JP7586829B2 - Tyk2阻害剤およびその使用 - Google Patents
Tyk2阻害剤およびその使用 Download PDFInfo
- Publication number
- JP7586829B2 JP7586829B2 JP2021552136A JP2021552136A JP7586829B2 JP 7586829 B2 JP7586829 B2 JP 7586829B2 JP 2021552136 A JP2021552136 A JP 2021552136A JP 2021552136 A JP2021552136 A JP 2021552136A JP 7586829 B2 JP7586829 B2 JP 7586829B2
- Authority
- JP
- Japan
- Prior art keywords
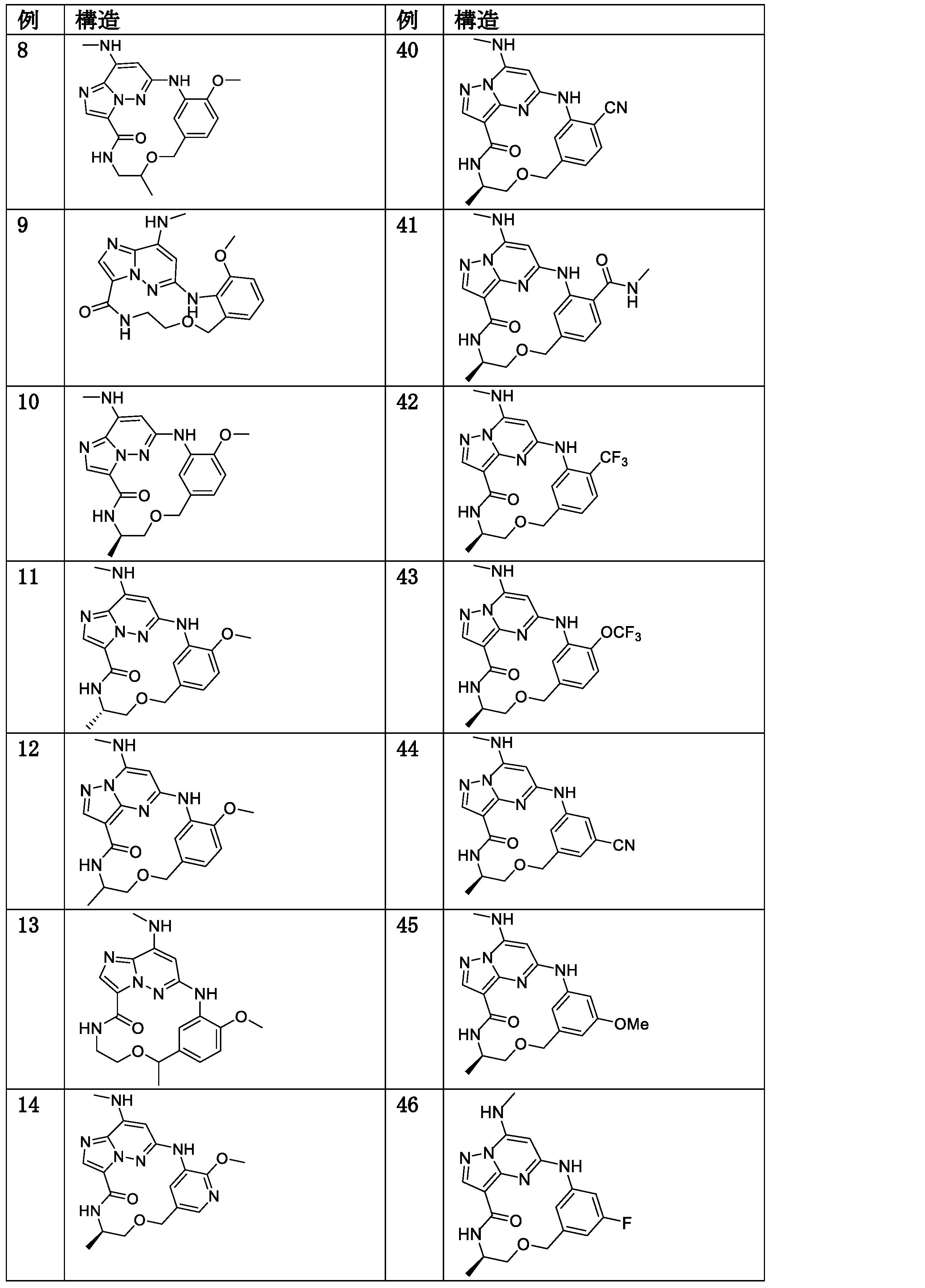
- alkyl
- mmol
- optionally substituted
- heterocycloalkyl
- haloalkyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
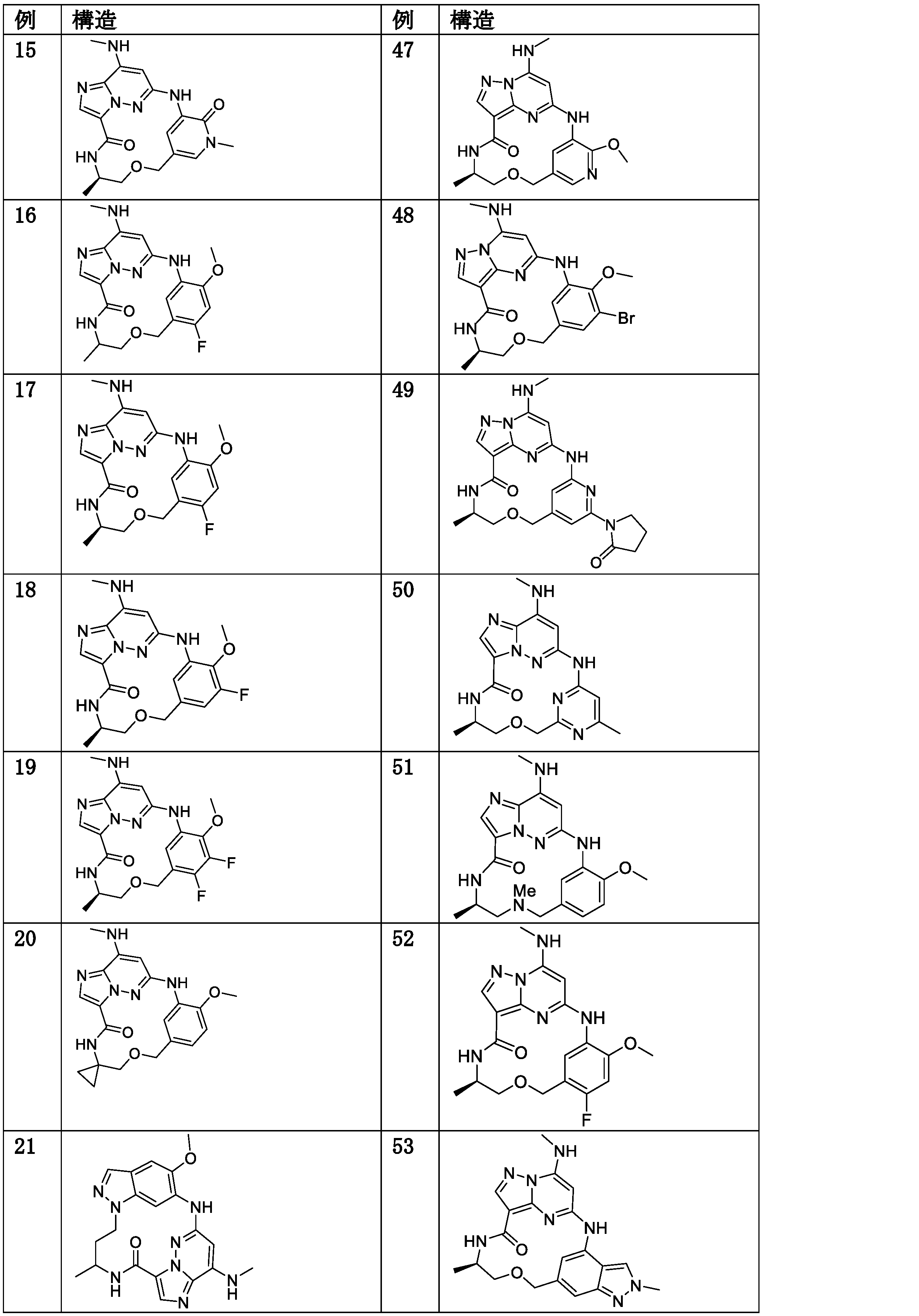
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/22—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains four or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/12—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains three hetero rings
- C07D498/18—Bridged systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/50—Pyridazines; Hydrogenated pyridazines
- A61K31/5025—Pyridazines; Hydrogenated pyridazines ortho- or peri-condensed with heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/50—Pyridazines; Hydrogenated pyridazines
- A61K31/504—Pyridazines; Hydrogenated pyridazines forming part of bridged ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/12—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains three hetero rings
- C07D491/18—Bridged systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/12—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains three hetero rings
- C07D498/16—Peri-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/22—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains four or more hetero rings
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Immunology (AREA)
- Epidemiology (AREA)
- Neurosurgery (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Transplantation (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
Description
本特許出願は、2019年3月11日に出願された米国仮特許出願第62/816,698号明細書、2019年4月17日に出願された米国仮特許出願第62/835,376号明細書、2019年7月23日に出願された米国仮特許出願第62/877,741号明細書、および2019年11月5日に出願された米国仮特許出願第62/931,119号明細書の利益を主張するものであり、それら各出願は、その全体で参照により本明細書に援用される。

Lは、4~10原子のリンカーであり、一つ以上のRLで任意に置換され、
各RLは独立して、重水素、ハロゲン、-CN,-ORb,-SRb,-S(=O)Ra,-S(=O)2Ra,-NO2,-NRcRd,-NHS(=O)2Ra,-S(=O)2NRcRd,-C(=O)Ra,-OC(=O)Ra,-C(=O)ORb,-OC(=O)ORb,-C(=O)NRcRd,-OC(=O)NRcRd,-NRbC(=O)NRcRd,-NRbC(=O)Ra,-NRbC(=O)ORb,C1-C6アルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、C2-C6アルケニル、C2-C6アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、もしくはヘテロアリールであり、または同じ炭素上の二つのRLが一緒になってオキソ、シクロアルキルもしくはヘテロシクロアルキルを形成し、または異なる炭素上の二つのRLが一緒になってシクロアルキルもしくはヘテロシクロアルキルを形成し、
環Aは、シクロアルキル、ヘテロシクロアルキル、アリール、またはヘテロアリールであり、
各RAは独立して、重水素、ハロゲン、-CN,-ORb,-SRb,-S(=O)Ra,-S(=O)2Ra,-NO2,-NRcRd,-NHS(=O)2Ra,-S(=O)2NRcRd,-C(=O)Ra,-OC(=O)Ra,-C(=O)ORb,-OC(=O)ORb,-C(=O)NRcRd,-OC(=O)NRcRd,-NRbC(=O)NRcRd,-NRbC(=O)Ra,-NRbC(=O)ORb,C1-C6アルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、C2-C6アルケニル、C2-C6アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、もしくはヘテロアリールであり、ここで各アルキル、アルケニル、アルキニル、シクロアルキル、ヘテロシクロアルキル、アリールおよびヘテロアリールは独立して一つ以上のRA1で任意に置換され、または同じ炭素上の二つのRAが一緒になってオキソを形成し、
各RA1は独立して、重水素、ハロゲン、-CN,-ORb,-SRb,-S(=O)Ra,-S(=O)2Ra,-NO2,-NRcRd,-NHS(=O)2Ra,-S(=O)2NRcRd,-C(=O)Ra,-OC(=O)Ra,-C(=O)ORb,-OC(=O)ORb,-C(=O)NRcRd,-OC(=O)NRcRd,-NRbC(=O)NRcRd,-NRbC(=O)Ra,-NRbC(=O)ORb,C1-C6アルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、C2-C6アルケニル、C2-C6アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、もしくはヘテロアリールであり、または同じ炭素上の二つのRA1が一緒になってオキソを形成し、
nは、0~4であり、
X1およびX2は、-N-または-C=であり、ただしX1またはX2のうちの一方が-N-で他方が-C=であり、
Y8は、CR8またはNであり、
Y6は、CR6またはNであり、
Y3は、CR3またはNであり、
Y9は、CR9またはNであり、
R3、R6、R8およびR9は独立して、水素、重水素、ハロゲン、-CN,-ORb,-SRb,-S(=O)Ra,-S(=O)2Ra,-NO2,-NRcRd,-NHS(=O)2Ra,-S(=O)2NRcRd,-C(=O)Ra,-OC(=O)Ra,-C(=O)ORb,-OC(=O)ORb,-C(=O)NRcRd,-OC(=O)NRcRd,-NRbC(=O)NRcRd,-NRbC(=O)Ra,-NRbC(=O)ORb,C1-C6アルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、C2-C6アルケニル、またはC2-C6アルキニルであり、
R4は、水素、C1-C6アルキル、C1-C6ヘテロアルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、C2-C6アルケニル、C2-C6アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、またはヘテロアリールであり、ここで各アルキル、アルケニル、アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、およびヘテロアリールは一つ以上のR4aで任意に置換され、
各R4aは独立して、重水素、ハロゲン、-CN,-ORb,-NRcRd,-C(=O)Ra,-C(=O)ORb,-C(=O)NRcRd,C1-C6アルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、シクロアルキル、ヘテロシクロアルキル、アリール、もしくはヘテロアリールであり、または同じ炭素上の二つのR4aが一緒になってオキソを形成し、
R5は、水素、C1-C6アルキル、C1-C6ハロアルキル、またはC1-C6ジュウテロアルキル(deuteroalkyl)であり、
R7は、水素、C1-C6アルキル、C1-C6ハロアルキル、またはC1-C6ジュウテロアルキル(deuteroalkyl)であり、
各Raは独立して、C1-C6アルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、C2-C6アルケニル、C2-C6アルキニル、シクロアルキル、ヘテロシクロアルキル、アリールまたはヘテロアリールであり、ここで各アルキル、アルケニル、アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、およびヘテロアリールは独立して、一つ以上のオキソ、重水素、ハロゲン、-CN、-OH、-OMe、-NH2、-C(=O)Me、-C(=O)OH、-C(=O)OMe、C1-C6アルキル、またはC1-C6ハロアルキルで任意に置換され、
各Rbは独立して、水素、C1-C6アルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、C2-C6アルケニル、C2-C6アルキニル、シクロアルキル、ヘテロシクロアルキル、アリールまたはヘテロアリールであり、ここで、各アルキル、アルケニル、アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、およびヘテロアリールは独立して、一つ以上のオキソ、重水素、ハロゲン、-CN、-OH、-OMe、-NH2、-C(=O)Me、-C(=O)OH、-C(=O)OMe、C1-C6アルキル、またはC1-C6ハロアルキルで任意に置換され、また
各RcおよびRdは独立して、水素、C1-C6アルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、C2-C6アルケニル、C2-C6アルキニル、シクロアルキル、ヘテロシクロアルキル、アリールもしくはヘテロアリールであり、ここで、各アルキル、アルケニル、アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、およびヘテロアリールは独立して、一つ以上のオキソ、重水素、ハロゲン、-CN,-OH,-OMe,-NH2,-C(=O)Me,-C(=O)OH,-C(=O)OMe,C1-C6アルキル、もしくはC1-C6ハロアルキルで任意に置換され、
またはRcおよびRdは、それらが結合される窒素原子と一緒になって一つ以上のオキソ、重水素、ハロゲン、-CN,-OH,-OMe,-NH2,-C(=O)Me,-C(=O)OH,-C(=O)OMe,C1-C6アルキル、もしくはC1-C6ハロアルキルで任意に置換されるヘテロシクロアルキルを形成する。

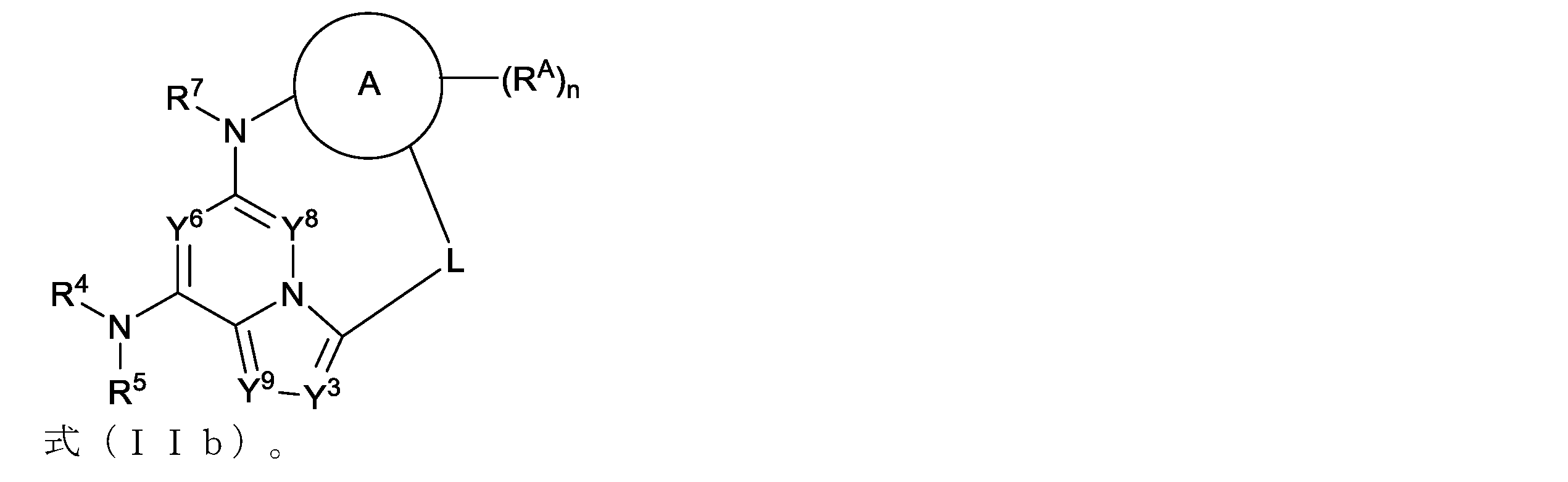
本明細書において言及される全ての公開文献、特許、および特許出願は、本明細書に特定される具体的な目的で、参照により本明細書に組み込まれる。
本明細書および添付の特許請求の範囲において使用される場合、文脈から別段であることが明白でない限り、単数形“a”、“an”および“the”は複数形の指示対象も含む。したがって例えば、「薬剤」への言及は、複数の当該薬剤を含み、「当該細胞」への言及は、一つ以上の細胞(または複数の細胞)および当業者に公知であるその均等物などへの言及を含む。例えば分子量などの物理的特性、または例えば化学式などの化学的特性に関し、本明細書において範囲が使用される場合、範囲のすべての組み合わせおよび副次的な組み合わせならびにその中の特定の実施形態が含まれることが意図される。用語「約」は、数値または数値範囲を指す場合、参照される数または数値範囲が、実験上の変動の範囲内(または統計的な実験誤差の範囲内)の近似値であることを指し、したがって、一部の例では当該数または当該数値範囲は、記載される数または数値範囲の1%~15%の間で変動することとなる。「含むこと」という用語(および例えば、「含む」、または「有すること」または「含有すること」などの関連用語)は、他の特定の実施形態では、例えば、本明細書に記載される任意の物質の組成、組成物、方法、またはプロセスなどの実施形態が、記載される特徴「からなる」または記載される特徴「から本質的になる」ことを除外することは意図されない。
本明細書において、TYK2介在性障害の治療に有用な化合物が記載される。一部の実施形態では、TYK2介在性障害は、自己免疫性障害、炎症性障害、増殖性障害、内分泌障害、神経学的障害、または移植に関連する障害である。

環Aは、任意に置換されたシクロアルキル、任意に置換されたヘテロシクロアルキル、任意に置換されたアリール、または任意に置換されたヘテロアリールであり、
Xは、CR8またはNであり、
R1は、-S(=O)R10,-S(=O)2R10,-S(=O)2NR12R13,-C(=O)R10,-C(=O)OR11,-C(=O)NR12R13,任意に置換されたC1-C6アルキル、任意に置換されたC1-C6ヘテロアルキル、任意に置換されたC1-C6ハロアルキル、任意に置換されたC1-C6ジュウテロアルキル(deuteroalkyl)、任意に置換されたC1-C6ヒドロキシアルキル、任意に置換されたC1-C6アミノアルキル、任意に置換されたC2-C6アルケニル、任意に置換されたC2-C6アルキニル、任意に置換されたシクロアルキル、任意に置換されたヘテロシクロアルキル、任意に置換されたアリール、または任意に置換されたヘテロアリールであり、
R2は、水素、任意に置換されたC1-C6アルキル、任意に置換されたC1-C6ハロアルキル、または任意に置換されたC1-C6ジュウテロアルキル(deuteroalkyl)であり、
R3、R6、およびR8は独立して、水素、重水素、ハロゲン、-CN,-ORb,-SRb,-S(=O)Ra,-S(=O)2Ra,-NO2,-NRcRd,-NHS(=O)2Ra,-S(=O)2NRcRd,-C(=O)Ra,-OC(=O)Ra,-C(=O)ORb,-OC(=O)ORb,-C(=O)NRcRd,-OC(=O)NRcRd,-NRbC(=O)NRcRd,-NRbC(=O)Ra,-NRbC(=O)ORb,任意に置換されたC1-C6アルキル、任意に置換されたC1-C6ハロアルキル、任意に置換されたC1-C6ジュウテロアルキル(deuteroalkyl)、任意に置換されたC1-C6ヒドロキシアルキル、任意に置換されたC1-C6アミノアルキル、任意に置換されたC2-C6アルケニル、または任意に置換されたC2-C6アルキニルであり、
R4は、水素、任意に置換されたC1-C6アルキル、任意に置換されたC1-C6ヘテロアルキル、任意に置換されたC1-C6ハロアルキル、任意に置換されたC1-C6ジュウテロアルキル(deuteroalkyl)、任意に置換されたC1-C6ヒドロキシアルキル、任意に置換されたC1-C6アミノアルキル、任意に置換されたC2-C6アルケニル、任意に置換されたC2-C6アルキニル、任意に置換されたシクロアルキル、任意に置換されたヘテロシクロアルキル、任意に置換されたアリール、または任意に置換されたヘテロアリールであり、
R5は、水素、任意に置換されたC1-C6アルキル、任意に置換されたC1-C6ハロアルキル、または任意に置換されたC1-C6ジュウテロアルキル(deuteroalkyl)であり、
R7は、水素、任意に置換されたC1-C6アルキル、任意に置換されたC1-C6ハロアルキル、または任意に置換されたC1-C6ジュウテロアルキル(deuteroalkyl)であり、
各R10は独立して、任意に置換されたC1-C6アルキル、任意に置換されたC1-C6ヘテロアルキル、任意に置換されたC1-C6ハロアルキル、任意に置換されたC1-C6ジュウテロアルキル(deuteroalkyl)、任意に置換されたC1-C6ヒドロキシアルキル、任意に置換されたC1-C6アミノアルキル、任意に置換されたC2-C6アルケニル、任意に置換されたC2-C6アルキニル、任意に置換されたシクロアルキル、任意に置換されたヘテロシクロアルキル、任意に置換されたアリール、または任意に置換されたヘテロアリールであり、
各R11は独立して、水素、任意に置換されたC1-C6アルキル、任意に置換されたC1-C6ハロアルキル、任意に置換されたC1-C6ジュウテロアルキル(deuteroalkyl)、任意に置換されたC1-C6ヒドロキシアルキル、任意に置換されたC1-C6アミノアルキル、任意に置換されたC2-C6アルケニル、任意に置換されたC2-C6アルキニル、任意に置換されたシクロアルキル、任意に置換されたヘテロシクロアルキル、任意に置換されたアリール、または任意に置換されたヘテロアリールであり、
各R12およびR13は独立して、水素、任意に置換されたC1-C6アルキル、任意に置換されたC1-C6ハロアルキル、任意に置換されたC1-C6ジュウテロアルキル(deuteroalkyl)、任意に置換されたC1-C6ヒドロキシアルキル、任意に置換されたC1-C6アミノアルキル、任意に置換されたC2-C6アルケニル、任意に置換されたC2-C6アルキニル、任意に置換されたシクロアルキル、任意に置換されたヘテロシクロアルキル、任意に置換されたアリール、もしくは任意に置換されたヘテロアリールであり、
またはR12およびR13は、それらが結合される窒素原子と一緒になって、任意に置換されるヘテロシクロアルキルを形成し;
各Raは独立して、C1-C6アルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、C2-C6アルケニル、C2-C6アルキニル、シクロアルキル、ヘテロシクロアルキル、アリールまたはヘテロアリールであり、ここで各アルキル、アルケニル、アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、およびヘテロアリールは独立して、一つ以上のオキソ、重水素、ハロゲン-CN,-OH,-OMe,-NH2,-C(=O)Me,-C(=O)OH,-C(=O)OMe,C1-C6アルキル、またはC1-C6ハロアルキルで任意に置換され、
各Rbは独立して、水素、C1-C6アルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、C2-C6アルケニル、C2-C6アルキニル、シクロアルキル、ヘテロシクロアルキル、アリールまたはヘテロアリールであり、ここで各アルキル、アルケニル、アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、およびヘテロアリールは独立して、一つ以上のオキソ、重水素、ハロゲン、-CN,-OH,-OMe,-NH2,-C(=O)Me,-C(=O)OH,-C(=O)OMe,C1-C6アルキル、またはC1-C6ハロアルキルで任意に置換され;また
各RcおよびRdは独立して、水素、C1-C6アルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、C2-C6アルケニル、C2-C6アルキニル、シクロアルキル、ヘテロシクロアルキル、アリールもしくはヘテロアリールであり、ここで各アルキル、アルケニル、アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、およびヘテロアリールは独立して、一つ以上のオキソ、重水素、ハロゲン、-CN,-OH,-OMe,-NH2,-C(=O)Me,-C(=O)OH,-C(=O)OMe,C1-C6アルキル、もしくはC1-C6ハロアルキルで任意に置換され、
またはRcおよびRdは、それらが結合される窒素原子と一緒になって一つ以上のオキソ、重水素、ハロゲン、-CN,-OH,-OMe,-NH2,-C(=O)Me,-C(=O)OH,-C(=O)OMe,C1-C6アルキル、もしくはC1-C6ハロアルキルで任意に置換されるヘテロシクロアルキルを形成する。

環Aは、シクロアルキル、ヘテロシクロアルキル、アリール、またはヘテロアリールであり、それら各々が一つ以上のRAで任意に置換され、
各RAは独立して、重水素、ハロゲン、-CN,-OR15,-SR15,-S(=O)R14,-S(=O)2R14,-NO2,-NR16R17,-NHS(=O)2R14,-S(=O)2NR16R17,-C(=O)R14,-OC(=O)R14,-C(=O)OR15,-OC(=O)OR15,-C(=O)NR16R17,-OC(=O)NR16R17,-NR15C(=O)NR16R17,-NR15C(=O)R14,-NR15C(=O)OR15,C1-C6アルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、C2-C6アルケニル、C2-C6アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、もしくはヘテロアリールであり、ここで各アルキル、アルケニル、アルキニル、シクロアルキル、ヘテロシクロアルキル、アリールおよびヘテロアリールは独立して一つ以上のRA1で任意に置換され、または同じ炭素上の二つのRAが一緒になってオキソを形成し、
各RA1は独立して、重水素、ハロゲン、-CN,-OR15,-SR15,-S(=O)R14,-S(=O)2R14,-NO2,-NR16R17,-NHS(=O)2R14,-S(=O)2NR16R17,-C(=O)R14,-OC(=O)R14,-C(=O)OR15,-OC(=O)OR15,-C(=O)NR16R17,-OC(=O)NR16R17,-NR15C(=O)NR16R17,-NR15C(=O)R14,-NR15C(=O)OR15,C1-C6アルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、C2-C6アルケニル、C2-C6アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、もしくはヘテロアリールであり、または同じ炭素上の二つのRA1が一緒になってオキソを形成し、
各R14は独立して、C1-C6アルキル、C1-C6ヘテロアルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、C2-C6アルケニル、C2-C6アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、またはヘテロアリールであり、ここで各アルキル、アルケニル、アルキニル、シクロアルキル、ヘテロシクロアルキル、アリールおよびヘテロアリールは独立して一つ以上のR14aで任意に置換され、
各R14aは独立して、重水素、ハロゲン、-CN,-ORb,-SRb,-S(=O)Ra,-S(=O)2Ra,-NO2,-NRcRd,-NHS(=O)2Ra,-S(=O)2NRcRd,-C(=O)Ra,-OC(=O)Ra,-C(=O)ORb,-OC(=O)ORb,-C(=O)NRcRd,-OC(=O)NRcRd,-NRbC(=O)NRcRd,-NRbC(=O)Ra,-NRbC(=O)ORb,C1-C6アルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、C2-C6アルケニル、C2-C6アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、もしくはヘテロアリールであり、または同じ炭素上の二つのR14aが一緒になってオキソを形成し、
各R15は独立して、水素、C1-C6アルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、C2-C6アルケニル、C2-C6アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、またはヘテロアリールであり、ここで各アルキル、アルケニル、アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、およびヘテロアリールは独立して、一つ以上のR15aで任意に置換され、
各R15aは独立して、重水素、ハロゲン、-CN,-ORb,-SRb,-S(=O)Ra,-S(=O)2Ra,-NO2,-NRcRd,-NHS(=O)2Ra,-S(=O)2NRcRd,-C(=O)Ra,-OC(=O)Ra,-C(=O)ORb,-OC(=O)ORb,-C(=O)NRcRd,-OC(=O)NRcRd,-NRbC(=O)NRcRd,-NRbC(=O)Ra,-NRbC(=O)ORb,C1-C6アルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、C2-C6アルケニル、C2-C6アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、もしくはヘテロアリールであり、または同じ炭素上の二つのR15aが一緒になってオキソを形成し、
各R16およびR17は独立して、水素、C1-C6アルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、C2-C6アルケニル、C2-C6アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、もしくはヘテロアリールであり、ここで各アルキル、アルケニル、アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、およびヘテロアリールは独立して、一つ以上のR16aで任意に置換され、
またはR16およびR17は、それらが結合される窒素原子と一緒になって、一つ以上のR16bで任意に置換されるヘテロシクロアルキルを形成し;
各R16aは独立して、重水素、ハロゲン、-CN,-ORb,-SRb,-S(=O)Ra,-S(=O)2Ra,-NO2,-NRcRd,-NHS(=O)2Ra,-S(=O)2NRcRd,-C(=O)Ra,-OC(=O)Ra,-C(=O)ORb,-OC(=O)ORb,-C(=O)NRcRd,-OC(=O)NRcRd,-NRbC(=O)NRcRd,-NRbC(=O)Ra,-NRbC(=O)ORb,C1-C6アルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、C2-C6アルケニル、C2-C6アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、もしくはヘテロアリールであり、または同じ炭素上の二つのR16aが一緒になってオキソを形成し、
各R16bは独立して、重水素、ハロゲン、-CN,-ORb,-SRb,-S(=O)Ra,-S(=O)2Ra,-NO2,-NRcRd,-NHS(=O)2Ra,-S(=O)2NRcRd,-C(=O)Ra,-OC(=O)Ra,-C(=O)ORb,-OC(=O)ORb,-C(=O)NRcRd,-OC(=O)NRcRd,-NRbC(=O)NRcRd,-NRbC(=O)Ra,-NRbC(=O)ORb,C1-C6アルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、C2-C6アルケニル、C2-C6アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、もしくはヘテロアリールであり、または同じ炭素上の二つのR16bが一緒になってオキソを形成し、
Xは、CR8またはNであり、
R1は、-S(=O)R10,-S(=O)2R10,-S(=O)2NR12R13,-C(=O)R10,-C(=O)OR11,-C(=O)NR12R13,C1-C6アルキル、C1-C6ヘテロアルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、C2-C6アルケニル、C2-C6アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、またはヘテロアリールであり、ここで各アルキル、アルケニル、アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、およびヘテロアリールは独立して、一つ以上のR1aで任意に置換され、
各R1aは独立して、重水素、ハロゲン、-CN,-ORb,-SRb,-S(=O)Ra,-S(=O)2Ra,-NO2,-NRcRd,-NHS(=O)2Ra,-S(=O)2NRcRd,-C(=O)Ra,-OC(=O)Ra,-C(=O)ORb,-OC(=O)ORb,-C(=O)NRcRd,-OC(=O)NRcRd,-NRbC(=O)NRcRd,-NRbC(=O)Ra,-NRbC(=O)ORb,C1-C6アルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、C2-C6アルケニル、C2-C6アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、もしくはヘテロアリールであり、ここで各アルキル、アルケニル、アルキニル、シクロアルキル、ヘテロシクロアルキル、アリールおよびヘテロアリールは独立して一つ以上のR1bで任意に置換され、または同じ炭素上の二つのR1aが一緒になってオキソを形成し、
各R1bは独立して、重水素、ハロゲン、-CN,-ORb,-SRb,-S(=O)Ra,-S(=O)2Ra,-NO2,-NRcRd,-NHS(=O)2Ra,-S(=O)2NRcRd,-C(=O)Ra,-OC(=O)Ra,-C(=O)ORb,-OC(=O)ORb,-C(=O)NRcRd,-OC(=O)NRcRd,-NRbC(=O)NRcRd,-NRbC(=O)Ra,-NRbC(=O)ORb,C1-C6アルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、C2-C6アルケニル、C2-C6アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、もしくはヘテロアリールであり、または同じ炭素上の二つのR1bが一緒になってオキソを形成し、
R2は、水素、C1-C6アルキル、C1-C6ハロアルキル、またはC1-C6ジュウテロアルキル(deuteroalkyl)であり、
R3、R6、およびR8は独立して、水素、重水素、ハロゲン、-CN,-ORb,-SRb,-S(=O)Ra,-S(=O)2Ra,-NO2,-NRcRd,-NHS(=O)2Ra,-S(=O)2NRcRd,-C(=O)Ra,-OC(=O)Ra,-C(=O)ORb,-OC(=O)ORb,-C(=O)NRcRd,-OC(=O)NRcRd,-NRbC(=O)NRcRd,-NRbC(=O)Ra,-NRbC(=O)ORb,C1-C6アルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、C2-C6アルケニル、またはC2-C6アルキニルであり、
R4は、水素、C1-C6アルキル、C1-C6ヘテロアルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、C2-C6アルケニル、C2-C6アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、またはヘテロアリールであり、ここで各アルキル、アルケニル、アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、およびヘテロアリールは一つ以上のR4aで任意に置換され、
各R4aは独立して、重水素、ハロゲン、-CN,-ORb,-NRcRd,-C(=O)Ra,-C(=O)ORb,-C(=O)NRcRd,C1-C6アルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、シクロアルキル、ヘテロシクロアルキル、アリール、もしくはヘテロアリールであり、または同じ炭素上の二つのR4aが一緒になってオキソを形成し、
R5は、水素、C1-C6アルキル、C1-C6ハロアルキル、またはC1-C6ジュウテロアルキル(deuteroalkyl)であり、
R7は、水素、C1-C6アルキル、C1-C6ハロアルキル、またはC1-C6ジュウテロアルキル(deuteroalkyl)であり、
各R10は独立して、C1-C6アルキル、C1-C6ヘテロアルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、C2-C6アルケニル、C2-C6アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、またはヘテロアリールであり、ここで各アルキル、アルケニル、アルキニル、シクロアルキル、ヘテロシクロアルキル、アリールおよびヘテロアリールは独立して一つ以上のR10aで任意に置換され、
各R10aは独立して、重水素、ハロゲン、-CN,-ORb,-SRb,-S(=O)Ra,-S(=O)2Ra,-NO2,-NRcRd,-NHS(=O)2Ra,-S(=O)2NRcRd,-C(=O)Ra,-OC(=O)Ra,-C(=O)ORb,-OC(=O)ORb,-C(=O)NRcRd,-OC(=O)NRcRd,-NRbC(=O)NRcRd,-NRbC(=O)Ra,-NRbC(=O)ORb,C1-C6アルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、C2-C6アルケニル、C2-C6アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、もしくはヘテロアリールであり、または同じ炭素上の二つのR10aが一緒になってオキソを形成し、
各R11は独立して、水素、C1-C6アルキル、C1-C6ヘテロアルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、C2-C6アルケニル、C2-C6アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、またはヘテロアリールであり、ここで各アルキル、アルケニル、アルキニル、シクロアルキル、ヘテロシクロアルキル、アリールおよびヘテロアリールは独立して、一つ以上のR11aで任意に置換され、
各R11aは独立して、水素、重水素、ハロゲン、-CN,-ORb,-SRb,-S(=O)Ra,-S(=O)2Ra,-NO2,-NRcRd,-NHS(=O)2Ra,-S(=O)2NRcRd,-C(=O)Ra,-OC(=O)Ra,-C(=O)ORb,-OC(=O)ORb,-C(=O)NRcRd,-OC(=O)NRcRd,-NRbC(=O)NRcRd,-NRbC(=O)Ra,-NRbC(=O)ORb,C1-C6アルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、C2-C6アルケニル、C2-C6アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、もしくはヘテロアリールであり、または同じ炭素上の二つのR11aが一緒になってオキソを形成し、
各R12およびR13は独立して、水素、C1-C6アルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、C2-C6アルケニル、C2-C6アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、またはヘテロアリールであり、ここで各アルキル、アルケニル、アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、およびヘテロアリールは独立して、一つ以上のR12aで任意に置換され、
各R12aは独立して、重水素、ハロゲン、-CN,-ORb,-SRb,-S(=O)Ra,-S(=O)2Ra,-NO2,-NRcRd,-NHS(=O)2Ra,-S(=O)2NRcRd,-C(=O)Ra,-OC(=O)Ra,-C(=O)ORb,-OC(=O)ORb,-C(=O)NRcRd,-OC(=O)NRcRd,-NRbC(=O)NRcRd,-NRbC(=O)Ra,-NRbC(=O)ORb,C1-C6アルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、C2-C6アルケニル、C2-C6アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、もしくはヘテロアリールであり、もしくは同じ炭素上の二つのR12aが一緒になってオキソを形成し、
またはR12およびR13は、それらが結合される窒素原子と一緒になって、一つ以上のR12bで任意に置換されるヘテロシクロアルキルを形成し、
各R12bは独立して、重水素、ハロゲン、-CN,-ORb,-SRb,-S(=O)Ra,-S(=O)2Ra,-NO2,-NRcRd,-NHS(=O)2Ra,-S(=O)2NRcRd,-C(=O)Ra,-OC(=O)Ra,-C(=O)ORb,-OC(=O)ORb,-C(=O)NRcRd,-OC(=O)NRcRd,-NRbC(=O)NRcRd,-NRbC(=O)Ra,-NRbC(=O)ORb,C1-C6アルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、C2-C6アルケニル、C2-C6アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、もしくはヘテロアリールであり、または同じ炭素上の二つのR12bが一緒になってオキソを形成し、
各Raは独立して、C1-C6アルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、C2-C6アルケニル、C2-C6アルキニル、シクロアルキル、ヘテロシクロアルキル、アリールまたはヘテロアリールであり、ここで各アルキル、アルケニル、アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、およびヘテロアリールは独立して、一つ以上のオキソ、重水素、ハロゲン、-CN、-OH、-OMe、-NH2、-C(=O)Me、-C(=O)OH、-C(=O)OMe、C1-C6アルキル、またはC1-C6ハロアルキルで任意に置換され、
各Rbは独立して、水素、C1-C6アルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、C2-C6アルケニル、C2-C6アルキニル、シクロアルキル、ヘテロシクロアルキル、アリールまたはヘテロアリールであり、ここで各アルキル、アルケニル、アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、およびヘテロアリールは独立して、一つ以上のオキソ、重水素、ハロゲン、-CN、-OH、-OMe、-NH2、-C(=O)Me、-C(=O)OH、-C(=O)OMe、C1-C6アルキル、またはC1-C6ハロアルキルで任意に置換され、また
各RcおよびRdは独立して、水素、C1-C6アルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、C2-C6アルケニル、C2-C6アルキニル、シクロアルキル、ヘテロシクロアルキル、アリールもしくはヘテロアリールであり、ここで各アルキル、アルケニル、アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、およびヘテロアリールは独立して、一つ以上のオキソ、重水素、ハロゲン、-CN、-OH、-OMe、-NH2、-C(=O)Me、-C(=O)OH、-C(=O)OMe、C1-C6アルキル、もしくはC1-C6ハロアルキルで任意に置換され、
またはRcおよびRdは、それらが結合される窒素原子と一緒になって、一つ以上のオキソ、重水素、ハロゲン、-CN、-OH、-OMe、-NH2、-C(=O)Me、-C(=O)OH、-C(=O)OMe、C1-C6アルキル、もしくはC1-C6ハロアルキルで任意に置換されるヘテロシクロアルキルを形成する。
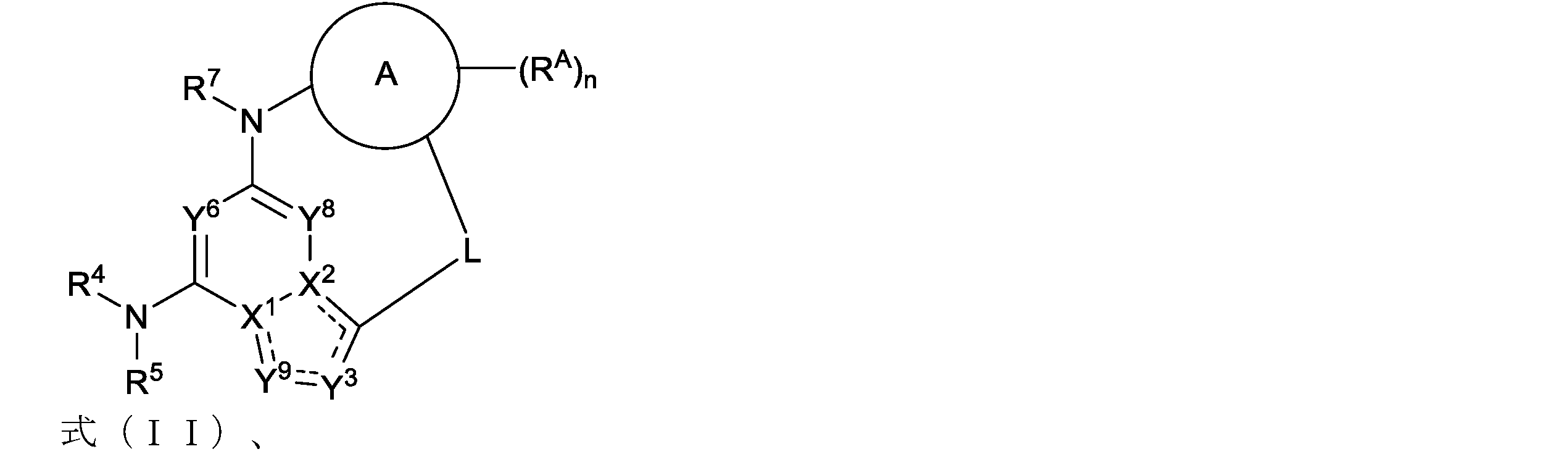
Lは、4~10原子の任意に置換されるリンカーであり、
環Aは、任意に置換されたシクロアルキル、任意に置換されたヘテロシクロアルキル、任意に置換されたアリール、または任意に置換されたヘテロアリールであり、
X1およびX2は、-N-または-C=であり、ただしX1またはX2のうちの一方が-N-で他方が-C=であり、
Y8は、CR8またはNであり、
Y6は、CR6またはNであり、
Y3は、CR3またはNであり、
Y9は、CR9またはNであり、
R3、R6、R8、およびR9は独立して、水素、重水素、ハロゲン、-CN,-ORb,-SRb,-S(=O)Ra,-S(=O)2Ra,-NO2,-NRcRd,-NHS(=O)2Ra,-S(=O)2NRcRd,-C(=O)Ra,-OC(=O)Ra,-C(=O)ORb,-OC(=O)ORb,-C(=O)NRcRd,-OC(=O)NRcRd,-NRbC(=O)NRcRd,-NRbC(=O)Ra,-NRbC(=O)ORb,任意に置換されたC1-C6アルキル、任意に置換されたC1-C6ハロアルキル、任意に置換されたC1-C6ジュウテロアルキル(deuteroalkyl)、任意に置換されたC1-C6ヒドロキシアルキル、任意に置換されたC1-C6アミノアルキル、任意に置換されたC2-C6アルケニル、または任意に置換されたC2-C6アルキニルであり、
R4は、水素、任意に置換されたC1-C6アルキル、任意に置換されたC1-C6ヘテロアルキル、任意に置換されたC1-C6ハロアルキル、任意に置換されたC1-C6ジュウテロアルキル(deuteroalkyl)、任意に置換されたC1-C6ヒドロキシアルキル、任意に置換されたC1-C6アミノアルキル、任意に置換されたC2-C6アルケニル、任意に置換されたC2-C6アルキニル、任意に置換されたシクロアルキル、任意に置換されたヘテロシクロアルキル、任意に置換されたアリール、または任意に置換されたヘテロアリールであり、
R5は、水素、任意に置換されたC1-C6アルキル、任意に置換されたC1-C6ハロアルキル、または任意に置換されたC1-C6ジュウテロアルキル(deuteroalkyl)であり、
R7は、水素、任意に置換されたC1-C6アルキル、任意に置換されたC1-C6ハロアルキル、または任意に置換されたC1-C6ジュウテロアルキル(deuteroalkyl)であり、
各Raは独立して、C1-C6アルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、C2-C6アルケニル、C2-C6アルキニル、シクロアルキル、ヘテロシクロアルキル、アリールまたはヘテロアリールであり、ここで各アルキル、アルケニル、アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、およびヘテロアリールは独立して、一つ以上のオキソ、重水素、ハロゲン-CN,-OH,-OMe,-NH2,-C(=O)Me,-C(=O)OH,-C(=O)OMe,C1-C6アルキル、またはC1-C6ハロアルキルで任意に置換され、
各Rbは独立して、水素、C1-C6アルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、C2-C6アルケニル、C2-C6アルキニル、シクロアルキル、ヘテロシクロアルキル、アリールまたはヘテロアリールであり、ここで各アルキル、アルケニル、アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、およびヘテロアリールは独立して、一つ以上のオキソ、重水素、ハロゲン、-CN,-OH,-OMe,-NH2,-C(=O)Me,-C(=O)OH,-C(=O)OMe,C1-C6アルキル、またはC1-C6ハロアルキルで任意に置換され;また
各RcおよびRdは独立して、水素、C1-C6アルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、C2-C6アルケニル、C2-C6アルキニル、シクロアルキル、ヘテロシクロアルキル、アリールもしくはヘテロアリールであり、ここで各アルキル、アルケニル、アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、およびヘテロアリールは独立して、一つ以上のオキソ、重水素、ハロゲン、-CN,-OH,-OMe,-NH2,-C(=O)Me,-C(=O)OH,-C(=O)OMe,C1-C6アルキル、もしくはC1-C6ハロアルキルで任意に置換され、
またはRcおよびRdは、それらが結合される窒素原子と一緒になって一つ以上のオキソ、重水素、ハロゲン、-CN,-OH,-OMe,-NH2,-C(=O)Me,-C(=O)OH,-C(=O)OMe,C1-C6アルキル、もしくはC1-C6ハロアルキルで任意に置換されるヘテロシクロアルキルを形成する。
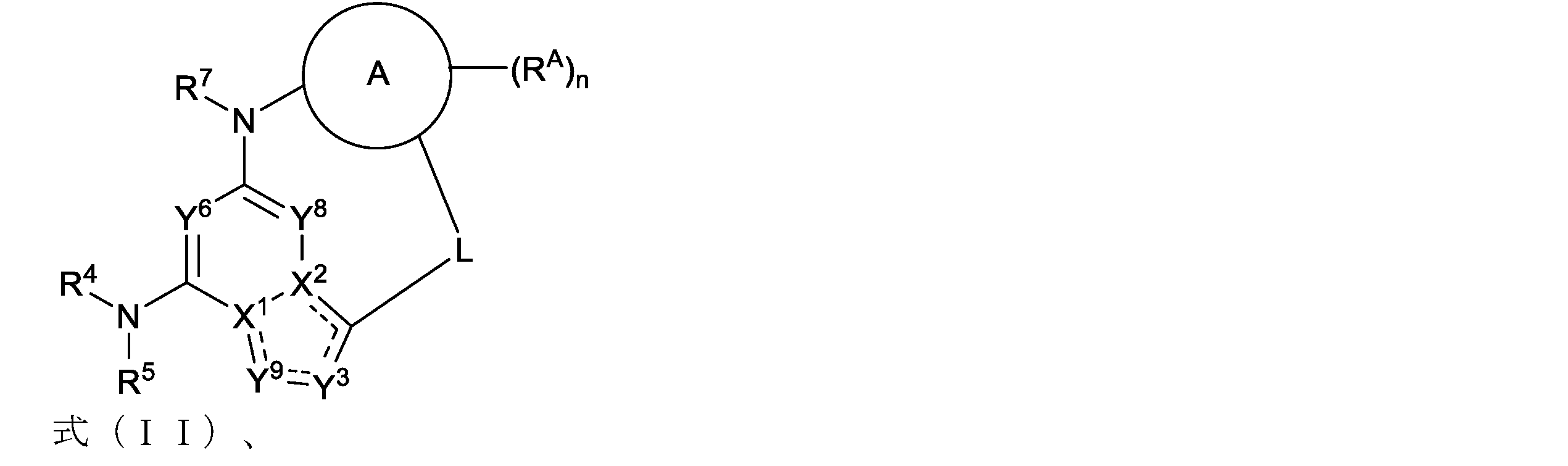
Lは、4~10原子のリンカーであり、一つ以上のRLで任意に置換され、
各RLは独立して、重水素、ハロゲン、-CN,-ORb,-SRb,-S(=O)Ra,-S(=O)2Ra,-NO2,-NRcRd,-NHS(=O)2Ra,-S(=O)2NRcRd,-C(=O)Ra,-OC(=O)Ra,-C(=O)ORb,-OC(=O)ORb,-C(=O)NRcRd,-OC(=O)NRcRd,-NRbC(=O)NRcRd,-NRbC(=O)Ra,-NRbC(=O)ORb,C1-C6アルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、C2-C6アルケニル、C2-C6アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、もしくはヘテロアリールであり、または同じ炭素上の二つのRLが一緒になってオキソ、シクロアルキルもしくはヘテロシクロアルキルを形成し、または異なる炭素上の二つのRLが一緒になってシクロアルキルもしくはヘテロシクロアルキルを形成し、
環Aは、シクロアルキル、ヘテロシクロアルキル、アリール、またはヘテロアリールであり、
各RAは独立して、重水素、ハロゲン、-CN,-ORb,-SRb,-S(=O)Ra,-S(=O)2Ra,-NO2,-NRcRd,-NHS(=O)2Ra,-S(=O)2NRcRd,-C(=O)Ra,-OC(=O)Ra,-C(=O)ORb,-OC(=O)ORb,-C(=O)NRcRd,-OC(=O)NRcRd,-NRbC(=O)NRcRd,-NRbC(=O)Ra,-NRbC(=O)ORb,C1-C6アルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、C2-C6アルケニル、C2-C6アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、もしくはヘテロアリールであり、ここで各アルキル、アルケニル、アルキニル、シクロアルキル、ヘテロシクロアルキル、アリールおよびヘテロアリールは独立して一つ以上のRA1で任意に置換され、または同じ炭素上の二つのRAが一緒になってオキソを形成し、
各RA1は独立して、重水素、ハロゲン、-CN,-ORb,-SRb,-S(=O)Ra,-S(=O)2Ra,-NO2,-NRcRd,-NHS(=O)2Ra,-S(=O)2NRcRd,-C(=O)Ra,-OC(=O)Ra,-C(=O)ORb,-OC(=O)ORb,-C(=O)NRcRd,-OC(=O)NRcRd,-NRbC(=O)NRcRd,-NRbC(=O)Ra,-NRbC(=O)ORb,C1-C6アルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、C2-C6アルケニル、C2-C6アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、もしくはヘテロアリールであり、または同じ炭素上の二つのRA1が一緒になってオキソを形成し、
nは、0~4であり、
X1およびX2は、-N-または-C=であり、ただしX1またはX2のうちの一方が-N-で他方が-C=であり、
Y8は、CR8またはNであり、
Y6は、CR6またはNであり、
Y3は、CR3またはNであり、
Y9は、CR9またはNであり、
R3、R6、R8およびR9は独立して、水素、重水素、ハロゲン、-CN,-ORb,-SRb,-S(=O)Ra,-S(=O)2Ra,-NO2,-NRcRd,-NHS(=O)2Ra,-S(=O)2NRcRd,-C(=O)Ra,-OC(=O)Ra,-C(=O)ORb,-OC(=O)ORb,-C(=O)NRcRd,-OC(=O)NRcRd,-NRbC(=O)NRcRd,-NRbC(=O)Ra,-NRbC(=O)ORb,C1-C6アルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、C2-C6アルケニル、またはC2-C6アルキニルであり、
R4は、水素、C1-C6アルキル、C1-C6ヘテロアルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、C2-C6アルケニル、C2-C6アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、またはヘテロアリールであり、ここで各アルキル、アルケニル、アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、およびヘテロアリールは一つ以上のR4aで任意に置換され、
各R4aは独立して、重水素、ハロゲン、-CN、-ORb、-NRcRd、-C(=O)Ra、-C(=O)ORb、-C(=O)NRcRd、C1-C6アルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、シクロアルキル、ヘテロシクロアルキル、アリール、もしくはヘテロアリールであり、または同じ炭素上の二つのR4aが一緒になってオキソを形成し、
R5は、水素、C1-C6アルキル、C1-C6ハロアルキル、またはC1-C6ジュウテロアルキル(deuteroalkyl)であり、
R7は、水素、C1-C6アルキル、C1-C6ハロアルキル、またはC1-C6ジュウテロアルキル(deuteroalkyl)であり、
各Raは独立して、C1-C6アルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、C2-C6アルケニル、C2-C6アルキニル、シクロアルキル、ヘテロシクロアルキル、アリールまたはヘテロアリールであり、ここで各アルキル、アルケニル、アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、およびヘテロアリールは独立して、一つ以上のオキソ、重水素、ハロゲン-CN,-OH,-OMe,-NH2,-C(=O)Me,-C(=O)OH,-C(=O)OMe,C1-C6アルキル、またはC1-C6ハロアルキルで任意に置換され、
各Rbは独立して、水素、C1-C6アルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、C2-C6アルケニル、C2-C6アルキニル、シクロアルキル、ヘテロシクロアルキル、アリールまたはヘテロアリールであり、ここで各アルキル、アルケニル、アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、およびヘテロアリールは独立して、一つ以上のオキソ、重水素、ハロゲン、-CN,-OH,-OMe,-NH2,-C(=O)Me,-C(=O)OH,-C(=O)OMe,C1-C6アルキル、またはC1-C6ハロアルキルで任意に置換され;また
各RcおよびRdは独立して、水素、C1-C6アルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、C2-C6アルケニル、C2-C6アルキニル、シクロアルキル、ヘテロシクロアルキル、アリールもしくはヘテロアリールであり、ここで各アルキル、アルケニル、アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、およびヘテロアリールは独立して、一つ以上のオキソ、重水素、ハロゲン、-CN,-OH,-OMe,-NH2,-C(=O)Me,-C(=O)OH,-C(=O)OMe,C1-C6アルキル、もしくはC1-C6ハロアルキルで任意に置換され、
またはRcおよびRdは、それらが結合される窒素原子と一緒になって一つ以上のオキソ、重水素、ハロゲン、-CN,-OH,-OMe,-NH2,-C(=O)Me,-C(=O)OH,-C(=O)OMe,C1-C6アルキル、もしくはC1-C6ハロアルキルで任意に置換されるヘテロシクロアルキルを形成する。
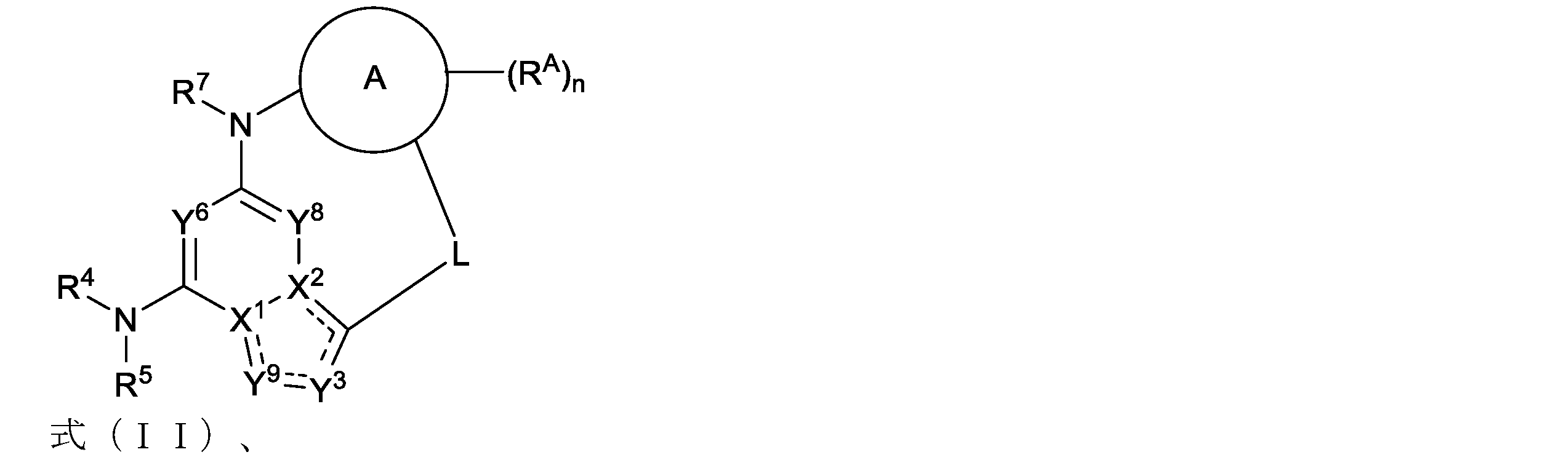
Lは、4~10原子のリンカーであり、一つ以上のRLで任意に置換され、
各RLは独立して、重水素、ハロゲン、-CN,-ORb,-SRb,-S(=O)Ra,-S(=O)2Ra,-NO2,-NRcRd,-NHS(=O)2Ra,-S(=O)2NRcRd,-C(=O)Ra,-OC(=O)Ra,-C(=O)ORb,-OC(=O)ORb,-C(=O)NRcRd,-OC(=O)NRcRd,-NRbC(=O)NRcRd,-NRbC(=O)Ra,-NRbC(=O)ORb,C1-C6アルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、C2-C6アルケニル、C2-C6アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、もしくはヘテロアリールであり、または同じ炭素上の二つのRLが一緒になってオキソ、シクロアルキルもしくはヘテロシクロアルキルを形成し、または隣接する炭素上の二つのRLが一緒になってシクロアルキルもしくはヘテロシクロアルキルを形成し、
環Aは、シクロアルキル、ヘテロシクロアルキル、アリール、またはヘテロアリールであり、
各RAは独立して、重水素、ハロゲン、-CN,-ORb,-SRb,-S(=O)Ra,-S(=O)2Ra,-NO2,-NRcRd,-NHS(=O)2Ra,-S(=O)2NRcRd,-C(=O)Ra,-OC(=O)Ra,-C(=O)ORb,-OC(=O)ORb,-C(=O)NRcRd,-OC(=O)NRcRd,-NRbC(=O)NRcRd,-NRbC(=O)Ra,-NRbC(=O)ORb,C1-C6アルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、C2-C6アルケニル、C2-C6アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、もしくはヘテロアリールであり、ここで各アルキル、アルケニル、アルキニル、シクロアルキル、ヘテロシクロアルキル、アリールおよびヘテロアリールは独立して一つ以上のRA1で任意に置換され、または同じ炭素上の二つのRAが一緒になってオキソを形成し、
各RA1は独立して、重水素、ハロゲン、-CN,-ORb,-SRb,-S(=O)Ra,-S(=O)2Ra,-NO2,-NRcRd,-NHS(=O)2Ra,-S(=O)2NRcRd,-C(=O)Ra,-OC(=O)Ra,-C(=O)ORb,-OC(=O)ORb,-C(=O)NRcRd,-OC(=O)NRcRd,-NRbC(=O)NRcRd,-NRbC(=O)Ra,-NRbC(=O)ORb,C1-C6アルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、C2-C6アルケニル、C2-C6アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、もしくはヘテロアリールであり、または同じ炭素上の二つのRA1が一緒になってオキソを形成し、
nは、0~4であり、
X1およびX2は、-N-または-C=であり、ただしX1またはX2のうちの一方が-N-で他方が-C=であり、
Y8は、CR8またはNであり、
Y6は、CR6またはNであり、
Y3は、CR3またはNであり、
Y9は、CR9またはNであり、
R3、R6、R8およびR9は独立して、水素、重水素、ハロゲン、-CN,-ORb,-SRb,-S(=O)Ra,-S(=O)2Ra,-NO2,-NRcRd,-NHS(=O)2Ra,-S(=O)2NRcRd,-C(=O)Ra,-OC(=O)Ra,-C(=O)ORb,-OC(=O)ORb,-C(=O)NRcRd,-OC(=O)NRcRd,-NRbC(=O)NRcRd,-NRbC(=O)Ra,-NRbC(=O)ORb,C1-C6アルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、C2-C6アルケニル、またはC2-C6アルキニルであり、
R4は、水素、C1-C6アルキル、C1-C6ヘテロアルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、C2-C6アルケニル、C2-C6アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、またはヘテロアリールであり、ここで各アルキル、アルケニル、アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、およびヘテロアリールは一つ以上のR4aで任意に置換され、
各R4aは独立して、重水素、ハロゲン、-CN,-ORb,-NRcRd,-C(=O)Ra,-C(=O)ORb,-C(=O)NRcRd,C1-C6アルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、シクロアルキル、ヘテロシクロアルキル、アリール、もしくはヘテロアリールであり、または同じ炭素上の二つのR4aが一緒になってオキソを形成し、
R5は、水素、C1-C6アルキル、C1-C6ハロアルキル、またはC1-C6ジュウテロアルキル(deuteroalkyl)であり、
R7は、水素、C1-C6アルキル、C1-C6ハロアルキル、またはC1-C6ジュウテロアルキル(deuteroalkyl)であり、
各Raは独立して、C1-C6アルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、C2-C6アルケニル、C2-C6アルキニル、シクロアルキル、ヘテロシクロアルキル、アリールまたはヘテロアリールであり、ここで各アルキル、アルケニル、アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、およびヘテロアリールは独立して、一つ以上のオキソ、重水素、ハロゲン-CN,-OH,-OMe,-NH2,-C(=O)Me,-C(=O)OH,-C(=O)OMe,C1-C6アルキル、またはC1-C6ハロアルキルで任意に置換され、
各Rbは独立して、水素、C1-C6アルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、C2-C6アルケニル、C2-C6アルキニル、シクロアルキル、ヘテロシクロアルキル、アリールまたはヘテロアリールであり、ここで各アルキル、アルケニル、アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、およびヘテロアリールは独立して、一つ以上のオキソ、重水素、ハロゲン、-CN,-OH,-OMe,-NH2,-C(=O)Me,-C(=O)OH,-C(=O)OMe,C1-C6アルキル、またはC1-C6ハロアルキルで任意に置換され;また
各RcおよびRdは独立して、水素、C1-C6アルキル、C1-C6ハロアルキル、C1-C6ジュウテロアルキル(deuteroalkyl)、C1-C6ヒドロキシアルキル、C1-C6アミノアルキル、C2-C6アルケニル、C2-C6アルキニル、シクロアルキル、ヘテロシクロアルキル、アリールもしくはヘテロアリールであり、ここで各アルキル、アルケニル、アルキニル、シクロアルキル、ヘテロシクロアルキル、アリール、およびヘテロアリールは独立して、一つ以上のオキソ、重水素、ハロゲン、-CN,-OH,-OMe,-NH2,-C(=O)Me,-C(=O)OH,-C(=O)OMe,C1-C6アルキル、もしくはC1-C6ハロアルキルで任意に置換され、
またはRcおよびRdは、それらが結合される窒素原子と一緒になって一つ以上のオキソ、重水素、ハロゲン、-CN,-OH,-OMe,-NH2,-C(=O)Me,-C(=O)OH,-C(=O)OMe,C1-C6アルキル、もしくはC1-C6ハロアルキルで任意に置換されるヘテロシクロアルキルを形成する。
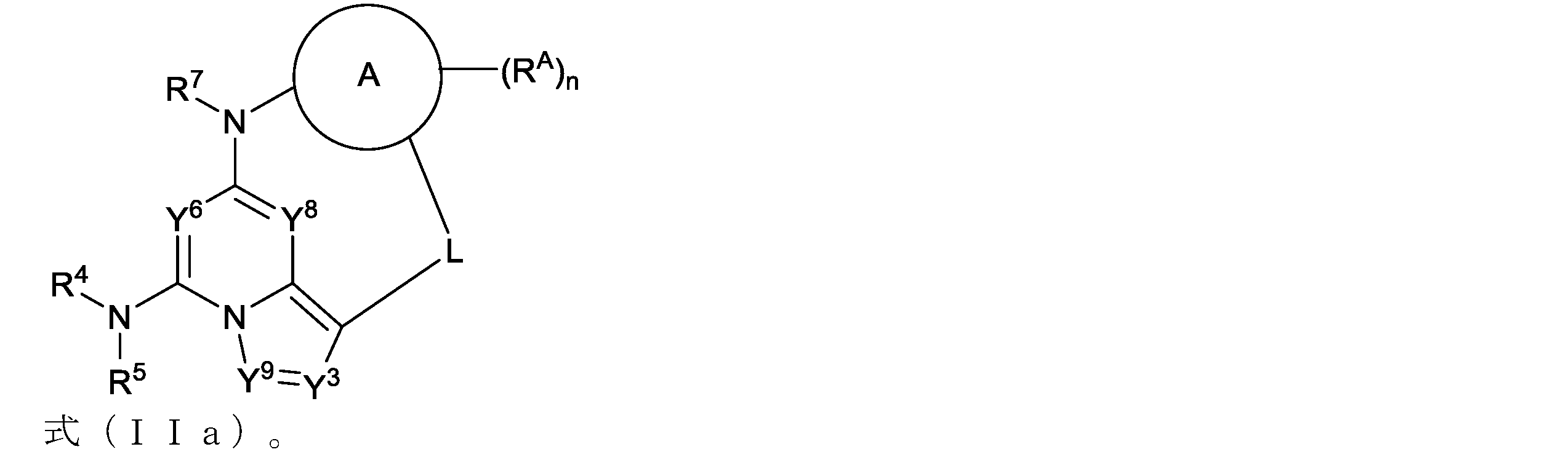
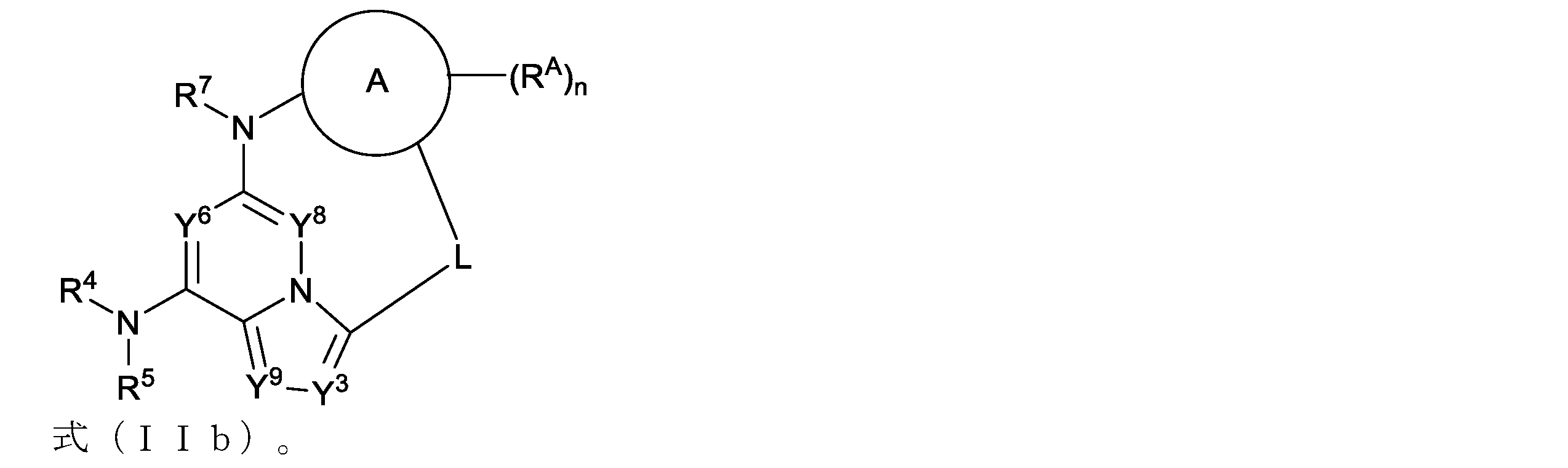





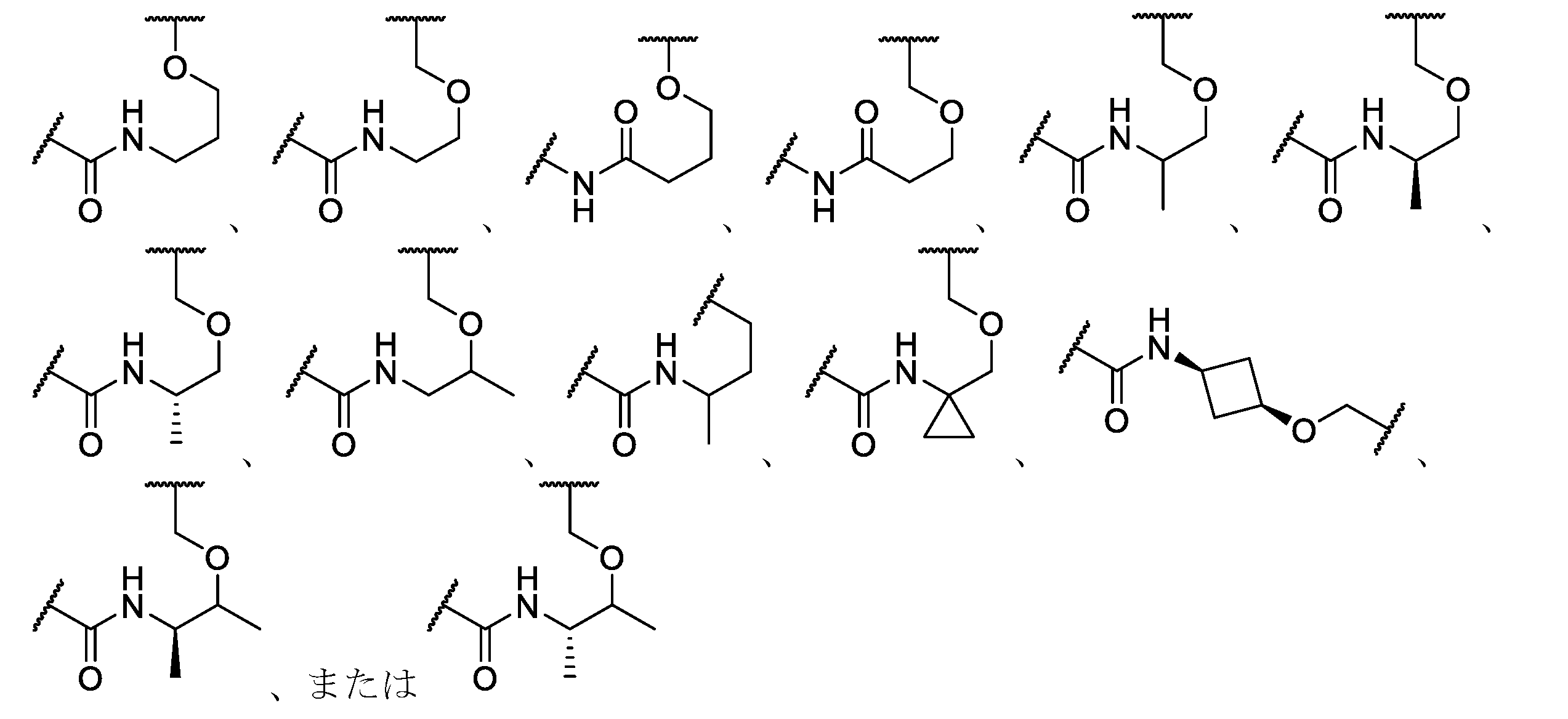
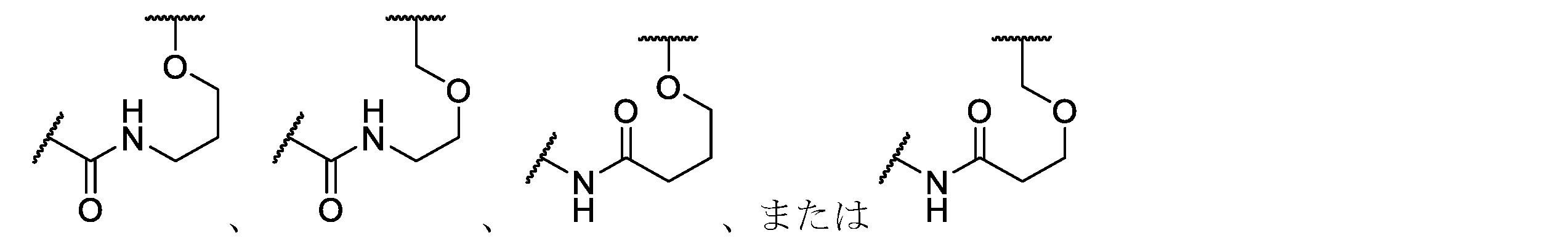
式(II)または(IIa)~(IId)の化合物の一部の実施形態では、Lは、





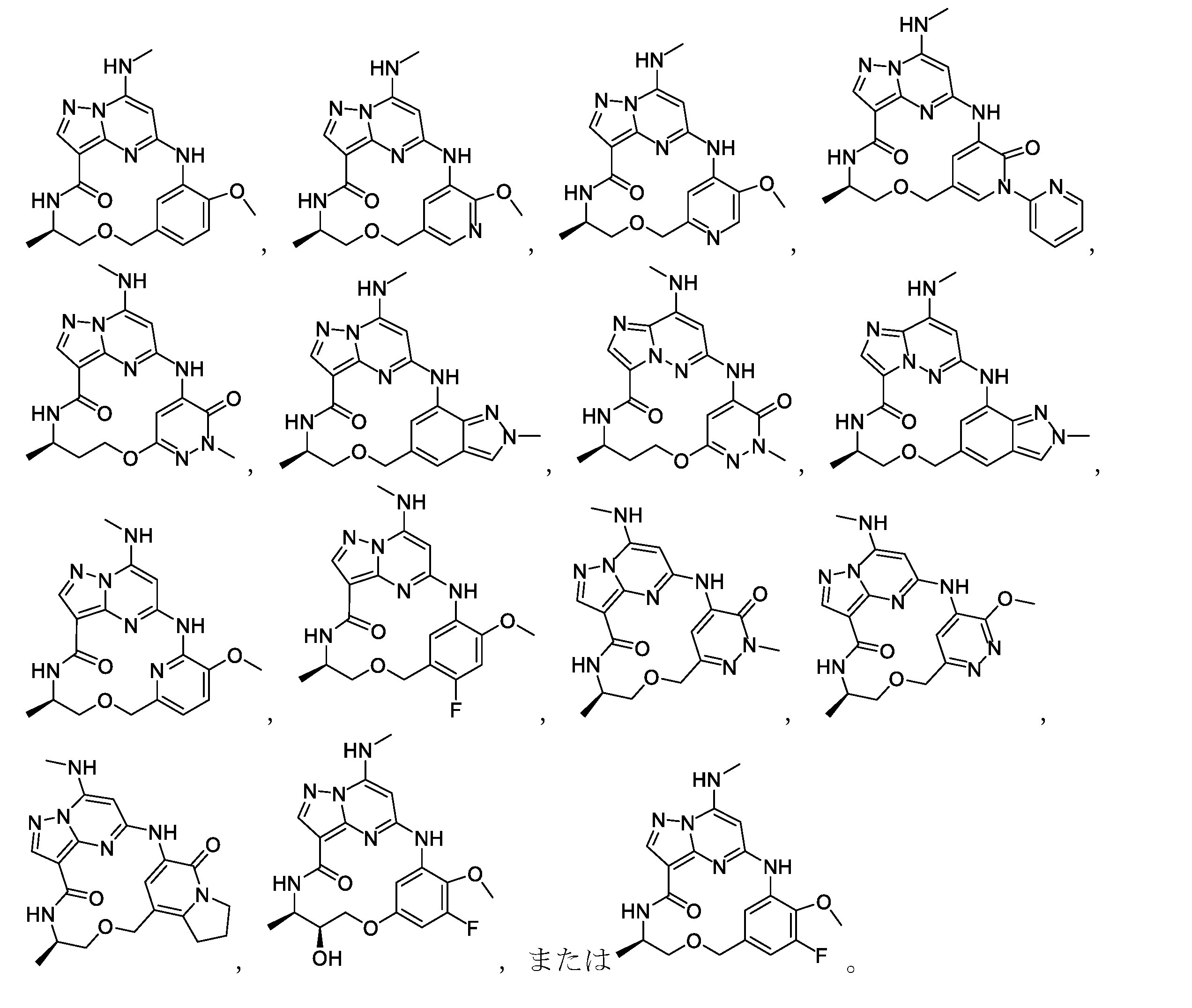
異性体/立体異性体
一部の実施形態では、本明細書に記載される化合物は、幾何異性体として存在する。一部の実施形態では、本明細書に記載される化合物は、一つ以上の二重結合を有する。本明細書に提示される化合物は、全てのシス(cis)、トランス(trans)、シン(syn)、アンチ(anti)、エントゲーゲン(entgegen)(E)、およびツザメン(zusammen)(Z)の異性体、ならびに対応するそれらの混合物を含む。一部の状況では、本明細書に記載の化合物は、一つ以上のキラル中心を有し、各中心は、R配置またはS配置で存在する。本明細書に記載される化合物は、全てのジアステレオマー、エナンチオマー、およびエピマーの形態、ならびに対応するそれらの混合物を含む。本明細書に提示される化合物および方法の追加の実施形態において、単一の分取工程、組み合わせ、または相互変換から生じるエナンチオマーおよび/またはジアステレオマーの混合物は、本明細書に記載される用途に有用である。一部の実施形態では、本明細書に記載の化合物は、化合物のラセミ混合物と、光学的に活性な分割剤とを反応させて、一対のジアステレオマー化合物を形成させ、当該ジアステレオマーを分離させ、そして光学的に純粋なエナンチオマーを回収することによって、それらの個々の立体異性体として調製される。一部の実施形態では、分離可能な複合体が好ましい。一部の実施形態では、ジアステレオマーは、別個の物理的特性(例えば、融点、沸点、溶解度、反応性など)を有し、これら相違点を利用することによって分離される。一部の実施形態では、ジアステレオマーは、キラルクロマトグラフィーによって、または好ましくは溶解度の差に基づく分離/分割技術によって分離される。一部の実施形態では、その後、光学的に純粋なエナンチオマーが分割剤と共に回収される。
一部の実施形態では、本明細書に記載の化合物は、その同位体標識された形態で存在する。一部の実施形態では、本明細書に開示される方法は、そのような同位体標識された化合物を投与することによって疾患を治療する方法を含む。一部の実施形態では、本明細書に開示される方法は、そのような同位体標識された化合物を医薬組成物として投与することによって疾患を治療する方法を含む。したがって一部の実施形態では、本明細書に開示される化合物は、本明細書で列挙したものと同一であるが、一つ以上の原子が、通常自然界で存在する原子質量または質量数とは異なる原子質量または質量数を有する原子によって置換されるという点では異なる、同位体標識化合物を含む。本明細書に記載される化合物、またはその溶媒和物もしくは立体異性体に組み込まれ得る同位体の例としては、水素、炭素、窒素、酸素、リン、硫黄、フッ素、および塩素の同位体が挙げられ、それぞれ例えば、2H、3H、13C、14C、l5N、18O、17O、31P、32P、35S、18F、および36Clなどがある。前述の同位体および/または他の原子の他の同位体を含有する本明細書に記載される化合物、ならびにその薬学的に許容可能な塩、溶媒和物、または立体異性体は、本開示の範囲内である。例えば、例えば3Hおよび14Cなどの放射性同位体が組み込まれた特定の同位体標識化合物は、薬剤および/または基質組織分布アッセイに有用である。トリチウム化、すなわち3H、および炭素-14、すなわち14Cといった同位体は、それらの調製および検出の容易さから特に好ましい。さらに例えば重水素、すなわち2Hなどの重い同位体を用いた置換は、代謝安定性の高さから生じる特定の治療上の有益性、例えばインビボ半減期の延長または必要用量の低下をもたらす。一部の実施形態では、同位体標識された化合物またはその薬学的に許容可能な塩、溶媒和物、もしくは立体異性体は、任意の適切な方法によって調製される。
一部の実施形態では、本明細書に記載の化合物は、その薬学的に許容可能な塩として存在する。一部の実施形態では、本明細書に開示される方法は、そのような薬学的に許容可能な塩を投与することによって疾患を治療する方法を含む。一部の実施形態では、本明細書に開示される方法は、そのような薬学的に許容可能な塩を医薬組成物として投与することによって疾患を治療する方法を含む。
一部の実施形態では、本明細書に記載される化合物は、溶媒和物として存在する。本開示は、そうした溶媒和物を投与することによって疾患を治療する方法を提供する。本開示はさらに、そうした溶媒和物を医薬組成物として投与することによって疾患を治療する方法を提供する。
一部の状況では、化合物は、互変異性体として存在する。本明細書に記載される化合物は、本明細書に記載される式内の全ての可能性のある互変異性体を含む。互変異性体は、水素原子の遊走によって相互交換が可能な化合物であり、一重結合と隣接する二重結合とのスイッチが付随する。互変異性化が可能である結合配置では、互変異性体の化学平衡が存在することとなる。本明細書に開示される化合物のすべての互変異性体が予期される。互変異性体の正確な比率は、温度、溶媒、およびpHを含むいくつかの要因に依存する。
本明細書に記載される反応で使用される化合物は、市販の化学物質および/または化学文献に記載される化合物から出発して、当業者に公知の有機合成技術に従って作製される。「市販の化学物質」は、Acros Organics社(ペンシルバニア州ピッツバーグ)、Aldrich Chemical社(ウィスコンシン州ミルウォーキー、Sigma Chemical社とFluka社を含む)、Apin Chemicals Ltd.(英国、ミルトンパーク)、Avocado Research社(英国、ランカシャー州)、BDH,Inc.(カナダ、トロント)、Bionet社(英国、コーンウォール)、Chem Service Inc.(ペンシルベニア州ウェストチェスター)、Crescent Chemical Co.(ニューヨーク州ハウパージュ)、Eastman Organic Chemicals社、Eastman Kodak Company(ニューヨーク州ロチェスター)、Fisher Scientific Co.(ペンシルバニア州ピッツバーグ)、Fisons Chemicals社(英国、レスターシャー)、Frontier Scientific社(ユタ州ローガン)、ICN Biomedicals,Inc.(カリフォルニア州コスタメサ)、Key Organics社(英国、コーンウォール)、Lancaster Synthesis社(ニューハンプシャー州ウィンドハム)、Maybridge Chemical Co.Ltd.(英国、コーンウォール)、Parish Chemical Co.(ユタ州オレム)、Pfaltz&Bauer,Inc.(コネチカット州ウォーターバリー)、Polyorganix社(テキサス州ヒューストン)、Pierce Chemical Co.(イリノイ州ロックフォード)、Riedel de Haen AG社(ドイツ、ハノーバー)、Spectrum Quality Product,Inc.(ニュージャージー州ニューブランズウィック)、TCI America社(オレゴン州ポートランド)、Trans World Chemicals,Inc.(メリーランド州ロックビル)、およびWako Chemicals USA,Inc.(バージニア州リッチモンド)をはじめとする標準的な商業的供給源から取得される。
特定の実施形態では、本明細書に記載の化合物は、純粋な化学物質として投与される。一部の実施形態では、本明細書に記載の化合物は、選択された投与経路、および例えば、Remington:The Science and Practice of Pharmacy(Gennaro、第21版、Mack Pub.Co.,Easton,PA(2005))に記載される標準的な薬学的手順に基づき選択される、薬学的に適切な、または許容可能な担体(本明細書において薬学的に適切な(または許容可能な)賦形剤、生理学的に適切な(または許容可能な)賦形剤、または生理学的に適切な(または許容可能な)担体とも呼称される)と組み合わせられる。
本明細書に開示される化合物、またはその薬学的に許容可能な塩、溶媒和物、もしくは立体異性体は、一つ以上の酵素のキナーゼ活性の阻害に有用である。一部の実施形態では、化合物および方法により阻害されるキナーゼは、TYK2である。
特定の例では、本明細書に記載される化合物、またはその薬学的に許容可能な塩、溶媒和物、もしくは立体異性体は、第二の治療剤と併用されて投与される。
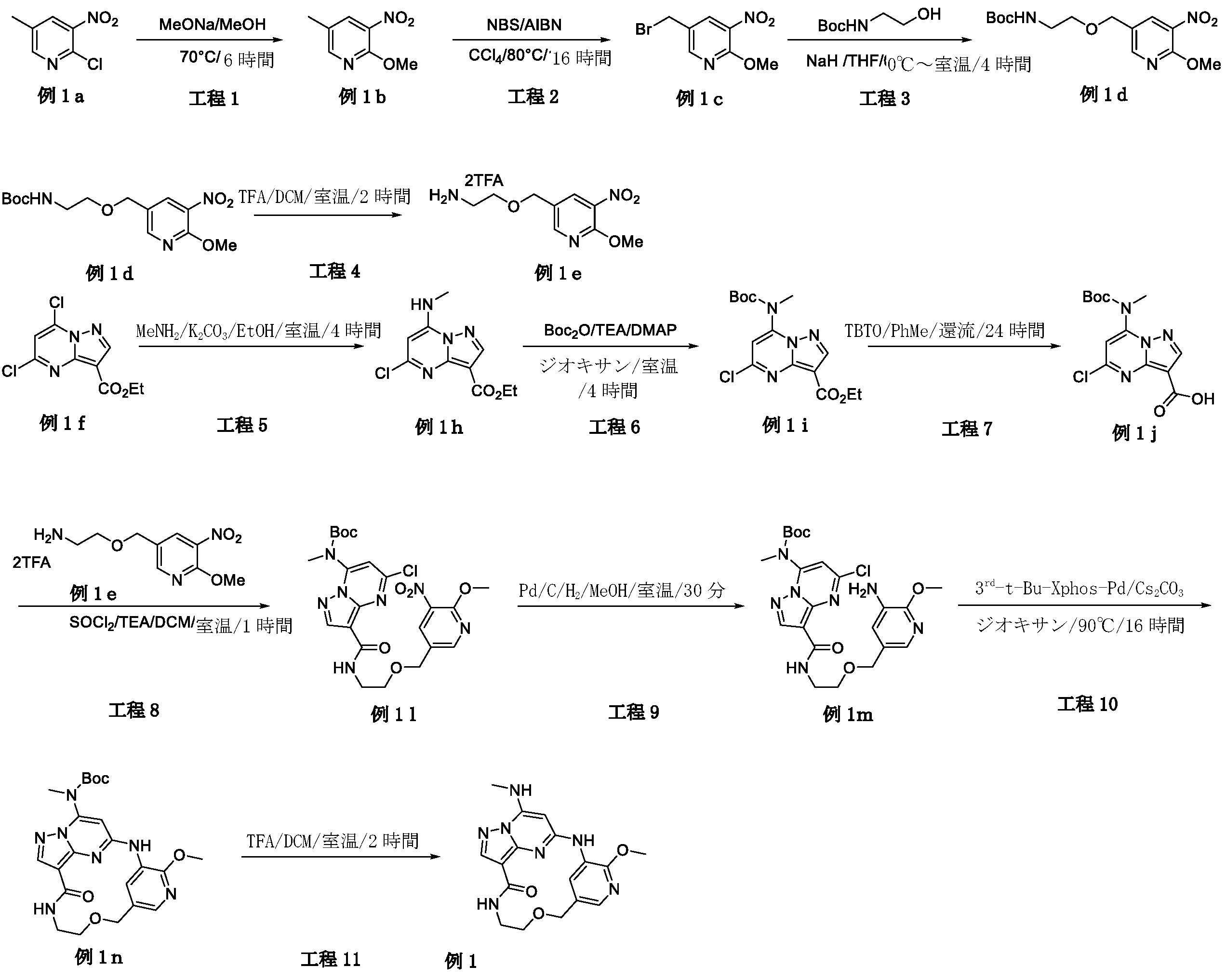
例1a(11.0g、63.74mmol)のメタノール(200mL)溶液に、NaOMe(6.88g、127.48mmol)を加えた。反応混合物を、70℃で、6時間攪拌した。混合物をH2O(1L)でクエンチし、ピンク色の沈殿物を形成した。固体をろ過により回収し、真空乾燥して、例1b(9.0g、収率84.0%)をピンク色の固体として得た。LCMS[M+1]+=169.1.
例1b(5.0g、29.76mmol)のCCl4(100mL)中の混合物に、NBS(6.36g、35.71mmol)およびAIBN(0.98g、5.95mmol)を加えた。反応混合物を、N2保護下、80℃で、16時間攪拌した。室温に冷却後、溶媒を除去し、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物である例1c(2.8g、収率38.2%)を黄色固体として得た。
LCMS[M+1]+=247.1.
tert-ブチル(2-ヒドロキシエチル)カルバメート(1.29g、8.02mmol)のTHF(30mL)溶液に、NaH(320mg、8.02mmol)を0℃で数回に分けて加えた。混合物を同温度で30分間攪拌し、次いでTHF中の例1c(1.8g、7.29mmol)を滴加した。反応混合物を室温で、4時間攪拌した。溶媒を除去し、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製し、生成物である例1d(600mg、収率25.2%)を黄色油状物として得た。LCMS[M+1-100]+=328.2.
例1d(600mg、3.3mmol)のDCM(5mL)溶液に、TFA(1mL)を加えた。反応混合物を室温で、2時間攪拌した。溶液を真空中で濃縮して、粗生成物である例1e(710mg、113.8%、未精製)を黄色油状物として得、これを精製することなく次の工程に直接使用した。LCMS[M+1]+=228.2.
例1f(2.0g、7.69mmol)のEtOH(40mL)溶液に、CH3NH2(7.7mL、MeOH中の2M、15.38mmol)およびK2CO3(2.12g、15.38mmol)を加えた。反応混合物を室温で、4時間攪拌した。混合物をH2O(200mL)中に注いで、白色の沈殿物を形成した。固体をろ過により回収して、真空乾燥して、生成物である例1h(1.85g、収率94.4%)を白色個体として得た。LCMS[M+1]+=255.2.
例1h(1.85g、7.26mmol)のジオキサン(40mL)溶液に、Boc2O(1.9g、8.72mmol)、TEA(1.09g、10.89mmol)およびDMAP(44mg、0.36mmol)を加えた。反応混合物を室温で、4時間攪拌した。溶媒を除去し、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製し、生成物である例1i(2.3g、収率89.3%)を灰白色固体として得た。LCMS[M+1]+=355.2.
トルエン(10mL)中の例1i(800mg、2.25mmol)の混合物に、TBTO(2.96g、4.51mmol)を加えた。反応混合物を、N2保護下、120℃で、24時間攪拌した。室温に冷却後、溶媒を除去し、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物である例1j(620mg、収率84.3%)を黄色固体として得た。LCMS[M+1]+=327.2.
例1j(500mg、1.53mmol)のDCM(10mL)溶液に、SOCl2(728mg、6.12mmol)を加え、反応混合物を室温で1時間攪拌した。溶媒を除去し、残渣をDCMで希釈し、これを、例1e(565mg、1.53mmol)およびTEA(773mg、7.65mmol)のDCM(10mL)溶液に0℃で滴加した。得られた混合物を、室温で1時間攪拌した後、濃縮した。残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製し、生成物である例1l(180mg、収率22.0%)を灰白色固体として得た。LCMS[M+1]+=536.3.
例1l(160mg、0.299mmol)のMeOH(30mL)中の混合物に、N2保護下で、Pd/C(16mg)を加えた。懸濁液を真空下で脱気し、H2でパージし、それをH2バルーン下で室温で30分間攪拌した。固体をろ過し、ろ液を濃縮した。残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物である例1m(82mg、収率54.2%)を黄色固体として得た。LCMS[M+1]+=506.2.
例1m(80mg、0.16mmol)の1,4-ジオキサン(5mL)溶液に、Cs2CO3(103mg、0.32mmol)および第3-t-Bu-Xphos-Pd(3rd-t-Bu-Xphos-Pd)(14mg、0.016mmol)を加えた。反応混合物を、N2下、90℃で、16時間攪拌した。室温に冷却後、溶媒を除去し、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物である例1n(50mg、収率67.4%)を灰白色固体として得た。LCMS[M+1]+=470.2。
例1n(50mg、0.11mmol)のDCM(2mL)溶液に、TFA(0.5mL)を加えた。反応混合物を室温で2時間攪拌し、次いで真空中で濃縮した。残渣を分取HPLCにより精製して、所望の生成物である例51(17.0mg、収率43.2%)を白色固体として得た。LCMS[M+1]+=370.2.1H NMR(300MHz,DMSO-d6)δ 9.14(s,1H),8.57(d,1H),8.14(s,1H),8.06(brs,1H),7.93(d,1H),7.75(d,1H),5.95(s,1H),4.53(s,2H),3.97(s,3H),3.59(t,2H),3.36-3.32(m,2H),2.92(d,3H).
実施例2:化合物例2の合成に関する全般的手順
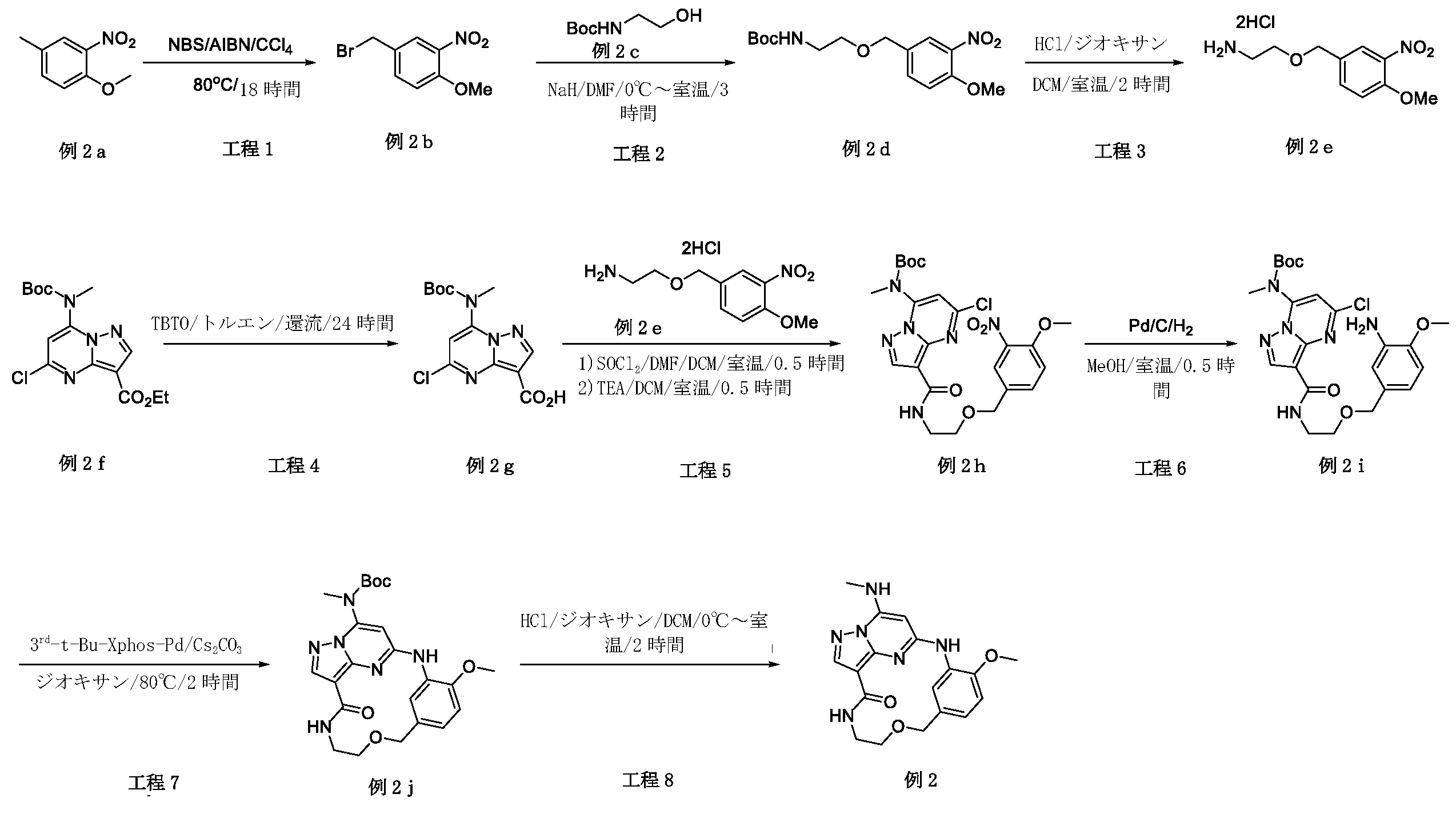
例2a(10.0g、59.8mmol、1.0当量)のCCl4(200mL)溶液に、NBS(10.8g、60.4mmol、1.01当量)およびAIBN(1.96g、12.0mmol、0.20当量)を加えた。反応混合物を、N2下、80℃で、18時間攪拌した。室温に冷却後、溶媒を除去し、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物である例2b(7.2g、収率49%)を黄色固体として得た。
LCMS[M+1]+=245.2.
例2b(1.0g、4.06mmol、1.0当量)および例2c(720mg、4.47mmol、1.1当量)のDMF(20mL)溶液に、NaH(244mg、鉱油中60%、6.1mmol、1.5当量)を0℃で数回に分けて加えた。反応混合物を、室温で4時間攪拌し、次いでNH4Clの飽和水溶液(40mL)中に注いで、これをEtOAc(50mL×3)で抽出した。一つに合わせた有機層をブラインで洗浄し、Na2SO4上で乾燥させ、濃縮した。粗生成物をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物である例2d(1.2g、収率90%)を黄色油状物として得た。LCMS[M+1]+=327.2.
例2d(800mg、2.5mmol、1.0当量)のDCM(8mL)溶液に、HCl/ジオキサン(1mL、ジオキサン中4M)を加えた。反応溶液を室温で2時間攪拌した。完了後、反応混合物を濃縮して、生成物である例2e(660mg、収率83%)を黄色固体として得た。LCMS[M+1]+=227.2
例2f(1.0g、2.8mmol、1.0当量、例1iより)のトルエン(15mL)溶液に、TBTO(3.3g、5.6mmol、2.0当量)を加えた。反応混合物を、N2下で、24時間、還流で攪拌した。濃縮後、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物である例2g(800mg、収率82%)を黄色固体として得た。LCMS[M+1]+=327.2.
例2g(452mg、1.7mmol、0.8当量)のDCM(10mL)溶液に、SOCl2(1.04g、8.8mmol、4.0当量)およびDMF(0.2mL)を加え、反応混合物を室温で0.5時間攪拌した。反応が完了した後、それを真空中で濃縮して粗生成物を得て、これをDCMで希釈し、次いで0℃で例2e(700mg、2.2mmol、1.0当量)およびTEA(1.1g、11.0mmol、5.0当量)のDCM(10mL)溶液に滴加した。得られた混合物を室温で0.5時間攪拌した。溶媒を除去し、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製し、生成物である例2h(300mg、収率3%)を黄色固体として得た。LCMS[M+1]+=535.3.
例2h(170mg、0.30mmol、1.0当量)のMeOH(30mL)溶液に、Pd/C(17mg)を加えた。懸濁液を真空下で脱気し、H2でパージし、それをH2バルーン下で室温で0.5時間攪拌した。固体をろ過し、ろ液を濃縮した。残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物である例2i(107mg、収率71%)を黄色油状物として得た。
LCMS[M+1]+=505.2.
例2i(100mg、0.20mmol、1.0当量)のジオキサン(5mL)溶液に、Cs2CO3(130.4mg、0.40mmol、2.0当量)および第3t-Bu-Xphos-Pd(17.8mg、0.02mmol、0.1当量)を加えた。反応混合物を、N2保護下、80℃で2時間攪拌した。固体をろ過し、ろ液を濃縮して、残渣を分取TLCにより精製して、例2j(50mg、収率53%)を黄色固体として得た。
LCMS[M+1]+=469.3。
例2j(50mg、0.10mmol、1.0当量)のDCM(2mL)溶液に、HCl/ジオキサン(1mL、THF中4M)を0℃で滴加した。反応混合物を室温で2時間攪拌した。完了後、反応混合物を濃縮し、残渣を分取HPLCにより精製して、所望の生成物である例2(17.0mg、収率46%)を淡黄色固体として得た。LCMS[M+1+=369.2.1H NMR(300MHz,DMSO-d6)δ 8.91(s,1H),8.32(d,1H),8.22(brs,1H),8.12(s,1H),7.82(q,1H),7.00(d,1H),6.92(dd,1H),5.95(s,1H),4.50(s,2H),3.88(s,3H),3.57(t,2H),3.44-3.32(m,2H),2.92(d,3H).
実施例3:化合物例3の合成に関する全般的手順

例3a(2.0g、11.69mmol、1.0当量)および例3b(2.45g、14.03mmol、1.2当量)の乾燥THF(20mL)溶液に、PPh3(3.69g、14.03mmol、1.2当量)およびDBAD(3.22g、14.03mmol、1.2当量)を、N2下で0℃で加え、これを室温で2時間攪拌した。溶媒を真空下で除去し、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製し、所望の生成物である例3c(2.5g、収率64.9%)を白色固体として得た。LCMS[M+1]+=327.3
例3c(1.0g、3.06mmol、1.0当量)のMeOH(10mL)溶液に、N2保護下で10%のPd/C(100mg)を加えた。懸濁液を真空下で脱気し、H2で3回パージした。混合物をH2バルーン下、室温で1時間攪拌した。懸濁液をセライトパッドを通してろ過し、ろ過ケーキをMeOHで洗浄した。一つに合わせたろ液を真空中で濃縮して、所望の生成物である例3d(900mg、収率99.1%)を無色油状物として得た。LCMS[M+1]+=297.3
例3d(100mg、0.28mmol、1.0当量)、例3e(108.7mg、0.40mmol、1.5当量、例1iより)、およびCs2CO3(183.6mg、0.56mmol、2.0当量)のジオキサン(5mL)中の混合物に、Pd(OAc)2(6.4mg、0.028mmol、0.1当量)、BINAP(35.1mg、0.056mmol、0.2当量)を加えた。混合物を、N2で3回脱気し、90℃で18時間攪拌した。反応物を、真空中で濃縮した。残渣を、シリカゲルフラッシュカラムクロマトグラフィーにより精製し、所望の生成物である例3f(130mg、収率75.9%)を淡褐色固体として得た。LCMS[M+1]+=615.4
EtOH(30mL)およびH2O(10mL)中の例3f(130mg、1.78mmol、1.0当量)の溶液に、NaOH(12.7mg、1.5mmol、1.0当量)を0℃で加えた。混合物を、80℃で16時間攪拌した。溶媒を除去して、粗生成物である例3g(160mg、定量)を白色固体として得た。LCMS[M+1]+=587.4
例3g(160mg、0.27mmol、1.0当量)のMeOH(2mL)溶液に、HCl/ジオキサン(1.0mL、ジオキサン中4M)を加え、これを室温で2時間攪拌した。混合物を濃縮し、残渣をEtOAc(30mL)で処理して、粗生成物である例3h(150mg、定量)を白色固体として得た。LCMS[M+1]+=387.4.
例3h(未精製、135mg、0.30mmol、1.0当量)、DIEA(196.7mg、1.52mmol、5.0当量)のDMF(10mL)溶液に、HATU(138.6mg、0.37mmol、1.2当量)を加えた。混合物を室温で1時間攪拌した。EtOAc(40mL)を反応混合物に加え、これをブライン(20mL×2)で洗浄し、Na2SO4上で乾燥して濃縮した。残渣を分取HPLCにより精製し、所望の生成物である例3(3.3mg、収率3.1%)を白色固体として得た。LCMS[M+1]+=369.1.1H NMR(300MHz,DMSO-d6)δ 8.87(s,1H),8.44(s,1H),8.25-8.11(m,2H),7.88(s,1H),6.98(s,1H),6.51(s,1H),6.07(s,1H),4.32-4.21(m,2H),3.84(s,3H),3.22-3.15(m,2H),2.92(s,3H),2.02-1.81(m,2H).
実施例4:化合物例4の合成に関する全般的手順

例4a(1g、5.92mmol)のTHF(10mL)溶液に、例4b(1.04g、5.92mmol)およびPPh3(1.86g、7.1mmol)を加えた。混合物を0℃に冷却し、DIAD(1.4g、7.1mmol)を滴加した。得られた混合物をN2下、室温で1時間攪拌した。反応混合物を、EtOAc(50mL×2)で抽出した。一つに合わせた有機相を、ブラインで洗浄し、Na2SO4上で乾燥し、ろ過し、ろ液を減圧下で濃縮した。残渣を、シリカゲルクロマトグラフィーにより精製して、例4c(3g、未精製)を黄色油状物として得た。
LCMS[M+1-100]+=227.1
例4c(未精製、3g、9.2mmol)のDCM(20mL)溶液に、TFA(10mL)を加えた。混合物を室温で、2時間攪拌した。反応混合物を濃縮した。残渣を、DCM(50mL×2)およびH2Oで抽出した。水層を、NaHCO3でアルカリ化し、DCM(50mL×2)で抽出した。一つに合わせた有機相を、ブラインで洗浄し、Na2SO4上で乾燥し、ろ過し、ろ液を減圧下で濃縮し、例4d(700mg、収率34%)を黄色油状物として得た。LCMS[M+1]+=227.1.1H NMR(400MHz,クロロホルム-d)δ 7.39(d,1H),7.10(dd,1H),7.01(d,1H),4.04(t,2H),3.90(s,3H),2.91(t,2H),1.92(p,2H).
例4e(10g、77.5mmol)のMeOH(100mL)溶液に、0℃で、NaHCO3(13g、155.0mmol)を加えた。次いで、Br2(18.6g、116.3mmol)を滴加し、得られた混合物を室温で一晩攪拌した。溶媒体積の半分を減圧下で除去した。残りを氷水に注いだ。形成した固体を回収し、乾燥させて、例4f(14.5g、収率90%)を赤色固体として得た。LCMS[M+1]+=209.9.1H NMR(400MHz,DMSO-d6)δ7.98(s,1H),6.96(s,2H).
例4f(14.5g、69.7mmol)のEtOH(100mL)溶液に、例4g(16.7g、111.5mmol)を加えた。混合物をN2下、80℃で一晩攪拌した。反応混合物を濃縮した。残渣をシリカゲルクロマトグラフィーにより精製して、例4h(7g、収率39%)(臭素化36%および塩素化64%)を黄色固体として得た。LCMS[M+1]+=260.0/306.0。1H NMR(400MHz,クロロホルム-d)δ 8.37(d,1H),7.57(s,0.36 H),7.38(s,0.64H),4.46(q,2H),1.43(t,3H)。
例4h(640mg、2.46mmol)のジオキサン(6mL)溶液に、例4i(409mg、2.71mmol)およびTEA(497mg、4.92mmol)を加えた。混合物を、N2下、90℃で2時間攪拌した。反応混合物を濃縮して、スラッジを得て、これをH2O(5mL)で粉砕して、固体にし、これをろ過して、H2Oで洗浄し、次いでDCM(20mL)で回収した。溶液をNa2SO4上で乾燥し、ろ過し、ろ液を減圧下で濃縮して、例4j(880mg、収率82%)を黄色固体として得た。LCMS[M+1]+=375.1.1H NMR(400MHz,クロロホルム-d)δ 8.10(s,1H),7.15(d,2H),6.85(d,2H),6.10(s,1H),5.48(s,2H),4.43(q,2H),3.78(s,3H),3.16(s,3H),1.41(t,3H).
例4j(680mg、1.81mmol)のTHF/MeOH/H2O(9mL/9mL/6mL)溶液に、LiOH.H2O(305mg、7.25mmol)を加えた。混合物を室温で一晩攪拌させた。THF/MeOHを真空中で除去し、得られた溶液を、1M HClを用いてpH=4に調整した。混合物を、DCM(30mL×2)で抽出した。一つに合わせた有機相を、ブラインで洗浄し、Na2SO4上で乾燥し、ろ過し、ろ液を減圧下で濃縮し、例4k(760mg、収率93%)を黄色固体として得た。LCMS[M+1]+=347.1。
例4k(660mg、1.9mmol)のDMF(6mL)溶液に、例4d(430mg、1.9mmol)、TEA(576mg、5.7mmol)およびHATU(867mg、2.28mmol)を加えた。混合物をN2下、室温で2時間攪拌した。反応混合物を、EtOAc(30mL×2)で抽出した。一つに合わせた有機相を、ブラインで洗浄し、Na2SO4上で乾燥し、ろ過し、ろ液を減圧下で濃縮した。残渣をシリカゲルクロマトグラフィーにより精製して、例4l(960mg、収率87%)を黄色固体として得た。LCMS[M+1]+=555.2。
例4l(30mg、0.054mmol)のTHF/HOAc(0.5mL/0.05mL)溶液に、Zn(35mg、0.54mmol)を加えた。混合物を室温で一晩攪拌させた。反応混合物を、NaHCO3水溶液でアルカリ化し、DCM(10mL×2)で抽出した。一つに合わせた有機相を、ブラインで洗浄し、Na2SO4上で乾燥し、ろ過し、ろ液を減圧下で濃縮し、例4m(30mg、未精製)を黄色油状物として得た。LCMS[M+1]+=525.2。
例4m(30mg、0.06mmol)のジオキサン(2mL)溶液に、Cs2CO3(37mg、0.11mmol)および第3-tBu-Xphos-Pd(5mg、0.006mmol)を加えた。混合物を、N2下、90℃で2時間攪拌した。反応混合物を、DCM(10mL×2)で抽出した。一つに合わせた有機相を、ブラインで洗浄し、Na2SO4上で乾燥し、ろ過し、ろ液を減圧下で濃縮し、例4n(40mg、未精製)を黄色油状物として得た。LCMS[M+1]+=489.2。
例4n(未精製、0.06mmol)のDCM(1mL)溶液に、HCl/EtOAc(0.3mL)を加えた。混合物を室温で、2時間攪拌した。混合物をろ過し、減圧下で濃縮した。残渣を分取HPLCにより精製し、例4(5.1mg、収率23%)を黄色固体として得た。LCMS[M+1]+=369.1.1H NMR(400MHz,クロロホルム-d)δ 8.82(s,1H),8.10-8.07(m,2H),7.00(s,1H),6.87(d,1H),6.65(s,1H),6.57(dd,1H),5.66(s,1H),4.33(t,2H),3.91(s,3H),3.50-3.44(m,2H),3.06(d,3H),2.12-2.03(m,2H).
実施例5:
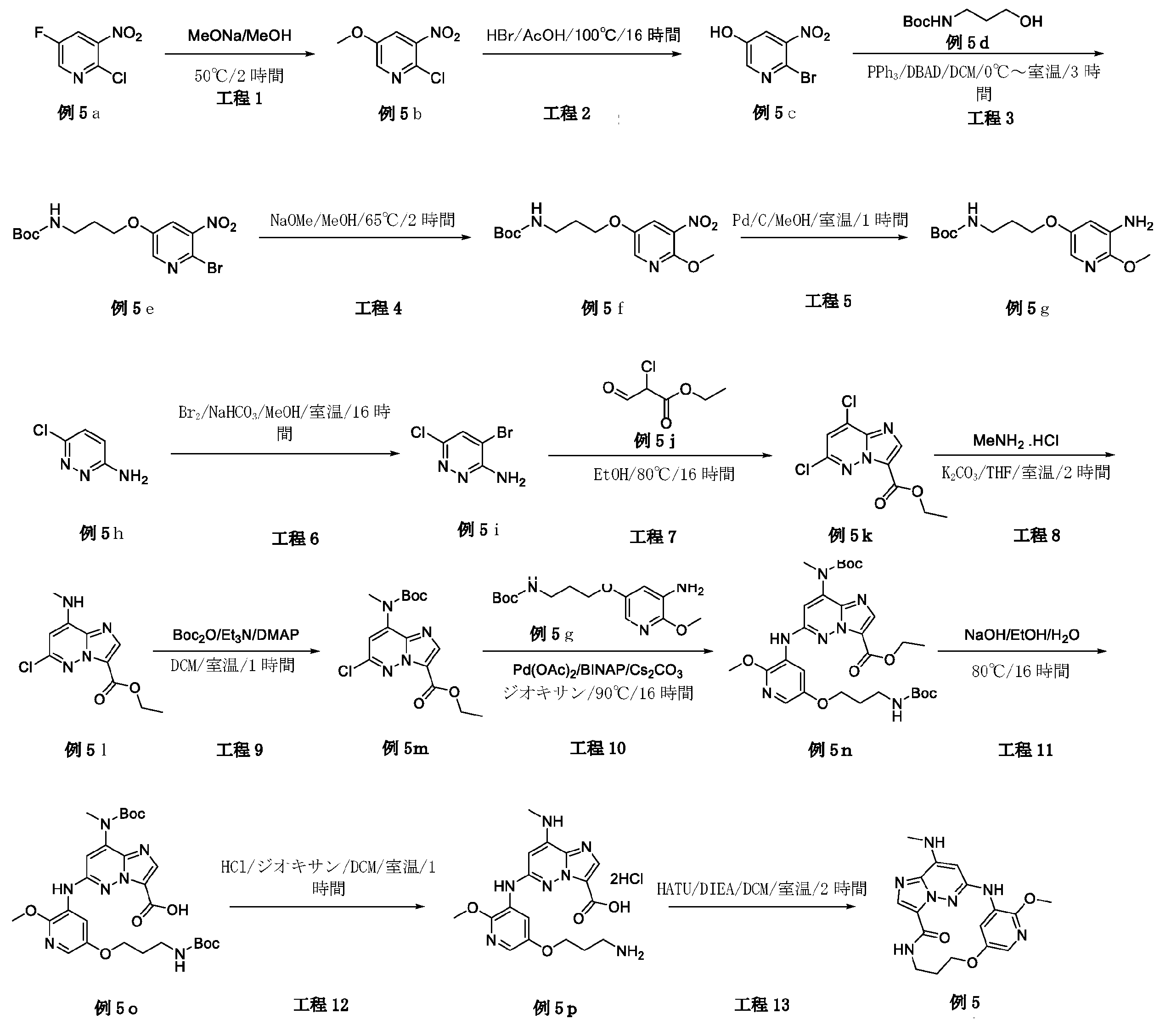
例5a(10.0g、56.8mmol、1.0当量)のMeOH(50mL)溶液に、0℃で、NaOMe(4.6g、85.2mmol、1.5当量)を加えた。反応混合物を50℃で2時間攪拌した。混合物を真空中で濃縮した。残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、所望の生成物である例5a(1.5g、収率14.1%)を黄色固体として得た。LCMS[M+1]+=189.1。
例5b(1.5g、13.58mmol、1.0当量)のHBr/AcOH(20mL)溶液を、100℃で16時間攪拌した。反応混合物を真空中で濃縮した。残渣を、H2O(20mL)で希釈し、飽和NaHCO3水溶液でpH約8に塩基性化した。この水溶液をEtOAc(50mL×3)で抽出した。一つに合わせた有機層をブラインで洗浄し、Na2SO4上で乾燥させ、真空中で濃縮した。残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、所望の生成物である例5c(1.0g、収率57.5%)を黄色固体として得た。LCMS[M+1]+=219.1。
例5c(900mg、4.13mmol、1.0当量)および例5d(867mg、4.95mmol、1.2当量)の乾燥DCM(20mL)溶液に、PPh3(1.3g、4.95mmol、1.2当量)、その後にDBAD(1.13g、4.95mmol、1.2当量)を、N2下で0℃で加えた。反応混合物を室温で3時間攪拌した。溶媒を真空下で除去し、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製し、所望の生成物である例5e(950mg、収率61.4%)を黄色固体として得た。LCMS[M+1]+=376.2。
例5e(950mg、2.54mmol、1.0当量)のMeOH(20mL)溶液に、NaOMe(412mg、7.62mmol、3.0当量)を加えた。反応混合物を、65℃で、2時間攪拌した。混合物を真空中で濃縮した。残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物である例5f(700mg、収率84.5%)を黄色固体として得た。LCMS[M+1]+=328.3。
例5f(650mg、1.98mmol、1.0当量)のMeOH(20mL)溶液に、N2保護下、10%のPd/C(60mg)を加えた。混合物をH2で3回脱気し、H2バルーン下で1時間、室温で攪拌した。固体をろ過した。ろ液を真空中で濃縮して、所望の生成物である例5g(550mg、収率93.2%)を無色油状物として得た。LCMS[M+1]+=298.3。
MeOH(30mL)中の例5f(5.0g、38.75mmol、1.0当量)およびNaHCO3(9.76g、116.2mmol、3.0当量)の混合物に、Br2(7.4g、46.51mmol、1.2当量)を0℃で滴加した。添加後、それを室温まで温め、16時間攪拌した。反応混合物を真空中で濃縮した。残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、所望の生成物である例5i(3.5g、収率43.6%)を黄色固体として得た。LCMS[M+1]+=208.1。
例5i(3.5g、16.9mmol、1.0当量)のEtOH(50mL)溶液に、例5j(5.07g、33.8mmol、2.0当量)を加え、これを80℃で16時間攪拌した。反応混合物を真空中で濃縮した。残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、所望の生成物である例5k(1.2g、収率34.2%)を白色固体として得た。LCMS[M+1]+=260.1。
THF(20mL)中の例5k(1.2g、4.61mmol、1.0当量)およびK2CO3(1.08g、13.8mmol、3.0当量)の混合物に、メタンアミン塩酸塩(467mg、6.91mmol、1.5当量)を加え、これを室温で2時間攪拌した。反応混合物は、真空中で濃縮した。残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、所望の生成物である例5l(1.05g、収率87.5%)を黄色固体として得た。LCMS[M+1]+=255.2。
例5l(1.05g、3.92mmol、1.0当量)、Et3N(1.19g、11.76mmol、3.0当量)およびDMAP(47.5mg、0.39mmol、0.1当量)のDCM(15mL)溶液に、Boc2O(1.27g、5.88mmol、1.5当量)を0℃で加え、これを室温で1時間攪拌した。反応混合物を真空中で濃縮した。残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、所望の生成物である例5m(1.1g、収率75.3%)を白色固体として得た。LCMS[M+1]+=355.2。
例5m(350mg、0.99mmol、1.0当量)、例5g(352mg、1.18mmol、1.2当量)、およびCs2CO3(643mg、20.0mmol、2.0当量)のジオキサン(10mL)中の混合物に、Pd(OAc)2(22mg、0.099mmol、0.1当量)およびBINAP(134mg、0.198mmol、0.2当量)を加えた。混合物を、N2で3回脱気し、次いで16時間で90℃に加熱した。反応物を真空中で濃縮した。残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、所望の生成物である例5n(290mg、収率47.7%)を淡褐色固体として得た。LCMS[M+1]+=616.4。
例5n(280mg、0.46mmol、1.0当量)のEtOH(2.5mL)とH2O(0.8mL)の溶液中に、0℃でNaOH(36.5mg、0.91mmol、2.0当量)を加えた。混合物を80℃に加熱し、16時間攪拌した。反応混合物を真空中で濃縮して、粗生成物である例5o(360mg、未精製、定量)を白色固体として得た。LCMS[M+1]+=588.4。
例5o(350mg、0.596mmol、1.0当量)のDCM(2mL)溶液に、HCl/ジオキサン(1.0mL、ジオキサン中4M)を加え、これを室温で1時間攪拌した。混合物を真空中で濃縮し、EtOAc(30mL)で処理して、粗生成物である例5p(160mg、収率58.4%)を白色固体として得た。LCMS[M+1]+=388.4。
例5p(160mg、0.35mmol、1.0当量)およびDIEA(135mg、1.04mmol、3.0当量)のDMF(5mL)溶液に、HATU(199mg、0.52mmol、1.5当量)を加えた。混合物を室温で2時間攪拌した。反応混合物にEtOAc(10mL)を加えて、ブライン(10mL×2)で洗浄した。有機層を、真空中で濃縮した。残渣を分取HPLCにより精製し、所望の生成物である例5(4.3mg、収率3.3%)を白色固体として得た。LCMS[M+1]+=370.2.1H NMR(300MHz,DMSO-d6)δ 8.71(s,1H),8.63(d,1H),8.60-8.57(m,1H),7.83(s,1H),7.58-7.55(m,1H),7.43(d,1H),6.33(s,1H),4.33-4.29(m,2H),3.94(s,3H),3.27-3.26(m,2H),2.88(d,3H),1.95-1.86(m,2H).
実施例6:
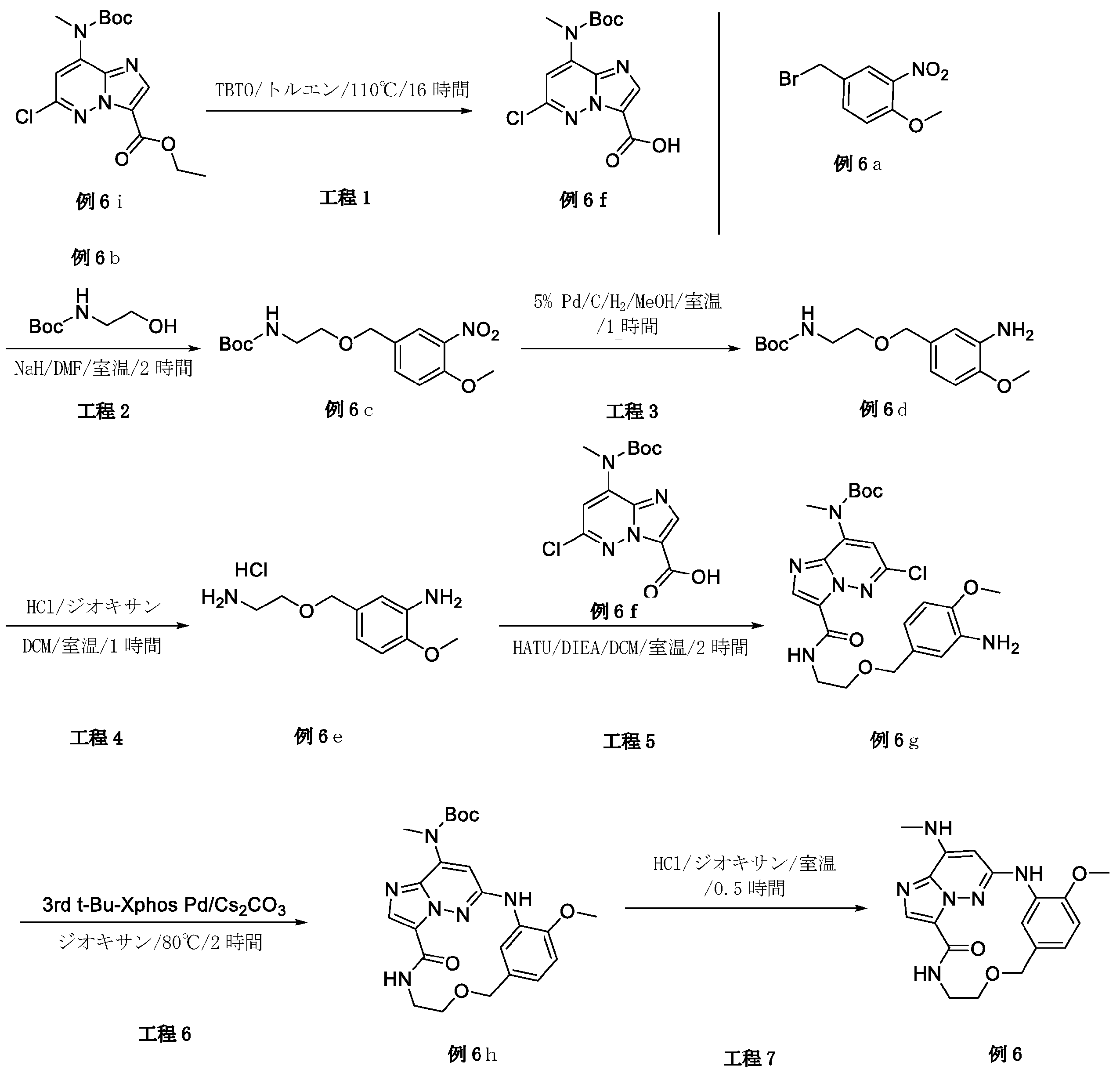
例6i(2.0g、5.6mmol、1.0当量)のトルエン(20mL)溶液に、TBTO(6.7g、11.2mmol、2.0当量)を加えた。混合物を、110℃に加熱し、16時間攪拌した。混合物を真空中で濃縮した。粗生成物をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、所望の生成物である例6f(1.7g、収率88.5%)を黄色固体として得た。LCMS[M+1]
+=327.2。
例6b(1.18g、7.34mmol、1.2当量)のDMF(10mL)溶液に、NaH(539mg、鉱油中60%、13.5mmol、2.2当量)を0℃で数回に分けて加えた。0.5時間攪拌した後、例6a(1.5g、6.12mmol、1.0当量)のDMF(20mL)溶液を滴加した。反応混合物を室温で、2時間攪拌した。反応を、0℃で飽和NH4Cl水溶液(50mL)でクエンチし、EtOAc(100mL×3)で抽出した。一つに合わせた有機層をブラインで洗浄し(50mL×3)、Na2SO4上で乾燥させ、真空中で濃縮した。粗生成物をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、所望の生成物である例6c(1.1g、収率55.2%)を黄色固体として得た。LCMS[M+1]
+=327.3。
例6c(500mg、1.53mmol、1.0当量)をMeOH(10mL)に溶解させて、N2保護下で、5%のPd/C(100mg)を加えた。系を真空にし、次いで水素で再充填した。混合溶液をH2バルーン下、室温で1時間攪拌した。反応混合物をろ過し、ろ液を濃縮して、無色油状物として所望の生成物である例6d(450mg、収率99.3%)を得た。LCMS[M+1]+=297.3。
例6d(450mg、1.52mmol、1.0当量)のDCM(10mL)溶液に、HCl/ジオキサン(3mL、4M)を加えた。反応混合物を室温で1時間攪拌した。反応溶液を真空中で濃縮して、所望の生成物である例6e(300mg、収率85.2%)を白色固体として得た。LCMS[M+1]+=197.3。
例6f(320mg、0.98mmol、1.0当量)のDCM(15mL)溶液に、DIEA(760mg、5.88mmol、6.0当量)およびHATU(448mg、1.17mmol、1.2当量)を加えた。0.5時間攪拌した後、例6e(316mg、1.17mmol、1.2当量)を加えた。反応溶液を室温で2時間攪拌した。溶媒を除去し、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製し、所望の生成物である例6g(220mg、収率44.4%)を黄色固体として得た。LCMS[M+1]+=505.4。
例6g(170mg、0.33mmol、1.0当量)のジオキサン(10mL)溶液に、Cs2CO3(219mg、0.67mmol、2.0当量)および第3-t-Bu-Xphos Pd(30mg、0.033mmol、0.1当量)を加えた。反応混合物を、N2下、80℃で2時間攪拌した。反応溶液を真空中で濃縮した。粗生成物を分取TLCにより精製して、所望の生成物である例6h(110mg、収率69.6%)を黄色固体として得た。LCMS[M+1]+=469.4。
例6h(110mg、0.17mmol、1.0当量)のDCM(5mL)溶液に、0℃でHCl/ジオキサン(1mL、ジオキサン中4M)を加えた。溶液を室温で0.5時間攪拌し、次いで濃縮した。粗生成物をMeOHに溶解させ、Na2CO3(水溶液)を加えてpH約8に塩基性化した。混合物を濃縮し、残渣を、分取TLCにより精製して、所望の生成物である例6(55mg、収率63.6%)を灰白色固体として得た。LCMS[M+1]+=369.4。1H NMR(300MHz,DMSO-d6)δ 8.76(brs,1H),8.42(s,1H),8.07(d,1H),7.81(s,1H),7.45-7.37(m,1H),7.01(d,1H),6.90(dd,1H),6.22(s,1H),4.50(s,2H),3.88(s,3H),3.57-3.54(m,2H),3.45-3.37(m,2H),2.89(d,3H).
実施例7:

例7a(21.0g、0.126mol)のCCl4(400mL)中の混合物に、NBS(23.5g、0.132mol)およびAIBN(4.1g、0.025mol)を加えた。反応混合物を、80℃で、16時間攪拌した。室温に冷却後、溶媒を除去し、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物である例7b(18.5g、収率59.8%)を黄色固体として得た。
例7c(2.13g、12.2mmol)のTHF(50mL)溶液に、NaH(0.81g、鉱油中60%、20.3mmol)を0℃で数回に分けて加えた。混合物を同温度で10分間攪拌し、次いでTHF中の例7b(2.0g、8.1mmol)を滴加した。反応混合物を室温で、3時間攪拌した。混合物を飽和NH4Cl水溶液(50mL)でクエンチし、EtOAc(50mL×2)で抽出した。一つに合わせた有機層をブラインで洗浄し、無水Na2SO4上で乾燥させ、真空中で濃縮した。残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物である例7d(1.1g、収率39.8)を黄色油状物として得た。LCMS[M+1-100]+=241.2.
例7d(1.0g、2.94mmol)のMeOH(50mL)溶液に、N2保護下で、5%のPd/C(100mg)を加えた。懸濁液を真空下で脱気し、H2で3回パージした。混合物をH2バルーン下、室温で2時間攪拌した。固体をろ過し、ろ液を濃縮して生成物である例7e(900mg、収率98.8%)を黄色油状物として得た。LCMS[M+Na]+=333.4.
例7e(500mg、1.6mmol)のDCM(10mL)溶液に、HCl/ジオキサン(2mL、ジオキサン中4M、8.0mmol)を加えた。反応混合物を室温で、1時間攪拌した。混合物を濃縮して、生成物である例7f(480mg、未精製、定量)を黄色油状物として得た。LCMS[M+1]+=211.2.
例7g(324mg、0.99mmol、例6fより)のDCM(20mL)溶液に、DIEA(1.0g、7.95mmol)およびHATU(415mg、1.1mmol)を加えた。10分間攪拌した後、例7f(450mg、2.14mmol)を混合物に加えた。反応混合物を室温で、2時間攪拌した。反応が完了した後、溶媒を除去し、粗生成物をシリカゲルクロマトグラフィーにより精製して、所望の生成物である例7h(200mg、収率24.3%)を黄色固体として得た。LCMS[M+1]+=519.2.
例7h(200mg、0.39mmol)のジオキサン(10mL)溶液に、Cs2CO3(251mg、0.77mmol)および第3-t-Bu-Xphos-Pd(34mg、0.04mmol)を加えた。反応混合物を、N2下、80℃で、3時間攪拌した。室温に冷却後、溶媒を除去し、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物である例7i(105mg、収率56.4%)を黄色固体として得た。LCMS[M+1]+=483.2.
例7i(100mg、0.2mmol)のDCM(5mL)溶液に、HCl/ジオキサン(1.0mL、ジオキサン中4M、4.0mmol)を加えた。反応混合物を室温で3時間攪拌し、次いで真空中で濃縮した。残渣をMeOH(5mL)に溶解させ、NaHCO3でpH約8まで塩基性化した。混合物にDCM(100mL)を加えた。混合物を、シリカゲルカラムを通してろ過した。ろ液を濃縮して、所望の生成物である例7(38.0mg、収率47.9%)を白色固体として得た。LCMS[M+1]+=383.3.1H NMR(300MHz,DMSO-d6)δ 8.78(d,1H),8.44(s,1H),8.15(d,1H),7.81(s,1H),7.41(d,1H),7.02(d,1H),6.89(dd,1H),6.22(s,1H),4.65(d,1H),4.38(d,1H),4.05-3.94(m,1H),3.89(s,3H),3.48(dd,1H),3.29-3.22(m,1H),2.88(d,3H),1.14(d,3H).
実施例8:
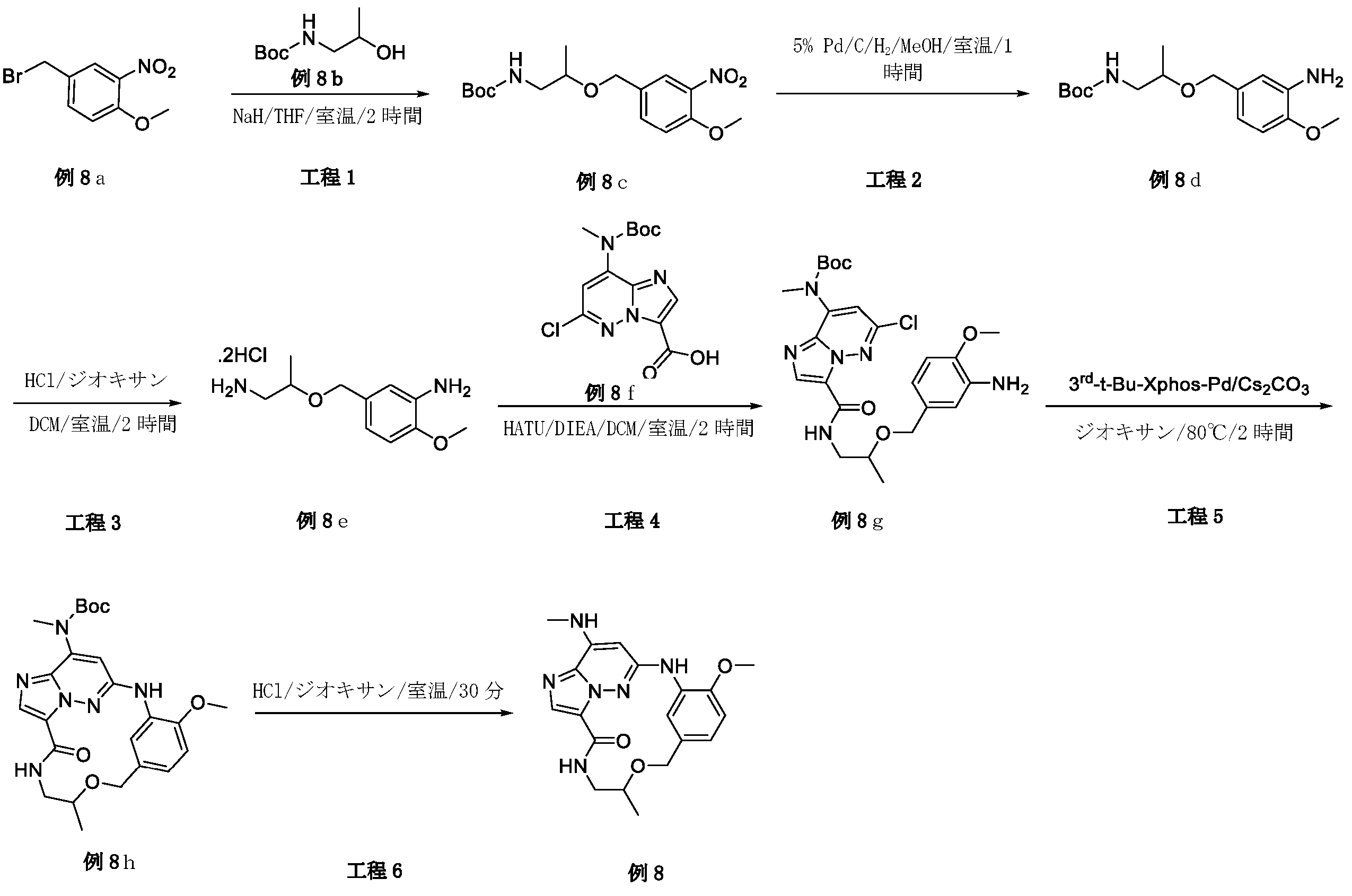
例8b(2.13g、12.20mmol、1.5当量)のTHF(50mL)溶液に、NaH(813mg、鉱油中60%、20.33mmol、2.5当量)を0℃で数回に分けて加えた。30分間攪拌した後、上記の溶液に、例8a(2.0g、8.13mmol、1.0当量)のTHF(10mL)溶液を加えた。反応混合物を室温で、2時間攪拌した。反応を、0℃で飽和NH4Cl水溶液(25mL)でクエンチし、EtOAc(50mL×3)で抽出した。一つに合わせた有機層をブラインで洗浄し、Na2SO4上で乾燥させ、濃縮した。粗生成物をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、所望の生成物である例8c(980mg、収率35.4%)を黄色固体として得た。LCMS[M+1]+=341.3.
例8c(980mg、2.88mmol、1.0当量)をMeOH(20mL)に溶解させて、N2保護下で、5%のPd/C(500mg)を加えた。懸濁液を真空にし、次いで水素で3回再充填した。混合物をH2バルーン下、室温で1時間攪拌した。固体をろ過し、ろ液を濃縮して所望の生成物である例8d(935mg、未精製、定量)を褐色固体として得た。LCMS[M+1]+=311.4.
工程3:例8e
例8d(835mg、2.69mmol、1.0当量)のDCM(12mL)溶液に、HCl/ジオキサン(3mL、ジオキサン中4M)を加えた。反応混合物を室温で、2時間攪拌した。溶媒を真空下で濃縮して、粗生成物である例8e(980mg、未精製、定量)を黄色固体として得た。LCMS[M+1]+=211.3
例8f(300mg、0.92mmol、1.0当量、例6fより)のDCM(10mL)溶液に、DIEA(947mg、7.34mmol、8.0当量)およびHATU(383mg、1.01mmol、1.1当量)を加えた。30分間攪拌した後、例8e(340mg、1.38mmol、1.5当量)を溶液に加えた。反応物を室温で2時間攪拌した。溶媒を濃縮し、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製し、所望の生成物である例8g(160mg、収率33.6%)を黄色油状物として得た。LCMS[M+1]+=519.3.
例8g(150mg、0.29mmol、1.0当量)のジオキサン(10mL)溶液に、Cs2CO3(188mg、0.58mmol、2.0当量)および第3-t-Bu-Xphos-Pd(27mg、0.029mmol、0.1当量)を加えた。反応混合物を、N2下、80℃で2時間攪拌した。反応溶液をろ過し、ろ液を濃縮した。粗生成物を分取TLCにより精製して、所望の生成物である例8h(90mg、収率64.5%)を黄色固体として得た。LCMS[M+1]+=483.4.
例8h(80mg、0.17mmol、1.0当量)のDCM(3mL)溶液に、0℃でHCl/ジオキサン(1mL、ジオキサン中4M)を加えた。反応物を室温で30分間攪拌し、次いで濃縮した。粗生成物をMeOHに溶解し、Na2CO3(過剰)を加え、室温で10分間攪拌した。固体をろ過し、ろ液を濃縮した。残渣をシリカゲルカラムクロマトグラフィーにより精製して、所望の生成物である例8(40.0mg、収率63.1%)を灰白色固体として得た。LCMS[M+1]+=383.3.1H NMR(300MHz,DMSO-d6)δ 8.73(s,1H),8.42(s,1H),8.10(d,1H),7.81(s,1H),7.40(d,1H),7.00(d,1H),6.92(dd,1H),6.21(s,1H),4.64(d,1H),4.42(d,1H),3.88(s,3H),3.65-3.49(m,1H),3.42-3.35(m,1H),3.27-3.14(m,1H),2.89(d,3H),1.20(d,3H).
実施例9:

例9a(10.0g、50.8mmol、1.0当量)の乾燥THF(100mL)溶液に、BH3.Me2S(6.1mL、DMS中10M、61.0mmol、1.2当量)を室温で滴加した。溶液を、70℃で3時間攪拌した。室温まで冷却した後、3MのHCl水溶液を、発泡がもはや観察されなくなるまで反応溶液中に滴加した。得られた混合物を、EtOAc(100mL×3)で抽出した。一つに合わせた有機層を、飽和Na2CO3水溶液で洗浄し、続いてブラインで洗浄し、Na2SO4上で乾燥し、真空中で濃縮して、生成物である例9b(8.7g、収率94%)を灰白色固体として得た。LCMS[M-18+1]+=166.2
例9b(2.6g、14.2mmol、1.0当量)の乾燥DCM(60mL)溶液に、PBr3(7.7g、28.4mmol、2.0当量)を滴加し、これを室温で2時間攪拌した。反応物をDCM(100mL)で希釈し、この溶液に、Na2CO3水溶液を中性pHが得られるまで加えた。得られた混合物を、DCM(100mL×2)で抽出した。一つに合わせた有機層を、ブラインで洗浄し、Na2SO4上で乾燥し、真空中で濃縮して、生成物である例9c(3.3g、収率95%)を灰白色固体として得た。LCMS[M+1]+=246.1.
例9d(1.47g、9.15mmol、1.5当量)の乾燥THF(10mL)溶液に、NaH(610mg、鉱油中60%、15.25mmol、2.5当量)を0℃で数回に分けて加え、これを30分間攪拌した。次いで例9c(1.50g、6.1mmol、1.0当量)のTHF(5mL)溶液を滴加した。混合物を、室温で1時間攪拌し、次いで水(15mL)でクエンチして、EtOAc(30mL×2)で抽出した。一つに合わせた有機層を、ブラインで洗浄し、Na2SO4上で乾燥し、真空中で濃縮し、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、所望の生成物である例9e(1.1g、収率55%)を黄色油状物として得た。LCMS[M+1]+=327.3.
例9e(1.1g、3.4mmol、1.0当量)のMeOH(25mL)溶液に、5%のPd/C(200mg)をN2保護下で加え、懸濁液を真空下で脱気し、H2で3回パージした。混合物をH2バルーン下、室温で2時間攪拌した。固体をろ過し、ろ液を濃縮して、所望の生成物である例9f(950mg、収率94%)を黄色油状物として得た。LCMS[M+1]+=297.3.
例9f(400mg、1.35mmol、1.0当量)のDCM(10mL)溶液に、HCl/ジオキサン(ジオキサン中4M、2mL)を加えた。溶液を、室温で2時間攪拌し、次いで濃縮して、生成物(650mg、未精製、定量)を黄色油状物として得た。LCMS[M+1]+=197.3
工程6:例9i
例9h(250mg、0.77mmol、1.0当量、例6fより)およびDIEA(695.3mg、5.39mmol、7.0当量)の溶液に、HATU(352mg、0.92mmol、1.2当量)を加え、それを室温で10分間攪拌した。次いで、例9g(452mg、2.31mmol、3.0当量)を加えた。混合物を、室温で2時間攪拌し、溶媒を除去した。残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、所望の生成物である例9i(280mg、収率72%)を黄色油状物として得た。LCMS[M+1]+=505.3.
例9i(100mg、0.20mmol、1.0当量)のジオキサン(5mL)溶液に、Cs2CO3(130mg、0.40mmol、2.0当量)および第3-t-Bu-Xphos-Pd(17.4mg、0.02mmol、0.1当量)を加えた。反応混合物を、N2下、110℃で4時間攪拌した。室温まで冷却した後、反応溶液をろ過し、ろ液を濃縮した。粗生成物を分取TLCにより精製して、所望の生成物である例9j(30mg、収率32%)を黄色固体として得た。LCMS[M+1]+=469.2.
例9j(20mg、0.043mmol、1.0当量)のDCM(6mL)溶液に、HCl/ジオキサン(2mL、ジオキサン中4M)を加え、これを室温で1時間攪拌し、次いで濃縮した。残渣を、MeOH(5mL)で希釈し、K2CO3(過剰)を加えた。混合物を室温で30分間攪拌した。固体をろ過し、ろ液を濃縮して、残渣を分取TLCにより精製して、所望の生成物である例9(10.5mg、収率66%)を白色固体として得た。LCMS[M+1]+=369.3.1H NMR(300MHz,DMSO-d6)δ 9.88(s,1H),7.78(s,1H),7.58(s,1H),7.48(d,1H),7.26(t,1H),7.16(d,1H),7.03(d,1H),6.08(s,1H),4.87(s,2H),3.80(s,5H),3.63 -3.53(m,2H),2.92(d,3H).
実施例10:

例10b(1.85g、10.6mmol)のTHF(20mL)溶液に、NaH(718mg、鉱油中60%、17.9mmol)を0℃で数回に分けて加えた。0.5時間攪拌した後、例10a(2.0g、8.16mmol、例7bから)のTHF(10mL)溶液を滴加した。反応混合物を室温で2時間攪拌した。反応を、0℃で飽和NH4Cl水溶液(50mL)でクエンチし、EtOAc(100mL×3)で抽出した。一つに合わせた有機層をブラインで洗浄し(50mL×3)、Na2SO4上で乾燥させ、真空中で濃縮した。粗生成物をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、所望の生成物である例10c(2.5g、収率89.9%)を黄色固体として得た。LCMS[M+1]+=341.3.
例10c(2.5g、7.35mmol)をMeOH(30mL)に溶解し、5%のPd/C(250mg)を、N2保護下で加えた。系を真空にし、次いで水素で3回再充填した。混合溶液をH2バルーン下、室温で1時間攪拌した。反応混合物をろ過し、ろ液を濃縮して、所望の生成物である例10d(1.5g、収率65.8%)を無色油状物として得た。LCMS[M+1]+=311.3.
工程3:例10e
例10d(1.0g、3.22mmol)のDCM(15mL)溶液に、HCl/ジオキサン(2mL、ジオキサン中4M、8mmol)を加えた。反応混合物を室温で1時間攪拌した。反応溶液を真空中で濃縮して、所望の生成物である例10e(700mg、収率79.3%)を白色固体として得た。LCMS[M+1]+=211.2.
例10f(519mg、1.84mmol、例6fより)のDCM(10mL)溶液に、DIEA(950mg、7.38mmol)およびHATU(559mg、1.47mmol)を加えた。0.5時間攪拌した後、例10e(400mg、1.23mmol)を加えた。反応溶液を室温で2時間攪拌した。溶媒を除去し、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、所望の生成物である例10g(210mg、収率32.9%)を黄色固体として得た。LCMS[M+1]+=519.3.
例10g(195mg、0.38mmol)のジオキサン(30mL)溶液に、Cs2CO3(245mg、0.75mmol)および第3-t-Bu-Xphos-Pd(33mg、0.04mmol)を加えた。反応混合物を、N2下、85℃で、5時間攪拌した。室温まで冷却した後、溶媒を除去し、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物である例10h(95mg、収率52.3%)を黄色固体として得た。LCMS[M+1]+=483.2.
例10h(95mg、0.2mmol)のDCM(5mL)溶液に、HCl/ジオキサン(1mL、ジオキサン中4M、4mmol)を加えた。反応混合物を室温で5時間攪拌し、次いで真空中で濃縮した。残渣をMeOH(5mL)に溶解させ、NaHCO3で塩基性化した(pH=8)。DCM(100mL)を混合物に加え、固体をろ過した。ろ液を濃縮して、所望の生成物である例10(50.0mg、収率66.4%)を白色固体として得た。LCMS[M+1]+=383.3.1H NMR(300MHz,DMSO-d6)δ 8.76(d,1H),8.45(s,1H),8.15(d,1H),7.82(s,1H),7.41(d,1H),7.01(d,1H),6.88(dd,1H),6.23(s,1H),4.62(d,1H),4.35(d,1H),4.05-3.93(m,1H),3.89(s,3H),3.54-3.46(m,1H),3.25(t,1H),2.88(d,3H),1.12(d,3H).
実施例11:

例11b(2.63g、15mmol)のTHF(50mL)溶液に、NaH(1.0g、鉱油中60%、25mmol)を0℃で数回に分けて加えた。10分間攪拌した後、例11a(2.46g、10mmol、例7bより)のTHF(10mL)溶液を滴加した。反応混合物を室温で、3時間攪拌した。反応を、0℃で飽和NH4Cl水溶液(20mL)でクエンチし、EtOAc(50mL)で抽出し、Na2SO4上で乾燥して、真空中で濃縮した。粗生成物をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、所望の生成物である例11c(2.6g、収率76.5%)を黄色固体として得た。LCMS[M+1]+=341.3.
例11c(1.5g、4.4mmol)をMeOH(30mL)に溶解し、次いで5%のPd/C(150mg)を、N2保護下で加えた。系を真空にし、次いで水素で3回再充填した。混合溶液をH2バルーン下、室温で2時間攪拌した。反応混合物をろ過し、ろ液を濃縮して、所望の生成物である例11d(1.35g、収率98.7%)を黄色油状物として得た。LCMS[M+1]+=311.3.
例11d(600mg、1.9mmol)のDCM(6mL)溶液に、HCl/ジオキサン(2mL、ジオキサン中4M、8mmol)を加えた。反応混合物を室温で、3時間攪拌した。反応溶液を真空中で濃縮して、所望の生成物である例11e(580mg、未精製、定量)を黄色油状物として得た。LCMS[M+1]+=211.2.
例11f(418mg、1.3mmol、例6fより)のDCM(30mL)溶液に、DIEA(1.3g、10.3mmol)およびHATU(730mg、1.9mmol)を加えた。0.5時間攪拌した後、例11e(580mg、2.0mmol)を加えた。反応混合物を室温で2時間攪拌した。溶媒を除去し、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、所望の生成物である例11g(240mg、収率36.1%)を黄色固体として得た。LCMS[M+1]+=519.3.
例11g(240mg、0.46mmol)のジオキサン(10mL)溶液に、Cs2CO3(302mg、0.92mmol)および第3-t-Bu-Xphos-Pd(41mg、0.05mmol)を加えた。反応混合物を、N2下、85℃で、5時間攪拌した。室温まで冷却した後、溶媒を除去し、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物である例11h(140mg、収率62.7%)を黄色固体として得た。
LCMS[M+1]+=483.2.
例11h(140mg、0.29mmol)のDCM(5mL)溶液に、HCl/ジオキサン(1mL、ジオキサン中4M、4mmol)を加えた。反応混合物を室温で5時間攪拌し、次いで真空中で濃縮した。残渣をMeOH(5mL)に溶解させ、NaHCO3で塩基性化した(pH=8)。混合物にDCM(100mL)を加えた。固体をろ過し、ろ液を濃縮して、所望の生成物である例11(70.0mg、収率66.4%)を灰白色固体として得た。LCMS[M+1]+=383.3.1H NMR(300MHz,DMSO-d6)δ 8.76(d,1H),8.45(s,1H),8.15(d,1H),7.82(s,1H),7.43(d,1H),7.01(d,1H),6.89(dd,1H),6.23(s,1H),4.64(d,1H),4.37(d,1H),4.05-3.92(m,1H),3.89(s,3H),3.46(dd,1H),3.25(t,1H),2.89(d,3H),1.13(d,3H).
実施例12:
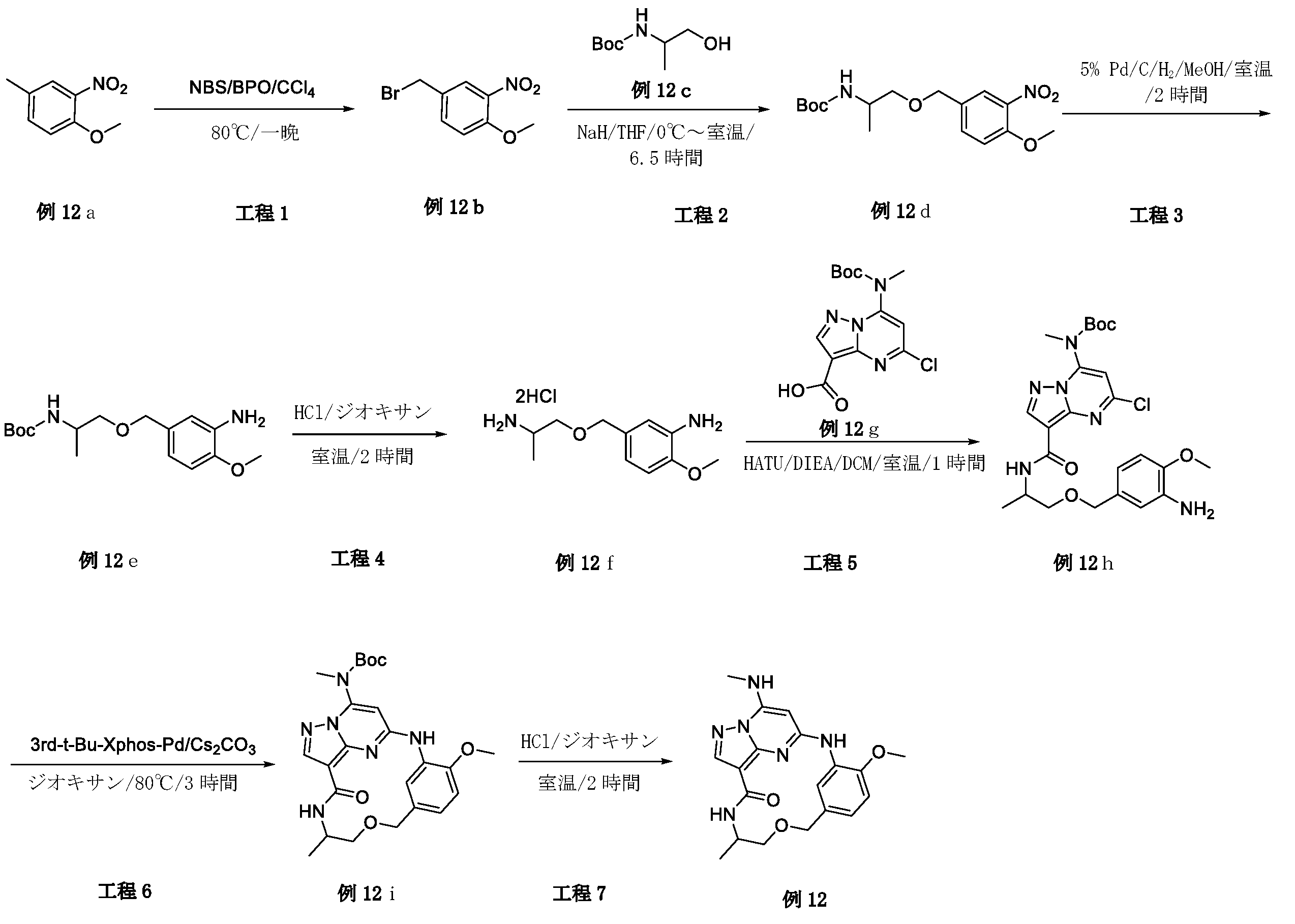
例12a(30.0g、179mmol)のCCl4(150mL)溶液に、BPO(4.4g、17.9mmol)、NBS(38.15g、216mmol)を加え、これを80℃で一晩攪拌した。冷却後、次いで混合物をDCMで希釈し、水で洗浄し、Na2SO4上で乾燥し、減圧下で濃縮して、例12b(37.0g、収率84.4%)を黄色固体として得、これを精製することなく次の工程に使用した。LCMS[M+1]+=246.0.1H NMR(400MHz,クロロホルム-d)δ 7.87(d,1H),7.57(dd,1H),7.07(d,1H),4.46(s,2H),3.96(d,3H).
例12b(2.46g、10.0mmol)のTHF(20mL)溶液に、NaH(400mg、鉱油中60%、10.0mmol)を0℃で加え、これを0.5時間攪拌した。次いで、例12c(1.75g、10.0mmol)を加え、得られた混合物を、室温で6時間攪拌した。混合物を、NH4Cl水溶液によりクエンチし、EtOAcにより抽出し、Na2SO4上で乾燥して、減圧下で濃縮した。残渣をシリカゲルカラムクロマトグラフィーにより精製して、例12d(3.3g、収率96.8%)を黄色固体として得た。LCMS[M+1-100]+=241.1.
MeOH(10mL)中の例12d(688mg、2.0mmol)および10%のPd/C(34mg)の懸濁液を、H2バルーン下で室温で2時間攪拌した。懸濁液をろ過し、ろ液を減圧下で濃縮して、例12e(640mg、粗収率103%)を黄色固体として得て、これを精製することなく次の工程に使用した。
例12e(400mg、1.3mmol)のジオキサン(2mL)溶液に、HCl/ジオキサン(1.0mL、ジオキサン中4M)を加え、これを室温で2時間攪拌した。混合物を濃縮し、残渣をEtOAc(30mL)で処理して、粗生成物である例12f(340mg、粗収率124%)を白色固体として得、これを精製することなく次の工程に使用した。
例12f(340mg、0.65mmol)、例12g(423mg、1.3mmol、例6fより)、およびTEA(810mg、8.1mmol)のDCM(10mL)溶液に、HATU(616mg、1.62mmol)を加えた。混合物を室温で1時間攪拌した。EtOAc(40mL)を反応混合物に加え、これをブライン(20mL×2)で洗浄し、Na2SO4上で乾燥して濃縮した。残渣をシリカゲルカラムクロマトグラフィーにより精製して所望の生成物である例12h(500mg、収率59%)を白色固体として得た。LCMS[M+1]+=519.2.
例12h(500mg、0.97mmol)、Cs2CO3(652mg、2.0mmol)のジオキサン(10mL)中の混合物に、第3-t-Bu-Xphos-Pd(89mg、0.1mmol)を加えた。混合物を、N2で3回脱気し、80℃で3時間攪拌した。混合物をDCMで希釈し、水で洗浄し、Na2SO4上で乾燥し、減圧下で濃縮して、例12i(450mg、粗収率93.3%)を白色固体として得、これを精製することなく次の工程に使用した。LCMS[M+1]+=483.3
例12i(200mg、0.42mmol)のジオキサン(2mL)溶液に、HCl/ジオキサン(1.0mL、ジオキサン中4M)を加え、これを室温で2時間攪拌した。混合物を濃縮し、残渣を、分取HPLCにより精製して、所望の生成物である例12(4.9mg、収率3.0%)を白色固体として得た。LCMS[M+1]+=383.3.1H NMR(400MHz,DMSO-d6)δ 8.90(s,1H),8.32(s,1H),8.22(d,1H),8.08(s,1H),7.80(d,1H),6.97(d,1H),6.88(d,1H),5.91(s,1H),4.54(d,1H),4.37(d,1H),3.85(s,3H),3.44(d,1H),2.89(d,3H),1.11(d,3H).
実施例13:
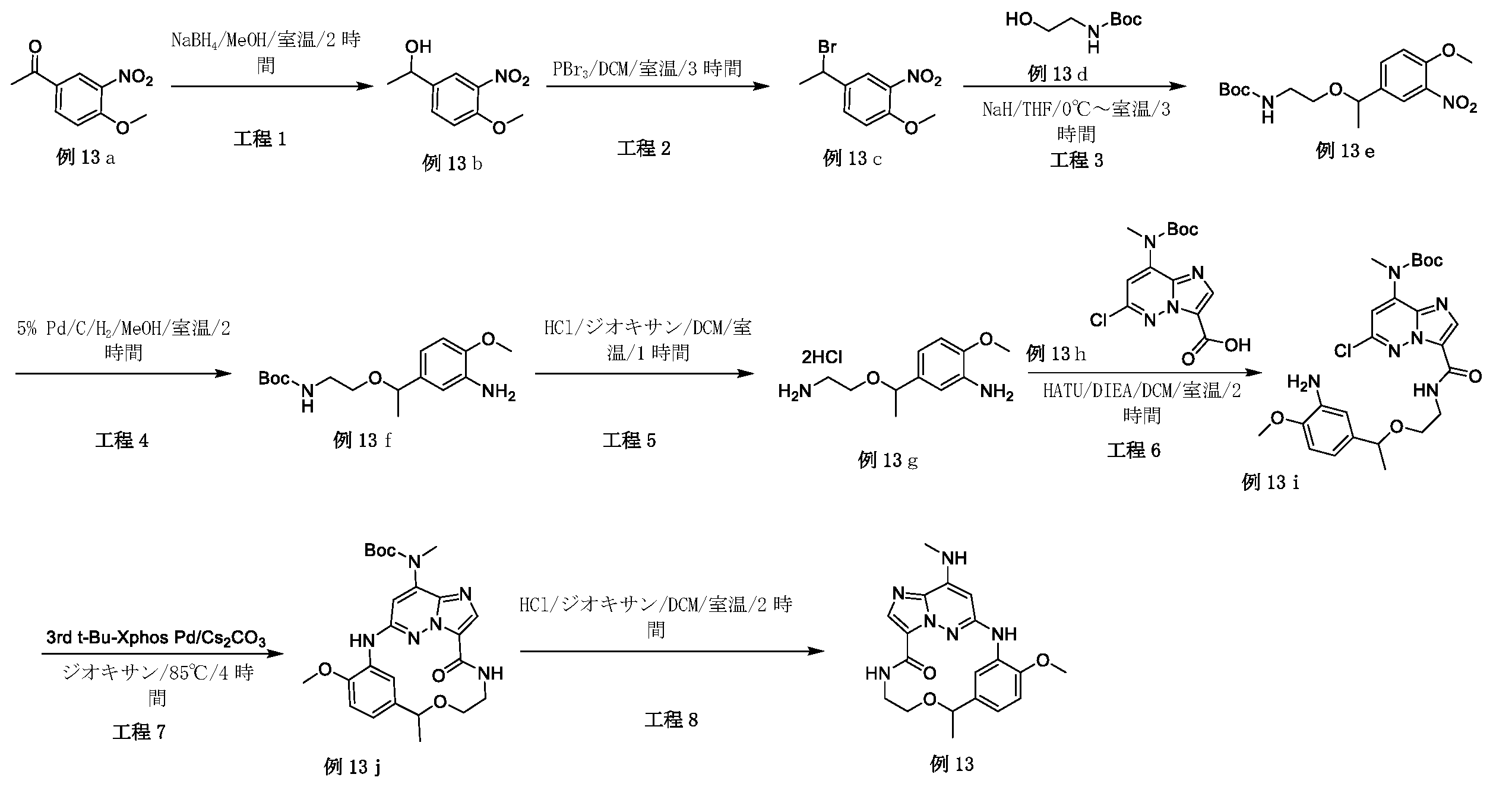
例13a(10.0g、0.05mol)のMeOH(150mL)溶液に、NaBH4(4.87g、0.13mol)を数回に分けて加えた。反応混合物を室温で、2時間攪拌した。溶媒を除去し、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製し、生成物である例13b(8.5g、収率84.1%)を黄色油状物として得た。LCMS[M+1]+=198.2.
例13b(1.97g、10.0mmol)のDCM(50mL)溶液に、PBr3(5.4g、20.0mmol)を加えた。反応混合物を室温で、3時間攪拌した。混合物を、DCM(100mL)で希釈し、飽和NaHCO3水溶液(50mL×2)で洗浄した。有機層を無水Na2SO4上で乾燥し、真空中で濃縮して、生成物である例13c(2.3g、収率88.5%)を黄色油状物として得た。
例13d(2.0g、12.5mmol)のTHF(50mL)溶液に、NaH(0.5g、鉱油中60%、12.5mmol)を0℃で数回に分けて加えた。混合物を同温度で10分間攪拌し、次いでTHF中の例13c(1.3g、5.0mmol)を滴加した。反応混合物を室温で、3時間攪拌した。混合物を飽和NH4Cl(30mL)でクエンチし、EtOAc(50mL)で抽出した。有機層を無水Na2SO4上で乾燥させ、真空中で濃縮した。残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物である例13e(810mg、収率47.6%)を黄色油状物として得た。LCMS[M+1-100]+=241.2.
例13e(800mg、2.4mmol)をMeOH(30mL)と混合し、N2保護下で、5%のPd/C(150mg)を加えた。系を真空にし、次いで水素で3回再充填した。混合物をH2バルーン下、室温で2時間攪拌した。反応混合物をろ過し、ろ液を濃縮した。残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物である例13f(420mg、収率57.6%)を黄色油状物として得た。LCMS[M+1]+=311.3.
例13f(400mg、1.29mmol)のDCM(5mL)溶液に、HCl/ジオキサン(2mL、ジオキサン中4M、8mmol)を加えた。反応混合物を室温で1時間攪拌した。反応溶液を真空中で濃縮して、所望の生成物である例13g(360mg、収率98.6%)を黄色油状物として得た。LCMS[M+1]+=211.2.
例13h(238mg、0.7mmol、例6fより)のDCM(20mL)溶液に、DIEA(752mg、5.8mmol)およびHATU(443mg、1.2mmol)を加えた。溶液を0.5時間攪拌した後、例13g(330mg、1.2mmol)を加えた。反応溶液を室温で2時間攪拌した。溶媒を除去し、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製し、所望の生成物である例13i(41mg、収率10.8%)を黄色固体として得た。LCMS[M+1]+=519.3.
例13i(41mg、0.08mmol)の1,4-ジオキサン(10mL)溶液に、Cs2CO3(51mg、0.16mmol)および第3-t-Bu-Xphos-Pd(7mg、0.01mmol)を加えた。反応混合物を、N2下、85℃で、4時間攪拌した。室温まで冷却した後、溶媒を除去し、残渣を分取TLCにより精製して、生成物である例13j(25mg、収率65.5%)を黄色固体として得た。LCMS[M+1]+=483.2.
例13j(25mg、0.05mmol)のDCM(3mL)溶液に、HCl/ジオキサン(0.2mL、ジオキサン中4M、0.8mmol)を加えた。反応混合物を室温で2時間攪拌し、次いで真空中で濃縮した。残渣をMeOH(5mL)に溶解し、次いで飽和NaHCO3水溶液を用いてpH弁(pH valve)を8に調整した。溶媒を除去し、残渣を分取TLCにより精製して、生成物である例13(11.8mg、収率59.6%)を灰白色固体として得た。LCMS[M+1]+=383.3.1H NMR(300MHz,DMSO-d6)δ 8.80(s,1H),8.42(s,1H),8.03(d,1H),7.80(s,1H),7.41(d,1H),7.00(d,1H),6.90(dd,1H),6.21(s,1H),4.45(q,1H),3.88(s,3H),3.75-3.71(m,1H),3.53-3.43(m,1H),3.42-3.37(m,1H),3.26-3.23(m,1H),2.89(d,3H),1.30(d,3H).
実施例14:

例14a(15.0g、87.2mmol)およびMeONa(14.1g、261.6mmol)のMeOH(100mL)溶液を、70℃で3時間攪拌した。混合物を減圧下で濃縮し、次いで水で希釈し、これを次いでEtOAcにより抽出し、無水Na2SO4上で乾燥し、濃縮して、粗生成物である例14b(13.4g、収率92.2%)を黄色固体として得た。残渣を、さらに精製することなく、次の工程で直接使用した。LCMS[M+1]+=169.1.
例14b(5.0g、29.8mmol)のCCl4(150mL)溶液に、BPO(720mg、2.98mmol)およびNBS(5.3g、29.8mmol)を加えた。反応混合物を、80℃で一晩攪拌し、次いでDCMで希釈し、水で洗浄し、無水Na2SO4上で乾燥した。ろ過後、ろ液を減圧下で濃縮し、これを次いでシリカゲルカラムクロマトグラフィーにより精製して、例14c(5.7g、収率77.6%)を黄色固体として得た。LCMS[M+1]+=247.0
例14d(2.1g、12.1mmol)のTHF(40mL)溶液に、0℃でNaH(1.46g、36.4mmol)を加えた。反応混合物を室温に温め、室温で0.5時間攪拌した。次いで、例14c(3.0g、12.1mmol)を加えた。混合物を、室温で6時間攪拌した後、次いで NH4Cl水溶液でクエンチし、EtOAcにより抽出し、無水Na2SO4上で乾燥した。ろ過後、ろ液を減圧下で濃縮し、これを次いでシリカゲルカラムクロマトグラフィーにより精製して、例14e(1.0g、収率:24.4%)を黄色固体として得た。LCMS[M-174]+=167.1.
例14e(1.0g、2.93mmol)およびPd/C(200mg)のMeOH(5mL)溶液を、室温で2時間、1気圧のH2下で攪拌した。ろ過後、ろ液を減圧下で濃縮して、例14f(850mg、収率:93.2%)を黄色固体として得、これを次の工程で直接使用した。LCMS[M-174]+=137.1
例14f(800mg未精製、1.3mmol)のDCM(4mL)溶液に、TFA(1.0mL)を加え、これを室温で2時間攪拌した。混合物を濃縮して、粗生成物である例14g(700mg、未精製、収率:定量)を黒色油状物として得た。LCMS[M-74]+=137.1.
例14g(30mg、0.1mmol)、例14h(33mg、0.1mmol、例6fより)、TEA(202mg、1.0mmol)のDCM(2mL)溶液に、HATU(38mg、0.1mmol)を加えた。反応混合物を室温で、2時間攪拌した。次いで、EtOAc(40mL)を反応混合物に加え、これをブライン(20mL×2)で洗浄し、無水Na2SO4上で乾燥して、濃縮した。残渣をシリカゲルカラムクロマトグラフィーにより精製して、所望の生成物である例14i(32mg、収率:62%)を褐色固体として得た。LCMS[M+1]+=520.2.
例14i(32mg、0.06mmol)、Cs2CO3(30mg、0.09mmol)のジオキサン(2mL)中の混合物に、第3-t-Bu-Xphos-Pd(5.5mg、0.006mmol)を加えた。混合物を、N2で3回脱気し、80℃で3時間攪拌した。次いで、反応混合物をDCMで希釈し、水で洗浄し、無水Na2SO4上で乾燥し、次いで減圧下で濃縮して、未精製の例14j(50mg、未精製、収率:定量)を白色固体として得、これをさらに精製することなく次の工程で使用した。LCMS[M+1]+=484.2
例14j(50mg、0.1mmol)のDCM(4mL)溶液に、TFA(1.0mL)を加え、これを室温で2時間攪拌した。混合物を濃縮し、残渣を、分取HPLCにより精製して、所望の生成物である例14(4.5mg、収率:31.4%)を白色固体として得た。LCMS[M+1]+=384.2.1H NMR(400MHz,クロロホルム-d)δ 8.58(s,1H),8.53(s,1H),8.08(s,1H),7.67(s,1H),6.69(s,1H),5.67(s,1H),4.68(d,1H),4.45(d,1H),4.24(br,1H),4.05(s,3H),3.57-3.54(m,1H),3.39-3.34(m,1H),3.03(d,3H),1.25(d,3H).
実施例15:
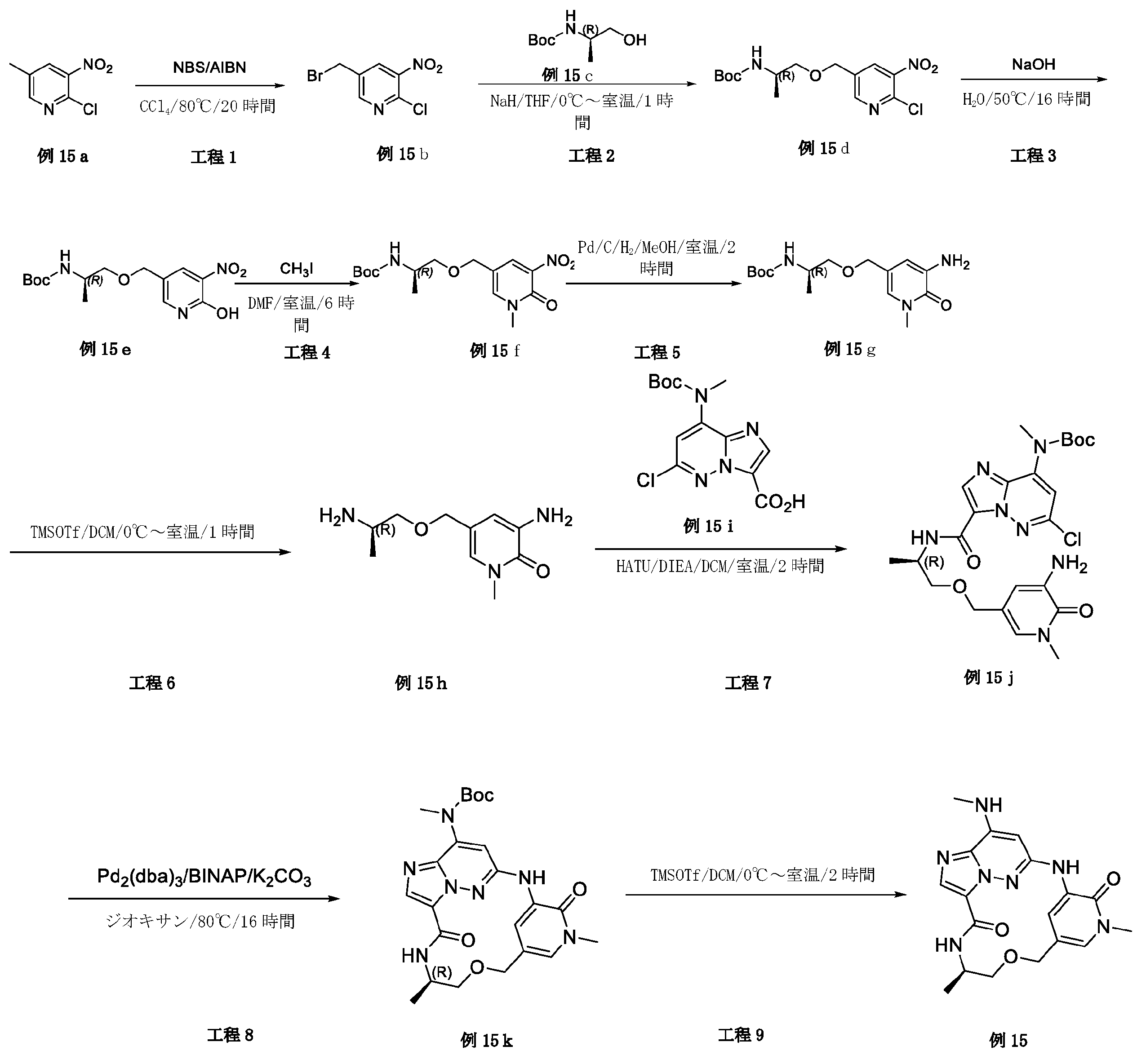
例15a(15.0g、87.2mmol)のCCl4(500mL)溶液に、NBS(31.0g、174.4mmol)およびAIBN(2.86g、17.4mmol)を加えた。反応混合物を、N2下、80℃で、20時間攪拌した。ろ過後、ろ液を濃縮し、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製し、生成物である例15b(7.5g、収率:34.2%)を黄色油状物として得た。LCMS[M+1]+=252.9.
例15c(5.8g、33.4mmol)のTHF(250mL)溶液に、NaH(1.3g、鉱油中60%、33.4mmol)を0℃で数回に分けて加えた。混合物を同温度で5分間攪拌し、次いでTHF中の例15b(7.0g、27.8mmol)を滴加した。反応混合物を室温で、1時間攪拌した。溶媒を濃縮後、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物である例15d(2.6g、収率:30.0%)を黄色油状物として得た。LCMS[M+1]+=346.2.
H2O(50mL)中の例15d(2.5g、7.2mmol)の混合物に、NaOH(1.2g、28.9mmol)を加えた。混合物を50℃で16時間攪拌した。室温まで冷却した後、反応溶液を真空中で濃縮して、所望の生成物である例15e(3.7g、未精製、収率:定量)を黄色固体として得た。LCMS[M+1]+=328.3.
例15e(3.7g、未精製、7.2mmol)のDMF(50mL)溶液に、CH3I(2.4g、17.0mmol)を加えた。反応混合物を室温で、6時間攪拌した。濃縮後、残渣をEtOAc(100mL)で希釈し、H2O(100mL)で洗浄し、無水Na2SO4上で乾燥して濃縮した。残渣をシリカゲルフラッシュカラムクロマトグラフィー(THF/石油エーテル=4/1)により精製し、生成物である例15f(860mg、収率:2工程で35.0%)を黄色油状物として得た。LCMS[M+1]+=342.2.
例15f(820mg、2.4mmol)をMeOH(20mL)に溶解させて、次いでN2保護下で、Pd/C(80mg)を数回に分けて加えた。混合物を真空下で脱気し、H2で3回パージした。混合物をH2バルーン下、室温で2時間攪拌した。固体をろ別し、ろ液を濃縮した。残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物である例15g(380mg、収率:50.8%)を黄色油状物として得た。LCMS[M+1]+=312.2.
例15g(370mg、1.2mmol)のDCM(10mL)溶液 に、0℃でTMSOTf(396mg、1.8mmol)を加えた。反応混合物を室温で1時間攪拌した。溶媒を真空中で濃縮して、所望の生成物である例15h(430mg、未精製)を黄色油状物として得た。LCMS[M+1]+=212.2.
例15i(260mg、0.8mmol、例6fより)のDCM(20mL)溶液に、DIEA(411mg、3.2mmol)およびHATU(303mg、0.8mmol)を加えた。混合物を5分間攪拌し、次いで例15h(420mg、未精製、2.0mmol)を加えた。得られた混合物を室温で2時間攪拌した。溶媒を除去し、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製し、所望の生成物である例15j(200mg、収率30.6%)を黄色固体として得た。LCMS[M+1]+=520.2.
例15j(190mg、0.37mmol)のジオキサン(20mL)溶液に、K2CO3(101mg、0.73mmol)、BINAP(228mg、0.37mmol)およびPd2(dba)3CHCl3(189mg、0.18mmol)を加えた。反応混合物を、N2下、80℃で、16時間攪拌した。室温まで冷却した後、溶媒を除去し、残渣を分取TLCにより精製して、生成物である例15k(50mg、収率28.3%)を黄色固体として得た。LCMS[M+1]+=484.4.
例15k(45mg、0.09mmol)のDCM(5mL)溶液に、0℃でTMSOTf(41mg、0.02mmol)を加えた。反応混合物を室温で2時間攪拌した。反応溶液を真空中で濃縮し、残渣を、分取TLCにより精製して、生成物である例15(15.3mg、収率:42.9%)を灰白色固体として得た。LCMS[M+1]+=384.3.1H NMR(300MHz,DMSO-d6)δ 8.73-8.63(m,2H),8.10(d,1H),7.83(s,1H),7.46(q,1H),7.33(d,1H),6.34(s,1H),4.47(d,1H),4.23(d,1H),4.05-4.03(m,1H),3.53(s,3H),3.48(d,1H),3.40(d,1H),2.87(d,3H),1.13(d,3H).
実施例16:

2-フルオロ-4-メトキシ-ベンズアルデヒド(16A)(5g、32.46mmol)を濃硫酸(30mL)に溶解し、-10℃に冷却した。濃硫酸(4mL)中の濃硝酸(2.1mL)を、20分間にわたって滴加した。-10℃未満でさらに1時間攪拌した後、混合物を粉砕氷中に注いだ。沈殿物をろ過により回収し、ジクロロメタン(40mL)と飽和炭酸水素ナトリウム(30mL)との間で分配した。有機層を乾燥(Na2SO4)し、真空中で蒸発させて、表題化合物(16B)(5.2g、80.50%)をクリーム色固体として得た。LC-MS(ESI):m/z=200.1[M+H]+.
水素化ホウ素ナトリウム(0.304g、8.04mmol)を、0℃で、メタノール(10mL)中の2-フルオロ-4-メトキシ-5-ニトロ-ベンズアルデヒド(16B)(0.8g、4.02mmol)の攪拌溶液に少しずつ加えた。2時間後、メタノールを真空中で除去した。残渣を、冷水で処理し、ジクロロメタンで抽出した。一つに合わせた有機層を、ブラインで洗浄し、乾燥(Na2SO4)し、次いで真空中で蒸発させて、表題化合物(16C)を未精製固体(0.79g、97.77%)として得た。LC-MS(ESI):m/z=202.1[M+H]+
無水ジエチルエーテル(5mL)中の四臭化炭素(2.64g、7.96mmol)を、無水ジエチルエーテル(15mL)中の(2-フルオロ-4-メトキシ-5-ニトロ-フェニル)メタノール(16C)(0.8g、3.98mmol)およびトリフェホスフィン(triphephosphine)(2.08g、7.96mmol)の攪拌溶液に滴加した。混合物を濃縮する前に一晩攪拌した。ヘキサン中の酢酸エチル(0~10%)を用いたクロマトグラフィーにより、表題化合物(16D)を淡黄色固体(0.69g、66.34%)として得た。LC-MS(ESI):m/z=264.1[M+H]+
水素化ナトリウム(105mg、2.62mmol)を、0℃でTHF(15mL)中のtert-ブチル N-(2-ヒドロキシ-1-メチル-エチル)カルバメート(0.46g、2.62mmol)の攪拌溶液に少しずつ加え、混合物を0℃で10分間攪拌し、次いで混合物に、0℃で1-(ブロモメチル)-2-フルオロ-4-メトキシ-5-ニトロ-ベンゼン(16D)(0.69g、2.62mmol)を加え、30分後、混合物を、冷水で処理し、酢酸エチルで抽出した。一つに合わせた有機層を、ブラインで洗浄し、乾燥(Na2SO4)し、次いで残渣をフラッシュクロマトグラフィーで精製して、表題化合物(16E)(0.1g、10.65%)を褐色固体として得た。LC-MS(ESI):m/z=381.1[M+23]+.1H NMR(400MHz,CDCl3)δ 8.02(d,1H),6.78(d,1H),4.60(s,1H),4.53(q,2H),3.96(s,3H),3.91-3.83(m,1H),3.49-3.43(m,2H),1.44(s,9H),1.18(d,3H).
トリフルオロ酢酸(1mL)を、N-[2-[(2-フルオロ-4-メトキシ-5-ニトロ-フェニル)メトキシ]-1-メチル-エチル]カルバメート(16E)(0.1g、0.28mmol)のDCM(3mL)溶液に加え、この混合物を2時間攪拌し、混合溶液を蒸発させて乾燥し、その後、表題化合物(16F)(0.1g.100%)を褐色液体として得、これをさらに精製することなく次の工程で使用した。LC-MS(ESI):m/z=259.2[M+H]+
[2-[(5-アミノ-2-フルオロ-4-メトキシ-フェニル)メトキシ]-1-メチル-エチル]アンモニウム;2,2,2-トリフルオロアセテート(16F)(0.1g、0.27mmol)をDMF(5mL)に溶解させ、この溶液に、HATU(0.153g、0.4mmol)、DIPEA(0.07g、0.54mmol)および中間体1(0.09g、0.27mmol)を室温で加えた。18時間後、溶液混合物を、EA(30mL)で希釈し、水(2×30mL)およびブライン(30mL)で洗浄し、Na2SO4で乾燥して濃縮した。粗生成物を、フラッシュクロマトグラフィー(PE/EA=3:1)により精製して、表題化合物(16G)(0.06g、39.47%)を白色固体として得た。LC-MS(ESI):m/z=567.2[M+H]+
tert-ブチル N-[6-クロロ-3-[[2-[(2-フルオロ-4-メトキシ-5-ニトロ-フェニル)メトキシ]-1-メチル-エチル]カルバモイル]イミダゾ[1,2-b]ピリダジン-8-イル]カルバメート(16G)(0.06g、0.1mmol)をエタノール(9mL)およびH2O(3mL)に溶解し、この溶液に、Fe粉末(60mg、1.06mmol)およびNH4Cl(34mg、0.64mmol)を加え、次いで反応混合物を3時間で85℃に加熱し、室温に冷却後、反応物をろ過し、ろ液を真空中で除去した。残渣を、フラッシュクロマトグラフィーにより精製して、表題化合物(16H)(0.044g、78.57%)を白色固体として得た。LC-MS(ESI):m/z=537.1[M+H]+
(16H)(44mg、0.082mmol)の1,4-ジオキサン(20mL)溶液に、Cs2CO3(80mg、0.25mmol)および第3-t-Bu-Xphos-Pd(30mg)を加えた。反応混合物を、N2下、85℃で、2時間攪拌した。室温に冷却後、溶媒を除去し、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物(例16I)(22mg、53.65%)を白色固体として得た。LC-MS(ESI):m/z=501.3[M+H]+
(16I)(22mg、0.044mmol)およびトリフルオロ酢酸(0.5mL)のDCM(4mL)溶液を、室温で2時間攪拌した。溶媒を蒸発させ、粗生成物を水とDCMとの間で分配した。水層を、NaHCO3で塩基性化し、DCMで抽出した。一つに合わせた有機層を、ブラインで洗浄し、硫酸ナトリウム上で乾燥し、ろ過し、蒸発させ、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物16(5mg、28.57%)を白色固体として得た。1H NMR(400MHz,CDCl3)δ 8.71(d,1H),8.28(d,1H),8.07(s,1H),6.74-6.60(m,3H),5.64(s,1H),4.69-2.59(m,2H),4.28-4.21(m,1H),3.93(d,3H),3.57(dd,1H),3.43-3.37(m,1H),3.05(d,3H),1.29(d,3H).LC-MS(ESI):m/z=401.2[M+H]+
実施例17:

水素化ナトリウム(228mg、5.70mmol)を、0℃でTHF(30mL)中のtert-ブチル N-[(1R)-2-ヒドロキシ-1-メチル-エチル]カルバメート(1g、5.70mmol)の攪拌溶液に少しずつ加え、混合物を0℃で10分間攪拌し、次いで混合物に、0℃で1-(ブロモメチル)-2-フルオロ-4-メトキシ-5-ニトロ-ベンゼン(16D)(1.5g、5.70mmol)を加え、30分後、混合物を、冷水で処理し、酢酸エチルで抽出した。一つに合わせた有機層を、ブラインで洗浄し、乾燥(Na2SO4)し、次いで残渣をフラッシュクロマトグラフィーで精製して、表題化合物(17E)(0.57g、27.94%)を白色固体として得た。LC-MS(ESI):m/z=359.1[M+H]+
トリフルオロ酢酸(3mL)を、tert-ブチル N-[(1R)-2-[(2-フルオロ-4-メトキシ-5-ニトロ-フェニル)メトキシ]-1-メチル-エチル]カルバメート(17E)(0.57g、1.53mmol)のDCM(8mL)溶液に加え、混合物を一晩攪拌し、混合溶液を蒸発させて乾燥し、その後、表題化合物(17F)(0.54g.91.21%)を褐色液体として得、これをさらに精製することなく次の工程で使用した。LC-MS(ESI):m/z=259.2[M+H]+
[(1R)-2-[(2-フルオロ-4-メトキシ-5-ニトロ-フェニル)メトキシ]-1-メチル-エチル]アンモニウム;2,2,2-トリフルオロアセテート(17F)(0.54g、1.45mmol)をDMF(15mL)に溶解させ、この溶液に、HATU(0.827g、2.17mmol)、DIPEA(0.374g、2.9mmol)および中間体1(0.473g、1.45mmol)を室温で加えた。18時間後、溶液混合物を、EA(50mL)で希釈し、水(2×50mL)およびブライン(50mL)で洗浄し、Na2SO4で乾燥して濃縮した。粗生成物を、フラッシュクロマトグラフィーにより精製して、表題化合物(17G)(0.512g、62.36%)を白色固体として得た。LC-MS(ESI):m/z=567.2[M+H]+
tert-ブチル N-[6-クロロ-3-[[(1R)-2-[(2-フルオロ-4-メトキシ-5-ニトロ-フェニル)メトキシ]-1-メチル-エチル]カルバモイル]イミダゾ[1,2-b]ピリダジン-8-イル]カルバメート(17G)(0.512g、0.9mmol)をエタノール(50mL)およびH2O(15mL)に溶解し、Fe(506mg、9.04mmol)およびNH4Cl(290mg、5.42mmol)を加え、次いで反応混合物を、3時間で85℃に加熱し、室温に冷却後、ろ過し、ろ液を真空中で除去した。残渣を、フラッシュクロマトグラフィー(PE/EA=3:1)により精製して、表題化合物(17H)(0.44g、90.90%)を白色固体として得た。LC-MS(ESI):m/z=537.1[M+H]+
(17H)(440mg、0.82mmol)の1,4-ジオキサン(80mL)溶液に、Cs2CO3(802mg、2.46mmol)および第3-t-Bu-Xphos-Pd(280mg)を加えた。反応混合物を、N2下、80℃で、2時間攪拌した。室温に冷却後、溶媒を除去し、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、所望の生成物(例17I)(230mg、56.09%)を白色固体として得た。LC-MS(ESI):m/z=501.3[M+H]+.
(17)(230mg、0.46mmol)およびトリフルオロ酢酸(2mL)のDCM(10mL)溶液を、室温で2時間攪拌した。溶媒を蒸発させ、粗生成物を水とDCMとの間で分配した。水層を、NaHCO3でクエンチし、DCMで抽出した。一つに合わせた有機層を、ブラインで洗浄し、硫酸ナトリウム上で乾燥し、ろ過し、蒸発させ、残渣をシリカゲルフラッシュカラムクロマトグラフィー(PE/EA=2:1)により精製して、生成物17(65mg、35.32%)を白色固体として得た。1H NMR(400MHz,CDCl3)δ 8.74(d,1H),8.30(d,1H),8.05(s,1H),6.70-6.55(m,2H),6.21(s,1H),5.59(s,1H),4.72-4.55(m,2H),4.31-4.21(m,1H),3.92(s,3H),3.59-3.55(dd,1H),3.44-3.38(m,1H),3.04(d,3H),1.29(d,3H).LC-MS(ESI):m/z=401.2[M+H]+.
実施例18:

3-フルオロ-4-メトキシ-ベンズアルデヒド(18A)(3.6g、23.37mmol)を濃硫酸(30mL)に溶解し、-10℃に冷却した。濃硫酸(4mL)中の濃硝酸(2.5mL)を、20分間にわたって滴加した。-10℃未満でさらに1時間攪拌した後、混合物を砕氷に注いだ。沈殿物をろ過により回収し、ジクロロメタン(40mL)と飽和炭酸水素ナトリウム(30mL)との間で分配した。有機層を乾燥(Na2SO4)し、真空中で蒸発させて、表題化合物(18B)(2.5g、53.76%)を油状物として得た。LC-MS(ESI):m/z=200.1[M+H]+.
水素化ホウ素ナトリウム(0.38g、10.04mmol)を、0℃で、メタノール(20mL)中の3-フルオロ-4メトキシ-5-ニトロ-ベンズアルデヒド(1g、5.02mmol)の攪拌溶液に少しずつ加えた。2時間後、メタノールを真空中で除去した。残渣を、冷水で処理し、ジクロロメタンで抽出した。一つに合わせた有機層を、ブラインで洗浄し、乾燥(Na2SO4)し、次いで真空中で蒸発させて、表題化合物(18C)を未精製固体(1g、99.0%)として得た。LC-MS(ESI):m/z=202.1[M+H]+.
(3-フルオロ-4-メトキシ-5-ニトロ-フェニル)メタノール(1g、4.97mmol)およびトリフェホスフィン(2.61g、9.95mmol)の無水ジエチルエーテル(30mL)溶液に、無水ジエチルエーテル(5mL)中の四臭化炭素(3.3g、9.95mmol)を滴加した。混合物を一晩攪拌した後、粘着性油状物まで濃縮した。シリカゲルクロマトグラフィーにより、表題化合物(18D)を淡黄色固体(0.95g、73.07%)として得た。LC-MS(ESI):m/z=264.1[M+H]+
水素化ナトリウム(144mg、3.61mmol)を、0℃でTHF(15mL)中のtert-ブチル N-(2-ヒドロキシ-1-メチル-エチル)カルバメート(0.63g、3.61mmol)の攪拌溶液に少しずつ加え、混合物を0℃で10分間攪拌し、次いで混合物に、0℃で1-(ブロモメチル)-3-フルオロ-4-メトキシ-5-ニトロ-ベンゼン(18D)(0.95g、3.61mmol)を加え、30分後、混合物を、冷水でクエンチし、酢酸エチルで抽出した。一つに合わせた有機層を、ブラインで洗浄し、乾燥(Na2SO4)し、次いで残渣をフラッシュクロマトグラフィーで精製して、表題化合物(18E)(0.63g、48.83%)を褐色固体として得た。LC-MS(ESI):m/z=359.1[M+H]+.
トリフルオロ酢酸(1.5mL)を、N-[2-[(3-フルオロ-4-メトキシ-5-ニトロ-フェニル)メトキシ]-1-メチル-エチル]カルバメート(18E)(0.63g、1.76mmol)のDCM(5mL)溶液に加え、この混合物を2時間攪拌し、混合溶液を蒸発させて乾燥し、その後、表題化合物(18F)(0.6g.91.46%)を褐色液体として得、これをさらに精製することなく次の工程で使用した。LC-MS(ESI):m/z=259.2[M+H]+
[(1R)-2-[(3-フルオロ-4-メトキシ-5-ニトロ-フェニル)メトキシ]-1-メチル-エチル]アンモニウム;2,2,2-トリフルオロアセテート(18F)(0.6g、1.6mmol)をDMF(10mL)に溶解させ、この溶液に、HATU(0.91g、2.41mmol)、DIPEA(0.41g、3.2mmol)および中間体1(0.52g、1.6mmol)を室温で加えた。18時間後、溶液混合物を、EA(50mL)で希釈し、水(2×50mL)およびブライン(50mL)で洗浄し、Na2SO4で乾燥して濃縮した。粗生成物を、フラッシュクロマトグラフィーにより精製して、表題化合物(18G)(545mg、59.89%)を白色固体として得た。LC-MS(ESI):m/z=567.2[M+H]+
tert-ブチル N-[6-クロロ-3-[[2-[(2-フルオロ-4-メトキシ-5-ニトロ-フェニル)メトキシ]-1-メチル-エチル]カルバモイル]イミダゾ[1,2-b]ピリダジン-8-イル]カルバメート(18G)(545mg、0.96mmol)をエタノール(45mL)およびH2O(15mL)に溶解し、鉄粉末(540mg、9.62mmol)およびNH4Cl(310mg、5.77mmol)を溶液に加え、次いで、反応混合物を3時間で85℃に加熱し、室温まで冷却した後、反応物をろ過し、ろ液を真空中で除去し、残渣を、フラッシュクロマトグラフィー(PE/EA=2:1)により精製し、表題化合物(18H)(450mg、87.2%)を白色固体として得た。LC-MS(ESI):m/z=537.1[M+H]+
(18H)(450mg、0.84mmol)の1,4-ジオキサン(100mL)溶液に、Cs2CO3(820mg、2.51mmol)および第3-t-Bu-Xphos-Pd(250mg)を加えた。反応混合物を、N2下、80℃で、2時間攪拌した。室温に冷却後、溶媒を除去し、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物(18I)(220mg、52.50%)を白色固体として得た。LC-MS(ESI):m/z=501.3[M+H]+.
(18I)(220mg、0.44mmol)およびトリフルオロ酢酸(1mL)のDCM(5mL)溶液を、室温で2時間攪拌した。溶媒を蒸発させ、粗生成物を水とDCMとの間で分配した。水層を、NaHCO3で塩基性化し、DCMで抽出した。一つに合わせた有機層を、ブラインで洗浄し、硫酸ナトリウム上で乾燥し、ろ過し、蒸発させ、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物18(71mg、40.34%)を白色固体として得た。1H NMR(400MHz,CDCl3)δ 8.76(d,1H),8.20(s,1H),8.07(s,1H),6.87(s,1H),6.63-6.58(m,1H),6.40(s,1H),5.63(s,1H),4.67(d,1H),4.38(d,1H),4.30-4.20(m,1H),4.05(d,3H),3.60-3.56(m,1H),3.48-3.38(m,1H),3.06(d,3H),1.29(d,3H).LC-MS(ESI):m/z=401.2[M+H]+.
実施例19:
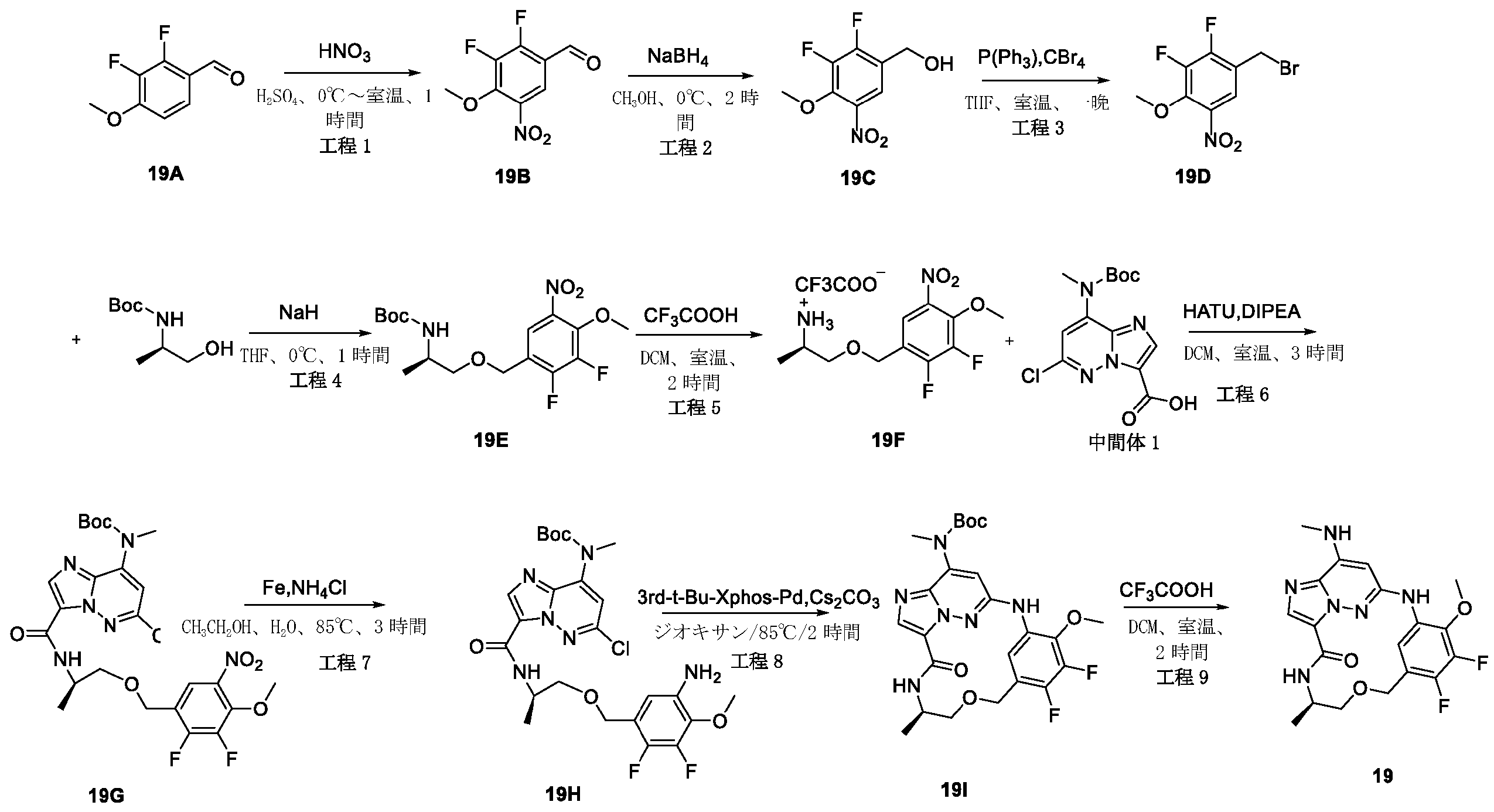
2,3-ジフルオロ-4-メトキシ-ベンズアルデヒド(19A)(3g、17.43mmol)を濃硫酸(18mL)に溶解し、-10℃に冷却した。濃硫酸(3mL)中の濃硝酸(1.5mL)を、10分間にわたって滴加した。-10℃未満でさらに1時間攪拌した後、混合物を砕氷に注いだ。沈殿物をろ過により回収し、ジクロロメタン(30mL)と飽和炭酸水素ナトリウム(30mL)との間で分配した。有機層を乾燥(Na2SO4)し、真空中で蒸発させて、表題化合物(19B)(3.1g、82.01%)を白色固体として得た。LC-MS(ESI):m/z=218.1[M+H]+
水素化ホウ素ナトリウム(1.08g、28.56mmol)を、0℃で、メタノール(60mL)中の2,3-ジフルオロ-4-メトキシ-5-ニトロ-ベンズアルデヒド(19B)(3.1g、14.28mmol)の攪拌溶液に少しずつ加えた。2時間後、メタノールを真空中で除去した。残渣を、冷水で処理し、ジクロロメタンで抽出した。一つに合わせた有機層を、ブラインで洗浄し、乾燥(Na2SO4)し、次いで真空中で蒸発させて、表題化合物(19C)を淡黄色固体(2.8g、89.51%)として得た。LC-MS(ESI):m/z=220.1[M+H]+
無水ジエチルエーテル(30mL)中の四臭化炭素(8.47g、25.56mmol)を、無水ジエチルエーテル(100mL)中の(2,3-ジフルオロ-4-メトキシ-5-ニトロ-フェニル)メタノール(19C)(2.8g、12.78mmol)およびトリフェホスフィン(triphephosphine)(6.7g、25.56mmol)の攪拌溶液に滴加した。混合物を一晩攪拌した後、粘着性油状物まで濃縮した。ヘキサン中の酢酸エチル(0~10%)によるシリカゲルクロマトグラフィーにより、表題化合物(19D)を淡黄色固体(2.12g、59.05%)として得た。LC-MS(ESI):m/z=281.9[M+H]+
水素化ナトリウム(92mg、2.31mmol)を、0℃でTHF(15mL)中のtert-ブチル N-[(1R)-2-ヒドロキシ-1-メチル-エチル]カルバメート(0.405g、2.31mmol)の攪拌溶液に少しずつ加え、混合物を0℃で10分間攪拌し、次いで混合物に、0℃で1-(ブロモメチル)-2,3-ジフルオロ-4-メトキシ-5-ニトロ-ベンゼン(19D)(0.65g、2.31mmol)を加え、10分後、混合物を、冷水で処理し、酢酸エチルで抽出した。一つに合わせた有機層を、ブラインで洗浄し、乾燥(Na2SO4)し、次いで残渣をフラッシュクロマトグラフィーで精製して、表題化合物(19E)(0.3g、34.48%)を褐色固体として得た。LC-MS(ESI):m/z=377.1[M+H]+
トリフルオロ酢酸(1.5mL)を、tert-ブチル N-[(1R)-2-[(2,3-ジフルオロ-4-メトキシ-5-ニトロ-フェニル)メトキシ]-1-メチル-エチル]カルバメート(19E)(0.3g、0.8mmol)のDCM(5mL)溶液に加え、この混合物を2時間攪拌し、混合溶液を蒸発させて乾燥し、その後、表題化合物(19F)(0.28g.90.03%)を褐色液体として得、これをさらに精製することなく次の工程で使用した。LC-MS(ESI):m/z=277.2[M+H]+
[(1R)-2-[(2,3-ジフルオロ-4-メトキシ-5-ニトロ-フェニル)メトキシ]-1-メチル-エチル]アンモニウム;2,2,2-トリフルオロアセテート(19F)(0.28g、0.71mmol)をDMF(5mL)に溶解させ、この溶液に、HATU(0.41g、1.07mmol)、DIPEA(0.185g、1.43mmol)および中間体1(0.24g、0.71mmol)を室温で加えた。18時間後、溶液混合物を、EA(50mL)で希釈し、水(2×50mL)およびブライン(50mL)で洗浄し、Na2SO4で乾燥して濃縮した。粗生成物を、フラッシュクロマトグラフィー(PE/EA=2:1)により精製して、表題化合物(19G)(0.18g、42.95%)を白色固体として得た。LC-MS(ESI):m/z=585.2[M+H]+
tert-ブチルN-[6-クロロ-3-[[(1R)-2-[(2,3-ジフルオロ-4-メトキシ-5-ニトロ-フェニル)メトキシ]-1-メチル-エチル]カルバモイル]イミダゾ[1,2-b]ピリダジン-8-イル]カルバメート(19G)(0.18g、0.3mmol)をエタノール(30mL)およびH2O(10mL)に溶解し、Fe粉末(172mg、3.08mmol)およびNH4Cl(100mg、1.85mmol)を溶液に加え、次いで、反応混合物を3時間で85℃に加熱し、室温まで冷却した後、反応物をろ過し、ろ液を真空中で除去し、残渣を、フラッシュクロマトグラフィーにより精製し、表題化合物(19H)(80mg、47.05%)を白色固体として得た。LC-MS(ESI):m/z=555.2[M+H]+
(19H)(80mg、0.14mmol)の1,4-ジオキサン(40mL)溶液に、Cs2CO3(141mg、0.43mmol)および第3-t-Bu-Xphos-Pd(50mg)を加えた。反応混合物を、N2下、80℃で、2時間攪拌した。室温に冷却後、溶媒を除去し、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物(19I)(42mg、47.05%)を白色固体として得た。LC-MS(ESI):m/z=519.3[M+H]+
19I(42mg、0.081mmol)およびトリフルオロ酢酸(0.3mL)のDCM(4mL)溶液を、室温で2時間攪拌した。溶媒を蒸発させ、粗生成物を水とDCMとの間で分配した。水層を、NaHCO3で塩基性化し、DCMで抽出した。一つに合わせた有機層を、ブラインで洗浄し、硫酸ナトリウム上で乾燥し、ろ過し、蒸発させ、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物19(12mg、36.36%)を白色固体として得た。1H NMR(400MHz,CDCl3)δ 8.67(d,1H),8.14(dd,1H),8.08(s,1H),6.72(s,1H),6.58(s,1H),5.63(s,1H),4.71-4.60(m,2H),4.31-4.21(m,1H),4.10(d,3H),3.61(dd,1H),3.46-3.39(m,1H),3.06(d,3H),1.31(d,3H).LC-MS(ESI):m/z=419.2[M+H]+.
実施例20:
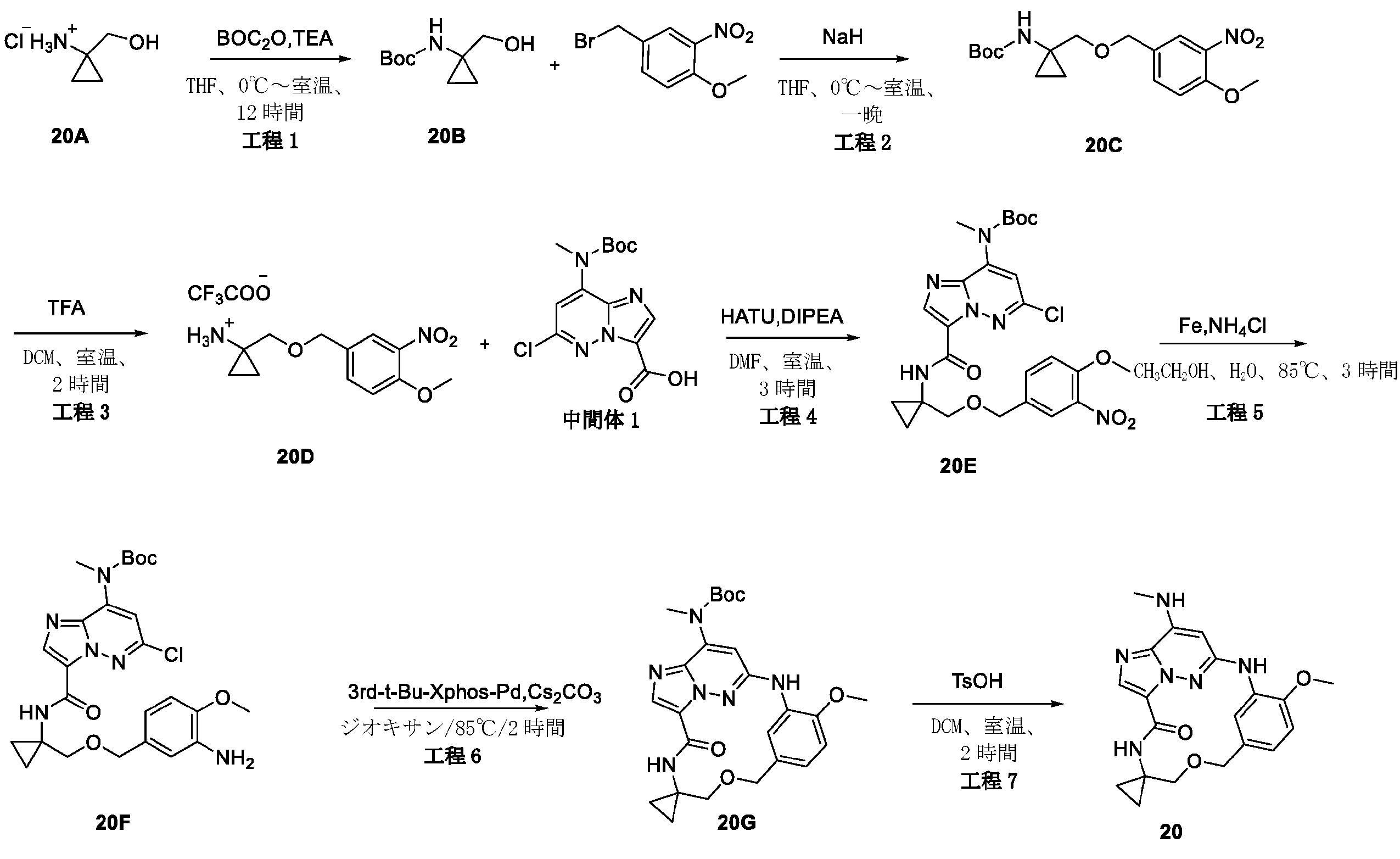
トリエチルアミン(4.93g、48.76mmol)を、0℃で、THF(50mL)中の(1-アミノシクロプロピル)メタノール塩酸塩(20A)(2g、16.25mmol)の攪拌溶液に滴加した。10分間の攪拌後、混合物に、THF(5mL)中のジ-tert-ブチル ジカルボネート(7.09g、32.50mmol)を0℃で滴加した。混合物を室温で一晩攪拌し、溶媒を真空中で除去した。残渣を酢酸エチル(60mL)で希釈し、水(2×60mL)およびブライン(50mL)で洗浄し、Na2SO4で乾燥して濃縮した。次いで、表題化合物(20B)(3g.100%)を白色固体として得、これをさらに精製することなく次の工程で使用した。1H NMR(400MHz,CDCl3)δ 5.05(s,1H),3.59(s,2H),2.40(s,1H),1.44(s,9H),0.83(m,4H).
水素化ナトリウム(480mg、12.02mmol)を、0℃でTHF(60mL)中のtert-ブチル N-[1-(ヒドロキシメチル)シクロプロピル]カルバメート(20B)(1.5g、8.01mmol)の攪拌溶液に少しずつ加え、混合物を0℃で30分間攪拌し、次いで混合物に、0℃で4-(ブロモメチル)-1-メトキシ-2-ニトロベンゼン(1.96g、8.01mmol)を加え、混合物を室温で一晩攪拌し、混合物を、冷水(80mL)で処理し、酢酸エチル(2×100mL)で抽出した。一つに合わせた有機層を、ブラインで洗浄し、Na2SO4で乾燥して濃縮し、次いで残渣をフラッシュクロマトグラフィーにより精製して、表題化合物(20C)(2.4g、85.40%)を淡黄色固体として得た。1H NMR(400MHz,CDCl3)δ 7.76(d,1H),7.46(dd,1H),7.02(d,1H),5.13(s,1H),4.46(s,2H),3.89(s,3H),3.44(s,2H),1.34(s,9H),0.81-0.66(m,4H).
トリフルオロ酢酸(1.5mL)を、tert-ブチル N-[1-[(4-メトキシ-3-ニトロ-フェニル)メトキシメチル]シクロプロピル]カルバメート(20C)(0.55g、1.56mmol)のDCM(5mL)溶液に加え、混合物を一晩攪拌し、混合溶液を蒸発させて乾燥し、その後、表題化合物(20D)(0.54g.94.57%)を褐色液体として得、これをさらに精製することなく次の工程で使用した。LC-MS(ESI):m/z=253.2[M+H]+
[1-[(4-メトキシ-3-ニトロ-フェニル)メトキシメチル]シクロプロピル]アンモニウム;2,2,2-トリフルオロアセテート(20D)(0.54g、1.47mmol)をDMF(10mL)に溶解させ、この溶液に、HATU(0.84g、2.21mmol)、DIPEA(0.38g、2.95mmol)および中間体1(0.1g、1.47mmol)を室温で加えた。18時間後、溶液混合物を、EA(50mL)で希釈し、水(2×50mL)およびブライン(50mL)で洗浄し、Na2SO4で乾燥して濃縮した。粗生成物を、フラッシュクロマトグラフィーにより精製して、表題化合物(20E)(0.52g、62.95%)を淡色(pale)固体として得た。LC-MS(ESI):m/z=561.3[M+H]+
tert-ブチル N-[6-クロロ-3-[[1-[(4-メトキシ-3-ニトロフェニル)メトキシメチル]シクロプロピル]カルバモイル]イミダゾ[1,2-b]ピリダジン-8-イル]-N-メチル-カルバメート(20E)(0.52g、0.93mmol)をエタノール(60mL)およびH2O(15mL)に溶解し、この溶液に、鉄粉末(520mg、9.28mmol)およびNH4Cl(0.3g、5.57mmol)を加え、次いで反応混合物を3時間で85℃に加熱し、室温に冷却後、反応物をろ過し、ろ液を真空中で除去した。残渣を、フラッシュクロマトグラフィー(PE/EA=2:1)により精製して、表題化合物(20F)(0.32g、65.04%)を白色固体として得た。LC-MS(ESI):m/z=531.3[M+H]+
(20F)(320mg、0.6mmol)の1,4-ジオキサン(40mL)溶液に、Cs2CO3(590mg、1.81mmol)および第3-t-Bu-Xphos-Pd(180mg)を加えた。反応混合物を、N2下、85℃で、2時間攪拌した。室温に冷却後、溶媒を除去し、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物(20G)(175mg、58.72%)を白色固体として得た。LC-MS(ESI):m/z=495.3[M+H]+
20G(175mg、0.35mmol)およびp-トルエンスルホン酸一水和物(101mg、0.53mmol)のDCM(4mL)溶液を、室温で2時間攪拌した。溶媒を蒸発させ、粗生成物を水とDCMとの間で分配した。水層を、NaHCO3で塩基性化し、DCMで抽出した。一つに合わせた有機層を、ブラインで洗浄し、硫酸ナトリウム上で乾燥し、ろ過し、蒸発させ、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物20(35mg、25.17%)を白色固体として得た。1H NMR(400MHz,CDCl3)δ 8.99(s,1H),8.36(d,1H),7.97(s,1H),6.90-6.73(m,3H),6.34(s,1H),5.63(s,1H),4.60(s,2H),3.93(s,3H),3.49(s,2H),3.04(d,3H),1.82(q,2H),0.68(q,2H).LC-MS(ESI):m/z=395.2[M+H]+
実施例21:

5-メトキシ-6-ニトロ-1H-インダゾール(1.0g、5.18mmol)、tert-ブチル(4-ブロモブタン-2-イル)カルバメート(1.7g、6.73mmol)、およびK2CO3(1.4g、10.36mmol)のDMF(30mL)溶液を、60℃で一晩攪拌した。反応完了後、反応物を氷浴中で冷却し、EA(100mL)で希釈し、溶液を水(3×20mL)で抽出した。一つにまとめた有機層をNa2SO4上で乾燥し、ろ過し、減圧下で濃縮し、粗成生物を得て、これをカラムクロマトグラフィー(PE/EtOAc=3/1)により精製して、所望の生成物(0.8g、42%)を白色固体として得た。LM-MS:m/z=365.4[M+H]+
tert-ブチル(4-(5-メトキシ-6-ニトロ-1H-インダゾール-1-イル)ブタン-2-イル)
カルバメート(0.7g、1.92mmol)およびトリフルオロ酢酸(0.5mL)のDCM(4mL)溶液を、室温で2時間攪拌した。溶媒を蒸発させ、粗生成物を水とDCMとの間で分配した。水層を、NaHCO3でクエンチし、DCMで抽出した。一つに合わせた有機層を、ブラインで洗浄し、硫酸ナトリウム上で乾燥し、ろ過し、蒸発させ、粗生成物を得て、これをカラムクロマトグラフィー(PE/EtOAc=3/1)により精製して、所望の生成物(0.4g、79%)を白色固体として得た。LM-MS:m/z=265.3[M+H]+
4-(5-メトキシ-6-ニトロ-1H-インダゾール-1-イル)ブタン-2-アミン(0.2g、0.76mmol)を、DCM(5mL)に溶解し、この溶液に、HATU(0.58g、1.515mmol)、DIPEA(0.15g、1.14mmol)および8-((tert-ブトキシカルボニル)(メチル)アミノ)-6-クロロイミダゾ[1,2-b]ピリダジン-3
-カルボン酸(0.25g、0.76mmol)を室温で加えた。混合物を室温で1時間攪拌し、次いでEA(20mL)で希釈し、水(10mL)およびブライン(10mL)で洗浄し、Na2SO4で乾燥して濃縮した。粗生成物を、フラッシュクロマトグラフィー(PE/EtOAc=3/1)により精製して、表題化合物(0.3g、69%)を白色固体として得た。LM-MS:m/z=574.0[M+H]+
カルバモイル)イミダゾ[1,2-b]ピリダジン-8-イル)(メチル)カルバメート(0.3g、0.52mmol)およびNH4Cl(330mg、6.24mmol)を、室温でMeOH(5mL)に溶解し、この溶液にZn粉末(405mg、6.24mmol)を加え、次いで反応混合物を室温で1時間攪拌した。反応が完了した後、反応物をろ過し、ろ液を真空中で除去した。残渣を、フラッシュクロマトグラフィーにより精製して、表題化合物(0.27g、96%)を白色固体として得た。LM-MS:m/z=543.0[M+H]+
ブタン-2-イル)カルバモイル)-6-クロロイミダゾ[1,2-b]ピリダジン-8-イル)(メチル)カルバメート(270mg、0.5mmol)の1,4-ジオキサン(20mL)溶液に、Cs2CO3(325mg、1.0mmol)および第3-t-Bu-Xphos-Pd(120mg)を加えた。反応混合物を、N2下、80℃で、3時間攪拌した。室温に冷却後、反応物をろ過し、溶媒を除去し、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、所望の化合物(150mg、59%)を白色固体として得た。LM-MS:m/z=507.7[M+H]+
-イミダゾ[1,2-b]ピリダジナ-3(6,1)-インダゾラシクロオクタファン(indazolacyclooctaphane)-18-イル)(メチル)カルバメート(0.15g、0.3mmol)およびトリフルオロ酢酸(0.2mL)のDCM(4mL)溶液を、室温で2時間攪拌した。溶媒を蒸発させ、粗生成物を水とDCMとの間で分配した。水層を、NaHCO3でクエンチし、DCMで抽出した。一つに合わせた有機層を、ブラインで洗浄し、硫酸ナトリウム上で乾燥し、ろ過した。ろ液を分取HPLCにより直接精製し、所望の生成物21(125mg、収率:42%)を褐色固体として得た。LM-MS:m/z=407.5[M+H]+.1H NMR(400MHz,CD3OD)δ 8.88(s,1H),7.93(s,1H),7.85(s,1H),7.22(s,1H),6.19(s,1H),4.61-4.48(m,3H),4.00(s,3H),3.04(s,3H),2.14(d,2H),1.01(d,3H).
実施例22:
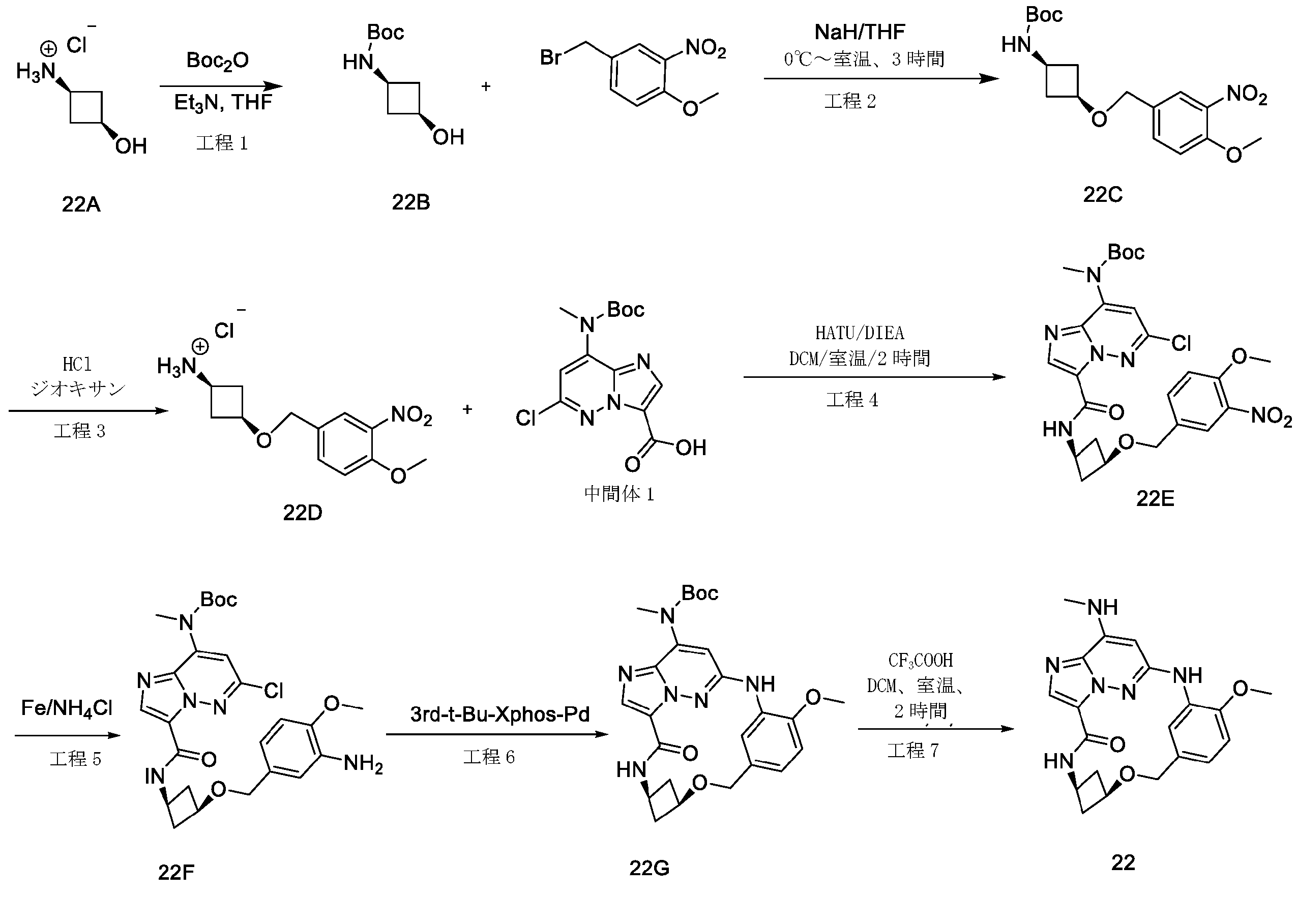
トリエチルアミン(4.93g、48.76mmol)を、0℃で、THF(50mL)中の(1s,3s)-3-ヒドロキシシクロブタン-1-アミニウムクロリド(aminium chloride)(22A)(2g、16.25mmol)の攪拌溶液に滴加した。10分間の攪拌後、THF(5mL)中のジtertブチル ジカルボネート(7.09g、32.50mmol)を0℃で混合物に滴加した。混合物を室温で一晩攪拌し、溶媒を真空中で除去した。残渣を酢酸エチル(60mL)で希釈し、水(2×60mL)およびブライン(50mL)で洗浄し、Na2SO4で乾燥して濃縮した。次いで、表題化合物(22B)(3g.100%)を無色油状物として得、これをさらに精製することなく次の工程で使用した。LC-MS(ESI):m/z=188.1[M+H]+
工程2:tert-ブチル((1s,3s)-3-((4-メトキシ-3-ニトロベンジル)オキシ)シクロブチル)カルバメート(22C)
水素化ナトリウム(577mg、14.43mmol)を、0℃でTHF(60mL)中のtert-ブチル((1S,3S)-3-ヒドロキシシクロブチル)カルバメート(22B)(1.5g、8.02mmol)の攪拌溶液に少しずつ加え、混合物を0℃で30分間攪拌し、次いで混合物に、0℃で4-(ブロモメチル)-1-メトキシ-2-ニトロベンゼン(0.69g、2.62mmol)を加え、混合物を室温で一晩攪拌し、混合物を、冷水(80mL)で処理し、酢酸エチル(2×100mL)で抽出した。一つに合わせた有機層を、ブラインで洗浄し、Na2SO4で乾燥して濃縮し、次いで残渣をフラッシュクロマトグラフィーにより精製して、表題化合物(22C)(0.2g、21.30%)を白色固体として得た。LC-MS(ESI):m/z=353.1[M+H]+
工程3:(1s,3s)-3-((4-メトキシ-3-ニトロベンジル)オキシ)シクロブタン-1-アミニウムクロリド(aminium chloride)(22D)
tert-ブチル((1s,3s)-3-((4-メトキシ-3-ニトロベンジル)オキシ)シクロブチル)カルバメート(222C)(0.55g、2.79mmol)のDCM(5mL)溶液に、トリフルオロ酢酸(1.5mL)を加え、混合物を室温で一晩攪拌し、混合溶液を蒸発させて乾燥し、次に表題化合物(22D)(0.39g.100%)を白色固体として得、さらに精製せずに次の工程に用いた。LC-MS(ESI):m/z=253.1[M+H]+
(1s,3s)-3-((4-メトキシ-3-ニトロベンジル)オキシ)シクロブタン-1-アミニウムクロリド(22D)(200mg、0.79mmol)をDMF(5mL)に溶解し、この溶液に、HATU(360mg、0.94mmol)、DIPEA(180mg、1.18mmol)および中間体1(257mg、0.79mmol)を室温で加えた。混合物を室温で18時間攪拌し、次いでEA(50mL)で希釈し、水(2×50mL)およびブライン(50mL)で洗浄し、Na2SO4で乾燥して濃縮した。粗生成物を、フラッシュクロマトグラフィー(PE/EA=2:1)により精製して、表題化合物(22E)(400mg、90.00%)を白色固体として得た。LC-MS(ESI):m/z=561.2[M+H]+
(6-クロロ-3-(((1s,3s)-3-((4-メトキシ-3-ニトロベンジル)オキシ)シクロブチル)カルバモイル)イミダゾ[1,2-b]ピリダジン-8-イル)(メチル)カルバメート(22E)(280mg、0.5mmol)をエタノール(9mL)およびH2O(3mL)に溶解し、この溶液に、鉄粉末(560mg、10mmol)およびNH4Cl(530mg、10mmol)を加え、次いで反応混合物を3時間で85℃に加熱し、室温に冷却後、反応物をろ過し、ろ液を真空中で除去した。残渣を、フラッシュクロマトグラフィーにより精製して、表題化合物(22H)(200mg、75.47%)を白色固体として得た。LC-MS(ESI):m/z=531.2[M+H]+.
(6-クロロ-3-(((1s,3s)-3-((4-メトキシ-3-ニトロベンジル)オキシ)シクロブチル)カルバモイル)イミダゾ[1,2-b]ピリダジン-8-イル)(メチル)カルバメート(22F)(265mg、0.5mmol)の1,4-ジオキサン溶液(10mL)に、Cs2CO3(326mg、1.0mmol)および第3-t-Bu-Xphos-Pd(35mg、0.04mmol)を加えた。反応混合物を、N2下、80℃で、3時間攪拌した。室温に冷却後、溶媒を除去し、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成化合物(22G)(50mg、20.24%)を白色固体として得た。LC-MS(ESI):m/z=495.1[M+H]+
tert-ブチル tert-ブチル((61s,63s,E)-36-メトキシ-8-オキソ-5-オキサ-2,7-ジアザ-1(6,3)-イミダゾ[1,2-b]ピリダジナ-3(1,3)-ベンゼナ-6(1,3)-シクロブタナシクロオクタファン(cyclobutanacyclooctaphane)-18-イル)(メチル)カルバメート(22G)(50mg、1.02mmol)およびp-トルエンスルホン酸一水和物(50mg、0.29mol)のDCM(4mL)溶液を室温で2時間攪拌した。溶媒を蒸発させ、粗生成物を水とDCMとの間で分配した。水層を、NaHCO3で塩基性化し、DCMで抽出した。一つに合わせた有機層を蒸発させ、残渣をTLCにより精製して、22(2mg、5.01%)を得た。LC-MS(ESI):m/z=395.1[M+H]+.1H NMR(400MHz,DMSO-d6)δ 9.15(d,1H),8.29(s,1H),8.18(d,1H),7.82(s,1H),7.38(d,1H),7.00(d,1H),6.85(dd,1H),6.23(d,1H),4.50(s,2H),4.34(dd,1H),4.21(s,1H),3.88(s,3H),2.89(d,3H),2.72-2.65(m,2H),1.75(d,2H).
実施例24:

例24a(10.0g、60.24mmol、1.0当量)のAcOH(150mL)溶液に、NaNO2(4.99g、72.29mmol、1.2当量)のH2O(10mL)溶液を室温で滴加した。反応混合物を室温で2時間攪拌した。次いで、この混合物に水(100mL)を加え、これを30分間攪拌した。沈殿した固体をろ過により回収し、これをH2OおよびMTBEで洗浄した。粗生成物をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、所望の生成物である例24b(3.9g、収率36.6%)を黄色固体として得た。LCMS[M+1]+=178.2.
例24b(3.9g、22.03mmol、1.0当量)のCH3CN(50mL)溶液に、CH3I(15.6g、110.17mmol、5.0当量)およびK2CO3(7.6g、55.08mmol、2.5当量)を加えた。反応混合物を80℃で16時間攪拌した。溶媒を濃縮し、未精製物を、シリカゲルフラッシュカラムクロマトグラフィーにより精製して、所望の生成物である例24c(3.17g、収率75.3%)を褐色固体として、その異性体(200mg)を褐色固体として得た。LCMS[M+1]+=192.2.例24c:1H NMR(300MHz,DMSO-d6)δ 8.21(d,1H),8.05(s,1H),7.79(s,1H),4.33(s,3H),2.51(s,3H).異性体:1H NMR(300MHz,DMSO-d6)δ 8.04(s,1H),7.96(s,1H),7.79(s,1H),4.23(s,3H),2.51(s,3H).
例24c(500mg、2.62mmol、1.0当量)のCCl4(12mL)溶液を80℃に加熱し、そこにNBS(559mg、3.14mmol、1.2当量)およびAIBN(429mg、2.62mmol、1.0当量)を加えた。反応混合物を80℃で4時間攪拌した。室温まで冷却した後、反応混合物を濃縮し、未精製物をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、所望の生成物の例24d(577mg、収率81.7%)を黄色固体として得た。1H NMR(300MHz,DMSO-d6)δ 8.78(s,1H),8.39(d,1H),8.36(d,1H),4.95(s,2H),4.28(s,3H).
例24e(681mg、3.89mmol、2.0当量)のTHF(10mL)溶液に、NaH(117mg、鉱油中60%、2.92mmol、1.5当量)を0℃で加えた。30分間攪拌した後、この混合物に例24d(525mg、1.94mmol、1.0当量)を加え、これを室温でさらに2時間攪拌した。混合物を飽和NH4Cl水溶液でクエンチし、EtOAc(30mL×3)で抽出した。一つに合わせた有機層をブラインで洗浄し、Na2SO4上で乾燥させ濃縮した。残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、所望の生成物である例24f(306mg、収率43.2%)を黄色固体として得た。LCMS[M+1-56]+=309.2.
例24f(306mg、0.84mmol、1.0当量)をMeOH(10mL)に溶解させて、N2保護下で、10%のPd/C(200mg)を数回に分けてゆっくりと加えた。系を真空にし、次いで水素で再充填した。混合溶液をH2バルーン下、室温で1時間攪拌した。反応混合物をセライトパッドを介してろ過し、ろ液を濃縮した。残渣を分取TLCにより精製して、所望の生成物である例24g(150mg、収率53.3%)を淡黄色固体として得た。LCMS[M+1+22]+=357.3.
例24g(130mg、0.39mmol、1.0当量)のDCM(6mL)溶液に、HCl/ジオキサン(2mL、ジオキサン中4M)を加えた。反応溶液を、室温で0.5時間攪拌し、濃縮して、所望の生成物である例24h(240mg、未精製)を黄色固体として得た。LCMS[M+1]+=235.3.
例24i(173mg、0.53mmol、1.0当量)のDCM(10mL)溶液に、DIEA(545mg、4.23mmol、8.0当量)およびHATU(221mg、0.58mmol、1.1当量)を加えた。30分間攪拌した後、例24h(215mg、0.79mmol、1.5当量)を加えた。反応混合物を室温で2時間攪拌した。溶媒を除去し、未精製物を分取TLC(EtOAc)により精製して、所望の生成物である例24j(250mg、収率87.0)を黄色油状物として得た。LCMS[M+1]+=543.3.
例24j(150mg、0.28mmol、1.0当量)のジオキサン(20mL)溶液に、Cs2CO3(180mg、0.55mmol、2.0当量)および第3-t-Bu-Xphos-Pd(25mg、0.028mmol、0.1当量)を加えた。反応混合物を、N2下、80℃で16時間攪拌した。反応混合物を濃縮し、分取TLCにより精製して、所望の生成物である例24k(50mg、収率35.7%)を黄色固体として得た。LCMS[M+1]+=507.3.
例24k(45mg、0.089mmol、1.0当量)のDCM(3mL)溶液に、HCl/ジオキサン(1mL、ジオキサン中4M)を加えた。反応溶液を室温で1時間攪拌し、溶媒を濃縮した。粗生成物をMeOHに溶解させ、Na2CO3を加えた。混合物を室温で10分間攪拌し、次いでろ過した。ろ液を濃縮して、残渣を分取TLCにより精製して、所望の生成物である例24(20.0mg、収率55.4%)を灰白色固体として得た。LCMS[M+1]+=407.3.1H NMR(300MHz,DMSO-d6)δ 9.19(s,1H),8.95(d,1H),8.32(s,1H),7.95(s,1H),7.83(s,1H),7.44(d,1H),7.20(s,1H),6.36(s,1H),4.73(d,1H),4.45(d,1H),4.21(s,3H),4.08-3.96(m,1H),3.48(d,1H),3.26-3.20(m,1H),2.90(d,3H),1.11(d,3H).
実施例25:

例25b(2.3g、13.4mmol)のTHF(40mL)溶液に、NaH(0.9g、鉱油中60%、22.4mmol)を0℃で数回に分けて加えた。10分間攪拌した後、例25a(2.2g、8.9mmol)のTHF(10mL)溶液を滴加した。反応混合物を室温で3時間攪拌した。反応を、0℃で飽和NH4Cl水溶液(20mL)でクエンチし、EtOAc(50mL×2)で抽出した。一つに合わせた有機層を、Na2SO4上で乾燥させ、真空中で濃縮した。粗生成物をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、所望の生成物である例25c(2.1g、収率69.4%)を黄色固体として得た。LCMS[M+1]+=341.3.
例25c(1.1g、3.2mmol)をMeOH(50mL)に溶解させて、N2保護下で、10%のPd/C(110mg)を数回に分けて加えた。系を真空にし、次いで水素で再充填した。混合物をH2バルーン下、室温で1時間攪拌し、次いでろ過した。ろ液を濃縮し、粗生成物をシリカゲルフラッシュカラムクロマトグラフィーにより精製し、所望の生成物である例25d(850mg、収率85.7%)を無色油状物として得た。LCMS[M+1]+=311.3.
DCM(5mL)およびMeOH(1mL)中の例25d(840mg、2.7mmol)の溶液に、HCl/ジオキサン(1mL、ジオキサン中4M、4mmol)を加えた。反応混合物を室温で1時間攪拌した。反応溶液を真空中で濃縮して、所望の生成物である例25e(800mg、未精製)を白色固体として得た。LCMS[M+1]+=211.2.
例25f(607mg、1.9mmol)のDCM(30mL)溶液に、DIEA(1.92g、14.9mmol)およびHATU(1.06g、2.8mmol)を加えた。0.5時間攪拌した後、例25e(790mg、2.8mmol)を加えた。反応混合物を室温で2時間攪拌した。溶媒を除去し、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製し、所望の生成物である例25g(310mg、収率21.4%)を黄色固体として得た。LCMS[M+1]+=519.3.
例25g(300mg、0.6mmol)のジオキサン(50mL)溶液に、Cs2CO3(377mg、1.2mmol)および第3-t-Bu-Xphos-Pd(154mg、0.2mmol)を加えた。反応混合物を、N2下、90℃で、6時間攪拌した。室温に冷却後、溶媒を除去し、残渣をシリカゲルカラムクロマトグラフィーにより精製して、生成物である例25h(110mg、収率39.4%)を黄色固体として得た。LCMS[M+1]+=483.2.
例25h(110mg、0.23mmol)のDCM(5mL)溶液に、HCl/ジオキサン(1mL、ジオキサン中4M、4mmol)を加えた。反応混合物を室温で5時間攪拌し、次いで真空中で濃縮した。残渣をMeOH(5mL)に溶解させ、NaHCO3で塩基性化した(pH=8)。沈殿物をろ過し、ろ液を濃縮した。残渣を分取TLCにより精製して、所望の生成物である例25(57.3mg、収率65.7%)を灰白色固体として得た。LCMS[M+1]+=383.2.1H NMR(300MHz,DMSO-d6)δ 8.92(s,1H),8.36(d,1H),8.24(d,1H),8.11(s,1H),7.81(d,1H),6.99(d,1H),6.92(d,1H),5.94(s,1H),4.57(d,1H),4.40(d,1H),3.94-3.83(m,4H),3.47(d,1H),3.29-3.25(m,1H),2.92(d,3H),1.14(d,3H).
実施例26:
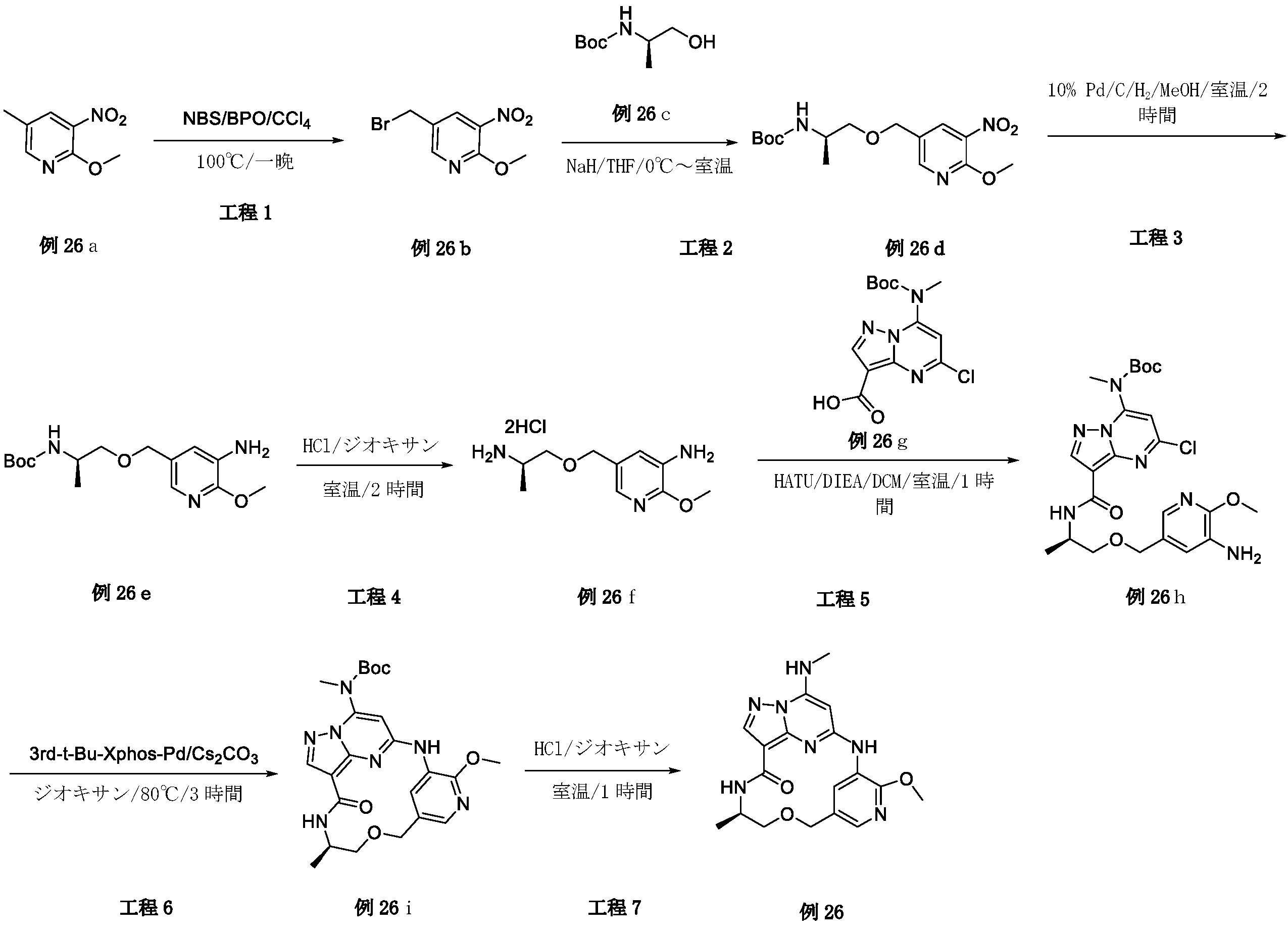
例26a(30.0g、179.0mmol)のCCl4(150mL)溶液に、BPO(4.4g、17.9mmol)、NBS(38.2g、216.0mmol)を加えた。反応混合物を、100℃で一晩攪拌し、次いでDCMで希釈し、水で洗浄し、無水Na2SO4上で乾燥した。溶液を減圧下で濃縮して、未精製残渣である例26b(37.0g、収率84.4%)を黄色固体として得、これをさらに精製することなく次の工程で直接使用した。LCMS[M+1]+=246.0.1H NMR(400MHz,クロロホルム-d)δ 7.87(d,1H),7.57(dd,1H),7.07(d,1H),4.46(s,2H),3.96(d,3H).
例26b(2.4g、9.8mmol)のTHF(20mL)溶液に、NaH(400mg、鉱油中60%、10.0mmol)を0℃で加えた。反応混合物を室温に温め、室温で0.5時間攪拌した。次いで、例26c(1.7g、9.7mmol)を加え、混合物を室温で6時間攪拌した。反応混合物を、飽和 NH4Cl(水溶液)でクエンチし、EtOAcにより抽出し、無水Na2SO4上で乾燥した。溶液を、減圧下で濃縮した。残渣をシリカゲルカラムクロマトグラフィーにより精製して、例26d(3.3g、収率96.8%)を黄色固体として得た。LCMS[M+1-100]+=241.1.
例26d(688mg、2.0mmol)および10%のPd/C(34mg)のMeOH(10mL)溶液を、室温で2時間、1気圧のH2下で攪拌した。混合物をろ過し、ろ液を減圧下で濃縮した。残渣である例26e(640mg、収率:定量)を、黄色固体として得、これを次の工程で直接使用した。LCMS[M-174]+=136.1
例26e(未精製550mg、1.77mmol)のDCM(10mL)溶液に、TFA(2.0mL)を加え、これを室温で2時間攪拌した。混合物を濃縮し、残渣をEtOAc(30mL)で処理して、粗生成物である例26f(340mg、収率:定量)を白色固体として得た。LCMS[M-74]+=137.1.
例26f(未精製300mg、1.42mmol)、例26g(464mg、1.42mmol)DIPEA(916mg、7.1mmol)のDCM(10mL)溶液に、HATU(538mg、1.42mmol)を加えた。反応混合物を室温で、1時間攪拌した。次いで、EtOAc(40mL)を反応混合物に加え、これをブライン(20mL×2)で洗浄し、無水Na2SO4上で乾燥して、濃縮した。残渣をシリカゲルカラムクロマトグラフィーにより精製して、所望の生成物である例26h(600mg、収率81.4%)を白色固体として得た。LCMS[M+1]+=520.2.
例26h(330mg、0.97mmol)、Cs2CO3(652mg、2.0mmol)のジオキサン(10mL)中の混合物に、第3-t-Bu-Xphos-Pd(89mg、0.1mmol)を加えた。混合物を、N2で3回脱気し、80℃で3時間攪拌した。次いで、反応混合物をDCMで希釈し、水で洗浄し、無水Na2SO4上で乾燥し、次いで減圧下で濃縮して、未精製の例26i(400mg、粗収率>100%)を白色固体として得、これをさらに精製することなく次の工程で使用した。LCMS[M+1]+=484.2
例26i(400mg、0.82mmol)のDCM(4mL)溶液に、TFA(1.0mL)を加え、これを室温で1時間攪拌した。混合物を濃縮し、残渣を、分取HPLCにより精製して、所望の生成物である例26(18.3mg、2工程で収率5.8%)を白色固体として得た。LCMS[M+1]+=384.1.1H NMR(400MHz,DMSO-d6)δ 9.11(s,1H),8.60-8.54(m,1H),8.10(s,1H),8.04(d,1H),7.89(d,1H),7.71(s,1H),5.90(s,1H),4.53(d,1H),4.43(d,1H),3.94(s,3H),3.86(s,1H),3.46(d,1H),3.25(s,1H),2.89(d,3H),1.11(d,3H).
実施例27:
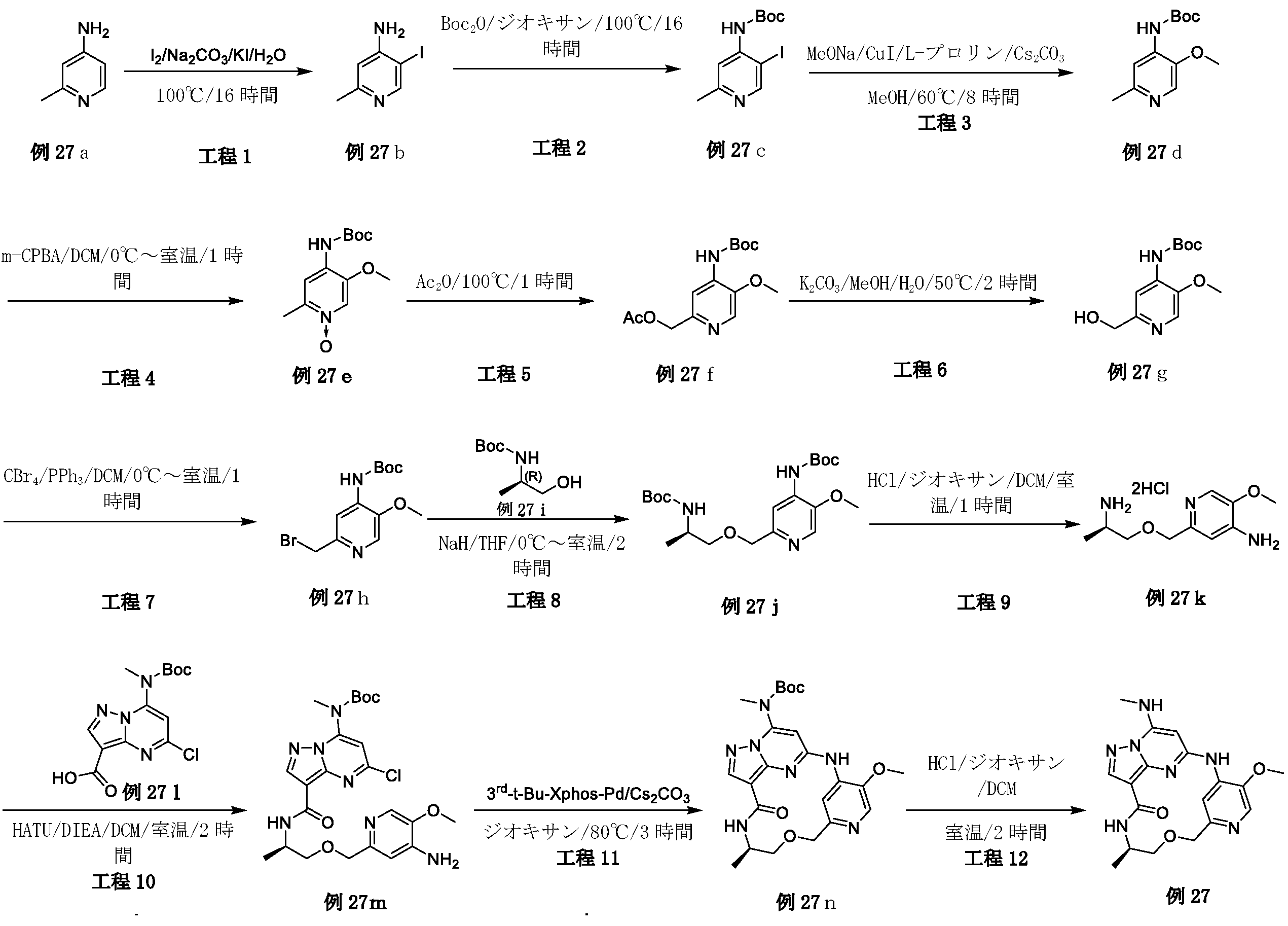
例27a(30.0g、277.8mmol、1.0当量)、Na2CO3(20.6g、194.5mmol、0.7当量)のH2O(150mL)溶液に、KI(59.9g、361.1mmol、1.3当量)およびI2(56.4g、222.2mmol、0.8当量)のH2O(50mL)溶液を100℃で加え、それを16時間攪拌した。室温まで冷却した後、反応混合物を、Na2SO3(35.0g、277.8mmol、1.0当量)でクエンチし、DCM(300mL×2)で抽出した。一つに合わせた有機層をブラインで洗浄し、Na2SO4上で乾燥させ濃縮した。残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物である例27b(8.4g、収率12.9%)を黄色固体として得た。LCMS[M+1]+=235.1
例27b(7.0g、50.7mmol、1.0当量)のジオキサン(50mL)溶液に、Boc2O(33.2g、152.1mmol、3.0当量)を加え、これを100℃で16時間攪拌した。反応混合物を濃縮し、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製し、生成物である例27c(3.5g、収率35%)を白色固体として得た。LCMS[M+1]+=335.1.
例27c(3.5g、10.5mmol、1.0当量)およびMeONa(2.82g、52.25mmol、5.0当量)のMeOH(30mL)溶液に、Cs2CO3(10.2g、21.0mmol、2.0当量)、CuI(199mg、1.05mmol、0.1当量)、およびL-プロリン(343mg、2.1mmol、0.2当量)を加えた。反応混合物を、N2保護下、60℃で8時間攪拌した。反応混合物を濃縮した。残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物である例27d(750mg、収率30.1%)を白色固体として得た。LCMS[M+1]+=239.3.
0℃(氷水浴)での例27d(550mg、2.3mmol、1.0当量)のDCM(10mL)溶液に、m-CPBA(596mg、3.45mmol、1.5当量)を少しずつ加えた。加えた後、反応物を室温で1時間攪拌した。溶液を、Na2SO3(150mg、1.15mmol、0.5当量)でクエンチし、DCM(30mL×2)で抽出した。一つに合わせた有機層をブラインで洗浄し、Na2SO4上で乾燥し、濃縮し、例27e(550mg、収率93.8%)を黄色固体として得た。LCMS[M+1]+=255.3.
例27e(400mg、1.65mmol、1.0当量)のAc2O(10mL)溶液を100℃で1時間攪拌した。混合物を濃縮して、粗生成物である例27f(550mg、収率:定量)を褐色油状物として得た。LCMS[M+1]+=297.3.
例27f(450mg、1.52mmol、1.0当量)のMeOH(15mL)およびH2O(5mL)中の溶液に、0℃でK2CO3(418.2mg、3.04mmol、2.0当量)を加えた。混合物を2時間、50℃で攪拌した。反応が完了した後、混合物を真空中で濃縮した。残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、所望の生成物である例27g(250mg、収率64.6%)を白色固体として得た。LCMS[M+1]+=255.2.
例27g(230mg、0.90mmol、1.0当量)、CBr4(597mg、1.80mmol、2.0当量)のDCM(15mL)溶液に、0℃で、DCM(5mL)中のPPh3(355mg、1.35mmol、1.5当量)を加え、これをN2保護下で室温で1時間攪拌した。混合物を真空中で濃縮した。残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物である例27h(150mg、収率52.7%)を黄色油状物として得た。LCMS[M+1]+=317.2.
例27i(165.6mg、0.94mmol、1.5当量)のTHF(5mL)溶液に、NaH(75.7mg、鉱油中60%、1.89mmol、3.0当量)を0℃で数回に分けて加えた。0.5時間攪拌した後、例27h(200mg、0.63mmol、1.0当量)のTHF(1mL)溶液を滴加した。反応混合物を室温で1.5時間攪拌した。反応を、0℃で飽和NH4Cl水溶液(10mL)でクエンチし、EtOAc(20mL×3)で抽出した。一つに合わせた有機層をブライン(10mL×2)で洗浄し、Na2SO4上で乾燥させ、真空中で濃縮した。粗生成物をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、所望の生成物である例27j(105mg、収率40.4%)を淡黄色固体として得た。LCMS[M+1]+=412.4.
工程9:例27k
例27j(105mg、0.25mmol、1.0当量)のDCM(15mL)溶液に、0℃でHCl/ジオキサン(0.2mL、ジオキサン中4mol/L)を加えた。反応混合物を室温で1時間攪拌した。反応溶液を真空中で濃縮して、所望の生成物である例27k(150mg、収率:定量)を白色固体として得た。LCMS[M+1]+=212.3.
例27l(121mg、0.37mmol、1.0当量)およびDIEA(239mg、1.85mmol、5.0当量)のDCM(30mL)溶液に、HATU(167mg、0.44mmol、1.2当量)を加えた。10分間攪拌した後、例27k(未精製105mg、0.37mmol、1.0当量)を加え、これを室温で2時間攪拌した。混合物を真空中で濃縮した。残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物である例27m(65mg、収率33.8%)を白色固体として得た。LCMS[M+1]+=520.3.
例27m(60mg、0.12mmol、1.0当量)のジオキサン(2mL)溶液に、Cs2CO3(75.1mg、0.24mmol、2.0当量)および第3t-Bu-XphosPd(10.2mg、0.012mmol、0.1当量)を加えた。反応混合物を、N2下、80℃で3時間攪拌した。反応溶液をろ過し、ろ液を真空中で濃縮した。粗生成物を分取TLCにより精製して、所望の生成物である例27n(25mg、収率44.7%)を白色固体として得た。LCMS[M+1]+=484.4.
例27n(25mg、0.052mmol、1.0当量)のDCM(2mL)溶液に、0℃でHCl/ジオキサン(0.2mL、4mol/Lジオキサン)を加え、これを室温で2時間攪拌した。反応溶液を真空中で濃縮した。粗生成物をMeOHに溶解させ、Na2CO3(過剰)を加えた。得られた混合物を、室温で10分間攪拌し、沈殿物をろ過した。ろ液を濃縮して、残渣を分取TLCにより精製して、所望の生成物である例27(7.2mg、収率36.4%)を白色固体として得た。LCMS[M+1]+=384.3.1H NMR(300MHz,DMSO-d6)δ 9.31(s,1H),8.67(s,1H),8.25(d,1H),8.20(d,1H),8.02-8.04(m,1H),6.13(s,1H),4.62(d,2H),4.47(d,2H),3.98(s,3H),3.89-3.96(m,1H),3.54-3.62(m,1H),3.32-3.45(m,1H),2.93(d,3H),1.19(d,3H).
実施例28:

例28a(20.0g、0.12mol、1.0当量)のCCl4(400mL)溶液に、NBS(72.4g、0.41mol、3.5当量)およびAIBN(13.4g、0.08mol、0.7当量)を加えた。反応混合物を、80℃で、16時間攪拌した。室温まで冷却した後、固体をろ過し、ろ液を濃縮した。残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物である例28b(15.1g、収率52%)を黄色固体として得た。LCMS[M+1]+=251.2.
例28c(12.7g、72.5mmol、1.2当量)のTHF(400mL)溶液に、NaH(2.9g、鉱油中60%、72.5mmol、1.2当量)を0℃で数回に分けて加えた。混合物を同温度で5分間攪拌し、次いでTHF(50mL)中の例28b(15.1g、60.4mmol、1.0当量)を滴加した。反応混合物を室温で、1時間攪拌した。反応をH2O(100mL)でクエンチし、EtOAc(200mL×3)で抽出した。一つに合わせた有機層をブラインで洗浄し、Na2SO4上で乾燥させ濃縮した。残渣をシリカゲルクロマトグラフィーにより精製して、所望の生成物である例28d(5.2g、収率25%)を黄色油状物として得た。LCMS[M+1]+=346.3.
例28d(5.1g、14.7mmol、1.0当量)のt-BuOH/H2O(100mL/30mL)溶液に、NaOH(2.9g、73.7mmol、5.0当量)を加えた。混合物を、80℃で16時間攪拌した。室温に冷却した後、混合物を0.2MのHCl水溶液で酸性化し、これを次いでDCM/MeOHの混合溶媒(200mL×3、v/v=10/1)で抽出した。一つに合わせた有機層をブラインで洗浄し、Na2SO4上で乾燥させ濃縮した。残渣をシリカゲルクロマトグラフィーにより精製して、所望の生成物である例28e(650mg、収率14%)を黄色固体として得た。LCMS[M+1]+=328.3.
例28e(600mg、1.84mmol、1.0当量)および例28f(580mg、3.67mmol、2.0当量)のDMF(12mL)溶液に、CuI(348.6mg、1.84mmol、1.0当量)、1,10-フェナントロリン(182mg、0.92mmol、0.5当量)、およびK3PO4(778mg、3.67mmol、2.0当量)を加えた。混合物を、N2下、110℃で、16時間攪拌した。反応溶液をEtOAc(100mL)で希釈し、ブライン(100mL×3)で洗浄して、Na2SO4上で乾燥させ、濃縮した。残渣をシリカゲルクロマトグラフィーにより精製して、所望の生成物である例28g(120mg、収率16.2%)を黄色固体として得た。LCMS[M+1]+=405.3.
例28g(115mg、0.284mmol、1.0当量)のEtOH(2.2mL)および水(0.7mL)の溶液に、Zn(92.5mg、1.423mmol、5.0当量)およびNH4Cl(76.8mg、1.423mmol、5.0当量)を加えた。反応混合物を80℃で1時間攪拌した。室温まで冷却した後、混合物をろ過し、ろ液を濃縮した。粗生成物をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物である例28h(85mg、収率80%)を黄色固体として得た。LCMS[M+1]+=375.3.
例28h(80mg、0.214mmol、1.0当量)のDCM(1mL)溶液に、HCl/ジオキサン(0.3mL、ジオキサン中4M)を加え、これを室温で1時間攪拌した。反応が完了した後、溶媒を濃縮して、例28i(70mg、未精製)を黄色固体として得た。未精製物を、さらに精製することなく、次の工程で直接使用した。LCMS[M+1]+=275.3.
例28j(57mg、0.175mmol、0.8当量)のDCM(1mL)溶液に、HATU(99.8mg、0.263mmol、1.2当量)およびDIEA(113mg、0.876mmol、4.0当量)を加えた。混合物を20分間攪拌し、次いで例28i(60mg、0.219mmol、1.0当量)を加えた。反応混合物を室温で、2時間攪拌した。溶液を真空中で濃縮し、未精製物を分取TLCにより精製して、生成物である例28k(70mg、収率69%)を黄色固体として得た。LCMS[M+1]+=583.3.
例28k(70mg、0.12mmol、1.0当量)のジオキサン(1mL)溶液に、Cs2CO3(78mg、0.24mmol、2.0当量)および第3t-Bu-Xphos-Pd(11mg、0.012mmol、0.1当量)を加えた。反応混合物を、N2保護下、80℃で4時間攪拌した。固体をろ過し、ろ液を濃縮した。残渣を分取TLCにより精製し、例28l(45mg、収率69%)を黄色固体として得た。LCMS[M+1]+=547.3.
例28l(40mg、0.073mmol、1.0当量)のDCM(1mL)溶液に、TFA(0.3mL)を0℃で滴加した。反応混合物を室温で2時間攪拌した。完了後、反応混合物を濃縮した。粗生成物をMeOH(2mL)に溶解し、NaHCO3で塩基性化した。固体をろ過し、ろ液を濃縮した。残渣を分取TLCにより精製し、例28(4.2mg、収率13%)を黄色固体として得た。LCMS[M+1]+=447.1.1H NMR(300MHz,DMSO-d6)δ 9.19(s,1H),8.64(d,1H),8.48(d,1H),8.22-8.15(m,2H),8.08-8.01(m,1H),7.96-7.90(m,1H),7.86(d,1H),7.59-7.51(m,2H),6.11(s,1H),4.43(s,2H),4.06-3.93(m,1H),3.63-3.48(m,2H),2.92(d,3H),1.19(d,3H).
実施例29:
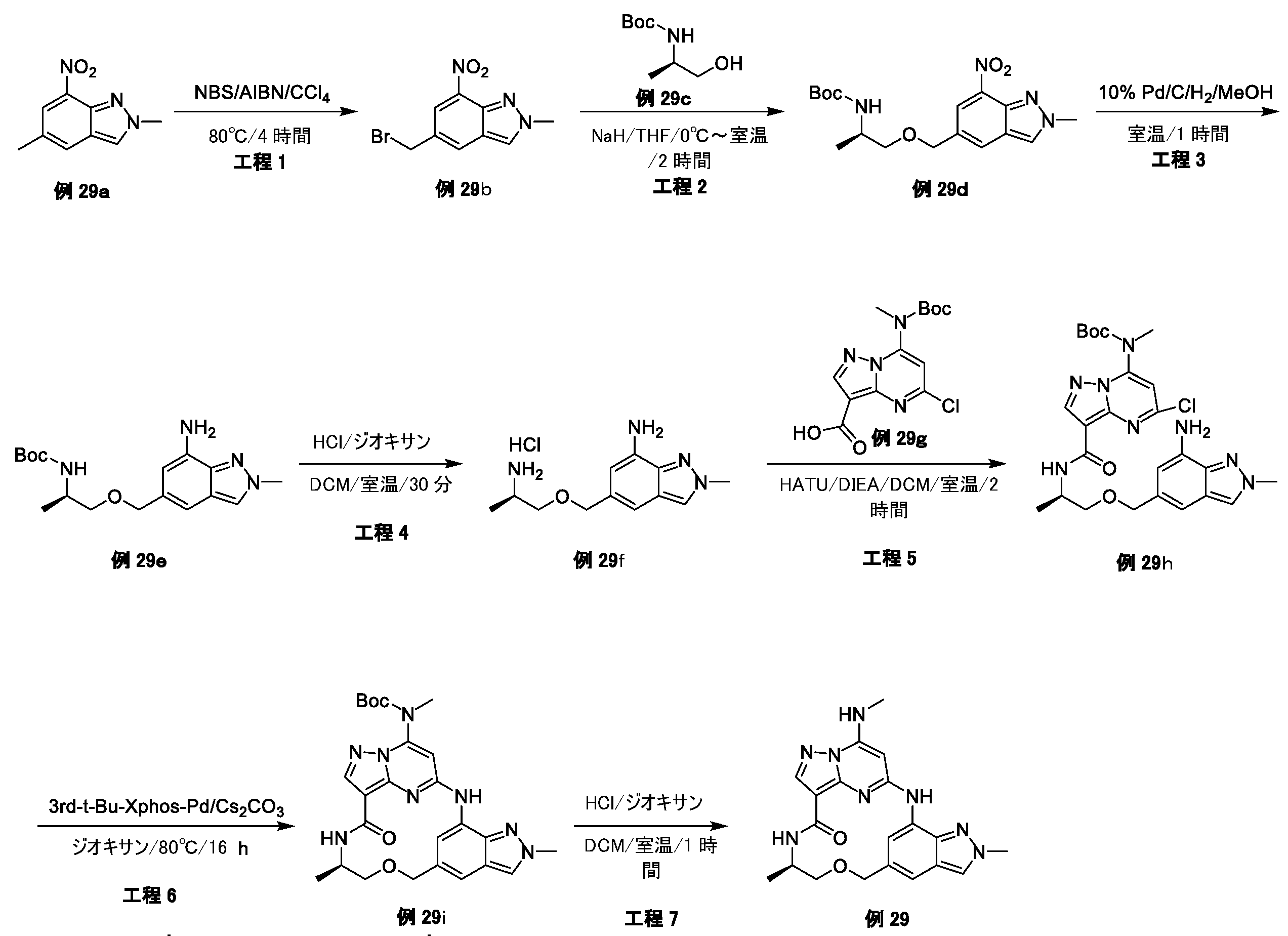
例29a(2.40g、12.57mmol、1.0当量)のCCl4(100mL)溶液を80℃に加熱し、次いでNBS(2.68g、15.08mmol、1.2当量)およびAIBN(2.06g、12.57mmol、1.0当量)を加えた。反応混合物を80℃で4時間攪拌した。反応溶液を濃縮し、シリカゲルフラッシュカラムクロマトグラフィーにより精製して、所望の生成物である例29b(2.16g、収率63.7%)を黄色固体として得た。
例29c(2.80g、16.00mmol、2.0当量)のTHF(100mL)溶液に、NaH(480mg、鉱油中60%、12.00mmol、1.5当量)を0℃で数回に分けて加えた。30分間攪拌した後、例29b(2.16g、8.00mmol、1.0当量)を加え、これを室温でさらに2時間攪拌した。反応を飽和NH4Cl水溶液でクエンチし、EtOAc(100mL×3)で抽出した。一つに合わせた有機層をブラインで洗浄し、Na2SO4上で乾燥させ濃縮した。残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、所望の生成物である例29d(1.27g、収率43.6%)を黄色固体として得た。LCMS[M+1-56]+=309.2.
例29d(1.27g、0.84mmol、1.0当量)をMeOH(30mL)に溶解させて、N2保護下で、10%のPd/C(500mg)を数回に分けて加えた。系を真空にし、次いで水素で再充填した。混合溶液をH2バルーン下、室温で1時間攪拌した。反応混合物をセライトパッドを介してろ過し、ろ液を濃縮して、所望の生成物である例29e(1.14g、収率97.8%)を黄色油状物として得た。LCMS[M+1+22]+=357.3.
例29e(1.14g、3.41mmol、1.0当量)のDCM(30mL)溶液に、HCl/ジオキサン(10mL、ジオキサン中4M)を加えた。反応溶液を、室温で0.5時間攪拌し、溶媒を除去して、所望の粗生成物である例29f(1.64g、収量:定量)を黄色固体として得た。LCMS[M+1]+=235.3.
例29g(300mg、0.92mmol、1.0当量)のDCM(15mL)溶液に、DIEA(947mg、7.34mmol、8.0当量)およびHATU(383mg、1.01mmol、1.1当量)を加えた。30分間攪拌した後、例29f(373mg、1.38mmol、1.5当量)を加えた。反応溶液を室温で2時間攪拌した。反応混合物を濃縮し、シリカゲルフラッシュカラムクロマトグラフィーにより精製して、所望の生成物である例29h(280mg、収率56.2%)を黄色固体として得た。LCMS[M+1]+=543.3.
例29h(240mg、0.44mmol、1.0当量)のジオキサン(10mL)溶液に、Cs2CO3(288mg、0.88mmol、2.0当量)および第3-t-Bu-Xphos-Pd(39mg、0.044mmol、0.1当量)を加えた。反応混合物を、N2下、80℃で16時間攪拌した。反応混合物を濃縮し、分取TLCにより精製して、所望の生成物である例29i(75mg、収率33.5%)を黄色固体として得た。LCMS[M+1]+=507.3.
例29i(65mg、0.13mmol、1.0当量)のDCM(6mL)溶液に、HCl/ジオキサン(3mL、ジオキサン中4M)を加えた。反応溶液を室温で1時間攪拌し、濃縮した。粗生成物をMeOHに溶解し、Na2CO3固体(過剰)を混合物に加え、これを室温で10分間攪拌した。混合物をろ過し、ろ液を濃縮した。残渣を分取TLCにより精製して、所望の生成物である例29(33.8mg、収率64.8%)を灰白色固体として得た。LCMS[M+1]+=407.3.1H NMR(300MHz,DMSO-d6)δ 9.60(s,1H),8.39(d,1H),8.31(s,1H),8.23(d,1H),8.13(s,1H),7.84(d,1H),7.22(s,1H),6.08(s,1H),4.65(d,1H),4.51(d,1H),4.20(s,3H),4.01-3.90(m,1H),3.51(d,1H),3.38(d,1H),2.93(d,3H),1.13(d,3H).
実施例30および実施例31

例30a(2.6g、15.0mmol)のTHF(30mL)溶液に、N2保護下、-78℃でMeLi(18.7mL、1.6moL/L)を滴加した。反応混合物を-78℃で2時間攪拌した。次いで、混合物を、NH4Cl(水溶液)によりクエンチし、DCMにより希釈し、水で洗浄し、無水Na2SO4上で乾燥させた。溶液を減圧下で濃縮し、残渣をシリカゲルカラムクロマトグラフィーにより精製して、例30b(1.8g、収率63%)を白色固体として得た。
例30b(380mg、2.0mmol)、TBAI(75mg、0.2mmol)のTHF(5mL)溶液に、NaH(173mg、鉱油中60%、3.0mmol)を0℃で加えた。反応混合物を室温に温め、0.5時間攪拌した。次いで、例30c(492mg、2.0mmol)を加えた。混合物を室温でさらに6時間攪拌した。反応混合物を、NH4Cl水溶液でクエンチし、EtOAcにより抽出し、無水Na2SO4上で乾燥した。溶液を、減圧下で濃縮した。残渣をシリカゲルカラムクロマトグラフィーにより精製して、例30d(230mg、収率:32%)を黄色固体として得た。LCMS[M+1-100]+=255.1.
例30d(230mg、0.65mmol)および10%のPd/C(50mg)のMeOH(10mL)溶液を、室温で2時間、1気圧のH2下で攪拌した。完了後、混合物をセライトでろ過し、ろ液を減圧下で濃縮した。残渣である例30e(178mg、収率:定量)を、黄色固体として得、これを次の工程で直接使用した。LCMS[M-188]+=137.1
工程4:例30f
例30e(178mg、0.75mmol)のDCM(4mL)溶液に、TFA(1.0mL)を加え、これを室温で2時間攪拌した。混合物を濃縮して、粗生成物である例30f(138mg、未精製、収率:82%)を黒色油状物として得た。LCMS[M+1]+=225.1.
例30f(138mg、0.62mmol)、例30g(200mg、0.62mmol)、およびTEA(311mg、3.1mmol)のDCM(5mL)溶液に、HATU(236mg、0.62mmol)を加えた。反応混合物を室温で、2時間攪拌した。次いで、DCM(40mL)を反応混合物に加え、これをブライン(20mL×2)で洗浄し、無水Na2SO4上で乾燥して、濃縮した。残渣をシリカゲルカラムクロマトグラフィーにより精製して、所望の生成物である例30h(150mg、収率45%)を褐色固体として得た。LCMS[M+1]+=533.2
例30h(150mg、0.28mmol)、Cs2CO3(137mg、0.42mmol)のジオキサン(2mL)中の混合物に、第3-t-Bu-Xphos-Pd(25mg、0.028mmol)を加えた。混合物を、N2で3回脱気し、80℃で3時間攪拌した。次いで、反応混合物をDCMで希釈し、水で洗浄し、無水Na2SO4上で乾燥し、次いで減圧下で濃縮して、未精製の例30i(170mg、未精製、収率:定量)を白色固体として得、これをさらに精製することなく次の工程で使用した。LCMS[M+1]+=497.2.
例30i(120mg、未精製、0.34mmol)のDCM(4mL)溶液に、TFA(1.0mL)を加え、これを室温で2時間攪拌した。混合物を濃縮し、残渣を分取HPLCにより精製して、以下の所望の生成物を得た:
例30(6.7mg、R.T.=1.743分、収率:5%)、白色固体LCMS[M+1]+=397.2.1H NMR(400MHz,DMSO-d6)δ 8.81(s,1H),8.54(s,1H),8.15(d,1H),8.10(s,1H),7.77(d,1H),6.89(q,2H),5.98(s,1H),4.58(d,1H),4.31(d,1H),3.84(s,3H),3.81(d,1H),3.73(d,1H),3.28(s,1H),2.88(d,3H),1.17(d,3H),1.08(d,3H).
および例31(3.1mg、R.T.=1.652分、収率:2%)、白色固体LCMS[M+1]+=397.2。
1H NMR(400MHz,DMSO-d6)δ 8.94(s,1H),8.33(s,1H),8.15(s,1H),8.07(s,1H),7.79(s,1H),6.92(d,2H),5.85(s,1H),4.56(d,1H),4.39(d,1H),3.84(s,3H),3.59(s,1H),2.88(d,3H),1.12(d,3H),1.03(d,3H).
実施例32および実施例33
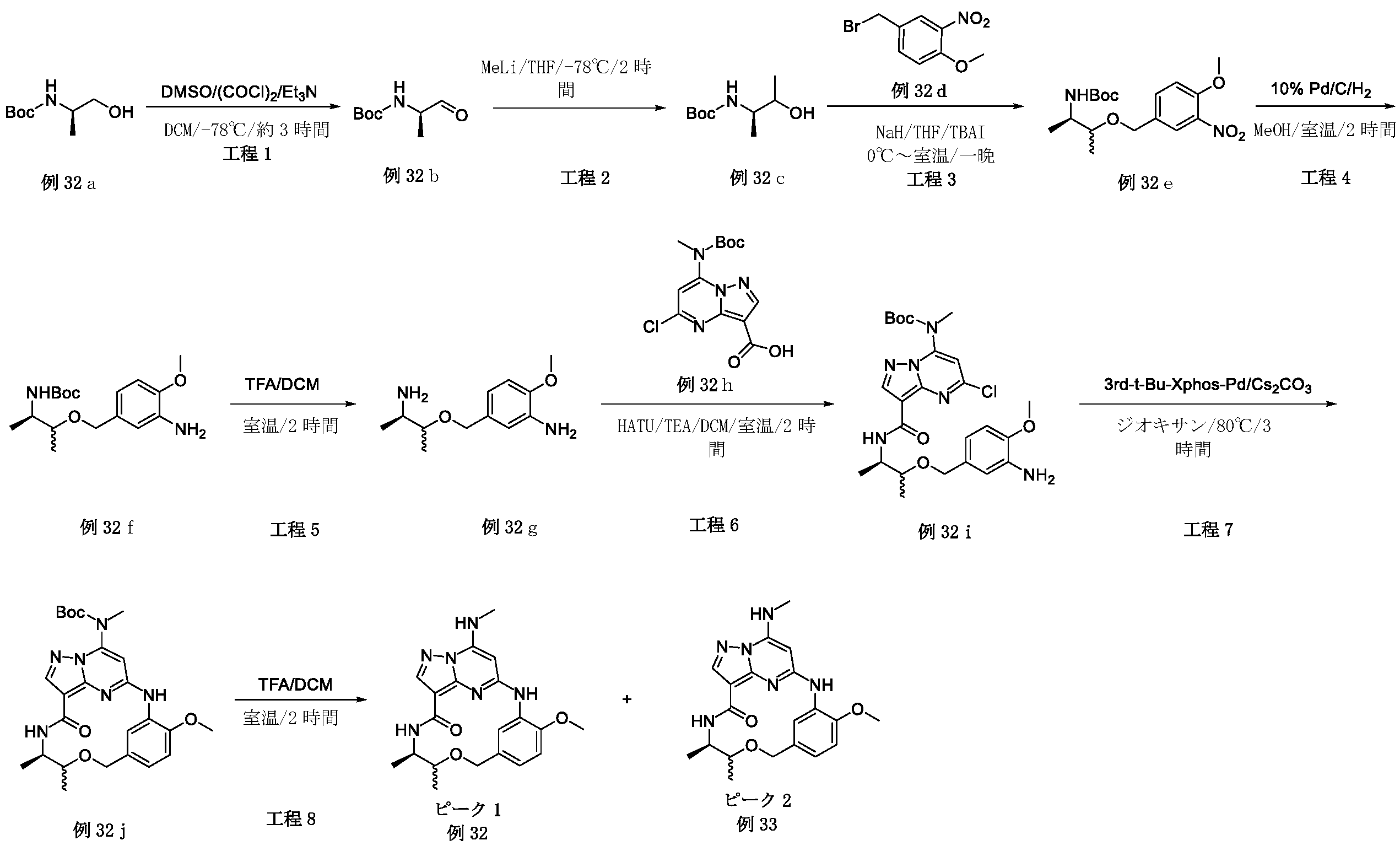
DMSO(3.3mL、47.9mmol)のDCM(10mL)溶液に、-78℃で(COCl)2(3.0mL、34.2mmol)を加え、これを-78℃で15分間攪拌した。次いで、THF(2mL)中の例32a(3.0g、17.1mmol)を滴加し、-78℃で2時間攪拌した。次いで、TEA(3.3mL、85.6mmol)を滴加し、得られた混合物を-78℃で0.5時間攪拌した。反応混合物をブラインでクエンチし、DCMで希釈し、水で洗浄し、無水Na2SO4上で乾燥させた。溶液を減圧下で濃縮し、これをシリカゲルカラムクロマトグラフィーにより精製して、例32b(2.1g、収率71%)を白色固体として得た。
例32b(1.2g、6.9mmol)のTHF(10mL)溶液に、N2保護下、-78℃でMeLi(10.8mL、17.3mmol、1.6moL/L)を滴加した。反応混合物を-78℃で2時間攪拌した。次いで反応混合物を、NH4Cl水溶液でクエンチし、DCMで希釈し、水で洗浄し、無水Na2SO4上で乾燥した。溶液を減圧下で濃縮し、これをシリカゲルカラムクロマトグラフィーにより精製して、例32c(600mg、収率46%)を白色固体として得た。
例32c(500mg、2.64mmol)、TBAI(96mg、0.26mmol)のTHF(5mL)溶液に、NaH(127mg、鉱油中60%、5.28mmol)を0℃で数回に分けて加えた。反応混合物を室温に温め、室温で0.5時間攪拌した。次いで、例32d(780mg、3.17mmol)を加えた。混合物を室温で一晩攪拌した。反応混合物を、NH4Cl水溶液でクエンチし、次いでEtOAcにより抽出し、無水Na2SO4上で乾燥した。溶液を減圧下で濃縮し、シリカゲルカラムクロマトグラフィーにより精製して、例32e(160mg、収率:18%)を黄色固体として得た。LCMS[M+1-100]+=255.1.
例32e(160mg、0.45mmol)および10%のPd/C(20mg)のMeOH(10mL)中の混合物を、室温で2時間、1気圧のH2下で攪拌した。次いで、懸濁液をろ過し、有機相を減圧下で濃縮して、未精製の例32f(140mg、収率:定量)を黄色固体として得、これを次の工程で直接使用した。LCMS[M-188]+=137.1.
例32f(140mg、0.43mmol)のDCM(4mL)溶液に、TFA(2.0mL)を加え、これを室温で2時間攪拌した。混合物を濃縮して、粗生成物である例32g(100mg、未精製、収率:定量)を黒色油状物として得た。LCMS[M+1]+=225.1.
例32g(97mg、0.43mmol)、例32h(141mg、0.43mmol)、TEA(112mg、0.86mmol)のDCM(5mL)溶液に、HATU(246mg、0.65mmol)を加えた。反応混合物を室温で、2時間攪拌した。次いで、DCM(40mL)を反応混合物に加え、これをブライン(20mL×2)で洗浄し、無水Na2SO4上で乾燥して、濃縮した。残渣をシリカゲルカラムクロマトグラフィーにより精製して、所望の生成物である例32i(110mg、収率48%)を褐色固体として得た。LCMS[M+1]+=533.2.
例32i(110mg、0.21mmol)、Cs2CO3(134mg、0.41mmol)のジオキサン(2mL)中の混合物に、第3-t-Bu-Xphos-Pd(17mg、0.021mmol)を加えた。混合物を、N2で3回脱気し、80℃で3時間攪拌した。次いで、反応混合物をDCMで希釈し、水で洗浄し、無水Na2SO4上で乾燥し、次いで減圧下で濃縮して、未精製の例32j(100mg未精製、収率:97%)を白色固体として得、これをさらに精製することなく次の工程で使用した。LCMS[M+1]+=497.2.
例32j(100mg、0.201mmol)のDCM(4mL)溶液に、TFA(1.0mL)を加え、これを室温で2時間攪拌した。混合物を濃縮し、NaHCO3水溶液により中和し、DCMにより抽出し、水で洗浄し、無水Na2SO4上で乾燥し、次いで減圧下で濃縮した。残渣を分取HPLCにより精製し、所望の生成物である例32(5.7mg、収率16%)を白色固体として得た。ピーク1として:LCMS[M+1]+=397.2;Rt=1.609分 1H NMR(400MHz,DMSO-d6)δ 8.97(s,1H),8.34(d,1H),8.16(d,1H),8.08(s,1H),7.81(d,1H),6.98-6.88(m,2H),5.86(s,1H),4.57(d,1H),4.40(d,1H),3.85(s,3H),3.59(q,1H),3.25-3.15(m,1H),1.13(d,3H),1.04(d,3H).
および例33(5.1mg、収率:13%)、白色固体として。ピーク2として:LCMS[M+1]+=397.2;Rt=1.675分。1H NMR(400MHz,メタノール-d4)δ 8.64-8.56(m,1H),8.42(s,1H),8.15(s,1H),6.89(t,2H),5.77(s,1H),5.49(s,1H),4.65(d,1H),4.36(d,1H),3.91(s,4H),3.82(d,1H),3.02(s,3H),1.23(d,6H).
実施例34:

例34a(15.0g、54.0mmol)のDMF(225mL)溶液に、SnCl2(35.8g、189.0mmol)を加えた。混合物を、N2で3回脱気し、25℃で1時間攪拌した。次いで水を加え、有機物をEtOAcで3回抽出した。一つに合わせた有機物を、無水Na2SO4上で乾燥し、次いで減圧下で濃縮し、これをシリカゲルカラムクロマトグラフィーにより精製し、所望の生成物である例34b(8.1g、収率61%)を赤色固体として得た。LCMS[M+1]+=246.0.
例34a(8.1g、32.5mmol)の例34c(100mL)中の溶液に、TsOH.H2O(627mg、3.3mmol)を加えた。混合物を、N2で3回脱気し、80℃で1時間攪拌した。混合物を、減圧下で濃縮し、シリカゲルカラムクロマトグラフィーにより精製し、所望の生成物である例34d(5.5g、収率:61%)を黄色固体として得た。LCMS[M+1]+=269.9.
例34d(5.97g、22.10mmol)のTHF(90mL)溶液に、LiAlH4(1.00g、26.47mmol)を-20℃で加えた。混合物を、N2で3回脱気し、-20℃で1時間攪拌した。次いで、反応を、水(1.2mL)の-20℃での添加によりクエンチした。得られた溶液を、NaOH水溶液(15%、3.6mL)およびEtOAc(1.2mL)で室温で希釈した。固体をろ過した。減圧下でろ液を濃縮し、シリカゲルカラムクロマトグラフィーにより精製して、所望の生成物である例34e(2.31g、収率46%)を黄色固体として得た。LCMS[M+1]+=241.9.
例34e(2.31g、9.79mmol)のDCM(45mL)溶液に、PPh3(3.30g、12.70mmol)を加えた。混合物を0℃に冷却した。次いで、CBr4(4.20g、12.70mmol)のDCM(5mL)溶液を滴加した。加えた後、反応混合物を、0℃で2時間攪拌した。混合物を、減圧下で濃縮し、シリカゲルカラムクロマトグラフィーにより精製し、所望の生成物である例34f(2.51g、収率:82%)を黄色固体として得た。LCMS[M+1]+=305.9
例34f(2.20g、7.26mmol)、例34g(1.52g、8.59mmol)のTHF(40mL)溶液に、NaH(348mg、鉱油中60%、8.59mmol)を加えた。混合物を、N2で3回脱気し、25℃で2時間攪拌した。次いで、反応をNH4Cl水溶液(10mL)の添加によりクエンチし、有機物をEtOAcで3回抽出した。一つに合わせた有機物を、無水Na2SO4上で乾燥し、減圧下で濃縮し、シリカゲルカラムクロマトグラフィーにより精製し、所望の生成物である例34h(1.20g、収率:75%)を黄色油状物として得た。LCMS[M+1-100]+=299.0.
例34h(820mg、2.06mmol)、NH2Boc(480mg、4.12mmol)、Cs2CO3(1340mg、4.12mmol)のジオキサン(5.0mL)溶液に、Pd2(dba)3(189mg、0.21mmol)、およびキサントホス(Xantphos)(123mg、0.21mmol)を加えた。混合物を、N2で3回脱気し、95℃で一晩攪拌した。次いで、反応混合物を、EtOAcにより希釈し、水で洗浄し、無水Na2SO4上で乾燥し、次いで減圧下で濃縮した。残渣をシリカゲルカラムクロマトグラフィーにより精製して、所望の生成物である例34i(520mg、収率58%)を白色固体として得た。LCMS[M+1-100]+=336.2.
例34i(170mg、0.55mmol)のDCM(5.0mL)溶液に、TFA(1.0mL)を加え、これを室温で2時間攪拌した。混合物を濃縮して、粗生成物である例34j(348mg未精製、収率:定量)を黒色油状物として得た。LCMS[M+1]+=236.1
例34j(348mg未精製、0.75mmol)、例34k(245mg、0.75mmol)、TEA(755mg、7.50mmol)のDCM(4mL)溶液に、HATU(284mg、0.75mmol)を加えた。反応混合物を室温で、2時間攪拌した。次いで、DCM(40mL)を反応混合物に加え、これをブライン(20mL×2)で洗浄し、無水Na2SO4上で乾燥して、濃縮した。残渣をシリカゲルカラムクロマトグラフィーにより精製して、所望の生成物である例34l(70mg、収率17%)を褐色固体として得た。LCMS[M+1]+=544.2.
例34l(70mg、0.13mmol)、Cs2CO3(65mg、0.20mmol)のジオキサン(2.0mL)中の混合物に、第3-t-Bu-Xphos-Pd(12mg、0.013mmol)を加えた。混合物を、N2で3回脱気し、80℃で3時間攪拌した。次いで、反応混合物をEtOAcで希釈し、水で洗浄し、無水Na2SO4上で乾燥し、次いで減圧下で濃縮して、未精製の例34m(60mg未精製、収率:定量)を白色固体として得、これをさらに精製することなく次の工程で使用した。LCMS[M+1]+=508.2
例34m(60mg未精製、0.49mmol)のTHF(1.4mL)溶液に、MeOH/HCl(2.0mL、6.0moL/L)を加え、これを室温で2時間攪拌した。混合物を濃縮し、残渣を、分取HPLCにより精製して、所望の生成物である例34(1.2mg、収率:3%)を白色固体として得た。LCMS[M+1]+=408.1.1H NMR(400MHz,DMSO-d6)δ 9.93(s,1H),8.38(d,1H),8.22(d,1H),8.13(s,1H),7.97(d,1H),7.21(s,1H),5.82(s,1H),4.68(d,1H),4.54(d,1H),3.89(d,1H),3.49(d,1H),3.29(s,1H),2.92(d,3H),2.63(s,3H),1.12(d,3H).
実施例35:

例35a(20.0g、86.6mmol、1.0当量)のEtOH(120mL)および濃HCl(40mL)中の溶液に、SnCl2(97.4g、433mmol、5.0当量)を加えた。反応混合物を、N2下、60℃で、3時間攪拌した。室温まで冷却した後、混合物を、0℃で、2MのNaOH水溶液(750mL)中に注いだ。DCM(800mL)を混合物に加え、白色固体をろ過により除去した。有機層を分離し、水相をDCM(500mL×2)で抽出した。一つに合わせた有機抽出物をブラインで洗浄し、Na2SO4上で乾燥させ濃縮した。未精製物をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物である例35b(16.8g、収率97%)を黄色固体として得た。LCMS[M+1]+=201.2.
例35b(12.0g、59.7mmol、1.0当量)のAcOH(115mL)およびH2O(33mL)中の溶液を0℃に冷却し、続いて水(20mL)中のNaNO2(4.9g、71.6mmol、1.2当量)を加えた。反応混合物を室温で2時間攪拌した。完了後、黄色の沈殿物の段階的形成が観察された。固体をろ過により回収し、濃縮して、生成物である例35c(12.5g、収率99%)を黄色固体として得た。
LCMS[M+1]+=212.2.
例35c(12.7g、60.0mmol、1.0当量)のACN(130mL)溶液に、K2CO3(16.6g、120.0mmol、2.0当量)およびMeI(25.6g、180.0mmol、3.0当量)を加えた。反応混合物を、60℃で、16時間攪拌した。室温まで冷却した後、固体をろ過し、ろ液を濃縮した。粗生成物を、シリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物である例35d(3.2g、収率24%、保持時間:1.48分)を白色固体として、例35d1(2.5g、収率18%、保持時間:1.42分)を白色固体として、および例35d2(3.4g、収率25%、保持時間:1.33分)を白色固体として得た。LCMS[M+1]+=226.2.
例35d(1.5g、6.6mmol、1.0当量)のCCl4(30mL)溶液に、NBS(1.76g、9.9mmol、1.5当量)およびAIBN(541.2mg、3.3mmol、0.5当量)を加えた。反応混合物を、80℃で、6時間攪拌した。室温まで冷却した後、溶媒を除去し、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物である例35e(1.5g、収率74%)を白色固体として得た。LCMS[M+1]+=306.2.
例35f(875mg、5.0mmol、1.5当量)のTHF(20mL)溶液に、NaH(160mg、4.0mmol、1.2当量)を0℃で数回に分けて加えた。混合物を同温度で30分間攪拌し、次いでTHF(15mL)中の例35e(1.0g、3.3mmol、1.0当量)を滴加した。反応混合物を室温で、16時間攪拌した。混合物を飽和NH4Cl(50mL)水溶液に注いで、EtOAc(50mL×3)で抽出した。一つに合わせた有機層をブラインで洗浄し、Na2SO4上で乾燥させ濃縮した。粗生成物をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物である例35g(860mg、収率65%)を白色固体として得た。LCMS[M+1]+=399.3.
例35g(860mg、2.16mmol、1.0当量)のジオキサン(10mL)溶液に、Cs2CO3(1.4g、4.32mmol、2.0当量)、NH2-Boc(505.4mg、4.32mmol、2.0当量)、キサントホス(250.0mg、0.43mmol、0.2当量)およびPd2(dba)3.CHCl3(227.7mg、0.22mmol、0.1当量)を加えた。反応混合物を、N2保護下、110℃で2時間攪拌した。室温まで冷却した後、溶媒を除去した。粗生成物をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、精製物である例35h(670mg、収率71%)を黄色油状物として得た。LCMS[M+1]+=436.4.
例35h(670mg、1.54mmol、1.0当量)のDCM(5mL)溶液に、TFA(2.5mL)を0℃で滴加した。反応混合物を室温で2時間攪拌した。溶液を真空中で濃縮して、粗生成物である例35i(1.2g、未精製)を黄色油状物として得、これを精製することなく次の工程に直接使用した。LCMS[M+1]+=236.2.
例35j(817.5mg、2.5mmol、0.8当量)のDCM(20mL)溶液に、HATU(2.28g、6.0mmol、1.2当量)およびDIEA(5.16g、40.0mmol、8.0当量)を加えた。混合物を20分間攪拌し、次いで例35i(1.2g、5.0mmol、1.0当量)を加えた。反応混合物を室温で、2時間攪拌した。溶液を真空中で濃縮して粗生成物を得て、これをシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物である例35k(260mg、収率19%)を黄色固体として得た。LCMS[M+1]+=544.4.
例35k(260mg、0.48mmol、1.0当量)のジオキサン(3mL)溶液に、Cs2CO3(313.0mg、0.96mmol、2.0当量)および第3-t-Bu-Xphos-Pd(44.1mg、0.05mmol、0.1当量)を加えた。反応混合物を、N2保護下、80℃で12時間攪拌した。固体をろ過し、ろ液を濃縮した。残渣を分取TLCにより精製し、例35l(75mg、収率31%)を黄色固体として得た。LCMS[M+1]+=508.3.
例35l(75mg、0.15mmol、1.0当量)のDCM(3mL)溶液に、HCl/ジオキサン(3mL、ジオキサン中4M)を0℃で滴加した。反応混合物を室温で2時間攪拌した。完了後、反応混合物を濃縮した。粗生成物をMeOH(2mL)に溶解させ、NaHCO3(過剰)を加えた。混合物を、室温で20分間攪拌し、次いでDCM(20mL)を加えた。固体をろ過し、ろ液を濃縮した。残渣を分取TLCにより精製して、例35(22.7mg、収率37%)を灰白色固体として得た。LCMS[M+1]+=408.2.1H NMR(300MHz,DMSO-d6)δ 10.05(s,1H),8.47(s,1H),8.36(d,1H),8.17(s,1H),7.96(d,1H),7.41(d,1H),6.06(s,1H),4.76(d,1H),4.65(d,1H),4.53(s,3H),3.99-3.95(m,1H),3.59-3.56(m,1H),3.44-3.38(m,1H),2.95(d,3H),1.16(d,3H).
実施例36:
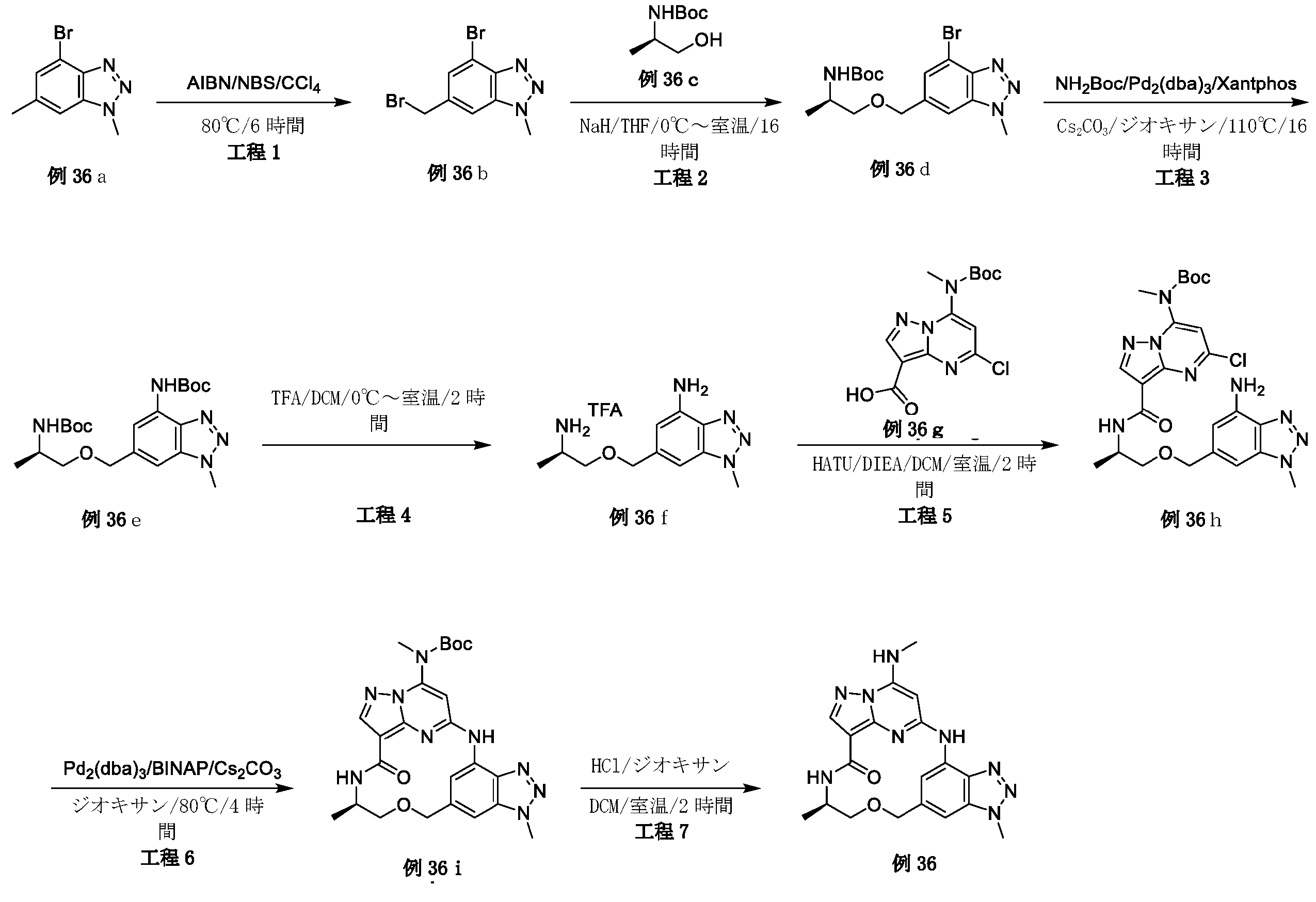
例36a(1.2g、5.0mmol、1.0当量)のCCl4(12mL)溶液に、NBS(1.42g、8.0mmol、1.5当量)およびAIBN(262mg、1.6mmol、0.3当量)を加えた。反応混合物を、80℃で、6時間攪拌した。室温まで冷却した後、溶媒を除去し、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物である例36b(850mg、収率52%)を黄色固体として得た。LCMS[M+1]+=306.2.
例36c(2.15g、12.0mmol、1.5当量)のTHF(25mL)溶液に、NaH(490mg、鉱油中60%、12.0mmol、1.5当量)を0℃で数回に分けて加えた。混合物を同温度で30分間攪拌し、次いでTHF(20mL)中の例36b(2.5g、8.0mmol、1.0当量)を滴加した。反応混合物を室温で、16時間攪拌した。次いで混合物を飽和NH4Cl(50mL)水溶液に注いで、EtOAc(50mL×3)で抽出した。一つに合わせた有機層をブラインで洗浄し、Na2SO4上で乾燥させ濃縮した。粗生成物をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物である例36d(1.8g、収率55%)を灰白色固体として得た。LCMS[M+1]+=399.3.
例36d(1.8g、4.5mmol、1.0当量)のジオキサン(36mL)溶液に、Cs2CO3(2.95g、9.0mmol、2.0当量)、NH2-Boc(4.23g、36.0mmol、8.0当量)、キサントホス(130mg、0.23mmol、0.05当量)およびPd2(dba)3(470mg、0.45mmol、0.1当量)を加えた。反応混合物を、N2保護下、110℃で16時間攪拌した。室温まで冷却した後、溶媒を除去した。粗生成物をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物である例36e(1.1g、収率56%)を黄色固体として得た。LCMS[M+1]+=436.3.
例36e(1.1g、3.0mmol、1.0当量)のDCM(11mL)溶液に、TFA(33mL)を0℃で滴加した。反応混合物を室温で2時間攪拌した。溶液を真空中で濃縮して、粗生成物である例36f(1.7g、未精製)を褐色油状物として得、これを精製することなく次の工程に直接使用した。LCMS[M+1]+=236.4.
例36g(485mg、1.489mmol、0.7当量)のDCM(10mL)溶液に、HATU(808mg、2.127mmol、1.0当量)およびDIEA(2.2g、17.021mmol、8.0当量)を加えた。混合物を20分間攪拌し、次いで例36f(500mg、2.127mmol、1.0当量)を加えた。反応混合物を室温で、2時間攪拌した。溶液を真空中で濃縮し、未精製物を分取TLCにより精製して、生成物である例36h(250mg、収率22%)を黄色固体として得た。LCMS[M+1]+=544.3.
例36h(250mg、0.46mmol、1.0当量)のジオキサン(2mL)溶液に、Cs2CO3(300mg、0.921mmol、2.0当量)、Pd2(dba)3(47mg、0.046mmol、0.1当量)およびBINAP(14mg、0.023mmol、0.05当量)を加えた。反応混合物を、N2保護下、80℃で4時間攪拌した。固体をろ過し、ろ液を濃縮した。残渣を分取TLCにより精製し、例36i(120mg、収率52%)を黄色固体として得た。LCMS[M+1]+=508.3.
例36i(120mg、0.236mmol、1.0当量)のDCM(1.2mL)溶液に、HCl/ジオキサン(6mL、ジオキサン中4M)を0℃で滴加した。反応混合物を室温で2時間攪拌した。完了後、反応混合物を濃縮した。粗生成物をMeOH(2mL)で処理し、この溶液にNaHCO3(過剰)を加え、これを室温で20分間攪拌した。次いで、混合物にDCM(20mL)を加え、固体をろ過した。ろ液を濃縮して、残渣を分取TLCにより精製して、例36(56.8mg、収率59%)を灰白色固体として得た。LCMS[M+1]+=408.2.1H NMR(300MHz,DMSO-d6)δ 10.31(s,1H),8.54(s,1H),8.38(d,1H),8.19(s,1H),7.98(d,1H),7.33(s,1H),6.13(s,1H),4.81(d,1H),4.68(d,1H),4.29(s,3H),3.99-3.95(m,1H),3.99-3.95(m,1H),3.45-3.29(m,1H),2.96(d,3H),1.15(d,3H).
実施例37:

例37a(1.9g、8.4mmol、1.0当量)のCCl4(20mL)溶液に、NBS(1.64g、9.24mmol、1.1当量)およびAIBN(137.8mg、0.84mmol、0.1当量)を加えた。反応混合物を、80℃で、6時間攪拌した。室温まで冷却した後、溶媒を除去し、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物である例37b(1.9g、収率74%)を白色固体として得た。LCMS[M+1]+=306.2.
例37c(1.6g、9.3mmol、1.5当量)のTHF(20mL)溶液に、NaH(372mg、鉱油中60%、9.3mmol、1.5当量)を0℃で数回に分けて加えた。混合物を同温度で30分間攪拌し、次いでTHF(20mL)中の例37b(1.9g、6.2mmol、1.0当量)を滴加した。反応混合物を室温で、16時間攪拌した。次いで混合物を飽和NH4Cl(50mL)水溶液に注いで、これをEtOAc(50mL×3)で抽出した。一つに合わせた有機層をブラインで洗浄し、Na2SO4上で乾燥させ濃縮した。粗生成物をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物である例37d(1.5g、収率61%)を白色固体として得た。LCMS[M+1]+=399.3.
例37d(1.48g、3.7mmol、1.0当量)のジオキサン(14mL)溶液に、Cs2CO3(2.4g、7.4mmol、2.0当量)、例37e(2.16g、18.5mmol、5.0当量)、キサントホス(428.5mg、0.74mmol、0.2当量)、およびPd2(dba)3(383mg、0.37mmol、0.1当量)を加えた。反応混合物を、N2保護下、110℃で2時間攪拌した。室温まで冷却した後、溶媒を除去した。粗生成物をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物である例37f(1.04g、収率64%)を黄色油状物として得た。LCMS[M+1]+=436.3.
例37f(1.04g、2.4mmol、1.0当量)のDCM(5mL)溶液に、TFA(5mL)を0℃で滴加した。反応混合物を室温で2時間攪拌した。溶液を真空中で濃縮して、粗生成物である例37g(930mg、未精製)を黄色油状物として得、これを精製することなく次の工程に直接使用した。LCMS[M+1]+=236.4
例37h(222mg、0.68mmol、0.8当量)のDCM(10mL)溶液に、HATU(323mg、0.85mmol、1.0当量)およびDIEA(438.6mg、3.4mmol、4.0当量)を加えた。混合物を20分間攪拌し、次いで例37g(320mg、0.85mmol、1.0当量)を加えた。反応混合物を室温で、2時間攪拌した。溶液を真空中で濃縮して、粗生成物をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物である例37i(180mg、収率39%)を黄色固体として得た。LCMS[M+1]+=544.3.
例37i(100mg、0.18mmol、1.0当量)のジオキサン(2mL)溶液に、Cs2CO3(117.4mg、0.36mmol、2.0当量)、Pd2(dba)3(18.6mg、0.018mmol、0.1当量)およびBINAP(22.4mg、0.036mmol、0.2当量)を加えた。反応混合物を、N2保護下、80℃で3時間攪拌した。固体をろ過し、ろ液を濃縮した。残渣を分取TLCにより精製し、例37j(80mg、収率88%)を黄色固体として得た。LCMS[M+1]+=508.3.
例37j(80mg、0.157mmol、1.0当量)のDCM(2mL)溶液に、HCl/ジオキサン(2mL、ジオキサン中4M)を0℃で滴加した。反応混合物を室温で2時間攪拌した。完了後、反応混合物を濃縮した。粗生成物をMeOH(2mL)で処理し、NaHCO3(過剰)を溶液に加え、混合物を室温で20分間攪拌し、次いでDCM(20mL)を加えた。固体をろ過し、ろ液を濃縮した。残渣を分取TLCにより精製して、例37(30.5mg、収率48%)を灰白色固体として得た。LCMS[M+1]+=408.2.1H NMR(300MHz,DMSO-d6)δ 9.55(s,1H),8.21(s,1H),8.14(s,1H),8.09(d,1H),7.99(d,1H),7.74(d,1H),5.78(s,1H),4.76(d,1H),4.58(d,1H),4.53(s,3H),3.86-3.84(m,1H),3.44-3.39(m,1H),3.16-3.09(m,1H),2.98(d,3H),1.10(d,3H).
実施例38:

例38a(5.08g、30.0mmol)、例38b(6.76g、38.6mmol)、PPh3(10.1g、38.5mmol)のTHF(100mL)溶液に、DIAD(8.30g、41.0mmol)を加えた。反応混合物を25℃で10時間攪拌した。次いで、EtOAc(400mL)を反応混合物に加え、これをブライン(100mL×2)で洗浄し、無水Na2SO4上で乾燥して、濃縮した。残渣をシリカゲルカラムクロマトグラフィーにより精製して、所望の生成物である例38c(4.8g、収率:49%)を黄色固体として得た。LCMS[M+1]+=327.15
例38c(4.8g、14.7mmol)のEtOH(79mL)中の混合物に、10%のPd-C(500mg)を加えた。混合物を、H2雰囲気下、25℃で4時間攪拌した。次いで、混合物を減圧下で濃縮して、未精製の例38d(4.44g未精製、収率:定量)を黄色固体として得た。残渣を、さらに精製することなく、次の工程で使用した。LCMS[M+1]+=297.18.
例38d(2.0g、6.76mmol)のHCl-MeOH(26mL、3N)溶液を、室温で2時間攪拌した。次いで、混合物を減圧下で濃縮して、未精製の例38e(1.6g未精製、収率:定量)を白色固体として得た。残渣を、さらに精製することなく、次の工程で使用した。LCMS[M+1]+=197.12.
例38e(707mg、3.6mmol)、例38f(1.4g、4.3mmol)、TEA(935mg、9.3mmol)のDCM(12mL)溶液に、HATU(2.25g、5.9mmol)を加え、これを室温で15時間攪拌した。次いで、EtOAc(400mL)を反応混合物に加え、これをブライン(100mL×2)で洗浄し、無水Na2SO4上で乾燥して、濃縮した。残渣をシリカゲルカラムクロマトグラフィーにより精製して、所望の生成物である例38g(428mg、収率:3工程で24%)を白色固体として得た。LCMS[M+1]+=506.19.
例38g(144mg、0.29mmol)、Cs2CO3(291mg、0.89mmol)のジオキサン(3mL)中の溶液に、第3-t-Bu-Xphos-Pd(16mg、0.02mmol)を加え、これをN2雰囲気下で80℃で15時間攪拌した。次いで、EtOAc(400mL)を反応混合物に加え、これをブライン(100mL×2)で洗浄し、無水Na2SO4上で乾燥して、濃縮した。残渣をシリカゲルカラムクロマトグラフィーにより精製して、所望の生成物である例38h(24mg、収率:18%)を白色固体として得た。LCMS[M+1]+=469.22
例38h(24mg、0.05mmol)のHCl-MeOH(2mL、3N)溶液を、氷浴中で0℃で攪拌し、次いで25℃まで6時間で温めた。次いで、反応混合物にEtOAc(400mL)を加え、これをブライン(100mL×2)で洗浄し、無水Na2SO4上で乾燥して、濃縮した。残渣をシリカゲルカラムカラムクロマトグラフィーにより精製して、例38(13mg、収率63%)を白色固体として得た。LCMS[M+1]+=369.16.1H NMR(400MHz,クロロホルム-d)δ 8.24(s,1H),7.05(s,1H),6.85(d,1H),6.75(dd,1H),5.39(s,1H),4.46-4.29(m,3H),3.86(s,3H),3.08(d,3H),1.35(d,3H).
実施例39:
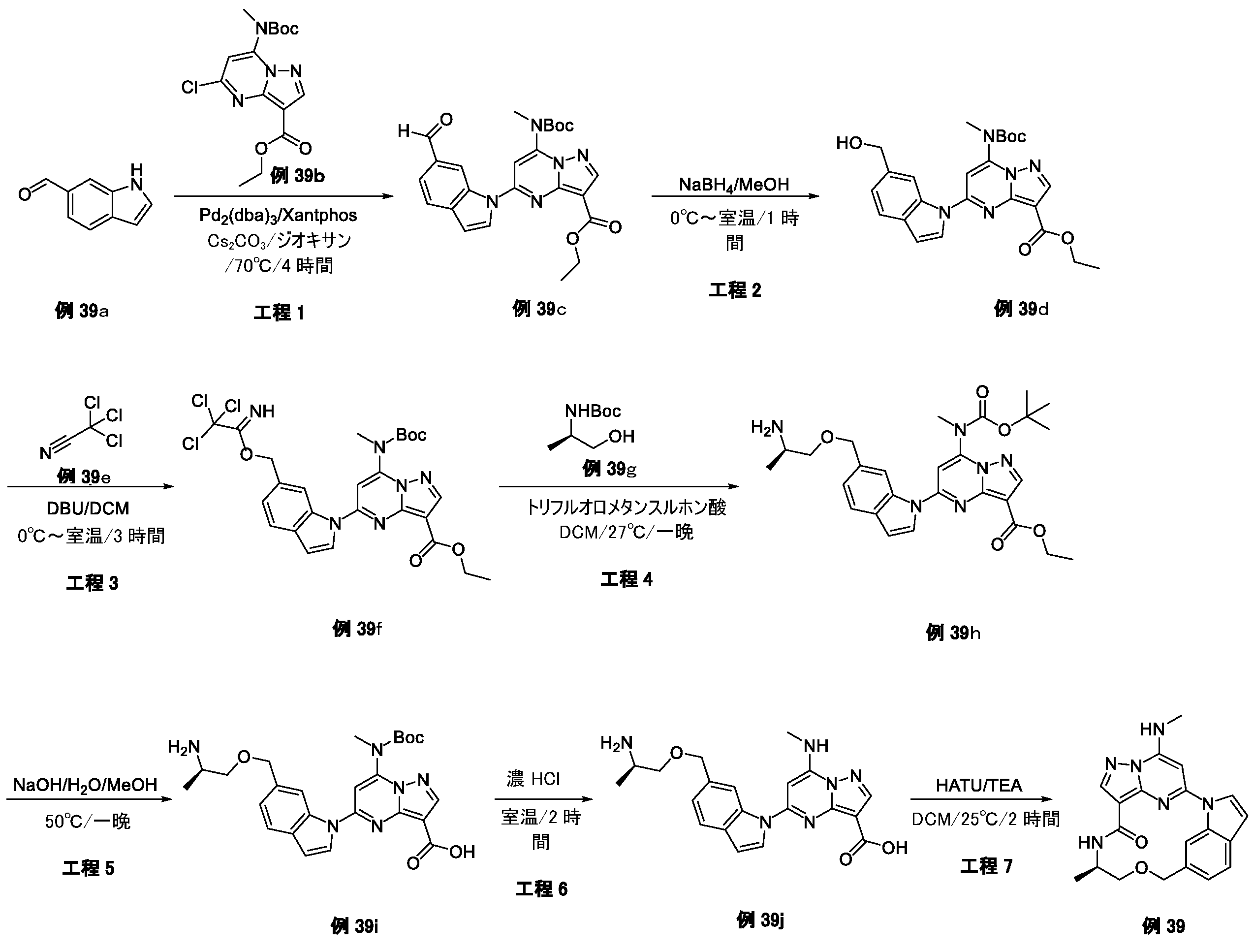
例39a(1.45g、10.0mmol)、例39b(3.55g、10.0mmol)、Pd2(dba)3(558mg、0.5mmol)、キサントホス(298mg、0.5mmol)、Cs2CO3(4.88g、15.0mmol)のジオキサン(30mL)中の混合物に、N2で3回脱気し、70℃で4時間攪拌した。次いで、反応混合物を室温に冷却し、DCMで希釈し、水で洗浄し、無水Na2SO4上で乾燥し、次いで減圧下で濃縮し、これをシリカゲルカラムクロマトグラフィーにより精製して、例39c(3.0g、収率:70%)を白色固体として得た。LCMS[M+1]+=464.1.1H NMR(400MHz、クロロホルム-d)δ 10.21(s,1H),9.64(s,1H),8.56(s,1H),7.91-7.85(m,2H),7.75(d,1H),7.08(s,1H),6.88(d,1H),4.54(q,2H),4.11(q,2H),3.46(s,3H),2.04(s,3H),1.58(s,3H),1.43(s,12H),1.24(d,3H).
例39c(3.1g、6.7mmol)のMeOH(20mL)溶液に、0℃で、NaBH4(254mg、6.7mol)を加えた。反応混合物を室温で1時間攪拌し、これを次いで水でクエンチし、DCMで希釈し、ブラインで洗浄し、無水Na2SO4上で乾燥させた。溶液を減圧下で濃縮し、シリカゲルカラムクロマトグラフィーにより精製して、例39d(2.8g、収率:90%)を白色固体として得た。LCMS[M+1-17]+=448.2.1H NMR(400MHz,クロロホルム-d)δ 9.26(s,1H),8.50(s,1H),7.62(t,2H),7.30(d,1H),7.02(s,1H),6.78(s,1H),4.92(s,2H),4.77(s,1H),4.49(q,2H),3.44(s,3H),1.46(d,3H),1.41(s,9H).
例39d(2.70g、5.8mmol)、例39e(2.52g、17.5mmol)のDCM(20mL)溶液に、0℃で、DBU(2.64g、17.5mmol)を加えた。反応混合物を室温に温め、3時間攪拌した。混合物を減圧下で濃縮し、シリカゲルカラムクロマトグラフィーにより精製して、例39f(2.3g、収率:65%)を褐色固体として得た。
例39f(2.30g、3.79mmol)、例39g(795mg、4.54mmol)のDCM(20mL)溶液に、0℃で、DCM(20mL)中のCF3SO3H(285mg、1.9mmol)を加えた。反応混合物を27℃に温め、一晩攪拌した。混合物を減圧下で濃縮し、C-18ゲルカラムクロマトグラフィーにより精製して、例39h(200mg、収率10%)を褐色固体として得た。LCMS[M+1]+=523.2.
例39h(200mg、0.38mmol)のMeOH(5mL)溶液に、H2O(5mL)中のNaOH(156mg、3.9mmol)を加え、これを50℃で一晩攪拌した。混合物を濃縮して、粗生成物である例39i(700mg未精製、収率100%)を白色固体として得た。
例39i(700mg未精製、0.38mmol)の濃HCl(5mL)溶液を、2時間、室温で攪拌した。残渣を逆相カラムにより精製し、所望の生成物である例39j(105mg、2工程の収率:75%)を褐色固体として得た。
LCMS[M+1]+=395.2.
例39i(100mg、0.254mmol)、TEA(51mg、0.51mmol)のDCM(10mL)溶液に、HATU(144mg、0.38mmol)を加えた。反応混合物を25℃で2時間攪拌した。次いで、反応混合物にDCM(20mL)を加え、これをブライン(20mL×2)で洗浄し、無水Na2SO4上で乾燥して、濃縮した。残渣を分取HPLCにより精製し、所望の生成物である例39(4.1mg、収率4%)を白色固体として得た。LCMS[M+1]+=377.1.1H NMR(400MHz,DMSO-d6)δ 9.07(s,1H),8.50(d,1H),8.35(s,1H),8.24(dd,2H),7.60(d,1H),7.03(d,1H),6.80(d,1H),6.61(s,1H),4.85(d,1H),4.60(d,1H),4.04(d,1H),3.63(d,1H),3.54(t,1H),3.30(s,1H),3.09(d,3H),1.15(d,3H).
実施例40:
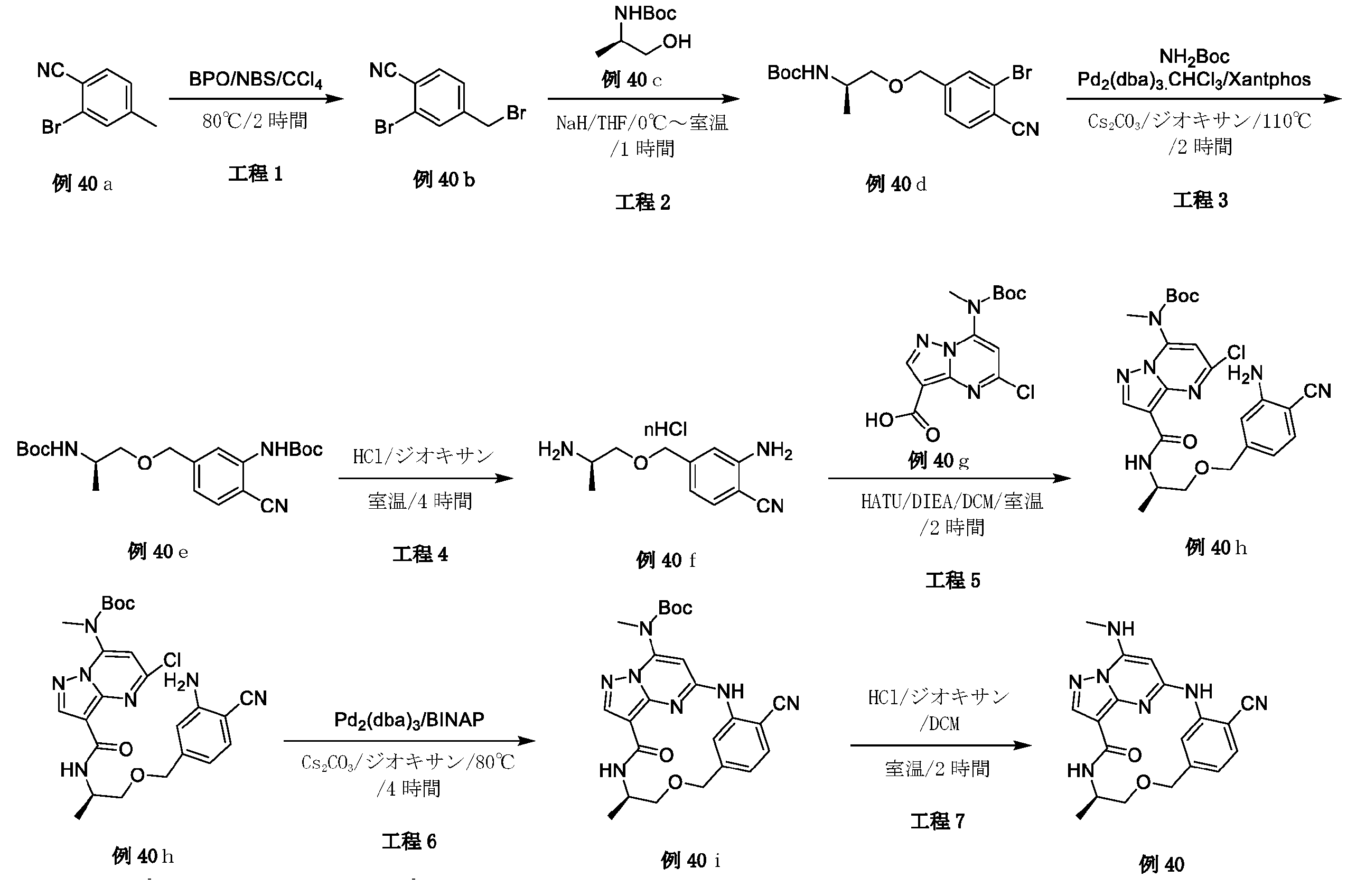
例40a(10.0g、51.3mmol、1.0当量)のCCl4(100mL)溶液に、NBS(10.04g、56.4mmol、1.1当量)およびBPO(1.24g、5.13mmol、0.1当量)を室温で加えた。混合物を、80℃で2時間攪拌した。反応が完了した後、混合物を室温に冷却した。懸濁液をEtOAc(150mL)で希釈し、これをセライトのパッドを通してろ過し、ろ過ケーキをEtOAc(150mL)で洗浄した。ろ液を真空中で濃縮して、未精製物をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物である例40b(12.5g、収率88%)を黄色固体として得た。1H NMR(300MHz,DMSO-d6)δ 8.00(d,1H),7.95(d,1H),7.66(dd,1H),4.75(s,2H).
例40c(3.53g、20.15mmol、1.1当量)のTHF(50mL)溶液を0℃まで冷却し、NaH(1.47g、鉱油中60%、36.64mmol、2.0当量)を数回に分けて加えた。混合物を0℃で30分間攪拌し、次いで例40b(5.0g、18.32mmol、1.0当量)を0℃で加え、これを室温で1時間攪拌した。混合物をNH4Cl水溶液(100mL)でクエンチし、EtOAc(100mL×3)で抽出した。一つに合わせた有機層をブラインで洗浄し、Na2SO4で乾燥させ、真空濃縮した。粗生成物をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物である例40d(3.3g、収率50%)を灰白色固体として得た。LCMS[M+1-100]+=269.1.
例40d(2.0g、5.43mmol、1.0当量)の乾燥ジオキサン(20mL)溶液に、Cs2CO3(5.30g、16.3mmol、3.0当量)、NH2-Boc(700mg、5.98mmol、1.1当量)、キサントホス(628mg、1.1mmol、0.2当量)およびPd2(dba)3.CHCl3(560mg、0.543mmol、0.1当量)を加えた。反応混合物を、N2保護下、110℃で2時間攪拌した。反応が完了した後、溶媒を除去し、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物である例40e(2.0g、収率91%)を黄色固体として得た。LCMS[M+23]+=428.3.
例40e(1.7g、4.2mmol、1.0当量)のHCl/ジオキサン(4M、20mL)溶液を、室温で4時間攪拌した。反応が完了した後、混合物を真空中で濃縮して、生成物である例40f(860mg、未精製、収率100%)を黄色固体として得た。LCMS[M+1]+=206.3.
例40g(830mg、2.54mmol、0.8当量)のDCM(8mL)溶液に、DIEA(1.65g、12.68mmol、4.0当量)およびHATU(964mg、2.54mmol、0.8当量)を加えた。混合物を室温で20分間攪拌した。次いで、例40f(650mg、3.17mmol、1.0当量)を加え、混合物を室温で2時間攪拌した。反応が完了した後、これを真空中で濃縮した。残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物である例40h(150mg、収率9%)を黄色固体として得た。LCMS[M+1]+=514.2.
例40h(200mg、0.39mmol、1.0当量)の乾燥ジオキサン(20mL)溶液に、Cs2CO3(253.4mg、0.78mmol、2.0当量)、BINAP(48.5mg、0.078mmol、0.2当量)およびPd2(dba)3(40.4mg、0.039mmol、0.1当量)を加えた。反応混合物を、N2保護下、80℃で4時間攪拌した。反応が完了した後、溶媒を除去し、残渣を分取TLCにより精製して、生成物である例40i(110mg、収率59%)を灰白色固体として得た。LCMS[M+1]+=478.3.
例40i(90mg、0.189mmol、1.0当量)のDCM(2mL)溶液に、HCl/ジオキサン(1mL、ジオキサン中4M)を加えた。反応物を室温で、2時間攪拌した。反応が完了した後、反応物をNaHCO3(過剰)で塩基性化した。固体をろ過し、ろ液を濃縮した。粗生成物を分取TLCにより精製して、生成物である例40(52.0mg、収率73%)を灰白色固体として得た。LCMS[M+1]+=378.1.1H NMR(300MHz,DMSO-d6)δ 9.78(s,1H),8.60(s,1H),8.18(s,1H),8.11(d,1H),8.06(d,1H),7.76(d,1H),7.12(dd,1H),5.97(s,1H),4.70(d,1H),4.53(d,1H),3.94-3.83(m,1H),3.53(dd,1H),3.28-3.20(m,1H),2.96(d,3H),1.15(d,3H).
実施例41:
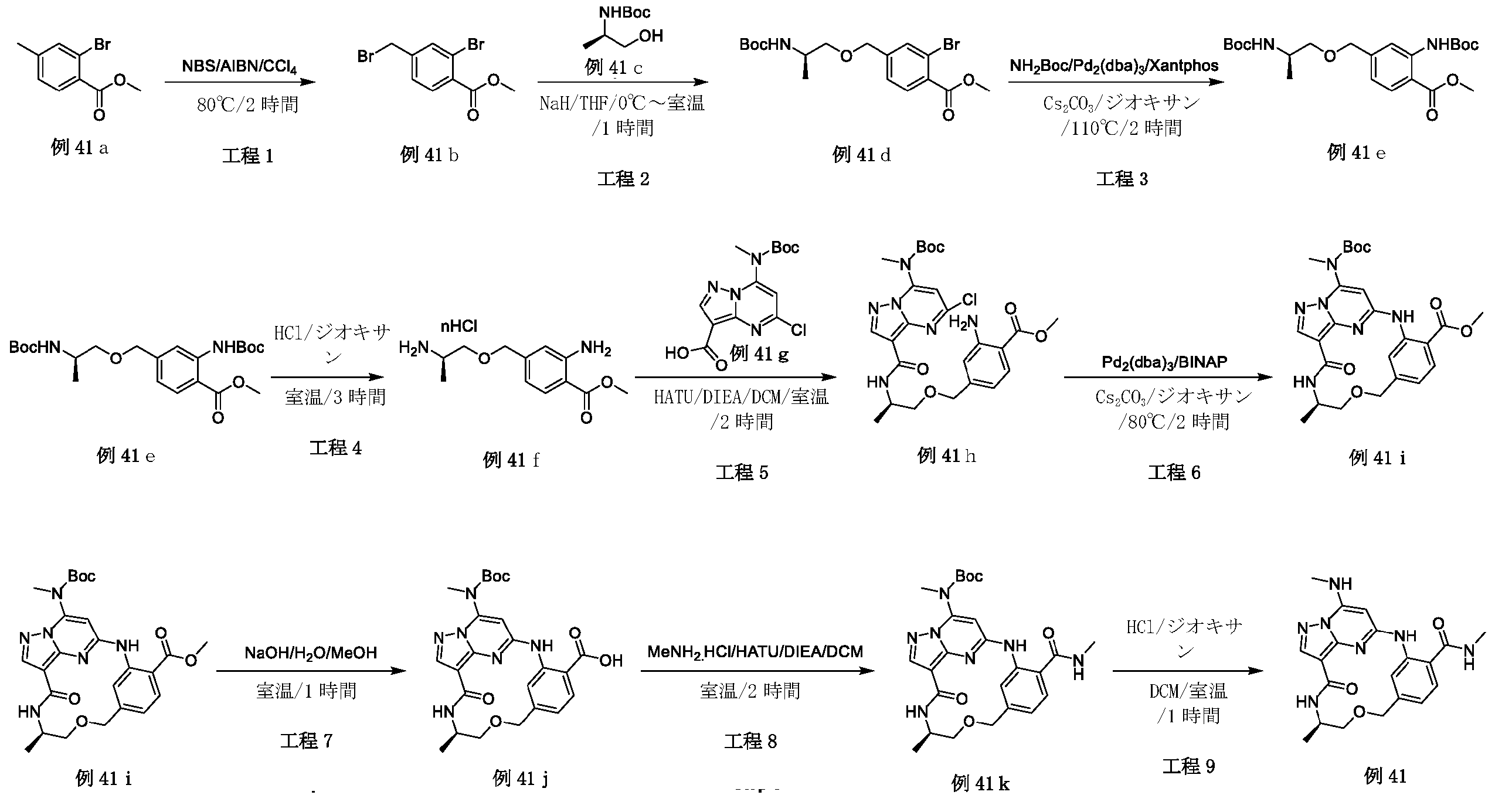
例41a(10.0g、43.7mmol、1.0当量)のCCl4(100mL)溶液に、NBS(11.66g、65.5mmol、1.5当量)およびAIBN(1.4g、8.7mmol、0.2当量)を室温で加えた。混合物を、80℃で2時間攪拌した。反応が完了した後、固体をろ過し、ろ液を濃縮した。残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物である例41b(11.8g、収率88%)を黄色固体として得た。
例41c(3.13g、17.86mmol、1.1当量)のTHF(50mL)溶液に、NaH(1.30g、32.47mmol、2.0当量)を0℃で数回に分けて加えた。20分間攪拌した後、0℃で例41b(5.0g、16.23mmol、1.0当量)を加えた。混合物をN2下、室温で1時間攪拌した。混合物をNH4Cl水溶液(100mL)でクエンチし、これをEtOAc(200mL×3)で抽出した。一つに合わせた有機層をブラインで洗浄し、Na2SO4上で乾燥させ濃縮した。粗生成物をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物である例41d(1.4g、収率22%)を灰白色固体として得た。LCMS[M+1-100]+=302.1.
例41d(1.4g、3.5mmol、1.0当量)のジオキサン(20mL)溶液に、Cs2CO3(3.4g、10.5mmol、3.0当量)、NH2-Boc(450mg、3.84mmol、1.1当量)、キサントホス(404mg、0.7mmol、0.2当量)およびPd2(dba)3(362mg、0.35mmol、0.1当量)を加えた。反応混合物を窒素により3回脱気し、110℃で2時間攪拌した。反応物を室温に冷却し、真空中で濃縮した。粗生成物をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物である例41e(1.42g、収量93%)を黄色固体として得た。LCMS[M+1+22]+=461.2
例41e(1.42g、3.24mmol、1.0当量)のHCl/ジオキサン(4M、20mL)溶液を、室温で3時間攪拌した。反応が完了した後、混合物を真空中で濃縮して、生成物である例41f(800mg、未精製、収率100%)を黄色固体として得た。LCMS[M+1]+=239.2.
例41g(789mg、2.42mmol、0.8当量)のDCM(20mL)溶液に、DIEA(1.57g、12.1mmol、4.0当量)およびHATU(1.72g、4.51mmol、1.5当量)を加えた。15分間攪拌した後、例41f(720mg、3.03mmol、1.0当量)を加えた。反応混合物を室温で、2時間攪拌した。反応が完了した後、これを真空中で濃縮した。残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物である例41h(520mg、収率39%)を黄色固体として得た。LCMS[M+1]+=547.3.
例41h(440mg、0.81mmol、1.0当量)のジオキサン(20mL)溶液に、Cs2CO3(523mg、1.61mmol、2.0当量)、BINAP(100mg、0.16mmol、0.2当量)およびPd2(dba)3(83.3mg、0.08mmol、0.1当量)を加えた。反応混合物を、N2保護下、80℃で2時間攪拌した。反応混合物を室温に冷却し、ろ過し、真空中で濃縮した。粗生成物を分取TLCにより精製して、生成物である例41i(220mg、収率54%)を灰白色固体として得た。LCMS[M+1]+=511.2.
例41i(200mg、0.39mmol、1.0当量)のMeOH(4mL)溶液に、2MのNaOH水溶液(0.4mL)を加えた。反応混合物を室温で、1時間攪拌した。反応が完了した後、混合物をHCl水溶液(1M)で酸性化し、次いでこれをEtOAc(20mL×3)で抽出した。一つに合わせた有機層を、ブラインで洗浄し、真空中で濃縮して、生成物である例41j(190mg、収率98%)を灰白色固体として得た。LCMS[M+1]+=497.2.
例41j(170mg、0.34mmol、1.0当量)のDCM(8mL)溶液に、DIEA(178mg、1.37mmol、4.0当量)およびHATU(195mg、0.51mmol、1.5当量)を加えた。15分間攪拌した後、メチルアミン塩酸塩(46mg、0.69mmol、2.0当量)を反応混合物に加え、これを室温で2時間攪拌した。反応が完了した後、これを真空中で濃縮した。残渣を分取TLCにより精製して、生成物である例41k(170mg、収率97%)を灰白色固体として得た。LCMS[M+1]+=510.2.
例41k(100mg、0.196mmol、1.0当量)のDCM(2mL)溶液に、室温でHCl/ジオキサン(2mL、ジオキサン中4M)を加えた。反応物を室温で、1時間攪拌した。反応が完了した後、反応物をNaHCO3(過剰)で塩基性化した。固体をろ過し、ろ液を濃縮した。粗生成物を分取TLCにより精製して、所望の生成物である例41(46.7mg、収率58%)を白色固体として得た。LCMS[M+1]+=410.2
1H NMR(300MHz,DMSO-d6)δ 10.95(s,1H),8.81(d,1H),8.69(d,1H),8.30(d,1H),8.17(s,1H),7.95(d,1H),7.69(d,1H),7.00-6.90(m,1H),5.74(s,1H),4.65(d,1H),4.54(d,1H),4.00-3.88(m,1H),3.61-3.54(m,1H),3.44-3.37(m,1H),2.97(d,3H),2.83(d,3H),1.18(d,3H).
実施例42:
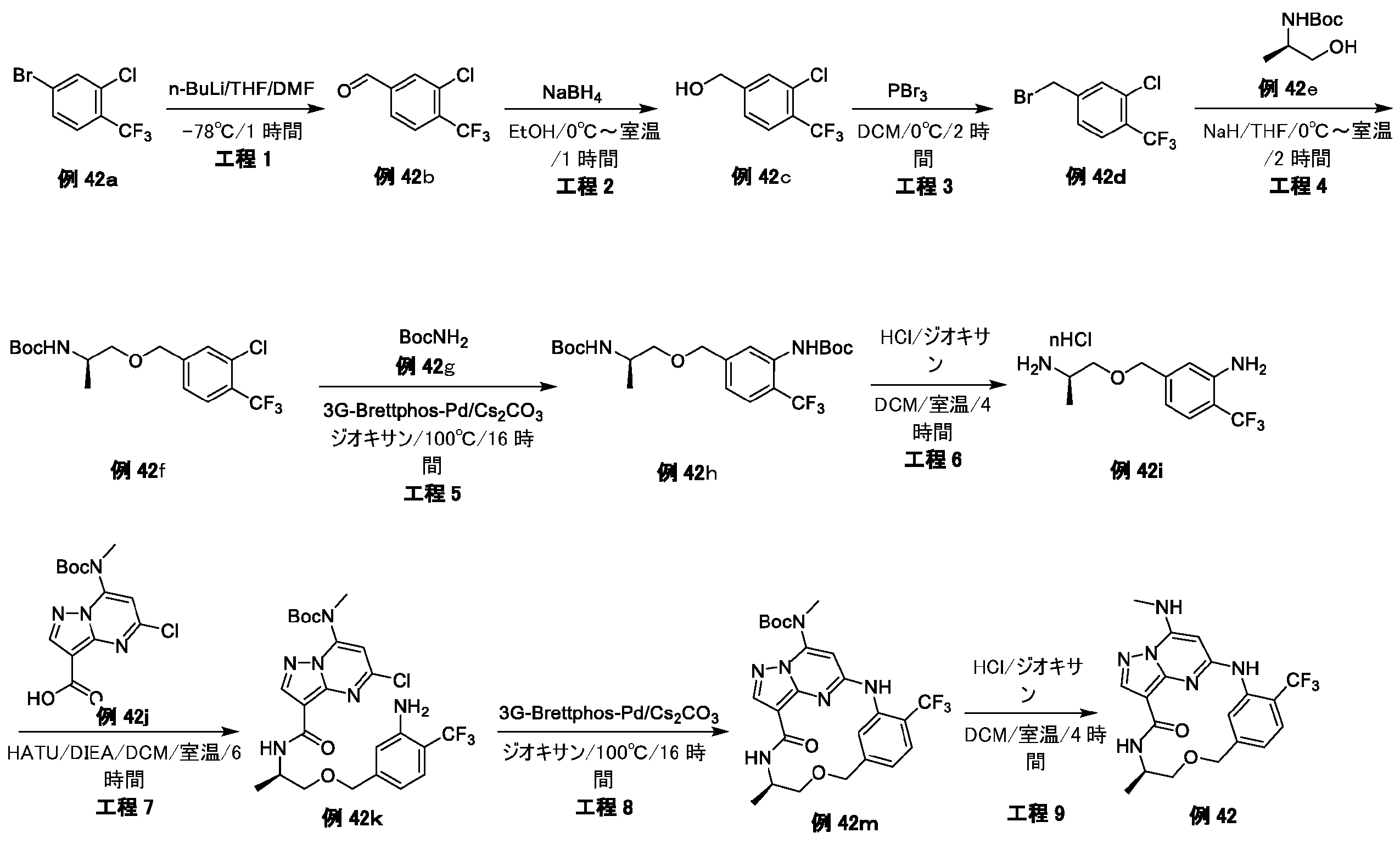
例42a(10.0g、38.7mmol、1.0当量)の乾燥THF(193mL)溶液に、n-BuLi(17mL、ヘキサン中2.5M、42.6mmol、1.1当量)を、15分間にわたって-78℃で滴加し、続いて20分間攪拌した。この混合物にDMF(28.3g、387mmol、10.0当量)を-78℃で滴加し、得られた混合物を-78℃でN2下でさらに1時間攪拌した。反応混合物を、1NのNH4Cl水溶液(100mL)でクエンチし、これを0℃で30分間攪拌した。次いで反応混合物を、EtOAc(100mL×3)で抽出した。一つに合わせた有機層を、ブラインで洗浄し、Na2SO4上で乾燥し、濃縮した。残渣をシリカカラムクロマトグラフィー(石油エーテル)により精製して、所望の生成物である例42b(6.1g、収率71%)を黄色油状物として得た。
1H NMR(300MHz,CDCl3-d)δ 10.06(s,1H),8.01(s,1H),7.91-7.85(m,2H).
例42b(4.9g、23.6mmol、1.0当量)のエタノール(100mL)溶液に、NaBH4(985mg、25.9mmol、1.1当量)を0℃で数回に分けて加えた。反応物を室温で、1時間攪拌した。反応混合物を、1NのNH4Cl水溶液(100mL)でクエンチし、これを0℃で30分間攪拌した。反応混合物を、EtOAc(100mL×3)で抽出した。一つに合わせた有機層をブラインで洗浄し、Na2SO4上で乾燥させ濃縮した。残渣をシリカカラムクロマトグラフィー(石油エーテル)により精製して、所望の生成物である例42c(3.7g、収率75%)を黄色油状物として得た。
例42c(3.7g、17.6mmol、1.0当量)のDCM(50mL)溶液に、0℃で、PBr3(5.2g、19.4mmol、1.1当量)を加えた。反応物を、0℃で2時間攪拌した。反応混合物をH2O(40mL)で希釈し、EtOAc(40mL×3)で抽出した。一つに合わせた有機層をNaHCO3水溶液で洗浄し、Na2SO4上で乾燥させ濃縮した。残渣をシリカカラムクロマトグラフィー(石油エーテル)により精製して、所望の生成物である例42d(2.1g、収率44%)を黄色油状物として得た。
例42e(1.5g、8.49mmol、1.1当量)のTHF(50mL)溶液に、NaH(864mg、鉱油中60%、21.6mmol、2.8当量)を0℃で数回に分けて加えた。15分間攪拌した後、0℃で例42d(2.1g、7.72mmol、1.0当量)を加えた。混合物を室温で2時間攪拌した。反応物をH2O(50mL)でクエンチし、EtOAc(50mL×3)で抽出した。一つに合わせた有機層をブラインで洗浄し、Na2SO4上で乾燥させ濃縮した。残渣をシリカゲルクロマトグラフィーにより精製して、所望の生成物である例42f(850mg、収率27%)を白色固体として得た。LCMS[M+1-100]+=268.0.
例42f(854mg、2.3mmol、1.0当量)および例42g(523mg、3.0mmol、1.3当量)のジオキサン(20mL)溶液に、3G-Brettphos-Pd(211mg、0.23mmol、0.1当量)およびCs2CO3(1.13g、3.54mmol、1.5当量)を加えた。混合反応物を窒素により3回脱気し、100℃で16時間攪拌した。反応物を室温に冷却し、濃縮した。残渣をシリカゲルクロマトグラフィーにより精製して、所望の生成物である例42h(720mg、収率70%)を黄色固体として得た。LCMS[M+1-100]+=349.2.
例42h(720mg、1.6mmol、1.0当量)のDCM(10mL)溶液に、HCl/ジオキサン(8mL、ジオキサン中4M、32mmol、20.0当量)を加えた。混合物を室温で、4時間攪拌した。反応が完了した後、溶媒を濃縮して、例42i(700mg、未精製)を黄色固体としてを得た。未精製物を、さらに精製することなく、次の工程で直接使用した。LCMS[M+1]+=249.2.
例42j(791mg、2.42mmol、1.0当量)のDCM(15mL)溶液に、DIEA(780mg、6.05mmol、2.5当量)およびHATU(1.01g、2.66mmol、1.1当量)を加えた。15分間攪拌した後、例42i(600mg、2.42mmol、1.0当量)を混合物に加えた。反応溶液を室温で6時間攪拌した。反応が完了した後、溶媒を除去し、未精製物をシリカゲルクロマトグラフィーにより精製して、所望の生成物である例42k(421mg、収率31%)を黄色固体として得た。LCMS[M+1]+=557.3.
例42k(200mg、0.36mmol、1.0当量)のジオキサン(5mL)溶液に、Cs2CO3(234mg、0.72mmol、2.0当量)、3G-Brettphos-Pd(33.0mg、0.036mmol、0.1当量)を加えた。反応混合物を、N2下、100℃で16時間攪拌した。反応混合物を室温に冷却し、ろ過し、真空中で濃縮した。粗生成物を分取TLCにより精製して、所望の生成物である例42m(110mg、収率59%)を淡黄色油状物として得た。LCMS[M+1]+=521.1.
例42m(100mg、3.1mmol)のDCM(2mL)溶液に、室温でHCl/ジオキサン(1mL、ジオキサン中4M)を加えた。反応物を室温で、4時間攪拌した。完了後、反応物をNaHCO3で塩基性化した。固体をろ過し、ろ液を濃縮した。粗生成物を、分取TLC(EtOAc)により精製して、所望の生成物である例42(42.2mg、収率53%)を灰白色固体として得た。LCMS[M+1]+=421.1.1H NMR(300MHz,DMSO-d6)δ 8.81(s,1H),8.42(s,1H),8.15(s,1H),8.00-7.96(m,2H),7.69(d,1H),7.22(d,1H),6.08(s,1H),4.67(d,1H),4.49(d,1H),3.86-3.84(m,1H),3.49-3.45(m,1H),3.21-3.15(m,1H),2.95(d,3H),1.14(d,3H).
実施例43:

例43a(2.0g、7.84mmol、1.0当量)のCCl4(40mL)溶液に、NBS(1.54g、8.63mmol、1.10当量)、AIBN(129mg、0.78mmol、0.1当量)を加えた。反応物を、N2下、80℃で3時間攪拌した。混合物を室温に冷却した。懸濁液を、EtOAc(50mL)で希釈し、次いでセライトのパッドを通してろ過した。ろ過ケーキを、EtOAc(50mL)で洗浄した。ろ液を真空中で濃縮し、粗生成物をシリカカラムクロマトグラフィー(石油エーテル)により精製して、所望の生成物である例43b(2.2g、収率84%)を黄色油状物として得た。1H NMR(300MHz,CDCl3-d)δ 7.86(d,1H),7.68(s,1H),7.57(d,1H),4.42(s,2H).
例43c(1.3g、7.29mmol、1.1当量)のTHF(50mL)溶液に、NaH(742mg、鉱油中60%、18.6mmol、2.8当量)を0℃で数回に分けて加えた。15分間攪拌した後、この混合物に0℃で例43b(2.2g、6.62mmol、1.0当量)を加え、これをN2下で室温でさらに2時間攪拌した。反応物をH2O(50mL)でクエンチし、EtOAc(50mL×3)で抽出した。一つに合わせた有機層をブラインで洗浄し、Na2SO4で乾燥させ、真空中で濃縮した。粗生成物をシリカゲルクロマトグラフィーにより精製して、所望の生成物である例43d(1.3g、収率41%)を黄色油状物として得た。LCMS[M+1-100]+=328.0.
例43d(800mg、1.87mmol、1.0当量)および例43e(328mg、2.8mmol、1.5当量)のジオキサン(10mL)溶液に、Pd2(dba)3(171mg、0.187mmol、0.1当量)、キサントホス(324mg、0.561mmol、0.3当量)およびCs2CO3(1.22g、3.74mmol、2.0当量)を加えた。反応混合物を窒素により3回脱気し、100℃で6時間攪拌した。反応物を室温に冷却し、真空中で濃縮した。粗生成物をシリカゲルクロマトグラフィーにより精製して、所望の粗生成物である例43f(612mg、収率70%)を黄色固体として得た。LCMS[M+1-100]+=365.2.
例43f(612mg、1.31mmol、1.0当量)のDCM(5mL)溶液に、HCl/ジオキサン(7mL、ジオキサン中4M、26.2mmol、20.0当量)を加えた。混合物を室温で、4時間攪拌した。反応が完了した後、溶媒を濃縮して、例43g(520mg、未精製)を白色固体として得た。LCMS[M+1]+=265.2.
例43h(572mg、1.75mmol、1.1当量)のDCM(20mL)溶液に、DIEA(513mg、3.98mmol、2.5当量)およびHATU(725mg、1.91mmol、1.2当量)を加えた。15分間攪拌した後、この混合物に例43g(420mg、1.59mmol、1.0当量)を加え、これを室温で6時間攪拌した。反応が完了した後、溶媒を除去し、粗生成物を分取TLCにより精製して、所望の生成物である例43i(198mg、収率22%)を黄色固体として得た。LCMS[M+1]+=573.1.
例43i(100mg、0.175mmol、1.0当量)のジオキサン(10mL)溶液に、Cs2CO3(114mg、0.35mmol、2.0当量)および第3-t-Bu-Xphos-Pd(27mg、0.035mmol、0.1当量)を加えた。反応混合物を、N2下、100℃で6時間攪拌した。反応混合物を室温に冷却し、ろ過し、真空中で濃縮した。粗生成物を分取TLCにより精製して、所望の生成物である例43j(53mg、収率56%)を淡黄色固体として得た。LCMS[M+1]+=537.4.
例43j(78mg、0.145mmol、1.0当量)のDCM(1mL)溶液に、室温で、HCl/ジオキサン(0.7mL、ジオキサン中4M、2.91mmol、20.0当量)を加えた。反応物を室温で、4時間攪拌した。反応が完了した後、反応物をNaHCO3(過剰)で塩基性化した。固体をろ過し、ろ液を濃縮した。粗生成物を、分取TLC(EtOAc)により精製して、所望の生成物である例43(33.2mg、収率52%)を灰白色固体として得た。LCMS[M+1]+=437.1.1H NMR(300MHz,DMSO-d6)δ 9.32(s,1H),8.64(d,1H),8.17(s,1H),8.15(d,1H),7.97(d,1H),7.37(dd,1H),7.03(dd,1H),5.99(s,1H),4.62(d,1H),4.49(d,1H),3.92-3.90(m,1H),3.52(dd,1H),3.35-3.30(m,1H),2.95(d,1H),1.16(d,1H).
実施例44:
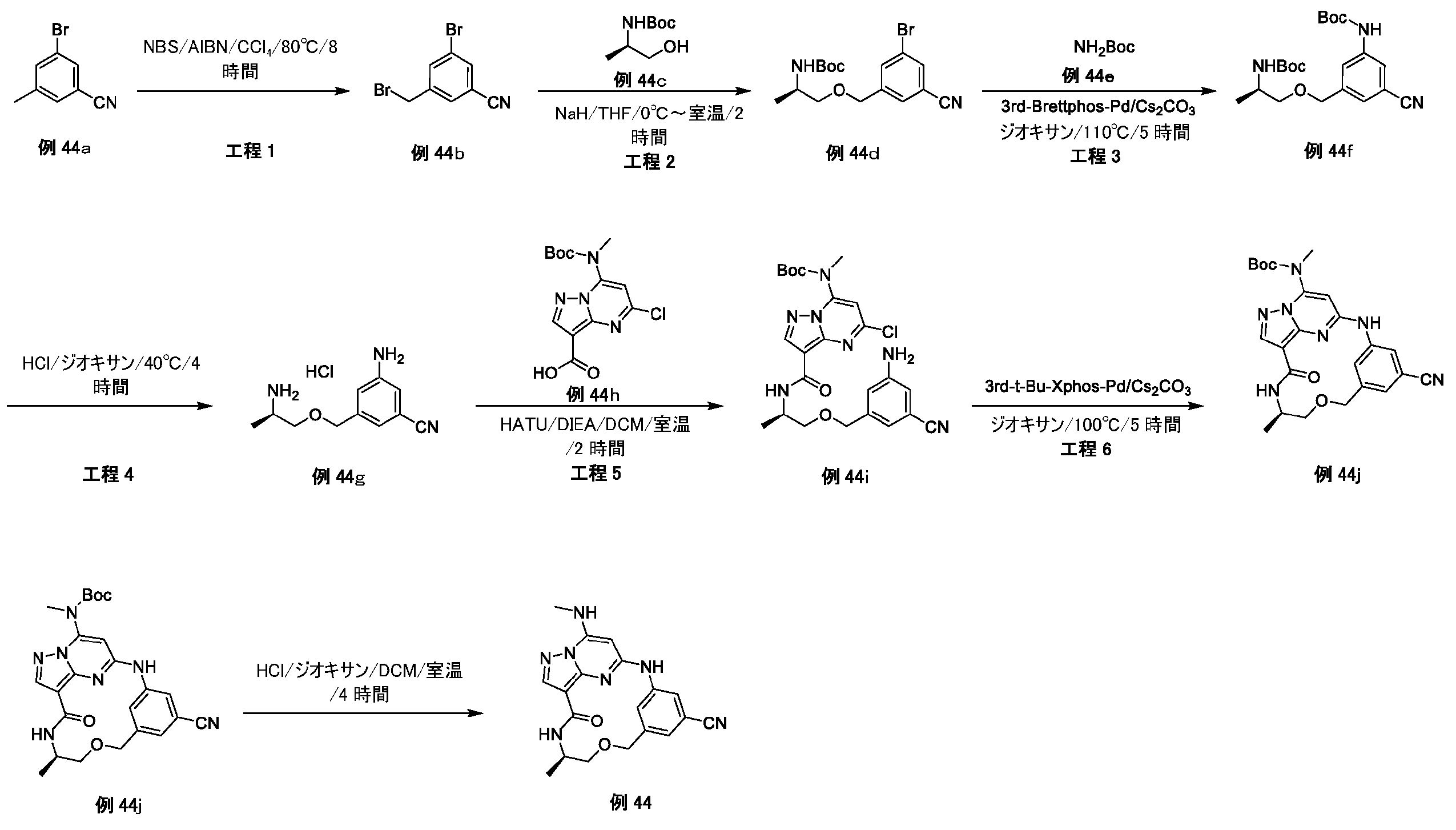
例44a(11.0g、56.1mmol、1.0当量)のCCl4(500mL)溶液に、NBS(15.0g、84.2mmol、1.5当量)およびAIBN(4.6g、28.1mmol、0.5当量)を加えた。反応混合物を、80℃で、8時間攪拌した。不溶性固体を除去した後、ろ液を濃縮し、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物である例44b(8.1g、収率53%)を黄色固体として得た。LCMS[M+1]+=275.9.
例44c(2.9g、16.4mmol、1.5当量)のTHF(40mL)溶液に、NaH(567mg、鉱油中60%、14.2mmol、1.3当量)を0℃で数回に分けて加えた。10分間攪拌した後、例44b(3.0g、10.9mmol、1.0当量)のTHF(10mL)溶液を滴加した。反応混合物を0℃~室温で2時間攪拌し、次いで溶媒を真空中で濃縮した。粗生成物をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、所望の生成物である例44d(1.9g、収率47%)を黄色固体として得た。LCMS[M+1]+=369.3.
例44d(1.8g、4.9mmol、1.0当量)のジオキサン(50mL)溶液に、Cs2CO3(3.2g、9.8mmol、2.0当量)、第3-Brettphos-Pd(442mg、0.5mmol、0.1当量)を加えた。反応混合物を、N2下、110℃で、5時間攪拌した。室温まで冷却した後、溶媒を除去し、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物である例44f(1.4g、収率71%)を黄色油状物として得た。LCMS[M+1]+=406.2.
工程4:例44g
例44f(700mg、1.7mmol、1.0当量)のHCl/ジオキサン(15mL、ジオキサン中4M)溶液を、40℃で4時間攪拌した。反応混合物を真空中で濃縮して、所望の生成物である例44g(640mg、未精製)を白色固体として得た。LCMS[M+1]+=206.2.
例44h(465mg、1.4mmol、1.0当量)のDCM(20mL)溶液に、DIEA(1.8g、14.2mmol、10.0当量)、HATU(649mg、1.7mmol、1.2当量)、および例44g(620mg、2.6mmol、1.8当量)を加えた。反応混合物を室温で2時間攪拌した。溶媒を除去し、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製し、所望の生成物である例44i(185mg、収率25%)を黄色油状物として得た。LCMS[M+1]+=514.3.
工程6:例44j
例44j(90mg、0.19mmol、1.0当量)のDCM(2mL)溶液に、HCl/ジオキサン(1mL、ジオキサン中4M)を加えた。反応混合物を室温で4時間攪拌し、次いで真空中で濃縮した。残渣をMeOH(5mL)に溶解させ、NaHCO3で塩基性化した。濃縮後、残渣を分取TLCにより精製して、所望の生成物である例44(41.5mg、収率58%)を灰白色固体として得た。LCMS[M+1]+=378.2.1H NMR(300MHz,DMSO-d6)δ 9.96(s,1H),8.90(s,1H),8.19-8.18(m,2H),8.03(d,1H),7.36(s,1H),7.30(s,1H),5.52(s,1H),4.65(d,1H),4.55(d,1H),3.95-3.93(m,1H),3.59-3.55(m,1H),3.42- 3.37(m,1H),2.95(d,3H),1.18(d,3H).
実施例45:
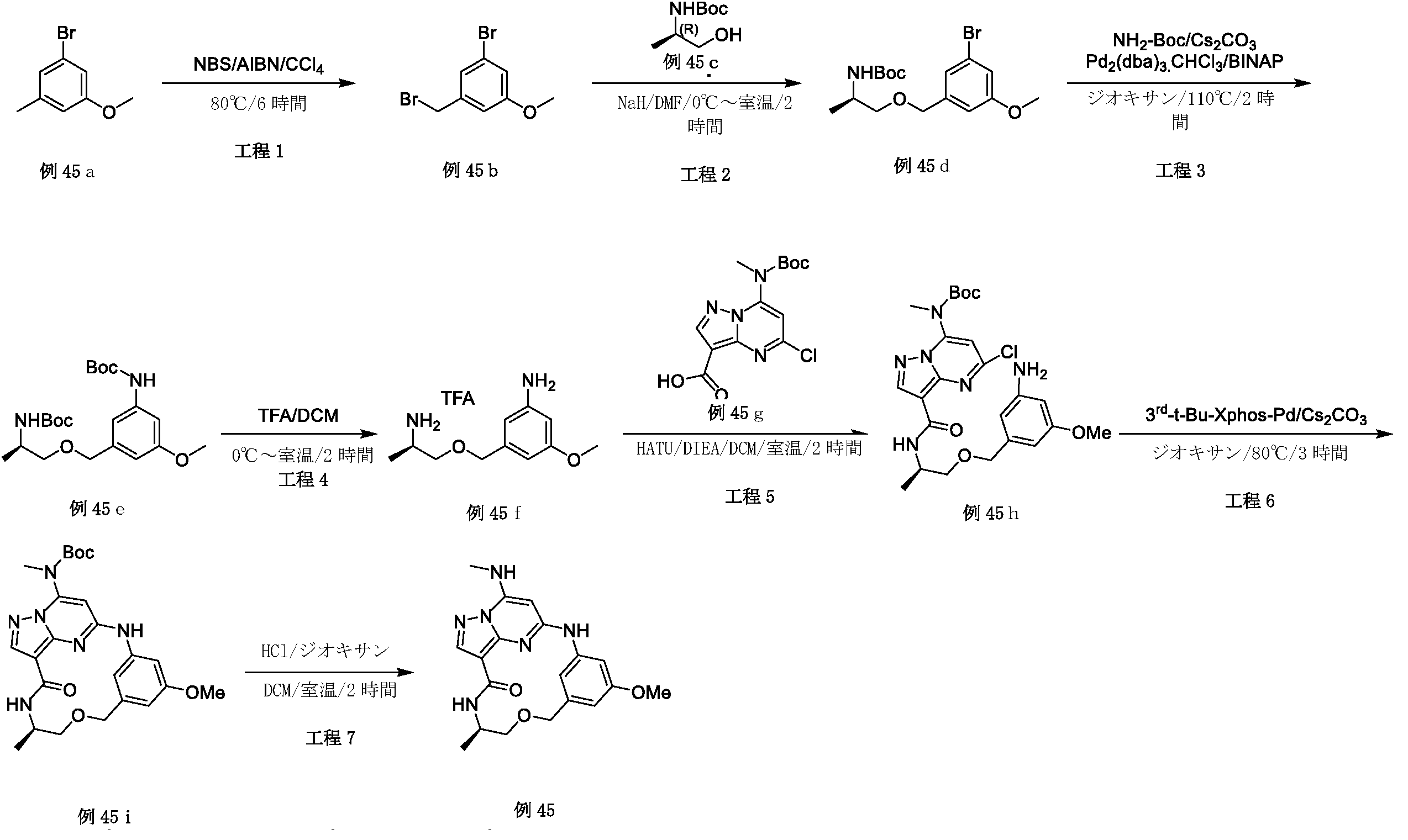
例45a(5.0g、24.9mmol、1.0当量)のCCl4(50mL)溶液に、NBS(4.9g、27.4mmol、1.1当量)およびAIBN(410mg、2.5mmol、0.1当量)を加えた。反応混合物を、80℃で、6時間攪拌した。室温まで冷却した後、溶媒を除去し、残渣をシリカゲルフラッシュカラムクロマトグラフィー(石油エーテル)により精製して、生成物である例45b(4.5g、収率60%)を白色固体として得た。LCMS[M+1]+=281.2.
例45c(1.9g、10.7mmol、1.5当量)のDMF(20mL)溶液に、NaH(340mg、鉱油中60%、8.5mmol、1.2当量)を0℃で数回に分けて加えた。混合物を同温度で30分間攪拌し、次いでDMF(20mL)中の例45b(2.0g、7.1mmol、1.0当量)を滴加した。反応混合物を室温で、2時間攪拌した。混合物を飽和NH4Cl水溶液(50mL)に注いで、これを次いでEtOAc(70mL×3)で抽出した。一つに合わせた有機層をブラインで洗浄し、Na2SO4上で乾燥させ濃縮した。粗生成物をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物である例45d(1.3g、収率50%)を白色固体として得た。LCMS[M+1]+=374.3.
例45d(1.3g、3.5mmol、1.0当量)のジオキサン(15mL)溶液に、Cs2CO3(2.3g、7.0mmol、2.0当量)、NH2-Boc(1.2g、10.5mmol、3.0当量)、BINAP(436.1mg、0.7mmol、0.2当量)、およびPd2(dba)3.CHCl3(362.3mg、0.35mmol、0.1当量)を加えた。反応混合物を、N2保護下、110℃で2時間攪拌した。室温まで冷却した後、溶媒を除去した。粗生成物をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物である例45e(980mg、収率68%)を黄色固体として得た。LCMS[M+1]+=411.3.
例45e(980mg、2.4mmol、1.0当量)のDCM(5mL)溶液に、TFA(2.5mL)を0℃で滴加した。反応混合物を室温で2時間攪拌した。溶液を真空中で濃縮して、粗生成物である例45f(1.6g、未精製、定量)を黄色油状物として得、これを精製することなく次の工程に直接使用した。LCMS[M+1]+=211.3.
例45g(392mg、1.2mmol、0.5当量)のDCM(8mL)溶液に、HATU(1.0g、2.8mmol、1.2当量)およびDIEA(1.2g、9.2mmol、4.0当量)を加えた。混合物を20分間攪拌し、次いで例45f(700mg、2.3mmol、1.0当量)を加えた。反応混合物を室温で、2時間攪拌した。溶液を真空中で濃縮して、粗生成物をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物である例45h(280mg、収率23%)を黄色固体として得た。LCMS[M+1]+=519.4.
例45h(260mg、0.5mmol、1.0当量)のジオキサン(3mL)溶液に、Cs2CO3(326mg、1.0mmol、2.0当量)および第3t-Bu-Xphos-Pd(44.1mg、0.05mmol、0.1当量)を加えた。反応混合物を、N2保護下、80℃で3時間攪拌した。固体をろ過し、ろ液を濃縮した。残渣を分取TLCにより精製し、例45i(120mg、収率50%)を黄色固体として得た。LCMS[M+1]+=483.3.
例45i(100mg、0.20mmol、1.0当量)のDCM(3mL)溶液に、HCl/ジオキサン(3mL、ジオキサン中4M)を0℃で滴加した。反応混合物を室温で2時間攪拌した。完了後、反応混合物を濃縮した。粗生成物をMeOH(2mL)に溶解し、次いでこの混合物にNaHCO3(過剰)を加え、これを室温で20分間攪拌した。混合物にDCM(20mL)を加えた後、固体をろ過し、ろ液を濃縮した。残渣を分取TLCにより精製して、例45(42.8mg、収率54%)を灰白色固体として得た。LCMS[M+1]+=383.2.1H NMR(300MHz,DMSO-d6)δ 9.62(s,1H),8.39(d,1H),8.16-8.13(m,2H),7.89(d,1H),6.49(s,2H),5.48(s,1H),4.60(d,1H),4.49(d,1H),3.92-3.89(m,1H),3.76(s,3H),3.54-3.50(m,1H),3.42-3.36(m,1H),2.93(d,3H),1.18(d,3H).
実施例46:

例46a(5.0g、32.3mmol、1.0当量)のCCl4(30mL)溶液に、BPO(2.3g、9.7lmmol、0.3当量)を80℃で加えた。5分間攪拌した後、NBS(6.9g、38.76mmol、1.2当量)を加え、80℃で16時間攪拌した。反応が完了した後、この懸濁液にEtOAc(150mL)を加え、これを飽和NaHCO3水溶液(100mL×3)で洗浄した。有機層をNa2SO4上で乾燥し、濃縮して、生成物である例46b(4.2g、収率56%)を黄色油状物として得た。LCMS[M+1]+=234.1
例46c(2.7g、15.5mmol、1.2当量)のTHF(40mL)溶液に、NaH(770mg、鉱油中60%、19.4mmol、1.5当量)を0℃で数回に分けて加えた。10分間攪拌した後、例46b(3.0g、12.9mmol、1.0当量)のTHF(5mL)溶液を滴加した。反応混合物を室温で2時間攪拌した。反応を、0℃で飽和NH4Cl水溶液(50mL)でクエンチし、EtOAc(100mL×3)で抽出した。一つに合わせた有機層をブライン(100mL×2)で洗浄し、Na2SO4上で乾燥させ、真空中で濃縮した。粗生成物をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、所望の生成物である例46d(3.1g、収率73%)を黄色油状物として得た。LCMS[M+1]+=329.3.
例46d(2.5g、7.62mmol、3.0当量)のMeOH(30mL)溶液に、N2保護下で、10%のPd/C(1.0g)を数回に分けて加えた。混合物をH2で3回脱気し、これをH2バルーン下で2時間、室温で攪拌した。固体をろ過し、ろ液を真空中で濃縮して、所望の生成物である例46e(2.8g、定量)を灰色油状物として得た。LCMS[M+1]+=299.3.
例46e(1.5g、5.03mmol、1.0当量)のDCM(25mL)溶液に、0℃でHCl/ジオキサン(5mL、ジオキサン中4M)を加えた。反応混合物を室温で2時間攪拌した。反応溶液を真空中で濃縮した。粗生成物をMeOHに溶解し、Na2CO3(過剰)を加え、室温で10分間攪拌した。固体をろ過し、ろ液を濃縮した。粗生成物をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、所望の生成物である例46f(860mg、収率87%)を黄色油状物として得た。LCMS[M+1]+=199.2.
例46g(200mg、1.01mmol、1.0当量)およびDIEA(521mg、4.04mmol、4.0当量)のDCM(5mL)溶液に、HATU(460mg、1.21mmol、1.2当量)を加えた。10分間攪拌した後、例46f(329mg、1.01mmol、1.0当量)を加え、これを室温で2時間攪拌した。混合物を真空中で濃縮した。残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物である例46h(160mg、収率31%)を黄色固体として得た。LCMS[M+1]+=507.3.
例46h(160mg、0.32mmol、1.0当量)のジオキサン(8mL)溶液に、Cs2CO3(308mg、0.96mmol、3.0当量)および第3-t-Bu-Xphos-Pd(84mg、0.096mmol、0.3当量)を加えた。反応混合物を、N2下、80℃で3時間攪拌した。反応溶液をろ過し、ろ液を真空中で濃縮した。粗生成物を分取TLCにより精製して、所望の生成物である例46i(45mg、収率30%)を黄色固体として得た。LCMS[M+1]+=471.3.
例46i(66mg、0.14mmol、1.0当量)のDCM(4mL)溶液に、0℃でHCl/ジオキサン(2mL、ジオキサン中4mol/L)を加え、これを室温で2時間攪拌した。反応溶液を真空中で濃縮した。粗生成物をMeOHに溶解し、この混合物にNa2CO3(過剰)を加え、これを室温で10分間攪拌した。固体をろ過し、ろ液を濃縮した。粗生成物を分取TLCにより精製して、所望の生成物である例46(20mg、収率36%)を黄色固体として得た。LCMS[M+1]+=371.3.1H NMR(300MHz,DMSO-d6)δ 9.79(s,1H),8.46(s,1H),8.31(d,1H),8.16(s,1H),7.93-8.01(m,1H),6.66-6.75(m,2H),5.49(s,1H),4.60(d,1H),4.50(d,1H),3.87-3.90(m,1H),3.55(dd,1H),3.44-3.38(m,1H),2.93(d,3H),1.18(d,3H).
実施例47:

例47b(525mg、3.0mmol)のTHF(15mL)溶液に、NaH(172mg、鉱油中60%、4.5mmol)を0℃で加えた。反応混合物を室温に温め、0.5時間攪拌した。次いで、例47a(741mg、3.0mmol)を加えた。得られた混合物を室温で6時間攪拌した。混合物を、NH4Cl水溶液によりクエンチし、次いでEtOAcにより抽出し、無水Na2SO4上で乾燥させた。溶液を減圧下で濃縮し、シリカゲルカラムクロマトグラフィーにより精製して、例47c(200mg、収率20%)を黄色固体として得た。LCMS[M-174]+=167.0.
例47c(200mg、0.58mmol)および10%のPd/C(30mg)のMeOH(5mL)中の混合物を、室温で2時間、1気圧のH2下で攪拌した。次いで、混合物をろ過し、ろ液を減圧下で濃縮して、未精製の例47d(200mg未精製、収率:約100%)を黄色固体として得、これを次の工程で直接使用した。LCMS[M-174]+=137.1
例47d(170mg、0.55mmol)のDCM(5.0mL)溶液に、TFA(1.0mL)を加え、これを室温で2時間攪拌した。混合物を濃縮して、粗生成物である例168g(403.5mg未精製、収率:約100%)を黒色油状物として得た。
例47e(403mg未精製、0.61mmol)、例47f(197mg、0.61mmol)、TEA(900mg、9.0mmol)のDCM(10mL)溶液に、HATU(230mg、0.605mmol)を加えた。反応混合物を室温で、2時間攪拌した。次いで、反応混合物にDCM(40mL)を加え、これをブライン(20mL×2)で洗浄し、無水Na2SO4上で乾燥して、濃縮した。残渣をシリカゲルカラムクロマトグラフィーにより精製して、所望の生成物である例47g(200mg、収率64%)を褐色固体として得た。LCMS[M+1]+=520.2
例47g(200mg、0.39mmol)、Cs2CO3(190mg、0.59mmol)のジオキサン(10.0mL)中の混合物に、第3-t-Bu-Xphos-Pd(35mg、0.039mmol)を加えた。混合物を、N2で3回脱気し、80℃で3時間攪拌した。次いで、反応混合物をEtOAcで希釈し、水で洗浄し、無水Na2SO4上で乾燥し、次いで減圧下で濃縮して、未精製の例47h(240mg未精製、収率:約100%)を白色固体として得、これをさらに精製することなく次の工程で使用した。LCMS[M+1]+=484.2
例47h(240mg未精製、0.49mmol)のTHF(1.4mL)溶液に、HCl/MeOH(2.0mL、6.0moL/L)を加え、これを室温で2時間攪拌した。混合物を濃縮し、残渣を、分取HPLCにより精製して、所望の生成物である例47(13.3mg、収率:2工程で7%)を白色固体として得た。LCMS[M+1]+=384.2.1H NMR(400MHz,DMSO-d6)δ 9.13(s,1H),8.57(d,1H),8.11(s,1H),8.05(d,1H),7.90(d,1H),7.71(d,1H),5.90(s,1H),4.54(d,1H),4.43(d,1H),3.95(s,3H),3.86(t,1H),3.46(dd,1H),3.28(dd,1H),2.89(d,3H),1.12(d,3H).
実施例48:
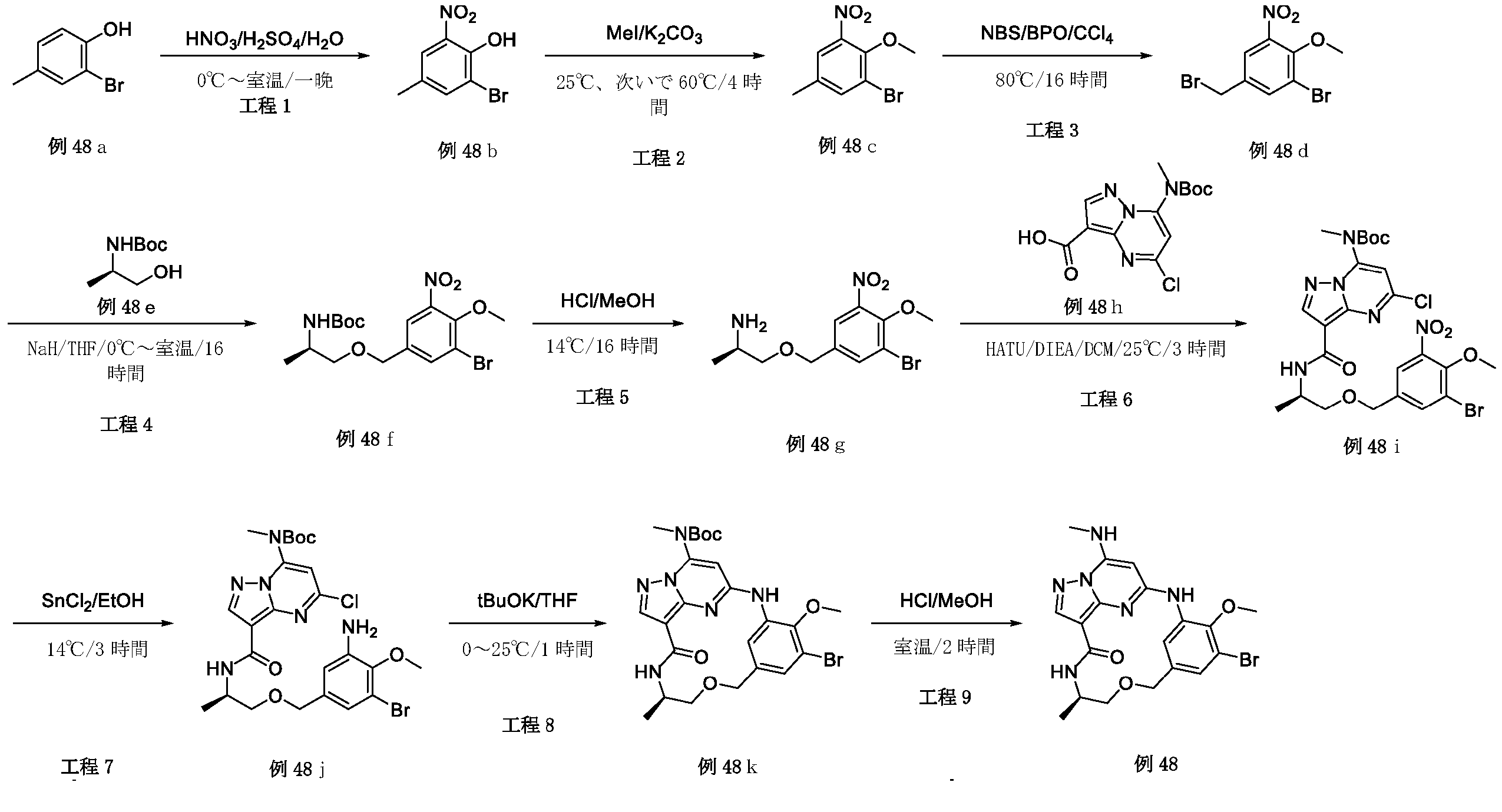
H2SO4(200mL)のH2O(620mL)溶液に、HNO3(56g、889mmol)を0℃で加えた。次いで、例48a(88g、471mmol)を加え、得られた混合物を室温で一晩攪拌した。完了後、混合物をEtOAcにより抽出し、無水Na2SO4上で乾燥させた。溶液を減圧下で濃縮し、これをシリカゲルカラムクロマトグラフィーにより精製して、例48b(81g、収率74%)を黄色固体として得た。
例48b(20g、86.6mmol)およびK2CO3(23.6g、171mmol)のDMF(70mL)中の攪拌溶液に、CH3I(17g、111.8mmol)を25℃で加えた。次いで反応混合物を60℃で4時間攪拌した。反応混合物を、EtOAcにより抽出した。有機層を、ブラインで洗浄し、無水Na2SO4上で乾燥させた。溶液を、減圧下で濃縮した。残渣をシリカゲルカラムカラムクロマトグラフィーにより精製して、例48c(22.5g、収率:定量)を黄色固体として得た。
例48c(10.0g、40.64mmol)のCCl4(10mL)溶液に、NBS(9.4g、52.84mmol)およびBPO(3.94g、16.26mmol)を加え、これを80℃で16時間攪拌した。混合物を減圧下で濃縮し、これをシリカゲルカラムクロマトグラフィーにより精製して、例48d(7.8g、収率:59%)を黄色固体として得た。
NaH(1.0g、25.1mmol)のTHF(70mL)溶液に、0℃で、例48d(6.8g、20.9mmol)および例48e(4.4g、25.1mmol)を加えた。混合物を室温で16時間攪拌した。反応混合物をH2Oでクエンチし、次いで減圧下で濃縮し、これをシリカゲルカラムクロマトグラフィーにより精製して、所望の生成物である例48f(6.25g、収率:71%)を黄色油状物として得た。LCMS[M-100+1]+=319.0/321.0.
HCl/MeOH(4M、70mL)の溶液に、例48f(6.25g、14.9mmol)を加えた。次いで反応混合物を14℃で2時間攪拌した。混合物を濃縮して、未精製の例48g(5.17g、未精製)を黄色固体として得、これをさらに精製することなく次の工程に使用した。LCMS[M+1]+=319.0/321.0
例48g(5g未精製、15.67mmol)のDCM(50mL)中の攪拌溶液に、HATU(7.68g、23.5mmol)、DIEA(4.57g、47.0mmol)および例48h(7.68g、23.5mmol)を加えた。混合物を25℃で3時間攪拌した。次いで反応混合物を減圧下で濃縮し、これをシリカゲルカラムクロマトグラフィーにより精製して、所望の生成物である例48i(8.4g、収率:85%)を黄色固体として得た。LCMS[M+1]+=627.1/629.1.
例48i(1g、1.59mmol)のEtOH(10mL)溶液に、SnCl2(0.91g、4.78mmol)を加え、これを14℃で2時間攪拌した。混合物を濃縮し、シリカゲルカラムクロマトグラフィーにより精製して、所望の生成物である例48j(1g、未精製)を黄色固体として得た。LCMS[M+1]+=597.1/599.1.
例232i(700mg、未精製)のTHF(30mL)溶液に、t-BuOK(394mg、3.51mmol)を0℃で加えた。次いで混合溶液を25℃で1時間攪拌した。混合物を濃縮し、残渣をシリカゲルカラムクロマトグラフィーにより精製し、所望の生成物である例48k(450mg、収率:69%)を黄色油状物として得た。LCMS[M+1]+=561.1/563.1.
例48k(100mg、0.34mmol)のMeOH(1mL)溶液に、HCl/MeOH(1.0mL、6.0moL/L)を加え、これを室温で2時間攪拌した。混合物を濃縮し、残渣を、分取HPLCにより精製して、所望の生成物である例48(36.5mg、収率:44%)を白色固体として得た。LCMS[M+1]+=461.1.1H NMR(400MHz,DMSO-d6)δ 9.17(s,1H),8.51(s,1H),8.16(s,1H),8.13(s,1H),7.92(d,1H),7.17(d,1H),6.03(s,1H),4.54(d,1H),4.42(d,1H),3.89-3.87(m,1H),3.77(s,3H),3.49(d,1H),3.33(d,1H),2.92(d,3H),1.14(d,3H).
実施例49:
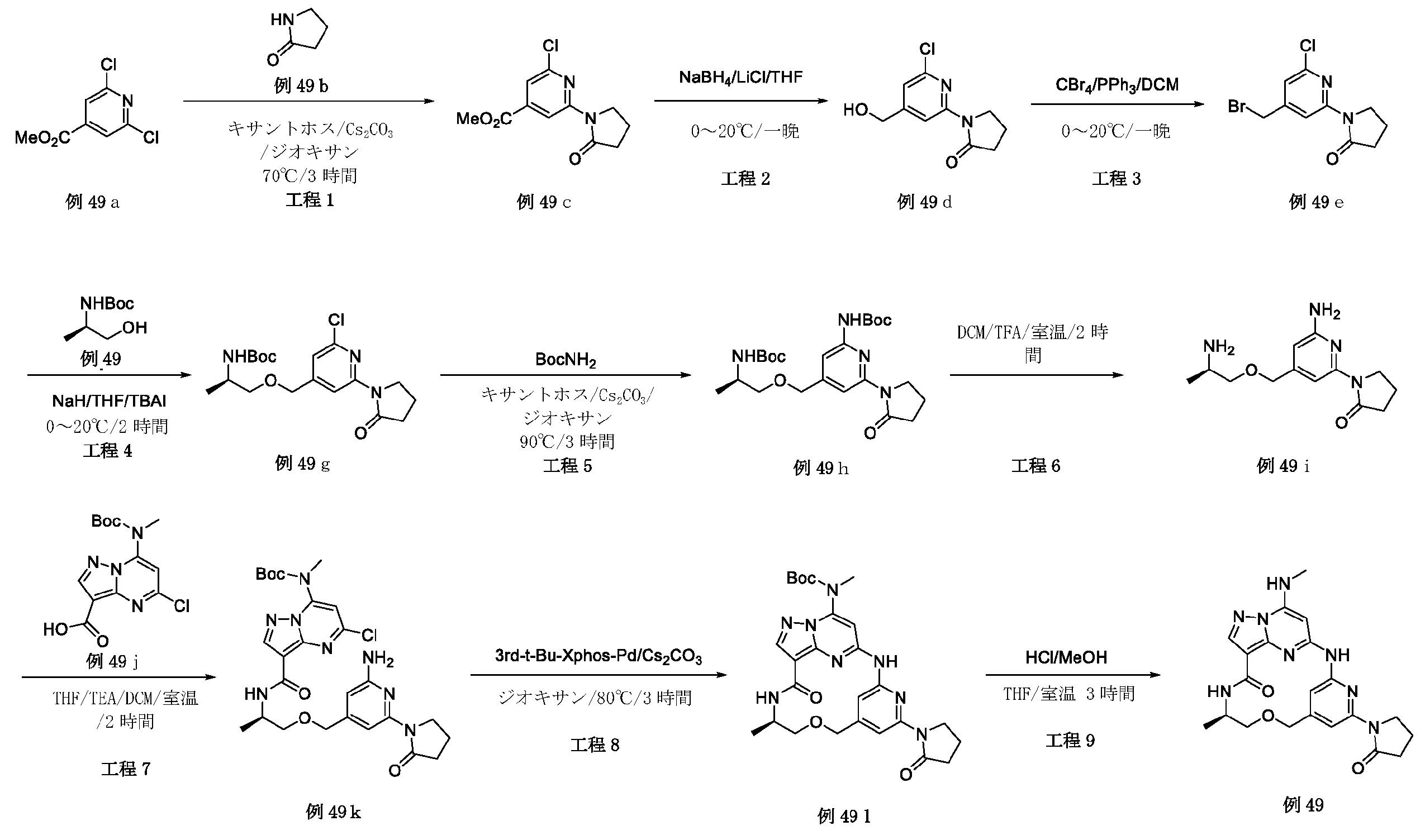
例49a(2.04g、10.0mmol)、例49b(850mg、10.0mmol)、およびCs2CO3(4.89g、15.0mmol)のジオキサン(30mL)溶液に、Pd2(dba)3(458mg、0.5mmol)、およびキサントホス(298mg、0.5mmol)を加えた。混合物を、N2で3回脱気し、70℃で3時間攪拌した。次いで、反応混合物を、EtOAcにより希釈し、水で洗浄し、無水Na2SO4上で乾燥し、次いで減圧下で濃縮した。残渣をシリカゲルカラムクロマトグラフィーにより精製して、所望の生成物である例49c(1.9g、収率:75%)を白色固体として得た。LCMS[M+1]+=255.0
例49c(1.1g、4.33mmol)のTHF(30mL)溶液に、NaBH4(165mg、4.33mmol)およびLiCl(1.3g、34.64mmol)を0℃で加えた。混合物を、N2で3回脱気し、20℃で一晩攪拌した。次いで反応混合物を、水(1.2mL)の0℃での添加によりクエンチした。得られた溶液を、室温で、NaOH水溶液(15%、3.6mL)、次いでEtOAc(1.2mL)で、希釈した。固体をろ別した。得られたろ液を減圧下で濃縮し、これをシリカゲルカラムクロマトグラフィーにより精製して、所望の生成物である例49d(400mg、収率:41%)を黄色固体として得た。LCMS[M+1]+=227.0
例49d(400mg、1.77mmol)のDCM(5mL)溶液に、PPh3(696mg、2.66mmol)を加えた。混合物を0℃に冷却し、次いで、CBr4(701mg、2.12mmol)のDCM(5mL)溶液を滴加した。加えた後、反応混合物を20℃で一晩攪拌した。次いでこの溶液を減圧下で濃縮し、シリカゲルカラムクロマトグラフィーにより精製して、所望の生成物である例49e(460mg、収率:90%)を黄色固体として得た。LCMS[M+1]+=288.9.
例49e(460mg、1.59mmol)および例49f(332mg、1.89mmol)のTHF(10mL)溶液に、NaH(87mg、鉱油中60%、2.18mmol)およびTBAI(60mg、0.16mmol)を0℃で加えた。次いで反応混合物を、20℃に温め、2時間攪拌した。次いで、反応を、NH4Cl水溶液(10mL)の添加によりクエンチし、これをEtOAcで3回抽出した。一つに合わせた有機層を無水Na2SO4上で乾燥させ、次いで減圧下で濃縮した。残渣をシリカゲルカラムカラムクロマトグラフィーにより精製して、所望の生成物である例49g(520mg、収率:85%)を黄色油状物として得た。LCMS[M+1]+=406.1.
例49g(520mg、1.35mmol)、NH2Boc(224mg、1.91mmol)、Cs2CO3(625mg、1.92mmol)のジオキサン(10mL)溶液に、Pd2(dba)3(114mg、0.12mmol)およびキサントホス(76mg、0.13mmol)を加えた。混合物を、N2で3回脱気し、次いで90℃で一晩攪拌した。次いで、反応混合物をEtOAcで希釈し、水で洗浄し、無水Na2SO4上で乾燥し、次いで減圧下で濃縮した。残渣をシリカゲルカラムクロマトグラフィーにより精製して、所望の生成物である例49h(650mg未精製、収率:定量)を白色固体として得た。LCMS[M+1]+=465.2.
例49h(410mg、0.88mmol)のDCM(8mL)溶液に、TFA(2mL)を加え、これを室温で2時間攪拌した。混合物を濃縮して、粗生成物である例49i(350mg、未精製、収率:定量)を黒色油状物として得た。LCMS[M+1]+=265.1.
例49i(350mg、0.76mmol)、例49j(248mg、0.76mmol)、およびTEA(760mg、7.6mmol)のDCM(15mL)溶液に、HATU(289mg、0.76mmol)を加えた。反応混合物を室温で、2時間攪拌した。次いで、DCM(40mL)を反応混合物に加え、これをブライン(20mL×2)で洗浄し、無水Na2SO4上で乾燥して、濃縮した。残渣をシリカゲルカラムクロマトグラフィーにより精製して、所望の生成物である例49k(304mg、収率:70%)を褐色固体として得た。LCMS[M+1]+=573.2.
例49k(304mg、0.43mmol)、およびCs2CO3(260mg、0.80mmol)のジオキサン(20mL)中の混合物に、第3-t-Bu-Xphos-Pd(46.3mg、0.053mmol)を加えた。混合物を、N2で3回脱気し、次いで80℃で3時間攪拌した。次いで、反応混合物を、EtOAcで希釈し、水で洗浄し、無水Na2SO4上で乾燥し、次いで減圧下で濃縮して、未精製の例49l(200mg、粗収率:70%)を褐色固体として得、これをさらに精製することなく次の工程で使用した。LCMS[M+1]+=537.2.
例49l(200mg未精製、0.37mmol)のTHF(1.0mL)溶液に、HCl/MeOH(1.0mL、6.0moL/L)を加え、これを室温で3時間攪拌した。混合物を濃縮し、残渣を、分取HPLCにより精製して、所望の生成物である例49(7.8mg、収率:5%)を白色固体として得た。LCMS[M+1]+=437.2.1H NMR(400MHz,DMSO-d6)δ 9.95(s,1H),8.32(d,1H),8.26(s,1H),8.16(s,1H),8.00(d,1H),7.77(s,1H),5.81(s,1H),4.58(q,2H),4.00(t,2H),3.90(s,1H),3.56(d,1H),3.43(t,1H),2.90(d,3H),2.56(t,2H),2.09-1.99(m,2H),1.18(d,3H).
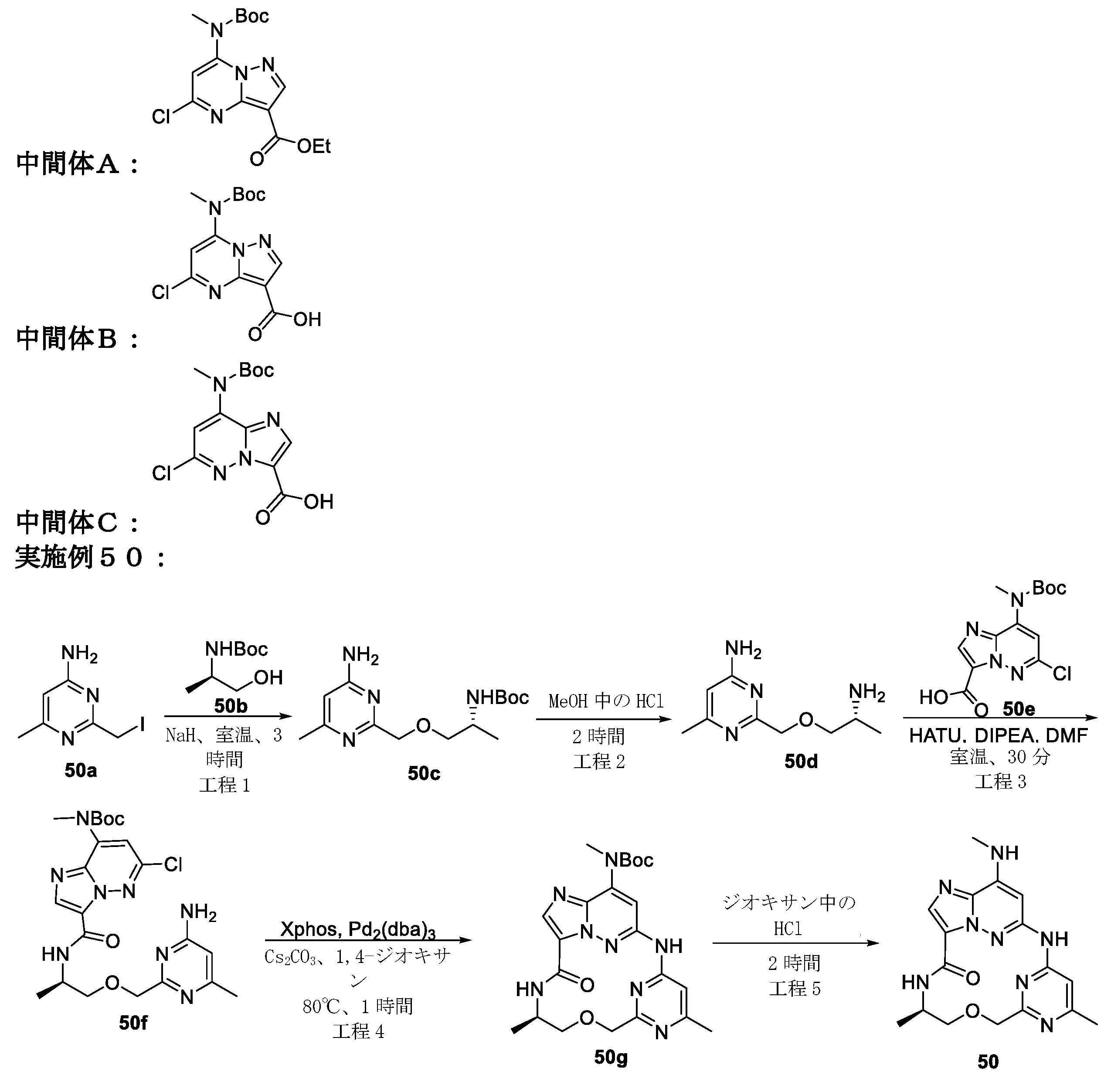
50b(90mg、0.5mmol)のTHF(4mL)溶液に、NaH(40mg、60%、2当量、1.0mmol)を0℃で加えた。20分後、50a(80mg、0.32mmol)(Studies on the Iodination of 4-Amino-2,6-dimethylpyrimidine-A Possibility of the Regiospecific Functionalization.Journal f.prakt.Chemie.Band 329,Heft 3,1987,S.400-408)を加え、次いで反応混合物を室温に温めた。室温で3時間攪拌した後、反応混合物を水中に注いで、次いで生成物をEA(2×20mL)で抽出し、Na2SO4上で乾燥し、真空中で濃縮した。残渣を、フラッシュクロマトグラフィーにより精製し、1c(56mg)を黄色固体として得た。LC-MS(ESI):m/z=297.3[M+H]+.
50cおよび塩酸(MeOH中4M)(3mL)の溶液を、室温で2時間攪拌した。溶媒を蒸発させ、粗生成物を水とDCMとの間で分配した。水層を、NaHCO3で塩基性化し、DCMで抽出した。一つに合わせた有機層を、ブラインで洗浄し、硫酸ナトリウム上で乾燥し、ろ過し、蒸発させて50dを得、これをさらに精製することなく次の工程で使用した。
50e(66mg、0.2mmol)のDMF(3mL)溶液に、HATU(76mg、0.2mmol)およびEt3N(36mg、0.36mmol)を連続的に加えた。反応混合物を、室温で0.5時間攪拌し、次いで、混合物に50d(1mLのDMF中の溶液)を加え、室温で0.5時間攪拌した。混合物を、水(10mL)で希釈し、DCM(10mL×3)で抽出した。次いで一つに合わせた有機層を水(10mL×2)およびブライン(5mL×1)で洗浄し、Na2SO4上で乾燥させ、ろ過し、真空中で濃縮した。残渣を、フラッシュクロマトグラフィーにより精製して、表題の50f(81mg、2工程で85%)を黄色固体として得た。LC-MS(ESI):m/z=505.3[M+H]+.
アルゴン下の50f(81mg、0.16mmol)の1,4-ジオキサン(4mL)溶液に、炭酸セシウム(0.13g、0.4mmol)、XPhos(24mg、0.05mmol)、およびPd2(dba)3(23mg、0.025mmol)を連続的に加えた。反応混合物を、80℃で1時間加熱した。室温まで冷却した後、混合物を水およびEtOAcで希釈した。有機層を分離し、水層をEtOAcで抽出した。次いで一つに合わせた有機層を水およびブラインで洗浄し、Na2SO4上で乾燥させ、ろ過し、真空中で濃縮した。残渣を、フラッシュクロマトグラフィーにより精製し、50g(40mg、53%)を黄色固体として得た。LC-MS(ESI):m/z=469.3[M+H]+.
50g(40mg、0.085mmol)および塩酸(1,4-ジオキサン中2M)(3mL)の溶液を、室温で2時間攪拌した。溶媒を蒸発させ、粗生成物を水とDCMとの間で分配した。水層を、NaHCO3で塩基性化し、DCMで抽出した。一つに合わせた有機層をブラインで洗浄し、Na2SO4上で乾燥させ、ろ過し、真空中で濃縮した。残渣を、フラッシュクロマトグラフィーにより精製し、例50(12mg、38%)を黄色固体として得た。LC-MS(ESI):m/z=369.2[M+H]+.1H NMR(400MHz,DMSO-d6)δ 10.11(s,1H),9.98(d,1H),7.88(s,1H),7.65(d,1H),6.63(s,1H),5.85(s,1H),4.56(d,2H),3.98-3.84(m,1H),3.68-3.63(m,1H),3.53-3.43(m,1H),2.91(d,3H),2.35(s,3H),1.23(s,3H).
実施例51:
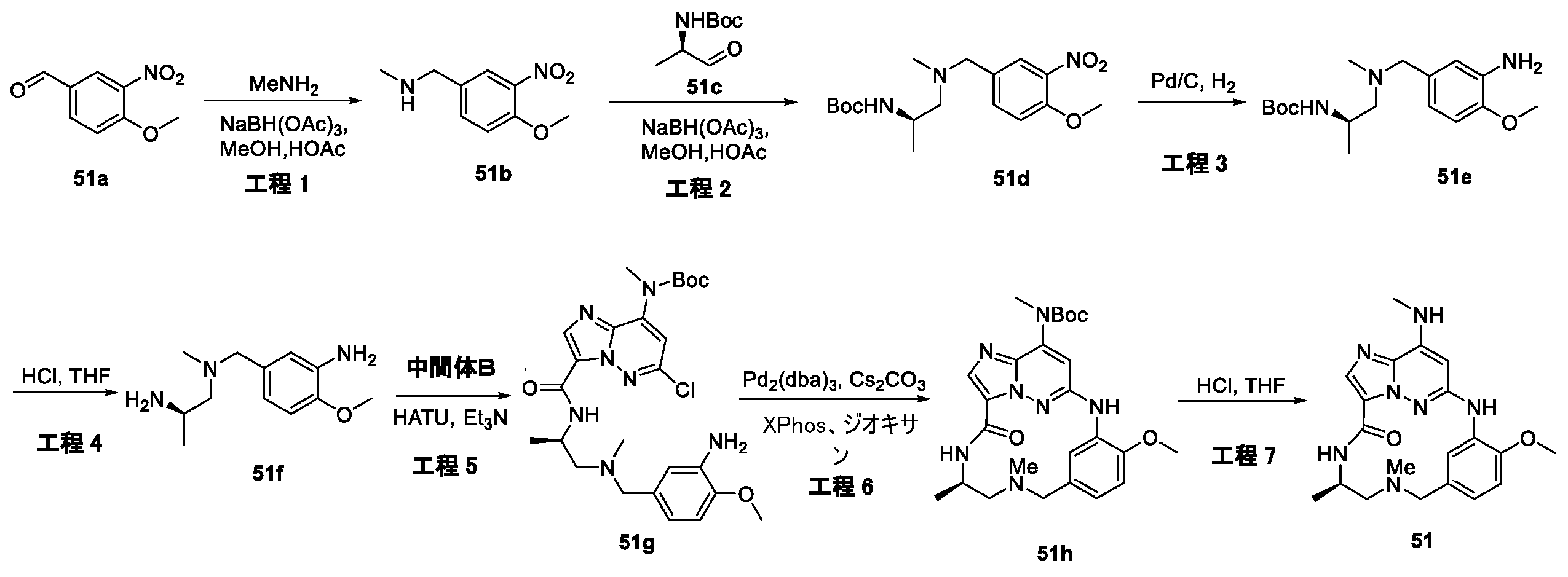
4-メトキシ-3-ニトロベンズアルデヒド(5g、27.6mmol)、CH3NH2(2.0g、64.5mmol)およびAcOH(3mL)をMeOH(50mL)に加えた。反応混合物を0℃で1時間攪拌した。NaBH(OAc)3(2.0g、64.5mmol)を加え、次いで室温で一晩攪拌した。次いで、反応混合物を水(200mL)中に注いで、酢酸エチルで抽出した。有機層をHCl(1M)で洗浄し、水層をNaHCO3で塩基性化し、酢酸エチルで抽出し、有機層を硫酸マグネシウム上で乾燥させ、ろ過し、真空中で濃縮して、表題化合物51b.(2g、44%)を得た。LC-MS(ESI):m/z=197.3[M+H]+
51b(1.6g、8.16mmol)、tert-ブチル(R)-(1-オキソプロパン-2-イル)カルバメート(1.73g、10.0mmol)およびAcOH(1mL)をメタノール(10mL)に溶解し、混合物を1時間攪拌した。NaBH(OAc)3(2.0g、64.5mmol)を加え、次いで室温で一晩攪拌した。次いで、反応混合物を水(100mL)中に注いで、水層をK2CO3で塩基性化し、酢酸エチルで抽出した。有機層を、硫酸マグネシウム上で乾燥し、ろ過し、真空中で濃縮して、表題化合物51d(1.05g、36.5%)を得た。LC-MS(ESI):m/z=354.3[M+H]+
51d(1g、0.40mmol)、Pd/C(37mg)をMeOH(20mL)に加え、混合物をH2ボール下で一晩攪拌し、この懸濁液をジクロロメタンで希釈し、セライトを通してろ過した。溶媒を除去して褐色の残渣を得、これをシリカゲルでのカラムクロマトグラフィーにより精製して、表題化合物51e(500mg、77.4%)を得た。LC-MS(ESI):m/z=324.3[M+H]+
化合物51e(500mg、1.55mmol)のTHF(10mL)溶液に、HCl/ジオキサン(5mL)を加え、混合物を室温で一晩攪拌し、次いで減圧下で濃縮し、残渣を精製することなく次の工程に直接使用した。LC-MS(ESI):m/z=224.2[M+H]+
化合物2e(345mg、1.55mmol)および8-((tert-ブトキシカルボニル)(メチル)アミノ)-6-クロロイミダゾ[1,2-b]ピリダジン-3-カルボン酸(中間体B、600mg、1.84mmol)のDMF(10mL)溶液に、TEA(3mL)およびHATU(1.52g、4mmol)を室温で加え、反応混合物を一晩攪拌して、次いで砕氷に注ぎ、酢酸エチルで抽出した。有機層を、硫酸マグネシウム上で乾燥し、ろ過し、真空中で濃縮した。残渣を、シリカゲルでのカラムクロマトグラフィーにより精製して、表題化合物51g(550mg、67%)を得た。LC-MS(ESI):m/z=533.3[M+H]+
51g(300mg、0.56mmol)の1,4-ジオキサン(10mL)溶液に、Pd2(dba)3(50mg、0.054mmol)、Cs2CO3(400mg、1.22mmol)およびXphos(30mg、0.05mmol)を加えた。反応混合物を95℃に加熱し、次いでN2下で3.5時間攪拌した。室温に冷却した後、混合物をろ過した。次いで、ろ液を50mLの水中に懸濁し、酢酸エチルで抽出し、有機層を硫酸マグネシウム上で乾燥させ、ろ過し、真空中で濃縮した。残渣を、シリカゲルでのカラムクロマトグラフィーで精製して、表題化合物51h(30mg、10.8%を得た。LC-MS(ESI):m/z=496.3[M+H]+
51h(30mg、0.06mmol)のMeOH(2mL)溶液にHCl/ジオキサン(5mL)を加え、混合物を室温で一晩攪拌し、次いで減圧下で濃縮し、残渣を砕氷に注ぎ、そして次いでK2CO3をpH>10まで加え、酢酸エチルで抽出し、有機層を硫酸マグネシウム上で乾燥し、ろ過し、真空中で濃縮した。残渣を、シリカゲルでのカラムクロマトグラフィーにより精製して、例51(5mg、21.0%)を得た。1H NMR(400MHz,DMSO-d6)δ 8.73(d,1H),8.36(s,1H),8.18(d,1H),7.78(s,1H),7.37(d,1H),6.95(d,1H),6.80(d,1H),6.17(s,1H),4.04-4.00(m,1H),3.86(s,3H),3.74(d,1H),3.12(d,1H),2.87(d,3H),2.46-2.34(m,1H),2.36(s,1H),1.96-1.92(m,1H),1.02(d,3H).LC-MS(ESI):m/z=396.3[M+H]+.
実施例52:

中間体B(0.24g、0.74mmol)のDMF(10mL)溶液に、HATU(0.31g、0.81mmol)およびEt3N(0.15g、1.5mmol)を連続的に加えた。反応混合物を、室温で0.5時間攪拌し、次いで、混合物に52a(0.19g、0.74mmol)(3mLのDMF中の溶液)を加え、室温で0.5時間攪拌した。混合物を、水で希釈し、DCMで抽出した。次いで一つに合わせた有機層をブラインで洗浄し、Na2SO4上で乾燥させ、ろ過し、真空中で濃縮した。残渣を、フラッシュクロマトグラフィーにより精製し、52c(0.33g、79%)を黄色固体として得た。LC-MS(ESI):m/z=567.2[M+H]+.
52c(0.33g、0.58mmol)のEtOH(20mL)および水(5mL)中の溶液に、鉄粉末(0.35g、6mmol)、NH4Cl(18mg、0.3mmol)を連続的に加えた。反応混合物を70℃で1時間加熱した。室温まで冷却した後、次いで、反応混合物をろ過し、溶媒を除去した。混合物を、水およびEtOAcで希釈した。有機層を分離し、水層をEtOAcで抽出した。次いで一つに合わせた有機層を水およびブラインで洗浄し、Na2SO4上で乾燥させ、ろ過し、真空中で濃縮した。残渣を、フラッシュクロマトグラフィーにより精製し、52d(0.19g、61%)を黄色固体として得た。LC-MS(ESI):m/z=537.3[M+H]+.
工程3:tert-ブチル((R,13E,14E)-34-フルオロ-36-メトキシ-7-メチル-9-オキソ-5-オキサ-2,8-ジアザ-1(5,3)-ピラゾロ[1,5-a]ピリミジナ-3(1,3)-ベンゼナシクロノナファン(benzenacyclononaphane)-17-イル)(メチル)カルバメート(52e)
52e(0.13g、0.26mmol)(4mL)のDCMおよび塩酸(1,4-ジオキサン中2M)(6mL)中の溶液を、室温で2時間攪拌した。溶媒を蒸発させ、粗生成物を水とDCMとの間で分配した。水層を、NaHCO3で塩基性化し、DCMで抽出した。一つに合わせた有機層を、ブラインで洗浄し、硫酸ナトリウム上で乾燥し、ろ過し、真空中で蒸発させた。残渣を、フラッシュクロマトグラフィーにより精製し、例52(31mg、30%)を白色固体として得た。LC-MS(ESI):m/z=401.3.[M+H]+.1H NMR(400MHz,DMSO)δ 8.88(s,1H),8.36(d,1H),8.18(d,1H),8.10(s,1H),7.80(d,1H),6.96(d,1H),5.90(s,1H),4.56(d,2H),3.89(s,3H),3.54-3.47(m,1H),3.35-3.29(m,1H),2.91(d,3H),1.30-1.19(m,1H),1.15(d,3H).
実施例53:
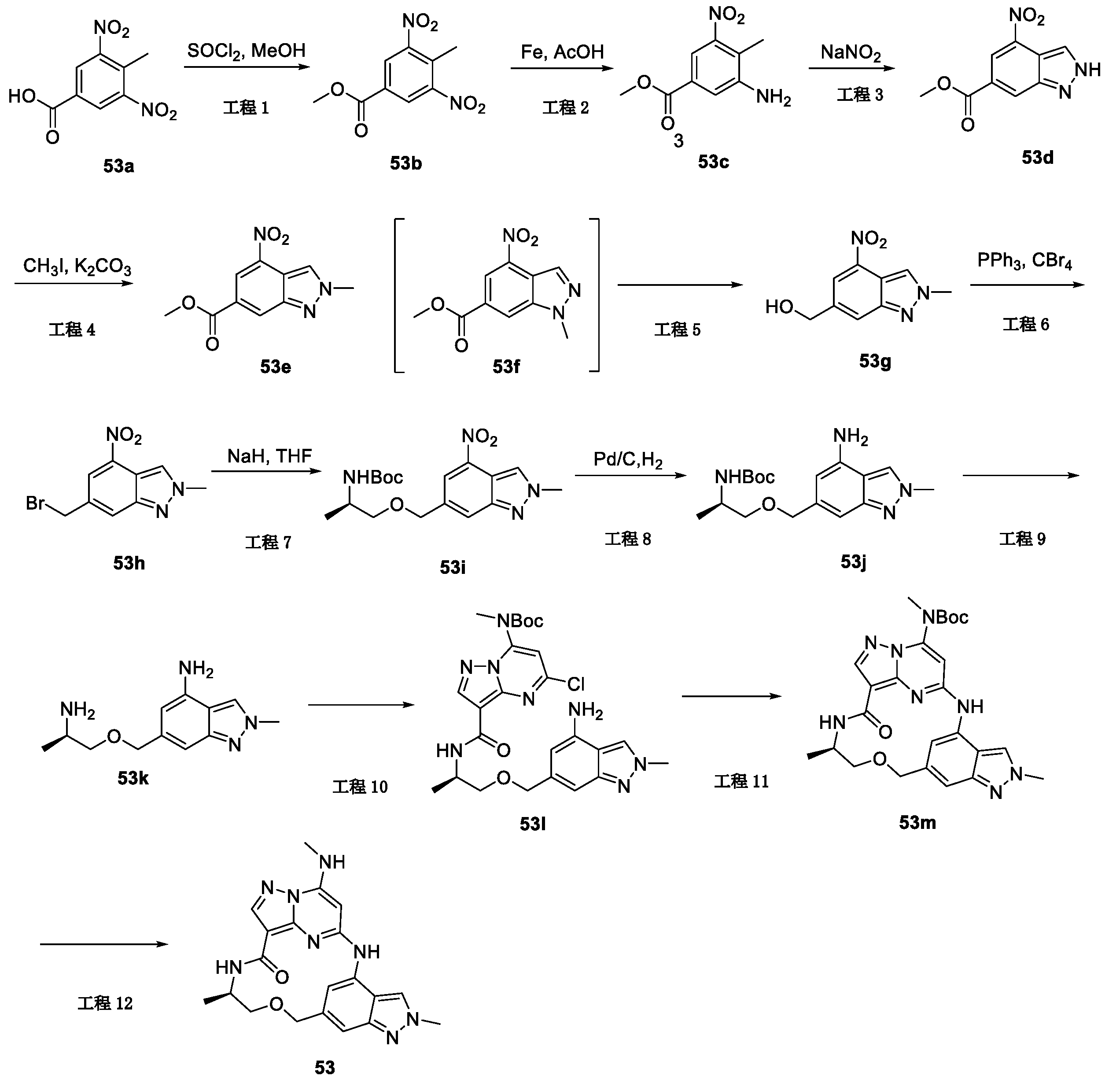
4-メチル-3,5-ジニトロ安息香酸(5g、22.1mmol)をMeOH(50mL)に加えた。SOCl2(6.6g、3.0mmol)を、0~20℃で滴加した。次いで反応混合物を60℃に加熱して、2時間攪拌した。室温に冷却した後、反応混合物を真空中で濃縮し、残渣をMTBEで洗浄し、乾燥させて53b(5g、94.2%)を得た。LC-MS(ESI):m/z=241.3[M+H]+
53b(5g、20.8mmol)を、AcOH(50mL)に加えた。Fe(1.68g、3.0mmol)をバッチで加え、次いで混合物を1時間攪拌した。次いで、反応混合物を水(200mL)中に注いで、酢酸エチルで抽出し、有機層を硫酸マグネシウム上で乾燥し、ろ過し、真空中で濃縮して、表題の53c(4g、91.6%)を得た。LC-MS(ESI):m/z=211.3[M+H]+
53c(4g、19mmol)を、AcOH(50mL)に加えた。NaNO2(10mLのH2O中で1.72g、2.5mmol)を滴加して、次いで混合物を1時間で40℃に加熱した。次いで、反応混合物を水(200mL)中に注いで、酢酸エチルで抽出し、有機層を硫酸マグネシウム上で乾燥し、ろ過し、真空中で濃縮して、表題の4d(3g、71.5%)を得た。LC-MS(ESI):m/z=222.3[M+H]+
53d(3g、13mmol)およびK2CO3(2.8g、20mmol)をDMF(50mL)に加えた。CH3I(3.1g、22mmol)を滴加し、次いで混合物を室温で1時間攪拌した。次いで、反応混合物を水(200mL)中に注いで、酢酸エチルで抽出し、有機層を硫酸マグネシウム上で乾燥し、ろ過し、真空中で濃縮した。残渣を、シリカゲルでのカラムクロマトグラフィー(PE:EA=2:1)により精製して、53e(0.7g、23%)および4f(0.7g、23%)を得た。LC-MS(ESI):m/z=236.3[M+H]+
53e(0.7g、3.0mmol)をN2下でTHF(20mL)に加えた。-60℃に冷却した後、DIBAL-H(トルエン中1mol/L、6mL、6mmol)を滴加し、次いで2時間攪拌した。次いで、反応混合物を水(100mL)中に注いで、酢酸エチルで抽出し、有機層を硫酸マグネシウム上で乾燥し、ろ過し、真空中で濃縮した。残渣を、シリカゲルでのカラムクロマトグラフィー(PE:EA=1:1)で精製し、53g(0.4g、64.5%)を得た。LC-MS(ESI):m/z=208.2[M+H]+
53g(400mg、1.92mmol)をDCM(10mL)に溶解し、CBr4(760mg、2.3mmol)およびPPh3(600mg、2.3mmol)を加えて、次いで室温で2時間攪拌した。次いで、反応混合物を水(100mL)中に注いで、酢酸エチルで抽出し、有機層を硫酸マグネシウム上で乾燥し、ろ過し、真空中で濃縮した。残渣を、シリカゲルでのカラムクロマトグラフィー(n-ヘキサン:酢酸エチル=5:1)により精製し、表題の53h(300mg、58.5%)を得た。LC-MS(ESI):m/z=271.2[M+H]+
tert-ブチル(R)-(1-ヒドロキシプロパン-2-イル)カルバメート(200mg、1.15mmol)をN2下でTHF(20mL)に加え、NaH(50mg、1.3mmol)を0℃で加え、この懸濁液を室温で0.5時間攪拌し、53h(300mg、1.10mmol)を加え、次いで混合物を室温で4時間攪拌し、次いで反応混合物を、水(100mL)に注ぎ、酢酸エチルで抽出し、有機層を硫酸マグネシウム上で乾燥し、ろ過して、真空中で濃縮した。残渣を、シリカゲルでのカラムクロマトグラフィー(n-ヘキサン:酢酸エチル=2:1)により精製し、53i(300mg、74.9%)を得た。LC-MS(ESI):m/z=365.2[M+H]+
53i(60mg、0.16mmol)およびPd/C(10mg)をMeOH(10mL)に加え、混合物をH2ボール下で一晩攪拌し、この懸濁液をジクロロメタンで希釈し、セライトを通してろ過した。溶媒を除去して褐色の残渣を得、これをシリカゲルでのカラムクロマトグラフィー(n-ヘキサン:酢酸エチル=1:1)により精製して、53j(50mg、93.56%)を得た。LC-MS(ESI):m/z=335.2[M+H]+
53j(50mg、0.15mmol)のTHF(2mL)溶液に、HCl/ジオキサン(6mol/L、1mL)を加え、混合物を、室温で一晩攪拌し、次いで減圧下で濃縮し、残渣を精製することなく次の工程に直接使用した。LC-MS(ESI):m/z=235.2[M+H]+
カルバモイル)-5-クロロピラゾロ[1,5-a]ピリミジン-7-イル)(メチル)カルバメート(53l)
53k(30mg、0.13mmol)および7-((tert-ブトキシカルボニル)(メチル)アミノ)-5-クロロピラゾロ[1,5-a]ピリミジン-3-カルボン酸(42mg、0.13mmol)のDMF(2mL)溶液に、TEA(0.1mL)およびHATU(50mg、0.13mmol)を室温で加え、反応混合物を一晩攪拌して、次いで砕氷に注ぎ、酢酸エチルで抽出した。有機層を、硫酸マグネシウム上で乾燥し、ろ過し、真空中で濃縮した。残渣を、シリカゲルでのカラムクロマトグラフィー(DCM:MeOH=20:1)で精製して、53l(20mg、28.3%)を得た。LC-MS(ESI):m/z=544.3[M+H]+
53l(20mg、0.036mmol)の1,4-ジオキサン(2mL)溶液に、Pd2(dba)3(10mg、0.01mmol)、Cs2CO3(20mg、0.06mmol)およびX-phos(6mg、0.01mmol)を加えた。反応混合物を95℃に加熱し、次いでN2下で3.5時間攪拌した。室温に冷却後、混合物をろ過し、次いでろ液を20mLの水中に懸濁し、酢酸エチルで抽出し、有機層を硫酸マグネシウム上で乾燥させ、ろ過し、真空中で濃縮した。残渣を、シリカゲルでのカラムクロマトグラフィー(DCM:MeOH=10:1)で精製して、53m(15mg、82.2%)を得た。LC-MS(ESI):m/z=507.3[M+H]+
工程12:(R,13E,14E,34E)-32,7-ジメチル-17-(メチルアミノ)-32H-5-オキサ-2,8-ジアザ-1(5,3)-ピラゾロ[1,5-a]ピリミジナ-3(4,6)-インダゾラシクロノナファン(indazolacyclononaphan)-9-オン
53m(15mg、0.03mmol)のMeOH(1mL)溶液にHCl/ジオキサン(1mL)を加え、混合物を室温で一晩攪拌し、次いで減圧下で濃縮し、残渣を砕氷に注ぎ、そして次いでK2CO3をpH>10まで加え、酢酸エチルで抽出し、有機層を硫酸マグネシウム上で乾燥し、ろ過し、真空中で濃縮した。残渣を、シリカゲルでのカラムクロマトグラフィー(DCM:MeOH=10:1)で精製して、生成物(5mg、41%)を得た。LC-MS(ESI):m/z=407.3[M+H]+
実施例54:
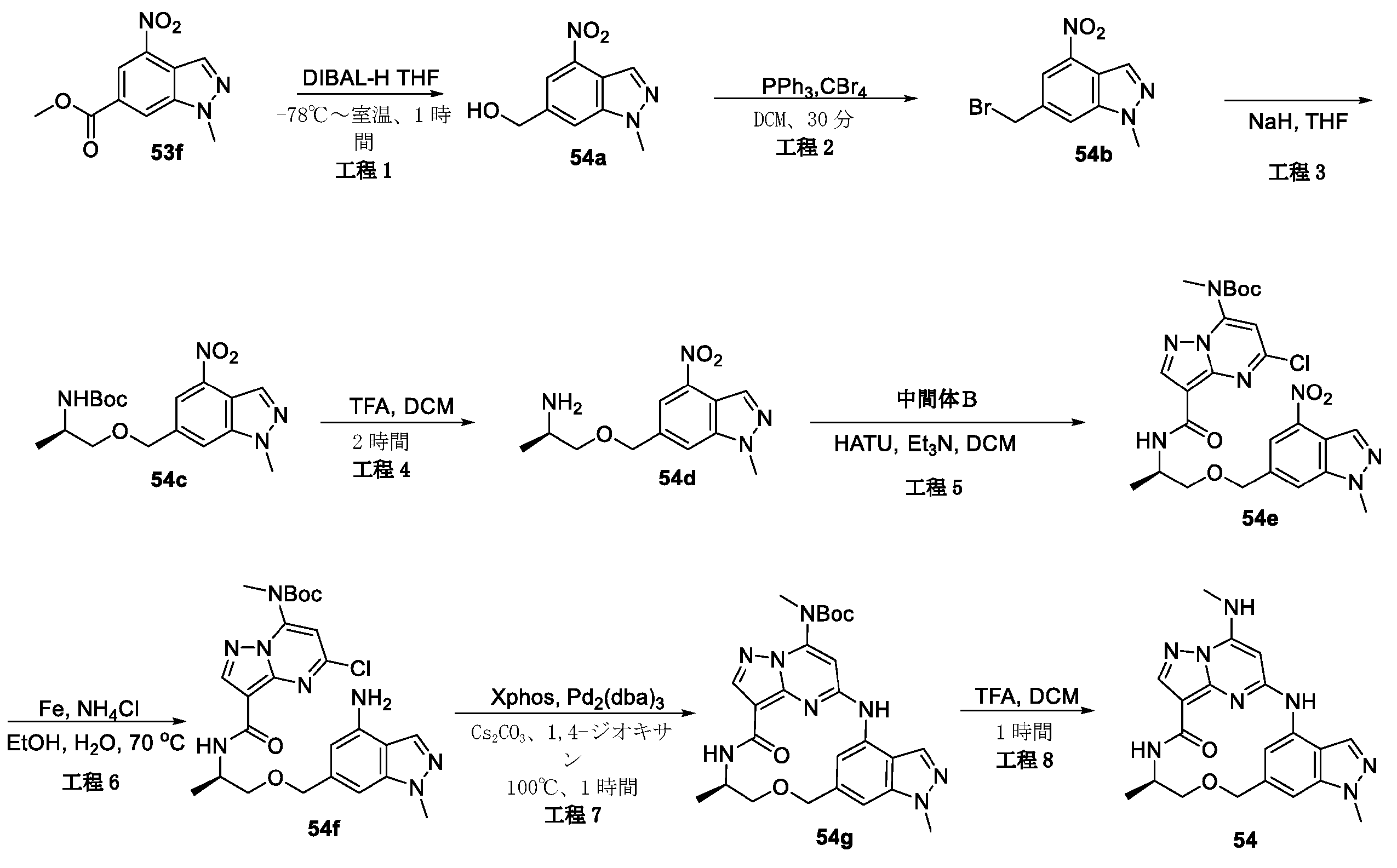
メチル53f(1.5g、6.4mmol)の無水テトラヒドロフラン(40mL)溶液を、窒素雰囲気下で-78℃に冷却した。次いで、テトラヒドロフラン(13mL)中の1Mの水素化ジイソブチルアルミニウムを滴加し、反応物を-78℃で30分間攪拌し、次いで室温に温め、1時間攪拌した。この時間後、反応物を10%のNH4Cl(20mL)で注意深く処理し、25℃未満で温度を維持した。添加が完了した後、層を分離し、水相は塩化メチレンで抽出し、一つに合わせた有機層は硫酸ナトリウム上で乾燥させた。乾燥剤をろ過により除去し、ろ液を減圧下で濃縮して、54aを黄色固体として得た:これをさらに精製することなく次の工程で使用した。
54a(1.32g、6.4mmol)およびテトラブロモメタン(3.18g、9.6mmol)のDCM(20ml)溶液に、トリフェニルホスフィン(2.52g、9.6mmol)を0℃で加え、混合物を同温度で1時間攪拌した。混合物を、減圧下で蒸発させた。残渣を、シリカゲルカラムクロマトグラフィーにより精製して、54b(1.2g、69%)を得た。1H NMR(400MHz,CDCl3)δ 8.58(s,1H),8.18(s,1H),7.77(s,1H),4.68(s,2H),4.17(s,3H).
54b(0.53g、3mmol)のTHF(20mL)溶液に、0℃でNaH(0.26g、60%、2.2当量、6.6mmol)を加えた。20分後、50b(0.68g、2.5mmol)を加え、次いで反応混合物を室温に温めた。室温で2時間攪拌した後、反応混合物を水中に注いで、次いで生成物をEA(2×50mL)で抽出し、Na2SO4上で乾燥し、真空中で濃縮した。残渣を、フラッシュクロマトグラフィーにより精製し、54c(0.63g、69%)を黄色固体として得た。
54c(0.63g、1.7mmol)のDCM(20mL)溶液にTFA(6mL)を加え、室温で2時間攪拌した。溶媒を蒸発させ、粗生成物を水(30mL)とDCM(50mL)との間で分配した。水層を、NaHCO3で塩基性化し、DCM(40mL)で抽出した。一つに合わせた有機層を、ブラインで洗浄し、硫酸ナトリウム上で乾燥し、ろ過し、蒸発させて54dを得、これをさらに精製することなく次の工程で使用した。
54d(0.45g、1.7mmol)および中間体B(0.53g、1.7mmol)のDCM(20mL)溶液に、Et3N(0.3g、3mmol)およびHATU(0.76g、2mmol)を連続的に加えた。反応混合物を、室温で0.5時間攪拌した。次いで混合物を、水(30mL)で希釈し、DCM(40mL×2)で抽出した。次いで一つに合わせた有機層をブライン(50mL)で洗浄し、Na2SO4上で乾燥させ、ろ過し、真空中で濃縮した。残渣を、フラッシュクロマトグラフィーにより精製し、表題の54e(0.82g、84%)を黄色固体として得た。LC-MS(ESI):m/z=573.3[M+H]+.
54e(0.82g、1.43mmol)のEtOH(20mL)および水(5mL)中の溶液に、Fe(0.67g、12mmol)、NH4Cl(54mg、1mmol)を連続的に加えた。反応混合物を80℃で40分間加熱した。室温まで冷却した後、次いで、反応混合物をろ過し、溶媒を除去した。混合物を、水(40mL)およびEtOAc(40mL)で希釈した。有機層を分離し、水層をEtOAc(40mL)で抽出した。次いで一つに合わせた有機層を水およびブラインで洗浄し、Na2SO4上で乾燥させ、ろ過し、真空中で濃縮した。残渣を、フラッシュクロマトグラフィーにより精製し、54f(0.53g、68%)を黄色固体として得た。LC-MS(ESI):m/z=543.3[M+H]+.
アルゴン下の5f(0.53g、0.98mmol)の1,4-ジオキサン(20mL)溶液に、炭酸セシウム(0.65g、2mmol)、XPhos(98mg、0.2mmol)、およびPd2(dba)3(46mg、0.5mmol)を連続的に加えた。反応混合物を、80℃で1時間加熱した。室温まで冷却した後、混合物を水(30mL)およびEtOAc(30mL)で希釈した。有機層を分離し、水層をEtOAc(40mL×2)で抽出した。次いで一つに合わせた有機層をブライン(40mL)で洗浄し、Na2SO4上で乾燥させ、ろ過し、真空中で濃縮した。残渣を、フラッシュクロマトグラフィーにより精製し、54g(0.31g、53%)を黄色固体として得た。LC-MS(ESI):m/z=469.3[M+H]+.
54g(0.31g、0.61mmol)のDCM(10mL)溶液にTFA(4mL)を加え、室温で1時間攪拌した。溶媒を蒸発させ、粗生成物を水(40mL)とDCM(40mL)との間で分配した。水層を、NaHCO3で塩基性化し、DCM(40mL×2)で抽出した。一つに合わせた有機層を、ブラインで洗浄し、硫酸ナトリウム上で乾燥し、ろ過し、蒸発させ、フラッシュクロマトグラフィーにより精製して、例54(0.13g、52%)を白色固体として得た。LC-MS(ESI):m/z=407.3[M+H]+.1H NMR(400MHz,DMSO-d6)δ 9.80(s,1H),8.40(d,1H),8.27(s,1H),8.24(s,1H),8.16(s,1H),7.97-7.92m,1H),7.16(s,1H),5.75(s,1H),4.75(d,1H),4.62(d,1H)),4.01(s,3H),3.58-3.53(m,1H),3.44-3.37(m,1H),2.97(d,3H),1.24(s,1H),1.14(d,3H).
実施例55:
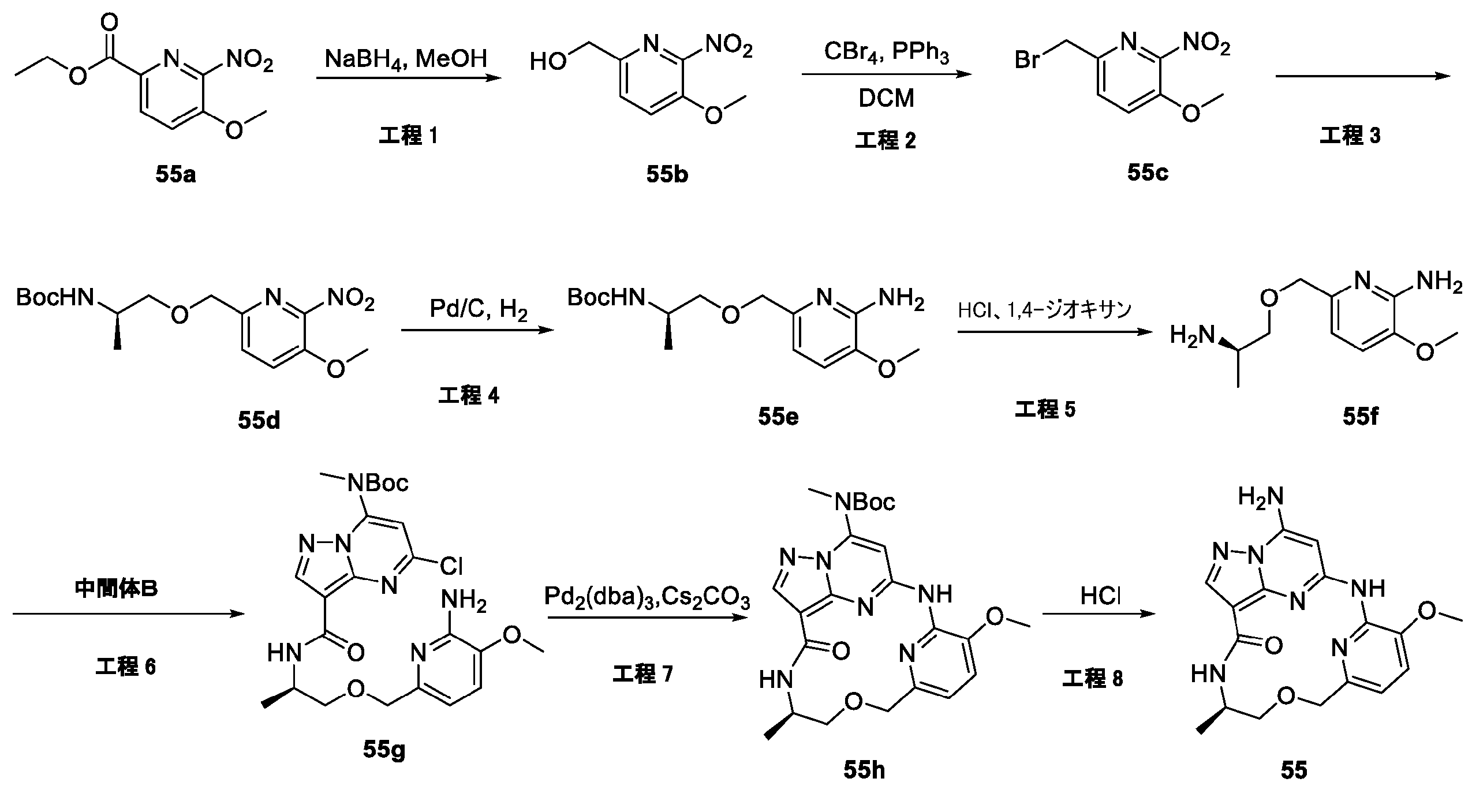
5-メトキシ-6-ニトロピコリン酸エチル(1g、4.42mmol)をMeOH(20mL)に加えた。NaBH4(2.0g、64.5mmol)を加え、次いで室温で2時間攪拌した。次いで、反応混合物を水(100mL)中に注いで、酢酸エチルで抽出し、有機層を硫酸マグネシウム上で乾燥し、ろ過し、真空中で濃縮して、55b(600mg、74%)を得た。LC-MS(ESI):m/z=195.3[M+H]+
55b(600mg、3.26mmol)をDCM(10mL)に溶解し、CBr4(1.3g、3.9mmol)およびPPh3(1.0g、3.9mmol)を加えて、次いで室温で2時間攪拌した。次いで、反応混合物を水(100mL)中に注いで、酢酸エチルで抽出し、有機層を硫酸マグネシウム上で乾燥し、ろ過し、真空中で濃縮した。残渣を、シリカゲルでのカラムクロマトグラフィー(PE:EA=5:1)により精製して、55c(350mg、43.7%)を得た。LC-MS(ESI):m/z=248.2[M+H]+
50b(200mg、1.15mmol)をN2ボール下でTHF(20mL)に加え、NaH(100mg、2.5mmol)を0℃で加え、この懸濁液を室温で0.5時間攪拌し、55c(250mg、1.00mmol)を加え、次いで混合物を室温で4時間攪拌し、次いで反応混合物を、水(100mL)に注ぎ、酢酸エチルで抽出し、有機層を硫酸マグネシウム上で乾燥し、ろ過して、真空中で濃縮した。残渣を、シリカゲルでのカラムクロマトグラフィー(PE:EA=3:1)により精製して、55d(300mg、64.6%)を得た。LC-MS(ESI):m/z=342.2[M+H]+
55d(250mg、0.73mmol)、およびPd/C(30mg)をMeOH(10mL)に加え、混合物をH2ボール下で一晩攪拌し、この懸濁液をジクロロメタンで希釈し、セライトを通してろ過した。溶媒を除去して褐色の残渣を得、これをシリカゲルでのカラムクロマトグラフィー(n-ヘキサン:酢酸エチル=3:1)により精製して、55e(200mg、88.3%)を得た。LC-MS(ESI):m/z=312.3[M+H]+
55e(200mg、1.55mmol)のTHF(10mL)溶液に、HCl/ジオキサン(4M、5mL)を加え、混合物を室温で一晩攪拌し、次いで減圧下で濃縮し、残渣を精製することなく次の工程に直接使用した。LC-MS(ESI):m/z=212.2[M+H]+
55f(100mg、0.47mmol)および中間体B(186mg、0.57mmol)のDMF(3mL)溶液に、TEA(0.5mL)およびHATU(216mg、0.57mmol)を室温で加えた。反応混合物を一晩攪拌し、次いで砕氷に注いで、酢酸エチルで抽出した。有機層を、硫酸マグネシウム上で乾燥し、ろ過し、真空中で濃縮した。残渣を、シリカゲルでのカラムクロマトグラフィー(DCM:MeOH=20:1)で精製して、55g(100mg、41%)を得た。LC-MS(ESI):m/z=520.3[M+H]+.
55g(100mg、0.19mmol)の1,4-ジオキサン(30mL)溶液に、Pd2(dba)3(30mg、0.03mmol)、Cs2CO3(200mg、0.61mmol)およびX-Phos(20mg、0.03mmol)を加えた。反応混合物を95℃に加熱し、次いでN2下で3.5時間攪拌した。室温に冷却後、混合物をろ過し、次いでろ液を20mLの水中に懸濁し、酢酸エチルで抽出し、有機層を硫酸マグネシウム上で乾燥させ、ろ過し、真空中で濃縮した。残渣を、シリカゲルでのカラムクロマトグラフィー(DCM:MeOH=10:1)で精製して、55h(80mg、84.2%)を得た。LC-MS(ESI):m/z=484.3[M+H]+
55h(50mg、0.10mmol)のMeOH(2mL)溶液にHCl/ジオキサン(2M、5mL)を加え、混合物を室温で一晩攪拌し、次いで減圧下で濃縮し、残渣を砕氷に注ぎ、そして次いでK2CO3をpH>10まで加え、酢酸エチルで抽出し、有機層を硫酸マグネシウム上で乾燥し、ろ過し、真空中で濃縮した。残渣を、シリカゲルでのカラムクロマトグラフィー(DCM:MeOH=10:1)により精製し、例55(20mg、52.2%)を得た。1H NMR(400MHz,CDCl3)δ 8.73(d,1H),8.36(s,1H),8.18(d,1H),7.78(s,1H),7.37(d,1H),6.95(d,1H),6.80(d,1H),6.17(s,1H),4.04-4.00(m,1H),3.86(s,3H),3.74(d,1H),3.12(d,1H),2.87(d,3H),2.46-2.34(m,1H),2.36(s,1H),1.96-1.92(m,1H),1.02(d,3H).LC-MS(ESI):m/z=384.3[M+H]+.
実施例56:
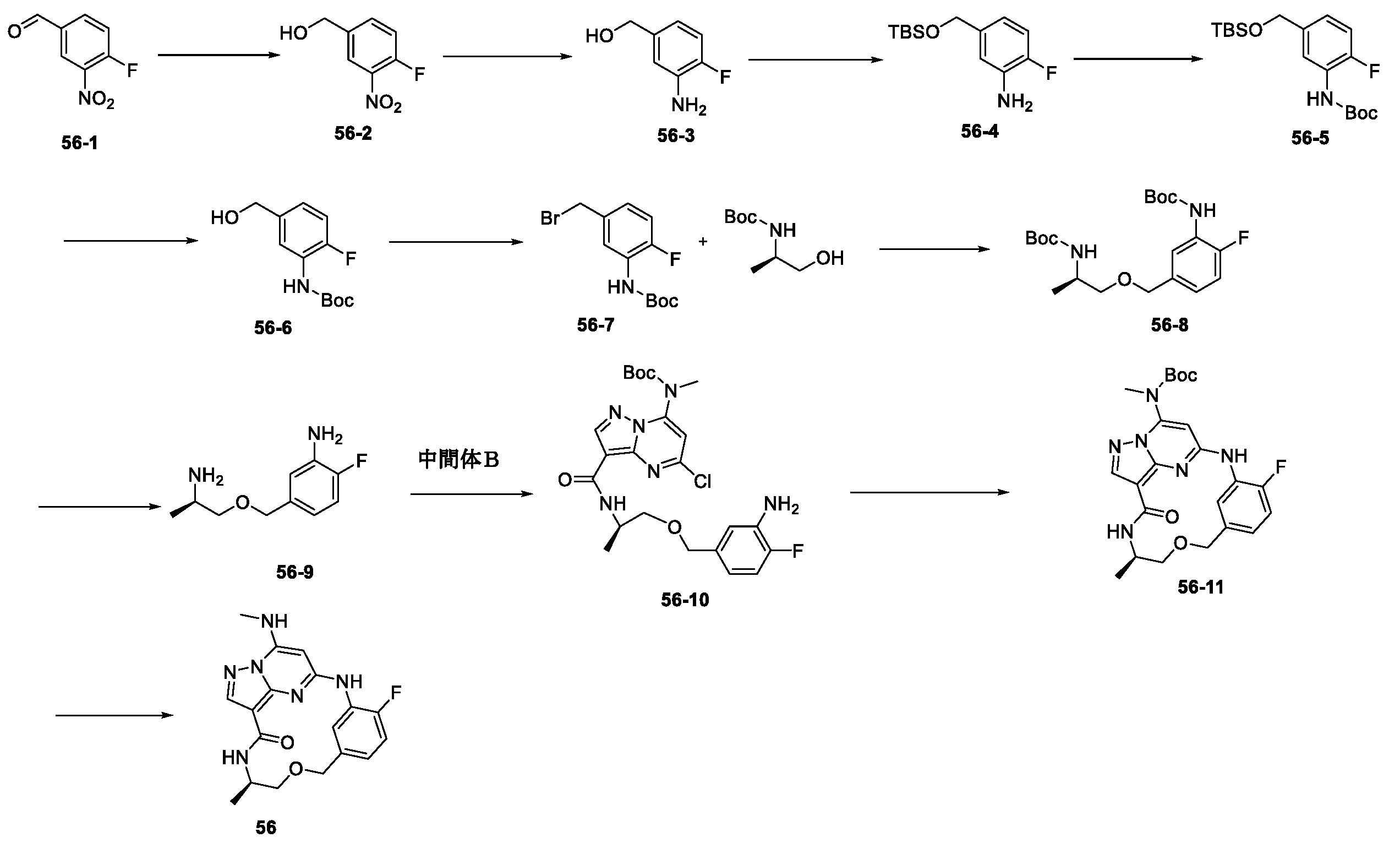
水素化ホウ素ナトリウム(1.9g、35.5mmol)を、0℃で、メタノール(100mL)中の4-フルオロ-3-ニトロ-ベンズアルデヒド(56-1)(3.0g、17.75mmol)の攪拌溶液に少しずつ加えた。室温での30分の攪拌後、メタノールを真空中で除去した。残渣を、冷水で処理し、ジクロロメタンで抽出した。一つに合わせた有機層を、ブラインで洗浄し、乾燥(Na2SO4)し、次いで真空中で蒸発させて、表題化合物(56-2)を未精製固体(2.5g、82%)として得た。LC-MS(ESI):m/z=172.1[M+H]+
(4-フルオロ-3-ニトロ-フェニル)メタノール(56-2)(1.0g、5.84mmol)をエタノール(9mL)およびH2O(3mL)に溶解し、Fe粉末(3.3g、58.4mmol)およびNH4Cl(4.06g、58.4mmol)を溶液に加え、次いで反応混合物を3時間で85℃に加熱し、室温に冷却した後、反応物をろ過し、ろ液を真空中で除去した。残渣を、フラッシュクロマトグラフィーにより精製して、表題化合物(56-3)(0.7g、80%)を白色固体として得た。LC-MS(ESI):m/z=142.2[M+H]+
(3-アミノ-4-フルオロ-フェニル)メタノール(56-3)(1.5g、10.6mmol)のDCM溶液に、TBSCl(2.4g、15.9mmol)およびイミダゾール(1.22g、18.0mmol)を0℃で加え、混合物を室温で一晩攪拌し、混合物を冷水で処理し、酢酸エチルで抽出した。一つに合わせた有機層を、ブラインで洗浄し、乾燥(Na2SO4)し、次いで残渣をフラッシュクロマトグラフィーにより精製して、表題化合物(56-4)(2.24g、83%)を得た。LC-MS(ESI):m/z=256.2[M+H]+
56-4のDCM溶液(2.24g、8.78mmol)に、Boc2O(3.8g、17.56mmol)、トリエチルアミン(2.66g、26.35mmol)およびDMAP(110mg、0.9mmol)を0℃で加え、混合物を室温で2時間攪拌し、混合物を冷水で処理し、酢酸エチルで抽出した。一つに合わせた有機層を、ブラインで洗浄し、乾燥(Na2SO4)し、次いで残渣をフラッシュクロマトグラフィーにより精製して、表題化合物(56-5)(2.1g、67%)を得た。LC-MS(ESI):m/z=356.1[M+H]+
56-5(2.0g、5.63mmol)のTHF溶液に、TBAF(2.9g、11.27mmol)を0℃で加え、混合物を室温で3時間攪拌し、混合物を冷水で処理し、酢酸エチルで抽出した。一つに合わせた有機層を、ブラインで洗浄し、乾燥(Na2SO4)し、次いで残渣をフラッシュクロマトグラフィーで精製して、表題化合物(56-6)(1.06g、78%)を褐色固体として得た。
LC-MS(ESI):m/z=242.2[M+H]+
無水ジエチルエーテル(5mL)中の四臭化炭素(2.2g、6.64mmol)を、無水ジエチルエーテル(15mL)中のtert-ブチル N-[2-フルオロ-5-(ヒドロキシメチル)フェニル]カルバメート(56-6)(0.8g、3.32mmol)およびトリフェニルホスフィン(1.74g、6.64mmol)の攪拌溶液に滴加した。混合物を濃縮する前に一晩攪拌した。ヘキサン中の酢酸エチル(0~10%)を用いたクロマトグラフィーにより、表題化合物(56-7)を淡黄色固体(0.73g、72%)として得た。LC-MS(ESI):m/z=304.2[M+H]+
カリウムtert-ブトキシド(220mg、2.0mmol)を、0℃で、tert-ブチル N-[(1R)-2-ヒドロキシ-1-メチル-エチル]カルバメート(350mg、2.0mmol)およびtert-ブチル N-[5-(ブロモメチル)-2-フルオロ-フェニル]カルバメート(56-7)(400mg、1.3mmol)のTHF(15mL)中の攪拌溶液に加え、混合物を、マイクロ波下で75℃で5分間攪拌した。混合物を、冷水で処理し、酢酸エチルで抽出した。一つに合わせた有機層を、ブラインで洗浄し、乾燥(Na2SO4)し、次いで残渣をフラッシュクロマトグラフィーで精製して、表題化合物(56-8)(40mg、7.6%)を褐色固体として得た。LC-MS(ESI):m/z=399.3[M+1]+
トリフルオロ酢酸(1mL)を、tert-ブチル N-[(1R)-2-[[3-(tert-ブトキシカルボニルアミノ)-4-フルオロ-フェニル]メトキシ]-1-メチル-エチル]カルバメート(56-8)(40mg、0.1mmol)のDCM(3mL)溶液に加え、この混合物を2時間攪拌し、混合溶液を蒸発させて乾燥し、その後、表題化合物(56-9)(18mg.90%)を褐色液体として得、これをさらに精製することなく次の工程で使用した。LC-MS(ESI):m/z=199.3[M+H]+
5-[[(2R)-2-アミノプロポキシ]メチル]-2-フルオロ-アニリン(56-9)(20mg、0.1mmol)をDMF(5mL)に溶解して、この溶液に、TCFH(42mg、0.15mmol)、1-メチルイミダゾール(41mg、0.5mmol)および中間体B(国際公開第2019023468号)(33mg、0.1mmol)を室温で加えた。室温での1時間の攪拌後、溶液混合物を、EA(30mL)で希釈し、水(2×30mL)およびブライン(30mL)で洗浄し、Na2SO4で乾燥して濃縮した。粗生成物を、フラッシュクロマトグラフィー(PE/EA=3:1)により精製して、表題化合物(56-10)(30mg、59%)を白色固体として得た。LC-MS(ESI):m/z=508.2[M+H]+
tert-ブチル N-[3-[[(1R)-2-[(3-アミノ-4-フルオロ-フェニル)メトキシ]-1-メチル-エチル]カルバモイル]-5-クロロ-ピラゾロ[1,5-a]ピリミジン-7-イル]-N-メチル-カルバメート(56-10)(30mg、0.06mmol)の1,4-ジオキサン(3mL)溶液に、Cs2CO3(40mg、0.12mmol)および第3-t-Bu-Xphos-Pd(5mg)を加えた。反応混合物を、N2下、85℃で、2時間攪拌した。室温に冷却後、溶媒を除去し、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物(56-11)(9mg、30%)を白色固体として得た。LC-MS(ESI):m/z=471.3[M+H]+
tert-ブチル((R,13E,14E)-36-フルオロ-7-メチル-9-オキソ-5-オキサ-2,8-ジアザ-1(5,3)-ピラゾロ[1,5-a]ピリミジナ-3(1,3)-ベンゼナシクロノナファン(benzenacyclononaphane)-17-イル)(メチル)カルバメート(56-11)(9mg、0.02mmol)およびトリフルオロ酢酸(0.5mL)のDCM(2mL)溶液を、室温で3時間攪拌した。溶媒を蒸発させ、粗生成物を水とDCMとの間で分配した。水層を、NaHCO3で塩基性化し、DCMで抽出した。一つに合わせた有機層を、ブラインで洗浄し、硫酸ナトリウム上で乾燥し、ろ過し、蒸発させ、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物である例56(3mg、40%)を白色固体として得た。LC-MS(ESI):m/z=371.2[M+H]+.1H NMR(400MHz,CDCl3)δ 8.68(d,1H),8.55(s,1H),8.43-8.39(m,1H),8.11(s,1H),7.08(dd,1H),6.88-6.81(m,1H),6.52-6.46(m,1H),5.42(s,1H),4.58(dd,2H),4.21-4.16(m,1H),3.63-3.57(m,1H),3.51-3.47(m,1H),3.11(d,3H),1.34(d,3H).
実施例57:

6-オキソ-1H-ピリダジン-3-カルボン酸メチル(57-1)(15g、97.3mmol)のAcOH(200mL)溶液に、AcOK(34g、346mmol)を-10℃で加え、混合物を20分間攪拌し、臭素(34.2g、214mmol)を20分間にわたって滴加した。80℃でさらに1時間攪拌した後、残渣を冷水で処理し、ジクロロメタンで抽出した。一つに合わせた有機層を、ブラインで洗浄し、乾燥(Na2SO4)し、次いで真空中で蒸発させて、表題化合物(57-2)を未精製固体(10.5g、46.3%)として得た。LC-MS(ESI):m/z=233.1[M+H]+.
5-ブロモ-6-オキソ-1H-ピリダジン-3-カルボン酸メチル(57-2)(6g、26mmol)のDMF(30mL)溶液に、Cs2CO3(17g、51mmol)およびヨードメタン(4.4g、31mmol)を0℃で加え、混合物を室温で4時間攪拌した。残渣を、冷水で処理し、EtOAcで抽出した。一つに合わせた有機層を、ブラインで洗浄し、乾燥(Na2SO4)し、次いで真空中で蒸発させて、表題化合物(57-3)を未精製固体(4.5g、71%)として得た。LC-MS(ESI):m/z=247.0[M+H]+.
5-ブロモ-1-メチル-6-オキソ-ピリダジン-3-カルボン酸メチル(57-3)(2.5g、10.1mmol)およびフェニルメタンアミン(1.08g、10.1mmol)の1,4-ジオキサン(30mL)溶液に、Pd2(dba)3(2.78g、3.04mmol)、Cs2CO3(6.6g、20.2mmol)、およびキサントホス(3.51g、6.07mmol)を、N2雰囲気下で加え、混合物を100℃で4時間攪拌し、残渣を冷水で処理し、EtOAcで抽出した。一つに合わせた有機層を、ブラインで洗浄し、乾燥(Na2SO4)し、次いで真空中で蒸発させて、表題化合物(57-4)(1.6g、58%)を得た。LC-MS(ESI):m/z=274.0[M+H]+.
5-(ベンジルアミノ)-1-メチル-6-オキソ-ピリダジン-3-カルボン酸メチル(57-4)(2.2g、8.0mmol)のメタノール(30mL)溶液に、10%のPd/C(2.0g)を室温で加えた。混合物は、3回水素置換し、水素雰囲気下で50℃で6時間攪拌し、ろ過し、EAで洗浄し、次いで真空中で蒸発させて、表題化合物(57-5)を未精製固体(1.3g、88%)として得た。LC-MS(ESI):m/z=184.2[M+H]+.
5-アミノ-1-メチル-6-オキソ-ピリダジン-3-カルボン酸メチル(57-5)のDCM(1.3g、7.1mmol)溶液に、Boc2O(3.9g、18mmol)およびDMAP(0.87g、7.1mmol)を0℃で加え、混合物を60℃で4時間攪拌し、混合物を冷水で処理し、酢酸エチルで抽出した。一つに合わせた有機層を、ブラインで洗浄し、乾燥(Na2SO4)し、次いで残渣をフラッシュクロマトグラフィーにより精製して、表題化合物(57-6)(2.3g、85%)を得た。LC-MS(ESI):m/z=384.1[M+H]+
水素化ホウ素ナトリウム(0.41g、11.0mmol)を、メタノール(20mL)中の5-[ビス(tert-ブトキシカルボニル)アミノ]-1-メチル-6-オキソ-ピリダジン-3-カルボン酸メチル(57-6)(2.1g、5.48mmol)の攪拌溶液に0℃で少しずつ加えた。2時間後、メタノールを真空中で除去した。混合物を、冷水で処理し、ジクロロメタンで抽出した。一つに合わせた有機層を、ブラインで洗浄し、乾燥(Na2SO4)し、次いで真空中で蒸発させ、ヘキサン中の酢酸エチル(0~10%)でのクロマトグラフィーにより、表題化合物(57-7)を淡黄色固体(1.6g、82%)として得た。
LC-MS(ESI):m/z=356.3[M+H]+
無水DCM(30mL)中の四臭化炭素(3.0g、9.0mmol)を、DCM(15mL)中のtert-ブチル N-tert-ブトキシカルボニル-N-[6-(ヒドロキシメチル)-2-メチル-3-オキソ-ピリダジン-4-イル]カルバメート(57-7)(1.6g、4.5mmol)およびトリフェニルホスフィン(2.36g、9.0mmol)の攪拌溶液に滴加した。混合物を濃縮する前に一晩攪拌した。ヘキサン中の酢酸エチル(0~10%)を用いたクロマトグラフィーにより、表題化合物(57-8)を淡黄色固体(1.2g、64%)として得た。
LC-MS(ESI):m/z=418.2[M+H]+
水素化ナトリウム(95mg、3.94mmol)を、0℃でTHF(15mL)中のtert-ブチル N-[(1R)-2-ヒドロキシ-1-メチル-エチル]カルバメート(0.69g、3.94mmol)の攪拌溶液に少しずつ加え、混合物を0℃で20分間攪拌し、次いで混合物に、0℃でtert-ブチル N-tert-ブトキシカルボニル-N-[6-(ヒドロキシメチル)-2-メチル-3-オキソ-ピリダジン-4-イル]カルバメート(57-8)(1.1g、2.63mmol)を加え、室温で4時間攪拌後、混合物を、冷水で処理し、酢酸エチルで抽出した。一つに合わせた有機層を、ブラインで洗浄し、乾燥(Na2SO4)し、次いで残渣をフラッシュクロマトグラフィーで精製して、表題化合物(57-9)(0.9g、80%)を褐色固体として得た。LC-MS(ESI):m/z=413.1[M+1]+
トリフルオロ酢酸(2mL)を、tert-ブチル N-[(1R)-2-[[5-(tert-ブトキシカルボニルアミノ)-1-メチル-6-オキソ-ピリダジン-3-イル]メトキシ]-1-メチル-エチル]カルバメート(57-9)(0.6g、1.5mmol)のDCM(5mL)溶液に加え、この混合物を2時間攪拌し、混合溶液を蒸発させて乾燥し、その後、表題化合物(57-10)(0.28g.91%)を褐色液体として得、これをさらに精製することなく次の工程で使用した。LC-MS(ESI):m/z=213.2[M+H]+
4-アミノ-6-[[(2R)-2-アミノプロポキシ]メチル]-2-メチル-ピリダジン-3-オン(57-10)(200mg、0.94mmol)および中間体B(308mg、0.94mmol)をDMF(5mL)に溶解し、この溶液に、N,N,N’,N’-テトラメチルクロロホルムアミジニウムヘキサフルオロホスフェート(397mg、1.41mmol)および1-メチルイミダゾール(387mg、4.71mmol)を室温で加えた。室温での1時間の攪拌後、溶液混合物を、EA(30mL)で希釈し、水(2×30mL)およびブライン(30mL)で洗浄し、Na2SO4で乾燥して濃縮した。粗生成物を、フラッシュクロマトグラフィー(PE/EA=3:1)により精製して、表題化合物(57-11)(210mg、43%)を白色固体として得た。
LC-MS(ESI):m/z=521.1[M+H]+
tert-ブチル N-[3-[[(1R)-2-[(5-アミノ-1-メチル-6-オキソ-ピリダジン-3-イル)メトキシ]-1-メチル-エチル]カルバモイル]-5-クロロ-ピラゾロ[1,5-a]ピリミジン-7-イル]-N-メチル-カルバメート(57-11)(200mg、0.38mmol)の1,4-ジオキサン(20mL)溶液に、Cs2CO3(250mg、0.77mmol)および第3-t-Bu-Xphos-Pd(20mg)を加えた。反応混合物を、N2下、85℃で、2時間攪拌した。室温に冷却後、溶媒を除去し、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物(57-12)(130mg、70%)を白色固体として得た。
LC-MS(ESI):m/z=485.0[M+H]+
tert-ブチル((R,13E,14E,34E)-31,7-ジメチル-36,9-ジオキソ-31,36-ジヒドロ-5-オキサ-2,8-ジアザ-1(5,3)-ピラゾロ[1,5-a]ピリミジナ-3(5,3)-ピリダジナシクロノナファン(pyridazinacyclononaphane)-17-イル)(メチル)カルバメート(57-12)(120mg、0.25mmol)およびトリフルオロ酢酸(1mL)のDCM(4mL)溶液を、室温で3時間攪拌した。溶媒を蒸発させ、粗生成物を水とDCMとの間で分配した。水層を、NaHCO3で塩基性化し、DCMで抽出した。一つに合わせた有機層を、ブラインで洗浄し、硫酸ナトリウム上で乾燥し、ろ過し、蒸発させ、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物(50mg、53%)を白色固体として得た。LC-MS(ESI):m/z=385.2[M+H]+.1H NMR(400MHz,DMSO-d6)δ 9.75(s,1H),8.48(s,1H),8.23(s,1H),8.17-8.07(m,2H),6.30(s,1H),4.45(dd,2H),4.05-3.96(m,1H),3.71(s,3H),3.68-3.62(m,1H),3.58-3.53(m,1H),2.92(d,3H),1.18(d,3H).
実施例58:
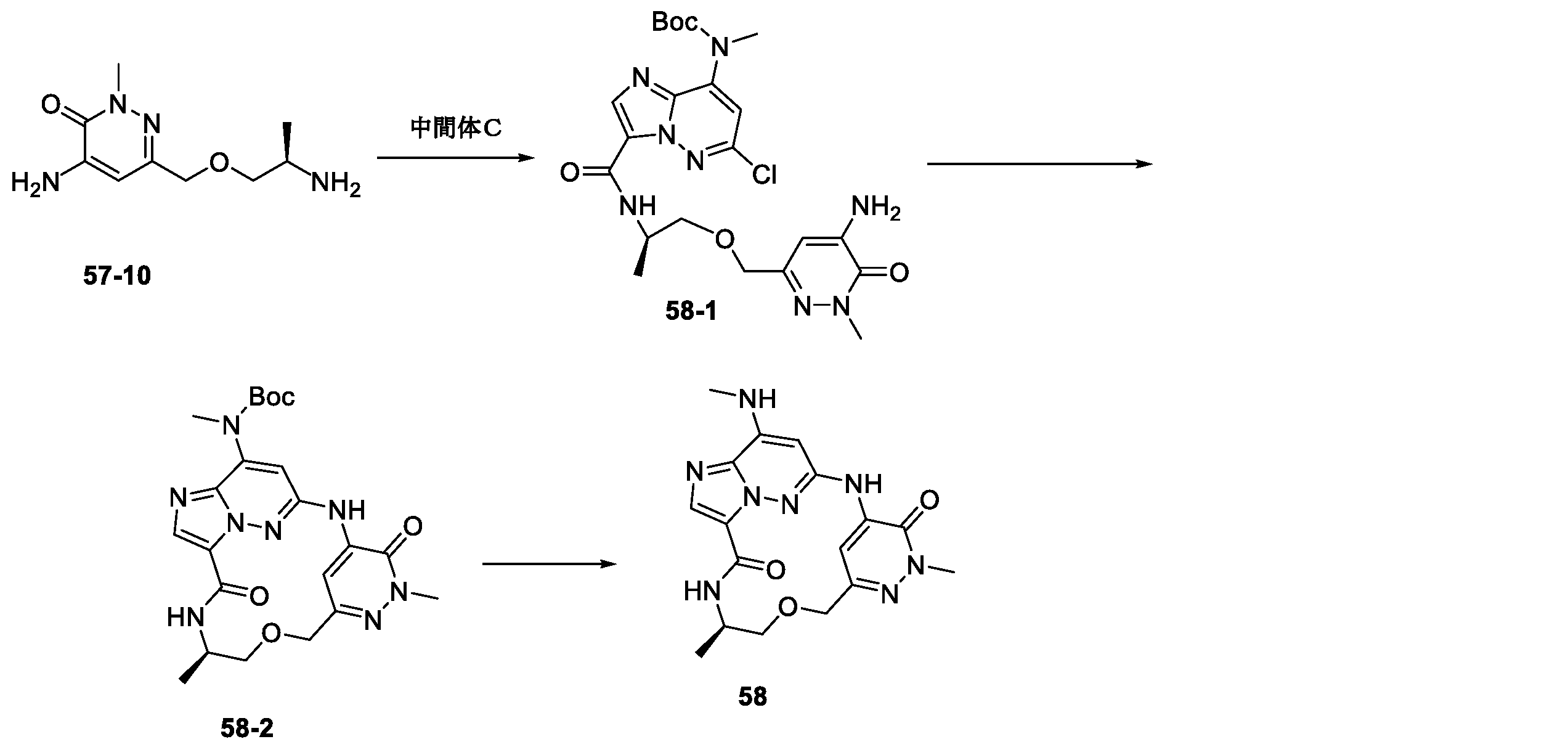
4-アミノ-6-[[(2R)-2-アミノプロポキシ]メチル]-2-メチル-ピリダジン-3-オン(57-10)(220mg、1.04mmol)および中間体C(340mg、1.04mmol)をDMF(5mL)に溶解し、この溶液に、DIPEA(0.67g、5.18mmol)およびHATU(0.6g、1.55mmol)を室温で加えた。6時間後、溶液混合物を、EA(30mL)で希釈し、水(2×30mL)およびブライン(30mL)で洗浄し、Na2SO4で乾燥して濃縮した。粗生成物を、フラッシュクロマトグラフィー(PE/EA=3:1)により精製して、表題化合物(58-1)(0.31g、57.4%)を白色固体として得た。LC-MS(ESI):m/z=521.2[M+H]+
tert-ブチル N-[3-[[(1R)-2-[(5-アミノ-1-メチル-6-オキソ-ピリダジン-3-イル)メトキシ]-1-メチル-エチル]カルバモイル]-6-クロロ-イミダゾ[1,2-b]ピリダジン-8-イル]-N-メチル-カルバメート(58-1)(220mg、0.42mmol)の1,4-ジオキサン(20mL)溶液に、Cs2CO3(275mg、0.84mmol)および第3-t-Bu-Xphos-Pd(30mg)を加えた。反応混合物を、N2雰囲気下、80℃で、2時間攪拌した。室温に冷却後、溶媒を除去し、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物(58-2)(160mg、78.2%)を白色固体として得た。LC-MS(ESI):m/z=485.2[M+H]+
tert-ブチル((15E,34E,7R)-31,7-ジメチル-36,9-ジオキソ-31,36-ジヒドロ-5-オキサ-2,8-ジアザ-1(6,3)-イミダゾ[1,2-a]ピリダジナ-3(5,3)-ピリダジナシクロノナファン(pyridazinacyclononaphane)-18-イル)(メチル)カルバメート(58-2)(180mg、0.372mmol)およびp-TsOH(192mg、1.11mmol)のDCM(10mL)溶液を、40℃で1時間攪拌した。溶媒を蒸発させ、粗生成物を水とDCMとの間で分配した。水層を、NaHCO3で塩基性化し、DCMで抽出した。一つに合わせた有機層を、ブラインで洗浄し、硫酸ナトリウム上で乾燥し、ろ過し、蒸発させ、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物である例58(110mg、77%)を白色固体として得た。LC-MS(ESI):m/z=385.2[M+H]+.1H NMR(400MHz,CDCl3)δ 9.50(s,1H),8.58(d,1H),8.14(s,1H),7.89(s,1H),7.72-7.64(m,1H),6.49(s,1H),4.58(d,1H),4.26(d,1H),4.15-4.03(m,1H),3.71(s,3H),3.64-3.58(m,1H),3.49-3.41(m,1H),2.89(d,3H),1.13(d,3H).
実施例59:
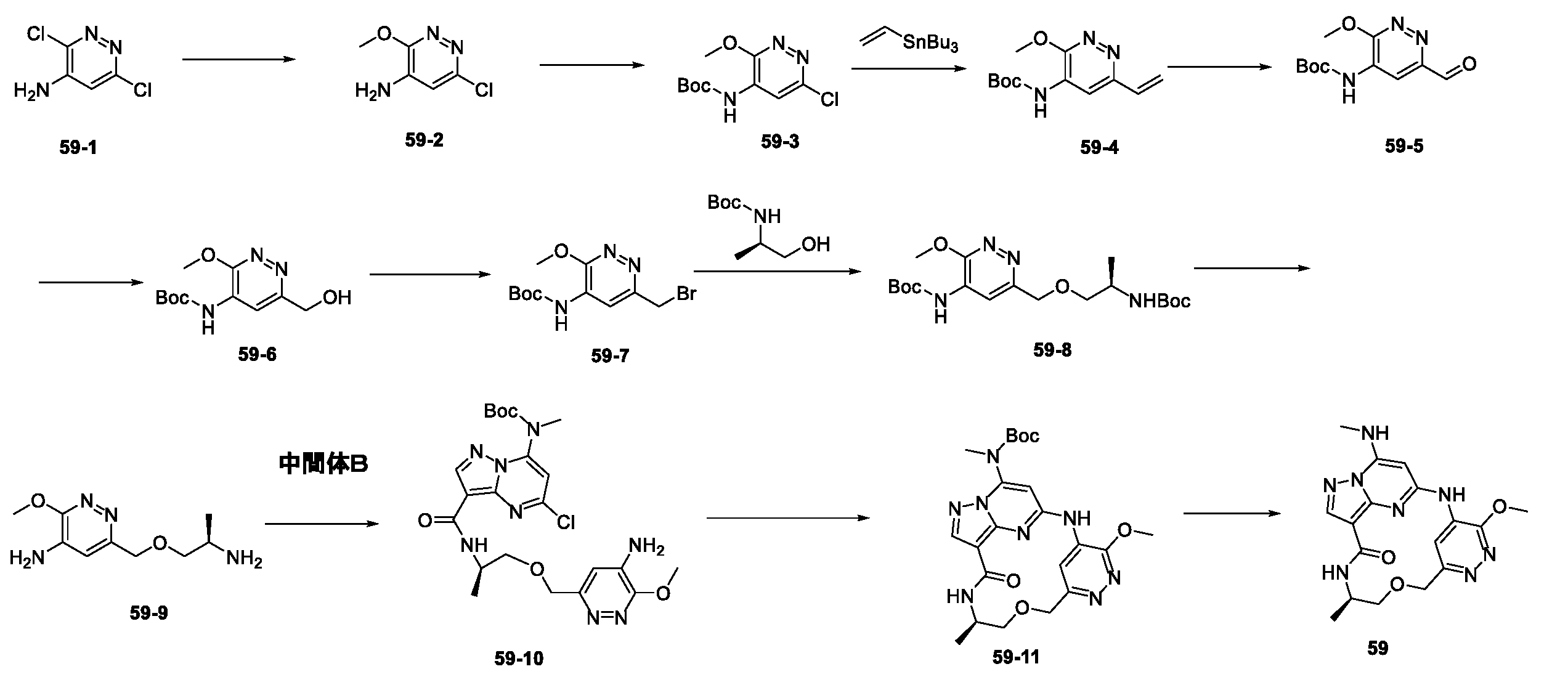
59-1(5.0g、30.5mmol)のDMSO(20mL)溶液に、LiOH(1.46g、61mmol)およびメタノール(30mL)を室温で加え、反応混合物を80℃で12時間攪拌した。室温に冷却した後、混合物を冷水で処理し、酢酸エチルで抽出した。一つに合わせた有機層を、ブラインで洗浄し、乾燥(Na2SO4)し、次いで残渣をフラッシュクロマトグラフィーにより精製して、表題化合物(59-2)(4.1g、85%)を褐色固体として得た。LC-MS(ESI):m/z=160.1[M+H]+
6-クロロ-3-メトキシ-ピリダジン-4-アミン(59-2)(4.0g、25.16mmol)のDCM溶液に、Boc2O(11.0g、50.31mmol)、トリエチルアミン(7.6g、75.5mmol)およびDMAP(307mg、2.52mmol)を0℃で加え、混合物を室温で3時間攪拌し、混合物を冷水で処理し、DCMで抽出した。一つに合わせた有機層を、ブラインで洗浄し、乾燥(Na2SO4)し、次いで残渣をフラッシュクロマトグラフィーにより精製して、表題化合物(59-3)(3.71g、57%)を褐色固体として得た。LC-MS(ESI):m/z=260.2[M+H]+
tert-ブチル N-(6-クロロ-3-メトキシ-ピリダジン-4-イル)カルバメート(59-3)(3.5g、13.51mmol)およびトリブチル(ビニル)すず(8.57g、27.03mmol)のDMF(30mL)溶液に、CuCl(4.01g、40.53mmol)、Pd(PPh3)4(1.56g、1.35mmol)を、N2雰囲気下、室温で加え、混合物を80℃で4時間攪拌し、次いで冷水で処理し、EtOAcで抽出した。一つに合わせた有機層を、ブラインで洗浄し、乾燥(Na2SO4)し、次いで残渣をフラッシュクロマトグラフィーにより精製して、表題化合物(59-4)(2.58g、76%)を得た。LC-MS(ESI):m/z=252.1[M+H]+
tert-ブチル N-(3-メトキシ-6-ビニル-ピリダジン-4-イル)カルバメート(59-4)(2.5g、10mmol)のDCM(30mL)溶液に、RuCl3(225mg、1mmol)および4-メチルモルホリン N-オキシド(3.51g、30mmol)を0℃で加え、室温に温め、1時間攪拌し、次いで冷水で処理し、EtOAcで抽出した。一つに合わせた有機層を、ブラインで洗浄し、乾燥(Na2SO4)し、次いで残渣をフラッシュクロマトグラフィーにより精製して、表題化合物(59-5)(1.87g、74%)を得た。LC-MS(ESI):m/z=254.1[M+H]+
水素化ホウ素ナトリウム(537mg、14.2mmol)を、THF(10mL)中のtert-ブチル N-(6-ホルミル-3-メトキシ-ピリダジン-4-イル)カルバメート(59-5)(1.8g、7.1mmol)の攪拌溶液に0℃で少しずつ加えた。室温での2時間の攪拌後、THFを真空中で除去した。残渣を、冷水で処理し、ジクロロメタンで抽出した。一つに合わせた有機層を、ブラインで洗浄し、乾燥(Na2SO4)し、次いで真空中で蒸発させて、表題化合物(59-6)を未精製固体(1.7g、94%)として得た。LC-MS(ESI):m/z=256.1[M+H]+
無水ジエチルエーテル(5mL)中の四臭化炭素(4.42g、13.34mmol)を、無水ジエチルエーテル(15mL)中のtert-ブチル N-[6-(ヒドロキシメチル)-3-メトキシ-ピリダジン-4-イル]カルバメート(59-6)(1.7g、6.67mmol)およびトリフェニルホスフィン(3.50g、13.34mmol)の攪拌溶液に滴加した。混合物を濃縮する前に一晩攪拌した。ヘキサン中の酢酸エチル(0~10%)を用いたクロマトグラフィーにより、表題化合物(59-7)を淡黄色固体(1.08g、51%)として得た。LC-MS(ESI):m/z=318.1[M+H]+
水素化ナトリウム(273mg、6.82mmol、60%)を、0℃でTHF(15mL)中のtert-ブチル N-(2-ヒドロキシ-1-メチル-エチル)カルバメート(0.9g、5.11mmol)の攪拌溶液に少しずつ加え、混合物を0℃で10分間攪拌し、次いで混合物に、0℃でtert-ブチル N-[6-(ブロモメチル)-3-メトキシ-ピリダジン-4-イル]カルバメート(59-7)(1.08g、3.41mmol)を加え、30分後、混合物を、冷水で処理し、酢酸エチルで抽出した。一つに合わせた有機層を、ブラインで洗浄し、乾燥(Na2SO4)し、次いで残渣をフラッシュクロマトグラフィーにより精製して、表題化合物(59-8)(930mg、66%)を褐色固体として得た。LC-MS(ESI):m/z=413.2[M+1]+
トリフルオロ酢酸(2mL)を、tert-ブチル N-[(1R)-2-[[5-(tert-ブトキシカルボニルアミノ)-6-メトキシ-ピリダジン-3-イル]メトキシ]-1-メチル-エチル]カルバメート(59-8)(200mg、0.48mmol)のDCM(8mL)溶液に加え、この混合物を2時間攪拌し、混合溶液を蒸発させて乾燥し、その後、表題化合物(59-9)(95mg.92%)を褐色液体として得、これをさらに精製することなく次の工程で使用した。LC-MS(ESI):m/z=213.2[M+H]+
6-[[(2R)-2-アミノプロポキシ]メチル]-3-メトキシ-ピリダジン-4-アミン(59-9)(95mg、0.45mmol)および中間体B(145mg、0.45mmol)をDMF(5mL)に溶解し、この溶液に、HATU(256mg、0.67mmol)およびDIPEA(116mg、0.90mmol)を室温で加えた。3時間後、溶液混合物を、EA(30mL)で希釈し、水(2×30mL)およびブライン(30mL)で洗浄し、Na2SO4で乾燥して濃縮した。粗生成物を、フラッシュクロマトグラフィー(PE/EA=3:1)により精製して、表題化合物(59-10)(108mg、46%)を白色固体として得た。LC-MS(ESI):m/z=521.0[M+H]+.
tert-ブチル N-[3-[[(1R)-2-[(5-アミノ-6-メトキシ-ピリダジン-3-イル)メトキシ]-1-メチル-エチル]カルバモイル]-5-クロロ-ピラゾロ[1,5-a]ピリミジン-7-イル]-N-メチル-カルバメート(59-10)(108mg、0.21mmol)の1,4-ジオキサン(10mL)溶液に、Cs2CO3(136mg、0.42mmol)および第3-t-Bu-Xphos-Pd(40mg)を加えた。反応混合物を、N2下、80℃で、2時間攪拌した。室温に冷却後、溶媒を除去し、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物(59-11)(22mg、21.7%)を白色固体として得た。LC-MS(ESI):m/z=485.1[M+H]+
tert-ブチル((R,13E,14E)-36-メトキシ-7-メチル-9-オキソ-5-オキサ-2,8-ジアザ-1(5,3)-ピラゾロ[1,5-a]ピリミジナ-3(5,3)-ピリダジナシクロノナファン(pyridazinacyclononaphane)-17-イル)(メチル)カルバメート(59-11)(14mg、0.044mmol)およびトリフルオロ酢酸(0.25mL)のDCM(2mL)溶液を、室温で3時間攪拌した。溶媒を蒸発させ、粗生成物を水とDCMとの間で分配した。水層を、NaHCO3で塩基性化し、DCMで抽出した。一つに合わせた有機層を、ブラインで洗浄し、硫酸ナトリウム上で乾燥し、ろ過し、蒸発させ、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物である例59(5mg、50%)を白色固体として得た。LC-MS(ESI):m/z=385.1[M+H]+.1H NMR(400MHz,CDCl3)δ 8.85(s,1H),8.38(s,1H),8.09(s,1H),7.22(s,1H),6.39(s,1H),5.51(s,1H),4.80(dd,2H),4.24(s,3H),4.22-4.18(m,1H),3.73-3.68(m,1H),3.62-3.55(m,1H),3.13(d,3H),1.35(d,3H).
実施例60:
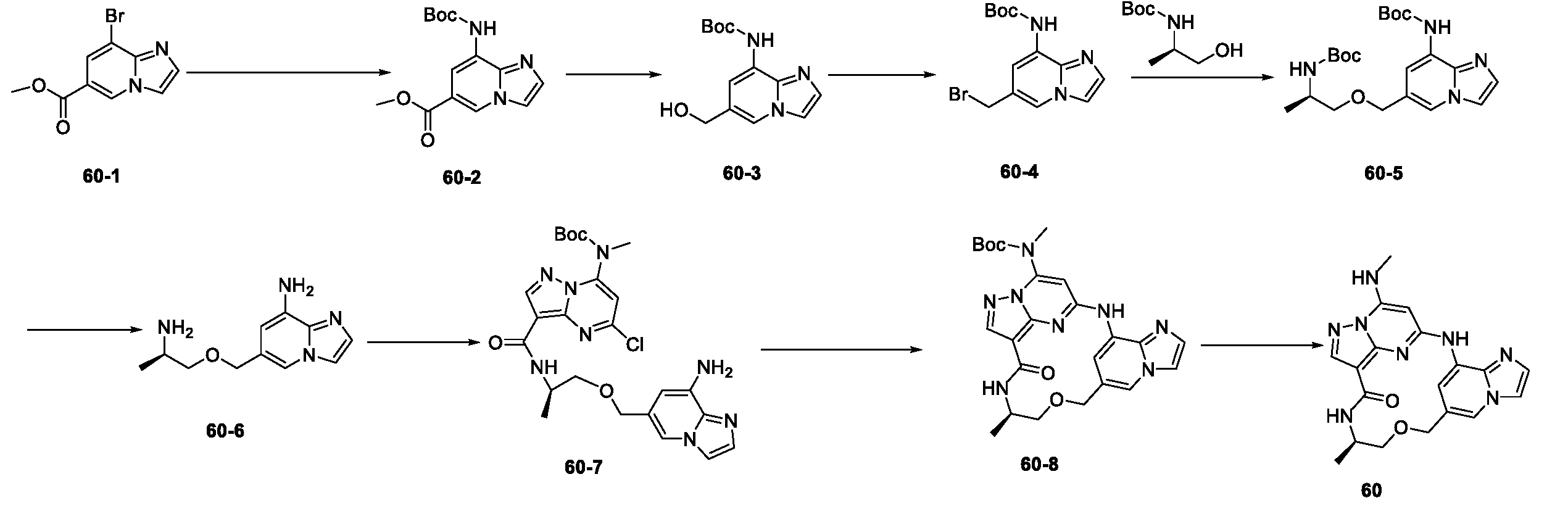
(60-1)(2.0g、7.84mmol)およびtert-ブチルカルバメート(1.41g、11.76mmol)の1,4-ジオキサン(100mL)溶液に、t-BuONa(1.2g、11.76mmol)、Pd2(dba)3(1.44g、1.57mmol)およびDpephos(1.7g、3.14mmol)をN2雰囲気下で加えた。反応混合物を、N2下、80℃で、2時間攪拌した。室温に冷却後、溶媒を除去し、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物(60-2)(450mg、20%)を白色固体として得た。LC-MS(ESI):m/z=292.3[M+H]+
水素化ホウ素ナトリウム(176mg、4.64mmol)を、メタノール(10mL)中の8-(tert-ブトキシカルボニルアミノ)イミダゾ[1,2-a]ピリジン-6-カルボン酸メチル(60-2)(0.54g、1.86mmol)の攪拌溶液に室温で少しずつ加えた。次いでこの混合物を50℃に温め、1時間攪拌し、メタノールを真空中で除去した。残渣を、冷水で処理し、ジクロロメタンで抽出した。一つに合わせた有機層を、ブラインで洗浄し、乾燥(Na2SO4)し、次いで真空中で蒸発させて、表題化合物(60-3)を未精製固体(0.27g、55%)として得た。LC-MS(ESI):m/z=264.3[M+H]+
無水ジエチルエーテル(5mL)中の四臭化炭素(0.68g、2.06mmol)を、無水ジエチルエーテル(15mL)中のtert-ブチル N-[6-(ヒドロキシメチル)イミダゾ[1,2-a]ピリジン-8-イル]カルバメート(60-3)(0.27g、1.03mmol)およびトリフェホスフィン(0.54g、2.06mmol)の攪拌溶液に滴加した。混合物を濃縮する前に一晩攪拌した。ヘキサン中の酢酸エチル(0~10%)を用いたクロマトグラフィーにより、表題化合物(60-4)を淡黄色固体(0.18g、54%)として得た。LC-MS(ESI):m/z=327.2[M+H]+
水素化ナトリウム(44mg、1.1mmol)を、0℃でTHF(15mL)中のtert-ブチル N-(2-ヒドロキシ-1-メチル-エチル)カルバメート(0.19g、1.1mmol)の攪拌溶液に少しずつ加え、この混合物を0℃で10分間攪拌し、次いで混合物に、0℃でtert-ブチル N-[6-(ブロモメチル)イミダゾ[1,2-a]ピリジン-8-イル]カルバメート(60-4)(0.18g、0.55mmol)を加え、室温に温め、1時間攪拌し、冷水でクエンチし、酢酸エチルで抽出した。一つに合わせた有機層を、ブラインで洗浄し、乾燥(Na2SO4)し、次いで残渣をフラッシュクロマトグラフィーで精製して、表題化合物(60-5)(0.2g、86%)を褐色固体として得た。LC-MS(ESI):m/z=421.5[M+H]+
トリフルオロ酢酸(1mL)を、tert-ブチル N-[(1R)-2-[[8-(tert-ブトキシカルボニルアミノ)イミダゾ[1,2-a]ピリジン-6-イル]メトキシ]-1-メチル-エチル]カルバメート(60-5)(0.2g、0.48mmol)のDCM(5mL)溶液に加え、この混合物を室温で2時間攪拌し、混合溶液を蒸発させて乾燥し、その後、表題化合物(60-6)(0.11g.100%)を褐色液体として得、これをさらに精製することなく次の工程で使用した。LC-MS(ESI):m/z=221.3[M+H]+
tert-ブチル N-[3-[[(1R)-2-[(8-アミノイミダゾ[1,2-a]ピリジン-6-イル)メトキシ]-1-メチル-エチル]カルバモイル]-5-クロロ-ピラゾロ[1,5-a]ピリミジン-7-イル]-N-メチル-カルバメート(60-6)(0.11g、0.5mmol)および中間体B(0.32g、0.5mmol)を、DMF(5mL)に溶解し、この溶液に、N,N,N’,N’-テトラメチルクロロホルムアミジニウムヘキサフルオロホスフェート(0.21g、0.75mmol)および1-メチルイミダゾール(0.08g、1.0mmol)を室温で加えた。3時間攪拌後、溶液混合物を、EA(30mL)で希釈し、水(2×10mL)およびブライン(10mL)で洗浄し、Na2SO4で乾燥して濃縮した。粗生成物を、フラッシュクロマトグラフィー(PE/EA=3:1)により精製して、表題化合物(60-7)(0.12g、45%)を白色固体として得た。LC-MS(ESI):m/z=529.0[M+H]+
(60-7)(120mg、0.23mmol)の1,4-ジオキサン(20mL)溶液に、Cs2CO3(150mg、0.46mmol)および第3-t-Bu-Xphos-Pd(120mg)を加えた。反応混合物を、N2下、80℃で、2時間攪拌した。室温に冷却後、溶媒を除去し、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物(60-8)(80mg、72%)を白色固体として得た。LC-MS(ESI):m/z=493.5[M+H]+
tert-ブチル メチル((R,13E,14E,37E)-7-メチル-9-オキソ-5-オキサ-2,8-ジアザ-1(5,3)-ピラゾロ[1,5-a]ピリミジナ-3(8,6)-イミダゾ[1,2-a]ピリジナシクロノナファン(pyridinacyclononaphane)-17-イル)カルバメート(60-8)(80mg、0.16mmol)およびトリフルオロ酢酸(0.5mL)のDCM(4mL)溶液を、室温で2時間攪拌した。溶媒を蒸発させ、粗生成物を水とDCMとの間で分配した。水層を、NaHCO3で塩基性化し、DCMで抽出した。一つに合わせた有機層を、ブラインで洗浄し、硫酸ナトリウム上で乾燥し、ろ過し、蒸発させ、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物である例60(20mg、29.5%)を白色固体として得た。1H NMR(400MHz,CD3OD)δ 8.50(d,1H),8.29(d,1H),8.14(s,1H),7.99(s,1H),7.77(d,1H),7.50(d,1H),4.52(q,2H),4.13-4.03(m,1H),3.59(dd,1H),3.49(dd,1H),2.99(s,3H),1.22(d,3H).LC-MS(ESI):m/z=393.5[M+H]+
実施例62:

3-フルオロ-4-メトキシ-ベンズアルデヒド(62-1)(3.6g、23.37mmol)を濃硫酸(30mL)に溶解し、-10℃に冷却した。濃硫酸(4mL)中の濃硝酸(2.5mL)を、20分間にわたって滴加した。-10℃未満でさらに1時間攪拌した後、混合物を砕氷に注いだ。沈殿物をろ過により回収し、ジクロロメタン(40mL)と飽和炭酸水素ナトリウム(30mL)との間で分配した。有機層を乾燥(Na2SO4)し、真空中で蒸発させて、表題化合物(62-2)(1.6g、34.23%)を油状物として得た。LC-MS(ESI):m/z=200.1[M+H]+.
水素化ホウ素ナトリウム(0.38g、10.04mmol)を、0℃で、メタノール(20mL)中の3-フルオロ-4-メトキシ-5-ニトロ-ベンズアルデヒド(62-2)(1.6g、8.0mmol)の攪拌溶液に少しずつ加えた。2時間後、メタノールを真空中で除去した。残渣を、冷水で処理し、ジクロロメタンで抽出した。一つに合わせた有機層を、ブラインで洗浄し、乾燥(Na2SO4)し、次いで真空中で蒸発させて、表題化合物(62-3)を未精製固体(1.4g、87.06%)として得た。LC-MS(ESI):m/z=202.1[M+H]+
(3-フルオロ-4-メトキシ-5-ニトロ-フェニル)メタノール(62-3)(1.4g、6.96mmol)およびトリフェホスフィン(2.61g、9.95mmol)の無水ジエチルエーテル(30mL)溶液に、無水ジエチルエーテル(5mL)中の四臭化炭素(3.3g、9.95mmol)を滴加した。混合物を一晩攪拌した後、粘着性油状物まで濃縮した。シリカゲルクロマトグラフィーにより、表題化合物(62-4)を淡黄色固体(1.3g、70.97%)として得た。LC-MS(ESI):m/z=264.1[M+H]+
水素化ナトリウム(144mg、3.61mmol)を、0℃でTHF(15mL)中のtert-ブチル N-(2-ヒドロキシ-1-メチル-エチル)カルバメート(0.63g、3.61mmol)の攪拌溶液に少しずつ加え、この混合物を0℃で10分間攪拌し、次いで混合物に、0℃で5-(ブロモメチル)-1-フルオロ-2-メトキシ-3-ニトロ-ベンゼン(62-4)(0.95g、3.61mmol)を加え、30分後、混合物を、冷水でクエンチし、酢酸エチルで抽出した。一つに合わせた有機層を、ブラインで洗浄し、乾燥(Na2SO4)し、次いで残渣をフラッシュクロマトグラフィーにより精製して、表題化合物(62-5)(0.63g、48.83%)を褐色固体として得た。LC-MS(ESI):m/z=359.1[M+H]+
トリフルオロ酢酸(1.5mL)を、tert-ブチルN-[(1R)-2-[(3-フルオロ-4-メトキシ-5-ニトロ-フェニル)メトキシ]-1-メチル-エチル]カルバメート(62-5)(0.63g、1.76mmol)のDCM(5mL)溶液に加え、この混合物を2時間攪拌し、混合溶液を蒸発させて乾燥し、その後、表題化合物(62-6)(0.6g.91.46%)を褐色液体として得、これをさらに精製することなく次の工程で使用した。LC-MS(ESI):m/z=259.2[M+H]+
(2R)-1-[(3-フルオロ-4-メトキシ-5-ニトロ-フェニル)メトキシ]プロパン-2-アミン(62-6)(0.6g、1.6mmol)をDMF(10mL)に溶解させ、この溶液に、HATU(0.91g、2.41mmol)、DIPEA(0.41g、3.2mmol)および中間体B(0.52g、1.6mmol)を室温で加えた。18時間後、溶液混合物を、EA(50mL)で希釈し、水(2×50mL)およびブライン(50mL)で洗浄し、Na2SO4で乾燥して濃縮した。粗生成物を、フラッシュクロマトグラフィーにより精製して、表題化合物(62-7)(545mg、59.89%)を白色固体として得た。LC-MS(ESI):m/z=567.2[M+H]+
tert-ブチルN-[5-クロロ-3-[[(1R)-2-[(3-フルオロ-4-メトキシ-5-ニトロ-フェニル)メトキシ]-1-メチル-エチル]カルバモイル]ピラゾロ[1,5-a]ピリミジン-7-イル]-N-メチル-カルバメート(62-7)(545mg、0.96mmol)をエタノール(45mL)およびH2O(15mL)に溶解し、鉄粉末(540mg、9.62mmol)およびNH4Cl(310mg、5.77mmol)を溶液に加え、次いで、反応混合物を3時間で85℃に加熱し、室温まで冷却した後、反応物をろ過し、ろ液を真空中で除去し、残渣を、フラッシュクロマトグラフィー(PE/EA=2:1)により精製し、表題化合物(62-8)(450mg、87.2%)を白色固体として得た。LC-MS(ESI):m/z=537.1[M+H]+
(62-8)(450mg、0.84mmol)の1,4-ジオキサン(100mL)溶液に、Cs2CO3(820mg、2.51mmol)および第3-t-Bu-Xphos-Pd(250mg)を加えた。反応混合物を、N2下、80℃で、2時間攪拌した。室温に冷却後、溶媒を除去し、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物(62-9)(220mg、52.50%)を白色固体として得た。LC-MS(ESI):m/z=501.3[M+H]+
(62-9)(220mg、0.44mmol)およびトリフルオロ酢酸(1mL)のDCM(5mL)溶液を、室温で2時間攪拌した。溶媒を蒸発させ、粗生成物を水とDCMとの間で分配した。水層を、NaHCO3で塩基性化し、DCMで抽出した。一つに合わせた有機層を、ブラインで洗浄し、硫酸ナトリウム上で乾燥し、ろ過し、蒸発させ、残渣をシリカゲルフラッシュカラムクロマトグラフィーにより精製して、生成物(71mg、40.34%)を白色固体として得た。LC-MS(ESI):m/z=401.3[M+H]+.1H NMR(400MHz,CDCl3)δ 8.56(s,1H),8.40-8.14(m,2H),7.14(s,1H),6.62(d,1H),6.22(d,1H),5.38(s,1H),4.53(dd,2H),4.18(dd,1H),4.03(d,3H),3.57(m,2H),3.10(d,3H),1.34(d,3H).
JH2ドメインに対する本明細書に記載の化合物の結合定数を、KINOMEscan(登録商標)アッセイ(DiscoveRx社)の以下のプロトコルにより決定した。ヒトTYK2(JH2ドメイン-シュードキナーゼ)の部分長構築物(参照配列NP_003322.3に基づくアミノ酸G556からD888)とNFkBのDNA結合ドメインとの融合タンパク質を、一過的にトランスフェクトされたHEK293細胞において発現する。これらのHEK293細胞から、製造業者の指示に従って、プロテアーゼ阻害剤カクテルコンプリート(Roche社)およびホスファターゼ阻害剤カクテルセットII(Merck社)の存在下で、M-PER抽出緩衝液(Pierce社)中で、抽出物を調製する。TYK2(JH2ドメイン-シュードキナーゼ)融合タンパク質は、発現抽出物に直接付加する、qPCR読み出し用のアンプリコンに融合したNFkB結合部位を含有するキメラ二本鎖DNAタグで標識される(結合反応におけるDNAタグの最終濃度は0.1nMである)。
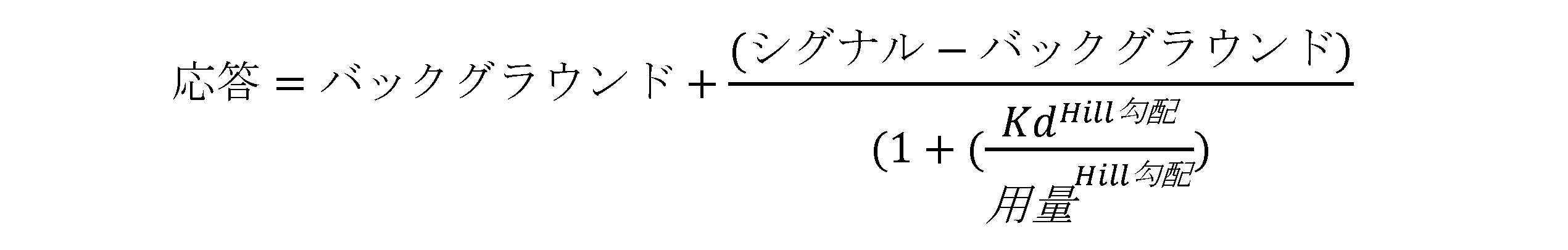

新鮮なヒトPBMCを、10%FBSを含むRPMI 1640培地中に再懸濁した。細胞を、200,000細胞/ウェルの濃度で丸底96ウェルプレートに播種した。被験化合物の10点希釈系列(最高用量は10uM、1:5希釈)を、液体ディスペンサー(Tecan D300e)を使用して当該ウェルに加え、37℃で1時間インキュベートした。次いで、ヒトIL-12組み換えタンパク質(R&D Systems社)を、最終濃度10ng/mlで当該ウェルに加え、37℃で15分間インキュベートした。細胞溶解物を調製し、メーカーのプロトコルに従って、Phospho STAT4(Tyr693)キット(Meso Scale Discovery社)により分析した。

新鮮なヒトPBMCを、10%FBSを含むRPMI 1640培地中に再懸濁した。細胞を、200,000細胞/ウェルの濃度で丸底96ウェルプレートに播種した。被験化合物の10点希釈系列(最高用量は10uM、1:5希釈)を、液体ディスペンサー(Tecan D300e)を使用して当該ウェルに加え、37℃で1時間インキュベートした。次いで、ヒトINFα組み換えタンパク質(R&D Systems社)を、最終濃度5000単位/mlで当該ウェルに加え、37℃で15分間インキュベートした。細胞溶解物を調製し、メーカーのプロトコルに従って、Phospho STAT3(Tyr705)細胞キット(Cisbio社)、またはPhospho STAT5(Tyr693)キット(Meso Scale Discovery社)により分析した。
対照はBMS-986165である:



上述のTYK2 JH2結合の方法と同様に、JAK1 JH2ドメインおよびJAK2 JH1ドメインの結合アッセイを、DiscoverX社のKINOMEscan(商標)を使用して行ったが、キナーゼドメインを変更した。これらのアッセイを実施して、被験化合物のJAK1 JH2ドメインとJAK2 JH1ドメインに対する結合選択性を比較した。結果を表4に示す。

上述のヒトPBMCにおけるINFα誘導型pSTAT5に対する方法と同様に、これらのアッセイを実施して、被験化合物がヒトPBMCにおけるJAK1.JAK2、およびJAK3の経路に対して交差活性を有するか否かを確認した。手順は記載されるとおりであったが、刺激を10ng/mlのGM-CSF、または20ng/mlのIL-2へ変更した。データを表5に示す。


実施例G1:非経口組成物
注射による投与に適した非経口医薬組成物を調製するために、本明細書に記載の化合物の水溶性塩100mgをDMSOに溶解し、次いで10mLの0.9%滅菌生理食塩水と混合する。当該混合物は、注射による投与に適した単位剤形に組み込まれる。
経口送達用の医薬組成物を調製するために、本明細書に記載の化合物100mgを、750mgのデンプンと混合する。当該混合物は、例えば硬質ゼラチンカプセルなどの経口投与に適した経口単位剤形に組み込まれる。
例えば硬質ロゼンジなどのバッカル送達用の医薬組成物を調製するために、420mgの粉末混合糖、1.6mLのライトコーンシロップ、2.4mLの蒸留水、および0.42mLのミント抽出物と、100mgの本明細書に記載の化合物を混合する。当該混合物を穏やかに混ぜ、型に注いで、バッカル投与に適したロゼンジを成形する。
Claims (13)
- 以下の式(IIa)または式(IIb)の化合物、またはその薬学的に許容可能な塩もしくは立体異性体:
式中:
Lは、
からなる群から選択され、
環Aは、フェニル、ピリジル、ピリミジル、ピリダジニル、インダゾリル、ベンゾキサゾリル、およびベンゾトリアゾリルからなる群から選択され、
各RAは独立して、重水素、ハロゲン、-CN、-OR b 、-NRcRd、-C(=O)NR c R d 、-C(=O)Ra 、C1-C6アルキル、C1-C6ハロアルキル、およびC1-C6ジュウテロアルキル(deuteroalkyl)からなる群から選択され、または同じ炭素上の二つのRAが一緒になってオキソを形成し、
nは、0~4であり、
Y8は、Nであり、
Y6は、CR 6 であり、
Y3は、CR 3 であり、
Y9は、Nであり、
R3 およびR 6 は、水素であり、
R4は、水素、C1-C6アルキル、C1-C6ヘテロアルキル、C1-C6ハロアルキル、およびC1-C6ジュウテロアルキル(deuteroalkyl)からなる群から選択され、
R 5は、水素であり、
R7は、水素またはC1-C6アルキルであり、
各Raは独立して、C1-C6アルキル、C1-C6ハロアルキル、およびC1-C6ジュウテロアルキル(deuteroalkyl)からなる群から選択され、
各Rbは独立して、水素、C1-C6アルキル、C1-C6ハロアルキル、およびC1-C6ジュウテロアルキル(deuteroalkyl)からなる群から選択され、また
各RcおよびRdは独立して、水素、C1-C6アルキル、C1-C6ハロアルキル、およびC1-C6ジュウテロアルキル(deuteroalkyl)からなる群から選択される。 - R4はC1-C6アルキルまたはC1-C6ジュウテロアルキル(deuteroalkyl)である、請求項1に記載の化合物、またはその薬学的に許容可能な塩もしくは立体異性体。
- 環Aはフェニルである、請求項1または2に記載の化合物、またはその薬学的に許容可能な塩もしくは立体異性体。
- 環Aはピリジル、ピリミジル、ピリダジニル、インダゾリル、ベンゾキサゾリル、およびベンゾトリアゾリルからなる群から選択される、請求項1または2に記載の化合物、またはその薬学的に許容可能な塩もしくは立体異性体。
- 各RAは独立して、重水素、ハロゲン、-CN、-ORb 、C 1-C6アルキル、C1-C6ハロアルキル、およびC1-C6ジュウテロアルキル(deuteroalkyl)からなる群から選択される、請求項1~4のいずれか一項に記載の化合物、またはその薬学的に許容可能な塩もしくは立体異性体。
- Lは、
からなる群から選択される、請求項1~5のいずれか一項に記載の化合物、またはその薬学的に許容可能な塩もしくは立体異性体。 - Lは、
である、請求項1~6のいずれか一項に記載の化合物、またはその薬学的に許容可能な塩もしくは立体異性体。 - 前記化合物は、
からなる群から選択される、請求項1に記載の化合物、またはその薬学的に許容可能な塩もしくは立体異性体。 - 請求項1~8のいずれか一項に記載される化合物、またはその薬学的に許容可能な塩もしくは立体異性体の治療有効量、および薬学的に許容可能な賦形剤を含む、医薬組成物。
- 生物学的サンプルにおいてTYK2酵素を阻害する方法であって、前記生物学的サンプルと、請求項1~8のいずれか一項に記載される化合物、またはその薬学的に許容可能な塩もしくは立体異性体を接触させることを含む、方法。
- TYK2介在性の障害を治療する方法における使用のための、請求項9に記載の医薬組成物であって、前記方法が、その必要のある患者に、請求項1~8のいずれか一項に記載の化合物、またはその薬学的に許容可能な塩もしくは立体異性体を投与することを含む、医薬組成物。
- 前記TYK2介在性の障害は、自己免疫性障害、炎症性障害、増殖性障害、内分泌障害、神経学的障害、または移植に関連する障害である、請求項11に記載の医薬組成物。
- 前記障害は、I型インターフェロン、IL-10、IL-12、またはIL-23のシグナル伝達と関連している、請求項11に記載の医薬組成物。
Applications Claiming Priority (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201962816698P | 2019-03-11 | 2019-03-11 | |
| US62/816,698 | 2019-03-11 | ||
| US201962835376P | 2019-04-17 | 2019-04-17 | |
| US62/835,376 | 2019-04-17 | ||
| US201962877741P | 2019-07-23 | 2019-07-23 | |
| US62/877,741 | 2019-07-23 | ||
| US201962931119P | 2019-11-05 | 2019-11-05 | |
| US62/931,119 | 2019-11-05 | ||
| PCT/US2020/021850 WO2020185755A1 (en) | 2019-03-11 | 2020-03-10 | Tyk2 inhibitors and uses thereof |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2022524974A JP2022524974A (ja) | 2022-05-11 |
| JP2022524974A5 JP2022524974A5 (ja) | 2023-03-20 |
| JP7586829B2 true JP7586829B2 (ja) | 2024-11-19 |
Family
ID=72427699
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2021552136A Active JP7586829B2 (ja) | 2019-03-11 | 2020-03-10 | Tyk2阻害剤およびその使用 |
Country Status (10)
| Country | Link |
|---|---|
| US (2) | US20220177486A1 (ja) |
| EP (1) | EP3938369A4 (ja) |
| JP (1) | JP7586829B2 (ja) |
| KR (1) | KR102899985B1 (ja) |
| CN (1) | CN113811534B (ja) |
| AU (1) | AU2020239026B2 (ja) |
| BR (1) | BR112021017996A2 (ja) |
| CA (1) | CA3132632A1 (ja) |
| IL (1) | IL286248B2 (ja) |
| WO (1) | WO2020185755A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2023541203A (ja) * | 2020-09-16 | 2023-09-28 | アルミス インコーポレイテッド | Tyk2阻害剤およびその使用 |
Families Citing this family (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP7631193B2 (ja) | 2018-10-22 | 2025-02-18 | アルミス インコーポレイテッド | Tyk2阻害剤およびその使用 |
| CA3134814A1 (en) * | 2019-03-26 | 2020-10-01 | Ventyx Biosciences, Inc. | Tyk2 pseudokinase ligands |
| WO2020233645A1 (zh) | 2019-05-21 | 2020-11-26 | 浙江海正药业股份有限公司 | 大环类衍生物、及其制备方法和用途 |
| JP7635228B2 (ja) | 2019-11-08 | 2025-02-25 | ベンティックス バイオサイエンシーズ,インク. | Tyk2偽キナーゼリガンド |
| CN114591351B (zh) * | 2020-12-03 | 2023-12-05 | 成都科岭源医药技术有限公司 | 一种多环化合物及其制备方法和用途 |
| EP4423086A1 (en) | 2021-10-25 | 2024-09-04 | Kymera Therapeutics, Inc. | Tyk2 degraders and uses thereof |
| CA3245808A1 (en) * | 2022-03-16 | 2023-09-21 | Alumis Inc. | TYK2 INHIBITORS AND THEIR USES |
| CN119487037A (zh) * | 2022-03-16 | 2025-02-18 | 阿鲁米斯股份有限公司 | Tyk2抑制剂及其用途 |
| WO2023208244A1 (zh) * | 2022-04-29 | 2023-11-02 | 南京明德新药研发有限公司 | 大环类化合物及其应用 |
| JP2026512838A (ja) * | 2023-03-31 | 2026-04-21 | 北京普祺医薬科技股▲分▼有限公司 | 大環化合物、医薬組成物及びその使用 |
| WO2024222807A1 (en) * | 2023-04-27 | 2024-10-31 | Hangzhou Highlightll Pharmaceutical Co., Ltd | Novel macrocyclic heteroaryl derivatives as kinase inhibitors |
| WO2025194074A1 (en) * | 2024-03-15 | 2025-09-18 | Neuron23, Inc. | Kinase modulators and methods of use thereof |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2010503690A (ja) | 2006-09-18 | 2010-02-04 | ポラリス・グループ | タンパク質キナーゼ阻害剤として有用なピラゾロ(1,5−a)(1,3,5)トリアジン及びピラゾロ(1,5−a)ピリミジン誘導体 |
| JP2017500304A (ja) | 2013-12-10 | 2017-01-05 | ブリストル−マイヤーズ スクイブ カンパニーBristol−Myers Squibb Company | IL−12、IL−23および/またはIFNα応答のモジュレーターとして有用なイミダゾピリダジン化合物 |
| JP2019501125A (ja) | 2015-11-18 | 2019-01-17 | ブリストル−マイヤーズ スクイブ カンパニーBristol−Myers Squibb Company | IL−12、IL−23および/またはIFNα応答のモジュレーターとして有用なイミダゾピリダジン化合物 |
| WO2019023468A1 (en) | 2017-07-28 | 2019-01-31 | Nimbus Lakshmi, Inc. | TYK2 INHIBITORS AND USES THEREOF |
| WO2020019376A1 (zh) | 2018-07-27 | 2020-01-30 | 东南大学 | 一种tdo小分子抑制剂衍生物及其抗肿瘤偶联物和制备方法 |
| WO2021092246A1 (en) | 2019-11-08 | 2021-05-14 | Ventyx Biosciences, Inc. | Tyk2 pseudokinase ligands |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013001310A1 (en) * | 2011-06-30 | 2013-01-03 | Centro Nacional De Investigaciones Oncológicas (Cnio) | Macrocyclic compounds and their use as cdk8 inhibitors |
| ES2933350T3 (es) * | 2014-01-24 | 2023-02-06 | Turning Point Therapeutics Inc | Macrociclos de diarilo como moduladores de proteína quinasas |
| AU2016287568B2 (en) * | 2015-07-02 | 2020-08-20 | Turning Point Therapeutics, Inc. | Chiral diaryl macrocycles as modulators of protein kinases |
| JOP20190092A1 (ar) * | 2016-10-26 | 2019-04-25 | Array Biopharma Inc | عملية لتحضير مركبات بيرازولو[1، 5-a]بيريميدين وأملاح منها |
| JOP20190213A1 (ar) * | 2017-03-16 | 2019-09-16 | Array Biopharma Inc | مركبات حلقية ضخمة كمثبطات لكيناز ros1 |
| CN111182903A (zh) * | 2017-07-28 | 2020-05-19 | 特普医药公司 | 巨环化合物及其用途 |
| CA3134814A1 (en) * | 2019-03-26 | 2020-10-01 | Ventyx Biosciences, Inc. | Tyk2 pseudokinase ligands |
-
2020
- 2020-03-10 CA CA3132632A patent/CA3132632A1/en active Pending
- 2020-03-10 US US17/438,329 patent/US20220177486A1/en not_active Abandoned
- 2020-03-10 EP EP20769748.3A patent/EP3938369A4/en active Pending
- 2020-03-10 BR BR112021017996A patent/BR112021017996A2/pt unknown
- 2020-03-10 AU AU2020239026A patent/AU2020239026B2/en active Active
- 2020-03-10 WO PCT/US2020/021850 patent/WO2020185755A1/en not_active Ceased
- 2020-03-10 IL IL286248A patent/IL286248B2/en unknown
- 2020-03-10 JP JP2021552136A patent/JP7586829B2/ja active Active
- 2020-03-10 CN CN202080034800.9A patent/CN113811534B/zh active Active
- 2020-03-10 KR KR1020217032541A patent/KR102899985B1/ko active Active
-
2025
- 2025-05-20 US US19/213,418 patent/US20250276977A1/en active Pending
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2010503690A (ja) | 2006-09-18 | 2010-02-04 | ポラリス・グループ | タンパク質キナーゼ阻害剤として有用なピラゾロ(1,5−a)(1,3,5)トリアジン及びピラゾロ(1,5−a)ピリミジン誘導体 |
| JP2017500304A (ja) | 2013-12-10 | 2017-01-05 | ブリストル−マイヤーズ スクイブ カンパニーBristol−Myers Squibb Company | IL−12、IL−23および/またはIFNα応答のモジュレーターとして有用なイミダゾピリダジン化合物 |
| JP2019501125A (ja) | 2015-11-18 | 2019-01-17 | ブリストル−マイヤーズ スクイブ カンパニーBristol−Myers Squibb Company | IL−12、IL−23および/またはIFNα応答のモジュレーターとして有用なイミダゾピリダジン化合物 |
| WO2019023468A1 (en) | 2017-07-28 | 2019-01-31 | Nimbus Lakshmi, Inc. | TYK2 INHIBITORS AND USES THEREOF |
| WO2020019376A1 (zh) | 2018-07-27 | 2020-01-30 | 东南大学 | 一种tdo小分子抑制剂衍生物及其抗肿瘤偶联物和制备方法 |
| WO2021092246A1 (en) | 2019-11-08 | 2021-05-14 | Ventyx Biosciences, Inc. | Tyk2 pseudokinase ligands |
Non-Patent Citations (1)
| Title |
|---|
| Bioorganic & Medicinal Chemistry Letters,2008年,Vol.18,pp.619-623 |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2023541203A (ja) * | 2020-09-16 | 2023-09-28 | アルミス インコーポレイテッド | Tyk2阻害剤およびその使用 |
| JP7824938B2 (ja) | 2020-09-16 | 2026-03-05 | アルミス インコーポレイテッド | Tyk2阻害剤およびその使用 |
Also Published As
| Publication number | Publication date |
|---|---|
| IL286248A (en) | 2021-10-31 |
| KR20210141973A (ko) | 2021-11-23 |
| US20250276977A1 (en) | 2025-09-04 |
| CN113811534B (zh) | 2024-10-29 |
| US20220177486A1 (en) | 2022-06-09 |
| JP2022524974A (ja) | 2022-05-11 |
| CN113811534A (zh) | 2021-12-17 |
| IL286248B1 (en) | 2025-08-01 |
| IL286248B2 (en) | 2025-12-01 |
| WO2020185755A1 (en) | 2020-09-17 |
| KR102899985B1 (ko) | 2025-12-15 |
| AU2020239026B2 (en) | 2025-10-09 |
| CA3132632A1 (en) | 2020-09-17 |
| EP3938369A4 (en) | 2023-01-25 |
| EP3938369A1 (en) | 2022-01-19 |
| AU2020239026A1 (en) | 2021-09-23 |
| BR112021017996A2 (pt) | 2021-11-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7586829B2 (ja) | Tyk2阻害剤およびその使用 | |
| JP7631193B2 (ja) | Tyk2阻害剤およびその使用 | |
| JP7824938B2 (ja) | Tyk2阻害剤およびその使用 | |
| CN115043836B (zh) | 一种咪唑并吡啶衍生物的p2x3受体选择性调节剂及其药物用途 | |
| JP2025509596A (ja) | Tyk2阻害剤およびその使用 | |
| JP2025509594A (ja) | Tyk2阻害剤およびその使用 | |
| HK40063819A (en) | Tyk2 inhibitors and uses thereof | |
| RU2813233C2 (ru) | Ингибиторы tyk2 и пути их применения | |
| HK40063819B (zh) | Tyk2抑制剂和其用途 | |
| HK40105557B (zh) | Tyk2抑制剂及其用途 | |
| HK40105557A (zh) | Tyk2抑制剂及其用途 | |
| HK40065275A (zh) | Tyk2抑制剂和其用途 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20230310 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20230310 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20240220 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20240222 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20240515 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20240722 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20240820 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20241016 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20241107 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 7586829 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |