JP6232073B2 - 生体由来カルボン酸エステルからのバイオ燃料の製造 - Google Patents
生体由来カルボン酸エステルからのバイオ燃料の製造 Download PDFInfo
- Publication number
- JP6232073B2 JP6232073B2 JP2015549446A JP2015549446A JP6232073B2 JP 6232073 B2 JP6232073 B2 JP 6232073B2 JP 2015549446 A JP2015549446 A JP 2015549446A JP 2015549446 A JP2015549446 A JP 2015549446A JP 6232073 B2 JP6232073 B2 JP 6232073B2
- Authority
- JP
- Japan
- Prior art keywords
- acid
- ester
- reaction
- carboxylic acid
- fermentation
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images

Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C67/00—Preparation of carboxylic acid esters
- C07C67/08—Preparation of carboxylic acid esters by reacting carboxylic acids or symmetrical anhydrides with the hydroxy or O-metal group of organic compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C29/00—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring
- C07C29/132—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring by reduction of an oxygen containing functional group
- C07C29/136—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring by reduction of an oxygen containing functional group of >C=O containing groups, e.g. —COOH
- C07C29/147—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring by reduction of an oxygen containing functional group of >C=O containing groups, e.g. —COOH of carboxylic acids or derivatives thereof
- C07C29/149—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring by reduction of an oxygen containing functional group of >C=O containing groups, e.g. —COOH of carboxylic acids or derivatives thereof with hydrogen or hydrogen-containing gases
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C67/00—Preparation of carboxylic acid esters
- C07C67/28—Preparation of carboxylic acid esters by modifying the hydroxylic moiety of the ester, such modification not being an introduction of an ester group
- C07C67/297—Preparation of carboxylic acid esters by modifying the hydroxylic moiety of the ester, such modification not being an introduction of an ester group by splitting-off hydrogen or functional groups; by hydrogenolysis of functional groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/02—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings
- C07D307/04—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
- C07D307/06—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having no double bonds between ring members or between ring members and non-ring members with only hydrogen atoms or radicals containing only hydrogen and carbon atoms, directly attached to ring carbon atoms
- C07D307/08—Preparation of tetrahydrofuran
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G3/00—Production of liquid hydrocarbon mixtures from oxygen-containing organic materials, e.g. fatty oils, fatty acids
- C10G3/50—Production of liquid hydrocarbon mixtures from oxygen-containing organic materials, e.g. fatty oils, fatty acids in the presence of hydrogen, hydrogen donors or hydrogen generating compounds
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10L—FUELS NOT OTHERWISE PROVIDED FOR; NATURAL GAS; SYNTHETIC NATURAL GAS OBTAINED BY PROCESSES NOT COVERED BY SUBCLASSES C10G OR C10K; LIQUIFIED PETROLEUM GAS; USE OF ADDITIVES TO FUELS OR FIRES; FIRE-LIGHTERS
- C10L1/00—Liquid carbonaceous fuels
- C10L1/02—Liquid carbonaceous fuels essentially based on components consisting of carbon, hydrogen, and oxygen only
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10L—FUELS NOT OTHERWISE PROVIDED FOR; NATURAL GAS; SYNTHETIC NATURAL GAS OBTAINED BY PROCESSES NOT COVERED BY SUBCLASSES C10G OR C10K; LIQUIFIED PETROLEUM GAS; USE OF ADDITIVES TO FUELS OR FIRES; FIRE-LIGHTERS
- C10L1/00—Liquid carbonaceous fuels
- C10L1/02—Liquid carbonaceous fuels essentially based on components consisting of carbon, hydrogen, and oxygen only
- C10L1/023—Liquid carbonaceous fuels essentially based on components consisting of carbon, hydrogen, and oxygen only for spark ignition
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10L—FUELS NOT OTHERWISE PROVIDED FOR; NATURAL GAS; SYNTHETIC NATURAL GAS OBTAINED BY PROCESSES NOT COVERED BY SUBCLASSES C10G OR C10K; LIQUIFIED PETROLEUM GAS; USE OF ADDITIVES TO FUELS OR FIRES; FIRE-LIGHTERS
- C10L1/00—Liquid carbonaceous fuels
- C10L1/04—Liquid carbonaceous fuels essentially based on blends of hydrocarbons
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10L—FUELS NOT OTHERWISE PROVIDED FOR; NATURAL GAS; SYNTHETIC NATURAL GAS OBTAINED BY PROCESSES NOT COVERED BY SUBCLASSES C10G OR C10K; LIQUIFIED PETROLEUM GAS; USE OF ADDITIVES TO FUELS OR FIRES; FIRE-LIGHTERS
- C10L1/00—Liquid carbonaceous fuels
- C10L1/04—Liquid carbonaceous fuels essentially based on blends of hydrocarbons
- C10L1/06—Liquid carbonaceous fuels essentially based on blends of hydrocarbons for spark ignition
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C29/00—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring
- C07C29/132—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring by reduction of an oxygen containing functional group
- C07C29/136—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring by reduction of an oxygen containing functional group of >C=O containing groups, e.g. —COOH
- C07C29/147—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring by reduction of an oxygen containing functional group of >C=O containing groups, e.g. —COOH of carboxylic acids or derivatives thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/02—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings
- C07D307/26—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member
- C07D307/30—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D307/32—Oxygen atoms
- C07D307/33—Oxygen atoms in position 2, the oxygen atom being in its keto or unsubstituted enol form
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10L—FUELS NOT OTHERWISE PROVIDED FOR; NATURAL GAS; SYNTHETIC NATURAL GAS OBTAINED BY PROCESSES NOT COVERED BY SUBCLASSES C10G OR C10K; LIQUIFIED PETROLEUM GAS; USE OF ADDITIVES TO FUELS OR FIRES; FIRE-LIGHTERS
- C10L2200/00—Components of fuel compositions
- C10L2200/04—Organic compounds
- C10L2200/0461—Fractions defined by their origin
- C10L2200/0469—Renewables or materials of biological origin
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10L—FUELS NOT OTHERWISE PROVIDED FOR; NATURAL GAS; SYNTHETIC NATURAL GAS OBTAINED BY PROCESSES NOT COVERED BY SUBCLASSES C10G OR C10K; LIQUIFIED PETROLEUM GAS; USE OF ADDITIVES TO FUELS OR FIRES; FIRE-LIGHTERS
- C10L2290/00—Fuel preparation or upgrading, processes or apparatus therefore, comprising specific process steps or apparatus units
- C10L2290/26—Composting, fermenting or anaerobic digestion fuel components or materials from which fuels are prepared
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P7/00—Preparation of oxygen-containing organic compounds
- C12P7/40—Preparation of oxygen-containing organic compounds containing a carboxyl group including Peroxycarboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P7/00—Preparation of oxygen-containing organic compounds
- C12P7/40—Preparation of oxygen-containing organic compounds containing a carboxyl group including Peroxycarboxylic acids
- C12P7/44—Polycarboxylic acids
- C12P7/46—Dicarboxylic acids having four or less carbon atoms, e.g. fumaric acid, maleic acid
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P7/00—Preparation of oxygen-containing organic compounds
- C12P7/40—Preparation of oxygen-containing organic compounds containing a carboxyl group including Peroxycarboxylic acids
- C12P7/44—Polycarboxylic acids
- C12P7/48—Tricarboxylic acids, e.g. citric acid
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E50/00—Technologies for the production of fuel of non-fossil origin
- Y02E50/10—Biofuels, e.g. bio-diesel
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/54—Improvements relating to the production of bulk chemicals using solvents, e.g. supercritical solvents or ionic liquids
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P30/00—Technologies relating to oil refining and petrochemical industry
- Y02P30/20—Technologies relating to oil refining and petrochemical industry using bio-feedstock
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Wood Science & Technology (AREA)
- Zoology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Health & Medical Sciences (AREA)
- Biotechnology (AREA)
- Microbiology (AREA)
- Biochemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Liquid Carbonaceous Fuels (AREA)
- Emergency Medicine (AREA)
Description
本特許出願は、2012年12月20日に出願された米国仮特許出願第61/739,790号に基づく優先権を主張するものであり、当該明細書の内容を本明細書の一部を構成するものとしてここに援用する。
本発明は、特定のバイオ燃料を製造するためのプロセスに関する。特に本発明は、水素化または水素化分解のいずれかにより生体由来のカルボン酸のエステルからバイオ燃料製品を製造するための方法に関する。
環境問題に対する認識の高まりと共に、石油をはじめとした化石由来資源の供給の不安定化に伴う経済的圧力や燃料コストの増大に起因して、代替燃料の開発が重要視されるようになっている。政策担当者らもその重要性を認識しており、バイオ燃料の生産および使用を奨励するようになった。石油化学または天然ガス由来の炭化水素資源依存からの脱却に関心が集まるようになり、再生可能かつ持続可能な「環境負荷の少ない(green process)」炭素資源の探索に注力する者もいる。初期のいわゆる第1世代バイオ燃料の製造は、食料生産物(例えば、デンプンおよび糖類を含むトウモロコシ、コムギ、サトウキビ等の植物)を発酵させることにより短鎖アルコール(例えば、エタノール、ブタノール)を得ることによるかまたは植物油(例えば、菜種、大豆)をグリセロール分解に付し、遊離した脂肪酸をエステル交換することによって、バイオディーゼルとして使用される脂肪酸メチルエステルを得ることにより行われていた。(例えば、Cesar B. Grandaら、“Sustainable Liquid Biofuels and Their Environmental Impact,” ENVIRONMENTAL PROGRESS, Vol. 26, No. 3, pp. 233- 250 (Oct. 2007)参照。当該文献を本明細書の一部を構成するものとしてここに援用する。)
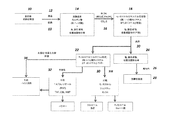
本発明は、バイオ燃料を製造するためのプロセスに関する。このプロセスは:a)少なくとも1種の遊離カルボン酸またはカルボン酸の混合物または少なくとも1種のカルボン酸のカルボン酸およびこれに付随する(associated)アルカリ金属塩もしくはアルカリ土類金属塩の混合物を含む発酵培養液を得る工程と;b)遊離カルボン酸を含む上記発酵培養液を粉末に乾燥させる工程と;c)対応する1種または複数種のエステルを合成するために、上記粉末中の上記カルボン酸と、アルコール溶媒または少なくとも1種のアルコールの混合物とを、CO2雰囲気中、他の酸触媒の実質的な不在下に、このアルコールまたはCO2の超臨界、臨界または臨界近傍条件に相当する反応温度および圧力下で反応させる工程と;d)1種以上のバイオ燃料を生成するために、生成したエステルを水素化分解または水素化のいずれかに供する工程と;e)1種以上のバイオ燃料を回収する工程とを含む。このエステルは、モノエステル、ジエステルまたはトリエステルとすることができる。好ましくは、エステルはジエステルまたはトリエステルである。モノエステルはモノアルコールに、ジエステルはジオールに、トリエステルはトリオールに変換することができる。バイオ燃料製品はアルカンまたはアルコールのいずれかとすることができる。
第I項−説明
A.
本開示の一部においては、糖類や他の植物系炭水化物等の生物体由来の炭素資源から水素化または水素化分解のいずれかを行うことによって様々なバイオ燃料製品を製造するためのプロセスについて記載する。このプロセスは、カルボン酸を発酵培養液から回収する機能と、バイオ燃料を生成する反応の原料として酸を使用する機能とを効率的な流れで結びつけたものである。本プロセスは、カルボン酸をその対応するエステル(例えば、モノエステル、ジエステルまたはトリエステル)に、比較的効率的かつ費用対効果の高い形で変換する方法を含む。
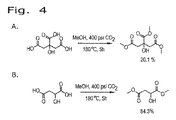
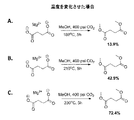
従来、エステルは、カルボン酸を酸触媒の存在下にアルコールと一緒に加熱することにより生成する。エステルが酸およびアルコールから生成する機構は、酸を触媒としてエステルが加水分解する工程の逆である。この反応は、用いた条件に応じてどちらの方向にも進行し得る。通常のエステル化プロセスにおいては、触媒として強酸が用いられない限り、カルボン酸はアルコールと反応しない。通常、触媒は濃硫酸または塩化水素である。プロトン化によりカルボニル基の求電子性が強くなり、弱い求核試薬であるアルコールと反応することが可能になる。
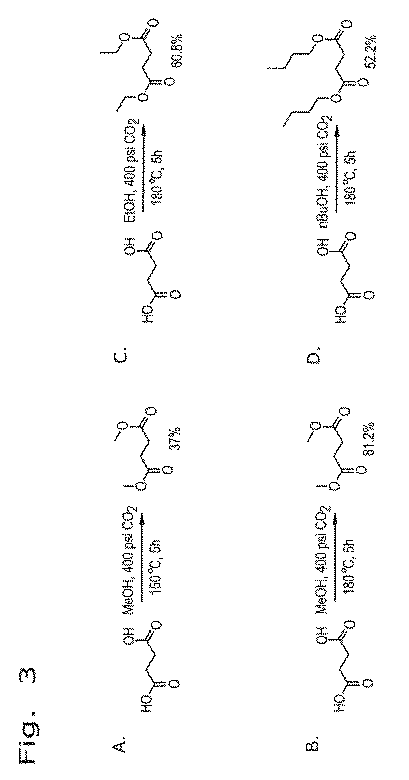
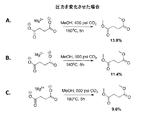
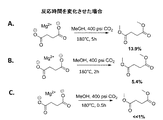
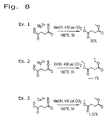
上述のエステル化プロセスは、発酵をベースとする炭素鎖原料の製造に統合することができ、これは、再生可能資源由来のカルボン酸からエステルを生成するより好都合な方法を提供するものである。本発明は、生物体由来のカルボン酸を、発酵培養液から簡素かつ費用対効果の高いプロセスで回収し、エステルに変換し、次いで水素化または水素化分解に供することによりバイオ燃料を製造することができる、直接的な経路を提供する。添付の図面に、エステルをさらなる下流プロセスに統合する方法を例示する。
A.
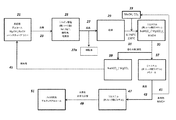
遊離カルボン酸を発酵培養液から単離するためのプロセスに、本エステル化方法に従い調製を行う実施例を統合する。この方法は、概して、次の工程:a)発酵培養液から細胞塊および他の生物片を除去するために、未精製の発酵培養液を濾過する工程と;b)発酵培養液を乾燥させる工程と;c)モノエステルおよびジエステルならびに炭酸塩(NaHCO3/MgCO3)の混合物を生成するために、乾燥した発酵培養液を過剰のメタノール(CH3OH)またはエタノール(C2H5OH)および二酸化炭素(CO2)と、約150℃〜アルコールおよび/またはCO2試薬の臨界近傍または臨界温度までの温度および臨界近傍または臨界圧力で反応させる工程と;d)副生成物を除去するために反応生成物を濾過する工程と;e)エステルを蒸留することにより精製する工程とを含む。
実施例:水素化
上に引用した参考文献に記載されているもの等のプロセスを用いて、上述のエステル化プロセスから回収したカルボン酸エステルを直接水素化することができる。例えば、コハク酸ジアルキルエステルをBDO、GBLおよびTHFに水素化するために金属銅触媒を使用することができる。次に水素化プロセスの例を記載する。
上述した発酵抽出により得られるエステルは、次いで触媒(例えば、還元CuO/ZnO)上で水素化分解される。これにより高い転化率(>98%)および選択性が達成されるはずである(例えば、国際公開第82/03854号)。
Claims (21)
- a)少なくとも1種の遊離カルボン酸を含み、且つ、pHが5未満である発酵培養液を得る工程と;b)前記遊離カルボン酸を含む前記発酵培養液を粉末に乾燥させる工程と;c)前記粉末中の前記カルボン酸を、アルコール溶媒と、CO2雰囲気中、他の酸触媒の不在下に、前記アルコールまたはCO2の少なくとも一方の超臨界、臨界または臨界近傍条件にある反応温度もしくは反応圧力のいずれかまたは両方で反応させることにより、エステルを合成する工程と;d)バイオ燃料を生成するために、前記エステルを水素化分解または水素化のいずれかに供する工程を含む、バイオ燃料を製造するためのプロセス。
- e)前記バイオ燃料を回収する工程をさらに含む、請求項1に記載のプロセス。
- 前記バイオ燃料がアルカンまたはアルコールである、請求項1に記載のプロセス。
- 前記バイオ燃料が、エタン、エタノール、プロパン、プロパノール、ブタン、1−ブタノール、オクタンおよびオクタノールの少なくとも1種を含む、請求項3に記載のプロセス。
- 前記発酵培養液が細胞塊および不溶な化合物を含み、前記発酵培養液から前記細胞塊および不溶な化合物を乾燥工程前またはエステル合成後のいずれかにおいて濾過する工程をさらに含む、請求項1に記載のプロセス。
- 水素化分解または水素化のいずれかの前に前記エステルを濃縮する工程をさらに含む、請求項5に記載のプロセス。
- 水素化分解または水素化のいずれかの前に濾過を行うことによって前記不溶な化合物を分離する工程をさらに含む、請求項5に記載のプロセス。
- 前記発酵培養液が連続発酵プロセスの一部であり、前記不溶な化合物を前記発酵培養液に戻して再利用する工程をさらに含む、請求項5に記載のプロセス。
- 前記発酵培養液がバッチ式発酵プロセスの一部であり、前記不溶な化合物を第2の発酵反応器で再利用する工程をさらに含む、請求項5に記載のプロセス。
- 前記アルコール溶媒がC1〜C20のR基を有し、少なくとも1種の飽和、不飽和、環状または芳香族種である、請求項1に記載のプロセス。
- 前記カルボン酸が、ギ酸、酢酸、プロピオン酸、乳酸、酪酸、イソ酪酸、吉草酸、ヘキサン酸、ヘプタン酸、デカン酸、ラウリン酸、ミリスチン酸、およびC15〜C18脂肪酸、フマル酸、イタコン酸、リンゴ酸、コハク酸、マレイン酸、マロン酸、グルタル酸、グルカル酸、シュウ酸、アジピン酸、ピメリン酸、スベリン酸、アゼライン酸、セバシン酸、ドデカン二酸、グルタコン酸、オルトフタル酸、イソフタル酸、テレフタル酸、クエン酸、イソクエン酸、アコニット酸、トリカルバリル酸およびトリメシン酸からなる群から選択される、請求項1に記載のプロセス。
- 前記カルボン酸が多価カルボン酸である、請求項1に記載のプロセス。
- 前記多価カルボン酸がジカルボン酸またはトリカルボン酸である、請求項12に記載のプロセス。
- 前記カルボン酸が多価カルボン酸である場合、前記エステルは少なくともジエステルである、請求項12に記載のプロセス。
- 前記反応温度が150℃〜250℃であり、前記圧力が400psi〜3,000psiの範囲にある、請求項1に記載のプロセス。
- 前記遊離カルボン酸が、ハロゲン化アシルを生成するためのハロゲン化物による活性化に付されない、請求項1に記載のプロセス。
- 前記発酵培養液のpHが1.5〜4.5の範囲にある、請求項1に記載のプロセス。
- 前記乾燥工程が、噴霧乾燥、ドラム乾燥または凍結乾燥の少なくとも1つによる、請求項1に記載のプロセス。
- 前記遊離カルボン酸の混合物が、少なくとも2価の酸、3価の酸または多価酸を含み、アルコールとの前記反応による、前記遊離カルボン酸の対応するエステルへの前記2価の酸、3価の酸または多価酸の転化率が最低でも50%である、請求項1に記載のプロセス。
- 前記エステルを少なくとも90%の純度に精製する工程をさらに含む、請求項1に記載のプロセス。
- 前記精製工程が、結晶化、クロマトグラフィーまたは蒸留の少なくとも1つによる、請求項20に記載のプロセス。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201261739790P | 2012-12-20 | 2012-12-20 | |
| US61/739,790 | 2012-12-20 | ||
| PCT/US2013/073793 WO2014099433A1 (en) | 2012-12-20 | 2013-12-09 | Biofuels production from bio-derived carboxylic-acid esters |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2016501964A JP2016501964A (ja) | 2016-01-21 |
| JP6232073B2 true JP6232073B2 (ja) | 2017-11-15 |
Family
ID=50979030
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2015549446A Active JP6232073B2 (ja) | 2012-12-20 | 2013-12-09 | 生体由来カルボン酸エステルからのバイオ燃料の製造 |
Country Status (11)
| Country | Link |
|---|---|
| US (2) | US20150368574A1 (ja) |
| EP (1) | EP2935532B1 (ja) |
| JP (1) | JP6232073B2 (ja) |
| KR (1) | KR102046209B1 (ja) |
| CN (1) | CN104854222B (ja) |
| AU (1) | AU2013363496B2 (ja) |
| BR (1) | BR112015014315B1 (ja) |
| CA (1) | CA2895316C (ja) |
| MX (1) | MX374839B (ja) |
| SG (1) | SG11201503746VA (ja) |
| WO (1) | WO2014099433A1 (ja) |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU2013338397B2 (en) * | 2012-10-29 | 2017-11-16 | Archer Daniels Midland Company | Alcohol-mediated esterification of carboxylic acids with carbonates |
| US9464026B2 (en) * | 2012-12-20 | 2016-10-11 | Archer-Daniels Midland Company | Recovering and using carboxylic acids from a fermentation broth |
| TWI485242B (zh) * | 2012-12-22 | 2015-05-21 | Univ Nat Pingtung Sci & Tech | 生質柴油製造方法 |
| KR101766956B1 (ko) | 2015-05-14 | 2017-08-23 | 한양대학교 산학협력단 | 고압 이산화탄소 기체를 이용한 미생물 발효공정으로부터의 에스테르 제조방법 |
| KR102368472B1 (ko) * | 2019-02-28 | 2022-02-25 | 서울대학교산학협력단 | 바이오디젤의 제조 방법 |
Family Cites Families (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| IT1190783B (it) * | 1981-04-29 | 1988-02-24 | Davy Mckee Oil & Chem | Processo per l'idrogenolisi di esteri di acidi carbossilici |
| GB8518576D0 (en) * | 1985-07-23 | 1985-08-29 | Bp Chem Int Ltd | Hydrogenation of carboxylic acid esters to alcohols |
| IL117232A0 (en) * | 1996-02-22 | 1996-06-18 | Yissum Res Dev Co | A process for the production of an ester or polyester of carboxylic acid from a concentrated fermentation liquor |
| ATE405665T1 (de) * | 1999-03-11 | 2008-09-15 | Zeachem Inc | Verfahren zur herstellung von äthanol |
| BR0012953A (pt) * | 1999-08-03 | 2002-09-24 | Archer Daniels Midland Co | Processo para a recupepação de ácidos organicos |
| US6902917B1 (en) * | 1999-08-03 | 2005-06-07 | Archer-Daniels-Midland Company | Process for recovery of organic acids from fermentration broths |
| JP4204926B2 (ja) * | 2003-08-07 | 2009-01-07 | 花王株式会社 | 脂肪酸エステルの製造方法 |
| KR101163807B1 (ko) * | 2004-06-16 | 2012-07-09 | 더 텍사스 에이 & 엠 유니버시티 시스템 | 카르복실산 및 알콜로의 생체물질 전환용 방법 및 시스템 |
| US20070161095A1 (en) * | 2005-01-18 | 2007-07-12 | Gurin Michael H | Biomass Fuel Synthesis Methods for Increased Energy Efficiency |
| US7772414B1 (en) * | 2006-11-14 | 2010-08-10 | Aerophase, Inc. | Process for producing biodiesel fuel products |
| US20080248540A1 (en) * | 2007-04-03 | 2008-10-09 | The Ohio State University | Methods of producing butanol |
| WO2009038864A1 (en) * | 2007-09-19 | 2009-03-26 | Endicott Biofuels Ii, Llc | Method for obtaining biodiesel, alternative fuels and renewable fuels tax credits and treatment |
| CA2625511A1 (en) * | 2008-03-13 | 2009-09-13 | Dilum Dunuwila | Process for the direct production of esters of carboxylic acids from fermentation broths |
| WO2009150145A1 (en) * | 2008-06-12 | 2009-12-17 | Basf Se | Use of solubilizers for homogenizing additive concentrates |
| US20110151528A1 (en) * | 2008-06-30 | 2011-06-23 | Toyota Jidosha Kabushiki Kaisha | Process for producing organic acid |
| WO2011015918A2 (en) * | 2009-08-03 | 2011-02-10 | Avesthagen Limited | Vectors and compounds for expression of recombinant cetuximab |
| CA2773301C (en) * | 2009-09-07 | 2018-02-13 | Council Of Scientific & Industrial Research | Process for preparation of pure alkyl esters from alkali metal salt of carboxylic acid |
| US20110177564A1 (en) * | 2010-01-15 | 2011-07-21 | Massachusetts Institute Of Technology | Bioprocess and microbe engineering for total carbon utilization in biofuel production |
| BR112012027661B1 (pt) * | 2010-04-27 | 2020-12-08 | Kiverdi, Inc. | método biológico e químico para a captura e a conversão de um composto de carbono inorgânico e/ou um composto orgânico contendo apenas um átomo de carbono em um produto químico orgânico |
| AU2011274301B2 (en) * | 2010-06-28 | 2015-06-11 | Nuseed Global Innovation Ltd | Methods of producing lipids |
| ES2570382T3 (es) * | 2010-11-02 | 2016-05-18 | Codexis Inc | Composiciones y métodos para la producción de azúcares fermentables |
| WO2012128788A1 (en) * | 2011-03-24 | 2012-09-27 | Elevance Renewable Sciences, Inc. | Functionalized monomers and polymers |
| CN102557870A (zh) * | 2011-12-15 | 2012-07-11 | 北京金骄生物质化工有限公司 | 一种利用醋酸和甲醇制备燃料乙醇的方法 |
-
2013
- 2013-12-09 MX MX2015007907A patent/MX374839B/es active IP Right Grant
- 2013-12-09 US US14/650,475 patent/US20150368574A1/en not_active Abandoned
- 2013-12-09 EP EP13866239.0A patent/EP2935532B1/en not_active Not-in-force
- 2013-12-09 JP JP2015549446A patent/JP6232073B2/ja active Active
- 2013-12-09 BR BR112015014315-6A patent/BR112015014315B1/pt active IP Right Grant
- 2013-12-09 KR KR1020157019671A patent/KR102046209B1/ko active Active
- 2013-12-09 CN CN201380065924.3A patent/CN104854222B/zh active Active
- 2013-12-09 AU AU2013363496A patent/AU2013363496B2/en active Active
- 2013-12-09 WO PCT/US2013/073793 patent/WO2014099433A1/en not_active Ceased
- 2013-12-09 CA CA2895316A patent/CA2895316C/en active Active
- 2013-12-09 SG SG11201503746VA patent/SG11201503746VA/en unknown
-
2016
- 2016-10-19 US US15/297,610 patent/US10118886B2/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| SG11201503746VA (en) | 2015-07-30 |
| BR112015014315A2 (pt) | 2017-07-11 |
| BR112015014315B1 (pt) | 2020-10-13 |
| US20170044086A1 (en) | 2017-02-16 |
| EP2935532A4 (en) | 2016-09-07 |
| KR102046209B1 (ko) | 2019-11-18 |
| JP2016501964A (ja) | 2016-01-21 |
| AU2013363496A1 (en) | 2015-06-04 |
| US10118886B2 (en) | 2018-11-06 |
| CN104854222B (zh) | 2018-04-10 |
| KR20150096521A (ko) | 2015-08-24 |
| EP2935532B1 (en) | 2019-06-19 |
| MX2015007907A (es) | 2015-10-05 |
| CN104854222A (zh) | 2015-08-19 |
| MX374839B (es) | 2025-03-06 |
| CA2895316C (en) | 2019-12-03 |
| WO2014099433A1 (en) | 2014-06-26 |
| EP2935532A1 (en) | 2015-10-28 |
| US20150368574A1 (en) | 2015-12-24 |
| AU2013363496B2 (en) | 2017-08-31 |
| CA2895316A1 (en) | 2014-06-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6230616B2 (ja) | 発酵培養液由来のカルボン酸の回収および使用 | |
| JP6232073B2 (ja) | 生体由来カルボン酸エステルからのバイオ燃料の製造 | |
| JP6230617B2 (ja) | 生物学的に誘導されたカルボン酸エステルからの水素化生成物 | |
| Almena et al. | Integrated biodiesel facilities: Review of glycerol-based production of fuels and chemicals | |
| US9834531B2 (en) | Use of carboxylic acids and furanic molecules for esterification | |
| TW201527274A (zh) | 方法 | |
| TW201542517A (zh) | 方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20161121 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20170615 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20170623 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20170912 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20170921 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20171020 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 6232073 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |