RU2561873C2 - Применение антагонистов опиоидных рецепторов при заболеваниях желудочно-кишечного тракта - Google Patents
Применение антагонистов опиоидных рецепторов при заболеваниях желудочно-кишечного тракта Download PDFInfo
- Publication number
- RU2561873C2 RU2561873C2 RU2012115450/15A RU2012115450A RU2561873C2 RU 2561873 C2 RU2561873 C2 RU 2561873C2 RU 2012115450/15 A RU2012115450/15 A RU 2012115450/15A RU 2012115450 A RU2012115450 A RU 2012115450A RU 2561873 C2 RU2561873 C2 RU 2561873C2
- Authority
- RU
- Russia
- Prior art keywords
- compound
- opioid
- dose
- day
- pharmaceutically acceptable
- Prior art date
Links
- 102000003840 Opioid Receptors Human genes 0.000 title abstract description 27
- 108090000137 Opioid Receptors Proteins 0.000 title abstract description 27
- 201000010099 disease Diseases 0.000 title description 6
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 title description 6
- 239000005557 antagonist Substances 0.000 title description 4
- 210000001035 gastrointestinal tract Anatomy 0.000 title description 3
- 150000001875 compounds Chemical class 0.000 claims abstract description 197
- 238000011282 treatment Methods 0.000 claims abstract description 49
- 150000003839 salts Chemical class 0.000 claims abstract description 24
- 238000000034 method Methods 0.000 claims abstract description 21
- 206010010774 Constipation Diseases 0.000 claims abstract description 11
- 208000030053 Opioid-Induced Constipation Diseases 0.000 claims abstract description 8
- 239000000014 opioid analgesic Substances 0.000 claims description 17
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 14
- 239000002552 dosage form Substances 0.000 claims description 10
- 239000003085 diluting agent Substances 0.000 claims description 3
- 239000003937 drug carrier Substances 0.000 claims description 3
- 239000008194 pharmaceutical composition Substances 0.000 claims 5
- 239000003814 drug Substances 0.000 abstract description 26
- 230000000694 effects Effects 0.000 abstract description 24
- 238000011161 development Methods 0.000 abstract description 17
- 229940005483 opioid analgesics Drugs 0.000 abstract description 16
- 208000024891 symptom Diseases 0.000 abstract description 10
- ZGCYVRNZWGUXNQ-UHFFFAOYSA-N Bevenopran Chemical compound C=1C=C(OC=2N=CC(=NC=2)C(N)=O)C(OC)=CC=1CNCCC1CCOCC1 ZGCYVRNZWGUXNQ-UHFFFAOYSA-N 0.000 abstract description 6
- 230000004913 activation Effects 0.000 abstract description 6
- 230000002265 prevention Effects 0.000 abstract description 6
- 239000000126 substance Substances 0.000 abstract description 5
- 206010002091 Anaesthesia Diseases 0.000 abstract description 3
- 230000037005 anaesthesia Effects 0.000 abstract description 3
- 230000001404 mediated effect Effects 0.000 abstract description 3
- 238000001949 anaesthesia Methods 0.000 abstract 1
- 230000005993 intestine dysfunction Effects 0.000 abstract 1
- BQJCRHHNABKAKU-KBQPJGBKSA-N morphine Chemical compound O([C@H]1[C@H](C=C[C@H]23)O)C4=C5[C@@]12CCN(C)[C@@H]3CC5=CC=C4O BQJCRHHNABKAKU-KBQPJGBKSA-N 0.000 description 42
- 210000003169 central nervous system Anatomy 0.000 description 32
- 239000000203 mixture Substances 0.000 description 32
- 102000051367 mu Opioid Receptors Human genes 0.000 description 29
- 108020001612 μ-opioid receptors Proteins 0.000 description 29
- 230000036470 plasma concentration Effects 0.000 description 22
- 229960005181 morphine Drugs 0.000 description 21
- 230000036592 analgesia Effects 0.000 description 19
- 230000008485 antagonism Effects 0.000 description 19
- 235000002639 sodium chloride Nutrition 0.000 description 18
- USPVLEIQIUNQGE-DBFLIVQGSA-N 2-[[(2s)-2-benzyl-3-[(3r,4r)-4-(3-hydroxyphenyl)-3,4-dimethylpiperidin-1-yl]propanoyl]amino]acetic acid;dihydrate Chemical compound O.O.C([C@@H](CN1C[C@@H]([C@](CC1)(C)C=1C=C(O)C=CC=1)C)C(=O)NCC(O)=O)C1=CC=CC=C1 USPVLEIQIUNQGE-DBFLIVQGSA-N 0.000 description 17
- 230000002093 peripheral effect Effects 0.000 description 17
- 229960004516 alvimopan Drugs 0.000 description 16
- 241000282412 Homo Species 0.000 description 15
- 229940079593 drug Drugs 0.000 description 15
- 241000699670 Mus sp. Species 0.000 description 14
- 241000282414 Homo sapiens Species 0.000 description 13
- 230000000144 pharmacologic effect Effects 0.000 description 13
- 210000002381 plasma Anatomy 0.000 description 13
- 238000013459 approach Methods 0.000 description 11
- 230000002441 reversible effect Effects 0.000 description 11
- 229940123257 Opioid receptor antagonist Drugs 0.000 description 10
- 241000700159 Rattus Species 0.000 description 10
- 239000002775 capsule Substances 0.000 description 10
- 239000003401 opiate antagonist Substances 0.000 description 10
- 239000003826 tablet Substances 0.000 description 10
- 238000004364 calculation method Methods 0.000 description 9
- 238000002347 injection Methods 0.000 description 9
- 239000007924 injection Substances 0.000 description 9
- DQCKKXVULJGBQN-XFWGSAIBSA-N naltrexone Chemical compound N1([C@@H]2CC3=CC=C(C=4O[C@@H]5[C@](C3=4)([C@]2(CCC5=O)O)CC1)O)CC1CC1 DQCKKXVULJGBQN-XFWGSAIBSA-N 0.000 description 9
- 229960003086 naltrexone Drugs 0.000 description 9
- 102100033350 ATP-dependent translocase ABCB1 Human genes 0.000 description 8
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 8
- 210000001072 colon Anatomy 0.000 description 8
- 230000005764 inhibitory process Effects 0.000 description 8
- 238000004519 manufacturing process Methods 0.000 description 8
- 208000002193 Pain Diseases 0.000 description 7
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 6
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 6
- 239000002253 acid Substances 0.000 description 6
- 210000004556 brain Anatomy 0.000 description 6
- 238000013270 controlled release Methods 0.000 description 6
- 238000002474 experimental method Methods 0.000 description 6
- 102000048260 kappa Opioid Receptors Human genes 0.000 description 6
- 239000007788 liquid Substances 0.000 description 6
- 230000036407 pain Effects 0.000 description 6
- 239000000243 solution Substances 0.000 description 6
- 239000000725 suspension Substances 0.000 description 6
- 230000009885 systemic effect Effects 0.000 description 6
- 108020001588 κ-opioid receptors Proteins 0.000 description 6
- -1 1,1'-methylene Chemical group 0.000 description 5
- 101100244562 Pseudomonas aeruginosa (strain ATCC 15692 / DSM 22644 / CIP 104116 / JCM 14847 / LMG 12228 / 1C / PRS 101 / PAO1) oprD gene Proteins 0.000 description 5
- 230000000202 analgesic effect Effects 0.000 description 5
- 108700023159 delta Opioid Receptors Proteins 0.000 description 5
- 102000048124 delta Opioid Receptors Human genes 0.000 description 5
- 238000009826 distribution Methods 0.000 description 5
- 239000000839 emulsion Substances 0.000 description 5
- 230000002496 gastric effect Effects 0.000 description 5
- 239000000843 powder Substances 0.000 description 5
- 238000011321 prophylaxis Methods 0.000 description 5
- 239000007787 solid Substances 0.000 description 5
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 5
- 102000004506 Blood Proteins Human genes 0.000 description 4
- 108010017384 Blood Proteins Proteins 0.000 description 4
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 4
- 230000001154 acute effect Effects 0.000 description 4
- JYGXADMDTFJGBT-VWUMJDOOSA-N hydrocortisone Chemical compound O=C1CC[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 JYGXADMDTFJGBT-VWUMJDOOSA-N 0.000 description 4
- 238000001802 infusion Methods 0.000 description 4
- 208000003243 intestinal obstruction Diseases 0.000 description 4
- 238000001990 intravenous administration Methods 0.000 description 4
- 239000008141 laxative Substances 0.000 description 4
- UZHSEJADLWPNLE-GRGSLBFTSA-N naloxone Chemical compound O=C([C@@H]1O2)CC[C@@]3(O)[C@H]4CC5=CC=C(O)C2=C5[C@@]13CCN4CC=C UZHSEJADLWPNLE-GRGSLBFTSA-N 0.000 description 4
- 229960004127 naloxone Drugs 0.000 description 4
- 239000003887 narcotic antagonist Substances 0.000 description 4
- 229940124531 pharmaceutical excipient Drugs 0.000 description 4
- 229920000642 polymer Polymers 0.000 description 4
- 239000011780 sodium chloride Substances 0.000 description 4
- 238000012360 testing method Methods 0.000 description 4
- 230000001225 therapeutic effect Effects 0.000 description 4
- 238000002560 therapeutic procedure Methods 0.000 description 4
- 239000000275 Adrenocorticotropic Hormone Substances 0.000 description 3
- 102400000739 Corticotropin Human genes 0.000 description 3
- 101800000414 Corticotropin Proteins 0.000 description 3
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 3
- 208000018522 Gastrointestinal disease Diseases 0.000 description 3
- 108010010803 Gelatin Proteins 0.000 description 3
- 108010047230 Member 1 Subfamily B ATP Binding Cassette Transporter Proteins 0.000 description 3
- 241001465754 Metazoa Species 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 206010057190 Respiratory tract infections Diseases 0.000 description 3
- 229920002472 Starch Polymers 0.000 description 3
- 206010047700 Vomiting Diseases 0.000 description 3
- 238000010521 absorption reaction Methods 0.000 description 3
- 239000002671 adjuvant Substances 0.000 description 3
- 239000000443 aerosol Substances 0.000 description 3
- 239000007864 aqueous solution Substances 0.000 description 3
- 210000004369 blood Anatomy 0.000 description 3
- 239000008280 blood Substances 0.000 description 3
- IDLFZVILOHSSID-OVLDLUHVSA-N corticotropin Chemical compound C([C@@H](C(=O)N[C@@H](CO)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C(C)C)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CC(N)=O)C(=O)NCC(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C=CC=CC=1)C(O)=O)NC(=O)[C@@H](N)CO)C1=CC=C(O)C=C1 IDLFZVILOHSSID-OVLDLUHVSA-N 0.000 description 3
- 229960000258 corticotropin Drugs 0.000 description 3
- 239000008273 gelatin Substances 0.000 description 3
- 229920000159 gelatin Polymers 0.000 description 3
- 235000019322 gelatine Nutrition 0.000 description 3
- 235000011852 gelatine desserts Nutrition 0.000 description 3
- 239000008187 granular material Substances 0.000 description 3
- 238000000338 in vitro Methods 0.000 description 3
- 210000000936 intestine Anatomy 0.000 description 3
- 230000002475 laxative effect Effects 0.000 description 3
- 239000002502 liposome Substances 0.000 description 3
- 239000000463 material Substances 0.000 description 3
- 210000004379 membrane Anatomy 0.000 description 3
- 239000012528 membrane Substances 0.000 description 3
- JVLBPIPGETUEET-GAAHOAFPSA-O methylnaltrexone Chemical compound C[N+]1([C@@H]2CC=3C4=C(C(=CC=3)O)O[C@@H]3[C@]4([C@@]2(O)CCC3=O)CC1)CC1CC1 JVLBPIPGETUEET-GAAHOAFPSA-O 0.000 description 3
- 239000006186 oral dosage form Substances 0.000 description 3
- 230000035515 penetration Effects 0.000 description 3
- 238000009520 phase I clinical trial Methods 0.000 description 3
- 239000006187 pill Substances 0.000 description 3
- 230000002980 postoperative effect Effects 0.000 description 3
- 239000003755 preservative agent Substances 0.000 description 3
- 238000011084 recovery Methods 0.000 description 3
- 210000000813 small intestine Anatomy 0.000 description 3
- 239000007909 solid dosage form Substances 0.000 description 3
- 239000002904 solvent Substances 0.000 description 3
- 239000008107 starch Substances 0.000 description 3
- 235000019698 starch Nutrition 0.000 description 3
- 210000001519 tissue Anatomy 0.000 description 3
- 238000005303 weighing Methods 0.000 description 3
- RZVAJINKPMORJF-UHFFFAOYSA-N Acetaminophen Chemical compound CC(=O)NC1=CC=C(O)C=C1 RZVAJINKPMORJF-UHFFFAOYSA-N 0.000 description 2
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 2
- 206010052105 Gastrointestinal hypomotility Diseases 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- 201000005081 Intestinal Pseudo-Obstruction Diseases 0.000 description 2
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 2
- 241000699666 Mus <mouse, genus> Species 0.000 description 2
- 206010028813 Nausea Diseases 0.000 description 2
- 229940127450 Opioid Agonists Drugs 0.000 description 2
- 229910019142 PO4 Inorganic materials 0.000 description 2
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 2
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 2
- 206010000059 abdominal discomfort Diseases 0.000 description 2
- 239000004480 active ingredient Substances 0.000 description 2
- 239000013543 active substance Substances 0.000 description 2
- 239000000556 agonist Substances 0.000 description 2
- 239000003708 ampul Substances 0.000 description 2
- BLFLLBZGZJTVJG-UHFFFAOYSA-N benzocaine Chemical compound CCOC(=O)C1=CC=C(N)C=C1 BLFLLBZGZJTVJG-UHFFFAOYSA-N 0.000 description 2
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 2
- 210000004978 chinese hamster ovary cell Anatomy 0.000 description 2
- 230000001684 chronic effect Effects 0.000 description 2
- 230000007012 clinical effect Effects 0.000 description 2
- 239000003086 colorant Substances 0.000 description 2
- 239000007891 compressed tablet Substances 0.000 description 2
- 238000000354 decomposition reaction Methods 0.000 description 2
- 230000001419 dependent effect Effects 0.000 description 2
- XYYVYLMBEZUESM-UHFFFAOYSA-N dihydrocodeine Natural products C1C(N(CCC234)C)C2C=CC(=O)C3OC2=C4C1=CC=C2OC XYYVYLMBEZUESM-UHFFFAOYSA-N 0.000 description 2
- 231100000673 dose–response relationship Toxicity 0.000 description 2
- 239000000975 dye Substances 0.000 description 2
- 239000003995 emulsifying agent Substances 0.000 description 2
- 235000003599 food sweetener Nutrition 0.000 description 2
- 238000009472 formulation Methods 0.000 description 2
- YQEMORVAKMFKLG-UHFFFAOYSA-N glycerine monostearate Natural products CCCCCCCCCCCCCCCCCC(=O)OC(CO)CO YQEMORVAKMFKLG-UHFFFAOYSA-N 0.000 description 2
- 235000011187 glycerol Nutrition 0.000 description 2
- SVUQHVRAGMNPLW-UHFFFAOYSA-N glycerol monostearate Natural products CCCCCCCCCCCCCCCCC(=O)OCC(O)CO SVUQHVRAGMNPLW-UHFFFAOYSA-N 0.000 description 2
- OROGSEYTTFOCAN-UHFFFAOYSA-N hydrocodone Natural products C1C(N(CCC234)C)C2C=CC(O)C3OC2=C4C1=CC=C2OC OROGSEYTTFOCAN-UHFFFAOYSA-N 0.000 description 2
- 229960000890 hydrocortisone Drugs 0.000 description 2
- 239000004615 ingredient Substances 0.000 description 2
- 230000000968 intestinal effect Effects 0.000 description 2
- 238000011813 knockout mouse model Methods 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- 210000003750 lower gastrointestinal tract Anatomy 0.000 description 2
- 239000000314 lubricant Substances 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 210000004400 mucous membrane Anatomy 0.000 description 2
- 230000008693 nausea Effects 0.000 description 2
- 239000003921 oil Substances 0.000 description 2
- 235000019198 oils Nutrition 0.000 description 2
- 239000003402 opiate agonist Substances 0.000 description 2
- 210000000056 organ Anatomy 0.000 description 2
- 238000007911 parenteral administration Methods 0.000 description 2
- 230000003285 pharmacodynamic effect Effects 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 2
- 239000010452 phosphate Substances 0.000 description 2
- 239000000902 placebo Substances 0.000 description 2
- 229940068196 placebo Drugs 0.000 description 2
- 230000003389 potentiating effect Effects 0.000 description 2
- 102000005962 receptors Human genes 0.000 description 2
- 108020003175 receptors Proteins 0.000 description 2
- 238000002271 resection Methods 0.000 description 2
- CVHZOJJKTDOEJC-UHFFFAOYSA-N saccharin Chemical compound C1=CC=C2C(=O)NS(=O)(=O)C2=C1 CVHZOJJKTDOEJC-UHFFFAOYSA-N 0.000 description 2
- 239000003381 stabilizer Substances 0.000 description 2
- 238000007920 subcutaneous administration Methods 0.000 description 2
- 239000000829 suppository Substances 0.000 description 2
- 238000001356 surgical procedure Methods 0.000 description 2
- 239000000375 suspending agent Substances 0.000 description 2
- 239000003765 sweetening agent Substances 0.000 description 2
- 230000008961 swelling Effects 0.000 description 2
- 239000000454 talc Substances 0.000 description 2
- 229910052623 talc Inorganic materials 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 230000008673 vomiting Effects 0.000 description 2
- IFGIYSGOEZJNBE-NQMNLMSRSA-N (3r,4r,4as,7ar,12bs)-3-(cyclopropylmethyl)-4a,9-dihydroxy-3-methyl-2,4,5,6,7a,13-hexahydro-1h-4,12-methanobenzofuro[3,2-e]isoquinoline-3-ium-7-one;bromide Chemical compound [Br-].C([N@+]1(C)[C@@H]2CC=3C4=C(C(=CC=3)O)O[C@@H]3[C@]4([C@@]2(O)CCC3=O)CC1)C1CC1 IFGIYSGOEZJNBE-NQMNLMSRSA-N 0.000 description 1
- WRRSFOZOETZUPG-FFHNEAJVSA-N (4r,4ar,7s,7ar,12bs)-9-methoxy-3-methyl-2,4,4a,7,7a,13-hexahydro-1h-4,12-methanobenzofuro[3,2-e]isoquinoline-7-ol;hydrate Chemical compound O.C([C@H]1[C@H](N(CC[C@@]112)C)C3)=C[C@H](O)[C@@H]1OC1=C2C3=CC=C1OC WRRSFOZOETZUPG-FFHNEAJVSA-N 0.000 description 1
- GHOKWGTUZJEAQD-ZETCQYMHSA-N (D)-(+)-Pantothenic acid Chemical compound OCC(C)(C)[C@@H](O)C(=O)NCCC(O)=O GHOKWGTUZJEAQD-ZETCQYMHSA-N 0.000 description 1
- TVYLLZQTGLZFBW-ZBFHGGJFSA-N (R,R)-tramadol Chemical compound COC1=CC=CC([C@]2(O)[C@H](CCCC2)CN(C)C)=C1 TVYLLZQTGLZFBW-ZBFHGGJFSA-N 0.000 description 1
- WXTMDXOMEHJXQO-UHFFFAOYSA-N 2,5-dihydroxybenzoic acid Chemical compound OC(=O)C1=CC(O)=CC=C1O WXTMDXOMEHJXQO-UHFFFAOYSA-N 0.000 description 1
- BZMADPOGYCRPAI-UHFFFAOYSA-N 2-(oxan-4-yl)ethanamine Chemical compound NCCC1CCOCC1 BZMADPOGYCRPAI-UHFFFAOYSA-N 0.000 description 1
- ALKYHXVLJMQRLQ-UHFFFAOYSA-N 3-Hydroxy-2-naphthoate Chemical compound C1=CC=C2C=C(O)C(C(=O)O)=CC2=C1 ALKYHXVLJMQRLQ-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-M 3-carboxy-2,3-dihydroxypropanoate Chemical compound OC(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-M 0.000 description 1
- ZNPAVDCRBSDRKH-UHFFFAOYSA-N 5-(4-formyl-2-methoxyphenoxy)pyrazine-2-carboxamide Chemical compound COC1=CC(C=O)=CC=C1OC1=CN=C(C(N)=O)C=N1 ZNPAVDCRBSDRKH-UHFFFAOYSA-N 0.000 description 1
- USSIQXCVUWKGNF-UHFFFAOYSA-N 6-(dimethylamino)-4,4-diphenylheptan-3-one Chemical compound C=1C=CC=CC=1C(CC(C)N(C)C)(C(=O)CC)C1=CC=CC=C1 USSIQXCVUWKGNF-UHFFFAOYSA-N 0.000 description 1
- 206010000060 Abdominal distension Diseases 0.000 description 1
- 244000215068 Acacia senegal Species 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 108010011485 Aspartame Proteins 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- 241000167854 Bourreria succulenta Species 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- 206010058019 Cancer Pain Diseases 0.000 description 1
- 241000700199 Cavia porcellus Species 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- 208000000094 Chronic Pain Diseases 0.000 description 1
- 208000035473 Communicable disease Diseases 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 1
- AEMOLEFTQBMNLQ-AQKNRBDQSA-N D-glucopyranuronic acid Chemical compound OC1O[C@H](C(O)=O)[C@@H](O)[C@H](O)[C@H]1O AEMOLEFTQBMNLQ-AQKNRBDQSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 241000305071 Enterobacterales Species 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 241000283073 Equus caballus Species 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- 229930091371 Fructose Natural products 0.000 description 1
- 239000005715 Fructose Substances 0.000 description 1
- RFSUNEUAIZKAJO-ARQDHWQXSA-N Fructose Chemical compound OC[C@H]1O[C@](O)(CO)[C@@H](O)[C@@H]1O RFSUNEUAIZKAJO-ARQDHWQXSA-N 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 1
- 241000287828 Gallus gallus Species 0.000 description 1
- 208000017228 Gastrointestinal motility disease Diseases 0.000 description 1
- 229920000084 Gum arabic Polymers 0.000 description 1
- 108010076876 Keratins Proteins 0.000 description 1
- 102000011782 Keratins Human genes 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- JAQUASYNZVUNQP-USXIJHARSA-N Levorphanol Chemical compound C1C2=CC=C(O)C=C2[C@]23CCN(C)[C@H]1[C@@H]2CCCC3 JAQUASYNZVUNQP-USXIJHARSA-N 0.000 description 1
- 241000282553 Macaca Species 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 244000246386 Mentha pulegium Species 0.000 description 1
- 235000016257 Mentha pulegium Nutrition 0.000 description 1
- 235000004357 Mentha x piperita Nutrition 0.000 description 1
- XADCESSVHJOZHK-UHFFFAOYSA-N Meperidine Chemical compound C=1C=CC=CC=1C1(C(=O)OCC)CCN(C)CC1 XADCESSVHJOZHK-UHFFFAOYSA-N 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- 241001529936 Murinae Species 0.000 description 1
- IDBPHNDTYPBSNI-UHFFFAOYSA-N N-(1-(2-(4-Ethyl-5-oxo-2-tetrazolin-1-yl)ethyl)-4-(methoxymethyl)-4-piperidyl)propionanilide Chemical compound C1CN(CCN2C(N(CC)N=N2)=O)CCC1(COC)N(C(=O)CC)C1=CC=CC=C1 IDBPHNDTYPBSNI-UHFFFAOYSA-N 0.000 description 1
- GXCLVBGFBYZDAG-UHFFFAOYSA-N N-[2-(1H-indol-3-yl)ethyl]-N-methylprop-2-en-1-amine Chemical compound CN(CCC1=CNC2=C1C=CC=C2)CC=C GXCLVBGFBYZDAG-UHFFFAOYSA-N 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- 206010028980 Neoplasm Diseases 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 240000007594 Oryza sativa Species 0.000 description 1
- 235000007164 Oryza sativa Nutrition 0.000 description 1
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 1
- BRUQQQPBMZOVGD-XFKAJCMBSA-N Oxycodone Chemical compound O=C([C@@H]1O2)CC[C@@]3(O)[C@H]4CC5=CC=C(OC)C2=C5[C@@]13CCN4C BRUQQQPBMZOVGD-XFKAJCMBSA-N 0.000 description 1
- UQCNKQCJZOAFTQ-ISWURRPUSA-N Oxymorphone Chemical compound O([C@H]1C(CC[C@]23O)=O)C4=C5[C@@]12CCN(C)[C@@H]3CC5=CC=C4O UQCNKQCJZOAFTQ-ISWURRPUSA-N 0.000 description 1
- 241000282577 Pan troglodytes Species 0.000 description 1
- 241001504519 Papio ursinus Species 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- 241000009328 Perro Species 0.000 description 1
- 241000286209 Phasianidae Species 0.000 description 1
- 208000031649 Postoperative Nausea and Vomiting Diseases 0.000 description 1
- 208000003251 Pruritus Diseases 0.000 description 1
- 241000283984 Rodentia Species 0.000 description 1
- 206010039897 Sedation Diseases 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 241000282898 Sus scrofa Species 0.000 description 1
- 229920002253 Tannate Polymers 0.000 description 1
- DTQVDTLACAAQTR-UHFFFAOYSA-M Trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-M 0.000 description 1
- 210000001015 abdomen Anatomy 0.000 description 1
- 239000000205 acacia gum Substances 0.000 description 1
- 235000010489 acacia gum Nutrition 0.000 description 1
- 238000009825 accumulation Methods 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 229960001391 alfentanil Drugs 0.000 description 1
- VREFGVBLTWBCJP-UHFFFAOYSA-N alprazolam Chemical compound C12=CC(Cl)=CC=C2N2C(C)=NN=C2CN=C1C1=CC=CC=C1 VREFGVBLTWBCJP-UHFFFAOYSA-N 0.000 description 1
- 230000003872 anastomosis Effects 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 208000022531 anorexia Diseases 0.000 description 1
- 230000003042 antagnostic effect Effects 0.000 description 1
- 239000012062 aqueous buffer Substances 0.000 description 1
- 239000008365 aqueous carrier Substances 0.000 description 1
- 229940072107 ascorbate Drugs 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 239000000605 aspartame Substances 0.000 description 1
- 235000010357 aspartame Nutrition 0.000 description 1
- IAOZJIPTCAWIRG-QWRGUYRKSA-N aspartame Chemical compound OC(=O)C[C@H](N)C(=O)N[C@H](C(=O)OC)CC1=CC=CC=C1 IAOZJIPTCAWIRG-QWRGUYRKSA-N 0.000 description 1
- 229960003438 aspartame Drugs 0.000 description 1
- 229940077388 benzenesulfonate Drugs 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- 229960005274 benzocaine Drugs 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 208000024330 bloating Diseases 0.000 description 1
- 230000008499 blood brain barrier function Effects 0.000 description 1
- 210000001218 blood-brain barrier Anatomy 0.000 description 1
- RMRJXGBAOAMLHD-IHFGGWKQSA-N buprenorphine Chemical compound C([C@]12[C@H]3OC=4C(O)=CC=C(C2=4)C[C@@H]2[C@]11CC[C@]3([C@H](C1)[C@](C)(O)C(C)(C)C)OC)CN2CC1CC1 RMRJXGBAOAMLHD-IHFGGWKQSA-N 0.000 description 1
- 229960001736 buprenorphine Drugs 0.000 description 1
- IFKLAQQSCNILHL-QHAWAJNXSA-N butorphanol Chemical compound N1([C@@H]2CC3=CC=C(C=C3[C@@]3([C@]2(CCCC3)O)CC1)O)CC1CCC1 IFKLAQQSCNILHL-QHAWAJNXSA-N 0.000 description 1
- 229960001113 butorphanol Drugs 0.000 description 1
- 201000011510 cancer Diseases 0.000 description 1
- 239000007894 caplet Substances 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- 235000019693 cherries Nutrition 0.000 description 1
- 229940001468 citrate Drugs 0.000 description 1
- 238000011260 co-administration Methods 0.000 description 1
- 239000003245 coal Substances 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 229960004126 codeine Drugs 0.000 description 1
- OROGSEYTTFOCAN-DNJOTXNNSA-N codeine Natural products C([C@H]1[C@H](N(CC[C@@]112)C)C3)=C[C@H](O)[C@@H]1OC1=C2C3=CC=C1OC OROGSEYTTFOCAN-DNJOTXNNSA-N 0.000 description 1
- 229940075614 colloidal silicon dioxide Drugs 0.000 description 1
- 239000012141 concentrate Substances 0.000 description 1
- 235000008504 concentrate Nutrition 0.000 description 1
- 239000000599 controlled substance Substances 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 206010061428 decreased appetite Diseases 0.000 description 1
- 230000002950 deficient Effects 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 229960004193 dextropropoxyphene Drugs 0.000 description 1
- XLMALTXPSGQGBX-GCJKJVERSA-N dextropropoxyphene Chemical compound C([C@](OC(=O)CC)([C@H](C)CN(C)C)C=1C=CC=CC=1)C1=CC=CC=C1 XLMALTXPSGQGBX-GCJKJVERSA-N 0.000 description 1
- 239000008121 dextrose Substances 0.000 description 1
- RBOXVHNMENFORY-DNJOTXNNSA-N dihydrocodeine Chemical compound C([C@H]1[C@H](N(CC[C@@]112)C)C3)C[C@H](O)[C@@H]1OC1=C2C3=CC=C1OC RBOXVHNMENFORY-DNJOTXNNSA-N 0.000 description 1
- 229960000920 dihydrocodeine Drugs 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- 239000008298 dragée Substances 0.000 description 1
- 229940126534 drug product Drugs 0.000 description 1
- 230000004064 dysfunction Effects 0.000 description 1
- 239000008144 emollient laxative Substances 0.000 description 1
- 238000005538 encapsulation Methods 0.000 description 1
- 229940028816 entereg Drugs 0.000 description 1
- 239000002662 enteric coated tablet Substances 0.000 description 1
- 210000004955 epithelial membrane Anatomy 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 238000013213 extrapolation Methods 0.000 description 1
- 229960002428 fentanyl Drugs 0.000 description 1
- PJMPHNIQZUBGLI-UHFFFAOYSA-N fentanyl Chemical compound C=1C=CC=CC=1N(C(=O)CC)C(CC1)CCN1CCC1=CC=CC=C1 PJMPHNIQZUBGLI-UHFFFAOYSA-N 0.000 description 1
- 239000007941 film coated tablet Substances 0.000 description 1
- 235000013312 flour Nutrition 0.000 description 1
- NBVXSUQYWXRMNV-UHFFFAOYSA-N fluoromethane Chemical compound FC NBVXSUQYWXRMNV-UHFFFAOYSA-N 0.000 description 1
- 235000013305 food Nutrition 0.000 description 1
- 229960002737 fructose Drugs 0.000 description 1
- 229940050411 fumarate Drugs 0.000 description 1
- 230000006870 function Effects 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 208000021302 gastroesophageal reflux disease Diseases 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 229940050410 gluconate Drugs 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 229940097042 glucuronate Drugs 0.000 description 1
- 229930195712 glutamate Natural products 0.000 description 1
- 235000001050 hortel pimenta Nutrition 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- LLPOLZWFYMWNKH-CMKMFDCUSA-N hydrocodone Chemical compound C([C@H]1[C@H](N(CC[C@@]112)C)C3)CC(=O)[C@@H]1OC1=C2C3=CC=C1OC LLPOLZWFYMWNKH-CMKMFDCUSA-N 0.000 description 1
- 229960000240 hydrocodone Drugs 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- 229910052739 hydrogen Inorganic materials 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- WVLOADHCBXTIJK-YNHQPCIGSA-N hydromorphone Chemical compound O([C@H]1C(CC[C@H]23)=O)C4=C5[C@@]12CCN(C)[C@@H]3CC5=CC=C4O WVLOADHCBXTIJK-YNHQPCIGSA-N 0.000 description 1
- 229960001410 hydromorphone Drugs 0.000 description 1
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 1
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 1
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 1
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 description 1
- 208000008384 ileus Diseases 0.000 description 1
- 239000012729 immediate-release (IR) formulation Substances 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 239000007972 injectable composition Substances 0.000 description 1
- 230000008991 intestinal motility Effects 0.000 description 1
- 238000000185 intracerebroventricular administration Methods 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 208000002551 irritable bowel syndrome Diseases 0.000 description 1
- 230000000622 irritating effect Effects 0.000 description 1
- TWBYWOBDOCUKOW-UHFFFAOYSA-M isonicotinate Chemical compound [O-]C(=O)C1=CC=NC=C1 TWBYWOBDOCUKOW-UHFFFAOYSA-M 0.000 description 1
- 230000007803 itching Effects 0.000 description 1
- 230000003907 kidney function Effects 0.000 description 1
- 229940125722 laxative agent Drugs 0.000 description 1
- 229960003406 levorphanol Drugs 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 230000003908 liver function Effects 0.000 description 1
- 239000003589 local anesthetic agent Substances 0.000 description 1
- 230000001050 lubricating effect Effects 0.000 description 1
- 239000003580 lung surfactant Substances 0.000 description 1
- 239000008176 lyophilized powder Substances 0.000 description 1
- ZLNQQNXFFQJAID-UHFFFAOYSA-L magnesium carbonate Chemical compound [Mg+2].[O-]C([O-])=O ZLNQQNXFFQJAID-UHFFFAOYSA-L 0.000 description 1
- 239000001095 magnesium carbonate Substances 0.000 description 1
- 229910000021 magnesium carbonate Inorganic materials 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 229940049920 malate Drugs 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N malic acid Chemical compound OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 229960001797 methadone Drugs 0.000 description 1
- OSWPMRLSEDHDFF-UHFFFAOYSA-N methyl salicylate Chemical compound COC(=O)C1=CC=CC=C1O OSWPMRLSEDHDFF-UHFFFAOYSA-N 0.000 description 1
- 230000000813 microbial effect Effects 0.000 description 1
- 239000003094 microcapsule Substances 0.000 description 1
- 239000011859 microparticle Substances 0.000 description 1
- 239000002480 mineral oil Substances 0.000 description 1
- 235000010446 mineral oil Nutrition 0.000 description 1
- 230000003020 moisturizing effect Effects 0.000 description 1
- 210000000214 mouth Anatomy 0.000 description 1
- 230000003533 narcotic effect Effects 0.000 description 1
- 239000006199 nebulizer Substances 0.000 description 1
- 125000004433 nitrogen atom Chemical group N* 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- 229940021182 non-steroidal anti-inflammatory drug Drugs 0.000 description 1
- 239000012457 nonaqueous media Substances 0.000 description 1
- 235000016709 nutrition Nutrition 0.000 description 1
- 230000035764 nutrition Effects 0.000 description 1
- 229940049964 oleate Drugs 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- 229940126701 oral medication Drugs 0.000 description 1
- 239000007935 oral tablet Substances 0.000 description 1
- 229960002085 oxycodone Drugs 0.000 description 1
- 229960005118 oxymorphone Drugs 0.000 description 1
- 239000006179 pH buffering agent Substances 0.000 description 1
- 238000002638 palliative care Methods 0.000 description 1
- 229940014662 pantothenate Drugs 0.000 description 1
- 235000019161 pantothenic acid Nutrition 0.000 description 1
- 239000011713 pantothenic acid Substances 0.000 description 1
- 229960005489 paracetamol Drugs 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- VOKSWYLNZZRQPF-GDIGMMSISA-N pentazocine Chemical compound C1C2=CC=C(O)C=C2[C@@]2(C)[C@@H](C)[C@@H]1N(CC=C(C)C)CC2 VOKSWYLNZZRQPF-GDIGMMSISA-N 0.000 description 1
- 229960005301 pentazocine Drugs 0.000 description 1
- 230000010412 perfusion Effects 0.000 description 1
- 229940079358 peripheral opioid receptor antagonist Drugs 0.000 description 1
- 230000008855 peristalsis Effects 0.000 description 1
- 230000002572 peristaltic effect Effects 0.000 description 1
- 230000002085 persistent effect Effects 0.000 description 1
- 229960000482 pethidine Drugs 0.000 description 1
- 239000003208 petroleum Substances 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- 238000002360 preparation method Methods 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- MVFGUOIZUNYYSO-UHFFFAOYSA-N prilocaine Chemical compound CCCNC(C)C(=O)NC1=CC=CC=C1C MVFGUOIZUNYYSO-UHFFFAOYSA-N 0.000 description 1
- 229960001807 prilocaine Drugs 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- 108090000765 processed proteins & peptides Proteins 0.000 description 1
- 102000004196 processed proteins & peptides Human genes 0.000 description 1
- 239000000651 prodrug Substances 0.000 description 1
- 229940002612 prodrug Drugs 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- 230000001141 propulsive effect Effects 0.000 description 1
- 230000002685 pulmonary effect Effects 0.000 description 1
- 210000001747 pupil Anatomy 0.000 description 1
- 239000000018 receptor agonist Substances 0.000 description 1
- 229940044601 receptor agonist Drugs 0.000 description 1
- 210000000664 rectum Anatomy 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 229940105899 relistor Drugs 0.000 description 1
- 230000000241 respiratory effect Effects 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- 235000009566 rice Nutrition 0.000 description 1
- 235000019204 saccharin Nutrition 0.000 description 1
- 229940081974 saccharin Drugs 0.000 description 1
- 239000000901 saccharin and its Na,K and Ca salt Substances 0.000 description 1
- YGSDEFSMJLZEOE-UHFFFAOYSA-M salicylate Chemical compound OC1=CC=CC=C1C([O-])=O YGSDEFSMJLZEOE-UHFFFAOYSA-M 0.000 description 1
- 229960001860 salicylate Drugs 0.000 description 1
- 230000036280 sedation Effects 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- 235000020183 skimmed milk Nutrition 0.000 description 1
- RYYKJJJTJZKILX-UHFFFAOYSA-M sodium octadecanoate Chemical compound [Na+].CCCCCCCCCCCCCCCCCC([O-])=O RYYKJJJTJZKILX-UHFFFAOYSA-M 0.000 description 1
- 239000012453 solvate Substances 0.000 description 1
- 239000003549 soybean oil Substances 0.000 description 1
- 235000012424 soybean oil Nutrition 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 230000000087 stabilizing effect Effects 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 230000003068 static effect Effects 0.000 description 1
- 239000008174 sterile solution Substances 0.000 description 1
- 239000008223 sterile water Substances 0.000 description 1
- 239000008227 sterile water for injection Substances 0.000 description 1
- 238000010254 subcutaneous injection Methods 0.000 description 1
- 239000007929 subcutaneous injection Substances 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 229940086735 succinate Drugs 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 229960004739 sufentanil Drugs 0.000 description 1
- GGCSSNBKKAUURC-UHFFFAOYSA-N sufentanil Chemical compound C1CN(CCC=2SC=CC=2)CCC1(COC)N(C(=O)CC)C1=CC=CC=C1 GGCSSNBKKAUURC-UHFFFAOYSA-N 0.000 description 1
- 239000007940 sugar coated tablet Substances 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- 230000008719 thickening Effects 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 229960004380 tramadol Drugs 0.000 description 1
- TVYLLZQTGLZFBW-GOEBONIOSA-N tramadol Natural products COC1=CC=CC([C@@]2(O)[C@@H](CCCC2)CN(C)C)=C1 TVYLLZQTGLZFBW-GOEBONIOSA-N 0.000 description 1
- LLPOLZWFYMWNKH-UHFFFAOYSA-N trans-dihydrocodeinone Natural products C1C(N(CCC234)C)C2CCC(=O)C3OC2=C4C1=CC=C2OC LLPOLZWFYMWNKH-UHFFFAOYSA-N 0.000 description 1
- 150000003626 triacylglycerols Chemical class 0.000 description 1
- 210000002438 upper gastrointestinal tract Anatomy 0.000 description 1
- 235000013311 vegetables Nutrition 0.000 description 1
- 239000009637 wintergreen oil Substances 0.000 description 1
Images






Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4965—Non-condensed pyrazines
- A61K31/497—Non-condensed pyrazines containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0053—Mouth and digestive tract, i.e. intraoral and peroral administration
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/08—Drugs for disorders of the alimentary tract or the digestive system for nausea, cinetosis or vertigo; Antiemetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/10—Laxatives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2121/00—Preparations for use in therapy
Abstract
Группа изобретений относится к медицине, а именно к способу лечения или профилактике опиоид-индуцированного запора или опиоид-индуцированной дисфункции кишечника у пациентов без снижения центрально опосредованного опиоидного обезболивания или развития симптомов центральной отмены опиоидов, ассоциированного с активацией опиоидных рецепторов на периферии. Для этого пациенту вводят эффективное количество 5-(2-метокси-4-{[2-(тетрагидропиран-4-ил)-этиламино]метил}-фенокси)пиразин-2-карбоксамида (Соединение I) или его фармацевтически приемлемой соли. Группа изобретений обеспечивает лечение или профилактику частых побочных эффектов у пациентов, которые принимают опиоиды в течение несколько дней. 3 н. и 9 з.п. ф-лы, 9 ил., 1 табл., 3 пр.
Description
1. ПЕРЕКРЕСТНЫЕ ССЫЛКИ НА РОДСТВЕННЫЕ ЗАЯВКИ
Настоящая заявка испрашивает приоритет 35 U.S.C. § 119(e) предварительной заявки № 61/243616, поданной 18 сентября 2009, содержание которой включено в настоящее описание в виде ссылки.
2. УРОВЕНЬ ТЕХНИКИ
Боль является наиболее частой причиной, по которой люди обращаются за медицинской помощью. Боль от легкой до умеренной обычно лечат ацетаминофеном и нестероидными противовоспалительными средствами. Опиоидные анальгетики, агонисты эндогенных μ-(мю), δ-(дельта) и/или κ-(каппа) опиоидных рецепторов главным образом в центральной нервной системе ("ЦНС") назначают при умеренной до сильной острой и хронической боли. Длительное применение опиоидных анальгетиков может приводить к физической зависимости и обладает побочными эффектами, такими как седативный эффект, помутнение сознания, тошнота, рвота, запор и зуд.
Опиоид-индуцированный запор, (ОИЗ ("OIC")) и ассоциированные состояния, называемые опиоид-индуцированной дисфункцией кишечника (ОДК ("OBD")), являются частыми побочными эффектами у пациентов, которые принимают опиоиды в течение более чем нескольких дней. Агонисты μ-опиоидных рецепторов в кишке и ЦНС ингибируют пропульсивную перистальтику в кишечнике, что приводит к запору. Другие симптомы ОИЗ или ОДК могут включать неполное опорожнение, тяжесть в животе, вздутие, дискомфорт в животе и желудочно-пищеводный рефлюкс. Вторичные осложнения включают псевдообструкцию кишечника, анорексию, тошноту, рвоту и влияние на введение и абсорбцию пероральных лекарственных средств. См. P. Holzer (2009) Regulatory Peptides 155: 11-17.
ОИЗ и ОДК можно лечить с помощью совместного назначения слабительных препаратов, но такой подход часто имеет ограниченную эффективность и требует частой коррекции дозы и смены слабительных. Другие варианты лечения включают совместное введение селективных антагонистов опиоидных рецепторов, таких как налоксон, налтрексон и N-метилналтрексон. С целью эффективного лечения опиоид-индуцированных заболеваний желудочно-кишечного тракта без снижения обезболивающего эффекта или развития неприятных и потенциально опасных центральных симптомов отмены опиоидов критично, чтобы в ЦНС не устанавливались пороговые фармакологически значимые концентрации опиоидных антагонистов. Суб-фармакологические пороговые концентрации опиоидных антагонистов в ЦНС на сегодняшний день были достигнуты или тщательным титрованием низких доз центрально-активных антагонистов (например, налоксона), или созданием антагониста с низкой системной биодоступностью (например, налоксон с контролируемым высвобождением), или путем использования опиоидных антагонистов, которые ограничены периферией, т.е. имеют ограниченный доступ в ЦНС (например, N-метилналтрексон).
Комбинированная композиция с фиксированным соотношением налоксона с контролируемым высвобождением и оксикодона с контролируемым высвобождением (TARGIN®) была одобрена в Европе в качестве пути снижения тяжести ОИЗ.
ENTEREG® (алвимопан) был одобрен для продажи в Соединенных Штатах и показан для ускорения времени до восстановления верхнего и нижнего желудочно-кишечного тракта после операции по частичной резекции толстой или тонкой кишки с первичным анастомозом. Такое применение ограничено кратковременным лечением послеоперационной непроходимости кишечника после резекции кишечника у госпитализированных пациентов в возрасте 18 лет или старше. Считают, что послеоперационная непроходимость кишечника, по меньшей мере, частично вызывается или ухудшается экзогенными опиоидными анальгетиками.
RELISTOR® (н-метилналтрексон), продукт, который вводят путем подкожной инъекции, был одобрен в Соединенных Штатах для лечения ОИЗ у пациентов с запущенными заболеваниями, которые получают паллиативный уход, когда ответ на терапию слабительными средствами недостаточен.
Так что, следовательно, существует необходимость в более эффективном пероральном лечении ОИЗ или ОДК, которые не влияют на обезболивание и не вызывают центральной отмены опиоидов у пациентов.
3. СУЩНОСТЬ
Настоящее описание обеспечивает способ лечения или профилактики ОИЗ или ОДК у пациента, которым может быть животное или человек, путем введения пациенту некоторого количества 5-(2-метокси-4-{[2-(тетрагидропиран-4-ил)этиламино]метил}фенокси)пиразин-2-карбоксамида (Соединение I) и/или его фармацевтически приемлемой соли, эффективных для облегчения симптомов ОИЗ или ОДК без снижения опиоидного обезболивания или формирования центральной отмены опиоидов. Настоящее описание дополнительно обеспечивает применение 5-(2-метокси-4-{[2-(тетрагидропиран-4-ил)этиламино]метил}фенокси)пиразин-2-карбоксамида (Соединение I) и/или его фармацевтически приемлемой соли для получения лекарственного средства для облегчения симптомов ОИЗ или ОДК без снижения опиоидного обезболивания или развития центральной отмены опиоидов. Изобретение, описанное в настоящем описании, частично основано на неожиданном открытии авторов, что соединение I, антагонист опиоидных рецепторов с эффективностью, сравнимой с известными антагонистами опиоидов налтрексоном и алвимопаном, достаточно распределяется у пациентов на периферии, так что им можно эффективно лечить ОИЗ или ОДК, не влияя на обезболивание или развитие центральной отмены опиоидов.
Авторы изобретения изучали результаты экспериментов в известных моделях на животных, созданных для определения эффективности и фармакокинетических свойств антагонистов опиоидных рецепторов, и определили, что данные для Соединения I согласовывались с сильным антагонистом опиоидных рецепторов, который в низких дозах значительно распределяется по периферии. В частности авторы изобретения получили из данных, что Соединение I является, по меньшей мере, в 100 раз менее сильным антагонистом опиоидных рецепторов в центральной нервной системе, чем налтрексон (Пример 1, Фиг. 1). Авторы изобретения также отметили, что Соединение I было приблизительно в 20 раз более сильным в обратном развитии морфин-индуцированного замедления моторики кишечника у мышей, когда морфин вводили системно (т.е., он действовал периферически), чем когда морфин вводили непосредственно в ЦНС. (Пример 1, фиг. 2)
Авторы изобретения дополнительно исследовали данные по распределению Соединения I в организме крыс и мышей и отметили, что имела место гораздо более высокая концентрация (в 17 раз больше) в плазме крови, чем в головном мозге. Исследование распределения у мышей, нокаутированных по переносчику P-гликопротеина ("P-gp") выявило, что проникновение из крови в головной мозг повышалось от около 7 до около 19-раз. (Пример 1). Полученные данные показали авторам изобретения, что Соединение I переносилось из ЦНС посредством P-gp переносчика, и что соединение у грызунов по существу распределялось по периферии.
Важно, что авторы изобретения дополнительно определили эффективную дозу Соединения I для лечения ОИЗ или ОДК у людей с использованием множественных подходов, описанных ниже, на основании фармакокинетических и фармакологических данных для Соединения I и для высоко распределяемого по периферии антагониста опиоидных рецепторов алвимопана. Во всех случаях рассчитанная эффективная доза Соединения I для лечения ОИЗ или ОДК была неожиданно существенно ниже дозы, которая влияет на ЦНС, что определяли измеряемыми эффектами на человеческие фармакодинамические маркеры в отношении антагонизма к опиоидным рецепторам в ЦНС. Следовательно, авторы изобретения обнаружили, что Соединение I является эффективным опиоидным антагонистом, который в очень низких дозах достаточно распределяется у людей на периферии, чтобы быть применимым для лечения ОИЗ или ОДК и других состояний, описанных в настоящем описании, где очень желательно селективное ингибирование опиоидных рецепторов на периферии, в противоположность ингибирования и периферических опиоидных рецепторов и рецепторов в ЦНС.
Предварительные результаты клинического исследования I фазы многократных нарастающих доз, проводимого с Соединением I у пациентов с ОИЗ, возникающим в результате хронической терапии опиоидами, показало, что такие низкие дозы, как 0,10 мг и 0,25 мг хорошо переносились и давали желаемые фармакологические эффекты.
4. КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Фиг. 1: Результаты формалинового теста у крыс. Антагонист опиоидных рецепторов центрального действия налтрексон (вводимый подкожно) оказался более чем в 100 раз более эффективным, чем Соединение I (2136231, вводимое перорально) в обратном развитии опиоидного обезболивания у крыс.
Фиг. 2: Результаты перорального введения Соединения I на обратное развитие ингибирования желудочно-кишечного (ЖК) транзита у мышей, вызываемого интрацеребровентрикулярным (непосредственно в ЦНС) введением морфина. Минимальная эффективная доза Соединения I, требуемая для начала обратного развития ингибирующего эффекта вводимого центрально морфина на транзит ЖК составила около 3,0 мг/кг.
Фиг. 3: Моделированный профиль свободного Соединения I, даваемого в виде 0,15 мг пероральной дозы, в плазме относительно времени у людей. "MOR Ki” (сплошная горизонтальная линия) представляет собой измеренную Ki Соединения I для человеческого μ-опиоидного рецептора.
Фиг. 4: Измеренный (3 мг и 10 мг) и смоделированный (0,15 мг и 0,05 мг) свободного Соединения I, даваемого людям в указанных дозах, в плазме относительно времени. MOR Ki” (сплошная горизонтальная линия) представляет собой измеренную Ki Соединения I для человеческого μ-опиоидного рецептора.
Фиг. 5: (Расширение Y-оси Фиг. 4): Измеренный (3 мг и 10 мг) и смоделированный (0,15 мг и 0,05 мг) профиль свободного Соединения I, даваемого людям в указанных дозах, в плазме относительно времени. “MOR Ki” (сплошная горизонтальная линия) представляет собой измеренную Ki Соединения I для человеческого μ-опиоидного рецептора.
Фиг. 6: Профиль свободного Алвимопана в плазме относительно времени (моделирование на основании данных) у пациентов с ОИЗ для пероральной дозы 0,5 мг, т.е. дозы, ассоциированной с эффективностью в клинических исследованиях.
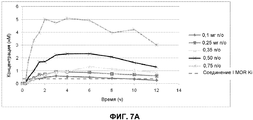
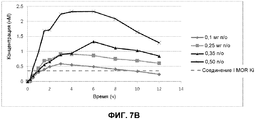
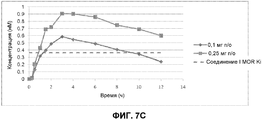
Фиг. 7A-7C: Профили концентрации Соединения I в плазме относительно времени, измеренные в клиническом исследовании I фазы пациентов с ОИЗ в течение интервала введения 12 часов. Уровни Соединения I в плазме измеряли через 15 мин, 30 мин, 60 мин, 90 мин, 2 ч, 3 ч, 4 ч, 6 ч, 8 ч, 10 ч и 12 ч после первого введения. Измеренная MOR Ki Соединения I показана пунктирной линией. Фиг. 7A показывает уровни Соединения I в плазме пациентов для каждой из исследуемых пяти доз. На фиг. 7B показаны уровни Соединения I в плазме пациентов, которым вводили 0,1 мг, 0,25 мг, 0,35 мг и 0,50 мг Соединения I. На фиг. 7C показаны уровни Соединения I в плазме пациентов, которым вводили 0,1 мг и 0,25 мг Соединения I.
5. ПОДРОБНОЕ ОПИСАНИЕ
Настоящее описание относится к способам селективной модуляции активности μ- , δ- и/или κ-опиоидных рецепторов на периферии по сравнению с ЦНС, включающим контакт μ-, δ- и/или κ-опиоидных рецепторов с эффективным количеством 5-(2-метокси-4-{[2-(тетрагидропиран-4-ил)этиламино]метил}фенокси)пиразин-2-карбоксамида (Соединение I). Соединение I ранее было описано как сильный центрально активный опиоидный антагонист. См. Патент США No. 7381719, который таким образом включен в виде ссылки полностью. В различных вариантах осуществления изобретения настоящее описание относится к способам лечения или профилактики состояния, ассоциированного с активацией μ-, δ- и/или κ-опиоидных рецепторов на периферии. В определенных вариантах осуществления изобретения описание относится к способам лечения или профилактики состояния, ассоциированного с активацией μ-опиоидных рецепторов на периферии.
В некоторых вариантах осуществления изобретения настоящее описание относится к способам лечения или профилактики расстройств желудочно-кишечного тракта путем введения пациенту терапевтически эффективного количества Соединения I, или его фармацевтически приемлемой соли. В частности, настоящее описание относится к способам лечения или профилактики расстройств желудочно-кишечного тракта у пациента, возникающих в результате применения опиоидных анальгетиков, без уменьшения обезболивания или развития центральной отмены опиоидов, которые включают введение пациенту терапевтически эффективного количества Соединения I, или его фармацевтически приемлемой соли. В некоторых специфических вариантах осуществления изобретения настоящее описание относится к способам лечения или профилактики ОИЗ у пациента без уменьшения обезболивания, обеспечиваемого опиоидным агонистом, или развития центральной отмены опиоидов, включающим введение пациенту терапевтически эффективного количества Соединения I или его фармацевтически приемлемой соли. В различных вариантах осуществления изобретения настоящее описание относится к способам лечения или профилактики ОДК у пациента без уменьшения обезболивания, обеспечиваемого опиоидным агонистом, или развития центральной отмены опиоидов, включающим введение пациенту терапевтически эффективного количества Соединения I, или его фармацевтически приемлемой соли.
В различных вариантах осуществления изобретения настоящее описание относится к применению Соединения I для получения лекарственного средства для селективной модуляции активности μ-, δ- и/или κ-опиоидных рецепторов на периферии, по сравнению с ЦНС. В различных вариантах осуществления изобретения настоящее описание относится к применению Соединения I для получения лекарственного средства для лечения или профилактики состояния, ассоциированного с активацией μ-, δ- и/или κ-опиоидных рецепторов на периферии. В определенных вариантах осуществления изобретения описание относится к применению Соединения I для получения лекарственного средства для лечения или профилактики состояния, ассоциированного с активацией μ-опиоидных рецепторов на периферии.
В определенных вариантах осуществления изобретения настоящее описание относится к применению эффективного количества Соединения I или его фармацевтически приемлемой соли для получения лекарственного средства для лечения или профилактики заболеваний желудочно-кишечного тракта у пациента. В частности настоящее описание относится к применению Соединения I или его фармацевтически приемлемой соли для получения лекарственного средства для лечения или профилактики желудочно-кишечных расстройств у пациента, возникающих в результате применения опиоидных анальгетиков, без уменьшения обезболивания или развития центральной отмены опиоидов. В некоторых специфических вариантах осуществления изобретения настоящее описание относится к применению Соединения I для получения лекарственного средства для лечения или профилактики ОИЗ у пациента без уменьшения обезболивания, обеспечиваемого опиоидными агонистами, или развития центральной отмены опиоидов. В некоторых специфических вариантах осуществления изобретения настоящее описание относится к применению Соединения I для получения лекарственного средства для лечения или профилактики ОДК у пациента без уменьшения обезболивания, обеспечиваемого опиоидными агонистами, или развития центральной отмены опиоидов.
Фразы "лечение", "процесс лечения", и подобные включают облегчение или прекращение состояния или его симптома, а также ускорение выздоровления от состояния. В одном варианте осуществления изобретения лечение включает ингибирование, например, снижение общей частоты эпизодов состояния или его симптомов.
Фразы "предотвращение", "профилактика", и подобные включают предупреждение развития состояния или его симптомов.
Термин "пациент" включает, но не ограничивается, человека или нечеловекообразное животное, такое как, например, корова, мартышка, павиан, шимпанзе, лошадь, овца, свинья, курица, индейка, перепелка, кошка, собака, мышь, крыса, кролик или морская свинка.
Термин "опиоид", как используется в настоящем описании, относится к классу веществ для лечения боли, который включает натуральные или синтетические соединения, которые связываются с и являются агонистами одного или более μ-, δ- и κ-опиоидных рецепторов.
5.1. Соединение по формуле I
В определенных вариантах осуществления изобретения настоящее описание относится к способу лечения или профилактики состояния у пациента, ассоциированного с активацией периферических опиоидных рецепторов, включающему введение пациенту эффективного количества 5-(2-метокси-4-{[2-(тетрагидропиран-4-ил)этиламино]метил}фенокси)пиразин-2-карбоксамида, имеющего структуру, показанную на формуле (I):

или его фармацевтически приемлемой соли.
Фраза "фармацевтически приемлемая соль", как используется в настоящем описании, представляет собой любую соль, которая может быть получена из Соединения I, включая соль, образованную из основной функциональной группы Соединения I, такой как азотная группа. Иллюстративные соли включают, но не ограничиваются, сульфат, цитрат, ацетат, трифторацетат, оксалат, хлорид, бромид, йодид, нитрат, бисульфат, фосфат, кислый фосфат, изоникотинат, лактат, салицилат, кислый цитрат, тартрат, олеат, таннат, пантотенат, битартрат, аскорбат, сукцинат, малат, малеат, гентисинат, фумарат, глюконат, глюкуронат, сахарат, формат, бензоат, глютамат, метансульфонат, этансульфонат, бензолсульфонат, п-толуолсульфонат, и памоат (т.е. 1,1'-метилен-бис-(2-гидрокси-3-нафтоат)). Специалист в области техники понимает, что аддитивные соли кислот Соединения I могут быть получены путем реакции соединений с соответствующей кислотой посредством множества известных способов.
В определенных вариантах осуществления изобретения описание относится к другим формам Соединения I, таким как пролекарства, радиоактивно меченные формы, сольваты (например, гидраты, или аддитивные соединения, которые содержат кислоту в избытке количества основных атомов азота из-за водородных связей), аморфные солидные формы и кристаллические солидные формы.
Соединение I может быть получено любым способом, известным в области техники. В определенных вариантах осуществления изобретения Соединение I может быть синтезировано с использованием исходных материалов 5-(4-формил-2-метоксифенокси)пиразин-2-карбоксамида и 2-(тетрагидропиран-4-ил)этиламина и протокола и реагентов, указанных в Примере 720 патента США No. 7381719, который таким образом включен в виде ссылки полностью.
5.2. Терапевтическое применение
Благодаря своей активности в качестве периферически-селективного антагониста опиоидных рецепторов, Соединение I преимущественно, применяют у людей и в ветеринарии для лечения или профилактики состояния, которое может быть облегчено селективным антагонизмом в отношении опиоидных рецепторов на периферии. Соединение I можно вводить любому пациенту, требующему ингибирования периферических опиоидных рецепторов. В некоторых вариантах осуществления изобретения Соединение I преимущественно применяют для лечения или профилактики состояния, которое может быть облегчено селективным антагонизмом в отношении периферических μ-опиоидных рецепторов. В различных вариантах осуществления изобретения Соединение I применяют для лечения или профилактики состояния, выбираемого из, но не ограничиваясь, послеоперационной непроходимости кишечника, послеоперационной тошноты и рвоты, опиоид-индуцированной тошноты и рвоты, опиоид-индуцированного подавления дыхания, опиоид-индуцированного запора, опиоид-индуцированной дисфункции кишечника, хронического идиопатического запора, синдрома раздраженного кишечника с преобладанием запора, псевдообструкции кишечника, отложенного опорожнения желудка, непереносимости энтерального питания, наркотической непроходимости кишечника, кишечной непроходимости, ускорения желудочно-кишечного восстановления после операции и опиоид-индуцированного вздутия. В некоторых предпочтительных вариантах осуществления изобретения Соединение I применяют для лечения или профилактики расстройств моторики желудочно-кишечного тракта у пациента. В определенных вариантах осуществления изобретения пациентом является человек.
В некоторых вариантах осуществления изобретения благодаря его сильного и селективного антагонизма в отношении периферических опиоидных рецепторов по сравнению с центральными опиоидными рецепторами, Соединение I применяют для лечения или профилактики состояния у пациента, возникающего в результате применения опиоидов, без уменьшения их обезболивающего эффекта или развития центральной отмены. В определенных вариантах осуществления изобретения, Соединение I применяют для лечения или профилактики ОИЗ без уменьшения обезболивающего эффекта опиоидов или развития центральной отмены опиоидов. В некоторых вариантах осуществления изобретения, Соединение I применяют для лечения или профилактики ОДК без уменьшения обезболивающих эффектов опиоидов или развития центральной отмены опиоидов.
5.3. Терапевтические композиции и способы введения
При введении пациенту Соединение I можно вводить как компонент композиции, которая включает фармацевтически приемлемый носитель или вспомогательное вещество. Композиции, включающие Соединение I, можно вводить перорально. Композиции также можно вводить любым другим удобным путем, например, путем инфузии или болюсной инъекции, путем абсорбции через эпителиальные или кожно-слизистые покровы (например, слизистую полости рта, прямой кишки и кишечника и др.) и можно вводить вместе со вторым терапевтически активным веществом (например, слабительным, размягчителем стула или опиоидным анальгетиком). Введение может быть системным или локальным. Известны различные системы доставки, например, инкапсуляция в липосомы, микрочастицы, микрокапсулы, множества частиц, капсулы и др. могут использоваться для введения Соединения I.
Способы введения включают, но не ограничиваются, внутрикожный, внутримышечный, интраперитонеальный, парентеральный, внутривенный, подкожный, интраназальный, пероральный, сублингвальный, трансдермальный, ректальный, путем ингаляции или местный. В большинстве случаев введение приводит к высвобождению Соединения I в кровоток. В предпочтительных вариантах осуществления изобретения введение не приводит к высвобождению в ЦНС в фармакологически значимых концентрациях.
Легочное введение также может использоваться, например, с использованием ингалятора или небулайзера, и композиции с аэрозольным веществом или посредством перфузии во фторуглероде или синтетическом легочном сурфактанте. В определенных вариантах осуществления изобретения, Соединение I может быть рецептировано в виде суппозитория, с традиционными наполнителями и вспомогательными веществами, такими как триглицериды.
Когда Соединение I включают для парентерального введения путем инъекции (например, непрерывной инфузии или болюсной инъекции), композиция для парентерального ведения может быть в форме суспензии, раствора, эмульсии в масляном или водном носителе и такие композиции могут дополнительно включать фармацевтически необходимые добавки, такие как один или более стабилизирующих агентов, диспергирующих агентов и подобных. Соединение I также может находиться в форме порошка для восстановления в виде инъекционной композиции.
В другом варианте осуществления изобретения Соединение I можно доставлять в пузырьке, в частности липосоме (см. Langer, Science 249: 1527-1533 (1990); и Treat et al., Liposomes in the Therapy of Infectious Disease and Cancer 317-327 and 353-365 (1989)).
В еще одном варианте осуществления изобретения Соединение I можно доставлять в системе с контролируемым высвобождением или системе с непрерывным высвобождением (см., например, Goodson, "Dental Applications" (pp. 115-138) in Medical Applications of Controlled Release, Vol. 2, Applications and Evaluation, R.S. Langer and D.L. Wise eds., CRC Press (1984)). Могут быть использованы другие системы с контролируемым или непрерывным высвобождением, обсуждаемые в обзоре Langer, Science 249: 1527-1533 (1990). В одном варианте осуществления изобретения может быть использован насос (Langer, Science 249: 1527-1533 (1990); Sefton, CRC Crit. Ref. Biomed. Eng. 14:201 (1987); Buchwald et al, Surgery 88:507 (1980); и Saudek et al., N. Engl. J. Med. 321:574 (1989)). В другом варианте осуществления изобретения могут быть использованы полимерные материалы (см. Medical Applications of Controlled Release (Langer and Wise eds., 1974); Controlled Drug Bioavailability, Drug Product Design and Performance (Smolen and Ball eds., 1984); Ranger and Peppas, J. Macromol. Sci. Rev. Macromol. Chem. 23:61 (1983); Levy et al, Science 228: 190 (1985); During et al, Ann. Neurol. 25:351 (1989); и Howard et al, J. Neurosurg. 71: 105 (1989)).
Композиции могут необязательно включать подходящее количество фармацевтически приемлемого вспомогательного вещества, чтобы обеспечить форму для соответствующего введения пациенту. Таким фармацевтическим вспомогательным веществом может быть разбавитель, суспендирующий агент, растворитель, вяжущее вещество, дезинтегрирующее вещество, консервант, краситель, смазывающее вещество и подобные. Фармацевтическое вспомогательное вещество может быть жидкостью, такой как вода или масло, включая таковые нефтяного, животного, растительного или синтетического происхождения, такие как арахисовое масло, соевое масло, минеральное масло, кунжутное масло и подобные. Фармацевтическим вспомогательным веществом может быть солевой раствор, гуммиарабик, желатин, паста крахмала, тальк, кератин, коллоидный диоксид кремния, мочевина и подобные. Кроме того, могут быть использованы добавочные, стабилизирующие, загущающие, смазывающие и красящие средства. В одном варианте осуществления изобретения фармацевтически приемлемое вспомогательное вещество является стерильным при введении пациенту. Вода является особенно применимым вспомогательным веществом, когда Соединение по формуле (I) вводят внутривенно. Солевые растворы и водный раствор декстрозы и глицерин также могут использоваться в качестве жидких вспомогательных веществ, особенно для инъекционных растворов. Подходящие фармацевтические вспомогательные вещества также включают крахмал, глюкозу, лактозу, сахарозу, желатин, солод, рис, муку, мел, силикагель, стеарат натрия, моностеарат глицерина, тальк, хлорид натрия, сухое снятое молоко, глицерин, пропиленгликоль, воду, этанол и подобные. Композиции, при желании, также могут содержать минорные количества увлажняющих или эмульгирующих веществ, или рН буферных веществ. Специфические примеры фармацевтически приемлемых носителей и вспомогательных веществ, которые могут быть использованы для рецептирования пероральных лекарственных форм, описаны в Handbook of Pharmaceutical Excipients, American Pharmaceutical Association (1986).
Композиции, включающие Соединение I, могут иметь форму растворов, суспензий, эмульсий, таблеток, пилюль, гранул, капсул, капсул, содержащих жидкости, порошков, композиций с непрерывным высвобождением, суппозиториев, эмульсий, аэрозолей, спреев, суспензий, сублингвальных разлагающихся лекарственных форм или любых других форм, подходящих для применения. В одном варианте осуществления изобретения композиция находится в форме капсулы (см., например, патент США No. 5698155). Другие примеры подходящих фармацевтических вспомогательных веществ описаны в Remington's Pharmaceutical Sciences 1447-1676 (Alfonso R. Gennaro ed., 19th ed. 1995), включенном в настоящее описание в виде ссылки.
В некоторых вариантах осуществления изобретения Соединение I рецептируют в соответствии с обычными методиками, как композицию, адаптированную для перорального введения людям. Соединение I для перорального введения может быть в форме таблеток, капсул, гелевых капсул, каплетов, пастилок, сублингвальных рассасывающихся твердых лекарственных форм, водных или масляных растворов, суспензий, гранул, порошков, эмульсий, сиропов или эликсиров, например. Когда Соединение I включают в пероральные таблетки, такие таблетки могут быть прессованными таблетками, растертыми таблетками (например, порошковые или разрушенные таблетки), таблетками, покрытыми кишечнорастворимой оболочкой, таблетками, покрытыми сахаром, таблетками, покрытыми пленкой, множественно прессованными таблетками или многослойными таблетками. Методики и композиции для получения твердых лекарственных форм описаны в Pharmaceutical Dosage Forms: Tablets (Lieberman, Lachman and Schwartz, eds., 2nd ed.) опубликованном Marcel Dekker, Inc. Методики и композиции для получения таблеток (прессованных и литых), капсул (твердых и мягких желатиновых) и пилюль также описаны в Remington's Pharmaceutical Sciences 1553-1593 (Arthur Osol, ed., 16th ed., Mack Publishing, Easton, PA 1980).
Жидкие пероральные лекарственные формы включают водные и неводные растворы, эмульсии, суспензии и растворы и/или суспензии, восстанавливаемые из нешипучих гранул, необязательно содержащие один или более подходящих растворителей, консервантов, эмульгирующих средств, суспендирующих средств, разбавителей, подсластителей, красителей, ароматизаторов и подобных. Методики и композиции для получения жидких пероральных лекарственных форм описаны в Pharmaceutical Dosage Forms: Disperse Systems, (Lieberman, Rieger and Banker, eds.), опубликованном Marcel Dekker, Inc.
Перорально вводимая композиция может содержать одно или более средств, например, подсластителей, таких как фруктоза, аспартам или сахарин; ароматизаторов, таких как мята перечная, винтергриновое масло или вишня; красителей; и консервантов для обеспечения фармацевтически приятной на вкус композиции. Более того, имея форму таблеток или пилюль, композиции могут быть покрыты оболочкой для замедления разложения и абсорбции в желудочно-кишечном тракте, таким образом обеспечивая непрерывное действие в течение длительного периода времени. Селективно проницаемые мембраны, окружающие осмотически активное движущееся соединение, также являются подходящими для перорально вводимых композиций. В таких последних случаях жидкость из окружающей капсулу среды пропитывает движущееся соединение, которое набухает, вытесняя вещество или композицию вещества через отверстия. Такие системы доставки могут обеспечивать профиль доставки по существу нулевого порядка в противоположность пиковым профилям композиций с немедленным высвобождением. Задерживающий по времени материал, такой как моностеарат глицерина или стеарат глицерина, также может быть использован. Пероральные композиции могут включать стандартные вспомогательные вещества, такие как маннит, лактоза, крахмал, стеарат магния, сахарин натрия, целлюлоза и карбонат магния. В одном варианте осуществления изобретения вспомогательные средства имеют фармацевтическую категорию.
Перорально вводимые композиции могут быть рецептированы так, чтобы высвобождать активный ингредиент в нижнем отделе желудочно-кишечного тракта и, особенно, в толстой кишке. Такие лекарственные формы для высвобождения активного ингредиента в толстой кишке могут включать полимеры, имеющие pH-зависимую растворимость, которые растворяются при pH толстой кишки, но не при рН тонкой кишки; полимеры, которые имеют медленную или pH-зависимую скорость набухания, растворения или разложения, так что лекарственная форма растворяется в толстой кишке скорее, чем в тонкой кишке; полимеры, которые разлагаются микробными ферментами в толстой кишке; или полимеры, которые образуют слой, который разрушается повышенным давлением в просвете, вызванным перистальтическими волнами. В других вариантах осуществления изобретения композиции включают капсулы гидроксипропилметилцеллюлозы, покрытые оболочкой из материалов, которые обеспечивают высвобождение в толстой кишке, такие как описанные в патенте США No. 7094425. Еще одни композиции для высвобождения в толстой кишке описаны в патенте США No. 6368629 и включают лекарственные формы, которые покрыты кислотолабильной оболочкой, которая растворяется энтеробактериями.
Когда Соединение I необходимо вводить парентерально, оно может находиться, например, в форме изотонического стерильного раствора. Альтернативно, когда Соединение I предназначено для ингаляций, оно может быть рецептировано в сухом аэрозоле или может быть рецептировано в водном или частично водном растворе.
В другом варианте осуществления изобретения Соединение I может быть рецептировано для внутривенного введения. Обычно композиции для внутривенного введения включают стерильный изотонический водный буфер. Когда необходимо, композиции также могут включать растворитель. Композиция для внутривенного введения может необязательно включать местный анестетик, такой как бензокаин или прилокаин, для уменьшения боли в месте инъекции. Обычно, ингредиенты поставляются или раздельно или смешанными вместе в стандартной лекарственной форме, например, в виде сухого лиофилизированного порошка или безводного концентрата в герметически закупоренном контейнере, таком как ампула или саше, показывающих количество активного вещества. Когда Соединение I необходимо вводить путем инфузии, оно может поставляться, например, с инфузионной бутылью, содержащей стерильную воду или солевой раствор фармакологического качества. Когда Соединение I вводят путем инъекции, может быть обеспечена ампула стерильной воды для инъекций или солевого раствора, так что ингредиенты могут быть смешаны перед введением.
В определенных вариантах осуществления изобретения, Соединение I находится в лекарственной форме вместе с опиоидным анальгетиком. Опиоидные анальгетики, которые могут быть рецептированы с Соединением I, включают, но не ограничиваются, алфентанил, бупренорфин, буторфанол, кодеин, дезоцин, дигидрокодеин, фентанил, гидрокодон, гидроморфон, леворфанол, меперидин (петидин), метадон, морфин, налбуфин, оксикодон, оксиморфон, пентазоцин, пропирам, пропоксифен, суфентанил и трамадол. Другие опиоидные анальгетики для включения в лекарственную форму, содержащую Соединение I, очевидны специалисту в области техники.
5.4. Терапевтические дозировки
Количество Соединения I, которое является эффективным для лечения ОИЗ или ОДК у людей, экстраполировано из фармакокинетических и фармакологических данных с использованием расчетов, основанных на множестве подходов - например, (i) сравнение фармакокинетических и фармакологических данных для Соединения I с фармакокинетическими и фармакологическими данными для распределяемого по периферии антагониста опиоидных рецепторов алвимопана из исследований эффективности у людей при ОИЗ, или (ii) расчеты, основанные на дозировке Соединения I, которая обеспечит максимальные свободные концентрации в плазме (Cmax), равные или более чем Ki Соединения I для μ-опиоидного рецептора, как указано ниже в примере 2. С использованием множества подходов, общая дозировка Соединения I, которая является эффективной в лечении ОИЗ или ОДК без антагонизма в отношении μ-опиоидных рецепторов в ЦНС и, следовательно, уменьшения опиоидного обезболивания или развития центральной отмены опиоидов, рассчитывали как приблизительно 0,1 мг/день, для введения или один или два раза в сутки. Количество Соединения I, эффективное для лечения ОИЗ или ОДК, которое экстраполируется из фармакокинетических и фармакологических данных, как описано выше, подтверждали в клинических исследованиях I фазы у пациентов с ОИЗ как приблизительно 0,1 мг/сутки.
В определенных вариантах осуществления изобретения крайняя доза, т.е. кратность различия между рассчитанной эффективной дозой Соединения I для лечения ОИЗ или ОДК и средней эффективной дозой, определяемой путем экспериментов, требуемых для противодействия эффектам морфина в ЦНС, составляет от около 25 до около 83. См. пример 2. Соответственно было обнаружено, что в низких дозах, требуемых для лечения ОИЗ или ОДК, Соединение I значительно распределяется на периферии и, следовательно, маловероятно, чтобы оно влияло на опиоидное обезболивание или вызывало центральную отмену опиоидов.
Подходящие эффективные дозировки для лечении ОИЗ или ОДК или других состояний, описанных в настоящем описании, у среднего мужчины (например, массой около 70 кг), варьируются от около 0,01 мг до около 1,5 мг дозы, полученной всем организмом, в сутки. В определенных вариантах осуществления изобретения подходящие эффективные дозировки варьируются от около 0,02 мг до около 1,25 мг дозы, полученной всем организмом, в сутки, например, от около 0,03 мг до около 1,0 мг дозы, полученной всем организмом, в сутки, например, от около 0,04 мг до около 0,75 мг дозы, полученной всем организмом, в сутки, например, от около 0,05 до около 0,5 мг дозы, полученной всем организмом, в сутки.
В определенных вариантах осуществления изобретения подходящая эффективная дозировка варьируется от около 0,1 до около 1 мг дозы, полученной всем организмом, в сутки. В различных вариантах осуществления изобретения подходящая эффективная дозировка варьируется от около 0,1 до около 0,75 мг дозы, полученной всем организмом в сутки, например, от около 0,1 до около 0,5 мг дозы, полученной всем организмом в сутки, например, от около 0,1 до около 0,35 мг дозы, полученной всем организмом в сутки, например, от около 0,1 до около 0,25 мг дозы, полученной всем организмом в сутки.
В различных вариантах осуществления изобретения подходящие эффективные дозировки варьируются от около 0,01 мг до около 1,0 мг дозы, полученной всем организмом в сутки, например, от около 0,02 мг до около 0,9 мг дозы, полученной всем организмом в сутки, например, от около 0,03 мг до около 0,8 мг дозы, полученной всем организмом в сутки, например, от около 0,04 мг до около 0,7 мг дозы, полученной всем организмом в сутки, например от около 0,05 мг до около 0,6 мг дозы, полученной всем организмом в сутки, например, от около 0,06 мг до около 0,5 мг дозы, полученной всем организмом в сутки, например, от около 0,07 мг до около 0,4 мг дозы, полученной всем организмом в сутки, например от около 0,08 мг до около 0,3 мг дозы, полученной всем организмом в сутки.
В еще дополнительных вариантах осуществления изобретения подходящие эффективные дозировки для лечения ОИЗ или ОДК или других состояний, описанных в настоящем описании у среднего мужчины (например, массой около 70 кг) варьируются от около 0,05 мг до около 0,5 мг дозы, полученной всем организмом в сутки, например, от около 0,06 мг до около 0,25 мг дозы, полученной всем организмом в сутки, например, от около 0,07 мг до около 0,2 мг дозы, полученной всем организмом в сутки, например, от около 0,08 мг до около 0,15 мг дозы, полученной всем организмом в сутки.
В различных вариантах осуществления изобретения подходящая эффективная дозировка для лечения ОИЗ или ОДК или других состояний, описанных в настоящем описании, у среднего мужчины (например, массой около 70 кг) варьируется от около 0,01 мг до около 0,5 мг дозы, полученной всем организмом два раза в сутки (BID), например, от около 0,02 мг до около 0,45 мг дозы, полученной всем организмом два раза в сутки, например, от около 0,03 мг до около 0,4 мг дозы, полученной всем организмом два раза в сутки, например, от около 0,04 мг до около 0,35 мг дозы, полученной всем организмом два раза в сутки, например, от около 0,05 мг до около 0,3 мг дозы, полученной всем организмом два раза в сутки, например, от около 0,06 мг до около 0,25 мг дозы, полученной всем организмом два раза в сутки, например, от около 0,07 мг до около 0,2 мг дозы, полученной всем организмом два раза в сутки, например, от около 0,08 мг до около 0,15 мг дозы, полученной всем организмом два раза в сутки.
Подходящие эффективные дозировки для лечения ОИЗ или ОДК или других состояний, описанных в настоящем описании, варьируются от около 0,0001 мг/кг/сутки до около 0,02 мг/кг/сутки. В определенных вариантах осуществления изобретения подходящие эффективные дозировки варьируются от около 0,0003 мг/кг/сутки до около 0,017 мг/кг/сутки, например, от около 0,0004 мг/кг/сутки до около 0,014 мг/кг/сутки, например, от около 0,0005 мг/кг/сутки до около 0,01 мг/кг/сутки, например, от около 0,0007 мг/кг/сутки до около 0,007 мг/кг/сутки.
В различных вариантах осуществления изобретения подходящие эффективные дозировки варьируются от около 0,0001 мг/кг/сутки до около 0,014 мг/кг/сутки, например, от около 0,0003 мг/кг/сутки до около 0,013 мг/кг/сутки, например, от около 0,0004 мг/кг/сутки до около 0,011 мг/кг/сутки, например, от около 0,0006 мг/кг/сутки до около 0,01 мг/кг/сутки, например, от около 0,0007 мг/кг/сутки до около 0,009 мг/кг/сутки, например, от около 0,0009 мг/кг/сутки до около 0,007 мг/кг/сутки, например, от около 0,001 мг/кг/сутки до около 0,006 мг/кг/сутки, например, от около 0,001 мг/кг/сутки до около 0,004 мг/кг/сутки.
В других вариантах осуществления изобретения подходящие дозировки для лечения ОИЗ или ОДК или других состояний, описанных в настоящем описании, варьируются от около 0,0007 мг/кг/сутки до около 0,007 мг/кг/сутки, например, от около 0,0009 мг/кг/сутки до около 0,004 мг/кг/сутки, например, от около 0,001 мг/кг/сутки до около 0,003 мг/кг/сутки, например, от около 0,001 мг/кг/сутки до около 0,002 мг/кг/сутки.
В различных вариантах осуществления изобретения подходящие эффективные дозировки для лечения ОИЗ или ОДК или других состояний, описанных в настоящем описании, варьируются от около 0,00015 мг/кг до около 0,007 мг/кг два раза в сутки, например, от около 0,0003 мг/кг до около 0,006 мг/кг два раза в сутки, например, от около 0,0004 мг/кг до около 0,004 мг/кг два раза в сутки, например, от около 0,0006 мг/кг до около 0,005 мг/кг два раза в сутки, например, от около 0,0007 мг/кг до около 0,004 мг/кг два раза в сутки, например, от около 0,0009 мг/кг до около 0,004 мг/кг два раза в сутки, например, от около 0,001 мг/кг до около 0,003 мг/кг два раза в сутки, например, от около 0,001 мг/кг до около 0,002 мг/кг два раза в сутки.
В определенных вариантах осуществления изобретения Соединение I вводят пациенту, который чувствителен к его эффектам и, следовательно, таким образом требует более низкой дозы для получения эффекта в лечении состояния, как описано в настоящем описании. В некоторых вариантах осуществления изобретения более низкая эффективная доза дает свободную концентрацию Соединения I в плазме, которая составляет около 0,75Ki. В других вариантах осуществления изобретения более низкая эффективная доза дает свободную концентрацию в плазме, которая составляет около 0,5 Ki. В дополнительных вариантах осуществления изобретения более низкая эффективная доза дает свободную концентрацию в плазме, которая составляет около 0,33Ki. В еще дополнительных вариантах осуществления изобретения более низкая эффективная дозировка дает свободную концентрацию в плазме Соединения I, которая составляет около 0,75Kb. В еще дополнительных вариантах осуществления изобретения более низкая эффективная доза дает свободную концентрацию в плазме, которая составляет около 0,5Kb. В различных вариантах осуществления изобретения более низкая эффективная доза дает концентрацию в плазме, которая составляет около 0,33Kb.
Понимают, что количество Соединения I, которое является эффективным для лечения или профилактики состояния, описанного в настоящем описании, может быть определено стандартными клиническими методиками. Кроме того, исследования in vitro и/или in vivo необязательно могут быть использованы для улучшения идентификации оптимальных диапазонов дозировок. Точная используемая дозировка также будет зависеть, например, от пути введения, композиции, выраженности заболевания, и уровня наркотического обезболивания, и может быть определена в соответствии с мнением практикующего врача и/или обстоятельств каждого пациента. В других примерах варианты обязательно возникают в зависимости от, среди других факторов, массы и физического состояния (например, функции печени и почек, тяжести основного заболевания) пациенту, получающего лечение, заболевания, подвергаемого лечению, тяжести симптомов, частоты интервала введения и наличия каких-либо опасных побочных эффектов.
Понятно, что в некоторых вариантах осуществления изобретения дозировку в сутки можно вводить в виде разовой дозы. В других вариантах осуществления изобретения суточную дозировку можно вводить в виде поделенной дозы. В других вариантах осуществления изобретения дозу Соединения I можно вводить по необходимости (PRN).
В определенных вариантах осуществления изобретения Соединение I вводят перед или после введения опиоидного анальгетика. В других вариантах осуществления изобретения Соединение I вводят одновременно с введением опиоидного анальгетика. В некоторых вариантах осуществления изобретения Соединение I и опиоидный анальгетик можно вводить тем же путем введения (например, перорально). В других вариантах осуществления изобретения Соединение I и опиоидный анальгетик вводят различными путями введения. В определенных вариантах осуществления изобретения, когда Соединение I и опиоидный анальгетик вводят вместе, опиоидный анальгетик и Соединение I вводят в лекарственной форме, содержащей разовую дозу (например, в капсуле, совместно рецептированной таблетке или рассасываемой сублингвальной твердой лекарственной форме).
6. ПРИМЕРЫ
Пример 1: Фармакокинетика и фармакология Соединения I в неклинических моделях
Эффективность Соединения I в ЦНС по сравнению с налтрексоном
Крысам вводили раздражающий формалин и 10 мг/кг морфина подкожно. Степень обезболивания определяли по снижению количества раз, которое крыса лизала или обращала внимание на место введения формалина (события). Крысам вводили или 0,02 мг/кг налтрексона (подкожная доза) или 1, 3 или 10 мг/кг Соединения I (пероральная дозировка) для определения способности соединений отменять обезболивающий эффект морфина. Было обнаружено, что налтрексон приблизительно в 100 раз более эффективен, чем Соединение I в отмене эффектов морфина в ЦНС крыс. (Фиг. 1) Налтрексон и Соединение I имели сравнимое сродство к μ-опиоидным рецепторам. Отдельные эксперименты показали, что Соединение I имеет высокую пероральную биодоступность у крыс. Обратное развитие индуцированного морфином обезболивания посредством антагониста опиоидных рецепторов в формалиновом тесте, вероятно, происходит преимущественно за счет центрально-опосредованного эффекта. Несоответствие эффективности предполагает, что Соединение I имеет существенно более низкий уровень проникновения в ЦНС, чем налтрексон.
Эффект Соединения I в ЦНС и на периферии
Мышам вводили или 10 мкг/кг морфина в желудочки головного мозга или 1 мг/кг морфина подкожно. Степень желудочно-кишечного транзита угольной пищи у мышей измеряли в отсутствие Соединения I и в присутствии различных доз Соединения I, чтобы определить способность Соединения I вызывать обратное развитие ингибирования желудочно-кишечного (ЖК) транзита, индуцированного морфином. Минимальную эффективную дозу Соединения I, требуемую для обратного развития ингибирования ЖК транзита у мышей, которым вводили морфин непосредственно в ЦНС, определяли как пероральную дозу около 3,0 мг/кг. (Фиг.2). Наоборот, эффективная доза Соединения I, требуемая для обратного развития ингибирования ЖК транзита у мышей, которым вводили морфин системно, определяли как пероральную дозу около 0,16 мг/кг. Следовательно, Соединение I было приблизительно в 20 раз более сильным в обратном развитии периферических эффектов морфина в замедлении ЖК транзита по сравнению с центральными эффектами морфина в замедлении ЖК транзита. Это предполагает, что Соединение I было около 20 раз более эффективным в антагонизме в отношении опиоидных рецепторов на периферии, чем в ЦНС.
Распределение Соединения I в плазме и головном мозге мышей дикого типа и нокаутированных по P-гликопротеиновому переносчику
Мышам дикого типа или нокаутированным по P-гликопротеину (P-gp) вводили 2,5 мг/кг Соединения I внутривенно. Мышей умерщвляли и количество Соединения I в крови и в головном мозге оценивали с использованием стандартных методик. Результаты проведенных экспериментов показали, что в плазме крови мышей дикого типа было обнаружено в 17 раз больше Соединения I, чем в головном мозге. Более того, проникновение Соединения I через гематоэнцефалический барьер увеличивалось от 7 до 19 раз у нокаутированных мышей, которые были дефицитны по P-gp переносчику. Результаты проведенных экспериментов показали, что Соединение I активно переносилось из ЦНС посредством P-gp, приводя к статическому выведению Соединения I из ЦНС, и что Соединение I, следовательно, у мышей существенно распределялось на периферии.
Пример 2: Рассчитанная эффективная доза Соединения I для лечения ОИЗ или ОДК у людей и определение достаточности распределения по периферии, при отсутствии влияния на центральное обезболивание или развития центральной отмены опиоидов
Неклинические исследования на животных, описанные выше, показали, что Соединение I проявляет некоторую степень распределения по периферии у мышей и крыс, и дополнительно предварительно обозначили основной механизм(ы), посредством которых Соединение I выводится из ЦНС, т.е. Соединение I является субстратом для выводящего переносчика P-gp. Однако ключевые вопросы представляют собой следующие, являются ли полученные данные значимыми для людей, и какой вклад они могут оказывать на терапевтическое применение Соединения I для лечения состояний у людей.
Главный вопрос представляет собой следующий, является ли Соединение I у людей достаточно распределяемым по периферии, чтобы быть целесообразным для лечения состояний, в частности ОИЗ или ОДК, полагаясь на селективный антагонизм в отношении опиоидных рецепторов на периферии по сравнению с антагонизмом в отношении опиоидных рецепторов в ЦНС, что будет мешать центрально-опосредованному обезболиванию и вызывать неприятные и потенциально опасные симптомы отмены опиоидов. Для решения этой проблемы, необходимо определить следующее: (i) наименьшую дозу ("пороговую дозу") Соединения I, которая дает антагонизм в отношении опиоидных рецепторов в ЦНС; и (ii) дозы Соединения I, которые, вероятно, дают достаточный уровень антагонизма в отношении опиоидных рецепторов в периферических тканях для эффективного лечения ОИЗ или ОДК.
Расчет наименьшей дозы Соединения I, которая дает антагонизм в отношении опиоидных рецепторов в ЦНС
Определение наименьшей дозы Соединения I, которая дает антагонизм в отношении опиоидных рецепторов, осуществляли непосредственно путем изучения сообщаемого эффекта Соединения I на фармакодинамические маркеры антагонизма в отношении центральных опиоидных рецепторов, который оценивали в клинических исследованиях Фазы I с разовой и множеством доз, т.е., обратного развития индуцированного морфином миоза (действие на диаметр зрачка) и увеличение адренокортикотропного гормона (АКТГ) и кортизола. Наблюдаемая доза Соединения I, полученная всем организмом, ассоциированная с пороговым обращением индуцированного морфином миоза, составила 10 мг. Более того, наблюдаемая доза Соединения I, полученная всем организмом, ассоциированная с пороговым увеличением АКТГ и кортизола, составила между 10 и 25 мг. Затем проводили консервативную оценку дозы, полученной всем организмом, для антагонизма в отношении центральных опиоидных рецепторов путем уменьшения таких наблюдаемых пороговых доз с помощью полулогарифмического фактор {т.е., 3-кратно). Следовательно, минимальные дозы Соединения I, ассоциированные с фармакологически значимым антагонизмом в отношении опиоидных рецепторов в ЦНС, были консервативно оценены как находящиеся в диапазоне от около 3 мг до около 10 мг.
Расчет дозы Соединения I, которая ожидаемо даст антагонизм в отношении опиоидных рецепторов в периферических тканях, достаточный для эффективного лечения ОИЗ или ОДК
Существует ряд подходов к определению эффективной дозы Соединения I для лечения ОИЗ или ОДК. Четыре перечислены ниже. В зависимости от подхода делают ряд основных предположений в расчете эффективной дозы Соединения I для лечения ОИЗ или ОДК: (i) μ-опиоидный рецептор является основной мишенью Соединения I для получения эффективности в лечении ОИЗ или ОДК, хотя другие опиоидные рецепторы также могут играть некоторую роль, (ii) Системное присутствие Соединения I на периферии (но не в ЦНС) достаточно для получения устойчивой эффективности в лечении ОИЗ или ОДК. Следовательно, расчеты основаны на системных концентрациях в плазме в противоположность возможным местным или локальным эффектам в ткани кишечника, хотя это также может вносить некоторый вклад в эффективность, (iii) Фармакокинетические свойства, особенно пероральная биодоступность Соединения I будут приблизительно сходными у здоровых добровольцев и у пациентов с ОИЗ или ОДК. (iv) Фармакокинетические параметры (в частности Cmax, период полужизни (t1/2) и площадь под кривой (AUC) для Соединения I продолжают быть дозозависимыми при нисходящем диапазоне доз приблизительно от 1 мг до 0,05 мг. Допущения (iii) и (iv) были целесообразными, т.к. было показано, что Соединение I имеет высоко согласованную и дозозависимую пероральную фармакокинетику в диапазоне доз от 1 мг до 100 мг. Более того, последующие эксперименты показали, что Соединение I имеет приблизительно 100% пероральную биодоступность у зеленых макак в диапазоне доз от 0,1 мг/кг до 0,5 мг/кг.
Первым подходом был подсчет, основанный на сравнении фармакокинетических и фармакологических данных для высоко распределяемого на периферии антагониста опиоидных рецепторов алвимопана. В частности, фармакокинетические и фармакологические данные, определенные для Соединения I, сравнивали с фармакокинетическими и фармакологическими данными для алвимопана, полученными в исследованиях эффективности 2 фазы для лечения пациентов с ОИЗ или ОДК. Webster et al. (2008) Pain 137:428-440. Вопросом, требующим ответа, было, какая доза Соединения I даст сравнимые системные уровни антагонизма в отношении μ-опиоидных рецепторов на периферии, что является функцией афинности двух лекарственных препаратов (Ki) в отношении μ-опиоидных рецепторов, концентраций лекарственных средств (оцениваемых по значениям Cmax) в плазме, и выраженности связывания с белками плазмы. Ki алвимопана и Соединения I определяли путем связывания рецептора in vitro в мембранах клеток CHO, экспрессирующих клонированные человеческие μ-опиоидные рецепторы. Как показано в таблице 1, Ki алвимопана к μ-опиоидным рецепторам составила 0,27 нМ и Ki Соединения I к μ-опиоидным рецепторам составила 0,36 нМ. Средний уровень в плазме свободного алвимопана Cmax после эффективной дозы 0,5 мг два раза в сутки алвимопана пациентам, страдающим от ОИЗ или ОДК в клинических исследованиях 2 фазы измеряли как приблизительно 0,1 нг/мл. Средний свободный уровень в плазме Cmax после 1 мг разовой пероральной дозы Соединения I здоровым лицам измеряли как 1,15 нг/мл. Для таких исходных расчетов свободное лекарственное средство оценивали с использованием связывания с мышиными белками плазмы, как замену связывания с человеческими белками плазмы; последующие экспериментальные данные подтвердили, что такие значения были действительно сравнимыми (см. Таблицу 1).
| Таблица 1 Фармакологические и фармакокинетические данные алвимопана и Соединения I |
||
| Алвимопан | Соединение I | |
| μ-опиоидный рецептор К1 (нМ) | 0,27 | 0,36 |
| Средний свободный уровень в плазме Cmax (нг/мл) | 0,11 | 1,152 |
| Связывание с белками плазмы (% свободного лекарственного средства) | 20,3 (человек) | 643 (мышь) 73,8 (человек) |
| 1 После 0,5 мг 2 раза в сутки (ДИ 0,03-0,25) 2 После дозы 1 мг (ДИ 1,66-2,16) 3 Были проведены исходные расчеты с использованием данных у мышей, предполагая, что человеческие значения будут сравнимыми. |
||
Исследования 1 фазы показали, что Соединение I имеет приблизительно линейные взаимоотношения между дозой и Cmax в плазме и площадью под кривой (AUC) в диапазоне доз от 1 до 100 мг. Доза Соединения I, необходимая для достижения Cmax равной 0,1 нг/мл (т.е. свободной концентрации алвимопана в плазме, эффективной для лечения ОИЗ или ОДК), следовательно, может быть оценена путем линейного экстраполирования на более низкие дозы, т.е. предполагая линейное взаимоотношение между дозой и Cmax в дозах менее чем 1 мг. С использованием такого подхода доза Соединения I, необходимая для получения Cmax свободного лекарственного средства в плазме 0,1 нг/мл, составила 0,087 мг. Относительную Ki двух Соединений (таблица 1) учитывали в определении дозы Соединения I, требуемой для достижения фармакологически эквивалентных уровней антагонизма в отношении μ-опиоидных рецепторов до такового, ассоциированного с клинически эффективной дозой алвимопана. Посредством простой пропорции полученное указывает, что неожиданно низкая доза Соединения I 0,12 мг будет эффективной в лечении ОИЗ или ОДК. Важно, что такая доза является в 25-83 раза ниже, чем дозы, ассоциированные с антагонизмом в отношении центральных опиоидных рецепторов, ясно показывая, что, при наличии центральной активности в более высоких дозах, Соединение I является все еще достаточно распределенным по периферии для лечения ОИЗ или ОДК в очень низких дозах, не влияя на центральное опиоидное обезболивание или не вызывая центральной отмены опиоидов.
Вторым подходом был расчет, основанный на эффективности (Κi) Соединения I, в качестве антагониста в отношении μ-опиоидных рецепторов. В таком случае фармакокинетические и фармакологические данные для Соединения I использовали для определения дозы Соединения, которая даст системную концентрацию свободного лекарственного средства в плазме (Cmax) на периферии, равную Κi для μ-опиоидных рецепторов (т.е., 0,36 нМ, как определено посредством связывания in vitro человеческого μ-опиоидного рецептора в клетках CHO - таблица 1). Как обсуждается выше, средний свободный уровень в плазме Cmax после 1 мг разовой пероральной дозы Соединения I здоровым людям измеряли как 1,15 нг/мл, или 2,98 нМ (молекулярная масса Соединения I: 386,4 г/моль). Предполагая линейное взаимоотношение между дозой и Cmax для доз ниже 1 мг, дозу Соединения I, необходимую для достижения 0,36 нМ свободного лекарственного средства, рассчитывали как неожиданно низкую дозу, полученную всем организмом 0,12 мг. Также полученная доза была в 25-83 раза ниже, чем дозы, ассоциированные с антагонизмом в отношении центральных опиоидных рецепторов, аналогично показывая, что при наличии центральной активности в более высоких дозах, Соединение I все еще достаточно распределяется по периферии для лечения ОИЗ или ОДК в очень низких дозах, не влияя на центральное опиоидное обезболивание или не вызывая центральной отмены опиоидов.
Третьим подходом было создание фармакокинетической модели для Соединения I в низких дозах. Это было осуществлено путем предположения линейного взаимоотношения между дозой и всеми параметрами времени в плазме для дозы Соединения I 1 мг, и экстраполируя их на более низкие дозы. Такая модель показывает, что после введения людям 0,15 мг Соединения I свободные концентрации в плазме Соединения были около или выше Ki к μ-опиоидным рецепторам (0,36 нМ) в течение около 6 часов (Фиг. 3), т.е., достаточно времени для получения эффективного сигнала. Полученная доза является от 20 до 67 раз ниже, чем дозы, ассоциированные с антагонизмом в отношении центральных опиоидных рецепторов, снова показывая, что, при наличии центральной активности в более высоких дозах, Соединение I является достаточно распределенным по периферии для лечения ОИЗ или ОДК в очень низких дозах, не влияя на центральное опиоидное обезболивание или не вызывая центральную отмену опиоидов (Фиг. 4 и фиг. 5).
Четвертым подходом было оценить клиническую фармакокинетическую модель для алвимопана, которая предполагает период воздействия концентрации свободного лекарственного средства, которая составляет только приблизительно 0,33 Ki для μ-опиоидных рецепторов, может быть достаточной для получения эффективности у пациентов с ОИЗ. (Фиг. 6) С использованием фармакокинетической модели и предположений, описанных выше, доза Соединения I 0,5 мг приводила к свободным концентрациям в плазме около или выше 0,33 Ki μ-опиоидного рецептора (0,36 нМ) в течение более чем 4 часов, т.е. достаточного времени для получения сигнала эффективности. Такая доза была в 60-200 раз ниже, чем дозы, ассоциированные с антагонизмом в отношении центральных опиоидных рецепторов, снова показывая, что, при наличии центральной активности в более высоких дозах, Соединение I все еще является достаточно распределяемым по периферии для лечения ОИЗ или ОДК не влияя на центральное опиоидное обезболивание или не вызывая центральной отмены опиоидов.
В дозах 1-3 мг, Соединение I имеет измеряемый период полужизни в плазме у людей приблизительно 12 часов. Если существует возможность в некоторой степени накопления лекарственного средства при схемах повторного введения, дозировки, ассоциированные с эффективностью для лечения ОИЗ или ОДК могут быть ощутимо ниже, чем дозы, рассчитанные выше, т.е. такими как от 0,01 до 0,02 мг дозы, полученной всем организмом один или два раза в сутки (для мужчины весом приблизительно 70 кг).
Пример 3: Клиническое исследование I фазы с многократно нарастающими дозами, проводимое с Соединением I; предварительные результаты
Целью клинического исследования I фазы с множеством нарастающих доз было оценить безопасность, переносимость, фармакокинетику и клинический эффект многократно нарастающих доз Соединения I, вводимых два раза в сутки (BID), пациентам, которые имеют опиоид-индуцированные запоры, (ОИЗ), как результат хронической терапии опиоидами для постоянной нераковой боли. Первой частью исследования было рандомизированное, двойное слепое, плацебо контролируемое исследование с многократно нарастающими дозами в течение которого пациенты получали 4 пероральных дозы исследуемого средства в течение 2 дней, относящихся к единице исследования. Каждая из групп доз 0,10 мг, 0,25 мг, 0,35 мг, 0,50 мг и 0,75 мг два раза в сутки включала 4 пациентов, и рандомизация была несбалансированна, с соотношением 3:1 лечения Соединением I (n=3) к лечению плацебо (n=1). Группа 0,75 мг два раза в сутки включала только 2 пациентов. Оценки клинических эффектов включали следующие показатели, которые оценивали начиная непосредственно после первой дозы исследуемого средства до отмены в день 3: (i) время до первого опорожнения кишечника после первой дозы исследуемого средства; (ii) количество опорожнений кишечника; и (iii) баллы комфорта опорожнений кишечника, масса стула и консистенция стула, измеренные по Bristol Stool Scale для каждого опорожнения кишечника, которые происходили, когда пациент относился к группе исследования.
Для фармакокинетических анализов образцы крови получали от каждого пациента в день 1 приблизительно за 1 час до первого введения средства исследования (время 0) и в каждый из последующих моментов времени после первого введения средства исследования: 15 мин, 30 мин, 60 мин, 90 мин, 2 ч, 3 ч, 4 ч, 6 ч, 8 ч, 10 ч, и 12 ч. Фиг. 7 A, 7B и 7C показывают уровень в плазме Соединения I, полученный после первой дозы в течение периода введения 12 ч и для каждой из 5 исследуемых доз (0,10 мг, 0,25 мг, 0,35 мг, 0,50 мг и 0,75 мг два раза в сутки). Как показано на фиг. 7A, все дозировки обеспечивали концентрации в плазме, которые были выше MOR Ki Соединения I [Ki (MOR) = 0,36 нМ], что показано пунктирной линией. Только в наименьшей дозе (0,1 мг) концентрация падала ниже MOR Ki перед окончанием интервала введения через 12 часов после первого введения; и даже в этой дозе концентрации были выше MOR Ki в течение почти 9 часов {см. фиг. 7C). Предварительные данные показывают, что дозы 0,10 мг и 0,25 мг хорошо переносились и давали желательные фармакологические эффекты.
Все публикации, патенты, патентные заявки и другие документы, представленные в настоящей заявке, таким образом включены в виде ссылки полностью для любых целей в той степени, как будто каждая отдельная публикация, патент, патентная заявка или другой документ отдельно предназначены для включения в виде ссылки для любых целей.
Тогда как были проиллюстрированы и описаны различные специфические варианты осуществления изобретения, понимают, что различные изменения могут быть сделаны без отклонения от рамок и тенденции изобретения(й).
Claims (12)
1. Фармацевтическая композиция в стандартной лекарственной форме для лечения опиоид-индуцированного запора, содержащая от около 0,01 мг до около 0,5 мг соединения формулы (I):

или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель, вспомогательное вещество или разбавитель.
или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель, вспомогательное вещество или разбавитель.
2. Фармацевтическая композиция по п. 1, которая пригодна для перорального введения.
3. Фармацевтическая композиция по п. 1, которая представляет собой таблетку.
4. Фармацевтическая композиция по любому одному из пп. 1-3, которая содержит около 0,25 мг соединения формулы (I) или его фармацевтически приемлемой соли.
5. Фармацевтическая композиция по любому одному из пп. 1-3, которая содержит около 0,5 мг соединения формулы (I) или его фармацевтически приемлемой соли.
6. Способ лечения опиоид-индуцированного запора, включающий введение пациенту терапевтически эффективного количества соединения формулы (I):

или его фармацевтически приемлемой соли в количестве в диапазоне от около 0,01 мг до около 0,5 мг в сутки.
или его фармацевтически приемлемой соли в количестве в диапазоне от около 0,01 мг до около 0,5 мг в сутки.
7. Способ по п. 6, где соединение формулы (I) вводят вместе с опиоидным анальгетиком.
8. Способ по п. 6 или 7, где соединение формулы (I) или его фармацевтически приемлемую соль вводят пациенту в количестве около 0,5 мг в сутки.
9. Применение соединения формулы (I):

или его фармацевтически приемлемой соли для лечения опиоид-индуцированного запора, где соединение формулы (I) или его фармацевтически приемлемую соль вводят перорально два раза в сутки в общем количестве от 0,1 до 0,5 мг в сутки.
или его фармацевтически приемлемой соли для лечения опиоид-индуцированного запора, где соединение формулы (I) или его фармацевтически приемлемую соль вводят перорально два раза в сутки в общем количестве от 0,1 до 0,5 мг в сутки.
10. Применение по п. 9, где соединение формулы (I) или его фармацевтически приемлемую соль вводят пациенту в количестве 0,5 мг в сутки.
11. Применение по п. 9, где соединение формулы (I) или его фармацевтически приемлемую соль вводят пациенту в количестве 0,25 мг.
12. Применение по любому одному из пп. 9-11, где пациент одновременно получает опиоидный анальгетик.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US24361609P | 2009-09-18 | 2009-09-18 | |
| US61/243,616 | 2009-09-18 | ||
| PCT/US2010/049311 WO2011035142A1 (en) | 2009-09-18 | 2010-09-17 | Use of opioid receptor antagonist for gastrointestinal tract disorders |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| RU2012115450A RU2012115450A (ru) | 2013-10-27 |
| RU2561873C2 true RU2561873C2 (ru) | 2015-09-10 |
Family
ID=43216894
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| RU2012115450/15A RU2561873C2 (ru) | 2009-09-18 | 2010-09-17 | Применение антагонистов опиоидных рецепторов при заболеваниях желудочно-кишечного тракта |
Country Status (19)
| Country | Link |
|---|---|
| US (1) | US20110071163A1 (ru) |
| EP (1) | EP2477627B1 (ru) |
| JP (1) | JP5797655B2 (ru) |
| KR (1) | KR20120092592A (ru) |
| CN (2) | CN104958298A (ru) |
| AU (1) | AU2010295464B2 (ru) |
| BR (1) | BR112012006069A8 (ru) |
| CA (1) | CA2774021A1 (ru) |
| CL (1) | CL2012000676A1 (ru) |
| CO (1) | CO6531453A2 (ru) |
| HK (1) | HK1210959A1 (ru) |
| IL (1) | IL218679A0 (ru) |
| MX (1) | MX342548B (ru) |
| NZ (1) | NZ598783A (ru) |
| PE (1) | PE20120991A1 (ru) |
| RU (1) | RU2561873C2 (ru) |
| TW (1) | TW201118084A (ru) |
| WO (1) | WO2011035142A1 (ru) |
| ZA (1) | ZA201201954B (ru) |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DK2760880T3 (en) * | 2012-12-28 | 2015-09-28 | Inst Medycyny Doswiadczalnej I Klinicznej | Peptidomimetics and use thereof |
| KR20190009792A (ko) | 2016-05-20 | 2019-01-29 | 에스테베 파마슈티칼스 에스에이 | 통증에 대한 다중 활성을 갖는 테트라하이드로피란 및 티오피란 유도체 |
| CN106117190B (zh) * | 2016-06-21 | 2019-03-29 | 山东川成医药股份有限公司 | 一种倍福普兰的合成方法 |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20080255152A1 (en) * | 2002-09-19 | 2008-10-16 | Maria-Jesus Blanco-Pillado | Diaryl ethers as opioid receptor antagonists |
Family Cites Families (42)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4191771A (en) * | 1974-09-06 | 1980-03-04 | Eli Lilly And Company | Analgesic method using 1,3,4-trisubstituted-4-arylpiperidines |
| US5166174A (en) * | 1987-01-28 | 1992-11-24 | K.K. Ueno Seiyaku Oyo Kenkyujo | Prostaglandins E and anti-ulcers containing same |
| US4992450A (en) * | 1987-04-16 | 1991-02-12 | Eli Lilly And Company | Piperidine opioid antagonists |
| US5319087A (en) * | 1987-04-16 | 1994-06-07 | Eli Lilly And Company | Piperidine opioid antagonists |
| US4891379A (en) * | 1987-04-16 | 1990-01-02 | Kabushiki Kaisha Kobe Seikosho | Piperidine opioid antagonists |
| US5064834A (en) * | 1987-04-16 | 1991-11-12 | Eli Lilly And Company | Piperidine opioid antagonists |
| US5250542A (en) * | 1991-03-29 | 1993-10-05 | Eli Lilly And Company | Peripherally selective piperidine carboxylate opioid antagonists |
| US5270328A (en) * | 1991-03-29 | 1993-12-14 | Eli Lilly And Company | Peripherally selective piperidine opioid antagonists |
| US5159081A (en) * | 1991-03-29 | 1992-10-27 | Eli Lilly And Company | Intermediates of peripherally selective n-carbonyl-3,4,4-trisubstituted piperidine opioid antagonists |
| US5698155A (en) | 1991-05-31 | 1997-12-16 | Gs Technologies, Inc. | Method for the manufacture of pharmaceutical cellulose capsules |
| WO1995028963A1 (fr) | 1994-04-22 | 1995-11-02 | Yamanouchi Pharmaceutical Co., Ltd. | Systeme de liberation de medicament specifique au colon |
| US5378448A (en) * | 1994-06-07 | 1995-01-03 | Lin; Hsing K. | Process for removing selenium from sulfur |
| TW490465B (en) * | 1994-11-24 | 2002-06-11 | Janssen Pharmaceutica Nv | Enterokinetic benzamide, the preparation process and the pharmaceutical compositions thereof |
| ATE215369T1 (de) * | 1996-09-05 | 2002-04-15 | Lilly Co Eli | Carbazolanaloge als selektive beta3-adrenergische agonisten |
| US6559158B1 (en) * | 1997-11-03 | 2003-05-06 | Ur Labs, Inc. | Use of methylnaltrexone and related compounds to treat chronic opioid use side affects |
| US6022884A (en) * | 1997-11-07 | 2000-02-08 | Amgen Inc. | Substituted pyridine compounds and methods of use |
| ES2234306T3 (es) | 1998-09-28 | 2005-06-16 | Warner-Lambert Company Llc | Administracion enterica y via colon usando capsulas de hpmc. |
| US6436959B1 (en) * | 1998-12-23 | 2002-08-20 | Ortho-Mcneil Pharmaceutical, Inc. | 4-[aryl(piperidin-4-yl)]aminobenzamides |
| US6194382B1 (en) * | 1999-03-03 | 2001-02-27 | Albert Einstein College Of Medicine Of Yeshiva University | Method and composition for treating irritable bowel syndrome using low doses of opioid receptor antagonists |
| DK1220849T3 (da) * | 1999-10-15 | 2004-09-27 | Sucampo Ag | Sammensætninger med bicykliske forbindelser og fremgangsmåde til stabilisering heraf |
| ATE275402T1 (de) * | 1999-11-01 | 2004-09-15 | John Rhodes | Arzneimittel zur behandlung von darmverstopfung und reizkolon |
| US6469030B2 (en) * | 1999-11-29 | 2002-10-22 | Adolor Corporation | Methods for the treatment and prevention of ileus |
| US6414016B1 (en) * | 2000-09-05 | 2002-07-02 | Sucampo, A.G. | Anti-constipation composition |
| ES2572332T3 (es) * | 2000-10-31 | 2016-05-31 | Rensselaer Polytech Inst | 2,6-metano-3-benzazocinas sustituidas en posición 8 y morfinanos sustituidos en posición 3 como ligandos de receptores opioides |
| CA2458471C (en) * | 2001-08-31 | 2012-07-31 | Sucampo Ag | Prostaglandin analogs as chloride channel opener |
| KR101009309B1 (ko) * | 2001-10-18 | 2011-01-18 | 넥타르 테라퓨틱스 | 아편양 길항제의 중합체 공액 |
| AR037524A1 (es) * | 2001-11-14 | 2004-11-17 | Sucampo Ag | Unidad de dosificacion que comprende un analogo de prostaglandina para el tratamiento de la constipacion |
| NZ541110A (en) * | 2002-12-27 | 2008-11-28 | Sucampo Ag | Derivatives of prostaglandins for treating abdominal discomfort |
| CA2513791A1 (en) * | 2003-03-07 | 2004-09-23 | Eli Lilly And Company | Opioid receptor antagonists |
| BRPI0417156A (pt) * | 2003-12-12 | 2007-03-06 | Lilly Co Eli | composto, composição farmacêutica, e, métodos para bloquear receptor mu, capa, delta ou combinação (heterodìmero) dos mesmos em mamìferos, para tratar e/ou prevenir doenças relacionadas com obesidade e obesidade, para suprimir apetite em um paciente, para efetuar perda de peso em um paciente obeso |
| EP1699783B1 (en) * | 2003-12-22 | 2012-07-25 | Eli Lilly And Company | Opioid receptor antagonists |
| ES2308471T3 (es) * | 2004-03-12 | 2008-12-01 | Eli Lilly And Company | Antagonistas de receptores de opioides. |
| ATE475640T1 (de) * | 2004-03-12 | 2010-08-15 | Lilly Co Eli | Antagonisten des opioidrezeptors |
| US8524731B2 (en) * | 2005-03-07 | 2013-09-03 | The University Of Chicago | Use of opioid antagonists to attenuate endothelial cell proliferation and migration |
| WO2006126529A1 (ja) * | 2005-05-25 | 2006-11-30 | Shionogi & Co., Ltd. | 6,7-不飽和-7-カルバモイル置換モルヒナン誘導体 |
| CN101410097A (zh) * | 2006-01-24 | 2009-04-15 | 株式会社·R-技术上野 | 软明胶胶囊制剂 |
| MY145633A (en) * | 2006-03-01 | 2012-03-15 | Theravance Inc | 8-azabicyclo[3.2.1]octane compounds as mu opioid receptor antagonists |
| TWI409067B (zh) * | 2007-02-28 | 2013-09-21 | Theravance Inc | 8-氮雜雙環〔3.2.1〕辛烷化合物之結晶型 |
| CA2702680A1 (en) * | 2007-10-18 | 2009-04-23 | Aiko Biotechnology | Combination analgesic employing opioid and neutral antagonist |
| KR101610165B1 (ko) * | 2007-12-11 | 2016-04-08 | 세라밴스 바이오파마 알앤디 아이피, 엘엘씨 | 뮤 오피오이드 수용체 길항제로서의 3-카르복시프로필-아미노테트랄린 유도체 |
| SG172016A1 (en) * | 2008-12-10 | 2011-07-28 | Theravance Inc | Crystalline forms of a 3-carboxypropyl-aminotetralin compound |
| CA2782529C (en) * | 2009-12-04 | 2015-05-26 | Alkermes, Inc. | Morphinan derivatives for the treatment of drug overdose |
-
2010
- 2010-09-17 CA CA2774021A patent/CA2774021A1/en not_active Abandoned
- 2010-09-17 MX MX2012003310A patent/MX342548B/es active IP Right Grant
- 2010-09-17 BR BR112012006069A patent/BR112012006069A8/pt not_active Application Discontinuation
- 2010-09-17 KR KR1020127008846A patent/KR20120092592A/ko not_active Application Discontinuation
- 2010-09-17 RU RU2012115450/15A patent/RU2561873C2/ru not_active IP Right Cessation
- 2010-09-17 US US12/884,985 patent/US20110071163A1/en not_active Abandoned
- 2010-09-17 PE PE2012000347A patent/PE20120991A1/es not_active Application Discontinuation
- 2010-09-17 TW TW099131769A patent/TW201118084A/zh unknown
- 2010-09-17 EP EP10757144.0A patent/EP2477627B1/en not_active Not-in-force
- 2010-09-17 NZ NZ598783A patent/NZ598783A/xx not_active IP Right Cessation
- 2010-09-17 AU AU2010295464A patent/AU2010295464B2/en not_active Ceased
- 2010-09-17 WO PCT/US2010/049311 patent/WO2011035142A1/en active Application Filing
- 2010-09-17 CN CN201510256187.6A patent/CN104958298A/zh active Pending
- 2010-09-17 CN CN2010800522092A patent/CN102695508A/zh active Pending
- 2010-09-17 JP JP2012529934A patent/JP5797655B2/ja not_active Expired - Fee Related
-
2012
- 2012-03-15 IL IL218679A patent/IL218679A0/en unknown
- 2012-03-15 ZA ZA2012/01954A patent/ZA201201954B/en unknown
- 2012-03-16 CL CL2012000676A patent/CL2012000676A1/es unknown
- 2012-04-18 CO CO12064152A patent/CO6531453A2/es not_active Application Discontinuation
-
2015
- 2015-12-02 HK HK15111842.0A patent/HK1210959A1/xx unknown
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20080255152A1 (en) * | 2002-09-19 | 2008-10-16 | Maria-Jesus Blanco-Pillado | Diaryl ethers as opioid receptor antagonists |
Non-Patent Citations (1)
| Title |
|---|
| FRIEDMAN J.D., Opioid antagonists in the treatment of opioid-induced consitipation and pruritus, Ann. Pharmacotherapy, 2001, 35, pp 85-91. CHANG HY et al.,Opioid-induced bowel dysfunction.,Current Treat Options. Gastroenterol., 2008, 11, pp. 1-8 * |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2011035142A1 (en) | 2011-03-24 |
| AU2010295464A1 (en) | 2012-04-12 |
| CO6531453A2 (es) | 2012-09-28 |
| CA2774021A1 (en) | 2011-03-24 |
| JP5797655B2 (ja) | 2015-10-21 |
| EP2477627A1 (en) | 2012-07-25 |
| ZA201201954B (en) | 2012-11-28 |
| US20110071163A1 (en) | 2011-03-24 |
| KR20120092592A (ko) | 2012-08-21 |
| BR112012006069A2 (pt) | 2016-03-29 |
| CN104958298A (zh) | 2015-10-07 |
| BR112012006069A8 (pt) | 2017-10-10 |
| AU2010295464B2 (en) | 2015-11-26 |
| JP2013505260A (ja) | 2013-02-14 |
| CL2012000676A1 (es) | 2012-10-19 |
| NZ598783A (en) | 2013-07-26 |
| CN102695508A (zh) | 2012-09-26 |
| IL218679A0 (en) | 2012-05-31 |
| MX342548B (es) | 2016-10-04 |
| HK1210959A1 (en) | 2016-05-13 |
| RU2012115450A (ru) | 2013-10-27 |
| PE20120991A1 (es) | 2012-08-01 |
| MX2012003310A (es) | 2012-07-17 |
| EP2477627B1 (en) | 2018-08-15 |
| TW201118084A (en) | 2011-06-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2009244805B2 (en) | Oral administration of peripherally-acting opioid antagonists | |
| DK2574167T3 (en) | LIQUID NOSE SPRAY CONTAINING low-dose naltrexone | |
| TWI473614B (zh) | Anti-analgesic inhibitors | |
| KR102082529B1 (ko) | 오피오이드-유도 유해 약역학 반응을 치료하기 위한 시스템 및 방법 | |
| TW200418474A (en) | Method of treating post-operative nausea and vomiting | |
| WO2007120485A2 (en) | Methods of treating pain with alkylxanthines and antiepileptics and compositions for use therefor | |
| WO2017066488A1 (en) | Treating pain using a composition comprising an opioid and an antiemetic | |
| RU2561873C2 (ru) | Применение антагонистов опиоидных рецепторов при заболеваниях желудочно-кишечного тракта | |
| US20100010029A1 (en) | Acute Pain Medications Based on Fast Acting Diclofenac-Opioid Combinations | |
| KR101971412B1 (ko) | 정맥 내 투여용 이부프로펜의 투여 | |
| JP2006131545A (ja) | 神経因性疼痛治療剤 | |
| CA2859203C (en) | Methods for treatment and prevention of opioid induced constipation using oral compositions of methylnaltrexone | |
| US10874658B2 (en) | Sublingual opioid formulations containing naloxone | |
| US9808431B2 (en) | Immediate release oral guaifenesin solution | |
| JP2013040206A (ja) | 消化器系の運動をイパモレリンを用いて刺激する方法 | |
| WO2015191686A1 (en) | Methods of administering methylnaltrexone | |
| Roberson et al. | Methylnaltrexone for opioid-induced constipation |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PC41 | Official registration of the transfer of exclusive right |
Effective date: 20160219 |
|
| MM4A | The patent is invalid due to non-payment of fees |
Effective date: 20190918 |