RU2335503C2 - Кристаллы сольвата производного хинолинкарбоновой кислоты - Google Patents
Кристаллы сольвата производного хинолинкарбоновой кислоты Download PDFInfo
- Publication number
- RU2335503C2 RU2335503C2 RU2005137156/04A RU2005137156A RU2335503C2 RU 2335503 C2 RU2335503 C2 RU 2335503C2 RU 2005137156/04 A RU2005137156/04 A RU 2005137156/04A RU 2005137156 A RU2005137156 A RU 2005137156A RU 2335503 C2 RU2335503 C2 RU 2335503C2
- Authority
- RU
- Russia
- Prior art keywords
- crystals
- compound
- methyl
- oxo
- type
- Prior art date
Links
- 239000013078 crystal Substances 0.000 title claims abstract description 191
- 239000012453 solvate Substances 0.000 title claims abstract description 15
- LOAUVZALPPNFOQ-UHFFFAOYSA-N quinaldic acid Chemical class C1=CC=CC2=NC(C(=O)O)=CC=C21 LOAUVZALPPNFOQ-UHFFFAOYSA-N 0.000 title 1
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 claims abstract description 102
- 238000002425 crystallisation Methods 0.000 claims abstract description 41
- PWNMXPDKBYZCOO-UHFFFAOYSA-N Prulifloxacin Chemical compound C1=C2N3C(C)SC3=C(C(O)=O)C(=O)C2=CC(F)=C1N(CC1)CCN1CC=1OC(=O)OC=1C PWNMXPDKBYZCOO-UHFFFAOYSA-N 0.000 claims abstract description 15
- 238000004807 desolvation Methods 0.000 claims abstract description 13
- 230000008025 crystallization Effects 0.000 claims description 40
- 230000015572 biosynthetic process Effects 0.000 claims description 19
- 238000000634 powder X-ray diffraction Methods 0.000 claims description 14
- 238000001228 spectrum Methods 0.000 claims description 14
- 230000002269 spontaneous effect Effects 0.000 claims description 12
- 238000000034 method Methods 0.000 claims description 9
- -1 5-methyl-2-oxo-1,3-dioxolen-4-yl Chemical group 0.000 claims 1
- 229940126062 Compound A Drugs 0.000 abstract description 78
- NLDMNSXOCDLTTB-UHFFFAOYSA-N Heterophylliin A Natural products O1C2COC(=O)C3=CC(O)=C(O)C(O)=C3C3=C(O)C(O)=C(O)C=C3C(=O)OC2C(OC(=O)C=2C=C(O)C(O)=C(O)C=2)C(O)C1OC(=O)C1=CC(O)=C(O)C(O)=C1 NLDMNSXOCDLTTB-UHFFFAOYSA-N 0.000 abstract description 78
- 150000001875 compounds Chemical class 0.000 abstract description 60
- 239000002904 solvent Substances 0.000 abstract description 46
- 239000000126 substance Substances 0.000 abstract description 5
- 230000000694 effects Effects 0.000 abstract description 2
- 238000000113 differential scanning calorimetry Methods 0.000 description 11
- 230000009466 transformation Effects 0.000 description 10
- 238000001035 drying Methods 0.000 description 9
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Natural products CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 8
- ZMXDDKWLCZADIW-UHFFFAOYSA-N Vilsmeier-Haack reagent Natural products CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 8
- 238000010438 heat treatment Methods 0.000 description 8
- 238000009835 boiling Methods 0.000 description 7
- 238000004519 manufacturing process Methods 0.000 description 7
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 6
- 230000001404 mediated effect Effects 0.000 description 6
- 238000002844 melting Methods 0.000 description 6
- 230000008018 melting Effects 0.000 description 6
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 6
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N N-phenyl amine Natural products NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 5
- 238000001816 cooling Methods 0.000 description 5
- 238000000354 decomposition reaction Methods 0.000 description 5
- 239000000843 powder Substances 0.000 description 5
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 4
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 4
- 230000008859 change Effects 0.000 description 4
- 238000001914 filtration Methods 0.000 description 4
- 238000001556 precipitation Methods 0.000 description 4
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 3
- 238000004090 dissolution Methods 0.000 description 3
- 235000019253 formic acid Nutrition 0.000 description 3
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 3
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 2
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Natural products CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 2
- LRHPLDYGYMQRHN-UHFFFAOYSA-N Butanol Natural products CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 2
- RENMDAKOXSCIGH-UHFFFAOYSA-N Chloroacetonitrile Chemical compound ClCC#N RENMDAKOXSCIGH-UHFFFAOYSA-N 0.000 description 2
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 2
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Natural products CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 2
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Natural products CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 2
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerol Natural products OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 2
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N Methyl ethyl ketone Natural products CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 2
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 2
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Natural products CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 2
- 238000002441 X-ray diffraction Methods 0.000 description 2
- WFDIJRYMOXRFFG-UHFFFAOYSA-N acetic acid anhydride Natural products CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- 229910052786 argon Inorganic materials 0.000 description 2
- 238000006243 chemical reaction Methods 0.000 description 2
- 239000007857 degradation product Substances 0.000 description 2
- 239000011261 inert gas Substances 0.000 description 2
- OKKJLVBELUTLKV-UHFFFAOYSA-N methanol Natural products OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 2
- VLKZOEOYAKHREP-UHFFFAOYSA-N methyl pentane Natural products CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 2
- 239000000203 mixture Substances 0.000 description 2
- UHOVQNZJYSORNB-UHFFFAOYSA-N monobenzene Natural products C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- LYGJENNIWJXYER-UHFFFAOYSA-N nitromethane Chemical compound C[N+]([O-])=O LYGJENNIWJXYER-UHFFFAOYSA-N 0.000 description 2
- 238000002360 preparation method Methods 0.000 description 2
- 238000001953 recrystallisation Methods 0.000 description 2
- 238000002411 thermogravimetry Methods 0.000 description 2
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 2
- 239000013585 weight reducing agent Substances 0.000 description 2
- HTSGKJQDMSTCGS-UHFFFAOYSA-N 1,4-bis(4-chlorophenyl)-2-(4-methylphenyl)sulfonylbutane-1,4-dione Chemical compound C1=CC(C)=CC=C1S(=O)(=O)C(C(=O)C=1C=CC(Cl)=CC=1)CC(=O)C1=CC=C(Cl)C=C1 HTSGKJQDMSTCGS-UHFFFAOYSA-N 0.000 description 1
- 229940093475 2-ethoxyethanol Drugs 0.000 description 1
- UAEPNZWRGJTJPN-UHFFFAOYSA-N Methylcyclohexane Natural products CC1CCCCC1 UAEPNZWRGJTJPN-UHFFFAOYSA-N 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- KXKVLQRXCPHEJC-UHFFFAOYSA-N acetic acid trimethyl ester Natural products COC(C)=O KXKVLQRXCPHEJC-UHFFFAOYSA-N 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 239000008186 active pharmaceutical agent Substances 0.000 description 1
- IMNFDUFMRHMDMM-UHFFFAOYSA-N anhydrous n-heptane Natural products CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 230000000844 anti-bacterial effect Effects 0.000 description 1
- VZGDMQKNWNREIO-UHFFFAOYSA-N carbon tetrachloride Substances ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- HEDRZPFGACZZDS-UHFFFAOYSA-N chloroform Substances ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 229930003836 cresol Natural products 0.000 description 1
- USIUVYZYUHIAEV-UHFFFAOYSA-N diphenyl ether Natural products C=1C=CC=CC=1OC1=CC=CC=C1 USIUVYZYUHIAEV-UHFFFAOYSA-N 0.000 description 1
- 239000003814 drug Substances 0.000 description 1
- 229940088679 drug related substance Drugs 0.000 description 1
- 239000000706 filtrate Substances 0.000 description 1
- ZHNUHDYFZUAESO-UHFFFAOYSA-N formamide Substances NC=O ZHNUHDYFZUAESO-UHFFFAOYSA-N 0.000 description 1
- 238000004817 gas chromatography Methods 0.000 description 1
- ZXEKIIBDNHEJCQ-UHFFFAOYSA-N iso-butyl alcohol Natural products CC(C)CO ZXEKIIBDNHEJCQ-UHFFFAOYSA-N 0.000 description 1
- 229940035429 isobutyl alcohol Drugs 0.000 description 1
- JMMWKPVZQRWMSS-UHFFFAOYSA-N isopropanol acetate Natural products CC(C)OC(C)=O JMMWKPVZQRWMSS-UHFFFAOYSA-N 0.000 description 1
- 229940011051 isopropyl acetate Drugs 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 238000004811 liquid chromatography Methods 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- GYNNXHKOJHMOHS-UHFFFAOYSA-N methyl-cycloheptane Natural products CC1CCCCCC1 GYNNXHKOJHMOHS-UHFFFAOYSA-N 0.000 description 1
- 235000016709 nutrition Nutrition 0.000 description 1
- 230000035764 nutrition Effects 0.000 description 1
- HFPZCAJZSCWRBC-UHFFFAOYSA-N p-cymene Natural products CC(C)C1=CC=C(C)C=C1 HFPZCAJZSCWRBC-UHFFFAOYSA-N 0.000 description 1
- 239000003208 petroleum Substances 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 239000000047 product Substances 0.000 description 1
- 229960001224 prulifloxacin Drugs 0.000 description 1
- 239000002994 raw material Substances 0.000 description 1
- 238000003303 reheating Methods 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 238000009738 saturating Methods 0.000 description 1
- 230000003595 spectral effect Effects 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- 229950011008 tetrachloroethylene Drugs 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 238000002076 thermal analysis method Methods 0.000 description 1
- 239000008096 xylene Substances 0.000 description 1
Images
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D513/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00
- C07D513/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00 in which the condensed system contains two hetero rings
- C07D513/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Communicable Diseases (AREA)
- Pharmacology & Pharmacy (AREA)
- Oncology (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Основной целью является предоставление кристаллов ацетонитрильного сольвата 6-фтор-1-метил-7-[4-(5-метил-2-оксо-1,3-диоксолен-4-ил)метил-1-пиперазинил]-4-оксо-4Н-[1,3]тиазето[3,2-а]хинолин-3-карбоновой кислоты (соединение В), которое является промежуточным соединением для преимущественного получения кристаллов 6-фтор-1-метил-7-[4-(5-метил-2-оксо-1,3-диоксолен-4-ил)метил-1-пиперазинил]-4-оксо-4Н-[1,3]тиазето[3,2-а]хинолин-3-карбоновой кислоты типа III (соединение А). Кристаллы соединения В можно преимущественно осадить путем регуляции концентрации сверхнасыщения при проведении кристаллизации с использованием ацетонитрила в качестве растворителя. Затем можно получить кристаллы соединения А типа III путем десольвации кристаллов. 4 н. и 2 з.п. ф-лы, 4 ил., 4 табл.
Description
Настоящее изобретение относится к кристаллу 6-фтор-1-метил-7-[4-(5-метил-2-оксо-1,3-диоксолен-4-ил)метил-1-пиперазинил]-4-оксо-4Н-[1,3]тиазето[3,2-а]хинолин-3-карбоновой кислоты (далее называется соединение А) ацетонитрильного сольвата (далее называется соединение В), к способу получения кристаллов соединения В и к способу получения кристаллов соединения А типа III с использованием кристаллов соединения В.
Соединение А обладает превосходной противобактериальной активностью (см., например, патентный документ 1) и продается на рынке как синтетическое противобактериальное средство. Известно, что существует 3 типа кристаллов (тип I, тип II и тип III) соединения А, которые обозначаются как тип I, тип II и тип III в соответствии с уменьшением температуры плавления, измеряемой с помощью дифференциальной сканирующей калориметрии (далее обозначается DSC) (см., например, не патентный документ 1). Кроме того, определенные растворимость, абсорбционная способность, терапевтическое действие и т. п. обуславливают наличие на рынке кристаллов типа III (см., например, не патентный документ 1).
Известно, что кристаллы соединения А типа I, типа II и типа III получают кристаллизацией из ацетонитрила, однако, условия кристаллизации не известны и существование соединения В тоже не было доказано (см., например, не патентный документ 1).
Патентный документ 1: JP-A-1-294680.
Не патентный документ 1: Kazuro Kakemi and others 7, "Chemical Structure, physicochemical properties and stability of Prulifloxacin", IYAKUHIN KENKYU, vol. 28 (1), pp 1-11 (1997).
Целью настоящего изобретения, в основном, является предоставление сырого вещества для получения кристаллов соединения А типа III, которые обладают замечательным фармацевтическим и фармакологическим действием, а также способа их получения.
До настоящего времени считалось, что кристаллы соединения А типа III получают непосредственно из раствора соединения А в ацетонитриле, так же, как и кристаллы типа I и типа II. Однако авторы настоящего изобретения обнаружили, что кристаллы типа III можно получить не прямой перекристаллизацией, как кристаллы типа I и типа II, а путем десольвации кристалла соединения В (см. экспериментальные примеры 1-3, описанные ниже). Авторы настоящего изобретения обнаружили, что кристаллы соединения В представляют собой важное промежуточное соединение для получения лекарственного средства (кристаллы соединения А типа III).
Кроме того, авторы настоящего изобретения провели интенсивные исследования по способу избирательного осаждения кристаллов соединения В, в результате чего было обнаружено, что указанную цель можно достичь путем регуляции концентрации сверхнасыщения (см. экспериментальный пример 1, описанный ниже).
То есть, авторы настоящего изобретения обнаружили, что кристаллы соединения В можно избирательно осадить путем регуляции концентрации сверхнасыщения при кристаллизации с использованием ацетонитрила в качестве растворителя и затем после десольвации кристаллов можно получить кристаллы соединения А типа III, что составляет предмет настоящего изобретения.
Настоящее изобретение включает в себя:
(1) кристаллы соединения В, имеющие пики дифракции, по меньшей мере, при 7,3°, 14,7°, 19,2° и 22,3° в спектре порошковой рентгенодифракции;
(2) способ получения кристаллов соединения В, отличающийся тем, что кристаллизацию проводят из раствора соединения А в ацетонитриле, причем во время образования центров кристаллизации концентрация сверхнасыщения (г/100 г) находится в интервале от 2,15 до 2,36;
(3) способ получения кристаллов соединения В, отличающийся тем, что кристаллизацию проводят из раствора соединения А в ацетонитриле, причем во время образования центров кристаллизации концентрация сверхнасыщения (г/100 г) находится в интервале от 0,41 до 2,36, и
(4) способ получения, описанный в пункте (3), где температура раствора во время добавления затравочного кристалла составляет 70°С или ниже.
В настоящем изобретении термин "спонтанное образование центров кристаллизации" означает образование центров кристаллизации, которое происходит спонтанно, когда кристаллизацию проводят без затравочного кристалла.
В настоящем изобретении термин "концентрация сверхнасыщения: Сх (г/100 г)" означает степень сверхнасыщения и соответствует следующей формуле:
Сх=С-Cs
где С (г/100 г) представляет собой массу соединения В (в пересчете на десольват), растворенного в 100 г растворителя.
Cs (г/100 г) представляет собой насыщающую массу (в пересчете на десольват) соединения В, растворенного в 100 г растворителя при температуре во время спонтанного образования центров кристаллизации или добавления затравочного кристалла.
То есть, если Сх>0, раствор находится в состоянии сверхнасыщения, а если Сх<0, концентрация раствора не достигает насыщения.
В настоящем изобретении термин "количество в пересчете на десольват" относится к массе, получающейся в результате преобразования массы соединения В (сольвата) в массу десольвата. Например, если масса соединения В составляет 502,5 г, то количество в пересчете на десольват составляет 461,5 г.
В настоящем изобретении термин "десольвация" означает удаление растворителя из сольвата. Например, если растворителем является вода, то в качестве примера можно привести превращение гидрата в безводную форму путем удаления молекул воды.
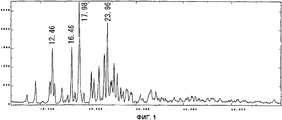
На фиг.1 приведен спектр порошковой рентгенодифракции кристаллов соединения А типа I. На вертикальной оси указана интенсивность (имп./сек. (cps)), а на горизонтальной оси указан угол диффракции (2Θ ± 0,2°).
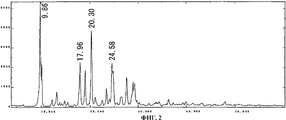
На фиг.2 приведен спектр порошковой рентгенодифракции кристаллов соединения А типа II. На вертикальной оси указана интенсивность (имп./сек. (cps)), а на горизонтальной оси указан угол диффракции (2Θ ± 0,2°).
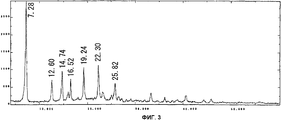
На фиг.3 приведен спектр порошковой рентгенодифракции кристаллов соединения В. На вертикальной оси указана интенсивность (имп./сек. (cps)), а на горизонтальной оси указан угол диффракции (2Θ ± 0,2°).
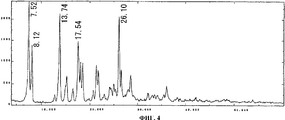
На фиг.4 приведен спектр порошковой рентгенодифракции кристаллов соединения А типа III. На вертикальной оси указана интенсивность (имп./сек. (cps)), а на горизонтальной оси указан угол диффракции (2Θ ± 0,2°).
Кристаллы соединения В можно получить путем осаждения из раствора, в котором концентрация сверхнасыщения (г/100 г) во время спонтанного образования центров кристаллизации находится в интервале от 2,15 до 2,36, и проведения кристаллизации из раствора соединения А в ацетонитриле при подавлении образования кристаллов соединения А типа I и типа II.
С другой стороны, если кристаллизацию проводят в условиях добавления затравочных кристаллов, кристаллы получают в зависимости от вида затравочных кристаллов. Так, при проведении кристаллизации путем добавления затравочных кристаллов, в отличие от кристаллизации, происходящей в результате спонтанного образования центров кристаллизации, кристаллы соединения В можно получить, даже если концентрация сверхнасыщения (г/100 г) находится в интервале от 0,41 до 2,36. Предпочтительно добавлять затравочные кристаллы в большем количестве (не менее чем 0,004 г/100 г растворителя), чем обычно (менее чем 0,004 г/100 г растворителя). Если используется маленькое количество затравочных кристаллов, добавляемые затравочные кристаллы становятся стимулом и наблюдается спонтанное образование новых центров кристаллизации. Однако, если затравочные кристаллы добавляют в большом количестве, преимущественно происходит рост добавленных затравочных кристаллов, а спонтанное образование центров кристаллизации подавляется, посредством чего образование кристаллов соединения А типа I или типа II может быть сведено к минимальному уровню.
Кристаллы соединения В подвергаются опосредованной растворителем трансформации, следовательно, температуру раствора во время спонтанного образования центров кристаллизации и во время добавления затравочных кристаллов поддерживают при 70°С или ниже, предпочтительно 67°С или ниже, более предпочтительно 55°С или ниже.
В настоящем изобретении термин "опосредованная растворителем трансформация" относится к преобразованию кристалла в другой вид в присутствии растворителя. Например, это означает, что при заранее определенной температуре кристаллы соединения В в растворителе трансформируются в кристаллы соединения А типа I.
Кристаллы соединения А типа III можно получить путем десольвации кристаллов соединения В. Десольвацию можно проводить путем сушки кристаллов сольвата в соответствии с традиционным методом; однако ее предпочтительно проводить при 80°С или ниже при пониженном давлении. Кроме того, как описано выше, кристаллы соединения А типа III подвергаются опосредованной растворителем трансформации, следовательно, температуру сушки поддерживают при 70°С или ниже, предпочтительно 67°С или ниже, более предпочтительно 55°С или ниже.
Более конкретно, кристаллы соединения В можно получить, например, следующим образом.
(1) Стадия растворения
Соединение А растворяют в ацетонитриле. Используют такие количества соединения А и ацетонитрила, чтобы получить заданную концентрацию сверхнасыщения. Растворение предпочтительно проводят при нагревании. Температура нагревания не ограничивается, однако растворение предпочтительно проводить при температуре, близкой к точке кипения ацетонитрила. Кроме того, данную стадию предпочтительно проводить в потоке инертного газа, такого как азот или аргон.
Чтобы удалить нерастворенные вещества, раствор можно отфильтровать. Чтобы предотвратить образование кристаллов в процессе фильтрации, фильтрацию предпочтительно проводить с использованием фильтра, снабженного обогревающим устройством, и при повышенном давлении. Если кристаллы образуются в фильтрате, их можно снова растворить путем повторного нагревания раствора после фильтрации.
(2) Стадия охлаждения
Раствор охлаждают до осаждения кристаллов. Необходимо контролировать температуру, при которой начинают осаждаться кристаллы, следовательно, кристаллизация, проводимая без добавления затравочных кристаллов, требует особого внимания. После осаждения кристаллов скорость охлаждения не ограничивается, однако охлаждение предпочтительно проводить со скоростью приблизительно 0,04°С/мин или быстрее, более предпочтительно со скоростью приблизительно 0,22°С/мин или быстрее. Не существует особого ограничения по температуре охлаждения (температура во время сбора осажденных кристаллов), однако она предпочтительно находится в интервале от 0 до 45°С, более предпочтительно от 0 до 25°С. Время выдержки после достижения температуры охлаждения не ограничивается, однако оно предпочтительно составляет 30 минут или больше, более предпочтительно 90 минут или больше. Кроме того, данную стадию предпочтительно проводить в потоке инертного газа, такого как азот или аргон.
(3) Стадия сбора кристаллов
Осажденные кристаллы можно собрать известным способом, таким как фильтрация или центрифугирование, их также можно высушить. Сушку осажденных кристаллов можно проводить традиционным способом. Чтобы предотвратить опосредованную растворителем трансформацию, температуру в процессе сушки устанавливают при 70°С или ниже, предпочтительно при 67°С или ниже, более предпочтительно при 55°С или ниже. Кроме того, может произойти десольвация кристаллов и в некоторых случаях наблюдается образование десольватов. Чтобы предотвратить десольвацию, предпочтительно сушить кристаллы при обычной температуре или ниже и при пониженном давлении. Кристаллы используются в качестве сырого материала для получения кристаллов соединения А типа III, как описано ниже, следовательно, их можно использовать без особой сушки.
(4) Способ получения кристаллов соединения А типа III
Кристаллы соединения А типа III можно получить путем дезольвации кристаллов соединения В с помощью традиционного способа. Условия сушки не ограничиваются, при условии, что растворитель может быть удален из кристалла сольвата, однако, сушку предпочтительно проводят при 80°С или ниже и при пониженном давлении. Кроме того, чтобы предотвратить опосредованную растворителем трансформацию, сушку проводят в течение периода от нескольких часов до нескольких десятков часов при температуре 70°С или ниже, предпочтительно 67°С или ниже, более предпочтительно 55°С или ниже.
Далее настоящее изобретение будет разъясняться более подробно со ссылкой на примеры сравнения, примеры и экспериментальные примеры. Излишне говорить, что настоящее изобретение не ограничивается нижеследующими примерами.
В данном описании термические анализы (анализ DSC и анализ TG) проводят, используя дифференциальный сканирующий калориметр с тепловым потоком DSC-50, термический гравиметрический анализатор TGA-50 и систему термического анализа TA-50, которые производятся Shimadzu, со скоростью нагревания 10°С/мин, а анализ порошковой рентгенодифракции проводят на порошковом рентгенодифрактометре, который производится Rigaku Denki Co. Ошибка измерения данного аппарата составляет ±0,2°.
Пример сравнения 1
Кристаллы 6-фтор-1-метил-7-[4-(5-метил-2-оксо-1,3-диоксолен-4-ил)метил-1-пиперазинил]-4-оксо-4Н-[1,3]тиазето[3,2-a]хинолин-3-карбоновой кислоты типа I (соединение А)
Соединение А получают в соответствии с описанием патентного документа 1. Соединение (7,0 г) растворяют при нагревании в 560 г ацетонитрила. Раствор постепенно охлаждают и когда температура раствора достигнет 25°С, добавляют 0,022 г кристаллов типа I в качестве затравочных кристаллов, чтобы вызвать осаждение кристаллов, в результате чего получают 1,80 г кристаллов соединения А типа I. Кристаллы подвергают анализу DSC и в результате определяют температуру плавления (эндотермический пик), которая находится в интервале от 213 до 225°С (разложение).
В не патентном документе 1 формы кристаллов обозначаются как тип I, тип II и тип III в соответствии с уменьшением температуры плавления, измеряемой с помощью анализа DSC. Сравнение результатов анализа DSC для кристаллов, полученных в примере сравнения 1, примере сравнения 2 и примере 3, показало, что кристаллы, полученные в данном примере сравнения, соответствуют кристаллам соединения А типа I.
Спектр порошковой рентгенодифракции для полученных кристаллов приведен на фиг. 1. Кристаллы соединения А типа I имеют отличительные пики при 12,5°, 16,5°, 18,0° и 24,0°.
Пример сравнения 2
Кристаллы 6-фтор-1-метил-7-[4-(5-метил-2-оксо-1,3-диоксолен-4-ил)метил-1-пиперазинил]-4-оксо-4Н-[1,3]тиазето[3,2-a]хинолин-3-карбоновой кислоты типа II (соединение А)
Соединение А получают в соответствии с описанием патентного документа 1. Соединение (14,4 г) растворяют при нагревании в 560 г ацетонитрила. Раствор постепенно охлаждают, и когда температура раствора достигает 25°С, добавляют 0,02 г кристаллов типа II в качестве затравочных кристаллов, чтобы вызвать осаждение кристаллов, в результате чего получают 10,8 г кристаллов соединения А типа II. Кристаллы подвергают анализу DSC и в результате определяют температуру плавления (эндотермический пик), которая находится в интервале от 179 до 189°С (трансформация кристаллов типа I) и от 213 до 225°С (разложение).
Сравнение результатов анализа DSC для кристаллов, полученных в примере сравнения 1, примере сравнения 2 и примере 3, показало, что кристаллы, полученные в данном примере сравнения, соответствуют кристаллам соединения А типа II.
Спектр порошковой рентгенодифракции для полученных кристаллов приведен на фиг. 2. Кристаллы соединения А типа II имеют отличительные пики при 9,9°, 18,0°, 20,3° и 24,6°.
Пример 1
Кристаллы ацетонитрильного сольвата 6-фтор-1-метил-7-[4-(5-метил-2-оксо-1,3-диоксолен-4-ил)метил-1-пиперазинил]-4-оксо-4Н-[1,3]тиазето[3,2-a]хинолин-3-карбоновой кислоты (соединение В)
Соединение А получают в соответствии с описанием патентного документа 1. Соединение (15,0 г) растворяют при нагревании в 560 г ацетонитрила и проводят кристаллизацию без добавления затравочных кристаллов, в результате чего получают 11,99 г кристаллов соединения В (в пересчете на десольват). Кристаллы подвергают анализу DSC и в результате определяют температуру плавления (эндотермический пик), которая находится в интервале от обычной температуры до 130°С (десольвация), от 134 до 149°С (трансформация) и от 213 до 225°С (разложение).
(1) Уменьшение массы при десольвации по данным анализа DSC и анализа TG показывает, что в состав сольвата входит одна молекула ацетонитрила и одна молекула соединения А. После уменьшения массы кристаллы анализируют методом порошковой рентгенодифракции и получают спектр, идентичный спектру кристаллов соединения А типа III. (2) Результаты анализа кристаллов, полученных путем выдерживания кристаллов соединения А типа III в насыщенных парах ацетонитрила, методом порошковой рентгенодифракции совпадают со спектральными данными, полученными для кристаллов примера 1. Кроме того, (3) кристаллы, из которых тщательным высушиванием полностью удален присоединенный растворитель, анализируют методом газовой хроматографии и обнаруживают ацетонитрил. Далее, (4) для кристаллизации не используют растворители, отличные от ацетонитрила. Результаты, описанные выше и т. п., демонстрируют, что кристаллы, полученные в примере 1, представляют собой ацетонитрильный сольват соединения А (соединение В).
Спектр порошковой рентгенодифракции для полученных кристаллов приведен на фиг. 3. Кристаллы соединения В имеют отличительные пики при 7,3°, 12,6°, 14,7°, 16,5°, 19,2°, 22,3° и 25,8°. В особенности, характеристичными являются пики при 7,3°, 14,7°, 19,2° и 22,3°.
Пример 2
Кристаллы ацетонитрильного сольвата 6-фтор-1-метил-7-[4-(5-метил-2-оксо-1,3-диоксолен-4-ил)метил-1-пиперазинил]-4-оксо-4Н-[1,3]тиазето[3,2-a]хинолин-3-карбоновой кислоты (соединение В)
Соединение А получают в соответствии с описанием патентного документа 1. Соединение (3,93 г) растворяют при нагревании в 561,5 г ацетонитрила. Раствор постепенно охлаждают, и когда температура раствора достигает 25°С, добавляют 0,449 г соединения В в качестве затравочных кристаллов, чтобы вызвать осаждение кристаллов, в результате чего получают 0,70 г кристаллов соединения В (в пересчете на десольват). Физические данные (результаты анализов DSC и рентгенодифракции) совпадают с данными, полученными для кристаллов примера 1.
Пример 3
Кристаллы 6-фтор-1-метил-7-[4-(5-метил-2-оксо-1,3-диоксолен-4-ил)метил-1-пиперазинил]-4-оксо-4Н-[1,3]тиазето[3,2-a]хинолин-3-карбоновой кислоты типа III (соединение А)
Кристаллы соединения В (9,8 г) сушат в течение 24 часов при 50°С и пониженном давлении (20 мм рт. ст.) с целью десольвации (выход 9,0 г). Кристаллы подвергают анализу DSC и в результате определяют температуру плавления (эндотермический пик), которая находится в интервале от 134 до 149°С (трансформация) и от 213 до 225°С (разложение).
Сравнение результатов анализа DSC для кристаллов, полученных в примере сравнения 1, примере сравнения 2 и примере 3, показало, что кристаллы, полученные в данном примере сравнения, соответствуют кристаллам соединения А типа III.
Спектр порошковой рентгенодифракции для полученных кристаллов приведен на фиг. 4. Кристаллы соединения В имеют отличительные пики при 7,5°, 8,1°, 13,7°, 17,5° и 26,1°. Данный спектр совпадает со спектром рентгенодифракции кристаллов соединения А типа III, приведенном в не патентном документе 1.
Экспериментальный пример 1
Влияние концентрации сверхнасыщения на кристаллизацию
Заданное количество соединения А растворяют в заданном количестве ацетонитрила, кристаллизацию проводят при разных концентрациях сверхнасыщения и полученные кристаллы анализируют с помощью порошкового рентгенодифрактометра. Результаты приведены в таблице 1.
| Таблица 1 | |||||
| Концен-трация питания (C) | Затра-вочные кристаллы | Температура во время спонтанного образования центров кристаллизации или добавления затравочных кристаллов (°C) | Растворимость во время спонтанного образования центров кристаллизации или добавления затравочных кристаллов (Cs) | Концентрация сверхнасыщения (Cx) | Вид осаж-денных кристаллов |
| 2,14 | Отсут. | 25 | 0,29 | 1,85 | B+I |
| 2,44 | Отсут. | 25 | 0,29 | 2,15 | В |
| 2,40 | Отсут. | 17 | 0,19 | 2,21 | В |
| 2,40 | Отсут. | 14,3 | 0,16 | 2,24 | В |
| 2,68 | Отсут. | 27 | 0,32 | 2,36 | В |
| 3,66 | Отсут. | 0 | 0,07 | 3,59 | II |
| 2,32 | III | 50 | 1,00 | 1,32 | B+I |
| 1,80 | III | 25 | 0,29 | 1,51 | B+I |
| 1,89 | III | 25 | 0,29 | 1,60 | В |
| 2,00 | III | 25 | 0,29 | 1,71 | В |
| 2,14 | III | 25 | 0,29 | 1,85 | В |
| 2,21 | III | 25 | 0,29 | 1,92 | В |
| 2,39 | III | 30 | 0,38 | 2,01 | В |
| 2,41 | III | 25 | 0,29 | 2,12 | В |
| 2,68 | III | 25 | 0,29 | 2,39 | В |
| 2,73 | III | 25 | 0,29 | 2,44 | В |
| 2,36 | В | 70 | 2,36 | 0,00 | B+I |
| 0,70 | В | 25 | 0,29 | 0,41 | В |
| 2,36 | В | 65 | 1,92 | 0,44 | В |
| 1,34 | В | 45 | 0,79 | 0,55 | В |
| 1,07 | В | 30 | 0,38 | 0,69 | В |
| 1,61 | В | 45 | 0,79 | 0,82 | В |
| 1,25 | В | 25 | 0,29 | 0,96 | В |
| 1,43 | В | 30 | 0,38 | 1,05 | В |
| 2,36 | В | 55 | 1,25 | 1,11 | В |
| 1,52 | В | 25 | 0,29 | 1,23 | В |
| 1,75 | В | 30 | 0,38 | 1,37 | В |
| 1,75 | В | 25 | 0,29 | 1,46 | В |
| 2,36 | В | 45 | 0,79 | 1,57 | В |
| 1,96 | В | 25 | 0,29 | 1,67 | В |
| 2,10 | В | 30 | 0,38 | 1,72 | В |
| 2,14 | В | 25 | 0,29 | 1,85 | В |
| 2,36 | В | 30 | 0,38 | 1,98 | В |
| 2,36 | В | 25 | 0,29 | 2,07 | В |
| 2,41 | В | 25 | 0,29 | 2,12 | В |
В таблице 1 I обозначает кристаллы соединения А типа I, II обозначает кристаллы соединения А типа II, III обозначает кристаллы соединения А типа III, а В обозначает кристаллы соединения В.
Как показано в таблице 1, кристаллы соединения А типа III не получают путем кристаллизации со спонтанным образованием центров кристаллизации или даже с добавлением кристаллов соединения А типа III в качестве затравочных кристаллов. Таким способом получают только кристаллы соединения А типа I и типа II и кристаллы соединения В. Следовательно, было обнаружено, что кристаллы соединения А типа III не могут быть получены непосредственно перекристаллизацией.
Кроме того, если затравочные кристаллы не добавляют, кристаллы соединения В получают, если концентрация сверхнасыщения (г/100 г) во время спонтанного образования центров кристаллизации находится в интервале от 2,15 до 2,36. Однако, если концентрация сверхнасыщения превышает верхнюю границу интервала, наблюдается примесь кристаллов соединения А типа II, а если она ниже нижнего предела интервала, наблюдается примесь кристаллов соединения А типа I.
Кроме того, если добавляют кристаллы соединения В в качестве затравочных кристаллов, то получают кристаллы соединения В, даже если концентрация сверхнасыщения (г/100 г) во время добавления затравочных кристаллов находится в интервале от 0,41 до 2,12. Полагают, что преимущественное осаждение кристаллов соединения В происходит в результате подавления образования других центров кристаллизации (кристаллы соединения А типа I) вследствие добавления затравочных кристаллов.
Экспериментальный пример 2
Десольват кристаллов соединения В
Кристаллы соединения В подвергают десольвации путем сушки при 80°С в течение 24 часов при пониженном давлении и полученные кристаллы анализируют с помощью порошкового рентгенодифрактометра. Физические характеристики полученных кристаллов совпадают с характеристиками кристаллов, полученных в примере 3. Следовательно, было обнаружено, что кристаллы соединения А типа III можно получить путем десольвации кристаллов соединения В.
Экспериментальный пример 3
Подбор растворителя для кристаллизации
(1) Путем добавления 3 мл растворителя к 50 мг соединения А определяют, растворяется соединение А в данном растворителе или нет.
(2) Чтобы исследовать стабильность соединения А в растворителе, к соединению А добавляют 2-кратный объем (объем растворителя (мл)/масса растворенного вещества (г)) растворителя, смесь держат 1000 минут при 50°С и затем исследуют стабильность соединения методом высокоэффективной жидкостной хроматографии.
Результаты описанных выше исследований (1) приведены в колонке *1 таблицы 2, а результаты описанных выше исследований (2) приведены в колонке *2 таблицы 2.
| Таблица 2 | ||
| Растворитель | Растворимость (*1) | Химическая стабильность (*2) |
| ацетонитрил | о | |
| петролейный эфир | х | - |
| лигроин | х | - |
| гексан | х | - |
| бензол | х | - |
| гептан | х | - |
| метилциклогексан | х | - |
| толуол | х | - |
| ксилол | х | - |
| п-цимол | х | - |
| четыреххлористый углерод | х | - |
| хлороформ | х | - |
| трихлорэтилен | х | - |
| тетрахлорэтилен | х | - |
| диизопропиловый эфир | х | - |
| тетрагидрофуран | х | - |
| диоксан | х | - |
| дибутиловый эфир | х | - |
| дифениловый эфир | х | - |
| этилацетат | х | - |
| метилацетат | х | - |
| изопропилацетат | х | - |
| ацетон | х | - |
| метилэтилкетон | х | - |
| этанол | х | - |
| метанол | х | х |
| 2-пропанол | х | - |
| изобутиловый спирт | х | - |
| 1-бутанол | х | - |
| глицерин | х | - |
| крезол | х | - |
| формамид | х | - |
| пиридин | △ - | - |
| нитрометан | △ - | - |
| хлорацетонитрил | △ - | - |
| N,N-диметилформамид | △ - | - |
| муравьиная кислота | △ х | - |
| уксусная кислота | △ х | - |
| анилин | △ х | - |
| 2-этоксиэтанол | ο х | - |
| фенол | ο х | - |
| уксусный ангидрид | ο х | - |
В колонке *1 таблицы 2 "ο", в случае, если растворитель имеет точку кипения 130°С или выше, соответствует растворителю, в котором соединение А растворяется при 130°С, а в случае, если растворитель имеет точку кипения 130°С или ниже, соответствует растворителю, в котором соединение А растворяется при температуре кипения (растворители, которые считаются подходящими для кристаллизации), "△" соответствует растворителю, в котором соединение А растворяется при обычной температуре (растворители, которые можно использовать для кристаллизации), а "х", в случае, если растворитель имеет точку кипения 130°С или выше, соответствует растворителю, в котором соединение А не растворяется при 130°С, а в случае, если растворитель имеет точку кипения 130°С или ниже, соответствует растворителю, в котором соединение А не растворяется при температуре кипения (растворители, которые не являются подходящими для кристаллизации). Кроме того, в колонке *2 таблицы 2 "ο" означает, что отсутствуют пики, не свойственные соединению А (продукт деградации отсутствует), "х" означает, что присутствует пик, отличный от пиков соединения А (присутствует продукт деградации), а "-" означает, что тестирование не проводилось.
Как показано в колонке *1 таблицы 2, для кристаллизации соединения В кроме ацетонитрила можно использовать еще 7 растворителей, а именно пиридин, нитрометан, хлорацетонитрил, N,N-диметилформамид, муравьиную кислоту, уксусную кислоту и анилин. Однако в 3 видах растворителей, а именно в муравьиной кислоте, уксусной кислоте и анилине, присутствует продукт разложения соединения А, следовательно, 3 вышеуказанных вида растворителей не подходят для кристаллизации.
Соответственно, используют другие 4 вида растворителей, при этом заданное количество соединения А добавляют к заданному количеству растворителя, растворяют, повышая температуру до 78°С или выше и смесь охлаждают до 25°С. Осажденные кристаллы отфильтровывают и анализируют с помощью порошкового рентгенодифрактометра. Результаты приведены в таблице 3.
| Таблица 3 | |||
| Растворитель | Подаваемое количество | ||
| Количество растворенного вещества (г) | Количество растворителя (мл) | Вид кристаллов | |
| пиридин | 4 | 67 | I |
| нитрометан | 3 | 50 | II |
| хлорацетонитрил | 1 | 5 | I |
| N,N-диметилформамид | 6 | 50 | I |
В таблице 3 I обозначает кристаллы соединения А типа I, II обозначает кристаллы соединения А типа II.
Как показано в таблице 3, при использовании 4 исследуемых видов растворителей нельзя получить кристаллы, отличные от кристаллов соединения А типа I и типа II.
Экспериментальный пример 4
Исследование опосредованной растворителем трансформации
Кристаллы соединения В добавляют к ацетонитрилу в количестве, которое не меньше, чем количество, необходимое для получения концентрации сверхнасыщения при заданной температуре (в состоянии, когда не все добавленные кристаллы растворяются и некоторое количество кристаллов находится в нерастворенном виде), перемешивают 30 минут, после чего кристаллы отфильтровывают и анализируют с помощью порошкового рентгенодифрактометра. Результаты приведены в таблице 4.
| Таблица 4 | |
| температура (°С) | Вид кристалла |
| 25 | Не изменился |
| 40 | Не изменился |
| 55 | Не изменился |
| 67 | Не изменился |
| 80 | Примесь кристаллов соединения А типа I |
Как показано в таблице 4, было обнаружено, что при 67°С или ниже трансформация кристаллов соединения В в другие кристаллы не наблюдается, однако при 80°С часть кристаллов соединения В трансформируется в кристаллы соединения А типа I.
Следовательно, считается, что предпочтительно выбирать такие условия кристаллизации, при которых кристаллы соединения В не оставляют в ацетонитриле при температуре 70°С или выше, насколько это возможно.
Промышленное применение
Кристаллы соединения В в соответствии с настоящим изобретением представляют собой важное промежуточное соединение для получения кристаллов соединения А типа III. Используя кристаллы соединения В можно преимущественно получать кристаллы соединения А типа III.
Кроме того, кристаллы соединения В можно получить путем регуляции концентрации сверхнасыщения, следовательно, способ получения данного соединения представляет собой превосходный способ получения больших количеств лекарственного вещества (т.е. кристаллов соединения А типа III) высокого качества.
Claims (6)
1. Кристалл ацетонитрильного сольвата 6-фтор-1-метил-7-[4-(5-метил-2-оксо-1,3-диоксолен-4-ил)-метил-1-пиперазинил]-4-оксо-4Н-[1,3]тиазето[3,2-а]хинолин-3-карбоновой кислоты, имеющий пики дифракции, по меньшей мере, при 7,3°, 14,7°, 19,2° и 22,3° в спектре порошковой рентгенодифракции.
2. Кристалл по п.1, имеющий пики дифракции, по меньшей мере, при 7,3°, 12,6°, 14,7°, 16,5°, 19,2°, 22,3° и 25,8° в спектре порошковой рентгенодифракции.
3. Способ получения кристалла ацетонитрильного сольвата 6-фтор-1-метил-7-[4-(5-метил-2-оксо-1,3-диоксолен-4-ил)метил-1-пиперазинил]-4-оксо-4Н-[1,3]тиазето[3,2-а]хинолин-3-карбоновой кислоты, отличающийся тем, что кристаллизацию проводят из ацетонитрильного раствора 6-фтор-1-метил-7-[4-(5-метил-2-оксо-1,3-диоксолен-4-ил)метил-1-пиперазинил]-4-оксо-4Н-[1,3]тиазето[3,2-а]хинолин-3-карбоновой кислоты, поддерживая концентрацию сверхнасыщения (г/100 г) в интервале от 2,15 до 2,36 во время спонтанного образования центров кристаллизации.
4. Способ получения кристалла ацетонитрильного сольвата 6-фтор-1-метил-7-[4-(5-метил-2-оксо-1,3-диоксолен-4-ил)метил-1-пиперазинил]-4-оксо-4Н-[1,3]тиазето[3,2-а]хинолин-3-карбоновой кислоты, отличающийся тем, что кристаллизацию проводят из ацетонитрильного раствора 6-фтор-1-метил-7-[4-(5-метил-2-оксо-1,3-диоксолен-4-ил)метил-1-пиперазинил]-4-оксо-4Н-[1,3]тиазето[3,2-а]хинолин-3-карбоновой кислоты, поддерживая концентрацию сверхнасыщения (г/100 г) в интервале от 0,41 до 2,36 во время добавления затравочных кристаллов.
5. Способ по п.4, где температура раствора во время добавления затравочных кристаллов составляет 70°С или ниже.
6. Способ получения кристалла 6-фтор-1-метил-7-[4-(5-метил-2-оксо-1,3-диоксолен-4-ил)метил-1-пиперазинил]-4-оксо-4Н-[1,3]тиазето[3,2-а]хинолин-3-карбоновой кислоты типа III, отличающийся тем, что проводят десольвацию кристаллов ацетонитрильного сольвата 6-фтор-1-метил-7-[4-(5-метил-2-оксо-1,3-диоксолен-4-ил)метил-1-пиперазинил]-4-оксо-4Н-[1,3]тиазето[3,2-а]хинолин-3-карбоновой кислоты.
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2003124643 | 2003-04-30 | ||
| JP2003-124643 | 2003-04-30 | ||
| JP2004-006057 | 2004-01-13 | ||
| JP2004006057 | 2004-01-13 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| RU2005137156A RU2005137156A (ru) | 2006-06-10 |
| RU2335503C2 true RU2335503C2 (ru) | 2008-10-10 |
Family
ID=33422075
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| RU2005137156/04A RU2335503C2 (ru) | 2003-04-30 | 2004-04-28 | Кристаллы сольвата производного хинолинкарбоновой кислоты |
Country Status (13)
| Country | Link |
|---|---|
| US (1) | US7612205B2 (ru) |
| EP (2) | EP2354144A1 (ru) |
| JP (1) | JP4655930B2 (ru) |
| KR (1) | KR101088988B1 (ru) |
| AU (2) | AU2004234287A1 (ru) |
| BR (1) | BRPI0409930A (ru) |
| CA (1) | CA2523854C (ru) |
| ES (1) | ES2388921T3 (ru) |
| MX (1) | MXPA05011525A (ru) |
| PL (1) | PL1626051T3 (ru) |
| PT (1) | PT1626051E (ru) |
| RU (1) | RU2335503C2 (ru) |
| WO (1) | WO2004096815A1 (ru) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2484768C1 (ru) * | 2012-02-07 | 2013-06-20 | Государственное бюджетное образовательное учреждение высшего профессионального образования "Воронежская государственная медицинская академия им. Н.Н. Бурденко" Министерства здравоохранения и социального развития Российской Федерации | Способ прогнозирования течения глаукомной оптической нейропатии |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| IN2012MN02925A (ru) | 2010-06-30 | 2015-05-22 | Cipla Ltd | |
| EP3773579A4 (en) | 2018-03-26 | 2022-03-09 | Clear Creek Bio, Inc. | COMPOSITIONS AND METHODS FOR INHIBITING DIHYDROOROTATE DEHYDROGENASE |
| AU2021236757A1 (en) * | 2020-03-20 | 2022-10-13 | Clear Creek Bio, Inc. | Stable polymorphic compositions of brequinar sodium and methods of use and manufacture thereof |
| WO2021189018A1 (en) | 2020-03-20 | 2021-09-23 | Clear Creek Bio, Inc. | Methods of treating viral infections using inhibitors of nucleotide synthesis pathways |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| SU1558915A1 (ru) * | 1988-07-29 | 1990-04-23 | Пермский фармацевтический институт | 2-(5 @ -Нитрофурил)-2,3-дигидро-5Н-1,3,4-тиадиазоло[2,3-в]-хиназолинон-5, про вл ющий противомикробную активность |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0751579B2 (ja) * | 1987-11-07 | 1995-06-05 | 日本新薬株式会社 | キノリンカルボン酸誘導体 |
-
2004
- 2004-04-28 MX MXPA05011525A patent/MXPA05011525A/es active IP Right Grant
- 2004-04-28 WO PCT/JP2004/006216 patent/WO2004096815A1/ja not_active Ceased
- 2004-04-28 CA CA2523854A patent/CA2523854C/en not_active Expired - Lifetime
- 2004-04-28 RU RU2005137156/04A patent/RU2335503C2/ru active
- 2004-04-28 KR KR1020057020448A patent/KR101088988B1/ko not_active Expired - Lifetime
- 2004-04-28 AU AU2004234287A patent/AU2004234287A1/en not_active Abandoned
- 2004-04-28 JP JP2005505937A patent/JP4655930B2/ja not_active Expired - Lifetime
- 2004-04-28 PT PT04730102T patent/PT1626051E/pt unknown
- 2004-04-28 US US10/555,039 patent/US7612205B2/en not_active Expired - Lifetime
- 2004-04-28 PL PL04730102T patent/PL1626051T3/pl unknown
- 2004-04-28 EP EP11161811A patent/EP2354144A1/en not_active Withdrawn
- 2004-04-28 BR BRPI0409930-3A patent/BRPI0409930A/pt not_active Application Discontinuation
- 2004-04-28 ES ES04730102T patent/ES2388921T3/es not_active Expired - Lifetime
- 2004-04-28 EP EP04730102A patent/EP1626051B1/en not_active Expired - Lifetime
-
2010
- 2010-04-01 AU AU2010201309A patent/AU2010201309B2/en not_active Expired
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| SU1558915A1 (ru) * | 1988-07-29 | 1990-04-23 | Пермский фармацевтический институт | 2-(5 @ -Нитрофурил)-2,3-дигидро-5Н-1,3,4-тиадиазоло[2,3-в]-хиназолинон-5, про вл ющий противомикробную активность |
Non-Patent Citations (1)
| Title |
|---|
| Kazuro Kakemi at al., "Chemical Structure, physicochemical properties and stability of Prulifloxacin", IYAKUHIN KENKYU, vol. 28 (1), pp. 1-11, 1997. * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2484768C1 (ru) * | 2012-02-07 | 2013-06-20 | Государственное бюджетное образовательное учреждение высшего профессионального образования "Воронежская государственная медицинская академия им. Н.Н. Бурденко" Министерства здравоохранения и социального развития Российской Федерации | Способ прогнозирования течения глаукомной оптической нейропатии |
Also Published As
| Publication number | Publication date |
|---|---|
| RU2005137156A (ru) | 2006-06-10 |
| MXPA05011525A (es) | 2005-12-12 |
| US20070149540A1 (en) | 2007-06-28 |
| WO2004096815A1 (ja) | 2004-11-11 |
| EP1626051A1 (en) | 2006-02-15 |
| EP1626051A4 (en) | 2008-06-25 |
| AU2010201309B2 (en) | 2011-10-13 |
| AU2010201309A1 (en) | 2010-04-22 |
| ES2388921T3 (es) | 2012-10-19 |
| BRPI0409930A (pt) | 2006-04-25 |
| AU2004234287A1 (en) | 2004-11-11 |
| PL1626051T3 (pl) | 2012-10-31 |
| KR20060008930A (ko) | 2006-01-27 |
| JPWO2004096815A1 (ja) | 2006-07-13 |
| PT1626051E (pt) | 2012-08-01 |
| EP2354144A1 (en) | 2011-08-10 |
| KR101088988B1 (ko) | 2011-12-01 |
| CA2523854A1 (en) | 2004-11-11 |
| JP4655930B2 (ja) | 2011-03-23 |
| EP1626051B1 (en) | 2012-06-13 |
| CA2523854C (en) | 2012-10-16 |
| US7612205B2 (en) | 2009-11-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| RU2125052C1 (ru) | Ангидрат пароксетин гидрохлорида, сольваты пароксетин гидрохлорида и способы их получения | |
| JP2000512992A (ja) | 多形性化合物 | |
| AU2010201309B2 (en) | Crystals of quinolinecarboxylic acid derivative solvate | |
| KR20070110128A (ko) | 퀴누클리딘 유도체의 신규한 염 | |
| KR20130086534A (ko) | 익사베필론의 고체 형태 | |
| US6703410B1 (en) | Crystal forms of 3-(2,4-dichlorobenzyl)-2-methyl-n-(pentylsulfonyl)-3h-benzimidazole-5-carboxamide | |
| KR101703202B1 (ko) | 3환성 벤조피란 화합물의 신규한 결정형태 및 그 제조방법 | |
| KR100659927B1 (ko) | 다형태를갖는독사조신메실레이트의새로운형태(제iii형) | |
| CN116120347B (zh) | 芳杂环类化合物的晶型、其组合物、制备方法及其应用 | |
| US20100113550A1 (en) | New Crystal Forms | |
| US7388096B2 (en) | Crystalline forms of a factor Xa inhibitor | |
| CN116583278B (zh) | 一种吡唑并[3,4-c]吡啶化合物的晶型及其制备方法和用途 | |
| TWI707851B (zh) | 哌嗪化合物的新穎結晶 | |
| JP2008524228A (ja) | Xa因子阻害剤の結晶フォーム | |
| CA3127469C (en) | Polymorphic forms of a substituted-quinoxaline-type bridged-piperidine compound | |
| CN119118911A (zh) | 一种苯甲酰胺类化合物的新晶型 | |
| EP2154137A1 (en) | Crystalline form of moxifloxacin base | |
| NO861369L (no) | Fremgangsmaate for fremstilling av et krystallinsk, vannfritt natriumsalt av 19-deoksyaglykon dianemycin. | |
| JP2008524253A (ja) | ケモカインレセプターアンタゴニストの固体形態およびその使用方法 | |
| HK1186174A (en) | Novel crystals of substituted phenylalkanoic acid and production process | |
| HK1199027B (en) | Crystalline form i of tyrosine kinase inhibitor dimaleate and preparation methods thereof |