JP6088063B2 - ブルトン型チロシンキナーゼの阻害剤 - Google Patents
ブルトン型チロシンキナーゼの阻害剤 Download PDFInfo
- Publication number
- JP6088063B2 JP6088063B2 JP2015542251A JP2015542251A JP6088063B2 JP 6088063 B2 JP6088063 B2 JP 6088063B2 JP 2015542251 A JP2015542251 A JP 2015542251A JP 2015542251 A JP2015542251 A JP 2015542251A JP 6088063 B2 JP6088063 B2 JP 6088063B2
- Authority
- JP
- Japan
- Prior art keywords
- fluoro
- tert
- butyl
- formula
- mmol
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- IYPATQYBZFOYNW-UHFFFAOYSA-N CC(C)(C)c(cc1C=NN2Cc(cc3)cc(CO)c3-c3cc(OC)ncc3)cc(F)c1C2=O Chemical compound CC(C)(C)c(cc1C=NN2Cc(cc3)cc(CO)c3-c3cc(OC)ncc3)cc(F)c1C2=O IYPATQYBZFOYNW-UHFFFAOYSA-N 0.000 description 1
- SPWHVZKPOYTIFN-UHFFFAOYSA-N CC(C)(C)c(cc1C=NN2c3cccc(Br)c3CO)cc(F)c1C2=O Chemical compound CC(C)(C)c(cc1C=NN2c3cccc(Br)c3CO)cc(F)c1C2=O SPWHVZKPOYTIFN-UHFFFAOYSA-N 0.000 description 1
- ZIOOXJFZMBQVJJ-UHFFFAOYSA-N CC(C)(C)c1cc(C)c(C(OC)=O)[s]1 Chemical compound CC(C)(C)c1cc(C)c(C(OC)=O)[s]1 ZIOOXJFZMBQVJJ-UHFFFAOYSA-N 0.000 description 1
- KNBJDKDSQTXWNG-UHFFFAOYSA-N CC(C)(C)c1cc(CNCc(ccc(-c2cc(-c3c[n](C)nc3)nc(OC)c2)c2)c2F)c(C(OC)=O)[s]1 Chemical compound CC(C)(C)c1cc(CNCc(ccc(-c2cc(-c3c[n](C)nc3)nc(OC)c2)c2)c2F)c(C(OC)=O)[s]1 KNBJDKDSQTXWNG-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/62—Oxygen or sulfur atoms
- C07D213/63—One oxygen atom
- C07D213/64—One oxygen atom attached in position 2 or 6
- C07D213/643—2-Phenoxypyridines; Derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
- A61P29/02—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID] without antiinflammatory effect
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/80—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having two double bonds between ring members or between ring members and non-ring members
- C07D211/82—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having two double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/10—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D495/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms
- C07D495/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D495/04—Ortho-condensed systems
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Immunology (AREA)
- Rheumatology (AREA)
- Pulmonology (AREA)
- Pain & Pain Management (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Physical Education & Sports Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Epidemiology (AREA)
- Plural Heterocyclic Compounds (AREA)
Description
本発明は、Btkを阻害し、異常なB細胞活性化により引き起こされる自己免疫疾患及び炎症性疾患の処置に有用である、新規化合物の使用に関する。
タンパク質キナーゼは、ヒト酵素の最大ファミリーの1つを構成し、リン酸塩基をタンパク質に加えることにより、多くの異なるシグナル伝達過程を制御する(T. Hunter, Cell 1987 50:823-829)。具体的には、チロシンキナーゼは、チロシン残基のフェノール類部分のタンパク質をリン酸化する。チロシンキナーゼファミリーは、細胞成長、遊走、及び分化を制御するメンバーを含む。異常なキナーゼ活性は、癌、自己免疫疾患、及び炎症性疾患を含む、様々なヒト疾患において結び付けられてきた。タンパク質キナーゼは、細胞のシグナル伝達の鍵となる制御因子であるので、細胞機能を、低分子キナーゼ阻害剤で調節するための標的を提供し、これにより、良好な薬物設計標的を作成する。キナーゼにより仲介される疾患過程の処置に加えて、キナーゼ活性の選択的阻害剤、及び有効な阻害剤はまた、細胞シグナル伝達過程の調査、及び治療対象の他の細胞標的の同定に有用である。
本出願は、本明細書において後述される、式IのBtk阻害剤化合物、その使用方法を提供する。
(式中:
Aは、場合により1個以上のA’で置換されている、不飽和又は部分的に飽和の単環式又は二環式ヘテロアリール又はフェニルであり;
A’は、ハロ、低級アルキル、又はオキソであり;
それぞれのR1は、独立して、ハロ、ヒドロキシル低級アルキル、又は低級アルキルであり;
mは、0、1、又は2であり;
R2は、メチルピラゾリルであり;
nは、0、又は1であり;
Xは、結合、CH2、又はNHC(=O)である)
の化合物、又はその薬学的に許容し得る塩を提供する。
(式中:
Aは、場合により1個以上のA’で置換されている、不飽和又は部分的に飽和の単環式又は二環式ヘテロアリール又はフェニルであり;
A’は、ハロ、低級アルキル、又はオキソであり;
それぞれのR1は、独立して、ハロ、ヒドロキシル低級アルキル、又は低級アルキルであり;
mは、0、1、又は2であり;
R2は、メチルピラゾリルであり;
nは、0、又は1であり;
Xは、CH2、又はNHC(=O)である)
の化合物、又はその薬学的に許容し得る塩を提供する。
定義
本明細書で用いられる語句「a」、又は「an」実体は、1個以上の該実体に言及する;例えば、1個の化合物は、1個以上の化合物、又は少なくとも1個の化合物に言及する。しかるが故に、用語「a」(又は「an」)、「1個以上」、及び「少なくとも1個」は、本明細書において互換的に用いることができる。
である。
本出願は、式I
(式中:
Aは、場合により1個以上のA’で置換されている、不飽和又は部分的に飽和の単環式又は二環式ヘテロアリール又はフェニルであり;
A’は、ハロ、低級アルキル、又はオキソであり;
それぞれのR1は、独立して、ハロ、ヒドロキシル低級アルキル、又は低級アルキルであり;
mは、0、1、又は2であり;
R2は、メチルピラゾリルであり;
nは、0、又は1であり;
Xは、結合、CH2、又はNHC(=O)である)
の化合物、又はその薬学的に許容し得る塩を提供する。
(式中:
Aは、場合により1個以上のA’で置換されている、不飽和又は部分的に飽和の単環式又は二環式ヘテロアリール又はフェニルであり;
A’は、ハロ、低級アルキル、又はオキソであり;
それぞれのR1は、独立して、ハロ、ヒドロキシル低級アルキル、又は低級アルキルであり;
mは、0、1、又は2であり;
R2は、メチルピラゾリルであり;
nは、0、又は1であり;
Xは、CH2、又はNHC(=O)である)
の化合物、又はその薬学的に許容し得る塩を提供する。
6−tert−ブチル−8−フルオロ−2−[4−(2−オキソ−1,2−ジヒドロ−ピリジン−4−イル)−ベンジル]−2H−フタラジン−1−オン;
6−tert−ブチル−8−フルオロ−2−[2−フルオロ−4−(2−オキソ−1,2−ジヒドロ−ピリジン−4−イル)−ベンジル]−2H−フタラジン−1−オン;
6−tert−ブチル−8−フルオロ−2−[2−ヒドロキシメチル−3−(2−オキソ−1,2−ジヒドロ−ピリジン−4−イル)−フェニル]−2H−フタラジン−1−オン;
6−tert−ブチル−2−[2,6−ジフルオロ−4−(2−オキソ−1,2−ジヒドロ−ピリジン−4−イル)−ベンジル]−8−フルオロ−2H−フタラジン−1−オン;
6−tert−ブチル−8−フルオロ−2−[2−ヒドロキシメチル−4−(2−オキソ−1,2−ジヒドロ−ピリジン−4−イル)−ベンジル]−2H−フタラジン−1−オン;
6−tert−ブチル−8−フルオロ−2−{2−フルオロ−4−[6−(1−メチル−1H−ピラゾール−4−イル)−2−オキソ−1,2−ジヒドロ−ピリジン−4−イル]−ベンジル}−2H−フタラジン−1−オン;
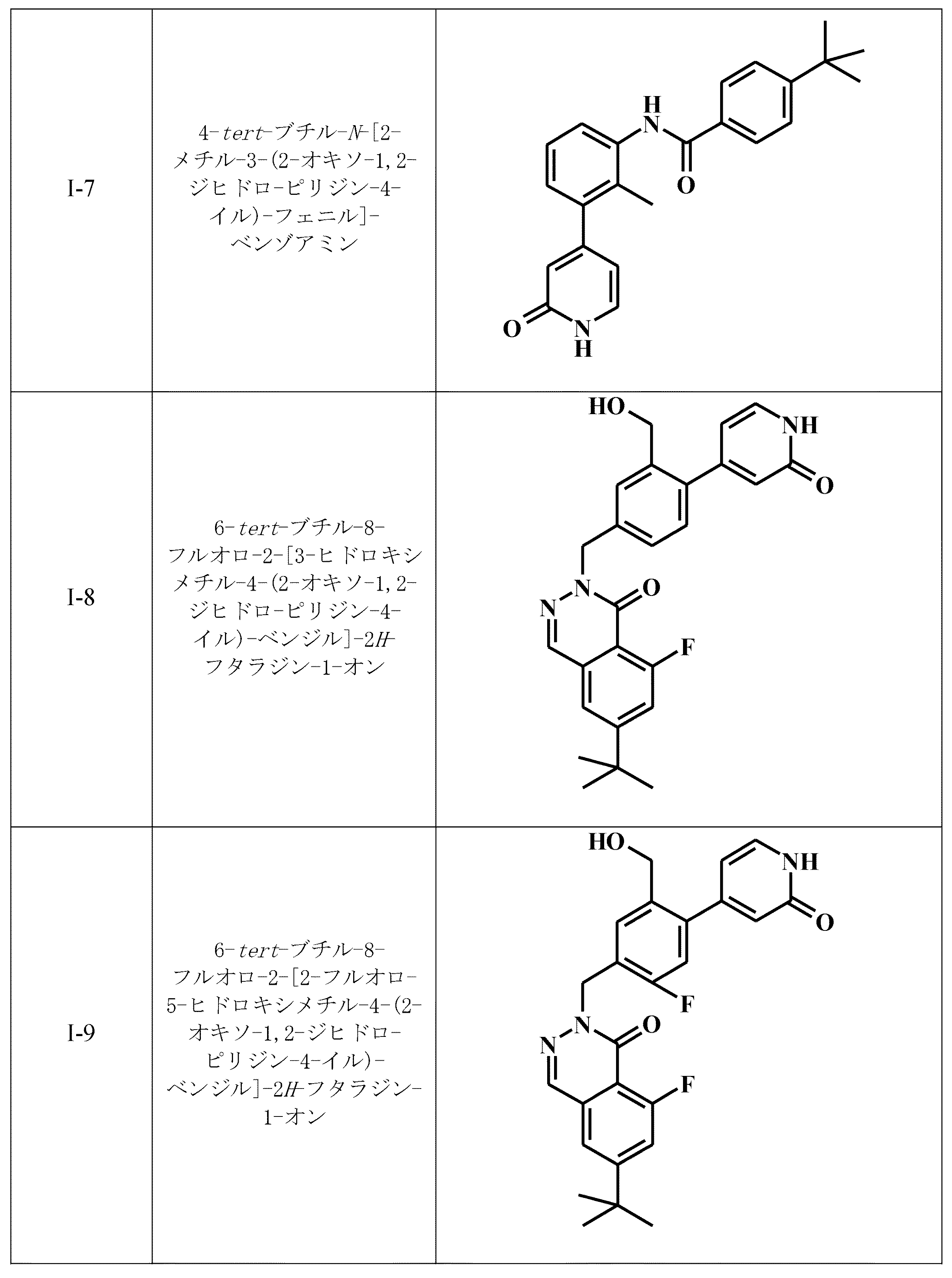
4−tert−ブチル−N−[2−メチル−3−(2−オキソ−1,2−ジヒドロ−ピリジン−4−イル)−フェニル]−ベンズアミド;
6−tert−ブチル−8−フルオロ−2−[3−ヒドロキシメチル−4−(2−オキソ−1,2−ジヒドロ−ピリジン−4−イル)−ベンジル]−2H−フタラジン−1−オン;
6−tert−ブチル−8−フルオロ−2−[2−フルオロ−5−ヒドロキシメチル−4−(2−オキソ−1,2−ジヒドロ−ピリジン−4−イル)−ベンジル]−2H−フタラジン−1−オン;及び
2−tert−ブチル−5−{2−フルオロ−4−[6−(1−メチル−1H−ピラゾール−4−イル)−2−オキソ−1,2−ジヒドロ−ピリジン−4−イル]−ベンジル}−4,5−ジヒドロ−チエノ[2,3−c]ピロール−6−オン
からなる群より選択される、式Iの化合物を提供する。
本発明により包含され、本発明の範囲内にある、代表的な化合物の例は、以下の表に提供される。以下のこれらの実施例及び調製は、当業者が、本発明をより明確に理解し、実施することを可能にするために提供される。これらは、本発明の範囲を制限すると見なされるべきではなく、単にその説明及び代表であると見なされるべきである。
本発明の化合物は、任意の従来手段により調製され得る。これらの化合物の合成に適した方法は、実施例において提供される。一般的に、本発明の化合物は、以下のスキームに従い調製され得る。
本発明の化合物は、多種多様な経口投与投薬形態、及び担体に製剤化され得る。経口投与は、錠剤、被覆錠剤、ドラジェ、硬ゼラチンカプセル剤及び軟ゼラチンカプセル剤、液剤、乳剤、シロップ剤、又は懸濁剤の形態であり得る。本発明の化合物は、他の投与経路のうち、継続(静脈内点滴)局所非経口、筋肉内、静脈内、皮下、経皮(透過増強剤を含み得る)、口腔、経鼻、吸入、及び坐剤投与を含む、他の投与経路により投与されるとき、有効である。好ましい投与方法は、一般的に、苦痛の程度、及び有効成分に対する患者の応答により調節され得る、便宜的な1日投薬処方計画を用いた経口である。
一般式Iの化合物は、ブルトン型チロシンキナーゼ(Btk)を阻害する。上流キナーゼによるBtkの活性化は、順に、前炎症性メディエーターの放出を刺激する、ホスホリパーゼ−Cγの活性化を生じる。式Iの化合物は、関節炎、並びに他の抗炎症性疾患、及び自己免疫疾患の処置において有用である。したがって、式I記載の化合物は、関節炎の処置に有用である。式Iの化合物は、細胞においてBtkを阻害するのに有用であり、B細胞発生を調節するのに有用である。本発明は、更に、薬学的に許容し得る担体、賦形剤、又は希釈剤と混合された、式Iの化合物を含有する医薬組成物を含む。
本出願は、必要な患者に、治療上有効量の式Iの化合物を投与することを含む、炎症状態、及び/又は自己免疫状態の処置方法を提供する。
一般的略語
一般的に用いられる略語は、アセチル(Ac)、azo−ビス−イソブチリルニトリル(AIBN)、雰囲気(Atm)、9−ボラビシクロ[3.3.1]ノナン(9−BBN、又はBBN)、2,2'−ビス(ジフェニルホスフィノ)−1,1'−ビナフチル(BINAP)、tert−ブトキシカルボニル(Boc)、ジ−tert−ブチルピロカルボナート、又はboc無水物(BOC2O)、ベンジル(Bn)、ブチル(Bu)、Chemical Abstracts Registration Number(CASRN)、ベンジルオキシカルボニル(CBZ、又はZ)、カルボニルジイミダゾール(CDI)、1,4−ジアザビシクロ[2.2.2]オクタン(DABCO)、ジエチルアミノ硫黄トリフルオリド(DAST)、ジベンジリデンアセトン(dba)、1,5−ジアザビシクロ[4.3.0]ノン−5−エン(DBN)、1,8−ジアザビシクロ[5.4.0]ウンデク−7−エン(DBU)、N,N'−ジシクロヘキシルカルボジイミド(DCC)、1,2−ジクロロエタン(DCE)、ジクロロメタン(DCM)、2,3−ジクロロ−5,6−ジシアノ−1,4−ベンゾキノン(DDQ)、アゾジカルボン酸ジエチル(DEAD)、ジ−イソ−プロピルアゾジカルボキシレート(DIAD)、ジ−イソ−ブチルアルミニウムヒドリド(DIBAL、又はDIBAL−H)、ジ−イソ−プロピルエチルアミン(DIPEA)、N,N−ジメチルアセトアミド(DMA)、4−N,N−ジメチルアミノピリジン(DMAP)、N,N−ジメチルホルムアミド(DMF)、ジメチルスルホキシド(DMSO)、1,1'−ビス−(ジフェニルホスフィノ)エタン(dppe)、1,1'−ビス−(ジフェニルホスフィノ)フェロセン(dppf)、1−(3−ジメチルアミノプロピル)−3−エチルカルボジイミドヒドロクロライド(EDCI)、2−エトキシ−1−エトキシカルボニル−1,2−ジヒドロキノリン(EEDQ)、エチル(Et)、酢酸エチル(EtOAc)、エタノール(EtOH)、2−エトキシ−2H−キノリン−1−カルボン酸エチルエステル(EEDQ)、ジエチルエーテル(Et2O)、エチルイソプロピルエーテル(EtOiPr)、O−(7−アザベンゾトリアゾール−1−イル)−N,N,N’N’−テトラメチルウロニウムヘキサフルオロホスファート酢酸(HATU)、酢酸(HOAc)、1−N−ヒドロキシベンゾトリアゾール(HOBt)、高圧液体クロマトグラフィー(HPLC)、イソ−プロパノール(IPA)、イソプロピルマグネシウムクロライド(iPrmgCl)、ヘキサメチルジシラザン(HMDS)、液体クロマトグラフィー質量分析(LCMS)、リチウムヘキサメチルジシラザン(LiHMDS)、メタ−クロロペルオキシ安息香酸(m−CPBA)、メタノール(MeOH)、融点(mp)、MeSO2−(メシル、又はMs)、メチル(Me)、アセトニトリル(MeCN)、m−クロロ過安息香酸(MCPBA)、質量スペクトル(ms)、メチルt−ブチルエーテル(MTBE)、メチルテトラヒドロフラン(MeTHF)、N−ブロモスクシンイミド(NBS)、n−ブチルリチウム(nBuLi)、N−カルボキシ無水物(NCA)、N−クロロスクシンイミド(NCS)、N−メチルモルホリン(NMM)、N−メチルピロリドン(NMP)、ピリジニウムクロロクロマート(PCC)、ジクロロ−((ビス−ジフェニルホスフィノ)フェロセニル)パラジウム(II)(Pd(dppf)Cl2)、パラジウム(II)アセタート(Pd(OAc)2)、トリス(ジベンジリデンアセトン)ジパラジウム(0)(Pd2(dba)3)、ピリジニウムジクロマート(PDC)、フェニル(Ph)、プロピル(Pr)、イソ−プロピル(i−Pr)、重量ポンド毎平方インチ(psi)、ピリジン(pyr)、1,2,3,4,5−ペンタフェニル−1'−(ジ−tert−ブチルホスフィノ)フェロセン(Q−Phos)、室温(周囲温度、rt、又はRT)、sec−ブチルリチウム(sBuLi)、tert−ブチルジメチルシリル、又はt−BuMe2Si(TBDMS)、テトラ−n−ブチルアンモニウムフルオライド(TBAF)、トリエチルアミン(TEA、又はEt3N)、2,2,6,6−テトラメチルピペリジネル−オキシル(TEMPO)、トリメチルシリルエトキシメチル(SEM)、トリフレート、又はCF3SO2−(Tf)、トリフルオロ酢酸(TFA)、1,1'−ビス−2,2,6,6−テトラメチルヘプタン−2,6−ジオン(TMHD)、O−ベンゾトリアゾール−1−イル−N,N,N',N'−テトラメチルウロニウムテトラフルオロボレート(TBTU)、薄層クロマトグラフィー(TLC)、テトラヒドロフラン(THF)、トリメチルシリル、又はMe3Si(TMS)、p−トルエンスルホン酸一水和物(TsOH、又はpTsOH)、4−Me−C6H4SO2−、又はトシル(Ts)、及びN−ウレタン−N−カルボキシ無水物(UNCA)を含む。接頭辞、標準(n)、イソ(i−)、2級(sec−)、3級(tert−)、及びneoを含む通常の命名法は、アルキル部分と一緒に用いられるとき、その慣習的意味を有する(J. Rigaudy and D. P. Klesney, Nomenclature in Organic Chemistry, IUPAC 1979 Pergamon Press, Oxford.)。
本発明の化合物は、市販の出発材料で出発して、当業者に既知の一般的な合成技術及び合成方法を用いることにより、調製され得る。以下の概略は、かかる化合物の調製に適した反応スキームである。更なる例証を、具体的な実施例において見ることができる。
具体的な略語
AIBN アゾビスイソブチロニトリル
AlCl3 三塩化アルミニウム
BBr3 三臭化ホウ素
BH3−DMS ボラン−ジメチルスルフィド複合体
CCl4 四塩化炭素
CH2Cl2 ジクロロメタン
CH3CN アセトニトリル
Cs2CO3 炭酸セシウム
DIPEA ジイソプロピルエチルアミン
DME ジメトキシエタン
DMF N,N−ジメチルホルムアミド
EtOAc 酢酸エチル
EtOH エタノール
HATU O−(7−アザベンゾトリアゾール−1−イル)−N,N,N’,N’−テトラメチルウロニウムヘキサフルオロホスファート
K2CO3 炭酸カリウム
KH2PO4 カリウムジホスファート
KOAc 酢酸カリウム
MeOH メタノール
Na2CO3 炭酸ナトリウム
Na2S2O3 チオ硫酸ナトリウム
Na2SO4 硫酸ナトリウム
NaBH4 水素化ホウ素ナトリウム
NaH 水素化ナトリウム
NaHCO3 炭酸水素ナトリウム
NaI ヨウ化ナトリウム
NaOMe ナトリウムメトキシド
NH4OAc 酢酸アンモニウム
PBr3 三臭化リン
Pd(dba)2 ビス(ジベンジリデンアセトン)パラジウム(0)
Pd2(dba)3 トリス(ジベンジリデンアセトン)ジパラジウム(0)
Pd(OAc)2 酢酸パラジウム(II)
Pd(PPh3)4 テトラキス(トリフェニルホスフィン)パラジウム(0)
iPrOH イソプロパノール
PYBOP (ベンゾトリアゾール−1−イル−オキシトリピロリジノホスホニウムヘキサフルオロホスファート)
THF テトラヒドロフラン
X−Phos 2−ジシクロヘキシルホスフィノ−2’,4’,6’−トリイソプロピルビフェニル
試薬を、Aldrich、Oakwood、Matrix、又は他の供給源から購入し、更に精製することなく用いた。加熱のためのマイクロ波照射を用いた反応は、Personal Chemistry Emrys Optimizer System、又はCEM Discovery Systemのいずれかを用いて、行った。数mg〜数gのスケールの精製を、当業者に公知の方法、例えば、シリカゲルフラッシュカラムの溶出により、行った;いくつかの場合で、CombiFlashシステムで溶出する、使い捨ての予め包装された数gのシリカゲルカラム(RediSep)の使用により、分取フラッシュカラム精製も行った。Biotage(商標)、及びISCO(商標)もまた、中間体の精製のため、本発明で用い得るフラッシュカラム器具である。
2−フルオロ−4−[2−メトキシ−6−(1−メチル−1H−ピラゾール−4−イル)−ピリジン−4−イル]−ベンジルアミン
圧力管中で、4−ブロモ−2−フルオロベンジルカルバミン酸tert−ブチル(5g、16.4mmol)、ビス(ピナコラト)ジボロン(6.26g、24.7mmol)及び酢酸カリウム(4.84g、49.3mmol)をNMP(75.0mL)と合わせて明黄色の溶液を与えた。反応混合物を窒素下に10分間脱気した。[1,1′−ビス(ジフェニルホスフィノ)フェロセン]ジクロロ−パラジウム(II)(722mg、0.986mmol)を加えた。反応混合物を100℃で20時間加熱した。反応混合物を、水を用いてクエンチし、CH2Cl2(3×100mL)で抽出した。合わせた有機層を、水、ブラインで洗浄し、Na2SO4で乾燥させ、濾過し、濃縮した。粗物質をフラッシュクロマトグラフィー(シリカゲル、120g、ヘキサン中の0%〜30%の酢酸エチル)によって精製した。[2−フルオロ−4−(4,4,5,5−テトラメチル−[1,3,2]ジオキサボロラン−2−イル)−ベンジル]−カルバミン酸tert−ブチルエステル(5.8g、100%)を黄色の油状物として得た。
マイクロ波管中で、2,6−ジクロロ−4−ヨードピリジン(4.49g、16.4mmol)及びテトラキス(トリフェニルホスフィン)パラジウム(0)(948mg、820μmmol)をDMF(60mL)と合わせて黄色の溶液を与えた。炭酸カリウム(6.8g、49.2mmol)及び[2−フルオロ−4−(4,4,5,5−テトラメチル−[1,3,2]ジオキサボロラン−2−イル)−ベンジル]−カルバミン酸tert−ブチルエステル(5.76g、16.4mmol)を加えた。反応物をマイクロ波反応器中で150℃にて90分間加熱した。反応混合物を水でクエンチし、次にCH2Cl2(3×)で抽出した。合わせたCH2Cl2層を、水、ブラインで洗浄し、Na2SO4で乾燥させ、濾過し、濃縮した。粗物質をフラッシュクロマトグラフィー(シリカゲル、40g、ヘキサン中の0〜25%の酢酸エチル)によって精製した。生成物[4−(2,6−ジクロロ−ピリジン−4−イル)−2−フルオロ−ベンジル]−カルバミン酸tert−ブチルエステル(4.0g、59%)を黄色の固体として得た。
圧力管中で、[4−(2,6−ジクロロ−ピリジン−4−イル)−2−フルオロ−ベンジル]−カルバミン酸tert−ブチルエステル(3.99g、10.7mmol)をMeOH(46mL)と合わせて無色の溶液を与えた。ナトリウムメトキシド(メタノール溶液中25重量%NaOMe)(6.97g、7.37mL、32.2mmol)を加えた。反応混合物を還流温度で一晩加熱した。反応混合物を濃縮し、粗物質をフラッシュクロマトグラフィー(シリカゲル、ヘキサン中の0%〜25%の酢酸エチル)によって精製した。[4−(2−クロロ−6−メトキシ−ピリジン−4−イル)−2−フルオロ−ベンジル]−カルバミン酸tert−ブチルエステル(2.72g、69%)をオフホワイトの固体として得た。
圧力管中で、[4−(2−クロロ−6−メトキシ−ピリジン−4−イル)−2−フルオロ−ベンジル]−カルバミン酸tert−ブチルエステル(120mg、0.330mmol)、及びテトラキス(トリフェニルホスフィン)パラジウム(0)(38mg、0.033mmol)をDMF(4mL)と合わせて明黄色の溶液を与えた。炭酸カリウム及び1−メチル−4−(4,4,5,5,−テトラメチル−1,3,2−ジオキサボロラン−2−イル)−1H−ピラゾール(68mg、0.330mmol)を加えた。反応混合物を撹拌し、100℃で一晩加熱した。翌朝、更なるテトラキス(トリフェニルホスフィン)パラジウム(0)(38mg、0.033mmol)及び1−メチル−4−(4,4,5,5,−テトラメチル−1,3,2−ジオキサボロラン−2−イル)−1H−ピラゾール(68mg、0.330mmol)を加えた。反応混合物を、マイクロ波照射を2時間使用して150℃で加熱した。反応混合物を室温に冷やし、次に水で希釈した。混合物をCH2Cl2(3×20mL)で抽出した。合わせた有機層を水、続いてブラインで洗浄した。次に有機抽出物をNa2SO4で乾燥させ、濾過し、濃縮した。粗物質を、フラッシュクロマトグラフィー(シリカゲル12g、ヘキサン中の0〜20%のEtOAc)を使用して精製して、{2−フルオロ−4−[2−メトキシ−6−(1−メチル−1H−ピラゾール−4−イル)−ピリジン−4−イル]−ベンジル}−カルバミン酸tert−ブチルエステル(85mg、52%)を白色の固体として生成した。
10mLの丸底フラスコ中で、{2−フルオロ−4−[2−メトキシ−6−(1−メチル−1H−ピラゾール−4−イル)−ピリジン−4−イル]−ベンジル}−カルバミン酸tert−ブチルエステル(45mg、109μmol)をCH2Cl2(560μL)及びTFAと合わせて明黄色の溶液を与えた。混合物を室温で2時間撹拌した。この後、反応混合物を濃縮し、生成物を高真空で2時間更に乾燥させた。得られた生成物2−フルオロ−4−[2−メトキシ−6−(1−メチル−1H−ピラゾール−4−イル)−ピリジン−4−イル]−ベンジルアミン(32mg、73%)を、更に精製することなくその後の反応に使用した。
実施例I−1
6−tert−ブチル−8−フルオロ−2−[4−(2−オキソ−1,2−ジヒドロ−ピリジン−4−イル)−ベンジル]−2H−フタラジン−1−オン
実施例I−2工程1に記載したものと類似の条件を使用して、6−tert−ブチル−8−フルオロ−2H−フタラジン−1−オン(これは、Berthel, S. et al. US 20100222325 Column 139に記載されているように調製してもよい;50mg、0.23mmol)と1−ブロモ−4−クロロメチル−ベンゼン(Aldrichから入手可能;51.4mg、0.25mmol)とを反応させて、2−(4−ブロモ−ベンジル)−6−tert−ブチル−8−フルオロ−2H−フタラジン−1−オン(80mg、90%)を黄色の油状物として与えた。
DME中の2−(4−ブロモ−ベンジル)−6−tert−ブチル−8−フルオロ−2H−フタラジン−1−オン(80mg、0.21mmol)とPd(PPh3)4(1mol%)との混合物をアルゴンで10分間でパージした。Na2CO3水溶液(2M;2当量)を加え、管をアルゴンでもう一度パージした。溶液を室温で5分間撹拌し、EtOH中の2−メトキシ−ピリジン−4−ボロン酸(Oakwood Products, Inc., 1741 Old Dunbar Road, West Columbia, SC 29172, USAから入手可能;39.3mg、0.26mmol)の溶液を加えた。混合物をアルゴンでパージし、蓋をし、90℃で1時間加熱した。混合物をセライトwを通して濾過し、セライトをCH2Cl2で洗浄した。濾液を乾燥させ(Na2SO4)、濾過し蒸発させた。残留物をクロマトグラフィー(シリカゲル)によって精製して6−tert−ブチル−8−フルオロ−2−[4−(2−メトキシ−ピリジン−4−イル)−ベンジル]−2H−フタラジン−1−オン(50mg、58%)をガム状物として与えた。MS:C25H25FN3O2[(M+H)+]の理論値418、実測値418。
6−tert−ブチル−8−フルオロ−2−[4−(2−メトキシ−ピリジン−4−イル)−ベンジル]−2H−フタラジン−1−オン(100.0mg、0.24mmol)をPBr3と、実施例I−7工程4に記載した条件を使用して反応させて、6−tert−ブチル−8−フルオロ−2−[4−(2−オキソ−1,2−ジヒドロ−ピリジン−4−イル)−ベンジル]−2H−フタラジン−1−オン(50mg、52%)を白色の固体として与えた。MS:C24H23FN3O[(M+H)+]の理論値404、実測値404。1H NMR(400 MHz, DMSO-d6) δ ppm 11.59(br. s. , 1 H), 8.45(d, J=2.4 Hz, 1 H), 7.80(d, J=1.5 Hz, 1 H), 7.72(dd, J=13.2, 1.5 Hz, 1 H), 7.65(d, J=8.3 Hz, 2 H), 7.34 - 7.46(m, 3 H), 6.55(d, J=1.5 Hz, 1 H), 6.46(dd, J=6.8, 2.0 Hz, 1 H), 5.32(s, 2 H), 1.35(s, 9 H)
6−tert−ブチル−8−フルオロ−2−[2−フルオロ−4−(2−オキソ−1,2−ジヒドロ−ピリジン−4−イル)−ベンジル]−2H−フタラジン−1−オン
DMF(5mL)中のNaH(60%、55mg、1.36mmol)の撹拌した懸濁液に、DMF(10mL)中の6−tert−ブチル−8−フルオロ−2H−フタラジン−1−オン(これは、Berthel, S. et al. US 20100222325 Column 139に記載されているように調製してもよい;150.0mg、0.68mmol)の溶液を0℃で滴下した。混合物を70℃に加熱し、30分間撹拌した。混合物を室温に冷やし、DMF(5mL)中の4−ブロモ−1−クロロメチル−2−フルオロ−ベンゼン(Aldrichから入手可能;167.3mg、0.75mmol)の溶液を加え、混合物を室温で4時間撹拌した。NH4Cl水溶液を加えた。混合物をEtOAcで抽出し、EtOAc抽出物を乾燥させ、蒸発させた。残留物をクロマトグラフィー(シリカゲル、5%EtOAc/ヘキサン)によって精製して、2−(4−ブロモ−2−フルオロ−ベンジル)−6−tert−ブチル−8−フルオロ−2H−フタラジン−1−オン(160mg、58%)を白色の固体として与えた。MS:C19H18BrF2N2O[(M+H)+]の理論値408、実測値408。
2−(4−ブロモ−2−フルオロ−ベンジル)−6−tert−ブチル−8−フルオロ−2H−フタラジン−1−オン(160mg、0.39mmol)を2−メトキシ−ピリジン−4−ボロン酸(Oakwood Products, Inc., 1741 Old Dunbar Road, West Columbia, SC 29172, USAから入手可能;60.4mg、0.49mmol)と、実施例I−1工程2に記載されているものと類似した条件を使用して反応させて6−tert−ブチル−8−フルオロ−2−[2−フルオロ−4−(2−メトキシ−ピリジン−4−イル)−ベンジル]−2H−フタラジン−1−オン(80mg、47%)を白色の固体として与えた。
6−tert−ブチル−8−フルオロ−2−[4−(2−メトキシ−ピリジン−4−イル)−ベンジル]−2H−フタラジン−1−オン(100.0mg、0.24mmol)をPBr3と、実施例I−7工程4に記載した条件を使用して反応させて、6−tert−ブチル−8−フルオロ−2−[4−(2−オキソ−1,2−ジヒドロ−ピリジン−4−イル)−ベンジル]−2H−フタラジン−1−オン(50mg、52%)を白色の固体として与えた。MS:C24H22F2N3O2[(M+H)+]の理論値422、実測値422。1H NMR(400 MHz, DMSO-d6) δ ppm 11.64(br. s. , 1 H), 8.45(d, J=2.4 Hz, 1 H), 7.80(d, J=1.5 Hz, 1 H), 7.73(dd, J=13.2, 1.5 Hz, 1 H), 7.59(d, J=11.2 Hz, 1 H), 7.42 - 7.50(m, 2 H), 7.32(t, J=8.1 Hz, 1 H), 6.62(d, J=1.5 Hz, 1 H), 6.50(dd, J=6.8, 1.5 Hz, 1 H), 5.36(s, 2 H), 1.35(s, 9 H)
6−tert−ブチル−8−フルオロ−2−[2−ヒドロキシメチル−3−(2−オキソ−1,2−ジヒドロ−ピリジン−4−イル)−フェニル]−2H−フタラジン−1−オン
DMF(5mL)中の6−tert−ブチル−8−フルオロ−2H−フタラジン−1−オン(これは、Berthel, S. et al. US 20100222325 Column 139に記載されているように調製してもよい;100mg、0.45mmol)の撹拌した溶液に、2−ブロモ−6−フルオロ−ベンズアルデヒド(Aldrichから入手可能;101.5mg、0.5mmol)、炭酸セシウム(325mg、0.27mmol)及びメトキシトリメチルシラン(0.1mL、0.91mmol)を加えた。混合物を60℃で4時間加熱し、次に室温に冷やし、水(25mL)で希釈した。得られた混合物を酢酸エチルで抽出し、乾燥させ(Na2SO4)、濾過し、蒸発させた。残留物をクロマトグラフィー(シリカゲル、10%のEtOAc/ヘキサン)によって精製して、2−ブロモ−6−(6−tert−ブチル−8−フルオロ−1−オキソ−1H−フタラジン−2−イル)−ベンズアルデヒド(105mg、57%)を黄色のガム状物として与えた。MS:C19H17BrFN2O2[(M+H)+]の理論値404、実測値404。
CH2Cl2とiPrOH(2:1;7.5mL)との混合物中の撹拌しかつ冷却した(約0℃)2−ブロモ−6−(6−tert−ブチル−8−フルオロ−1−オキソ−1H−フタラジン−2−イル)−ベンズアルデヒド(100mg、0.25mmol)の溶液に、NaBH4(4.5mg、0.12mmol)を加えた。混合物を約0℃で30分間撹拌し、次に水を加え、混合物をEtOAcで抽出した。EtOAc抽出物を乾燥させ(Na2SO4)、濾過し、蒸発させた。残留物をクロマトグラフィー(シリカゲル、20%のEtOAc/ヘキサン)によって精製して、2−(3−ブロモ−2−ヒドロキシメチル−フェニル)−6−tert−ブチル−8−フルオロ−2H−フタラジン−1−オン(90mg、84%)を白色の固体として与えた。MS:C19H19BrFN2O2[(M+H)+]の理論値406、実測値406。
20%の水性ジオキサン(20mL)中の2−(3−ブロモ−2−ヒドロキシメチル−フェニル)−6−tert−ブチル−8−フルオロ−2H−フタラジン−1−オン(90mg、0.22mmol)の撹拌した溶液に、2−メトキシ−ピリジン−4−ボロン酸(Oakwood Products, Inc., 1741 Old Dunbar Road, West Columbia, SC 29172, USAから入手可能;40.8mg、0.27mmol)、K2CO3(61.3mg、0.44mmol)、及びトリシクロヘキシルホスフィン(3.7mg、0.01mmol)を加えた。フラスコを排気し、窒素で3回充填戻した。Pd(dba)2(6.1mg、0.01mmol)を加え、混合物を96℃で5時間加熱した。混合物を室温に冷やし、溶媒を蒸発させた。残留物をクロマトグラフィー(シリカゲル、20%のEtOAc/ヘキサン)によって精製して、6−tert−ブチル−8−フルオロ−2−[2−ヒドロキシメチル−3−(2−メトキシ−ピリジン−4−イル)−フェニル]−2H−フタラジン−1−オン(60mg、62%)を白色の固体として与えた。MS:C25H25FN3O3[(M+H)+]の理論値434、実測値434。
CH3CN中の6−tert−ブチル−8−フルオロ−2−[2−ヒドロキシメチル−3−(2−メトキシ−ピリジン−4−イル)−フェニル]−2H−フタラジン−1−オン(50mg、0.12mmol)、トリメチルシリルクロリド(2当量)及びNaI(1当量)の混合物を還流温度で2時間加熱し、次に室温に冷やした。10%Na2S2O3水溶液を加え、混合物を飽和NaHCO3水溶液の添加によって塩基性にした。混合物を10%のMeOH/CH2Cl2で抽出し、有機抽出物を乾燥させ(Na2SO4)、濾過し、蒸発させた。残留物をクロマトグラフィー(シリカゲル、MeOH/CH2Cl2)によって精製して、6−tert−ブチル−8−フルオロ−2−[2−ヒドロキシメチル−3−(2−オキソ−1,2−ジヒドロ−ピリジン−4−イル)−フェニル]−2H−フタラジン−1−オン(23mg、48%)を白色の固体として与えた。MS:C24H23FN3O3[(M+H)+]の理論値420、実測値420.4。1H NMR(400 MHz, DMSO-d6) δ ppm 11.66(br. s. , 1 H), 8.51(s, 1 H), 7.87(s, 1 H), 7.74(d, J=13.7 Hz, 1 H), 7.50 - 7.56(m, 1 H), 7.36 - 7.48(m, 3 H), 6.38(s, 1 H), 6.30(d, J=6.8 Hz, 1 H), 4.71(t, J=5.1 Hz, 1 H), 4.29(br. s., 2 H), 1.38(s, 9 H).
6−tert−ブチル−2−[2,6−ジフルオロ−4−(2−オキソ−1,2−ジヒドロ−ピリジン−4−イル)−ベンジル]−8−フルオロ−2H−フタラジン−1−オン
CCl4(15mL)中の5−ブロモ−1,3−ジフルオロ−2−メチルベンゼン(Oakwood Products, Inc., 1741 Old Dunbar Road, West Columbia, SC 29172, USA米国から入手可能;1.0g、4.83mmol)、N−ブロモスクシンイミド(0.86g、4.83mmol)及びAIBN(40mg、0.24mmol)の混合物を還流温度で一晩加熱した。混合物をセライトを通して濾過し、溶媒を蒸発させた。残留物をクロマトグラフィー(シリカゲル、2%のEtOAc/ヘキサン)によって精製して、5−ブロモ−2−ブロモメチル−1,3−ジフルオロ−ベンゼン(1.1g、79%)を無色の液体として与えた。
DMF(1mL)中のNaH(60%、182mg、4.54mmol)の撹拌した懸濁液に、DMF(1.5mL)中の6−tert−ブチル−8−フルオロ−2H−フタラジン−1−オン(これは、Berthel, S. et al. US 20100222325 Column 139に記載されているように調製してもよい;500mg、2.27mmol)の溶液を0℃で滴下した。混合物を70℃に加熱し、30分間撹拌した。混合物を室温に冷やし、DMF(1.5mL)中の5−ブロモ−2−ブロモメチル−1,3−ジフルオロ−ベンゼン(715mg、2.5mmol)の溶液を加え、混合物を室温で4時間撹拌した。混合物を冷やし、冷水(5mL)を加えた。混合物をEtOAcで抽出し、有機抽出物を乾燥させ、蒸発させた。残留物をクロマトグラフィー(シリカゲル、5〜10%のEtOAc/ヘキサン)によって精製して、2−(4−ブロモ−2,6−ジフルオロ−ベンジル)−6−tert−ブチル−8−フルオロ−2H−フタラジン−1−オン(555mg、57%)を黄色の固体として与えた。MS:C19H17BrF3N2O[(M+H)+]の理論値425、実測値425。
DME(1.6mL)中の2−(4−ブロモ−2,6−ジフルオロ−ベンジル)−6−tert−ブチル−8−フルオロ−2H−フタラジン−1−オン(550mg、1.29mmol)の溶液を、アルゴンで10分間パージした。Pd(PPh3)4(14mg、0.01mmol)を加え、混合物をアルゴンで10分間パージした。Na2CO3水溶液(2M;1.3mL、2.6mmol)を加え、管をアルゴンで5分間パージした。溶液を室温で5分間撹拌し、EtOH(1.6mL)中の2−メトキシ−ピリジン−4−ボロン酸(Oakwood Products, Inc., 1741 Old Dunbar Road, West Columbia, SC 29172, USAから入手可能;245mg、1.61mmol)の溶液を加えた。混合物をアルゴンで10分間パージし、蓋をし、90℃で1時間加熱した。混合物をセライトを通して濾過し、セライトをCH2Cl2で洗浄した。濾液を乾燥させ(Na2SO4)、濾過し、蒸発させた。残留物をクロマトグラフィー(シリカゲル、15%のEtOAc/ヘキサン)によって精製して、6−tert−ブチル−2−[2,6−ジフルオロ−4−(2−メトキシ−ピリジン−4−イル)−ベンジル]−8−フルオロ−2H−フタラジン−1−オン(420mg、71%)を黄色の固体として与えた。MS:C25H23F3N3O2[(M+H)+]の理論値454、実測値454。
CH2Cl2(15mL)中の6−tert−ブチル−2−[2,6−ジフルオロ−4−(2−メトキシ−ピリジン−4−イル)−ベンジル]−8−フルオロ−2H−フタラジン−1−オン(200mg、0.44mmol)の溶液を0℃に冷却した。BBr3(CH2Cl2中1M;4.4mL、4.4mmol)を加え、混合物を室温で4時間撹拌した。混合物をもう一度0℃に冷却し、BBr3の更なる部分(CH2Cl2中1M;4.4mL、4.4mmol)を加えた。混合物を室温で48時間撹拌し、CH2Cl2(25mL)を加えた。混合物を飽和NaHCO3水溶液で洗浄し、有機層を乾燥させ(Na2SO4)、濾過し、蒸発させた。残留物をクロマトグラフィー(シリカゲル、2〜5%のMeOH/CH2Cl2)によって精製して、6−tert−ブチル−2−[2,6−ジフルオロ−4−(2−オキソ−1,2−ジヒドロ−ピリジン−4−イル)−ベンジル]−8−フルオロ−2H−フタラジン−1−オン(62mg、32%)をオフホワイトの固体として与えた。MS:C24H21F3N3O2[(M+H)+]の理論値440、実測値440。1H NMR(400 MHz, CDCl3) δ ppm 11.67(br. s., 1 H), 8.05(d, J=2.4 Hz, 1 H), 7.37 - 7.42(m, 3 H), 7.11(d, J=7.8 Hz, 2 H), 6.71(s, 1 H), 6.37 - 6.47(m, 1 H), 5.49(s, 2 H), 1.56(s, 18 H), 1.36(s, 9 H).
6−tert−ブチル−8−フルオロ−2−[2−ヒドロキシメチル−4−(2−オキソ−1,2−ジヒドロ−ピリジン−4−イル)−ベンジル]−2H−フタラジン−1−オン
塩化チオニル(10mL)を5−ブロモ−2−メチル−安息香酸(2.0g、9.3mmol)に0℃で加え、次にDMF(1滴)を加えた。混合物を窒素下に還流温度で3時間加熱した。反応混合物を蒸発させ、乾燥MeOH(5mL)を残留物に加えた。混合物を濃縮し、EtOAcを加えた。混合物を飽和NaHCO3水溶液及びブラインで洗浄した。有機相を乾燥させ(Na2SO4)、濾過し、蒸発させて、5−ブロモ−2−メチル−安息香酸メチルエステル(2.1g、98%)をオフホワイトの固体として与えた。
CCl4(10mL)中の5−ブロモ−2−メチル−安息香酸メチルエステル(500mg、2.19mmol)、N−ブロモスクシンイミド(389mg、2.18mmol)及びAIBN(18mg、0.11mmol)の混合物を還流温度で一晩加熱した。反応混合物を冷やし、5%Na2S2O3水溶液(5mL)を加え、混合物を濃縮した。残留物をEtOAc(30mL)で希釈し、得られた混合物を水(5mL)及びブライン(5mL)で洗浄した。有機相を乾燥させ(Na2SO4)、濾過し、蒸発させた。残留物をクロマトグラフィー(シリカゲル、0〜1%のEtOAc/ヘキサン)によって精製して、5−ブロモ−2−ブロモメチル−安息香酸メチルエステル(600mg、89%)を与えた。
トルエン(1.4mmol)中の5−ブロモ−2−ブロモメチル−安息香酸メチルエステル(200mg、0.65mmol)の溶液を、トルエン(1.4mL)中のDIBAL−H(トルエン中25%w/w;0.87mL、1.3mmol)の溶液に窒素下、0℃で滴下した。反応混合物を0℃で2時間撹拌し、次に1M HClの添加によってpH1に酸性化した。混合物をEtOAc(3×25mL)で抽出し、合わせた有機抽出物をブラインで洗浄し、乾燥させ(Na2SO4)、濾過し、蒸発させて、(5−ブロモ−2−ブロモメチル−フェニル)−メタノール(143mg、78%)をオフホワイトの固体として与えた。
NaH(60%、80mg、2mmol)を、DMF(2mL)中の6−tert−ブチル−8−フルオロ−2H−フタラジン−1−オン(これは、Berthel, S. et al. US 20100222325 Column 139に記載されているように調製してもよい;220mg、1mmol)の溶液に0℃で加えた。混合物を0℃で5分間撹拌し、次に70℃で30分間加熱した。混合物を室温に冷やし、(5−ブロモ−2−ブロモメチル−フェニル)−メタノール(140mg、0.5mmol)の溶液を加え、混合物を室温で1時間撹拌した。氷水(2mL)を加えた。混合物をEtOAc(3×25mL)で抽出した。EtOAc抽出物を水(3×5mL)及びブライン(5mL)で洗浄し、乾燥させ(Na2SO4)、濾過し、蒸発させた。残留物を分取HPLCによって精製して、2−(4−ブロモ−2−ヒドロキシメチル−ベンジル)−6−tert−ブチル−8−フルオロ−2H−フタラジン−1−オン(62mg、15%)を黄色のガム状物として与えた。MS:C20H21BrFN2O2[(M+H)+]の理論値419、実測値418.8。
20%水性ジオキサン(11mL)中の2−(4−ブロモ−2−ヒドロキシメチル−ベンジル)−6−tert−ブチル−8−フルオロ−2H−フタラジン−1−オン(60mg、0.143mmol)の撹拌した溶液に、2−メトキシ−ピリジン−4−ボロン酸(Oakwood Products, Inc., 1741 Old Dunbar Road, West Columbia, SC 29172, USAから入手可能;26mg、0.17mmol)、K2CO3(39mg、0.29mmol)、及びトリシクロヘキシルホスフィン(2mg、0.006mmol)を加えた。混合物をアルゴンで20分間パージし、Pd2(dba)3(5mg、0.006mmol)を加えた。混合物を100℃で4時間加熱した。混合物を濃縮し、EtOAc(30mL)を加えた。得られた混合物を水(3×5mL)及びブライン(5mL)で洗浄した。有機層を乾燥させ(Na2SO4)、濾過し、蒸発させた。残留物をクロマトグラフィー(シリカゲル、10%のEtOAc/ヘキサン)によって精製して、6−tert−ブチル−8−フルオロ−2−[2−ヒドロキシメチル−4−(2−メトキシ−ピリジン−4−イル)−ベンジル]−2H−フタラジン−1−オン(54mg、84%)を黄色のガム状物として与えた。MS:C26H26FN3O3[(M+H)+]の理論値448、実測値447.8。
CH3CN(15mL)中の6−tert−ブチル−8−フルオロ−2−[2−ヒドロキシメチル−4−(2−メトキシ−ピリジン−4−イル)−ベンジル]−2H−フタラジン−1−オン(260mg、0.58mmol)、トリメチルシリルクロリド(0.12g、1.1mmol)及びNaI(122mg、0.81mmol)の混合物を窒素下に還流温度で3時間加熱し、次に室温に冷やした。5%Na2S2O3水溶液(5mL)を加え、混合物を飽和NaHCO3水溶液(5mL)の添加によって塩基性にした。混合物を濃縮し、EtOAc(50mL)を加えた。得られた混合物を水(5mL)及びブラインで洗浄し、乾燥させ(Na2SO4)、濾過し、蒸発させた。残留物をクロマトグラフィー(シリカゲル、2〜5%のMeOH/CH2Cl2)によって精製して、6−tert−ブチル−8−フルオロ−2−[2−ヒドロキシメチル−4−(2−オキソ−1,2−ジヒドロ−ピリジン−4−イル)−ベンジル]−2H−フタラジン−1−オン(60mg、24%)をオフホワイトの固体として与えた。MS:C24H23FN3O3[(M+H)+]の理論値434、実測値434.4。1H NMR(400 MHz, DMSO-d6) δ ppm 11.59(br. s. , 1 H), 8.46(d, J=2.4 Hz, 1 H), 7.81(d, J=1.5 Hz, 1 H), 7.69 - 7.76(m, 2 H), 7.50(dd, J=8.3, 2.0 Hz, 1 H), 7.44(d, J=6.8 Hz, 1 H), 7.10(d, J=7.8 Hz, 1 H), 6.53(s, 1 H), 6.46(d, J=7.3 Hz, 1 H), 5.36(s, 2 H), 5.29(t, J=5.4 Hz, 1 H), 4.72(d, J=5.4 Hz, 2 H), 1.36(s, 9 H).
6−tert−ブチル−8−フルオロ−2−{2−フルオロ−4−[6−(1−メチル−1H−ピラゾール−4−イル)−2−オキソ−1,2−ジヒドロ−ピリジン−4−イル]−ベンジル}−2H−フタラジン−1−オン
CCl4(30mL)中の4−ブロモ−2−フルオロ−1−メチルベンゼン(Aldrichから入手可能;2.0g、10.58mmol)、N−ブロモスクシンイミド(1.88g、10.58mmol)及びAIBN(87mg、0.53mmol)の混合物を窒素下に還流温度で4時間加熱した。反応混合物を室温に冷やし、5%Na2S2O3水溶液を加え、混合物を濃縮した。水(10mL)を加え、混合物をEtOAc(3×50mL)で抽出した。合わせた有機相を乾燥させ(Na2SO4)、濾過し、蒸発させた。残留物をクロマトグラフィー(シリカゲル、ヘキサン)によって精製して、4−ブロモ−1−ブロモメチル−2−フルオロ−ベンゼン(1.5g、53%)を無色の液体として与えた。
DMF(5mL)中のNaH(60%、473mg、11.8mmol)の懸濁液に、6−tert−ブチル−8−フルオロ−2H−フタラジン−1−オン(これは、Berthel, S. et al. US 20100222325 Column 139に記載されているように調製してもよい;1.3g、5.91mmol)の溶液を0℃で加えた。混合物を0℃で5分間撹拌し、窒素下、70℃で30分間加熱した。混合物を室温に冷やし、DMF(3mL)中の4−ブロモ−1−ブロモメチル−2−フルオロ−ベンゼン(1.74g、6.5mmol)の溶液を加え、混合物を室温で1.5時間撹拌した。冷水(5mL)を加えた。混合物をEtOAcで抽出し、有機抽出物を乾燥させ(Na2SO4)、蒸発させた。残留物をクロマトグラフィー(シリカゲル、20%のEtOAc/ヘキサン)によって精製して、2−(4−ブロモ−2−フルオロ−ベンジル)−6−tert−ブチル−8−フルオロ−2H−フタラジン−1−オン(1.0g、41%)を黄色の固体として与えた。MS:C19H18BrF2N2O[(M+H)+]の理論値407、実測値407.2。
ジオキサン(16mL)中の2−(4−ブロモ−2−フルオロ−ベンジル)−6−tert−ブチル−8−フルオロ−2H−フタラジン−1−オン(500mg、1.22mmol)、4,4,5,5,4’,4’,5’,5’−オクタメチル−[2,2’]ビ[[1,3,2]ジオキサボロラニル](Aldrichから入手可能;405mg、1.58mmol)、及び乾燥KOAc(361mg、3.67mmol)の溶液をアルゴン下に脱気した。X−Phos(Aldrichから入手可能;87mg、0.18mmol)及びPd(OAc)2(13mg、0.061mmol)を加え、混合物を95℃で40分間加熱し、浴温度を80℃に下げ、反応フラスコを、撹拌を維持しながら加熱浴から引き上げた。2,6−ジクロロ−4−ヨード−ピリジン(Aldrichから入手可能;300mg、1.09mmol)及びK2CO3(507mg、3.67mmol)を加えた。アルゴンで脱気しておいた水(3.4mL)を加えた。反応混合物をアルゴンで脱気した。トリシクロヘキシルホスフィン(51mg、0.18mmol)及びPd2(dba)3(56mg、0.061mmol)を加え、混合物を80℃で14時間撹拌した。混合物をセライトを通して濾過し、セライトをEtOAc(3×25mL)で洗浄した。濾液を蒸発させCH2Cl2(50mL)を加えた。得られた混合物を水及びブラインで洗浄した。有機層を乾燥させ(Na2SO4)、濾過し、蒸発させた。残留物をクロマトグラフィー(シリカゲル、30%のEtOAc/ヘキサン)によって精製して、6−tert−ブチル−2−[4−(2,6−ジクロロ−ピリジン−4−イル)−2−フルオロ−ベンジル]−8−フルオロ−2H−フタラジン−1−オン(260mg、44%)を黄色の固体として与えた。MS:C24H20Cl2F2N3O[(M+H)+]の理論値474、実測値474。
密閉可能な管中の、6−tert−ブチル−2−[4−(2,6−ジクロロ−ピリジン−4−イル)−2−フルオロ−ベンジル]−8−フルオロ−2H−フタラジン−1−オン(160mg、0.34mmol)、1−メチル−4−(4,4,5,5−テトラメチル−[1,3,2]ジオキサボロラン−2−イル)−1H−ピラゾール(Aldrichから入手可能;56mg、0.27mmol)、K2CO3(140mg、1.01mmol)及びDMF(3mL)の混合物をアルゴンで30分間パージした。Pd(PPh3)4(39mg、0.03mmol)を加え、混合物を10分間パージした。管を密閉し、100℃で16時間加熱した。反応混合物を冷やし、水(5mL)で希釈し、EtOAc(3×20mL)で抽出した。有機層を乾燥させ(Na2SO4)、濾過し、蒸発させた。残留物をクロマトグラフィー(シリカゲル、20〜50%のEtOAc/ヘキサン)によって精製して、6−tert−ブチル−2−{4−[2−クロロ−6−(1−メチル−1H−ピラゾール−4−イル)−ピリジン−4−イル]−2−フルオロ−ベンジル}−8−フルオロ−2H−フタラジン−1−オン(65mg、37%)を黄色のガム状物として与えた。MS:C28H25ClF2N5O[(M+H)+]の理論値520、実測値520。
6−tert−ブチル−2−{4−[2−クロロ−6−(1−メチル−1H−ピラゾール−4−イル)−ピリジン−4−イル]−2−フルオロ−ベンジル}−8−フルオロ−2H−フタラジン−1−オン(65mg、0.125mmol)と10M HCl(7mL)との混合物を130℃で96時間加熱した。反応混合物を濃縮し、氷水(5mL)を残留物に加えた。得られた混合物を飽和NaHCO3水溶液の添加によって中和した。混合物をEtOAc(3×25mL)で抽出した。合わせた有機層を乾燥させ(Na2SO4)、濾過し、蒸発させた。残留物を、分取HPLC(カラム=XTerra C18(250x19mm)10μ、流速=14.0mL/分、水、CH3CN中の5mM NH4OAc)によって精製して、6−tert−ブチル−8−フルオロ−2−{2−フルオロ−4−[6−(1−メチル−1H−ピラゾール−4−イル)−2−オキソ−1,2−ジヒドロ−ピリジン−4−イル]−ベンジル}−2H−フタラジン−1−オン(17mg、27%)をオフホワイトの固体として与えた。MS:C28H26F2N5O2[(M+H)+]の理論値502、実測値502.4。1H NMR(400 MHz, DMSO-d6) δ ppm 11.72(s, 1 H), 8.45(d, J=2.4 Hz, 1 H), 8.41(s, 1 H), 8.15(s, 1 H), 7.80(s, 1 H), 7.63 - 7.76(m, 2 H), 7.55(d, J=8.3 Hz, 1 H), 7.35(t, J=8.1 Hz, 1 H), 6.86(br. s., 1 H), 6.46(s, 1 H), 5.38(s, 2 H), 3.87(s, 3 H), 1.36(s, 9 H).
4−tert−ブチル−N−[2−メチル−3−(2−オキソ−1,2−ジヒドロ−ピリジン−4−イル)−フェニル]−ベンズアミド
実施例I−1工程2に記載したものと類似の条件を使用して、4−ブロモ−2−メトキシ−ピリジン(Oakwood Products, Inc., 1741 Old Dunbar Road, West Columbia, SC 29172, USAから入手可能;500mg、2.26mmol)を2−メチル−3−ニトロフェニルボロン酸(Combi-Blocks Inc., 7949 Silverton Avenue, Suite 915, San Diego, CA 92126, USAから入手可能;60.4mg、0.49mmol)と反応させて、2−メトキシ−4−(2−メチル−3−ニトロ−フェニル)−ピリジン(420mg、65%)を白色の固体として与えた。
MeOH(10mL)中の2−メトキシ−4−(2−メチル−3−ニトロ−フェニル)−ピリジン(100mg、0.41mmol)と10%パラジウム−炭素(10mg)との混合物を、水素風船を室温で2時間使用して水素化した。混合物をセライトを通して濾過し、セライトをMeOHで洗浄した。合わせた濾液を蒸発させて、3−(2−メトキシ−ピリジン−4−イル)−2−メチル−フェニルアミン(80mg、91%)を白色の固体として与えた。MS:C13H15N2O[(M+H)+]の理論値215、実測値215。
CH2Cl2(15mL)中の4−tert−ブチル−安息香酸(Aldrichから入手可能;197mg、1.12mmol)の溶液、トリエチルアミン(0.14mL、2.8mmol)及びHATU(Aldrichから入手可能;532mg、1.4mmol)を室温で30分間撹拌した。3−(2−メトキシ−ピリジン−4−イル)−2−メチル−フェニルアミン(200mg、0.93mmol)を加え、混合物を室温で18時間撹拌した。飽和NaHCO3水溶液を加えた。有機相を分離し、乾燥させ(Na2SO4)、濾過し、蒸発させた。残留物をクロマトグラフィー(シリカゲル、10%のEtOAc/ヘキサン)によって精製して、4−tert−ブチル−N−[3−(2−メトキシ−ピリジン−4−イル)−2−メチル−フェニル]−ベンズアミド(110mg、31%)を黄色のガム状物として与えた。MS:C24H27N2O2[(M+H)+]の理論値375、実測値375。
0℃の1,2−ジクロロエタン(15mL)中の4−tert−ブチル−N−[3−(2−メトキシ−ピリジン−4−イル)−2−メチル−フェニル]−ベンズアミド(115mg、0.3mmol)の撹拌した溶液に、PBr3(97.7mg、0.89mmol)をゆっくりと加えた。混合物を還流温度で4時間加熱した。氷水を、続いて10%NaHCO3水溶液(5mL)を加えた。混合物を5%MeOH/CH2Cl2で抽出し、有機抽出物を乾燥させ(Na2SO4)、濾過し、蒸発させた。残留物をクロマトグラフィー(シリカゲル、5%のMeOH/CH2Cl2)によって精製して、4−tert−ブチル−N−[2−メチル−3−(2−オキソ−1,2−ジヒドロ−ピリジン−4−イル)−フェニル]−ベンズアミド(15mg、14%)を白色の固体として与えた。MS:C23H25N2O2[(M+H)+]の理論値361、実測値361。1H NMR(400 MHz, CDCl3) δ ppm 11.81(br. s. , 1 H), 7.96(d, J=7.8 Hz, 1 H), 7.84(d, J=8.3 Hz, 2 H), 7.71(s, 1 H), 7.53(d, J=8.8 Hz, 2 H), 7.30 - 7.39(m, 2 H), 7.08(d, J=7.3 Hz, 1 H), 6.52(s, 1 H), 6.26(dd, J=6.6, 1.7 Hz, 1 H), 2.25(s, 3 H), 1.36(s, 9 H).
6−tert−ブチル−8−フルオロ−2−[3−ヒドロキシメチル−4−(2−オキソ−1,2−ジヒドロ−ピリジン−4−イル)−ベンジル]−2H−フタラジン−1−オン
CH3CN(30mL)中の2−ブロモ−5−メチル−安息香酸(Aldrichから入手可能;1.0g、4.65mmol)、N−ブロモスクシンイミド(1.24g、6.98mmol)及び過酸化ベンゾイル(23mg、0.09mmol)の混合物を還流温度で一晩加熱した。反応混合物を濃縮し、残留物をクロマトグラフィー(シリカゲル、10%のEtOAc/ヘキサン)によって精製して、2−ブロモ−5−ブロモメチル−安息香酸(1.2g、87%)を明黄色の固体として与えた。MS:C8H7Br2O2[(M+H)+]の理論値293、実測値293。NMRはわずかな不純物の存在を示しているが、この物質を次の工程で直接使用した。
BH3−DMS(THF中2M溶液、6.1mL、12.2mmol)を、窒素下、THF(15mL)中の2−ブロモ−5−ブロモメチル−安息香酸(1.2g、4.08mmol)の0℃の溶液に加えた。混合物を室温で一晩撹拌した。MeOH及び氷水を加え、混合物を濃縮した。EtOAc(100mL)を加え、混合物を水(10mL)及びブライン(10mL)で洗浄した。合わせた有機層を乾燥させ(Na2SO4)、濾過し、蒸発させて、粗の(2−ブロモ−5−ブロモメチル−フェニル)−メタノール(1.2g)を白色の固体として与え、これを精製することなく次の工程で直接使用した。
CH2Cl2(30mL)中の粗の(2−ブロモ−5−ブロモメチル−フェニル)−メタノール(工程2から;0.95g、〜3.4mmol)の冷却した(0℃)溶液に、DIPEA(0.66g、5.1mmol)及びメチルクロロメチルエーテル(0.34g、4.24mmol)を窒素下で加えた。混合物を室温で一晩撹拌した。CH2Cl2(50mL)を加え、混合物をNaHCO3水溶液で洗浄した。合わせた有機抽出物を乾燥させ(Na2SO4)、濾過し、蒸発させた。残留物をクロマトグラフィー(シリカゲル、2〜5%のEtOAc/ヘキサン)によって精製して、1−ブロモ−4−ブロモメチル−2−メトキシメトキシメチル−ベンゼン(275mg、25%)を無色の液体として与えた。
DMF(3mL)中のNaH(60%、145mg、3.63mmol)の懸濁液に、DMF(2mL)中の6−tert−ブチル−8−フルオロ−2H−フタラジン−1−オン(これは、Berthel, S. et al. US 20100222325 Column 139に記載されているように調製してもよい;400.0mg、1.82mmol)の溶液を0℃で滴下した。混合物を室温で5分間撹拌し、次に70℃で30分間加熱した。混合物を室温に冷やし、DMF(2mL)中の1−ブロモ−4−ブロモメチル−2−メトキシメトキシメチル−ベンゼン(684mg、2mmol)の溶液を加え、混合物を室温で4時間撹拌した。水(5mL)を加え、混合物をEtOAc(3×20mL)で抽出した。合わせた有機層を水(3×5mL)及びブライン(5mL)で洗浄し、乾燥させ(Na2SO4)、濾過し、蒸発させた。残留物をクロマトグラフィー(シリカゲル、12%のEtOAc/ヘキサン)によって精製して、2−(4−ブロモ−3−メトキシメトキシメチル−ベンジル)−6−tert−ブチル−8−フルオロ−2H−フタラジン−1−オン(230mg、27%)を黄色のガム状物として与えた。MS:C22H25BrFN2O3[(M+H)+]の理論値463、実測値462.8。
密閉可能な管中のDME(1.5mL)中の2−(4−ブロモ−3−メトキシメトキシメチル−ベンジル)−6−tert−ブチル−8−フルオロ−2H−フタラジン−1−オン(185mg、0.4mmol)の溶液をアルゴンで10分間パージした。Pd(PPh3)4(5mg、0.004mmol)を加え、混合物をアルゴンで10分間パージした。Na2CO3水溶液(0.4mL、0.8mmol)を加え、管をアルゴンで5分間パージした。溶液を室温で5分間撹拌し、EtOH(1.5mL)中の2−メトキシ−ピリジン−4−ボロン酸(Oakwood Products, Inc., 1741 Old Dunbar Road, West Columbia, SC 29172, USAから入手可能;75mg、0.49mmol)の溶液を加えた。混合物をアルゴンで5分間パージし、蓋をし、90℃で1時間加熱した。混合物をセライトを通して濾過し、セライトをCH2Cl2で洗浄した。濾液を乾燥させ(Na2SO4)、濾過し、蒸発させた。残留物をクロマトグラフィー(シリカゲル、15%のEtOAc/ヘキサン)によって精製して、6−tert−ブチル−8−フルオロ−2−[3−メトキシメトキシメチル−4−(2−メトキシ−ピリジン−4−イル)−ベンジル]−2H−フタラジン−1−オン(65mg、33%)を黄色のガム状物として与えた。MS:C28H31FN3O4[(M+H)+]の理論値492、実測値492.2。
ジオキサン(10mL)中の6−tert−ブチル−8−フルオロ−2−[3−メトキシメトキシメチル−4−(2−メトキシ−ピリジン−4−イル)−ベンジル]−2H−フタラジン−1−オン(68mg、0.138mmol)の溶液を0℃に冷却し、ジオキサン中の4M HClの溶液(0.7mL、2.8mmol)を滴下した。混合物を室温で4時間撹拌し、次に溶媒を蒸発させた。飽和NaHCO3水溶液を加えてpHを8にし、混合物をEtOAc(3×15mL)で抽出した。合わせたEtOAc抽出物を乾燥させ(Na2SO4)、濾過し、蒸発させて、粗の6−tert−ブチル−8−フルオロ−2−[3−ヒドロキシメチル−4−(2−メトキシ−ピリジン−4−イル)−ベンジル]−2H−フタラジン−1−オン(37mg)をガム状物として与え、これを更に精製することなく次の工程で直接使用した。MS:C26H27FN3O3[(M+H)+]の理論値448、実測値448.0。
CH3CN(5mL)中の粗の6−tert−ブチル−8−フルオロ−2−[3−ヒドロキシメチル−4−(2−メトキシ−ピリジン−4−イル)−ベンジル]−2H−フタラジン−1−オン(工程6から;37mg、0.08mmol)、トリメチルシリルクロリド(0.017g、0.157mmol)及びNaI(17.4mg、0.12mmol)の混合物を還流温度で3時間加熱し、次に濃縮した。Na2S2O3水溶液(1mL)を、続いて飽和NaHCO3水溶液(2mL)を加えた。混合物をCH2Cl2(3×15mL)で抽出し、合わせた有機層を乾燥させ(Na2SO4)、濾過し、蒸発させた。残留物をクロマトグラフィー(シリカゲル、1〜2%のMeOH/CH2Cl2)によって精製して、6−tert−ブチル−8−フルオロ−2−[3−ヒドロキシメチル−4−(2−オキソ−1,2−ジヒドロ−ピリジン−4−イル)−ベンジル]−2H−フタラジン−1−オン(15mg、25%)をオフホワイトの固体として与えた。MS:C25H25FN3O3[(M+H)+]の理論値434、実測値434.0。1H NMR(400 MHz, CDCl3) δ ppm 10.78(s, 1 H), 8.11(d, J=2.4 Hz, 1 H), 7.64(s, 1 H), 7.39 - 7.49(m, 3 H), 6.53(s, 1 H), 6.30(d, J=5.9 Hz, 1 H), 5.39(s, 2 H), 4.61(s, 2 H), 1.37(s, 9 H).
6−tert−ブチル−8−フルオロ−2−[2−フルオロ−5−ヒドロキシメチル−4−(2−オキソ−1,2−ジヒドロ−ピリジン−4−イル)−ベンジル]−2H−フタラジン−1−オン
50%水性ジオキサン(96mL)中の2−ブロモ−4−フルオロ−5−メチル−ベンズアルデヒド(3B Scientific Corporation, 1840 Industrial Drive, Suite 160, Libertyville, IL 60048, USAから入手可能;1.0g、4.61mmol)、スルファミン酸(2.68g、27.65mmol)、亜塩素酸ナトリウム(539mg、6mmol)及びKH2PO4(7.52g、55.3mmol)の混合物を室温で一晩撹拌した。反応混合物を濃縮し、水(20mL)を加えた。得られた混合物をEtOAc(3×250mL)で抽出した。合わせた有機層を乾燥させ(Na2SO4)、濾過し、蒸発させて2−ブロモ−4−フルオロ−5−メチル−安息香酸(1.0g、93%)をオフホワイトの固体として与えた。MS:C8H7BrFO2[(M−H)−]の理論値231、実測値231.0。
CH3CN(10mL)中の2−ブロモ−4−フルオロ−5−メチル−安息香酸(0.5g、2.14mmol)、N−ブロモスクシンイミド(381mg、2.15mmol)及び過酸化ベンゾイル(10mg、0.04mmol)の混合物を還流温度で4時間加熱した。反応混合物を0℃に冷却し、5%の水性Na2S2O3を加えた。CH3CNを蒸発させ、残留物をEtOAc(3×100mL)で抽出した。合わせた有機相を乾燥させ(Na2SO4)、濾過し、蒸発させた。残留物をクロマトグラフィー(シリカゲル、2〜20%のEtOAc/ヘキサン)によって精製して、2−ブロモ−5−ブロモメチル−4−フルオロ−安息香酸(500mg、74%)を与えた。MS:C8H6Br2FO2[(M−H)−]の理論値309、実測値308.6。
BH3−DMS(THF中2M溶液、1.9mL、3.8mmol)を、THF(5mL)中の2−ブロモ−5−ブロモメチル−4−フルオロ−安息香酸(400mg、1.28mmol)の0℃の溶液に窒素下で加えた。混合物を室温で一晩撹拌した。MeOH及び氷水を加え、混合物を濃縮した。EtOAc(50mL)を加え、混合物を水(5mL)及びブライン(5mL)で洗浄した。合わせた有機層を乾燥させ(Na2SO4)、濾過し、蒸発させて(2−ブロモ−5−ブロモメチル−4−フルオロ−フェニル)−メタノール(320mg、84%)を白色の固体として与え、これを精製することなく次の工程で直接使用した。
NaH(60%、108mg、2.72mmol)を、DMF(3mL)中の6−tert−ブチル−8−フルオロ−2H−フタラジン−1−オン(これは、Berthel, S. et al. US 20100222325 Column 139に記載されているように調製してもよい;300.0mg、1.36mmol)の溶液に0℃で少しずつ加えた。混合物を70℃で30分間加熱した。混合物を室温に冷却し、DMF(2mL)中の(2−ブロモ−5−ブロモメチル−4−フルオロ−フェニル)−メタノール(203mg、0.68mmol)の溶液を加え、混合物を室温で2.5時間撹拌した。水(5mL)を加え、混合物をEtOAc(3×15mL)で抽出した。合わせた有機層を乾燥させ(Na2SO4)、濾過し、蒸発させた。残留物を分取HPLCによって精製して、2−(4−ブロモ−2−フルオロ−5−ヒドロキシメチル−ベンジル)−6−tert−ブチル−8−フルオロ−2H−フタラジン−1−オン(59mg、10%)を与えた。MS:C20H20BrF2N2O2[(M+H)+]の理論値437、実測値436.8。
20%水性ジオキサン(11mL)中の2−(4−ブロモ−2−フルオロ−5−ヒドロキシメチル−ベンジル)−6−tert−ブチル−8−フルオロ−2H−フタラジン−1−オン(55mg、0.13mmol)の撹拌した溶液に、2−メトキシ−ピリジン−4−ボロン酸(Oakwood Products, Inc., 1741 Old Dunbar Road, West Columbia, SC 29172, USA米国から入手可能;23mg、0.153mmol)、K2CO3(35mg、0.25mmol)、及びトリシクロヘキシルホスフィン(1mg、0.005mmol)を加えた。混合物をアルゴンで20分間パージし、Pd2(dba)3(5mg、0.005mmol)を加えた。混合物を100℃で4時間加熱した。混合物を濃縮し、EtOAc(30mL)を加えた。得られた混合物を水(3×5mL)及びブライン(5mL)で洗浄した。合わせた有機層を乾燥させ(Na2SO4)、濾過し、蒸発させた。残留物をクロマトグラフィー(シリカゲル、10%のEtOAc/ヘキサン)によって精製して、6−tert−ブチル−8−フルオロ−2−[2−フルオロ−5−ヒドロキシメチル−4−(2−メトキシ−ピリジン−4−イル)−ベンジル]−2H−フタラジン−1−オン(56mg、95%)を黄色のガム状物として与えた。MS:C26H26F2N3O3[(M+H)+]の理論値466、実測値466.0。
CH3CN(5mL)中の6−tert−ブチル−8−フルオロ−2−[2−フルオロ−5−ヒドロキシメチル−4−(2−メトキシ−ピリジン−4−イル)−ベンジル]−2H−フタラジン−1−オン(55mg、0.12mmol)、トリメチルシリルクロリド(24mg、0.22mmol)及びNaI(25mg、0.17mmol)の混合物を還流温度で3時間加熱し、次に蒸発させた。5%Na2S2O3水溶液(1mL)、続いて飽和NaHCO3水溶液(2mL)を加えた。混合物をCH2Cl2(3×15mL)で抽出した。合わせた有機抽出物を乾燥させ(Na2SO4)、濾過し、蒸発させた。残留物をクロマトグラフィー(シリカゲル、1〜5%のMeOH/CH2Cl2)によって精製して、6−tert−ブチル−8−フルオロ−2−[2−フルオロ−5−ヒドロキシメチル−4−(2−オキソ−1,2−ジヒドロ−ピリジン−4−イル)−ベンジル]−2H−フタラジン−1−オン(33mg、62%)をオフホワイトの固体として与えた。MS:C25H24F2N3O3[(M+H)+]の理論値452、実測値452.0。1H NMR(400 MHz, DMSO-d6) δ ppm 11.67(br. s. , 1 H), 8.46(d, J=2.4 Hz, 1 H), 7.81(s, 1 H), 7.74(d, J=13.2 Hz, 1 H), 7.37 - 7.44(m, 2 H), 7.12(d, J=10.3 Hz, 1 H), 6.28(s, 1 H), 6.19(d, J=6.4 Hz, 1 H), 5.36(s, 2 H), 5.20(t, J=5.4 Hz, 1 H), 4.32(d, J=5.4 Hz, 2 H), 1.36(s, 9 H).
2−tert−ブチル−5−{2−フルオロ−4−[6−(1−メチル−1H−ピラゾール−4−イル)−2−オキソ−1,2−ジヒドロ−ピリジン−4−イル]−ベンジル}−4,5−ジヒドロ−チエノ[2,3−c]ピロール−6−オン
MeOH(211mL)中の3−メチルチオフェン−2−カルボン酸(Aldrichから入手可能;15g、106mmol)の懸濁液を0℃に冷却した。濃硫酸(6mL、113mmol)を滴下し、混合物を室温で3日間撹拌した。反応混合物を濃縮し、残留物をEtOAcと飽和NaHCO3水溶液とに分配した。有機相を乾燥させ(Na2SO4)、濾過し、蒸発させて褐色の油状物を得たが、これは、NMRによると、所望のメチルエステル(84%)及び出発物質(16%)の混合物を含んでいた。粗生成物をEtOAcに溶解し、溶液を1M 水性NaOHで洗浄した。有機相を乾燥させ(Na2SO4)、濾過し、蒸発させて、3−メチル−チオフェン−2−カルボン酸メチルエステル(13.6g、82%)を明褐色の油状物として与えた。1H NMR(300 MHz, CDCl3) δ ppm 7.39(d, J=5.09 Hz, 1 H), 6.92(d, J=5.20 Hz, 1 H), 3.87(s, 3 H), 2.57(s, 3 H).
アルゴン下のAlCl3(17.3g、130mmol)とCH2Cl2(20mL)との混合物を−78℃に冷却した。CH2Cl2(10mL)中の3−メチルチオフェン−2−カルボン酸メチル(13.5g、86.4mmol)の溶液を5分間かけて滴下した。反応混合物を−78℃で5分間撹拌した。CH2Cl2(10mL)中の2−クロロ−2−メチルプロパン(Aldrichから入手可能;9.9mL、90.7mmol)の溶液を30分間かけて冷反応混合物に滴下した。反応混合物を室温温まるにまかせ、週末にかけて撹拌した。次に反応混合物を氷水に注いだ。有機相を分離し、乾燥させ(Na2SO4)、濾過し、濃縮した。残留物をクロマトグラフィー(シリカゲル、0〜5%のEtOAc/ヘキサン)によって精製して、5−tert−ブチル−3−メチル−チオフェン−2−カルボン酸メチルエステル(7.05g、38%)を黄色の油状物として与えた。1H NMR(300 MHz, CDCl3) δ ppm 6.68(s, 1 H), 3.84(s, 3 H), 2.50(s, 3 H), 1.38(s, 9 H).
3−メチル−チオフェン−2−カルボン酸メチルエステル(6.06g、28.5mmol)、N−ブロモスクシンイミド(6.1g、34.3mmol)、AIBN(234mg、1.43mmol)及びCCl4(80mL)の混合物を90℃で一晩加熱した。反応混合物を冷やし、濾過した。濾液を濃縮し、残留物をクロマトグラフィー(シリカゲル、5%のEtOAc/ヘキサン)によって精製して3−ブロモメチル−5−tert−ブチル−チオフェン−2−カルボン酸メチルエステル(2.65g、32%)を黄色の油状物として与えた。第1カラムからの混合画分を濃縮し、クロマトグラフィー(シリカゲル、5%のEtOAc/ヘキサン)によって精製して、3−ブロモメチル−5−tert−ブチル−チオフェン−2−カルボン酸メチルエステル(2.54g、30%)を与えた。1H NMR(300 MHz, CDCl3) δ ppm 6.93(s, 1 H), 4.87(s, 2 H), 3.88(s, 3 H), 1.39(s, 8 H).
3−ブロモメチル−5−tert−ブチル−チオフェン−2−カルボン酸メチルエステル(130mg、446μmol)、2−フルオロ−4−[2−メトキシ−6−(1−メチル−1H−ピラゾール−4−イル)−ピリジン−4−イル]−ベンジルアミン(これは、上記中間体の部の合成に記載したように調製してもよい;0.375g、1.2mmol)、Cs2CO3(390mg、1.2mmol)、及びアセトニトリル(7mL)の混合物を室温で週末にかけて撹拌した。反応混合物を濾過し、混合物をシリカゲルで濃縮した。残留物を80gのシリカゲルカラムに装填し、カラムを50〜100%のEtOAc/ヘキサンで溶離して、5−tert−ブチル−3−({2−フルオロ−4−[2−メトキシ−6−(1−メチル−1H−ピラゾール−4−イル)−ピリジン−4−イル]−ベンジルアミノ}−メチル)−チオフェン−2−カルボン酸メチルエステル(60mg、26%)を無色の油状物として与えた。MS:C28H32FN4O3S[(M+H)+]の理論値523、実測値523.3。
5−tert−ブチル−3−({2−フルオロ−4−[2−メトキシ−6−(1−メチル−1H−ピラゾール−4−イル)−ピリジン−4−イル]−ベンジルアミノ}−メチル)−チオフェン−2−カルボン酸メチルエステル(60mg、115μmol)、水酸化リチウム一水和物(25.3mg、574μmol)、THF(1mL)及び水(1mL)の混合物を60℃で18時間加熱した。反応混合物を室温に冷却し、THFを蒸発させた。水(10mL)を加えた。水性1M HClを、白色の懸濁液が形成されるまで加えた。混合物をEtOAcで抽出し、有機抽出物を乾燥させ(Na2SO4)、濾過し、蒸発させて、5−tert−ブチル−3−({2−フルオロ−4−[2−メトキシ−6−(1−メチル−1H−ピラゾール−4−イル)−ピリジン−4−イル]−ベンジルアミノ}−メチル)−チオフェン−2−カルボン酸(45mg、77%)を白色の固体として与えた。MS:C27H30FN4O3S[(M+H)+]の理論値509、実測値509.3。粗生成物を、更に精製することなく次の工程に用いた。
5−tert−ブチル−3−({2−フルオロ−4−[2−メトキシ−6−(1−メチル−1H−ピラゾール−4−イル)−ピリジン−4−イル]−ベンジルアミノ}−メチル)−チオフェン−2−カルボン酸(45mg、88.5μmol)、PYBOP(60mg、115μmol)、DIPEA(50μL、286μmol)、及びDMF(2.0mL)の混合物を室温で一晩撹拌した。溶媒を窒素流下で蒸発させた。EtOAc(10mL)を加え、溶液を飽和NaHCO3水溶液で洗浄した。有機相を乾燥させ(Na2SO4)、濾過し、蒸発させて2−tert−ブチル−5−{2−フルオロ−4−[2−メトキシ−6−(1−メチル−1H−ピラゾール−4−イル)−ピリジン−4−イル]−ベンジル}−4,5−ジヒドロ−チエノ[2,3−c]ピロール−6−オン(77mg、177%)を与えた。MS:C27H28FN4O2S[(M+H)+]の理論値491、実測値491.3。粗生成物を更に精製することなく次の工程で使用した。
CH3CN(2mL)中の粗の2−tert−ブチル−5−{2−フルオロ−4−[2−メトキシ−6−(1−メチル−1H−ピラゾール−4−イル)−ピリジン−4−イル]−ベンジル}−4,5−ジヒドロ−チエノ[2,3−c]ピロール−6−オン(粗、工程6から;77mg、およそ88.5μmol)、トリメチルシリルクロリド(40μL、0.32mmol)及びNaI(47mg、0.31mmol)の混合物を80℃で2時間加熱し、次に室温に冷やした。EtOAc(10mL)を加え、続いて1M Na2S2O3水溶液(5mL)を、続いて飽和NaHCO3水溶液(2mL)を加えた。有機相を乾燥させ(Na2SO4)、濾過し、蒸発させた。残留物をクロマトグラフィー(シリカゲル、1%NH4OAcを含む0〜5%のMeOH/CH2Cl2)によって精製して、2−tert−ブチル−5−{2−フルオロ−4−[6−(1−メチル−1H−ピラゾール−4−イル)−2−オキソ−1,2−ジヒドロ−ピリジン−4−イル]−ベンジル}−4,5−ジヒドロ−チエノ[2,3−c]ピロール−6−オン(35mg、47%)を明黄色の粉末として与えた。MS:C26H26FN4O2S[(M+1)+]の理論値477、実測値477.2。1H NMR(300 MHz, DMSO-d6) δ ppm 11.75(s, 2 H), 8.42(s, 1 H), 8.16(s, 1 H), 7.57 - 7.73(m, 3 H), 7.37(t, J=8.1 Hz, 1 H), 7.03(s, 1 H), 6.47(br. s., 1 H), 5.76(s, 1 H), 4.75(s, 2 H), 4.33(s, 2 H), 3.88(s, 3 H), 2.96 - 3.06(m, 3 H), 1.73(t, J=6.5 Hz, 3 H), 1.37(s, 9 H).
ブルトン型チロシンキナーゼ(Btk)阻害アッセイ
アッセイは、濾過による、放射活性33Pリン酸化製品の捕獲である。Btk、ビオチン化SH2ペプチド基質(Src相同性)、とATPとの相互作用は、ペプチド基質のリン酸化を導く。ビオチン化製品は、ストレプトアビジンセファロースビーズに結合する。全ての結合した放射標識製品を、シンチレーションカウンターにより検出する。
1)試料調製:試験化合物を、アッセイバッファー(イミダゾール、グリセロール−2−ホスファート、EGTA、MnCl2、MgCl2、BSA)中でhalf-log増加で希釈した。
2)ビーズ調製
a.)500gで遠心分離することにより、ビーズを洗浄する。
b.)ビーズを、PBS、及びEDTAと再構成して、20% ビーズスラリーを生成する。
3)基質を含まない反応混合物(アッセイバッファー、DTT、ATP、33P ATP)、及び基質を含む混合物(アッセイバッファー、DTT、ATP、33P ATP、ペプチド基質)を、30℃で15分間予めインキュベートする。
4)酵素バッファー(イミダゾール、グリセロール−2−ホスファート、BSA)中のBtk 10μL、及び試験化合物 10μLを、室温で10分間予めインキュベートして、アッセイを開始させる。
5)反応混合物 30μLを、基質と共に、又はなしで、Btk、及び化合物に加える。
6)トータルアッセイ混合物 50μLを、30℃で30分間インキュベートする。
7)アッセイ 40μLを、フィルタープレート中のビーズスラリー 150μLに移して、反応を止める。
8)30分後、フィルタープレートを、以下の工程で洗浄する。
a.NaCl 3×250μL
b.1% リン酸を含有するNaCl 3×250μL
c.H2O 1×250μL
9)プレートを、65℃で1時間、又は室温で一晩、乾燥させる。
10)マイクロシンチ−20 50μLを加え、33P(cpm)をシンチレーションカウンターにてカウントする。
生データ(cpm)からパーセント活性を計算する。
パーセント活性=(試料−bkg)/(トータル活性−bkg)×100
1部位用量応答S字状モデルを用いて、パーセント活性からIC50を計算する。
y=A+((B−A)/(1+((x/C)D))))
x=化合物濃度、y=%活性、A=分、B=最大、C=IC50、D=1(ヒル傾斜)。
このBTK競合アッセイは、FRET(Forster/Fluorescence Resonance Energy Transfer)技術を用いて、ブルトン型チロシンキナーゼの不活性化状態について、化合物の能力(IC50)を測定する。BTK−Eu複合体を、氷上で1時間インキュベートし、その後、開始濃度50nM BTK-Bioease(商標):10nM Eu−ストレプトアビジン(Perkin- Elmer、カタログ番号AD0062)で用いた。アッセイバッファーは、20mM HEPES(pH7.15)、0.1mM DTT、10mM MgCl2、3% キナーゼ安定剤を含む、0.5mg/ml BSA(Fremont Biosolutions、カタログ番号STB−K02)からなっていた。1時間後、上記由来の反応混合物を、アッセイバッファー中10倍希釈して、5nM BTK:1nM Eu−ストレプトアビジン複合体(ドナーフルオロフォア)を作成した。次に、陰性対照でないBTK−Euのみを含む、0.11nM BTK−Euと0.11nM Kinase Tracer 178(Invitrogen、カタログ番号PV5593)の混合物18μlを、384ウェルの平底プレート(Greiner、784076)に文注した。アッセイで試験すべき化合物を、10×濃度として調製し、half-log増加の連続希釈をDMSO中にて行い、10カ所曲線を作成した。FRET反応を開始するために、DMSO中の10×ストックとして調製した化合物をプレートに加え、プレートを、14℃で18〜24時間インキュベートした。
%最大FRET=100×[(FSR化合物−FSR平均最小値)/(FSR平均最大値−FSR平均最小値)]
(式中、FSR=FRETシグナル比)。%最大FRET曲線を、活性ベースでプロットし(Excel)、IC50(%)、ヒル傾斜、z’、及び%CVを決定した。平均IC50、及び標準偏差を、デュプリケート曲線(2つの独立した希釈由来の一重項阻害曲線)から、Microsoft Excelを用いて、もたらす。
ヒト血液中のB細胞のB細胞受容体により仲介される活性化を抑制する、Btk阻害剤の能力を試験する方法は、以下の通りである。
以下の制限:24時間薬物なし、非喫煙者の、健常ボランティアから、ヒト全血(HWB)を得る。静脈穿刺により、ヘパリンナトリウムで抗凝固剤処理したVacutainer管に血液を集める。試験化合物を10倍希釈し、PBS中の所望の出発薬物濃度(20×)、続いて連続3倍PBS中の10% DMSOを希釈して、9カ所の用量応答曲線を作成する。各化合物希釈液 5.5μlを、デュプリケートで、2mlの96ウェルV底プレート(Analytical Sales and Services、番号59623−23)に加え;PBS中の10% DMSO 5.5μlを、対照ウェル、及び非刺激ウェルに加える。HWB(100μl)をそれぞれのウェルに加え、混合後、プレートを、37℃、5% CO2、湿度 100%で、30分間、インキュベートする。ヤギF(ab’)2抗ヒトIgM(Southern Biotech、番号2022−14)(500μg/ml 溶液 10μl、終濃度50μg/ml)を、混合しながら、それぞれのウェル(非刺激ウェル以外)に加え、プレートを更に20時間インキュベートする。
本発明の化合物によるB細胞の活性化の阻害を、抗IgMにより刺激されるB細胞応答に対する試験化合物の作用を決定することにより、示す。
成長培地:L−グルタミン(Invitrogen、カタログ番号61870−010)、10% ウシ胎児血清(FBS、Summit Biotechnology、カタログ番号FP−100−05);1mM ピルビン酸ナトリウム(Invitrogen、カタログ番号11360−070)を含むRPMI1640培地。
最高の最終アッセイ濃度 100μMを達成するために、10mM 化合物ストック溶液(DMSO中に作成) 24μLを、FLIPRバッファー576μLに直接加える。試験化合物を、FLIPRバッファー(Biomek 2000 robotic pipettorを用いる)中に希釈し、以下の希釈スキーム:ビークル、1.00×10−4、1.00×10−5、3.16×10−6、1.00×10−6、3.16×10−7、1.00×10−7、3.16×10−8Mで得る。
カルシウムの細胞内増加を、最大−最小統計(Molecular Devices FLIPR対照を用いて、刺激抗体の添加により引き起こされるピークから、休止ベースラインを引くこと)、及び統計的エクスポートソフトウェアを用いて報告する。non-linear curve fit(GraphPad Prism software)を用いて、IC50を決定した。
0日目に、マウスの尾部、又は背中に、フロイント完全アジュバント(CFA)中のII型コラーゲン(i.d.)の乳剤を注射する。コラーゲン免疫後、動物は、およそ21〜35日目に、関節炎を発症する。21日目に、フロイント不完全アジュバント(IFA;i.d.)中のコラーゲンの全身投与により、関節炎の発症はを同期した(ブーストした)。ブーストに対するシグナルである、中程度の関節炎(スコア1又は2;以下のスコア記載を参照)の任意の発症について、20日後、動物を毎日調べた。ブースト後、マウスをスコア化し、候補治療剤で予め規定した時間(典型的には、2〜3週間)投与し、投与頻度は、毎日(QD)、又は1日2回(BID)である。
0日目に、フロイント不完全アジュバント(IFA)中のウシII型コラーゲンの乳剤を、背中のいくつかの位置に、皮内注射(i.d.)した。およそ7日目に、尾部又は背中の別の部位に、コラーゲン乳剤のブースター注射を行った。一般的に、最初のコラーゲン注射の12〜14日後に、関節炎を観察する。後述(関節炎の評価)の通り、14日以降、関節炎の発症について、動物を評価する。2回目の投与時に開始する予防的方法で、及び予め規定した時間(典型的には、2〜3週)、毎日(QD)、又は1日2回(BID)の投与頻度で、動物に、候補治療剤を投与する。
両方のモデルにおいて、足及び下肢関節の炎症を、後述の基準に従い、4本の足の評価を含む、スコア決定システムを用いて、定量する。
スコア決定: 1=足又は1つの指の腫れ、及び/又は赤み
2=2つより多い関節での腫れ
3=2つより多い病変関節を伴った、足のひどい腫れ
4=足及び指全体の重篤な関節炎。
ベースラインの測定のため、評価を0日目に行い、評価を、最初の兆候又は腫れの時点で再開し、実験の終わりまで、1週間当たり最大3回行う。それぞれのマウスの関節炎指標を、個々の足の4つのスコアを加えること(1匹の動物あたり最大16となる)により、得る。
オスBrown-Norwaymラットを、ミョウバン0.2ml中のOA(オブアルブミン)100μgで、1週間に1回、3週間(0、7、及び14日)腹腔内感作する。21日目(最終感作の1週間後)に、ラットに、ビークル、又は化合物製剤のいずれかを1日4回皮下投与し、0.5時間後、OAエアロゾル投与(1%OA、45分間)し、投与の4又は24時間後に終えた。屠殺時に、血清、及び血漿を、それぞれ、血清学、及びPKのため、全ての動物から採取した。気管カニューレを挿入し、肺を、3×PBSで洗浄する。BAL体液を、トータルの白血球数、及び特異な白血球カウントについて分析する。細胞アリコート(20〜100μl)中のトータルの白血球数を、Coulter Counterにより決定する。特異な白血球カウントのため、試料 50〜200μlを、Cytospinにて遠心分離し、Diff-Quikでスライド染色する。単球、好酸球、好中球、及びリンパ球の割合を、標準的形態学的基準を用いて、光学顕微鏡検査下でカウントし、パーセントとして表す。Btkの代表的阻害剤は、対照レベルと比較して、OA感作及び投与ラットのBAL中のトータル白血球カウントの低減を示す。
Claims (9)
- 式I
(式中:
Aは、6−tert−ブチル−8−フルオロ−1−オキソ−2H−フタラジン−2-イルであり;
それぞれのR1は、独立して、F、又はヒドロキシメチルであり;
mは、1であり;
R2は、メチルピラゾリルであり;
nは、0であり;
Xは、CH2である)
の化合物、又はその薬学的に許容し得る塩。 - 6−tert−ブチル−8−フルオロ−2−[4−(2−オキソ−1,2−ジヒドロ−ピリジン−4−イル)−ベンジル]−2H−フタラジン−1−オン;
6−tert−ブチル−8−フルオロ−2−[2−フルオロ−4−(2−オキソ−1,2−ジヒドロ−ピリジン−4−イル)−ベンジル]−2H−フタラジン−1−オン;
6−tert−ブチル−8−フルオロ−2−[2−ヒドロキシメチル−3−(2−オキソ−1,2−ジヒドロ−ピリジン−4−イル)−フェニル]−2H−フタラジン−1−オン;
6−tert−ブチル−2−[2,6−ジフルオロ−4−(2−オキソ−1,2−ジヒドロ−ピリジン−4−イル)−ベンジル]−8−フルオロ−2H−フタラジン−1−オン;
6−tert−ブチル−8−フルオロ−2−[2−ヒドロキシメチル−4−(2−オキソ−1,2−ジヒドロ−ピリジン−4−イル)−ベンジル]−2H−フタラジン−1−オン;
6−tert−ブチル−8−フルオロ−2−{2−フルオロ−4−[6−(1−メチル−1H−ピラゾール−4−イル)−2−オキソ−1,2−ジヒドロ−ピリジン−4−イル]−ベンジル}−2H−フタラジン−1−オン;
6−tert−ブチル−8−フルオロ−2−[3−ヒドロキシメチル−4−(2−オキソ−1,2−ジヒドロ−ピリジン−4−イル)−ベンジル]−2H−フタラジン−1−オン;及び
6−tert−ブチル−8−フルオロ−2−[2−フルオロ−5−ヒドロキシメチル−4−(2−オキソ−1,2−ジヒドロ−ピリジン−4−イル)−ベンジル]−2H−フタラジン−1−オン
からなる群より選択される化合物、又はその薬学的に許容し得る塩。 - 少なくとも1個の薬学的に許容し得る担体、賦形剤、又は希釈剤と混合された、請求項1又は2記載の化合物を含む、医薬組成物。
- 炎症状態、及び/又は自己免疫状態を処置するための請求項3記載の医薬組成物。
- 炎症状態を処置するための請求項3記載の医薬組成物。
- 関節リウマチを処置するための請求項3記載の医薬組成物。
- 喘息を処置するための請求項3記載の医薬組成物。
- 炎症状態、及び/又は自己免疫状態の処置用医薬の製造のための、請求項1又は2記載の化合物の使用。
- 炎症状態、及び/又は自己免疫状態の処置において使用するための、請求項1又は2記載の化合物。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201261727130P | 2012-11-16 | 2012-11-16 | |
| US61/727,130 | 2012-11-16 | ||
| PCT/EP2013/073667 WO2014076104A1 (en) | 2012-11-16 | 2013-11-13 | Inhibitors of bruton's tyrosine kinase |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2015537016A JP2015537016A (ja) | 2015-12-24 |
| JP6088063B2 true JP6088063B2 (ja) | 2017-03-01 |
Family
ID=49584724
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2015542251A Expired - Fee Related JP6088063B2 (ja) | 2012-11-16 | 2013-11-13 | ブルトン型チロシンキナーゼの阻害剤 |
Country Status (12)
| Country | Link |
|---|---|
| US (1) | US9458105B2 (ja) |
| EP (1) | EP2920162B1 (ja) |
| JP (1) | JP6088063B2 (ja) |
| KR (1) | KR101713465B1 (ja) |
| CN (1) | CN104812746B (ja) |
| AR (1) | AR093478A1 (ja) |
| BR (1) | BR112015010693A2 (ja) |
| CA (1) | CA2890671A1 (ja) |
| MX (1) | MX2015006162A (ja) |
| RU (1) | RU2015120216A (ja) |
| TW (1) | TW201425297A (ja) |
| WO (1) | WO2014076104A1 (ja) |
Families Citing this family (26)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20190330226A1 (en) * | 2018-04-26 | 2019-10-31 | Vanderbilt University | Positive allosteric modulators of the muscarinic acetylcholine receptor m1 |
| CN109336864A (zh) * | 2018-10-04 | 2019-02-15 | 南京先进生物材料与过程装备研究院有限公司 | 酞嗪酮类化合物晶型n及其制备方法 |
| CN109180643A (zh) * | 2018-10-04 | 2019-01-11 | 南京先进生物材料与过程装备研究院有限公司 | 酞嗪酮类化合物晶型a及其制备方法 |
| CN109485636A (zh) * | 2018-10-04 | 2019-03-19 | 南京先进生物材料与过程装备研究院有限公司 | 一种新型btk激酶抑制剂的盐酸盐及其制备方法与用途 |
| CN109180642A (zh) * | 2018-10-04 | 2019-01-11 | 南京先进生物材料与过程装备研究院有限公司 | 酞嗪酮类btk抑制剂及其应用 |
| CN109369619A (zh) * | 2018-10-04 | 2019-02-22 | 南京先进生物材料与过程装备研究院有限公司 | 酞嗪酮类化合物晶型b及其制备方法 |
| CN109180644A (zh) * | 2018-10-04 | 2019-01-11 | 南京先进生物材料与过程装备研究院有限公司 | 一种新型btk激酶抑制剂的甲磺酸盐及其制备方法与用途 |
| CN109172592A (zh) * | 2018-10-04 | 2019-01-11 | 南京先进生物材料与过程装备研究院有限公司 | 一种抗肿瘤药物组合物 |
| CN109293635A (zh) * | 2018-10-04 | 2019-02-01 | 南京先进生物材料与过程装备研究院有限公司 | 一种新型btk激酶抑制剂的p晶型及其制备方法 |
| CN109336863A (zh) * | 2018-10-04 | 2019-02-15 | 南京先进生物材料与过程装备研究院有限公司 | 一种新型酞嗪酮类btk抑制剂、制备及其应用 |
| CN109288830A (zh) * | 2018-10-04 | 2019-02-01 | 南京先进生物材料与过程装备研究院有限公司 | 一种紫杉醇和新型酞嗪酮类化合物联合用药物组合物 |
| CN109276571A (zh) * | 2018-10-31 | 2019-01-29 | 南京先进生物材料与过程装备研究院有限公司 | 一种紫杉醇和新型硝基酞嗪酮btk抑制剂联合用药物组合物及其应用 |
| CN109288841A (zh) * | 2018-10-31 | 2019-02-01 | 南京先进生物材料与过程装备研究院有限公司 | 一种紫杉醇和对苯氟酞嗪酮类btk抑制剂联合用药物组合物及其应用 |
| CN109331018A (zh) * | 2018-10-31 | 2019-02-15 | 南京先进生物材料与过程装备研究院有限公司 | 一种紫杉醇和硝基酞嗪酮btk抑制剂联合用药物组合物及其应用 |
| CN109481442A (zh) * | 2018-10-31 | 2019-03-19 | 南京先进生物材料与过程装备研究院有限公司 | 一种紫杉醇和邻苯基酞嗪酮类btk抑制剂联合用药物组合物及其应用 |
| CN109172562A (zh) * | 2018-10-31 | 2019-01-11 | 南京先进生物材料与过程装备研究院有限公司 | 一种紫杉醇和甲氧基酞嗪酮类btk抑制剂联合用药物组合物及其应用 |
| CN109481441A (zh) * | 2018-10-31 | 2019-03-19 | 南京先进生物材料与过程装备研究院有限公司 | 一种紫杉醇和新型甲氧基酞嗪酮类btk抑制剂联合用药物组合物及其应用 |
| CN109223759A (zh) * | 2018-10-31 | 2019-01-18 | 南京先进生物材料与过程装备研究院有限公司 | 一种紫杉醇和新型酞嗪酮类btk抑制剂联合用药物组合物及其应用 |
| CN109394766A (zh) * | 2018-10-31 | 2019-03-01 | 南京先进生物材料与过程装备研究院有限公司 | 一种紫杉醇和对苯硝基酞嗪酮类btk抑制剂联合用药物组合物及其应用 |
| CN109331005A (zh) * | 2018-10-31 | 2019-02-15 | 南京先进生物材料与过程装备研究院有限公司 | 一种紫杉醇和氟酞嗪类btk抑制剂联合用药物组合物及其应用 |
| CN109394765A (zh) * | 2018-10-31 | 2019-03-01 | 南京先进生物材料与过程装备研究院有限公司 | 一种紫杉醇和对苯基酞嗪酮类btk抑制剂联合用药物组合物及其应用 |
| CN109481444A (zh) * | 2018-10-31 | 2019-03-19 | 南京先进生物材料与过程装备研究院有限公司 | 一种紫杉醇和酞嗪类btk抑制剂联合用药物组合物及其应用 |
| CN109481443A (zh) * | 2018-10-31 | 2019-03-19 | 南京先进生物材料与过程装备研究院有限公司 | 一种紫杉醇和邻苯氟酞嗪酮类btk抑制剂联合用药物组合物及其应用 |
| ES3055177T3 (en) | 2019-10-30 | 2026-02-10 | Biogen Ma Inc | Condensed pyridazine or pyrimidine as btk inhibitors |
| CN116917279A (zh) * | 2020-08-07 | 2023-10-20 | 渤健马萨诸塞州股份有限公司 | Btk抑制剂 |
| US20240100172A1 (en) | 2020-12-21 | 2024-03-28 | Hangzhou Jijing Pharmaceutical Technology Limited | Methods and compounds for targeted autophagy |
Family Cites Families (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH1045750A (ja) * | 1995-09-20 | 1998-02-17 | Takeda Chem Ind Ltd | アゾール化合物、その製造方法及び用途 |
| GB0111078D0 (en) * | 2001-05-04 | 2001-06-27 | Novartis Ag | Organic compounds |
| WO2009098144A1 (en) * | 2008-02-05 | 2009-08-13 | F. Hoffmann-La Roche Ag | Novel pyridinones and pyridazinones |
| WO2009156284A1 (en) * | 2008-06-24 | 2009-12-30 | F. Hoffmann-La Roche Ag | Novel substituted pyridin-2-ones and pyridazin-3-ones |
| US8299077B2 (en) * | 2009-03-02 | 2012-10-30 | Roche Palo Alto Llc | Inhibitors of Bruton's tyrosine kinase |
| AR082590A1 (es) * | 2010-08-12 | 2012-12-19 | Hoffmann La Roche | Inhibidores de la tirosina-quinasa de bruton |
| US9249123B2 (en) * | 2010-09-01 | 2016-02-02 | Genentech, Inc. | Pyridinones/pyrazinones, method of making, and method of use thereof |
| CA2834077A1 (en) * | 2011-05-17 | 2012-11-22 | F. Hoffmann-La Roche Ag | Inhibitors of bruton's tyrosine kinase |
| BR112014003582A2 (pt) * | 2011-08-17 | 2017-03-14 | Hoffmann La Roche | inibidores da tirosina quinase de bruton |
| RU2622391C2 (ru) * | 2011-11-03 | 2017-06-15 | Ф. Хоффманн-Ля Рош Аг | Соединения 8-фторфталазин-1(2н)-она в качестве ингибиторов тирозинкиназы брутона |
| US8742098B2 (en) * | 2011-12-09 | 2014-06-03 | Hoffmann-La Roche Inc. | Inhibitors of Bruton's tyrosine kinase |
| RU2015111133A (ru) * | 2012-09-13 | 2016-11-10 | Ф. Хоффманн-Ля Рош Аг | Ингибиторы тирозинкиназы брутона |
| HK1218416A1 (zh) * | 2013-03-05 | 2017-02-17 | F. Hoffmann-La Roche Ag | 布鲁顿氏酪氨酸激酶抑制剂 |
| MX2015010820A (es) * | 2013-03-05 | 2015-11-30 | Hoffmann La Roche | Inhibidores de tirosina cinasa de bruton. |
| WO2015000949A1 (en) * | 2013-07-03 | 2015-01-08 | F. Hoffmann-La Roche Ag | Heteroaryl pyridone and aza-pyridone amide compounds |
-
2013
- 2013-11-13 MX MX2015006162A patent/MX2015006162A/es unknown
- 2013-11-13 CN CN201380059533.0A patent/CN104812746B/zh not_active Expired - Fee Related
- 2013-11-13 BR BR112015010693A patent/BR112015010693A2/pt not_active Application Discontinuation
- 2013-11-13 CA CA2890671A patent/CA2890671A1/en not_active Abandoned
- 2013-11-13 RU RU2015120216A patent/RU2015120216A/ru not_active Application Discontinuation
- 2013-11-13 KR KR1020157015815A patent/KR101713465B1/ko not_active Expired - Fee Related
- 2013-11-13 EP EP13791795.1A patent/EP2920162B1/en not_active Not-in-force
- 2013-11-13 US US14/441,711 patent/US9458105B2/en not_active Expired - Fee Related
- 2013-11-13 WO PCT/EP2013/073667 patent/WO2014076104A1/en not_active Ceased
- 2013-11-13 JP JP2015542251A patent/JP6088063B2/ja not_active Expired - Fee Related
- 2013-11-14 AR ARP130104183A patent/AR093478A1/es unknown
- 2013-11-15 TW TW102141777A patent/TW201425297A/zh unknown
Also Published As
| Publication number | Publication date |
|---|---|
| AR093478A1 (es) | 2015-06-10 |
| MX2015006162A (es) | 2015-08-14 |
| CN104812746A (zh) | 2015-07-29 |
| US20150291525A1 (en) | 2015-10-15 |
| JP2015537016A (ja) | 2015-12-24 |
| BR112015010693A2 (pt) | 2017-07-11 |
| CA2890671A1 (en) | 2014-05-22 |
| KR20150084061A (ko) | 2015-07-21 |
| KR101713465B1 (ko) | 2017-03-07 |
| CN104812746B (zh) | 2017-03-08 |
| WO2014076104A1 (en) | 2014-05-22 |
| HK1212689A1 (zh) | 2016-06-17 |
| EP2920162B1 (en) | 2017-04-19 |
| US9458105B2 (en) | 2016-10-04 |
| TW201425297A (zh) | 2014-07-01 |
| RU2015120216A (ru) | 2017-01-10 |
| EP2920162A1 (en) | 2015-09-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6088063B2 (ja) | ブルトン型チロシンキナーゼの阻害剤 | |
| KR101585753B1 (ko) | 브루톤 티로신 키나아제의 억제제 | |
| JP6139690B2 (ja) | ブルートンチロシンキナーゼの阻害剤 | |
| KR101763504B1 (ko) | 브루톤 티로신 키나아제의 억제제 | |
| KR20150054994A (ko) | 브루톤 티로신 키나아제의 억제제 | |
| JP6219523B2 (ja) | ブルトンチロシンキナーゼの阻害剤 | |
| KR20150113195A (ko) | 브루톤 티로신 키나아제의 억제제 | |
| US20180305340A1 (en) | Inhibitors of bruton's tyrosine kinase | |
| KR101707761B1 (ko) | 브루톤 티로신 키나아제 억제제로서의 티아졸 유도체 | |
| CA2902038C (en) | Inhibitors of bruton's tyrosine kinase | |
| HK1212689B (en) | Inhibitors of bruton's tyrosine kinase |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A529 | Written submission of copy of amendment under article 34 pct |
Free format text: JAPANESE INTERMEDIATE CODE: A529 Effective date: 20150714 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20150714 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20160719 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20160927 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20170110 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20170202 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 6088063 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| LAPS | Cancellation because of no payment of annual fees |