JP4965776B2 - 新規生理活性ペプチドおよびその用途 - Google Patents
新規生理活性ペプチドおよびその用途 Download PDFInfo
- Publication number
- JP4965776B2 JP4965776B2 JP2001216683A JP2001216683A JP4965776B2 JP 4965776 B2 JP4965776 B2 JP 4965776B2 JP 2001216683 A JP2001216683 A JP 2001216683A JP 2001216683 A JP2001216683 A JP 2001216683A JP 4965776 B2 JP4965776 B2 JP 4965776B2
- Authority
- JP
- Japan
- Prior art keywords
- peptide
- seq
- protein
- present
- amino acid
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 108090000765 processed proteins & peptides Proteins 0.000 title claims description 578
- 102000004196 processed proteins & peptides Human genes 0.000 title description 19
- 230000000975 bioactive effect Effects 0.000 title description 2
- 108090000623 proteins and genes Proteins 0.000 claims description 510
- 102000004169 proteins and genes Human genes 0.000 claims description 459
- 125000003275 alpha amino acid group Chemical group 0.000 claims description 390
- 150000003839 salts Chemical class 0.000 claims description 275
- 238000000034 method Methods 0.000 claims description 214
- 150000001875 compounds Chemical class 0.000 claims description 201
- 230000027455 binding Effects 0.000 claims description 130
- 238000012216 screening Methods 0.000 claims description 123
- 239000013598 vector Substances 0.000 claims description 34
- 238000004519 manufacturing process Methods 0.000 claims description 20
- 108091033319 polynucleotide Proteins 0.000 claims description 17
- 102000040430 polynucleotide Human genes 0.000 claims description 17
- 239000002157 polynucleotide Substances 0.000 claims description 17
- 230000003213 activating effect Effects 0.000 claims description 12
- 235000018102 proteins Nutrition 0.000 description 441
- 210000004027 cell Anatomy 0.000 description 254
- 108020004414 DNA Proteins 0.000 description 165
- 239000002585 base Substances 0.000 description 97
- 230000000694 effects Effects 0.000 description 90
- 238000012360 testing method Methods 0.000 description 72
- 239000000243 solution Substances 0.000 description 57
- 239000003446 ligand Substances 0.000 description 55
- 230000036961 partial effect Effects 0.000 description 50
- 102100030364 Prokineticin receptor 1 Human genes 0.000 description 49
- 235000001014 amino acid Nutrition 0.000 description 48
- 150000001413 amino acids Chemical class 0.000 description 45
- 239000000126 substance Substances 0.000 description 44
- 239000002609 medium Substances 0.000 description 41
- 108020003175 receptors Proteins 0.000 description 41
- 238000006243 chemical reaction Methods 0.000 description 40
- 102000005962 receptors Human genes 0.000 description 40
- 230000003834 intracellular effect Effects 0.000 description 36
- 239000007788 liquid Substances 0.000 description 36
- -1 α-naphthyl Chemical group 0.000 description 36
- 239000003814 drug Substances 0.000 description 34
- 210000000170 cell membrane Anatomy 0.000 description 31
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 30
- 239000000872 buffer Substances 0.000 description 30
- 239000013615 primer Substances 0.000 description 30
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 28
- 239000012528 membrane Substances 0.000 description 28
- 241001465754 Metazoa Species 0.000 description 26
- 239000000556 agonist Substances 0.000 description 26
- 150000007523 nucleic acids Chemical class 0.000 description 25
- 102000039446 nucleic acids Human genes 0.000 description 25
- 108020004707 nucleic acids Proteins 0.000 description 25
- 210000001519 tissue Anatomy 0.000 description 25
- 239000000427 antigen Substances 0.000 description 24
- YZXBAPSDXZZRGB-DOFZRALJSA-N arachidonic acid Chemical compound CCCCC\C=C/C\C=C/C\C=C/C\C=C/CCCC(O)=O YZXBAPSDXZZRGB-DOFZRALJSA-N 0.000 description 24
- 230000014509 gene expression Effects 0.000 description 24
- 102000036639 antigens Human genes 0.000 description 23
- 108091007433 antigens Proteins 0.000 description 23
- 230000008859 change Effects 0.000 description 23
- 239000003795 chemical substances by application Substances 0.000 description 23
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 22
- 235000014680 Saccharomyces cerevisiae Nutrition 0.000 description 22
- 239000000203 mixture Substances 0.000 description 22
- 125000003729 nucleotide group Chemical group 0.000 description 22
- 208000010643 digestive system disease Diseases 0.000 description 21
- 108090000045 G-Protein-Coupled Receptors Proteins 0.000 description 20
- 239000002773 nucleotide Substances 0.000 description 20
- 102000004190 Enzymes Human genes 0.000 description 19
- 108090000790 Enzymes Proteins 0.000 description 19
- 102000003688 G-Protein-Coupled Receptors Human genes 0.000 description 19
- 239000011575 calcium Substances 0.000 description 19
- 239000002299 complementary DNA Substances 0.000 description 19
- 229940088598 enzyme Drugs 0.000 description 19
- 238000002347 injection Methods 0.000 description 19
- 239000007924 injection Substances 0.000 description 19
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 18
- 229940098773 bovine serum albumin Drugs 0.000 description 18
- 201000010099 disease Diseases 0.000 description 18
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 18
- 239000000284 extract Substances 0.000 description 18
- 150000002500 ions Chemical class 0.000 description 18
- 125000006239 protecting group Chemical group 0.000 description 18
- 238000000746 purification Methods 0.000 description 18
- 239000011347 resin Substances 0.000 description 18
- 229920005989 resin Polymers 0.000 description 18
- 230000004936 stimulating effect Effects 0.000 description 18
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 18
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 17
- 150000002148 esters Chemical class 0.000 description 17
- 230000000069 prophylactic effect Effects 0.000 description 17
- OHCQJHSOBUTRHG-KGGHGJDLSA-N FORSKOLIN Chemical compound O=C([C@@]12O)C[C@](C)(C=C)O[C@]1(C)[C@@H](OC(=O)C)[C@@H](O)[C@@H]1[C@]2(C)[C@@H](O)CCC1(C)C OHCQJHSOBUTRHG-KGGHGJDLSA-N 0.000 description 16
- 230000009471 action Effects 0.000 description 16
- 210000004556 brain Anatomy 0.000 description 16
- 230000006957 competitive inhibition Effects 0.000 description 16
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 15
- 238000005259 measurement Methods 0.000 description 15
- 239000000047 product Substances 0.000 description 15
- 239000012085 test solution Substances 0.000 description 15
- 108060001084 Luciferase Proteins 0.000 description 14
- 241000124008 Mammalia Species 0.000 description 14
- IQFYYKKMVGJFEH-XLPZGREQSA-N Thymidine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 IQFYYKKMVGJFEH-XLPZGREQSA-N 0.000 description 14
- 210000004102 animal cell Anatomy 0.000 description 14
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 14
- 238000012258 culturing Methods 0.000 description 14
- 238000005516 engineering process Methods 0.000 description 14
- 230000001965 increasing effect Effects 0.000 description 14
- 239000000523 sample Substances 0.000 description 14
- XOFLBQFBSOEHOG-UUOKFMHZSA-N γS-GTP Chemical compound C1=2NC(N)=NC(=O)C=2N=CN1[C@@H]1O[C@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=S)[C@@H](O)[C@H]1O XOFLBQFBSOEHOG-UUOKFMHZSA-N 0.000 description 14
- 239000005557 antagonist Substances 0.000 description 13
- 238000002372 labelling Methods 0.000 description 13
- 239000013612 plasmid Substances 0.000 description 13
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 12
- FWMNVWWHGCHHJJ-SKKKGAJSSA-N 4-amino-1-[(2r)-6-amino-2-[[(2r)-2-[[(2r)-2-[[(2r)-2-amino-3-phenylpropanoyl]amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]hexanoyl]piperidine-4-carboxylic acid Chemical compound C([C@H](C(=O)N[C@H](CC(C)C)C(=O)N[C@H](CCCCN)C(=O)N1CCC(N)(CC1)C(O)=O)NC(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 FWMNVWWHGCHHJJ-SKKKGAJSSA-N 0.000 description 12
- 239000007995 HEPES buffer Substances 0.000 description 12
- 108010076504 Protein Sorting Signals Proteins 0.000 description 12
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 12
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 12
- 239000012634 fragment Substances 0.000 description 12
- 229920001223 polyethylene glycol Polymers 0.000 description 12
- 239000011535 reaction buffer Substances 0.000 description 12
- 230000001225 therapeutic effect Effects 0.000 description 12
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 11
- 241000699666 Mus <mouse, genus> Species 0.000 description 11
- 239000002202 Polyethylene glycol Substances 0.000 description 11
- 241000700159 Rattus Species 0.000 description 11
- 238000004020 luminiscence type Methods 0.000 description 11
- 238000002360 preparation method Methods 0.000 description 11
- 230000001737 promoting effect Effects 0.000 description 11
- 238000000926 separation method Methods 0.000 description 11
- 239000002904 solvent Substances 0.000 description 11
- 206010010774 Constipation Diseases 0.000 description 10
- 206010012735 Diarrhoea Diseases 0.000 description 10
- 208000004232 Enteritis Diseases 0.000 description 10
- 108091006027 G proteins Proteins 0.000 description 10
- 241000238631 Hexapoda Species 0.000 description 10
- 239000005089 Luciferase Substances 0.000 description 10
- 241000283973 Oryctolagus cuniculus Species 0.000 description 10
- 230000004913 activation Effects 0.000 description 10
- 150000001408 amides Chemical class 0.000 description 10
- 230000000692 anti-sense effect Effects 0.000 description 10
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 description 10
- 239000007790 solid phase Substances 0.000 description 10
- 239000006228 supernatant Substances 0.000 description 10
- 229940124597 therapeutic agent Drugs 0.000 description 10
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 9
- 241000282693 Cercopithecidae Species 0.000 description 9
- 108091000058 GTP-Binding Proteins 0.000 description 9
- 206010025476 Malabsorption Diseases 0.000 description 9
- 208000004155 Malabsorption Syndromes Diseases 0.000 description 9
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 9
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 9
- 238000003556 assay Methods 0.000 description 9
- 239000002775 capsule Substances 0.000 description 9
- 210000004978 chinese hamster ovary cell Anatomy 0.000 description 9
- 238000009833 condensation Methods 0.000 description 9
- 230000005494 condensation Effects 0.000 description 9
- 238000000855 fermentation Methods 0.000 description 9
- 230000004151 fermentation Effects 0.000 description 9
- 238000003018 immunoassay Methods 0.000 description 9
- 238000001727 in vivo Methods 0.000 description 9
- 238000010647 peptide synthesis reaction Methods 0.000 description 9
- 238000004007 reversed phase HPLC Methods 0.000 description 9
- 230000000638 stimulation Effects 0.000 description 9
- 229940104230 thymidine Drugs 0.000 description 9
- SUZLHDUTVMZSEV-UHFFFAOYSA-N Deoxycoleonol Natural products C12C(=O)CC(C)(C=C)OC2(C)C(OC(=O)C)C(O)C2C1(C)C(O)CCC2(C)C SUZLHDUTVMZSEV-UHFFFAOYSA-N 0.000 description 8
- 241000588722 Escherichia Species 0.000 description 8
- 102000030782 GTP binding Human genes 0.000 description 8
- 239000012981 Hank's balanced salt solution Substances 0.000 description 8
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 8
- 239000002671 adjuvant Substances 0.000 description 8
- 229940114079 arachidonic acid Drugs 0.000 description 8
- 235000021342 arachidonic acid Nutrition 0.000 description 8
- OHCQJHSOBUTRHG-UHFFFAOYSA-N colforsin Natural products OC12C(=O)CC(C)(C=C)OC1(C)C(OC(=O)C)C(O)C1C2(C)C(O)CCC1(C)C OHCQJHSOBUTRHG-UHFFFAOYSA-N 0.000 description 8
- 239000012153 distilled water Substances 0.000 description 8
- 239000002552 dosage form Substances 0.000 description 8
- 230000006870 function Effects 0.000 description 8
- 230000012010 growth Effects 0.000 description 8
- 235000013336 milk Nutrition 0.000 description 8
- 239000008267 milk Substances 0.000 description 8
- 210000004080 milk Anatomy 0.000 description 8
- 230000009871 nonspecific binding Effects 0.000 description 8
- 238000005406 washing Methods 0.000 description 8
- 108020004491 Antisense DNA Proteins 0.000 description 7
- 241000193830 Bacillus <bacterium> Species 0.000 description 7
- DWRXFEITVBNRMK-UHFFFAOYSA-N Beta-D-1-Arabinofuranosylthymine Natural products O=C1NC(=O)C(C)=CN1C1C(O)C(O)C(CO)O1 DWRXFEITVBNRMK-UHFFFAOYSA-N 0.000 description 7
- 241000283690 Bos taurus Species 0.000 description 7
- 101150074155 DHFR gene Proteins 0.000 description 7
- 241000588724 Escherichia coli Species 0.000 description 7
- 102000043136 MAP kinase family Human genes 0.000 description 7
- 108091054455 MAP kinase family Proteins 0.000 description 7
- 241000699670 Mus sp. Species 0.000 description 7
- 102000016978 Orphan receptors Human genes 0.000 description 7
- 108070000031 Orphan receptors Proteins 0.000 description 7
- 241001494479 Pecora Species 0.000 description 7
- 239000002253 acid Substances 0.000 description 7
- 239000003816 antisense DNA Substances 0.000 description 7
- IQFYYKKMVGJFEH-UHFFFAOYSA-N beta-L-thymidine Natural products O=C1NC(=O)C(C)=CN1C1OC(CO)C(O)C1 IQFYYKKMVGJFEH-UHFFFAOYSA-N 0.000 description 7
- 230000005540 biological transmission Effects 0.000 description 7
- 230000003491 cAMP production Effects 0.000 description 7
- 238000010367 cloning Methods 0.000 description 7
- 230000000295 complement effect Effects 0.000 description 7
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 7
- 210000004408 hybridoma Anatomy 0.000 description 7
- 239000000546 pharmaceutical excipient Substances 0.000 description 7
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N phenol group Chemical group C1(=CC=CC=C1)O ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 7
- 239000002243 precursor Substances 0.000 description 7
- 238000011160 research Methods 0.000 description 7
- 239000003826 tablet Substances 0.000 description 7
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 6
- 241000282472 Canis lupus familiaris Species 0.000 description 6
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 6
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 6
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 6
- 241000700605 Viruses Species 0.000 description 6
- 235000011054 acetic acid Nutrition 0.000 description 6
- SESFRYSPDFLNCH-UHFFFAOYSA-N benzyl benzoate Chemical compound C=1C=CC=CC=1C(=O)OCC1=CC=CC=C1 SESFRYSPDFLNCH-UHFFFAOYSA-N 0.000 description 6
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 6
- 230000008602 contraction Effects 0.000 description 6
- 238000002474 experimental method Methods 0.000 description 6
- 239000012091 fetal bovine serum Substances 0.000 description 6
- 210000001035 gastrointestinal tract Anatomy 0.000 description 6
- 230000016784 immunoglobulin production Effects 0.000 description 6
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 6
- 108010093708 mamba intestinal toxin 1 Proteins 0.000 description 6
- 108020004999 messenger RNA Proteins 0.000 description 6
- 230000003449 preventive effect Effects 0.000 description 6
- 229960003081 probenecid Drugs 0.000 description 6
- 238000011002 quantification Methods 0.000 description 6
- 229940044601 receptor agonist Drugs 0.000 description 6
- 239000000018 receptor agonist Substances 0.000 description 6
- 230000004044 response Effects 0.000 description 6
- 239000011780 sodium chloride Substances 0.000 description 6
- 235000000346 sugar Nutrition 0.000 description 6
- 230000001629 suppression Effects 0.000 description 6
- 230000014616 translation Effects 0.000 description 6
- UMCMPZBLKLEWAF-BCTGSCMUSA-N 3-[(3-cholamidopropyl)dimethylammonio]propane-1-sulfonate Chemical compound C([C@H]1C[C@H]2O)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC(=O)NCCC[N+](C)(C)CCCS([O-])(=O)=O)C)[C@@]2(C)[C@@H](O)C1 UMCMPZBLKLEWAF-BCTGSCMUSA-N 0.000 description 5
- 102000002260 Alkaline Phosphatase Human genes 0.000 description 5
- 108020004774 Alkaline Phosphatase Proteins 0.000 description 5
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 5
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 5
- 241000620209 Escherichia coli DH5[alpha] Species 0.000 description 5
- 241000282326 Felis catus Species 0.000 description 5
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 5
- 241000282412 Homo Species 0.000 description 5
- 102100040125 Prokineticin-2 Human genes 0.000 description 5
- 239000004480 active ingredient Substances 0.000 description 5
- 125000002252 acyl group Chemical group 0.000 description 5
- 238000005273 aeration Methods 0.000 description 5
- 238000013019 agitation Methods 0.000 description 5
- 125000003277 amino group Chemical group 0.000 description 5
- VZTDIZULWFCMLS-UHFFFAOYSA-N ammonium formate Chemical compound [NH4+].[O-]C=O VZTDIZULWFCMLS-UHFFFAOYSA-N 0.000 description 5
- 238000004458 analytical method Methods 0.000 description 5
- 102000005936 beta-Galactosidase Human genes 0.000 description 5
- 108010005774 beta-Galactosidase Proteins 0.000 description 5
- 230000037396 body weight Effects 0.000 description 5
- 239000001569 carbon dioxide Substances 0.000 description 5
- 229910002092 carbon dioxide Inorganic materials 0.000 description 5
- 239000012228 culture supernatant Substances 0.000 description 5
- 230000002950 deficient Effects 0.000 description 5
- 229940079593 drug Drugs 0.000 description 5
- 230000032050 esterification Effects 0.000 description 5
- 238000005886 esterification reaction Methods 0.000 description 5
- 238000005194 fractionation Methods 0.000 description 5
- 125000000524 functional group Chemical group 0.000 description 5
- 239000008103 glucose Substances 0.000 description 5
- 238000009396 hybridization Methods 0.000 description 5
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 5
- 230000000984 immunochemical effect Effects 0.000 description 5
- 230000002401 inhibitory effect Effects 0.000 description 5
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 5
- 230000020477 pH reduction Effects 0.000 description 5
- 239000008363 phosphate buffer Substances 0.000 description 5
- DBABZHXKTCFAPX-UHFFFAOYSA-N probenecid Chemical compound CCCN(CCC)S(=O)(=O)C1=CC=C(C(O)=O)C=C1 DBABZHXKTCFAPX-UHFFFAOYSA-N 0.000 description 5
- 238000010517 secondary reaction Methods 0.000 description 5
- 230000035945 sensitivity Effects 0.000 description 5
- 239000008159 sesame oil Substances 0.000 description 5
- 235000011803 sesame oil Nutrition 0.000 description 5
- 239000011734 sodium Substances 0.000 description 5
- 208000024891 symptom Diseases 0.000 description 5
- 239000011534 wash buffer Substances 0.000 description 5
- APIXJSLKIYYUKG-UHFFFAOYSA-N 3 Isobutyl 1 methylxanthine Chemical compound O=C1N(C)C(=O)N(CC(C)C)C2=C1N=CN2 APIXJSLKIYYUKG-UHFFFAOYSA-N 0.000 description 4
- ZOOGRGPOEVQQDX-UUOKFMHZSA-N 3',5'-cyclic GMP Chemical compound C([C@H]1O2)OP(O)(=O)O[C@H]1[C@@H](O)[C@@H]2N1C(N=C(NC2=O)N)=C2N=C1 ZOOGRGPOEVQQDX-UUOKFMHZSA-N 0.000 description 4
- KDCGOANMDULRCW-UHFFFAOYSA-N 7H-purine Chemical compound N1=CNC2=NC=NC2=C1 KDCGOANMDULRCW-UHFFFAOYSA-N 0.000 description 4
- 229920001817 Agar Polymers 0.000 description 4
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 4
- 241000700198 Cavia Species 0.000 description 4
- 108010035563 Chloramphenicol O-acetyltransferase Proteins 0.000 description 4
- 229920002261 Corn starch Polymers 0.000 description 4
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 4
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 4
- 108010010803 Gelatin Proteins 0.000 description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 4
- 206010035226 Plasma cell myeloma Diseases 0.000 description 4
- 239000004698 Polyethylene Substances 0.000 description 4
- WCUXLLCKKVVCTQ-UHFFFAOYSA-M Potassium chloride Chemical compound [Cl-].[K+] WCUXLLCKKVVCTQ-UHFFFAOYSA-M 0.000 description 4
- 102000007568 Proto-Oncogene Proteins c-fos Human genes 0.000 description 4
- 108010071563 Proto-Oncogene Proteins c-fos Proteins 0.000 description 4
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 4
- 108700008625 Reporter Genes Proteins 0.000 description 4
- 108091081024 Start codon Proteins 0.000 description 4
- 229930006000 Sucrose Natural products 0.000 description 4
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 4
- 241000282887 Suidae Species 0.000 description 4
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 4
- 239000007983 Tris buffer Substances 0.000 description 4
- INAPMGSXUVUWAF-GCVPSNMTSA-N [(2r,3s,5r,6r)-2,3,4,5,6-pentahydroxycyclohexyl] dihydrogen phosphate Chemical compound OC1[C@H](O)[C@@H](O)C(OP(O)(O)=O)[C@H](O)[C@@H]1O INAPMGSXUVUWAF-GCVPSNMTSA-N 0.000 description 4
- 238000010521 absorption reaction Methods 0.000 description 4
- OIPILFWXSMYKGL-UHFFFAOYSA-N acetylcholine Chemical compound CC(=O)OCC[N+](C)(C)C OIPILFWXSMYKGL-UHFFFAOYSA-N 0.000 description 4
- 229960004373 acetylcholine Drugs 0.000 description 4
- 238000010306 acid treatment Methods 0.000 description 4
- 239000008272 agar Substances 0.000 description 4
- 125000000217 alkyl group Chemical group 0.000 description 4
- 210000000628 antibody-producing cell Anatomy 0.000 description 4
- 239000008346 aqueous phase Substances 0.000 description 4
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 4
- 239000011230 binding agent Substances 0.000 description 4
- 210000004899 c-terminal region Anatomy 0.000 description 4
- 150000007942 carboxylates Chemical class 0.000 description 4
- 238000005119 centrifugation Methods 0.000 description 4
- 239000003153 chemical reaction reagent Substances 0.000 description 4
- 239000008120 corn starch Substances 0.000 description 4
- 238000010168 coupling process Methods 0.000 description 4
- 238000001514 detection method Methods 0.000 description 4
- 239000003085 diluting agent Substances 0.000 description 4
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 4
- 235000013601 eggs Nutrition 0.000 description 4
- 239000013604 expression vector Substances 0.000 description 4
- 239000012894 fetal calf serum Substances 0.000 description 4
- 239000000796 flavoring agent Substances 0.000 description 4
- MKXKFYHWDHIYRV-UHFFFAOYSA-N flutamide Chemical compound CC(C)C(=O)NC1=CC=C([N+]([O-])=O)C(C(F)(F)F)=C1 MKXKFYHWDHIYRV-UHFFFAOYSA-N 0.000 description 4
- 235000013355 food flavoring agent Nutrition 0.000 description 4
- 239000007789 gas Substances 0.000 description 4
- 239000008273 gelatin Substances 0.000 description 4
- 229920000159 gelatin Polymers 0.000 description 4
- 235000019322 gelatine Nutrition 0.000 description 4
- 235000011852 gelatine desserts Nutrition 0.000 description 4
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 4
- 238000000338 in vitro Methods 0.000 description 4
- 239000007791 liquid phase Substances 0.000 description 4
- 230000004048 modification Effects 0.000 description 4
- 238000012986 modification Methods 0.000 description 4
- 201000000050 myeloid neoplasm Diseases 0.000 description 4
- 239000003921 oil Substances 0.000 description 4
- 235000019198 oils Nutrition 0.000 description 4
- 210000000287 oocyte Anatomy 0.000 description 4
- VLTRZXGMWDSKGL-UHFFFAOYSA-N perchloric acid Chemical compound OCl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-N 0.000 description 4
- YPJUNDFVDDCYIH-UHFFFAOYSA-N perfluorobutyric acid Chemical compound OC(=O)C(F)(F)C(F)(F)C(F)(F)F YPJUNDFVDDCYIH-UHFFFAOYSA-N 0.000 description 4
- 239000008194 pharmaceutical composition Substances 0.000 description 4
- 229920000642 polymer Polymers 0.000 description 4
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 4
- 229920000053 polysorbate 80 Polymers 0.000 description 4
- 239000002987 primer (paints) Substances 0.000 description 4
- 230000035755 proliferation Effects 0.000 description 4
- 230000009822 protein phosphorylation Effects 0.000 description 4
- 239000002994 raw material Substances 0.000 description 4
- 108091008146 restriction endonucleases Proteins 0.000 description 4
- FGDZQCVHDSGLHJ-UHFFFAOYSA-M rubidium chloride Chemical compound [Cl-].[Rb+] FGDZQCVHDSGLHJ-UHFFFAOYSA-M 0.000 description 4
- 210000002966 serum Anatomy 0.000 description 4
- 230000019491 signal transduction Effects 0.000 description 4
- 239000003381 stabilizer Substances 0.000 description 4
- KDYFGRWQOYBRFD-UHFFFAOYSA-N succinic acid Chemical compound OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 4
- 239000005720 sucrose Substances 0.000 description 4
- 239000000725 suspension Substances 0.000 description 4
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 4
- 230000014621 translational initiation Effects 0.000 description 4
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 4
- 241000701161 unidentified adenovirus Species 0.000 description 4
- 239000003981 vehicle Substances 0.000 description 4
- OISVCGZHLKNMSJ-UHFFFAOYSA-N 2,6-dimethylpyridine Chemical compound CC1=CC=CC(C)=N1 OISVCGZHLKNMSJ-UHFFFAOYSA-N 0.000 description 3
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 3
- 108010000239 Aequorin Proteins 0.000 description 3
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 3
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 3
- 244000063299 Bacillus subtilis Species 0.000 description 3
- 235000014469 Bacillus subtilis Nutrition 0.000 description 3
- 102000014914 Carrier Proteins Human genes 0.000 description 3
- 108010078791 Carrier Proteins Proteins 0.000 description 3
- 235000019750 Crude protein Nutrition 0.000 description 3
- 238000000018 DNA microarray Methods 0.000 description 3
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 3
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 3
- QMMFVYPAHWMCMS-UHFFFAOYSA-N Dimethyl sulfide Chemical compound CSC QMMFVYPAHWMCMS-UHFFFAOYSA-N 0.000 description 3
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 3
- 102220623679 G-protein coupled receptor 15_R10A_mutation Human genes 0.000 description 3
- 208000018522 Gastrointestinal disease Diseases 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 3
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 3
- 102400001132 Melanin-concentrating hormone Human genes 0.000 description 3
- 101800002739 Melanin-concentrating hormone Proteins 0.000 description 3
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 3
- 101710163270 Nuclease Proteins 0.000 description 3
- 108700026244 Open Reading Frames Proteins 0.000 description 3
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 3
- 108091034057 RNA (poly(A)) Proteins 0.000 description 3
- 239000012980 RPMI-1640 medium Substances 0.000 description 3
- 241000269370 Xenopus <genus> Species 0.000 description 3
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 3
- 229960000723 ampicillin Drugs 0.000 description 3
- AVKUERGKIZMTKX-NJBDSQKTSA-N ampicillin Chemical compound C1([C@@H](N)C(=O)N[C@H]2[C@H]3SC([C@@H](N3C2=O)C(O)=O)(C)C)=CC=CC=C1 AVKUERGKIZMTKX-NJBDSQKTSA-N 0.000 description 3
- 239000003708 ampul Substances 0.000 description 3
- 239000007864 aqueous solution Substances 0.000 description 3
- 235000019445 benzyl alcohol Nutrition 0.000 description 3
- 229960002903 benzyl benzoate Drugs 0.000 description 3
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 3
- 230000015572 biosynthetic process Effects 0.000 description 3
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 3
- 150000001718 carbodiimides Chemical class 0.000 description 3
- 230000015556 catabolic process Effects 0.000 description 3
- 239000001913 cellulose Substances 0.000 description 3
- 229920002678 cellulose Polymers 0.000 description 3
- 238000006482 condensation reaction Methods 0.000 description 3
- 230000037020 contractile activity Effects 0.000 description 3
- 230000001276 controlling effect Effects 0.000 description 3
- 230000008878 coupling Effects 0.000 description 3
- 238000005859 coupling reaction Methods 0.000 description 3
- 238000006731 degradation reaction Methods 0.000 description 3
- 238000011161 development Methods 0.000 description 3
- 230000018109 developmental process Effects 0.000 description 3
- 229940039227 diagnostic agent Drugs 0.000 description 3
- 239000000032 diagnostic agent Substances 0.000 description 3
- 239000003623 enhancer Substances 0.000 description 3
- 235000019253 formic acid Nutrition 0.000 description 3
- 210000004602 germ cell Anatomy 0.000 description 3
- 239000011521 glass Substances 0.000 description 3
- 239000003365 glass fiber Substances 0.000 description 3
- 125000002883 imidazolyl group Chemical group 0.000 description 3
- 230000003053 immunization Effects 0.000 description 3
- 238000010348 incorporation Methods 0.000 description 3
- 239000003112 inhibitor Substances 0.000 description 3
- 238000010253 intravenous injection Methods 0.000 description 3
- 238000004255 ion exchange chromatography Methods 0.000 description 3
- 239000000644 isotonic solution Substances 0.000 description 3
- 239000008101 lactose Substances 0.000 description 3
- 150000002632 lipids Chemical class 0.000 description 3
- 230000002934 lysing effect Effects 0.000 description 3
- 235000019359 magnesium stearate Nutrition 0.000 description 3
- 230000013011 mating Effects 0.000 description 3
- 238000000691 measurement method Methods 0.000 description 3
- 230000001404 mediated effect Effects 0.000 description 3
- 229910052751 metal Inorganic materials 0.000 description 3
- 239000002184 metal Substances 0.000 description 3
- 150000002739 metals Chemical class 0.000 description 3
- 229940098779 methanesulfonic acid Drugs 0.000 description 3
- 150000007522 mineralic acids Chemical class 0.000 description 3
- 238000002156 mixing Methods 0.000 description 3
- 238000010369 molecular cloning Methods 0.000 description 3
- 239000002858 neurotransmitter agent Substances 0.000 description 3
- 239000002736 nonionic surfactant Substances 0.000 description 3
- 238000010899 nucleation Methods 0.000 description 3
- 150000007524 organic acids Chemical class 0.000 description 3
- 238000007911 parenteral administration Methods 0.000 description 3
- 239000012071 phase Substances 0.000 description 3
- 239000002504 physiological saline solution Substances 0.000 description 3
- 229920002401 polyacrylamide Polymers 0.000 description 3
- 239000000244 polyoxyethylene sorbitan monooleate Substances 0.000 description 3
- 229940068968 polysorbate 80 Drugs 0.000 description 3
- 239000002244 precipitate Substances 0.000 description 3
- 238000001556 precipitation Methods 0.000 description 3
- 239000003755 preservative agent Substances 0.000 description 3
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 3
- 238000001243 protein synthesis Methods 0.000 description 3
- 238000003127 radioimmunoassay Methods 0.000 description 3
- 238000003757 reverse transcription PCR Methods 0.000 description 3
- 229910052708 sodium Inorganic materials 0.000 description 3
- 210000001082 somatic cell Anatomy 0.000 description 3
- 239000003549 soybean oil Substances 0.000 description 3
- 235000012424 soybean oil Nutrition 0.000 description 3
- 125000001424 substituent group Chemical group 0.000 description 3
- 150000005846 sugar alcohols Polymers 0.000 description 3
- 239000000829 suppository Substances 0.000 description 3
- 210000001550 testis Anatomy 0.000 description 3
- RMVRSNDYEFQCLF-UHFFFAOYSA-N thiophenol Chemical compound SC1=CC=CC=C1 RMVRSNDYEFQCLF-UHFFFAOYSA-N 0.000 description 3
- 231100000331 toxic Toxicity 0.000 description 3
- 230000002588 toxic effect Effects 0.000 description 3
- 238000001890 transfection Methods 0.000 description 3
- 238000013519 translation Methods 0.000 description 3
- 241001515965 unidentified phage Species 0.000 description 3
- 241001430294 unidentified retrovirus Species 0.000 description 3
- NPWMTBZSRRLQNJ-VKHMYHEASA-N (3s)-3-aminopiperidine-2,6-dione Chemical compound N[C@H]1CCC(=O)NC1=O NPWMTBZSRRLQNJ-VKHMYHEASA-N 0.000 description 2
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 2
- 125000003088 (fluoren-9-ylmethoxy)carbonyl group Chemical group 0.000 description 2
- VYMPLPIFKRHAAC-UHFFFAOYSA-N 1,2-ethanedithiol Chemical compound SCCS VYMPLPIFKRHAAC-UHFFFAOYSA-N 0.000 description 2
- 125000001917 2,4-dinitrophenyl group Chemical group [H]C1=C([H])C(=C([H])C(=C1*)[N+]([O-])=O)[N+]([O-])=O 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 2
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 2
- 244000215068 Acacia senegal Species 0.000 description 2
- 241000416162 Astragalus gummifer Species 0.000 description 2
- 239000005711 Benzoic acid Substances 0.000 description 2
- 241000255789 Bombyx mori Species 0.000 description 2
- 241000167854 Bourreria succulenta Species 0.000 description 2
- 241000283707 Capra Species 0.000 description 2
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- 241000699802 Cricetulus griseus Species 0.000 description 2
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 2
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 2
- 102000053602 DNA Human genes 0.000 description 2
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 2
- 102000017914 EDNRA Human genes 0.000 description 2
- 108010090549 Endothelin A Receptor Proteins 0.000 description 2
- 241000283086 Equidae Species 0.000 description 2
- 241001302160 Escherichia coli str. K-12 substr. DH10B Species 0.000 description 2
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 2
- 101150102561 GPA1 gene Proteins 0.000 description 2
- 108010081952 Galanin-Like Peptide Proteins 0.000 description 2
- 102100031689 Galanin-like peptide Human genes 0.000 description 2
- SXRSQZLOMIGNAQ-UHFFFAOYSA-N Glutaraldehyde Chemical compound O=CCCCC=O SXRSQZLOMIGNAQ-UHFFFAOYSA-N 0.000 description 2
- 102000002068 Glycopeptides Human genes 0.000 description 2
- 108010015899 Glycopeptides Proteins 0.000 description 2
- 229920000084 Gum arabic Polymers 0.000 description 2
- 101001061518 Homo sapiens RNA-binding protein FUS Proteins 0.000 description 2
- 101000659267 Homo sapiens Tumor suppressor candidate 2 Proteins 0.000 description 2
- 108091006905 Human Serum Albumin Proteins 0.000 description 2
- 102000008100 Human Serum Albumin Human genes 0.000 description 2
- 108060003951 Immunoglobulin Proteins 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 2
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 2
- 244000246386 Mentha pulegium Species 0.000 description 2
- 235000016257 Mentha pulegium Nutrition 0.000 description 2
- 235000004357 Mentha x piperita Nutrition 0.000 description 2
- 101100018717 Mus musculus Il1rl1 gene Proteins 0.000 description 2
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 2
- 125000001429 N-terminal alpha-amino-acid group Chemical group 0.000 description 2
- 102400001111 Nociceptin Human genes 0.000 description 2
- 108090000622 Nociceptin Proteins 0.000 description 2
- 108091005804 Peptidases Proteins 0.000 description 2
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- 206010036790 Productive cough Diseases 0.000 description 2
- 102000009087 Prolactin-Releasing Hormone Human genes 0.000 description 2
- 108010087786 Prolactin-Releasing Hormone Proteins 0.000 description 2
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 2
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 2
- 102100028469 RNA-binding protein FUS Human genes 0.000 description 2
- 238000010240 RT-PCR analysis Methods 0.000 description 2
- 102000007056 Recombinant Fusion Proteins Human genes 0.000 description 2
- 108010008281 Recombinant Fusion Proteins Proteins 0.000 description 2
- 108091027981 Response element Proteins 0.000 description 2
- 101150006985 STE2 gene Proteins 0.000 description 2
- 229920005654 Sephadex Polymers 0.000 description 2
- 239000012507 Sephadex™ Substances 0.000 description 2
- 239000000877 Sex Attractant Substances 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- 102100023802 Somatostatin receptor type 2 Human genes 0.000 description 2
- 229920002472 Starch Polymers 0.000 description 2
- 241000282898 Sus scrofa Species 0.000 description 2
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 2
- 108020005038 Terminator Codon Proteins 0.000 description 2
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 2
- RYYWUUFWQRZTIU-UHFFFAOYSA-N Thiophosphoric acid Chemical class OP(O)(S)=O RYYWUUFWQRZTIU-UHFFFAOYSA-N 0.000 description 2
- 229920001615 Tragacanth Polymers 0.000 description 2
- 241000255993 Trichoplusia ni Species 0.000 description 2
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 2
- 102000050488 Urotensin II Human genes 0.000 description 2
- 108010018369 Urotensin II Proteins 0.000 description 2
- MMWCIQZXVOZEGG-HOZKJCLWSA-N [(1S,2R,3S,4S,5R,6S)-2,3,5-trihydroxy-4,6-diphosphonooxycyclohexyl] dihydrogen phosphate Chemical compound O[C@H]1[C@@H](O)[C@H](OP(O)(O)=O)[C@@H](OP(O)(O)=O)[C@H](O)[C@H]1OP(O)(O)=O MMWCIQZXVOZEGG-HOZKJCLWSA-N 0.000 description 2
- 235000010489 acacia gum Nutrition 0.000 description 2
- 239000000205 acacia gum Substances 0.000 description 2
- AMJRSUWJSRKGNO-UHFFFAOYSA-N acetyloxymethyl 2-[n-[2-(acetyloxymethoxy)-2-oxoethyl]-2-[2-[2-[bis[2-(acetyloxymethoxy)-2-oxoethyl]amino]-5-(2,7-dichloro-3-hydroxy-6-oxoxanthen-9-yl)phenoxy]ethoxy]-4-methylanilino]acetate Chemical compound CC(=O)OCOC(=O)CN(CC(=O)OCOC(C)=O)C1=CC=C(C)C=C1OCCOC1=CC(C2=C3C=C(Cl)C(=O)C=C3OC3=CC(O)=C(Cl)C=C32)=CC=C1N(CC(=O)OCOC(C)=O)CC(=O)OCOC(C)=O AMJRSUWJSRKGNO-UHFFFAOYSA-N 0.000 description 2
- 108020002494 acetyltransferase Proteins 0.000 description 2
- 102000005421 acetyltransferase Human genes 0.000 description 2
- 150000008065 acid anhydrides Chemical class 0.000 description 2
- 230000002378 acidificating effect Effects 0.000 description 2
- DZBUGLKDJFMEHC-UHFFFAOYSA-N acridine Chemical compound C1=CC=CC2=CC3=CC=CC=C3N=C21 DZBUGLKDJFMEHC-UHFFFAOYSA-N 0.000 description 2
- 239000013543 active substance Substances 0.000 description 2
- 239000000654 additive Substances 0.000 description 2
- PYMYPHUHKUWMLA-LMVFSUKVSA-N aldehydo-D-ribose Chemical compound OC[C@@H](O)[C@@H](O)[C@@H](O)C=O PYMYPHUHKUWMLA-LMVFSUKVSA-N 0.000 description 2
- 235000010443 alginic acid Nutrition 0.000 description 2
- 239000000783 alginic acid Substances 0.000 description 2
- 229920000615 alginic acid Polymers 0.000 description 2
- 229960001126 alginic acid Drugs 0.000 description 2
- 150000004781 alginic acids Chemical class 0.000 description 2
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 2
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 2
- 150000003863 ammonium salts Chemical class 0.000 description 2
- RDOXTESZEPMUJZ-UHFFFAOYSA-N anisole Chemical compound COC1=CC=CC=C1 RDOXTESZEPMUJZ-UHFFFAOYSA-N 0.000 description 2
- 239000003963 antioxidant agent Substances 0.000 description 2
- 230000003078 antioxidant effect Effects 0.000 description 2
- 125000003710 aryl alkyl group Chemical group 0.000 description 2
- 238000003149 assay kit Methods 0.000 description 2
- 239000005441 aurora Substances 0.000 description 2
- 229960000686 benzalkonium chloride Drugs 0.000 description 2
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 2
- 229940092714 benzenesulfonic acid Drugs 0.000 description 2
- 235000010233 benzoic acid Nutrition 0.000 description 2
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 2
- CADWTSSKOVRVJC-UHFFFAOYSA-N benzyl(dimethyl)azanium;chloride Chemical compound [Cl-].C[NH+](C)CC1=CC=CC=C1 CADWTSSKOVRVJC-UHFFFAOYSA-N 0.000 description 2
- 230000033228 biological regulation Effects 0.000 description 2
- 210000004369 blood Anatomy 0.000 description 2
- 239000008280 blood Substances 0.000 description 2
- 238000006664 bond formation reaction Methods 0.000 description 2
- 229910052796 boron Inorganic materials 0.000 description 2
- 238000009395 breeding Methods 0.000 description 2
- 230000001488 breeding effect Effects 0.000 description 2
- 239000007853 buffer solution Substances 0.000 description 2
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 2
- 239000001506 calcium phosphate Substances 0.000 description 2
- 229910000389 calcium phosphate Inorganic materials 0.000 description 2
- 229910052799 carbon Inorganic materials 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- 230000025084 cell cycle arrest Effects 0.000 description 2
- 230000007910 cell fusion Effects 0.000 description 2
- 239000013592 cell lysate Substances 0.000 description 2
- 235000019693 cherries Nutrition 0.000 description 2
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 2
- 235000012000 cholesterol Nutrition 0.000 description 2
- 238000004587 chromatography analysis Methods 0.000 description 2
- 239000003240 coconut oil Substances 0.000 description 2
- 235000019864 coconut oil Nutrition 0.000 description 2
- 210000001072 colon Anatomy 0.000 description 2
- 230000002860 competitive effect Effects 0.000 description 2
- 239000000470 constituent Substances 0.000 description 2
- 239000006059 cover glass Substances 0.000 description 2
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 2
- PAFZNILMFXTMIY-UHFFFAOYSA-N cyclohexylamine Chemical compound NC1CCCCC1 PAFZNILMFXTMIY-UHFFFAOYSA-N 0.000 description 2
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 2
- 230000000593 degrading effect Effects 0.000 description 2
- 238000003745 diagnosis Methods 0.000 description 2
- 210000004921 distal colon Anatomy 0.000 description 2
- 238000003379 elimination reaction Methods 0.000 description 2
- 239000006274 endogenous ligand Substances 0.000 description 2
- 238000006266 etherification reaction Methods 0.000 description 2
- 150000002170 ethers Chemical class 0.000 description 2
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 2
- 239000003925 fat Substances 0.000 description 2
- 235000019197 fats Nutrition 0.000 description 2
- 238000001914 filtration Methods 0.000 description 2
- 235000003599 food sweetener Nutrition 0.000 description 2
- 239000001530 fumaric acid Substances 0.000 description 2
- 235000011087 fumaric acid Nutrition 0.000 description 2
- 230000004927 fusion Effects 0.000 description 2
- 230000002496 gastric effect Effects 0.000 description 2
- 239000000499 gel Substances 0.000 description 2
- 238000002523 gelfiltration Methods 0.000 description 2
- 239000001963 growth medium Substances 0.000 description 2
- 125000000623 heterocyclic group Chemical group 0.000 description 2
- 229940088597 hormone Drugs 0.000 description 2
- 239000005556 hormone Substances 0.000 description 2
- 235000001050 hortel pimenta Nutrition 0.000 description 2
- 239000000017 hydrogel Substances 0.000 description 2
- 230000002209 hydrophobic effect Effects 0.000 description 2
- FDGQSTZJBFJUBT-UHFFFAOYSA-N hypoxanthine Chemical compound O=C1NC=NC2=C1NC=N2 FDGQSTZJBFJUBT-UHFFFAOYSA-N 0.000 description 2
- 102000018358 immunoglobulin Human genes 0.000 description 2
- 238000011534 incubation Methods 0.000 description 2
- 125000001041 indolyl group Chemical group 0.000 description 2
- 150000007529 inorganic bases Chemical class 0.000 description 2
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical group N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 2
- 230000003993 interaction Effects 0.000 description 2
- 230000000968 intestinal effect Effects 0.000 description 2
- 238000002955 isolation Methods 0.000 description 2
- 238000011813 knockout mouse model Methods 0.000 description 2
- 238000011068 loading method Methods 0.000 description 2
- 231100000053 low toxicity Toxicity 0.000 description 2
- 239000000314 lubricant Substances 0.000 description 2
- HWYHZTIRURJOHG-UHFFFAOYSA-N luminol Chemical compound O=C1NNC(=O)C2=C1C(N)=CC=C2 HWYHZTIRURJOHG-UHFFFAOYSA-N 0.000 description 2
- 210000004072 lung Anatomy 0.000 description 2
- 239000006166 lysate Substances 0.000 description 2
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 2
- 239000011976 maleic acid Substances 0.000 description 2
- 239000001630 malic acid Substances 0.000 description 2
- 235000011090 malic acid Nutrition 0.000 description 2
- 235000010355 mannitol Nutrition 0.000 description 2
- 239000003550 marker Substances 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- 125000001360 methionine group Chemical group N[C@@H](CCSC)C(=O)* 0.000 description 2
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 2
- 239000003094 microcapsule Substances 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- PULGYDLMFSFVBL-SMFNREODSA-N nociceptin Chemical compound C([C@@H](C(=O)N[C@H](C(=O)NCC(=O)N[C@@H](C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CO)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCC(N)=O)C(O)=O)[C@@H](C)O)NC(=O)CNC(=O)CNC(=O)[C@@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 PULGYDLMFSFVBL-SMFNREODSA-N 0.000 description 2
- 239000002777 nucleoside Substances 0.000 description 2
- 210000004940 nucleus Anatomy 0.000 description 2
- 210000000056 organ Anatomy 0.000 description 2
- 235000005985 organic acids Nutrition 0.000 description 2
- 150000007530 organic bases Chemical class 0.000 description 2
- IZUPBVBPLAPZRR-UHFFFAOYSA-N pentachlorophenol Chemical compound OC1=C(Cl)C(Cl)=C(Cl)C(Cl)=C1Cl IZUPBVBPLAPZRR-UHFFFAOYSA-N 0.000 description 2
- 229940021222 peritoneal dialysis isotonic solution Drugs 0.000 description 2
- 239000013034 phenoxy resin Substances 0.000 description 2
- 229920006287 phenoxy resin Polymers 0.000 description 2
- WVDDGKGOMKODPV-ZQBYOMGUSA-N phenyl(114C)methanol Chemical compound O[14CH2]C1=CC=CC=C1 WVDDGKGOMKODPV-ZQBYOMGUSA-N 0.000 description 2
- YBYRMVIVWMBXKQ-UHFFFAOYSA-N phenylmethanesulfonyl fluoride Chemical compound FS(=O)(=O)CC1=CC=CC=C1 YBYRMVIVWMBXKQ-UHFFFAOYSA-N 0.000 description 2
- 150000003904 phospholipids Chemical class 0.000 description 2
- 230000001766 physiological effect Effects 0.000 description 2
- 239000006187 pill Substances 0.000 description 2
- 239000001103 potassium chloride Substances 0.000 description 2
- 235000011164 potassium chloride Nutrition 0.000 description 2
- 239000002877 prolactin releasing hormone Substances 0.000 description 2
- 235000019260 propionic acid Nutrition 0.000 description 2
- 229940076372 protein antagonist Drugs 0.000 description 2
- 238000000159 protein binding assay Methods 0.000 description 2
- ZCCUUQDIBDJBTK-UHFFFAOYSA-N psoralen Chemical compound C1=C2OC(=O)C=CC2=CC2=C1OC=C2 ZCCUUQDIBDJBTK-UHFFFAOYSA-N 0.000 description 2
- 150000003212 purines Chemical class 0.000 description 2
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 2
- 150000003230 pyrimidines Chemical class 0.000 description 2
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 2
- 239000000941 radioactive substance Substances 0.000 description 2
- 229940044551 receptor antagonist Drugs 0.000 description 2
- 239000002464 receptor antagonist Substances 0.000 description 2
- 230000002829 reductive effect Effects 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- 238000004366 reverse phase liquid chromatography Methods 0.000 description 2
- 230000002441 reversible effect Effects 0.000 description 2
- CVHZOJJKTDOEJC-UHFFFAOYSA-N saccharin Chemical compound C1=CC=C2C(=O)NS(=O)(=O)C2=C1 CVHZOJJKTDOEJC-UHFFFAOYSA-N 0.000 description 2
- 235000019204 saccharin Nutrition 0.000 description 2
- 229940081974 saccharin Drugs 0.000 description 2
- 239000000901 saccharin and its Na,K and Ca salt Substances 0.000 description 2
- 238000005185 salting out Methods 0.000 description 2
- CDAISMWEOUEBRE-UHFFFAOYSA-N scyllo-inosotol Natural products OC1C(O)C(O)C(O)C(O)C1O CDAISMWEOUEBRE-UHFFFAOYSA-N 0.000 description 2
- 230000001953 sensory effect Effects 0.000 description 2
- 239000003998 snake venom Substances 0.000 description 2
- 239000007974 sodium acetate buffer Substances 0.000 description 2
- 238000010532 solid phase synthesis reaction Methods 0.000 description 2
- 229960002920 sorbitol Drugs 0.000 description 2
- 210000000952 spleen Anatomy 0.000 description 2
- 210000004989 spleen cell Anatomy 0.000 description 2
- 210000003802 sputum Anatomy 0.000 description 2
- 208000024794 sputum Diseases 0.000 description 2
- 230000010473 stable expression Effects 0.000 description 2
- 239000012086 standard solution Substances 0.000 description 2
- 239000008107 starch Substances 0.000 description 2
- 235000019698 starch Nutrition 0.000 description 2
- 210000000130 stem cell Anatomy 0.000 description 2
- UCSJYZPVAKXKNQ-HZYVHMACSA-N streptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O UCSJYZPVAKXKNQ-HZYVHMACSA-N 0.000 description 2
- 239000000758 substrate Substances 0.000 description 2
- 238000009495 sugar coating Methods 0.000 description 2
- 150000008163 sugars Chemical class 0.000 description 2
- 239000004094 surface-active agent Substances 0.000 description 2
- 239000003765 sweetening agent Substances 0.000 description 2
- 208000011580 syndromic disease Diseases 0.000 description 2
- 238000003786 synthesis reaction Methods 0.000 description 2
- 239000011975 tartaric acid Substances 0.000 description 2
- 235000002906 tartaric acid Nutrition 0.000 description 2
- 238000010998 test method Methods 0.000 description 2
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 2
- 238000011144 upstream manufacturing Methods 0.000 description 2
- HFNHAPQMXICKCF-USJMABIRSA-N urotensin-ii Chemical compound N([C@@H](CC(O)=O)C(=O)N[C@H]1CSSC[C@H](NC(=O)[C@H](CC=2C=CC(O)=CC=2)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CC=2C3=CC=CC=C3NC=2)NC(=O)[C@H](CC=2C=CC=CC=2)NC1=O)C(=O)N[C@@H](C(C)C)C(O)=O)C(=O)[C@@H]1CCCN1C(=O)[C@@H](NC(=O)[C@@H](N)CCC(O)=O)[C@@H](C)O HFNHAPQMXICKCF-USJMABIRSA-N 0.000 description 2
- 235000015112 vegetable and seed oil Nutrition 0.000 description 2
- 239000008158 vegetable oil Substances 0.000 description 2
- 239000008215 water for injection Substances 0.000 description 2
- KYRUKRFVOACELK-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 3-(4-hydroxyphenyl)propanoate Chemical compound C1=CC(O)=CC=C1CCC(=O)ON1C(=O)CCC1=O KYRUKRFVOACELK-UHFFFAOYSA-N 0.000 description 1
- DIGQNXIGRZPYDK-WKSCXVIASA-N (2R)-6-amino-2-[[2-[[(2S)-2-[[2-[[(2R)-2-[[(2S)-2-[[(2R,3S)-2-[[2-[[(2S)-2-[[2-[[(2S)-2-[[(2S)-2-[[(2R)-2-[[(2S,3S)-2-[[(2R)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[2-[[(2S)-2-[[(2R)-2-[[2-[[2-[[2-[(2-amino-1-hydroxyethylidene)amino]-3-carboxy-1-hydroxypropylidene]amino]-1-hydroxy-3-sulfanylpropylidene]amino]-1-hydroxyethylidene]amino]-1-hydroxy-3-sulfanylpropylidene]amino]-1,3-dihydroxypropylidene]amino]-1-hydroxyethylidene]amino]-1-hydroxypropylidene]amino]-1,3-dihydroxypropylidene]amino]-1,3-dihydroxypropylidene]amino]-1-hydroxy-3-sulfanylpropylidene]amino]-1,3-dihydroxybutylidene]amino]-1-hydroxy-3-sulfanylpropylidene]amino]-1-hydroxypropylidene]amino]-1,3-dihydroxypropylidene]amino]-1-hydroxyethylidene]amino]-1,5-dihydroxy-5-iminopentylidene]amino]-1-hydroxy-3-sulfanylpropylidene]amino]-1,3-dihydroxybutylidene]amino]-1-hydroxy-3-sulfanylpropylidene]amino]-1,3-dihydroxypropylidene]amino]-1-hydroxyethylidene]amino]-1-hydroxy-3-sulfanylpropylidene]amino]-1-hydroxyethylidene]amino]hexanoic acid Chemical compound C[C@@H]([C@@H](C(=N[C@@H](CS)C(=N[C@@H](C)C(=N[C@@H](CO)C(=NCC(=N[C@@H](CCC(=N)O)C(=NC(CS)C(=N[C@H]([C@H](C)O)C(=N[C@H](CS)C(=N[C@H](CO)C(=NCC(=N[C@H](CS)C(=NCC(=N[C@H](CCCCN)C(=O)O)O)O)O)O)O)O)O)O)O)O)O)O)O)N=C([C@H](CS)N=C([C@H](CO)N=C([C@H](CO)N=C([C@H](C)N=C(CN=C([C@H](CO)N=C([C@H](CS)N=C(CN=C(C(CS)N=C(C(CC(=O)O)N=C(CN)O)O)O)O)O)O)O)O)O)O)O)O DIGQNXIGRZPYDK-WKSCXVIASA-N 0.000 description 1
- SBKVPJHMSUXZTA-MEJXFZFPSA-N (2S)-2-[[(2S)-2-[[(2S)-1-[(2S)-5-amino-2-[[2-[[(2S)-1-[(2S)-6-amino-2-[[(2S)-2-[[(2S)-5-amino-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-amino-3-(1H-indol-3-yl)propanoyl]amino]-3-(1H-imidazol-4-yl)propanoyl]amino]-3-(1H-indol-3-yl)propanoyl]amino]-4-methylpentanoyl]amino]-5-oxopentanoyl]amino]-4-methylpentanoyl]amino]hexanoyl]pyrrolidine-2-carbonyl]amino]acetyl]amino]-5-oxopentanoyl]pyrrolidine-2-carbonyl]amino]-4-methylsulfanylbutanoyl]amino]-3-(4-hydroxyphenyl)propanoic acid Chemical compound C([C@@H](C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCN)C(=O)N1CCC[C@H]1C(=O)NCC(=O)N[C@@H](CCC(N)=O)C(=O)N1CCC[C@H]1C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(O)=O)NC(=O)[C@@H](N)CC=1C2=CC=CC=C2NC=1)C1=CNC=N1 SBKVPJHMSUXZTA-MEJXFZFPSA-N 0.000 description 1
- XNRRDBJRLNJVMF-GKAPJAKFSA-N (2s)-3-(dimethylamino)-n-(ethyliminomethylidene)pyrrolidine-2-carboxamide Chemical compound CCN=C=NC(=O)[C@H]1NCCC1N(C)C XNRRDBJRLNJVMF-GKAPJAKFSA-N 0.000 description 1
- BHQCQFFYRZLCQQ-UHFFFAOYSA-N (3alpha,5alpha,7alpha,12alpha)-3,7,12-trihydroxy-cholan-24-oic acid Natural products OC1CC2CC(O)CCC2(C)C2C1C1CCC(C(CCC(O)=O)C)C1(C)C(O)C2 BHQCQFFYRZLCQQ-UHFFFAOYSA-N 0.000 description 1
- UHPQFNXOFFPHJW-UHFFFAOYSA-N (4-methylphenyl)-phenylmethanamine Chemical compound C1=CC(C)=CC=C1C(N)C1=CC=CC=C1 UHPQFNXOFFPHJW-UHFFFAOYSA-N 0.000 description 1
- 102000040650 (ribonucleotides)n+m Human genes 0.000 description 1
- BDNKZNFMNDZQMI-UHFFFAOYSA-N 1,3-diisopropylcarbodiimide Chemical compound CC(C)N=C=NC(C)C BDNKZNFMNDZQMI-UHFFFAOYSA-N 0.000 description 1
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- LHJGJYXLEPZJPM-UHFFFAOYSA-N 2,4,5-trichlorophenol Chemical compound OC1=CC(Cl)=C(Cl)C=C1Cl LHJGJYXLEPZJPM-UHFFFAOYSA-N 0.000 description 1
- UFBJCMHMOXMLKC-UHFFFAOYSA-N 2,4-dinitrophenol Chemical compound OC1=CC=C([N+]([O-])=O)C=C1[N+]([O-])=O UFBJCMHMOXMLKC-UHFFFAOYSA-N 0.000 description 1
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 1
- ASJSAQIRZKANQN-CRCLSJGQSA-N 2-deoxy-D-ribose Chemical compound OC[C@@H](O)[C@@H](O)CC=O ASJSAQIRZKANQN-CRCLSJGQSA-N 0.000 description 1
- BFSVOASYOCHEOV-UHFFFAOYSA-N 2-diethylaminoethanol Chemical compound CCN(CC)CCO BFSVOASYOCHEOV-UHFFFAOYSA-N 0.000 description 1
- CFMZSMGAMPBRBE-UHFFFAOYSA-N 2-hydroxyisoindole-1,3-dione Chemical compound C1=CC=C2C(=O)N(O)C(=O)C2=C1 CFMZSMGAMPBRBE-UHFFFAOYSA-N 0.000 description 1
- XWKFPIODWVPXLX-UHFFFAOYSA-N 2-methyl-5-methylpyridine Natural products CC1=CC=C(C)N=C1 XWKFPIODWVPXLX-UHFFFAOYSA-N 0.000 description 1
- BSKHPKMHTQYZBB-UHFFFAOYSA-N 2-methylpyridine Chemical compound CC1=CC=CC=N1 BSKHPKMHTQYZBB-UHFFFAOYSA-N 0.000 description 1
- 125000000094 2-phenylethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])([H])* 0.000 description 1
- VXGRJERITKFWPL-UHFFFAOYSA-N 4',5'-Dihydropsoralen Natural products C1=C2OC(=O)C=CC2=CC2=C1OCC2 VXGRJERITKFWPL-UHFFFAOYSA-N 0.000 description 1
- OEBIVOHKFYSBPE-UHFFFAOYSA-N 4-Benzyloxybenzyl alcohol Chemical compound C1=CC(CO)=CC=C1OCC1=CC=CC=C1 OEBIVOHKFYSBPE-UHFFFAOYSA-N 0.000 description 1
- TVZGACDUOSZQKY-LBPRGKRZSA-N 4-aminofolic acid Chemical compound C1=NC2=NC(N)=NC(N)=C2N=C1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 TVZGACDUOSZQKY-LBPRGKRZSA-N 0.000 description 1
- BTJIUGUIPKRLHP-UHFFFAOYSA-N 4-nitrophenol Chemical compound OC1=CC=C([N+]([O-])=O)C=C1 BTJIUGUIPKRLHP-UHFFFAOYSA-N 0.000 description 1
- BZTDTCNHAFUJOG-UHFFFAOYSA-N 6-carboxyfluorescein Chemical compound C12=CC=C(O)C=C2OC2=CC(O)=CC=C2C11OC(=O)C2=CC=C(C(=O)O)C=C21 BZTDTCNHAFUJOG-UHFFFAOYSA-N 0.000 description 1
- 102400000069 Activation peptide Human genes 0.000 description 1
- 101800001401 Activation peptide Proteins 0.000 description 1
- 229920000936 Agarose Polymers 0.000 description 1
- 102000018746 Apelin Human genes 0.000 description 1
- 108010052412 Apelin Proteins 0.000 description 1
- KWKQGHSSNHPGOW-BQBZGAKWSA-N Arg-Ala-Gly Chemical compound [H]N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C)C(=O)NCC(O)=O KWKQGHSSNHPGOW-BQBZGAKWSA-N 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 206010003445 Ascites Diseases 0.000 description 1
- 241000271566 Aves Species 0.000 description 1
- 108010028006 B-Cell Activating Factor Proteins 0.000 description 1
- 102000016605 B-Cell Activating Factor Human genes 0.000 description 1
- 210000002237 B-cell of pancreatic islet Anatomy 0.000 description 1
- 241000409811 Bombyx mori nucleopolyhedrovirus Species 0.000 description 1
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N Boron Chemical compound [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 description 1
- 101000800130 Bos taurus Thyroglobulin Proteins 0.000 description 1
- 102000055006 Calcitonin Human genes 0.000 description 1
- 108060001064 Calcitonin Proteins 0.000 description 1
- BHPQYMZQTOCNFJ-UHFFFAOYSA-N Calcium cation Chemical compound [Ca+2] BHPQYMZQTOCNFJ-UHFFFAOYSA-N 0.000 description 1
- UXVMQQNJUSDDNG-UHFFFAOYSA-L Calcium chloride Chemical compound [Cl-].[Cl-].[Ca+2] UXVMQQNJUSDDNG-UHFFFAOYSA-L 0.000 description 1
- 241000700199 Cavia porcellus Species 0.000 description 1
- 239000004380 Cholic acid Substances 0.000 description 1
- 108090000317 Chymotrypsin Proteins 0.000 description 1
- 102000029816 Collagenase Human genes 0.000 description 1
- 108060005980 Collagenase Proteins 0.000 description 1
- 108020004635 Complementary DNA Proteins 0.000 description 1
- 229940126062 Compound A Drugs 0.000 description 1
- IGXWBGJHJZYPQS-SSDOTTSWSA-N D-Luciferin Chemical compound OC(=O)[C@H]1CSC(C=2SC3=CC=C(O)C=C3N=2)=N1 IGXWBGJHJZYPQS-SSDOTTSWSA-N 0.000 description 1
- 108020001019 DNA Primers Proteins 0.000 description 1
- 230000009946 DNA mutation Effects 0.000 description 1
- 239000003155 DNA primer Substances 0.000 description 1
- CYCGRDQQIOGCKX-UHFFFAOYSA-N Dehydro-luciferin Natural products OC(=O)C1=CSC(C=2SC3=CC(O)=CC=C3N=2)=N1 CYCGRDQQIOGCKX-UHFFFAOYSA-N 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- 239000004375 Dextrin Substances 0.000 description 1
- 229920001353 Dextrin Polymers 0.000 description 1
- XBPCUCUWBYBCDP-UHFFFAOYSA-N Dicyclohexylamine Chemical compound C1CCCCC1NC1CCCCC1 XBPCUCUWBYBCDP-UHFFFAOYSA-N 0.000 description 1
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 1
- QRLVDLBMBULFAL-UHFFFAOYSA-N Digitonin Natural products CC1CCC2(OC1)OC3C(O)C4C5CCC6CC(OC7OC(CO)C(OC8OC(CO)C(O)C(OC9OCC(O)C(O)C9OC%10OC(CO)C(O)C(OC%11OC(CO)C(O)C(O)C%11O)C%10O)C8O)C(O)C7O)C(O)CC6(C)C5CCC4(C)C3C2C QRLVDLBMBULFAL-UHFFFAOYSA-N 0.000 description 1
- LTLYEAJONXGNFG-DCAQKATOSA-N E64 Chemical compound NC(=N)NCCCCNC(=O)[C@H](CC(C)C)NC(=O)[C@H]1O[C@@H]1C(O)=O LTLYEAJONXGNFG-DCAQKATOSA-N 0.000 description 1
- 238000011891 EIA kit Methods 0.000 description 1
- 241001452028 Escherichia coli DH1 Species 0.000 description 1
- 241001646716 Escherichia coli K-12 Species 0.000 description 1
- 241000701959 Escherichia virus Lambda Species 0.000 description 1
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 1
- 108060002716 Exonuclease Proteins 0.000 description 1
- 101150106375 Far1 gene Proteins 0.000 description 1
- BJGNCJDXODQBOB-UHFFFAOYSA-N Fivefly Luciferin Natural products OC(=O)C1CSC(C=2SC3=CC(O)=CC=C3N=2)=N1 BJGNCJDXODQBOB-UHFFFAOYSA-N 0.000 description 1
- KRHYYFGTRYWZRS-UHFFFAOYSA-N Fluorane Chemical compound F KRHYYFGTRYWZRS-UHFFFAOYSA-N 0.000 description 1
- 102000034286 G proteins Human genes 0.000 description 1
- 101800001586 Ghrelin Proteins 0.000 description 1
- 102400000442 Ghrelin-28 Human genes 0.000 description 1
- DIXKFOPPGWKZLY-CIUDSAMLSA-N Glu-Arg-Asp Chemical compound [H]N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(O)=O)C(O)=O DIXKFOPPGWKZLY-CIUDSAMLSA-N 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- 108090000288 Glycoproteins Proteins 0.000 description 1
- 102000003886 Glycoproteins Human genes 0.000 description 1
- 102000005744 Glycoside Hydrolases Human genes 0.000 description 1
- 108010031186 Glycoside Hydrolases Proteins 0.000 description 1
- 101150009006 HIS3 gene Proteins 0.000 description 1
- NLDMNSXOCDLTTB-UHFFFAOYSA-N Heterophylliin A Natural products O1C2COC(=O)C3=CC(O)=C(O)C(O)=C3C3=C(O)C(O)=C(O)C=C3C(=O)OC2C(OC(=O)C=2C=C(O)C(O)=C(O)C=2)C(O)C1OC(=O)C1=CC(O)=C(O)C(O)=C1 NLDMNSXOCDLTTB-UHFFFAOYSA-N 0.000 description 1
- SQUHHTBVTRBESD-UHFFFAOYSA-N Hexa-Ac-myo-Inositol Natural products CC(=O)OC1C(OC(C)=O)C(OC(C)=O)C(OC(C)=O)C(OC(C)=O)C1OC(C)=O SQUHHTBVTRBESD-UHFFFAOYSA-N 0.000 description 1
- 101001132878 Homo sapiens Motilin receptor Proteins 0.000 description 1
- 101001122499 Homo sapiens Nociceptin receptor Proteins 0.000 description 1
- 101000829127 Homo sapiens Somatostatin receptor type 2 Proteins 0.000 description 1
- 101000644251 Homo sapiens Urotensin-2 receptor Proteins 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- 206010062767 Hypophysitis Diseases 0.000 description 1
- UGQMRVRMYYASKQ-UHFFFAOYSA-N Hypoxanthine nucleoside Natural products OC1C(O)C(CO)OC1N1C(NC=NC2=O)=C2N=C1 UGQMRVRMYYASKQ-UHFFFAOYSA-N 0.000 description 1
- 102000004877 Insulin Human genes 0.000 description 1
- 108090001061 Insulin Proteins 0.000 description 1
- 108010047761 Interferon-alpha Proteins 0.000 description 1
- 102000006992 Interferon-alpha Human genes 0.000 description 1
- 241000235058 Komagataella pastoris Species 0.000 description 1
- 238000012218 Kunkel's method Methods 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 description 1
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 1
- GDBQQVLCIARPGH-UHFFFAOYSA-N Leupeptin Natural products CC(C)CC(NC(C)=O)C(=O)NC(CC(C)C)C(=O)NC(C=O)CCCN=C(N)N GDBQQVLCIARPGH-UHFFFAOYSA-N 0.000 description 1
- DDWFXDSYGUXRAY-UHFFFAOYSA-N Luciferin Natural products CCc1c(C)c(CC2NC(=O)C(=C2C=C)C)[nH]c1Cc3[nH]c4C(=C5/NC(CC(=O)O)C(C)C5CC(=O)O)CC(=O)c4c3C DDWFXDSYGUXRAY-UHFFFAOYSA-N 0.000 description 1
- 239000006142 Luria-Bertani Agar Substances 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- 102000013460 Malate Dehydrogenase Human genes 0.000 description 1
- 108010026217 Malate Dehydrogenase Proteins 0.000 description 1
- PEEHTFAAVSWFBL-UHFFFAOYSA-N Maleimide Chemical compound O=C1NC(=O)C=C1 PEEHTFAAVSWFBL-UHFFFAOYSA-N 0.000 description 1
- 241000555303 Mamestra brassicae Species 0.000 description 1
- 108010038049 Mating Factor Proteins 0.000 description 1
- 101150106280 Mchr1 gene Proteins 0.000 description 1
- 102100027375 Melanin-concentrating hormone receptor 1 Human genes 0.000 description 1
- 108700036626 Melanin-concentrating hormone receptor 1 Proteins 0.000 description 1
- 108010052285 Membrane Proteins Proteins 0.000 description 1
- 102000003792 Metallothionein Human genes 0.000 description 1
- 108090000157 Metallothionein Proteins 0.000 description 1
- 102100033818 Motilin receptor Human genes 0.000 description 1
- 102000016943 Muramidase Human genes 0.000 description 1
- 108010014251 Muramidase Proteins 0.000 description 1
- 241000711408 Murine respirovirus Species 0.000 description 1
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 1
- 108010062010 N-Acetylmuramoyl-L-alanine Amidase Proteins 0.000 description 1
- NQTADLQHYWFPDB-UHFFFAOYSA-N N-Hydroxysuccinimide Chemical compound ON1C(=O)CCC1=O NQTADLQHYWFPDB-UHFFFAOYSA-N 0.000 description 1
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 1
- VIHYIVKEECZGOU-UHFFFAOYSA-N N-acetylimidazole Chemical compound CC(=O)N1C=CN=C1 VIHYIVKEECZGOU-UHFFFAOYSA-N 0.000 description 1
- 229930182474 N-glycoside Natural products 0.000 description 1
- 125000000729 N-terminal amino-acid group Chemical group 0.000 description 1
- 101150064037 NGF gene Proteins 0.000 description 1
- 229930193140 Neomycin Natural products 0.000 description 1
- 206010028980 Neoplasm Diseases 0.000 description 1
- 102100028646 Nociceptin receptor Human genes 0.000 description 1
- 108010079246 OMPA outer membrane proteins Proteins 0.000 description 1
- 208000008589 Obesity Diseases 0.000 description 1
- 108091034117 Oligonucleotide Proteins 0.000 description 1
- 108020005187 Oligonucleotide Probes Proteins 0.000 description 1
- 102000002512 Orexin Human genes 0.000 description 1
- 239000008118 PEG 6000 Substances 0.000 description 1
- 101150012394 PHO5 gene Proteins 0.000 description 1
- 241000282579 Pan Species 0.000 description 1
- 229930182555 Penicillin Natural products 0.000 description 1
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 1
- 102000035195 Peptidases Human genes 0.000 description 1
- 239000001888 Peptone Substances 0.000 description 1
- 108010080698 Peptones Proteins 0.000 description 1
- 102000003992 Peroxidases Human genes 0.000 description 1
- 108010002724 Pheromone Receptors Proteins 0.000 description 1
- 108010022181 Phosphopyruvate Hydratase Proteins 0.000 description 1
- 108091000080 Phosphotransferase Proteins 0.000 description 1
- 241000276498 Pollachius virens Species 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- 229920002584 Polyethylene Glycol 6000 Polymers 0.000 description 1
- 101710182846 Polyhedrin Proteins 0.000 description 1
- 108010039918 Polylysine Proteins 0.000 description 1
- 239000004743 Polypropylene Substances 0.000 description 1
- 239000004793 Polystyrene Substances 0.000 description 1
- HCBIBCJNVBAKAB-UHFFFAOYSA-N Procaine hydrochloride Chemical compound Cl.CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 HCBIBCJNVBAKAB-UHFFFAOYSA-N 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- 239000004365 Protease Substances 0.000 description 1
- 102000001253 Protein Kinase Human genes 0.000 description 1
- 108010079005 RDV peptide Proteins 0.000 description 1
- 238000010802 RNA extraction kit Methods 0.000 description 1
- 108020005091 Replication Origin Proteins 0.000 description 1
- 102100037486 Reverse transcriptase/ribonuclease H Human genes 0.000 description 1
- 101100394989 Rhodopseudomonas palustris (strain ATCC BAA-98 / CGA009) hisI gene Proteins 0.000 description 1
- 102000006382 Ribonucleases Human genes 0.000 description 1
- 108010083644 Ribonucleases Proteins 0.000 description 1
- 239000012891 Ringer solution Substances 0.000 description 1
- 229910003797 SPO1 Inorganic materials 0.000 description 1
- 229910003798 SPO2 Inorganic materials 0.000 description 1
- 101150014136 SUC2 gene Proteins 0.000 description 1
- 101100150136 Saccharomyces cerevisiae (strain ATCC 204508 / S288c) SPO1 gene Proteins 0.000 description 1
- 241000235347 Schizosaccharomyces pombe Species 0.000 description 1
- 101100478210 Schizosaccharomyces pombe (strain 972 / ATCC 24843) spo2 gene Proteins 0.000 description 1
- 108091081021 Sense strand Proteins 0.000 description 1
- 238000012300 Sequence Analysis Methods 0.000 description 1
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 1
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 1
- 108020004682 Single-Stranded DNA Proteins 0.000 description 1
- 244000061456 Solanum tuberosum Species 0.000 description 1
- 235000002595 Solanum tuberosum Nutrition 0.000 description 1
- 229940124269 Somatostatin receptor 2 agonist Drugs 0.000 description 1
- 235000019764 Soybean Meal Nutrition 0.000 description 1
- 241000256251 Spodoptera frugiperda Species 0.000 description 1
- 108090000787 Subtilisin Proteins 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- 210000001744 T-lymphocyte Anatomy 0.000 description 1
- 108010022394 Threonine synthase Proteins 0.000 description 1
- 101710120037 Toxin CcdB Proteins 0.000 description 1
- 108700019146 Transgenes Proteins 0.000 description 1
- GSEJCLTVZPLZKY-UHFFFAOYSA-N Triethanolamine Chemical compound OCCN(CCO)CCO GSEJCLTVZPLZKY-UHFFFAOYSA-N 0.000 description 1
- RHQDFWAXVIIEBN-UHFFFAOYSA-N Trifluoroethanol Chemical compound OCC(F)(F)F RHQDFWAXVIIEBN-UHFFFAOYSA-N 0.000 description 1
- 239000013504 Triton X-100 Substances 0.000 description 1
- 229920004890 Triton X-100 Polymers 0.000 description 1
- 108090000631 Trypsin Proteins 0.000 description 1
- 102000004142 Trypsin Human genes 0.000 description 1
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 description 1
- 108091023045 Untranslated Region Proteins 0.000 description 1
- 102100020942 Urotensin-2 receptor Human genes 0.000 description 1
- 101710205907 Urotensin-2 receptor Proteins 0.000 description 1
- 241000700618 Vaccinia virus Species 0.000 description 1
- APQIVBCUIUDSMB-OSUNSFLBSA-N Val-Ile-Thr Chemical compound CC[C@H](C)[C@@H](C(=O)N[C@@H]([C@@H](C)O)C(=O)O)NC(=O)[C@H](C(C)C)N APQIVBCUIUDSMB-OSUNSFLBSA-N 0.000 description 1
- 102100038344 Vomeronasal type-1 receptor 2 Human genes 0.000 description 1
- 241000269368 Xenopus laevis Species 0.000 description 1
- 240000008042 Zea mays Species 0.000 description 1
- 235000005824 Zea mays ssp. parviglumis Nutrition 0.000 description 1
- 235000002017 Zea mays subsp mays Nutrition 0.000 description 1
- QNEPTKZEXBPDLF-JDTILAPWSA-N [(3s,8s,9s,10r,13r,14s,17r)-10,13-dimethyl-17-[(2r)-6-methylheptan-2-yl]-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1h-cyclopenta[a]phenanthren-3-yl] carbonochloridate Chemical compound C1C=C2C[C@@H](OC(Cl)=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 QNEPTKZEXBPDLF-JDTILAPWSA-N 0.000 description 1
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Chemical class Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 1
- XAKBSHICSHRJCL-UHFFFAOYSA-N [CH2]C(=O)C1=CC=CC=C1 Chemical group [CH2]C(=O)C1=CC=CC=C1 XAKBSHICSHRJCL-UHFFFAOYSA-N 0.000 description 1
- 230000005856 abnormality Effects 0.000 description 1
- 238000009825 accumulation Methods 0.000 description 1
- KXKVLQRXCPHEJC-UHFFFAOYSA-N acetic acid trimethyl ester Natural products COC(C)=O KXKVLQRXCPHEJC-UHFFFAOYSA-N 0.000 description 1
- 125000003670 adamantan-2-yl group Chemical group [H]C1([H])C(C2([H])[H])([H])C([H])([H])C3([H])C([*])([H])C1([H])C([H])([H])C2([H])C3([H])[H] 0.000 description 1
- 125000005076 adamantyloxycarbonyl group Chemical group C12(CC3CC(CC(C1)C3)C2)OC(=O)* 0.000 description 1
- 210000001789 adipocyte Anatomy 0.000 description 1
- 210000004100 adrenal gland Anatomy 0.000 description 1
- 239000003463 adsorbent Substances 0.000 description 1
- 238000001042 affinity chromatography Methods 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 239000002168 alkylating agent Substances 0.000 description 1
- 229940100198 alkylating agent Drugs 0.000 description 1
- 102000004139 alpha-Amylases Human genes 0.000 description 1
- 108090000637 alpha-Amylases Proteins 0.000 description 1
- 229940024171 alpha-amylase Drugs 0.000 description 1
- AZDRQVAHHNSJOQ-UHFFFAOYSA-N alumane Chemical class [AlH3] AZDRQVAHHNSJOQ-UHFFFAOYSA-N 0.000 description 1
- 150000001412 amines Chemical group 0.000 description 1
- 150000003862 amino acid derivatives Chemical class 0.000 description 1
- 125000000539 amino acid group Chemical group 0.000 description 1
- 238000003277 amino acid sequence analysis Methods 0.000 description 1
- 125000004202 aminomethyl group Chemical group [H]N([H])C([H])([H])* 0.000 description 1
- 229960003896 aminopterin Drugs 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 230000003321 amplification Effects 0.000 description 1
- 239000000883 anti-obesity agent Substances 0.000 description 1
- 230000001147 anti-toxic effect Effects 0.000 description 1
- 229940125710 antiobesity agent Drugs 0.000 description 1
- BWVPHIKGXQBZPV-QKFDDRBGSA-N apelin Chemical compound NCC(=O)N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCSC)C(=O)N1CCC[C@H]1C(=O)N[C@@H](CC(C)C)C(=O)N1[C@H](C(=O)N[C@@H](CC(O)=O)C(=O)NCC(=O)N[C@@H](CC(N)=O)C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(O)=O)C(=O)NCC(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=2NC=NC=2)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N2[C@@H](CCC2)C(=O)N[C@@H](CCCNC(N)=N)C(=O)NCC(=O)N[C@@H](CO)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(N)=O)C(=O)NCC(=O)N2[C@@H](CCC2)C(=O)NCC(=O)N2[C@@H](CCC2)C(=O)N[C@@H](CC=2C3=CC=CC=C3NC=2)C(=O)N[C@@H](CCC(N)=O)C(=O)NCC(=O)NCC(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N2[C@@H](CCC2)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC=2NC=NC=2)C(=O)N[C@@H](CCCCN)C(=O)NCC(=O)N2[C@@H](CCC2)C(=O)N[C@@H](CCSC)C(=O)N2[C@@H](CCC2)C(=O)N[C@@H](CC=2C=CC=CC=2)C(O)=O)CCC1 BWVPHIKGXQBZPV-QKFDDRBGSA-N 0.000 description 1
- PYMYPHUHKUWMLA-UHFFFAOYSA-N arabinose Natural products OCC(O)C(O)C(O)C=O PYMYPHUHKUWMLA-UHFFFAOYSA-N 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 108010030518 arginine endopeptidase Proteins 0.000 description 1
- 125000003435 aroyl group Chemical group 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- 235000003704 aspartic acid Nutrition 0.000 description 1
- 239000012752 auxiliary agent Substances 0.000 description 1
- 150000001540 azides Chemical class 0.000 description 1
- 210000003719 b-lymphocyte Anatomy 0.000 description 1
- 210000004227 basal ganglia Anatomy 0.000 description 1
- 210000003651 basophil Anatomy 0.000 description 1
- 239000011324 bead Substances 0.000 description 1
- JUHORIMYRDESRB-UHFFFAOYSA-N benzathine Chemical compound C=1C=CC=CC=1CNCCNCC1=CC=CC=C1 JUHORIMYRDESRB-UHFFFAOYSA-N 0.000 description 1
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 1
- RXUBZLMIGSAPEJ-UHFFFAOYSA-N benzyl n-aminocarbamate Chemical compound NNC(=O)OCC1=CC=CC=C1 RXUBZLMIGSAPEJ-UHFFFAOYSA-N 0.000 description 1
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 1
- SRBFZHDQGSBBOR-UHFFFAOYSA-N beta-D-Pyranose-Lyxose Natural products OC1COC(O)C(O)C1O SRBFZHDQGSBBOR-UHFFFAOYSA-N 0.000 description 1
- 102000006995 beta-Glucosidase Human genes 0.000 description 1
- 108010047754 beta-Glucosidase Proteins 0.000 description 1
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 1
- 230000008827 biological function Effects 0.000 description 1
- 230000001851 biosynthetic effect Effects 0.000 description 1
- 210000000601 blood cell Anatomy 0.000 description 1
- 210000004204 blood vessel Anatomy 0.000 description 1
- 210000001124 body fluid Anatomy 0.000 description 1
- 239000010839 body fluid Substances 0.000 description 1
- 210000000988 bone and bone Anatomy 0.000 description 1
- 210000002449 bone cell Anatomy 0.000 description 1
- 210000001185 bone marrow Anatomy 0.000 description 1
- 210000002798 bone marrow cell Anatomy 0.000 description 1
- 239000012888 bovine serum Substances 0.000 description 1
- 239000001273 butane Substances 0.000 description 1
- 125000004106 butoxy group Chemical group [*]OC([H])([H])C([H])([H])C(C([H])([H])[H])([H])[H] 0.000 description 1
- BBBFJLBPOGFECG-VJVYQDLKSA-N calcitonin Chemical compound N([C@H](C(=O)N[C@@H](CC(C)C)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)NCC(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H]([C@@H](C)O)C(=O)N1[C@@H](CCC1)C(N)=O)C(C)C)C(=O)[C@@H]1CSSC[C@H](N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CO)C(=O)N[C@@H]([C@@H](C)O)C(=O)N1 BBBFJLBPOGFECG-VJVYQDLKSA-N 0.000 description 1
- 229960004015 calcitonin Drugs 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 239000001110 calcium chloride Substances 0.000 description 1
- 229910001628 calcium chloride Inorganic materials 0.000 description 1
- 229910001424 calcium ion Inorganic materials 0.000 description 1
- 159000000007 calcium salts Chemical class 0.000 description 1
- 201000011510 cancer Diseases 0.000 description 1
- 229940041514 candida albicans extract Drugs 0.000 description 1
- 150000004657 carbamic acid derivatives Chemical class 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 210000000748 cardiovascular system Anatomy 0.000 description 1
- 239000005018 casein Substances 0.000 description 1
- BECPQYXYKAMYBN-UHFFFAOYSA-N casein, tech. Chemical compound NCCCCC(C(O)=O)N=C(O)C(CC(O)=O)N=C(O)C(CCC(O)=N)N=C(O)C(CC(C)C)N=C(O)C(CCC(O)=O)N=C(O)C(CC(O)=O)N=C(O)C(CCC(O)=O)N=C(O)C(C(C)O)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=O)N=C(O)C(CCC(O)=O)N=C(O)C(COP(O)(O)=O)N=C(O)C(CCC(O)=N)N=C(O)C(N)CC1=CC=CC=C1 BECPQYXYKAMYBN-UHFFFAOYSA-N 0.000 description 1
- 235000021240 caseins Nutrition 0.000 description 1
- 239000004359 castor oil Substances 0.000 description 1
- 235000019438 castor oil Nutrition 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 238000010531 catalytic reduction reaction Methods 0.000 description 1
- 150000001768 cations Chemical class 0.000 description 1
- 210000001159 caudate nucleus Anatomy 0.000 description 1
- 239000006285 cell suspension Substances 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 210000001638 cerebellum Anatomy 0.000 description 1
- 210000003710 cerebral cortex Anatomy 0.000 description 1
- 239000013522 chelant Substances 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 125000004218 chloromethyl group Chemical group [H]C([H])(Cl)* 0.000 description 1
- 150000001841 cholesterols Chemical class 0.000 description 1
- 235000019416 cholic acid Nutrition 0.000 description 1
- BHQCQFFYRZLCQQ-OELDTZBJSA-N cholic acid Chemical compound C([C@H]1C[C@H]2O)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC(O)=O)C)[C@@]2(C)[C@@H](O)C1 BHQCQFFYRZLCQQ-OELDTZBJSA-N 0.000 description 1
- 229960002471 cholic acid Drugs 0.000 description 1
- 210000001612 chondrocyte Anatomy 0.000 description 1
- 210000000349 chromosome Anatomy 0.000 description 1
- 229960002376 chymotrypsin Drugs 0.000 description 1
- 235000015165 citric acid Nutrition 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 229960002424 collagenase Drugs 0.000 description 1
- 238000004440 column chromatography Methods 0.000 description 1
- 239000007859 condensation product Substances 0.000 description 1
- 230000021615 conjugation Effects 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 235000005822 corn Nutrition 0.000 description 1
- 239000007822 coupling agent Substances 0.000 description 1
- 238000004132 cross linking Methods 0.000 description 1
- 210000004748 cultured cell Anatomy 0.000 description 1
- 125000000753 cycloalkyl group Chemical group 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 230000006378 damage Effects 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 239000003398 denaturant Substances 0.000 description 1
- 238000000432 density-gradient centrifugation Methods 0.000 description 1
- 229940009976 deoxycholate Drugs 0.000 description 1
- KXGVEGMKQFWNSR-LLQZFEROSA-N deoxycholic acid Chemical compound C([C@H]1CC2)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC(O)=O)C)[C@@]2(C)[C@@H](O)C1 KXGVEGMKQFWNSR-LLQZFEROSA-N 0.000 description 1
- KXGVEGMKQFWNSR-UHFFFAOYSA-N deoxycholic acid Natural products C1CC2CC(O)CCC2(C)C2C1C1CCC(C(CCC(O)=O)C)C1(C)C(O)C2 KXGVEGMKQFWNSR-UHFFFAOYSA-N 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 235000019425 dextrin Nutrition 0.000 description 1
- 238000000502 dialysis Methods 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- MTHSVFCYNBDYFN-UHFFFAOYSA-N diethylene glycol Chemical compound OCCOCCO MTHSVFCYNBDYFN-UHFFFAOYSA-N 0.000 description 1
- 230000029087 digestion Effects 0.000 description 1
- 210000002249 digestive system Anatomy 0.000 description 1
- UVYVLBIGDKGWPX-KUAJCENISA-N digitonin Chemical compound O([C@@H]1[C@@H]([C@]2(CC[C@@H]3[C@@]4(C)C[C@@H](O)[C@H](O[C@H]5[C@@H]([C@@H](O)[C@@H](O[C@H]6[C@@H]([C@@H](O[C@H]7[C@@H]([C@@H](O)[C@H](O)CO7)O)[C@H](O)[C@@H](CO)O6)O[C@H]6[C@@H]([C@@H](O[C@H]7[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O7)O)[C@@H](O)[C@@H](CO)O6)O)[C@@H](CO)O5)O)C[C@@H]4CC[C@H]3[C@@H]2[C@@H]1O)C)[C@@H]1C)[C@]11CC[C@@H](C)CO1 UVYVLBIGDKGWPX-KUAJCENISA-N 0.000 description 1
- UVYVLBIGDKGWPX-UHFFFAOYSA-N digitonine Natural products CC1C(C2(CCC3C4(C)CC(O)C(OC5C(C(O)C(OC6C(C(OC7C(C(O)C(O)CO7)O)C(O)C(CO)O6)OC6C(C(OC7C(C(O)C(O)C(CO)O7)O)C(O)C(CO)O6)O)C(CO)O5)O)CC4CCC3C2C2O)C)C2OC11CCC(C)CO1 UVYVLBIGDKGWPX-UHFFFAOYSA-N 0.000 description 1
- 102000004419 dihydrofolate reductase Human genes 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 239000013024 dilution buffer Substances 0.000 description 1
- ZZVUWRFHKOJYTH-UHFFFAOYSA-N diphenhydramine Chemical group C=1C=CC=CC=1C(OCCN(C)C)C1=CC=CC=C1 ZZVUWRFHKOJYTH-UHFFFAOYSA-N 0.000 description 1
- MGHPNCMVUAKAIE-UHFFFAOYSA-N diphenylmethanamine Chemical compound C=1C=CC=CC=1C(N)C1=CC=CC=C1 MGHPNCMVUAKAIE-UHFFFAOYSA-N 0.000 description 1
- 229940042399 direct acting antivirals protease inhibitors Drugs 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 150000004662 dithiols Chemical class 0.000 description 1
- NAGJZTKCGNOGPW-UHFFFAOYSA-N dithiophosphoric acid Chemical class OP(O)(S)=S NAGJZTKCGNOGPW-UHFFFAOYSA-N 0.000 description 1
- 239000008298 dragée Substances 0.000 description 1
- 238000009510 drug design Methods 0.000 description 1
- 238000009509 drug development Methods 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 238000001962 electrophoresis Methods 0.000 description 1
- 238000004520 electroporation Methods 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000010828 elution Methods 0.000 description 1
- 230000001804 emulsifying effect Effects 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 210000002889 endothelial cell Anatomy 0.000 description 1
- 239000002702 enteric coating Substances 0.000 description 1
- 238000009505 enteric coating Methods 0.000 description 1
- 210000003979 eosinophil Anatomy 0.000 description 1
- 210000001339 epidermal cell Anatomy 0.000 description 1
- 210000002919 epithelial cell Anatomy 0.000 description 1
- 239000003797 essential amino acid Substances 0.000 description 1
- 235000020776 essential amino acid Nutrition 0.000 description 1
- 125000003754 ethoxycarbonyl group Chemical group C(=O)(OCC)* 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 230000005284 excitation Effects 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 102000013165 exonuclease Human genes 0.000 description 1
- 239000013613 expression plasmid Substances 0.000 description 1
- 239000000835 fiber Substances 0.000 description 1
- 210000002950 fibroblast Anatomy 0.000 description 1
- 239000007941 film coated tablet Substances 0.000 description 1
- ZFKJVJIDPQDDFY-UHFFFAOYSA-N fluorescamine Chemical compound C12=CC=CC=C2C(=O)OC1(C1=O)OC=C1C1=CC=CC=C1 ZFKJVJIDPQDDFY-UHFFFAOYSA-N 0.000 description 1
- MHMNJMPURVTYEJ-UHFFFAOYSA-N fluorescein-5-isothiocyanate Chemical compound O1C(=O)C2=CC(N=C=S)=CC=C2C21C1=CC=C(O)C=C1OC1=CC(O)=CC=C21 MHMNJMPURVTYEJ-UHFFFAOYSA-N 0.000 description 1
- 230000037406 food intake Effects 0.000 description 1
- 235000010855 food raising agent Nutrition 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 238000007710 freezing Methods 0.000 description 1
- 230000008014 freezing Effects 0.000 description 1
- 239000012737 fresh medium Substances 0.000 description 1
- 210000001652 frontal lobe Anatomy 0.000 description 1
- YFHXZQPUBCBNIP-UHFFFAOYSA-N fura-2 Chemical compound CC1=CC=C(N(CC(O)=O)CC(O)=O)C(OCCOC=2C(=CC=3OC(=CC=3C=2)C=2OC(=CN=2)C(O)=O)N(CC(O)=O)CC(O)=O)=C1 YFHXZQPUBCBNIP-UHFFFAOYSA-N 0.000 description 1
- VPSRLGDRGCKUTK-UHFFFAOYSA-N fura-2-acetoxymethyl ester Chemical compound CC(=O)OCOC(=O)CN(CC(=O)OCOC(C)=O)C1=CC=C(C)C=C1OCCOC(C(=C1)N(CC(=O)OCOC(C)=O)CC(=O)OCOC(C)=O)=CC2=C1OC(C=1OC(=CN=1)C(=O)OCOC(C)=O)=C2 VPSRLGDRGCKUTK-UHFFFAOYSA-N 0.000 description 1
- 108020001507 fusion proteins Proteins 0.000 description 1
- 102000037865 fusion proteins Human genes 0.000 description 1
- 210000000232 gallbladder Anatomy 0.000 description 1
- 230000005861 gene abnormality Effects 0.000 description 1
- 238000001415 gene therapy Methods 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 238000010353 genetic engineering Methods 0.000 description 1
- GNKDKYIHGQKHHM-RJKLHVOGSA-N ghrelin Chemical compound C([C@H](NC(=O)[C@@H](NC(=O)[C@H](CO)NC(=O)CN)COC(=O)CCCCCCC)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CO)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1N=CNC=1)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)C1=CC=CC=C1 GNKDKYIHGQKHHM-RJKLHVOGSA-N 0.000 description 1
- 230000002518 glial effect Effects 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- 239000004220 glutamic acid Substances 0.000 description 1
- 125000000291 glutamic acid group Chemical group N[C@@H](CCC(O)=O)C(=O)* 0.000 description 1
- ZEMPKEQAKRGZGQ-XOQCFJPHSA-N glycerol triricinoleate Natural products CCCCCC[C@@H](O)CC=CCCCCCCCC(=O)OC[C@@H](COC(=O)CCCCCCCC=CC[C@@H](O)CCCCCC)OC(=O)CCCCCCCC=CC[C@H](O)CCCCCC ZEMPKEQAKRGZGQ-XOQCFJPHSA-N 0.000 description 1
- 150000002334 glycols Chemical class 0.000 description 1
- VPZXBVLAVMBEQI-UHFFFAOYSA-N glycyl-DL-alpha-alanine Natural products OC(=O)C(C)NC(=O)CN VPZXBVLAVMBEQI-UHFFFAOYSA-N 0.000 description 1
- 210000002149 gonad Anatomy 0.000 description 1
- 239000006451 grace's insect medium Substances 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 229960000789 guanidine hydrochloride Drugs 0.000 description 1
- PJJJBBJSCAKJQF-UHFFFAOYSA-N guanidinium chloride Chemical compound [Cl-].NC(N)=[NH2+] PJJJBBJSCAKJQF-UHFFFAOYSA-N 0.000 description 1
- 125000002795 guanidino group Chemical group C(N)(=N)N* 0.000 description 1
- 150000008282 halocarbons Chemical class 0.000 description 1
- 229910052736 halogen Inorganic materials 0.000 description 1
- 150000002367 halogens Chemical class 0.000 description 1
- 210000002216 heart Anatomy 0.000 description 1
- 210000003494 hepatocyte Anatomy 0.000 description 1
- 210000001320 hippocampus Anatomy 0.000 description 1
- 210000003630 histaminocyte Anatomy 0.000 description 1
- 230000013632 homeostatic process Effects 0.000 description 1
- 210000005260 human cell Anatomy 0.000 description 1
- 235000003642 hunger Nutrition 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- 229910052739 hydrogen Inorganic materials 0.000 description 1
- 229910000040 hydrogen fluoride Inorganic materials 0.000 description 1
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 description 1
- 230000002267 hypothalamic effect Effects 0.000 description 1
- 210000003016 hypothalamus Anatomy 0.000 description 1
- 210000003405 ileum Anatomy 0.000 description 1
- 210000002865 immune cell Anatomy 0.000 description 1
- 230000001900 immune effect Effects 0.000 description 1
- 210000000987 immune system Anatomy 0.000 description 1
- 238000002649 immunization Methods 0.000 description 1
- 238000001114 immunoprecipitation Methods 0.000 description 1
- 230000001976 improved effect Effects 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 229940042040 innovative drug Drugs 0.000 description 1
- CDAISMWEOUEBRE-GPIVLXJGSA-N inositol Chemical compound O[C@H]1[C@H](O)[C@@H](O)[C@H](O)[C@H](O)[C@@H]1O CDAISMWEOUEBRE-GPIVLXJGSA-N 0.000 description 1
- 229960000367 inositol Drugs 0.000 description 1
- 238000003780 insertion Methods 0.000 description 1
- 230000037431 insertion Effects 0.000 description 1
- 229940125396 insulin Drugs 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 238000001155 isoelectric focusing Methods 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 108010045069 keyhole-limpet hemocyanin Proteins 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 238000000021 kinase assay Methods 0.000 description 1
- 210000001821 langerhans cell Anatomy 0.000 description 1
- 210000002429 large intestine Anatomy 0.000 description 1
- 231100001231 less toxic Toxicity 0.000 description 1
- GDBQQVLCIARPGH-ULQDDVLXSA-N leupeptin Chemical compound CC(C)C[C@H](NC(C)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@H](C=O)CCCN=C(N)N GDBQQVLCIARPGH-ULQDDVLXSA-N 0.000 description 1
- 108010052968 leupeptin Proteins 0.000 description 1
- 125000005647 linker group Chemical group 0.000 description 1
- 239000002502 liposome Substances 0.000 description 1
- 238000004811 liquid chromatography Methods 0.000 description 1
- 239000008297 liquid dosage form Substances 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 230000033001 locomotion Effects 0.000 description 1
- KNJDBYZZKAZQNG-UHFFFAOYSA-N lucigenin Chemical compound [O-][N+]([O-])=O.[O-][N+]([O-])=O.C12=CC=CC=C2[N+](C)=C(C=CC=C2)C2=C1C1=C(C=CC=C2)C2=[N+](C)C2=CC=CC=C12 KNJDBYZZKAZQNG-UHFFFAOYSA-N 0.000 description 1
- 210000001165 lymph node Anatomy 0.000 description 1
- 239000004325 lysozyme Substances 0.000 description 1
- 229960000274 lysozyme Drugs 0.000 description 1
- 235000010335 lysozyme Nutrition 0.000 description 1
- RLSSMJSEOOYNOY-UHFFFAOYSA-N m-cresol Chemical compound CC1=CC=CC(O)=C1 RLSSMJSEOOYNOY-UHFFFAOYSA-N 0.000 description 1
- 210000002540 macrophage Anatomy 0.000 description 1
- 229910001629 magnesium chloride Inorganic materials 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- 210000004962 mammalian cell Anatomy 0.000 description 1
- 210000001161 mammalian embryo Anatomy 0.000 description 1
- 210000004216 mammary stem cell Anatomy 0.000 description 1
- 235000013372 meat Nutrition 0.000 description 1
- 230000010534 mechanism of action Effects 0.000 description 1
- 210000003593 megakaryocyte Anatomy 0.000 description 1
- ORRDHOMWDPJSNL-UHFFFAOYSA-N melanin concentrating hormone Chemical compound N1C(=O)C(C(C)C)NC(=O)C(CCCNC(N)=N)NC(=O)CNC(=O)C(C(C)C)NC(=O)C(CCSC)NC(=O)C(NC(=O)C(CCCNC(N)=N)NC(=O)C(NC(=O)C(NC(=O)C(N)CC(O)=O)C(C)O)CCSC)CSSCC(C(=O)NC(CC=2C3=CC=CC=C3NC=2)C(=O)NC(CCC(O)=O)C(=O)NC(C(C)C)C(O)=O)NC(=O)C2CCCN2C(=O)C(CCCNC(N)=N)NC(=O)C1CC1=CC=C(O)C=C1 ORRDHOMWDPJSNL-UHFFFAOYSA-N 0.000 description 1
- 210000003584 mesangial cell Anatomy 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 229940100630 metacresol Drugs 0.000 description 1
- UKVIEHSSVKSQBA-UHFFFAOYSA-N methane;palladium Chemical compound C.[Pd] UKVIEHSSVKSQBA-UHFFFAOYSA-N 0.000 description 1
- 229960000485 methotrexate Drugs 0.000 description 1
- UZKWTJUDCOPSNM-UHFFFAOYSA-N methoxybenzene Substances CCCCOC=C UZKWTJUDCOPSNM-UHFFFAOYSA-N 0.000 description 1
- YACKEPLHDIMKIO-UHFFFAOYSA-N methylphosphonic acid Chemical class CP(O)(O)=O YACKEPLHDIMKIO-UHFFFAOYSA-N 0.000 description 1
- 238000000520 microinjection Methods 0.000 description 1
- 239000004005 microsphere Substances 0.000 description 1
- 239000012046 mixed solvent Substances 0.000 description 1
- 239000003068 molecular probe Substances 0.000 description 1
- 210000001616 monocyte Anatomy 0.000 description 1
- 150000002772 monosaccharides Chemical class 0.000 description 1
- 229910000403 monosodium phosphate Inorganic materials 0.000 description 1
- 235000019799 monosodium phosphate Nutrition 0.000 description 1
- 210000003205 muscle Anatomy 0.000 description 1
- 210000000663 muscle cell Anatomy 0.000 description 1
- 230000035772 mutation Effects 0.000 description 1
- 208000031225 myocardial ischemia Diseases 0.000 description 1
- VMGAPWLDMVPYIA-HIDZBRGKSA-N n'-amino-n-iminomethanimidamide Chemical compound N\N=C\N=N VMGAPWLDMVPYIA-HIDZBRGKSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- ZUSSTQCWRDLYJA-UHFFFAOYSA-N n-hydroxy-5-norbornene-2,3-dicarboximide Chemical compound C1=CC2CC1C1C2C(=O)N(O)C1=O ZUSSTQCWRDLYJA-UHFFFAOYSA-N 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 210000000822 natural killer cell Anatomy 0.000 description 1
- 229960004927 neomycin Drugs 0.000 description 1
- 210000000653 nervous system Anatomy 0.000 description 1
- 210000002569 neuron Anatomy 0.000 description 1
- 230000003472 neutralizing effect Effects 0.000 description 1
- 210000000440 neutrophil Anatomy 0.000 description 1
- 239000002547 new drug Substances 0.000 description 1
- FEMOMIGRRWSMCU-UHFFFAOYSA-N ninhydrin Chemical compound C1=CC=C2C(=O)C(O)(O)C(=O)C2=C1 FEMOMIGRRWSMCU-UHFFFAOYSA-N 0.000 description 1
- 150000002823 nitrates Chemical class 0.000 description 1
- 150000002825 nitriles Chemical class 0.000 description 1
- 238000003199 nucleic acid amplification method Methods 0.000 description 1
- 150000003833 nucleoside derivatives Chemical class 0.000 description 1
- 235000020824 obesity Nutrition 0.000 description 1
- 210000000869 occipital lobe Anatomy 0.000 description 1
- 210000000956 olfactory bulb Anatomy 0.000 description 1
- 239000002751 oligonucleotide probe Substances 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 108060005714 orexin Proteins 0.000 description 1
- 230000008520 organization Effects 0.000 description 1
- 210000004798 organs belonging to the digestive system Anatomy 0.000 description 1
- 210000000963 osteoblast Anatomy 0.000 description 1
- 210000002997 osteoclast Anatomy 0.000 description 1
- 210000001672 ovary Anatomy 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 230000001590 oxidative effect Effects 0.000 description 1
- IWDCLRJOBJJRNH-UHFFFAOYSA-N p-cresol Chemical compound CC1=CC=C(O)C=C1 IWDCLRJOBJJRNH-UHFFFAOYSA-N 0.000 description 1
- 230000008058 pain sensation Effects 0.000 description 1
- 210000000496 pancreas Anatomy 0.000 description 1
- 235000020200 pasteurised milk Nutrition 0.000 description 1
- 230000007170 pathology Effects 0.000 description 1
- 101150019841 penP gene Proteins 0.000 description 1
- 229940049954 penicillin Drugs 0.000 description 1
- 125000001148 pentyloxycarbonyl group Chemical group 0.000 description 1
- 108010091212 pepstatin Proteins 0.000 description 1
- 229950000964 pepstatin Drugs 0.000 description 1
- FAXGPCHRFPCXOO-LXTPJMTPSA-N pepstatin A Chemical compound OC(=O)C[C@H](O)[C@H](CC(C)C)NC(=O)[C@H](C)NC(=O)C[C@H](O)[C@H](CC(C)C)NC(=O)[C@H](C(C)C)NC(=O)[C@H](C(C)C)NC(=O)CC(C)C FAXGPCHRFPCXOO-LXTPJMTPSA-N 0.000 description 1
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 1
- 235000019319 peptone Nutrition 0.000 description 1
- 210000005259 peripheral blood Anatomy 0.000 description 1
- 239000011886 peripheral blood Substances 0.000 description 1
- 210000004976 peripheral blood cell Anatomy 0.000 description 1
- 230000002093 peripheral effect Effects 0.000 description 1
- 230000035699 permeability Effects 0.000 description 1
- 108040007629 peroxidase activity proteins Proteins 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- 125000002467 phosphate group Chemical group [H]OP(=O)(O[H])O[*] 0.000 description 1
- 150000008298 phosphoramidates Chemical class 0.000 description 1
- PTMHPRAIXMAOOB-UHFFFAOYSA-N phosphoramidic acid Chemical compound NP(O)(O)=O PTMHPRAIXMAOOB-UHFFFAOYSA-N 0.000 description 1
- 102000020233 phosphotransferase Human genes 0.000 description 1
- 125000000612 phthaloyl group Chemical group C(C=1C(C(=O)*)=CC=CC1)(=O)* 0.000 description 1
- 230000001817 pituitary effect Effects 0.000 description 1
- 210000003635 pituitary gland Anatomy 0.000 description 1
- 210000002826 placenta Anatomy 0.000 description 1
- 239000000419 plant extract Substances 0.000 description 1
- 239000013600 plasmid vector Substances 0.000 description 1
- 229920001983 poloxamer Polymers 0.000 description 1
- 229920000729 poly(L-lysine) polymer Polymers 0.000 description 1
- 229920002523 polyethylene Glycol 1000 Polymers 0.000 description 1
- 229920000656 polylysine Polymers 0.000 description 1
- 238000003752 polymerase chain reaction Methods 0.000 description 1
- 229920001184 polypeptide Polymers 0.000 description 1
- 229920001155 polypropylene Polymers 0.000 description 1
- 229920001282 polysaccharide Polymers 0.000 description 1
- 239000005017 polysaccharide Substances 0.000 description 1
- 150000004804 polysaccharides Chemical class 0.000 description 1
- 229920002223 polystyrene Polymers 0.000 description 1
- XAEFZNCEHLXOMS-UHFFFAOYSA-M potassium benzoate Chemical compound [K+].[O-]C(=O)C1=CC=CC=C1 XAEFZNCEHLXOMS-UHFFFAOYSA-M 0.000 description 1
- 229910001414 potassium ion Inorganic materials 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 229960004919 procaine Drugs 0.000 description 1
- MFDFERRIHVXMIY-UHFFFAOYSA-N procaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 MFDFERRIHVXMIY-UHFFFAOYSA-N 0.000 description 1
- 229960001309 procaine hydrochloride Drugs 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- 229940043274 prophylactic drug Drugs 0.000 description 1
- FVSKHRXBFJPNKK-UHFFFAOYSA-N propionitrile Chemical compound CCC#N FVSKHRXBFJPNKK-UHFFFAOYSA-N 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 210000002307 prostate Anatomy 0.000 description 1
- 235000019833 protease Nutrition 0.000 description 1
- 235000019419 proteases Nutrition 0.000 description 1
- 108060006633 protein kinase Proteins 0.000 description 1
- 210000002637 putamen Anatomy 0.000 description 1
- 230000006340 racemization Effects 0.000 description 1
- 239000002516 radical scavenger Substances 0.000 description 1
- 230000002285 radioactive effect Effects 0.000 description 1
- 101150079601 recA gene Proteins 0.000 description 1
- 238000001525 receptor binding assay Methods 0.000 description 1
- 238000003259 recombinant expression Methods 0.000 description 1
- 230000006798 recombination Effects 0.000 description 1
- 238000005215 recombination Methods 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 238000006722 reduction reaction Methods 0.000 description 1
- 230000003578 releasing effect Effects 0.000 description 1
- BOLDJAUMGUJJKM-LSDHHAIUSA-N renifolin D Natural products CC(=C)[C@@H]1Cc2c(O)c(O)ccc2[C@H]1CC(=O)c3ccc(O)cc3O BOLDJAUMGUJJKM-LSDHHAIUSA-N 0.000 description 1
- 230000010076 replication Effects 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 238000012163 sequencing technique Methods 0.000 description 1
- 230000007781 signaling event Effects 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- 239000010703 silicon Substances 0.000 description 1
- 229910052710 silicon Inorganic materials 0.000 description 1
- 210000002027 skeletal muscle Anatomy 0.000 description 1
- 210000003491 skin Anatomy 0.000 description 1
- 210000000813 small intestine Anatomy 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- AJPJDKMHJJGVTQ-UHFFFAOYSA-M sodium dihydrogen phosphate Chemical compound [Na+].OP(O)([O-])=O AJPJDKMHJJGVTQ-UHFFFAOYSA-M 0.000 description 1
- 238000002415 sodium dodecyl sulfate polyacrylamide gel electrophoresis Methods 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 239000007901 soft capsule Substances 0.000 description 1
- 239000007787 solid Substances 0.000 description 1
- 239000007909 solid dosage form Substances 0.000 description 1
- 238000000638 solvent extraction Methods 0.000 description 1
- 108090000586 somatostatin receptor 2 Proteins 0.000 description 1
- 238000001179 sorption measurement Methods 0.000 description 1
- 238000002336 sorption--desorption measurement Methods 0.000 description 1
- 239000004455 soybean meal Substances 0.000 description 1
- 230000009870 specific binding Effects 0.000 description 1
- 210000000278 spinal cord Anatomy 0.000 description 1
- 238000010186 staining Methods 0.000 description 1
- 230000037351 starvation Effects 0.000 description 1
- 125000004079 stearyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 238000003756 stirring Methods 0.000 description 1
- 210000002784 stomach Anatomy 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 229960005322 streptomycin Drugs 0.000 description 1
- 210000002536 stromal cell Anatomy 0.000 description 1
- 238000012916 structural analysis Methods 0.000 description 1
- 239000007929 subcutaneous injection Substances 0.000 description 1
- 238000010254 subcutaneous injection Methods 0.000 description 1
- 210000001913 submandibular gland Anatomy 0.000 description 1
- 210000003523 substantia nigra Anatomy 0.000 description 1
- 239000001384 succinic acid Substances 0.000 description 1
- 239000007940 sugar coated tablet Substances 0.000 description 1
- 150000003462 sulfoxides Chemical class 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- 239000013589 supplement Substances 0.000 description 1
- 239000002511 suppository base Substances 0.000 description 1
- 238000002198 surface plasmon resonance spectroscopy Methods 0.000 description 1
- 230000008961 swelling Effects 0.000 description 1
- 210000002437 synoviocyte Anatomy 0.000 description 1
- 238000001308 synthesis method Methods 0.000 description 1
- 239000000057 synthetic resin Substances 0.000 description 1
- 229920003002 synthetic resin Polymers 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 210000003478 temporal lobe Anatomy 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- UWHCKJMYHZGTIT-UHFFFAOYSA-N tetraethylene glycol Chemical compound OCCOCCOCCOCCO UWHCKJMYHZGTIT-UHFFFAOYSA-N 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 1
- 210000001103 thalamus Anatomy 0.000 description 1
- 238000010257 thawing Methods 0.000 description 1
- 229940126585 therapeutic drug Drugs 0.000 description 1
- HNKJADCVZUBCPG-UHFFFAOYSA-N thioanisole Chemical compound CSC1=CC=CC=C1 HNKJADCVZUBCPG-UHFFFAOYSA-N 0.000 description 1
- 125000003396 thiol group Chemical group [H]S* 0.000 description 1
- 108010061238 threonyl-glycine Proteins 0.000 description 1
- 210000001541 thymus gland Anatomy 0.000 description 1
- 210000001685 thyroid gland Anatomy 0.000 description 1
- 238000012090 tissue culture technique Methods 0.000 description 1
- 239000003053 toxin Substances 0.000 description 1
- 231100000765 toxin Toxicity 0.000 description 1
- 108700012359 toxins Proteins 0.000 description 1
- 235000010487 tragacanth Nutrition 0.000 description 1
- 239000000196 tragacanth Substances 0.000 description 1
- 229940116362 tragacanth Drugs 0.000 description 1
- 108091006106 transcriptional activators Proteins 0.000 description 1
- 230000002463 transducing effect Effects 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 230000009261 transgenic effect Effects 0.000 description 1
- 230000010474 transient expression Effects 0.000 description 1
- YNJBWRMUSHSURL-UHFFFAOYSA-N trichloroacetic acid Chemical compound OC(=O)C(Cl)(Cl)Cl YNJBWRMUSHSURL-UHFFFAOYSA-N 0.000 description 1
- 125000004044 trifluoroacetyl group Chemical group FC(C(=O)*)(F)F 0.000 description 1
- 239000001226 triphosphate Substances 0.000 description 1
- 235000011178 triphosphate Nutrition 0.000 description 1
- 125000002221 trityl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C([*])(C1=C(C(=C(C(=C1[H])[H])[H])[H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 239000012588 trypsin Substances 0.000 description 1
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 1
- 238000000108 ultra-filtration Methods 0.000 description 1
- 238000005199 ultracentrifugation Methods 0.000 description 1
- 238000002604 ultrasonography Methods 0.000 description 1
- 241000701447 unidentified baculovirus Species 0.000 description 1
- 210000004291 uterus Anatomy 0.000 description 1
- 229940088594 vitamin Drugs 0.000 description 1
- 239000011782 vitamin Substances 0.000 description 1
- 235000013343 vitamin Nutrition 0.000 description 1
- 229930003231 vitamin Natural products 0.000 description 1
- 239000003643 water by type Substances 0.000 description 1
- 210000005253 yeast cell Anatomy 0.000 description 1
- 239000012138 yeast extract Substances 0.000 description 1
Images






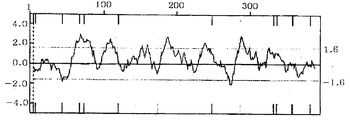









Landscapes
- Investigating Or Analysing Biological Materials (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Medicines Containing Material From Animals Or Micro-Organisms (AREA)
- Peptides Or Proteins (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Description
【発明の属する技術分野】
本発明は、▲1▼配列番号:20または配列番号:21で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有することを特徴とするペプチドまたはその塩、および▲2▼オーファン受容体タンパク質である配列番号:1または配列番号:で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質またはその塩と、配列番号:20または配列番号:21で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有することを特徴とするペプチドまたはその塩を用いることを特徴とする、消化器疾患の予防・治療剤として有用な化合物またはその塩などのスクリーニング方法などに関する。
【0002】
【従来の技術】
生体のホメオスタシスの維持、生殖、個体の発達、代謝、成長、神経系、循環器系、免疫系、消化器系、代謝系の調節、感覚受容などの重要な機能調節は、様々なホルモンや神経伝達物質のような内在性因子あるいは光や匂いなどの感覚刺激をこれらに対して生体が備えている細胞膜に存在する特異的な受容体を介して細胞が受容し、それに応じた反応をすることによって行われている。このような機能調節に与るホルモンや神経伝達物質の受容体の多くは guanine nucleotide-binding protein(以下、Gタンパク質と略称する場合がある)と共役しており、このGタンパク質の活性化によって細胞内にシグナルを伝達して様々な機能を発現させることを特徴とする。また、これらの受容体タンパク質は共通して7個の膜貫通領域を有する。これらのことからこうした受容体はGタンパク質共役型受容体あるいは7回膜貫通型受容体と総称される。このように生体機能の調節には様々なホルモンや神経伝達物質およびそれに対する受容体タンパク質が存在して相互作用し、重要な役割を果たしていることがわかっているが、未知の作用物質(ホルモンや神経伝達物質など)およびそれに対する受容体が存在するかどうかについてはいまだ不明なことが多い。
近年、ヒトゲノムDNAあるいは各種ヒト組織由来のcDNAのランダムな配列決定による配列情報の蓄積および遺伝子解析技術の急速な進歩によってヒトの遺伝子が加速度的に解明されてきている。それにともない、機能未知のタンパク質をコードすると予想される多くの遺伝子の存在が明らかになっている。Gタンパク質共役型受容体は、7個の膜貫通領域を有するのみでなくその核酸あるいはアミノ酸に多くの共通配列が存在するためそのようなタンパク質の中から明確にGタンパク質共役型受容体として区分することができる。一方でこうした構造の類似性を利用したポリメラーゼ・チェーン・リアクション(Polymerase Chain Reaction:以下、PCRと略称する)法によってもこうしたGタンパク質共役型受容体遺伝子が得られている(Nature Cell Biology、Vol. 2、703-708 (2000))。このようにしてこれまでに得られたGタンパク質共役型受容体のうちには既知の受容体との構造の相同性が高いサブタイプであって容易にそのリガンドを予測することが可能な場合もあるが、ほとんどの場合その内因性リガンドは予測不能であり、これらの受容体は対応するリガンドが見いだされていない。このことからこれらの受容体はオーファン受容体と呼ばれている。このようなオーファン受容体の未同定の内因性リガンドは、リガンドが知られていなかったために十分な解析がなされていなかった生物現象に関与している可能性がある。そして、このようなリガンドが重要な生理作用や病態と関連している場合には、その受容体作動薬あるいは拮抗薬の開発が革新的な医薬品の創製に結びつくことが期待される(Stadel, J. et al.、TiPS、18巻、430-437頁、1997年、Marchese, A. et al.、TiPS、20巻、370-375頁、1999年、Civelli, O. et al.、Brain Res.、848巻、63-65頁、1999年)。しかし、これまで実際にオーファンGタンパク質共役型受容体のリガンドを同定した例はそれほど多くない。
最近、幾つかのグループによってこうしたオーファン受容体のリガンド探索の試みがなされ、新たな生理活性ペプチドであるリガンドの単離・構造決定が報告されている。ReinsheidらおよびMeunierらは独立に、動物細胞にオーファンGタンパク質共役型受容体LC132あるいはORL1をコードするcDNAを導入して受容体を発現させ、その応答を指標としてorphanin FQあるいはnociceptinと名付けられた新規ペプチドをブタ脳あるいはラット脳の抽出物より単離し、配列を決定した(Reinsheid, R. K. et al.、Science、270巻、792-794頁、1995年、Meunier, J.-C. et al.、Nature、377巻、532-535頁、1995年)。このペプチドは痛覚に関与していることが報告されたが、さらに、受容体のノックアウトマウスの研究により記憶に関与していることが明らかにされた(Manabe, T. et al.、Nature、394巻、577-581頁、1998年)。
その後これまでに上記と同様な方法によりPrRP(prolactin releasing peptide)、orexin、apelin、ghrelinおよびGALP(galanin-like peptide)などの新規ペプチドがオーファンGタンパク質共役型受容体のリガンドとして単離された(Hinuma, S. et al.、Nature、393巻、272-276頁、1998年、Sakurai, T. et al.、Cell、92巻、573-585頁、1998年、Tatemoto, K. et al.、Bichem. Biophys. Res. Commun.、251巻、471-476頁、1998年、Kojima, M. et al.、Nature、402巻、656-660頁、1999年、Ohtaki, T. et al.、J. Biol. Chem.、274巻、37041-37045頁、1999年)。
一方、これまで明らかでなかった生理活性ペプチドの受容体が同様な方法によって解明される場合もある。腸管収縮に関与するmotilinの受容体がGPR38であることが明らかにされた(Feighner, S. D. et al.、Science、284巻、2184-2188頁、1999年)ほか、SLC−1がメラニン凝集ホルモン(MCH)の受容体として同定され(Chambers, J. et al.、Nature、400巻、261-265頁、1999年、Saito, Y. et al.、Nature、400巻、265-269頁、1999年、Shimomura, Y. et al.、Biochem. Biophys. Res. Commun.、261巻、622-626頁、1999年、Lembo, P. M. C. et al.、Nature Cell Biol.、1巻、267-271頁、1999年、Bachner, D. et al.、FEBS Lett.、457巻、522-524頁、1999年)、またGPR14(SENR)が urotensin II の受容体であることが報告された(Ames, R. S. et al.、Nature、401巻、282-286頁、1999年、Mori, M. et al.、Biochem. Biophys. Res. Commun.、265巻、123-129頁、1999年、Nothacker, H.-P. et al.、Nature Cell Biol.、1巻、383-385頁、1999年、Liu, Q. et al.、Biochem. Biophys. Res. Commun.、266巻、174-178頁、1999年)。MCHはそのノックアウトマウスが羸痩の phenotype を示すことから肥満に関与することが示されていたが(Shimada, M. et al.、Nature、396巻、670-674頁、1998年)、その受容体が明らかにされたことにより抗肥満薬としての可能性を有する受容体拮抗薬の探索が可能となった。また、urotensin IIはサルに静脈内投与することによって心虚血を惹起することから心循環系に強力な作用を示すことも報告されている(Ames, R. S. et al.、Nature、401巻、282-286頁、1999年)。
このように、オーファン受容体およびそのリガンドは新たな生理作用に関与する場合が多く、その解明は新たな医薬品開発に結びつくことが期待される。しかし、オーファン受容体のリガンド探索においては多くの困難さが伴い、これまでに数多くのオーファン受容体の存在が明らかにされながらそのリガンドが明らかにされた受容体はごく一部に過ぎない。
本発明者らは、オーファンGタンパク質共役型受容体である新規受容体ZAQ(本願明細書の配列番号:1で表されるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質:以下、本明細書において、単にZAQと称する場合がある)を見出したが、そのリガンドが何であるのかはこれまで不明であった。
【0003】
【発明が解決しようとする課題】
オーファン受容体タンパク質であるZAQに対するリガンドの探索と、ZAQおよびそのリガンドを用いることを特徴とする化合物などのスクリーニング方法の確立が課題とされていた。
【0004】
【課題を解決するための手段】
本発明者らは、牛乳抽出液中にZAQ特異的なリガンド活性を有する物質が存在することを見出し、当該物質を分離し、構造決定をおこなった。さらに、本活性成分のヒト型ペプチドをコードする遺伝子を見出し、これを動物細胞に発現させたところ、培養上清中にZAQ発現細胞を活性化するペプチド性物質が分泌されていることを確認した。
本発明者らは、かかる知見に基づいて、ZAQおよびZAQリガンドペプチドを用いたスクリーニング系を用いて、ZAQの介在する疾患の治療薬(ZAQ拮抗薬あるいは作動薬など、具体的には消化器疾患の予防・治療薬など)のスクリーニングができることを見出した。
【0005】
すなわち、本発明は、
(1)配列番号:20または配列番号:21で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するペプチドまたはその塩、
(2)配列番号:21で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するペプチドである前記(1)記載のペプチドまたはその塩、
(3)配列番号:20で表わされるアミノ酸配列を含有する前記(1)記載のペプチドまたはその塩、
(4)配列番号:21で表わされるアミノ酸配列を含有する前記(1)記載のペプチドまたはその塩、
(5)配列番号:22または配列番号:23で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有することを特徴とする前記(1)記載のペプチドまたはその塩、
(6)前記(1)記載のペプチドをコードするポリヌクレオチドを含有するポリヌクレオチド、
(7)DNAである前記(6)記載のポリヌクレオチド、
(8)配列番号:26または配列番号:27で表される塩基配列を含有する前記(7)記載のDNA、
(9)配列番号:28または配列番号:29で表される塩基配列を含有する前記(7)記載のDNA、
(10)前記(6)記載のポリヌクレオチドを含有する組換えベクター、
(11)前記(10)記載の組換えベクターで形質転換された形質転換体、
(12)前記(11)記載の形質転換体を培養し、前記(1)記載のペプチドを生成・蓄積せしめることを特徴とする前記(1)記載のペプチドまたはその塩の製造法、
(13)前記(1)記載のペプチドまたはその塩に対する抗体、
(14)前記(1)記載のペプチドまたはその塩および配列番号:1で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質もしくはその部分ペプチドまたはその塩を用いることを特徴とする、前記(1)記載のペプチドまたはその塩と配列番号:1で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質またはその塩との結合性を変化させる化合物またはその塩のスクリーニング方法、
(15)前記(1)記載のペプチドまたはその塩および配列番号:36で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質もしくはその部分ペプチドまたはその塩を用いることを特徴とする、前記(1)記載のペプチドまたはその塩と配列番号:36で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質またはその塩との結合性を変化させる化合物またはその塩のスクリーニング方法、
(16)前記(1)記載のペプチドまたはその塩および配列番号:1で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質もしくはその部分ペプチドまたはその塩を含有することを特徴とする、前記(1)記載のペプチドまたはその塩と配列番号:1で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質またはその塩との結合性を変化させる化合物またはその塩のスクリーニング用キット、
(17)前記(1)記載のペプチドまたはその塩および配列番号:36で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質もしくはその部分ペプチドまたはその塩を含有することを特徴とする、前記(1)記載のペプチドまたはその塩と配列番号:36で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質またはその塩との結合性を変化させる化合物またはその塩のスクリーニング用キット、
(18)前記(14)記載のスクリーニング方法または前記(16)記載のスクリーニング用キットを用いて得られうる、前記(1)記載のペプチドまたはその塩と配列番号:1で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質またはその塩との結合性を変化させる化合物またはその塩、
(19)前記(15)記載のスクリーニング方法または前記(17)記載のスクリーニング用キットを用いて得られうる、前記(1)記載のペプチドまたはその塩と配列番号:36で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質またはその塩との結合性を変化させる化合物またはその塩、
(20)前記(18)または(19)記載の化合物またはその塩を含有してなる医薬、
(21)消化器疾患の予防・治療剤である前記(20)記載の医薬、
(22)前記(1)記載のペプチドまたはその塩を含有してなる医薬、
(23)消化器疾患の予防・治療剤である前記(22)記載の医薬、
(24)哺乳動物に対して、前記(14)記載のスクリーニング方法または前記(16)記載のスクリーニング用キットを用いて得られうる、前記(1)記載のペプチドまたはその塩と配列番号:1で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質またはその塩との結合性を変化させる化合物またはその塩の有効量を投与することを特徴とする消化器疾患の予防・治療方法、
(25)哺乳動物に対して、前記(15)記載のスクリーニング方法または前記(17)記載のスクリーニング用キットを用いて得られうる、前記(1)記載のペプチドまたはその塩と配列番号:36で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質またはその塩との結合性を変化させる化合物またはその塩の有効量を投与することを特徴とする消化器疾患の予防・治療方法、
(26)消化器疾患の予防・治療剤を製造するための前記(14)記載のスクリーニング方法または前記(16)記載のスクリーニング用キットを用いて得られうる、前記(1)記載のペプチドまたはその塩と配列番号:1で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質またはその塩との結合性を変化させる化合物またはその塩の使用、
(27)消化器疾患の予防・治療剤を製造するための前記(15)記載のスクリーニング方法または前記(17)記載のスクリーニング用キットを用いて得られうる、前記(1)記載のペプチドまたはその塩と配列番号:36で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質またはその塩との結合性を変化させる化合物またはその塩の使用などを提供するものである。
【0006】
さらには、本発明は、
(28)配列番号:20または配列番号:21で表されるアミノ酸配列と実質的に同一のアミノ酸配列が、配列番号:20または配列番号:21で表されるアミノ酸配列と約60%以上(好ましくは約70%以上、さらに好ましくは約80%以上、より好ましくは約85%以上、特に好ましくは約90%以上、最も好ましくは約95%以上)の相同性を有するアミノ酸配列である前記(1)記載のペプチドまたはその塩、
(29)配列番号:20または配列番号:21で表されるアミノ酸配列と実質的に同一のアミノ酸配列が、▲1▼配列番号:20または配列番号:21で表されるアミノ酸配列中の1または2個以上(好ましくは、1〜30個程度、より好ましくは1〜20個程度)のアミノ酸が欠失したアミノ酸配列、▲2▼配列番号:20または配列番号:21で表されるアミノ酸配列に1または2個以上(好ましくは、1〜40個程度、より好ましくは1〜30個程度、なかでも好ましくは1〜20個程度)のアミノ酸が付加したアミノ酸配列、▲3▼配列番号:20または配列番号:21で表されるアミノ酸配列中の1または2個以上(好ましくは、1〜30個程度、より好ましくは1〜20個程度)のアミノ酸が他のアミノ酸で置換されたアミノ酸配列、または▲4▼それらを組み合わせたアミノ酸配列である前記(1)記載のペプチドまたはその塩、
【0007】
(30)(i)前記(1)記載のペプチドまたはその塩と配列番号:1で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質もしくはその部分ペプチドまたはその塩とを接触させた場合と、(ii)前記(1)記載のペプチドまたはその塩および試験化合物と配列番号:1で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質もしくはその部分ペプチドまたはその塩とを接触させた場合との比較を行なうことを特徴とする前記(14)記載のスクリーニング方法、
(31)(i)標識した前記(1)記載のペプチドまたはその塩を配列番号:1で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質もしくはその部分ペプチドまたはその塩に接触させた場合と、(ii)標識した前記(1)記載のペプチドまたはその塩および試験化合物を配列番号:1で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質もしくはその部分ペプチドまたはその塩に接触させた場合における、標識した前記(1)記載のペプチドまたはその塩の配列番号:1で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質もしくはその部分ペプチドまたはその塩に対する結合量を測定し、比較することを特徴とする前記(1)記載のペプチドまたはその塩と配列番号:1で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質またはその塩との結合性を変化させる化合物またはその塩のスクリーニング方法、
(32)(i)標識した前記(1)記載のペプチドまたはその塩を配列番号:1で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質を含有する細胞に接触させた場合と、(ii)標識した前記(1)記載のペプチドまたはその塩および試験化合物を配列番号:1で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質を含有する細胞に接触させた場合における、標識した前記(1)記載のペプチドまたはその塩の当該細胞に対する結合量を測定し、比較することを特徴とする前記(1)記載のペプチドまたはその塩と配列番号:1で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質またはその塩との結合性を変化させる化合物またはその塩のスクリーニング方法、
(33)(i)標識した前記(1)記載のペプチドまたはその塩を配列番号:1で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質を含有する細胞の膜画分に接触させた場合と、(ii)標識した前記(1)記載のペプチドまたはその塩および試験化合物を配列番号:1で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質を含有する細胞の膜画分に接触させた場合における、標識した前記(1)記載のペプチドまたはその塩の当該細胞の膜画分に対する結合量を測定し、比較することを特徴とする前記(1)記載のペプチドまたはその塩と配列番号:1で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質またはその塩との結合性を変化させる化合物またはその塩のスクリーニング方法、
(34)(i)標識した前記(1)記載のペプチドまたはその塩を、配列番号:1で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質をコードするDNAを含有するDNAを含有する組換えベクターで形質転換された形質転換体を培養することによって当該形質転換体の細胞膜に発現したタンパク質に接触させた場合と、(ii)標識した前記(1)記載のペプチドまたはその塩および試験化合物を、配列番号:1で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質をコードするDNAを含有するDNAを含有する組換えベクターで形質転換された形質転換体を培養することによって当該形質転換体の細胞膜に発現したタンパク質に接触させた場合における、標識した前記(1)記載のペプチドまたはその塩の当該タンパク質に対する結合量を測定し、比較することを特徴とする、前記(1)記載のペプチドまたはその塩と配列番号:1で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質またはその塩との結合性を変化させる化合物またはその塩のスクリーニング方法、
(35)(i)前記(1)記載のペプチドまたはその塩と配列番号:36で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質もしくはその部分ペプチドまたはその塩とを接触させた場合と、(ii)前記(1)記載のペプチドまたはその塩および試験化合物と配列番号:36で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質もしくはその部分ペプチドまたはその塩とを接触させた場合との比較を行なうことを特徴とする前記(15)記載のスクリーニング方法、
(36)(i)標識した前記(1)記載のペプチドまたはその塩を配列番号:36で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質もしくはその部分ペプチドまたはその塩に接触させた場合と、(ii)標識した前記(1)記載のペプチドまたはその塩および試験化合物を配列番号:36で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質もしくはその部分ペプチドまたはその塩に接触させた場合における、標識した前記(1)記載のペプチドまたはその塩の配列番号:36で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質もしくはその部分ペプチドまたはその塩に対する結合量を測定し、比較することを特徴とする前記(1)記載のペプチドまたはその塩と配列番号:36で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質またはその塩との結合性を変化させる化合物またはその塩のスクリーニング方法、
(37)(i)標識した前記(1)記載のペプチドまたはその塩を配列番号:36で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質を含有する細胞に接触させた場合と、(ii)標識した前記(1)記載のペプチドまたはその塩および試験化合物を配列番号:36で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質を含有する細胞に接触させた場合における、標識した前記(1)記載のペプチドまたはその塩の当該細胞に対する結合量を測定し、比較することを特徴とする前記(1)記載のペプチドまたはその塩と配列番号:36で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質またはその塩との結合性を変化させる化合物またはその塩のスクリーニング方法、
(38)(i)標識した前記(1)記載のペプチドまたはその塩を配列番号:36で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質を含有する細胞の膜画分に接触させた場合と、(ii)標識した前記(1)記載のペプチドまたはその塩および試験化合物を配列番号:36で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質を含有する細胞の膜画分に接触させた場合における、標識した前記(1)記載のペプチドまたはその塩の当該細胞の膜画分に対する結合量を測定し、比較することを特徴とする前記(1)記載のペプチドまたはその塩と配列番号:36で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質またはその塩との結合性を変化させる化合物またはその塩のスクリーニング方法、
(39)(i)標識した前記(1)記載のペプチドまたはその塩を、配列番号:36で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質をコードするDNAを含有するDNAを含有する組換えベクターで形質転換された形質転換体を培養することによって当該形質転換体の細胞膜に発現したタンパク質に接触させた場合と、(ii)標識した前記(1)記載のペプチドまたはその塩および試験化合物を、配列番号:36で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質をコードするDNAを含有するDNAを含有する組換えベクターで形質転換された形質転換体を培養することによって当該形質転換体の細胞膜に発現したタンパク質に接触させた場合における、標識した前記(1)記載のペプチドまたはその塩の当該タンパク質に対する結合量を測定し、比較することを特徴とする、前記(1)記載のペプチドまたはその塩と配列番号:36で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質またはその塩との結合性を変化させる化合物またはその塩のスクリーニング方法、
【0008】
(40)前記(30)〜(34)記載のスクリーニング方法で得られうる、(i)前記(1)記載のペプチドまたはその塩と配列番号:1で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質またはその塩との結合性を変化させる化合物またはその塩、
(41)前記(35)〜(39)記載のスクリーニング方法で得られうる、(i)前記(1)記載のペプチドまたはその塩と配列番号:36で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質またはその塩との結合性を変化させる化合物またはその塩、
(42)前記(40)または(41)記載の化合物またはその塩を含有する医薬、
【0009】
(43)配列番号:1で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質を含有する細胞を含有することを特徴とする前記(16)記載のスクリーニング用キット、
(44)配列番号:1で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質を含有する細胞の膜画分を含有することを特徴とする前記(16)記載のスクリーニング用キット、
(45)配列番号:1で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質をコードするDNAを含有するDNAを含有する組換えベクターで形質転換された形質転換体を培養することによって当該形質転換体の細胞膜に発現したタンパク質を含有することを特徴とする前記(16)記載のスクリーニング用キット、
(46)配列番号:36で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質を含有する細胞を含有することを特徴とする前記(17)記載のスクリーニング用キット、
(47)配列番号:36で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質を含有する細胞の膜画分を含有することを特徴とする前記(17)記載のスクリーニング用キット、
(48)配列番号:36で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質をコードするDNAを含有するDNAを含有する組換えベクターで形質転換された形質転換体を培養することによって当該形質転換体の細胞膜に発現したタンパク質を含有することを特徴とする前記(17)記載のスクリーニング用キット、
(49)前記(43)〜(45)記載のスクリーニング用キットを用いて得られうる、前記(1)記載のペプチドまたはその塩と配列番号:1で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質またはその塩との結合性を変化させる化合物またはその塩、
(50)前記(46)〜(48)記載のスクリーニング用キットを用いて得られうる、前記(1)記載のペプチドまたはその塩と配列番号:36で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質またはその塩との結合性を変化させる化合物またはその塩、
(51)前記(49)または(50)記載の化合物またはその塩を含有する医薬、
(52)消化器疾患の予防・治療剤である前記(42)または(51)記載の医薬、
【0010】
(53)前記(13)記載の抗体と、前記(1)記載のペプチドまたはその塩とを接触させることを特徴とする、前記(1)記載のペプチドまたはその塩の定量法、
(54)前記(13)記載の抗体と、被検液および標識化された前記(1)記載のペプチドまたはその塩とを競合的に反応させ、当該抗体に結合した標識化された前記(1)記載のペプチドまたはその塩の割合を測定することを特徴とする被検液中の前記(1)記載のペプチドまたはその塩の定量法、
(54)被検液と担体上に不溶化した前記(11)記載の抗体および標識化された前記(11)項記載の抗体とを同時あるいは連続的に反応させたのち、不溶化担体上の標識剤の活性を測定することを特徴とする被検液中の前記(1)記載のペプチドまたはその塩の定量法、
(55)前記(7)記載のDNAとハイストリンジェントな条件でハイブリダイズするポリヌクレオチド、
(56)前記(7)記載のDNAの塩基配列と相補的な塩基配列またはその一部を含有するポリヌクレオチド、
(57)配列番号:28または配列番号:29で表される塩基配列と相補的な塩基配列またはその一部を含有する前記(56)記載のポリヌクレオチド、
(58)前記(1)記載のペプチドまたはその塩および配列番号:40、配列番号:47、配列番号:48もしくは配列番号:49で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質もしくはその部分ペプチドまたはその塩を用いることを特徴とする、前記(1)記載のペプチドまたはその塩と配列番号:40、配列番号:47、配列番号:48もしくは配列番号:49で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質またはその塩との結合性を変化させる化合物またはその塩のスクリーニング方法、
(59)前記(1)記載のペプチドまたはその塩および配列番号:40、配列番号:47、配列番号:48もしくは配列番号:49で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質もしくはその部分ペプチドまたはその塩を含有することを特徴とする、前記(1)記載のペプチドまたはその塩と配列番号:40、配列番号:47、配列番号:48もしくは配列番号:49で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質またはその塩との結合性を変化させる化合物またはその塩のスクリーニング用キット、
(60)前記(58)記載のスクリーニング方法または前記(59)記載のスクリーニング用キットを用いて得られうる、前記(1)記載のペプチドまたはその塩と配列番号:40、配列番号:47、配列番号:48もしくは配列番号:49で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質またはその塩との結合性を変化させる化合物またはその塩、
(61)前記(60)記載の化合物またはその塩を含有してなる医薬、
(62)消化器疾患の予防・治療剤である前記(61)記載の医薬、
(63)配列番号:34で表されるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するペプチドまたはその塩および配列番号:1、配列番号:36、配列番号:40、配列番号:47、配列番号:48もしくは配列番号:49で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質もしくはその部分ペプチドまたはその塩を用いることを特徴とする、配列番号:34で表されるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するペプチドまたはその塩と配列番号:1、配列番号:36、配列番号:40、配列番号:47、配列番号:48もしくは配列番号:49で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質またはその塩との結合性を変化させる化合物またはその塩のスクリーニング方法、
(64)配列番号:34で表されるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するペプチドまたはその塩および配列番号:1、配列番号:36、配列番号:40、配列番号:47、配列番号:48もしくは配列番号:49で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質もしくはその部分ペプチドまたはその塩を含有することを特徴とする、配列番号:34で表されるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するペプチドまたはその塩と配列番号:1、配列番号:36、配列番号:40、配列番号:47、配列番号:48もしくは配列番号:49で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質またはその塩との結合性を変化させる化合物またはその塩のスクリーニング用キット、
(65)前記(63)記載のスクリーニング方法または前記(64)記載のスクリーニング用キットを用いて得られうる、配列番号:34で表されるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するペプチドまたはその塩と配列番号:1、配列番号:36、配列番号:40、配列番号:47、配列番号:48もしくは配列番号:49で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質またはその塩との結合性を変化させる化合物またはその塩、
(66)前記(65)記載の化合物またはその塩を含有してなる医薬、
(67)消化器疾患の予防・治療剤である前記(66)記載の医薬などを提供する。
【0011】
【発明の実施の形態】
本発明のペプチドまたはその塩は、配列番号:1で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質またはその塩(以下、本発明のタンパク質と略記することがある)と結合する能力を有するペプチドまたはその塩であり、本発明のタンパク質と結合し、活性化する能力を有するペプチドまたはその塩である。
さらに、本発明のペプチドまたはその塩は、配列番号:36、配列番号:40、配列番号:47、配列番号:48または配列番号:49で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質またはその塩と結合する能力を有するペプチドまたはその塩であり、当該タンパク質と結合し、活性化する能力も有するペプチドまたはその塩である。
なかでも好ましくは、本発明のペプチドまたはその塩は、配列番号:20または配列番号:21で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有し、本発明のタンパク質と結合し、活性化する能力を有することを特徴とするペプチドまたはその塩である。
本発明のペプチドまたはその塩の本発明のタンパク質と結合する能力および本発明のタンパク質を活性化する能力は後述の方法により測定することができる。
以下、本発明のペプチドまたはその塩を本発明のペプチドと略記する場合がある。
【0012】
本発明のタンパク質(Gタンパク質共役型受容体タンパク質)は、配列番号:1で表わされるアミノ酸配列(図1〜図3または図4〜図6中のアミノ酸配列)と同一もしくは実質的に同一のアミノ酸配列を含有する受容体タンパク質である(以下、本発明のタンパク質またはその塩を本発明のタンパク質と略記する場合がある)。
【0013】
本発明のペプチドおよび本発明のタンパク質は、例えば、ヒトや哺乳動物(例えば、モルモット、ラット、マウス、ウサギ、ブタ、ヒツジ、ウシ、サルなど)のあらゆる細胞(例えば、脾細胞、神経細胞、グリア細胞、膵臓β細胞、骨髄細胞、メサンギウム細胞、ランゲルハンス細胞、表皮細胞、上皮細胞、内皮細胞、繊維芽細胞、繊維細胞、筋細胞、脂肪細胞、免疫細胞(例、マクロファージ、T細胞、B細胞、ナチュラルキラー細胞、肥満細胞、好中球、好塩基球、好酸球、単球)、巨核球、滑膜細胞、軟骨細胞、骨細胞、骨芽細胞、破骨細胞、乳腺細胞、肝細胞もしくは間質細胞、またはこれら細胞の前駆細胞、幹細胞もしくはガン細胞など)や血球系の細胞(例えば、MEL,M1,CTLL−2,HT−2,WEHI−3,HL−60,JOSK−1,K562,ML−1,MOLT−3,MOLT−4,MOLT−10,CCRF−CEM,TALL−1,Jurkat,CCRT−HSB−2,KE−37,SKW−3,HUT−78,HUT−102,H9,U937,THP−1,HEL,JK−1,CMK,KO−812,MEG−01など)、またはそれらの細胞が存在するあらゆる組織、例えば、脳、脳の各部位(例、嗅球、扁頭核、大脳基底球、海馬、視床、視床下部、視床下核、大脳皮質、延髄、小脳、後頭葉、前頭葉、側頭葉、被殻、尾状核、脳染、黒質)、脊髄、下垂体、胃、膵臓、腎臓、肝臓、生殖腺、甲状腺、胆のう、骨髄、副腎、皮膚、筋肉、肺、消化管(例、大腸、小腸)、血管、心臓、胸腺、脾臓、顎下腺、末梢血、末梢血球、前立腺、睾丸、精巣、卵巣、胎盤、子宮、骨、関節、骨格筋など(特に、脳や脳の各部位)に由来するペプチド・タンパク質であってもよく、また合成ペプチド・合成タンパク質であってもよい。
本発明のペプチドまたは本発明のタンパク質がシグナル配列を有している場合は当該ペプチドまたはタンパク質を効率良く細胞外に分泌させることができる。
【0014】
配列番号:20または配列番号:21で表わされるアミノ酸配列と実質的に同一のアミノ酸配列としては、例えば、配列番号:20または配列番号:21で表されるアミノ酸配列と約60%以上(好ましくは約70%以上、さらに好ましくは約80%以上、より好ましくは約85%以上、特に好ましくは約90%以上、最も好ましくは約95%以上)の相同性を有するアミノ酸配列などが挙げられる。
配列番号:20または配列番号:21で表わされるアミノ酸配列と実質的に同一のアミノ酸配列を含有するペプチドとしては、例えば、配列番号:20または配列番号:21で表わされるアミノ酸配列と実質的に同一のアミノ酸配列を含有し、配列番号:20または配列番号:21で表わされるアミノ酸配列を含有するペプチドと実質的に同質の性質を有するペプチドなどが好ましい。
以下、配列番号:20または配列番号:21で表わされるアミノ酸配列を含有するペプチドをヒト型ZAQリガンド成熟体ペプチドと記載することがある。
【0015】
配列番号:20または配列番号:21で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するペプチドとしては、例えば、配列番号:20または配列番号:21で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有し、配列番号:20または配列番号:21で表わされるアミノ酸配列を含有するペプチドと実質的に同質の活性を有するペプチドなどが好ましく、具体的には配列番号:20、配列番号:21、配列番号:22または配列番号:23で表わされるアミノ酸配列を含有するペプチド等が挙げられる。
以下、配列番号:22または配列番号:23で表わされるアミノ酸配列を含有するペプチドをヒト型ZAQリガンド前駆体ペプチドと記載することがある。
実質的に同質の活性としては、例えば、本発明のタンパク質に対する結合活性、本発明のタンパク質を介するシグナル情報伝達作用、消化管(例、腸管)収縮活性などが挙げられる。実質的に同質とは、それらの活性が性質的に同質であることを示す。したがって、本発明のタンパク質に対する結合活性、本発明のタンパク質を介するシグナル情報伝達作用、消化管収縮活性などの活性が同等(例、約0.5〜2倍)であることが好ましいが、これらの活性の程度やペプチドの分子量などの量的要素は異なっていてもよい。
これらの活性の測定は、公知の方法に準じて行なうことができるが、例えば、後述するスクリーニング方法などに従っても測定することができる。
【0016】
また、本発明のペプチドとしては、▲1▼配列番号:20または配列番号:21で表されるアミノ酸配列中の1または2個以上(好ましくは、1〜30個程度、より好ましくは1〜20個程度)のアミノ酸が欠失したアミノ酸配列、▲2▼配列番号:20または配列番号:21で表されるアミノ酸配列に1または2個以上(好ましくは、1〜40個程度、より好ましくは1〜30個程度、なかでも好ましくは1〜20個程度)のアミノ酸が付加したアミノ酸配列、▲3▼配列番号:20または配列番号:21で表されるアミノ酸配列中の1または2個以上(好ましくは、1〜30個程度、より好ましくは1〜20個程度)のアミノ酸が他のアミノ酸で置換されたアミノ酸配列、または▲4▼それらを組み合わせたアミノ酸配列を含有するペプチドなども用いられる。
本発明のペプチドとして好ましくはヒトまたはヒト以外の哺乳動物、さらに好ましくはヒト由来のペプチドである。
【0017】
配列番号:1で表わされるアミノ酸配列と実質的に同一のアミノ酸配列としては、例えば、配列番号:1で表わされるアミノ酸配列と約90%以上、好ましくは約95%以上、より好ましくは約98%以上の相同性を有するアミノ酸配列などが挙げられる。
配列番号:1で表わされるアミノ酸配列と実質的に同一のアミノ酸配列を含有するタンパク質としては、例えば、配列番号:1で表わされるアミノ酸配列と実質的に同一のアミノ酸配列を含有し、配列番号:1で表わされるアミノ酸配列を含有するタンパク質と実質的に同質の性質を有するタンパク質などが好ましい。
本発明の配列番号:1で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質としては、例えば、配列番号:1で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有し、配列番号:1で表わされるアミノ酸配列を含有するタンパク質と実質的に同質の活性を有するタンパク質などが好ましい。
実質的に同質の活性としては、例えば、リガンド結合活性、シグナル情報伝達作用などが挙げられる。実質的に同質とは、それらの活性が性質的に同質であることを示す。したがって、リガンド結合活性やシグナル情報伝達作用などの活性が同等(例、約0.5〜2倍)であることが好ましいが、これらの活性の程度やタンパク質の分子量などの量的要素は異なっていてもよい。
リガンド結合活性やシグナル情報伝達作用などの活性の測定は、公知の方法に準じて行なうことができるが、例えば、後述の決定方法やスクリーニング方法に従っても測定することができる。
以下、配列番号:1で表わされるアミノ酸配列を含有するタンパク質をZAQと記載することがある。
また、本発明のタンパク質としては、▲1▼配列番号:1で表わされるアミノ酸配列中の1または2個以上(好ましくは、1〜30個程度、より好ましくは1〜10個程度、さらに好ましくは数個(1または2個))のアミノ酸が欠失したアミノ酸配列、▲2▼配列番号:1で表わされるアミノ酸配列に1または2個以上(好ましくは、1〜30個程度、より好ましくは1〜10個程度、さらに好ましくは数個(1または2個))のアミノ酸が付加したアミノ酸配列、▲3▼配列番号:1で表わされるアミノ酸配列中の1または2個以上(好ましくは、1〜30個程度、より好ましくは1〜10個程度、さらに好ましくは数個(1または2個))のアミノ酸が他のアミノ酸で置換されたアミノ酸配列、または▲4▼それらを組み合わせたアミノ酸配列を含有するタンパク質なども用いられる。
【0018】
配列番号:36で表わされるアミノ酸配列と実質的に同一のアミノ酸配列としては、例えば、配列番号:36で表わされるアミノ酸配列と約90%以上、好ましくは約95%以上、より好ましくは約98%以上の相同性を有するアミノ酸配列などが挙げられる。
配列番号:36で表わされるアミノ酸配列と実質的に同一のアミノ酸配列を含有するタンパク質としては、例えば、配列番号:36で表わされるアミノ酸配列と実質的に同一のアミノ酸配列を含有し、配列番号:36で表わされるアミノ酸配列を含有するタンパク質と実質的に同質の性質を有するタンパク質などが好ましい。
配列番号:36で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質としては、例えば、配列番号:36で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有し、配列番号:36で表わされるアミノ酸配列を含有するタンパク質と実質的に同質の活性を有するタンパク質などが好ましく、具体的にはWO 98/46620に記載のタンパク質などが挙げられる。
配列番号:40で表わされるアミノ酸配列と実質的に同一のアミノ酸配列としては、例えば、配列番号:40で表わされるアミノ酸配列と約97%以上、好ましくは約98%以上、より好ましくは約99%以上、最も好ましくは約99.5%以上の相同性を有するアミノ酸配列などが挙げられる。
配列番号:40で表わされるアミノ酸配列と実質的に同一のアミノ酸配列を含有するタンパク質としては、例えば、配列番号:40で表わされるアミノ酸配列と実質的に同一のアミノ酸配列を含有し、配列番号:40で表わされるアミノ酸配列を含有するタンパク質と実質的に同質の性質を有するタンパク質などが好ましい。
配列番号:40で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質としては、例えば、配列番号:40で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有し、配列番号:40で表わされるアミノ酸配列を含有するタンパク質と実質的に同質の活性を有するタンパク質などが好ましい。
配列番号:47で表わされるアミノ酸配列と実質的に同一のアミノ酸配列としては、例えば、配列番号:47で表わされるアミノ酸配列と約95%以上、好ましくは約96%以上、より好ましくは約97%以上、最も好ましくは約98%以上の相同性を有するアミノ酸配列などが挙げられる。
配列番号:47で表わされるアミノ酸配列と実質的に同一のアミノ酸配列を含有するタンパク質としては、例えば、配列番号:47で表わされるアミノ酸配列と実質的に同一のアミノ酸配列を含有し、配列番号:47で表わされるアミノ酸配列を含有するタンパク質と実質的に同質の性質を有するタンパク質などが好ましい。
配列番号:47で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質としては、例えば、配列番号:47で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有し、配列番号:47で表わされるアミノ酸配列を含有するタンパク質と実質的に同質の活性を有するタンパク質などが好ましい。
配列番号:48で表わされるアミノ酸配列と実質的に同一のアミノ酸配列としては、例えば、配列番号:48で表わされるアミノ酸配列と約95%以上、好ましくは約96%以上、より好ましくは約97%以上、最も好ましくは約98%以上の相同性を有するアミノ酸配列などが挙げられる。
配列番号:48で表わされるアミノ酸配列と実質的に同一のアミノ酸配列を含有するタンパク質としては、例えば、配列番号:48で表わされるアミノ酸配列と実質的に同一のアミノ酸配列を含有し、配列番号:48で表わされるアミノ酸配列を含有するタンパク質と実質的に同質の性質を有するタンパク質などが好ましい。
配列番号:48で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質としては、例えば、配列番号:48で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有し、配列番号:48で表わされるアミノ酸配列を含有するタンパク質と実質的に同質の活性を有するタンパク質などが好ましく、具体的には、Biochem. Biophys. Acta、1491巻、369-375頁、2000年に記載のタンパク質などが挙げられる。
配列番号:49で表わされるアミノ酸配列と実質的に同一のアミノ酸配列としては、例えば、配列番号:49で表わされるアミノ酸配列と約95%以上、好ましくは約96%以上、より好ましくは約97%以上、最も好ましくは約98%以上の相同性を有するアミノ酸配列などが挙げられる。
配列番号:49で表わされるアミノ酸配列と実質的に同一のアミノ酸配列を含有するタンパク質としては、例えば、配列番号:49で表わされるアミノ酸配列と実質的に同一のアミノ酸配列を含有し、配列番号:49で表わされるアミノ酸配列を含有するタンパク質と実質的に同質の性質を有するタンパク質などが好ましい。
配列番号:49で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質としては、例えば、配列番号:49で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有し、配列番号:49で表わされるアミノ酸配列を含有するタンパク質と実質的に同質の活性を有するタンパク質などが好ましく、具体的にはWO 98/46620に記載のタンパク質などが挙げられる。
実質的に同質の活性としては、例えば、リガンド結合活性、シグナル情報伝達作用などが挙げられる。実質的に同質とは、それらの活性が性質的に同質であることを示す。したがって、リガンド結合活性やシグナル情報伝達作用などの活性が同等(例、約0.5〜2倍)であることが好ましいが、これらの活性の程度やタンパク質の分子量などの量的要素は異なっていてもよい。
リガンド結合活性やシグナル情報伝達作用などの活性の測定は、公知の方法に準じて行なうことができる。
配列番号:34で表されるアミノ酸配列と実質的に同一のアミノ酸配列としては、配列番号:34で表わされるアミノ酸配列と約60%以上、好ましくは約70%以上、より好ましくは約80%以上、最も好ましくは約90%以上の相同性を有するアミノ酸配列などが挙げられる。
配列番号:34で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するペプチドとしては、例えば、配列番号:34で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有し、配列番号:34で表わされるアミノ酸配列を含有するペプチドと実質的に同質の活性(例、回腸収縮作用、遠位大腸収縮作用、近位大腸弛緩作用など)を有するペプチドなどが好ましく、具体的には後述のMIT1などが挙げられる。
【0019】
本明細書におけるペプチドおよびタンパク質は、ペプチド標記の慣例に従って左端がN末端(アミノ末端)、右端がC末端(カルボキシル末端)である。配列番号:1で表わされるアミノ酸配列を含有するタンパク質をはじめとする本発明のタンパク質は、C末端がカルボキシル基(−COOH)、カルボキシレート(−COO-)、アミド(−CONH2)またはエステル(−COOR)の何れであってもよい。
ここでエステルにおけるRとしては、例えば、メチル、エチル、n−プロピル、イソプロピルもしくはn−ブチルなどのC1-6アルキル基、例えば、シクロペンチル、シクロヘキシルなどのC3-8シクロアルキル基、例えば、フェニル、α−ナフチルなどのC6-12アリール基、例えば、ベンジル、フェネチルなどのフェニル−C1-2アルキル基もしくはα−ナフチルメチルなどのα−ナフチル−C1-2アルキル基などのC7-14アラルキル基のほか、経口用エステルとして汎用されるピバロイルオキシメチル基などが用いられる。
本発明のペプチド・タンパク質がC末端以外にカルボキシル基(またはカルボキシレート)を有している場合、カルボキシル基がアミド化またはエステル化されているものも本発明のペプチド・タンパク質に含まれる。この場合のエステルとしては、例えば上記したC末端のエステルなどが用いられる。
さらに、本発明のペプチド・タンパク質には、上記したペプチド・タンパク質において、N末端のメチオニン残基のアミノ基が保護基(例えば、ホルミル基、アセチル基などのC2-6アルカノイル基などのC1-6アシル基など)で保護されているもの、N端側が生体内で切断され生成したグルタミル基がピログルタミン酸化したもの、分子内のアミノ酸の側鎖上の置換基(例えば、−OH、−SH、アミノ基、イミダゾール基、インドール基、グアニジノ基など)が適当な保護基(例えば、ホルミル基、アセチル基などのC2-6アルカノイル基などのC1-6アシル基など)で保護されているもの、あるいは糖鎖が結合したいわゆる糖ペプチド・糖タンパク質などの複合ペプチド・複合タンパク質なども含まれる。
本発明のペプチドの具体例としては、例えば、配列番号:20で表わされるアミノ酸配列を含有するヒト由来(より好ましくはヒト脳由来)のペプチド、または配列番号:21で表わされるアミノ酸配列を含有するヒト由来(より好ましくはヒト脳由来)のペプチドなどがあげられる。さらに好ましくは、配列番号:21で表わされるアミノ酸配列を含有するヒト由来のペプチドなどがあげられる。
本発明のタンパク質の具体例としては、例えば、配列番号:1で表わされるアミノ酸配列を含有するヒト由来(より好ましくはヒト脳由来)のタンパク質などがあげられる。
【0020】
本発明のタンパク質の部分ペプチド(以下、本発明の部分ペプチドと略記する場合がある)としては、上記した本発明のタンパク質の部分ペプチドであれば何れのものであってもよいが、例えば、本発明のタンパク質分子のうち、細胞膜の外に露出している部位であって、実質的に同質のリガンド結合活性を有するものなどが用いられる。
具体的には、配列番号:1で表わされるアミノ酸配列を含有するタンパク質の部分ペプチドとしては、図7で示される疎水性プロット解析において細胞外領域(親水性(Hydrophilic)部位)であると分析された部分を含むペプチドである。また、疎水性(Hydrophobic)部位を一部に含むペプチドも同様に用いることができる。個々のドメインを個別に含むペプチドも用い得るが、複数のドメインを同時に含む部分のペプチドでも良い。
本発明の部分ペプチドのアミノ酸の数は、前記した本発明のタンパク質の構成アミノ酸配列のうち少なくとも20個以上、好ましくは50個以上、より好ましくは100個以上のアミノ酸配列を含有するペプチドなどが好ましい。
実質的に同一のアミノ酸配列とは、これらアミノ酸配列と約50%以上、好ましくは約70%以上、より好ましくは約80%以上、さらに好ましくは約90%以上、最も好ましくは約95%以上の相同性を有するアミノ酸配列を示す。
ここで、「実質的に同質のリガンド結合活性」とは、前記と同意義を示す。「実質的に同質のリガンド結合活性」の測定は公知の方法に準じて行なうことができる。
【0021】
また、本発明の部分ペプチドは、前記アミノ酸配列中の1または2個以上(好ましくは、1〜10個程度、さらに好ましくは数個(1または2個))のアミノ酸が欠失し、または、そのアミノ酸配列に1または2個以上(好ましくは、1〜20個程度、より好ましくは1〜10個程度、さらに好ましくは数個(1または2個))のアミノ酸が付加し、または、そのアミノ酸配列中の1または2個以上(好ましくは、1〜10個程度、より好ましくは1〜5個程度、さらに好ましくは数個(1または2個))のアミノ酸が他のアミノ酸で置換されていてもよい。
また、本発明の部分ペプチドはC末端がカルボキシル基(−COOH)、カルボキシレート(−COO-)、アミド(−CONH2)またはエステル(−COOR)の何れであってもよい。本発明の部分ペプチドがC末端以外にカルボキシル基(またはカルボキシレート)を有している場合、カルボキシル基がアミド化またはエステル化されているものも本発明の部分ペプチドに含まれる。この場合のエステルとしては、例えば前記したC末端のエステルなどが用いられる。
さらに、本発明の部分ペプチドには、前記した本発明のタンパク質と同様に、N末端のメチオニン残基のアミノ基が保護基で保護されているもの、N端側が生体内で切断され生成したGlnがピログルタミン酸化したもの、分子内のアミノ酸の側鎖上の置換基が適当な保護基で保護されているもの、あるいは糖鎖が結合したいわゆる糖ペプチドなどの複合ペプチドなども含まれる。
配列番号:36、配列番号:40、配列番号:47、配列番号:48または配列番号:49で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質の部分ペプチドとしては、前記した配列番号:36、配列番号:40、配列番号:47、配列番号:48または配列番号:49で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質の部分ペプチドであれば何れのものであってもよいが、例えば、タンパク質分子のうち、細胞膜の外に露出している部位であって、実質的に同質のリガンド結合活性を有するものなどが用いられる。当該部分ペプチドのアミノ酸の数は、前記タンパク質の構成アミノ酸配列のうち少なくとも20個以上、好ましくは50個以上、より好ましくは100個以上のアミノ酸配列を含有するペプチドなどが好ましい。実質的に同一のアミノ酸配列とは、これらアミノ酸配列と約50%以上、好ましくは約70%以上、より好ましくは約80%以上、さらに好ましくは約90%以上、最も好ましくは約95%以上の相同性を有するアミノ酸配列を示す。
【0022】
本発明のペプチドまたは本発明のタンパク質もしくはその部分ペプチド等の塩としては、とりわけ生理学的に許容される酸付加塩が好ましい。この様な塩としては、例えば無機酸(例えば、塩酸、リン酸、臭化水素酸、硫酸)との塩、あるいは有機酸(例えば、酢酸、ギ酸、プロピオン酸、フマル酸、マレイン酸、コハク酸、酒石酸、クエン酸、リンゴ酸、蓚酸、安息香酸、メタンスルホン酸、ベンゼンスルホン酸)との塩などが用いられる。
【0023】
本発明のペプチドもしくはタンパク質またはその塩、および配列番号:36、配列番号:40もしくは配列番号:47で表されるアミノ酸配列を含有するタンパク質またはその塩は、前述したヒトや哺乳動物の細胞または組織から公知のペプチド・タンパク質の精製方法によって製造することもできるし、後述する本発明のペプチド、本発明のタンパク質、配列番号:36、配列番号:40または配列番号:47で表されるアミノ酸配列を含有するタンパク質をコードするDNAを含有する形質転換体を培養することによっても製造することができる。また、後述のペプチド・タンパク質合成法またはこれに準じて製造することもできる。さらに、配列番号:48で表されるアミノ酸配列を含有するタンパク質またはその塩は、Biochem. Biophys. Acta、1491巻、369-375頁、2000年に記載の方法に準じて製造できる。配列番号:49で表されるアミノ酸配列を含有するタンパク質またはその塩は、WO 98/46620に記載の方法に準じて製造できる。ヒトや哺乳動物の組織または細胞から製造する場合、ヒトや哺乳動物の組織または細胞をホモジナイズした後、酸などで抽出を行ない、当該抽出液を逆相クロマトグラフィー、イオン交換クロマトグラフィーなどのクロマトグラフィーを組み合わせることにより精製単離することができる。
【0024】
本発明のペプチドまたは本発明のタンパク質もしくはその部分ペプチドまたはそれらのアミド体またはそれらの塩の合成には、通常市販のペプチド・タンパク質合成用樹脂を用いることができる。そのような樹脂としては、例えば、クロロメチル樹脂、ヒドロキシメチル樹脂、ベンズヒドリルアミン樹脂、アミノメチル樹脂、4−ベンジルオキシベンジルアルコール樹脂、4−メチルベンズヒドリルアミン樹脂、PAM樹脂、4−ヒドロキシメチルメチルフェニルアセトアミドメチル樹脂、ポリアクリルアミド樹脂、4−(2',4'−ジメトキシフェニル−ヒドロキシメチル)フェノキシ樹脂、4−(2',4'−ジメトキシフェニル−Fmocアミノエチル)フェノキシ樹脂などを挙げることができる。このような樹脂を用い、α−アミノ基と側鎖官能基を適当に保護したアミノ酸を、目的とするペプチド・タンパク質の配列通りに、公知の各種縮合方法に従い、樹脂上で縮合させる。反応の最後に樹脂からペプチド・タンパク質を切り出すと同時に各種保護基を除去し、さらに高希釈溶液中で分子内ジスルフィド結合形成反応を実施し、目的のペプチド・タンパク質またはそれらのアミド体を取得する。
前記した保護アミノ酸の縮合に関しては、ペプチド・タンパク質合成に使用できる各種活性化試薬を用いることができるが、特に、カルボジイミド類がよい。カルボジイミド類としては、DCC、N,N'−ジイソプロピルカルボジイミド、N−エチル−N'−(3−ジメチルアミノプロリル)カルボジイミドなどが用いられる。これらによる活性化にはラセミ化抑制添加剤(例えば、HOBt、HOOBt)とともに保護アミノ酸を直接樹脂に添加するかまたは、対称酸無水物またはHOBtエステルあるいはHOOBtエステルとしてあらかじめ保護アミノ酸の活性化を行なった後に樹脂に添加することができる。
【0025】
保護アミノ酸の活性化や樹脂との縮合に用いられる溶媒としては、ペプチド・タンパク質縮合反応に使用しうることが知られている溶媒から適宜選択されうる。例えば、N,N−ジメチルホルムアミド,N,N−ジメチルアセトアミド,N−メチルピロリドンなどの酸アミド類、塩化メチレン,クロロホルムなどのハロゲン化炭化水素類、トリフルオロエタノールなどのアルコール類、ジメチルスルホキシドなどのスルホキシド類、ピリジン,ジオキサン,テトラヒドロフランなどのエーテル類、アセトニトリル,プロピオニトリルなどのニトリル類、酢酸メチル,酢酸エチルなどのエステル類あるいはこれらの適宜の混合物などが用いられる。反応温度はタンパク質結合形成反応に使用され得ることが知られている範囲から適宜選択され、通常約−20℃〜50℃の範囲から適宜選択される。活性化されたアミノ酸誘導体は通常1.5〜4倍過剰で用いられる。ニンヒドリン反応を用いたテストの結果、縮合が不十分な場合には保護基の脱離を行うことなく縮合反応を繰り返すことにより十分な縮合を行なうことができる。反応を繰り返しても十分な縮合が得られないときには、無水酢酸またはアセチルイミダゾールを用いて未反応アミノ酸をアセチル化することができる。
【0026】
原料のアミノ基の保護基としては、例えば、Z、Boc、ターシャリーペンチルオキシカルボニル、イソボルニルオキシカルボニル、4−メトキシベンジルオキシカルボニル、Cl−Z、Br−Z、アダマンチルオキシカルボニル、トリフルオロアセチル、フタロイル、ホルミル、2−ニトロフェニルスルフェニル、ジフェニルホスフィノチオイル、Fmocなどが用いられる。
カルボキシル基は、例えば、アルキルエステル化(例えば、メチル、エチル、プロピル、ブチル、ターシャリーブチル、シクロペンチル、シクロヘキシル、シクロヘプチル、シクロオクチル、2−アダマンチルなどの直鎖状、分枝状もしくは環状アルキルエステル化)、アラルキルエステル化(例えば、ベンジルエステル、4−ニトロベンジルエステル、4−メトキシベンジルエステル、4−クロロベンジルエステル、ベンズヒドリルエステル化)、フェナシルエステル化、ベンジルオキシカルボニルヒドラジド化、ターシャリーブトキシカルボニルヒドラジド化、トリチルヒドラジド化などによって保護することができる。
セリンの水酸基は、例えば、エステル化またはエーテル化によって保護することができる。このエステル化に適する基としては、例えば、アセチル基などの低級アルカノイル基、ベンゾイル基などのアロイル基、ベンジルオキシカルボニル基、エトキシカルボニル基などの炭酸から誘導される基などが用いられる。また、エーテル化に適する基としては、例えば、ベンジル基、テトラヒドロピラニル基、t-ブチル基などである。
チロシンのフェノール性水酸基の保護基としては、例えば、Bzl、Cl2−Bzl、2−ニトロベンジル、Br−Z、ターシャリーブチルなどが用いられる。
ヒスチジンのイミダゾールの保護基としては、例えば、Tos、4−メトキシ−2,3,6−トリメチルベンゼンスルホニル、DNP、ベンジルオキシメチル、Bum、Boc、Trt、Fmocなどが用いられる。
【0027】
原料のカルボキシル基の活性化されたものとしては、例えば、対応する酸無水物、アジド、活性エステル〔アルコール(例えば、ペンタクロロフェノール、2,4,5−トリクロロフェノール、2,4−ジニトロフェノール、シアノメチルアルコール、パラニトロフェノール、HONB、N−ヒドロキシスクシミド、N−ヒドロキシフタルイミド、HOBt)とのエステル〕などが用いられる。原料のアミノ基の活性化されたものとしては、例えば、対応するリン酸アミドが用いられる。
保護基の除去(脱離)方法としては、例えば、Pd−黒あるいはPd−炭素などの触媒の存在下での水素気流中での接触還元や、また、無水フッ化水素、メタンスルホン酸、トリフルオロメタンスルホン酸、トリフルオロ酢酸あるいはこれらの混合液などによる酸処理や、ジイソプロピルエチルアミン、トリエチルアミン、ピペリジン、ピペラジンなどによる塩基処理、また液体アンモニア中ナトリウムによる還元なども用いられる。上記酸処理による脱離反応は、一般に約−20℃〜40℃の温度で行なわれるが、酸処理においては、例えば、アニソール、フェノール、チオアニソール、メタクレゾール、パラクレゾール、ジメチルスルフィド、1,4−ブタンジチオール、1,2−エタンジチオールなどのようなカチオン捕捉剤の添加が有効である。また、ヒスチジンのイミダゾール保護基として用いられる2,4−ジニトロフェニル基はチオフェノール処理により除去され、トリプトファンのインドール保護基として用いられるホルミル基は上記の1,2−エタンジチオール、1,4−ブタンジチオールなどの存在下の酸処理による脱保護以外に、希水酸化ナトリウム溶液、希アンモニアなどによるアルカリ処理によっても除去される。
【0028】
原料の反応に関与すべきでない官能基の保護、保護基の脱離、および反応に関与する官能基の活性化などは公知の手段から、官能基の保護に用いる保護基は公知の基からそれぞれ適宜選択され、適用される。
ペプチド・タンパク質のアミド体を得る別の方法としては、例えば、まず、カルボキシ末端アミノ酸のα−カルボキシル基をアミド化して保護した後、アミノ基側にペプチド鎖を所望の鎖長まで延ばした後、当該ペプチド鎖のN末端のα−アミノ基の保護基のみを除いたペプチド・タンパク質とC末端のカルボキシル基の保護基のみを除去したペプチド・タンパク質とを製造し、この両ペプチド・両タンパク質を上記したような混合溶媒中で縮合させる。縮合反応の詳細については上記と同様である。縮合により得られた保護ペプチド・保護タンパク質を精製した後、上記方法によりすべての保護基を除去し、所望の粗ペプチド・粗タンパク質を得ることができる。この粗ペプチド・粗タンパク質は既知の各種精製手段を駆使して精製し、主要画分を凍結乾燥することで所望のペプチド・タンパク質のアミド体を得ることができる。
ペプチド・タンパク質のエステル体を得るには、例えば、カルボキシ末端アミノ酸のα−カルボキシル基を所望のアルコール類と縮合しアミノ酸エステルとした後、ペプチド・タンパク質のアミド体と同様にして、所望のペプチド・タンパク質のエステル体を得ることができる。
【0029】
本発明のペプチドは、公知のペプチドの合成法に従って製造することができる。また、本発明のタンパク質の部分ペプチドまたはその塩は、公知のペプチドの合成法に従って、あるいは本発明のタンパク質を適当なペプチダーゼで切断することによって製造することができる。
ペプチドの合成法としては、例えば、固相合成法、液相合成法のいずれによっても良い。すなわち、本発明のペプチドもしくは本発明のタンパク質を構成し得る部分ペプチドまたはアミノ酸と残余部分とを縮合させ、生成物が保護基を有する場合は保護基を脱離することにより目的のペプチドを製造することができる。
公知の縮合方法や保護基の脱離としては、例えば、以下の▲1▼〜▲5▼に記載された方法が挙げられる。
▲1▼M. Bodanszky および M.A. Ondetti、ペプチド シンセシス (Peptide Synthesis), Interscience Publishers, New York (1966年)
▲2▼SchroederおよびLuebke、ザ ペプチド(The Peptide), Academic Press, New York (1965年)
▲3▼泉屋信夫他、ペプチド合成の基礎と実験、 丸善(株) (1975年)
▲4▼矢島治明 および榊原俊平、生化学実験講座 1、 タンパク質の化学IV、 205、(1977年)
▲5▼矢島治明監修、続医薬品の開発 第14巻 ペプチド合成 広川書店
また、反応後は通常の精製法、たとえば、溶媒抽出・蒸留・カラムクロマトグラフィー・液体クロマトグラフィー・再結晶などを組み合わせて本発明のペプチドまたは本発明の部分ペプチドを精製単離することができる。上記方法で得られるペプチドまたは部分ペプチドが遊離体である場合は、公知の方法によって適当な塩に変換することができるし、逆に塩で得られた場合は、公知の方法によって遊離体に変換することができる。
【0030】
本発明のペプチド・タンパク質をコードするDNAとしては、前述した本発明のペプチド・タンパク質をコードする塩基配列を含有するものであればいかなるものであってもよい。また、ゲノムDNA、ゲノムDNAライブラリー、前記した細胞・組織由来のcDNA、前記した細胞・組織由来のcDNAライブラリー、合成DNAのいずれでもよい。ライブラリーに使用するベクターは、バクテリオファージ、プラスミド、コスミド、ファージミドなどいずれであってもよい。また、前記した細胞・組織よりtotalRNAまたはmRNA画分を調製したものを用いて直接Reverse Transcriptase Polymerase Chain Reaction(以下、RT−PCR法と略称する)によって増幅することもできる。
【0031】
具体的には、本発明のペプチドをコードするDNAとしては、例えば、配列番号:26または配列番号:27で表わされる塩基配列を含有するDNA、または配列番号:26または配列番号:27で表わされる塩基配列を含有するDNAとハイストリンジェントな条件下でハイブリダイズするDNAを含有し、本発明のペプチドと実質的に同質の活性(例、本発明のタンパク質に対する結合活性、本発明のタンパク質を介するシグナル情報伝達作用、消化管(例、腸管)収縮活性など)を有するペプチドをコードするDNAであれば何れのものでもよい。
配列番号:26で表わされる塩基配列を含有するDNAとしては、配列番号:28で表わされる塩基配列を含有するDNA等が挙げられる。
配列番号:26で表わされる塩基配列を含有するDNAとハイストリンジェントな条件下でハイブリダイズするDNAとしては、例えば、配列番号:26で表わされる塩基配列と約60%以上、好ましくは約70%以上、より好ましくは約80%以上の相同性を有する塩基配列を含有するDNAなどが用いられる。
配列番号:27で表わされる塩基配列を含有するDNAとしては、配列番号:29で表わされる塩基配列を含有するDNA等が挙げられる。
配列番号:27で表わされる塩基配列を含有するDNAとハイストリンジェントな条件下でハイブリダイズするDNAとしては、例えば、配列番号:27で表わされる塩基配列と約60%以上、好ましくは約70%以上、より好ましくは約80%以上の相同性を有する塩基配列を含有するDNAなどが用いられる。
【0032】
また、本発明のタンパク質をコードするDNAとしては、例えば、配列番号:2または配列番号:3で表わされる塩基配列を含有するDNA、または配列番号:2または配列番号:3で表わされる塩基配列を含有するDNAとハイストリンジェントな条件下でハイブリダイズするDNAを含有し、本発明のタンパク質と実質的に同質の活性(例、リガンド結合活性、シグナル情報伝達作用など)を有するタンパク質をコードするDNAであれば何れのものでもよい。
配列番号:2または配列番号:3で表わされる塩基配列を含有するDNAとハイストリンジェントな条件下でハイブリダイズするDNAとしては、例えば、配列番号:2または配列番号:3で表わされる塩基配列と約90%以上、好ましくは約95%以上、より好ましくは約98%以上の相同性を有する塩基配列を含有するDNAなどが用いられる。
【0033】
ハイブリダイゼーションは、公知の方法あるいはそれに準じる方法、例えば、モレキュラー・クローニング(Molecular Cloning)2nd(J. Sambrook et al., Cold Spring Harbor Lab. Press, 1989)に記載の方法などに従って行なうことができる。また、市販のライブラリーを使用する場合、添付の使用説明書に記載の方法に従って行なうことができる。より好ましくは、ハイストリンジェントな条件に従って行なうことができる。
ハイストリンジェントな条件とは、例えば、ナトリウム濃度が約19〜40mM、好ましくは約19〜20mMで、温度が約50〜70℃、好ましくは約60〜65℃の条件を示す。特に、ナトリウム濃度が約19mMで温度が約65℃の場合が最も好ましい。
【0034】
より具体的には、配列番号:20で表わされるアミノ酸配列を含有するペプチドをコードするDNAとしては、配列番号:26で表わされる塩基配列を含有するDNAがあげられ、配列番号:21で表わされるアミノ酸配列を含有するペプチドをコードするDNAとしては、配列番号:27で表わされる塩基配列を含有するDNAがあげられ、配列番号:22で表わされるアミノ酸配列を含有するペプチドをコードするDNAとしては、配列番号:28で表わされる塩基配列を含有するDNAがあげられ、配列番号:23で表わされるアミノ酸配列を含有するペプチドをコードするDNAとしては、配列番号:29で表わされる塩基配列を含有するDNAがあげられる。
また、配列番号:1で表わされるアミノ酸配列を含有するタンパク質をコードするDNAとしては、配列番号:2または配列番号:3で表わされる塩基配列を含有するDNAがあげられる。
【0035】
本発明のペプチドをコードするDNAの塩基配列と相補的な塩基配列またはその一部を含有してなるヌクレオチド(オリゴヌクレオチド)とは、本発明のペプチドをコードするDNAを包含するだけではなく、RNAをも包含する意味で用いられる。
本発明に従えば、本発明のペプチドの遺伝子の複製又は発現を阻害することのできるアンチセンス・(オリゴ)ヌクレオチド(核酸)を、クローン化したあるいは決定されたペプチドをコードするDNAの塩基配列の塩基配列情報に基づき設計し、合成しうる。そうした(オリゴ)ヌクレオチド(核酸)は、本発明のペプチドの遺伝子のRNAとハイブリダイズすることができ、当該RNAの合成又は機能を阻害することができるか、あるいは本発明のペプチド関連RNAとの相互作用を介して本発明のペプチドの遺伝子の発現を調節・制御することができる。本発明のペプチド関連RNAの選択された配列に相補的な(オリゴ)ヌクレオチド、及び本発明のペプチド関連RNAと特異的にハイブリダイズすることができる(オリゴ)ヌクレオチドは、生体内及び生体外で本発明のペプチドの遺伝子の発現を調節・制御するのに有用であり、また病気などの治療又は診断に有用である。
用語「対応する」とは、遺伝子を含めたヌクレオチド、塩基配列又は核酸の特定の配列に相同性を有するあるいは相補的であることを意味する。ヌクレオチド、塩基配列又は核酸とペプチドとの間で「対応する」とは、ヌクレオチド(核酸)の配列又はその相補体から誘導される指令にあるペプチドのアミノ酸を通常指している。本発明のペプチドの遺伝子の5’端ヘアピンループ、5’端6−ベースペア・リピート、5’端非翻訳領域、ポリペプチド翻訳開始コドン、タンパク質コード領域、ORF翻訳開始コドン、3’端非翻訳領域、3’端パリンドローム領域、及び3’端ヘアピンループは好ましい対象領域として選択しうるが、本発明のペプチドの遺伝子内の如何なる領域も対象として選択しうる。
【0036】
目的核酸と対象領域の少なくとも一部に相補的な(オリゴ)ヌクレオチドとの関係、すなわち対象物とハイブリダイズすることができる(オリゴ)ヌクレオチドとの関係は、「アンチセンス」であるということができる。アンチセンス・(オリゴ)ヌクレオチドは、2−デオキシ−D−リボースを含有しているポリデオキシヌクレオチド、D−リボースを含有しているポリヌクレオチド、プリン又はピリミジン塩基のN−グリコシドであるその他のタイプのポリヌクレオチド、あるいは非ヌクレオチド骨格を有するその他のポリマー(例えば、市販のタンパク質核酸及び合成配列特異的な核酸ポリマー)又は特殊な結合を含有するその他のポリマー(但し、当該ポリマーはDNAやRNA中に見出されるような塩基のペアリナグや塩基の付着を許容する配置をもつヌクレオチドを含有する)などが挙げられる。それらは、2本鎖DNA、1本鎖DNA、2本鎖RNA、1本鎖RNA、さらにDNA:RNAハイブリッドであることができ、さらに非修飾ポリヌクレオチド又は非修飾オリゴヌクレオチド、さらには公知の修飾の付加されたもの、例えば当該分野で知られた標識のあるもの、キャップの付いたもの、メチル化されたもの、1個以上の天然のヌクレオチドを類縁物で置換したもの、分子内ヌクレオチド修飾のされたもの、例えば非荷電結合(例えば、メチルホスホネート、ホスホトリエステル、ホスホルアミデート、カルバメートなど)を持つもの、電荷を有する結合又は硫黄含有結合(例えば、ホスホロチオエート、ホスホロジチオエートなど)を持つもの、例えばタンパク質(ヌクレアーゼ、ヌクレアーゼ・インヒビター、トキシン、抗体、シグナルペプチド、ポリ−L−リジンなど)や糖(例えば、モノサッカライドなど)などの側鎖基を有しているもの、インターカレント化合物(例えば、アクリジン、プソラレンなど)を持つもの、キレート化合物(例えば、金属、放射活性をもつ金属、ホウ素、酸化性の金属など)を含有するもの、アルキル化剤を含有するもの、修飾された結合を持つもの(例えば、αアノマー型の核酸など)であってもよい。ここで「ヌクレオシド」、「ヌクレオチド」及び「核酸」とは、プリン及びピリミジン塩基を含有するのみでなく、修飾されたその他の複素環型塩基をもつようなものを含んでいて良い。こうした修飾物は、メチル化されたプリン及びピリミジン、アシル化されたプリン及びピリミジン、あるいはその他の複素環を含むものであってよい。修飾されたヌクレオチド及び修飾されたヌクレオチドはまた糖部分が修飾されていてよく、例えば1個以上の水酸基がハロゲンとか、脂肪族基などで置換されていたり、あるいはエーテル、アミンなどの官能基に変換されていてよい。
【0037】
本発明のアンチセンス核酸は、RNA、DNA、あるいは修飾された核酸である。修飾された核酸の具体例としては核酸の硫黄誘導体やチオホスフェート誘導体、そしてポリヌクレオシドアミドやオリゴヌクレオシドアミドの分解に抵抗性のものが挙げられるが、それに限定されるものではない。本発明のアンチセンス核酸は次のような方針で好ましく設計されうる。すなわち、細胞内でのアンチセンス核酸をより安定なものにする、アンチセンス核酸の細胞透過性をより高める、目標とするセンス鎖に対する親和性をより大きなものにする、そしてもし毒性があるならアンチセンス核酸の毒性をより小さなものにする。
こうして修飾は当該分野で数多く知られており、例えば J. Kawakami et al., Pharm Tech Japan, Vol. 8, pp.247, 1992; Vol. 8, pp.395, 1992; S. T. Crooke et al. ed., Antisense Research and Applications, CRC Press, 1993 などに開示がある。
本発明のアンチセンス核酸は、変化せしめられたり、修飾された糖、塩基、結合を含有していて良く、リポゾーム、ミクロスフェアのような特殊な形態で供与されたり、遺伝子治療により適用されたり、付加された形態で与えられることができうる。こうして付加形態で用いられるものとしては、リン酸基骨格の電荷を中和するように働くポリリジンのようなポリカチオン体、細胞膜との相互作用を高めたり、核酸の取込みを増大せしめるような脂質(例えば、ホスホリピッド、コレステロールなど)といった疎水性のものが挙げられる。付加するに好ましい脂質としては、コレステロールやその誘導体(例えば、コレステリルクロロホルメート、コール酸など)が挙げられる。こうしたものは、核酸の3’端あるいは5’端に付着させることができ、塩基、糖、分子内ヌクレオシド結合を介して付着させることができうる。その他の基としては、核酸の3’端あるいは5’端に特異的に配置されたキャップ用の基で、エキソヌクレアーゼ、RNaseなどのヌクレアーゼによる分解を阻止するためのものが挙げられる。こうしたキャップ用の基としては、ポリエチレングリコール、テトラエチレングリコールなどのグリコールをはじめとした当該分野で知られた水酸基の保護基が挙げられるが、それに限定されるものではない。
アンチセンス核酸の阻害活性は、本発明の形質転換体、本発明の生体内や生体外の遺伝子発現系、あるいはタンパク質の生体内や生体外の翻訳系を用いて調べることができる。当該核酸は公知の各種の方法で細胞に適用できる。
【0038】
本発明の部分ペプチドをコードするDNAとしては、前述した本発明の部分ペプチドをコードする塩基配列を含有するものであればいかなるものであってもよい。また、ゲノムDNA、ゲノムDNAライブラリー、前記した細胞・組織由来のcDNA、前記した細胞・組織由来のcDNAライブラリー、合成DNAのいずれでもよい。ライブラリーに使用するベクターは、バクテリオファージ、プラスミド、コスミド、ファージミドなどいずれであってもよい。また、前記した細胞・組織よりmRNA画分を調製したものを用いて直接Reverse Transcriptase Polymerase Chain Reaction(以下、RT−PCR法と略称する)によって増幅することもできる。
具体的には、本発明の部分ペプチドをコードするDNAとしては、例えば、配列番号:2または配列番号:3で表わされる塩基配列を含有するDNAの部分塩基配列を含有するDNA、または▲2▼配列番号:2または配列番号:3で表わされる塩基配列を含有するDNAとハイストリンジェントな条件下でハイブリダイズするDNAを含有し、本発明のタンパク質ペプチドと実質的に同質の活性(例、リガンド結合活性、シグナル情報伝達作用など)を有するタンパク質をコードするDNAの部分塩基配列を含有するDNAなどが用いられる。
配列番号:2または配列番号:3で表わされる塩基配列を含有するDNAとハイストリンジェントな条件下でハイブリダイズするDNAとしては、例えば、配列番号:2または配列番号:3で表わされる塩基配列と約90%以上、好ましくは約95%以上、より好ましくは約98%以上の相同性を有する塩基配列を含有するDNAなどが用いられる。
【0039】
本発明のペプチドを完全にコードするDNAのクローニングの手段としては、本発明のペプチドをコードするDNAの塩基配列の部分塩基配列を含有する合成DNAプライマーを用いてPCR法によって増幅するか、または適当なベクターに組み込んだDNAを本発明のペプチドの一部あるいは全領域をコードするDNA断片もしくは合成DNAを用いて標識したものとのハイブリダイゼーションによって選別することができる。ハイブリダイゼーションの方法は、例えば、モレキュラー・クローニング(Molecular Cloning)2nd(J. Sambrook et al., Cold Spring Harbor Lab. Press, 1989)に記載の方法などに従って行なうことができる。また、市販のライブラリーを使用する場合、添付の使用説明書に記載の方法に従って行なうことができる。
本発明のタンパク質またはその部分ペプチド(以下、本発明のタンパク質と略記する)を完全にコードするDNAのクローニングも本発明のペプチドを完全にコードするDNAのクローニングと同様にして行うことができる。
【0040】
DNAの塩基配列の変換は、PCRや公知のキット、例えば、MutanTM-super Express Km(宝酒造(株))、MutanTM-K(宝酒造(株))等を用いて、ODA-LA PCR法やGapped duplex法やKunkel法等の公知の方法あるいはそれらに準じる方法に従って行なうことができる。
クローン化されたペプチド・タンパク質をコードするDNAは目的によりそのまま、または所望により制限酵素で消化したり、リンカーを付加したりして使用することができる。当該DNAはその5’末端側に翻訳開始コドンとしてのATGを有し、また3’末端側には翻訳終止コドンとしてのTAA、TGAまたはTAGを有していてもよい。これらの翻訳開始コドンや翻訳終止コドンは、適当な合成DNAアダプターを用いて付加することもできる。
本発明のペプチド・タンパク質の発現ベクターは、例えば、(イ)本発明のペプチド・タンパク質をコードするDNAから目的とするDNA断片を切り出し、(ロ)当該DNA断片を適当な発現ベクター中のプロモーターの下流に連結することにより製造することができる。
【0041】
ベクターとしては、大腸菌由来のプラスミド(例、pBR322,pBR325,pUC12,pUC13)、枯草菌由来のプラスミド(例、pUB110,pTP5,pC194)、酵母由来プラスミド(例、pSH19,pSH15)、λファージなどのバクテリオファージ、レトロウイルス,ワクシニアウイルス,バキュロウイルスなどの動物ウイルスなどの他、pA1−11、pXT1、pRc/CMV、pRc/RSV、pcDNAI/Neo、pcDNA3.1、pRc/CMV2、pRc/RSV(Invitrogen社)などが用いられる。
本発明で用いられるプロモーターとしては、遺伝子の発現に用いる宿主に対応して適切なプロモーターであればいかなるものでもよい。例えば、動物細胞を宿主として用いる場合は、SRαプロモーター、SV40プロモーター、HIV-LTRプロモーター、CMVプロモーター、HSV-TKプロモーターなどが挙げられる。
これらのうち、CMVプロモーター、SRαプロモーターなどを用いるのが好ましい。宿主がエシェリヒア属菌である場合は、trpプロモーター、lacプロモーター、recAプロモーター、λPLプロモーター、lppプロモーターなどが、宿主がバチルス属菌である場合は、SPO1プロモーター、SPO2プロモーター、penPプロモーターなど、宿主が酵母である場合は、PHO5プロモーター、PGKプロモーター、GAPプロモーター、ADHプロモーターなどが好ましい。宿主が昆虫細胞である場合は、ポリヘドリンプロモーター、P10プロモーターなどが好ましい。
【0042】
発現ベクターには、以上の他に、所望によりエンハンサー、スプライシングシグナル、ポリA付加シグナル、選択マーカー、SV40複製オリジン(以下、SV40oriと略称する場合がある)などを含有しているものを用いることができる。選択マーカーとしては、例えば、ジヒドロ葉酸還元酵素(以下、dhfrと略称する場合がある)遺伝子〔メソトレキセート(MTX)耐性〕、アンピシリン耐性遺伝子(以下、Amprと略称する場合がある)、ネオマイシン耐性遺伝子(以下、Neorと略称する場合がある、G418耐性)等が挙げられる。特に、CHO(dhfr-)細胞を用いてdhfr遺伝子を選択マーカーとして使用する場合、目的遺伝子をチミジンを含まない培地によっても選択できる。
また、必要に応じて、宿主に合ったシグナル配列を、本発明のタンパク質のN端末側に付加する。宿主がエシェリヒア属菌である場合は、PhoA・シグナル配列、OmpA・シグナル配列などが、宿主がバチルス属菌である場合は、α−アミラーゼ・シグナル配列、サブチリシン・シグナル配列などが、宿主が酵母である場合は、MFα・シグナル配列、SUC2・シグナル配列など、宿主が動物細胞である場合には、インシュリン・シグナル配列、α−インターフェロン・シグナル配列、抗体分子・シグナル配列などがそれぞれ利用できる。
このようにして構築された本発明のペプチド・タンパク質をコードするDNAを含有するベクターを用いて、形質転換体を製造することができる。
【0043】
宿主としては、例えば、エシェリヒア属菌、バチルス属菌、酵母、昆虫細胞、昆虫、動物細胞などが用いられる。
エシェリヒア属菌の具体例としては、エシェリヒア・コリ(Escherichia coli)K12・DH1〔プロシージングズ・オブ・ザ・ナショナル・アカデミー・オブ・サイエンシイズ・オブ・ザ・ユーエスエー(Proc. Natl. Acad. . Sci. USA),60巻,160(1968)〕,JM103〔ヌクイレック・アシッズ・リサーチ,(Nucleic Acids Research),9巻,309(1981)〕,JA221〔ジャーナル・オブ・モレキュラー・バイオロジー(Journal of Molecular Biology),120巻,517(1978)〕,HB101〔ジャーナル・オブ・モレキュラー・バイオロジー,41巻,459(1969)〕,C600〔ジェネティックス(Genetics),39巻,440(1954)〕などが用いられる。
バチルス属菌としては、例えば、バチルス・サチルス(Bacillus subtilis)MI114〔ジーン,24巻,255(1983)〕,207−21〔ジャーナル・オブ・バイオケミストリー(Journal of Biochemistry),95巻,87(1984)〕などが用いられる。
酵母としては、例えば、サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)AH22,AH22R-,NA87−11A,DKD−5D,20B−12、シゾサッカロマイセス・ポンベ(Schizosaccharomyces pombe)NCYC1913,NCYC2036、ピキア・パストリス(Pichia pastoris)などが用いられる。
【0044】
昆虫細胞としては、例えば、ウイルスがAcNPVの場合は、夜盗蛾の幼虫由来株化細胞(Spodoptera frugiperda cell;Sf細胞)、Trichoplusia niの中腸由来のMG1細胞、Trichoplusia niの卵由来のHigh FiveTM細胞、Mamestra brassicae由来の細胞またはEstigmena acrea由来の細胞などが用いられる。ウイルスがBmNPVの場合は、蚕由来株化細胞(Bombyx mori N;BmN細胞)などが用いられる。当該Sf細胞としては、例えば、Sf9細胞(ATCC CRL1711)、Sf21細胞(以上、Vaughn, J.L.ら、イン・ヴィボ(In Vivo),13, 213-217,(1977))などが用いられる。
昆虫としては、例えば、カイコの幼虫などが用いられる〔前田ら、ネイチャー(Nature),315巻,592(1985)〕。
動物細胞としては、例えば、サル細胞COS−7,Vero,チャイニーズハムスター細胞CHO(以下、CHO細胞と略記),dhfr遺伝子欠損チャイニーズハムスター細胞CHO(以下、CHO(dhfr-)細胞と略記),マウスL細胞,マウスAtT−20,マウスミエローマ細胞,ラットGH3,ヒトFL細胞などが用いられる。
【0045】
エシェリヒア属菌を形質転換するには、例えば、プロシージングズ・オブ・ザ・ナショナル・アカデミー・オブ・サイエンジイズ・オブ・ザ・ユーエスエー(Proc. Natl. Acad. . Sci. USA),69巻,2110(1972)やジーン(Gene),17巻,107(1982)などに記載の方法に従って行なうことができる。
バチルス属菌を形質転換するには、例えば、モレキュラー・アンド・ジェネラル・ジェネティックス(Molecular & General Genetics),168巻,111(1979)などに記載の方法に従って行なうことができる。
酵母を形質転換するには、例えば、メッソズ・イン・エンザイモロジー(Methods in Enzymology),194巻,182−187(1991)、プロシージングズ・オブ・ザ・ナショナル・アカデミー・オブ・サイエンシイズ・オブ・ザ・ユーエスエー(Proc. Natl. Acad. Sci. USA),75巻,1929(1978)などに記載の方法に従って行なうことができる。
昆虫細胞または昆虫を形質転換するには、例えば、バイオ/テクノロジー(Bio/Technology)、6、47−55(1988))などに記載の方法に従って行なうことができる。
動物細胞を形質転換するには、例えば、細胞工学別冊8 新 細胞工学実験プロトコール.263−267(1995)(秀潤社発行)、ヴィロロジー(Virology),52巻,456(1973)に記載の方法に従って行なうことができる。
このようにして、Gタンパク質共役型タンパク質をコードするDNAを含有する発現ベクターで形質転換された形質転換体が得られる。
宿主がエシェリヒア属菌、バチルス属菌である形質転換体を培養する際、培養に使用される培地としては液体培地が適当であり、その中には当該形質転換体の生育に必要な炭素源、窒素源、無機物その他が含有せしめられる。炭素源としては、例えば、グルコース、デキストリン、可溶性澱粉、ショ糖など、窒素源としては、例えば、アンモニウム塩類、硝酸塩類、コーンスチープ・リカー、ペプトン、カゼイン、肉エキス、大豆粕、バレイショ抽出液などの無機または有機物質、無機物としては、例えば、塩化カルシウム、リン酸二水素ナトリウム、塩化マグネシウムなどが挙げられる。また、酵母エキス、ビタミン類、生長促進因子などを添加してもよい。培地のpHは約5〜8が望ましい。
【0046】
エシェリヒア属菌を培養する際の培地としては、例えば、グルコース、カザミノ酸を含むM9培地〔ミラー(Miller),ジャーナル・オブ・エクスペリメンツ・イン・モレキュラー・ジェネティックス(Journal of Experiments in Molecular Genetics),431−433,Cold Spring Harbor Laboratory, New York 1972〕が好ましい。ここに必要によりプロモーターを効率よく働かせるために、例えば、3β−インドリルアクリル酸のような薬剤を加えることができる。
宿主がエシェリヒア属菌の場合、培養は通常約15〜43℃で約3〜24時間行ない、必要により、通気や撹拌を加えることもできる。
宿主がバチルス属菌の場合、培養は通常約30〜40℃で約6〜24時間行ない、必要により通気や撹拌を加えることもできる。
宿主が酵母である形質転換体を培養する際、培地としては、例えば、バークホールダー(Burkholder)最小培地〔Bostian, K. L. ら、「プロシージングズ・オブ・ザ・ナショナル・アカデミー・オブ・サイエンシイズ・オブ・ザ・ユーエスエー(Proc. Natl. Acad. . Sci. USA),77巻,4505(1980)〕や0.5%カザミノ酸を含有するSD培地〔Bitter, G. A. ら、「プロシージングズ・オブ・ザ・ナショナル・アカデミー・オブ・サイエンシイズ・オブ・ザ・ユーエスエー(Proc. Natl. Acad. . Sci. USA),81巻,5330(1984)〕が挙げられる。培地のpHは約5〜8に調整するのが好ましい。培養は通常約20℃〜35℃で約24〜72時間行ない、必要に応じて通気や撹拌を加える。
【0047】
宿主が昆虫細胞または昆虫である形質転換体を培養する際、培地としては、Grace's Insect Medium(Grace, T.C.C.,ネイチャー(Nature),195,788(1962))に非動化した10%ウシ血清等の添加物を適宜加えたものなどが用いられる。培地のpHは約6.2〜6.4に調整するのが好ましい。培養は通常約27℃で約3〜5日間行ない、必要に応じて通気や撹拌を加える。
宿主が動物細胞である形質転換体を培養する際、培地としては、例えば、約5〜20%の胎児牛血清を含むMEM培地〔サイエンス(Science),122巻,501(1952)〕,DMEM培地〔ヴィロロジー(Virology),8巻,396(1959)〕,RPMI 1640培地〔ジャーナル・オブ・ザ・アメリカン・メディカル・アソシエーション(The Journal of the American Medical Association)199巻,519(1967)〕,199培地〔プロシージング・オブ・ザ・ソサイエティ・フォー・ザ・バイオロジカル・メディスン(Proceeding of the Society for the Biological Medicine),73巻,1(1950)〕などが用いられる。pHは約6〜8であるのが好ましい。培養は通常約30℃〜40℃で約15〜60時間行ない、必要に応じて通気や撹拌を加える。
以上のようにして、形質転換体の細胞内、細胞膜または細胞外に本発明のペプチド・タンパク質を生成せしめることができる。
【0048】
上記培養物から本発明のペプチド・タンパク質を分離精製するには、例えば、下記の方法により行なうことができる。
本発明のペプチド・タンパク質を培養菌体あるいは細胞から抽出するに際しては、培養後、公知の方法で菌体あるいは細胞を集め、これを適当な緩衝液に懸濁し、超音波、リゾチームおよび/または凍結融解などによって菌体あるいは細胞を破壊したのち、遠心分離やろ過によりタンパク質の粗抽出液を得る方法などが適宜用いられる。緩衝液の中に尿素や塩酸グアニジンなどのタンパク質変性剤や、トリトンX−100TMなどの界面活性剤が含まれていてもよい。培養液中にペプチド・タンパク質が分泌される場合には、培養終了後、公知の方法で菌体あるいは細胞と上清とを分離し、上清を集める。
このようにして得られた培養上清、あるいは抽出液中に含まれるペプチド・タンパク質の精製は、公知の分離・精製法を適切に組み合わせて行なうことができる。これらの公知の分離、精製法としては、塩析や溶媒沈澱法などの溶解度を利用する方法、透析法、限外ろ過法、ゲルろ過法、およびSDS−ポリアクリルアミドゲル電気泳動法などの主として分子量の差を利用する方法、イオン交換クロマトグラフィーなどの荷電の差を利用する方法、アフィニティークロマトグラフィーなどの特異的親和性を利用する方法、逆相高速液体クロマトグラフィーなどの疎水性の差を利用する方法、等電点電気泳動法などの等電点の差を利用する方法などが用いられる。
【0049】
このようにして得られるペプチド・タンパク質が遊離体で得られた場合には、公知の方法あるいはそれに準じる方法によって塩に変換することができ、逆に塩で得られた場合には公知の方法あるいはそれに準じる方法により、遊離体または他の塩に変換することができる。
なお、組換え体が産生するペプチド・タンパク質を、精製前または精製後に適当なタンパク質修飾酵素を作用させることにより、任意に修飾を加えたり、ペプチド・ポリペプチドを部分的に除去することもできる。タンパク質修飾酵素としては、例えば、トリプシン、キモトリプシン、アルギニルエンドペプチダーゼ、プロテインキナーゼ、グリコシダーゼなどが用いられる。
このようにして生成する本発明のペプチドの活性は、標識した本発明のペプチドと本発明のタンパク質との結合実験および特異抗体を用いたエンザイムイムノアッセイなどにより測定することができる。
また、生成する本発明のタンパク質の活性は、標識した本発明のペプチドとの結合実験および特異抗体を用いたエンザイムイムノアッセイなどにより測定することができる。
【0050】
本発明のペプチドまたは本発明のタンパク質もしくはその部分ペプチドまたはそれらの塩に対する抗体は、本発明のペプチドまたは本発明のタンパク質もしくはその部分ペプチドまたはそれらの塩を認識し得る抗体であれば、ポリクローナル抗体、モノクローナル抗体の何れであってもよい。
本発明のペプチドまたは本発明のタンパク質もしくはその部分ペプチドまたはそれらの塩(以下、本発明のペプチド・タンパク質等と略記する場合がある)に対する抗体は、本発明のペプチド・タンパク質等を抗原として用い、公知の抗体または抗血清の製造法に従って製造することができる。
【0051】
〔モノクローナル抗体の作製〕
(a)モノクローナル抗体産生細胞の作製
本発明のペプチド・タンパク質等は、哺乳動物に対して投与により抗体産生が可能な部位にそれ自体あるいは担体、希釈剤とともに投与される。投与に際して抗体産生能を高めるため、完全フロイントアジュバントや不完全フロイントアジュバントを投与してもよい。投与は通常2〜6週毎に1回ずつ、計2〜10回程度行なわれる。用いられる哺乳動物としては、例えば、サル、ウサギ、イヌ、モルモット、マウス、ラット、ヒツジ、ヤギが挙げられるが、マウスおよびラットが好ましく用いられる。
モノクローナル抗体産生細胞の作製に際しては、抗原を免疫された温血動物、例えば、マウスから抗体価の認められた個体を選択し最終免疫の2〜5日後に脾臓またはリンパ節を採取し、それらに含まれる抗体産生細胞を骨髄腫細胞と融合させることにより、モノクローナル抗体産生ハイブリドーマを調製することができる。抗血清中の抗体価の測定は、例えば、後記の標識化した本発明のペプチド・タンパク質等と抗血清とを反応させたのち、抗体に結合した標識剤の活性を測定することにより行なうことができる。融合操作は既知の方法、例えば、ケーラーとミルスタインの方法〔ネイチャー(Nature)、256巻、495頁(1975年)〕に従い実施することができる。融合促進剤としては、例えば、ポリエチレングリコール(PEG)やセンダイウィルスなどが挙げられるが、好ましくはPEGが用いられる。
骨髄腫細胞としては、例えば、NS−1、P3U1、SP2/0などが挙げられるが、P3U1が好ましく用いられる。用いられる抗体産生細胞(脾臓細胞)数と骨髄腫細胞数との好ましい比率は1:1〜20:1程度であり、PEG(好ましくは、PEG1000〜PEG6000)が10〜80%程度の濃度で添加され、約20〜40℃、好ましくは約30〜37℃で約1〜10分間インキュベートすることにより効率よく細胞融合を実施できる。
【0052】
モノクローナル抗体産生ハイブリドーマのスクリーニングには種々の方法が使用できるが、例えば、本発明のペプチド・タンパク質等抗原を直接あるいは担体とともに吸着させた固相(例、マイクロプレート)にハイブリドーマ培養上清を添加し、次に放射性物質や酵素などで標識した抗免疫グロブリン抗体(細胞融合に用いられる細胞がマウスの場合、抗マウス免疫グロブリン抗体が用いられる)またはプロテインAを加え、固相に結合したモノクローナル抗体を検出する方法、抗免疫グロブリン抗体またはプロテインAを吸着させた固相にハイブリドーマ培養上清を添加し、放射性物質や酵素などで標識した本発明のペプチド・タンパク質等を加え、固相に結合したモノクローナル抗体を検出する方法などが挙げられる。
モノクローナル抗体の選別は、公知あるいはそれに準じる方法に従って行なうことができるが、通常はHAT(ヒポキサンチン、アミノプテリン、チミジン)を添加した動物細胞用培地などで行なうことができる。選別および育種用培地としては、ハイブリドーマが生育できるものならばどのような培地を用いても良い。例えば、1〜20%、好ましくは10〜20%の牛胎児血清を含むRPMI 1640培地、1〜10%の牛胎児血清を含むGIT培地(和光純薬工業(株))またはハイブリドーマ培養用無血清培地(SFM−101、日水製薬(株))などを用いることができる。培養温度は、通常20〜40℃、好ましくは約37℃である。培養時間は、通常5日〜3週間、好ましくは1週間〜2週間である。培養は、通常5%炭酸ガス下で行なうことができる。ハイブリドーマ培養上清の抗体価は、上記の抗血清中の抗体価の測定と同様にして測定できる。
【0053】
(b)モノクローナル抗体の精製
モノクローナル抗体の分離精製は、通常のポリクローナル抗体の分離精製と同様に免疫グロブリンの分離精製法〔例、塩析法、アルコール沈殿法、等電点沈殿法、電気泳動法、イオン交換体(例、DEAE)による吸脱着法、超遠心法、ゲルろ過法、抗原結合固相またはプロテインAあるいはプロテインGなどの活性吸着剤により抗体のみを採取し、結合を解離させて抗体を得る特異的精製法〕に従って行なうことができる。
【0054】
〔ポリクローナル抗体の作製〕
本発明のポリクローナル抗体は、公知あるいはそれに準じる方法にしたがって製造することができる。例えば、免疫抗原(本発明のペプチド・タンパク質等の抗原)とキャリアータンパク質との複合体をつくり、上記のモノクローナル抗体の製造法と同様に哺乳動物に免疫を行ない、当該免疫動物から本発明のペプチド・タンパク質等に対する抗体含有物を採取して、抗体の分離精製を行なうことにより製造できる。
哺乳動物を免疫するために用いられる免疫抗原とキャリアータンパク質との複合体に関し、キャリアータンパク質の種類およびキャリアーとハプテンとの混合比は、キャリアーに架橋させて免疫したハプテンに対して抗体が効率良くできれば、どの様なものをどの様な比率で架橋させてもよいが、例えば、ウシ血清アルブミン、ウシサイログロブリン、キーホール・リンペット・ヘモシアニン等を重量比でハプテン1に対し、約0.1〜20、好ましくは約1〜5の割合でカプルさせる方法が用いられる。
また、ハプテンとキャリアーのカプリングには、種々の縮合剤を用いることができるが、グルタルアルデヒドやカルボジイミド、マレイミド活性エステル、チオール基、ジチオビリジル基を含有する活性エステル試薬等が用いられる。
縮合生成物は、温血動物に対して、抗体産生が可能な部位にそれ自体あるいは担体、希釈剤とともに投与される。投与に際して抗体産生能を高めるため、完全フロイントアジュバントや不完全フロイントアジュバントを投与してもよい。投与は、通常約2〜6週毎に1回ずつ、計約3〜10回程度行なうことができる。
ポリクローナル抗体は、上記の方法で免疫された哺乳動物の血液、腹水など、好ましくは血液から採取することができる。
抗血清中のポリクローナル抗体価の測定は、上記の血清中の抗体価の測定と同様にして測定できる。ポリクローナル抗体の分離精製は、上記のモノクローナル抗体の分離精製と同様の免疫グロブリンの分離精製法に従って行なうことができる。
【0055】
本発明のペプチド、本発明のペプチドをコードするDNA(以下、本発明のDNAと略記する場合がある)および本発明のペプチドに対する抗体(以下、本発明の抗体と略記する場合がある)は、▲1▼本発明のペプチドが関与する各種疾病の予防・治療剤、▲2▼本発明のペプチドと本発明のタンパク質との結合性を変化させる化合物またはその塩のスクリーニング、▲3▼本発明のペプチドまたはその塩の定量、▲4▼遺伝子診断剤、▲5▼アンチセンスDNAを含有する医薬、▲6▼本発明の抗体を含有する医薬、▲7▼本発明のDNAを含有する非ヒト動物の作出、▲8▼構造的に類似したリガンド・受容体との比較にもとづいたドラッグデザイン、などの実施のために有用である。
特に、本発明の組換えタンパク質の発現系を用いた受容体結合アッセイ系を用いることによって、ヒトや哺乳動物に特異的な本発明のタンパク質に対するリガンドの結合性を変化させる化合物(例、ZAQアゴニスト、ZAQアンタゴニストなど)をスクリーニングすることができ、当該アゴニストまたはアンタゴニストを各種疾病の予防・治療剤などとして使用することができる。
本発明のペプチド、本発明のDNAおよび本発明の抗体の用途について、以下に具体的に説明する。
【0056】
(1)本発明のペプチドが関与する各種疾病の治療・予防剤
後述の実施例に記載したように、本発明のペプチドは、生体内で液性因子として存在し、本発明のタンパク質を活性化し、本発明のタンパク質を発現した細胞の細胞内Ca2+イオン濃度を上昇させることから、本発明のタンパク質(Gタンパク質共役型受容体)のリガンドであることが明らかとなった。
また、本発明のペプチドは、ヘビ毒 Mamba Intestinal Toxin 1(MIT1と略称することもある;配列番号:34;Toxicon、28巻、847-856頁、1990年;FEBS Letters、 461巻、183-188頁、1999年)とアミノ酸レベルで約56%の相同性が認められる。
MIT1は回腸や遠位大腸の収縮、あるいは近位大腸の弛緩を引き起こし、その程度は40 mM塩化カリウムに匹敵するほど強いことが報告されている(FEBS Letters、461巻、183-188頁、1999年)が、その作用点や作用メカニズムは解明されていなかった。本発明者らはMIT1の作用も本発明のタンパク質を介して発現されていることを明らかにした。
以上のことから、本発明のペプチドは、腸管全体の収縮あるいは弛緩を引き起こし、腸管の運動(消化活動)などを制御する活性を有する(例、後述の実施例11)。したがって、本発明のDNA等が欠損している場合あるいは発現量が異常に減少している場合、例えば、消化器疾患(例えば腸炎、下痢、便秘、吸収不良性症候群など)等、種々の疾病が発症する。
したがって、本発明のペプチドおよび本発明のDNAは例えば、消化器疾患(例えば腸炎、下痢、便秘、吸収不良性症候群など)等、種々の疾病の治療・予防剤等の医薬として使用することができる。
例えば、生体内において本発明のペプチドが減少あるいは欠損しているために、本発明のタンパク質が発現している細胞における情報伝達が十分に、あるいは正常に発揮されない患者がいる場合に、(イ)本発明のDNAを当該患者に投与し、生体内で本発明のペプチドを発現させることによって、(ロ)細胞に本発明のDNAを挿入し、本発明のペプチドを発現させた後に、当該細胞を患者に移植することによって、または(ハ)本発明のペプチドを当該患者に投与すること等によって、当該患者における本発明のペプチドの役割を十分に、あるいは正常に発揮させることができる。
本発明のDNAを上記の治療・予防剤として使用する場合は、当該DNAを単独あるいはレトロウイルスベクター、アデノウイルスベクター、アデノウイルスアソシエーテッドウイルスベクター等の適当なベクターに挿入した後、常套手段に従って、ヒトまたは温血動物に投与することができる。本発明のDNAは、そのままで、あるいは摂取促進のための補助剤等の生理学的に認められる担体とともに製剤化し、遺伝子銃やハイドロゲルカテーテルのようなカテーテルによって投与できる。
本発明のペプチドを上記の治療・予防剤として使用する場合は、少なくとも90%、好ましくは95%以上、より好ましくは98%以上、さらに好ましくは99%以上に精製されたものを使用するのが好ましい。
【0057】
本発明のペプチドは、例えば、必要に応じて糖衣を施した錠剤、カプセル剤、エリキシル剤、マイクロカプセル剤等として経口的に、あるいは水もしくはそれ以外の薬学的に許容し得る液との無菌性溶液、または懸濁液剤等の注射剤の形で非経口的に使用できる。例えば、本発明のペプチドを生理学的に認められる担体、香味剤、賦形剤、ベヒクル、防腐剤、安定剤、結合剤等とともに一般に認められた製剤実施に要求される単位用量形態で混和することによって製造することができる。これら製剤における有効成分量は指示された範囲の適当な用量が得られるようにするものである。
錠剤、カプセル剤等に混和することができる添加剤としては、例えば、ゼラチン、コーンスターチ、トラガント、アラビアゴムのような結合剤、結晶性セルロースのような賦形剤、コーンスターチ、ゼラチン、アルギン酸等のような膨化剤、ステアリン酸マグネシウムのような潤滑剤、ショ糖、乳糖またはサッカリンのような甘味剤、ペパーミント、アカモノ油またはチェリーのような香味剤等が用いられる。調剤単位形態がカプセルである場合には、前記タイプの材料にさらに油脂のような液状担体を含有することができる。注射のための無菌組成物は注射用水のようなベヒクル中の活性物質、胡麻油、椰子油等のような天然産出植物油等を溶解または懸濁させる等の通常の製剤実施に従って処方することができる。
注射用の水性液としては、例えば、生理食塩水、ブドウ糖やその他の補助薬を含む等張液(例えば、D−ソルビトール、D−マンニトール、塩化ナトリウム等)等が挙げられ、適当な溶解補助剤、例えば、アルコール(例えば、エタノール等)、ポリアルコール(例えば、プロピレングリコール、ポリエチレングリコール等)、非イオン性界面活性剤(例えば、ポリソルベート80TM、HCO−50等)等と併用してもよい。油性液としては、例えば、ゴマ油、大豆油等が挙げられ、溶解補助剤として安息香酸ベンジル、ベンジルアルコール等と併用してもよい。また、緩衝剤(例えば、リン酸塩緩衝液、酢酸ナトリウム緩衝液等)、無痛化剤(例えば、塩化ベンザルコニウム、塩酸プロカイン等)、安定剤(例えば、ヒト血清アルブミン、ポリエチレングリコール等)、保存剤(例えば、ベンジルアルコール、フェノール等)、酸化防止剤等と配合してもよい。調製された注射液は、通常、適当なアンプルに充填される。
本発明のDNAが挿入されたベクターも上記と同様に製剤化され、通常、非経口的に使用される。
【0058】
このようにして得られる製剤は、安全で低毒性であるので、例えば、哺乳動物(例えばヒト、ラット、マウス、モルモット、ウサギ、ヒツジ、ブタ、ウシ、ウマ、ネコ、イヌ、サル等)に対して投与することができる。
本発明のペプチドの投与量は、対象疾患、投与対象、投与ルート等により差異はあるが、例えば、消化器疾患の治療目的で本発明のペプチドを経口投与する場合、一般的に成人(60kgとして)においては、一日につき本発明のペプチドを約1mg〜1000mg、好ましくは約10〜500mg、より好ましくは約10〜200mg投与する。非経口的に投与する場合は、本発明のペプチドの1回投与量は投与対象、対象疾患等によっても異なるが、例えば、消化器疾患の治療目的で本発明のペプチドを注射剤の形で成人(体重60kgとして)に投与する場合、一日につき当該ペプチドを約1〜1000mg程度、好ましくは約1〜200mg程度、より好ましくは約10〜100mg程度を患部に注射することにより投与するのが好都合である。他の動物の場合も、60kg当たりに換算した量を投与することができる。
【0059】
(2)本発明のペプチドと本発明のタンパク質との結合性を変化させる化合物またはその塩のスクリーニング
本発明のペプチドおよび本発明のタンパク質ならびに部分ペプチドを用いることを特徴とする、本発明のペプチドと本発明のタンパク質との結合性を変化させる化合物またはその塩のスクリーニング方法、または本発明のペプチドおよび本発明のタンパク質を用いることを特徴とする本発明のペプチドと本発明のタンパク質との結合性を変化させる化合物またはその塩のスクリーニング用キット(以下、本発明のスクリーニング方法、本発明のスクリーニング用キットと略記する)について以下に詳述する。
【0060】
本発明のタンパク質を用いるか、または組換え型本発明のタンパク質の発現系を構築し、当該発現系を用いた本発明のペプチドとの結合アッセイ系(リガンド・受容体アッセイ系)を用いることによって、本発明のペプチドと本発明のタンパク質との結合性を変化させる化合物、本発明のペプチドと本発明のタンパク質との結合性を変化させる化合物(例えば、ペプチド、タンパク質、非ペプチド性化合物、合成化合物、発酵生産物など)またはその塩をスクリーニングすることができる。
このような化合物には、本発明のタンパク質を介して細胞刺激活性(例えば、アラキドン酸遊離、アセチルコリン遊離、細胞内Ca2+遊離、細胞内cAMP生成、細胞内cGMP生成、イノシトールリン酸産生、細胞膜電位変動、細胞内タンパク質のリン酸化、c−fosの活性化、pHの低下などを促進する活性または抑制する活性など)を有する化合物(ZAQアゴニスト)と当該細胞刺激活性を有しない化合物(ZAQアンタゴニスト)などが含まれる。「本発明のペプチドと本発明のタンパク質との結合性を変化させる」とは、本発明のペプチドと本発明のタンパク質との結合を阻害する場合と促進する場合の両方を包含するものである。
すなわち、本発明は、(i)本発明のタンパク質に、本発明のペプチドを接触させた場合と(ii)上記した本発明のタンパク質に、本発明のペプチドおよび試験化合物を接触させた場合との比較を行なうことを特徴とする本発明のペプチドと本発明のタンパク質との結合性を変化させる化合物またはその塩のスクリーニング方法を提供する。
本発明のスクリーニング方法においては、(i)上記した本発明のタンパク質に本発明のペプチドを接触させた場合と(ii)上記した本発明のタンパク質に本発明のペプチドおよび試験化合物を接触させた場合における、例えば当該本発明のタンパク質に対する本発明のペプチドの結合量、細胞刺激活性などを測定して比較する。
【0061】
本発明のスクリーニング方法は具体的には、
▲1▼標識した本発明のペプチドを、上記した本発明のタンパク質に接触させた場合と、標識した本発明のペプチドおよび試験化合物を本発明のタンパク質に接触させた場合における、標識した本発明のペプチドの本発明のタンパク質に対する結合量を測定し、比較することを特徴とする本発明のペプチドと本発明のタンパク質との結合性を変化させる化合物またはその塩のスクリーニング方法、
▲2▼標識した本発明のペプチドを本発明のタンパク質を含有する細胞または当該細胞の膜画分に接触させた場合と、標識した本発明のペプチドおよび試験化合物を本発明のタンパク質を含有する細胞または当該細胞の膜画分に接触させた場合における、標識した本発明のペプチドの当該細胞または当該膜画分に対する結合量を測定し比較することを特徴とする、本発明のペプチドと本発明のタンパク質との結合性を変化させる化合物またはその塩のスクリーニング方法、
▲3▼標識した本発明のペプチドを、本発明のタンパク質をコードするDNAを含有する形質転換体を培養することによって細胞膜上に発現した本発明のタンパク質に接触させた場合と、標識した本発明のペプチドおよび試験化合物を本発明のタンパク質をコードするDNAを含有する形質転換体を培養することによって細胞膜上に発現した本発明のタンパク質に接触させた場合における、標識した本発明のペプチドの本発明のタンパク質に対する結合量を測定し比較することを特徴とする、本発明のペプチドと本発明のタンパク質との結合性を変化させる化合物またはその塩のスクリーニング方法、
▲4▼本発明のペプチドを本発明のタンパク質を含有する細胞に接触させた場合と、本発明のペプチドおよび試験化合物を本発明のタンパク質を含有する細胞に接触させた場合における、本発明のタンパク質を介した細胞刺激活性(例えば、アラキドン酸遊離、アセチルコリン遊離、細胞内Ca2+遊離、細胞内cAMP生成、細胞内cGMP生成、イノシトールリン酸産生、細胞膜電位変動、細胞内タンパク質のリン酸化、c−fosの活性化、pHの低下などを促進する活性または抑制する活性など)を測定し、比較することを特徴とする本発明のペプチドと本発明のタンパク質との結合性を変化させる化合物またはその塩のスクリーニング方法、および
▲5▼本発明のペプチドを、本発明のタンパク質をコードするDNAを含有する形質転換体を培養することによって細胞膜上に発現した本発明のタンパク質に接触させた場合と、本発明のペプチドおよび試験化合物を、本発明のタンパク質をコードするDNAを含有する形質転換体を培養することによって細胞膜上に発現した本発明のタンパク質に接触させた場合における、本発明のタンパク質を介する細胞刺激活性(例えば、アラキドン酸遊離、アセチルコリン遊離、細胞内Ca2+遊離、細胞内cAMP生成、細胞内cGMP生成、イノシトールリン酸産生、細胞膜電位変動、細胞内タンパク質のリン酸化、c−fosの活性化、pHの低下などを促進する活性または抑制する活性など)を測定し、比較することを特徴とする本発明のペプチドと本発明のタンパク質との結合性を変化させる化合物またはその塩のスクリーニング方法などである。
【0062】
本発明のスクリーニング方法の具体的な説明を以下にする。
まず、本発明のスクリーニング方法に用いる本発明のタンパク質としては、上記の本発明のタンパク質を含有するものであれば何れのものであってもよい。しかし、特にヒト由来の臓器は入手が極めて困難なことから、スクリーニングに用いられるものとしては、組換え体を用いて大量発現させた本発明のタンパク質などが適している。
本発明のタンパク質を製造するには、前述の方法などが用いられる。
本発明のスクリーニング方法において、本発明のタンパク質を含有する細胞あるいは当該細胞膜画分などを用いる場合、後述の調製法に従えばよい。
本発明のタンパク質を含有する細胞を用いる場合、当該細胞をグルタルアルデヒド、ホルマリンなどで固定化してもよい。固定化方法は公知の方法に従って行うことができる。
本発明のタンパク質を含有する細胞としては、本発明のタンパク質を発現した宿主細胞をいうが、当該宿主細胞としては、前述の大腸菌、枯草菌、酵母、昆虫細胞、動物細胞などがあげられる。
膜画分としては、細胞を破砕した後、公知の方法で得られる細胞膜が多く含まれる画分のことをいう。細胞の破砕方法としては、Potter−Elvehjem型ホモジナイザーで細胞を押し潰す方法、ワーリングブレンダーやポリトロン(Kinematica社製)による破砕、超音波による破砕、フレンチプレスなどで加圧しながら細胞を細いノズルから噴出させることによる破砕などがあげられる。細胞膜の分画には、分画遠心分離法や密度勾配遠心分離法などの遠心力による分画法が主として用いられる。例えば、細胞破砕液を低速(500rpm〜3000rpm)で短時間(通常、約1分〜10分)遠心し、上清をさらに高速(15000rpm〜30000rpm)で通常30分〜2時間遠心し、得られる沈澱を膜画分とする。当該膜画分中には、発現した本発明のタンパク質と細胞由来のリン脂質や膜タンパク質などの膜成分が多く含まれる。
当該本発明のタンパク質を含有する細胞や膜画分中の本発明のタンパク質の量は、1細胞当たり103〜108分子であるのが好ましく、105〜107分子であるのが好適である。なお、発現量が多いほど膜画分当たりのリガンド結合活性(比活性)が高くなり、高感度なスクリーニング系の構築が可能になるばかりでなく、同一ロットで大量の試料を測定できるようになる。
本発明のペプチドと本発明のタンパク質との結合性を変化させる化合物をスクリーニングする前記の▲1▼〜▲3▼を実施するためには、適当な本発明のタンパク質画分と、標識した本発明のペプチドが用いられる。本発明のタンパク質を含む画分としては、天然型の本発明のタンパク質を含む画分か、またはそれと同等の活性を有する組換え型の本発明のタンパク質を含む画分などが望ましい。ここで、同等の活性とは、同等のリガンド結合活性などを示す。標識した本発明のペプチドとしては、例えば〔3H〕、〔125I〕、〔14C〕、〔35S〕などで標識された本発明のペプチドなどを利用することができる。特に、ボルトン−ハンター試薬を用いて公知の方法で調製した本発明のペプチドの標識体を利用することもできる。
具体的には、本発明のペプチドと本発明のタンパク質との結合性を変化させる化合物のスクリーニングを行うには、まず本発明のタンパク質を含有する細胞または細胞の膜画分を、スクリーニングに適したバッファーに懸濁することにより受容体標品を調製する。バッファーには、pH4〜10(望ましくはpH6〜8)のリン酸バッファー、トリス−塩酸バッファーなどのリガンドと受容体との結合を阻害しないバッファーであればいずれでもよい。また、非特異的結合を低減させる目的で、CHAPS、Tween−80TM(花王−アトラス社)、ジギトニン、デオキシコレートなどの界面活性剤をバッファーに加えることもできる。さらに、プロテアーゼによる本発明のタンパク質や本発明のペプチドの分解を抑える目的でPMSF、ロイペプチン、E−64(ペプチド研究所製)、ペプスタチンなどのプロテアーゼ阻害剤を添加することもできる。0.01ml〜10mlの当該受容体溶液に、一定量(5000cpm〜500000cpm)の標識した本発明のペプチドを添加し、同時に10-4〜10-1μMの試験化合物を共存させる。非特異的結合量(NSB)を知るために大過剰の未標識の本発明のペプチドを加えた反応チューブも用意する。反応は0℃から50℃、望ましくは4℃から37℃で20分から24時間、望ましくは30分から3時間行う。反応後、ガラス繊維濾紙等で濾過し、適量の同バッファーで洗浄した後、ガラス繊維濾紙に残存する放射活性を液体シンチレーションカウンターまたはγ−カウンターで計測する。拮抗する物質がない場合のカウント(B0)から非特異的結合量(NSB)を引いたカウント(B0−NSB)を100%とした時、特異的結合量(B−NSB)が例えば50%以下になる試験化合物を拮抗阻害能力のある候補物質として選択することができる。
また、本発明のタンパク質と本発明のペプチドとの結合を測定する方法として、BIAcore(アマシャムファルマシアバイオテク社製)を用いることもできる。この方法では、本発明のペプチドを装置に添付のプロトコールに従ったアミノカップリング法によってセンサーチップに固定し、本発明のタンパク質を含有する細胞または本発明のタンパク質をコードするDNAを含有する形質変換体から精製した本発明のタンパク質または本発明のタンパク質を含む膜画分、あるいは精製した本発明のタンパク質または本発明のタンパク質を含む膜画分および試験化合物を含むリン酸バッファーまたはトリスバッファーなどの緩衝液をセンサーチップ上を毎分2−20μlの流量で通過させる。 センサーチップ上の本発明のペプチドと本発明のタンパク質とが結合することによって生じる表面プラズモン共鳴の変化を共存する試験化合物が変化させることを観察することによって本発明のタンパク質と本発明のペプチドとの結合を変化させる化合物のスクリーニングを行なうことができる。この方法は、本発明のタンパク質をセンサーチップに固定し、本発明のペプチドまたは本発明のペプチドおよび試験化合物を含むリン酸バッファーまたはトリスバッファーなどの緩衝液をセンサーチップ上を通過させる方法を用いても同様に測定することができる。試験化合物としては、上記と同様のものなどがあげられる。
本発明のペプチドと本発明のタンパク質との結合性を変化させる化合物をスクリーニングする前記の▲4▼〜▲5▼の方法を実施するためには、本発明のタンパク質を介する細胞刺激活性(例えば、アラキドン酸遊離、アセチルコリン遊離、細胞内Ca2+遊離、細胞内cAMP生成、細胞内cGMP生成、イノシトールリン酸産生、細胞膜電位変動、細胞内タンパク質のリン酸化、c−fosの活性化、pHの低下などを促進する活性または抑制する活性など)を公知の方法または市販の測定用キットを用いて測定することができる。具体的には、まず、本発明のタンパク質を含有する細胞をマルチウェルプレート等に培養する。スクリーニングを行うにあたっては前もって新鮮な培地あるいは細胞に毒性を示さない適当なバッファーに交換し、試験化合物などを添加して一定時間インキュベートした後、細胞を抽出あるいは上清液を回収して、生成した産物をそれぞれの方法に従って定量する。細胞刺激活性の指標とする物質(例えば、アラキドン酸など)の生成が、細胞が含有する分解酵素によって検定困難な場合は、当該分解酵素に対する阻害剤を添加してアッセイを行なってもよい。また、cAMP産生抑制などの活性については、フォルスコリンなどで細胞の基礎的産生量を増大させておいた細胞に対する産生抑制作用として検出することができる。
【0063】
細胞刺激活性を測定してスクリーニングを行なうには、適当な本発明のタンパク質を発現した細胞が用いられる。本発明のタンパク質を発現した細胞としては、前述の組換え型本発明のタンパク質発現細胞株などが望ましい。形質転換体である本発明のタンパク質発現細胞は安定発現株でも一過性発現株でも構わない。
また、動物細胞の種類は上記と同様のものが用いられる。
試験化合物としては、例えばペプチド、タンパク質、非ペプチド性化合物、合成化合物、発酵生産物、細胞抽出液、植物抽出液、動物組織抽出液などがあげられる。
【0064】
上記のリガンド・レセプターアッセイ系について、さらに具体的に記載すると以下のような(1)〜(12)のアッセイ系が用いられる。
(1)受容体発現細胞が受容体アゴニストによって刺激されると細胞内のGタンパクが活性化されてGTPが結合する。この現象は受容体発現細胞の膜画分においても観察される。通常、GTPは加水分解されてGDPへと変化するが、このとき反応液中にGTPγSを添加しておくとGTPγSはGTPと同様にGタンパクに結合するが、加水分解されずにGタンパクを含む細胞膜に結合した状態が維持される。標識したGTPγSを用いると細胞膜に残存した放射活性を測定することによって受容体アゴニストの受容体発現細胞刺激活性を測定することができる。この反応を利用して本発明のペプチドの本発明のタンパク質発現細胞に対する刺激活性を測定することができる。この方法は、前記▲4▼〜▲5▼のように本発明のタンパク質を含む細胞を用いるものではなく、▲1▼〜▲3▼のように本発明のタンパク質を含む膜画分を用いるアッセイ法であるが、▲4▼〜▲5▼のように細胞刺激活性を測定するものであり、本測定法において本発明のタンパク質を含む膜画分へのGTPγS結合促進活性を示す物質はアゴニストである。ここにおいて、本発明のペプチドあるいは本発明のペプチドおよび試験化合物を添加し、本発明のペプチドの単独投与に比べて本発明のタンパク質を含む細胞膜画分へのGTPγS結合促進活性に変化が生じることを観察することによって本発明のペプチドと本発明のタンパク質との結合性を変化させる化合物をスクリーニングすることができる。このとき、本発明のペプチドによる本発明のタンパク質を含む細胞膜画分へのGTPγS結合促進活性を抑制する活性を示す化合物を拮抗阻害能力のある候補物質として選択することができる。一方、試験化合物のみを投与し、本発明のタンパク質を含む細胞膜画分へのGTPγS結合促進活性を観察することによりアゴニストのスクリーニングを行なうこともできる。
スクリーニング法の一例についてより具体的に以下に述べる。本発明のタンパク質を含む細胞膜画分を、膜希釈緩衝液(50 mM Tris, 5 mM MgCl2, 150 mM NaCl, 1 μM GDP, 0.1% BSA pH 7.4)で希釈する。希釈率は、本発明のタンパク質の発現量により異なる。これをFalcon2053に0.2mlずつ分注し、本発明のペプチドあるいは本発明のペプチドおよび試験化合物を加え、さらに終濃度200 pMとなるように[35S]GTPγSを加える。25℃で1時間保温した後、氷冷した洗浄用緩衝液(50 mM Tris, 5 mM MgCl2, 150 mM NaCl, 0.1% BSA , 0.05% CHAPS pH 7.4 1.5ml)を加えて、ガラス繊維ろ紙GF/Fでろ過する。65℃、30分保温して乾燥後、液体シンチレーションカウンターでろ紙上に残った膜画分に結合した[35S]GTPγSの放射活性を測定する。本発明のペプチドのみを加えた実験区の放射活性を100%、本発明のペプチドを加えなかった実験区の放射活性を0%とし、本発明のペプチドによるGTPγS結合促進活性に対する試験化合物の影響を算出する。GTPγS結合促進活性が例えば50%以下になる試験化合物を拮抗阻害能力のある候補物質として選択することができる。
【0065】
(2)本発明のタンパク質の発現細胞は本発明のペプチドの刺激によって細胞内cAMP量が減少する。この反応を利用して本発明のペプチドの本発明のタンパク質の発現細胞に対する刺激活性を測定することができる。
本発明のタンパク質を発現させた種々の動物細胞のcAMP産生量はマウス、ラット、ウサギ、ヤギ、ウシなどを免疫して得られた抗cAMP抗体と125I標識cAMP(ともに市販品)を使用することによってRIAあるいは抗cAMP抗体と標識cAMPとを組み合わせた他のEIA系でも測定することができる。また抗cAMP抗体をプロテインAあるいは抗cAMP抗体産生に用いた動物のIgGなどに対する抗体などを使用して固定したシンチラントを含むビーズと125I標識cAMPとを使用するSPA法による定量も可能である(アマシャムファルマシアバイオテク製のキットを使用する)。
cAMP産生抑制のアッセイにおいて、フォルスコリンまたはcalcitoninなど細胞内cAMP量を増加させるようなリガンドなどによって細胞内cAMP量を上昇させ、本発明のペプチドまたは本発明のペプチドおよび試験化合物を添加することによって本発明のペプチドの単独投与による細胞内cAMP量の抑制が変化することを観察し、本発明のペプチドと本発明のタンパク質の結合を変化させる化合物のスクリーニングを行なうことができる。このとき、本発明のペプチドによる本発明のタンパク質の発現細胞のcAMP産生抑制活性を阻害する活性を示す化合物を拮抗阻害能力のある候補物質として選択することができる。一方、試験化合物のみを添加してcAMP産生抑制活性を調べることによりアゴニスト活性を示す化合物のスクリーニングを行なうことができる。
スクリーニング法をより具体的に以下に記載する。本発明のタンパク質発現CHO細胞(ZAQC−B1細胞;後述の実施例3(3−5))を24穴プレートに5x104 cell/wellで播種し、48時間培養する。細胞を0.2mM 3−イソブチル−メチルキサンチンと0.05% BSAと20mM HEPESを含むハンクスバッファー(pH7.4)で洗浄する(以下、0.2mM 3−イソブチル−メチルキサンチンと0.05% BSAと20mM HEPESを含むハンクスバッファー(pH7.4)を、反応用バッファーと呼ぶ)。その後0.5mlの反応用バッファーを加えて30分間培養器で保温する。反応用バッファーを除き、新たに0.25mlの反応用バッファーを細胞に加えた後、2μMフォルスコリンを含む0.25mlの反応用バッファーに1 nMの本発明のペプチドあるいは1 nMの本発明のペプチドおよび試験化合物を添加したものを細胞に加え、37℃で24分間反応させる。100μlの20%過塩素酸を加えて反応を停止させ、次に氷上で1時間置くことにより細胞内cAMPを抽出する。抽出液中のcAMP量は、cAMP EIAキット(アマシャムファルマシアバイオテク)を用いて測定する。フォルスコリン刺激によって産生されたcAMP量を100%とし、1 nMの本発明のペプチドの添加によって抑制されたcAMP量を0%として、本発明のペプチドによるcAMP産生抑制活性に対する試験化合物の影響を算出する。本発明のペプチドの活性を阻害してcAMP産生活性が例えば50%以上になる試験化合物を拮抗阻害能力のある候補物質として選択することができる。
cAMP産生促進活性を測定するには、フォルスコリンを添加せずに本発明のタンパク質発現CHO細胞に試験化合物を添加して産生されたcAMPを上記の方法で定量する。
【0066】
(3)CRE(cAMP response element)を含むDNAを、ピッカジーン・ベイシックベクターまたはピッカジーン・エンハンサーベクター(東洋インキ製造(株))のルシフェラーゼ遺伝子上流のマルチクローニングサイトに挿入し、これをCRE−レポーター遺伝子ベクターとする。CRE−レポーター遺伝子ベクターをトランスフェクションした細胞において、cAMP上昇を伴う刺激は、CREを介したルシフェラーゼ遺伝子発現とそれに引き続くルシフェラーゼタンパク質の産生を誘導する。つまり、ルシフェラーゼ活性を測定することにより、CRE−レポーター遺伝子ベクター導入細胞内のcAMP量の変動を検出することができる。CRE−レポーター遺伝子ベクターを本発明のタンパク質発現細胞にトランスフェクションした細胞を利用して本発明のペプチドと本発明のタンパク質の結合を変化させる化合物のスクリーニングを行なうことができる。具体的なスクリーニング法を以下に記す。
CRE−レポーター遺伝子を導入した本発明のタンパク質発現細胞を24穴プレートに5x103 cell/wellで播種し、48時間培養する。細胞を0.2mM 3−イソブチル−メチルキサンチンと0.05% BSAと20mM HEPESを含むハンクスバッファー(pH7.4)で洗浄する(以下、0.2mM 3−イソブチル−メチルキサンチンと0.05% BSAと20mM HEPESを含むハンクスバッファー(pH7.4)を、反応用バッファーと呼ぶ)。その後0.5mlの反応用バッファーを加えて30分間培養器で保温する。反応用バッファーを除き、新たに0.25mlの反応用バッファーを細胞に加えた後、1 nMの本発明のペプチドあるいは1 nMの本発明のペプチドおよび試験化合物と2μMフォルスコリンを含む0.25mlの反応用バッファーを細胞に加え、37℃で24分間反応させる。細胞をピッカジーン用細胞溶解剤(東洋インキ製造(株))で溶かし、溶解液に発光基質(東洋インキ製造(株))を添加する。ルシフェラーゼによる発光は、ルミノメーター、液体シンチレーションカウンターまたはトップカウンターにより測定する。本発明のペプチドと本発明のタンパク質の結合を変化させる化合物の影響はルシフェラーゼによる発光量を本発明のペプチドを単独で投与した場合と比較することによって測定することができる。このとき、本発明のペプチドの投与によりフォルスコリン刺激による発光量の増加が抑制されるが、この抑制を回復させる化合物を拮抗阻害能力のある候補物質として選択することができる。一方、試験化合物のみを投与し、フォルスコリン刺激によって上昇した発光量の本発明のペプチドと同様な抑制を観察することによりアゴニストのスクリーニングを行なうこともできる。
レポーター遺伝子として、ルシフェラーゼ以外に例えばアルカリフォスファターゼ、クロラムフェニコール・アセチルトランスフェラーゼあるいはβ−ガラクトシダーゼを用いることもできる。これらのレポーター遺伝子の遺伝子産物の酵素活性は以下のように市販の測定キットを用いて容易に測定することができる。アルカリフォスファターゼ活性は、例えば和光純薬製Lumi-Phos 530によって、クロラムフェニコール・アセチルトランスフェラーゼ(chloramphenicol acetyltransferase)活性は、例えば和光純薬製FAST CAT chrolamphenicol Acetyltransferase Assay Kitによって、β−ガラクトシダーゼ活性は、例えば和光純薬製Aurora Gal-XEによって測定することができる。
【0067】
(4)本発明のタンパク質発現細胞は、本発明のペプチドによる刺激の結果、アラキドン酸代謝物を細胞外に放出する。あらかじめ、放射活性を有するアラキドン酸を細胞に取り込ませておくことによって、この活性を細胞外に放出された放射活性を測定することによって測定することができる。このとき、本発明のペプチドあるいは本発明のペプチドおよび試験化合物を添加して、本発明のペプチドのアラキドン酸代謝物放出活性に対する影響を調べることにより、本発明のペプチドと本発明のタンパク質の結合に影響を与える化合物のスクリーニングを行なうことができる。このとき、本発明のペプチドによるアラキドン酸代謝物放出活性を阻害する化合物を拮抗阻害能力のある候補物質として選択することができる。また、試験化合物のみを添加し、本発明のタンパク質発現細胞のアラキドン酸代謝物放出活性を調べることによりアゴニスト活性を示す化合物のスクリーニングを行なうこともできる。本発明のペプチドと本発明のタンパク質の結合に影響を与える化合物のスクリーニング法より具体的に以下に述べる。
本発明のタンパク質発現CHO細胞を24穴プレートに5x104 cell/wellで播種し、24時間培養後、[3H]アラキドン酸を0.25 μCi/wellとなるよう添加する。[3H]アラキドン酸添加16時間後、細胞を0.05% BSAと20mM HEPESを含むハンクスバッファー(pH7.4)で洗浄し、各wellに0.05% BSAと20mM HEPESを含むハンクスバッファー(pH7.4)に溶解した終濃度10 nMの本発明のペプチドあるいは10 nMの本発明のペプチドおよび試験化合物を含むバッファー500 μlを添加する。以降、0.05% BSAと20mM HEPESを含むハンクスバッファー(pH7.4)を反応用バッファーと呼ぶ。37℃で60分間インキュベートした後に、反応液400 μlをシンチレーターに加え、反応液中に遊離した[3H]アラキドン酸代謝物の量をシンチレーションカウンターにより測定する。本発明のペプチドの非添加反応バッファーによる培地中の[3H]アラキドン酸代謝物の量を0%とし、10 nMの本発明のペプチドを添加したときの培地中の[3H]アラキドン酸代謝物の量を100%として試験化合物の本発明のペプチドと本発明のタンパク質の結合に対する影響を算出する。アラキドン酸代謝物産生活性が例えば50%以下になる試験化合物を拮抗阻害能力のある候補物質として選択することができる。
【0068】
(5)本発明のタンパク質発現細胞を、本発明のペプチドにより刺激することによって細胞内のCa2+イオン濃度が上昇する。これを利用することによって本発明のペプチドと本発明のタンパク質の結合に対する試験化合物の影響を調べることができる。具体的には、後述の実施例3(3−5)および実施例5(5−3−4)に準じた方法で調べることができる。
本発明のタンパク質発現細胞を、滅菌した顕微鏡用カバーグラス上に播き、2日後、培養液を4 mM Fura-2 AM(同仁化学研究所)を縣濁したHBSSに置換し、室温で2時間30分おく。HBSSで洗浄した後、キュベットにカバーグラスをセットし、蛍光測定器で、本発明のペプチドあるいは本発明のペプチドおよび試験化合物を加えたときの励起波長340nm及び380nmでの505nmの蛍光強度の比の上昇を測定する。このとき、本発明のペプチドを単独で投与したときに比べて試験化合物の添加によって生じる蛍光強度の変化を測定することにより本発明のペプチドと本発明のタンパク質の結合に対して影響を与える化合物のスクリーニングを行なうことができる。
また、以下のようにFLIPR(モレキュラーデバイス社製)を使うこともできる。すなわち、細胞縣濁液にFluo-3 AM(同仁化学研究所製)を添加し、細胞に取り込ませた後、上清を遠心により数度洗浄後、96穴プレートに細胞を播く。FLIPR装置にセットし、Fura-2の場合と同様に本発明のペプチドあるいは本発明のペプチドおよび試験化合物を加え、本発明のペプチドを単独で投与したときに比べて試験化合物の添加によって観測される蛍光強度が変化することを測定することにより、本発明のペプチドと本発明のタンパク質の結合に対して影響を与える化合物のスクリーニングを行なうことができる。
これらにおいて、本発明のペプチドによる蛍光強度の上昇を抑制する化合物を拮抗阻害能力のある候補物質として選択することができる。一方、試験化合物のみの添加による蛍光強度の上昇を観察することによってアゴニストのスクリーニングを行なうこともできる。
本発明のタンパク質発現細胞に、細胞内Ca2+イオンの上昇によって発光するようなタンパク質の遺伝子(例、aequorinなど)を共発現させておき、細胞内Ca2+イオン濃度の上昇によって、当該遺伝子(例、aequorinなど)がCa2+結合型となり発光することを利用して、本発明のペプチドあるいは本発明のペプチドおよび試験化合物を加え、本発明のペプチドを単独で投与したときに比べて試験化合物の添加によって観測される発光強度が変化することを測定することにより、本発明のペプチドと本発明のタンパク質の結合に対して影響を与える化合物のスクリーニングを行なうことができる。方法は、蛍光物質を取り込ませないこと以外は上記と同様である。
【0069】
(6)受容体を発現する細胞に受容体アゴニストを添加すると、細胞内イノシトール三リン酸濃度が上昇することが知られている。本発明のペプチドによって生じる本発明のタンパク質細胞におけるこの反応を観察することにより本発明のペプチドと本発明のタンパク質の結合に影響を与える化合物のスクリーニングを行なうことができる。24穴プレートに播いて1日目の細胞にmyo-[2-3H] inositol(2.5マイクロCi/well)を添加した培地中で1日培養した細胞を、よく洗浄後、本発明のペプチドあるいは本発明のペプチドおよび試験化合物を添加した後、10%過塩素酸を加え反応を止める。1.5 M KOH、60mM HEPES溶液で中和し、0.5mlのAG1x8樹脂(Bio-Rad)を詰めたカラムに通し、5mM Na2BO3 60mM HCOONH4で洗浄した後、1M HCOONH4 、0.1M HCOOHで溶出した放射活性を液体シンチレーションカウンターで測定する。本発明のペプチドの非添加反応バッファーによる培地中の放射活性を0%とし、本発明のペプチドを添加したときの培地中の放射活性を100%として試験化合物の本発明のペプチドと本発明のタンパク質の結合に対する影響を算出する。イノシトール三リン酸産生活性が例えば50%以下になる試験化合物を拮抗阻害能力のある候補物質として選択することができる。一方、試験化合物のみの添加によるイノシトール三リン酸産生上昇を観察することによってアゴニストのスクリーニングを行なうこともできる。
【0070】
(7)TRE(TPA response element)を含むDNAを、ピッカジーン・ベイシックベクターまたはピッカジーン・エンハンサーベクター(東洋インキ製造(株))のルシフェラーゼ遺伝子上流のマルチクローニングサイトに挿入し、これをTRE−レポーター遺伝子ベクターとする。TRE−レポーター遺伝子ベクターをトランスフェクションした細胞において、細胞内Ca2+イオン濃度上昇を伴う刺激は、TREを介したルシフェラーゼ遺伝子発現とそれに引き続くルシフェラーゼタンパク質の産生を誘導する。つまり、ルシフェラーゼ活性を測定することにより、TRE−レポーター遺伝子ベクター導入細胞内のカルシウムイオン量の変動を検出することができる。TRE−レポーター遺伝子ベクターを本発明のタンパク質発現細胞にトランスフェクトした細胞を利用した本発明のペプチドと本発明のタンパク質の結合を変化させる化合物の具体的なスクリーニング法を以下に記す。
TRE−レポーター遺伝子を導入した本発明のタンパク質発現細胞を24穴プレートに5x103 cell/wellで播種し、48時間培養する。細胞を0.05% BSAと20mM HEPESを含むハンクスバッファー(pH7.4)で洗浄した後、10 nMの本発明のペプチドあるいは10 nMの本発明のペプチドおよび試験化合物を添加し、37℃で60分間反応させる。細胞をピッカジーン用細胞溶解剤(東洋インキ製造(株))で溶かし、溶解液に発光基質(東洋インキ製造(株))を添加する。ルシフェラーゼによる発光は、ルミノメーター、液体シンチレーションカウンターまたはトップカウンターにより測定する。本発明のペプチドと本発明のタンパク質の結合を変化させる化合物の影響は、ルシフェラーゼによる発光量を本発明のペプチドを単独で投与した場合と比較することによって測定することができる。このとき、本発明のペプチドの投与により細胞内Ca2+イオン濃度の上昇によって発光量が増加するが、この増加を抑制する化合物を拮抗阻害能力のある候補物質として選択することができる。一方、試験化合物のみを投与し、本発明のペプチドと同様な発光量の増加を観察することによりアゴニストのスクリーニングを行なうこともできる。
レポーター遺伝子として、ルシフェラーゼ以外に例えばアルカリフォスファターゼ、クロラムフェニコール・アセチルトランスフェラーゼあるいはβ−ガラクトシダーゼを用いることもできる。これらのレポーター遺伝子の遺伝子産物の酵素活性は以下のように市販の測定キットを用いて容易に測定することができる。アルカリフォスファターゼ活性は、例えば和光純薬製Lumi-Phos 530によって、クロラムフェニコール・アセチルトランスフェラーゼ(chloramphenicol acetyltransferase)活性は、例えば和光純薬製FAST CAT chrolamphenicol Acetyltransferase Assay Kitによって、β−ガラクトシダーゼ活性は、例えば和光純薬製Aurora Gal-XEによって測定することができる。
【0071】
(8)本発明のペプチドに応答した本発明のタンパク質発現細胞はMAPキナーゼ活性化によって増殖が観察される。この増殖をMAPキナーゼ活性、チミジン取り込み、細胞数測定(MTTなど)によって測定することができる。これを利用して本発明のペプチドと本発明のタンパク質の結合を変化させる化合物のスクリーニングを行なうことができる。
MAPキナーゼ活性は、本発明のペプチドあるいは本発明のペプチドおよび試験化合物を細胞に添加した後、細胞溶解液から抗MAPキナーゼ抗体を用いた免疫沈降によってMAPキナーゼ分画を得た後、例えば和光純薬製MAP Kinase Assay Kitとγ-[32P]-ATPを使用して容易に測定できる。チミジン取り込み活性は、本発明のタンパク質発現細胞を播き、本発明のペプチドあるいは本発明のペプチドおよび試験化合物を添加した後、[methyl-3H]-チミジンを加え、その後、細胞内に取り込まれた標識チミジンの放射活性を細胞を溶解して液体シンチレーションカウンターで計数することによって測定することができる。
本発明のタンパク質発現細胞の増殖は、発現細胞を播き、本発明のペプチドあるいは本発明のペプチドおよび試験化合物を添加した後にMTT (3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide) を添加し、細胞内に取り込まれてMTTが変化したMTTホルマザンを塩酸酸性としたイソプロパノールで細胞を溶解した後、570nmの吸収を測定することによっても測定できる。
本発明のペプチドと本発明のタンパク質の結合を変化させる化合物の、標識チミジン取り込み活性を利用した具体的なスクリーニング法を以下に記す。
本発明のタンパク質発現細胞を24穴プレートにウェル当たり5000個まき一日間培養する。次に血清を含まない培地で2日間培養し、細胞を飢餓状態にする。本発明のペプチドあるいは本発明のペプチドおよび試験化合物を細胞に添加して24時間培養した後、[methyl-3H]-チミジンをウェル当たり0.015MBq添加し6時間培養する。細胞をPBSで洗った後、メタノールを添加して10分間放置する。次に5%トリクロロ酢酸を添加して15分間放置後、固定された細胞を蒸留水で4回洗う。0.3N水酸化ナトリウム溶液で細胞を溶解し、溶解液中の放射活性を液体シンチレーションカウンターで測定する。本発明のペプチドと本発明のタンパク質の結合を変化させる化合物の影響は、チミジン取り込みによる放射活性の上昇を本発明のペプチドを単独で投与した場合と比較することによって測定することができる。このとき、本発明のペプチドの投与による放射活性の増加を抑制する化合物を拮抗阻害能力のある候補物質として選択することができる。一方、試験化合物のみを投与し、本発明のペプチドと同様な放射活性の増加を観察することによりアゴニストのスクリーニングを行なうこともできる。
【0072】
(9)本発明のタンパク質発現細胞に本発明のペプチドを添加すると、Kチャンネルが活性化し、細胞内にあるK+イオンが、細胞外に流出する。K+イオンと同族元素であるRb+イオンは、K+イオンと区別無くKチャンネルを通って細胞外に流出するので、細胞に標識Rb ([86Rb])を添加して取り込ませておいた後、本発明のペプチドの刺激によって流出する[86Rb]の流れを測定することで本発明のペプチドの作用を測定できる。本発明のペプチドと本発明のタンパク質の結合を変化させる化合物の、[86Rb]流出活性を利用した具体的なスクリーニング法を以下に記す。
24穴にまいて2日後の本発明のタンパク質発現細胞を1mCi/mlの86RbClを含む培地中で2時間保温する。培地をよく洗浄し、外液中の86RbClを完全に除く。本発明のペプチドあるいは本発明のペプチドおよび試験化合物を細胞に添加して30分後の外液を回収し、γカウンターで放射活性を測定する。本発明のペプチドと本発明のタンパク質の結合を変化させる化合物の影響は、[86Rb]流出による放射活性の上昇を本発明のペプチドを単独で投与した場合と比較することによって測定することができる。このとき、本発明のペプチドの投与による放射活性の上昇を抑制する化合物を拮抗阻害能力のある候補物質として選択することができる。一方、試験化合物のみを投与し、本発明のペプチドと同様な放射活性の上昇を観察することによりアゴニストのスクリーニングを行なうこともできる。
【0073】
(10)本発明のタンパク質発現細胞が本発明のペプチドに反応して変化する細胞外のpH(acidification rate)をCytosensor装置(モレキュラーデバイス社)を使用して測定することによって、本発明のペプチドの活性を測定することができる。Cytosensor装置を利用した、細胞外pH変化の測定をすることによる本発明のペプチドと本発明のタンパク質の結合を変化させる化合物の具体的なスクリーニング法を以下に記す。
本発明のタンパク質発現細胞をCytosensor装置用のカプセル内で終夜培養し、装置のチャンバーにセットして細胞外pHが安定するまで約2時間0.1% BSAを含むRPMI 1640培地(モレキュラーデバイス社製)を灌流させる。pHが安定した後、本発明のペプチドあるいは本発明のペプチドおよび試験化合物を含む培地を細胞上に灌流させることによって生じる培地のpH変化を測定する。本発明のペプチドと本発明のタンパク質の結合を変化させる化合物の影響は、本発明のタンパク質発現細胞の細胞外pH変化を本発明のペプチドを単独で投与した場合と比較することによって測定することができる。このとき、本発明のペプチドの投与による細胞外pH変化を抑制する化合物を拮抗阻害能力のある候補物質として選択することができる。一方、試験化合物のみを投与し、本発明のペプチドと同様な細胞外pH変化を観察することによりアゴニストのスクリーニングを行なうこともできる。
【0074】
(11)酵母(Saccharomyces cerevisiae)のhaploidα-mating Type(MATα)の性フェロモン受容体Ste2はGタンパク質Gpa1とカップルしており、性フェロモンα−mating factorに応答してMAPキナーゼを活性化し、以下、Far1(cell-cycle arrest)および転写活性化因子Ste12が活性化される。Ste12は接合に関与するFUS1を含む種々のタンパク質の発現を誘導する。一方、制御因子Sst2は以上の過程に抑制的に機能する。この系において、受容体遺伝子を導入した酵母を作製し、受容体アゴニスト刺激によって酵母細胞内のシグナル伝達系を活性化し、その結果生じる増殖などの指標を用いた、受容体アゴニストと受容体との反応の測定系の試みが行なわれている(Pausch, M. H., Trends in Biotechnology, vol. 15, pp. 487-494 (1997))。このような受容体遺伝子導入酵母の系を利用して本発明のペプチドおよび本発明のタンパク質の結合を変化させる化合物のスクリーニングを行なうことができる。
MATα酵母のSte2およびGpa1をコードする遺伝子を除去し、代わりに本発明のタンパク質の遺伝子およびGpa1−Gai2融合タンパク質をコードする遺伝子を導入する。Farをコードする遺伝子を除去してcell-cycle arrestが生じないようにし、また、Sstをコードする遺伝子を除去することによって本発明のペプチドに対する応答の感度を向上させておく。さらに、FUS1にヒスチジン生合成遺伝子HIS3をつなげたFUS1−HIS3遺伝子を導入する。以上の遺伝子組換え操作は例えば、Priceら(Price, L. A. et al., Molecular and Cellular Biology, vol. 15, pp. 6188-6195 (1995))の報告に記載の方法において、ソマトスタチン受容体タイプ2(SSTR2)遺伝子を本発明のタンパク質の遺伝子に置き換えて実施することによって容易に行なうことができる。こうして構築された形質変換酵母は本発明のタンパク質のリガンドである本発明のペプチドに高感度で反応し、その結果MAPキナーゼの活性化が起きてヒスチジン生合成酵素が合成されるようになって、ヒスチジン欠乏培地で生育可能になる。これを利用して、ヒスチジン欠乏培地での酵母の生育を指標として本発明のペプチドによる本発明のタンパク質発現酵母の応答を観察することができる。
上記のようにして作製された形質変換酵母を完全合成培地の液体培地で終夜培養し、2x104 cell/mlの濃度でヒスチジンを除去した溶解寒天培地に加え、9x9 cmの角形シャーレに播く。寒天が固化した後、本発明のペプチドあるいは本発明のペプチドおよび試験化合物をしみこませた滅菌濾紙を寒天表面におき、30℃で3日間培養する。本発明のペプチドと本発明のタンパク質の結合を変化させる化合物の影響は、濾紙の周囲の酵母の生育を本発明のペプチドを単独で投与した場合と比較することによって測定することができる。このとき、本発明のペプチドの投与による酵母の生育を抑制する化合物を拮抗阻害能力のある候補物質として選択することができる。一方、試験化合物のみを投与し、本発明のペプチドと同様な酵母の生育を観察することによりアゴニストのスクリーニングを行なうこともできる。また、あらかじめ、寒天培地に本発明のペプチドを添加しておいて滅菌濾紙に試験化合物のみをしみこませて培養し、シャーレ全面での酵母の生育が濾紙の周囲で影響を受けることを観察することによっても本発明のペプチドと本発明のタンパク質の結合を変化させる化合物の影響を調べることができる。
【0075】
(12)本発明のタンパク質の遺伝子RNAをアフリカツメガエル卵母細胞に注入し、本発明のペプチドによって刺激すると細胞内Ca2+イオン濃度が上昇して、calcium-activated chloride currentが生じる。これを膜電位の変化としてとらえることが出来る(K+イオン濃度勾配に変化がある場合も同様)。本発明のペプチドによって生じる本発明のタンパク質導入アフリカツメガエル卵母細胞におけるこの反応を観察することにより本発明のペプチドと本発明のタンパク質の結合に影響を与える化合物のスクリーニングを行なうことができる。
氷冷して動けなくなった雌のアフリカツメガエルから取り出した、卵母細胞塊を、MBS液(88mM NaCl, 1mM KCl, 0.41mM CaCl2, 0.33mM Ca(NO3)2, 0.82mM MgSO4、2.4mM NaHCO3、10mM HEPES、pH7.4)に溶かしたコラーゲナーゼ(0.5mg/ml)で卵塊がほぐれるまで19℃、1−6時間、150rpmで処理する。外液をMBS液に置換することで3度洗浄し、マイクロマニピュレーターでpoly(A)+ SLT cRNA (50ng/50nl)をマイクロインジェクションする。本発明のタンパク質 mRNAは、組織や細胞から調製しても、プラスミドからin vitroで転写してもよい。これをMBS液中で20℃で3日培養する。これをRinger液を流しているvoltage clamp装置のくぼみに置き、電位固定用ガラス微小電極、電位測定用ガラス微小電極を細胞内に刺入し、(−)極は、細胞外に置く。電位が安定したら、本発明のペプチドまたは本発明のペプチドおよび試験化合物を含むRinger液を流して電位変化を記録する。本発明のペプチドと本発明のタンパク質の結合を変化させる化合物の影響は、本発明のタンパク質導入アフリカツメガエル卵母細胞の細胞膜電位変化を本発明のペプチドを単独で投与した場合と比較することによって測定することができる。このとき、本発明のペプチドの投与による細胞膜電位変化を抑制する化合物を拮抗阻害能力のある候補物質として選択することができる。一方、試験化合物のみを投与し本発明のペプチドと同様な細胞膜電位変化を観察することによりアゴニストのスクリーニングを行なうこともできる。
この系において、反応を変化量を増大して測定しやすいように各種のGタンパク質遺伝子のpoly(A)+ RNAを導入することもできる。またaequorinのようなCa2+イオン存在下で発光を生じるようなタンパクの遺伝子のpoly(A)+ RNAを共インジェクションすることにより膜電位変化ではなく発光を観察してこの反応を測定することもできる。
【0076】
本発明のペプチドと本発明のタンパク質との結合性を変化させる化合物またはその塩のスクリーニング用キットは、本発明のタンパク質、本発明のタンパク質を含有する細胞、あるいは本発明のタンパク質を含有する細胞の膜画分、および本発明のペプチドを含有するものである。
本発明のスクリーニング用キットの例としては、次のものがあげられる。
1.スクリーニング用試薬
▲1▼測定用緩衝液および洗浄用緩衝液
Hanks' Balanced Salt Solution(ギブコ社製)に、0.05%のウシ血清アルブミン(シグマ社製)を加えたもの。
孔径0.45μmのフィルターで濾過滅菌し、4℃で保存するか、あるいは用時調製しても良い。
▲2▼本発明のタンパク質の標品
本発明のタンパク質を発現させたCHO細胞を、12穴プレートに5×105個/穴で継代し、37℃、5%CO2、95%airで2日間培養したもの。
▲3▼標識リガンド
〔3H〕、〔125I〕、〔14C〕、〔35S〕などで標識した本発明のペプチド。
適当な溶媒または緩衝液に溶解したものを4℃あるいは−20℃にて保存し、用時に測定用緩衝液にて1μMに希釈する。
▲4▼リガンド標準液
本発明のペプチドを0.1%ウシ血清アルブミン(シグマ社製)を含むPBSで1mMとなるように溶解し、−20℃で保存する。
2.測定法
▲1▼12穴組織培養用プレートにて培養した本発明のタンパク質を発現させた細胞を、測定用緩衝液1mlで2回洗浄した後、490μlの測定用緩衝液を各穴に加える。
▲2▼10-3〜10-10Mの試験化合物溶液を5μl加えた後、標識した本発明のペプチドを5μl加え、室温にて1時間反応させる。非特異的結合量を知るためには試験化合物のかわりに10-3Mの本発明のペプチドを5μl加えておく。
▲3▼反応液を除去し、1mlの洗浄用緩衝液で3回洗浄する。細胞に結合した標識した本発明のペプチドを0.2N NaOH−1%SDSで溶解し、4mlの液体シンチレーターA(和光純薬製)と混合する。
▲4▼液体シンチレーションカウンター(ベックマン社製)を用いて放射活性を測定し、Percent Maximum Binding(PMB)を次の式〔数1〕で求める。
〔数1〕
PMB=[(B−NSB)/(B0−NSB)]×100
PMB:Percent Maximum Binding
B :検体を加えた時の値
NSB:Non-specific Binding(非特異的結合量)
B0 :最大結合量
【0077】
本発明のスクリーニング方法またはスクリーニング用キットを用いて得られうる化合物またはその塩は、本発明のペプチドと本発明のタンパク質との結合を変化させる(結合を阻害あるいは促進する)化合物であり、具体的には本発明のタンパク質を介して細胞刺激活性を有する化合物またはその塩(いわゆる本発明のタンパク質のアゴニスト(ZAQアゴニスト))、あるいは当該刺激活性を有しない化合物(いわゆる本発明のタンパク質のアンタゴニスト(ZAQアンタゴニスト))である。当該化合物としては、ペプチド、タンパク、非ペプチド性化合物、合成化合物、発酵生産物などがあげられ、これら化合物は新規な化合物であってもよいし、公知の化合物であってもよい。
上記本発明のタンパク質のアゴニストであるかアンタゴニストであるかの具体的な評価方法は以下の(i)または(ii)に従えばよい。
(i)前記▲1▼〜▲3▼のスクリーニング方法で示されるバインディング・アッセイを行い、本発明のペプチドと本発明のタンパク質との結合性を変化させる(特に、結合を阻害する)化合物を得た後、当該化合物が上記した本発明のタンパク質を介する細胞刺激活性を有しているか否かを測定する。細胞刺激活性を有する化合物またはその塩は本発明のタンパク質のアゴニストであり、当該活性を有しない化合物またはその塩は本発明のタンパク質のアンタゴニストである。
(ii)(a)試験化合物を本発明のタンパク質を含有する細胞に接触させ、上記本発明のタンパク質を介した細胞刺激活性を測定する。細胞刺激活性を有する化合物またはその塩は、本発明のタンパク質のアゴニストである。
(b)本発明のタンパク質を活性化する化合物(例えば、本発明のペプチドまたは本発明のタンパク質のアゴニストなど)を本発明のタンパク質を含有する細胞に接触させた場合と、本発明のタンパク質を活性化する化合物および試験化合物を本発明のタンパク質を含有する細胞に接触させた場合における、本発明のタンパク質を介した細胞刺激活性を測定し、比較する。本発明のタンパク質を活性化する化合物による細胞刺激活性を減少させ得る化合物またはその塩は本発明のタンパク質のアンタゴニストである。
当該本発明のタンパク質のアゴニストは、本発明のタンパク質に対する本発明のペプチドが有する生理活性と同様の作用を有しているので、本発明のペプチドと同様に安全で低毒性な医薬として有用である。
本発明のタンパク質アンタゴニストは、本発明のタンパク質に対する本発明のペプチドが有する生理活性を抑制することができるので、当該受容体活性を抑制する安全で低毒性な医薬として有用である。
【0078】
上述のように、本発明のペプチドは腸管の収縮などを制御する活性を有することから、消化器疾患(例えば腸炎、下痢、便秘、吸収不良性症候群など)などの疾病の治療・予防剤等の医薬として使用することができるため、上記のスクリーニング方法またはスクリーニング用キットを用いて得られる化合物のうち、本発明のタンパク質のアゴニスト(ZAQアゴニスト)は消化器疾患(例えば腸炎、下痢、便秘、吸収不良性症候群など)などの疾病の治療・予防剤などとして用いることができる。また、本発明のタンパク質のアンタゴニスト(ZAQアンタゴニスト)は消化器疾患(例えば腸炎、下痢、便秘、吸収不良性症候群など)などの疾病の治療・予防剤などとして用いることができる。
上記のスクリーニング方法またはスクリーニング用キットを用いて得られうる化合物の塩としては、例えば、薬学的に許容可能な塩などが用いられる。例えば、無機塩基との塩、有機塩基との塩、無機酸との塩、有機酸との塩、塩基性または酸性アミノ酸との塩などがあげられる。
無機塩基との塩の好適な例としては、例えばナトリウム塩、カリウム塩などのアルカリ金属塩、カルシウム塩、マグネシウム塩などのアルカリ土類金属塩、ならびにアルミニウム塩、アンモニウム塩などがあげられる。
有機塩基との塩の好適な例としては、例えばトリメチルアミン、トリエチルアミン、ピリジン、ピコリン、2,6−ルチジン、エタノールアミン、ジエタノールアミン、トリエタノールアミン、シクロヘキシルアミン、ジシクロヘキシルアミン、N,N’−ジベンジルエチレンジアミンなどとの塩などがあげられる。
無機酸との塩の好適な例としては、例えば塩酸、臭化水素酸、硫酸、リン酸などとの塩があげられる。
有機酸との塩の好適な例としては、例えばギ酸、酢酸、プロピオン酸、フマル酸、シュウ酸、酒石酸、マレイン酸、クエン酸、コハク酸、リンゴ酸、メタンスルホン酸、ベンゼンスルホン酸、安息香酸などとの塩があげられる。
塩基性アミノ酸との塩の好適な例としては、例えばアルギニン、リジン、オルチニンなどとの塩があげられ、酸性アミノ酸との好適な例としては、例えばアスパラギン酸、グルタミン酸などとの塩があげられる。
本発明のスクリーニング方法またはスクリーニング用キットを用いて得られる化合物またはその塩を上述の医薬として使用する場合、上記の本発明のペプチドを医薬として実施する場合と同様にして実施することができる。
【0079】
本発明のスクリーニング方法またはスクリーニング用キットを用いて得られる化合物またはその塩を上述の医薬として使用する場合、常套手段に従って実施することができる。例えば、必要に応じて糖衣や腸溶性被膜を施した錠剤、カプセル剤、エリキシル剤、マイクロカプセル剤などとして経口的に、あるいは水もしくはそれ以外の薬学的に許容し得る液との無菌性溶液、または懸濁液剤などの注射剤の形で非経口的に使用できる。例えば、当該化合物またはその塩を生理学的に認められる担体、香味剤、賦形剤、ベヒクル、防腐剤、安定剤、結合剤などとともに一般に認められた単位用量形態で混和することによって製造することができる。これら製剤における有効成分量は指示された範囲の適当な用量が得られるようにするものである。
錠剤、カプセル剤などに混和することができる添加剤としては、例えばゼラチン、コーンスターチ、トラガントガム、アラビアゴムのような結合剤、結晶性セルロースのような賦形剤、コーンスターチ、ゼラチン、アルギン酸などのような膨化剤、ステアリン酸マグネシウムのような潤滑剤、ショ糖、乳糖またはサッカリンのような甘味剤、ペパーミント、アカモノ油またはチェリーのような香味剤などが用いられる。調剤単位形態がカプセルである場合には、前記タイプの材料にさらに油脂のような液状担体を含有することができる。注射のための無菌組成物は注射用水のようなベヒクル中の活性物質、胡麻油、椰子油などのような天然産出植物油などを溶解または懸濁させるなどの通常の製剤実施にしたがって処方することができる。
注射用の水性液としては、例えば、生理食塩水、ブドウ糖やその他の補助薬を含む等張液(例えば、D−ソルビトール、D−マンニトール、塩化ナトリウムなど)などがあげられ、適当な溶解補助剤、たとえばアルコール(たとえばエタノール)、ポリアルコール(たとえばプロピレングリコール、ポリエチレングリコール)、非イオン性界面活性剤(たとえばポリソルベート80TM、HCO−50)などと併用してもよい。油性液としてはゴマ油、大豆油などがあげられ、溶解補助剤として安息香酸ベンジル、ベンジルアルコールなどと併用してもよい。
また、緩衝剤(例えば、リン酸塩緩衝液、酢酸ナトリウム緩衝液)、無痛化剤(例えば、塩化ベンザルコニウム、塩酸プロカインなど)、安定剤(例えば、ヒト血清アルブミン、ポリエチレングリコールなど)、保存剤(例えば、ベンジルアルコール、フェノールなど)、酸化防止剤などと配合してもよい。調製された注射液は通常、適当なアンプルに充填される。
【0080】
このようにして得られる製剤は安全で低毒性であるので、例えば哺乳動物(例えば、ヒト、マウス、ラット、モルモット、ウサギ、ヒツジ、ブタ、ウシ、ネコ、イヌ、サル、チンパンジーなど)に対して投与することができる。
本発明のスクリーニング方法またはスクリーニング用キットを用いて得られる化合物またはその塩の投与量は、症状などにより差異はあるが、経口投与の場合、一般的に成人(体重60kgとして)においては、一日につき約0.1から1000mg、好ましくは約1.0から300mg、より好ましくは約3.0から50mgである。非経口的に投与する場合は、その1回投与量は投与対象、対象臓器、症状、投与方法などによっても異なるが、たとえば注射剤の形では成人の消化器疾患患者(体重60kgとして)への投与においては、ZAQアンタゴニストを一日につき約0.01から30mg程度、好ましくは約0.1から20mg程度、より好ましくは約0.1から10mg程度を静脈注射により投与するのが好都合である。他の動物の場合も、60kg当たりに換算した量を投与することができる。
【0081】
(3)本発明のペプチドまたはその塩の定量
本発明の抗体は、本発明のペプチドを特異的に認識することができるので、被検液中の本発明のペプチドの定量、特にサンドイッチ免疫測定法による定量等に使用することができる。
すなわち、本発明は、
(i)本発明の抗体と、被検液および標識化された本発明のペプチドとを競合的に反応させ、当該抗体に結合した標識化された本発明のペプチドの割合を測定することを特徴とする被検液中の本発明のペプチドの定量法、および
(ii)被検液と担体上に不溶化した本発明の抗体および標識化された本発明の別の抗体とを同時あるいは連続的に反応させたのち、不溶化担体上の標識剤の活性を測定することを特徴とする被検液中の本発明のペプチドの定量法を提供する。
【0082】
また、本発明のペプチドに対するモノクローナル抗体(以下、本発明のモノクローナル抗体と称する場合がある)を用いて本発明のペプチドの定量を行なえるほか、組織染色等による検出を行なうこともできる。これらの目的には、抗体分子そのものを用いてもよく、また、抗体分子のF(ab')2 、Fab'、あるいはFab画分を用いてもよい。
本発明の抗体を用いる本発明のペプチドの定量法は、特に制限されるべきものではなく、被測定液中の抗原量(例えば、本発明のペプチド量)に対応した抗体、抗原もしくは抗体−抗原複合体の量を化学的または物理的手段により検出し、これを既知量の抗原を含む標準液を用いて作製した標準曲線より算出する測定法であれば、いずれの測定法を用いてもよい。例えば、ネフロメトリー、競合法、イムノメトリック法およびサンドイッチ法が好適に用いられるが、感度、特異性の点で、後述するサンドイッチ法を用いるのが特に好ましい。
標識物質を用いる測定法に用いられる標識剤としては、例えば、放射性同位元素、酵素、蛍光物質、発光物質等が用いられる。放射性同位元素としては、例えば、〔125I〕、〔131I〕、〔3H〕、〔14C〕等が用いられる。上記酵素としては、安定で比活性の大きなものが好ましく、例えば、β−ガラクトシダーゼ、β−グルコシダーゼ、アルカリフォスファターゼ、パーオキシダーゼ、リンゴ酸脱水素酵素等が用いられる。蛍光物質としては、例えば、フルオレスカミン、フルオレッセンイソチオシアネート等が用いられる。発光物質としては、例えば、ルミノール、ルミノール誘導体、ルシフェリン、ルシゲニン等が用いられる。さらに、抗体あるいは抗原と標識剤との結合にビオチン−アビジン系を用いることもできる。
【0083】
抗原あるいは抗体の不溶化にあたっては、物理吸着を用いてもよく、また通常ペプチドあるいは酵素等を不溶化、固定化するのに用いられる化学結合を用いる方法でもよい。担体としては、アガロース、デキストラン、セルロース等の不溶性多糖類、ポリスチレン、ポリアクリルアミド、シリコン等の合成樹脂、あるいはガラス等が挙げられる。
サンドイッチ法においては不溶化した本発明のモノクローナル抗体に被検液を反応させ(1次反応)、さらに標識化した別の本発明のモノクローナル抗体を反応させ(2次反応)たのち、不溶化担体上の標識剤の活性を測定することにより被検液中の本発明のペプチド量を定量することができる。1次反応と2次反応は逆の順序に行っても、また、同時に行なってもよいし時間をずらして行なってもよい。標識化剤および不溶化の方法は前記のそれらに準じることができる。また、サンドイッチ法による免疫測定法において、固相用抗体あるいは標識用抗体に用いられる抗体は必ずしも1種類である必要はなく、測定感度を向上させる等の目的で2種類以上の抗体の混合物を用いてもよい。
本発明のサンドイッチ法による本発明のペプチドの測定法においては、1次反応と2次反応に用いられる本発明のモノクローナル抗体は、本発明のペプチドの結合する部位が相異なる抗体が好ましく用いられる。すなわち、1次反応および2次反応に用いられる抗体は、例えば、2次反応で用いられる抗体が、本発明のペプチドのC端部を認識する場合、1次反応で用いられる抗体は、好ましくはC端部以外、例えばN端部を認識する抗体が用いられる。
【0084】
本発明のモノクローナル抗体をサンドイッチ法以外の測定システム、例えば、競合法、イムノメトリック法あるいはネフロメトリー等に用いることができる。
競合法では、被検液中の抗原と標識抗原とを抗体に対して競合的に反応させたのち、未反応の標識抗原(F)と、抗体と結合した標識抗原(B)とを分離し(B/F分離)、B,Fいずれかの標識量を測定し、被検液中の抗原量を定量する。本反応法には、抗体として可溶性抗体を用い、B/F分離をポリエチレングリコール、前記抗体に対する第2抗体等を用いる液相法、および、第1抗体として固相化抗体を用いるか、あるいは、第1抗体は可溶性のものを用い第2抗体として固相化抗体を用いる固相化法とが用いられる。
イムノメトリック法では、被検液中の抗原と固相化抗原とを一定量の標識化抗体に対して競合反応させた後固相と液相を分離するか、あるいは、被検液中の抗原と過剰量の標識化抗体とを反応させ、次に固相化抗原を加え未反応の標識化抗体を固相に結合させたのち、固相と液相を分離する。次に、いずれかの相の標識量を測定し被検液中の抗原量を定量する。
また、ネフロメトリーでは、ゲル内あるいは溶液中で抗原抗体反応の結果生じた不溶性の沈降物の量を測定する。被検液中の抗原量が僅かであり、少量の沈降物しか得られない場合にもレーザーの散乱を利用するレーザーネフロメトリー等が好適に用いられる。
【0085】
これら個々の免疫学的測定法を本発明の定量方法に適用するにあたっては、特別の条件、操作等の設定は必要とされない。それぞれの方法における通常の条件、操作法に当業者の通常の技術的配慮を加えて本発明のペプチドの測定系を構築すればよい。これらの一般的な技術手段の詳細については、総説、成書等を参照することができる。
例えば、入江 寛編「ラジオイムノアッセイ」(講談社、昭和49年発行)、入江 寛編「続ラジオイムノアッセイ」(講談社、昭和54年発行)、石川栄治ら編「酵素免疫測定法」(医学書院、昭和53年発行)、石川栄治ら編「酵素免疫測定法」(第2版)(医学書院、昭和57年発行)、石川栄治ら編「酵素免疫測定法」(第3版)(医学書院、昭和62年発行)、「Methods in ENZYMOLOGY」 Vol. 70(Immunochemical Techniques(Part A))、同書 Vol. 73(Immunochemical Techniques(Part B))、 同書 Vol. 74(Immunochemical Techniques(Part C))、同書 Vol. 84(Immunochemical Techniques(Part D:Selected Immunoassays))、同書 Vol. 92(Immunochemical Techniques(Part E:Monoclonal Antibodies and General Immunoassay Methods))、 同書 Vol. 121(Immunochemical Techniques(Part I:Hybridoma Technology and Monoclonal Antibodies))(以上、アカデミックプレス社発行)等を参照することができる。
以上のようにして、本発明の抗体を用いることによって、本発明のペプチドを感度良く定量することができる。
さらには、本発明の抗体を用いて本発明のペプチドの濃度を定量することによって、(1)本発明のペプチドの濃度の増多が検出された場合、例えば、消化器疾患(例えば腸炎、下痢、便秘、吸収不良性症候群など)に罹患している、または将来罹患する可能性が高いと診断することができる。また、(2)本発明のペプチドの濃度の減少が検出された場合、例えば、消化器疾患(例えば腸炎、下痢、便秘、吸収不良性症候群など)に罹患している、または将来罹患する可能性が高いと診断することができる。
また、本発明の抗体は、体液や組織等の被検体中に存在する本発明のペプチドを検出するために使用することができる。また、本発明のペプチドを精製するために使用する抗体カラムの作製、精製時の各分画中の本発明のペプチドの検出、被検細胞内における本発明のペプチドの挙動の分析等のために使用することができる。
【0086】
(4)遺伝子診断剤
本発明のDNAは、例えば、プローブとして使用することにより、ヒトまたは温血動物(例えば、ラット、マウス、モルモット、ウサギ、トリ、ヒツジ、ブタ、ウシ、ウマ、ネコ、イヌ、サル等)における本発明のペプチドをコードするDNAまたはmRNAの異常(遺伝子異常)を検出することができるので、例えば、当該DNAまたはmRNAの損傷、突然変異あるいは発現低下や、当該DNAまたはmRNAの増加あるいは発現過多等の遺伝子診断剤として有用である。
本発明のDNAを用いる上記の遺伝子診断は、例えば、公知のノーザンハイブリダイゼーションやPCR−SSCP法(ゲノミックス(Genomics),第5巻,874〜879頁(1989年)、プロシージングズ・オブ・ザ・ナショナル・アカデミー・オブ・サイエンシイズ・オブ・ユーエスエー(Proceedings of the National Academy of Sciences of the USA),第86巻,2766〜2770頁(1989年))、DNAマイクロアレイ等により実施することができる。
例えば、ノーザンハイブリダイゼーションやDNAマイクロアレイにより発現低下が検出された場合やPCR−SSCP法やDNAマイクロアレイによりDNAの突然変異が検出された場合は、例えば、消化器疾患(例えば腸炎、下痢、便秘、吸収不良性症候群など)に罹患している可能性が高いと診断することができる。
(5)アンチセンスDNAを含有する医薬
本発明のDNAに相補的に結合し、当該DNAの発現を抑制することができるアンチセンスDNAは、生体内における本発明のペプチドまたは本発明のDNAの機能を抑制することができるので、例えば、本発明のペプチドの発現過多に起因する疾患(例えば、消化器疾患(例えば腸炎、下痢、便秘、吸収不良性症候群など))の治療・予防剤として使用することができる。
上記アンチセンスDNAを上記の治療・予防剤として、前記した本発明のDNAを含有する各種疾病の治療・予防剤と同様に使用することができる。
例えば、当該アンチセンスDNAを単独あるいはレトロウイルスベクター、アデノウイルスベクター、アデノウイルスアソシエーテッドウイルスベクター等の適当なベクターに挿入した後、常套手段に従って投与することができる。当該アンチセンスDNAは、そのままで、あるいは摂取促進のために補助剤等の生理学的に認められる担体とともに製剤化し、遺伝子銃やハイドロゲルカテーテルのようなカテーテルによって投与できる。
さらに、当該アンチセンスDNAは、組織や細胞における本発明のDNAの存在やその発現状況を調べるための診断用オリゴヌクレオチドプローブとして使用することもできる。
【0087】
(6)本発明の抗体を含有する医薬
本発明のペプチドの活性を中和する作用を有する本発明の抗体は、例えば、本発明のペプチドの発現過多に起因する疾患(例えば、消化器疾患(例えば腸炎、下痢、便秘、吸収不良性症候群など))の治療・予防剤等の医薬として使用することができる。
本発明の抗体を含有する上記疾患の治療・予防剤は、そのまま液剤として、または適当な剤型の医薬組成物として、ヒトまたは哺乳動物(例、ラット、ウサギ、ヒツジ、ブタ、ウシ、ネコ、イヌ、サル等)に対して経口的または非経口的に投与することができる。投与量は、投与対象、対象疾患、症状、投与ルート等によっても異なるが、例えば、消化器疾患治療の目的で本発明の抗体を1回量として、通常0.01〜20mg/kg体重程度、好ましくは0.1〜10mg/kg体重程度、さらに好ましくは0.1〜5mg/kg体重程度を、1日1〜5回程度、好ましくは1日1〜3回程度、静脈注射により投与するのが好都合である。他の非経口投与および経口投与の場合もこれに準ずる量を投与することができる。症状が特に重い場合には、その症状に応じて増量してもよい。
本発明の抗体は、それ自体または適当な医薬組成物として投与することができる。上記投与に用いられる医薬組成物は、上記またはその塩と薬理学的に許容され得る担体、希釈剤もしくは賦形剤とを含むものである。かかる組成物は、経口または非経口投与に適する剤形として提供される。
すなわち、例えば、経口投与のための組成物としては、固体または液体の剤形、具体的には錠剤(糖衣錠、フィルムコーティング錠を含む)、丸剤、顆粒剤、散剤、カプセル剤(ソフトカプセル剤を含む)、シロップ剤、乳剤、懸濁剤等があげられる。かかる組成物は公知の方法によって製造され、製剤分野において通常用いられる担体、希釈剤もしくは賦形剤を含有するものである。例えば、錠剤用の担体、賦形剤としては、乳糖、でんぷん、蔗糖、ステアリン酸マグネシウム等が用いられる。
【0088】
非経口投与のための組成物としては、例えば、注射剤、坐剤等が用いられ、注射剤は静脈注射剤、皮下注射剤、皮内注射剤、筋肉注射剤、点滴注射剤等の剤形を包含する。かかる注射剤は、公知の方法に従って、例えば、上記抗体またはその塩を通常注射剤に用いられる無菌の水性もしくは油性液に溶解、懸濁または乳化することによって調製する。注射用の水性液としては、例えば、生理食塩水、ブドウ糖やその他の補助薬を含む等張液等が用いられ、適当な溶解補助剤、例えば、アルコール(例、エタノール)、ポリアルコール(例、プロピレングリコール、ポリエチレングリコール)、非イオン界面活性剤〔例、ポリソルベート80、HCO−50(polyoxyethylene(50mol)adduct of hydrogenated castor oil)〕等と併用してもよい。油性液としては、例えば、ゴマ油、大豆油等が用いられ、溶解補助剤として安息香酸ベンジル、ベンジルアルコール等を併用してもよい。調製された注射液は、通常、適当なアンプルに充填される。直腸投与に用いられる坐剤は、上記抗体またはその塩を通常の坐薬用基剤に混合することによって調製される。
上記の経口用または非経口用医薬組成物は、活性成分の投与量に適合するような投薬単位の剤形に調製されることが好都合である。かかる投薬単位の剤形としては、錠剤、丸剤、カプセル剤、注射剤(アンプル)、坐剤等が例示され、それぞれの投薬単位剤形当たり通常5〜500mg程度、とりわけ注射剤では5〜100mg程度、その他の剤形では10〜250mg程度の上記抗体が含有されていることが好ましい。
なお前記した各組成物は、上記抗体との配合により好ましくない相互作用を生じない限り他の活性成分を含有してもよい。
【0089】
(7)本発明のDNAを含有する非ヒト動物の作出
本発明のDNAを用いて、本発明のタンパク質等を発現するトランスジェニック非ヒト動物を作出することができる。非ヒト動物としては、哺乳動物(例えば、ラット、マウス、ウサギ、ヒツジ、ブタ、ウシ、ネコ、イヌ、サルなど)など(以下、動物と略記する)が挙げられるが、特に、マウス、ウサギなどが好適である。
本発明のDNAを対象動物に導入させるにあたっては、当該DNAを動物細胞で発現させうるプロモーターの下流に結合した遺伝子コンストラクトとして用いるのが一般に有利である。例えば、ウサギ由来の本発明のDNAを導入させる場合、これと相同性が高い動物由来の本発明のDNAを動物細胞で発現させうる各種プロモーターの下流に結合した遺伝子コンストラクトを、例えば、ウサギ受精卵へマイクロインジェクションすることによって本発明のタンパク質等を高産生するDNA導入動物を作出できる。このプロモーターとしては、例えば、ウイルス由来プロモーター、メタロチオネイン等のユビキアスな発現プロモーターも使用しうるが、好ましくは脳で特異的に発現するNGF遺伝子プロモーターやエノラーゼ遺伝子プロモーターなどが用いられる。
【0090】
受精卵細胞段階における本発明のDNAの導入は、対象動物の胚芽細胞および体細胞の全てに存在するように確保される。DNA導入後の作出動物の胚芽細胞において本発明のタンパク質等が存在することは、作出動物の子孫が全てその胚芽細胞及び体細胞の全てに本発明のタンパク質等を含有することを意味する。遺伝子を受け継いだこの種の動物の子孫はその胚芽細胞および体細胞の全てに本発明のタンパク質等を含有する。
本発明のDNA導入動物は、交配により遺伝子を安定に保持することを確認して、当該DNA保有動物として通常の飼育環境で飼育継代を行うことができる。さらに、目的DNAを保有する雌雄の動物を交配することにより、導入遺伝子を相同染色体の両方に持つホモザイゴート動物を取得し、この雌雄の動物を交配することによりすべての子孫が当該DNAを含有するように繁殖継代することができる。
本発明のDNAが導入された動物は、本発明のタンパク質等が高発現させられているので、本発明のタンパク質等に対するアゴニストまたはアンタゴニストのスクリーニング用の動物などとして有用である。
本発明のDNA導入動物を、組織培養のための細胞源として使用することもできる。例えば、本発明のDNA導入マウスの組織中のDNAもしくはRNAを直接分析するか、あるいは遺伝子により発現された本発明のタンパク質が存在する組織を分析することにより、本発明のタンパク質等について分析することができる。本発明のタンパク質等を含有する組織の細胞を標準組織培養技術により培養し、これらを使用して、例えば、脳や末梢組織由来のような一般に培養困難な組織からの細胞の機能を研究することができる。また、その細胞を用いることにより、例えば、各種組織の機能を高めるような医薬の選択も可能である。また、高発現細胞株があれば、そこから、本発明のタンパク質等を単離精製することも可能である。
【0091】
(8)本発明のペプチドの用途など
本発明のペプチドおよび配列番号:36、配列番号:40、配列番号:47、配列番号:48もしくは配列番号:49で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質もしくはその部分ペプチドまたはその塩を用いることを特徴とする、本発明のペプチドと配列番号:36、配列番号:40、配列番号:47、配列番号:48もしくは配列番号:49で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質またはその塩との結合性を変化させる化合物またはその塩のスクリーニング方法は、前記(2)記載の本発明のスクリーニング方法と同様に実施することができる。
本発明のペプチドおよび配列番号:36、配列番号:40、配列番号:47、配列番号:48もしくは配列番号:49で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質もしくはその部分ペプチドまたはその塩を含有することを特徴とする、本発明のペプチドと配列番号:36、配列番号:40、配列番号:47、配列番号:48もしくは配列番号:49で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質またはその塩との結合性を変化させる化合物またはその塩のスクリーニング用キットは、前記(2)記載の本発明のスクリーニング用キットと同様に実施することができる。
配列番号:34で表されるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するペプチドまたはその塩および配列番号:1、配列番号:36、配列番号:40、配列番号:47、配列番号:48もしくは配列番号:49で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質もしくはその部分ペプチドまたはその塩を用いることを特徴とする、配列番号:34で表されるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するペプチドまたはその塩と配列番号:1、配列番号:36、配列番号:40、配列番号:47、配列番号:48もしくは配列番号:49で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質またはその塩との結合性を変化させる化合物またはその塩のスクリーニング方法は、前記(2)記載の本発明のスクリーニング方法と同様に実施することができる。
配列番号:34で表されるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するペプチドまたはその塩および配列番号:1、配列番号:36、配列番号:40、配列番号:47、配列番号:48もしくは配列番号:49で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質もしくはその部分ペプチドまたはその塩を含有することを特徴とする、配列番号:34で表されるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するペプチドまたはその塩と配列番号:1、配列番号:36、配列番号:40、配列番号:47、配列番号:48もしくは配列番号:49で表わされるアミノ酸配列と同一または実質的に同一のアミノ酸配列を含有するタンパク質またはその塩との結合性を変化させる化合物またはその塩のスクリーニング用キットは、前記(2)記載の本発明のスクリーニング用キットと同様に実施することができる。
上記スクリーニング方法またはスクリーニング用キットを用いて得られうる化合物またはその塩は、前記(2)記載の本発明のスクリーニング方法または本発明のスクリーニング用キットを用いて得られうる化合物またはその塩と同様に使用することができる。
【0092】
本明細書および図面において、塩基やアミノ酸などを略号で表示する場合、IUPAC−IUB Commission on Biochemical Nomenclatureによる略号あるいは当該分野における慣用略号に基づくものであり、その例を下記する。またアミノ酸に関し光学異性体があり得る場合は、特に明示しなければL体を示すものとする。

また、本明細書中で繁用される置換基、保護基および試薬等を下記の記号で表記する。
本明細書の配列表の配列番号は、以下の配列を示す。
〔配列番号:1〕
本発明のヒト脳由来タンパク質のアミノ酸配列を示す。
〔配列番号:2〕
配列番号:1で表わされるアミノ酸配列を含有する本発明のヒト脳由来タンパク質をコードするDNAの塩基配列を示す(ZAQC)。
〔配列番号:3〕
配列番号:1で表わされるアミノ酸配列を含有する本発明のヒト脳由来タンパク質をコードするDNAの塩基配列を示す(ZAQT)。
〔配列番号:4〕
後述の実施例1で用いられたプライマー1の塩基配列を示す。
〔配列番号:5〕
後述の実施例1で用いられたプライマー2の塩基配列を示す。
〔配列番号:6〕
後述の実施例2で用いられたプライマー3の塩基配列を示す。
〔配列番号:7〕
後述の実施例2で用いられたプライマー4の塩基配列を示す。
〔配列番号:8〕
後述の実施例2で用いられたZAQprobeの塩基配列を示す。
〔配列番号:9〕
後述の実施例2で用いられたプライマーZAQC Salの塩基配列を示す。
〔配列番号:10〕
後述の実施例2で用いられたプライマーZAQC Speの塩基配列を示す。
〔配列番号:11〕
後述の実施例3(3−8)で精製されたZAQ活性化ペプチドのN末端のアミノ酸配列を示す。
〔配列番号:12〕
後述の実施例4で用いられたプライマーZF1の塩基配列を示す。
〔配列番号:13〕
後述の実施例4で用いられたプライマーZF2の塩基配列を示す。
〔配列番号:14〕
後述の実施例4で用いられたプライマーZF3の塩基配列を示す。
〔配列番号:15〕
後述の実施例4で得られたヒト型ZAQリガンドペプチドをコードするDNAの3’端塩基配列を示す。
〔配列番号:16〕
後述の実施例4で用いられたプライマーZAQL−CFの塩基配列を示す。
〔配列番号:17〕
後述の実施例4で用いられたプライマーZAQL−XR1の塩基配列を示す。
〔配列番号:18〕
後述の実施例4で得られたDNA断片の塩基配列を示す。
〔配列番号:19〕
後述の実施例4で得られたDNA断片の塩基配列を示す。
〔配列番号:20〕
ヒト型ZAQリガンド成熟体ペプチドのアミノ酸配列を示す。
〔配列番号:21〕
ヒト型ZAQリガンド成熟体ペプチドのアミノ酸配列を示す。
〔配列番号:22〕
ヒト型ZAQリガンド前駆体ペプチドのアミノ酸配列を示す。
〔配列番号:23〕
ヒト型ZAQリガンド前駆体ペプチドのアミノ酸配列を示す。
〔配列番号:24〕
配列番号:28で表わされるヒト型ZAQリガンド前駆体ペプチドをコードするDNAを含有するDNAの塩基配列を示す。
〔配列番号:25〕
配列番号:29で表わされるヒト型ZAQリガンド前駆体ペプチドをコードするDNAを含有するDNAの塩基配列を示す。
〔配列番号:26〕
配列番号:20で表わされるヒト型ZAQリガンド成熟体ペプチドをコードするDNAの塩基配列を示す。
〔配列番号:27〕
配列番号:21で表わされるヒト型ZAQリガンド成熟ペプチドをコードするDNAの塩基配列を示す。
〔配列番号:28〕
配列番号:22で表わされるヒト型ZAQリガンド前駆体ペプチドをコードするDNAの塩基配列を示す。
〔配列番号:29〕
配列番号:23で表わされるヒト型ZAQリガンド前駆体ペプチドをコードするDNAの塩基配列を示す。
〔配列番号:30〕
後述の実施例5(5−1)で用いられたDNA断片の塩基配列を示す。
〔配列番号:31〕
後述の実施例6(6−2)で分析された、ヒト型ZAQリガンドペプチドのN末端アミノ酸配列を示す。
〔配列番号:32〕
後述の実施例7で用いられたプライマーの塩基配列を示す。
〔配列番号:33〕
後述の実施例7で用いられたプライマーの塩基配列を示す。
〔配列番号:34〕
後述の実施例8で精製されたヘビ毒MIT1のアミノ酸配列を示す。
〔配列番号:35〕
ヒト型I5E受容体タンパク質をコードするcDNAの塩基配列を示す。
〔配列番号:36〕
ヒト型I5E受容体タンパク質のアミノ酸配列を示す。
〔配列番号:37〕
後述の実施例9で用いられたプライマーの塩基配列を示す。
〔配列番号:38〕
後述の実施例9で用いられたプライマーの塩基配列を示す。
〔配列番号:39〕
新規Gタンパク質共役型受容体タンパク質(rZAQ1)をコードするcDNAの塩基配列を示す。
〔配列番号:40〕
新規Gタンパク質共役型受容体タンパク質(rZAQ1)のアミノ酸配列を示す。
〔配列番号:41〕
後述の実施例10で用いられたプローブの塩基配列を示す。
〔配列番号:42〕
後述の実施例10で用いられたプローブの塩基配列を示す。
〔配列番号:43〕
後述の実施例10で用いられたプライマーの塩基配列を示す。
〔配列番号:44〕
後述の実施例10で用いられたプライマーの塩基配列を示す。
〔配列番号:45〕
後述の実施例10で用いられたプライマーの塩基配列を示す。
〔配列番号:46〕
新規Gタンパク質共役型受容体タンパク質(rZAQ2)をコードするcDNAの塩基配列を示す。
〔配列番号:47〕
新規Gタンパク質共役型受容体タンパク質(rZAQ2)のアミノ酸配列を示す。
〔配列番号:48〕
マウス由来Gタンパク質共役型受容体タンパク質(GPR73)のアミノ酸配列を示す。
〔配列番号:49〕
マウス由来Gタンパク質共役型受容体タンパク質(mI5E)のアミノ酸配列を示す。
〔配列番号:50〕
マウス由来Gタンパク質共役型受容体タンパク質(GPR73)をコードするcDNAの塩基配列を示す。
〔配列番号:51〕
マウス由来Gタンパク質共役型受容体タンパク質(mI5E)コードするcDNAの塩基配列を示す。
〔配列番号:52〕
参考例1で用いられたDNA断片#1の塩基配列を示す。
〔配列番号:53〕
参考例1で用いられたDNA断片#2の塩基配列を示す。
〔配列番号:54〕
参考例1で用いられたDNA断片#3の塩基配列を示す。
〔配列番号:55〕
参考例1で用いられたDNA断片#4の塩基配列を示す。
〔配列番号:56〕
参考例1で用いられたDNA断片#5の塩基配列を示す。
〔配列番号:57〕
参考例1で用いられたDNA断片#6の塩基配列を示す。
〔配列番号:58〕
配列番号:21で表わされるヒト型ZAQリガンドをコードする合成DNAの塩基配列を示す。
【0095】
後述の実施例1で得られた形質転換体エシェリヒア・コリ(Escherichia coli)DH5α/pCR2.1−ZAQCは、平成11年8月23日から、日本国茨城県つくば市東1−1−1 中央第6 独立行政法人産業技術総合研究所 特許生物寄託センター(旧 通商産業省工業技術院生命工学工業技術研究所(NIBH))に寄託番号FERM BP−6855として、平成11年8月4日から、日本国大阪府大阪市淀川区十三本町2−17−85 財団法人・発酵研究所(IFO)に寄託番号IFO 16301として寄託されている。
後述の実施例1で得られた形質転換体エシェリヒア・コリ(Escherichia coli)DH5α/pCR2.1−ZAQTは、平成11年8月23日から独立行政法人産業技術総合研究所 特許生物寄託センター(旧 通商産業省工業技術院生命工学工業技術研究所(NIBH))に寄託番号FERM BP−6856として、平成11年8月4日から財団法人・発酵研究所(IFO)に寄託番号IFO 16302として寄託されている。
後述の実施例4で得られた形質転換体エシェリヒア・コリ(Escherichia coli)TOP10/pHMITAは、平成12年7月13日から独立行政法人産業技術総合研究所特許生物寄託センター(旧 通商産業省工業技術院生命工学工業技術研究所(NIBH))に寄託番号FERM BP−7219として、平成12年5月26日から財団法人・発酵研究所(IFO)に寄託番号IFO 16440として寄託されている。
後述の実施例4で得られた形質転換体エシェリヒア・コリ(Escherichia coli)TOP10/pHMITGは、平成12年7月13日から独立行政法人産業技術総合研究所特許生物寄託センター(旧 通商産業省工業技術院生命工学工業技術研究所(NIBH))に寄託番号FERM BP−7220として、平成12年5月26日から財団法人・発酵研究所(IFO)に寄託番号IFO 16441として寄託されている。
後述の実施例9で得られた形質転換体エシェリヒア・コリ(Escherichia coli)DH5α/pCR2.1‐rZAQ1は、平成12年8月21日から独立行政法人産業技術総合研究所 特許生物寄託センター(旧 通商産業省工業技術院生命工学工業技術研究所(NIBH))に寄託番号FERM BP−7275として、平成12年8月1日から財団法人・発酵研究所(IFO)に寄託番号IFO16459として寄託されている。
後述の実施例10で得られた形質転換体エシェリヒア・コリ (Escherichia coli)DH10B/pCMV−rZAQ2は、平成12年8月21日から独立行政法人産業技術総合研究所 特許生物寄託センター(旧 通商産業省工業技術院生命工学工業技術研究所(NIBH))に寄託番号FERM BP−7276として、平成12年8月1日から財団法人・発酵研究所(IFO)に寄託番号IFO16460として寄託されている。
後述の参考例1で取得されたエシェリヒア・コリ(Escherichia coli)MM294(DE3)/pTCh1ZAQは、平成13(2001)年4月27日から、独立行政法人産業技術総合研究所 特許生物寄託センターに寄託番号FERMBP−7571として、平成13年1月16日から受託番号IFO 16527として財団法人発酵研究所(IFO)に寄託されている。
【0096】
【実施例】
以下に実施例および参考例を示して、本発明をより詳細に説明するが、これらは本発明の範囲を限定するものではない。なお、大腸菌を用いての遺伝子操作法は、モレキュラー・クローニング(Molecular cloning)に記載されている方法に従った。
【0097】
実施例1 Gタンパク質共役型受容体タンパク質ZAQをコードするcDNAのクローニングと塩基配列の決定
ヒト脳下垂体cDNA(CLONTECH社)を鋳型とし、2個のプライマー、プライマー1(5'-GTCGACATGGAGACCACCATGGGGTTCATGG-3';配列番号:4)及びプライマー2(5'-ACTAGTTTATTTTAGTCTGATGCAGTCCACCTCTTC-3';配列番号:5)を用いてPCR反応を行った。当該反応における反応液の組成は上記cDNAの10分の1量を鋳型として使用し、Advantage2 Polymerase Mix(CLONTECH社)1/50量、プライマー1及びプライマー2を各0.2μM, dNTPs 200μM、及び酵素に添付のバッファーを加え、25μlの液量とした。PCR反応は、94℃・2分の後、94℃・20秒、72℃・100秒のサイクルを3回、94℃・20秒、68℃・100秒のサイクルを3回、94℃・20秒、64℃・20秒、68℃・100秒のサイクルを38回繰り返し、最後に68℃・7分の伸長反応を行った。当該PCR反応後の反応産物をTAクローニングキット(Invitrogen社)の処方に従いプラスミドベクターpCR2.1(Invitrogen社)へサブクローニングした。これを大腸菌DH5αに導入し、cDNAをもつクローンをアンピシリンを含むLB寒天培地中で選択した後、個々のクローンの配列を解析した結果、新規Gタンパク質共役型受容体タンパク質をコードする2種類のcDNA配列ZAQC(配列番号:2)及びZAQT(配列番号:3)を得た。このcDNAより導き出されるアミノ酸配列を含有するタンパク質はいずれも同一配列(配列番号:1)を有したためZAQと命名し、配列番号:2で表されるDNAを含有する形質転換体を大腸菌(Escherichia coli)DH5α/pCR2.1−ZAQCと、配列番号:3で表されるDNAを含有する形質転換体を大腸菌DH5α/pCR2.1−ZAQTと命名した。
【0098】
実施例2 Taqman PCRによるZAQの発現分布の解析
Taqman PCRに用いるプライマー及びプローブは、Primer Express ver.1.0(PEバイオシステムスジャパン)を用いて検索し、プライマー3(5'-TCATGTTGCTCCACTGGAAGG-3'(配列番号:6))、プライマー4(5'-CCAATTGTCTTGAGGTCCAGG-3'(配列番号:7))、ZAQprobe(5'-TTCTTACAATGGCGGTAAGTCCAGTGCAG-3'(配列番号:8))を選択した。プローブのリポーター色素として、FAM( 6-carboxyfluorescein )を付加した。
スタンダードDNAとして、後述の実施例3で得られたpAK−ZAQCを鋳型に、プライマーZAQC Sal(5'-GTCGACATGGAGACCACCATGGGGTTCATGG-3'(配列番号:9))およびZAQC Spe(5'-ACTAGTTTATTTTAGTCTGATGCAGTCCACCTCTTC-3'(配列番号:10))を用いて増幅したPCR断片を、CHROMA SPIN200(CLONTECH Laboratories,Inc.(CA,USA))を用いて精製し、100−106コピ−/μlに調整して使用した。各組識のcDNAソースとして、Human Multiple Tissue cDNA Panel IおよびPanel II(CLONTECH Laboratories, Inc.)を使用した。プライマー、プローブ、鋳型に、Taqman Universal PCR Master Mix(PEバイオシステムスジャパン)を添付書類記載の規定量加え、ABI PRISM 7700 Sequence Detection System(PEバイオシステムズジャパン)でPCR反応および解析をおこなった。
結果を図8および表1に示した。主に精巣、ついで肺、脳等の部位でZAQの発現がみられた。
【表1】
実施例3 ZAQを活性化するペプチドの単離
(3−1)牛乳抽出液の調製
市販の低温殺菌牛乳を用いて、以下の操作を行い抽出液を調製した。牛乳2 literを高速遠心機(CR26H、R10A型ローター:日立株式会社)を用いて、10,000 rpm、15分間、4℃で遠心し、得られた上清をガーゼでろ過し、脂質片を取り除いた。上清に最終濃度1 Mになるように酢酸を加え、4℃にて30分間攪拌し、次いで高速遠心機(CR26H、R10A型ローター:日立株式会社)を用いて10,000 rpm、15分間遠心し上清をガーゼでろ過し不溶物を除去した。上清に撹拌しながら2倍容のアセトンを加え4℃にて3時間攪拌した。次いで高速遠心機(CR26H、R10A型ローター:日立株式会社)を用いて10,000 rpm、15分間遠心後、得られた上清をガーゼでろ過し不溶物を除去した。得られた上清をロータリーエバポレーターにかけ、アセトンを除去し、最終的に1350mlまで濃縮した。得られた濃縮液を、675mlごとに338mlのジエチルエーテルと混合し、分液ロート中にて激しく混和し、2相分離後、水相を得た。得られた水相について同じ操作をさらに1回繰り返し、清澄な水相を得た。得られた水相を、ロータリーエバポレーターを用いて800mlまで濃縮し、最終的な抽出液を得た。
【0100】
(3−2)牛乳抽出液のC18逆相クロマトグラフィーによる粗分画
オクタデシル基を固定したシリカゲルを充填したカラムSep-Pak C18(Waters社)10gをメタノールで膨潤後、1 M 酢酸で平衡化した。このカラムに、(3−1)で調製した抽出液(牛乳2 liter分)を添着した。続いて、このカラムに、100mlの1 M 酢酸を流しゲルを洗浄した。次に、このカラムに200mlの60%アセトニトリル/0.1%トリフルオロ酢酸を流し、目的とする粗ペプチド成分を溶出した。得られた溶出液を、エバポレーターを用いて濃縮した後、凍結乾燥機(12EL; VirTis社)にて凍結乾燥した。
【0101】
(3−3)牛乳抽出液のスルホプロピルイオン交換クロマトグラフィーによる粗分画
ポリプロピレン製のカラムに100 mM塩酸中で膨潤させたSP Sephadex C-25(Amersham Pharmacia Biotech 社)を、容量が2mlになるよう充填し、蒸留水及び2 M ギ酸アンモニウム(pH 4.0)で洗浄した後、I液(2 M ギ酸アンモニウム:アセトニトリル:水=1:25:74)で平衡化した。上記(3−2)で得られた凍結乾燥物をI液20mlに溶解し、SP Sephadex C-25 2 mlにロードした。I液10mlで洗浄後、II液(2 M ギ酸アンモニウム:アセトニトリル:水=1:2.5:6.5)、III液(2 M ギ酸アンモニウム:アセトニトリル:水=1:1:2)、IV液(2 M ギ酸アンモニウム:アセトニトリル:水=1:0.5:0.5)各10mlで順次溶出した。得られたI液からIV液を、それぞれ凍結乾燥機(12EL; VirTis社)にて凍結乾燥した。
【0102】
(3−4)牛乳抽出液のTSKgel ODS80Ts逆相高速液体クロマトグラフィーによる分画
TSKgel ODS-80Ts逆相高速液体クロマトグラフィー用カラム(東ソー株式会社、4.6 mm x 25 cm)を、40℃にて、流速1ml/minでA液(0.1% トリフルオロ酢酸/蒸留水)容量91.7%/B液(0.1%トリフルオロ酢酸/60%アセトニトリル)容量8.3%を流し、平衡化した。上記(3−3)で得られたI液からIV液の凍結乾燥物を、それぞれ1 M 酢酸4mlに溶解しクロマトグラフィー操作に処した。即ち、凍結乾燥物の溶液4mlを当該カラムに添着した後、流速1ml/minで、1分間かけてA液容量67%/B液容量33%まで上昇させ、次いで40分間かけてA液容量67%/B液容量33%からA液容量0%/B液容量100%まで、B液濃度を直線的グラジエントで上昇させた。
溶出液を、1mlずつフラクション番号をつけて分取し、各フラクション2 μlを150 μl の0.2% Bovine Serum Albumin(BSA)/蒸留水と混合し凍結乾燥した。この乾燥物を後述の(3−5)に記した細胞内Ca2+イオン濃度上昇活性測定用のアッセイ用サンプルとした。
【0103】
(3−5)FLIPRを用いた細胞内Ca2+イオン濃度上昇活性の測定
ZAQ安定発現細胞株は以下のようにして調製した。すなわち、実施例1で得たDH5α/pCR2.1−ZAQCの1クローンを、アンピシリンを含むLB培地で振とう培養し、プラスミドpCR2.1−ZAQCを得た。これを制限酵素SalIおよびSpeIで処理し、ZAQCをコードするインサート部分を切り出した。同様に制限酵素SalIおよびSpeIで処理したpAKKO−1.11H(Biochemica et Biophysica Acta 1219 (1994) 251-259)と、当該インサート部分をLigation Express Kit (CLONTECH Laboratories, Inc.(CA, USA ))を用いて連結し、大腸菌DH10Bにエレクトロポーレーション法にて導入した。得られたクローンの有するプラスミドの構造を、制限酵素処理ならびに配列解析で確認し、正しい構築のものをCHO細胞発現用プラスミドpAK−ZAQCとして使用した。
このプラスミドpAK−ZAQCをCHO/dhfr-細胞(American Type Culture Collection)にCellPhect Transfection kit(Amersham Pharmacia Biotech社)を用いて形質導入することにより取得した。まず、蒸留水120 μlに溶解したプラスミドDNA 4 μgに対してBuffer A(CellPhect Transfection Kitに添付)120 μlを添加し、撹拌し、10分間静置後、Buffer B(CellPhect Transfection Kitに添付)240 μlを添加し、激しく撹拌し当該DNAを含有するDNA−リン酸カルシウム複合体を形成させた。 5 x 105個のCHO/ dhfr- 細胞を60mmシャーレに播き、10%のウシ胎児血清(BIO WHITTAKER 社)を含むHam's F-12培地(日水製薬株式会社)中で37℃、5%炭酸ガス中で1日間培養した後、当該DNA−リン酸カルシウム複合体の懸濁液480 μl をシャーレの当該細胞上に滴下させた。これを、37℃、5%炭酸ガス中にて6時間培養した後、血清を含まないHam's F-12培地で2回細胞を洗浄し、シャーレの当該細胞上に15%グリセロールを含む緩衝液(140 mM NaCl, 25 mM HEPES, 1.4 mM Na2HPO4, pH7.1) 1.2mlを添加し2分間処理した。これを、再度、血清を含まないHam's F-12培地で2回洗浄した後、10%のウシ胎児血清を含むHam's F-12培地中で37℃、5%炭酸ガス中で一晩培養した。当該細胞をトリプシン処理により分散させてシャーレから回収し、2 x 104 個ずつ6-well plateに植え込み、透析済み10%ウシ胎児血清(JRH BIOSCIENCES 社)、1 mM MEM非必須アミノ酸溶液(大日本製薬株式会社)、100 units/ml Penicillin、100 μg/ml Streptomycinを含むDulbecco's modified Eagle medium(DMEM)培地(日水製薬株式会社)中にて37℃、5%炭酸ガス中にて培養を開始した。プラスミドの導入された形質転換CHO細胞は当該培地中で生育するが、非導入細胞は次第に死滅していくので、培養開始後2日毎に培地を交換して死滅細胞を除去した。培養開始8−10日後に生育してきた形質転換CHO細胞のコロニーを約21個選んだ。それぞれ選択された細胞からRNAを市販のRNA単離用キットを用いて回収し、以降公知のRT−PCR法によりZAQを高発現するZAQ発現CHO細胞B−1番クローン(以後、ZAQC−B1細胞と略称する)を選別した。
また、対照としてETA(エンドセリンA受容体)発現CHO細胞24番クローン(以後ETA24細胞と略称する。Journal of Pharmacology and Experimental Therapeutics, 279巻、675-685頁、1996年参照)を用いた。上記(3−4)で得られたアッセイ用サンプルについて、ZAQC−B1細胞及びETA24細胞における細胞内Ca2+イオン濃度上昇活性の測定をFLIPR(Molecular Devices社)を用いて行った。ZAQC−B1細胞、ETA24細胞共に10%透析処理済ウシ胎児血清(以後d FBSとする)を加えたDMEMで継代培養しているものを用いた。ZAQC−B1細胞、ETA24細胞をそれぞれ15×104cells/mlとなるように培地(10% d FBS-DMEM)に懸濁し、FLIPR用96穴プレート(Black plate clear bottom、Coster社)に分注器を用いて各ウェルに200 μlずつ植え込み(3.0×104cells/200μl/ウェル)、5% CO2インキュベーター中にて37℃で一晩培養した後用いた(以後細胞プレートとする)。H/HBSS(ニッスイハンクス2(日水製薬株式会社) 9.8g、炭酸水素ナトリウム 0.35g、HEPES 4.77 g 、水酸化ナトリウム溶液で pH7.4に合わせた後、フィルター滅菌処理)20 ml、250 mM Probenecid 200 μl、ウシ胎児血清(FBS) 200 μlを混合した。また、Fluo 3-AM(同仁化学研究所)2バイアル(50 μg)をジメチルスルフォキサイド 40 μl、20% Pluronic acid(Molecular Probes社) 40 μlに溶解し、これを上記H/HBSS−Probenecid−FBS に加え、混和後、8連ピペットを用いて培養液を除いた細胞プレートに各ウェル 100 μlずつ分注し、5% CO2インキュベーター中にて37℃で1時間インキュベートした(色素ローディング)。上記(3−4)で得られたアッセイ用サンプルについて、各フラクションに、2.5 mM Probenecid、0.1% CHAPSを含むH/HBSS 150 μlを加えて希釈し、FLIPR用96穴プレート(V-Bottomプレート、Coster社)へ移した(以後、サンプルプレートとする)。細胞プレートの色素ローディング終了後、H/HBSSに2.5 mM Probenecidを加えた洗浄バッファーでプレートウォッシャー(Molecular Devices社)を用いて細胞プレートを4回洗浄し、洗浄後100 μlの洗浄バッファーを残した。この細胞プレートとサンプルプレートをFLIPRにセットしアッセイを行った(FLIPRにより、サンプルプレートから50 μlのサンプルが細胞プレートへと移される)。
その結果、上記(3−3)IV液を上記(3−4)逆相高速液体クロマトグラフィー分離して得られたフラクションNo.53にZAQC−B1細胞に特異的な細胞内Ca2+イオン濃度上昇活性が見られた。
【0104】
(3−6)TSKgel Super−Phenyl逆相高速液体クロマトグラフィーによる精製
TSKgel Super-Phenyl逆相高速液体クロマトグラフィー用カラム(東ソー株式会社、 0.46 cm x 10 cm)を、40℃にて、流速1ml/minでA液(0.1% トリフルオロ酢酸/蒸留水)容量91.7%/B液(0.1% トリフルオロ酢酸/60% アセトニトリル)容量8.3%を流し平衡化した。上記(3−4)で得られたフラクションNo.53についてクロマトグラフィー操作を行った。即ち、フラクションNo.53の溶液1mlを当該カラムに添着した後、流速1ml/minで、1分間かけてA液容量75%/B液容量25%まで上昇させ、次いで75分間かけてA液容量67%/B液容量33%まで、B液濃度を直線的グラジエントで上昇させた。
溶出液を、500 μlずつフラクションNo.をつけて分取した。分取フラクションより各25 μlづつ0.2% BSA 150 μlと混合し凍結乾燥機(12EL; VirTis社)で凍結乾燥させた。この乾燥物に、2.5 mM Probenecid、 0.1% CHAPSを含むH/HBSS 150 μl を加えて溶解し、この溶液50 μlを用いて上記(3−5)の試験法により、細胞内Ca2+イオン濃度上昇活性を測定することにより、ZAQC−B1細胞に対する受容体活性化作用を測定した。その結果、目的とするZAQC−B1細胞に対する受容体活性化作用を有する成分、すなわち、ZAQ活性化成分は、主としてフラクションNo.103-105に溶出されていることが判明した。
【0105】
(3−7)μRPC C2/C18 ST 4.6/100逆相高速液体クロマトグラフィーによる精製
μRPC C2/C18 ST 4.6/100逆相高速液体クロマトグラフィー用カラム(Amersham Pharmacia Biotech社、 0.46 cm x 10 cm)を、40℃にて、流速1ml/minでA液(ヘプタフルオロ酪酸/蒸留水)容量95%/B液(0.1%ヘプタフルオロ酪酸/100% アセトニトリル)容量5%を流し平衡化した。
上記(3−6)で得られたTSKgel Super-Phenyl逆相高速液体クロマトグラフィー分取フラクションのうちフラクションNo.103-105をそのままμRPC C2/C18 ST 4.6/100逆相カラムに添着した後、流速1ml/minで1分間でA液(0.1%ヘプタフルオロ酪酸/蒸留水)容量95%/B液(0.1% ヘプタフルオロ酪酸/100% アセトニトリル)容量5%からA液容量65%/B液容量35%まで急速に上昇させ、これを次に、流速1ml/minで、60分間かけてA液容量50%/B液容量50%まで直線的グラジエントで上昇させ溶出液を回収した。溶出液は、210nmの紫外吸収では単一なピークとして検出された。
溶出液を、500 μlずつフラクション番号をつけて分取し、分取フラクションより各10 μlづつを0.2% BSA 150 μlと混合し凍結乾燥機(12EL;VirTis社)で凍結乾燥させた。この乾燥物に、2.5 mM Probenecid、 0.1% CHAPSを含むH/HBSS 75 μlを加えて溶解し、この溶液50 μlを用いて上記(3−5)の試験法により、ZAQC−B1細胞に対する受容体活性化作用を測定した。その結果、目的とするZAQC−B1細胞に対する受容体活性化作用を有する成分、すなわち、ZAQ活性化成分は、フラクションNo.82-84に溶出されていることが判明した。この活性ピークは、210nmの紫外吸収ピークに完全に一致し、単一ペプチドにまで精製されたものと判断した。
【0106】
(3−8)精製されたZAQ活性化ペプチドの構造解析
上記(3−7)で得られたZAQ活性化成分について以下の方法で構造決定を実施した。ZAQ活性化成分精製標品を凍結乾燥し、得られた凍結乾燥物を溶媒DMSO(ジメチルサルフォキシド)に溶解した。この溶液の一部をプロテインシークエンサー(パーキンエルマー社、PE Biosystems Procise 491cLC)を用いたN末端からのアミノ酸配列解析に供した。その結果、N末端のアミノ酸残基から16番目のアミノ酸残基のうち、14残基を同定することができた(Ala Val Ile Thr Gly Ala Xaa Glu Arg Asp Val Gln Xaa Arg Ala Gly (配列番号:11;Xaaは未同定残基))。
【0107】
実施例4 ヒト型ZAQリガンドぺプチドのcDNAのクローニング
実施例3で得られた牛乳から精製されたZAQを活性化するペプチドのN末端アミノ酸配列(配列番号:11)をクエリーとしてデータベースをBlast検索したところ、配列番号:11で表わされるアミノ酸配列を有するペプチドをコードするDNAの塩基配列と同等な配列を含むヒトEST(X40467)を見出した。本配列は完全長のオープンリーディング・フレームを有していなかったので、以下にRACE法により未確定部分の配列を明らかにし、引き続いて完全長のオープンリーディング・フレームを有すcDNAクローンを取得した。
EST(X40467)の情報よりプライマーZF1(配列番号:12)、ZF2(配列番号:13)とZF3(配列番号:14)を作成し、ヒト精巣Marathon-Ready cDNA (CLONTECH社)を鋳型として以下に記した3'RACE実験を実施した。
ZF1: 5'-GGTGCCACGCGAGTCTCAATCATGCTCC-3' (配列番号:12)
ZF2: 5'-GGGGCCTGTGAGCGGGATGTCCAGTGTG-3' (配列番号:13)
ZF3: 5'-CTTCTTCAGGAAACGCAAGCACCACACC-3' (配列番号:14)
3'RACEのPCR反応液は50 x Advantage 2 Polymerase Mix (CLONTECH社)を1 μl、添付の10 x Advantage 2 PCR buffer (400 mM Tricine-KOH, 150 mM KOAc, 35 mM Mg(OAc)2 , 37.5μg/ml BSA, 0.05%Tween-20, 0.05% Nonidet-P40)を5 μl、dNTP mixture (2.5 mM each, 宝酒造)を4 μl、10 μMプライマーZF1を 1 μl、10 μMプライマーAP1(プライマーAP1はCLONTECH社のMarathon-Ready cDNA Kitに添付のもの)を1 μl、鋳型cDNA(CLONTECH社、ヒト精巣Marathon-Ready cDNA)を5 μl、及び蒸留水を33 μlを混合して作製した。反応条件は94℃・60秒の初期変性後、94℃・30秒-72℃・4分のサイクル反応を5回、94℃・30秒-70℃・4分のサイクル反応を5回、94℃・30秒-68℃・44分のサイクル反応を25回行った。
続いて、当該PCR反応の反応液を鋳型としてnested PCRを実施した。反応液は50 x Advantage 2 Polymerase Mix (CLONTECH社)を1 μl、添付の10 x Advantage 2 PCR buffer (400 mM Tricine-KOH, 150 mM KOAc, 35 mM Mg(OAc)2, 37.5μg/ml BSA, 0.05%Tween-20, 0.05% Nonidet-P40)を5 μl、dNTP mixture (2.5 mM each, 宝酒造)を4 μl、10 μMプライマーZF2を 1 μl、10 μMプライマーAP2(プライマーAP2はCLONTECH社のMarathon-Ready cDNA Kitに添付のもの)を1 μl、鋳型DNA(当該PCR反応液50倍希釈液)を5 μl、及び蒸留水を33 μlを混合して作製した。反応条件は94℃・60秒の初期変性後、94℃・30秒-72℃・4分のサイクル反応を5回、94℃・30秒-70℃・4分のサイクル反応を5回、94℃・30秒-68℃・44分のサイクル反応を25回行った。
さらに続いて、当該PCR反応の反応液を鋳型として2回目のnested PCRを実施した。反応液は50 x Advantage 2 Polymerase Mix (CLONTECH社)を1 μl、添付の10 x Advantage 2 PCR buffer (400 mM Tricine-KOH, 150 mM KOAc, 35 mM Mg(OAc)2 , 37.5μg/ml BSA, 0.05%Tween-20, 0.05% Nonidet-P40)を5 μl、dNTP mixture (2.5 mM each, 宝酒造)を4 μl、10 μMプライマーZF3を 1 μl、10 μMプライマーAP2(プライマーAP2はCLONTECH社のMarathon-Ready cDNA Kitに添付のものを用いた。)を1 μl、鋳型DNA(当該PCR反応液50倍希釈液)を5 μl、及び蒸留水を33 μlを混合して作製した。反応条件は94℃・60秒の初期変性後、94℃・30秒-72℃・4分のサイクル反応を5回、94℃・30秒-70℃・4分のサイクル反応を5回、94℃・30秒-68℃・44分のサイクル反応を25回行った。得られたDNA断片をTOPO TA Cloning Kit (Invitrogen社)を用いて添付のマニュアルに記載された方法に従ってクローニングした。クローニングされたDNAの塩基配列をABI377DNA sequencerを用いて解読し、3'端配列(配列番号:15)を得た。
配列番号:15で表わされる塩基配列及びEST(X40467)の情報によりプライマーZAQL-CF(配列番号:16)及びZAQL-XR1(配列番号:17)を作成した。ヒト精巣Marathon-Ready cDNA (CLONTECH社)を鋳型としてプライマーZAQL-CF とZAQL-XR1を用いてPCRを実施した。
ZAQL-CF: 5'-CCACCATGAGAGGTGCCACG-3' (配列番号:16)
ZAQL-XR1: 5'-CTCGAGCTCAGGAAAAGGATGGTG-3' (配列番号:17)
PCR反応液はPfuTurbo DNA polymerase(Stratagene社)を1 μl、添付の10 x PCR bufferを5 μl、2.5 mM dNTP mixtureを4 μl、10 μMプライマーZAQL-CF及びZAQL-XR1を各2.5 μl、鋳型DNAを5μl、及び蒸留水を30 μlを混合して作製した。反応条件は95℃・1分の初期変性後、95℃・1分-60℃・1分-72℃・1分のサイクル反応を40回、および72℃・10分の最終伸長反応とした。得られたDNA断片をTOPO TA Cloning Kit (Invitrogen社)を用いて添付のマニュアルに記載された方法に従ってクローニングした。クローニングされたDNA断片の塩基配列をABI 377 DNA sequencerを用いて解読した結果、371bpの、それぞれ配列番号:18および配列番号:19で表わされる塩基配列を有していることが明らかとなった。配列番号:18で表わされる塩基配列を含有するDNA断片を有するプラスミドをpHMITAと、配列番号:19で表わされる塩基配列を含有するDNA断片を有するプラスミドをpHMITGと命名した。
プラスミドpHMITA及びpHMITGにより大腸菌(Escherichia coli)を形質転換し、それぞれエシェリヒア・コリ(Escherichia coli)TOP10/pHMITAおよびエシェリヒア・コリ(Escherichia coli)TOP10/pHMITGと命名した。
これらのDNA断片の塩基配列を解析した結果、配列番号:18で表わされるDNA断片は、配列番号:22で表わされるヒト型ZAQリガンド前駆体ペプチド(Aタイプ、105アミノ酸残基)をコードするDNA(配列番号:28)を含んでおり、配列番号:19で表わされるDNA断片は、配列番号:23で表わされるヒト型ZAQリガンド前駆体ペプチド(Gタイプ、105アミノ酸残基)をコードするDNA(配列番号:29)を含んでいることが明らかとなった。
また、配列番号:28および配列番号:29で表わされる塩基配列は典型的なシグナル配列を有しており、配列番号:28で表わされる塩基配列を含有するDNAは、配列番号:20で表わされるヒト型ZAQリガンド成熟体ペプチド(Aタイプ、86アミノ酸残基)をコードする258塩基対からなるDNA(配列番号:26)を含んでおり、配列番号:29で表わされる塩基配列を含有するDNAは、配列番号:21で表わされるヒト型ZAQリガンド成熟体ペプチド(Gタイプ、86アミノ酸残基)をコードする258塩基対からなるDNA(配列番号:27)を含んでいることが明らかとなった。
【0108】
実施例5 ヒト型ZAQリガンドペプチドの哺乳動物細胞での産生(1)
(5−1)ヒト型ZAQリガンド前駆体ペプチド哺乳動物細胞発現ベクターの構築
実施例4において取得したプラスミドpHMITGからEcoRI、XhoI制限酵素消化によってヒト型ZAQリガンド前駆体ペプチドをコードするcDNAを含む382bpのDNA断片(配列番号:30)を切出した。
すなわち、プラスミドpHMITGをEcoRIおよびXhoIで酵素消化し、得られたDNAを1.5%アガロースゲルを用いて電気泳動し、サイバーグリーン染色される約382bpのバンドを含むゲル片を剃刀で切り取った。当該ゲル片よりGene Clean spin DNA抽出キット(BIO 101社)を用いてDNA断片を回収した。得られたDNA断片をCMV-IEエンハンサーおよびchicken beta-actin promoterを発現プロモーターとする哺乳動物細胞発現ベクターpCAN618(図11)に対してEcoRI、XhoI制限酵素切断部位に定法に従ってクローニングした。クローニングされたDNA断片の塩基配列を前述の方法により解読した結果、配列番号:30で表わされる塩基配列を有していることが確認された。このヒト型ZAQリガンド前駆体ペプチドをコードするDNAを含有する哺乳動物細胞発現ベクターをpCANZAQLg2と命名した。
【0109】
(5-2)COS7細胞への発現ベクターの導入
COS7細胞はATCCより購入し、DMEM培地(10% FBSを加えたもの)を用いて継代培養しているものを用いた。DMEM培地を用いてCOS7細胞を1.5×106cells/dishとなるよう10cmシャーレにまき、37℃、5% CO2インキュベーター中で一晩培養した。ヒト型ZAQリガンド前駆体ペプチド発現プラスミド(pCANZAQLg2)2μg(2μlのTEバッファーに溶解)にバッファーEC(Effectene transfection reagent、QIAGEN)298μlを加え、さらにEnhancer 16μlを加え、1秒間混和後室温で3分間放置した。さらにEffectene Transfection Reagent 60μlを加え、10秒間混和後室温で10分間放置した。前日にまいた細胞の上清を除き、DMEM培地10mlで1回洗浄し、DMEM培地を9mlを加えた。プラスミド溶液にDMEM培地1mlを加えて混和後細胞に滴下し、全体を混ぜた後37℃、5% CO2インキュベーター中で一晩培養した。DMEM培地10mlで2回洗浄し、DMEM培地10mlを加え、37℃、5% CO2インキュベーター中で一晩培養した。2日後、培養上清を回収した。
【0110】
(5−3)ヒト型ZAQリガンド前駆体ペプチド発現COS7細胞培養上清からのZAQを活性化するペプチドの部分精製
(5−3−1)ヒト型ZAQリガンド前駆体ペプチド発現COS7細胞培養上清抽出液の調製
ヒト型ZAQリガンド前駆体ペプチド発現COS7細胞培養上清を回収し、以下の操作を行い抽出液を調製した。先ず、細胞培養上清(約18.5ml)に終濃度が1 Mになるように酢酸1.1mlを滴下し、一時間攪拌した。さらにその2倍容量のアセトンを加え、4℃にて30分間攪拌し、次いで高速遠心機(CR26H、23型ローター:日立株式会社)を用いて15,000 rpm、30分間遠心し上清を得た。得られた上清をエバポレーターにかけ、アセトンを除去した後、凍結乾燥機(12EL; VirTis社)にて凍結乾燥した。
【0111】
(5−3−2)ヒト型ZAQリガンド前駆体ペプチド発現COS7細胞培養上清のSephadex G50ゲルろ過クロマトグラフィー及びSepPakカラムクロマトグラフィー
上記(5−3−1)で得られた凍結乾燥粉末を1M酢酸2mlに溶解後、1M酢酸で平衡化したSephadex G15 (直径3cm、35ml、Pharmacia Biotech 社)カラムに吸着させた後、1M酢酸をカラムに流し、溶出液を5mlづつフラクションNo.をつけて分取し、凍結乾燥機(12EL; VirTis社)で凍結乾燥させた。
SepPak C18-5gカラム(10ml)を、メタノールにて膨潤後、0.1% トリフルオロ酢酸/蒸留水を流し、平衡化した。Sephadex G50ゲルろ過クロマトグラフィー分取フラクションのうちフラクションNo.1-16の凍結乾燥品をまとめて0.1%トリフルオロ酢酸/蒸留水 3mlに溶解し、SepPak C18-5gカラムに添着した後、0.1% トリフルオロ酢酸/蒸留水 24mlで洗浄後、0.1% トリフルオロ酢酸/60% アセトニトリル20mlで溶出した。得られた溶出液をサーバントにかけた。
【0112】
(5−3−3)Super ODS逆相高速液体クロマトグラフィーによる精製
TSKgel Super ODS逆相高速液体クロマトグラフィー用カラム(東ソー株式会社、 0.46 cm x 10 cm)を、40℃にて、流速1 ml/minでA液(0.1% トリフルオロ酢酸/蒸留水)を流し、平衡化した。(5−3−2)で得られたSepPak C18-5gカラムフラクションをサーバントにかけた後、Super ODS逆相高速液体クロマトグラフィーに添着し、流速 1ml/minで60分間で A液(0.1% トリフルオロ酢酸/蒸留水)容量100%/B液(0.1% トリフルオロ酢酸/60% アセトニトリル)容量0%からA液容量0%/B液容量100%まで直線的グラジエントで上昇させ、溶出液を回収した。
溶出液を、1 mlずつフラクションNo.をつけて分取し、分取フラクション全量を凍結乾燥機(12EL; VirTis社)で凍結乾燥させた。この乾燥物にH/HBSSに2.5mM Probenecid 、0.2% BSAを加えたもの150μlを加えて溶解し、この溶液を用いて下記(5−3−4)の試験法により、ZAQC−B1細胞に対する受容体活性化作用を測定した。
【0113】
(5−3−4)FLIPRを用いた細胞内Ca2+イオン濃度上昇活性の測定
上記(5−3−3)で得られたサンプルについて、実施例3(3−5)で得られたZAQ発現細胞(ZAQC−B1)における細胞内Ca2+イオン濃度上昇活性の測定をFLIPRを用いて行った。また、対照としてhOT7T175発現細胞(hOT7T175-16;WO00/24890に記載)を用いた。
ZAQC−B1細胞、hOT7T175-16細胞共に10%透析処理済ウシ胎児血清(以後d FBSとする)を加えたDMEMで継代培養しているものを用いた。ZAQC−B1細胞、hOT7T175-16細胞をそれぞれ15×104cells/mlとなるように培地(10%dFBS-DMEM)に懸濁し、FLIPR用96穴プレート(Black plate clear bottom、Coster社)に分注器を用いて各ウェルに200μlずつ播き(3.0×104cells/200μl/ウェル)、5% CO2インキュベーター中で37℃で一晩培養した後、用いた(以後細胞プレートとする)。H/HBSS(HANKS' 9.8g、炭酸水素ナトリウム 0.35g、HEPES 4.77 g 、水酸化ナトリウムで pH7.4に合わせた後、フィルター滅菌処理)21ml、250mM Probenecid 210μl、ウシ胎児血清(FBS) 210μlを混合した。また、Fluo3-AM 2バイアル(50μg)をジメチルスルフォキサイド 42μl、20% Pluronic acid 42μlに溶解し、これを上記H/HBSS−Probenecid−FBS に加え、混和後、8連ピペットを用いて培養液を除いた細胞プレートに各ウェル 100μlずつ分注し、5% CO2インキュベーター中で37℃で1時間インキュベートした(色素ローディング)。上記(5−3−3)で得られたアッセイ用サンプルについて、各フラクションにH/HBSSに2.5mM Probenecid 、0.2% BSAを加えたもの150μlを加えて溶解し、FLIPR用96穴プレート(V-Bottomプレート、Coster社)へ移した(以後、サンプルプレートとする)。細胞プレートの色素ローディング終了後、H/HBSSに2.5mM Probenecidを加えた洗浄バッファーでプレートウォッシャー(Molecular Devices社)を用いて細胞プレートを4回洗浄し、洗浄後100μlの洗浄バッファーを残した。この細胞プレートとサンプルプレートをFLIPRにセットし、アッセイを行った(FLIPRにより、サンプルプレートから0.05mlのサンプルが細胞プレートへと移される)。フラクションNo.48-68にZAQC−B1細胞特異的な細胞内Ca2+イオン濃度上昇活性が見られた。このことから、目的とするZAQC−B1細胞に対する受容体活性化作用を有する成分、すなわち、ZAQ活性化成分は、フラクションNo.48-68に溶出されていることが判明した。
【0114】
実施例6 ヒト型ZAQリガンドペプチドの哺乳動物細胞での産生(2)
(6−1)培養上清の調製
実施例5に記載した方法でCOS7細胞にヒト型ZAQリガンド前駆体ペプチド発現プラスミド(pCANZAQLg2)を導入した。すなわち、DMEM培地を用いてCOS7細胞を3.0×106cells/dishとなるよう15cmシャーレにまき、37℃、5% CO2インキュベーター中で一晩培養した。ヒト型ZAQリガンド前駆体ペプチド発現プラスミド(pCANZAQLg2)4μg(4μlのTEバッファーに溶解)にバッファーEC(Effectene transfection reagent、QIAGEN)600μlを加え、さらにEnhancer 32μlを加え、1秒間混和後室温で3分間放置した。さらにEffectene Transfection Reagent 120μlを加え、10秒間混和後室温で10分間放置した。前日にまいた細胞の上清を除き、DMEM培地10mlで1回洗浄し、DMEM培地を30mlを加えた。プラスミド溶液にDMEM培地1mlを加えて混和後細胞に滴下し、全体を混ぜた後37℃、5% CO2インキュベーター中で一晩培養した。DMEM培地10mlで1回洗浄し、DMEM培地20mlを加え、37℃、5% CO2インキュベーター中で一晩培養した。1日後、培養上清を回収し、さらにDMEM培地20mlを加え、37℃、5% CO2インキュベーター中で一晩培養した後培養上清を回収した。
【0115】
(6−2)培養上清からのヒト型ZAQリガンドペプチドの精製
(6−1)に記載した方法で15cmシャーレ80枚分の培養上清を回収し、これに酢酸を終濃度1Mになるように添加した。1時間攪拌した後、2倍容のアセトンを添加しタンパク質を析出させた。4℃にて30分間攪拌し、次いで高速遠心機(CR26H、RR10A 型ローター:日立株式会社)を用いて10,000 rpm、30分間遠心し上清を得た。得られた上清をエバポレーターにかけアセトンを除去し、あらかじめ0.1%トリフルオロ酢酸/蒸留水で平衡化した逆相カラム(Waters社C18、100 g)に流した。0.1%トリフルオロ酢酸/蒸留水 1000ml、次いで0.1%トリフルオロ酢酸/20%アセトニトリル1000mlでカラムを洗浄した後、0.1%トリフルオロ酢酸/60%アセトニトリル1000mlでペプチドを溶出した。得られた溶出液をエバポレーターにかけた後、凍結乾燥器(12EL; VirTis社)にて凍結乾燥した。
TSKgel ODS80TM逆相高速液体クロマトグラフィー用カラム(東ソー株式会社、21.5 mm x 30 cm)を、40℃にて、流速4ml/minでA液(0.1%トリフルオロ酢酸/蒸留水)を流し、平衡化した。得られた凍結乾燥粉末をA液に溶解した後、当該ODS80TMカラムに添着し、流速4ml/minで120分間に A液(0.1%トリフルオロ酢酸/蒸留水)容量60%/B液(0.1%トリフルオロ酢酸/60%アセトニトリル)容量40%からA液容量0%/B液容量100%まで直線的グラジエントで上昇させて、ペプチドを溶出させた。
溶出液を、8mlずつフラクションNo.をつけて分取し、分取フラクションから50 μlを取り凍結乾燥機(12EL; VirTis社)で凍結乾燥させた。この乾燥物にH/HBSSに2.5mM Probenecid 、0.2% BSAを加えたもの200μlを加えて溶解し、この溶液を用いて上記(5−3−4)の試験法により、ZAQC−B1細胞に対する受容体活性化作用を測定した。その結果、目的とするZAQC−B1細胞に対する受容体活性化作用を有する成分、すなわち、ZAQ活性化成分は、フラクションNo. 32に溶出されていることが判った。
TSKgel CM-2SWイオン交換高速液体クロマトグラフィー用カラム(東ソー株式会社、4.6 mm x 25 cm)を、25℃にて、流速1ml/minでA液(10 mMぎ酸アンモニウム/10%アセトニトリル)を流し、平衡化した。上記フラクションNo.32を当該CM-2SWカラムに添着し、流速1ml/minで60分間にA液(10 mMぎ酸アンモニウム/10% アセトニトリル)容量100%/B液(1000 mMぎ酸アンモニウム/10% アセトニトリル)容量0%からA液容量0%/B液容量100%まで直線的グラジエントで上昇させて、ペプチドを溶出させた。
溶出液を、1mlずつフラクションNo.をつけて分取し、分取フラクションから1.5 μlを取り、これをH/HBSSに2.5mM Probenecid 、0.2% BSA 200μl希釈し、この溶液を用いて上記(5−3−4)の試験法により、ZAQC−B1細胞に対する受容体活性化作用を測定した。その結果、目的とするZAQC−B1細胞に対する受容体活性化作用を有する成分、すなわち、ZAQ活性化成分は、フラクションNo. 56および57に溶出されていることが判った。
TSKgel Super phenyl逆相高速液体クロマトグラフィー用カラム(東ソー株式会社、4.6 mm x 10 cm)を、40℃にて、流速1ml/minでA液(0.1% トリフルオロ酢酸/蒸留水)を流し、平衡化した。上記フラクション No.56および57を当該Super phenylカラムに添着し、流速1ml/minで60分間にA液(0.1% トリフルオロ酢酸/蒸留水)容量70%/B液(0.1% トリフルオロ酢酸/60% アセトニトリル)容量30%からA液容量50%/B液容量50%まで直線的グラジエントで上昇させて、ペプチドを溶出させた。
溶出液を、1mlずつフラクションNo.をつけて分取し、分取フラクションから1.5 μlを取り、これをH/HBSSに2.5mM Probenecid 、0.2% BSA 200μl希釈し、この溶液を用いて上記(5−3−4)の試験法により、ZAQC−B1細胞に対する受容体活性化作用を測定した。その結果、目的とするZAQC−B1細胞に対する受容体活性化作用を有する成分、すなわち、ZAQ活性化成分は、フラクションNo. 54、 55および56に溶出されていることが判った。本活性は単一な紫外吸収ピークと一致し、活性成分が単一にまで精製されたものと判断した。
ZAQ活性化成分精製標品中の溶媒を凍結乾燥して除去し、得られた凍結乾燥物を溶媒DMSO(ジメチルサルフォキシド)に溶解した。この溶液の一部(約7.5 pmol)をプロテインシークエンサー(パーキンエルマー社、PE Biosystems Procise 491cLC)を用いたN末端アミノ酸配列解析に供した。その結果、N末端のアミノ酸残基から10番目のアミノ酸残基のうち、9残基を同定することができた(Ala Val Ile Thr Gly Ala Xaa Glu Arg Asp (配列番号:31;Xaaは未同定残基))。得られたアミノ酸配列は、予想されるヒト型ZAQリガンド成熟体ペプチドのN端アミノ酸配列と一致した。また、ZAQ活性化成分精製標品の質量分析を Finnigan LCQ LC/MS装置(Thermoquest, San Jose, CA)を用いて、エレクトロスプレーイオン化法により実施し、分子量が9657.6であることを確認した。これは10個のシステイン残基がすべてジスルフィド結合を形成した86残基のヒト型ZAQリガンド成熟体ペプチド(配列番号:21)の理論値9657.3に良く一致し、ZAQ活性化成分精製標品が、配列番号:21で表わされるアミノ酸配列を有するヒト型ZAQリガンド成熟体ペプチドを有していることが確認された。
【0116】
(6−3)精製ヒト型ZAQリガンドペプチドのZAQ活性化作用の測定
上記(6−2)で精製したヒト型ZAQリガンド成熟体ペプチドのZAQC−B1細胞に対する受容体活性化作用を上記(5−3−4)の試験法により測定した。その結果、ZAQ発現CHO細胞(ZAQC−B1細胞)においてヒト型ZAQリガンド成熟体ペプチドは濃度依存的に細胞内カルシウム濃度の上昇を惹起した。EC50値は96 pMで、ヒト型ZAQリガンド成熟体ペプチドは非常に強いアゴニスト活性を示すことが明らかとなった。結果を図10に示す。
【0117】
実施例7 ヒト型ZAQリガンドペプチドの哺乳動物細胞での産生(3)
(7−1)ヒト型ZAQリガンドペプチド安定発現CHO細胞株の樹立
実施例4に記載したプラスミドpHMITGを鋳型として下記のプライマー
5' GTCGACCACCATGAGAGGTGCCACGC 3' (配列番号:32)
5' ACTAGTCGCAGAACTGGTAGGTATGG 3' (配列番号:33)
を用いてヒト型ZAQリガンドcDNAをPCR増幅し、pCR Blunt IIベクター(Invitrogen社)にクローニングした。得られた正しい配列を有するクローンからインサートcDNAをSalI及びSpeI制限酵素を用いて切り出し、pAKKO1.11H発現ベクターに組み込んだ。当該プラスミドをCHO/dhFr-細胞(American Type Culture Collection)に、上記(3−5)に記載したCellPhect Transfection kit(Amersham Pharmacia Biotech社)を用いた方法に従って形質導入した。形質導入株数クローンから培養上清を回収し、(3−5)に記載した試験方法により、ZAQC−B1細胞の細胞内Ca2+イオン濃度を上昇させるZAQリガンド活性を測定した。これによりZAQリガンドを高発現するZAQL−1発現CHO細胞クローンNo.4を選別した。
(7−2)ヒト型ZAQリガンド(ZAQL−1)発現CHO細胞無血清培養上清の調製
透析済み10%ウシ胎児血清(JRH BIOSCIENCES 社)、1 mM MEM非必須アミノ酸溶液、100 units/ml Penicillin、100 μg/ml Streptomycinを含む Dulbecco's Modified Eagle Medium (DMEM) 培地(日水製薬株式会社)でSingle Tray(Nunc社)4枚コンフルエントまで培養したZAQL−1発現CHO細胞クローンNo.4を、トリプシン処理して分散後、遠心して回収した。上記Single Tray 1枚分の細胞を上記培地1.5 Lに懸濁後Cell Factories 10(Nunc社)に植え込み、4基のCell Factories 10について37℃で5%炭酸ガス中にて3日間培養した。培養上清を除いた後、前述のH/HBSS 1 Lで1基のCell Factories 10の細胞を洗浄した。H/HBSSを除いた後、1基のCell Factories 10あたり2 Lの無血清培地(1 mM MEM非必須アミノ酸溶液、100 units/ml Penicillin、100 μg/ml Streptomycinを含む Dulbecco's Modified Eagle Medium培地)を加えさらに2日間培養した。回収した培養上清を日立高速遠心機で1,000 rpmで10分間遠心後、ガーゼを用いてろ過し清澄な上清を得た。これに酢酸を最終濃度1 Mになるように添加し以下の操作を行った。
【0118】
(7−3)ZAQL−1発現CHO細胞無血清培養上清のオクタドデシル逆相クロマトグラフィーによる粗分画
オクタドデシル基を固定したシリカゲルを充填したPrepC18(Waters社)をメタノールで膨潤後、ガラス製カラムに充填した(50 mm×100 mm)。その後、1 M酢酸で平衡化したカラムに、(6−2)で調製した抽出液を添着した。次にこのカラムを800mlの1M酢酸で洗浄した。次に、このカラムに1000mlの60%アセトニトリル/0.1%トリフルオロ酢酸を流し、目的とする粗ペプチド成分を溶出した。得られた溶出液を、エバポレーターを用いて濃縮した後、凍結乾燥機(12EL; VirTis社)にて凍結乾燥した。
(7−4)Wakosil−II 5C18HG Prep逆相高速液体クロマトグラフィーによる分離
Wakosil-II 5C18HG Prep逆相高速液体クロマトグラフィー用カラム(和光純薬、20mm×250mm)を、40℃にて、流速5ml/minでA液(0.1%トリフルオロ酢酸/蒸留水)容量91.7%/B液(0.1%トリフルオロ酢酸/60%アセトニトリル)容量8.3%を流し平衡化した。上記(7−3)で得られた凍結乾燥物についてクロマトグラフィー操作を行った。即ち、凍結乾燥物を1M酢酸36mlを加えて溶解し遠心後、その内の1/3を当該カラムに添着した後、流速5ml/mlで、1分間かけてA液容量66.7%/B液容量33.3% まで上昇させ、次いで120分間かけてA液容量16.7%B液容量83.3%まで、B液濃度を直線的グラジエントで上昇させた。溶出液を5mlずつフラクション番号をつけて分取した。分取フラクションより各3 μlづつ0.2% BSA 150 μl と混合し凍結乾燥機(12EL; VirTis社)で凍結乾燥させた。この乾燥物に、アッセイバッファー〔H/HBSS(ニッスイハンクス2(日水製薬株式会社) 9.8g、炭酸水素ナトリウム 0.35g、HEPES 4.77 g 、水酸化ナトリウム溶液で pH7.4に合わせた後、フィルター滅菌処理)に、2.5 mM Probenecid および0.1% CHAPS を添加したもの〕150 μlを加えて溶解し、この溶液50 μl を用いて上記(3−5)の試験法に従い、ZAQ−B1受容体活性化作用を測定した。その結果、目的とするZAQ−B1受容体活性化作用を有する成分は、主としてフラクションNo.73-75に溶出されていることが判明した。
【0119】
(7−5)TSKgel CM−2SWイオン交換高速液体クロマトグラフィーによる分離
TSKgel CM-2SWイオン交換高速液体クロマトグラフィー用カラム(東ソー、7.8×300mm)を25℃にて、流速2 ml/minでA液(4 Mギ酸アンモニウム:蒸留水:アセトニトリル = 1 : 299 : 100)容量100%、B液(4 Mギ酸アンモニウム:蒸留水:アセトニトリル = 1 : 2 : 1)容量0%を流し平衡化した。(7−4)で得られたWakosil-II 5C18HG Prep逆相高速液体クロマトグラフィー分取フラクションのうちフラクションNo.73-75を凍結乾燥したものをA液4 mlに溶解しTSKgel CM-2SWイオン交換カラムに添着した後、流速1ml/minで120分間かけてA液容量25%/B液容量75% まで直線的グラジエントで上昇させ溶出液を回収した。溶出液を2mlずつフラクション番号をつけて分取し、分取フラクションより各10 μlづつを0.2% BSA 100 μl 混合し凍結乾燥機(12EL; VirTis社)で凍結乾燥させた。この乾燥物に上記アッセイバッファー100 μl を加えて溶解しこれをさらに同バッファーで100倍希釈し上記(3−5)の試験法に従い、ZAQ受容体活性化作用を測定した。その結果、目的とするZAQ受容体活性化作用を有する成分はフラクションNo.95-100に溶出されていることが判明した。
(7−6)TSKgel ODS−80Ts逆相高速液体クロマトグラフィーによる精製
TSKgel ODS-80Ts 逆相高速液体クロマトグラフィー用カラム(東ソー、4.6 mm x 100 mm)を、40℃にて、流速1ml/minでA液(0.1% トリフルオロ酢酸/蒸留水)容量91.7%/B液(0.1% トリフルオロ酢酸/60% アセトニトリル)容量8.3%を流し平衡化した。上記(7−5)で得られたフラクションNo.95-100の凍結乾燥物についてクロマトグラフィー操作を行った。即ち、凍結乾燥物を1 M酢酸4 mlを加えて溶解し当該カラムに添着した後、流速1ml/minで、1分間かけてA液容量75%/B液容量25%まで上昇させ、次いで60分間かけてA液容量25%B液容量75%までB液濃度を直線的グラジエントで上昇させ溶出液を回収した。溶出液を1mlずつフラクション番号をつけて分取した。(3−5)に記載した方法に従いZAQリガンド活性を測定し、当該活性が単一な紫外吸収ピークと一致するフラクションに溶出されていることを確認した。これよりZAQリガンドが単一に精製されたものと判断した。
【0120】
(7−7)精製されたZAQリガンドペプチドの構造解析
上記(7−6)で得られたZAQリガンドペプチドについて以下の方法で構造決定を実施した。ZAQ活性化主成分精製標品中の凍結乾燥機(12EL; VirTis社)にて凍結乾燥した。得られた凍結乾燥物を溶媒DMSO(ジメチルサルフォキシド)に溶解した。この溶液の一部をプロテインシークエンサー(パーキンエルマー社、PE Biosystems Procise 491cLC)を用いたN末端からのアミノ酸配列解析に供した。その結果、予想されるヒト方ZAQリガンドペプチド成熟体(配列番号:21)と一致するN端アミノ酸配列を得た。また、Finnigan LCQ LC/MS装置を用いて、エレクトロンスプレーイオン化法により質量分析を行い、分子量が9658.0であると算定した。本測定値はヒト型ZAQリガンド成熟体ペプチド(配列番号:21)の理論値9657.3にと良く一致し、目的とする配列番号:21で表わされるアミノ酸配列を含有するペプチドが取得できたことが確認された。
【0121】
実施例8 ヘビ毒MIT1およびヒト型ZAQリガンドとZAQおよびI5Eとの反応性
(8−1)ヘビ毒MIT1の単離
(8−1−1)Wakosil−II 5C18HG Prep逆相高速液体クロマトグラフィーによる分離
Wakosil-II 5C18HG Prep逆相高速液体クロマトグラフィー用カラム(和光純薬、 20mm×250mm)を、40℃にて、流速5 ml/minでA液(0.1% トリフルオロ酢酸/蒸留水)容量91.7%/B液(0.1% トリフルオロ酢酸/60% アセトニトリル)容量8.3%を流し平衡化した。Black Mamba毒液(Sigma社)凍結乾燥品50 mgに1 M AcOH 4 mlを加え溶解し、15,000 rpmで10分間遠心して得られた上清についてクロマトグラフィー操作を行った。サンプルを当該カラムに添着した後、流速5 ml/minで、1分間かけてA液容量91.7%/B液容量8.3% まで上昇させ、次いで120分間かけてA液容量33.4%B液容量66.6%まで、B液濃度を直線的グラジエントで上昇させた。溶出液を20分後から10 mlずつフラクション番号をつけて分取した。各フラクションから1 μlを採取し、前述のアッセイバッファーで10,000倍希釈後、上記(3−5)の試験法に従い、ZAQ−B1受容体活性化作用を測定した。その結果、目的とするZAQ-B1受容体活性化作用を有する成分は、主としてフラクションNo.21-23に溶出されていることが判明した。
(8−1−2)TSKgel CM−2SWイオン交換高速液体クロマトグラフィーによる分離
TSKgel CM-2SWイオン交換高速液体クロマトグラフィー用カラム(東ソー、4.6×250 mm)を25℃にて、流速1 ml/minでA液(4 Mギ酸アンモニウム:蒸留水:アセトニトリル = 1 : 299 : 100)容量100%、B液(4 Mギ酸アンモニウム:蒸留水:アセトニトリル = 1 : 2 : 1)容量0%を流し平衡化した。(7−1)で得られたWakosil-II 5C18HG Prep逆相高速液体クロマトグラフィー分取フラクションのうちフラクションNo.21-23を凍結乾燥したものをA液4 mlに溶解しTSKgel CM-2SWイオン交換カラムに添着した後、流速 1 ml/minで90分間かけてA液容量0%/B液容量100%まで直線的グラジエントで上昇させ溶出液を回収した。溶出液を1 mlずつフラクション番号をつけて分取した。各フラクションから1 μlを採取し、前述のアッセイバッファーで10,000倍希釈後、上記(3−5)の試験法に従い、 ZAQ-B1受容体活性化作用を測定した。その結果、目的とするZAQ-B1受容体活性化作用を有する成分は、主としてフラクションNo.50-51に溶出されていることが判明した。
【0122】
(8−1−3)Vydac238TP3410逆相高速液体クロマトグラフィーによる分離
Vydac238TP3410逆相高速液体クロマトグラフィー用カラム(Vydac、 4.6mm×100 mm)を、40℃にて、流速1 ml/minでA液(0.1% トリフルオロ酢酸/蒸留水)容量91.7%/B液(0.1% トリフルオロ酢酸/60% アセトニトリル)容量8.3%を流し平衡化した。(8−1−2)で得られたTSKgel CM-2SWイオン交換高速液体クロマトグラフィー分取フラクションのうちフラクションNo.50-51を直接当該カラムに添着した後、流速1 ml/minで、1分間かけてA液容量75%/B液容量25% まで上昇させ、次いで75分間かけてA液容量41.7%B液容量58.3%まで、B液濃度を直線的グラジエントで上昇させた。溶出液を0.5 mlずつフラクション番号をつけて分取した。各フラクションから1 μlを採取し、前述のアッセイバッファーで10,000倍希釈後、上記(3−5)の試験法により、ZAQ−B1受容体活性化作用を測定した。その結果、目的とするZAQ−B1受容体活性化作用を有する成分は、主としてフラクションNo.108-115に溶出されていることが判明した。
(8−1−4)精製されたヘビ毒MIT1の構造解析
上記(8−1−3)で得られたヘビ毒MIT1について以下の方法で構造決定を実施した。 精製したMIT1を凍結乾燥機(12EL;VirTis社)にて凍結乾燥した。得られたペプチド凍結乾燥物を溶媒DMSO(ジメチルサルフォキシド)に溶解した。この溶液の一部をプロテインシークエンサー(パーキンエルマー社、PE Biosystems Procise 491cLC)を用いたN末端からのアミノ酸配列解析に供した。その結果、予想されるヘビ毒MIT1(配列番号:34)と一致するN端アミノ酸配列を得た。また、Finnigan LCQ LC/MS装置を用いて、エレクトロンスプレーイオン化法により質量分析を行い、分子量が8506.8であると算定した。本測定値はヘビ毒MIT1の理論値8506.4に良く一致し、目的とする配列番号:34で表わされるアミノ酸配列を含有するペプチドが取得できたことが確認された。
【0123】
(8−2)I5E安定発現細胞株の樹立
ヒト型I5E受容体cDNA(配列番号:35)を公知のPCR法によりクローニングし、pAKKO1.11H発現ベクターに組み込んだ。当該発現ベクターを(3−5)に記載した方法に従い、CHO/dhFr-細胞(American Type Culture Collection)に形質導入した。培養開始10-14日後に生育してきた形質転換CHO細胞のコロニーを約20個選んだ。 選択した細胞を3×104個/wellで96穴プレートに植え込み、(3−5)に記載した試験法に従い、MIT1に対する反応性を検討した。反応性の良いI5E発現CHO細胞4番クローン(I5E−4細胞と略称する)を選別した。配列番号:35で表される塩基配列がコードするアミノ酸配列を配列番号:36に示す。
(8−3)ヒト型ZAQリガンドペプチドおよびMIT1のZAQ受容体およびI5E受容体活性化作用の測定
実施例7に記載した方法で精製したヒト型ZAQリガンドぺプチド、及び上記(8−1)に記載した方法で精製したヘビ毒MIT1について、それらのZAQ受容体活性化作用、及びI5E受容体活性化作用を(3−5)に記載した方法に従い測定した。結果を〔図12〕および〔図13〕に示す。
その結果、ヒト型ZAQリガンドぺプチドおよびヘビ毒MIT1は濃度依存的に細胞内カルシウム濃度の上昇を惹起した。ヘビ毒MIT1のZAQ受容体活性化作用はヒト型ZAQリガンドペプチドのそれに比べて10倍強かった。また、ヘビ毒MIT1のヒト型I5E受容体活性化作用はヒト型ZAQリガンドペプチドのそれに比べて100倍程度強いものであった。
【0124】
実施例9 ラット脳cDNA由来新規Gタンパク質共役型受容体タンパク質(rZAQ1)をコードするcDNAのクローニングと塩基配列の決定
ラット全脳cDNAライブラリー(CLONTECH社)を鋳型とし、2種類のプライマー(配列番号:37および配列番号:38)を用いてPCR反応を行った。当該反応における反応液の組成は上記cDNAを10分の1量鋳型として使用し、 Advantage-2 cDNApolymerase Mix (CLONTECH社)1/50量、プライマー各0.2μM、dNTPs 200μM、および酵素に添付のバッファーを加え、25μlの液量とした。PCR反応は、▲1▼94℃・2分の後、▲2▼94℃・20秒、72℃・1分30秒のサイクルを3回、▲3▼94℃・20秒、68℃・1分30秒のサイクルを3回、▲4▼94℃・20秒、62℃・20秒、68℃1分のサイクルを36回繰り返し、最後に68℃・7分の伸長反応を行った。当該PCR反応後の反応産物をTOPO−TAクローニングキット(Invitrogen社)の処方に従いプラスミドベクターpCR2.1‐TOPO(Invitrogen社)へサブクローニングした。これを大腸菌DH5αに導入し、cDNAを持つクローンを,アンピシリンを含むLB寒天培地中で選択した。個々のクローンの配列を解析した結果、新規Gタンパク質共役型受容体タンパク質をコードするcDNA(配列番号:39)を得た。当該cDNAの塩基配列から導き出されるアミノ酸配列(配列番号:40)には、配列番号:1で表されるアミノ酸配列と83.7%の相同性がみられた。このアミノ酸配列を含有する新規Gタンパク質共役型受容体タンパク質をrZAQ1と命名した。また配列番号:39で表わされる塩基配列を含有するDNAを含有する形質転換体(大腸菌)については、エシェリヒア・コリ(Escherichia coli)DH5α/pCR2.1‐rZAQ1と命名した。
【0125】
実施例10 ラット脳cDNA由来 新規Gタンパク質共役型受容体タンパク質(rZAQ2)をコードするcDNAのクローニングと塩基配列の決定
rZAQ2をコードするクローンは、genetrapper法で取得した。すなわち、プローブ(配列番号:41および配列番号:42)をビオチン化したのち、一本鎖にしたラット全脳cDNAライブラリー(GIBCO−BRL社)とハイブリダイゼーションし、得られた一本鎖遺伝子をプライマー(配列番号:43および配列番号:44)を用いて二本鎖に修復した。この遺伝子を大腸菌DH10Bにエレクトロポーレーションし、アンピシリン耐性を指標として形質転換体を得た。さらに、プローブ(配列番号:41)とプライマー(配列番号:45)を用いたコロニーPCRで、目的とする塩基配列をコードするクローンを選択した。このクローンの塩基配列から予測されるORF(open reading frame)の塩基配列(配列番号:46)より導き出されるアミノ酸配列(配列番号:47)は、rZAQ1と80.6%の相同性がみられた。このアミノ酸配列を含有する新規Gタンパク質共役型受容体タンパク質をrZAQ2と命名した。また、このgenetrapper法で取得した形質転換体(大腸菌)を、エシェリヒア・コリ(Escherichia coli)DH10B/pCMV-rZAQ2と命名した。
【0126】
実施例11 モルモット回腸摘出標本を用いた収縮実験
モルモット(std:Hartley、7-8週令、雄性、体重450 g程度)の頸動脈をはさみで切断し失血死させた後、開腹し回腸を摘出した。回腸を、95% O2-5% CO2ガスを吹き込んだTyrode液(組成:138 mM NaCl、2.7 mM KCl、1.8 mM CaCl2、0.5 mM MgCl2、1.1 mM NaH2PO4、11.9 mM NaHCO3、5.6 mM glucose)を入れたガラスシャーレに入れ、手術用はさみとピンセットを用いて回腸に付着している脂肪片や結合組織を除去した後に、約1.5 cmの長さに切り標本として用いた。この標本を37℃に暖めた上記Tyrode液を満たしたオーガンバス(20ml)中に懸垂し、0.5gの負荷をかけて30分以上かけて安定させた。
回腸標本の収縮反応は、NEC三栄のAMPLIFIER CASE 7747を用いて測定した。
アセチルコリン1 μMを投与し最大収縮反応を測定した後に、参考例1の方法に準じて得られたヒト型ZAQリガンドペプチド(ZAQL-1)または上記(8−1−3)で得られたヘビ毒MIT1を累積投与して収縮反応を測定した。ZAQL−1とMIT1の希釈には0.05% bovine serum albuminを含む生理食塩水を用いた。
結果を〔図14〕に示す。ZAQL−1およびMIT1により惹起される収縮反応は、アセチルコリン1 μMにより惹起される収縮反応を100%としてパーセントで示した。
その結果、ZAQL−1とMIT1は容量依存的に強力な収縮反応を惹起した。容量反応曲線から算出したZAQL−1およびMIT1のEC50値は、それぞれ1.79 nMおよび0.40 nMであった。
【0127】
実施例12 ZAQおよびI5E発現CHO細胞膜画分を用いたバインディングアッセイ
(12−1)125I−MIT1の調製
[125I]-Bolton-Hunter Reagent (monoiodinated、37 MBq、NEN社NEX120)に含まれるベンゼンを窒素ガスで留去後、DMSO20 μlを添加し、ピペッティングして乾固物を溶解した。ここにBorateバッファー(組成;100 mM H3BO3、pH 8.5)で希釈した4 nmol のMIT1を含む溶液80 μlを添加し、直ちに混合後、室温で2時間インキュベーションした。その後、10%アセトニトリル-0.1%トリフルオロ酢酸200 μlを添加し、HPLC分離用サンプルとした。これを室温にて流速1 ml/minでA液(10%アセトニトリル/0.1%トリフルオロ酢酸)容量100%/B液(0.1% トリフルオロ酢酸/40% アセトニトリル)容量0%を流し、平衡化したTSKgel Super-ODS逆相高速液体クロマトグラフィー用カラム(東ソー株式会社、 0.46 cm x 10 cm)に添着した後、流速1ml/mlで、1分間かけてA液容量60%/B液容量40%まで上昇させ、次いで60分間かけてA液容量40%/B液容量60%まで、B液濃度を直線的グラジエントで上昇させた。溶出される[125I]-Bolton-Hunter Reagentが導入されたMIT1を手動で分取し、バッファー(組成;20 mM Tris、1 mM EDTA、0.2% BSA、0.1% CHAPS、0.03% NaN3、pH 7.4)で希釈し分注後、−80℃で保存した。
【0128】
(12−2)細胞膜画分の調製
細胞膜画分は、Journal of Pharmacology and Experimental Therapeutics、279巻、675-685頁、1996年記載の方法に従い調製した。10%透析済みウシ胎児血清(JRH BIOSCIENCES 社)、1 mM MEM非必須アミノ酸溶液、100 units/ml Penicillin、および100 μg/ml Streptomycinを含む Dulbecco's modified Eagle medium (DMEM) 培地(日水製薬株式会社)中、37℃、5%炭酸ガス中にて受容体発現CHO細胞、すなわち、ZAQC-B1細胞またはI5E−4細胞をSingletray(Nunc)にて培養した。80-90%の細胞密度になったところで培養上清を捨て、トレイに1 g/l EDTAを含むPBS(宝酒造)を30 ml加え、細胞を洗浄後、上清を捨て上記PBS-EDTAを30 ml添加し、室温で放置した。トレイをしんとうし、細胞をトレイからはがし、細胞懸濁液を回収した。さらにトレイに上記PBS-EDTA 10 mlを再度加えて洗浄し、トレイに残った細胞をはがし、最初に得られた細胞懸濁液細胞と混合した。混合した細胞懸濁液を、高速遠心機(TOMY RL601)を用いて1,000 rpmで5分間遠心した。上清を捨て、沈殿した細胞を−80℃で保存した。この沈殿(Singletray 1枚分)に破砕用バッファー(組成;10 mM NaHCO3、5 mM EDTA、0.5 mM phenylmethylsulfonylfluoride (PMSF)、10 μg/ml pepstatin-A、20μg/ml leupeptin、10 μg/ml E-64)4mlを添加し、ピペッティングして沈殿を懸濁し、次いでポリトロン(刃型PTA7)を用いて、目盛り設定4、20秒間、3回の条件で破砕した。次に、日立高速冷却遠心機CR26HでNo.26ローターを用い、2,500 rpmで10分間遠心した。得られた上清をさらに超遠心機(日立工機SCP70H)でSRP70ATローターを用いて、35,000 rpmで1時間遠心した。得られた沈殿にバインディングアッセイバッファー(組成;20 mM Tris、1 mM EDTA、0.5 mM PMSF、10 μg/ml pepstatin-A、40 μg/ml leupeptin、10μg/ml E-64、0.03% NaN3、pH 7.4)5 mlを添加し、懸濁後、Cell Strainer(FALCON 2350)でろ過し、ZAQC−B1細胞膜画分(すなわち、ZAQ膜画分)あるいはI5E−4細胞膜画分(すなわち、I5E膜画分)を得た。得られたそれぞれの膜画分のタンパク濃度をCoomassie Protein Assay Reagent(PIERCE)を用いて測定後、100 μlずつ分注し、−80℃で保存した。
【0129】
(12−3)バインディングアッセイ
0.1% BSAを含む上記(12−2)記載のバインディングアッセイバッファーにて、ZAQ膜画分は10 μg/mlに、I5E膜画分は20 μg/mlにそれぞれ希釈し、200 μlずつチューブ(FALCON 2053)に分注した。この希釈した各膜画分に、試験化合物2 μl および10 nM 125I-MIT1 2 μlを添加し、25℃で1時間インキュベーションした。試験化合物としては、参考例1の方法に準じて得られたヒト型ZAQリガンドペプチド(ZAQL−1)または上記(8−1−3)で得られたヘビ毒MIT1(非標識MIT1)を用いた。
次に、反応液にろ過バッファー(組成;20 mM Tris、EDTA、0.1% BSA、0.05% CHAPS、0.03% NaN3、pH 7.4)1.5 mlを加え、バッファー(組成;20 mM Tris、0.3% polyethyleneimine、pH 7.4)で予め処理したガラス繊維ろ紙GF/F(Whatman)で直ちにろ過し、反応チューブに再度ろ過バッファー1.5 mlを添加し同様にろ過した。ガラス繊維ろ紙の放射活性を、γカウンター(COBRA、Packard)を用いて測定し、125I-MIT1 結合量を測定した。試験化合物として非標識MIT1 (終濃度1 μM) を用いた場合の 125I-MIT1結合量を非特異的結合量(NSB)、試験化合物を全く添加しない場合の125I-MIT1結合量を総結合量(TB)と定め、最大特異的結合量(TB-NSB)を算出した。また、試験化合物を添加した際に得られた結合量(B)についても、特異的結合量(B-NSB)を算出し、最大特異的結合量に対するパーセント{ %SPB=(B-NSB)/(TB-NSB) × 100 }を以って試験化合物存在下の結合量を表わした。
ZAQ膜画分を用いた結果を〔図15〕に、I5E膜画分を用いた結果を〔図16〕に示す。
このバインディングアッセイ系で算出した非標識MIT1のIC50値は、38 pM(ZAQ膜画分)、32 pM(I5E膜画分)であった。ZAQL-1のIC50値は、35 nM(ZAQ膜画分)、93 nM(I5E膜画分)であった。
【0130】
参考例1 大腸菌でのヒトZAQリガンド(配列番号:21)の製造
(参考例1−1) 大腸菌でのヒトZAQリガンド発現プラスミドの構築
(a)以下に示す6種のDNA断片#1〜#6を用いて、ZAQリガンドの構造DNAを調製した。
#1 :
5'-TATGGCGGTGATTACCGGTGCGTGCGAACGTGATGTGCAGTGCGGTGCGGGTACCTGCTGCGCGATTAGCCTGTGGCTGCGTGGTCTG-3' (配列番号:52)、
#2:
5'-CGTATGTGCACCCCGCTGGGTCGTGAAGGTGAAGAATGCCATCCGGGTAGCCATAAAGTGCCGTTCTTCCGTAAACGTAAACATCATACCTG-3' (配列番号:53)、
#3:
5'-CCCGTGCCTGCCGAACCTGCTGTGCAGCCGTTTCCCGGATGGTCGTTATCGTTGCAGCATGGATCTGAAAAACATTAACTTTTAGG-3' (配列番号:54)、
#4:
5'-CACATACGCAGACCACGCAGCCACAGGCTAATCGCGCAGCAGGTACCCGCACCGCACTGCACATCACGTTCGCACGCACCGGTAATCACCGCCA-3' (配列番号:55)、
#5:
5'-AGGCACGGGCAGGTATGATGTTTACGTTTACGGAAGAACGGCACTTTATGGCTACCCGGATGGCATTCTTCACCTTCACGACCCAGCGGGGTG-3' (配列番号:56)、
#6:
5'-GATCCCTAAAAGTTAATGTTTTTCAGATCCATGCTGCAACGATAACGACCATCCGGGAAACGGCTGCACAGCAGGTTCGGC-3' (配列番号:57)。
【0131】
(b)DNAオリゴマーのリン酸化
5’側になるべき上記#1および#6を除いた4種のDNAオリゴマー(#2〜#5)各々を、25μlのリン酸化反応液〔DNAオリゴマー10μg,50mM Tris−HCl,pH7.6, 10mM MgCl2, 1mMスペルミジン,10mM ジチオスレイトール(以後DTTと略記),0.1mg/mlウシ血清アルブミン(以後BSAと略記),1mM ATP,10ユニットT4ポリヌクレオチドキナーゼ(宝酒造)〕中で37℃・1時間反応させ、各オリゴマーの5’末端をリン酸化した。フェノール処理を行った後、2倍量のエタノールを加え、−70℃に冷却した後、遠心でDNAを沈殿させた。
(c)DNAフラグメントの連結
上記(b)で得られたDNAフラグメントと上記#1および#6を合わせ、120μlとした。この混合液を90℃で10分間保持した後、室温まで徐冷しアニーリングを行った後、TaKaRa DNA Ligation Kit ver.2(宝酒造)を用いてライゲーション反応を行った。アニーリング液30μlにキットに付属のII液30μlを加え、よく混合した後、キットに付属のI液60μlを加え、37℃・1時間反応させ、ライゲーションを行った。その後、フェノール処理を行ない、水層を回収して2倍量のエタノールを加え、−70℃に冷却した後、遠心でDNAを沈殿させた。この様にして得られたDNAフラグメントをT4ポリヌクレオチドキナーゼ(宝酒造)によるリン酸化を行った後、以下の工程(d)に供した。
【0132】
(d) ZAQリガンド発現ベクターの構築
発現用ベクターとしてはpTCII(特開2000-178297号に記載)をNdeIおよびBamHI(宝酒造)で37℃・2時間消化した後、1%アガロースゲル電気泳動により4.3kbのDNA断片をQIAquick Gel Extraction Kit (キアゲン社)を用いて抽出し、25μlのTE緩衝液に溶解した。このpTCIIのNdeI、BamHI断片と上記により調製したZAQリガンドの構造遺伝子(配列番号:58)をTaKaRa DNA ligation kit ver.2 (宝酒造)を用いてライゲーション反応を行った。この反応液を10μl用いて大腸菌JM109コンピテントセル(東洋紡)を形質転換し、10μg/mlのテトラサイクリンを含むLB寒天培地上に播き、37℃で1晩培養し、生じたテトラサイクリン耐性コロニー選んだ。この形質転換体をLB培地で一晩培養し、QIAprep8 Miniprep Kit(キアゲン社)を用いてプラスミドpTCh1ZAQを調製した。このZAQリガンドDNAの塩基配列をアプライドバイオシステムズ社モデル377DNAシーケンサーを用いて確認した。プラスミドpTCh1ZAQを大腸菌(Escherichia coli)MM294(DE3)に形質転換し、ZAQリガンド発現株Escherichia coli MM294(DE3)/ pTCh1ZAQを得た。
【0133】
(参考例1−2)ZAQリガンドの製造
上記のEscherichia coli MM294(DE3)/ pTCh1ZAQを5.0mg/Lのテトラサイクリンを含むLB培地・1L(1%ペプトン、0.5%酵母エキス、0.5%塩化ナトリウム)を用いて2L容フラスコ中で37℃、8時間振とう培養した。得られた培養液を19Lの主発酵培地(1.68%リン酸1水素ナトリウム、0.3%リン酸2水素カリウム、0.1%塩化アンモニウム、0.05%塩化ナトリウム、0.05%硫酸マグネシウム、0.02%消泡剤、0.00025%硫酸第1鉄、0.0005%塩酸チアミン、1.5%ブドウ糖、1.5%ハイケースアミノ)を仕込んだ50L容発酵槽へ移植して、30℃で通気攪拌を開始した。培養液の濁度が500クレット単位になったところで、イソプロピル−β−D−チオガラクトピラノシドの最終濃度が12mg/Lになるように添加し、さらに4時間培養を行った。培養終了後、培養液を遠心分離し、約200gの湿菌体を取得し、−80℃で保存した。
この形質転換大腸菌MM294(DE3)/pTCh1ZAQは、受託番号IFO 16527として財団法人発酵研究所(IFO)に寄託されている。
(参考例1−3)ZAQリガンドの活性化
参考例(1−2)で得られた菌体200gに、200mMトリス/HCl、7Mグアニジン塩酸塩(pH8.0)400mlを加えて菌体を溶解した後、遠心分離(10000rpm、1時間)を行った。上澄液に0.4Mアルギニン、50mMトリス/HCl、0.2mM GSSG、1mM GSH(pH8.0)10リットルを加えて、4℃で一晩活性化を行った。
【0134】
(参考例1−4)ZAQリガンドの精製
参考例(1−3)で活性化の終了した再生液をpH6.0に調整し、50mMリン酸緩衝液(pH6.0)で平衡化したSP−セファロースカラム(11.3cm×15cm)に吸着させた後、600mM NaCl/50mMリン酸緩衝液(pH6.0)で溶出し、ZAQリガンドを含むフラクションをプールした。この画分を50mM リン酸緩衝液(pH6.0)で平衡化したCM−5PW(21.5mm×150mmL)に通液し、吸着、洗浄した後、0−100%B(B=50mM リン酸緩衝液+1M NaCl、pH6.05)の段階勾配(60分)で溶出を行いZAQリガンド画分(溶出時間約40分)を得た。この画分を、さらに0.1%トリフルオロ酢酸で平衡化したC4P−50(21.5mmID×300mmL、昭和電工)に通液し、吸着、洗浄した後、25−50%B(B:80%アセトニトリル/0.1%トリフルオロ酢酸)の段階勾配(60分)で溶出を行い、ZAQリガンド画分(溶出時間約40分)をプールした後、凍結乾燥を行い、ZAQリガンド凍結乾燥粉末約80mgを得た。
【0135】
(参考例1−5)ZAQリガンドの特徴決定
(a)SDSポリアクリルアミドゲル電気泳動を用いた分析
参考例(1−4)で得られたZAQリガンドを100mM DTTを添加したSample buffer[Laemmli, Nature, 227, 680 (1979)]に懸濁し、95℃で1分間加熱した後、マルチゲル15/25(第一化学薬品)で電気泳動を行った。泳動後のゲルをクーマシー・ブリリアント・ブルー(Coomassie brilliant blue)で染色した結果、実施例5で得られたCOS7細胞由来の組換え型ZAQリガンド標品と同じ位置に、単一のタンパク質バンドが認められた。このことから、参考例(1−4)で得られた大腸菌由来の組換え型ZAQリガンド標品は極めて高純度であり、COS7細胞から調製した組換え型ZAQリガンドと分子量的に同一であることが分かった。
【0136】
(b)アミノ酸組成分析
アミノ酸組成をアミノ酸分析計(日立L−8500A Amino Acid Analyzer)を用いて決定した。その結果、ZAQリガンド(配列番号:21で表されるアミノ酸配列からなるペプチド)のDNAの塩基配列から推定されるアミノ酸組成と一致した(表2)。
【表2】
1)0時間に外挿した値
2)未検出
【0137】
(c)N末端アミノ酸配列分析
N末端アミノ酸配列を気相プロテインシーケンサー(PEアプライドバイオシステムズ モデル492)を用いて決定した。その結果、得られたZAQリガンドのDNAの塩基配列から推定されたZAQリガンドのN末端アミノ酸配列と一致した(表3)。
【表3】
N.D.は未検出を示す。
【0138】
(d)C末端アミノ酸分析
C末端アミノ酸をアミノ酸分析計(日立L−8500A Amino Acid Analyzer)を用いて決定した。得られたZAQリガンドはDNAの塩基配列から推定されたC末端アミノ酸と一致した(表4)。
【表4】
質量分析をnanoESIイオン源を装着したLCQイオントラップ質量分析計(サーモクエスト社製)を用いて行った。その結果、分子量9657.55±0.89が得られ、配列番号:21の、しかも配列番号:21が含有する10残基のCysが5対のジスルフィド結合を形成したZAQリガンドの理論分子量(9657.3)と良く一致していた。
【0139】
(参考例1−6)ZAQリガンドの活性測定(FLIPRを用いた細胞内Caイオン濃度上昇活性の測定)
参考例(1−4)で得られた純化された大腸菌由来の組換え型ZAQリガンド標品を実施例5の方法を用いて、活性測定(FLIPRを用いた細胞内Caイオン濃度上昇活性の測定)を行った。その結果、実施例5で得られたCOS7細胞由来の組換え型標品(ZAQリガンドの精製品)と同等の活性を有していた。
【0140】
【発明の効果】
本発明のペプチド、本発明のペプチドをコードするDNA(以下、本発明のDNAと略記する場合がある)および本発明のペプチドに対する抗体(以下、本発明の抗体と略記する場合がある)は、▲1▼本発明のペプチドが関与する各種疾病の治療・予防剤、▲2▼本発明のペプチドと本発明のタンパク質との結合性を変化させる化合物またはその塩のスクリーニング、▲3▼本発明のペプチドまたはその塩の定量、▲4▼遺伝子診断剤、▲5▼アンチセンスDNAを含有する医薬、▲6▼本発明の抗体を含有する医薬、▲7▼本発明のDNAを含有する非ヒト動物の作出、▲8▼構造的に類似したリガンド・受容体との比較にもとづいたドラッグデザイン、などの実施のために有用である。
【0141】
【配列表】
【図面の簡単な説明】
【図1】実施例1で得られた本発明のヒト脳由来タンパク質をコードするDNAの塩基配列(ZAQC)、およびそれから推定されるアミノ酸配列を示す(図2に続く)。
【図2】実施例1で得られた本発明のヒト脳由来タンパク質をコードするDNAの塩基配列(ZAQC)、およびそれから推定されるアミノ酸配列を示す(図1の続き、図3に続く)。
【図3】実施例1で得られた本発明のヒト脳由来タンパク質をコードするDNAの塩基配列(ZAQC)、およびそれから推定されるアミノ酸配列を示す(図2の続き)。
【図4】実施例1で得られた本発明のヒト脳由来タンパク質をコードするDNAの塩基配列(ZAQT)、およびそれから推定されるアミノ酸配列を示す(図5に続く)。
【図5】実施例1で得られた本発明のヒト脳由来タンパク質をコードするDNAの塩基配列(ZAQT)、およびそれから推定されるアミノ酸配列を示す(図4の続き、図6に続く)。
【図6】実施例1で得られた本発明のヒト脳由来タンパク質をコードするDNAの塩基配列(ZAQT)、およびそれから推定されるアミノ酸配列を示す(図5の続き)。
【図7】本発明のヒト脳由来タンパク質の疎水性プロットを示す。
【図8】実施例2で行われたZAQの発現分布の解析結果を示す。
【図9】MIT1、ヒト型ZAQリガンド前駆体ペプチド(Aタイプ)およびヒト型ZAQリガンド前駆体ペプチド(Gタイプ)のアミノ酸配列を示す。
図中、「MIT1」はMIT1のアミノ酸配列を、「Human(A type)」はヒト型ZAQリガンド成熟体ペプチド(Aタイプ)のアミノ酸配列を、「Human(G type)」はヒト型ZAQリガンド成熟体ペプチド(Gタイプ)のアミノ酸配列を、それぞれ示す。
【図10】実施例6(6−3)で行われた、精製ZAQリガンドペプチドのZAQ活性化作用の測定結果を示す。
【図11】実施例5(5−1)で用いたプラスミドpCAN618の制限酵素地図を示す。
【図12】実施例8(8−3)で行われたヒト型ZAQリガンドペプチドおよびMIT1のZAQ受容体活性化作用の測定結果を示す。図中、○はヒト型ZAQリガンドペプチドを、●はMIT1を示す。
【図13】実施例8(8−3)で行われたヒト型ZAQリガンドペプチドおよびMIT1のI5E受容体活性化作用の測定結果を示す。図中、○はヒト型ZAQリガンドペプチドを、●はMIT1を示す。
【図14】実施例11で行われた収縮作用の測定結果を示す。図中、黒四角はヒト型ZAQリガンドペプチドを、●はMIT1を示す。
【図15】実施例12で行われたZAQ膜画分を用いたバインディングアッセイの測定結果を示す。図中、黒四角は可変濃度(横軸記載)のヒト型ZAQリガンドペプチドを試験化合物として加えた場合の125I-MIT1 特異的結合量を、●は同様にMIT1を試験化合物として加えた場合の125I-MIT1 特異的結合量を示す。
【図16】実施例12で行われたI5E膜画分を用いたバインディングアッセイの測定結果を示す。図中、□は可変濃度(横軸記載)のヒト型ZAQリガンドペプチドを試験化合物として加えた場合の125I-MIT1 特異的結合量を、○は可変濃度(横軸記載)のMIT1を試験化合物として加えた場合の125I-MIT1 特異的結合量を示す。
Claims (16)
- 配列番号:20または配列番号:21で表わされるアミノ酸配列からなるペプチドまたはその塩。
- 配列番号:20で表わされるアミノ酸配列からなる請求項1記載のペプチドまたはその塩。
- 配列番号:21で表わされるアミノ酸配列からなる請求項1記載のペプチドまたはその塩。
- 配列番号:22で表わされるアミノ酸配列からなるペプチドまたはその塩。
- 請求項1記載のペプチドをコードするポリヌクレオチドからなるポリヌクレオチド。
- 請求項4記載のペプチドをコードするポリヌクレオチドからなるポリヌクレオチド。
- 配列番号:26または配列番号:27で表される塩基配列からなるDNAである請求項5記載のポリヌクレオチド。
- 配列番号:28で表される塩基配列からなるDNAである請求項6記載のポリヌクレオチド。
- 請求項5または6記載のポリヌクレオチドを含有する組換えベクター。
- 請求項9記載の組換えベクターで形質転換された形質転換体。
- 請求項10記載の形質転換体を培養し、請求項1記載のペプチドを生成・蓄積せしめることを特徴とする請求項1記載のペプチドまたはその塩の製造法。
- 請求項1記載のペプチドまたはその塩に対する抗体。
- ペプチドまたはその塩および配列番号:1で表わされるアミノ酸配列を含むタンパク質またはその塩を用いることを特徴とする、前記ペプチドまたはその塩と配列番号:1で表わされるアミノ酸配列を含むタンパク質またはその塩との結合性を変化させる化合物またはその塩のスクリーニング方法であって、
前記ペプチドが、
(i) 配列番号:20もしくは配列番号:21で表わされるアミノ酸配列からなるペプチド、または
(ii) 配列番号:20もしくは配列番号:21で表されるアミノ酸配列と90%以上の相同性を有するアミノ酸配列からなり、配列番号:1で表されるアミノ酸配列を含むタンパク質に結合し、該タンパク質を活性化しうるペプチド、
のいずれかである、スクリーニング方法。 - ペプチドまたはその塩および配列番号:36で表わされるアミノ酸配列を含むタンパク質またはその塩を用いることを特徴とする、前記ペプチドまたはその塩と配列番号:36で表わされるアミノ酸配列を含むタンパク質またはその塩との結合性を変化させる化合物またはその塩のスクリーニング方法であって、
前記ペプチドが、
(i) 配列番号:20もしくは配列番号:21で表わされるアミノ酸配列からなるペプチド、または
(ii) 配列番号:20もしくは配列番号:21で表されるアミノ酸配列と90%以上の相同性を有するアミノ酸配列からなり、配列番号:1で表されるアミノ酸配列を含むタンパク質に結合し、該タンパク質を活性化しうるペプチド、
のいずれかである、スクリーニング方法。 - ペプチドまたはその塩および配列番号:1で表わされるアミノ酸配列を含むタンパク質またはその塩を含有することを特徴とする、前記ペプチドまたはその塩と配列番号:1で表わされるアミノ酸配列を含むタンパク質またはその塩との結合性を変化させる化合物またはその塩のスクリーニング用キットであって、
前記ペプチドが、
(i) 配列番号:20もしくは配列番号:21で表わされるアミノ酸配列からなるペプチド、または
(ii) 配列番号:20もしくは配列番号:21で表されるアミノ酸配列と90%以上の相同性を有するアミノ酸配列からなり、配列番号:1で表されるアミノ酸配列を含むタンパク質に結合し、該タンパク質を活性化しうるペプチド、
のいずれかである、スクリーニング用キット。 - ペプチドまたはその塩および配列番号:36で表わされるアミノ酸配列を含むタンパク質またはその塩を含有することを特徴とする、前記ペプチドまたはその塩と配列番号:36で表わされるアミノ酸配列を含むタンパク質またはその塩との結合性を変化させる化合物またはその塩のスクリーニング用キットであって、
前記ペプチドが、
(i) 配列番号:20もしくは配列番号:21で表わされるアミノ酸配列からなるペプチド、または
(ii) 配列番号:20もしくは配列番号:21で表されるアミノ酸配列と90%以上の相同性を有するアミノ酸配列からなり、配列番号:1で表されるアミノ酸配列を含むタンパク質に結合し、該タンパク質を活性化しうるペプチド、
のいずれかである、スクリーニング用キット。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2001216683A JP4965776B2 (ja) | 2000-07-18 | 2001-07-17 | 新規生理活性ペプチドおよびその用途 |
Applications Claiming Priority (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2000-217442 | 2000-07-18 | ||
| JP2000217442 | 2000-07-18 | ||
| JP2000217442 | 2000-07-18 | ||
| JP2001026779 | 2001-02-02 | ||
| JP2001-26779 | 2001-02-02 | ||
| JP2001026779 | 2001-02-02 | ||
| JP2001216683A JP4965776B2 (ja) | 2000-07-18 | 2001-07-17 | 新規生理活性ペプチドおよびその用途 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2002335976A JP2002335976A (ja) | 2002-11-26 |
| JP2002335976A5 JP2002335976A5 (ja) | 2008-08-14 |
| JP4965776B2 true JP4965776B2 (ja) | 2012-07-04 |
Family
ID=27344096
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2001216683A Expired - Fee Related JP4965776B2 (ja) | 2000-07-18 | 2001-07-17 | 新規生理活性ペプチドおよびその用途 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP4965776B2 (ja) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| SG11202011845XA (en) * | 2018-08-03 | 2020-12-30 | Laboratory Of Cell Applied Technologies Co | Production method for culture supernatant preparation |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2328895A1 (en) * | 1998-06-02 | 1999-12-09 | Genentech, Inc. | Secreted and transmembrane polypeptides and nucleic acids encoding the same |
| JP2002095474A (ja) * | 1999-08-27 | 2002-04-02 | Takeda Chem Ind Ltd | 新規g蛋白質共役型レセプター蛋白質およびそのdna |
-
2001
- 2001-07-17 JP JP2001216683A patent/JP4965776B2/ja not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| JP2002335976A (ja) | 2002-11-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US7419956B2 (en) | Isolated physiologically active peptide and use thereof | |
| JP4486187B2 (ja) | 新規g蛋白質共役型レセプター蛋白質、そのdnaおよびそのリガンド | |
| US7354726B2 (en) | Screening method | |
| US20050240008A1 (en) | Novel protein, dna thereof and process for producing the same | |
| EP1207198A1 (en) | Novel g protein-coupled receptor protein and dna thereof | |
| US7268212B2 (en) | Ligand of G protein-coupled receptor protein and DNA thereof | |
| US20050176632A1 (en) | Angiogenesis inhibitors | |
| EP1357129A2 (en) | Novel physiologically active peptide and use thereof | |
| EP1273657A1 (en) | Novel protein, process for producing the same and use thereof | |
| JP4965776B2 (ja) | 新規生理活性ペプチドおよびその用途 | |
| JP2012167017A (ja) | ペプチドおよびその用途 | |
| US20060035835A1 (en) | Novel protein, production and use thereof | |
| JPWO2006118328A1 (ja) | 脱顆粒抑制剤 | |
| JP4128030B2 (ja) | 新規リガンドおよびそのdna | |
| JP4772684B2 (ja) | スクリーニング方法 | |
| JP4004767B2 (ja) | 新規g蛋白質共役型レセプター蛋白質およびそのdna | |
| US20040101956A1 (en) | Novel g protein-coupled receptor protein | |
| JP4535670B2 (ja) | 新規ポリペプチド、そのdnaおよびそれらの用途 | |
| JP2000175691A (ja) | 新規g蛋白質共役型レセプタ―蛋白質およびそのdna | |
| JP4959880B2 (ja) | Gpr8に対するリガンドおよびそのdna | |
| JP2002095474A (ja) | 新規g蛋白質共役型レセプター蛋白質およびそのdna | |
| US20030082648A1 (en) | Novel mass receptor-analogous protein and dnas thereof | |
| JP2004073179A (ja) | 新規タンパク質、そのdnaおよび用途 | |
| JP2004051622A (ja) | 新規リガンドおよびそのdna | |
| US20040072293A1 (en) | Novel physiologically active peptide and use thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20080701 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20080701 |
|
| RD03 | Notification of appointment of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7423 Effective date: 20080701 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20110322 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20110523 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20110920 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20111118 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20120306 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20120330 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20150406 Year of fee payment: 3 |
|
| LAPS | Cancellation because of no payment of annual fees |