JP6974349B2 - ヘモグロビン異常症の処置のための材料及び方法 - Google Patents
ヘモグロビン異常症の処置のための材料及び方法 Download PDFInfo
- Publication number
- JP6974349B2 JP6974349B2 JP2018555646A JP2018555646A JP6974349B2 JP 6974349 B2 JP6974349 B2 JP 6974349B2 JP 2018555646 A JP2018555646 A JP 2018555646A JP 2018555646 A JP2018555646 A JP 2018555646A JP 6974349 B2 JP6974349 B2 JP 6974349B2
- Authority
- JP
- Japan
- Prior art keywords
- cells
- sequence
- dna
- cell
- gene
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 238000000034 method Methods 0.000 title claims description 402
- 102000001554 Hemoglobins Human genes 0.000 title description 60
- 108010054147 Hemoglobins Proteins 0.000 title description 60
- 238000011282 treatment Methods 0.000 title description 41
- 239000000463 material Substances 0.000 title description 14
- 230000005856 abnormality Effects 0.000 title description 6
- 210000004027 cell Anatomy 0.000 claims description 369
- 108091033409 CRISPR Proteins 0.000 claims description 193
- 101100493741 Homo sapiens BCL11A gene Proteins 0.000 claims description 183
- 150000007523 nucleic acids Chemical group 0.000 claims description 179
- 108090000765 processed proteins & peptides Proteins 0.000 claims description 123
- 230000001105 regulatory effect Effects 0.000 claims description 123
- 102000004196 processed proteins & peptides Human genes 0.000 claims description 122
- 229920001184 polypeptide Polymers 0.000 claims description 119
- 108090000623 proteins and genes Proteins 0.000 claims description 119
- 230000004048 modification Effects 0.000 claims description 106
- 238000012986 modification Methods 0.000 claims description 106
- 108020004414 DNA Proteins 0.000 claims description 99
- 230000005782 double-strand break Effects 0.000 claims description 96
- 108020005004 Guide RNA Proteins 0.000 claims description 90
- 102000004533 Endonucleases Human genes 0.000 claims description 81
- 108010042407 Endonucleases Proteins 0.000 claims description 81
- 108091028043 Nucleic acid sequence Proteins 0.000 claims description 77
- 102000040430 polynucleotide Human genes 0.000 claims description 70
- 108091033319 polynucleotide Proteins 0.000 claims description 70
- 239000002157 polynucleotide Substances 0.000 claims description 70
- 230000037430 deletion Effects 0.000 claims description 68
- 238000012217 deletion Methods 0.000 claims description 68
- 230000002103 transcriptional effect Effects 0.000 claims description 62
- 230000005783 single-strand break Effects 0.000 claims description 54
- 108010077850 Nuclear Localization Signals Proteins 0.000 claims description 51
- 238000010362 genome editing Methods 0.000 claims description 47
- 102000004169 proteins and genes Human genes 0.000 claims description 45
- 108010008532 Deoxyribonuclease I Proteins 0.000 claims description 44
- 102000007260 Deoxyribonuclease I Human genes 0.000 claims description 44
- 230000022532 regulation of transcription, DNA-dependent Effects 0.000 claims description 39
- 230000037431 insertion Effects 0.000 claims description 35
- 238000003780 insertion Methods 0.000 claims description 35
- 239000013598 vector Substances 0.000 claims description 35
- 210000003743 erythrocyte Anatomy 0.000 claims description 29
- 150000002632 lipids Chemical class 0.000 claims description 29
- 241000193996 Streptococcus pyogenes Species 0.000 claims description 28
- 230000002779 inactivation Effects 0.000 claims description 27
- 102000004389 Ribonucleoproteins Human genes 0.000 claims description 23
- 108010081734 Ribonucleoproteins Proteins 0.000 claims description 23
- 239000002299 complementary DNA Substances 0.000 claims description 23
- 229920002477 rna polymer Polymers 0.000 claims description 22
- 241000829100 Macaca mulatta polyomavirus 1 Species 0.000 claims description 21
- 239000002105 nanoparticle Substances 0.000 claims description 20
- 210000005260 human cell Anatomy 0.000 claims description 19
- 239000013611 chromosomal DNA Substances 0.000 claims description 18
- 238000004520 electroporation Methods 0.000 claims description 12
- 206010020751 Hypersensitivity Diseases 0.000 claims description 10
- 241000702421 Dependoparvovirus Species 0.000 claims description 8
- 208000003950 B-cell lymphoma Diseases 0.000 claims description 5
- 239000003623 enhancer Substances 0.000 claims description 5
- 230000004913 activation Effects 0.000 claims description 3
- 210000004899 c-terminal region Anatomy 0.000 claims description 3
- 238000005520 cutting process Methods 0.000 claims description 3
- 108091027544 Subgenomic mRNA Proteins 0.000 claims 4
- 108091092724 Noncoding DNA Proteins 0.000 claims 1
- 241000194017 Streptococcus Species 0.000 claims 1
- 102000039446 nucleic acids Human genes 0.000 description 155
- 108020004707 nucleic acids Proteins 0.000 description 155
- 239000002773 nucleotide Substances 0.000 description 129
- 125000003729 nucleotide group Chemical group 0.000 description 126
- 102000053602 DNA Human genes 0.000 description 97
- 210000000130 stem cell Anatomy 0.000 description 89
- 210000003958 hematopoietic stem cell Anatomy 0.000 description 78
- 102100031573 Hematopoietic progenitor cell antigen CD34 Human genes 0.000 description 77
- 101000777663 Homo sapiens Hematopoietic progenitor cell antigen CD34 Proteins 0.000 description 77
- 125000006850 spacer group Chemical group 0.000 description 76
- 238000003776 cleavage reaction Methods 0.000 description 65
- 108020004999 messenger RNA Proteins 0.000 description 65
- 230000007017 scission Effects 0.000 description 65
- 230000009368 gene silencing by RNA Effects 0.000 description 64
- 108091030071 RNAI Proteins 0.000 description 59
- 230000027455 binding Effects 0.000 description 54
- 238000009739 binding Methods 0.000 description 54
- 230000014509 gene expression Effects 0.000 description 52
- 101710163270 Nuclease Proteins 0.000 description 50
- 208000009869 Neu-Laxova syndrome Diseases 0.000 description 48
- 230000035772 mutation Effects 0.000 description 48
- 210000004263 induced pluripotent stem cell Anatomy 0.000 description 45
- 230000004064 dysfunction Effects 0.000 description 41
- 235000018102 proteins Nutrition 0.000 description 41
- 108091005886 Hemoglobin subunit gamma Proteins 0.000 description 38
- 102100038617 Hemoglobin subunit gamma-2 Human genes 0.000 description 38
- 230000000694 effects Effects 0.000 description 38
- 150000001413 amino acids Chemical class 0.000 description 36
- 108091079001 CRISPR RNA Proteins 0.000 description 35
- 239000000203 mixture Substances 0.000 description 33
- 230000008672 reprogramming Effects 0.000 description 33
- -1 NANOG Proteins 0.000 description 32
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 32
- 230000006780 non-homologous end joining Effects 0.000 description 32
- 108091034117 Oligonucleotide Proteins 0.000 description 31
- 230000004069 differentiation Effects 0.000 description 30
- 239000002679 microRNA Substances 0.000 description 30
- 235000001014 amino acid Nutrition 0.000 description 29
- 229940024606 amino acid Drugs 0.000 description 29
- 230000006870 function Effects 0.000 description 29
- 230000008439 repair process Effects 0.000 description 28
- 230000000295 complement effect Effects 0.000 description 25
- 108091070501 miRNA Proteins 0.000 description 24
- 238000001727 in vivo Methods 0.000 description 23
- 230000001965 increasing effect Effects 0.000 description 23
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 22
- 125000005647 linker group Chemical group 0.000 description 22
- 108091023040 Transcription factor Proteins 0.000 description 21
- 102000040945 Transcription factor Human genes 0.000 description 21
- 210000002901 mesenchymal stem cell Anatomy 0.000 description 21
- 210000001082 somatic cell Anatomy 0.000 description 21
- 230000008901 benefit Effects 0.000 description 20
- 230000000925 erythroid effect Effects 0.000 description 20
- 230000008685 targeting Effects 0.000 description 20
- 230000037361 pathway Effects 0.000 description 19
- 210000001185 bone marrow Anatomy 0.000 description 18
- 201000010099 disease Diseases 0.000 description 18
- 239000013612 plasmid Substances 0.000 description 18
- 230000003612 virological effect Effects 0.000 description 18
- 241000700605 Viruses Species 0.000 description 17
- 238000004458 analytical method Methods 0.000 description 17
- 238000013459 approach Methods 0.000 description 17
- 238000002347 injection Methods 0.000 description 17
- 239000007924 injection Substances 0.000 description 17
- 241000894006 Bacteria Species 0.000 description 16
- 108700011259 MicroRNAs Proteins 0.000 description 16
- 108010017070 Zinc Finger Nucleases Proteins 0.000 description 16
- 238000002054 transplantation Methods 0.000 description 15
- 230000003827 upregulation Effects 0.000 description 15
- 230000004568 DNA-binding Effects 0.000 description 14
- 208000035475 disorder Diseases 0.000 description 14
- 230000003993 interaction Effects 0.000 description 14
- 150000001875 compounds Chemical class 0.000 description 13
- 238000004519 manufacturing process Methods 0.000 description 13
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 12
- 230000034431 double-strand break repair via homologous recombination Effects 0.000 description 12
- 239000003814 drug Substances 0.000 description 12
- 230000001404 mediated effect Effects 0.000 description 12
- 208000024891 symptom Diseases 0.000 description 12
- 102100027685 Hemoglobin subunit alpha Human genes 0.000 description 11
- 108091005902 Hemoglobin subunit alpha Proteins 0.000 description 11
- 102100021519 Hemoglobin subunit beta Human genes 0.000 description 11
- 108020004459 Small interfering RNA Proteins 0.000 description 11
- 230000015572 biosynthetic process Effects 0.000 description 11
- OPTASPLRGRRNAP-UHFFFAOYSA-N cytosine Natural products NC=1C=CNC(=O)N=1 OPTASPLRGRRNAP-UHFFFAOYSA-N 0.000 description 11
- 102000018146 globin Human genes 0.000 description 11
- 108060003196 globin Proteins 0.000 description 11
- 230000001939 inductive effect Effects 0.000 description 11
- 230000003834 intracellular effect Effects 0.000 description 11
- 230000002829 reductive effect Effects 0.000 description 11
- 235000000346 sugar Nutrition 0.000 description 11
- 238000013518 transcription Methods 0.000 description 11
- 230000035897 transcription Effects 0.000 description 11
- 108010017080 Granulocyte Colony-Stimulating Factor Proteins 0.000 description 10
- 102000004269 Granulocyte Colony-Stimulating Factor Human genes 0.000 description 10
- 108091005904 Hemoglobin subunit beta Proteins 0.000 description 10
- 239000002253 acid Substances 0.000 description 10
- UYTPUPDQBNUYGX-UHFFFAOYSA-N guanine Chemical class O=C1NC(N)=NC2=C1N=CN2 UYTPUPDQBNUYGX-UHFFFAOYSA-N 0.000 description 10
- 230000001976 improved effect Effects 0.000 description 10
- 230000009437 off-target effect Effects 0.000 description 10
- 239000002243 precursor Substances 0.000 description 10
- 230000008569 process Effects 0.000 description 10
- 238000012163 sequencing technique Methods 0.000 description 10
- RYVNIFSIEDRLSJ-UHFFFAOYSA-N 5-(hydroxymethyl)cytosine Chemical compound NC=1NC(=O)N=CC=1CO RYVNIFSIEDRLSJ-UHFFFAOYSA-N 0.000 description 9
- 230000007018 DNA scission Effects 0.000 description 9
- 241000699670 Mus sp. Species 0.000 description 9
- 239000000872 buffer Substances 0.000 description 9
- 239000013604 expression vector Substances 0.000 description 9
- 238000002955 isolation Methods 0.000 description 9
- 230000000670 limiting effect Effects 0.000 description 9
- 239000002777 nucleoside Substances 0.000 description 9
- 239000004055 small Interfering RNA Substances 0.000 description 9
- 230000001225 therapeutic effect Effects 0.000 description 9
- 238000002560 therapeutic procedure Methods 0.000 description 9
- 230000014616 translation Effects 0.000 description 9
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 8
- 102000040650 (ribonucleotides)n+m Human genes 0.000 description 8
- 241000282412 Homo Species 0.000 description 8
- 108091092195 Intron Proteins 0.000 description 8
- 206010028980 Neoplasm Diseases 0.000 description 8
- 238000007792 addition Methods 0.000 description 8
- 230000033228 biological regulation Effects 0.000 description 8
- 238000012239 gene modification Methods 0.000 description 8
- 230000006801 homologous recombination Effects 0.000 description 8
- 238000002744 homologous recombination Methods 0.000 description 8
- 238000002513 implantation Methods 0.000 description 8
- 239000002245 particle Substances 0.000 description 8
- 210000005259 peripheral blood Anatomy 0.000 description 8
- 239000011886 peripheral blood Substances 0.000 description 8
- 230000002441 reversible effect Effects 0.000 description 8
- 238000006467 substitution reaction Methods 0.000 description 8
- 210000001519 tissue Anatomy 0.000 description 8
- 238000013519 translation Methods 0.000 description 8
- 239000003981 vehicle Substances 0.000 description 8
- GFFGJBXGBJISGV-UHFFFAOYSA-N Adenine Chemical compound NC1=NC=NC2=C1N=CN2 GFFGJBXGBJISGV-UHFFFAOYSA-N 0.000 description 7
- 229930024421 Adenine Natural products 0.000 description 7
- 230000033616 DNA repair Effects 0.000 description 7
- 108091093037 Peptide nucleic acid Proteins 0.000 description 7
- 229960000643 adenine Drugs 0.000 description 7
- 125000000217 alkyl group Chemical group 0.000 description 7
- 230000000740 bleeding effect Effects 0.000 description 7
- 210000000601 blood cell Anatomy 0.000 description 7
- 239000003153 chemical reaction reagent Substances 0.000 description 7
- 239000003795 chemical substances by application Substances 0.000 description 7
- 238000001514 detection method Methods 0.000 description 7
- 238000000684 flow cytometry Methods 0.000 description 7
- 230000004927 fusion Effects 0.000 description 7
- 230000006698 induction Effects 0.000 description 7
- 230000015788 innate immune response Effects 0.000 description 7
- 239000002609 medium Substances 0.000 description 7
- 238000005457 optimization Methods 0.000 description 7
- 229910052760 oxygen Inorganic materials 0.000 description 7
- 229920001223 polyethylene glycol Polymers 0.000 description 7
- 208000007056 sickle cell anemia Diseases 0.000 description 7
- 230000009885 systemic effect Effects 0.000 description 7
- 238000001890 transfection Methods 0.000 description 7
- 239000002202 Polyethylene glycol Substances 0.000 description 6
- ISAKRJDGNUQOIC-UHFFFAOYSA-N Uracil Chemical compound O=C1C=CNC(=O)N1 ISAKRJDGNUQOIC-UHFFFAOYSA-N 0.000 description 6
- 238000003556 assay Methods 0.000 description 6
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 6
- 229940104302 cytosine Drugs 0.000 description 6
- 238000013461 design Methods 0.000 description 6
- 238000011161 development Methods 0.000 description 6
- 230000018109 developmental process Effects 0.000 description 6
- 229940079593 drug Drugs 0.000 description 6
- 238000002474 experimental method Methods 0.000 description 6
- 238000006062 fragmentation reaction Methods 0.000 description 6
- 230000030279 gene silencing Effects 0.000 description 6
- 238000001415 gene therapy Methods 0.000 description 6
- 230000002068 genetic effect Effects 0.000 description 6
- 230000003394 haemopoietic effect Effects 0.000 description 6
- 230000028993 immune response Effects 0.000 description 6
- 230000000415 inactivating effect Effects 0.000 description 6
- 230000010354 integration Effects 0.000 description 6
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 6
- 239000001301 oxygen Substances 0.000 description 6
- 210000001778 pluripotent stem cell Anatomy 0.000 description 6
- 238000011160 research Methods 0.000 description 6
- 150000003384 small molecules Chemical class 0.000 description 6
- RWQNBRDOKXIBIV-UHFFFAOYSA-N thymine Chemical compound CC1=CNC(=O)NC1=O RWQNBRDOKXIBIV-UHFFFAOYSA-N 0.000 description 6
- 239000013603 viral vector Substances 0.000 description 6
- 241000701022 Cytomegalovirus Species 0.000 description 5
- 102220605874 Cytosolic arginine sensor for mTORC1 subunit 2_D10A_mutation Human genes 0.000 description 5
- 238000010442 DNA editing Methods 0.000 description 5
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 5
- 241001465754 Metazoa Species 0.000 description 5
- 238000012228 RNA interference-mediated gene silencing Methods 0.000 description 5
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 5
- YIJVOACVHQZMKI-JXOAFFINSA-N [[(2r,3s,4r,5r)-5-(4-amino-5-methyl-2-oxopyrimidin-1-yl)-3,4-dihydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl] phosphono hydrogen phosphate Chemical compound O=C1N=C(N)C(C)=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)O1 YIJVOACVHQZMKI-JXOAFFINSA-N 0.000 description 5
- 239000004480 active ingredient Substances 0.000 description 5
- 230000003321 amplification Effects 0.000 description 5
- 238000000137 annealing Methods 0.000 description 5
- 238000001574 biopsy Methods 0.000 description 5
- 210000002798 bone marrow cell Anatomy 0.000 description 5
- 230000015556 catabolic process Effects 0.000 description 5
- 235000012000 cholesterol Nutrition 0.000 description 5
- 230000003247 decreasing effect Effects 0.000 description 5
- 238000006731 degradation reaction Methods 0.000 description 5
- 230000012010 growth Effects 0.000 description 5
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 5
- 235000014304 histidine Nutrition 0.000 description 5
- 238000009396 hybridization Methods 0.000 description 5
- 238000000338 in vitro Methods 0.000 description 5
- 239000002502 liposome Substances 0.000 description 5
- 230000007246 mechanism Effects 0.000 description 5
- 238000003199 nucleic acid amplification method Methods 0.000 description 5
- 125000003835 nucleoside group Chemical group 0.000 description 5
- 230000036961 partial effect Effects 0.000 description 5
- 239000000546 pharmaceutical excipient Substances 0.000 description 5
- 239000000047 product Substances 0.000 description 5
- 150000003212 purines Chemical class 0.000 description 5
- 230000006798 recombination Effects 0.000 description 5
- 238000005215 recombination Methods 0.000 description 5
- 150000003839 salts Chemical class 0.000 description 5
- 238000003786 synthesis reaction Methods 0.000 description 5
- WAEXFXRVDQXREF-UHFFFAOYSA-N vorinostat Chemical compound ONC(=O)CCCCCCC(=O)NC1=CC=CC=C1 WAEXFXRVDQXREF-UHFFFAOYSA-N 0.000 description 5
- 229910052725 zinc Inorganic materials 0.000 description 5
- 239000011701 zinc Substances 0.000 description 5
- OVONXEQGWXGFJD-UHFFFAOYSA-N 4-sulfanylidene-1h-pyrimidin-2-one Chemical compound SC=1C=CNC(=O)N=1 OVONXEQGWXGFJD-UHFFFAOYSA-N 0.000 description 4
- LRSASMSXMSNRBT-UHFFFAOYSA-N 5-methylcytosine Chemical compound CC1=CNC(=O)N=C1N LRSASMSXMSNRBT-UHFFFAOYSA-N 0.000 description 4
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 4
- 108090000994 Catalytic RNA Proteins 0.000 description 4
- 102000053642 Catalytic RNA Human genes 0.000 description 4
- 108020004705 Codon Proteins 0.000 description 4
- 102000004127 Cytokines Human genes 0.000 description 4
- 108090000695 Cytokines Proteins 0.000 description 4
- 108010093099 Endoribonucleases Proteins 0.000 description 4
- 102000002494 Endoribonucleases Human genes 0.000 description 4
- 108700024394 Exon Proteins 0.000 description 4
- 108090000353 Histone deacetylase Proteins 0.000 description 4
- 102100038720 Histone deacetylase 9 Human genes 0.000 description 4
- 101100220044 Homo sapiens CD34 gene Proteins 0.000 description 4
- 101000687905 Homo sapiens Transcription factor SOX-2 Proteins 0.000 description 4
- FDGQSTZJBFJUBT-UHFFFAOYSA-N Hypoxanthine Natural products O=C1NC=NC2=C1NC=N2 FDGQSTZJBFJUBT-UHFFFAOYSA-N 0.000 description 4
- 241000124008 Mammalia Species 0.000 description 4
- 241000699666 Mus <mouse, genus> Species 0.000 description 4
- 240000002853 Nelumbo nucifera Species 0.000 description 4
- 235000006508 Nelumbo nucifera Nutrition 0.000 description 4
- 235000006510 Nelumbo pentapetala Nutrition 0.000 description 4
- 229910019142 PO4 Inorganic materials 0.000 description 4
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 4
- 108091028113 Trans-activating crRNA Proteins 0.000 description 4
- 102100024270 Transcription factor SOX-2 Human genes 0.000 description 4
- 150000001408 amides Chemical group 0.000 description 4
- 238000003491 array Methods 0.000 description 4
- 230000001580 bacterial effect Effects 0.000 description 4
- 208000005980 beta thalassemia Diseases 0.000 description 4
- 210000004369 blood Anatomy 0.000 description 4
- 239000008280 blood Substances 0.000 description 4
- 201000011510 cancer Diseases 0.000 description 4
- 230000003197 catalytic effect Effects 0.000 description 4
- 125000002091 cationic group Chemical group 0.000 description 4
- 230000024245 cell differentiation Effects 0.000 description 4
- 230000003833 cell viability Effects 0.000 description 4
- 230000001413 cellular effect Effects 0.000 description 4
- 238000006243 chemical reaction Methods 0.000 description 4
- 239000000539 dimer Substances 0.000 description 4
- 239000012636 effector Substances 0.000 description 4
- 238000001917 fluorescence detection Methods 0.000 description 4
- 238000009472 formulation Methods 0.000 description 4
- 238000013467 fragmentation Methods 0.000 description 4
- 239000000833 heterodimer Substances 0.000 description 4
- 238000004128 high performance liquid chromatography Methods 0.000 description 4
- 238000001802 infusion Methods 0.000 description 4
- 239000007788 liquid Substances 0.000 description 4
- 230000009438 off-target cleavage Effects 0.000 description 4
- 235000021317 phosphate Nutrition 0.000 description 4
- 150000003904 phospholipids Chemical class 0.000 description 4
- YIQPUIGJQJDJOS-UHFFFAOYSA-N plerixafor Chemical compound C=1C=C(CN2CCNCCCNCCNCCC2)C=CC=1CN1CCCNCCNCCCNCC1 YIQPUIGJQJDJOS-UHFFFAOYSA-N 0.000 description 4
- 229960002169 plerixafor Drugs 0.000 description 4
- 230000010076 replication Effects 0.000 description 4
- 108091092562 ribozyme Proteins 0.000 description 4
- 239000000243 solution Substances 0.000 description 4
- RYYWUUFWQRZTIU-UHFFFAOYSA-K thiophosphate Chemical compound [O-]P([O-])([O-])=S RYYWUUFWQRZTIU-UHFFFAOYSA-K 0.000 description 4
- 230000005030 transcription termination Effects 0.000 description 4
- 238000011144 upstream manufacturing Methods 0.000 description 4
- 229940035893 uracil Drugs 0.000 description 4
- 229960000237 vorinostat Drugs 0.000 description 4
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 4
- WCAWZARVUUEHRX-FRKDHNBESA-N (2e,4e,6r)-7-[4-(dimethylamino)phenyl]-n-hydroxy-6-methyl-7-oxohepta-2,4-dienamide Chemical compound ONC(=O)/C=C/C=C/[C@@H](C)C(=O)C1=CC=C(N(C)C)C=C1 WCAWZARVUUEHRX-FRKDHNBESA-N 0.000 description 3
- MSSXOMSJDRHRMC-UHFFFAOYSA-N 9H-purine-2,6-diamine Chemical compound NC1=NC(N)=C2NC=NC2=N1 MSSXOMSJDRHRMC-UHFFFAOYSA-N 0.000 description 3
- 239000013607 AAV vector Substances 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- 108091023037 Aptamer Proteins 0.000 description 3
- 108091060290 Chromatid Proteins 0.000 description 3
- 101100239628 Danio rerio myca gene Proteins 0.000 description 3
- 208000000059 Dyspnea Diseases 0.000 description 3
- 206010013975 Dyspnoeas Diseases 0.000 description 3
- 102100031690 Erythroid transcription factor Human genes 0.000 description 3
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 3
- 241000206602 Eukaryota Species 0.000 description 3
- 108010044495 Fetal Hemoglobin Proteins 0.000 description 3
- 108060003760 HNH nuclease Proteins 0.000 description 3
- 102000029812 HNH nuclease Human genes 0.000 description 3
- 101001139134 Homo sapiens Krueppel-like factor 4 Proteins 0.000 description 3
- 101001000998 Homo sapiens Protein phosphatase 1 regulatory subunit 12C Proteins 0.000 description 3
- 101000669402 Homo sapiens Toll-like receptor 7 Proteins 0.000 description 3
- 101000800483 Homo sapiens Toll-like receptor 8 Proteins 0.000 description 3
- 102100020677 Krueppel-like factor 4 Human genes 0.000 description 3
- 108700021430 Kruppel-Like Factor 4 Proteins 0.000 description 3
- 101710126211 POU domain, class 5, transcription factor 1 Proteins 0.000 description 3
- 239000004952 Polyamide Substances 0.000 description 3
- 102100035620 Protein phosphatase 1 regulatory subunit 12C Human genes 0.000 description 3
- 230000026279 RNA modification Effects 0.000 description 3
- 101100247004 Rattus norvegicus Qsox1 gene Proteins 0.000 description 3
- 102000006382 Ribonucleases Human genes 0.000 description 3
- 108010083644 Ribonucleases Proteins 0.000 description 3
- 206010039491 Sarcoma Diseases 0.000 description 3
- 108020004682 Single-Stranded DNA Proteins 0.000 description 3
- 108091036066 Three prime untranslated region Proteins 0.000 description 3
- 102100039390 Toll-like receptor 7 Human genes 0.000 description 3
- 102100033110 Toll-like receptor 8 Human genes 0.000 description 3
- 108700019146 Transgenes Proteins 0.000 description 3
- 150000007513 acids Chemical class 0.000 description 3
- 239000011543 agarose gel Substances 0.000 description 3
- 230000004075 alteration Effects 0.000 description 3
- 210000003719 b-lymphocyte Anatomy 0.000 description 3
- 238000009583 bone marrow aspiration Methods 0.000 description 3
- 210000000170 cell membrane Anatomy 0.000 description 3
- 238000002659 cell therapy Methods 0.000 description 3
- 230000008859 change Effects 0.000 description 3
- 210000004756 chromatid Anatomy 0.000 description 3
- 210000000349 chromosome Anatomy 0.000 description 3
- 230000003111 delayed effect Effects 0.000 description 3
- 238000009826 distribution Methods 0.000 description 3
- 239000003937 drug carrier Substances 0.000 description 3
- 238000005516 engineering process Methods 0.000 description 3
- 210000003527 eukaryotic cell Anatomy 0.000 description 3
- 206010016256 fatigue Diseases 0.000 description 3
- 230000002349 favourable effect Effects 0.000 description 3
- OVBPIULPVIDEAO-LBPRGKRZSA-N folic acid Chemical compound C=1N=C2NC(N)=NC(=O)C2=NC=1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 OVBPIULPVIDEAO-LBPRGKRZSA-N 0.000 description 3
- 108020001507 fusion proteins Proteins 0.000 description 3
- 102000037865 fusion proteins Human genes 0.000 description 3
- 238000012226 gene silencing method Methods 0.000 description 3
- 230000005017 genetic modification Effects 0.000 description 3
- 235000013617 genetically modified food Nutrition 0.000 description 3
- 210000001654 germ layer Anatomy 0.000 description 3
- 230000011132 hemopoiesis Effects 0.000 description 3
- 125000005842 heteroatom Chemical group 0.000 description 3
- 239000000411 inducer Substances 0.000 description 3
- 239000003112 inhibitor Substances 0.000 description 3
- 238000001990 intravenous administration Methods 0.000 description 3
- 101150111214 lin-28 gene Proteins 0.000 description 3
- 230000007774 longterm Effects 0.000 description 3
- 230000004060 metabolic process Effects 0.000 description 3
- 229910052751 metal Inorganic materials 0.000 description 3
- 239000002184 metal Substances 0.000 description 3
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 3
- 230000011987 methylation Effects 0.000 description 3
- 238000007069 methylation reaction Methods 0.000 description 3
- 108091027963 non-coding RNA Proteins 0.000 description 3
- 102000042567 non-coding RNA Human genes 0.000 description 3
- 210000004940 nucleus Anatomy 0.000 description 3
- 230000002093 peripheral effect Effects 0.000 description 3
- 229920002401 polyacrylamide Polymers 0.000 description 3
- 229920002647 polyamide Polymers 0.000 description 3
- 229920000768 polyamine Polymers 0.000 description 3
- 229920000642 polymer Polymers 0.000 description 3
- 230000002062 proliferating effect Effects 0.000 description 3
- 230000035755 proliferation Effects 0.000 description 3
- 239000002213 purine nucleotide Substances 0.000 description 3
- 230000022983 regulation of cell cycle Effects 0.000 description 3
- 230000004044 response Effects 0.000 description 3
- 238000003757 reverse transcription PCR Methods 0.000 description 3
- 238000012552 review Methods 0.000 description 3
- OHRURASPPZQGQM-GCCNXGTGSA-N romidepsin Chemical compound O1C(=O)[C@H](C(C)C)NC(=O)C(=C/C)/NC(=O)[C@H]2CSSCC\C=C\[C@@H]1CC(=O)N[C@H](C(C)C)C(=O)N2 OHRURASPPZQGQM-GCCNXGTGSA-N 0.000 description 3
- 239000000523 sample Substances 0.000 description 3
- 208000013220 shortness of breath Diseases 0.000 description 3
- 239000011780 sodium chloride Substances 0.000 description 3
- 241000894007 species Species 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- 239000000758 substrate Substances 0.000 description 3
- 229940124597 therapeutic agent Drugs 0.000 description 3
- 150000003568 thioethers Chemical class 0.000 description 3
- 238000010361 transduction Methods 0.000 description 3
- 230000026683 transduction Effects 0.000 description 3
- 230000005945 translocation Effects 0.000 description 3
- 241000701161 unidentified adenovirus Species 0.000 description 3
- 241001529453 unidentified herpesvirus Species 0.000 description 3
- 241001430294 unidentified retrovirus Species 0.000 description 3
- 230000035899 viability Effects 0.000 description 3
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 description 2
- BHQCQFFYRZLCQQ-UHFFFAOYSA-N (3alpha,5alpha,7alpha,12alpha)-3,7,12-trihydroxy-cholan-24-oic acid Natural products OC1CC2CC(O)CCC2(C)C2C1C1CCC(C(CCC(O)=O)C)C1(C)C(O)C2 BHQCQFFYRZLCQQ-UHFFFAOYSA-N 0.000 description 2
- QGVQZRDQPDLHHV-DPAQBDIFSA-N (3s,8s,9s,10r,13r,14s,17r)-10,13-dimethyl-17-[(2r)-6-methylheptan-2-yl]-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1h-cyclopenta[a]phenanthrene-3-thiol Chemical compound C1C=C2C[C@@H](S)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 QGVQZRDQPDLHHV-DPAQBDIFSA-N 0.000 description 2
- PFDHVDFPTKSEKN-YOXFSPIKSA-N 2-Amino-8-oxo-9,10-epoxy-decanoic acid Chemical compound OC(=O)[C@H](N)CCCCCC(=O)C1CO1 PFDHVDFPTKSEKN-YOXFSPIKSA-N 0.000 description 2
- FZWGECJQACGGTI-UHFFFAOYSA-N 2-amino-7-methyl-1,7-dihydro-6H-purin-6-one Chemical compound NC1=NC(O)=C2N(C)C=NC2=N1 FZWGECJQACGGTI-UHFFFAOYSA-N 0.000 description 2
- GBPSCCPAXYTNMB-UHFFFAOYSA-N 4-(1,3-dioxo-2-benzo[de]isoquinolinyl)-N-hydroxybutanamide Chemical compound C1=CC(C(N(CCCC(=O)NO)C2=O)=O)=C3C2=CC=CC3=C1 GBPSCCPAXYTNMB-UHFFFAOYSA-N 0.000 description 2
- ZLAQATDNGLKIEV-UHFFFAOYSA-N 5-methyl-2-sulfanylidene-1h-pyrimidin-4-one Chemical compound CC1=CNC(=S)NC1=O ZLAQATDNGLKIEV-UHFFFAOYSA-N 0.000 description 2
- UJBCLAXPPIDQEE-UHFFFAOYSA-N 5-prop-1-ynyl-1h-pyrimidine-2,4-dione Chemical compound CC#CC1=CNC(=O)NC1=O UJBCLAXPPIDQEE-UHFFFAOYSA-N 0.000 description 2
- PEHVGBZKEYRQSX-UHFFFAOYSA-N 7-deaza-adenine Chemical compound NC1=NC=NC2=C1C=CN2 PEHVGBZKEYRQSX-UHFFFAOYSA-N 0.000 description 2
- LOSIULRWFAEMFL-UHFFFAOYSA-N 7-deazaguanine Chemical compound O=C1NC(N)=NC2=C1CC=N2 LOSIULRWFAEMFL-UHFFFAOYSA-N 0.000 description 2
- HCGHYQLFMPXSDU-UHFFFAOYSA-N 7-methyladenine Chemical compound C1=NC(N)=C2N(C)C=NC2=N1 HCGHYQLFMPXSDU-UHFFFAOYSA-N 0.000 description 2
- KDCGOANMDULRCW-UHFFFAOYSA-N 7H-purine Chemical compound N1=CNC2=NC=NC2=C1 KDCGOANMDULRCW-UHFFFAOYSA-N 0.000 description 2
- LPXQRXLUHJKZIE-UHFFFAOYSA-N 8-azaguanine Chemical compound NC1=NC(O)=C2NN=NC2=N1 LPXQRXLUHJKZIE-UHFFFAOYSA-N 0.000 description 2
- 229960005508 8-azaguanine Drugs 0.000 description 2
- LRFVTYWOQMYALW-UHFFFAOYSA-N 9H-xanthine Chemical compound O=C1NC(=O)NC2=C1NC=N2 LRFVTYWOQMYALW-UHFFFAOYSA-N 0.000 description 2
- 108010044267 Abnormal Hemoglobins Proteins 0.000 description 2
- 102100037435 Antiviral innate immune response receptor RIG-I Human genes 0.000 description 2
- 241000203069 Archaea Species 0.000 description 2
- 102000005427 Asialoglycoprotein Receptor Human genes 0.000 description 2
- DCXYFEDJOCDNAF-UHFFFAOYSA-N Asparagine Natural products OC(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-N 0.000 description 2
- 108091032955 Bacterial small RNA Proteins 0.000 description 2
- KXDAEFPNCMNJSK-UHFFFAOYSA-N Benzamide Chemical compound NC(=O)C1=CC=CC=C1 KXDAEFPNCMNJSK-UHFFFAOYSA-N 0.000 description 2
- 206010006002 Bone pain Diseases 0.000 description 2
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 2
- 101150018129 CSF2 gene Proteins 0.000 description 2
- 101150069031 CSN2 gene Proteins 0.000 description 2
- 101100285688 Caenorhabditis elegans hrg-7 gene Proteins 0.000 description 2
- 102000014914 Carrier Proteins Human genes 0.000 description 2
- 108010078791 Carrier Proteins Proteins 0.000 description 2
- 206010008479 Chest Pain Diseases 0.000 description 2
- 239000004380 Cholic acid Substances 0.000 description 2
- 108010077544 Chromatin Proteins 0.000 description 2
- 101150074775 Csf1 gene Proteins 0.000 description 2
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 2
- 101000889900 Enterobacteria phage T4 Intron-associated endonuclease 1 Proteins 0.000 description 2
- 102000004190 Enzymes Human genes 0.000 description 2
- 108090000790 Enzymes Proteins 0.000 description 2
- 238000012413 Fluorescence activated cell sorting analysis Methods 0.000 description 2
- 241000589599 Francisella tularensis subsp. novicida Species 0.000 description 2
- 101150106478 GPS1 gene Proteins 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- 102100036646 Glutamyl-tRNA(Gln) amidotransferase subunit A, mitochondrial Human genes 0.000 description 2
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 2
- 101000952099 Homo sapiens Antiviral innate immune response receptor RIG-I Proteins 0.000 description 2
- 101001066268 Homo sapiens Erythroid transcription factor Proteins 0.000 description 2
- 101001072655 Homo sapiens Glutamyl-tRNA(Gln) amidotransferase subunit A, mitochondrial Proteins 0.000 description 2
- 101000615488 Homo sapiens Methyl-CpG-binding domain protein 2 Proteins 0.000 description 2
- 101000831496 Homo sapiens Toll-like receptor 3 Proteins 0.000 description 2
- 241000725303 Human immunodeficiency virus Species 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 2
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 2
- 206010023126 Jaundice Diseases 0.000 description 2
- DCXYFEDJOCDNAF-REOHCLBHSA-N L-asparagine Chemical compound OC(=O)[C@@H](N)CC(N)=O DCXYFEDJOCDNAF-REOHCLBHSA-N 0.000 description 2
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 2
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 2
- 241001112693 Lachnospiraceae Species 0.000 description 2
- 241000713666 Lentivirus Species 0.000 description 2
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 2
- 239000004472 Lysine Substances 0.000 description 2
- 102100027754 Mast/stem cell growth factor receptor Kit Human genes 0.000 description 2
- 201000009906 Meningitis Diseases 0.000 description 2
- 102100021299 Methyl-CpG-binding domain protein 2 Human genes 0.000 description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 2
- 108010085220 Multiprotein Complexes Proteins 0.000 description 2
- 102000007474 Multiprotein Complexes Human genes 0.000 description 2
- 241000699660 Mus musculus Species 0.000 description 2
- 101100219625 Mus musculus Casd1 gene Proteins 0.000 description 2
- 102100038895 Myc proto-oncogene protein Human genes 0.000 description 2
- 101710135898 Myc proto-oncogene protein Proteins 0.000 description 2
- 101100385413 Neurospora crassa (strain ATCC 24698 / 74-OR23-1A / CBS 708.71 / DSM 1257 / FGSC 987) csm-3 gene Proteins 0.000 description 2
- REYJJPSVUYRZGE-UHFFFAOYSA-N Octadecylamine Chemical compound CCCCCCCCCCCCCCCCCCN REYJJPSVUYRZGE-UHFFFAOYSA-N 0.000 description 2
- 102100035423 POU domain, class 5, transcription factor 1 Human genes 0.000 description 2
- 102000004160 Phosphoric Monoester Hydrolases Human genes 0.000 description 2
- 108090000608 Phosphoric Monoester Hydrolases Proteins 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- 229920002873 Polyethylenimine Polymers 0.000 description 2
- WCUXLLCKKVVCTQ-UHFFFAOYSA-M Potassium chloride Chemical compound [Cl-].[K+] WCUXLLCKKVVCTQ-UHFFFAOYSA-M 0.000 description 2
- 108020005067 RNA Splice Sites Proteins 0.000 description 2
- 102000000574 RNA-Induced Silencing Complex Human genes 0.000 description 2
- 108010016790 RNA-Induced Silencing Complex Proteins 0.000 description 2
- 101100047461 Rattus norvegicus Trpm8 gene Proteins 0.000 description 2
- 108091081062 Repeated sequence (DNA) Proteins 0.000 description 2
- 102000003661 Ribonuclease III Human genes 0.000 description 2
- 108010057163 Ribonuclease III Proteins 0.000 description 2
- PYMYPHUHKUWMLA-LMVFSUKVSA-N Ribose Natural products OC[C@@H](O)[C@@H](O)[C@@H](O)C=O PYMYPHUHKUWMLA-LMVFSUKVSA-N 0.000 description 2
- 108020004422 Riboswitch Proteins 0.000 description 2
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 2
- 210000000173 T-lymphoid precursor cell Anatomy 0.000 description 2
- 206010043276 Teratoma Diseases 0.000 description 2
- 102000002689 Toll-like receptor Human genes 0.000 description 2
- 108020000411 Toll-like receptor Proteins 0.000 description 2
- 102100024324 Toll-like receptor 3 Human genes 0.000 description 2
- 101710150448 Transcriptional regulator Myc Proteins 0.000 description 2
- DRTQHJPVMGBUCF-XVFCMESISA-N Uridine Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C=C1 DRTQHJPVMGBUCF-XVFCMESISA-N 0.000 description 2
- 241000700618 Vaccinia virus Species 0.000 description 2
- NRLNQCOGCKAESA-KWXKLSQISA-N [(6z,9z,28z,31z)-heptatriaconta-6,9,28,31-tetraen-19-yl] 4-(dimethylamino)butanoate Chemical compound CCCCC\C=C/C\C=C/CCCCCCCCC(OC(=O)CCCN(C)C)CCCCCCCC\C=C/C\C=C/CCCCC NRLNQCOGCKAESA-KWXKLSQISA-N 0.000 description 2
- LCQWKKZWHQFOAH-UHFFFAOYSA-N [[3,4-dihydroxy-5-[6-(methylamino)purin-9-yl]oxolan-2-yl]methoxy-hydroxyphosphoryl] phosphono hydrogen phosphate Chemical compound C1=NC=2C(NC)=NC=NC=2N1C1OC(COP(O)(=O)OP(O)(=O)OP(O)(O)=O)C(O)C1O LCQWKKZWHQFOAH-UHFFFAOYSA-N 0.000 description 2
- 239000000370 acceptor Substances 0.000 description 2
- 125000000218 acetic acid group Chemical group C(C)(=O)* 0.000 description 2
- XVIYCJDWYLJQBG-UHFFFAOYSA-N acetic acid;adamantane Chemical compound CC(O)=O.C1C(C2)CC3CC1CC2C3 XVIYCJDWYLJQBG-UHFFFAOYSA-N 0.000 description 2
- DZBUGLKDJFMEHC-UHFFFAOYSA-N acridine Chemical compound C1=CC=CC2=CC3=CC=CC=C3N=C21 DZBUGLKDJFMEHC-UHFFFAOYSA-N 0.000 description 2
- 235000004279 alanine Nutrition 0.000 description 2
- 125000001931 aliphatic group Chemical group 0.000 description 2
- HMFHBZSHGGEWLO-UHFFFAOYSA-N alpha-D-Furanose-Ribose Natural products OCC1OC(O)C(O)C1O HMFHBZSHGGEWLO-UHFFFAOYSA-N 0.000 description 2
- 125000005122 aminoalkylamino group Chemical group 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 108010006523 asialoglycoprotein receptor Proteins 0.000 description 2
- 235000009582 asparagine Nutrition 0.000 description 2
- 229960001230 asparagine Drugs 0.000 description 2
- 235000003704 aspartic acid Nutrition 0.000 description 2
- 230000009286 beneficial effect Effects 0.000 description 2
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 2
- 230000008436 biogenesis Effects 0.000 description 2
- 210000000988 bone and bone Anatomy 0.000 description 2
- GYKLFBYWXZYSOW-UHFFFAOYSA-N butanoyloxymethyl 2,2-dimethylpropanoate Chemical compound CCCC(=O)OCOC(=O)C(C)(C)C GYKLFBYWXZYSOW-UHFFFAOYSA-N 0.000 description 2
- 239000001506 calcium phosphate Substances 0.000 description 2
- 229910000389 calcium phosphate Inorganic materials 0.000 description 2
- 235000011010 calcium phosphates Nutrition 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- 101150055766 cat gene Proteins 0.000 description 2
- 150000001768 cations Chemical class 0.000 description 2
- 230000022131 cell cycle Effects 0.000 description 2
- 229930002875 chlorophyll Natural products 0.000 description 2
- 235000019804 chlorophyll Nutrition 0.000 description 2
- 239000001752 chlorophylls and chlorophyllins Substances 0.000 description 2
- BHQCQFFYRZLCQQ-OELDTZBJSA-N cholic acid Chemical compound C([C@H]1C[C@H]2O)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC(O)=O)C)[C@@]2(C)[C@@H](O)C1 BHQCQFFYRZLCQQ-OELDTZBJSA-N 0.000 description 2
- 235000019416 cholic acid Nutrition 0.000 description 2
- 229960002471 cholic acid Drugs 0.000 description 2
- 210000003483 chromatin Anatomy 0.000 description 2
- 230000002759 chromosomal effect Effects 0.000 description 2
- 238000004590 computer program Methods 0.000 description 2
- 239000013068 control sample Substances 0.000 description 2
- 101150055601 cops2 gene Proteins 0.000 description 2
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 2
- 235000018417 cysteine Nutrition 0.000 description 2
- 230000006378 damage Effects 0.000 description 2
- 230000005860 defense response to virus Effects 0.000 description 2
- 230000007812 deficiency Effects 0.000 description 2
- 238000000432 density-gradient centrifugation Methods 0.000 description 2
- KXGVEGMKQFWNSR-UHFFFAOYSA-N deoxycholic acid Natural products C1CC2CC(O)CCC2(C)C2C1C1CCC(C(CCC(O)=O)C)C1(C)C(O)C2 KXGVEGMKQFWNSR-UHFFFAOYSA-N 0.000 description 2
- 230000001419 dependent effect Effects 0.000 description 2
- NIJJYAXOARWZEE-UHFFFAOYSA-N di-n-propyl-acetic acid Natural products CCCC(C(O)=O)CCC NIJJYAXOARWZEE-UHFFFAOYSA-N 0.000 description 2
- 238000003745 diagnosis Methods 0.000 description 2
- 238000006471 dimerization reaction Methods 0.000 description 2
- GTZOYNFRVVHLDZ-UHFFFAOYSA-N dodecane-1,1-diol Chemical compound CCCCCCCCCCCC(O)O GTZOYNFRVVHLDZ-UHFFFAOYSA-N 0.000 description 2
- 230000009977 dual effect Effects 0.000 description 2
- 230000002255 enzymatic effect Effects 0.000 description 2
- 230000008995 epigenetic change Effects 0.000 description 2
- 230000001973 epigenetic effect Effects 0.000 description 2
- 239000012091 fetal bovine serum Substances 0.000 description 2
- 230000001605 fetal effect Effects 0.000 description 2
- 210000002950 fibroblast Anatomy 0.000 description 2
- 235000019152 folic acid Nutrition 0.000 description 2
- 239000011724 folic acid Substances 0.000 description 2
- 239000000499 gel Substances 0.000 description 2
- 210000004602 germ cell Anatomy 0.000 description 2
- 239000008103 glucose Substances 0.000 description 2
- 235000011187 glycerol Nutrition 0.000 description 2
- 125000003827 glycol group Chemical group 0.000 description 2
- 125000001475 halogen functional group Chemical group 0.000 description 2
- 210000003494 hepatocyte Anatomy 0.000 description 2
- 239000000710 homodimer Substances 0.000 description 2
- 230000002209 hydrophobic effect Effects 0.000 description 2
- 230000001900 immune effect Effects 0.000 description 2
- 238000003364 immunohistochemistry Methods 0.000 description 2
- 239000007943 implant Substances 0.000 description 2
- 238000010348 incorporation Methods 0.000 description 2
- 239000000543 intermediate Substances 0.000 description 2
- 238000007912 intraperitoneal administration Methods 0.000 description 2
- 239000007791 liquid phase Substances 0.000 description 2
- 230000004807 localization Effects 0.000 description 2
- 210000002540 macrophage Anatomy 0.000 description 2
- 238000000520 microinjection Methods 0.000 description 2
- 210000003470 mitochondria Anatomy 0.000 description 2
- 230000007935 neutral effect Effects 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- 150000003833 nucleoside derivatives Chemical class 0.000 description 2
- 238000011580 nude mouse model Methods 0.000 description 2
- 239000003921 oil Substances 0.000 description 2
- ONTNXMBMXUNDBF-UHFFFAOYSA-N pentatriacontane-17,18,19-triol Chemical compound CCCCCCCCCCCCCCCCC(O)C(O)C(O)CCCCCCCCCCCCCCCC ONTNXMBMXUNDBF-UHFFFAOYSA-N 0.000 description 2
- 230000002688 persistence Effects 0.000 description 2
- RDOWQLZANAYVLL-UHFFFAOYSA-N phenanthridine Chemical compound C1=CC=C2C3=CC=CC=C3C=NC2=C1 RDOWQLZANAYVLL-UHFFFAOYSA-N 0.000 description 2
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 2
- 239000010452 phosphate Substances 0.000 description 2
- 150000004713 phosphodiesters Chemical group 0.000 description 2
- 230000008488 polyadenylation Effects 0.000 description 2
- 238000001556 precipitation Methods 0.000 description 2
- 238000002360 preparation method Methods 0.000 description 2
- 108091007428 primary miRNA Proteins 0.000 description 2
- 210000001236 prokaryotic cell Anatomy 0.000 description 2
- 230000001737 promoting effect Effects 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- 238000003259 recombinant expression Methods 0.000 description 2
- 230000008263 repair mechanism Effects 0.000 description 2
- 230000000717 retained effect Effects 0.000 description 2
- 108010091666 romidepsin Proteins 0.000 description 2
- 229960003452 romidepsin Drugs 0.000 description 2
- 210000000468 rubriblast Anatomy 0.000 description 2
- 210000002966 serum Anatomy 0.000 description 2
- 239000012679 serum free medium Substances 0.000 description 2
- 239000011734 sodium Substances 0.000 description 2
- 229910052708 sodium Inorganic materials 0.000 description 2
- 210000000952 spleen Anatomy 0.000 description 2
- 238000003860 storage Methods 0.000 description 2
- 238000007920 subcutaneous administration Methods 0.000 description 2
- 150000008163 sugars Chemical class 0.000 description 2
- 229910052717 sulfur Inorganic materials 0.000 description 2
- 230000004083 survival effect Effects 0.000 description 2
- 239000000725 suspension Substances 0.000 description 2
- 238000007910 systemic administration Methods 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- 229940113082 thymine Drugs 0.000 description 2
- 108091006106 transcriptional activators Proteins 0.000 description 2
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 2
- ZMANZCXQSJIPKH-UHFFFAOYSA-O triethylammonium ion Chemical compound CC[NH+](CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-O 0.000 description 2
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 2
- 238000009966 trimming Methods 0.000 description 2
- 241000701447 unidentified baculovirus Species 0.000 description 2
- MSRILKIQRXUYCT-UHFFFAOYSA-M valproate semisodium Chemical compound [Na+].CCCC(C(O)=O)CCC.CCCC(C([O-])=O)CCC MSRILKIQRXUYCT-UHFFFAOYSA-M 0.000 description 2
- 229960000604 valproic acid Drugs 0.000 description 2
- 238000001262 western blot Methods 0.000 description 2
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical compound O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 1
- MTCFGRXMJLQNBG-REOHCLBHSA-N (2S)-2-Amino-3-hydroxypropansäure Chemical compound OC[C@H](N)C(O)=O MTCFGRXMJLQNBG-REOHCLBHSA-N 0.000 description 1
- GNYCTMYOHGBSBI-SVZOTFJBSA-N (3s,6r,9s,12r)-6,9-dimethyl-3-[6-[(2s)-oxiran-2-yl]-6-oxohexyl]-1,4,7,10-tetrazabicyclo[10.3.0]pentadecane-2,5,8,11-tetrone Chemical compound C([C@H]1C(=O)N2CCC[C@@H]2C(=O)N[C@H](C(N[C@H](C)C(=O)N1)=O)C)CCCCC(=O)[C@@H]1CO1 GNYCTMYOHGBSBI-SVZOTFJBSA-N 0.000 description 1
- LLOKIGWPNVSDGJ-AFBVCZJXSA-N (3s,6s,9s,12r)-3,6-dibenzyl-9-[6-[(2s)-oxiran-2-yl]-6-oxohexyl]-1,4,7,10-tetrazabicyclo[10.3.0]pentadecane-2,5,8,11-tetrone Chemical compound C([C@H]1C(=O)N2CCC[C@@H]2C(=O)N[C@H](C(N[C@@H](CC=2C=CC=CC=2)C(=O)N1)=O)CCCCCC(=O)[C@H]1OC1)C1=CC=CC=C1 LLOKIGWPNVSDGJ-AFBVCZJXSA-N 0.000 description 1
- BWDQBBCUWLSASG-MDZDMXLPSA-N (e)-n-hydroxy-3-[4-[[2-hydroxyethyl-[2-(1h-indol-3-yl)ethyl]amino]methyl]phenyl]prop-2-enamide Chemical compound C=1NC2=CC=CC=C2C=1CCN(CCO)CC1=CC=C(\C=C\C(=O)NO)C=C1 BWDQBBCUWLSASG-MDZDMXLPSA-N 0.000 description 1
- FYADHXFMURLYQI-UHFFFAOYSA-N 1,2,4-triazine Chemical class C1=CN=NC=N1 FYADHXFMURLYQI-UHFFFAOYSA-N 0.000 description 1
- KILNVBDSWZSGLL-KXQOOQHDSA-N 1,2-dihexadecanoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCCCCCCCCCCCCCC KILNVBDSWZSGLL-KXQOOQHDSA-N 0.000 description 1
- RVHYPUORVDKRTM-UHFFFAOYSA-N 1-[2-[bis(2-hydroxydodecyl)amino]ethyl-[2-[4-[2-[bis(2-hydroxydodecyl)amino]ethyl]piperazin-1-yl]ethyl]amino]dodecan-2-ol Chemical compound CCCCCCCCCCC(O)CN(CC(O)CCCCCCCCCC)CCN(CC(O)CCCCCCCCCC)CCN1CCN(CCN(CC(O)CCCCCCCCCC)CC(O)CCCCCCCCCC)CC1 RVHYPUORVDKRTM-UHFFFAOYSA-N 0.000 description 1
- QAOBBBBDJSWHMU-WMBBNPMCSA-N 16,16-dimethylprostaglandin E2 Chemical compound CCCCC(C)(C)[C@H](O)\C=C\[C@H]1[C@H](O)CC(=O)[C@@H]1C\C=C/CCCC(O)=O QAOBBBBDJSWHMU-WMBBNPMCSA-N 0.000 description 1
- UHUHBFMZVCOEOV-UHFFFAOYSA-N 1h-imidazo[4,5-c]pyridin-4-amine Chemical compound NC1=NC=CC2=C1N=CN2 UHUHBFMZVCOEOV-UHFFFAOYSA-N 0.000 description 1
- LDGWQMRUWMSZIU-LQDDAWAPSA-M 2,3-bis[(z)-octadec-9-enoxy]propyl-trimethylazanium;chloride Chemical compound [Cl-].CCCCCCCC\C=C/CCCCCCCCOCC(C[N+](C)(C)C)OCCCCCCCC\C=C/CCCCCCCC LDGWQMRUWMSZIU-LQDDAWAPSA-M 0.000 description 1
- MUPNITTWEOEDNT-TWMSPMCMSA-N 2,3-bis[[(Z)-octadec-9-enoyl]oxy]propyl-trimethylazanium (3S,8S,9S,10R,13R,14S,17R)-10,13-dimethyl-17-[(2R)-6-methylheptan-2-yl]-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3-ol Chemical compound CC(C)CCC[C@@H](C)[C@H]1CC[C@H]2[C@@H]3CC=C4C[C@@H](O)CC[C@]4(C)[C@H]3CC[C@]12C.CCCCCCCC\C=C/CCCCCCCC(=O)OCC(C[N+](C)(C)C)OC(=O)CCCCCCC\C=C/CCCCCCCC MUPNITTWEOEDNT-TWMSPMCMSA-N 0.000 description 1
- KSXTUUUQYQYKCR-LQDDAWAPSA-M 2,3-bis[[(z)-octadec-9-enoyl]oxy]propyl-trimethylazanium;chloride Chemical compound [Cl-].CCCCCCCC\C=C/CCCCCCCC(=O)OCC(C[N+](C)(C)C)OC(=O)CCCCCCC\C=C/CCCCCCCC KSXTUUUQYQYKCR-LQDDAWAPSA-M 0.000 description 1
- WALUVDCNGPQPOD-UHFFFAOYSA-M 2,3-di(tetradecoxy)propyl-(2-hydroxyethyl)-dimethylazanium;bromide Chemical compound [Br-].CCCCCCCCCCCCCCOCC(C[N+](C)(C)CCO)OCCCCCCCCCCCCCC WALUVDCNGPQPOD-UHFFFAOYSA-M 0.000 description 1
- VEPOHXYIFQMVHW-XOZOLZJESA-N 2,3-dihydroxybutanedioic acid (2S,3S)-3,4-dimethyl-2-phenylmorpholine Chemical compound OC(C(O)C(O)=O)C(O)=O.C[C@H]1[C@@H](OCCN1C)c1ccccc1 VEPOHXYIFQMVHW-XOZOLZJESA-N 0.000 description 1
- QSHACTSJHMKXTE-UHFFFAOYSA-N 2-(2-aminopropyl)-7h-purin-6-amine Chemical compound CC(N)CC1=NC(N)=C2NC=NC2=N1 QSHACTSJHMKXTE-UHFFFAOYSA-N 0.000 description 1
- PIINGYXNCHTJTF-UHFFFAOYSA-N 2-(2-azaniumylethylamino)acetate Chemical group NCCNCC(O)=O PIINGYXNCHTJTF-UHFFFAOYSA-N 0.000 description 1
- MIJDSYMOBYNHOT-UHFFFAOYSA-N 2-(ethylamino)ethanol Chemical compound CCNCCO MIJDSYMOBYNHOT-UHFFFAOYSA-N 0.000 description 1
- LRFJOIPOPUJUMI-KWXKLSQISA-N 2-[2,2-bis[(9z,12z)-octadeca-9,12-dienyl]-1,3-dioxolan-4-yl]-n,n-dimethylethanamine Chemical compound CCCCC\C=C/C\C=C/CCCCCCCCC1(CCCCCCCC\C=C/C\C=C/CCCCC)OCC(CCN(C)C)O1 LRFJOIPOPUJUMI-KWXKLSQISA-N 0.000 description 1
- 125000004200 2-methoxyethyl group Chemical group [H]C([H])([H])OC([H])([H])C([H])([H])* 0.000 description 1
- YNFSUOFXEVCDTC-UHFFFAOYSA-N 2-n-methyl-7h-purine-2,6-diamine Chemical compound CNC1=NC(N)=C2NC=NC2=N1 YNFSUOFXEVCDTC-UHFFFAOYSA-N 0.000 description 1
- GOLORTLGFDVFDW-UHFFFAOYSA-N 3-(1h-benzimidazol-2-yl)-7-(diethylamino)chromen-2-one Chemical compound C1=CC=C2NC(C3=CC4=CC=C(C=C4OC3=O)N(CC)CC)=NC2=C1 GOLORTLGFDVFDW-UHFFFAOYSA-N 0.000 description 1
- HXVVOLDXHIMZJZ-UHFFFAOYSA-N 3-[2-[2-[2-[bis[3-(dodecylamino)-3-oxopropyl]amino]ethyl-[3-(dodecylamino)-3-oxopropyl]amino]ethylamino]ethyl-[3-(dodecylamino)-3-oxopropyl]amino]-n-dodecylpropanamide Chemical compound CCCCCCCCCCCCNC(=O)CCN(CCC(=O)NCCCCCCCCCCCC)CCN(CCC(=O)NCCCCCCCCCCCC)CCNCCN(CCC(=O)NCCCCCCCCCCCC)CCC(=O)NCCCCCCCCCCCC HXVVOLDXHIMZJZ-UHFFFAOYSA-N 0.000 description 1
- OBKXEAXTFZPCHS-UHFFFAOYSA-N 4-phenylbutyric acid Chemical compound OC(=O)CCCC1=CC=CC=C1 OBKXEAXTFZPCHS-UHFFFAOYSA-N 0.000 description 1
- LQLQRFGHAALLLE-UHFFFAOYSA-N 5-bromouracil Chemical compound BrC1=CNC(=O)NC1=O LQLQRFGHAALLLE-UHFFFAOYSA-N 0.000 description 1
- JDBGXEHEIRGOBU-UHFFFAOYSA-N 5-hydroxymethyluracil Chemical compound OCC1=CNC(=O)NC1=O JDBGXEHEIRGOBU-UHFFFAOYSA-N 0.000 description 1
- JTDYUFSDZATMKU-UHFFFAOYSA-N 6-(1,3-dioxo-2-benzo[de]isoquinolinyl)-N-hydroxyhexanamide Chemical compound C1=CC(C(N(CCCCCC(=O)NO)C2=O)=O)=C3C2=CC=CC3=C1 JTDYUFSDZATMKU-UHFFFAOYSA-N 0.000 description 1
- KXBCLNRMQPRVTP-UHFFFAOYSA-N 6-amino-1,5-dihydroimidazo[4,5-c]pyridin-4-one Chemical compound O=C1NC(N)=CC2=C1N=CN2 KXBCLNRMQPRVTP-UHFFFAOYSA-N 0.000 description 1
- DCPSTSVLRXOYGS-UHFFFAOYSA-N 6-amino-1h-pyrimidine-2-thione Chemical compound NC1=CC=NC(S)=N1 DCPSTSVLRXOYGS-UHFFFAOYSA-N 0.000 description 1
- QNNARSZPGNJZIX-UHFFFAOYSA-N 6-amino-5-prop-1-ynyl-1h-pyrimidin-2-one Chemical compound CC#CC1=CNC(=O)N=C1N QNNARSZPGNJZIX-UHFFFAOYSA-N 0.000 description 1
- CKOMXBHMKXXTNW-UHFFFAOYSA-N 6-methyladenine Chemical compound CNC1=NC=NC2=C1N=CN2 CKOMXBHMKXXTNW-UHFFFAOYSA-N 0.000 description 1
- HRYKDUPGBWLLHO-UHFFFAOYSA-N 8-azaadenine Chemical compound NC1=NC=NC2=NNN=C12 HRYKDUPGBWLLHO-UHFFFAOYSA-N 0.000 description 1
- 241000580270 Adeno-associated virus - 4 Species 0.000 description 1
- 108700028369 Alleles Proteins 0.000 description 1
- 241000710929 Alphavirus Species 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- 108091093088 Amplicon Proteins 0.000 description 1
- 241001550224 Apha Species 0.000 description 1
- 101100410782 Arabidopsis thaliana PXG1 gene Proteins 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 208000006820 Arthralgia Diseases 0.000 description 1
- RJUHZPRQRQLCFL-IMJSIDKUSA-N Asn-Asn Chemical compound NC(=O)C[C@H](N)C(=O)N[C@@H](CC(N)=O)C(O)=O RJUHZPRQRQLCFL-IMJSIDKUSA-N 0.000 description 1
- KLKHFFMNGWULBN-VKHMYHEASA-N Asn-Gly Chemical compound NC(=O)C[C@H](N)C(=O)NCC(O)=O KLKHFFMNGWULBN-VKHMYHEASA-N 0.000 description 1
- MQLZLIYPFDIDMZ-HAFWLYHUSA-N Asn-Ile Chemical compound CC[C@H](C)[C@@H](C(O)=O)NC(=O)[C@@H](N)CC(N)=O MQLZLIYPFDIDMZ-HAFWLYHUSA-N 0.000 description 1
- RFLHBLWLFUFFDZ-UHFFFAOYSA-N BML-210 Chemical compound NC1=CC=CC=C1NC(=O)CCCCCCC(=O)NC1=CC=CC=C1 RFLHBLWLFUFFDZ-UHFFFAOYSA-N 0.000 description 1
- 108010087504 Beta-Globulins Proteins 0.000 description 1
- 206010006187 Breast cancer Diseases 0.000 description 1
- 208000026310 Breast neoplasm Diseases 0.000 description 1
- 102100022002 CD59 glycoprotein Human genes 0.000 description 1
- 238000010356 CRISPR-Cas9 genome editing Methods 0.000 description 1
- 101100257372 Caenorhabditis elegans sox-3 gene Proteins 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 101000709520 Chlamydia trachomatis serovar L2 (strain 434/Bu / ATCC VR-902B) Atypical response regulator protein ChxR Proteins 0.000 description 1
- 108091026890 Coding region Proteins 0.000 description 1
- 108020004635 Complementary DNA Proteins 0.000 description 1
- 108091029523 CpG island Proteins 0.000 description 1
- 241000192700 Cyanobacteria Species 0.000 description 1
- ZZZCUOFIHGPKAK-UHFFFAOYSA-N D-erythro-ascorbic acid Natural products OCC1OC(=O)C(O)=C1O ZZZCUOFIHGPKAK-UHFFFAOYSA-N 0.000 description 1
- HMFHBZSHGGEWLO-SOOFDHNKSA-N D-ribofuranose Chemical compound OC[C@H]1OC(O)[C@H](O)[C@@H]1O HMFHBZSHGGEWLO-SOOFDHNKSA-N 0.000 description 1
- 108010060248 DNA Ligase ATP Proteins 0.000 description 1
- 102100033195 DNA ligase 4 Human genes 0.000 description 1
- 230000007067 DNA methylation Effects 0.000 description 1
- 229940126190 DNA methyltransferase inhibitor Drugs 0.000 description 1
- 230000004543 DNA replication Effects 0.000 description 1
- 102000052510 DNA-Binding Proteins Human genes 0.000 description 1
- 108700020911 DNA-Binding Proteins Proteins 0.000 description 1
- XULFJDKZVHTRLG-JDVCJPALSA-N DOSPA trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F.CCCCCCCC\C=C/CCCCCCCCOCC(C[N+](C)(C)CCNC(=O)C(CCCNCCCN)NCCCN)OCCCCCCCC\C=C/CCCCCCCC XULFJDKZVHTRLG-JDVCJPALSA-N 0.000 description 1
- 102000016911 Deoxyribonucleases Human genes 0.000 description 1
- 108010053770 Deoxyribonucleases Proteins 0.000 description 1
- 108010002156 Depsipeptides Proteins 0.000 description 1
- 102100037124 Developmental pluripotency-associated 5 protein Human genes 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- 239000004375 Dextrin Substances 0.000 description 1
- 229920001353 Dextrin Polymers 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 206010061818 Disease progression Diseases 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- 241000196324 Embryophyta Species 0.000 description 1
- 241000709661 Enterovirus Species 0.000 description 1
- 241000991587 Enterovirus C Species 0.000 description 1
- 101710100588 Erythroid transcription factor Proteins 0.000 description 1
- 102000004678 Exoribonucleases Human genes 0.000 description 1
- 108010002700 Exoribonucleases Proteins 0.000 description 1
- 108010088742 GATA Transcription Factors Proteins 0.000 description 1
- 102000009041 GATA Transcription Factors Human genes 0.000 description 1
- 108010028165 GATA1 Transcription Factor Proteins 0.000 description 1
- 102000016669 GATA1 Transcription Factor Human genes 0.000 description 1
- 101100264215 Gallus gallus XRCC6 gene Proteins 0.000 description 1
- 108700028146 Genetic Enhancer Elements Proteins 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- 108010043121 Green Fluorescent Proteins Proteins 0.000 description 1
- 102000004144 Green Fluorescent Proteins Human genes 0.000 description 1
- 108010051041 HC toxin Proteins 0.000 description 1
- 108050008753 HNH endonucleases Proteins 0.000 description 1
- 102000000310 HNH endonucleases Human genes 0.000 description 1
- 101710154606 Hemagglutinin Proteins 0.000 description 1
- HTTJABKRGRZYRN-UHFFFAOYSA-N Heparin Chemical compound OC1C(NC(=O)C)C(O)OC(COS(O)(=O)=O)C1OC1C(OS(O)(=O)=O)C(O)C(OC2C(C(OS(O)(=O)=O)C(OC3C(C(O)C(O)C(O3)C(O)=O)OS(O)(=O)=O)C(CO)O2)NS(O)(=O)=O)C(C(O)=O)O1 HTTJABKRGRZYRN-UHFFFAOYSA-N 0.000 description 1
- 206010019842 Hepatomegaly Diseases 0.000 description 1
- MDCTVRUPVLZSPG-BQBZGAKWSA-N His-Asp Chemical compound OC(=O)C[C@@H](C(O)=O)NC(=O)[C@@H](N)CC1=CNC=N1 MDCTVRUPVLZSPG-BQBZGAKWSA-N 0.000 description 1
- 102000008157 Histone Demethylases Human genes 0.000 description 1
- 108010074870 Histone Demethylases Proteins 0.000 description 1
- 102000011787 Histone Methyltransferases Human genes 0.000 description 1
- 108010036115 Histone Methyltransferases Proteins 0.000 description 1
- 102000003893 Histone acetyltransferases Human genes 0.000 description 1
- 108090000246 Histone acetyltransferases Proteins 0.000 description 1
- 108010033040 Histones Proteins 0.000 description 1
- 101000897400 Homo sapiens CD59 glycoprotein Proteins 0.000 description 1
- 101000881848 Homo sapiens Developmental pluripotency-associated 5 protein Proteins 0.000 description 1
- 101000899111 Homo sapiens Hemoglobin subunit beta Proteins 0.000 description 1
- 101001109685 Homo sapiens Nuclear receptor subfamily 5 group A member 2 Proteins 0.000 description 1
- 101000984042 Homo sapiens Protein lin-28 homolog A Proteins 0.000 description 1
- 101000574648 Homo sapiens Retinoid-inducible serine carboxypeptidase Proteins 0.000 description 1
- 101000800116 Homo sapiens Thy-1 membrane glycoprotein Proteins 0.000 description 1
- 101000835093 Homo sapiens Transferrin receptor protein 1 Proteins 0.000 description 1
- 206010020880 Hypertrophy Diseases 0.000 description 1
- 206010021143 Hypoxia Diseases 0.000 description 1
- XQFRJNBWHJMXHO-RRKCRQDMSA-N IDUR Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(I)=C1 XQFRJNBWHJMXHO-RRKCRQDMSA-N 0.000 description 1
- 229930010555 Inosine Natural products 0.000 description 1
- UGQMRVRMYYASKQ-KQYNXXCUSA-N Inosine Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C2=NC=NC(O)=C2N=C1 UGQMRVRMYYASKQ-KQYNXXCUSA-N 0.000 description 1
- 102100034343 Integrase Human genes 0.000 description 1
- 101710203526 Integrase Proteins 0.000 description 1
- 102100034170 Interferon-induced, double-stranded RNA-activated protein kinase Human genes 0.000 description 1
- 101150059802 KU80 gene Proteins 0.000 description 1
- 101150072501 Klf2 gene Proteins 0.000 description 1
- XUJNEKJLAYXESH-REOHCLBHSA-N L-Cysteine Chemical compound SC[C@H](N)C(O)=O XUJNEKJLAYXESH-REOHCLBHSA-N 0.000 description 1
- ONIBWKKTOPOVIA-BYPYZUCNSA-N L-Proline Chemical compound OC(=O)[C@@H]1CCCN1 ONIBWKKTOPOVIA-BYPYZUCNSA-N 0.000 description 1
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 1
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 1
- AGPKZVBTJJNPAG-WHFBIAKZSA-N L-isoleucine Chemical compound CC[C@H](C)[C@H](N)C(O)=O AGPKZVBTJJNPAG-WHFBIAKZSA-N 0.000 description 1
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 1
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 description 1
- AYFVYJQAPQTCCC-GBXIJSLDSA-N L-threonine Chemical compound C[C@@H](O)[C@H](N)C(O)=O AYFVYJQAPQTCCC-GBXIJSLDSA-N 0.000 description 1
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 description 1
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 1
- KZSNJWFQEVHDMF-BYPYZUCNSA-N L-valine Chemical compound CC(C)[C@H](N)C(O)=O KZSNJWFQEVHDMF-BYPYZUCNSA-N 0.000 description 1
- 108091026898 Leader sequence (mRNA) Proteins 0.000 description 1
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 1
- 229940124647 MEK inhibitor Drugs 0.000 description 1
- 108091030146 MiRBase Proteins 0.000 description 1
- 101100355655 Mus musculus Eras gene Proteins 0.000 description 1
- 101100446513 Mus musculus Fgf4 gene Proteins 0.000 description 1
- 101000969137 Mus musculus Metallothionein-1 Proteins 0.000 description 1
- 101100293261 Mus musculus Naa15 gene Proteins 0.000 description 1
- 101100310650 Mus musculus Sox18 gene Proteins 0.000 description 1
- 101100257376 Mus musculus Sox3 gene Proteins 0.000 description 1
- 101100369076 Mus musculus Tdgf1 gene Proteins 0.000 description 1
- 108091057508 Myc family Proteins 0.000 description 1
- VQAYFKKCNSOZKM-IOSLPCCCSA-N N(6)-methyladenosine Chemical compound C1=NC=2C(NC)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O VQAYFKKCNSOZKM-IOSLPCCCSA-N 0.000 description 1
- HRNLUBSXIHFDHP-UHFFFAOYSA-N N-(2-aminophenyl)-4-[[[4-(3-pyridinyl)-2-pyrimidinyl]amino]methyl]benzamide Chemical compound NC1=CC=CC=C1NC(=O)C(C=C1)=CC=C1CNC1=NC=CC(C=2C=NC=CC=2)=N1 HRNLUBSXIHFDHP-UHFFFAOYSA-N 0.000 description 1
- OVBPIULPVIDEAO-UHFFFAOYSA-N N-Pteroyl-L-glutaminsaeure Natural products C=1N=C2NC(N)=NC(=O)C2=NC=1CNC1=CC=C(C(=O)NC(CCC(O)=O)C(O)=O)C=C1 OVBPIULPVIDEAO-UHFFFAOYSA-N 0.000 description 1
- BHUZLJOUHMBZQY-YXQOSMAKSA-N N-[4-[(2R,4R,6S)-4-[[(4,5-diphenyl-2-oxazolyl)thio]methyl]-6-[4-(hydroxymethyl)phenyl]-1,3-dioxan-2-yl]phenyl]-N'-hydroxyoctanediamide Chemical compound C1=CC(CO)=CC=C1[C@H]1O[C@@H](C=2C=CC(NC(=O)CCCCCCC(=O)NO)=CC=2)O[C@@H](CSC=2OC(=C(N=2)C=2C=CC=CC=2)C=2C=CC=CC=2)C1 BHUZLJOUHMBZQY-YXQOSMAKSA-N 0.000 description 1
- 101150082943 NAT1 gene Proteins 0.000 description 1
- 101150063042 NR0B1 gene Proteins 0.000 description 1
- VQAYFKKCNSOZKM-UHFFFAOYSA-N NSC 29409 Natural products C1=NC=2C(NC)=NC=NC=2N1C1OC(CO)C(O)C1O VQAYFKKCNSOZKM-UHFFFAOYSA-N 0.000 description 1
- 241000588650 Neisseria meningitidis Species 0.000 description 1
- 229930193140 Neomycin Natural products 0.000 description 1
- 102100022669 Nuclear receptor subfamily 5 group A member 2 Human genes 0.000 description 1
- 108091007494 Nucleic acid- binding domains Proteins 0.000 description 1
- 108091005461 Nucleic proteins Proteins 0.000 description 1
- JWOGUUIOCYMBPV-UHFFFAOYSA-N OT-Key 11219 Natural products N1C(=O)C(CCCCCC(=O)CC)NC(=O)C2CCCCN2C(=O)C(C(C)CC)NC(=O)C1CC1=CN(OC)C2=CC=CC=C12 JWOGUUIOCYMBPV-UHFFFAOYSA-N 0.000 description 1
- 101710093908 Outer capsid protein VP4 Proteins 0.000 description 1
- 101710135467 Outer capsid protein sigma-1 Proteins 0.000 description 1
- 238000010222 PCR analysis Methods 0.000 description 1
- 108091081548 Palindromic sequence Proteins 0.000 description 1
- 108010077524 Peptide Elongation Factor 1 Proteins 0.000 description 1
- 241000710778 Pestivirus Species 0.000 description 1
- PCNDJXKNXGMECE-UHFFFAOYSA-N Phenazine Natural products C1=CC=CC2=NC3=CC=CC=C3N=C21 PCNDJXKNXGMECE-UHFFFAOYSA-N 0.000 description 1
- 102000006335 Phosphate-Binding Proteins Human genes 0.000 description 1
- 108010058514 Phosphate-Binding Proteins Proteins 0.000 description 1
- 102100028251 Phosphoglycerate kinase 1 Human genes 0.000 description 1
- 101710139464 Phosphoglycerate kinase 1 Proteins 0.000 description 1
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 1
- 108091000080 Phosphotransferase Proteins 0.000 description 1
- 101000658568 Planomicrobium okeanokoites Type II restriction enzyme FokI Proteins 0.000 description 1
- 229920000954 Polyglycolide Polymers 0.000 description 1
- 239000004721 Polyphenylene oxide Substances 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- ONIBWKKTOPOVIA-UHFFFAOYSA-N Proline Natural products OC(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-N 0.000 description 1
- 101710176177 Protein A56 Proteins 0.000 description 1
- 102000001253 Protein Kinase Human genes 0.000 description 1
- 101710150336 Protein Rex Proteins 0.000 description 1
- 102100025460 Protein lin-28 homolog A Human genes 0.000 description 1
- 102000016971 Proto-Oncogene Proteins c-kit Human genes 0.000 description 1
- 108010014608 Proto-Oncogene Proteins c-kit Proteins 0.000 description 1
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Natural products OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 1
- 101150010363 REM2 gene Proteins 0.000 description 1
- 102000014450 RNA Polymerase III Human genes 0.000 description 1
- 108010078067 RNA Polymerase III Proteins 0.000 description 1
- 230000007022 RNA scission Effects 0.000 description 1
- 101100127230 Rattus norvegicus Khdc3 gene Proteins 0.000 description 1
- 102100025483 Retinoid-inducible serine carboxypeptidase Human genes 0.000 description 1
- 101150052594 SLC2A3 gene Proteins 0.000 description 1
- RJFAYQIBOAGBLC-BYPYZUCNSA-N Selenium-L-methionine Chemical compound C[Se]CC[C@H](N)C(O)=O RJFAYQIBOAGBLC-BYPYZUCNSA-N 0.000 description 1
- RJFAYQIBOAGBLC-UHFFFAOYSA-N Selenomethionine Natural products C[Se]CCC(N)C(O)=O RJFAYQIBOAGBLC-UHFFFAOYSA-N 0.000 description 1
- 208000018020 Sickle cell-beta-thalassemia disease syndrome Diseases 0.000 description 1
- 241000700584 Simplexvirus Species 0.000 description 1
- 108091027967 Small hairpin RNA Proteins 0.000 description 1
- 101150001847 Sox15 gene Proteins 0.000 description 1
- 241000713896 Spleen necrosis virus Species 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- 210000001744 T-lymphocyte Anatomy 0.000 description 1
- 108010006785 Taq Polymerase Proteins 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 239000004098 Tetracycline Substances 0.000 description 1
- 208000035199 Tetraploidy Diseases 0.000 description 1
- 206010043391 Thalassaemia beta Diseases 0.000 description 1
- 206010043395 Thalassaemia sickle cell Diseases 0.000 description 1
- 208000002903 Thalassemia Diseases 0.000 description 1
- 241000589499 Thermus thermophilus Species 0.000 description 1
- RYYWUUFWQRZTIU-UHFFFAOYSA-N Thiophosphoric acid Chemical group OP(O)(S)=O RYYWUUFWQRZTIU-UHFFFAOYSA-N 0.000 description 1
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 description 1
- 239000004473 Threonine Substances 0.000 description 1
- 102100033523 Thy-1 membrane glycoprotein Human genes 0.000 description 1
- IQFYYKKMVGJFEH-XLPZGREQSA-N Thymidine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 IQFYYKKMVGJFEH-XLPZGREQSA-N 0.000 description 1
- RTAQQCXQSZGOHL-UHFFFAOYSA-N Titanium Chemical compound [Ti] RTAQQCXQSZGOHL-UHFFFAOYSA-N 0.000 description 1
- 108700009124 Transcription Initiation Site Proteins 0.000 description 1
- 102100037116 Transcription elongation factor 1 homolog Human genes 0.000 description 1
- 102100026144 Transferrin receptor protein 1 Human genes 0.000 description 1
- 206010052779 Transplant rejections Diseases 0.000 description 1
- LLOKIGWPNVSDGJ-UHFFFAOYSA-N Trapoxin B Natural products C1OC1C(=O)CCCCCC(C(NC(CC=1C=CC=CC=1)C(=O)N1)=O)NC(=O)C2CCCN2C(=O)C1CC1=CC=CC=C1 LLOKIGWPNVSDGJ-UHFFFAOYSA-N 0.000 description 1
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 description 1
- KZSNJWFQEVHDMF-UHFFFAOYSA-N Valine Natural products CC(C)C(N)C(O)=O KZSNJWFQEVHDMF-UHFFFAOYSA-N 0.000 description 1
- 208000036142 Viral infection Diseases 0.000 description 1
- 229930003268 Vitamin C Natural products 0.000 description 1
- 208000027418 Wounds and injury Diseases 0.000 description 1
- 102100036973 X-ray repair cross-complementing protein 5 Human genes 0.000 description 1
- 102100036976 X-ray repair cross-complementing protein 6 Human genes 0.000 description 1
- 101000929049 Xenopus tropicalis Derriere protein Proteins 0.000 description 1
- 102000038627 Zinc finger transcription factors Human genes 0.000 description 1
- 108091007916 Zinc finger transcription factors Proteins 0.000 description 1
- FHHZHGZBHYYWTG-INFSMZHSSA-N [(2r,3s,4r,5r)-5-(2-amino-7-methyl-6-oxo-3h-purin-9-ium-9-yl)-3,4-dihydroxyoxolan-2-yl]methyl [[[(2r,3s,4r,5r)-5-(2-amino-6-oxo-3h-purin-9-yl)-3,4-dihydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-hydroxyphosphoryl] phosphate Chemical compound N1C(N)=NC(=O)C2=C1[N+]([C@H]1[C@@H]([C@H](O)[C@@H](COP([O-])(=O)OP(O)(=O)OP(O)(=O)OC[C@@H]3[C@H]([C@@H](O)[C@@H](O3)N3C4=C(C(N=C(N)N4)=O)N=C3)O)O1)O)=CN2C FHHZHGZBHYYWTG-INFSMZHSSA-N 0.000 description 1
- ISXSJGHXHUZXNF-LXZPIJOJSA-N [(3s,8s,9s,10r,13r,14s,17r)-10,13-dimethyl-17-[(2r)-6-methylheptan-2-yl]-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1h-cyclopenta[a]phenanthren-3-yl] n-[2-(dimethylamino)ethyl]carbamate;hydrochloride Chemical compound Cl.C1C=C2C[C@@H](OC(=O)NCCN(C)C)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 ISXSJGHXHUZXNF-LXZPIJOJSA-N 0.000 description 1
- 230000001594 aberrant effect Effects 0.000 description 1
- 102000005421 acetyltransferase Human genes 0.000 description 1
- 108020002494 acetyltransferase Proteins 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 210000004504 adult stem cell Anatomy 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 230000032683 aging Effects 0.000 description 1
- 125000003295 alanine group Chemical group N[C@@H](C)C(=O)* 0.000 description 1
- 150000001336 alkenes Chemical class 0.000 description 1
- 125000005083 alkoxyalkoxy group Chemical group 0.000 description 1
- 125000005600 alkyl phosphonate group Chemical group 0.000 description 1
- 208000026935 allergic disease Diseases 0.000 description 1
- 230000000735 allogeneic effect Effects 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 208000007502 anemia Diseases 0.000 description 1
- 230000033115 angiogenesis Effects 0.000 description 1
- 125000000129 anionic group Chemical group 0.000 description 1
- PYKYMHQGRFAEBM-UHFFFAOYSA-N anthraquinone Natural products CCC(=O)c1c(O)c2C(=O)C3C(C=CC=C3O)C(=O)c2cc1CC(=O)OC PYKYMHQGRFAEBM-UHFFFAOYSA-N 0.000 description 1
- 150000004056 anthraquinones Chemical class 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 230000000692 anti-sense effect Effects 0.000 description 1
- 239000004599 antimicrobial Substances 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- 230000006907 apoptotic process Effects 0.000 description 1
- 239000008365 aqueous carrier Substances 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 125000003710 aryl alkyl group Chemical group 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 229960002756 azacitidine Drugs 0.000 description 1
- 230000037429 base substitution Effects 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- DRTQHJPVMGBUCF-PSQAKQOGSA-N beta-L-uridine Natural products O[C@H]1[C@@H](O)[C@H](CO)O[C@@H]1N1C(=O)NC(=O)C=C1 DRTQHJPVMGBUCF-PSQAKQOGSA-N 0.000 description 1
- 239000012867 bioactive agent Substances 0.000 description 1
- 230000008236 biological pathway Effects 0.000 description 1
- 230000031018 biological processes and functions Effects 0.000 description 1
- 239000012472 biological sample Substances 0.000 description 1
- 230000005540 biological transmission Effects 0.000 description 1
- 229960002685 biotin Drugs 0.000 description 1
- 235000020958 biotin Nutrition 0.000 description 1
- 239000011616 biotin Substances 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- 210000005208 blood dendritic cell Anatomy 0.000 description 1
- 210000001124 body fluid Anatomy 0.000 description 1
- 239000010839 body fluid Substances 0.000 description 1
- 229910000085 borane Inorganic materials 0.000 description 1
- 239000000337 buffer salt Substances 0.000 description 1
- OJEQKLURWBVCGF-UHFFFAOYSA-N butanoyl 2,2-dimethylpropaneperoxoate Chemical compound C(C(C)(C)C)(=O)OOC(CCC)=O OJEQKLURWBVCGF-UHFFFAOYSA-N 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 229940087373 calcium oxide Drugs 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- 235000014633 carbohydrates Nutrition 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 230000032823 cell division Effects 0.000 description 1
- 230000009134 cell regulation Effects 0.000 description 1
- 230000007248 cellular mechanism Effects 0.000 description 1
- 230000033077 cellular process Effects 0.000 description 1
- 230000036755 cellular response Effects 0.000 description 1
- 230000004700 cellular uptake Effects 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 239000002738 chelating agent Substances 0.000 description 1
- 238000007385 chemical modification Methods 0.000 description 1
- 238000002512 chemotherapy Methods 0.000 description 1
- 230000010428 chromatin condensation Effects 0.000 description 1
- 238000010367 cloning Methods 0.000 description 1
- 238000003169 complementation method Methods 0.000 description 1
- 230000000536 complexating effect Effects 0.000 description 1
- 239000003636 conditioned culture medium Substances 0.000 description 1
- 230000003750 conditioning effect Effects 0.000 description 1
- 230000021615 conjugation Effects 0.000 description 1
- 229920001577 copolymer Polymers 0.000 description 1
- 235000012343 cottonseed oil Nutrition 0.000 description 1
- 239000002385 cottonseed oil Substances 0.000 description 1
- 230000002338 cryopreservative effect Effects 0.000 description 1
- 238000012258 culturing Methods 0.000 description 1
- 125000000753 cycloalkyl group Chemical group 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000596 cyclohexenyl group Chemical group C1(=CCCCC1)* 0.000 description 1
- UHDGCWIWMRVCDJ-ZAKLUEHWSA-N cytidine Chemical group O=C1N=C(N)C=CN1[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O1 UHDGCWIWMRVCDJ-ZAKLUEHWSA-N 0.000 description 1
- 230000007423 decrease Effects 0.000 description 1
- 230000002939 deleterious effect Effects 0.000 description 1
- 238000002716 delivery method Methods 0.000 description 1
- 210000005258 dental pulp stem cell Anatomy 0.000 description 1
- 238000012938 design process Methods 0.000 description 1
- UREBDLICKHMUKA-CXSFZGCWSA-N dexamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-CXSFZGCWSA-N 0.000 description 1
- 229960003957 dexamethasone Drugs 0.000 description 1
- 235000019425 dextrin Nutrition 0.000 description 1
- 238000010586 diagram Methods 0.000 description 1
- 230000029087 digestion Effects 0.000 description 1
- 238000007865 diluting Methods 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- NAGJZTKCGNOGPW-UHFFFAOYSA-K dioxido-sulfanylidene-sulfido-$l^{5}-phosphane Chemical compound [O-]P([O-])([S-])=S NAGJZTKCGNOGPW-UHFFFAOYSA-K 0.000 description 1
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 1
- 230000005750 disease progression Effects 0.000 description 1
- 208000002173 dizziness Diseases 0.000 description 1
- 239000003968 dna methyltransferase inhibitor Substances 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 230000005684 electric field Effects 0.000 description 1
- 210000001671 embryonic stem cell Anatomy 0.000 description 1
- 238000004945 emulsification Methods 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 210000000222 eosinocyte Anatomy 0.000 description 1
- 210000003979 eosinophil Anatomy 0.000 description 1
- CHNUOJQWGUIOLD-NFZZJPOKSA-N epalrestat Chemical compound C=1C=CC=CC=1\C=C(/C)\C=C1/SC(=S)N(CC(O)=O)C1=O CHNUOJQWGUIOLD-NFZZJPOKSA-N 0.000 description 1
- 230000006718 epigenetic regulation Effects 0.000 description 1
- 210000000267 erythroid cell Anatomy 0.000 description 1
- 102000015694 estrogen receptors Human genes 0.000 description 1
- 108010038795 estrogen receptors Proteins 0.000 description 1
- 230000000763 evoking effect Effects 0.000 description 1
- 230000029142 excretion Effects 0.000 description 1
- 229960004887 ferric hydroxide Drugs 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- GNBHRKFJIUUOQI-UHFFFAOYSA-N fluorescein Chemical compound O1C(=O)C2=CC=CC=C2C21C1=CC=C(O)C=C1OC1=CC(O)=CC=C21 GNBHRKFJIUUOQI-UHFFFAOYSA-N 0.000 description 1
- 229940014144 folate Drugs 0.000 description 1
- 229960000304 folic acid Drugs 0.000 description 1
- 230000037406 food intake Effects 0.000 description 1
- 239000012634 fragment Substances 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 108010074605 gamma-Globulins Proteins 0.000 description 1
- 238000012246 gene addition Methods 0.000 description 1
- 230000009395 genetic defect Effects 0.000 description 1
- 238000012268 genome sequencing Methods 0.000 description 1
- 230000037442 genomic alteration Effects 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- 239000004220 glutamic acid Substances 0.000 description 1
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 1
- 235000004554 glutamine Nutrition 0.000 description 1
- 150000004676 glycans Chemical class 0.000 description 1
- 210000003714 granulocyte Anatomy 0.000 description 1
- 239000003102 growth factor Substances 0.000 description 1
- 239000001963 growth medium Substances 0.000 description 1
- 230000009931 harmful effect Effects 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- GNYCTMYOHGBSBI-UHFFFAOYSA-N helminthsporium carbonum toxin Natural products N1C(=O)C(C)NC(=O)C(C)NC(=O)C2CCCN2C(=O)C1CCCCCC(=O)C1CO1 GNYCTMYOHGBSBI-UHFFFAOYSA-N 0.000 description 1
- 239000000185 hemagglutinin Substances 0.000 description 1
- 201000005787 hematologic cancer Diseases 0.000 description 1
- 208000024200 hematopoietic and lymphoid system neoplasm Diseases 0.000 description 1
- 229960002897 heparin Drugs 0.000 description 1
- 229920000669 heparin Polymers 0.000 description 1
- 125000000592 heterocycloalkyl group Chemical group 0.000 description 1
- 125000001245 hexylamino group Chemical group [H]N([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C(C([H])([H])[H])([H])[H] 0.000 description 1
- 210000003630 histaminocyte Anatomy 0.000 description 1
- 239000000017 hydrogel Substances 0.000 description 1
- 229910052739 hydrogen Inorganic materials 0.000 description 1
- 229920013821 hydroxy alkyl cellulose Polymers 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- 230000007954 hypoxia Effects 0.000 description 1
- 230000005934 immune activation Effects 0.000 description 1
- 210000000987 immune system Anatomy 0.000 description 1
- 230000002163 immunogen Effects 0.000 description 1
- 230000005847 immunogenicity Effects 0.000 description 1
- 230000002055 immunohistochemical effect Effects 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 230000028709 inflammatory response Effects 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- 208000014674 injury Diseases 0.000 description 1
- 108091005434 innate immune receptors Proteins 0.000 description 1
- 210000005007 innate immune system Anatomy 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- 229960003786 inosine Drugs 0.000 description 1
- 230000003914 insulin secretion Effects 0.000 description 1
- 239000000138 intercalating agent Substances 0.000 description 1
- 238000001361 intraarterial administration Methods 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000007914 intraventricular administration Methods 0.000 description 1
- IEECXTSVVFWGSE-UHFFFAOYSA-M iron(3+);oxygen(2-);hydroxide Chemical compound [OH-].[O-2].[Fe+3] IEECXTSVVFWGSE-UHFFFAOYSA-M 0.000 description 1
- 230000002427 irreversible effect Effects 0.000 description 1
- AGPKZVBTJJNPAG-UHFFFAOYSA-N isoleucine Natural products CCC(C)C(N)C(O)=O AGPKZVBTJJNPAG-UHFFFAOYSA-N 0.000 description 1
- 229960000310 isoleucine Drugs 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- JJWLVOIRVHMVIS-UHFFFAOYSA-N isopropylamine Chemical compound CC(C)N JJWLVOIRVHMVIS-UHFFFAOYSA-N 0.000 description 1
- 101150085005 ku70 gene Proteins 0.000 description 1
- 238000011031 large-scale manufacturing process Methods 0.000 description 1
- 208000032839 leukemia Diseases 0.000 description 1
- 238000001638 lipofection Methods 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 230000001926 lymphatic effect Effects 0.000 description 1
- 210000003738 lymphoid progenitor cell Anatomy 0.000 description 1
- 229920002521 macromolecule Polymers 0.000 description 1
- 210000004962 mammalian cell Anatomy 0.000 description 1
- 229960002510 mandelic acid Drugs 0.000 description 1
- 239000003550 marker Substances 0.000 description 1
- 238000004949 mass spectrometry Methods 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 230000034217 membrane fusion Effects 0.000 description 1
- 229930182817 methionine Natural products 0.000 description 1
- YACKEPLHDIMKIO-UHFFFAOYSA-N methylphosphonic acid Chemical class CP(O)(O)=O YACKEPLHDIMKIO-UHFFFAOYSA-N 0.000 description 1
- 239000000693 micelle Substances 0.000 description 1
- 108091007426 microRNA precursor Proteins 0.000 description 1
- 230000000813 microbial effect Effects 0.000 description 1
- 230000003278 mimic effect Effects 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 230000000116 mitigating effect Effects 0.000 description 1
- 239000002829 mitogen activated protein kinase inhibitor Substances 0.000 description 1
- 230000009456 molecular mechanism Effects 0.000 description 1
- 238000009126 molecular therapy Methods 0.000 description 1
- 210000003205 muscle Anatomy 0.000 description 1
- 230000001114 myogenic effect Effects 0.000 description 1
- GZCNJTFELNTSAB-UHFFFAOYSA-N n'-(7h-purin-6-yl)hexane-1,6-diamine Chemical compound NCCCCCCNC1=NC=NC2=C1NC=N2 GZCNJTFELNTSAB-UHFFFAOYSA-N 0.000 description 1
- FMURUEPQXKJIPS-UHFFFAOYSA-N n-(1-benzylpiperidin-4-yl)-6,7-dimethoxy-2-(4-methyl-1,4-diazepan-1-yl)quinazolin-4-amine;trihydrochloride Chemical compound Cl.Cl.Cl.C=12C=C(OC)C(OC)=CC2=NC(N2CCN(C)CCC2)=NC=1NC(CC1)CCN1CC1=CC=CC=C1 FMURUEPQXKJIPS-UHFFFAOYSA-N 0.000 description 1
- 108091008800 n-Myc Proteins 0.000 description 1
- 239000002077 nanosphere Substances 0.000 description 1
- 210000000822 natural killer cell Anatomy 0.000 description 1
- 230000032965 negative regulation of cell volume Effects 0.000 description 1
- 230000031990 negative regulation of inflammatory response Effects 0.000 description 1
- 229960004927 neomycin Drugs 0.000 description 1
- 230000009826 neoplastic cell growth Effects 0.000 description 1
- 230000004766 neurogenesis Effects 0.000 description 1
- 210000000440 neutrophil Anatomy 0.000 description 1
- 210000003924 normoblast Anatomy 0.000 description 1
- 235000015097 nutrients Nutrition 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 235000019198 oils Nutrition 0.000 description 1
- 230000005868 ontogenesis Effects 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 125000001181 organosilyl group Chemical group [SiH3]* 0.000 description 1
- 230000003204 osmotic effect Effects 0.000 description 1
- 101800002712 p27 Proteins 0.000 description 1
- 239000006174 pH buffer Substances 0.000 description 1
- 238000004806 packaging method and process Methods 0.000 description 1
- IPCSVZSSVZVIGE-UHFFFAOYSA-N palmitic acid group Chemical group C(CCCCCCCCCCCCCCC)(=O)O IPCSVZSSVZVIGE-UHFFFAOYSA-N 0.000 description 1
- 125000000913 palmityl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 210000004197 pelvis Anatomy 0.000 description 1
- 230000035699 permeability Effects 0.000 description 1
- 230000003285 pharmacodynamic effect Effects 0.000 description 1
- 229950009215 phenylbutanoic acid Drugs 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000002953 phosphate buffered saline Substances 0.000 description 1
- 125000002467 phosphate group Chemical group [H]OP(=O)(O[H])O[*] 0.000 description 1
- WTJKGGKOPKCXLL-RRHRGVEJSA-N phosphatidylcholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCCCCCCC=CCCCCCCCC WTJKGGKOPKCXLL-RRHRGVEJSA-N 0.000 description 1
- UEZVMMHDMIWARA-UHFFFAOYSA-M phosphonate Chemical compound [O-]P(=O)=O UEZVMMHDMIWARA-UHFFFAOYSA-M 0.000 description 1
- 150000008298 phosphoramidates Chemical class 0.000 description 1
- 150000003013 phosphoric acid derivatives Chemical group 0.000 description 1
- 125000004437 phosphorous atom Chemical group 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 239000011574 phosphorus Substances 0.000 description 1
- 102000020233 phosphotransferase Human genes 0.000 description 1
- 230000007180 physiological regulation Effects 0.000 description 1
- 239000000049 pigment Substances 0.000 description 1
- 239000013600 plasmid vector Substances 0.000 description 1
- 229920003023 plastic Polymers 0.000 description 1
- 239000004033 plastic Substances 0.000 description 1
- 229920000747 poly(lactic acid) Polymers 0.000 description 1
- 229920000728 polyester Polymers 0.000 description 1
- 229920000570 polyether Polymers 0.000 description 1
- 210000002729 polyribosome Anatomy 0.000 description 1
- 229920001282 polysaccharide Polymers 0.000 description 1
- 239000005017 polysaccharide Substances 0.000 description 1
- 230000001124 posttranscriptional effect Effects 0.000 description 1
- 230000032361 posttranscriptional gene silencing Effects 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000001103 potassium chloride Substances 0.000 description 1
- 235000011164 potassium chloride Nutrition 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 230000000750 progressive effect Effects 0.000 description 1
- 230000000069 prophylactic effect Effects 0.000 description 1
- 230000004952 protein activity Effects 0.000 description 1
- 238000002331 protein detection Methods 0.000 description 1
- 108060006633 protein kinase Proteins 0.000 description 1
- 210000001938 protoplast Anatomy 0.000 description 1
- ZJFJVRPLNAMIKH-UHFFFAOYSA-N pseudo-u Chemical compound O=C1NC(=O)C(C)=CN1C1OC(COP(O)(=S)OC2C(OC(C2)N2C(N=C(N)C=C2)=O)COP(O)(=S)OC2C(OC(C2)N2C(N=C(N)C=C2)=O)COP(O)(=S)OC2C(OC(C2)N2C(N=C(N)C=C2)=O)COP(O)(=S)OC2C(OC(C2)N2C(NC(=O)C(C)=C2)=O)COP(O)(=S)OC2C(OC(C2)N2C3=C(C(NC(N)=N3)=O)N=C2)COP(O)(=S)OC2C(OC(C2)N2C3=NC=NC(N)=C3N=C2)COP(O)(=S)OC2C(OC(C2)N2C3=NC=NC(N)=C3N=C2)COP(O)(=S)OC2C(OC(C2)N2C(N=C(N)C=C2)=O)COP(O)(=S)OC2C(OC(C2)N2C(NC(=O)C(C)=C2)=O)COP(O)(=S)OC2C(OC(C2)N2C(NC(=O)C(C)=C2)=O)COP(O)(=S)OC2C(OC(C2)N2C3=C(C(NC(N)=N3)=O)N=C2)COP(O)(=S)OC2C(OC(C2)N2C3=C(C(NC(N)=N3)=O)N=C2)COP(O)(=S)OC2C(OC(C2)N2C3=C(C(NC(N)=N3)=O)N=C2)COP(O)(=S)OC2C(OC(C2)N2C3=NC=NC(N)=C3N=C2)CO)C(O)C1 ZJFJVRPLNAMIKH-UHFFFAOYSA-N 0.000 description 1
- 238000011002 quantification Methods 0.000 description 1
- 239000002096 quantum dot Substances 0.000 description 1
- 230000005855 radiation Effects 0.000 description 1
- 238000001959 radiotherapy Methods 0.000 description 1
- 230000008707 rearrangement Effects 0.000 description 1
- 102000005962 receptors Human genes 0.000 description 1
- 108020003175 receptors Proteins 0.000 description 1
- 238000002407 reforming Methods 0.000 description 1
- 230000026267 regulation of growth Effects 0.000 description 1
- 230000012121 regulation of immune response Effects 0.000 description 1
- 125000006853 reporter group Chemical group 0.000 description 1
- 230000028617 response to DNA damage stimulus Effects 0.000 description 1
- 108091008146 restriction endonucleases Proteins 0.000 description 1
- PYWVYCXTNDRMGF-UHFFFAOYSA-N rhodamine B Chemical compound [Cl-].C=12C=CC(=[N+](CC)CC)C=C2OC2=CC(N(CC)CC)=CC=C2C=1C1=CC=CC=C1C(O)=O PYWVYCXTNDRMGF-UHFFFAOYSA-N 0.000 description 1
- 125000000548 ribosyl group Chemical group C1([C@H](O)[C@H](O)[C@H](O1)CO)* 0.000 description 1
- 244000195895 saibo Species 0.000 description 1
- 108010038379 sargramostim Proteins 0.000 description 1
- 238000012216 screening Methods 0.000 description 1
- 238000010187 selection method Methods 0.000 description 1
- 229960002718 selenomethionine Drugs 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 230000035939 shock Effects 0.000 description 1
- 235000021391 short chain fatty acids Nutrition 0.000 description 1
- 150000004666 short chain fatty acids Chemical class 0.000 description 1
- 210000003491 skin Anatomy 0.000 description 1
- 210000004927 skin cell Anatomy 0.000 description 1
- 210000001626 skin fibroblast Anatomy 0.000 description 1
- MFBOGIVSZKQAPD-UHFFFAOYSA-M sodium butyrate Chemical compound [Na+].CCCC([O-])=O MFBOGIVSZKQAPD-UHFFFAOYSA-M 0.000 description 1
- 229960002232 sodium phenylbutyrate Drugs 0.000 description 1
- VPZRWNZGLKXFOE-UHFFFAOYSA-M sodium phenylbutyrate Chemical compound [Na+].[O-]C(=O)CCCC1=CC=CC=C1 VPZRWNZGLKXFOE-UHFFFAOYSA-M 0.000 description 1
- 239000001488 sodium phosphate Substances 0.000 description 1
- 229910000162 sodium phosphate Inorganic materials 0.000 description 1
- 239000002904 solvent Substances 0.000 description 1
- 230000000392 somatic effect Effects 0.000 description 1
- 210000001988 somatic stem cell Anatomy 0.000 description 1
- 230000002269 spontaneous effect Effects 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 238000012289 standard assay Methods 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 150000003431 steroids Chemical class 0.000 description 1
- 230000000638 stimulation Effects 0.000 description 1
- 210000002536 stromal cell Anatomy 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- IIACRCGMVDHOTQ-UHFFFAOYSA-N sulfamic acid Chemical group NS(O)(=O)=O IIACRCGMVDHOTQ-UHFFFAOYSA-N 0.000 description 1
- 125000000565 sulfonamide group Chemical group 0.000 description 1
- BDHFUVZGWQCTTF-UHFFFAOYSA-M sulfonate Chemical compound [O-]S(=O)=O BDHFUVZGWQCTTF-UHFFFAOYSA-M 0.000 description 1
- 150000003457 sulfones Chemical group 0.000 description 1
- 150000003462 sulfoxides Chemical class 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 238000002636 symptomatic treatment Methods 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 230000002123 temporal effect Effects 0.000 description 1
- 229960002180 tetracycline Drugs 0.000 description 1
- 229930101283 tetracycline Natural products 0.000 description 1
- 235000019364 tetracycline Nutrition 0.000 description 1
- 150000003522 tetracyclines Chemical class 0.000 description 1
- 229930003799 tocopherol Natural products 0.000 description 1
- 239000011732 tocopherol Substances 0.000 description 1
- 125000002640 tocopherol group Chemical class 0.000 description 1
- 235000019149 tocopherols Nutrition 0.000 description 1
- 231100000027 toxicology Toxicity 0.000 description 1
- 239000003053 toxin Substances 0.000 description 1
- 231100000765 toxin Toxicity 0.000 description 1
- 230000001052 transient effect Effects 0.000 description 1
- 230000007704 transition Effects 0.000 description 1
- 230000014621 translational initiation Effects 0.000 description 1
- 108010060596 trapoxin B Proteins 0.000 description 1
- 150000003626 triacylglycerols Chemical class 0.000 description 1
- RTKIYFITIVXBLE-QEQCGCAPSA-N trichostatin A Chemical compound ONC(=O)/C=C/C(/C)=C/[C@@H](C)C(=O)C1=CC=C(N(C)C)C=C1 RTKIYFITIVXBLE-QEQCGCAPSA-N 0.000 description 1
- UORVGPXVDQYIDP-UHFFFAOYSA-N trihydridoboron Substances B UORVGPXVDQYIDP-UHFFFAOYSA-N 0.000 description 1
- RYFMWSXOAZQYPI-UHFFFAOYSA-K trisodium phosphate Chemical compound [Na+].[Na+].[Na+].[O-]P([O-])([O-])=O RYFMWSXOAZQYPI-UHFFFAOYSA-K 0.000 description 1
- 230000005747 tumor angiogenesis Effects 0.000 description 1
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 1
- ZDPHROOEEOARMN-UHFFFAOYSA-N undecanoic acid Chemical group CCCCCCCCCCC(O)=O ZDPHROOEEOARMN-UHFFFAOYSA-N 0.000 description 1
- 125000002948 undecyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 241001515965 unidentified phage Species 0.000 description 1
- DRTQHJPVMGBUCF-UHFFFAOYSA-N uracil arabinoside Natural products OC1C(O)C(CO)OC1N1C(=O)NC(=O)C=C1 DRTQHJPVMGBUCF-UHFFFAOYSA-N 0.000 description 1
- 229940045145 uridine Drugs 0.000 description 1
- 229960005486 vaccine Drugs 0.000 description 1
- 239000004474 valine Substances 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 230000029812 viral genome replication Effects 0.000 description 1
- 230000009385 viral infection Effects 0.000 description 1
- 235000019154 vitamin C Nutrition 0.000 description 1
- 239000011718 vitamin C Substances 0.000 description 1
- 239000011534 wash buffer Substances 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
Images








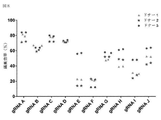


































Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K48/00—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy
- A61K48/005—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy characterised by an aspect of the 'active' part of the composition delivered, i.e. the nucleic acid delivered
- A61K48/0066—Manipulation of the nucleic acid to modify its expression pattern, e.g. enhance its duration of expression, achieved by the presence of particular introns in the delivered nucleic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/43—Enzymes; Proenzymes; Derivatives thereof
- A61K38/46—Hydrolases (3)
- A61K38/465—Hydrolases (3) acting on ester bonds (3.1), e.g. lipases, ribonucleases
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K35/00—Medicinal preparations containing materials or reaction products thereof with undetermined constitution
- A61K35/12—Materials from mammals; Compositions comprising non-specified tissues or cells; Compositions comprising non-embryonic stem cells; Genetically modified cells
- A61K35/28—Bone marrow; Haematopoietic stem cells; Mesenchymal stem cells of any origin, e.g. adipose-derived stem cells
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K48/00—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy
- A61K48/0008—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy characterised by an aspect of the 'non-active' part of the composition delivered, e.g. wherein such 'non-active' part is not delivered simultaneously with the 'active' part of the composition
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K48/00—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy
- A61K48/005—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy characterised by an aspect of the 'active' part of the composition delivered, i.e. the nucleic acid delivered
- A61K48/0058—Nucleic acids adapted for tissue specific expression, e.g. having tissue specific promoters as part of a contruct
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K48/00—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy
- A61K48/0075—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy characterised by an aspect of the delivery route, e.g. oral, subcutaneous
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/46—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates
- C07K14/47—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates from mammals
- C07K14/4701—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates from mammals not used
- C07K14/4702—Regulators; Modulating activity
- C07K14/4705—Regulators; Modulating activity stimulating, promoting or activating activity
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/10—Processes for the isolation, preparation or purification of DNA or RNA
- C12N15/102—Mutagenizing nucleic acids
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/113—Non-coding nucleic acids modulating the expression of genes, e.g. antisense oligonucleotides; Antisense DNA or RNA; Triplex- forming oligonucleotides; Catalytic nucleic acids, e.g. ribozymes; Nucleic acids used in co-suppression or gene silencing
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0634—Cells from the blood or the immune system
- C12N5/0647—Haematopoietic stem cells; Uncommitted or multipotent progenitors
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/14—Hydrolases (3)
- C12N9/16—Hydrolases (3) acting on ester bonds (3.1)
- C12N9/22—Ribonucleases RNAses, DNAses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/06—Antianaemics
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/10—Type of nucleic acid
- C12N2310/20—Type of nucleic acid involving clustered regularly interspaced short palindromic repeats [CRISPRs]
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/30—Chemical structure
- C12N2310/31—Chemical structure of the backbone
- C12N2310/315—Phosphorothioates
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2506/00—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells
- C12N2506/11—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells from blood or immune system cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2506/00—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells
- C12N2506/13—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells from connective tissue cells, from mesenchymal cells
- C12N2506/1346—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells from connective tissue cells, from mesenchymal cells from mesenchymal stem cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2506/00—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells
- C12N2506/45—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells from artificially induced pluripotent stem cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2510/00—Genetically modified cells
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Genetics & Genomics (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Organic Chemistry (AREA)
- Biomedical Technology (AREA)
- Zoology (AREA)
- Biotechnology (AREA)
- General Health & Medical Sciences (AREA)
- Wood Science & Technology (AREA)
- Molecular Biology (AREA)
- General Engineering & Computer Science (AREA)
- Medicinal Chemistry (AREA)
- Biochemistry (AREA)
- Microbiology (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Epidemiology (AREA)
- Biophysics (AREA)
- Physics & Mathematics (AREA)
- Plant Pathology (AREA)
- Immunology (AREA)
- Hematology (AREA)
- Gastroenterology & Hepatology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Crystallography & Structural Chemistry (AREA)
- Cell Biology (AREA)
- Developmental Biology & Embryology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Diabetes (AREA)
- Virology (AREA)
- Toxicology (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Description
本出願は、2016年4月18日に出願された米国仮出願第62/324,024号;2016年9月1日に出願された米国仮出願第62/382,522号;及び2016年12月2日に出願された米国仮特許出願第62/429,428号の利益を主張する。これらの全ての出願は、その全体が参照により本明細書に組み込まれる。
本出願は、コンピューター可読形態の配列表(ファイル名:160077PCT Sequence Listing;14,446,299バイト(ASCIIテキストファイル);2017年4月7日作成)を含み、当該配列表は、その全体が参照により本明細書に援用され、また本開示の一部を形成する。
配列番号1〜29,482は、BCL11A遺伝子またはBCL11A遺伝子の調節エレメントをコードする他のDNA配列の中またはその付近で、S.pyogenes Cas9エンドヌクレアーゼを用いて標的化するための20bpスペーサー配列である。
胎児ヘモグロビン

BCL11A遺伝子における12.4kbの転写制御配列の領域を標的部位のためにスキャンした。各エリアについて、配列NRGを有するプロトスペーサー隣接モチーフ(PAM)をスキャンした。配列表の配列番号1〜29,482に示すように、PAMに対応するgRNA 20bpスペーサー配列を特定した。
BCL11A遺伝子における12.4kbの転写制御配列の領域を標的部位のためにスキャンした。各エリアについて、配列NNGRRTを有するプロトスペーサー隣接モチーフ(PAM)をスキャンした。配列表の配列番号29,843〜32,387に示すように、PAMに対応するgRNA 20bpスペーサー配列を特定した。
BCL11A遺伝子における12.4kbの転写制御配列の領域を標的部位のためにスキャンした。各エリアについて、配列NNAGAAWを有するプロトスペーサー隣接モチーフ(PAM)をスキャンした。配列表の配列番号32,388〜33,420に示すように、PAMに対応するgRNA 20bpスペーサー配列を特定した。
BCL11A遺伝子における12.4kbの転写制御配列の領域を標的部位のためにスキャンした。各エリアについて、配列NAAAACを有するプロトスペーサー隣接モチーフ(PAM)をスキャンした。配列表の配列番号33,421〜33,851に示すように、PAMに対応するgRNA 20bpスペーサー配列を特定した。
BCL11A遺伝子における12.4kbの転写制御配列の領域を標的部位のためにスキャンした。各エリアについて、配列NNNNGHTTを有するプロトスペーサー隣接モチーフ(PAM)をスキャンした。配列表の配列番号33,852〜36,731に示すように、PAMに対応するgRNA 20bpスペーサー配列を特定した。
BCL11A遺伝子における12.4kbの転写制御配列の領域を標的部位のためにスキャンした。各エリアについて、配列YTNを有するプロトスペーサー隣接モチーフ(PAM)をスキャンした。配列表の配列番号36,732〜71,947に示すように、PAMに対応するgRNA 22bpスペーサー配列を特定した。
理論上の結合及び実験で評価された活性の両方を伴うマルチステッププロセスにおいて、候補ガイドをスクリーニング及び選択する。例示として、PAMが隣接する特定のオンターゲット部位(例えば、BCL11A遺伝子の転写制御配列中の部位)にマッチする配列を有する候補ガイドに対し、同様の配列を有するオフターゲット部位を切断する可能性を評価することができ、これには、以下により詳細に記載し例示するようなオフターゲット結合の評価に利用できる様々なバイオインフォマティクスツールのうちの1つ以上を用いて、意図される位置以外の染色体位置における作用の可能性を評価する。オフターゲット活性が比較的低いと予測される候補は、次に実験的評価を受けてそのオンターゲット活性を測定し、次に様々な部位におけるオフターゲット活性を測定することができる。好ましいガイドは、選択された座位で所望レベルの遺伝子編集を達成するのに十分に高いオンターゲット活性と、他の染色体座位における変更の可能性を低減する比較的低いオフターゲット活性とを有する。オンターゲット活性:オフターゲット活性の比は、しばしばガイドの「特異性」と呼ばれる。
最も低いオフターゲット活性を有すると予測されたgRNAは、次にK562細胞におけるオンターゲット活性を試験し、TIDEを用いてインデルの出現頻度を評価することになる。
上記実施例におけるTIDE及び次世代シークエンシング研究から得られた最良のオンターゲット活性を有するgRNAは、次に全ゲノムシークエンシングを用いてオフターゲット活性を試験することになる。候補gRNAをCD34+細胞またはiPSCでより徹底的に評価する。
TIDE及び次世代シークエンシング研究から得られた最良のオンターゲット活性及び最も低いオフターゲット活性を有するgRNAは、組み合わせて試験を行って、各gRNAの組合せの使用から得られる欠失のサイズを評価することになる。有望なgRNAの組合せは、初代ヒトCD34+細胞で評価する。
オンターゲット活性及びオフターゲット活性の両方におけるgRNAの試験をした後は、修飾/不活性化及びノックイン戦略をHDR遺伝子編集に対して試験することになる。
HDR遺伝子編集に対する異なる戦略を試験した後は、初代ヒト細胞においてリードCRISPR−Cas9/DNAドナーの組合せの欠失の効率、組換え、及びオフターゲット特異性を再評価することになる。Cas9 mRNAまたはRNPは送達のために脂質ナノ粒子に製剤化し、sgRNAはナノ粒子に製剤化するかまたはAAVとして送達させ、ドナーDNAはナノ粒子に製剤化するかAAVとして送達させる。
CRISPR−Cas9/DNAドナーの組合せを再評価した後は、リード製剤を動物モデルにおいてin vivoで試験することになる。
ヒトドナー1〜3からのヒト動員末梢血CD34+細胞を、CD34+増大サプリメントを含む無血清StemSpan培地中で2日間培養した。100,000細胞を洗浄し、Corfu Large(CLO)gRNA、Corfu Small(CSO)gRNA、HPFH5 gRNA、Kenya gRNA、SD2 sgRNA、またはSPY101 sgRNAと共にCas9 mRNAを用いてエレクトロポレーションした。細胞を2日間回復させてから赤血球系分化培地(IMDM+Glutamaxに5%のヒト血清、10μg/mlのインスリン、20ng/mlのSCF、5ng/mlのIL−3、3U/mlのEPO、1uMのデキサメタゾン、1uMのβ−エストラジオール、330ug/mlのホロ−トランスフェリン、及び2U/mlのヘパリンを追加したもの)に切り替えた。本明細書に記載の「シークエンシング(sequence)によるオンターゲット及びオフターゲットの変異検出」及び「変異検出アッセイ」のセクションで説明されているように、Corfu Large(CLO)gRNAでエレクトロポレーションした細胞、Corfu Small(CSO)gRNAでエレクトロポレーションした細胞、HPFH5 gRNAでエレクトロポレーションした細胞、Kenya gRNAでエレクトロポレーションした細胞、SD2 sgRNAでエレクトロポレーションした細胞、及びSPY101 sgRNAでエレクトロポレーションした細胞の各々について挿入/欠失(「インデル」)のパーセンテージを決定した(図3)。これらの細胞を赤血球系分化培地中で12日間分化させた後、RNAを収集して定量的リアルタイムPCRによりヘモグロビンレベルを評価した(図4A〜4C)。
SPY101 sgRNAを使用した場合にBCL11A遺伝子のイントロン2中で生じ得る遺伝子編集結果は3つ考えられる。SPY101 sgRNAを使用した場合に生じ得る第1の遺伝子編集結果は、両方のアレルにおいてインデルのみをもたらす(インデル/インデル、図6)。SPY101 sgRNAを使用した場合に生じ得る第2の遺伝子編集結果は、2つのアレルにおいて両方のインデル及び野生型配列によるクローンをもたらす(インデル/WT、図6)。SPY101 sgRNAを使用した場合に生じ得る第3の遺伝子編集結果は、両方のアレルにおいて野生型配列によるコロニーをもたらす(WT/WT、図6)。
以下の表(表4)は、実施例16〜17で使用したgRNAに関する情報を示している。
a、g、u:2’−O−メチル残基
s:ホスホロチオアート
A、C、G、U:RNA残基
ヒトmPB CD34+細胞における6つの異なるHPFHバリアントの再形成、すなわち「標的」の編集から得られた結果が図16A〜B及び図17に示されている。CRISPR/Cas9を用いてCD34+細胞を処置し、赤血球に分化させ、次に、図15に示されている実験プロセスを用いて、バルク(図16A〜B)及びコロニー(図17)におけるHBF mRNA及びタンパク質発現をアッセイした。
4名の独立したドナーからのヒト動員末梢血(mPB)CD34+細胞を、100ng/mlの組換えヒト幹細胞因子(SCF)、100ng/mlの組換えヒトFit3−リガンド(FLT3L)、及び100ng/mlのトロンボポエチン(TPO)を含む無血清CellGro(登録商標)培地中で培養した。ドナー当たり200,000細胞を洗浄し、Lonzaエレクトロポレーション装置を用いて、いずれのCRISPR/Cas9編集構成要素もなし(模擬エレクトロポレーション試料)、陰性対照としてのGFP gRNA及びCas9タンパク質(GFP)、SPY101 gRNA及びCas9タンパク質(SPY)、SD2 gRNA及びCas9タンパク質(SD2)、または二重BCL11Aエキソン2 gRNA及びCas9タンパク質(Ex2)を伴ったものに対しエレクトロポレーションした。組換えCas9タンパク質は、2つのSV40核局在化配列(NLS)に隣接するS.pyogenes Cas9をコードする。これらの実験は、gRNA:Cas9の重量比が1:1のリボ核タンパク質(RNP)を用いて実施した。SPY101 gRNAは、BCL11a座位のイントロン2内でDHS+58 Gata1結合部位のインデル分断を創出する。SD2 gRNAは、ヒトベータグロビン座位内でインデル及び4.9kb欠失を創出する。4.9kb欠失はHBG1の上流に位置し、HBG2配列全体を含む。4.9kb欠失は、HBG2コード配列に対し5’側の168bpで開始し、HBG1コード配列に対し5’側の168bpで終了する。エキソン2 gRNAはBCL11A座位のエキソン2上に196bpの欠失を創出し、陽性対照としての役割を果たした。エレクトロポレーションしなかったヒトmPB CD34+細胞は陰性対照(EPなし)としての役割を果たした。
ゲノムのオンターゲット編集が治療の成功に必須である一方で、任意のオフターゲット事象の検出は、製品安全性を保証する重要な構成要素である。オフターゲット部位における改変を検出する1つの方法は、オンターゲット部位に最も類似しているゲノムの領域をハイブリッドキャプチャーシークエンシングによって豊富化し、検出される任意のインデルを定量化することを伴う。
CliniMACS CD34マイクロビーズ及びCliniMACS Prodigy(Miltenyi Biotec)を用いて健康なドナーからヒト動員末梢血(mPB)CD34+細胞を単離し、100ng/mlの組換えヒト幹細胞因子(SCF)、100ng/mlの組換えヒトFit3−リガンド(FLT3L)、及び100ng/mlのトロンボポエチン(TPO)を含む無血清CellGro(登録商標)培地中で培養した。次に、Maxcyte(登録商標)デバイスを用いて、製造業者の指示に従って、以下のうちの1つを用いて細胞にエレクトロポレーションした:いずれのCRISPR/Cas9編集構成要素も含有しない空のベクター(模擬エレクトロポレーション試料)、陰性対照としてのGFP gRNA及びCas9タンパク質(GFP)、SPY101 gRNA及びCas9タンパク質(SPY101)、またはSD2 gRNA及びCas9タンパク質(SD2)。組換えCas9タンパク質は、2つのSV40核局在化配列(NLS)に隣接するS.pyogenes Cas9をコードする。これらの実験は、gRNA:Cas9の重量比が1:1のリボ核タンパク質(RNP)を用いて実施した。
SPY101を用いたSCD及びβ−サラセミアの処置に対しCRISPR−Cas9を用いて最も高い有効性を達成するため、発明者らは、2つの異なるCas9フォーマット、Cas9 mRNAまたはCas9タンパク質について、動員末梢血(mPB)からのヒトCD34+細胞における編集効率、有効性、及び毒性を評価した。Cas9タンパク質に対するCas9 mRNAについての様々なソースの比較を、Cas9 mRNA及びSPY101 gRNAまたはSPY101 gRNAと複合体化されたCas9タンパク質(リボ核タンパク質(RNP)複合体として)をヒトmPB CD34+細胞にエレクトロポレーションし、エレクトロポレーション後48時間時におけるこれらの編集効率及び細胞生存率を評価することによって行った。Cas9タンパク質に対するCas9 mRNAの様々なソースを比較し、一部のCas9 mRNAはCas9タンパク質に対し同様のレベルの編集効率を達成できる(図31)一方で、図32A〜Bに示されるように、ほとんどは対照試料(エレクトロポレーションなし(EPなし)または基質なしのエレクトロポレーション(模擬EP)の対照)と比較して有意に低い細胞生存率を有することを見いだした。このことは、Cas9 RNPが、ヒトmPB CD34+細胞へのCas9及びgRNAの効率的送達のために使用する上で最良のフォーマットであることを示している。
Claims (12)
- ヒト細胞におけるB細胞リンパ腫11A(BCL11A)遺伝子をゲノム編集によって編集する方法であって、
前記ヒト細胞にS.pyogenes Cas9エンドヌクレアーゼ及び1つ以上のガイドRNA(gRNA)を導入して、前記BCL11A遺伝子または前記BCL11A遺伝子の調節エレメントをコードする他のDNA配列の中またはその付近で1つ以上の1本鎖切断(SSB)または2本鎖切断(DSB)を起こし、その結果前記BCL11A遺伝子の転写制御配列の永続的な欠失、修飾、または不活性化をもたらすステップを含み、任意に、前記転写制御配列が前記BCL11A遺伝子の2番目のイントロンまたは前記BCL11A遺伝子の赤血球エンハンサーの+58 DNA高感受性部位(DHS)中に位置し、前記1つ以上のgRNAの1つが配列番号71,959の核酸配列を含む、前記方法。 - (i)前記細胞に、前記S.pyogenes Cas9エンドヌクレアーゼ及び前記1つ以上のgRNAをコードする1つ以上のポリヌクレオチドを導入すること;または
(ii)前記細胞に、前記S.pyogenes Cas9エンドヌクレアーゼ及び前記1つ以上のgRNAをコードする1つ以上のリボ核酸(RNA)を導入することを含み、任意には、前記1つ以上のポリヌクレオチドまたは1つ以上のRNAが、1つ以上の改変ポリヌクレオチドまたは1つ以上の改変RNAである、請求項1に記載の方法。 - 前記1つ以上のタンパク質またはポリペプチドが、N末端、C末端、またはN末端及びC末端の両方で1つ以上の核局在化シグナル(NLS)に隣接しており、任意には、前記1つ以上のNLSがSV40 NLSである、請求項1または2に記載の方法。
- 前記1つ以上のgRNAが単一分子ガイドRNA(sgRNA)である、請求項1〜3のいずれか1項に記載の方法。
- 前記S.pyogenes Cas9エンドヌクレアーゼが前記1つ以上のgRNAまたは前記1つ以上のsgRNAと予め複合体化されて1つ以上のリボ核タンパク質(RNP)を形成し、任意には、前記RNP内のsgRNA:エンドヌクレアーゼの重量比が1:1である、請求項1〜4のいずれか1項に記載の方法。
- 前記S.pyogenes Cas9エンドヌクレアーゼがN末端SV40 NLS及びC末端SV40 NLSを含み、sgRNA:DNAエンドヌクレアーゼの重量比が1:1である、請求項5に記載の方法。
- 前記方法が、前記細胞に、改変転写制御配列を含む野生型BCL11A遺伝子またはcDNAを含むポリヌクレオチドドナーテンプレートを導入することをさらに含む、請求項1〜6のいずれか1項に記載の方法。
- (i)前記方法が、前記細胞に、1つのガイドリボ核酸(gRNA)と、改変転写制御配列を含む野生型BCL11A遺伝子またはcDNAを含むポリヌクレオチドドナーテンプレートとを導入することを含み、ここで、前記Cas9エンドヌクレアーゼが、前記BCL11A遺伝子または前記BCL11A遺伝子の調節エレメントをコードする他のDNA配列の中またはその付近の座位に1本鎖切断(SSB)または2本鎖切断(DSB)を起こし、前記座位で前記ポリヌクレオチドドナーテンプレートからの新たな配列を染色体DNAに挿入することを容易にして、その結果、前記座位に近接する前記染色体DNAの前記転写制御配列の永続的な挿入、修飾、または不活性化をもたらす;または
(ii)前記方法が、前記細胞に、前記1つ以上のgRNAと、改変転写制御配列を含む野生型BCL11A遺伝子またはcDNAを含むポリヌクレオチドドナーテンプレートとを導入することを含み、ここで、前記Cas9エンドヌクレアーゼが、前記BCL11A遺伝子または前記BCL11A遺伝子の調節エレメントをコードする他のDNA配列の中またはその付近で、一対の1本鎖切断(SSB)または2本鎖切断(DSB)を、第1の切断を5’座位で、第2の切断を3’座位で起こしまたは創出し、前記5’座位と前記3’座位との間で前記ポリヌクレオチドドナーテンプレートからの新たな配列を染色体DNAに挿入することを容易にして、その結果、前記5’座位と前記3’座位との間で前記染色体DNAの前記転写制御配列における永続的な挿入、修飾、または不活性化をもたらす、
請求項1〜7のいずれか1項に記載の方法。 - 前記Cas9エンドヌクレアーゼが1つもしくは2つのgRNAまたは1つもしくは2つのsgRNAと予め複合体化されて1つ以上のリボ核タンパク質(RNP)を形成し、任意には、前記RNP内のsgRNA:Cas9エンドヌクレアーゼの重量比が1:1である、請求項8に記載の方法。
- (i)前記ドナーテンプレートが1本鎖または2本鎖であり、及び/または
(ii)前記改変転写制御配列が前記BCL11A遺伝子の2番目のイントロン中に位置するか、または、前記改変転写制御配列が前記BCL11A遺伝子の赤血球エンハンサーの+58 DNA高感受性部位(DHS)中に位置する、請求項7〜9のいずれか1項に記載の方法。 - (i)前記Cas9、gRNA、及びドナーテンプレートが、それぞれが別々の脂質ナノ粒子に製剤化されるか、または全てが1つの脂質ナノ粒子に同時に製剤化されるか;または
(ii)前記Cas9が1つの脂質ナノ粒子に製剤化され、前記gRNA及びドナーテンプレートの両方がアデノ随伴ウイルス(AAV)ベクターによって前記細胞に送達されるか;または
(iii)前記Cas9が1つの脂質ナノ粒子に製剤化され、前記gRNAがエレクトロポレーションによって前記細胞に送達され、ドナーテンプレートがアデノ随伴ウイルス(AAV)ベクターによって前記細胞に送達される、請求項7〜10のいずれか1項に記載の方法。 - 前記1つ以上のRNPがエレクトロポレーションによって前記細胞に送達される、請求項5〜11のいずれか1項に記載の方法。
Applications Claiming Priority (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201662324024P | 2016-04-18 | 2016-04-18 | |
| US62/324,024 | 2016-04-18 | ||
| US201662382522P | 2016-09-01 | 2016-09-01 | |
| US62/382,522 | 2016-09-01 | ||
| US201662429428P | 2016-12-02 | 2016-12-02 | |
| US62/429,428 | 2016-12-02 | ||
| PCT/IB2017/000577 WO2017182881A2 (en) | 2016-04-18 | 2017-04-18 | Materials and methods for treatment of hemoglobinopathies |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2019513407A JP2019513407A (ja) | 2019-05-30 |
| JP2019513407A5 JP2019513407A5 (ja) | 2020-05-28 |
| JP6974349B2 true JP6974349B2 (ja) | 2021-12-01 |
Family
ID=58993163
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2018555646A Active JP6974349B2 (ja) | 2016-04-18 | 2017-04-18 | ヘモグロビン異常症の処置のための材料及び方法 |
Country Status (21)
| Country | Link |
|---|---|
| US (4) | US20200330609A1 (ja) |
| EP (2) | EP3445388B1 (ja) |
| JP (1) | JP6974349B2 (ja) |
| KR (1) | KR102351329B1 (ja) |
| CN (1) | CN109715198B (ja) |
| AU (1) | AU2017252023B2 (ja) |
| BR (1) | BR112018071321A2 (ja) |
| CA (1) | CA3021467A1 (ja) |
| DK (1) | DK3445388T3 (ja) |
| FI (1) | FI3445388T3 (ja) |
| HR (1) | HRP20240657T1 (ja) |
| IL (1) | IL262416B2 (ja) |
| LT (1) | LT3445388T (ja) |
| MD (1) | MD3445388T2 (ja) |
| MX (1) | MX2018012729A (ja) |
| PL (1) | PL3445388T3 (ja) |
| PT (1) | PT3445388T (ja) |
| RS (1) | RS65592B1 (ja) |
| SG (2) | SG11201809144XA (ja) |
| SI (1) | SI3445388T1 (ja) |
| WO (1) | WO2017182881A2 (ja) |
Families Citing this family (26)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2016135558A2 (en) | 2015-02-23 | 2016-09-01 | Crispr Therapeutics Ag | Materials and methods for treatment of hemoglobinopathies |
| EP3294873B2 (en) * | 2015-05-08 | 2024-09-18 | The Children's Medical Center Corporation | Targeting bcl11a enhancer functional regions for fetal hemoglobin reinduction |
| JP7030522B2 (ja) | 2015-05-11 | 2022-03-07 | エディタス・メディシン、インコーポレイテッド | 幹細胞における遺伝子編集のための最適化crispr/cas9システムおよび方法 |
| SG10202112057QA (en) | 2015-05-12 | 2021-12-30 | Sangamo Therapeutics Inc | Nuclease-mediated regulation of gene expression |
| WO2016201047A1 (en) | 2015-06-09 | 2016-12-15 | Editas Medicine, Inc. | Crispr/cas-related methods and compositions for improving transplantation |
| EP3371306B8 (en) | 2015-11-04 | 2023-02-22 | Vertex Pharmaceuticals Incorporated | Materials and methods for treatment of hemoglobinopathies |
| TW201839136A (zh) | 2017-02-06 | 2018-11-01 | 瑞士商諾華公司 | 治療血色素異常症之組合物及方法 |
| WO2018218135A1 (en) | 2017-05-25 | 2018-11-29 | The Children's Medical Center Corporation | Bcl11a guide delivery |
| US11866726B2 (en) | 2017-07-14 | 2024-01-09 | Editas Medicine, Inc. | Systems and methods for targeted integration and genome editing and detection thereof using integrated priming sites |
| EP3701029A1 (en) * | 2017-10-26 | 2020-09-02 | Vertex Pharmaceuticals Incorporated | Materials and methods for treatment of hemoglobinopathies |
| CN109722414A (zh) * | 2017-10-27 | 2019-05-07 | 博雅辑因(北京)生物科技有限公司 | 一种高效制备成熟红细胞的方法以及用于制备成熟红细胞的培养基 |
| BR112020011255A2 (pt) * | 2017-12-05 | 2020-11-24 | Vertex Pharmaceuticals Incorporated | células-tronco e progenitoras hematopoiéticas cd34+ humanas modificadas por crispr-cas9 e usos das mesmas |
| CA3085338A1 (en) * | 2017-12-11 | 2019-06-20 | Editas Medicine, Inc. | Cpf1-related methods and compositions for gene editing |
| MA51788A (fr) | 2018-02-05 | 2020-12-16 | Vertex Pharma | Substances et méthodes pour traiter des hémoglobinopathies |
| US11566236B2 (en) | 2018-02-05 | 2023-01-31 | Vertex Pharmaceuticals Incorporated | Materials and methods for treatment of hemoglobinopathies |
| WO2020205838A1 (en) * | 2019-04-02 | 2020-10-08 | Sangamo Therapeutics, Inc. | Methods for the treatment of beta-thalassemia |
| CN110117659B (zh) * | 2019-06-18 | 2022-10-11 | 上海奕谱生物科技有限公司 | 一种新型的肿瘤标记物stamp-ep10及其应用 |
| CN114364799B (zh) * | 2019-08-28 | 2024-07-19 | 甘李药业股份有限公司 | 编辑造血干/祖细胞中bcl11a基因的方法 |
| EP4237558A1 (en) * | 2020-10-30 | 2023-09-06 | Arbor Biotechnologies, Inc. | Compositions comprising an rna guide targeting bcl11a and uses thereof |
| GB202017555D0 (en) * | 2020-11-06 | 2020-12-23 | Harbour Antibodies Bv | Antibody-conjugated nanoparticles |
| US20240026386A1 (en) * | 2020-12-03 | 2024-01-25 | Scribe Therapeutics Inc. | Compositions and methods for the targeting of bcl11a |
| US20240189457A1 (en) * | 2021-04-02 | 2024-06-13 | Shanghaitech University | Gene therapy for treating beta-hemoglobinopathies |
| JP2024533313A (ja) * | 2021-09-08 | 2024-09-12 | フラッグシップ パイオニアリング イノベーションズ シックス,エルエルシー | Hbbを調節する組成物及び方法 |
| WO2024050450A1 (en) | 2022-08-31 | 2024-03-07 | Gigamune, Inc. | Engineered enveloped vectors and methods of use thereof |
| WO2024073751A1 (en) | 2022-09-29 | 2024-04-04 | Vor Biopharma Inc. | Methods and compositions for gene modification and enrichment |
| WO2024095213A1 (en) * | 2022-11-02 | 2024-05-10 | Incisive Genetics, Inc. | Compositions and methods for preventing, ameliorating, or treating sickle cell disease and compositions and methods for disrupting genes and gene segments |
Family Cites Families (139)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3687808A (en) | 1969-08-14 | 1972-08-29 | Univ Leland Stanford Junior | Synthetic polynucleotides |
| US4469863A (en) | 1980-11-12 | 1984-09-04 | Ts O Paul O P | Nonionic nucleic acid alkyl and aryl phosphonates and processes for manufacture and use thereof |
| US5023243A (en) | 1981-10-23 | 1991-06-11 | Molecular Biosystems, Inc. | Oligonucleotide therapeutic agent and method of making same |
| US4476301A (en) | 1982-04-29 | 1984-10-09 | Centre National De La Recherche Scientifique | Oligonucleotides, a process for preparing the same and their application as mediators of the action of interferon |
| JPS5927900A (ja) | 1982-08-09 | 1984-02-14 | Wakunaga Seiyaku Kk | 固定化オリゴヌクレオチド |
| FR2540122B1 (fr) | 1983-01-27 | 1985-11-29 | Centre Nat Rech Scient | Nouveaux composes comportant une sequence d'oligonucleotide liee a un agent d'intercalation, leur procede de synthese et leur application |
| US4605735A (en) | 1983-02-14 | 1986-08-12 | Wakunaga Seiyaku Kabushiki Kaisha | Oligonucleotide derivatives |
| US4948882A (en) | 1983-02-22 | 1990-08-14 | Syngene, Inc. | Single-stranded labelled oligonucleotides, reactive monomers and methods of synthesis |
| US4824941A (en) | 1983-03-10 | 1989-04-25 | Julian Gordon | Specific antibody to the native form of 2'5'-oligonucleotides, the method of preparation and the use as reagents in immunoassays or for binding 2'5'-oligonucleotides in biological systems |
| US4587044A (en) | 1983-09-01 | 1986-05-06 | The Johns Hopkins University | Linkage of proteins to nucleic acids |
| US5118802A (en) | 1983-12-20 | 1992-06-02 | California Institute Of Technology | DNA-reporter conjugates linked via the 2' or 5'-primary amino group of the 5'-terminal nucleoside |
| US5550111A (en) | 1984-07-11 | 1996-08-27 | Temple University-Of The Commonwealth System Of Higher Education | Dual action 2',5'-oligoadenylate antiviral derivatives and uses thereof |
| US5258506A (en) | 1984-10-16 | 1993-11-02 | Chiron Corporation | Photolabile reagents for incorporation into oligonucleotide chains |
| US5430136A (en) | 1984-10-16 | 1995-07-04 | Chiron Corporation | Oligonucleotides having selectably cleavable and/or abasic sites |
| US5367066A (en) | 1984-10-16 | 1994-11-22 | Chiron Corporation | Oligonucleotides with selectably cleavable and/or abasic sites |
| US4828979A (en) | 1984-11-08 | 1989-05-09 | Life Technologies, Inc. | Nucleotide analogs for nucleic acid labeling and detection |
| FR2575751B1 (fr) | 1985-01-08 | 1987-04-03 | Pasteur Institut | Nouveaux nucleosides de derives de l'adenosine, leur preparation et leurs applications biologiques |
| US5405938A (en) | 1989-12-20 | 1995-04-11 | Anti-Gene Development Group | Sequence-specific binding polymers for duplex nucleic acids |
| US5034506A (en) | 1985-03-15 | 1991-07-23 | Anti-Gene Development Group | Uncharged morpholino-based polymers having achiral intersubunit linkages |
| US5166315A (en) | 1989-12-20 | 1992-11-24 | Anti-Gene Development Group | Sequence-specific binding polymers for duplex nucleic acids |
| US5185444A (en) | 1985-03-15 | 1993-02-09 | Anti-Gene Deveopment Group | Uncharged morpolino-based polymers having phosphorous containing chiral intersubunit linkages |
| US5235033A (en) | 1985-03-15 | 1993-08-10 | Anti-Gene Development Group | Alpha-morpholino ribonucleoside derivatives and polymers thereof |
| US4762779A (en) | 1985-06-13 | 1988-08-09 | Amgen Inc. | Compositions and methods for functionalizing nucleic acids |
| US5317098A (en) | 1986-03-17 | 1994-05-31 | Hiroaki Shizuya | Non-radioisotope tagging of fragments |
| JPS638396A (ja) | 1986-06-30 | 1988-01-14 | Wakunaga Pharmaceut Co Ltd | ポリ標識化オリゴヌクレオチド誘導体 |
| US5264423A (en) | 1987-03-25 | 1993-11-23 | The United States Of America As Represented By The Department Of Health And Human Services | Inhibitors for replication of retroviruses and for the expression of oncogene products |
| US5276019A (en) | 1987-03-25 | 1994-01-04 | The United States Of America As Represented By The Department Of Health And Human Services | Inhibitors for replication of retroviruses and for the expression of oncogene products |
| US4904582A (en) | 1987-06-11 | 1990-02-27 | Synthetic Genetics | Novel amphiphilic nucleic acid conjugates |
| WO1988010264A1 (en) | 1987-06-24 | 1988-12-29 | Howard Florey Institute Of Experimental Physiology | Nucleoside derivatives |
| US5585481A (en) | 1987-09-21 | 1996-12-17 | Gen-Probe Incorporated | Linking reagents for nucleotide probes |
| US4924624A (en) | 1987-10-22 | 1990-05-15 | Temple University-Of The Commonwealth System Of Higher Education | 2,',5'-phosphorothioate oligoadenylates and plant antiviral uses thereof |
| US5188897A (en) | 1987-10-22 | 1993-02-23 | Temple University Of The Commonwealth System Of Higher Education | Encapsulated 2',5'-phosphorothioate oligoadenylates |
| US5525465A (en) | 1987-10-28 | 1996-06-11 | Howard Florey Institute Of Experimental Physiology And Medicine | Oligonucleotide-polyamide conjugates and methods of production and applications of the same |
| DE3738460A1 (de) | 1987-11-12 | 1989-05-24 | Max Planck Gesellschaft | Modifizierte oligonukleotide |
| US5082830A (en) | 1988-02-26 | 1992-01-21 | Enzo Biochem, Inc. | End labeled nucleotide probe |
| WO1989009221A1 (en) | 1988-03-25 | 1989-10-05 | University Of Virginia Alumni Patents Foundation | Oligonucleotide n-alkylphosphoramidates |
| US5278302A (en) | 1988-05-26 | 1994-01-11 | University Patents, Inc. | Polynucleotide phosphorodithioates |
| US5109124A (en) | 1988-06-01 | 1992-04-28 | Biogen, Inc. | Nucleic acid probe linked to a label having a terminal cysteine |
| US5216141A (en) | 1988-06-06 | 1993-06-01 | Benner Steven A | Oligonucleotide analogs containing sulfur linkages |
| US5175273A (en) | 1988-07-01 | 1992-12-29 | Genentech, Inc. | Nucleic acid intercalating agents |
| US5262536A (en) | 1988-09-15 | 1993-11-16 | E. I. Du Pont De Nemours And Company | Reagents for the preparation of 5'-tagged oligonucleotides |
| US5512439A (en) | 1988-11-21 | 1996-04-30 | Dynal As | Oligonucleotide-linked magnetic particles and uses thereof |
| US5599923A (en) | 1989-03-06 | 1997-02-04 | Board Of Regents, University Of Tx | Texaphyrin metal complexes having improved functionalization |
| US5457183A (en) | 1989-03-06 | 1995-10-10 | Board Of Regents, The University Of Texas System | Hydroxylated texaphyrins |
| US5391723A (en) | 1989-05-31 | 1995-02-21 | Neorx Corporation | Oligonucleotide conjugates |
| US4958013A (en) | 1989-06-06 | 1990-09-18 | Northwestern University | Cholesteryl modified oligonucleotides |
| US5451463A (en) | 1989-08-28 | 1995-09-19 | Clontech Laboratories, Inc. | Non-nucleoside 1,3-diol reagents for labeling synthetic oligonucleotides |
| US5134066A (en) | 1989-08-29 | 1992-07-28 | Monsanto Company | Improved probes using nucleosides containing 3-dezauracil analogs |
| US5254469A (en) | 1989-09-12 | 1993-10-19 | Eastman Kodak Company | Oligonucleotide-enzyme conjugate that can be used as a probe in hybridization assays and polymerase chain reaction procedures |
| US5399676A (en) | 1989-10-23 | 1995-03-21 | Gilead Sciences | Oligonucleotides with inverted polarity |
| US5264564A (en) | 1989-10-24 | 1993-11-23 | Gilead Sciences | Oligonucleotide analogs with novel linkages |
| US5264562A (en) | 1989-10-24 | 1993-11-23 | Gilead Sciences, Inc. | Oligonucleotide analogs with novel linkages |
| US5292873A (en) | 1989-11-29 | 1994-03-08 | The Research Foundation Of State University Of New York | Nucleic acids labeled with naphthoquinone probe |
| US5177198A (en) | 1989-11-30 | 1993-01-05 | University Of N.C. At Chapel Hill | Process for preparing oligoribonucleoside and oligodeoxyribonucleoside boranophosphates |
| US5130302A (en) | 1989-12-20 | 1992-07-14 | Boron Bilogicals, Inc. | Boronated nucleoside, nucleotide and oligonucleotide compounds, compositions and methods for using same |
| US5486603A (en) | 1990-01-08 | 1996-01-23 | Gilead Sciences, Inc. | Oligonucleotide having enhanced binding affinity |
| US5587470A (en) | 1990-01-11 | 1996-12-24 | Isis Pharmaceuticals, Inc. | 3-deazapurines |
| US5578718A (en) | 1990-01-11 | 1996-11-26 | Isis Pharmaceuticals, Inc. | Thiol-derivatized nucleosides |
| US5587361A (en) | 1991-10-15 | 1996-12-24 | Isis Pharmaceuticals, Inc. | Oligonucleotides having phosphorothioate linkages of high chiral purity |
| US5459255A (en) | 1990-01-11 | 1995-10-17 | Isis Pharmaceuticals, Inc. | N-2 substituted purines |
| US5681941A (en) | 1990-01-11 | 1997-10-28 | Isis Pharmaceuticals, Inc. | Substituted purines and oligonucleotide cross-linking |
| AU7579991A (en) | 1990-02-20 | 1991-09-18 | Gilead Sciences, Inc. | Pseudonucleosides and pseudonucleotides and their polymers |
| US5214136A (en) | 1990-02-20 | 1993-05-25 | Gilead Sciences, Inc. | Anthraquinone-derivatives oligonucleotides |
| US5321131A (en) | 1990-03-08 | 1994-06-14 | Hybridon, Inc. | Site-specific functionalization of oligodeoxynucleotides for non-radioactive labelling |
| US5470967A (en) | 1990-04-10 | 1995-11-28 | The Dupont Merck Pharmaceutical Company | Oligonucleotide analogs with sulfamate linkages |
| ES2116977T3 (es) | 1990-05-11 | 1998-08-01 | Microprobe Corp | Soportes solidos para ensayos de hibridacion de acidos nucleicos y metodos para inmovilizar oligonucleotidos de modo covalente. |
| US5614617A (en) | 1990-07-27 | 1997-03-25 | Isis Pharmaceuticals, Inc. | Nuclease resistant, pyrimidine modified oligonucleotides that detect and modulate gene expression |
| US5618704A (en) | 1990-07-27 | 1997-04-08 | Isis Pharmacueticals, Inc. | Backbone-modified oligonucleotide analogs and preparation thereof through radical coupling |
| US5489677A (en) | 1990-07-27 | 1996-02-06 | Isis Pharmaceuticals, Inc. | Oligonucleoside linkages containing adjacent oxygen and nitrogen atoms |
| US5608046A (en) | 1990-07-27 | 1997-03-04 | Isis Pharmaceuticals, Inc. | Conjugated 4'-desmethyl nucleoside analog compounds |
| US5688941A (en) | 1990-07-27 | 1997-11-18 | Isis Pharmaceuticals, Inc. | Methods of making conjugated 4' desmethyl nucleoside analog compounds |
| US5541307A (en) | 1990-07-27 | 1996-07-30 | Isis Pharmaceuticals, Inc. | Backbone modified oligonucleotide analogs and solid phase synthesis thereof |
| US5218105A (en) | 1990-07-27 | 1993-06-08 | Isis Pharmaceuticals | Polyamine conjugated oligonucleotides |
| US5138045A (en) | 1990-07-27 | 1992-08-11 | Isis Pharmaceuticals | Polyamine conjugated oligonucleotides |
| US5602240A (en) | 1990-07-27 | 1997-02-11 | Ciba Geigy Ag. | Backbone modified oligonucleotide analogs |
| US5610289A (en) | 1990-07-27 | 1997-03-11 | Isis Pharmaceuticals, Inc. | Backbone modified oligonucleotide analogues |
| US5623070A (en) | 1990-07-27 | 1997-04-22 | Isis Pharmaceuticals, Inc. | Heteroatomic oligonucleoside linkages |
| US5677437A (en) | 1990-07-27 | 1997-10-14 | Isis Pharmaceuticals, Inc. | Heteroatomic oligonucleoside linkages |
| US5245022A (en) | 1990-08-03 | 1993-09-14 | Sterling Drug, Inc. | Exonuclease resistant terminally substituted oligonucleotides |
| ES2083593T3 (es) | 1990-08-03 | 1996-04-16 | Sterling Winthrop Inc | Compuestos y metodos para inhibir la expresion de genes. |
| US5177196A (en) | 1990-08-16 | 1993-01-05 | Microprobe Corporation | Oligo (α-arabinofuranosyl nucleotides) and α-arabinofuranosyl precursors thereof |
| US5512667A (en) | 1990-08-28 | 1996-04-30 | Reed; Michael W. | Trifunctional intermediates for preparing 3'-tailed oligonucleotides |
| US5214134A (en) | 1990-09-12 | 1993-05-25 | Sterling Winthrop Inc. | Process of linking nucleosides with a siloxane bridge |
| US5561225A (en) | 1990-09-19 | 1996-10-01 | Southern Research Institute | Polynucleotide analogs containing sulfonate and sulfonamide internucleoside linkages |
| WO1992005186A1 (en) | 1990-09-20 | 1992-04-02 | Gilead Sciences | Modified internucleoside linkages |
| US5432272A (en) | 1990-10-09 | 1995-07-11 | Benner; Steven A. | Method for incorporating into a DNA or RNA oligonucleotide using nucleotides bearing heterocyclic bases |
| US5173414A (en) | 1990-10-30 | 1992-12-22 | Applied Immune Sciences, Inc. | Production of recombinant adeno-associated virus vectors |
| CA2095212A1 (en) | 1990-11-08 | 1992-05-09 | Sudhir Agrawal | Incorporation of multiple reporter groups on synthetic oligonucleotides |
| US5714331A (en) | 1991-05-24 | 1998-02-03 | Buchardt, Deceased; Ole | Peptide nucleic acids having enhanced binding affinity, sequence specificity and solubility |
| US5719262A (en) | 1993-11-22 | 1998-02-17 | Buchardt, Deceased; Ole | Peptide nucleic acids having amino acid side chains |
| US5539082A (en) | 1993-04-26 | 1996-07-23 | Nielsen; Peter E. | Peptide nucleic acids |
| US5371241A (en) | 1991-07-19 | 1994-12-06 | Pharmacia P-L Biochemicals Inc. | Fluorescein labelled phosphoramidites |
| US5571799A (en) | 1991-08-12 | 1996-11-05 | Basco, Ltd. | (2'-5') oligoadenylate analogues useful as inhibitors of host-v5.-graft response |
| TW393513B (en) | 1991-11-26 | 2000-06-11 | Isis Pharmaceuticals Inc | Enhanced triple-helix and double-helix formation with oligomers containing modified pyrimidines |
| US5484908A (en) | 1991-11-26 | 1996-01-16 | Gilead Sciences, Inc. | Oligonucleotides containing 5-propynyl pyrimidines |
| US5595726A (en) | 1992-01-21 | 1997-01-21 | Pharmacyclics, Inc. | Chromophore probe for detection of nucleic acid |
| US5565552A (en) | 1992-01-21 | 1996-10-15 | Pharmacyclics, Inc. | Method of expanded porphyrin-oligonucleotide conjugate synthesis |
| US5633360A (en) | 1992-04-14 | 1997-05-27 | Gilead Sciences, Inc. | Oligonucleotide analogs capable of passive cell membrane permeation |
| US5434257A (en) | 1992-06-01 | 1995-07-18 | Gilead Sciences, Inc. | Binding compentent oligomers containing unsaturated 3',5' and 2',5' linkages |
| US5272250A (en) | 1992-07-10 | 1993-12-21 | Spielvogel Bernard F | Boronated phosphoramidate compounds |
| US5574142A (en) | 1992-12-15 | 1996-11-12 | Microprobe Corporation | Peptide linkers for improved oligonucleotide delivery |
| US5476925A (en) | 1993-02-01 | 1995-12-19 | Northwestern University | Oligodeoxyribonucleotides including 3'-aminonucleoside-phosphoramidate linkages and terminal 3'-amino groups |
| GB9304618D0 (en) | 1993-03-06 | 1993-04-21 | Ciba Geigy Ag | Chemical compounds |
| AU6412794A (en) | 1993-03-31 | 1994-10-24 | Sterling Winthrop Inc. | Oligonucleotides with amide linkages replacing phosphodiester linkages |
| US5502177A (en) | 1993-09-17 | 1996-03-26 | Gilead Sciences, Inc. | Pyrimidine derivatives for labeled binding partners |
| JP3952312B2 (ja) | 1993-11-09 | 2007-08-01 | メディカル カレッジ オブ オハイオ | アデノ関連ウイルス複製遺伝子を発現可能な安定な細胞株 |
| PT733103E (pt) | 1993-11-09 | 2004-07-30 | Targeted Genetics Corp | Criacao de elevados titulos de vectores de aav recombinantes |
| US5457187A (en) | 1993-12-08 | 1995-10-10 | Board Of Regents University Of Nebraska | Oligonucleotides containing 5-fluorouracil |
| US5596091A (en) | 1994-03-18 | 1997-01-21 | The Regents Of The University Of California | Antisense oligonucleotides comprising 5-aminoalkyl pyrimidine nucleotides |
| US5625050A (en) | 1994-03-31 | 1997-04-29 | Amgen Inc. | Modified oligonucleotides and intermediates useful in nucleic acid therapeutics |
| US5525711A (en) | 1994-05-18 | 1996-06-11 | The United States Of America As Represented By The Secretary Of The Department Of Health And Human Services | Pteridine nucleotide analogs as fluorescent DNA probes |
| US5658785A (en) | 1994-06-06 | 1997-08-19 | Children's Hospital, Inc. | Adeno-associated virus materials and methods |
| US5597696A (en) | 1994-07-18 | 1997-01-28 | Becton Dickinson And Company | Covalent cyanine dye oligonucleotide conjugates |
| US5580731A (en) | 1994-08-25 | 1996-12-03 | Chiron Corporation | N-4 modified pyrimidine deoxynucleotides and oligonucleotide probes synthesized therewith |
| US5856152A (en) | 1994-10-28 | 1999-01-05 | The Trustees Of The University Of Pennsylvania | Hybrid adenovirus-AAV vector and methods of use therefor |
| WO1996017947A1 (en) | 1994-12-06 | 1996-06-13 | Targeted Genetics Corporation | Packaging cell lines for generation of high titers of recombinant aav vectors |
| FR2737730B1 (fr) | 1995-08-10 | 1997-09-05 | Pasteur Merieux Serums Vacc | Procede de purification de virus par chromatographie |
| US6143548A (en) | 1995-08-30 | 2000-11-07 | Genzyme Corporation | Chromatographic purification of adeno-associated virus (AAV) |
| ES2317646T3 (es) | 1995-09-08 | 2009-04-16 | Genzyme Corporation | Vectores aav mejorados para terapia genica. |
| US5910434A (en) | 1995-12-15 | 1999-06-08 | Systemix, Inc. | Method for obtaining retroviral packaging cell lines producing high transducing efficiency retroviral supernatant |
| PT1944362E (pt) | 1997-09-05 | 2016-01-27 | Genzyme Corp | Métodos de produção de preparações de alto título de vetores aav recombinantes desprovidos de adjuvantes |
| US6258595B1 (en) | 1999-03-18 | 2001-07-10 | The Trustees Of The University Of Pennsylvania | Compositions and methods for helper-free production of recombinant adeno-associated viruses |
| US6287860B1 (en) | 2000-01-20 | 2001-09-11 | Isis Pharmaceuticals, Inc. | Antisense inhibition of MEKK2 expression |
| AU2001255575B2 (en) | 2000-04-28 | 2006-08-31 | The Trustees Of The University Of Pennsylvania | Recombinant aav vectors with aav5 capsids and aav5 vectors pseudotyped in heterologous capsids |
| US20030158403A1 (en) | 2001-07-03 | 2003-08-21 | Isis Pharmaceuticals, Inc. | Nuclease resistant chimeric oligonucleotides |
| WO2013080784A1 (ja) | 2011-11-30 | 2013-06-06 | シャープ株式会社 | メモリ回路とその駆動方法、及び、これを用いた不揮発性記憶装置、並びに、液晶表示装置 |
| EP3272356A1 (en) * | 2012-02-24 | 2018-01-24 | Fred Hutchinson Cancer Research Center | Compositions and methods for the treatment of hemoglobinopathies |
| DE102012007232B4 (de) | 2012-04-07 | 2014-03-13 | Susanne Weller | Verfahren zur Herstellung von rotierenden elektrischen Maschinen |
| DK2800811T3 (en) | 2012-05-25 | 2017-07-17 | Univ Vienna | METHODS AND COMPOSITIONS FOR RNA DIRECTIVE TARGET DNA MODIFICATION AND FOR RNA DIRECTIVE MODULATION OF TRANSCRIPTION |
| CN109554350B (zh) * | 2012-11-27 | 2022-09-23 | 儿童医疗中心有限公司 | 用于胎儿血红蛋白再诱导的靶向bcl11a远端调控元件 |
| US20150044772A1 (en) * | 2013-08-09 | 2015-02-12 | Sage Labs, Inc. | Crispr/cas system-based novel fusion protein and its applications in genome editing |
| JP2015092462A (ja) | 2013-09-30 | 2015-05-14 | Tdk株式会社 | 正極及びそれを用いたリチウムイオン二次電池 |
| JP6202701B2 (ja) | 2014-03-21 | 2017-09-27 | 株式会社日立国際電気 | 基板処理装置、半導体装置の製造方法及びプログラム |
| CA2946881A1 (en) * | 2014-04-28 | 2015-11-05 | Recombinetics, Inc. | Multiplex gene editing in swine |
| US20170037431A1 (en) * | 2014-05-01 | 2017-02-09 | University Of Washington | In vivo Gene Engineering with Adenoviral Vectors |
| US20170191123A1 (en) * | 2014-05-28 | 2017-07-06 | Toolgen Incorporated | Method for Sensitive Detection of Target DNA Using Target-Specific Nuclease |
| JP6197169B2 (ja) | 2014-09-29 | 2017-09-20 | 東芝メモリ株式会社 | 半導体装置の製造方法 |
| EP3701029A1 (en) * | 2017-10-26 | 2020-09-02 | Vertex Pharmaceuticals Incorporated | Materials and methods for treatment of hemoglobinopathies |
| BR112020011255A2 (pt) * | 2017-12-05 | 2020-11-24 | Vertex Pharmaceuticals Incorporated | células-tronco e progenitoras hematopoiéticas cd34+ humanas modificadas por crispr-cas9 e usos das mesmas |
-
2017
- 2017-04-18 SG SG11201809144XA patent/SG11201809144XA/en unknown
- 2017-04-18 SG SG10202010261YA patent/SG10202010261YA/en unknown
- 2017-04-18 US US16/094,408 patent/US20200330609A1/en not_active Abandoned
- 2017-04-18 MD MDE20190261T patent/MD3445388T2/ro unknown
- 2017-04-18 AU AU2017252023A patent/AU2017252023B2/en active Active
- 2017-04-18 EP EP17727373.7A patent/EP3445388B1/en active Active
- 2017-04-18 JP JP2018555646A patent/JP6974349B2/ja active Active
- 2017-04-18 HR HRP20240657TT patent/HRP20240657T1/hr unknown
- 2017-04-18 CN CN201780037255.7A patent/CN109715198B/zh active Active
- 2017-04-18 IL IL262416A patent/IL262416B2/en unknown
- 2017-04-18 LT LTEPPCT/IB2017/000577T patent/LT3445388T/lt unknown
- 2017-04-18 EP EP24170225.7A patent/EP4424829A2/en active Pending
- 2017-04-18 FI FIEP17727373.7T patent/FI3445388T3/fi active
- 2017-04-18 BR BR112018071321A patent/BR112018071321A2/pt unknown
- 2017-04-18 MX MX2018012729A patent/MX2018012729A/es unknown
- 2017-04-18 KR KR1020187033195A patent/KR102351329B1/ko active IP Right Grant
- 2017-04-18 SI SI201731520T patent/SI3445388T1/sl unknown
- 2017-04-18 DK DK17727373.7T patent/DK3445388T3/da active
- 2017-04-18 CA CA3021467A patent/CA3021467A1/en active Pending
- 2017-04-18 RS RS20240622A patent/RS65592B1/sr unknown
- 2017-04-18 PT PT177273737T patent/PT3445388T/pt unknown
- 2017-04-18 PL PL17727373.7T patent/PL3445388T3/pl unknown
- 2017-04-18 WO PCT/IB2017/000577 patent/WO2017182881A2/en active Application Filing
-
2019
- 2019-03-18 US US16/356,373 patent/US20190201553A1/en not_active Abandoned
-
2022
- 2022-02-02 US US17/591,563 patent/US20220211874A1/en not_active Abandoned
-
2024
- 2024-03-21 US US18/612,729 patent/US20240226339A1/en active Pending
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6974349B2 (ja) | ヘモグロビン異常症の処置のための材料及び方法 | |
| JP7240456B2 (ja) | アルファ1アンチトリプシン欠乏症の治療のための材料および方法 | |
| US20240229078A1 (en) | Materials and methods for treatment of glycogen storage disease type 1a | |
| WO2017191503A1 (en) | Materials and methods for treatment of hemoglobinopathies | |
| US20210180091A1 (en) | Materials and methods for treatment of hemoglobinopathies | |
| EP3516058A1 (en) | Compositions and methods for gene editing | |
| US11083799B2 (en) | Materials and methods for treatment of hereditary haemochromatosis | |
| US20190038771A1 (en) | Materials and methods for treatment of severe combined immunodeficiency (scid) or omenn syndrome | |
| US11268077B2 (en) | Materials and methods for treatment of hemoglobinopathies | |
| US11566236B2 (en) | Materials and methods for treatment of hemoglobinopathies |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20200420 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20200420 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20210608 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20210906 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20211005 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20211104 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 6974349 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |