KR20180098675A - Hiv 성숙 억제 활성을 갖는 루판의 아민 유도체 - Google Patents
Hiv 성숙 억제 활성을 갖는 루판의 아민 유도체 Download PDFInfo
- Publication number
- KR20180098675A KR20180098675A KR1020187023717A KR20187023717A KR20180098675A KR 20180098675 A KR20180098675 A KR 20180098675A KR 1020187023717 A KR1020187023717 A KR 1020187023717A KR 20187023717 A KR20187023717 A KR 20187023717A KR 20180098675 A KR20180098675 A KR 20180098675A
- Authority
- KR
- South Korea
- Prior art keywords
- mmol
- alkyl
- compound
- salt
- give
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Withdrawn
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J63/00—Steroids in which the cyclopenta(a)hydrophenanthrene skeleton has been modified by expansion of only one ring by one or two atoms
- C07J63/008—Expansion of ring D by one atom, e.g. D homo steroids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Virology (AREA)
- General Chemical & Material Sciences (AREA)
- Tropical Medicine & Parasitology (AREA)
- Communicable Diseases (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Molecular Biology (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Oncology (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- AIDS & HIV (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Steroid Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Abstract
본 발명은 하기 화학식 I에 따른 구조에 의해 특징되는 화합물, 또는 이의 약제학적으로 허용되는 염에 관한 것이다. 본 발명의 화합물은 HIV의 치료 또는 예방을 위해 유용하다:
Description
관련 출원에 대한 상호 참조문헌
본 출원은 2016년 1월 20일에 출원된 미국가출원일련번호 제62/28089호의 이점을 주장한다.
본 발명의 분야
본 발명은 (i) HIV로 감염된 대상체에서 HIV 복제를 억제하거나, (ii) HIV로 감염된 대상체를 치료하기 위한, 화합물, 약제 조성물, 및 이의 사용 방법에 관한 것이다.
인간 면역결핍 바이러스 타입 1(HIV-1)은 후천성 면역 결핍 질병(AIDS)의 발병을 유도한다. HIV 병증의 수는 계속 증가하고 있으며, 현재 전 세계적으로 2500만 명이 넘는 사람들이 상기 바이러스로 고통받고 있다. 현재, 항레트로바이러스 약물을 이용한 바이러스 복제의 장기간 억제가 HIV-1 감염증을 치료하기 위한 유일한 옵션(option)이다. 실제로, 미국 식품의약국은 환자의 생존과 삶의 질을 크게 향상시키는 것으로 밝혀진 6개의 상이한 억제제 부류에 걸쳐 25종의 약물을 승인하였다. 그러나, 바람직하지 않은 약물-약물 상호작용; 약물-식품 상호작용; 치료법에 대한 불순응(non-adherence to therapy); 및 효소 타겟의 돌연변이로 인한 약물 내성으로 인해 추가 치료법이 여전히 요구된다.
현재, 거의 모든 HIV 양성인 환자들은 고활성 항레트로바이러스 요법("HAART")으로 언급되는 항레트로바이러스 약물 조합물의 치료 요법으로 치료된다. 그러나, HAART 요법은 종종 약물-내성 HIV-1 변이체의 신속한 출현을 피하기 위해 환자에게 매일 상이한 약물들의 조합물이 투여되어야 하기 때문에, 흔히 복잡하다. 환자 생존에 대한 HAART의 긍정적인 영향에도 불구하고, 약물 내성이 여전히 발생할 수 있다. 다중약물-내성 HIV-1 분리균(isolate)의 출현은 심각한 임상적 결과를 나타내고, 구제 요법(salvage therapy)으로서 공지된, 신규한 약물 요법으로 억제되어야 한다.
현재의 가이드라인은, 구제 요법이 적어도 두 개, 및 바람직하게는 세 개의 완전 활성 약물을 포함할 것을 권장한다. 통상적으로, 제1선 치료법은 바이러스 효소 역전사효소(reverse transcriptase) 및 프로테아제(protease)를 타겟으로 하는 3개 내지 4개의 약물들을 조합한다. 구제 요법에 대한 한 가지 옵션은 내성 분리균에 대해 활성을 유지하는 동일한 작용기작 부류(mechanistic class)로부터의 약물들의 상이한 조합물을 투여하는 것이다. 그러나, 이러한 방법에 대한 옵션은, 내성 돌연변이가 동일한 부류 내의 상이한 약물에 종종 광범위한 교차 내성을 부여하기 때문에, 흔히 제한적이다. 대안적인 치료 전략이 최근 융합, 진입, 및 인테그라아제(integrase) 억제제의 개발과 함께 이용가능하게 되었다. 그러나, 세 가지 신규한 약물 부류에 대한 모든 내성은 실험 및 환자 둘 모두에서 이미 보고되었다. 따라서, 항레트로바이러스 약물에 의한 HIV-1-감염된 환자의 지속된 성공적인 치료는 새로운 타겟 및 작용 메커니즘을 갖는 신규하고 개선된 약물의 지속적인 개발을 필요로 할 것이다.
현재, 항레트로바이러스 약물로의 바이러스 복제의 장기간 억제는 HIV-1 감염증을 치료하기 위한 유일한 옵션이다. 지금까지, 많은 승인된 약물들이 환자의 생존을 크게 증가시키는 것으로 나타났다. 그러나, 고활성 항레트로바이러스 치료법(HAART)으로서 공지된 치료 요법은 약물-내성 HIV-1 변이체의 신속한 출현을 피하기 위해 환자에게 상이한 약물들의 조합물이 투여되어야 하기 때문에, 흔히 복잡하다. 환자 생존에 대한 HAART의 긍정적인 영향에도 불구하고, 약물 내성이 여전히 발생할 수 있다.
HIV Gag 폴리단백질 전구체(Pr55Gag)는 4개의 단백질 도메인, 즉, 기질(matrix: MA), 캡시드(capsid: CA), 뉴클레오캡시드(nucleocapsid: NC)와 p6, 및 2개의 스페이서 펩티드, 즉, SP1 및 SP2로 이루어진 것으로서, 이는 신규한 치료 타겟을 나타낸다. Gag 폴리단백질의 분해는 감염성 바이러스 입자 생산의 진행에서 중요한 역할을 하지만, 지금까지, 이러한 메커니즘에 대하여 항레트로바이러스 약물이 승인된 적이 없다.
대부분의 세포 타입에서, 어셈블리(assembly)는 원형질 막(plasma membrane)에서 발생하며, Gag의 MA 도메인은 막 결합을 매개한다. 어셈블리는 세포로부터 미성숙한 입자의 발아(budding)에 의해 완료된다. 입자 방출을 수반하면서, 바이러스 엔코딩된 PR은 Gag를 4개의 성숙한 단백질 도메인, 즉, MA, CA, NC와 p6, 및 2개의 스페이서 펩티드, 즉 SP1 및 SP2로 절단한다. Gag-Pol은 또한 PR에 의해 절단되어, 바이러스 효소, 즉, PR, RT 및 IN을 유리시킨다. Gag 단백질분해 과정은 성숙(maturation)으로서 알려진, 입자 내에서 형태학적 재배열을 유도한다. 성숙은 미성숙한 도넛-형상 입자를 성숙한 비리온(virion)으로 전환시키는데, 이는 NC 및 바이러스 효소인 RT 및 IN과의 복합물에서 바이러스 RNA 게놈을 둘러싸는 CA 쉘(shell)로 이루어진 응축된 원뿔 코어를 함유한다. 성숙은 신규한 세포의 감염을 위한 바이러스를 제조하고, 입자 감염성에 대해 절대적으로 필수적이다.
베비리맷(Bevirimat)(PA-457)은 Gag의 과정에서의 최종 단계, 즉 감염성 바이러스 입자의 형성에 필요한, 캡시드-SP1(p25)의 캡시드로의 전환을 억제하는 성숙 억제제이다. 베비리맷은 ART-내성 및 야생형 HIV에 대해 활성을 가지고, 모든 부류로부터 항레트로바이러스와의 상승효과를 나타낸다. 베비리맷은, 20 ㎍/mL 이상의 최저치(trough level)를 달성하고 Q369, V370 또는 T371에서 주요 기준선 Gag 다형태 중 임의의 다형태를 지니지 않는 환자에서 HIV 바이러스 로드(load)를 평균 1.3 log10/mL까지 감소시켰다. 그러나, Q369, V370 또는 T371에서 Gag 다형태를 지니는 베비리맷 사용자는 이러한 부위에서 Gag 다형태를 지니지 않는 환자보다 훨씬 낮은 로드 감소로 입증되었다.
성숙 억제제의 다른 예는 PCT 특허출원번호 WO2011/100308호, PCT 특허출원번호 PCT/US2012/024288호, 중국 PCT 출원번호 PCT/CN2011/001302호, 중국 PCT 출원번호 PCT/CN2011/001303호, 중국 PCT 출원번호 PCT/CN2011/002105호, PCT/CN2011/002159호, WO2013/090664호, WO2013/123019호, WO 2013/043778호, WO 2014/123889호, WO 2011/153315호, WO 2011/153319호, WO 2012/106188호, WO 2012/106190호, WO 2013/169578호, WO 2014/13081호에서 확인될 수 있다. 종래 기술에서 성숙 억제제는 다형태 범위(polymorphism coverage)의 영역에서 빈틈을 남겼으며, 이에 의해, 광범위한 임상적 관련 gag 서열에 대한 효능은 장기간 지속성 시험에서 강력한 효능을 위해 요구되는 임상적 관련 단백질 조정 항바이러스 활성을 포함하는 전반적인 효능과 함께 매우 중요하다. 현재까지, 성숙 억제제는 이러한 성질들의 최적의 균형을 달성하지 못하였다.
이에 따라, HIV 감염증의 예방 및/또는 치료를 위한 상술된 성질들의 효과적인 균형을 이루는 대안적인 화합물을 발견하는 것이 기술 분야의 진보일 것이다.
간단하게, 일 양태에서, 본 발명은 하기 화학식 I의 화합물, 또는 이의 약제학적으로 허용되는 염을 제공한다:

상기 식에서,
R1은 H 또는 C1-4알킬이며;
L1은 결합, C1-8알킬렌, 또는 C0-3알킬렌C(O)C0-3알킬렌이며;
R2는 N(R3)R4, 또는 -OR3이며, 여기서, 각 R3 및 R4는 독립적으로, H, C1-3알킬, C1-3알킬렌-C3-6사이클로알킬, C1-3알킬렌-C5-7헤테로사이클, C1-3알킬렌N(C1-3알킬)2, C1-3알킬렌C5-7아릴, C(O)C5-7아릴, C(O)C1-4알킬, C1-3알킬렌C5-7헤테로아릴이며, 여기서, 각 R3 및 R4는 할로겐, C1-4알킬, SO2C1-3알킬로부터 독립적으로 선택된 1 또는 2개의 치환체로 치환될 수 있거나;
R2가 N(R3)R4일 때, R3 및 R4는 함께 결합할 수 있고, 이러한 것이 결합된 N과 함께 4원 내지 7원 헤테로사이클을 형성할 수 있으며, 상기 헤테로사이클은 N, S, 및 O로부터 선택된 하나의 다른 헤테로원자를 함유할 수 있으며, 상기 S 원자는 2개 이하의 옥소 기에 의해 치환될 수 있으며, 상기 헤테로사이클은 할로겐, C1-4알킬, SO2C1-3알킬로부터 독립적으로 선택된 1 또는 2개의 치환체로 치환될 수 있으며;
V는 C4-8사이클로알킬렌, C4-8사이클로알케닐렌, C5-8아릴, 또는 5원 내지 8원 헤테로아릴 고리이며, 여기서, V는 할로겐 및 C1-3알킬로부터 독립적으로 선택된 1 또는 2개의 치환체로 치환될 수 있으며, 상기 알킬은 1 또는 2개의 할로겐으로 치환될 수 있으며;
A는 -CO2C1-3알킬 또는 CO2H이다.
다른 양태에서, 본 발명은 본 발명의 화합물 또는 염을 포함하는 약제 조성물을 제공한다.
다른 양태에서, 본 발명은 (i) HIV로 감염된 대상체에서 HIV 복제를 억제하거나, (ii) HIV로 감염된 대상체를 치료하거나, (iii) 이러한 감염증의 위험이 있는 대상체에서 HIV 감염증을 예방하는 방법을 제공한다.
다른 양태에서, 본 발명은 치료에서, 및 특히, 인간 약제에서 사용하기 위한 본 발명의 화합물 또는 염을 제공한다.
다른 양태에서, 본 발명은 (i) HIV로 감염된 대상체에서 HIV 복제를 억제하거나, (ii) HIV로 감염된 대상체를 치료하거나, (iii) 이러한 감염증의 위험이 있는 대상체에서 HIV 감염증을 예방하기 위한 약제의 제조를 위한 본 발명의 화합물의 용도를 제공한다.
바람직하게, R1은 H이다.
바람직하게, L1은 C1-4알킬렌, C(O), 또는 C(O)C1-3알킬렌이다.
바람직하게, R2는 N(R3)(R4)이다.
바람직하게, R3 및 R4는 독립적으로, C1-3알킬렌N(C1-3알킬)2, 또는 C1-3알킬렌페닐이며, 여기서, 각 R3 및 R4는 할로겐 또는 SO2C1-3알킬로부터 독립적으로 선택된 1개의 치환체로 치환될 수 있거나; R3 및 R4는 함께 결합되고, 이러한 것이 결합된 N과 함께 6원 헤테로사이클을 형성하며, 상기 헤테로사이클은 1개의 S 원자를 함유할 수 있으며, 상기 S 원자는 2개의 옥소 기에 의해 치환될 수 있으며; 각 R3 및 R4는 할로겐, C1-4알킬, SO2C1-3알킬로부터 독립적으로 선택된 1 또는 2개의 치환체로 치환될 수 있다.
바람직하게, V는 할로겐 또는 C1-3알킬로 임의적으로 치환된 페닐렌 또는 사이클로헥세닐렌이며, 상기 알킬은 1 또는 2개의 할로겐으로 치환될 수 있다.
바람직하게, A는 -CO2H이다.
바람직하게, 본 발명의 방법에서, 대상체는 인간이다.
약제학적으로 허용되는 염은 당해 분야에 널리 공지된 다양한 유기 및 무기 반대 이온으로부터 유도될 수 있고, 단지 일 예로서, 소듐, 포타슘, 칼슘, 마그네슘, 암모늄, 및 테트라알킬암모늄을 포함하고, 분자가 염기성 작용성을 함유할 때, 유기산 또는 무기산의 염, 예를 들어, 하이드로클로라이드, 하이드로브로마이드, 타르트레이트, 메실레이트, 아세테이트, 말레에이트, 라이신 및 옥살레이트를 함유한다. 적합한 염은 문헌[P. Heinrich Stahl, Camille G. Wermuth (Eds.), Handbook of Pharmaceutical Salts Properties, Selection, and Use; 2002]에 기술된 염들을 포함한다.
본 발명은 약리학적 증강제로서 유용한 하나 이상의 제제와 조합하여 사용될 수 있을 뿐만 아니라 HIV의 예방 또는 치료를 위한 추가 화합물과 함께 또는 HIV의 예방 또는 치료를 위한 추가 화합물 없이 사용될 수 있다. 이러한 약리학적 증강제(또는 약동학적 부스터(pharmakinetic booster))의 예는 리토나버(ritonavir) 및 코비시스타트(Cobicistat)(종래에는, GS-9350)를 포함하지만, 이로 제한되지는 않는다.
실시예
하기 실시예는 상기 기술된 발명을 제조하고 사용하는 방식을 보다 충분히 설명하기 위해 제공된다. 이러한 실시예는 결코 본 발명의 진정한 범위를 제한하고자 하는 것이 아니라 오히려 예시적인 목적으로 위해 제공되는 것으로 이해된다. 하기 실시예 및 상기 합성 반응식에서, 하기 약어는 하기 의미를 갖는다. 약어가 정의되지 않는 경우, 이는 이의 일반적으로 허용되는 의미를 갖는다.
aq. = 수성
μL = 마이크로리터
μM = 마이크로몰
NMR = 핵 자기 공명
boc = 3차-부톡시카보닐
br = 브로드(broad)
Cbz = 벤질옥시카보닐
d = 더블렛(doublet)
δ = 화학적 이동
℃ = 섭씨 온도
DCE = 1,2-디클로로에탄
DCM = 디클로로메탄
dd = 더블렛의 더블렛
DIEA 또는 DIPEA = N,N-디이소프로필에틸아민
DMEM = 둘베코 변형 이글 배지
DMF = N,N-디메틸포름아미드
DMP = Dess-Martin 페리오디난
DMSO = 디메틸설폭사이드
DPPA = 디페녹시포스포릴 아지드
FA = 포름산
EtOAc = 에틸 아세테이트
g = 그램
h 또는 hr = 시
HBTU = 2-(1H-벤조트리아졸-1-일)-1,1,3,3-테트라메틸우로늄 헥사플루오로포스페이트
HCV = C형 간염 바이러스
HPLC = 고성능 액체 크로마토그래피
Hz = 헤르츠
IU = 국제 단위
IC50 = 50% 억제에서의 억제 농도
J = 결합 상수(달리 명시하지 않는 한 Hz로 제공됨)
K-HMDS = 포타슘 비스(트리메틸실릴)아미드
m = 멀티플렛(multiplet)
M = 몰
M+H+ = 모 질량 스펙트럼 피크 플러스 H+
mg = 밀리그램
min = 분
mL = 밀리리터
mM = 밀리몰(millimolar)
mmol = 밀리몰(millimole)
MS = 질량 스펙트럼
N = 노르말
nm = 나노몰
PE = 석유 에테르
PCC = 피리디늄 클로로크로메이트
ppm = 백만분율(parts per million)
q.s. = 충분한 양
s = 싱글렛(singlet)
RT = 실온
sat. = 포화
t = 트리플렛(triplet)
TBAF = 테트라-n-부틸암모늄 플루오라이드
TBSCI = 3차-부틸디메틸실릴 클로라이드
TEA = 트리에틸아민
테트라키스 = 테트라키스(트리페닐포스핀)팔라듐(0)
TFA = 트리플루오로아세트산
THF = 테트라하이드로푸란
UPLC = 초고성능 액체 크로마토그래피
장비 설명
1H NMR 스펙트럼을 Bruker Ascend 400 분광계 상에서 기록하였다. 화학적 이동을 백만분율(ppm, δ 단위)로 표시한다. 결합 상수는 헤르츠 단위이다(Hz). 분할 패턴은 겉보기 다중도를 기술하고, s(싱글렛), d(더블렛), t(트리플렛), q(쿼테트), quint(퀸테트), m(멀티플렛), br(브로드)로서 지정한다.
분석적 저-해상도 질량 스펙트럼(MS)을 구배 용리 방법을 이용하여 Waters BEH C18, 2.1 × 50 mm, 1.7 ㎛를 이용한 SQ Detector를 구비한 Waters ACQUITY UPLC 상에서 기록하였다.
용매 A: 물 중 0.1% 포름산(FA);
용매 B: 아세토니트릴 중 0.1% FA;
0.5분 동안 30% B, 이후 2.5분에 걸쳐 30 내지 100% B.
반응식 및 실험 절차
하기 반응식 및 절차는 본 발명의 화합물이 어떻게 제조될 수 있는 지를 설명한다. 언급된 특정 용매 및 반응 조건도 역시 예시적인 것으로서 제한하려는 것은 아니다. 기술되지 않은 화합물은 시판되거나 이용가능한 출발 물질을 이용하여 당업자에 의해 용이하게 제조된다. 본원에 개시된 실시예는 단지 예시가 목적이며 본 발명의 범위를 제한하려는 것은 아니다. 모든 실시예는 본원에 개시된 검정을 이용하여 21 μM 내지 1 nM의 LHIV IC50 값을 나타내었다.
실시예 중 몇몇의 실시예의 경우에, 존재하는 경우의 C28 2차 알코올의 입체 화학이 이의 절대 배열에 대해서 명확하게 확인되지 않았다. 달리 언급되지 않는 한, 본 출원에 예시된 화합물은 광학적으로 순수한 입체이성질체로서 분리되었으며, 초기에 작도된 대로의 배열로 지정되었다. 이들 중 일부는 도시된 바와 같은 그 단일의 C28 위치에서 반대 입체화학으로서 나열될 수 있는 가능성이 있다. 이는 결코 발명의 범위 또는 화학식 I의 화합물의 이용성을 제한하는 것을 의미하지 않는다. 본원에 포함된 추가의 실시예는, 이로 한정되는 것은 아니지만, 1D 및 2D NMR 방법, 진동 원편광 이색성(vibrational circular dichroism) 및 X-선 결정학을 포함한 당업자에게는 널리 공지된 분광분석 방법에 의해서 도시된 형태를 지니는 것으로 측정되었다. 부분입체이성질체 둘 모두를 제조하기 위한 이들 실시예 및 방법은 C28 위치에서의 R 및 S 형태 둘 모두의 순수한 입체이성질체가 용이하게 얻어지고, 분리되며, 특징분석됨을 명확히 예시하는 것으로 제공되어야 하며, 어떠한 나머지 한정되지 않은 실시예는 당업자에게는 잘 공지된 유사한 방법에 의해서 용이하게 확인될 수 있다.

단계 A: 중간체 1:
(3aR,5aR,5bR,7aR,9S,11aR,11bR,13aS)-9-하이드록시-3a-(하이드록시메틸)-1-이소프로필-5a,5b,8,8,11a-펜타메틸-3,3a,4,5,5a,5b,6,7,7a,8,9,10,11,11a,11b,12,13,13a-옥타데카하이드로-2H-사이클로펜타[a]크리센-2-온

EtOH(200 mL) 및 톨루엔(200 mL) 중 중간체 1A, WO2013/090664호(40 g, 74 mmol) 및 KOH(16.6 g, 296 mmol)의 혼합물을 실온에서 밤새 교반하였다. 얻어진 혼합물을 6 N HCl로 중화시키고, 감압 하에서 농축하여 휘발성 물질을 제거하였다. 잔부를 DCM과 H2O 사이로 분배하고, 층들을 분리하였다. 유기층을 염수로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 감압 하에서 농축하여 중간체 1(27.4 g, 81% 수율)을 수득하고, 이를 추가 정제 없이 다음 단계에서 바로 사용하였다. LC/MS: m/z 이론치 456.4, 실험치 457.5(M + 1)+.
단계 B: 중간체 2:
(3aR,5aR,5bR,7aR,11aR,11bR,13aS)-1-이소프로필-5a,5b,8,8,11a-펜타메틸-2,9-디옥소-3,3a,4,5,5a,5b,6,7,7a,8,9,10,11,11a,11b,12,13,13a-옥타데카하이드로-2H-사이클로펜타[a]크리센-3a-카르브알데하이드

DCM(20 mL) 중 중간체 1(1 g, 2.2 mmol) 및 PCC(940 mg, 4.4 mmol)의 혼합물을 실온에서 밤새 교반하였다. 얻어진 혼합물을 DCM으로 희석시키고, 셀라이트 패드를 통해 여과하였다. 여액을 감압 하에서 농축하여 생성물을 수득하고, 이를 플래시 크로마토그래피(실리카 겔, PE 중 0 내지 30% EtOAc)에 의해 정제하여 중간체 2(398 mg, 40% 수율)를 백색 고체로서 수득하였다. LC/MS: m/z 이론치 452.3, 실험치 453.5 (M + 1)+.
단계 C: 중간체 3:
(3aR,5aR,5bR,7aR,11aR,11bR,13aS)-1-이소프로필-5a,5b,8,8,11a-펜타메틸-2,9-디옥소-3,3a,4,5,5a,5b,6,7,7a,8,9,10,11,11a,11b,12,13,13a-옥타데카하이드로-2H-사이클로펜타[a]크리센-3a-카복실산

t-BuOH(20 mL), H2O(30 mL), 및 THF(25 mL) 중 중간체 2(3 g, 6.6 mmol), NaH2PO4(4.8 g, 40 mmol), NaClO2(3.6 g, 40 mmol)의 혼합물을 이소부틴(15 mL)으로 처리하였다. 실온에서 2시간 동안 교반한 후에, 얻어진 혼합물을 H2O로 희석시키고, EtOAc로 추출하였다. 유기층을 포화 Na2S2O3 및 염수로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 감압 하에서 농축하여 미정제 생성물을 수득하고, 이를 플래시 크로마토그래피(실리카 겔, PE 중 0 내지 50% EtOAc)에 의해 정제하여 중간체 3(2.3 g, 74% 수율)을 백색 고체로서 수득하였다. 1H NMR (400 MHz, CDCl3) δ 10.37 (br, 1H), 3.27 - 3.15 (m, 1H), 2.79 (dd, J = 12.7, 3.0 Hz, 1H), 2.66 - 2.41 (m, 4H), 2.22 (d, J = 18.7 Hz, 1H), 2.09 - 1.86 (m, 4H), 1.65 - 1.21 (m, 18H), 1.11 - 0.96 (m, 14H). LC/MS: m/z 이론치 468.3, 실험치 469.4 (M + 1)+.
단계 D: 중간체 4:
(3aR,5aR,5bR,7aR,11aR,11bR,13aS)-3a-아미노-1-이소프로필-5a,5b,8,8,11a-펜타메틸-3a,4,5,5a,5b,6,7,7a,8,10,11,11a,11b,12,13,13a-헥사데카하이드로-2H-사이클로펜타[a]크리센-2,9(3H)-디온

톨루엔(6 mL) 중 중간체 3(600 mg, 1.28 mmol) 및 TEA(0.21 mL, 1.53 mmol)의 용액에 DPPA(422 mg, 1.53 mmol)를 첨가하였다. 반응을 실온에서 30분 동안 교반하고, 이후에, 감압 하에서 농축하였다. 잔부를 EtOAc와 H2O 사이로 분배하였다. 유기층을 염수로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 감압 하에서 농축하여 미정제 아실 아지드 중간체를 수득하고, 이를 톨루엔(10 mL)에 용해시키고, 80℃에서 30분 동안 가열하여 이소시아네이트를 수득하였다. 얻어진 혼합물을 농축하고, 잔부를 DCM(5 mL) 중 진한 HCl(5 mL)로 처리하였다. 혼합물을 실온에서 밤새 교반하고, 감압 하에서 농축하여 잔부를 수득하고, 이를 EtOH에서 분쇄하여 여과 후 중간체 4(278 mg, 49% 수율)를 백색 고체로서 수득하였다. LC/MS: m/z 이론치 439.4, 실험치 440.3 (M + 1)+.
단계 E: 중간체 5:
3차-부틸 ((3aR,5aR,5bR,7aR,11aR,11bR,13aS)-1-이소프로필-5a,5b,8,8,11a-펜타메틸-2,9-디옥소-3,3a,4,5,5a,5b,6,7,7a,8,9,10,11,11a,11b,12,13,13a-옥타데카하이드로-2H-사이클로펜타[a]크리센-3a-일)카바메이트
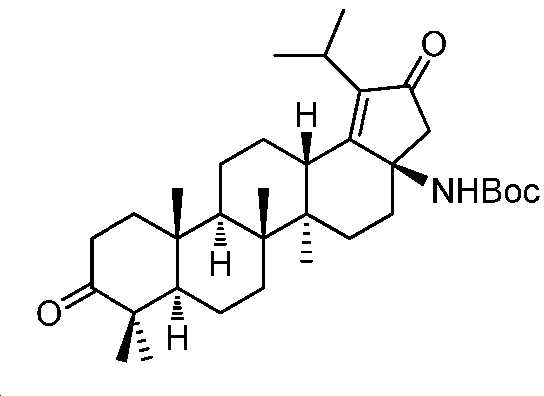
디옥산(20 mL) 및 1 N NaOH(9 mL) 중 중간체 4(2 g, 4.5 mmol)의 용액에 Boc2O(1.7 g, 7.7 mmol)를 첨가하였다. 반응을 실온에서 밤새 교반하고, 이후에, 1 N HCl로 중화시키고, EtOAc로 추출하였다. 유기층을 염수로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 감압 하에서 농축하여 잔부를 수득하고, 실리카겔 크로마토그래피(0 내지 50% EtOAc/PE)에 의해 정제하여 중간체 5(1.9 g, 77% 수율)를 백색 고체로서 수득하였다. LC/MS: m/z 이론치 539.4, 실험치 540.4 (M + 1)+.
단계 F: 중간체 6:
(3aR,5aR,5bR,7aR,11aR,11bR,13aS)-3a-((3차-부톡시카보닐)아미노)-1-이소프로필-5a,5b,8,8,11a-펜타메틸-2-옥소-3,3a,4,5,5a,5b,6,7,7a,8,11,11a,11b,12,13,13a-헥사데카하이드로-2H-사이클로펜타[a]크리센-9-일 트리플루오로메탄설포네이트

-78℃에서, 무수 THF(16 mL) 중 중간체 5(1.6 g, 3.0 mmol)의 용액에 K-HMDS(1 M, 13.5 mL, 13.5 mmol)를 N2 분위기 하에서 적가하였다. 반응을 -78℃에서 2시간 동안 교반하고, 이후에, 무수 THF(8 mL) 중 PhNTf2(1.3 g, 3.7 mmol)의 용액을 적가하였다. 반응을 -78℃에서 다른 2시간 동안 교반하고, 실온까지 서서히 가온시켰다. 얻어진 혼합물을 NH4Cl 포화수용액으로 켄칭시키고, EtOAc로 추출하였다. 유기층을 염수로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 감압 하에서 농축하여 잔부를 수득하고, 이를 플래시 크로마토그래피(실리카겔, PE 중 0 내지 15% EtOAc)에 의해 정제하여 중간체 6(1.5 g, 75% 수율)을 백색 고체로서 수득하였다. LC/MS: m/z 이론치 671.4, 실험치 672.3 (M + 1)+. 1H NMR (400 MHz, CDCl3) δ 5.59 (dd, J = 6.7, 1.9 Hz, 1H), 4.60 (s, 1H), 3.12 (t, J = 13.9, 7.0 Hz, 1H), 2.90 (d, J = 11.9 Hz, 1H), 2.66 (d, J = 18.6 Hz, 1H), 2.37 - 2.20 (m, 3H), 2.06 - 1.80 (m, 5H), 1.58 - 1.40 (m, 17H), 1.29 - 1.13 (m, 14H), 1.05 - 0.91 (m, 8H).
중간체 8의 합성.
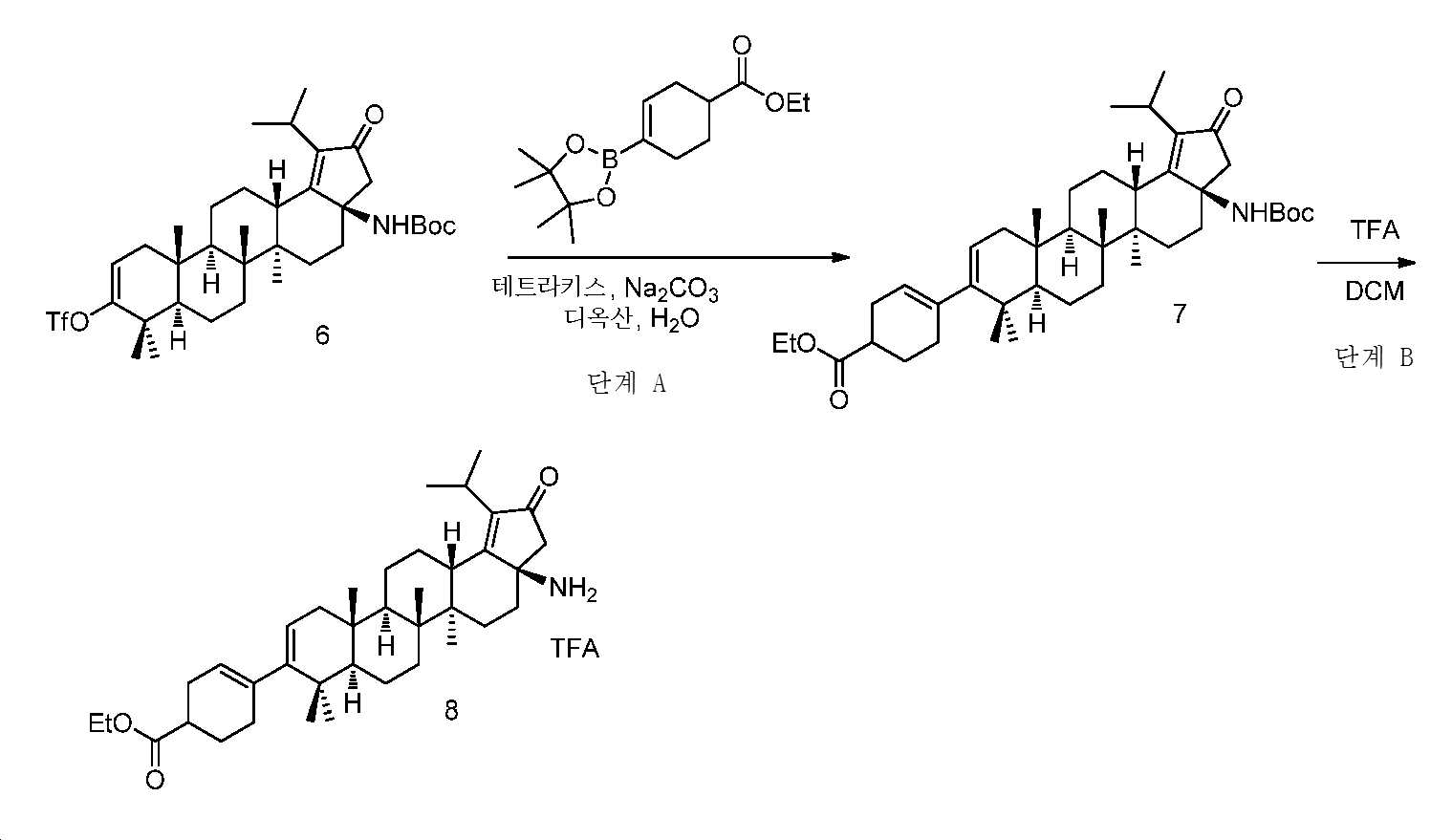
단계 A: 중간체 7:
에틸 4-((3aR,5aR,5bR,7aR,11aS,11bR,13aS)-3a-((3차-부톡시카보닐)아미노)-1-이소프로필-5a,5b,8,8,11a-펜타메틸-2-옥소-3,3a,4,5,5a,5b,6,7,7a,8,11,11a,11b,12,13,13a-헥사데카하이드로-2H-사이클로펜타[a]크리센-9-일)사이클로헥스-3-엔카복실레이트

디옥산(2 mL) 및 H2O(0.5 mL) 중 중간체 6(50 mg, 0.07 mmol), 에틸 4-(4,4,5,5-테트라메틸-1,3,2-디옥사보롤란-2-일)사이클로헥스-3-엔-1-카복실레이트(22 mg, 0.08 mmol), 테트라키스(5 mg, 0.0037 mmol) 및 Na2CO3(24 mg, 0.22 mmol)의 혼합물을 N2로 3회 퍼징하였다. 혼합물을 85℃에서 4시간 동안 가열하고, 얻어진 혼합물을 셀라이트 패드를 통해 여과하고, 여액을 EtOAc와 H2O 사이로 분배하였다. 유기층을 염수로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 감압 하에서 농축하여 잔부를 수득하고, 이를 실리카겔 크로마토그래피(0 내지 15% EtOAc/PE)에 의해 정제하여 중간체 7(45 mg, 89% 수율)을 백색 고체로서 수득하였다. LC/MS: m/z 이론치 675.5, 실험치 676.4 (M + 1)+.
단계 B: 중간체 8:
에틸 4-((3aR,5aR,5bR,7aR,11aS,11bR,13aS)-3a-아미노-1-이소프로필-5a,5b,8,8,11a-펜타메틸-2-옥소-3,3a,4,5,5a,5b,6,7,7a,8,11,11a,11b,12,13,13a-헥사데카하이드로-2H-사이클로펜타[a]크리센-9-일)사이클로헥스-3-엔카복실레이트 트리플루오로아세트산 염

TFA(2 mL) 및 DCM(2 mL) 중 중간체 7(70 mg, 0.10 mmol)의 용액을 실온에서 2.5시간 동안 교반하였다. 얻어진 혼합물을 감압 하에서 농축하여 중간체 8(정량 수율)을 백색 고체로서 수득하고, 이를 정제 없이 다음 단계에서 사용하였다. LC/MS: m/z 이론치 575.4, 실험치 576.4 (M + 1)+.
실시예 1
(화합물 10):
4-((3aR,5aR,5bR,7aR,11aS,11bR,13aS)-1-이소프로필-5a,5b,8,8,11a-펜타메틸-3a-((2-(4-(메틸설포닐)피페리딘-1-일)에틸)아미노)-2-옥소-3,3a,4,5,5a,5b,6,7,7a,8,11,11a,11b,12,13,13a-헥사데카하이드로-2H-사이클로펜타[a]크리센-9-일)사이클로헥스-3-엔카복실산
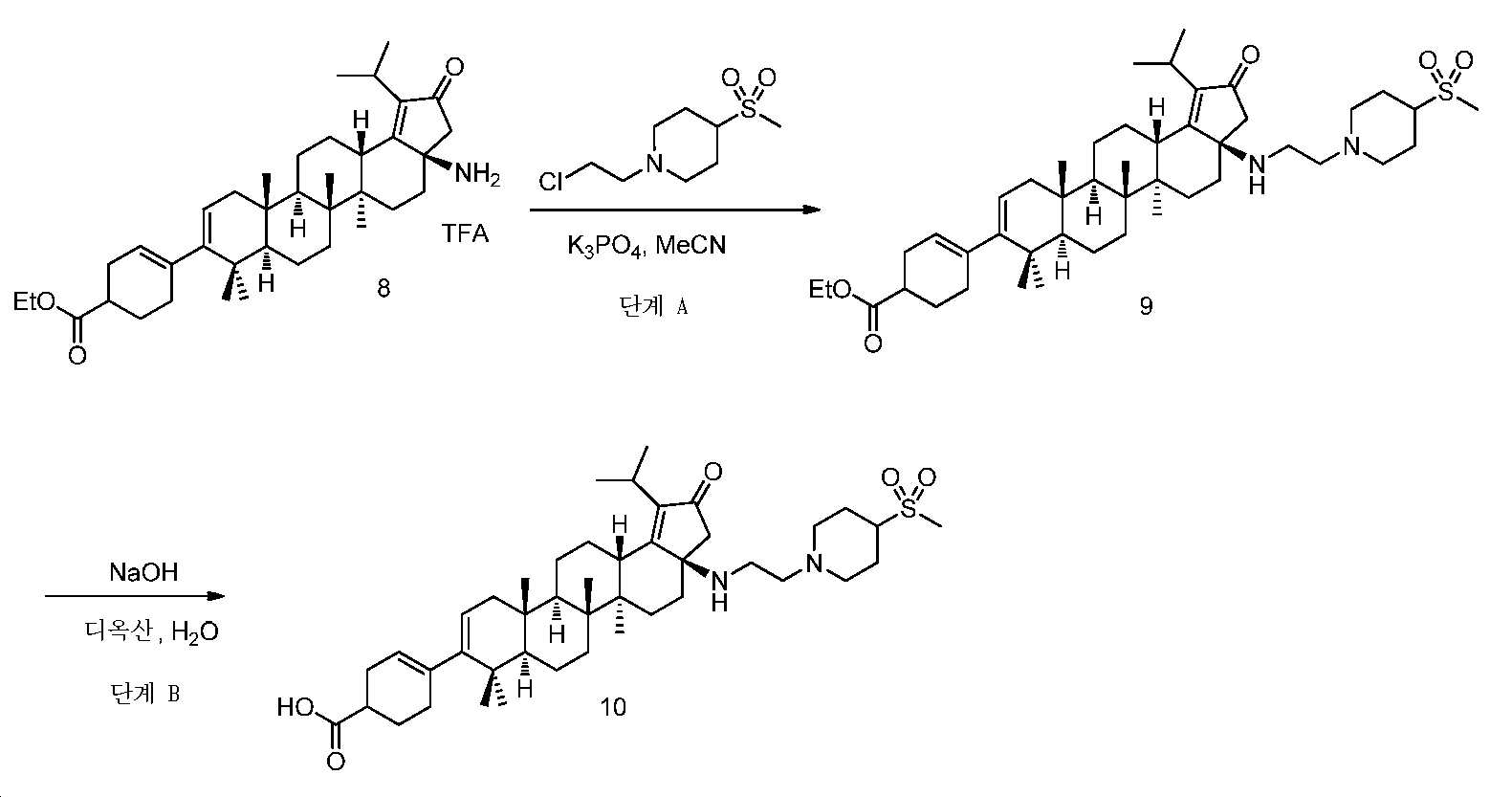
단계 A: 중간체 9:
에틸 4-((3aR,5aR,5bR,7aR,11aS,11bR,13aS)-1-이소프로필-5a,5b,8,8,11a-펜타메틸-3a-((2-(4-(메틸설포닐)피페리딘-1-일)에틸)아미노)-2-옥소-3,3a,4,5,5a,5b,6,7,7a,8,11,11a,11b,12,13,13a-헥사데카하이드로-2H-사이클로펜타[a]크리센-9-일)사이클로헥스-3-엔카복실레이트
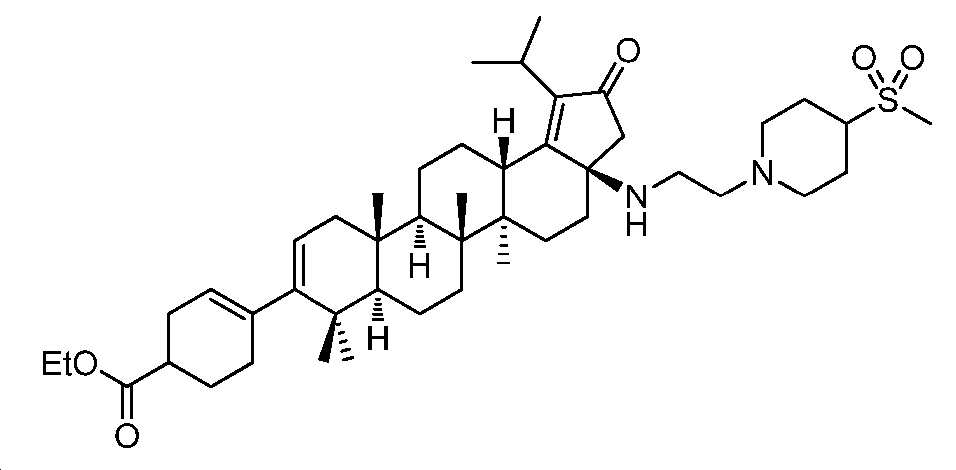
무수 MeCN(1.0 mL) 중 중간체 8(35 mg, 0.06 mmol), 1-(2-클로로에틸)-4-(메틸설포닐)피페리딘(55 mg, 0.12 mmol) 및 K3PO4(64 mg, 0.3 mmol)의 혼합물을 최대 100℃까지 가열하고, N2 분위기 하에서 밤새 교반하였다. 얻어진 혼합물을 EtOAc와 H2O 사이로 분배하였다. 유기층을 염수로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 감압 하에서 농축하여 잔부를 수득하고, 이를 실리카겔 크로마토그래피(0 내지 5% MeOH/DCM)에 의해 정제하여 중간체 9(16 mg, 35% 수율)를 황색 오일로서 수득하였다. LC/MS: m/z 이론치 764.5, 실험치 765.5 (M + 1)+.
단계 B: 화합물 10(실시예 1:
4-((3aR,5aR,5bR,7aR,11aS,11bR,13aS)-1-이소프로필-5a,5b,8,8,11a-펜타메틸-3a-((2-(4-(메틸설포닐)피페리딘-1-일)에틸)아미노)-2-옥소-3,3a,4,5,5a,5b,6,7,7a,8,11,11a,11b,12,13,13a-헥사데카하이드로-2H-사이클로펜타[a]크리센-9-일)사이클로헥스-3-엔카복실산
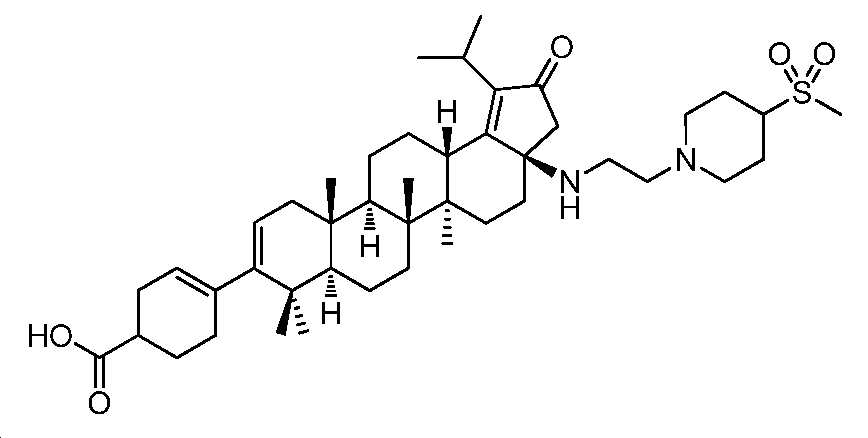
디옥산(1 mL) 및 1 N NaOH(0.1 mL) 중 중간체 9(16 mg, 0.02 mmol)의 혼합물을 75℃에서 밤새 교반하였다. 반응을 냉각시키고, 이후에, EtOAc로 희석시키고, 1 N HCl로 pH 3 내지 4까지 산성화시켰다. 층들을 분리하고, 유기층을 염수로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 감압 하에서 농축하여 잔부를 수득하고, 이를 역상 크로마토그래피(5 내지 100% MeCN/H2O + 0.1% FA)에 의해 정제하여 화합물 10(4.4 mg, 30% 수율)을 수득하였다. LC/MS: m/z 이론치 736.5, 실험치 737.4 (M + 1)+. 1H NMR (400 MHz, DMSO) δ 8.23 (s, 1H), 5.50 - 5.16 (m, J = 91.8, 40.0 Hz, 2H), 3.23 - 2.76 (m, 8H), 2.50 - 0.71 (m, 57H).
실시예 2
(화합물 12): 4
-((3aR,5aR,5bR,7aR,11aS,11bR,13aS)-3a-((2-(1,1-디옥시도티오모르폴리노)에틸)아미노)-1-이소프로필-5a,5b,8,8,11a-펜타메틸-2-옥소-3,3a,4,5,5a,5b,6,7,7a,8,11,11a,11b,12,13,13a-헥사데카하이드로-2H-사이클로펜타[a]크리센-9-일)사이클로헥스-3-엔카복실산

단계 A: 중간체 11:
에틸 4-((3aR,5aR,5bR,7aR,11aS,11bR,13aS)-3a-((2-(1,1-디옥시도티오모르폴리노)에틸)아미노)-1-이소프로필-5a,5b,8,8,11a-펜타메틸-2-옥소-3,3a,4,5,5a,5b,6,7,7a,8,11,11a,11b,12,13,13a-헥사데카하이드로-2H-사이클로펜타[a]크리센-9-일)사이클로헥스-3-엔카복실레이트

무수 MeCN(2.0 mL) 중 중간체 8(35 mg, 0.06 mmol), 4-(2-클로로에틸)티오모르폴린 1,1-디옥사이드(18 mg, 0.1 mmol) 및 K3PO4(64 mg, 0.3 mmol)의 혼합물을 최대 100℃까지 가열하고, N2 분위기 하에서 밤새 교반하였다. 얻어진 혼합물을 EtOAc와 H2O 사이로 분배하였다. 유기층을 염수로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 감압 하에서 농축하여 잔부를 수득하고, 이를 실리카겔 크로마토그래피(0 내지 5% MeOH/DCM)에 의해 정제하여 중간체 11(8 mg, 18% 수율)을 황색 오일로서 수득하였다. LC/MS: m/z 이론치 736.5, 실험치 737.5 (M + 1)+.
단계 B: 화합물 12:
4-((3aR,5aR,5bR,7aR,11aS,11bR,13aS)-3a-((2-(1,1-디옥시도티오모르폴리노)에틸)아미노)-1-이소프로필-5a,5b,8,8,11a-펜타메틸-2-옥소-3,3a,4,5,5a,5b,6,7,7a,8,11,11a,11b,12,13,13a-헥사데카하이드로-2H-사이클로펜타[a]크리센-9-일)사이클로헥스-3-엔카복실산

디옥산(1 mL) 및 1 N NaOH(0.05 mL) 중 중간체 11(8 mg, 0.01 mmol)의 혼합물을 75℃에서 밤새 교반하였다. 반응을 실온까지 냉각시키고, EtOAc로 희석시키고, 이후에, 1 N HCl로 pH 3 내지 4까지 산성화시켰다. 층들을 분리하고, 유기층을 염수로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 감압 하에서 농축하여 잔부를 수득하고, 이를 역상 크로마토그래피(5 내지 100% MeCN/H2O + 0.1% FA)에 의해 정제하여 화합물 12(5 mg, 65% 수율)를 백색 분말로서 수득하였다. LC/MS: m/z 이론치 708.5, 실험치 709.3 (M + 1)+. 1H NMR (400 MHz, DMSO) δ 8.33 (s, 1H), 5.55 - 5.13 (m, J = 91.5, 40.0 Hz, 2H), 3.18 - 3.00 (m, J = 16.2 Hz, 6H), 2.99 - 2.83 (m, 4H), 2.50 - 0.71 (m, 51H).
중간체 15의 합성

단계 A: 중간체 13:
3차-부틸 (2-(디메틸아미노)에틸)카바메이트
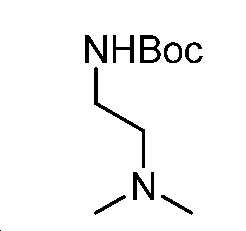
DCM(30 mL) 중 N1,N1-디메틸에탄-1,2-디아민(2 g, 23 mmol)의 용액을 Boc2O(5.9 g, 27 mmol)로 처리하였다. 실온에서 1시간 동안 교반한 후에, 얻어진 혼합물을 감압 하에서 농축하여 잔부를 수득하고, 이를 실리카겔 크로마토그래피(0 내지 10% MeOH/DCM)에 의해 정제하여 중간체 13(4.2 g, 98% 수율)을 무색 오일로서 수득하였다. LC/MS: m/z 이론치 188.2, 실험치 189.2 (M + 1)+.
단계 B: 중간체 14:
3차-부틸 4-클로로벤질(2-(디메틸아미노)에틸)카바메이트
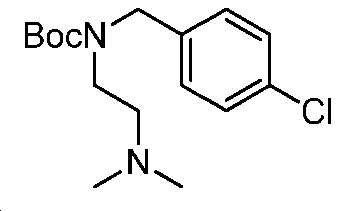
0℃에서, DMF(20 mL) 중 중간체 13(1 g, 5.3 mmol)의 용액에 NaH(60%, 255 mg, 6.4 mmol)를 첨가하였다. 얻어진 혼합물을 실온에서 1시간 동안 교반하고, 이후에, 1-(브로모메틸)-4-클로로벤젠(1.4 g, 6.9 mmol)으로 처리하였다. 실온에서 30분 동안 교반한 후에, 반응 혼합물을 포화 NH4Cl로 켄칭시키고, EtOAc로 추출하였다. 층들을 분리하고, 유기층을 염수로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 감압 하에서 농축하여 중간체 14를 수득하고, 이를 추가 정제 없이 다음 단계에서 사용하였다. LC/MS: m/z 이론치 312.2, 실험치 313.2 (M + 1)+.
단계 C: 중간체 15:
N
1
-(4-클로로벤질)-N
2
,N
2
-디메틸에탄-1,2-디아민 디하이드로클로라이드
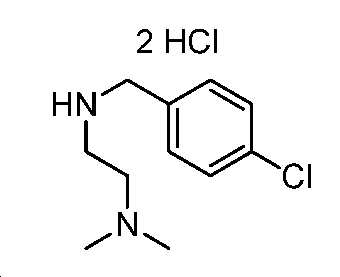
중간체 14를 디옥산(10 mL) 중 4 N HCl로 처리하였다. 실온에서 밤새 교반한 후에, 반응 혼합물을 감압 하에서 농축하여 잔부를 수득하고, 이를 에테르로 분쇄하고, 여과하여 중간체 15(600 mg, 2 단계에 걸쳐 40% 수율, 2HCl 염)를 백색 고체로서 수득하였다. 1H NMR (400 MHz, DMSO) δ 11.10 (s, 1H), 10.05 (s, 2H), 7.65 (d, J = 8.4 Hz, 2H), 7.51 (d, J = 8.4 Hz, 2H), 4.20 (s, 2H), 3.56 - 3.39 (m, 4H), 2.83 (s, 6H). LC/MS: m/z 이론치 212.1, 실험치 213.2 (M + 1)+.
실시예 3
(화합물 18):
4-((3aR,5aR,5bR,7aR,11aS,11bR,13aS)-3a-((2-((4-클로로벤질)(2-(디메틸아미노)에틸)아미노)에틸)아미노)-1-이소프로필-5a,5b,8,8,11a-펜타메틸-2-옥소-3,3a,4,5,5a,5b,6,7,7a,8,11,11a,11b,12,13,13a-헥사데카하이드로-2H-사이클로펜타[a]크리센-9-일)사이클로헥스-3-엔카복실산
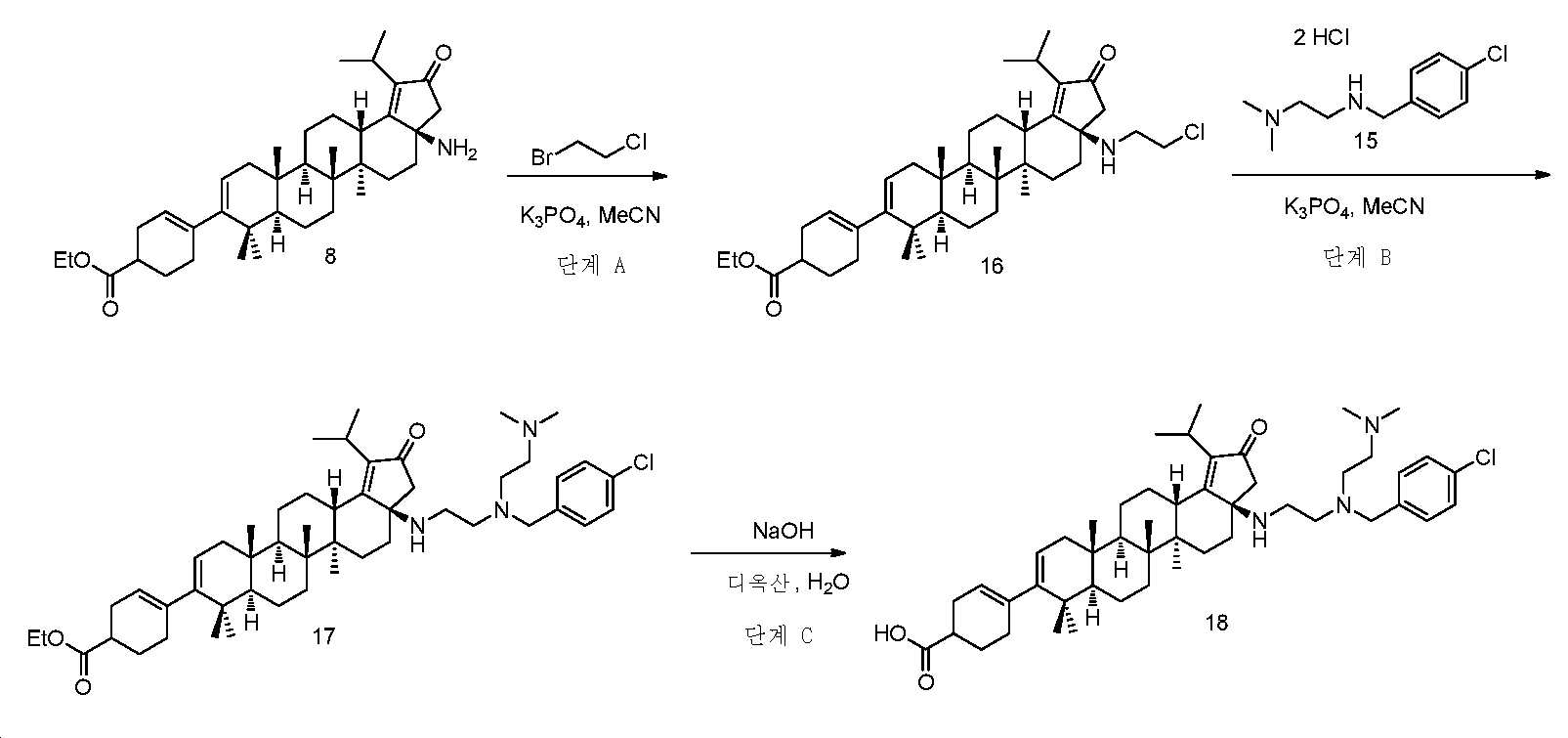
단계 A: 중간체 16:
에틸 4-((3aR,5aR,5bR,7aR,11aS,11bR,13aS)-3a-((2-클로로에틸)아미노)-1-이소프로필-5a,5b,8,8,11a-펜타메틸-2-옥소-3,3a,4,5,5a,5b,6,7,7a,8,11,11a,11b,12,13,13a-헥사데카하이드로-2H-사이클로펜타[a]크리센-9-일)사이클로헥스-3-엔카복실레이트

1-브로모-2-클로로에탄(8 mL) 및 무수 MeCN(1 mL) 중 중간체 8(860 mg, 1.5 mmol) 및 K3PO4(1.9 g, 9.0 mmol)의 현탁액을 N2 분위기 하, 100℃에서 밤새 교반하였다. 반응을 실온까지 냉각시키고, DCM과 H2O 사이로 분배하였다. 유기층을 염수로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 감압 하에서 농축하여 잔부를 수득하고, 이를 실리카겔 크로마토그래피(0 내지 50% EtOAc/PE)에 의해 정제하여 중간체 16(200 mg, 21% 수율)을 백색 고체로서 수득하였다. LC/MS: m/z 이론치 637.4, 실험치 638.8 (M + 1)+.
단계 B: 중간체 17: 에틸 4-((3aR,5aR,5bR,7aR,11aS,11bR,13aS)-3a-((2-((4-클로로벤질)(2-(디메틸아미노)에틸)아미노)에틸)아미노)-1-이소프로필-5a,5b,8,8,11a-펜타메틸-2-옥소-3,3a,4,5,5a,5b,6,7,7a,8,11,11a,11b,12,13,13a-헥사데카하이드로-2H-사이클로펜타[a]크리센-9-일)사이클로헥스-3-엔카복실레이트

무수 MeCN(4 mL) 중 중간체 16(200 mg, 0.31 mmol), 중간체 15(167 mg, 0.78 mmol) 및 K3PO4(532 mg, 2.5 mmol)의 현탁액을 N2 분위기 하, 100℃에서 밤새 교반하였다. 반응을 실온까지 냉각시키고, EtOAc와 H2O 사이로 분배하였다. 유기층을 염수로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 감압 하에서 농축하여 잔부를 수득하고, 이를 실리카겔 크로마토그래피(0 내지 10% MeOH/DCM)에 의해 정제하여 중간체 17(83 mg, 32% 수율)을 백색 고체로서 수득하였다. LC/MS: m/z 이론치 813.6, 실험치 814.9 (M + 1)+.
단계 C: 화합물 18:
4-((3aR,5aR,5bR,7aR,11aS,11bR,13aS)-3a-((2-((4-클로로벤질)(2-(디메틸아미노)에틸)아미노)에틸)아미노)-1-이소프로필-5a,5b,8,8,11a-펜타메틸-2-옥소-3,3a,4,5,5a,5b,6,7,7a,8,11,11a,11b,12,13,13a-헥사데카하이드로-2H-사이클로펜타[a]크리센-9-일)사이클로헥스-3-엔카복실산

디옥산(1 mL) 및 1 N NaOH(1 mL) 중 중간체 17(80 mg, 0.1 mmol)의 혼합물을 75℃에서 밤새 교반하였다. 반응을 실온까지 냉각시키고, EtOAc로 희석시키고, 이후에, 1 N HCl로 pH 3 내지 4까지 산성화시켰다. 층들을 분리하고, 유기층을 염수로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 감압 하에서 농축하여 잔부를 수득하고, 이를 역상 크로마토그래피(60 내지 100% MeCN/H2O + 0.1% FA)에 의해 정제하여 화합물 18(18 mg, 23% 수율)을 백색 분말로서 수득하였다. LC/MS: m/z 이론치 785.5, 실험치 786.9 (M + 1)+. 1H NMR (400 MHz, 몇 방울의 MeOH를 함유한 CDCl3) δ 7.30 - 7.19 (m, 4H), 5.37 (s, 1H), 5.21 (d, J = 5.5 Hz, 1H), 3.67 - 3.52 (m, J = 7.3 Hz, 2H), 3.42 - 3.39 (m, 1H), 3.20 - 3.10 (m, 1H), 2.95 (d, J = 12.2 Hz, 1H), 2.61 - 0.91 (m, 59H).
중간체 20의 합성
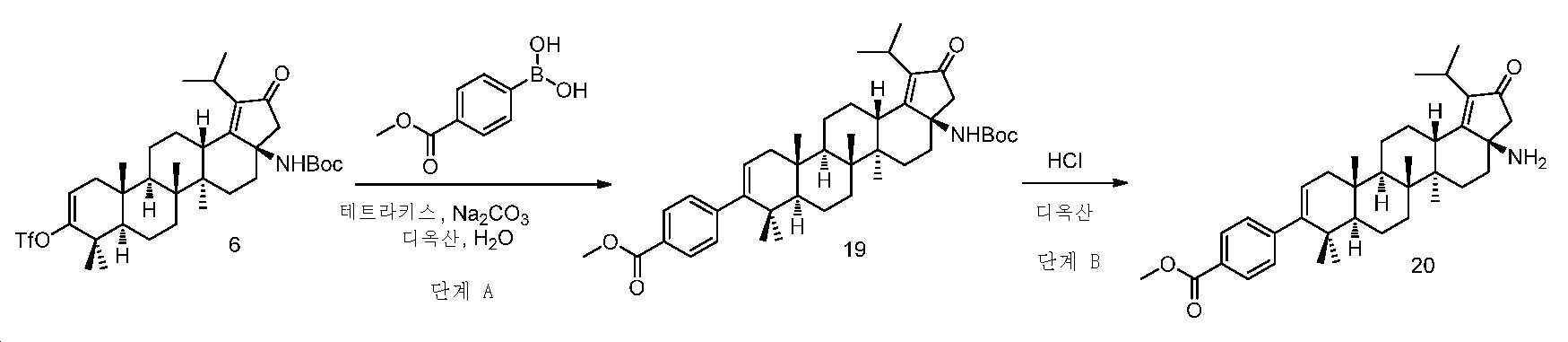
단계 A: 중간체 19:
메틸 4-((3aR,5aR,5bR,7aR,11aS,11bR,13aS)-3a-((3차-부톡시카보닐)아미노)-1-이소프로필-5a,5b,8,8,11a-펜타메틸-2-옥소-3,3a,4,5,5a,5b,6,7,7a,8,11,11a,11b,12,13,13a-헥사데카하이드로-2H-사이클로펜타[a]크리센-9-일)벤조에이트
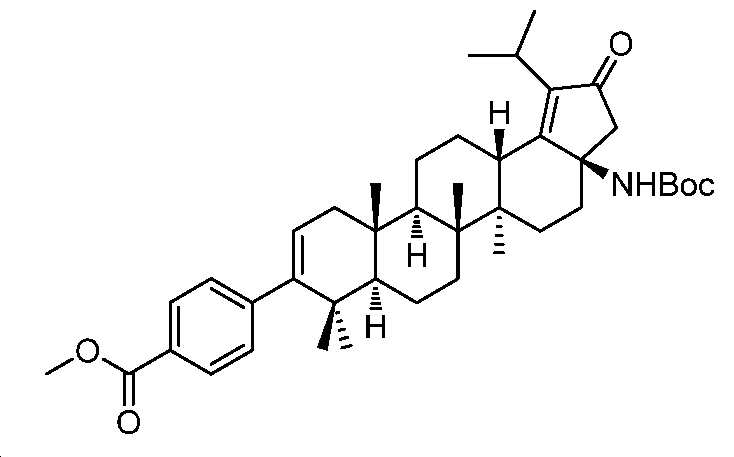
디옥산(10 mL) 및 H2O(2.5 mL) 중 중간체 6(1.5 g, 2.2 mmol), (4-(메톡시카보닐)페닐)보론산(603 mg, 3.3 mmol), 테트라키스(516 mg, 0.44 mmol) 및 Na2CO3(710 mg, 6.7 mmol)의 혼합물을 N2로 3회 퍼징하였다. 반응을 85℃에서 밤새 교반하고, 실온까지 냉각시키고, 셀라이트 패드를 통해 여과하고, 여액을 EtOAc와 H2O 사이로 분배하였다. 유기층을 염수로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 감압 하에서 농축하여 잔부를 수득하고, 이를 실리카겔 크로마토그래피(0 내지 40% EtOAc/PE)에 의해 정제하여 중간체 19(700 mg, 48% 수율)를 백색 고체로서 수득하였다. LC/MS: m/z 이론치 657.4, 실험치 658.8 (M + 1)+.
단계 B: 중간체 20:
메틸 4-((3aR,5aR,5bR,7aR,11aS,11bR,13aS)-3a-아미노-1-이소프로필-5a,5b,8,8,11a-펜타메틸-2-옥소-3,3a,4,5,5a,5b,6,7,7a,8,11,11a,11b,12,13,13a-헥사데카하이드로-2H-사이클로펜타[a]크리센-9-일)벤조에이트 하이드로클로라이드

4 N HCl/디옥산(7 mL) 및 DCM(7 mL) 중 중간체 19(700 mg, 1.1 mmol)의 혼합물을 실온에서 밤새 교반하였다. 얻어진 혼합물을 감압 하에서 농축하여 중간체 20(873 mg, 정량 수율)을 백색 고체로서 수득하고, 이를 추가 정제 없이 다음 단계에서 사용하였다. LC/MS: m/z 이론치 557.4, 실험치 558.7 (M + 1)+.
실시예 4
(화합물 21):
4-((3aR,5aR,5bR,7aR,11aS,11bR,13aS)-3a-((2-((4-클로로벤질)(2-(디메틸아미노)에틸)아미노)에틸)아미노)-1-이소프로필-5a,5b,8,8,11a-펜타메틸-2-옥소-3,3a,4,5,5a,5b,6,7,7a,8,11,11a,11b,12,13,13a-헥사데카하이드로-2H-사이클로펜타[a]크리센-9-일)벤조산 하이드로클로라이드 염

표제 화합물, 화합물 21을 중간체 20을 사용하여 실시예 3과 유사한 방식으로 제조하였다. 미정제 생성물을 역상 크로마토그래피(50 내지 100% MeCN/H2O + 0.1% FA)에 의해 정제하고, 이후에, 몇 방울의 디옥산 중 HCl로 처리하여 화합물 21(32 mg, 36% 수율)을 백색 분말로서 수득하였다. LC/MS: m/z 이론치 781.5, 실험치 782.7 (M + 1)+. 1H NMR (400 MHz, 몇 방울의 MeOH를 함유한 CDCl3) δ 7.94 (d, J = 7.9 Hz, 2H), 7.32 - 7.13 (m, J = 24.7, 16.6, 8.3 Hz, 6H), 5.32 (d, J = 5.0 Hz, 1H), 3.61 (q, J = 13.7 Hz, 2H), 3.27 - 3.07 (m, 10H), 3.01 - 2.57 (m, 7H), 2.47 - 0.94 (m, 38H).
실시예 5
(화합물 22):
4-((3aR,5aR,5bR,11aS)-3a-((2-(1,1-디옥시도티오모르폴리노)에틸)아미노)-1-이소프로필-5a,5b,8,8,11a-펜타메틸-2-옥소-3,3a,4,5,5a,5b,6,7,7a,8,11,11a,11b,12,13,13a-헥사데카하이드로-2H-사이클로펜타[a]크리센-9-일)벤조산 하이드로클로라이드
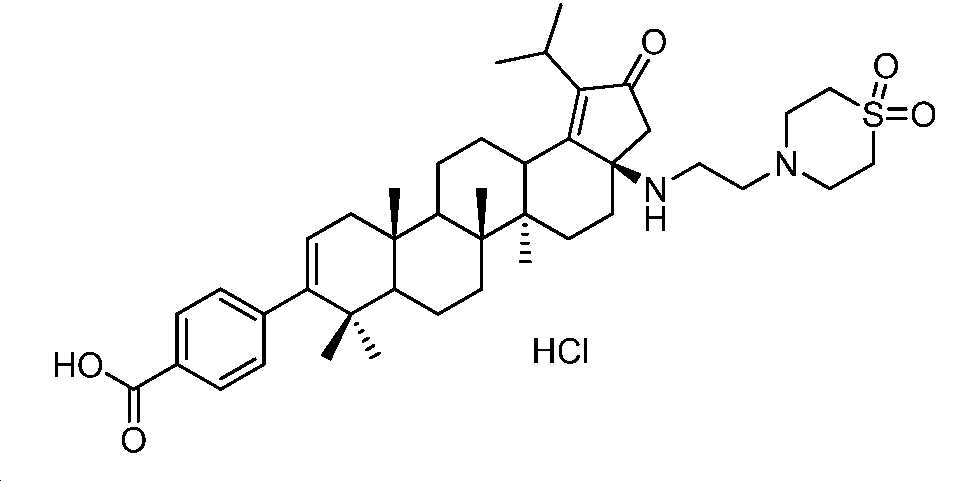
표제 화합물, 화합물 22를 중간체 20을 사용하여 실시예 2와 유사한 방식으로 제조하였다. 미정제 생성물을 역상 크로마토그래피(50 내지 100% MeCN/H2O + 0.1% FA)에 의해 정제하고, 이후에, 몇 방울의 디옥산 중 HCl로 처리하여 화합물 22(31 mg, 45% 수율)를 백색 분말로서 수득하였다. LC/MS: m/z 이론치 704.4, 실험치 705.2 (M + 1)+. 1H NMR (400 MHz, 몇 방울의 MeOH를 함유한 CDCl3) δ 7.96 (d, J = 8.2 Hz, 2H), 7.21 (d, J = 8.2 Hz, 2H), 5.32 (d, J = 4.9 Hz, 1H), 3.47 - 3.15 (m, 10H), 2.69 - 0.90 (m, 43H).
중간체 24의 합성

단계 A: 중간체 23:
3차-부틸 2-(1,1-디옥시도티오모르폴리노)아세테이트

무수 MeCN(5 mL) 중 티오모르폴린 1,1-디옥사이드(500 mg, 3.7 mmol), 3차-부틸 2-브로모아세테이트(1.1 g, 5.5 mmol), K3PO4(2.5 g, 1.07 mmol)의 혼합물을 N2 분위기 하, 85℃에서 밤새 교반하였다. 얻어진 혼합물을 EtOAc와 H2O 사이로 분배하였다. 유기층을 염수로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 감압 하에서 농축하여 잔부를 수득하고, 이를 실리카겔 크로마토그래피(0 내지 50% EtOAc/PE)에 의해 정제하여 중간체 23(485 mg, 53% 수율)을 무색 오일로서 수득하였다. LC/MS: m/z 이론치 249.1, 실험치 250.4 (M + 1)+.
단계 B: 중간체 24:
2-(1,1-디옥시도티오모르폴리노)아세트산 하이드로클로라이드 염

4 N HCl/디옥산(5 mL) 및 DCM(5 mL) 중 중간체 23(485 mg, 1.9 mmol)의 혼합물을 실온에서 밤새 교반하였다. 얻어진 혼합물을 감압 하에서 농축하여 중간체 24(432 mg, 정량 수율)를 백색 고체로서 수득하고, 이를 추가 정제 없이 사용하였다. LC/MS: m/z 이론치 193.0, 실험치 194.0 (M + 1)+.
실시예 6
(화합물 26):
4-((3aR,5aR,5bR,11aS)-3a-(2-(1,1-디옥시도티오모르폴리노)아세트아미도)-1-이소프로필-5a,5b,8,8,11a-펜타메틸-2-옥소-3,3a,4,5,5a,5b,6,7,7a,8,11,11a,11b,12,13,13a-헥사데카하이드로-2H-사이클로펜타[a]크리센-9-일)벤조산 하이드로클로라이드 염
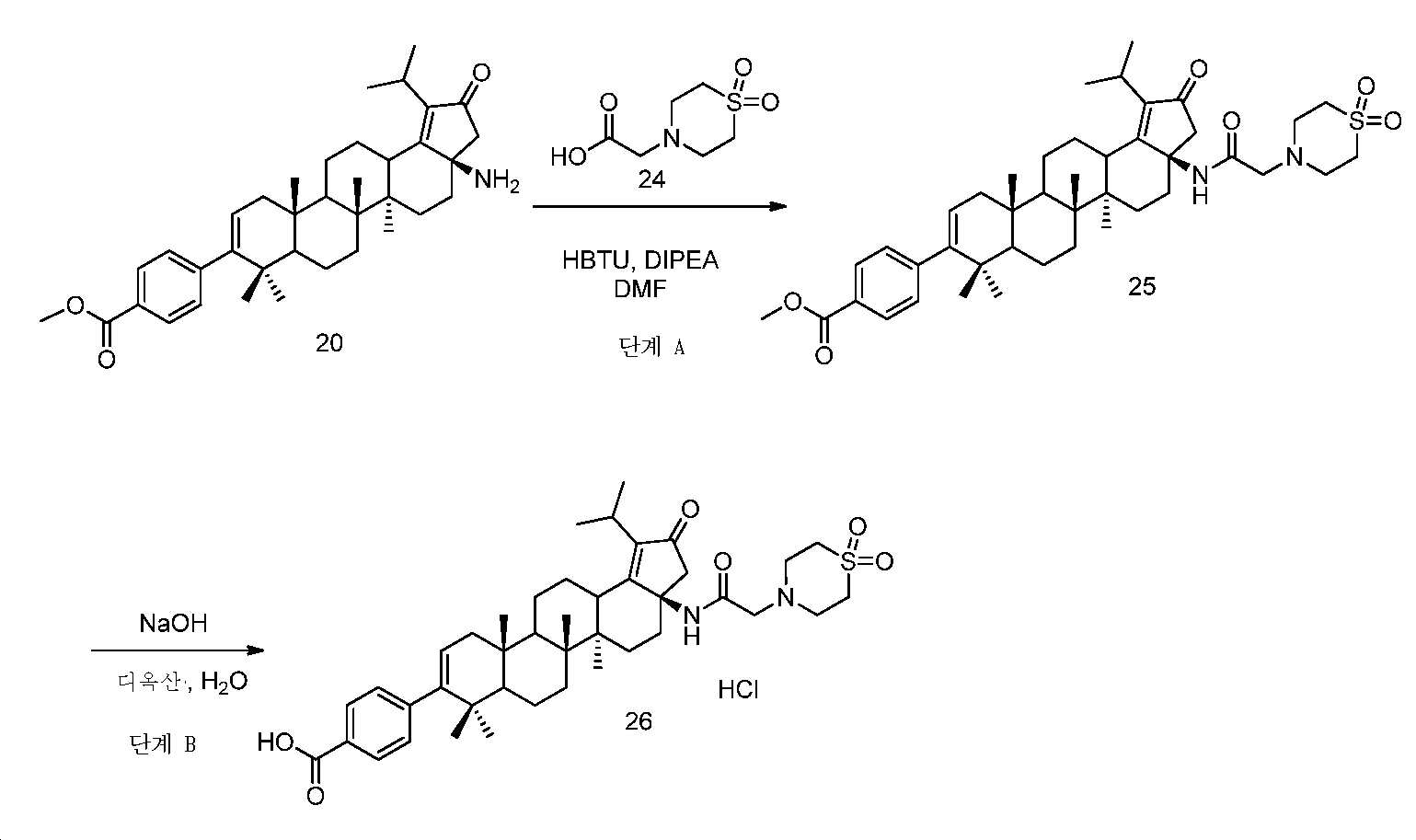
단계 A: 중간체 25:
메틸 4-((3aR,5aR,5bR,11aS)-3a-(2-(1,1-디옥시도티오모르폴리노)아세트아미도)-1-이소프로필-5a,5b,8,8,11a-펜타메틸-2-옥소-3,3a,4,5,5a,5b,6,7,7a,8,11,11a,11b,12,13,13a-헥사데카하이드로-2H-사이클로펜타[a]크리센-9-일)벤조에이트
DCM(2 mL) 중 중간체 20(100 mg, 0.18 mmol), 중간체 24, HBTU(104 mg, 0.27 mg) 및 DIPEA(0.23 mL, 1.25 mmol)의 혼합물을 실온에서 밤새 교반하였다. 얻어진 혼합물을 DCM과 H2O 사이로 분배하였다. 유기층을 염수로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 감압 하에서 농축하여 잔부를 실리카겔 크로마토그래피(0 내지 10% MeOH/DCM)에 의해 정제하여 중간체 25(70 mg, 53% 수율)를 백색 고체로서 수득하였다. LC/MS: m/z 이론치 732.4, 실험치 733.5 (M + 1)+.
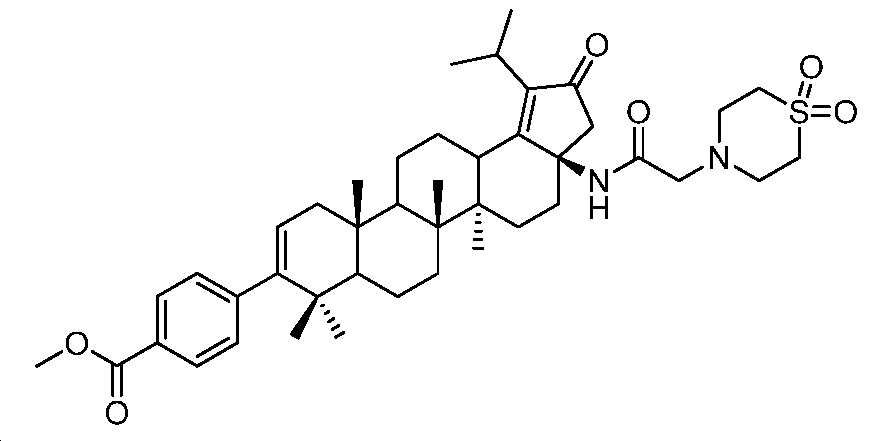
단계 B: 화합물 26:
4-((3aR,5aR,5bR,11aS)-3a-(2-(1,1-디옥시도티오모르폴리노)아세트아미도)-1-이소프로필-5a,5b,8,8,11a-펜타메틸-2-옥소-3,3a,4,5,5a,5b,6,7,7a,8,11,11a,11b,12,13,13a-헥사데카하이드로-2H-사이클로펜타[a]크리센-9-일)벤조산 하이드로클로라이드 염

표제 화합물, 화합물 26을 중간체 25(100 mg, 0.14 mmol)를 사용하여 실시예 1과 유사한 방식으로 제조하였다. 미정제 잔부를 역상 크로마토그래피(50 내지 100% MeCN/H2O + 0.1% FA)에 의해 정제하고, 몇 방울의 디옥산 중 HCl로 처리하여 화합물 26(37 mg, 38% 수율)을 백색 분말로서 수득하였다. LC/MS: m/z 이론치 718.4, 실험치 719.2 (M + 1)+. 1H NMR (400 MHz, 몇 방울의 MeOH를 함유한 CDCl3) δ 7.96 (d, J = 8.2 Hz, 2H), 7.21 (d, J = 8.2 Hz, 2H), 5.33 (d, J = 5.0 Hz, 1H), 3.74 - 3.40 (m, 8H), 3.19 (dt, J = 13.9, 6.9 Hz, 1H), 2.90 - 2.79 (m, 1H), 2.66 (d, J = 18.4 Hz, 1H), 2.34 - 0.89 (m, 40H).
실시예 7
(화합물 29)

단계 A: 중간체 27:
메틸 4-((3aR,5aR,5bR,11aS)-3a-이소시아네이토-1-이소프로필-5a,5b,8,8,11a-펜타메틸-2-옥소-3,3a,4,5,5a,5b,6,7,7a,8,11,11a,11b,12,13,13a-헥사데카하이드로-2H-사이클로펜타[a]크리센-9-일)벤조에이트
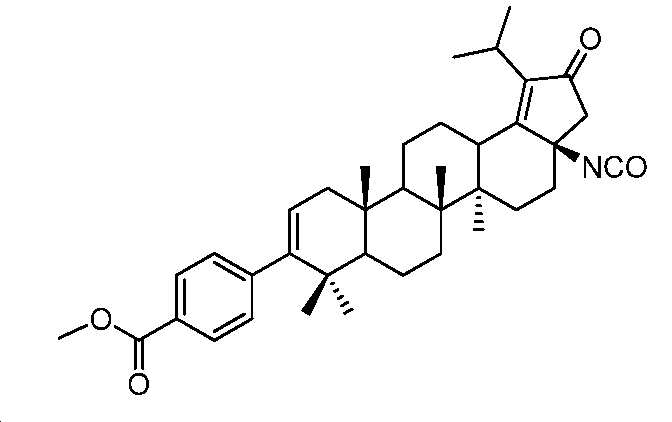
0℃에서, THF(1.2 mL) 중 메틸 4-((3aR,5aR,5bR,11aS)-3a-아미노-1-이소프로필-5a,5b,8,8,11a-펜타메틸-2-옥소-3,3a,4,5,5a,5b,6,7,7a,8,11,11a,11b,12,13,13a-헥사데카하이드로-2H-사이클로펜타[a]크리센-9-일)벤조에이트(120 mg, 0.215 mmol) 및 DIPEA(0.095 mL, 0.538 mmol)의 용액에 트리포스겐(127 mg, 0.43 mmol)을 첨가하였다. 실온에서 1시간 동안 교반한 후에, 얻어진 혼합물을 H2O로 켄칭시키고, EtOAc로 추출하였다. 유기층을 염수로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 감압 하에서 농축하여 미정제 생성물을 수득하고, 이를 플래시 크로마토그래피(실리카겔, PE 중 0 내지 10% EtOAc)에 의해 정제하여 표제 화합물(91 mg, 72% 수율)을 백색 고체로서 수득하였다. LC-MS (ESI): m/z (M+1) = 584.3.
단계 B: 중간체 28:
메틸 4-((3aR,5aR,5bR,11aS)-3a-(3-(4-클로로벤질)-3-(2-(디메틸아미노)에틸)우레이도)-1-이소프로필-5a,5b,8,8,11a-펜타메틸-2-옥소-3,3a,4,5,5a,5b,6,7,7a,8,11,11a,11b,12,13,13a-헥사데카하이드로-2H-사이클로펜타[a]크리센-9-일)벤조에이트

THF(1 mL) 중 메틸 4-((3aR,5aR,5bR,11aS)-3a-이소시아네이토-1-이소프로필-5a,5b,8,8,11a-펜타메틸-2-옥소-3,3a,4,5,5a,5b,6,7,7a,8,11,11a,11b,12,13,13a-헥사데카하이드로-2H-사이클로펜타[a]크리센-9-일)벤조에이트(91 mg, 0.156 mmol), N 1 -(4-클로로벤질)-N 2 ,N 2 -디메틸에탄-1,2-디아민(133 mg, 0.468 mmol) 및 DIPEA(0.21 mL, 1.25 mmol)의 용액을 실온에서 밤새 교반하였다. 얻어진 혼합물을 EtOAc와 H2O 사이로 분배하였다. 유기층을 염수로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 감압 하에서 농축하여 미정제 생성물을 수득하고, 이를 플래시 크로마토그래피(실리카겔, DCM 중 0 내지 10% MeOH)에 의해 정제하여 표제 화합물(70 mg, 57% 수율)을 백색 고체로서 수득하였다. LC-MS (ESI): m/z (M/M+2) = 796.5/798.5.
단계 C: 화합물 29:
4-((3aR,5aR,5bR,11aS)-3a-(3-(4-클로로벤질)-3-(2-(디메틸아미노)에틸)우레이도)-1-이소프로필-5a,5b,8,8,11a-펜타메틸-2-옥소-3,3a,4,5,5a,5b,6,7,7a,8,11,11a,11b,12,13,13a-헥사데카하이드로-2H-사이클로펜타[a]크리센-9-일)벤조산
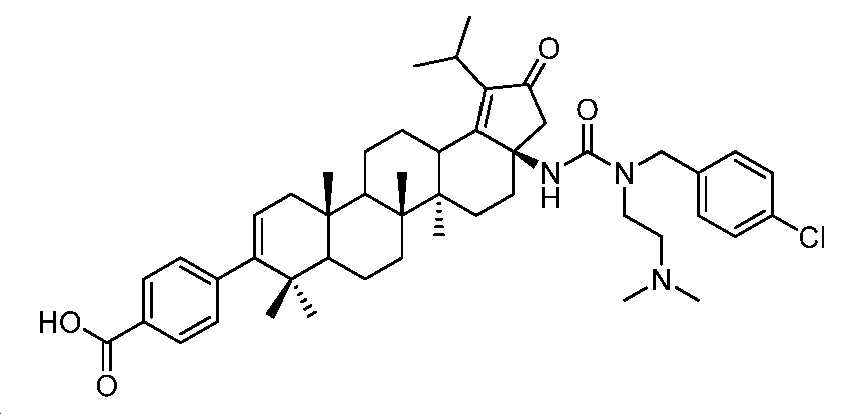
1 N NaOH(0.88 mL) 및 디옥산(1 mL) 중 메틸 4-((3aR,5aR,5bR,11aS)-3a-(3-(4-클로로벤질)-3-(2-(디메틸아미노)에틸)우레이도)-1-이소프로필-5a,5b,8,8,11a-펜타메틸-2-옥소-3,3a,4,5,5a,5b,6,7,7a,8,11,11a,11b,12,13,13a-헥사데카하이드로-2H-사이클로펜타[a]크리센-9-일)벤조에이트(70 mg, 0.088 mmol)의 혼합물을 75℃에서 1.5시간 동안 교반하였다. 얻어진 혼합물을 1 N HCl로 pH 3 내지 4까지 산성화시키고, EtOAc로 추출하였다. 유기층을 염수로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 감압 하에서 농축하여 미정제 생성물을 수득하고, 이를 HPLC(C18, 0.1% 포름산을 함유한 H2O 중 50 내지 100% MeCN)에 의해 정제하여 동결건조 후 표제 화합물(38 mg, 53% 수율)을 백색 분말로서 수득하였다(몇 방울의 디옥산 중 HCl을 LCMS에 의해 수집된 분획에 첨가하여 HCl 염을 수득함). LC-MS (ESI): m/z (M/M+2) = 782.4/784.4. 1H NMR (400 MHz, 몇 방울의 MeOH를 함유한 CDCl3) δ 7.95 (d, J = 8.2 Hz, 2H), 7.37 (d, J = 8.3 Hz, 2H), 7.20 (t, J = 8.9 Hz, 4H), 5.32 (d, J = 4.4 Hz, 1H), 4.60 (dd, J = 59.7, 17.4 Hz, 2H), 4.05 - 3.91 (m, 1H), 3.68 (dd, J = 14.1, 7.0 Hz, 1H), 3.60 - 3.48 (m, 1H), 3.42 - 3.34 (m, 2H), 3.29 - 3.11 (m, 3H), 2.95 - 2.59 (m, 8H), 2.40 - 0.52 (m, 35H).
실시예 8
(화합물 33)

단계 A: 중간체 30:
에틸 N-(4-클로로벤질)-N-(2-(디메틸아미노)에틸)글리시네이트

DCE(8 mL) 중 N 1 -(4-클로로벤질)-N 2 ,N 2 -디메틸에탄-1,2-디아민(800 mg, 3.76 mmol), 에틸 2-옥소아세테이트(50% 톨루엔 용액, 422 mg, 4.14 mmol) 및 NaBH3CN(331 mg, 5.27 mmol)의 혼합물을 실온에서 4시간 동안 교반하였다. 얻어진 혼합물을 NH4Cl 포화수용액으로 켄칭시키고, DCM/i-PrOH(85:15)로 추출하였다. 유기층을 염수로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 감압 하에서 농축하여 표제 화합물(200 mg, 18% 수율)을 황색 오일로서 수득하고, 이를 추가 정제 없이 다음 단계에서 사용하였다. LC-MS (ESI): m/z (M/M+2) = 299.3/301.4.
단계 B: 중간체 31:
N-(4-클로로벤질)-N-(2-(디메틸아미노)에틸)글리신
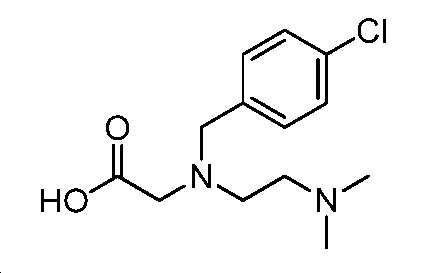
10 N NaOH(0.67 mL, 6.7 mmol) 및 디옥산(1 mL) 중 에틸 N-(4-클로로벤질)-N-(2-(디메틸아미노)에틸)글리시네이트(200 mg, 0.67 mmol)의 혼합물을 120℃에서 밤새 교반하였다. 얻어진 혼합물을 1 N HCl로 중화시키고, 감압 하에서 농축하여 미정제 생성물을 수득하고, 이를 HPLC(C18, 0.3% 포름산을 함유한 H2O 중 0 내지 100% MeOH)에 의해 정제하여 표제 화합물(65 mg, 36% 수율)을 무색 오일로서 수득하였다. LC-MS (ESI): m/z (M/M+2) = 271.2/273.1.
단계 C: 중간체 32:
메틸 4-((3aR,5aR,5bR,11aS)-3a-(2-((4-클로로벤질)(2-(디메틸아미노)에틸)아미노)아세트아미도)-1-이소프로필-5a,5b,8,8,11a-펜타메틸-2-옥소-3,3a,4,5,5a,5b,6,7,7a,8,11,11a,11b,12,13,13a-헥사데카하이드로-2H-사이클로펜타[a]크리센-9-일)벤조에이트
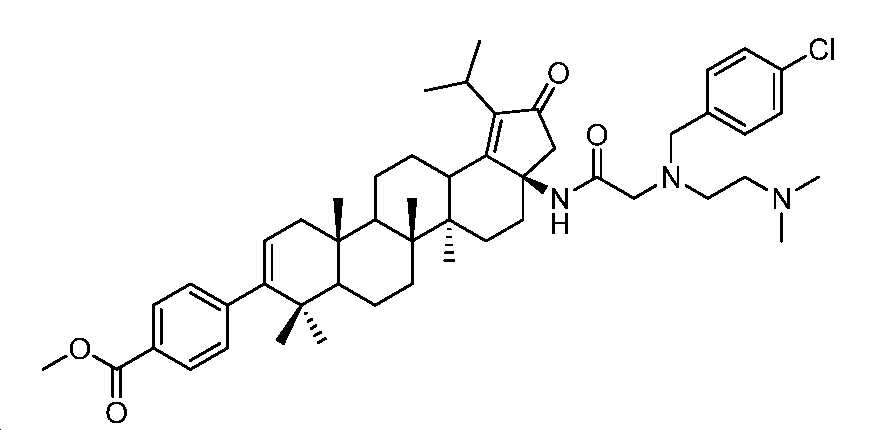
DCM(1 mL) 중 메틸 4-((3aR,5aR,5bR,11aS)-3a-아미노-1-이소프로필-5a,5b,8,8,11a-펜타메틸-2-옥소-3,3a,4,5,5a,5b,6,7,7a,8,11,11a,11b,12,13,13a-헥사데카하이드로-2H-사이클로펜타[a]크리센-9-일)벤조에이트(60 mg, 0.108 mmol), N-(4-클로로벤질)-N-(2-(디메틸아미노)에틸)글리신(116 mg, 0.43 mmol), HBTU(61 mg, 0.16 mmol) 및 DIPEA(76.3 mg, 0.592 mmol)의 혼합물을 실온에서 밤새 교반하였다. 얻어진 혼합물을 DCM과 H2O 사이로 분배하였다. 유기층을 염수로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 감압 하에서 농축하여 미정제 생성물을 수득하고, 이를 플래시 크로마토그래피(실리카겔, DCM 중 0 내지 10% MeOH)에 의해 정제하여 표제 화합물(60 mg, 69% 수율)을 백색 고체로서 수득하였다. LC-MS (ESI): m/z (M/M+2) = 810.5/812.5.
단계 C: 화합물 33:
4-((3aR,5aR,5bR,11aS)-3a-(2-((4-클로로벤질)(2-(디메틸아미노)에틸)아미노)아세트아미도)-1-이소프로필-5a,5b,8,8,11a-펜타메틸-2-옥소-3,3a,4,5,5a,5b,6,7,7a,8,11,11a,11b,12,13,13a-헥사데카하이드로-2H-사이클로펜타[a]크리센-9-일)벤조산
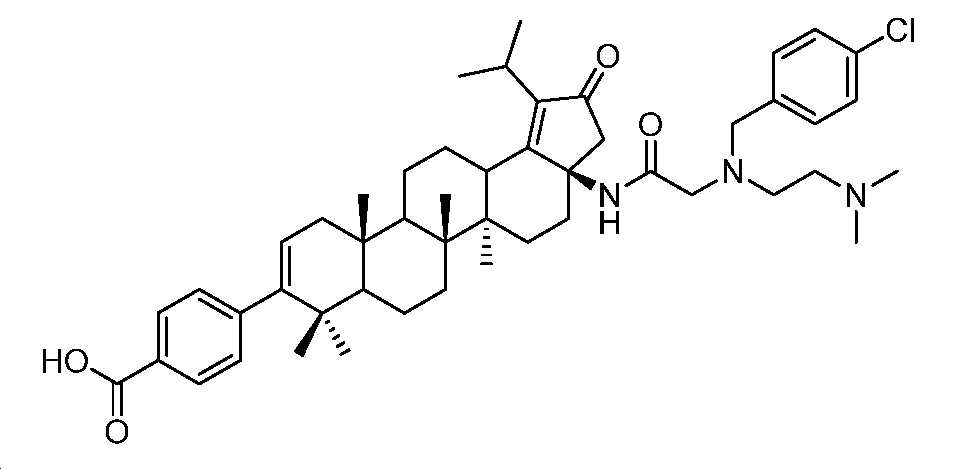
2.5 N LiOH(1 mL) 및 디옥산(1 mL) 중 메틸 4-((3aR,5aR,5bR,11aS)-3a-(2-((4-클로로벤질)(2-(디메틸아미노)에틸)아미노)아세트아미도)-1-이소프로필-5a,5b,8,8,11a-펜타메틸-2-옥소-3,3a,4,5,5a,5b,6,7,7a,8,11,11a,11b,12,13,13a-헥사데카하이드로-2H-사이클로펜타[a]크리센-9-일)벤조에이트(60 mg, 0.074 mmol)의 혼합물을 75℃에서 2.5시간 동안 교반하였다. 얻어진 혼합물을 1 N HCl로 pH 3 내지 4까지 산성화시키고, EtOAc로 추출하였다. 유기층을 염수로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 감압 하에서 농축하여 미정제 생성물을 수득하고, 이를 HPLC(C18, 0.1% 포름산을 함유한 H2O 중 50 내지 100% MeCN)에 의해 정제하여 동결건조 후 표제 화합물(8.2 mg, 14% 수율)을 백색 분말로서 수득하였다(몇 방울의 디옥산 중 HCl을 LCMS에 의해 수집된 분획에 첨가하여 HCl 염을 수득함). LC-MS (ESI): m/z (M/M+2) = 796.5/798.4. 1H NMR (400 MHz, 몇 방울의 MeOH를 함유한 CDCl3) δ 7.95 (d, J = 8.1 Hz, 2H), 7.60 (d, J = 7.4 Hz, 2H), 7.41 (d, J = 7.9 Hz, 2H), 7.21 (d, J = 8.1 Hz, 2H), 5.32 (d, J = 5.0 Hz, 1H), 4.30 (s, 2H), 4.15 - 3.27 (m, 8H), 3.18 (dt, J = 14.1, 7.2 Hz, 1H), 2.84 (s, 6H), 2.63 - 2.32 (m, 6H), 2.18 - 0.95 (m, 32H).
실시예 9
(화합물 37)
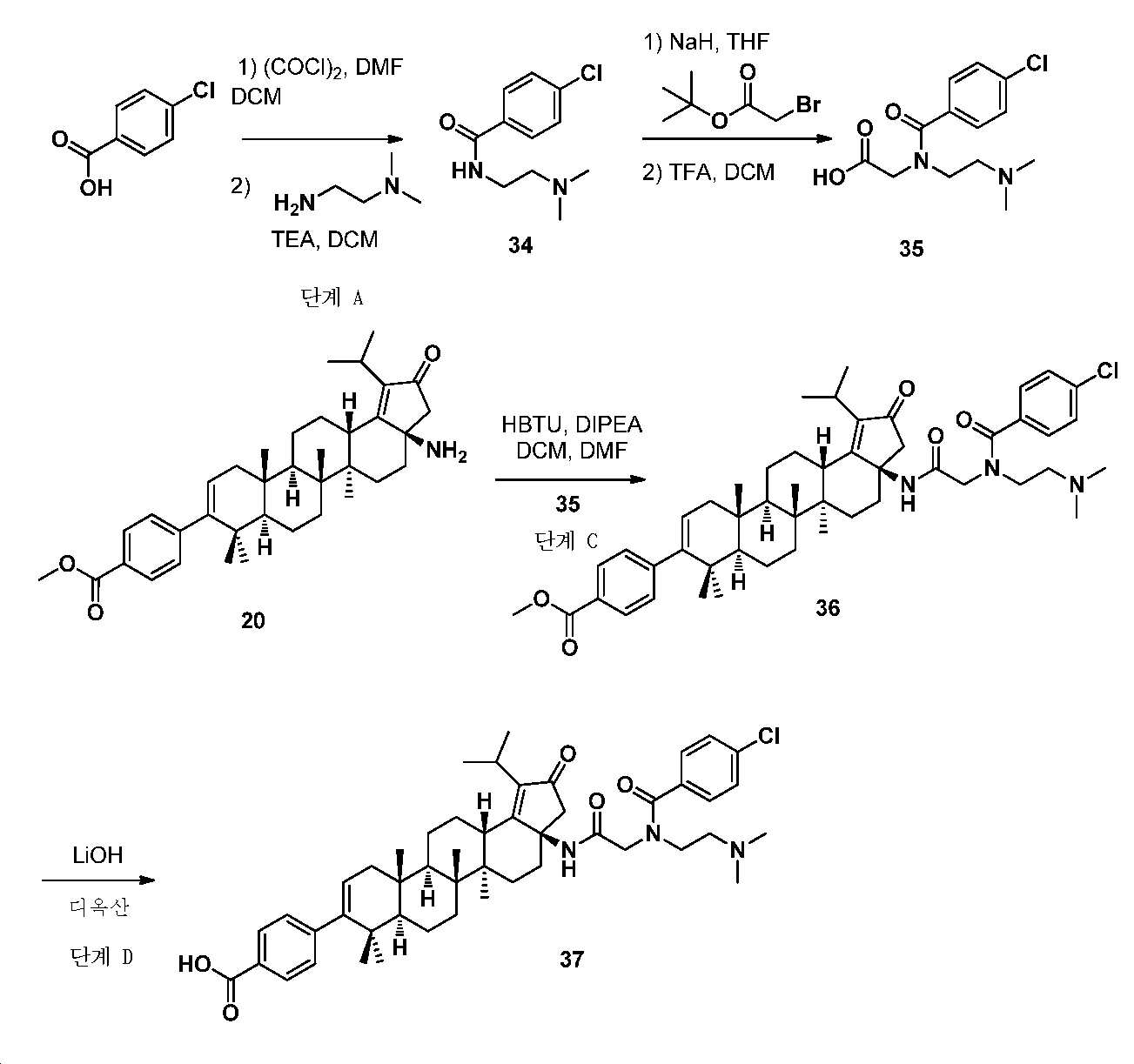
단계 A: 중간체 34:
4-클로로-N-(2-(디메틸아미노)에틸)벤즈아미드
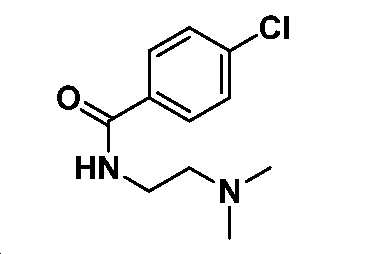
0℃에서, DCM(20 mL) 중 4-클로로벤조산(1 g, 6.4 mmol)의 용액에 옥살릴 클로라이드(10 mL) 및 DMF(1 방울)를 첨가하였다. 실온까지 가온시키고, 30분 동안 교반시킨 후에, 얻어진 혼합물을 감압 하에서 농축하여 미정제 아실 클로라이드를 수득하고, 이를 DCM(10 mL)으로 희석하고, DCM 중 N 1 ,N 1 -디메틸에탄-1,2-디아민(0.62 g, 7.0 mmol) 및 TEA(0.97 g, 9.6 mmol)의 사전-냉각 용액에 도입하였다. 실온에서 2시간 동안 교반한 후에, 얻어진 혼합물을 NaHCO3 포화수용액으로 켄칭시키고, DCM으로 추출하였다. 유기층을 염수로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 감압 하에서 농축하여 미정제 생성물을 수득하고, 이를 ISCO(실리카겔, DCM 중 0 내지 10% MeOH)에 의해 정제하여 4-클로로벤조일 클로라이드(1.3 g, 90% 수율)를 황색 오일로서 수득하였다. LC-MS (ESI): m/z (M/M+2) = 227.3/229.3. 1H NMR (400 MHz, CDCl3) δ 7.73 (d, J = 8.6 Hz, 2H), 7.39 (d, J = 8.6 Hz, 2H), 6.83 (s, 1H), 3.50 (dd, J = 11.3, 5.4 Hz, 2H), 2.59 - 2.47 (m, 2H), 2.27 (s, 6H).
단계 B: 중간체 35:
2-(4-c]클로로-N-(2-(디메틸아미노)에틸)벤즈아미도)아세트산

0℃에서, THF(20 mL) 중 4-클로로-N-(2-(디메틸아미노)에틸)벤즈아미드(1.3 g, 5.75 mmol)의 용액에 NaH(277 mg, 6.9 mmol)를 첨가하였다. 얻어진 혼합물을 실온까지 가온시키고, 3차-부틸 2-브로모아세테이트(1.24 g, 6.3 mmol)의 첨가 전 30분 동안 교반하였다. 실온에서 밤새 교반한 후에, 반응 혼합물을 NH4Cl 포화수용액으로 켄칭시키고, EtOAc로 추출하였다. 유기층을 염수로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 감압 하에서 농축하여 미정제 생성물을 수득하고, 이를 역상 크로마토그래피(C18, 0.1% 포름산을 함유한 H2O 중 0 내지 100% MeCN)에 의해 정제하여 3차-부틸 2-(4-클로로-N-(2-(디메틸아미노)에틸)벤즈아미도)아세테이트를 수득하고, 이를 DCM(10 mL)에 용해시키고, TFA(5 mL)로 처리하여 2-(4-클로로-N-(2-(디메틸아미노)에틸)벤즈아미도)아세트산(280 mg, 17% 수율)을 밝은 황색 고체로서 수득하였다. LC-MS (ESI): m/z (M/M+2) = 285.0/287.0. 1H NMR (400 MHz, DMSO) δ 7.49 (d, J = 8.4 Hz, 2H), 7.39 (d, J = 8.4 Hz, 2H), 3.83 - 3.73 (m, 2H), 3.67 (s, 2H), 3.25 - 3.11 (m, J = 6.7, 3.7 Hz, 2H), 2.71 (s, 6H). (산 양성자(acid proton)는 관찰되지 않음).
단계 C: 중간체 36:
메틸 4-((3aR,5aR,5bR,7aR,11aS,11bR,13aS)-3a-(2-(4-클로로-N-(2-(디메틸아미노)에틸)벤즈아미도)아세트아미도)-1-이소프로필-5a,5b,8,8,11a-펜타메틸-2-옥소-3,3a,4,5,5a,5b,6,7,7a,8,11,11a,11b,12,13,13a-헥사데카하이드로-2H-사이클로펜타[a]크리센-9-일)벤조에이트

DCM(5 mL) 및 DMF(2 mL) 중 메틸 4-((3aR,5aR,5bR,7aR,11aS,11bR,13aS)-3a-아미노-1-이소프로필-5a,5b,8,8,11a-펜타메틸-2-옥소-3,3a,4,5,5a,5b,6,7,7a,8,11,11a,11b,12,13,13a-헥사데카하이드로-2H-사이클로펜타[a]크리센-9-일)벤조에이트(80 mg, 0.14 mmol), 2-(4-c]클로로-N-(2-(디메틸아미노)에틸)벤즈아미도)아세트산(100 mg, 0.35 mmol), HBTU(250 mg, 0.65 mmol) 및 DIPEA(270 mg, 2.1 mmol)의 혼합물을 40℃에서 밤새 교반하였다. 얻어진 혼합물을 NaHCO3 포화수용액으로 켄칭시키고, DCM으로 추출하였다. 유기층을 염수로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 감압 하에서 농축하여 미정제 생성물을 수득하고, 이를 ISCO(실리카겔, DCM 중 0 내지 10% MeOH)에 의해 정제하여 메틸 4-((3aR,5aR,5bR,7aR,11aS,11bR,13aS)-3a-(2-(4-클로로-N-(2-(디메틸아미노)에틸)벤즈아미도)아세트아미도)-1-이소프로필-5a,5b,8,8,11a-펜타메틸-2-옥소-3,3a,4,5,5a,5b,6,7,7a,8,11,11a,11b,12,13,13a-헥사데카하이드로-2H-사이클로펜타[a]크리센-9-일)벤조에이트(40 mg, 34% 수율)를 밝은 황색 고체로서 수득하였다. LC-MS (ESI): m/z (M/M+2) = 824.2/826.4 . 1H NMR (400 MHz, CDCl3) δ 7.94 (d, J = 8.3 Hz, 2H), 7.69 (s, 1H), 7.51 - 7.33 (m, 4H), 7.21 (d, J = 8.3 Hz, 2H), 5.38 - 5.23 (m, 3H), 4.20 - 3.95 (m, J = 15.6 Hz, 2H), 3.91 (s, 3H), 3.71 - 3.57 (m, 1H), 3.56 - 3.36 (m, 1H), 3.23 - 3.03 (m, 2H), 2.63 - 1.00 (m, 45H).
단계 D: 화합물 37:
4-((3aR,5aR,5bR,7aR,11aS,11bR,13aS)-3a-(2-(4-클로로-N-(2-(디메틸아미노)에틸)벤즈아미도)아세트아미도)-1-이소프로필-5a,5b,8,8,11a-펜타메틸-2-옥소-3,3a,4,5,5a,5b,6,7,7a,8,11,11a,11b,12,13,13a-헥사데카하이드로-2H-사이클로펜타[a]크리센-9-일)벤조산
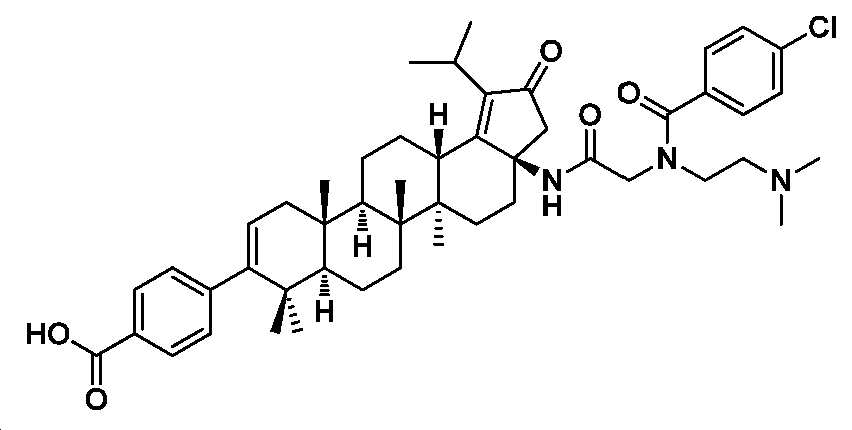
2.5 N LiOH(0.4 mL) 및 디옥산(2 mL) 중 메틸 4-((3aR,5aR,5bR,7aR,11aS,11bR,13aS)-3a-(2-(4-클로로-N-(2-(디메틸아미노)에틸)벤즈아미도)아세트아미도)-1-이소프로필-5a,5b,8,8,11a-펜타메틸-2-옥소-3,3a,4,5,5a,5b,6,7,7a,8,11,11a,11b,12,13,13a-헥사데카하이드로-2H-사이클로펜타[a]크리센-9-일)벤조에이트(40 mg, 0.05 mmol)의 혼합물을 60℃에서 2시간 동안 교반하였다. 얻어진 혼합물을 1 N HCl로 pH 6 내지 7까지 산성화시키고, DCM으로 추출하였다. 유기층을 염수로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 감압 하에서 농축하여 미정제 생성물을 수득하고, 이를 역상 크로마토그래피(C18, 0.1% 포름산을 함유한 H2O 중 0 내지 100% MeCN)에 의해 정제하여 4-((3aR,5aR,5bR,7aR,11aS,11bR,13aS)-3a-(2-(4-클로로-N-(2-(디메틸아미노)에틸)벤즈아미도)아세트아미도)-1-이소프로필-5a,5b,8,8,11a-펜타메틸-2-옥소-3,3a,4,5,5a,5b,6,7,7a,8,11,11a,11b,12,13,13a-헥사데카하이드로-2H-사이클로펜타[a]크리센-9-일)벤조산(20 mg, 51% 수율)을 백색 분말로서 수득하였다. 몇 방울의 디옥산 중 4 N HCl을 HPLC에 의해 수집된 분획에 첨가하여 상응하는 HCl 염을 수득하였다. LC-MS (ESI): m/z (M/M+2) = 810.5/812.4. 1H NMR (400 MHz, MeOD) δ 7.93 (d, J = 8.3 Hz, 2H), 7.47 (dd, J = 26.7, 8.2 Hz, 4H), 7.24 (d, J = 8.2 Hz, 2H), 5.34 (d, J = 4.8 Hz, 1H), 4.21 - 3.87 (m, 4H), 3.44 - 3.34 (m, 2H), 3.28 - 3.20 (m, 1H), 2.99 (d, J = 9.1 Hz, 6H), 2.80 - 2.44 (m, 3H), 2.38 - 2.15 (m, 3H), 2.04 - 0.96 (m, 34H).
실시예 10
(화합물 51)
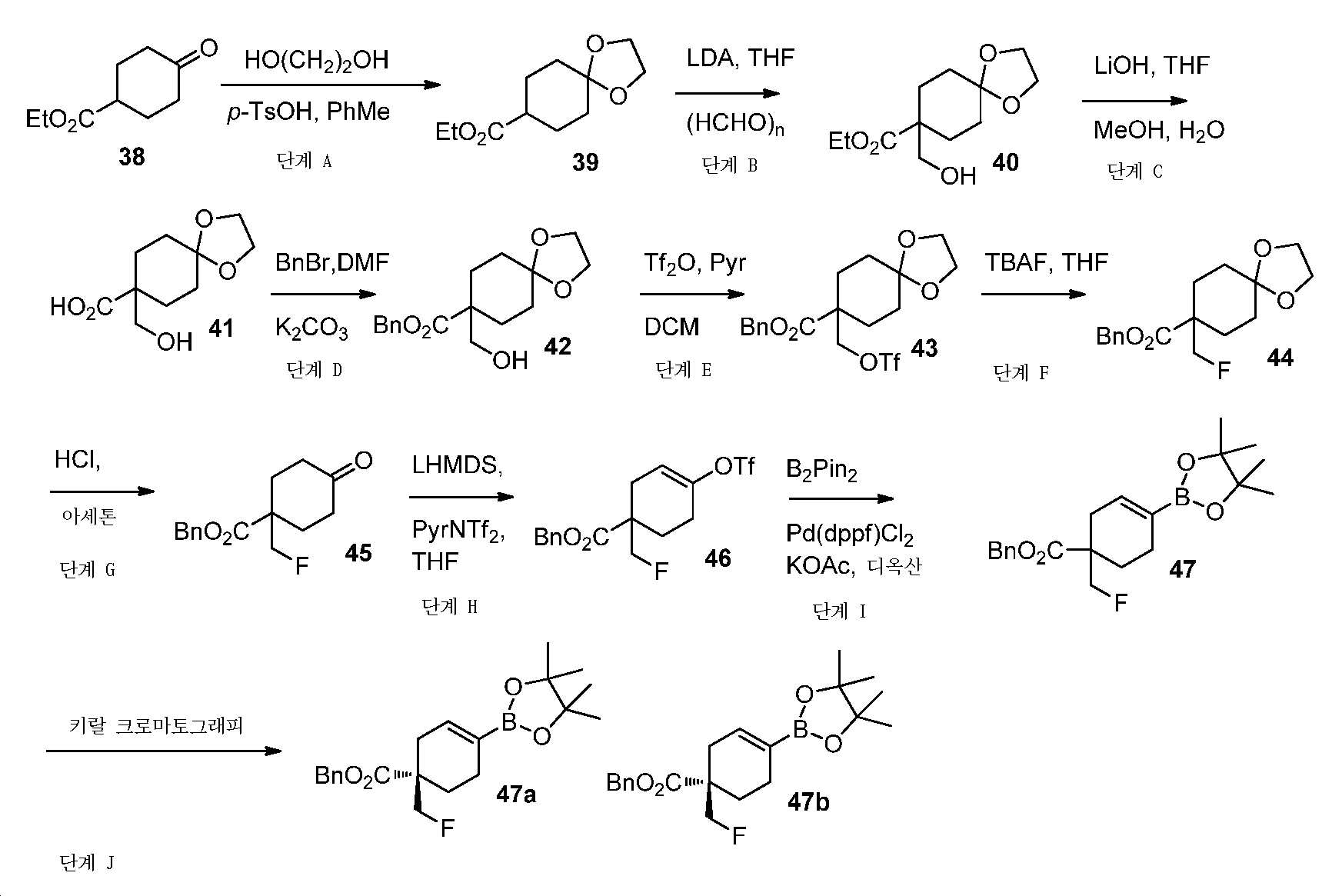
단계 A: 에틸 1,4-디옥사스피로[4.5]데칸-8-카복실레이트

톨루엔 중 에틸 4-옥소사이클로헥산-1-카복실레이트(62 g, 0.36 mol), 에탄-1,2-디올(22.3 mL, 0.4 mol) 및 p-TsOH(0.7 g, 3.6 mmol)의 혼합물을 딘-스탁 물 트랩(Dean-Stark water trap) 및 콘덴서를 이용하여 24시간 동안 가열 환류시켰다. 실온까지 냉각시킨 후에, 얻어진 혼합물을 EtOAc로 희석시키고, NaHCO3 포화수용액 및 염수로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 감압 하에서 농축하여 미정제 생성물을 플래시 크로마토그래피(실리카겔, PE 중 0 내지 20% EtOAc)에 의해 정제하여 표제 화합물을 무색 오일(70 g, 91% 수율)로서 수득하였다. LC-MS (ESI): m/z (M+1) = 215.05. 1H NMR (400 MHz, CDCl3) δ 4.13 (q, J = 7.1 Hz, 2H), 3.94 (s, 4H), 2.38 - 2.28 (m, 1H), 1.99 - 1.89 (m, 2H), 1.86 - 1.70 (m, 4H), 1.63 - 1.50 (m, 2H), 1.25 (t, J = 7.1 Hz, 3H).
단계 B: 에틸 8-(하이드록시메틸)-1,4-디옥사스피로[4.5]데칸-8-카복실레이트
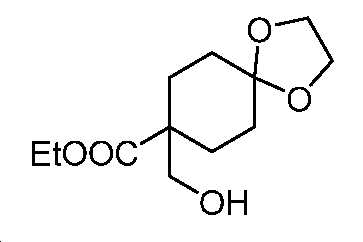
-78℃에서, 무수 THF(700 mL) 중 에틸 1,4-디옥사스피로[4.5]데칸-8-카복실레이트(70 g, 326 mmol)의 용액에, 내부 온도를 -60℃ 미만으로 유지될 때 N2 분위기 하에서 LDA(THF 중 2 M, 196 mL, 391 mmol)를 첨가하였다. 파라포름알데하이드(14.8 g, 490 mmol)를 한번에 첨가하기 전에, 혼합물을 -78℃에서 30분 동안 교반하였다. 실온에서 밤새 교반한 후에, 얻어진 혼합물을 NH4Cl 포화수용액으로 켄칭시키고, EtOAc로 추출하였다. 유기층을 염수로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 감압 하에서 농축하여 미정제 생성물을 수득하고, 이를 플래시 크로마토그래피(실리카겔, PE 중 0 내지 40% EtOAc)에 의해 정제하여 표제 화합물을 무색 오일(54 g, 68% 수율)로서 수득하였다. LC-MS (ESI): m/z (M+1) = 245.23. 1H NMR (400 MHz, CDCl3) δ 4.20 (q, J = 7.1 Hz, 2H), 3.94 (t, J = 2.7 Hz, 4H), 3.64 (d, J = 6.3 Hz, 2H), 2.19 - 2.11 (m, J = 9.5, 3.8 Hz, 2H), 2.04 (t, J = 6.4 Hz, 1H), 1.69 - 1.52 (m, 6H), 1.28 (t, J = 7.1 Hz, 3H).
단계 C: 8-(하이드록시메틸)-1,4-디옥사스피로[4.5]데칸-8-카복실산

THF(320 mL) 및 MeOH(105 mL) 중 에틸 8-(하이드록시메틸)-1,4-디옥사스피로[4.5]데칸-8-카복실레이트(54 g, 221 mmol)의 용액에 LiOH(3 N 수용액, 147 mL, 441 mmol)를 첨가하였다. 60℃에서 밤새 교반한 후에, 얻어진 혼합물을 디옥산 중 4 N HCl로 pH 3 내지 4까지 산성화시키고, 감압 하에서 농축하여 LiCl과 혼합된 표제 화합물(정량 수율)을 수득하고, 이를 정제 없이 다음 단계에서 직접적으로 사용하였다. LC-MS (ESI): m/z (M+1) = 217.30. LC-MS (ESI): m/z (M-1) = 215.01. 1H NMR (400 MHz, MeOD) δ 3.90 (s, 4H), 3.48 (s, 2H), 2.09 - 2.00 (m, 2H), 1.80 - 1.70 (m, 2H), 1.65 - 1.55 (m, 2H), 1.53 - 1.43 (m, 2H).
단계 D: 벤질 8-(하이드록시메틸)-1,4-디옥사스피로[4.5]데칸-8-카복실레이트

DMF(250 mL) 중 8-(하이드록시메틸)-1,4-디옥사스피로[4.5]데칸-8-카복실산 (36 g, 이전 단계로부터 얻어진 미정제 생성물), 벤질 브로마이드(19.7 mL, 166 mmol) 및 K2CO3(34.5 g, 250 mmol)의 혼합물을 실온에서 밤새 교반하였다. 얻어진 혼합물을 여과하고, 여액을 EtOAc와 H2O 사이로 분배하였다. 층들을 분리하고, 유기층을 염수로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 감압 하에서 농축하여 미정제 생성물을 수득하고, 이를 플래시 크로마토그래피(실리카겔, PE 중 0 내지 30% EtOAc)에 의해 정제하여 표제 화합물을 무색 오일(33 g, 2 단계에 걸쳐 65% 수율)로서 수득하였다. LC-MS (ESI): m/z (M+1) = 307.51. 1H NMR (400 MHz, CDCl3) δ 7.42 - 7.28 (m, 5H), 5.18 (s, 2H), 4.03 - 3.83 (m, 4H), 3.66 (d, J = 6.4 Hz, 2H), 2.20 - 2.14 (m, 2H), 1.90 (t, J = 6.5 Hz, 1H), 1.69 - 1.58 (m, 6H).
단계 E: 벤질 8-((((트리플루오로메틸)설포닐)옥시)메틸)-1,4-디옥사스피로[4.5]데칸-8-카복실레이트

-10℃에서, 무수 DCM(500 mL) 중 벤질 8-(하이드록시메틸)-1,4-디옥사스피로[4.5]데칸-8-카복실레이트(33 g, 108 mmol) 및 피리딘(15.8 mL, 145 mmol)의 용액에 N2 분위기 하에서 Tf2O(24.5 mL, 145 mmol)를 적가하였다. 0℃에서 2시간 동안 교반한 후에, 얻어진 혼합물을 감압 하에서 농축하고, 잔부를 DCM과 H2O 사이로 분배하였다. 층들을 분리하고, 유기층을 염수로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 감압 하에서 농축하여 미정제 생성물을 수득하고, 이를 플래시 크로마토그래피(실리카겔, PE 중 0 내지 30% EtOAc)에 의해 정제하여 표제 화합물을 황색 오일(40 g, 85% 수율)로서 수득하였다. LC-MS (ESI): m/z (M+1) = 439.10. 1H NMR (400 MHz, CDCl3) δ 7.41 - 7.29 (m, 5H), 5.18 (s, 2H), 4.51 (s, 2H), 3.97 - 3.87 (m, 4H), 2.28 - 2.16 (m, 2H), 1.72 - 1.61 (m, 6H).
단계 F: 벤질 8-(플루오로메틸)-1,4-디옥사스피로[4.5]데칸-8-카복실레이트
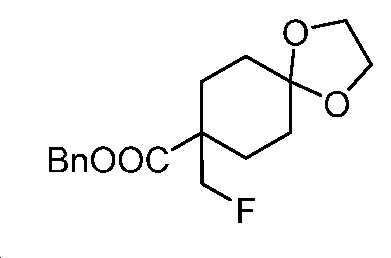
무수 THF(300 mL) 중 벤질 8-((((트리플루오로메틸)설포닐)옥시)메틸)-1,4-디옥사스피로[4.5]데칸-8-카복실레이트(40 g, 91 mmol)의 용액에 TBAF(THF 중 1 M, 137 mL, 137 mmol)를 적가하였다. 실온에서 30분 동안 교반한 후에, 얻어진 혼합물을 EtOAc와 H2O 사이로 분배하였다. 층들을 분리하고, 유기층을 염수로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 감압 하에서 농축하여 미정제 생성물을 수득하고, 이를 플래시 크로마토그래피(실리카겔, PE 중 0 내지 30% EtOAc)에 의해 정제하여 표제 화합물을 밝은 황색 오일(25 g, 89% 수율)로서 수득하였다. LC-MS (ESI): m/z (M+1) = 309.10. 1H NMR (400 MHz, CDCl3) δ 7.41 - 7.29 (m, 5H), 5.18 (s, 2H), 4.42 (d, J = 47.2 Hz, 2H), 3.98 - 3.87 (m, 4H), 2.26 - 2.15 (m, 2H), 1.70 - 1.58 (m, 6H).
단계 G: 벤질 1-(플루오로메틸)-4-옥소사이클로헥산-1-카복실레이트
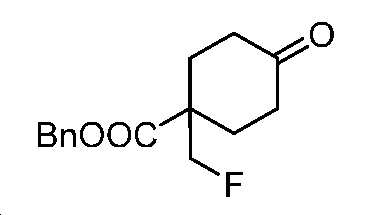
HCl(1.5 N 수용액, 216 mL, 324 mmol) 및 아세톤(250 mL) 중 벤질 8-(플루오로메틸)-1,4-디옥사스피로[4.5]데칸-8-카복실레이트(25 g, 81 mmol)의 혼합물을 실온에서 밤새 교반하였다. 얻어진 혼합물을 감압 하에서 농축시키고, 잔부를 EtOAc와 H2O 사이로 분배하였다. 층들을 분리하고, 유기층을 염수로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 감압 하에서 농축하여 미정제 생성물을 수득하고, 이를 플래시 크로마토그래피(실리카겔, PE 중 0 내지 30% EtOAc)에 의해 정제하여 표제 화합물을 밝은 황색 오일(20 g, 93% 수율)로서 수득하였다. LC-MS (ESI): m/z (M+1) = 265.24. 1H NMR (400 MHz, CDCl3) δ 7.41 - 7.31 (m, 5H), 5.25 (s, 2H), 4.50 (d, J = 47.0 Hz, 2H), 2.50 - 2.30 (m, 6H), 1.84 - 1.73 (m, 2H).
단계 H: 벤질 1-(플루오로메틸)-4-(((트리플루오로메틸)설포닐)옥시)사이클로헥스-3-엔-1-카복실레이트

-78℃에서, 무수 THF(200 mL) 중 벤질 1-(플루오로메틸)-4-옥소사이클로헥산-1-카복실레이트(20 g, 76 mmol) 및 1,1,1-트리플루오로-N-(피리딘-2-일)-N-((트리플루오로메틸)설포닐)메탄 설폰아미드(35 g, 98 mmol)의 용액에 K-HMDS(THF 중 1 M, 113 mL, 113 mmol)를 N2 분위기 하에서 적가하였다. 실온에서 밤새 교반한 후에, 얻어진 혼합물을 NH4Cl 포화수용액으로 켄칭시키고, EtOAc로 추출하였다. 유기층을 염수로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 감압 하에서 농축하여 미정제 생성물을 수득하고, 이를 플래시 크로마토그래피(실리카겔, PE 중 0 내지 10% EtOAc)에 의해 정제하여 표제 화합물을 황색 오일(24 g, 86% 수율)로서 수득하였다. LC-MS (ESI): m/z (M+1) = 397.01. 1H NMR (400 MHz, CDCl3) δ 7.44 - 7.27 (m, 5H), 5.75 (t, J = 4.0 Hz, 1H), 5.24 - 5.11 (m, 2H), 4.62 - 4.37 (m, 2H), 2.83 - 2.70 (m, 1H), 2.45 - 2.15 (m, 4H), 1.98 - 1.85 (m, 1H).
단계 I: 벤질 1-(플루오로메틸)-4-(4,4,5,5-테트라메틸-1,3,2-디옥사보롤란-2-일)사이클로헥스-3-엔-1-카복실레이트
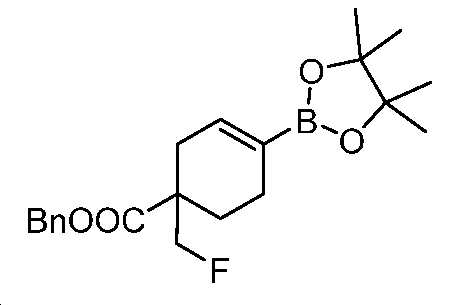
디옥산(200 mL) 중 벤질 1-(플루오로메틸)-4-(((트리플루오로메틸)설포닐)옥시)사이클로헥스-3-엔-1-카복실레이트(24 g, 61 mmol), 4,4,4',4',5,5,5',5'-옥타메틸-2,2'-바이(1,3,2-디옥사보롤란)(17 g, 68 mmol), Pd(dppf)Cl2(5.3 g, 6.5 mmol) 및 KOAc(19 g, 195 mmol)의 혼합물을 N2 분위기 하, 80℃에서 밤새 교반하였다. 얻어진 혼합물을 셀라이트 패드를 통해 여과하고, 여액을 EtOAc와 H2O 사이로 분배하였다. 층들을 분리하고, 유기층을 염수로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 감압 하에서 농축하여 미정제 생성물을 수득하고, 이를 플래시 크로마토그래피(실리카겔, PE 중 0 내지 15% EtOAc)에 의해 정제하여 표제 화합물을 무색 오일(18 g, 79% 수율)로서 수득하였다. LC-MS (ESI): m/z (M+1) = 375.40. 1H NMR (400 MHz, CDCl3) δ 7.41 - 7.26 (m, 5H), 6.51 (s, 1H), 5.16 (s, 2H), 4.63 - 4.37 (m, 2H), 2.70 - 2.58 (m, 1H), 2.23 - 2.10 (m, 3H), 1.95 - 1.75 (m, 2H), 1.25 (s, 12H).
단계 J: 벤질 (R)-1-(플루오로메틸)-4-(4,4,5,5-테트라메틸-1,3,2-디옥사보롤란-2-일)사이클로헥스-3-엔-1-카복실레이트 및 벤질 (S)-1-(플루오로메틸)-4-(4,4,5,5-테트라메틸-1,3,2-디옥사보롤란-2-일)사이클로헥스-3-엔-1-카복실레이트
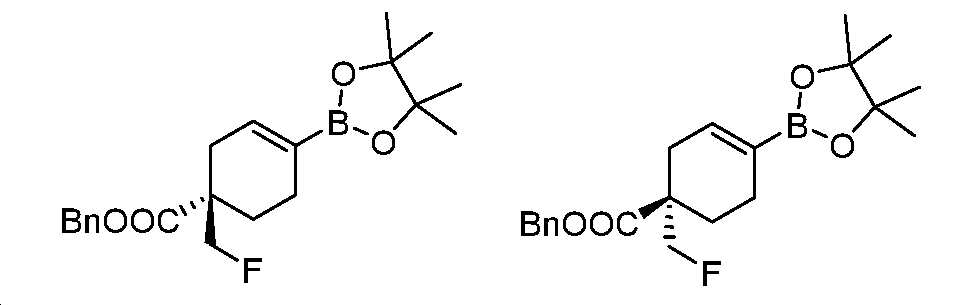
벤질 1-(플루오로메틸)-4-(4,4,5,5-테트라메틸-1,3,2-디옥사보롤란-2-일)사이클로헥스-3-엔-1-카복실레이트(9.4 g, 25 mmol)를 조건(컬럼: Regis (S,S)-whelk-O1; 컬럼 크기: 0.46 cm I.D. × 25 cm L; 주입: 5 ㎕; 이동상: CO2/[헥산:IPA=4:1, (v/v)] = 80/20; 유량: 2.0 ml/분; 파장: UV 220 nm; 온도: 35℃)을 이용하여 키랄 크로마토그래피에 의해 분리하여 벤질 (R)-1-(플루오로메틸)-4-(4,4,5,5-테트라메틸-1,3,2-디옥사보롤란-2-일)사이클로헥스-3-엔-1-카복실레이트(제2 용리액, 피크 B, 2.8 g, 7.5 mmol) 및 벤질 (S)-1-(플루오로메틸)-4-(4,4,5,5-테트라메틸-1,3,2-디옥사보롤란-2-일)사이클로헥스-3-엔-1-카복실레이트(제1 용리액, 피크 A, 2.9 g, 7.7 mmol)를 수득하였다. 이성질체들은 명시된 절대 입체화학으로 실험적으로 지정되었다. 이러한 지정을 이용한 하기에 기술되는 화합물은 이러한 실험적 지정에 따른다.

단계 A: (3aR,5aR,5bR,7aR,11aR,11bR,13aS)-3a-((2-(1,1-디옥시도티오모르폴리노)에틸)아미노)-1-이소프로필-5a,5b,8,8,11a-펜타메틸-3,3a,4,5,5a,6,7,7a,8,10,11,11a,11b,12,13,13a-헥사데카하이드로-2H-사이클로펜타[a]크리센-2,9(5bH)-디온
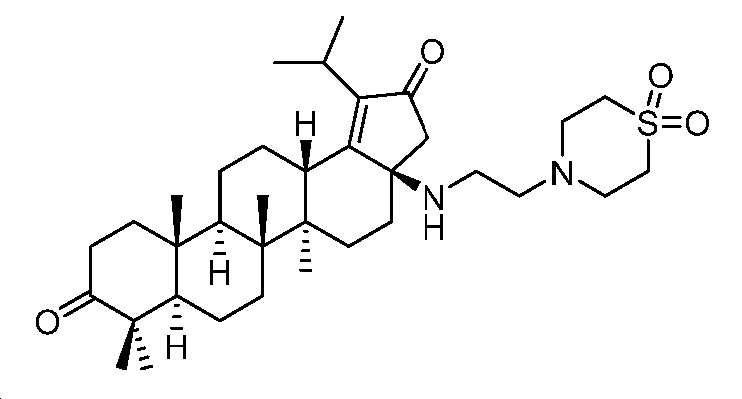
MeCN(4 mL) 중 (3aR,5aR,5bR,7aR,11aR,11bR,13aS)-3a-아미노-1-이소프로필-5a,5b,8,8,11a-펜타메틸-3,3a,4,5,5a,6,7,7a,8,10,11,11a,11b,12,13,13a-헥사데카하이드로-2H-사이클로펜타[a]크리센-2,9(5bH)-디온(400 mg, 0.9 mmol), 4-(2-클로로에틸)티오모르폴린 1,1-디옥사이드(198 mg, 1.0 mmol), K3PO4(579 mg, 2.7 mmol) 및 KI(226 mg, 1.4 mmol)의 혼합물을 N2 분위기 하, 100℃에서 밤새 교반하였다. 얻어진 혼합물을 EtOAc로 희석하고, 여과하였다. 여액을 EtOAc와 H2O 사이로 분배하고, 유기층을 염수로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 감압 하에서 농축하여 미정제 생성물을 수득하고, 이를 플래시 크로마토그래피(실리카겔, DCM 중 0 내지 10% MeOH)에 의해 정제하여 표제 화합물을 밝은 황색 고체(300 mg, 56% 수율)로서 수득하였다. LC-MS (ESI): m/z (M+1) = 601.39. 1H NMR (400 MHz, CDCl3) δ 3.17 - 3.12 (m, 1H), 3.11 - 2.81 (m, J = 17.4 Hz, 12H), 2.69 - 2.59 (m, 2H), 2.58 - 2.38 (m, 4H), 2.37 - 2.24 (m, 2H), 2.13 - 1.85 (m, 6H), 1.54 - 1.37 (m, 8H), 1.27 - 1.16 (m, 9H), 1.11 - 0.92 (m, 12H).
단계 B: (3aR,5aR,5bR,7aR,11aR,11bR,13aS)-3a-((2-(1,1-디옥시도티오모르폴리노)에틸)아미노)-1-이소프로필-5a,5b,8,8,11a-펜타메틸-2-옥소-3,3a,4,5,5a,5b,6,7,7a,8,11,11a,11b,12,13,13a-헥사데카하이드로-2H-사이클로펜타[a]크리센-9-일트리플루오로메탄설포네이트

-78℃에서, 무수 THF(5 mL) 중 (3aR,5aR,5bR,7aR,11aR,11bR,13aS)-3a-((2-(1,1-디옥시도티오모르폴리노)에틸)아미노)-1-이소프로필-5a,5b,8,8,11a-펜타메틸-3,3a,4,5,5a,6,7,7a,8,10,11,11a,11b,12,13,13a-헥사데카하이드로-2H-사이클로펜타[a]크리센-2,9(5bH)-디온(300 mg, 0.5 mmol) 및 1,1,1-트리플루오로-N-페닐-N-((트리플루오로메틸)설포닐)메탄 설폰아미드(267 mg, 0.75 mmol)의 용액에 K-HMDS(THF 중 1 M, 1 mL, 1 mmol)를 N2 분위기 하에서 적가하였다. 실온에서 밤새 교반한 후에, 얻어진 혼합물을 NH4Cl 포화수용액으로 켄칭시키고, DCM으로 추출하였다. 유기층을 염수로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 감압 하에서 농축하여 미정제 생성물을 수득하고, 이를 플래시 크로마토그래피(실리카겔, DCM 중 0 내지 10% MeOH)에 의해 정제하여 표제 화합물을 밝은 황색 고체(130 mg, 36% 수율)로서 수득하였다. LC-MS (ESI): m/z (M+1) = 733.26.
단계 C: 벤질 (R)-4-((3aR,5aR,5bR,7aR,11aS,11bR,13aS)-3a-((2-(1,1-디옥시도티오모르폴리노)에틸)아미노)-1-이소프로필-5a,5b,8,8,11a-펜타메틸-2-옥소-3,3a,4,5,5a,5b,6,7,7a,8,11,11a,11b,12,13,13a-헥사데카하이드로-2H-사이클로펜타[a]크리센-9-일)-1-(플루오로메틸)사이클로헥스-3-엔-1-카복실레이트
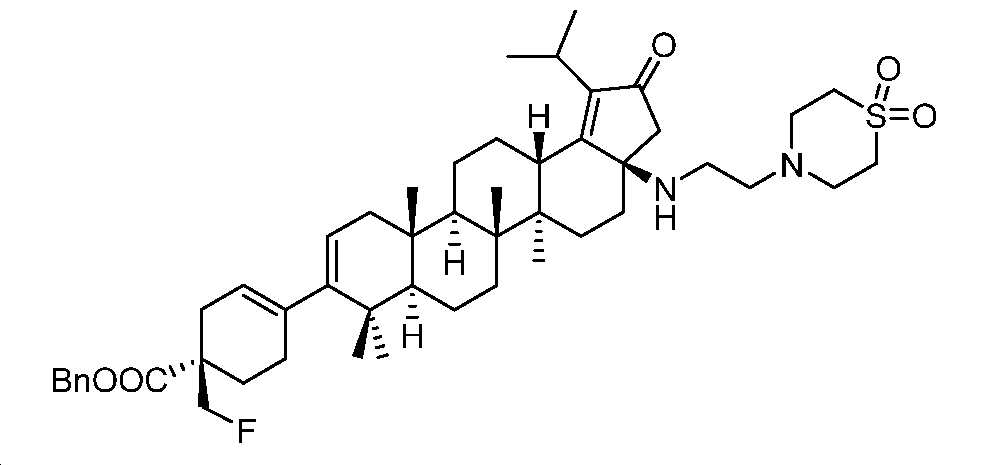
디옥산(1.5 mL) 및 H2O(0.5 mL) 중 (3aR,5aR,5bR,7aR,11aR,11bR,13aS)-3a-((2-(1,1-디옥시도티오모르폴리노)에틸)아미노)-1-이소프로필-5a,5b,8,8,11a-펜타메틸-2-옥소-3,3a,4,5,5a,5b,6,7,7a,8,11,11a,11b,12,13,13a-헥사데카하이드로-2H-사이클로펜타[a]크리센-9-일트리플루오로메탄설포네이트(60 mg, 0.08 mmol), 벤질 (R)-1-(플루오로메틸)-4-(4,4,5,5-테트라메틸-1,3,2-디옥사보롤란-2-일)사이클로헥스-3-엔-1-카복실레이트(37 mg, 0.1 mmol), Pd(PPh3)4(20 mg, 0.018 mmol) 및 Na2CO3(28 mg, 0.27 mmol)의 혼합물을 N2 분위기 하, 80℃에서 3시간 동안 교반하였다. 얻어진 혼합물을 DCM과 H2O 사이로 분배하고, 유기층을 염수로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 감압 하에서 농축하여 미정제 생성물을 수득하고, 이를 플래시 크로마토그래피(실리카겔, DCM 중 0 내지 10% MeOH)에 의해 정제하여 표제 화합물을 밝은 황색 고체(40 mg, 60% 수율)로서 수득하였다. LC-MS (ESI): m/z (M+1) = 831.52.
단계 D: (R)-4-((3aR,5aR,5bR,7aR,11aS,11bR,13aS)-3a-((2-(1,1-디옥시도티오모르폴리노)에틸)아미노)-1-이소프로필-5a,5b,8,8,11a-펜타메틸-2-옥소-3,3a,4,5,5a,5b,6,7,7a,8,11,11a,11b,12,13,13a-헥사데카하이드로-2H-사이클로펜타[a]크리센-9-일)-1-(플루오로메틸)사이클로헥스-3-엔-1-카복실산

1 N NaOH(0.5 mL), MeOH(0.1 mL) 및 디옥산(0.5 mL) 중 벤질 (R)-4-((3aR,5aR,5bR,7aR,11aS,11bR,13aS)-3a-((2-(1,1-디옥시도티오모르폴리노)에틸)아미노)-1-이소프로필-5a,5b,8,8,11a-펜타메틸-2-옥소-3,3a,4,5,5a,5b,6,7,7a,8,11,11a,11b,12,13,13a-헥사데카하이드로-2H-사이클로펜타[a]크리센-9-일)-1-(플루오로메틸)사이클로헥스-3-엔-1-카복실레이트(40 mg, 0.048 mmol)의 혼합물을 80℃에서 2시간 동안 교반하였다. 얻어진 혼합물을 1 N HCl로 pH 3 내지 4까지 산성화시키고, DCM으로 추출하였다. 유기층을 염수로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 감압 하에서 농축하여 미정제 생성물을 수득하고, 이를 HPLC(C18, 0.1% 포름산을 함유한 H2O 중 0 내지 100% MeCN)에 의해 정제하였다. 몇 방울의 디옥산 중 4 N HCl을 HPLC에 의해 수집된 분획에 첨가하여 표제 화합물을 HCl 염(8 mg, 22% 수율)으로서 수득하였다. LC-MS (ESI): m/z (M+1) = 741.40. 1H NMR (400 MHz, MeOD) δ 5.35 (s, 1H), 5.23 (d, J = 4.6 Hz, 1H), 4.54 (dd, J = 18.8, 8.8 Hz, 1H), 4.42 (dd, J = 18.6, 8.8 Hz, 1H), 3.30 - 3.21 (m, 8H), 3.13 - 1.34 (m, 30H), 1.30 - 0.91 (m, 21H).
실시예 11
(화합물 53)
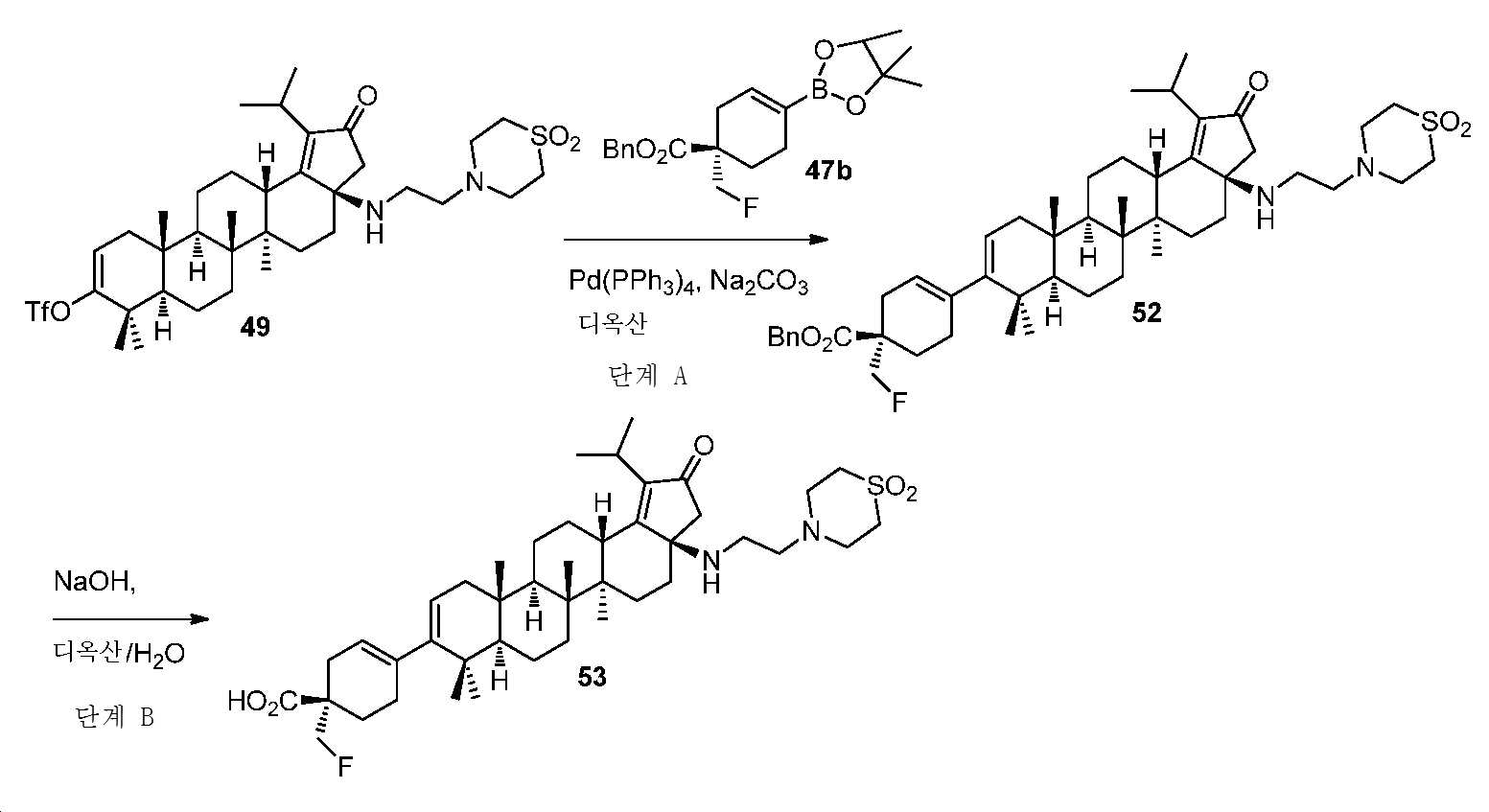
단계 A: 벤질 (S)-4-((3aR,5aR,5bR,7aR,11aS,11bR,13aS)-3a-((2-(1,1-디옥시도티오모르폴리노)에틸)아미노)-1-이소프로필-5a,5b,8,8,11a-펜타메틸-2-옥소-3,3a,4,5,5a,5b,6,7,7a,8,11,11a,11b,12,13,13a-헥사데카하이드로-2H-사이클로펜타[a]크리센-9-일)-1-(플루오로메틸)사이클로헥스-3-엔-1-카복실레이트

디옥산(1.5 mL) 및 H2O(0.5 mL) 중 (3aR,5aR,5bR,7aR,11aR,11bR,13aS)-3a-((2-(1,1-디옥시도티오모르폴리노)에틸)아미노)-1-이소프로필-5a,5b,8,8,11a-펜타메틸-2-옥소-3,3a,4,5,5a,5b,6,7,7a,8,11,11a,11b,12,13,13a-헥사데카하이드로-2H-사이클로펜타[a]크리센-9-일트리플루오로메탄설포네이트(60 mg, 0.08 mmol), 벤질 (S)-1-(플루오로메틸)-4-(4,4,5,5-테트라메틸-1,3,2-디옥사보롤란-2-일)사이클로헥스-3-엔-1-카복실레이트(37 mg, 0.1 mmol), Pd(PPh3)4(20 mg, 0.018 mmol) 및 Na2CO3(28 mg, 0.27 mmol)의 혼합물을 N2 분위기 하, 80℃에서 2시간 동안 교반하였다. 얻어진 혼합물을 DCM과 H2O 사이로 분배하고, 유기층을 염수로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 감압 하에서 농축하여 미정제 생성물을 수득하고, 이를 플래시 크로마토그래피(실리카겔, DCM 중 0 내지 10% MeOH)에 의해 정제하여 표제 화합물을 밝은 황색 고체(45 mg, 68% 수율)로서 수득하였다. LC-MS (ESI): m/z (M+1) = 831.53.
단계 B: (S)-4-((3aR,5aR,5bR,7aR,11aS,11bR,13aS)-3a-((2-(1,1-디옥시도티오모르폴리노)에틸)아미노)-1-이소프로필-5a,5b,8,8,11a-펜타메틸-2-옥소-3,3a,4,5,5a,5b,6,7,7a,8,11,11a,11b,12,13,13a-헥사데카하이드로-2H-사이클로펜타[a]크리센-9-일)-1-(플루오로메틸)사이클로헥스-3-엔-1-카복실산
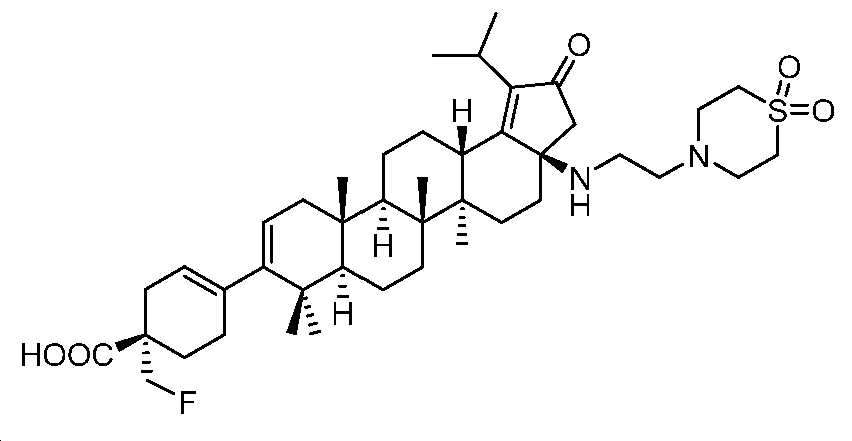
1 N NaOH(0.5 mL), MeOH(0.1 mL) 및 디옥산(0.5 mL) 중 벤질 (S)-4-((3aR,5aR,5bR,7aR,11aS,11bR,13aS)-3a-((2-(1,1-디옥시도 티오모르폴리노)에틸)아미노)-1-이소프로필-5a,5b,8,8,11a-펜타메틸-2-옥소-3,3a,4,5,5a,5b,6,7,7a,8,11,11a,11b,12,13,13a-헥사데카하이드로-2H-사이클로펜타[a]크리센-9-일)-1-(플루오로메틸)사이클로헥스-3-엔-1-카복실레이트(45 mg, 0.054 mmol)의 혼합물을 80℃에서 2시간 동안 교반하였다. 얻어진 혼합물을 1 N HCl로 pH 3 내지 4까지 산성화시키고, DCM으로 추출하였다. 유기층을 염수로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 감압 하에서 농축하여 미정제 생성물을 수득하고, 이를 HPLC(C18, 0.1% 포름산을 함유한 H2O 중 0 내지 100% MeCN)에 의해 정제하였다. 몇 방울의 디옥산 중 4 N HCl을 HPLC에 의해 수집된 분획에 첨가하여 표제 화합물을 HCl 염(13 mg, 32% 수율)으로서 수득하였다. LC-MS (ESI): m/z (M+1) = 741.52. 1H NMR (400 MHz, MeOD) δ 5.34 (s, 1H), 5.23 (d, J = 4.7 Hz, 1H), 4.55 (dd, J = 18.5, 8.8 Hz, 1H), 4.43 (dd, J = 18.4, 8.8 Hz, 1H), 3.30 - 3.14 (m, 8H), 3.07 - 1.41 (m, 30H), 1.31 - 1.17 (m, 9H), 1.15 - 0.81 (m, 12H).
MT4 세포 항바이러스 검정
실험 절차:
항바이러스 HIV 활성 및 화합물-유래 세포독성을 인간 T-세포 림프친화 바이러스 형질전환 세포주 MT4에서 프로피듐 요오다이드 기반 절차(propidium iodide based procedure)에 의해서 동시에 측정하였다. 시험 화합물의 분취액을 Cetus Pro/Pette을 사용하여 96-웰 플레이트(Costar 3598) 내의 배지(RPMI 1640, 10% 우태아혈청(FCS), 및 젠타마이신)에 순차적으로 희석시켰다. 기하급수적으로 성장하는 MT4 세포를 수확하고, Jouan 원심분리기(모델 CR4 12)에서 10분 동안 1000 rpm으로 원심분리하였다. 세포 펠릿을 신선한 배지(RPMI 1640, 20% FCS, 20% IL-2, 및 젠타마이신)에 5 × 105 세포/ml의 밀도로 재현탁시켰다. 세포 분취액을 100 × TCID50의 바이러스 감염 다중도를 얻도록 희석된 HIV-1(균주 IIIB)의 첨가에 의해서 감염시켰다. 유사한 세포 분취액을 배지로 희석시켜 모의-감염된 대조군(mock-infected control)을 제공하였다. 세포 감염은 가습된 5% CO2 대기를 지니는 조직 배양 인큐베이터에서 37℃에서 1시간 동안 진행시켰다. 1시간 동안 인큐베이션(incubation)한 후에, 바이러스/세포 현탁액을 신선한 배지로 6-배 희석시키고, 125 ㎕의 세포 현탁액을 사전-희석된 화합물을 함유하는 플레이트의 각각의 웰에 첨가하였다. 이어서, 플레이트를 가습된 5% CO2를 지니는 조직 배양 인큐베이터에 5일 동안 넣어 두었다. 인큐베이션 기간의 마지막에, 세포의 수 및 그에 따른 HIV-유도된 세포병증을 (A) 프로피듐 요오다이드 염색 또는 (B) MTS 테트라졸륨 염색 방법 중 어느 하나에 의해서 추정하였다.
프로피듐 요오다이드 판독을 위해, 27 ㎕의 5% Nonidet-40을 인큐베이션 플레이트의 각각의 웰에 첨가하였다. 코스타르 멀티팁 피페터(Costar multitip pipetter)로 완전히 혼합한 후에, 60 ㎕의 혼합물을 필터-바닥구성 96-웰 플레이트에 옮겼다. 플레이트를 자동화된 검정 기구(Screen Machine, Idexx Laboratories)에서 분석하였다. 사용된 대조군 및 표준은 모든 검정에서 0.01 내지 1 μM의 농도 범위에 걸쳐서 시험된 3'-아지도-3'-데옥시티미딘이었다. 3'-아지도-3'-데옥시티미딘에 대한 IC50 값의 예상 범위는 0.04 내지 0.12 μM이다. 이러한 검정은 프로피듐 요오다이드 염료를 사용하여 각각의 웰의 DNA 함량을 추정하게 한다.
MTS 판독을 위해, 20 ㎕ CellTiter 96 AQ One Solution 시약(Promega #G3582)을 각각의 웰에 첨가하였다. MTS 시약 첨가 후 75분 후에, 흡광도를 Tecan Sunrise96-웰 플레이트 판독기를 사용하여 492 nM에서 판독하였다.
분석:
시험 화합물의 항바이러스 효과는 EC50, 즉, HIV-유도된 세포병증 효과의 50% 감소를 생성시키는 억제 농도로서 보고된다. 이러한 효과는 비감염된 MT4 세포 대조군과 비교하여 HIV-감염된 MT4 세포의 세포 성장의 50%를 회복시키기 위해서 요구되는 시험 화합물의 양에 의해서 측정된다. IC50은 RoboSage, Automated Curve Fitting Program, version 5.00, 10-Jul-1995에 의해서 계산되었다.
각각의 검정 플레이트에 대해서, 화합물이 없는 비감염된 세포 또는 감염된 세포를 함유한 웰의 결과(상대적인 형광 단위, rfU 또는 OD 값)를 각각 평균하였다. 화합물-유래 세포독성의 측정을 위해서, 다양한 화합물 농도 및 비감염된 세포를 함유하는 웰로부터의 결과를 화합물 처리 없는 비감염된 세포의 평균과 비교하였다. 세포 잔류 백분율이 하기 식에 의해서 측정된다:
세포 잔류 백분율 = (화합물-처리된 비감염된 세포, rfU, 또는 OD 값 / 비처리된 비감염된 세포) × 100
79% 또는 그 미만의 세포 잔류 백분율의 수준은 그 농도에서의 화합물에 대한 직접적인 화합물-유래 세포독성의 유의 수준을 나타낸다. 이러한 상태가 발생하는 경우에 그러한 농도에서의 화합물-처리된 감염된 웰로부터의 결과는 EC50의 계산에 포함되지 않는다.
화합물 항바이러스 활성의 측정을 위해서, 다양한 화합물 농도 및 감염된 세포를 함유하는 웰로부터의 결과가 화합물 처리 없는 비감염된 및 감염된 세포의 평균과 비교된다. 바이러스의 억제 백분율은 하기 식에 의해서 측정된다:
바이러스 억제 백분율 = (1-((평균 비처리된 비감염된 세포 -처리된 감염된 세포) / (평균 비처리된 비감염된 세포 -평균 비처리된 감염된 세포))) × 100.
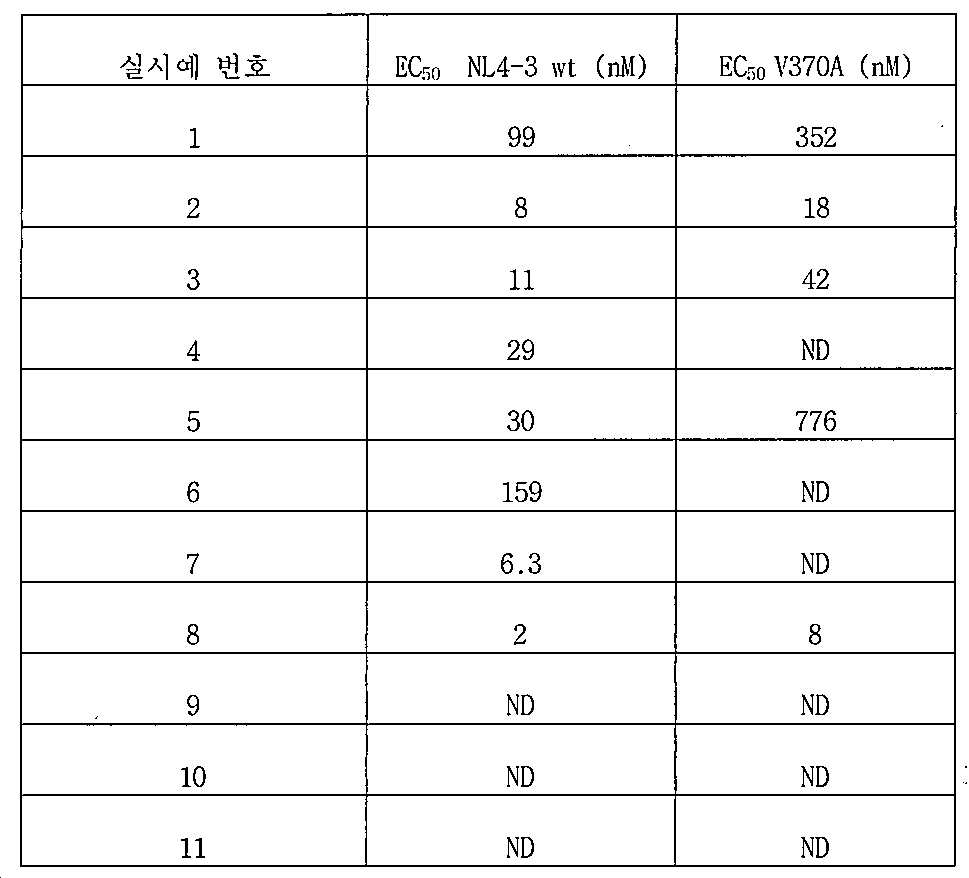
하기 표는 상기 실시예의 화합물에 대한 EC50 값을 요약한 것이다.
Claims (14)
- 하기 화학식 I의 화합물, 또는 이의 약제학적으로 허용되는 염:

상기 식에서,
R1은 H 또는 C1-4알킬이며;
L1은 결합, C1-8알킬렌, 또는 C0-3알킬렌C(O)C0-3알킬렌이며;
R2는 N(R3)R4, 또는 -OR3이며, 여기서, 각 R3 및 R4는 독립적으로, H, C1-3알킬, C1-3알킬렌-C3-6사이클로알킬, C1-3알킬렌-C5-7헤테로사이클, C1-3알킬렌N(C1-3알킬)2, C1-3알킬렌C5-7아릴, C(O)C5-7아릴, C(O)C1-4알킬, C1-3알킬렌C5-7헤테로아릴이며, 각 R3 및 R4는 할로겐, C1-4알킬, SO2C1-3알킬로부터 독립적으로 선택된 1 또는 2개의 치환체로 치환될 수 있거나;
R2가 N(R3)R4일 때, R3 및 R4는 함께 결합할 수 있고 이러한 것이 결합된 N과 함께 4원 내지 7원 헤테로사이클을 형성할 수 있으며, 상기 헤테로사이클은 N, S, 및 O로부터 선택된 1개의 다른 헤테로원자를 함유할 수 있으며, 상기 S 원자는 2개 이하의 옥소 기에 의해 치환될 수 있으며, 상기 헤테로사이클은 할로겐, C1-4알킬, SO2C1-3알킬로부터 독립적으로 선택된 1 또는 2개의 치환체로 치환될 수 있으며;
V는 C4-8사이클로알킬렌, C4-8사이클로알케닐렌, C5-8아릴, 또는 5원 내지 8원 헤테로아릴 고리이며, 여기서, V는 할로겐 및 C1-3알킬로부터 독립적으로 선택된 1 또는 2개의 치환체로 치환될 수 있으며, 상기 알킬은 1 또는 2개의 할로겐으로 치환될 수 있으며;
A는 -CO2C1-3알킬 또는 CO2H이다. - 제1항에 있어서, R1이 H인 화합물 또는 염.
- 제1항 또는 제2항에 있어서, L1이 C1-4알킬렌, C(O), 또는 C(O)C1-4알킬렌인 화합물 또는 염.
- 제1항 내지 제3항 중 어느 한 항에 있어서, R2가 N(R3)(R4)인 화합물 또는 염.
- 제1항 내지 제4항 중 어느 한 항에 있어서, R3 및 R4가 독립적으로, C1-3알킬렌N(C1-3알킬)2, 또는 C1-3알킬렌페닐이며, 여기서, 각 R3 및 R4가 할로겐 또는 SO2C1-3알킬로부터 독립적으로 선택된 1개의 치환체로 치환될 수 있거나; R3 및 R4가 함께 결합되고 이러한 것이 결합된 N과 함께 6원 헤테로사이클을 형성하며, 상기 헤테로사이클이 1개의 S 원자를 함유할 수 있으며, 상기 S 원자가 2개의 옥소 기에 의해 치환될 수 있으며; 각 R3 및 R4가 할로겐, C1-4알킬, SO2C1-3알킬로부터 독립적으로 선택된 1 또는 2개의 치환체로 치환될 수 있는 화합물 또는 염.
- 제1항 내지 제5항 중 어느 한 항에 있어서, V가 할로겐 또는 C1-3알킬로 임의적으로 치환된 페닐렌 또는 사이클로헥세닐렌이며, 여기서, 상기 알킬이 1 또는 2개의 할로겐으로 치환될 수 있는 화합물 또는 염.
- 제1항 내지 제6항 중 어느 한 항에 있어서, A가 -CO2H인 화합물 또는 염.
- 제1항 내지 제7항 중 어느 한 항에 있어서, 염이 염기 염인 염.
- 제8항에 있어서, 염이 라이신 염인 염.
- 제1항 내지 제9항 중 어느 한 항에 따른 화합물 또는 염을 포함하는 약제 조성물.
- 대상체에 제10항에 따른 약제 조성물을 투여하는 것을 포함하는, 인간에서 HIV 감염증을 치료하는 방법.
- 대상체에 제10항에 따른 약제 조성물을 투여하는 것을 포함하는, HIV 감염증이 발달할 위험이 있는 대상체에서 HIV 감염증을 예방하는 방법.
- 인간에서 HIV 감염증의 치료에 사용하기 위한 약제의 제조에서의 제1항 내지 제9항 중 어느 한 항에서 규정된 바와 같은 화합물 또는 염의 용도.
- 치료(therapy)에 사용하기 위한 약제의 제조에서의 제1항 내지 제9항 중 어느 한 항에서 규정된 바와 같은 화합물 또는 염의 용도.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201662280809P | 2016-01-20 | 2016-01-20 | |
| US62/280,809 | 2016-01-20 | ||
| PCT/IB2017/050279 WO2017125870A1 (en) | 2016-01-20 | 2017-01-19 | Amine derivatives of lupanes with hiv maturation inhibitory activity |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| KR20180098675A true KR20180098675A (ko) | 2018-09-04 |
Family
ID=57944462
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| KR1020187023717A Withdrawn KR20180098675A (ko) | 2016-01-20 | 2017-01-19 | Hiv 성숙 억제 활성을 갖는 루판의 아민 유도체 |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US20190023733A1 (ko) |
| EP (1) | EP3405474A1 (ko) |
| JP (1) | JP2019502728A (ko) |
| KR (1) | KR20180098675A (ko) |
| CN (1) | CN108699103A (ko) |
| AU (1) | AU2017208874A1 (ko) |
| BR (1) | BR112018014622A2 (ko) |
| CA (1) | CA3011616A1 (ko) |
| WO (1) | WO2017125870A1 (ko) |
Family Cites Families (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2711424A1 (en) * | 2008-01-03 | 2009-07-09 | Virochem Pharma Inc. | Novel lupane derivatives |
| WO2011100308A1 (en) | 2010-02-11 | 2011-08-18 | Glaxosmithkline Llc | Derivatives of betulin |
| US8754068B2 (en) | 2010-06-04 | 2014-06-17 | Bristol-Myers Squibb Company | Modified C-3 betulinic acid derivatives as HIV maturation inhibitors |
| RS54239B1 (sr) | 2010-06-04 | 2015-12-31 | Bristol-Myers Squibb Company | C-28 amidi modifikovanih derivata c-3 betulinske kiseline kao inhibitori sazrevanja hiv-a |
| US8846647B2 (en) * | 2011-01-31 | 2014-09-30 | Bristol-Myers Squibb Company | C-17 and C-3 modified triterpenoids with HIV maturation inhibitory activity |
| HUE026371T2 (en) | 2011-01-31 | 2016-05-30 | Bristol Myers Squibb Co | C-28 amines from modified C-3 betulinic acid derivatives as anti-HIV matrices |
| MX2014002936A (es) | 2011-09-21 | 2014-04-25 | Bristol Myers Squibb Co | Nuevos derivados de acido betulinico con actividad antiviral. |
| JO3387B1 (ar) | 2011-12-16 | 2019-03-13 | Glaxosmithkline Llc | مشتقات بيتولين |
| US8906889B2 (en) * | 2012-02-15 | 2014-12-09 | Bristol-Myers Squibb Company | C-3 cycloalkenyl triterpenoids with HIV maturation inhibitory activity |
| US8889854B2 (en) | 2012-05-07 | 2014-11-18 | Bristol-Myers Squibb Company | C-17 bicyclic amines of triterpenoids with HIV maturation inhibitory activity |
| GB201212937D0 (en) | 2012-07-20 | 2012-09-05 | Dupont Nutrition Biosci Aps | Method |
| JP6186010B2 (ja) | 2013-02-06 | 2017-08-23 | ブリストル−マイヤーズ スクイブ カンパニーBristol−Myers Squibb Company | Hiv成熟阻害活性を有するc−19修飾トリテルペノイド類 |
| US9969767B2 (en) * | 2014-11-14 | 2018-05-15 | VIIV Healthcare UK (No.5) Limited | Oxolupene derivatives |
-
2017
- 2017-01-19 EP EP17702433.8A patent/EP3405474A1/en not_active Withdrawn
- 2017-01-19 BR BR112018014622A patent/BR112018014622A2/pt not_active Application Discontinuation
- 2017-01-19 CA CA3011616A patent/CA3011616A1/en not_active Abandoned
- 2017-01-19 CN CN201780014642.9A patent/CN108699103A/zh active Pending
- 2017-01-19 KR KR1020187023717A patent/KR20180098675A/ko not_active Withdrawn
- 2017-01-19 JP JP2018537865A patent/JP2019502728A/ja active Pending
- 2017-01-19 WO PCT/IB2017/050279 patent/WO2017125870A1/en not_active Ceased
- 2017-01-19 AU AU2017208874A patent/AU2017208874A1/en not_active Abandoned
- 2017-01-19 US US16/070,325 patent/US20190023733A1/en not_active Abandoned
Also Published As
| Publication number | Publication date |
|---|---|
| CN108699103A (zh) | 2018-10-23 |
| US20190023733A1 (en) | 2019-01-24 |
| WO2017125870A1 (en) | 2017-07-27 |
| CA3011616A1 (en) | 2017-07-27 |
| JP2019502728A (ja) | 2019-01-31 |
| EP3405474A1 (en) | 2018-11-28 |
| BR112018014622A2 (pt) | 2018-12-11 |
| AU2017208874A1 (en) | 2018-08-02 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP0708085B1 (en) | Antiviral ethers of aspartate protease substrate isosteres | |
| KR20180134875A (ko) | B형 간염 항바이러스제 | |
| KR20070057936A (ko) | 질소 함유 복소환 유도체 및 이들을 유효 성분으로 하는약제 | |
| KR101419320B1 (ko) | 2-(치환된-아미노)-벤조티아졸 설폰아미드 hiv 프로테아제 저해제 | |
| KR20170024115A (ko) | 바이러스 감염의 치료에 사용하기 위한 이소인돌린 유도체 | |
| Mizuguchi et al. | A minimally cytotoxic CD4 mimic as an HIV entry inhibitor | |
| KR20070007759A (ko) | 바이러스 복제 억제제로서 유용한 치환된 아릴티오우레아유도체 | |
| JP2012516333A (ja) | Hivインテグラーゼ阻害剤としての架橋化合物 | |
| WO2015149607A1 (zh) | 杂环构建的三肽环氧酮类化合物、其制备方法和应用 | |
| EP3291810A1 (en) | Histone deacetylase inhibitors and compositions and methods of use thereof | |
| JP2008540529A (ja) | Hivインテグラ−ゼ阻害剤 | |
| JP2012528160A (ja) | Hivプロテアーゼ阻害薬 | |
| CN109836477A (zh) | 含有苯并噻二嗪-3-酮1,1-二氧化物的苯丙氨酸衍生物及其制备方法与应用 | |
| EP3172189A1 (en) | Phenyl and tertbutylacetic acid substituted pyridinones having anti-hiv effects | |
| WO2013016441A1 (en) | Hiv integrase inhibitory oxoisoindoline sulfonamides | |
| KR20070108856A (ko) | Hiv-1 캡시드 형성의 억제제: 치환된 아릴 아미노메틸티아졸 우레아 및 그것의 유사체 | |
| JP6710376B2 (ja) | Hiv感染阻害剤 | |
| KR20180098675A (ko) | Hiv 성숙 억제 활성을 갖는 루판의 아민 유도체 | |
| EP4052726B1 (en) | Cd4 mimic compound with anti-hiv activity | |
| CN101296901A (zh) | Hiv-1蛋白酶抑制剂 | |
| KR20180054826A (ko) | Hiv 성숙 억제 활성을 갖는 화합물 | |
| US11542288B2 (en) | Antiviral compounds | |
| WO2025114399A1 (en) | Flavivirus inhibitors | |
| WO2021231782A1 (en) | Perk inhibitors for treating viral infections | |
| WO2014028051A1 (en) | Inhibitors of protein phosphatase-1 and uses thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PA0105 | International application |
Patent event date: 20180817 Patent event code: PA01051R01D Comment text: International Patent Application |
|
| PG1501 | Laying open of application | ||
| PC1203 | Withdrawal of no request for examination | ||
| WITN | Application deemed withdrawn, e.g. because no request for examination was filed or no examination fee was paid |