JP6357586B2 - 熱除去プロセス - Google Patents
熱除去プロセス Download PDFInfo
- Publication number
- JP6357586B2 JP6357586B2 JP2017522533A JP2017522533A JP6357586B2 JP 6357586 B2 JP6357586 B2 JP 6357586B2 JP 2017522533 A JP2017522533 A JP 2017522533A JP 2017522533 A JP2017522533 A JP 2017522533A JP 6357586 B2 JP6357586 B2 JP 6357586B2
- Authority
- JP
- Japan
- Prior art keywords
- reaction
- coolant
- reactor
- reactant
- train
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images




Classifications
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G2/00—Production of liquid hydrocarbon mixtures of undefined composition from oxides of carbon
- C10G2/30—Production of liquid hydrocarbon mixtures of undefined composition from oxides of carbon from carbon monoxide with hydrogen
- C10G2/32—Production of liquid hydrocarbon mixtures of undefined composition from oxides of carbon from carbon monoxide with hydrogen with the use of catalysts
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J19/00—Chemical, physical or physico-chemical processes in general; Their relevant apparatus
- B01J19/0006—Controlling or regulating processes
- B01J19/0013—Controlling the temperature of the process
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J19/00—Chemical, physical or physico-chemical processes in general; Their relevant apparatus
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J8/00—Chemical or physical processes in general, conducted in the presence of fluids and solid particles; Apparatus for such processes
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J8/00—Chemical or physical processes in general, conducted in the presence of fluids and solid particles; Apparatus for such processes
- B01J8/001—Controlling catalytic processes
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C1/00—Preparation of hydrocarbons from one or more compounds, none of them being a hydrocarbon
- C07C1/02—Preparation of hydrocarbons from one or more compounds, none of them being a hydrocarbon from oxides of a carbon
- C07C1/04—Preparation of hydrocarbons from one or more compounds, none of them being a hydrocarbon from oxides of a carbon from carbon monoxide with hydrogen
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C1/00—Preparation of hydrocarbons from one or more compounds, none of them being a hydrocarbon
- C07C1/02—Preparation of hydrocarbons from one or more compounds, none of them being a hydrocarbon from oxides of a carbon
- C07C1/04—Preparation of hydrocarbons from one or more compounds, none of them being a hydrocarbon from oxides of a carbon from carbon monoxide with hydrogen
- C07C1/0455—Reaction conditions
- C07C1/047—Processes in which one or more parameters are changed during the process; Starting-up of the process
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C1/00—Preparation of hydrocarbons from one or more compounds, none of them being a hydrocarbon
- C07C1/02—Preparation of hydrocarbons from one or more compounds, none of them being a hydrocarbon from oxides of a carbon
- C07C1/04—Preparation of hydrocarbons from one or more compounds, none of them being a hydrocarbon from oxides of a carbon from carbon monoxide with hydrogen
- C07C1/0455—Reaction conditions
- C07C1/048—Temperature controlling measures
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C29/00—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring
- C07C29/15—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring by reduction of oxides of carbon exclusively
- C07C29/151—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring by reduction of oxides of carbon exclusively with hydrogen or hydrogen-containing gases
- C07C29/1512—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring by reduction of oxides of carbon exclusively with hydrogen or hydrogen-containing gases characterised by reaction conditions
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C31/00—Saturated compounds having hydroxy or O-metal groups bound to acyclic carbon atoms
- C07C31/02—Monohydroxylic acyclic alcohols
- C07C31/04—Methanol
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G2/00—Production of liquid hydrocarbon mixtures of undefined composition from oxides of carbon
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G2/00—Production of liquid hydrocarbon mixtures of undefined composition from oxides of carbon
- C10G2/30—Production of liquid hydrocarbon mixtures of undefined composition from oxides of carbon from carbon monoxide with hydrogen
- C10G2/32—Production of liquid hydrocarbon mixtures of undefined composition from oxides of carbon from carbon monoxide with hydrogen with the use of catalysts
- C10G2/34—Apparatus, reactors
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2208/00—Processes carried out in the presence of solid particles; Reactors therefor
- B01J2208/00008—Controlling the process
- B01J2208/00017—Controlling the temperature
- B01J2208/00106—Controlling the temperature by indirect heat exchange
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2208/00—Processes carried out in the presence of solid particles; Reactors therefor
- B01J2208/00008—Controlling the process
- B01J2208/00539—Pressure
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2208/00—Processes carried out in the presence of solid particles; Reactors therefor
- B01J2208/00008—Controlling the process
- B01J2208/00548—Flow
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2208/00—Processes carried out in the presence of solid particles; Reactors therefor
- B01J2208/00008—Controlling the process
- B01J2208/00628—Controlling the composition of the reactive mixture
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2208/00—Processes carried out in the presence of solid particles; Reactors therefor
- B01J2208/00008—Controlling the process
- B01J2208/00628—Controlling the composition of the reactive mixture
- B01J2208/00637—Means for stopping or slowing down the reaction
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2219/00—Chemical, physical or physico-chemical processes in general; Their relevant apparatus
- B01J2219/00002—Chemical plants
- B01J2219/00018—Construction aspects
- B01J2219/0002—Plants assembled from modules joined together
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2219/00—Chemical, physical or physico-chemical processes in general; Their relevant apparatus
- B01J2219/00002—Chemical plants
- B01J2219/00027—Process aspects
- B01J2219/00038—Processes in parallel
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2219/00—Chemical, physical or physico-chemical processes in general; Their relevant apparatus
- B01J2219/00002—Chemical plants
- B01J2219/00027—Process aspects
- B01J2219/0004—Processes in series
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2219/00—Chemical, physical or physico-chemical processes in general; Their relevant apparatus
- B01J2219/00049—Controlling or regulating processes
- B01J2219/00051—Controlling the temperature
- B01J2219/00074—Controlling the temperature by indirect heating or cooling employing heat exchange fluids
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2219/00—Chemical, physical or physico-chemical processes in general; Their relevant apparatus
- B01J2219/00049—Controlling or regulating processes
- B01J2219/00162—Controlling or regulating processes controlling the pressure
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2219/00—Chemical, physical or physico-chemical processes in general; Their relevant apparatus
- B01J2219/00049—Controlling or regulating processes
- B01J2219/00164—Controlling or regulating processes controlling the flow
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2219/00—Chemical, physical or physico-chemical processes in general; Their relevant apparatus
- B01J2219/00049—Controlling or regulating processes
- B01J2219/00186—Controlling or regulating processes controlling the composition of the reactive mixture
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/582—Recycling of unreacted starting or intermediate materials
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Engineering & Computer Science (AREA)
- General Chemical & Material Sciences (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Physical Or Chemical Processes And Apparatus (AREA)
- Production Of Liquid Hydrocarbon Mixture For Refining Petroleum (AREA)
- Yarns And Mechanical Finishing Of Yarns Or Ropes (AREA)
- Investigation Of Foundation Soil And Reinforcement Of Foundation Soil By Compacting Or Drainage (AREA)
- Processing And Handling Of Plastics And Other Materials For Molding In General (AREA)
Description
(a)反応物質供給ストリームを、少なくとも2つの別個の反応物質サブストリームに分割するステップと、
(b)反応器を含む別個の反応列内に各反応物質サブストリームを供給するステップと、
(c)共通冷却剤リザーバから各反応器内に冷却剤ストリームを供給するステップと、
(d)反応器内で発熱反応を実行して、反応生成物と、熱が伝わった冷却剤とを生成するステップと、
(e)各反応列からの熱が伝わった冷却剤を単一の共通リザーバに供給して冷却剤から熱を除去するステップと、
(f)ステップ(e)において熱が除去された冷却剤をステップ(c)に戻すステップと、
を含み、
ステップ(b)における各反応器は、同じ温度及び圧力で動作し、
各反応器における発熱反応の進行は、反応器が一部を成す反応列を通る反応物質サブストリームの流量を調整することによって、及び/又は各反応列内に供給される反応物質サブストリームの組成を調整することによって制御される、
方法を提供する。
ステップ(a)
本発明の方法の第1のステップ(ステップ(a))では、反応物質供給ストリームを少なくとも2つの反応物質サブストリームに分割し、各サブストリームを別個の反応列に供給する。このように反応物質供給ストリームを分離すると、生じる反応の規模が最大になることが保証される。
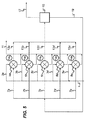
本発明の方法のステップ(b)では、各反応物質サブストリームが別個の反応列内に供給される。各反応列は、少なくとも1つの反応器を含む。反応度を最大化するには、各反応列が複数の反応器を含むことが有利になり得る。複数の反応器が存在する場合には、これらを直列又は並列に配置することができる。複数の反応器は、並列に配置されることが好ましい。いくつかの実施形態では、例えば発熱反応がFT反応である場合、複数の反応器が並列に配置される。
反応器は、触媒を含むことができる。触媒は、不均一系触媒であることが好ましい。1つの実施形態では、触媒が粒子状固体の形を取ることができる。
本発明の方法のステップ(c)では、各反応列の反応器内に冷却剤が供給される。各反応列内に供給される冷却剤は、共通冷却剤リザーバからのものである。この点、全ての反応列内に供給を行う単一の冷却剤循環ネットワークが存在する。冷却剤は、新鮮な冷却剤と、ステップ(e)において蒸気ドラムから再循環される冷却剤とで構成される。
反応物質サブストリームは、反応列に沿って1又は2以上の反応器を流れると、触媒に接触して発熱反応が生じる(ステップ(d))。発熱反応によって発生する熱は冷却剤に伝わり、したがって反応から熱が除去される。したがって、ステップ(d)では、反応生成物と、熱が伝わった冷却剤とが生じる。冷却剤が熱油である場合、蒸発ではなく膨張によって熱を吸収する。冷却剤が、発熱反応によって発生する熱よりも低い沸点を有する流体である場合、部分的相変化を受けることによって反応から熱を除去し、例えば、水が冷却剤である場合には蒸気が発生する。
発熱反応がフィッシャートロプシュプロセスである場合、反応生成物は、(主に脂肪族である)炭化水素及び水を含む。「脂肪族炭化水素」という用語は、2又は3以上の炭素原子、又は3又は4以上の炭素原子、又は4又は5以上の炭素原子、又は5又は6以上の炭素原子、又は6又は7以上の炭素原子を有する脂肪族炭化水素を意味するために使用するものである。高分子量の脂肪族炭化水素は、最大約200個の炭素原子、最大約150個の炭素原子、最大約100個の炭素原子、最大約90個の炭素原子、最大約80個の炭素原子、最大約70個の炭素原子、最大約60個の炭素原子、最大約50個の炭素原子、最大約40個の炭素原子又は最大約30個の炭素原子を有することができる。例としては、エタン、プロパン、ブタン、ペンタン、ヘキサン、オクタン、デカン及びドデカンなどを挙げることができる。
ステップ(e)では、各反応列からの熱が伝わった冷却剤が、共通の単一冷却剤リザーバに供給される。このリザーバでは、発熱反応からの吸収熱を除去し、冷却剤を元の状態に戻す。その後、冷却剤を再循環させてステップ(c)に戻すことができる。
単一の共通冷却剤リザーバを使用するということは、(使用する通常の変数である)個々の反応列の温度を用いて、各反応器で生じている発熱反応の性能を制御することができないことを意味する。この場合、驚くべきことに、本発明者らは、各反応列を通る反応物質サブストリームの流量(又は毎時ガス空間速度(GHSV))を制御することによって異なる反応列を容認可能な程度に制御できることを発見した。
上述したように、本発明の方法の利点は、特に反応物質供給ストリームが少なくとも3つの反応物質サブストリームに分割される場合、個々の反応列をシステムから分離すると同時に残りの反応列を動作状態に維持し、これらの動作に対する影響を最小化できる点である。このことは、発熱プロセスにおいて不均一系触媒が使用され、この触媒の性能が時間と共に低下して何らかの時点で再生が必要になる場合に特に有利である。触媒の再装填が必要な場合にも有用となり得る。
反応器内で発熱反応を実行して、反応生成物と、熱が伝わった第1の冷却剤とを生成するステップと、
第2の冷却剤リザーバに関連する第2の冷却剤循環システムを準備するステップと、
分離すべき反応列からの熱が伝わった第1の冷却剤を第2の冷却剤リザーバに向け直すステップと、
分離すべき反応列への第1の冷却剤の供給を停止すると同時に、第2の冷却剤リザーバから分離すべき反応列に第2の冷却剤の供給を開始するステップと、
を含む方法を提供する。
各反応列の反応器内で発熱反応が実行されて、反応生成物と、熱が伝わった第1の冷却剤とが生成され、
分離された反応列に、第2の冷却剤リザーバから第2の冷却剤が供給され、上記方法は、
分離された反応列への第2の冷却剤の供給を停止すると同時に、分離された反応列への第1の冷却剤の供給を開始するステップと、
分離された反応列の反応器の動作条件が分離されなかった反応器の動作条件に一致するまでプロセスを実行するステップと、
分離された反応列からの熱が伝わった第1の冷却剤を共通冷却剤リザーバに向け直すステップと、
を含む方法を提供する。
したがって、1つの態様では、本発明は、発熱反応を開始する方法であって、
(a)それぞれが少なくとも1つの反応器を含む少なくとも2つの別個の反応列を準備するステップと、
(b)各反応列内に供給される冷却剤を含む単一の共通リザーバを含む共通冷却剤循環システムを準備するステップと、
(c)各反応列への冷却剤の循環を開始するステップと、
(d)反応器の圧力を所望の反応圧に上昇させるステップと、
(e)各反応列内に反応物質供給ストリームを供給するステップと、
(f)単一の共通リザーバの温度を上昇させる一方で、各反応列を通る反応物質供給ストリームのGHSVを、所望の範囲の発熱反応が得られるように調整するステップと、
を含む方法を提供する。
(a)それぞれが少なくとも1つの反応器を含む複数の反応列を準備するステップと、
(b)発熱反応を開始すべき開始反応器を含む反応列を除く各反応列内に供給される第1の冷却剤を含む単一の共通リザーバを含む共通冷却剤循環システムを準備するステップと、
(c)開始反応器を含む反応列内に供給される第2の冷却剤を含む第2の冷却剤リザーバに関連する第2の冷却剤循環システムを準備するステップと、
(d)開始反応器内の圧力を所望の反応圧に上昇させるステップと、
(e)開始反応器を含む反応列内に反応物質供給ストリームを供給するステップと、
(f)開始反応器の動作条件が、開始反応器から排出された冷却剤を共通冷却剤循環システムに再導入できるようになるまでプロセスを実行するステップと、
(g)開始反応器を含む反応列への第1の冷却剤の供給を開始すると同時に、開始反応器を含む反応列への第2の冷却剤の供給を停止するステップと、
(h)開始反応器を含む反応列から単一の共通リザーバに第1の冷却剤を向け直すステップと、
を含む方法を提供する。
別の実施形態では、本明細書で説明する単一の共通冷却剤リザーバ方法を、各反応列における冷却剤温度を個別に制御することが可能なシステム内に提供することができる。この実施形態は、規模の経済性を活用して生産単価を最小化するために反応列の生産能力を最大化する能力などの、上述した単一の共通冷却剤リザーバ方法に関連する利点を全てもたらす。しかしながら、この実施形態は、これに加えて反応列内の個々の反応器の個別温度制御も可能にする。
(a)反応物質供給ストリームを少なくとも2つの別個の反応物質サブストリームに分割するステップと、
(b)反応器を含む別個の反応列内に各反応物質サブストリームを供給するステップと、
(c)共通冷却剤リザーバから各反応器内に冷却剤ストリームを供給するステップと、
(d)反応器内で発熱反応を実行して、反応生成物と、熱が伝わった冷却剤とを生成するステップと、
(e)各反応列からの熱が伝わった冷却剤を、単一の共通リザーバに供給して冷却剤から熱を除去するステップと、
(f)ステップ(e)において熱が除去された冷却剤をステップ(c)に戻すステップと、
を含み、
冷却剤は、発熱反応温度よりも低い沸点を有する流体であり、
ステップ(d)及び(e)における熱が伝わった冷却剤は二相冷却剤であり、
各反応器における発熱反応の進行が、二相冷却剤の圧力を調整することによって制御される、
方法を提供する。
合成ガス(CO及びH2)を含む反応物質供給ストリーム(1)を5つの別個の反応物質サブストリームに分割して、5つの別個の反応列(3a、3b、3c、3d、3e)に供給されるようにする。各反応列は、並列に配置された5つの反応器を含み、これらの各々は、約40重量パーセントのコバルトを含むフィッシャートロプシュ触媒を含む。反応物質供給ストリーム内のH2に対するCOの比率は、0.5である。水を含む冷却剤循環システムを準備する。単一の共通蒸気ドラム(15)から別個の各反応列の反応器の冷却剤側に水が供給されるように循環を開始する。水は、部分的に蒸発して水と蒸気との混合物になった後に、再循環されて単一の共通蒸気ドラム(15)に戻る。単一の共通蒸気ドラムの温度及び圧力が200℃の温度及び14.5バール(g)の圧力に上昇し、この時点で5つの反応物質サブストリームを15,000hr-1の流量でそれぞれの反応列に供給して、各反応器でフィッシャートロプシュ反応が開始されるようにする。各反応器では、合成ガスが反応して炭化水素生成物と水を生成する。反応によって生じる熱は、循環水を部分的に蒸発させ、反応器から排出された冷却剤は、水と蒸気の混合物を含むようになる。水と蒸気は、単一の共通蒸気ドラムに移動すると分離される。上述したように、蒸気は除去され、水は反応列に再循環される。再循環水にさらなる水(9)を加えて、蒸気の除去を補償する。このプロセスは、70%のCO2変換で動作する。各反応器内の触媒の活性は時間と共に低下するので、このプロセスを可能にして同レベルの変換を維持するために、各反応列への反応物質供給ストリームのGHSVを低減する必要がある。
合成ガス(CO及びH2)を含む反応物質供給ストリーム(1)を5つの別個の反応物質サブストリームに分割して、5つの別個の反応列(3a、3b、3c、3d、3e)(図3を参照)に供給されるようにする。各反応列は、それぞれがCu/ZnO/Al2O3触媒の固定床を含む1つのマイクロチャネルリアクタを含む。反応物質供給ストリームは、5mol%のCO2、26mol%のCO、64mol%のH2及び5mol%のN2を含んでいた。この反応物質供給ストリームを、250℃及び50バール(g)で1,500hr-1で反応器に供給する。水を含む冷却剤循環システムを準備する。単一の共通蒸気ドラム(15)から別個の各反応列の反応器の冷却剤側に水が供給されるように循環を開始する。反応器では、水が部分的に蒸発して水と蒸気の混合物になった後に、再循環されて単一の共通蒸気ドラム(15)に戻される。単一の共通蒸気ドラムの温度及び圧力は、250℃の温度及び39バール(g)の圧力に上昇する。各反応物質サブストリームの流量は、反応列に入ると、自動流量調整弁(4a、4b、4c、4d、4e)を用いて、この反応列に存在する反応器内の触媒の不活性化などの因子を考慮するように個別に調整することができる。
合成ガス(CO及びH2)を含む反応物質供給ストリーム(1)を4つの別個の反応物質サブストリームに分割して、4つの別個の反応列(3b、3c、3d、3e)(図1を参照)に供給されるようにする。各反応列は、約40重量パーセントのコバルトを含むフィッシャートロプシュ触媒の固定床を含む1つの反応器を含む。通常、反応物質供給ストリーム内のH2に対するCOの比率は、0.5〜0.6である。水を含む冷却剤循環システムを準備する。循環中には、単一の共通蒸気ドラム(15)から別個の各反応列の反応器の冷却剤側に水が供給される。水は、部分的に蒸発して水と蒸気との混合物になった後に、再循環されて単一の共通蒸気ドラム(15)に戻る。単一の共通蒸気ドラムは、約205℃の温度で動作し、反応器出口における冷却剤温度は、205℃〜214℃とすることができる。単一の共通蒸気ドラムは、反応器冷却剤出口において19.7バール(g)(300psi又は2068kPa)の最大圧力をもたらす。各反応器において同様のCO変換のフィッシャートロプシュ反応が起きるように、4つの反応物質サブストリームを12,000hr-1〜15,000hr-1の流量でそれぞれの反応列に供給する。各反応器では、合成ガスが反応して炭化水素生成物と水を生成する。反応によって生じる熱は、循環水を部分的に蒸発させ、反応器から排出された冷却剤は、水と蒸気の混合物を含むようになる。水と蒸気は、単一の共通蒸気ドラムに移動すると分離される。上述したように、蒸気は除去され、水は反応列に再循環される。再循環水にさらなる水(9)を加えて、蒸気の除去を補償する。水は、蒸気ドラムと反応器との間で任意に加熱することができる。プロセスは、通常は68%〜72%の狭い範囲のCO変換で行われる。
Claims (15)
- 発熱反応からの熱を除去する方法であって、
(a)反応物質供給ストリームを少なくとも2つの別個の反応物質サブストリームに分割するステップと、
(b)反応器を含む別個の反応列内に各反応物質サブストリームを供給するステップと、
(c)共通冷却剤リザーバから各反応器内に冷却剤ストリームを供給するステップと、 (d)前記各反応器において発熱反応を実行して、反応生成物を生成し、前記発熱反応からの熱が前記冷却剤に伝えられるステップと、
(e)前記各反応列からの熱が伝わった冷却剤を単一の共通リザーバに供給して前記冷却剤から前記熱を除去するステップと、
(f)ステップ(e)において熱が除去された前記冷却剤をステップ(c)に戻すステップと、
を含み、
前記冷却剤は、前記発熱反応温度よりも低い沸点を有する流体であり、
ステップ(d)及び(e)における熱が伝わった前記冷却剤は、二相冷却剤であり、
各反応器における前記発熱反応の進行は、前記二相冷却剤の圧力を調整することによって制御される、
ことを特徴とする方法。 - 前記反応器は、マイクロチャネルリアクタである、請求項1に記載の方法。
- 前記冷却剤は、水を含む、請求項1に記載の方法。
- 前記共通冷却剤リザーバは、蒸気ドラムである、請求項3に記載の方法。
- 前記蒸気ドラムは、100〜300℃の温度、及び100〜3400kPaの圧力で動作する、請求項4に記載の方法。
- 前記発熱反応は、フィッシャートロプシュプロセスであり、前記蒸気ドラムは、200〜225℃の温度及び1200〜2600kPaの圧力で動作する、請求項4に記載の方法。
- 前記発熱反応は、フィッシャートロプシュプロセスであり、前記蒸気ドラムは、200〜220℃の温度及び1700〜1900kPaの圧力で動作する、請求項4に記載の方法。
- 前記発熱反応は、フィッシャートロプシュ反応、メタノール生成及びエチレンオキシド生成から成る群から選択される、請求項1に記載の方法。
- 前記反応器は、固体触媒を含む、請求項1に記載の方法。
- 前記発熱反応はフィッシャートロプシュ反応であり、前記反応物質供給ストリームはシンガスを含み、前記反応生成物は炭化水素生成物である、請求項1に記載の方法。
- 前記二相冷却剤の圧力は、弁を使用して制御される、請求項1に記載の方法。
- 前記冷却剤が前記単一の共通蒸気ドラムに移動すると、前記水及び前記蒸気は分離され、前記蒸気は除去され、前記水は前記反応列に再循環され、前記再循環された水にさらなる水を加えて前記蒸気の除去を補償する、請求項4に記載の方法。
- 前記各反応器における前記発熱反応の進行を、前記反応器が一部を成す前記反応列を通る前記反応物質サブストリームの流量を調整することによって制御するステップを含む、請求項1に記載の方法。
- 前記各反応器における前記発熱反応の進行を、各反応列内に供給される前記反応物質サブストリームの組成を調整することによって制御するステップを含む、請求項1に記載の方法。
- 前記二相冷却剤の圧力調整は、前記二相冷却剤が依然として前記反応列内に存在する間に、前記反応器の下流で行われる、請求項1に記載の方法。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201462066233P | 2014-10-20 | 2014-10-20 | |
| US62/066,233 | 2014-10-20 | ||
| PCT/GB2015/053131 WO2016063043A2 (en) | 2014-10-20 | 2015-10-20 | Process of removing heat |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2017537773A JP2017537773A (ja) | 2017-12-21 |
| JP2017537773A5 JP2017537773A5 (ja) | 2018-02-08 |
| JP6357586B2 true JP6357586B2 (ja) | 2018-07-11 |
Family
ID=54545382
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2017522533A Active JP6357586B2 (ja) | 2014-10-20 | 2015-10-20 | 熱除去プロセス |
Country Status (9)
| Country | Link |
|---|---|
| US (4) | US9822049B2 (ja) |
| JP (1) | JP6357586B2 (ja) |
| KR (1) | KR102425630B1 (ja) |
| CN (2) | CN106999899A (ja) |
| AU (1) | AU2015334662B2 (ja) |
| CA (1) | CA2964526C (ja) |
| DE (1) | DE112015004756T5 (ja) |
| GB (1) | GB2550677B (ja) |
| WO (1) | WO2016063043A2 (ja) |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN106478383B (zh) * | 2015-08-28 | 2019-07-09 | 中国石油化工股份有限公司 | 甲醇制二甲醚的方法和反应设备及甲醇制烯烃的方法和系统 |
| FR3050123B1 (fr) * | 2016-04-15 | 2021-01-22 | Engie | Dispositif et procede d'hydrogenation du co2 pour produire du methanol et dispositif et procede de cogeneration de methanol et de methane de synthese |
| US10258953B2 (en) * | 2016-08-05 | 2019-04-16 | Covestro Llc | Systems and processes for producing polyether polyols |
| CN110787755B (zh) * | 2019-11-08 | 2020-10-20 | 李德祥 | 一种反应釜汽包与相关管道的布置方法 |
| CN113813989B (zh) * | 2020-06-18 | 2023-05-26 | 中国石油天然气股份有限公司 | 丙烯醛的制备方法 |
| WO2023012121A1 (en) | 2021-08-02 | 2023-02-09 | Velocys Technologies Limited | Process for operating a plant facility during catalyst regeneration |
| GB2609508B (en) * | 2021-08-02 | 2023-10-18 | Velocys Tech Ltd | Process |
Family Cites Families (36)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE1252662B (ja) | 1965-06-25 | |||
| CH534005A (de) | 1968-02-01 | 1973-02-28 | Bayer Ag | Verfahren zur Herstellung eines Palladium und Gold enthaltenden Katalysators |
| IL84232A (en) | 1986-10-31 | 1992-06-21 | Shell Int Research | Catalyst and process for the catalytic production of ethylene oxide |
| US5557014A (en) | 1990-03-05 | 1996-09-17 | Catalytica, Inc. | Catalytic system for olefin oxidation to carbonyl products |
| US5418202A (en) | 1993-12-30 | 1995-05-23 | Shell Oil Company | Ethylene oxide catalyst and process |
| DE19523271A1 (de) | 1995-06-27 | 1997-01-02 | Hoechst Ag | Katalysator und Verfahren zur Herstellung von Vinylacetat |
| US5705661A (en) | 1995-09-25 | 1998-01-06 | Mitsubishi Chemical Corporation | Catalyst for production of ethylene oxide |
| US6022823A (en) | 1995-11-07 | 2000-02-08 | Millennium Petrochemicals, Inc. | Process for the production of supported palladium-gold catalysts |
| DE19651287A1 (de) | 1996-12-10 | 1998-06-18 | Wacker Chemie Gmbh | Ionische Organosiliciumverbindungen, deren Herstellung und Verwendung |
| US6072078A (en) | 1997-12-12 | 2000-06-06 | Celanese International Corporation | Vinyl acetate production using a catalyst comprising palladium, gold, copper and any of certain fourth metals |
| GB9810928D0 (en) | 1998-05-22 | 1998-07-22 | Bp Chem Int Ltd | Catalyst and process |
| US6106695A (en) * | 1999-03-22 | 2000-08-22 | Uop Llc | Catalytic hydrocracking process |
| US6312586B1 (en) | 1999-09-27 | 2001-11-06 | Uop Llc | Multireactor parallel flow hydrocracking process |
| US6512017B1 (en) * | 1999-10-14 | 2003-01-28 | Sasol Technology (Proprietary) Limited | Handling of a catalyst |
| WO2001083105A1 (en) | 2000-05-01 | 2001-11-08 | Scientific Design Company, Inc. | Ethylene oxide catalyst |
| US7012103B2 (en) * | 2003-03-24 | 2006-03-14 | Conocophillips Company | Commercial fischer-tropsch reactor |
| US7294734B2 (en) | 2003-05-02 | 2007-11-13 | Velocys, Inc. | Process for converting a hydrocarbon to an oxygenate or a nitrile |
| US20050175519A1 (en) * | 2004-02-06 | 2005-08-11 | Rogers William A.Jr. | Microchannel compression reactor |
| US8747805B2 (en) | 2004-02-11 | 2014-06-10 | Velocys, Inc. | Process for conducting an equilibrium limited chemical reaction using microchannel technology |
| JP5731092B2 (ja) | 2004-08-12 | 2015-06-10 | ヴェロシス,インク. | マイクロチャネルプロセス技術を用いてエチレンをエチレンオキシドに変換するためのマイクロチャネル反応器及びそれを用いた方法 |
| US7468455B2 (en) | 2004-11-03 | 2008-12-23 | Velocys, Inc. | Process and apparatus for improved methods for making vinyl acetate monomer (VAM) |
| US8383872B2 (en) * | 2004-11-16 | 2013-02-26 | Velocys, Inc. | Multiphase reaction process using microchannel technology |
| DE102007024934B4 (de) * | 2007-05-29 | 2010-04-29 | Man Dwe Gmbh | Rohrbündelreaktoren mit Druckflüssigkeitskühlung |
| CN100523132C (zh) * | 2007-06-13 | 2009-08-05 | 中国石油天然气集团公司 | 一种利用固定床装置进行费托合成的方法 |
| JP5159245B2 (ja) * | 2007-10-24 | 2013-03-06 | 月島機械株式会社 | パラジクロロベンゼンの製造方法 |
| CN101480592B (zh) * | 2008-01-12 | 2012-01-11 | 杭州林达化工技术工程有限公司 | 一种固定床组合反应设备 |
| US7745667B2 (en) | 2008-04-07 | 2010-06-29 | Velocys | Microchannel apparatus comprising structured walls, chemical processes, methods of making formaldehyde |
| US8137476B2 (en) * | 2009-04-06 | 2012-03-20 | Synfuels International, Inc. | Secondary reaction quench device and method of use |
| JP5802397B2 (ja) * | 2011-01-31 | 2015-10-28 | 独立行政法人石油天然ガス・金属鉱物資源機構 | 温度制御システム |
| JP5793325B2 (ja) * | 2011-03-30 | 2015-10-14 | 独立行政法人石油天然ガス・金属鉱物資源機構 | 温度制御システム、炭化水素合成反応装置、炭化水素合成反応システム |
| EP2723485B1 (en) * | 2011-06-23 | 2020-06-03 | Dow Technology Investments LLC | System and method for producing oxidized olefins |
| CA2842176C (en) | 2011-07-19 | 2019-10-15 | Velocys, Inc. | Microchannel reactors and fabrication processes |
| US8889747B2 (en) | 2011-10-11 | 2014-11-18 | Bp Corporation North America Inc. | Fischer Tropsch reactor with integrated organic rankine cycle |
| GB201214122D0 (en) | 2012-08-07 | 2012-09-19 | Oxford Catalysts Ltd | Treating of catalyst support |
| KR20140080661A (ko) * | 2012-12-13 | 2014-07-01 | 재단법인 포항산업과학연구원 | 고발열량의 합성천연가스 제조장치 및 그 제조방법 |
| MY190621A (en) * | 2014-03-17 | 2022-04-27 | Shell Int Research | A method for start-up and operation of a fischer-tropsch reactor |
-
2015
- 2015-10-20 KR KR1020177013399A patent/KR102425630B1/ko active IP Right Grant
- 2015-10-20 CA CA2964526A patent/CA2964526C/en active Active
- 2015-10-20 CN CN201580066675.9A patent/CN106999899A/zh active Pending
- 2015-10-20 AU AU2015334662A patent/AU2015334662B2/en not_active Ceased
- 2015-10-20 US US14/918,314 patent/US9822049B2/en active Active
- 2015-10-20 WO PCT/GB2015/053131 patent/WO2016063043A2/en active Application Filing
- 2015-10-20 CN CN202110562305.1A patent/CN113289561A/zh active Pending
- 2015-10-20 DE DE112015004756.5T patent/DE112015004756T5/de active Pending
- 2015-10-20 GB GB1706468.4A patent/GB2550677B/en active Active
- 2015-10-20 JP JP2017522533A patent/JP6357586B2/ja active Active
-
2017
- 2017-11-20 US US15/817,308 patent/US10065911B2/en active Active
-
2018
- 2018-09-04 US US16/121,106 patent/US10851035B2/en active Active
-
2020
- 2020-11-17 US US17/099,804 patent/US11247953B2/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| GB201706468D0 (en) | 2017-06-07 |
| GB2550677A (en) | 2017-11-29 |
| CA2964526C (en) | 2024-02-13 |
| CA2964526A1 (en) | 2016-04-28 |
| WO2016063043A2 (en) | 2016-04-28 |
| DE112015004756T5 (de) | 2017-09-28 |
| US11247953B2 (en) | 2022-02-15 |
| GB2550677B (en) | 2018-07-25 |
| KR20170078702A (ko) | 2017-07-07 |
| JP2017537773A (ja) | 2017-12-21 |
| AU2015334662B2 (en) | 2018-06-07 |
| CN113289561A (zh) | 2021-08-24 |
| US20160107962A1 (en) | 2016-04-21 |
| US20210070681A1 (en) | 2021-03-11 |
| US20190016654A1 (en) | 2019-01-17 |
| KR102425630B1 (ko) | 2022-07-27 |
| US20180072643A1 (en) | 2018-03-15 |
| US9822049B2 (en) | 2017-11-21 |
| CN106999899A (zh) | 2017-08-01 |
| WO2016063043A3 (en) | 2017-03-02 |
| US10851035B2 (en) | 2020-12-01 |
| US10065911B2 (en) | 2018-09-04 |
| AU2015334662A1 (en) | 2017-05-04 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6357586B2 (ja) | 熱除去プロセス | |
| JP5051924B2 (ja) | メタン転換製造プロセス | |
| US10138182B2 (en) | Oxygen transfer agents for the oxidative dehydrogenation of hydrocarbons and systems and processes using the same | |
| RU2478426C2 (ru) | Катализатор и способ конвертации природного газа в высокоуглеродистые соединения | |
| JP5305486B2 (ja) | メタンからの芳香族炭化水素の製造 | |
| US8052938B2 (en) | Aromatics co-production in a methanol-to-propylene unit | |
| JP5235414B2 (ja) | 多帯域を有し、再生または新鮮な触媒の各帯域への追加を伴う移動床反応装置 | |
| US7807114B2 (en) | Reactor system with interstage product removal | |
| US8796496B2 (en) | Process for preparing benzene from methane | |
| US10087124B2 (en) | Production of aromatic hydrocarbons | |
| Nederlof et al. | Application of staged O2 feeding in the oxidative dehydrogenation of ethylbenzene to styrene over Al2O3 and P2O5/SiO2 catalysts | |
| JP2016540632A (ja) | 反応管内で流動層を形成しながら吸熱反応を実施する方法及び装置 | |
| Regel-Rosocka | 3 Technology of simple hydrocarbon intermediates |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20171116 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20171116 |
|
| A871 | Explanation of circumstances concerning accelerated examination |
Free format text: JAPANESE INTERMEDIATE CODE: A871 Effective date: 20171116 |
|
| A975 | Report on accelerated examination |
Free format text: JAPANESE INTERMEDIATE CODE: A971005 Effective date: 20171128 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20180129 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20180427 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20180528 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20180618 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 6357586 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |