JP6025018B2 - 新規のアルギン酸資化菌、その細菌が産生するアルギン酸を分解する酵素を含む菌抽出液、それらを用いてオリゴ糖、不飽和単糖、ないしα−ケト酸を製造する方法 - Google Patents
新規のアルギン酸資化菌、その細菌が産生するアルギン酸を分解する酵素を含む菌抽出液、それらを用いてオリゴ糖、不飽和単糖、ないしα−ケト酸を製造する方法 Download PDFInfo
- Publication number
- JP6025018B2 JP6025018B2 JP2012067226A JP2012067226A JP6025018B2 JP 6025018 B2 JP6025018 B2 JP 6025018B2 JP 2012067226 A JP2012067226 A JP 2012067226A JP 2012067226 A JP2012067226 A JP 2012067226A JP 6025018 B2 JP6025018 B2 JP 6025018B2
- Authority
- JP
- Japan
- Prior art keywords
- alginic acid
- alginate
- choh
- acid
- chemical formula
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 229920000615 alginic acid Polymers 0.000 title claims description 236
- 235000010443 alginic acid Nutrition 0.000 title claims description 176
- 229960001126 alginic acid Drugs 0.000 title claims description 143
- 239000000783 alginic acid Substances 0.000 title claims description 143
- 102000004190 Enzymes Human genes 0.000 title claims description 141
- 108090000790 Enzymes Proteins 0.000 title claims description 141
- 150000004781 alginic acids Chemical class 0.000 title claims description 138
- 241000894006 Bacteria Species 0.000 title claims description 93
- 239000000284 extract Substances 0.000 title claims description 71
- 230000001580 bacterial effect Effects 0.000 title claims description 51
- 229920001542 oligosaccharide Polymers 0.000 title claims description 31
- 150000002482 oligosaccharides Chemical class 0.000 title claims description 30
- 150000002772 monosaccharides Chemical class 0.000 title description 74
- 150000004716 alpha keto acids Chemical class 0.000 title description 50
- 238000004519 manufacturing process Methods 0.000 title description 24
- 239000000126 substance Substances 0.000 claims description 100
- 238000000034 method Methods 0.000 claims description 80
- 230000000593 degrading effect Effects 0.000 claims description 48
- 108010004131 poly(beta-D-mannuronate) lyase Proteins 0.000 claims description 27
- 238000006731 degradation reaction Methods 0.000 claims description 16
- 230000000813 microbial effect Effects 0.000 claims description 16
- 230000015556 catabolic process Effects 0.000 claims description 13
- 229940088598 enzyme Drugs 0.000 description 135
- 239000000243 solution Substances 0.000 description 112
- 230000000694 effects Effects 0.000 description 101
- 229940072056 alginate Drugs 0.000 description 93
- 125000003275 alpha amino acid group Chemical group 0.000 description 75
- 238000006243 chemical reaction Methods 0.000 description 67
- 108090000856 Lyases Proteins 0.000 description 63
- 102000004317 Lyases Human genes 0.000 description 62
- 241000589565 Flavobacterium Species 0.000 description 60
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 52
- 108090000623 proteins and genes Proteins 0.000 description 50
- 235000010413 sodium alginate Nutrition 0.000 description 50
- 238000000354 decomposition reaction Methods 0.000 description 46
- IXPNQXFRVYWDDI-UHFFFAOYSA-N 1-methyl-2,4-dioxo-1,3-diazinane-5-carboximidamide Chemical compound CN1CC(C(N)=N)C(=O)NC1=O IXPNQXFRVYWDDI-UHFFFAOYSA-N 0.000 description 45
- 239000000661 sodium alginate Substances 0.000 description 45
- 229940005550 sodium alginate Drugs 0.000 description 45
- FHVDTGUDJYJELY-UHFFFAOYSA-N 6-{[2-carboxy-4,5-dihydroxy-6-(phosphanyloxy)oxan-3-yl]oxy}-4,5-dihydroxy-3-phosphanyloxane-2-carboxylic acid Chemical compound O1C(C(O)=O)C(P)C(O)C(O)C1OC1C(C(O)=O)OC(OP)C(O)C1O FHVDTGUDJYJELY-UHFFFAOYSA-N 0.000 description 33
- 238000010828 elution Methods 0.000 description 33
- 238000002835 absorbance Methods 0.000 description 32
- 235000001014 amino acid Nutrition 0.000 description 32
- 239000002609 medium Substances 0.000 description 30
- 235000018102 proteins Nutrition 0.000 description 30
- 102000004169 proteins and genes Human genes 0.000 description 30
- 229940024606 amino acid Drugs 0.000 description 29
- 239000002253 acid Substances 0.000 description 27
- 150000001413 amino acids Chemical class 0.000 description 26
- 239000011780 sodium chloride Substances 0.000 description 26
- 150000004804 polysaccharides Chemical class 0.000 description 24
- BFNBIHQBYMNNAN-UHFFFAOYSA-N ammonium sulfate Chemical compound N.N.OS(O)(=O)=O BFNBIHQBYMNNAN-UHFFFAOYSA-N 0.000 description 23
- 239000000758 substrate Substances 0.000 description 23
- 238000011534 incubation Methods 0.000 description 21
- 239000000047 product Substances 0.000 description 21
- 108020004465 16S ribosomal RNA Proteins 0.000 description 20
- FWMNVWWHGCHHJJ-SKKKGAJSSA-N 4-amino-1-[(2r)-6-amino-2-[[(2r)-2-[[(2r)-2-[[(2r)-2-amino-3-phenylpropanoyl]amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]hexanoyl]piperidine-4-carboxylic acid Chemical compound C([C@H](C(=O)N[C@H](CC(C)C)C(=O)N[C@H](CCCCN)C(=O)N1CCC(N)(CC1)C(O)=O)NC(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 FWMNVWWHGCHHJJ-SKKKGAJSSA-N 0.000 description 19
- 238000011161 development Methods 0.000 description 19
- 229920000642 polymer Polymers 0.000 description 19
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 18
- 150000002016 disaccharides Chemical class 0.000 description 17
- 239000007983 Tris buffer Substances 0.000 description 16
- 238000004458 analytical method Methods 0.000 description 16
- 238000002415 sodium dodecyl sulfate polyacrylamide gel electrophoresis Methods 0.000 description 16
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 16
- IAJILQKETJEXLJ-SQOUGZDYSA-N L-guluronic acid Chemical compound O=C[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)=O IAJILQKETJEXLJ-SQOUGZDYSA-N 0.000 description 15
- 238000000746 purification Methods 0.000 description 15
- 238000004255 ion exchange chromatography Methods 0.000 description 14
- AEMOLEFTQBMNLQ-UHFFFAOYSA-N beta-D-galactopyranuronic acid Natural products OC1OC(C(O)=O)C(O)C(O)C1O AEMOLEFTQBMNLQ-UHFFFAOYSA-N 0.000 description 13
- AEMOLEFTQBMNLQ-YBSDWZGDSA-N d-mannuronic acid Chemical compound O[C@@H]1O[C@@H](C(O)=O)[C@H](O)[C@@H](O)[C@H]1O AEMOLEFTQBMNLQ-YBSDWZGDSA-N 0.000 description 13
- 239000003480 eluent Substances 0.000 description 13
- 239000006870 ms-medium Substances 0.000 description 13
- 150000003839 salts Chemical class 0.000 description 13
- 238000004809 thin layer chromatography Methods 0.000 description 13
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 12
- 229920001519 homopolymer Polymers 0.000 description 12
- 239000012064 sodium phosphate buffer Substances 0.000 description 12
- RVBUGGBMJDPOST-UHFFFAOYSA-N 2-thiobarbituric acid Chemical compound O=C1CC(=O)NC(=S)N1 RVBUGGBMJDPOST-UHFFFAOYSA-N 0.000 description 11
- 238000007796 conventional method Methods 0.000 description 11
- 229920001282 polysaccharide Polymers 0.000 description 11
- 239000005017 polysaccharide Substances 0.000 description 11
- 108020004414 DNA Proteins 0.000 description 10
- 241001052616 Flavobacterium limicola Species 0.000 description 10
- 238000004440 column chromatography Methods 0.000 description 10
- 239000007857 degradation product Substances 0.000 description 10
- 238000000605 extraction Methods 0.000 description 10
- 230000012010 growth Effects 0.000 description 10
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 9
- -1 alginic acid ester Chemical class 0.000 description 9
- 238000010586 diagram Methods 0.000 description 9
- 239000008103 glucose Substances 0.000 description 9
- 235000000346 sugar Nutrition 0.000 description 9
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 9
- 238000007447 staining method Methods 0.000 description 8
- 229920002472 Starch Polymers 0.000 description 7
- 238000004587 chromatography analysis Methods 0.000 description 7
- 238000001641 gel filtration chromatography Methods 0.000 description 7
- 239000008107 starch Substances 0.000 description 7
- 235000019698 starch Nutrition 0.000 description 7
- 239000006228 supernatant Substances 0.000 description 7
- 238000012360 testing method Methods 0.000 description 7
- 150000004044 tetrasaccharides Chemical class 0.000 description 7
- 150000004043 trisaccharides Chemical class 0.000 description 7
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 6
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 6
- 238000012258 culturing Methods 0.000 description 6
- 238000002955 isolation Methods 0.000 description 6
- 238000005259 measurement Methods 0.000 description 6
- 239000002244 precipitate Substances 0.000 description 6
- 238000002360 preparation method Methods 0.000 description 6
- 241000894007 species Species 0.000 description 6
- 241000199919 Phaeophyceae Species 0.000 description 5
- 238000012300 Sequence Analysis Methods 0.000 description 5
- 150000007513 acids Chemical class 0.000 description 5
- 229910052921 ammonium sulfate Inorganic materials 0.000 description 5
- 235000011130 ammonium sulphate Nutrition 0.000 description 5
- 238000004364 calculation method Methods 0.000 description 5
- 239000012295 chemical reaction liquid Substances 0.000 description 5
- 239000003153 chemical reaction reagent Substances 0.000 description 5
- 230000007423 decrease Effects 0.000 description 5
- 238000000502 dialysis Methods 0.000 description 5
- 239000007788 liquid Substances 0.000 description 5
- 239000000463 material Substances 0.000 description 5
- 230000000877 morphologic effect Effects 0.000 description 5
- 239000008188 pellet Substances 0.000 description 5
- 230000001766 physiological effect Effects 0.000 description 5
- 238000006467 substitution reaction Methods 0.000 description 5
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 4
- 125000001429 N-terminal alpha-amino-acid group Chemical group 0.000 description 4
- 239000012505 Superdex™ Substances 0.000 description 4
- 239000000872 buffer Substances 0.000 description 4
- 238000005119 centrifugation Methods 0.000 description 4
- 239000012153 distilled water Substances 0.000 description 4
- 238000004043 dyeing Methods 0.000 description 4
- 239000000706 filtrate Substances 0.000 description 4
- 244000005700 microbiome Species 0.000 description 4
- 238000013081 phylogenetic analysis Methods 0.000 description 4
- 230000035484 reaction time Effects 0.000 description 4
- 239000011734 sodium Substances 0.000 description 4
- 239000013598 vector Substances 0.000 description 4
- MTCFGRXMJLQNBG-REOHCLBHSA-N (2S)-2-Amino-3-hydroxypropansäure Chemical compound OC[C@H](N)C(O)=O MTCFGRXMJLQNBG-REOHCLBHSA-N 0.000 description 3
- 241001179699 Algicola Species 0.000 description 3
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 3
- 102000016938 Catalase Human genes 0.000 description 3
- 108010053835 Catalase Proteins 0.000 description 3
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 3
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 3
- AYFVYJQAPQTCCC-GBXIJSLDSA-N L-threonine Chemical compound C[C@@H](O)[C@H](N)C(O)=O AYFVYJQAPQTCCC-GBXIJSLDSA-N 0.000 description 3
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 description 3
- KZSNJWFQEVHDMF-BYPYZUCNSA-N L-valine Chemical compound CC(C)[C@H](N)C(O)=O KZSNJWFQEVHDMF-BYPYZUCNSA-N 0.000 description 3
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 3
- 229920000161 Locust bean gum Polymers 0.000 description 3
- 229910002651 NO3 Inorganic materials 0.000 description 3
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 3
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 3
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 description 3
- 239000004473 Threonine Substances 0.000 description 3
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 description 3
- KZSNJWFQEVHDMF-UHFFFAOYSA-N Valine Natural products CC(C)C(N)C(O)=O KZSNJWFQEVHDMF-UHFFFAOYSA-N 0.000 description 3
- 239000007864 aqueous solution Substances 0.000 description 3
- 239000007853 buffer solution Substances 0.000 description 3
- 239000001768 carboxy methyl cellulose Substances 0.000 description 3
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 3
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 3
- 238000002474 experimental method Methods 0.000 description 3
- 238000000855 fermentation Methods 0.000 description 3
- 230000004151 fermentation Effects 0.000 description 3
- 238000013418 fluorescence derivatisation Methods 0.000 description 3
- 238000005194 fractionation Methods 0.000 description 3
- 239000007789 gas Substances 0.000 description 3
- 239000000499 gel Substances 0.000 description 3
- 239000001963 growth medium Substances 0.000 description 3
- 150000004687 hexahydrates Chemical class 0.000 description 3
- 230000003834 intracellular effect Effects 0.000 description 3
- 235000005772 leucine Nutrition 0.000 description 3
- 235000010420 locust bean gum Nutrition 0.000 description 3
- 239000000711 locust bean gum Substances 0.000 description 3
- 238000004949 mass spectrometry Methods 0.000 description 3
- 230000003647 oxidation Effects 0.000 description 3
- 238000007254 oxidation reaction Methods 0.000 description 3
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 3
- 239000013612 plasmid Substances 0.000 description 3
- 235000004400 serine Nutrition 0.000 description 3
- 125000001424 substituent group Chemical group 0.000 description 3
- 150000008163 sugars Chemical class 0.000 description 3
- 235000008521 threonine Nutrition 0.000 description 3
- 235000002374 tyrosine Nutrition 0.000 description 3
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 3
- 239000004474 valine Substances 0.000 description 3
- 235000014393 valine Nutrition 0.000 description 3
- GFNJWVBJKYYUIN-CMUOTRNOSA-N (2-dodecyl-3-hydroxy-5-methylphenyl) (2e,4e,6e,8e,10e,12e,14e,16e)-17-(4-hydroxy-3-methylphenyl)heptadeca-2,4,6,8,10,12,14,16-octaenoate Chemical compound CCCCCCCCCCCCC1=C(O)C=C(C)C=C1OC(=O)\C=C\C=C\C=C\C=C\C=C\C=C\C=C\C=C\C1=CC=C(O)C(C)=C1 GFNJWVBJKYYUIN-CMUOTRNOSA-N 0.000 description 2
- 241000186361 Actinobacteria <class> Species 0.000 description 2
- 239000004475 Arginine Substances 0.000 description 2
- DCXYFEDJOCDNAF-UHFFFAOYSA-N Asparagine Natural products OC(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-N 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- 108010077805 Bacterial Proteins Proteins 0.000 description 2
- 241001474374 Blennius Species 0.000 description 2
- 241000193417 Brevibacillus laterosporus Species 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- 241001260848 Coccophora langsdorfii Species 0.000 description 2
- 240000005109 Cryptomeria japonica Species 0.000 description 2
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 2
- WQZGKKKJIJFFOK-QTVWNMPRSA-N D-mannopyranose Chemical compound OC[C@H]1OC(O)[C@@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-QTVWNMPRSA-N 0.000 description 2
- 108010014303 DNA-directed DNA polymerase Proteins 0.000 description 2
- 102000016928 DNA-directed DNA polymerase Human genes 0.000 description 2
- 101100029163 Dictyostelium discoideum pefA gene Proteins 0.000 description 2
- 241000777654 Flavobacterium omnivorum Species 0.000 description 2
- 241000589564 Flavobacterium sp. Species 0.000 description 2
- MGSGAJRNDILWPP-VHNQBQQVSA-N Flexirubin Natural products CCCCCCCCCCCCc1cc(C)cc(C(=O)C=C/C=C/C=C/C=C/C=C/C=C/C=C/C=C/c2ccc(O)c(C)c2)c1O MGSGAJRNDILWPP-VHNQBQQVSA-N 0.000 description 2
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 2
- 239000004471 Glycine Substances 0.000 description 2
- SIKJAQJRHWYJAI-UHFFFAOYSA-N Indole Chemical compound C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 description 2
- XUJNEKJLAYXESH-REOHCLBHSA-N L-Cysteine Chemical compound SC[C@H](N)C(O)=O XUJNEKJLAYXESH-REOHCLBHSA-N 0.000 description 2
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 description 2
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 2
- DCXYFEDJOCDNAF-REOHCLBHSA-N L-asparagine Chemical compound OC(=O)[C@@H](N)CC(N)=O DCXYFEDJOCDNAF-REOHCLBHSA-N 0.000 description 2
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 2
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 2
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 2
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 2
- AGPKZVBTJJNPAG-WHFBIAKZSA-N L-isoleucine Chemical compound CC[C@H](C)[C@H](N)C(O)=O AGPKZVBTJJNPAG-WHFBIAKZSA-N 0.000 description 2
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 2
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 description 2
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 2
- 108090001060 Lipase Proteins 0.000 description 2
- 239000006142 Luria-Bertani Agar Substances 0.000 description 2
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 2
- 239000004472 Lysine Substances 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- 235000002492 Rungia klossii Nutrition 0.000 description 2
- 244000117054 Rungia klossii Species 0.000 description 2
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 2
- 241001228709 Suruga Species 0.000 description 2
- 238000011481 absorbance measurement Methods 0.000 description 2
- 230000002378 acidificating effect Effects 0.000 description 2
- WNLRTRBMVRJNCN-UHFFFAOYSA-N adipic acid Chemical compound OC(=O)CCCCC(O)=O WNLRTRBMVRJNCN-UHFFFAOYSA-N 0.000 description 2
- 235000004279 alanine Nutrition 0.000 description 2
- 125000000539 amino acid group Chemical group 0.000 description 2
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 2
- 235000009582 asparagine Nutrition 0.000 description 2
- 229960001230 asparagine Drugs 0.000 description 2
- 235000003704 aspartic acid Nutrition 0.000 description 2
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 2
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 2
- 239000005018 casein Substances 0.000 description 2
- BECPQYXYKAMYBN-UHFFFAOYSA-N casein, tech. Chemical compound NCCCCC(C(O)=O)N=C(O)C(CC(O)=O)N=C(O)C(CCC(O)=N)N=C(O)C(CC(C)C)N=C(O)C(CCC(O)=O)N=C(O)C(CC(O)=O)N=C(O)C(CCC(O)=O)N=C(O)C(C(C)O)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=O)N=C(O)C(CCC(O)=O)N=C(O)C(COP(O)(O)=O)N=C(O)C(CCC(O)=N)N=C(O)C(N)CC1=CC=CC=C1 BECPQYXYKAMYBN-UHFFFAOYSA-N 0.000 description 2
- 235000021240 caseins Nutrition 0.000 description 2
- 150000001875 compounds Chemical class 0.000 description 2
- 235000018417 cysteine Nutrition 0.000 description 2
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 2
- 238000006911 enzymatic reaction Methods 0.000 description 2
- 238000012869 ethanol precipitation Methods 0.000 description 2
- 238000010353 genetic engineering Methods 0.000 description 2
- 235000013922 glutamic acid Nutrition 0.000 description 2
- 239000004220 glutamic acid Substances 0.000 description 2
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 2
- 235000004554 glutamine Nutrition 0.000 description 2
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 2
- 230000007062 hydrolysis Effects 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- 238000005342 ion exchange Methods 0.000 description 2
- PHTQWCKDNZKARW-UHFFFAOYSA-N isoamylol Chemical compound CC(C)CCO PHTQWCKDNZKARW-UHFFFAOYSA-N 0.000 description 2
- AGPKZVBTJJNPAG-UHFFFAOYSA-N isoleucine Natural products CCC(C)C(N)C(O)=O AGPKZVBTJJNPAG-UHFFFAOYSA-N 0.000 description 2
- 229960000310 isoleucine Drugs 0.000 description 2
- 235000014705 isoleucine Nutrition 0.000 description 2
- 150000004715 keto acids Chemical class 0.000 description 2
- 238000001840 matrix-assisted laser desorption--ionisation time-of-flight mass spectrometry Methods 0.000 description 2
- 229930182817 methionine Natural products 0.000 description 2
- 239000000203 mixture Substances 0.000 description 2
- DNIAPMSPPWPWGF-UHFFFAOYSA-N monopropylene glycol Natural products CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 2
- 230000004899 motility Effects 0.000 description 2
- 239000002773 nucleotide Substances 0.000 description 2
- 125000003729 nucleotide group Chemical group 0.000 description 2
- 238000001139 pH measurement Methods 0.000 description 2
- 239000000049 pigment Substances 0.000 description 2
- 239000013600 plasmid vector Substances 0.000 description 2
- 238000002264 polyacrylamide gel electrophoresis Methods 0.000 description 2
- 235000010408 potassium alginate Nutrition 0.000 description 2
- 239000011541 reaction mixture Substances 0.000 description 2
- 238000011084 recovery Methods 0.000 description 2
- 238000012827 research and development Methods 0.000 description 2
- 238000012163 sequencing technique Methods 0.000 description 2
- 239000000741 silica gel Substances 0.000 description 2
- 229910002027 silica gel Inorganic materials 0.000 description 2
- 238000010186 staining Methods 0.000 description 2
- 238000010561 standard procedure Methods 0.000 description 2
- 238000010257 thawing Methods 0.000 description 2
- 238000009210 therapy by ultrasound Methods 0.000 description 2
- 238000013519 translation Methods 0.000 description 2
- GZCWLCBFPRFLKL-UHFFFAOYSA-N 1-prop-2-ynoxypropan-2-ol Chemical compound CC(O)COCC#C GZCWLCBFPRFLKL-UHFFFAOYSA-N 0.000 description 1
- OWEGMIWEEQEYGQ-UHFFFAOYSA-N 100676-05-9 Natural products OC1C(O)C(O)C(CO)OC1OCC1C(O)C(O)C(O)C(OC2C(OC(O)C(O)C2O)CO)O1 OWEGMIWEEQEYGQ-UHFFFAOYSA-N 0.000 description 1
- 108010051457 Acid Phosphatase Proteins 0.000 description 1
- 102000013563 Acid Phosphatase Human genes 0.000 description 1
- 229920001817 Agar Polymers 0.000 description 1
- 108020004774 Alkaline Phosphatase Proteins 0.000 description 1
- 102000002260 Alkaline Phosphatase Human genes 0.000 description 1
- 241000239290 Araneae Species 0.000 description 1
- 108010082340 Arginine deiminase Proteins 0.000 description 1
- 241000512259 Ascophyllum nodosum Species 0.000 description 1
- 241000589151 Azotobacter Species 0.000 description 1
- 101710180684 Beta-hexosaminidase Proteins 0.000 description 1
- 101710124976 Beta-hexosaminidase A Proteins 0.000 description 1
- 235000014653 Carica parviflora Nutrition 0.000 description 1
- 244000132059 Carica parviflora Species 0.000 description 1
- 108090000317 Chymotrypsin Proteins 0.000 description 1
- 108020004638 Circular DNA Proteins 0.000 description 1
- 102000000634 Cytochrome c oxidase subunit IV Human genes 0.000 description 1
- 108050008072 Cytochrome c oxidase subunit IV Proteins 0.000 description 1
- LEVWYRKDKASIDU-QWWZWVQMSA-N D-cystine Chemical compound OC(=O)[C@H](N)CSSC[C@@H](N)C(O)=O LEVWYRKDKASIDU-QWWZWVQMSA-N 0.000 description 1
- AEMOLEFTQBMNLQ-VANFPWTGSA-N D-mannopyranuronic acid Chemical compound OC1O[C@H](C(O)=O)[C@@H](O)[C@H](O)[C@@H]1O AEMOLEFTQBMNLQ-VANFPWTGSA-N 0.000 description 1
- 102000012410 DNA Ligases Human genes 0.000 description 1
- 108010061982 DNA Ligases Proteins 0.000 description 1
- 238000007400 DNA extraction Methods 0.000 description 1
- 239000004278 EU approved seasoning Substances 0.000 description 1
- 241000620209 Escherichia coli DH5[alpha] Species 0.000 description 1
- 108090000371 Esterases Proteins 0.000 description 1
- 241000233866 Fungi Species 0.000 description 1
- IAJILQKETJEXLJ-UHFFFAOYSA-N Galacturonsaeure Natural products O=CC(O)C(O)C(O)C(O)C(O)=O IAJILQKETJEXLJ-UHFFFAOYSA-N 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 102000053187 Glucuronidase Human genes 0.000 description 1
- 108010060309 Glucuronidase Proteins 0.000 description 1
- 238000003794 Gram staining Methods 0.000 description 1
- 241000588748 Klebsiella Species 0.000 description 1
- ONIBWKKTOPOVIA-BYPYZUCNSA-N L-Proline Chemical compound OC(=O)[C@@H]1CCCN1 ONIBWKKTOPOVIA-BYPYZUCNSA-N 0.000 description 1
- 239000004367 Lipase Substances 0.000 description 1
- 102000004882 Lipase Human genes 0.000 description 1
- 102100024295 Maltase-glucoamylase Human genes 0.000 description 1
- GUBGYTABKSRVRQ-PICCSMPSSA-N Maltose Natural products O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@@H](CO)OC(O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-PICCSMPSSA-N 0.000 description 1
- OVRNDRQMDRJTHS-UHFFFAOYSA-N N-acelyl-D-glucosamine Natural products CC(=O)NC1C(O)OC(CO)C(O)C1O OVRNDRQMDRJTHS-UHFFFAOYSA-N 0.000 description 1
- OVRNDRQMDRJTHS-FMDGEEDCSA-N N-acetyl-beta-D-glucosamine Chemical compound CC(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O OVRNDRQMDRJTHS-FMDGEEDCSA-N 0.000 description 1
- 241000588652 Neisseria gonorrhoeae Species 0.000 description 1
- BPCLKHIDDIVGBM-UHFFFAOYSA-N OC(C1O)C(O)OC(C(O)=O)=C1O Chemical compound OC(C1O)C(O)OC(C(O)=O)=C1O BPCLKHIDDIVGBM-UHFFFAOYSA-N 0.000 description 1
- 102000004316 Oxidoreductases Human genes 0.000 description 1
- 108090000854 Oxidoreductases Proteins 0.000 description 1
- 238000012408 PCR amplification Methods 0.000 description 1
- HLCFGWHYROZGBI-JJKGCWMISA-M Potassium gluconate Chemical compound [K+].OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O HLCFGWHYROZGBI-JJKGCWMISA-M 0.000 description 1
- ONIBWKKTOPOVIA-UHFFFAOYSA-N Proline Natural products OC(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-N 0.000 description 1
- 102100030122 Protein O-GlcNAcase Human genes 0.000 description 1
- 101710081801 Protein O-GlcNAcase Proteins 0.000 description 1
- 108010076504 Protein Sorting Signals Proteins 0.000 description 1
- 241000589516 Pseudomonas Species 0.000 description 1
- 241000589517 Pseudomonas aeruginosa Species 0.000 description 1
- 101710199095 Putative beta-hexosaminidase Proteins 0.000 description 1
- 241000206572 Rhodophyta Species 0.000 description 1
- 101000841440 Saccharophagus degradans (strain 2-40 / ATCC 43961 / DSM 17024) Exo-oligoalginate lyase Proteins 0.000 description 1
- 229910000831 Steel Inorganic materials 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- 108010006785 Taq Polymerase Proteins 0.000 description 1
- 239000007984 Tris EDTA buffer Substances 0.000 description 1
- 108090000631 Trypsin Proteins 0.000 description 1
- 102000004142 Trypsin Human genes 0.000 description 1
- 241001261506 Undaria pinnatifida Species 0.000 description 1
- 108010046334 Urease Proteins 0.000 description 1
- 241000607598 Vibrio Species 0.000 description 1
- 239000006096 absorbing agent Substances 0.000 description 1
- 238000009825 accumulation Methods 0.000 description 1
- 239000001361 adipic acid Substances 0.000 description 1
- 235000011037 adipic acid Nutrition 0.000 description 1
- 238000001042 affinity chromatography Methods 0.000 description 1
- 239000008272 agar Substances 0.000 description 1
- 125000000304 alkynyl group Chemical group 0.000 description 1
- 102000005840 alpha-Galactosidase Human genes 0.000 description 1
- 108010030291 alpha-Galactosidase Proteins 0.000 description 1
- 108010028144 alpha-Glucosidases Proteins 0.000 description 1
- 102000012086 alpha-L-Fucosidase Human genes 0.000 description 1
- 108010061314 alpha-L-Fucosidase Proteins 0.000 description 1
- 102000019199 alpha-Mannosidase Human genes 0.000 description 1
- 108010012864 alpha-Mannosidase Proteins 0.000 description 1
- 150000001370 alpha-amino acid derivatives Chemical class 0.000 description 1
- 235000008206 alpha-amino acids Nutrition 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 235000010407 ammonium alginate Nutrition 0.000 description 1
- 239000000728 ammonium alginate Substances 0.000 description 1
- KPGABFJTMYCRHJ-YZOKENDUSA-N ammonium alginate Chemical compound [NH4+].[NH4+].O1[C@@H](C([O-])=O)[C@@H](OC)[C@H](O)[C@H](O)[C@@H]1O[C@@H]1[C@@H](C([O-])=O)O[C@@H](O)[C@@H](O)[C@H]1O KPGABFJTMYCRHJ-YZOKENDUSA-N 0.000 description 1
- 230000003321 amplification Effects 0.000 description 1
- 238000005349 anion exchange Methods 0.000 description 1
- 239000000883 anti-obesity agent Substances 0.000 description 1
- 239000003472 antidiabetic agent Substances 0.000 description 1
- 229940030225 antihemorrhagics Drugs 0.000 description 1
- 239000003524 antilipemic agent Substances 0.000 description 1
- 229940125710 antiobesity agent Drugs 0.000 description 1
- 239000008346 aqueous phase Substances 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 230000000721 bacterilogical effect Effects 0.000 description 1
- 239000011324 bead Substances 0.000 description 1
- 102000005936 beta-Galactosidase Human genes 0.000 description 1
- 108010005774 beta-Galactosidase Proteins 0.000 description 1
- 102000006995 beta-Glucosidase Human genes 0.000 description 1
- 108010047754 beta-Glucosidase Proteins 0.000 description 1
- 238000010876 biochemical test Methods 0.000 description 1
- 229920000704 biodegradable plastic Polymers 0.000 description 1
- 235000014121 butter Nutrition 0.000 description 1
- 235000010410 calcium alginate Nutrition 0.000 description 1
- 239000000648 calcium alginate Substances 0.000 description 1
- 229960002681 calcium alginate Drugs 0.000 description 1
- OKHHGHGGPDJQHR-YMOPUZKJSA-L calcium;(2s,3s,4s,5s,6r)-6-[(2r,3s,4r,5s,6r)-2-carboxy-6-[(2r,3s,4r,5s,6r)-2-carboxylato-4,5,6-trihydroxyoxan-3-yl]oxy-4,5-dihydroxyoxan-3-yl]oxy-3,4,5-trihydroxyoxane-2-carboxylate Chemical compound [Ca+2].O[C@@H]1[C@H](O)[C@H](O)O[C@@H](C([O-])=O)[C@H]1O[C@H]1[C@@H](O)[C@@H](O)[C@H](O[C@H]2[C@H]([C@@H](O)[C@H](O)[C@H](O2)C([O-])=O)O)[C@H](C(O)=O)O1 OKHHGHGGPDJQHR-YMOPUZKJSA-L 0.000 description 1
- 239000003560 cancer drug Substances 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- 235000014633 carbohydrates Nutrition 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 239000003638 chemical reducing agent Substances 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- YTRQFSDWAXHJCC-UHFFFAOYSA-N chloroform;phenol Chemical compound ClC(Cl)Cl.OC1=CC=CC=C1 YTRQFSDWAXHJCC-UHFFFAOYSA-N 0.000 description 1
- 229960002376 chymotrypsin Drugs 0.000 description 1
- 238000010367 cloning Methods 0.000 description 1
- 238000004040 coloring Methods 0.000 description 1
- NKLPQNGYXWVELD-UHFFFAOYSA-M coomassie brilliant blue Chemical compound [Na+].C1=CC(OCC)=CC=C1NC1=CC=C(C(=C2C=CC(C=C2)=[N+](CC)CC=2C=C(C=CC=2)S([O-])(=O)=O)C=2C=CC(=CC=2)N(CC)CC=2C=C(C=CC=2)S([O-])(=O)=O)C=C1 NKLPQNGYXWVELD-UHFFFAOYSA-M 0.000 description 1
- 239000002537 cosmetic Substances 0.000 description 1
- 238000012136 culture method Methods 0.000 description 1
- 229960003067 cystine Drugs 0.000 description 1
- 238000004042 decolorization Methods 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 239000005548 dental material Substances 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 235000013325 dietary fiber Nutrition 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- 239000003814 drug Substances 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 235000013305 food Nutrition 0.000 description 1
- 239000002778 food additive Substances 0.000 description 1
- 235000011194 food seasoning agent Nutrition 0.000 description 1
- 230000008014 freezing Effects 0.000 description 1
- 238000007710 freezing Methods 0.000 description 1
- 235000013376 functional food Nutrition 0.000 description 1
- 229920001002 functional polymer Polymers 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 238000002523 gelfiltration Methods 0.000 description 1
- 239000003349 gelling agent Substances 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 239000002874 hemostatic agent Substances 0.000 description 1
- 125000000623 heterocyclic group Chemical group 0.000 description 1
- 230000002209 hydrophobic effect Effects 0.000 description 1
- 229940126904 hypoglycaemic agent Drugs 0.000 description 1
- 230000006303 immediate early viral mRNA transcription Effects 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- PZOUSPYUWWUPPK-UHFFFAOYSA-N indole Natural products CC1=CC=CC2=C1C=CN2 PZOUSPYUWWUPPK-UHFFFAOYSA-N 0.000 description 1
- RKJUIXBNRJVNHR-UHFFFAOYSA-N indolenine Natural products C1=CC=C2CC=NC2=C1 RKJUIXBNRJVNHR-UHFFFAOYSA-N 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 230000000968 intestinal effect Effects 0.000 description 1
- 230000006799 invasive growth in response to glucose limitation Effects 0.000 description 1
- 125000000468 ketone group Chemical group 0.000 description 1
- 235000019421 lipase Nutrition 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N malic acid Chemical compound OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 229910021645 metal ion Inorganic materials 0.000 description 1
- 239000004570 mortar (masonry) Substances 0.000 description 1
- 229950006780 n-acetylglucosamine Drugs 0.000 description 1
- 229910052757 nitrogen Inorganic materials 0.000 description 1
- 238000003199 nucleic acid amplification method Methods 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 238000006395 oxidase reaction Methods 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 230000020477 pH reduction Effects 0.000 description 1
- 150000004686 pentahydrates Chemical class 0.000 description 1
- KHIWWQKSHDUIBK-UHFFFAOYSA-N periodic acid Chemical compound OI(=O)(=O)=O KHIWWQKSHDUIBK-UHFFFAOYSA-N 0.000 description 1
- 150000002978 peroxides Chemical class 0.000 description 1
- 239000012071 phase Substances 0.000 description 1
- IPBVNPXQWQGGJP-UHFFFAOYSA-N phenyl acetate Chemical compound CC(=O)OC1=CC=CC=C1 IPBVNPXQWQGGJP-UHFFFAOYSA-N 0.000 description 1
- 229920002401 polyacrylamide Polymers 0.000 description 1
- 239000002861 polymer material Substances 0.000 description 1
- 108091022901 polysaccharide lyase Proteins 0.000 description 1
- 102000020244 polysaccharide lyase Human genes 0.000 description 1
- 239000000737 potassium alginate Substances 0.000 description 1
- MZYRDLHIWXQJCQ-YZOKENDUSA-L potassium alginate Chemical compound [K+].[K+].O1[C@@H](C([O-])=O)[C@@H](OC)[C@H](O)[C@H](O)[C@@H]1O[C@@H]1[C@@H](C([O-])=O)O[C@@H](O)[C@@H](O)[C@H]1O MZYRDLHIWXQJCQ-YZOKENDUSA-L 0.000 description 1
- 239000004224 potassium gluconate Substances 0.000 description 1
- 229960003189 potassium gluconate Drugs 0.000 description 1
- 235000013926 potassium gluconate Nutrition 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 235000013406 prebiotics Nutrition 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 230000035755 proliferation Effects 0.000 description 1
- 238000003908 quality control method Methods 0.000 description 1
- 125000001453 quaternary ammonium group Chemical group 0.000 description 1
- 239000002994 raw material Substances 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 108091008146 restriction endonucleases Proteins 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 239000013535 sea water Substances 0.000 description 1
- 230000003248 secreting effect Effects 0.000 description 1
- 230000028327 secretion Effects 0.000 description 1
- 235000015170 shellfish Nutrition 0.000 description 1
- 239000001509 sodium citrate Substances 0.000 description 1
- NLJMYIDDQXHKNR-UHFFFAOYSA-K sodium citrate Chemical compound O.O.[Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O NLJMYIDDQXHKNR-UHFFFAOYSA-K 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 239000002689 soil Substances 0.000 description 1
- 238000000527 sonication Methods 0.000 description 1
- 238000001179 sorption measurement Methods 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 239000010959 steel Substances 0.000 description 1
- 238000003756 stirring Methods 0.000 description 1
- 125000005017 substituted alkenyl group Chemical group 0.000 description 1
- 125000000547 substituted alkyl group Chemical group 0.000 description 1
- 125000005346 substituted cycloalkyl group Chemical group 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- 238000001308 synthesis method Methods 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- 235000013619 trace mineral Nutrition 0.000 description 1
- 239000011573 trace mineral Substances 0.000 description 1
- 239000012588 trypsin Substances 0.000 description 1
Images























Landscapes
- Enzymes And Modification Thereof (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
Description

[化3]HOOC−COH=CH−CHOH−CHOH−CHO。
[化6]HOOC−COH=CH−CHOH−CHOH−CHO。
[化9]HOOC−COH=CH−CHOH−CHOH−CHO。
[化12]HOOC−COH=CH−CHOH−CHOH−CHO。
[化15]HOOC−COH=CH−CHOH−CHOH−CHO。
[化18]HOOC−COH=CH−CHOH−CHOH−CHO。
[化21]HOOC−COH=CH−CHOH−CHOH−CHO
について、本明細書では、上記式(化19)の不飽和単糖、式(化20)のα−ケト酸および式(化21)のウロン酸をまとめて「単糖」という場合があり、式(化20)のα−ケト酸および式(化21)のウロン酸をまとめて「α−ケト酸」という場合がある。さらに、本発明における上記式(化19)、式(化20)および式(化21)の物質の−COOH、−OH、−CHOは、それぞれ、−COOR1、−OR2、−OR3H(R1、R2、R3は、それぞれ、置換基を有していてもよいアルキル基、置換基を有していてもよいアルケニル基、置換基を有していてもよいシクロアルキル基、置換基を有していてもよいアルキニル基、置換基を有していてもよいアリール基または置換基を有していてもよい複素環のいずれかである)であってもよく、すなわち、−COOH、−OH、−CHOとなり得るものであればよい。
(a)グルコースから酸を産生しない、
(b)デンプンを加水分解できる、
(c)硝酸塩を還元しない。
[化24]HOOC−COH=CH−CHOH−CHOH−CHO。
[化27]HOOC−COH=CH−CHOH−CHOH−CHO。
(f)分子量が約30kDaである、
(g)アルギン酸分解反応における至適pHが約8である、
(h)最も安定なpHが7<pH<8である、
(i)アルギン酸分解反応における至適温度が約54℃である、
(j)60℃で30分間アルギン酸分解反応を行った後にアルギン酸分解酵素活性が消失する、
(k)アルギン酸分解反応における至適NaCl濃度Cが0.05<C<0.3(mol/L)である、
(l)アルギン酸の構造中、D−マンヌロン酸のホモポリマーである部分(Mブロック)、L−グルロン酸のホモポリマーである部分(Gブロック)、MブロックとGブロックとが交互に結合した部分(MGブロック)およびD−マンヌロン酸とL−グルロン酸とが交互に結合した部分(交互ポリマー)のいずれも分解することができる。
(m)配列番号2、配列番号12または配列番号13のアミノ酸配列、
(n)配列番号2、配列番号12または配列番号13のアミノ酸配列において1もしくは数個のアミノ酸が欠失、置換もしくは付加されたアミノ酸配列。なお、配列番号2は本発明に係る新規のFlavobacterium属細菌の一態様であるUMI−01が産生する、新規のエンド型アルギン酸リアーゼの一態様であるFlAly−1の成熟型におけるN末端40残基と同一のアミノ酸配列である。また、配列番号12はFlAly−1の翻訳型の全アミノ酸配列288残基と、配列番号13はFlAly−1の成熟型の全アミノ酸配列260残基とそれぞれ同一のアミノ酸配列である。
[化30]HOOC−COH=CH−CHOH−CHOH−CHO。なお、本発明に係るオリゴ糖、不飽和単糖、ないしα−ケト酸を製造する方法において、上述した本発明に係るFlavobacterium属細菌または分解酵素の構成と同等または相当する構成については再度の説明を省略する。
(i)分子量が約30kDaである、
(ii)アルギン酸分解反応における至適pHが約8である、
(iii)最も安定なpHが7<pH<8である、
(iv)アルギン酸分解反応における至適温度が約54℃である、
(v)60℃で30分間アルギン酸分解反応を行った後にアルギン酸分解酵素活性が消失する、
(vi)アルギン酸分解反応における至適NaCl濃度Cが0.05<C<0.3(mol/L)である、
(vii)アルギン酸の構造中、D−マンヌロン酸のホモポリマーである部分(Mブロック)、L−グルロン酸のホモポリマーである部分(Gブロック)、MブロックとGブロックとが交互に結合した部分(MGブロック)およびD−マンヌロン酸とL−グルロン酸とが交互に結合した部分(交互ポリマー)のいずれも分解することができる。
(viii)配列番号2のアミノ酸配列、
(ix)配列番号2のアミノ酸配列において1もしくは数個のアミノ酸が欠失、置換もしくは付加されたアミノ酸配列。
(x)配列番号12または配列番号13のアミノ酸配列、
(xi)配列番号12または配列番号13のアミノ酸配列において1もしくは数個のアミノ酸が欠失、置換もしくは付加されたアミノ酸配列。
(xii)配列番号12または配列番号13のアミノ酸配列からなるタンパク質、
(xiii)配列番号12または配列番号13のアミノ酸配列において1もしくは数個のアミノ酸が欠失、置換もしくは付加されたアミノ酸配列からなり、かつエンド型アルギン酸リアーゼ活性を有するタンパク質。
(1)アルギン酸資化菌の単離
腐敗したスギモク(Coccophora langsdorfii、褐藻類の一種)を採取して集積培養法を行い、アルギン酸を資化する菌を単離し、これをアルギン酸資化菌とした。
下記の試薬を下記の終濃度となるよう蒸留水に溶解し、これをMS培地とした。
Na2HPO4 3.6 g/L
KH2PO4 0.75g/L
NH4Cl 0.5 g/L
MgSO4 0.2 g/L
B;デンプン+MS培地
C;カルボキシメチルセルロース+MS培地
D;ローカストビーンガム+MS培地
E;多糖類を加えないLB培地
A ;0、5、10、15、20、30、40、60、70
B、C、D;0、5、10、20、30、40、60、70
E ;0、5、10、15、20、30、40、60
実施例1(1)のアルギン酸資化菌について、株式会社テクノスルガ・ラボにおいて16S rDNA塩基配列解析および菌学的性状試験を行った。
16S rDNA塩基配列解析は、下記の試薬、装置およびプログラムを用いて、添付の仕様書に従って行った。
PCR;PrimeSTAR HS DNA Polymerase(タカラバイオ社)
サイクルシークエンス;BigDye Terminator v3.1 Cycle Sequencing Kit(Applied Biosystems社)
使用プライマー(中川恭好ら、日本放線菌学会、 放線菌の分類と同定、第88−117頁、2001年);PCR増幅:9F、1510R、シークエンス:9F、785F、802R、1510R
シークエンス;ABI PRISM 3130 xl Genetic Analyzar System(Applied Biosystems社)
配列決定;ChromasPro 1.4(Technelysium Pty Ltd.社)
相同性検索および簡易分子系統解析;ソフトウェア:アポロン2.0(テクノスルガ・ラボ社)、データベース:アポロンDB−BA 6.0(テクノスルガ・ラボ社)、国際塩基配列データベース(GenBank/DDBJ/EMBL)
菌学的性状試験は、下記の試薬、機器および方法を用いて行った。
グラム染色;フェイバーG「ニッスイ」(日水製薬社)
カタラーゼ反応、グルコースからの酸/ガス産生、グルコースの酸化/発酵(O/F);既報(BARROWら、Cowan and Steel’s Manual for the dentification of Medical Bacteria.3rd edition、1993年)に記載の方法
生化学試験、資化性試験;API20NE Ver.7(bioMerieux社)
酵素反応試験;APIZYM(bioMerieux社)
生育試験、フレキシルビン色素の産生、カゼインおよびデンプンの加水分解;NCIMB Ltd.社(http://www.ncimb.co.uk/)との技術提携事項および分類・同定の関連文献に従った方法
培養温度30℃での形態的性状についての結果を下記に示す。
1.細胞形態 桿菌(0.7−0.8×1.0−1.5μm)
2.運動性 なし
3.グラム染色 陰性
4.胞子の有無 なし
5.コロニー形態(LB寒天培地上、24時間)
直径:1.0mm以下
色調:黄色
形:円形
隆起状態:レンズ状
周縁:全縁
表面の形状:スムーズ
透明度:不透明
粘調度:バター様
生理学的性状についての結果を下記に示す。+は陽性を、−は陰性をそれぞれ示す。
1.グルコースからの酸/ガス産生(酸産生/ガス産生) −/−
2.グルコースの酸化/発酵(酸化/発酵) −/−
3.グルコースの酸性化 −
4.フレキシルビン色素の産生 −
5.インドール産生 −
6.硝酸塩の還元 −
7.MB2216寒天での生育 +
8.生育温度
8−1. 5℃ +(反応弱い)
8−2.15℃ +
8−3.37℃ +
8−4.45℃ −
9.加水分解
9−1.カゼイン −
9−2.デンプン +
9−3.エスクリン +
9−4.ゼラチン −
10.酵素反応
10−1.カタラーゼ +
10−2.オキシダーゼ +
10−3.アルギニンジヒドロラーゼ −
10−4.ウレアーゼ −
10−5.β−ガラクトシダーゼ +
10−6.チトクロームオキシダーゼ +
10−7.アルカリフォスファターゼ +
10−8.エステラーゼ(C4) −
10−9.エステラーゼ リパーゼ(C8) −
10−10.リパーゼ(C14) −
10−11.ロイシン アリルアミダーゼ +
10−12.バリン アリルアミダーゼ −
10−13.シスチン アリルアミダーゼ −
10−14.トリプシン −
10−15.キモトリプシン −
10−16.酸性ホスファターゼ +
10−17.ナフトール−AS−BI−ホスホハイドロラーゼ −
10−18.α−ガラクトシダーゼ −
10−19.β−グルクロニダーゼ −
10−20.α−グルコシダーゼ −
10−21.β−グルコシダーゼ −
10−22.N−アセチル−β−グルコサミニダーゼ −
10−23.α−マンノシダーゼ −
10−24.α−フコシダーゼ −
11.資化性試験
11−1.グルコース −
11−2.L−アラビノース −
11−3.D−マンノース +
11−4.D−マンニトール +
11−5.N−アセチル−D−グルコサミン −
11−6.マルトース −
11−7.グルコン酸カリウム −
11−8.n−カプリン酸 −
11−9.アジピン酸 −
11−10.dl−リンゴ酸 −
11−11.クエン酸ナトリウム −
11−12.酢酸フェニル −
(1)培地における至適アルギン酸ナトリウム濃度の検討
[1−1]UMI−01の培養
実施例1(2)のMS培地に0.5%(w/v)、1%(w/v)、1%(w/v)および2%(w/v)となるようアルギン酸ナトリウムを加え、a、b、cおよびdとした。a、b、cおよびdに、UMI−01を植菌して、600nmにおける吸光度が1.5となるまで、実施例1(2)に記載の条件下で前培養し、前培養菌液を得た。
本実施例(1)[1−1]のA、B、CおよびDについて、培養中、下記に示す培養時間の時点で、サンプラーを用いて10mLの培地を分取して、pHおよび600nmにおける吸光度を測定した。pHの測定結果に基づき、CおよびDについては、シリンジを用いて5%(v/v)リン酸水溶液を滴下して、培地のpHを7.5に調整することにより、培養中の培地のpHを7.5に維持した。AおよびBについては、pHの調整は行わなかった。
A:0、11、20、28.5、40、48.5、58、77.5、89
B:0、4、22、30、40、62、68、74、89
C:0、4、6、14、22、30、40、47、57、66,76、83、90
D:0、4、8.5、14、23.5、28.5、32、35、42、48.5、57、68、77、90
[2−1]菌抽出液の調製
本実施例(1)[1−1]のDについて、培養中、培養時間が23.5、32、42、50、62、74および90時間の時点で、サンプラーを用いて10mLの培地を分取し、d1、d2、d3、d4、d5、d6およびd7とした。これを10,000×g、4℃の条件下で10分間遠心分離を行って、菌体ペレットを回収した。
10mmol/Lのリン酸ナトリウム緩衝液(pH7.0)に、終濃度0.15%(w/v)となるようアルギン酸ナトリウムを加え、1/20容量の本実施例(2)[2−1]の菌抽出液を加えた後、30℃にて15分間インキュベートした。その間、記録計により235nmにおける吸光度上昇を記録した。
(1)UMI−01の培養
実施例1(2)のMS培地に、1%(w/v)、1%(w/v)および2%(w/v)となるようアルギン酸ナトリウムを加えた培地を用意し、A、BおよびCとして、実施例3(1)[1−1]に記載の方法により、UMI−01を培養した。ただし、本培養は800mLに代えて100mLのスケールで行い、本培養の時間は90時間に代えて、48時間とした。
本実施例(1)のA、BおよびCについて、実施例3(2)[2−1]に記載の方法により菌抽出液を得た。
本実施例(2)のA、A2、A3、BおよびCについて、既報(Porzioら、Biochim.Biophys. Acta.、第490巻、第27−34頁、1977年)に記載の方法によりSDS−PAGEを行った。なお、ゲルは10%ポリアクリルアミドスラブゲル(1mm厚、10×12cm)を用い、サンプルは1ウェル当たり10μLをアプライした。また、染色には、終濃度0.5%(w/v)のCoomassie Brilliant Blue R−250(ThermoSCIENTIFIC社)を溶解したメタノール/酢酸水溶液{メタノール:酢酸:水=50:10:40(v:v:v)}を、脱色には、メタノール/酢酸水溶液{メタノール:酢酸:水=5:7:88(v:v:v)}をそれぞれ用いた。その結果を図4上図に示す。
[4−1]精製菌抽出液の調製
本実施例(2)の菌抽出液Cについて、定法に従って硫安分画を行い、40〜60%飽和硫安画分および60〜90%飽和硫安画分を得た。これを10mmol/Lリン酸ナトリウム緩衝液(pH7.0)に溶解した後、10mmol/Lリン酸ナトリウム緩衝液(pH7.0)を外液として透析し、内液を回収して精製菌抽出液とした。40〜60%飽和硫安画分を透析したものをC40−60、60〜90%飽和硫安画分を透析したものをC60−90とした。
本実施例(4)[4−1]のC60−90について、TOYOPEARL CM−650 M(東ソー社)を用いて、添付の仕様書に従ってイオン交換クロマトグラフィーを行い、溶出液を得て、これを精製菌抽出液のイオン交換クロマトグラフィー画分とした。なお、溶出液として、0〜0.3mol/Lの範囲で直線的に濃度を変化させたNaCl水溶液を用いた。
本実施例(4)[4−1]のC60−90、および本実施例(4)[4−2]のイオン交換クロマトグラフィー画分について、本実施例(3)に記載の方法によりSDS−PAGEを行った。その結果を図4下図に示す。
[5−1]アルギン酸分解酵素活性の算出
10mmol/Lのリン酸ナトリウム緩衝液(pH7.0)に、NaClおよびアルギン酸ナトリウムをそれぞれ、0.1mol/Lおよび0.5%(w/v)となるよう加えたものを2つ用意し、反応液pおよび反応液qとした。反応液pおよび反応液qに、1/20容量の本実施例(4)[4−1]のC60−90およびC40−60をそれぞれ加えた後、30℃にて6時間インキュベートした。その後、実施例3(2)[2−2]に記載の方法によりアルギン酸分解酵素活性を算出したところ、反応液pは239U/mLであり、反応液qは201U/mLであった。
本実施例(5)[5−1]の反応液pおよび反応液qについて、インキュベート開始から、1分間、5分間、10分間、30分間、1時間、2時間、4時間および6時間経過時に、2μLの反応液を分取した。分取した反応液を1μLずつ、2枚のTLC60シリカゲルプレート(Merck社)にアプライし、1−ブタノール:酢酸:水=2:1:1(v:v:v)で混合した展開溶液を用いて展開した。その後、一方のプレートについてはチオバルビツール酸発色法を行うことにより、不飽和糖やα−ケト酸を検出し、もう一方のプレートについては硫酸発色法を行うことにより、全糖質を検出した。その結果を図5に示す。
[化33]HOOC−COH=CH−CHOH−CHOH−CHO。
(1)UMI−01の培養
実施例1(2)のMS培地に、1%(w/v)となるようアルギン酸ナトリウムを加えた培地を用意し、実施例3(1)[1−1]に記載の方法により、UMI−01を培養した。ただし、本培養は800mLに代えて1000mLのスケールで行い、本培養の時間は90時間に代えて48時間とし、本培養の培養温度は30℃に代えて25℃とした。
本実施例(1)のUMI−01について、実施例3(2)[2−1]に記載の方法により、約5gの菌体ペレットを得て、約50mLの菌抽出液を得た。ただし、遠心分離の時間は10分間に代えて15分間とした。
本実施例(2)の精製菌抽出液について実施例3(2)[2−2]に記載の方法により、アルギン酸分解酵素活性を算出したところ、500U/mLであった。すなわち、500U/mL×20mL=計10,000Uの酵素を得ることができた。
[4−1]吸光度測定
10mmol/Lのリン酸ナトリウム緩衝液(pH7.0)に、下記のアルギン酸塩を終濃度0.15%(w/v)となるよう加えたものを用意し、A、B、
CおよびDとした。
B:主としてMブロックを含むアルギン酸のナトリウム塩(M−rich)
C:主としてMGブロックを含むアルギン酸のナトリウム塩(MG−rich)
D:主としてGブロックを含むアルギン酸のナトリウム塩(G−rich)
本実施例(2)[2−1]のBおよびDについて、インキュベート開始から、0分間、5分間、10分間、30分間、60分間、120分間および180分間経過時に、2μLの反応液を分取し、実施例4(5)[5−2]に記載の方法により薄層クロマトグラフィーおよびチオバルビツール酸発色法を行った。その結果を図7に示す。
10mmol/Lのリン酸ナトリウム緩衝液(pH7.0)に、アルギン酸ナトリウムを1%(w/v)となるよう加えたものを2つ用意し、反応液pおよび反応液qとした。反応液pには、本実施例(2)の精製菌抽出液を50U/mL相当量加え、反応液qには、市販のFlavobacterium由来アルギン酸リアーゼ(Sigma社)を50U/mL相当量加えた後、いずれも30℃にて90分間インキュベートした。
(1)精製菌抽出液の調製
実施例1(2)のMS培地に、1/10000量のTrace elements溶液 (0.1NのHCl 1LにFeCl39.7g、CaCl27.8g、CoCl26水和物0.218g、CuSO45水和物0.156g、NiCl36水和物0.118gおよびCrCl36水和物0.105gを溶解したもの)および1%(w/v)となるようアルギン酸ナトリウムを加えた培地(アルギン酸ナトリウム入りMS最少培地)を用意し、実施例3(1)[1−1]に記載の方法により、UMI−01を培養した。ただし、前培養は100mL、本培養は1000mLのスケールで行い、本培養の培地に加える前培養菌液は20mLとした。続いて、実施例3(2)[2−1]に記載の方法において、10mmol/Lリン酸ナトリウム緩衝液(pH7.0)に代えて10mmol/LTris−HCl緩衝液(pH7.6;以下「トリス緩衝液」という。)を用いて抽出操作を行い、菌抽出液を得た。
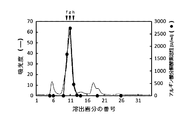
トリス緩衝液で平衡化したToyopearl DEAE−650Mカラム(直径2.5×長さ17.5cm;東ソー社)に本実施例6(1)の精製菌抽出液を供した。その後、0〜0.3mol/Lの直線濃度勾配および0.5mol/LのNaClを含むトリス緩衝液(NaCl/トリス緩衝液)を用いてカラムに吸着させたタンパク質の溶出を行い、溶出液を10mLずつ分取して、分取開始から順に、1〜120番の溶出画分とした。各溶出画分について、含有タンパク質量の指標として280nmにおける吸光度を測定した。また、各溶出画分について、実施例3(2)[2−2]に記載の方法によりアルギン酸分解酵素活性を測定した。その結果を図9に示す。図9に示すように、アルギン酸分解酵素活性は、5〜7番の溶出画分(トリス緩衝液による溶出液;図9においてピークaで示す)、および、31〜45番の溶出画分(0.05〜0.15mol/L付近のNaCl水溶液による溶出液;図9においてピークbおよびcで示す)にピークが確認された。
本実施例6(2)の第一酵素液を凍結乾燥した。続いて、これをトリス緩衝液に溶解し、トリス緩衝液を外液として透析して、内液を回収した。次に、遠心濾過フィルターUltrafree MC/CL(Millipore社)を用いて濾過し、濾液を回収した。濾液をMono Q 5/50 GL陰イオン交換カラム(5×50mm)を装着したAKTA−FPLC (GEヘルスケア社)に供した。その後、0〜0.3mol/Lの直線濃度勾配および0.5mol/LのNaCl/トリス緩衝液を用いて、流速0.8mL/分でカラムに吸着させたタンパク質の溶出を行い、溶出液を1mLずつ分取して、分取開始から順に、1〜35番の溶出画分とした。各溶出画分について、280nmにおける吸光度を測定し、また、実施例3(2)[2−2]に記載の方法によりアルギン酸分解酵素活性を測定した。その結果を図11に示す。図11に示すように、アルギン酸分解酵素活性は、2および3番の溶出画分(トリス緩衝液による溶出液;図11においてピークdで示す)にピークが確認された。また、吸光度は、2および3番の溶出画分(トリス緩衝液による溶出画分;図11においてピークdで示す)および11〜15番の溶出画分(0.1mol/L付近のNaCl/トリス緩衝液による溶出液;図11においてピークeで示す)にピークが確認された。
本実施例6(3)の第二酵素液を0.3mol/LのNaCl/トリス緩衝液に溶解し、0.3mol/LのNaCl/トリス緩衝液を外液として透析して、内液を回収した。次に、遠心濾過フィルターUltrafree MC/CL(Millipore社)を用いて濾過し、濾液を回収した。濾液を、Superdex 75 10/300 GLカラム(0.1×30cm)を装着したAKTA−FPLCゲル濾過カラムクロマトグラフィーに供した。その後、0.3mol/LのNaCl/トリス緩衝液を用いて流速1.0mL/分でカラムに吸着させたタンパク質の溶出を行い、溶出液を1mLずつ分取して、分取開始から順に、1〜35番の溶出画分とした。各溶出画分について、280nmにおける吸光度を測定し、また、実施例3(2)[2−2]に記載の方法によりアルギン酸分解酵素活性を測定した。その結果を図13に示す。図13に示すように、アルギン酸分解酵素活性は、10〜12番の溶出画分;図13においてピークf〜hで示す)にピークが確認された。
(1)アルギン酸の分解様式の検討
10mmol/Lのリン酸ナトリウム緩衝液(pH7.0)に、終濃度0.15%(w/v)となるようアルギン酸ナトリウムを加えて解析用基質液とした。オストワルド粘度計を用いて解析用基質液の30℃における粘度を測定した。続いて、解析用基質液に精製FlAly−1液0.1mL(60U)を加えて反応液とし、30℃にて30分間インキュベートすることによりアルギン酸分解反応を行った。その間、235nmにおける吸光度を測定するとともに、オストワルド粘度計を用いて粘度を測定した。反応液の粘度の測定値について、解析用基質液の測定値を1として、相対値(相対粘度)を算出した。その結果を図15に示す。図15に示すように、相対粘度はアルギン酸分解反応の初期に急激に低下した後、穏やかに低下を続け、30分後には1.08となった。一方、吸光度の増加速度、すなわちアルギン酸の分解産物である不飽和糖の生成速度は、反応時間を通じてほぼ一定であった。これらの結果から、FlAly−1は、アルギン酸の分子内部をランダムに分解する酵素であること、すなわち、エンド型のアルギン酸リアーゼであることが明らかになった。
本実施例7(1)の解析用基質液を調製し、8.3Uの精製FlAly−1液を加えて反応液とした。ただし、10mmol/Lのリン酸ナトリウム緩衝液のpHを変化させることにより、種々のpHの反応液を調製した。反応液を30℃にて15分間インキュベートすることによりアルギン酸分解反応を行い、実施例3(2)[2−2]に記載の方法によりアルギン酸分解酵素活性を測定した。その後、測定されたアルギン酸分解酵素活性の最高値を100%として、相対的活性値を算出した。その結果を図16左図に示す。図16左図に示すように、相対的活性値は、反応液のpHが約8の場合に100%となった。この結果から、FlAly−1のアルギン酸分解反応における至適pHは約8であることが明らかになった。
本実施例7(2)に記載の方法において、反応時間を15分間に代えて3時間としてアルギン酸分解反応を行い、これを当初反応液とした。当初反応液を急冷した後、本実施例7(1)の解析用基質液に加えて、30℃で10分間インキュベートすることにより二度目のアルギン酸分解反応を行った。その後、実施例3(2)[2−2]に記載の方法によりアルギン酸分解酵素活性を測定し、本実施例7(2)に記載の方法により相対的活性値を算出した。その結果を図16右図に示す。図16右図に示すように、相対的活性値は、当初反応液のpHが7.6の場合に100%であり、pH7〜8の場合に80%以上であった。この結果から、pH7〜8でアルギン酸分解反応を行った後に、アルギン酸分解酵素活性が最もよく残存すること、すなわち、FlAly−1はpHが7<pH<8において最も安定であることが明らかになった。
本実施例7(1)の解析用基質液に、11.3Uの精製FlAly−1液を加えて反応液とし、種々の温度にて15分間インキュベートすることによりアルギン酸分解反応を行った。その後、実施例3(2)[2−2]に記載の方法によりアルギン酸分解酵素活性を測定し、本実施例7(2)に記載の方法により相対的活性値を算出した。その結果を図17左図に示す。図17左図に示すように、相対的活性値は、反応液の温度が約54℃の場合に100%であった。この結果から、FlAly−1のアルギン酸分解反応における至適温度は約54℃であることが明らかになった。
本実施例7(5)に記載の方法において、反応時間を15分間に代えて30分間としてアルギン酸分解反応を行い、これを当初反応液とした。当初反応液について、本実施例7(3)に記載の方法により、相対的活性値を求めた。その結果を図17右図に示す。図17右図に示すように、相対的活性値は、当初反応液のアルギン酸分解反応時の温度が、20℃〜47℃の場合に50%以上であり、60℃の場合に0%であった。この結果から、FlAly−1は、47℃で30分間アルギン酸分解反応を行った後ではアルギン酸分解酵素活性が50%以上残存しているのに対し、60℃で30分間アルギン酸分解反応を行った後ではアルギン酸分解酵素活性が消失することが明らかになった。
本実施例7(1)の解析用基質液に、8.8Uの精製FlAly−1液を加え、種々の終濃度になるようにNaClを添加して反応液とした。反応液を30℃にて10分間インキュベートすることによりアルギン酸分解反応を行った。その後、実施例3(2)[2−2]に記載の方法によりアルギン酸分解酵素活性を測定し、本実施例7(2)に記載の方法により相対的活性値を算出した。その結果を図18に示す。図18に示すように、相対的活性値は、反応液におけるNaClの終濃度が0.1および0.2mol/Lの場合に100%であり、0.05mol/Lおよび0.4mol/Lの場合に90%以上であった。この結果から、FlAly−1のアルギン酸分解反応における至適NaCl濃度Cは、0.05<C<0.3(mol/L)であることが明らかとなった。
基質を、アルギン酸ナトリウムに代えてアルギン酸ナトリウムならびに実施例5(4)[4−1]のM−rich、MG−richおよびG−richとして、本実施例7(1)の解析用基質液を調製した。解析用基質液に2.1Uの精製FlAly−1液を加えて反応液とし、30℃にて5分間インキュベートすることによりアルギン酸分解反応を行った。その間、235nmにおける吸光度を測定した。その結果を図19に示す。図19に示すように、アルギン酸ナトリウム、M−rich、MG−richおよびG−richのいずれが基質である場合も、同程度の吸光度が測定された。この結果から、FlAly−1は、Mブロック、MGブロック、Gブロックおよび交互ポリマーをランダムに含むアルギン酸(アルギン酸ナトリウム)、主としてMブロックを含むアルギン酸(M−rich)、主としてMGブロックを含むアルギン酸(MG−rich)、および主としてGブロックを含むアルギン酸(G−rich)のいずれも同様に分解することができることが明らかになった。すなわち、FlAly−1は、アルギン酸の構造中、D−マンヌロン酸のホモポリマーである部分(Mブロック)、L−グルロン酸のホモポリマーである部分(Gブロック)、MブロックとGブロックとが交互に結合した部分(MGブロック)およびD−マンヌロン酸とL−グルロン酸とが交互に結合した部分(交互ポリマー)のいずれも同様に分解することができることが示された。
pHを7.0に代えて7.2とし、基質をアルギン酸ナトリウムに代えて二価金属イオンが共存した場合にゲル化し難いM−richとして、本実施例7(1)の解析用基質液を調製した。解析用基質液に表4の左欄に示す試薬を表4の中欄に示す終濃度となるよう添加し、8.69Uの精製FlAly−1液を加えて反応液とした。反応液を、30℃にて5分間インキュベートすることによりアルギン酸分解反応を行った。また、コントロールとして、試薬を添加せずに、同様にアルギン酸分解反応を行った。その後、実施例3(2)[2−2]に記載の方法によりアルギン酸分解酵素活性を測定し、コントロールのアルギン酸分解酵素活性の値を100%として、相対的活性値を算出した。その結果を表4右欄に示す。表4右欄に示すように、アルギン酸分解酵素活性は、反応液中にNa+やNH4+などが存在した場合に増大する一方で、Co2+やCd2+、Cu2+などが存在した場合に低下する傾向であること、およびDTTなどの還元剤が存在した場合には、ほとんど変化しないことが明らかになった。
アルギン酸ナトリウムの終濃度を0.15(w/v)に代えて0.75%(w/v)として、本実施例7(1)の解析用基質液を調製した。解析用基質液に、500Uの精製FlAly−1液を加えて反応液とし、30℃にて0〜24時間インキュベートすることによりアルギン酸分解反応を行った。その後、実施例4(5)[5−2]に記載の方法により、分解産物の検討を行った。その結果を図20に示す。図20に示すように、硫酸発色法およびチオバルビツール酸発色法のいずれにおいても、2〜4糖を示すバンドが主に確認された。その一方で、単糖(不飽和単糖やα−ケト酸)を示すバンドは確認されなかった。これらの結果から、FlAly−1はアルギン酸を分解して2〜4糖のオリゴ糖を生成させること、および、単糖は生成させないことが明らかになった。なお、UMI−01の菌抽出液や精製菌抽出液により単糖(不飽和単糖やα−ケト酸)が生成したのは、アルギン酸オリゴ糖を単糖(不飽和単糖やα−ケト酸)にまで分解する、FlAly−1とは別の二糖分解酵素をUMI−01が有するためと考えられた。
(1)FlAly−1のN末端アミノ酸配列
FlAly−1のN末端アミノ酸配列を、プロテインシーケンサー(473A型;アプライドバイオシステムズ社)を用いて分析した。その結果、N末端40残基のアミノ酸配列はSKTAKIDWSHWTVTVPEENPDKPGKPYSLGYPEILNYAED(配列番号2)であることが明らかになった。次に、配列番号2のアミノ酸配列について、BLAST検索を行った。その結果を表5に示す。表5に示すように、配列番号2のアミノ酸配列と、100%の相同性を示す既知のアミノ酸配列は検出されなかった。また、配列番号2のアミノ酸配列と46〜51%の相同性を示す既知のアミノ酸配列が検出された。これらは、多糖リアーゼファミリー7(PL−7)に属する微生物由来アルギン酸リアーゼおよび類似の配列を有するタンパク質であった。この結果から、FlAly−1は新規のアルギン酸リアーゼであることが明らかになった。
UMI−01を、実施例6(1)のアルギン酸ナトリウム入りMS最少培地10mLで培養した後、8000rpmで20分間遠心分離を行い、菌体ペレットを回収した。菌体ペレットをマイクロピペットの先で1μL程度取ってマイクロチューブに入れ、ISOHAIR(NIPPON GENE社)を用いて、添付の使用書に従いゲノムDNAを抽出して、抽出液を得た。抽出液に、フェノール/クロロホルム/イソアミルアルコール(25:24:1/v:v:v)200μLを加えて5分間穏やかに転倒混和することによりタンパク質を不溶化させ、11000×gで5分間遠心分離を行った後、ゲノムDNAを含む水相を回収した。その後、エタノール沈殿を行ってゲノムDNAを回収し、TE緩衝液(pH8.0)に溶解した。ゲノムDNAの濃度は、ナノドロップ(NanoDrop 1000)を用いて測定した。
下記条件により一次PCRおよび二次PCRを行って、二次PCR産物を得た。二次PCR産物を、定法に従い、pTaq1プラスミドベクターに挿入した後、大腸菌DH5αにトランスフォーメーションしてクローニングし、クローンプラスミドを得た。
DNAポリメラーゼ;Ex Taq polymerase(TaKaRa社)
プライマー;
配列番号2のアミノ酸配列に基づき設計した縮重プライマーとして1Fおよび2F
1F:GGNAARACNGCNAARATHGA(配列番号3)
2F:CAYWSNCAYTGGACNGTNAC(配列番号4)
PL−7の高度保存領域のアミノ酸配列に基づき設計した縮重プライマーとして1Rおよび2R
1R:TCNGCNGCRTARTTNARDAT(配列番号5)
2R:TANARNCCNGCNGCYTTRAARTA(配列番号6)
[なお、プライマーの塩基配列中、NはA、C、GまたはTを、RはAまたはGを、HはA、TまたはCを、YはCまたはTを、WはAまたはTを、SはCまたはGを、DはA、GまたはTをそれぞれ示す。]
鋳型DNA;本実施例8(2)のゲノムDNA
使用機器;PCR Thermal Cycler DiceR mini(TaKaRa社)
反応条件;96℃で2分間の後、96℃で30秒、45℃で30秒および72℃で1分の反応を1サイクルとして、40サイクル行い、最後に72℃で7分間インキュベートすることにより、一次PCR産物を得た。
鋳型DNA;一次PCR産物
プライマー;(TA PCR Cloning Kit;ダイナエクスプレス社)
M13 BD−Fw Primer
M13 BD−Rev Primer
反応条件;40サイクルを30サイクルとした他は一次PCRと同条件で行い、二次PCR産物を得た。
鋳型DNA;本実施例8(2)のゲノムDNAを各種の制限酵素により消化し、フェノール−クロロホルム処理およびエタノール沈殿を行って精製したものをT4 DNA Ligase(TaKaRa社)により自己ライゲーションさせて得られた環状DNA。
プライマー;
inv−1F:AATGAGAGGTACGTATGCTATTGACGA(配列番号7)
inv−2F:GCCGCGTTATTATTGCGCAAATTCACGG(配列番号8)
inv−1R:CAACAGACTTGTCTTTTGGGTCATCGTA(配列番号9)
inv−2R:GGATGCGATTTTATCCTCAGCATAATTT(配列番号10)
反応条件;96℃で2分間の後、96℃で30秒、60℃で30秒および72℃で5分の反応を1サイクルとして、30サイクル行い、最後に72℃で7分間。
本実施例8(3)のクローンプラスミドについて、定法に従い塩基配列のシークエンスを行って、FlAly−1遺伝子の翻訳領域の全塩基配列(配列番号11)を決定した。また、配列番号11の塩基配列から、FlAly−1の全アミノ酸配列(配列番号12)を演繹した。その結果を図21に示す。なお、図21において、本実施例8(1)で決定したN末端アミノ酸配列(配列番号2)に相当する部分を下線で示す。図21に示すように、FlAly−1遺伝子の翻訳領域の塩基配列(配列番号11)は867bpであり、FlAly−1のアミノ酸配列(配列番号12)は288残基であった。ここで、本実施例8(1)で決定したN末端アミノ酸配列(配列番号2)よりN末端側の28アミノ酸残基は、FlAly−1には存在していなかったことから、分泌シグナル配列と推定された。このことから、成熟型のFlAly−1は、配列番号12のアミノ酸配列(翻訳型のアミノ酸配列)のうち、配列番号2のアミノ酸配列をN末端として、そこからC末端までの全部で260アミノ酸残基(配列番号13)から成り、その理論分子量は29669.4Daと算定された。この理論分子量は、実施例6で行ったSDS−PAGEにより推定された分子量である約30kDaと一致しており、整合性のある結果が得られたことが明らかになった。
(1)ゲル濾過クロマトグラフィーによる精製
[1−1]アルギン酸の分解およびアルギン酸分解産物の回収
蒸留水50mLに、アルギン酸ナトリウム500mgを溶解した後、実施例5(2)の精製菌抽出液を60U/mL相当量加えて、これを反応液とした。反応液を30℃にて12時間インキュベートした後、透析用セルロースチューブUC36−32(三光純薬)を用いて、蒸留水450mLを外液として透析して、外液を回収した。ロータリーエバポレーターを用いて回収した外液を濃縮乾固し、375mgの濃縮乾固物を得て、これをアルギン酸分解産物とした。
本実施例(1)[1−1]のアルギン酸分解産物350mgを5mLの50mmol/Lリン酸ナトリウム緩衝液(pH7.0)に溶解し、バイオゲルP2カラム(4.6×70cm;分画分子量100−1800;BioRad社)を用いて、添付の仕様書に従ってゲル濾過クロマトグラフィーを行い、溶出液を10mLずつ分取した。その後、各溶出液の235nmにおける吸光度を測定した。また、各溶出液から2μLをとり、実施例4(5)[5−2]に記載の方法により薄層クロマトグラフィーおよびチオバルビツール酸発色法を行った。その結果を図23に示す。
本実施例(1)[1−1]に記載の方法により、アルギン酸の分解およびアルギン酸分解産物の回収を行った。続いて、アルギン酸分解産物350mgを5mLの蒸留水に溶解し、第4級アンモニウム基をイオン交換基とするTOYOPEARL SuperQ−650 M(2×20cm;東ソー社)を用いて、添付の仕様書に従ってイオン交換クロマトグラフィーを行い、溶出液を5mLずつ分取した。なお、溶出液には直線濃度勾配0〜0.15mol/LのNaCl水溶液を用いた。その後、各溶出液の235nmにおける吸光度を測定した。また、各溶出液から2μLをとり、実施例4(5)[5−2]に記載の方法により薄層クロマトグラフィーおよびチオバルビツール酸発色法を行った。その結果を図24に示す。
[化36]HOOC−COH=CH−CHOH−CHOH−CHO。
上述の実施例6の実験を、UMI−01に代えてFlavobacterium limicola F31株を用いて行ったところ、UMI−01を用いた場合と同様の結果が得られた。
Claims (6)
- 受託番号NITE P−1076であるアルギン酸資化菌。
- アルギン酸を分解して次式(化4)、次式(化5)および次式(化6)からなる群から選択される1または2以上の物質を製造することができる分解酵素を産生する請求項1に記載のアルギン酸資化菌;
[化6]HOOC−COH=CH−CHOH−CHOH−CHO。 - 請求項1または請求項2に記載のアルギン酸資化菌の菌抽出液であって、アルギン酸を分解して次式(化7)、次式(化8)および次式(化9)からなる群から選択される1または2以上の物質を製造することができる分解酵素を含む前記菌抽出液;
[化9]HOOC−COH=CH−CHOH−CHOH−CHO。 - 分解酵素として、エンド型アルギン酸リアーゼおよび二糖分解酵素を含む、請求項3に記載の菌抽出液。
- アルギン酸を分解してオリゴ糖、ならびに次式(化10)、次式(化11)および次式(化12)からなる群から選択される1または2以上の物質を製造する方法であって、請求項1または請求項2に記載のアルギン酸資化菌が産生する分解酵素をアルギン酸に作用させる工程を有する前記方法;
[化12]HOOC−COH=CH−CHOH−CHOH−CHO。 - アルギン酸を分解して次式(化13)、次式(化14)および次式(化15)からなる群から選択される1または2以上の物質を製造するための、請求項1または請求項2に記載のアルギン酸資化菌の使用;
[化15]HOOC−COH=CH−CHOH−CHOH−CHO。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012067226A JP6025018B2 (ja) | 2011-03-24 | 2012-03-23 | 新規のアルギン酸資化菌、その細菌が産生するアルギン酸を分解する酵素を含む菌抽出液、それらを用いてオリゴ糖、不飽和単糖、ないしα−ケト酸を製造する方法 |
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011066721 | 2011-03-24 | ||
| JP2011066721 | 2011-03-24 | ||
| JP2012067226A JP6025018B2 (ja) | 2011-03-24 | 2012-03-23 | 新規のアルギン酸資化菌、その細菌が産生するアルギン酸を分解する酵素を含む菌抽出液、それらを用いてオリゴ糖、不飽和単糖、ないしα−ケト酸を製造する方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2012210208A JP2012210208A (ja) | 2012-11-01 |
| JP6025018B2 true JP6025018B2 (ja) | 2016-11-16 |
Family
ID=47264739
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2012067226A Active JP6025018B2 (ja) | 2011-03-24 | 2012-03-23 | 新規のアルギン酸資化菌、その細菌が産生するアルギン酸を分解する酵素を含む菌抽出液、それらを用いてオリゴ糖、不飽和単糖、ないしα−ケト酸を製造する方法 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP6025018B2 (ja) |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102994407B (zh) * | 2011-12-16 | 2014-10-29 | 中国科学院大连化学物理研究所 | 黄杆菌属菌株与内切褐藻胶裂解酶编码基因及制备与应用 |
| US10927363B2 (en) | 2016-04-04 | 2021-02-23 | Mie University | DNA sequence and expression vector for alginate lyase |
| MX2023004262A (es) | 2020-10-29 | 2023-04-26 | Procter & Gamble | Composiciones de limpieza que contienen enzimas alginasas. |
| CN117730984A (zh) * | 2024-01-05 | 2024-03-22 | 山东海之宝海洋科技有限公司 | 海带酶解产物及其应用 |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH04169189A (ja) * | 1990-11-01 | 1992-06-17 | Kibun Food Chemifa Co Ltd | アルギン酸、その塩類又はその誘導体の分解物の製造法 |
| JP3076856B2 (ja) * | 1991-07-05 | 2000-08-14 | 大塚化学株式会社 | 細菌によるアルギン酸の分解法 |
| JP3309220B2 (ja) * | 2000-11-14 | 2002-07-29 | 大塚化学株式会社 | アルギン酸リアーゼ |
| JP5364352B2 (ja) * | 2008-11-20 | 2013-12-11 | 国立大学法人鳥取大学 | 新規なフコイダン資化性微生物 |
-
2012
- 2012-03-23 JP JP2012067226A patent/JP6025018B2/ja active Active
Also Published As
| Publication number | Publication date |
|---|---|
| JP2012210208A (ja) | 2012-11-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Zhu et al. | Preparation of trisaccharides from alginate by a novel alginate lyase Alg7A from marine bacterium Vibrio sp. W13 | |
| Cardozo et al. | Bioconversion of α-chitin into N-acetyl-glucosamine using chitinases produced by marine-derived Aeromonas caviae isolates | |
| JP6025018B2 (ja) | 新規のアルギン酸資化菌、その細菌が産生するアルギン酸を分解する酵素を含む菌抽出液、それらを用いてオリゴ糖、不飽和単糖、ないしα−ケト酸を製造する方法 | |
| Chi et al. | Production and characterization of a novel thermostable extracellular agarase from Pseudoalteromonas hodoensis newly isolated from the west sea of South Korea | |
| Siddiqi et al. | Complete genome sequencing of Arachidicoccus ginsenosidimutans sp. nov., and its application for production of minor ginsenosides by finding a novel ginsenoside-transforming β-glucosidase | |
| Chen et al. | Muricauda amphidinii sp. nov., a novel marine bacterium isolated from the phycosphere of dinoflagellate Amphidinium carterae | |
| WO2012002450A1 (ja) | D-サクシニラーゼ、およびこれを用いたd-アミノ酸の製造方法 | |
| Liu et al. | Purification, characterization, and hydrolysate analysis of dextranase from Arthrobacter oxydans G6-4B | |
| KR100876662B1 (ko) | 알지네이트 가수분해 활성을 갖는 스트렙토마이세스 속균주 및 알지네이트 분해효소 | |
| Duhsaki et al. | Improving the efficiency and sustainability of chitin bioconversion through a combination of Streptomyces chitin-active-secretomes and mechanical-milling | |
| Kang et al. | Mucilaginibacter sabulilitoris sp. nov., isolated from marine sand in a firth | |
| Xu et al. | Purification and characterization of cold-adapted and salt-tolerant dextranase from Cellulosimicrobium sp. THN1 and its potential application for treatment of dental plaque | |
| CN102311930B (zh) | 一种由Pseudomonas.sp.HZJ216生产的褐藻胶裂解酶 | |
| Singh et al. | Flavobacterium vireti sp. nov., isolated from soil | |
| KR20150101789A (ko) | 복합 효소 생산능 및 항균 활성을 갖는 바실러스 서브틸리스 bk418 균주 및 이의 용도 | |
| AU2018333356B2 (en) | Bacterial strain Clostridium histolyticum and its use | |
| CN101245325B (zh) | 一株产新型碱性蛋白酶的嗜麦芽寡养单胞菌 | |
| JP4073787B2 (ja) | 硫酸化フコグルクロノマンナン | |
| Muhammad et al. | Three novel marine species of the genus Reichenbachiella exhibiting degradation of complex polysaccharides | |
| Chen et al. | Muricauda chongwuensis sp. nov., isolated from coastal seawater of China | |
| JP2010022280A (ja) | 新規微生物、イヌリナーゼ、イヌリン分解剤、イヌロオリゴ糖の製造方法及びイヌリナーゼの製造方法 | |
| CN108676738A (zh) | 一株产碱性蛋白酶嗜盐菌及其应用 | |
| CN103865865B (zh) | 一种海参肠道产碱性蛋白酶菌株及其应用 | |
| JP5364352B2 (ja) | 新規なフコイダン資化性微生物 | |
| 茶木貴光 et al. | Purification and characterization of alginate lyase from Pseudoalteromonas sp. strain no. 1786 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20150323 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A821 Effective date: 20150323 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20160210 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20160408 |
|
| RD03 | Notification of appointment of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7423 Effective date: 20160525 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20160607 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A821 Effective date: 20160525 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20160914 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20160929 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 6025018 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |