JP5144244B2 - 電極触媒およびその用途、ならびに電極触媒の製造方法 - Google Patents
電極触媒およびその用途、ならびに電極触媒の製造方法 Download PDFInfo
- Publication number
- JP5144244B2 JP5144244B2 JP2007328807A JP2007328807A JP5144244B2 JP 5144244 B2 JP5144244 B2 JP 5144244B2 JP 2007328807 A JP2007328807 A JP 2007328807A JP 2007328807 A JP2007328807 A JP 2007328807A JP 5144244 B2 JP5144244 B2 JP 5144244B2
- Authority
- JP
- Japan
- Prior art keywords
- transition metal
- metal element
- titanium
- electrode catalyst
- niobium
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 239000003054 catalyst Substances 0.000 title claims description 155
- 238000004519 manufacturing process Methods 0.000 title claims description 26
- 239000010411 electrocatalyst Substances 0.000 title claims description 11
- 229910052723 transition metal Inorganic materials 0.000 claims description 161
- 229910044991 metal oxide Inorganic materials 0.000 claims description 94
- 150000004706 metal oxides Chemical class 0.000 claims description 94
- 239000000463 material Substances 0.000 claims description 84
- 239000001301 oxygen Substances 0.000 claims description 79
- 229910052760 oxygen Inorganic materials 0.000 claims description 79
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 claims description 78
- 239000010936 titanium Substances 0.000 claims description 68
- 229910052751 metal Inorganic materials 0.000 claims description 66
- 239000002184 metal Substances 0.000 claims description 66
- 229910052719 titanium Inorganic materials 0.000 claims description 66
- RTAQQCXQSZGOHL-UHFFFAOYSA-N Titanium Chemical compound [Ti] RTAQQCXQSZGOHL-UHFFFAOYSA-N 0.000 claims description 65
- 239000010955 niobium Substances 0.000 claims description 60
- 229910052758 niobium Inorganic materials 0.000 claims description 59
- GUCVJGMIXFAOAE-UHFFFAOYSA-N niobium atom Chemical compound [Nb] GUCVJGMIXFAOAE-UHFFFAOYSA-N 0.000 claims description 59
- 238000010438 heat treatment Methods 0.000 claims description 56
- 229910052715 tantalum Inorganic materials 0.000 claims description 54
- GUVRBAGPIYLISA-UHFFFAOYSA-N tantalum atom Chemical compound [Ta] GUVRBAGPIYLISA-UHFFFAOYSA-N 0.000 claims description 54
- QCWXUUIWCKQGHC-UHFFFAOYSA-N Zirconium Chemical compound [Zr] QCWXUUIWCKQGHC-UHFFFAOYSA-N 0.000 claims description 44
- 229910052726 zirconium Inorganic materials 0.000 claims description 44
- 150000002736 metal compounds Chemical class 0.000 claims description 42
- 239000000446 fuel Substances 0.000 claims description 39
- 238000000034 method Methods 0.000 claims description 37
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 claims description 36
- 150000002902 organometallic compounds Chemical class 0.000 claims description 30
- 150000003839 salts Chemical class 0.000 claims description 30
- 239000012528 membrane Substances 0.000 claims description 29
- 150000004696 coordination complex Chemical class 0.000 claims description 28
- 239000002245 particle Substances 0.000 claims description 28
- 239000012298 atmosphere Substances 0.000 claims description 25
- 229910052697 platinum Inorganic materials 0.000 claims description 18
- 239000003792 electrolyte Substances 0.000 claims description 17
- 230000001965 increasing effect Effects 0.000 claims description 15
- 230000003647 oxidation Effects 0.000 claims description 14
- 238000007254 oxidation reaction Methods 0.000 claims description 14
- 238000006460 hydrolysis reaction Methods 0.000 claims description 10
- 238000007353 oxidative pyrolysis Methods 0.000 claims description 10
- 230000007062 hydrolysis Effects 0.000 claims description 9
- 239000005518 polymer electrolyte Substances 0.000 claims description 7
- 230000009467 reduction Effects 0.000 description 30
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 25
- 239000007787 solid Substances 0.000 description 22
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 21
- 229910052799 carbon Inorganic materials 0.000 description 19
- 239000000843 powder Substances 0.000 description 17
- 239000002994 raw material Substances 0.000 description 15
- 239000000243 solution Substances 0.000 description 15
- 238000005979 thermal decomposition reaction Methods 0.000 description 13
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Chemical compound O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 13
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 12
- 239000000126 substance Substances 0.000 description 12
- 150000004703 alkoxides Chemical class 0.000 description 11
- 230000007547 defect Effects 0.000 description 11
- 238000001228 spectrum Methods 0.000 description 11
- 150000007942 carboxylates Chemical class 0.000 description 9
- 238000001035 drying Methods 0.000 description 9
- 239000006104 solid solution Substances 0.000 description 9
- OGIDPMRJRNCKJF-UHFFFAOYSA-N titanium oxide Inorganic materials [Ti]=O OGIDPMRJRNCKJF-UHFFFAOYSA-N 0.000 description 9
- 229920000557 Nafion® Polymers 0.000 description 8
- 238000002441 X-ray diffraction Methods 0.000 description 8
- 230000003197 catalytic effect Effects 0.000 description 8
- 230000008859 change Effects 0.000 description 8
- 238000006243 chemical reaction Methods 0.000 description 8
- 239000007788 liquid Substances 0.000 description 8
- 238000005259 measurement Methods 0.000 description 8
- BFRGSJVXBIWTCF-UHFFFAOYSA-N niobium monoxide Chemical compound [Nb]=O BFRGSJVXBIWTCF-UHFFFAOYSA-N 0.000 description 8
- 239000002904 solvent Substances 0.000 description 8
- 230000002378 acidificating effect Effects 0.000 description 7
- 239000007789 gas Substances 0.000 description 7
- 239000004570 mortar (masonry) Substances 0.000 description 7
- -1 titanium halide Chemical class 0.000 description 7
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 6
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 6
- 238000011156 evaluation Methods 0.000 description 6
- 230000003301 hydrolyzing effect Effects 0.000 description 6
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 6
- 229920000642 polymer Polymers 0.000 description 6
- 229920000128 polypyrrole Polymers 0.000 description 6
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 5
- 125000003545 alkoxy group Chemical group 0.000 description 5
- 125000004429 atom Chemical group 0.000 description 5
- 239000006185 dispersion Substances 0.000 description 5
- 238000001914 filtration Methods 0.000 description 5
- 239000001257 hydrogen Substances 0.000 description 5
- 229910052739 hydrogen Inorganic materials 0.000 description 5
- 230000035484 reaction time Effects 0.000 description 5
- 238000003756 stirring Methods 0.000 description 5
- 238000005406 washing Methods 0.000 description 5
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 4
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 4
- 125000002843 carboxylic acid group Chemical group 0.000 description 4
- 229920001940 conductive polymer Polymers 0.000 description 4
- 238000001816 cooling Methods 0.000 description 4
- 238000001704 evaporation Methods 0.000 description 4
- 239000012535 impurity Substances 0.000 description 4
- 239000011261 inert gas Substances 0.000 description 4
- HFLAMWCKUFHSAZ-UHFFFAOYSA-N niobium dioxide Chemical compound O=[Nb]=O HFLAMWCKUFHSAZ-UHFFFAOYSA-N 0.000 description 4
- 239000012299 nitrogen atmosphere Substances 0.000 description 4
- 230000001590 oxidative effect Effects 0.000 description 4
- 229920000767 polyaniline Polymers 0.000 description 4
- 229920000123 polythiophene Polymers 0.000 description 4
- 230000002441 reversible effect Effects 0.000 description 4
- 239000000725 suspension Substances 0.000 description 4
- NJLQUTOLTXWLBV-UHFFFAOYSA-N 2-ethylhexanoic acid titanium Chemical compound [Ti].CCCCC(CC)C(O)=O.CCCCC(CC)C(O)=O.CCCCC(CC)C(O)=O.CCCCC(CC)C(O)=O NJLQUTOLTXWLBV-UHFFFAOYSA-N 0.000 description 3
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 3
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 3
- 239000006227 byproduct Substances 0.000 description 3
- 230000007797 corrosion Effects 0.000 description 3
- 238000005260 corrosion Methods 0.000 description 3
- 238000009792 diffusion process Methods 0.000 description 3
- 239000002270 dispersing agent Substances 0.000 description 3
- 230000008020 evaporation Effects 0.000 description 3
- 238000010304 firing Methods 0.000 description 3
- 229910021397 glassy carbon Inorganic materials 0.000 description 3
- 229910001507 metal halide Inorganic materials 0.000 description 3
- 150000005309 metal halides Chemical class 0.000 description 3
- URLJKFSTXLNXLG-UHFFFAOYSA-N niobium(5+);oxygen(2-) Chemical compound [O-2].[O-2].[O-2].[O-2].[O-2].[Nb+5].[Nb+5] URLJKFSTXLNXLG-UHFFFAOYSA-N 0.000 description 3
- 125000004430 oxygen atom Chemical group O* 0.000 description 3
- 238000000197 pyrolysis Methods 0.000 description 3
- 238000004062 sedimentation Methods 0.000 description 3
- 239000002002 slurry Substances 0.000 description 3
- 238000000859 sublimation Methods 0.000 description 3
- 230000008022 sublimation Effects 0.000 description 3
- 239000004408 titanium dioxide Substances 0.000 description 3
- GEYOCULIXLDCMW-UHFFFAOYSA-N 1,2-phenylenediamine Chemical compound NC1=CC=CC=C1N GEYOCULIXLDCMW-UHFFFAOYSA-N 0.000 description 2
- 125000001140 1,4-phenylene group Chemical group [H]C1=C([H])C([*:2])=C([H])C([H])=C1[*:1] 0.000 description 2
- VWBVCOPVKXNMMZ-UHFFFAOYSA-N 1,5-diaminoanthracene-9,10-dione Chemical compound O=C1C2=C(N)C=CC=C2C(=O)C2=C1C=CC=C2N VWBVCOPVKXNMMZ-UHFFFAOYSA-N 0.000 description 2
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 2
- 239000004215 Carbon black (E152) Substances 0.000 description 2
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 2
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 2
- SMWDFEZZVXVKRB-UHFFFAOYSA-N Quinoline Chemical class N1=CC=CC2=CC=CC=C21 SMWDFEZZVXVKRB-UHFFFAOYSA-N 0.000 description 2
- YRKCREAYFQTBPV-UHFFFAOYSA-N acetylacetone Chemical compound CC(=O)CC(C)=O YRKCREAYFQTBPV-UHFFFAOYSA-N 0.000 description 2
- 239000002253 acid Substances 0.000 description 2
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 2
- 238000009835 boiling Methods 0.000 description 2
- 239000006229 carbon black Substances 0.000 description 2
- 238000000576 coating method Methods 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- 239000011521 glass Substances 0.000 description 2
- 239000010439 graphite Substances 0.000 description 2
- 229910002804 graphite Inorganic materials 0.000 description 2
- 229930195733 hydrocarbon Natural products 0.000 description 2
- 150000002430 hydrocarbons Chemical class 0.000 description 2
- DEIVNMVWRDMSMJ-UHFFFAOYSA-N hydrogen peroxide;oxotitanium Chemical compound OO.[Ti]=O DEIVNMVWRDMSMJ-UHFFFAOYSA-N 0.000 description 2
- AMWRITDGCCNYAT-UHFFFAOYSA-L hydroxy(oxo)manganese;manganese Chemical compound [Mn].O[Mn]=O.O[Mn]=O AMWRITDGCCNYAT-UHFFFAOYSA-L 0.000 description 2
- 230000001976 improved effect Effects 0.000 description 2
- 238000000691 measurement method Methods 0.000 description 2
- 239000000203 mixture Substances 0.000 description 2
- KELHQGOVULCJSG-UHFFFAOYSA-N n,n-dimethyl-1-(5-methylfuran-2-yl)ethane-1,2-diamine Chemical compound CN(C)C(CN)C1=CC=C(C)O1 KELHQGOVULCJSG-UHFFFAOYSA-N 0.000 description 2
- ZKATWMILCYLAPD-UHFFFAOYSA-N niobium pentoxide Inorganic materials O=[Nb](=O)O[Nb](=O)=O ZKATWMILCYLAPD-UHFFFAOYSA-N 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- QGLKJKCYBOYXKC-UHFFFAOYSA-N nonaoxidotritungsten Chemical compound O=[W]1(=O)O[W](=O)(=O)O[W](=O)(=O)O1 QGLKJKCYBOYXKC-UHFFFAOYSA-N 0.000 description 2
- 125000000962 organic group Chemical group 0.000 description 2
- 229920001197 polyacetylene Polymers 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- 239000012266 salt solution Substances 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 125000000542 sulfonic acid group Chemical group 0.000 description 2
- 230000007704 transition Effects 0.000 description 2
- 229910001930 tungsten oxide Inorganic materials 0.000 description 2
- 238000003828 vacuum filtration Methods 0.000 description 2
- POILWHVDKZOXJZ-ARJAWSKDSA-M (z)-4-oxopent-2-en-2-olate Chemical compound C\C([O-])=C\C(C)=O POILWHVDKZOXJZ-ARJAWSKDSA-M 0.000 description 1
- CYWDDBNPXTUVNN-UHFFFAOYSA-I 2-ethylhexanoate;niobium(5+) Chemical compound [Nb+5].CCCCC(CC)C([O-])=O.CCCCC(CC)C([O-])=O.CCCCC(CC)C([O-])=O.CCCCC(CC)C([O-])=O.CCCCC(CC)C([O-])=O CYWDDBNPXTUVNN-UHFFFAOYSA-I 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 238000004438 BET method Methods 0.000 description 1
- FIPWRIJSWJWJAI-UHFFFAOYSA-N Butyl carbitol 6-propylpiperonyl ether Chemical compound C1=C(CCC)C(COCCOCCOCCCC)=CC2=C1OCO2 FIPWRIJSWJWJAI-UHFFFAOYSA-N 0.000 description 1
- XMWRBQBLMFGWIX-UHFFFAOYSA-N C60 fullerene Chemical compound C12=C3C(C4=C56)=C7C8=C5C5=C9C%10=C6C6=C4C1=C1C4=C6C6=C%10C%10=C9C9=C%11C5=C8C5=C8C7=C3C3=C7C2=C1C1=C2C4=C6C4=C%10C6=C9C9=C%11C5=C5C8=C3C3=C7C1=C1C2=C4C6=C2C9=C5C3=C12 XMWRBQBLMFGWIX-UHFFFAOYSA-N 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- 229910010413 TiO 2 Inorganic materials 0.000 description 1
- XHCLAFWTIXFWPH-UHFFFAOYSA-N [O-2].[O-2].[O-2].[O-2].[O-2].[V+5].[V+5] Chemical compound [O-2].[O-2].[O-2].[O-2].[O-2].[V+5].[V+5] XHCLAFWTIXFWPH-UHFFFAOYSA-N 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 229910052786 argon Inorganic materials 0.000 description 1
- 239000002585 base Substances 0.000 description 1
- FPCJKVGGYOAWIZ-UHFFFAOYSA-N butan-1-ol;titanium Chemical compound [Ti].CCCCO.CCCCO.CCCCO.CCCCO FPCJKVGGYOAWIZ-UHFFFAOYSA-N 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 229910002092 carbon dioxide Inorganic materials 0.000 description 1
- 239000002134 carbon nanofiber Substances 0.000 description 1
- 229910021393 carbon nanotube Inorganic materials 0.000 description 1
- 239000002041 carbon nanotube Substances 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 150000001735 carboxylic acids Chemical class 0.000 description 1
- 239000000919 ceramic Substances 0.000 description 1
- 229910000428 cobalt oxide Inorganic materials 0.000 description 1
- IVMYJDGYRUAWML-UHFFFAOYSA-N cobalt(ii) oxide Chemical compound [Co]=O IVMYJDGYRUAWML-UHFFFAOYSA-N 0.000 description 1
- 150000001875 compounds Chemical class 0.000 description 1
- 239000004020 conductor Substances 0.000 description 1
- 230000008602 contraction Effects 0.000 description 1
- 238000000354 decomposition reaction Methods 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- HTXDPTMKBJXEOW-UHFFFAOYSA-N dioxoiridium Chemical compound O=[Ir]=O HTXDPTMKBJXEOW-UHFFFAOYSA-N 0.000 description 1
- 238000007598 dipping method Methods 0.000 description 1
- 238000003487 electrochemical reaction Methods 0.000 description 1
- 238000003411 electrode reaction Methods 0.000 description 1
- HHFAWKCIHAUFRX-UHFFFAOYSA-N ethoxide Chemical compound CC[O-] HHFAWKCIHAUFRX-UHFFFAOYSA-N 0.000 description 1
- 239000004744 fabric Substances 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- UQSQSQZYBQSBJZ-UHFFFAOYSA-N fluorosulfonic acid Chemical compound OS(F)(=O)=O UQSQSQZYBQSBJZ-UHFFFAOYSA-N 0.000 description 1
- 239000011888 foil Substances 0.000 description 1
- 238000004108 freeze drying Methods 0.000 description 1
- 229910003472 fullerene Inorganic materials 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 230000005484 gravity Effects 0.000 description 1
- 238000007602 hot air drying Methods 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 230000001939 inductive effect Effects 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- 229910052809 inorganic oxide Inorganic materials 0.000 description 1
- 229910000457 iridium oxide Inorganic materials 0.000 description 1
- ZXEKIIBDNHEJCQ-UHFFFAOYSA-N isobutanol Chemical compound CC(C)CO ZXEKIIBDNHEJCQ-UHFFFAOYSA-N 0.000 description 1
- 239000003446 ligand Substances 0.000 description 1
- 239000011244 liquid electrolyte Substances 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 150000002739 metals Chemical class 0.000 description 1
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Chemical compound C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 1
- 239000012982 microporous membrane Substances 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 239000000178 monomer Substances 0.000 description 1
- 239000002116 nanohorn Substances 0.000 description 1
- 229910000480 nickel oxide Inorganic materials 0.000 description 1
- 150000002821 niobium Chemical class 0.000 description 1
- 229910000484 niobium oxide Inorganic materials 0.000 description 1
- ZTILUDNICMILKJ-UHFFFAOYSA-N niobium(v) ethoxide Chemical compound CCO[Nb](OCC)(OCC)(OCC)OCC ZTILUDNICMILKJ-UHFFFAOYSA-N 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- GNRSAWUEBMWBQH-UHFFFAOYSA-N oxonickel Chemical compound [Ni]=O GNRSAWUEBMWBQH-UHFFFAOYSA-N 0.000 description 1
- DCKVFVYPWDKYDN-UHFFFAOYSA-L oxygen(2-);titanium(4+);sulfate Chemical compound [O-2].[Ti+4].[O-]S([O-])(=O)=O DCKVFVYPWDKYDN-UHFFFAOYSA-L 0.000 description 1
- YHBDIEWMOMLKOO-UHFFFAOYSA-I pentachloroniobium Chemical compound Cl[Nb](Cl)(Cl)(Cl)Cl YHBDIEWMOMLKOO-UHFFFAOYSA-I 0.000 description 1
- 229960005235 piperonyl butoxide Drugs 0.000 description 1
- UQWLEJDCBWVKSN-UHFFFAOYSA-N platinum zirconium Chemical compound [Zr].[Pt] UQWLEJDCBWVKSN-UHFFFAOYSA-N 0.000 description 1
- 239000011148 porous material Substances 0.000 description 1
- 230000001376 precipitating effect Effects 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 239000000047 product Substances 0.000 description 1
- IKNCGYCHMGNBCP-UHFFFAOYSA-N propan-1-olate Chemical compound CCC[O-] IKNCGYCHMGNBCP-UHFFFAOYSA-N 0.000 description 1
- OGHBATFHNDZKSO-UHFFFAOYSA-N propan-2-olate Chemical compound CC(C)[O-] OGHBATFHNDZKSO-UHFFFAOYSA-N 0.000 description 1
- 238000010298 pulverizing process Methods 0.000 description 1
- 238000005086 pumping Methods 0.000 description 1
- 239000012495 reaction gas Substances 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 230000000630 rising effect Effects 0.000 description 1
- 238000005096 rolling process Methods 0.000 description 1
- 229910001925 ruthenium oxide Inorganic materials 0.000 description 1
- WOCIAKWEIIZHES-UHFFFAOYSA-N ruthenium(iv) oxide Chemical compound O=[Ru]=O WOCIAKWEIIZHES-UHFFFAOYSA-N 0.000 description 1
- 238000007650 screen-printing Methods 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 229910001220 stainless steel Inorganic materials 0.000 description 1
- 239000010935 stainless steel Substances 0.000 description 1
- TXEYQDLBPFQVAA-UHFFFAOYSA-N tetrafluoromethane Chemical compound FC(F)(F)F TXEYQDLBPFQVAA-UHFFFAOYSA-N 0.000 description 1
- LLZRNZOLAXHGLL-UHFFFAOYSA-J titanic acid Chemical compound O[Ti](O)(O)O LLZRNZOLAXHGLL-UHFFFAOYSA-J 0.000 description 1
- 229910000348 titanium sulfate Inorganic materials 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 229910001935 vanadium oxide Inorganic materials 0.000 description 1
- 239000013585 weight reducing agent Substances 0.000 description 1
Images






Classifications
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E60/00—Enabling technologies; Technologies with a potential or indirect contribution to GHG emissions mitigation
- Y02E60/30—Hydrogen technology
- Y02E60/50—Fuel cells
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P70/00—Climate change mitigation technologies in the production process for final industrial or consumer products
- Y02P70/50—Manufacturing or production processes characterised by the final manufactured product
Description
電池用触媒が提案されている。しかし、該燃料電池用触媒は、白金を併用することを想定しており、未だ改善の余地があった。
(1)
ニオブ、チタン、タンタルおよびジルコニウムからなる群から選択される少なくとも二種以上の遷移金属元素を含み、且つ白金を含まない金属酸化物材料からなることを特徴と
する電極触媒。
粉末であることを特徴とする(1)に記載の電極触媒。
BET比表面積が1〜1000m2/gの範囲であることを特徴とする(1)または(
2)に記載の電極触媒。
ニオブ、チタン、タンタルおよびジルコニウムからなる群から選択される少なくとも一種の遷移金属元素(a)を含む金属化合物(A)と、
ニオブ、チタン、タンタルおよびジルコニウムからなる群から選択される少なくとも一種の遷移金属元素(b)を含む金属化合物(B)(ただし、遷移金属元素(a)と遷移金属元素(b)とは異なる)とを、
前記遷移金属元素(a)および/または前記遷移金属元素(b)の価数が大きくなるように、酸素含有雰囲気下で熱処理することにより得られる白金を含まない金属酸化物材料からなることを特徴とする(1)〜(3)のいずれかに記載の電極触媒。
ニオブ、チタン、タンタルおよびジルコニウムからなる群から選択される少なくとも一種の遷移金属元素(c)を含む、金属塩または金属錯体(C)と、
ニオブ、チタン、タンタルおよびジルコニウムからなる群から選択される少なくとも一種の遷移金属元素(d)を含む、金属塩または金属錯体(D)(ただし、遷移金属元素(c)と遷移金属元素(d)とは異なる)とを、
加水分解することにより得られる白金を含まない金属酸化物材料からなることを特徴とする(1)〜(3)のいずれかに記載の電極触媒。
ニオブ、チタン、タンタルおよびジルコニウムからなる群から選択される少なくとも一種の遷移金属元素(e)を含む金属有機化合物(E)と、
ニオブ、チタン、タンタルおよびジルコニウムからなる群から選択される少なくとも一種の遷移金属元素(f)を含む金属有機化合物(F)(ただし、遷移金属元素(e)と遷移金属元素(f)とは異なる)とを、
酸化熱分解することにより得られる白金を含まない金属酸化物材料からなることを特徴とする(1)〜(3)のいずれかに記載の電極触媒。
(1)〜(6)のいずれかに記載の電極触媒を含むことを特徴とする電極触媒層。
さらに電子伝導性粒子を含むことを特徴とする(7)に記載の電極触媒層。
カソードとアノードと前記カソードおよび前記アノードの間に配置された電解質膜とを有する膜電極接合体であって、前記カソードが(7)または(8)に記載の電極触媒層を有することを特徴とする膜電極接合体。
(9)に記載の膜電極接合体を備えることを特徴とする燃料電池。
固体高分子型燃料電池であることを特徴とする(10)に記載の燃料電池。
ニオブ、チタン、タンタルおよびジルコニウムからなる群から選択される少なくとも一種の遷移金属元素(a)を含む金属化合物(A)と、
ニオブ、チタン、タンタルおよびジルコニウムからなる群から選択される少なくとも一種の遷移金属元素(b)を含む金属化合物(B)(ただし、遷移金属元素(a)と遷移金属元素(b)とは異なる)とを、
前記遷移金属元素(a)および/または前記遷移金属元素(b)の価数が大きくなるように、酸素含有雰囲気下で熱処理することにより金属酸化物材料を得る工程を含むことを特徴とする(1)〜(3)のいずれかに記載の電極触媒の製造方法。
熱処理の温度が400〜1200℃の範囲であることを特徴とする(12)に記載の製造方法。
ニオブ、チタン、タンタルおよびジルコニウムからなる群から選択される少なくとも一種の遷移金属元素(c)を含む、金属塩または金属錯体(C)と、
ニオブ、チタン、タンタルおよびジルコニウムからなる群から選択される少なくとも一種の遷移金属元素(d)を含む、金属塩または金属錯体(D)(ただし、遷移金属元素(c)と遷移金属元素(d)とは異なる)とを、
加水分解することにより金属酸化物材料を得る工程を含むことを特徴とする(1)〜(3)のいずれかに記載の電極触媒の製造方法。
さらに酸素含有雰囲気下で熱処理する工程を含むことを特徴とする(14)に記載の製造方法。
熱処理の温度が400〜1200℃の範囲であることを特徴とする(15)に記載の製造方法。
ニオブ、チタン、タンタルおよびジルコニウムからなる群から選択される少なくとも一種の遷移金属元素(e)を含む金属有機化合物(E)と、
ニオブ、チタン、タンタルおよびジルコニウムからなる群から選択される少なくとも一種の遷移金属元素(f)を含む金属有機化合物(F)(ただし、遷移金属元素(e)と遷移金属元素(f)とは異なる)とを、
酸化熱分解することにより金属酸化物材料を得る工程を含むことを特徴とする(1)〜(3)のいずれかに記載の電極触媒の製造方法。
金属酸化物材料を解砕する工程をさらに含むことを特徴とする(12)〜(17)のいずれかに記載の製造方法。
も腐蝕し難く安定である。
本発明の電極触媒は、ニオブ、チタン、タンタルおよびジルコニウムからなる群から選択される少なくとも二種以上の遷移金属元素を含み、且つ白金を含まないことを特徴としている。
前記電極触媒は、粉末であることが好ましい。粉末であると、触媒面積が大きく、触媒能に優れるため好ましい。
しくは10〜100m2/gである。前記電極触媒のBET比表面積が1m2/gより小さいと、触媒面積が小さく、1000m2/gよりと大きいと凝集しやすく扱いにくい。
前記電極触媒の粉末の粒径は、BET法で求めた比表面積に基づいて、粉末を球形に換算して、下記式(1)より求めることができる。
電極触媒の粉末の粒径:D(μm)
電極触媒の粉末の比重:ρ(g/cm3)
電極触媒の粉末のBET比表面積:S(m2/g)
本発明の電極触媒は、下記(I)〜(III)のいずれかであることが好ましい。
を酸化熱分解することにより得られる白金を含まない金属酸化物材料からなる電極触媒(III)。
[電極触媒(I)]
本発明の電極触媒は、ニオブ、チタン、タンタルおよびジルコニウムからなる群から選択される少なくとも一種の遷移金属元素(a)を含む金属化合物(A)と、ニオブ、チタン、タンタルおよびジルコニウムからなる群から選択される少なくとも一種の遷移金属元素(b)を含む金属化合物(B)(ただし、遷移金属元素(a)と遷移金属元素(b)とは異なる)とを、前記遷移金属元素(a)および/または前記遷移金属元素(b)の価数が大きくなるように、酸素含有雰囲気下で熱処理することにより得られる白金を含まない金属酸化物材料からなることが好ましい。
しかしながら、遷移金属元素(a)と遷移金属元素(b)とのモル比(遷移金属元素(a):遷移金属元素(b))が0.01:99.99〜5:95の範囲である場合、遷移金属元素(a)のモル比が小さいさいため、遷移金属元素(a)の価数変化を電極触媒のXRDスペクトルにより特定することが困難な場合がある。そこで、遷移金属元素(a)のモル比を5より大きくする以外は同一条件で電極触媒を製造して、遷移金属元素(a)の価数変化を電極触媒のXRDスペクトルにより特定する。前記特定した価数変化から、モル比(遷移金属元素(a):遷移金属元素(b))が0.01:99.99〜5:95の範囲である場合の遷移金属元素(a)の価数変化を推定する。
例えば、遷移金属元素(a)がニオブであり、遷移金属元素(b)がチタンである場合には、ニオブとチタンとのモル比(ニオブ:チタン)は、通常0.2:99.8〜20:80の範囲であり、好ましくは0.5:99.5〜10:90の範囲であり、より好ましくは1:99〜10:90の範囲である。
、三酸化チタン(Ti2O3)、一酸化チタン(TiO)等が挙げられ、二酸化ニオブ(NbO2)、三酸化チタン(Ti2O3)が好ましい。
熱処理は、金属化合物(A)および金属化合物(B)から金属酸化物材料が得られるような、酸化雰囲気で熱処理する条件であればよく、熱処理温度、熱処理時間、酸素濃度は、金属化合物(A)および金属化合物(B)と金属酸化物材料の種類により異なる。
また、炉内の雰囲気を制御することにより、反応速度をコントロールすることができる。例えば、窒素、アルゴン等の不活性ガスを炉内に流し、炉内の雰囲気を制御しながら焼成することにより、反応速度をコントロールすることができる。真空炉においては、必要な酸素量まで減圧後、その減圧を維持して熱処理する。
本発明の電極触媒は、ニオブ、チタン、タンタルおよびジルコニウムからなる群から選択される少なくとも一種の遷移金属元素(c)を含む、金属塩または金属錯体(C)と、ニオブ、チタン、タンタルおよびジルコニウムからなる群から選択される少なくとも一種の遷移金属元素(d)を含む、金属塩または金属錯体(D)(ただし、遷移金属元素(c)と遷移金属元素(d)とは異なる)とを加水分解することにより得られる白金を含まない金属酸化物材料からなることが好ましい。
例えば、遷移金属元素(c)がニオブであり、遷移金属元素(d)がチタンである場合には、ニオブとチタンとのモル比(ニオブ:チタン)は、通常0.2:99.8〜20:80の範囲であり、好ましくは0.5:99.5〜10:90の範囲であり、より好ましくは1:99〜10:90の範囲である。
酸塩、プロピオン酸塩などの低級脂肪酸塩が好ましい。また金属ハロゲン化物としては、塩化物が好ましい。
反応は、室温で行っても、冷却、加熱して行ってもよい。加熱は、得られる金属酸化物材料の結晶性を高めることになる。また、水酸基が脱離し表面に欠陥のある金属酸化物材料になりやすく好ましい。冷却は、反応が均一になり、得られる金属酸化物材料の比表面積が大きくなり好ましい。
固液分離には、粒子の沈降、濃縮、ろ過、洗浄、乾燥等の工程により行われるが、前記工程を全て行う必要は必ずしも無く、スラリーの性状等によっても必要な工程は異なる。沈降、濃縮、ろ過、洗浄により、液中に溶解する不純物を除去することができる。沈降速度、あるいはろ過速度を変えるために、凝集剤や分散剤を用いてもよい。該凝集剤あるいは分散剤は、蒸発、昇華、熱分解等により気体として除去可能なものが好ましい。ろ過、洗浄により、溶剤、溶剤に溶解している上記金属塩または金属錯体の加水分解副生成物を除去することができる。
熱処理は、金属酸化物材料の結晶性を向上させるために行われるが、同時に、不純物を、蒸発、昇華、熱分解等により気体として除去することができる。また、熱処理温度によっては粒子表面に有する水酸基や、原料に由来するアルコキシ基やカルボン酸基等を脱離し、表面欠陥の多い金属酸化物材料とすることができる。この方法により除去できる不純物としては、上記金属塩または金属錯体の種類によって異なるが、加水分解副生成物などが挙げられる。通常、熱処理温度は400〜1200℃で行われる。また、熱処理時間は、原料として用いる上記金属塩または金属錯体、金属酸化物材料の種類や熱処理温度、酸素濃度により適宜時間を決定することができるが、通常は、10分〜5時間である。なお、熱処理時間は、昇温および降温の時間を含める。焼成雰囲気は特に制限はなく、通常、大気中、不活性ガス中、もしくは減圧中で行われる。焼成温度が高くなるほど、また焼成時間が長くなるほど、金属酸化物材料の結晶性が高くなるが、比表面積が小さくなる。最適条件は、そのバランスで決定する。
本発明の電極触媒は、ニオブ、チタン、タンタルおよびジルコニウムからなる群から選択される少なくとも一種の遷移金属元素(e)を含む金属有機化合物(E)と、ニオブ、チタン、タンタルおよびジルコニウムからなる群から選択される少なくとも一種の遷移金属元素(f)を含む金属有機化合物(F)(ただし、遷移金属元素(e)と遷移金属元素(f)とは異なる)とを酸化熱分解することにより得られる白金を含まない金属酸化物材料からなることが好ましい。
例えば、遷移金属元素(e)がニオブであり、遷移金属元素(f)がチタンである場合には、ニオブとチタンとのモル比(ニオブ:チタン)は、通常0.2:99.8〜20:80の範囲であり、好ましくは0.5:99.5〜10:90の範囲であり、より好ましくは1:99〜10:90の範囲である。
50:50の範囲であり、好ましくは2:98〜30:70の範囲であり、より好ましくは5:95〜20:80の範囲である。
程度であり、より好ましくは1〜18である。
前記温度が200℃未満であると、酸化熱分解が不十分で灰分が残る傾向がある。
酸化熱分解は、酸素含有雰囲気下で行われてもよく、その際の酸素濃度としては、空気中の酸素濃度で充分である。
素(e)および/または前記遷移金属元素(f)の価数を熱処理前と比べて大きくすることができる。価数を大きくすることにより、触媒能が向上する傾向があるため好ましい。
本発明に用いる電極触媒の、下記測定法(A)に従って測定される酸素還元開始電位は、可逆水素電極を基準として好ましくは0.4V(vs.NHE)以上である。
〔測定法(A):
電子伝導性粒子であるカーボンに分散させた電極触媒が1重量%となるように、該電極触媒およびカーボンを溶剤中に入れ、超音波で攪拌し懸濁液を得る。なお、カーボンとしては、カーボンブラック(比表面積:100〜300m2/g)(例えばキャボット社製
XC−72)を用い、電極触媒とカーボンとが重量比で95:5になるように分散させる。また、溶剤としては、イソプロピルアルコール:水(重量比)=2:1を用いる。
電極とし、5mV/秒の電位走査速度で分極することにより電流−電位曲線を測定した際の、酸素雰囲気での還元電流と窒素雰囲気での還元電流とに0.2μA/cm2以上の差
が現れ始める電位を酸素還元開始電位とする。〕
上記酸素還元開始電位が0.7V(vs.NHE)未満であると、前記電極触媒を燃料電池のカソード用の電極触媒として用いた際に過酸化水素が発生することがある。また酸素還元開始電位は0.85V(vs.NHE)以上であることが、好適に酸素を還元するために好ましい。また、酸素還元開始電位は高い程好ましく、特に上限は無いが、理論値の1.23V(vs.NHE)である。
まで使用可能である。
本発明の電極触媒の製造方法は、下記工程(I)〜(III)のいずれかを含むことを特徴としている。
(III)の詳細な説明と同様である。
また、上記工程(I)〜(III)により得られる金属酸化物材料を、さらに解砕し、より微細な粉末にすることが好ましい。より微細な粉末の金属酸化物材料からなる電極触媒は、電極触媒層中に好適に分散する傾向にある。
本発明の電極触媒層は、前記電極触媒を含むことを特徴としている。
本発明の電極触媒層には、さらに電子伝導性粒子を含むことが好ましい。電極触媒を含む電極触媒層にさらに電子伝導性粒子を含むと還元電流を高めることができる。電子伝導性粒子は、電極触媒に、電気化学的反応を誘起させるための電気的接点を生じさせるため、還元電流を高めることができる。
電子伝導性粒子としては、炭素、導電性高分子、導電性セラミクス、金属または酸化タングステンもしくは酸化イリジウムなどの導電性無機酸化物が挙げられ、それらを単独ま
たは組み合わせて用いることができる。特に、炭素は比表面積が大きいため、炭素単独または炭素とその他の電子伝導性粒子との混合物が好ましい。電極触媒と炭素とを含む電極触媒層は、還元電流をより高めることができる。
また、本発明の電極触媒層には通常、さらに電解質として高分子電解質または導電性高分子を含む。
電極触媒層の形成方法としては、特に制限はないが、例えば、前記電極触媒と電子伝導性粒子と電解質とを含む懸濁液を、後述する電解質膜またはガス拡散層に塗布する方法が挙げられる。前記塗布する方法としては、ディッピング法、スクリーン印刷法、ロールコーティング法、スプレー法などが挙げられる。また、前記電極触媒と電子伝導性粒子と電解質とを含む懸濁液を、塗布法またはろ過法により基材に電極触媒層を形成した後、転写法で電解質膜に電極触媒層を形成する方法が挙げられる。
本発明の膜電極接合体は、カソードとアノードと前記カソードおよびアノードの間に配置された電解質膜とを有する膜電極接合体であって、前記カソードが、前述の電極触媒層を有することを特徴としている。
ガス拡散層としては、電子伝導性を有し、ガスの拡散性が高く、耐食性の高いものであれば何であっても構わないが、一般的にはカーボンペーパー、カーボンクロスなどの炭素系多孔質材料や、軽量化のためにステンレス、耐食材を被服したアルミニウム箔が用いられる。
燃料電池の電極反応はいわゆる3相界面(電解質‐電極触媒‐反応ガス)で起こる。燃料電池は、使用される電解質などの違いにより数種類に分類され、溶融炭酸塩型(MCFC)、リン酸型(PAFC)、固体酸化物型(SOFC)、固体高分子型(PEFC)等がある。中でも本発明の膜電極接合体は、固体高分子型燃料電池に使用することが好ましい。
(電極触媒の製造)
2−エチルヘキサン酸チタン(IV)(和光純薬製)5.0gと2−エチルヘキサン酸ニオブ(IV)(和光純薬製)0.1gをアルミナ製坩堝に入れ、電気炉(株式会社デンケン製 卓上マッフル炉 KDF P90)中で、窒素を50NL/分の流量で流しながら下記条件で酸化熱分解した。固形分0.71gを回収した。さらに、回収した固形分を乳鉢で充分に解砕を行い、電極触媒(1)を得た。
酸化熱分解温度:600℃
酸化熱分解時間(保持時間):2時間
(燃料電池用電極の製造)
酸素還元能の測定は、次のように行った。上記酸化熱分解で得られた電極触媒(1)0.95gとカーボン(キャボット社製 XC−72)0.5gをイソプロピルアルコール
:純水=2:1の重量比で混合した溶液10gに入れ、超音波で撹拌、縣濁して混合した。これをグラッシーカーボン電極(径:5.2mm)に30μl塗布し、50℃で2時間乾燥した。ナフィオン(登録商標)(デュポン社 5%ナフィオン溶液(DE521))10μlをさらに塗布し、120℃で1時間乾燥し、燃料電池用電極(1)を得た。
このようにして作製した燃料電池用電極(1)の触媒能(酸素還元能)を以下の方法で評価した。
を測定した。その際、同濃度の硫酸溶液中での可逆水素電極を参照電極とした。
た。
すなわち、酸素還元開始電位が高いほど、また、酸素還元電流が大きいほど、燃料電池用電極(1)の触媒能(酸素還元能)が高いことを示す。
実施例1で作製した燃料電池用電極(1)は、酸素還元開始電位が0.9V(vs.NHE)であり、高い酸素還元能を示すことがわかった。
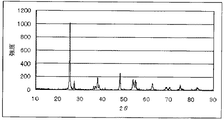
理学電機株式会社製 ロータフレックスを用いて、得られた電極触媒(1)のX線回折を行った。図2に、電極触媒(1)のXRDスペクトルを示す。アナターゼ型の酸化チタン(ニオブが約2モル%含有)であることがわかった。
島津製作所株式会社製 マイクロメリティクス ジェミニ2360を用いて電極触媒(1)のBET比表面積を測定した。
(電極触媒の製造)
チタン(IV)テトラブトキシドモノマー(和光純薬製)5.0gとニオブ(V)エトキシド(和光純薬製)0.05gをエタノール(和光純薬製)100mlに溶解した。撹拌しながら、この溶液にイオン交換水1.2mlを滴下した。その後、1時間撹拌を継続した。冷却後、この溶液を減圧ろ過し、固形分を得た。この固形分を100mlのイオン交換水で洗浄した後、減圧ろ過した。この洗浄、減圧ろ過の操作を5回繰り返し、固形分を得た。この固形分の全量をガラス製シャーレーに入れ、120℃で1時間乾燥し、固形
分1.2gを得た。回収した固形分を乳鉢で充分に解砕を行った。
熱処理温度:600℃
熱処理時間(保持時間):2時間
熱処理後、自然冷却し、固形分1.0gを回収した。さらに、回収した固形分を乳鉢で充分に解砕を行い、電極触媒(2)を得た。
電極触媒(1)に替えて電極触媒(2)を用いた以外は実施例1と同様にして燃料電池用電極(2)を得た。
燃料電池用電極(1)に替えて燃料電池用電極(2)を用いた以外は実施例1と同様にして酸素還元能の評価を行った。
実施例2で作製した燃料電池用電極(2)は、酸素還元開始電位が1.0V(vs.NHE)であり、高い酸素還元能を示すことがわかった。
電極触媒(1)に替えて電極触媒(2)を用いた以外は実施例1と同様にしてX線回折を行った。図4に、電極触媒(2)のXRDスペクトルを示す。アナターゼ型の酸化チタン(ニオブが約1モル%含有)であることがわかった。
電極触媒(1)に替えて電極触媒(2)を用いた以外は実施例1と同様にしてBET比表面積を測定した。
(電極触媒の製造)
還流冷却管を具備した容器に、硫酸酸化チタン(和光純薬製)5.0gおよび純水125mlを入れ、撹拌した。さらに、該容器に五塩化ニオブ(和光純薬製)0.9gおよびエタノール(和光純薬製)0.9gを混合した溶液を滴下した。その後、沸点まで加熱し、5時間沸点を維持した。冷却後、この溶液を減圧ろ過し、固形分を得た。この固形分を100mlのイオン交換水で洗浄した後、減圧ろ過した。この洗浄、減圧ろ過の操作を5回繰り返し、固形分を得た。この固形分の全量をガラス製シャーレーに入れ、120℃で1時間乾燥し、固形分2.0gを得た。回収した固形分を乳鉢で充分に解砕を行った。
熱処理温度:1000℃
熱処理時間(保持時間):2時間
熱処理後、自然冷却し、固形分1.8gを回収した。さらに、回収した固形分を乳鉢で充分に解砕を行い、電極触媒(3)を得た。
電極触媒(1)に替えて電極触媒(3)を用いた以外は実施例1と同様にして燃料電池用電極(3)を得た。
燃料電池用電極(1)に替えて燃料電池用電極(3)を用いた以外は実施例1と同様にして酸素還元能の評価を行った。
実施例3で作製した燃料電池用電極(3)は、酸素還元開始電位が1.0V(vs.NHE)であり、高い酸素還元能を示すことがわかった。
電極触媒(1)に替えて電極触媒(3)を用いた以外は実施例1と同様にしてX線回折を行った。図6に、電極触媒(3)のXRDスペクトルを示す。ルチル型の酸化チタン(ニオブが約10モル%含有)であることがわかった。電極触媒(3)のXRDパターンは、市販のルチル型の酸化チタン(添川理化学製)に比べて低角度側へシフトしていること、およびルチル型の酸化チタン以外のXRDパターンがみられないことから、約10モル%のニオブが酸化チタンに固溶していると考えられる。参考として図7に、市販のルチル型の酸化チタン(添川理化学製)のXRDスペクトルを示す。
電極触媒(1)に替えて電極触媒(3)を用いた以外は実施例1と同様にしてBET比表面積を測定した。
Claims (10)
- カソードとアノードと前記カソードおよび前記アノードの間に配置された電解質膜とを有する膜電極接合体であって、
前記カソードが電極触媒層を有し、
前記電極触媒層が電極触媒を含み、
前記電極触媒が、ニオブ、チタン、タンタルおよびジルコニウムからなる群から選択される少なくとも二種以上の遷移金属元素を含み、且つ白金を含まない金属酸化物材料からなることを特徴とする膜電極接合体。 - 前記金属酸化物材料が、
ニオブ、チタン、タンタルおよびジルコニウムからなる群から選択される少なくとも一種の遷移金属元素(a)を含む金属化合物(A)と、
ニオブ、チタン、タンタルおよびジルコニウムからなる群から選択される少なくとも一種の遷移金属元素(b)を含む金属化合物(B)(ただし、遷移金属元素(a)と遷移金属元素(b)とは異なる)とを、
前記遷移金属元素(a)および/または前記遷移金属元素(b)の価数が大きくなるように、酸素含有雰囲気下で熱処理することにより得られる白金を含まない金属酸化物材料である請求項1に記載の膜電極接合体。 - 前記金属酸化物材料が、
ニオブ、チタン、タンタルおよびジルコニウムからなる群から選択される少なくとも一種の遷移金属元素(c)を含む、金属塩または金属錯体(C)と、
ニオブ、チタン、タンタルおよびジルコニウムからなる群から選択される少なくとも一種の遷移金属元素(d)を含む、金属塩または金属錯体(D)(ただし、遷移金属元素(c)と遷移金属元素(d)とは異なる)とを、
加水分解することにより得られる白金を含まない金属酸化物材料である請求項1に記載の膜電極接合体。 - 前記金属酸化物材料が、
ニオブ、チタン、タンタルおよびジルコニウムからなる群から選択される少なくとも一種の遷移金属元素(e)を含む金属有機化合物(E)と、
ニオブ、チタン、タンタルおよびジルコニウムからなる群から選択される少なくとも一種の遷移金属元素(f)を含む金属有機化合物(F)(ただし、遷移金属元素(e)と遷移金属元素(f)とは異なる)とを、
酸化熱分解することにより得られる白金を含まない金属酸化物材料である請求項1に記載の膜電極接合体。 - 前記電極触媒層が、
さらに電子伝導性粒子を含むことを特徴とする請求項1〜4のいずれか一項に記載の膜電極接合体。 - 請求項1〜5のいずれか一項に記載の膜電極接合体を備えることを特徴とする燃料電池。
- 固体高分子型燃料電池であることを特徴とする請求項6に記載の燃料電池。
- ニオブ、チタン、タンタルおよびジルコニウムからなる群から選択される少なくとも一種の遷移金属元素(a)を含む金属化合物(A)と、
ニオブ、チタン、タンタルおよびジルコニウムからなる群から選択される少なくとも一種の遷移金属元素(b)を含む金属化合物(B)(ただし、遷移金属元素(a)と遷移金属元素(b)とは異なる)とを、
前記遷移金属元素(a)および/または前記遷移金属元素(b)の価数が大きくなるように、酸素含有雰囲気下で熱処理することにより金属酸化物材料を得る工程を含むことを特徴とする請求項1または2に記載の膜電極接合体を製造する方法。 - ニオブ、チタン、タンタルおよびジルコニウムからなる群から選択される少なくとも一種の遷移金属元素(c)を含む、金属塩または金属錯体(C)と、
ニオブ、チタン、タンタルおよびジルコニウムからなる群から選択される少なくとも一種の遷移金属元素(d)を含む、金属塩または金属錯体(D)(ただし、遷移金属元素(c)と遷移金属元素(d)とは異なる)とを、
加水分解することにより金属酸化物材料を得る工程を含むことを特徴とする請求項1または3に記載の膜電極接合体を製造する方法。 - ニオブ、チタン、タンタルおよびジルコニウムからなる群から選択される少なくとも一種の遷移金属元素(e)を含む金属有機化合物(E)と、
ニオブ、チタン、タンタルおよびジルコニウムからなる群から選択される少なくとも一種の遷移金属元素(f)を含む金属有機化合物(F)(ただし、遷移金属元素(e)と遷移金属元素(f)とは異なる)とを、
酸化熱分解することにより金属酸化物材料を得る工程を含むことを特徴とする請求項1または4に記載の膜電極接合体を製造する方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2007328807A JP5144244B2 (ja) | 2007-12-20 | 2007-12-20 | 電極触媒およびその用途、ならびに電極触媒の製造方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2007328807A JP5144244B2 (ja) | 2007-12-20 | 2007-12-20 | 電極触媒およびその用途、ならびに電極触媒の製造方法 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2012256196A Division JP5559858B2 (ja) | 2012-11-22 | 2012-11-22 | 電極触媒およびその用途、ならびに電極触媒の製造方法 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2009148706A JP2009148706A (ja) | 2009-07-09 |
| JP2009148706A5 JP2009148706A5 (ja) | 2011-01-27 |
| JP5144244B2 true JP5144244B2 (ja) | 2013-02-13 |
Family
ID=40918535
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2007328807A Expired - Fee Related JP5144244B2 (ja) | 2007-12-20 | 2007-12-20 | 電極触媒およびその用途、ならびに電極触媒の製造方法 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP5144244B2 (ja) |
Families Citing this family (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP4932183B2 (ja) * | 2005-06-10 | 2012-05-16 | 株式会社Adeka | 有機酸金属塩組成物及び該組成物を用いた薄膜の製造方法 |
| JP2006342138A (ja) * | 2005-06-10 | 2006-12-21 | Adeka Corp | 2−エチルヘキサン酸ニオブ誘導体及びその製造方法 |
| JP5353287B2 (ja) * | 2008-03-21 | 2013-11-27 | 住友化学株式会社 | 電極触媒の製造方法および電極触媒 |
| JP5381639B2 (ja) * | 2009-11-20 | 2014-01-08 | 富士電機株式会社 | 固体電解質形燃料電池およびその製造方法 |
| US20140004444A1 (en) * | 2010-09-28 | 2014-01-02 | Isotta Cerri | Fuel cell electrocatalyst |
| EP2658018B1 (en) * | 2010-12-22 | 2020-06-24 | Showa Denko K.K. | Production process for electrode catalyst for fuel cell and uses thereof |
| JP5797435B2 (ja) | 2011-03-24 | 2015-10-21 | 国立大学法人横浜国立大学 | 酸素還元触媒 |
| JP6241214B2 (ja) * | 2013-11-08 | 2017-12-06 | 東洋インキScホールディングス株式会社 | 燃料電池電極形成用組成物、およびそれを用いた燃料電池 |
| JP6570515B2 (ja) | 2014-03-25 | 2019-09-04 | 国立大学法人横浜国立大学 | 酸素還元触媒及びその製造方法 |
| WO2020250898A1 (ja) | 2019-06-12 | 2020-12-17 | 国立大学法人横浜国立大学 | 酸素還元触媒、燃料電池、空気電池及び酸素還元触媒の製造方法 |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP3236686B2 (ja) * | 1992-12-25 | 2001-12-10 | ペルメレック電極株式会社 | ガス電極とその製造方法 |
| JPH09147876A (ja) * | 1995-11-28 | 1997-06-06 | Mitsubishi Heavy Ind Ltd | 固体電解質型電気化学セルの燃料極材料 |
| EP1858108A4 (en) * | 2005-01-27 | 2012-08-29 | Nippon Kayaku Kk | MODIFIED TITANIUM OXIDE PARTICLE AND THIS USING PHOTOELECTRIC CONVERTER |
| JP2006351405A (ja) * | 2005-06-17 | 2006-12-28 | Nippon Telegr & Teleph Corp <Ntt> | Sofc燃料極およびその製造方法 |
-
2007
- 2007-12-20 JP JP2007328807A patent/JP5144244B2/ja not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| JP2009148706A (ja) | 2009-07-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5144244B2 (ja) | 電極触媒およびその用途、ならびに電極触媒の製造方法 | |
| JP5254975B2 (ja) | 金属酸化物電極触媒およびその用途、ならびに金属酸化物電極触媒の製造方法 | |
| JP5474250B2 (ja) | 電極触媒層、膜電極接合体および燃料電池 | |
| JP5578849B2 (ja) | 触媒およびその製造方法ならびにその用途 | |
| US20130330659A1 (en) | Method for producing fuel cell electrode catalyst | |
| JP5537433B2 (ja) | 触媒およびその製造方法ならびにその用途 | |
| JP5106342B2 (ja) | 触媒及びその製造方法ならびにその用途 | |
| JP5559858B2 (ja) | 電極触媒およびその用途、ならびに電極触媒の製造方法 | |
| JP5650254B2 (ja) | 酸素還元触媒 | |
| CN103199273B (zh) | 催化剂层、膜电极接合体和燃料电池 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20101208 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20101208 |
|
| RD02 | Notification of acceptance of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7422 Effective date: 20101208 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20120319 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20120508 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20120706 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20121023 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20121122 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20151130 Year of fee payment: 3 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 5144244 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| LAPS | Cancellation because of no payment of annual fees |