JP7263473B2 - Ttk阻害剤の固体形態 - Google Patents
Ttk阻害剤の固体形態 Download PDFInfo
- Publication number
- JP7263473B2 JP7263473B2 JP2021167806A JP2021167806A JP7263473B2 JP 7263473 B2 JP7263473 B2 JP 7263473B2 JP 2021167806 A JP2021167806 A JP 2021167806A JP 2021167806 A JP2021167806 A JP 2021167806A JP 7263473 B2 JP7263473 B2 JP 7263473B2
- Authority
- JP
- Japan
- Prior art keywords
- cancer
- compound
- hydrobromide
- crystal
- crystalline
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images







Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C30—CRYSTAL GROWTH
- C30B—SINGLE-CRYSTAL GROWTH; UNIDIRECTIONAL SOLIDIFICATION OF EUTECTIC MATERIAL OR UNIDIRECTIONAL DEMIXING OF EUTECTOID MATERIAL; REFINING BY ZONE-MELTING OF MATERIAL; PRODUCTION OF A HOMOGENEOUS POLYCRYSTALLINE MATERIAL WITH DEFINED STRUCTURE; SINGLE CRYSTALS OR HOMOGENEOUS POLYCRYSTALLINE MATERIAL WITH DEFINED STRUCTURE; AFTER-TREATMENT OF SINGLE CRYSTALS OR A HOMOGENEOUS POLYCRYSTALLINE MATERIAL WITH DEFINED STRUCTURE; APPARATUS THEREFOR
- C30B29/00—Single crystals or homogeneous polycrystalline material with defined structure characterised by the material or by their shape
- C30B29/54—Organic compounds
- C30B29/58—Macromolecular compounds
-
- C—CHEMISTRY; METALLURGY
- C30—CRYSTAL GROWTH
- C30B—SINGLE-CRYSTAL GROWTH; UNIDIRECTIONAL SOLIDIFICATION OF EUTECTIC MATERIAL OR UNIDIRECTIONAL DEMIXING OF EUTECTOID MATERIAL; REFINING BY ZONE-MELTING OF MATERIAL; PRODUCTION OF A HOMOGENEOUS POLYCRYSTALLINE MATERIAL WITH DEFINED STRUCTURE; SINGLE CRYSTALS OR HOMOGENEOUS POLYCRYSTALLINE MATERIAL WITH DEFINED STRUCTURE; AFTER-TREATMENT OF SINGLE CRYSTALS OR A HOMOGENEOUS POLYCRYSTALLINE MATERIAL WITH DEFINED STRUCTURE; APPARATUS THEREFOR
- C30B7/00—Single-crystal growth from solutions using solvents which are liquid at normal temperature, e.g. aqueous solutions
- C30B7/02—Single-crystal growth from solutions using solvents which are liquid at normal temperature, e.g. aqueous solutions by evaporation of the solvent
-
- C—CHEMISTRY; METALLURGY
- C30—CRYSTAL GROWTH
- C30B—SINGLE-CRYSTAL GROWTH; UNIDIRECTIONAL SOLIDIFICATION OF EUTECTIC MATERIAL OR UNIDIRECTIONAL DEMIXING OF EUTECTOID MATERIAL; REFINING BY ZONE-MELTING OF MATERIAL; PRODUCTION OF A HOMOGENEOUS POLYCRYSTALLINE MATERIAL WITH DEFINED STRUCTURE; SINGLE CRYSTALS OR HOMOGENEOUS POLYCRYSTALLINE MATERIAL WITH DEFINED STRUCTURE; AFTER-TREATMENT OF SINGLE CRYSTALS OR A HOMOGENEOUS POLYCRYSTALLINE MATERIAL WITH DEFINED STRUCTURE; APPARATUS THEREFOR
- C30B7/00—Single-crystal growth from solutions using solvents which are liquid at normal temperature, e.g. aqueous solutions
- C30B7/14—Single-crystal growth from solutions using solvents which are liquid at normal temperature, e.g. aqueous solutions the crystallising materials being formed by chemical reactions in the solution
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/13—Crystalline forms, e.g. polymorphs
Description
本出願は、2016年7月18に出願された米国仮特許出願第62/363,424号の恩典を主張する。前記出願の全教示は参照により本明細書に組み込まれる。

〔1〕下記構造式により表される化合物(I)の臭化水素酸塩:

またはその溶媒和物であって、化合物(I)と臭化水素酸との間のモル比は1:1である、臭化水素酸塩またはその溶媒和物、
〔2〕前記塩は非溶媒和形態である、〔1〕記載の臭化水素酸塩、
〔3〕前記臭化水素酸塩は結晶性である、〔1〕または〔2〕記載の臭化水素酸塩、
〔4〕前記結晶性臭化水素酸塩は、2θにおける5.9°、11.9°、21.6°、および22.0°±0.2のX線粉末回折パターンピークの3つまたは4つにより特徴付けられる、〔3〕記載の臭化水素酸塩、
〔5〕前記結晶性臭化水素酸塩は、2θにおける5.9°、10.0°、11.9°、13.8°、17.3°、19.4°、21.3°、21.6°、および22.0°±0.2のX線粉末回折パターンピークのうち、任意の3つ、4つ、5つ、6つ、7つ、または8つにより特徴付けられる、〔3〕記載の臭化水素酸塩、
〔6〕前記結晶性臭化水素酸塩は、2θにおける5.9°、10.0°、11.9°、13.8°、17.3°、19.4°、21.3°、21.6°、および22.0°±0.2のX線粉末回折パターンピークにより特徴付けられる、〔3〕記載の臭化水素酸塩、
〔7〕前記臭化水素酸塩は、図7に示されるX線粉末回折パターンピークにより特徴付けられる、〔3〕~〔6〕のいずれか一項記載の臭化水素酸塩、
〔8〕前記臭化水素酸塩は221±2℃の示差走査熱量計(DSC)ピーク相転移温度により特徴付けられる、〔1〕~〔7〕のいずれか一項記載の臭化水素酸塩、
〔9〕〔1〕~〔8〕のいずれか一項記載の臭化水素酸塩、および薬学的に許容される担体または希釈剤を含む、医薬組成物、
〔10〕有効量の〔1〕~〔8〕のいずれか一項記載の臭化水素酸塩を含む、TTKを過剰発現する癌を有する被験体を治療するための医薬組成物、
〔11〕前記癌は膵臓癌、前立腺癌、肺癌、メラノーマ、乳癌、結腸癌、または卵巣癌である、〔10〕記載の医薬組成物、
〔12〕前記癌は肺癌、乳癌および結腸癌である、〔11〕記載の医薬組成物、
〔13〕前記癌は乳癌である、〔12〕記載の医薬組成物
に関する。
発明の1つの実施形態では、およそ1:2のモル比を有する化合物(I):リン酸共結晶が提供される(本明細書の実施例を参照されたい)。発明はまた、化合物(I):リン酸の他のモル比の共結晶を包含する。
本発明の1つの実施形態では、およそ1:1のモル比を有する化合物(I)臭化水素酸が提供される(本明細書の実施例を参照されたい)。1つの実施形態では、1:1化合物(I)臭化水素酸塩の少なくとも特定の重量パーセンテージは結晶性である。
1:1化合物(I)臭化水素酸塩ならびに化合物(I)およびリン酸の共結晶の特定の固体形態は、例えば、遅蒸発、徐冷、およびアンチソルベント沈殿により調製することができる。
本教示の別の態様は、被験体に、有効量の、本明細書で記載される化合物(I)およびリン酸の共結晶(または化合物(I)の臭化水素酸塩)を投与することを含む、癌を有する被験体を治療する方法に関する。1つの実施形態では、本明細書で記載される共結晶(または化合物(I)の臭化水素酸塩)は腫瘍の増殖を阻害する。例えば、本明細書で記載される共結晶(または化合物(I)の臭化水素酸塩)はTTKを過剰発現する腫瘍の増殖を阻害する。
本明細書で開示される1:1化合物(I)臭化水素酸塩(または化合物(I)およびリン酸共結晶)は、被験体に投与するための医薬組成物に好適に製剤化することができる。
略語:
BSA ベンゼンスルホン酸
DSC 示差走査熱量測定
Equiv 当量
NA 入手不可能
NMP N-メチル-2-ピロリドン
pTSA パラ-トルエンスルホン酸
RH 相対湿度
rt 室温
temp 温度
TGA 熱重量分析
THF テトラヒドロフラン
wt% 重量パーセント
XRPD X線粉末回折
X線粉末回折(XRPD)
XRPD分析を、DAVINCI構成を有するBruker D8 Discover回折計を用いて、試料を1.5~45°2θ角で走査して実施した。およそ、1-2mgの各スクリーニング試料を使用した。
SC-XRD分析を、293KでOxford Diffractions製のX-Calibur回折計で、Mo Kα源およびグラファイトモノクロメータを用いて実施した。構造解析および精密化を、それぞれ、SHELXS-97およびSHELXL-97を使用して実施した。
3-10mgの材料を空のアルミニウムパンに秤量し、同時Setaram LABSYS EVO温度-重量測定/示差走査熱量計(TGA-DTA/DSC)にロボットを用いてロードし、室温で保持した。試料をそれから、10℃/分の速度で30℃から350℃まで加熱し、その時間の間、試料重量の変化を任意の示差熱イベントと共に記録した。窒素をパージガスとして100cm3/分の流速で使用した。分析前に、機器を秤量し、温度を、それぞれ、100mg参照重量およびインジウム参照標準を用いて較正した。試料分析をCALISTOソフトウェアの助けにより実施し、この場合、熱イベントの対応する質量損失および温度をオンセット温度とし、製造者の仕様書に従い測定した。全ての分析を10℃/分の加熱速度を用いて実施し、バックグラウンドを減算した。
1-4mgの材料をアルミニウムDSCパンに秤量し、アルミニウム蓋で密封せずに封印した。試料パンをそれからTA Instruments Q2000(冷却器が取り付けられている)にロードした。安定な熱流応答が35℃で得られるとすぐに、試料および基準を300℃まで10℃/分の速度で加熱し、得られた熱流応答をモニタした。分析前に、インジウム参照標準を使用して機器を温度および熱流較正した。試料分析を TA Universal Analysis2000ソフトウェアの助けにより実施し、この場合、熱イベントの温度をオンセットおよびピーク温度とし、製造者の仕様書に従い測定した。
核磁気共鳴測定を、Bruker Avance DRX400機器で400MHzおよび室温で、DMSO-d6またはCD3ODを溶媒として内部標準なしで使用して、記録した。
15N固体核磁気共鳴分析を500MHz Bruker Avance III固体分光計で実施した。15NスペクトルをCP-MAS(マジック角回転下における交差分極)によりBruker 4mmプローブヘッド、2つのRFチャネル上で獲得し、試料回転数は7kHzとした。
塩スクリーンを、6つの溶媒(H2O、DMSO、1,4-ジオキサン、酢酸、MeOH/THF(1:1)、1-BuOH/MEK1:1、およびNMP)ならびに12の薬学的に許容される酸(HCl、HBr、H3PO4、H2SO4、pTSA、BSA、ナフタレンスルホン酸、エタンスルホン酸、メタンスルホン酸、エタンジスルホン酸、L-マレイン酸、および2-アミノスルホン酸)を用いて実施した。
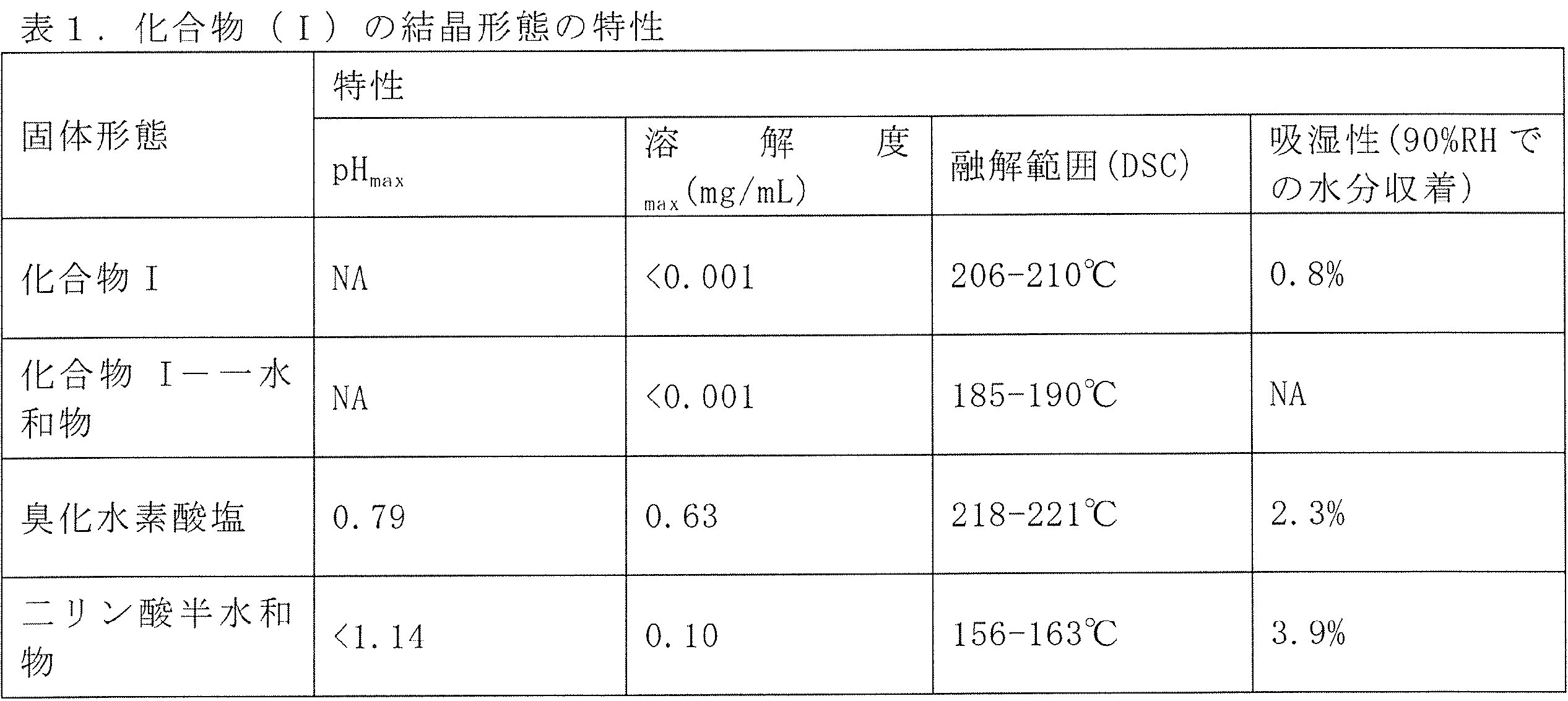
方法A:化合物(I)(605.5mg、1当量)をアセトン(14mL)中でスラリー化した。スラリーを室温で5分間撹拌し均質化させた。次に、H3PO4(459.46mg、4当量)のアセトン溶液(6mL)を迅速に添加した。黄色ゲルが得られ、混合物を室温で撹拌しながら2日間維持した。沈殿物をその後濾過し、アセトン(20mL)で洗浄し、4℃/10mbarで乾燥させた。得られた固体をさらに、TGA、XRPD、DSC、1H NMR、固体15N NMR、IR、Raman、および単結晶X線結晶解析により二リン酸半水和物として特徴付けた。


MeOH/THF(9.7mL、1:1V/V)の混合物に、ガラスバイアル中、室温で化合物(I)(500mg、1当量)を添加した。H3PO4(238mg、2当量)をそれから添加し、得られた混合物を室温で2時間の間撹拌した。溶液は変色したが、沈殿は生じなかった。透明溶液をそれから、1日間5℃で、それから1日間-20℃で放置させた。固体は得られなかった。溶媒をその後、30℃/10mbarで除去し、得られた固体をXRPDにより分析し、アモルファス固体であることが示された。1H NMR(400MHz、CD3OD)δ8.53-8.58(m、1H)、8.46-8.50(m、1H)、8.36(s、1H)、7.80-7.86(m、1H)、7.72-7.76(m、1H)、7.55-7.61(m、2H)、7.18(d、J=8.0Hz、1H)、5.92(s、1H)、3.52(d、J=6.8Hz、2H)、2.77-2.86(m、1H)、2.28-2.38(m、1H)、2.25(s、3H)、2.18-2.24(m、2H)、1.88-1.99(m、2H)、1.37(s、3H)、0.75-0.84(m、2H)、0.54-0.64(m、2H)。
1-ブタノールおよびMEK(60mL、1:1V/V)の混合物に化合物(I)(502mg、1当量)を添加した。スラリーを60℃で加熱し、HBr(1-ブタノールおよびMEK中4.8%溶液(2.5mL、2当量)を添加した。混合物を60℃で2時間の間、それから5℃で一晩撹拌した。それから、固体を濾過により回収し、ジエチルエーテルで洗浄し、XRPD、TGA、IR、RamanおよびDSCにより臭化水素酸塩として特徴付けた。

方法:
各形態の1回経口、カプセル中粉末用量を、3(n=3)匹の雌スプラーグドーリーラットおよび3匹の雄ビーグル犬(n=3)に5mg有効活性成分/kg体重で投与した。血漿をその後、LC/MSにより化合物血漿レベルについて分析した。
化合物を乳鉢と乳棒を用いて粉砕し、注意深く、化学天秤で風袋の重さを量ったゼラチンカプセル中に分注した。所望の化合物質量が達成されるまでロードし続けた。サイズ9カプセルをラット投与に使用し、サイズ3カプセルをイヌに使用した。注:化合物ロードは各形態の生物学的同等性比に対して補正した。
血漿試料(20μL)および添加済み血漿標準試料を、100ng/mLベラパミルを内部標準として含む氷冷アセトニトリルで5倍希釈した。試料および標準を、96ウェルフォーマットで0.22μmメンブランに通して濾過した。濾液をその後、水~30%アセトニトリルで希釈した。

血漿試料(20μL)および添加済み血漿標準試料を、内部標準として40ng/mLジクロフェナクを含む氷冷アセトニトリルで11倍希釈した。混合物を2分間ボルテックスし、遠心分離した。0.5μL上清をWaters Acquity BEH1.7μm 2.1×50mmカラムに0.6mL/分で、Acquity UPLCにより注入した。カラムを10%メタノールを含む水で平衡化させた。化合物を95%メタノールまでの勾配を用いて溶離させた。移動相は0.025%(v/v)ギ酸および1mM NH4OAcを含んだ。


[1]化合物(I)および共形成剤分子の共結晶

または前記共結晶の溶媒和物であって、前記共形成剤分子はリン酸である、共結晶またはその溶媒和物。
[2]前記共結晶中の化合物(I):リン酸のモル比は1:2である、[1]記載の共結晶。
[3]前記共結晶は水和物である、[1]または[2]記載の共結晶。
[4]前記共結晶中の化合物(I):リン酸:H2Oのモル比は1:2:1/2である、[1]~[3]のいずれか一項記載の共結晶。
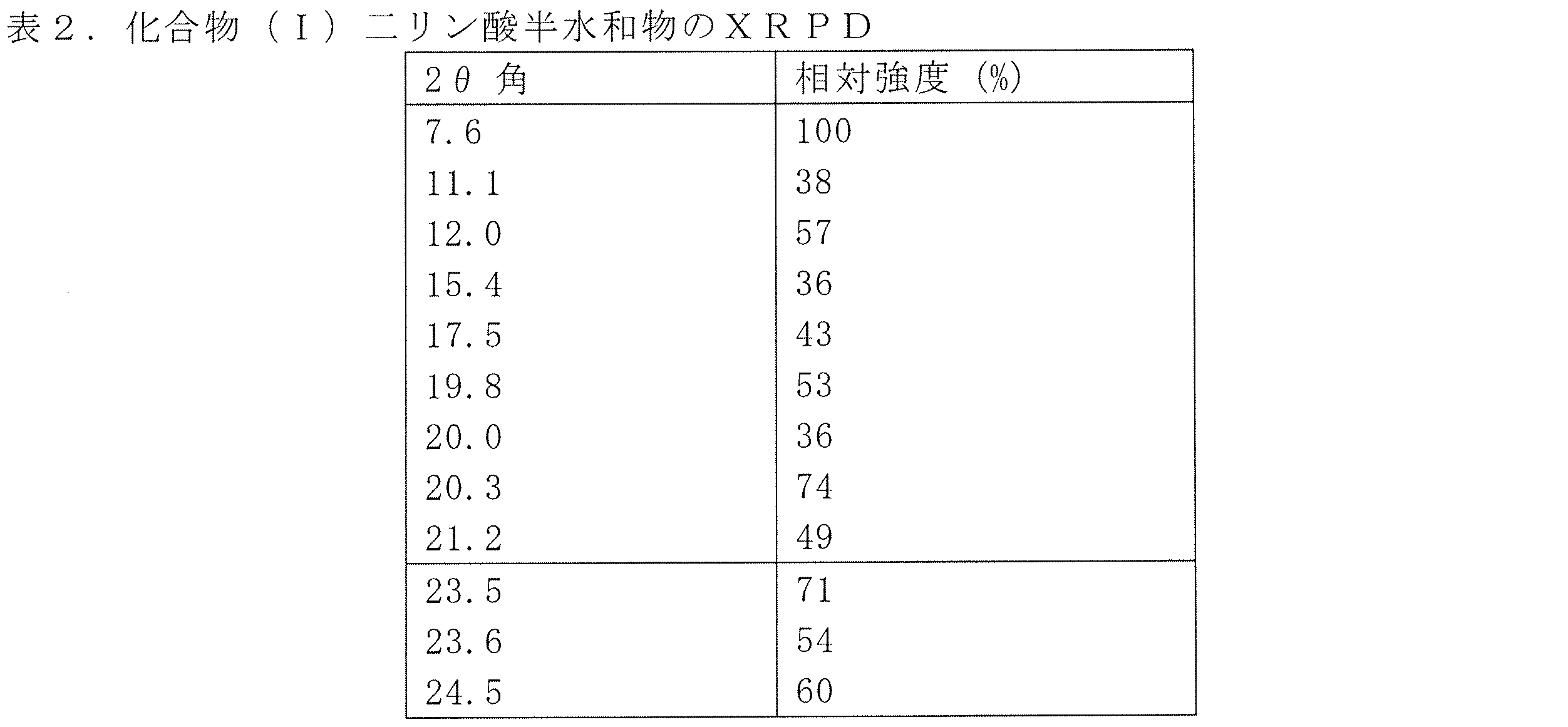
[5]前記共結晶は、2θにおける7.6°、12.0°、20.3°、23.5°、および24.5°±0.2のX線粉末回折パターンピークのうち、任意の3つ、4つまたは5つにより特徴付けられる、[1]~[4]のいずれか一項記載の共結晶。
[6]前記共結晶は、2θにおける7.6°、11.1°、12.0°、15.4°、17.5°、19.8°、20.0°、20.3°、21.2°、23.5°、23.6°、および24.5°±0.2のX線粉末回折パターンピークのうち、任意の3つ、4つ、5つ、6つ、7つ、8つ、9つ、10、または11により特徴付けられる、[1]~[4]のいずれか一項記載の共結晶。
[7]前記共結晶は、2θにおける7.6°、11.1°、12.0°、15.4°、17.5°、19.8°、20.0°、20.3°、21.2°、23.5°、23.6°、および24.5°±0.2のX線粉末回折パターンピークにより特徴付けられる、[1]~[4]のいずれか一項記載の共結晶。
[8]前記共結晶は、図1にて示されるX線粉末回折パターンにより特徴付けられる、[1]~[4]のいずれか一項記載の共結晶。
[9]前記共結晶は、160±4℃の示差走査熱量計(DSC)ピーク相転移温度により特徴付けられる、[1]~[8]のいずれか一項記載の共結晶。
[10]水素結合がリン酸の水素と化合物(I)のピリジン環の窒素原子との間で形成され、前記水素結合は2.5~2.9Åの距離を有する、[1]~[9]のいずれか一項記載の共結晶。
[11]前記共結晶は-122.5±2ppmの15N-固体核磁気共鳴分光法(15N-ssNMR)ピークにより特徴付けられ、-122.5ppmのピークの強度は、-302ppmのピークよりも少なくとも40-70%小さい、[1]~[10]のいずれか一項記載の共結晶。
[12][1]~[11]のいずれか一項記載の共結晶、および薬学的に許容される担体または希釈剤を含む、医薬組成物。
[13]下記構造式により表される化合物(I)の臭化水素酸塩:

またはその溶媒和物であって、化合物(I)と臭化水素酸との間のモル比は1:1である、臭化水素酸塩またはその溶媒和物。
[14]前記塩は非溶媒和形態である、[13]記載の臭化水素酸塩。
[15]前記臭化水素酸塩は結晶性である、[13]または[14]記載の臭化水素酸塩。
[16]前記結晶性臭化水素酸塩は、2θにおける5.9°、11.9°、21.6°、および22.0°±0.2のX線粉末回折パターンピークの3つまたは4つにより特徴付けられる、[15]記載の臭化水素酸塩。
[17]前記結晶性臭化水素酸塩は、2θにおける5.9°、10.0°、11.9°、13.8°、17.3°、19.4°、21.3°、21.6°、および22.0°±0.2のX線粉末回折パターンピークのうち、任意の3つ、4つ、5つ、6つ、7つ、または8つにより特徴付けられる、[15]記載の臭化水素酸塩。
[18]前記結晶性臭化水素酸塩は、2θにおける5.9°、10.0°、11.9°、13.8°、17.3°、19.4°、21.3°、21.6°、および22.0°±0.2のX線粉末回折パターンピークにより特徴付けられる、[15]記載の臭化水素酸塩。
[19]前記臭化水素酸塩は、図7に示されるX線粉末回折パターンピークにより特徴付けられる、[15]~[18]のいずれか一項記載の臭化水素酸塩。
[20]前記臭化水素酸塩は221±2℃の示差走査熱量計(DSC)ピーク相転移温度により特徴付けられる、[13]~[19]のいずれか一項記載の臭化水素酸塩。
[21][13]~[20]のいずれか一項記載の臭化水素酸塩、および薬学的に許容される担体または希釈剤を含む、医薬組成物。
[22][1]記載の化合物(I)およびリン酸の共結晶を調製する方法であって、好適な溶媒中のリン酸および化合物(I)の懸濁液を混合すること、ならびに前記溶媒を除去することを含む、方法。
[23]化合物(I)とリン酸との間のモル比が1:2~1:8である、[22]記載の方法。
[24]前記溶媒はアセトン、ジエチルエーテル、MeOH/THF、酢酸および1,4-ジオキサンである、[22]または[23]記載の方法。
[25]前記溶媒はTHF/MeOHであり、前記2つの溶媒間の比は3:2~2:3(V:V)である、[22]記載の方法。
[26]被験体に、有効量の、[1]~[11]のいずれか一項記載の共結晶、または有効量の、[13]~[20]のいずれか一項記載の臭化水素酸塩を投与することを含む、癌を有する被験体を治療する方法。
[27]前記癌は膵臓癌、前立腺癌、肺癌、メラノーマ、乳癌、結腸癌、または卵巣癌である、[26]記載の方法。
[28]前記癌は肺癌、乳癌および結腸癌である、[27]記載の方法。
[29]前記癌は乳癌である、[28]記載の方法。
Claims (11)
- 下記構造式により表される化合物(I)の臭化水素酸塩:

またはその溶媒和物であって、化合物(I)と臭化水素酸との間のモル比は1:1である、臭化水素酸塩またはその溶媒和物。 - 前記塩は非溶媒和形態である、請求項1記載の臭化水素酸塩。
- 前記臭化水素酸塩は結晶性である、請求項1または2記載の臭化水素酸塩。
- 前記結晶性臭化水素酸塩は、2θにおける5.9°、10.0°、11.9°、13.8°、17.3°、19.4°、21.3°、21.6°、および22.0°±0.2のX線粉末回折パターンピークのうち、任意の7つまたは8つにより特徴付けられる、請求項3記載の臭化水素酸塩。
- 前記結晶性臭化水素酸塩は、2θにおける5.9°、10.0°、11.9°、13.8°、17.3°、19.4°、21.3°、21.6°、および22.0°±0.2のX線粉末回折パターンピークにより特徴付けられる、請求項3記載の臭化水素酸塩。
- 前記臭化水素酸塩は221±2℃の示差走査熱量計(DSC)ピーク相転移温度により特徴付けられる、請求項1~5のいずれか一項記載の臭化水素酸塩。
- 請求項1~6のいずれか一項記載の臭化水素酸塩、および薬学的に許容される担体または希釈剤を含む、医薬組成物。
- 有効量の請求項1~6のいずれか一項記載の臭化水素酸塩を含む、癌を有する被験体を治療するための医薬組成物。
- 前記癌は膵臓癌、前立腺癌、肺癌、メラノーマ、乳癌、結腸癌、または卵巣癌である、請求項8記載の医薬組成物。
- 前記癌は肺癌、乳癌または結腸癌である、請求項9記載の医薬組成物。
- 前記癌は乳癌である、請求項10記載の医薬組成物。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201662363424P | 2016-07-18 | 2016-07-18 | |
| US62/363,424 | 2016-07-18 | ||
| JP2019501928A JP6961675B2 (ja) | 2016-07-18 | 2017-07-13 | Ttk阻害剤の固体形態 |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2019501928A Division JP6961675B2 (ja) | 2016-07-18 | 2017-07-13 | Ttk阻害剤の固体形態 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2022001598A JP2022001598A (ja) | 2022-01-06 |
| JP7263473B2 true JP7263473B2 (ja) | 2023-04-24 |
Family
ID=60991755
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2019501928A Active JP6961675B2 (ja) | 2016-07-18 | 2017-07-13 | Ttk阻害剤の固体形態 |
| JP2021167806A Active JP7263473B2 (ja) | 2016-07-18 | 2021-10-13 | Ttk阻害剤の固体形態 |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2019501928A Active JP6961675B2 (ja) | 2016-07-18 | 2017-07-13 | Ttk阻害剤の固体形態 |
Country Status (26)
| Country | Link |
|---|---|
| US (3) | US10584130B2 (ja) |
| EP (1) | EP3484888B1 (ja) |
| JP (2) | JP6961675B2 (ja) |
| KR (2) | KR102537088B1 (ja) |
| CN (2) | CN109476667B (ja) |
| AU (2) | AU2017299850B2 (ja) |
| BR (1) | BR112019000813A2 (ja) |
| CA (1) | CA3030230A1 (ja) |
| DK (1) | DK3484888T3 (ja) |
| ES (1) | ES2945108T3 (ja) |
| FI (1) | FI3484888T5 (ja) |
| HR (1) | HRP20230481T1 (ja) |
| HU (1) | HUE061872T2 (ja) |
| IL (1) | IL264173A (ja) |
| LT (1) | LT3484888T (ja) |
| MA (1) | MA45691A (ja) |
| MX (2) | MX2019000744A (ja) |
| NZ (1) | NZ749844A (ja) |
| PL (1) | PL3484888T3 (ja) |
| PT (1) | PT3484888T (ja) |
| RS (1) | RS64210B1 (ja) |
| RU (2) | RU2021124795A (ja) |
| SG (2) | SG11201900113UA (ja) |
| SI (1) | SI3484888T1 (ja) |
| TW (2) | TWI745400B (ja) |
| WO (1) | WO2018014116A1 (ja) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2021124795A (ru) | 2016-07-18 | 2021-09-14 | Юниверсити Хелс Нетуорк | Твердые формы ингибитора ttk |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015070349A1 (en) | 2012-11-16 | 2015-05-21 | University Health Network | Pyrazolopyrimidine compounds |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP4429604B2 (ja) | 2001-02-21 | 2010-03-10 | ノバルティス バクシンズ アンド ダイアグノスティックス,インコーポレーテッド | 診断におけるttkおよび癌における治療標的としてのttk |
| PT1812440E (pt) | 2004-11-04 | 2011-01-25 | Vertex Pharma | Pirazolo[1,5-a]pirimidinas úteis enquanto inibidores de proteínas cinases |
| TW201107329A (en) * | 2009-07-30 | 2011-03-01 | Oncotherapy Science Inc | Fused imidazole derivative having ttk inhibitory action |
| TWI541243B (zh) * | 2010-09-10 | 2016-07-11 | 拜耳知識產權公司 | 經取代咪唑并嗒 |
| US20130303532A1 (en) * | 2010-12-17 | 2013-11-14 | Bayer Intellectual Property Gmbh | Imidazopyrazines for use as mps-1 and tkk inhibitors in the treatment hyperproliferative disorders |
| JP6095857B2 (ja) * | 2013-11-15 | 2017-03-15 | ユニバーシティ・ヘルス・ネットワーク | ピラゾロピリミジン化合物 |
| RU2021124795A (ru) | 2016-07-18 | 2021-09-14 | Юниверсити Хелс Нетуорк | Твердые формы ингибитора ttk |
-
2017
- 2017-07-13 RU RU2021124795A patent/RU2021124795A/ru unknown
- 2017-07-13 KR KR1020197001829A patent/KR102537088B1/ko active IP Right Grant
- 2017-07-13 SG SG11201900113UA patent/SG11201900113UA/en unknown
- 2017-07-13 MX MX2019000744A patent/MX2019000744A/es unknown
- 2017-07-13 PL PL17830139.6T patent/PL3484888T3/pl unknown
- 2017-07-13 HU HUE17830139A patent/HUE061872T2/hu unknown
- 2017-07-13 US US16/318,426 patent/US10584130B2/en active Active
- 2017-07-13 SG SG10202103332UA patent/SG10202103332UA/en unknown
- 2017-07-13 RS RS20230383A patent/RS64210B1/sr unknown
- 2017-07-13 KR KR1020237017039A patent/KR20230074839A/ko not_active Application Discontinuation
- 2017-07-13 SI SI201731352T patent/SI3484888T1/sl unknown
- 2017-07-13 BR BR112019000813-6A patent/BR112019000813A2/pt unknown
- 2017-07-13 NZ NZ749844A patent/NZ749844A/en unknown
- 2017-07-13 FI FIEP17830139.6T patent/FI3484888T5/fi active
- 2017-07-13 CN CN201780043473.1A patent/CN109476667B/zh active Active
- 2017-07-13 HR HRP20230481TT patent/HRP20230481T1/hr unknown
- 2017-07-13 PT PT178301396T patent/PT3484888T/pt unknown
- 2017-07-13 RU RU2019101109A patent/RU2753905C2/ru active
- 2017-07-13 CA CA3030230A patent/CA3030230A1/en active Pending
- 2017-07-13 DK DK17830139.6T patent/DK3484888T3/da active
- 2017-07-13 EP EP17830139.6A patent/EP3484888B1/en active Active
- 2017-07-13 ES ES17830139T patent/ES2945108T3/es active Active
- 2017-07-13 AU AU2017299850A patent/AU2017299850B2/en active Active
- 2017-07-13 CN CN202210644487.1A patent/CN115093416B/zh active Active
- 2017-07-13 WO PCT/CA2017/050848 patent/WO2018014116A1/en unknown
- 2017-07-13 MA MA045691A patent/MA45691A/fr unknown
- 2017-07-13 JP JP2019501928A patent/JP6961675B2/ja active Active
- 2017-07-13 LT LTEPPCT/CA2017/050848T patent/LT3484888T/lt unknown
- 2017-07-18 TW TW106123975A patent/TWI745400B/zh active
- 2017-07-18 TW TW110137560A patent/TWI824313B/zh active
-
2019
- 2019-01-09 IL IL264173A patent/IL264173A/en unknown
- 2019-01-17 MX MX2021008658A patent/MX2021008658A/es unknown
-
2020
- 2020-03-02 US US16/806,392 patent/US11104681B2/en active Active
-
2021
- 2021-08-24 AU AU2021221447A patent/AU2021221447B2/en active Active
- 2021-08-27 US US17/459,725 patent/US11878980B2/en active Active
- 2021-10-13 JP JP2021167806A patent/JP7263473B2/ja active Active
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015070349A1 (en) | 2012-11-16 | 2015-05-21 | University Health Network | Pyrazolopyrimidine compounds |
Non-Patent Citations (3)
| Title |
|---|
| 平山令明,有機化合物結晶作製ハンドブック,2008年,pp. 17-23,37-40,45-51,57-65 |
| 長瀬博監訳,最新 創薬化学 下巻,株式会社テクノミック,1999年09月25日,pp. 347-365 |
| 高田則幸,創薬段階における原薬Formスクリーニングと選択,PHARM STAGE,Vol.6, No.10,2007年01月15日,p.20-25 |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN108349891B (zh) | 用于s1p1受体相关病症的化合物的结晶l-精氨酸盐 | |
| AU2014336929B2 (en) | Salt and crystal forms of PLK-4 inhibitor | |
| AU2014336929A1 (en) | Salt and crystal forms of PLK-4 inhibitor | |
| TW201335161A (zh) | 二氫吡咯并[1,2-c]咪唑基皮質醛酮素合成酶或芳族酶抑制劑之新穎型式及鹽 | |
| JP7263473B2 (ja) | Ttk阻害剤の固体形態 | |
| KR20230061431A (ko) | 아데노신 a2b 수용체 길항제의 공결정 | |
| NZ790209A (en) | Solid forms of ttk inhibitor | |
| CA3137191A1 (en) | Crystal form s4 of the plk4 inhibitor (ir,2s)-(e)-2-(3-(4-((cis-2,6-dimethylmorpholino)methyl)styryl)- 1 h-imidazol-6-yl)-5'-methoxyspiro[cyclopropane-l,3'-indolin]-2'-one fumarate | |
| AU2016315396A1 (en) | 1-(4-(2-((1-(3,4-difluorophenyl)-1 H-pyrazol-3-yl)methoxy)ethyl)piperazin-1-yl)ethanone salts | |
| TW201125861A (en) | CDC7 inhibitor salts | |
| WO2023116895A1 (zh) | Kras抑制剂的多晶型物及其制备方法和用途 | |
| WO2020222189A1 (en) | Crystalline form of 6-[4-[1-(propan-2-yl)piperidin-4-yl]-1,4-diazepan-1-yl]-n-(pyrdin-4-yl)pyridine-2-carboxamide |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20211015 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20221003 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20221229 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20230404 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20230412 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 7263473 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |