(1-(3-甲氧基苯基)己烷-2-基)胺基甲酸第三丁基酯(12.0 g, 39.033 mmol, 1當量)於二氯甲烷(50 mL)中之溶液中緩慢添加於1,4-二噁烷中之4M HCl (150 mL)。將混合物在室溫下攪拌5 h。藉由TLC監測反應進展,在反應完成之後,在減壓下濃縮反應混合物。藉由NaHCO3
飽和水溶液鹼化所獲得粗製物。使用EtOAc (3 × 200 mL)萃取化合物。使用鹽水(100 mL)洗滌有機層,藉由無水硫酸鈉乾燥,過濾並濃縮以得到(S)-1-(3-甲氧基苯基)己烷-2-胺。LCMS (ES) m/z = 208.1 [M+H]+
程序 1 : 化合物 1 之合成  在-10℃及氮氣氛下,向(R)-2-甲基環氧乙烷(10 g, 172 mmol, 1當量)於無水四氫呋喃(100 mL)中之經攪拌混合物中添加苯基溴化鎂(3M於二乙醚中) (63 mL, 183 mmol, 1.1當量)。將所得混合物逐漸升溫至室溫並攪拌16 h。藉由TLC (於己烷中之15%乙酸乙酯)監測反應進展。使用飽和氯化銨溶液淬滅反應混合物。使用乙酸乙酯(3 × 300 mL)萃取粗產物,使用水(200 mL)、鹽水(100 mL)洗滌合併之有機物,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以提供粗產物,藉由急速管柱層析使用於己烷中之15%乙酸乙酯作為洗脫劑進行純化以獲得(R)-1-苯基丙烷-2-醇。1
H NMR (400 MHz, CDCl3
) δ ppm 1.23 (d, J = 2.0 Hz, 3 H), 1.48 (s, 1 H), 2.66 – 2.71 (m, 1 H), 2.77 – 2.80 (m, 1 H), 4.02 (bs, 1 H), 7.21 – 7.31 (m, 5 H)。
在0℃及氮氣氛下,向(R)-1-苯基丙烷-2-醇(0.5 g, 3.671 mmol, 1當量)及三乙胺(1.54 mL, 11 mmol, 3當量)於二氯甲烷(10 mL)中之經攪拌混合物中添加甲磺醯氯(0.42 mL, 5.5 mmol, 1.5當量)。將所得混合物逐漸升溫至室溫並攪拌2 h。藉由TLC (於己烷中之50%二氯甲烷)監測反應進展。使用水淬滅反應混合物。使用二氯甲烷(3 × 30 mL)萃取粗產物,使用水(50 mL)、鹽水(20 mL)洗滌合併之有機物,藉由無水Na2
SO4
乾燥,在減壓下濃縮以提供粗產物(R)-甲烷磺酸1-苯基丙烷-2-基酯,其未經純化即原樣用於下一步驟中。1H NMR (400 MHz, CDCl3
) δ ppm 1.47 (d, J = 6.4 Hz, 3 H), 2.51 (s, 3 H), 2.89 - 2.99 (m, 2 H), 4.85 - 4.95 (m, 1 H), 7.16 - 7.40 (m, 5 H)。
在室溫及氮氣氛下,向(R)-甲烷磺酸1-苯基丙烷-2-基酯(0.5 g粗製物,2 mmol, 1當量)於N,N-二甲基甲醯胺(10 mL)中之經攪拌溶液中添加疊氮化鈉(0.18 g, 2.80 mmol, 1.2當量)。將所得混合物加熱至80℃並攪拌16 h。藉由TLC (於己烷中之5%乙酸乙酯)監測反應進展。在反應完成之後,將反應混合物冷卻至室溫並使用水淬滅。使用乙酸乙酯(3 × 30 mL)萃取混合物,使用水(4 × 30 mL)、鹽水(20 mL)洗滌合併之有機物,藉由無水Na2
SO4
乾燥,在減壓下濃縮成粗產物,藉由急速管柱層析使用於己烷中之5%乙酸乙酯作為洗脫劑純化以獲得(S)-(2-疊氮基丙基)苯。1
H NMR (400 MHz, CDCl3
) δ ppm 1.29 (d, J = 6.8 Hz, 3 H), 2.69 - 2.74 (m, 1 H), 2.81 - 2.86 (m, 1 H), 3.66 - 3.72 (m, 1 H), 7.18 - 7.32 (m, 5 H)。
在室溫及氮氣氛下,向(S)-(2-疊氮基丙基)苯(0.24 g, 1.5 mmol, 1當量)於乙酸乙酯(10 mL)中之經攪拌溶液中添加鈀(10%於活性碳粉上,50%水潤濕) (0.05 g)。藉由使用氫壓(氣囊,若大規模帕爾裝置(parr apparatus)適宜)攪拌8 h來對所得混合物實施氫化。藉由TLC (於己烷中之10%乙酸乙酯)監測反應進展。在反應完成之後,經由矽藻土墊過濾反應混合物,使用乙酸乙酯洗滌矽藻土墊,在減壓下濃縮濾液以獲得(S)-1-苯基丙烷-2-胺。1
H NMR (400 MHz, CDCl3
) δ ppm 1.12 (d, J = 6.8 Hz, 3 H), 2.49 - 2.59 (m, 1 H), 2.68 - 2.77 (m, 1 H), 3.12 - 3.20 (m, 1 H), 4.68 (s, 2 H), 7.09 - 7.31 (m, 5 H)。
在室溫下,向(S)-1-苯基丙烷-2-胺(0.2 g, 1.5 mmol, 1當量)及4-嗎啉基苯甲醛(0.28 g, 1.5 mmol, 1當量)於甲苯(10 mL)中之溶液中添加無水MgSO4
(0.2 g)。將反應混合物在110℃下攪拌4 h。TLC (於己烷中之70%乙酸乙酯)顯示反應已完成。藉由過濾自反應混合物去除固體部分且在減壓下濃縮濾液。所獲得粗製(S)-1-(4-嗎啉基苯基)-N-(1-苯基丙烷-2-基)甲亞胺未經進一步純化即用於下一步驟中。
將(S)-1-(4-嗎啉基苯基)-N-(1-苯基丙烷-2-基)甲亞胺(0.5 g粗製物)於三氟甲磺酸(0.5 mL)中之溶液在130℃下攪拌24 h。TLC 5% (於二氯甲烷中之甲醇)顯示反應已完成。將反應液冷卻至室溫且使用冰冷水(5 mL)稀釋,且然後使用10%氫氧化鈉水溶液鹼化至最高pH = 12。將產物萃取至乙酸乙酯(30 mL)中,藉由無水硫酸鈉乾燥有機層,過濾並在減壓下濃縮以獲得粗產物。藉由急速層析使用於二氯甲烷中之5%甲醇作為洗脫劑純化所獲得粗產物。藉由製備型HPLC [分析條件:管柱:Inertsil ODS 3V (250 mm × 4.6 mm × 5 µm),移動相(A):於水中之0.1%氨,移動相(B):CH3
CN,流速:1.0 mL/min,B之組成:0/10, 12/80, 25/90, 27/10, 30/10]再純化所分離產物以獲得4-(4-((3S)-3-甲基-1,2,3,4-四氫異喹啉-1-基)苯基)嗎啉。LCMS (ES) m/z = 309.2 [M+H]+;1H NMR (400 MHz, CDCl3): δ ppm 1.24 (d, J = 4.4 Hz, 3 H), 2.69 - 2.81 (m, 2 H), 3.14 - 3.20 (m, 5 H), 3.85 (bs, 4 H), 5.05 (s, 1 H), 6.69 (d, J = 8.0 Hz, 1 H), 6.87 (d, J = 6.8 Hz, 2 H), 6.99 (s, 1 H), 7.09 (s, 2 H), 7.21 - 7.25 (m, 2 H)。
在0℃下,向4-(4-((3S)-3-甲基-1,2,3,4-四氫異喹啉-1-基)苯基)嗎啉(0.04 g, 0.129 mmol, 1當量)於氯仿(5 mL)中之溶液中添加碳酸氫鈉(0.021 g, 0.259 mmol, 2.0當量),隨後添加2-氯乙醯氯(0.015 mL, 0.194 mmol, 1.5當量)。將混合物升溫至室溫並在N2
氣氛下攪拌3 h。TLC (於己烷中之40%乙酸乙酯)顯示反應已完成。然後,使用二氯甲烷(20 mL)稀釋反應液並使用水(20 mL)、鹽水(20 mL)洗滌,藉由無水Na2
SO4
乾燥,過濾並在減壓下濃縮以獲得粗產物。藉由急速管柱層析使用於正己烷中之50% EtOAc作為移動相純化粗產物以獲得2-氯-1-((3S)-3-甲基-1-(4-嗎啉基苯基)-3,4-二氫異喹啉-2(1H)-基)乙烷-1-酮。LCMS (ES) m/z = 385.3 [M+H]+
;1
H NMR (400 MHz, CDCl3
): δ ppm 1.25 (bs, 3 H), 2.3 - 2.5 (m, 1 H), 2.75 - 2.95 (m, 1 H), 3.14 (bs, 4 H), 3.85 (bs, 4 H), 4.16 - 4.26 (m, 3 H), 5.97 (bs, 1 H), 6.83 (bs, 2 H), 7.07 (bs, 2 H), 7.19 - 7.25 (m, 4 H)。對掌性HPLC純度:48.93 (反式): 47.12% (順式)。
在-10℃及氮氣氛下,向(R)-2-甲基環氧乙烷(10 g, 172 mmol, 1當量)於無水四氫呋喃(100 mL)中之經攪拌混合物中添加苯基溴化鎂(3M於二乙醚中) (63 mL, 183 mmol, 1.1當量)。將所得混合物逐漸升溫至室溫並攪拌16 h。藉由TLC (於己烷中之15%乙酸乙酯)監測反應進展。使用飽和氯化銨溶液淬滅反應混合物。使用乙酸乙酯(3 × 300 mL)萃取粗產物,使用水(200 mL)、鹽水(100 mL)洗滌合併之有機物,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以提供粗產物,藉由急速管柱層析使用於己烷中之15%乙酸乙酯作為洗脫劑進行純化以獲得(R)-1-苯基丙烷-2-醇。1
H NMR (400 MHz, CDCl3
) δ ppm 1.23 (d, J = 2.0 Hz, 3 H), 1.48 (s, 1 H), 2.66 – 2.71 (m, 1 H), 2.77 – 2.80 (m, 1 H), 4.02 (bs, 1 H), 7.21 – 7.31 (m, 5 H)。
在0℃及氮氣氛下,向(R)-1-苯基丙烷-2-醇(0.5 g, 3.671 mmol, 1當量)及三乙胺(1.54 mL, 11 mmol, 3當量)於二氯甲烷(10 mL)中之經攪拌混合物中添加甲磺醯氯(0.42 mL, 5.5 mmol, 1.5當量)。將所得混合物逐漸升溫至室溫並攪拌2 h。藉由TLC (於己烷中之50%二氯甲烷)監測反應進展。使用水淬滅反應混合物。使用二氯甲烷(3 × 30 mL)萃取粗產物,使用水(50 mL)、鹽水(20 mL)洗滌合併之有機物,藉由無水Na2
SO4
乾燥,在減壓下濃縮以提供粗產物(R)-甲烷磺酸1-苯基丙烷-2-基酯,其未經純化即原樣用於下一步驟中。1H NMR (400 MHz, CDCl3
) δ ppm 1.47 (d, J = 6.4 Hz, 3 H), 2.51 (s, 3 H), 2.89 - 2.99 (m, 2 H), 4.85 - 4.95 (m, 1 H), 7.16 - 7.40 (m, 5 H)。
在室溫及氮氣氛下,向(R)-甲烷磺酸1-苯基丙烷-2-基酯(0.5 g粗製物,2 mmol, 1當量)於N,N-二甲基甲醯胺(10 mL)中之經攪拌溶液中添加疊氮化鈉(0.18 g, 2.80 mmol, 1.2當量)。將所得混合物加熱至80℃並攪拌16 h。藉由TLC (於己烷中之5%乙酸乙酯)監測反應進展。在反應完成之後,將反應混合物冷卻至室溫並使用水淬滅。使用乙酸乙酯(3 × 30 mL)萃取混合物,使用水(4 × 30 mL)、鹽水(20 mL)洗滌合併之有機物,藉由無水Na2
SO4
乾燥,在減壓下濃縮成粗產物,藉由急速管柱層析使用於己烷中之5%乙酸乙酯作為洗脫劑純化以獲得(S)-(2-疊氮基丙基)苯。1
H NMR (400 MHz, CDCl3
) δ ppm 1.29 (d, J = 6.8 Hz, 3 H), 2.69 - 2.74 (m, 1 H), 2.81 - 2.86 (m, 1 H), 3.66 - 3.72 (m, 1 H), 7.18 - 7.32 (m, 5 H)。
在室溫及氮氣氛下,向(S)-(2-疊氮基丙基)苯(0.24 g, 1.5 mmol, 1當量)於乙酸乙酯(10 mL)中之經攪拌溶液中添加鈀(10%於活性碳粉上,50%水潤濕) (0.05 g)。藉由使用氫壓(氣囊,若大規模帕爾裝置(parr apparatus)適宜)攪拌8 h來對所得混合物實施氫化。藉由TLC (於己烷中之10%乙酸乙酯)監測反應進展。在反應完成之後,經由矽藻土墊過濾反應混合物,使用乙酸乙酯洗滌矽藻土墊,在減壓下濃縮濾液以獲得(S)-1-苯基丙烷-2-胺。1
H NMR (400 MHz, CDCl3
) δ ppm 1.12 (d, J = 6.8 Hz, 3 H), 2.49 - 2.59 (m, 1 H), 2.68 - 2.77 (m, 1 H), 3.12 - 3.20 (m, 1 H), 4.68 (s, 2 H), 7.09 - 7.31 (m, 5 H)。
在室溫下,向(S)-1-苯基丙烷-2-胺(0.2 g, 1.5 mmol, 1當量)及4-嗎啉基苯甲醛(0.28 g, 1.5 mmol, 1當量)於甲苯(10 mL)中之溶液中添加無水MgSO4
(0.2 g)。將反應混合物在110℃下攪拌4 h。TLC (於己烷中之70%乙酸乙酯)顯示反應已完成。藉由過濾自反應混合物去除固體部分且在減壓下濃縮濾液。所獲得粗製(S)-1-(4-嗎啉基苯基)-N-(1-苯基丙烷-2-基)甲亞胺未經進一步純化即用於下一步驟中。
將(S)-1-(4-嗎啉基苯基)-N-(1-苯基丙烷-2-基)甲亞胺(0.5 g粗製物)於三氟甲磺酸(0.5 mL)中之溶液在130℃下攪拌24 h。TLC 5% (於二氯甲烷中之甲醇)顯示反應已完成。將反應液冷卻至室溫且使用冰冷水(5 mL)稀釋,且然後使用10%氫氧化鈉水溶液鹼化至最高pH = 12。將產物萃取至乙酸乙酯(30 mL)中,藉由無水硫酸鈉乾燥有機層,過濾並在減壓下濃縮以獲得粗產物。藉由急速層析使用於二氯甲烷中之5%甲醇作為洗脫劑純化所獲得粗產物。藉由製備型HPLC [分析條件:管柱:Inertsil ODS 3V (250 mm × 4.6 mm × 5 µm),移動相(A):於水中之0.1%氨,移動相(B):CH3
CN,流速:1.0 mL/min,B之組成:0/10, 12/80, 25/90, 27/10, 30/10]再純化所分離產物以獲得4-(4-((3S)-3-甲基-1,2,3,4-四氫異喹啉-1-基)苯基)嗎啉。LCMS (ES) m/z = 309.2 [M+H]+;1H NMR (400 MHz, CDCl3): δ ppm 1.24 (d, J = 4.4 Hz, 3 H), 2.69 - 2.81 (m, 2 H), 3.14 - 3.20 (m, 5 H), 3.85 (bs, 4 H), 5.05 (s, 1 H), 6.69 (d, J = 8.0 Hz, 1 H), 6.87 (d, J = 6.8 Hz, 2 H), 6.99 (s, 1 H), 7.09 (s, 2 H), 7.21 - 7.25 (m, 2 H)。
在0℃下,向4-(4-((3S)-3-甲基-1,2,3,4-四氫異喹啉-1-基)苯基)嗎啉(0.04 g, 0.129 mmol, 1當量)於氯仿(5 mL)中之溶液中添加碳酸氫鈉(0.021 g, 0.259 mmol, 2.0當量),隨後添加2-氯乙醯氯(0.015 mL, 0.194 mmol, 1.5當量)。將混合物升溫至室溫並在N2
氣氛下攪拌3 h。TLC (於己烷中之40%乙酸乙酯)顯示反應已完成。然後,使用二氯甲烷(20 mL)稀釋反應液並使用水(20 mL)、鹽水(20 mL)洗滌,藉由無水Na2
SO4
乾燥,過濾並在減壓下濃縮以獲得粗產物。藉由急速管柱層析使用於正己烷中之50% EtOAc作為移動相純化粗產物以獲得2-氯-1-((3S)-3-甲基-1-(4-嗎啉基苯基)-3,4-二氫異喹啉-2(1H)-基)乙烷-1-酮。LCMS (ES) m/z = 385.3 [M+H]+
;1
H NMR (400 MHz, CDCl3
): δ ppm 1.25 (bs, 3 H), 2.3 - 2.5 (m, 1 H), 2.75 - 2.95 (m, 1 H), 3.14 (bs, 4 H), 3.85 (bs, 4 H), 4.16 - 4.26 (m, 3 H), 5.97 (bs, 1 H), 6.83 (bs, 2 H), 7.07 (bs, 2 H), 7.19 - 7.25 (m, 4 H)。對掌性HPLC純度:48.93 (反式): 47.12% (順式)。
程序 2 :化合物 2 及 3 之合成  將D-苯基丙胺酸甲酯鹽酸鹽(0.3 g, 1.391 mmol, 1當量)分配於乙酸乙酯與碳酸氫鈉溶液之間並攪拌15 min,分離乙酸乙酯層且使用乙酸乙酯(2 × 50 mL)萃取水層。使用水(50 mL)、鹽水(50 mL)洗滌合併之乙酸乙酯部分,藉由無水硫酸鈉乾燥,並在減壓下濃縮以獲得D-苯基丙胺酸甲酯。LCMS (ES) m/z = 180.1 [M+H]+
;1
H NMR (400 MHz, CDCl3
) δ ppm, 2.83 - 2.88 (m, 1 H), 3.07 - 3.11 (m, 1 H), 3.71 - 3.75 (m, 4 H), 7.18 - 7.32 (m, 5 H)。
向D-苯基丙胺酸甲酯(0.1 g, 0.557 mmol, 1當量)於甲苯(10 mL)中之溶液中添加4-甲醯基苯甲酸甲酯(0.0.09 g, 0.557 mmol, 1當量)。將反應混合物加熱至120℃並攪拌1 h。在減壓下濃縮混合物以獲得 (R,E)-4-(((1-甲氧基-1-側氧基-3-苯基丙烷-2-基)亞胺基)甲基)苯甲酸甲酯且所獲得粗產物未經任何進一步純化即用於下一步驟中。
將(R,E)-4-(((1-甲氧基-1-側氧基-3-苯基丙烷-2-基)亞胺基)甲基)苯甲酸甲酯(0.3 g粗製物)與三氟甲磺酸(2 mL)混合且將混合物加熱至130℃並攪拌18 h,且藉由LCMS分析混合物(LCMS展示水解產物(3R)-1-(4-羧基苯基)-1,2,3,4-四氫異喹啉-3-甲酸)。將混合物冷卻至0℃且添加10 mL無水甲醇。將所得混合物加熱至80℃並攪拌2 h。將混合物冷卻至0℃並使用三乙胺中和,且在減壓下濃縮。使用乙酸乙酯溶解所獲得粗製物,使用水(3 × 20 mL)、鹽水(20 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮。藉由急速層析使用於己烷中之20%乙酸乙酯作為洗脫劑純化粗產物以提供(3R)-1-(4-(甲氧基羰基)苯基)-1,2,3,4-四氫異喹啉-3-甲酸甲酯。LCMS (ES) m/z = 326.2 [M+H]+
。
在0℃及氮氣氛下,向(3R)-1-(4-(甲氧基羰基)苯基)-1,2,3,4-四氫異喹啉-3-甲酸甲酯(0.12 g, 0.368 mmol, 1當量)及碳酸氫鈉(0.06 g, 0.737 mmol, 2當量)於氯仿(5 mL)中之經攪拌混合物中添加2-氯乙醯氯(0.044 mL, 0.553 mmol, 1.5當量)。將所得混合物升溫至室溫並攪拌2 h。藉由TLC (於己烷中之20%乙酸乙酯)監測反應進展。在反應完成之後,使用二氯甲烷(50 mL)稀釋混合物,使用水(2 × 20 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物。藉由製備型HPLC [分析條件:-管柱:Inertsil ODS 3V (250 mm × 4.6 mm × 5 µm),移動相(A):於水中之0.1%氨,移動相(B):CH3
CN,流速:1.0 mL/min,B之組成:0/20, 12/80, 25/90, 27/20, 30/20]純化粗產物以獲得(1S,3R)-2-(2-氯乙醯基)-1-(4-(甲氧基羰基)苯基)-1,2,3,4-四氫異喹啉-3-甲酸甲酯(3)。在TLC中,極性斑點視為對應於另一異構體。LCMS (ES) m/z = 402 [M+H]+;1H NMR (400 MHz, DMSO-d6) δ ppm 3.04 - 3.15 (m, 1 H), 3.30 (m, 1 H), 3.46 (m, 3 H), 3.78 - 3.79 (s, 3 H), 3.98, 4.30 (m, 0.5 H, 0.5 H), 4.67 - 4.73 (m, 1 H), 5.21, 5.37 (m, 0.5 H, 0.5 H), 6.28, 6.52 (s, 0.5 H, 0.5 H), 7.11 - 7.21 (m, 3 H), 7.51 - 7.60 (m, 3 H), 7.88 - 7.89 (m, 2 H)。2個峰之對掌性HPLC純度為61.1%及36.7%;
及(1R,3R)-2-(2-氯乙醯基)-1-(4-(甲氧基羰基)苯基)-1,2,3,4-四氫異喹啉-3-甲酸甲酯(2)。在TLC中,非極性斑點視為對應於另一異構體。LCMS (ES) m/z = 402 [M+H]+;1H NMR (400 MHz, DMSO-d6) δ ppm 2.98 - 3.02 (m, 0.5 H), 3.07 - 3.12 (m, 1H), 3.21 - 3.22 (m, 0.5 H), 3.27 (s, 1 H), 3.66 (s, 2 H), 3.80 (s, 3 H), 4.22 - 4.26 (m, 1 H), 4.31 - 4.40 (m, 1 H), 4.65 - 4.75, 5.02 (m, 1 H), 6.42, 6.77 (m, 0.8 H, 0.3 H), 7.11 - 7.38 (m, 4 H), 7.63 (d, J = 7.2 Hz, 2 H), 7.83 - 7.89 (m, 2 H)。2個峰之對掌性HPLC純度為74.37%及25.6%。
將D-苯基丙胺酸甲酯鹽酸鹽(0.3 g, 1.391 mmol, 1當量)分配於乙酸乙酯與碳酸氫鈉溶液之間並攪拌15 min,分離乙酸乙酯層且使用乙酸乙酯(2 × 50 mL)萃取水層。使用水(50 mL)、鹽水(50 mL)洗滌合併之乙酸乙酯部分,藉由無水硫酸鈉乾燥,並在減壓下濃縮以獲得D-苯基丙胺酸甲酯。LCMS (ES) m/z = 180.1 [M+H]+
;1
H NMR (400 MHz, CDCl3
) δ ppm, 2.83 - 2.88 (m, 1 H), 3.07 - 3.11 (m, 1 H), 3.71 - 3.75 (m, 4 H), 7.18 - 7.32 (m, 5 H)。
向D-苯基丙胺酸甲酯(0.1 g, 0.557 mmol, 1當量)於甲苯(10 mL)中之溶液中添加4-甲醯基苯甲酸甲酯(0.0.09 g, 0.557 mmol, 1當量)。將反應混合物加熱至120℃並攪拌1 h。在減壓下濃縮混合物以獲得 (R,E)-4-(((1-甲氧基-1-側氧基-3-苯基丙烷-2-基)亞胺基)甲基)苯甲酸甲酯且所獲得粗產物未經任何進一步純化即用於下一步驟中。
將(R,E)-4-(((1-甲氧基-1-側氧基-3-苯基丙烷-2-基)亞胺基)甲基)苯甲酸甲酯(0.3 g粗製物)與三氟甲磺酸(2 mL)混合且將混合物加熱至130℃並攪拌18 h,且藉由LCMS分析混合物(LCMS展示水解產物(3R)-1-(4-羧基苯基)-1,2,3,4-四氫異喹啉-3-甲酸)。將混合物冷卻至0℃且添加10 mL無水甲醇。將所得混合物加熱至80℃並攪拌2 h。將混合物冷卻至0℃並使用三乙胺中和,且在減壓下濃縮。使用乙酸乙酯溶解所獲得粗製物,使用水(3 × 20 mL)、鹽水(20 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮。藉由急速層析使用於己烷中之20%乙酸乙酯作為洗脫劑純化粗產物以提供(3R)-1-(4-(甲氧基羰基)苯基)-1,2,3,4-四氫異喹啉-3-甲酸甲酯。LCMS (ES) m/z = 326.2 [M+H]+
。
在0℃及氮氣氛下,向(3R)-1-(4-(甲氧基羰基)苯基)-1,2,3,4-四氫異喹啉-3-甲酸甲酯(0.12 g, 0.368 mmol, 1當量)及碳酸氫鈉(0.06 g, 0.737 mmol, 2當量)於氯仿(5 mL)中之經攪拌混合物中添加2-氯乙醯氯(0.044 mL, 0.553 mmol, 1.5當量)。將所得混合物升溫至室溫並攪拌2 h。藉由TLC (於己烷中之20%乙酸乙酯)監測反應進展。在反應完成之後,使用二氯甲烷(50 mL)稀釋混合物,使用水(2 × 20 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物。藉由製備型HPLC [分析條件:-管柱:Inertsil ODS 3V (250 mm × 4.6 mm × 5 µm),移動相(A):於水中之0.1%氨,移動相(B):CH3
CN,流速:1.0 mL/min,B之組成:0/20, 12/80, 25/90, 27/20, 30/20]純化粗產物以獲得(1S,3R)-2-(2-氯乙醯基)-1-(4-(甲氧基羰基)苯基)-1,2,3,4-四氫異喹啉-3-甲酸甲酯(3)。在TLC中,極性斑點視為對應於另一異構體。LCMS (ES) m/z = 402 [M+H]+;1H NMR (400 MHz, DMSO-d6) δ ppm 3.04 - 3.15 (m, 1 H), 3.30 (m, 1 H), 3.46 (m, 3 H), 3.78 - 3.79 (s, 3 H), 3.98, 4.30 (m, 0.5 H, 0.5 H), 4.67 - 4.73 (m, 1 H), 5.21, 5.37 (m, 0.5 H, 0.5 H), 6.28, 6.52 (s, 0.5 H, 0.5 H), 7.11 - 7.21 (m, 3 H), 7.51 - 7.60 (m, 3 H), 7.88 - 7.89 (m, 2 H)。2個峰之對掌性HPLC純度為61.1%及36.7%;
及(1R,3R)-2-(2-氯乙醯基)-1-(4-(甲氧基羰基)苯基)-1,2,3,4-四氫異喹啉-3-甲酸甲酯(2)。在TLC中,非極性斑點視為對應於另一異構體。LCMS (ES) m/z = 402 [M+H]+;1H NMR (400 MHz, DMSO-d6) δ ppm 2.98 - 3.02 (m, 0.5 H), 3.07 - 3.12 (m, 1H), 3.21 - 3.22 (m, 0.5 H), 3.27 (s, 1 H), 3.66 (s, 2 H), 3.80 (s, 3 H), 4.22 - 4.26 (m, 1 H), 4.31 - 4.40 (m, 1 H), 4.65 - 4.75, 5.02 (m, 1 H), 6.42, 6.77 (m, 0.8 H, 0.3 H), 7.11 - 7.38 (m, 4 H), 7.63 (d, J = 7.2 Hz, 2 H), 7.83 - 7.89 (m, 2 H)。2個峰之對掌性HPLC純度為74.37%及25.6%。
程序 3 : 化合物 4 及 5 之合成 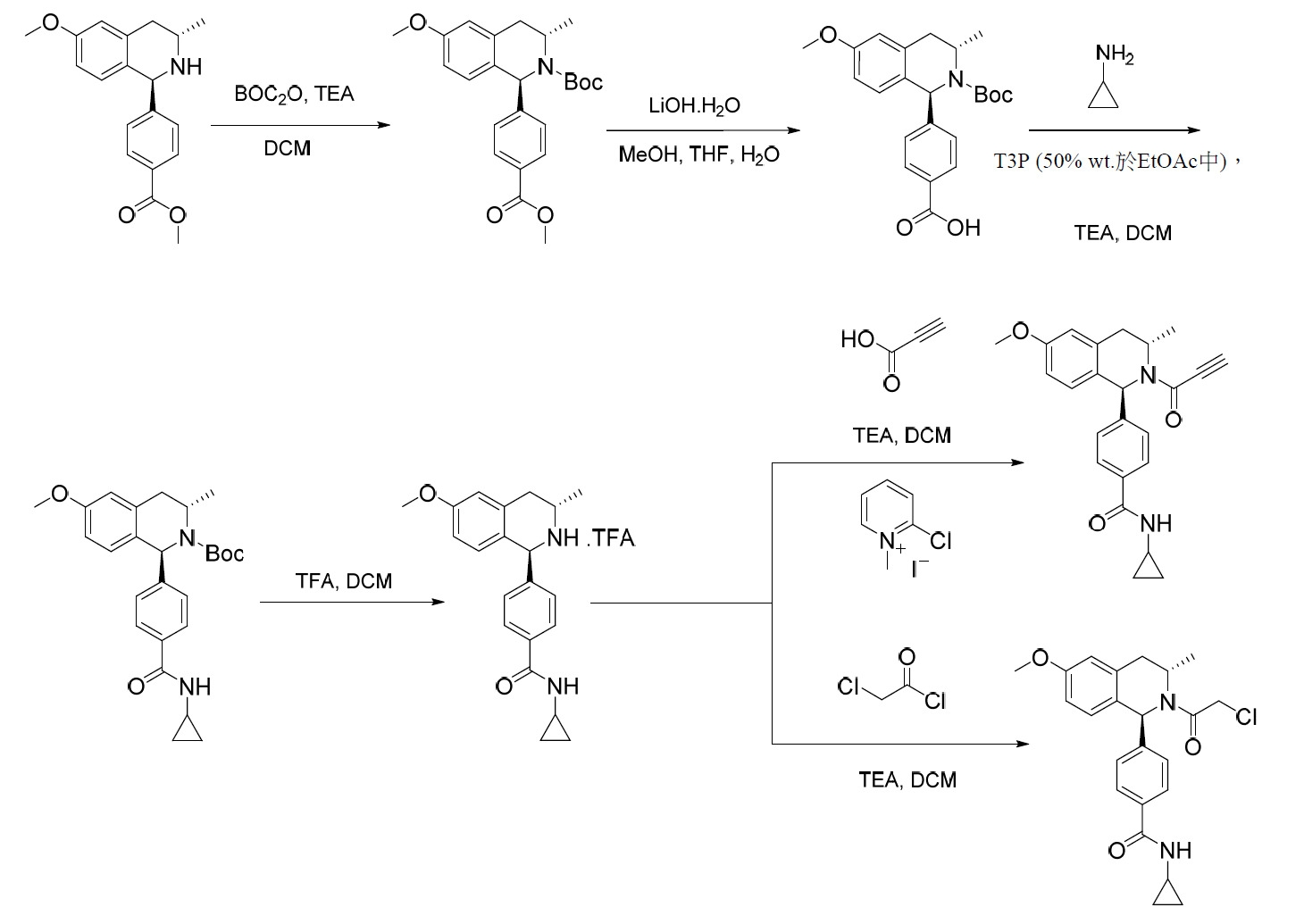 在室溫下,向化合物4-((1S,3S)-6-甲氧基-3-甲基-1,2,3,4-四氫異喹啉-1-基)苯甲酸甲酯(0.35 g, 1.12 mmol, 1.0當量)於DCM (10 mL)中之溶液中添加三乙胺(0.45 g, 4.49 mmol, 4.0當量)及二碳酸二-第三丁基酯(0.715 g, 2.24 mmol, 2.0當量)且將混合物攪拌16 h。TLC (於己烷中之50% EtOAc)顯示反應已完成。在減壓下濃縮反應混合物,且使用EtOAc (50 mL)稀釋粗製物,使用水(2 × 50 mL)洗滌。藉由無水Na2
SO4
乾燥有機層,在減壓下濃縮成粗產物(1S,3S)-6-甲氧基-1-(4-(甲氧基羰基)苯基)-3-甲基-3,4-二氫異喹啉-2(1H)-甲酸第三丁基酯。LC-MS (m/z): 356.0 [M-t
Bu+H]+
。
向化合物(1S,3S)-6-甲氧基-1-(4-(甲氧基羰基)苯基)-3-甲基-3,4-二氫異喹啉-2(1H)-甲酸第三丁基酯(0.650 g, 1.57 mmol, 1.0當量)於THF: MeOH: H2
O (9 mL:1 mL)之混合物中之溶液中添加氫氧化鋰(0.331 g, 7.89 mmol, 5.0當量)並在室溫下攪拌16 h。TLC (於己烷中之50% EtOAc)顯示反應已完成。在減壓下濃縮反應混合物,且使用5%檸檬酸溶液(pH=9)酸化粗製物。使用EtOAc (50 mL)稀釋反應混合物且分離有機層,並藉由無水Na2
SO4
乾燥,在減壓下濃縮以得到粗製4-((1S,3S)-2-(第三丁氧基羰基)-6-甲氧基-3-甲基-1,2,3,4-四氫異喹啉-1-基)苯甲酸。LC-MS (m/z): 396.0 [M+H]+
。
在0℃下,向化合物4-((1S,3S)-2-(第三丁氧基羰基)-6-甲氧基-3-甲基-1,2,3,4-四氫異喹啉-1-基)苯甲酸(0.38 g, 0.957 mmol, 1.0當量)於DCM (10 mL)中之溶液中添加三乙胺(0.4 mL, 2.87 mmol, 3.0當量)及環丙胺(0.65 g, 1.14 mmol, 1.2當量)且將混合物攪拌15 min。在相同溫度下向上述反應混合物中添加T3P (50% wt in EtOAc) (1.4 mL, 1.97 mmol, 1.2當量)並攪拌16 h。TLC (於己烷中之50% EtOAc)顯示反應已完成。在減壓下濃縮反應混合物,且使用EtOAc (50 mL)稀釋粗製物,使用水(2 × 50 mL)洗滌。藉由無水Na2
SO4
乾燥有機層,並在減壓下濃縮以得到粗製(1S,3S)-1-(4-(環丙基胺甲醯基)苯基)-6-甲氧基-3-甲基-3,4-二氫異喹啉-2(1H)-甲酸第三丁基酯。LC-MS (m/z): 381.0 [M-t
Bu+H]+
。
在0℃下,向化合物(1S,3S)-1-(4-(環丙基胺甲醯基)苯基)-6-甲氧基-3-甲基-3,4-二氫異喹啉-2(1H)-甲酸第三丁基酯(0.27 g, 0.61 mmol, 1.0當量)於DCM (10 mL)中之溶液中添加三氟乙酸(0.084 g, 0.74 mmol, 1.2當量)且將混合物攪拌2 h。TLC (於己烷中之50% EtOAc)顯示反應已完成。在減壓下濃縮反應混合物以得到粗製N-環丙基-4-((1S,3S)-6-甲氧基-3-甲基-2-(2,2,2-三氟乙醯基)-1,2,3,4-四氫-2λ4
-異喹啉-1-基)苯甲醯胺。LC-MS (m/z): 337.0 [M+H]+
。
在0℃下,向N-環丙基-4-((1S,3S)-6-甲氧基-3-甲基-2-(2,2,2-三氟乙醯基)-1,2,3,4-四氫-2λ4
-異喹啉-1-基)苯甲醯胺(0.2 g, 0.59 mmol, 1.0當量)於DCM (8.0 mL)中之溶液中添加TEA (0.12 g, 1.18 mmol, 2.0當量),攪拌15 min且然後在0℃下添加2-氯乙醯氯(0.08 g, 0.71 mmol, 1.2當量)。將混合物在室溫下攪拌1 hr。LCMS及TLC (於己烷中之50% EtOAc)顯示反應已完成。使用飽和NaHCO3
溶液(10 mL)稀釋反應混合物且使用DCM (2 × 50 mL)萃取。藉由Na2
SO4
乾燥有機層並濃縮以得到粗產物。藉由急速管柱層析使用15% EtOAc/己烷作為洗脫劑純化粗產物,隨後藉由製備型TLC使用於己烷中之30% EtOAc作為洗脫劑進行純化以得到4-((1S,3S)-2-(2-氯乙醯基)-6-甲氧基-3-甲基-1,2,3,4-四氫異喹啉-1-基)-N-環丙基苯甲醯胺。LC-MS (m/z): 413.3 [M+H]+
,1
H NMR (400 MHz, DMSO-d6
): δ 0.49 - 0.52 (m, 2H), 0.52 - 0.64 (m, 2 H), 0.66 (bs, 3 H), 2.65-2.66 (m, 1H), 2.77 - 2.81 (m, 1H), 3.71 (s, 3H), 4.37 - 4.74 (bs, 3H), 6.13 (s, 1H), 6.74 - 6.75 (m, 1H), 6.79 - 6.81 (m, 1H), 7.320-7.302 (m, 2H), 7.48 (d, J=8.4 Hz, 1H), 7.64 (s, 2H), 8.13 (s, 1H)。
向N-環丙基-4-((1S,3S)-6-甲氧基-3-甲基-2-(2,2,2-三氟乙醯基)-1,2,3,4-四氫-2λ4
-異喹啉-1-基)苯甲醯胺(0.2 g, 0.59 mmol, 1.0當量)於DCM (10.0 mL)中之溶液中添加三乙胺(0.162 g, 1.42 mmol, 2.4當量)及丙炔酸(0.041 mL, 0.59 mmol, 1.0當量)並攪拌15分鐘。向上述反應混合物中添加2-氯-1-甲基吡啶-1-碘化鎓(0.182 g, 0.71 mmol, 1.2當量)並攪拌16小時。LCMS及TLC (於DCM中之5% MeOH)顯示反應已完成。使用水(10 mL)稀釋反應混合物且分離有機層,藉由Na2
SO4
乾燥並在減壓下濃縮以得到粗產物。藉由製備型TLC使用於己烷中之70% EtOAc作為洗脫劑純化粗產物以得到N-環丙基-4-((1S,3S)-6-甲氧基-3-甲基-2-丙炔醯基-1,2,3,4-四氫異喹啉-1-基)苯甲醯胺。LC-MS (m/z): 389.0 [M+H]+
,1
H NMR (400 MHz, DMSO-d6
): δ 0.49 - 0.51 (m, 2H), 0.52 - 0.65 (m, 2H), 0.66 (bs, 3H), 2.65 - 2.66 (m, 1H), 2.78 - 2.82 (m, 1H), 3.71 (s, 3H), 4.37 - 4.74 (bs, 3H), 6.13 (s, 1H), 6.75 - 6.78 (m, 1H), 6.79 - 6.81 (m, 1H), 7.32 - 7.30 (m, 2H), 7.48 (d, J=8.4 Hz, 1H), 7.64 (s, 2H), 8.13 (s, 1H)。
在室溫下,向化合物4-((1S,3S)-6-甲氧基-3-甲基-1,2,3,4-四氫異喹啉-1-基)苯甲酸甲酯(0.35 g, 1.12 mmol, 1.0當量)於DCM (10 mL)中之溶液中添加三乙胺(0.45 g, 4.49 mmol, 4.0當量)及二碳酸二-第三丁基酯(0.715 g, 2.24 mmol, 2.0當量)且將混合物攪拌16 h。TLC (於己烷中之50% EtOAc)顯示反應已完成。在減壓下濃縮反應混合物,且使用EtOAc (50 mL)稀釋粗製物,使用水(2 × 50 mL)洗滌。藉由無水Na2
SO4
乾燥有機層,在減壓下濃縮成粗產物(1S,3S)-6-甲氧基-1-(4-(甲氧基羰基)苯基)-3-甲基-3,4-二氫異喹啉-2(1H)-甲酸第三丁基酯。LC-MS (m/z): 356.0 [M-t
Bu+H]+
。
向化合物(1S,3S)-6-甲氧基-1-(4-(甲氧基羰基)苯基)-3-甲基-3,4-二氫異喹啉-2(1H)-甲酸第三丁基酯(0.650 g, 1.57 mmol, 1.0當量)於THF: MeOH: H2
O (9 mL:1 mL)之混合物中之溶液中添加氫氧化鋰(0.331 g, 7.89 mmol, 5.0當量)並在室溫下攪拌16 h。TLC (於己烷中之50% EtOAc)顯示反應已完成。在減壓下濃縮反應混合物,且使用5%檸檬酸溶液(pH=9)酸化粗製物。使用EtOAc (50 mL)稀釋反應混合物且分離有機層,並藉由無水Na2
SO4
乾燥,在減壓下濃縮以得到粗製4-((1S,3S)-2-(第三丁氧基羰基)-6-甲氧基-3-甲基-1,2,3,4-四氫異喹啉-1-基)苯甲酸。LC-MS (m/z): 396.0 [M+H]+
。
在0℃下,向化合物4-((1S,3S)-2-(第三丁氧基羰基)-6-甲氧基-3-甲基-1,2,3,4-四氫異喹啉-1-基)苯甲酸(0.38 g, 0.957 mmol, 1.0當量)於DCM (10 mL)中之溶液中添加三乙胺(0.4 mL, 2.87 mmol, 3.0當量)及環丙胺(0.65 g, 1.14 mmol, 1.2當量)且將混合物攪拌15 min。在相同溫度下向上述反應混合物中添加T3P (50% wt in EtOAc) (1.4 mL, 1.97 mmol, 1.2當量)並攪拌16 h。TLC (於己烷中之50% EtOAc)顯示反應已完成。在減壓下濃縮反應混合物,且使用EtOAc (50 mL)稀釋粗製物,使用水(2 × 50 mL)洗滌。藉由無水Na2
SO4
乾燥有機層,並在減壓下濃縮以得到粗製(1S,3S)-1-(4-(環丙基胺甲醯基)苯基)-6-甲氧基-3-甲基-3,4-二氫異喹啉-2(1H)-甲酸第三丁基酯。LC-MS (m/z): 381.0 [M-t
Bu+H]+
。
在0℃下,向化合物(1S,3S)-1-(4-(環丙基胺甲醯基)苯基)-6-甲氧基-3-甲基-3,4-二氫異喹啉-2(1H)-甲酸第三丁基酯(0.27 g, 0.61 mmol, 1.0當量)於DCM (10 mL)中之溶液中添加三氟乙酸(0.084 g, 0.74 mmol, 1.2當量)且將混合物攪拌2 h。TLC (於己烷中之50% EtOAc)顯示反應已完成。在減壓下濃縮反應混合物以得到粗製N-環丙基-4-((1S,3S)-6-甲氧基-3-甲基-2-(2,2,2-三氟乙醯基)-1,2,3,4-四氫-2λ4
-異喹啉-1-基)苯甲醯胺。LC-MS (m/z): 337.0 [M+H]+
。
在0℃下,向N-環丙基-4-((1S,3S)-6-甲氧基-3-甲基-2-(2,2,2-三氟乙醯基)-1,2,3,4-四氫-2λ4
-異喹啉-1-基)苯甲醯胺(0.2 g, 0.59 mmol, 1.0當量)於DCM (8.0 mL)中之溶液中添加TEA (0.12 g, 1.18 mmol, 2.0當量),攪拌15 min且然後在0℃下添加2-氯乙醯氯(0.08 g, 0.71 mmol, 1.2當量)。將混合物在室溫下攪拌1 hr。LCMS及TLC (於己烷中之50% EtOAc)顯示反應已完成。使用飽和NaHCO3
溶液(10 mL)稀釋反應混合物且使用DCM (2 × 50 mL)萃取。藉由Na2
SO4
乾燥有機層並濃縮以得到粗產物。藉由急速管柱層析使用15% EtOAc/己烷作為洗脫劑純化粗產物,隨後藉由製備型TLC使用於己烷中之30% EtOAc作為洗脫劑進行純化以得到4-((1S,3S)-2-(2-氯乙醯基)-6-甲氧基-3-甲基-1,2,3,4-四氫異喹啉-1-基)-N-環丙基苯甲醯胺。LC-MS (m/z): 413.3 [M+H]+
,1
H NMR (400 MHz, DMSO-d6
): δ 0.49 - 0.52 (m, 2H), 0.52 - 0.64 (m, 2 H), 0.66 (bs, 3 H), 2.65-2.66 (m, 1H), 2.77 - 2.81 (m, 1H), 3.71 (s, 3H), 4.37 - 4.74 (bs, 3H), 6.13 (s, 1H), 6.74 - 6.75 (m, 1H), 6.79 - 6.81 (m, 1H), 7.320-7.302 (m, 2H), 7.48 (d, J=8.4 Hz, 1H), 7.64 (s, 2H), 8.13 (s, 1H)。
向N-環丙基-4-((1S,3S)-6-甲氧基-3-甲基-2-(2,2,2-三氟乙醯基)-1,2,3,4-四氫-2λ4
-異喹啉-1-基)苯甲醯胺(0.2 g, 0.59 mmol, 1.0當量)於DCM (10.0 mL)中之溶液中添加三乙胺(0.162 g, 1.42 mmol, 2.4當量)及丙炔酸(0.041 mL, 0.59 mmol, 1.0當量)並攪拌15分鐘。向上述反應混合物中添加2-氯-1-甲基吡啶-1-碘化鎓(0.182 g, 0.71 mmol, 1.2當量)並攪拌16小時。LCMS及TLC (於DCM中之5% MeOH)顯示反應已完成。使用水(10 mL)稀釋反應混合物且分離有機層,藉由Na2
SO4
乾燥並在減壓下濃縮以得到粗產物。藉由製備型TLC使用於己烷中之70% EtOAc作為洗脫劑純化粗產物以得到N-環丙基-4-((1S,3S)-6-甲氧基-3-甲基-2-丙炔醯基-1,2,3,4-四氫異喹啉-1-基)苯甲醯胺。LC-MS (m/z): 389.0 [M+H]+
,1
H NMR (400 MHz, DMSO-d6
): δ 0.49 - 0.51 (m, 2H), 0.52 - 0.65 (m, 2H), 0.66 (bs, 3H), 2.65 - 2.66 (m, 1H), 2.78 - 2.82 (m, 1H), 3.71 (s, 3H), 4.37 - 4.74 (bs, 3H), 6.13 (s, 1H), 6.75 - 6.78 (m, 1H), 6.79 - 6.81 (m, 1H), 7.32 - 7.30 (m, 2H), 7.48 (d, J=8.4 Hz, 1H), 7.64 (s, 2H), 8.13 (s, 1H)。
程序 4 : 化合物 6 及 7 之合成  在0℃下,向(R)-2-甲基環氧乙烷(3.0 g, 51.72 mmol, 1.0當量)於THF (30 mL)中之溶液中逐滴添加(3-甲氧基苯基)溴化鎂(62 mL, 62.06 mmol, 1.2當量)。將反應混合物在室溫下攪拌6小時。此後,使用氯化銨水溶液(10 mL)淬滅反應混合物且將產物萃取至乙酸乙酯(100 mL)中。使用水(2 × 20 mL)、鹽水(15 mL)洗滌有機層,藉由無水MgSO4
乾燥,過濾並在減壓下濃縮以獲得粗產物,藉由矽膠急速管柱層析(正己烷/ EtOAc = 8:1, Rf
= 0.24)純化以得到(R)-1-(3-甲氧基苯基)丙烷-2-醇。1
H NMR (400 MHz, DMSO-d6
) δ ppm 1.00 (d, J = 6.4 Hz, 3H), 2.49 - 2.51 (m, 1H), 2.61 - 2.66 (m, 1H), 3.70 (s, 3H), 3.76 - 3.81 (m, 1H), 4.51 (d, J = 4.8 Hz, 1H), 6.71 - 6.74 (m, 3H), 7.14 (t, J = 7.8 Hz, 1H)。
在0℃下,向(R)-1-(3-甲氧基苯基)丙烷-2-醇(2.8 g, 16.86 mmol, 1.0當量)於DCM (30 mL)中之溶液中添加三乙胺(5.1 g, 50.58 mmol, 3.0當量),隨後添加甲磺醯氯(2.8 g, 25.30 mmol, 1.5當量)。將混合物在0℃及N2
氣氛下攪拌1.0 h。TLC (於正己烷中之30% EtOAc)顯示反應已完成。使用NaHCO3
飽和水溶液(15 mL)稀釋反應液且使用DCM (50 mL)萃取。藉由無水Na2
SO4
乾燥有機層,過濾並在減壓下濃縮以得到(R)-甲烷磺酸1-(3-甲氧基苯基)丙烷-2-基酯。1
H NMR (400 MHz, CDCl3
) δ ppm 1.47 (d, J = 6.0 Hz, 3H), 2.56 (s, 3H), 2.84 - 2.89 (m, 1H), 2.93 - 2.99 (m, 1H), 3.79 (s, 3H), 4.87 - 4.92 (m, 1H), 6.77 - 6.82 (m, 3H), 7.21 - 7.23 (m, 1H)。
在室溫下,向(R)-甲烷磺酸1-(3-甲氧基苯基)丙烷-2-基酯(3.8 g, 15.57 mmol, 1.0當量)於DMF (38 mL)中之溶液中添加疊氮化鈉(1.2 g, 18.68 mmol, 1.2當量)。將混合物在80℃下攪拌16 h。TLC (於正己烷中之5% EtOAc)顯示反應已完成。使用水(15 mL)及EtOAc (50 mL)稀釋反應液,分離有機層,使用水(5 × 25 mL)、鹽水(10 mL)洗滌,藉由無水MgSO4
乾燥,過濾並在減壓下濃縮以提供粗產物。藉由矽膠急速管柱層析(正己烷/ EtOAc = 9.7: 0.2, Rf
= 0.6)純化粗產物以得到(S)-1-(2-疊氮基丙基)-3-甲氧基苯。1
H NMR (400 MHz, CDCl3
) δ ppm 1.26 (d, J = 6.0 Hz, 3H), 2.66 - 2.71 (m, 1H), 2.78 - 2.83 (m, 1H), 3.62 - 3.71 (m, 1H), 3.80 (s, 3H), 6.74 - 6.79 (m, 3H), 7.20 - 7.22 (m, 1H)。
在室溫下,向(S)-1-(2-疊氮基丙基)-3-甲氧基苯(2.37 g, 12.40 mmol, 1.0當量)於乙酸乙酯(23 mL)中之溶液中添加Pd/C (150 mg 10% Pd)。將所得反應混合物在100 PSI及室溫下於帕爾振盪器(parr shaker)中氫化20 h。此後,藉由經由矽藻土過濾來去除觸媒,在減壓下濃縮濾液以提供(S)-1-(3-甲氧基苯基)丙烷-2-胺。LCMS (ES) m/z = 343.3 [M+H]+
。1
H NMR (400 MHz, CDCl3
) δ ppm 1.13 (d, J = 6.0 Hz, 3H), 2.44 - 2.54 (m, 1H), 2.67 - 2.72 (m, 1H), 3.16 - 3.21 (m, 1H), 3.79 (s, 3H), 6.74 - 6.78 (m, 3H), 7.21 (t, J = 7.8 Hz, 1H)。在1
H NMR中未觀察到NH2
質子。
將(S)-1-(3-甲氧基苯基)丙烷-2-胺(0.3 g, 1.81 mmol, 1.0當量)及4-甲醯基苯甲酸甲酯(0.36 g, 2.18 mmol, 1.2當量)於甲苯(4 mL)中之溶液在90℃下於微波中輻照20 min。此後,在減壓下濃縮揮發性部分以提供(S)-4-(((1-(3-甲氧基苯基)丙烷-2-基)亞胺基)甲基)苯甲酸甲酯。此產物原樣用於環化步驟中。
將(S)-4-(((1-(3-甲氧基苯基)丙烷-2-基)亞胺基)甲基)苯甲酸甲酯(前一步驟產物)於TFA (2 mL)中之溶液在140℃下於微波中輻照45 min。此後,在減壓下濃縮揮發性部分並使用NaHCO3
飽和水溶液(10 mL)及EtOAc (40 mL)稀釋。分離有機層,使用鹽水(10 mL)洗滌,藉由無水MgSO4
乾燥,過濾並在減壓下濃縮以獲得粗產物。藉由矽膠急速管柱層析(正己烷/ EtOAc = 3:2, Rf
= 0.4 -對於非極性斑點,Rf
= 0.3 -對於極性斑點)純化所獲得粗產物以得到4-((1R,3S)-6-甲氧基-3-甲基-1,2,3,4-四氫異喹啉-1-基)苯甲酸甲酯(較小極性之斑點)及4-((1S,3S)-6-甲氧基-3-甲基-1,2,3,4-四氫異喹啉-1-基)苯甲酸甲酯(極性斑點)。
4-((1R,3S)-6-甲氧基-3-甲基-1,2,3,4-四氫異喹啉-1-基)苯甲酸甲酯:LCMS (ES) m/z = 312.2 [M+H]+
,1
H NMR (400 MHz, CDCl3
) δ ppm 1.26 (d, J = 6.0 Hz, 3 H), 2.74 - 2.76 (m, 2 H), 3.19 - 3.20 (m, 1 H), 3.76 (s, 3 H), 3.90 (s, 3 H), 5.12 (s, 1 H), 6.49 - 6.64 (m, 3 H), 7.41 (d, J = 8.0 Hz, 2 H), 8.00 (d, J = 7.6 Hz, 2 H)。在1
H NMR中未觀察到NH質子。
4-((1S,3S)-6-甲氧基-3-甲基-1,2,3,4-四氫異喹啉-1-基)苯甲酸甲酯: LCMS (ES) m/z = 312.2 [M+H]+
。1
H NMR (400 MHz, CDCl3
) δ ppm 1.11 (d, J = 6.4 Hz, 3 H), 2.53 - 2.60 (m, 1 H), 2.82 - 2.87 (m, 1 H), 3.04 - 3.07 (m, 1 H), 3.80 (s, 3 H), 3.89 (s, 3 H), 5.24 (s, 1 H), 6.67 - 6.69 (m, 2 H), 6.79 (d, J = 8.8 Hz, 1 H), 7.20 (d, J = 8.4 Hz, 2 H), 7.94 (d, J = 8.0 Hz, 2 H)。在1
H NMR中未觀察到NH質子。
在0℃下,向4-((1R,3S)-6-甲氧基-3-甲基-1,2,3,4-四氫異喹啉-1-基)苯甲酸甲酯(0.2 g, 0.64 mmol, 1當量)於DCM (5.0 mL)中之溶液中添加三乙胺(0.19 g, 1.92 mmol, 3.0當量),隨後添加2-氯乙醯氯(0.095 g, 0.83 mmol, 1.3當量)。將混合物在0℃及N2
氣氛下攪拌2.0 h。TLC (於正己烷中之35% EtOAc)顯示反應已完成。然後,使用NaHCO3
飽和水溶液(5 mL)稀釋反應液且使用DCM (25 mL)萃取。藉由無水Na2
SO4
乾燥有機層,過濾並在減壓下濃縮。藉由製備型TLC使用於正己烷中之40% EtOAc作為移動相純化粗產物以得到4-((1R,3S)-2-(2-氯乙醯基)-6-甲氧基-3-甲基-1,2,3,4-四氫異喹啉-1-基)苯甲酸甲酯(6
): LCMS (ES) m/z = 388.1 [M+H]+
。1
H NMR (400 MHz, DMSO-d6
) (在70℃下) δ ppm 1.10 (d, J = 5.6 Hz, 3H), 3.00 (bs, 2H), 3.77 (m, 3H), 3.82 (s, 3H), 4.21 (bs, 1H), 4.47 (q, J = 13.6 Hz, 2H), 6.45 (bs, 1H), 6.83 - 6.85 (m, 2H), 7.16 (bs, 1H), 7.26 (d, J = 8.0 Hz, 2H), 7.88 (d, J = 8.0 Hz, 2H)。
在0℃下,向4-((1S,3S)-6-甲氧基-3-甲基-1,2,3,4-四氫異喹啉-1-基)苯甲酸甲酯(0.11 g, 0.35 mmol, 1當量)於DCM (4.0 mL)中之溶液中添加三乙胺(0.1 g, 1.05 mmol, 3.0當量),隨後添加2-氯乙醯氯(0.05 g, 0.45 mmol, 1.3當量)。將混合物在0℃及N2
氣氛下攪拌2.0 h。TLC (於正己烷中之35% EtOAc)顯示反應已完成。然後,使用NaHCO3
飽和水溶液(5 mL)稀釋反應液且使用DCM (25 mL)萃取。藉由無水Na2
SO4
乾燥有機層,過濾並在減壓下濃縮。藉由製備型TLC使用於正己烷中之40% EtOAc作為移動相純化粗產物以得到4-((1S,3S)-2-(2-氯乙醯基)-6-甲氧基-3-甲基-1,2,3,4-四氫異喹啉-1-基)苯甲酸甲酯(7
): LCMS (ES) m/z = 388.0 [M+H]+
。1
H NMR (400 MHz, DMSO-d6
) (在70℃下) δ ppm 0.97 (bs, 3H), 2.65 (bs, 2H), 3.71 (s, 3H), 3.79 (s, 3H), 4.40 (bs, 1H), 4.75 (bs, 2H), 6.17 (bs, 1H), 6.76 - 6.82 (m, 2H), 7.40 -7.49 (m, 3H), 7.81 (bs, 2H)。
在0℃下,向(R)-2-甲基環氧乙烷(3.0 g, 51.72 mmol, 1.0當量)於THF (30 mL)中之溶液中逐滴添加(3-甲氧基苯基)溴化鎂(62 mL, 62.06 mmol, 1.2當量)。將反應混合物在室溫下攪拌6小時。此後,使用氯化銨水溶液(10 mL)淬滅反應混合物且將產物萃取至乙酸乙酯(100 mL)中。使用水(2 × 20 mL)、鹽水(15 mL)洗滌有機層,藉由無水MgSO4
乾燥,過濾並在減壓下濃縮以獲得粗產物,藉由矽膠急速管柱層析(正己烷/ EtOAc = 8:1, Rf
= 0.24)純化以得到(R)-1-(3-甲氧基苯基)丙烷-2-醇。1
H NMR (400 MHz, DMSO-d6
) δ ppm 1.00 (d, J = 6.4 Hz, 3H), 2.49 - 2.51 (m, 1H), 2.61 - 2.66 (m, 1H), 3.70 (s, 3H), 3.76 - 3.81 (m, 1H), 4.51 (d, J = 4.8 Hz, 1H), 6.71 - 6.74 (m, 3H), 7.14 (t, J = 7.8 Hz, 1H)。
在0℃下,向(R)-1-(3-甲氧基苯基)丙烷-2-醇(2.8 g, 16.86 mmol, 1.0當量)於DCM (30 mL)中之溶液中添加三乙胺(5.1 g, 50.58 mmol, 3.0當量),隨後添加甲磺醯氯(2.8 g, 25.30 mmol, 1.5當量)。將混合物在0℃及N2
氣氛下攪拌1.0 h。TLC (於正己烷中之30% EtOAc)顯示反應已完成。使用NaHCO3
飽和水溶液(15 mL)稀釋反應液且使用DCM (50 mL)萃取。藉由無水Na2
SO4
乾燥有機層,過濾並在減壓下濃縮以得到(R)-甲烷磺酸1-(3-甲氧基苯基)丙烷-2-基酯。1
H NMR (400 MHz, CDCl3
) δ ppm 1.47 (d, J = 6.0 Hz, 3H), 2.56 (s, 3H), 2.84 - 2.89 (m, 1H), 2.93 - 2.99 (m, 1H), 3.79 (s, 3H), 4.87 - 4.92 (m, 1H), 6.77 - 6.82 (m, 3H), 7.21 - 7.23 (m, 1H)。
在室溫下,向(R)-甲烷磺酸1-(3-甲氧基苯基)丙烷-2-基酯(3.8 g, 15.57 mmol, 1.0當量)於DMF (38 mL)中之溶液中添加疊氮化鈉(1.2 g, 18.68 mmol, 1.2當量)。將混合物在80℃下攪拌16 h。TLC (於正己烷中之5% EtOAc)顯示反應已完成。使用水(15 mL)及EtOAc (50 mL)稀釋反應液,分離有機層,使用水(5 × 25 mL)、鹽水(10 mL)洗滌,藉由無水MgSO4
乾燥,過濾並在減壓下濃縮以提供粗產物。藉由矽膠急速管柱層析(正己烷/ EtOAc = 9.7: 0.2, Rf
= 0.6)純化粗產物以得到(S)-1-(2-疊氮基丙基)-3-甲氧基苯。1
H NMR (400 MHz, CDCl3
) δ ppm 1.26 (d, J = 6.0 Hz, 3H), 2.66 - 2.71 (m, 1H), 2.78 - 2.83 (m, 1H), 3.62 - 3.71 (m, 1H), 3.80 (s, 3H), 6.74 - 6.79 (m, 3H), 7.20 - 7.22 (m, 1H)。
在室溫下,向(S)-1-(2-疊氮基丙基)-3-甲氧基苯(2.37 g, 12.40 mmol, 1.0當量)於乙酸乙酯(23 mL)中之溶液中添加Pd/C (150 mg 10% Pd)。將所得反應混合物在100 PSI及室溫下於帕爾振盪器(parr shaker)中氫化20 h。此後,藉由經由矽藻土過濾來去除觸媒,在減壓下濃縮濾液以提供(S)-1-(3-甲氧基苯基)丙烷-2-胺。LCMS (ES) m/z = 343.3 [M+H]+
。1
H NMR (400 MHz, CDCl3
) δ ppm 1.13 (d, J = 6.0 Hz, 3H), 2.44 - 2.54 (m, 1H), 2.67 - 2.72 (m, 1H), 3.16 - 3.21 (m, 1H), 3.79 (s, 3H), 6.74 - 6.78 (m, 3H), 7.21 (t, J = 7.8 Hz, 1H)。在1
H NMR中未觀察到NH2
質子。
將(S)-1-(3-甲氧基苯基)丙烷-2-胺(0.3 g, 1.81 mmol, 1.0當量)及4-甲醯基苯甲酸甲酯(0.36 g, 2.18 mmol, 1.2當量)於甲苯(4 mL)中之溶液在90℃下於微波中輻照20 min。此後,在減壓下濃縮揮發性部分以提供(S)-4-(((1-(3-甲氧基苯基)丙烷-2-基)亞胺基)甲基)苯甲酸甲酯。此產物原樣用於環化步驟中。
將(S)-4-(((1-(3-甲氧基苯基)丙烷-2-基)亞胺基)甲基)苯甲酸甲酯(前一步驟產物)於TFA (2 mL)中之溶液在140℃下於微波中輻照45 min。此後,在減壓下濃縮揮發性部分並使用NaHCO3
飽和水溶液(10 mL)及EtOAc (40 mL)稀釋。分離有機層,使用鹽水(10 mL)洗滌,藉由無水MgSO4
乾燥,過濾並在減壓下濃縮以獲得粗產物。藉由矽膠急速管柱層析(正己烷/ EtOAc = 3:2, Rf
= 0.4 -對於非極性斑點,Rf
= 0.3 -對於極性斑點)純化所獲得粗產物以得到4-((1R,3S)-6-甲氧基-3-甲基-1,2,3,4-四氫異喹啉-1-基)苯甲酸甲酯(較小極性之斑點)及4-((1S,3S)-6-甲氧基-3-甲基-1,2,3,4-四氫異喹啉-1-基)苯甲酸甲酯(極性斑點)。
4-((1R,3S)-6-甲氧基-3-甲基-1,2,3,4-四氫異喹啉-1-基)苯甲酸甲酯:LCMS (ES) m/z = 312.2 [M+H]+
,1
H NMR (400 MHz, CDCl3
) δ ppm 1.26 (d, J = 6.0 Hz, 3 H), 2.74 - 2.76 (m, 2 H), 3.19 - 3.20 (m, 1 H), 3.76 (s, 3 H), 3.90 (s, 3 H), 5.12 (s, 1 H), 6.49 - 6.64 (m, 3 H), 7.41 (d, J = 8.0 Hz, 2 H), 8.00 (d, J = 7.6 Hz, 2 H)。在1
H NMR中未觀察到NH質子。
4-((1S,3S)-6-甲氧基-3-甲基-1,2,3,4-四氫異喹啉-1-基)苯甲酸甲酯: LCMS (ES) m/z = 312.2 [M+H]+
。1
H NMR (400 MHz, CDCl3
) δ ppm 1.11 (d, J = 6.4 Hz, 3 H), 2.53 - 2.60 (m, 1 H), 2.82 - 2.87 (m, 1 H), 3.04 - 3.07 (m, 1 H), 3.80 (s, 3 H), 3.89 (s, 3 H), 5.24 (s, 1 H), 6.67 - 6.69 (m, 2 H), 6.79 (d, J = 8.8 Hz, 1 H), 7.20 (d, J = 8.4 Hz, 2 H), 7.94 (d, J = 8.0 Hz, 2 H)。在1
H NMR中未觀察到NH質子。
在0℃下,向4-((1R,3S)-6-甲氧基-3-甲基-1,2,3,4-四氫異喹啉-1-基)苯甲酸甲酯(0.2 g, 0.64 mmol, 1當量)於DCM (5.0 mL)中之溶液中添加三乙胺(0.19 g, 1.92 mmol, 3.0當量),隨後添加2-氯乙醯氯(0.095 g, 0.83 mmol, 1.3當量)。將混合物在0℃及N2
氣氛下攪拌2.0 h。TLC (於正己烷中之35% EtOAc)顯示反應已完成。然後,使用NaHCO3
飽和水溶液(5 mL)稀釋反應液且使用DCM (25 mL)萃取。藉由無水Na2
SO4
乾燥有機層,過濾並在減壓下濃縮。藉由製備型TLC使用於正己烷中之40% EtOAc作為移動相純化粗產物以得到4-((1R,3S)-2-(2-氯乙醯基)-6-甲氧基-3-甲基-1,2,3,4-四氫異喹啉-1-基)苯甲酸甲酯(6
): LCMS (ES) m/z = 388.1 [M+H]+
。1
H NMR (400 MHz, DMSO-d6
) (在70℃下) δ ppm 1.10 (d, J = 5.6 Hz, 3H), 3.00 (bs, 2H), 3.77 (m, 3H), 3.82 (s, 3H), 4.21 (bs, 1H), 4.47 (q, J = 13.6 Hz, 2H), 6.45 (bs, 1H), 6.83 - 6.85 (m, 2H), 7.16 (bs, 1H), 7.26 (d, J = 8.0 Hz, 2H), 7.88 (d, J = 8.0 Hz, 2H)。
在0℃下,向4-((1S,3S)-6-甲氧基-3-甲基-1,2,3,4-四氫異喹啉-1-基)苯甲酸甲酯(0.11 g, 0.35 mmol, 1當量)於DCM (4.0 mL)中之溶液中添加三乙胺(0.1 g, 1.05 mmol, 3.0當量),隨後添加2-氯乙醯氯(0.05 g, 0.45 mmol, 1.3當量)。將混合物在0℃及N2
氣氛下攪拌2.0 h。TLC (於正己烷中之35% EtOAc)顯示反應已完成。然後,使用NaHCO3
飽和水溶液(5 mL)稀釋反應液且使用DCM (25 mL)萃取。藉由無水Na2
SO4
乾燥有機層,過濾並在減壓下濃縮。藉由製備型TLC使用於正己烷中之40% EtOAc作為移動相純化粗產物以得到4-((1S,3S)-2-(2-氯乙醯基)-6-甲氧基-3-甲基-1,2,3,4-四氫異喹啉-1-基)苯甲酸甲酯(7
): LCMS (ES) m/z = 388.0 [M+H]+
。1
H NMR (400 MHz, DMSO-d6
) (在70℃下) δ ppm 0.97 (bs, 3H), 2.65 (bs, 2H), 3.71 (s, 3H), 3.79 (s, 3H), 4.40 (bs, 1H), 4.75 (bs, 2H), 6.17 (bs, 1H), 6.76 - 6.82 (m, 2H), 7.40 -7.49 (m, 3H), 7.81 (bs, 2H)。
程序 5 :化合物 8 、 9 及 10 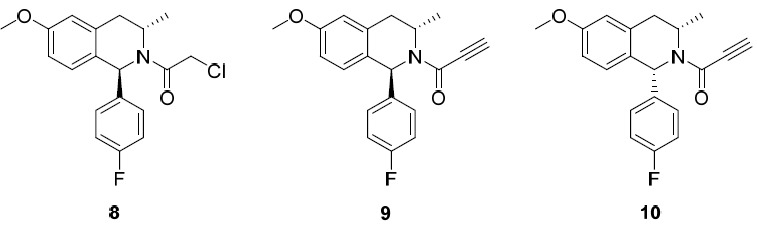 根據實例6及7中所提供之程序使用適當起始材料來合成化合物8、9及10。
化合物8: LC-MS (m/z): 348.3 [M+H]+
。1
H NMR (400 MHz, DMSO-d6
) δ ppm 0.95 (d, J = 5.2 Hz, 3H), 2.58 - 2.65 (m, 1H), 3.02 - 3.12 (m, 1H), 3.71 (s, 3H), 4.41 - 4.72 (m, 3H), 6.11 (s, 1H), 6.76 (s, 1H), 6.80 (d, J = 8.4 Hz, 1H), 7.04 (bs, 2H), 7.27 (dd, J = 8.4, 5.6 Hz, 2H), 7.45 (d, J = 8.4 Hz, 1H)。在60℃下記錄此NMR。
化合物9: LC-MS (m/z): 324.0 [M+H]+
。1
H NMR (400 MHz, DMSO-d6
) δ ppm 0.92 (d, J = 6.0 Hz, 1H), 1.07 (d, J = 6.0 Hz, 2H), 2.57 - 2.73 (m, 1H), 2.82 - 2.90 (m, 1H), 3.72 - 3.73 (m, 3H), 4.22 (s, 0.4H), 4.49 (s, 0.6H), 4.70 (bs, 0.5H), 4.92 (bs, 0.5H), 6.09 (s, 0.6H), 6.31 (s, 0.4H), 6.78 - 6.85 (m, 2H), 7.01 (t, J = 9.0 Hz, 1H), 7.08 (t, J = 8.4 Hz, 1H), 7.21 - 7.23 (m, 2H), 7.40 (d, J = 8.0 Hz, 0.6H), 7.57 (d, J = 8.4 Hz, 0.4H)。在60℃下記錄此NMR。
化合物10: LC-MS (m/z): 324.3 [M+H]+
。1
H NMR (400 MHz, DMSO-d6
) δ ppm 1.09 - 1.13 (m, 3H), 2.21 - 2.28 (m, 0.5H), 2.90 - 2.94 (m, 0.5H), 3.07 - 3.08 (m, 1H), 3.77 (s, 3H), 4.07 (bs, 0.5H), 4.49 - 4.57 (m, 1.5H), 6.53 - 6.56 (m, 1H), 6.82 - 6.88 (m, 2H), 7.02 - 7.14 (m, 4.5H), 7.32 - 7.36 (m, 0.5H)。在60℃下記錄此NMR。
根據實例6及7中所提供之程序使用適當起始材料來合成化合物8、9及10。
化合物8: LC-MS (m/z): 348.3 [M+H]+
。1
H NMR (400 MHz, DMSO-d6
) δ ppm 0.95 (d, J = 5.2 Hz, 3H), 2.58 - 2.65 (m, 1H), 3.02 - 3.12 (m, 1H), 3.71 (s, 3H), 4.41 - 4.72 (m, 3H), 6.11 (s, 1H), 6.76 (s, 1H), 6.80 (d, J = 8.4 Hz, 1H), 7.04 (bs, 2H), 7.27 (dd, J = 8.4, 5.6 Hz, 2H), 7.45 (d, J = 8.4 Hz, 1H)。在60℃下記錄此NMR。
化合物9: LC-MS (m/z): 324.0 [M+H]+
。1
H NMR (400 MHz, DMSO-d6
) δ ppm 0.92 (d, J = 6.0 Hz, 1H), 1.07 (d, J = 6.0 Hz, 2H), 2.57 - 2.73 (m, 1H), 2.82 - 2.90 (m, 1H), 3.72 - 3.73 (m, 3H), 4.22 (s, 0.4H), 4.49 (s, 0.6H), 4.70 (bs, 0.5H), 4.92 (bs, 0.5H), 6.09 (s, 0.6H), 6.31 (s, 0.4H), 6.78 - 6.85 (m, 2H), 7.01 (t, J = 9.0 Hz, 1H), 7.08 (t, J = 8.4 Hz, 1H), 7.21 - 7.23 (m, 2H), 7.40 (d, J = 8.0 Hz, 0.6H), 7.57 (d, J = 8.4 Hz, 0.4H)。在60℃下記錄此NMR。
化合物10: LC-MS (m/z): 324.3 [M+H]+
。1
H NMR (400 MHz, DMSO-d6
) δ ppm 1.09 - 1.13 (m, 3H), 2.21 - 2.28 (m, 0.5H), 2.90 - 2.94 (m, 0.5H), 3.07 - 3.08 (m, 1H), 3.77 (s, 3H), 4.07 (bs, 0.5H), 4.49 - 4.57 (m, 1.5H), 6.53 - 6.56 (m, 1H), 6.82 - 6.88 (m, 2H), 7.02 - 7.14 (m, 4.5H), 7.32 - 7.36 (m, 0.5H)。在60℃下記錄此NMR。
程序 6 : 化合物 11 之合成 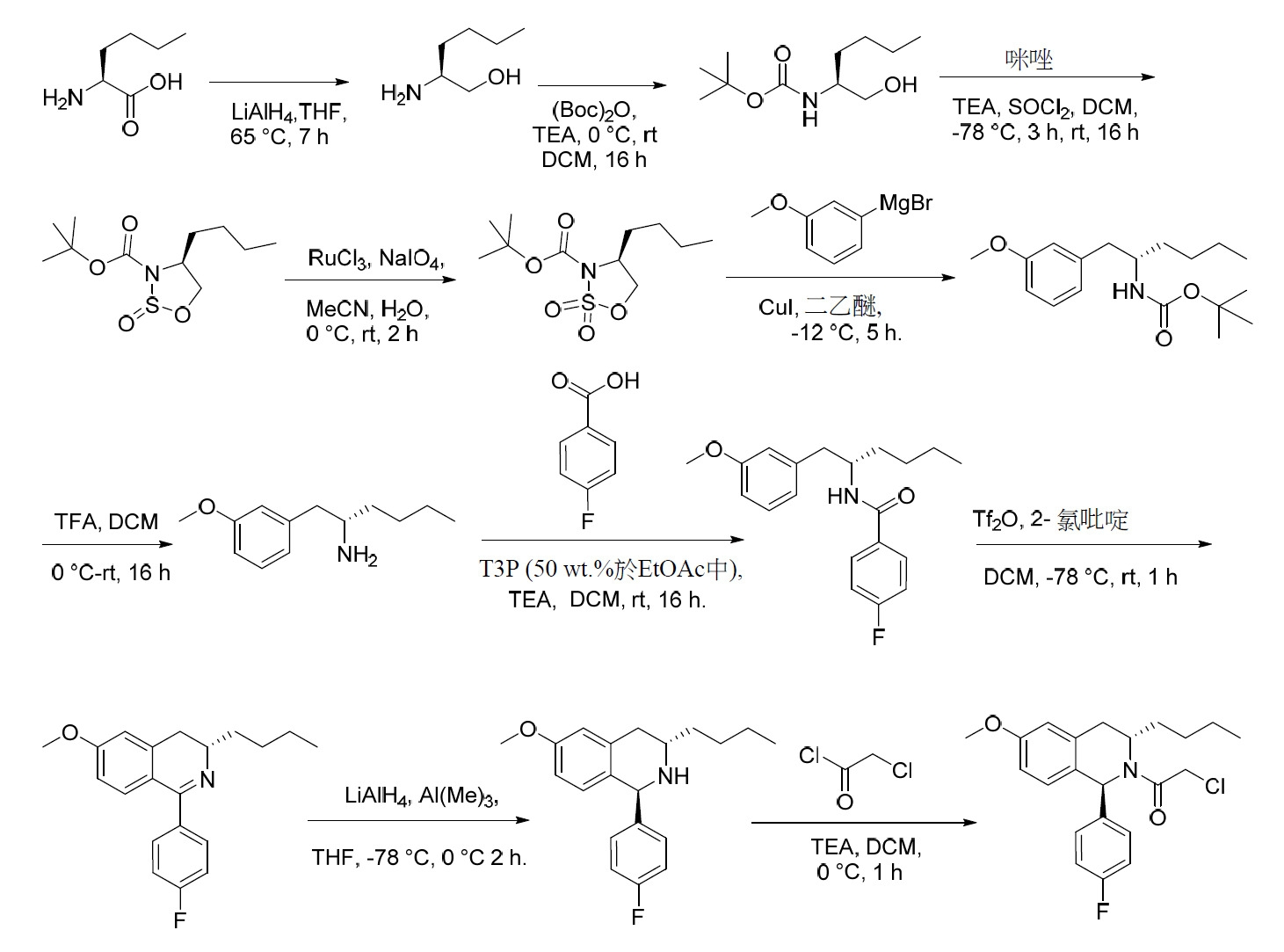 在0℃下,向(S)-2-胺基己酸(5 g, 38.14 mmol, 1當量)於THF (140 mL)中之溶液中添加於THF中之1 M LAH溶液(76.28 mL, 76.28 mmol, 2當量)。將反應混合物升溫至室溫,然後將混合物在65℃及N2氣氛下攪拌7 h。TLC (於DCM中之10% MeOH)顯示反應已完成。將反應混合物冷卻至室溫,使用二乙醚(50 mL)稀釋反應液,在費希爾後處理之後,經由燒結漏斗使用二乙醚過濾反應混合物,在減壓下濃縮濾液且粗製(S)-2-胺基己烷-1-醇未經進一步純化即用於下一步驟中。1
H NMR (400 MHz, CDCl3
) δ ppm 0.83 - 0.91 (m, 3H), 1.2 - 1.42 (m, 6H), 2.82 - 2.83 (m, 1H), 3.24 - 3.29 (m, 1H), 3.57 - 3.61 (m, 1H)。
在0℃下,向(S)-2-胺基己烷-1-醇(4.2 g, 35.83 mmol, 1當量)於DCM (40 mL)中之溶液中逐滴添加TEA (10 mL, 71.67 mmol, 2當量),將其攪拌5 mins,然後添加二碳酸二-第三丁基酯(9.86 mL, 43.00 mmol, 1.2當量)。在室溫下攪拌18 h之後,使用水(75 mL)及鹽水(75 mL)洗滌,藉由Na2
SO4
乾燥,並在真空中濃縮。對殘餘物實施combiflash矽膠層析(使用於DCM中之MeOH作為洗脫劑)以得到(S)-(1-羥基己烷-2-基)胺基甲酸第三丁基酯。1
H NMR (400 MHz, CDCl3
) δ ppm 0.89 (s, 3H), 1.32 - 1.43 (m, 6H), 1.44 (s, 9H), 3.50 - 3.54 (m, 1H), 3.61 - 3.67 (m, 2H), 4.59 (bs, 1H)。
在-78℃下,向1H-咪唑(5.1 g, 75.57 mmol, 4當量)及三乙胺(7.9 mL, 56.68 mmol, 3當量)於無水二氯甲烷(30 mL)中之溶液中逐滴添加亞硫醯氯(1.5 mL, 20.78 mmol, 1.1當量)。將反應混合物攪拌5 min且同時在-78℃下冷卻,且經30 min逐滴添加於無水二氯甲烷(30 mL)中之(S)-(1-羥基己烷-2-基)胺基甲酸第三丁基酯(4.1 g, 18.89 mmol, 1當量)。將反應混合物在-78℃下攪拌3小時。攪拌反應混合物,同時升溫至室溫過夜。添加水(100 mL)並分離各相。將水相進一步萃取至二氯甲烷(150 mL)中,使用水(100 mL)洗滌合併之有機物,藉由無水Na2
SO4
乾燥,過濾並在減壓下濃縮。(4S)-4-丁基-1,2,3-氧雜噻唑啶-3-甲酸第三丁基酯2-氧化物未經進一步純化即用於下一步驟中。
在0℃下,將水合氯化釕(III) (0.002 g, 0.013 mmol, 0.007當量)添加至(4S)-4-丁基-1,2,3-氧雜噻唑啶-3-甲酸第三丁基酯2-氧化物(5 g, 19 mmol, 1當量)於乙腈(50 mL)及水(50 mL)中之經攪拌溶液中,隨後逐份添加過碘酸鈉(4.4 g, 20.91 mmol, 1.1當量)。將雙相混合物在20℃下攪拌2小時。添加水(250 mL)且將混合物萃取至乙酸乙酯(2 × 150 mL)中。使用水(150 mL)、鹽水(150 mL)洗滌合併之有機物,藉由Na2
SO4
乾燥,過濾並在減壓下濃縮以獲得粗產物,藉由管柱層析使用於己烷中之10%乙酸乙酯作為洗脫劑純化粗產物以得到(S)-4-丁基-1,2,3-氧雜噻唑啶-3-甲酸第三丁基酯2,2-二氧化物。1
H NMR (400 MHz, CDCl3
) δ ppm 0.90 - 1.25 (m, 3H), 1.31 - 1.38 (m, 6H), 1.48 (s, 9H), 1.75 - 1.95 (m, 2H), 4.27 - 4.32 (m, 2H), 4.61 - 4.65 (m, 1H)。
在-12℃下,以10min時間向碘化銅(0.238 g, 1.25 mmol, 0.1當量)於二乙醚(25 mL)中之溶液中逐滴添加(3-甲氧基苯基)溴化鎂(1M於THF中) (25 mL, 25.08 mmol, 2.0當量)。將反應混合物在-12℃下攪拌30 min。此後,在-12℃下向反應液中逐滴添加(S)-4-丁基-1,2,3-氧雜噻唑啶-3-甲酸第三丁基酯2,2-二氧化物(3.5 g, 12.54 mmol, 1.0當量)於二乙醚(15 mL)中之溶液。將所得混合物在-12℃下攪拌4 h。最後,使用10%檸檬酸水溶液(15 mL)在-12℃下終止反應並使用乙酸乙酯(100 mL)稀釋。分離有機層並使用鹽水(20 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並濃縮以得到粗產物,藉由急速管柱層析使用於正己烷中之15%乙酸乙酯作為洗脫劑純化以獲得(S)-(1-(3-甲氧基苯基)己烷-2-基)胺基甲酸第三丁基酯。LC-MS (m/z) = 252.0 [M+H]+
。1
H NMR (400 MHz, CDCl3
) δ ppm 0.86 - 0.87 (m, 3H), 1.23 - 1.35 (m, 6H), 1.40 (s, 9H), 2.73 (bs, 2H), 3.78 (s, 3H), 4.29 (bs, 1H), 6.71 - 6.76 (m, 3H), 7.19 (t, J = 7.8 Hz, 1H)。未觀察到醯胺NH。
在0℃下,向(S)-(1-(3-甲氧基苯基)己烷-2-基)胺基甲酸第三丁基酯(3.7 g, 12.05 mmol, 1當量)於二氯甲烷(30 mL)中之溶液中添加三氟乙酸(2.7 g, 24.10 mmol, 2當量)。將混合物在室溫下攪拌16 h。藉由TLC監測反應進展,在反應完成之後,在減壓下濃縮反應混合物。使用冰冷水(10 mL)溶解所獲得產物並使用NaHCO3
飽和水溶液鹼化。使用EtOAc (100 mL)萃取化合物。使用鹽水(10 mL)洗滌有機層,藉由無水硫酸鈉乾燥,過濾並濃縮以獲得(S)-1-(3-甲氧基苯基)己烷-2-胺。LC-MS (m/z) = 208.1 [M+H]+
。1
H NMR (400 MHz, CDCl3
) δ ppm 0.82 - 0.90 (m, 3H), 1.23 - 1.42 (m, 4H), 1.57 - 1.62 (m, 2H), 2.69 - 2.91 (m, 2H), 3.25 - 3.58 (m, 1H), 3.80 (s, 3H), 6.69 - 6.78 (m, 3H), 7.21 (t, J = 8.0 Hz, 1H)。未觀察到NH2
質子。
向4-氟苯甲酸(1.62 g, 11.59 mmol, 1.2當量)於DCM (25 mL)中之溶液中添加TEA (3.9 g, 38.64 mmol, 4當量),將反應液攪拌15 min且然後在0℃下添加T3P (50 wt%於EtOAc中) (4.6 g, 14.49 mmol, 1.5當量)並再攪拌5 min。將(S)-1-(3-甲氧基苯基)己烷-2-胺(2.0 g, 9.66 mmol, 1當量)添加至反應混合物中且然後在室溫下攪拌反應混合物。藉由TLC (於己烷中之20%乙酸乙酯)監測反應進展。在16 h之後,使用DCM (50 mL)及飽和碳酸氫鈉溶液(20 mL)稀釋反應混合物。分離有機層,使用鹽水溶液(20 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得(S)-4-氟-N-(1-(3-甲氧基苯基)己烷-2-基)苯甲醯胺。LC-MS (m/z) = 330.0 [M+H]+
。1
H NMR (400 MHz, CDCl3
) δ ppm 0.88 - 0.89 (m, 3H), 1.26 - 1.48 (m, 6H), 2.83 - 2.93 (m, 2H), 3.76 (s, 3H), 4.36 - 4.38 (m, 1H), 5.74 (d, J = 7.6 Hz, 1H), 6.69 - 6.80 (m, 3H), 7.08 (t, J = 8.4 Hz, 2H), 7.21 (t, J = 7.6 Hz, 1H), 7.68 (t, J = 6.4 Hz, 2H)。
在-78℃下,經由注射器經1 min將三氟甲烷磺酸酐(2.5 mL, 14.89 mmol, 2.0當量)添加至醯胺(S)-4-氟-N-(1-(3-甲氧基苯基)己烷-2-基)苯甲醯胺(2.45 g, 7.44 mmol, 1當量)及2-氯吡啶(1.4 mL, 14.89 mmol, 2.0當量)於二氯甲烷(25 mL)中之經攪拌混合物中。在5 min之後,將反應混合物置於冰-水浴中並升溫至0℃。在5 min之後,將所得溶液升溫至23℃。在1 h之後,引入氫氧化鈉水溶液(5 mL, 1N)以中和三氟甲烷磺酸鹽。添加二氯甲烷(50 mL)以稀釋混合物且分離各層。使用鹽水(2 mL)洗滌有機層,藉由無水硫酸鈉乾燥,並過濾。在減壓下去除揮發物以得到(S)-3-丁基-1-(4-氟苯基)-6-甲氧基-3,4-二氫異喹啉。LC-MS (m/z) = 312.0 [M+H]+
。
在-78℃及氮氣下,將(S)-3-丁基-1-(4-氟苯基)-6-甲氧基-3,4-二氫異喹啉(0.8 g, 2.57 mmol, 1當量)於無水THF (5 mL)中之溶液逐滴添加至1M氫化鋰鋁(於THF中,25.7 mL, 25.72 mmol, 10當量)及25%w/w三甲基鋁(於己烷中,3.7 mL, 12.85 mmol, 5當量)於THF (20 mL)中之混合物中。將懸浮液在-78℃下攪拌1 h,並經3 h升溫至0℃。使用氯化鈉飽和水溶液(5 mL)淬滅反應混合物,隨後使用EtOAc (30 mL)稀釋且濾除沈澱物。最後,藉由無水硫酸鈉乾燥濾液,過濾並在減壓下蒸發。藉由矽膠急速層析(EtOAc/正己烷 = 75/25)純化殘餘物以得到(1S,3S)-3-丁基-1-(4-氟苯基)-6-甲氧基-1,2,3,4-四氫異喹啉(藉由nOe實驗可證實反式)。
使用金屬清除劑Quadrasil® TA處理所分離純產物(將化合物溶於THF (5 mL)中且添加Quadrasil® TA (100 mg),將混合物攪拌0.5 h,過濾。再重複一次並濃縮)。LC-MS (m/z) = 314.0 [M+H]+
。1
H NMR (400 MHz, CDCl3
) δ ppm 0.85 (bs, 3H), 1.26 (bs, 4H), 1.45 (bs, 2H), 2.60 - 2.69 (m, 1H), 2.87 - 2.92 (m, 2H), 3.80 (s, 3H), 5.20 (s, 1 H), 6.69 (s, 2H), 6.79 - 6.81 (m, 1H), 6.97 - 7.05 (m, 2H), 7.13 (s, 2H)。
在0℃下,向(1S,3S)-3-丁基-1-(4-氟苯基)-6-甲氧基-1,2,3,4-四氫異喹啉(0.1 g, 0.31 mmol, 1當量)於DCM (4 mL)中之溶液中添加三乙胺(0.08 g, 0.77 mmol, 2.5當量),隨後添加2-氯乙醯氯(0.054 g, 0.47 mmol, 1.5當量)。將混合物在0℃及N2
氣氛下攪拌1 h。TLC (於己烷中之25% EtOAc)顯示反應已完成。然後,使用NaHCO3
飽和水溶液(5 mL)稀釋反應液且使用DCM (25 mL)萃取產物。藉由無水Na2
SO4
乾燥有機層,過濾並在減壓下濃縮以獲得粗產物。藉由製備型TLC使用於正己烷中之25% EtOAc作為移動相純化所獲得粗產物以得到1-((1S,3S)-3-丁基-1-(4-氟苯基)-6-甲氧基-3,4-二氫異喹啉-2(1H)-基)-2-氯乙烷-1-酮。LC-MS (m/z) = 390.1 [M+H]+
。1
H NMR (400 MHz, DMSO-d6
): (在70℃下記錄,此乃因使用RT NMR看到旋轉異構行為) δ 0.79 - 0.81 (m, 3H), 1.00 (bs, 1H), 1.20 (bs, 4H), 1.40 (bs, 1H), 2.78 - 2.83 (m, 1H), 2.87 - 2.90 (m, 2H), 3.71 (s, 3H), 4.50 (bs, 2H), 6.09 (s, 1H), 6.77 - 6.80 (m, 2H), 7.03 (bs, 2H), 7.28 (t, J = 6.6 Hz, 2H), 7.41 (d, J = 8.0 Hz, 1H)。
在0℃下,向(S)-2-胺基己酸(5 g, 38.14 mmol, 1當量)於THF (140 mL)中之溶液中添加於THF中之1 M LAH溶液(76.28 mL, 76.28 mmol, 2當量)。將反應混合物升溫至室溫,然後將混合物在65℃及N2氣氛下攪拌7 h。TLC (於DCM中之10% MeOH)顯示反應已完成。將反應混合物冷卻至室溫,使用二乙醚(50 mL)稀釋反應液,在費希爾後處理之後,經由燒結漏斗使用二乙醚過濾反應混合物,在減壓下濃縮濾液且粗製(S)-2-胺基己烷-1-醇未經進一步純化即用於下一步驟中。1
H NMR (400 MHz, CDCl3
) δ ppm 0.83 - 0.91 (m, 3H), 1.2 - 1.42 (m, 6H), 2.82 - 2.83 (m, 1H), 3.24 - 3.29 (m, 1H), 3.57 - 3.61 (m, 1H)。
在0℃下,向(S)-2-胺基己烷-1-醇(4.2 g, 35.83 mmol, 1當量)於DCM (40 mL)中之溶液中逐滴添加TEA (10 mL, 71.67 mmol, 2當量),將其攪拌5 mins,然後添加二碳酸二-第三丁基酯(9.86 mL, 43.00 mmol, 1.2當量)。在室溫下攪拌18 h之後,使用水(75 mL)及鹽水(75 mL)洗滌,藉由Na2
SO4
乾燥,並在真空中濃縮。對殘餘物實施combiflash矽膠層析(使用於DCM中之MeOH作為洗脫劑)以得到(S)-(1-羥基己烷-2-基)胺基甲酸第三丁基酯。1
H NMR (400 MHz, CDCl3
) δ ppm 0.89 (s, 3H), 1.32 - 1.43 (m, 6H), 1.44 (s, 9H), 3.50 - 3.54 (m, 1H), 3.61 - 3.67 (m, 2H), 4.59 (bs, 1H)。
在-78℃下,向1H-咪唑(5.1 g, 75.57 mmol, 4當量)及三乙胺(7.9 mL, 56.68 mmol, 3當量)於無水二氯甲烷(30 mL)中之溶液中逐滴添加亞硫醯氯(1.5 mL, 20.78 mmol, 1.1當量)。將反應混合物攪拌5 min且同時在-78℃下冷卻,且經30 min逐滴添加於無水二氯甲烷(30 mL)中之(S)-(1-羥基己烷-2-基)胺基甲酸第三丁基酯(4.1 g, 18.89 mmol, 1當量)。將反應混合物在-78℃下攪拌3小時。攪拌反應混合物,同時升溫至室溫過夜。添加水(100 mL)並分離各相。將水相進一步萃取至二氯甲烷(150 mL)中,使用水(100 mL)洗滌合併之有機物,藉由無水Na2
SO4
乾燥,過濾並在減壓下濃縮。(4S)-4-丁基-1,2,3-氧雜噻唑啶-3-甲酸第三丁基酯2-氧化物未經進一步純化即用於下一步驟中。
在0℃下,將水合氯化釕(III) (0.002 g, 0.013 mmol, 0.007當量)添加至(4S)-4-丁基-1,2,3-氧雜噻唑啶-3-甲酸第三丁基酯2-氧化物(5 g, 19 mmol, 1當量)於乙腈(50 mL)及水(50 mL)中之經攪拌溶液中,隨後逐份添加過碘酸鈉(4.4 g, 20.91 mmol, 1.1當量)。將雙相混合物在20℃下攪拌2小時。添加水(250 mL)且將混合物萃取至乙酸乙酯(2 × 150 mL)中。使用水(150 mL)、鹽水(150 mL)洗滌合併之有機物,藉由Na2
SO4
乾燥,過濾並在減壓下濃縮以獲得粗產物,藉由管柱層析使用於己烷中之10%乙酸乙酯作為洗脫劑純化粗產物以得到(S)-4-丁基-1,2,3-氧雜噻唑啶-3-甲酸第三丁基酯2,2-二氧化物。1
H NMR (400 MHz, CDCl3
) δ ppm 0.90 - 1.25 (m, 3H), 1.31 - 1.38 (m, 6H), 1.48 (s, 9H), 1.75 - 1.95 (m, 2H), 4.27 - 4.32 (m, 2H), 4.61 - 4.65 (m, 1H)。
在-12℃下,以10min時間向碘化銅(0.238 g, 1.25 mmol, 0.1當量)於二乙醚(25 mL)中之溶液中逐滴添加(3-甲氧基苯基)溴化鎂(1M於THF中) (25 mL, 25.08 mmol, 2.0當量)。將反應混合物在-12℃下攪拌30 min。此後,在-12℃下向反應液中逐滴添加(S)-4-丁基-1,2,3-氧雜噻唑啶-3-甲酸第三丁基酯2,2-二氧化物(3.5 g, 12.54 mmol, 1.0當量)於二乙醚(15 mL)中之溶液。將所得混合物在-12℃下攪拌4 h。最後,使用10%檸檬酸水溶液(15 mL)在-12℃下終止反應並使用乙酸乙酯(100 mL)稀釋。分離有機層並使用鹽水(20 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並濃縮以得到粗產物,藉由急速管柱層析使用於正己烷中之15%乙酸乙酯作為洗脫劑純化以獲得(S)-(1-(3-甲氧基苯基)己烷-2-基)胺基甲酸第三丁基酯。LC-MS (m/z) = 252.0 [M+H]+
。1
H NMR (400 MHz, CDCl3
) δ ppm 0.86 - 0.87 (m, 3H), 1.23 - 1.35 (m, 6H), 1.40 (s, 9H), 2.73 (bs, 2H), 3.78 (s, 3H), 4.29 (bs, 1H), 6.71 - 6.76 (m, 3H), 7.19 (t, J = 7.8 Hz, 1H)。未觀察到醯胺NH。
在0℃下,向(S)-(1-(3-甲氧基苯基)己烷-2-基)胺基甲酸第三丁基酯(3.7 g, 12.05 mmol, 1當量)於二氯甲烷(30 mL)中之溶液中添加三氟乙酸(2.7 g, 24.10 mmol, 2當量)。將混合物在室溫下攪拌16 h。藉由TLC監測反應進展,在反應完成之後,在減壓下濃縮反應混合物。使用冰冷水(10 mL)溶解所獲得產物並使用NaHCO3
飽和水溶液鹼化。使用EtOAc (100 mL)萃取化合物。使用鹽水(10 mL)洗滌有機層,藉由無水硫酸鈉乾燥,過濾並濃縮以獲得(S)-1-(3-甲氧基苯基)己烷-2-胺。LC-MS (m/z) = 208.1 [M+H]+
。1
H NMR (400 MHz, CDCl3
) δ ppm 0.82 - 0.90 (m, 3H), 1.23 - 1.42 (m, 4H), 1.57 - 1.62 (m, 2H), 2.69 - 2.91 (m, 2H), 3.25 - 3.58 (m, 1H), 3.80 (s, 3H), 6.69 - 6.78 (m, 3H), 7.21 (t, J = 8.0 Hz, 1H)。未觀察到NH2
質子。
向4-氟苯甲酸(1.62 g, 11.59 mmol, 1.2當量)於DCM (25 mL)中之溶液中添加TEA (3.9 g, 38.64 mmol, 4當量),將反應液攪拌15 min且然後在0℃下添加T3P (50 wt%於EtOAc中) (4.6 g, 14.49 mmol, 1.5當量)並再攪拌5 min。將(S)-1-(3-甲氧基苯基)己烷-2-胺(2.0 g, 9.66 mmol, 1當量)添加至反應混合物中且然後在室溫下攪拌反應混合物。藉由TLC (於己烷中之20%乙酸乙酯)監測反應進展。在16 h之後,使用DCM (50 mL)及飽和碳酸氫鈉溶液(20 mL)稀釋反應混合物。分離有機層,使用鹽水溶液(20 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得(S)-4-氟-N-(1-(3-甲氧基苯基)己烷-2-基)苯甲醯胺。LC-MS (m/z) = 330.0 [M+H]+
。1
H NMR (400 MHz, CDCl3
) δ ppm 0.88 - 0.89 (m, 3H), 1.26 - 1.48 (m, 6H), 2.83 - 2.93 (m, 2H), 3.76 (s, 3H), 4.36 - 4.38 (m, 1H), 5.74 (d, J = 7.6 Hz, 1H), 6.69 - 6.80 (m, 3H), 7.08 (t, J = 8.4 Hz, 2H), 7.21 (t, J = 7.6 Hz, 1H), 7.68 (t, J = 6.4 Hz, 2H)。
在-78℃下,經由注射器經1 min將三氟甲烷磺酸酐(2.5 mL, 14.89 mmol, 2.0當量)添加至醯胺(S)-4-氟-N-(1-(3-甲氧基苯基)己烷-2-基)苯甲醯胺(2.45 g, 7.44 mmol, 1當量)及2-氯吡啶(1.4 mL, 14.89 mmol, 2.0當量)於二氯甲烷(25 mL)中之經攪拌混合物中。在5 min之後,將反應混合物置於冰-水浴中並升溫至0℃。在5 min之後,將所得溶液升溫至23℃。在1 h之後,引入氫氧化鈉水溶液(5 mL, 1N)以中和三氟甲烷磺酸鹽。添加二氯甲烷(50 mL)以稀釋混合物且分離各層。使用鹽水(2 mL)洗滌有機層,藉由無水硫酸鈉乾燥,並過濾。在減壓下去除揮發物以得到(S)-3-丁基-1-(4-氟苯基)-6-甲氧基-3,4-二氫異喹啉。LC-MS (m/z) = 312.0 [M+H]+
。
在-78℃及氮氣下,將(S)-3-丁基-1-(4-氟苯基)-6-甲氧基-3,4-二氫異喹啉(0.8 g, 2.57 mmol, 1當量)於無水THF (5 mL)中之溶液逐滴添加至1M氫化鋰鋁(於THF中,25.7 mL, 25.72 mmol, 10當量)及25%w/w三甲基鋁(於己烷中,3.7 mL, 12.85 mmol, 5當量)於THF (20 mL)中之混合物中。將懸浮液在-78℃下攪拌1 h,並經3 h升溫至0℃。使用氯化鈉飽和水溶液(5 mL)淬滅反應混合物,隨後使用EtOAc (30 mL)稀釋且濾除沈澱物。最後,藉由無水硫酸鈉乾燥濾液,過濾並在減壓下蒸發。藉由矽膠急速層析(EtOAc/正己烷 = 75/25)純化殘餘物以得到(1S,3S)-3-丁基-1-(4-氟苯基)-6-甲氧基-1,2,3,4-四氫異喹啉(藉由nOe實驗可證實反式)。
使用金屬清除劑Quadrasil® TA處理所分離純產物(將化合物溶於THF (5 mL)中且添加Quadrasil® TA (100 mg),將混合物攪拌0.5 h,過濾。再重複一次並濃縮)。LC-MS (m/z) = 314.0 [M+H]+
。1
H NMR (400 MHz, CDCl3
) δ ppm 0.85 (bs, 3H), 1.26 (bs, 4H), 1.45 (bs, 2H), 2.60 - 2.69 (m, 1H), 2.87 - 2.92 (m, 2H), 3.80 (s, 3H), 5.20 (s, 1 H), 6.69 (s, 2H), 6.79 - 6.81 (m, 1H), 6.97 - 7.05 (m, 2H), 7.13 (s, 2H)。
在0℃下,向(1S,3S)-3-丁基-1-(4-氟苯基)-6-甲氧基-1,2,3,4-四氫異喹啉(0.1 g, 0.31 mmol, 1當量)於DCM (4 mL)中之溶液中添加三乙胺(0.08 g, 0.77 mmol, 2.5當量),隨後添加2-氯乙醯氯(0.054 g, 0.47 mmol, 1.5當量)。將混合物在0℃及N2
氣氛下攪拌1 h。TLC (於己烷中之25% EtOAc)顯示反應已完成。然後,使用NaHCO3
飽和水溶液(5 mL)稀釋反應液且使用DCM (25 mL)萃取產物。藉由無水Na2
SO4
乾燥有機層,過濾並在減壓下濃縮以獲得粗產物。藉由製備型TLC使用於正己烷中之25% EtOAc作為移動相純化所獲得粗產物以得到1-((1S,3S)-3-丁基-1-(4-氟苯基)-6-甲氧基-3,4-二氫異喹啉-2(1H)-基)-2-氯乙烷-1-酮。LC-MS (m/z) = 390.1 [M+H]+
。1
H NMR (400 MHz, DMSO-d6
): (在70℃下記錄,此乃因使用RT NMR看到旋轉異構行為) δ 0.79 - 0.81 (m, 3H), 1.00 (bs, 1H), 1.20 (bs, 4H), 1.40 (bs, 1H), 2.78 - 2.83 (m, 1H), 2.87 - 2.90 (m, 2H), 3.71 (s, 3H), 4.50 (bs, 2H), 6.09 (s, 1H), 6.77 - 6.80 (m, 2H), 7.03 (bs, 2H), 7.28 (t, J = 6.6 Hz, 2H), 7.41 (d, J = 8.0 Hz, 1H)。
程序 7 :化合物 12 之合成  在室溫下,向3-(三甲基矽烷基)丙炔酸(0.1 g, 0.7 mmol, 1.0當量)於DMF (0.003 mL, 0.0038 mmol, 0.04當量)中之溶液中添加草醯氯(0.066 mL, 0.77 mmol, 1.1當量)並攪拌30分鐘。此後,在減壓下濃縮反應混合物以提供3-(三甲基矽烷基)丙炔醯氯,其未經任何進一步純化即用於下一步驟中。
在0℃下,向(1S,3S)-3-丁基-1-(4-氟苯基)-6-甲氧基-1,2,3,4-四氫異喹啉(0.15 g, 0.47 mmol, 1.0當量)於乙腈(3.5 mL)中之溶液中添加碳酸氫鈉(0.3 g, 3.57 mmol, 7.5當量)。在攪拌5分鐘之後,添加3-(三甲基矽烷基)丙炔醯氯(0.113 g, 0.69 mmol, 1.5當量)於乙腈(1.5 mL)中之溶液。將所得混合物在0℃下攪拌15 min,且藉由TLC (於正己烷中之20%乙酸乙酯)監測反應進展。此後,藉由通過矽藻土墊來去除來自反應物質之固體部分,使用乙腈洗滌。在減壓下濃縮所獲得濾液以提供1-((1S,3S)-3-丁基-1-(4-氟苯基)-6-甲氧基-3,4-二氫異喹啉-2(1H)-基)-3-(三甲基矽烷基)丙-2-炔-1-酮,其其未經任何進一步純化即用於下一步驟中。LC-MS (m/z) = 438.2 [M+H]+
。
在-78℃下,向1-((1S,3S)-3-丁基-1-(4-氟苯基)-6-甲氧基-3,4-二氫異喹啉-2(1H)-基)-3-(三甲基矽烷基)丙-2-炔-1-酮(0.2 g, 0.45 mmol, 1當量)於THF (4.0 mL)中之溶液中添加四丁基氟化銨(於THF中之1M溶液) (0.5 mL, 0.5 mmol, 1.1當量)。將此反應混合物在-78℃下攪拌15分鐘。藉由TLC (於正己烷中之20%乙酸乙酯)監測反應進展。此後,使用水(5 mL)稀釋反應混合物且使用乙酸乙酯(25 mL)萃取產物。藉由無水硫酸鈉乾燥合併之有機層,過濾並在減壓下濃縮以獲得粗產物,藉由製備型TLC使用於正己烷中之20%乙酸乙酯作為洗脫劑純化以提供1-((1S,3S)-3-丁基-1-(4-氟苯基)-6-甲氧基-3,4-二氫異喹啉-2(1H)-基)丙-2-炔-1-酮。LC-MS (m/z) = 366.1 [M+H]+
。1
H NMR (400 MHz, DMSO-d6
) δ ppm 0.81 - 0.88 (m, 3H), 1.20 - 1.24 (m, 5H), 1.50 (bs, 1H), 2.78 - 2.89 (s, 2H), 3.72 (s, 3H), 4.18 (s, 0.3H), 4.54 (s, 0.5H), 4.50 (bs, 0.5H), 4.70 (bs, 0.7H), 6.05 (s, 0.6H), 6.31 (s, 0.4H), 6.77 - 6.84 (m, 2H), 6.98 - 7.10 (m, 2H), 7.23 - 7.24 (m, 2H), 7.36 (d,J
= 8.0 Hz, 0.7H), 7.53 (d,J
= 7.6 Hz, 0.3H)。
在室溫下,向3-(三甲基矽烷基)丙炔酸(0.1 g, 0.7 mmol, 1.0當量)於DMF (0.003 mL, 0.0038 mmol, 0.04當量)中之溶液中添加草醯氯(0.066 mL, 0.77 mmol, 1.1當量)並攪拌30分鐘。此後,在減壓下濃縮反應混合物以提供3-(三甲基矽烷基)丙炔醯氯,其未經任何進一步純化即用於下一步驟中。
在0℃下,向(1S,3S)-3-丁基-1-(4-氟苯基)-6-甲氧基-1,2,3,4-四氫異喹啉(0.15 g, 0.47 mmol, 1.0當量)於乙腈(3.5 mL)中之溶液中添加碳酸氫鈉(0.3 g, 3.57 mmol, 7.5當量)。在攪拌5分鐘之後,添加3-(三甲基矽烷基)丙炔醯氯(0.113 g, 0.69 mmol, 1.5當量)於乙腈(1.5 mL)中之溶液。將所得混合物在0℃下攪拌15 min,且藉由TLC (於正己烷中之20%乙酸乙酯)監測反應進展。此後,藉由通過矽藻土墊來去除來自反應物質之固體部分,使用乙腈洗滌。在減壓下濃縮所獲得濾液以提供1-((1S,3S)-3-丁基-1-(4-氟苯基)-6-甲氧基-3,4-二氫異喹啉-2(1H)-基)-3-(三甲基矽烷基)丙-2-炔-1-酮,其其未經任何進一步純化即用於下一步驟中。LC-MS (m/z) = 438.2 [M+H]+
。
在-78℃下,向1-((1S,3S)-3-丁基-1-(4-氟苯基)-6-甲氧基-3,4-二氫異喹啉-2(1H)-基)-3-(三甲基矽烷基)丙-2-炔-1-酮(0.2 g, 0.45 mmol, 1當量)於THF (4.0 mL)中之溶液中添加四丁基氟化銨(於THF中之1M溶液) (0.5 mL, 0.5 mmol, 1.1當量)。將此反應混合物在-78℃下攪拌15分鐘。藉由TLC (於正己烷中之20%乙酸乙酯)監測反應進展。此後,使用水(5 mL)稀釋反應混合物且使用乙酸乙酯(25 mL)萃取產物。藉由無水硫酸鈉乾燥合併之有機層,過濾並在減壓下濃縮以獲得粗產物,藉由製備型TLC使用於正己烷中之20%乙酸乙酯作為洗脫劑純化以提供1-((1S,3S)-3-丁基-1-(4-氟苯基)-6-甲氧基-3,4-二氫異喹啉-2(1H)-基)丙-2-炔-1-酮。LC-MS (m/z) = 366.1 [M+H]+
。1
H NMR (400 MHz, DMSO-d6
) δ ppm 0.81 - 0.88 (m, 3H), 1.20 - 1.24 (m, 5H), 1.50 (bs, 1H), 2.78 - 2.89 (s, 2H), 3.72 (s, 3H), 4.18 (s, 0.3H), 4.54 (s, 0.5H), 4.50 (bs, 0.5H), 4.70 (bs, 0.7H), 6.05 (s, 0.6H), 6.31 (s, 0.4H), 6.77 - 6.84 (m, 2H), 6.98 - 7.10 (m, 2H), 7.23 - 7.24 (m, 2H), 7.36 (d,J
= 8.0 Hz, 0.7H), 7.53 (d,J
= 7.6 Hz, 0.3H)。
程序 8 :化合物 13 之合成 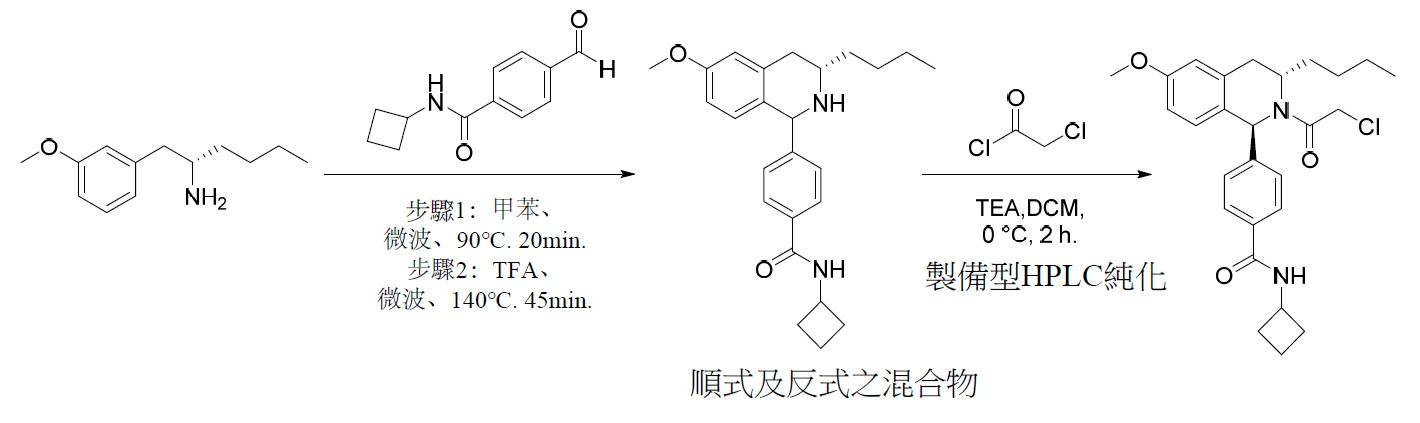 將(S)-1-(3-甲氧基苯基)己烷-2-胺(1.0 g, 1.27 mmol, 1當量)及N-環丁基-4-甲醯基苯甲醯胺(1.17 g, 5.79 mmol, 1.2當量)於甲苯(4 mL)在之溶液在90℃下於微波中輻照20 min。此後,在減壓下濃縮揮發性部分並在TFA (4 mL)中用於環化步驟中,且在140℃下於微波中輻照45 min。此後,在減壓下濃縮揮發性部分且使用NaHCO3
飽和水溶液(10 mL)及EtOAc (40 mL)稀釋所獲得粗製物。分離有機層,使用鹽水(10 mL)洗滌,藉由無水MgSO4
乾燥,過濾並在減壓下濃縮以獲得粗產物。藉由矽膠急速管柱層析(正己烷/EtOAc)純化所獲得粗產物以得到4-((1S,3S)-3-丁基-2-(2-氯乙醯基)-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)-N-環丁基苯甲醯胺。LC-MS (m/z): 393.3 [M+H]+
。
向4-((3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)-N-環丁基苯甲醯胺(0.5 g, 1.27 mmol, 1當量)於DCM (10 mL)中之溶液中添加TEA (0.4 g, 3.18 mmol, 2.5當量),隨後添加2-氯乙醯氯(0.091 mL, 1.14 mmol, 0.9當量),在0℃下攪拌6 h。TLC (於己烷中之30%乙酸乙酯)顯示反應已完成。在減壓下濃縮反應混合物,且使用EtOAc (50 mL)稀釋粗製物,使用水(2 × 50 mL)洗滌。藉由無水Na2
SO4
乾燥有機層,在減壓下濃縮以得到粗產物。藉由製備型HPLC純化[分析條件:管柱:Inertsil ODS 3V (250 mm × 4.6 mm × 5µm),移動相(A):於水中之0.1%氨,移動相(B):CH3
CN,流速:1.0 mL/min,B之組成:0/10,12/80,25/90,27/10,30/10]純化所獲得粗產物以獲得4-((1S,3S)-3-丁基-2-(2-氯乙醯基)-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)-N-環丁基苯甲醯胺。LCMS (ES) m/z: 469.0 [M+H]+
, HPLC純度:99.8%,對掌性HPLC純度:99.92%。1
H NMR (400 MHz, CDCl3
): δ ppm 0.78 - 0.81 (m, 3 H), 1.22 - 1.41 (m, 6 H), 1.60 - 1.69 (m, 2 H), 1.99 - 2.06 (m, 2 H), 2.18 - 2.20 (m, 2 H), 2.80 - 2.84 (m, 2 H), 3.09 - 3.10 (m, 1 H), 3.71 (s, 3 H), 4.31 - 4.37 (m, 1 H), 4.55 (bs, 2 H), 6.13 (s, 1 H), 6.77 - 6.81 (m, 2 H), 7.32 - 7.34 (m, 2 H), 7.43 (m, 1 H), 7.66 (s, 1 H)。
將(S)-1-(3-甲氧基苯基)己烷-2-胺(1.0 g, 1.27 mmol, 1當量)及N-環丁基-4-甲醯基苯甲醯胺(1.17 g, 5.79 mmol, 1.2當量)於甲苯(4 mL)在之溶液在90℃下於微波中輻照20 min。此後,在減壓下濃縮揮發性部分並在TFA (4 mL)中用於環化步驟中,且在140℃下於微波中輻照45 min。此後,在減壓下濃縮揮發性部分且使用NaHCO3
飽和水溶液(10 mL)及EtOAc (40 mL)稀釋所獲得粗製物。分離有機層,使用鹽水(10 mL)洗滌,藉由無水MgSO4
乾燥,過濾並在減壓下濃縮以獲得粗產物。藉由矽膠急速管柱層析(正己烷/EtOAc)純化所獲得粗產物以得到4-((1S,3S)-3-丁基-2-(2-氯乙醯基)-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)-N-環丁基苯甲醯胺。LC-MS (m/z): 393.3 [M+H]+
。
向4-((3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)-N-環丁基苯甲醯胺(0.5 g, 1.27 mmol, 1當量)於DCM (10 mL)中之溶液中添加TEA (0.4 g, 3.18 mmol, 2.5當量),隨後添加2-氯乙醯氯(0.091 mL, 1.14 mmol, 0.9當量),在0℃下攪拌6 h。TLC (於己烷中之30%乙酸乙酯)顯示反應已完成。在減壓下濃縮反應混合物,且使用EtOAc (50 mL)稀釋粗製物,使用水(2 × 50 mL)洗滌。藉由無水Na2
SO4
乾燥有機層,在減壓下濃縮以得到粗產物。藉由製備型HPLC純化[分析條件:管柱:Inertsil ODS 3V (250 mm × 4.6 mm × 5µm),移動相(A):於水中之0.1%氨,移動相(B):CH3
CN,流速:1.0 mL/min,B之組成:0/10,12/80,25/90,27/10,30/10]純化所獲得粗產物以獲得4-((1S,3S)-3-丁基-2-(2-氯乙醯基)-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)-N-環丁基苯甲醯胺。LCMS (ES) m/z: 469.0 [M+H]+
, HPLC純度:99.8%,對掌性HPLC純度:99.92%。1
H NMR (400 MHz, CDCl3
): δ ppm 0.78 - 0.81 (m, 3 H), 1.22 - 1.41 (m, 6 H), 1.60 - 1.69 (m, 2 H), 1.99 - 2.06 (m, 2 H), 2.18 - 2.20 (m, 2 H), 2.80 - 2.84 (m, 2 H), 3.09 - 3.10 (m, 1 H), 3.71 (s, 3 H), 4.31 - 4.37 (m, 1 H), 4.55 (bs, 2 H), 6.13 (s, 1 H), 6.77 - 6.81 (m, 2 H), 7.32 - 7.34 (m, 2 H), 7.43 (m, 1 H), 7.66 (s, 1 H)。
程序 9 :化合物 14 之合成 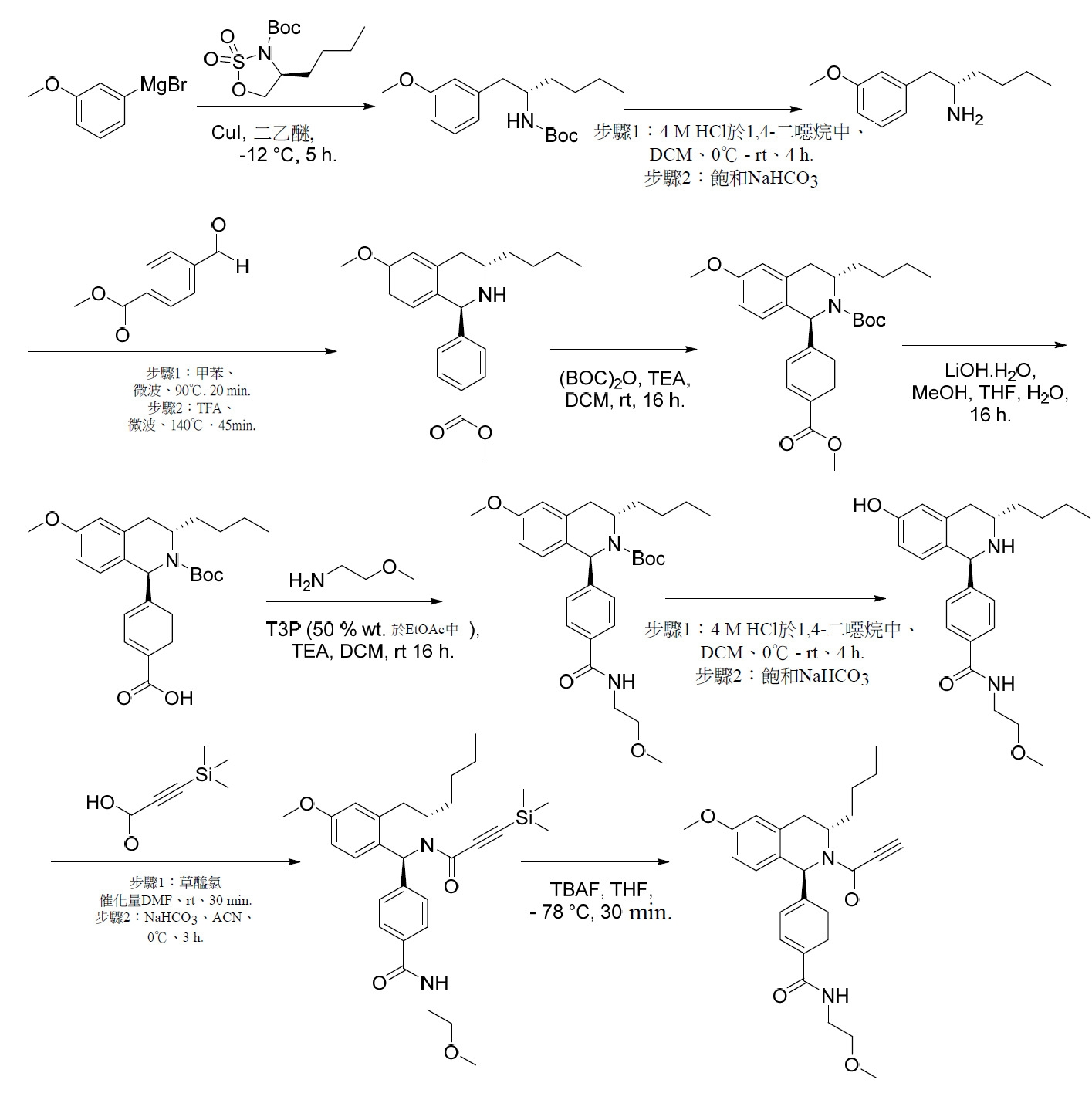 在-12℃下,以10 min時間向碘化銅(0.510 g, 2.68 mmol, 0.1當量)於二乙醚(50 mL)中之溶液中逐滴添加(3-甲氧基苯基)溴化鎂(1M於THF中) (53 mL, 53.76 mmol, 2當量)。將反應混合物在-12℃下攪拌30 min。此後,將(S)-4-丁基-1,2,3-氧雜噻唑啶-3-甲酸第三丁基酯2,2-二氧化物(7.5 g, 26.88 mmol, 1當量)於二乙醚(15 mL)中之溶液在-12℃下逐滴添加至反應物質中。將所得混合物在-12℃下攪拌4 h。最後,使用10%檸檬酸水溶液(15 mL)在-12℃下終止反應並使用乙酸乙酯(100 mL)稀釋。分離有機層並使用鹽水(50 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並濃縮以得到粗產物,藉由急速管柱層析使用於正己烷中之15%乙酸乙酯作為洗脫劑純化以獲得(S)-(1-(3-甲氧基苯基)己烷-2-基)胺基甲酸第三丁基酯。LC-MS (m/z): 252.0 [M+H]+
。1
H NMR (400 MHz, CDCl3
): δ ppm 0.86 - 0.87 (m, 3 H), 1.23 - 1.35 (m, 6 H), 1.40 (s, 9 H), 2.73 (bs, 2 H), 3.78 (s, 3 H), 4.29 (bs, 1 H), 6.71 - 6.76 (m, 3 H), 7.19 (t,J
= 7.8 Hz, 1 H)。未觀察到醯胺NH。
在0℃下,向(S)-(1-(3-甲氧基苯基)己烷-2-基)胺基甲酸第三丁基酯(10 g, 32.57 mmol, 1當量)於二氯甲烷(50 mL)中之溶液中添加於1,4-二噁烷中之4M HCl (20 mL, 64.10 mmol, 2當量)。將混合物在室溫下攪拌16 h。藉由TLC監測反應進展,在反應完成之後,在減壓下濃縮反應混合物。使用冰冷水(10 mL)溶解所獲得粗製物且藉由NaHCO3
飽和水溶液鹼化。使用EtOAc (100 mL)萃取化合物。使用鹽水(10 mL)洗滌有機層,藉由無水硫酸鈉乾燥,過濾並濃縮以獲得(S)-1-(3-甲氧基苯基)己烷-2-胺。LC-MS (m/z): 208.1 [M+H]+
。1
H NMR (400 MHz, CDCl3
): δ ppm 0.82 - 0.90 (m, 3 H), 1.23 - 1.42 (m, 4 H), 1.57 - 1.62 (m, 2 H), 2.69 - 2.91 (m, 2 H), 3.25 - 3.58 (m, 1 H), 3.80 (s, 3 H), 6.69 - 6.78 (m, 3 H), 7.21 (t,J
= 8.0 Hz, 1 H) (未觀察到NH2
質子)。
將(S)-1-(3-甲氧基苯基)己烷-2-胺(0.7 g, 3.37 mmol, 1當量)及4-甲醯基苯甲酸甲酯(0.664 g, 4.05 mmol, 1當量)於甲苯(4 mL)中之溶液在90℃下於微波中輻照20 min。此後,在減壓下濃縮揮發性部分並在TFA (4 mL)中原樣用於環化步驟中,且在140℃下於微波中輻照45 min。此後,在減壓下濃縮揮發性部分且使用NaHCO3
飽和水溶液(10 mL)及EtOAc (40 mL)稀釋所獲得粗製物。分離有機層,使用鹽水(10 mL)洗滌,藉由無水MgSO4
乾燥,過濾並在減壓下濃縮以獲得粗產物。藉由矽膠急速管柱層析(正己烷/EtOAc)純化所獲得粗產物以得到4-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)苯甲酸甲酯。LC-MS (m/z): 208.1 [M+H]+
。1
H NMR (400 MHz, CDCl3
): δ ppm 0.84 - 0.86 (m, 3 H), 1.23 - 1.24 (m, 6 H), 2.55 - 2.59 (m, 1 H), 2.85 - 2.88 (m, 2 H), 3.75 (s, 3 H), 3.80 (s, 3 H), 5.29 (s, 3 H), 6.67 - 6.69 (m, 2 H), 6.70 - 6.80 (m, 1 H), 7.21 - 7.26 (s, 2 H), 7.94 - 8.0 (m, 2 H)。
在室溫下,向化合物4-((1S,3S)-6-甲氧基-3-甲基-1,2,3,4-四氫異喹啉-1-基)苯甲酸甲酯(0.35 g, 1.12 mmol, 1當量)於DCM (10 mL)中之溶液中添加三乙胺(0.45 g, 4.49 mmol, 4當量)及二碳酸二-第三丁基酯(0.715 g, 2.24 mmol, 2當量)且將混合物攪拌16 h。TLC (於己烷中之50% EtOAc)顯示反應已完成。在減壓下濃縮反應混合物,且使用EtOAc (50 mL)稀釋粗製物,使用水(2 × 50 mL)洗滌。藉由無水Na2
SO4
乾燥有機層,在減壓下濃縮成粗產物(1S,3S)-6-甲氧基-1-(4-(甲氧基羰基)苯基)-3-甲基-3,4-二氫異喹啉-2(1H)-甲酸第三丁基酯。LC-MS (m/z): 356.0 [M-t
Bu+H]+
。
向化合物(1S,3S)-6-甲氧基-1-(4-(甲氧基羰基)苯基)-3-甲基-3,4-二氫異喹啉-2(1H)-甲酸第三丁基酯(0.650 g, 1.57 mmol, 1當量)於THF: MeOH: H2
O (9 mL:1 mL:1 mL)之混合物中之溶液中添加氫氧化鋰(0.331 g, 7.89 mmol, 5當量)並在室溫下攪拌16 h。TLC (於己烷中之50% EtOAc)顯示反應已完成。在減壓下濃縮反應混合物,且使用5%檸檬酸溶液(pH=9)酸化粗製物。使用EtOAc (50 mL)稀釋反應混合物且分離有機層,並藉由無水Na2
SO4
乾燥,在減壓下濃縮以得到粗產物4-((1S,3S)-2-(第三丁氧基羰基)-6-甲氧基-3-甲基-1,2,3,4-四氫異喹啉-1-基)苯甲酸。LC-MS (m/z): 396.0 [M+H]+
。
在0℃下,向化合物4-((1S,3S)-2-(第三丁氧基羰基)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)苯甲酸(0.520 g, 1.18 mmol, 1當量)於DCM (10 mL)中之溶液中添加三乙胺(0.6 mL, 4.73 mmol, 4當量)及2-甲氧基乙烷-1-胺(0.106 g, 1.42 mmol, 1.2當量)且將混合物攪拌15 min。在相同溫度下向上述反應混合物中添加T3P (50% wt in EtOAc) (1.4 mL, 1.7 mmol, 1.5當量)並攪拌16 h。TLC (於己烷中之30% EtOAc)顯示反應已完成。在減壓下濃縮反應混合物,且使用EtOAc (50 mL)稀釋粗製物,使用水(2 × 50 mL)洗滌。藉由無水Na2
SO4
乾燥有機層,在減壓下濃縮以得到粗產物(1S,3S)-3-丁基-6-甲氧基-1-(4-((2-甲氧基乙基)胺甲醯基)苯基)-3,4-二氫異喹啉-2(1H)-甲酸第三丁基酯。LC-MS (m/z): 497.0 [M-t
Bu+H]+
。
在0℃下,向(1S,3S)-3-丁基-6-甲氧基-1-(4-((2-甲氧基乙基)胺甲醯基)苯基)-3,4-二氫異喹啉-2(1H)-甲酸第三丁基酯(0.380 g, 0.76 mmol, 1當量)於二氯甲烷(50 mL)中之溶液中添加於1,4-二噁烷中之4M HCl (10 mL, 1.52 mmol, 2當量)。將混合物在室溫下攪拌16 h。藉由TLC監測反應進展,在反應完成之後,在減壓下濃縮反應混合物。使用冰冷水(20 mL)溶解所獲得粗製物且藉由NaHCO3
飽和水溶液鹼化。使用EtOAc (100 mL)萃取化合物。使用鹽水(10 mL)洗滌有機層,藉由無水硫酸鈉乾燥,過濾並濃縮以獲得4-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)-N-(2-甲氧基乙基)苯甲醯胺。LC-MS (m/z): 397.0 [M+H]+
。
向3-(三甲基矽烷基)丙炔酸(0.172 g, 1.20 mmol, 1當量)中添加DMF (0.003 g, 0.048 mmol, 0.04當量)及草醯氯(0.114 mL, 1.33 mmol, 1.1當量)並攪拌30 min。此後,在減壓下濃縮反應混合物以獲得粗製3-(三甲基矽烷基)丙炔醯氯且使用ACN (1 mL)稀釋此粗製物,且在0℃下添加至含有4-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)-N-(2-甲氧基乙基)苯甲醯胺(0.320 g, 0.807 mmol, 1當量)及NaHCO3
(0.508 g, 6.05 mmol, 7.5當量)於ACN (5 mL)中之經攪拌溶液之反應混合物中並攪拌15 min。LCMS及TLC (於己烷中之70% EtOAc)顯示反應已完成。過濾反應液並在減壓下濃縮以得到粗產物4-((1S,3S)-3-丁基-6-甲氧基-2-(3-(三甲基矽烷基)丙炔醯基)-1,2,3,4-四氫異喹啉-1-基)-N-(2甲氧基乙基)苯甲醯胺,其未經進一步純化即用於下一步驟中。LC-MS (m/z): 521.0 [M+H]+
。
向於THF (10.0 mL)中之4-((1S,3S)-3-丁基-6-甲氧基-2-(3-(三甲基矽烷基)丙炔醯基)-1,2,3,4-四氫異喹啉-1-基)-N-(2-甲氧基乙基)苯甲醯胺(0.360 g, 0.69 mmol, 1當量)中添加TBAF(於THF中之1M溶液) (0.48 mL, 0.48 mmol, 2當量)並攪拌30 min。此後,在減壓下濃縮反應混合物,使用乙酸乙酯(100 mL)稀釋且使用水(2 × 10 mL)洗滌。藉由Na2
SO4
乾燥有機層並濃縮以得到粗產物,藉由製備型TLC層析使用於己烷中之70% EtOAc作為洗脫劑進一步純化以獲得4-((1S,3S)-3-丁基-6-甲氧基-2-丙炔醯基-1,2,3,4-四氫異喹啉-1-基)-N-(2甲氧基乙基)苯甲醯胺。LC-MS (m/z): 449.2 [M+H]+
;HPLC純度:98.6%,對掌性HPLC純度:99.98%。1
H NMR (400 MHz, DMSO-d6
): δ 0.80 - 0.83 (m, 3H), 0.92 - 1.24 (m, 5H), 1.50 (bs, 1H), 2.80 - 2.90 (m, 2H), 3.10 (s, 1H), 3.24 (s, 3H), 3.38 - 3.42 (m, 4H), 3.71 - 3.72 (m, 2H), 4.16 - 4.46 (s, 1H), 4.58 - 4.75 (bs, 1 H), 6.07 - 6.34 (s, 1H), 6.77 - 6.84 (m, 2H), 7.29 - 7.31 (m, 2H), 7.38 - 7.58 (m, 1H), 7.64 - 7.73 (m, 2H), 8.13 - 8.19 (m, 1H)。
在-12℃下,以10 min時間向碘化銅(0.510 g, 2.68 mmol, 0.1當量)於二乙醚(50 mL)中之溶液中逐滴添加(3-甲氧基苯基)溴化鎂(1M於THF中) (53 mL, 53.76 mmol, 2當量)。將反應混合物在-12℃下攪拌30 min。此後,將(S)-4-丁基-1,2,3-氧雜噻唑啶-3-甲酸第三丁基酯2,2-二氧化物(7.5 g, 26.88 mmol, 1當量)於二乙醚(15 mL)中之溶液在-12℃下逐滴添加至反應物質中。將所得混合物在-12℃下攪拌4 h。最後,使用10%檸檬酸水溶液(15 mL)在-12℃下終止反應並使用乙酸乙酯(100 mL)稀釋。分離有機層並使用鹽水(50 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並濃縮以得到粗產物,藉由急速管柱層析使用於正己烷中之15%乙酸乙酯作為洗脫劑純化以獲得(S)-(1-(3-甲氧基苯基)己烷-2-基)胺基甲酸第三丁基酯。LC-MS (m/z): 252.0 [M+H]+
。1
H NMR (400 MHz, CDCl3
): δ ppm 0.86 - 0.87 (m, 3 H), 1.23 - 1.35 (m, 6 H), 1.40 (s, 9 H), 2.73 (bs, 2 H), 3.78 (s, 3 H), 4.29 (bs, 1 H), 6.71 - 6.76 (m, 3 H), 7.19 (t,J
= 7.8 Hz, 1 H)。未觀察到醯胺NH。
在0℃下,向(S)-(1-(3-甲氧基苯基)己烷-2-基)胺基甲酸第三丁基酯(10 g, 32.57 mmol, 1當量)於二氯甲烷(50 mL)中之溶液中添加於1,4-二噁烷中之4M HCl (20 mL, 64.10 mmol, 2當量)。將混合物在室溫下攪拌16 h。藉由TLC監測反應進展,在反應完成之後,在減壓下濃縮反應混合物。使用冰冷水(10 mL)溶解所獲得粗製物且藉由NaHCO3
飽和水溶液鹼化。使用EtOAc (100 mL)萃取化合物。使用鹽水(10 mL)洗滌有機層,藉由無水硫酸鈉乾燥,過濾並濃縮以獲得(S)-1-(3-甲氧基苯基)己烷-2-胺。LC-MS (m/z): 208.1 [M+H]+
。1
H NMR (400 MHz, CDCl3
): δ ppm 0.82 - 0.90 (m, 3 H), 1.23 - 1.42 (m, 4 H), 1.57 - 1.62 (m, 2 H), 2.69 - 2.91 (m, 2 H), 3.25 - 3.58 (m, 1 H), 3.80 (s, 3 H), 6.69 - 6.78 (m, 3 H), 7.21 (t,J
= 8.0 Hz, 1 H) (未觀察到NH2
質子)。
將(S)-1-(3-甲氧基苯基)己烷-2-胺(0.7 g, 3.37 mmol, 1當量)及4-甲醯基苯甲酸甲酯(0.664 g, 4.05 mmol, 1當量)於甲苯(4 mL)中之溶液在90℃下於微波中輻照20 min。此後,在減壓下濃縮揮發性部分並在TFA (4 mL)中原樣用於環化步驟中,且在140℃下於微波中輻照45 min。此後,在減壓下濃縮揮發性部分且使用NaHCO3
飽和水溶液(10 mL)及EtOAc (40 mL)稀釋所獲得粗製物。分離有機層,使用鹽水(10 mL)洗滌,藉由無水MgSO4
乾燥,過濾並在減壓下濃縮以獲得粗產物。藉由矽膠急速管柱層析(正己烷/EtOAc)純化所獲得粗產物以得到4-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)苯甲酸甲酯。LC-MS (m/z): 208.1 [M+H]+
。1
H NMR (400 MHz, CDCl3
): δ ppm 0.84 - 0.86 (m, 3 H), 1.23 - 1.24 (m, 6 H), 2.55 - 2.59 (m, 1 H), 2.85 - 2.88 (m, 2 H), 3.75 (s, 3 H), 3.80 (s, 3 H), 5.29 (s, 3 H), 6.67 - 6.69 (m, 2 H), 6.70 - 6.80 (m, 1 H), 7.21 - 7.26 (s, 2 H), 7.94 - 8.0 (m, 2 H)。
在室溫下,向化合物4-((1S,3S)-6-甲氧基-3-甲基-1,2,3,4-四氫異喹啉-1-基)苯甲酸甲酯(0.35 g, 1.12 mmol, 1當量)於DCM (10 mL)中之溶液中添加三乙胺(0.45 g, 4.49 mmol, 4當量)及二碳酸二-第三丁基酯(0.715 g, 2.24 mmol, 2當量)且將混合物攪拌16 h。TLC (於己烷中之50% EtOAc)顯示反應已完成。在減壓下濃縮反應混合物,且使用EtOAc (50 mL)稀釋粗製物,使用水(2 × 50 mL)洗滌。藉由無水Na2
SO4
乾燥有機層,在減壓下濃縮成粗產物(1S,3S)-6-甲氧基-1-(4-(甲氧基羰基)苯基)-3-甲基-3,4-二氫異喹啉-2(1H)-甲酸第三丁基酯。LC-MS (m/z): 356.0 [M-t
Bu+H]+
。
向化合物(1S,3S)-6-甲氧基-1-(4-(甲氧基羰基)苯基)-3-甲基-3,4-二氫異喹啉-2(1H)-甲酸第三丁基酯(0.650 g, 1.57 mmol, 1當量)於THF: MeOH: H2
O (9 mL:1 mL:1 mL)之混合物中之溶液中添加氫氧化鋰(0.331 g, 7.89 mmol, 5當量)並在室溫下攪拌16 h。TLC (於己烷中之50% EtOAc)顯示反應已完成。在減壓下濃縮反應混合物,且使用5%檸檬酸溶液(pH=9)酸化粗製物。使用EtOAc (50 mL)稀釋反應混合物且分離有機層,並藉由無水Na2
SO4
乾燥,在減壓下濃縮以得到粗產物4-((1S,3S)-2-(第三丁氧基羰基)-6-甲氧基-3-甲基-1,2,3,4-四氫異喹啉-1-基)苯甲酸。LC-MS (m/z): 396.0 [M+H]+
。
在0℃下,向化合物4-((1S,3S)-2-(第三丁氧基羰基)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)苯甲酸(0.520 g, 1.18 mmol, 1當量)於DCM (10 mL)中之溶液中添加三乙胺(0.6 mL, 4.73 mmol, 4當量)及2-甲氧基乙烷-1-胺(0.106 g, 1.42 mmol, 1.2當量)且將混合物攪拌15 min。在相同溫度下向上述反應混合物中添加T3P (50% wt in EtOAc) (1.4 mL, 1.7 mmol, 1.5當量)並攪拌16 h。TLC (於己烷中之30% EtOAc)顯示反應已完成。在減壓下濃縮反應混合物,且使用EtOAc (50 mL)稀釋粗製物,使用水(2 × 50 mL)洗滌。藉由無水Na2
SO4
乾燥有機層,在減壓下濃縮以得到粗產物(1S,3S)-3-丁基-6-甲氧基-1-(4-((2-甲氧基乙基)胺甲醯基)苯基)-3,4-二氫異喹啉-2(1H)-甲酸第三丁基酯。LC-MS (m/z): 497.0 [M-t
Bu+H]+
。
在0℃下,向(1S,3S)-3-丁基-6-甲氧基-1-(4-((2-甲氧基乙基)胺甲醯基)苯基)-3,4-二氫異喹啉-2(1H)-甲酸第三丁基酯(0.380 g, 0.76 mmol, 1當量)於二氯甲烷(50 mL)中之溶液中添加於1,4-二噁烷中之4M HCl (10 mL, 1.52 mmol, 2當量)。將混合物在室溫下攪拌16 h。藉由TLC監測反應進展,在反應完成之後,在減壓下濃縮反應混合物。使用冰冷水(20 mL)溶解所獲得粗製物且藉由NaHCO3
飽和水溶液鹼化。使用EtOAc (100 mL)萃取化合物。使用鹽水(10 mL)洗滌有機層,藉由無水硫酸鈉乾燥,過濾並濃縮以獲得4-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)-N-(2-甲氧基乙基)苯甲醯胺。LC-MS (m/z): 397.0 [M+H]+
。
向3-(三甲基矽烷基)丙炔酸(0.172 g, 1.20 mmol, 1當量)中添加DMF (0.003 g, 0.048 mmol, 0.04當量)及草醯氯(0.114 mL, 1.33 mmol, 1.1當量)並攪拌30 min。此後,在減壓下濃縮反應混合物以獲得粗製3-(三甲基矽烷基)丙炔醯氯且使用ACN (1 mL)稀釋此粗製物,且在0℃下添加至含有4-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)-N-(2-甲氧基乙基)苯甲醯胺(0.320 g, 0.807 mmol, 1當量)及NaHCO3
(0.508 g, 6.05 mmol, 7.5當量)於ACN (5 mL)中之經攪拌溶液之反應混合物中並攪拌15 min。LCMS及TLC (於己烷中之70% EtOAc)顯示反應已完成。過濾反應液並在減壓下濃縮以得到粗產物4-((1S,3S)-3-丁基-6-甲氧基-2-(3-(三甲基矽烷基)丙炔醯基)-1,2,3,4-四氫異喹啉-1-基)-N-(2甲氧基乙基)苯甲醯胺,其未經進一步純化即用於下一步驟中。LC-MS (m/z): 521.0 [M+H]+
。
向於THF (10.0 mL)中之4-((1S,3S)-3-丁基-6-甲氧基-2-(3-(三甲基矽烷基)丙炔醯基)-1,2,3,4-四氫異喹啉-1-基)-N-(2-甲氧基乙基)苯甲醯胺(0.360 g, 0.69 mmol, 1當量)中添加TBAF(於THF中之1M溶液) (0.48 mL, 0.48 mmol, 2當量)並攪拌30 min。此後,在減壓下濃縮反應混合物,使用乙酸乙酯(100 mL)稀釋且使用水(2 × 10 mL)洗滌。藉由Na2
SO4
乾燥有機層並濃縮以得到粗產物,藉由製備型TLC層析使用於己烷中之70% EtOAc作為洗脫劑進一步純化以獲得4-((1S,3S)-3-丁基-6-甲氧基-2-丙炔醯基-1,2,3,4-四氫異喹啉-1-基)-N-(2甲氧基乙基)苯甲醯胺。LC-MS (m/z): 449.2 [M+H]+
;HPLC純度:98.6%,對掌性HPLC純度:99.98%。1
H NMR (400 MHz, DMSO-d6
): δ 0.80 - 0.83 (m, 3H), 0.92 - 1.24 (m, 5H), 1.50 (bs, 1H), 2.80 - 2.90 (m, 2H), 3.10 (s, 1H), 3.24 (s, 3H), 3.38 - 3.42 (m, 4H), 3.71 - 3.72 (m, 2H), 4.16 - 4.46 (s, 1H), 4.58 - 4.75 (bs, 1 H), 6.07 - 6.34 (s, 1H), 6.77 - 6.84 (m, 2H), 7.29 - 7.31 (m, 2H), 7.38 - 7.58 (m, 1H), 7.64 - 7.73 (m, 2H), 8.13 - 8.19 (m, 1H)。
程序 10 :化合物 15 之合成 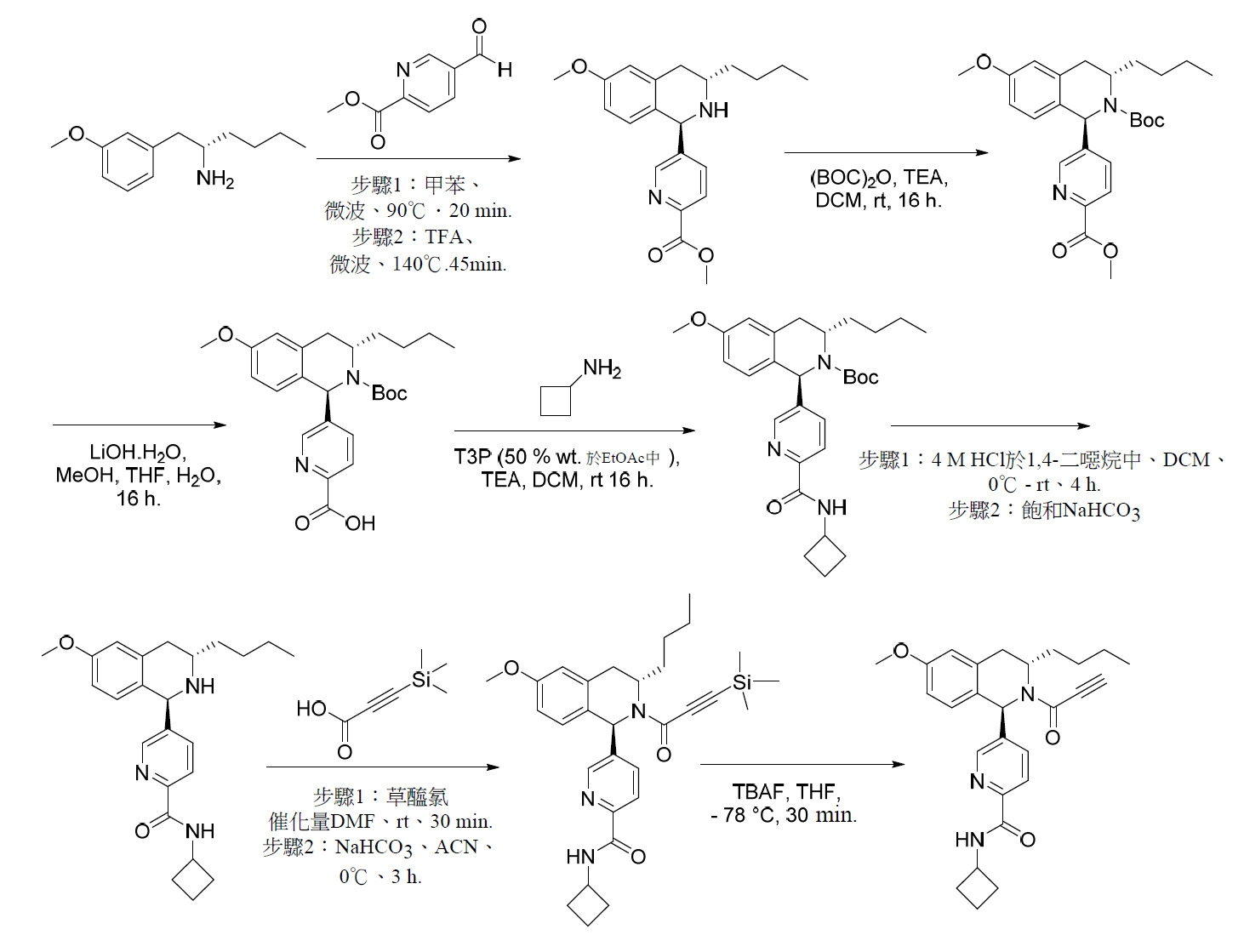 將(S)-1-(3-甲氧基苯基)己烷-2-胺(0.890 g, 4.29 mmol, 1當量)及5-甲醯基吡啶甲酸甲酯(0.850 g, 5.15 mmol, 1.2當量)於甲苯(4 mL)中之溶液在90℃下於微波中輻照20 min。此後,在減壓下濃縮揮發性部分並在TFA (4 mL)中原樣用於環化步驟中,且在140℃下於微波中輻照45 min。此後,在減壓下濃縮揮發性部分且使用NaHCO3
飽和水溶液(10 mL)及EtOAc (40 mL)稀釋所獲得粗製物。分離有機層,使用鹽水(10 mL)洗滌,藉由無水MgSO4
乾燥,過濾並在減壓下濃縮以獲得粗產物。藉由矽膠急速管柱層析(正己烷/EtOAc)純化所獲得粗產物以得到5-((1R,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)吡啶甲酸甲酯。LC-MS (m/z): 355.4 [M+H]+
。1
H NMR (400 MHz, CDCl3
): δ ppm 0.83 - 0.87 (s, 3H), 1.23 - 1.25 (m, 6H), 2.58 - 2.62 (m, 1H), 2.81 - 2.91 (m, 2H), 3.80 (s, 3H), 3.99 (s, 3H), 5.31 (s, 1H), 6.69 - 6.71 (m, 2H), 6.67 - 6.80 (m, 1H), 7.54 - 7.55 (m, 1H), 8.01 - 8.03 (m, 1H), 8.67 (s, 1H)。
在室溫下,向化合物5-((1R,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)吡啶甲酸甲酯(0.320 g, 0.18 mmol, 1當量)於DCM (10 mL)中之溶液中添加三乙胺(0.50 g, 3.61 mmol, 4當量)及二碳酸二-第三丁基酯(0.394 g, 1.80 mmol, 2當量)且將混合物攪拌16 h。TLC (於己烷中之50% EtOAc)顯示反應已完成。在減壓下濃縮反應混合物,且使用EtOAc (50 mL)稀釋粗製物,使用水(2 × 50 mL)洗滌。藉由無水Na2
SO4
乾燥有機層,在減壓下濃縮成粗產物(1R,3S)-3-丁基-6-甲氧基-1-(6-(甲氧基羰基)吡啶-3-基)-3,4-二氫異喹啉-2(1H)-甲酸第三丁基酯。LC-MS (m/z): 455.0 [M-t
Bu+H]+
。
向化合物(1R,3S)-3-丁基-6-甲氧基-1-(6-(甲氧基羰基)吡啶-3-基)-3,4-二氫異喹啉-2(1H)-甲酸第三丁基酯(0.450 g, 0.99 mmol, 1當量)於THF: MeOH: H2
O (9 mL:1 mL:1 mL)之混合物中之溶液中添加氫氧化鋰(0.208 g, 4.96 mmol, 5當量)並在室溫下攪拌16 h。TLC (於己烷中之50% EtOAc)顯示反應已完成。在減壓下濃縮反應混合物,且使用5%檸檬酸溶液(pH=9)酸化粗製物。使用EtOAc (50 mL)稀釋反應混合物且分離有機層,並藉由無水Na2
SO4
乾燥,在減壓下濃縮以得到粗產物5-((1R,3S)-2-(第三丁氧基羰基)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)吡啶甲酸。LC-MS (m/z): 441.0 [M+H]+
。
在0℃下,向化合物5-((1R,3S)-2-(第三丁氧基羰基)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)吡啶甲酸(0.300 g, 0.68 mmol, 1當量)於DCM (10 mL)中之溶液中添加三乙胺(0.38 mL, 2.72 mmol, 4當量)及環丁胺(0.058 g, 0.817 mmol, 1.2當量)且將混合物攪拌15 min。在相同溫度下,向上述反應混合物中添加T3P (50% wt於EtOAc中) (0.72 mL, 0.81 mmol, 1.5當量)並攪拌16 h。TLC (於己烷中之30% EtOAc)顯示反應已完成。在減壓下濃縮反應混合物,且使用EtOAc (50 mL)稀釋粗製物,使用水(2 × 50 mL)洗滌。藉由無水Na2
SO4
乾燥有機層,在減壓下濃縮以得到粗產物(1R,3S)-3-丁基-1-(6-(環丁基胺甲醯基)吡啶-3-基)-6-甲氧基-3,4-二氫異喹啉-2(1H)-甲酸第三丁基酯。LC-MS (m/z): 494.0 [M-t
Bu+H]+
。
在0℃下,向(1R,3S)-3-丁基-1-(6-(環丁基胺甲醯基)吡啶-3-基)-6-甲氧基-3,4-二氫異喹啉-2(1H)-甲酸第三丁基酯(0.220 g, 0.445 mmol, 1當量)於二氯甲烷(50 mL)中之溶液中添加於1,4-二噁烷中之4M HCl (10 mL, 0.89 mmol, 2當量)。將混合物在室溫下攪拌16 h。藉由TLC監測反應進展,在反應完成之後,在減壓下濃縮反應混合物。使用冰冷水(20 mL)溶解所獲得粗製物且藉由NaHCO3
飽和水溶液鹼化。使用EtOAc (100 mL)萃取化合物。使用鹽水(10 mL)洗滌有機層,藉由無水硫酸鈉乾燥,過濾並濃縮以獲得5-((1R,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)-N-環丁基吡啶醯胺。LC-MS (m/z): 394.0 [M+H]+
。
向3-(三甲基矽烷基)丙炔酸(0.084 g, 0.59 mmol, 1當量)中添加DMF (0.003 g, 0.048 mmol, 0.04當量)及草醯氯(0.055 mL, 0.64 mmol, 1.1當量)並攪拌30 min。此後,在減壓下濃縮反應混合物以獲得粗製3-(三甲基矽烷基)丙炔醯氯,使用ACN (1 mL)稀釋並在0℃下添加至含有5-((1R,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)-N-環丁基吡啶醯胺(0.155 g, 0.394 mmol, 1當量)及NaHCO3
(0.248 g, 2.95 mmol, 7.5當量)於ACN (5 mL)中之經攪拌溶液之反應混合物中,且攪拌15 min。LCMS及TLC (於己烷中之70% EtOAc)顯示反應已完成。過濾反應液並在減壓下濃縮以得到粗產物5-((1R,3S)-3-丁基-6-甲氧基-2-(3-(三甲基矽烷基)丙炔醯基)-1,2,3,4-四氫異喹啉-1-基)-N-環丁基吡啶醯胺,其未經進一步純化即用於下一步驟中。LC-MS (m/z): 518.0 [M+H]+
。
向於THF (10.0 mL)中之5-((1R,3S)-3-丁基-6-甲氧基-2-(3-(三甲基矽烷基)丙炔醯基)-1,2,3,4-四氫異喹啉-1-基)-N-環丁基吡啶醯胺(0.210 g, 0.405 mmol, 1當量)中添加TBAF(於THF中之1M溶液) (0.81 mL, 0.81 mmol, 2當量)並攪拌30 min。此後,在減壓下濃縮反應混合物,使用乙酸乙酯(100 mL)稀釋且使用水(2 × 10 mL)洗滌。藉由Na2
SO4
乾燥有機層並濃縮以得到粗產物,藉由製備型TLC層析使用於己烷中之70% EtOAc作為洗脫劑進一步純化以獲得5-((1R,3S)-3-丁基-6-甲氧基-2-丙炔醯基-1,2,3,4-四氫異喹啉-1-基)-N-環丁基吡啶醯胺。LC-MS (m/z): 446.2 [M+H]+
;HPLC純度:99.45%,對掌性HPLC純度:99.8%。1
H NMR (400 MHz, DMSO-d6
): δ 0.79 - 0.83 (m, 3H), 1.20 - 1.24 (m, 5H), 1.46 (bs, 1H), 1.57 - 1.63 (m, 2H), 2.09 - 2.16 (m, 4H), 2.81- 2.95 (m, 1H), 3.15 - 3.19 (m, 1H), 4.35 - 4.41 (m, 1H), 4.63 (s, 1H), 4.77 (bs, 1 H), 6.15 (s, 0.7 H), 6.47 (s, 0.39 H), 6.78 - 6.83 (m, 2H), 7.42 - 7.44 (m, 1H), 7.78 - 7.88 (m, 3H), 8.56 - 8.60 (m, 1H), 8.68 - 8.70 (m, 1H)。
將(S)-1-(3-甲氧基苯基)己烷-2-胺(0.890 g, 4.29 mmol, 1當量)及5-甲醯基吡啶甲酸甲酯(0.850 g, 5.15 mmol, 1.2當量)於甲苯(4 mL)中之溶液在90℃下於微波中輻照20 min。此後,在減壓下濃縮揮發性部分並在TFA (4 mL)中原樣用於環化步驟中,且在140℃下於微波中輻照45 min。此後,在減壓下濃縮揮發性部分且使用NaHCO3
飽和水溶液(10 mL)及EtOAc (40 mL)稀釋所獲得粗製物。分離有機層,使用鹽水(10 mL)洗滌,藉由無水MgSO4
乾燥,過濾並在減壓下濃縮以獲得粗產物。藉由矽膠急速管柱層析(正己烷/EtOAc)純化所獲得粗產物以得到5-((1R,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)吡啶甲酸甲酯。LC-MS (m/z): 355.4 [M+H]+
。1
H NMR (400 MHz, CDCl3
): δ ppm 0.83 - 0.87 (s, 3H), 1.23 - 1.25 (m, 6H), 2.58 - 2.62 (m, 1H), 2.81 - 2.91 (m, 2H), 3.80 (s, 3H), 3.99 (s, 3H), 5.31 (s, 1H), 6.69 - 6.71 (m, 2H), 6.67 - 6.80 (m, 1H), 7.54 - 7.55 (m, 1H), 8.01 - 8.03 (m, 1H), 8.67 (s, 1H)。
在室溫下,向化合物5-((1R,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)吡啶甲酸甲酯(0.320 g, 0.18 mmol, 1當量)於DCM (10 mL)中之溶液中添加三乙胺(0.50 g, 3.61 mmol, 4當量)及二碳酸二-第三丁基酯(0.394 g, 1.80 mmol, 2當量)且將混合物攪拌16 h。TLC (於己烷中之50% EtOAc)顯示反應已完成。在減壓下濃縮反應混合物,且使用EtOAc (50 mL)稀釋粗製物,使用水(2 × 50 mL)洗滌。藉由無水Na2
SO4
乾燥有機層,在減壓下濃縮成粗產物(1R,3S)-3-丁基-6-甲氧基-1-(6-(甲氧基羰基)吡啶-3-基)-3,4-二氫異喹啉-2(1H)-甲酸第三丁基酯。LC-MS (m/z): 455.0 [M-t
Bu+H]+
。
向化合物(1R,3S)-3-丁基-6-甲氧基-1-(6-(甲氧基羰基)吡啶-3-基)-3,4-二氫異喹啉-2(1H)-甲酸第三丁基酯(0.450 g, 0.99 mmol, 1當量)於THF: MeOH: H2
O (9 mL:1 mL:1 mL)之混合物中之溶液中添加氫氧化鋰(0.208 g, 4.96 mmol, 5當量)並在室溫下攪拌16 h。TLC (於己烷中之50% EtOAc)顯示反應已完成。在減壓下濃縮反應混合物,且使用5%檸檬酸溶液(pH=9)酸化粗製物。使用EtOAc (50 mL)稀釋反應混合物且分離有機層,並藉由無水Na2
SO4
乾燥,在減壓下濃縮以得到粗產物5-((1R,3S)-2-(第三丁氧基羰基)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)吡啶甲酸。LC-MS (m/z): 441.0 [M+H]+
。
在0℃下,向化合物5-((1R,3S)-2-(第三丁氧基羰基)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)吡啶甲酸(0.300 g, 0.68 mmol, 1當量)於DCM (10 mL)中之溶液中添加三乙胺(0.38 mL, 2.72 mmol, 4當量)及環丁胺(0.058 g, 0.817 mmol, 1.2當量)且將混合物攪拌15 min。在相同溫度下,向上述反應混合物中添加T3P (50% wt於EtOAc中) (0.72 mL, 0.81 mmol, 1.5當量)並攪拌16 h。TLC (於己烷中之30% EtOAc)顯示反應已完成。在減壓下濃縮反應混合物,且使用EtOAc (50 mL)稀釋粗製物,使用水(2 × 50 mL)洗滌。藉由無水Na2
SO4
乾燥有機層,在減壓下濃縮以得到粗產物(1R,3S)-3-丁基-1-(6-(環丁基胺甲醯基)吡啶-3-基)-6-甲氧基-3,4-二氫異喹啉-2(1H)-甲酸第三丁基酯。LC-MS (m/z): 494.0 [M-t
Bu+H]+
。
在0℃下,向(1R,3S)-3-丁基-1-(6-(環丁基胺甲醯基)吡啶-3-基)-6-甲氧基-3,4-二氫異喹啉-2(1H)-甲酸第三丁基酯(0.220 g, 0.445 mmol, 1當量)於二氯甲烷(50 mL)中之溶液中添加於1,4-二噁烷中之4M HCl (10 mL, 0.89 mmol, 2當量)。將混合物在室溫下攪拌16 h。藉由TLC監測反應進展,在反應完成之後,在減壓下濃縮反應混合物。使用冰冷水(20 mL)溶解所獲得粗製物且藉由NaHCO3
飽和水溶液鹼化。使用EtOAc (100 mL)萃取化合物。使用鹽水(10 mL)洗滌有機層,藉由無水硫酸鈉乾燥,過濾並濃縮以獲得5-((1R,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)-N-環丁基吡啶醯胺。LC-MS (m/z): 394.0 [M+H]+
。
向3-(三甲基矽烷基)丙炔酸(0.084 g, 0.59 mmol, 1當量)中添加DMF (0.003 g, 0.048 mmol, 0.04當量)及草醯氯(0.055 mL, 0.64 mmol, 1.1當量)並攪拌30 min。此後,在減壓下濃縮反應混合物以獲得粗製3-(三甲基矽烷基)丙炔醯氯,使用ACN (1 mL)稀釋並在0℃下添加至含有5-((1R,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)-N-環丁基吡啶醯胺(0.155 g, 0.394 mmol, 1當量)及NaHCO3
(0.248 g, 2.95 mmol, 7.5當量)於ACN (5 mL)中之經攪拌溶液之反應混合物中,且攪拌15 min。LCMS及TLC (於己烷中之70% EtOAc)顯示反應已完成。過濾反應液並在減壓下濃縮以得到粗產物5-((1R,3S)-3-丁基-6-甲氧基-2-(3-(三甲基矽烷基)丙炔醯基)-1,2,3,4-四氫異喹啉-1-基)-N-環丁基吡啶醯胺,其未經進一步純化即用於下一步驟中。LC-MS (m/z): 518.0 [M+H]+
。
向於THF (10.0 mL)中之5-((1R,3S)-3-丁基-6-甲氧基-2-(3-(三甲基矽烷基)丙炔醯基)-1,2,3,4-四氫異喹啉-1-基)-N-環丁基吡啶醯胺(0.210 g, 0.405 mmol, 1當量)中添加TBAF(於THF中之1M溶液) (0.81 mL, 0.81 mmol, 2當量)並攪拌30 min。此後,在減壓下濃縮反應混合物,使用乙酸乙酯(100 mL)稀釋且使用水(2 × 10 mL)洗滌。藉由Na2
SO4
乾燥有機層並濃縮以得到粗產物,藉由製備型TLC層析使用於己烷中之70% EtOAc作為洗脫劑進一步純化以獲得5-((1R,3S)-3-丁基-6-甲氧基-2-丙炔醯基-1,2,3,4-四氫異喹啉-1-基)-N-環丁基吡啶醯胺。LC-MS (m/z): 446.2 [M+H]+
;HPLC純度:99.45%,對掌性HPLC純度:99.8%。1
H NMR (400 MHz, DMSO-d6
): δ 0.79 - 0.83 (m, 3H), 1.20 - 1.24 (m, 5H), 1.46 (bs, 1H), 1.57 - 1.63 (m, 2H), 2.09 - 2.16 (m, 4H), 2.81- 2.95 (m, 1H), 3.15 - 3.19 (m, 1H), 4.35 - 4.41 (m, 1H), 4.63 (s, 1H), 4.77 (bs, 1 H), 6.15 (s, 0.7 H), 6.47 (s, 0.39 H), 6.78 - 6.83 (m, 2H), 7.42 - 7.44 (m, 1H), 7.78 - 7.88 (m, 3H), 8.56 - 8.60 (m, 1H), 8.68 - 8.70 (m, 1H)。
程序 11 : 化合物 40 及 41 之合成  在40℃下,向40-1
(200 mg, 1.02 mmol, 1當量)於MeOH (8 mL)中之溶液中添加於DCM (1 mL)中之SOCl2
(609.43 mg, 5.12 mmol, 371.60 µL, 5當量)。然後將混合物在40℃下攪拌3h以得到黃色溶液。TLC (使用水淬滅,使用PE/ MeOH=20/1洗脫)顯示反應已完成。在減壓下濃縮混合物以得到40-2
。
在20℃下,向40-2
(80 mg, 382.33 umol, 1當量)之溶液中添加於DCM (15 mL)中之4A MS (700 mg, 382.33 umol, 1當量)及4-甲醯基苯甲酸甲酯(62.76 mg, 382.33 umol, 1當量)並攪拌0.5h以得到黃色溶液。TLC (使用PE/EA=3/1洗脫)顯示反應已完成。使用DCM (10 mL)稀釋反應溶液,使用水(10 mL*3)洗滌。藉由無水Na2
SO4
乾燥有機層,在減壓下濃縮以得到40-3
。
將40-3
(150 mg, 422.08 umol, 1當量)於TFA (4.81 g, 42.21 mmol, 3.13 mL, 100當量)中之溶液在80℃下攪拌16h以得到黃色溶液。TLC (使用水淬滅,使用PE/EA=3/1洗脫)顯示反應已完成。使用DCM (10 mL)稀釋反應溶液,使用NaHCO3
溶液洗滌直至pH=8為止。藉由無水Na2
SO4
乾燥有機層,在減壓下濃縮以得到40-4
。
在0℃下,向40-4
(150 mg, 422.08 umol, 1當量)及Et3
N (85.42 mg, 844.16 umol, 117.50 µL, 2當量)於DCM (5 mL)中之溶液中添加2-氯乙醯氯(71.51 mg, 633.12 umol, 50.36 µL, 1.5當量)並保持1h以得到黃色溶液。TLC (使用水淬滅,使用PE/EA=3/1洗脫)顯示反應已完成。藉由製備型TLC純化反應液以得到40
及41
。
化合物40: LC-MS (m/z): 432.0[M]+
。1
H NMR (400 MHz,氯仿-d
) δ ppm 3.08 - 3.13 (m, 1 H) 3.28 (br s, 1 H) 3.59 (s, 4 H) 3.77 (s, 4 H) 3.85 - 3.90 (m, 5 H) 3.93 - 3.99 (m, 1 H) 4.07 (br s, 1 H) 4.11 - 4.18 (m, 1 H) 5.17 (br s, 1 H) 5.28 (br s, 1 H) 6.13 (s, 1 H) 6.42 (s, 1 H) 6.61 - 6.69 (m, 2 H) 6.77 - 6.86 (m, 2 H) 7.28 - 7.36 (m, 5 H) 7.91 (br d,J
=8.28 Hz, 1 H) 7.98 (br d,J
=8.03 Hz, 2 H)。
化合物41: LC-MS (m/z): 432.0[M]+
。1
H NMR (400 MHz,氯仿-d
) δ ppm 2.59 - 2.71 (m, 1 H) 2.99 (br dd,J
=14.81, 4.52 Hz, 1 H) 3.20 (br s, 1 H) 3.33 (br s, 1 H) 3.79 (s, 3 H) 3.77 - 3.80 (m, 1 H) 3.83 (s, 4 H) 3.89 (s, 4 H) 4.07 - 4.26 (m, 3 H) 4.40 (br dd,J
=12.92, 4.64 Hz, 1 H) 4.68 - 4.77 (m, 1 H) 4.73 (br s, 1 H) 6.10 (s, 1 H) 6.76 - 6.85 (m, 2 H) 6.89 (br d,J
=8.53 Hz, 1 H) 7.00 (br d,J
=7.28 Hz, 1 H) 7.35 (br d,J
=8.28 Hz, 1 H) 7.63 (br d,J
=8.28 Hz, 2 H) 7.89 (br d,J
=7.53 Hz, 1 H) 7.99 (br d,J
=8.28 Hz, 2 H)。
在40℃下,向40-1
(200 mg, 1.02 mmol, 1當量)於MeOH (8 mL)中之溶液中添加於DCM (1 mL)中之SOCl2
(609.43 mg, 5.12 mmol, 371.60 µL, 5當量)。然後將混合物在40℃下攪拌3h以得到黃色溶液。TLC (使用水淬滅,使用PE/ MeOH=20/1洗脫)顯示反應已完成。在減壓下濃縮混合物以得到40-2
。
在20℃下,向40-2
(80 mg, 382.33 umol, 1當量)之溶液中添加於DCM (15 mL)中之4A MS (700 mg, 382.33 umol, 1當量)及4-甲醯基苯甲酸甲酯(62.76 mg, 382.33 umol, 1當量)並攪拌0.5h以得到黃色溶液。TLC (使用PE/EA=3/1洗脫)顯示反應已完成。使用DCM (10 mL)稀釋反應溶液,使用水(10 mL*3)洗滌。藉由無水Na2
SO4
乾燥有機層,在減壓下濃縮以得到40-3
。
將40-3
(150 mg, 422.08 umol, 1當量)於TFA (4.81 g, 42.21 mmol, 3.13 mL, 100當量)中之溶液在80℃下攪拌16h以得到黃色溶液。TLC (使用水淬滅,使用PE/EA=3/1洗脫)顯示反應已完成。使用DCM (10 mL)稀釋反應溶液,使用NaHCO3
溶液洗滌直至pH=8為止。藉由無水Na2
SO4
乾燥有機層,在減壓下濃縮以得到40-4
。
在0℃下,向40-4
(150 mg, 422.08 umol, 1當量)及Et3
N (85.42 mg, 844.16 umol, 117.50 µL, 2當量)於DCM (5 mL)中之溶液中添加2-氯乙醯氯(71.51 mg, 633.12 umol, 50.36 µL, 1.5當量)並保持1h以得到黃色溶液。TLC (使用水淬滅,使用PE/EA=3/1洗脫)顯示反應已完成。藉由製備型TLC純化反應液以得到40
及41
。
化合物40: LC-MS (m/z): 432.0[M]+
。1
H NMR (400 MHz,氯仿-d
) δ ppm 3.08 - 3.13 (m, 1 H) 3.28 (br s, 1 H) 3.59 (s, 4 H) 3.77 (s, 4 H) 3.85 - 3.90 (m, 5 H) 3.93 - 3.99 (m, 1 H) 4.07 (br s, 1 H) 4.11 - 4.18 (m, 1 H) 5.17 (br s, 1 H) 5.28 (br s, 1 H) 6.13 (s, 1 H) 6.42 (s, 1 H) 6.61 - 6.69 (m, 2 H) 6.77 - 6.86 (m, 2 H) 7.28 - 7.36 (m, 5 H) 7.91 (br d,J
=8.28 Hz, 1 H) 7.98 (br d,J
=8.03 Hz, 2 H)。
化合物41: LC-MS (m/z): 432.0[M]+
。1
H NMR (400 MHz,氯仿-d
) δ ppm 2.59 - 2.71 (m, 1 H) 2.99 (br dd,J
=14.81, 4.52 Hz, 1 H) 3.20 (br s, 1 H) 3.33 (br s, 1 H) 3.79 (s, 3 H) 3.77 - 3.80 (m, 1 H) 3.83 (s, 4 H) 3.89 (s, 4 H) 4.07 - 4.26 (m, 3 H) 4.40 (br dd,J
=12.92, 4.64 Hz, 1 H) 4.68 - 4.77 (m, 1 H) 4.73 (br s, 1 H) 6.10 (s, 1 H) 6.76 - 6.85 (m, 2 H) 6.89 (br d,J
=8.53 Hz, 1 H) 7.00 (br d,J
=7.28 Hz, 1 H) 7.35 (br d,J
=8.28 Hz, 1 H) 7.63 (br d,J
=8.28 Hz, 2 H) 7.89 (br d,J
=7.53 Hz, 1 H) 7.99 (br d,J
=8.28 Hz, 2 H)。
程序 12: 化合物 42 及 43 之合成  向42-1
(150 mg, 552.83 umol, 1當量)於MeOH (4 mL)中之溶液中添加於DCM (1 mL)中之SOCl2
(65.77 mg, 552.83 umol, 40.10 µL, 1當量)。將混合物在30℃下攪拌16h以得到無色溶液。TLC顯示反應已完成。蒸餾(40℃)反應混合物以得到42-3
(HCl鹽)。1
H NMR (400 MHz,氯仿-d
) δ ppm 2.25 (br s, 5 H) 3.44 (br d,J
=4.52 Hz, 2 H) 3.75 (s, 3 H) 4.40 (br s, 1 H) 6.97 (br d,J
=4.52 Hz, 1 H) 7.28 (br s, 1 H) 7.33 (br s, 1 H) 8.61 (br s, 3 H)。
在20℃下,向42-3
(50 mg, 269.92 umol, 1當量)及4-甲醯基苯甲酸甲酯(44.31 mg, 269.92 umol, 1當量)於甲苯(3 mL)中之溶液中添加TFA (15.39 mg, 134.96 umol, 9.99 µL, 0.5當量)。將混合物在80℃下攪拌16h以得到黃色溶液。TLC顯示反應已完成。藉由製備型TLC純化反應混合物以得到42-4
及42-5
。1
H NMR (400 MHz,氯仿-d
) δ ppm 2.96 - 3.05 (m, 1 H) 3.10 - 3.18 (m, 1 H) 3.73 (s, 3 H) 3.83 - 3.88 (m, 1 H) 3.86 (dd,J
=7.15, 5.65 Hz, 1 H) 3.91 (s, 3 H) 5.44 (s, 1 H) 6.85 (d,J
=5.02 Hz, 1 H) 7.19 (d,J
=5.02 Hz, 1 H) 7.40 (d,J
=8.28 Hz, 2 H) 8.00 (d,J
=8.28 Hz, 2H)。
在0℃下,向42-4
(40 mg, 120.71 umol, 1當量)及TEA (18.32 mg, 181.06 umol, 25.20 µL, 1.5當量)於DCM (3 mL)中之溶液中添加2-氯乙醯氯(20.45 mg, 181.06 umol, 14.40 µL, 1.5當量)。將混合物在30℃下攪拌1h以得到黃色溶液。TLC (使用水淬滅,使用PE/EA=0/1洗脫)顯示反應已完成。藉由製備型TLC純化反應液以得到42
。1
H NMR (400 MHz,氯仿-d
) δ ppm 3.02 - 3.59 (m, 2 H) 3.60 - 3.70 (m, 3 H) 3.88 (br d,J
=5.52 Hz, 3 H) 4.06 (br d,J
=13.30 Hz, 1 H) 5.03 - 5.27 (m, 1 H) 6.27 (br s, 1 H) 6.77 (d,J
=5.02 Hz, 1 H) 7.09 - 7.20 (m, 1 H) 7.32 - 7.52 (m, 2 H) 7.88 - 8.10 (m, 2 H)。LC-MS (m/z): 407.9[M]+
。
在0℃下,向42-5
(60.00 mg, 181.06 umol, 1當量)及TEA (27.48 mg, 271.59 umol, 37.80 µL, 1.5當量)於DCM (3 mL)中之溶液中添加2-氯乙醯氯(30.67 mg, 271.59 umol, 21.60 µL, 1.5當量)。將混合物在30℃下攪拌1h以得到黃色溶液。TLC (使用水淬滅,使用PE/EA=3/1洗脫)顯示反應已完成。藉由製備型TLC再純化混合物以得到43
。1
H NMR (400 MHz,氯仿-d
) δ ppm 2.01 (s, 1 H) 2.99 - 3.17 (m, 5 H) 3.56 (br d,J
=16.06 Hz, 1 H) 3.89 (s, 4 H) 4.18 (d,J
=12.30 Hz, 1 H) 4.30 (br s, 1 H) 4.87 (br s, 1 H) 6.88 - 7.05 (m, 2 H) 7.28 (br s, 1 H) 7.39 (br s, 2 H) 7.94 (br s, 2 H)。LC-MS (m/z): 407.9[M]+
。
向42-1
(150 mg, 552.83 umol, 1當量)於MeOH (4 mL)中之溶液中添加於DCM (1 mL)中之SOCl2
(65.77 mg, 552.83 umol, 40.10 µL, 1當量)。將混合物在30℃下攪拌16h以得到無色溶液。TLC顯示反應已完成。蒸餾(40℃)反應混合物以得到42-3
(HCl鹽)。1
H NMR (400 MHz,氯仿-d
) δ ppm 2.25 (br s, 5 H) 3.44 (br d,J
=4.52 Hz, 2 H) 3.75 (s, 3 H) 4.40 (br s, 1 H) 6.97 (br d,J
=4.52 Hz, 1 H) 7.28 (br s, 1 H) 7.33 (br s, 1 H) 8.61 (br s, 3 H)。
在20℃下,向42-3
(50 mg, 269.92 umol, 1當量)及4-甲醯基苯甲酸甲酯(44.31 mg, 269.92 umol, 1當量)於甲苯(3 mL)中之溶液中添加TFA (15.39 mg, 134.96 umol, 9.99 µL, 0.5當量)。將混合物在80℃下攪拌16h以得到黃色溶液。TLC顯示反應已完成。藉由製備型TLC純化反應混合物以得到42-4
及42-5
。1
H NMR (400 MHz,氯仿-d
) δ ppm 2.96 - 3.05 (m, 1 H) 3.10 - 3.18 (m, 1 H) 3.73 (s, 3 H) 3.83 - 3.88 (m, 1 H) 3.86 (dd,J
=7.15, 5.65 Hz, 1 H) 3.91 (s, 3 H) 5.44 (s, 1 H) 6.85 (d,J
=5.02 Hz, 1 H) 7.19 (d,J
=5.02 Hz, 1 H) 7.40 (d,J
=8.28 Hz, 2 H) 8.00 (d,J
=8.28 Hz, 2H)。
在0℃下,向42-4
(40 mg, 120.71 umol, 1當量)及TEA (18.32 mg, 181.06 umol, 25.20 µL, 1.5當量)於DCM (3 mL)中之溶液中添加2-氯乙醯氯(20.45 mg, 181.06 umol, 14.40 µL, 1.5當量)。將混合物在30℃下攪拌1h以得到黃色溶液。TLC (使用水淬滅,使用PE/EA=0/1洗脫)顯示反應已完成。藉由製備型TLC純化反應液以得到42
。1
H NMR (400 MHz,氯仿-d
) δ ppm 3.02 - 3.59 (m, 2 H) 3.60 - 3.70 (m, 3 H) 3.88 (br d,J
=5.52 Hz, 3 H) 4.06 (br d,J
=13.30 Hz, 1 H) 5.03 - 5.27 (m, 1 H) 6.27 (br s, 1 H) 6.77 (d,J
=5.02 Hz, 1 H) 7.09 - 7.20 (m, 1 H) 7.32 - 7.52 (m, 2 H) 7.88 - 8.10 (m, 2 H)。LC-MS (m/z): 407.9[M]+
。
在0℃下,向42-5
(60.00 mg, 181.06 umol, 1當量)及TEA (27.48 mg, 271.59 umol, 37.80 µL, 1.5當量)於DCM (3 mL)中之溶液中添加2-氯乙醯氯(30.67 mg, 271.59 umol, 21.60 µL, 1.5當量)。將混合物在30℃下攪拌1h以得到黃色溶液。TLC (使用水淬滅,使用PE/EA=3/1洗脫)顯示反應已完成。藉由製備型TLC再純化混合物以得到43
。1
H NMR (400 MHz,氯仿-d
) δ ppm 2.01 (s, 1 H) 2.99 - 3.17 (m, 5 H) 3.56 (br d,J
=16.06 Hz, 1 H) 3.89 (s, 4 H) 4.18 (d,J
=12.30 Hz, 1 H) 4.30 (br s, 1 H) 4.87 (br s, 1 H) 6.88 - 7.05 (m, 2 H) 7.28 (br s, 1 H) 7.39 (br s, 2 H) 7.94 (br s, 2 H)。LC-MS (m/z): 407.9[M]+
。
程序 13: 化合物 44 及 45 之合成  向2-甲基丙烷-2-亞磺醯胺(6.24 g, 51.49 mmol, 2當量)於THF (120 mL)中之此溶液中添加Ti(OEt)4
(58.72 g, 257.43 mmol, 53.38 mL, 10當量)、於THF (100 mL)中之44-1
(5 g, 25.74 mmol, 1當量)。將褐色溶液加熱至75℃並藉由TLC監測。在5 hr之後,將反應液冷卻至25℃以得到粗製44-2
。然後將反應液冷卻至- 20℃,添加NaBH4
(973.93 mg, 25.74 mmol, 1當量)且將反應液在-20℃下攪拌3 hr,然後升溫至25℃並攪拌12 hr。LCMS顯示反應已完成。添加等體積之NaCl飽和水溶液(60 mL)以沈澱鈦鹽。在攪拌5 min之後,經由矽藻土過濾懸浮液且使用EtOAc (100 mL × 2)洗滌濾餅。分離有機層,且使用EtOAc (2 × 100 mL)萃取水層。藉由無水硫酸鈉乾燥合併之有機層並濃縮以產生殘餘物。藉由使用於石油醚中之乙酸乙酯(0%至80%)洗脫之急速層析(二氧化矽)純化殘餘物以得到44-3
。1
H NMR (400MHz, CDCl3
) δ = 6.77 - 6.62 (m, 3H), 3.80 (d,J
= 4.8 Hz, 6H), 3.64 - 3.49 (m, 1H), 3.18 (br d,J
= 4.8 Hz, 1H), 2.78 - 2.59 (m, 2H), 1.14 - 1.05 (m, 12H)。
向44-3
(2 g, 6.68 mmol, 1當量)於MeOH (20 mL)中之混合物中逐滴添加HCl/二噁烷(4 M, 20 mL, 11.98當量)。將混合物在20℃下攪拌 12 h以得到褐色混合物。LCMS顯示反應已完成。使用60 mL HCl (0.1 M)稀釋反應混合物。使用乙酸乙酯(30 mL × 2)萃取所得混合物。將水相調節至pH = 8。使用乙酸乙酯(30 mL × 3)萃取水相。使用無水Na2
SO4
乾燥合併之有機相,過濾並在真空中濃縮以提供44-4
。1
H NMR (400MHz, CDCl3
) δ = 6.78 - 6.83 (m, 1H), 6.70 - 6.75 (m, 2H), 3.86 (d,J
= 4.4 Hz, 6H), 3.07 - 3.21 (m, 1H), 2.78 - 2.59 (m, 2H), 2.67 (m, 1H), 2.44 (m, 1H), 1.64 (brs, 2H), 1.12 (d,J
= 6.4 Hz, 3H)。
將4A分子篩(3 g, 2.56 mmol, 1當量)添加至44-4
(500 mg, 2.56 mmol, 1當量)及4-嗎啉基苯甲醛(489.68 mg, 2.56 mmol, 1當量)於甲苯(20 mL)中之溶液中,將混合物在120℃下攪拌4 hr。LCMS展示起始材料並未完全消耗。將反應混合物在120℃下再攪拌4 h。LCMS展示起始材料已完全消耗。過濾反應混合物且在真空中濃縮濾液。將殘餘物溶於TFA (32.58 g, 285.73 mmol, 21.16 mL, 111.58當量)中且將溶液在120℃下加熱20 h。LC-MS展示起始材料已完全消耗。在減壓下濃縮反應混合物。藉由2N NaOH水溶液將混合物調節至pH=9並使用EtOAc (30 mL × 3)萃取。使用鹽水(30 mL)洗滌合併之有機層,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以得到殘餘物。藉由管柱層析(SiO2
,二氯甲烷:甲醇= 100: 1至100: 5)純化殘餘物以提供N136-6及44-5
。1
H NMR (400 MHz,氯仿-d
) δ ppm 1.25 (d,J
= 6.4 Hz, 3H), 2.58 - 2.80 (m, 2H), 3.12 - 3.22 (m, 5H), 3.57 - 3.64 (m, 3H), 3.84 - 3.91 (m, 7H), 5.01 (s, 1H), 6.18 - 6.25 (m, 1H), 6.57 - 6.66 (m, 1H), 6.89 (d,J
= 8.8 Hz, 2H), 7.23 (d,J
= 8.8 Hz, 2H)。
在0℃下,向44-6
(122 mg, 331.10 umol, 1當量)及TEA (335.04 mg, 3.31 mmol, 460.85 µL, 10當量)於DCM (6 mL)中之混合物中添加2-氯乙醯氯(112.19 mg, 993.30 umol, 79.00 µL, 3當量)。將混合物在0℃下攪拌30 min以得到褐色混合物。LCMS展示起始材料已完全消耗。在減壓下濃縮反應混合物以得到殘餘物。藉由製備型TLC純化殘餘物且然後藉由凍乾乾燥以提供44
及45
。
化合物44: LC-MS (m/z): 445.0 [M+H]+ 1
H NMR (400 MHz,氯仿-d
) δ 0.99 (br s, 3H), 2.41 (br d,J
= 14.8 Hz, 1H), 2.80 - 3.28 (m, 5H), 3.60 - 4.57 (m, 12H), 4.83 (br s, 1H), 5.74 (br s, 1H), 6.57 (s, 1H), 6.65 - 6.90 (m, 3H), 7.03 (d,J
= 8.8 Hz, 2H)
化合物45: LC-MS (m/z): 445.0 [M+H]+ 1
H NMR (400 MHz,氯仿-d
) δ 0.99 (br s, 3H), 2.41 (br d,J
= 14.8 Hz, 1H), 2.80 - 3.28 (m, 5H), 3.60 - 4.57 (m, 12H), 4.83 (br s, 1H), 5.74 (br s, 1H), 6.57 (s, 1H), 6.65 - 6.90 (m, 3H), 7.03 (d,J
= 8.8 Hz, 2H)
向2-甲基丙烷-2-亞磺醯胺(6.24 g, 51.49 mmol, 2當量)於THF (120 mL)中之此溶液中添加Ti(OEt)4
(58.72 g, 257.43 mmol, 53.38 mL, 10當量)、於THF (100 mL)中之44-1
(5 g, 25.74 mmol, 1當量)。將褐色溶液加熱至75℃並藉由TLC監測。在5 hr之後,將反應液冷卻至25℃以得到粗製44-2
。然後將反應液冷卻至- 20℃,添加NaBH4
(973.93 mg, 25.74 mmol, 1當量)且將反應液在-20℃下攪拌3 hr,然後升溫至25℃並攪拌12 hr。LCMS顯示反應已完成。添加等體積之NaCl飽和水溶液(60 mL)以沈澱鈦鹽。在攪拌5 min之後,經由矽藻土過濾懸浮液且使用EtOAc (100 mL × 2)洗滌濾餅。分離有機層,且使用EtOAc (2 × 100 mL)萃取水層。藉由無水硫酸鈉乾燥合併之有機層並濃縮以產生殘餘物。藉由使用於石油醚中之乙酸乙酯(0%至80%)洗脫之急速層析(二氧化矽)純化殘餘物以得到44-3
。1
H NMR (400MHz, CDCl3
) δ = 6.77 - 6.62 (m, 3H), 3.80 (d,J
= 4.8 Hz, 6H), 3.64 - 3.49 (m, 1H), 3.18 (br d,J
= 4.8 Hz, 1H), 2.78 - 2.59 (m, 2H), 1.14 - 1.05 (m, 12H)。
向44-3
(2 g, 6.68 mmol, 1當量)於MeOH (20 mL)中之混合物中逐滴添加HCl/二噁烷(4 M, 20 mL, 11.98當量)。將混合物在20℃下攪拌 12 h以得到褐色混合物。LCMS顯示反應已完成。使用60 mL HCl (0.1 M)稀釋反應混合物。使用乙酸乙酯(30 mL × 2)萃取所得混合物。將水相調節至pH = 8。使用乙酸乙酯(30 mL × 3)萃取水相。使用無水Na2
SO4
乾燥合併之有機相,過濾並在真空中濃縮以提供44-4
。1
H NMR (400MHz, CDCl3
) δ = 6.78 - 6.83 (m, 1H), 6.70 - 6.75 (m, 2H), 3.86 (d,J
= 4.4 Hz, 6H), 3.07 - 3.21 (m, 1H), 2.78 - 2.59 (m, 2H), 2.67 (m, 1H), 2.44 (m, 1H), 1.64 (brs, 2H), 1.12 (d,J
= 6.4 Hz, 3H)。
將4A分子篩(3 g, 2.56 mmol, 1當量)添加至44-4
(500 mg, 2.56 mmol, 1當量)及4-嗎啉基苯甲醛(489.68 mg, 2.56 mmol, 1當量)於甲苯(20 mL)中之溶液中,將混合物在120℃下攪拌4 hr。LCMS展示起始材料並未完全消耗。將反應混合物在120℃下再攪拌4 h。LCMS展示起始材料已完全消耗。過濾反應混合物且在真空中濃縮濾液。將殘餘物溶於TFA (32.58 g, 285.73 mmol, 21.16 mL, 111.58當量)中且將溶液在120℃下加熱20 h。LC-MS展示起始材料已完全消耗。在減壓下濃縮反應混合物。藉由2N NaOH水溶液將混合物調節至pH=9並使用EtOAc (30 mL × 3)萃取。使用鹽水(30 mL)洗滌合併之有機層,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以得到殘餘物。藉由管柱層析(SiO2
,二氯甲烷:甲醇= 100: 1至100: 5)純化殘餘物以提供N136-6及44-5
。1
H NMR (400 MHz,氯仿-d
) δ ppm 1.25 (d,J
= 6.4 Hz, 3H), 2.58 - 2.80 (m, 2H), 3.12 - 3.22 (m, 5H), 3.57 - 3.64 (m, 3H), 3.84 - 3.91 (m, 7H), 5.01 (s, 1H), 6.18 - 6.25 (m, 1H), 6.57 - 6.66 (m, 1H), 6.89 (d,J
= 8.8 Hz, 2H), 7.23 (d,J
= 8.8 Hz, 2H)。
在0℃下,向44-6
(122 mg, 331.10 umol, 1當量)及TEA (335.04 mg, 3.31 mmol, 460.85 µL, 10當量)於DCM (6 mL)中之混合物中添加2-氯乙醯氯(112.19 mg, 993.30 umol, 79.00 µL, 3當量)。將混合物在0℃下攪拌30 min以得到褐色混合物。LCMS展示起始材料已完全消耗。在減壓下濃縮反應混合物以得到殘餘物。藉由製備型TLC純化殘餘物且然後藉由凍乾乾燥以提供44
及45
。
化合物44: LC-MS (m/z): 445.0 [M+H]+ 1
H NMR (400 MHz,氯仿-d
) δ 0.99 (br s, 3H), 2.41 (br d,J
= 14.8 Hz, 1H), 2.80 - 3.28 (m, 5H), 3.60 - 4.57 (m, 12H), 4.83 (br s, 1H), 5.74 (br s, 1H), 6.57 (s, 1H), 6.65 - 6.90 (m, 3H), 7.03 (d,J
= 8.8 Hz, 2H)
化合物45: LC-MS (m/z): 445.0 [M+H]+ 1
H NMR (400 MHz,氯仿-d
) δ 0.99 (br s, 3H), 2.41 (br d,J
= 14.8 Hz, 1H), 2.80 - 3.28 (m, 5H), 3.60 - 4.57 (m, 12H), 4.83 (br s, 1H), 5.74 (br s, 1H), 6.57 (s, 1H), 6.65 - 6.90 (m, 3H), 7.03 (d,J
= 8.8 Hz, 2H)
程序 14 : 化合物 46 之合成 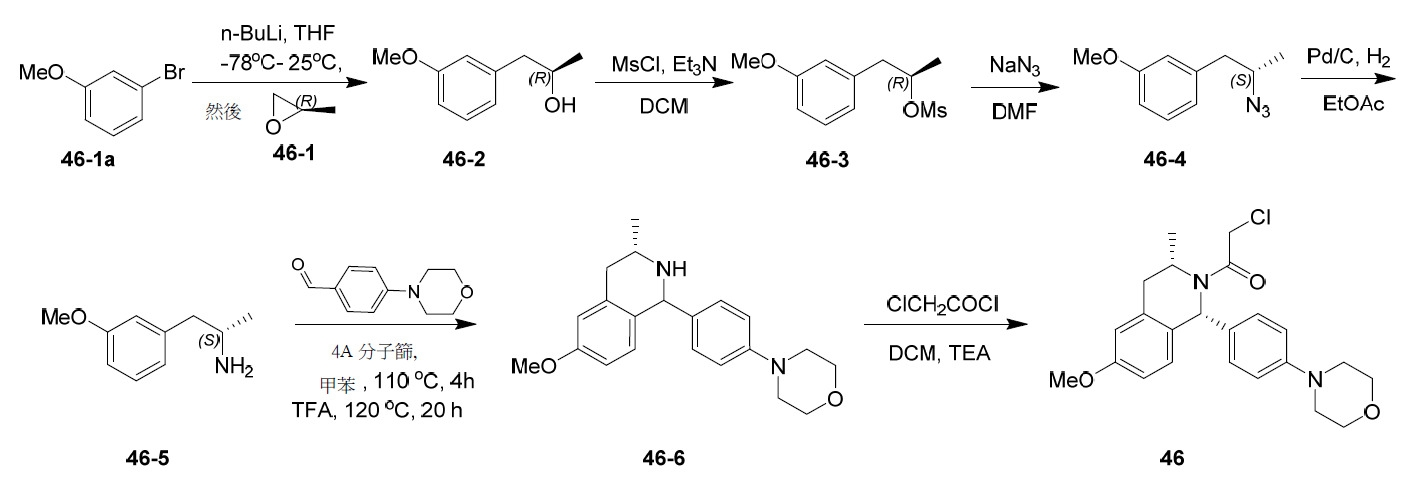 在-78℃下,向46-1a
(5 g, 26.73 mmol, 3.38 mL, 1當量)於THF (150 mL)中之溶液中逐滴添加n
-BuLi (2.5 M, 16.04 mL, 1.5當量)。在攪拌30 min之後,一次性添加46-1
(1.71 g, 29.41 mmol, 2.06 mL, 1.1當量)。將混合物在-78℃下攪拌1 hr,然後升溫至25℃並攪拌12 hr。TLC顯示反應已完成。藉由添加飽和NH4
Cl (50 mL)水溶液來終止反應。使用乙酸乙酯(3 × 50 mL)萃取水相且使用鹽水(100 mL)洗滌合併之有機層,藉由無水Na2
SO4
乾燥,過濾並在減壓下濃縮。藉由使用於石油醚中之乙酸乙酯(0%至30%)洗脫之急速層析(二氧化矽)純化粗產物以得到46-2
。1
H NMR (400MHz, CDCl3
) δ = 7.15 (t,J
= 8.0 Hz, 1H), 6.79 - 6.59 (m, 3H), 3.99 - 3.89 (m, 1H), 3.72 (s, 3H), 2.74 - 2.53 (m, 2H), 1.63 (br s, 1H), 1.19 - 1.14 (m, 1H), 1.17 (d,J
= 6.0 Hz, 2H)。
在0℃下,向46-2
(1 g, 6.02 mmol, 1當量)及Et3
N (1.83 g, 18.05 mmol, 2.51 mL, 3當量)於DCM (20 mL)中之溶液中添加MsCl (1.03 g, 9.02 mmol, 698.48 µL, 1.5當量)。將混合物在25℃下攪拌1 h。TLC顯示反應已完成。將反應液傾倒至NaHCO3
飽和水溶液(50 mL)中,使用CH2
Cl2
(3 × 50 mL)萃取混合物。使用鹽水(50 mL)洗滌合併之有機層,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以得到46-3
,其未經進一步純化即直接用於下一步驟中。1
H NMR (400MHz, CDCl3
) δ = 7.22 - 7.12 (m, 1H), 6.82 - 6.62 (m, 3H), 4.89 - 4.73 (m, 1H), 3.73 (s, 3H), 2.95 - 2.74 (m, 2H), 2.49 (s, 3H), 1.40 (d,J
= 6.0 Hz, 3H)。
向46-3
(1.5 g, 6.14 mmol,1當量)於DMF (8 mL)中之溶液中添加NaN3
(798.30 mg, 12.28 mmol,2當量)。將混合物在80℃下加熱2 h。TLC顯示反應已完成。添加90 mL水,使用120 mL EtOAc/己烷(1:1)混合物萃取混合物。藉由無水Na2
SO4
乾燥萃取物並蒸發以得到粗製46-4
,其未經進一步純化即直接用於下一步驟中。
在N2
下,向46-4
(1.1 g, 5.75 mmol, 1當量)於EtOAc (100 mL)中之溶液中添加Pd/C (500 mg, 10%純度)。將懸浮液在真空下脫氣並使用H2
吹掃若干次。將混合物在H2
(15 psi)及25℃下攪拌1小時。TLC顯示反應已完成。過濾反應混合物且濃縮濾液。藉由使用於CH2
Cl2
中之MeOH (0%至20%)洗脫之急速層析(二氧化矽)純化粗產物以得到46-5
。1
H NMR (400MHz, CDCl3
) δ = 7.20 - 7.08 (m, 1H), 6.74 - 6.61 (m, 3H), 3.72 (s, 3H), 3.17 - 3.03 (m, 1H), 2.62 (dd,J
= 5.6, 13.6 Hz, 1H), 2.42 (dd,J
= 8.0, 13.2 Hz, 1H), 1.42 (s, 2H), 1.05 (d,J
= 6.4 Hz, 3H)。
將4A MS (600 mg)添加至46-5
(100 mg, 605.21 umol, 1當量)及4-嗎啉基苯甲醛(115.73 mg, 605.21 umol, 1當量)於甲苯(4 mL)中之溶液中且將混合物在120℃下攪拌4 hr。過濾反應混合物且在真空中濃縮濾液。將殘餘物溶於TFA (5.00 mL)中且將溶液在120℃下加熱20 hr。TLC顯示反應已完成。在減壓下濃縮反應混合物。藉由2N NaOH水溶液鹼化混合物並使用EtOAc (30 mL × 3)萃取。使用鹽水(30 mL)洗滌合併之有機層,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以得到46-6
,其未經進一步純化即直接用於下一步驟中。1
H NMR (400MHz, CDCl3
) δ = 7.19 - 7.13 (m, 2H), 6.80 (d,J
= 8.4 Hz, 2H), 6.58 - 6.47 (m, 3H), 4.93 (s, 1H), 3.80 - 3.77 (m, 4H), 3.69 (s, 3H), 3.16 - 3.11 (m, 1H), 3.10 - 3.05 (m, 4H), 2.71 - 2.63 (m, 2H), 1.97 (s, 1H), 1.16 (d,J
= 6.0 Hz, 3H)。
在0℃下,向46-6
(250.00 mg, 738.68 umol, 1當量)及Et3
N (224.24 mg, 2.22 mmol, 308.45 µL, 3當量)於DCM (5 mL)中之溶液中添加2-氯乙醯氯(166.86 mg, 1.48 mmol, 117.51 µL, 2當量)。將混合物在25℃下攪拌1 h。LCMS顯示反應已完成。將反應液傾倒至飽和NaHCO3
(50 mL)溶液中,使用CH2
Cl2
(3 × 50 mL)萃取混合物。使用鹽水(50 mL)洗滌合併之有機層,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮。藉由使用於石油醚中之乙酸乙酯(0%至40%)洗脫之急速層析(二氧化矽)純化殘餘物以得到46
。1
H NMR (400MHz, DMSO-d6
) δ = 7.15 (br d,J
= 8.8 Hz, 1H), 7.00 (d,J
= 8.4 Hz, 2H), 6.90 - 6.79 (m, 4H), 6.31 (br s, 1H), 4.53 - 4.35 (m, 2H), 4.28 - 4.18 (m, 1H), 3.79 (s, 3H), 3.76 - 3.70 (m, 2H), 3.76 - 3.70 (m, 1H), 3.76 - 3.70 (m, 1H), 3.12 - 3.08 (m, 4H), 3.01 - 2.93 (m, 1H), 2.49 - 2.37 (m, 1H), 1.14 (d,J
= 6.4 Hz, 3H)。
在-78℃下,向46-1a
(5 g, 26.73 mmol, 3.38 mL, 1當量)於THF (150 mL)中之溶液中逐滴添加n
-BuLi (2.5 M, 16.04 mL, 1.5當量)。在攪拌30 min之後,一次性添加46-1
(1.71 g, 29.41 mmol, 2.06 mL, 1.1當量)。將混合物在-78℃下攪拌1 hr,然後升溫至25℃並攪拌12 hr。TLC顯示反應已完成。藉由添加飽和NH4
Cl (50 mL)水溶液來終止反應。使用乙酸乙酯(3 × 50 mL)萃取水相且使用鹽水(100 mL)洗滌合併之有機層,藉由無水Na2
SO4
乾燥,過濾並在減壓下濃縮。藉由使用於石油醚中之乙酸乙酯(0%至30%)洗脫之急速層析(二氧化矽)純化粗產物以得到46-2
。1
H NMR (400MHz, CDCl3
) δ = 7.15 (t,J
= 8.0 Hz, 1H), 6.79 - 6.59 (m, 3H), 3.99 - 3.89 (m, 1H), 3.72 (s, 3H), 2.74 - 2.53 (m, 2H), 1.63 (br s, 1H), 1.19 - 1.14 (m, 1H), 1.17 (d,J
= 6.0 Hz, 2H)。
在0℃下,向46-2
(1 g, 6.02 mmol, 1當量)及Et3
N (1.83 g, 18.05 mmol, 2.51 mL, 3當量)於DCM (20 mL)中之溶液中添加MsCl (1.03 g, 9.02 mmol, 698.48 µL, 1.5當量)。將混合物在25℃下攪拌1 h。TLC顯示反應已完成。將反應液傾倒至NaHCO3
飽和水溶液(50 mL)中,使用CH2
Cl2
(3 × 50 mL)萃取混合物。使用鹽水(50 mL)洗滌合併之有機層,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以得到46-3
,其未經進一步純化即直接用於下一步驟中。1
H NMR (400MHz, CDCl3
) δ = 7.22 - 7.12 (m, 1H), 6.82 - 6.62 (m, 3H), 4.89 - 4.73 (m, 1H), 3.73 (s, 3H), 2.95 - 2.74 (m, 2H), 2.49 (s, 3H), 1.40 (d,J
= 6.0 Hz, 3H)。
向46-3
(1.5 g, 6.14 mmol,1當量)於DMF (8 mL)中之溶液中添加NaN3
(798.30 mg, 12.28 mmol,2當量)。將混合物在80℃下加熱2 h。TLC顯示反應已完成。添加90 mL水,使用120 mL EtOAc/己烷(1:1)混合物萃取混合物。藉由無水Na2
SO4
乾燥萃取物並蒸發以得到粗製46-4
,其未經進一步純化即直接用於下一步驟中。
在N2
下,向46-4
(1.1 g, 5.75 mmol, 1當量)於EtOAc (100 mL)中之溶液中添加Pd/C (500 mg, 10%純度)。將懸浮液在真空下脫氣並使用H2
吹掃若干次。將混合物在H2
(15 psi)及25℃下攪拌1小時。TLC顯示反應已完成。過濾反應混合物且濃縮濾液。藉由使用於CH2
Cl2
中之MeOH (0%至20%)洗脫之急速層析(二氧化矽)純化粗產物以得到46-5
。1
H NMR (400MHz, CDCl3
) δ = 7.20 - 7.08 (m, 1H), 6.74 - 6.61 (m, 3H), 3.72 (s, 3H), 3.17 - 3.03 (m, 1H), 2.62 (dd,J
= 5.6, 13.6 Hz, 1H), 2.42 (dd,J
= 8.0, 13.2 Hz, 1H), 1.42 (s, 2H), 1.05 (d,J
= 6.4 Hz, 3H)。
將4A MS (600 mg)添加至46-5
(100 mg, 605.21 umol, 1當量)及4-嗎啉基苯甲醛(115.73 mg, 605.21 umol, 1當量)於甲苯(4 mL)中之溶液中且將混合物在120℃下攪拌4 hr。過濾反應混合物且在真空中濃縮濾液。將殘餘物溶於TFA (5.00 mL)中且將溶液在120℃下加熱20 hr。TLC顯示反應已完成。在減壓下濃縮反應混合物。藉由2N NaOH水溶液鹼化混合物並使用EtOAc (30 mL × 3)萃取。使用鹽水(30 mL)洗滌合併之有機層,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以得到46-6
,其未經進一步純化即直接用於下一步驟中。1
H NMR (400MHz, CDCl3
) δ = 7.19 - 7.13 (m, 2H), 6.80 (d,J
= 8.4 Hz, 2H), 6.58 - 6.47 (m, 3H), 4.93 (s, 1H), 3.80 - 3.77 (m, 4H), 3.69 (s, 3H), 3.16 - 3.11 (m, 1H), 3.10 - 3.05 (m, 4H), 2.71 - 2.63 (m, 2H), 1.97 (s, 1H), 1.16 (d,J
= 6.0 Hz, 3H)。
在0℃下,向46-6
(250.00 mg, 738.68 umol, 1當量)及Et3
N (224.24 mg, 2.22 mmol, 308.45 µL, 3當量)於DCM (5 mL)中之溶液中添加2-氯乙醯氯(166.86 mg, 1.48 mmol, 117.51 µL, 2當量)。將混合物在25℃下攪拌1 h。LCMS顯示反應已完成。將反應液傾倒至飽和NaHCO3
(50 mL)溶液中,使用CH2
Cl2
(3 × 50 mL)萃取混合物。使用鹽水(50 mL)洗滌合併之有機層,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮。藉由使用於石油醚中之乙酸乙酯(0%至40%)洗脫之急速層析(二氧化矽)純化殘餘物以得到46
。1
H NMR (400MHz, DMSO-d6
) δ = 7.15 (br d,J
= 8.8 Hz, 1H), 7.00 (d,J
= 8.4 Hz, 2H), 6.90 - 6.79 (m, 4H), 6.31 (br s, 1H), 4.53 - 4.35 (m, 2H), 4.28 - 4.18 (m, 1H), 3.79 (s, 3H), 3.76 - 3.70 (m, 2H), 3.76 - 3.70 (m, 1H), 3.76 - 3.70 (m, 1H), 3.12 - 3.08 (m, 4H), 3.01 - 2.93 (m, 1H), 2.49 - 2.37 (m, 1H), 1.14 (d,J
= 6.4 Hz, 3H)。
程序 15 :化合物 47 之合成  在-78℃下,向47-1a
(5 g, 26.73 mmol, 3.38 mL, 1當量)於THF (150 mL)中之溶液中逐滴添加n
-BuLi (2.5 M, 16.04 mL, 1.5當量)。在攪拌30 min之後,一次性添加47-1
(1.71 g, 29.41 mmol, 2.06 mL, 1.1當量)。將混合物在-78℃下攪拌1 hr,然後升溫至25℃並攪拌12 hr。TLC顯示反應已完成。藉由添加飽和NH4
Cl (50 mL)水溶液來終止反應。使用乙酸乙酯(3 × 50 mL)萃取水相且使用鹽水(100 mL)洗滌合併之有機層,藉由無水Na2
SO4
乾燥,過濾並在減壓下濃縮。藉由使用於石油醚中之乙酸乙酯(0%至30%)洗脫之急速層析(二氧化矽)純化粗產物以得到47-2
。1
H NMR (400MHz, CDCl3
) δ = 7.15 (t,J
= 8.0 Hz, 1H), 6.79 - 6.59 (m, 3H), 3.99 - 3.89 (m, 1H), 3.72 (s, 3H), 2.74 - 2.53 (m, 2H), 1.63 (br s, 1H), 1.19 - 1.14 (m, 1H), 1.17 (d,J
= 6.0 Hz, 2H)。
在0℃下,向47-2
(1 g, 6.02 mmol, 1當量)及Et3
N (1.83 g, 18.05 mmol, 2.51 mL, 3當量)於DCM (20 mL)中之溶液中添加MsCl (1.03 g, 9.02 mmol, 698.48 µL, 1.5當量)。將混合物在25℃下攪拌1 h。TLC顯示反應已完成。將反應液傾倒至NaHCO3
飽和水溶液(50 mL)中,使用CH2
Cl2
(3 × 50 mL)萃取混合物。使用鹽水(50 mL)洗滌合併之有機層,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以得到47-3
,其未經進一步純化即直接用於下一步驟中。1
H NMR (400MHz, CDCl3
) δ = 7.22 - 7.12 (m, 1H), 6.82 - 6.62 (m, 3H), 4.89 - 4.73 (m, 1H), 3.73 (s, 3H), 2.95 - 2.74 (m, 2H), 2.49 (s, 3H), 1.40 (d,J
= 6.0 Hz, 3H)。
向47-3
(1.5 g, 6.14 mmol,1當量)於DMF (8 mL)中之溶液中添加NaN3
(798.30 mg, 12.28 mmol,2當量)。將混合物在80℃下加熱2 h。TLC顯示反應已完成。添加90 mL水,使用120 mL EtOAc/己烷(1:1)混合物萃取混合物。藉由無水Na2
SO4
乾燥萃取物並蒸發以得到粗製47-4
,其未經進一步純化即直接用於下一步驟中。
在N2
下,向47-4
(1.1 g, 5.75 mmol, 1當量)於EtOAc (100 mL)中之溶液中添加Pd/C (500 mg, 10%純度)。將懸浮液在真空下脫氣並使用H2
吹掃若干次。將混合物在H2
(15 psi)及25℃下攪拌1小時。TLC顯示反應已完成。過濾反應混合物且濃縮濾液。藉由使用於CH2
Cl2
中之MeOH (0%至20%)洗脫之急速層析(二氧化矽)純化粗產物以得到47-5
。1
H NMR (400MHz, CDCl3
) δ = 7.20 - 7.08 (m, 1H), 6.74 - 6.61 (m, 3H), 3.72 (s, 3H), 3.17 - 3.03 (m, 1H), 2.62 (dd,J
= 5.6, 13.6 Hz, 1H), 2.42 (dd,J
= 8.0, 13.2 Hz, 1H), 1.42 (s, 2H), 1.05 (d,J
= 6.4 Hz, 3H)。
將4A分子篩(3 g, 3.03 mmol, 1.00當量)添加至47-5
(500 mg, 3.03 mmol, 1當量)及4-嗎啉基苯甲醛(578.66 mg, 3.03 mmol, 1當量)於甲苯(25 mL)中之溶液中,將混合物在120℃下攪拌4 hr。LCMS顯示反應已完成。在冷卻至室溫之,過濾混合物且將濾液在真空下濃縮至較小體積以得到粗製N135-6A
,其未經進一步純化即直接用於下一步驟中。
將47-6A
(1 g, 2.95 mmol, 1當量)溶於TFA (46.20 g, 405.19 mmol, 30.00 mL, 137.13當量)中,將溶液在120℃下加熱20 hr以得到褐色溶液。LCMS顯示反應已完成。在減壓下濃縮反應混合物。藉由2N NaOH水溶液鹼化混合物並使用EtOAc (50 mL × 3)萃取。使用鹽水(50 mL)洗滌合併之有機層,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮。藉由使用於CH2
Cl2
中之MeOH
(0%至5%)洗脫之急速層析(二氧化矽)純化殘餘物。藉由製備型TLC (DCM/MeOH=10/1, Rf1=0.5, Rf2=0.45)純化粗產物以得到順式產物(作為主要產物)。
藉由進一步之製備型TLC純化獲得痕量反式產物(95%純度)且藉由2D NMR可明確證實結構。
在0℃下,向47-6B
(10 mg, 29.55 umol, 1當量)及Et3
N (8.97 mg, 88.64 umol, 12.34 µL, 3當量)於DCM (2 mL)中之溶液中添加2-氯乙醯氯(6.67 mg, 59.09 umol, 4.70 µL, 2當量)。將混合物在25℃下攪拌1 h以得到褐色溶液。LCMS顯示反應已完成。在減壓下濃縮反應液。藉由製備型TLC (PE/EA=2/1, Rf=0.4)純化殘餘物以得到47
。1
H NMR (400MHz, CDCl3
) δ = 7.24 (br d,J
= 8.0 Hz, 1H), 7.02 (d,J
= 8.4 Hz, 2H), 6.78 - 6.69 (m, 3H), 6.61 (s, 1H), 5.78 (br s, 1H), 4.84 (br s, 1H), 4.04 (br d,J
= 12.4 Hz, 1H), 3.81 (br d,J
= 12.4 Hz, 1H), 3.77 - 3.73 (m, 4H), 3.72 (s, 3H), 3.06 - 2.99 (m, 4H), 2.96 (br s, 1H), 2.44 (br d,J
= 16.0 Hz, 1H), 0.97 (br d,J
= 5.6 Hz, 3H)。
在-78℃下,向47-1a
(5 g, 26.73 mmol, 3.38 mL, 1當量)於THF (150 mL)中之溶液中逐滴添加n
-BuLi (2.5 M, 16.04 mL, 1.5當量)。在攪拌30 min之後,一次性添加47-1
(1.71 g, 29.41 mmol, 2.06 mL, 1.1當量)。將混合物在-78℃下攪拌1 hr,然後升溫至25℃並攪拌12 hr。TLC顯示反應已完成。藉由添加飽和NH4
Cl (50 mL)水溶液來終止反應。使用乙酸乙酯(3 × 50 mL)萃取水相且使用鹽水(100 mL)洗滌合併之有機層,藉由無水Na2
SO4
乾燥,過濾並在減壓下濃縮。藉由使用於石油醚中之乙酸乙酯(0%至30%)洗脫之急速層析(二氧化矽)純化粗產物以得到47-2
。1
H NMR (400MHz, CDCl3
) δ = 7.15 (t,J
= 8.0 Hz, 1H), 6.79 - 6.59 (m, 3H), 3.99 - 3.89 (m, 1H), 3.72 (s, 3H), 2.74 - 2.53 (m, 2H), 1.63 (br s, 1H), 1.19 - 1.14 (m, 1H), 1.17 (d,J
= 6.0 Hz, 2H)。
在0℃下,向47-2
(1 g, 6.02 mmol, 1當量)及Et3
N (1.83 g, 18.05 mmol, 2.51 mL, 3當量)於DCM (20 mL)中之溶液中添加MsCl (1.03 g, 9.02 mmol, 698.48 µL, 1.5當量)。將混合物在25℃下攪拌1 h。TLC顯示反應已完成。將反應液傾倒至NaHCO3
飽和水溶液(50 mL)中,使用CH2
Cl2
(3 × 50 mL)萃取混合物。使用鹽水(50 mL)洗滌合併之有機層,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以得到47-3
,其未經進一步純化即直接用於下一步驟中。1
H NMR (400MHz, CDCl3
) δ = 7.22 - 7.12 (m, 1H), 6.82 - 6.62 (m, 3H), 4.89 - 4.73 (m, 1H), 3.73 (s, 3H), 2.95 - 2.74 (m, 2H), 2.49 (s, 3H), 1.40 (d,J
= 6.0 Hz, 3H)。
向47-3
(1.5 g, 6.14 mmol,1當量)於DMF (8 mL)中之溶液中添加NaN3
(798.30 mg, 12.28 mmol,2當量)。將混合物在80℃下加熱2 h。TLC顯示反應已完成。添加90 mL水,使用120 mL EtOAc/己烷(1:1)混合物萃取混合物。藉由無水Na2
SO4
乾燥萃取物並蒸發以得到粗製47-4
,其未經進一步純化即直接用於下一步驟中。
在N2
下,向47-4
(1.1 g, 5.75 mmol, 1當量)於EtOAc (100 mL)中之溶液中添加Pd/C (500 mg, 10%純度)。將懸浮液在真空下脫氣並使用H2
吹掃若干次。將混合物在H2
(15 psi)及25℃下攪拌1小時。TLC顯示反應已完成。過濾反應混合物且濃縮濾液。藉由使用於CH2
Cl2
中之MeOH (0%至20%)洗脫之急速層析(二氧化矽)純化粗產物以得到47-5
。1
H NMR (400MHz, CDCl3
) δ = 7.20 - 7.08 (m, 1H), 6.74 - 6.61 (m, 3H), 3.72 (s, 3H), 3.17 - 3.03 (m, 1H), 2.62 (dd,J
= 5.6, 13.6 Hz, 1H), 2.42 (dd,J
= 8.0, 13.2 Hz, 1H), 1.42 (s, 2H), 1.05 (d,J
= 6.4 Hz, 3H)。
將4A分子篩(3 g, 3.03 mmol, 1.00當量)添加至47-5
(500 mg, 3.03 mmol, 1當量)及4-嗎啉基苯甲醛(578.66 mg, 3.03 mmol, 1當量)於甲苯(25 mL)中之溶液中,將混合物在120℃下攪拌4 hr。LCMS顯示反應已完成。在冷卻至室溫之,過濾混合物且將濾液在真空下濃縮至較小體積以得到粗製N135-6A
,其未經進一步純化即直接用於下一步驟中。
將47-6A
(1 g, 2.95 mmol, 1當量)溶於TFA (46.20 g, 405.19 mmol, 30.00 mL, 137.13當量)中,將溶液在120℃下加熱20 hr以得到褐色溶液。LCMS顯示反應已完成。在減壓下濃縮反應混合物。藉由2N NaOH水溶液鹼化混合物並使用EtOAc (50 mL × 3)萃取。使用鹽水(50 mL)洗滌合併之有機層,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮。藉由使用於CH2
Cl2
中之MeOH
(0%至5%)洗脫之急速層析(二氧化矽)純化殘餘物。藉由製備型TLC (DCM/MeOH=10/1, Rf1=0.5, Rf2=0.45)純化粗產物以得到順式產物(作為主要產物)。
藉由進一步之製備型TLC純化獲得痕量反式產物(95%純度)且藉由2D NMR可明確證實結構。
在0℃下,向47-6B
(10 mg, 29.55 umol, 1當量)及Et3
N (8.97 mg, 88.64 umol, 12.34 µL, 3當量)於DCM (2 mL)中之溶液中添加2-氯乙醯氯(6.67 mg, 59.09 umol, 4.70 µL, 2當量)。將混合物在25℃下攪拌1 h以得到褐色溶液。LCMS顯示反應已完成。在減壓下濃縮反應液。藉由製備型TLC (PE/EA=2/1, Rf=0.4)純化殘餘物以得到47
。1
H NMR (400MHz, CDCl3
) δ = 7.24 (br d,J
= 8.0 Hz, 1H), 7.02 (d,J
= 8.4 Hz, 2H), 6.78 - 6.69 (m, 3H), 6.61 (s, 1H), 5.78 (br s, 1H), 4.84 (br s, 1H), 4.04 (br d,J
= 12.4 Hz, 1H), 3.81 (br d,J
= 12.4 Hz, 1H), 3.77 - 3.73 (m, 4H), 3.72 (s, 3H), 3.06 - 2.99 (m, 4H), 2.96 (br s, 1H), 2.44 (br d,J
= 16.0 Hz, 1H), 0.97 (br d,J
= 5.6 Hz, 3H)。
程序 16 :化合物 39 之合成 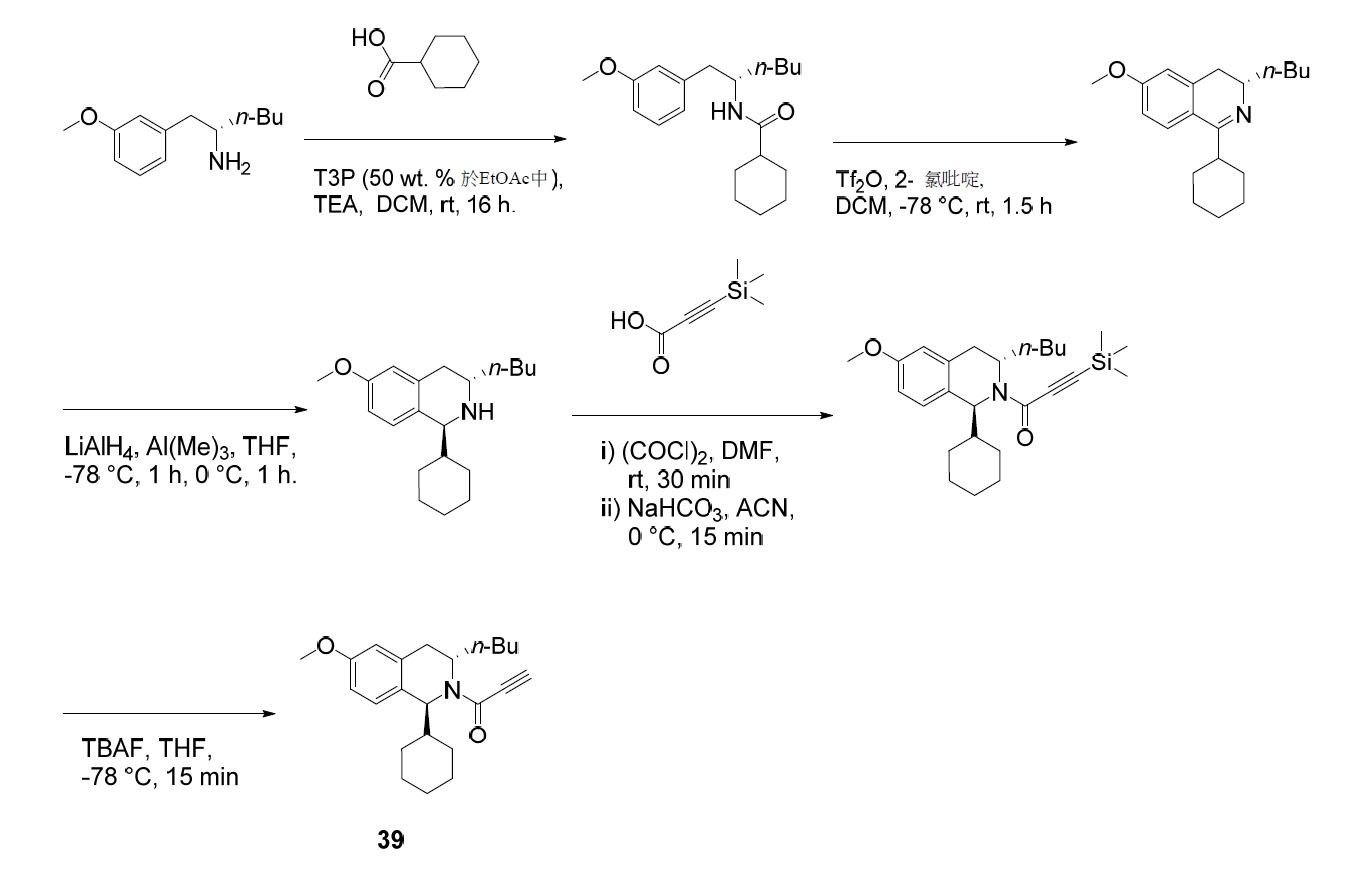 (S)-N-(1-(3-甲氧基苯基)己烷-2-基)環己烷甲醯胺:向環己烷甲酸(0.78 g, 6.11 mmol, 1.15當量)於DCM (15 mL)中之溶液中添加TEA (2.14 g, 21.24 mmol, 4當量),攪拌5分鐘,且然後在0℃下添加T3P (50 wt.%於EtOAc中) (2.53 g, 7.96 mmol, 1.5當量)並再攪拌5分鐘。向反應混合物中添加(S)-1-(3-甲氧基苯基)己烷-2-胺(1.1 g, 5.31 mmol, 1當量)且將反應混合物在室溫下攪拌16小時。藉由TLC (於己烷中之20%乙酸乙酯)監測反應進展。使用DCM (50 mL)及飽和碳酸氫鈉溶液(20 mL)稀釋反應混合物,分離有機層,使用鹽水溶液(20 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得(S)-N-(1-(3-甲氧基苯基)己烷-2-基)環己烷甲醯胺。LC-MS (m/z) = 318.3 [M+H]+
。1
H NMR (400 MHz, CDCl3
) δ ppm 0.84 - 0.92 (m, 3 H), 1.16 - 1.38 (m, 9 H), 1.49 - 1.51 (m, 1 H), 1.65 - 1.80 (m, 6 H), 1.92 - 2.03 (m, 1 H), 2.70 - 2.79 (m, 2 H), 3.78 (s, 3 H), 4.12 - 4.17 (m, 1 H), 5.11 (d,J
= 8.4 Hz, 1 H), 6.70 - 6.75 (m, 3 H), 7.18 (t,J
= 7.8 Hz, 1 H)。
(S)-3-丁基-1-環己基-6-甲氧基-3,4-二氫異喹啉:在-78℃下,經由注射器經1分鐘時段將三氟甲烷磺酸酐(1.45 mL, 8.64 mmol, 2.0當量)添加至(S)-N-(1-(3-甲氧基苯基)己烷-2-基)環己烷甲醯胺(1.37 g, 4.32 mmol, 1當量)及2-氯吡啶(0.81 mL, 8.64 mmol, 2.0當量)於二氯甲烷(13 mL)中之經攪拌混合物中。在5分鐘之後,將反應混合物置於冰-水浴中並升溫至0℃。在5分鐘之後,將所得溶液升溫至23℃。在1 h之後,引入氫氧化鈉水溶液(5 mL, 1N)以中和三氟甲烷磺酸鹽。添加二氯甲烷(50 mL)以稀釋混合物且分離各層。使用鹽水(10 mL)洗滌有機層,藉由無水硫酸鈉乾燥,並過濾。在減壓下去除揮發物以得到粗產物。所獲得粗產物未經進一步純化即用於下一步驟中。LC-MS (m/z) = 300.3 [M+H]+
。
(1S,3S)-3-丁基-1-環己基-6-甲氧基-1,2,3,4-四氫異喹啉:在-78℃及氮氣氛下,將(S)-3-丁基-1-環己基-6-甲氧基-3,4-二氫異喹啉(1.37 g, 4.58 mmol, 1當量)於無水THF (20 mL)中之溶液逐滴添加至1M氫化鋰鋁(於THF中,22.9 mL, 22.90 mmol, 5.0當量)及2M三甲基鋁溶液(於甲苯中,11.45 mL, 22.90 mmol, 5當量)於THF (20 mL)中之混合物中。將懸浮液在-78℃下攪拌1 h,並經30分鐘升溫至0℃。使用氯化鈉飽和水溶液(5 mL)淬滅反應混合物,隨後使用EtOAc (30 mL)稀釋且濾除所獲得沈澱物。最後,藉由無水硫酸鈉乾燥濾液,過濾並在減壓下蒸發。藉由急速層析(EtOAc/正己烷 = 20/80)純化殘餘物以得到(1S,3S)-3-丁基-1-環己基-6-甲氧基-1,2,3,4-四氫異喹啉(藉由nOe實驗可證實反式異構體)。使用金屬清除劑QuadraSil® AP處理所分離純產物(將化合物溶於THF (10 mL)中且添加QuadraSil® AP (1 g),將混合物攪拌0.5 h,過濾。再重複一次並濃縮)。LC-MS (m/z) = 302.3 [M+H]+ 1
H NMR (400 MHz, CDCl3
) δ ppm 0.90 - 0.93 (m, 3 H), 1.02 - 1.04 (m, 1 H), 1.17 (bs, 3 H), 1.25 - 1.29 (m, 1 H), 1.34 - 1.35 (s, 3 H), 1.36 - 1.42 (m, 3 H), 1.66 - 1.68 (m, 3 H), 1.68 - 1.77 (m, 3 H), 2.41 - 2.47 (m, 1 H), 2.80 - 2.85 (m, 1 H), 3.10 (bs, 1 H), 3.58 (d,J
= 6.4 Hz, 1 H), 3.76 (s, 3 H), 6.60 (s, 1 H), 6.66 (d,J
= 8.4 Hz, 1 H), 6.98 (d,J
= 8.4 Hz, 1 H)。
1-((1S,3S)-3-丁基-1-環己基-6-甲氧基-3,4-二氫異喹啉-2(1H)-基)-3-(三甲基矽烷基)丙-2-炔-1-酮:第一步驟:-在室溫下,向3-(三甲基矽烷基)丙炔酸(141 mg, 0.99 mmol, 1.5當量)於DMF (1.9 mg, 0.026 mmol, 0.04當量)中之溶液中添加草醯氯(0.13 g, 1.05 mmol, 1.6當量)並攪拌30分鐘。此後,在減壓下濃縮反應混合物以得到3-(三甲基矽烷基)丙炔醯氯。此醯氯未經任何進一步純化即用於下一步驟中。
第二步驟:-在0℃下,向(1S,3S)-3-丁基-1-環己基-6-甲氧基-1,2,3,4-四氫異喹啉(0.2 g, 0.66 mmol, 1.0當量)於乙腈(5.0 mL)中之溶液中添加碳酸氫鈉(0.42 g, 4.98 mmol, 7.5當量)。在攪拌5分鐘之後,向上述反應物質中添加3-(三甲基矽烷基)丙炔醯氯於乙腈(2.0 mL)中之溶液。將所得混合物在0℃下攪拌15 min,藉由TLC (於正己烷中之15%乙酸乙酯)監測反應進展。此後,使用EtOAc (30 mL)及水(5 mL)稀釋反應物質。分離有機層,使用鹽水溶液(10 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物。此粗產物未經任何進一步純化即用於下一步驟中。LC-MS (m/z) = 426.7 [M+H]+
1-((1S,3S)-3-丁基-1-環己基-6-甲氧基-3,4-二氫異喹啉-2(1H)-基)丙-2-炔-1-酮:在-78℃下,向1-((1S,3S)-3-丁基-1-環己基-6-甲氧基-3,4-二氫異喹啉-2(1H)-基)-3-(三甲基矽烷基)丙-2-炔-1-酮(0.29 g, 0.68 mmol, 1.0當量)於THF (5.0 mL)中之溶液中添加TBAF (於THF中之1M溶液) (0.75 mL, 0.75 mmol, 1.1當量)。將此反應混合物在-78℃下攪拌15分鐘。藉由TLC (於正己烷中之15%乙酸乙酯)監測反應進展。此後,使用NaHCO3
飽和水溶液(5 mL)淬滅反應混合物且使用乙酸乙酯(30 mL)萃取產物。藉由無水硫酸鈉乾燥有機層,過濾並在減壓下濃縮。藉由製備型TLC使用於正己烷中之15%乙酸乙酯作為洗脫劑純化所獲得粗產物以得到1-((1S,3S)-3-丁基-1-環己基-6-甲氧基-3,4-二氫異喹啉-2(1H)-基)丙-2-炔-1-酮。LC-MS (m/z) = 354.6 [M+H]+ 1
H NMR (400 MHz, DMSO-d6
) δ ppm 0.54 - 0.68 (m, 1 H), 0.76 - 0.77 (m, 3 H), 0.82 - 0.99 (m, 3 H), 1.08 - 1.14 (m, 6 H), 1.38 - 1.66 (m, 7 H), 2.74 - 2.78 (m, 1 H), 2.96 - 3.08 (m, 1 H), 3.72 (s, 3 H), 4.16 (bs, 0.5 H), 4.35 - 4.38 (m, 0.5 H), 4.50 - 4.52 (m, 1 H), 4.75 - 4.78 (m, 1 H), 6.74 (d,J
= 8.4 Hz, 1 H), 6.84 (d,J
= 12.8 Hz, 1 H), 7.01 - 7.08 (m, 1 H)。
(S)-N-(1-(3-甲氧基苯基)己烷-2-基)環己烷甲醯胺:向環己烷甲酸(0.78 g, 6.11 mmol, 1.15當量)於DCM (15 mL)中之溶液中添加TEA (2.14 g, 21.24 mmol, 4當量),攪拌5分鐘,且然後在0℃下添加T3P (50 wt.%於EtOAc中) (2.53 g, 7.96 mmol, 1.5當量)並再攪拌5分鐘。向反應混合物中添加(S)-1-(3-甲氧基苯基)己烷-2-胺(1.1 g, 5.31 mmol, 1當量)且將反應混合物在室溫下攪拌16小時。藉由TLC (於己烷中之20%乙酸乙酯)監測反應進展。使用DCM (50 mL)及飽和碳酸氫鈉溶液(20 mL)稀釋反應混合物,分離有機層,使用鹽水溶液(20 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得(S)-N-(1-(3-甲氧基苯基)己烷-2-基)環己烷甲醯胺。LC-MS (m/z) = 318.3 [M+H]+
。1
H NMR (400 MHz, CDCl3
) δ ppm 0.84 - 0.92 (m, 3 H), 1.16 - 1.38 (m, 9 H), 1.49 - 1.51 (m, 1 H), 1.65 - 1.80 (m, 6 H), 1.92 - 2.03 (m, 1 H), 2.70 - 2.79 (m, 2 H), 3.78 (s, 3 H), 4.12 - 4.17 (m, 1 H), 5.11 (d,J
= 8.4 Hz, 1 H), 6.70 - 6.75 (m, 3 H), 7.18 (t,J
= 7.8 Hz, 1 H)。
(S)-3-丁基-1-環己基-6-甲氧基-3,4-二氫異喹啉:在-78℃下,經由注射器經1分鐘時段將三氟甲烷磺酸酐(1.45 mL, 8.64 mmol, 2.0當量)添加至(S)-N-(1-(3-甲氧基苯基)己烷-2-基)環己烷甲醯胺(1.37 g, 4.32 mmol, 1當量)及2-氯吡啶(0.81 mL, 8.64 mmol, 2.0當量)於二氯甲烷(13 mL)中之經攪拌混合物中。在5分鐘之後,將反應混合物置於冰-水浴中並升溫至0℃。在5分鐘之後,將所得溶液升溫至23℃。在1 h之後,引入氫氧化鈉水溶液(5 mL, 1N)以中和三氟甲烷磺酸鹽。添加二氯甲烷(50 mL)以稀釋混合物且分離各層。使用鹽水(10 mL)洗滌有機層,藉由無水硫酸鈉乾燥,並過濾。在減壓下去除揮發物以得到粗產物。所獲得粗產物未經進一步純化即用於下一步驟中。LC-MS (m/z) = 300.3 [M+H]+
。
(1S,3S)-3-丁基-1-環己基-6-甲氧基-1,2,3,4-四氫異喹啉:在-78℃及氮氣氛下,將(S)-3-丁基-1-環己基-6-甲氧基-3,4-二氫異喹啉(1.37 g, 4.58 mmol, 1當量)於無水THF (20 mL)中之溶液逐滴添加至1M氫化鋰鋁(於THF中,22.9 mL, 22.90 mmol, 5.0當量)及2M三甲基鋁溶液(於甲苯中,11.45 mL, 22.90 mmol, 5當量)於THF (20 mL)中之混合物中。將懸浮液在-78℃下攪拌1 h,並經30分鐘升溫至0℃。使用氯化鈉飽和水溶液(5 mL)淬滅反應混合物,隨後使用EtOAc (30 mL)稀釋且濾除所獲得沈澱物。最後,藉由無水硫酸鈉乾燥濾液,過濾並在減壓下蒸發。藉由急速層析(EtOAc/正己烷 = 20/80)純化殘餘物以得到(1S,3S)-3-丁基-1-環己基-6-甲氧基-1,2,3,4-四氫異喹啉(藉由nOe實驗可證實反式異構體)。使用金屬清除劑QuadraSil® AP處理所分離純產物(將化合物溶於THF (10 mL)中且添加QuadraSil® AP (1 g),將混合物攪拌0.5 h,過濾。再重複一次並濃縮)。LC-MS (m/z) = 302.3 [M+H]+ 1
H NMR (400 MHz, CDCl3
) δ ppm 0.90 - 0.93 (m, 3 H), 1.02 - 1.04 (m, 1 H), 1.17 (bs, 3 H), 1.25 - 1.29 (m, 1 H), 1.34 - 1.35 (s, 3 H), 1.36 - 1.42 (m, 3 H), 1.66 - 1.68 (m, 3 H), 1.68 - 1.77 (m, 3 H), 2.41 - 2.47 (m, 1 H), 2.80 - 2.85 (m, 1 H), 3.10 (bs, 1 H), 3.58 (d,J
= 6.4 Hz, 1 H), 3.76 (s, 3 H), 6.60 (s, 1 H), 6.66 (d,J
= 8.4 Hz, 1 H), 6.98 (d,J
= 8.4 Hz, 1 H)。
1-((1S,3S)-3-丁基-1-環己基-6-甲氧基-3,4-二氫異喹啉-2(1H)-基)-3-(三甲基矽烷基)丙-2-炔-1-酮:第一步驟:-在室溫下,向3-(三甲基矽烷基)丙炔酸(141 mg, 0.99 mmol, 1.5當量)於DMF (1.9 mg, 0.026 mmol, 0.04當量)中之溶液中添加草醯氯(0.13 g, 1.05 mmol, 1.6當量)並攪拌30分鐘。此後,在減壓下濃縮反應混合物以得到3-(三甲基矽烷基)丙炔醯氯。此醯氯未經任何進一步純化即用於下一步驟中。
第二步驟:-在0℃下,向(1S,3S)-3-丁基-1-環己基-6-甲氧基-1,2,3,4-四氫異喹啉(0.2 g, 0.66 mmol, 1.0當量)於乙腈(5.0 mL)中之溶液中添加碳酸氫鈉(0.42 g, 4.98 mmol, 7.5當量)。在攪拌5分鐘之後,向上述反應物質中添加3-(三甲基矽烷基)丙炔醯氯於乙腈(2.0 mL)中之溶液。將所得混合物在0℃下攪拌15 min,藉由TLC (於正己烷中之15%乙酸乙酯)監測反應進展。此後,使用EtOAc (30 mL)及水(5 mL)稀釋反應物質。分離有機層,使用鹽水溶液(10 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物。此粗產物未經任何進一步純化即用於下一步驟中。LC-MS (m/z) = 426.7 [M+H]+
1-((1S,3S)-3-丁基-1-環己基-6-甲氧基-3,4-二氫異喹啉-2(1H)-基)丙-2-炔-1-酮:在-78℃下,向1-((1S,3S)-3-丁基-1-環己基-6-甲氧基-3,4-二氫異喹啉-2(1H)-基)-3-(三甲基矽烷基)丙-2-炔-1-酮(0.29 g, 0.68 mmol, 1.0當量)於THF (5.0 mL)中之溶液中添加TBAF (於THF中之1M溶液) (0.75 mL, 0.75 mmol, 1.1當量)。將此反應混合物在-78℃下攪拌15分鐘。藉由TLC (於正己烷中之15%乙酸乙酯)監測反應進展。此後,使用NaHCO3
飽和水溶液(5 mL)淬滅反應混合物且使用乙酸乙酯(30 mL)萃取產物。藉由無水硫酸鈉乾燥有機層,過濾並在減壓下濃縮。藉由製備型TLC使用於正己烷中之15%乙酸乙酯作為洗脫劑純化所獲得粗產物以得到1-((1S,3S)-3-丁基-1-環己基-6-甲氧基-3,4-二氫異喹啉-2(1H)-基)丙-2-炔-1-酮。LC-MS (m/z) = 354.6 [M+H]+ 1
H NMR (400 MHz, DMSO-d6
) δ ppm 0.54 - 0.68 (m, 1 H), 0.76 - 0.77 (m, 3 H), 0.82 - 0.99 (m, 3 H), 1.08 - 1.14 (m, 6 H), 1.38 - 1.66 (m, 7 H), 2.74 - 2.78 (m, 1 H), 2.96 - 3.08 (m, 1 H), 3.72 (s, 3 H), 4.16 (bs, 0.5 H), 4.35 - 4.38 (m, 0.5 H), 4.50 - 4.52 (m, 1 H), 4.75 - 4.78 (m, 1 H), 6.74 (d,J
= 8.4 Hz, 1 H), 6.84 (d,J
= 12.8 Hz, 1 H), 7.01 - 7.08 (m, 1 H)。
程序 17 :化合物 28 之合成 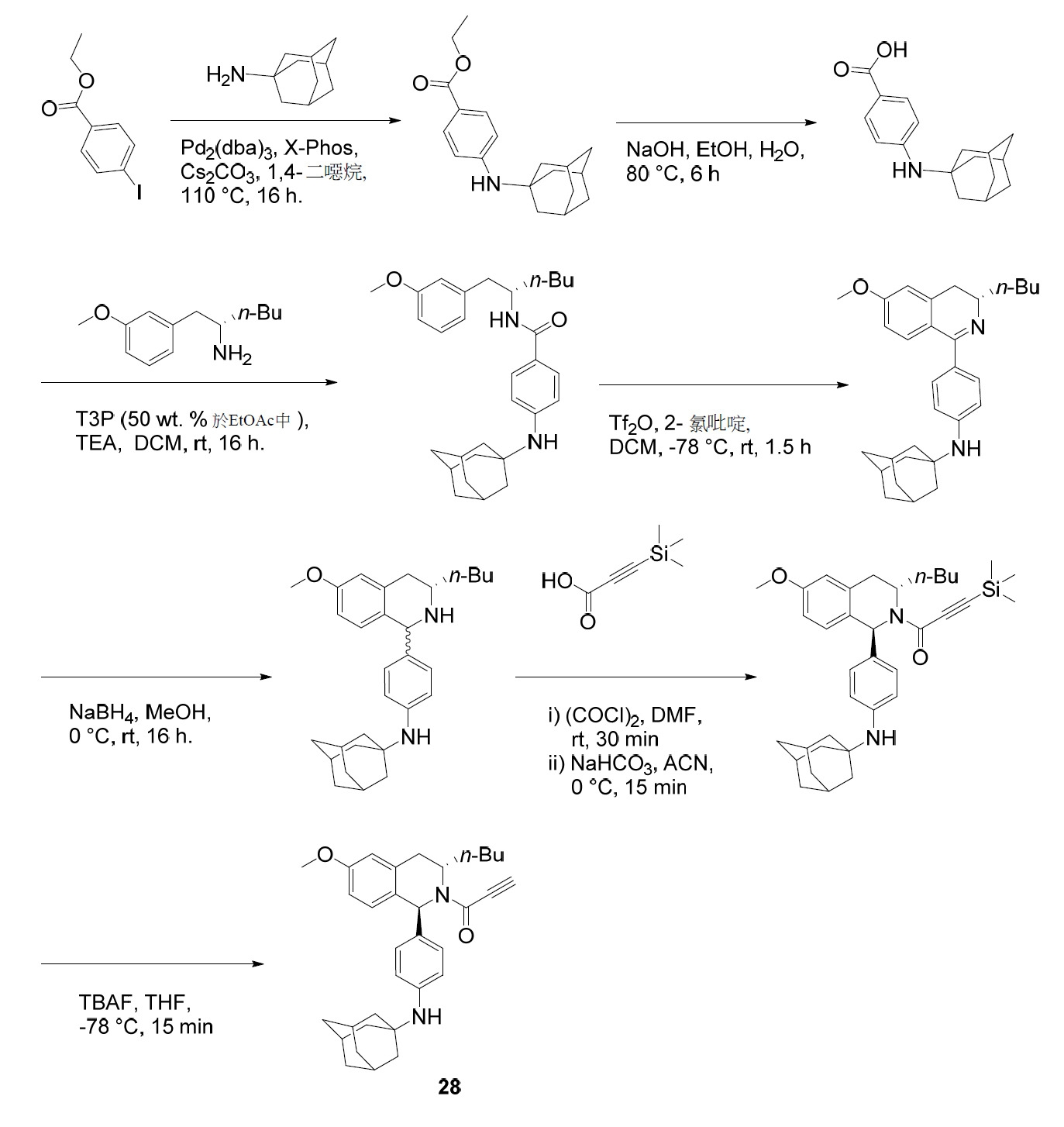 4-(((3s,5s,7s)-金剛烷-1-基)胺基)苯甲酸乙酯:在室溫下,向4-碘苯甲酸乙酯(6.0 g, 21.73 mmol, 1當量)及(3s,5s,7s)-金剛烷-1-胺(3.93 g, 26.08 mmol, 1.2當量)於1,4-二噁烷中之經攪拌溶液中添加XPhos (0.51 g, 1.08 mmol, 0.05當量)及Cs2
CO3
(14.26 g, 43.26 mmol, 2當量)。將反應混合物在氬下吹掃15 min,然後向反應液中添加Pd2
(dba)3
(0.516 g, 0.65 mmol, 0.03當量)並在110℃下攪拌16 h。藉由TLC (於己烷中之15%乙酸乙酯)監測反應進展。在反應完成之後,經由矽藻土床過濾反應混合物且使用乙酸乙酯(150 mL)洗滌矽藻土床。在減壓下濃縮濾液以獲得粗製物。藉由急速層析使用乙酸乙酯之己烷溶液作為洗脫劑純化所獲得粗產物以獲得4-(((3s,5s,7s)-金剛烷-1-基)胺基)苯甲酸乙酯。LC-MS (m/z) = 300.0 [M+H]+ 1
H NMR (400 MHz, CDCl3
) δ ppm 1.34 (t,J
= 7.0 Hz, 3 H), 1.67 - 1.74 (m, 6 H), 1.97 (s, 6 H), 2.14 (s, 3 H), 4.29 (q,J
= 6.9 Hz, 2 H), 6.67 (d,J
= 8.4 Hz, 2 H), 7.80 (d,J
= 8.4 Hz, 2 H)。
4-(((3s,5s,7s)-金剛烷-1-基)胺基)苯甲酸:向4-(((3s,5s,7s)-金剛烷-1-基)胺基)苯甲酸乙酯(1.6 g, 5.37 mmol, 1當量)於EtOH (29 mL)及水(11 mL)中之溶液中添加氫氧化鈉(0.43 g, 10.70 mmol, 2當量)並在80℃下攪拌6 h。TLC (於己烷中之15%乙酸乙酯)顯示反應已完成。在減壓下濃縮反應混合物且使用5%檸檬酸水溶液(pH=4)酸化所獲得粗製物。最後,使用EtOAc (75 mL)自水層萃取產物,藉由無水Na2
SO4
乾燥,過濾並在減壓下濃縮以得到產物4-(((3s,5s,7s)-金剛烷-1-基)胺基)苯甲酸。LC-MS (m/z) = 272.0 [M+H]+ 1
H NMR (400 MHz, DMSO-d6
) δ ppm 1.61 - 1.68 (m, 6 H), 1.91 (s, 6 H), 2.06 (s, 3 H), 5.87 (bs, 1 H), 6.72 (d,J
= 8.8 Hz, 2 H), 7.58 (d,J
= 8.8 Hz, 2 H), 12.00 (bs, 1 H)。
4-(((3R,5R,7R)-金剛烷-1-基)胺基)-N-((S)-1-(3-甲氧基苯基)己烷-2-基)苯甲醯胺:向4-(((3s,5s,7s)-金剛烷-1-基)胺基)苯甲酸(1.05 g, 3.88 mmol, 1.2當量)於DCM (20 mL)中之溶液中添加TEA (1.3 g, 12.92 mmol, 4當量),攪拌5 min且然後在0℃下添加T3P (50 wt.%於EtOAc中) (1.53 g, 4.84 mmol, 1.5當量)並再攪拌5 min。然後,向反應混合物中添加(S)-1-(3-甲氧基苯基)己烷-2-胺(0.67 g, 3.23 mmol, 1當量)且然後將反應混合物在室溫下攪拌16 h。藉由TLC (於己烷中之30%乙酸乙酯)監測反應進展。使用DCM (50 mL)及飽和碳酸氫鈉溶液(20 mL)稀釋反應混合物,分離有機層,使用鹽水溶液(20 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物。藉由急速層析使用乙酸乙酯之己烷溶液作為洗脫劑純化所獲得粗產物以得到4-(((3R,5R,7R)-金剛烷-1-基)胺基)-N-((S)-1-(3-甲氧基苯基)己烷-2-基)苯甲醯胺。LC-MS (m/z) = 461.0 [M+H]+ 1
H NMR (400 MHz, DMSO-d6
) δ ppm 0.80 (bs, 3 H), 1.22 (bs, 4 H), 1.45 (bs, 2 H), 1.64 (s, 6 H), 1.89 (s, 6 H), 2.05 (s, 3 H), 2.65 - 2.76 (m, 2 H), 3.16 (d,J
= 4.0 Hz, 1 H), 3.66 (s, 3 H), 4.07 (bs, 1 H), 5.49 (s, 1 H), 6.68 - 6.75 (m, 4 H), 7.11 - 7.12 (m, 1 H), 7.50 (d,J
= 7.2 Hz, 2 H), 7.57 - 7.64 (m, 1 H)。
(3R,5R,7R)-N-(4-((S)-3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基)苯基)金剛烷-1-胺:在-78℃下,經由注射器,以1 min時間將三氟甲烷磺酸酐(0.547 mL, 3.26 mmol, 3.0當量)添加至4-(((3R,5R,7R)-金剛烷-1-基)胺基)-N-((S)-1-(3-甲氧基苯基)己烷-2-基)苯甲醯胺(0.5 g, 1.08 mmol, 1當量)及2-氯吡啶(0.3 mL, 3.26 mmol, 3.0當量)於二氯甲烷(3.6 mL)中之經攪拌混合物中。在5 min之後,將反應混合物置於冰-水浴中並升溫至0℃。在5 min之後,將所得溶液升溫至23℃。在1 h之後,引入氫氧化鈉水溶液(5 mL, 1N)以中和三氟甲烷磺酸鹽。添加二氯甲烷(50 mL)以稀釋混合物且分離各層。使用鹽水(7 mL)洗滌有機層,藉由無水硫酸鈉乾燥,並過濾。在減壓下去除揮發物以得到粗產物。藉由急速層析使用乙酸乙酯之己烷溶液作為洗脫劑純化所獲得粗產物以得到期望產物((3R,5R,7R)-N-(4-((S)-3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基)苯基)金剛烷-1-胺。LC-MS (m/z) = 443.3 [M+H]+
(3R,5R,7R)-N-(4-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)苯基)金剛烷-1-胺:在0℃下,向(3R,5R,7R)-N-(4-((S)-3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基)苯基)金剛烷-1-胺(0.5 g, 1.13 mmol, 1當量)於甲醇(9 mL)中之溶液中添加硼氫化鈉(0.128 g, 33.93 mmol, 3當量)。將懸浮液在室溫下攪拌16 h。此後,濃縮反應混合物且使用EtOAc (30 mL)及水(10 mL)稀釋所獲得粗製物。分離有機層,使用鹽水溶液(10 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物。藉由急速層析使用乙酸乙酯之己烷溶液作為洗脫劑純化所獲得粗產物以得到(3R,5R,7R)-N-(4-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)苯基)金剛烷-1-胺。使用金屬清除劑QuadraSil® AP處理所分離純產物(將化合物溶於THF (5 mL)中且添加QuadraSil® AP (50 mg),將混合物攪拌0.5 h,過濾。再重複一次並濃縮)。LC-MS (m/z) = 445.3 [M+H]+ 1
H NMR (400 MHz, CDCl3
) δ ppm 0.84 - 088 (m, 3 H), 1.22 - 1.30 (m, 4 H), 1.42 - 1.43 (m, 2 H), 1.60 - 1.70 (m, 6 H), 1.85 (s, 6 H), 2.09 (bs, 3 H), 2.53 - 2.60 (m, 1 H), 2.82 - 2.86 (m, 1 H), 2.87 - 2.97 (m, 1 H), 3.78 (s, 3 H), 5.13 (s, 1 H), 6.66 - 6.70 (m, 4 H), 6.84 - 6.90 (m, 3 H)。
1-((1S,3S)-1-(4-(((3R,5R,7R)-金剛烷-1-基)胺基)苯基)-3-丁基-6-甲氧基-3,4-二氫異喹啉-2(1H)-基)-3-(三甲基矽烷基)丙-2-炔-1-酮:第一步驟:-在室溫下,向3-(三甲基矽烷基)丙炔酸(11.8 mg, 0.083 mmol, 1.0當量)於DMF (0.24 mg, 0.003 mmol, 0.04當量)中之溶液中添加草醯氯(11.5 mg, 0.091 mmol, 1.1當量)並攪拌30分鐘。此後,在減壓下濃縮反應混合物以得到3-(三甲基矽烷基)丙炔醯氯。此醯氯未經任何進一步純化即用於下一步驟中。
第二步驟:-在0℃下,向(3R,5R,7R)-N-(4-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)苯基)金剛烷-1-胺(37 mg, 0.083 mmol, 1.0當量)於乙腈(1.0 mL)中之溶液中添加碳酸氫鈉(52.5 mg, 0.62 mmol, 7.5當量)。在攪拌5分鐘之後,向上述反應物質中添加3-(三甲基矽烷基)丙炔醯氯於乙腈(1.0 mL)中之溶液。將所得混合物在0℃下攪拌15 min,藉由TLC (於正己烷中之70%乙酸乙酯)監測反應進展。此後,使用EtOAc (30 mL)及水(10 mL)稀釋反應物質。分離有機層,使用鹽水溶液(10 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物。此粗產物未經任何進一步純化即用於下一步驟中。LC-MS (m/z) = 569.4 [M+H]+
1-((1S,3S)-1-(4-(((3R,5R,7R)-金剛烷-1-基)胺基)苯基)-3-丁基-6-甲氧基-3,4-二氫異喹啉-2(1H)-基)丙-2-炔-1-酮:在-78℃下,向1-((1S,3S)-1-(4-(((3R,5R,7R)-金剛烷-1-基)胺基)苯基)-3-丁基-6-甲氧基-3,4-二氫異喹啉-2(1H)-基)-3-(三甲基矽烷基)丙-2-炔-1-酮(48 mg, 0.084 mmol, 1.0當量)於THF (1.5 mL)中之溶液中添加TBAF (於THF中之1M溶液) (0.092 mL, 0.092 mmol, 1.1當量)。將此反應混合物在-78℃下攪拌15分鐘。藉由TLC (於正己烷中之25%乙酸乙酯)監測反應進展。此後,使用NaHCO3
飽和水溶液(5 mL)淬滅反應混合物且使用乙酸乙酯(25 mL)萃取產物。藉由無水硫酸鈉乾燥有機層,過濾並在減壓下濃縮。藉由製備型TLC使用於正己烷中之25%乙酸乙酯作為洗脫劑純化所獲得粗產物以得到1-((1S,3S)-1-(4-(((3R,5R,7R)-金剛烷-1-基)胺基)苯基)-3-丁基-6-甲氧基-3,4-二氫異喹啉-2(1H)-基)丙-2-炔-1-酮。LC-MS (m/z) = 497.3 ([M+H]+ 1
H NMR (400 MHz, DMSO-d6
) δ ppm 0.79 - 0.80 (m, 3 H), 1.18 - 1.24 (m, 6 H), 1.51 (bs, 1 H), 1.61 (s, 5 H), 1.79 (s, 6 H), 2.01 (s, 3 H), 2.71 - 2.83 (m, 1 H), 3.00 - 3.09 (m, 2 H), 3.72 - 3.73 (m, 3 H), 4.15 (bs, 0.3 H), 4.40 (bs, 1 H), 4.61 (bs, 0.7 H), 5.95 (s, 0.5 H), 6.16 (s, 0.5 H), 6.59 - 6.66 (m, 2 H), 6.76 - 6.85 (m, 4 H), 7.29 (d,J
= 8.4 Hz, 0.5 H), 7.40 (d,J
= 7.6 Hz, 0.5 H)。
4-(((3s,5s,7s)-金剛烷-1-基)胺基)苯甲酸乙酯:在室溫下,向4-碘苯甲酸乙酯(6.0 g, 21.73 mmol, 1當量)及(3s,5s,7s)-金剛烷-1-胺(3.93 g, 26.08 mmol, 1.2當量)於1,4-二噁烷中之經攪拌溶液中添加XPhos (0.51 g, 1.08 mmol, 0.05當量)及Cs2
CO3
(14.26 g, 43.26 mmol, 2當量)。將反應混合物在氬下吹掃15 min,然後向反應液中添加Pd2
(dba)3
(0.516 g, 0.65 mmol, 0.03當量)並在110℃下攪拌16 h。藉由TLC (於己烷中之15%乙酸乙酯)監測反應進展。在反應完成之後,經由矽藻土床過濾反應混合物且使用乙酸乙酯(150 mL)洗滌矽藻土床。在減壓下濃縮濾液以獲得粗製物。藉由急速層析使用乙酸乙酯之己烷溶液作為洗脫劑純化所獲得粗產物以獲得4-(((3s,5s,7s)-金剛烷-1-基)胺基)苯甲酸乙酯。LC-MS (m/z) = 300.0 [M+H]+ 1
H NMR (400 MHz, CDCl3
) δ ppm 1.34 (t,J
= 7.0 Hz, 3 H), 1.67 - 1.74 (m, 6 H), 1.97 (s, 6 H), 2.14 (s, 3 H), 4.29 (q,J
= 6.9 Hz, 2 H), 6.67 (d,J
= 8.4 Hz, 2 H), 7.80 (d,J
= 8.4 Hz, 2 H)。
4-(((3s,5s,7s)-金剛烷-1-基)胺基)苯甲酸:向4-(((3s,5s,7s)-金剛烷-1-基)胺基)苯甲酸乙酯(1.6 g, 5.37 mmol, 1當量)於EtOH (29 mL)及水(11 mL)中之溶液中添加氫氧化鈉(0.43 g, 10.70 mmol, 2當量)並在80℃下攪拌6 h。TLC (於己烷中之15%乙酸乙酯)顯示反應已完成。在減壓下濃縮反應混合物且使用5%檸檬酸水溶液(pH=4)酸化所獲得粗製物。最後,使用EtOAc (75 mL)自水層萃取產物,藉由無水Na2
SO4
乾燥,過濾並在減壓下濃縮以得到產物4-(((3s,5s,7s)-金剛烷-1-基)胺基)苯甲酸。LC-MS (m/z) = 272.0 [M+H]+ 1
H NMR (400 MHz, DMSO-d6
) δ ppm 1.61 - 1.68 (m, 6 H), 1.91 (s, 6 H), 2.06 (s, 3 H), 5.87 (bs, 1 H), 6.72 (d,J
= 8.8 Hz, 2 H), 7.58 (d,J
= 8.8 Hz, 2 H), 12.00 (bs, 1 H)。
4-(((3R,5R,7R)-金剛烷-1-基)胺基)-N-((S)-1-(3-甲氧基苯基)己烷-2-基)苯甲醯胺:向4-(((3s,5s,7s)-金剛烷-1-基)胺基)苯甲酸(1.05 g, 3.88 mmol, 1.2當量)於DCM (20 mL)中之溶液中添加TEA (1.3 g, 12.92 mmol, 4當量),攪拌5 min且然後在0℃下添加T3P (50 wt.%於EtOAc中) (1.53 g, 4.84 mmol, 1.5當量)並再攪拌5 min。然後,向反應混合物中添加(S)-1-(3-甲氧基苯基)己烷-2-胺(0.67 g, 3.23 mmol, 1當量)且然後將反應混合物在室溫下攪拌16 h。藉由TLC (於己烷中之30%乙酸乙酯)監測反應進展。使用DCM (50 mL)及飽和碳酸氫鈉溶液(20 mL)稀釋反應混合物,分離有機層,使用鹽水溶液(20 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物。藉由急速層析使用乙酸乙酯之己烷溶液作為洗脫劑純化所獲得粗產物以得到4-(((3R,5R,7R)-金剛烷-1-基)胺基)-N-((S)-1-(3-甲氧基苯基)己烷-2-基)苯甲醯胺。LC-MS (m/z) = 461.0 [M+H]+ 1
H NMR (400 MHz, DMSO-d6
) δ ppm 0.80 (bs, 3 H), 1.22 (bs, 4 H), 1.45 (bs, 2 H), 1.64 (s, 6 H), 1.89 (s, 6 H), 2.05 (s, 3 H), 2.65 - 2.76 (m, 2 H), 3.16 (d,J
= 4.0 Hz, 1 H), 3.66 (s, 3 H), 4.07 (bs, 1 H), 5.49 (s, 1 H), 6.68 - 6.75 (m, 4 H), 7.11 - 7.12 (m, 1 H), 7.50 (d,J
= 7.2 Hz, 2 H), 7.57 - 7.64 (m, 1 H)。
(3R,5R,7R)-N-(4-((S)-3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基)苯基)金剛烷-1-胺:在-78℃下,經由注射器,以1 min時間將三氟甲烷磺酸酐(0.547 mL, 3.26 mmol, 3.0當量)添加至4-(((3R,5R,7R)-金剛烷-1-基)胺基)-N-((S)-1-(3-甲氧基苯基)己烷-2-基)苯甲醯胺(0.5 g, 1.08 mmol, 1當量)及2-氯吡啶(0.3 mL, 3.26 mmol, 3.0當量)於二氯甲烷(3.6 mL)中之經攪拌混合物中。在5 min之後,將反應混合物置於冰-水浴中並升溫至0℃。在5 min之後,將所得溶液升溫至23℃。在1 h之後,引入氫氧化鈉水溶液(5 mL, 1N)以中和三氟甲烷磺酸鹽。添加二氯甲烷(50 mL)以稀釋混合物且分離各層。使用鹽水(7 mL)洗滌有機層,藉由無水硫酸鈉乾燥,並過濾。在減壓下去除揮發物以得到粗產物。藉由急速層析使用乙酸乙酯之己烷溶液作為洗脫劑純化所獲得粗產物以得到期望產物((3R,5R,7R)-N-(4-((S)-3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基)苯基)金剛烷-1-胺。LC-MS (m/z) = 443.3 [M+H]+
(3R,5R,7R)-N-(4-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)苯基)金剛烷-1-胺:在0℃下,向(3R,5R,7R)-N-(4-((S)-3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基)苯基)金剛烷-1-胺(0.5 g, 1.13 mmol, 1當量)於甲醇(9 mL)中之溶液中添加硼氫化鈉(0.128 g, 33.93 mmol, 3當量)。將懸浮液在室溫下攪拌16 h。此後,濃縮反應混合物且使用EtOAc (30 mL)及水(10 mL)稀釋所獲得粗製物。分離有機層,使用鹽水溶液(10 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物。藉由急速層析使用乙酸乙酯之己烷溶液作為洗脫劑純化所獲得粗產物以得到(3R,5R,7R)-N-(4-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)苯基)金剛烷-1-胺。使用金屬清除劑QuadraSil® AP處理所分離純產物(將化合物溶於THF (5 mL)中且添加QuadraSil® AP (50 mg),將混合物攪拌0.5 h,過濾。再重複一次並濃縮)。LC-MS (m/z) = 445.3 [M+H]+ 1
H NMR (400 MHz, CDCl3
) δ ppm 0.84 - 088 (m, 3 H), 1.22 - 1.30 (m, 4 H), 1.42 - 1.43 (m, 2 H), 1.60 - 1.70 (m, 6 H), 1.85 (s, 6 H), 2.09 (bs, 3 H), 2.53 - 2.60 (m, 1 H), 2.82 - 2.86 (m, 1 H), 2.87 - 2.97 (m, 1 H), 3.78 (s, 3 H), 5.13 (s, 1 H), 6.66 - 6.70 (m, 4 H), 6.84 - 6.90 (m, 3 H)。
1-((1S,3S)-1-(4-(((3R,5R,7R)-金剛烷-1-基)胺基)苯基)-3-丁基-6-甲氧基-3,4-二氫異喹啉-2(1H)-基)-3-(三甲基矽烷基)丙-2-炔-1-酮:第一步驟:-在室溫下,向3-(三甲基矽烷基)丙炔酸(11.8 mg, 0.083 mmol, 1.0當量)於DMF (0.24 mg, 0.003 mmol, 0.04當量)中之溶液中添加草醯氯(11.5 mg, 0.091 mmol, 1.1當量)並攪拌30分鐘。此後,在減壓下濃縮反應混合物以得到3-(三甲基矽烷基)丙炔醯氯。此醯氯未經任何進一步純化即用於下一步驟中。
第二步驟:-在0℃下,向(3R,5R,7R)-N-(4-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)苯基)金剛烷-1-胺(37 mg, 0.083 mmol, 1.0當量)於乙腈(1.0 mL)中之溶液中添加碳酸氫鈉(52.5 mg, 0.62 mmol, 7.5當量)。在攪拌5分鐘之後,向上述反應物質中添加3-(三甲基矽烷基)丙炔醯氯於乙腈(1.0 mL)中之溶液。將所得混合物在0℃下攪拌15 min,藉由TLC (於正己烷中之70%乙酸乙酯)監測反應進展。此後,使用EtOAc (30 mL)及水(10 mL)稀釋反應物質。分離有機層,使用鹽水溶液(10 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物。此粗產物未經任何進一步純化即用於下一步驟中。LC-MS (m/z) = 569.4 [M+H]+
1-((1S,3S)-1-(4-(((3R,5R,7R)-金剛烷-1-基)胺基)苯基)-3-丁基-6-甲氧基-3,4-二氫異喹啉-2(1H)-基)丙-2-炔-1-酮:在-78℃下,向1-((1S,3S)-1-(4-(((3R,5R,7R)-金剛烷-1-基)胺基)苯基)-3-丁基-6-甲氧基-3,4-二氫異喹啉-2(1H)-基)-3-(三甲基矽烷基)丙-2-炔-1-酮(48 mg, 0.084 mmol, 1.0當量)於THF (1.5 mL)中之溶液中添加TBAF (於THF中之1M溶液) (0.092 mL, 0.092 mmol, 1.1當量)。將此反應混合物在-78℃下攪拌15分鐘。藉由TLC (於正己烷中之25%乙酸乙酯)監測反應進展。此後,使用NaHCO3
飽和水溶液(5 mL)淬滅反應混合物且使用乙酸乙酯(25 mL)萃取產物。藉由無水硫酸鈉乾燥有機層,過濾並在減壓下濃縮。藉由製備型TLC使用於正己烷中之25%乙酸乙酯作為洗脫劑純化所獲得粗產物以得到1-((1S,3S)-1-(4-(((3R,5R,7R)-金剛烷-1-基)胺基)苯基)-3-丁基-6-甲氧基-3,4-二氫異喹啉-2(1H)-基)丙-2-炔-1-酮。LC-MS (m/z) = 497.3 ([M+H]+ 1
H NMR (400 MHz, DMSO-d6
) δ ppm 0.79 - 0.80 (m, 3 H), 1.18 - 1.24 (m, 6 H), 1.51 (bs, 1 H), 1.61 (s, 5 H), 1.79 (s, 6 H), 2.01 (s, 3 H), 2.71 - 2.83 (m, 1 H), 3.00 - 3.09 (m, 2 H), 3.72 - 3.73 (m, 3 H), 4.15 (bs, 0.3 H), 4.40 (bs, 1 H), 4.61 (bs, 0.7 H), 5.95 (s, 0.5 H), 6.16 (s, 0.5 H), 6.59 - 6.66 (m, 2 H), 6.76 - 6.85 (m, 4 H), 7.29 (d,J
= 8.4 Hz, 0.5 H), 7.40 (d,J
= 7.6 Hz, 0.5 H)。
程序 18 :化合物 55 之合成 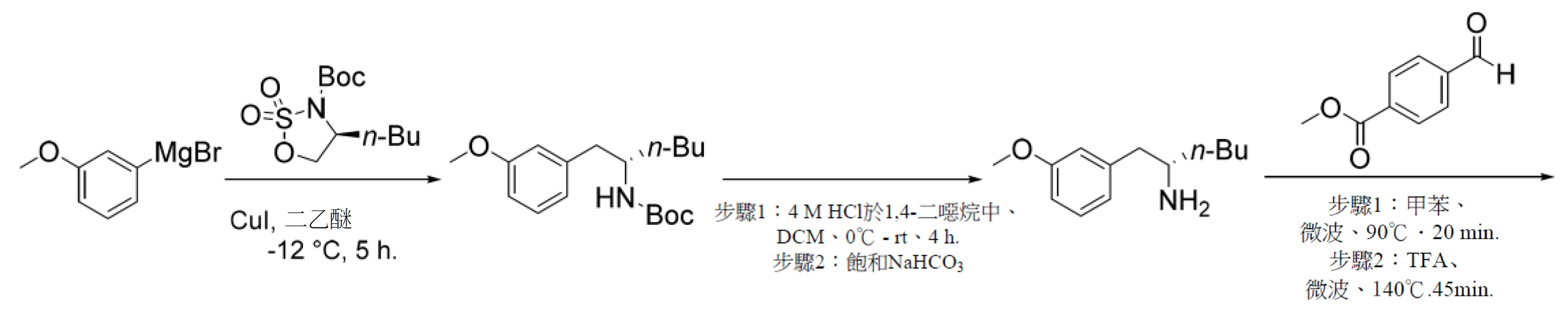
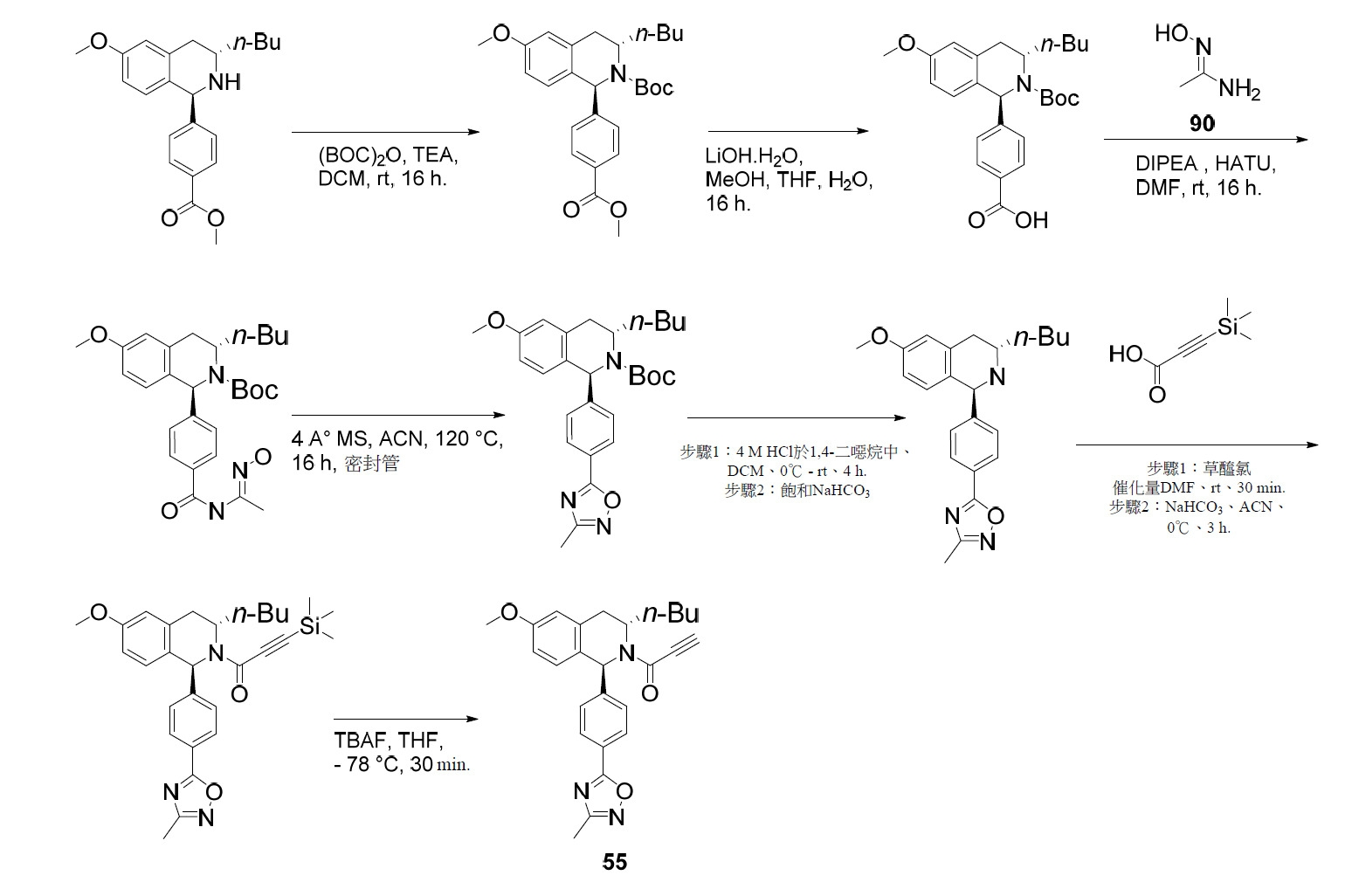 (S)-(1-(3-甲氧基苯基)己烷-2-基)胺基甲酸第三丁基酯:在-12℃下,以10 min時間向碘化銅(0.510 g, 2.68 mmol, 0.1當量)於二乙醚(50 mL)中之溶液中逐滴添加(3-甲氧基苯基)溴化鎂(1M於THF中) (53 mL, 53.76 mmol, 2當量)。將反應混合物在-12℃下攪拌30 min。此後,將(S)-4-丁基-1,2,3-氧雜噻唑啶-3-甲酸第三丁基酯2,2-二氧化物(7.5 g, 26.88 mmol, 1當量)於二乙醚(15 mL)中之溶液在-12℃下逐滴添加至反應物質中。將所得混合物在-12℃下攪拌4 h。最後,使用10%檸檬酸水溶液(15 mL)在-12℃下終止反應並使用乙酸乙酯(100 mL)稀釋。分離有機層並使用鹽水(50 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並濃縮以得到粗產物,藉由急速管柱層析使用於正己烷中之15%乙酸乙酯作為洗脫劑純化以獲得(S)-(1-(3-甲氧基苯基)己烷-2-基)胺基甲酸第三丁基酯。LC-MS (m/z): 252.0 [M+H]+ 1
H NMR (400 MHz, CDCl3
): δ ppm 0.86 - 0.87 (m, 3 H), 1.23 - 1.35 (m, 6 H), 1.40 (s, 9 H), 2.73 (bs, 2 H), 3.78 (s, 3 H), 4.29 (bs, 1 H), 6.71 - 6.76 (m, 3 H), 7.19 (t,J
= 7.8 Hz, 1 H)。未觀察到醯胺NH。
(S)-1-(3-甲氧基苯基)己烷-2-胺:在0℃下,向(S)-(1-(3-甲氧基苯基)己烷-2-基)胺基甲酸第三丁基酯(10 g, 32.57 mmol, 1當量)於二氯甲烷(50 mL)中之溶液中添加於1,4-二噁烷中之4M HCl (20 mL, 64.10 mmol, 2當量)。將混合物在室溫下攪拌16 h。藉由TLC監測反應進展,在反應完成之後,在減壓下濃縮反應混合物。使用冰冷水(10 mL)溶解所獲得粗製物且藉由NaHCO3
飽和水溶液鹼化。使用EtOAc (100 mL)萃取化合物。使用鹽水(10 mL)洗滌有機層,藉由無水硫酸鈉乾燥,過濾並濃縮以獲得(S)-1-(3-甲氧基苯基)己烷-2-胺。LC-MS (m/z): 208.1 [M+H]+
。1
H NMR (400 MHz, CDCl3
): δ ppm 0.82 - 0.90 (m, 3 H), 1.23 - 1.42 (m, 4 H), 1.57 - 1.62 (m, 2 H), 2.69 - 2.91 (m, 2 H), 3.25 - 3.58 (m, 1 H), 3.80 (s, 3 H), 6.69 - 6.78 (m, 3 H), 7.21 (t,J
= 8.0 Hz, 1 H)。未觀察到NH2
質子。
4-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)苯甲酸甲酯:將(S)-1-(3-甲氧基苯基)己烷-2-胺(0.7 g, 3.37 mmol, 1當量)及4-甲醯基苯甲酸甲酯(0.664 g, 4.05 mmol, 1當量)於甲苯(4 mL)中之溶液在90℃下於微波中輻照20 min。此後,在減壓下濃縮揮發性部分並在TFA (4 mL)中原樣用於環化步驟中,且在140℃下於微波中輻照45 min。此後,在減壓下濃縮揮發性部分且使用NaHCO3
飽和水溶液(10 mL)及EtOAc (40 mL)稀釋所獲得粗製物。分離有機層,使用鹽水(10 mL)洗滌,藉由無水MgSO4
乾燥,過濾並在減壓下濃縮以獲得粗產物。藉由矽膠急速管柱層析(正己烷/EtOAc)純化所獲得粗產物以得到4-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)苯甲酸甲酯。LC-MS (m/z): 208.1 [M+H]+
。1
H NMR (400 MHz, CDCl3
): δ ppm 0.84 - 0.86 (m, 3 H), 1.23 - 1.24 (m, 6 H), 2.55 - 2.59 (m, 1 H), 2.85 - 2.88 (m, 2 H), 3.75 (s, 3 H), 3.80 (s, 3 H), 5.29 (s, 3 H), 6.67 - 6.69 (m, 2 H), 6.70 - 6.80 (m, 1 H), 7.21 - 7.26 (s, 2 H), 7.94 - 8.0 (m, 2 H)。
(1S,3S)-6-甲氧基-1-(4-(甲氧基羰基)苯基)-3-甲基-3,4-二氫異喹啉-2(1H)-甲酸第三丁基酯:在室溫下,向化合物4-((1S,3S)-6-甲氧基-3-甲基-1,2,3,4-四氫異喹啉-1-基)苯甲酸甲酯(0.35 g, 1.12 mmol, 1當量)於DCM (10 mL)中之溶液中添加三乙胺(0.45 g, 4.49 mmol, 4當量)及二碳酸二-第三丁基酯(0.715 g, 2.24 mmol, 2當量)且將混合物攪拌16 h。TLC (於己烷中之50% EtOAc)顯示反應已完成。在減壓下濃縮反應混合物,且使用EtOAc (50 mL)稀釋粗製物,使用水(2 × 50 mL)洗滌。藉由無水Na2
SO4
乾燥有機層,在減壓下濃縮成粗產物(1S,3S)-6-甲氧基-1-(4-(甲氧基羰基)苯基)-3-甲基-3,4-二氫異喹啉-2(1H)-甲酸第三丁基酯。LC-MS (m/z): 356.0 [M-t
Bu+H]+
。
4-((1S,3S)-2-(第三丁氧基羰基)-6-甲氧基-3-甲基-1,2,3,4-四氫異喹啉-1-基)苯甲酸:向化合物(1S,3S)-6-甲氧基-1-(4-(甲氧基羰基)苯基)-3-甲基-3,4-二氫異喹啉-2(1H)-甲酸第三丁基酯(0.650 g, 1.57 mmol, 1當量)於THF: MeOH: H2
O (9 mL:1 mL:1 mL)之混合物中之溶液中添加氫氧化鋰(0.331 g, 7.89 mmol, 5當量)並在室溫下攪拌16 h。TLC (於己烷中之50% EtOAc)顯示反應已完成。在減壓下濃縮反應混合物,且使用5%檸檬酸溶液(pH=9)酸化粗製物。使用EtOAc (50 mL)稀釋反應混合物且分離有機層,並藉由無水Na2
SO4
乾燥,在減壓下濃縮以得到粗產物4-((1S,3S)-2-(第三丁氧基羰基)-6-甲氧基-3-甲基-1,2,3,4-四氫異喹啉-1-基)苯甲酸。LC-MS (m/z): 396.0 [M+H]。
(1S,3S)-3-丁基-1-(4-(((E)-1-(羥基亞胺基)乙基)胺甲醯基)苯基)-6-甲氧基-3,4-二氫異喹啉-2(1H)-甲酸第三丁基酯:在室溫下,向4-((1S,3S)-2-(第三丁氧基羰基)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)苯甲酸(0.2 g, 0.45 mmol, 1當量)於DMF (10 mL)中之溶液中添加DIPEA (0.15 mL, 0.91 mmol, 2當量)及HATU (0.207 g, 5.46 mmol, 1.2當量),攪拌15 min且然後添加(E)-N'-羥基乙脒(0.043 g, 0.591 mmol, 1.3當量),且將反應混合物在室溫下攪拌3 h。藉由TLC (於己烷中之30% EtOAc)監測反應進展。在反應完成之後,將反應混合物傾倒至碎冰中且然後使用EtOAc (2 × 50 mL)萃取。藉由無水硫酸鈉乾燥合併之有機層,過濾並在減壓下濃縮以獲得(1S,3S)-3-丁基-1-(4-(((E)-1-(羥基亞胺基)乙基)胺甲醯基)苯基)-6-甲氧基-3,4-二氫異喹啉-2(1H)-甲酸第三丁基酯。LC-MS (m/z) = 496.0 [M+H]+
。
(1S,3S)-3-丁基-6-甲氧基-1-(4-(3-甲基-1,2,4-噁二唑-5-基)苯基)-3,4-二氫異喹啉-2(1H)-甲酸第三丁基酯:向(1S,3S)-3-丁基-1-(4-(((E)-1-(羥基亞胺基)乙基)胺甲醯基)苯基)-6-甲氧基-3,4-二氫異喹啉-2(1H)-甲酸第三丁基酯(0.3 g, 0.606 mmol, 1當量)於ACN (10 mL)中之溶液中添加4 Å MS (0.1 g)且將反應混合物在120℃下於密封管中攪拌3 h。藉由TLC (於己烷中之50% EtOAc)監測反應進展。在反應完成之後,經由燒結漏斗過濾反應混合物且在減壓下濃縮所獲得濾液以獲得粗製物。藉由矽膠管柱層析使用於己烷中之25 - 30% EtOAc作為洗脫劑純化以提供(1S,3S)-3-丁基-6-甲氧基-1-(4-(3-甲基-1,2,4-噁二唑-5-基)苯基)-3,4-二氫異喹啉-2(1H)-甲酸第三丁基酯。LC-MS (m/z) = 478.0 [M+H]+
。
5-(4-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)苯基)-3-甲基-1,2,4-噁二唑:在0℃下,向(1S,3S)-3-丁基-6-甲氧基-1-(4-(3-甲基-1,2,4-噁二唑-5-基)苯基)-3,4-二氫異喹啉-2(1H)-甲酸第三丁基酯(0.185 g, 0.387 mmol, 1當量)於二氯甲烷(20 mL)中之溶液中添加於1,4-二噁烷中之4 M HCl (10 mL, 1.52 mmol, 2當量)。將混合物在室溫下攪拌16 h。藉由TLC監測反應進展,在反應完成之後,在減壓下濃縮反應混合物。使用冰冷水(20 mL)溶解所獲得粗製物且藉由NaHCO3
飽和水溶液鹼化。使用EtOAc (100 mL)萃取化合物。使用鹽水(10 mL)洗滌有機層,藉由無水硫酸鈉乾燥,過濾並濃縮以獲得5-(4-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)苯基)-3-甲基-1,2,4-噁二唑。LC-MS (m/z): 378.0 [M+H]+
。
1-((1S,3S)-3-丁基-6-甲氧基-1-(4-(3-甲基-1,2,4-噁二唑-5-基)苯基)-3,4-二氫異喹啉-2(1H)-基)-3-(三甲基矽烷基)丙-2-炔-1-酮:向3-(三甲基矽烷基)丙炔酸(0.090 g, 0.260 mmol, 1當量)中添加DMF (0.0008 g, 0.010 mmol, 0.04當量)及草醯氯(0.05 mL, 0.969 mmol, 1.1當量)並攪拌30 min。此後,在減壓下濃縮反應混合物以獲得粗製3-(三甲基矽烷基)丙炔醯氯且使用ACN (1 mL)稀釋此粗製物,且在0℃下添加至含有5-(4-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)苯基)-3-甲基-1,2,4-噁二唑(0.160 g, 0.424 mmol, 1當量)及NaHCO3
(0.267 g, 3.181 mmol, 7.5當量)於ACN (5 mL)中之經攪拌溶液之反應混合物中並攪拌15 min。LCMS及TLC (於己烷中之30 % EtOAc)顯示反應已完成。過濾反應液並在減壓下濃縮以得到粗產物1-((1S,3S)-3-丁基-6-甲氧基-1-(4-(3-甲基-1,2,4-噁二唑-5-基)苯基)-3,4-二氫異喹啉-2(1H)-基)-3-(三甲基矽烷基)丙-2-炔-1-酮,其未經進一步純化即用於下一步驟中。LC-MS (m/z): 502.0 [M+H]+
。
1-((1S,3S)-3-丁基-6-甲氧基-1-(4-(3-甲基-1,2,4-噁二唑-5-基)苯基)-3,4-二氫異喹啉-2(1H)-基)丙-2-炔-1-酮:向於THF (10.0 mL)中之1-((1S,3S)-3-丁基-6-甲氧基-1-(4-(3-甲基-1,2,4-噁二唑-5-基)苯基)-3,4-二氫異喹啉-2(1H)-基)-3-(三甲基矽烷基)丙-2-炔-1-酮(0.100 g, 0.199 mmol, 1當量)中添加TBAF (於THF之1M溶液) (0.104 mL, 0.399 mmol, 2當量)並攪拌30 min。此後,在減壓下濃縮反應混合物,使用乙酸乙酯(100 mL)稀釋且使用水(2 × 10 mL)洗滌。藉由Na2
SO4
乾燥有機層並濃縮以得到粗產物,藉由製備型TLC層析使用於己烷中之20% EtOAc作為洗脫劑進一步純化以獲得1-((1S,3S)-3-丁基-6-甲氧基-1-(4-(3-甲基-1,2,4-噁二唑-5-基)苯基)-3,4-二氫異喹啉-2(1H)-基)丙-2-炔-1-酮。LC-MS (m/z): 430.1 [M+H]+
。1
H NMR (400 MHz, DMSO-d6
): δ 0.80 - 0.81 (m, 3 H), 0.83 - 1.49 (m, 6 H), 2.31 (s, 3 H), 2.81 - 2.92 (m, 1 H), 2.93 - 3.13 (m, 1 H), 3.71 (s, 3 H), 4.35 (s, 0.5 H), 4.59 (s, 0.5 H), 4.65 - 4.77 (bs, 1 H), 6.13 (s, 0.7 H), 6.41 (s, 0.5 H), 6.78 - 6.86 (m, 2 H), 7.49 - 7.53 (m, 2 H), 7.68 - 7.70 (m, 1 H), 7.92 - 8.00 (m, 2 H)。
(S)-(1-(3-甲氧基苯基)己烷-2-基)胺基甲酸第三丁基酯:在-12℃下,以10 min時間向碘化銅(0.510 g, 2.68 mmol, 0.1當量)於二乙醚(50 mL)中之溶液中逐滴添加(3-甲氧基苯基)溴化鎂(1M於THF中) (53 mL, 53.76 mmol, 2當量)。將反應混合物在-12℃下攪拌30 min。此後,將(S)-4-丁基-1,2,3-氧雜噻唑啶-3-甲酸第三丁基酯2,2-二氧化物(7.5 g, 26.88 mmol, 1當量)於二乙醚(15 mL)中之溶液在-12℃下逐滴添加至反應物質中。將所得混合物在-12℃下攪拌4 h。最後,使用10%檸檬酸水溶液(15 mL)在-12℃下終止反應並使用乙酸乙酯(100 mL)稀釋。分離有機層並使用鹽水(50 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並濃縮以得到粗產物,藉由急速管柱層析使用於正己烷中之15%乙酸乙酯作為洗脫劑純化以獲得(S)-(1-(3-甲氧基苯基)己烷-2-基)胺基甲酸第三丁基酯。LC-MS (m/z): 252.0 [M+H]+ 1
H NMR (400 MHz, CDCl3
): δ ppm 0.86 - 0.87 (m, 3 H), 1.23 - 1.35 (m, 6 H), 1.40 (s, 9 H), 2.73 (bs, 2 H), 3.78 (s, 3 H), 4.29 (bs, 1 H), 6.71 - 6.76 (m, 3 H), 7.19 (t,J
= 7.8 Hz, 1 H)。未觀察到醯胺NH。
(S)-1-(3-甲氧基苯基)己烷-2-胺:在0℃下,向(S)-(1-(3-甲氧基苯基)己烷-2-基)胺基甲酸第三丁基酯(10 g, 32.57 mmol, 1當量)於二氯甲烷(50 mL)中之溶液中添加於1,4-二噁烷中之4M HCl (20 mL, 64.10 mmol, 2當量)。將混合物在室溫下攪拌16 h。藉由TLC監測反應進展,在反應完成之後,在減壓下濃縮反應混合物。使用冰冷水(10 mL)溶解所獲得粗製物且藉由NaHCO3
飽和水溶液鹼化。使用EtOAc (100 mL)萃取化合物。使用鹽水(10 mL)洗滌有機層,藉由無水硫酸鈉乾燥,過濾並濃縮以獲得(S)-1-(3-甲氧基苯基)己烷-2-胺。LC-MS (m/z): 208.1 [M+H]+
。1
H NMR (400 MHz, CDCl3
): δ ppm 0.82 - 0.90 (m, 3 H), 1.23 - 1.42 (m, 4 H), 1.57 - 1.62 (m, 2 H), 2.69 - 2.91 (m, 2 H), 3.25 - 3.58 (m, 1 H), 3.80 (s, 3 H), 6.69 - 6.78 (m, 3 H), 7.21 (t,J
= 8.0 Hz, 1 H)。未觀察到NH2
質子。
4-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)苯甲酸甲酯:將(S)-1-(3-甲氧基苯基)己烷-2-胺(0.7 g, 3.37 mmol, 1當量)及4-甲醯基苯甲酸甲酯(0.664 g, 4.05 mmol, 1當量)於甲苯(4 mL)中之溶液在90℃下於微波中輻照20 min。此後,在減壓下濃縮揮發性部分並在TFA (4 mL)中原樣用於環化步驟中,且在140℃下於微波中輻照45 min。此後,在減壓下濃縮揮發性部分且使用NaHCO3
飽和水溶液(10 mL)及EtOAc (40 mL)稀釋所獲得粗製物。分離有機層,使用鹽水(10 mL)洗滌,藉由無水MgSO4
乾燥,過濾並在減壓下濃縮以獲得粗產物。藉由矽膠急速管柱層析(正己烷/EtOAc)純化所獲得粗產物以得到4-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)苯甲酸甲酯。LC-MS (m/z): 208.1 [M+H]+
。1
H NMR (400 MHz, CDCl3
): δ ppm 0.84 - 0.86 (m, 3 H), 1.23 - 1.24 (m, 6 H), 2.55 - 2.59 (m, 1 H), 2.85 - 2.88 (m, 2 H), 3.75 (s, 3 H), 3.80 (s, 3 H), 5.29 (s, 3 H), 6.67 - 6.69 (m, 2 H), 6.70 - 6.80 (m, 1 H), 7.21 - 7.26 (s, 2 H), 7.94 - 8.0 (m, 2 H)。
(1S,3S)-6-甲氧基-1-(4-(甲氧基羰基)苯基)-3-甲基-3,4-二氫異喹啉-2(1H)-甲酸第三丁基酯:在室溫下,向化合物4-((1S,3S)-6-甲氧基-3-甲基-1,2,3,4-四氫異喹啉-1-基)苯甲酸甲酯(0.35 g, 1.12 mmol, 1當量)於DCM (10 mL)中之溶液中添加三乙胺(0.45 g, 4.49 mmol, 4當量)及二碳酸二-第三丁基酯(0.715 g, 2.24 mmol, 2當量)且將混合物攪拌16 h。TLC (於己烷中之50% EtOAc)顯示反應已完成。在減壓下濃縮反應混合物,且使用EtOAc (50 mL)稀釋粗製物,使用水(2 × 50 mL)洗滌。藉由無水Na2
SO4
乾燥有機層,在減壓下濃縮成粗產物(1S,3S)-6-甲氧基-1-(4-(甲氧基羰基)苯基)-3-甲基-3,4-二氫異喹啉-2(1H)-甲酸第三丁基酯。LC-MS (m/z): 356.0 [M-t
Bu+H]+
。
4-((1S,3S)-2-(第三丁氧基羰基)-6-甲氧基-3-甲基-1,2,3,4-四氫異喹啉-1-基)苯甲酸:向化合物(1S,3S)-6-甲氧基-1-(4-(甲氧基羰基)苯基)-3-甲基-3,4-二氫異喹啉-2(1H)-甲酸第三丁基酯(0.650 g, 1.57 mmol, 1當量)於THF: MeOH: H2
O (9 mL:1 mL:1 mL)之混合物中之溶液中添加氫氧化鋰(0.331 g, 7.89 mmol, 5當量)並在室溫下攪拌16 h。TLC (於己烷中之50% EtOAc)顯示反應已完成。在減壓下濃縮反應混合物,且使用5%檸檬酸溶液(pH=9)酸化粗製物。使用EtOAc (50 mL)稀釋反應混合物且分離有機層,並藉由無水Na2
SO4
乾燥,在減壓下濃縮以得到粗產物4-((1S,3S)-2-(第三丁氧基羰基)-6-甲氧基-3-甲基-1,2,3,4-四氫異喹啉-1-基)苯甲酸。LC-MS (m/z): 396.0 [M+H]。
(1S,3S)-3-丁基-1-(4-(((E)-1-(羥基亞胺基)乙基)胺甲醯基)苯基)-6-甲氧基-3,4-二氫異喹啉-2(1H)-甲酸第三丁基酯:在室溫下,向4-((1S,3S)-2-(第三丁氧基羰基)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)苯甲酸(0.2 g, 0.45 mmol, 1當量)於DMF (10 mL)中之溶液中添加DIPEA (0.15 mL, 0.91 mmol, 2當量)及HATU (0.207 g, 5.46 mmol, 1.2當量),攪拌15 min且然後添加(E)-N'-羥基乙脒(0.043 g, 0.591 mmol, 1.3當量),且將反應混合物在室溫下攪拌3 h。藉由TLC (於己烷中之30% EtOAc)監測反應進展。在反應完成之後,將反應混合物傾倒至碎冰中且然後使用EtOAc (2 × 50 mL)萃取。藉由無水硫酸鈉乾燥合併之有機層,過濾並在減壓下濃縮以獲得(1S,3S)-3-丁基-1-(4-(((E)-1-(羥基亞胺基)乙基)胺甲醯基)苯基)-6-甲氧基-3,4-二氫異喹啉-2(1H)-甲酸第三丁基酯。LC-MS (m/z) = 496.0 [M+H]+
。
(1S,3S)-3-丁基-6-甲氧基-1-(4-(3-甲基-1,2,4-噁二唑-5-基)苯基)-3,4-二氫異喹啉-2(1H)-甲酸第三丁基酯:向(1S,3S)-3-丁基-1-(4-(((E)-1-(羥基亞胺基)乙基)胺甲醯基)苯基)-6-甲氧基-3,4-二氫異喹啉-2(1H)-甲酸第三丁基酯(0.3 g, 0.606 mmol, 1當量)於ACN (10 mL)中之溶液中添加4 Å MS (0.1 g)且將反應混合物在120℃下於密封管中攪拌3 h。藉由TLC (於己烷中之50% EtOAc)監測反應進展。在反應完成之後,經由燒結漏斗過濾反應混合物且在減壓下濃縮所獲得濾液以獲得粗製物。藉由矽膠管柱層析使用於己烷中之25 - 30% EtOAc作為洗脫劑純化以提供(1S,3S)-3-丁基-6-甲氧基-1-(4-(3-甲基-1,2,4-噁二唑-5-基)苯基)-3,4-二氫異喹啉-2(1H)-甲酸第三丁基酯。LC-MS (m/z) = 478.0 [M+H]+
。
5-(4-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)苯基)-3-甲基-1,2,4-噁二唑:在0℃下,向(1S,3S)-3-丁基-6-甲氧基-1-(4-(3-甲基-1,2,4-噁二唑-5-基)苯基)-3,4-二氫異喹啉-2(1H)-甲酸第三丁基酯(0.185 g, 0.387 mmol, 1當量)於二氯甲烷(20 mL)中之溶液中添加於1,4-二噁烷中之4 M HCl (10 mL, 1.52 mmol, 2當量)。將混合物在室溫下攪拌16 h。藉由TLC監測反應進展,在反應完成之後,在減壓下濃縮反應混合物。使用冰冷水(20 mL)溶解所獲得粗製物且藉由NaHCO3
飽和水溶液鹼化。使用EtOAc (100 mL)萃取化合物。使用鹽水(10 mL)洗滌有機層,藉由無水硫酸鈉乾燥,過濾並濃縮以獲得5-(4-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)苯基)-3-甲基-1,2,4-噁二唑。LC-MS (m/z): 378.0 [M+H]+
。
1-((1S,3S)-3-丁基-6-甲氧基-1-(4-(3-甲基-1,2,4-噁二唑-5-基)苯基)-3,4-二氫異喹啉-2(1H)-基)-3-(三甲基矽烷基)丙-2-炔-1-酮:向3-(三甲基矽烷基)丙炔酸(0.090 g, 0.260 mmol, 1當量)中添加DMF (0.0008 g, 0.010 mmol, 0.04當量)及草醯氯(0.05 mL, 0.969 mmol, 1.1當量)並攪拌30 min。此後,在減壓下濃縮反應混合物以獲得粗製3-(三甲基矽烷基)丙炔醯氯且使用ACN (1 mL)稀釋此粗製物,且在0℃下添加至含有5-(4-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)苯基)-3-甲基-1,2,4-噁二唑(0.160 g, 0.424 mmol, 1當量)及NaHCO3
(0.267 g, 3.181 mmol, 7.5當量)於ACN (5 mL)中之經攪拌溶液之反應混合物中並攪拌15 min。LCMS及TLC (於己烷中之30 % EtOAc)顯示反應已完成。過濾反應液並在減壓下濃縮以得到粗產物1-((1S,3S)-3-丁基-6-甲氧基-1-(4-(3-甲基-1,2,4-噁二唑-5-基)苯基)-3,4-二氫異喹啉-2(1H)-基)-3-(三甲基矽烷基)丙-2-炔-1-酮,其未經進一步純化即用於下一步驟中。LC-MS (m/z): 502.0 [M+H]+
。
1-((1S,3S)-3-丁基-6-甲氧基-1-(4-(3-甲基-1,2,4-噁二唑-5-基)苯基)-3,4-二氫異喹啉-2(1H)-基)丙-2-炔-1-酮:向於THF (10.0 mL)中之1-((1S,3S)-3-丁基-6-甲氧基-1-(4-(3-甲基-1,2,4-噁二唑-5-基)苯基)-3,4-二氫異喹啉-2(1H)-基)-3-(三甲基矽烷基)丙-2-炔-1-酮(0.100 g, 0.199 mmol, 1當量)中添加TBAF (於THF之1M溶液) (0.104 mL, 0.399 mmol, 2當量)並攪拌30 min。此後,在減壓下濃縮反應混合物,使用乙酸乙酯(100 mL)稀釋且使用水(2 × 10 mL)洗滌。藉由Na2
SO4
乾燥有機層並濃縮以得到粗產物,藉由製備型TLC層析使用於己烷中之20% EtOAc作為洗脫劑進一步純化以獲得1-((1S,3S)-3-丁基-6-甲氧基-1-(4-(3-甲基-1,2,4-噁二唑-5-基)苯基)-3,4-二氫異喹啉-2(1H)-基)丙-2-炔-1-酮。LC-MS (m/z): 430.1 [M+H]+
。1
H NMR (400 MHz, DMSO-d6
): δ 0.80 - 0.81 (m, 3 H), 0.83 - 1.49 (m, 6 H), 2.31 (s, 3 H), 2.81 - 2.92 (m, 1 H), 2.93 - 3.13 (m, 1 H), 3.71 (s, 3 H), 4.35 (s, 0.5 H), 4.59 (s, 0.5 H), 4.65 - 4.77 (bs, 1 H), 6.13 (s, 0.7 H), 6.41 (s, 0.5 H), 6.78 - 6.86 (m, 2 H), 7.49 - 7.53 (m, 2 H), 7.68 - 7.70 (m, 1 H), 7.92 - 8.00 (m, 2 H)。
程序 19 :化合物 38 之合成 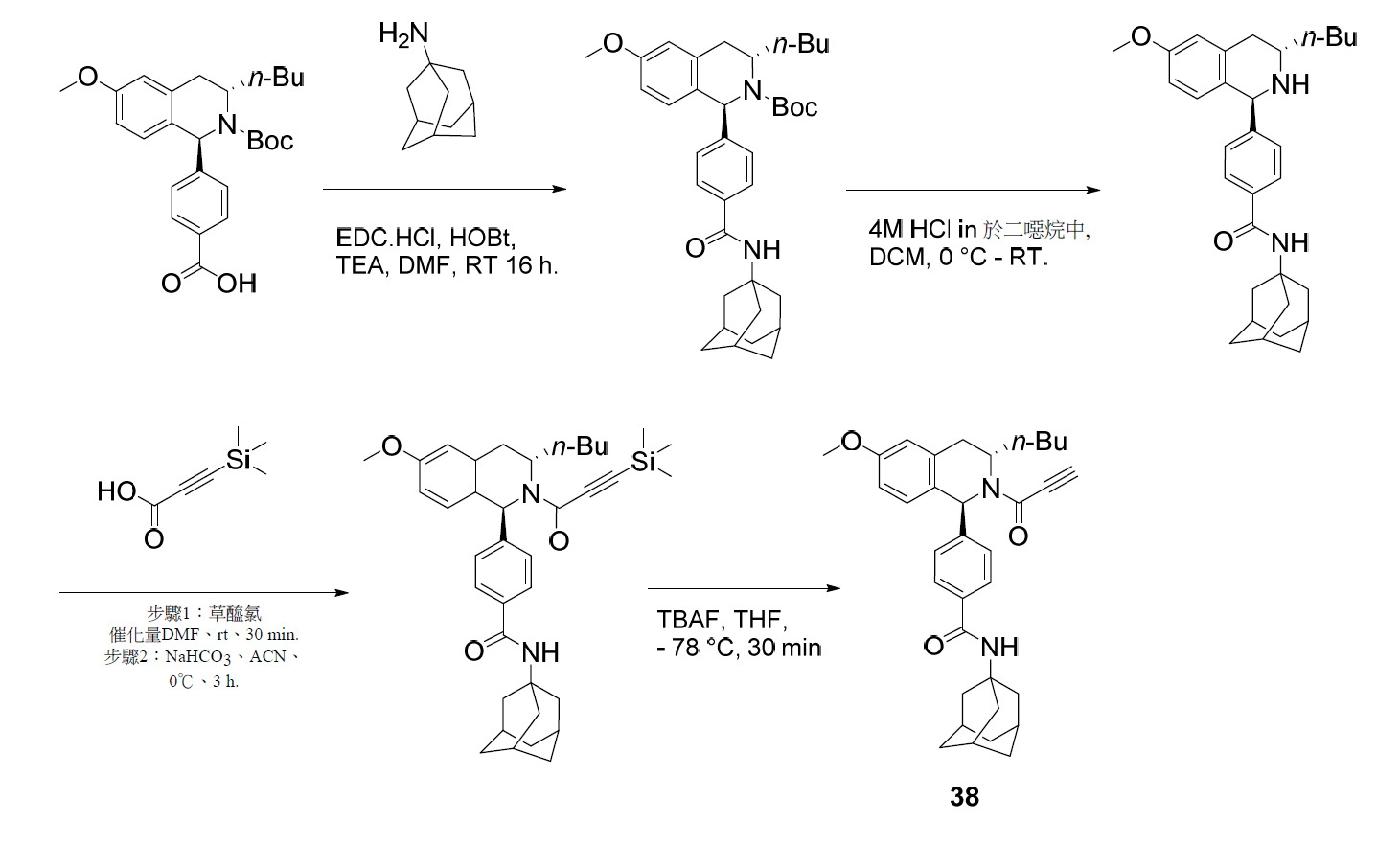 (1S,3S)-1-(4-(((3R,5R,7R)-金剛烷-1-基)胺甲醯基)苯基)-3-丁基-6-甲氧基-3,4-二氫異喹啉-2(1H)-甲酸第三丁基酯:在室溫下,向4-((1S,3S)-2-(第三丁氧基羰基)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)苯甲酸(0.2 g, 0.45 mmol, 1當量)於DCM (10 mL)中之溶液中添加TEA (0.2 mL, 1.36 mmol, 3當量)、(3s,5s,7s)-金剛烷-1-胺(0.068 g, 0.45 mmol, 1當量)及HOBt (0.092 g, 0.682 mmol, 1.5當量),攪拌15 min且然後添加EDC.HCl (0.13 g, 0.682 mmol, 1.5當量),且將反應混合物在室溫下攪拌16 h。藉由TLC (於己烷中之30% EtOAc)監測反應進展。在反應完成之後,使用水稀釋反應混合物且然後使用DCM (2 × 50 mL)萃取。藉由無水硫酸鈉乾燥合併之有機層,過濾並在減壓下濃縮以獲得(1S,3S)-1-(4-(((3R,5R,7R)-金剛烷-1-基)胺甲醯基)苯基)-3-丁基-6-甲氧基-3,4-二氫異喹啉-2(1H)-甲酸第三丁基酯。LC-MS (m/z): 517.3 [M+H]+
。1
H NMR (400 MHz, DMSO-d6
): δ ppm 0.81 (s, 3 H), 0.98 - 1.40 (m, 15 H), 1.62 (s, 6 H), 2.00 (s, 9 H), 2.75 (s, 1 H), 3.03 (s, 1 H), 3.68 (s, 3 H), 4.30 - 4.44 (m, 1 H), 5.87 (s, 1 H), 6.73 (s, 2 H), 7.28 (s, 2 H), 7.41 (s, 2 H), 7.59 (s, 2 H)。
N-((3R,5R,7R)-金剛烷-1-基)-4-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)苯甲醯胺:在0℃下,向(1S,3S)-1-(4-(((3R,5R,7R)-金剛烷-1-基)胺甲醯基)苯基)-3-丁基-6-甲氧基-3,4-二氫異喹啉-2(1H)-甲酸第三丁基酯(0.23 g, 0.401 mmol, 1當量)於二氯甲烷(10 mL)中之溶液中添加於1,4-二噁烷中之4M HCl (7 mL)。將混合物在室溫下攪拌3 h。藉由TLC監測反應進展,在反應完成之後,在減壓下濃縮反應混合物。使用冰冷水(10 mL)溶解所獲得粗製物且藉由NaHCO3
飽和水溶液鹼化。使用EtOAc (30 mL)萃取化合物。使用鹽水(10 mL)洗滌有機層,藉由無水硫酸鈉乾燥,過濾並濃縮以獲得N-((3R,5R,7R)-金剛烷-1-基)-4-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)苯甲醯胺。LC-MS (m/z): 473.7 [M+H]+
。
N-((3R,5R,7R)-金剛烷-1-基)-4-((1S,3S)-3-丁基-6-甲氧基-2-(3-(三甲基矽烷基)丙炔醯基)-1,2,3,4-四氫異喹啉-1-基)苯甲醯胺:向3-(三甲基矽烷基)丙炔酸(0.085 g, 0.59 mmol, 1當量)中添加DMF (0.001 mL, 0.023 mmol, 0.04當量)及草醯氯(0.061 mL, 0.717 mmol, 1.2當量)並攪拌30 min。此後,在減壓下濃縮反應混合物以獲得粗製3-(三甲基矽烷基)丙炔醯氯,使用ACN (1 mL)稀釋並在0℃下添加至含有N-((3R,5R,7R)-金剛烷-1-基)-4-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)苯甲醯胺(0.19 g, 0.401 mmol, 1當量)及NaHCO3
(0.253 g, 3.01 mmol, 7.5當量)於ACN (5 mL)之經攪拌溶液之反應混合物中,且攪拌15 min。LCMS及TLC (於己烷中之40% EtOAc)顯示反應已完成。過濾反應液並在減壓下濃縮以得到粗產物N-((3R,5R,7R)-金剛烷-1-基)-4-((1S,3S)-3-丁基-6-甲氧基-2-(3-(三甲基矽烷基)丙炔醯基)-1,2,3,4-四氫異喹啉-1-基)苯甲醯胺,其未經進一步純化即用於下一步驟中。LC-MS (m/z): 597.3 [M+H]+
。
N-((3R,5R,7R)-金剛烷-1-基)-4-((1S,3S)-3-丁基-6-甲氧基-2-丙炔醯基-1,2,3,4-四氫異喹啉-1-基)苯甲醯胺:向於THF (10.0 mL)中之N-((3R,5R,7R)-金剛烷-1-基)-4-((1S,3S)-3-丁基-6-甲氧基-2-(3-(三甲基矽烷基)丙炔醯基)-1,2,3,4-四氫異喹啉-1-基)苯甲醯胺(0.17 g, 0.284 mmol, 1當量)中添加TBAF(於THF中之1M溶液) (0.081 mL, 0.313 mmol, 1.1當量)並攪拌15 min。此後,在減壓下濃縮反應混合物,使用乙酸乙酯(30 mL)稀釋且使用水(2 × 10 mL)洗滌。藉由Na2
SO4
乾燥有機層並濃縮以得到粗產物,藉由急速管柱層析使用於己烷中之20% EtOAc作為洗脫劑進一步純化以得到N-((3R,5R,7R)-金剛烷-1-基)-4-((1S,3S)-3-丁基-6-甲氧基-2-丙炔醯基-1,2,3,4-四氫異喹啉-1-基)苯甲醯胺。LC-MS (m/z): 525.8 [M+H]+
。1
H NMR (400 MHz, DMSO-d6
): δ 0.81 (t,J
= 6.0 Hz, 3 H), 1.22 (s, 6 H), 1.48 - 1.61 (m, 7 H), 2.00 (s, 8 H), 2.85 - 2.89 (m, 1 H), 3.10 - 3.13 (m, 1 H), 3.69 (d,J
= 6.0 Hz, 3 H), 4.29 (s, 1 H), 4.59 (bs, 1 H), 6.06 (s, 1 H), 6.76 - 6.84 (m, 2 H), 7.25 - 7.29 (m, 2 H), 7.41 - 7.47 (m, 2 H), 7.55 (d,J
= 8.0 Hz, 1 H), 7.62 (d,J
= 8.0 Hz, 1 H)。
(1S,3S)-1-(4-(((3R,5R,7R)-金剛烷-1-基)胺甲醯基)苯基)-3-丁基-6-甲氧基-3,4-二氫異喹啉-2(1H)-甲酸第三丁基酯:在室溫下,向4-((1S,3S)-2-(第三丁氧基羰基)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)苯甲酸(0.2 g, 0.45 mmol, 1當量)於DCM (10 mL)中之溶液中添加TEA (0.2 mL, 1.36 mmol, 3當量)、(3s,5s,7s)-金剛烷-1-胺(0.068 g, 0.45 mmol, 1當量)及HOBt (0.092 g, 0.682 mmol, 1.5當量),攪拌15 min且然後添加EDC.HCl (0.13 g, 0.682 mmol, 1.5當量),且將反應混合物在室溫下攪拌16 h。藉由TLC (於己烷中之30% EtOAc)監測反應進展。在反應完成之後,使用水稀釋反應混合物且然後使用DCM (2 × 50 mL)萃取。藉由無水硫酸鈉乾燥合併之有機層,過濾並在減壓下濃縮以獲得(1S,3S)-1-(4-(((3R,5R,7R)-金剛烷-1-基)胺甲醯基)苯基)-3-丁基-6-甲氧基-3,4-二氫異喹啉-2(1H)-甲酸第三丁基酯。LC-MS (m/z): 517.3 [M+H]+
。1
H NMR (400 MHz, DMSO-d6
): δ ppm 0.81 (s, 3 H), 0.98 - 1.40 (m, 15 H), 1.62 (s, 6 H), 2.00 (s, 9 H), 2.75 (s, 1 H), 3.03 (s, 1 H), 3.68 (s, 3 H), 4.30 - 4.44 (m, 1 H), 5.87 (s, 1 H), 6.73 (s, 2 H), 7.28 (s, 2 H), 7.41 (s, 2 H), 7.59 (s, 2 H)。
N-((3R,5R,7R)-金剛烷-1-基)-4-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)苯甲醯胺:在0℃下,向(1S,3S)-1-(4-(((3R,5R,7R)-金剛烷-1-基)胺甲醯基)苯基)-3-丁基-6-甲氧基-3,4-二氫異喹啉-2(1H)-甲酸第三丁基酯(0.23 g, 0.401 mmol, 1當量)於二氯甲烷(10 mL)中之溶液中添加於1,4-二噁烷中之4M HCl (7 mL)。將混合物在室溫下攪拌3 h。藉由TLC監測反應進展,在反應完成之後,在減壓下濃縮反應混合物。使用冰冷水(10 mL)溶解所獲得粗製物且藉由NaHCO3
飽和水溶液鹼化。使用EtOAc (30 mL)萃取化合物。使用鹽水(10 mL)洗滌有機層,藉由無水硫酸鈉乾燥,過濾並濃縮以獲得N-((3R,5R,7R)-金剛烷-1-基)-4-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)苯甲醯胺。LC-MS (m/z): 473.7 [M+H]+
。
N-((3R,5R,7R)-金剛烷-1-基)-4-((1S,3S)-3-丁基-6-甲氧基-2-(3-(三甲基矽烷基)丙炔醯基)-1,2,3,4-四氫異喹啉-1-基)苯甲醯胺:向3-(三甲基矽烷基)丙炔酸(0.085 g, 0.59 mmol, 1當量)中添加DMF (0.001 mL, 0.023 mmol, 0.04當量)及草醯氯(0.061 mL, 0.717 mmol, 1.2當量)並攪拌30 min。此後,在減壓下濃縮反應混合物以獲得粗製3-(三甲基矽烷基)丙炔醯氯,使用ACN (1 mL)稀釋並在0℃下添加至含有N-((3R,5R,7R)-金剛烷-1-基)-4-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)苯甲醯胺(0.19 g, 0.401 mmol, 1當量)及NaHCO3
(0.253 g, 3.01 mmol, 7.5當量)於ACN (5 mL)之經攪拌溶液之反應混合物中,且攪拌15 min。LCMS及TLC (於己烷中之40% EtOAc)顯示反應已完成。過濾反應液並在減壓下濃縮以得到粗產物N-((3R,5R,7R)-金剛烷-1-基)-4-((1S,3S)-3-丁基-6-甲氧基-2-(3-(三甲基矽烷基)丙炔醯基)-1,2,3,4-四氫異喹啉-1-基)苯甲醯胺,其未經進一步純化即用於下一步驟中。LC-MS (m/z): 597.3 [M+H]+
。
N-((3R,5R,7R)-金剛烷-1-基)-4-((1S,3S)-3-丁基-6-甲氧基-2-丙炔醯基-1,2,3,4-四氫異喹啉-1-基)苯甲醯胺:向於THF (10.0 mL)中之N-((3R,5R,7R)-金剛烷-1-基)-4-((1S,3S)-3-丁基-6-甲氧基-2-(3-(三甲基矽烷基)丙炔醯基)-1,2,3,4-四氫異喹啉-1-基)苯甲醯胺(0.17 g, 0.284 mmol, 1當量)中添加TBAF(於THF中之1M溶液) (0.081 mL, 0.313 mmol, 1.1當量)並攪拌15 min。此後,在減壓下濃縮反應混合物,使用乙酸乙酯(30 mL)稀釋且使用水(2 × 10 mL)洗滌。藉由Na2
SO4
乾燥有機層並濃縮以得到粗產物,藉由急速管柱層析使用於己烷中之20% EtOAc作為洗脫劑進一步純化以得到N-((3R,5R,7R)-金剛烷-1-基)-4-((1S,3S)-3-丁基-6-甲氧基-2-丙炔醯基-1,2,3,4-四氫異喹啉-1-基)苯甲醯胺。LC-MS (m/z): 525.8 [M+H]+
。1
H NMR (400 MHz, DMSO-d6
): δ 0.81 (t,J
= 6.0 Hz, 3 H), 1.22 (s, 6 H), 1.48 - 1.61 (m, 7 H), 2.00 (s, 8 H), 2.85 - 2.89 (m, 1 H), 3.10 - 3.13 (m, 1 H), 3.69 (d,J
= 6.0 Hz, 3 H), 4.29 (s, 1 H), 4.59 (bs, 1 H), 6.06 (s, 1 H), 6.76 - 6.84 (m, 2 H), 7.25 - 7.29 (m, 2 H), 7.41 - 7.47 (m, 2 H), 7.55 (d,J
= 8.0 Hz, 1 H), 7.62 (d,J
= 8.0 Hz, 1 H)。
程序 20 :化合物 48 之合成 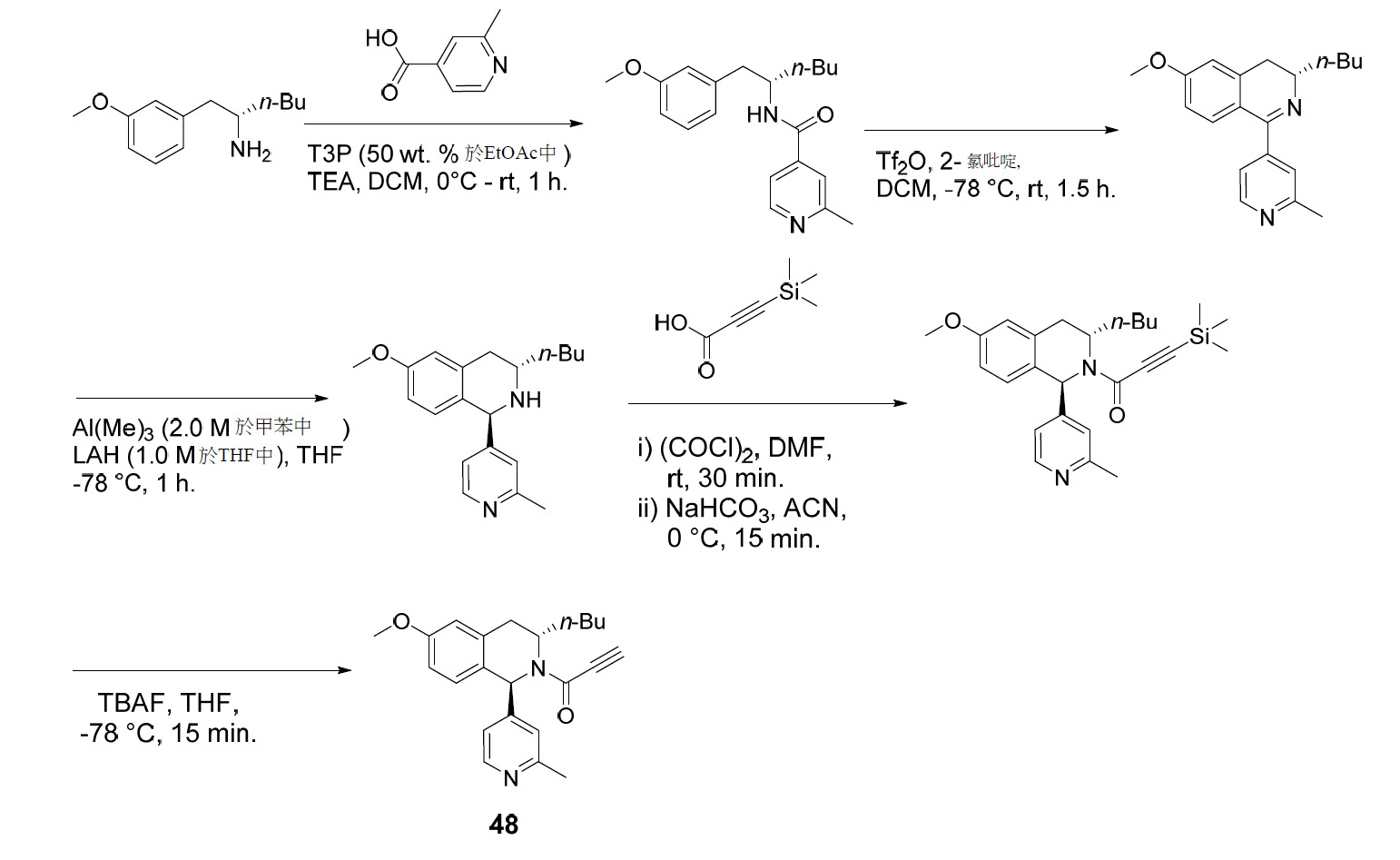 (S)-N-(1-(3-甲氧基苯基)己烷-2-基)-2-甲基異菸鹼醯胺:向2-甲基異菸鹼酸(0.654 g, 4.77 mmol, 1.1當量)於DCM (15 mL)中之溶液中添加TEA (2.5 mL, 17.36 mmol, 4當量),攪拌15 min且然後在0℃下添加T3P (50 wt.% in EtOAc) (4.2 mL, 6.51 mmol, 1.5當量)並再攪拌5 min。然後,向反應混合物中添加(S)-1-(3-甲氧基苯基)己烷-2-胺(0.900 g, 4.34 mmol, 1當量)且然後將反應混合物在室溫下攪拌16 h。藉由TLC (於己烷中之40%乙酸乙酯)監測反應進展。使用DCM (30 mL)及飽和碳酸氫鈉溶液(15 mL)稀釋反應混合物,分離有機層,使用鹽水溶液(12 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得(S)-N-(1-(3-甲氧基苯基)己烷-2-基)-2-甲基異菸鹼醯胺。LCMS (ES) (m/z) = 327.3 [M+H]+ 1
H NMR (400 MHz, CDCl3
) δ ppm 0.87 - 0.89 (m, 3 H), 1.36 - 1.46 (m, 4 H), 1.60 - 1.69 (m, 2 H), 2.59 (s, 3 H), 2.81 - 2.93 (m, 2 H), 3.76 (s, 3 H), 4.36 - 4.37 (m, 1 H), 5.83 (d,J
= 7.2 Hz, 1 H), 6.74 - 6.78 (m, 3 H), 7.21 (t,J
= 7.6 Hz, 2 H), 7.37 (s, 1 H), 8.57 (d,J
= 4.8 Hz, 1 H)。
(S)-3-丁基-6-甲氧基-1-(2-甲基吡啶-4-基)-3,4-二氫異喹啉:在-78℃下,經由注射器,以1 min時間將三氟甲烷磺酸酐(1.28 mL, 7.65 mmol, 2當量)添加至(S)-N-(1-(3-甲氧基苯基)己烷-2-基)-2-甲基異菸鹼醯胺(1.25 g, 3.82 mmol, 1當量)及2-氯吡啶(0.72 mL, 7.65 mmol, 2當量)於二氯甲烷(10 mL)中之經攪拌混合物中。在5 min之後,將反應混合物置於冰-水浴中並升溫至0℃。在5 min之後,將所得溶液升溫至23℃。TLC (於DCM中之5% MeOH)顯示反應已完成。在1 h之後,引入氫氧化鈉水溶液(12 mL, 1N)以中和三氟甲烷磺酸鹽。添加二氯甲烷(70 mL)以稀釋混合物且分離各層。使用鹽水(10 mL)洗滌有機層,藉由無水硫酸鈉乾燥,並過濾。在減壓下去除揮發物以得到粗產物。藉由急速層析使用乙酸乙酯之己烷溶液作為洗脫劑純化所獲得粗產物以得到期望產物(S)-3-丁基-6-甲氧基-1-(2-甲基吡啶-4-基)-3,4-二氫異喹啉。LCMS (ES) m/z = 309.4 [M+H]+
(1S,3S)-3-丁基-6-甲氧基-1-(2-甲基吡啶-4-基)-1,2,3,4-四氫異喹啉:在-78℃及氮氣下,將(S)-3-丁基-6-甲氧基-1-(2-甲基吡啶-4-基)-3,4-二氫異喹啉 (
0.54 g, 1.78 mmol, 1當量)於無水THF (4 mL)中之溶液逐滴添加至於THF中之1M氫化鋰鋁(17.8 mL, 17.8 mmol, 10當量)及三甲基鋁(2M於THF中) (4.46 mL, 12.85 mmol, 5當量)之混合物中。將懸浮液在-78℃下攪拌1 h,並經1 h升溫至0℃。TLC (於DCM中之5% MeOH)顯示反應已完成。使用氯化鈉飽和水溶液(8 mL)淬滅反應混合物,隨後使用EtOAc (30 mL)稀釋且濾除沈澱物。最後,藉由無水硫酸鈉乾燥濾液,過濾並在減壓下蒸發。藉由矽膠急速層析使用乙酸乙酯之己烷溶液作為洗脫劑純化殘餘物以得到(1S,3S)-3-丁基-6-甲氧基-1-(2-甲基吡啶-4-基)-1,2,3,4-四氫異喹啉。LCMS (ES) m/z = 311.3 [M+H]+
1-((1S,3S)-3-丁基-6-甲氧基-1-(2-甲基吡啶-4-基)-3,4-二氫異喹啉-2(1H)-基)-3-(三甲基矽烷基)丙-2-炔-1-酮:第一步驟:-在室溫下,向3-(三甲基矽烷基)丙炔酸(206 mg, 1.44 mmol, 3當量)於DMF (0.014 mL, 0.05 mmol, 0.04當量)中之溶液中添加草醯氯(0.13 mL, 1.59 mmol, 1.1當量)並攪拌30分鐘。此後,在減壓下濃縮反應混合物以得到3-(三甲基矽烷基)丙炔醯氯。此醯氯未經任何進一步純化即用於下一步驟中。
第二步驟:-在0℃下,向(1S,3S)-3-丁基-6-甲氧基-1-(2-甲基吡啶-4-基)-1,2,3,4-四氫異喹啉(0.15 g, 0.48 mmol, 1.0當量)於乙腈(5.0 mL)中之溶液中添加碳酸氫鈉(0.30 g, 3.62 mmol, 7.5當量)。在攪拌5分鐘之後,向上述反應物質中添加3-(三甲基矽烷基)丙炔醯氯於乙腈(3.0 mL)中之溶液。將所得混合物在0℃下攪拌15 min,藉由TLC (於正己烷中之55%乙酸乙酯)監測反應進展。此後,使用EtOAc (15 mL)及水(5 mL)稀釋反應物質。分離有機層,使用鹽水溶液(5 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物。此粗產物未經任何進一步純化即用於下一步驟中。LCMS (ES) m/z = 435.3 [M+H]+
1-((1S,3S)-3-丁基-6-甲氧基-1-(2-甲基吡啶-4-基)-3,4-二氫異喹啉-2(1H)-基)丙-2-炔-1-酮:在-78℃下,向1-((1S,3S)-3-丁基-6-甲氧基-1-(2-甲基吡啶-4-基)-3,4-二氫異喹啉-2(1H)-基)-3-(三甲基矽烷基)丙-2-炔-1-酮(0.25 g, 0.576 mmol, 1當量)於THF (5.0 mL)中之溶液中添加TBAF (於THF中之1M溶液) (0.63 mL, 0.63 mmol, 1.1當量)。將此反應混合物在-78℃下攪拌15分鐘。藉由TLC (於正己烷中之60%乙酸乙酯)監測反應進展。此後,使用NaHCO3
飽和水溶液(8 mL)淬滅反應混合物且使用乙酸乙酯(25 mL)萃取產物。藉由無水硫酸鈉乾燥有機層,過濾並在減壓下濃縮。藉由製備型TLC使用於正己烷中之55%乙酸乙酯作為洗脫劑純化所獲得粗產物以獲得1-((1S,3S)-3-丁基-6-甲氧基-1-(2-甲基吡啶-4-基)-3,4-二氫異喹啉-2(1H)-基)丙-2-炔-1-酮。LCMS (ES) m/z = 363.4 [M+H]+
。1
H NMR (400MHz, DMSO-d6
) ppm δ 0.80 (d, 3 H), 1.24 (s, 4 H), 1.50 (s, 2 H), 2.38 (s, 3 H), 2.80 - 2.88 (m, 1 H), 3.12 - 3.15 (m, 1 H), 3.72 (s, 3 H), 4.38 (s, 1 H), 4.57 - 4.74 (m, 1 H), 5.97 (s, 1 H), 6.78 (s, 2 H), 6.99 - 7.07 (m, 2 H), 7.40 - 7.55 (m, 1 H), 8.23 - 8.29 (m, 1 H)。
(S)-N-(1-(3-甲氧基苯基)己烷-2-基)-2-甲基異菸鹼醯胺:向2-甲基異菸鹼酸(0.654 g, 4.77 mmol, 1.1當量)於DCM (15 mL)中之溶液中添加TEA (2.5 mL, 17.36 mmol, 4當量),攪拌15 min且然後在0℃下添加T3P (50 wt.% in EtOAc) (4.2 mL, 6.51 mmol, 1.5當量)並再攪拌5 min。然後,向反應混合物中添加(S)-1-(3-甲氧基苯基)己烷-2-胺(0.900 g, 4.34 mmol, 1當量)且然後將反應混合物在室溫下攪拌16 h。藉由TLC (於己烷中之40%乙酸乙酯)監測反應進展。使用DCM (30 mL)及飽和碳酸氫鈉溶液(15 mL)稀釋反應混合物,分離有機層,使用鹽水溶液(12 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得(S)-N-(1-(3-甲氧基苯基)己烷-2-基)-2-甲基異菸鹼醯胺。LCMS (ES) (m/z) = 327.3 [M+H]+ 1
H NMR (400 MHz, CDCl3
) δ ppm 0.87 - 0.89 (m, 3 H), 1.36 - 1.46 (m, 4 H), 1.60 - 1.69 (m, 2 H), 2.59 (s, 3 H), 2.81 - 2.93 (m, 2 H), 3.76 (s, 3 H), 4.36 - 4.37 (m, 1 H), 5.83 (d,J
= 7.2 Hz, 1 H), 6.74 - 6.78 (m, 3 H), 7.21 (t,J
= 7.6 Hz, 2 H), 7.37 (s, 1 H), 8.57 (d,J
= 4.8 Hz, 1 H)。
(S)-3-丁基-6-甲氧基-1-(2-甲基吡啶-4-基)-3,4-二氫異喹啉:在-78℃下,經由注射器,以1 min時間將三氟甲烷磺酸酐(1.28 mL, 7.65 mmol, 2當量)添加至(S)-N-(1-(3-甲氧基苯基)己烷-2-基)-2-甲基異菸鹼醯胺(1.25 g, 3.82 mmol, 1當量)及2-氯吡啶(0.72 mL, 7.65 mmol, 2當量)於二氯甲烷(10 mL)中之經攪拌混合物中。在5 min之後,將反應混合物置於冰-水浴中並升溫至0℃。在5 min之後,將所得溶液升溫至23℃。TLC (於DCM中之5% MeOH)顯示反應已完成。在1 h之後,引入氫氧化鈉水溶液(12 mL, 1N)以中和三氟甲烷磺酸鹽。添加二氯甲烷(70 mL)以稀釋混合物且分離各層。使用鹽水(10 mL)洗滌有機層,藉由無水硫酸鈉乾燥,並過濾。在減壓下去除揮發物以得到粗產物。藉由急速層析使用乙酸乙酯之己烷溶液作為洗脫劑純化所獲得粗產物以得到期望產物(S)-3-丁基-6-甲氧基-1-(2-甲基吡啶-4-基)-3,4-二氫異喹啉。LCMS (ES) m/z = 309.4 [M+H]+
(1S,3S)-3-丁基-6-甲氧基-1-(2-甲基吡啶-4-基)-1,2,3,4-四氫異喹啉:在-78℃及氮氣下,將(S)-3-丁基-6-甲氧基-1-(2-甲基吡啶-4-基)-3,4-二氫異喹啉 (
0.54 g, 1.78 mmol, 1當量)於無水THF (4 mL)中之溶液逐滴添加至於THF中之1M氫化鋰鋁(17.8 mL, 17.8 mmol, 10當量)及三甲基鋁(2M於THF中) (4.46 mL, 12.85 mmol, 5當量)之混合物中。將懸浮液在-78℃下攪拌1 h,並經1 h升溫至0℃。TLC (於DCM中之5% MeOH)顯示反應已完成。使用氯化鈉飽和水溶液(8 mL)淬滅反應混合物,隨後使用EtOAc (30 mL)稀釋且濾除沈澱物。最後,藉由無水硫酸鈉乾燥濾液,過濾並在減壓下蒸發。藉由矽膠急速層析使用乙酸乙酯之己烷溶液作為洗脫劑純化殘餘物以得到(1S,3S)-3-丁基-6-甲氧基-1-(2-甲基吡啶-4-基)-1,2,3,4-四氫異喹啉。LCMS (ES) m/z = 311.3 [M+H]+
1-((1S,3S)-3-丁基-6-甲氧基-1-(2-甲基吡啶-4-基)-3,4-二氫異喹啉-2(1H)-基)-3-(三甲基矽烷基)丙-2-炔-1-酮:第一步驟:-在室溫下,向3-(三甲基矽烷基)丙炔酸(206 mg, 1.44 mmol, 3當量)於DMF (0.014 mL, 0.05 mmol, 0.04當量)中之溶液中添加草醯氯(0.13 mL, 1.59 mmol, 1.1當量)並攪拌30分鐘。此後,在減壓下濃縮反應混合物以得到3-(三甲基矽烷基)丙炔醯氯。此醯氯未經任何進一步純化即用於下一步驟中。
第二步驟:-在0℃下,向(1S,3S)-3-丁基-6-甲氧基-1-(2-甲基吡啶-4-基)-1,2,3,4-四氫異喹啉(0.15 g, 0.48 mmol, 1.0當量)於乙腈(5.0 mL)中之溶液中添加碳酸氫鈉(0.30 g, 3.62 mmol, 7.5當量)。在攪拌5分鐘之後,向上述反應物質中添加3-(三甲基矽烷基)丙炔醯氯於乙腈(3.0 mL)中之溶液。將所得混合物在0℃下攪拌15 min,藉由TLC (於正己烷中之55%乙酸乙酯)監測反應進展。此後,使用EtOAc (15 mL)及水(5 mL)稀釋反應物質。分離有機層,使用鹽水溶液(5 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物。此粗產物未經任何進一步純化即用於下一步驟中。LCMS (ES) m/z = 435.3 [M+H]+
1-((1S,3S)-3-丁基-6-甲氧基-1-(2-甲基吡啶-4-基)-3,4-二氫異喹啉-2(1H)-基)丙-2-炔-1-酮:在-78℃下,向1-((1S,3S)-3-丁基-6-甲氧基-1-(2-甲基吡啶-4-基)-3,4-二氫異喹啉-2(1H)-基)-3-(三甲基矽烷基)丙-2-炔-1-酮(0.25 g, 0.576 mmol, 1當量)於THF (5.0 mL)中之溶液中添加TBAF (於THF中之1M溶液) (0.63 mL, 0.63 mmol, 1.1當量)。將此反應混合物在-78℃下攪拌15分鐘。藉由TLC (於正己烷中之60%乙酸乙酯)監測反應進展。此後,使用NaHCO3
飽和水溶液(8 mL)淬滅反應混合物且使用乙酸乙酯(25 mL)萃取產物。藉由無水硫酸鈉乾燥有機層,過濾並在減壓下濃縮。藉由製備型TLC使用於正己烷中之55%乙酸乙酯作為洗脫劑純化所獲得粗產物以獲得1-((1S,3S)-3-丁基-6-甲氧基-1-(2-甲基吡啶-4-基)-3,4-二氫異喹啉-2(1H)-基)丙-2-炔-1-酮。LCMS (ES) m/z = 363.4 [M+H]+
。1
H NMR (400MHz, DMSO-d6
) ppm δ 0.80 (d, 3 H), 1.24 (s, 4 H), 1.50 (s, 2 H), 2.38 (s, 3 H), 2.80 - 2.88 (m, 1 H), 3.12 - 3.15 (m, 1 H), 3.72 (s, 3 H), 4.38 (s, 1 H), 4.57 - 4.74 (m, 1 H), 5.97 (s, 1 H), 6.78 (s, 2 H), 6.99 - 7.07 (m, 2 H), 7.40 - 7.55 (m, 1 H), 8.23 - 8.29 (m, 1 H)。
程序 21 :化合物 49 之合成 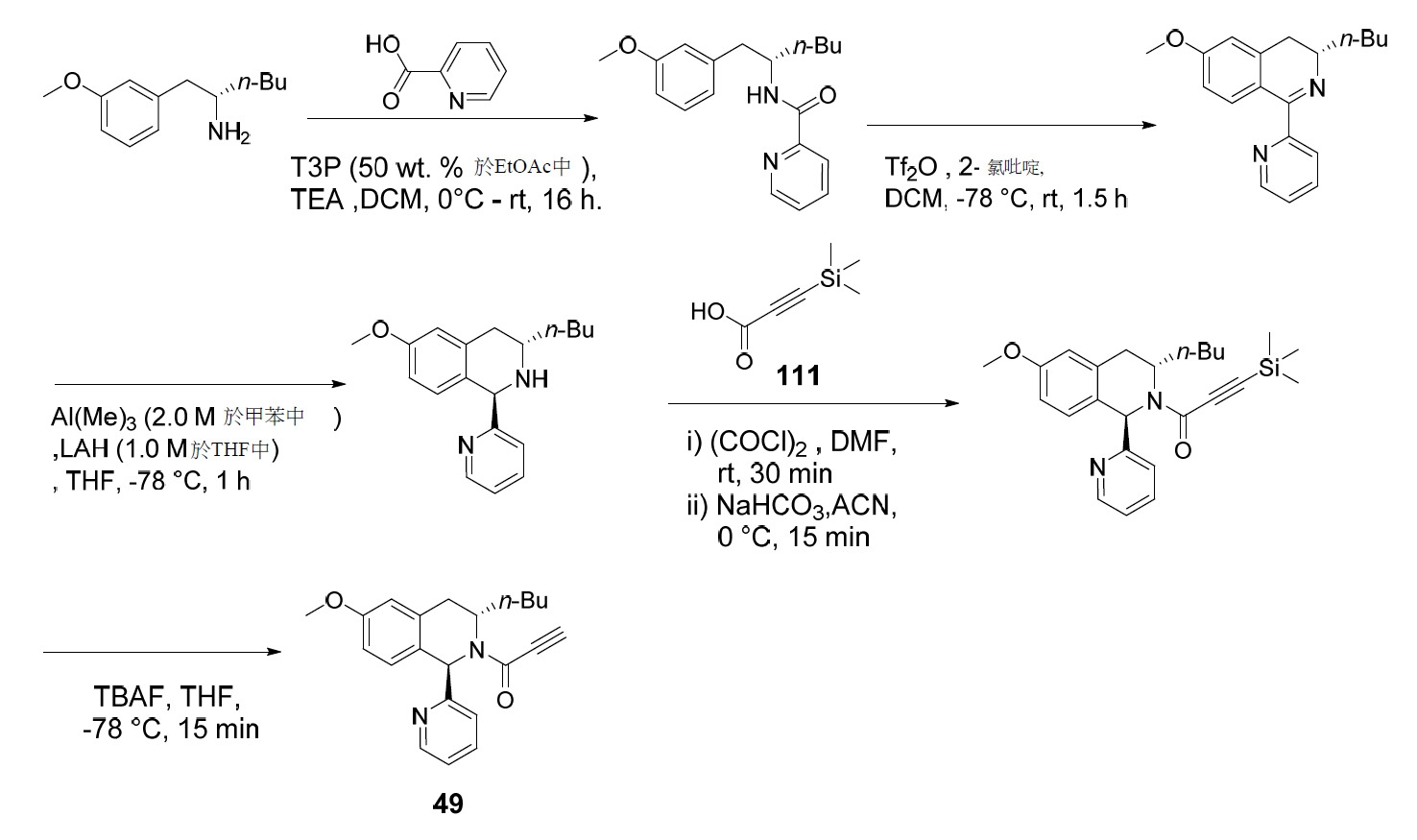 (S)-N-(1-(3-甲氧基苯基)己烷-2-基)吡啶醯胺:向吡啶甲酸(0.68 g, 5.54 mmol, 1.15當量)於DCM (15 mL)中之溶液中添加TEA (2.7 mL, 19.29 mmol, 4當量),攪拌15 min且然後在0℃下添加T3P (50 wt.%於EtOAc中) (4.6 mL, 7.23 mmol, 1.5當量)並再攪拌5 min。然後,向反應混合物中添加(S)-1-(3-甲氧基苯基)己烷-2-胺(1.0 g, 4.82 mmol, 1當量)且然後將反應混合物在室溫下攪拌16 h。藉由TLC (於己烷中之40%乙酸乙酯)監測反應進展。使用DCM (30 mL)及飽和碳酸氫鈉溶液(15 mL)稀釋反應混合物,分離有機層,使用鹽水溶液(12 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得(S)-N-(1-(3-甲氧基苯基)己烷-2-基)-2-吡啶醯胺。LC-MS (m/z) = 313.4 [M+H]+ 1
H NMR (400 MHz, CDCl3
) δ ppm 0.84 (t,J
= 6.8 Hz, 3 H), 1.23 - 1.60 (m, 6 H), 2.81 - 2.94 (m, 2 H), 3.74 (s, 3 H), 4.35 - 4.36 (m, 1 H), 6.73 - 6.82 (m, 3 H), 7.17 (t,J
= 8.0 Hz, 1 H), 7.39 (t,J
= 6.0 Hz, 1 H), 7.82 (t,J
= 8.0 Hz, 1 H), 7.91 (d,J
= 8.0 Hz, 1 H), 8.17 (d,J
= 7.6 Hz, 1 H), 8.52 (d,J
= 4.0 Hz, 1 H)。
(S)-3-丁基-6-甲氧基-1-(吡啶-2-基)-3,4-二氫異喹啉:在-78℃下,經由注射器,以1 min時間將三氟甲烷磺酸酐(1.55 mL, 9.28 mmol, 2.0當量)添加至(S)-N-(1-(3-甲氧基苯基)己烷-2-基)吡啶醯胺(1.4 g, 4.64 mmol, 1當量)及2-氯吡啶(0.87 mL, 9.28 mmol, 2.0當量)於二氯甲烷(15 mL)中之攪拌混合物中。在5 min之後,將反應混合物置於冰-水浴中並升溫至0℃。在5 min之後,將所得溶液升溫至23℃。TLC (於正己烷中之40%乙酸乙酯)顯示反應已完成。在1 h之後,引入氫氧化鈉水溶液(12 mL, 1N)以中和三氟甲烷磺酸鹽。添加二氯甲烷(50 mL)以稀釋混合物且分離各層。使用鹽水(10 mL)洗滌有機層,藉由無水硫酸鈉乾燥,並過濾。在減壓下去除揮發物以得到粗產物。藉由急速層析使用乙酸乙酯之己烷溶液作為洗脫劑,純化所獲得粗產物,以得到期望產物(S)-3-丁基-6-甲氧基-1-(吡啶-2-基)-3,4-二氫異喹啉。LC-MS (m/z) = 295.1 [M+H]+ 1
H NMR (400 MHz, CDCl3
) δ ppm 0.93 (t,J
= 7.2 Hz, 3 H), 1.26 - 1.43 (m, 4 H), 1.65 - 1.88 (m, 2 H), 2.58 - 2.65 (m, 1 H), 2.79 - 2.87 (m, 1 H), 3.58 (bs, 1 H), 3.83 (s, 3 H), 6.73 - 6.75 (m, 2 H), 7.32 - 7.37 (m, 2 H), 7.77 - 7.83 (m, 2 H), 8.64 - 8.70 (m, 1 H)。
(1R,3S)-3-丁基-6-甲氧基-1-(吡啶-2-基)-1,2,3,4-四氫異喹啉:在-78℃及氮氣下,將(S)-3-丁基-6-甲氧基-1-(吡啶-2-基)-3,4-二氫異喹啉(0.1 g, 0.34 mmol, 1當量)於無水THF (4 mL)中之溶液逐滴添加至於THF中之1M氫化鋰鋁(3.4 mL, 3.40 mmol, 10當量)及三甲基鋁(2M甲苯溶液) (0.85 mL, 1.70 mmol, 5當量)之混合物中。將懸浮液在-78℃下攪拌1 h,並經1 h升溫至0℃。TLC (於DCM中之5% MeOH)顯示反應已完成。使用氯化鈉飽和水溶液(4 mL)淬滅反應混合物,隨後使用EtOAc (15 mL)稀釋且濾除沈澱物。最後,藉由無水硫酸鈉乾燥濾液,過濾並在減壓下蒸發。藉由矽膠急速層析使用乙酸乙酯之己烷溶液作為洗脫劑純化殘餘物以得到(1R,3S)-3-丁基-6-甲氧基-1-(吡啶-2-基)-1,2,3,4-四氫異喹啉。LC-MS (m/z) = 311.3 [M+H]+
1-((1R,3S)-3-丁基-6-甲氧基-1-(吡啶-2-基)-3,4-二氫異喹啉-2(1H)-基)-3-(三甲基矽烷基)丙-2-炔-1-酮:第一步驟:在室溫下,向3-(三甲基矽烷基)丙炔酸(0.10 g, 0.709 mmol, 3.0當量)於DMF (0.0007 mL 0.009 mmol, 0.04當量)中之溶液中添加草醯氯(0.02 mL, 0.26 mmol, 1.1當量)且將反應液攪拌30分鐘。此後,在減壓下濃縮反應混合物以產生3-(三甲基矽烷基)丙炔醯氯。此醯氯未經進一步純化即用於下一步驟中。
第二步驟:在0℃下,向(1S,3S)-3-丁基-6-甲氧基-1-(2-甲基吡啶-4-基)-1,2,3,4-四氫異喹啉(0.07 g, 0.236 mmol, 1.0當量)於乙腈(3.0 mL)中之溶液中添加碳酸氫鈉(0.149 g, 1.77 mmol, 7.5當量)。在攪拌5分鐘之後,向上述反應物質中添加3-(三甲基矽烷基)丙炔醯氯於乙腈(2.0 mL)中之溶液。將所得混合物在0℃下攪拌15 min,藉由TLC (於正己烷中之60%乙酸乙酯)監測反應進展。此後,使用EtOAc (15 mL)及水(5 mL)稀釋反應物質。分離有機層,使用鹽水溶液(5 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物。此粗產物未經任何進一步純化即用於下一步驟中。LC-MS (m/z) = 421.3 [M+H]+
1-((1R,3S)-3-丁基-6-甲氧基-1-(吡啶-2-基)-3,4-二氫異喹啉-2(1H)-基)丙-2-炔-1-酮:在-78℃下,向1-((1R,3S)-3-丁基-6-甲氧基-1-(吡啶-2-基)-3,4-二氫異喹啉-2(1H)-基)-3-(三甲基矽烷基)丙-2-炔-1-酮(0.07 g, 0.166 mmol, 1.0當量)於THF (4.0 mL)中之溶液中添加TBAF (於THF中之1M溶液) (0.183 mL, 0.183 mmol, 1.1當量)。將此反應混合物在-78℃下攪拌15分鐘。藉由TLC (於正己烷中之50%乙酸乙酯)監測反應進展。此後,使用NaHCO3
飽和水溶液(2 mL)淬滅反應混合物且使用乙酸乙酯(25 mL)萃取產物。藉由無水硫酸鈉乾燥有機層,過濾並在減壓下濃縮。藉由製備型TLC使用於正己烷中之50%乙酸乙酯作為洗脫劑純化所獲得粗產物以得到1-((1R,3S)-3-丁基-6-甲氧基-1-(吡啶-2-基)-3,4-二氫異喹啉-2(1H)-基)丙-2-炔-1-酮。LC-MS (m/z) = 349.4 [M+H]+ 1
H NMR (400MHz, DMSO-d6
): δ 0.80 (t,J
= 6.8 Hz, 3 H), 1.20 - 1.44 (m, 6 H), 2.71 - 2.83 (m, 1 H), 3.03 (s, 1 H), 3.43 - 3.47 (m, 1 H), 3.67 - 3.69 (m, 3 H), 4.56 - 4.70 (m, 2 H), 6.01 - 6.28 (m, 2 H), 6.72 - 6.79 (m, 1 H), 7.09 - 7.18 (m, 1 H), 7.35 - 7.44 (m, 1 H), 7.56 - 7.69 (m, 1 H), 8.32 - 8.42 (m, 1H)。
(S)-N-(1-(3-甲氧基苯基)己烷-2-基)吡啶醯胺:向吡啶甲酸(0.68 g, 5.54 mmol, 1.15當量)於DCM (15 mL)中之溶液中添加TEA (2.7 mL, 19.29 mmol, 4當量),攪拌15 min且然後在0℃下添加T3P (50 wt.%於EtOAc中) (4.6 mL, 7.23 mmol, 1.5當量)並再攪拌5 min。然後,向反應混合物中添加(S)-1-(3-甲氧基苯基)己烷-2-胺(1.0 g, 4.82 mmol, 1當量)且然後將反應混合物在室溫下攪拌16 h。藉由TLC (於己烷中之40%乙酸乙酯)監測反應進展。使用DCM (30 mL)及飽和碳酸氫鈉溶液(15 mL)稀釋反應混合物,分離有機層,使用鹽水溶液(12 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得(S)-N-(1-(3-甲氧基苯基)己烷-2-基)-2-吡啶醯胺。LC-MS (m/z) = 313.4 [M+H]+ 1
H NMR (400 MHz, CDCl3
) δ ppm 0.84 (t,J
= 6.8 Hz, 3 H), 1.23 - 1.60 (m, 6 H), 2.81 - 2.94 (m, 2 H), 3.74 (s, 3 H), 4.35 - 4.36 (m, 1 H), 6.73 - 6.82 (m, 3 H), 7.17 (t,J
= 8.0 Hz, 1 H), 7.39 (t,J
= 6.0 Hz, 1 H), 7.82 (t,J
= 8.0 Hz, 1 H), 7.91 (d,J
= 8.0 Hz, 1 H), 8.17 (d,J
= 7.6 Hz, 1 H), 8.52 (d,J
= 4.0 Hz, 1 H)。
(S)-3-丁基-6-甲氧基-1-(吡啶-2-基)-3,4-二氫異喹啉:在-78℃下,經由注射器,以1 min時間將三氟甲烷磺酸酐(1.55 mL, 9.28 mmol, 2.0當量)添加至(S)-N-(1-(3-甲氧基苯基)己烷-2-基)吡啶醯胺(1.4 g, 4.64 mmol, 1當量)及2-氯吡啶(0.87 mL, 9.28 mmol, 2.0當量)於二氯甲烷(15 mL)中之攪拌混合物中。在5 min之後,將反應混合物置於冰-水浴中並升溫至0℃。在5 min之後,將所得溶液升溫至23℃。TLC (於正己烷中之40%乙酸乙酯)顯示反應已完成。在1 h之後,引入氫氧化鈉水溶液(12 mL, 1N)以中和三氟甲烷磺酸鹽。添加二氯甲烷(50 mL)以稀釋混合物且分離各層。使用鹽水(10 mL)洗滌有機層,藉由無水硫酸鈉乾燥,並過濾。在減壓下去除揮發物以得到粗產物。藉由急速層析使用乙酸乙酯之己烷溶液作為洗脫劑,純化所獲得粗產物,以得到期望產物(S)-3-丁基-6-甲氧基-1-(吡啶-2-基)-3,4-二氫異喹啉。LC-MS (m/z) = 295.1 [M+H]+ 1
H NMR (400 MHz, CDCl3
) δ ppm 0.93 (t,J
= 7.2 Hz, 3 H), 1.26 - 1.43 (m, 4 H), 1.65 - 1.88 (m, 2 H), 2.58 - 2.65 (m, 1 H), 2.79 - 2.87 (m, 1 H), 3.58 (bs, 1 H), 3.83 (s, 3 H), 6.73 - 6.75 (m, 2 H), 7.32 - 7.37 (m, 2 H), 7.77 - 7.83 (m, 2 H), 8.64 - 8.70 (m, 1 H)。
(1R,3S)-3-丁基-6-甲氧基-1-(吡啶-2-基)-1,2,3,4-四氫異喹啉:在-78℃及氮氣下,將(S)-3-丁基-6-甲氧基-1-(吡啶-2-基)-3,4-二氫異喹啉(0.1 g, 0.34 mmol, 1當量)於無水THF (4 mL)中之溶液逐滴添加至於THF中之1M氫化鋰鋁(3.4 mL, 3.40 mmol, 10當量)及三甲基鋁(2M甲苯溶液) (0.85 mL, 1.70 mmol, 5當量)之混合物中。將懸浮液在-78℃下攪拌1 h,並經1 h升溫至0℃。TLC (於DCM中之5% MeOH)顯示反應已完成。使用氯化鈉飽和水溶液(4 mL)淬滅反應混合物,隨後使用EtOAc (15 mL)稀釋且濾除沈澱物。最後,藉由無水硫酸鈉乾燥濾液,過濾並在減壓下蒸發。藉由矽膠急速層析使用乙酸乙酯之己烷溶液作為洗脫劑純化殘餘物以得到(1R,3S)-3-丁基-6-甲氧基-1-(吡啶-2-基)-1,2,3,4-四氫異喹啉。LC-MS (m/z) = 311.3 [M+H]+
1-((1R,3S)-3-丁基-6-甲氧基-1-(吡啶-2-基)-3,4-二氫異喹啉-2(1H)-基)-3-(三甲基矽烷基)丙-2-炔-1-酮:第一步驟:在室溫下,向3-(三甲基矽烷基)丙炔酸(0.10 g, 0.709 mmol, 3.0當量)於DMF (0.0007 mL 0.009 mmol, 0.04當量)中之溶液中添加草醯氯(0.02 mL, 0.26 mmol, 1.1當量)且將反應液攪拌30分鐘。此後,在減壓下濃縮反應混合物以產生3-(三甲基矽烷基)丙炔醯氯。此醯氯未經進一步純化即用於下一步驟中。
第二步驟:在0℃下,向(1S,3S)-3-丁基-6-甲氧基-1-(2-甲基吡啶-4-基)-1,2,3,4-四氫異喹啉(0.07 g, 0.236 mmol, 1.0當量)於乙腈(3.0 mL)中之溶液中添加碳酸氫鈉(0.149 g, 1.77 mmol, 7.5當量)。在攪拌5分鐘之後,向上述反應物質中添加3-(三甲基矽烷基)丙炔醯氯於乙腈(2.0 mL)中之溶液。將所得混合物在0℃下攪拌15 min,藉由TLC (於正己烷中之60%乙酸乙酯)監測反應進展。此後,使用EtOAc (15 mL)及水(5 mL)稀釋反應物質。分離有機層,使用鹽水溶液(5 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物。此粗產物未經任何進一步純化即用於下一步驟中。LC-MS (m/z) = 421.3 [M+H]+
1-((1R,3S)-3-丁基-6-甲氧基-1-(吡啶-2-基)-3,4-二氫異喹啉-2(1H)-基)丙-2-炔-1-酮:在-78℃下,向1-((1R,3S)-3-丁基-6-甲氧基-1-(吡啶-2-基)-3,4-二氫異喹啉-2(1H)-基)-3-(三甲基矽烷基)丙-2-炔-1-酮(0.07 g, 0.166 mmol, 1.0當量)於THF (4.0 mL)中之溶液中添加TBAF (於THF中之1M溶液) (0.183 mL, 0.183 mmol, 1.1當量)。將此反應混合物在-78℃下攪拌15分鐘。藉由TLC (於正己烷中之50%乙酸乙酯)監測反應進展。此後,使用NaHCO3
飽和水溶液(2 mL)淬滅反應混合物且使用乙酸乙酯(25 mL)萃取產物。藉由無水硫酸鈉乾燥有機層,過濾並在減壓下濃縮。藉由製備型TLC使用於正己烷中之50%乙酸乙酯作為洗脫劑純化所獲得粗產物以得到1-((1R,3S)-3-丁基-6-甲氧基-1-(吡啶-2-基)-3,4-二氫異喹啉-2(1H)-基)丙-2-炔-1-酮。LC-MS (m/z) = 349.4 [M+H]+ 1
H NMR (400MHz, DMSO-d6
): δ 0.80 (t,J
= 6.8 Hz, 3 H), 1.20 - 1.44 (m, 6 H), 2.71 - 2.83 (m, 1 H), 3.03 (s, 1 H), 3.43 - 3.47 (m, 1 H), 3.67 - 3.69 (m, 3 H), 4.56 - 4.70 (m, 2 H), 6.01 - 6.28 (m, 2 H), 6.72 - 6.79 (m, 1 H), 7.09 - 7.18 (m, 1 H), 7.35 - 7.44 (m, 1 H), 7.56 - 7.69 (m, 1 H), 8.32 - 8.42 (m, 1H)。
程序 22 :化合物 50 之合成 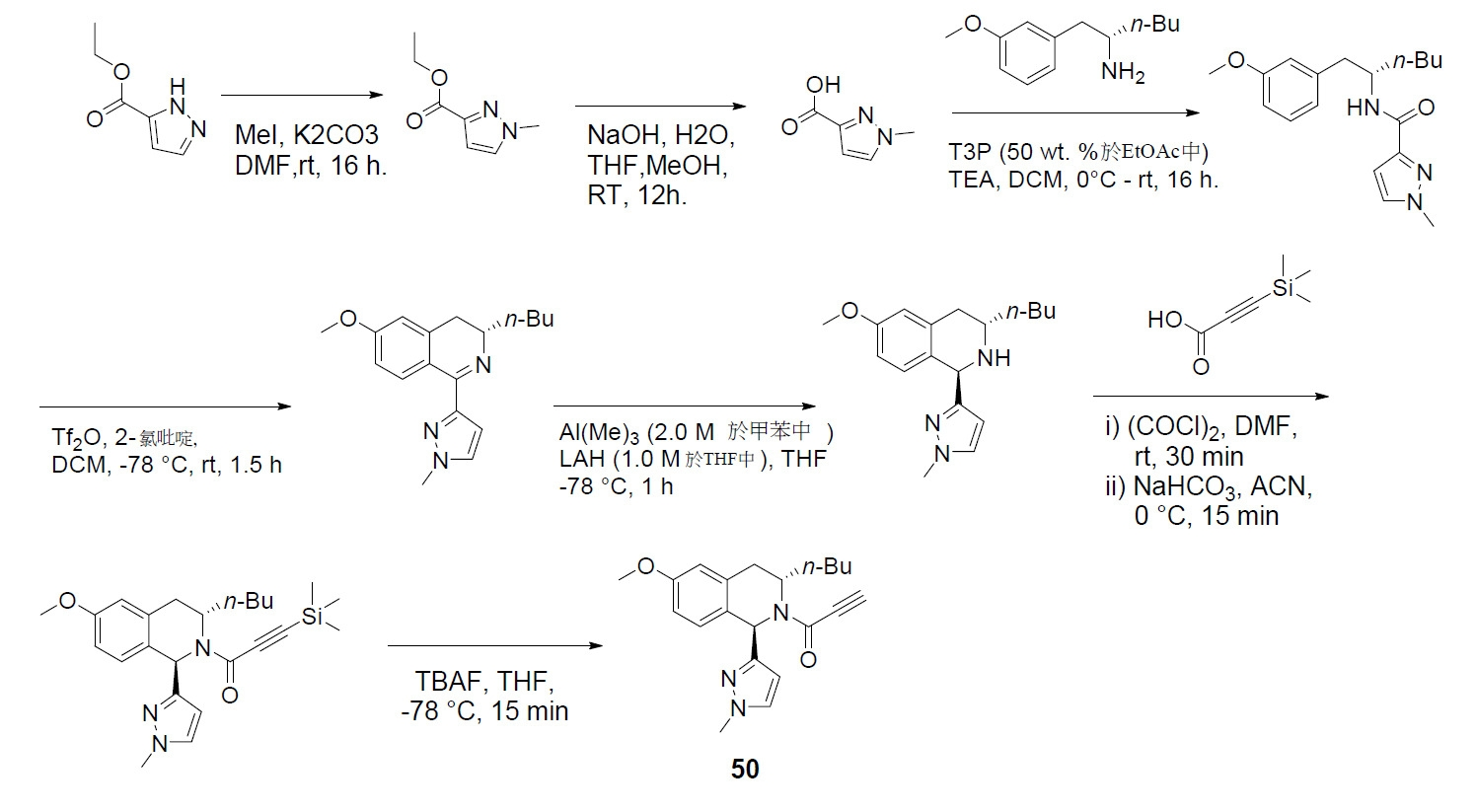 1-甲基-1H-吡唑-3-甲酸乙酯:在室溫下,向1H-吡唑-5-甲酸乙酯(4.0 g, 28.5 mmol, 1.0當量)於DMF (40 mL)中之溶液中添加碳酸鉀(7.89 g, 57.1 mmol, 2當量)及碘甲烷(3.55 mL, 57.1 mmol, 2當量)。將反應混合物在室溫下攪拌16 h。藉由TLC (於正己烷中之30%乙酸乙酯)監測反應進展 。此後, 使用冰冷水(30 mL)稀釋反應混合物並使用乙酸乙酯(100 mL)萃取。分離有機層,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以得到1-甲基-1H-吡唑-3-甲酸乙酯之粗產物。LCMS (ES) m/z = 155.1 [M+H]+ 1
H NMR (400 MHz, CDCl3
) δ ppm 1.38 (t,J
= 7.2 Hz, 3 H), 3.97 (s, 3 H), 4.38 (q,J
= 6.8 Hz, 2 H), 6.79 (d,J
= 1.6 Hz, 1 H), 7.37 (d,J
= 1.2 Hz, 1 H)。
1-甲基-1H-吡唑-3-甲酸:向 1-甲基-1H-吡唑-3-甲酸乙酯(2.0 g, 13.0 mmol, 1當量)於THF (10 mL)及甲醇(10 mL)中之溶液中添加2 M氫氧化鈉溶液(15 mL)。將反應混合物在室溫下攪拌16 h。藉由TLC (於正己烷中之70%乙酸乙酯)監測反應進展。在反應完成之後,濃縮反應混合物以去除溶劑。使用1N HCl溶液(pH 3)酸化反應混合物並使用乙酸乙酯(120 mL)萃取。藉由無水硫酸鈉乾燥有機層,過濾並在減壓下濃縮以產生粗製1-甲基-1H-吡唑-5-甲酸。LCMS (m/z) = 127.1 [M+H]+ 1
H NMR (400 MHz, DMSO-d6
) δ ppm 3.87 (s, 3 H), 6.63 - 6.64 (m, 1 H), 7.75 (s, 1 H), 12.54 (s,1 H)。
(S)-N-(1-(3-甲氧基苯基)己烷-2-基)-1-甲基-1H-吡唑-3-甲醯胺:向(2S)-1-(3-甲氧基苯基)己烷-2-胺(1.6 g, 7.72 mmol, 1.0當量)於DCM (10 mL)中之溶液中添加1-甲基-1H-吡唑-3-甲酸(1.17 g, 9.26 mmol, 1.2當量)及三乙胺(4.3 mL, 30.9 mmol, 4.0當量)。在0℃下,向此混合物中添加丙烷膦酸酐(7.37 mL, 11.6 mmol, 1.5當量)。在添加之後,將反應混合物在室溫下攪拌16 h。藉由TLC (於正己烷中之70%乙酸乙酯)監測反應進展。在反應完成之後,使用NaHCO3
水溶液(15 mL)淬滅反應混合物並使用DCM (70 mL)萃取。藉由無水Na2
SO4
乾燥有機層,過濾並在減壓下濃縮以得到粗製N-[(2S)-1-(3-甲氧基苯基)己烷-2-基]-1-甲基-1H-吡唑-5-甲醯胺。LCMS (m/z) = 316.2 [M+H]+
(3S)-3-丁基-6-甲氧基-1-(1-甲基-1H-吡唑-3-基)-3,4-二氫異喹啉:在-78℃下,經由注射器經10分鐘時段將三氟甲烷磺酸酐(3.19 mL, 19.0 mmol, 2.0當量)添加至N-[(2S)-1-(3-甲氧基苯基)己烷-2-基]-1-甲基-1H-吡唑-3-甲醯胺(3.0 g, 9.51 mmol, 1.0當量)及2-氯吡啶(1.8 mL, 19.0 mmol, 2.0當量)於二氯甲烷(20 mL)中之經攪拌混合物中。在5 min之後,將反應混合物置於冰-水浴中並升溫至0℃。在5 min之後,將所得溶液升溫至23℃。藉由TLC (於正己烷中之70%乙酸乙酯)監測反應進展。在15分鐘之後,將氫氧化鈉水溶液(12 mL, 1N)在0℃下添加至反應混合物中以中和三氟甲烷磺酸鹽。添加二氯甲烷(50 mL)以稀釋混合物且分離各層。使用鹽水(10 mL)洗滌有機層,藉由無水硫酸鈉乾燥,並過濾。在減壓下去除揮發物以得到粗產物。藉由急速層析使用乙酸乙酯之己烷溶液作為洗脫劑純化所獲得粗產物以得到期望產物(3S)-3-丁基-6-甲氧基-1-(1-甲基-1H-吡唑-3-基)-3,4-二氫異喹啉。LCMS (m/z) = 298.1 [M+H]+ 1
H NMR (400 MHz, CDCl3
) δ ppm: 0.88 (t,J
= 6.4 Hz, 3 H), 1.24 - 1.44 (m, 4 H), 1.54 - 2.03 (m, 2 H), 2.76 - 2.88 (m, 1 H), 3.01 - 3.20 (m, 1 H), 3.92 (s, 3 H), 4.04 (s, 4H), 6.85 (d,J
= 1.2Hz, 1 H), 6.91 (d,J
= 8.8 Hz, 1 H), 7.10 (s, 1 H), 7.56 (s, 1 H), 8.13 (d,J
= 8.8 Hz, 1 H)。
(1R,3S)-3-丁基-6-甲氧基-1-(1-甲基-1H-吡唑-3-基)-1,2,3,4-四氫異喹啉:在-78℃下,向三甲基鋁(7.82 mL, 15.6 mmol, 5當量)於四氫呋喃(8 mL)中之溶液中添加氫化鋰鋁(31.3 mL, 31.3 mmol, 10當量),隨後添加於THF (3 mL)中之(3S)-3-丁基-6-甲氧基-1-(1-甲基-1H-吡唑-3-基)-3,4-二氫異喹啉(0.93 g, 3.13 mmol, 1當量)。將反應液在-78℃下攪拌1 h。藉由TLC (於正己烷中之70%乙酸乙酯)監測反應進展。在1 h之後,反應完成。使用鹽水溶液(10 mL)在0℃下淬滅反應混合物,使用乙酸乙酯(15 mL)稀釋。然後經由矽藻土床過濾並使用乙酸乙酯(20 mL)洗滌。自水層分離有機層並在減壓下濃縮以得到粗製物。藉由急速管柱層析在矽膠上使用具有增加極性之於DCM中之2 - 3% MeOH作為溶劑純化粗製物以得到(1R,3S)-3-丁基-6-甲氧基-1-(1-甲基-1H-吡唑-3-基)-1,2,3,4-四氫異喹啉(順式及反式之混合物)。LCMS (m/z) = 300.2 [M+H]+
1-[(3S)-3-丁基-6-甲氧基-1-(1-甲基-1H-吡唑-3-基)-1,2,3,4-四氫異喹啉-2-基]-3-(三甲基矽烷基)丙-2-炔-1-酮:第一步驟:在室溫下,向3-(三甲基矽烷基)丙炔酸(0.2 g, 1.41 mmol, 1當量)於DMF (0.004 mL, 0.056 mmol, 0.04當量)中之溶液中添加草醯氯(0.13 mL, 1.55 mmol, 1.1當量)且將反應液攪拌30分鐘。此後,在減壓下濃縮反應混合物以產生3-(三甲基矽烷基)丙炔醯氯。此醯氯未經進一步純化即用於下一步驟中。
第二步驟:在0℃下,向(1S,3S)-3-丁基-6-甲氧基-1-(2-甲基吡啶-4-基)-1,2,3,4-四氫異喹啉(0.42 g, 1.40 mmol, 1當量)於乙腈(3.0 mL)中之溶液中添加碳酸氫鈉(0.88 g, 10.5 mmol, 7.5當量)。在攪拌5分鐘之後,向上述反應物質中添加3-(三甲基矽烷基)丙炔醯氯於乙腈(2 mL)中之溶液。將所得混合物在0℃下攪拌15 min,藉由TLC (於正己烷中之60%乙酸乙酯)監測反應進展。此後,使用EtOAc (20 mL)及水(5 mL)稀釋反應物質。分離有機層,使用鹽水溶液(5 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物。此粗產物未經任何進一步純化即用於下一步驟中。LCMS (m/z) = 424.3 [M+H]+
。
1-[(1R,3S)-3-丁基-6-甲氧基-1-(1-甲基-1H-吡唑-3-基)-1,2,3,4-四氫異喹啉-2-基]丙-2-炔-1-酮:在-78℃下,向-[(3S)-3-丁基-6-甲氧基-1-(1-甲基-1H-吡唑-3-基)-1,2,3,4-四氫異喹啉-2-基]-3-(三甲基矽烷基)丙-2-炔-1-酮(0.5 g, 1.18 mmol, 1當量)於THF (8.0 mL)中之溶液中添加TBAF (於THF中之1M溶液,1.30 mL, 1.30 mmol, 1.1當量)。將此反應混合物在-78℃下攪拌15分鐘。藉由TLC (於正己烷中之50%乙酸乙酯)監測反應進展。此後,使用NaHCO3
飽和水溶液(2 mL)淬滅反應混合物且使用乙酸乙酯(25 mL)萃取產物。藉由無水硫酸鈉乾燥有機層,過濾並在減壓下濃縮。藉由製備型TLC使用於正己烷中之50%乙酸乙酯作為洗脫劑純化所獲得粗產物以得到1-[(1R,3S)-3-丁基-6-甲氧基-1-(1-甲基-1H-吡唑-3-基)-1,2,3,4-四氫異喹啉-2-基]丙-2-炔-1-酮。LCMS (m/z) = 352.4 [M+H]+
。1
H NMR (400MHz, DMSO-d6
): δ 0.79 (t,J
= 6.0 Hz, 3 H), 0.94 - 1.43 (m, 6 H), 2.71 - 2.83 (m, 1 H), 2.93 - 2.98 (m, 0.5 H), 3.25 - 3.26 (m, 0.5 H), 3.64 - 3.70 (m, 6 H), 4.30 - 4.56 (m, 2 H), 5.91 - 6.20 (m, 2 H), 6.73 - 6.79 (m, 2 H), 7.26 - 7.47 (m, 2 H)。
1-甲基-1H-吡唑-3-甲酸乙酯:在室溫下,向1H-吡唑-5-甲酸乙酯(4.0 g, 28.5 mmol, 1.0當量)於DMF (40 mL)中之溶液中添加碳酸鉀(7.89 g, 57.1 mmol, 2當量)及碘甲烷(3.55 mL, 57.1 mmol, 2當量)。將反應混合物在室溫下攪拌16 h。藉由TLC (於正己烷中之30%乙酸乙酯)監測反應進展 。此後, 使用冰冷水(30 mL)稀釋反應混合物並使用乙酸乙酯(100 mL)萃取。分離有機層,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以得到1-甲基-1H-吡唑-3-甲酸乙酯之粗產物。LCMS (ES) m/z = 155.1 [M+H]+ 1
H NMR (400 MHz, CDCl3
) δ ppm 1.38 (t,J
= 7.2 Hz, 3 H), 3.97 (s, 3 H), 4.38 (q,J
= 6.8 Hz, 2 H), 6.79 (d,J
= 1.6 Hz, 1 H), 7.37 (d,J
= 1.2 Hz, 1 H)。
1-甲基-1H-吡唑-3-甲酸:向 1-甲基-1H-吡唑-3-甲酸乙酯(2.0 g, 13.0 mmol, 1當量)於THF (10 mL)及甲醇(10 mL)中之溶液中添加2 M氫氧化鈉溶液(15 mL)。將反應混合物在室溫下攪拌16 h。藉由TLC (於正己烷中之70%乙酸乙酯)監測反應進展。在反應完成之後,濃縮反應混合物以去除溶劑。使用1N HCl溶液(pH 3)酸化反應混合物並使用乙酸乙酯(120 mL)萃取。藉由無水硫酸鈉乾燥有機層,過濾並在減壓下濃縮以產生粗製1-甲基-1H-吡唑-5-甲酸。LCMS (m/z) = 127.1 [M+H]+ 1
H NMR (400 MHz, DMSO-d6
) δ ppm 3.87 (s, 3 H), 6.63 - 6.64 (m, 1 H), 7.75 (s, 1 H), 12.54 (s,1 H)。
(S)-N-(1-(3-甲氧基苯基)己烷-2-基)-1-甲基-1H-吡唑-3-甲醯胺:向(2S)-1-(3-甲氧基苯基)己烷-2-胺(1.6 g, 7.72 mmol, 1.0當量)於DCM (10 mL)中之溶液中添加1-甲基-1H-吡唑-3-甲酸(1.17 g, 9.26 mmol, 1.2當量)及三乙胺(4.3 mL, 30.9 mmol, 4.0當量)。在0℃下,向此混合物中添加丙烷膦酸酐(7.37 mL, 11.6 mmol, 1.5當量)。在添加之後,將反應混合物在室溫下攪拌16 h。藉由TLC (於正己烷中之70%乙酸乙酯)監測反應進展。在反應完成之後,使用NaHCO3
水溶液(15 mL)淬滅反應混合物並使用DCM (70 mL)萃取。藉由無水Na2
SO4
乾燥有機層,過濾並在減壓下濃縮以得到粗製N-[(2S)-1-(3-甲氧基苯基)己烷-2-基]-1-甲基-1H-吡唑-5-甲醯胺。LCMS (m/z) = 316.2 [M+H]+
(3S)-3-丁基-6-甲氧基-1-(1-甲基-1H-吡唑-3-基)-3,4-二氫異喹啉:在-78℃下,經由注射器經10分鐘時段將三氟甲烷磺酸酐(3.19 mL, 19.0 mmol, 2.0當量)添加至N-[(2S)-1-(3-甲氧基苯基)己烷-2-基]-1-甲基-1H-吡唑-3-甲醯胺(3.0 g, 9.51 mmol, 1.0當量)及2-氯吡啶(1.8 mL, 19.0 mmol, 2.0當量)於二氯甲烷(20 mL)中之經攪拌混合物中。在5 min之後,將反應混合物置於冰-水浴中並升溫至0℃。在5 min之後,將所得溶液升溫至23℃。藉由TLC (於正己烷中之70%乙酸乙酯)監測反應進展。在15分鐘之後,將氫氧化鈉水溶液(12 mL, 1N)在0℃下添加至反應混合物中以中和三氟甲烷磺酸鹽。添加二氯甲烷(50 mL)以稀釋混合物且分離各層。使用鹽水(10 mL)洗滌有機層,藉由無水硫酸鈉乾燥,並過濾。在減壓下去除揮發物以得到粗產物。藉由急速層析使用乙酸乙酯之己烷溶液作為洗脫劑純化所獲得粗產物以得到期望產物(3S)-3-丁基-6-甲氧基-1-(1-甲基-1H-吡唑-3-基)-3,4-二氫異喹啉。LCMS (m/z) = 298.1 [M+H]+ 1
H NMR (400 MHz, CDCl3
) δ ppm: 0.88 (t,J
= 6.4 Hz, 3 H), 1.24 - 1.44 (m, 4 H), 1.54 - 2.03 (m, 2 H), 2.76 - 2.88 (m, 1 H), 3.01 - 3.20 (m, 1 H), 3.92 (s, 3 H), 4.04 (s, 4H), 6.85 (d,J
= 1.2Hz, 1 H), 6.91 (d,J
= 8.8 Hz, 1 H), 7.10 (s, 1 H), 7.56 (s, 1 H), 8.13 (d,J
= 8.8 Hz, 1 H)。
(1R,3S)-3-丁基-6-甲氧基-1-(1-甲基-1H-吡唑-3-基)-1,2,3,4-四氫異喹啉:在-78℃下,向三甲基鋁(7.82 mL, 15.6 mmol, 5當量)於四氫呋喃(8 mL)中之溶液中添加氫化鋰鋁(31.3 mL, 31.3 mmol, 10當量),隨後添加於THF (3 mL)中之(3S)-3-丁基-6-甲氧基-1-(1-甲基-1H-吡唑-3-基)-3,4-二氫異喹啉(0.93 g, 3.13 mmol, 1當量)。將反應液在-78℃下攪拌1 h。藉由TLC (於正己烷中之70%乙酸乙酯)監測反應進展。在1 h之後,反應完成。使用鹽水溶液(10 mL)在0℃下淬滅反應混合物,使用乙酸乙酯(15 mL)稀釋。然後經由矽藻土床過濾並使用乙酸乙酯(20 mL)洗滌。自水層分離有機層並在減壓下濃縮以得到粗製物。藉由急速管柱層析在矽膠上使用具有增加極性之於DCM中之2 - 3% MeOH作為溶劑純化粗製物以得到(1R,3S)-3-丁基-6-甲氧基-1-(1-甲基-1H-吡唑-3-基)-1,2,3,4-四氫異喹啉(順式及反式之混合物)。LCMS (m/z) = 300.2 [M+H]+
1-[(3S)-3-丁基-6-甲氧基-1-(1-甲基-1H-吡唑-3-基)-1,2,3,4-四氫異喹啉-2-基]-3-(三甲基矽烷基)丙-2-炔-1-酮:第一步驟:在室溫下,向3-(三甲基矽烷基)丙炔酸(0.2 g, 1.41 mmol, 1當量)於DMF (0.004 mL, 0.056 mmol, 0.04當量)中之溶液中添加草醯氯(0.13 mL, 1.55 mmol, 1.1當量)且將反應液攪拌30分鐘。此後,在減壓下濃縮反應混合物以產生3-(三甲基矽烷基)丙炔醯氯。此醯氯未經進一步純化即用於下一步驟中。
第二步驟:在0℃下,向(1S,3S)-3-丁基-6-甲氧基-1-(2-甲基吡啶-4-基)-1,2,3,4-四氫異喹啉(0.42 g, 1.40 mmol, 1當量)於乙腈(3.0 mL)中之溶液中添加碳酸氫鈉(0.88 g, 10.5 mmol, 7.5當量)。在攪拌5分鐘之後,向上述反應物質中添加3-(三甲基矽烷基)丙炔醯氯於乙腈(2 mL)中之溶液。將所得混合物在0℃下攪拌15 min,藉由TLC (於正己烷中之60%乙酸乙酯)監測反應進展。此後,使用EtOAc (20 mL)及水(5 mL)稀釋反應物質。分離有機層,使用鹽水溶液(5 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物。此粗產物未經任何進一步純化即用於下一步驟中。LCMS (m/z) = 424.3 [M+H]+
。
1-[(1R,3S)-3-丁基-6-甲氧基-1-(1-甲基-1H-吡唑-3-基)-1,2,3,4-四氫異喹啉-2-基]丙-2-炔-1-酮:在-78℃下,向-[(3S)-3-丁基-6-甲氧基-1-(1-甲基-1H-吡唑-3-基)-1,2,3,4-四氫異喹啉-2-基]-3-(三甲基矽烷基)丙-2-炔-1-酮(0.5 g, 1.18 mmol, 1當量)於THF (8.0 mL)中之溶液中添加TBAF (於THF中之1M溶液,1.30 mL, 1.30 mmol, 1.1當量)。將此反應混合物在-78℃下攪拌15分鐘。藉由TLC (於正己烷中之50%乙酸乙酯)監測反應進展。此後,使用NaHCO3
飽和水溶液(2 mL)淬滅反應混合物且使用乙酸乙酯(25 mL)萃取產物。藉由無水硫酸鈉乾燥有機層,過濾並在減壓下濃縮。藉由製備型TLC使用於正己烷中之50%乙酸乙酯作為洗脫劑純化所獲得粗產物以得到1-[(1R,3S)-3-丁基-6-甲氧基-1-(1-甲基-1H-吡唑-3-基)-1,2,3,4-四氫異喹啉-2-基]丙-2-炔-1-酮。LCMS (m/z) = 352.4 [M+H]+
。1
H NMR (400MHz, DMSO-d6
): δ 0.79 (t,J
= 6.0 Hz, 3 H), 0.94 - 1.43 (m, 6 H), 2.71 - 2.83 (m, 1 H), 2.93 - 2.98 (m, 0.5 H), 3.25 - 3.26 (m, 0.5 H), 3.64 - 3.70 (m, 6 H), 4.30 - 4.56 (m, 2 H), 5.91 - 6.20 (m, 2 H), 6.73 - 6.79 (m, 2 H), 7.26 - 7.47 (m, 2 H)。
程序 23 :化合物 76 之合成 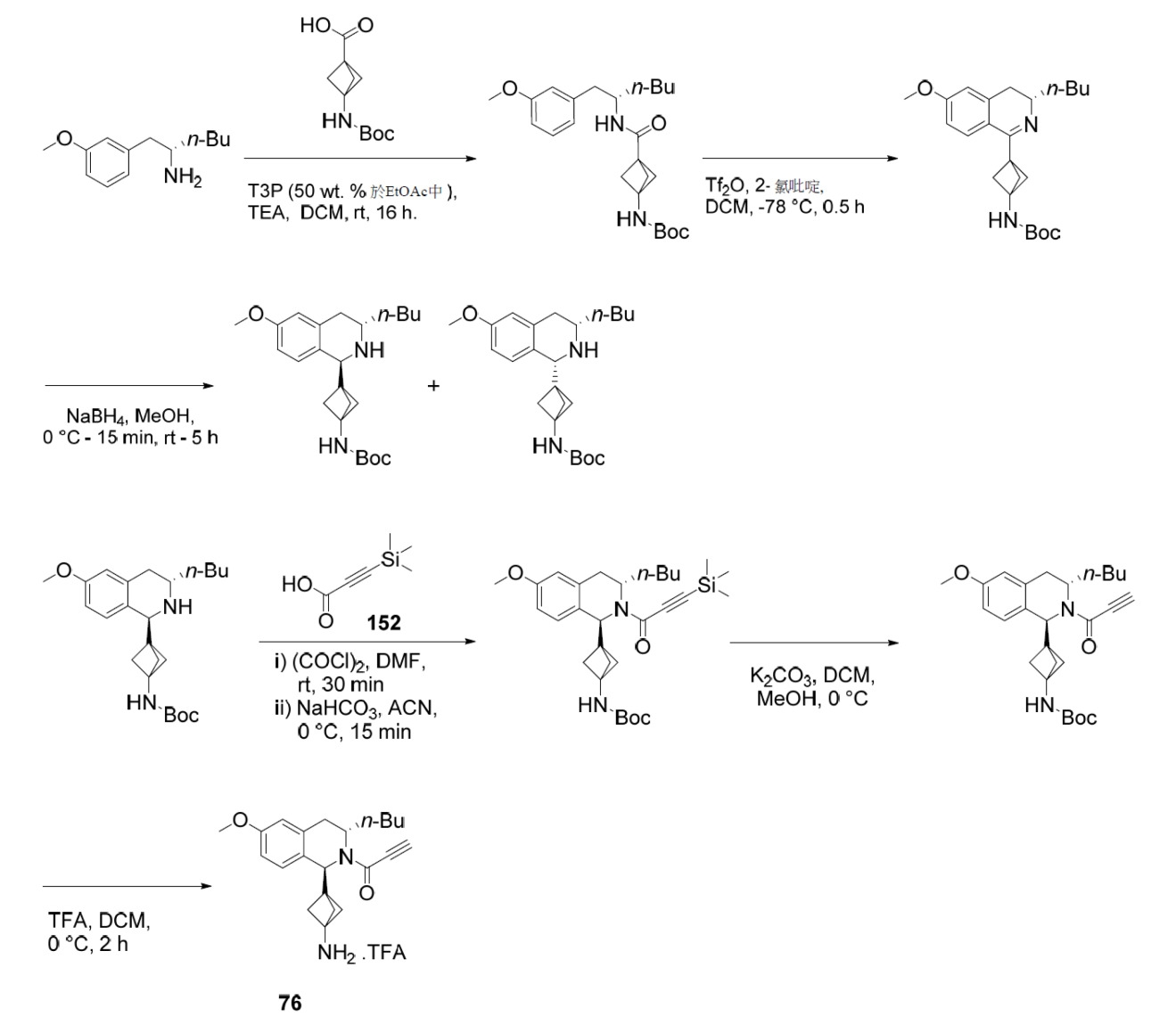 (S)-(3-((1-(3-甲氧基苯基)己烷-2-基)胺甲醯基)雙環[1.1.1]戊烷-1-基)胺基甲酸第三丁基酯:向3-((第三丁氧基羰基)胺基)雙環[1.1.1]戊烷-1-甲酸(2.19 g, 9.62 mmol, 1.05當量)於DCM (10.0 mL)中之溶液中添加TEA (3.71 g, 36.7 mmol, 4當量),攪拌5 min且然後在0℃下添加T3P (50 wt.%於EtOAc中) (4.37 g, 13.70 mmol, 1.5當量)並再攪拌30 min。然後在0℃下向反應混合物中添加(S)-1-(3-甲氧基苯基)己烷-2-胺(1.90 g, 9.16 mmol, 1當量)於DCM (5.0 mL)中之溶液且然後將反應混合物在室溫下攪拌16 h。藉由TLC (於正己烷中之40%乙酸乙酯)監測反應進展。此後,使用DCM (75 mL)稀釋反應混合物,分離有機層,使用碳酸氫鈉飽和水溶液(2 × 15 mL)、水(15 mL)、鹽水溶液(15 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物。所得殘餘物未經任何進一步純化即用於下一反應中。LCMS (ES) m/z = 417.3 [M+H]+。1
H NMR (400 MHz, CDCl3
) δ ppm 0.86 (bs, 3 H), 1.25 - 1.28 (m, 6 H), 1.43 (s, 9 H), 2.17 (s, 6 H), 2.68 - 2.81 (m, 2 H), 3.86 (s, 3 H), 4.11 - 4.13 (m 1 H), 4.93 (bs, 1 H), 5.16 (d,J
= 8.4 Hz, 1 H), 6.67 - 6.76 (m, 3 H), 7.19 (t,J
= 8.0 Hz, 1 H)。
(S)-(3-(3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基)雙環[1.1.1]戊烷-1-基)胺基甲酸第三丁基酯:在-78℃下,經由注射器向(S)-(3-((1-(3-甲氧基苯基)己烷-2-基)胺甲醯基)雙環[1.1.1]戊烷-1-基)胺基甲酸第三丁基酯(3.2 g, 7.68 mmol, 1當量)及2-氯吡啶(1.31 g, 11.50 mmol, 1.5當量)於二氯甲烷(32 mL)中之經攪拌溶液中緩慢逐滴添加三氟甲烷磺酸酐(1.94 mL, 11.50 mmol, 1.5當量)。在10分鐘之後,引入氫氧化鈉水溶液(10 mL, 1N)以中和三氟甲烷磺酸鹽。添加二氯甲烷(75 mL)以稀釋混合物且分離各層。使用鹽水(15 mL)洗滌有機層,藉由無水硫酸鈉乾燥,並過濾。在減壓下去除揮發物以得到粗產物。所得殘餘物未經任何進一步純化即用於下一反應中。LCMS (ES) m/z = 399.3 [M+H]+。
(3-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)雙環[1.1.1]戊烷-1-基)胺基甲酸第三丁基酯及(3-((1R,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)雙環[1.1.1]戊烷-1-基)胺基甲酸第三丁基酯:在0℃下向(S)-(3-(3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基)雙環[1.1.1]戊烷-1-基)胺基甲酸第三丁基酯(3.0 g, 7.53 mmol, 1當量)於甲醇(30 mL)中之溶液中逐份添加硼氫化鈉(0.854 mg, 22.60 mmol, 3當量)。將懸浮液在室溫下攪拌1 h。藉由TLC (於正己烷中之50% EtOAc)監測反應進展。此後,在減壓下濃縮反應混合物且使用EtOAc (100 mL)及水(20 mL)稀釋所獲得粗製物。分離有機層,使用鹽水溶液(20 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物。藉由急速管柱層析在矽膠上使用於正己烷中之乙酸乙酯作為洗脫劑純化所獲得粗產物。使用於正己烷中之30 - 70%乙酸乙酯洗脫期望產物。合併含有產物之部分並在減壓下濃縮以得到(3-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)雙環[1.1.1]戊烷-1-基)胺基甲酸第三丁基酯(1,3-反式異構體)及(3-((1R,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)雙環[1.1.1]戊烷-1-基)胺基甲酸第三丁基酯(1,3-順式異構體)。
順式異構體:LCMS (ES) m/z = 401.3 [M+H]+。1
H NMR (400 MHz, CDCl3
) δ ppm 0.92 (t,J
= 6.8 Hz, 3 H), 1.23 - 1.29 (m, 3 H), 1.36 - 1.37 (m, 3 H), 1.43 (s, 9 H), 1.93 - 2.04 (m, 6 H), 2.37 - 2.44 (m, 1 H), 2.64 - 2.68 (m, 1 H), 2.76 (bs, 1 H), 3.77 (s, 3 H), 4.20 (s, 1 H), 4.91 (bs, 1 H), 6.59 (s, 1 H), 6.68 (d,J
= 8.4 Hz, 1 H), 7.05 (d,J
= 8.8 Hz, 1 H)。
反式異構體:LCMS (ES) m/z = 401.3 [M+H]+。1
H NMR (400 MHz, CDCl3
) δ ppm 0.93 - 0.94 (m, 3 H), 1.25 - 1.28 (m, 5 H), 1.36 - 1.47 (m, 10 H), 1.93 - 1.98 (m, 6 H), 2.43 - 2.50 (m, 1 H), 2.69 - 2.72 (m, 1 H), 3.13 (bs, 1 H), 3.77 (s, 3 H), 4.13 (s, 1 H), 4.90 (bs, 1 H), 6.60 (s, 1 H), 6.65 (d,J
= 8.4 Hz, 1 H), 6.89 (d,J
= 8.8 Hz, 1 H)。
在室溫下,向(3-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)雙環[1.1.1]戊烷-1-基)胺基甲酸第三丁基酯(0.3 g, 0.749 mmol, 1當量)於THF (4 mL)中之溶液中添加QuadraSil® AP (標記程度:1.5-2.0 mmol/g載量(480 mg),藉由所需QuadraSil®之量(針對1.0當量) = mmol先前步驟中所使用之銅觸媒/mmol清除劑載量來計算)並攪拌1 h。此後,藉由通過過濾紙來去除固體部分且在減壓下濃縮濾液以得到(3-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)雙環[1.1.1]戊烷-1-基)胺基甲酸第三丁基1酯。LCMS (ES) m/z = 401.3 [M+H]+。1
H NMR (400 MHz, CDCl3
) δ ppm 0.93 - 0.94 (m, 3 H), 1.25 - 1.28 (m, 5 H), 1.36 - 1.47 (m, 10 H), 1.93 - 1.98 (m, 6 H), 2.43 - 2.50 (m, 1 H), 2.69 - 2.72 (m, 1 H), 3.13 (bs, 1 H), 3.77 (s, 3 H), 4.13 (s, 1 H), 4.90 (bs, 1 H), 6.60 (s, 1 H), 6.65 (d,J
= 8.4 Hz, 1 H), 6.89 (d,J
= 8.8 Hz, 1 H)。
(3-((1S,3S)-3-丁基-6-甲氧基-2-(3-(三甲基矽烷基)丙炔醯基)-1,2,3,4-四氫異喹啉-1-基)雙環[1.1.1]戊烷-1-基)胺基甲酸第三丁基酯:第一步驟:在室溫下,向3-(三甲基矽烷基)丙炔酸(155 mg, 1.09 mmol, 1.0當量)於DMF (3.2 mg, 0.04 mmol, 0.04當量)中之溶液中添加草醯氯(152 mg, 1.2 mmol, 1.1當量)並攪拌30分鐘。此後,在減壓下濃縮反應混合物以得到3-(三甲基矽烷基)丙炔醯氯。此醯氯未經任何進一步純化即用於下一步驟中。
第二步驟:在0℃下,向(3-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)雙環[1.1.1]戊烷-1-基)胺基甲酸第三丁基酯(285 mg, 0.711 mmol, 1.0當量)於乙腈(3.0 mL)中之溶液中添加碳酸氫鈉(448 mg, 5.34 mmol, 7.5當量)。在攪拌5分鐘之後,在0℃下向上述反應物質中添加3-(三甲基矽烷基)丙炔醯氯(171 mg, 1.07 mmol, 1.5當量)於乙腈(1 mL)中之溶液。將所得混合物在0℃下攪拌15 min,藉由TLC (於正己烷中之50%乙酸乙酯)監測反應進展。此後,使用EtOAc (40 mL)及水(10 mL)稀釋反應物質。分離有機層,使用鹽水溶液(7 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物。藉由製備型TLC藉由使用於正己烷中之25% EtOAc作為移動相純化所獲得粗產物以得到(3-((1S,3S)-3-丁基-6-甲氧基-2-(3-(三甲基矽烷基)丙炔醯基)-1,2,3,4-四氫異喹啉-1-基)雙環[1.1.1]戊烷-1-基)胺基甲酸第三丁基酯。LCMS (ES) m/z = 525.3 [M+H]+。1
H NMR (400 MHz, CDCl3
) δ ppm 0.25 - 0.28 (m, 9 H), 0.80 - 0.84 (m, 3 H), 1.16 - 1.26 (m, 6 H), 1.39 (s, 9 H), 1.73 - 1.82 (m, 6 H), 2.67 - 2.71 (m, 1 H), 2.92 - 2.96 (m, 1 H), 3.02 - 3.11 (m, 1 H), 3.80 - 3.81 (m, 3 H), 4.44 (bs, 1 H), 5.28 (s, 1 H), 6.67 - 6.74 (m, 2 H), 6.92 (d,J
= 8.4 Hz, 1 H)。
(3-((1S,3S)-3-丁基-6-甲氧基-2-丙炔醯基-1,2,3,4-四氫異喹啉-1-基)雙環[1.1.1]戊烷-1-基)胺基甲酸第三丁基酯:在0℃下,向(3-((1S,3S)-3-丁基-6-甲氧基-2-(3-(三甲基矽烷基)丙炔醯基)-1,2,3,4-四氫異喹啉-1-基)雙環[1.1.1]戊烷-1-基)胺基甲酸第三丁基酯(145 mg, 0.276 mmol, 1當量)於DCM (16 mL) / MeOH (3.3 mL)中之溶液中添加K2
CO3
(232 mg, 1.66 mmol, 6.0當量)。將反應混合物在0℃下攪拌3 hr以得到白色溶液。藉由TLC (於正己烷中之30 %乙酸乙酯)監測反應進展。此後,使用DCM (50 mL)及水(10 mL)稀釋反應混合物。分離有機層,藉由Na2
SO4
乾燥並在減壓下濃縮以得到3-((1S,3S)-3-丁基-6-甲氧基-2-丙炔醯基-1,2,3,4-四氫異喹啉-1-基)雙環[1.1.1]戊烷-1-基)胺基甲酸第三丁基酯。所得殘餘物未經任何進一步純化即用於下一反應中。LCMS (ES) m/z = 397.3 [M+H]+ - 56。1
H NMR (400 MHz, CDCl3
) δ ppm 0.80 - 0.82 (m, 3 H), 1.18 - 1.25 (m, 6 H), 1.38 - 1.39 (m, 9 H), 1.75 - 1.84 (m, 6 H), 2.68 - 2.72 (m, 1 H), 2.92 - 3.11 (m, 2 H), 3.80 - 3.81 (m, 3 H), 4.45 (bs, 1 H), 4.84 (s, 1 H), 5.27 (s, 1 H), 6.67 - 6.73 (m, 2 H), 6.92 (d,J
= 8.4 Hz, 1 H)。
1-((1S,3S)-1-(3-胺基雙環[1.1.1]戊烷-1-基)-3-丁基-6-甲氧基-3,4-二氫異喹啉-2(1H)-基)丙-2-炔-1-酮:在0℃下,向(3-((1S,3S)-3-丁基-6-甲氧基-2-丙炔醯基-1,2,3,4-四氫異喹啉-1-基)雙環[1.1.1]戊烷-1-基)胺基甲酸第三丁基酯(120 mg, 0.265 mmol, 1.0當量)於DCM (10 mL)中之溶液中添加TFA (2.42 g, 21.2 mmol, 80當量)。將混合物在0℃下攪拌1.5 hr。LCMS顯示反應已完成。然後,在減壓下濃縮反應混合物。將所獲得殘餘物溶於水(5 mL)中,隨後凍乾以得到1-((1S,3S)-1-(3-胺基雙環[1.1.1]戊烷-1-基)-3-丁基-6-甲氧基-3,4-二氫異喹啉-2(1H)-基)丙-2-炔-1-酮。 LCMS (ES) m/z = 353.3 [M+H]+。1
H NMR (400 MHz, DMSO-d6
) δ ppm 0.77 (bs, 3 H), 0.88 - 0.95 (m, 1 H), 1.16 - 1.17 (m, 4 H), 1.34 (bs, 1 H), 1.58 - 1.77 (m, 6 H), 2.78 - 2.89 (m, 2 H), 3.73 (s, 3 H), 4.32 - 4.43 (m, 1 H), 4.57 - 4.62 (m, 1 H), 5.19 - 5.29 (m, 1 H), 6.79 (d,J
= 8.0 Hz, 1 H), 6.87 (s, 1 H), 6.05 - 7.14 (m, 1 H), 8.37 - 8.44 (m, 3 H)。
(S)-(3-((1-(3-甲氧基苯基)己烷-2-基)胺甲醯基)雙環[1.1.1]戊烷-1-基)胺基甲酸第三丁基酯:向3-((第三丁氧基羰基)胺基)雙環[1.1.1]戊烷-1-甲酸(2.19 g, 9.62 mmol, 1.05當量)於DCM (10.0 mL)中之溶液中添加TEA (3.71 g, 36.7 mmol, 4當量),攪拌5 min且然後在0℃下添加T3P (50 wt.%於EtOAc中) (4.37 g, 13.70 mmol, 1.5當量)並再攪拌30 min。然後在0℃下向反應混合物中添加(S)-1-(3-甲氧基苯基)己烷-2-胺(1.90 g, 9.16 mmol, 1當量)於DCM (5.0 mL)中之溶液且然後將反應混合物在室溫下攪拌16 h。藉由TLC (於正己烷中之40%乙酸乙酯)監測反應進展。此後,使用DCM (75 mL)稀釋反應混合物,分離有機層,使用碳酸氫鈉飽和水溶液(2 × 15 mL)、水(15 mL)、鹽水溶液(15 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物。所得殘餘物未經任何進一步純化即用於下一反應中。LCMS (ES) m/z = 417.3 [M+H]+。1
H NMR (400 MHz, CDCl3
) δ ppm 0.86 (bs, 3 H), 1.25 - 1.28 (m, 6 H), 1.43 (s, 9 H), 2.17 (s, 6 H), 2.68 - 2.81 (m, 2 H), 3.86 (s, 3 H), 4.11 - 4.13 (m 1 H), 4.93 (bs, 1 H), 5.16 (d,J
= 8.4 Hz, 1 H), 6.67 - 6.76 (m, 3 H), 7.19 (t,J
= 8.0 Hz, 1 H)。
(S)-(3-(3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基)雙環[1.1.1]戊烷-1-基)胺基甲酸第三丁基酯:在-78℃下,經由注射器向(S)-(3-((1-(3-甲氧基苯基)己烷-2-基)胺甲醯基)雙環[1.1.1]戊烷-1-基)胺基甲酸第三丁基酯(3.2 g, 7.68 mmol, 1當量)及2-氯吡啶(1.31 g, 11.50 mmol, 1.5當量)於二氯甲烷(32 mL)中之經攪拌溶液中緩慢逐滴添加三氟甲烷磺酸酐(1.94 mL, 11.50 mmol, 1.5當量)。在10分鐘之後,引入氫氧化鈉水溶液(10 mL, 1N)以中和三氟甲烷磺酸鹽。添加二氯甲烷(75 mL)以稀釋混合物且分離各層。使用鹽水(15 mL)洗滌有機層,藉由無水硫酸鈉乾燥,並過濾。在減壓下去除揮發物以得到粗產物。所得殘餘物未經任何進一步純化即用於下一反應中。LCMS (ES) m/z = 399.3 [M+H]+。
(3-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)雙環[1.1.1]戊烷-1-基)胺基甲酸第三丁基酯及(3-((1R,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)雙環[1.1.1]戊烷-1-基)胺基甲酸第三丁基酯:在0℃下向(S)-(3-(3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基)雙環[1.1.1]戊烷-1-基)胺基甲酸第三丁基酯(3.0 g, 7.53 mmol, 1當量)於甲醇(30 mL)中之溶液中逐份添加硼氫化鈉(0.854 mg, 22.60 mmol, 3當量)。將懸浮液在室溫下攪拌1 h。藉由TLC (於正己烷中之50% EtOAc)監測反應進展。此後,在減壓下濃縮反應混合物且使用EtOAc (100 mL)及水(20 mL)稀釋所獲得粗製物。分離有機層,使用鹽水溶液(20 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物。藉由急速管柱層析在矽膠上使用於正己烷中之乙酸乙酯作為洗脫劑純化所獲得粗產物。使用於正己烷中之30 - 70%乙酸乙酯洗脫期望產物。合併含有產物之部分並在減壓下濃縮以得到(3-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)雙環[1.1.1]戊烷-1-基)胺基甲酸第三丁基酯(1,3-反式異構體)及(3-((1R,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)雙環[1.1.1]戊烷-1-基)胺基甲酸第三丁基酯(1,3-順式異構體)。
順式異構體:LCMS (ES) m/z = 401.3 [M+H]+。1
H NMR (400 MHz, CDCl3
) δ ppm 0.92 (t,J
= 6.8 Hz, 3 H), 1.23 - 1.29 (m, 3 H), 1.36 - 1.37 (m, 3 H), 1.43 (s, 9 H), 1.93 - 2.04 (m, 6 H), 2.37 - 2.44 (m, 1 H), 2.64 - 2.68 (m, 1 H), 2.76 (bs, 1 H), 3.77 (s, 3 H), 4.20 (s, 1 H), 4.91 (bs, 1 H), 6.59 (s, 1 H), 6.68 (d,J
= 8.4 Hz, 1 H), 7.05 (d,J
= 8.8 Hz, 1 H)。
反式異構體:LCMS (ES) m/z = 401.3 [M+H]+。1
H NMR (400 MHz, CDCl3
) δ ppm 0.93 - 0.94 (m, 3 H), 1.25 - 1.28 (m, 5 H), 1.36 - 1.47 (m, 10 H), 1.93 - 1.98 (m, 6 H), 2.43 - 2.50 (m, 1 H), 2.69 - 2.72 (m, 1 H), 3.13 (bs, 1 H), 3.77 (s, 3 H), 4.13 (s, 1 H), 4.90 (bs, 1 H), 6.60 (s, 1 H), 6.65 (d,J
= 8.4 Hz, 1 H), 6.89 (d,J
= 8.8 Hz, 1 H)。
在室溫下,向(3-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)雙環[1.1.1]戊烷-1-基)胺基甲酸第三丁基酯(0.3 g, 0.749 mmol, 1當量)於THF (4 mL)中之溶液中添加QuadraSil® AP (標記程度:1.5-2.0 mmol/g載量(480 mg),藉由所需QuadraSil®之量(針對1.0當量) = mmol先前步驟中所使用之銅觸媒/mmol清除劑載量來計算)並攪拌1 h。此後,藉由通過過濾紙來去除固體部分且在減壓下濃縮濾液以得到(3-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)雙環[1.1.1]戊烷-1-基)胺基甲酸第三丁基1酯。LCMS (ES) m/z = 401.3 [M+H]+。1
H NMR (400 MHz, CDCl3
) δ ppm 0.93 - 0.94 (m, 3 H), 1.25 - 1.28 (m, 5 H), 1.36 - 1.47 (m, 10 H), 1.93 - 1.98 (m, 6 H), 2.43 - 2.50 (m, 1 H), 2.69 - 2.72 (m, 1 H), 3.13 (bs, 1 H), 3.77 (s, 3 H), 4.13 (s, 1 H), 4.90 (bs, 1 H), 6.60 (s, 1 H), 6.65 (d,J
= 8.4 Hz, 1 H), 6.89 (d,J
= 8.8 Hz, 1 H)。
(3-((1S,3S)-3-丁基-6-甲氧基-2-(3-(三甲基矽烷基)丙炔醯基)-1,2,3,4-四氫異喹啉-1-基)雙環[1.1.1]戊烷-1-基)胺基甲酸第三丁基酯:第一步驟:在室溫下,向3-(三甲基矽烷基)丙炔酸(155 mg, 1.09 mmol, 1.0當量)於DMF (3.2 mg, 0.04 mmol, 0.04當量)中之溶液中添加草醯氯(152 mg, 1.2 mmol, 1.1當量)並攪拌30分鐘。此後,在減壓下濃縮反應混合物以得到3-(三甲基矽烷基)丙炔醯氯。此醯氯未經任何進一步純化即用於下一步驟中。
第二步驟:在0℃下,向(3-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)雙環[1.1.1]戊烷-1-基)胺基甲酸第三丁基酯(285 mg, 0.711 mmol, 1.0當量)於乙腈(3.0 mL)中之溶液中添加碳酸氫鈉(448 mg, 5.34 mmol, 7.5當量)。在攪拌5分鐘之後,在0℃下向上述反應物質中添加3-(三甲基矽烷基)丙炔醯氯(171 mg, 1.07 mmol, 1.5當量)於乙腈(1 mL)中之溶液。將所得混合物在0℃下攪拌15 min,藉由TLC (於正己烷中之50%乙酸乙酯)監測反應進展。此後,使用EtOAc (40 mL)及水(10 mL)稀釋反應物質。分離有機層,使用鹽水溶液(7 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物。藉由製備型TLC藉由使用於正己烷中之25% EtOAc作為移動相純化所獲得粗產物以得到(3-((1S,3S)-3-丁基-6-甲氧基-2-(3-(三甲基矽烷基)丙炔醯基)-1,2,3,4-四氫異喹啉-1-基)雙環[1.1.1]戊烷-1-基)胺基甲酸第三丁基酯。LCMS (ES) m/z = 525.3 [M+H]+。1
H NMR (400 MHz, CDCl3
) δ ppm 0.25 - 0.28 (m, 9 H), 0.80 - 0.84 (m, 3 H), 1.16 - 1.26 (m, 6 H), 1.39 (s, 9 H), 1.73 - 1.82 (m, 6 H), 2.67 - 2.71 (m, 1 H), 2.92 - 2.96 (m, 1 H), 3.02 - 3.11 (m, 1 H), 3.80 - 3.81 (m, 3 H), 4.44 (bs, 1 H), 5.28 (s, 1 H), 6.67 - 6.74 (m, 2 H), 6.92 (d,J
= 8.4 Hz, 1 H)。
(3-((1S,3S)-3-丁基-6-甲氧基-2-丙炔醯基-1,2,3,4-四氫異喹啉-1-基)雙環[1.1.1]戊烷-1-基)胺基甲酸第三丁基酯:在0℃下,向(3-((1S,3S)-3-丁基-6-甲氧基-2-(3-(三甲基矽烷基)丙炔醯基)-1,2,3,4-四氫異喹啉-1-基)雙環[1.1.1]戊烷-1-基)胺基甲酸第三丁基酯(145 mg, 0.276 mmol, 1當量)於DCM (16 mL) / MeOH (3.3 mL)中之溶液中添加K2
CO3
(232 mg, 1.66 mmol, 6.0當量)。將反應混合物在0℃下攪拌3 hr以得到白色溶液。藉由TLC (於正己烷中之30 %乙酸乙酯)監測反應進展。此後,使用DCM (50 mL)及水(10 mL)稀釋反應混合物。分離有機層,藉由Na2
SO4
乾燥並在減壓下濃縮以得到3-((1S,3S)-3-丁基-6-甲氧基-2-丙炔醯基-1,2,3,4-四氫異喹啉-1-基)雙環[1.1.1]戊烷-1-基)胺基甲酸第三丁基酯。所得殘餘物未經任何進一步純化即用於下一反應中。LCMS (ES) m/z = 397.3 [M+H]+ - 56。1
H NMR (400 MHz, CDCl3
) δ ppm 0.80 - 0.82 (m, 3 H), 1.18 - 1.25 (m, 6 H), 1.38 - 1.39 (m, 9 H), 1.75 - 1.84 (m, 6 H), 2.68 - 2.72 (m, 1 H), 2.92 - 3.11 (m, 2 H), 3.80 - 3.81 (m, 3 H), 4.45 (bs, 1 H), 4.84 (s, 1 H), 5.27 (s, 1 H), 6.67 - 6.73 (m, 2 H), 6.92 (d,J
= 8.4 Hz, 1 H)。
1-((1S,3S)-1-(3-胺基雙環[1.1.1]戊烷-1-基)-3-丁基-6-甲氧基-3,4-二氫異喹啉-2(1H)-基)丙-2-炔-1-酮:在0℃下,向(3-((1S,3S)-3-丁基-6-甲氧基-2-丙炔醯基-1,2,3,4-四氫異喹啉-1-基)雙環[1.1.1]戊烷-1-基)胺基甲酸第三丁基酯(120 mg, 0.265 mmol, 1.0當量)於DCM (10 mL)中之溶液中添加TFA (2.42 g, 21.2 mmol, 80當量)。將混合物在0℃下攪拌1.5 hr。LCMS顯示反應已完成。然後,在減壓下濃縮反應混合物。將所獲得殘餘物溶於水(5 mL)中,隨後凍乾以得到1-((1S,3S)-1-(3-胺基雙環[1.1.1]戊烷-1-基)-3-丁基-6-甲氧基-3,4-二氫異喹啉-2(1H)-基)丙-2-炔-1-酮。 LCMS (ES) m/z = 353.3 [M+H]+。1
H NMR (400 MHz, DMSO-d6
) δ ppm 0.77 (bs, 3 H), 0.88 - 0.95 (m, 1 H), 1.16 - 1.17 (m, 4 H), 1.34 (bs, 1 H), 1.58 - 1.77 (m, 6 H), 2.78 - 2.89 (m, 2 H), 3.73 (s, 3 H), 4.32 - 4.43 (m, 1 H), 4.57 - 4.62 (m, 1 H), 5.19 - 5.29 (m, 1 H), 6.79 (d,J
= 8.0 Hz, 1 H), 6.87 (s, 1 H), 6.05 - 7.14 (m, 1 H), 8.37 - 8.44 (m, 3 H)。
程序 24 :化合物 94 之合成 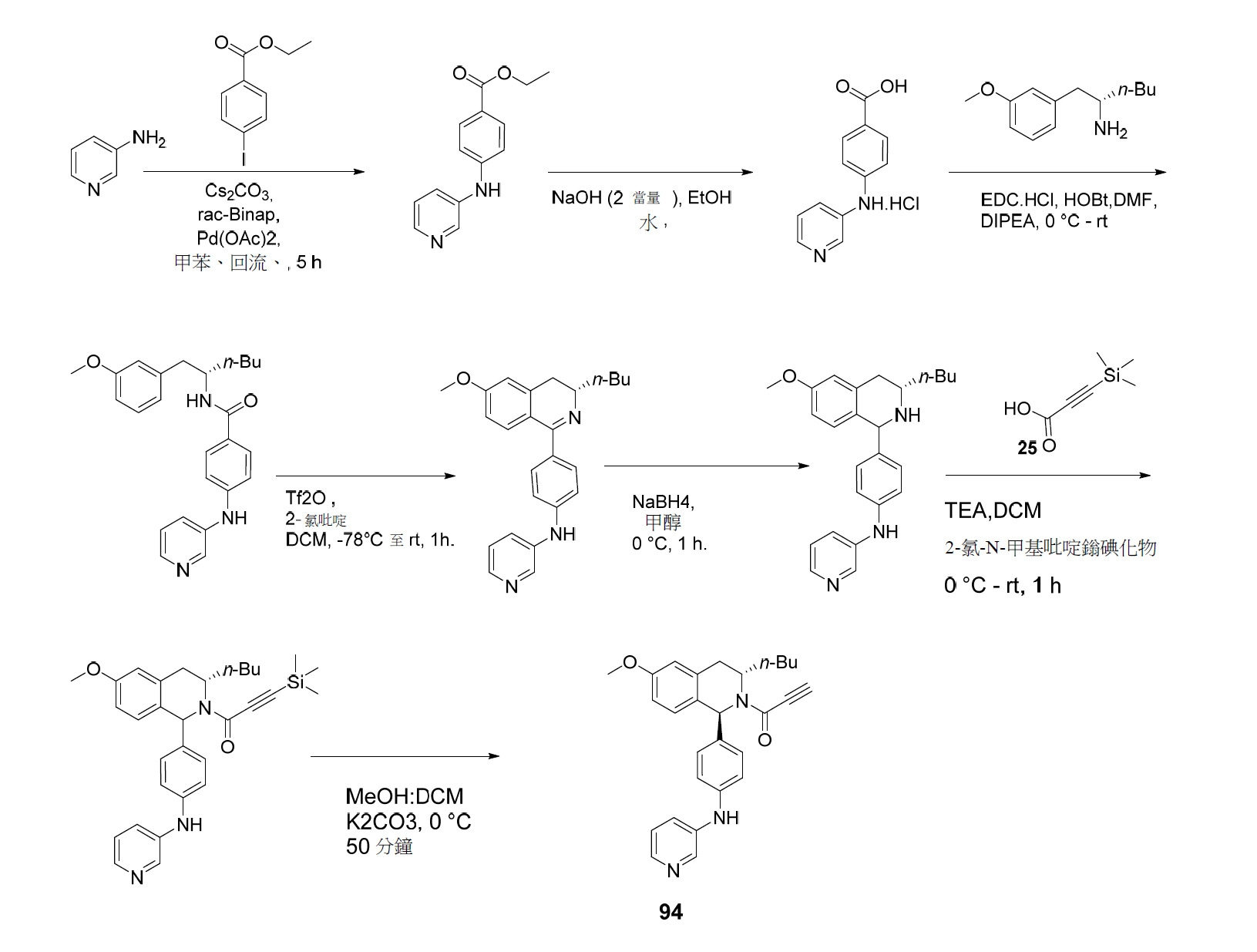 4-[(吡啶-3-基)胺基]苯甲酸乙酯:在室溫下,向4-碘苯甲酸乙酯(5.0 g, 18.1 mmol, 1.0當量)及吡啶-3-胺(2.05 g, 21.7 mmol, 1.2當量)於甲苯(100 mL)中之溶液中添加碳酸銫(11.8 g, 36.2 mmol, 2當量)及binap (0.226 g, 0.362 mmol, 0.02當量),使用氮將反應混合物吹掃20 min且然後添加Pd(OAc)2
(0.081 g, 0.362 mmol, 0.02當量)。然後,將反應混合物在回流下條件攪拌30 h。將反應混合物冷卻至室溫,經由矽藻土床過濾,使用EtOAc (200 mL)洗滌床且在減壓下濃縮濾液以得到粗產物。藉由急速管柱層析在矽膠上使用於正己烷中之EtOAc純化所獲得粗產物。使用於己烷中之40 - 45%乙酸乙酯作為洗脫劑分離產物以提供4-(吡啶-3-基胺基)苯甲酸乙酯。LCMS (ES) m/z = 243.1 [M+H]+。1
H NMR (400 MHz, DMSO-d6
): δ ppm 1.27 (t,J
= 6.8 Hz, 3 H), 4.23 (q,J
= 7.2 Hz, 2 H), 7.07 (d,J
= 8.8 Hz, 2 H), 7.29 - 7.32 (m, 1 H), 7.59 (d,J
= 6.8 Hz, 1 H), 7.81 (d,J
= 8.4 Hz, 2 H), 8.15 - 8.16 (m, 1 H), 8.41 - 8.42 (m, 1 H), 8.91 (s, 1 H)。
4-[(吡啶-3-基)胺基]苯甲酸鹽酸鹽:向4-[(吡啶-3-基)胺基]苯甲酸乙酯(2.5 g, 10.3 mmol, 1.0當量)於乙醇(30 mL)中之經攪拌溶液中添加於水(10 mL)中之氫氧化鈉(0.84 g, 20.6 mmol, 2.0當量)。將反應混合物在75℃下攪拌16 h。藉由TLC (於正己烷中之70%乙酸乙酯)監測反應進展。在反應完成之後,濃縮反應混合物。使用1N HCl (pH ~ 2)酸化合併之粗製物並在減壓下蒸發以得到粗產物之鹽酸鹽。乾燥產物並以鹽酸鹽形式用於下一步驟中。LCMS (ES) m/z = 215.1 [M+H]+游離胺質量。
N-[(2S)-1-(3-甲氧基苯基)己烷-2-基]-4-[(吡啶-3-基)胺基]苯甲醯胺:向4-(吡啶-3-基胺基)苯甲酸鹽酸鹽(3.10 g, 12.3 mmol, 1.6當量)於DMF (30 mL)中之經攪拌溶液中添加DIPEA (8.1 mL, 46.3 mmol, 6當量)並攪拌5分鐘,然後在0℃下添加EDC.HCl (2.2 g, 11.6 mmol, 1.5當量),隨後添加(S)-1-(3-甲氧基苯基)己烷-2-胺(1.6 g, 7.72 mmol, 1當量)。在攪拌5分鐘之後,在0℃下向反應混合物中添加HOBt (1.56 g, 11.6 mmol, 1.5當量)且然後將反應混合物在室溫下攪拌16 h。此後,使用EtOAc (60 mL)稀釋反應混合物,使用飽和碳酸氫鈉溶液(20 mL)、水(10 mL)、鹽水溶液(10 mL)洗滌有機層,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮。藉由急速管柱層析使用於DCM中之5% MeOH作為洗脫劑純化粗產物以獲得N-[(2S)-1-(3-甲氧基苯基)己烷-2-基]-4-[(吡啶-3-基)胺基]苯甲醯胺。LCMS (ES) m/z = 404.4 [M+H]+。1
H NMR (400MHz, DMSO-d6
) δ ppm: 0.91 (bs, 3 H), 1.23 (s, 4 H), 1.49 (s, 2 H), 2.71 - 2.81 (m, 2 H), 3.66 (s, 3 H), 4.11 - 4.12 (m, 1 H), 6.69 (d,J
= 8.0 Hz, 1 H), 6.76 (s, 2 H), 7.04 (d,J
= 8.0 Hz, 2 H), 7.11 - 7.13 (m, 1 H), 7.26 - 7.28 (m, 1 H), 7.52 - 7.53 (m, 1 H), 7.71 (d,J
= 8.0 Hz, 2 H), 7.92 (d,J
= 8.4 Hz, 1 H), 8.07 (d,J
= 8.0 Hz, 1 H), 8.37 (s, 1 H), 8.66 (s, 1 H)。
N-{4-[(3S)-3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基]苯基}吡啶-3-胺:在-78℃下,經由注射器向N-[(2S)-1-(3-甲氧基苯基)己烷-2-基]-4-[(吡啶-3-基)胺基]苯甲醯胺(2.4 g, 5.95 mmol, 1當量)及2-氯吡啶(1.69 mL, 17.8 mmol, 3.0當量)於二氯甲烷(30 mL)中之經攪拌溶液中緩慢逐滴添加三氟甲烷磺酸酐(3.0 mL, 17.8 mmol, 3.0當量)。在5 min之後,將反應混合物置於冰-水浴中並升溫至0℃。在5 min之後,將所得溶液升溫至23℃。藉由TLC (於DCM中之5% MeOH)監測反應進展。在1 h之後,使用氫氧化鈉水溶液(25 mL, 1N)終止反應以中和三氟甲烷磺酸鹽。添加二氯甲烷(150 mL)以稀釋混合物且分離各層。使用DCM (100 mL)萃取水層。使用鹽水(25 mL)洗滌合併之有機層,藉由無水硫酸鈉乾燥,並過濾。在減壓下去除揮發物以得到粗產物。藉由急速層析使用於DCM中之5% MeOH作為洗脫劑純化所獲得粗產物以得到期望產物N-{4-[(3S)-3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基]苯基}吡啶-3-胺。LCMS (ES) m/z = 386.2 [M+H]+。
N-{4-[(3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基]苯基}吡啶-3-胺:在0℃下,向N-{4-[(3S)-3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基]苯基}吡啶-3-胺(1.9 g, 4.93 mmol, 1當量)於甲醇(45 mL)中之經攪拌溶液中逐份添加硼氫化鈉(0.559 g, 14.8 mmol, 3當量)。將懸浮液在室溫下攪拌1 h。濃縮反應混合物且使用EtOAc (300 mL)及水(100 mL)稀釋所獲得粗製物。分離有機層,使用鹽水溶液(100 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物。藉由急速層析使用於DCM中之5%甲醇作為洗脫劑純化所獲得粗產物以得到N-{4-[(3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基]苯基}吡啶-3-胺。LCMS (ES) m/z = 388.4 [M+H]+。
1-[(3S)-3-丁基-6-甲氧基-1-{4-[(吡啶-3-基)胺基]苯基}-1,2,3,4-四氫異喹啉-2-基]-3-(三甲基矽烷基)丙-2-炔-1-酮:在室溫下,向N-{4-[(3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基]苯基}吡啶-3-胺(0.400 g, 1.03 mmol, 1.0當量)於DCM (12.0 mL)中之經攪拌溶液中添加三乙胺(0.363 mL, 2.58 mmol, 2.5當量)、3-(三甲基矽烷基)丙炔酸(0.176 g, 1.24 mmol, 1.2當量)及2-氯-1-甲基吡啶鎓碘化物(0.316 g, 1.24 mmol, 1.2當量)且將反應液在室溫下攪拌1 h。使用DCM (25 mL)稀釋反應混合物,使用水(10 mL)及鹽水(10 mL)洗滌,藉由無水硫酸鈉乾燥。過濾有機層並在減壓下濃縮以得到1-[(3S)-3-丁基-6-甲氧基-1-{4-[(吡啶-3-基)胺基]苯基}-1,2,3,4-四氫異喹啉-2-基]-3-(三甲基矽烷基)丙-2-炔-1-酮。LCMS (ES) m/z = 512.3 [M+H]+。
1-[(1S,3S)-3-丁基-6-甲氧基-1-{4-[(吡啶-3-基)胺基]苯基}-1,2,3,4-四氫異喹啉-2-基]丙-2-炔-1-酮:在0℃下,向1-[(3S)-3-丁基-6-甲氧基-1-{4-[(吡啶-3-基)胺基]苯基}-1,2,3,4-四氫異喹啉-2-基]-3-(三甲基矽烷基)丙-2-炔-1-酮(0.640 g, 1.25 mmol, 1.0當量)於MeOH:DCM (1:5) (12 mL)混合物中之經攪拌溶液中添加碳酸鉀(1.04 g, 7.50 mmol, 6.0當量)。然後,將反應混合物在0℃下攪拌50分鐘。然後,使用DCM (25.0 mL)稀釋反應混合物,使用水(10.0 mL)洗滌。分離有機層並藉由無水硫酸鈉乾燥。過濾有機層並在減壓下濃縮以得到粗產物,藉由急速管柱層析使用乙酸乙酯之己烷溶液作為洗脫劑進行純化。藉由製備型TLC使用於己烷中之60%乙酸乙酯作為洗脫劑藉由運行兩次來進一步純化所分離混合物。收集產物部分並在減壓下濃縮以得到純產物。藉由溶於乙腈(1.0 mL)及水(2.0 mL)混合物中來使所獲得純產物保持於凍乾狀態下16 h以得到1-[(1S,3S)-3-丁基-6-甲氧基-1-{4-[(吡啶-3-基)胺基]苯基}-1,2,3,4-四氫異喹啉-2-基]丙-2-炔-1-酮。LCMS (ES) m/z = 440.5 [M+H]+。1
H NMR (400 MHz, DMSO-d 6
) δ ppm 0.79 - 0.80 (m, 3 H), 1.21 (bs, 4 H), 1.49 (s, 2 H), 2.77 - 2.87 (m, 1.5 H), 3.06 - 3.10 (m, 0.5 H), 3.70 - 3.72 (m, 3 H), 4.31 (s, 0.5 H), 4.48 (s, 0.5 H), 4.58 (s, 0.5 H), 4.66 (s, 0.5 H), 6.00 (s, 0.5 H), 6.24 (s, 0.5 H), 6.77 - 6.83 (m, 2 H), 6.92 - 6.99 (m, 2 H), 7.05 - 7.13 (m, 2 H), 7.15 - 7.17 (m, 1 H), 7.35 - 7.39 (m, 1.5 H), 7.52 - 7.54 (m, 0.5 H), 7.94 - 7.98 (m, 1 H), 8.24 - 8.29 (m, 2 H)。
4-[(吡啶-3-基)胺基]苯甲酸乙酯:在室溫下,向4-碘苯甲酸乙酯(5.0 g, 18.1 mmol, 1.0當量)及吡啶-3-胺(2.05 g, 21.7 mmol, 1.2當量)於甲苯(100 mL)中之溶液中添加碳酸銫(11.8 g, 36.2 mmol, 2當量)及binap (0.226 g, 0.362 mmol, 0.02當量),使用氮將反應混合物吹掃20 min且然後添加Pd(OAc)2
(0.081 g, 0.362 mmol, 0.02當量)。然後,將反應混合物在回流下條件攪拌30 h。將反應混合物冷卻至室溫,經由矽藻土床過濾,使用EtOAc (200 mL)洗滌床且在減壓下濃縮濾液以得到粗產物。藉由急速管柱層析在矽膠上使用於正己烷中之EtOAc純化所獲得粗產物。使用於己烷中之40 - 45%乙酸乙酯作為洗脫劑分離產物以提供4-(吡啶-3-基胺基)苯甲酸乙酯。LCMS (ES) m/z = 243.1 [M+H]+。1
H NMR (400 MHz, DMSO-d6
): δ ppm 1.27 (t,J
= 6.8 Hz, 3 H), 4.23 (q,J
= 7.2 Hz, 2 H), 7.07 (d,J
= 8.8 Hz, 2 H), 7.29 - 7.32 (m, 1 H), 7.59 (d,J
= 6.8 Hz, 1 H), 7.81 (d,J
= 8.4 Hz, 2 H), 8.15 - 8.16 (m, 1 H), 8.41 - 8.42 (m, 1 H), 8.91 (s, 1 H)。
4-[(吡啶-3-基)胺基]苯甲酸鹽酸鹽:向4-[(吡啶-3-基)胺基]苯甲酸乙酯(2.5 g, 10.3 mmol, 1.0當量)於乙醇(30 mL)中之經攪拌溶液中添加於水(10 mL)中之氫氧化鈉(0.84 g, 20.6 mmol, 2.0當量)。將反應混合物在75℃下攪拌16 h。藉由TLC (於正己烷中之70%乙酸乙酯)監測反應進展。在反應完成之後,濃縮反應混合物。使用1N HCl (pH ~ 2)酸化合併之粗製物並在減壓下蒸發以得到粗產物之鹽酸鹽。乾燥產物並以鹽酸鹽形式用於下一步驟中。LCMS (ES) m/z = 215.1 [M+H]+游離胺質量。
N-[(2S)-1-(3-甲氧基苯基)己烷-2-基]-4-[(吡啶-3-基)胺基]苯甲醯胺:向4-(吡啶-3-基胺基)苯甲酸鹽酸鹽(3.10 g, 12.3 mmol, 1.6當量)於DMF (30 mL)中之經攪拌溶液中添加DIPEA (8.1 mL, 46.3 mmol, 6當量)並攪拌5分鐘,然後在0℃下添加EDC.HCl (2.2 g, 11.6 mmol, 1.5當量),隨後添加(S)-1-(3-甲氧基苯基)己烷-2-胺(1.6 g, 7.72 mmol, 1當量)。在攪拌5分鐘之後,在0℃下向反應混合物中添加HOBt (1.56 g, 11.6 mmol, 1.5當量)且然後將反應混合物在室溫下攪拌16 h。此後,使用EtOAc (60 mL)稀釋反應混合物,使用飽和碳酸氫鈉溶液(20 mL)、水(10 mL)、鹽水溶液(10 mL)洗滌有機層,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮。藉由急速管柱層析使用於DCM中之5% MeOH作為洗脫劑純化粗產物以獲得N-[(2S)-1-(3-甲氧基苯基)己烷-2-基]-4-[(吡啶-3-基)胺基]苯甲醯胺。LCMS (ES) m/z = 404.4 [M+H]+。1
H NMR (400MHz, DMSO-d6
) δ ppm: 0.91 (bs, 3 H), 1.23 (s, 4 H), 1.49 (s, 2 H), 2.71 - 2.81 (m, 2 H), 3.66 (s, 3 H), 4.11 - 4.12 (m, 1 H), 6.69 (d,J
= 8.0 Hz, 1 H), 6.76 (s, 2 H), 7.04 (d,J
= 8.0 Hz, 2 H), 7.11 - 7.13 (m, 1 H), 7.26 - 7.28 (m, 1 H), 7.52 - 7.53 (m, 1 H), 7.71 (d,J
= 8.0 Hz, 2 H), 7.92 (d,J
= 8.4 Hz, 1 H), 8.07 (d,J
= 8.0 Hz, 1 H), 8.37 (s, 1 H), 8.66 (s, 1 H)。
N-{4-[(3S)-3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基]苯基}吡啶-3-胺:在-78℃下,經由注射器向N-[(2S)-1-(3-甲氧基苯基)己烷-2-基]-4-[(吡啶-3-基)胺基]苯甲醯胺(2.4 g, 5.95 mmol, 1當量)及2-氯吡啶(1.69 mL, 17.8 mmol, 3.0當量)於二氯甲烷(30 mL)中之經攪拌溶液中緩慢逐滴添加三氟甲烷磺酸酐(3.0 mL, 17.8 mmol, 3.0當量)。在5 min之後,將反應混合物置於冰-水浴中並升溫至0℃。在5 min之後,將所得溶液升溫至23℃。藉由TLC (於DCM中之5% MeOH)監測反應進展。在1 h之後,使用氫氧化鈉水溶液(25 mL, 1N)終止反應以中和三氟甲烷磺酸鹽。添加二氯甲烷(150 mL)以稀釋混合物且分離各層。使用DCM (100 mL)萃取水層。使用鹽水(25 mL)洗滌合併之有機層,藉由無水硫酸鈉乾燥,並過濾。在減壓下去除揮發物以得到粗產物。藉由急速層析使用於DCM中之5% MeOH作為洗脫劑純化所獲得粗產物以得到期望產物N-{4-[(3S)-3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基]苯基}吡啶-3-胺。LCMS (ES) m/z = 386.2 [M+H]+。
N-{4-[(3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基]苯基}吡啶-3-胺:在0℃下,向N-{4-[(3S)-3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基]苯基}吡啶-3-胺(1.9 g, 4.93 mmol, 1當量)於甲醇(45 mL)中之經攪拌溶液中逐份添加硼氫化鈉(0.559 g, 14.8 mmol, 3當量)。將懸浮液在室溫下攪拌1 h。濃縮反應混合物且使用EtOAc (300 mL)及水(100 mL)稀釋所獲得粗製物。分離有機層,使用鹽水溶液(100 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物。藉由急速層析使用於DCM中之5%甲醇作為洗脫劑純化所獲得粗產物以得到N-{4-[(3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基]苯基}吡啶-3-胺。LCMS (ES) m/z = 388.4 [M+H]+。
1-[(3S)-3-丁基-6-甲氧基-1-{4-[(吡啶-3-基)胺基]苯基}-1,2,3,4-四氫異喹啉-2-基]-3-(三甲基矽烷基)丙-2-炔-1-酮:在室溫下,向N-{4-[(3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基]苯基}吡啶-3-胺(0.400 g, 1.03 mmol, 1.0當量)於DCM (12.0 mL)中之經攪拌溶液中添加三乙胺(0.363 mL, 2.58 mmol, 2.5當量)、3-(三甲基矽烷基)丙炔酸(0.176 g, 1.24 mmol, 1.2當量)及2-氯-1-甲基吡啶鎓碘化物(0.316 g, 1.24 mmol, 1.2當量)且將反應液在室溫下攪拌1 h。使用DCM (25 mL)稀釋反應混合物,使用水(10 mL)及鹽水(10 mL)洗滌,藉由無水硫酸鈉乾燥。過濾有機層並在減壓下濃縮以得到1-[(3S)-3-丁基-6-甲氧基-1-{4-[(吡啶-3-基)胺基]苯基}-1,2,3,4-四氫異喹啉-2-基]-3-(三甲基矽烷基)丙-2-炔-1-酮。LCMS (ES) m/z = 512.3 [M+H]+。
1-[(1S,3S)-3-丁基-6-甲氧基-1-{4-[(吡啶-3-基)胺基]苯基}-1,2,3,4-四氫異喹啉-2-基]丙-2-炔-1-酮:在0℃下,向1-[(3S)-3-丁基-6-甲氧基-1-{4-[(吡啶-3-基)胺基]苯基}-1,2,3,4-四氫異喹啉-2-基]-3-(三甲基矽烷基)丙-2-炔-1-酮(0.640 g, 1.25 mmol, 1.0當量)於MeOH:DCM (1:5) (12 mL)混合物中之經攪拌溶液中添加碳酸鉀(1.04 g, 7.50 mmol, 6.0當量)。然後,將反應混合物在0℃下攪拌50分鐘。然後,使用DCM (25.0 mL)稀釋反應混合物,使用水(10.0 mL)洗滌。分離有機層並藉由無水硫酸鈉乾燥。過濾有機層並在減壓下濃縮以得到粗產物,藉由急速管柱層析使用乙酸乙酯之己烷溶液作為洗脫劑進行純化。藉由製備型TLC使用於己烷中之60%乙酸乙酯作為洗脫劑藉由運行兩次來進一步純化所分離混合物。收集產物部分並在減壓下濃縮以得到純產物。藉由溶於乙腈(1.0 mL)及水(2.0 mL)混合物中來使所獲得純產物保持於凍乾狀態下16 h以得到1-[(1S,3S)-3-丁基-6-甲氧基-1-{4-[(吡啶-3-基)胺基]苯基}-1,2,3,4-四氫異喹啉-2-基]丙-2-炔-1-酮。LCMS (ES) m/z = 440.5 [M+H]+。1
H NMR (400 MHz, DMSO-d 6
) δ ppm 0.79 - 0.80 (m, 3 H), 1.21 (bs, 4 H), 1.49 (s, 2 H), 2.77 - 2.87 (m, 1.5 H), 3.06 - 3.10 (m, 0.5 H), 3.70 - 3.72 (m, 3 H), 4.31 (s, 0.5 H), 4.48 (s, 0.5 H), 4.58 (s, 0.5 H), 4.66 (s, 0.5 H), 6.00 (s, 0.5 H), 6.24 (s, 0.5 H), 6.77 - 6.83 (m, 2 H), 6.92 - 6.99 (m, 2 H), 7.05 - 7.13 (m, 2 H), 7.15 - 7.17 (m, 1 H), 7.35 - 7.39 (m, 1.5 H), 7.52 - 7.54 (m, 0.5 H), 7.94 - 7.98 (m, 1 H), 8.24 - 8.29 (m, 2 H)。
程序 25 :化合物 100 之合成 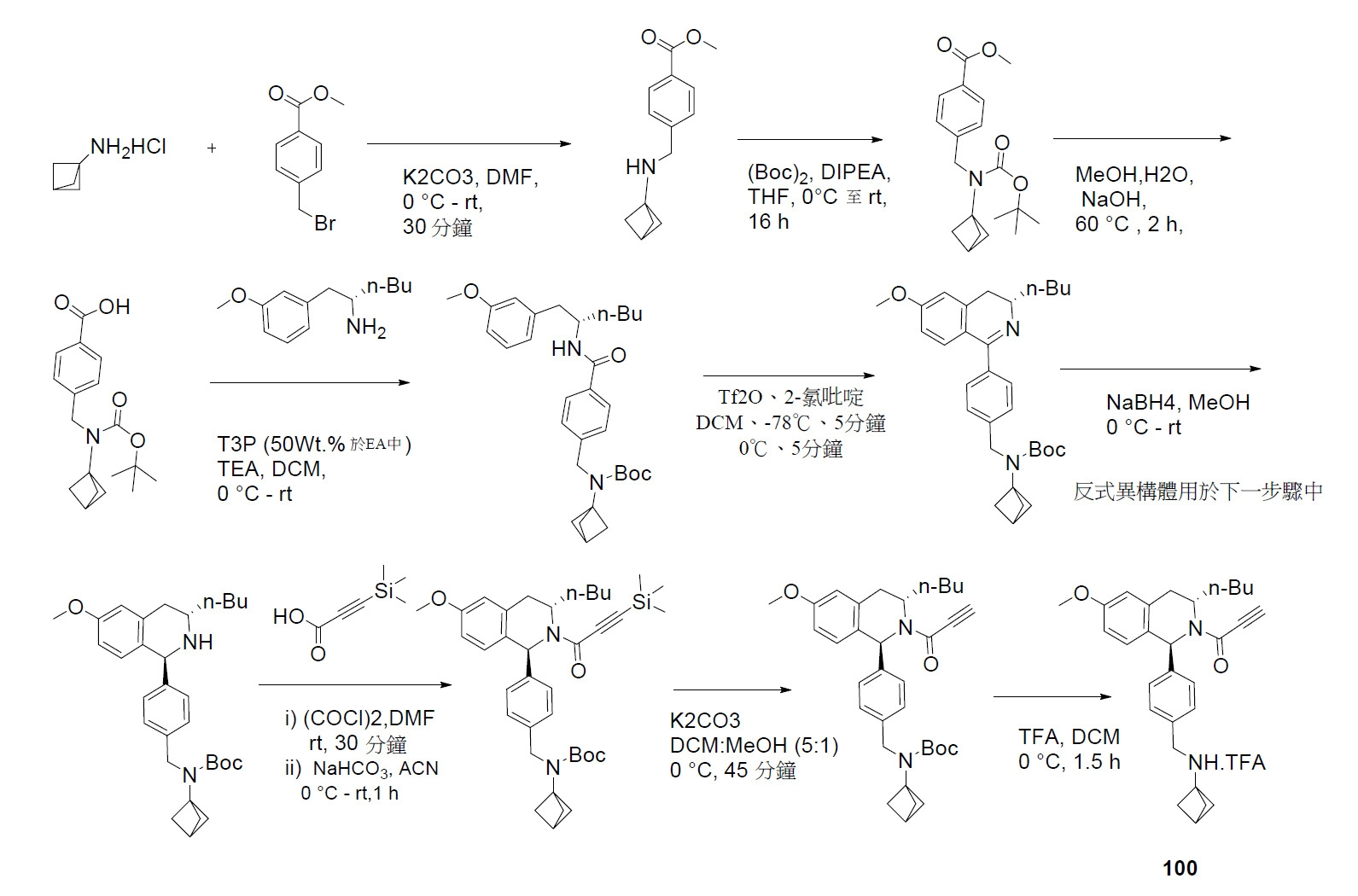 4-[({雙環[1.1.1]戊烷-1-基}胺基)甲基]苯甲酸甲酯:在0℃下,向雙環[1.1.1]戊烷-1-胺鹽酸鹽(2.1 g, 17.6 mmol, 1.0當量)於DMF (30.0 mL)中之經攪拌溶液中添加碳酸鉀(7.3 g, 52.7 mmol, 3.0當量)。在攪拌5分鐘之後,添加4-(溴甲基)苯甲酸甲酯(3.22 g, 14.0 mmol, 0.8當量)。然後將反應混合物在室溫下攪拌30分鐘。使用水(30 mL)稀釋反應混合物,萃取至乙酸乙酯(2 × 30 mL)中。使用冷卻水(40 mL)、鹽水(20 mL)洗滌合併之有機層,藉由無水硫酸鈉乾燥。過濾有機層並在減壓下濃縮以得到粗產物,藉由急速管柱層析在矽膠上使用乙酸乙酯之己烷溶液作為洗脫劑進行純化。使用於己烷中之15 -18%乙酸乙酯分離產物。收集產物部分並在減壓下濃縮以得到4-((雙環[1.1.1]戊烷-1-基胺基)甲基)苯甲酸甲酯。LCMS (ES) m/z = 232.2 [M+H]+。1
H NMR (400 MHz, DMSO-d6
): δ ppm 1.64 (s, 6 H), 2.28 (s, 1 H), 3.69 (s, 2 H), 3.74 (s, 3 H), 7.45 - 7.48 (m, 2 H), 7.86 - 7.88 (m, 2 H)。
4-[({雙環[1.1.1]戊烷-1-基}[(第三丁氧基)羰基]胺基)甲基]苯甲酸甲酯:在0℃下,向4-((雙環[1.1.1]戊烷-1-基胺基)甲基)苯甲酸甲酯(2.2 g, 9.51 mmol, 1當量)於THF (30 mL)中之經攪拌溶液中添加DIPEA (5 mL, 28.5 mmol, 3當量),隨後添加boc酐(6.6 mL, 28.5 mmol, 3當量)。然後,將反應混合物在室溫下攪拌16 h。使用水(25 mL)稀釋反應混合物,使用乙酸乙酯(2 × 30 mL)萃取。使用水(20 mL)、鹽水(20 mL)洗滌合併之有機層,藉由無水硫酸鈉乾燥。過濾有機層並在減壓下濃縮以得到粗產物,藉由急速管柱層析在矽膠上使用乙酸乙酯之己烷溶液作為洗脫劑進行純化。使用於己烷中之12 -16 %乙酸乙酯分離產物。收集產物部分並在減壓下濃縮以得到4-((雙環[1.1.1]戊烷-1-基(第三丁氧基羰基)胺基)甲基)苯甲酸甲酯。LCMS (ES) m/z = 332.2 [M+H] +,但觀察到276.2 (不含第三丁基)。
4-[({雙環[1.1.1]戊烷-1-基}[(第三丁氧基)羰基]胺基)甲基]苯甲酸:在室溫下,向4-((雙環[1.1.1]戊烷-1-基(第三丁氧基羰基)胺基)甲基)苯甲酸甲酯(2.75 g, 8.30 mmol, 1當量)於MeOH (20 mL)及水(10 mL)中之溶液中添加氫氧化鈉(0.680 g, 16.6 mmol, 2當量)且將反應混合物在60℃下攪拌2 h。藉由TLC (於DCM中之5% MeOH)監測反應進展。在反應完成之後,在減壓下濃縮反應混合物以自反應物質去除甲醇且使用5%檸檬酸(pH ~ 4)酸化剩餘水層且然後使用EtOAc (100 mL)萃取產物。藉由無水硫酸鈉乾燥有機層,過濾並在減壓下濃縮以得到4-((雙環[1.1.1]戊烷-1-基(第三丁氧基羰基)胺基)甲基)苯甲酸。LCMS (ES) m/z = 318.1 [M+H] +,但觀察到262.1 (不含第三丁基)。1
H NMR (400 MHz, DMSO-d6
): δ ppm 1.38 (s, 9 H), 1.92(s, 6 H), 2.33 (s, 1 H), 4.41 (s, 2 H), 7.26 (d,J
= 8.4 Hz, 2 H), 7.88 (d,J
= 8.4 Hz, 2 H), 12.40 (bs, 1 H)。
N-{雙環[1.1.1]戊烷-1-基}-N-[(4-{[(2S)-1-(3-甲氧基苯基)己烷-2-基]胺甲醯基}苯基)甲基]胺基甲酸第三丁基酯:向4-((雙環[1.1.1]戊烷-1-基(第三丁氧基羰基)胺基)甲基)苯甲酸(1.99 g, 6.27 mmol, 1當量)於DCM (20 mL)中之溶液中添加TEA (2.64 mL, 18.8 mmol, 3當量),攪拌5 min且然後在0℃下添加T3P (50 wt.%於EtOAc中) (6 mL, 9.41 mmol, 1.5當量)並再攪拌30 min。然後在0℃下向反應混合物中添加(S)-1-(3-甲氧基苯基)己烷-2-胺(1.3 g, 6.27 mmol, 1當量)於DCM (10 mL)中之溶液且然後將反應混合物在室溫下攪拌2 h。藉由TLC (於己烷中之20%乙酸乙酯)監測反應進展。此後,使用DCM (100 mL)稀釋反應混合物,使用飽和碳酸氫鈉溶液(40 mL)、水(40 mL)、鹽水溶液(20 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物。藉由急速層析在矽膠上純化所獲得粗產物。使用於正己烷中之16 - 18%乙酸乙酯洗脫期望產物。合併含有產物之部分並在減壓下濃縮以得到(S)-雙環[1.1.1]戊烷-1-基(4-((1-(3-甲氧基苯基)己烷-2-基)胺甲醯基)苄基)胺基甲酸第三丁基酯。LCMS (ES) m/z = 507.3 [M+H]+。1
H NMR (400 MHz, DMSO-d6
): δ ppm 0.81 (s, 3 H), 1.25 - 1.26 (m, 4 H), 1.39 (s, 9 H), 1.50 (s, 2 H), 1.93 (s, 6 H), 2.33 (s, 1 H), 2.75 (bs, 2 H), 3.65 (s, 3 H), 4.12 (bs, 1 H), 4.39 (s, 2 H), 6.69 (d,J
= 8.0 Hz, 1 H), 6.77 (s, 2 H), 7.11 - 7.15 (m, 1 H), 7.20 - 7.21 (m, 2 H), 7.71 (d,J
= 7.2 Hz, 2 H), 8.04 (bs, 1 H)。
N-{雙環[1.1.1]戊烷-1-基}-N-({4-[(3S)-3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基]苯基}甲基)胺基甲酸第三丁基酯:在-78℃下,經由注射器向(S)-雙環[1.1.1]戊烷-1-基(4-((1-(3-甲氧基苯基)己烷-2-基)胺甲醯基)苄基)胺基甲酸第三丁基酯(2.3 g, 4.54 mmol, 1當量)
及2-氯吡啶(1.30 mL, 13.6 mmol, 3.0當量)於二氯甲烷(25 mL)中之經攪拌溶液中緩慢逐滴添加三氟甲烷磺酸酐(2.3 mL, 13.6 mmol, 3當量)。在5 min之後,將反應混合物置於冰-水浴中並升溫至0℃。在5分鐘之後,使用氫氧化鈉水溶液(1N, 20 mL)淬滅反應混合物以中和三氟甲烷磺酸鹽。添加二氯甲烷(150 mL)以稀釋混合物且分離各層。使用DCM (100 mL)萃取水層。使用鹽水(25 mL)洗滌合併之有機層,藉由無水硫酸鈉乾燥,過濾並在壓力下濃縮以獲得粗產物,藉由急速管柱層析在矽膠上使用乙酸乙酯之己烷溶液作為洗脫劑進行純化。使用於己烷中之12 -15%乙酸乙酯分離產物。收集產物部分並在減壓下濃縮以得到N-{雙環[1.1.1]戊烷-1-基}-N-({4-[(3S)-3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基]苯基}甲基)胺基甲酸第三丁基酯。在管柱純化中,亦分離出N-({4-[(3S)-3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基]苯基}甲基)雙環[1.1.1]戊烷-1-胺。LCMS (ES) m/z = 489.3 [M+H]+。1
H NMR (400 MHz, DMSO-d6
) δ ppm 0.87 - 0.91 (m, 3 H), 1.32 - 1.40 (m, 12 H), 1.56 - 1.67 (m, 3 H), 1.95 (s, 6 H), 2.30 - 2.34 (m, 1 H), 2.41 - 2.48 (m, 2 H), 2.74 - 2.78 (m, 1 H), 3.78 (s, 3 H), 4.42 (s, 2 H), 6.80 (d,J
= 8.4 Hz, 1 H), 6.90 (s, 1 H), 7.07 (d,J
= 8.4 Hz, 1 H), 7.23 (d,J
= 7.6 Hz, 2 H), 7.45 - 7.46 (m, 2 H)。
N-{雙環[1.1.1]戊烷-1-基}-N-({4-[(1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基]苯基}甲基)胺基甲酸第三丁基酯:在0℃下,向(S)-雙環[1.1.1]戊烷-1-基(4-(3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基)苄基)胺基甲酸第三丁基酯(0.850 g, 1.74 mmol, 1當量)於甲醇(10 mL)中之溶液中逐份添加硼氫化鈉(0.197 g, 5.22 mmol, 3當量)。將懸浮液在室溫下攪拌1 h。藉由TLC (於己烷中之20% EA)監測反應進展。此後,濃縮反應混合物且使用EtOAc (20 mL)及水(10 mL)稀釋所獲得粗製物。分離有機層,使用鹽水溶液(10 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物。藉由製備型TLC使用於正己烷中之20%乙酸乙酯純化所獲得粗產物。收集產物部分並在減壓下濃縮以得到N-{雙環[1.1.1]戊烷-1-基}-N-({4-[(1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基]苯基}甲基)胺基甲酸第三丁基酯。LCMS (ES) m/z = 491.3 [M+H]+。1
H NMR (400 MHz, DMSO-d6
) δ ppm 0.75 - 0.78 (m, 3 H), 1.08 - 1.17 (m, 6 H), 1.36 (bs, 9 H), 1.90 (s, 6 H), 2.30 (s, 1 H), 2.37 (s, 1 H), 2.73 - 2.75 (m, 2 H), 3.70 (s, 3 H), 4.30 (s, 2 H), 5.04 (bs, 1 H), 6.64 - 6.69 (m, 2 H), 6.75 - 6.77 (m, 1 H), 7.04 (s, 4 H)。
N-{雙環[1.1.1]戊烷-1-基}-N-({4-[(1S,3S)-3-丁基-6-甲氧基-2-[3-(三甲基矽烷基)丙-2-炔醯基]-1,2,3,4-四氫異喹啉-1-基]苯基}甲基)胺基甲酸第三丁基酯:第一步驟:在室溫下,向3-(三甲基矽烷基)丙炔酸(35 mg, 0.246 mmol, 1.0當量)於DMF (0.001 mL, 0.009 mmol, 0.04當量)中之溶液中添加草醯氯(0.023 mL, 0.271 mmol, 1.1當量)並攪拌30分鐘。然後,在減壓下濃縮反應混合物以得到3-(三甲基矽烷基)丙炔醯氯。此醯氯未經任何進一步純化即用於下一步驟中。
第二步驟:在0℃下,向N-{雙環[1.1.1]戊烷-1-基}-N-({4-[(1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基]苯基}甲基)胺基甲酸第三丁基酯(0.085 g, 0.173 mmol, 1當量)於乙腈(3 mL)中之溶液中添加碳酸氫鈉(0.118 g, 1.39 mmol, 8.0當量)。在攪拌5分鐘之後,向上述反應物質中添加3-(三甲基矽烷基)丙炔醯氯(0.031 g, 0.191 mmol, 1.1當量)於乙腈(2.0 mL)中之溶液。將所得混合物在0℃下攪拌1 h。然後使用EtOAc (10 mL)及水(5 mL)稀釋反應混合物。分離有機層,使用鹽水溶液(5 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物。此粗產物未經任何進一步純化即用於下一步驟中。 LCMS (ES) m/z = 615.3 [M+H]+,但觀察到559.3 (不含第三丁基)。
N-{雙環[1.1.1]戊烷-1-基}-N-({4-[(1S,3S)-3-丁基-6-甲氧基-2-(丙-2-炔醯基)-1,2,3,4-四氫異喹啉-1-基]苯基}甲基)胺基甲酸第三丁基酯:在0℃下,向雙環[1.1.1]戊烷-1-基(4-((1S,3S)-3-丁基-6-甲氧基-2-(3-(三甲基矽烷基)丙炔醯基)-1,2,3,4-四氫異喹啉-1-基)苄基)胺基甲酸第三丁基酯(85 mg, 0.138 mmol, 1當量)於DCM (5 mL) / MeOH (1 mL)中之溶液中添加K2CO3 (115 mg, 0.829 mmol, 6當量)。將混合物在0℃下攪拌45分鐘。然後使用DCM (10 mL)稀釋反應混合物並添加水(2 mL)。使用DCM (2 × 5 mL)萃取有機層並藉由無水Na2
SO4
乾燥。過濾有機層並濃縮以獲得粗產物,藉由製備型TLC使用於己烷中之20%乙酸乙酯作為移動相純化產物。收集產物部分並在減壓下濃縮以得到雙環[1.1.1]戊烷-1-基(4-((1S,3S)-3-丁基-6-甲氧基-2-丙炔醯基-1,2,3,4-四氫異喹啉-1-基)苄基)胺基甲酸第三丁基酯。LCMS (ES) m/z = 543.3 [M+H]+,但觀察到不含boc基團者。
N-{雙環[1.1.1]戊烷-1-基}-N-({4-[(1S,3S)-3-丁基-6-甲氧基-2-(丙-2-炔醯基)-1,2,3,4-四氫異喹啉-1-基]苯基}甲基)-2,2,2-三氟乙醯胺:在0℃下,向N-{雙環[1.1.1]戊烷-1-基}-N-({4-[(1S,3S)-3-丁基-6-甲氧基-2-(丙-2-炔醯基)-1,2,3,4-四氫異喹啉-1-基]苯基}甲基)胺基甲酸第三丁基酯(0.040 g, 0.073 mmol, 1.0當量)於DCM (1.0 mL)中之經攪拌溶液中添加TFA (0.150 mL)。然後,將反應混合物在0℃下攪拌1.5 h。在減壓下蒸發反應混合物且將水浴溫度保持於30℃。藉由添加水(1.5 mL)及乙腈(0.5 mL)混合物來使所獲得粗產物保持於凍乾狀態下16 h。將所獲得產物用於分析中。LCMS (ES) m/z = 443.3 [M+H]+ (所觀察到游離胺質量)。1
H NMR (400 MHz, DMSO-d6
) δ ppm 0.79 - 0.82 (m, 3 H), 1.13 - 1.21 (m, 4 H), 1.47 (bs, 2 H), 1.97 - 1.98 (m, 6 H), 2.65 (s, 1 H), 2.77 (bs, 1 H), 2.85 - 2.89 (m, 0.5 H), 3.14 - 3.15 (m, 0.5 H), 3.69 - 3.70 (m, 3 H), 3.98 (s, 2 H), 4.29 (s, 0.5 H), 4.60 (s, 1 H), 4.74 (s, 0.5 H), 6.04 (s, 0.5 H), 6.33(s, 0.5 H), 6.75 - 6.83(m, 2 H), 7.28 - 7.32 (m, 3 H), 7.37 - 7.42 (m, 1.5 H), 7.60 - 7.62 (m, 0.5 H), 9.42 (bs, 2 H)。
4-[({雙環[1.1.1]戊烷-1-基}胺基)甲基]苯甲酸甲酯:在0℃下,向雙環[1.1.1]戊烷-1-胺鹽酸鹽(2.1 g, 17.6 mmol, 1.0當量)於DMF (30.0 mL)中之經攪拌溶液中添加碳酸鉀(7.3 g, 52.7 mmol, 3.0當量)。在攪拌5分鐘之後,添加4-(溴甲基)苯甲酸甲酯(3.22 g, 14.0 mmol, 0.8當量)。然後將反應混合物在室溫下攪拌30分鐘。使用水(30 mL)稀釋反應混合物,萃取至乙酸乙酯(2 × 30 mL)中。使用冷卻水(40 mL)、鹽水(20 mL)洗滌合併之有機層,藉由無水硫酸鈉乾燥。過濾有機層並在減壓下濃縮以得到粗產物,藉由急速管柱層析在矽膠上使用乙酸乙酯之己烷溶液作為洗脫劑進行純化。使用於己烷中之15 -18%乙酸乙酯分離產物。收集產物部分並在減壓下濃縮以得到4-((雙環[1.1.1]戊烷-1-基胺基)甲基)苯甲酸甲酯。LCMS (ES) m/z = 232.2 [M+H]+。1
H NMR (400 MHz, DMSO-d6
): δ ppm 1.64 (s, 6 H), 2.28 (s, 1 H), 3.69 (s, 2 H), 3.74 (s, 3 H), 7.45 - 7.48 (m, 2 H), 7.86 - 7.88 (m, 2 H)。
4-[({雙環[1.1.1]戊烷-1-基}[(第三丁氧基)羰基]胺基)甲基]苯甲酸甲酯:在0℃下,向4-((雙環[1.1.1]戊烷-1-基胺基)甲基)苯甲酸甲酯(2.2 g, 9.51 mmol, 1當量)於THF (30 mL)中之經攪拌溶液中添加DIPEA (5 mL, 28.5 mmol, 3當量),隨後添加boc酐(6.6 mL, 28.5 mmol, 3當量)。然後,將反應混合物在室溫下攪拌16 h。使用水(25 mL)稀釋反應混合物,使用乙酸乙酯(2 × 30 mL)萃取。使用水(20 mL)、鹽水(20 mL)洗滌合併之有機層,藉由無水硫酸鈉乾燥。過濾有機層並在減壓下濃縮以得到粗產物,藉由急速管柱層析在矽膠上使用乙酸乙酯之己烷溶液作為洗脫劑進行純化。使用於己烷中之12 -16 %乙酸乙酯分離產物。收集產物部分並在減壓下濃縮以得到4-((雙環[1.1.1]戊烷-1-基(第三丁氧基羰基)胺基)甲基)苯甲酸甲酯。LCMS (ES) m/z = 332.2 [M+H] +,但觀察到276.2 (不含第三丁基)。
4-[({雙環[1.1.1]戊烷-1-基}[(第三丁氧基)羰基]胺基)甲基]苯甲酸:在室溫下,向4-((雙環[1.1.1]戊烷-1-基(第三丁氧基羰基)胺基)甲基)苯甲酸甲酯(2.75 g, 8.30 mmol, 1當量)於MeOH (20 mL)及水(10 mL)中之溶液中添加氫氧化鈉(0.680 g, 16.6 mmol, 2當量)且將反應混合物在60℃下攪拌2 h。藉由TLC (於DCM中之5% MeOH)監測反應進展。在反應完成之後,在減壓下濃縮反應混合物以自反應物質去除甲醇且使用5%檸檬酸(pH ~ 4)酸化剩餘水層且然後使用EtOAc (100 mL)萃取產物。藉由無水硫酸鈉乾燥有機層,過濾並在減壓下濃縮以得到4-((雙環[1.1.1]戊烷-1-基(第三丁氧基羰基)胺基)甲基)苯甲酸。LCMS (ES) m/z = 318.1 [M+H] +,但觀察到262.1 (不含第三丁基)。1
H NMR (400 MHz, DMSO-d6
): δ ppm 1.38 (s, 9 H), 1.92(s, 6 H), 2.33 (s, 1 H), 4.41 (s, 2 H), 7.26 (d,J
= 8.4 Hz, 2 H), 7.88 (d,J
= 8.4 Hz, 2 H), 12.40 (bs, 1 H)。
N-{雙環[1.1.1]戊烷-1-基}-N-[(4-{[(2S)-1-(3-甲氧基苯基)己烷-2-基]胺甲醯基}苯基)甲基]胺基甲酸第三丁基酯:向4-((雙環[1.1.1]戊烷-1-基(第三丁氧基羰基)胺基)甲基)苯甲酸(1.99 g, 6.27 mmol, 1當量)於DCM (20 mL)中之溶液中添加TEA (2.64 mL, 18.8 mmol, 3當量),攪拌5 min且然後在0℃下添加T3P (50 wt.%於EtOAc中) (6 mL, 9.41 mmol, 1.5當量)並再攪拌30 min。然後在0℃下向反應混合物中添加(S)-1-(3-甲氧基苯基)己烷-2-胺(1.3 g, 6.27 mmol, 1當量)於DCM (10 mL)中之溶液且然後將反應混合物在室溫下攪拌2 h。藉由TLC (於己烷中之20%乙酸乙酯)監測反應進展。此後,使用DCM (100 mL)稀釋反應混合物,使用飽和碳酸氫鈉溶液(40 mL)、水(40 mL)、鹽水溶液(20 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物。藉由急速層析在矽膠上純化所獲得粗產物。使用於正己烷中之16 - 18%乙酸乙酯洗脫期望產物。合併含有產物之部分並在減壓下濃縮以得到(S)-雙環[1.1.1]戊烷-1-基(4-((1-(3-甲氧基苯基)己烷-2-基)胺甲醯基)苄基)胺基甲酸第三丁基酯。LCMS (ES) m/z = 507.3 [M+H]+。1
H NMR (400 MHz, DMSO-d6
): δ ppm 0.81 (s, 3 H), 1.25 - 1.26 (m, 4 H), 1.39 (s, 9 H), 1.50 (s, 2 H), 1.93 (s, 6 H), 2.33 (s, 1 H), 2.75 (bs, 2 H), 3.65 (s, 3 H), 4.12 (bs, 1 H), 4.39 (s, 2 H), 6.69 (d,J
= 8.0 Hz, 1 H), 6.77 (s, 2 H), 7.11 - 7.15 (m, 1 H), 7.20 - 7.21 (m, 2 H), 7.71 (d,J
= 7.2 Hz, 2 H), 8.04 (bs, 1 H)。
N-{雙環[1.1.1]戊烷-1-基}-N-({4-[(3S)-3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基]苯基}甲基)胺基甲酸第三丁基酯:在-78℃下,經由注射器向(S)-雙環[1.1.1]戊烷-1-基(4-((1-(3-甲氧基苯基)己烷-2-基)胺甲醯基)苄基)胺基甲酸第三丁基酯(2.3 g, 4.54 mmol, 1當量)
及2-氯吡啶(1.30 mL, 13.6 mmol, 3.0當量)於二氯甲烷(25 mL)中之經攪拌溶液中緩慢逐滴添加三氟甲烷磺酸酐(2.3 mL, 13.6 mmol, 3當量)。在5 min之後,將反應混合物置於冰-水浴中並升溫至0℃。在5分鐘之後,使用氫氧化鈉水溶液(1N, 20 mL)淬滅反應混合物以中和三氟甲烷磺酸鹽。添加二氯甲烷(150 mL)以稀釋混合物且分離各層。使用DCM (100 mL)萃取水層。使用鹽水(25 mL)洗滌合併之有機層,藉由無水硫酸鈉乾燥,過濾並在壓力下濃縮以獲得粗產物,藉由急速管柱層析在矽膠上使用乙酸乙酯之己烷溶液作為洗脫劑進行純化。使用於己烷中之12 -15%乙酸乙酯分離產物。收集產物部分並在減壓下濃縮以得到N-{雙環[1.1.1]戊烷-1-基}-N-({4-[(3S)-3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基]苯基}甲基)胺基甲酸第三丁基酯。在管柱純化中,亦分離出N-({4-[(3S)-3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基]苯基}甲基)雙環[1.1.1]戊烷-1-胺。LCMS (ES) m/z = 489.3 [M+H]+。1
H NMR (400 MHz, DMSO-d6
) δ ppm 0.87 - 0.91 (m, 3 H), 1.32 - 1.40 (m, 12 H), 1.56 - 1.67 (m, 3 H), 1.95 (s, 6 H), 2.30 - 2.34 (m, 1 H), 2.41 - 2.48 (m, 2 H), 2.74 - 2.78 (m, 1 H), 3.78 (s, 3 H), 4.42 (s, 2 H), 6.80 (d,J
= 8.4 Hz, 1 H), 6.90 (s, 1 H), 7.07 (d,J
= 8.4 Hz, 1 H), 7.23 (d,J
= 7.6 Hz, 2 H), 7.45 - 7.46 (m, 2 H)。
N-{雙環[1.1.1]戊烷-1-基}-N-({4-[(1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基]苯基}甲基)胺基甲酸第三丁基酯:在0℃下,向(S)-雙環[1.1.1]戊烷-1-基(4-(3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基)苄基)胺基甲酸第三丁基酯(0.850 g, 1.74 mmol, 1當量)於甲醇(10 mL)中之溶液中逐份添加硼氫化鈉(0.197 g, 5.22 mmol, 3當量)。將懸浮液在室溫下攪拌1 h。藉由TLC (於己烷中之20% EA)監測反應進展。此後,濃縮反應混合物且使用EtOAc (20 mL)及水(10 mL)稀釋所獲得粗製物。分離有機層,使用鹽水溶液(10 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物。藉由製備型TLC使用於正己烷中之20%乙酸乙酯純化所獲得粗產物。收集產物部分並在減壓下濃縮以得到N-{雙環[1.1.1]戊烷-1-基}-N-({4-[(1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基]苯基}甲基)胺基甲酸第三丁基酯。LCMS (ES) m/z = 491.3 [M+H]+。1
H NMR (400 MHz, DMSO-d6
) δ ppm 0.75 - 0.78 (m, 3 H), 1.08 - 1.17 (m, 6 H), 1.36 (bs, 9 H), 1.90 (s, 6 H), 2.30 (s, 1 H), 2.37 (s, 1 H), 2.73 - 2.75 (m, 2 H), 3.70 (s, 3 H), 4.30 (s, 2 H), 5.04 (bs, 1 H), 6.64 - 6.69 (m, 2 H), 6.75 - 6.77 (m, 1 H), 7.04 (s, 4 H)。
N-{雙環[1.1.1]戊烷-1-基}-N-({4-[(1S,3S)-3-丁基-6-甲氧基-2-[3-(三甲基矽烷基)丙-2-炔醯基]-1,2,3,4-四氫異喹啉-1-基]苯基}甲基)胺基甲酸第三丁基酯:第一步驟:在室溫下,向3-(三甲基矽烷基)丙炔酸(35 mg, 0.246 mmol, 1.0當量)於DMF (0.001 mL, 0.009 mmol, 0.04當量)中之溶液中添加草醯氯(0.023 mL, 0.271 mmol, 1.1當量)並攪拌30分鐘。然後,在減壓下濃縮反應混合物以得到3-(三甲基矽烷基)丙炔醯氯。此醯氯未經任何進一步純化即用於下一步驟中。
第二步驟:在0℃下,向N-{雙環[1.1.1]戊烷-1-基}-N-({4-[(1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基]苯基}甲基)胺基甲酸第三丁基酯(0.085 g, 0.173 mmol, 1當量)於乙腈(3 mL)中之溶液中添加碳酸氫鈉(0.118 g, 1.39 mmol, 8.0當量)。在攪拌5分鐘之後,向上述反應物質中添加3-(三甲基矽烷基)丙炔醯氯(0.031 g, 0.191 mmol, 1.1當量)於乙腈(2.0 mL)中之溶液。將所得混合物在0℃下攪拌1 h。然後使用EtOAc (10 mL)及水(5 mL)稀釋反應混合物。分離有機層,使用鹽水溶液(5 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物。此粗產物未經任何進一步純化即用於下一步驟中。 LCMS (ES) m/z = 615.3 [M+H]+,但觀察到559.3 (不含第三丁基)。
N-{雙環[1.1.1]戊烷-1-基}-N-({4-[(1S,3S)-3-丁基-6-甲氧基-2-(丙-2-炔醯基)-1,2,3,4-四氫異喹啉-1-基]苯基}甲基)胺基甲酸第三丁基酯:在0℃下,向雙環[1.1.1]戊烷-1-基(4-((1S,3S)-3-丁基-6-甲氧基-2-(3-(三甲基矽烷基)丙炔醯基)-1,2,3,4-四氫異喹啉-1-基)苄基)胺基甲酸第三丁基酯(85 mg, 0.138 mmol, 1當量)於DCM (5 mL) / MeOH (1 mL)中之溶液中添加K2CO3 (115 mg, 0.829 mmol, 6當量)。將混合物在0℃下攪拌45分鐘。然後使用DCM (10 mL)稀釋反應混合物並添加水(2 mL)。使用DCM (2 × 5 mL)萃取有機層並藉由無水Na2
SO4
乾燥。過濾有機層並濃縮以獲得粗產物,藉由製備型TLC使用於己烷中之20%乙酸乙酯作為移動相純化產物。收集產物部分並在減壓下濃縮以得到雙環[1.1.1]戊烷-1-基(4-((1S,3S)-3-丁基-6-甲氧基-2-丙炔醯基-1,2,3,4-四氫異喹啉-1-基)苄基)胺基甲酸第三丁基酯。LCMS (ES) m/z = 543.3 [M+H]+,但觀察到不含boc基團者。
N-{雙環[1.1.1]戊烷-1-基}-N-({4-[(1S,3S)-3-丁基-6-甲氧基-2-(丙-2-炔醯基)-1,2,3,4-四氫異喹啉-1-基]苯基}甲基)-2,2,2-三氟乙醯胺:在0℃下,向N-{雙環[1.1.1]戊烷-1-基}-N-({4-[(1S,3S)-3-丁基-6-甲氧基-2-(丙-2-炔醯基)-1,2,3,4-四氫異喹啉-1-基]苯基}甲基)胺基甲酸第三丁基酯(0.040 g, 0.073 mmol, 1.0當量)於DCM (1.0 mL)中之經攪拌溶液中添加TFA (0.150 mL)。然後,將反應混合物在0℃下攪拌1.5 h。在減壓下蒸發反應混合物且將水浴溫度保持於30℃。藉由添加水(1.5 mL)及乙腈(0.5 mL)混合物來使所獲得粗產物保持於凍乾狀態下16 h。將所獲得產物用於分析中。LCMS (ES) m/z = 443.3 [M+H]+ (所觀察到游離胺質量)。1
H NMR (400 MHz, DMSO-d6
) δ ppm 0.79 - 0.82 (m, 3 H), 1.13 - 1.21 (m, 4 H), 1.47 (bs, 2 H), 1.97 - 1.98 (m, 6 H), 2.65 (s, 1 H), 2.77 (bs, 1 H), 2.85 - 2.89 (m, 0.5 H), 3.14 - 3.15 (m, 0.5 H), 3.69 - 3.70 (m, 3 H), 3.98 (s, 2 H), 4.29 (s, 0.5 H), 4.60 (s, 1 H), 4.74 (s, 0.5 H), 6.04 (s, 0.5 H), 6.33(s, 0.5 H), 6.75 - 6.83(m, 2 H), 7.28 - 7.32 (m, 3 H), 7.37 - 7.42 (m, 1.5 H), 7.60 - 7.62 (m, 0.5 H), 9.42 (bs, 2 H)。
程序 26 :化合物 98 之合成 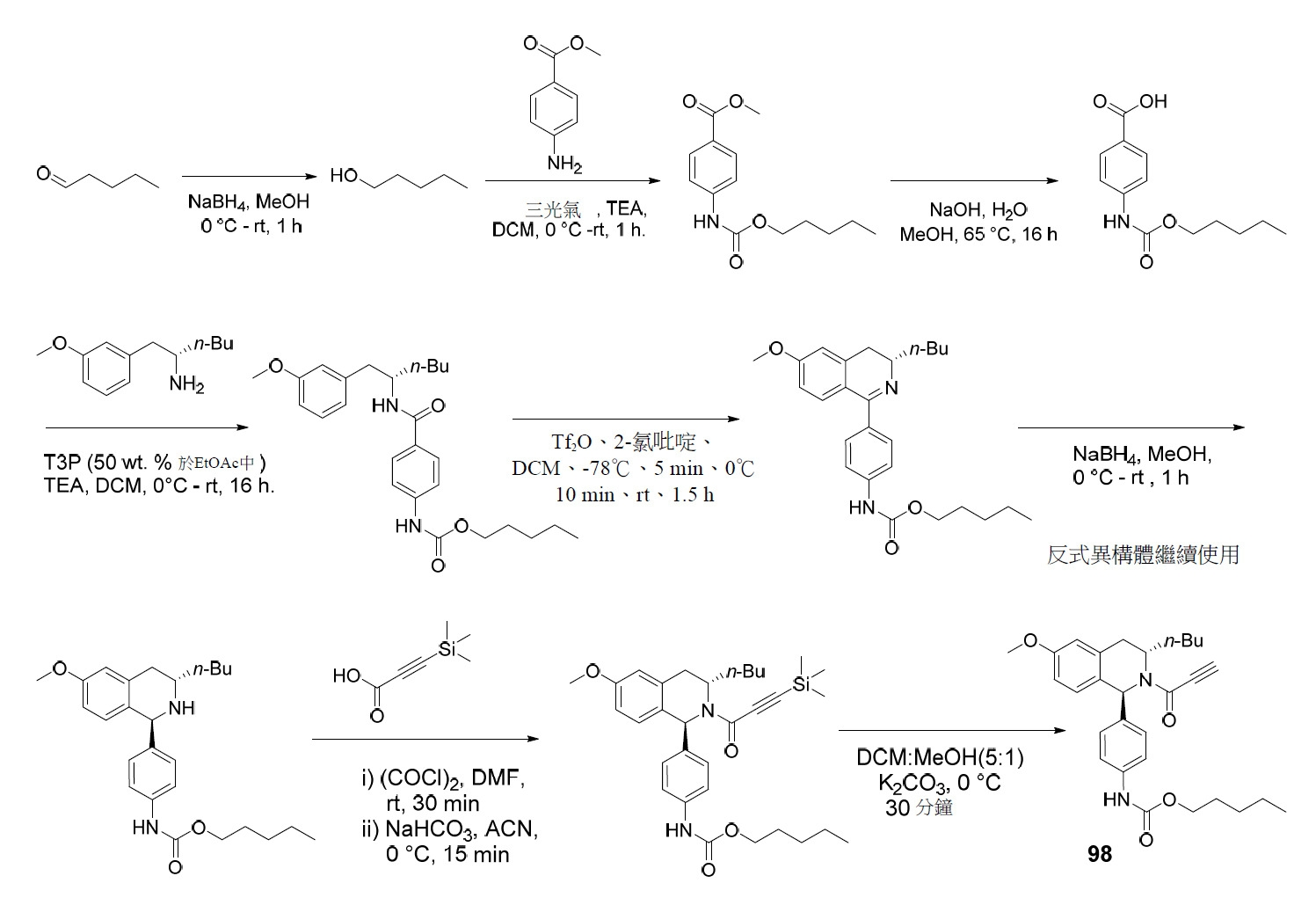 戊烷-1-醇:在0℃下,向戊醛(8.0 g, 92.9 mmol, 1.0當量)於甲醇(50 mL)中之溶液中逐份添加硼氫化鈉(10.5 g, 279 mmol, 3.0當量)。將反應混合物在室溫下攪拌1 h。藉由TLC (於正己烷中之20%乙酸乙酯)監測反應進展。在反應完成之後,使用幾滴丙酮淬滅反應混合物,濃縮,使用水(35 mL)稀釋並使用乙酸乙酯(2 × 50 mL)萃取。藉由無水硫酸鈉乾燥有機層,過濾並在減壓下濃縮以得到粗製戊烷-1-醇。1
H NMR (400 MHz, DMSO-d6
) δ ppm 0.84 (t,J
= 6.0 Hz, 3 H), 1.13 - 1.24 (m, 4 H), 1.37 - 1.42 (m, 2 H), 3.31 - 3.37 (m, 2 H), 4.30 (t,J
= 4.8 Hz, 1 H)。
4-{[(戊基氧基)羰基]胺基}苯甲酸甲酯:向戊烷-1-醇(5.0 g, 56.7 mmol, 1.0當量)於DCM (75 mL)中之溶液中添加4-胺基苯甲酸甲酯(10.3 g, 68.1 mmol, 1.2當量)及三乙胺(39.5 mL, 284 mmol, 5.0當量)。在0℃下,向此混合物中逐份添加三光氣(11.8 g, 23.8 mmol, 0.7當量)。將反應混合物在室溫下攪拌1 h。藉由TLC (於正己烷中之15%乙酸乙酯)監測反應進展。在反應完成之後,使用碳酸氫鈉水溶液(80 mL)淬滅反應混合物並使用乙酸乙酯(140 mL)萃取。藉由無水硫酸鈉乾燥有機層,過濾並在減壓下濃縮以得到粗製物。藉由急速管柱層析在矽膠上使用具有增加極性之於正己烷中之5 - 10%乙酸乙酯純化粗製物以得到4-(((戊基氧基)羰基)胺基)苯甲酸甲酯。1
H NMR (400 MHz, DMSO-d6
) δ ppm 0.86 (s, 3 H), 1.31 (s, 4 H), 1.60 (s, 2 H), 3.78 (s, 3 H), 4.07 (t,J
= 6.0 Hz, 2 H), 7.57 (d,J
= 8.0 Hz, 2 H), 7.85 (d,J
= 8.0 Hz, 2 H), 10.0 (s, 1 H)。
4-{[(戊基氧基)羰基]胺基}苯甲酸:向4-(((戊基氧基)羰基)胺基)苯甲酸甲酯(6.6 g, 24.9 mmol, 1當量)於甲醇(70 mL)中之溶液中添加氫氧化鈉(1.53 g, 37.3 mmol, 1.5當量)及水(35 mL)。將反應混合物在65℃下攪拌16 h。藉由TLC (於正己烷中之40%乙酸乙酯)監測反應進展。在反應完成之後,使用10%檸檬酸溶液將反應混合物酸化至最高pH = 4並使用乙酸乙酯(160 mL)萃取。藉由無水硫酸鈉乾燥有機層,過濾並在減壓下濃縮以得到粗製4-(((戊基氧基)羰基)胺基)苯甲酸。1
H NMR (400 MHz, DMSO-d6
) δ ppm 0.86 (t,J
= 7.2 Hz, 3 H), 1.30 - 1.38 (m, 4 H), 1.59 - 1.62 (m, 2 H), 4.07 (t,J
= 6.4 Hz, 2 H), 7.54 (d,J
= 8.8 Hz, 2 H), 7.83 (d,J
= 8.4 Hz, 2 H), 9.97 (s, 1 H), 12.54 (s, 1 H)。
(S)-(4-((1-(3-甲氧基苯基)己烷-2-基)胺甲醯基)苯基)胺基甲酸戊酯:向4-(((戊基氧基)羰基)胺基)苯甲酸(2.06 g, 8.20 mmol, 1.0當量)於DCM (35 mL)中之溶液中添加(4.61 mL, 32.8 mmol, 4當量),攪拌15 min且然後在0℃下添加T3P (50 wt.%於EtOAc中) (7.8 mL, 12.3 mmol, 1.5當量)並再攪拌5 min。然後,向反應混合物中添加(S)-1-(3-甲氧基苯基)己烷-2-胺(1.70 g, 8.20 mmol, 1.0當量)且然後將反應混合物在室溫下攪拌16 h。藉由TLC (於己烷中之40%乙酸乙酯)監測反應進展。使用DCM (70 mL)及飽和碳酸氫鈉溶液(30 mL)稀釋反應混合物,分離有機層,使用鹽水溶液(12 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得(S)-(4-((1-(3-甲氧基苯基)己烷-2-基)胺甲醯基)苯基)胺基甲酸戊酯。LCMS (ES) m/z = 441.2 [M+H]+ 1
H NMR (400 MHz, DMSO-d6
) δ ppm 0.80 - 0.86 (m, 6 H), 1.21 - 1.31 (m, 8 H), 1.47- 1.72 (m, 4 H), 2.65 - 2.79 (m, 2 H), 3.65 (s, 3 H), 4.06 - 4.13 (m, 3 H), 6.67 - 6.77 (m, 3 H), 7.12 (t,J
= 7.6 Hz, 1 H), 7.47 (d,J
= 8.4 Hz, 2 H), 7.70 (d,J
= 8.4 Hz, 2 H), 7.98 (d,J
= 8.0 Hz, 1 H), 9.81 (s, 1 H)。
(S)-(4-(3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基)苯基)胺基甲酸戊酯:在-78℃下,經由注射器,以1 min時間將三氟甲烷磺酸酐(1.91 mL, 11.3 mmol, 2當量)添加至(S)-(4-((1-(3-甲氧基苯基)己烷-2-基)胺甲醯基)苯基)胺基甲酸戊酯(2.50 g, 5.67 mmol, 1當量)及2-氯吡啶(1.07 mL, 11.3 mmol, 2當量)於二氯甲烷(20 mL)中之經攪拌混合物中。在5 min之後,將反應混合物置於冰-水浴中並升溫至0℃。在5 min之後,將所得溶液升溫至23℃。TLC (於正己烷中之20%乙酸乙酯)顯示反應已完成。在1 h之後,引入氫氧化鈉水溶液(30 mL, 1N)以中和三氟甲烷磺酸鹽。添加二氯甲烷(120 mL)以稀釋混合物且分離各層。使用鹽水(20 mL)洗滌有機層,藉由無水硫酸鈉乾燥,並過濾。在減壓下去除揮發物以得到粗產物。藉由急速層析使用乙酸乙酯之己烷溶液作為洗脫劑純化所獲得粗產物以得到期望產物(S)-(4-(3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基)苯基)胺基甲酸戊酯。LCMS (ES) m/z = 423.3 [M+H]+
。1
H NMR (400 MHz, DMSO-d6
) δ ppm 0.79 - 0.89 (m, 6 H), 1.32 - 1.60 (m, 8 H), 1.61 - 1.96 (m, 4 H), 2.30 - 2.48 (m, 2 H), 2.65 - 2.76 (m, 1 H), 3.78 (s, 3 H), 4.07 (t,J
= 6.4 Hz, 2 H), 6.80 (d,J
= 8.4 Hz, 1 H), 6.89 (s, 1 H), 7.11 (d,J
= 8.4 Hz, 1 H), 7.42 (d,J
= 8.8 Hz, 2 H), 7.51 (d,J
= 8.8 Hz, 2 H), 9.74 (s, 1 H)。
N-{4-[(1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基]苯基}胺基甲酸戊酯:在0℃下,向(S)-(4-(3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基)苯基)胺基甲酸戊酯(0.300 g, 0.710 mmol, 1當量)於甲醇(6 mL)中之溶液中逐份添加硼氫化鈉(0.081 g, 2.13 mmol, 3當量)。將懸浮液在室溫下攪拌1 h。此後,濃縮反應混合物且使用EtOAc (30 mL)及水(10 mL)稀釋所獲得粗製物。分離有機層,使用鹽水溶液(10 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物。藉由製備型TLC使用於正己烷中之30%乙酸乙酯作為洗脫劑純化所獲得粗產物。收集產物部分並在減壓下濃縮以得到(4-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)苯基)胺基甲酸戊酯。LCMS (ES) m/z = 425.3 [M+H]+1
H NMR (400 MHz, DMSO-d6
) δ ppm 0.79 - 0.85 (m, 6 H), 1.17 - 1.35 (m, 10 H), 1.58 (s, 2 H), 2.77 (bs, 2 H), 3.14 - 3.15 (m,1 H), 3.70 (s, 3 H), 4.00 - 4.02 (m, 2 H), 5.04 (s, 1 H), 6.64 - 6.74 (m , 3 H), 6.99 (d,J
= 8.0 Hz, 2 H), 7.31 (d,J
= 7.6 Hz, 2 H), 9.50 (s, 1 H)。
N-{4-[(1S,3S)-3-丁基-6-甲氧基-2-[3-(三甲基矽烷基)丙-2-炔醯基]-1,2,3,4-四氫異喹啉-1-基]苯基}胺基甲酸戊酯:第一步驟:在室溫下,向3-(三甲基矽烷基)丙炔酸(15 mg, 0.105 mmol, 1.0當量)於DMF (0.0003 mL, 0.004 mmol, 0.04當量)中之溶液中添加草醯氯(0.010 mL, 0.116 mmol, 1.1當量)並攪拌30分鐘。然後,在減壓下濃縮反應混合物以得到3-(三甲基矽烷基)丙炔醯氯。此醯氯未經任何進一步純化即用於下一步驟中。
第二步驟:在0℃下,向N-{4-[(1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基]苯基}胺基甲酸戊酯(0.035 g, 0.082 mmol, 1.0當量)於乙腈(3 mL)中之溶液中添加碳酸氫鈉(0.057 g, 0.659 mmol, 8.0當量)。在攪拌5分鐘之後,向上述反應物質中添加3-(三甲基矽烷基)丙炔醯氯(0.015 g, 0.090 mmol, 1.1當量)於乙腈(2.0 mL)中之溶液。將所得混合物在0℃下攪拌30分鐘,藉由TLC (於正己烷中之20%乙酸乙酯)監測反應進展。此後,使用EtOAc (15 mL)及水(5 mL)稀釋反應物質。分離有機層,使用鹽水溶液(5 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物。此粗產物未經任何進一步純化即用於下一步驟中。LC-MS (ES) m/z = 549.4 [M+H]+。
N-{4-[(1S,3S)-3-丁基-6-甲氧基-2-(丙-2-炔醯基)-1,2,3,4-四氫異喹啉-1-基]苯基}胺基甲酸戊酯:在0℃下,向N-{4-[(1S,3S)-3-丁基-6-甲氧基-2-[3-(三甲基矽烷基)丙-2-炔醯基]-1,2,3,4-四氫異喹啉-1-基]苯基}胺基甲酸戊酯(0.045 g, 0.082 mmol, 1當量)於MeOH:DCM (1:5) (6 mL)混合物中之經攪拌溶液中添加碳酸鉀(0.068 g, 0.492 mmol, 6當量)。然後,將反應混合物在0℃下攪拌30分鐘。然後,使用DCM (15.0 mL)稀釋反應混合物,使用水(3.0 mL)洗滌。分離有機層並藉由無水硫酸鈉乾燥。過濾有機層並在減壓下濃縮以得到粗產物,藉由製備型TLC使用於己烷中之20%乙酸乙酯作為洗脫劑進行純化。收集產物部分並在減壓下濃縮以得到純產物。藉由溶於乙腈(1.0 mL)及水(2.0 mL)混合物中來使所獲得純產物保持於凍乾狀態下16 h以得到N-{4-[(1S,3S)-3-丁基-6-甲氧基-2-(丙-2-炔醯基)-1,2,3,4-四氫異喹啉-1-基]苯基}胺基甲酸戊酯。LCMS (ES) m/z: 477.3 [M+H]+。1
H NMR (400 MHz, DMSO-d 6
) δ ppm: 0.78 - 0.85 (m, 6 H), 1.21 - 1.29 (m, 9 H), 1.46 - 1.56 (m, 3 H), 2.65 (s, 1 H), 2.82 - 2.86 (m, 0.5 H), 3.06 - 3.09 (m, 0.5 H), 3.69 - 3.70 (m, 3 H), 3.98 - 4.01 (m, 2 H), 4.30 (s, 0.5 H), 4.49 (s, 0.5 H), 4.60 (s, 0.5 H), 4.68 (s, 0.5 H), 5.97 (s, 0.5 H), 6.22 (s, 0.5 H), 6.75 - 6.81 (m, 2 H), 7.07 - 7.12 (m, 2 H), 7.25 - 7.27 (m, 1 H), 7.31 - 7.36 (m, 1.5 H), 7.53 -7.55 (m, 0.5 H), 9.46 (s, 0.5 H), 9.53 (s, 0.5 H)。
戊烷-1-醇:在0℃下,向戊醛(8.0 g, 92.9 mmol, 1.0當量)於甲醇(50 mL)中之溶液中逐份添加硼氫化鈉(10.5 g, 279 mmol, 3.0當量)。將反應混合物在室溫下攪拌1 h。藉由TLC (於正己烷中之20%乙酸乙酯)監測反應進展。在反應完成之後,使用幾滴丙酮淬滅反應混合物,濃縮,使用水(35 mL)稀釋並使用乙酸乙酯(2 × 50 mL)萃取。藉由無水硫酸鈉乾燥有機層,過濾並在減壓下濃縮以得到粗製戊烷-1-醇。1
H NMR (400 MHz, DMSO-d6
) δ ppm 0.84 (t,J
= 6.0 Hz, 3 H), 1.13 - 1.24 (m, 4 H), 1.37 - 1.42 (m, 2 H), 3.31 - 3.37 (m, 2 H), 4.30 (t,J
= 4.8 Hz, 1 H)。
4-{[(戊基氧基)羰基]胺基}苯甲酸甲酯:向戊烷-1-醇(5.0 g, 56.7 mmol, 1.0當量)於DCM (75 mL)中之溶液中添加4-胺基苯甲酸甲酯(10.3 g, 68.1 mmol, 1.2當量)及三乙胺(39.5 mL, 284 mmol, 5.0當量)。在0℃下,向此混合物中逐份添加三光氣(11.8 g, 23.8 mmol, 0.7當量)。將反應混合物在室溫下攪拌1 h。藉由TLC (於正己烷中之15%乙酸乙酯)監測反應進展。在反應完成之後,使用碳酸氫鈉水溶液(80 mL)淬滅反應混合物並使用乙酸乙酯(140 mL)萃取。藉由無水硫酸鈉乾燥有機層,過濾並在減壓下濃縮以得到粗製物。藉由急速管柱層析在矽膠上使用具有增加極性之於正己烷中之5 - 10%乙酸乙酯純化粗製物以得到4-(((戊基氧基)羰基)胺基)苯甲酸甲酯。1
H NMR (400 MHz, DMSO-d6
) δ ppm 0.86 (s, 3 H), 1.31 (s, 4 H), 1.60 (s, 2 H), 3.78 (s, 3 H), 4.07 (t,J
= 6.0 Hz, 2 H), 7.57 (d,J
= 8.0 Hz, 2 H), 7.85 (d,J
= 8.0 Hz, 2 H), 10.0 (s, 1 H)。
4-{[(戊基氧基)羰基]胺基}苯甲酸:向4-(((戊基氧基)羰基)胺基)苯甲酸甲酯(6.6 g, 24.9 mmol, 1當量)於甲醇(70 mL)中之溶液中添加氫氧化鈉(1.53 g, 37.3 mmol, 1.5當量)及水(35 mL)。將反應混合物在65℃下攪拌16 h。藉由TLC (於正己烷中之40%乙酸乙酯)監測反應進展。在反應完成之後,使用10%檸檬酸溶液將反應混合物酸化至最高pH = 4並使用乙酸乙酯(160 mL)萃取。藉由無水硫酸鈉乾燥有機層,過濾並在減壓下濃縮以得到粗製4-(((戊基氧基)羰基)胺基)苯甲酸。1
H NMR (400 MHz, DMSO-d6
) δ ppm 0.86 (t,J
= 7.2 Hz, 3 H), 1.30 - 1.38 (m, 4 H), 1.59 - 1.62 (m, 2 H), 4.07 (t,J
= 6.4 Hz, 2 H), 7.54 (d,J
= 8.8 Hz, 2 H), 7.83 (d,J
= 8.4 Hz, 2 H), 9.97 (s, 1 H), 12.54 (s, 1 H)。
(S)-(4-((1-(3-甲氧基苯基)己烷-2-基)胺甲醯基)苯基)胺基甲酸戊酯:向4-(((戊基氧基)羰基)胺基)苯甲酸(2.06 g, 8.20 mmol, 1.0當量)於DCM (35 mL)中之溶液中添加(4.61 mL, 32.8 mmol, 4當量),攪拌15 min且然後在0℃下添加T3P (50 wt.%於EtOAc中) (7.8 mL, 12.3 mmol, 1.5當量)並再攪拌5 min。然後,向反應混合物中添加(S)-1-(3-甲氧基苯基)己烷-2-胺(1.70 g, 8.20 mmol, 1.0當量)且然後將反應混合物在室溫下攪拌16 h。藉由TLC (於己烷中之40%乙酸乙酯)監測反應進展。使用DCM (70 mL)及飽和碳酸氫鈉溶液(30 mL)稀釋反應混合物,分離有機層,使用鹽水溶液(12 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得(S)-(4-((1-(3-甲氧基苯基)己烷-2-基)胺甲醯基)苯基)胺基甲酸戊酯。LCMS (ES) m/z = 441.2 [M+H]+ 1
H NMR (400 MHz, DMSO-d6
) δ ppm 0.80 - 0.86 (m, 6 H), 1.21 - 1.31 (m, 8 H), 1.47- 1.72 (m, 4 H), 2.65 - 2.79 (m, 2 H), 3.65 (s, 3 H), 4.06 - 4.13 (m, 3 H), 6.67 - 6.77 (m, 3 H), 7.12 (t,J
= 7.6 Hz, 1 H), 7.47 (d,J
= 8.4 Hz, 2 H), 7.70 (d,J
= 8.4 Hz, 2 H), 7.98 (d,J
= 8.0 Hz, 1 H), 9.81 (s, 1 H)。
(S)-(4-(3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基)苯基)胺基甲酸戊酯:在-78℃下,經由注射器,以1 min時間將三氟甲烷磺酸酐(1.91 mL, 11.3 mmol, 2當量)添加至(S)-(4-((1-(3-甲氧基苯基)己烷-2-基)胺甲醯基)苯基)胺基甲酸戊酯(2.50 g, 5.67 mmol, 1當量)及2-氯吡啶(1.07 mL, 11.3 mmol, 2當量)於二氯甲烷(20 mL)中之經攪拌混合物中。在5 min之後,將反應混合物置於冰-水浴中並升溫至0℃。在5 min之後,將所得溶液升溫至23℃。TLC (於正己烷中之20%乙酸乙酯)顯示反應已完成。在1 h之後,引入氫氧化鈉水溶液(30 mL, 1N)以中和三氟甲烷磺酸鹽。添加二氯甲烷(120 mL)以稀釋混合物且分離各層。使用鹽水(20 mL)洗滌有機層,藉由無水硫酸鈉乾燥,並過濾。在減壓下去除揮發物以得到粗產物。藉由急速層析使用乙酸乙酯之己烷溶液作為洗脫劑純化所獲得粗產物以得到期望產物(S)-(4-(3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基)苯基)胺基甲酸戊酯。LCMS (ES) m/z = 423.3 [M+H]+
。1
H NMR (400 MHz, DMSO-d6
) δ ppm 0.79 - 0.89 (m, 6 H), 1.32 - 1.60 (m, 8 H), 1.61 - 1.96 (m, 4 H), 2.30 - 2.48 (m, 2 H), 2.65 - 2.76 (m, 1 H), 3.78 (s, 3 H), 4.07 (t,J
= 6.4 Hz, 2 H), 6.80 (d,J
= 8.4 Hz, 1 H), 6.89 (s, 1 H), 7.11 (d,J
= 8.4 Hz, 1 H), 7.42 (d,J
= 8.8 Hz, 2 H), 7.51 (d,J
= 8.8 Hz, 2 H), 9.74 (s, 1 H)。
N-{4-[(1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基]苯基}胺基甲酸戊酯:在0℃下,向(S)-(4-(3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基)苯基)胺基甲酸戊酯(0.300 g, 0.710 mmol, 1當量)於甲醇(6 mL)中之溶液中逐份添加硼氫化鈉(0.081 g, 2.13 mmol, 3當量)。將懸浮液在室溫下攪拌1 h。此後,濃縮反應混合物且使用EtOAc (30 mL)及水(10 mL)稀釋所獲得粗製物。分離有機層,使用鹽水溶液(10 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物。藉由製備型TLC使用於正己烷中之30%乙酸乙酯作為洗脫劑純化所獲得粗產物。收集產物部分並在減壓下濃縮以得到(4-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)苯基)胺基甲酸戊酯。LCMS (ES) m/z = 425.3 [M+H]+1
H NMR (400 MHz, DMSO-d6
) δ ppm 0.79 - 0.85 (m, 6 H), 1.17 - 1.35 (m, 10 H), 1.58 (s, 2 H), 2.77 (bs, 2 H), 3.14 - 3.15 (m,1 H), 3.70 (s, 3 H), 4.00 - 4.02 (m, 2 H), 5.04 (s, 1 H), 6.64 - 6.74 (m , 3 H), 6.99 (d,J
= 8.0 Hz, 2 H), 7.31 (d,J
= 7.6 Hz, 2 H), 9.50 (s, 1 H)。
N-{4-[(1S,3S)-3-丁基-6-甲氧基-2-[3-(三甲基矽烷基)丙-2-炔醯基]-1,2,3,4-四氫異喹啉-1-基]苯基}胺基甲酸戊酯:第一步驟:在室溫下,向3-(三甲基矽烷基)丙炔酸(15 mg, 0.105 mmol, 1.0當量)於DMF (0.0003 mL, 0.004 mmol, 0.04當量)中之溶液中添加草醯氯(0.010 mL, 0.116 mmol, 1.1當量)並攪拌30分鐘。然後,在減壓下濃縮反應混合物以得到3-(三甲基矽烷基)丙炔醯氯。此醯氯未經任何進一步純化即用於下一步驟中。
第二步驟:在0℃下,向N-{4-[(1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基]苯基}胺基甲酸戊酯(0.035 g, 0.082 mmol, 1.0當量)於乙腈(3 mL)中之溶液中添加碳酸氫鈉(0.057 g, 0.659 mmol, 8.0當量)。在攪拌5分鐘之後,向上述反應物質中添加3-(三甲基矽烷基)丙炔醯氯(0.015 g, 0.090 mmol, 1.1當量)於乙腈(2.0 mL)中之溶液。將所得混合物在0℃下攪拌30分鐘,藉由TLC (於正己烷中之20%乙酸乙酯)監測反應進展。此後,使用EtOAc (15 mL)及水(5 mL)稀釋反應物質。分離有機層,使用鹽水溶液(5 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物。此粗產物未經任何進一步純化即用於下一步驟中。LC-MS (ES) m/z = 549.4 [M+H]+。
N-{4-[(1S,3S)-3-丁基-6-甲氧基-2-(丙-2-炔醯基)-1,2,3,4-四氫異喹啉-1-基]苯基}胺基甲酸戊酯:在0℃下,向N-{4-[(1S,3S)-3-丁基-6-甲氧基-2-[3-(三甲基矽烷基)丙-2-炔醯基]-1,2,3,4-四氫異喹啉-1-基]苯基}胺基甲酸戊酯(0.045 g, 0.082 mmol, 1當量)於MeOH:DCM (1:5) (6 mL)混合物中之經攪拌溶液中添加碳酸鉀(0.068 g, 0.492 mmol, 6當量)。然後,將反應混合物在0℃下攪拌30分鐘。然後,使用DCM (15.0 mL)稀釋反應混合物,使用水(3.0 mL)洗滌。分離有機層並藉由無水硫酸鈉乾燥。過濾有機層並在減壓下濃縮以得到粗產物,藉由製備型TLC使用於己烷中之20%乙酸乙酯作為洗脫劑進行純化。收集產物部分並在減壓下濃縮以得到純產物。藉由溶於乙腈(1.0 mL)及水(2.0 mL)混合物中來使所獲得純產物保持於凍乾狀態下16 h以得到N-{4-[(1S,3S)-3-丁基-6-甲氧基-2-(丙-2-炔醯基)-1,2,3,4-四氫異喹啉-1-基]苯基}胺基甲酸戊酯。LCMS (ES) m/z: 477.3 [M+H]+。1
H NMR (400 MHz, DMSO-d 6
) δ ppm: 0.78 - 0.85 (m, 6 H), 1.21 - 1.29 (m, 9 H), 1.46 - 1.56 (m, 3 H), 2.65 (s, 1 H), 2.82 - 2.86 (m, 0.5 H), 3.06 - 3.09 (m, 0.5 H), 3.69 - 3.70 (m, 3 H), 3.98 - 4.01 (m, 2 H), 4.30 (s, 0.5 H), 4.49 (s, 0.5 H), 4.60 (s, 0.5 H), 4.68 (s, 0.5 H), 5.97 (s, 0.5 H), 6.22 (s, 0.5 H), 6.75 - 6.81 (m, 2 H), 7.07 - 7.12 (m, 2 H), 7.25 - 7.27 (m, 1 H), 7.31 - 7.36 (m, 1.5 H), 7.53 -7.55 (m, 0.5 H), 9.46 (s, 0.5 H), 9.53 (s, 0.5 H)。
程序 27 :化合物 101 之合成  1-((1S,3S)-1-(4-(((3R,5R,7R)-金剛烷-1-基)胺基)苯基)-3-丁基-6-甲氧基-3,4-二氫異喹啉-2(1H)-基)丙-2-炔-1-酮鹽酸鹽:在0℃下,向1-((1S,3S)-1-(4-(((3R,5R,7R)-金剛烷-1-基)胺基)苯基)-3-丁基-6-甲氧基-3,4-二氫異喹啉-2(1H)-基)丙-2-炔-1-酮(10 mg, 0.02 mmol, 1當量)於ACN (1.0 mL)中之溶液中添加1M HCl (水溶液) (0.04 mL, 0.04 mmol, 2當量)且將混合物在0℃下攪拌15 min。此後,使用水(2 mL)稀釋反應混合物,隨後冷卻至-78℃並凍乾以得到1-((1S,3S)-1-(4-(((3R,5R,7R)-金剛烷-1-基)胺基)苯基)-3-丁基-6-甲氧基-3,4-二氫異喹啉-2(1H)-基)丙-2-炔-1-酮鹽酸鹽。LCMS (ES) m/z = 497.3 [M+H]+,不包含HCl鹽質量。1
H NMR (400 MHz, DMSO-d6
) δ ppm 0.80 (t,J
= 6.4 Hz, 3 H), 1.19 - 1.21 (m, 6 H), 1.47 - 1.60 (m, 7 H), 1.75 (s, 5 H), 2.07 (s, 3 H), 2.64 (bs, 1 H), 2.86 - 2.90 (m, 1 H), 3.06 - 3.09 (m, 1 H), 3.69 - 3.71 (m, 3 H), 4.35 (s, 0.5 H), 4.55 (bs, 0.5 H), 4.65 (s, 0.5 H), 4.72 (bs, 0.5 H), 6.09 (s, 1 H), 6.35 (bs, 0.5 H), 6.78 - 6.84 (m, 2 H), 7.15 - 7.30 (m, 2 H), 7.41 - 7.47 (m, 2 H), 7.60 (bs, 0.5 H), 10.50 - 10.60 (m, 1 H)。
1-((1S,3S)-1-(4-(((3R,5R,7R)-金剛烷-1-基)胺基)苯基)-3-丁基-6-甲氧基-3,4-二氫異喹啉-2(1H)-基)丙-2-炔-1-酮鹽酸鹽:在0℃下,向1-((1S,3S)-1-(4-(((3R,5R,7R)-金剛烷-1-基)胺基)苯基)-3-丁基-6-甲氧基-3,4-二氫異喹啉-2(1H)-基)丙-2-炔-1-酮(10 mg, 0.02 mmol, 1當量)於ACN (1.0 mL)中之溶液中添加1M HCl (水溶液) (0.04 mL, 0.04 mmol, 2當量)且將混合物在0℃下攪拌15 min。此後,使用水(2 mL)稀釋反應混合物,隨後冷卻至-78℃並凍乾以得到1-((1S,3S)-1-(4-(((3R,5R,7R)-金剛烷-1-基)胺基)苯基)-3-丁基-6-甲氧基-3,4-二氫異喹啉-2(1H)-基)丙-2-炔-1-酮鹽酸鹽。LCMS (ES) m/z = 497.3 [M+H]+,不包含HCl鹽質量。1
H NMR (400 MHz, DMSO-d6
) δ ppm 0.80 (t,J
= 6.4 Hz, 3 H), 1.19 - 1.21 (m, 6 H), 1.47 - 1.60 (m, 7 H), 1.75 (s, 5 H), 2.07 (s, 3 H), 2.64 (bs, 1 H), 2.86 - 2.90 (m, 1 H), 3.06 - 3.09 (m, 1 H), 3.69 - 3.71 (m, 3 H), 4.35 (s, 0.5 H), 4.55 (bs, 0.5 H), 4.65 (s, 0.5 H), 4.72 (bs, 0.5 H), 6.09 (s, 1 H), 6.35 (bs, 0.5 H), 6.78 - 6.84 (m, 2 H), 7.15 - 7.30 (m, 2 H), 7.41 - 7.47 (m, 2 H), 7.60 (bs, 0.5 H), 10.50 - 10.60 (m, 1 H)。
程序 28 :化合物 97 之合成 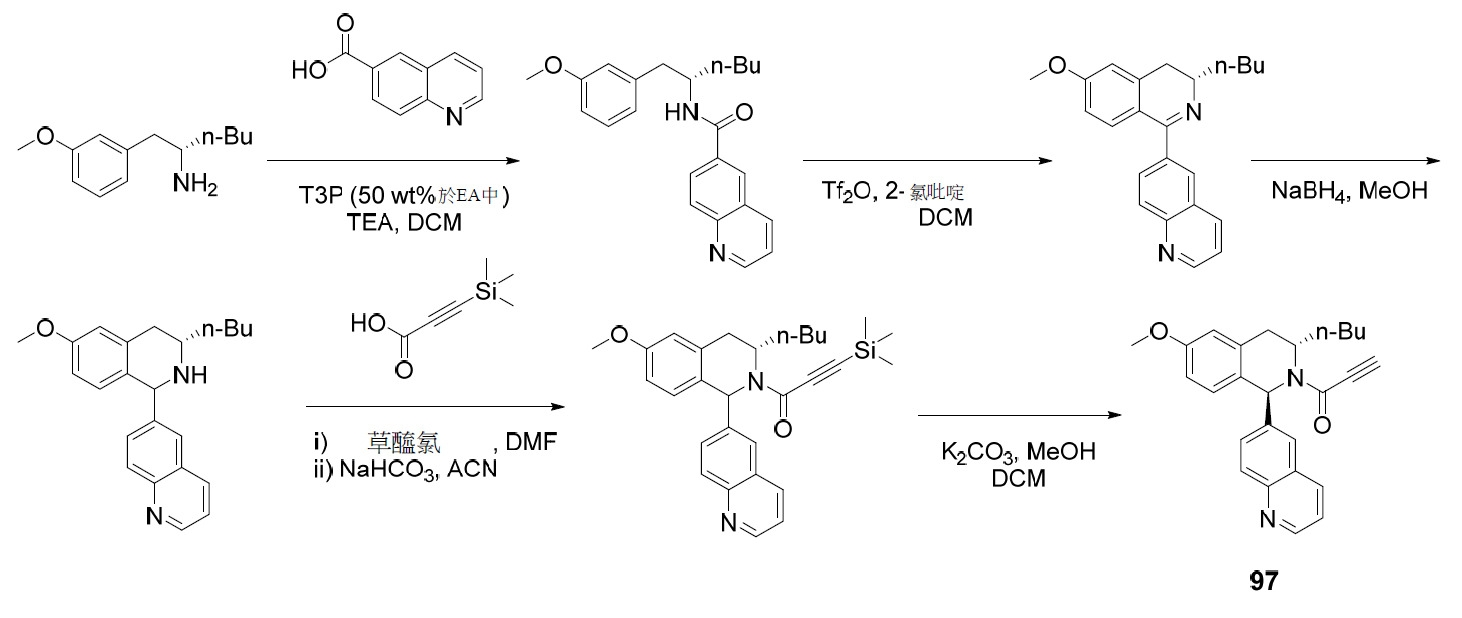 (S)-N-(1-(3-甲氧基苯基)己烷-2-基)喹啉-6-甲醯胺:在0℃及氮氣氛下,向異喹啉-6-甲酸(0.735 g, 4.24 mmol, 1當量)於DCM (10 mL)中之溶液中添加1-(3-甲氧基苯基)己烷-2-胺(0.968 g, 4.67 mmol, 1.1當量),攪拌10 min且然後在0℃下向反應混合物中添加丙烷膦酸酐(4.18 mL, 6.37 mmol, 1.5當量),在0℃下攪拌15 min且然後在0℃下向反應混合物中添加溶於DCM (10 mL)中之三乙胺(2.20 mL, 17 mmol, 4當量),且然後將反應混合物在室溫下攪拌2 h。TLC (於己烷中之40% EtOAc)顯示反應在2 h之後完成。使用乙酸乙酯(100 mL)稀釋反應混合物,使用飽和碳酸氫鈉溶液(20 mL)及水(30 mL)洗滌。藉由無水Na2
SO4
乾燥有機層,過濾並在減壓下濃縮以得到(S)-N-(1-(3-甲氧基苯基)己烷-2-基)喹啉-6-甲醯胺粗製物。LCMS (ES) m/z = 363 [M+H]+。
(S)-6-(3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基)喹諾酮:在室溫及氮氣氛下,向N-[(2S)-1-(3-甲氧基苯基)己烷-2-基]喹啉-6-甲醯胺(1 g, 2.76 mmol, 1當量)於DCM (10 mL)中之溶液中添加2-氯吡啶(0.783 mL, 8.28 mmol, 3當量)。然後在-78℃下添加三氟甲烷磺酸酐(1.39 mL, 8.28 mmol, 3當量),攪拌5 min,然後升溫至0℃,在0℃下攪拌30 min且然後將反應混合物在室溫下攪拌1 h。TLC (於DCM中之5% MeOH)展示起始材料以及新斑點。藉由LCMS監測反應。在減壓下濃縮反應物質以獲得粗製殘餘物,使用10%氫氧化鈉溶液(15 mL)淬滅所獲得殘餘物,使用DCM (2 × 150 mL)萃取,使用無水Na2SO4乾燥合併之有機層,過濾並在減壓下濃縮以得到粗產物,藉由急速管柱層析使用於DCM中之5% MeOH作為洗脫劑純化以獲得6-[(3S)-3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基]喹啉。LCMS (ES) m/z = 345 [M+H]+。1
H NMR (400 MHz, DMSO-d6
) δ ppm 0.90 (t,J
= 7.2 Hz, 3 H), 1.46 (s, 1 H), 1.61 (t,J
= 6.4 Hz, 3 H), 1.73 (s, 2 H), 2.65 (s, 1 H), 2.80 - 2.83 (m, 1 H), 3.42 (d,J
= 11.2 Hz, 1 H), 3.80 (s, 3 H), 6.83 (d,J
= 8.8 Hz, 1 H), 6.95 (s, 1 H), 7.17 (d,J
= 8.8 Hz, 1 H), 7.54 - 7.57 (m, 1 H), 7.91 -8.10 (m, 3 H), 8.45 (d,J
= 7.6 Hz, 1 H), 8.93 (d,J
= 2.8 Hz, 1 H)。
6-((3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)喹諾酮:在0℃下,向(S)-6-(3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基)喹啉(0.5 g, 1.45 mmol, 1當量)於MeOH (10 mL)中之經攪拌溶液中添加硼氫化鈉(0.165 g, 4.35 mmol, 3當量)且將反應液在室溫下攪拌1 h。藉由TLC (於己烷中之70% EtOAc)監測反應。此後,完成完成。在減壓下濃縮反應液以去除甲醇且將所獲得粗製物溶於EtOAc (100 mL)中,並使用水(10 mL)洗滌。藉由無水硫酸鈉乾燥有機層,過濾並在減壓下濃縮以提供6-((3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)喹啉。LCMS (ES) m/z = 347.2 [M+H]+。
1-((3S)-3-丁基-6-甲氧基-1-(喹啉-6-基)-3,4-二氫異喹啉-2(1H)-基)-3-(三甲基矽烷基)丙-2-炔-1-酮:步驟1:在室溫下,向於DMF (0.004 mL, 0.054 mmol, 0.04當量)中之3-(三甲基矽烷基)丙炔酸(0.195 g, 1.37 mmol, 1當量)中添加草醯氯(0.13 mL, 1.51 mmol, 1.1當量)且將反應液在室溫下攪拌30 min。此後,在減壓下濃縮反應混合物以提供3-(三甲基矽烷基)丙炔醯氯且將其用於下一步驟中。
步驟2:在0℃下,向6-((3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)喹啉(0.35 g, 1.01 mmol, 1當量)於ACN (8.0 mL)中之經攪拌溶液中添加碳酸氫鈉(0.64 g, 7.58 mmol, 7.5當量),在0℃下攪拌15 min且然後在0℃下添加於ACN (2.0 mL)中之3-(三甲基矽烷基)丙炔醯氯(0.19 g, 1.21 mmol, 1.2當量)
,並將反應液在室溫下攪拌30 min。藉由TLC (於己烷中之70% EtOAc)監測反應。此後,使用EtOAc (100 mL)稀釋反應混合物且使用水(10 mL)洗滌。藉由無水硫酸鈉乾燥有機層,過濾並在減壓下濃縮以提供1-((3S)-3-丁基-6-甲氧基-1-(喹啉-6-基)-3,4-二氫異喹啉-2(1H)-基)-3-(三甲基矽烷基)丙-2-炔-1-酮。LCMS (ES) m/z = 471.3 [M+H]+。
1-((1S,3S)-3-丁基-6-甲氧基-1-(喹啉-6-基)-3,4-二氫異喹啉-2(1H)-基)丙-2-炔-1-酮:在0℃下,向1-((3S)-3-丁基-6-甲氧基-1-(喹啉-6-基)-3,4-二氫異喹啉-2(1H)-基)-3-(三甲基矽烷基)丙-2-炔-1-酮(0.53 g, 1.13 mmol, 1當量)於甲醇(4 mL)及DCM (30 mL)中之經攪拌溶液中添加碳酸鉀(0.93 g, 6.76 mmol, 6當量)且將反應液在0℃下攪拌1 h。藉由TLC (於己烷中之50% EtOAc)監測反應。此後,使用水(10 mL)稀釋反應混合物並使用DCM (150 mL)萃取。藉由無水硫酸鈉乾燥有機層,過濾並在減壓下濃縮以得到粗製物。藉由製備型TLC使用於己烷中之50% EtOAc作為洗脫劑(洗脫兩次)將粗製物純化兩次以提供1-((1S,3S)-3-丁基-6-甲氧基-1-(喹啉-6-基)-3,4-二氫異喹啉-2(1H)-基)丙-2-炔-1-酮。LCMS (ES) m/z = 399.2 [M+H]+。1
H NMR (400 MHz, DMSO-d6
) δ ppm 0.82 (t,J
= 7.0 Hz, 3 H), 1.20 - 1.21 (m, 4 H), 1.53 (bs, 2 H), 2.80 - 2.84 (m, 1 H), 2.90 - 2.94 (m, 1H), 3.68 - 3.70 (m, 3 H), 4.25 (s, 0.38 H), 4.62 (s, 0.73 H), 4.68 (bs, 0.6 H), 4.82 (bs, 0.8 H), 6.21 (s, 0.6 H), 6.49 (s, 0.4 H), 6.77 - 6.86 (m, 2 H), 7.44 - 7.52 (m, 2 H), 7.71 (t,J
= 7.2 Hz, 1.5 H), 7.79 - 7.91 (m, 2.5 H), 8.28 - 8.36 (m, 1 H), 8.78 - 8.82 (m, 1H)。
(S)-N-(1-(3-甲氧基苯基)己烷-2-基)喹啉-6-甲醯胺:在0℃及氮氣氛下,向異喹啉-6-甲酸(0.735 g, 4.24 mmol, 1當量)於DCM (10 mL)中之溶液中添加1-(3-甲氧基苯基)己烷-2-胺(0.968 g, 4.67 mmol, 1.1當量),攪拌10 min且然後在0℃下向反應混合物中添加丙烷膦酸酐(4.18 mL, 6.37 mmol, 1.5當量),在0℃下攪拌15 min且然後在0℃下向反應混合物中添加溶於DCM (10 mL)中之三乙胺(2.20 mL, 17 mmol, 4當量),且然後將反應混合物在室溫下攪拌2 h。TLC (於己烷中之40% EtOAc)顯示反應在2 h之後完成。使用乙酸乙酯(100 mL)稀釋反應混合物,使用飽和碳酸氫鈉溶液(20 mL)及水(30 mL)洗滌。藉由無水Na2
SO4
乾燥有機層,過濾並在減壓下濃縮以得到(S)-N-(1-(3-甲氧基苯基)己烷-2-基)喹啉-6-甲醯胺粗製物。LCMS (ES) m/z = 363 [M+H]+。
(S)-6-(3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基)喹諾酮:在室溫及氮氣氛下,向N-[(2S)-1-(3-甲氧基苯基)己烷-2-基]喹啉-6-甲醯胺(1 g, 2.76 mmol, 1當量)於DCM (10 mL)中之溶液中添加2-氯吡啶(0.783 mL, 8.28 mmol, 3當量)。然後在-78℃下添加三氟甲烷磺酸酐(1.39 mL, 8.28 mmol, 3當量),攪拌5 min,然後升溫至0℃,在0℃下攪拌30 min且然後將反應混合物在室溫下攪拌1 h。TLC (於DCM中之5% MeOH)展示起始材料以及新斑點。藉由LCMS監測反應。在減壓下濃縮反應物質以獲得粗製殘餘物,使用10%氫氧化鈉溶液(15 mL)淬滅所獲得殘餘物,使用DCM (2 × 150 mL)萃取,使用無水Na2SO4乾燥合併之有機層,過濾並在減壓下濃縮以得到粗產物,藉由急速管柱層析使用於DCM中之5% MeOH作為洗脫劑純化以獲得6-[(3S)-3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基]喹啉。LCMS (ES) m/z = 345 [M+H]+。1
H NMR (400 MHz, DMSO-d6
) δ ppm 0.90 (t,J
= 7.2 Hz, 3 H), 1.46 (s, 1 H), 1.61 (t,J
= 6.4 Hz, 3 H), 1.73 (s, 2 H), 2.65 (s, 1 H), 2.80 - 2.83 (m, 1 H), 3.42 (d,J
= 11.2 Hz, 1 H), 3.80 (s, 3 H), 6.83 (d,J
= 8.8 Hz, 1 H), 6.95 (s, 1 H), 7.17 (d,J
= 8.8 Hz, 1 H), 7.54 - 7.57 (m, 1 H), 7.91 -8.10 (m, 3 H), 8.45 (d,J
= 7.6 Hz, 1 H), 8.93 (d,J
= 2.8 Hz, 1 H)。
6-((3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)喹諾酮:在0℃下,向(S)-6-(3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基)喹啉(0.5 g, 1.45 mmol, 1當量)於MeOH (10 mL)中之經攪拌溶液中添加硼氫化鈉(0.165 g, 4.35 mmol, 3當量)且將反應液在室溫下攪拌1 h。藉由TLC (於己烷中之70% EtOAc)監測反應。此後,完成完成。在減壓下濃縮反應液以去除甲醇且將所獲得粗製物溶於EtOAc (100 mL)中,並使用水(10 mL)洗滌。藉由無水硫酸鈉乾燥有機層,過濾並在減壓下濃縮以提供6-((3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)喹啉。LCMS (ES) m/z = 347.2 [M+H]+。
1-((3S)-3-丁基-6-甲氧基-1-(喹啉-6-基)-3,4-二氫異喹啉-2(1H)-基)-3-(三甲基矽烷基)丙-2-炔-1-酮:步驟1:在室溫下,向於DMF (0.004 mL, 0.054 mmol, 0.04當量)中之3-(三甲基矽烷基)丙炔酸(0.195 g, 1.37 mmol, 1當量)中添加草醯氯(0.13 mL, 1.51 mmol, 1.1當量)且將反應液在室溫下攪拌30 min。此後,在減壓下濃縮反應混合物以提供3-(三甲基矽烷基)丙炔醯氯且將其用於下一步驟中。
步驟2:在0℃下,向6-((3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)喹啉(0.35 g, 1.01 mmol, 1當量)於ACN (8.0 mL)中之經攪拌溶液中添加碳酸氫鈉(0.64 g, 7.58 mmol, 7.5當量),在0℃下攪拌15 min且然後在0℃下添加於ACN (2.0 mL)中之3-(三甲基矽烷基)丙炔醯氯(0.19 g, 1.21 mmol, 1.2當量)
,並將反應液在室溫下攪拌30 min。藉由TLC (於己烷中之70% EtOAc)監測反應。此後,使用EtOAc (100 mL)稀釋反應混合物且使用水(10 mL)洗滌。藉由無水硫酸鈉乾燥有機層,過濾並在減壓下濃縮以提供1-((3S)-3-丁基-6-甲氧基-1-(喹啉-6-基)-3,4-二氫異喹啉-2(1H)-基)-3-(三甲基矽烷基)丙-2-炔-1-酮。LCMS (ES) m/z = 471.3 [M+H]+。
1-((1S,3S)-3-丁基-6-甲氧基-1-(喹啉-6-基)-3,4-二氫異喹啉-2(1H)-基)丙-2-炔-1-酮:在0℃下,向1-((3S)-3-丁基-6-甲氧基-1-(喹啉-6-基)-3,4-二氫異喹啉-2(1H)-基)-3-(三甲基矽烷基)丙-2-炔-1-酮(0.53 g, 1.13 mmol, 1當量)於甲醇(4 mL)及DCM (30 mL)中之經攪拌溶液中添加碳酸鉀(0.93 g, 6.76 mmol, 6當量)且將反應液在0℃下攪拌1 h。藉由TLC (於己烷中之50% EtOAc)監測反應。此後,使用水(10 mL)稀釋反應混合物並使用DCM (150 mL)萃取。藉由無水硫酸鈉乾燥有機層,過濾並在減壓下濃縮以得到粗製物。藉由製備型TLC使用於己烷中之50% EtOAc作為洗脫劑(洗脫兩次)將粗製物純化兩次以提供1-((1S,3S)-3-丁基-6-甲氧基-1-(喹啉-6-基)-3,4-二氫異喹啉-2(1H)-基)丙-2-炔-1-酮。LCMS (ES) m/z = 399.2 [M+H]+。1
H NMR (400 MHz, DMSO-d6
) δ ppm 0.82 (t,J
= 7.0 Hz, 3 H), 1.20 - 1.21 (m, 4 H), 1.53 (bs, 2 H), 2.80 - 2.84 (m, 1 H), 2.90 - 2.94 (m, 1H), 3.68 - 3.70 (m, 3 H), 4.25 (s, 0.38 H), 4.62 (s, 0.73 H), 4.68 (bs, 0.6 H), 4.82 (bs, 0.8 H), 6.21 (s, 0.6 H), 6.49 (s, 0.4 H), 6.77 - 6.86 (m, 2 H), 7.44 - 7.52 (m, 2 H), 7.71 (t,J
= 7.2 Hz, 1.5 H), 7.79 - 7.91 (m, 2.5 H), 8.28 - 8.36 (m, 1 H), 8.78 - 8.82 (m, 1H)。
程序 29 :化合物 27 之合成 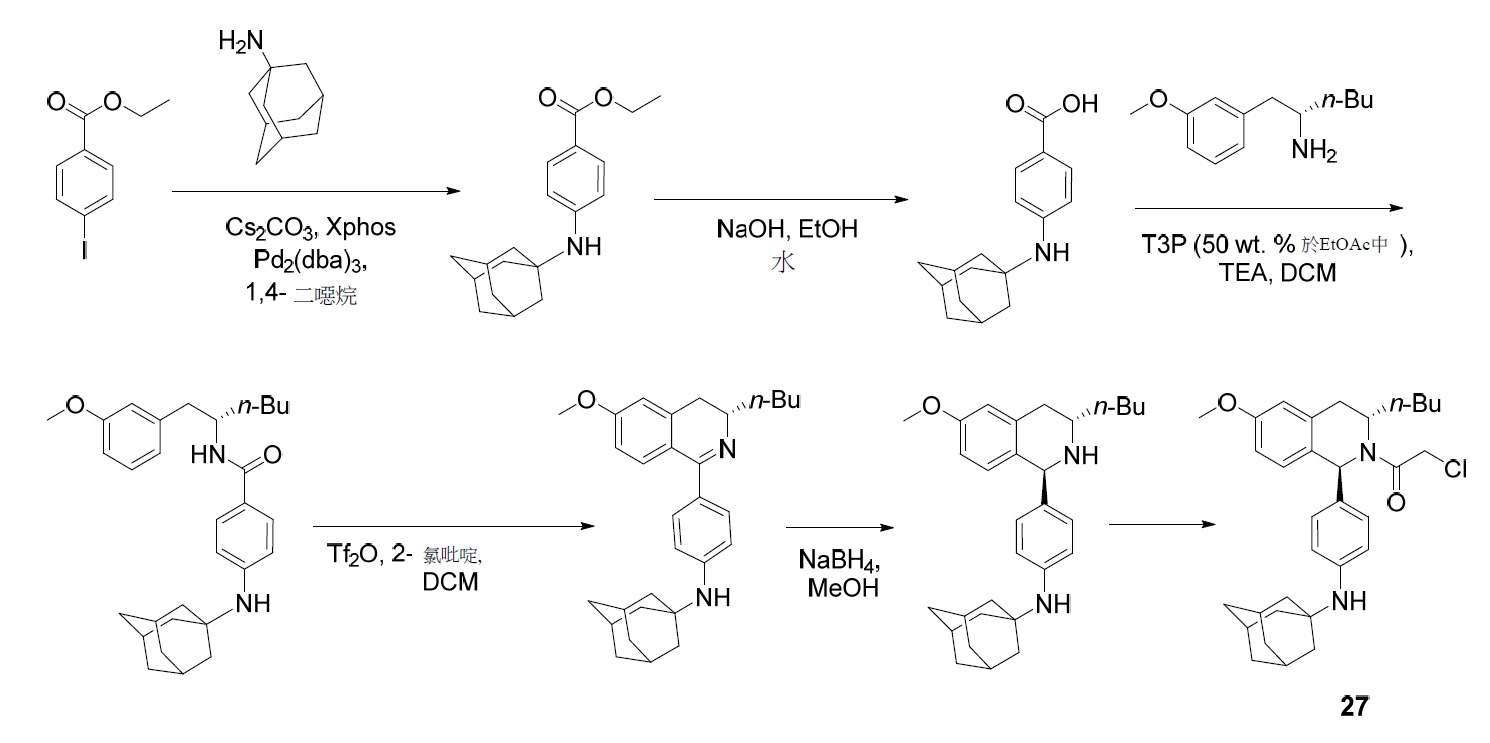 4-(((3s,5s,7s)-金剛烷-1-基)胺基)苯甲酸乙酯:在室溫下,向4-碘苯甲酸乙酯(30 g, 109 mmol, 1當量)於1,4-二噁烷(500 mL)中之溶液中添加金剛烷-1-胺(19.7 g, 130 mmol, 1.2當量)、碳酸銫(70.8 g, 217 mmol, 2當量)、二環己基[2',4',6'-參(丙烷-2-基)-[1,1'-聯苯]-2-基]磷烷(2.59 g, 5.43 mmol, 0.05當量)且將反應混合物在氮氣氛下吹掃30 min。然後在室溫下向混合物中添加參(二亞苄基丙酮)二鈀(2.99 g, 3.26 mmol, 0.03當量)。然後,將反應混合物升溫至110℃並在密封管中保持16 h。藉由TLC (於正己烷中之10% EtOAc)監測反應進展。在反應完成之後,將反應混合物冷卻至室溫並通過矽藻土床,且在減壓下濃縮濾液以得到粗製物。使用EtOAc (500 mL)萃取所獲得粗製物並使用水(100 mL)及鹽水溶液(100 mL)洗滌。藉由無水硫酸鈉乾燥有機層,過濾並在減壓下濃縮以得到粗產物。藉由急速管柱層析在矽膠上使用於己烷中之6-7%乙酸乙酯純化所獲得粗產物。合併含有產物之部分並在減壓下濃縮以得到4-[(金剛烷-1-基)胺基]苯甲酸乙酯。LCMS (ES) m/z = 300.2 [M+H]+。1
H NMR (400 MHz, DMSO-d6
) δ ppm 1.25 (t,J
= 8 Hz, 3 H), 1.48 (s, 6 H), 1.90 (s, 6 H), 2.04 (s, 3 H), 4.15 - 4.22 (m, 2 H), 5.95 (s, 1 H), 6.73 (d,J
= 8 Hz, 2 H), 7.59 (d,J
= 8 Hz, 2 H)。
4-(((3s,5s,7s)-金剛烷-1-基)胺基)苯甲酸:在0℃下,向4-[(金剛烷-1-基)胺基]苯甲酸乙酯(8.20 g, 27.4 mmol, 1當量)於乙醇(140 mL)中之溶液中添加於水(52 mL)中之氫氧化鈉(2.25 g, 54.8 mmol, 2當量)。然後將反應混合物升溫至80℃並攪拌6 h。藉由TLC (於DCM中之5%甲醇)監測反應進展。在反應完成之後,將反應混合物冷卻至室溫且在減壓下去除有機溶劑。然後,使用1 N HCl (pH = 2)酸化所得殘餘物並使用DCM (200 mL)萃取。藉由無水硫酸鈉乾燥有機層,過濾並在減壓下濃縮以得到4-[(金剛烷-1-基)胺基]苯甲酸。LCMS (ES) m/z = 272.2 [M+H]+。1
H NMR (400 MHz, DMSO-d6
) δ ppm 1.60 - 1.67 (m, 6 H), 1.89 (d,J
= 9.2 Hz, 6 H), 2.04 (s, 3 H), 5.91 (s, 1 H), 6.71 (d,J
= 8 Hz 2 H), 7.57 (d,J
= 8.8 Hz, 2 H), 12.21 (s, 1 H)。
4-(((3R,5R,7R)-金剛烷-1-基)胺基)-N-((S)-1-(3-甲氧基苯基)己烷-2-基)苯甲醯胺:向4-[(金剛烷-1-基)胺基]苯甲酸(3.6 g, 13.5 mmol, 1當量)於DCM (25 mL)中之溶液中添加三乙胺(7.5 mL, 54.0 mmol, 4當量)且將反應混合物冷卻至0℃。在0℃下向反應混合物中添加三丙基-1,3,5,2λ⁵,4λ⁵,6λ⁵-三氧雜三磷雜環己烷-2,4,6-三酮(12.0 mL, 20.3 mmol, 1.5當量)並攪拌30 min。然後向反應混合物中添加(2S)-1-(3-甲氧基苯基)己烷-2-胺(2.8 g, 13.5 mmol, 1當量)於DCM (5 mL)中之溶液。在添加後,將反應混合物升溫至室溫然後攪拌16 h。藉由TLC (於DCM中之5% MeOH)監測反應進展。在反應完成之後,使用DCM (400 mL)及飽和碳酸氫鈉(50 mL)稀釋反應混合物。分離有機層,使用鹽水溶液(25 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮。藉由急速管柱層析在矽膠上使用於己烷中之20-27%乙酸乙酯純化所獲得粗產物。合併含有產物之部分並在減壓下濃縮以得到4-[(金剛烷-1-基)胺基]-N-[(2S)-1-(3-甲氧基苯基)己烷-2-基]苯甲醯胺。LCMS (ES) m/z = 461.3 [M+H]+。1
H NMR (400 MHz, DMSO-d6
) δ ppm 0.80 (s, 3 H), 1.13 - 1.22 (m, 4 H), 1.46 (s, 2 H), 1.64 (s, 5 H), 1.89 (s, 5 H), 2.05 (s, 4 H), 2.65 - 2.78 (m, 2 H), 3.57 (s, 3 H), 4.00 - 4.09 (m, 2 H), 5.53 (s, 1 H), 5.74 (s, 1 H), 6.71 (t,J
= 4 Hz, 4 H), 7.13 (d,J
= 8 Hz, 1 H), 7.57 (d,J
= 4 Hz, 2 H), 7.65 (d,J
= 8 Hz, 1 H)。
(3R,5R,7R)-N-(4-((S)-3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基)苯基)金剛烷-1-胺:在室溫下,向4-[(金剛烷-1-基)胺基]-N-[(2S)-1-(3-甲氧基苯基)己烷-2-基]苯甲醯胺(1.92 g, 4.17 mmol, 1當量)於DCM (25 mL)中之溶液中添加2-氯吡啶(1.18 mL, 12.5 mmol, 3當量)並將反應混合物冷卻至-78℃,且在-78℃下向混合物中添加三氟甲烷磺酸三氟甲烷磺醯基酯(2.97 mL, 12.5 mmol, 3當量)並攪拌。在10 min之後,將反應混合物置於冰-水浴中並升溫至0℃保持10 min。然後將所得溶液升溫至室溫並攪拌1 hr。藉由TLC (於正己烷中之70% EtOAc)監測反應進展。在反應完成之後,使用1M NaOH (10 mL)在0℃下淬滅反應混合物且然後使用DCM (50 mL)稀釋,並使用DCM (100 mL)萃取且使用水(5 mL)洗滌。藉由無水硫酸鈉乾燥有機層,過濾並在減壓下濃縮以得到粗產物。藉由急速管柱層析在矽膠上使用於己烷中之56-65%乙酸乙酯純化所獲得粗產物。合併含有產物之部分並在減壓下濃縮以提供N-{4-[(3S)-3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基]苯基}金剛烷-1-胺。LCMS (ES) m/z = 443.3 [M+H]+。
(3R,5R,7R)-N-(4-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)苯基)金剛烷-1-胺:在0℃下,向N-{4-[(3S)-3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基]苯基}金剛烷-1-胺(0.4 g, 0.904 mmol, 1當量)於甲醇(10 mL)中之溶液中逐份添加硼氫化鈉(0.103 g, 2.71 mmol, 3當量)。將懸浮液在室溫下攪拌30 min。藉由TLC (於DCM中之5% MeOH)監測反應進展。然後,濃縮反應混合物且使用EtOAc (50 mL)及水(10 mL)稀釋所獲得粗製物。分離有機層,使用鹽水溶液(10 mL)洗滌,且藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物。藉由製備型TLC使用於正己烷中之60%乙酸乙酯作為洗脫劑純化所獲得粗產物以得到N-{4-[(1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基]環己基}金剛烷-1-胺。LCMS (ES) m/z = 445.3 [M+H]+。1
H NMR (400 MHz, DMSO-d6
) δ ppm 1.12 - 1.31 (m, 9 H), 1.59 (s, 6 H), 1.80 (s, 6 H), 2.03 (d,J
= 3 H), 3.27 (s, 1H), 3.69 (s, 3 H), 6.62 (t,J
= 8 Hz 4 H), 6.74 (t,J
= 8.8 Hz, 3 H)。
1-((1S,3S)-1-(4-(((3R,5R,7R)-金剛烷-1-基)胺基)苯基)-3-丁基-6-甲氧基-3,4-二氫異喹啉-2(1H)-基)-2-氯乙烷-1-酮:向N-{4-[(1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基]苯基}金剛烷-1-胺(0.030 g, 0.067 mmol, 1當量)於DCM (5 mL)中之溶液中添加碳酸氫鈉(8.5 mg, 0.101 mmol, 1.5當量)且冷卻至0℃,並攪拌10分鐘。然後,在0℃下添加2-氯乙醯氯(0.003 mL, 0.067 mmol, 1當量)並升溫至室溫,攪拌30分鐘。藉由TLC (於正己烷中之70% EtOAc)監測反應進展。在反應完成之後,使用1M NaOH (5 mL)在0℃下淬滅反應混合物且然後使用DCM (5 mL)稀釋,並使用DCM (100 mL)萃取且使用水(5 mL)洗滌。藉由無水硫酸鈉乾燥有機層,過濾並在減壓下濃縮以得到粗產物。藉由製備型TLC使用於正己烷中之70%乙酸乙酯作為洗脫劑純化所獲得粗產物。收集產物部分並在減壓下濃縮以得到1-[(1S,3S)-1-{4-[(金剛烷-1-基)胺基]苯基}-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-2-基]-2-氯乙烷-1-酮。LCMS (ES) m/z = 521.5 [M+H]+。1
H NMR (400 MHz, DMSO-d6
) δ ppm 0.78 (s, 3 H), 1.29 (d,J
= 6.8 Hz, 6 H), 1.40 (bs, 2 H), 1.58 (s, 3 H), 1.78 (s, 3 H), 1.99 (s, 2 H), 2.15 (bs, 1 H), 2.65 - 2.83 (m, 3 H), 3.69 (s, 3 H), 3.79 - 3.82 (m, 1 H), 4.47 - 4.93 (m, 5 H), 5.89 - 5.93 (m, 1 H), 6.56 - 6.65 (m, 2 H), 6.76 (d,J
= 8 Hz, 2 H), 6.87 (s, 2 H), 7.36 (d,J
= 8 Hz, 1 H)。
4-(((3s,5s,7s)-金剛烷-1-基)胺基)苯甲酸乙酯:在室溫下,向4-碘苯甲酸乙酯(30 g, 109 mmol, 1當量)於1,4-二噁烷(500 mL)中之溶液中添加金剛烷-1-胺(19.7 g, 130 mmol, 1.2當量)、碳酸銫(70.8 g, 217 mmol, 2當量)、二環己基[2',4',6'-參(丙烷-2-基)-[1,1'-聯苯]-2-基]磷烷(2.59 g, 5.43 mmol, 0.05當量)且將反應混合物在氮氣氛下吹掃30 min。然後在室溫下向混合物中添加參(二亞苄基丙酮)二鈀(2.99 g, 3.26 mmol, 0.03當量)。然後,將反應混合物升溫至110℃並在密封管中保持16 h。藉由TLC (於正己烷中之10% EtOAc)監測反應進展。在反應完成之後,將反應混合物冷卻至室溫並通過矽藻土床,且在減壓下濃縮濾液以得到粗製物。使用EtOAc (500 mL)萃取所獲得粗製物並使用水(100 mL)及鹽水溶液(100 mL)洗滌。藉由無水硫酸鈉乾燥有機層,過濾並在減壓下濃縮以得到粗產物。藉由急速管柱層析在矽膠上使用於己烷中之6-7%乙酸乙酯純化所獲得粗產物。合併含有產物之部分並在減壓下濃縮以得到4-[(金剛烷-1-基)胺基]苯甲酸乙酯。LCMS (ES) m/z = 300.2 [M+H]+。1
H NMR (400 MHz, DMSO-d6
) δ ppm 1.25 (t,J
= 8 Hz, 3 H), 1.48 (s, 6 H), 1.90 (s, 6 H), 2.04 (s, 3 H), 4.15 - 4.22 (m, 2 H), 5.95 (s, 1 H), 6.73 (d,J
= 8 Hz, 2 H), 7.59 (d,J
= 8 Hz, 2 H)。
4-(((3s,5s,7s)-金剛烷-1-基)胺基)苯甲酸:在0℃下,向4-[(金剛烷-1-基)胺基]苯甲酸乙酯(8.20 g, 27.4 mmol, 1當量)於乙醇(140 mL)中之溶液中添加於水(52 mL)中之氫氧化鈉(2.25 g, 54.8 mmol, 2當量)。然後將反應混合物升溫至80℃並攪拌6 h。藉由TLC (於DCM中之5%甲醇)監測反應進展。在反應完成之後,將反應混合物冷卻至室溫且在減壓下去除有機溶劑。然後,使用1 N HCl (pH = 2)酸化所得殘餘物並使用DCM (200 mL)萃取。藉由無水硫酸鈉乾燥有機層,過濾並在減壓下濃縮以得到4-[(金剛烷-1-基)胺基]苯甲酸。LCMS (ES) m/z = 272.2 [M+H]+。1
H NMR (400 MHz, DMSO-d6
) δ ppm 1.60 - 1.67 (m, 6 H), 1.89 (d,J
= 9.2 Hz, 6 H), 2.04 (s, 3 H), 5.91 (s, 1 H), 6.71 (d,J
= 8 Hz 2 H), 7.57 (d,J
= 8.8 Hz, 2 H), 12.21 (s, 1 H)。
4-(((3R,5R,7R)-金剛烷-1-基)胺基)-N-((S)-1-(3-甲氧基苯基)己烷-2-基)苯甲醯胺:向4-[(金剛烷-1-基)胺基]苯甲酸(3.6 g, 13.5 mmol, 1當量)於DCM (25 mL)中之溶液中添加三乙胺(7.5 mL, 54.0 mmol, 4當量)且將反應混合物冷卻至0℃。在0℃下向反應混合物中添加三丙基-1,3,5,2λ⁵,4λ⁵,6λ⁵-三氧雜三磷雜環己烷-2,4,6-三酮(12.0 mL, 20.3 mmol, 1.5當量)並攪拌30 min。然後向反應混合物中添加(2S)-1-(3-甲氧基苯基)己烷-2-胺(2.8 g, 13.5 mmol, 1當量)於DCM (5 mL)中之溶液。在添加後,將反應混合物升溫至室溫然後攪拌16 h。藉由TLC (於DCM中之5% MeOH)監測反應進展。在反應完成之後,使用DCM (400 mL)及飽和碳酸氫鈉(50 mL)稀釋反應混合物。分離有機層,使用鹽水溶液(25 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮。藉由急速管柱層析在矽膠上使用於己烷中之20-27%乙酸乙酯純化所獲得粗產物。合併含有產物之部分並在減壓下濃縮以得到4-[(金剛烷-1-基)胺基]-N-[(2S)-1-(3-甲氧基苯基)己烷-2-基]苯甲醯胺。LCMS (ES) m/z = 461.3 [M+H]+。1
H NMR (400 MHz, DMSO-d6
) δ ppm 0.80 (s, 3 H), 1.13 - 1.22 (m, 4 H), 1.46 (s, 2 H), 1.64 (s, 5 H), 1.89 (s, 5 H), 2.05 (s, 4 H), 2.65 - 2.78 (m, 2 H), 3.57 (s, 3 H), 4.00 - 4.09 (m, 2 H), 5.53 (s, 1 H), 5.74 (s, 1 H), 6.71 (t,J
= 4 Hz, 4 H), 7.13 (d,J
= 8 Hz, 1 H), 7.57 (d,J
= 4 Hz, 2 H), 7.65 (d,J
= 8 Hz, 1 H)。
(3R,5R,7R)-N-(4-((S)-3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基)苯基)金剛烷-1-胺:在室溫下,向4-[(金剛烷-1-基)胺基]-N-[(2S)-1-(3-甲氧基苯基)己烷-2-基]苯甲醯胺(1.92 g, 4.17 mmol, 1當量)於DCM (25 mL)中之溶液中添加2-氯吡啶(1.18 mL, 12.5 mmol, 3當量)並將反應混合物冷卻至-78℃,且在-78℃下向混合物中添加三氟甲烷磺酸三氟甲烷磺醯基酯(2.97 mL, 12.5 mmol, 3當量)並攪拌。在10 min之後,將反應混合物置於冰-水浴中並升溫至0℃保持10 min。然後將所得溶液升溫至室溫並攪拌1 hr。藉由TLC (於正己烷中之70% EtOAc)監測反應進展。在反應完成之後,使用1M NaOH (10 mL)在0℃下淬滅反應混合物且然後使用DCM (50 mL)稀釋,並使用DCM (100 mL)萃取且使用水(5 mL)洗滌。藉由無水硫酸鈉乾燥有機層,過濾並在減壓下濃縮以得到粗產物。藉由急速管柱層析在矽膠上使用於己烷中之56-65%乙酸乙酯純化所獲得粗產物。合併含有產物之部分並在減壓下濃縮以提供N-{4-[(3S)-3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基]苯基}金剛烷-1-胺。LCMS (ES) m/z = 443.3 [M+H]+。
(3R,5R,7R)-N-(4-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)苯基)金剛烷-1-胺:在0℃下,向N-{4-[(3S)-3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基]苯基}金剛烷-1-胺(0.4 g, 0.904 mmol, 1當量)於甲醇(10 mL)中之溶液中逐份添加硼氫化鈉(0.103 g, 2.71 mmol, 3當量)。將懸浮液在室溫下攪拌30 min。藉由TLC (於DCM中之5% MeOH)監測反應進展。然後,濃縮反應混合物且使用EtOAc (50 mL)及水(10 mL)稀釋所獲得粗製物。分離有機層,使用鹽水溶液(10 mL)洗滌,且藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物。藉由製備型TLC使用於正己烷中之60%乙酸乙酯作為洗脫劑純化所獲得粗產物以得到N-{4-[(1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基]環己基}金剛烷-1-胺。LCMS (ES) m/z = 445.3 [M+H]+。1
H NMR (400 MHz, DMSO-d6
) δ ppm 1.12 - 1.31 (m, 9 H), 1.59 (s, 6 H), 1.80 (s, 6 H), 2.03 (d,J
= 3 H), 3.27 (s, 1H), 3.69 (s, 3 H), 6.62 (t,J
= 8 Hz 4 H), 6.74 (t,J
= 8.8 Hz, 3 H)。
1-((1S,3S)-1-(4-(((3R,5R,7R)-金剛烷-1-基)胺基)苯基)-3-丁基-6-甲氧基-3,4-二氫異喹啉-2(1H)-基)-2-氯乙烷-1-酮:向N-{4-[(1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基]苯基}金剛烷-1-胺(0.030 g, 0.067 mmol, 1當量)於DCM (5 mL)中之溶液中添加碳酸氫鈉(8.5 mg, 0.101 mmol, 1.5當量)且冷卻至0℃,並攪拌10分鐘。然後,在0℃下添加2-氯乙醯氯(0.003 mL, 0.067 mmol, 1當量)並升溫至室溫,攪拌30分鐘。藉由TLC (於正己烷中之70% EtOAc)監測反應進展。在反應完成之後,使用1M NaOH (5 mL)在0℃下淬滅反應混合物且然後使用DCM (5 mL)稀釋,並使用DCM (100 mL)萃取且使用水(5 mL)洗滌。藉由無水硫酸鈉乾燥有機層,過濾並在減壓下濃縮以得到粗產物。藉由製備型TLC使用於正己烷中之70%乙酸乙酯作為洗脫劑純化所獲得粗產物。收集產物部分並在減壓下濃縮以得到1-[(1S,3S)-1-{4-[(金剛烷-1-基)胺基]苯基}-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-2-基]-2-氯乙烷-1-酮。LCMS (ES) m/z = 521.5 [M+H]+。1
H NMR (400 MHz, DMSO-d6
) δ ppm 0.78 (s, 3 H), 1.29 (d,J
= 6.8 Hz, 6 H), 1.40 (bs, 2 H), 1.58 (s, 3 H), 1.78 (s, 3 H), 1.99 (s, 2 H), 2.15 (bs, 1 H), 2.65 - 2.83 (m, 3 H), 3.69 (s, 3 H), 3.79 - 3.82 (m, 1 H), 4.47 - 4.93 (m, 5 H), 5.89 - 5.93 (m, 1 H), 6.56 - 6.65 (m, 2 H), 6.76 (d,J
= 8 Hz, 2 H), 6.87 (s, 2 H), 7.36 (d,J
= 8 Hz, 1 H)。
程序 30 :化合物 102 之合成 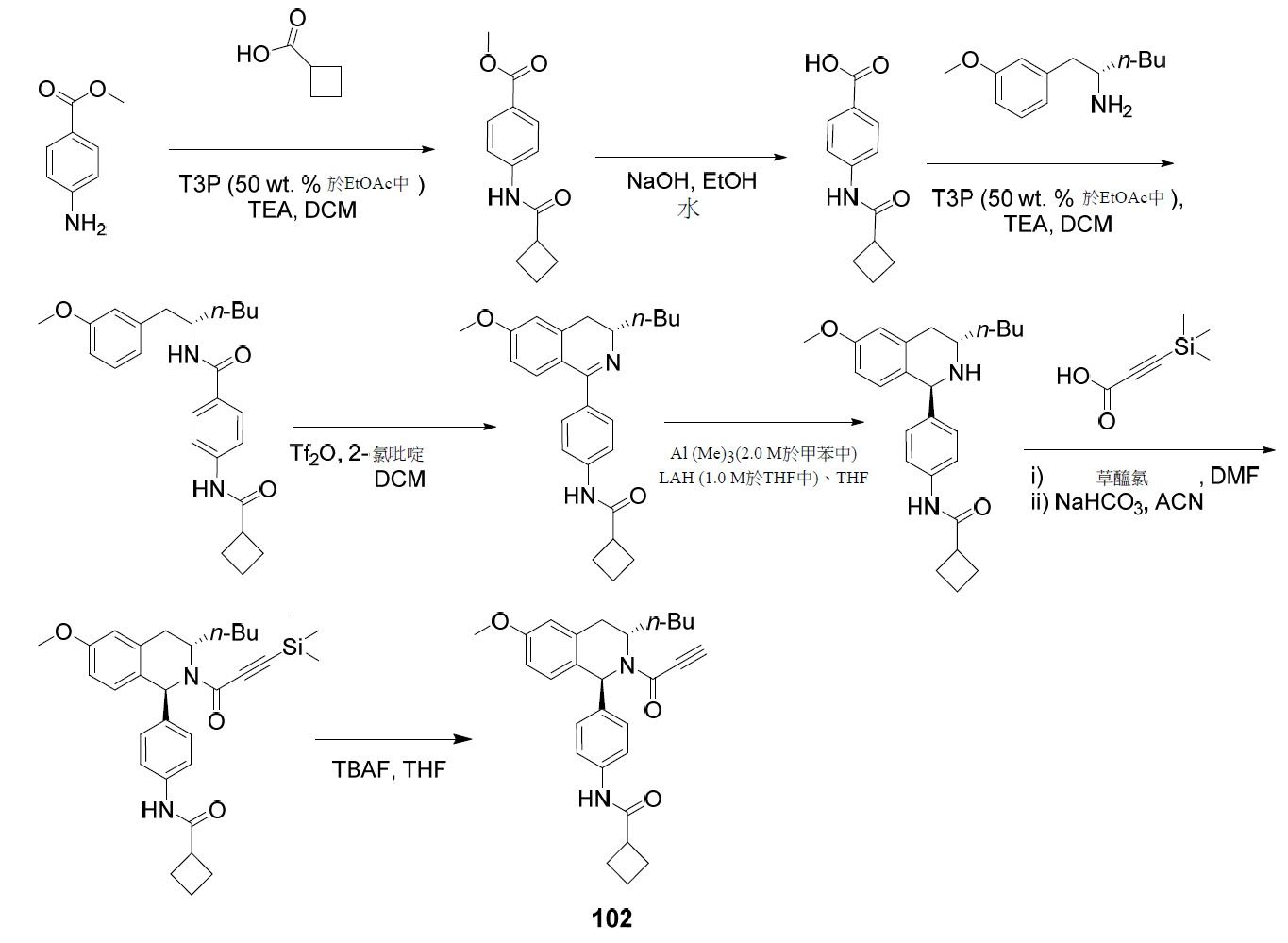 4-(環丁烷甲醯胺基)苯甲酸甲酯:在0℃下,向環丁烷甲酸(3.64 g, 36.4 mmol, 1.1當量)於DCM (15 mL)中之溶液中添加TEA (18.4 mL, 132 mmol, 4當量),攪拌5 min且然後添加T3P (50 wt.%於EtOAc中) (15.0 mL, 49.6 mmol, 1.5當量),並再攪拌30 min。然後在0℃下向反應混合物中添加4-胺基苯甲酸甲酯(5.0 g, 33.1 mmol, 1當量)於DCM (10 mL)中之溶液且然後將反應混合物在室溫下攪拌16 h。藉由TLC (於正己烷中之30%乙酸乙酯)監測反應進展。此後,使用DCM (2 × 100 mL)及飽和碳酸氫鈉溶液(15 mL)稀釋反應混合物。分離有機層,使用水(10 mL)、鹽水溶液(10 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以得到4-環丁烷醯胺基苯甲酸甲酯。LCMS (ES) m/z = 234.1 [M+H]+。1
H NMR (400 MHz, DMSO-d6
) δ ppm 1.77 - 1.97 (m, 2 H), 2.07 - 2.17 (m, 2 H), 2.22 - 2.47 (m, 2 H), 3.10 - 3.22 (m, 1 H), 3.78 (s, 3 H), 7.10 (d,J
= 8.0 Hz, 2 H), 7.86 (d,J
= 8.4 Hz, 2 H), 10.05 (s, 1H)。
4-(環丁烷甲醯胺基)苯甲酸:在室溫下,向4-環丁烷醯胺基苯甲酸甲酯(3.50 g, 15 mmol, 1當量)於EtOH (15.0 mL)及水(7.5 mL)中之溶液中添加氫氧化鈉(1.20 g, 30.0 mmol, 2當量)且將反應混合物在80℃下攪拌16 h。藉由TLC (於己烷中之30%乙酸乙酯)監測反應進展。在反應完成之後,在減壓下濃縮反應混合物以自反應物質去除乙醇且使用EtOAc (20 mL)萃取剩餘水層。最後,使用5%檸檬酸(pH ~ 4)酸化水層且然後使用EtOAc (2 × 80 mL)萃取產物。藉由無水硫酸鈉乾燥有機層,過濾並在減壓下濃縮以得到產物,使用正戊烷(10 mL)研磨5分鐘,傾析戊烷層並在高真空下乾燥以提供4-環丁烷醯胺基苯甲酸。LCMS (ES) m/z = 220.3 [M+H]+。
(S)-4-(環丁烷甲醯胺基)-N-(1-(3-甲氧基苯基)己烷-2-基)苯甲醯胺:在0℃及氮氣氛下,向4-環丁烷醯胺基苯甲酸(2.12 g, 9.65 mmol, 1當量)於DCM (15 mL)中之溶液中添加三乙胺(5.42 mL, 38.6 mmol, 4當量),攪拌10 min且然後在0℃下向反應混合物中添加丙烷膦酸酐(50 wt.%於乙酸乙酯中) (4.6 g, 14.5 mmol, 1.5當量),在0℃下攪拌15 min且然後在0℃下向反應混合物中添加溶於DCM (5 mL)中之(2S)-1-(3-甲氧基苯基)己烷-2-胺(2 g, 9.65 mmol, 1當量),且然後將反應混合物在室溫下攪拌16 h。TLC (於己烷中之50% EtOAc)顯示反應在16 h之後完成。使用DCM (80 mL)稀釋反應混合物,使用飽和碳酸氫鈉溶液(20 mL)及水(10 mL)洗滌。藉由無水Na2
SO4
乾燥有機層,過濾並在減壓下濃縮以得到4-環丁烷醯胺基-N-[(2S)-1-(3-甲氧基苯基)己烷-2-基]苯甲醯胺。LCMS (ES) m/z = 409.3 [M+H]+。
(S)-N-(4-(3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基)苯基)環丁烷甲醯胺:在室溫下,向4-環丁烷醯胺基-N-[(2S)-1-(3-甲氧基苯基)己烷-2-基]苯甲醯胺(0.1 g, 0.245 mmol, 1當量)於DCM (5 mL)中之溶液中添加2-氯吡啶(0.046 mL, 0.490 mmol, 2當量)並將反應混合物冷卻至-78℃,且在-78℃下向混合物中添加三氟甲烷磺酸三氟甲烷磺醯基酯(0.082 mL, 0.49 mmol, 2當量)並攪拌。在10 min之後,將反應混合物置於冰-水浴中並升溫至0℃保持10 min。然後將所得溶液升溫至室溫並攪拌1 h。藉由TLC (於正己烷中之70% EtOAc)監測反應進展。在反應完成之後,使用1M NaOH (5 mL)在0℃下淬滅反應混合物且然後使用DCM (5 mL)稀釋,並使用DCM (25 mL)萃取且使用水(5 mL)洗滌。藉由無水硫酸鈉乾燥有機層,過濾並在減壓下濃縮以提供(S)-N-(4-(3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基)苯基)環丁烷甲醯胺。LCMS (ES) m/z = 391.3 [M+H]+。
N-(4-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)苯基)環丁烷甲醯胺:在0℃及氮氣氛下,向N-{4-[(3S)-3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基]苯基}環丁烷甲醯胺(1.40 g, 3.58 mmol, 1當量)於MeOH (20 mL)中之溶液中添加硼氫化鈉(0.396 g, 10.8 mmol, 3當量)且然後將反應混合物在室溫下攪拌1 h。藉由TLC (於己烷中之70% EtOAc)及LC-MS監測反應。在反應完成之後,使用丙酮淬滅反應混合物並在減壓下濃縮,然後使用EtOAc (50 mL)萃取並使用水(10 mL)洗滌。藉由無水硫酸鈉乾燥有機層,過濾並在減壓下濃縮以得到粗產物。藉由急速管柱層析使用於己烷中之45-55%乙酸乙酯作為洗脫劑純化所獲得粗產物。合併含有產物之部分並在減壓下濃縮以得到順式/反式混合物之粗製物。同樣,藉由製備型TLC使用於正己烷中之60%乙酸乙酯作為洗脫劑純化粗製物以得到N-{4-[(1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基]苯基}環丁烷甲醯胺。LCMS (ES) m/z = 393.3 [M+H]+。
N-(4-((1S,3S)-3-丁基-6-甲氧基-2-(3-(三甲基矽烷基)丙炔醯基)-1,2,3,4-四氫異喹啉-1-基)苯基)環丁烷甲醯胺:步驟1:在室溫下,向3-(三甲基矽烷基)丙炔酸(0.1 g, 0.703 mmol, 1當量)於DMF (0.002 mL, 0.028 mmol, 0.04當量)中之溶液中添加草醯氯(0.072 mL, 0.844 mmol,當量)並攪拌30分鐘。然後,在減壓下濃縮反應混合物以得到3 (三甲基矽烷基)丙炔醯氯。
步驟2:在0℃下,向N-{4-[(1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基]苯基}環丁烷甲醯胺(0.2 g, 0.509 mmol, 1當量)於乙腈(7.0 mL)中之溶液中添加碳酸氫鈉(0.325 g, 3.82 mmol, 7.5當量)。在攪拌5分鐘之後,在0℃下向上述反應物質中添加3-(三甲基矽烷基)丙炔醯氯(0.123 g, 0.764 mmol, 1.5當量)於乙腈(3 mL)中之溶液。將所得混合物在0℃下攪拌45 min,藉由TLC (於正己烷中之25%乙酸乙酯)監測反應進展。此後,使用EtOAc (20 mL)及水(5 mL)稀釋反應物質。分離有機層,使用鹽水溶液(7.0 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物。LCMS (ES) m/z = 517.3 [M+H]+。
N-(4-((1S,3S)-3-丁基-6-甲氧基-2-丙炔醯基-1,2,3,4-四氫異喹啉-1-基)苯基)環丁烷甲醯胺:在-78℃下,向N-{4-[(1S,3S)-3-丁基-6-甲氧基-2-[3-(三甲基矽烷基)丙-2-炔醯基]-1,2,3,4-四氫異喹啉-1-基]苯基}環丁烷甲醯胺(0.2 g, 0.387 mmol, 1當量)於THF (5 mL)中之溶液中添加TBAF (於THF中之1M溶液) (0.968 mL, 0.968 mmol, 2.5當量)。將此反應混合物在-78℃下攪拌15分鐘。藉由TLC (於正己烷中之25%乙酸乙酯)監測反應進展。此後,使用NaHCO3飽和水溶液(10 mL)在-78℃下淬滅反應混合物且使用乙酸乙酯(2 × 50 mL)萃取產物。藉由無水硫酸鈉乾燥合併之有機層,過濾並在減壓下濃縮。藉由製備型TLC使用於正己烷中之30%乙酸乙酯作為洗脫劑純化所獲得粗產物。收集產物部分並在減壓下濃縮以得到N-{4-[(1S,3S)-3-丁基-6-甲氧基-2-(丙-2-炔醯基)-1,2,3,4-四氫異喹啉-1-基]苯基}環丁烷甲醯胺。LCMS (ES) m/z = 445.2 [M+H]+。1
H NMR (400 MHZ, DMSO-d6
) δ ppm:觀察到旋轉異構體圖案以及其他峰(少量雜質)。1
H NMR (400MHz, DMSO-d6
) δ ppm 0.65 - 0.68 (m, 3 H), 0.75 (bs, 1 H), 1.09 - 1.11 (m, 4 H), 1.25 (s, 1 H), 1.54 (s, 1 H), 1.78 - 1.86 (m, 1 H), 1.89 - 1.93 (m, 1 H), 2.05 - 2.07 (m, 2 H), 2.20 - 2.25 (m, 2 H), 2.63 - 2.66 (m, 0.5 H), 2.99 - 3.01 (m, 0.5 H), 3.13 - 3.18 (m, 1 H), 3.74 - 3.76 (m, 3 H), 4.04 (s, 0.5 H), 4.53 - 4.59 (m, 1.5 H), 6.50 - 6.55 (m, 1 H), 6.77 - 6.79 (m, 0.5 H), 6.84 (s, 1 H), 6.90 - 6.94 (m, 2 H), 7.03 (d,J
= 8.0 Hz, 1 H), 7.26 (d,J
= 8.4 Hz, 0.5 H), 7.47 -7.54 (m, 2 H), 9.67 - 9.72 (m, 1 H)。
4-(環丁烷甲醯胺基)苯甲酸甲酯:在0℃下,向環丁烷甲酸(3.64 g, 36.4 mmol, 1.1當量)於DCM (15 mL)中之溶液中添加TEA (18.4 mL, 132 mmol, 4當量),攪拌5 min且然後添加T3P (50 wt.%於EtOAc中) (15.0 mL, 49.6 mmol, 1.5當量),並再攪拌30 min。然後在0℃下向反應混合物中添加4-胺基苯甲酸甲酯(5.0 g, 33.1 mmol, 1當量)於DCM (10 mL)中之溶液且然後將反應混合物在室溫下攪拌16 h。藉由TLC (於正己烷中之30%乙酸乙酯)監測反應進展。此後,使用DCM (2 × 100 mL)及飽和碳酸氫鈉溶液(15 mL)稀釋反應混合物。分離有機層,使用水(10 mL)、鹽水溶液(10 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以得到4-環丁烷醯胺基苯甲酸甲酯。LCMS (ES) m/z = 234.1 [M+H]+。1
H NMR (400 MHz, DMSO-d6
) δ ppm 1.77 - 1.97 (m, 2 H), 2.07 - 2.17 (m, 2 H), 2.22 - 2.47 (m, 2 H), 3.10 - 3.22 (m, 1 H), 3.78 (s, 3 H), 7.10 (d,J
= 8.0 Hz, 2 H), 7.86 (d,J
= 8.4 Hz, 2 H), 10.05 (s, 1H)。
4-(環丁烷甲醯胺基)苯甲酸:在室溫下,向4-環丁烷醯胺基苯甲酸甲酯(3.50 g, 15 mmol, 1當量)於EtOH (15.0 mL)及水(7.5 mL)中之溶液中添加氫氧化鈉(1.20 g, 30.0 mmol, 2當量)且將反應混合物在80℃下攪拌16 h。藉由TLC (於己烷中之30%乙酸乙酯)監測反應進展。在反應完成之後,在減壓下濃縮反應混合物以自反應物質去除乙醇且使用EtOAc (20 mL)萃取剩餘水層。最後,使用5%檸檬酸(pH ~ 4)酸化水層且然後使用EtOAc (2 × 80 mL)萃取產物。藉由無水硫酸鈉乾燥有機層,過濾並在減壓下濃縮以得到產物,使用正戊烷(10 mL)研磨5分鐘,傾析戊烷層並在高真空下乾燥以提供4-環丁烷醯胺基苯甲酸。LCMS (ES) m/z = 220.3 [M+H]+。
(S)-4-(環丁烷甲醯胺基)-N-(1-(3-甲氧基苯基)己烷-2-基)苯甲醯胺:在0℃及氮氣氛下,向4-環丁烷醯胺基苯甲酸(2.12 g, 9.65 mmol, 1當量)於DCM (15 mL)中之溶液中添加三乙胺(5.42 mL, 38.6 mmol, 4當量),攪拌10 min且然後在0℃下向反應混合物中添加丙烷膦酸酐(50 wt.%於乙酸乙酯中) (4.6 g, 14.5 mmol, 1.5當量),在0℃下攪拌15 min且然後在0℃下向反應混合物中添加溶於DCM (5 mL)中之(2S)-1-(3-甲氧基苯基)己烷-2-胺(2 g, 9.65 mmol, 1當量),且然後將反應混合物在室溫下攪拌16 h。TLC (於己烷中之50% EtOAc)顯示反應在16 h之後完成。使用DCM (80 mL)稀釋反應混合物,使用飽和碳酸氫鈉溶液(20 mL)及水(10 mL)洗滌。藉由無水Na2
SO4
乾燥有機層,過濾並在減壓下濃縮以得到4-環丁烷醯胺基-N-[(2S)-1-(3-甲氧基苯基)己烷-2-基]苯甲醯胺。LCMS (ES) m/z = 409.3 [M+H]+。
(S)-N-(4-(3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基)苯基)環丁烷甲醯胺:在室溫下,向4-環丁烷醯胺基-N-[(2S)-1-(3-甲氧基苯基)己烷-2-基]苯甲醯胺(0.1 g, 0.245 mmol, 1當量)於DCM (5 mL)中之溶液中添加2-氯吡啶(0.046 mL, 0.490 mmol, 2當量)並將反應混合物冷卻至-78℃,且在-78℃下向混合物中添加三氟甲烷磺酸三氟甲烷磺醯基酯(0.082 mL, 0.49 mmol, 2當量)並攪拌。在10 min之後,將反應混合物置於冰-水浴中並升溫至0℃保持10 min。然後將所得溶液升溫至室溫並攪拌1 h。藉由TLC (於正己烷中之70% EtOAc)監測反應進展。在反應完成之後,使用1M NaOH (5 mL)在0℃下淬滅反應混合物且然後使用DCM (5 mL)稀釋,並使用DCM (25 mL)萃取且使用水(5 mL)洗滌。藉由無水硫酸鈉乾燥有機層,過濾並在減壓下濃縮以提供(S)-N-(4-(3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基)苯基)環丁烷甲醯胺。LCMS (ES) m/z = 391.3 [M+H]+。
N-(4-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)苯基)環丁烷甲醯胺:在0℃及氮氣氛下,向N-{4-[(3S)-3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基]苯基}環丁烷甲醯胺(1.40 g, 3.58 mmol, 1當量)於MeOH (20 mL)中之溶液中添加硼氫化鈉(0.396 g, 10.8 mmol, 3當量)且然後將反應混合物在室溫下攪拌1 h。藉由TLC (於己烷中之70% EtOAc)及LC-MS監測反應。在反應完成之後,使用丙酮淬滅反應混合物並在減壓下濃縮,然後使用EtOAc (50 mL)萃取並使用水(10 mL)洗滌。藉由無水硫酸鈉乾燥有機層,過濾並在減壓下濃縮以得到粗產物。藉由急速管柱層析使用於己烷中之45-55%乙酸乙酯作為洗脫劑純化所獲得粗產物。合併含有產物之部分並在減壓下濃縮以得到順式/反式混合物之粗製物。同樣,藉由製備型TLC使用於正己烷中之60%乙酸乙酯作為洗脫劑純化粗製物以得到N-{4-[(1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基]苯基}環丁烷甲醯胺。LCMS (ES) m/z = 393.3 [M+H]+。
N-(4-((1S,3S)-3-丁基-6-甲氧基-2-(3-(三甲基矽烷基)丙炔醯基)-1,2,3,4-四氫異喹啉-1-基)苯基)環丁烷甲醯胺:步驟1:在室溫下,向3-(三甲基矽烷基)丙炔酸(0.1 g, 0.703 mmol, 1當量)於DMF (0.002 mL, 0.028 mmol, 0.04當量)中之溶液中添加草醯氯(0.072 mL, 0.844 mmol,當量)並攪拌30分鐘。然後,在減壓下濃縮反應混合物以得到3 (三甲基矽烷基)丙炔醯氯。
步驟2:在0℃下,向N-{4-[(1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基]苯基}環丁烷甲醯胺(0.2 g, 0.509 mmol, 1當量)於乙腈(7.0 mL)中之溶液中添加碳酸氫鈉(0.325 g, 3.82 mmol, 7.5當量)。在攪拌5分鐘之後,在0℃下向上述反應物質中添加3-(三甲基矽烷基)丙炔醯氯(0.123 g, 0.764 mmol, 1.5當量)於乙腈(3 mL)中之溶液。將所得混合物在0℃下攪拌45 min,藉由TLC (於正己烷中之25%乙酸乙酯)監測反應進展。此後,使用EtOAc (20 mL)及水(5 mL)稀釋反應物質。分離有機層,使用鹽水溶液(7.0 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物。LCMS (ES) m/z = 517.3 [M+H]+。
N-(4-((1S,3S)-3-丁基-6-甲氧基-2-丙炔醯基-1,2,3,4-四氫異喹啉-1-基)苯基)環丁烷甲醯胺:在-78℃下,向N-{4-[(1S,3S)-3-丁基-6-甲氧基-2-[3-(三甲基矽烷基)丙-2-炔醯基]-1,2,3,4-四氫異喹啉-1-基]苯基}環丁烷甲醯胺(0.2 g, 0.387 mmol, 1當量)於THF (5 mL)中之溶液中添加TBAF (於THF中之1M溶液) (0.968 mL, 0.968 mmol, 2.5當量)。將此反應混合物在-78℃下攪拌15分鐘。藉由TLC (於正己烷中之25%乙酸乙酯)監測反應進展。此後,使用NaHCO3飽和水溶液(10 mL)在-78℃下淬滅反應混合物且使用乙酸乙酯(2 × 50 mL)萃取產物。藉由無水硫酸鈉乾燥合併之有機層,過濾並在減壓下濃縮。藉由製備型TLC使用於正己烷中之30%乙酸乙酯作為洗脫劑純化所獲得粗產物。收集產物部分並在減壓下濃縮以得到N-{4-[(1S,3S)-3-丁基-6-甲氧基-2-(丙-2-炔醯基)-1,2,3,4-四氫異喹啉-1-基]苯基}環丁烷甲醯胺。LCMS (ES) m/z = 445.2 [M+H]+。1
H NMR (400 MHZ, DMSO-d6
) δ ppm:觀察到旋轉異構體圖案以及其他峰(少量雜質)。1
H NMR (400MHz, DMSO-d6
) δ ppm 0.65 - 0.68 (m, 3 H), 0.75 (bs, 1 H), 1.09 - 1.11 (m, 4 H), 1.25 (s, 1 H), 1.54 (s, 1 H), 1.78 - 1.86 (m, 1 H), 1.89 - 1.93 (m, 1 H), 2.05 - 2.07 (m, 2 H), 2.20 - 2.25 (m, 2 H), 2.63 - 2.66 (m, 0.5 H), 2.99 - 3.01 (m, 0.5 H), 3.13 - 3.18 (m, 1 H), 3.74 - 3.76 (m, 3 H), 4.04 (s, 0.5 H), 4.53 - 4.59 (m, 1.5 H), 6.50 - 6.55 (m, 1 H), 6.77 - 6.79 (m, 0.5 H), 6.84 (s, 1 H), 6.90 - 6.94 (m, 2 H), 7.03 (d,J
= 8.0 Hz, 1 H), 7.26 (d,J
= 8.4 Hz, 0.5 H), 7.47 -7.54 (m, 2 H), 9.67 - 9.72 (m, 1 H)。
程序 31 :化合物 96 之合成 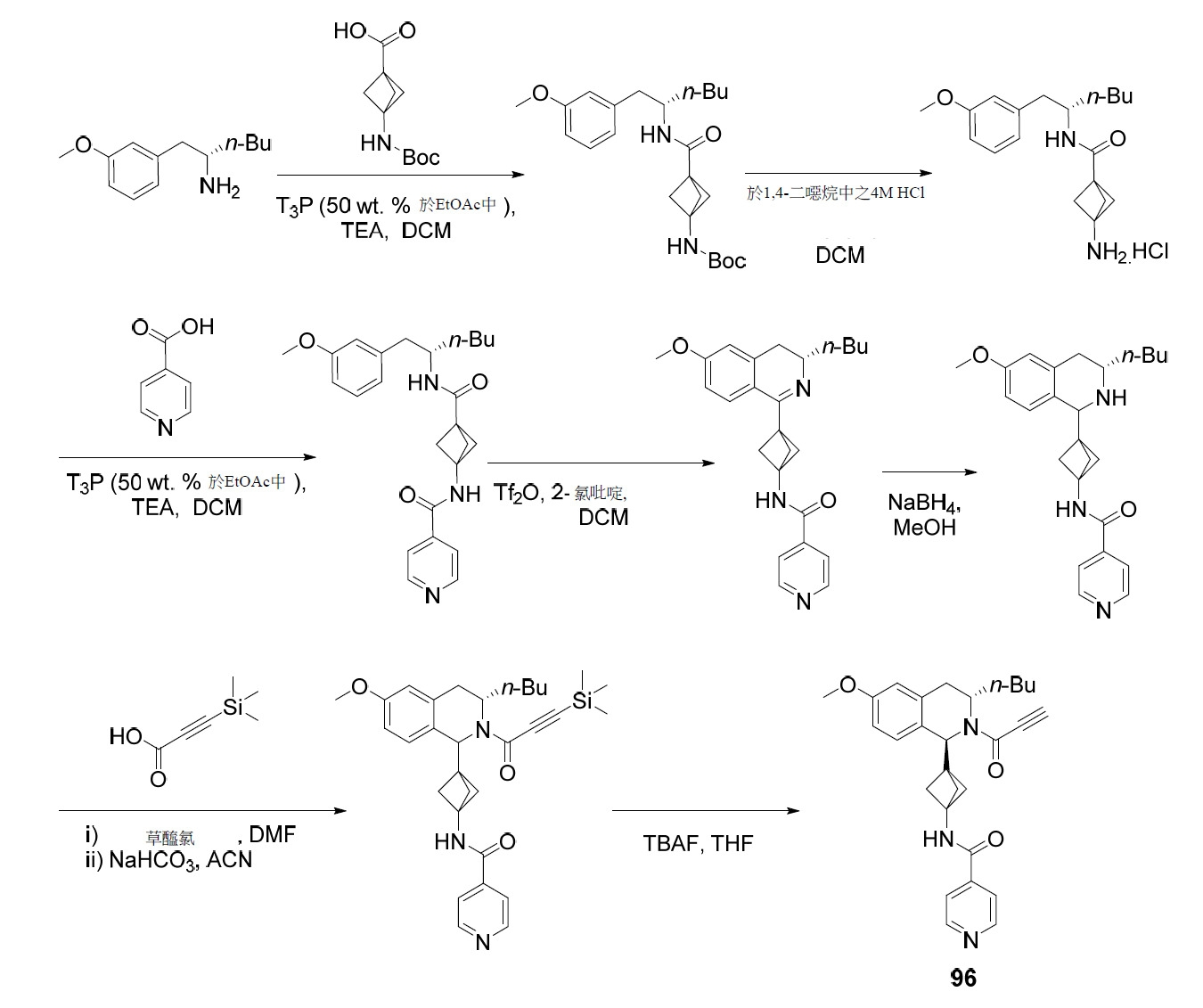 (S)-(3-((1-(3-甲氧基苯基)己烷-2-基)胺甲醯基)雙環[1.1.1]戊烷-1-基)胺基甲酸第三丁基酯:向3-((第三丁氧基羰基)胺基)雙環[1.1.1]戊烷-1-甲酸(2.3 g, 10.1 mmol, 1.05當量)於DCM (30 mL)中之溶液中添加TEA (4.04 mL, 28.9 mmol, 3當量),攪拌5 min且然後在0℃下添加T3P (50 wt.%於EtOAc中) (4.85 mL, 14.5 mmol, 1.5當量),並再攪拌30 min。然後在0℃下向反應混合物中添加 (S)-1-(3-甲氧基苯基)己烷-2-胺(2 g, 9.65 mmol, 1當量)於DCM (10 mL)中之溶液且然後將反應混合物在室溫下攪拌16 h。藉由TLC (於正己烷中之40%乙酸乙酯)監測反應進展。此後,使用DCM (50 mL)稀釋反應混合物,使用碳酸氫鈉飽和水溶液(2 × 10 mL)、水(10 mL)、鹽水溶液(10 mL)洗滌有機層,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物。藉由急速層析使用於正己烷中之0 - 30% EtOAc作為洗脫劑純化所獲得粗製物以得到(S)-(3-((1-(3-甲氧基苯基)己烷-2-基)胺甲醯基)雙環[1.1.1]戊烷-1-基)胺基甲酸第三丁基酯。LCMS (ES) m/z = 417.5 [M+H]+。1
H NMR (400 MHz, CDCl3
) δ ppm 0.84 (bs, 3 H), 1.27 (bs, 6 H), 1.42 (s, 9 H), 2.16 (s, 6 H), 2.67 - 2.79 (m, 2 H), 3.77 (s, 3 H), 4.11 (bs, 1 H), 4.92 (bs, 1 H), 5.15 (d,J
= 8.4 Hz, 1 H), 6.66 - 6.57 (m, 3 H), 7.18 (t,J
= 8.0 Hz, 1 H)。
(S)-3-胺基-N-(1-(3-甲氧基苯基)己烷-2-基)雙環[1.1.1]戊烷-1-甲醯胺鹽酸鹽:在0℃下,向(S)-(3-((1-(3-甲氧基苯基)己烷-2-基)胺甲醯基)雙環[1.1.1]戊烷-1-基)胺基甲酸第三丁基酯(3 g, 7.20 mmol, 1當量)於二氯甲烷(50 mL)中之溶液中添加於1,4 -二噁烷中之4 M HCl (5 mL)。將混合物升溫至室溫並攪拌12 h。藉由TLC (於己烷中之60% EtOAc)監測反應進展,在反應完成之後,在減壓下濃縮反應混合物以獲得粗製物,使用二乙醚(10 mL)及正戊烷(10 mL)之混合物研磨並傾析溶劑,在減壓下乾燥以獲得(S)-3-胺基-N-(1-(3-甲氧基苯基)己烷-2-基)雙環[1.1.1]戊烷-1-甲醯胺鹽酸鹽。LCMS (ES) m/z = 317 [M+H]+。1
H NMR (400 MHz, DMSO-d6
) δ ppm 0.81 (s, 3 H), 1.09 - 1.21 (m, 4 H), 1.37 - 1.40 (m, 2 H), 2.05 (s, 6 H), 2.56 - 2.64 (m, 2 H), 3.7 (s, 3 H), 3.84 (bs, 1 H), 6.70 - 6.72 (m, 3 H), 7.13 (t,J
= 7.6Hz
, 1 H), 7.56 (d,J
= 8.8Hz
, 1 H), 8.73 (bs, 3 H)。
(S)-N-(3-((1-(3-甲氧基苯基)己烷-2-基)胺甲醯基)雙環[1.1.1]戊烷-1-基)異菸鹼醯胺:向3-胺基-N-[(2S)-1-(3-甲氧基苯基)己烷-2-基]雙環[1.1.1]戊烷-1-甲醯胺鹽酸鹽(0.6 g, 1.70 mmol, 1當量)及吡啶-4-甲酸(0.251 g, 2.04 mmol, 1.2當量)於DCM (10 mL)中之溶液中添加三乙胺(1.32 mL, 10.2 mmol, 6當量),攪拌5 min且然後在0℃下添加T3P (50 wt.%於EtOAc中) (2.03 mL, 3.40 mmol, 2當量),並再攪拌30 min且然後將反應混合物在室溫下攪拌3 h。藉由TLC (於正己烷中之60%乙酸乙酯)監測反應進展。此後,使用DCM (20 mL)稀釋反應混合物,使用碳酸氫鈉飽和水溶液(2 × 10 mL)、水(10 mL)、鹽水溶液(10 mL)洗滌有機層,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物。藉由矽膠管柱層析使用於己烷中之35 - 40% EtOAc作為洗脫劑純化粗製物以提供N-(3-{[(2S)-1-(3-甲氧基苯基)己烷-2-基]胺甲醯基}雙環[1.1.1]戊烷-1-基)吡啶-4-甲醯胺。LCMS (ES) m/z = 422.3 [M+H]+
(S)-N-(3-(3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基)雙環[1.1.1]戊烷-1-基)異菸鹼醯胺:在室溫及氮氣氛下,向N-(3-{[(2S)-1-(3-甲氧基苯基)己烷-2-基]胺甲醯基}雙環[1.1.1]戊烷-1-基)吡啶-4-甲醯胺(0.4 g, 0.949 mmol, 1當量)於DCM (10 mL)在之經攪拌溶液中添加2-氯吡啶(0.18 mL, 1.90 mmol, 2當量)且將所得反應混合物冷卻至-78℃,且然後逐滴添加三氟甲烷磺酸酐(0.32 mL, 1.90 mmol, 2當量)。然後將反應混合物在-78℃下攪拌5 min,然後升溫至0℃並攪拌10 min,然後升溫至室溫並攪拌1.5 h。藉由TLC (5% MeOH/DCM)監測反應。此後,使用1N NaOH溶液中和反應液且使用DCM (2 × 50 mL)萃取。使用水(10 mL)洗滌合併之有機層,分離各層。藉由無水硫酸鈉乾燥有機層,過濾並在減壓下濃縮以獲得粗製物。藉由急速層析使用於正己烷中之15-20% EtOAc作為洗脫劑純化所獲得粗製物以得到N-{3-[(3S)-3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基]雙環[1.1.1]戊烷-1-基}吡啶-4-甲醯胺。LCMS (ES) m/z = 404.3 [M+H]+。
N-(3-((3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)雙環[1.1.1]戊烷-1-基)異菸鹼醯胺:在0℃下,向N-{3-[(3S)-3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基]雙環[1.1.1]戊烷-1-基}吡啶-4-甲醯胺(0.27 g, 0.669 mmol, 1當量)於甲醇(10 mL)中之溶液中逐份添加硼氫化鈉(0.076 g, 2.01 mmol, 3當量)。將懸浮液在0℃下攪拌3 h。此後,濃縮反應混合物且使用EtOAc (50 mL)及水(10 mL)稀釋所獲得粗製物。分離有機層,使用鹽水溶液(5 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以提供N-(3-((3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)雙環[1.1.1]戊烷-1-基)異菸鹼醯胺。LCMS (ES) m/z = 406.2 [M+H]+。
N-(3-((3S)-3-丁基-6-甲氧基-2-(3-(三甲基矽烷基)丙炔醯基)-1,2,3,4-四氫異喹啉-1-基)雙環[1.1.1]戊烷-1-基)異菸鹼醯胺:步驟1:在室溫下,向3-(三甲基矽烷基)丙炔酸(0.05 g, 0.352 mmol, 1當量)於DMF (0.001 mL, 0.014 mmol, 0.04當量)中之溶液中添加草醯氯(0.033 mL, 0.387 mmol, 1.1當量)並攪拌30分鐘。此後,在減壓下濃縮反應混合物以得到3-(三甲基矽烷基)丙炔醯氯。
步驟2:在0℃下,向N-{3-[(1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基]雙環[1.1.1]戊烷-1-基}吡啶-4-甲醯胺(0.220 g, 0.542 mmol, 1當量)於乙腈(3.0 mL)中之溶液中添加碳酸氫鈉(0.346 g, 4.07 mmol, 7.5當量)。在攪拌5分鐘之後,在0℃下向上述反應物質中添加3-(三甲基矽烷基)丙炔醯氯(0.131 g, 0.814 mmol, 1.5當量)於乙腈(1.0 mL)中之溶液。將所得混合物在0℃下攪拌15 min,藉由TLC (5% MeOH/DCM)監測反應進展。在LCMS中觀察到期望產物質量。此後,使用飽和碳酸氫鈉溶液(10 ml)終止反應,使用EtOAc (20 mL)稀釋,在室溫下攪拌5 min。然後分離各層。使用EtOAc (2 × 50 mL)萃取水層。使用水(15 mL)洗滌合併之有機層,分離各層。然後,藉由無水硫酸鈉乾燥有機層,過濾並在減壓下濃縮以獲得產物N-{3-[(1S,3S)-3-丁基-6-甲氧基-2-[3-(三甲基矽烷基)丙-2-炔醯基]-1,2,3,4-四氫異喹啉-1-基]雙環[1.1.1]戊烷-1-基}吡啶-4-甲醯胺。LCMS (ES) m/z = 530.2 [M+H]+。
N-(3-((1S,3S)-3-丁基-6-甲氧基-2-丙炔醯基-1,2,3,4-四氫異喹啉-1-基)雙環[1.1.1]戊烷-1-基)異菸鹼醯胺:在0℃下,向N-(3-((1S,3S)-3-丁基-6-甲氧基-2-(3-(三甲基矽烷基)丙炔醯基)-1,2,3,4-四氫異喹啉-1-基)雙環[1.1.1]戊烷-1-基)異菸鹼醯胺(0.25 g, 0.47 mmol, 1當量)於甲醇(3 mL)及DCM (20 mL)中之經攪拌溶液中添加碳酸鉀(0.39 g, 2.83 mmol, 6當量)且將反應液在0℃下攪拌1 h。藉由TLC (5% MeOH - DCM)監測反應。此後,使用水(10.0 mL)稀釋反應混合物並使用DCM (150 mL)萃取。藉由無水硫酸鈉乾燥有機層,過濾並在減壓下濃縮以得到粗製物。藉由製備型TLC使用4% MeOH - DCM作為洗脫劑(洗脫三次)純化粗製物兩次以分離期望產物。藉由製備型HPLC使用下列條件再純化。(分析條件:管柱:X-BridgeC-18 (250mm × 4.6mm × 5µm);移動相(A):於水中之0.1%氨;移動相(B):乙腈;流速:1.0 mL/min;梯度B:0/10, 12/60, 22/95, 25/95, 27/10, 30/10)。在減壓下濃縮自製備型HPLC獲得之部分並凍乾以提供N-(3-((1S,3S)-3-丁基-6-甲氧基-2-丙炔醯基-1,2,3,4-四氫異喹啉-1-基)雙環[1.1.1]戊烷-1-基)異菸鹼醯胺。LCMS (ES) m/z = 458.3 [M+H]+。
(S)-(3-((1-(3-甲氧基苯基)己烷-2-基)胺甲醯基)雙環[1.1.1]戊烷-1-基)胺基甲酸第三丁基酯:向3-((第三丁氧基羰基)胺基)雙環[1.1.1]戊烷-1-甲酸(2.3 g, 10.1 mmol, 1.05當量)於DCM (30 mL)中之溶液中添加TEA (4.04 mL, 28.9 mmol, 3當量),攪拌5 min且然後在0℃下添加T3P (50 wt.%於EtOAc中) (4.85 mL, 14.5 mmol, 1.5當量),並再攪拌30 min。然後在0℃下向反應混合物中添加 (S)-1-(3-甲氧基苯基)己烷-2-胺(2 g, 9.65 mmol, 1當量)於DCM (10 mL)中之溶液且然後將反應混合物在室溫下攪拌16 h。藉由TLC (於正己烷中之40%乙酸乙酯)監測反應進展。此後,使用DCM (50 mL)稀釋反應混合物,使用碳酸氫鈉飽和水溶液(2 × 10 mL)、水(10 mL)、鹽水溶液(10 mL)洗滌有機層,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物。藉由急速層析使用於正己烷中之0 - 30% EtOAc作為洗脫劑純化所獲得粗製物以得到(S)-(3-((1-(3-甲氧基苯基)己烷-2-基)胺甲醯基)雙環[1.1.1]戊烷-1-基)胺基甲酸第三丁基酯。LCMS (ES) m/z = 417.5 [M+H]+。1
H NMR (400 MHz, CDCl3
) δ ppm 0.84 (bs, 3 H), 1.27 (bs, 6 H), 1.42 (s, 9 H), 2.16 (s, 6 H), 2.67 - 2.79 (m, 2 H), 3.77 (s, 3 H), 4.11 (bs, 1 H), 4.92 (bs, 1 H), 5.15 (d,J
= 8.4 Hz, 1 H), 6.66 - 6.57 (m, 3 H), 7.18 (t,J
= 8.0 Hz, 1 H)。
(S)-3-胺基-N-(1-(3-甲氧基苯基)己烷-2-基)雙環[1.1.1]戊烷-1-甲醯胺鹽酸鹽:在0℃下,向(S)-(3-((1-(3-甲氧基苯基)己烷-2-基)胺甲醯基)雙環[1.1.1]戊烷-1-基)胺基甲酸第三丁基酯(3 g, 7.20 mmol, 1當量)於二氯甲烷(50 mL)中之溶液中添加於1,4 -二噁烷中之4 M HCl (5 mL)。將混合物升溫至室溫並攪拌12 h。藉由TLC (於己烷中之60% EtOAc)監測反應進展,在反應完成之後,在減壓下濃縮反應混合物以獲得粗製物,使用二乙醚(10 mL)及正戊烷(10 mL)之混合物研磨並傾析溶劑,在減壓下乾燥以獲得(S)-3-胺基-N-(1-(3-甲氧基苯基)己烷-2-基)雙環[1.1.1]戊烷-1-甲醯胺鹽酸鹽。LCMS (ES) m/z = 317 [M+H]+。1
H NMR (400 MHz, DMSO-d6
) δ ppm 0.81 (s, 3 H), 1.09 - 1.21 (m, 4 H), 1.37 - 1.40 (m, 2 H), 2.05 (s, 6 H), 2.56 - 2.64 (m, 2 H), 3.7 (s, 3 H), 3.84 (bs, 1 H), 6.70 - 6.72 (m, 3 H), 7.13 (t,J
= 7.6Hz
, 1 H), 7.56 (d,J
= 8.8Hz
, 1 H), 8.73 (bs, 3 H)。
(S)-N-(3-((1-(3-甲氧基苯基)己烷-2-基)胺甲醯基)雙環[1.1.1]戊烷-1-基)異菸鹼醯胺:向3-胺基-N-[(2S)-1-(3-甲氧基苯基)己烷-2-基]雙環[1.1.1]戊烷-1-甲醯胺鹽酸鹽(0.6 g, 1.70 mmol, 1當量)及吡啶-4-甲酸(0.251 g, 2.04 mmol, 1.2當量)於DCM (10 mL)中之溶液中添加三乙胺(1.32 mL, 10.2 mmol, 6當量),攪拌5 min且然後在0℃下添加T3P (50 wt.%於EtOAc中) (2.03 mL, 3.40 mmol, 2當量),並再攪拌30 min且然後將反應混合物在室溫下攪拌3 h。藉由TLC (於正己烷中之60%乙酸乙酯)監測反應進展。此後,使用DCM (20 mL)稀釋反應混合物,使用碳酸氫鈉飽和水溶液(2 × 10 mL)、水(10 mL)、鹽水溶液(10 mL)洗滌有機層,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物。藉由矽膠管柱層析使用於己烷中之35 - 40% EtOAc作為洗脫劑純化粗製物以提供N-(3-{[(2S)-1-(3-甲氧基苯基)己烷-2-基]胺甲醯基}雙環[1.1.1]戊烷-1-基)吡啶-4-甲醯胺。LCMS (ES) m/z = 422.3 [M+H]+
(S)-N-(3-(3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基)雙環[1.1.1]戊烷-1-基)異菸鹼醯胺:在室溫及氮氣氛下,向N-(3-{[(2S)-1-(3-甲氧基苯基)己烷-2-基]胺甲醯基}雙環[1.1.1]戊烷-1-基)吡啶-4-甲醯胺(0.4 g, 0.949 mmol, 1當量)於DCM (10 mL)在之經攪拌溶液中添加2-氯吡啶(0.18 mL, 1.90 mmol, 2當量)且將所得反應混合物冷卻至-78℃,且然後逐滴添加三氟甲烷磺酸酐(0.32 mL, 1.90 mmol, 2當量)。然後將反應混合物在-78℃下攪拌5 min,然後升溫至0℃並攪拌10 min,然後升溫至室溫並攪拌1.5 h。藉由TLC (5% MeOH/DCM)監測反應。此後,使用1N NaOH溶液中和反應液且使用DCM (2 × 50 mL)萃取。使用水(10 mL)洗滌合併之有機層,分離各層。藉由無水硫酸鈉乾燥有機層,過濾並在減壓下濃縮以獲得粗製物。藉由急速層析使用於正己烷中之15-20% EtOAc作為洗脫劑純化所獲得粗製物以得到N-{3-[(3S)-3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基]雙環[1.1.1]戊烷-1-基}吡啶-4-甲醯胺。LCMS (ES) m/z = 404.3 [M+H]+。
N-(3-((3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)雙環[1.1.1]戊烷-1-基)異菸鹼醯胺:在0℃下,向N-{3-[(3S)-3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基]雙環[1.1.1]戊烷-1-基}吡啶-4-甲醯胺(0.27 g, 0.669 mmol, 1當量)於甲醇(10 mL)中之溶液中逐份添加硼氫化鈉(0.076 g, 2.01 mmol, 3當量)。將懸浮液在0℃下攪拌3 h。此後,濃縮反應混合物且使用EtOAc (50 mL)及水(10 mL)稀釋所獲得粗製物。分離有機層,使用鹽水溶液(5 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以提供N-(3-((3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)雙環[1.1.1]戊烷-1-基)異菸鹼醯胺。LCMS (ES) m/z = 406.2 [M+H]+。
N-(3-((3S)-3-丁基-6-甲氧基-2-(3-(三甲基矽烷基)丙炔醯基)-1,2,3,4-四氫異喹啉-1-基)雙環[1.1.1]戊烷-1-基)異菸鹼醯胺:步驟1:在室溫下,向3-(三甲基矽烷基)丙炔酸(0.05 g, 0.352 mmol, 1當量)於DMF (0.001 mL, 0.014 mmol, 0.04當量)中之溶液中添加草醯氯(0.033 mL, 0.387 mmol, 1.1當量)並攪拌30分鐘。此後,在減壓下濃縮反應混合物以得到3-(三甲基矽烷基)丙炔醯氯。
步驟2:在0℃下,向N-{3-[(1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基]雙環[1.1.1]戊烷-1-基}吡啶-4-甲醯胺(0.220 g, 0.542 mmol, 1當量)於乙腈(3.0 mL)中之溶液中添加碳酸氫鈉(0.346 g, 4.07 mmol, 7.5當量)。在攪拌5分鐘之後,在0℃下向上述反應物質中添加3-(三甲基矽烷基)丙炔醯氯(0.131 g, 0.814 mmol, 1.5當量)於乙腈(1.0 mL)中之溶液。將所得混合物在0℃下攪拌15 min,藉由TLC (5% MeOH/DCM)監測反應進展。在LCMS中觀察到期望產物質量。此後,使用飽和碳酸氫鈉溶液(10 ml)終止反應,使用EtOAc (20 mL)稀釋,在室溫下攪拌5 min。然後分離各層。使用EtOAc (2 × 50 mL)萃取水層。使用水(15 mL)洗滌合併之有機層,分離各層。然後,藉由無水硫酸鈉乾燥有機層,過濾並在減壓下濃縮以獲得產物N-{3-[(1S,3S)-3-丁基-6-甲氧基-2-[3-(三甲基矽烷基)丙-2-炔醯基]-1,2,3,4-四氫異喹啉-1-基]雙環[1.1.1]戊烷-1-基}吡啶-4-甲醯胺。LCMS (ES) m/z = 530.2 [M+H]+。
N-(3-((1S,3S)-3-丁基-6-甲氧基-2-丙炔醯基-1,2,3,4-四氫異喹啉-1-基)雙環[1.1.1]戊烷-1-基)異菸鹼醯胺:在0℃下,向N-(3-((1S,3S)-3-丁基-6-甲氧基-2-(3-(三甲基矽烷基)丙炔醯基)-1,2,3,4-四氫異喹啉-1-基)雙環[1.1.1]戊烷-1-基)異菸鹼醯胺(0.25 g, 0.47 mmol, 1當量)於甲醇(3 mL)及DCM (20 mL)中之經攪拌溶液中添加碳酸鉀(0.39 g, 2.83 mmol, 6當量)且將反應液在0℃下攪拌1 h。藉由TLC (5% MeOH - DCM)監測反應。此後,使用水(10.0 mL)稀釋反應混合物並使用DCM (150 mL)萃取。藉由無水硫酸鈉乾燥有機層,過濾並在減壓下濃縮以得到粗製物。藉由製備型TLC使用4% MeOH - DCM作為洗脫劑(洗脫三次)純化粗製物兩次以分離期望產物。藉由製備型HPLC使用下列條件再純化。(分析條件:管柱:X-BridgeC-18 (250mm × 4.6mm × 5µm);移動相(A):於水中之0.1%氨;移動相(B):乙腈;流速:1.0 mL/min;梯度B:0/10, 12/60, 22/95, 25/95, 27/10, 30/10)。在減壓下濃縮自製備型HPLC獲得之部分並凍乾以提供N-(3-((1S,3S)-3-丁基-6-甲氧基-2-丙炔醯基-1,2,3,4-四氫異喹啉-1-基)雙環[1.1.1]戊烷-1-基)異菸鹼醯胺。LCMS (ES) m/z = 458.3 [M+H]+。
程序 32 :化合物 54 之合成 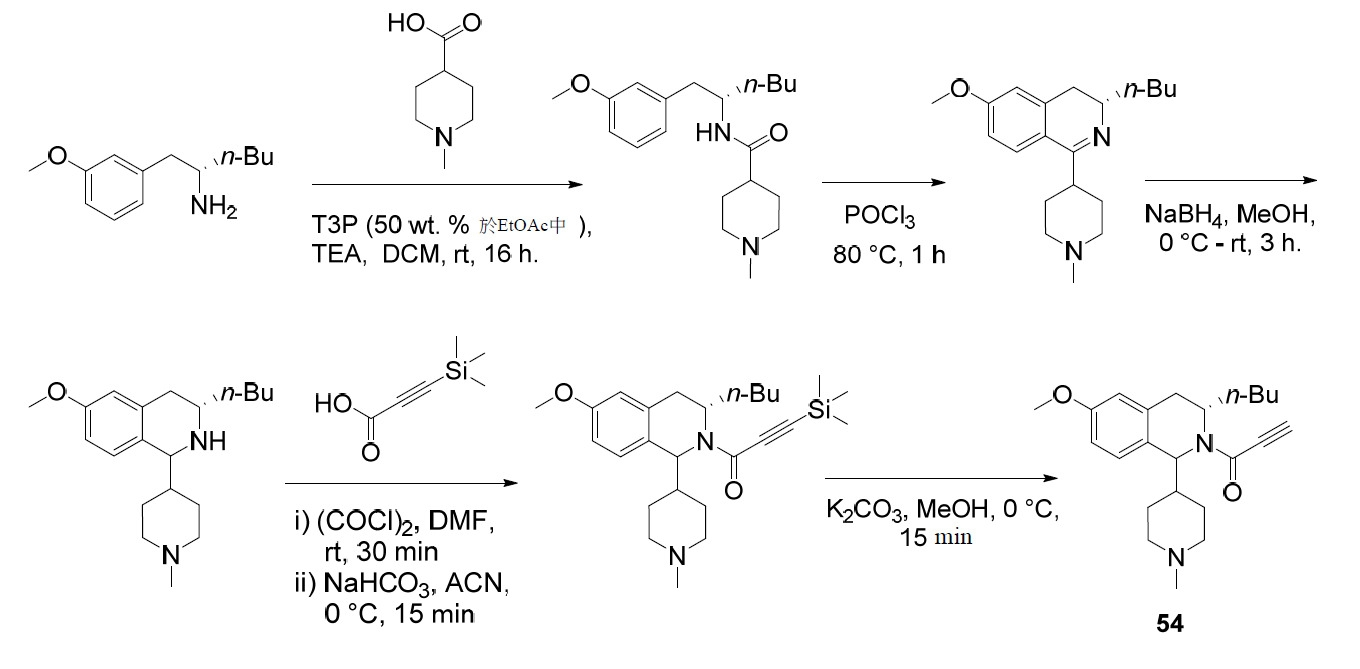 (S)-N-(1-(3-甲氧基苯基)己烷-2-基)-1-甲基六氫吡啶-4-甲醯胺:向1-甲基六氫吡啶-4-甲酸(0.64 g, 3.76 mmol, 1.2當量)於DCM (20 mL)中之溶液中添加TEA (1.75 mL, 12.56 mmol, 4當量),攪拌15 min且然後在0℃下添加T3P (50 wt.%於EtOAc中) (9.9 mL, 4.71 mmol, 1.5當量),並再攪拌5 min。然後,向反應混合物中添加(S)-1-(3-甲氧基苯基)己烷-2-胺(0.65 g, 3.14 mmol, 1當量)且然後將反應混合物在室溫下攪拌16 h。藉由TLC (於己烷中之70%乙酸乙酯)監測反應進展。使用DCM (50 mL)及飽和碳酸氫鈉溶液(20 mL)稀釋反應混合物,分離有機層,使用鹽水溶液(20 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物,藉由矽膠急速層析使用70% EtOAc/正己烷作為洗脫劑純化以得到(S)-N-(1-(3-甲氧基苯基)己烷-2-基)-1-甲基六氫吡啶-4-甲醯胺。LCMS (ES) m/z = 333 [M+H]+ 1
H NMR (400 MHz, DMSO-d6
) δ ppm 0.80 - 0.90 (m, 3 H), 1.18 - 1.30 (m, 6 H), 1.39 - 1.52 (m, 4 H), 1.73 - 1.82 (m, 2 H), 1.92 - 1.96 (m, 1 H), 2.12 (s, 3 H), 2.58 - 2.65 (m, 2 H), 2.70 - 2.75 (m, 2 H), 3.69 (s, 3 H), 3.86 (bs, 1 H), 6.70 - 6.71 (m, 3 H), 7.11 - 7.15 (m, 1 H), 7.48 (d,J
= 7.6 Hz, 1 H)。
(S)-3-丁基-6-甲氧基-1-(1-甲基六氫吡啶-4-基)-3,4-二氫異喹啉:將(S)-N-(1-(3-甲氧基苯基)己烷-2-基)-1-甲基六氫吡啶-4-甲醯胺(0.3 g, 0.90 mmol, 1當量)於POCl3 (0.1 mL, 1.08 mmol, 1.2當量)中之經攪拌溶液中在80℃下攪拌1小時,藉由TLC監測檢查反應進展,在反應完成之後,將反應液冷卻至室溫,在減壓下濃縮反應物質以獲得粗製殘餘物,使用10% NaOH水溶液(pH = 8)鹼化所獲得殘餘物,使用(2 × 20) mL乙酸乙酯萃取水層,使用無水Na2SO4乾燥合併之有機層,過濾並在減壓下濃縮以得到(S)-3-丁基-6-甲氧基-1-(1-甲基六氫吡啶-4-基)-3,4-二氫異喹啉。LC-MS (m/z) = 315 [M+H]+
(3S)-3-丁基-6-甲氧基-1-(1-甲基六氫吡啶-4-基)-1,2,3,4-四氫異喹啉:在0℃下,向第三丁基(S)-3-丁基-6-甲氧基-1-(1-甲基六氫吡啶-4-基)-3,4-二氫異喹啉(0.2 g, 0.63 mmol, 1.0當量)於甲醇(10 mL)中之溶液中逐份添加硼氫化鈉(0.07 g, 1.91 mmol, 3當量)。將懸浮液在室溫下攪拌15 min。藉由TLC (於DCM中之10% MeOH)監測反應進展。此後,濃縮反應混合物且使用EtOAc (15 mL)及水(8 mL)稀釋所獲得粗製物。分離有機層,使用鹽水溶液(7 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物,藉由急速管柱層析使用於DCM中之8% MeOH作為移動相純化以得到(3S)-3-丁基-6-甲氧基-1-(1-甲基六氫吡啶-4-基)-1,2,3,4-四氫異喹啉。LCMS (ES) m/z = 317 [M+H]+。1
H NMR (400 MHz, DMSO-d6
) δ ppm 0.87 (bs, 3 H), 1.21 - 1.45 (m, 10 H), 2.11 - 2.14 (m, 3 H), 1.76 (bs, 2 H), 2.60 - 2.80 (m, 4 H), 3.48 (bs, 1 H), 3.67 (s, 3 H), 3.91 (s, 1 H), 6.30 (bs, 1 H), 6.59 - 6.69 (m, 2 H), 7.02 - 7.04 (m, 2 H)。
1-((3S)-3-丁基-6-甲氧基-1-(1-甲基六氫吡啶-4-基)-3,4-二氫異喹啉-2(1H)-基)-3-(三甲基矽烷基)丙-2-炔-1-酮:第一步驟:在室溫下,向3-(三甲基矽烷基)丙炔酸(0.1 g, 0.70 mmol, 1.0當量)於DMF (0.002 mL, 0.028 mmol, 0.04當量)中之溶液中添加草醯氯(0.065 mL, 0.77 mmol, 1.1當量)並攪拌30分鐘。此後,在減壓下濃縮反應混合物以得到3-(三甲基矽烷基)丙炔醯氯。所獲得醯氯未經任何進一步純化即用於下一步驟中。
第二步驟:在0℃下,向(3S)-3-丁基-6-甲氧基-1-(1-甲基六氫吡啶-4-基)-1,2,3,4-四氫異喹啉(0.18 g, 0.5696 mmol, 1.0當量)於乙腈(10 mL)中之溶液中添加碳酸氫鈉(0.35 g, 4.27 mmol, 7.5當量)。在攪拌5分鐘之後,在0℃下向上述反應物質中添加3-(三甲基矽烷基)丙炔醯氯(0.109 g, 0.62 mmol, 1.2當量)於乙腈(5 mL)中之溶液。將所得混合物在0℃下攪拌15 min,藉由TLC (於DCM中之5% MeOH)監測反應進展。此後,使用EtOAc (40 mL)及水(8 mL)稀釋反應物質。分離有機層,使用鹽水溶液(7 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物,其未經任何進一步純化即用於下一步驟中。LCMS (ES) m/z = 441 [M+H]+。
1-((3S)-3-丁基-6-甲氧基-1-(1-甲基六氫吡啶-4-基)-3,4-二氫異喹啉-2(1H)-基)丙-2-炔-1-酮:在0℃下,向1-((3S)-3-丁基-6-甲氧基-1-(1-甲基六氫吡啶-4-基)-3,4-二氫異喹啉-2(1H)-基)-3-(三甲基矽烷基)丙-2-炔-1-酮(0.2 g, 0.45 mmol, 1.0當量)於MeOH (15 mL)中之溶液中添加K2
CO3
(0.187 g, 1.36 mmol, 3當量)。將反應混合物在0℃下攪拌15 min以得到白色溶液。藉由TLC (於DCM中之10% MeOH)監測反應進展。此後,使用DCM (5 mL)及水(5 mL)稀釋反應混合物。分離有機層,藉由Na2
SO4
乾燥並在減壓下濃縮以獲得粗產物,藉由下列製備型HPLC條件進行純化。分析條件:管柱:X-BridgeC-18 (250mm × 4.6mm × 5µm);移動相(A):於水中之0.1%氨;移動相(B):CAN;流速:1.0 mL/min。
收集產物部分並在減壓下濃縮以得到1-((3S)-3-丁基-6-甲氧基-1-(1-甲基六氫吡啶-4-基)-3,4-二氫異喹啉-2(1H)-基)丙-2-炔-1-酮。LCMS (ES) m/z = 369 [M+H]+。1
H NMR (400 MHz, CDCl3
) δ ppm 0.80 - 0.87 (m, 3 H), 1.15 - 1.20 (m, 2 H), 1.22 - 1.32 (m, 4 H), 1.60 - 1.66 (m, 3 H), 1.72 - 1.77 (m, 2 H), 1.95 - 2.04 (m, 1 H), 2.07 - 2.08 (m, 3 H), 2.73 - 2.80 (m, 2 H), 3.06 - 3.17 (m, 2 H), 3.71 (s, 3 H), 3.90 (bs, 1 H), 4.08 (bs, 1 H), 4.53 - 4.55 (m, 1 H), 4.91 - 4.93 (m, 1 H), 6.67 - 6.72 (m, 1 H), 6.81 (s, 1 H), 7.00 - 7.05 (m, 1 H)。
(S)-N-(1-(3-甲氧基苯基)己烷-2-基)-1-甲基六氫吡啶-4-甲醯胺:向1-甲基六氫吡啶-4-甲酸(0.64 g, 3.76 mmol, 1.2當量)於DCM (20 mL)中之溶液中添加TEA (1.75 mL, 12.56 mmol, 4當量),攪拌15 min且然後在0℃下添加T3P (50 wt.%於EtOAc中) (9.9 mL, 4.71 mmol, 1.5當量),並再攪拌5 min。然後,向反應混合物中添加(S)-1-(3-甲氧基苯基)己烷-2-胺(0.65 g, 3.14 mmol, 1當量)且然後將反應混合物在室溫下攪拌16 h。藉由TLC (於己烷中之70%乙酸乙酯)監測反應進展。使用DCM (50 mL)及飽和碳酸氫鈉溶液(20 mL)稀釋反應混合物,分離有機層,使用鹽水溶液(20 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物,藉由矽膠急速層析使用70% EtOAc/正己烷作為洗脫劑純化以得到(S)-N-(1-(3-甲氧基苯基)己烷-2-基)-1-甲基六氫吡啶-4-甲醯胺。LCMS (ES) m/z = 333 [M+H]+ 1
H NMR (400 MHz, DMSO-d6
) δ ppm 0.80 - 0.90 (m, 3 H), 1.18 - 1.30 (m, 6 H), 1.39 - 1.52 (m, 4 H), 1.73 - 1.82 (m, 2 H), 1.92 - 1.96 (m, 1 H), 2.12 (s, 3 H), 2.58 - 2.65 (m, 2 H), 2.70 - 2.75 (m, 2 H), 3.69 (s, 3 H), 3.86 (bs, 1 H), 6.70 - 6.71 (m, 3 H), 7.11 - 7.15 (m, 1 H), 7.48 (d,J
= 7.6 Hz, 1 H)。
(S)-3-丁基-6-甲氧基-1-(1-甲基六氫吡啶-4-基)-3,4-二氫異喹啉:將(S)-N-(1-(3-甲氧基苯基)己烷-2-基)-1-甲基六氫吡啶-4-甲醯胺(0.3 g, 0.90 mmol, 1當量)於POCl3 (0.1 mL, 1.08 mmol, 1.2當量)中之經攪拌溶液中在80℃下攪拌1小時,藉由TLC監測檢查反應進展,在反應完成之後,將反應液冷卻至室溫,在減壓下濃縮反應物質以獲得粗製殘餘物,使用10% NaOH水溶液(pH = 8)鹼化所獲得殘餘物,使用(2 × 20) mL乙酸乙酯萃取水層,使用無水Na2SO4乾燥合併之有機層,過濾並在減壓下濃縮以得到(S)-3-丁基-6-甲氧基-1-(1-甲基六氫吡啶-4-基)-3,4-二氫異喹啉。LC-MS (m/z) = 315 [M+H]+
(3S)-3-丁基-6-甲氧基-1-(1-甲基六氫吡啶-4-基)-1,2,3,4-四氫異喹啉:在0℃下,向第三丁基(S)-3-丁基-6-甲氧基-1-(1-甲基六氫吡啶-4-基)-3,4-二氫異喹啉(0.2 g, 0.63 mmol, 1.0當量)於甲醇(10 mL)中之溶液中逐份添加硼氫化鈉(0.07 g, 1.91 mmol, 3當量)。將懸浮液在室溫下攪拌15 min。藉由TLC (於DCM中之10% MeOH)監測反應進展。此後,濃縮反應混合物且使用EtOAc (15 mL)及水(8 mL)稀釋所獲得粗製物。分離有機層,使用鹽水溶液(7 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物,藉由急速管柱層析使用於DCM中之8% MeOH作為移動相純化以得到(3S)-3-丁基-6-甲氧基-1-(1-甲基六氫吡啶-4-基)-1,2,3,4-四氫異喹啉。LCMS (ES) m/z = 317 [M+H]+。1
H NMR (400 MHz, DMSO-d6
) δ ppm 0.87 (bs, 3 H), 1.21 - 1.45 (m, 10 H), 2.11 - 2.14 (m, 3 H), 1.76 (bs, 2 H), 2.60 - 2.80 (m, 4 H), 3.48 (bs, 1 H), 3.67 (s, 3 H), 3.91 (s, 1 H), 6.30 (bs, 1 H), 6.59 - 6.69 (m, 2 H), 7.02 - 7.04 (m, 2 H)。
1-((3S)-3-丁基-6-甲氧基-1-(1-甲基六氫吡啶-4-基)-3,4-二氫異喹啉-2(1H)-基)-3-(三甲基矽烷基)丙-2-炔-1-酮:第一步驟:在室溫下,向3-(三甲基矽烷基)丙炔酸(0.1 g, 0.70 mmol, 1.0當量)於DMF (0.002 mL, 0.028 mmol, 0.04當量)中之溶液中添加草醯氯(0.065 mL, 0.77 mmol, 1.1當量)並攪拌30分鐘。此後,在減壓下濃縮反應混合物以得到3-(三甲基矽烷基)丙炔醯氯。所獲得醯氯未經任何進一步純化即用於下一步驟中。
第二步驟:在0℃下,向(3S)-3-丁基-6-甲氧基-1-(1-甲基六氫吡啶-4-基)-1,2,3,4-四氫異喹啉(0.18 g, 0.5696 mmol, 1.0當量)於乙腈(10 mL)中之溶液中添加碳酸氫鈉(0.35 g, 4.27 mmol, 7.5當量)。在攪拌5分鐘之後,在0℃下向上述反應物質中添加3-(三甲基矽烷基)丙炔醯氯(0.109 g, 0.62 mmol, 1.2當量)於乙腈(5 mL)中之溶液。將所得混合物在0℃下攪拌15 min,藉由TLC (於DCM中之5% MeOH)監測反應進展。此後,使用EtOAc (40 mL)及水(8 mL)稀釋反應物質。分離有機層,使用鹽水溶液(7 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物,其未經任何進一步純化即用於下一步驟中。LCMS (ES) m/z = 441 [M+H]+。
1-((3S)-3-丁基-6-甲氧基-1-(1-甲基六氫吡啶-4-基)-3,4-二氫異喹啉-2(1H)-基)丙-2-炔-1-酮:在0℃下,向1-((3S)-3-丁基-6-甲氧基-1-(1-甲基六氫吡啶-4-基)-3,4-二氫異喹啉-2(1H)-基)-3-(三甲基矽烷基)丙-2-炔-1-酮(0.2 g, 0.45 mmol, 1.0當量)於MeOH (15 mL)中之溶液中添加K2
CO3
(0.187 g, 1.36 mmol, 3當量)。將反應混合物在0℃下攪拌15 min以得到白色溶液。藉由TLC (於DCM中之10% MeOH)監測反應進展。此後,使用DCM (5 mL)及水(5 mL)稀釋反應混合物。分離有機層,藉由Na2
SO4
乾燥並在減壓下濃縮以獲得粗產物,藉由下列製備型HPLC條件進行純化。分析條件:管柱:X-BridgeC-18 (250mm × 4.6mm × 5µm);移動相(A):於水中之0.1%氨;移動相(B):CAN;流速:1.0 mL/min。
收集產物部分並在減壓下濃縮以得到1-((3S)-3-丁基-6-甲氧基-1-(1-甲基六氫吡啶-4-基)-3,4-二氫異喹啉-2(1H)-基)丙-2-炔-1-酮。LCMS (ES) m/z = 369 [M+H]+。1
H NMR (400 MHz, CDCl3
) δ ppm 0.80 - 0.87 (m, 3 H), 1.15 - 1.20 (m, 2 H), 1.22 - 1.32 (m, 4 H), 1.60 - 1.66 (m, 3 H), 1.72 - 1.77 (m, 2 H), 1.95 - 2.04 (m, 1 H), 2.07 - 2.08 (m, 3 H), 2.73 - 2.80 (m, 2 H), 3.06 - 3.17 (m, 2 H), 3.71 (s, 3 H), 3.90 (bs, 1 H), 4.08 (bs, 1 H), 4.53 - 4.55 (m, 1 H), 4.91 - 4.93 (m, 1 H), 6.67 - 6.72 (m, 1 H), 6.81 (s, 1 H), 7.00 - 7.05 (m, 1 H)。
程序 33 :化合物 99 之合成 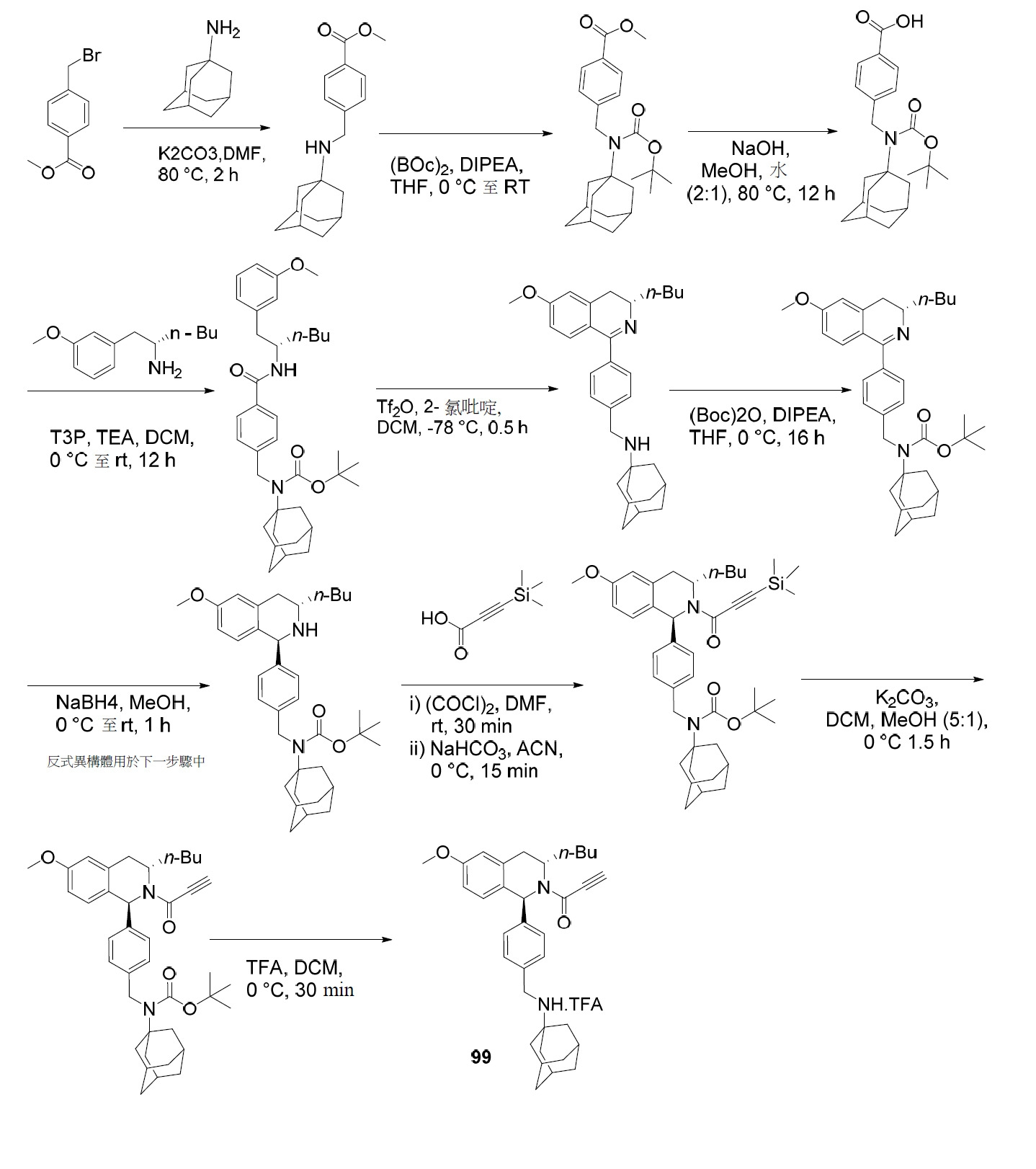 4-((((3s,5s,7s)-金剛烷-1-基)胺基)甲基)苯甲酸甲酯:在0℃下,向(3s,5s,7s)-金剛烷-1-胺(1.98 g, 13.1 mmol, 1.0當量)於DMF (30.0 mL)中之經攪拌溶液中添加碳酸鉀(2.71 g, 19.6 mmol, 1.5當量)。在攪拌5分鐘之後,添加4-(溴甲基)苯甲酸甲酯(2.7 g, 11.8 mmol, 0.9當量)。然後,將反應混合物在80℃下攪拌2 h。在反應完成之後,將反應混合物冷卻至室溫,使用水(30 mL)稀釋反應混合物,萃取至乙酸乙酯(2 × 30 mL)中。使用冷卻水(40 mL)、鹽水(20 mL)洗滌合併之有機層,藉由無水硫酸鈉乾燥。過濾有機層並在減壓下濃縮以得到粗產物,藉由急速管柱層析在矽膠上使用乙酸乙酯之己烷溶液作為洗脫劑進行純化。使用於己烷中之15 - 20 %乙酸乙酯分離產物。收集產物部分並在減壓下濃縮以得到4-((((3s,5s,7s)-金剛烷-1-基)胺基)甲基)苯甲酸甲酯。LCMS (ES) m/z = 300 [M+H]+。
4-{[(金剛烷-1-基)[(第三丁氧基)羰基]胺基]甲基}苯甲酸甲酯:在0℃下,向4-{[(金剛烷-1-基)胺基]甲基}苯甲酸甲酯(3 g, 10.0 mmol, 1當量)於THF (30 mL)中之溶液中添加N,N-二異丙基乙基胺(5.25 mL, 30.1 mmol, 3當量)及二碳酸二-第三丁基酯(6.91 mL, 30.1 mmol, 3當量)。將反應液在室溫下攪拌16 h。TLC (於己烷中之30% EtOAc)顯示反應已完成。此後,在減壓下濃縮反應混合物且使用NaHCO3
飽和水溶液(15 mL)及乙酸乙酯(30 mL)稀釋所獲得粗製物。分離有機層,使用水(20 mL)、鹽水溶液(10 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以得到粗製物(9 g),藉由急速層析使用於己烷中之0 - 25% EtOAc作為洗脫劑純化以得到4-{[(金剛烷-1-基)[(第三丁氧基)羰基]胺基]甲基}苯甲酸甲酯。LCMS (ES) m/z = 300 [M+H]+,觀察到Boc基團裂解質量。1
H NMR (400 MHz, CDCl3
) δ ppm 1.42 (s, 9 H), 1.52 - 1.72 (m, 6 H), 1.98 - 2.03 (m, 3 H), 1.52 - 1.72 (m, 6 H), 2.16 - 2.19 (m, 3 H), 4.62 - 4.67 (m, 2 H), 7.27 (d,J
= 8.8 Hz, 2 H), 7.97 (d,J
= 6.8 Hz, 2 H)。
4-((((3s,5s,7s)-金剛烷-1-基)(第三丁氧基羰基)胺基)甲基)苯甲酸:在室溫下,向4-((((3s,5s,7s)-金剛烷-1-基)(第三丁氧基羰基)胺基)甲基)苯甲酸甲酯(3 g, 7.51 mmol, 1當量)於MeOH (30 mL)及水(15 mL)中之溶液中添加氫氧化鈉(0.616 g, 15 mmol, 2.0當量)且將反應混合物在80℃下攪拌12 h。藉由TLC (於己烷中之60% E.A)監測反應進展。在反應完成之後,在減壓下濃縮反應混合物以自反應物質去除甲醇且使用EtOAc (50 mL)萃取剩餘水層。最後,使用5%檸檬酸(pH = 4)酸化水層且然後使用EtOAc (250 mL)萃取產物。藉由無水硫酸鈉乾燥有機層,過濾並在減壓下濃縮以得到產物,使用正戊烷(20 mL)研磨,傾析戊烷層並在高真空下乾燥以提供4-((((3s,5s,7s)-金剛烷-1-基)(第三丁氧基羰基)胺基)甲基)苯甲酸。LCMS (ES) m/z = 286 [M+H]+ (未觀察到boc質量)。1
H NMR (400 MHz, DMSO-d6
): δ ppm 1.33 (s, 9 H),1.88 - 2.20 (m, 9 H), 4.58 (s, 2 H), 7.28 (d,J
= 8 Hz, 2 H), 7.87 (d,J
= 8 Hz, 2 H), 12.79 (bs, 1 H)。
4-((((1R,3R)-金剛烷-1-基)胺基)甲基)-N-((S)-1-(3-甲氧基苯基)己烷-2-基)苯甲醯胺:向4-((((3s,5s,7s)-金剛烷-1-基)(第三丁氧基羰基)胺基)甲基)苯甲酸(2.66 g, 6.27 mmol, 1.1當量)於DCM (20 mL)中之溶液中添加TEA (2.63 mL, 18.8 mmol, 3當量),攪拌5 min且然後在0℃下添加T3P (50 wt.%於EtOAc中) (3.15 mL, 9.41 mmol, 1.5當量)並再攪拌30 min。然後在0℃下向反應混合物中添加(S)-1-(3-甲氧基苯基)己烷-2-胺(1.3 g, 6.27 mmol, 1當量)於DCM (10 mL)中之溶液且然後將反應混合物在室溫下攪拌12 h。藉由TLC (於正己烷中之60%乙酸乙酯)監測反應進展。此後,使用DCM (50 mL)稀釋反應混合物,使用碳酸氫鈉飽和水溶液(2 × 10 mL)、水(10 mL)、鹽水溶液(10 mL)洗滌有機層,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物,藉由急速層析使用於正己烷中之25 - 30% EtOAc作為洗脫劑純化以得到4-((((1R,3R)-金剛烷-1-基)胺基)甲基)-N-((S)-1-(3-甲氧基苯基)己烷-2-基)苯甲醯胺。LCMS (ES) m/z = 576 [M+H]+。1
H NMR (400 MHz, DMSO-d6
) δ ppm 0.80 - 0.87 (m, 3 H), 1.12 - 1.35 (m, 9 H), 1.52 (bs, 5 H), 1.97 - 2.03 (m, 6 H), 2.74 (bs, 1 H), 3.64 (s, 3 H), 4.11 (bs, 1 H), 3.8 (bs, 1 H), 4.11(bs, 1 H), 4.55 (bs, 2 H), 6.70 - 6.76 (m, 2 H), 7.13 - 7.22 (m, 3 H), 7.71 (m, 1 H), 8.09 (bs, 1 H)。
N-({4-[(3S)-3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基]苯基}甲基)金剛烷-1-胺:在-78℃下,經由注射器向4-((((1R,3R)-金剛烷-1-基)胺基)甲基)-N-((S)-1-(3-甲氧基苯基)己烷-2-基)苯甲醯胺(2.2 g, 3.83 mmol, 1當量)及2-氯吡啶(3.6 mL, 38.32 mmol, 10.0當量)於二氯甲烷(5 mL)中之經攪拌溶液中緩慢逐滴添加三氟甲烷磺酸酐(1.9 mL, 11.49 mmol, 3.0當量)。在5 min之後,將反應混合物置於冰-水浴中並升溫至0℃。在5 min之後,將所得溶液升溫至23℃並保持1 h。藉由TLC (於己烷中之30% EA)監測反應進展。在30分鐘之後,使用氫氧化鈉水溶液(3 mL, 1N)終止反應以中和三氟甲烷磺酸鹽。添加二氯甲烷(15 mL)以稀釋混合物且分離各層。使用DCM (2 × 50 mL)萃取水層。使用鹽水(5 mL)洗滌合併之有機層,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以得到粗產物。LCMS (ES) m/z = 457 [M+H]+。
N-(金剛烷-1-基)-N-({4-[(3S)-3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基]苯基}甲基)胺基甲酸第三丁基酯:在0℃下,向(1R,3R)-N-(4-((S)-3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基)苄基)金剛烷-1-胺(2.1 g, 4.60 mmol, 1.0當量)於THF (20.0 mL)中之經攪拌溶液中添加DIPEA (2.46 mL, 13.8 mmol, 3.0當量),隨後添加boc酐(3.17 mL, 13.8 mmol, 3.0當量)。然後,將反應混合物在室溫下攪拌16 h。使用水(25 mL)稀釋反應混合物,使用乙酸乙酯(2 × 30 mL)萃取。使用水(20 mL)、鹽水(20 mL)洗滌合併之有機層,藉由無水硫酸鈉乾燥。過濾有機層並在減壓下濃縮以得到粗產物,藉由矽膠管柱層析使用於己烷中之30%乙酸乙酯作為洗脫劑純化以得到((1R,3R)-金剛烷-1-基)(4-((S)-3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基)苄基)胺基甲酸第三丁基酯。LCMS (ES) m/z = 557 [M+H]+1
H NMR (400 MHz, DMSO-d6
) δ ppm 0.89 (t,J
= 7.2 Hz, 3 H), 1.3 (s, 9 H), 1.54 - 1.65 (m, 9 H), 1.99 - 2.10 (m, 9 H), 2.4 - 2.6 (m, 2 H), 2.74 - 2.77 (m, 1 H), 3.78 (s,3 H), 4.58 (s, 2 H), 6.79 - 6.81 (m, 1 H), 6.90 (s, 1 H), 7.07 - 7.09 (m, 1 H), 7.23 - 7.25 (m, 2 H), 7.44 - 7.46 (m, 1 H)。
N-(金剛烷-1-基)-N-({4-[(1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基]苯基}甲基)胺基甲酸第三丁基酯:在0℃下,向((1R,3R)-金剛烷-1-基)(4-((S)-3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基)苄基)胺基甲酸第三丁基酯(0.8 g, 1.44 mmol, 1當量)於甲醇(15 mL)中之溶液中逐份添加硼氫化鈉(0.159 g, 4.31 mmol, 3當量)。將懸浮液在室溫下攪拌30 min。藉由TLC (於己烷中之20% EA)監測反應進展。此後,使用丙酮(10 mL)淬滅反應混合物,濃縮且使用EtOAc (20 mL)及水(5 mL)稀釋所獲得粗製物。分離有機層,使用鹽水溶液(10 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物。藉由製備型TLC使用於正己烷中之20%乙酸乙酯作為洗脫劑純化所獲得粗產物以得到((1R,3R)-金剛烷-1-基)(4-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)苄基)胺基甲酸第三丁基酯(1, 3反式異構體)。LCMS (ES) m/z = 559 [M+H]+。1
H NMR (400 MHz, CDCl3
) δ ppm 0.8 - 0.9 (m, 3 H), 1.1 - 1.3 (m, 5 H), 1.39 (s, 9 H), 1.59 - 1.70 (m, 9 H), 2.02 (bs, 3 H), 2.10 (s, 5 H), 2.73 (bs, 2 H), 2.88 - 2.99 (m, 2 H), 3.79 (s, 3 H), 4.55 (s, 2 H), 5.29 (s, 2 H), 6.68 (s, 2 H), 6.84 (bs, 1 H), 7.11 (bs, 3 H)。
N-(金剛烷-1-基)-N-({4-[(1S,3S)-3-丁基-6-甲氧基-2-[3-(三甲基矽烷基)丙-2-炔醯基]-1,2,3,4-四氫異喹啉-1-基]苯基}甲基)胺基甲酸第三丁基酯:第一步驟:在室溫下,向3-(三甲基矽烷基)丙炔酸(0.043 g, 0.302 mmol, 1.0當量)於DMF (0.00094 mL, 0.012 mmol, 0.04當量)中之溶液中添加草醯氯(0.028 mL, 0.33 mmol, 1.1當量)並攪拌30分鐘。然後,在減壓下濃縮反應混合物以得到3-(三甲基矽烷基)丙炔醯氯。此醯氯粗製物未經任何進一步純化即用於下一步驟中。
第二步驟:在0℃下,向((1R,3R)-金剛烷-1-基)(4-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)苄基)胺基甲酸第三丁基酯(0.080 g, 0.143 mmol, 1.0當量)於乙腈(10 mL)中之溶液中添加碳酸氫鈉(0.091 g, 1.07 mmol, 7.5當量)。在攪拌5分鐘之後,向上述反應物質中添加3-(三甲基矽烷基)丙炔醯氯(0.025 g, 0.157 mmol, 1.1當量)於乙腈(5.0 mL)中之溶液。將所得混合物在0℃下攪拌1 h,藉由TLC (於正己烷中之20%乙酸乙酯)監測反應進展。此後,使用EtOAc (100 mL)及水(50 mL)稀釋反應物質。分離有機層,使用鹽水溶液(10 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮。此粗產物未經任何進一步純化即用於下一步驟中。LCMS (ES) m/z = 583 [M+H]+ (未觀察到Boc基團質量)。
N-(金剛烷-1-基)-N-({4-[(1S,3S)-3-丁基-6-甲氧基-2-(丙-2-炔醯基)-1,2,3,4-四氫異喹啉-1-基]苯基}甲基)胺基甲酸第三丁基酯:在0℃下,向((1R,3R)-金剛烷-1-基)(4-((1S,3S)-3-丁基-6-甲氧基-2-(3-(三甲基矽烷基)丙炔醯基)-1,2,3,4-四氫異喹啉-1-基)苄基)胺基甲酸第三丁基酯(171 mg, 0.25 mmol, 1當量)於DCM (10 mL) / MeOH (2 mL)中之溶液中添加K2
CO3
(207 mg, 1.50 mmol, 6當量)。將混合物在0℃下攪拌2 h,在反應完成之後,使用DCM (5 mL)稀釋反應混合物並添加H2O (3 mL)。使用DCM (3 × 10 mL)萃取有機層並藉由Na2
SO4
乾燥,且濃縮。藉由製備型TLC使用於己烷中之20% E.A純化殘餘物以得到((1R,3R)-金剛烷-1-基)(4-((1S,3S)-3-丁基-6-甲氧基-2-丙炔醯基-1,2,3,4-四氫異喹啉-1-基)苄基)胺基甲酸第三丁基酯。LCMS (ES) m/z = 511.2 [M+H]+。(未展示基團質量)。1
H NMR (400 MHz, CDCl3
) δ ppm 0.81 - 0.85 (m, 3 H), 0.93 - 0.95 (m, 1 H), 1.2 - 1.2 (m, 6 H), 1.38 - 1.56 (m, 9 H), 1.68 - 1.71 (m, 2 H), 1.91 - 2.0 (m, 3 H), 2.01 (bs, 5 H), 2.17 (s, 1 H), 2.67 - 2.82 (m, 2 H), 2.88 (s, 1 H), 3.80 - 3.81 (m, 3 H), 4.48 - 4.61 (m, 4 H), 6.29 (d,J
= 14 Hz,1 H), 6.69 (s, 1 H), 6.78 - 6.84 (m, 1 H), 6.93 - 7.08 (m, 4 H), 7.33 (d,J
= 8.4 Hz, 1 H)。
N-(金剛烷-1-基)-N-({4-[(1S,3S)-3-丁基-6-甲氧基-2-(丙-2-炔醯基)-1,2,3,4-四氫異喹啉-1-基]苯基}甲基)-2,2,2-三氟乙醯胺:在冷卻(0℃)條件,向((1R,3R)-金剛烷-1-基)(4-((1S,3S)-3-丁基-6-甲氧基-2-丙炔醯基-1,2,3,4-四氫異喹啉-1-基)苄基)胺基甲酸第三丁基酯(0.06 g, 0.098 mmol, 1當量)於DCM (15 mL)中之經攪拌溶液中逐滴添加TFA (0.2 mL),將反應混合物在0℃下攪拌2 h,藉由TLC (於己烷中之70% EA)檢查反應進展,在反應完成之後,在減壓下濃縮反應混合物以獲得粗產物,藉由製備型HPLC純化藉由使用下列分析條件進一步純化以得到1-((1S,3S)-1-(4-((((1R,3R)-金剛烷-1-基)(2,2,2-三氟乙醯基)-l4-氮烷基)甲基)苯基)-3-丁基-6-甲氧基-3,4-二氫異喹啉-2(1H)-基)丙-2-炔-1-酮。分析條件:管柱:X-BridgeC-18 (250 mm × 4.6 mm × 5 µm);移動相(A):於水中之0.1%TFA;移動相(B):乙腈;流速:1.0 mL/min;梯度B:0/20, 12/60, 22/95, 25/95, 27/20, 30/20。LCMS (ES) m/z = 511.1 [M+H]+。1
H NMR (400 MHz, CDCl3
) δ ppm 0.80 - 0.84 (m, 3 H), 0.90 - 1.2 (m, 7 H), 1.5 - 1.67 (m, 6 H), 1.86 (s, 4 H), 1.90 - 2.1 (m, 2 H), 2.76 - 2.90 (m, 2 H), 3.0- 3.16 (m, 1 H), 3.70 - 3.80 (m, 4 H), 4.01 (bs, 2 H), 4.59 - 4.75 (m, 2 H), 6.75 - 6.81 (m, 2 H), 7.07 - 7.12 (m, 1 H), 7.27 - 7.44 (m, 4 H), 8.46 (bs, 2 H)。
4-((((3s,5s,7s)-金剛烷-1-基)胺基)甲基)苯甲酸甲酯:在0℃下,向(3s,5s,7s)-金剛烷-1-胺(1.98 g, 13.1 mmol, 1.0當量)於DMF (30.0 mL)中之經攪拌溶液中添加碳酸鉀(2.71 g, 19.6 mmol, 1.5當量)。在攪拌5分鐘之後,添加4-(溴甲基)苯甲酸甲酯(2.7 g, 11.8 mmol, 0.9當量)。然後,將反應混合物在80℃下攪拌2 h。在反應完成之後,將反應混合物冷卻至室溫,使用水(30 mL)稀釋反應混合物,萃取至乙酸乙酯(2 × 30 mL)中。使用冷卻水(40 mL)、鹽水(20 mL)洗滌合併之有機層,藉由無水硫酸鈉乾燥。過濾有機層並在減壓下濃縮以得到粗產物,藉由急速管柱層析在矽膠上使用乙酸乙酯之己烷溶液作為洗脫劑進行純化。使用於己烷中之15 - 20 %乙酸乙酯分離產物。收集產物部分並在減壓下濃縮以得到4-((((3s,5s,7s)-金剛烷-1-基)胺基)甲基)苯甲酸甲酯。LCMS (ES) m/z = 300 [M+H]+。
4-{[(金剛烷-1-基)[(第三丁氧基)羰基]胺基]甲基}苯甲酸甲酯:在0℃下,向4-{[(金剛烷-1-基)胺基]甲基}苯甲酸甲酯(3 g, 10.0 mmol, 1當量)於THF (30 mL)中之溶液中添加N,N-二異丙基乙基胺(5.25 mL, 30.1 mmol, 3當量)及二碳酸二-第三丁基酯(6.91 mL, 30.1 mmol, 3當量)。將反應液在室溫下攪拌16 h。TLC (於己烷中之30% EtOAc)顯示反應已完成。此後,在減壓下濃縮反應混合物且使用NaHCO3
飽和水溶液(15 mL)及乙酸乙酯(30 mL)稀釋所獲得粗製物。分離有機層,使用水(20 mL)、鹽水溶液(10 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以得到粗製物(9 g),藉由急速層析使用於己烷中之0 - 25% EtOAc作為洗脫劑純化以得到4-{[(金剛烷-1-基)[(第三丁氧基)羰基]胺基]甲基}苯甲酸甲酯。LCMS (ES) m/z = 300 [M+H]+,觀察到Boc基團裂解質量。1
H NMR (400 MHz, CDCl3
) δ ppm 1.42 (s, 9 H), 1.52 - 1.72 (m, 6 H), 1.98 - 2.03 (m, 3 H), 1.52 - 1.72 (m, 6 H), 2.16 - 2.19 (m, 3 H), 4.62 - 4.67 (m, 2 H), 7.27 (d,J
= 8.8 Hz, 2 H), 7.97 (d,J
= 6.8 Hz, 2 H)。
4-((((3s,5s,7s)-金剛烷-1-基)(第三丁氧基羰基)胺基)甲基)苯甲酸:在室溫下,向4-((((3s,5s,7s)-金剛烷-1-基)(第三丁氧基羰基)胺基)甲基)苯甲酸甲酯(3 g, 7.51 mmol, 1當量)於MeOH (30 mL)及水(15 mL)中之溶液中添加氫氧化鈉(0.616 g, 15 mmol, 2.0當量)且將反應混合物在80℃下攪拌12 h。藉由TLC (於己烷中之60% E.A)監測反應進展。在反應完成之後,在減壓下濃縮反應混合物以自反應物質去除甲醇且使用EtOAc (50 mL)萃取剩餘水層。最後,使用5%檸檬酸(pH = 4)酸化水層且然後使用EtOAc (250 mL)萃取產物。藉由無水硫酸鈉乾燥有機層,過濾並在減壓下濃縮以得到產物,使用正戊烷(20 mL)研磨,傾析戊烷層並在高真空下乾燥以提供4-((((3s,5s,7s)-金剛烷-1-基)(第三丁氧基羰基)胺基)甲基)苯甲酸。LCMS (ES) m/z = 286 [M+H]+ (未觀察到boc質量)。1
H NMR (400 MHz, DMSO-d6
): δ ppm 1.33 (s, 9 H),1.88 - 2.20 (m, 9 H), 4.58 (s, 2 H), 7.28 (d,J
= 8 Hz, 2 H), 7.87 (d,J
= 8 Hz, 2 H), 12.79 (bs, 1 H)。
4-((((1R,3R)-金剛烷-1-基)胺基)甲基)-N-((S)-1-(3-甲氧基苯基)己烷-2-基)苯甲醯胺:向4-((((3s,5s,7s)-金剛烷-1-基)(第三丁氧基羰基)胺基)甲基)苯甲酸(2.66 g, 6.27 mmol, 1.1當量)於DCM (20 mL)中之溶液中添加TEA (2.63 mL, 18.8 mmol, 3當量),攪拌5 min且然後在0℃下添加T3P (50 wt.%於EtOAc中) (3.15 mL, 9.41 mmol, 1.5當量)並再攪拌30 min。然後在0℃下向反應混合物中添加(S)-1-(3-甲氧基苯基)己烷-2-胺(1.3 g, 6.27 mmol, 1當量)於DCM (10 mL)中之溶液且然後將反應混合物在室溫下攪拌12 h。藉由TLC (於正己烷中之60%乙酸乙酯)監測反應進展。此後,使用DCM (50 mL)稀釋反應混合物,使用碳酸氫鈉飽和水溶液(2 × 10 mL)、水(10 mL)、鹽水溶液(10 mL)洗滌有機層,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物,藉由急速層析使用於正己烷中之25 - 30% EtOAc作為洗脫劑純化以得到4-((((1R,3R)-金剛烷-1-基)胺基)甲基)-N-((S)-1-(3-甲氧基苯基)己烷-2-基)苯甲醯胺。LCMS (ES) m/z = 576 [M+H]+。1
H NMR (400 MHz, DMSO-d6
) δ ppm 0.80 - 0.87 (m, 3 H), 1.12 - 1.35 (m, 9 H), 1.52 (bs, 5 H), 1.97 - 2.03 (m, 6 H), 2.74 (bs, 1 H), 3.64 (s, 3 H), 4.11 (bs, 1 H), 3.8 (bs, 1 H), 4.11(bs, 1 H), 4.55 (bs, 2 H), 6.70 - 6.76 (m, 2 H), 7.13 - 7.22 (m, 3 H), 7.71 (m, 1 H), 8.09 (bs, 1 H)。
N-({4-[(3S)-3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基]苯基}甲基)金剛烷-1-胺:在-78℃下,經由注射器向4-((((1R,3R)-金剛烷-1-基)胺基)甲基)-N-((S)-1-(3-甲氧基苯基)己烷-2-基)苯甲醯胺(2.2 g, 3.83 mmol, 1當量)及2-氯吡啶(3.6 mL, 38.32 mmol, 10.0當量)於二氯甲烷(5 mL)中之經攪拌溶液中緩慢逐滴添加三氟甲烷磺酸酐(1.9 mL, 11.49 mmol, 3.0當量)。在5 min之後,將反應混合物置於冰-水浴中並升溫至0℃。在5 min之後,將所得溶液升溫至23℃並保持1 h。藉由TLC (於己烷中之30% EA)監測反應進展。在30分鐘之後,使用氫氧化鈉水溶液(3 mL, 1N)終止反應以中和三氟甲烷磺酸鹽。添加二氯甲烷(15 mL)以稀釋混合物且分離各層。使用DCM (2 × 50 mL)萃取水層。使用鹽水(5 mL)洗滌合併之有機層,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以得到粗產物。LCMS (ES) m/z = 457 [M+H]+。
N-(金剛烷-1-基)-N-({4-[(3S)-3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基]苯基}甲基)胺基甲酸第三丁基酯:在0℃下,向(1R,3R)-N-(4-((S)-3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基)苄基)金剛烷-1-胺(2.1 g, 4.60 mmol, 1.0當量)於THF (20.0 mL)中之經攪拌溶液中添加DIPEA (2.46 mL, 13.8 mmol, 3.0當量),隨後添加boc酐(3.17 mL, 13.8 mmol, 3.0當量)。然後,將反應混合物在室溫下攪拌16 h。使用水(25 mL)稀釋反應混合物,使用乙酸乙酯(2 × 30 mL)萃取。使用水(20 mL)、鹽水(20 mL)洗滌合併之有機層,藉由無水硫酸鈉乾燥。過濾有機層並在減壓下濃縮以得到粗產物,藉由矽膠管柱層析使用於己烷中之30%乙酸乙酯作為洗脫劑純化以得到((1R,3R)-金剛烷-1-基)(4-((S)-3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基)苄基)胺基甲酸第三丁基酯。LCMS (ES) m/z = 557 [M+H]+1
H NMR (400 MHz, DMSO-d6
) δ ppm 0.89 (t,J
= 7.2 Hz, 3 H), 1.3 (s, 9 H), 1.54 - 1.65 (m, 9 H), 1.99 - 2.10 (m, 9 H), 2.4 - 2.6 (m, 2 H), 2.74 - 2.77 (m, 1 H), 3.78 (s,3 H), 4.58 (s, 2 H), 6.79 - 6.81 (m, 1 H), 6.90 (s, 1 H), 7.07 - 7.09 (m, 1 H), 7.23 - 7.25 (m, 2 H), 7.44 - 7.46 (m, 1 H)。
N-(金剛烷-1-基)-N-({4-[(1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基]苯基}甲基)胺基甲酸第三丁基酯:在0℃下,向((1R,3R)-金剛烷-1-基)(4-((S)-3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基)苄基)胺基甲酸第三丁基酯(0.8 g, 1.44 mmol, 1當量)於甲醇(15 mL)中之溶液中逐份添加硼氫化鈉(0.159 g, 4.31 mmol, 3當量)。將懸浮液在室溫下攪拌30 min。藉由TLC (於己烷中之20% EA)監測反應進展。此後,使用丙酮(10 mL)淬滅反應混合物,濃縮且使用EtOAc (20 mL)及水(5 mL)稀釋所獲得粗製物。分離有機層,使用鹽水溶液(10 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物。藉由製備型TLC使用於正己烷中之20%乙酸乙酯作為洗脫劑純化所獲得粗產物以得到((1R,3R)-金剛烷-1-基)(4-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)苄基)胺基甲酸第三丁基酯(1, 3反式異構體)。LCMS (ES) m/z = 559 [M+H]+。1
H NMR (400 MHz, CDCl3
) δ ppm 0.8 - 0.9 (m, 3 H), 1.1 - 1.3 (m, 5 H), 1.39 (s, 9 H), 1.59 - 1.70 (m, 9 H), 2.02 (bs, 3 H), 2.10 (s, 5 H), 2.73 (bs, 2 H), 2.88 - 2.99 (m, 2 H), 3.79 (s, 3 H), 4.55 (s, 2 H), 5.29 (s, 2 H), 6.68 (s, 2 H), 6.84 (bs, 1 H), 7.11 (bs, 3 H)。
N-(金剛烷-1-基)-N-({4-[(1S,3S)-3-丁基-6-甲氧基-2-[3-(三甲基矽烷基)丙-2-炔醯基]-1,2,3,4-四氫異喹啉-1-基]苯基}甲基)胺基甲酸第三丁基酯:第一步驟:在室溫下,向3-(三甲基矽烷基)丙炔酸(0.043 g, 0.302 mmol, 1.0當量)於DMF (0.00094 mL, 0.012 mmol, 0.04當量)中之溶液中添加草醯氯(0.028 mL, 0.33 mmol, 1.1當量)並攪拌30分鐘。然後,在減壓下濃縮反應混合物以得到3-(三甲基矽烷基)丙炔醯氯。此醯氯粗製物未經任何進一步純化即用於下一步驟中。
第二步驟:在0℃下,向((1R,3R)-金剛烷-1-基)(4-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)苄基)胺基甲酸第三丁基酯(0.080 g, 0.143 mmol, 1.0當量)於乙腈(10 mL)中之溶液中添加碳酸氫鈉(0.091 g, 1.07 mmol, 7.5當量)。在攪拌5分鐘之後,向上述反應物質中添加3-(三甲基矽烷基)丙炔醯氯(0.025 g, 0.157 mmol, 1.1當量)於乙腈(5.0 mL)中之溶液。將所得混合物在0℃下攪拌1 h,藉由TLC (於正己烷中之20%乙酸乙酯)監測反應進展。此後,使用EtOAc (100 mL)及水(50 mL)稀釋反應物質。分離有機層,使用鹽水溶液(10 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮。此粗產物未經任何進一步純化即用於下一步驟中。LCMS (ES) m/z = 583 [M+H]+ (未觀察到Boc基團質量)。
N-(金剛烷-1-基)-N-({4-[(1S,3S)-3-丁基-6-甲氧基-2-(丙-2-炔醯基)-1,2,3,4-四氫異喹啉-1-基]苯基}甲基)胺基甲酸第三丁基酯:在0℃下,向((1R,3R)-金剛烷-1-基)(4-((1S,3S)-3-丁基-6-甲氧基-2-(3-(三甲基矽烷基)丙炔醯基)-1,2,3,4-四氫異喹啉-1-基)苄基)胺基甲酸第三丁基酯(171 mg, 0.25 mmol, 1當量)於DCM (10 mL) / MeOH (2 mL)中之溶液中添加K2
CO3
(207 mg, 1.50 mmol, 6當量)。將混合物在0℃下攪拌2 h,在反應完成之後,使用DCM (5 mL)稀釋反應混合物並添加H2O (3 mL)。使用DCM (3 × 10 mL)萃取有機層並藉由Na2
SO4
乾燥,且濃縮。藉由製備型TLC使用於己烷中之20% E.A純化殘餘物以得到((1R,3R)-金剛烷-1-基)(4-((1S,3S)-3-丁基-6-甲氧基-2-丙炔醯基-1,2,3,4-四氫異喹啉-1-基)苄基)胺基甲酸第三丁基酯。LCMS (ES) m/z = 511.2 [M+H]+。(未展示基團質量)。1
H NMR (400 MHz, CDCl3
) δ ppm 0.81 - 0.85 (m, 3 H), 0.93 - 0.95 (m, 1 H), 1.2 - 1.2 (m, 6 H), 1.38 - 1.56 (m, 9 H), 1.68 - 1.71 (m, 2 H), 1.91 - 2.0 (m, 3 H), 2.01 (bs, 5 H), 2.17 (s, 1 H), 2.67 - 2.82 (m, 2 H), 2.88 (s, 1 H), 3.80 - 3.81 (m, 3 H), 4.48 - 4.61 (m, 4 H), 6.29 (d,J
= 14 Hz,1 H), 6.69 (s, 1 H), 6.78 - 6.84 (m, 1 H), 6.93 - 7.08 (m, 4 H), 7.33 (d,J
= 8.4 Hz, 1 H)。
N-(金剛烷-1-基)-N-({4-[(1S,3S)-3-丁基-6-甲氧基-2-(丙-2-炔醯基)-1,2,3,4-四氫異喹啉-1-基]苯基}甲基)-2,2,2-三氟乙醯胺:在冷卻(0℃)條件,向((1R,3R)-金剛烷-1-基)(4-((1S,3S)-3-丁基-6-甲氧基-2-丙炔醯基-1,2,3,4-四氫異喹啉-1-基)苄基)胺基甲酸第三丁基酯(0.06 g, 0.098 mmol, 1當量)於DCM (15 mL)中之經攪拌溶液中逐滴添加TFA (0.2 mL),將反應混合物在0℃下攪拌2 h,藉由TLC (於己烷中之70% EA)檢查反應進展,在反應完成之後,在減壓下濃縮反應混合物以獲得粗產物,藉由製備型HPLC純化藉由使用下列分析條件進一步純化以得到1-((1S,3S)-1-(4-((((1R,3R)-金剛烷-1-基)(2,2,2-三氟乙醯基)-l4-氮烷基)甲基)苯基)-3-丁基-6-甲氧基-3,4-二氫異喹啉-2(1H)-基)丙-2-炔-1-酮。分析條件:管柱:X-BridgeC-18 (250 mm × 4.6 mm × 5 µm);移動相(A):於水中之0.1%TFA;移動相(B):乙腈;流速:1.0 mL/min;梯度B:0/20, 12/60, 22/95, 25/95, 27/20, 30/20。LCMS (ES) m/z = 511.1 [M+H]+。1
H NMR (400 MHz, CDCl3
) δ ppm 0.80 - 0.84 (m, 3 H), 0.90 - 1.2 (m, 7 H), 1.5 - 1.67 (m, 6 H), 1.86 (s, 4 H), 1.90 - 2.1 (m, 2 H), 2.76 - 2.90 (m, 2 H), 3.0- 3.16 (m, 1 H), 3.70 - 3.80 (m, 4 H), 4.01 (bs, 2 H), 4.59 - 4.75 (m, 2 H), 6.75 - 6.81 (m, 2 H), 7.07 - 7.12 (m, 1 H), 7.27 - 7.44 (m, 4 H), 8.46 (bs, 2 H)。
程序 34 :化合物 18 之合成 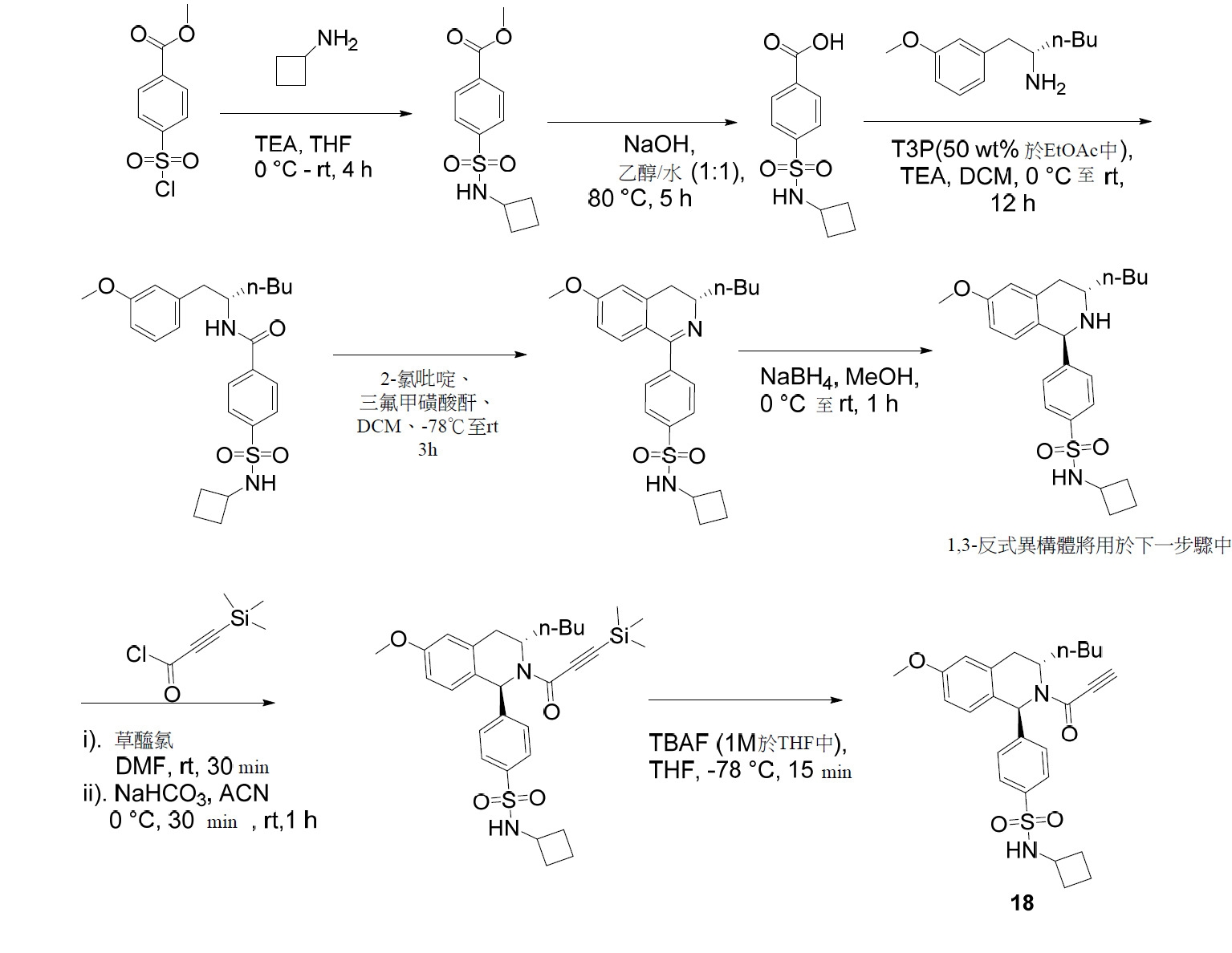 4-(環丁基胺磺醯基)苯甲酸甲酯:在室溫下,向4-(氯磺醯基)苯甲酸甲酯(5.0 g, 21.3 mmol, 1.0當量)於THF (50 mL)中之溶液中添加三乙胺(8.92 mL, 63.9 mmol, 3.0當量)及環丁胺(1.83 mL, 21.3 mmol, 1.0當量)且將混合物在室溫下攪拌3 h,TLC (於己烷中之30% EtOAc)顯示反應已完成。在減壓下濃縮反應混合物且使用EtOAc (250 mL)稀釋所獲得粗製物,使用水(2 × 50 mL)洗滌。藉由無水Na2
SO4
乾燥有機層,在減壓下濃縮以得到粗產物。藉由急速層析使用於己烷中之15-20% EtOAc作為洗脫劑純化粗製物以得到4-(N-環丁基胺磺醯基)苯甲酸甲酯。LCMS (ES) m/z: 270.1 [M+H]+。1
H NMR (400MHz, DMSO-d6
) δ 1.56 - 1.67 (m, 2 H), 1.71 - 1.76 (m, 2 H), 2.10 - 2.14 (m, 2 H), 3.78-3.86 (m, 1 H), 3.95 (s, 3 H), 4.67 - 4.69 (m,1 H), 7.92 (d,J
= 8.4 Hz, 2 H), 8.15 (d,J
= 8.4 Hz, 2 H)。
4-(環丁基胺磺醯基)苯甲酸:在室溫下,向4-(環丁基胺磺醯基)苯甲酸甲酯(5 g, 18.6 mmol, 1當量)於EtOH (25 mL)及水(25 mL)中之經攪拌溶液中添加氫氧化鈉(1.49 g, 37.1 mmol, 2當量)且將反應液在80℃下攪拌6 h。TLC (於己烷中之70% EtOAc)顯示反應在6 h之後完成。將反應混合物冷卻至室溫,在減壓下濃縮反應混合物。使用EtOAc (2 × 20 mL)萃取所獲得水層且然後使用2 N HCl (pH ~ 5)酸化水層,且然後使用EtOAc (50 mL)萃取。藉由無水硫酸鈉乾燥有機層,過濾並在減壓下濃縮以提供標題化合物4-(環丁基胺磺醯基)苯甲酸。LCMS (ES) m/z = 254 [M-H]-。1
H NMR (400 MHz, DMSO-d6
) δ ppm 1.20 - 1.45 (m, 2 H), 1.68 - 1.72 (m, 2 H), 1.60 - 1.88 (m, 2 H), 1.74 - 1.78 (m, 4 H), 3.60 - 3.66 (m, 1 H), 7.86 - 7.88 (m, 2 H), 8.09 - 8.13 (m, 3 H), 4.37 (bs, 1 H), 13.5 (bs, 1 H)。
4-(環丁基胺磺醯基)-N-[(2S)-1-(3-甲氧基苯基)己烷-2-基]苯甲醯胺:在0℃及氮氣氛下,向4-(環丁基胺磺醯基)苯甲酸(3.69 g, 14.5 mmol, 1當量)於DCM (30 mL)中之溶液中添加三乙胺(6.06 mL, 43.4 mmol, 3.0當量),攪拌10 min且然後在0℃下向反應混合物中添加丙烷膦酸酐(50 wt.%於乙酸乙酯中) (9.69 mL, 8.57 mmol, 2.0當量),在0℃下攪拌15 min且然後在0℃下向反應混合物中添加溶於DCM (20.0 mL)中之(2S)-1-(3-甲氧基苯基)己烷-2-胺(3.0 g, 14.5 mmol, 1當量),且然後將反應混合物在室溫下攪拌16 h。TLC (於己烷中之40% EtOAc)顯示反應在16 h之後完成。使用DCM (150 mL)稀釋反應混合物,使用飽和碳酸氫鈉溶液(20 mL)及水(30 mL)洗滌。藉由無水Na2
SO4
乾燥有機層,過濾並在減壓下濃縮以得到4-(環丁基胺磺醯基)-N-[(2S)-1-(3-甲氧基苯基)己烷-2-基]苯甲醯胺。LCMS (ES) m/z = 445.2 [M+H]+。1
H NMR (400 MHz,CDCl3
) δ ppm 0.87 - 0.89 (m, 3 H), 1.32 - 1.36 (m, 4 H), 1.49 - 1.54 (m, 1 H), 1.74 - 1.78 (m, 4 H), 2.1 (bs, 2 H), 2.86 - 2.94 (m, 2 H), 3.75 - 3.78 (m, 4 H), 4.37 (bs, 1 H), 4.76 (bs, 3 H), 5.58 - 5.89 (m, 1 H), 6.74 - 6.67 (m, 3 H), 7.19 - 7.25 (m, 1 H), 7.74 - 7.75 (m, 2 H), 7.84 - 7.86 (m, 2 H)。
(S)-4-(3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基)-N-環丁基苯磺醯胺:在室溫及氮氣氛下,向(S)-4-(N-環丁基胺磺醯基)-N-(1-(3-甲氧基苯基)己烷-2-基)苯甲醯胺(3 g, 6.75 mmol, 1當量)於DCM (30 mL)中之溶液中添加2-氯吡啶(1.28 mL, 13.5 mmol, 2當量)。然後在-78℃下添加三氟甲烷磺酸酐(2.27 mL, 13.5 mmol, 2當量),攪拌5 min,然後升溫至0℃,在0℃下攪拌30 min且然後將反應混合物在室溫下攪拌3 h。TLC (100% EtOAc)展示起始材料以及新斑點。藉由LC-MS監測反應。在減壓下濃縮反應物質以獲得粗製殘餘物,使用10%氫氧化鈉溶液(15 mL)淬滅所得殘餘物,使用(2 × 150 mL)乙酸乙酯萃取,使用無水Na2
SO4
乾燥合併之有機層,過濾並在減壓下濃縮以得到粗產物,藉由急速管柱層析使用於己烷中之100%乙酸乙酯作為洗脫劑純化以獲得(S)-4-(3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基)-N-環丁基苯磺醯胺。LCMS (ES) m/z = 427.1 [M+H]+。1
H NMR (400MHz, CDCl3
) δ ppm: 7.88 (d,J
= 7.6 Hz, 2 H), 7.70 (d,J
= 8 Hz, 2 H), 7.05 (d,J
= 8.8 Hz, 1 H), 6.78 (s, 1 H), 6.72 (d,J
= 8 Hz, 1 H), 4.68 - 4.67 (m, 1 H), 3.89 (s, 3 H), 3.56 - 3.52 (m, 1 H), 2.82 - 2.71 (m, 1 H), 2.82 - 2.71 (m, 1 H), 2.62 - 2.46 (m, 1 H), 2.18 - 2.16 (m, 1 H), 1.83 - 1.75 (m, 4 H), 1.61 - 1.56 (m, 3 H), 1.49 - 1.35 (m, 2 H), 0.95 - 0.91 (m, 3 H)。
4-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)-N-環丁基苯磺醯胺:在0℃下,向(S)-4-(3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基)-N-環丁基苯磺醯胺(1.5 g, 3.52 mmol, 1當量)於甲醇(15 mL)中之溶液中逐份添加硼氫化鈉(0.388 g, 10.5 mmol, 3當量)。將懸浮液在室溫下攪拌1 h。藉由TLC (於己烷中之50% EA)監測反應進展。此後,使用丙酮(10 mL)淬滅反應混合物,濃縮且使用EtOAc (30 mL)及水(20 mL)稀釋所獲得粗產物。分離有機層,使用鹽水溶液(10 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物。藉由製備型TLC使用於正己烷中之50%乙酸乙酯作為洗脫劑純化所獲得粗產物以得到4-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)-N-環丁基苯磺醯胺(1, 3反式異構體)。LCMS (ES) (m/z) = 429 [M+H]+。
4-((1S,3S)-3-丁基-6-甲氧基-2-(3-(三甲基矽烷基)丙炔醯基)-1,2,3,4-四氫異喹啉-1-基)-N-環丁基苯磺醯胺:第一步驟:在室溫下,向3-(三甲基矽烷基)丙炔酸(0.2 g, 1.41 mmol, 1當量)於DMF (0.04 mL, 0.56 mmol, 0.04當量)中之溶液中添加草醯氯(0.15 mL, 1.69 mmol, 1.2當量)並攪拌30分鐘。然後,在減壓下濃縮反應混合物以得到3-(三甲基矽烷基)丙炔醯氯。此醯氯粗製物未經任何進一步純化即用於下一步驟中。
第二步驟:在0℃下,向4-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)-N-環丁基苯磺醯胺(0.2 g, 0.467 mmol, 1當量)於乙腈(10 mL)中之溶液中添加碳酸氫鈉(0.298 g, 3.5 mmol, 7.5當量),隨後添加於乙腈(5 mL)中之3-(三甲基矽烷基)丙炔醯氯(0.09 g, 0.56 mmol, 1.2當量)。將混合物在0℃下攪拌5分鐘,然後在室溫下攪拌1 h。然後使用水(5 mL)稀釋反應混合物且使用乙酸乙酯(2 × 15 mL)萃取。藉由無水Na2
SO4
乾燥有機層,過濾並在減壓下濃縮以獲得粗製4-((1S,3S)-3-丁基-6-甲氧基-2-(3-(三甲基矽烷基)丙炔醯基)-1,2,3,4-四氫異喹啉-1-基)-N-環丁基苯磺醯胺。LCMS (ES) (m/z) = 553 [M+H]+。
4-((1S,3S)-3-丁基-6-甲氧基-2-丙炔醯基-1,2,3,4-四氫異喹啉-1-基)-N-環丁基苯磺醯胺:反式異構體:在-78℃下,向4-((1S,3S)-3-丁基-6-甲氧基-2-(3-(三甲基矽烷基)丙炔醯基)-1,2,3,4-四氫異喹啉-1-基)-N-環丁基苯磺醯胺(0.200 g, 0.362 mmol, 1.0當量)於THF (5.0 mL)中之經攪拌溶液中添加四丁基氟化銨(1M於THF中) (0.43 mL, 0.43 mmol, 1.2當量)。將反應混合物在相同溫度下攪拌30分鐘。使用飽和碳酸氫鈉溶液(5 mL)淬滅反應混合物,使用乙酸乙酯(2 × 20 mL)萃取。使用水(10 mL)、鹽水(10 mL)洗滌合併之有機層並藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以得到粗製物。藉由急速層析使用於正己烷中之60%乙酸乙酯作為洗脫劑純化所獲得粗產物以得到期望產物4-((1S,3S)-3-丁基-6-甲氧基-2-丙炔醯基-1,2,3,4-四氫異喹啉-1-基)-N-環丁基苯磺醯胺。LCMS (ES) m/z = 481.3 [M+H]+。1
H NMR (400 MHz, DMSO-d6
) δ ppm 0.80 - 0.81 (m, 3 H), 0.90 - 0.95 (m, 1 H), 1.20 - 1.39 (m, 4 H), 1.40 - 1.49 (m, 3 H), 1.71 - 1.76 (m, 2 H), 1.88 - 1.90 (m, 2 H), 2.81 - 2.94 (m, 1 H), 3.06 - 3.17 (m, 1 H), 3.55 - 3.57 (m, 1 H), 3.77 (s, 3 H), 4.44 (s, 1 H), 4.75 (s, 1 H), 6.06 (s, 1 H), 6.77 - 6.83 (m, 2 H), 7.37 - 7.43 (m, 2 H), 7.60 - 7.69 (m, 3 H)。
4-(環丁基胺磺醯基)苯甲酸甲酯:在室溫下,向4-(氯磺醯基)苯甲酸甲酯(5.0 g, 21.3 mmol, 1.0當量)於THF (50 mL)中之溶液中添加三乙胺(8.92 mL, 63.9 mmol, 3.0當量)及環丁胺(1.83 mL, 21.3 mmol, 1.0當量)且將混合物在室溫下攪拌3 h,TLC (於己烷中之30% EtOAc)顯示反應已完成。在減壓下濃縮反應混合物且使用EtOAc (250 mL)稀釋所獲得粗製物,使用水(2 × 50 mL)洗滌。藉由無水Na2
SO4
乾燥有機層,在減壓下濃縮以得到粗產物。藉由急速層析使用於己烷中之15-20% EtOAc作為洗脫劑純化粗製物以得到4-(N-環丁基胺磺醯基)苯甲酸甲酯。LCMS (ES) m/z: 270.1 [M+H]+。1
H NMR (400MHz, DMSO-d6
) δ 1.56 - 1.67 (m, 2 H), 1.71 - 1.76 (m, 2 H), 2.10 - 2.14 (m, 2 H), 3.78-3.86 (m, 1 H), 3.95 (s, 3 H), 4.67 - 4.69 (m,1 H), 7.92 (d,J
= 8.4 Hz, 2 H), 8.15 (d,J
= 8.4 Hz, 2 H)。
4-(環丁基胺磺醯基)苯甲酸:在室溫下,向4-(環丁基胺磺醯基)苯甲酸甲酯(5 g, 18.6 mmol, 1當量)於EtOH (25 mL)及水(25 mL)中之經攪拌溶液中添加氫氧化鈉(1.49 g, 37.1 mmol, 2當量)且將反應液在80℃下攪拌6 h。TLC (於己烷中之70% EtOAc)顯示反應在6 h之後完成。將反應混合物冷卻至室溫,在減壓下濃縮反應混合物。使用EtOAc (2 × 20 mL)萃取所獲得水層且然後使用2 N HCl (pH ~ 5)酸化水層,且然後使用EtOAc (50 mL)萃取。藉由無水硫酸鈉乾燥有機層,過濾並在減壓下濃縮以提供標題化合物4-(環丁基胺磺醯基)苯甲酸。LCMS (ES) m/z = 254 [M-H]-。1
H NMR (400 MHz, DMSO-d6
) δ ppm 1.20 - 1.45 (m, 2 H), 1.68 - 1.72 (m, 2 H), 1.60 - 1.88 (m, 2 H), 1.74 - 1.78 (m, 4 H), 3.60 - 3.66 (m, 1 H), 7.86 - 7.88 (m, 2 H), 8.09 - 8.13 (m, 3 H), 4.37 (bs, 1 H), 13.5 (bs, 1 H)。
4-(環丁基胺磺醯基)-N-[(2S)-1-(3-甲氧基苯基)己烷-2-基]苯甲醯胺:在0℃及氮氣氛下,向4-(環丁基胺磺醯基)苯甲酸(3.69 g, 14.5 mmol, 1當量)於DCM (30 mL)中之溶液中添加三乙胺(6.06 mL, 43.4 mmol, 3.0當量),攪拌10 min且然後在0℃下向反應混合物中添加丙烷膦酸酐(50 wt.%於乙酸乙酯中) (9.69 mL, 8.57 mmol, 2.0當量),在0℃下攪拌15 min且然後在0℃下向反應混合物中添加溶於DCM (20.0 mL)中之(2S)-1-(3-甲氧基苯基)己烷-2-胺(3.0 g, 14.5 mmol, 1當量),且然後將反應混合物在室溫下攪拌16 h。TLC (於己烷中之40% EtOAc)顯示反應在16 h之後完成。使用DCM (150 mL)稀釋反應混合物,使用飽和碳酸氫鈉溶液(20 mL)及水(30 mL)洗滌。藉由無水Na2
SO4
乾燥有機層,過濾並在減壓下濃縮以得到4-(環丁基胺磺醯基)-N-[(2S)-1-(3-甲氧基苯基)己烷-2-基]苯甲醯胺。LCMS (ES) m/z = 445.2 [M+H]+。1
H NMR (400 MHz,CDCl3
) δ ppm 0.87 - 0.89 (m, 3 H), 1.32 - 1.36 (m, 4 H), 1.49 - 1.54 (m, 1 H), 1.74 - 1.78 (m, 4 H), 2.1 (bs, 2 H), 2.86 - 2.94 (m, 2 H), 3.75 - 3.78 (m, 4 H), 4.37 (bs, 1 H), 4.76 (bs, 3 H), 5.58 - 5.89 (m, 1 H), 6.74 - 6.67 (m, 3 H), 7.19 - 7.25 (m, 1 H), 7.74 - 7.75 (m, 2 H), 7.84 - 7.86 (m, 2 H)。
(S)-4-(3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基)-N-環丁基苯磺醯胺:在室溫及氮氣氛下,向(S)-4-(N-環丁基胺磺醯基)-N-(1-(3-甲氧基苯基)己烷-2-基)苯甲醯胺(3 g, 6.75 mmol, 1當量)於DCM (30 mL)中之溶液中添加2-氯吡啶(1.28 mL, 13.5 mmol, 2當量)。然後在-78℃下添加三氟甲烷磺酸酐(2.27 mL, 13.5 mmol, 2當量),攪拌5 min,然後升溫至0℃,在0℃下攪拌30 min且然後將反應混合物在室溫下攪拌3 h。TLC (100% EtOAc)展示起始材料以及新斑點。藉由LC-MS監測反應。在減壓下濃縮反應物質以獲得粗製殘餘物,使用10%氫氧化鈉溶液(15 mL)淬滅所得殘餘物,使用(2 × 150 mL)乙酸乙酯萃取,使用無水Na2
SO4
乾燥合併之有機層,過濾並在減壓下濃縮以得到粗產物,藉由急速管柱層析使用於己烷中之100%乙酸乙酯作為洗脫劑純化以獲得(S)-4-(3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基)-N-環丁基苯磺醯胺。LCMS (ES) m/z = 427.1 [M+H]+。1
H NMR (400MHz, CDCl3
) δ ppm: 7.88 (d,J
= 7.6 Hz, 2 H), 7.70 (d,J
= 8 Hz, 2 H), 7.05 (d,J
= 8.8 Hz, 1 H), 6.78 (s, 1 H), 6.72 (d,J
= 8 Hz, 1 H), 4.68 - 4.67 (m, 1 H), 3.89 (s, 3 H), 3.56 - 3.52 (m, 1 H), 2.82 - 2.71 (m, 1 H), 2.82 - 2.71 (m, 1 H), 2.62 - 2.46 (m, 1 H), 2.18 - 2.16 (m, 1 H), 1.83 - 1.75 (m, 4 H), 1.61 - 1.56 (m, 3 H), 1.49 - 1.35 (m, 2 H), 0.95 - 0.91 (m, 3 H)。
4-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)-N-環丁基苯磺醯胺:在0℃下,向(S)-4-(3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基)-N-環丁基苯磺醯胺(1.5 g, 3.52 mmol, 1當量)於甲醇(15 mL)中之溶液中逐份添加硼氫化鈉(0.388 g, 10.5 mmol, 3當量)。將懸浮液在室溫下攪拌1 h。藉由TLC (於己烷中之50% EA)監測反應進展。此後,使用丙酮(10 mL)淬滅反應混合物,濃縮且使用EtOAc (30 mL)及水(20 mL)稀釋所獲得粗產物。分離有機層,使用鹽水溶液(10 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物。藉由製備型TLC使用於正己烷中之50%乙酸乙酯作為洗脫劑純化所獲得粗產物以得到4-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)-N-環丁基苯磺醯胺(1, 3反式異構體)。LCMS (ES) (m/z) = 429 [M+H]+。
4-((1S,3S)-3-丁基-6-甲氧基-2-(3-(三甲基矽烷基)丙炔醯基)-1,2,3,4-四氫異喹啉-1-基)-N-環丁基苯磺醯胺:第一步驟:在室溫下,向3-(三甲基矽烷基)丙炔酸(0.2 g, 1.41 mmol, 1當量)於DMF (0.04 mL, 0.56 mmol, 0.04當量)中之溶液中添加草醯氯(0.15 mL, 1.69 mmol, 1.2當量)並攪拌30分鐘。然後,在減壓下濃縮反應混合物以得到3-(三甲基矽烷基)丙炔醯氯。此醯氯粗製物未經任何進一步純化即用於下一步驟中。
第二步驟:在0℃下,向4-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)-N-環丁基苯磺醯胺(0.2 g, 0.467 mmol, 1當量)於乙腈(10 mL)中之溶液中添加碳酸氫鈉(0.298 g, 3.5 mmol, 7.5當量),隨後添加於乙腈(5 mL)中之3-(三甲基矽烷基)丙炔醯氯(0.09 g, 0.56 mmol, 1.2當量)。將混合物在0℃下攪拌5分鐘,然後在室溫下攪拌1 h。然後使用水(5 mL)稀釋反應混合物且使用乙酸乙酯(2 × 15 mL)萃取。藉由無水Na2
SO4
乾燥有機層,過濾並在減壓下濃縮以獲得粗製4-((1S,3S)-3-丁基-6-甲氧基-2-(3-(三甲基矽烷基)丙炔醯基)-1,2,3,4-四氫異喹啉-1-基)-N-環丁基苯磺醯胺。LCMS (ES) (m/z) = 553 [M+H]+。
4-((1S,3S)-3-丁基-6-甲氧基-2-丙炔醯基-1,2,3,4-四氫異喹啉-1-基)-N-環丁基苯磺醯胺:反式異構體:在-78℃下,向4-((1S,3S)-3-丁基-6-甲氧基-2-(3-(三甲基矽烷基)丙炔醯基)-1,2,3,4-四氫異喹啉-1-基)-N-環丁基苯磺醯胺(0.200 g, 0.362 mmol, 1.0當量)於THF (5.0 mL)中之經攪拌溶液中添加四丁基氟化銨(1M於THF中) (0.43 mL, 0.43 mmol, 1.2當量)。將反應混合物在相同溫度下攪拌30分鐘。使用飽和碳酸氫鈉溶液(5 mL)淬滅反應混合物,使用乙酸乙酯(2 × 20 mL)萃取。使用水(10 mL)、鹽水(10 mL)洗滌合併之有機層並藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以得到粗製物。藉由急速層析使用於正己烷中之60%乙酸乙酯作為洗脫劑純化所獲得粗產物以得到期望產物4-((1S,3S)-3-丁基-6-甲氧基-2-丙炔醯基-1,2,3,4-四氫異喹啉-1-基)-N-環丁基苯磺醯胺。LCMS (ES) m/z = 481.3 [M+H]+。1
H NMR (400 MHz, DMSO-d6
) δ ppm 0.80 - 0.81 (m, 3 H), 0.90 - 0.95 (m, 1 H), 1.20 - 1.39 (m, 4 H), 1.40 - 1.49 (m, 3 H), 1.71 - 1.76 (m, 2 H), 1.88 - 1.90 (m, 2 H), 2.81 - 2.94 (m, 1 H), 3.06 - 3.17 (m, 1 H), 3.55 - 3.57 (m, 1 H), 3.77 (s, 3 H), 4.44 (s, 1 H), 4.75 (s, 1 H), 6.06 (s, 1 H), 6.77 - 6.83 (m, 2 H), 7.37 - 7.43 (m, 2 H), 7.60 - 7.69 (m, 3 H)。
程序 35 :化合物 95 之合成 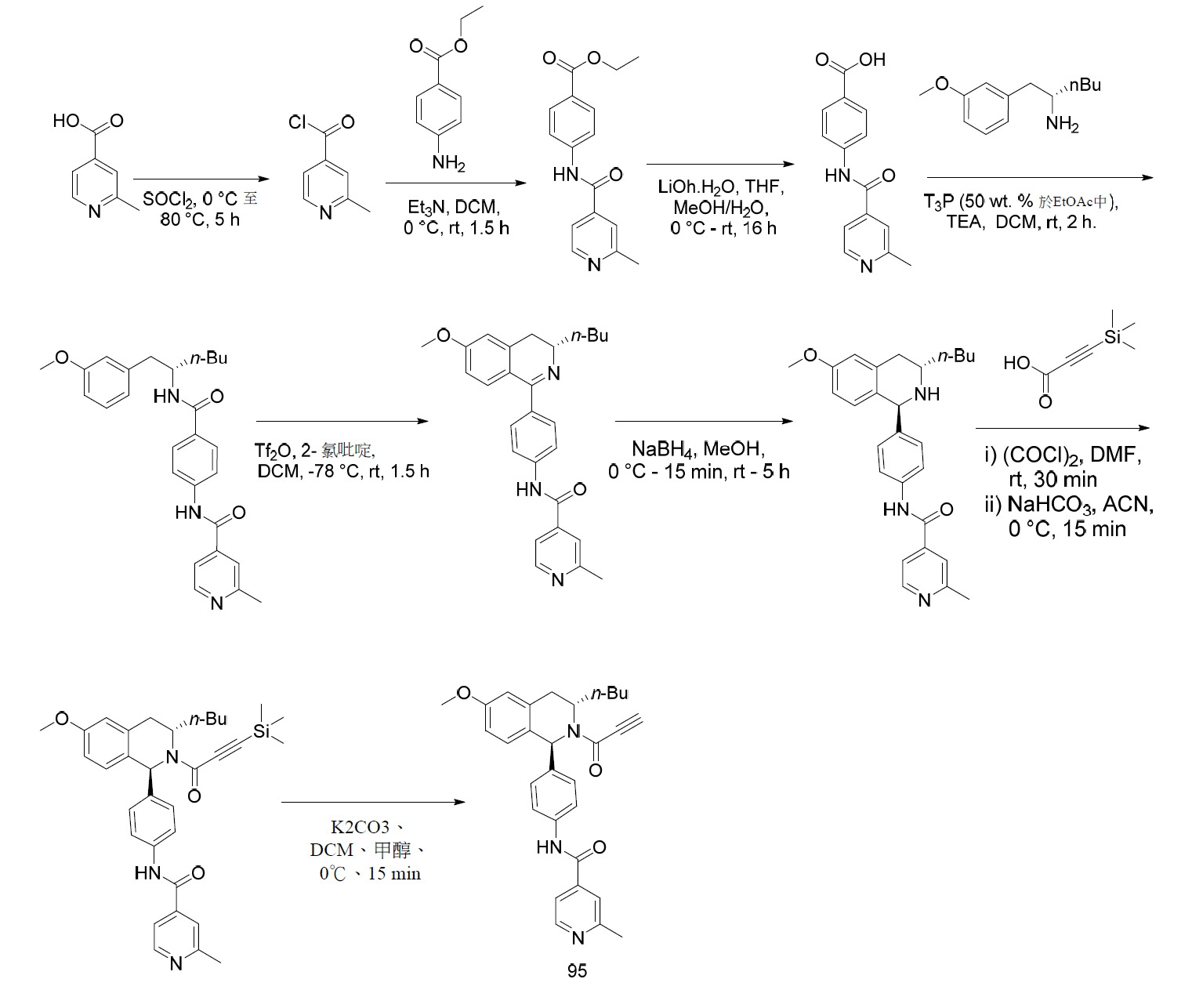 2-甲基吡啶-4-羰基氯:在0℃下,向2-甲基吡啶-4-甲酸(3.00 g, 21.9 mmol, 1.0當量)於N,N-二甲基甲醯胺(0.067, 0.87 mmol, 0.04當量)中之溶液中添加SOCl2 (40.0 mL),將此混合物在80℃下回流3 h,且在減壓下濃縮過量亞硫醯氯以得到2-甲基吡啶-4-羰基氯。此粗產物未經任何進一步純化即用於下一步驟中。
4-(2-甲基吡啶-4-醯胺基)苯甲酸乙酯:在0℃下,向4-胺基苯甲酸乙酯(3.19 g, 19.3 mmo, l,1.0當量)於DCM (40.0 mL)中之經攪拌溶液中添加三乙胺(15.0 mL, 116 mmol, 6當量)且將反應液攪拌15 min。然後,緩慢添加2-甲基吡啶-4-羰基氯(20 ml DCM) (3.00 g, 19.3 mmol, 1當量)。此後,將反應液在室溫下攪拌12 h。在反應完成之後,使用水將反應混合物緩慢淬滅,使用DCM (100 mL)稀釋,在室溫下攪拌5 min。然後分離各層。使用DCM (2 × 100.0 mL)萃取水層。使用水(50.0 mL)洗滌合併之有機層,分離各層。然後藉由無水硫酸鈉乾燥有機層,過濾並在減壓下濃縮以獲得粗製物。藉由管柱層析使用10% MeOH/DCM作為洗脫劑純化粗製物以提供4-(2-甲基吡啶-4-醯胺基)苯甲酸乙酯。LCMS (ES) m/z = 285.2 [M+H]+。1
H NMR (400MHz, DMSO-d6
) δ ppm 1.28 - 1.31 (m, 3 H), 2.48 (s, 3 H), 4.25 - 4.30 (m, 2 H), 7.62 (d,J
= 4.8 Hz, 1 H), 7.76 (s, 1 H), 7.89 - 7.97 (m, 4 H), 8.61 (d,J
= 5.2 Hz, 1 H), 10.72 (s, 1 H),
4-(2-甲基吡啶-4-醯胺基)苯甲酸:向4-(2-甲基吡啶-4-醯胺基)苯甲酸乙酯(2.50 g, 8.79 mmol, 1.0當量)於MeOH (20.0 mL)、THF (20.0 mL)及水(15.0 mL)中之經攪拌溶液中添加LiOH.H2O (0.76 g, 17.6 mmol, 2.0當量)且將反應液在室溫下攪拌16 h。藉由TLC (5% MeOH/DCM)監測反應。此後,在減壓及室溫下濃縮反應混合物且使用EtOAc (2 × 50 mL)萃取水層。然後使用5%檸檬酸(Ph = 5)酸化水層,過濾並在減壓下乾燥以得到4-(2-甲基吡啶-4-醯胺基)苯甲酸。LC-MS (ES) (m/z) = 257.2 [M+H]+。1
H NMR (400 MHz, DMSO-d6
) δ ppm 7.57 - 7.64(m, 1 H), 7.71(s, 1 H), 7.87 - 7.95(m, 4 H), 8.63(d,J
= 4.8 Hz, 1 H), 10.69 (s, 1 H), 12.71 (s, 1 H)。
N-(4-{[(2S)-1-(3-甲氧基苯基)己烷-2-基]胺甲醯基}苯基)-2-甲基吡啶-4-甲醯胺:向(2S)-1-(3-甲氧基苯基)己烷-2-胺(1.0 g, 4.82 mmol, 1.0當量)、4-(2-甲基吡啶-4-醯胺基)苯甲酸(1.48 g, 5.79 mmol, 1.2當量)於DCM (20.0 mL)中之溶液中添加三乙胺(2.69 mL, 19.3 mmol, 4當量),攪拌5 min且然後在0℃下添加T3P (50 wt.%於EtOAc中) (2.15 ml, 7.24 mmol, 1.5當量)並再攪拌30 min。藉由TLC (5% MeOH/DCM)監測反應進展。在反應完成之後,使用水(20.0 mL)淬滅反應混合物並使用DCM (2 × 25 mL)萃取。使用水(15 mL)及鹽水溶液(15 mL)洗滌合併之有機萃取物並藉由無水Na2
SO4
乾燥,在減壓下濃縮以獲得粗製化合物。藉由急速管柱層析使用於DCM中之3 - 5%甲醇作為洗脫劑純化粗製物以產生乙基(S)-N-(4-((1-(3-甲氧基苯基)己烷-2-基)胺甲醯基)苯基)-2-甲基異菸鹼醯胺。LC-MS (m/z) = 446.3 [M+H]+。1
H NMR (400 MHz, DMSO-d6
) δ ppm: 0.82 (d,J
= 6.8 Hz, 3 H), 1.13-1.31 (m, 5 H), 1.50 (d,J
= 6.0 Hz, 2 H), 2.65 (s, 3 H), 2.71-2.81 (m, 2 H), 3.67 (s, 3 H), 4.08 - 4.15 (m,1 H), 6.69 (d,J
= 8.8 Hz, 1 H), 6.78 (d,J
=6.4 Hz, 2 H), 7.13 (t,J
= 8.0 Hz, 1 H), 7.63 (d,J
= 4.8 Hz, 1 H), 7.72 (s,1 H), 7.80 (s, 4 H), 8.09 (d,J
=8.8 Hz, 1 H), 8.62 (d,J
= 5.2 Hz, 1 H), 10.59 (s,1 H)。
N-{4-[(3S)-3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基]苯基}-2-甲基吡啶-4-甲醯胺:在-78℃下,向(S)-N-(4-((1-(3-甲氧基苯基)己烷-2-基)胺甲醯基)苯基)-2-甲基異菸鹼醯胺(1.2 g, 2.69 mmol, 1當量)及2-氯吡啶(1.02 mL, 10.8 mmol, 4.0當量)於二氯甲烷(25.0 mL)中之經攪拌溶液中緩慢逐滴添加三氟甲烷磺酸酐(1.81 mL, 10.8 mmol, 4.0當量)。在5 min之後,將反應混合物置於冰-水浴中並升溫至0℃,且在0℃下攪拌所得溶液。在1.5 h之後,使用1.0 N氫氧化鈉水溶液(15 mL)終止反應以中和三氟甲烷磺酸鹽。添加二氯甲烷(2 × 15 mL)以稀釋反應混合物且分離各層。使用鹽水(5 mL)洗滌合併之有機層,藉由無水硫酸鈉乾燥,並過濾。在減壓下去除揮發物以得到(S)-N-(4-(3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基)苯基)-2-甲基異菸鹼醯胺之粗產物。LCMS (ES) m/z = 428.3 [M+H]+。
N-{4-[(1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基]苯基}-2-甲基吡啶-4-甲醯胺:在0℃下,向N-{4-[(3S)-3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基]苯基}-2-甲基吡啶-4-甲醯胺(260 mg, 0.608 mmol, 1當量)於甲醇(10 mL)中之溶液中逐份添加硼氫化鈉(69.0 mg, 1.82 mmol, 3當量)。將懸浮液在室溫下攪拌1 h。藉由TLC (於DCM中之5%甲醇)監測反應進展。此後,濃縮反應混合物且使用EtOAc (20 mL)及水(10 mL)稀釋所獲得粗製物。分離有機層,使用鹽水溶液(10 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物。藉由製備型TLC使用於DCM中之5%甲醇純化所獲得粗產物。收集產物部分並在減壓下濃縮以得到N-{雙環[1.1.1]戊烷-1-基}-N-({4-[(1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基]苯基}甲基)胺基甲酸第三丁基酯。
N-{4-[(1S,3S)-3-丁基-6-甲氧基-2-[3-(三甲基矽烷基)丙-2-炔醯基]-1,2,3,4-四氫異喹啉-1-基]苯基}-2-甲基吡啶-4-甲醯胺:第一步驟:在室溫下,向3-(三甲基矽烷基)丙-2-炔酸(20 mg, 0.0141 mmol, 1.0當量)於DMF (0.00043 mL, 0.00562 mmol, 0.04當量)中之溶液中添加草醯氯(0.013 mL, 0.155 mmol, 1.1當量)並攪拌30分鐘。此後,在減壓下濃縮反應混合物以得到3-(三甲基矽烷基)丙-2-炔醯基氯。此醯氯未經任何進一步純化即用於下一步驟中。
第二步驟:在0℃下,向N-(4-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)苯基)-2-甲基異菸鹼醯胺(40 mg, 0.093 mmol, 1當量)於乙腈(4.0 mL)中之溶液中添加碳酸氫鈉(59.4 mg, 0.698 mmol, 7.5當量)。在攪拌5分鐘之後,在0℃下向上述反應物質中添加3-(三甲基矽烷基)丙-2-炔醯基氯(22.4 mg, 0.140 mmol, 1.5當量)於乙腈(2.0 mL)中之溶液。將所得混合物在0℃下攪拌15 min。藉由TLC (於正己烷中之50%乙酸乙酯)監測反應進展。此後,使用EtOAc (20 mL)及水(10 mL)稀釋反應物質。分離有機層,使用鹽水溶液(7.0 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗製N-(4-((1S,3S)-3-丁基-6-甲氧基-2-(3-(三甲基矽烷基)丙炔醯基)-1,2,3,4-四氫異喹啉-1-基)苯基)-2-甲基異菸鹼醯胺。LCMS (ES) m/z = 554.4 [M+H]+。
N-{4-[(1S,3S)-3-丁基-6-甲氧基-2-(丙-2-炔醯基)-1,2,3,4-四氫異喹啉-1-基]苯基}-2-甲基吡啶-4-甲醯胺:在0℃下,向N-(4-((1S,3S)-3-丁基-6-甲氧基-2-(3-(三甲基矽烷基)丙炔醯基)-1,2,3,4-四氫異喹啉-1-基)苯基)-2-甲基異菸鹼醯胺(0.14 g, 0.253 mmol, 1當量)於二氯甲烷(10 mL)及甲醇(2 mL)中之溶液中添加碳酸鉀(0.213 g, 1.52 mmol, 6.0當量)。將此反應混合物在0℃下攪拌30分鐘。藉由LC-MS監測反應進展。在起始材料完成之後,使用二氯甲烷(2 × 10.0 mL)稀釋反應混合物並使用水(10.0 mL)分離,且藉由無水硫酸鈉乾燥合併之有機層,過濾並在減壓下濃縮以獲得粗製化合物。藉由製備型HPLC純化方法藉由使用下列分析條件純化所獲得粗產物以提供N-(4-((1S,3S)-3-丁基-6-甲氧基-2-丙炔醯基-1,2,3,4-四氫異喹啉-1-基)苯基)-2-甲基異菸鹼醯胺。分析條件:管柱:X-BridgeC-18 (250mm × 4.6mm × 5µm);移動相(A):於水中之0.1%氨;移動相(B):乙腈;流速:1.0 mL/min;梯度B:0/10, 12/60, 22/95, 25/95, 27/10, 30/10。LCMS (ES) m/z = 482.5 [M+H]+。1
H NMR (400 MHz, DMSO-d6
) δ ppm: 0.81 (t,J
= 6.8 Hz, 3 H), 1.22 (bs, 5 H), 1.50 (bs, 2 H), 2.53 (s, 3 H), 2.79 (s, 1 H), 3.12 (s, 1 H), 3.70 (d,J
= 5.6 Hz, 2 H), 4.30 (s, 1 H), 4.59 (s, 1 H), 6.03 (s, 1 H), 6.28 (s, 1 H), 6.80 (t,J
= 8.0 Hz, 1 H), 7.21 (t,J
= 8.4 Hz, 2 H), 7.41 (d,J
= 7.6 Hz, 1 H), 7.57 (d,J
= 8.4 Hz, 2 H), 7.64 (d,J
= 8.8 Hz, 2 H), 8.58 (d,J
= 4.8 Hz, 1 H),10.32 (s, 1 H)。
2-甲基吡啶-4-羰基氯:在0℃下,向2-甲基吡啶-4-甲酸(3.00 g, 21.9 mmol, 1.0當量)於N,N-二甲基甲醯胺(0.067, 0.87 mmol, 0.04當量)中之溶液中添加SOCl2 (40.0 mL),將此混合物在80℃下回流3 h,且在減壓下濃縮過量亞硫醯氯以得到2-甲基吡啶-4-羰基氯。此粗產物未經任何進一步純化即用於下一步驟中。
4-(2-甲基吡啶-4-醯胺基)苯甲酸乙酯:在0℃下,向4-胺基苯甲酸乙酯(3.19 g, 19.3 mmo, l,1.0當量)於DCM (40.0 mL)中之經攪拌溶液中添加三乙胺(15.0 mL, 116 mmol, 6當量)且將反應液攪拌15 min。然後,緩慢添加2-甲基吡啶-4-羰基氯(20 ml DCM) (3.00 g, 19.3 mmol, 1當量)。此後,將反應液在室溫下攪拌12 h。在反應完成之後,使用水將反應混合物緩慢淬滅,使用DCM (100 mL)稀釋,在室溫下攪拌5 min。然後分離各層。使用DCM (2 × 100.0 mL)萃取水層。使用水(50.0 mL)洗滌合併之有機層,分離各層。然後藉由無水硫酸鈉乾燥有機層,過濾並在減壓下濃縮以獲得粗製物。藉由管柱層析使用10% MeOH/DCM作為洗脫劑純化粗製物以提供4-(2-甲基吡啶-4-醯胺基)苯甲酸乙酯。LCMS (ES) m/z = 285.2 [M+H]+。1
H NMR (400MHz, DMSO-d6
) δ ppm 1.28 - 1.31 (m, 3 H), 2.48 (s, 3 H), 4.25 - 4.30 (m, 2 H), 7.62 (d,J
= 4.8 Hz, 1 H), 7.76 (s, 1 H), 7.89 - 7.97 (m, 4 H), 8.61 (d,J
= 5.2 Hz, 1 H), 10.72 (s, 1 H),
4-(2-甲基吡啶-4-醯胺基)苯甲酸:向4-(2-甲基吡啶-4-醯胺基)苯甲酸乙酯(2.50 g, 8.79 mmol, 1.0當量)於MeOH (20.0 mL)、THF (20.0 mL)及水(15.0 mL)中之經攪拌溶液中添加LiOH.H2O (0.76 g, 17.6 mmol, 2.0當量)且將反應液在室溫下攪拌16 h。藉由TLC (5% MeOH/DCM)監測反應。此後,在減壓及室溫下濃縮反應混合物且使用EtOAc (2 × 50 mL)萃取水層。然後使用5%檸檬酸(Ph = 5)酸化水層,過濾並在減壓下乾燥以得到4-(2-甲基吡啶-4-醯胺基)苯甲酸。LC-MS (ES) (m/z) = 257.2 [M+H]+。1
H NMR (400 MHz, DMSO-d6
) δ ppm 7.57 - 7.64(m, 1 H), 7.71(s, 1 H), 7.87 - 7.95(m, 4 H), 8.63(d,J
= 4.8 Hz, 1 H), 10.69 (s, 1 H), 12.71 (s, 1 H)。
N-(4-{[(2S)-1-(3-甲氧基苯基)己烷-2-基]胺甲醯基}苯基)-2-甲基吡啶-4-甲醯胺:向(2S)-1-(3-甲氧基苯基)己烷-2-胺(1.0 g, 4.82 mmol, 1.0當量)、4-(2-甲基吡啶-4-醯胺基)苯甲酸(1.48 g, 5.79 mmol, 1.2當量)於DCM (20.0 mL)中之溶液中添加三乙胺(2.69 mL, 19.3 mmol, 4當量),攪拌5 min且然後在0℃下添加T3P (50 wt.%於EtOAc中) (2.15 ml, 7.24 mmol, 1.5當量)並再攪拌30 min。藉由TLC (5% MeOH/DCM)監測反應進展。在反應完成之後,使用水(20.0 mL)淬滅反應混合物並使用DCM (2 × 25 mL)萃取。使用水(15 mL)及鹽水溶液(15 mL)洗滌合併之有機萃取物並藉由無水Na2
SO4
乾燥,在減壓下濃縮以獲得粗製化合物。藉由急速管柱層析使用於DCM中之3 - 5%甲醇作為洗脫劑純化粗製物以產生乙基(S)-N-(4-((1-(3-甲氧基苯基)己烷-2-基)胺甲醯基)苯基)-2-甲基異菸鹼醯胺。LC-MS (m/z) = 446.3 [M+H]+。1
H NMR (400 MHz, DMSO-d6
) δ ppm: 0.82 (d,J
= 6.8 Hz, 3 H), 1.13-1.31 (m, 5 H), 1.50 (d,J
= 6.0 Hz, 2 H), 2.65 (s, 3 H), 2.71-2.81 (m, 2 H), 3.67 (s, 3 H), 4.08 - 4.15 (m,1 H), 6.69 (d,J
= 8.8 Hz, 1 H), 6.78 (d,J
=6.4 Hz, 2 H), 7.13 (t,J
= 8.0 Hz, 1 H), 7.63 (d,J
= 4.8 Hz, 1 H), 7.72 (s,1 H), 7.80 (s, 4 H), 8.09 (d,J
=8.8 Hz, 1 H), 8.62 (d,J
= 5.2 Hz, 1 H), 10.59 (s,1 H)。
N-{4-[(3S)-3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基]苯基}-2-甲基吡啶-4-甲醯胺:在-78℃下,向(S)-N-(4-((1-(3-甲氧基苯基)己烷-2-基)胺甲醯基)苯基)-2-甲基異菸鹼醯胺(1.2 g, 2.69 mmol, 1當量)及2-氯吡啶(1.02 mL, 10.8 mmol, 4.0當量)於二氯甲烷(25.0 mL)中之經攪拌溶液中緩慢逐滴添加三氟甲烷磺酸酐(1.81 mL, 10.8 mmol, 4.0當量)。在5 min之後,將反應混合物置於冰-水浴中並升溫至0℃,且在0℃下攪拌所得溶液。在1.5 h之後,使用1.0 N氫氧化鈉水溶液(15 mL)終止反應以中和三氟甲烷磺酸鹽。添加二氯甲烷(2 × 15 mL)以稀釋反應混合物且分離各層。使用鹽水(5 mL)洗滌合併之有機層,藉由無水硫酸鈉乾燥,並過濾。在減壓下去除揮發物以得到(S)-N-(4-(3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基)苯基)-2-甲基異菸鹼醯胺之粗產物。LCMS (ES) m/z = 428.3 [M+H]+。
N-{4-[(1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基]苯基}-2-甲基吡啶-4-甲醯胺:在0℃下,向N-{4-[(3S)-3-丁基-6-甲氧基-3,4-二氫異喹啉-1-基]苯基}-2-甲基吡啶-4-甲醯胺(260 mg, 0.608 mmol, 1當量)於甲醇(10 mL)中之溶液中逐份添加硼氫化鈉(69.0 mg, 1.82 mmol, 3當量)。將懸浮液在室溫下攪拌1 h。藉由TLC (於DCM中之5%甲醇)監測反應進展。此後,濃縮反應混合物且使用EtOAc (20 mL)及水(10 mL)稀釋所獲得粗製物。分離有機層,使用鹽水溶液(10 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗產物。藉由製備型TLC使用於DCM中之5%甲醇純化所獲得粗產物。收集產物部分並在減壓下濃縮以得到N-{雙環[1.1.1]戊烷-1-基}-N-({4-[(1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基]苯基}甲基)胺基甲酸第三丁基酯。
N-{4-[(1S,3S)-3-丁基-6-甲氧基-2-[3-(三甲基矽烷基)丙-2-炔醯基]-1,2,3,4-四氫異喹啉-1-基]苯基}-2-甲基吡啶-4-甲醯胺:第一步驟:在室溫下,向3-(三甲基矽烷基)丙-2-炔酸(20 mg, 0.0141 mmol, 1.0當量)於DMF (0.00043 mL, 0.00562 mmol, 0.04當量)中之溶液中添加草醯氯(0.013 mL, 0.155 mmol, 1.1當量)並攪拌30分鐘。此後,在減壓下濃縮反應混合物以得到3-(三甲基矽烷基)丙-2-炔醯基氯。此醯氯未經任何進一步純化即用於下一步驟中。
第二步驟:在0℃下,向N-(4-((1S,3S)-3-丁基-6-甲氧基-1,2,3,4-四氫異喹啉-1-基)苯基)-2-甲基異菸鹼醯胺(40 mg, 0.093 mmol, 1當量)於乙腈(4.0 mL)中之溶液中添加碳酸氫鈉(59.4 mg, 0.698 mmol, 7.5當量)。在攪拌5分鐘之後,在0℃下向上述反應物質中添加3-(三甲基矽烷基)丙-2-炔醯基氯(22.4 mg, 0.140 mmol, 1.5當量)於乙腈(2.0 mL)中之溶液。將所得混合物在0℃下攪拌15 min。藉由TLC (於正己烷中之50%乙酸乙酯)監測反應進展。此後,使用EtOAc (20 mL)及水(10 mL)稀釋反應物質。分離有機層,使用鹽水溶液(7.0 mL)洗滌,藉由無水硫酸鈉乾燥,過濾並在減壓下濃縮以獲得粗製N-(4-((1S,3S)-3-丁基-6-甲氧基-2-(3-(三甲基矽烷基)丙炔醯基)-1,2,3,4-四氫異喹啉-1-基)苯基)-2-甲基異菸鹼醯胺。LCMS (ES) m/z = 554.4 [M+H]+。
N-{4-[(1S,3S)-3-丁基-6-甲氧基-2-(丙-2-炔醯基)-1,2,3,4-四氫異喹啉-1-基]苯基}-2-甲基吡啶-4-甲醯胺:在0℃下,向N-(4-((1S,3S)-3-丁基-6-甲氧基-2-(3-(三甲基矽烷基)丙炔醯基)-1,2,3,4-四氫異喹啉-1-基)苯基)-2-甲基異菸鹼醯胺(0.14 g, 0.253 mmol, 1當量)於二氯甲烷(10 mL)及甲醇(2 mL)中之溶液中添加碳酸鉀(0.213 g, 1.52 mmol, 6.0當量)。將此反應混合物在0℃下攪拌30分鐘。藉由LC-MS監測反應進展。在起始材料完成之後,使用二氯甲烷(2 × 10.0 mL)稀釋反應混合物並使用水(10.0 mL)分離,且藉由無水硫酸鈉乾燥合併之有機層,過濾並在減壓下濃縮以獲得粗製化合物。藉由製備型HPLC純化方法藉由使用下列分析條件純化所獲得粗產物以提供N-(4-((1S,3S)-3-丁基-6-甲氧基-2-丙炔醯基-1,2,3,4-四氫異喹啉-1-基)苯基)-2-甲基異菸鹼醯胺。分析條件:管柱:X-BridgeC-18 (250mm × 4.6mm × 5µm);移動相(A):於水中之0.1%氨;移動相(B):乙腈;流速:1.0 mL/min;梯度B:0/10, 12/60, 22/95, 25/95, 27/10, 30/10。LCMS (ES) m/z = 482.5 [M+H]+。1
H NMR (400 MHz, DMSO-d6
) δ ppm: 0.81 (t,J
= 6.8 Hz, 3 H), 1.22 (bs, 5 H), 1.50 (bs, 2 H), 2.53 (s, 3 H), 2.79 (s, 1 H), 3.12 (s, 1 H), 3.70 (d,J
= 5.6 Hz, 2 H), 4.30 (s, 1 H), 4.59 (s, 1 H), 6.03 (s, 1 H), 6.28 (s, 1 H), 6.80 (t,J
= 8.0 Hz, 1 H), 7.21 (t,J
= 8.4 Hz, 2 H), 7.41 (d,J
= 7.6 Hz, 1 H), 7.57 (d,J
= 8.4 Hz, 2 H), 7.64 (d,J
= 8.8 Hz, 2 H), 8.58 (d,J
= 4.8 Hz, 1 H),10.32 (s, 1 H)。
表1中所展示之化合物可或係根據上述程序使用適當試劑及起始材料來合成。所選數據展示於表2中。表 2
| 編號 | MS [M+H]+ | |
| 1 | 385.3 | |
| 2 | 402 | |
| 3 | 402 | |
| 4 | 389 | |
| 5 | 413.3 | |
| 6 | 388.1 | |
| 7 | 388.1 | |
| 8 | 348.3 | |
| 9 | 324 | |
| 10 | 324.3 | |
| 11 | 390 | |
| 12 | 365 | |
| 13 | 469 | |
| 14 | 448 | |
| 15 | 445 | |
| 18 | 481.3 | |
| 27 | 521.5 | |
| 28 | 497 | |
| 33 | 415.93 | |
| 34 | 418.89 | |
| 35 | 358.83 | |
| 36 | 428.93 | |
| 38 | 525.8 | |
| 39 | 354.6 | |
| 40 | 432.83 | |
| 42 | 408.87 | |
| 48 | 363.4 | |
| 49 | 349.4 | |
| 50 | 352.4 | |
| 54 | 369 | |
| 55 | 429.5 | |
| 57 | 349 | |
| 76 | 353.3 | |
| 93 | 434.95 | |
| 94 | 440 | |
| 95 | 482 | |
| 96 | 458 | |
| 97 | 399 | |
| 98 | 477 | |
| 99 | 610 | |
| 100 | 541 | |
| 101 | 498 | |
| 102 | 445 |
生物實例 實例 1 : 細胞增殖 (Alamar Blue) 分析
實施細胞活力分析以評價化合物在人類癌細胞系786-O (腎細胞癌)、SJSA-1 (骨肉瘤)及/或A431 (表皮樣癌)中之功效。可在類似方法中測試其他細胞系,例如胰臟癌細胞系(例如Panc 02.13、BxPC-3、Panc 12、Panc 02.03、Panc 6.03、PSN-1、HPAC及Capan-1)、前列腺癌細胞系(例如PC-3、DU145、22Rv1、NCI-H660、BPH1、LNCaP、BM-1604及MDA PCa 2b)等。
將細胞(SJSA-1、786-O及/或A431)接種(5000個細胞/100 µL/孔)於96孔組織培養板中並在37℃/ 5% CO2
下培育16-24小時。然後使用化合物(25 µL,5×)處理細胞。化合物濃度為10-0.0005 µM,製成3倍連續稀釋液且最終DMSO濃度為1%。然後將板在37℃/ 5% CO2
下於潮濕環境中培育24h。然後,向每一孔中添加Alamar藍色™試劑(最終濃度為1X-12.5 µL)並在37℃/ 5% CO2
下培育1.5小時。在螢光讀數儀上於540 nm激發及590 nm發射波長下讀取板。隨後使用S形劑量-反應曲線(可變斜率)在GraphPad Prism® 5軟體中測定IC50
值。表3展示如本文所闡述之實例性化合物之細胞增殖數據。表 3
| 編號 | IC50 (µM) | ||
| 786-O | SJSA-1 | A431 | |
| 1 | 2 | 2.8 | - |
| 2 | 0.742 | 0.855 | 8.7 |
| 3 | 0.512 | 0.736 | >10 |
| 4 | 0.588 | 1 | - |
| 5 | 0.212 | 0.382 | - |
| 6 | 3 | 3.2 | >10 |
| 7 | 0.137 | 0.22 | 7.8 |
| 8 | 0.181 | 0.183 | 7.9 |
| 9 | 0.223 | 0.436 | 4.9 |
| 10 | 0.164 | 0.278 | 8.6 |
| 11 | 0.016 | 0.018 | >10 |
| 12 | 0.014 | 0.025 | 3.5 |
| 13 | 0.002 | 0.005 | 3.6 |
| 14 | 0.024 | 0.07 | 7.6 |
| 15 | 0.010 | 0.018 | 3.5 |
| 18 | 0.004 | 0.008 | 8.9 |
| 27 | 0.004 | 0.006 | 8.4 |
| 28 | 0.004 | 0.016 | 2.9 |
| 33 | 0.2591 | - | - |
| 34 | 0.5917 | - | - |
| 35 | - | 2.56 | - |
| 38 | 0.005 | 0.010 | 10 |
| 39 | 0.158 | 0.170 | >10 |
| 44 | 0.592 | 0.599 | - |
| 45 | 5.891 | >3 | - |
| 46 | - | 2.56 | - |
| 47 | 0.259 | 0.329 | - |
| 48 | 0.053 | 0.050 | 5 |
| 49 | 0.023 | 0.040 | 6.6 |
| 50 | 0.065 | 0.101 | >10 |
| 54 | 0.743 | 0.659 | 5.8 |
| 55 | 0.027 | 0.032 | 2.3 |
| 57 | 0.023 | 0.040 | 6.6 |
| 76 | 0.187 | 0.623 | >10 |
| 94 | 0.108 | 0.047 | >10 |
| 95 | 0.037 | 0.2 | >10 |
| 96 | 0.163 | 0.368 | >10 |
| 97 | 0.026 | 0.068 | >10 |
| 98 | 0.024 | 0.053 | 8.92 |
| 99 | 0.002 | 0.002 | 2.77 |
| 100 | 0.013 | 0.021 | 4.06 |
| 101 | 0.008 | 0.0115 | 6.87 |
| 102 | 0.021 | 0.021 | 6.13 |
亦在人類肺癌細胞系A549 (GPX4抑制劑之抗性/不敏感細胞系,作為對照)分析(反篩選)所選化合物以評價差異性活性。將細胞以800-2,000個細胞/孔之密度接種於96孔板中並在37℃下培育過夜。藉由在DMSO中進行3倍連續稀釋來產生9個不同濃度之化合物儲備液(500×)之系列。將該等化合物進一步稀釋於培養基中且然後添加至細胞中,從而最終DMSO濃度等於0.25%或更小。在培育96小時之後,向每一孔中添加50μL CellTiter Glo試劑(Promega)且在10分鐘之後使用EnVision (PerkinElmer)量測發光。使用RSL3 (原型GPX4抑制劑,亦稱為RSL-3)作為自最高濃度一直滴定至30 μM之參考化合物。首先自30 μM (作為最高濃度)一式兩份(4.6 nM -30 μM之範圍)來測試所有化合物。對於展示初始範圍以外之功效之化合物而言,則將最高濃度調節至更高(自最高1000 μM)或更低。將來自僅使用DMSO處理之細胞之發光設定為Max且抑制%計算如下:抑制% = (Max-試樣值)/Max*100。使用XL-擬合軟體(ID Business Solutions Ltd.)分析數據。計算IC50
、相對IC50
或最大抑制%。數據展示於表4中。表 4
| 編號 | IC50 (µM) | ||
| A549 | KP4 | ||
| 40 | 6.238 (RSL3 3.078) | 0.019 (RSL3 0.002) | |
| 41 | 1.315 (RSL3 3.078) | 0.042 (RSL3 0.002) | |
| 42 | 5.05 (RSL3 3.078) | 0.0618 (RSL3 0.002) | |
| 43 | 10.3 (RSL3 3.078) | 0.525 (RSL3 0.003) | |
| 44 | 6.575 (RSL3 2.735) | - | |
| 45 | 4.742 (RSL3 2.735) | - | |
| 46 | 3.825 (RSL3 3.667) | - | |
| 47 | 6.575 (RSL3 2.735) | - |
實例 2 : GPX4 抑制分析
表5展示,本文所提供之化合物係GPX4抑制劑。研究已展示,親脂性抗氧化劑(例如菲羅他仃)可拯救細胞以免受GPX4抑制誘導之鐵依賴型細胞死亡。舉例而言,間質態GPX4敲除細胞可在菲羅他仃存在下存活,然而,在菲羅他仃之供應終止時,該等細胞發生鐵依賴型細胞死亡(例如參見Viswanathan等人,Nature 547:453-7, 2017)。亦已以實驗方式測得,可藉由阻斷鐵依賴型細胞死亡路徑之其他組分來恢復GPX4i (例如脂質ROS清除劑(菲羅他仃、利普羅他仃)、脂氧合酶抑制劑、鐵螯合劑及半胱天冬酶抑制劑),而細胞凋亡抑制劑則不能恢復。該等發現指示了非細胞凋亡性、鐵依賴性、氧化性細胞死亡(亦即鐵依賴型細胞死亡)。因此,分子能夠誘導鐵依賴型細胞死亡性癌細胞死亡且該能力可藉由添加菲羅他仃來減弱可明確指示,該分子係GPX4抑制劑。表5中之數據展示,本文所提供之化合物在菲羅他仃存在下損失抑制活性且由此係有效GPX4抑制劑。表 5
| 化合物 編號 | 786-O (IC50 , µM) | SJSA-1 (IC50 , µM) | ||
| 無菲羅他仃 | 2 µM 菲羅他仃 | 無菲羅他仃 | 2 µM 菲羅他仃 | |
| 4 | 0.588 | 5.731 | 1 | 3.563 |
| 5 | 0.212 | > 10.00 | 0.382 | > 10.00 |
| 8 | 0.181 | > 10.00 | 0.183 | > 10.00 |
| 9 | 0.223 | > 10.00 | 0.436 | 3.744 |
實例 3 :西方印漬之方法及結果 - GPX4 之凝膠遷移率變動
確立GPX4西方印漬分析之遷移率變動以在與化合物一起培育之後於基於細胞之分析中且於來自化合物治療小鼠的腫瘤中直接評價靶咬合。可使用遷移率變動作為GPX4不可逆抑制劑之藥效動力學標記。在基於細胞之分析中,將對GPX4抑制劑敏感之細胞(例如MiaPaCa-2)以10 cm (2-8 × 106
個細胞)接種並生長過夜。若使用較小盤,則可基於表面積相應地調節細胞接種數量。第二天,使用DMSO及各種化合物在指示濃度下處理細胞一定時間段(例如0.5、1、2、4、6或最多72小時)。然後在0.3-0.5 mL補充有蛋白酶抑制劑(Roche)及磷酸酶抑制劑(Sigma)之RIPA緩衝液(Sigma)中裂解細胞。使用BCA套組(Pierce)分析裂解物之蛋白質濃度。在4-12%或12% NuPage凝膠(Life Technologies)上運行正規化量之溶解物(20-40 μg蛋白質/泳道)且使用iBlot®轉移模組(Life Technologies)將蛋白質轉移至聚二氟亞乙烯(PVDF)或硝基纖維素膜上。在使用含有5%非脂乳之1×TBST阻斷一小時之後,使用表6中所展示之一級抗體在4℃下於室溫下過夜探測膜。來自其他供應商之類似抗體亦可用於西方印漬分析中。在使用含有0.1% Tween20之1X TBS洗滌5次之後,藉由二次施加抗體(例如抗小鼠-HRP、抗兔-HRP、抗山羊-HRP、抗小鼠IgG Dylight 800偶聯物或抗兔IgG DyLight 680偶聯物) (1:10000;Cell signaling或與來自不同供應商之抗體類似之IR)在室溫下探測膜一小時。在洗滌5次之後,使用ImageQuant-LAS-4010 (化學發光) (GE Healthcare) (若使用HRP-偶聯之二級抗體)或Odyssey®成像系統(Licor Biosciences) (若使用紅外偶聯之二級抗體)掃描膜。表 6 :用於西方印漬分析之一級抗體
| 抗體名稱 | 供應商 | 目錄號 | 物種 | MW | 稀釋度 |
| β-肌動蛋白(加載對照) | Sigma | A5441 | 小鼠 | 43kd | 1:10000 |
| 紐蛋白(Vinculin) (加載對照) | Sigma | V9131 | 小鼠 | 116KD | 1:2000 |
| GPX4 | Abcam | ab125066 | 兔 | 22kd | 1:1000 |
| GPX4 | Abcam | ab41787 | 兔 | 22kd | 1:1000 |
在GPX4之基於細胞之西方印漬分析中評估化合物40且結果展示於圖1中。在DMSO處理試樣中,GPX4表現為雙聯體 -主下自由或未結合GPX4帶及次上帶(可能係結合麩胱甘肽之GPX4 (Cozza等人,Free Radical Biology and Medicine,第112卷,第1-11頁,2017))。若使試樣沸騰於過量還原劑二硫蘇糖醇(DTT)中,則上帶之量可有所減小。在使用GPX4之共價、不可逆抑制劑(例如 RSL-3及ML162)而非可逆抑制劑(例如ML210)處理時,SDS-PAGE還原凝膠中之GPX4移動地較為緩慢(表現為較大分子量蛋白質),此可能係由於添加了與GPX4共價連接之小分子。不同於結合麩胱甘肽之GPX4,結合不可逆抑制劑之GPX4上帶不能由過量DTT減少。另外, GPX4遷移率變動距離與不可逆GPX4抑制劑之分子量相關-不可逆抑制劑愈大,則變動距離愈大。因此,可使用GPX4西方印漬之此簡單遷移率變動來便利地直接評價在活體外、細胞中及腫瘤中不可逆抑制劑之靶咬合。如圖1中所展示,使用化合物40處理MiaPaCa-2細胞使得GPX4自下未結合帶劑量依賴性遷移率變動至上結合帶。在濃度大於50 nM時,化合物40使幾乎所有GPX4轉移至上帶。
實例 4 : GPX4 抑制劑之 Kinact/Ki 測定
下列實例展示,靶與GPX4之咬合極為快速。 第 1 天 - 種子細胞 :
將細胞以5×105
個Calu6細胞/孔接種至5 × 6孔板中。 第 2 天 - 使用化合物處理細胞 , 製備用於凝膠之試樣 :
使用1、0.75、0.5、0.25及0.1 µM抑制劑 + 2 µM菲羅他仃-1處理細胞0、10、20、30、45、60分鐘。針對每一化合物稀釋度(1、0.75、0.5、0.25、0.1 mM),製備10 µL 1000× DMSO儲備溶液。在2 µM菲羅他仃-1最終濃度下製備完全細胞培養基(EMEM + 10% FBS)。藉由將1000x抑制劑以1x最終濃度 (1、0.75、0.5、0.25、0.1 µM)添加至補充有菲羅他仃-1之培養基中來製備藥物溶液,且使用DMSO作為陰性對照。
藉由使用去離子水將5x細胞裂解緩衝液(Cell Signaling Technology 9803號)及100x蛋白酶/磷酸酶抑制劑混合劑(Cell Signaling Technology 5872號)稀釋至1x來製備細胞裂解緩衝液。
使用藥物溶液以1小時時程處理細胞。在t = 60、45、30、20、10、0分鐘時將一個濃度之藥物添加至每一6孔板中。自每一6孔板中之1個孔中之細胞抽吸培養基並添加1 mL含有藥物+菲羅他仃之培養基(t = 60 min)。在各時間點之間使細胞返回培育器中。抽吸培養基並在每一後續時間點下向細胞中添加藥物。在t = 10 min時,向額外孔中添加DMSO作為陰性對照。
在t = 0時,自細胞抽吸培養基,使用冰冷PBS洗滌細胞並抽吸,向每孔中添加75 µL 1x細胞裂解緩衝液,使用細胞刮擦器刮擦板底,並將溶解物轉移至1.5 mL埃彭道夫管(Eppendorf tube)中且儲存於-20℃下。
製備SDS-PAGE運行緩衝液(2 L 1x MES Bolt運行緩衝液(ThermoFisher Scientific B0002號),儲存於4℃下過夜以在下一天使用)。 第 3 天 - 實施 BCA 分析且運行凝膠 :
在冰上解凍溶解物,在4℃下以18,000 x g離心10分鐘,且在上清液上遵循製造商方案(ThermoFisher Scientific 23225號)實施BCA分析。藉由以10:1比率混合Bolt 4x LDS試樣緩衝液(ThermoFisher Scientific B0008號)與2-巰基乙醇來製備3.6x LDS/BME試樣緩衝液。在96孔PCR板中,添加19 µL 3.6× LDS/BME試樣緩衝液及50 µL溶解物試樣。使用1x LDS/BME將溶解物稀釋至1 mg/mL,將板在95℃下於PCR機器中加熱10 min,將15 µL/孔(15 µg總溶解物)加載至12% Bis- Tris Bolt凝膠中,且在200V下使用冷1x MES運行緩衝液運行凝膠約35分鐘(直至染料前沿到達凝膠底部為止)。此後,將凝膠在水中洗滌5分鐘,在20%乙醇/水中洗滌10分鐘,並使用iBlot2 (ThermoFisher Scientific)轉移至膜中。使用Licor TBS阻斷緩衝液(Licor 927-60001號)在室溫下將膜阻斷1h並與抗GPX4抗體(Abcam ab125066號)在Licor TBS阻斷緩衝液中之1:1000稀釋液一起在4℃及輕微搖動下培育過夜。 第 4 天 - 顯影印漬 , 量化凝膠變動 :
使用1x TBST將膜洗滌30分鐘(更換洗滌緩衝液3-4次),與Licor二級抗體(Licor 926-68021號,1:40,000,於Licor TBS阻斷緩衝液中)一起在室溫及輕微搖動下培育1h,使用1x TBST洗滌30分鐘,使用Licor成像儀刮擦且使用Image studio量化帶。
RSL3及化合物13及15之Kinact/Ki數據展示於圖2中。
實例 5 : 藥物動力學研究
Jubilant Biosys之Institutional Animal Ethical Committee (IAEC) (IAEC/JDC/2019/188R (對於小鼠)及IAEC/JDC/2019/189R (對於大鼠),由CPCSEA (Committee for the Purpose of Control and Supervision of Experiments on Animals)提名)批準小鼠及大鼠藥物動力學實驗。自印度之Vivo Biotech, Hyderabad獲得雄性Balb/c小鼠(約6-8週齡且體重範圍為22-25 g)及雄性SD大鼠(6-8週齡且體重範圍為200-250 g)。將動物隔離於Jubilant Biosys之動物屋中7天時段且使用12:12 h光:暗循環,且在研究之前將動物根據體重分級。
飼養:將動物分組飼養於標準聚碳酸酯籠中,該等籠具有放置粒化食物及引用水瓶之不銹鋼頂部格柵;使用作為玉米芯作為鋪墊材料且每週至少兩次或視需要進行更換。
隨意飲食:提供由Altromin Spezialfutter GmbH & Co. KG., ImSeelenkamp20. D-32791 Lage製得之齧齒類動物飼料。
隨意水:向具有不銹鋼吸管之聚碳酸酯瓶中之動物隨意提供純化水。A) 小鼠程序 :
分別以5、20及10 mg/kg之劑量進行靜脈內、口服及腹膜腔內藥物動力學研究,口服及腹膜腔內途徑之劑量體積為10 mL/Kg,而靜脈內途徑為5 mL/kg。進行稀疏採樣且在每一時間點下使用三隻小鼠進行採血(約100 µL),其中在0.083 (僅用於靜脈內)、0.25、0.5、1、2、4、8、10 (僅用於口服)及24 h時自眶後叢收集。將血樣收集於含有K2
.EDTA作為抗凝血劑之管中並在10,000 rpm下於維持於4℃之冷藏離心機(Biofuge, Heraeus,德國)中離心5 min以分離血漿。
組I (靜脈內)在尾部靜脈中以5 mg/Kg經靜脈內接受測試化合物之溶液調配物(使用30% Kolliphore EL在WFI中製得);劑量體積:5 mL/Kg;濃度:1 mg/mL。
組II (口服)藉由經口途徑使用口服胃管灌食針以20 mg/Kg接受測試化合物之溶液調配物(使用30% Kolliphore EL在WFI中製得);劑量體積:10 mL/Kg;濃度:2 mg/mL。
組III (腹膜腔內)藉由腹膜腔內途徑以10 mg/Kg接受測試化合物之溶液調配物(使用30% Kolliphore EL在WFI中製得);劑量體積:10 mL/Kg;濃度:1 mg/mL。B) 大鼠程序 :
以2 mg/kg及10 mg/kg之劑量及2 mL/Kg及10 mL/Kg之劑量體積進行靜脈內及口服藥物動力學研究。進行連續採血且在0.083 (僅用於靜脈內)、0.25、0.5、1、2、4、8、10 (僅用於口服)及24 h中之每一時間點下自眶後叢收集約200 µL。將血樣收集於含有K2
.EDTA作為抗凝血劑之管中並在10,000 rpm下於維持於4℃之冷藏離心機(Biofuge, Heraeus,德國)中離心5 min以分離血漿。
組I (靜脈內)在尾部靜脈中以2 mg/Kg經靜脈內接受測試化合物之溶液調配物(使用30% Kolliphore EL在WFI中製得);劑量體積:2 mL/Kg;濃度:1 mg/mL。
組II (口服)使用口服胃管灌食針以10 mg/Kg接受測試化合物之溶液調配物(使用30% Kolliphore EL在WFI中製得);劑量體積:10 mL/Kg;濃度:1 mg/mL。
藉由非房室方法使用Phoenix WinNonlin第8.1版來分析測試化合物之血液濃度-時間數據。數據展示於下文表7中。表 7
| 化合物 | 12 | 28 |
| 小鼠IV-PK (5 mg/kg) | T1/2 : 0.5 h, Cmax : 882 ng/mL, AUC: 181 ng*h/mL, CL: 454 mL/min/kg, Vd: 19.5 L/kg。 | T1/2 : 3.5 h, Cmax : 5446 ng/mL, AUC: 1635 ng*h/mL, CL: 49 mL/min/kg, Vd: 14.7 L/kg |
| 大鼠IV-PK (2 mg/kg): | T1/2 : 3.15 h, Cmax : 3529 ng/mL, AUC: 1082 ng*h/mL, CL: 30 mL/min/kg, Vd: 8.2 L/kg |
本申請案中所引用之所有出版物、專利、專利申請案及其他文件皆出於所有目的全文以引用方式併入本文中,其併入程度如同出於所有目的將每一個別出版物、專利、專利申請案或其他文件個別地指明以引用方式併入一般。
儘管已闡釋並闡述各個具體實施例,但應瞭解,可在不背離本發明之精神及範圍之情況下進行各種改變。
圖1展示在GPX4之基於細胞之西方印漬(Western blot)分析中測試之化合物40。
圖2展示如本文所闡述之化合物之Kinact/Ki數據。

Claims (38)
- 一種式I化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽,I 其中: 環A係C4 -C10 環烷基、雜環基、芳基或雜芳基; X係-O-、-S-、-NR9 -、-CR5 =CR5 -或-CR5 =N-; p為0、1或2; q為0、1、2或3; R1 係C1 -C6 烷基、C2 -C6 烯基、C2 -C6 炔基、C1 -C6 鹵代烷基、C3 -C10 環烷基、-CN、-OR7 、-C(O)OR6 、-C(O)N(R7 )2 、-OC(O)R6 、-S(O)2 R8 、-S(O)2 N(R7 )2 、-S(O)N(R7 )2 、-S(O)R8 、-N(R7 )2 、-NO2 、-C1 -C6 烷基-OR7 或-Si(R15 )3 ; R2 係-C1 -C2 鹵代烷基、-C2 -C3 烯基、-C2 -C3 鹵代烯基、C2 炔基或-CH2 OS(O)2 -苯基,其中該等C1 -C2 烷基鹵基及-C2 -C3 烯基鹵基視情況經一或兩個-CH3 取代,且該等C2 炔基及苯基視情況經一個-CH3 取代; 每一R3 獨立地係鹵基、-CN、-OH、-OR8 、-NH2 、-NHR8 、-N(R8 )2 、-S(O)2 R8 、-S(O)R8 、-S(O)2 N(R7 )2 、-S(O)N(R7 )2 、-NO2 、-Si(R12 )3 、-SF5 、-C(O)OR6 、-C(O)N(R7 )2 、-NR12 C(O)R8 、-NR12 C(O)OR8 、-OC(O)N(R7 )2 、-OC(O)R8 、-C(O)R6 、-OC(O)CHR8 N(R12 )2 、C1 -C6 烷基、C2 -C6 烯基、C2 -C6 炔基、C3 -C10 環烷基、雜環基、芳基、雜芳基、-C1 -C6 烷基C3 -C10 環烷基、-C2 -C6 烯基C3 -C10 環烷基、-C1 -C6 烷基雜環基、-C2 -C6 烯基雜環基、-C1 -C6 烷基芳基、-C2 -C6 烯基芳基、C1 -C6 烷基雜芳基或-C2 -C6 烯基雜芳基;其中R3 之每一C1 -C6 烷基、C2 -C6 烯基、C2 -C6 炔基、C3 -C10 環烷基、雜環基、芳基、雜芳基、-C1 -C6 烷基C3 -C10 環烷基、-C2 -C6 烯基C3 -C10 環烷基、-C1 -C6 烷基雜環基、-C2 -C6 烯基雜環基、-C1 -C6 烷基芳基、-C2 -C6 烯基芳基、C1 -C6 烷基雜芳基或-C2 -C6 烯基雜芳基獨立地視情況經一至三個R10 取代; 每一R4 獨立地係鹵基、-CN、-OH、-OR8 、-NH2 、-NHR8 、-N(R8 )2 、-S(O)2 R8 、-S(O)R8 、-S(O)2 N(R7 )2 、-S(O)N(R7 )2 、-NO2 、-Si(R15 )3 、-C(O)OR6 、-C(O)N(R7 )2 、-NR12 C(O)R8 、-OC(O)R8 、-C(O)R6 、-NR12 C(O)OR8 、-OC(O)N(R7 )2 、-OC(O)CHR8 N(R12 )2 、C1 -C6 烷基、C2 -C6 烯基、C2 -C6 炔基、C3 -C10 環烷基、雜環基、芳基、雜芳基、-C1 -C6 烷基C3 -C10 環烷基、-C2 -C6 烯基C3 -C10 環烷基、-C1 -C6 烷基雜環基、-C2 -C6 烯基雜環基、-C1 -C6 烷基芳基、-C2 -C6 烯基芳基、C1 -C6 烷基雜芳基或-C2 -C6 烯基雜芳基;其中R4 之每一C1 -C6 烷基、C2 -C6 烯基、C2 -C6 炔基、C3 -C10 環烷基、雜環基、芳基、雜芳基、-C1 -C6 烷基C3 -C10 環烷基、-C2 -C6 烯基C3 -C10 環烷基、-C1 -C6 烷基雜環基、-C2 -C6 烯基雜環基、-C1 -C6 烷基芳基、-C2 -C6 烯基芳基、C1 -C6 烷基雜芳基或-C2 -C6 烯基雜芳基視情況獨立地視情況經一至三個R10 取代; 每一R5 獨立地係氫、鹵基、-CN、-OH、-OR8 、-NH2 、-NHR8 、-N(R8 )2 、-S(O)2 R8 、-S(O)R8 、-S(O)2 N(R7 )2 、-S(O)N(R7 )2 、-NO2 、-Si(R15 )3 、-C(O)OR6 、-C(O)N(R7 )2 、-NR12 C(O)R8 、-OC(O)R8 、-C(O)R6 、-NR12 C(O)OR8 、-OC(O)N(R7 )2 、-OC(O)CHR8 N(R12 )2 、C1 -C6 烷基、C2 -C6 烯基、C2 -C6 炔基、C3 -C10 環烷基、雜環基、芳基、雜芳基、-C1 -C6 烷基C3 -C10 環烷基、-C2 -C6 烯基C3 -C10 環烷基、-C1 -C6 烷基雜環基、-C2 -C6 烯基雜環基、-C1 -C6 烷基芳基、-C2 -C6 烯基芳基、C1 -C6 烷基雜芳基或-C2 -C6 烯基雜芳基;其中R5 之每一C1 -C6 烷基、C2 -C6 烯基、C2 -C6 炔基、C3 -C10 環烷基、雜環基、芳基、雜芳基、-C1 -C6 烷基C3 -C10 環烷基、-C2 -C6 烯基C3 -C10 環烷基、-C1 -C6 烷基雜環基、-C2 -C6 烯基雜環基、-C1 -C6 烷基芳基、-C2 -C6 烯基芳基、C1 -C6 烷基雜芳基或-C2 -C6 烯基雜芳基視情況獨立地視情況經一至三個R10 取代; 每一R6 獨立地係氫、C1 -C6 烷基、C2 -C6 烯基、C2 -C6 炔基、C3 -C10 環烷基、雜環基、芳基、雜芳基、-C1 -C6 烷基C3 -C10 環烷基、-C2 -C6 烯基C3 -C10 環烷基、-C1 -C6 烷基雜環基、-C2 -C6 烯基雜環基、-C1 -C6 烷基芳基、-C2 -C6 烯基芳基、C1 -C6 烷基雜芳基或-C2 -C6 烯基雜芳基;其中每一R6 獨立地進一步經一至三個R11 取代; 每一R7 獨立地係氫、C1 -C6 烷基、C2 -C6 烯基、C2 -C6 炔基、C3 -C10 環烷基、雜環基、芳基、雜芳基、-C1 -C6 烷基C3 -C6 環烷基、-C2 -C6 烯基C3 -C6 環烷基、-C1 -C6 烷基雜環基、-C2 -C6 烯基雜環基、-C1 -C6 烷基芳基、-C2 -C6 烯基芳基、-C1 -C6 烷基雜芳基、-C2 -C6 烯基雜芳基,或兩個R7 與其所連接之氮原子一起形成4至7員雜環基;其中每一R7 或由此形成之環獨立地進一步經一至三個R11 取代; 每一R8 獨立地係C1 -C6 烷基、C2 -C6 烯基、C2 -C6 炔基、C3 -C10 環烷基、雜環基、芳基、雜芳基、-C1 -C6 烷基C3 -C10 環烷基、-C2 -C6 烯基C3 -C10 環烷基、-C1 -C6 烷基雜環基、-C2 -C6 烯基雜環基、-C1 -C6 烷基芳基、-C2 -C6 烯基芳基、-C1 -C6 烷基雜芳基或-C2 -C6 烯基雜芳基;其中每一R8 獨立地進一步經一至三個R11 取代; R9 係氫或C1 -C6 烷基; 每一R10 獨立地係鹵基、-CN、-OR12 、-NO2 、-N(R12 )2 、-S(O)R13 、-S(O)2 R13 、-S(O)N(R12 )2 、-S(O)2 N(R12 )2 、-Si(R12 )3 、-C(O)R12 、-C(O)OR12 、-C(O)N(R12 )2 、-NR12 C(O)R12 、-OC(O)R12 、-OC(O)OR12 、-OC(O)N(R12 )2 、-NR12 C(O)OR12 、-OC(O)CHR12 N(R12 )2 、C1 -C6 烷基、C1 -C6 鹵代烷基、C2 -C6 烯基、C2 -C6 炔基、C3 -C10 環烷基、雜環基、芳基或雜芳基,其中R10 之每一C1 -C6 烷基、C1 -C6 鹵代烷基、C2 -C6 烯基、C2 -C6 炔基、C3 -C10 環烷基、雜環基、芳基或雜芳基視情況獨立地經一至三個R11 取代; 每一R11 獨立地係鹵基、-CN、-OR12 、-NO2 、-N(R12 )2 、-S(O)R13 、-S(O)2 R13 、-S(O)N(R12 )2 、-S(O)2 N(R12 )2 、-Si(R12 )3 、-C(O)R12 、-C(O)OR12 、-C(O)N(R12 )2 、-NR12 C(O)R12 、-OC(O)R12 、-OC(O)OR12 、-OC(O)N(R12 )2 、-NR12 C(O)OR12 、-OC(O)CHR12 N(R12 )2 、C1 -C6 烷基、C1 -C6 鹵代烷基、C2 -C6 烯基、C2 -C6 炔基、C3 -C10 環烷基、雜環基、芳基或雜芳基; 每一R12 獨立地係氫、C1 -C6 烷基或C3 -C10 環烷基; 每一R13 獨立地係C1 -C6 烷基或C3 -C10 環烷基;且 每一R15 獨立地係C1 -C6 烷基、C2 -C6 烯基、芳基、雜芳基、-C1 -C6 烷基芳基、-C2 -C6 烯基芳基、-C1 -C6 烷基雜芳基及-C2 -C6 烯基雜芳基。
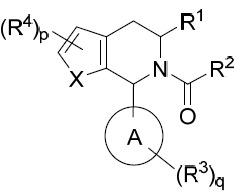
- 如請求項1之化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽,其由式IA化合物代表:IA。

- 如請求項1之化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽,其由式IB化合物代表:IB。
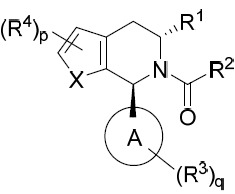
- 如請求項1之化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽,其由式II化合物代表:II。
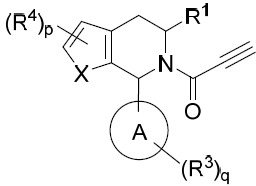
- 如請求項1之化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽,其由式IIA化合物代表:IIA。
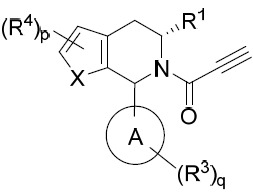
- 如請求項1之化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽,其由式IIB化合物代表:IIB。
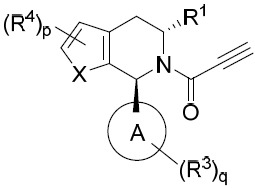
- 如請求項1之化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽,其由式III化合物代表:III 其中R14 係鹵基。

- 如請求項1之化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽,其由式IIIA化合物代表:IIIA 其中R14 係鹵基。
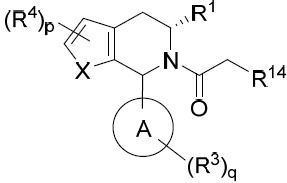
- 如請求項1之化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽,其由式IIIB化合物代表:IIIB 其中R14 係鹵基。
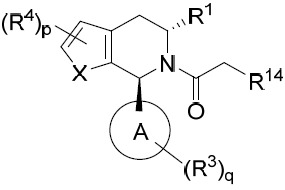
- 如請求項1至9中任一項之化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽,其中環A係C4 -C10 環烷基。
- 如請求項1至9中任一項之化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽,其中環A係雜環基。
- 如請求項1至9中任一項之化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽,其中環A係芳基。
- 如請求項1至9中任一項之化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽,其中環A係雜芳基。
- 如請求項1之化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽,其由式VIII化合物代表:VIII。
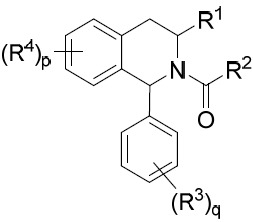
- 如請求項1之化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽,其由式VIIIA化合物代表:VIIIA。
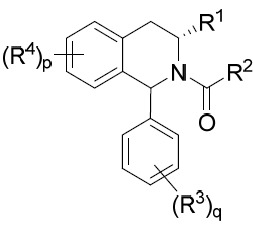
- 如請求項1之化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽,其由式VIIIB化合物代表:VIIIB。
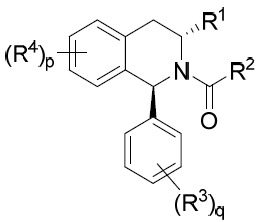
- 如請求項1至16中任一項之化合物,其中R1 係C1 -C6 烷基、C2 -C6 烯基、C2 -C6 炔基、C1 -C6 鹵代烷基、C3 -C10 環烷基、-CN、-C(O)OR6 、-C(O)N(R7 )2 、-N(R7 )2 、-OR7 或-C1 -C6 烷基-OR7 。
- 如請求項1至16中任一項之化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽,其中R1 係-C(O)OR6 或-C(O)N(R7 )2 。
- 如請求項1至16中任一項之化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽,其中R1 係C1 -C6 烷基。
- 如請求項1至16中任一項之化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽,其中p為0或1。
- 如請求項1之化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽,其由式V化合物代表:IX 其中R16 係氫或C2 -C5 烷基。

- 如請求項1之化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽,其由式IXA化合物代表:IXA 其中R16 係氫或C2 -C5 烷基。
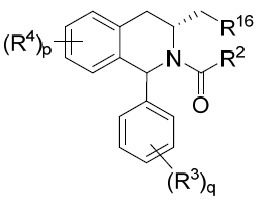
- 如請求項1之化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽,其由式IXB化合物代表:IXB 其中R16 係氫或C2 -C5 烷基。
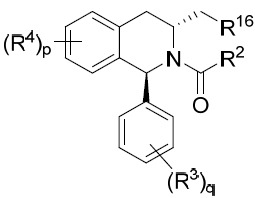
- 如請求項1至23中任一項之化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽,其中q為2或3。
- 如請求項1至24中任一項之化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽,其中每一R4 獨立地係鹵基、-CN、-OR7 、C1 -C6 烷基、C2 -C6 炔基或C3 -C10 環烷基;其中R4 之每一C1 -C6 烷基、C2 -C6 炔基或C3 -C10 環烷基獨立地視情況經一至三個R10 取代。
- 如請求項1至25中任一項之化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽,其中q為0。
- 一種化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽,該化合物選自由表1中所列示化合物組成之群。
- 一種醫藥組合物,其包含如請求項1至27中任一項之化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽及醫藥上可接受之載劑。
- 一種抑制細胞中之GPX4之方法,其包括使細胞與有效量之如請求項1至27中任一項之化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽接觸。
- 如請求項29之方法,其中該細胞係癌細胞。
- 一種治療個體之癌症之方法,其包括向患有癌症之個體投與治療有效量之如請求項1至27中任一項之化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或醫藥上可接受之鹽。
- 如請求項31之方法,其中該癌症係腎上腺皮質癌、肛門癌、膽管癌、膀胱癌、骨癌、腦癌、乳癌、子宮頸癌、結腸癌、子宮內膜癌、食道癌、頭頸癌、腸癌、肝癌、肺癌、口腔癌、卵巢癌、胰臟癌、腎癌、前列腺癌、唾液腺癌、皮膚癌、胃癌、睪丸癌、喉癌、甲狀腺癌、子宮癌、陰道癌、肉瘤或軟組織癌。
- 如請求項32之方法,其中該癌症係骨肉瘤、神經膠質瘤、星形細胞瘤、神經母細胞瘤、小腸癌、支氣管癌、小細胞肺癌、非小細胞肺癌、基底細胞癌或黑色素瘤。
- 如請求項33之方法,其中該癌症係血液學癌症。
- 如請求項33之方法,其中該血液學癌症係急性淋巴母細胞性白血病(ALL)、急性骨髓樣白血病(AML)、淋巴瘤(例如何傑金氏淋巴瘤(Hodgkin’s lymphoma)、非何傑金氏淋巴瘤(Non-Hodgkin’s lymphoma)、伯基特氏淋巴瘤(Burkitt's lymphoma))、慢性淋巴球性白血病(CLL)、慢性骨髓性白血病(CML)、毛細胞慢性骨髓性白血病(CML)或多發性骨髓瘤。
- 如請求項29至35中任一項之方法,其進一步包括投與治療有效量之第二治療劑。
- 如請求項36之方法,其中該第二治療劑係鉑類藥劑、烷基化劑、抗癌抗生素、抗代謝物、拓撲異構酶I抑制劑、拓撲異構酶II抑制劑或抗微管劑。
- 一種製備式I化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或鹽之方法:I 其包括使式1-5化合物與式1-6化合物:
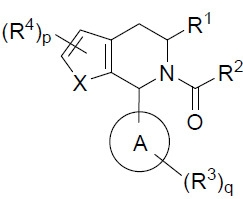 在足以提供該式I化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或鹽之反應條件下接觸;其中: 環A係C4 -C10 環烷基、雜環基、芳基或雜芳基; X係-O-、-S-、-NR9 -、-CR5 =CR5 -或-CR5 =N-; p為0、1或2; q為0、1、2或3; R1 係C1 -C6 烷基、C2 -C6 烯基、C2 -C6 炔基、C1 -C6 鹵代烷基、C3 -C10 環烷基、-CN、-OR7 、-C(O)OR6 、-C(O)N(R7 )2 、-OC(O)R6 、-S(O)2 R8 、-S(O)2 N(R7 )2 、-S(O)N(R7 )2 、-S(O)R8 、-N(R7 )2 、-NO2 、-C1 -C6 烷基-OR7 或-Si(R15 )3 ; R2 係-C1 -C2 鹵代烷基、-C2 -C3 烯基、-C2 -C3 鹵代烯基、C2 炔基或-CH2 OS(O)2 -苯基,其中該等C1 -C2 烷基鹵基及-C2 -C3 烯基鹵基視情況經一或兩個-CH3 取代,且該等C2 炔基及苯基視情況經一個-CH3 取代; 每一R3 獨立地係鹵基、-CN、-OH、-OR8 、-NH2 、-NHR8 、-N(R8 )2 、-S(O)2 R8 、-S(O)R8 、-S(O)2 N(R7 )2 、-S(O)N(R7 )2 、-NO2 、-Si(R12 )3 、-SF5 、-C(O)OR6 、-C(O)N(R7 )2 、-NR12 C(O)R8 、-NR12 C(O)OR8 、-OC(O)N(R7 )2 、-OC(O)R8 、-C(O)R6 、-OC(O)CHR8 N(R12 )2 、C1 -C6 烷基、C2 -C6 烯基、C2 -C6 炔基、C3 -C10 環烷基、雜環基、芳基、雜芳基、-C1 -C6 烷基C3 -C10 環烷基、-C2 -C6 烯基C3 -C10 環烷基、-C1 -C6 烷基雜環基、-C2 -C6 烯基雜環基、-C1 -C6 烷基芳基、-C2 -C6 烯基芳基、C1 -C6 烷基雜芳基或-C2 -C6 烯基雜芳基;其中R3 之每一C1 -C6 烷基、C2 -C6 烯基、C2 -C6 炔基、C3 -C10 環烷基、雜環基、芳基、雜芳基、-C1 -C6 烷基C3 -C10 環烷基、-C2 -C6 烯基C3 -C10 環烷基、-C1 -C6 烷基雜環基、-C2 -C6 烯基雜環基、-C1 -C6 烷基芳基、-C2 -C6 烯基芳基、C1 -C6 烷基雜芳基或-C2 -C6 烯基雜芳基獨立地視情況經一至三個R10 取代; 每一R4 獨立地係鹵基、-CN、-OH、-OR8 、-NH2 、-NHR8 、-N(R8 )2 、-S(O)2 R8 、-S(O)R8 、-S(O)2 N(R7 )2 、-S(O)N(R7 )2 、-NO2 、-Si(R15 )3 、-C(O)OR6 、-C(O)N(R7 )2 、-NR12 C(O)R8 、-OC(O)R8 、-C(O)R6 、-NR12 C(O)OR8 、-OC(O)N(R7 )2 、-OC(O)CHR8 N(R12 )2 、C1 -C6 烷基、C2 -C6 烯基、C2 -C6 炔基、C3 -C10 環烷基、雜環基、芳基、雜芳基、-C1 -C6 烷基C3 -C10 環烷基、-C2 -C6 烯基C3 -C10 環烷基、-C1 -C6 烷基雜環基、-C2 -C6 烯基雜環基、-C1 -C6 烷基芳基、-C2 -C6 烯基芳基、C1 -C6 烷基雜芳基或-C2 -C6 烯基雜芳基;其中R4 之每一C1 -C6 烷基、C2 -C6 烯基、C2 -C6 炔基、C3 -C10 環烷基、雜環基、芳基、雜芳基、-C1 -C6 烷基C3 -C10 環烷基、-C2 -C6 烯基C3 -C10 環烷基、-C1 -C6 烷基雜環基、-C2 -C6 烯基雜環基、-C1 -C6 烷基芳基、-C2 -C6 烯基芳基、C1 -C6 烷基雜芳基或-C2 -C6 烯基雜芳基視情況獨立地視情況經一至三個R10 取代; 每一R5 獨立地係氫、鹵基、-CN、-OH、-OR8 、-NH2 、-NHR8 、-N(R8 )2 、-S(O)2 R8 、-S(O)R8 、-S(O)2 N(R7 )2 、-S(O)N(R7 )2 、-NO2 、-Si(R15 )3 、-C(O)OR6 、-C(O)N(R7 )2 、-NR12 C(O)R8 、-OC(O)R8 、-C(O)R6 、-NR12 C(O)OR8 、-OC(O)N(R7 )2 、-OC(O)CHR8 N(R12 )2 、C1 -C6 烷基、C2 -C6 烯基、C2 -C6 炔基、C3 -C10 環烷基、雜環基、芳基、雜芳基、-C1 -C6 烷基C3 -C10 環烷基、-C2 -C6 烯基C3 -C10 環烷基、-C1 -C6 烷基雜環基、-C2 -C6 烯基雜環基、-C1 -C6 烷基芳基、-C2 -C6 烯基芳基、C1 -C6 烷基雜芳基或-C2 -C6 烯基雜芳基;其中R5 之每一C1 -C6 烷基、C2 -C6 烯基、C2 -C6 炔基、C3 -C10 環烷基、雜環基、芳基、雜芳基、-C1 -C6 烷基C3 -C10 環烷基、-C2 -C6 烯基C3 -C10 環烷基、-C1 -C6 烷基雜環基、-C2 -C6 烯基雜環基、-C1 -C6 烷基芳基、-C2 -C6 烯基芳基、C1 -C6 烷基雜芳基或-C2 -C6 烯基雜芳基視情況獨立地視情況經一至三個R10 取代; 每一R6 獨立地係氫、C1 -C6 烷基、C2 -C6 烯基、C2 -C6 炔基、C3 -C10 環烷基、雜環基、芳基、雜芳基、-C1 -C6 烷基C3 -C10 環烷基、-C2 -C6 烯基C3 -C10 環烷基、-C1 -C6 烷基雜環基、-C2 -C6 烯基雜環基、-C1 -C6 烷基芳基、-C2 -C6 烯基芳基、C1 -C6 烷基雜芳基或-C2 -C6 烯基雜芳基;其中每一R6 獨立地進一步經一至三個R11 取代; 每一R7 獨立地係氫、C1 -C6 烷基、C2 -C6 烯基、C2 -C6 炔基、C3 -C10 環烷基、雜環基、芳基、雜芳基、-C1 -C6 烷基C3 -C6 環烷基、-C2 -C6 烯基C3 -C6 環烷基、-C1 -C6 烷基雜環基、-C2 -C6 烯基雜環基、-C1 -C6 烷基芳基、-C2 -C6 烯基芳基、-C1 -C6 烷基雜芳基、-C2 -C6 烯基雜芳基,或兩個R7 與其所附接之氮原子一起形成4至7員雜環基;其中每一R7 或由此形成之環獨立地進一步經一至三個R11 取代; 每一R8 獨立地係C1 -C6 烷基、C2 -C6 烯基、C2 -C6 炔基、C3 -C10 環烷基、雜環基、芳基、雜芳基、-C1 -C6 烷基C3 -C10 環烷基、-C2 -C6 烯基C3 -C10 環烷基、-C1 -C6 烷基雜環基、-C2 -C6 烯基雜環基、-C1 -C6 烷基芳基、-C2 -C6 烯基芳基、-C1 -C6 烷基雜芳基或-C2 -C6 烯基雜芳基;其中每一R8 獨立地進一步經一至三個R11 取代; R9 係氫或C1 -C6 烷基; 每一R10 獨立地係鹵基、-CN、-OR12 、-NO2 、-N(R12 )2 、-S(O)R13 、-S(O)2 R13 、-S(O)N(R12 )2 、-S(O)2 N(R12 )2 、-Si(R12 )3 、-C(O)R12 、-C(O)OR12 、-C(O)N(R12 )2 、-NR12 C(O)R12 、-OC(O)R12 、-OC(O)OR12 、-OC(O)N(R12 )2 、-NR12 C(O)OR12 、-OC(O)CHR12 N(R12 )2 、C1 -C6 烷基、C1 -C6 鹵代烷基、C2 -C6 烯基、C2 -C6 炔基、C3 -C10 環烷基、雜環基、芳基或雜芳基,其中R10 之每一C1 -C6 烷基、C1 -C6 鹵代烷基、C2 -C6 烯基、C2 -C6 炔基、C3 -C10 環烷基、雜環基、芳基或雜芳基視情況獨立地經一至三個R11 取代; 每一R11 獨立地係鹵基、-CN、-OR12 、-NO2 、-N(R12 )2 、-S(O)R13 、-S(O)2 R13 、-S(O)N(R12 )2 、-S(O)2 N(R12 )2 、-Si(R12 )3 、-C(O)R12 、-C(O)OR12 、-C(O)N(R12 )2 、-NR12 C(O)R12 、-OC(O)R12 、-OC(O)OR12 、-OC(O)N(R12 )2 、-NR12 C(O)OR12 、-OC(O)CHR12 N(R12 )2 、C1 -C6 烷基、C1 -C6 鹵代烷基、C2 -C6 烯基、C2 -C6 炔基、C3 -C10 環烷基、雜環基、芳基或雜芳基; 每一R12 獨立地係氫、C1 -C6 烷基或C3 -C10 環烷基; 每一R13 獨立地係C1 -C6 烷基或C3 -C10 環烷基;且 每一R15 獨立地係C1 -C6 烷基、C2 -C6 烯基、芳基、雜芳基、-C1 -C6 烷基芳基、-C2 -C6 烯基芳基、-C1 -C6 烷基雜芳基及-C2 -C6 烯基雜芳基。
在足以提供該式I化合物或其互變異構體、立體異構體、立體異構體混合物、同位素富集之類似物或鹽之反應條件下接觸;其中: 環A係C4 -C10 環烷基、雜環基、芳基或雜芳基; X係-O-、-S-、-NR9 -、-CR5 =CR5 -或-CR5 =N-; p為0、1或2; q為0、1、2或3; R1 係C1 -C6 烷基、C2 -C6 烯基、C2 -C6 炔基、C1 -C6 鹵代烷基、C3 -C10 環烷基、-CN、-OR7 、-C(O)OR6 、-C(O)N(R7 )2 、-OC(O)R6 、-S(O)2 R8 、-S(O)2 N(R7 )2 、-S(O)N(R7 )2 、-S(O)R8 、-N(R7 )2 、-NO2 、-C1 -C6 烷基-OR7 或-Si(R15 )3 ; R2 係-C1 -C2 鹵代烷基、-C2 -C3 烯基、-C2 -C3 鹵代烯基、C2 炔基或-CH2 OS(O)2 -苯基,其中該等C1 -C2 烷基鹵基及-C2 -C3 烯基鹵基視情況經一或兩個-CH3 取代,且該等C2 炔基及苯基視情況經一個-CH3 取代; 每一R3 獨立地係鹵基、-CN、-OH、-OR8 、-NH2 、-NHR8 、-N(R8 )2 、-S(O)2 R8 、-S(O)R8 、-S(O)2 N(R7 )2 、-S(O)N(R7 )2 、-NO2 、-Si(R12 )3 、-SF5 、-C(O)OR6 、-C(O)N(R7 )2 、-NR12 C(O)R8 、-NR12 C(O)OR8 、-OC(O)N(R7 )2 、-OC(O)R8 、-C(O)R6 、-OC(O)CHR8 N(R12 )2 、C1 -C6 烷基、C2 -C6 烯基、C2 -C6 炔基、C3 -C10 環烷基、雜環基、芳基、雜芳基、-C1 -C6 烷基C3 -C10 環烷基、-C2 -C6 烯基C3 -C10 環烷基、-C1 -C6 烷基雜環基、-C2 -C6 烯基雜環基、-C1 -C6 烷基芳基、-C2 -C6 烯基芳基、C1 -C6 烷基雜芳基或-C2 -C6 烯基雜芳基;其中R3 之每一C1 -C6 烷基、C2 -C6 烯基、C2 -C6 炔基、C3 -C10 環烷基、雜環基、芳基、雜芳基、-C1 -C6 烷基C3 -C10 環烷基、-C2 -C6 烯基C3 -C10 環烷基、-C1 -C6 烷基雜環基、-C2 -C6 烯基雜環基、-C1 -C6 烷基芳基、-C2 -C6 烯基芳基、C1 -C6 烷基雜芳基或-C2 -C6 烯基雜芳基獨立地視情況經一至三個R10 取代; 每一R4 獨立地係鹵基、-CN、-OH、-OR8 、-NH2 、-NHR8 、-N(R8 )2 、-S(O)2 R8 、-S(O)R8 、-S(O)2 N(R7 )2 、-S(O)N(R7 )2 、-NO2 、-Si(R15 )3 、-C(O)OR6 、-C(O)N(R7 )2 、-NR12 C(O)R8 、-OC(O)R8 、-C(O)R6 、-NR12 C(O)OR8 、-OC(O)N(R7 )2 、-OC(O)CHR8 N(R12 )2 、C1 -C6 烷基、C2 -C6 烯基、C2 -C6 炔基、C3 -C10 環烷基、雜環基、芳基、雜芳基、-C1 -C6 烷基C3 -C10 環烷基、-C2 -C6 烯基C3 -C10 環烷基、-C1 -C6 烷基雜環基、-C2 -C6 烯基雜環基、-C1 -C6 烷基芳基、-C2 -C6 烯基芳基、C1 -C6 烷基雜芳基或-C2 -C6 烯基雜芳基;其中R4 之每一C1 -C6 烷基、C2 -C6 烯基、C2 -C6 炔基、C3 -C10 環烷基、雜環基、芳基、雜芳基、-C1 -C6 烷基C3 -C10 環烷基、-C2 -C6 烯基C3 -C10 環烷基、-C1 -C6 烷基雜環基、-C2 -C6 烯基雜環基、-C1 -C6 烷基芳基、-C2 -C6 烯基芳基、C1 -C6 烷基雜芳基或-C2 -C6 烯基雜芳基視情況獨立地視情況經一至三個R10 取代; 每一R5 獨立地係氫、鹵基、-CN、-OH、-OR8 、-NH2 、-NHR8 、-N(R8 )2 、-S(O)2 R8 、-S(O)R8 、-S(O)2 N(R7 )2 、-S(O)N(R7 )2 、-NO2 、-Si(R15 )3 、-C(O)OR6 、-C(O)N(R7 )2 、-NR12 C(O)R8 、-OC(O)R8 、-C(O)R6 、-NR12 C(O)OR8 、-OC(O)N(R7 )2 、-OC(O)CHR8 N(R12 )2 、C1 -C6 烷基、C2 -C6 烯基、C2 -C6 炔基、C3 -C10 環烷基、雜環基、芳基、雜芳基、-C1 -C6 烷基C3 -C10 環烷基、-C2 -C6 烯基C3 -C10 環烷基、-C1 -C6 烷基雜環基、-C2 -C6 烯基雜環基、-C1 -C6 烷基芳基、-C2 -C6 烯基芳基、C1 -C6 烷基雜芳基或-C2 -C6 烯基雜芳基;其中R5 之每一C1 -C6 烷基、C2 -C6 烯基、C2 -C6 炔基、C3 -C10 環烷基、雜環基、芳基、雜芳基、-C1 -C6 烷基C3 -C10 環烷基、-C2 -C6 烯基C3 -C10 環烷基、-C1 -C6 烷基雜環基、-C2 -C6 烯基雜環基、-C1 -C6 烷基芳基、-C2 -C6 烯基芳基、C1 -C6 烷基雜芳基或-C2 -C6 烯基雜芳基視情況獨立地視情況經一至三個R10 取代; 每一R6 獨立地係氫、C1 -C6 烷基、C2 -C6 烯基、C2 -C6 炔基、C3 -C10 環烷基、雜環基、芳基、雜芳基、-C1 -C6 烷基C3 -C10 環烷基、-C2 -C6 烯基C3 -C10 環烷基、-C1 -C6 烷基雜環基、-C2 -C6 烯基雜環基、-C1 -C6 烷基芳基、-C2 -C6 烯基芳基、C1 -C6 烷基雜芳基或-C2 -C6 烯基雜芳基;其中每一R6 獨立地進一步經一至三個R11 取代; 每一R7 獨立地係氫、C1 -C6 烷基、C2 -C6 烯基、C2 -C6 炔基、C3 -C10 環烷基、雜環基、芳基、雜芳基、-C1 -C6 烷基C3 -C6 環烷基、-C2 -C6 烯基C3 -C6 環烷基、-C1 -C6 烷基雜環基、-C2 -C6 烯基雜環基、-C1 -C6 烷基芳基、-C2 -C6 烯基芳基、-C1 -C6 烷基雜芳基、-C2 -C6 烯基雜芳基,或兩個R7 與其所附接之氮原子一起形成4至7員雜環基;其中每一R7 或由此形成之環獨立地進一步經一至三個R11 取代; 每一R8 獨立地係C1 -C6 烷基、C2 -C6 烯基、C2 -C6 炔基、C3 -C10 環烷基、雜環基、芳基、雜芳基、-C1 -C6 烷基C3 -C10 環烷基、-C2 -C6 烯基C3 -C10 環烷基、-C1 -C6 烷基雜環基、-C2 -C6 烯基雜環基、-C1 -C6 烷基芳基、-C2 -C6 烯基芳基、-C1 -C6 烷基雜芳基或-C2 -C6 烯基雜芳基;其中每一R8 獨立地進一步經一至三個R11 取代; R9 係氫或C1 -C6 烷基; 每一R10 獨立地係鹵基、-CN、-OR12 、-NO2 、-N(R12 )2 、-S(O)R13 、-S(O)2 R13 、-S(O)N(R12 )2 、-S(O)2 N(R12 )2 、-Si(R12 )3 、-C(O)R12 、-C(O)OR12 、-C(O)N(R12 )2 、-NR12 C(O)R12 、-OC(O)R12 、-OC(O)OR12 、-OC(O)N(R12 )2 、-NR12 C(O)OR12 、-OC(O)CHR12 N(R12 )2 、C1 -C6 烷基、C1 -C6 鹵代烷基、C2 -C6 烯基、C2 -C6 炔基、C3 -C10 環烷基、雜環基、芳基或雜芳基,其中R10 之每一C1 -C6 烷基、C1 -C6 鹵代烷基、C2 -C6 烯基、C2 -C6 炔基、C3 -C10 環烷基、雜環基、芳基或雜芳基視情況獨立地經一至三個R11 取代; 每一R11 獨立地係鹵基、-CN、-OR12 、-NO2 、-N(R12 )2 、-S(O)R13 、-S(O)2 R13 、-S(O)N(R12 )2 、-S(O)2 N(R12 )2 、-Si(R12 )3 、-C(O)R12 、-C(O)OR12 、-C(O)N(R12 )2 、-NR12 C(O)R12 、-OC(O)R12 、-OC(O)OR12 、-OC(O)N(R12 )2 、-NR12 C(O)OR12 、-OC(O)CHR12 N(R12 )2 、C1 -C6 烷基、C1 -C6 鹵代烷基、C2 -C6 烯基、C2 -C6 炔基、C3 -C10 環烷基、雜環基、芳基或雜芳基; 每一R12 獨立地係氫、C1 -C6 烷基或C3 -C10 環烷基; 每一R13 獨立地係C1 -C6 烷基或C3 -C10 環烷基;且 每一R15 獨立地係C1 -C6 烷基、C2 -C6 烯基、芳基、雜芳基、-C1 -C6 烷基芳基、-C2 -C6 烯基芳基、-C1 -C6 烷基雜芳基及-C2 -C6 烯基雜芳基。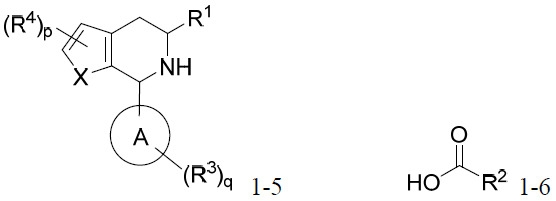
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| WOPCT/US19/19854 | 2019-02-27 | ||
| PCT/US2019/019854 WO2019168999A1 (en) | 2018-02-28 | 2019-02-27 | Compounds with ferroptosis inducing activity and methods of their use |
| US16/287,805 | 2019-02-27 | ||
| US16/287,805 US11098040B2 (en) | 2018-02-28 | 2019-02-27 | Compounds and methods of use |
| US201962893092P | 2019-08-28 | 2019-08-28 | |
| US62/893,092 | 2019-08-28 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| TW202045480A true TW202045480A (zh) | 2020-12-16 |
Family
ID=72238937
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| TW109106570A TW202045480A (zh) | 2019-02-27 | 2020-02-27 | 化合物及使用方法 |
Country Status (13)
| Country | Link |
|---|---|
| EP (1) | EP3931183A1 (zh) |
| JP (1) | JP2022522694A (zh) |
| KR (1) | KR20220012221A (zh) |
| CN (1) | CN114008024A (zh) |
| AU (1) | AU2020228056A1 (zh) |
| BR (1) | BR112021016833A2 (zh) |
| CA (1) | CA3131385A1 (zh) |
| EA (1) | EA202192122A1 (zh) |
| IL (1) | IL285730A (zh) |
| MX (1) | MX2021010173A (zh) |
| SG (1) | SG11202109035QA (zh) |
| TW (1) | TW202045480A (zh) |
| WO (1) | WO2020176757A1 (zh) |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10118890B2 (en) | 2014-10-10 | 2018-11-06 | The Research Foundation For The State University Of New York | Trifluoromethoxylation of arenes via intramolecular trifluoromethoxy group migration |
| CN112041301A (zh) | 2018-02-28 | 2020-12-04 | 费罗治疗公司 | 具有铁死亡诱导活性的化合物及其使用方法 |
| US11040964B2 (en) | 2019-02-27 | 2021-06-22 | Ferro Therapeutics, Inc. | Compounds and methods of use |
| WO2022042657A1 (en) * | 2020-08-26 | 2022-03-03 | Ferro Therapeutics, Inc. | Compounds and methods of use |
| KR20230110964A (ko) * | 2022-01-17 | 2023-07-25 | 포항공과대학교 산학협력단 | Marh6(marchf6) 조절제를 포함하는, 페로토시스 조절용 조성물 및 방법 |
| WO2024015637A1 (en) * | 2022-07-15 | 2024-01-18 | Ferro Therapeutics, Inc. | Glutathione peroxidase 4 (gpx4) inhibitors for the treatment of cancer |
| CN114984019B (zh) * | 2022-07-15 | 2023-08-22 | 山东中医药大学 | 一种铁死亡抑制剂化合物及在肝损伤修复领域的应用 |
| CN116983310B (zh) * | 2023-08-29 | 2024-04-30 | 南方医科大学南方医院 | 一种治疗肝癌的药物组合物及其应用 |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| MXPA03004023A (es) * | 2000-11-08 | 2004-02-12 | Lilly Icos Llc | Compuestos quimicos. |
| EP3384908B1 (en) * | 2013-12-02 | 2020-09-30 | The Trustees of Columbia University in the City of New York | Modulating ferroptosis and treating excitotoxic disorders |
| MX2018002407A (es) * | 2015-08-26 | 2018-11-29 | Blueprint Medicines Corp | Compuestos y composiciones utiles para tratar trastornos relacionados con ntrk. |
| US20190008961A1 (en) * | 2016-01-07 | 2019-01-10 | The Broad Institute, Inc. | Compounds and methods for increasing tumor infiltration by immune cells |
| WO2018118711A1 (en) * | 2016-12-19 | 2018-06-28 | The Trustees Of Columbia University In The City Of New York | Small molecule ferroptosis inducers |
| CN112041301A (zh) * | 2018-02-28 | 2020-12-04 | 费罗治疗公司 | 具有铁死亡诱导活性的化合物及其使用方法 |
| EP4076434A1 (en) * | 2019-12-17 | 2022-10-26 | Flagship Pioneering Innovations V, Inc. | Combination anti-cancer therapies with inducers of iron-dependent cellular disassembly |
| CN113336748B (zh) * | 2021-04-12 | 2022-03-25 | 北京大学 | 一种gpx4蛋白降解剂及其制备方法和应用、一种抗肿瘤细胞药物 |
-
2020
- 2020-02-27 MX MX2021010173A patent/MX2021010173A/es unknown
- 2020-02-27 EP EP20712811.7A patent/EP3931183A1/en active Pending
- 2020-02-27 CN CN202080028989.0A patent/CN114008024A/zh active Pending
- 2020-02-27 CA CA3131385A patent/CA3131385A1/en active Pending
- 2020-02-27 WO PCT/US2020/020150 patent/WO2020176757A1/en unknown
- 2020-02-27 SG SG11202109035QA patent/SG11202109035QA/en unknown
- 2020-02-27 KR KR1020217030619A patent/KR20220012221A/ko unknown
- 2020-02-27 JP JP2021549973A patent/JP2022522694A/ja active Pending
- 2020-02-27 TW TW109106570A patent/TW202045480A/zh unknown
- 2020-02-27 EA EA202192122A patent/EA202192122A1/ru unknown
- 2020-02-27 BR BR112021016833A patent/BR112021016833A2/pt not_active Application Discontinuation
- 2020-02-27 AU AU2020228056A patent/AU2020228056A1/en not_active Abandoned
-
2021
- 2021-08-19 IL IL285730A patent/IL285730A/en unknown
Also Published As
| Publication number | Publication date |
|---|---|
| JP2022522694A (ja) | 2022-04-20 |
| MX2021010173A (es) | 2021-12-10 |
| CN114008024A (zh) | 2022-02-01 |
| SG11202109035QA (en) | 2021-09-29 |
| EP3931183A1 (en) | 2022-01-05 |
| KR20220012221A (ko) | 2022-02-03 |
| IL285730A (en) | 2021-10-31 |
| WO2020176757A1 (en) | 2020-09-03 |
| BR112021016833A2 (pt) | 2021-11-23 |
| AU2020228056A1 (en) | 2021-09-16 |
| CA3131385A1 (en) | 2020-09-03 |
| EA202192122A1 (ru) | 2021-12-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| TW202045480A (zh) | 化合物及使用方法 | |
| JP7348906B2 (ja) | フェロトーシス誘導活性を有する化合物およびそれらの使用方法 | |
| US11691972B2 (en) | Compounds for targeted degradation of BRD9 | |
| US20220002280A1 (en) | Compounds and methods of use | |
| US20220194917A1 (en) | Benzothiophene-based selective estrogen receptor downregulator compounds | |
| BR112017023490B1 (pt) | Formas cristalinas de um composto modulador de quinases, composição farmacêutica e processos de preparação de uma cápsula e de um comprimido | |
| US20240092739A1 (en) | Compounds and methods of use | |
| WO2021041539A2 (en) | Compounds and methods of use | |
| WO2019183145A1 (en) | Compounds and methods for ido and tdo modulation, and indications therefor | |
| CA2882157A1 (en) | Substituted azaindole compounds, salts, pharmaceutical compositions thereof and methods of use | |
| US11013728B2 (en) | Cyclin-dependent kinase 8 and/or 19 inhibitor | |
| TW202328101A (zh) | 用於標靶降解brd9之經選擇的化合物 | |
| WO2024015637A1 (en) | Glutathione peroxidase 4 (gpx4) inhibitors for the treatment of cancer | |
| US20220227716A1 (en) | Benzimidazole and hydrogenated carbazole derivatives as gpx4 inhibitors | |
| TWI810547B (zh) | Pd-l1拮抗劑化合物 |