KR20120120907A - 신규한 아연 아지드 착물 및 이를 이용한 테트라졸 유도체의 제조방법 - Google Patents
신규한 아연 아지드 착물 및 이를 이용한 테트라졸 유도체의 제조방법 Download PDFInfo
- Publication number
- KR20120120907A KR20120120907A KR1020120042543A KR20120042543A KR20120120907A KR 20120120907 A KR20120120907 A KR 20120120907A KR 1020120042543 A KR1020120042543 A KR 1020120042543A KR 20120042543 A KR20120042543 A KR 20120042543A KR 20120120907 A KR20120120907 A KR 20120120907A
- Authority
- KR
- South Korea
- Prior art keywords
- zinc
- azide
- alkyl
- formula
- complex
- Prior art date
Links
Images

Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F3/00—Compounds containing elements of Groups 2 or 12 of the Periodic System
- C07F3/06—Zinc compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K33/00—Medicinal preparations containing inorganic active ingredients
- A61K33/24—Heavy metals; Compounds thereof
- A61K33/30—Zinc; Compounds thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D257/00—Heterocyclic compounds containing rings having four nitrogen atoms as the only ring hetero atoms
- C07D257/02—Heterocyclic compounds containing rings having four nitrogen atoms as the only ring hetero atoms not condensed with other rings
- C07D257/04—Five-membered rings
Abstract
본 발명은 신규한 아연 아지드 착물에 관한 것이다. 본 발명은 또한, 아연 아지드 착물을 사용하여 니트릴 유도체로부터 5-치환된-1H-테트라졸 유도체를 제조하는 방법에 관한 것이다. 본 발명에 따라, 특히, 고혈압 치료용 약제학적 활성 화합물 또는 그의 제조에 유용한 중간체를 효과적으로 제조할 수 있다.
Description
본 발명은 신규한 아연 아지드 착물에 관한 것이다. 본 발명은 또한, 아연 아지드 착물을 사용하여 니트릴 유도체로부터 5-치환된-1H-테트라졸 유도체를 제조하는 방법에 관한 것이다. 본 발명에 따라, 특히, 고혈압 치료용 약제학적 활성 화합물 또는 그의 제조에 유용한 중간체를 효과적으로 제조할 수 있다.
일반적으로 테트라졸기는 생체 내에서 약리학적으로 카르복실산의 대체 역할을 하면서 약동력학적으로는 카르복실산보다 안정하여 각종 질환을 대상으로 하는 치료제의 핵심 구성요소로서의 역할을 하는 등 의약화학 분야에서 매우 중요한 위치를 차지하고 있다. 특히, 고혈압 및 울혈성 심부전증의 치료에 사용되고 있는 안지오텐신 II 수용체 차단제(ARB)로 널리 알려진 하기 화학식의 로사탄-칼륨, 발사탄, 칸데사탄 실렉세틸, 이베사탄, 올메사탄 메독소밀 및 피마살탄 등의 화합물은 공통적으로 테트라졸기를 포함하고 있다.


상기 ARB 계열 화합물의 제조 방법에 대해서는 기존에 다양한 합성 경로의 제조 방법들이 수많은 문헌들에 이미 보고되어 있고, 그 중 니트릴 화합물로부터 테트라졸 화합물을 제조하는 방법이 하기 문헌에 개시되어 있다.
로사탄칼륨의 경우 미국특허 제5,138,069호, 국제특허공개 WO2007/020654호 및 WO2007/026375호, 발사탄의 경우 미국특허 제5,965,592호 및 제5,399,579호, 국제특허공개 WO2007/014412호, 칸데사탄 실렉세틸의 경우 미국특허 제5,705,517호, 유럽특허 제 459,136호, 국제특허공개 WO2006/015134호, 2007/094015호 및 2007/054965호, 이베사탄의 경우 미국특허 제5,270,317호, 제5,629,331호 및 제7,211,676호, 미국특허공개 제20090286990호 및 국제특허공개 WO2007/013101호, 올메사탄 메독소밀의 경우 유럽 특허 제 503,785호에 개시되어 있다.
한편, 니트릴 화합물로부터 테트라졸 화합물을 합성하는 고전적인 방법으로는 N,N-디메틸포름아미드 용매를 사용하고 반응 온도 120℃에서 염화암모늄 존재 하에 니트릴 화합물을 나트륨 아지드와 반응시키는 방법이 알려져 있지만 (참조: W.G. Fineegan 등, J. Am . Chem . Soc ., 1958, 80, 3908), 니트릴 화합물이 입체적으로 방해를 받는(sterically hindered) 경우에는 매우 낮은 수율, 순도 및 반응 중에 생성되는 승화성의 아지드화암모늄에 의한 폭발의 위험성 등의 문제점이 있어 대규모 생산 시설에 적용하기에 어려운 단점이 있다.
트리메틸틴 아지드 또는 트리부틸틴 아지드를 사용한 방법도 비교적 양호한 수율로 테트라졸 화합물을 제조할 수 있는 방법으로 기 공지되어 있다 (참조: J.V. Duncia등 J. Org . Chem., 1991, 56, 2395). 그러나, 트리알킬틴 아지드 화합물을 사용하는 경우 이들 화합물의 독성 문제로 제조과정에 특별한 주의를 요하며, 환경 문제를 야기시킬 수 있고, 발생되는 폐액으로부터 주석을 전체적으로 회수해야 하는 것과 같은 추가의 공정이 요구됨으로써 높은 제조 비용이 예상되는 문제점이 있다.
또한, 최근에는, 앞서 언급한 트리알킬틴 아지드 화합물의 대체용으로 국제특허공개 WO2005/014602호 및 대한민국특허공개 제2006/0038994호에서 유기붕소 아지드 또는 유기알루미늄 아지드를 사용하여 테트라졸 화합물을 높은 수율 및 낮은 제조 비용으로 제조할 수 있고, 독성이 없어 친환경적인 공정의 장점이 있다고 보고하였다. 그러나, 유기붕소 아지드 및 유기알루미늄 아지드의 취급은 상당한 무수(anhydrous) 상태와 같은 특별한 주의를 필요로 하고 반응은 질소 또는 아르곤 하에서 수행되어야 하는 문제가 있다. 또한, 대한민국특허공개 제2006/0038994호의 실시예 12의 발사탄 제조에서는 디이소부틸-Al-아지드를 사용한 결과 해당 Al-아지드를 1차(1.4당량) 및 2차(0.8당량) 2회에 걸쳐 투입하여 110~130℃ 범위의 고온에서 반응시켜도 출발물질이 약 23% 남고, 생성물로의 전환율이 약 77% 정도 되는 낮은 전환율을 보여주고 있다.
그 외에도, 아연염 (Zn salt), 예를 들면 염화아연 (ZnCl2) 또는 브롬화아연 (ZnBr2) 및 나트륨 아지드를 사용한 방법이 안전하고 효율적인 테트라졸 유도체 제조방법으로 알려져 있고 (참조: B.M. Sharpless 등 J. Org . Chem., 2001, 66, 7945), 관련 특허로서 국제특허공개 WO96/037481호 및 미국특허 제5,502,191호 등이 공지되었다. 그러나, 입체적으로 방해를 받는 니트릴 화합물, 예를 들어, 발사탄과 같은 비페닐 니트릴 화합물의 경우 반응 완결을 위해서는 고온에서의 긴 반응시간 및 과량의 아연염과 나트륨 아지드가 사용되어야 하는 문제점이 있어 아연염 및 나트륨 아지드를 사용한 방법이 앞에서 언급된 기존 방법들의 단점을 해결하는 대안이 되기에는 부족하다.
이에 본 발명자들은 언급된 기존의 ARB 계열 화합물의 제조 방법들이 가지고 있는 문제점, 예를 들면 긴 반응시간, 독성이 심한 틴금속의 사용, 과량의 아지드염의 사용, 낮은 반응 전환율 및 수율, 승화성 부생성물에 의한 폭발 위험 등을 획기적으로 개선하고, 대규모 상업 생산에 적합하며, 경제적이고, 환경 친화적인 제조 방법을 개발하기 위해 집중적인 연구를 수행하였으며, 그 결과 본 발명을 완성하였다.
본 발명은 하기 화학식 (3)의 신규한 아연 아지드 착물에 관한 것이다:
[화학식 3]
상기 식에서
X는 NO3, OH, Cl, Br, I 또는 이들의 조합을 나타내며,
L은 Zn과 결합할 수 있는 아민 리간드를 나타내고,
a, b 및 c는 각각 0<a<2, 0<b<4 및 0≤c<2의 조건을 만족한다.
L은 바람직하게는 하기 그룹 중에서 선택된 양쪽결합 리간드(bidentate ligand) 중의 하나를 나타낸다:

그 중에서도 N,N,N',N'-테트라메틸에틸렌디아민(TMEDA)이 가장 바람직하다.
화학식 (3)의 신규한 아연 아지드 착물을 제조하기 위해서는 아연 화합물, 예를 들어, 질산아연 6수화물, 디클로로아연, 디브로모아연 또는 디요오도아연을 물에 녹인 다음 알칼리금속 아지드, 예를 들어, 리튬 아지드, 나트륨 아지드 또는 칼륨 아지드를 함께 반응기에 넣고 교반시키면서 혼합물을 45~60℃ 온도로 승온시킨다. 여기에 아민 리간드, 예를 들어, N,N,N',N'-테트라메틸에틸렌디아민(TMEDA)을 천천히 적가한다. 생성된 고체를 여과하고 물로 세척한 후 질소하 또는 진공하에서 건조시켜 목적하는 아연 아지드 착물을 수득한다.
이렇게 하여 얻어진 아연 아지드 착물은 혼합물 형태이며, 화학식 3으로 나타내어진다. 이를 정제하여 사용할 수도 있다. 하지만 혼합물 형태로 사용하는 것과 정제를 하여 순수하게 얻어진 형태로 사용하는 것은 반응에 있어서 크게 차이가 없다. 정제를 하여 얻어진 아연 아지드 착물의 구조는 도 1에 나타낸 바와 같다.
구체적인 반응조건은 문헌(J. Rollin등 Synthesis, 1990, 130-132)을 참고할 수 있으며, 목적하는 착물의 구조에 따라 아연 화합물, 나트륨 아지드 및 아민 리간드를 적정 몰비로 사용하여 반응시킨다.
본 발명에 따른 아연 아지드 착물 중에서 대표적인 것으로는 화학식 (3)에서 a가 1이고, b가 2이며, c가 0이고, L이 N,N,N',N'-테트라메틸에틸렌디아민 (TMEDA)인 아연 아지드 착물 Zn(TMEDA)(N3)2를 언급할 수 있다. 또한, 상응하는 혼합물로는 [Zn(NO3)2?6H2O, NaN3 및 TMEDA] 혼합물 또는 [ZnCl2, NaN3 및 TMEDA]의 혼합물을 언급할 수 있다.
본 발명은 또한, 착물 형성 반응에 의해 제조된 화학식 (3)의 아연 아지드 착물을 분리된 상태로 또는 동일 반응기 내에서 (in situ) 화학식 (2)의 니트릴 화합물과 반응시켜 화학식 (1)의 치환된 테트라졸 유도체를 제조하는 방법에 관한 것이다.
[화학식 1]

[화학식 2]
[화학식 3]
상기 식에서
R은 유기 잔기를 나타내고,
X, L, a, b 및 c는 앞에서 정의한 바와 같다.
바람직하게는, R은
1) 각각 임의로 치환된, 바람직하게는 임의로 1 내지 4 치환된, 직쇄 또는 측쇄 C1-C6-알킬 또는 C3-C6-사이클로알킬을 나타내고, 여기에서 치환체는 할로겐; 하이드록시; C1-C6- 알킬; C1-C6-알콕시; 각각 C1-C6-알킬, 할로겐, 하이드록시, 니트로 및 C1-C6-알콕시로 구성된 그룹 중에서 선택된 치환체에 의해 임의로 치환된, 바람직하게는 임의로 1 내지 4 치환된, 페닐, 피리딘, 피리미딘, 이미다졸, 티오펜 및 퓨란; 중에서 선택되거나,
2) 하기 구조식의 페닐 또는 비페닐을 나타내며:

여기에서
R1 및 R2는 각각 독립적으로 수소; 할로겐; 하이드록시; 니트로; C1-C6-알킬, 예를 들어, 메틸, 에틸, 프로필, 이소프로필, 부틸, t-부틸, 헥실; C3-C6-사이클로알킬, 예를 들어, 사이클로프로필, 사이클로부틸, 사이클로펜틸, 사이클로헥실; C1-C6-알콕시; 각각 할로겐, 카복시, 옥소, C1-C6-알킬, C1-C6-알콕시, 하이드록시-C1-C6-알킬, C1-C6-알콕시카보닐, C1-C6-알콕시카보닐-C1-C6-알킬, C3-C6-알칸디일, 디(C1-C6-알킬)아미노티오카보닐-C1-C6-알킬, 실렉세틸옥시카보닐 및 메독소밀옥시카보닐로 구성된 그룹 중에서 선택된 치환체에 의해 임의로 1 내지 4 치환된 페닐메틸, 피리딜메틸, 피리미딜메틸, 이미다졸릴메틸, 벤즈이미다졸릴메틸, 티오페닐메틸, 퓨라닐메틸; 각각 C1-C6-알킬, C3-C6-사이클로알킬, C1-C6-알킬카보닐, 카복시-C1-C6-알킬 및 C1-C6-알콕시카보닐-C1-C6-알킬로 구성된 그룹 중에서 선택된 1 또는 2개의 치환체에 의해 임의로 1 또는 2 치환된 아미노 또는 아미노-C1-C6-알킬, 예를 들어, 메틸아미노, 에틸아미노, 프로필아미노, 디메틸아미노, 디에틸아미노, 디이소프로필아미노, 사이클로프로필아미노, 사이클로부틸아미노, 사이클로펜틸아미노, 사이클로헥실아미노로 구성된 그룹으로부터 선택된다.
본 발명에 따라 제조된 화학식 (1) 화합물에서 치환기 R의 몇 가지 구체적인 구조를 도시하면 다음과 같다:

구조 b 에서 R3는 수소 또는 메틸을 나타내고,
구조 c 에서 R4는 수소, 메틸, 에틸 또는 실렉세틸을 나타내며,
구조 e 에서 R5는 수소, 메틸, 에틸 또는 메독소밀을 나타낸다.
화학식 (1)의 테트라졸 유도체 제조에 있어서, 화학식 (3)의 아연 아지드 착물을 형성시키고 여과 및 건조를 통해 분리된 화합물을 사용할 수도 있고, 아연 아지드 착물 형성반응을 수행한 다음 여과 및 건조 공정 없이 동일 반응기 내에서(in situ ) 생성된 아연 아지드 착물에 화학식 (2)의 출발물질을 첨가하여 반응시킬 수도 있다. 바람직하게는 아연 아지드 착물을 분리하여 사용한다.
본 발명에 따른 화학식 (1) 화합물의 제조 반응에서 용매로는 알킬화 벤젠, 예를 들어, 톨루엔, 크실렌, 에틸벤젠; 할로겐화 방향족 용매, 예를 들어, 클로로벤젠, o-, m- 또는 p-클로로톨루엔, 디클로로벤젠, 트리플루오로메틸벤젠; 비양자성 극성용매, 예를 들어, 디메틸포름아미드, 디메틸아세트아미드, N-메틸피롤리디논, 디메틸설폭사이드; 양자성 극성용매, 예를 들어, 에탄올, 프로판올, 이소프로판올, 부탄올, t-부탄올, 펜탄올, 사이클로프로판올, 사이클로부탄올, 사이클로펜탄올, 사이클로헥산올로 구성된 그룹 중에서 선택된 단일 또는 혼합 용매를 사용한다.
반응은 90~130℃ 온도에서 수행하는 것이 기준 시간 안에 높은 전환율을 달성하는데 유리하며, 바람직하게는 100~120℃ 온도가 적당하다. 반응 시간은 10~30 시간, 바람직하게는 15~24 시간의 범위가 적당하며 사용하는 출발물질에 따라 차이가 있다.
본 발명의 방법은 화학식 (1) 화합물의 제조 반응에 사용되는 출발물질이 반응성 치환기 또는 아지드와 반응할 수 있는 치환기 등을 포함하고 있는 경우 관능기를 보호 및 탈보호하는 것에 대해 공지된 종래의 방법을 이용하여 먼저, 반응성 치환기를 보호하고, 테트라졸 환을 형성한 후 상응하는 보호기를 제거하는 단계를 추가로 포함할 수 있다.
본 발명에 따르면, 특히, 고혈압 치료용 약제학적 활성 화합물 또는 그의 제조에 유용한 중간체를 효과적이고, 경제적이며, 환경 친화적인 방법으로 제조할 수 있다.
도 1은 실시예 1에 따라 제조된 아연 아지드 착물을 정제하여 순수하게 얻어진 Zn(N3)2TMEDA의 X-선 구조를 나타낸다.
이하, 본 발명을 하기 실시예 및 비교예를 통해 좀더 구체적으로 설명한다. 그러나 이는 본 발명에 대한 이해를 돕기 위한 것일 뿐, 어떤 의미로든 본 발명의 범위가 이들 실시예로 한정되어 해석되는 것은 아니다.
실시예
1-1: 아연
아지드
착물의
제조
6.25kg의 질산아연 6수화물과 4.0L의 물을 반응기에 투입하고 상온에서 교반시켜 완전히 용해시켰다. 2.73kg의 나트륨 아지드를 7.5L의 물에 완전히 용해시킨 용액을 반응기에 투입하였다. 반응 혼합물 온도를 45?50℃로 승온한 후 2.5kg의 N,N,N',N'-테트라메틸에틸렌디아민을 1시간에 걸쳐 천천히 적가하고 동온도에서 3시간 동안 교반하였다. 반응 혼합물을 상온으로 냉각시킨 후 생성된 고체는 여과하여 11.0L의 물로 세척하고 질소하에서 건조시켜 표제화합물을 얻었다.
1H NMR (δ ppm, DMSO-d6 , 300MHz), 2.3 (s, 6H), 2.5 (s, 4H)
13C NMR (δ ppm, DMSO-d6 , 75MHz), 47, 57
원소분석: C 22.8%, H 5.3%, N 40.2%
Zn2 +-EDTA 적정: 31% wt/wt
TMEDA 1H-NMR 적정: 33% (외부 표준물질: 아세트산 나트륨)
수분함량: 5%
적외선 스펙트럼: 2981, 2915, 2856, 2064(N3 stretching), 1464, 947 cm-1
실시예
1-2: 아연
아지드
착물의
제조
2.86kg의 염화아연과 4.0L의 물을 반응기에 투입하고 상온에서 교반시켜 완전히 용해시켰다. 2.73kg의 나트륨 아지드를 7.5L의 물에 완전히 용해시킨 용액을 반응기에 투입하였다. 반응 혼합물 온도를 45?50℃로 승온한 후 2.5kg의 N,N,N',N'-테트라메틸에틸렌디아민을 1시간에 걸쳐 천천히 적가하고 동온도에서 3시간 동안 교반하였다. 반응 혼합물을 상온으로 냉각시킨 후 생성된 고체는 여과하여 11.0L의 물로 세척하고 질소하에서 건조시켜 표제화합물을 얻었다.
비교예
1: 5-(4'-
메틸
[1,1'-비페닐]-2-일)-1H-
테트라졸의
제조
비교예
1-1: 본 발명의 분리된 아연
아지드
착물을
사용한 방법
2-(p-톨릴)벤조니트릴 2.1g과 톨루엔 5.0ml을 반응 용기에 투입하고 혼합물을 교반하면서 실시예 1-1에서 얻어진 고체화합물 4.3g을 투입하였다. 반응 혼합물을 가열 환류하고 24시간 후에 HPLC로 반응 전환율 99.2%를 확인하였다. 반응 혼합물을 실온으로 냉각하여 1N-HCl과 에틸아세테이트로 층을 분리하였다. 얻어진 유기층을 농축하고 n-헥산으로 슬러리화하여 순도 98.6%, 수율 84%의 순수한 백색 결정성 표제화합물을 얻었다.
HPLC: Agilent 1100 series, 용매: TFA 0.1%, 아세토니트릴(30→100)/물(70→0), 유속:1.5ml/분, 파장:225nm, 컬럼: CAPCELL PAK C18 (4.6mm I.D X 250 mm, 5 ㎛, TYPE AQ)
비교예
1-2: (
CH
3
)
3
SnN
3
를 사용한 방법
2-(p-톨릴)벤조니트릴 2.1g과 톨루엔 5.0ml을 반응용기에 투입하고 혼합물을 교반하면서, (CH3)3SnN3 3.3g을 투입하였다. 반응 혼합물을 가열 환류하여 24시간 후에 HPLC로 반응 전환율 79.8%를 확인하였다.
비교예
1-3: (
CH
3
CH
2
)
2
AlN
3
를 사용한 방법
건조된 반응 용기에 톨루엔 5.0ml 및 나트륨 아지드 1.0g을 투입한 후 혼합물을 0℃로 냉각하였다. 디에틸알루미늄 클로라이드 1.0 M 용액 14.8ml를 천천히 적가하였다. 반응혼합물을 12시간 이상 실온에서 교반하였다. 이어서 2-(p-톨릴)벤조니트릴 2.1g과 톨루엔 5.0ml을 반응 용기에 투입하고 혼합물을 가열 환류하여 24시간 후에 HPLC로 반응 전환율 86.1%를 확인하였다.
실시예
2: 2-부틸-4-
클로로
-5-
하이드록시메틸
-1-[(2'-(1H-
테트라졸
-5-일)-비페닐-4-일)-
메틸
]-
이미다졸
(
로사탄
)의 제조 (방법 1)
2-부틸-4-클로로-5-하이드록시메틸-1-[(2'-시아노-비페닐-4-일)-메틸]-이미다졸 379.9g과 n-부탄올 1.5L를 반응기에 투입하고 혼합물을 교반하면서, 실시예 1-1에서 얻은 고체화합물 370.7g을 투입하였다. 반응 혼합물의 내부 온도를 100?120℃로 유지하면서 24시간 반응 혼합물을 교반하였다. 반응기 내부 온도를 실온으로 냉각하고, 여기에 28% 암모니아수 1.0L와 물 1.0L를 적가한 후 층 분리 하였다. 얻어진 유기층에 물 1.7L와 수산화나트륨 240.0g을 넣고 상온에서 2시간 교반 후 다시 층 분리 하였다. 얻어진 유기층에 물 1.5L, 수산화나트륨 64.0g 및 염화나트륨 400g을 투입하여 다시 층 분리 하였다. 마지막으로 유기층에 물 1.8L를 넣고 교반하면서 황산을 사용하여 pH 3~4로 맞춘 후 15~17시간 교반하였다. 생성된 고체를 여과하여 n-부탄올 0.9L로 세척한 후 질소로 건조시켜 표제화합물 361.2g (수율 85.4%)을 얻었다.

실시예
3: 2-부틸-4-
클로로
-5-
하이드록시메틸
-1-[(2'-(1H-
테트라졸
-5-일)-비페닐-4-일)-
메틸
]-
이미다졸
(
로사탄
)의 제조 (방법 2)
n-부탄올 1.5L를 반응기에 투입하여 교반하면서 나트륨 아지드 189.0g과 염화아연 200.1g을 투입하였다. N,N,N,N'- 테트라메틸에틸렌디아민 168.5g을 천천히 적가하였다. 반응 혼합물을 100?120℃에서 3시간 이상 교반하고 실온으로 냉각 후에 2-부틸-4-클로로-5-하이드록시메틸-1-[(2'-시아노-비페닐-4-일)-메틸]-이미다졸 379.8g을 투입하고 다시 가열하여 내부 온도를 100?120℃로 유지하면서 24시간 반응시켰다. 내부 온도를 실온으로 냉각하고, 여기에 28% 암모니아수 1.0L와 물 1.0L을 적가한 후 층 분리 하였다. 얻어진 유기층에 물 1.7L와 수산화나트륨 240.0g을 넣고 상온에서 2시간 교반 후 다시 층 분리 하였다. 얻어진 유기층에 물 1.5L와 수산화나트륨 64.0g, 염화나트륨 400g을 투입하여 다시 층 분리 하였다. 마지막으로 유기층에 물 1.8L를 넣고 교반하면서 황산을 사용하여 pH 3~4로 맞춘 후 15~17시간 교반하였다. 생성된 고체를 여과하여 n-부탄올 0.9L로 세척한 후 질소로 건조하여 표제화합물 351.0g (수율 83.0%)을 얻었다.
실시예
4: 2-부틸-4-
클로로
-5-
하이드록시메틸
-1-[(2'-(1H-
테트라졸
-5-일)-비페닐-4-일)-
메틸
]-
이미다졸
(
로사탄
)의 제조 (방법 3)
n-부탄올 1.5L을 반응기에 투입하여 교반하면서 나트륨 아지드 189.0g과 질산아연 6수화물 445.3g을 투입하였다. N,N,N,N'-테트라메틸에틸렌디아민 168.5g을 천천히 적가하였다. 반응 혼합물을 100~120℃에서 3시간 이상 교반하고 실온으로 냉각시킨 후 2-부틸-4-클로로-5-하이드록시메틸-1-[(2'-시아노-비페닐-4-일)-메틸]-이미다졸 379.8g을 투입하였다. 다시 가열하여 내부 온도를 100~120℃로 유지하면서 24시간 반응시켰다. 내부 온도를 실온으로 냉각시키고, 여기에 28% 암모니아수 1.0L와 물 1.0L을 적가한 후 층 분리 하였다. 얻어진 유기층에 물 1.7L와 수산화나트륨 240.0g을 넣고 상온에서 2시간 교반한 후 다시 층 분리 하였다. 얻어진 유기층에 물 1.5L와 수산화나트륨 64.0g, 염화나트륨 400g 을 투입하여 층 분리 하였다. 마지막으로 유기층에 물 1.8L를 넣고 교반하면서 황산을 사용하여 pH 3~4로 맞춘 후 15~17시간 교반하였다. 생성된 고체를 여과하여 n-부탄올 0.9L로 세척한 후 질소로 건조하여 표제화합물 355.2g (수율 84.0%)을 얻었다.
실시예
5: 2-부틸-4-
클로로
-5-
하이드록시메틸
-1-[(2'-(1H-
테트라졸
-5-일)-비페닐-4-일)-
메틸
]-
이미다졸
칼륨염 (
로사탄
칼륨)의 제조
실시예 2 의 방법에 따라 제조된 2-부틸-4-클로로-5-하이드록시메틸-1-[(2'-(1H-테트라졸-5-일)-비페닐-4-일)-메틸]-이미다졸 122.6g을 이소프로판올 580mL에 현탁시켰다. 여기에 수산화칼륨 19.2g을 이소프로판올 580mL에 녹인 용액을 천천히 적가하였다. 혼합물이 모두 녹은 것을 확인한 후 온도를 올려 사용한 이소프로판올의 60~70%를 단순 증류시켰다. 증류가 완료되면 온도를 40~50℃로 낮춘 후 사이클로헥산 580mL를 약 1시간에 걸쳐서 천천히 적가하였다. 적가 후 서서히 실온으로 냉각시켜 2시간 교반하고 생성된 고체를 여과하여 이소프로판올과 사이클로헥산 혼합액으로 2회 세척하였다. 질소로 건조시켜 표제화합물 125.4g (수율 93.7%)을 수득하였다.

실시예
6: N-
펜타노일
-N-[(2'-
시아노비페닐
-4-일)
메틸
]-(L)-발린
메틸
에스테르의 제조
탄산칼륨 37.5g을 90mL의 물에 첨가하여 완전히 녹인 후 N-[(2'-시아노비페닐-4-일)메틸]-(L)-발린 메틸 에스테르 35.9g과 톨루엔 900mL을 투입하여 완전히 용해될 때까지 실온에서 교반하였다. 반응물이 녹은 것을 확인한 후 반응기에 18.0g의 발레릴 클로라이드를 천천히 투입하였다. 반응이 완료된 것을 확인한 후 900mL의 물을 추가 투입하여 층 분리 하였다. 얻어진 유기층을 900mL의 물로 세척하여 98.0% 순도의 표제화합물을 얻었으며, 별도의 정제과정없이 유기층을 다음 반응에 그대로 사용하였다.
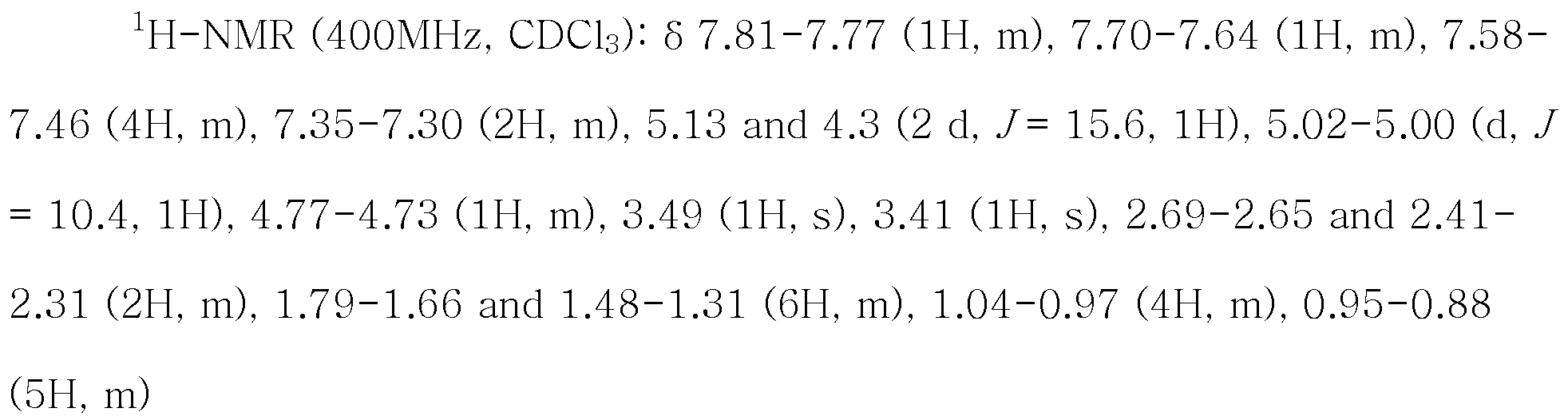
실시예
7: N-
펜타노일
-N-[(
2'-(1H-테트라졸-5-일)비페닐
-4-일)
메틸
]-(L)-발린 (
발사탄
)의 제조 (방법 1)
실시예 1-1에서 얻은 고체화합물 35.0g을 실시예 6에서 얻은 N-펜타노일-N-[(2'-시아노비페닐-4-일)메틸]-(L)-발린 메틸 에스테르의 톨루엔 용액에 가한 후 24시간 이상 가열하여 환류시켰다. 반응 혼합물을 45~55℃까지 냉각시키고, 반응 혼합물에 50mL의 물과 50mL의 28% 암모니아수를 투입한 후 층 분리하였다. 유기층을 5% 브라인용액 100mL를 사용하여 세척하고 50mL의 물과 황산 14.8g을 투입한 후 2시간 이상 실온에서 교반하였다. 혼합물을 층 분리하고 50mL의 물로 세척한 후 50mL의 물과 수산화나트륨 10.0g을 투입하여 실온에서 12시간 이상 교반하였다. 가수분해 반응 완결을 확인한 후 교반을 멈추고 층 분리를 통해서 얻어진 수용액 층에 디클로로메탄 100mL를 투입하였다. 여기에 황산 24.5g을 50mL의 물에 희석한 용액을 실온에서 투입하고 1시간 교반하였다. 층 분리하여 얻어진 유기층을 2회에 걸쳐 50mL의 물로 세척하고 노르말 헥산 150mL을 천천히 적가하여 실온에서 20시간 이상 교반하여 생성된 고체를 여과하였다. 여과한 고체를 노르말 헥산 50mL로 세척하고 건조시켜 38.7g의 표제화합물을 얻었다.
실시예
8: N-
펜타노일
-N-[(2'-(1H-
테트라졸
-5-일)비페닐-4-일)
메틸
]-(L)-발린 (
발사탄
)의 제조 (방법 2)
실시예 6에서 얻은 N-펜타노일-N-[(2'-시아노비페닐-4-일)메틸]-(L)-발린 메틸 에스테르의 톨루엔 용액에 나트륨 아지드 20.2g과 염화아연 21.8g을 투입하였다. 50~60℃까지 승온하여 1시간 교반한 후 N,N,N,N'-테트라메틸에틸렌디아민 18.6g을 천천히 적가하였다. 반응물을 가열 환류하여 24시간 이상 교반한 후 내부 온도를 45~55℃까지 냉각시키고 여기에 50mL의 물과 50mL의 28% 암모니아수를 첨가한 다음 층 분리 하였다. 유기층을 다시 100mL의 5%-브라인 용액으로 세척하고 100mL의 물과 황산 19.6g을 투입한 후 2시간 이상 실온에서 교반하였다. 혼합물을 층 분리하고 100mL의 물로 세척한 후 100mL의 물과 수산화나트륨 20.0g을 투입하여 실온에서 12시간 이상 교반하여 층 분리 하였다. 얻어진 수용액 층에 디클로로메탄 200mL와 황산 29.4g을 100mL의 물에 희석하여 실온에서 투입한 후 1시간 교반하였다. 층 분리하여 얻어진 유기층을 100mL의 물로 2회 세척하고 노르말 헥산 300mL를 실온에서 천천히 적가한 다음 실온에서 20시간 이상 교반하고 생성된 고체를 여과하였다. 여과한 고체를 노르말 헥산 100mL로 세척하고 건조시켜 표제화합물 36.9g을 얻었다.
실시예
9: 2-부틸-1-[(2'-(1H-
테트라졸
-5-일)비페닐-4-일)
메틸
]
스피로
[2-이미다졸린-4,1'-
사이클로펜탄
]-5-온 (
이베사탄
)의 제조 (방법 1)
2-부틸-3-[2'-{시아노비페닐-4-일}메틸]-1,3-디아자스피로[4,4]논-1-엔-4-온 5.0g과 톨루엔 15mL를 반응기에 투입하고 교반하면서 실시예 1-1에서 얻은 고체화합물 5.1g을 투입하였다. 반응기 내부 온도를 100?120℃로 유지하면서 20시간 이상 반응시켰다. 반응 완료를 확인한 후 내부 온도를 50~60℃로 냉각시키고, 28% 암모니아수 15mL와 물 15mL를 적가하여 층 분리하였다. 얻어진 유기층을 물 25mL로 세척한 후 25mL의 물과 35% 염산으로 pH2~3을 유지하면서 교반하였다. 생성된 고체를 여과하여 물 25mL로 세척하고 질소로 건조시켜 표제 화합물 4.8g 을 얻었다.

실시예
10: 2-부틸-1-[(2'-(1H-
테트라졸
-5-일)비페닐-4-일)
메틸
]
스피로
[2-이미다졸린-4,1'-
사이클로펜탄
]-5-온 (
이베사탄
)의 제조 (방법 2)
2-부틸-3-[2'-{시아노비페닐-4-일}메틸]-1,3-디아자스피로[4,4]논-1-엔-4-온 5.0g과 톨루엔 15mL를 반응기에 넣고 교반하면서 나트륨 아지드 2.7g, 염화아연 2.75g, N,N,N,N'-테트라메틸에틸렌디아민 2.3g을 투입하였다. 반응 혼합물의 내부 온도를 100?120℃로 유지하면서 24시간 이상 반응 혼합물을 교반하였다. 반응기 내부 온도를 50~60℃로 냉각시키고, 28% 암모니아수 15mL와 물 15mL를 적가하여 층 분리하였다. 얻어진 유기층을 물 25mL로 세척하고 25mL의 물과 35% 염산으로 pH2~3을 유지하면서 교반하였다. 생성된 고체를 여과하여 물 25mL로 세척하고 질소로 건조시켜 표제화합물 4.6g을 얻었다.
실시예
11: 4-(1-
하이드록시
-1-
메틸에틸
)-2-프로필-1-[2'-(1H-
테트라졸
-5-일)비페닐-4-
일메틸
]
이미다졸
-5-
카르복실산
(
올메사탄
)의 제조
1-[{2'-시아노비페닐-4-일}메틸]-4-(1-하이드록시-1-메틸에틸)-2-프로필이미다졸-5-카르본산 에틸 에스테르 4.3g, 에탄올 20mL 및 n-부탄올 20mL를 반응기에 투입하고 교반하면서, 실시예 1-1에서 얻은 고체 화합물 5.1g을 투입하였다. 반응 혼합물을 가열 환류하면서 24 시간 이상 반응시켰다. HPLC로 반응 완결을 확인한 후에 반응기 내부 온도를 실온으로 냉각시키고, 28% 암모니아수 20mL와 물 20mL를 적가하여 층 분리 하였다. 얻어진 유기층은 물 20mL와 황산으로 pH2~3을 유지하면서 1시간 이상 교반하였다. 층 분리하여 얻어진 유기층을 농축하고 여기에 THF 20mL와 물 10mL를 넣고 용해시킨 후 LiOH 0.85g을 투입하고 25℃에서 반응시켰다. HPLC로 반응 완결을 확인한 후에 1N-HCl로 중화하고 생성된 고체를 여과하여 표제화합물 3.6g을 얻었다.

Claims (14)
- 화학식 (3)의 아연 아지드 착물:
[화학식 3]

상기 식에서
X는 NO3, OH, Cl, Br, I 또는 이들의 조합을 나타내며,
L은 Zn과 결합할 수 있는 아민 리간드를 나타내고,
a, b 및 c는 각각 0<a<2, 0<b<4 및 0≤c<2의 조건을 만족한다. - 제1항에 있어서, L이 하기 그룹 중에서 선택된 양쪽결합 리간드(bidentate ligand) 중의 하나를 나타내는 아연 아지드 착물:

- 제2항에 있어서, L이 N,N,N,N'-테트라메틸에틸렌디아민 (TMEDA)인 아연 아지드 착물.
- 제1항에 있어서, 화학식 (3)에서 a가 1이고, b가 2이며, c가 0이고, L이 N,N,N,N'-테트라메틸에틸렌디아민 (TMEDA)인 아연 아지드 착물.
- 아연 화합물과 알칼리금속 아지드의 혼합물에 아민 리간드를 가하여 반응시킴을 특징으로 하여 제1항에 따른 화학식 (3)의 아연 아지드 착물을 제조하는 방법.
- 제5항에 있어서, 아연 화합물이 질산아연 6수화물, 디클로로아연, 디브로모아연 및 디요오도아연 중에서 선택되는 방법.
- 제5항에 있어서, 알칼리금속 아지드가 리튬 아지드, 나트륨 아지드 및 칼륨 아지드 중에서 선택되는 방법.
- 제5항의 착물 형성 반응에 의해 제조된 화학식 (3)의 아연 아지드 착물을 분리된 상태로 또는 동일 반응기 내에서 (in situ) 화학식 (2)의 니트릴 화합물과 반응시킴을 특징으로 하여 화학식 (1)의 치환된 테트라졸 유도체를 제조하는 방법:
[화학식 1]

[화학식 2]

상기 식에서
R은 유기 잔기를 나타낸다. - 제8항에 있어서, R이
1) 각각 임의로 치환된 직쇄 또는 측쇄 C1-C6-알킬 또는 C3-C6-사이클로알킬을 나타내고, 여기에서 치환체는 할로겐; 하이드록시; C1-C6- 알킬; C1-C6-알콕시; 각각 C1-C6-알킬, 할로겐, 하이드록시, 니트로 및 C1-C6-알콕시로 구성된 그룹 중에서 선택된 치환체에 의해 임의로 치환된 페닐, 피리딘, 피리미딘, 이미다졸, 티오펜 및 퓨란; 중에서 선택되거나,
2) 하기 구조식의 페닐 또는 비페닐을 나타내며:

여기에서
R1 및 R2는 각각 독립적으로 수소; 할로겐; 하이드록시; 니트로; C1-C6-알킬; C3-C6-사이클로알킬; C1-C6-알콕시; 각각 할로겐, 카복시, 옥소, C1-C6-알킬, C1-C6-알콕시, 하이드록시-C1-C6-알킬, C1-C6-알콕시카보닐, C1-C6-알콕시카보닐-C1-C6-알킬, C3-C6-알칸디일, 디(C1-C6-알킬)아미노티오카보닐-C1-C6-알킬, 실렉세틸옥시카보닐 및 메독소밀옥시카보닐로 구성된 그룹 중에서 선택된 치환체에 의해 임의로 1 내지 4 치환된 페닐메틸, 피리딜메틸, 피리미딜메틸, 이미다졸릴메틸, 벤즈이미다졸릴메틸, 티오페닐메틸, 퓨라닐메틸; 각각 C1-C6-알킬, C3-C6-사이클로알킬, C1-C6-알킬카보닐, 카복시-C1-C6-알킬 및 C1-C6-알콕시카보닐-C1-C6-알킬로 구성된 그룹 중에서 선택된 1 또는 2개의 치환체에 의해 임의로 1 또는 2 치환된 아미노 또는 아미노-C1-C6-알킬로 구성된 그룹으로부터 선택되는 방법. - 제8항에 있어서, R이 하기 구조 a 내지 f에서 선택되는 방법:

구조 b 에서 R3는 수소 또는 메틸을 나타내고,
구조 c 에서 R4는 수소, 메틸, 에틸 또는 실렉세틸을 나타내며,
구조 e 에서 R5는 수소, 메틸, 에틸 또는 메독소밀을 나타낸다. - 제8항에 있어서, 톨루엔, 크실렌, 에틸벤젠, 클로로벤젠, o-, m- 또는 p-클로로톨루엔, 디클로로벤젠, 트리플루오로메틸벤젠, 디메틸포름아미드, 디메틸아세트아미드, N-메틸피롤리디논, 디메틸설폭사이드, 에탄올, 프로판올, 이소프로판올, 부탄올, t-부탄올, 펜탄올, 사이클로프로판올, 사이클로부탄올, 사이클로펜탄올 및 사이클로헥산올로 구성된 그룹 중에서 선택된 단일 또는 혼합 용매를 사용하는 방법.
- 제8항에 있어서, 반응을 90~130℃ 온도에서 수행하는 방법.
- 제12항에 있어서, 반응을 100~120℃ 온도에서 수행하는 방법.
- 제8항에 있어서, 화학식 (2)의 화합물이 반응성 치환기 또는 아지드와 반응할 수 있는 치환기를 포함하는 경우, 반응성 치환기를 보호하고, 테트라졸 환을 형성한 후, 상응하는 보호기를 제거하는 단계를 추가로 포함하는 방법.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR20110038664 | 2011-04-25 | ||
| KR1020110038664 | 2011-04-25 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| KR20120120907A true KR20120120907A (ko) | 2012-11-02 |
| KR101942064B1 KR101942064B1 (ko) | 2019-01-24 |
Family
ID=47072884
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| KR1020120042543A KR101942064B1 (ko) | 2011-04-25 | 2012-04-24 | 신규한 아연 아지드 착물 및 이를 이용한 테트라졸 유도체의 제조방법 |
Country Status (2)
| Country | Link |
|---|---|
| KR (1) | KR101942064B1 (ko) |
| WO (1) | WO2012148148A2 (ko) |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN109761924B (zh) * | 2019-02-26 | 2020-09-01 | 安徽美诺华药物化学有限公司 | 一种改进的缬沙坦反应混合液的后处理方法 |
| CN112079788A (zh) * | 2019-06-13 | 2020-12-15 | 安徽美诺华药物化学有限公司 | 一种缬沙坦的制备方法 |
| CN110467604B (zh) * | 2019-08-29 | 2020-09-08 | 浙江天宇药业股份有限公司 | 一种氯沙坦的制备方法 |
| EP3939967A1 (en) | 2020-07-15 | 2022-01-19 | KRKA, d.d., Novo mesto | A continuous process for the preparation of (s)-methyl n-((2'-cyano-[1,1'-biphenyl]-4-yl)methyl)-n-pentanoylvalinate in a flow reactor |
| CN113501831B (zh) * | 2021-07-15 | 2022-11-15 | 西安近代化学研究所 | 一种5-氨基四氮唑锌配合物、合成方法及应用 |
| CN116375687A (zh) * | 2021-12-22 | 2023-07-04 | 浙江华海药业股份有限公司 | 一种高纯度的氯沙坦钾及其制备方法 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1996037481A1 (fr) * | 1995-05-26 | 1996-11-28 | Chugoku Kayaku Kabushiki Kaisha | Nouveau reactif de synthese de tetrazole et son emploi dans un procede de production de tetrazoles |
| CN101774975A (zh) * | 2009-12-25 | 2010-07-14 | 中国科学院过程工程研究所 | 一种离子液体催化的环合反应方法 |
| KR20120018826A (ko) * | 2003-07-15 | 2012-03-05 | 노파르티스 아게 | 유기 붕소 및 유기 알루미늄 아지드로부터 테트라졸 유도체의 제조 방법 |
-
2012
- 2012-04-24 KR KR1020120042543A patent/KR101942064B1/ko active IP Right Grant
- 2012-04-24 WO PCT/KR2012/003135 patent/WO2012148148A2/en active Application Filing
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1996037481A1 (fr) * | 1995-05-26 | 1996-11-28 | Chugoku Kayaku Kabushiki Kaisha | Nouveau reactif de synthese de tetrazole et son emploi dans un procede de production de tetrazoles |
| KR20120018826A (ko) * | 2003-07-15 | 2012-03-05 | 노파르티스 아게 | 유기 붕소 및 유기 알루미늄 아지드로부터 테트라졸 유도체의 제조 방법 |
| CN101774975A (zh) * | 2009-12-25 | 2010-07-14 | 中国科学院过程工程研究所 | 一种离子液体催化的环合反应方法 |
Non-Patent Citations (3)
| Title |
|---|
| M Montazerozohori et al, ‘Synthesis and spectral characterization of a new symmetric bidentate Schiff-base and its zinc complexes’, Journal of Coordination Chemistry, 2008, Vol.61, No.24, pp.3934-3942 * |
| M Montazerozohori et al, ‘Synthesis and spectral characterization of a new symmetric bidentate Schiff-base and its zinc complexes’, Journal of Coordination Chemistry, 2008, Vol.61, No.24, pp.3934-3942* * |
| 일본 재공표특허공보 WO09/637481 1부. * |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2012148148A9 (en) | 2013-02-14 |
| WO2012148148A3 (en) | 2013-01-03 |
| KR101942064B1 (ko) | 2019-01-24 |
| WO2012148148A2 (en) | 2012-11-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR101942064B1 (ko) | 신규한 아연 아지드 착물 및 이를 이용한 테트라졸 유도체의 제조방법 | |
| US8592474B2 (en) | Process for the preparation or purification of olmesartan medoxomil | |
| US8530506B2 (en) | Process for production of biphenyl derivative | |
| US20080076932A1 (en) | A process for the preparation of phenyltetrazole compounds | |
| US7741492B2 (en) | Method for obtaining a pharmaceutically active compound (Irbesartan) and its synthesis intermediate | |
| EP1885714B1 (en) | Process for the preparation of 2-alkyl-1-((2'-substituted-biphenyl-4-yl)methyl)-imidazole, dihydroimidazole or benzimidazole derivatives | |
| KR102443292B1 (ko) | 1-[5-(2-플루오로페닐)-1-(피리딘-3-일술포닐)-1h-피롤-3-일]-n-메틸메탄아민모노푸마르산염의 제조법 | |
| JP5925899B2 (ja) | ビアリール化合物の製造方法 | |
| CN102060798A (zh) | 2-(1-氢-4-四唑)-4′- 甲基联苯及其衍生物的合成方法 | |
| KR100662110B1 (ko) | 테트라졸 유도체의 제조방법 | |
| WO2006134078A1 (en) | Method for obtaining benzimidazole derivatives and intermediates thereof | |
| KR100809159B1 (ko) | 로사탄의 개선된 제조방법 | |
| US20060183916A1 (en) | Process for the preparation of phenyltetrazole derivatives | |
| Shuangxia et al. | An efficient and green synthetic route to losartan | |
| WO2010133909A2 (en) | Process for preparation of 5-substituted tetrazoles | |
| US20080281097A1 (en) | Process for Preparing an Angiotensin II Receptor Antagonist | |
| KR101009404B1 (ko) | (에스)-엔-(1-카르복시-2-메틸-프로-1-필)-엔-펜타노일-엔-[2'-(1에이취-테트라졸-5-일)비페닐-4-일-메틸]아민화합물의 고순도 제조방법 | |
| KR101012135B1 (ko) | 발사르탄 메틸 에스테르의 제조방법 | |
| WO2010065432A1 (en) | Nitrooxy derivatives as angiotensin ii receptor antagonists | |
| KR20070117381A (ko) | 로사탄의 새로운 제조방법 | |
| KR100995755B1 (ko) | 트리틸 칸데사르탄 실렉세틸의 개선된 제조방법 | |
| KR20110109638A (ko) | 올메사탄 실렉세틸의 제조 방법 | |
| KR20070087764A (ko) | 로사탄의 제조방법 | |
| JP2019196359A (ja) | ピリミジン誘導体およびそれらの中間体を調製する化学的方法 | |
| CN111233921A (zh) | 新化合物及其用于合成磷霉素杂质d的方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| N231 | Notification of change of applicant | ||
| A201 | Request for examination | ||
| E902 | Notification of reason for refusal | ||
| E701 | Decision to grant or registration of patent right |