RU2195312C2 - СПОСОБЫ И КОМПОЗИЦИИ, ИСПОЛЬЗУЕМЫЕ ДЛЯ ИНГИБИРОВАНИЯ αvβ5-ОПОСРЕДОВАННОГО АНГИОГЕНЕЗА - Google Patents
СПОСОБЫ И КОМПОЗИЦИИ, ИСПОЛЬЗУЕМЫЕ ДЛЯ ИНГИБИРОВАНИЯ αvβ5-ОПОСРЕДОВАННОГО АНГИОГЕНЕЗА Download PDFInfo
- Publication number
- RU2195312C2 RU2195312C2 RU98123834/14A RU98123834A RU2195312C2 RU 2195312 C2 RU2195312 C2 RU 2195312C2 RU 98123834/14 A RU98123834/14 A RU 98123834/14A RU 98123834 A RU98123834 A RU 98123834A RU 2195312 C2 RU2195312 C2 RU 2195312C2
- Authority
- RU
- Russia
- Prior art keywords
- angiogenesis
- tissue
- antagonist
- compound
- tumor
- Prior art date
Links
- 0 CCCCCOC1=CC=C(C[C@](*I(CC(CC*C2)(C(C)C)C2=O)(=C)=O)C(*)=O)CC1 Chemical compound CCCCCOC1=CC=C(C[C@](*I(CC(CC*C2)(C(C)C)C2=O)(=C)=O)C(*)=O)CC1 0.000 description 1
Images











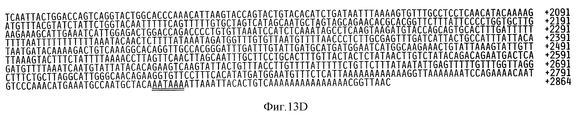







Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/04—Peptides having up to 20 amino acids in a fully defined sequence; Derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/43—Enzymes; Proenzymes; Derivatives thereof
- A61K38/46—Hydrolases (3)
- A61K38/48—Hydrolases (3) acting on peptide bonds (3.4)
- A61K38/4886—Metalloendopeptidases (3.4.24), e.g. collagenase
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C279/00—Derivatives of guanidine, i.e. compounds containing the group, the singly-bound nitrogen atoms not being part of nitro or nitroso groups
- C07C279/04—Derivatives of guanidine, i.e. compounds containing the group, the singly-bound nitrogen atoms not being part of nitro or nitroso groups having nitrogen atoms of guanidine groups bound to acyclic carbon atoms of a carbon skeleton
- C07C279/14—Derivatives of guanidine, i.e. compounds containing the group, the singly-bound nitrogen atoms not being part of nitro or nitroso groups having nitrogen atoms of guanidine groups bound to acyclic carbon atoms of a carbon skeleton being further substituted by carboxyl groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C311/00—Amides of sulfonic acids, i.e. compounds having singly-bound oxygen atoms of sulfo groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/01—Sulfonamides having sulfur atoms of sulfonamide groups bound to acyclic carbon atoms
- C07C311/02—Sulfonamides having sulfur atoms of sulfonamide groups bound to acyclic carbon atoms of an acyclic saturated carbon skeleton
- C07C311/03—Sulfonamides having sulfur atoms of sulfonamide groups bound to acyclic carbon atoms of an acyclic saturated carbon skeleton having the nitrogen atoms of the sulfonamide groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C311/06—Sulfonamides having sulfur atoms of sulfonamide groups bound to acyclic carbon atoms of an acyclic saturated carbon skeleton having the nitrogen atoms of the sulfonamide groups bound to hydrogen atoms or to acyclic carbon atoms to acyclic carbon atoms of hydrocarbon radicals substituted by carboxyl groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C311/00—Amides of sulfonic acids, i.e. compounds having singly-bound oxygen atoms of sulfo groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/01—Sulfonamides having sulfur atoms of sulfonamide groups bound to acyclic carbon atoms
- C07C311/10—Sulfonamides having sulfur atoms of sulfonamide groups bound to acyclic carbon atoms of a saturated carbon skeleton containing rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D263/00—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings
- C07D263/02—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings
- C07D263/30—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D263/34—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D263/36—One oxygen atom
- C07D263/38—One oxygen atom attached in position 2
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/745—Blood coagulation or fibrinolysis factors
- C07K14/75—Fibrinogen
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2839—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the integrin superfamily
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2839—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the integrin superfamily
- C07K16/2848—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the integrin superfamily against integrin beta3-subunit-containing molecules, e.g. CD41, CD51, CD61
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/14—Hydrolases (3)
- C12N9/48—Hydrolases (3) acting on peptide bonds (3.4)
- C12N9/50—Proteinases, e.g. Endopeptidases (3.4.21-3.4.25)
- C12N9/64—Proteinases, e.g. Endopeptidases (3.4.21-3.4.25) derived from animal tissue
- C12N9/6421—Proteinases, e.g. Endopeptidases (3.4.21-3.4.25) derived from animal tissue from mammals
- C12N9/6489—Metalloendopeptidases (3.4.24)
- C12N9/6491—Matrix metalloproteases [MMP's], e.g. interstitial collagenase (3.4.24.7); Stromelysins (3.4.24.17; 3.2.1.22); Matrilysin (3.4.24.23)
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2602/00—Systems containing two condensed rings
- C07C2602/36—Systems containing two condensed rings the rings having more than two atoms in common
- C07C2602/42—Systems containing two condensed rings the rings having more than two atoms in common the bicyclo ring system containing seven carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/73—Inducing cell death, e.g. apoptosis, necrosis or inhibition of cell proliferation
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10S—TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10S435/00—Chemistry: molecular biology and microbiology
- Y10S435/975—Kit
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Immunology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Genetics & Genomics (AREA)
- Gastroenterology & Hepatology (AREA)
- Molecular Biology (AREA)
- Biochemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Biophysics (AREA)
- Zoology (AREA)
- Epidemiology (AREA)
- Wood Science & Technology (AREA)
- Biomedical Technology (AREA)
- Hematology (AREA)
- General Engineering & Computer Science (AREA)
- Toxicology (AREA)
- Rheumatology (AREA)
- Microbiology (AREA)
- Biotechnology (AREA)
- Diabetes (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Physical Education & Sports Medicine (AREA)
- Pulmonology (AREA)
- Pain & Pain Management (AREA)
- Ophthalmology & Optometry (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
Abstract
Изобретение относится к медицине, в частности к способу ингибирования ангиогенеза в тканях с использованием антагонистов витронектина αvβ5. αvβ5-Опосредованный ангиогенез коррелирует с действием цитокинов, включая эндотелиальный фактор роста, трансформирующий фактор роста-α и эпидермальный фактор роста. Ингибирование αvβ5-опосредованного ангиогенеза особенно предпочтительно при эндотелиальных глазных болезнях с реваскуляризацией, при опухолевом росте и при воспалительных заболеваниях, где указанное ингибирование осуществляют с использованием терапевтических композиций, содержащих антагонисты интегрина αvβ5. Изобретение позволяет использовать композиции для лечения заболеваний, связанных с ангиогенезом. 5 с. и 31 з.п.ф-лы, 21 ил., 2 табл.
Description
Область техники, к которой относится изобретение
Изобретение относится к области медицины, более конкретно к способам и композициям для ингибирования αvβ5 -опосредованного ангиогенеза тканей с использованием антагонистов рецептора αvβ5 витронектина.
Изобретение относится к области медицины, более конкретно к способам и композициям для ингибирования αvβ5 -опосредованного ангиогенеза тканей с использованием антагонистов рецептора αvβ5 витронектина.
Уровень техники
Интегрины представляют собой класс клеточных рецепторов, которые, как известно, связываются с белками внеклеточного матрикса и тем самым опосредуют взаимодействие между клетками и между клеткой и внеклеточным матриксом, которое обычно называют событиями клеточной адгезии. Несмотря на то, что многие интегрины и их соответствующие лиганды описаны в литературе, биологическая функция многих интегринов остается невыясненной. Интегриновые рецепторы представляют собой семейство белков, которые имеют общие структурные характеристики нековалентных гетеродимерных гликопротеиновых комплексов, образованных α- и β-субъединицами.
Интегрины представляют собой класс клеточных рецепторов, которые, как известно, связываются с белками внеклеточного матрикса и тем самым опосредуют взаимодействие между клетками и между клеткой и внеклеточным матриксом, которое обычно называют событиями клеточной адгезии. Несмотря на то, что многие интегрины и их соответствующие лиганды описаны в литературе, биологическая функция многих интегринов остается невыясненной. Интегриновые рецепторы представляют собой семейство белков, которые имеют общие структурные характеристики нековалентных гетеродимерных гликопротеиновых комплексов, образованных α- и β-субъединицами.
В настоящее время известно, что рецептор витронектина, названный так благодаря присущей ему способности к преимущественному связыванию с витронектином, относится к классу, состоящему из трех различных интегринов, обозначаемых αvβ1, αvβ3 и αvβ5. Horton, Int. J. Exp. Pathol. , 71:741-759 (1990). αvβ1 связывается с фибронектином и витронектином, αvβ3 связывается с лигандами широкого ряда, включая фибрин, фибриноген, ламинин, тромбоспондин, витронектин, фактор фон Виллебранда, остеоспонтин, и костный сиалопротеин I. αvβ5 связывается с витронектином. Конкретная клеточно-адгезивная роль, которую эти три интегрина играют во многих клеточных взаимодействиях в тканях, пока еще исследуется. Однако очевидно, что существуют различные биологические функции, а также существуют различные интегрины и субъединицы, имеющие одинаковые биологические специфичности.
Одним из важных сайтов, которые интегрины распознают в лигандах, является трипептидная последовательность, аргинин - глицин - аспарагиновая кислота (RGD). RGD присутствует во всех лигандах, идентифицированных выше для интегриновых рецепторов витронектина. Этот сайт распознавания RGD может имитироваться полипептидами ("пептидами"), которые содержат последовательность RGD, и такие RGD-пептиды являются известными ингибиторами функции интегрина. Однако важно отметить, что в зависимости от последовательности и структуры RGD-пептида специфичность ингибирования конкретных интегринов-мишеней может быть изменена.
Обсуждение сайта распознавания RGD можно найти в работах Pierschbacher et al. , Nature, 309:30-33 (1984), Pierschbacher et al., Proc. Natl. Acad. Sci., USA, 81:5985-5988 (1984). Различные RGD-полипептиды с различной специфичностью по отношению к интегрину были также описаны Grant et al., Cell, 58: 933-943 (1989), Cheresh et al., Cell., 58:945-953 (1989), Aumailley et al. , FEBS Letts. , 291: 50-54 (1991), и Pfaff et al., J.Biol.Chem., 269: 20233-20238 (1994), и в патентах США 4517686, 4589881, 4578079, 4614517, 4661111, 4792525, 4683291, 4879237, 4988621, 5041380 и 5061693.
Ангиогенез также называют реваскуляризацией, то есть процессом васкуляризации тканей, который заключается в образовании новых развивающихся кровеносных сосудов в ткани. Этот процесс опосредуется инфильтрацией эндотелиальных клеток и клеток гладких мышц. Очевидно, этот процесс протекает в соответствии с одним из следующих механизмов: 1) сосуды могут разрастаться из уже существующих сосудов; 2) образование новых сосудов может быть вызвано клетками-предшественниками (сосудогенез); или 3) существующие мелкие сосуды могут увеличиваться в диаметре. Blood et al., Bioch. Biophys. Acta., 1032: 89-118 (1990). Известно, что эндотелиальные клетки сосудов содержат, по крайней мере, пять RGD-зависимых интегринов, включая рецептор витронектина (αvβ3 или αvβ5), рецептор коллагена Типов I и IV (α1β1), рецептор ламинина (α2β1), рецептор фибронектина/ламинина/коллагена (α3β1), и рецептор фибронектина (α5β1). Davis et al., J.Cell.Biochem., 51:206-218 (1993). Известно, что мышечные клетки желудка содержат, по крайней мере, шесть RGD-зависимых интегринов, включая α5β1, αvβ3 и αvβ5.
Ангиогенез является важным процессом при неонатальном росте, но он также играет важную роль в заживлении ран и в патогенезе широкого ряда клинически серьезных болезней, включая воспаление тканей, артрит, псориаз, рак, диабетическая ретинопатия, дегенерация желтого пятна, и другие реваскулярные заболевания глаз. Эти ассоциированные с ангиогенезом клинические состояния называются ангиогенными заболеваниями. Folkman et al., Science, 235:442-447 (1987). Ангиогенез, в основном, отсутствует в тканях взрослого индивидуума или в зрелых тканях, хотя он наблюдается при заживлении ран и в цикле роста желтого тела. См. например, Moses et al., Science, 248: 1408-1410 (1990).
Ангиогенез является важным процессом при неонатальном росте, но он также играет важную роль в заживлении ран и в патогенезе широкого ряда клинически серьезных болезней, включая воспаление тканей, артрит, псориаз, рак, диабетическая ретинопатия, дегенерация желтого пятна, и другие реваскулярные заболевания глаз. Эти ассоциированные с ангиогенезом клинические состояния называются ангиогенными заболеваниями. Folkman et al., Science, 235:442-447 (1987). Ангиогенез, в основном, отсутствует в тканях взрослого индивидуума или в зрелых тканях, хотя он наблюдается при заживлении ран и в цикле роста желтого тела. См. например, Moses et al., Science, 248: 1408-1410 (1990).
Ингибирование клеточной адгезии in vitro с использованием моноклональных антител, обладающих иммуноспецифичностью для различных α- или β-субъединиц интегрина, основано на участии рецептора αvβ3 витронектина в клеточной адгезии ряда типов клеток, включая эндотелиальные клетки микрососудов. Davis et al., J.Cell.Biol., 51:206-218 (1993). Кроме того, в работе Nicosia et al. , Am. J. Pathol., 138:829-833 (1991) было описано использование RGD-пептида, GRGDS, для ингибирования in vitro-образования "микрососудов" из крысиной аорты, культивированной в коллагеновом геле.
Однако ингибирование образования "микрососудов" in vitro в культуре коллагенового геля не является моделью для ингибирования ангиогенеза в тканях, поскольку эта модель не указывает на то, что микрососудистые структуры являются аналогичными капиллярными разрастаниям или что образование микрососудов в культуре коллагенового геля является аналогичным росту новых сосудов в интактных тканях, таких как артритная ткань, опухолевая ткань или патологическая ткань, в которой было бы желательным ингибировать ангиогенез.
Роль αvβ3 в ангиогенезе была недавно подтверждена. См. Brooks et al., Science, 264-571 (1994). Было показано, что интегрин экспрессируется на кровеносных сосудах в гранулированной ткани раны человека, но не экспрессируется в нормальной коже. Моноклональные антитела против рецептора αvβ5 ингибировали ангиогенез, индуцированный факторами роста (цитокинами), основным фактором роста фибробластов (bFGF) и фактором-α некроза опухоли (TNF-α), а также фрагментами меланомы. Однако антагонисты ингибируют лишь вновь образованные сосуды и не ингибируют уже сформировавшиеся сосуды. Кроме того, было также показано, что специфические линейные и циклические RGD-содержащие пептиды ингибируют реваскуляризацию.
Было высказано предположение, что ингибирование ангиогенеза может быть использовано в терапии для ограничения роста опухолей. Было предположено, что ингибирование ангиогенеза может быть осуществлено (1) путем ингибирования высвобождения "ангиогенных молекул", таких как как bFGF (основный фактор роста фибробластов); (2) путем нейтрализации ангиогенных молекул, например, с использованием антител против bFGF; и (3) путем ингибирования ответа эндотелиальных клеток на ангиогенные стимуляторы. Этот последний способ заслуживает особого внимания и был описан в работе Folcman et al., Cancer Biology, 3: 89-96 (1992), где было указано несколько ингибиторов клеточно-эндотелиального ответа, включая ингибитор коллагеназы, ингибиторы базально-мембранного метаболизма, ангиостатические стероиды, грибковые ингибиторы ангиогенеза, тромбоцитарный фактор 4, тромбоспондин, лекарственные средства против артрита, такие как D-пеницилламин и тиомалат золота, аналоги витамина D3, альфа-интерферон, и т.п., которые могут быть использованы для ингибирования ангиогенеза. Кроме того, были предложены и другие ингибиторы ангиогенеза, см. Blood et al., Bioch.Biophys.Acta., 1032:89-118 (1990), Moses et al. , Science, 248: 1408-1410 (1990), Ingber et al., Lab. Invest, 59:44-51 (1988), и патенты США 5092885, 5112946, 5192744 и 5202352.
Однако до появления настоящего изобретения не было высказано каких-либо предположений о роли интегрина αvβ5 в ангиогенезе и не было подтверждено, что какие-либо ингибиторы ангиогенеза, описанные в предшествующих работах, направлены на ингибирование αvβ5. Кроме того, помимо настоящего изобретения не было работ, в которых было бы описано участие интегрина αvβ5 в реваскуляризации, а в частности реваскуляризации, индуцированной факторами роста, сосудистым эндотелиальным фактором роста (VEGF), трансформирующим фактором роста-α (TGF-α), и эпидермальным фактором роста (EGF).
Хотя число факторов роста, участвующих в регулировании ангиогенеза, является ограниченным, однако для конверсии состояния покоя в состояние реваскуляризации существуют различные уровни регуляции. См., D'Amore, Investigative Ophthal, Visual Sci., 35:3974-3979 (1994). Некоторые из факторов роста, которые участвуют в ангиогенезе, регулируются на уровне синтеза, а другие регулируются в состоянии активации. Эти клеточные события происходят в том случае, когда сосуды, находящиеся в состоянии покоя, подвергаются реваскуляризации после ранения или ишемии.
В частности, VEGF является, очевидно, главным медиатором ангиогенеза в первичной опухоли и при ишемических заболеваниях глаз. См. обзор Folkman, Nature Medicine, 1:27-31 (1995). VEGF представляет собой гомодимер в 46 килодальтон (кДа), который является специфичным для эндотелиальных клеток ангиогенным (Ferrara et al., Endocrin Rev., 13:18-32 (1992)), и сосудопроницаемым фактором (Senger et al., Cancer Res., 46:5629-5632 (1986), который связывается с высокоаффинными мембраноассоциированными рецепторами с тирозинкиназной активностью (Jakeman et al., J.Clin Invest., 89-244-253 (1992)).
Недавно было показано, что активация тирозинкиназного рецептора стимулирует интегринзависимую миграцию клеток к белкам внеклеточного матрикса. В частности, Klemke et al., J.Cell.Biol., 127:859-866 (1994) считают, что EGF-рецептор (EGFR) тирозинкиназы участвует в стимуляции клеточной подвижности, но не участвует в адгезии клеток карциномы поджелудочной железы человека FG к витронектину с использованием интегрина αvβ5. Эти авторы непосредственно показали, что "захват" EGFR лиганда EGF стимулирует активацию EGFR тирозинкиназы, которая, в конечном счете, стимулирует РКС (протеинкиназа С)-зависимый метаболизм, приводящий к индуцированию αvβ5-зависимой клеточной миграции витронектинового субстрата, на который клетки обычно не способны мигрировать. Таким образом, исследования Klemke и др. свидетельствуют о корреляции присутствия цитокинов, в частности EGF, с интегринзависимой активностью миграции клеток. Было показано, что активация РКС приводит к регуляции ангиогенеза в системе модели хориоаллантоисной мембраны цыпленка. См. Tsopanoglou et al., J. Vasc. Res., 30:202-208 (1993). Эти авторы идентифицировали специфические активаторы и ингибиторы РКС, которые соответственно стимулируют и ингибируют ангиогенез в экспериментальной системе.
Однако ни у Klemke и др., ни у Tsopanoglou и др. не обсуждается роль цитокинов и экспрессии и/или активации интегрина αvβ5 в стимуляции ангиогенеза в различных условиях и при различных патологиях, а также их ингибирование αvβ5-специфическими антагонистами.
Недавно, в системе обезьяньей модели заболевания глаз было экспериментально показано, что ишемия сетчатки, индуцированная окклюзией вены сетчатки глаза, приводит к продуцированию VEGF в камере глазного яблока. Эти данные совпадают с реваскуляризацией радужной оболочки глаза, которая наблюдалась и описана Miller et al., Am. J. Path. 145:574-584 (1994). Кроме того, с использованием мышиной экспериментальной модели пролиферативной ретинопатии, в которой была индуцирована гипоксия, были получены данные, свидетельствующие о том, что уровень матричной РНК VEGF возрастает в течение 6-12 часов относительной гипоксии и остается повышенным вплоть до образования новых сосудов. По мере замедления процесса образования новых сосудов экспрессируется VEGF, Pierce et al., Proc. Natl. Acad. Sci., USA, 92:905-909 (1995).
Таким образом, недавно полученные данные, продемонстрированные на экспериментальных моделях ишемии, показали, что индуцирование VEGF коррелирует с ишемией и последующей реваскуляризацией. VEGF и другие факторы роста также участвуют в других состояниях и патологиях с образованием новых сосудов, как описано в обзоре Folkman, Nature Medicine, 1:27-31 (1995).
В работах Folkman и др. также систематизированы современные клинические методы, используемые для регуляции нежелательного ангиогенеза. Пациентам, участвующим в клинических испытаниях, проводили лечение ангиогенными ингибиторами, включая тромбоцитарный фактор 4, фумагиллиновое производное, карбокси-аминотриазол, и т.п. Однако ни в этих работах, ни в современной литературе по терапии нет каких-либо указаний на корреляцию экспрессии αvβ5 ангиогенезом и, в частности, указаний на то, что этот ангиогенез индуцируется VEGF. Таким образом, до появления настоящего изобретения ни в одной работе не было описано применение терапевтической схемы лечения с использованием антагонистов αvβ5 для регуляции ангиогенеза в тканях, подвергающихся ангиогенезу, который коррелирует с присутствием и активацией αvβ5.
Поэтому помимо исследований, упоминаемых в настоящем описании и касающихся αvβ5 и связи факторов роста с ангиогенезом, авторам настоящей заявки не известны какие-либо другие доказательства того, что ангиогенез в тканях может быть ингибирован с использованием ингибиторов αvβ5-опосредованной клеточной адгезии. В частности, нигде ранее не было продемонстрировано, что функция αvβ5 необходима для ангиогенеза в тканях или что антигонисты αvβ5 могут ингибировать ангиогенез в тканях, в частности, при заболеваниях глаз с реваскуляризацией.
Поэтому помимо исследований, упоминаемых в настоящем описании и касающихся αvβ5 и связи факторов роста с ангиогенезом, авторам настоящей заявки не известны какие-либо другие доказательства того, что ангиогенез в тканях может быть ингибирован с использованием ингибиторов αvβ5-опосредованной клеточной адгезии. В частности, нигде ранее не было продемонстрировано, что функция αvβ5 необходима для ангиогенеза в тканях или что антигонисты αvβ5 могут ингибировать ангиогенез в тканях, в частности, при заболеваниях глаз с реваскуляризацией.
Сущность изобретения
В настоящем изобретении было продемонстрировано, что помимо механизма αvβ5-опосредованного ангиогенеза в тканях существует также и другой новый зависимый механизм ангиогенеза. Таким образом, в настоящем изобретении описаны ингибиторы интегрина αvβ5, которые могут ингибировать ангиогенез. Кроме того, в настоящем изобретении показано, что αvβ5-опосредованная активность в стимулировании ангиогенеза коррелирует с активацией фактором роста (цитокином) рецепторов фактора роста тирозинкиназ и протеинкиназы С (РКС). Факторами роста (цитокинами), которые действуют подобным образом, являются эндотелиальный фактор роста (VEGF), трансформирующий фактор роста-α (TGF-α), эпидермальный фактор роста (EGF) и т.п.
зависимый механизм ангиогенеза. Таким образом, в настоящем изобретении описаны ингибиторы интегрина αvβ5, которые могут ингибировать ангиогенез. Кроме того, в настоящем изобретении показано, что αvβ5-опосредованная активность в стимулировании ангиогенеза коррелирует с активацией фактором роста (цитокином) рецепторов фактора роста тирозинкиназ и протеинкиназы С (РКС). Факторами роста (цитокинами), которые действуют подобным образом, являются эндотелиальный фактор роста (VEGF), трансформирующий фактор роста-α (TGF-α), эпидермальный фактор роста (EGF) и т.п.
В настоящем изобретении было продемонстрировано, что помимо механизма αvβ5-опосредованного ангиогенеза в тканях существует также и другой новый
В настоящем изобретении описаны способы ингибирования ангиогенеза в тканях, предусматривающие введение в эту ткань композиции, содержащей ангиогенез-ингибирующее количество антагониста интегрина αvβ5.
Тканью, подвергающейся лечению, может быть любая ткань, в которой желательно ингибировать ангиогенез, такая как патологическая ткань, где происходит образование новых сосудов. Примерами такой ткани являются глазная ткань, подвергаемая реваскуляризации; воспалительная ткань; твердые опухоли; метастазы; ткани, подвергающиеся рестенозу, и т.п. В предпочтительных вариантах осуществления изобретения образование новых сосудов ассоциируемое с экспрессией αvβ5, является результатом воздействия факторов роста, VEGF, TGF-α и EGF.
Тканью, подвергающейся лечению, может быть любая ткань, в которой желательно ингибировать ангиогенез, такая как патологическая ткань, где происходит образование новых сосудов. Примерами такой ткани являются глазная ткань, подвергаемая реваскуляризации; воспалительная ткань; твердые опухоли; метастазы; ткани, подвергающиеся рестенозу, и т.п. В предпочтительных вариантах осуществления изобретения образование новых сосудов ассоциируемое с экспрессией αvβ5, является результатом воздействия факторов роста, VEGF, TGF-α и EGF.
Особенно предпочтительными являются терапевтические способы, направленные на ингибирование VEGF-индуцированной васкуляризации в тканях, таких как ткани глаз, где ангиогенез особенно явно выражен при их заболевании, включая такие заболевания, как диабетическая ретинопатия (также называемая пролиферативной диабетической ретинопатией), возрастная дегенерация желтого пятна, предполагаемый гистоплазмоз глаз, ретинопатия при преждевременном развитии, серповидноклеточная ретинопатия и реваскулярная глаукома. В других предпочтительных вариантах терапевтические способы направлены на ингибирование ангиогенеза, который происходит при реваскулярных нарушениях роговицы, таких как трансплантация роговицы, герпетический кератит, сифилитический кератит, птеригий, неоваскулярный паннус (поверхностный диффузный кератит), ассоциированный с использованием контактных линз, и т.п.
Антагонист интегрина αvβ5, предназначенный для использования в целях настоящего изобретения, обладает способностью ингибировать связывание с αvβ5 и конкурентно ингибировать способность αvβ5 связываться с природным витронектиновым лигандом. Предпочтительно, чтобы этот антагонист обладал специфичностью к αvβ5, а не к другим интегринам. В конкретном предпочтительном варианте антагонист αvβ5 ингибирует связывание витронектина или других RGD-содержащих лигандов с αvβ5, но в основном не ингибирует связывание витронектина с αvβ5 или α11bβ3. Предпочтительным антагонистом αvβ5 может быть гибридный полипептид, линейный или циклический полипептид, дериватизированный полипептид, моноклональное антитело или его фрагменты, или неорганическая молекула, которая имитирует αvβ5-лиганд, и которую часто называют органическим миметиком, где все указанные антагонисты специфически взаимодействуют с αvβ5.
Введение антагонистов αvβ5 настоящего изобретения может быть внутриглазным, внутривенным, чрескожным, внутрисуставным, внутримышечным и пероральным. В других предпочтительных вариантах осуществления изобретения, такое введение координируют с химиотерапевтической схемой введения для подавления образования опухолей и метастазов рака.
Введение антагонистов αvβ5 настоящего изобретения может быть внутриглазным, внутривенным, чрескожным, внутрисуставным, внутримышечным и пероральным. В других предпочтительных вариантах осуществления изобретения, такое введение координируют с химиотерапевтической схемой введения для подавления образования опухолей и метастазов рака.
Краткое описание фигур
Фигуры являются частью настоящего описания.
Фигуры являются частью настоящего описания.
Фиг.1A-1D иллюстрируют ингибирование цитокин-индуцированного ангиогенеза в роговице глаза кролика антителами-антагонистами интегрина αv. Индуцирование ангиогенеза путем обработки либо bFGF, либо VEGF и эффекты их обработки антителами-антагонистами интегрина αv, PIF6 (αvβ5) и LM609 (αvβ5), описаны в примере 4. OD и OS соответствуют правому и левому глазу лабораторного кролика. Большие стрелки указывают на ангиогенез роговицы с отеком, а маленькие стрелки указывают на нормальные сосуды лимба конъюнктивы. На фиг.1А и 1В проиллюстрировано bFGF-индуцирование ангиогенеза, а на фиг.1С и 1D показано VEGF-индуцирование ангиогенеза. На фиг.1А и 1С показаны роговицы кролика, обработанные антителом PIG6, а на фиг.1В и 1D показаны роговицы кролика, обработанные антителом LM609.
На фиг. 2А и 2В изображены гистограммы, иллюстрирующие среднюю площадь реваскуляризации в мм2 ± стандартная ошибка (n=8 для каждой из двух серий) после индуцирования соответственно либо bFGF, либо VEGF с последующей обработкой моноклональным антителом (mAb) либо P1F6, либо LM609. Результаты обсуждаются в примере 4.
На фиг.3А и 3В приведены гистограммы, иллюстрирующие количественные результаты. Гистограммы иллюстрируют график индекса ангиогенеза (по оси Y) в зависимости от контроля и обработки антителом. Фиг. 3А и 3В соответственно иллюстрируют bFGF- и VEGF-индуцированный ангиогенез. Результаты обсуждаются в примере 4.
На фиг.4А и 4В приведены гистограммы, иллюстрирующие количественные результаты, показанные на фиг.3A-3F. На приведенных гистограммах индекс ангиогенеза отложен по оси Y в зависимости от контроля и обработки антителом. Фиг. 4А и 4В соответственно иллюстрируют bFGF- и VEGF-индуцированный ангиогенез. Результаты обсуждаются в примере 6.
На фиг.5А-5Е проиллюстрировано влияние моноклональных антител против интегрина и влияние кальфостина С на САМ-ангиогенез, индуцированный отдельно цитокинами bFGF, VEGF и TGF-α. Была также проведена оценка РМА. Анализы и результаты обсуждаются в примере 6. Результаты показаны в виде гистограммы, где на оси Y отложен индекс ангиогенеза, а на оси Х контроль или различные ингибиторы. На фиг.5А-5Е соответственно проиллюстрирован ангиогенез, индуцированный bFGF, TNF-α, VEGF, TGF-α и РМА.
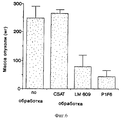
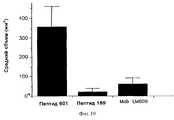
На фиг. 6 приведена гистограмма, иллюстрирующая влияние обработки антителом на рост меланомы CS1 в САМ куриного эмбриона, анализ которого осуществляли, как описано в примере 5С и 6D. На этой гистограмме по оси Y отложена масса опухолей в миллиграммах (мг) в зависимости от различных обработок, отложенных по оси X. CSAT - контрольное антитело, направленное против β1-субъединицы интегрина. LM609 и P1F6 были описаны выше.
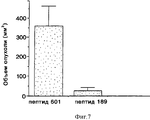
На фиг. 7 приводится гистограмма, иллюстрирующая влияние контроля по сравнению с влиянием пептида-антагониста интегрина αvβ5, меченого пептида 189 (SEQ ID No: 9), на опухолевый рост меланомы, измеренный в виде объема опухоли в мм3 (по оси Y). Анализ и результаты описаны в примере 8.
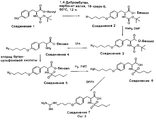
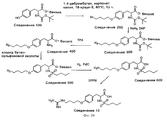
На фиг. 8 проиллюстрирован синтез Соединения 7, описанный в примере 10A-G.
На фиг. 9 проиллюстрирован синтез Соединения 9, описанный в примере 10А-С; Н-1.
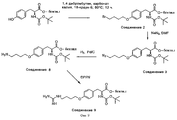
На фиг. 10 проиллюстрирован синтез Соединения 10, описанный в примере 10J.
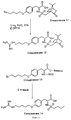
На фиг.11 проиллюстрирован синтез Соединений 12 и 14, описанный в примерах 10K-L и 10M-N соответственно.
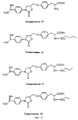
На фиг. 12 показаны химические структуры Соединения 15, Соединения 16, Соединения 17, и Соединения 18. Синтез этих соединений подробно описан в примере 100-R.
На фиг. 13А и 13В показана линейная кДНК-последовательность куриного ММР-2 вместе с выведенной аминокислотной последовательностью, показанной во второй строке. На третьей и четвертой строках соответственно показана выведенная аминокислотная последовательность ММР-2 человека и цыпленка, описанная в примере 7. кДНК-последовательность цыпленка представлена в SEQ ID No:23 вместе с кодированной аминокислотной последовательностью, которая также представлена отдельно в SEQ 1D No:24. Номер первого нуклеотида 5'-нетранслируемой области и области, кодирующей профермент, обозначен на фиг.13А отрицательным числом, а в Списке последовательностей, он обозначен числом 1, в результате чего кажется, что эта последняя последовательность длиннее, чем на фигуре, однако нуклеотидная последовательность, изображенная на фигуре, по своей длине идентична последовательности, представленной в Списке последовательностей, и отличается от нее только нумерацией. В соответствии с этим ссылки на положения нуклеотидов куриной последовательности или на последовательность ММР-2 в данном описании, например, на праймеры, используемые при амплификации фрагментов ММР-2, относятся к положениям нуклеотидов, указанных на фигуре, а не в Списке последовательностей.
На фиг. 14 показана последовательность аминокислотных остатков зрелого белка ММР-2, имеющего 631 остаток. Положения аминокислотных остатков фрагментов, происходящих от ММР-2 человека, соответствуют положениям, указанным на фиг. Последовательность аминокислотных остатков представлена в SEQ 1D N 25.
На фиг.15 показано влияние пептидов 85189 и инертной соли аналога 121974 на VEGF-индуцированный ангиогенез в СДМ-модели, описанной в примере 6А. Это влияние сравнивали с необработанными (отмеченными как NT) препаратами и препаратами, обработанными контрольным (отмеченным 69601) пептидом. Влияние на ангиогенез определяли путем измерения числа точек ветвления, как описано в примере 6А.
На фиг.16, 17 и 18 соответственно показано уменьшение массы опухоли для опухолей UCLAP-3, M21-L и FgM после внутривенного введения контрольного пептида 69601 и антагониста, как описано ниже в примере 6D. Данные выражали в виде кривой зависимости массы опухоли (ось Y) от обработок пептидом (ось X).
На фиг.19 проиллюстрировано воздействие пептидов и антител на опухолевый рост меланомы в химерной модели "мышь-человек", как описано ниже в примере 8. Была также проведена оценка контрольного пептида 69601 (отмечен 601) и антагониста 85189 (отмечен 189). Тестируемым антителом было антитело LM609. Объем опухоли в мм3 (ось Y) определяли в зависимости от различных обработок (ось X).
На фиг. 20А и 20В соответственно показано влияние антагониста 85189 (отмечен 189) по сравнению с контрольным пептидом (отмечен 601) на уменьшение объема и сырой массы опухоли M21-L в зависимости от интервала доз 10, 50 и 250 мкг на инъекцию, как описано ниже в примере 8.
На фиг. 21А и 21В показана эффективность пептида-антагониста 85189 (отмечен 189, сплошная линия и заштрихованные кружки), по сравнению с контрольным пептидом 69601 (отмечен 601, пунктирная линия и незаштрихованные квадраты) в уменьшении объема опухоли M21-L в модели "мышь-человек" при двух различных режимах обработки, как описано в примере 8. Была построена кривая зависимости объемов опухоли в мм3 (ось Y) от времени в днях (ось X).
Сведения, подтверждающие осуществимость изобретения
А. Определения
Аминокислотный остаток - аминокислота, образованная в результате химического расщепления (гидролиза) полипептида по его пептидным связям. Аминокислотными остатками, описанными в настоящей заявке, являются предпочтительно "L" изомерные формы. Однако любой L-аминокислотный остаток может быть заменен на остаток изомерной формы "D" при условии, что при этом будут сохранены нужные функциональные свойства полипептида.
А. Определения
Аминокислотный остаток - аминокислота, образованная в результате химического расщепления (гидролиза) полипептида по его пептидным связям. Аминокислотными остатками, описанными в настоящей заявке, являются предпочтительно "L" изомерные формы. Однако любой L-аминокислотный остаток может быть заменен на остаток изомерной формы "D" при условии, что при этом будут сохранены нужные функциональные свойства полипептида.
NH2 означает свободную группу, присутствующую на амино-конце полипептида. В соответствии со стандартной номенклатурой полипептидов (описанной в J. Biol. Chem. , 243:3552-59 (1969) и принятой в $. 822(b)(2) статьи 37 CFR) в таблице A приводятся сокращения, принятые для обозначения аминокислотных остатков.
Кроме того, ниже приведены следующие обозначения:
ВОС - Трет-бутилоксикарбонил
DCCl - Дициклогексилкарбодиимид
DMF - Диметилформамид
Оме - Метокси
HOBt - 1-Гидроксибензотриазол
Следует отметить, что все последовательности аминокислотных остатков представлены формулами, где левая и правая ориентация имеет общепринятое направление от амино-конца к карбокси-концу. Кроме того, следует отметить, что черточка в начале или в конце последовательности аминокислотных остатков указывает на пептидную связь с другой последовательностью в одном или нескольких аминокислотных остатках.
ВОС - Трет-бутилоксикарбонил
DCCl - Дициклогексилкарбодиимид
DMF - Диметилформамид
Оме - Метокси
HOBt - 1-Гидроксибензотриазол
Следует отметить, что все последовательности аминокислотных остатков представлены формулами, где левая и правая ориентация имеет общепринятое направление от амино-конца к карбокси-концу. Кроме того, следует отметить, что черточка в начале или в конце последовательности аминокислотных остатков указывает на пептидную связь с другой последовательностью в одном или нескольких аминокислотных остатках.
Полипептид - линейная последовательность аминокислотных остатков, связанных друг с другом посредством пептидной связи между альфа-аминогруппой и карбоксигруппой смежных аминокислотных остатков.
Пептид - линейная последовательность из не более чем около 50 аминокислотных остатков, связанных друг с другом так же, как и в полипептиде.
Циклический пептид означает соединение, имеющее гетероатомную кольцевую структуру, которая содержит несколько амидных связей, как и в обычном пептиде. Циклический пептид может быть цивилизованным линейным полипептидом "голова к хвосту", в котором N-конец линейного пептида связан амидной связью с концевым карбоксилом линейного пептида, либо он может содержать кольцевую структуру, в котором полимер является гомогенным или гетерогенным, и содержит амидные связи и/или другие связи для замыкания кольца, такие как дисульфидные мостики, сложные тиоэфиры, тиоамиды, гуанидиногруппы, и другие подобные связи.
Белок - линейная последовательность из более чем 50 аминокислотных остатков, связанных друг с другом так же, как и в полипептиде.
Гибридный белок - полипептид, содержащий, по крайней мере, два различных полипептидных домена, правильно сшитых посредством обычной пептидной связи ("слитые"), где два домена соответствуют пептидам, не являющимся слитыми по природе.
Синтетический пептид - полученная химическим способом цепь аминокислотных остатков, правильно сшитых посредством пептидных связей, где указанная цепь не содержит природных белков и их фрагментов.
В. Общие положения
Настоящее изобретение относится к открытию того факта, что ангиогенез опосредуется специфическим рецептором αvβ5 винтронектина и что ингибированные функции αvβ5 приводит к ингибированию ангиогенеза. Это открытие имеет важное значение из-за роли, которую играет ангиогенез в различных патологических процессах. Путем ингибирования ангиогенеза можно воздействовать на заболевание, ослабить его симптомы, а в некоторых случаях вылечить заболевание. В случае если рост новых сосудов является причиной патологии или вносит вклад в патологию, ассоциированную с заболеванием, то ингибирование ангиогенеза будет способствовать ослаблению неблагоприятных последствий данного заболевания. Примерами таких заболеваний являются ревматоидный артрит, диабетическая ретинопатия, воспалительные заболевания, рестеноз и т.п. В том случае, когда рост новых кровеносных сосудов требуется для поддержания роста нежелательной ткани, то ингибирование ангиогенеза будет способствовать снижению кровоснабжения тканей и тем самым способствовать снижению массы ткани, требующей соответствующего кровоснабжения. В качестве примера может служить рост новых кровеносных сосудов в ответ на ишемию, который приводит к индуцированному факторами роста ангиогенезу, росту опухолей, где реваскуляризация является постоянным и необходимым условием для поддержания роста опухоли
после того, как ее толщина достигнет нескольких миллиметров, а также для возникновения метастазов твердых опухолей.
Настоящее изобретение относится к открытию того факта, что ангиогенез опосредуется специфическим рецептором αvβ5 винтронектина и что ингибированные функции αvβ5 приводит к ингибированию ангиогенеза. Это открытие имеет важное значение из-за роли, которую играет ангиогенез в различных патологических процессах. Путем ингибирования ангиогенеза можно воздействовать на заболевание, ослабить его симптомы, а в некоторых случаях вылечить заболевание. В случае если рост новых сосудов является причиной патологии или вносит вклад в патологию, ассоциированную с заболеванием, то ингибирование ангиогенеза будет способствовать ослаблению неблагоприятных последствий данного заболевания. Примерами таких заболеваний являются ревматоидный артрит, диабетическая ретинопатия, воспалительные заболевания, рестеноз и т.п. В том случае, когда рост новых кровеносных сосудов требуется для поддержания роста нежелательной ткани, то ингибирование ангиогенеза будет способствовать снижению кровоснабжения тканей и тем самым способствовать снижению массы ткани, требующей соответствующего кровоснабжения. В качестве примера может служить рост новых кровеносных сосудов в ответ на ишемию, который приводит к индуцированному факторами роста ангиогенезу, росту опухолей, где реваскуляризация является постоянным и необходимым условием для поддержания роста опухоли
после того, как ее толщина достигнет нескольких миллиметров, а также для возникновения метастазов твердых опухолей.
Способы настоящего изобретения являются эффективными отчасти потому, что эта терапия является в высокой степени избирательной в отношении ангиогенеза, но не в отношении других биологических процессов. Как показано в примерах, лишь вновь образованные сосуды содержат αvβ5, а поэтому данные терапевтические методы не оказывают неблагоприятного воздействия на зрелые сосуды.
Обнаружение того факта, что ингибирование лишь αvβ5 может эффективно подавлять ангиогенез, позволило разработать терапевтические композиции, которые будут обладать высокой специфичностью, а поэтому относительно низкой токсичностью. Хотя в настоящей заявке описано использование пептидных реагентов, которые способны ингибировать один или несколько интегринов, однако можно использовать и другие реагенты, которые будут более избирательно ингибировать αvβ5. Поэтому некоторые реагенты, полученные на основе пептидов, не обладают побочным действием, направленным на ингибирование других биологических процессов, кроме тех, которые опосредованы интегрином αvβ5.
Так, например, как показано в описании настоящего изобретения, можно получить моноклональные антитела, которые будут в высокой степени избирательными в отношении иммунной реакции с αvβ5, а не с αvβ1, αvβ1 или α11bβ3, и которые будут в одинаковой степени избирательными для ингибирования функции αvβ5. Кроме того, могут быть сконструированы RGD-содержащие пептиды, которые являются избирательными в отношении ингибирования αvβ5, как описано ниже.
Так, например, как показано в описании настоящего изобретения, можно получить моноклональные антитела, которые будут в высокой степени избирательными в отношении иммунной реакции с αvβ5, а не с αvβ1, αvβ1 или α11bβ3, и которые будут в одинаковой степени избирательными для ингибирования функции αvβ5. Кроме того, могут быть сконструированы RGD-содержащие пептиды, которые являются избирательными в отношении ингибирования αvβ5, как описано ниже.
До разработки настоящего изобретения было известно, что ангиогенез и любой процесс, зависящий от ангиогенеза, может быть ингибирован in vivo с использованием реагентов, которые обладают антагонистическим действием на биологическую функцию αvβ5.
С. Способы ингибирования ангиогенеза
Настоящее изобретение относится к способу ингибирования ангиогенеза в ткани, и, тем самым, к ингибированию процессов в ткани, которые зависят от ангиогенеза. В основном этот способ предусматривает введение в ткань композиции, содержащей ангиогенез-ингибирующее количество антагониста αvβ5.
Ткань-мишень, на которую направлены способы настоящего изобретения, определяется как αvβ5-содержащая ткань, которая характеризуется детектируемым присутствием интегринового рецептора αvβ5. Другими словами, αvβ5-содержащая ткань определяется присутствием αvβ5-рецепторного комплекса в клеточных мембранах. Такие ткани включают эпителиальные и мезенхимальные клетки. Присутствие рецептора может быть определено различными способами, включая иммунореактивность рецептора с антителом против интегринового рецептора αvβ5, где указанная иммунная реакция в тканях обнаруживается с помощью микроскопии, иммунопреципитации, анализа на конкурентное связываание с лигандом и другими методами. Предпочтительные антитела, используемые для обнаружения присутствия αvβ5 в ткани, описаны ниже и в примере 1. Так, например, распределение αvβ5 в почках, коже, глазной ткани, определяемое с помощью иммунофлуоресцентной микроскопии, описано в примере 2.
С. Способы ингибирования ангиогенеза
Настоящее изобретение относится к способу ингибирования ангиогенеза в ткани, и, тем самым, к ингибированию процессов в ткани, которые зависят от ангиогенеза. В основном этот способ предусматривает введение в ткань композиции, содержащей ангиогенез-ингибирующее количество антагониста αvβ5.
Ткань-мишень, на которую направлены способы настоящего изобретения, определяется как αvβ5-содержащая ткань, которая характеризуется детектируемым присутствием интегринового рецептора αvβ5. Другими словами, αvβ5-содержащая ткань определяется присутствием αvβ5-рецепторного комплекса в клеточных мембранах. Такие ткани включают эпителиальные и мезенхимальные клетки. Присутствие рецептора может быть определено различными способами, включая иммунореактивность рецептора с антителом против интегринового рецептора αvβ5, где указанная иммунная реакция в тканях обнаруживается с помощью микроскопии, иммунопреципитации, анализа на конкурентное связываание с лигандом и другими методами. Предпочтительные антитела, используемые для обнаружения присутствия αvβ5 в ткани, описаны ниже и в примере 1. Так, например, распределение αvβ5 в почках, коже, глазной ткани, определяемое с помощью иммунофлуоресцентной микроскопии, описано в примере 2.
С точки зрения способов настоящего изобретения αvβ5-содержащая ткань характеризуется также тем, что она имеет признаки ангиогенеза. Как было описано ранее, ангиогенез включает ряд процессов, участвующих в образовании новых сосудов в ткани, включая такие процессы, как "разрастание", сосудогенез, или расширение сосудов, где все эти процессы ангиогенеза опосредуются экспрессией αvβ5 и зависят от этой экспрессии. За исключением заживления ран, полученных в результате травм, образования желтого тела и эмбриогенеза, очевидно, что большинство процессов ангиогенеза ассоциируется с патологическими процессами, а поэтому использование способов настоящего изобретения является избирательным в отношении данного заболевания и не оказывает неблагоприятных побочных эффектов.
Существует ряд заболеваний, при которых ангиогенез играет, очевидно, важную роль и которые называются ангиогенными заболеваниями, включая такие заболевания, но не ограничиваясь ими, как воспалительные состояния, например иммунные и неимаз; состояния, ассоциированные с нежелательной и несвоевременной инвазией сосудов, такие как рестеноз, капиллярная пролиферация в атеросклеротических бляшках и остеопороз; и состояния, ассоциированные с раком, такие как твердые опухоли, метастазы твердых опухолей, ангиофибромы, ретролентальная фиброплазия, гемангиомы, саркома Копоши и подобные раковые заболевания, при которых для поддержания опухолевого роста требуется образование новых сосудов.
Глазные болезни, характеризующиеся реваскуляризацией, являются особенно предпочтительной целью для данной терапии. Глазная реваскуляризация является одним из главных патологических изменений, наблюдаемых при подавляющем большинстве глазных болезней и приводит к катастрофической потере зрения. Рост новых кровеносных сосудов из уже существующих сосудов сосудистой оболочки глаз, сосудов сетчатки или паралимбальных сосудов может вызывать отеки, геморрагии или образование фиброваскулярной мембраны, что приводит к нарушению нормальной анатомической структуры глаза и сопровождается потерей нормальной зрительной функции.
Глазные болезни, характеризующиеся ангиогенезом, включают реваскулярные нарушения роговицы, к которым относятся трансплантация роговицы, герпетический кератит, сифилитический кератит, птеригий, реваскулярный паннус, ассоциированный с использованием контактных линз, и т.п. Другими глазными болезнями также являются диабетическая ретинопатия (DR), возрастная дегенерация желтого пятна (ARMD), предполагаемый гистоплазмоз глаз (POHS), ретинопатия при преждевременном развитии (ROP), реваскулярная глаукома и т.п. Хотя ингибирование ангиогенеза при некоторых заболеваниях не обязательно приводит к излечению основного заболевания, однако оно может значительно уменьшить наблюдаемую заболеваемость, ассоциированную с этим ангиогенезом.
Так, например, 90% из 300000 человек, страдающих диабетом в течение 25 лет, могут иметь форму DR, которая представляет собой заболевание сетчатки глаз, характеризующееся кровоточивостью и/или пролиферацией кровеносных сосудов. У тридцати процентов из этих пациентов это состояние может быть фактически улучшено благодаря терапевтическим способам настоящего изобретения. Что касается ARMD, то 25% населения из 65, приблизительно 630000 человек будут иметь определенную форму этого заболевания с прогнозом, что к 2030 году свыше 6,3 миллионов человек будут иметь ARMD. Поэтому возможность ингибировать αvβ5-ассоциированный ангиогенез с использованием терапевтических композиций и способов настоящего изобретения имеет громадное значение для медицины.
Таким образом, способы, которые приводят к ингибированию ангиогенеза в патологических тканях, способствуют ослаблению симптомов заболевания и в зависимости от конкретного заболевания, могут обеспечить излечение от этого заболевания. В одном из вариантов настоящего изобретения рассматривается ингибирование ангиогенеза, per se, в ткани. Степень ангиогенеза в ткани, а следовательно, степень достигаемого ингибирования способами настоящего изобретения может быть оценена различными методами, такими как методы, описанные в примерах и используемые для обнаружения αvβ5-иммуноположительной структуры вновь появившихся и незрелых сосудов посредством иммуногистохимического анализа.
Как описано в настоящей заявке, любые из различных тканей или органов, состоящих из организованных тканей, могут поддерживать ангиогенез при патологических состояниях, включая ткани кожи, мышцы, кишки, соединительную ткань, суставы, кости и другие ткани, в которых кровеносные сосуды могут разрастаться под действием ангиогенных стимуляторов.
В частности, способы и композиции настоящего изобретения, содержащие αvβ5-антагонист, могут быть использованы в терапевтических целях для ингибирования ангиогенеза, индуцированного факторами роста, называемыми также цитокинами. В физиологических условиях ангиогенез является в высокой степени регулируемым процессом, и, как было показано в опубликованной ранее работе Brooks et al., Science, 264:569-5761 (1994), он активируется специфическими ангиогенными молекулами, такими как основной фактор роста фибробластов (bFGF). Была также описана и негативная регуляция ангиогенеза. Таким образом, ангиогенез регулируется посредством сложного балланса между локальными стимуляторами и ингибиторами. См., D'Amore, Investigative Ophthal. Visual Sci., 35:3974-3979 (1994).
Если физиологический баланс ангиогенных стимуляторов и ингибиторов, который тщательно контролируется в нормальных и находящихся в состоянии покоя капиллярных сосудах, нарушается, что происходит при некоторых патологических состояниях, то эндотелиальные клетки капилляров начинают пролифирировать, мигрировать и, в конечном счете, дифференцироваться с образованием новых кровеносных сосудов.
Ангиогенеэ может быть охарактеризован как каскад событий, включающих серию ранних событий и последующую серию поздних событий, как было описано в обзоре Leibovich "Role of Cytokines in the Process of Tumor Angiogenesis", в "Human Cytokines: Their Role in Disease and Therapy", eds. Aggarwal and Puri, Chapter 35, Blackwell Science, Inc. (1995). Ранние события предшествуют доставке ангиогенных факторов роста и цитокинов, поставляемых из внесосудистого источника. Затем ранние события протекают в сети микрососудов-мишеней с нарушением внутриклеточных связей, с индуцированном экспрессии антигенов активации эндогениальных клеток и протеолитического фенотипа, и с инициацией миграции эндотелиальных клеток в определенном направлении. Поздние события характеризуются автокринной и паракринной экспрессией генов факторов роста и цитокинов в клетках, таких как эндотелиальные клетки, перициты и клетки гладких мышц, с развитием капиллярного утолщения. Эти клетки, в свою очередь, модулируют взаимодействия клеток с внеклеточным матриксом, что приводит к образованию петель новых функциональных капилляров из уже существующих зрелых сосудов.
Как уже обсуждалось в настоящем описании, в литературе приводятся указания на связь между появлением факторов роста, включая те факторы, которые ассоциируются с повышением экспрессии αvβ5, а именно VEGF, TGF-α и EGF, с увеличением массы опухоли и с началом ангиогенеза при пролиферативном реваскулярном заболевании глаз как у человека, так и у экспериментальных животных.
Таким образом, VEGF, EGF, TGF-α и многие другие цитокины рассматриваются как факторы роста, которые характеризуются своей способностью стимулировать клеточный рост. Такими факторами роста являются белки, секретируемые одной клеткой, которая действует на секретирующую клетку или другую клетку. Их способность действовать, таким образом, зависит от присутствия рецепторов факторов роста, которыми обычно являются трансмембранные белки. Факторы роста, такие как VEGF, также обычно именуются цитокинами, которые определяются как полипептидные гормоны, секретируемые клеткой и влияющие на рост и метаболизм либо той же самой (аутокринный гормон) или другой (паракринный гормон) клетки. Термин "цитокин" не ограничивается молекулами, продуцируемыми клетками иммунной системы и модификаторами иммунного ответа той же самой системы. Так, например, термин "цитокин" представляет собой широкую категорию, одна из субкатегорий которой, относящаяся к типу биологического ответа, представляет собой стимулирующие факторы роста или усилители роста, такие как VEGF, bFGF, EGF, TGF-α и т.п. См. обзор Aggarwal et al., "Common and Uncommon Features of Cytokines and Cytokine Receptors: An Overview", в "Human Cytokines; Their Role in Disease and Therapy", eds. Aggarwal and Puri, Chapter I, Blackwell Science, Inc. (1995).
В настоящем изобретении для ингибирования ангиогенеза в тканях рассматривается использование αvβ5-специфических антагонистов, но не антагонистов факторов роста, таких как антитела против VEGF. В предпочтительных вариантах изобретения антагонисты αvβ5, описанные в настоящей заявке, используются для ингибирования опосредованного факторами роста ангиогенеза, при котором индуцируется экспрессия интегринового αvβ5-рецептора. В этом контексте предпочтительными факторами роста являются VEGF, EGF, TGF-α и т.п.
Как обсуждалось в главе "Уровень техники", известно, что факторы роста EGF и VEGF связываются с их клеточными рецепторами, которые действуют как тирозинкиназы. Было показано, что активация рецептора EGF коррелирует с активацией протеинкиназы С, которая приводит к активации αvβ5, что способствует миграции специфических клеток на витронектиновый субстрат. Таким образом, механизм взаимодействия между действием цитокинов или факторов роста и координацией ответа на экспрессию или активацию интегрина является сложным биологическим процессом. Как показано в настоящем изобретении (см. пример 6А), обработка тканей в кроличьей модели глаза или куриной хориоаллантоистной модели цитокинами VEGF приводит к αvβ5-потенцированному ангиогенезу, который не зависит от активации протеинкиназы С.
В своем особенно предпочтительном варианте настоящее изобретение предусматривает использование антагонистов αvβ5 для ингибирования ангиогенеза в любой ткани, в которой этот ангиогенез индуцируется VEGF. Так, например, было показано, что ишемия сетчатки в различных системах моделей животных приводит к стимуляции VEGF, который секретируется из клеток Мюллера и продуцирование которого затем приводит к индуцированию реваскуляризации тканей глаза. См., Miller et al., Am. J. Pathol., 145:574-584 (1994), и Pierce et al., Proc. Natl. Acad. Sci., USA, 92:905-909 (1995).
Таким образом, в соответствии с настоящим изобретением тканью, на которую направлено лечение, является ткань пациента, страдающего диабетической ретинопатией, дегенерацией желтого пятна, реваскулярной глаукомой или подобными заболеваниями, указанными выше, а ингибируемым ангиогенезом является ангиогенез ткани сетчатки, при котором происходит реваскуляризация ткани. В качестве примеров могут служить ткани роговицы, полученные от пациентов с реваскуляризацией тканей глаза, или от пациентов с заболеваниями, описанными выше и в примерах. Примерам экспериментальной модели, используемой для оценки действия антагониста αvβ5 настоящего изобретения в целях лечения ангиогенеза сетчатки, является мышиная модель реваскуляризации сетчатки, описанная в примере 9.
В других родственных вариантах тканью, на которую направлено лечение, является воспалительная ткань, а ингибируемым ангиогенезом является ангиогенез воспалительной ткани, при котором происходит реваскуляризация воспалительной ткани. В этом варианте указанный способ предусматривает ингибирование ангиогенеза в артритных тканях, таких как ткани пациента, страдающего хроническим суставным ревматизмом, ткани, пораженные иммунным или неиммунным воспалением, ткани, пораженные псориазом, и т.п.
Считается, что цитокины, интерлейкин 1 и фактор некроза опухоли-α, ассоциируются с ревматоидным артритом и играют непосредственную роль в деструкции суставов посредством индуцирования экспрессии адгезивных молекул на эндотелиальных клетках и высвобождения ферментов. См., Arend et al., Arthritis & Rheumatism, 38: 151-160 (1995). Для блокирования цитокинов цитокин-специфическими ингибиторами, а также молекул, ответственных за адгезию клеток-мишеней, и экспрессируемых при данном состоянии, были предложены терапевтические схемы лечения. См., Haskard et al., Cell Adhesion Comm., 2:235-238 (1994).
Таким образом, ингибирование ангиогенеза при артритных заболеваниях посредством адресной терапии, направленной против адгезивных молекул αvβ5, является другим предпочтительным вариантом настоящего изобретения.
В еще одном родственном варианте осуществления изобретения тканью, подвергаемой обработке, является опухолевая ткань пациента, страдающего твердой опухолью, метастазами, раком кожи, раком молочной железы, гемангиомой, или ангиофибромой, а также другими формами рака; а ангиогенезом, подвергаемым ингибированию, является ангиогенез опухолевой ткани, при котором происходит реваскуляризация опухолевой ткани. Обычно тканями твердых опухолей, против которых направлены способы настоящего изобретения, являются ткани легких, поджелудочной железы, молочной железы, толстой кишки, гортани, яичников, и т.п.
Роль сложной сети цитокинов, которые присутствуют в твердых опухолях человека, описана в обзоре Leek et al., J. Leukocyte Biol., 56:423-435 (1994), который вводится в настоящее описание посредством ссылки. Очевидно, ряд цитокинов, включая VEGF, кислотный, а также основный FGF (bFGF), TGF-α и TGF-β, EGF, TNF- тромбоцитарный фактор роста эндотелиальных клеток, ангиогенин, интерфероны α и γ, интерлейкины 1, 6 и 8 и т.п. оказывают влияние на различные клеточные механизмы ангиогенеза в злокачественных тканях и клеточных линиях. Так, например, недавно было обнаружено, что VEGF, помимо его локализации в опухолях различных видов, также связан с ангиогенезом в карциноме молочной железы, как описано Brown et al., Human Path., 26:86-91 (1995).
тромбоцитарный фактор роста эндотелиальных клеток, ангиогенин, интерфероны α и γ, интерлейкины 1, 6 и 8 и т.п. оказывают влияние на различные клеточные механизмы ангиогенеза в злокачественных тканях и клеточных линиях. Так, например, недавно было обнаружено, что VEGF, помимо его локализации в опухолях различных видов, также связан с ангиогенезом в карциноме молочной железы, как описано Brown et al., Human Path., 26:86-91 (1995).
Опухоли, которые секретируют различные цитокины и в которых в ответ на это индуцируется локальный ангиогенез (в настоящем изобретении, в частности, цитокины VEGF, TGF-α и EGF и соответственно αvβ5-опосредованный ангиогенез) могут быть идентифицированы путем скрининга образцов опухолевой ткани с использованием антител против цитокинов. Эти методы известны каждому специалисту, имеющему дело с культивированными или полученными в результате биопсии образцами опухолевой ткани. Антитела против вышеуказанных цитокинов являются коммерчески доступными и поставляются Oncogene Sciences (Uniondale, NY). Таким образом, скрининг отобранных опухолевых тканей этими методами позволяет проводить оценку потенциальной ангиогенез-ингибирующей активности антагонистов αvβ5 настоящего изобретения.
Характерный ангиогенез в опухолевой ткани и его ингибирование описаны в примерах.
Благодаря важной роли, которую играет образование новых кровеносных сосудов в опухолевой ткани, ингибирование ангиогенеза в опухолевой ткани является еще одним предпочтительным вариантом настоящего изобретения. При отсутствии реваскуляризации опухолевой ткани эта опухолевая ткань не получает нужных питательных элементов, в результате чего ее рост замедляется, прекращается дополнительный рост, происходит регрессия опухоли и, в конечном счете, ее некроз, который приводит к уничтожению опухоли.
Другими словами, настоящее изобретение относится к способу ингибирования реваскуляризации опухоли путем ингибирования ангиогенеза в этой опухоли в соответствии с настоящим изобретением. Аналогично, настоящее изобретение относится к способу ингибирования роста опухоли с использованием способов ингибирования ангиогенеза.
Эти способы являются также особенно эффективными против образования метастазов, вследствие того, что (1) их образование требует васкуляризации первичной опухоли, для того чтобы раковые клетки могли покидать первичную опухоль, и (2) внедрение метастазов во вторичные участки требует реваскуляризации для поддержания их роста. В своем родственном варианте настоящее изобретение относится к использованию указанного способа в сочетании с другими методами терапии, такими как стандартная химиотерапия, направленная против твердых опухолей и применяемая для подавления возникновения метастазов. Введение ингибитора ангиогенеза обычно осуществляют во время или после химиотерапии, хотя предпочтительно, чтобы ингибирование ангиогенеза осуществлялось после проведения химиотерапии в то время, когда опухолевая ткань будет восприимчивой к токсической атаке благодаря индуцированию ангиогенеза для восстановления доставки крови и питательных веществ к опухолевой ткани. Кроме того, предпочтительно, чтобы способ ингибирования ангиогенеза осуществлялся после хирургической операции по удалению твердой опухоли, где указанная операция является профилактической мерой против образования метастазов.
Хотя способы настоящего изобретения применяются для ингибирования реваскуляризации опухоли, эти способы могут быть с таким же успехом использованы для ингибирования роста опухолевой ткани, для ингибирования метастазирования опухоли и для регрессии уже развившихся опухолей. В последнем случае уменьшение опухолевой массы оценивают с помощью аналитической модели глаза кролика, используемой в настоящем изобретении, или с помощью экспериментальной химерной модели "мышь-человек", в которой кожу мыши с тяжелым комбинированным иммунодефицитом (SCID) заменяли неонатальной крайней плотью человека, как описано в работе Yan et al., J. Clin. Invest., 91:986-996 (1993), которая вводится в настоящее описание посредством ссылки. Последняя модель представляет собой дополнительную in vivo-модель для исследования ангиогенеза и его ингибирования способами настоящего изобретения. Характерные результаты, полученные на кроличьей модели опухоли с использованием антагонистов αvβ5 настоящего изобретения, представлены в примерах 5С и 6D, а результаты ингибирования ангиогенеза, полученные на мышиной модели SCID, описаны в примере 8.
Рестеноз представляет собой процесс миграции и пролиферации клеток гладких мышц (SMC) в область чрескожной внутрипросветной коронарной ангиопластики, который препятствует осуществлению успешной ангиоластики. Такая миграция и пролиферация SMC во время рестеноза может рассматриваться как процесс ангиогенеза, который может быть ингибирован способами настоящего изобретения. Поэтому в настоящем изобретении также рассматривается ингибирование рестеноза у пациента, подвергнутого пластической операции на сосудах, путем ингибирования ангиогенеза методами настоящего изобретения. Для ингибирования рестеноза антагонист αvβ5 обычно вводят после пластической операции на сосудах примерно через 2-28 дней, а предпочтительно в течение примерно первых 14 дней после операции.
Во многих вариантах настоящего изобретения пациентом, подвергаемым лечению способами настоящего изобретения, является предпочтительно человек, хотя следует отметить, что принципы, на основе которых разработано настоящее изобретение, указывают на то, что это изобретение является эффективным для всех млекопитающих, которые могут быть включены в понятие "пациент". В этой связи следует отметить, что таким млекопитающим может быть млекопитающее любого вида, а в частности сельскохозяйственные или домашние животные, для лечения которых могут быть использованы способы настоящего изобретения.
В соответствии с настоящим изобретением способы ингибирования ангиогенеза в тканях, а следовательно, и применение этих способов для лечения заболеваний, опосредованных ангиогенезом, предусматривают контактирование ткани, в которой происходит ангиоенез, или ткани с риском возникновения такого ангиогенеза, с композицией, содержащей терапевтически эффективное количество антагониста αvβ5, способного ингибировать связывание αvβ5 с его природным лигандом. Таким образом, этот способ предусматривает введение пациенту терапевтически эффективного количества физиологически приемлемой композиции, содержащей терапевтически эффективное количество антагониста αvβ5 настоящего изобретения.
Интервал доз, подходящих для введения антагониста αvβ5, зависит от формы этого антагониста, и его действия, как будет описано ниже, и представляет собой количества, достаточные для продуцирования нужного эффекта, который приводит к ослаблению ангиогенеза и симптомов заболевания, опосредованных этим ангиогенезом. Во избежание побочных эффектов, таких как синдром повышенной вязкости крови, отек легких, застойная сердечная недостаточность и т. п. эти дозы не должны быть слишком большими. В основном эти дозы варьируются в зависимости от возраста, состояния, пола и тяжести заболевания пациента и могут быть определены специалистом. В случае какого-либо осложнения эти дозы могут быть также скорректированы самим лечащим врачом.
Антагонист αvβ5 представляет собой молекулу, которая блокирует или подавляет физиологическую или фармакологическую активность αvβ5 посредством ингибирования активности связывания рецептора с его лигандом, а именно с витронектином. Предпочтительными антагонистами αvβ5 могут быть моноклональные антитела, пептиды или органическая молекула, которая имитирует αvβ5-лиганд.
Терапевтически эффективное количество представляет собой такое количество антагониста αvβ5, которое является достаточным для обнаруживаемого ингибирования ангиогенеза в тканях-мишенях, то есть ангиогенез-ингибирующее количество. Ингибирование ангиогенеза может быть измерено in situ с помощью иммуногистохимического анализа, описанного ниже, или другими методами, известными специалистам.
Несмотря на то, что антагонист αvβ5 может иметь форму имитирующей αvβ5-лиганд органической молекулы, RGD-содержащего пептида, моноклонального антитела против αvβ5, его фрагмента или имитатора αvβ5-рецептора, однако следует отметить, что активность, а следовательно, и экспрессия "терапевтически эффективного" количества может варьироваться. Однако, как было показано с использованием аналитических способов настоящего изобретения, любой специалист может легко оценить эффективность кандидата-антагониста αvβ5 настоящего изобретения.
Эффективность антагониста αvβ5 может быть измерена разными способами, включая анализ ингибирование ангиогенеза в САМ, in vivo-анализ с использованием кроличьей модели глазных заболеваний и путем измерения ингибирования связывания природного лиганда с αvβ5 и т.п., как описано в настоящей заявке и подобными исследованиями.
Предпочтительный антагонист αvβ5 обладает способностью, в основном, ингибировать связывание природного лиганда, такого как витронектин, с αvβ5 в растворе при концентрации антагониста менее чем 0,5 микромоль (мкМ), предпочтительно менее чем 0,1 мкМ, а более предпочтительно менее чем 0,05 мкМ. Понятие "в основном" означает, что при ингибировании в присутствии антагониста αvβ5, наблюдается, по крайней мере, 50%-ное снижение уровня связывания витронектина, и это 50%-ное ингибирование определяется в настоящем описании как величина IС50.
Более предпочтительный антагонист αvβ5 обладает избирательностью преимущественно к αvβ5 по сравнению с другими интегринами. Так, например, предпочтительный антагонист αvβ5, в основном, ингибирует связывание витронектина с αvβ5, в основном, не ингибирует связывание витронектина с другими интегринами, такими как αvβ1, αvβ3 или α11bβ3. Особенно предпочтительным является антагонист αvβ5, активность которого в ингибировании связывания витронектина с αvβ5 в 10-100 раз ниже IС50-активности в ингибировании связывания витронектина с другими интегринами. Типичные анализы для измерения IС50-активности в ингибировании связывания витронектина с интегрином описаны в примерах.
Терапевтически эффективным количеством антагониста αvβ5 настоящего изобретения, используемого в форме моноклонального антитела, является обычно такое количество, которое при его введении в виде физиологически приемлемой композиции, является достаточным для достижения концентрации в плазме от около 0,01 микрограммов (мкг) на миллилитр (мл) до около 100 мкг/мл, предпочтительно от около 1 мкг/мл до около 5 мкг/мл, а обычно около 5 мкг/мл. Иначе говоря, эта доза может варьироваться от около 0,1 мг/кг, предпочтительно от около 0,2 мг/кг до около 200 мг/кг, а более предпочтительно от около 0,5 мг/кг до около 20 мг/кг при введении в виде разовой или дробной дозы ежедневно в течение одного или нескольких дней.
В случае если этот антагонист используется в форме фрагмента моноклонального антитела, то его количество может быть легко скорректировано исходя из массы этого фрагмента по отношению к массе целого антитела. Предпочтительная молярная концентрация в плазме составляет от около 2 микромоль (мкМ) до около 5 миллимоль (мМ), а предпочтительно от около 100 мкМ до 1 мМ антитела-антагониста.
Терапевтически эффективным количеством антагониста αvβ5 настоящего изобретения, используемого в форме полипептида или в форме другой небольшой молекулы аналогичного размера, имитирующей αvβ5-лиганд, является обычно такое количество, которое при его введении в виде физиологически приемлемой композиции является достаточным для достижения концентрации в плазме от около 0,1 до около 200 мкг/мл, предпочтительно от около 1 мкг/мл до около 150 мкг/мл. Если полипептид имеет массу около 500 граммов на моль, то предпочтительная молярная концентрация в плазме составляет от около 2 микромоль (мкМ) до около 5 миллимоль (мМ), а предпочтительно от около 100 мкМ до 1 мМ полипептида-антагониста. Иначе говоря, эта доза на массу тела может варьироваться от около 0,1 мг/кг до около 300 мг/кг, а
предпочтительно от около 0,2 мг/кг до 200 мг/кг при введении в виде разовой или дробной дозы ежедневно, в течение одного или нескольких дней.
предпочтительно от около 0,2 мг/кг до 200 мг/кг при введении в виде разовой или дробной дозы ежедневно, в течение одного или нескольких дней.
Моноклональные антитела, полипептиды или органические молекулы-миметики могут быть введены парентерально путем инъекции или путем постепенного вливания в течение определенного периода времени. Хотя ткань в организме, подвергаемая лечению, обычно доступна при системном введении, а поэтому такое лечение в большинстве случаев проводят путем внутривенного введения терапевтической композиции, однако в настоящем изобретении также рассматриваются другие ткани и способы доставки, где имеется вероятность, что эти ткани также содержат молекулу-мишень. Таким образом, моноклональные антитела, полипептиды или органические молекулы-миметики могут быть введены внутриглазным способом, внутривенно, внутрибрюшинно, внутримышечно, подкожно, внутриполостно, чрескожно и, кроме того, они могут быть введены с использованием перистальтических средств.
Терапевтические композиции, содержащие антагонист αvβ5 настоящего изобретения, обычно вводят внутривенно, например, в виде инъекции стандартной дозы. Термин "стандартная доза", используемый в связи с терапевтической композицией настоящего изобретения, означает физически дискретные лекарственные формы, подходящие для введения унифицированной дозы пациенту, где каждая такая лекарственная форма содержит предварительно определенное количество активного вещества, достаточного для продуцирования нужного терапевтического эффекта, в сочетании с требуемым разбавителем, то есть носителем или наполнителем.
В одном из предпочтительных вариантов настоящего изобретения, описанным в примерах, антагонист αvβ5 вводят внутривенно в виде разовой дозы.
Эти композиции вводят таким же способом, как и лекарственный препарат, и в терапевтически эффективном количестве. Вводимое количество и схема введения зависят от индивидуума, подвергаемого лечению, способности организма индивидуума усваивать активный ингредиент и от желаемого терапевтического эффекта. Точное количество активного ингредиента, необходимое для введения, определяется лечащим врачом и является индивидуальным для каждого пациента. Однако в настоящем описании приводятся интервалы доз, подходящие для системного введения в зависимости от способа введения. Подходящие схемы введения также варьируются, но в качестве примера можно указать первоначальное введение с последующим введением дробных доз через интервалы в один час или более путем последовательных инъекций или введение другим способом. Альтернативно, может быть использовано продолжительное внутривенное вливание, достаточное для поддержания концентрации лекарственного средства в крови в пределах, определенных для in vivo-терапии.
D. Терапевтические композиции
Настоящее изобретение относится к терапевтическим композициям, используемым для практического осуществления способов, описанных в настоящей заявке. Терапевтические композиции настоящего изобретения содержат физиологически приемлемый носитель в сочетании с антагонистом αvβ5, описанным выше, в котором этот активный ингредиент растворен или диспергирован. В предпочтительном варианте осуществления настоящего изобретения терапевтическая композиция, содержащая антагонист αvβ5, является неиммуногенной при ее введении млекопитающему или человеку в терапевтических целях.
Настоящее изобретение относится к терапевтическим композициям, используемым для практического осуществления способов, описанных в настоящей заявке. Терапевтические композиции настоящего изобретения содержат физиологически приемлемый носитель в сочетании с антагонистом αvβ5, описанным выше, в котором этот активный ингредиент растворен или диспергирован. В предпочтительном варианте осуществления настоящего изобретения терапевтическая композиция, содержащая антагонист αvβ5, является неиммуногенной при ее введении млекопитающему или человеку в терапевтических целях.
Используемые в настоящем описании термины "фармацевтически приемлемый", "физиологически приемлемый" и их грамматические варианты, относящиеся к композициям, носителям, разбавителям и реагентам, являются взаимозаменяемыми и означают, что эти материалы могут быть введены млекопитающим внутрь или наружно без продуцирования у них нежелательных побочных эффектов, таких как тошнота, головокружение, расстройства желудка и т.п.
Приготовление фармакологической композиции, которая содержит активные ингредиенты, растворенные или диспергированные в ней, хорошо известно специалистам, и не ограничивается лишь конкретными препаратами. Обычно такие композиции получают в виде инъецируемых жидких растворов или суспезий, однако эти композиции могут быть также получены и в виде твердых форм, подходящих для получения растворов или суспензий непосредственно перед их использованием. Этот препарат может быть также эмульгирован.
Активный ингредиент может быть смешан с наполнителями, которые являются фармацевтически приемлемыми и совместимыми с активным ингредиентом, в количествах, подходящих для использования в данных терапевтических способах, описанных в настоящей заявке. Подходящими наполнителями являются, например, вода, физиологический раствор, декстроза, глицерин, этанол, или т.п., и их комбинации. Кроме того, если необходимо, эта композиция может содержать небольшие количества вспомогательных веществ, таких как смачивающие или эмульгирующие агенты, рН-буферные агенты и т.п., которые повышают эффективность активного ингредиента.
Терапевтическая композиция настоящего изобретения может включать фармацевтически приемлемые соли указанных компонентов. Фармацевтически приемлемыми солями являются кислотно-аддитивные соли (образованные со свободными аминогруппами полипептида), которые образуются с неорганическими кислотами, такими как, например, соляная или фосфорная кислота, или с органическими кислотами, такими как уксусная, винная, миндальная кислота, и т.п. Соли, образованные со свободными карбоксильными группами, также могут быть получены из неорганических оснований, таких как, например, гидроксиды натрия, калия, аммония, кальция или железа, и из органических оснований, таких как изопропиламин, триметиламин, 2-этиламиноэтанол, гистидин, прокаин, и т.п.
При использовании в получении циклических полипептидных антагонистов αvβ5, особенно предпочтительной является соль НСl.
Физиологически приемлемые носители хорошо известны специалистам. Примерами жидких носителей являются стерильные водные растворы, которые не содержат других компонентов помимо активных ингредиентов и воды, либо содержат буфер, такой как фосфат натрия при физиологическом значении рН, физиологический раствор, либо то и другое, то есть забуференный фосфатом физиологический раствор. Кроме того, водные носители могут содержать более одной буферной соли, а также такие соли, как хлориды натрия и калия, декстроза, полиэтиленгликоль, и другие растворенные вещества.
Жидкие композиции могут также содержать жидкие фазы в сочетании с водой или в отсутствии воды. Примерами таких дополнительных жидких фаз являются глицерин, растительные масла, такие как масло из семян хлопчатника, и эмульсии типа "вода-масло".
Терапевтическая композиция содержит ангиогенезингибирующее количество антагониста αvβ5 настоящего изобретения, обычно полученного таким образом, что оно включает, по крайней мере, 0,1 мас.% антагониста на массу всей терапевтической композиции. "Мас.%" означает отношение массы ингибитора к массе всей композиции. Так, например, 0,1 мас.% означает 0,1 граммов ингибитора на 100 граммов всей композиции.
Е. Антагонисты интегрина αvβ5
Антагонисты αvβ5 используются в способах настоящего изобретения для ингибирования ангиогенеза в тканях и могут быть получены в различных формах, которые включают соединения, взаимодействующие с αvβ5 таким образом, что это взаимодействие препятствует функциональному взаимодействию αvβ5 с его природными лигандами. Примерами таких антагонистов является аналоги или имитаторы αvβ5, происходящие от сайта связывания с лигандом, который присутствует на αvβ5-миметиках природного αvβ5-лиганда и имитирует структурную область, участвующую в связывании с αvβ5-лигандом; полипептиды, имеющие последовательность, соответствующую домену функционального связывания с природным лигандом, специфичным для αvβ5, а в частности, последовательность, соответствующую RGD-содержащему домену природного αvβ5-лиганда; и антитела, обладающие иммунореактивностью по отношению либо к αvβ5, либо к природному лиганду; причем все указанные антагонисты обладают антагонистической активностью, определенной в настоящем описании.
Антагонисты αvβ5 используются в способах настоящего изобретения для ингибирования ангиогенеза в тканях и могут быть получены в различных формах, которые включают соединения, взаимодействующие с αvβ5 таким образом, что это взаимодействие препятствует функциональному взаимодействию αvβ5 с его природными лигандами. Примерами таких антагонистов является аналоги или имитаторы αvβ5, происходящие от сайта связывания с лигандом, который присутствует на αvβ5-миметиках природного αvβ5-лиганда и имитирует структурную область, участвующую в связывании с αvβ5-лигандом; полипептиды, имеющие последовательность, соответствующую домену функционального связывания с природным лигандом, специфичным для αvβ5, а в частности, последовательность, соответствующую RGD-содержащему домену природного αvβ5-лиганда; и антитела, обладающие иммунореактивностью по отношению либо к αvβ5, либо к природному лиганду; причем все указанные антагонисты обладают антагонистической активностью, определенной в настоящем описании.
1. Полипептиды
В одном из вариантов настоящего изобретения рассматриваются антагонисты αvβ5 в форме полипептидов. Полипептид (пептид)-антагонист αvβ5 может иметь характеристики последовательности природного αvβ5-лиганда или самого αvβ5 в области, участвующей во взаимодействии лиганда с αvβ5, и обладает αvβ5-антагонистической активностью, описанной в настоящей заявке. Предпочтительный пептид-антагонист αvβ5 содержит RGD-трипептид и соответствует последовательности природного лиганда в RGD-содержащей области.
В одном из вариантов настоящего изобретения рассматриваются антагонисты αvβ5 в форме полипептидов. Полипептид (пептид)-антагонист αvβ5 может иметь характеристики последовательности природного αvβ5-лиганда или самого αvβ5 в области, участвующей во взаимодействии лиганда с αvβ5, и обладает αvβ5-антагонистической активностью, описанной в настоящей заявке. Предпочтительный пептид-антагонист αvβ5 содержит RGD-трипептид и соответствует последовательности природного лиганда в RGD-содержащей области.
Предпочтительные RGD-содержащие полипептиды имеют последовательность, соответствующую последовательности аминокислотных остатков RGD-содержащей области природного αvβ5-лиганда, такого как витронектин, последовательность которого хорошо известна.
Особенно предпочтительный пептидный антагонист αvβ5 ингибирует преимущественно связывание αvβ5 с его природным лигандом (или лигандами) по сравнению с другими интегринами, как было описано выше. Эти αvβ5-специфические пептиды являются особенно предпочтительными, по крайней мере, потому, что их специфичность по отношению к αvβ5 способствует снижению нежелательных побочных эффектов, таких как ингибирование других интегринов. Идентификация предпочтительных пептидов-антагонистов αvβ5, обладающих избирательностью по отношению к αvβ5, может быть легко осуществлена с помощью типичных анализов на ингибирование связывания, таких как ELISA-анализ, описанный в примерах.
Полипептид настоящего изобретения, в основном, содержит не более чем около 100 аминокислотных остатков, предпочтительно не более чем около 60 остатков, а более предпочтительно не более чем около 30 остатков. Пептиды могут быть линейными или циклическими, хотя особенно предпочтительными являются циклические пептиды. Предпочтительные пептиды описаны в примерах.
Если пептид включает более чем около 100 остатков, то он обычно имеет форму гибридного белка или фрагмента белка, описанного в настоящей заявке.
При этом следует отметить, что рассматриваемый полипептид необязательно должен быть идентичным аминокислотной последовательности природного лиганда для αvβ5, при условии, что он включает последовательность, необходимую для ингибирования связывания αvβ5-лиганда с αvβ5, и способен действовать как αvβ5-антагонист в анализе, описанном в настоящей заявке.
Рассматриваемым полипептидом является любой аналог, фрагмент или химическое производное, аминокислотная последовательность которых определена в настоящем описании, при условии, что этот полипептид является антагонистом по отношению к αvβ5. Поэтому настоящее изобретение может также включать различные изменения, замены, инсерции и делеции, где указанные модификации обеспечивают определенные преимущества в использовании этого полипептида. В этой связи полипептид настоящего изобретения, который является антагонистом по отношению к  соответствует, а предпочтительно идентичен последовательности указанного пептида, в который было внесено одно или несколько модификаций и который при этом сохраняет способность действовать как антагонист по отношению к αvβ5 в одном или нескольких анализах, описанных в настоящей заявке.
соответствует, а предпочтительно идентичен последовательности указанного пептида, в который было внесено одно или несколько модификаций и который при этом сохраняет способность действовать как антагонист по отношению к αvβ5 в одном или нескольких анализах, описанных в настоящей заявке.
Таким образом, полипептид может иметь любую из различных форм пептидных производных, включая амиды, конъюгаты с белками, циклические пептиды, полимеризованные пептиды, аналоги, фрагменты, химически модифицированные пептиды и другие подобные производные.
Термин "аналог" означает любой полипептид, имеющий последовательность аминокислотных остатков, в основном идентичную последовательности, конкретно показанной в настоящем описании, где один или несколько остатков были консервативно заменены функционально аналогичными остатками и где указанная последовательность обладает антагонистической активностью по отношению к αvβ5, как описано в настоящей заявке. Примерами консервативных замен являются замена одного неполярного (гидрофобного) остатка на другой, такой как изолейцин, валин, лейцин или метионин; замена одного полярного (гидрофильного) остатка на другой, например такая замена, как аргинин-лизин, глутамин-аспарагин, глицин-серин; замена одного основного остатка на другой, такой как лизин, аргинин или гистидин; или замена одного кислотного остатка на другой, такой как аспарагиновая кислота или глутаминовая кислота.
Понятие "консервативная замена" также подразумевает использование химически дериватизированного остатка вместо недериватизированного остатка при условии, что полученный в результате такой замены полипептид будет обладать требуемой ингибирующей активностью.
Термин "химическое производное" относится к рассматриваемому полипептиду, имеющему один или несколько остатков, подвергнутых химической дериватизации путем реакции с функциональной боковой группой. Помимо производных по боковой группе химическое производное может иметь одну или несколько модификаций в каркасе полипептидной молекулы, включая α-аминозамены, такие как введение N-метильной, N-этильной, N-пропильной группы и т.п., и α-карбонильные замены, такие как введение сложнотиоэфирной, тиоамидной, гуанидиногруппы, и т.п. Такими дериватизированными молекулами являются, например, молекулы, в которых свободные аминогруппы были дериватизированы с образованием гидрохлоридов амина, п-толуолсульфонильных групп, карбобензоксигрупп, 1-бутоксикарбонильных групп, хлорацетильных групп, или формильных групп. Свободные карбоксильные группы могут быть дериватизированы с образованием солей, метиловых или этиловых сложных эфиров, или сложных эфиров других типов, или гидразидов. Свободные гидроксильные группы могут быть дериватизированы с образованием О-ацильных или О-алкильных производных. Имидазольная часть гистидина может быть дериватизирована у атома азота с образованием N-им-бензилгистидина. К химическим производным относятся также пептиды, которые содержат одно или несколько природных аминокислотных производных двадцати стандартных аминокислот. Так, например, пролин может быть заменен на 4-гидроксипролин; лизин может быть заменен на 5-гидроксилизин; гистидин может быть заменен на 3-метилгистидин; серин может быть заменен на гомосерин; и лизин может быть заменен на орнитин. К полипептидам настоящего изобретения также относится любой полипептид, имеющий один или несколько инсерций и/или делеций, или остатков, относящихся к последовательности полипептида, показанной в настоящем описании, при условии, что указанный полипептид сохраняет требуемую активность.
Особенно предпочтительным производным является циклически пептид, имеющий формулу цикло(Arg-Gly-Asp-D-Phe-NMe-Val), сокращенно с (RGDf-NMeV), где на валиновом остатке пептида имеется N-метил-замещенная α-аминогруппа, и где циклизация приводит к соединению первичных амино- и карбоксиконцов пептида.
Термин "фрагмент" относится к любому рассматриваемому полипептиду, аминокислотная последовательность которого является короче, чем аминокислотная последовательность полипептида, указанного в настоящем описании.
Если полипептид настоящего изобретения имеет последовательность, которая не является идентичной последовательности природного αvβ5-лиганда, то обычно это обусловлено тем, что было сделано одно или несколько консервативных или неконсервативных замен, то есть в основном было заменено не более чем около 30%, а предпочтительно не более чем 10% аминокислотных остатков. Кроме того, для создания "линкера", посредством которого полипептиды настоящего изобретения могут быть присоединены к метке, твердой матрице или к носителю, в любой конец полипептида могут быть также добавлены дополнительные остатки.
Метки, твердые матрицы и носители, которые могут быть использованы с полипептидами настоящего изобретения, описаны ниже.
Аминокислотные линкеры представляют собой, по крайней мере, один остаток и могут иметь 40 или более остатков, а в основном от 1 до 10 остатков, но они не образуют эпитопы αvβ5-лигандов. Типичными аминокислотыми остатками, используемыми для создания линкера, являются тирозин, цистеин, лизин, глутаминовая кислота и аспарагиновая кислота или т.п. Кроме того, если это не оговорено особо, то рассматриваемый полипептид может по своей последовательности отличаться от природной последовательности αvβ5-лиганда вследствие того, что эта последовательность была модифицирована путем -NH2-концевого ацилирования, например, путем ацетилирования или амидирования тригликолевой кислоты, путем карбоксиконцевого амидирования, например, с использованием аммиака, метиламина, и путем аналогичных концевых модификаций. Как хорошо известно, концевые модификации могут быть использованы для снижения восприимчивости к гидролизу протеиназой и, тем самым, для продления времени полужизни полипептидов в растворах, а особенно в биологических жидкостях, в которых могут присутствовать протеазы. В связи с этим циклизация полипептида также является полезной концевой модификацией и является, в частности, предпочтительной благодаря тому, что в результате такой циклизации образуются стабильные структуры, а также предпочтительной с точки зрения биологической активности, которая наблюдается в случае таких циклических пептидов и которая описана в настоящей заявке.
Любой пептид настоящего изобретения может быть использован в форме фармацевтически приемлемой соли. Подходящими кислотами, которые способны образовывать соли с пептидами настоящего изобретения, являются неорганические кислоты, такие как трифторуксусная кислота (TFA), хлористоводородная кислота (HCl), бромистоводородная кислота, перхлорная кислота, азотная кислота, тиоциановая кислота, серная кислота, метансульфоновая кислота, уксусная кислота, фосфороуксусная кислота, пропионовая кислота, гликолевая кислота, молочная кислота, пировиноградная кислота, щавелевая кислота, малоновая кислота, янтарная кислота, малеиновая кислота, фумаровая кислота, антранилиновая кислота, коричная кислота, нафталинсульфоновая кислота, сульфанилиновая кислота, и т.п. Особенно предпочтительной является соль НСl.
Подходящими основаниями, способными образовывать соли с пептидами настоящего изобретения, являются неорганические основания, такие как гидроксид натрия, гидроксид аммония, гидроксид калия, и т.п.; органические основания, такие как моно-, ди- и три-алкил- и ариламины (например, триэтиламин, диизопропиламин, метиламин, диметиламин, и т.п.) и необязательно замещенные этаноламины (например, этаноламин, диэтаноламин, и т.п.).
Кроме того, пептиды настоящего изобретения могут быть получены, как описано в примерах, за исключением свободной ионной соли, в которой заряженные кислотная или основная группы, присутствующие в боковых группах аминокислотных остатков (например, Arg, Asp и т.п.), ассоциируются и нейтрализуют друг друга с образованием соединения "внутренней соли".
Пептид настоящего изобретения, который также относится к рассматриваемому полипептиду, может быть синтезирован любыми методами, известными специалистам в этой области, включая технику рекомбинантных ДНК. С точки зрения чистоты, антигенной специфичности, отсутствия нежелательных побочных продуктов, легкости осуществления и т.п. предпочтительным методом является химический синтез, такой как твердофазный синтез типа Меррифилда. Прекрасный обзор многих подходящих способов приводится в работах Steward et al., "Solid Phase Peptide Synthesis", W.H. Freeman Co., San Francisco, 1969; Bodanszky et al. , "Peptide Synthesis", John Wiley & Sons, Second Edition, 1976; J. Meienhofer, "Hormonal Proteins and Peptides", Vol. 2, p. 46, Academic Press (New York), 1983; Merrifield, Adv. Enzymol., 32:221-96, 1969; Fields et al., Int. J. Peptide Protein Res., 35:161-214, 1990; и патент США 4244946 для твердофазного пептидного синтеза, и Schroder et al., "The Peptides", Vol. 1, Academic Press (New York), 1965 для классического синтеза в растворе; все указанные работы вводятся в настоящее описание посредством ссылки.
Подходящие защитные группы, используемые в таком синтезе, описаны в вышеуказанных работах и в работе J.F.W. McOmie "Protective Groups in Organic Chemistry", Plenum Press, New York, 1973, которая вводится в настоящее описание посредством ссылки.
В общих чертах рассматриваемые методы твердофазного синтеза предусматривают последовательное присоединение одного или нескольких аминокислотных остатков или соответствующим образом защищенных аминокислотных остатков к растущей пептидной цепи. Обычно амино- или карбоксигруппу первого аминокислотного остатка защищают подходящей избирательно удаляемой защитной группой. Для аминокислот, содержащих реакционноспособную боковую группу, такую как лизин, используется другая избирательно удаляемая защитная группа.
Так, например, с использованием твердофазного синтеза защищенную или дериватизированную аминокислоту связывают с инертным твердым носителем посредством незащищенной карбоксильной группы или аминогруппы. Затем защитную амино- или карбоксигруппу избирательно удаляют, после чего добавляют следующую аминокислоту в последовательности, имеющей комплиментарную и подходящим образом защищенную амино- или карбоксигруппу, и подвергают реакции в условиях, подходящих для образования амидной связи с остатком, уже присоединенным к твердому носителю. Затем от этого только что добавленного аминокислотного остатка отщепляют защитную амино- или карбоксигруппу, после чего добавляют следующую аминокислоту (соответствующим образом защищенную) и т.д. После того как все нужные аминокислоты будут присоединены с образованием последовательности, любые оставшиеся концевые и боковые защитные группы (и твердый носитель) последовательно или одновременно удаляют и получают конечный линейный полипептид.
Линейные полипептиды, полученные, например, как описано выше, могут быть подвергнуты реакции циклизации с образованием соответствующих циклических пептидов. Типичный метод получения циклического пептида описан Zimmer et al. . Peptides 1992, pp. 393-394, ESCOM Science Publishers. B.V., 1993. Обычно третбутоксикарбонилзащищенный метиловый сложный эфир пептида растворяют в метаноле и добавляют раствор гидроксида натрия, а затем полученную смесь оставляют для реакции при 20oС для гидролитического отщепления метилэфирной защитной группы. После выпаривания растворителя трет-бутоксикарбонилзащищенный пептид экстрагируют этилацетатом из подкисленного водного растворителя. Затем требутилкарбонильную защитную группу удаляют в слабых кислотных условиях в диоксановом сорастворителе. Полученный таким образом незащищенный линейный пептид со свободными амино- и карбоксиконцами превращают в его соответствующий циклический пептид посредством реакции разбавленного раствора линейного пептида в смеси дихлорметана и диметилформамида с дициклогексилкарбодиимидом в присутствии 1-гидроксибензотриазола и N-метилморфолина. Затем полученный циклический пептид очищают с помощью хроматографии.
Альтернативные методы синтеза циклического пептида описаны Gurrath et al., Eur. J. Biochem., 210-911-921 (1992), а также в примерах.
Кроме того, антагонист αvβ5 может быть получен в форме гибридного белка. Гибридные белки представляют собой белки, продуцируемые методами рекомбинантных ДНК, описанных в настоящей заявке, где рассматриваемый полипептид экспрессируется в виде гибрида со вторым белком-носителем, таким как глутатион-сульфгидрилтрансфераза (GST) или с другим хорошо известным носителем. Предпочтительные гибридные белки включают полипептид ММР-2, описанный ниже. Получение гибридного белка ММР-2 описано в примерах.
Особенно предпочтительные пептиды или производные пептидов, которые могут быть использованы в способах настоящего изобретения в тканях, в которых, в основном, происходит αvβ5-ассоциированный ангиогенез, описаны в примерах, и к таким пептидам относятся полипептиды, представленные в SEQ ID No:4, 6, 7, 8 и 9.
Предпочтительными также являются полипептиды, происходящие от гибридного белка ММР-2, описанного в настоящей заявке, и имеющие последовательности, представленные в SEQ ID No: 11-22.
2. Моноклональные антитела
В одном из своих вариантов настоящее изобретение относится к антагонистам αvβ5 в форме моноклональных антител, которые способны к иммунной реакции с αvβ5 и ингибируют связывание αvβ5 с его природным лигандом, описанным в настоящей заявке. В настоящей заявке также описаны клеточные линии, продуцирующие эти антитела, способы продуцирования этих клеточных линий и способы продуцирования моноклональных антител.
В одном из своих вариантов настоящее изобретение относится к антагонистам αvβ5 в форме моноклональных антител, которые способны к иммунной реакции с αvβ5 и ингибируют связывание αvβ5 с его природным лигандом, описанным в настоящей заявке. В настоящей заявке также описаны клеточные линии, продуцирующие эти антитела, способы продуцирования этих клеточных линий и способы продуцирования моноклональных антител.
Моноклональное антитело настоящего изобретения представляет собой молекулы антитела, которые 1) способны к иммунной реакции с выделенным αvβ5, 2) ингибируют связывание витронектина с αvβ5. Предпочтительными моноклональными антителами, которые преимущественно связываются с αvβ5, являются моноклональные антитела, обладающие иммунореактивными свойствами моноклональных антител (mAb) PIF6 и Р5Н9, описанных в примерах.
Термин "антитело или молекула антитела", используемый в настоящем описании в его различных грамматических формах, имеет собирательный смысл и относится к популяции иммуноглобулиновых молекул и/или иммунологически активных фрагментов иммуноглобулиновых молекул, то есть молекул, которые содержат антигенсвязывающий центр антитела или паратоп.
Понятие "антигенсвязывающий центр антитела" означает структурную часть молекулы антитела, состоящий из вариабельной и гипервариабельной областей тяжелой и легкой цепи, которые специфически связываются с антигеном.
Примерами антител, подходящих для использования в настоящем изобретении, являются молекулы иммуноглобулина, в основном интактные молекулы иммуноглобулина и те части молекулы иммуноглобулина, которые содержат паратоп, включая фрагменты, которые хорошо известны специалистам, такие как Fab, Fab', F(ab')2 и F(v), и которые также называются фрагментами антитела.
В другом своем предпочтительном варианте настоящее изобретение относится к усеченной молекуле иммуноглобулина, содержащей Fab-фрагмент, происходящий от моноклонального антитела настоящего изобретения. Fab-фрагмент, в котором отсутствует Fc-рецептор, является растворимым и имеет, с терапевтической точки зрения, преимущественное время полужизни в сыворотке, а с диагностической точки зрения он имеет то преимущество, что может быть использован в качестве растворимого Fab-фрагмента. Получение растворимого Fab-фрагмента, в основном, известно специалистам в области иммунологии и может быть осуществлено различными методами.
Так, например. Fab- и F(ab')2-части (фрагменты) антител получают посредством протеолитической реакции папаина и пепсина, соответственно проводимой, в основном, на интактных антителах хорошо известными методами. См., например, патент США 4342566 (Theofilopolous & Dixon). Fab'-фрагменты антитела также хорошо известны и могут быть получены из F(ab')2-фрагментов с последующим восстановлением дисульфидных связей, соединяющих две части тяжелой цепи, как и в случае с меркаптоэтанолом, и с последующим алкилированием полученного меркаптана белка с использованием такого реагента, как иодоацетамид. Предпочтительным является антитело, содержащее интактные молекулы иммуноглобулина, и это антитело используется в настоящем изобретении в качестве иллюстрации.
Понятие "моноклональное антитело", используемое в его различных грамматических формах, означает популяцию молекул антител, содержащих лишь один вид антигенсвязывающего центра антитела, способного к иммунной реакции с конкретным эпитопом. Таким образом, моноклональное антитело обычно обладает уникальной аффинностью связывания для данного эпитопа, с которым оно иммунно реагирует. Поэтому моноклональное антитело может содержать молекулу антитела, имеющего множество антигенсвязывающих центров, каждый из которых является иммуноспецифичным для каждого отдельного эпитопа, например биспецифичное моноклональное антитело.
Моноклональное антитело обычно состоит из антител, продуцируемых клонами одиночной клетки, называемой гибридомой, которая секретирует (продуцирует) только один вид молекулы антитела. Эта гибридомная клетка образуется благодаря слиянию антителопродуцирующей клетки с миеломной или другой самовоспроизводящейся клеточной линией. Получение таких антител было впервые описано Kohler и Milstein, Nature, 256:495-497 (1975), и это описание вводится в настоящее описание посредством ссылки. Другие методы описаны Zola, Monoclonal Antibodies: A Manual of Techniques, CRC Press, Inc. (1987). Затем супернатанты от полученных таким образом гибридом скринируют на присутствие молекул антител, способных к иммунной реакции с αvβ5, и на ингибирование связывания αvβ5 с его природными лигандами.
Короче говоря, для образования гибридомы, из которой продуцируется моноклональное антитело, миеломную или другую самопродуцирующуюся клеточную линию гибридизируют с лимфоцитатами, полученным из селезенки млекопитающего, гипериммунизированного источником αvβ5.
При этом предпочтительно, чтобы миеломная клеточная линия, используемая для получения гибридомы, происходила от того же самого вида, что и лимфоциты. Обычно предпочтительным штаммом млекопитающего является мышь линии 129 GIX+. Мышиными миеломами, подходящими для использования в целях настоящего изобретения, являются гипоксантин-аминоптеринтимидин-чувствительные (HAT) клеточные линии Р3Х63-Аg8.653, и sp2/0-Agl4, которые были депонированы в Американской коллекции типовых культур (Rockville, MD) под номером допуска CRL 1580 и CRL 1581 соответственно.
При этом предпочтительно, чтобы миеломная клеточная линия, используемая для получения гибридомы, происходила от того же самого вида, что и лимфоциты. Обычно предпочтительным штаммом млекопитающего является мышь линии 129 GIX+. Мышиными миеломами, подходящими для использования в целях настоящего изобретения, являются гипоксантин-аминоптеринтимидин-чувствительные (HAT) клеточные линии Р3Х63-Аg8.653, и sp2/0-Agl4, которые были депонированы в Американской коллекции типовых культур (Rockville, MD) под номером допуска CRL 1580 и CRL 1581 соответственно.
Спленоциты обычно подвергают слиянию с клетками миеломы с использованием полиэтиленгликоля (ПЭГ) 1500. Слитые гибриды отбирают по их селективности к HAT. Гибридомы, продуцирующие моноклональное антитело настоящего изобретения, идентифицируют с использованием твердофазного иммуноферментного анализа (ELISA), вариант которого описан в примерах.
Моноклональное антитело настоящего изобретения может быть также продуцировано путем инициации культуры моноклональной гибридомы, включающей питательную среду, содержащую гибридому, секретирующую молекулы антитела соответствующей специфичности. Эту культуру поддерживают в условиях и в течение периода времени, достаточных для того, чтобы данная гибридома секретировала молекулы антитела в эту среду. Затем антителосодержащую среду собирают. После этого молекулы антитела могут быть выделены хорошо известными методами.
Среды, используемые для получения этих композиций, хорошо известны специалистам, являются коммерчески доступными и включают синтетические культуральные среды, инбредные мыши и т.п. Примером синтетической среды является минимальная поддерживающая среда Дульбекко (DMEM; Dulbecco et al., Virol., 8: 396, 1959), в которую были добавлены 4,5 г/л глюкозы, 20 мМ глутамин, и 20% фетальная телячья сыворотка. Примером линии инбредной мыши может служить BALB/c.
Другие способы продуцирования моноклонального антитела, гибридомной клеточной линии или культуры гибридомных клеток также хорошо известны. См., например, метод выделения моноклональных антител из иммунологического спектра антител, описанного Sastry et al., Proc. Natl. Acad. Sci., USA, 86: 5728-5732 (1989), и Huse et al., Science, 246:1275-1281 (1989).
В настоящем изобретении рассматриваются также гибридомная клетка и культуры, содержащие гибридомную клетку, которая продуцирует моноклональное антитело настоящего изобретения. Особенно предпочтительной является гибридомная клеточная линия, которая секретирует моноклональное антитело (mAb) PIF6 и моноклональное антитело Р5Н9, получение которых описано в примерах.
В одном из своих вариантов настоящее изобретение относится к моноклональному антителу, которое обладает иммунореактивными свойствами (mAb) PIF6 или (mAb) P5H9.
Без излишнего экспериментирования можно также определить, имеет ли моноклональное антитело ту же самую (то есть эквивалентную) специфичность (иммунореактивные свойства), как и моноклональное антитело настоящего изобретения, что может быть осуществлено путем установления того факта, препятствует ли первое антитело связыванию последнего антитела с предварительно отобранной молекулой-мишенью. Если тестируемое моноклональное антитело конкурирует с моноклональным антителом настоящего изобретения, на что указывает снижение уровня связывания моноклонального антитела настоящего изобретения в стандартном анализе на конкурентное связывание с молекулой-мишенью, присутствующей в твердой фазе, то очевидно, что эти два моноклональных антитела связываются с тем же самым и/или близкородственным эпитопом.
Еще один способ определения того факта, имеет ли моноклональное антитело специфичность моноклонального антитела настоящего изобретения, заключается в том, что осуществляют предварительное инкубирование моноклонального антитела настоящего изобретения с молекулой-мишенью, с которой оно обычно реагирует, а затем добавляют тестируемое моноклональное антитело для того, чтобы определить, будет ли тестируемое моноклональное антитело ингибировать его способность связываться с молекулой-мишенью. Если тестируемое моноклональное антитело ингибирует эту способность, то, по всей вероятности, оно имеет ту же самую или функционально эквивалентную специфичность к эпитопу, что и моноклональное антитело настоящего изобретения.
Другая возможность определить, имеет ли моноклональное антитело специфичность моноклонального антитела настоящего изобретения, заключается в определении аминокислотной последовательности CDR-областей рассматриваемых антител. Молекулы антител, имеющие идентичные или функционально эквивалентные последовательности аминокислотных остатков в их CDR-областях, имеют ту же самую специфичность. Методы секвенирования полипептидов хорошо известны специалистам.
Иммуноспецифичность антител, их способность связываться с молекулами-мишенями и присущая им аффинность, которую антитело обнаруживает по отношению к эптитопу, определяют по эпитопу, с которым данное антитело иммунно реагирует. Специфичность по отношению к эпитопу определяется, по крайней мере частично, последовательностью аминокислотных остатков вариабельной области легкой цепи.
Использование термина "имеющий специфичность связывания" указывает на то, что эквивалентные моноклональные антитела обладают теми же самыми или аналогичными иммунореактивными (связывающими) характеристиками и конкурируют за связывание с предварительной выбранной молекулой-мишенью.
"Очеловеченные" (гуманизированные) моноклональные антитела часто имеют существенные преимущества по сравнению с мышиными моноклональными антителами, особенно в том случае, когда они используются для введения человеку в терапевтических целях. В частности, человеческие антитела не выводятся из кровотока так быстро, как "чужеродные" антитела. Кроме того, человеческие антитела не активируют иммунную систему так, как чужеродные антигены и чужеродные антитела. Методы получения "очеловеченных" антител, в основном, хорошо известны специалистам, и могут быть легко применимы к антителам настоящего изобретения.
Таким образом, в одном из своих вариантов настоящее изобретение относится к моноклональному антителу настоящего изобретения, которое является "очеловеченным" (гуманизированным) путем введения в него компонентов человеческой иммунной системы без какого-либо заметного воздействия на способность этого антитела связываться с антигеном.
3. αvβ5-Специфические имитаторы
В настоящем описании было показано, что антагонистами αvβ5, которые могут быть в основном использованы в настоящем изобретении, являются полипептиды, антитела и другие молекулы, называемые "миметиками", которые обладают способностью препятствовать функции αvβ5. Особенно предпочтительными являются антагонисты, которые влияют на функцию αvβ5, и не влияют на функции других интегринов.
В настоящем описании было показано, что антагонистами αvβ5, которые могут быть в основном использованы в настоящем изобретении, являются полипептиды, антитела и другие молекулы, называемые "миметиками", которые обладают способностью препятствовать функции αvβ5. Особенно предпочтительными являются антагонисты, которые влияют на функцию αvβ5, и не влияют на функции других интегринов.
В этой связи следует отметить, что для использования в способе настоящего изобретения может быть использован ряд таких реагентов при условии, что эти реагенты обладают необходимой биологической активностью. Эти реагенты обычно называют миметиками (имитаторами) из-за их способности "имитировать" αvβ5-лиганд, участвующий в функциональном взаимодействии рецептора и лиганда путем блокирования связывания домена в данном рецепторе с лигандом и тем самым препятствовать (то есть ингибировать) его нормальной функции. В альтернативном варианте антагонист αvβ5 может быть имитатором рецептора, а не его лиганда.
Миметик представляет собой любую молекулу, не являющуюся антителом или происходящим от лиганда пептидом, которая обладает вышеописанными свойствами. Такой молекулой может быть синтетический пептид, аналог или производное пептида, соединение, которое имеет такой же связывающий "карман", как и вышеописанный связывающий домен, например органическая молекула-миметик, или другая молекула.
Предпочтительным миметиком настоящего изобретения является органическая молекула, называемая органическим миметиком. Особенно предпочтительными органическими молекулами-миметиками, которые действуют как антагонисты αvβ5 благодаря сходству с лигандом для αvβ5, являются Соединения 7, 9, 10, 12, 14, 15, 16, 17, и 18, описанные в примере 10.
Получение αvβ5-миметика может быть осуществлено любыми из существующих методов структурного анализа, используемых для разработки лекарственных средств, и хорошо известных специалистам, включая методы молекулярного моделирования, анализ методом двухмерного ядерного магнитного резонанса (2-D-ЯМР), методы с использованием библиотек пептидных аналогов или других химических полимеров или соединений, а также другие методики разработки лекарственных средств.
Исходя из широкого структурного спектра молекул, представленных в настоящем изобретении, что свидетельствует о том, что антагонистами αvβ5 могут быть гибридный полипептид (например, гибридный белок ММР-2), небольшой полипептид, циклический пептид, производное пептида, органическая миметическая молекула или моноклональное антитело, очевидно, что существует разнообразие различных химических структур, которые обладают общей функциональной способностью к избирательному ингибированию αvβ5, а поэтому структура рассматриваемого антагониста αvβ5, используемого в методах настоящего изобретения, не должна рассматриваться как ограничение настоящего изобретения и может включать любой αvβ5-миметик, определенный в настоящем описании.
F. Способы идентификации антагонистов αvβ5
В настоящем изобретении также рассматриваются методы идентификации кандидата-антагониста αvβ5 для использования в способах настоящего изобретения. В этих методах анализа молекулы-кандидаты оценивают на их способность ингибировать связывание αvβ5 с их природными лигандами, а также оценивают на их способность ингибировать ангиогенез в тканях.
В настоящем изобретении также рассматриваются методы идентификации кандидата-антагониста αvβ5 для использования в способах настоящего изобретения. В этих методах анализа молекулы-кандидаты оценивают на их способность ингибировать связывание αvβ5 с их природными лигандами, а также оценивают на их способность ингибировать ангиогенез в тканях.
В первом анализе измеряют уровень ангиогенеза в хорионаллантоисной мембране цыпленка (САМ), и этот анализ называют СДМ-анализом. СДМ-анализ был подробно описан другими авторами, и далее он будет использован для измерения уровня ангиогенеза и реваскуляризации опухолевых тканей. См., Ausprunk et al. , Am. J. Pathol., 79-597-618 (1975): и Ossonski et al., Cancer Res., 40: 2300-2309 (1980).
САМ-анализ хорошо известен как модель анализа in vivo-ангиогенеза, поскольку в этой модели происходит реваскуляризация всей ткани. Истинные кровеносные сосуды эмбриона цыпленка выращивают в САМ или в ткани, растущей на САМ.
Как было показано в настоящем описании, САМ-анализ иллюстрирует ингибирование реваскуляризации как исходя из количества, так и исходя из уровня роста новых сосудов. Кроме того, в этой модели легко прослеживается рост любой ткани, трансплантированной в САМ, такой как опухолевая ткань. И наконец, этот анализ является особенно эффективным вследствие того, что он позволяет осуществлять внутренний контроль на токсичность аналитической системы. Куриный эмбрион обрабатывают любым тестируемым реагентом. Здоровье эмбриона как таковое является показателем токсичности.
Во втором анализе осуществляют измерение уровня ангиогенеза in vivo на глазной модели кролика, а поэтому этот анализ называется анализом глаза кроличьим. Этот анализ глаза кроличий был описан подробно другими авторами, и далее он будет использован для измерения уровня ангиогенеза и реваскуляризации в присутствии ингибиторов ангиогенеза, таких как талидомин. См. D' Amato et al., Proc. Natl. Acad. Sci., USA, 91:4082-4085 (1994).
Этот анализ хорошо известен как модель анализа на in vivo-ангиогенез, поскольку процесс реваскуляризации, иллюстрируемый благодаря росту кроличьих кровеносных сосудов от края роговицы в роговицу, легко визуализируется благодаря
природной прозрачности роговицы глаза. Кроме того, в этом анализе может быть легко осуществлен мониторинг как степени, так и количественного уровня стимуляции или ингибирования реваскуляризации или регрессии реваскуляризации.
природной прозрачности роговицы глаза. Кроме того, в этом анализе может быть легко осуществлен мониторинг как степени, так и количественного уровня стимуляции или ингибирования реваскуляризации или регрессии реваскуляризации.
И наконец, кролика обрабатывают любым тестируемым реагентом, и здоровье кролика как таковое является показателем токсичности тестируемого реагента.
В третьем анализе измеряют ингибирование прямого связывания природного лианда, витронектина, с αvβ5, и предпочтительный вариант этого анализа подробно описан в примерах. В этом анализе обычно измеряют степень ингибирования связывания природного лиганда, такого как витронектин, с выделенным αvβ5 в твердой фазе с помощью ELISA, где ингибирование этого связывания опосредуется αvβ5-специфическим ингибированием.
Таким образом, этот анализ может быть также использован для идентификации соединений, которые обладают специфичностью по отношению к αvβ5, и не ингибируют связывания природных лигандов с другими интегринами. Анализ на специфичность проводят путем проведения параллельных ELISA-анализов, где как αvβ5, так и другие интегрины подвергают конкурентному скринингу в отдельных аналитических камерах на их соответствующую способность связываться с природным лигандом, и на присутствие соединений-кандидатов, ингибирующих соответствующую способность интегринов связываться с предварительно выбранным лигандом. Предпочтительные схемы проведения анализов скрининга описаны в примерах.
G. Промышленные препараты
Настоящее изобретение также относится к промышленному изделию, которое представляет собой контейнер с этикеткой, предназначенный для хранения антагониста αvβ5 настоящего изобретения. Это промышленное изделие включает упаковочный материал и фармацевтический агент, содержащийся в этом упаковочном материале.
Настоящее изобретение также относится к промышленному изделию, которое представляет собой контейнер с этикеткой, предназначенный для хранения антагониста αvβ5 настоящего изобретения. Это промышленное изделие включает упаковочный материал и фармацевтический агент, содержащийся в этом упаковочном материале.
Фармацевтическим агентом, содержащимся в промышленном изделии, является любой антагонист αvβ5 настоящего изобретения, изготовленный в фармацевтически приемлемой форме, описанной в настоящей заявке. Это промышленное изделие содержит количество фармацевтического агента в разовой дозе или в дробных дозах, достаточное для лечения заболевания, указанного в настоящем описании.
Упаковочный материал включает этикетку, на которой описано использование содержащегося в нем фармацевтического агента, например, для лечения заболевания путем ингибирования ангиогенеза и подобных состояний, описанных в настоящей заявке. Эта этикетка может, кроме того, включать инструкцию по использованию и соответствующую информацию, которая может оказаться необходимой для потребителя. Упаковочный материал может включать контейнер (или контейнеры) для хранения фармацевтического агента.
Используемый в настоящем описании термин "упаковочный материал" относится к такому материалу, как стекло, пластик, бумага, фольга, и другие материалы, способные удерживать фармацевтический агент в фиксированном состоянии. Так, например, упаковочным материалом могут быть пластиковые или стеклянные сосуды, многослойные оберточные материалы и подобные контейнеры, используемые для хранения фармацевтической композиции, включающей фармацевтический агент.
В предпочтительном варианте настоящего изобретения упаковочный материал включает этикетку, представляющую собой фактическое описание, в котором указано содержимое промышленного изделия и использование содержащегося в нем фармацевтического агента.
Примеры
Нижеследующие примеры, относящиеся к настоящему изобретению, лишь иллюстрируют изобретение и не должны рассматриваться как ограничение его объема. Кроме того, в настоящее изобретение могут быть включены различные его варианты, которые известны в настоящее время, либо будут разработаны позже специалистами в этой области, но которые не выходят за рамки нижеследующей формулы изобретения.
Нижеследующие примеры, относящиеся к настоящему изобретению, лишь иллюстрируют изобретение и не должны рассматриваться как ограничение его объема. Кроме того, в настоящее изобретение могут быть включены различные его варианты, которые известны в настоящее время, либо будут разработаны позже специалистами в этой области, но которые не выходят за рамки нижеследующей формулы изобретения.
1. Получение αvβ5-специфических моноклональных антител
Моноклональные антитела, P1F6 и Р5Н9, были продуцированы с использованием стандартных гибридомных методов путем иммунизации мышей RBF/DnJ клетками карциномы легких А549, как описано в работе Wayner et al., J. Cell. Biol. , 113: 919-929 (1991), которая вводится в настоящее описание посредством ссылки. Из иммунизированных мышей удаляли селезенки и эти селезенки гибридизировали с клетками миеломы Ns-1/FOX-NY. Гибридомы, продуцирующие антитело, направленное против рецепторов витронектина, присутствующих в клетках карциномы, скринировали путем специфического ингибирования адгезии UCLA-P3 с сенсибилизированными витронектином поверхностями, как описано Wayner и др., и клонировали путем лимитирующего разведения на фидерных слоях тимоцитов.
Моноклональные антитела, P1F6 и Р5Н9, были продуцированы с использованием стандартных гибридомных методов путем иммунизации мышей RBF/DnJ клетками карциномы легких А549, как описано в работе Wayner et al., J. Cell. Biol. , 113: 919-929 (1991), которая вводится в настоящее описание посредством ссылки. Из иммунизированных мышей удаляли селезенки и эти селезенки гибридизировали с клетками миеломы Ns-1/FOX-NY. Гибридомы, продуцирующие антитело, направленное против рецепторов витронектина, присутствующих в клетках карциномы, скринировали путем специфического ингибирования адгезии UCLA-P3 с сенсибилизированными витронектином поверхностями, как описано Wayner и др., и клонировали путем лимитирующего разведения на фидерных слоях тимоцитов.
Было показано, что оба моноклональных антитела P1F6 и Р5Н9 вступают в специфическую иммунную реакцию с αvβ5-комплексом и не вступают в иммунную реакцию с α-субъединицей, β5-субъединией или с другими интегринами. Моноклональное антитело P1F6 поставлялось фирмой Gibco BRL (Life Technologies, Inc. , Gaithersburg, MD), а моноклональное антитело P5H9 поставлялось Dr. E. Wayner (Fred Hutchinson Cancer Research Institute, Seattle, WA).
Другие моноклональные αvβ5-антитела, используемые в настоящем изобретении, были аналогично получены и охарактеризованы, как описано ниже. Кроме того, моноклональные антитела против αvβ5 были продуцированы путем слияния клеток селезенки, выделенных из мышей, которых иммунизировали αvβ5-рецептором либо в очищенном, либо в неочищенном виде. Очистка αvβ5 является процедурой, хорошо известной любому специалисту в области биологии интегринов, и была описана в работе Smith et al., J.Biol.Chem., 265:11008-11013 (1990), которая вводится в настоящее описание посредством ссылки. После очистки выделенный рецептор использовали как иммуноген для иммунизации мышей, как описано в разделе Е2, и получали в основном, как описано в работе Kohler & Milstein, Nature, 256:495-497 (1975), которая вводится в настоящее описание посредством ссылки. Полученные клоны гибридомы скринировали на реакцию с иммуногеном, а затем характеризовали, как описано в нижеследующих примерах.
2. Характеризация специфичности моноклональных антител против αvβ5 и их использование при картировании распределения в тканях, экспрессирующих αvβ5
А. Специфичность к витронектину
Как было показано Wayner et al., J.Cell.Biol., 113:919-929 (1991), моноклональное антитело Р5Н9, полученное в примере 1, блокирует связывание клеток карциномы UCLA-P3 с витронектином, но при этом не влияет на связывание клеток с коллагеном или фибронектином. Было также показано, что те же самые клетки содержат только лишь αvβ5-рецептор витронектина и ни одна из них не обладает αvβ5-специфичностью, что приводит к иммунопреципитации гетеродимера, состоящего из α-цепи (160 кДа) и β-цепи (95 кДа) в невосстанавливающих условиях. Было также показано, что αvβ5-рецептор, распознаваемый антителом Р5Н9, опосредует адгезию клеток меланомы М21 и клеток карциномы Н2981 с витронектином. Моноклональное антитело P1F6 имеет тот же самый профиль иммунореактивности.
А. Специфичность к витронектину
Как было показано Wayner et al., J.Cell.Biol., 113:919-929 (1991), моноклональное антитело Р5Н9, полученное в примере 1, блокирует связывание клеток карциномы UCLA-P3 с витронектином, но при этом не влияет на связывание клеток с коллагеном или фибронектином. Было также показано, что те же самые клетки содержат только лишь αvβ5-рецептор витронектина и ни одна из них не обладает αvβ5-специфичностью, что приводит к иммунопреципитации гетеродимера, состоящего из α-цепи (160 кДа) и β-цепи (95 кДа) в невосстанавливающих условиях. Было также показано, что αvβ5-рецептор, распознаваемый антителом Р5Н9, опосредует адгезию клеток меланомы М21 и клеток карциномы Н2981 с витронектином. Моноклональное антитело P1F6 имеет тот же самый профиль иммунореактивности.
В. Иммунофлуоресценция с использованием антител против интегринового рецептора
В период заживления раны базальные мембраны кровеносных сосудов экспрессируют несколько адгезивных белков, включая фактор фон Виллебранда, фибронектин, и фибрин. Кроме того, некоторые члены интегринового семейства рецепторов адгезии экспрессируются на поверхности культивированных клеток гладкой мышцы и эндотелиальных клеток. См. Cheresh, Proc. Natl. Acad. Sci., USA, 84: 6471 (1987); Janat et al., J. Cell. Physiol. 151:588 (1992); и Cheng et al., J. Cell. Physiol., 139:275 (1989).
В период заживления раны базальные мембраны кровеносных сосудов экспрессируют несколько адгезивных белков, включая фактор фон Виллебранда, фибронектин, и фибрин. Кроме того, некоторые члены интегринового семейства рецепторов адгезии экспрессируются на поверхности культивированных клеток гладкой мышцы и эндотелиальных клеток. См. Cheresh, Proc. Natl. Acad. Sci., USA, 84: 6471 (1987); Janat et al., J. Cell. Physiol. 151:588 (1992); и Cheng et al., J. Cell. Physiol., 139:275 (1989).
Помимо структуры и функции β5-субъединицы интегрина распределение этой субъединицы в тканях путем картирования с использованием других моноклональных антител против β5, описано в работе Pasqualini et al., J.Cell.Sci., 105: 101-111 (1993), которая вводится в настоящее описание посредством ссылки.
Описанные выше моноклональные антитела против β5-субъединицы, аналогичные антителам, описанным в примере 1, были секретированы из гибридом, которые получали с использованием спленоцитов от мышей, иммунизированных клеточной линией карциномы легкого человека А549. Эти гибридомы отбирали путем позитивного поверхностного окрашивания клеток А549 супернатантом культуры гибридом и путем иммунопреципитации αvβ5-комплексов из поверхностно-меченых экстрактов А549. Затем моноклональные антитела использовали для картирования распределения β5-субъединицы в тканях тимуса, кожи и почках здорового человека. Толстые срезы, толщиной 4 микрона, вырезали из блоков ткани, замороженных на криостатном микротоме, для последующего стрептавидин-биотин-иммунопероксидазного окрашивания с использованием антител, обладающих специфичностью к интегринам β5, проведенного, как описано в работе Pasqualini и др., см. ссылку.
С помощью окрашивания тимусных срезов было выявлено распределение β5 на кровеносных сосудах, тельцах вилочковой железы (тельцах Гассаля), клетках коры надпочечника и медуллярных стромальных клетках, а также на базальных мембранах. Срезы кожи обнаруживали присутствие β5 на базальном слое эпидермиса и на стенках некоторых кожных кровеносных сосудов, а срезы почек обнаруживали окрашивание гломерулярных областей, юкстагломерулярного аппарата, проксимальных извитых почечных канальцев и прямых почечных канальцев. Таким образом, распределение β5 является однородным для различных типов клеток, а именно, что наиболее важно, на капиллярных эндотелиальных клетках, окрашивание которых соответствует окрашиванию культивированных эндотелиальных клеток пупочной вены.
С. Иммунофлуоресценция ткани сетчатки человека у пациентов, страдающих заболеваниями глаз, с использованием антител против интегринового рецептора
Реваскуляризация ткани глаза является наиболее распространенным патологическим изменением, которое наблюдается при глазных болезнях широкого ряда и которое приводит к катастрофической потере зрения. Рост новых кровеносных сосудов из уже существующих хороидальных, ретинальных или паралимбальных сосудов может вызывать отеки, гемморагии или образование фиброваскулярной мембраны, что приводит к нарушению нормальных анатомических функций глаза и сопровождается потерей нормальной зрительной функции.
Реваскуляризация ткани глаза является наиболее распространенным патологическим изменением, которое наблюдается при глазных болезнях широкого ряда и которое приводит к катастрофической потере зрения. Рост новых кровеносных сосудов из уже существующих хороидальных, ретинальных или паралимбальных сосудов может вызывать отеки, гемморагии или образование фиброваскулярной мембраны, что приводит к нарушению нормальных анатомических функций глаза и сопровождается потерей нормальной зрительной функции.
В физиологических условиях ангиогенез строго регулируется, и было показано, что он активируется специфическими ангиогенными цитокинами, такими как основной фактор роста фибробластов (bFGF) и фактор некроза опухоли-α (TNFα). Как описано Brooks et аl., Science, 264:569-571 (1994), было показано, что моноклональные антитела против αvβ5 блокируют как bFGF- и TNFα-индуцированный ангиогенез в экспериментальных системах, включая модель САМ, описанную ниже. Как описано в примерах 4-6, моноклональные антитела против αvβ5 блокируют отдельные механизмы ангиогенеза, который специфически индуцируется сосудистым эндотелиальным фактором роста (VEGF), трансформирующим фактором роста-α (TGF-α) и эпидермальным фактором роста (EGF).
Таким образом, как описано в контексте настоящего изобретения, два механизма ангиогенеза обусловлены действием различных интегринов  Для исследования экспрессии и роли этих интегринов в патологии глазных болезней эпиретинальные реваскулярные мембраны и субретинальные реваскулярные мембраны были получены целиком при витрэктомии пациентов с пролиферативной диабетической ретинопатией (PDR). Эти пациенты наблюдались в клинике и были отобраны на гистологический анализ на основании того, что они страдали прогрессирующим пролиферативным заболеванием с реваскуляризацией, которое было диагностировано путем клинического обследования и с помощью флуоресцентной ангиографии глазного дна. Полученную ткань сразу замораживали путем криоконсервации в Tissue Tek и делали срезы.
Для исследования экспрессии и роли этих интегринов в патологии глазных болезней эпиретинальные реваскулярные мембраны и субретинальные реваскулярные мембраны были получены целиком при витрэктомии пациентов с пролиферативной диабетической ретинопатией (PDR). Эти пациенты наблюдались в клинике и были отобраны на гистологический анализ на основании того, что они страдали прогрессирующим пролиферативным заболеванием с реваскуляризацией, которое было диагностировано путем клинического обследования и с помощью флуоресцентной ангиографии глазного дна. Полученную ткань сразу замораживали путем криоконсервации в Tissue Tek и делали срезы.
При исследовании тканей этих пациентов методами иммунофлуоресценции кровеносные сосуды обнаруживали позитивный результат на интегрин αvβ5, о чем свидетельствовала иммунная реактивность с мышиным моноклональным антителом LM609. Было выявлено, что распределение интегрина ограничено кровеносными сосудами и соответствует окрашиванию, проводимому для анализа на маркер кровеносных сосудов, фактор фон Виллебранда, картированный с использованием кроличьего антитела против этого фактора. Сайты иммунореактивности были визуализированы с помощью либо конъюгированного с родамином антитела против мышиных иммуноглобулинов, либо конъюгированного с флуоресцеином антитела против кроличьих иммуноглобулинов, использование которых позволило определять совместную локализацию интегрина и антител, специфичных для кровеносных сосудов.
Образцы, полученные от пациентов с нормальной глазной тканью или от пациентов с атрофическими мембранами, не содержащими активно пролиферирующих кровеносных сосудов, и исследованные с помощью иммунофлуоресценции, показали отрицательный результат на интегрин αvβ3.
Параллельно те же самые ткани анализировали с помощью иммуногистохимического анализа на присутствие и распределение αvβ5 с использованием моноклонального антитела против αvβ5, P1F6, полученного в примере 1. Окрашивание выявило присутствие αvβ5 на кровеносных сосудах, что соответствует распределению фактора фон Виллебранда. Однако несосудистая ткань также обнаруживала ограниченную флуоресценцию при мечении антителом P1F6, что указывало на более широкое распределение αvβ5. Этот результат отличался от распределения αvβ5, которое ограничивалось кровеносными сосудами.
Параллельно те же самые ткани анализировали с помощью иммуногистохимического анализа на присутствие и распределение αvβ5 с использованием моноклонального антитела против αvβ5, P1F6, полученного в примере 1. Окрашивание выявило присутствие αvβ5 на кровеносных сосудах, что соответствует распределению фактора фон Виллебранда. Однако несосудистая ткань также обнаруживала ограниченную флуоресценцию при мечении антителом P1F6, что указывало на более широкое распределение αvβ5. Этот результат отличался от распределения αvβ5, которое ограничивалось кровеносными сосудами.
При иммунофлуоресцентном окрашивании мембран проводили сравнение αvβ5 с использованием соответствующих антител LM609 и P1F6, и фактически одинаковый характер окрашивания на стенке кровеносных сосудов указывал на то, что αvβ3 и αvβ5 присутствуют на поверхности вновь пролифирирующих кровеносных сосудов человека, возникающих при реваскуляторных глазных болезнях, таких как диабетическая ретинопатия.
Таким образом, результаты, описанные в настоящей заявке, показали, что интегриновый рецептор αvβ5 селективно экспрессируется в специфических типах тканей, в которых происходит ангиогенез, что подтверждает исследование реваскуляторных мембран, взятых от пациентов, страдающих заболеванием с прогрессирующей пролиферативной реваскуляризацией. Поэтому эти ткани наряду с тканями, подвергнутыми воздействию факторов роста, как описано ниже в примерах 4-6, представляют собой идеальные мишени с точки зрения терапевтических целей настоящего изобретения.
3. Получение синтетических пептидов
а. Процедуры синтеза
Циклические полипептиды, используемые для осуществления способов настоящего изобретения, были синтезированы стандартными методами твердофазного синтеза, описанными, например, Marrifield, Adv. Enzymol., 32:221-296 (1969), и Fields, G.B. & Noble, R.L. Int.J.Peptide Protein Res., 35:161-214 (1990).
а. Процедуры синтеза
Циклические полипептиды, используемые для осуществления способов настоящего изобретения, были синтезированы стандартными методами твердофазного синтеза, описанными, например, Marrifield, Adv. Enzymol., 32:221-296 (1969), и Fields, G.B. & Noble, R.L. Int.J.Peptide Protein Res., 35:161-214 (1990).
Два грамма (г) BOC-Arg-Gly-Asp-D-Phe-Val-OMe (SEQ ID No:l) были сначала разведены в 60 миллилитрах (мл) метанола, в который было добавлено 1,5 мл 2н. раствора гидроксида натрия с образованием смеси. Затем эту смесь размешивали в течение 3 часов при 20oС. После выпаривания, остаток растворяли в воде и подкисляли до рН 3 разбавленной НС1 и экстрагировали этилацетатом. Экстракт сушили над Na2SО4, снова выпаривали, и полученный BOC-Arg-Gly-Asp-D-Phe-Val-OH (SEQ ID No:2) перемешивали в течение 2 часов при 20oС с 20 миллилитрами 2н. НСl в диоксане. Полученную смесь выпаривали и получали H-Arg-Gly-Asp-D-Phe-Val-OH (SEQ ID No: 3), который затем растворяли в смеси 1800 мл дихлорметана и 200 мл диметилформамида (ДМФ) с последующим охлаждением до 0oС. После этого последовательно при перемешивании добавляли 0,5 г дициклогексилкарбодиимида (DCCI), 0,3 г 1-гидроксибензотриазола (HOBt) и 0,23 мл N-метилморфолина.
Полученную смесь перемешивали еще 24 часа при 0oС, а затем еще 48 часов при 20oС. Раствор концентрировали и обрабатывали смешанной ионообменной смолой для удаления солей. После этого полученную смолу удаляли путем фильтрации, осветленный раствор выпаривали, а остаток очищали с помощью хроматографии, в результате чего получали цикло(Arg-Gly-Asp-D-Phe-Val) (также обозначенный однобуквенным кодом как c-RGDfV)(SEQ ID No:4). В случае если в обозначении пептида присутствуют строчные буквы, то они обозначают D-форму аминокислоты, а не L-форму, как в случае обозначения заглавными буквами.
Циклический контрольный пептид, цикло(Arg-Ala-Asp-D-Phe-Val) (также обозначенный однобуквенным кодом как RADfV) (SEQ ID N:5) был получен, как описано выше. Ранее было показано, что циклический пептид c-RGDfV (SEQ ID No:5) ингибирует связывание фибриногена с интегрином αvβ3, и не ингибирует связывание фибриногена с интегринами α11bβ3 или α5β1 (Pfaff et al., J.Biol. Chem. , 269:20233-20238, 1994).
Другие пептиды, которые специфически ингибируют связывание природных лигандов с αvβ5 и которые были получены аналогичным образом, тестировали на специфичность и диапазон активности, как описано в нижеследующих примерах. Такими пептидами являются следующие аналогичным образом полученные пептиды: цикло(Gly-D-Arg-Gly-Asp-Phe-Val) (SEQ ID N:6) и цикло (Arg-Gly-Asp-Phe-D-Val) (SEQ ID N:7). Пептиды, имеющие аминокислотную последовательность Tyr-Thr-Ala-Glu-Cys-Lys-Pro-Gln-Val-Thr-Arg-Gly-Asp-Val-Phe (SEQ ID No: 8) и цикло (Arg-Gly-Asp-D-Phe-NMeVal) (SEQ ID No:9) были также получены синтетически. В SEQ ID No:9, префикс "Me" в символах MeVal означает, что валин в положении 6 был модифицирован путем метилирования у азота альфа-аминогруппы в амидной связи валинового остатка.
b. Альтернативная процедура синтеза
i. Синтез TFA-соли цикло (Arg-Gly-Asp-D-Phe-NМeVal)
Fmoc-Arg(Mtr)-Gly-Asp(OBut)-DPhe-NMeVal-ONa синтезировали с использованием твердофазных методов типа Меррифилда путем последовательного постадийного добавления NМeVal, DPhe, ASP(OBut), Gly Fmoc-Arg(Mtr) к 4-гидроксиметил-феноксиметилполистироловой смоле (тип смолы Ванга) (обычные методы пептидного синтеза типа Меррифилда осуществляли, как описано в Houben-Weyl, 1. С. Volume 15/11, pages 1-806 (1974). Полистироловая смола и предшественники аминокислотных остатков поставлялись от коммерческих компаний Aldrich, Sigma или Fluka). Затем, после завершения последовательного добавления аминокислотных остатков, смолу отделяли от пептидной цепи с использованием смеси (1: 1) TFA/дихлорметана, в результате чего получали Fmoc-Arg(Mtr)-Gly-Asp(OBut)-DPhe-NMeVal-OH-продукт. Затем группу Fmoc удаляли с использованием 1: 1-смеси пиперидина/ДМФ, в результате чего получали неочищенный предшественник Arg(Mtr)-Gly-Asp(OBut)-DPhe-NMeVal-ОН, который затем очищали обычным способом с помощью ВЭЖХ.
i. Синтез TFA-соли цикло (Arg-Gly-Asp-D-Phe-NМeVal)
Fmoc-Arg(Mtr)-Gly-Asp(OBut)-DPhe-NMeVal-ONa синтезировали с использованием твердофазных методов типа Меррифилда путем последовательного постадийного добавления NМeVal, DPhe, ASP(OBut), Gly Fmoc-Arg(Mtr) к 4-гидроксиметил-феноксиметилполистироловой смоле (тип смолы Ванга) (обычные методы пептидного синтеза типа Меррифилда осуществляли, как описано в Houben-Weyl, 1. С. Volume 15/11, pages 1-806 (1974). Полистироловая смола и предшественники аминокислотных остатков поставлялись от коммерческих компаний Aldrich, Sigma или Fluka). Затем, после завершения последовательного добавления аминокислотных остатков, смолу отделяли от пептидной цепи с использованием смеси (1: 1) TFA/дихлорметана, в результате чего получали Fmoc-Arg(Mtr)-Gly-Asp(OBut)-DPhe-NMeVal-OH-продукт. Затем группу Fmoc удаляли с использованием 1: 1-смеси пиперидина/ДМФ, в результате чего получали неочищенный предшественник Arg(Mtr)-Gly-Asp(OBut)-DPhe-NMeVal-ОН, который затем очищали обычным способом с помощью ВЭЖХ.
Для циклизации раствор 0,6 г Arg(Mtr)-Gly-Asp(OBut)-DPhe-NMeVal-OH (синтезированного, как описано выше) в 15 мл ДМФ (диметилформамида; Aldrich), разводили 85 миллитрами дихлорметана (Aldrich) и добавляли 50 мг NаНСО3. После охлаждения в смеси сухой лед/ацетон, добавляли 40 мкл дифенилфосфорилазида (Aldrich). После выдерживания при комнатной температуре в течение 16 часов раствор концентрировали. Концентрат подвергали гель-фильтрации (на колонке с Сефадексом в смеси изопропанол/вода, 8:2), а затем очищали с помощью ВЭЖХ обычным способом. После обработки TFA (фторуксусная кислота/Н2O, 98: 2) получали цикло-(Arg-Gly-Asp-DPhe-NMeVal)•TFA, который затем очищали с помощью ВЭЖХ обычным способом; RT=19,5; FAB-MC (М+Н): 589.
ii. Синтез "внутренней соли"
Соль трифторуксусной кислоты (TFA) удаляли из вышепродуцированного циклического пептида путем суспендирования цикло (Arg-Gly-Asp-DPhe-NMeVal)•TFA в воде с последующим выпариванием под вакуумом для удаления TFA. Образованный таким образом циклический пептид именовали "внутренней солью" и обозначали цикло(Arg-Gly-Asp-DPhe-NMeVal). Термин "внутренняя соль" используется потому, что этот циклический пептид содержит два противоположно заряженных остатка, которые обеспечивают друг для друга внутриэлектрический баланс с образованием в целом незаряженной молекулы. Один из заряженных остатков содержит кислотную часть, а другой заряженный остаток содержит амино-часть. Если кислотная часть расположена в непосредственной близости от другой части, то эта кислотная часть может быть депротонирована амино-частью, которая образует карбоксилат/аммониевую соль с суммарным нейтральным зарядом.
Соль трифторуксусной кислоты (TFA) удаляли из вышепродуцированного циклического пептида путем суспендирования цикло (Arg-Gly-Asp-DPhe-NMeVal)•TFA в воде с последующим выпариванием под вакуумом для удаления TFA. Образованный таким образом циклический пептид именовали "внутренней солью" и обозначали цикло(Arg-Gly-Asp-DPhe-NMeVal). Термин "внутренняя соль" используется потому, что этот циклический пептид содержит два противоположно заряженных остатка, которые обеспечивают друг для друга внутриэлектрический баланс с образованием в целом незаряженной молекулы. Один из заряженных остатков содержит кислотную часть, а другой заряженный остаток содержит амино-часть. Если кислотная часть расположена в непосредственной близости от другой части, то эта кислотная часть может быть депротонирована амино-частью, которая образует карбоксилат/аммониевую соль с суммарным нейтральным зарядом.
iii. Обработка НСl с получением цикло (Arg-Gly-Asp-D-Phe-NMeVal)•НСl
80 мг цикло(Arg-Gly-Asp-D-Phe-NMeVal) растворяли в 0,01 М НСl 5-6 раз и после каждой процедуры растворения подвергали сушке вымораживанием. После очистки с помощью ВЭЖХ получали цикло(Arg-Gly-Asp-DPhe-NMeVal)•НСl; FAB-MC (М+Н):589.
80 мг цикло(Arg-Gly-Asp-D-Phe-NMeVal) растворяли в 0,01 М НСl 5-6 раз и после каждой процедуры растворения подвергали сушке вымораживанием. После очистки с помощью ВЭЖХ получали цикло(Arg-Gly-Asp-DPhe-NMeVal)•НСl; FAB-MC (М+Н):589.
iv. Обработка метансульфоновой кислотой с получением цикло(Arg-Gly-Asp-DPhe-NMeVal)•МеSО3Н
80 мг цикло(Arg-Gly-Asp-D-Phe-NMeVal) растворяли в 0,01 М МеSО3Н (метансульфоновой кислоте) 5-6 раз и после каждой процедуры растворения подвергали сушке вымораживанием. После очистки с помощью ВЭЖХ получали цикло(Arg-Gly-Asp-DPhe-NMeVal)•MeSО3Н; RT=17,8; FAB-MC (М+Н): 589.
80 мг цикло(Arg-Gly-Asp-D-Phe-NMeVal) растворяли в 0,01 М МеSО3Н (метансульфоновой кислоте) 5-6 раз и после каждой процедуры растворения подвергали сушке вымораживанием. После очистки с помощью ВЭЖХ получали цикло(Arg-Gly-Asp-DPhe-NMeVal)•MeSО3Н; RT=17,8; FAB-MC (М+Н): 589.
Альтернативные методы циклизации предусматривают дериватизацию цепей боковых групп ациклического пептидного предшественника сульфгидрильными группами, а в условиях рН, слегка превышающем физиологический рН (рН 7,5), дериватизацию внутримолекулярных форм дисульфидных связей другими сульфгидрильными группами, присутствующими в молекуле, с образованием циклического пептида. Кроме того, С-конец карбоксилатной части ациклического пептидного предшественника может быть подвергнут реакции со свободной сульфгидрильной частью, присутствующей в молекуле, с продуцированием тиоэфирных циклизованных пептидов.
4. Ингибирование индуцированного факторами роста ангиогенеза антагонистами αvβ5, которое было измерено в in vivo-анализе с использованием кроличьей модели глаза
Действие антагониста против αvβ5 на индуцированный фактором роста ангиогенез может наблюдаться в природных прозрачных структурах, например в роговице глаза. Новые кровеносные сосуды растут от края роговицы, который хорошо снабжается кровью, по направлению к центру роговицы, который обычно не имеет кровеносных сосудов. Обработка роговицы стимуляторами ангиогенеза, такими как VEGF и TGF-α индуцировала рост новых кровеносных сосудов от края роговицы. Антагонисты ангиогенеза, нанесенные на роговицу, ингибируют рост новых кровеносных сосудов от края роговицы. Таким образом, роговица подвергается ангиогенезу посредством инвазии эндотелиальных клеток от края роговицы в плотно упакованную коллагеном ткань роговицы, которую можно легко наблюдать. Поэтому кроличья модель глаза обеспечивает in vivo-модель для прямого наблюдения стимуляции и ингибирования ангиогенеза после имплантации соединений в роговицу глаза.
Действие антагониста против αvβ5 на индуцированный фактором роста ангиогенез может наблюдаться в природных прозрачных структурах, например в роговице глаза. Новые кровеносные сосуды растут от края роговицы, который хорошо снабжается кровью, по направлению к центру роговицы, который обычно не имеет кровеносных сосудов. Обработка роговицы стимуляторами ангиогенеза, такими как VEGF и TGF-α индуцировала рост новых кровеносных сосудов от края роговицы. Антагонисты ангиогенеза, нанесенные на роговицу, ингибируют рост новых кровеносных сосудов от края роговицы. Таким образом, роговица подвергается ангиогенезу посредством инвазии эндотелиальных клеток от края роговицы в плотно упакованную коллагеном ткань роговицы, которую можно легко наблюдать. Поэтому кроличья модель глаза обеспечивает in vivo-модель для прямого наблюдения стимуляции и ингибирования ангиогенеза после имплантации соединений в роговицу глаза.
A. in vivo-анализ, проведенный на кроличьей модели глаза
1) Ангиогенез, индуцированный факторами роста
В in vivo-анализе на кроличьей модели глаза ангиогенез был индуцирован факторами роста и описан ниже.
1) Ангиогенез, индуцированный факторами роста
В in vivo-анализе на кроличьей модели глаза ангиогенез был индуцирован факторами роста и описан ниже.
а. Получение гидроновых осадков, содержащих факторы роста и моноклональные антитела
Гранулы из гидронового полимера, содержащие фактор роста и моноклональные антитела (mAb) были получены, как описано D'Amato et al., Proc.Natl. Acad. Sci. , USA, 91:4082-4085 (1994), отдельные гранулы, содержащие 750 нг фактора роста (называемого также цитокином), и антитело, в частности либо bFGF, либо VEGF, связанное с сукрафлатом (карафатом) (Carafet, Marion Merrell Dow Corporation, Cincinnati, OH) для стабилизации цитокинов и для обеспечения их медленного высвобождения в окружающие ткани. Кроме того, были получены гидроновые гранулы, которые содержали либо 40 мкг mAb P1F6 (против αvβ5), либо контрольное антитело LM609 (против αvβ3) в PBS.
Гранулы из гидронового полимера, содержащие фактор роста и моноклональные антитела (mAb) были получены, как описано D'Amato et al., Proc.Natl. Acad. Sci. , USA, 91:4082-4085 (1994), отдельные гранулы, содержащие 750 нг фактора роста (называемого также цитокином), и антитело, в частности либо bFGF, либо VEGF, связанное с сукрафлатом (карафатом) (Carafet, Marion Merrell Dow Corporation, Cincinnati, OH) для стабилизации цитокинов и для обеспечения их медленного высвобождения в окружающие ткани. Кроме того, были получены гидроновые гранулы, которые содержали либо 40 мкг mAb P1F6 (против αvβ5), либо контрольное антитело LM609 (против αvβ3) в PBS.
Все тестированные моноклональные антитела были очищены из асцитной жидкости с использованием аффинной хроматографии на колонках с белком А-Сефарозой CL-4B хорошо известными методами. Затем проэлюированный иммуноглобулин диализовали против PBS и обрабатывали гелем Detoxi-gel (Pierce Chemicals, Rockford, IL) для удаления эндотоксина. Известно, что эндотоксин обладает сильным ангиогенным и воспалительным действием. Поэтому моноклональные антитела были протестированы на присутствие эндотоксина посредством анализа с использованием хромогенного лизата амебоцитов (Chromogenic Lumulus Amebocyte Lysate Assay (BioWhittaker, MD), и в анализе на модели кроличьего глаза были использованы только те антитела (mAb), которые не содержали обнаруживаемого эндотоксина.
Гранулы были сформованы в специально приготовленных тефлоновых формах в виде стержней, которые имели сердцевину размером 2,5 мм, высверленную в их поверхности. Приблизительно 12 мкл материала для формования помещали в каждую форму и полимеризовали в течение ночи в стерильном вытяжном шкафу. Затем гранулы стерилизовали путем ультрафиолетового облучения.
Для экспериментов на двух глазах были использованы группы из восьми животных, где каждому животному вводили гидроновый имплантат, содержащий предварительно отобранный цитокин с предварительно отобранным антителом или контрольным иммуноглобулином. Более конкретно, для каждого кролика в одну роговицу хирургически имплантировали гидроновую гранулу, содержащую либо bFGF, либо VEGF в сочетании с mAb, P1F6, а другую роговицу обрабатывали либо bFGF, либо VEGF в сочетании с P1F6 и LM609. Отдельные гранулы имплантировали в хирургически созданные "карманы", образованные средней стромой роговицы кроликов. Хирургическую процедуру проводили в условиях стерильности с использованием рабочего микроскопа модели М691 Wild, снабженного светоделителем, в который была вмонтирована камера для фотографического регистрирования отдельных роговиц. В строме роговицы был создан "карман" (3 мм • 5 мм) путем разреза глубиной 3 мм, составляющей половину толщины роговицы, лезвием Beaver 69. Строму рассекали по периферии с использованием шпателя для радужной оболочки глаза и гранулу имплантировали так, чтобы ее периферический край находился на расстоянии 2 мм от лимба.
В течение последующих 12 дней цитокины и mAb диффундировали из имплантированных гранул в окружающую ткань, стимулируя тем самым ангиогенез от края роговицы.
Левую и правую роговицы обозначали соответственно OS и OD. Затем роговицы наблюдали в течение 12 дней. Фотографии делали на 10-й день после операции, то есть, во время, когда образование новых сосудов является максимальным.
Репрезентативные фотографические результаты вышеупомянутых обработок смесями цитокинов/mAb показаны на фиг.1А-1D. Параллельный количественный анализ mAb-ингибирования цитокин-индуцированного ангиогенеза показан на фиг. 2А и 2В. На фиг.1А и 1D, где роговицы были соответственно обработаны комбинациями bFGF/P1F6 и VEGF/LM609, показан явно выраженный цитокин-индуцированный ангиогенез с отеком, как обозначено крупными стрелками. Поэтому антитело против αvβ5 P1F6, не оказывало эффективного ингибирующего действия на bFGF-индуцированный ангиогенез. Аналогично антитело против αvβ3, LM609 не оказывало эффективного ингибирующего действия на VEGF-индуцированный ангиогенез.
В отличие от этого в кроличьей модели, где использовали обработку цитокин/mАb-комбинациями bFGF/LM609 и VEGF/P1F6, цитокин-индуцированный ангиогенез ингибировался антителами, как показано на фиг.1В и 1С соответственно. На этих фиг. нормальные сосуды лимба конъюнктивы, обозначенные маленькими стрелками, указывают на эффективность антитела против интегрина в ингибировании одного типа цитокининдуцированного ангиогенеза.
Была также проведена количественная оценка влияния иммуннореактивности интегрин-специфических mAb на вышеуказанный цитокин-индуцированный ангиогенез, как показано на фиг.2А и 2В. Ангиогенез был стимулирован либо bFGF, либо VEGF, как показано соответственно на фиг.2А и 2В. Фотографирование обработанных глаз проводили ежедневно с помощью рабочего микроскопа Wild, оснащенного камерой Nikon. Фотографии получали на диапозитивной пленке Kodak Ektachrome 64Т и изображение преобразовывали для компьютерного количественного анализа с использованием программного обеспечения Biorad's Molecular Analyst 1.1 после обнаружения с помощью визуализирующего денситометра модели GS670. Гистограммы иллюстрируют среднюю площадь новообразованных сосудов ± стандартная ошибка (n=8 для каждой из двух групп) после обработки моноклональным антителом P1F6 или LM609.
Как показано на фиг.2А, антитело LM609 способствовало уменьшению bFGF-индуцированного ангиогенеза на 86% (р<0,005, парный t-критерий), по сравнению с обработкой двух глаз одного и того же животного антителом P1F6. В том случае когда для стимуляции ангиогенеза использовали VEGF, как показано на фиг. 2В, то наблюдался противоположный эффект, где антитело P1F6 способствовало уменьшению средней площади реваскуляризации на 60% (р<0,03, парный t-критерий), по сравнению с LМ609-обработанным глазом, который обнаруживал минимальный эффект после VEGF-индуцированного ангиогенеза.
Важно отметить, что обработка конкретным mAb влияла лишь на цитокин-индуцированный рост новых кровеносных сосудов, тогда как уже существующие перилимбальные сосуды не были подвержены воздействию mAb, что позволяет сделать вывод, что наблюдаемое воздействие mAb ограничено лишь воздействием на образование новых кровеносных сосудов в роговице.
Аналогичные анализы были осуществлены с использованием синтетических пептидов, полученных, как описано в примере 3 и как описано ниже для ингибирования цитокин-индуцированного ангиогенеза, который конкретно коррелирует с экспрессией αvβ5.
Для подтверждения этих результатов, свидетельствующих о том, что ангиогенез, индуцированный конкретным цитокином, ингибируется только одним типом интегрин-специфического антитела, а в частности, что интегриновый рецептор αvβ5 играет определенную роль в VEGF-индуцированном ангиогенеэе, был проведен анализ на другой модели реваскуляризации, представляющей собой хориоаллантоисную мембрану ципленка (САМ), с использованием комбинации цитокинов и антител против интегрина, как описано в следующем примере.
Для подтверждения этих результатов, свидетельствующих о том, что ангиогенез, индуцированный конкретным цитокином, ингибируется только одним типом интегрин-специфического антитела, а в частности, что интегриновый рецептор αvβ5 играет определенную роль в VEGF-индуцированном ангиогенеэе, был проведен анализ на другой модели реваскуляризации, представляющей собой хориоаллантоисную мембрану ципленка (САМ), с использованием комбинации цитокинов и антител против интегрина, как описано в следующем примере.
b. Обработка полипептидами
В каждом эксперименте использовали 8 кроликов, которым в один глаз вводили гранулу, содержащую 100 нанограммов (нг) bFGF, а в другой глаз вводили гранулу, содержащую 1 микрограмм (мкг) VEGF. Эти гранулы вводили в "карман" роговицы, как описано выше, после чего цитокины стимулировали рост новых кровеносных сосудов в роговице. Пептиды вводили подкожно (s.q.) в 1 мл PBS в начальной дозе 50 мкг на кг веса кролика в день введения гранул, а затем ежедневно вводили s. q. дозы 20 мкг/кг. Через 7 дней роговицу исследовали, как описано выше.
В каждом эксперименте использовали 8 кроликов, которым в один глаз вводили гранулу, содержащую 100 нанограммов (нг) bFGF, а в другой глаз вводили гранулу, содержащую 1 микрограмм (мкг) VEGF. Эти гранулы вводили в "карман" роговицы, как описано выше, после чего цитокины стимулировали рост новых кровеносных сосудов в роговице. Пептиды вводили подкожно (s.q.) в 1 мл PBS в начальной дозе 50 мкг на кг веса кролика в день введения гранул, а затем ежедневно вводили s. q. дозы 20 мкг/кг. Через 7 дней роговицу исследовали, как описано выше.
Кролики, которым был введен контрольный пептид 69601, обнаруживали значительный рост кровеносных сосудов в роговице через 7 дней как в bFGF-, так и в VEGF-стимулированном глазе. У кроликов, которым был введен пептид 85189, наблюдался менее чем 50% рост кровеносных сосудов в роговице по сравнению с контрольными bFGF-стимулированными глазами, а в VEGF-стимулированных глазах наблюдалось почти 100% ингибирование роста кровеносных сосудов.
5. Ангиогенез в препарате хориоаллантоисной мембраны цыпленка (САМ)
А. Характеризация необработанной САМ
1) Получение САМ
Ангиогенез может быть индуцирован на хориоаллантоисной мембране цыпленка (САМ) после того, как нормальный эмбриогенный ангиогенез приведен к образованию зрелых кровеносных сосудов. Было показано, что ангиогенез индуцируется в ответ на действие специфических цитокинов или опухолевых фрагментов, как описано Leibovich et al., Nature, 329:630 (1987) и Ausprunk et al., Am. J. Pathol., 79:597 (1975). CAM могут быть получены из куриных эмбрионов для последующего индуцирования ангиогенеза и его ингибирования антагонистом αvβ5 настоящего изобретения, как описано ниже и в примере 6.
А. Характеризация необработанной САМ
1) Получение САМ
Ангиогенез может быть индуцирован на хориоаллантоисной мембране цыпленка (САМ) после того, как нормальный эмбриогенный ангиогенез приведен к образованию зрелых кровеносных сосудов. Было показано, что ангиогенез индуцируется в ответ на действие специфических цитокинов или опухолевых фрагментов, как описано Leibovich et al., Nature, 329:630 (1987) и Ausprunk et al., Am. J. Pathol., 79:597 (1975). CAM могут быть получены из куриных эмбрионов для последующего индуцирования ангиогенеза и его ингибирования антагонистом αvβ5 настоящего изобретения, как описано ниже и в примере 6.
10-дневные куриные эмбрионы получали от фирмы McIntyre Poultry (Lakeside, CA) и инкубировали при 37oС в условиях 60%-ной влажности. В скорлупе на конце яйца как раз над воздушным мешком было проделано небольшое отверстие с использованием небольшой специальной дрели (Dremel, Division of Emerson Electrik Co., WI). Второе отверстие было просверлено на широкой боковой стороне яйца в области, в которой отсутствуют эмбриональные кровеносные сосуды, что было определено заранее путем просвечивания яйца. К первоначальному отверстию прилагали отрицательное давление, которое приводило к вытягиванию САМ (хориоаллантоисной мембраны) из оболочки скорлупы и создавало ложный воздушный мешок поверх САМ. Затем в скорлупе над нижерасположенной САМ было вырезано квадратное окно размером 1,0 сантиметров (см) • 1,0 см с использованием небольшого круглого резца (Dremel). Это небольшое окно давало прямой доступ к расположенной под ним САМ.
Полученный препарат САМ затем использовали на 10 день эмбриогенеза, когда ангиогенез уменьшался. Таким образом, этот препарат использовали в целях настоящего изобретения для индуцирования образования новых кровеносных сосудов в ответ на обработку цитокинами.
2) Гистология САМ
Для анализа микроскопической структуры эмбриона САМ из блоков, замороженных на криостатном микротоме, получали срезы толщиной 6 микрон (мк) для проведения иммунофлуоресцентного анализа.
Для анализа микроскопической структуры эмбриона САМ из блоков, замороженных на криостатном микротоме, получали срезы толщиной 6 микрон (мк) для проведения иммунофлуоресцентного анализа.
Типичные необработанные 10-дневные САМ представляли собой область, лишенную кровеносных сосудов. Поскольку ангиогенез в САМ-системе снижался на этой стадии эмбриогенеза, то эту систему использовали в настоящем изобретении для стимуляции различными цитокинами продуцирования новых кровеносных сосудов из сосудов, уже существующих в близлежащих областях, в тех областях САМ, в которых кровеносные сосуды обычно отсутствуют.
Как было показано на САМ-модели и в последующих примерах, при росте новых сосудов, происходящем при нормальном эмбриогенезе или продуцированном цитокинами, в кровеносных сосудах экспрессируются αvβ3 и αvβ5.
В. Ангиогенез, индуцированный факторами роста
Было показано, что ангиогенез индуцируется цитокинами или факторами роста, как описано в примере 4А на кроличьей модели глаза. В экспериментах, описанных в настоящей заявке, ангиогенез в препаратах кроличьей роговицы, описанный в примере 4, одинаково индуцировался факторами роста, которые были локально нанесены на кровеносные сосуды САМ, как описано в настоящей заявке.
В. Ангиогенез, индуцированный факторами роста
Было показано, что ангиогенез индуцируется цитокинами или факторами роста, как описано в примере 4А на кроличьей модели глаза. В экспериментах, описанных в настоящей заявке, ангиогенез в препаратах кроличьей роговицы, описанный в примере 4, одинаково индуцировался факторами роста, которые были локально нанесены на кровеносные сосуды САМ, как описано в настоящей заявке.
Ангиогенез индуцировали путем помещения диска фильтровальной бумаги размером 5 мм • 5 мм (фильтровальная бумага Whatman N 1), насыщенной сбалансированным солевым раствором Хэнкса (HBSS, GIBCO, Grand Island, NY) или HBSS, содержащим предварительно отобранные цитокины в предварительно определенной концентрации (то есть концентрации, необходимой для испытания их воздействия на ангиогенез), на САМ 10-дневного куриного эмбриона в область, лишенную кровеносных сосудов, после чего окна были заклеены липкой лентой. Ангиогенез прослеживали через 72 часа посредством фотомикроскопии. САМ быстро замораживали, а затем криостатные 6 мкм срезы фиксировали ацетоном и окрашивали посредством иммунофлуоресценции, как описано В примере 2В и 2С с использованием 10 мкг/мл отобранных антител против интегрина, включая антитела, направленные против αvβ5, как описано в примере 1.
Предварительные исследования Brooks et al., Science, 264 569-571 (1994), показали, что кровеносные сосуды легко наблюдаются как в bFGF-, так и в TGF-α-обработанных препаратах, но они отсутствуют в необработанных САМ. Авторы также показали, что после bFGF-индуцированного ангиогенеза, экспрессия αvβ3 усиливалась. Экспрессия интегрина β1 не отличалась от экспрессии, которая наблюдалась в необработанных САМ, причем, β1 также легко обнаруживался на стимулированных кровеносных сосудах.
Эти опубликованные данные показали, что в случае САМ человека и цыпленка, в кровеносных сосудах, участвующих в ангиогенезе, наблюдается усиленная экспрессия αvβ3. В соответствии с этим экспрессия αvβ5 на культивированных эндотелильных клетках была индуцирована различными цитокинами in vitro, как описано Janat et al., J.Cell.Physiol., 151:588 (1992); Enenstein et al., Exp. Cell Res., 203:499 (1992) и Swerlick et al., J. Invest. Perm., 99; 715 (1993).
В настоящем изобретении был определен отдельный цитокин-опосредованный механизм стимуляции ангиогенеза, который зависит от экспрессии и активации другого адгезивного интегринового рецептора αvβ5. Влияние обработки САМ цитокинами VEGF, TGF-α и EGF на ангиогенез, ассоциированный с экспрессией αvβ5, и их ингибирование антагонистами αvβ5 описано в примере 6.
С. Ангиогенез, индуцированный опухолями
Для исследования роли αvβ5 в опухоль-индуцированном ангиогенезе различные фрагменты αvβ5-негативной меланомы и карциномы человека были использованы в анализе САМ, которая была предварительно выращена и выделена из САМ 17-дневного куриного эмбриона, как описано Brooks et аl., J.Cell.Biol., 122: 1351 (1993) и в настоящей заявке.
Для исследования роли αvβ5 в опухоль-индуцированном ангиогенезе различные фрагменты αvβ5-негативной меланомы и карциномы человека были использованы в анализе САМ, которая была предварительно выращена и выделена из САМ 17-дневного куриного эмбриона, как описано Brooks et аl., J.Cell.Biol., 122: 1351 (1993) и в настоящей заявке.
Ангиогенез индуцировали в системе анализа САМ путем прямого нанесения фрагмента опухоли на САМ. САМ куриного
эмбриона получали методом, описанным выше. Вместо диска фильтровальной бумаги, 50-55 миллиграммов (мг) по массе фрагмента одной αvβ5-негативной опухоли, полученной в результате культивирования суспензии клеточной линии, описанной ниже, помещали на САМ в области, где обычно отсутствуют кровеносные сосуды.
эмбриона получали методом, описанным выше. Вместо диска фильтровальной бумаги, 50-55 миллиграммов (мг) по массе фрагмента одной αvβ5-негативной опухоли, полученной в результате культивирования суспензии клеточной линии, описанной ниже, помещали на САМ в области, где обычно отсутствуют кровеносные сосуды.
Для выращивания твердых опухолей человека на САМ куриных эмбрионов использовали клеточные линии миелоидной (HL-60 или KG-1) и лимфоидной (Т-клеточной линии Jurkat, HPB/ALL.PEER; и различные В-клеточные линии) рабдомиосаркомы, описанной Pasqualini et al., J.Cell Sci. 105:101-111 1993). Моноклеточную суспензию различных клеточных линий сначала наносили на САМ в полном объеме - 30 мкл стерильного HDSS. Окна заклеивали скотчем и эмбрионы инкубировали в течение 7 дней для продуцирования поражений человеческой опухоли. По истечении 7 дней уже у 17-дневного эмбриона вырезали опухоли из САМ и отсекали окружающую САМ ткань. Опухоли рассекали на 50-55 мг фрагменты для использования в ингиогенезе. Опухолевые фрагменты помещали на новую серию САМ 10-дневного эмбриона, как описано в примере 5А, в область, лишенную кровеносных сосудов.
Затем опухоли, выращенные in vivo на САМ куриных эмбрионов с локальным нанесением и без нанесения или с внутривенным введением αvβ5-индуцирующих цитокинов (VEGF, TGF-α или EGF), окрашивали mAb, P1F6 или Р5Н9 для выявления экспрессии αvβ5, как было описано ранее.
Затем эти САМ-опухолевые препараты последовательно обрабатывали, как описано в примерах 6С и 6D, для оценки влияния антител и пептидов, включая С-концевые фрагменты ММР-2, на опухоль-индуцированный ангиогенез.
В одном варианте клетки меланомы хомячка CS-1, полученные от Dr. Caroline Damsky от University of California at San Francisco, использовали в анализе САМ, как описано выше для образования опухолей меланомы. После переноса приблизительно 50 мг опухолевых фрагментов CS-1 на новую САМ 10-дневного куриного эмбриона в отдельные препараты вводили внутривенные инъекции либо 100 мкг, либо 300 мкг антитела P1F6, антитела LM609, или контрольного антитела CSAT (анти-β1). Дополнительный контроль представляет собой препарат, который не подвергали обработке. Результаты обсуждений приводятся ниже в примере 6D.
6. Ингибирование ангиогенеза, измеренного в анализе САМ
А. Ингибирование ангиогенеза, индуцированного фактором роста, путем внутривенного введения ингибиторов
Действие моноклональных антител, введенных внутривенно в препарат САМ, на ангиогенез, индуцированный фактором роста, оценивали на его использование в качестве in vitro-системы модели настоящего изобретения.
А. Ингибирование ангиогенеза, индуцированного фактором роста, путем внутривенного введения ингибиторов
Действие моноклональных антител, введенных внутривенно в препарат САМ, на ангиогенез, индуцированный фактором роста, оценивали на его использование в качестве in vitro-системы модели настоящего изобретения.
После активной реваскуляризации, то есть после прекращения роста сосудов, экспрессия αvβ5 снижалась до уровней, не обнаруживаемых путем иммунофлуоресцентного анализа. Эта регуляция экспрессии αvβ5 в кровеносных сосудах, подвергающихся ангиогенезу по сравнению с отсутствием экспрессии в зрелых сосудах дает уникальную возможность контролировать и ингибировать нижеописанный ангиогенез, которая предоставлена настоящим изобретением в качестве модели системы анализа на ангиогенез в САМ.
Получение САМ куриного эмбриона для внутривенных инъекций осуществляли в основном, как описано выше.
Ангиогенез сначала индуцировали на 10-дневных куриных эмбрионах с использованием фильтровальных дисков, насыщенных фактором роста. В частности, в первых анализах ангиогенез индуцировали путем обработки цитокином bFGF или VEGF, каждый из которых имел концентрацию 150 нг/мл.
Для нанесения факторов роста явно выраженные кровеносные сосуды отбирали в процессе просвечивания, и на скорлупе яйца делали отметки с указанием их положений. В скорлупе просверливали дыры, опускали туда САМ, а затем насыщенную фактором роста фильтровальную бумагу отдельно помещали на САМ, как описано выше. Окна заклеивали стерильной липкой лентой, и эмбрионы помещали в инкубатор.
Через 24 часа на боковой стороне яйца в скорлупе осторожно вырезали второе небольшое окно непосредственно над предварительно выбранными выступающими кровеносными сосудами. Внешнюю скорлупу яйца осторожно отделяли, сохраняя интактными мембраны эмбриона. Оболочку скорлупы делали прозрачной с использованием небольшой капли минерального масла (Perkin-Elmer Corp. Norwalk, CT), что позволяло легко визуализировать кровеносные сосуды. Затем забуференный фосфатом физиологический раствор (PBS), 75 мкг очищенных стерильных антител против интегрина или 75 мкг синтетических пептидов (циклический пептид RGDfV, SEQ 1D No:4 и контрольный циклический пептид RGDfV, SEQ ID No:5) в PBS инъецировали в кровеносные сосуды, присутствующие на САМ, индуцированной фактором роста. Окна заклеивали липкой лентой и эмбрионы оставляли на 72 часа для инкубирования.
Диски фильтровальной бумаги и репрезентативные ткани, окружающие САМ, фотографировали на стереомикроскопе и среднюю величину индекса ангиогенеза ± стандартная ошибка определяли для 12 САМ в соответствующих условиях (фиг. 3А-3В и фиг.4А-4В). Ангиогенез оценивали для каждого эмбриона двойным слепым методом путем анализа числа и степени разветвления кровеносных сосудов в области каждого диска. Оценки варьировались в диапазоне от 1 (низкая) до 4 (высокая), и индекс ангиогенеза определяли путем вычитания фона, соответствующего оценке 1, из всех значений.
Специфичность опосредованного антителом против интегрина ингибирования индуцированного фактором роста ангиогенеза в САМ-модели соответствовала специфичности, которая наблюдалась с использованием кроличьей модели роговицы, описанной выше. Как bFGF, так и VEGF индуцировали ангиогенез в контрольной PBS-обработанной САМ. Однако обработка αvβ5-специфическим антителом, P1F6 приводила к ингибированию VEGF-индуцированного ангиогенеза, но не оказывала ингибирующего действия на bFGF-индуцированный ангиогенез. В противоположном этому αvβ3-специфическое антитело LM609 ингибировало bFGF-индуцированный ангиогенез, но оказывало незначительное воздействие на ангиогенез в VEGF-индуцированной САМ.
Эти результаты также показаны на гистограммах на фиг.3А и 3В соответственно для bFGF- и VEGF-обработанных САМ, где представлена кривая зависимости индекса ангиогенеза от обработки либо антителом М609, либо антителом P1F6, а также показан контроль без обработки антителом. Таким образом, ингибирование индуцированного фактором роста ангиогенеза интегрин-специфическими антителами зависит от типа фактора роста.
Обработка RGD-содержащими пептидами подтверждает полученные выше результаты. В присутствии PBS обработка bFGF и VEGF приводила к индуцированию ангиогенеза у контрольных САМ. В противоположность этому циклический пептидный антагонист RGDfV (SEQ 1D No:4), направленный как против αvβ3, так и против αvβ5, ингибирует ангиогенез, индуцированный либо bFGF, либо VEGF. Циклический пептид RGDfV, (SEQ 1D No:5) не оказывает какого-либо воздействия ни на bFGF, ни на VEGF-обработанные САМ-препараты. Эти результаты также показаны на фиг.4А и 4В, где построена кривая индекса ангиогенеза для bFGF- и VEGF-стимулированных САМ, иллюстрирующая воздействие испытуемых пептидов и контрольного пептида. Таким образом, эти результаты вместе с результатами, полученными для кроличьей роговицы, указывают на то, что bFGF- и VEGF-индуцированный ангиогенез зависит от различных, но гомологичных αv-специфических интегринов, которые, однако, оба могут быть ингибированы циклическим пептидом RGDfV.
В другом анализе, осуществляемом на модели САМ, в которой индуцируют VEGF-опосредованный ангиогенез, 2 мкг пептида 85189 (SEQ 1D No:9) и аналога пептида в виде инертной соли 121974 отдельно инъецируют внутривенно, как было описано ранее. Действие пептидов оценивают по сравнению с действием контрольного пептида 69601 (SEQ 1D No:5) и необработанных (отмеченных как NT) препаратов.
Действие пептидов на VEGF-индуцированный ангиогенез измеряли путем определения числа точек ветвления кровеносных сосудов. Таким образом, ангиогенез или его отсутствие оценивали путем подсчета числа точек ветвления кровеносных сосудов, которые присутствуют в пределах фильтровальных дисков. Считается, что разветвленные кровеносные сосуды соответствуют, главным образом, новым ангиогенным разрастающимся кровеносным сосудам. Количественную оценку осуществляют двойным слепым методом с использованием, по крайней мере, двух независимых наблюдений. Результаты выражали как индекс ангиогенеза, где указанный индекс представляет собой число VEGF-стимулированных точек ветвления минус число точек ветвления нестимулированного контроля на один фильтровальный диск. Обычно в экспериментах использовали 6-10 эмбрионов на конкретный режим. Как показано на фиг.15, оба пептида 85189 и 121974 полностью ингибировали ангиогенез, на что указывало снижение измеряемых точек ветвления по сравнению с необработанным или контрольным обработанным пептидом препаратами.
Был проведен дополнительный аналогичный анализ с использованием синтетических пептидов, полученных, как описано в примере 3, для выявления тех пептидов, которые обладают специфичностью к αvβ5, но не к αvβ3-регулируемому ангиогенезу. Эти анализы также осуществляли с использованием С-концевых фрагментов ММР-2, полученных, как описано в примере 3 и 7 и с использованием органических молекул, полученных, как описано в примере 10.
Специфичность ингибирования интегрин-специфическими антителами индуцированного фактором роста ангиогенеза была, кроме того, подтверждена и подкреплена путем осуществления расширенного анализа на индуцирование ангиогенеза факторами роста с использованием фактора некроза опухоли-α (TNFα), трансформирующего фактора роста α (TGF-α) или форболового сложного эфира, 4-β-форбол-12-миристат-13-ацетата (РМА).
Вышеуказанные факторы роста (цитокины), включая bFGF и VEGF, были отдельно нанесены в концентрации 1,0 мкг/мл на 10-дневную модель САМ, как было описано ранее. РМА использовали в концентрации 20 нг/мл.
Через 24 часа после обработки фактором роста антитела, LM609 и P1F6 или ингибитор протеинкиназы С (РКС), кальфостин С отдельно наносили на САМ-модель, либо в виде разовой внутрисосудистой дозы, как описано выше, либо путем местного применения, как описано ниже в следующем примере. Для внутрисосудистых инъекций в течение следующего 3-дневного периода антитела использовали в концентрации 75 мкг на эмбрион, а кальфостин С использовали в дозе 100 нМ.
На 13-й день фильтровальные диски и ассоциированную с ними ткань САМ отсекали и анализировали на ангиогенез с использованием стереомикроскопа. Ангиогенез оценивали двойным слепым методом путем анализа числа и степени ветвления кровеносных сосудов в области дисков. Шкала оценок составляла от 1 (низкая) до 4 (высокая). Индекс ангиогенеза определяли путем вычитания из всех полученных данных фонового значения, соответствующего баллу 1 по шкале оценок. Эксперименты повторяли 2-4 раза с использованием 5-6 эмбрионов на данный режим.
Как показано соответственно на фиг.7а и 7В, антитело против αvβ3, LM609 блокировало ангиогенез в ответ на введение bFGF и TNFα, тогда как антитело против αvβ5, P1F6 оказывало незначительное ингибирующее действие. В противоположность этому, как показано на фиг.5С-5Е, антитело P1F6 эффективно ингибировало ангиогенез, индуцированный VEGF, TGF-α или РМА, тогда как антитело LM609 не оказывало такого действия.
РМА является сильным стимулятором ангиогенеза и обладает способностью активировать протеинкиназу С (РКС), принадлежащую к внутриклеточному семейству сериновых треонинкиназ. Поэтому мы также исследовали действие кальфостина С, ингибитора РКС, на ангиогенез САМ цыпленка. Кальфостин С блокировал ангиогенез, индуцированный РМА (фиг.5Е), а также VEGF и TGF-α (как показано на фиг. 5С и 5D соответственно), но имел минимальное действие на bFGF- или TNFα опосредованный ангиогенез (показанный соответственно на фиг. 5А и 5В).
Вместе эти результаты свидетельствуют о существовании двух различных механизмов ангиогенеза, где один механизм зависит от αvβ5-опосредованного сигнала, который, в основном, не зависит от РКС, как было ранее описано Brooks et al. , Science, 264:569-571 (1994), а второй механизм потенцируется αvβ5-опосредованной трансдукцией сигнала, который в высокой степени зависит от РКС-активации.
Помимо вышеописанных экспериментов для определения локализации антител P1F6 и LM609 в САМ-тканях, которые были инокулированы внутривенно LM609, фиксированные срезы блокировали 2,5% BSA в HBSS в течение 1 часа при комнатной температуре с последующим окрашиванием разведенными (1:250) меченными родамином козьими вторыми антителами против мыши. (Таgо). Затем эти срезы анализировали на микроскопе Цейсса с использованием иммунофлуоресцентного соединения.
В. Ингибирование индуцированного фактором роста ангиогенеза путем местного применения ингибитора
Для того чтобы определить, играет ли αvβ5 активную роль в ангиогенезе, фильтровальные диски, насыщенные факторами роста, описанными выше, помещали на САМ для индуцирования ангиогенеза с последующим нанесением либо P1F6, либо LM609.
Для того чтобы определить, играет ли αvβ5 активную роль в ангиогенезе, фильтровальные диски, насыщенные факторами роста, описанными выше, помещали на САМ для индуцирования ангиогенеза с последующим нанесением либо P1F6, либо LM609.
Затем диски обрабатывали 50 мл HBSS, содержащем 25 мг mAb в полном объеме 25 мкл стерильного HBSS, через 0,24 и 48 часов. Через 72 часа САМ собирали и помещали в 35 мм чашку Петри и промывали один раз 1 мл PBS. Затем нижнюю сторону фильтровальной бумаги и САМ-ткань анализировали под стереомикроскопом Olympus, причем эти исследования проводились двумя наблюдателями двойным слепым методом. Ингибирование ангиогенеза считалось значимым, если в САМ, непосредственно под диском, обнаруживалось более чем 50%-ное снижение инфильтрации кровеносных сосудов. Эти эксперименты повторяли 4 раза на одно антитело с использованием 6-7 эмбрионов на один режим.
Для оценки действия интегрин-специфических антител на уже существующие зрелые кровеносные сосуды, сформированные в результате нормального развития сосудистой ткани, смежной с участками, лишенными сосудов, фильтровальные диски, насыщенные антителами, помещали на реваскуляризованные области САМ, полученной от 10-дневных эмбрионов, которые не обрабатывали путем местного нанесения цитокина.
Анализ САМ также осуществляли с использованием синтетических пептидов настоящего изобретения для определения действия циклических и линеаризованных пептидов на индуцированный фактором роста ангиогенез. 8 мкг пептидов, полученных, как описано ранее, отдельно помещали в общий объем 25 мкл стерильного HBSS. Раствор пептида сразу наносили на САМ-препарат, а затем снова наносили через 24 и 48 часов. Через 72 часа фильтровальную бумагу и окружающие САМ ткани отсекали и наблюдали, как описано выше.
Аналогичные анализы проводили с использованием фрагментов ММР-2 и органических молекул, полученных, как описано в примерах 7 и 10.
С. Ингибирование опухоль-индуцированного ангиогенеза путем местного применения
1) Обработка моноклональными антителами
Помимо анализа на ангиогенез, описанного выше, где проводили оценку действия αvβ5-специфического антитела и пептидных антагонистов, была также исследована роль αvβ5 в опухоль-индуцированном ангиогенезе. В качестве индуктора ангиогенеза использовали негативные по αvβ5 ткани, предварительно культивированные и выделенные из САМ 17-дневного куриного эмбриона. Фрагменты получали, как описано в примере 5С.
1) Обработка моноклональными антителами
Помимо анализа на ангиогенез, описанного выше, где проводили оценку действия αvβ5-специфического антитела и пептидных антагонистов, была также исследована роль αvβ5 в опухоль-индуцированном ангиогенезе. В качестве индуктора ангиогенеза использовали негативные по αvβ5 ткани, предварительно культивированные и выделенные из САМ 17-дневного куриного эмбриона. Фрагменты получали, как описано в примере 5С.
Как описано выше, mAb отдельно локально наносили на фрагменты опухолей в концентрации 25 мкг в 25 мкл HDSS, после чего окна заклеивали липкой лентой. Затем через 24 часа и через 48 часов снова добавляли mAb таким же образом. Через 72 часа опухоли и окружающие ткани анализировали, как описано выше.
Как описано в примере 5С, опухоли сначала получали путем трансплантации клеточной линии человека, которая не экспрессирует интегрин αvβ5, в САМ 10-дневные куриные эмбрионы.
Для количественной оценки воздействия mAb на опухоль-индуцированный ангиогенез кровеносные сосуды, проросшие в опухоль в фокальной плоскости САМ, были подсчитаны под стереомикроскопом двумя наблюдателями двойным слепым методом.
Синтетические пептиды, полученные в примере 3, ММР-2-препараты, описанные в примере 7, и органические молекулы, полученные в примере 10, аналогичным образом наносили на опухоль-индуцированную ангиогенную САМ-аналитическую систему, как описано выше. Влияние пептидов, включая ММР-2-препараты, и органических молекул, описанных в настоящем изобретении, на жизнеспособность сосудов оценивали аналогичным образом.
D. Ингибирование опухоль-индуцированного ангиогенеза путем внутривенного введения
1) Обработка моноклональными антителами
Опухоль-индуцированные кровеносные сосуды, полученные, как описано выше, также обрабатывали mAb, введенными путем внутривенной инъекции. Опухоли меланомы CS-1 помещали на САМ, как описано в примере 5С, и окна заклеивали липкой лентой, а затем, через 24 часа, 100-300 мкг очищенных mAb инокулировали путем разовой внутривенной инъекции в кровеносные сосуды куриного эмбриона, как описано выше. После этого куриные эмбрионы инкубировали в течение 7 дней. Затем определяли степень ангиогенеза, как описано выше. По истечении этого периода опухоли вырезали и измеряли их массу для того, чтобы определить влияние антител на рост или суппрессию опухоли.
1) Обработка моноклональными антителами
Опухоль-индуцированные кровеносные сосуды, полученные, как описано выше, также обрабатывали mAb, введенными путем внутривенной инъекции. Опухоли меланомы CS-1 помещали на САМ, как описано в примере 5С, и окна заклеивали липкой лентой, а затем, через 24 часа, 100-300 мкг очищенных mAb инокулировали путем разовой внутривенной инъекции в кровеносные сосуды куриного эмбриона, как описано выше. После этого куриные эмбрионы инкубировали в течение 7 дней. Затем определяли степень ангиогенеза, как описано выше. По истечении этого периода опухоли вырезали и измеряли их массу для того, чтобы определить влияние антител на рост или суппрессию опухоли.
Результаты обработки опухолей CS-1 300 микрограммами αvβ5-специфического антитела P1F6 показаны на фиг.6. В этом случае при сравнении с необработанными и CSAT-обработанными опухолями масса опухоли резко снижалась до менее чем 50 мг. αvβ5-Специфическое антитело, LM609, также ингибировало рост опухоли, однако менее эффективно, чем антитело P1F6. Были получены также результаты сравнения с опухолями, обработанными 100 мкг P1F6. Например, P1F6 эффективно ингибировало αvβ5-опосредованный ангиогенез в опухолевой модели САМ-препарата, что приводило к снижению клеточной массы опухоли.
2) Обработка другими антагонистами αvβ5
Была также проведена оценка действия пептидов, ММР-2-препаратов или органических молекул на опухоль-индуцированную васкуляризацию в аналитической САМ-системе. Препарат опухолевой САМ использовали, как описано выше, за исключением того, что вместо внутривенного введения mAb в визуально обнаруживаемые кровеносные сосуды путем внутривенной инъекции отдельно вводили синтетические пептиды, содержащие препараты ММР-2, полученные, как описано в примере 7, и органические молекулы, полученные в примере 10.
Была также проведена оценка действия пептидов, ММР-2-препаратов или органических молекул на опухоль-индуцированную васкуляризацию в аналитической САМ-системе. Препарат опухолевой САМ использовали, как описано выше, за исключением того, что вместо внутривенного введения mAb в визуально обнаруживаемые кровеносные сосуды путем внутривенной инъекции отдельно вводили синтетические пептиды, содержащие препараты ММР-2, полученные, как описано в примере 7, и органические молекулы, полученные в примере 10.
В одной конкретной серии анализов проводили дополнительные анализы на регрессию опухоли с использованием αvβ5-реактивного пептида 85189 (SEQ ID No:9) по сравнению с пептидом 69601 (SEQ ID No:5), использованном в качестве контроля. Анализы осуществляли, как описано выше, за исключением того, что 100 мкг пептида внутривенно вводили в САМ через 18 часов после имплантирования различных опухолей, которые в данном случае представляют собой типы опухолей UCLAP-3, M21-L, и FgM. Еще через 48 часов опухоли вырезали и получали сырую массу.
Фиг. 16, 17 и 18 соответственно указывают на снижение массы опухолей UCLAP-3, M21-L, и FgM после внутривенной инъекции пептида 85189 и, в противоположность этому, указывают на отсутствие такого эффекта с использованием PBS или пептида 69601.
7. Идентификация αvβ5-специфических антагонистов, детектируемых с помощью анализа на ингибирование связывания с клетками и на связывание "лиганд-рецептор"
А. Ингибирование связывания с клетками
Анализ на ингибирование связывания с клетками, который является одним из способов определения интегрин-рецепторной специфичности и антагонистов настоящего изобретения, осуществляли, как описано ниже.
А. Ингибирование связывания с клетками
Анализ на ингибирование связывания с клетками, который является одним из способов определения интегрин-рецепторной специфичности и антагонистов настоящего изобретения, осуществляли, как описано ниже.
Для этого клетки меланомы хомячка CS-1, в которых отсутствует экспрессия αvβ5 и αvβ5, сначала трансфецировали плазмидой для экспрессии  субъединицы, как было описано ранее Filardo et al., J.Cell.Biol., 130: 441-450 (1995). Специфичность потенциальных αvβ5-антагонистов определяли по их способности блокировать связывание αvβ5-экспрессирующих клеток CS-1 с планшетами, сенсибилизированными VN или ламинином. В качестве примера типичного анализа лунки сначала покрывали на ночь 10 мкг/мл субстрата. После промывания и блокирования 1% термоденатурированным BSA и PBS при комнатной температуре в течение 30 минут пептид 85189 (SEQ ID No:9), взятый в интервале концентраций 0,0001 мкМ - 100 мкМ, отдельно смешивали с клетками CS-1 с нанесением этих клеток на лунки с плотностью 50000 кл./лун. После 10-15-минутного инкубирования при 37oС раствор, содержащий клетки и пептиды, удаляли. Затем, после окрашивания 1% кристаллическим фиолетовым, определяли число связанных клеток. Ассоциированный с клетками кристаллический фиолетовый элюировали путем добавления 100 микролитров (мкл) 10% уксусной кислоты. Клеточную адгезию оценивали количественно путем измерения оптической плотности элюированного кристаллического фиолетового на длине волны при 600 нм.
субъединицы, как было описано ранее Filardo et al., J.Cell.Biol., 130: 441-450 (1995). Специфичность потенциальных αvβ5-антагонистов определяли по их способности блокировать связывание αvβ5-экспрессирующих клеток CS-1 с планшетами, сенсибилизированными VN или ламинином. В качестве примера типичного анализа лунки сначала покрывали на ночь 10 мкг/мл субстрата. После промывания и блокирования 1% термоденатурированным BSA и PBS при комнатной температуре в течение 30 минут пептид 85189 (SEQ ID No:9), взятый в интервале концентраций 0,0001 мкМ - 100 мкМ, отдельно смешивали с клетками CS-1 с нанесением этих клеток на лунки с плотностью 50000 кл./лун. После 10-15-минутного инкубирования при 37oС раствор, содержащий клетки и пептиды, удаляли. Затем, после окрашивания 1% кристаллическим фиолетовым, определяли число связанных клеток. Ассоциированный с клетками кристаллический фиолетовый элюировали путем добавления 100 микролитров (мкл) 10% уксусной кислоты. Клеточную адгезию оценивали количественно путем измерения оптической плотности элюированного кристаллического фиолетового на длине волны при 600 нм.
Аналогичные анализы осуществляли с использованием гибридных белков или пар синтетических пептидов, содержащих различные области белка ММР-2. Происходящие от ММР-2 полипептиды содержат области С-конца ММР-2, обладающие активностью в реакции связывания с αvβ5, и способные ингибировать активацию ММР-2 и ассоциированные активности. Эти полипептиды получали либо в виде синтетических полипептидов, имеющих последовательность, происходящую от С-концевого домена ММР-2, как описано в примере 1, или в виде гибридных белков, включая весь С-концевой домен или часть С-концевого домена ММР-2, полученного, как описано ниже. С-концевые молекулы ММР-2 представляли специфические последовательности человека и цыпленка.
С-концевой домен ММР-2, происходящий от цыпленка, также называемый гемопексиновым доменом, непосредственно смежным с шарнирной областью, содержал аминокислотные остатки 445-637 ММР-2. Полная нуклеотидная и кодированная аминокислотная последовательность ММР-2 цыпленка описана ниже и показана на фиг. 13А и 13В; эти нуклеотидная последовательность и аминокислотная последовательности представлены как SEQ ID No:23 и SEQ ID No:24 соответственно. Нуклеотидная и аминокислотная последовательности ММР-2 человека также описаны ниже, причем аминокислотная последовательность представлена на фиг.14 и в SEQ ID No:25. С-концевой домен в ММР-2 человека, который соответствует области 445-637 ММР-2 цыпленка, начинается с остатка 439 и кончается остатком 631, что обусловлено отсутствием шести остатков в человеческой последовательности, как показано на фиг.13А и 13В. Оба С-концевых синтетических пептида, происходящих от ММР-2 человека и ММР-2 цыпленка, и используемых в целях настоящего изобретения, представлены в таблице 1. Аминокислотные последовательности синтетических пептидов являются такими же, как последовательности, генерированные рекомбинантными аналогами гибридного белка, но без GST-гибридного компонента. С-концевые гибридные белки, происходящие от ММР-2 цыпленка и ММР-2 человека, были получены, как описано ниже.
Гибридный белок ММР-2 представляет собой химерный полипептид, имеющий последовательность С-концевого домена ММР-2 или его часть, соединенную (соответствующим образом связанную посредством ковалентной пептидной связи) с белком-носителем (слитым белком), таким как глутатионсульфгидрилтрансфераза (GST).
Для амплификации различных областей ММР-2 цыпленка и ММР-2 человека были сконструированы праймерные последовательности на основе известных соответствующих кДНК-последовательностей ММР-2 цыпленка и ММР-2 человека. Полная верхняя цепь нуклеотидной последовательности кДНК непроцессированного ММР-2 цыпленка, называемого также прогелатиназой, показана на фиг.13А и 13В вместе с выведенной аминокислотной последовательностью, показанной на второй строке (Aimes et al., Biochem., 300:729-736, 1994). В третьей и четвертой строках фиг. 13 показаны соответственно выведенная аминокислотная последовательность ММР-2 человека (Collier et al., J.Biol.Chem., 263:6579-6587 (1988), и ММР-2 мыши (Reponen et al., J.Biol.Chem., 267:7856-7862 (1992). Идентичные остатки показаны точками, а отличающиеся остатки обозначены их однобуквенным кодом IUPAC. Отсутствие остатков обозначено черточкой. Нумерация аминокислотных остатков начинается с первого остатка профермента, причем остатки сигнального пептида обозначены отрицательными числами. Нуклеотидная последовательность пронумерована на фиг.13 соответствующим образом, хотя в Списке последовательностей, первый нуклеотид обозначен цифрой 1. Предполагаемый кодон инициации трансляции (ATG) обозначен тремя вперед смотрящими головками стрелок, а сигнал терминации трансляции (TGA) указан звездочкой. Аминоконцевые последовательности для куриного профермента и активного фермента обозначены ромбиками и одной головкой стрелки. Как было установлено ранее, куриные нуклеотидная и аминокислотная последовательности представлены вместе как SEQ ID No: 23, а кодированная аминокислотная последовательность представлена отдельно как SEQ ID No:24.
Матрицы для генерирования амплифицированных областей ММР-2 цыпленка представляли собой либо кДНК, кодирующую полноразмерный зрелый полипептид ММР-2 цыпленка и поставляемую от Dr.J.P.Quigley из State of New York at Stoney Brook, New York, или кДНК, генерированную с использованием стандартной техники из полной клеточной РНК-матрицы, полученной из выделенного образца ткани хориоаллантоисной мембраны цыпленка. В последнем случае кДНК получали с использованием обратной транскриптазы MuLV и "прямого" праймера, специфичного для 3'-концевых нуклеотидов 5'ATTGAATTCTTCTACAGTTCA3' (SEQ ID No: 26), 5' и 3'-концы которых были соответственно комплементарными нуклеотидам 1932-1912 опубликованной последовательности ММР-2 цыпленка. Как указано в надписях к фиг.13, положения нуклеотидов праймеров, описанных в настоящей заявке. соответствуют положениям, показанным на фиг.13 и не показанным в Списке последовательностей, где последовательность начинается с числа 1, а не отрицательным числом, как показано на фиг. Полимеразную цепную реакцию с обратной транскриптазой (RT-PCR) осуществляли в соответствии с инструкциями производителя, приложенными к набору для PCR (GeneAmp RNA PCR Kit, Perkin Elmer). Были также сконструированы праймеры, содержащие внутренний EcoRI-рестрикционный сайт.
Из любой из вышеописанных кДНК-матриц был получен ряд С-концевых областей ММР-2 цыпленка, каждый из которых имеет природный цистеиновый остаток в положении 637 в карбоксиконце, с помощью PCR с использованием 3'-праймера, указанного выше (SEQ ID No:26), в паре с одним из ряда 5'-праймеров, указанных ниже. Нижеследующие гибридные белки ММР-2, кодированные амплифицированные областями и имеющие соответствующие положения аминокислотных остатков, показаны на фиг.13А и 13В, а также представлены в SEQ ID No:24: 203-637; 2) 274-637; 3) 292-637; 4) 410-637; 5) 445-637. Для амплификации нуклеотидных областей, кодирующих вышеуказанные гибридные белки ММР-2, были сконструированы "обратные" или 5'-праймеры, которые кодируют старт-сайты полипептида 3' и создают путем PCR-встраивания, внутренний рестрикционный BamHI-сайт для направленного лигирования в экспрессирующие векторы либо в pGEX-1λT, либо в pGEX-3Х. 5'-праймеры включали нижеследующие последовательности, 5'- и 3'-концы которых соответствовали указанным 5' и 3'-положениям нуклеотидов последовательности ММР-2 цыпленка, показанной на фиг.13 (положения аминокислотных остатков старт-сайта также указаны для каждого праймера): 1) нуклеотиды 599-619, кодирующие старт-сайт 203, 5'ATGGGATCCACTGCAAATTTC 3' (SEQ ID No:27), 2) нуклеотиды 809-830, кодирующие старт-сайт 274 5'GCCGGATCCATGACCAGTGTA 3' (SEQ ID No:28); 3) нуклеотиды 863-883, кодирующие старт-сайт 292 5-GTGGGATCCCTGAAGACTATG 3' (SEQ ID No: 29); 4) нуклеотиды 1217-1237, кодирующие старт-сайт 410 5'AGGGGATCCTTAAGGGGATTC 3' (SEQ ID No:30) и 5) нуклеотиды 1325-1345, кодирующие старт-сайт 445 5-CTCGGATCCTCTGCAAGCACG 3' (SEQ ID No:31).
Указанные нуклеотидные области кДНК-матрицы были затем амплифицированы в 35 циклов (температура отжига 55oС) в соответствии с инструкциями производителя PCR-системы (Expand Hign Fidelity PCR System, Boehringer Mannhiem). Полученные PCR-продукты подвергали гель-фильтрации, гидролизовали рестриктирующими ферментами BamH1 и EcoRI и снова очищали, после чего лигировали либо в вектор pGEX-1λT, либо в вектор pGEX-3Х (Pharmacia Biotech, Uppsala, Sweden), которые перед проведением реакции лигирования были аналогичным образом гидролизованы, а также дефосфорилированы. Плазмиду выбирали исходя из нужной рамки считывания амплифицированного продукта. Штамм BSJ72 или BL21 компетентных клеток E.coli трансформировали отдельными конструкциями путем теплового шока. Полученные колонии скринировали с помощью PCR на включение плазмиды, кодирующей соответствующий гибридный белок ММР-2, а затем для проверки целостности введенной кодирующей последовательности секвенировали методом дидезокситерминации цепи. Кроме того, проверка встраивания плазмиды подтверждалась экспрессией гибридного белка GST-MMP-2 соответствующего размера.
Очистку каждого из рекомбинантных гибридных белков GST-MMP-2 осуществляли с использованием IPTG-индуцированных культур в фазе логарифмического роста, как, в основном, описано производителями системы GST Gene Fusion System (Pharmacia Biotech). Для этого выделенные бактерии подвергали лизису путем обработки ультразвуком и инкубировали с детергентом с последующим осветлением и иммобилизацией рекомбинантного белка на сефарозе 4В-глутатионе (Pharmacia Biotech). После интенсивного промывания иммобилизованные гибридные белки отдельно элюировали с аффинной матрицы 10 мМ восстановленным глутатионом в 50 мМ Трис-HCl, рН 8,0, а затем тщательно диализовали против PBS для удаления остаточного глутатиона непосредственно перед использованием.
Предварительные попытки продуцирования белков-гибридов между остатками 445 и 637 ММР-2 цыпленка, которые кодируют только один цистеиновый остаток, приводили к получению нерастворимых продуктов. Поэтому для продуцирования других растворимых ММР-2-гибридных белков, происходящих от С-концевой области, которые не включали бы эндогенной концевой цистеиновый остаток, присутствующий в предварительно описанном гибридном белке, в амплифицированные ММР-2-области были включены нуклеотидные последовательности, кодирующие цистеиновый остаток, если необходимо, в зависимости от конкретного гибридного белка. Обычно этот природный цистеиновый остаток присутствует в последовательности ММР-2 цыпленка в положении 446 и в положении 637. В последовательности человека эти положения соответствуют 440 и 631 соответственно. Поэтому были сконструированы гибридные белки, которые содержат встроенные концевые цистеиновые остатки в амино- или карбоксиконце нужных последовательностей ММР-2 цыпленка так, чтобы обеспечивалось связывание посредством дисульфидной связи с природным цистеином на другом конце, как этого требует конструкция. Синтетические фрагменты ММР-2 для белка цыпленка и человека получали способом, аналогичным описанному в примере 3.
Олигонуклеотидные праймеры были соответствующим образом сконструированы в целях амплификации С-концевых областей ММР-2 цыпленка для экспрессии растворимых гибридных белков MMP-2/GST. Амплифицированные С-концевые области ММР-2 цыпленка включали последовательности, кодирующие аминокислоты в положениях 445-518, 445-552, 516-657 и 549-637. Для гибридных белков, содержащих остаток 517, цистеиновый остаток заменяли на кодируемый природный тирозиновый остаток, что обеспечивало дисульфидное связывание с цистеиновым остатком в положении 446 или 637. Для гибридных белков, содержащих остаток 551, цистеин заменяли на кодируемый природный триптофановый остаток, что обеспечивало дисульфидное связывание с цистеиновым остатком в положении 446 или 637.
Вкратце, pGEX-3Х-плазмидную конструкцию, кодирующую гибридный белок GST/MMP-2 (410-637), полученный, как описано выше, использовали в качестве матрицы для амплификации в соответствии с инструкцией производителя PCR-набора Expand Hign Fidelity PCR Kit (Boehringer Mannhiem) с использованием серии олигонуклеотидных праймеров, которые были сконструированы на основе опубликованной последовательности ММР-2 цыпленка (показанной на фиг.13А и 13В, и в SEQ ID No:23). Один сконструированный "обратный" праймер, кодирующий старт-сайт белка ММР-2 цыпленка в положении 445, расположенном после сконструированного внутреннего сайта рестрикционной эндонуклеазы BamHI, встроенного для инсерции в вектор pGEX-3X-GST, имел нуклеотидную последовательность (5'CTCGGATCCTCTGCAAGCACG 3', SEQ ID No:32). 5'- и 3'-концы праймера соответствовали положениям 1325-1345 последовательности ММР-2 цыпленка, показанной соответственно на фиг. 13А и 13В. Другой сконструированный "обратный" праймер, кодирующий старт-сайт белка ММР-2 цыпленка в положении 516, расположенной после сконструированного внутреннего сайта рестрикционной эндонуклеазы BamHI, встроенного для инсерции в вектор pGEX-3X-GST, и кодирующий цистеиновый остаток в положении 517, имел нуклеотидную последовательность (5'GCAGGATCCGAGTGCTGGGTTTATAC 3', SEQ ID No:33). 5'- и 3'-концы праймера соответствовали положениям 1537-1562 последовательности ММР-2 цыпленка, показанной соответственно на фиг. 13. Третий сконструированный "обратный" праймер, кодирующий старт-сайт белка ММР-2 цыпленка в положении 549, расположенном после сконструированного внутреннего сайта рестрикционной эндонуклеазы EcoRI, встроенного для инсерции в вектор pGEX-1λT-GST, и кодирующий цистеиновый остаток в положении 551, имел нуклеотидную последовательность (5'GCAGAATTCAACTGTGGCAGAAACAAG 3', SEQ 1D No:34). 5' и 3'-концы праймера соответствовали положениям 1639-1665 последовательности ММР-2 цыпленка, показанной соответственно на фиг.13.
Эти "обратные" праймеры были отдельно использованы с одним из нижеуказанных "прямых" праймеров для продуцирования вышеуказанных областей из С-концевого домена ММР-2 цыпленка. Первый сконструированный прямой (антисмысловой) праймер, кодирующий стоп-сайт белка ММР-2 цыпленка в положении 518 и кодирующий цистеиновый остаток в положении 517, содержащий внутренний сайт рестрикционной эндонуклеазы EcoRI, встроенный для инсерции в вектор GST, имел нуклеотидную последовательность (5'GTAGAATTCCAGCACTCATTTCCTGC3', SEQ ID No:35). 5' и 3'-концы праймера соответствовали положениям 1562-1537, читаемым в направлении 5'-->3', были частично комплементарны соответственно положениям 1562-1537 последовательности ММР-2 цыпленка, показанной соответственно на фиг.13. Второй сконструированный "прямой" праймер, кодирующий стоп-сайт белка ММР-2 цыпленка в положении 552 и содержащий внутренний сайт рестрикционной эндонуклеазы EcoRI, встроенный для инсерции в вектор GST, имел нуклеотидную последовательность (5'TCTGAATTCTGCCACAGTTGAAGG 3', SEQ ID No:36). 5' и 3'-концы праймера, читаемые в направлении 5'-->3', были комплементарны соответственно частично положениям 1666-1643 последовательности ММР-2 цыпленка, показанной соответственно на фиг.13. Третий сконструированный прямой праймер, кодирующий стоп-сайт белка ММР-2 цыпленка в положении 637 и содержащий внутренний сайт рестрикционной эндонуклеазы EcoRI, встроенный для инсерции в вектор GST, имел нуклеотидную последовательность (5' ATTGAATTCTTCTACAGTTCA 3', SEQ 1D No: 37). 5' и 3'-концы праймера, читаемые в направлении 5'-->3', были частично комплементарны соответственно положениям 1932-1912 последовательности ММР-2 цыпленка, показанной соответственно на фиг.13.
Эти области карбоксиконцов ММР-2 цыпленка, соединенные с вышеуказанными прямым и обратным праймерами и использованные для конкретных комбинаций в целях продуцирования гибридных белков, содержащих, по крайней мере, один встроенный цистеиновый остаток, как было показано выше, были отдельно амплифицированы с использованием 30 циклов при температуре отжига 55oС в соответствии с инструкцией производителя PCR-набора Expand High Fidelity PCR System (Boehringer Mannhiem). Полученные продукты амплификации отдельно очищали, гидролизовали, если это необходимо, рестриктирующими ферментами BamH1 или EcoRI, а затем повторно очищали и лигировали в соответствующий вектор гибридного белка GST либо pGEX-3Х, либо pGEX-1λT, как указано выше, посредством рамки считывания обратного олигонуклеотидного праймера. Для лигирования амплифицированных продуктов ММР-2 векторы аналогичным способом гиролизовали, а также дефосфорилировали и проводили реакцию лигирования. Затем компетентные клетки BL21 штамма E.coli - отдельно трансформировали полученными ММР-2-содержащими векторными конструкциями путем теплового шока. Затем полученные колонии скринировали на включение соответствующей кодирующей гибридный белок плазмиды с помощью PCR и продуцировали GST-гибридный белок соответствующего размера с последующим дидезоксисеквенированием положительных клонов для подтверждения целостности введенной кодирующей последовательности. Очистку рекомбинантных GST-гибридных белков осуществляли с использованием IPTG-индуцированных культур в логарифмической фазе роста, в основном, как описано выше, для продуцирования других GSТ-ММР-2-гибридных белков.
Помимо гибридных белков "GST-MMP-2 цыпленка", описанных выше, были продуцированы два гибридных белка "GST-MMP-2 человека" для экспрессии аминокислотных областей 203-601 и 439-631 зрелого полипептида профермента ММР-2 человека. Указанные области соответствовали областям 203-637 и 443-637 ММР-2 цыпленка. Гибридные белки "GST-MMP-2 человека" продуцировали посредством PCR, как описано выше для гибридных белков "GST-MMP-2 цыпленка" с использованием матричной кДНК, которая кодирует полную открытую рамку считывания ММР-2 человека и которая была предоставлена Dr.W.G.Stetler-Stevenson at the National Cancer Institute, Bethesda, MD. 5'-праймерные последовательности, сконструированные исходя из опубликованной ранее последовательности ММР-2 человека (Collier et al. , J.Biol.Chem., 263:6579-6587 (1988), кодировали встроенный внутренний рестрикционный EcoRI-сайт для вставки амплифицированных продуктов в соответствующий экспрессионный вектор.
Один скоструированный "обратный" праймер, кодирующий старт-сайт белка ММР-2 человека в положении 203, расположенном после сконструированного внутреннего сайта рестрикционной эндонуклеазы EcoRI, встроенного для инсерции в вектор pGEX-1λT-GST, имел нуклеотидную последовательность 5'-GATGAATTCTACTGCAAGTT 3' (SEQ ID No:38), 5' и 3'-концы праймера соответствовали положениям 685-704 последовательности открытой рамки считывания ММР-2 человека. Другой "обратный" праймер, кодирующий старт-сайт белка ММР-2 человека в положении 439, расположенный после сконструированного внутреннего сайта рестрикционной эндонуклеазы EcoRI, встроенного для инсерции в вектор pGEX-1λT-GST имел нуклеотидную последовательность (5' CACTGAATTCATCTGCAAACA 3', SEQ ID No:39). 5' и 3'-концы праймера соответствовали положениям 1392-1412 последовательности открытой рамки считывания ММР-2 человека.
Каждый из вышеуказанных праймеров использовали отдельно с соответствующим прямым праймером, имеющим 5' и 3'-концы, соответственно комплементарные основаниям 1998 и 1978 последовательности ММР-2 человека, которая заканчивается задолго до открытой рамки считывания ММР-2 и осуществляет терминацию белка после аминокислотного остатка 631. Амплифицированные продукты экспрессировали гибридные белки, содержащие аминокислотные остатки 203-631 ММР-2 человека (SEQ ID No:40) и 439-631 (SEQ ID No:12).
Полученные PCR-продукты очищали, гидролизовали EcoRI и снова очищали для лигирования в плазмиду pGEX-1λT, которая перед реакцией лигирования была аналогичным образом гидролизована и дефосфорилирована. Клетки были трансформированы, как описано ранее.
Другие гибридные белки ММР-2 человека, содержащие аминокислотные остатки 410-631 (SEQ ID No: 11), 439-512 (SEQ ID No:13), 439-546 (SEQ ID No:14), 510-631 (SEQ ID No: 15) и 543-631 (SEQ ID No:16) были также получены, как описано выше с использованием способов настоящего изобретения.
В. Анализ на связывание лиганда-рецептора
αvβ5-иммунореактивные антитела и синтетические пептиды, полученные, как описано в примерах 1 и 3 соответственно, скринировали путем измерения их способности ингибировать активность связывания рецепторов αvβ5, αvβ3 и α11bβ3 в анализах на связывание очищенных лиганда и рецептора. Способ проведения таких анализов был описан Barbas et al., Proc.Natl. Acad. Sci. , USA, 90:10003-10007 (1993), Smith et al., J.Biol.Chem., 265:11008-110'13 (1990) и Pfaff et al., J.Biol.Chem., 269:20233-20238 (1994), описание этих работ вводится в настоящее описание посредством ссылки.
αvβ5-иммунореактивные антитела и синтетические пептиды, полученные, как описано в примерах 1 и 3 соответственно, скринировали путем измерения их способности ингибировать активность связывания рецепторов αvβ5, αvβ3 и α11bβ3 в анализах на связывание очищенных лиганда и рецептора. Способ проведения таких анализов был описан Barbas et al., Proc.Natl. Acad. Sci. , USA, 90:10003-10007 (1993), Smith et al., J.Biol.Chem., 265:11008-110'13 (1990) и Pfaff et al., J.Biol.Chem., 269:20233-20238 (1994), описание этих работ вводится в настоящее описание посредством ссылки.
Был описан метод идентификации антагонистов в анализе на связывание "лиганд-рецептор", в котором рецептор был иммобилизован на твердом носителе, а лиганд и антагонист были растворимыми. Был также описан анализ на связывание "лиганд-рецептор", в котором лиганд был иммобилизован на твердом носителе, а рецептор и антагонист были растворимыми.
Вкратце, выбранные очищенные интегрины отдельно иммобилизовали на лунках планшета для микротитрования Titertek при концентрации 50 нанограмм (нг) на лунку. Очистка рецепторов, проводимая в анализе на связывание "лиганд-рецептор", хорошо известна специалистам и легко осуществляется методами, известными каждому специалисту. После инкубирования в течение 18 часов при 4oС участки неспецифического связывания на планшете блокировали 10 миллиграммами/миллилитр (мг/мл) альбумина бычьей сыворотки (BSA) в забуференном трисом физиологическом растворе. Для исследования ингибирования различные концентрации выбранных антител или пептидов тестировали на их способность блокировать связывание 125I-витронектина или других меченых лигандов с интегриновыми рецепторами, αvβ5, αvβ3 и α11bβ3.
Хотя эти лиганды обнаруживают оптимальное связывание с конкретным интегрином, то есть связывание витронектина с αvβ5 и αvβ3, а фибриногена с α11bβ3, однако исследование ингибирования связывания с использованием антител или пептидов дл блокирования связывания витронектина с любым из рецепторов позволяет точно определить количество пептида в микромолях (мкМ), необходимое для полумаксимального ингибирования связывания рецептора с лигандом. Радиоактивно меченные лиганды были использованы в концентрациях 1 нМ, и связывание обнаруживали в сравнении с немечеными синтетическими пептидами. После трехчасового инкубирования свободные лиганды удаляли путем промывания, а связанные лиганды подсчитывали с помощью гамма-счетчика.
Хотя эти лиганды обнаруживают оптимальное связывание с конкретным интегрином, то есть связывание витронектина с αvβ5 и αvβ3, а фибриногена с α11bβ3, однако исследование ингибирования связывания с использованием антител или пептидов дл блокирования связывания витронектина с любым из рецепторов позволяет точно определить количество пептида в микромолях (мкМ), необходимое для полумаксимального ингибирования связывания рецептора с лигандом. Радиоактивно меченные лиганды были использованы в концентрациях 1 нМ, и связывание обнаруживали в сравнении с немечеными синтетическими пептидами. После трехчасового инкубирования свободные лиганды удаляли путем промывания, а связанные лиганды подсчитывали с помощью гамма-счетчика.
Таким образом, описанный здесь анализ на связывание лиганда с рецептором использовали для скрининга на кольцевые или линеаризованные синтетические пептиды, а также моноклональные антитела и органические молекулы, которые обладают избирательной специфичностью по отношению к конкретному интегриновому рецептору, в частности, к αvβ5, и которые использовались в качестве антагонистов рецептора витронектина (αvβ5) при осуществлении настоящего изобретения.
8. In vivo-Регрессия роста опухолевой ткани под действием антагонистов αvβ5, измеренная в анализе с использованием химерной модели "мышь-человек"
Химерную in vivo-модель "мышь-человек" генерировали путем замены участка кожи. взятой от мыши SCID, кожей неонатальной крайней плоти человека. Химерную in vivo-модель "мышь-человек" получали, в основном, как описано Yan et al. , J. Clin.Invest., 91:986-996 (1993). Для этого у мыши SCID (в возрасте 6-8 недель) хирургически удаляли 2 см2 - площадь квадрата кожи и заменяли кожей крайней плоти человека. Мышей анестезировали и на участке 5 см2 с каждой стороны латеральной области брюшины удаляли шерсть путем выбривания. Два круглых слоя имплантата 2 см2 получали путем удаления всей толщины кожи ниже фасции. Имплантируемую кожу человека полной толщины того же самого размера, полученную от крайней неонатальной плоти человека, помещали на раненую область мыши и зашивали. Имплантат покрывали Band-Aid и на кожу накладывали шов. Рану также покрывали микропористым лейкопластырем.
Химерную in vivo-модель "мышь-человек" генерировали путем замены участка кожи. взятой от мыши SCID, кожей неонатальной крайней плоти человека. Химерную in vivo-модель "мышь-человек" получали, в основном, как описано Yan et al. , J. Clin.Invest., 91:986-996 (1993). Для этого у мыши SCID (в возрасте 6-8 недель) хирургически удаляли 2 см2 - площадь квадрата кожи и заменяли кожей крайней плоти человека. Мышей анестезировали и на участке 5 см2 с каждой стороны латеральной области брюшины удаляли шерсть путем выбривания. Два круглых слоя имплантата 2 см2 получали путем удаления всей толщины кожи ниже фасции. Имплантируемую кожу человека полной толщины того же самого размера, полученную от крайней неонатальной плоти человека, помещали на раненую область мыши и зашивали. Имплантат покрывали Band-Aid и на кожу накладывали шов. Рану также покрывали микропористым лейкопластырем.
После вживления кожного имплантата человеческую крайнюю плоть инокулировали клетками меланомы. Для формирования твердых человеческих опухолей на коже человека, имплантированной мыши SCID, использовали клеточную линию меланомы человека M2IL. Моноклеточную суспензию 2•106 M2IL инъецировали внутрикожно в кожный имплантат человека. Затем за ростом измеряемых опухолей человека у мышей наблюдали в течение 2-4 недель.
После того как опухоль достигала измеряемых размеров, мышам 3 раза в неделю в течение 3 недель инъецировали внутрибрюшинно либо 250 мкг пептида (в объеме 100 мкл), имеющего SEQ ID No:9 (циклический RGD-содержащий пептид Arg-Gly-Asp-D-Phe-Asn-NMe-Val) либо контрольного пептида, цикло(Arg-βAla-Asp-D-Phe-Val). По окончании этого времени опухоль вырезали и анализировали ее массу и гистологию.
Результаты показаны на фиг.7, где приводится кривая зависимости объема в мм3 (по оси Y) от обработки пептидами (ось X). Испытуемый пептид, имеющий SEQ ID No: 9, отмеченный на графике как пептид 189, способствовал значительному снижению объема опухоли до приблизительно 25 мм3 по сравнению с контрольным пептидом (отмеченным как пептид 601), где объем опухоли превышал 300 мм3.
Таким образом, блокирование рецептора αvβ5 путем внутривенного введения пептида-антагониста αvβ5 189 приводило к регрессии меланомы в этой системе модели таким же образом, как это имело место в системах САМ-модели и кроличьей глазной модели, описанных ранее.
В других экспериментах, проводимых на опухолевых клетках меланомы M2IL в аналитической системе с использованием химерной модели "мышь-человек", ответ, продуцированный антителом LM609, сравнивали с ответом, продуцированным синтетическим пептидом 85189 (SEQ ID No:9), а также с ответом, продуцированным контрольным синтетическим пептидом 69601 (SEQ ID No:5). Этот анализ осуществляли, как описано выше. Результаты, показанные на фиг.21, продемонстрировали, что синтетический пептид 85189 способствовал снижению объема опухоли
до менее чем 25 мм3 по сравнению с контрольным пептидом, где объем опухоли превышал приблизительно 300 мм3. Антитело LM609 также способствовало значительному снижению объема опухоли до приблизительно 60 мм3.
до менее чем 25 мм3 по сравнению с контрольным пептидом, где объем опухоли превышал приблизительно 300 мм3. Антитело LM609 также способствовало значительному снижению объема опухоли до приблизительно 60 мм3.
Таким образом, блокирование рецептора αvβ5 путем внутривенного введения αvβ5-специфического антитела LM609 и пептидов приводит к регрессии карциномы в этой системе модели по сравнению с другими системами модели, описанными в настоящем изобретении.
Помимо анализа с использованием модели мышей SCID, имеющих измеряемые опухоли M2IL, в предварительном анализе была построена дозозависимая кривая для пептидов 69601 (контрольного) и 85189 (испытуемого), инъекцированных в концентрациях от 10 до 250 мкг/мл. Был определен средний объем и масса опухоли, вырезанной после обработки, и результаты этого определения показаны на фиг. 20А и 20В. Пептид 85189 оказывал эффективное ингибирующее действие на рост опухоли M2IL при всех испытуемых концентрациях по сравнению с контрольным пептидом, используемым в наиболее эффективной дозе 250 мкг/мл.
Для анализа эффективности пептида 85189 в течение всего цикла обработки была проведена оценка двух режимов обработки на той же самой модели опухоли SCID. В одном анализе обработку пептидом 85189 или 69601 начинали на 6-й день со дня 0, когда была проведена подкожная инъекция в кожу мыши 3•105 клеток опухоли M2I-L, при этом внутрибрюшинные инъекции 250 мкг/мл пептида 85189 или контрольного пептида 69601 вводили через день вплоть до дня 29. Другой анализ проводили идентичным образом за исключением того, что обработку начинали на 20-й день. По окончании анализов опухоли вырезали и определяли средний объем опухоли в мм3. На основании этих данных строили кривую зависимости этого объема ± стандартная ошибка среднего значения.
Результаты этих анализов, проиллюстрированные соответственно на фиг.21А и 21В, показали, что пептид 85189, но не пептид 69601, ингибирует рост опухоли в различные периоды времени после обработки в зависимости от конкретно выбранного режима. Таким образом, пептид 85189 является эффективным αvβ5-антагонистом ангиогенеэа, а следовательно, и роста опухоли.
Химерную модель SCID/человек, описанную выше, также использовали для оценки эффективности других антагонистов αvβ5 настоящего изобретения, а именно антител, ММР-2-препаратов, полученных, как описано ранее, и органических молекул, которые были получены, как описано в примере 10.
9. Получение мышиной модели для αvβ5-опосредованного ангиогенеза сетчатки и его ингибирование антагонистами αvβ5
Исходя из наблюдения экспресии αvβ3 и αvβ5 в реваскуляризованной ткани сетчатки, описанного в примере 2С, для исследования действия системно введенных пептидных антагонистов обоих интегринов на ангиогенез в сетчатке была использована новая мышиная модель. У новорожденной мыши сосуды сетчатки развиваются первые две недели после рождения, в течение которых поверхностная сосудистая система сетчатки образует широкую и в высокой степени разветвленную сеть сосудов, которая развивается в диске зрительного нерва и радиально распространяется к периферии, покрывая поверхность сетчатки, что наблюдается и у других млекопитающих и человека (Jiang et al., Glia, 15:1-10 (1995).
Исходя из наблюдения экспресии αvβ3 и αvβ5 в реваскуляризованной ткани сетчатки, описанного в примере 2С, для исследования действия системно введенных пептидных антагонистов обоих интегринов на ангиогенез в сетчатке была использована новая мышиная модель. У новорожденной мыши сосуды сетчатки развиваются первые две недели после рождения, в течение которых поверхностная сосудистая система сетчатки образует широкую и в высокой степени разветвленную сеть сосудов, которая развивается в диске зрительного нерва и радиально распространяется к периферии, покрывая поверхность сетчатки, что наблюдается и у других млекопитающих и человека (Jiang et al., Glia, 15:1-10 (1995).
Для этой модели новорожденной мыши инъекцировали подкожно два раза в день в течение 4 дней, начиная со дня 0, циклический пептид RGDfV (SEQ ID Nо:4) (обозначаемый также пептидом 203) или контрольный пептид RADfV (SEQ ID Nо: 5). На пятый день после рождения глазное яблоко удаляли и фиксировали в 4,0% параформальдегиде (PFA) при комнатной температуре.
Для количественной оценки ангиогенеза в мышиной сетчатке измеряли расстояние от диска зрительного нерва до более удаленной точки одного сосуда, выбранного в каждом из шести равных срезов, в направлении, приблизительно соответствующем расположению часовой стрелки, указывающей на 12 ч на циферблате часов. Величину этого расстояния вычисляли и усредняли с аналогичными данными, полученными от всего массива информации. Для измерения общего объема кровеносных сосудов сетчатки весь образец сканировали в 2,0 мкм оптических срезах и хранили отдельно друг от друга. Затем для пороговых и расчетных кубических пикселов в каждом срезе использовали функцию "подсева" (введение дополнительной информации) в программном обеспечении Lasersharp, Bio-Rad's. Макроинформацию записывали для суммирования объема всех срезов и определяли величину для всех сосудистых структур.
При прямом измерении роста сосудов по фотографиям в двух измерениях системно введенный пептидный антагонист 203 ингибировал ангиогенез в сетчатке по сравнению с контрольным пептидом на 44% (N=9, р<0,0000001, парный t-критерий). Между необработанной новорожденной мышью и 5-дневной мышью, которой был введен пептид 203, не наблюдалось какого-либо статистического различия, что свидетельствало о том, что этот пептид эффективно ингибирует ангиогенез. Кроме того, не было установлено какого-либо статистического различия между необработанной 5-дневной мышью и мышью того же возраста, обработанной контрольным пептидом. Таким образом, у RGDfV-обработанной новорожденной мыши по сравнению с ее необработанным аналогом наблюдалось 100% ингибирование ангиогенеза в сетчатке.
С использованием более количественных анализов, проводимых для определения роста опухоли в трехмерном пространстве, у животных, обработанных пептидом 203, по сравнению с контролем наблюдалось 78%-ное снижение объема кровеносных сосудов сетчатки. Средний объем сосудов у животных, обработанных пептидом 203, на 5-ый день после рождения составлял 3,6•106 мкм3, а у контрольного необработанного животного этот объем составлял 15,7•106 мкм3. Объем, занимаемый кровеносными сосудами сетчатки у необработанных новорожденных мышей, не отличался от объема для 5-дневных животных, обработанных пептидов 203.
Результаты, полученные выше, показали, что антагонисты специфически блокируют образование новых кровеносных сосудов, не оказывая при этом влияния на уже сформированные сосуды. Эти результаты показали, что патология реваскулярного заболевания сетчатки отличается от патологии, наблюдаемой при субретинальном реваскулярном заболевании и что антагонисты αvβ5 являются эффективными для лечения пациентов с заболеванием, сопровождающимся слепотой, ассоциированной с ангиогенезом.
Аналогичные анализы были осуществлены с использованием ММР-2 и органических имитаторов антагонистов αvβ5, полученных, как описано в примере 7 и примере 10.
10. Получение органических молекул-антагонистов αvβ5
Синтез органических антагонистов интегрина αvβ5, а именно Соединений 7, 9, 10, 12, 14, 15, 16, 17 и 18, описан ниже, и также показан в представленных фигурах. Полученные органические молекулы, а именно органические миметики настоящего изобретения, были затем использованы в способах ингибирования αvβ5-опосредованного ангиогенеза.
Синтез органических антагонистов интегрина αvβ5, а именно Соединений 7, 9, 10, 12, 14, 15, 16, 17 и 18, описан ниже, и также показан в представленных фигурах. Полученные органические молекулы, а именно органические миметики настоящего изобретения, были затем использованы в способах ингибирования αvβ5-опосредованного ангиогенеза.
Для каждого синтеза, описанного ниже, оптические вращения измеряли на УФ-спектрофотометре Perkin-Elmer 241, а видимые спектры регистрировали на спектрометре DU-70. 1H- и 13С-ЯМР-спектры регистрировали при 400 и 500 МГц на спектрометре Druker AMX-400 и ДМХ-500. Масс-спектры высокого разрешения (ВРМС) регистрировали на масс-спектрометре VG-ZAB-ZSE с ионизацией путем бомбардировки быстрыми атомами (FAB). Колоночную хроматографию проводили на силикагеле 70-230 меш. Препаративную ТСХ осуществляли на силикагеле Merck Art. 5744 (0,5 мм). Температуры плавления регистрировали на приборе Thomas Hoover.
А. Соединение 1: т-Вос-L-тирозина бензиловый эфир, проиллюстрированный на фиг.8.

Compound 1 = соединение 1; benzyl = бензил.
К раствору N-(трет-бутоксикарбонил)-L-тирозина (т-Вос-L-тирозина) (1,0 эквивалент; Aldrich) в 0,10 М (М) метиленхлориде добавляли дициклогексилкарбодиимид (DCC) (1,5 эквивалента) при 25oС и перемешивали в течение одного часа. Затем добавляли 1,5 эквивалента бензилового спирта и смесь перемешивали еще 12 часов при 25oС, после этого реакционную смесь разбавляли этилацетатом (0,10 М) и дважды (2Х) промывали водой, один раз (1X) насыщенным солевым раствором, а затем сушили сульфатом магния. Затем растворитель удаляли в вакууме и неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле. Соединение 1, бензиловый эфир т-Boc-L-тирозина может быть закуплен у фирмы Sigma.
В. Соединение 2: (S)-3-(4-(4-Бромбутилокси)фенил-2-N-трет-бутилоксикарбонилпропионовой кислоты бензиловый эфир, представленный на фиг.8 стадия i

Compound 2 = соединение 2; benzyl = бензил.
Compound 2 = соединение 2; benzyl = бензил.
Смесь бензилового эфира T-Boc-L-тирозина (2 г, 5,38 ммоль; синтезированного как описано выше), 1,4-дибромбутана (1,9 мл, 16,2 ммоль; Aldrich), карбоната калия (5 г), и смолы 18-краун-6 (0,1 г; Aldrich) нагревали при 80oС в течение 12 часов. После охлаждения осадок отфильтровывали, и реакционную смесь упаривали досуха в вакууме. Затем неочищенный продукт очищали путем кристаллизации с использованием 100% гексана, в результате чего получали 2,5 г (92%) Соединения 2.
С. Соединение 3: (S)-3-(4-(4-Азидобутилокси)фенил-2-N-трет-бутилоксикарбонил-пропионовой кислоты бензиловой эфир, представленный на фиг.8, стадия ii

Compound 3 = соединение 3; Benzyl = бензил.
Compound 3 = соединение 3; Benzyl = бензил.
Соединение 2 (2,5 г, 4,9 ммоль) перемешивали с азидом натрия (1,6 г, 25 ммоль) в диметилформамиде (ДМФ) (20 мл) при температуре 25oС в течение 12 часов. Затем растворитель выпаривали и остаток обрабатывали водой (приблизительно 10 мл) и дважды экстрагировали этилацетатом. Органические слои объединяли, сушили сульфатом магния и упаривали с получением 2,0 грамм (90%) Соединения 3 в виде бесцветного сиропа (FAB-MC: 469 (М+Н+).
D. Соединение 4: (S)-3-(4-(4-Азидобутилокси)фенил-2-аминопропионовой кислоты бензиловый эфир, проиллюстрированный на фиг.8, стадия iii

Compound 3 = соединение 3; Benzyl = бензил.
Compound 3 = соединение 3; Benzyl = бензил.
Соединение 3 (2,0 г, 4,4 ммоль) растворяли в трифторуксусной кислоте (TFA; 2 мл) и перемешивали в течение 3 часов при комнатной температуре. После упаривания в вакууме получали 1,6 г (количественный выход) Соединения 4 в виде бесцветого сиропа, которое было использовано в последующей стадии без дополнительной очистки. FAB-MC: 369 (М+H+).
Е. Соединение 5:(S)-3-(4-(4-Азидобутилокси)фенил-2-бутилсульфонамидо-пропионовой кислоты бензиловый эфир, проиллюстрированный на фиг.8, стадия iv

Compound 5 = соединение 5; Benzyl = бензил.
Compound 5 = соединение 5; Benzyl = бензил.
Смесь Соединения 4 (1,6 г, 4,3 ммоль), хлорида бутансульфоновой кислоты (0,84 мл, 6,6 ммоль) и триэтиламина (1,5 эквивалента) перемешивали в метиленхлориде (20 мл) в течение 12 часов при комнатной температуре. После этого реакционную смесь упаривали, а остаток растворяли в этилацетате и промывали разбавленной соляной кислотой, водным раствором бикарбоната натрия и водой. После упаривания досуха неочищенный продукт очищали с помощью флеш-хроматографии (силикагель, толуол/этилацетат, 15:1), в результате чего получали 1,4 г (67%) Соединения 5 в виде аморфного твердого вещества.
F. Соединение 6: (S)-3-(4-(4-Аминобутилокси)фенил-2-бутилсульфонамидопропионовая кислота, проиллюстрированная на фиг.8, стадия v

Compound 6 = соединение 6.
Compound 6 = соединение 6.
Соединение 5 (1,3 г, 2,6 ммоль) растворяли в 20 мл смеси этилацетат/метанол/вода (5/3/1) и 0,2 мл трифторуксусной кислоты (TFA), а затем гидрировали под давлением водорода в 1 атмосферу в аппарате Парра (Parr Shaker) при 25oС в присутствии 100 мг палладия (10% на угле). Через три часа катализатор отфильтровывали и растворитель выпаривали с получением Соединения 6 в виде маслянистого остатка. После лиофилизации из воды было получено 1,0 г (количественный выход) Соединения 6 в виде белого порошка. FAB-MC: 373 (М+Н+).
G. Соединение 7: (S)-3-(4-(4-Гуанидинобутилокси)фенил-2-бутилсульфонамидо-пропионовая кислота, проиллюстрированная на фиг.8, стадия vi

Compound 7 = соединение 7
Соединение 6 (200 мг; 0,5 ммоль), нитрат 3,5-диметилпиразол-1-карбоксамидина (DPFN) (170 мг, 0,8 ммоль; Aldrich Chemical Company) и триэтиламин (0,15 мл, 1,0 ммоль) в диметилформамиде (DMF; 5 мл) нагревали при 60oС в течение 12 часов. После охлаждения растворитель выпаривали в вакууме и остаток очищали с помощью ВЭЖХ (Lichrocart RP-18, градиент ацетонитрил/вода + 0,3% TFA = 99:1-->1:99), в результате чего после лиофилизации было получено 50 мг (25%) Соединения 7 в виде белого аморфного порошка. FAB-MC: 415 (М+Н+), т.пл. 70oС.
Compound 7 = соединение 7
Соединение 6 (200 мг; 0,5 ммоль), нитрат 3,5-диметилпиразол-1-карбоксамидина (DPFN) (170 мг, 0,8 ммоль; Aldrich Chemical Company) и триэтиламин (0,15 мл, 1,0 ммоль) в диметилформамиде (DMF; 5 мл) нагревали при 60oС в течение 12 часов. После охлаждения растворитель выпаривали в вакууме и остаток очищали с помощью ВЭЖХ (Lichrocart RP-18, градиент ацетонитрил/вода + 0,3% TFA = 99:1-->1:99), в результате чего после лиофилизации было получено 50 мг (25%) Соединения 7 в виде белого аморфного порошка. FAB-MC: 415 (М+Н+), т.пл. 70oС.
Н. Соединение 8: (S)-3-(4-(4-Аминобутилокси)фенил-2-N-третбутилоксикарбонил-пропионовая кислота, проиллюстрированная на фиг.9, стадия iii

Compound 8 = соединение 8; Benzyl = бензил.
Compound 8 = соединение 8; Benzyl = бензил.
Соединение 3 (0,5 г, 1,07 ммоль) растворяли в 10 мл смеси этилацетат/метанол/вода (5/3/1) и 0,1 трифторуксусной кислоты (TFA) и гидрировали под давлением водорода (1 атмосфера; аппарат Parr Shaker) при 25oС в присутствии 30 мг палладия (10% на угле). Через 3 часа, катализатор отфильтровывали, а растворитель выпаривали, в результате чего получали Соединение 8 в виде маслянистого остатка. После лиофилизации из воды получали 370 мг (количественный выход) Соединения 8 в виде белого порошка. FAB-MC: 353 (М+Н+).
I. Соединение 9: (S)-3-(4-(4-Гуанидинобутилокси)фенил-2-N-третбутилоксикарбонилпропионовая кислота, проиллюстрированная на фиг.9, стадия iv

Compound 9 = соединение 9; Benzyl = бензил
Соединение 8 (200 мг, 0,5 ммоль), нитрат 3,5-диметилпиразол-1-карбоксамидина (DPEN) (170 мг, 0,8 ммоль; Aldrich Chemical Company) и триэтиламин (0,15 мл, 1,0 ммоль) в диметилформамиде (DMF; 5 мл) нагревали при 60oС в течение 12 часов. После охлаждения растворитель выпаривали в вакууме, а остаток очищали с помощью ВЭЖХ (Lichrocart RP-18; в градиенте ацетонитрил/вода + 0,3% TFA = 99:1 --> 1:99) с получением после лиофилизации 160 мг (90%) Соединения 9 в виде белого аморфного порошка. FAB-MC: 395 (М+Н+).
Compound 9 = соединение 9; Benzyl = бензил
Соединение 8 (200 мг, 0,5 ммоль), нитрат 3,5-диметилпиразол-1-карбоксамидина (DPEN) (170 мг, 0,8 ммоль; Aldrich Chemical Company) и триэтиламин (0,15 мл, 1,0 ммоль) в диметилформамиде (DMF; 5 мл) нагревали при 60oС в течение 12 часов. После охлаждения растворитель выпаривали в вакууме, а остаток очищали с помощью ВЭЖХ (Lichrocart RP-18; в градиенте ацетонитрил/вода + 0,3% TFA = 99:1 --> 1:99) с получением после лиофилизации 160 мг (90%) Соединения 9 в виде белого аморфного порошка. FAB-MC: 395 (М+Н+).
J. Соединение 10:(R)-3-(4-(4-гуанидинобутилокси)фенил-2-бутилсульфонамидопропионовая кислота, проиллюстрированная на фиг.10, стадия i-vi

Compound 10 = соединение 10;
Идентичная последовательность реакции синтеза Соединения 7 была использована для получения 205 мг аналога D-тирозина 10 в виде белого аморфного вещества (FAB-MC: 415 (М+Н+) с использованием промежуточных соединений 100-600 с образованием Соединения 10.
Compound 10 = соединение 10;
Идентичная последовательность реакции синтеза Соединения 7 была использована для получения 205 мг аналога D-тирозина 10 в виде белого аморфного вещества (FAB-MC: 415 (М+Н+) с использованием промежуточных соединений 100-600 с образованием Соединения 10.
1) Соединение 100: т-Вос-D-тирозина бензиловый эфир, проиллюстрированный на фиг.10

Compound 100 = соединение 100; Benzyl = бензил
К раствору N-(трет-бутоксикарбонил)-D-тирозин-(т-Вос-L-тирозина)(1,0 эквивалент; Aldrich) в 0,10 М метиленхлориде добавляли дициклогексилкарбодиимид (DCC) (1,5 эквивалента) при 25oС и перемешивали в течение 1 часа. Затем добавляли 1,5 эквивалента бензилового спирта и смесь перемешивали еще 12 часов при 25oС. Реакционную смесь разбавляли этилацетатом (0,10 М), а затем промывали водой (2Х), насыщенным солевым раствором (1X) и сушили сульфатом магния. Растворитель удаляли в вакууме, и неочищеный продукт очищали с помощью колоночной хроматографии на силикагеле.
Compound 100 = соединение 100; Benzyl = бензил
К раствору N-(трет-бутоксикарбонил)-D-тирозин-(т-Вос-L-тирозина)(1,0 эквивалент; Aldrich) в 0,10 М метиленхлориде добавляли дициклогексилкарбодиимид (DCC) (1,5 эквивалента) при 25oС и перемешивали в течение 1 часа. Затем добавляли 1,5 эквивалента бензилового спирта и смесь перемешивали еще 12 часов при 25oС. Реакционную смесь разбавляли этилацетатом (0,10 М), а затем промывали водой (2Х), насыщенным солевым раствором (1X) и сушили сульфатом магния. Растворитель удаляли в вакууме, и неочищеный продукт очищали с помощью колоночной хроматографии на силикагеле.
2) Соединение 200: (R)-3-(4-(4-Бромбутилокси)фенил-2-N-трет-бутилоксикарбонилпропионовой кислоты бензиловый эфир, проиллюстрированный на фиг.10, стадия i

Compound 200 = соединение 200; Benzyl =бензил.
Compound 200 = соединение 200; Benzyl =бензил.
Смесь бензилового эфира т-Boc-D-тирозина (2 г, 5,38 ммоль; синтезированного, как описано выше), 1,4-дибромбутана (1,9 мл, 16,2 ммоль; Aldrich), карбоната калия (5 г) и 18-краун-6 (0,1 г, Aldrich) нагревали при температуре 80oС в течение 12 часов. После охлаждения осадок отфильтровывали и реакционную смесь упаривали досуха в вакууме. Затем неочищенный продукт очищали путем кристаллизации с использованием 100% гексана, в результате чего получали 2,5 г (92%) Соединения 200.
3) Соединение 300: (R)-3-(4-(4-Азидобутилокси)фенил-2-N-трет-бутилоксикарбонилпропионовой кислоты бензиловый эфир, проиллюстрированный на фиг. 10, стадия ii

Compound 300 = соединение 300; Benzyl = бензил
Соединение 200 (2,5 г, 4,9 ммоль) перемешивали с азидом натрия (1,6 г, 25 ммоль) в диметилформамиде (DMF) (20 мл) при 25oС в течение 12 часов. Затем растворитель выпаривали, а остаток обрабатывали водой (приблизительно 10 мл и дважды экстрагировали этилацетатом. Органические слои объединяли, сушили сульфатом магния и упаривали, в результате чего получали 2,0 г (90%) Соединения 300 в виде бесцветного сиропа (FAB-MC: 469 (М+Н+)
4) Соединение 400: (R)-3-(4-(4-Азидобутилокси)фенил-2-аминопропионовой кислоты бензиловый эфир, проиллюстрированный на фиг.10, стадия iii

Compound 400 = соединение 400; Benzyl = бензил
Соединение 300 (2,0 г, 4,4 ммоль) растворяли в трифторуксусной кислоте (TFA; 2 мл) и перемешивали в течение 3 часов при комнатной температуре. После упаривания в вакууме получали 1,6 г (количественный выход) Соединения 400 в виде бесцветного сиропа, который использовали в последующей стадии без дополнительной очистки. FAB-MC: 369 (М+Н+).
Compound 300 = соединение 300; Benzyl = бензил
Соединение 200 (2,5 г, 4,9 ммоль) перемешивали с азидом натрия (1,6 г, 25 ммоль) в диметилформамиде (DMF) (20 мл) при 25oС в течение 12 часов. Затем растворитель выпаривали, а остаток обрабатывали водой (приблизительно 10 мл и дважды экстрагировали этилацетатом. Органические слои объединяли, сушили сульфатом магния и упаривали, в результате чего получали 2,0 г (90%) Соединения 300 в виде бесцветного сиропа (FAB-MC: 469 (М+Н+)
4) Соединение 400: (R)-3-(4-(4-Азидобутилокси)фенил-2-аминопропионовой кислоты бензиловый эфир, проиллюстрированный на фиг.10, стадия iii
Compound 400 = соединение 400; Benzyl = бензил
Соединение 300 (2,0 г, 4,4 ммоль) растворяли в трифторуксусной кислоте (TFA; 2 мл) и перемешивали в течение 3 часов при комнатной температуре. После упаривания в вакууме получали 1,6 г (количественный выход) Соединения 400 в виде бесцветного сиропа, который использовали в последующей стадии без дополнительной очистки. FAB-MC: 369 (М+Н+).
5) Соединение 500: (R)-3-(4-(4-Азидобутилокси)фенил-2-бутилсульфонамидопропионовой кислоты бензиловый эфир, проиллюстрированный на фиг.10, стадия iv

Compound 500 = соединение 500; Benzyl = бензил
Смесь Соединения 400 (1,6 г, 4,3 ммоль), хлорида бутансульфоновой кислоты (0,84 мл, 6,6 ммоль) и триэтиламина (1,5 эквивалента) перемешивали в метиленхлориде (20 мл) в течение 12 часов при комнатной температуре. Реакционную смесь упаривали, а остаток растворяли в этилацетате и промывали разбавленной соляной кислотой, водным раствором бикарбоната натрия и водой. После упаривания досуха неочищенный продукт очищали с помощью флеш-хроматограции (силикагель; толуол/этилацетат, 15:1) и получали 1,4 г (67%) Соединения 500 в виде аморфного твердого вещества.
Compound 500 = соединение 500; Benzyl = бензил
Смесь Соединения 400 (1,6 г, 4,3 ммоль), хлорида бутансульфоновой кислоты (0,84 мл, 6,6 ммоль) и триэтиламина (1,5 эквивалента) перемешивали в метиленхлориде (20 мл) в течение 12 часов при комнатной температуре. Реакционную смесь упаривали, а остаток растворяли в этилацетате и промывали разбавленной соляной кислотой, водным раствором бикарбоната натрия и водой. После упаривания досуха неочищенный продукт очищали с помощью флеш-хроматограции (силикагель; толуол/этилацетат, 15:1) и получали 1,4 г (67%) Соединения 500 в виде аморфного твердого вещества.
6) Соединение 600: (R)-3-(4-(4-Аминобутилокси)фенил-2-бутилсульфонамидопропионовая кислота, проиллюстрированная на фиг.10, стадия v

Compound 600 = соединение 600.
Compound 600 = соединение 600.
Соединение 500 (1,3 г, 2,6 ммоль) растворяли в 20 мл смеси этилацетат/метанол/вода (5: 3: 1) и 0,2 мл трифторуксусной кислоты (TFA), а затем гидрировали под давлением водорода (1 атмосфера; аппарат Parr Shaker) при 25oС в присутствии 100 мг палладия (10% на угле). Через 3 часа катализатор отфильтровывали, а растворитель выпаривали, в результате чего получали Соединение 600 в виде маслянистого остатка. После лиофилизации из воды получали 1,0 г (количественный выход) Соединения 600 в виде белого порошка. FAB-MC: 373 (М+Н+).
7) Соединение 10:(R)-3-(4-(4-Гуанидинобутилокси)фенил-2-бутилсульфонамидопропионовая кислота, проиллюстрированная на фиг.10, стадия vi
Соединение 600 (200 мг, 0,5 ммоль), нитрат 3,5-диметилпиразол-1-карбоксамидина (DPFN) (170 мг, 0,8 ммоль; Aldrich Chemical Company) и триэтиламин (0,15 мл, 1,0 ммоль) в диметилформамиде (DMF; 5 мл) нагревали при 60oС в течение 12 часов. После охлаждения растворитель выпаривали в вакууме и остаток очищали с помощью ВЭЖХ (Lichrocart RP-18, в градиенте ацетонитрил/вода + 0,3% TFA, от 99:1 до 1:99), а затем после лиофилизации получали 50 мг (25%) Соединения 10 в виде белого аморфного порошка. FAB-MC: 415 (М+Н+), т.пл. 70oС.
Соединение 600 (200 мг, 0,5 ммоль), нитрат 3,5-диметилпиразол-1-карбоксамидина (DPFN) (170 мг, 0,8 ммоль; Aldrich Chemical Company) и триэтиламин (0,15 мл, 1,0 ммоль) в диметилформамиде (DMF; 5 мл) нагревали при 60oС в течение 12 часов. После охлаждения растворитель выпаривали в вакууме и остаток очищали с помощью ВЭЖХ (Lichrocart RP-18, в градиенте ацетонитрил/вода + 0,3% TFA, от 99:1 до 1:99), а затем после лиофилизации получали 50 мг (25%) Соединения 10 в виде белого аморфного порошка. FAB-MC: 415 (М+Н+), т.пл. 70oС.
К) Соединение 11: (S)-3-(4-(4-Азидобутилокси)фенил-2-(10-камфарсульфонамидо)-пропионовой кислоты бензиловый эфир, проиллюстрированный на фиг.3.

Compound 11 = соединение 11;
Смесь Соединения 4 (1,0 г, 2,7 ммоль), хлорида 10-камфарсульфоновой кислоты (6,6 ммоль; Aldrich Chemical Company) и триэтиламина (1,5 эквивалента) перемешивали в метиленхлориде (20 мл) в течение 12 часов при комнатной температуре. Затем реакционную смесь упаривали, а остаток растворяли в этилацетате и промывали разбавленной НСl, водным раствором бикарбоната натрия и водой. После упаривания досуха неочищенный продукт очищали с помощью флэш-хроматографии (силикагель; толуол/этилацетат = 15/1) и получали 1,4 г (67%) соединения 11 в виде аморфного твердого вещества.
L. Соединение 12: (S)-3-(4-(4-Гуанидинобутилокси)фенил-2-(10-камфарсульфонамидо)-пропионовая кислота, проиллюстрированная на фиг.3, стадии i-ii

Compound 12 = соединение 12.
Compound 12 = соединение 12.
Соединение 12 получали после реакции гидрирования и гуанилирования. Соединение 11 в соответствии с нижеследующими условиями.
Стадия i: Соединение 11 (1,3 г, 2,6 ммоль) растворяли в 20 мл смеси этилацетат/метанол/вода (5/3/1) и 0,2 мл трифторуксусной кислоты (TFA), а затем гидрировали под давлением водорода (1 атмосфера; аппарат Parr Shaker) при 25oС в присутствии 100 мг палладия (10% на угле). Через 3 часа катализатор отфильтровывали, а растворитель выпаривали, в результате чего получали промежуточный амин в виде маслянистого остатка. После лиофилизации из воды получали 1,0 г (количественный выход) промежуточного амина в виде белого порошка, который использовали в последующей стадии:
Стадия ii: Только что полученное промежуточное аминовое соединение (200 мг, 0,5 ммоль), нитрат 3,5-диметилпиразол-1-карбоксамидина (DPFN) (170 мг, 0,8 ммоль; Aldrich Chemical Company) и триэтиламин (0,15 мл, 1,0 ммоль) в диметилформамиде (DMF; 5 мл), нагревали при 60oС в течение 12 часов. После охлаждения растворитель выпаривали в вакууме, а остаток очищали с помощью ВЭЖХ (Lichrocart PR-18, градиент ацетонитрил/вода + 0,3% TFA, 99:1
-->1:99) и после лиофилизации получали соединение 12 в виде белого аморфного порошка. FAB-МС: 509,6 (М+Н+).
Стадия ii: Только что полученное промежуточное аминовое соединение (200 мг, 0,5 ммоль), нитрат 3,5-диметилпиразол-1-карбоксамидина (DPFN) (170 мг, 0,8 ммоль; Aldrich Chemical Company) и триэтиламин (0,15 мл, 1,0 ммоль) в диметилформамиде (DMF; 5 мл), нагревали при 60oС в течение 12 часов. После охлаждения растворитель выпаривали в вакууме, а остаток очищали с помощью ВЭЖХ (Lichrocart PR-18, градиент ацетонитрил/вода + 0,3% TFA, 99:1
-->1:99) и после лиофилизации получали соединение 12 в виде белого аморфного порошка. FAB-МС: 509,6 (М+Н+).
М. Соединение 13: (S)-3-(4-(5-Бромпентилокси)фенил-2-N-трет-бутилоксикарбонилпропионовой кислоты бензиловый эфир, проиллюстрированный на фиг.11.

Compound 13 = соединение 13; Benzyl = бензил
Смесь бензилового эфира т-Вос-L-тирозина (4,5 г, 12,1 ммоль; Соединение 1, синтезированное, как описано выше), 1,5-дибромпентана (5 мл, 36,7 ммоль; Aldrich), карбоната калия (10 г) и 18-краун-6 (0,25 г, Aldrich) нагревали при 80oС в течение 12 часов. После охлаждения осадок отфильтровывали и реакционную смесь упаривали досуха в вакууме. Затем неочищенный продукт очищали путем кристаллизации с использованием 100% гексана, в результате чего получали 5,35 г (85%) Соединения 13.
N. Соединение 14: (S)-3-(4-(5-Гуанидинопентилокси)фенил-2-бутилсульфонамидопропионовая кислота, проиллюстрированная на фиг.11, стадии i-v

Compound 14 = соединение 14
5-Стадийную последовательность реакций бром-азидного обмена, Вос-отщепления, сульфирования хлоридом бутансульфоновой кислоты, гидрирования и гуанилирования DPFN проводили аналогично вышеуказанным методам с использованием промежуточных Соединений 1-6 для получения Соединения 7, или методам с использованием Соединений 100-600 для получения Соединения 10, как описано выше.
Compound 14 = соединение 14
5-Стадийную последовательность реакций бром-азидного обмена, Вос-отщепления, сульфирования хлоридом бутансульфоновой кислоты, гидрирования и гуанилирования DPFN проводили аналогично вышеуказанным методам с использованием промежуточных Соединений 1-6 для получения Соединения 7, или методам с использованием Соединений 100-600 для получения Соединения 10, как описано выше.
Таким образом было получено Соединение 14 в виде белого порошка. FAB-MC: 429 (М+Н+).
О. Соединение 15: 3-(4-Амидинофенил)-5-(4-(2-карбокси-2-аминоэтил)фенокси)метил-2-оксазолидинона дигидрохлорид, показанный на фиг.12.
1) Синтез исходного продукта 2-N-ВОС-амино-3-(4-гидроксифенил)пропионата для Соединения 15

Compound 15 = соединение 15
Исходный продукт, 2-М-ВОС-амино-3-(4-гидроксифенил)пропионат получали путем этерификации (D- или L-) N-(третбутоксикарбонил)-L(D)-тирозина (т-Boc-L(D)-тирозина) (1,0 эквивалент; Sigma) в 0,10 М метаноле и разбавленной 1% НС1. Реакционную смесь перемешивали в течение 12 часов при 25oС, а затем нейтрализовали карбонатом калия, разбавляли этилацетатом (0,10 М), после чего промывали водой (2Х), солевым раствором (1X), и сушили сульфатом магния. Растворитель удаляли в вакууме и неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле с получением 2-N-ВОС-амино-3-(4-гидроксифенил)пропионата.
Compound 15 = соединение 15
Исходный продукт, 2-М-ВОС-амино-3-(4-гидроксифенил)пропионат получали путем этерификации (D- или L-) N-(третбутоксикарбонил)-L(D)-тирозина (т-Boc-L(D)-тирозина) (1,0 эквивалент; Sigma) в 0,10 М метаноле и разбавленной 1% НС1. Реакционную смесь перемешивали в течение 12 часов при 25oС, а затем нейтрализовали карбонатом калия, разбавляли этилацетатом (0,10 М), после чего промывали водой (2Х), солевым раствором (1X), и сушили сульфатом магния. Растворитель удаляли в вакууме и неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле с получением 2-N-ВОС-амино-3-(4-гидроксифенил)пропионата.
2) Синтез исходного продукта 3-п-N-ВОС-амидино-фенил-5-метансульфонилоксиметил-2-оксазолидинона для Соединения 15; 3-стадийная процедура описана ниже.
п-Амино-бензонитрил (1,0 эквивалент; Aldrich) в метиленхлориде (0,10 М) перемешивали с 2,3-эпоксипропанолом (1,0 эквивалент; Aldrich) в течение 12 часов при 25oС. Затем растворитель удаляли в вакууме и неочищенный 4-(2,3-дигидроксипропиламино)бензонитрил использовали в последующей стадии, представленной ниже.
4-(2,3-Дигидроксипропиламино)бензонитрил (1,0 эквивалент, полученный, как описано выше) в диметилформамиде (0,10 М) при 25oС перемешивали с диэтилкарбонатом (1,1 эквивалента, Aldrich) при температуре 110oС в течение 6 часов. Затем реакционную смесь разбавляли этилацетатом (0,10 М), после чего промывали водой (2Х) и насыщенным солевым раствором (1X) и сушили сульфатом магния. Растворитель удаляли в вакууме и неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле, в результате чего получали 3-(4-цианофенил)-5-гидроксиметил-2-оксазолидин, который использовали в последующей стадии, описанной ниже.
3-(4-Цианофенил)-5-гидроксиметил-2-оксазолидин (1,0 эквивалент, полученный, как описано выше) в метиленхлориде (0,10 М) при 25oС перемешивали с 1,1 эквивалента бисульфида, 1,1 эквивалента метилиодида, 1,1 эквивалента ацетата аммония. Реакционную смесь перемешивали в течение 6 часов, а затем разбавляли этилацетатом (0,10 М), промывали водой (2Х), насыщенным солевым раствором (1X) и сушили сульфатом магния. После этого растворитель удаляли в вакууме и неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле, в результате чего получали амидин, который использовали в последующей стадии, описанной ниже.
1,0 эквивалент амидина, синтезированного, как описано выше, защищали 1,1 эквивалента BOC-ON (2-(ВОС-оксиимино)-2-фенилацетонитрил; Aldrich) в метиленхлориде (0,10 М) при температуре 25oС и перемешивали в течение 6 часов. После этого реакционную смесь разбавляли этилацетатом (0,10 М), а затем промывали водой (2Х) и насыщенным солевым раствором (1X) и, наконец, сушили сульфатом магния. Затем растворитель удаляли в вакууме и неочищенный продукт подвергали реакции этерификации в 0,10 М метиленхлориде и 1,1 эквивалента метансульфонилхлорида. Реакционную смесь перемешивали при 0oС в течение 6 часов, а затем гасили водой (5 эквивалентов), разбавляли этилацетатом (0,10 М), после чего промывали водой (2Х), насыщенным солевым раствором (1X) и сушили сульфатом магния. Растворитель удаляли в вакууме и неочищенный продукт очищали с помощью колоночной хроматографими на силикагеле, в результате чего получали 3-пара-N-ВОС-амидинофенил-5-метан-сульфонилоксиметил-2-оксазолидинон.
3) Реакция взаимодействия промежуточных соединений 2-N-ВОС-амино-3-(4-гидроксифенил)пропионата с 3-пара-N-ВОС-амидинофенил-5-метансульфонилоксиметил-2-оксазолидиноном с образованием защищенной формы Соединения 15, а именно 3-(4-ВОС-амидинофенил)-5-(4-(2-метокси-карбонил-2N-ВОС-аминоэтил)фениоксилметил-2-оксазолидинона
Смесь 1,9 г 2-N-ВОС-амино-3-(4-гидроксифенил)пропионата (полученного, как описано выше), 20 мл диметилформамида (DMF) и NaH (1,0 эквивалент) перемешивали в течение 30 минут при комнатной температуре. После завершения перемешивания добавляли 1,8 г 3-п-N-ВОС-амидинофенил-5-метансульфонилоксиметил-2-оксазолидинона (полученного, как описано выше) в 10 мл диметилформамида (DMF) и перемешивание продолжали в течение 15 минут при комнатной температуре. Затем реакционную смесь разбавляли этилацетатом (0,10 М), после чего промывали водой (2Х) и насыщенным солевым раствором (1X) и сушили сульфатом магния. Растворитель удаляли в вакууме и неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле, в результате чего получали защищенную форму Соединения 15, а именно 3-(4-ВОС-амидинофенил)-5-(4-(2-метоксикарбонил-2N-ВОС-аминоэтил)фениоксилметил-2-оксазолидинон, который был использован в последующей стадии.
Смесь 1,9 г 2-N-ВОС-амино-3-(4-гидроксифенил)пропионата (полученного, как описано выше), 20 мл диметилформамида (DMF) и NaH (1,0 эквивалент) перемешивали в течение 30 минут при комнатной температуре. После завершения перемешивания добавляли 1,8 г 3-п-N-ВОС-амидинофенил-5-метансульфонилоксиметил-2-оксазолидинона (полученного, как описано выше) в 10 мл диметилформамида (DMF) и перемешивание продолжали в течение 15 минут при комнатной температуре. Затем реакционную смесь разбавляли этилацетатом (0,10 М), после чего промывали водой (2Х) и насыщенным солевым раствором (1X) и сушили сульфатом магния. Растворитель удаляли в вакууме и неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле, в результате чего получали защищенную форму Соединения 15, а именно 3-(4-ВОС-амидинофенил)-5-(4-(2-метоксикарбонил-2N-ВОС-аминоэтил)фениоксилметил-2-оксазолидинон, который был использован в последующей стадии.
4) Реакция снятия защиты с защищенной формы Соединения 15 с образованием Соединения 15, а именно дигидрохлорида 3-(4-амидинофенил)-5-(4-(2-карбокси-2-аминоэтил)фенокси) метил-2-оксазолидинона, показанного на фиг.12.
Защищенную форму Соединения 15, 3-(4-ВОС-амидинофенил)-5-(4-(2-метоксикарбонил-2N-ВОС-аминоэтил)фениоксилметил-2-оксазолидинон (1,0 эквивалент; синтезированный, как описано выше) обрабатывали 4 мл 2н. NaOH в течение 4 часов при комнатной температуре. К полученной смеси по каплям добавляли 40 мл раствора 2н. НСl в диоксане при температуре от 0 до 25oС в течение 3 часов. Затем реакционную смесь гасили путем добавления бикарбоната натрия (5 эквивалентов), после чего разбавляли этилацетатом (0,10 М), промывали водой (2Х), насыщенным солевым раствором (1X) и сушили сульфатом магния. Растворитель удаляли в вакууме и неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле, в результате чего получали Соединение 15: 3-(4-амидинофенил)-5-(4-(2-карбокси-2-аминоэтил)фенокси)метил-2-оксазолидинона дигидрохлорид; т.пл. 165oС (с разл.).
Р. Соединение 16: 3-(4-Амидинофенил)-5-(4-(2-карбокси-2-N-бутилсульфониламиноэтил)фенокси) метил-2-оксазолидинон, проиллюстрированный на фиг.12.
1) Синтез исходного продукта, а именно 2-N-бутилсульфониламино-3-(4-гидроксифенил)пропионата для Соединения 16

Compound 16 = соединение 16
Исходный продукт, а именно 2-N-бутилсульфониламино-3-(4-гидроксифенил)пропионат был получен путем реакции этерификации ((D или L)-тирозина) (1,0 эквивалент; Sigma) в 0,10 М метаноле и разбавленной 1% НСl. Реакционную смесь перемешивали в течение 12 часов при 25oС, а затем нейтрализовали карбонатом калия, разбавляли этилацетатом (0,10 М), после чего промывали водой (2Х), насыщенным солевым раствором (1X) и сушили сульфатом магния. После удаления растворителя в вакууме неочищенный продукт использовали в последующей стадии.
Compound 16 = соединение 16
Исходный продукт, а именно 2-N-бутилсульфониламино-3-(4-гидроксифенил)пропионат был получен путем реакции этерификации ((D или L)-тирозина) (1,0 эквивалент; Sigma) в 0,10 М метаноле и разбавленной 1% НСl. Реакционную смесь перемешивали в течение 12 часов при 25oС, а затем нейтрализовали карбонатом калия, разбавляли этилацетатом (0,10 М), после чего промывали водой (2Х), насыщенным солевым раствором (1X) и сушили сульфатом магния. После удаления растворителя в вакууме неочищенный продукт использовали в последующей стадии.
Смесь вышеуказанного соединения (4,3 ммоль), хлорида бутансульфоновой кислоты (6,6 ммоль) и триэтиламина (1,5 эквивалента) перемешивали в метиленхлориде (20 мл) в течение 12 часов при комнатной температуре. Затем реакционную смесь упаривали, а остаток растворяли в этилацетате, промывали разбавленной НСl, водным раствором бикарбоната натрия и водой. После упаривания досуха неочищенный продукт очищали с помощью Флэш-хроматографии (силикагель; толуол/этилацетат - 15:1), в результате чего получали целевое соединение.
2) Синтез исходного продукта, 3-пара-N-ВОС-амидинофенил-5-метансульфонилоксиметил-2-оксазолидинона для Соединения 16: 3-стадийная процедура описана ниже.
п-Амино-бензонитрил (1,0 эквивалент; Aldrich) в метиленхлориде (0,10 М) перемешивали с 2,3-эпоксипропанолом (1,0 эквивалент; Aldrich) в течение 12 часов при 25oС. После удаления растворителя в вакууме был получен неочищенный 4-(2,3-дигидроксипропиламино)бензонитрил, который использовали в последующей стадии, описанной ниже.
4-(2,3-Дигидроксипропиламино)бензонитрил (1,0 эквивалент, полученный, как описано выше) в диметилформамиде (0,10 М) при 25oС перемешивали с диэтилкарбонатом (1,1 эквивалент; Aldrich) и трет-бутилатом калия (1,1 эквивалента; Aldrich) при температуре 110oС в течение 6 часов. После этого реакционную смесь разбавляли этилацетатом (0,10 М) и промывали водой (2Х), насыщенным солевым раствором (1X), а затем сушили сульфатом магния. После удаления растворителя в вакууме неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле, в результате чего получали 3-(4-цианофенил)-5-гидроксиметил-2-оксазолидин, который использовали в последующей стадии.
3-(4-Цианофенил)-5-гидроксиметил-2-оксазолидин (1,0 эквивалент, полученный, как описано выше) в метиленхлориде (0,10 М) при 25oС перемешивали с 1,1 эквивалента бисульфида, 1,1 эквивалента метилиодида, и 1,1 эквивалента ацетата аммония. Реакционную смесь перемешивали в течение 6 часов, а затем разбавляли этилацетатом (0,10 М), промывали водой (2Х) и насыщенным солевым раствором (1X), после чего сушили сульфатом магния. Затем растворитель удаляли в вакууме и неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле, в результате чего получали амидин, который использовали в последующей стадии, описанной ниже.
1,0 эквивалент амидина, синтезированного, как описано выше, защищали 1,1 эквивалента BOC-ON (2-(ВОС-оксиимино)-2-фенилацетонитрил; Aldrich) в метиленхлориде (0,10 М) при температуре 25oС, и перемешивали в течение 6 часов. Затем реакционную смесь разбавляли этилацетатом (0,10 М), после чего промывали водой (2Х), насыщенным солевым раствором (1X) и сушили сульфатом магния. После удаления растворителя в вакууме неочищенный продукт этерифицировали в 0,10 М метиленхлориде и 1,1 эквиваленте метансульфонилхлорида. Реакционную смесь перемешивали при 0oС в течение 6 часов, а затем гасили путем добавления воды (5 эквивалентов), после чего разбавляли этилацетатом (0,10 М), промывали водой (2Х) и насыщенным солевым раствором (1X) и сушили сульфатом магния. Растворитель удаляли в вакууме и неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле, в результате чего получали 3-пара-N-ВОС-амидинофенил-5-метансульфонилоксиметил-2-оксазолидинон.
3) Реакция взаимодействия промежуточных соединений 2-N-бутилсульфониламино-3-(4-гидроксифенил)пропионата с 3-пара-N-ВОС-амидино-фенил-5-метансульфонилоксиметил-2-оксазолидиноном с образованием защищенной формы Соединения 16, а именно 3-(4-ВОС-амидинофенил)-5-(4-(2-метоксикарбонил-2-N-бутилсульфониламиноэтил)фениоксилметил-2-оксазолидинона
Смесь 1,9 г 2-N-бутилсульфониламино-3-(4-гидроксифенил)пропионата (полученного, как описано выше), 20 мл диметилформамида (DMF) и гидрида натрия (NaH) (1,0 эквивалент) перемешивали в течение 30 минут при комнатной температуре. После перемешивания добавляли 1,8 г 3-N-ВОС-амидинофенил-5-метансульфонилоксиметил-2-оксазолидинона (полученного, как описано выше) в 10 мл диметилформамида (DMF), и эту смесь перемешивали еще 15 минут при комнатной температуре. Затем реакционную смесь разбавляли этилацетатом (0,10 М), после чего промывали водой (2Х) и насыщенным солевым раствором (1X) и сушили сульфатом магния. После удаления растворителя в вакууме неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле, в результате чего получали защищенную форму Соединения 16, 3-(4-ВОС-амидинофенил)-5-(4-(2-метоксикарбонил-2-N-бутилульфониламиноэтил)фениоксилметил-2-оксазолидинон, который был использован в последующей стадии, описанной ниже.
Смесь 1,9 г 2-N-бутилсульфониламино-3-(4-гидроксифенил)пропионата (полученного, как описано выше), 20 мл диметилформамида (DMF) и гидрида натрия (NaH) (1,0 эквивалент) перемешивали в течение 30 минут при комнатной температуре. После перемешивания добавляли 1,8 г 3-N-ВОС-амидинофенил-5-метансульфонилоксиметил-2-оксазолидинона (полученного, как описано выше) в 10 мл диметилформамида (DMF), и эту смесь перемешивали еще 15 минут при комнатной температуре. Затем реакционную смесь разбавляли этилацетатом (0,10 М), после чего промывали водой (2Х) и насыщенным солевым раствором (1X) и сушили сульфатом магния. После удаления растворителя в вакууме неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле, в результате чего получали защищенную форму Соединения 16, 3-(4-ВОС-амидинофенил)-5-(4-(2-метоксикарбонил-2-N-бутилульфониламиноэтил)фениоксилметил-2-оксазолидинон, который был использован в последующей стадии, описанной ниже.
4) Реакция снятия защиты с защищенной формы Соединения 16 с образованием Соединения 16: 3-(4-амидинофенил)-5-(4-(2-карбокси-2-N-бутилсульфониламиноэтил)фенокси)метил-2-оксазолидинона, представленного на фиг.12.
Защищенную форму Соединения 16, 3-(4-ВОС-амидинофенил)-5-(4-(2-метоксикарбонил-2-N-бутилсульфониламиноэтил)-фениоксилметил-2-оксазолидинон (1,0 эквивалент, синтезированный, как описано выше) обрабатывали 4 мл 2н. NaOH в течение 4 часов при комнатной температуре. К полученной смеси в течение 3 часов по каплям добавляли 40 мл раствора 2н. НСl в диоксане при температуре от 0oС до 25oС. Затем реакционную смесь гасили бикарбонатом натрия (5 эквивалентов), а затем разбавляли этилацетатом (0,10 М), промывали водой (2Х), солевым раствором (1X), и сушили сульфатом магния. После удаления растворителя в вакууме неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле, в результате чего получали Соединение 16: 3-(4-амидинофенил)-5-(4-(2-карбокси-2-N-бутилсульфониламиноэтил)фенокси)метил-2-оксазолидинон, т.пл. 236-237oС.
Q. Соединение 17: 3-(4-амидинофенил)-5-(4-(2-карбокси-2-N-пропилсульфониламиноэтил)фенокси)метил-2-оксазолидинон, проиллюстрированный на фиг.12
1) Синтез исходного соединения 2-N-пропилсульфониламино-3-(4-гидрокси-фенил)пропионата для Соединения 17:

Compound 17 = соединение 17
Исходный материал 2-пропилсульфониламино-3-(4-гидроксифенил)пропионат получали путем этерификации ((D или L)-тирозина) (1,0 эквивалент) (Sigma) в 0,10 М метаноле и разбавленной 1% НСl. Реакционную смесь перемешивали при 25oС в течение 12 часов, а затем нейтрализовали карбонатом калия, разбавляли этилацетатом (0,10 М), промывали водой (2Х), солевым раствором (1X) и сушили сульфатом магния. После удаления растворителя в вакууме неочищенный продукт использовали в последующей стадии.
1) Синтез исходного соединения 2-N-пропилсульфониламино-3-(4-гидрокси-фенил)пропионата для Соединения 17:
Compound 17 = соединение 17
Исходный материал 2-пропилсульфониламино-3-(4-гидроксифенил)пропионат получали путем этерификации ((D или L)-тирозина) (1,0 эквивалент) (Sigma) в 0,10 М метаноле и разбавленной 1% НСl. Реакционную смесь перемешивали при 25oС в течение 12 часов, а затем нейтрализовали карбонатом калия, разбавляли этилацетатом (0,10 М), промывали водой (2Х), солевым раствором (1X) и сушили сульфатом магния. После удаления растворителя в вакууме неочищенный продукт использовали в последующей стадии.
Смесь вышеуказанного соединения (4,3 ммоль), хлорида пропилсульфоновой кислоты (6,6 ммоль; Aldrich) и триэтиламина (1,5 эквивалента) перемешивали в метиленхлориде (20 мл) в течение 12 часов при комнатной температуре. После упаривания реакционной смеси остаток растворяли в этилацетате, промывали разбавленной НСl, водным раствором бикарбоната натрия и водой. После упаривания досуха неочищенный продукт очищали с помощью флэш-хроматографии (силикагель, толуол/этилацетат, 15;1), и получали целевое соединение.
2) Синтез исходного материала 3-п-N-ВОС-амидинофенил-5-метансульфонилоксиметил-2-оксазолидинона для Соединения 17: 3-стадийная процедура представлена ниже.
п-Амино-бензонитрил (1,0 эквивалент, Aldrich) в метиленхлориде (0,10 М) перемешивали с 2,3-эпоксипропанолом (1,0 эквивалент; Aldrich) в течение 12 часов при 25oС. После удаления растворителя в вакууме неочищенный 4-(2,3-дигидроксипропиламино)бензонитрил использовали в стадии, описанной ниже.
4-(2,3-Дигидроксипропиламино)бензонитрил (1,0 эквивалент, полученный, как описано выше) в диметилформамиде (0,10 М) при 25oС перемешивали с диэтилкарбонатом (1,1 эквивалента; Aldrich) и трет-бутилатом калия (1,1 эквивалента; Aldrich) при температуре 110oС в течение 6 часов. Затем реакционную смесь разбавляли этилацетатом (0,10 М), промывали водой (2Х), солевым раствором (1X) и сушили сульфатом магния. После удаления растворителя в вакууме неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле и получали 3-(4-цианофенил)-5-гидроксиметил-2-оксазолидин, который использовали в последующей стадии.
3-(4-Цианофенил)-5-гидроксиметил-2-оксазолидин (1,0 эквивалент, полученный, как описано выше) в метиленхлориде (0,10 М) при 25oС перемешивали с 1,1 эквивалента бисульфида, 1,1 эквивалента метилиодида, и 1,1 эквивалента ацетата аммония. Реакционную смесь перемешивали в течение 6 часов, а затем разбавляли этилацетатом (0,10 М), промывали водой (2Х), солевым раствором (1X) и сушили сульфатом магния. После удаления растворителя в вакууме неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле и получали амидин, который использовали в последующей стадии.
1,0 эквивалент амидина, синтезированного, как описано выше, защищали 1,1 эквивалента BOC-ON (2-(ВОС-оксиимино)-2-фенилацетонитрил; Aldrich) в метиленхлориде (0,10 М) при 25oС и перемешивали в течение 6 часов. Полученную реакционную смесь разбавляли этилацетатом (0,10 М), промывали водой (2Х), солевым раствором (1X) и сушили сульфатом магния. После удаления растворителя в вакууме неочищенный продукт подвергали реакции этерификации в 0,10 М метиленхлориде и 1,1 эквивалента метансульфонилхлорида. Реакционную смесь перемешивали при 0oС в течение 6 часов, а затем гасили водой (5 эквивалентов), и разбавляли этилацетатом (0,10 М), после чего промывали водой (2Х), солевым раствором (1X) и сушили сульфатом магния. После удаления растворителя в вакууме неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле, в результате чего получали 3-п-N-ВОС-амидинофенил-5-метансульфонилокси-метил-2-оксазолидинон.
3) Реакция взаимодействия промежуточного 2-N-пропилсульфониламино-3-(4-гидроксифенил)пропионата с 3-п-N-BOC-амидинофенил-5-метансульфонилоксиметил-2-оксазолидиноном с образованием защищенной формы Соединения 17, а именно 3-(4-ВОС-амидинофенил)-5-(4-(2-метоксикарбонил-2-N-пропилсульфониламиноэтил)-фениоксилметил-2-оксазолидинона
Смесь 1,9 г 2-N-пропилсульфониламино-3-(4-гидроксифенил) пропионата (полученного, как описано выше), 20 мл диметилформамида (DMF) и NaH (1,0 эквивалент) перемешивали в течение 30 минут при комнатной температуре. После перемешивания добавляли 1,8 г 3-п-N-ВОС-амидинофенил-5-метансульфонилоксиметил-2-оксазолидинона (полученного, как описано выше) в 10 мл диметилформамида (DMF) и смесь снова перемешивали в течение 15 минут при комнатной температуре. Реакционную смесь разбавляли этилацетатом (0,10 М), промывали водой (2Х), солевым раствором (1X) и сушили сульфатом магния. После удаления растворителя в вакууме неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле, в результате чего получали защищенную форму Соединения 17, 3-(4-ВОС-амидинофенил)-5-(4-(2-метоксикарбонил-2-N-пропилсульфониламиноэтил)-фениоксилметил-2-оксазолидинон, который использовали в последующей стадии.
Смесь 1,9 г 2-N-пропилсульфониламино-3-(4-гидроксифенил) пропионата (полученного, как описано выше), 20 мл диметилформамида (DMF) и NaH (1,0 эквивалент) перемешивали в течение 30 минут при комнатной температуре. После перемешивания добавляли 1,8 г 3-п-N-ВОС-амидинофенил-5-метансульфонилоксиметил-2-оксазолидинона (полученного, как описано выше) в 10 мл диметилформамида (DMF) и смесь снова перемешивали в течение 15 минут при комнатной температуре. Реакционную смесь разбавляли этилацетатом (0,10 М), промывали водой (2Х), солевым раствором (1X) и сушили сульфатом магния. После удаления растворителя в вакууме неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле, в результате чего получали защищенную форму Соединения 17, 3-(4-ВОС-амидинофенил)-5-(4-(2-метоксикарбонил-2-N-пропилсульфониламиноэтил)-фениоксилметил-2-оксазолидинон, который использовали в последующей стадии.
4) Реакция деблокирования защищенной формы Соединения 17 с образованием Соединения 17: 3-(4-амидинофенил)-5-(4-(2-карбокси-2-N-пропилсульфониламиноэтил)фенокси)метил-2-оксазолидинона, фиг.12
Защищенную форму Соединения 17,3-(4-ВОС-амидинофенил)-5-(4-(2-метоксикарбонил-2-N-пропилсульфониламиноэтил)-фениоксилметил-2-оксазолидинона (1,0 эквивалент, синтезированного, как описано выше) обрабатывали 4 мл 2н. NaOH в течение 4 часов при комнатной температуре. К полученной смеси по каплям в течение 3 часов добавляли 40 мл раствора 2н. НСl в диоксане при температуре от 0 до 25oС. Реакционную смесь гасили бикарбонатом натрия (5 эквивалентов), а затем разбавляли этилацетатом (0,10 М), промывали водой (2Х), солевым раствором (1X) и сушили сульфатом магния. После удаления растворителя в вакууме неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле, и получали Соединение 17: 3-(4-амидинофенил)-5-(4-(2-карбокси-2-N-пропилсульфониламиноэтил)-фенокси)метил-2-оксазолидинон; т.пл. 200oС (с разл.).
Защищенную форму Соединения 17,3-(4-ВОС-амидинофенил)-5-(4-(2-метоксикарбонил-2-N-пропилсульфониламиноэтил)-фениоксилметил-2-оксазолидинона (1,0 эквивалент, синтезированного, как описано выше) обрабатывали 4 мл 2н. NaOH в течение 4 часов при комнатной температуре. К полученной смеси по каплям в течение 3 часов добавляли 40 мл раствора 2н. НСl в диоксане при температуре от 0 до 25oС. Реакционную смесь гасили бикарбонатом натрия (5 эквивалентов), а затем разбавляли этилацетатом (0,10 М), промывали водой (2Х), солевым раствором (1X) и сушили сульфатом магния. После удаления растворителя в вакууме неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле, и получали Соединение 17: 3-(4-амидинофенил)-5-(4-(2-карбокси-2-N-пропилсульфониламиноэтил)-фенокси)метил-2-оксазолидинон; т.пл. 200oС (с разл.).
R. Соединение 18: 3-(4-амидинофенил)-5-(4-(2-карбокси-2-N-этилсульфониламиноэтил)фенокси)метил-2-оксазолидинон, представленный на фиг.12
1) Синтез исходного продукта 2-N-этилсульфониламино-3-(4-гидрокси-фенил)пропионата для Соединения 18

Compound 18 = соединение 18
Исходный продукт 2-N-этилсульфониламино-3-(4-гидроксифенил)пропионат получали путем этерификации ((D или L) тирозина) (1,0 эквивалент; Sigma) в 0,10 М метаноле и разбавленной 1% НСl. Реакционную смесь перемешивали при 25oС в течение 12 часов, а затем нейтрализовали карбонатом калия, разбавляли этилацетатом (0,10 М), промывали водой (2Х), солевым раствором (1X) и сушили сульфатом магния. После удаления растворителя в вакууме неочищенный продукт использовали в последующей стадии.
1) Синтез исходного продукта 2-N-этилсульфониламино-3-(4-гидрокси-фенил)пропионата для Соединения 18
Compound 18 = соединение 18
Исходный продукт 2-N-этилсульфониламино-3-(4-гидроксифенил)пропионат получали путем этерификации ((D или L) тирозина) (1,0 эквивалент; Sigma) в 0,10 М метаноле и разбавленной 1% НСl. Реакционную смесь перемешивали при 25oС в течение 12 часов, а затем нейтрализовали карбонатом калия, разбавляли этилацетатом (0,10 М), промывали водой (2Х), солевым раствором (1X) и сушили сульфатом магния. После удаления растворителя в вакууме неочищенный продукт использовали в последующей стадии.
Смесь вышеуказанного соединения (4,3 ммоль), хлорида этилсульфоновой кислоты (6,6 ммоль: Aldrich) и триэтиламина (1,5 эквивалента) перемешивали в метиленхлориде (20 мл) в течение 12 часов при комнатной температуре. Затем реакционную смесь упаривали, а остаток растворяли в этилацетате, промывали разбавленной НСl, водным раствором бикарбоната натрия и водой. После упаривания досуха неочищенный продукт очищали с помощью флэш-хроматографии (силикагель; элюент: толуол/этилацетат, 15:1), в результате чего было получено целевое соединение.
2) Синтез исходного продукта 3-п-N-ВОС-амидинофенил-5-метансульфонилоксиметил-2-оксазолидинона для Соединения 18: 3-стадийная процедура описана ниже.
п-Амино-бензонитрил (1,0 эквивалент; Aldrich) в метиленхлориде (0,10 М) перемешивали с 2,3-эпоксипропанолом (1,0 эквивалент; Aldrich) в течение 12 часов при 25oС. Растворитель удаляли в вакууме и неочищенный 4-(2,3-дигидроксипропиламино)-бензонитрил использовали в последующей стадии.
4-(2,3-Дигидроксипропиламино)бензонитрил (1,0 эквивалент, полученный, как описано выше) в диметилформамиде (0,10 М) при 25oС перемешивали с диэтилкарбонатом (1,1 эквивалента; Aldrich) и трет-бутилатом калия (1,1 эквивалента; Aldrich) в течение 6 часов при 110oС. Реакционную смесь разбавляли этилацетатом (0,10 М), промывали водой (2Х), солевым раствором (1X) и сушили сульфатом магния. Растворитель удаляли в вакууме и неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле, в результате чего получали 3-(4-цианофенил)-5-гидроксиметил-2-оксазолидин, который использовали в последующей стадии.
3-(4-Цианофенил)-5-гидрокси-метил-2-оксазолидин (1,0 эквивалент, полученный, как описано выше) в метиленхлориде (0,10 М) при 25oС перемешивали с 1,1 эквивалента бисульфида, 1,1 эквивалента метилиодида, и 1,1 эквивалента ацетата аммония. Реакционную смесь перемешивали в течение 6 часов, разбавляли этилацетатом (0,10 М), а затем промывали водой (2Х), солевым раствором (1X) и сушили сульфатом магния. После удаления растворителя в вакууме неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле и получали амидин, который использовали в последующей стадии.
1,0 эквивалент амидина, синтезированного, как описано выше, защищали 1,1 эквивалента BOC-ON-(2-(ВОС-оксиимино)-2-фенилацетонитрил; Aldricn) в метиленхлориде (0,10 М) при 25oС и перемешивали в течение 6 часов. Затем реакционную смесь разбавляли этилацетатом (0,10 М), промывали водой (2Х), солевым раствором (1X) и сушили сульфатом магния. Растворитель удаляли в вакууме и неочищенный продукт подвергали реакции этерификации в 0,10 М метиленхлориде и 1,1 эквивалента метансульфонилхлориде. Реакционную смесь перемешивали в течение 6 часов при 0oС, а затем гасили водой (5 эквивалентов), разбавляли этилацетатом (0,10 М), промывали водой (2Х), солевым раствором (1X) и сушили сульфатом магния. Затем растворитель удаляли в вакууме и неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле, в результате чего получали 3-п-N-ВОС-амидинофенил-5-метансульфонилоксиметил-2-оксазолидинон.
3) Реакция взаимодействия промежуточного 2-N-этилсульфониламино-3-(4-гидроксифенил)пропионата с 3-п-N-BOC-амидинофенил-5-метансульфонилоксиметил-2-оксазолидиноном с образованием защищенной формы Соединения 18, а именно 3-(4-ВОС-амидинофенил)-5-(4-2-метоксикарбонил-2-N-этилсульфониламиноэтил)-феноксиоксилметил-2-оксазолидинона
Смесь 1,9 г 2-N-этилсульфониламино-3-(4-гидрокси-фенил)пропионата (синтезированного, как описано выше), 20 мл диметилформамида (DMF) и гидрида натрия (1,0 эквивалент) перемешивали в течение 30 минут при комнатной температуре. После перемешивания добавляли 1,8 г 3-п-N-ВОС-амидинофенил-5-метансульфонилоксиметил-2-оксазолидинона (полученного, как описано выше) в 10 мл диметилформамида (DMF) и смесь снова перемешивали в течение 15 минут при комнатной температуре. Реакционную смесь разбавляли этилацетатом (0,10 М), промывали водой (2Х), солевым раствором (1X) и сушили сульфатом магния. После удаления растворителя в вакууме неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле, в результате чего получали защищенную форму Соединения 18, а именно 3-(4-ВОС-амидинофенил)-5-(4-(2-метоксикарбонил-2-N-этилсульфониламиноэтил)фениоксилметил-2-оксазолидинон, который использовали в последующей стадии.
Смесь 1,9 г 2-N-этилсульфониламино-3-(4-гидрокси-фенил)пропионата (синтезированного, как описано выше), 20 мл диметилформамида (DMF) и гидрида натрия (1,0 эквивалент) перемешивали в течение 30 минут при комнатной температуре. После перемешивания добавляли 1,8 г 3-п-N-ВОС-амидинофенил-5-метансульфонилоксиметил-2-оксазолидинона (полученного, как описано выше) в 10 мл диметилформамида (DMF) и смесь снова перемешивали в течение 15 минут при комнатной температуре. Реакционную смесь разбавляли этилацетатом (0,10 М), промывали водой (2Х), солевым раствором (1X) и сушили сульфатом магния. После удаления растворителя в вакууме неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле, в результате чего получали защищенную форму Соединения 18, а именно 3-(4-ВОС-амидинофенил)-5-(4-(2-метоксикарбонил-2-N-этилсульфониламиноэтил)фениоксилметил-2-оксазолидинон, который использовали в последующей стадии.
4) Реакция деблокирования защищенной формы Соединения 18 с образованием Соединения 18: 3-(4-амидинофенил)-5-(4-(2-карбокси-2-N-этилсульфониламиноэтил)фенокси)метил-2-оксазолидинона, представленного на фиг.12.
Защищенную форму Соединения 18, а именно 3-(4-(ВОС-амидинофенил)-5-(4-(2-метоксикарбонил-2-N-этилсульфониламиноэтил)фениоксилметил-2-оксазолидинон (1,0 эквивалент, синтезированный, как описано выше) обрабатывали 4 миллилитрами 2н. NaOH в течение 4 часов при комнатной температуре. К полученной смеси в течение 3 часов по каплям добавляли 40 мл раствора 2н. НСl в диоксане при температуре от 0 до 25oС. Полученную реакционную смесь гасили бикарбонатом натрия (5 эквивалентов), а затем разбавляли этилацетатом (0,10 М), промывали водой (2Х) и солевым раствором (1X), после чего сушили сульфатом магния. Растворитель удаляли в вакууме и неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле, в результате чего получали Соединение 18: 3-(4-амидинофенил)-5-(4-(2-карбокси-2-N-этилсульфониламиноэтил)-фенокси)метил-2-оксазолидинон; т.пл. 212oС (с разл.).
Вышеуказанное описание считается достаточным для практического применения настоящего изобретения. Различные варианты настоящего изобретения помимо тех, которые указаны и описаны в настоящей заявке, будут понятны специалистам исходя из вышеприведенного описания и не выходят за рамки нижеследующей формулы изобретения.
Claims (35)
1. Средство (набор), включающее в себя упаковочный материал и фармацевтический агент, содержащийся в указанном упаковочном материале, где указанный фармацевтический агент оказывает эффективное ингибирующее действие на ангиогенез в ткани, а указанный упаковочный материал включает в себя этикетку, в которой указано, что упомянутый фармацевтический агент может быть использован для лечения заболеваний путем ингибирования ангиогенеза, и где упомянутый фармацевтический агент содержит количество антагониста интегрина αvβ5, ингибирующее ангиогенез, где указанным антагонистом является полипептид матриксной металлопротеиназы, который включает в себя последовательность аминокислотных остатков, показанную в SEQ ID No. 11, 12, 13, 14, 15, 16, 17, 19, 20, 21 или 22, где указанным антагонистом является слитый белок, содержащий указанный полипептид матриксной металлопротеиназы, или где указанным антагонистом является циклический пептид с последовательностью аминокислотных остатков, показанной в SEQ ID No. 9.
2. Средство по п. 1, где указанной тканью является воспаленная ткань, а указанным заболеванием является артрит или ревматоидный артрит.
3. Средство по п. 1, где указанной тканью является солидная опухоль или метастазы солидной опухоли.
4. Средство по п. 1, где указанной тканью является ткань сетчатки, а указанным заболеванием является ретинопатия, диабетическая ретинопатия или дегенерация желтого пятна.
5. Антагонист интегрина αvβ5, содержащий полипептид матриксной металлопротеиназы, который включает в себя последовательность аминокислотных остатков, показанную в SEQ ID No. 11, 12, 13, 14, 15, 16, 17, 19, 20, 21 или 22.
6. Антагонист по п. 5, где указанным полипептидом является слитый белок.
7. Антагонист по п. 5, где указанный полипептид имеет последовательность аминокислотных остатков, показанную в SEQ ID No. 11, 12, 13, 14, 15, 16, 17, 19, 20, 21 или 22.
8. Антагонист интегрина αvβ5, содержащий циклический пептид с последовательностью аминокислотных остатков, показанной в SEQ ID No. 9.
9. Фармацевтический агент, содержащий антагонист интегрина αvβ5, по п. 5 или 8 в фармацевтически приемлемом носителе в количестве, достаточном для ингибирования ангиогенеза в ткани.
10. Способ ингибирования ангиогенеза в ткани, предусматривающий введение в указанную ткань композиции, содержащей количество антагониста αvβ5, ингибирующее ангиогенез, где указанным антагонистом является полипептид матриксной металлопротеиназы, который включает в себя последовательность аминокислотных остатков, показанную в SEQ ID No. 11, 12, 13, 14, 15, 16, 17, 19, 20, 21 или 22, где указанным антагонистом является слитый белок, содержащий указанный полипептид матриксной металлопротеиназы, или где указанным антагонистом является циклический пептид с последовательностью аминокислотных остатков, показанной в SEQ ID No. 9.
11. Способ по п. 10, вызывающий у пациента регрессию солидной опухоли.
12. Способ по п. 10, вызывающий у пациента ингибирование роста ткани солидной опухоли, подверженной неоваскуляризации.
13. Способ по п. 10, предусматривающий лечение у пациента воспаленной ткани, в которой имеет место неоваскуляризация.
14. Способ по п. 10, предусматривающий лечение пациента, у которого имеет место неоваскуляризация ткани сетчатки.
15. Способ по п. 10, предусматривающий уменьшение у пациента кровоснабжения ткани, необходимого для поддержания нового роста указанной ткани.
16. Способ по пп. 10-15, где указанный антагонист интегрина αvβ5 предпочтительно ингибирует связывание фибриногена с αvβ5, по сравнению со связыванием фибриногена с α11bβ3.
17. Способ по пп. 10-15, где указанной тканью является ткань человека.
17. Способ по пп. 10-15, где указанной тканью является ткань человека.
18. Способ по п. 10 или 13, где указанной тканью является воспаленная ткань, а указанным ангиогенезом является ангиогенез воспаленной ткани.
19. Способ по п. 18, где указанной тканью является ткань, пораженная артритом.
20. Способ по п. 19, где указанная ткань, пораженная артритом, присутствует у млекопитающего с ревматоидным артритом.
21. Способ по пп. 10, 13 или 14, где указанной тканью является ткань сетчатки, а указанным ангиогенезом является ангиогенез сетчатки.
22. Способ по п. 21, где указанная ткань сетчатки присутствует у пациента с диабетической ретинопатией или дегенерацией желтого пятна.
23. Способ по пп. 10, 11, 12 или 15, где указанной тканью является солидная опухоль или метастазы солидной опухоли, а указанным ангиогенезом является опухолевый ангиогенез.
24. Способ по п. 23, где указанной тканью является карцинома.
25. Способ по п. 23, где указанной солидной опухолью является опухоль легких, поджелудочной железы, молочной железы, толстой кишки, гортани или яичников.
26. Способ по п. 23, где указанное введение осуществляют в сочетании с химиотерапией.
27. Способ по пп. 10-15, где указанное введение предусматривает внутривенное, чрескожное, внутрисиновиальное, внутримышечное или пероральное введение.
28. Способ по пп. 10-15, где указанное количество, ингибирующее ангиогенез, составляет примерно от 0,1 до 300 мг/кг.
29. Способ по пп. 10-15, где указанное терапевтически эффективное количество составляет примерно от 0,1 до 300 мг/кг.
30. Способ по пп. 10-15, где указанное введение предусматривает внутривенное введение разовой дозы.
31. Способ по пп. 10-15, где указанное введение предусматривает введение одной или нескольких доз ежедневно в течение одного или нескольких дней.
32. Способ по пп. 10, 13 или 14, где указанный ангиогенез присутствует у пациента, имеющего глазное заболевание, выбранное из группы глазных болезней, состоящей из диабетической ретинопатии, возрастной дегенерации желтого пятна, предположительного глазного гистоплазмоза, преждевременной ретинопатии и неоваскулярной глаукомы.
33. Способ по п. 10 или 13, где указанный ангиогенез присутствует у больного с неоваскулярным расстройством роговицы, выбранным из группы нарушений, состоящей из трансплантации роговицы, герпетического кератита, сифилитического кератита, птеригия и неоваскулярного паннуса, ассоциированного с применением контактных линз.
34. Способ по пп. 10, 13 или 14, где указанный ангиогенез индуцирован цитокином.
35. Способ по п. 34, где указанный цитокин выбран из группы, состоящей из фактора роста сосудистого эндотелия, трансформирующего фактора роста α и эпидермального фактора роста.
36. Способ по п. 34, где указанный цитокин является фактором роста сосудистого эндотелия, а указанный ангиогенез выбран из группы, состоящей из ангиогенеза сетчатки, ангиогенеза роговицы, опухолевого ангиогенеза и ангиогенеза воспаленной ткани.
Приоритет по пунктам:
30.05.1997 по пп. 1-6, 9-33;
31.05.1996 по пп. 7 и 8.
30.05.1997 по пп. 1-6, 9-33;
31.05.1996 по пп. 7 и 8.
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US1877396P | 1996-05-31 | 1996-05-31 | |
| US1586996P | 1996-05-31 | 1996-05-31 | |
| US60/018,773 | 1996-05-31 | ||
| US60/015,869 | 1996-05-31 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| RU98123834A RU98123834A (ru) | 2000-10-27 |
| RU2195312C2 true RU2195312C2 (ru) | 2002-12-27 |
Family
ID=26687900
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| RU98123834/14A RU2195312C2 (ru) | 1996-05-31 | 1997-05-30 | СПОСОБЫ И КОМПОЗИЦИИ, ИСПОЛЬЗУЕМЫЕ ДЛЯ ИНГИБИРОВАНИЯ αvβ5-ОПОСРЕДОВАННОГО АНГИОГЕНЕЗА |
| RU98123833/14A RU2194528C2 (ru) | 1996-05-31 | 1997-05-30 | Способы и композиции, используемые для ингибирования ангиогенеза |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| RU98123833/14A RU2194528C2 (ru) | 1996-05-31 | 1997-05-30 | Способы и композиции, используемые для ингибирования ангиогенеза |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US6500924B1 (ru) |
| JP (1) | JP2000516201A (ru) |
| CN (1) | CN1226172A (ru) |
| AU (2) | AU738782B2 (ru) |
| PT (1) | PT951295E (ru) |
| RU (2) | RU2195312C2 (ru) |
| SK (2) | SK163598A3 (ru) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2600794C2 (ru) * | 2010-10-07 | 2016-10-27 | Аэрпио Терапьютикс, Инк. | Композиции и способы лечения глазного отека, неоваскуляризации и связанных с ними заболеваний |
Families Citing this family (45)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7053041B1 (en) * | 1996-05-31 | 2006-05-30 | The Scripps Research Institute | Methods and compositions useful for inhibition of αvβ5mediated angiogenesis |
| CZ40998A3 (cs) * | 1995-08-14 | 1998-09-16 | The Scripps Research Institute | Použití antagonisty anb5 pro výrobu prostředků, schopných inhibovat angiogenezi v tkáních |
| US20040063790A1 (en) * | 1996-05-31 | 2004-04-01 | The Scripps Research Institute | Methods for inhibition of angiogenesis |
| US20050281821A1 (en) * | 1999-01-06 | 2005-12-22 | Flavia Pernasetti | Method and composition for angiogenesis inhibition |
| EP1260538B1 (en) * | 1999-10-06 | 2010-12-01 | Kaneka Corporation | Process for producing polyimide resin |
| ATE350660T1 (de) * | 2001-02-21 | 2007-01-15 | Eisai Co Ltd | Verfahren zur untersuchung der wirkung eines angionegesis-hemmers unter vermittlung durch hemmung der integrin-expression |
| US7019108B2 (en) * | 2001-06-01 | 2006-03-28 | University Of Southern California | Method and composition for inhibition of angiogenesis and tumor growth using compounds based on a sequence within MMP-2 |
| BR0211604A (pt) * | 2001-08-01 | 2004-08-24 | Merck Patent Gmbh | Inibidores de integrina para o tratamento de doenças do olho |
| US7301006B2 (en) * | 2002-07-16 | 2007-11-27 | Wisconsin Alumni Research Foundation | Methods and materials for the synthesis of modified peptides |
| FR2856598B1 (fr) * | 2003-06-25 | 2005-10-28 | Inst Nat Sante Rech Med | Peptides derives de la proteine mmp-2 et leur utilisation en immunotherapie antitumorale |
| WO2005090406A2 (en) | 2004-03-12 | 2005-09-29 | Vasgene Therapeutics, Inc. | Antibodies binding to ephb4 for inhibiting angiogenesis and tumor growth |
| US7973134B2 (en) * | 2004-07-07 | 2011-07-05 | Cell Signaling Technology, Inc. | Reagents for the detection of protein phosphorylation in anaplastic large cell lymphoma signaling pathways |
| WO2006034455A2 (en) | 2004-09-23 | 2006-03-30 | Vasgene Therapeutics, Inc. | Polipeptide compounds for inhibiting angiogenesis and tumor growth |
| US7935790B2 (en) * | 2004-10-04 | 2011-05-03 | Cell Singaling Technology, Inc. | Reagents for the detection of protein phosphorylation in T-cell receptor signaling pathways |
| US7807789B2 (en) * | 2004-12-21 | 2010-10-05 | Cell Signaling Technology, Inc. | Reagents for the detection of protein phosphorylation in EGFR-signaling pathways |
| JP2008526186A (ja) * | 2005-01-03 | 2008-07-24 | エフ.ホフマン−ラ ロシュ アーゲー | ポリペプチド足場としてのヘモペキシン様構造 |
| US20090099340A1 (en) * | 2007-10-12 | 2009-04-16 | Cell Signaling Technology, Inc. | Reagents for the detection of protein phosphorylation in carcinoma signaling pathways |
| US20060286148A1 (en) * | 2005-05-18 | 2006-12-21 | Ppd, Inc. | Method of forming implants |
| US20070048325A1 (en) * | 2005-08-24 | 2007-03-01 | Dennis Van Epps | Combination therapies for inhibiting integrin-extracellular matrix interactions |
| US20100151495A9 (en) * | 2005-08-31 | 2010-06-17 | Cell Signaling Technolgy, Inc. | Reagents for the detection of protein phosphorylation in carcinoma signaling pathways |
| EP1934867A2 (en) * | 2005-08-31 | 2008-06-25 | Cell Signaling Technology, Inc. | Reagents for the detection of protein phosphorylation in leukemia signaling pathways |
| JP2009513949A (ja) * | 2005-09-20 | 2009-04-02 | エンブレクス,インコーポレイテッド | 鳥卵の胚盤葉の位置を迅速かつ正確に特定する方法 |
| AU2007207764B2 (en) | 2006-01-12 | 2012-06-07 | Alexion Pharmaceuticals, Inc. | Antibodies to OX-2/CD200 and uses thereof |
| WO2007127335A2 (en) * | 2006-04-27 | 2007-11-08 | Cell Signaling Technology, Inc. | Reagents for the detection of protein phosphorylation in atm and atr kinase signaling pathways |
| EP1862541A1 (en) * | 2006-06-01 | 2007-12-05 | Institut National De La Sante Et De La Recherche Medicale (Inserm) | Polypeptides derived from the hemopexin-like domain of metalloproteinase MMP-2 |
| US7939636B2 (en) * | 2006-08-11 | 2011-05-10 | Cell Signaling Technology, Inc. | Reagents for the detection of protein phosphorylation in c-Src signaling pathways |
| US20090258442A1 (en) * | 2006-08-31 | 2009-10-15 | Cell Signaling Technology, Inc. | Reagents for the detection of protein phosphorylation in carcinoma signaling pathways |
| CN101835485B (zh) | 2007-02-01 | 2016-10-26 | 阿塞勒隆制药公司 | 活化素-actriia拮抗剂及在治疗或预防乳腺癌中的用途 |
| EP1972639A3 (en) | 2007-03-07 | 2008-12-03 | Cell Signaling Technology, Inc. | Reagents for the detection of protein phosphorylation in carcinoma signaling pathways |
| US20090068684A1 (en) * | 2007-03-26 | 2009-03-12 | Cell Signaling Technology, Inc. | Serine and threoninephosphorylation sites |
| US20080238709A1 (en) * | 2007-03-28 | 2008-10-02 | Faramarz Vaziri | One-way communication apparatus with dynamic key generation |
| EP1983003A3 (en) | 2007-04-19 | 2009-03-11 | Peter Hornbeck | Tyrosine phosphorylation sites and antibodies specific for them |
| US7977462B2 (en) | 2007-04-19 | 2011-07-12 | Cell Signaling Technology, Inc. | Tyrosine phosphorylation sites |
| EP1983002A3 (en) | 2007-04-19 | 2009-03-11 | Peter Hornbeck | Tyrosine phosphorylation sites and antibodies specific for them |
| US20090053831A1 (en) | 2007-05-01 | 2009-02-26 | Cell Signaling Technology, Inc. | Tyrosine phosphorylation sites |
| CN101861161B (zh) | 2007-09-18 | 2017-04-19 | 阿塞勒隆制药公司 | 活化素‑actriia拮抗剂和减少或抑制fsh分泌的用途 |
| US20090203043A1 (en) | 2007-11-21 | 2009-08-13 | Peter Hornbeck | Protein phosphorylation by basophilic serine/threonine kinases in insulin signaling pathways |
| US20090220991A1 (en) * | 2008-02-29 | 2009-09-03 | Cell Signaling Technology, Inc. | Reagents for the detection of protein phosphorylation in leukemia signaling pathways |
| CA2780243C (en) | 2009-11-10 | 2019-12-03 | Allegro Pharmaceuticals, Inc. | Compositions and methods for inhibiting cellular adhesion or directing diagnostic or therapeutic agents to rgd binding sites |
| US11673914B2 (en) | 2009-11-10 | 2023-06-13 | Allegro Pharmaceuticals, LLC | Peptide therapies for reduction of macular thickening |
| JP6106159B2 (ja) | 2011-05-09 | 2017-03-29 | アレグロ ファーマシューティカルズ インコーポレイテッドAllegro Pharmaceuticals,Inc. | R−g−システイン酸ペプチドを含む医薬組成物 |
| CA2868883C (en) | 2012-03-30 | 2022-10-04 | Sorrento Therapeutics Inc. | Fully human antibodies that bind to vegfr2 |
| EP3515495A4 (en) | 2016-09-26 | 2020-08-26 | Ensemble Group Holdings | METHOD OF EVALUATION AND TREATMENT OF CANCER IN HUMANS WITH DYSREGULATED LYMPHATIC SYSTEMS |
| EA201992326A1 (ru) * | 2017-03-31 | 2020-03-13 | Дзе Риджентс Оф Дзе Юниверсити Оф Калифорния | Композиции и способы оказания направленного воздействия на альфа-v бета-3-положительные раковые стволовые клетки (csc) и лечения (avb3) лекарственно-устойчивых видов рака |
| CN113663929A (zh) * | 2021-09-24 | 2021-11-19 | 深圳市广利达精密机械有限公司 | 一种自动化机械加工零件自动加工分拣设备及其方法 |
Family Cites Families (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5092885A (en) | 1987-02-12 | 1992-03-03 | The Government Of The United States Of America As Represented By The Secretary Of The Department Of Health And Human Services | Peptides with laminin activity |
| EP0341303A4 (en) | 1987-11-19 | 1990-06-27 | Scripps Clinic Res | MONOCLONAL ANTIBODY AGAINST THE RGD-DIRECTED ADHESION RECEPTOR OF ENDOTHELIAL CELLS. |
| JPH01169343A (ja) | 1987-12-25 | 1989-07-04 | Nippon Sheet Glass Co Ltd | ガラス板の切口欠点検出装置 |
| US5135919A (en) | 1988-01-19 | 1992-08-04 | Children's Medical Center Corporation | Method and a pharmaceutical composition for the inhibition of angiogenesis |
| EP0398925B1 (en) | 1988-01-19 | 1993-10-20 | Children's Hospital Corporation | Growth inhibiting agent and the use thereof |
| US5575815A (en) | 1988-08-24 | 1996-11-19 | Endoluminal Therapeutics, Inc. | Local polymeric gel therapy |
| US5686568A (en) | 1989-06-16 | 1997-11-11 | Cor Therapeutics, Inc. | Platelet aggregration inhibitors |
| US5112946A (en) | 1989-07-06 | 1992-05-12 | Repligen Corporation | Modified pf4 compositions and methods of use |
| US5192744A (en) | 1990-01-12 | 1993-03-09 | Northwestern University | Method of inhibiting angiogenesis of tumors |
| ATE130517T1 (de) | 1990-08-08 | 1995-12-15 | Takeda Chemical Industries Ltd | Intravaskulär embolisierendes mittel mit gehalt an einem die angiogenesis hemmenden stoff. |
| JP3353209B2 (ja) | 1992-04-03 | 2002-12-03 | ジェネンテク,インコーポレイテッド | αvβ3インテグリンに対する抗体 |
| SK57693A3 (en) | 1992-06-18 | 1994-07-06 | Merck Patent Gmbh | Linear peptides and pharmaceutical agents on their base |
| UA43823C2 (uk) | 1992-07-06 | 2002-01-15 | Мерк Патент Геселлшафт Міт Бесшренктер Хафтунг | ФАРМАЦЕВТИЧНА КОМПОЗИЦІЯ ДЛЯ ІНГІБУВАННЯ ІНТЕГРИН <font face="Symbol">a</font><sub>V</sub><font face="Symbol">b</font><sub>3</sub>-ОПОСЕРЕДКОВАНОЇ КЛІТИННОЇ АДГЕЗІЇ КЛІТИН ССАВЦІВ, СПОСІБ ЛІКУВАННЯ ТА ПРОФІЛАКТИКИ ЗАХВОРЮВАННЯ, АСОЦІЙОВАНОГО З ПОРУШЕННЯМ АДГЕЗІЇ КЛІТИН, СПОСІБ БЛОКУВАННЯ ЗВ'ЯЗУВАННЯ ФІБРИНОГЕНОМ ІНТЕГРИНУ, КОМПОЗИЦІЯ ДЛЯ ЗАГОЄННЯ РАН |
| DE4310643A1 (de) | 1993-04-01 | 1994-10-06 | Merck Patent Gmbh | Cyclische Adhäsionsinhibitoren |
| US5981478A (en) | 1993-11-24 | 1999-11-09 | La Jolla Cancer Research Foundation | Integrin-binding peptides |
| US5770565A (en) | 1994-04-13 | 1998-06-23 | La Jolla Cancer Research Center | Peptides for reducing or inhibiting bone resorption |
| DE19534177A1 (de) | 1995-09-15 | 1997-03-20 | Merck Patent Gmbh | Cyclische Adhäsionsinhibitoren |
| DE19538741A1 (de) | 1995-10-18 | 1997-04-24 | Merck Patent Gmbh | Cyclopeptidderivate |
-
1997
- 1997-05-30 RU RU98123834/14A patent/RU2195312C2/ru not_active IP Right Cessation
- 1997-05-30 AU AU32183/97A patent/AU738782B2/en not_active Ceased
- 1997-05-30 AU AU32893/97A patent/AU733303C/en not_active Ceased
- 1997-05-30 US US09/194,468 patent/US6500924B1/en not_active Expired - Lifetime
- 1997-05-30 JP JP09542941A patent/JP2000516201A/ja not_active Ceased
- 1997-05-30 CN CN97196822A patent/CN1226172A/zh active Pending
- 1997-05-30 SK SK1635-98A patent/SK163598A3/sk unknown
- 1997-05-30 PT PT97928698T patent/PT951295E/pt unknown
- 1997-05-30 RU RU98123833/14A patent/RU2194528C2/ru not_active IP Right Cessation
- 1997-05-30 SK SK1632-98A patent/SK163298A3/sk unknown
Non-Patent Citations (2)
| Title |
|---|
| FOLKMAN, Nature Medicine, 1995, 1:27-31. * |
| Технология лекарственных форм./Под ред. Т.С. Кондратьевой. - М.: Медицина, 1991, т.1, с. 65, 131. * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2600794C2 (ru) * | 2010-10-07 | 2016-10-27 | Аэрпио Терапьютикс, Инк. | Композиции и способы лечения глазного отека, неоваскуляризации и связанных с ними заболеваний |
Also Published As
| Publication number | Publication date |
|---|---|
| AU738782B2 (en) | 2001-09-27 |
| AU3289397A (en) | 1998-01-05 |
| SK163298A3 (en) | 1999-07-12 |
| PT951295E (pt) | 2010-07-29 |
| AU733303B2 (en) | 2001-05-10 |
| AU3218397A (en) | 1998-01-05 |
| JP2000516201A (ja) | 2000-12-05 |
| US6500924B1 (en) | 2002-12-31 |
| CN1226172A (zh) | 1999-08-18 |
| SK163598A3 (en) | 1999-06-11 |
| AU733303C (en) | 2002-08-08 |
| RU2194528C2 (ru) | 2002-12-20 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| RU2195312C2 (ru) | СПОСОБЫ И КОМПОЗИЦИИ, ИСПОЛЬЗУЕМЫЕ ДЛЯ ИНГИБИРОВАНИЯ αvβ5-ОПОСРЕДОВАННОГО АНГИОГЕНЕЗА | |
| JP4544639B2 (ja) | αVβ5仲介血管形成の阻止に有効な方法及び組成物 | |
| KR100327081B1 (ko) | 혈관형성억제에유용한조성물 | |
| CA2256543C (en) | Methods and compositions useful for inhibition of angiogenesis | |
| WO1997045447A9 (en) | METHODS AND COMPOSITIONS USEFUL FOR INHIBITION OF αvβ5 MEDIATED ANGIOGENESIS | |
| WO1997006791A9 (en) | METHODS AND COMPOSITIONS USEFUL FOR INHIBITION OF αvβ5 MEDIATED ANGIOGENESIS | |
| US20060165703A1 (en) | Methods and compositions useful for inhibition of alpha v beta 5 mediated angiogenesis | |
| US20060270710A1 (en) | Methods for inhibition of angiogenesis | |
| US20030176334A1 (en) | Methods and compositions useful for inhibition of angiogenesis | |
| MXPA98009944A (en) | Methods and useful compositions for medium angiogenesis inhibition by alfavbe | |
| MXPA98009945A (en) | Useful methods and compositions for angiogene inhibition |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| MM4A | The patent is invalid due to non-payment of fees |
Effective date: 20060531 |