JP4699693B2 - C−官能基化含窒素大環式化合物製造のための新規方法および得られる新規中間体 - Google Patents
C−官能基化含窒素大環式化合物製造のための新規方法および得られる新規中間体 Download PDFInfo
- Publication number
- JP4699693B2 JP4699693B2 JP2003532478A JP2003532478A JP4699693B2 JP 4699693 B2 JP4699693 B2 JP 4699693B2 JP 2003532478 A JP2003532478 A JP 2003532478A JP 2003532478 A JP2003532478 A JP 2003532478A JP 4699693 B2 JP4699693 B2 JP 4699693B2
- Authority
- JP
- Japan
- Prior art keywords
- group
- compound
- formula
- defined above
- groups
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 0 CN(CCN(C)*B1)C*CNCCN1C1*C1 Chemical compound CN(CCN(C)*B1)C*CNCCN1C1*C1 0.000 description 2
- FBZJILZAOVQRLE-UHFFFAOYSA-N CC12N3CCNC1(C)NCCN2CC3 Chemical compound CC12N3CCNC1(C)NCCN2CC3 FBZJILZAOVQRLE-UHFFFAOYSA-N 0.000 description 1
- GSMHMRGPPVLUJY-NOYMGPGASA-N C[C@]1([C@@](C)(NCCC2)N2CC2)N2C(COCC=C)CN1 Chemical compound C[C@]1([C@@](C)(NCCC2)N2CC2)N2C(COCC=C)CN1 GSMHMRGPPVLUJY-NOYMGPGASA-N 0.000 description 1
- QISSZEODMJTDCQ-UHFFFAOYSA-N OCC1NCCNCCNCCNC1 Chemical compound OCC1NCCNCCNCCNC1 QISSZEODMJTDCQ-UHFFFAOYSA-N 0.000 description 1
Images


Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/08—Bridged systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D257/00—Heterocyclic compounds containing rings having four nitrogen atoms as the only ring hetero atoms
- C07D257/02—Heterocyclic compounds containing rings having four nitrogen atoms as the only ring hetero atoms not condensed with other rings
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Description

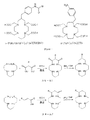
基AまたはBの一方は−CH2−基であり、他方の基は、−CH(R′)−CH2−基[式中、R′は、1〜12の炭素原子を含む飽和もしくは不飽和脂肪族基であるか、または−(CH2)n−O−R1′基[式中、nは0および4の間の整数であり、R1′は水素原子または1〜8の炭素原子を含む飽和もしくは不飽和脂肪族基である]であるか、または−(CH2)n−C(=O)−O−R1′基[式中、nおよびR1′は前記と同意義である]であるか、または−(CH2)n−R2′基[式中、R2′は非置換フェニル基であるか、またはアミノ、ニトロ、クロロ、ブロモ、ヨード、メトキシもしくはヒドロキシ基から選ばれる1またはそれ以上の基で置換されているフェニル基である]である]であるか、
で示される化合物の製造方法であって、
以下の連続反応工程:
工程(a):式(II):
で示される化合物を式(III):
で示される化合物を形成させる工程;
工程(b):工程(a)で得られた式(IV)の化合物を式(V):
で示される化合物を形成させる工程;および、
工程(c):工程(b)で得られた式(VI)の化合物を酸処理に付して前記式(I)の化合物を形成させる工程、を含む方法である。
で示される化合物へのアクセスを可能にする。
この式(VII)の化合物は本発明の他の主題を構成する。
具体的な態様によれば、本発明の他の主題は、式(Ia):
●基A、BおよびDは式(I)の化合物に関して上に述べた意義を有し;そして、
●基Z1、Z2、Z3およびZ4(これらは同じであっても異なっていてもよい)の各々は個別に、
−水素原子;または、
−1〜15の炭素原子を含む直線状もしくは分枝アルキル基または7〜12の炭素原子を含む[ヘテロ(アリール)]アルキル基;または、
−−(CH2)w−Y基[式中、
□ wは0より大きく6以下、より具体的には3以下の数で
あり;そして、
□ Yは、
−[−C(=O)]y−V基[式中、
●yは0または1に等しく;そして、
●VはOH基もしくはOR3基[式中、R3は、所望により1またはそれ以上のCOOH、SO3H、PO3H2またはCO(NH2)基で置換されていてもよい、1〜6の炭素原子、より具体的には1〜3の炭素原子を含むアルキル基である]、または
NH2、NHR4もしくはN(R4)(R5)基[式中、R4およびR5は1〜4の炭素原子を含むアルキルラジカルである]である];または、
−−P(=O)(OR6)(OR7)基[式中、R6およびR7は各々個別
に水素原子または1〜4の炭素原子を含むアルキルラジカルである];または、
−−SO3H基、である]を表す]
を有する化合物が得られる。
工程(d)の官能基化は当業者の範囲内にある適当な処理によって達成できる。
別の態様によれば、本発明の他の主題は、上に定義したさらなる工程(d)を含む式(I′)の化合物の製造方法である。
で示される化合物である。
以下の実施例は本発明を例示するものであるがこれを限定するものではない。
(a) 9a,9b−ジメチルオクタヒドロ−1,3a,6a,9−テトラアザフェナレンの製造(化合物1)
10.7、18.3、23.3、39.2、42.0、45.6、46.6、49.0、51.1、68.1、73.3。
m/z=210 M+●。
(b) メチル10b,10c−ジメチルデカヒドロ−3a,5a,8a,10a−テトラアザピレン−2−カルボキシラートの製造(化合物3)
9.9、10.6、17.9、35.4、44.7、46.3、46.4、46.6、47.9、48.9、50.2、50.4、52.1、73.2、73.8、174.6。
m/z=308 M+●。
(c) メチル1,4,8,11−テトラアザシクロテトラデカン−6−カルボキシラートの製造(化合物5)
1 H NMRスペクトル(500MHz、D 2 O、δ ppm):
2.22(qt,2H)、3.10−3.80(m,17H)、3.78(s,3H)。
21.9、38.3、41.7、42.4、44.5、46.9、56.5、173.5。
m/z=259(M+H)+●。
実施例B:1,4,8,11−テトラアザシクロテトラデカン−6−カルボン酸の合成(化合物6)
1 H NMRスペクトル(500MHz、D 2 O、δ ppm):
1.55(qt,2H)、2.64(qt,1H)、2.70−3.00(m,16H)。
22.3、38.6、42.3、42.7、44.8、46.9、175.7。
元素分析(C 11 H 24 N 4 O 2 ・3HCl・3H 2 Oとして):
理論値:C 32.40%;H 8.16%;N 13.74%
実測値:C 32.66%;H 8.03%;N 13.81%
(a) 10b,10c−ジメチルデカヒドロ−3a,5a,8a,10a−テトラアザピレン−2−オールの製造(化合物4)
m/z=267(M+H)+●。
(b) 1,4,8,11−テトラアザシクロデカン−6−オールの製造(化合物7)
1 H NMRスペクトル(200MHz、D 2 O、δ ppm):
2.11(qt,2H)、2.8−3.8(m,17H)。
13 C NMRスペクトル(50MHz、D 2 O、δ ppm):
23.8、43.8、45.3(×2)、46.1(×2)、46.7、50.3、59.5、61.7。
m/z=217(M+H)+●。
(a) 9b,9c−ジメチルデカヒドロ−2a,4a,7a,9a−テトラアザシクロペンタ[cd]フェナレン−1−イルメタノールの製造(化合物8)
m/z=267(M+H)+●。
(b) 1,4,7,10−テトラアザシクロトリデカ−5−イル−メタノールの製造(化合物9)
13 C NMRスペクトル(125MHz、D 2 O、δ ppm):
29.0、46.4、47.9、49.1、49.3、50.1、50.1、50.4、58.5、63.4。
m/z=217(M+H)+●。
(a) 5a,8b−ジメチルオクタヒドロ−2a,5,6,8a−テトラアザアセナフチレンの製造(化合物2)
m/z=197(M+H)+●。
(b) 8b,8c−ジメチルデカヒドロ−2a,4a,6a,8a−テトラアザシクロペンタ[fg]アセナフチレン−1−イルメタノールの製造(化合物10)
m/z=253 (M+H)+●。
(c) 1,4,7,10−テトラアザシクロドデカ−2−イルメタノールの製造(化合物11)
13 C NMRスペクトル(125MHz、D 2 O、δ ppm):
42.1、43.3、43.9、44.0、44.5、44.9、46.9、56.0、59.5。
m/z=203(M+H)+●。
(a) オクタヒドロ−1,3a,6a,9−テトラアザフェナレンの製造(化合物12)
19.8、54.0−55.5(ブロード)、67.3、77.3。
(b) メチル デカヒドロ−3a,5a,8a,10a−テトラアザピレン−2−カルボキシラートの製造(化合物13)
(a) オクタヒドロ−2a,5,6,8a−テトラアザアセナフチレンの製造(化合物14)
m/z=169(M+H)+●。
(b) デカヒドロ−2a,4a,6a,8a−テトラアザシクロペンタ[fg]アセナフチレン−1−イルメタノールの製造(化合物15)
(a) 3−アリルオキシ−1,2−プロパンジオール ジトシラートの製造(化合物16)
2.33(s,6H)、3.44(d,2H)、3.72(m,2H)、4.07(m,2H)、4.61(m,1H)、4.99−5.10(m,2H)、5.52−5.71(m,1H)、7.20(m,4H)、7.55−7.70(m,4H)。
21.7(×2)、67.5、67.7、72.3、77.0、117.5、127.9(×3)、129.9(×2)、133.8(×2)、145.3(×2)。
m/z=306(M+●)。
(c) 1−(1,4,7,10−テトラアザシクロトリデカ−5−イル)アリルオキシメタンの製造(化合物18)
1.34(m,2H)、1.96(s,4H)、2.16(m,1H)、2.25−2.55(m,14H)、3.01(m,1H)、3.10(m,1H)、3.63(m,2H)、4.81(dd,1H,J=1.0Hz,10.5Hz)、4.91(dd,1H,J=1.5Hz,15.5Hz)、5.55(m,1H,J=5.5Hz,10.5Hz,15.5Hz)。
(CH2−β)29.1、(CH2−α) 46.2、47.8、48.9、49.2、49.9、49.8、50.4、(CH−) 56.8、(CH2−O) 72.1、72.4、(CH2=C) 116.8、(CH=) 134.8。
m/z=256.89(M+●)。
元素分析(C 13 H 28 N 4 O・H 2 Oとして):
理論値:C 56.89;H 11.03;N 20.42
実測値:C 57.39;H 10.98;N 19.94
14.8(×4)、25.7、45.2、51.5(×2)、52.1(×2)、52.2、55.6(×2)、55.7(×2)、56.0(×2)、56.7(×2)、60.7(×4)、172.2(×4)、176.1。
1.56(m,2H,J=6.3Hz)、2.39(m,1H)、2.45−3.10(m,14H)、2.85(s,3H)、2.87(s,9H)、2.93(s,3H)、2.97(s,3H)、3.01(s,3H)、3.02(s,3H)、3.15−3.6(m,11H)。
(CH2−β)24.2、(CH3−N) 35.8(×3)、36.1、36.8、37.1、37.4(×2)、(CH2−α、CH−α)50.8(×2)、50.9、51.5、52.1(×2)、54.7、54.8、(CH2−CO) 57.4(×2)、58.0、60.2、(CH2−OH) 62.5、(C=O)170.9、171.0、171.0、172.8。
m/z=557.71(M+●)。
1.40(m,2H)、2.3−3.5(m,49H)、3.72(m,2H)、4.93(d,1H,J=10.3Hz)、5.05(dd,1H,J=1.8Hz,17Hz)、5.60(m,1H)。
(CH2−β)23.4、(CH3−N) 35.6(×2)、35.7、35.9、37.1、37.2、37.3、37.5、(CH2−α、CH−α)49.8、50.9、51.9、52.2(×2)、53.2、(×2)、55.4、(CH2−CO) 57.3、57.6、57.5、58.2、(CH2−O) 69.7、72.2、(CH2=) 116.5、(CH=) 135.2、(C=O) 170.5、170.8、(×2)、171.1。
m/z=597.41(M+●)。
(1)Tabushi, I.; Taniguchi, Y.; Kato, H.; Tetrahedron Lett., 1977, 12, 1049-1052。
(2)Tabushi, I.; Fujiyoshi, M.; Heterocycles, 1977, 7, 851-855。
(3)Kimura, E.; Koike, T.; Machida, R.; Nagai, R.; Kodama, M.; Inorg.Chem., 1984, 23, 4181-4188。
(5)Meares, C.F.; DeNardot, S.J.; Cole, W.C.; Mol, M.K., US 4678667, 1987。
(6)Morphy, J.R.; Parker, D.; Alexander, R.; Bains, A.; Carne, A.F.; Eaton, M.A.W.; Harrison, A.; Millican, A.; Phipps, A.; Rhind, S.K.; Titmas, R.; Weatherby, D.; J, Chem, Soc, Chem, Commun., 1988, 156-158。
(8) Kruper, W.J.; Pollock, D.K.; Fordyce, W.A.; Fazio, M.J.; Inbasekaran, M.N.; Muthyala, R., US 5489425, 1996。
(9) Zhu, S.; Kou, F.; Lin, H.; Lin, C.; Lin, M.; Chen, Y.; Inorg, Chem., 1996, 35, 5851-5859。
(11) McAuley, A.; Subramanian, S.; Inorg, Chim, Acta, 2000, 300, 477-486。
(12) Buttafava, A.; Fabbrizzi, L.; Perotti, A.; Seghi, B.; J, Chem, Soc, Chem, Commun., 1982, 1166-1167。
(13) Inouye, Y.; Kanamori, T.; Sugiyama, M.; Yoshida, T.; Koike, T.; Shionoya, M.; Enomoto, K.; Suehiro, K.; Kimura, E.; Antivir, Chem, Chemother., 1995, 6, 337-344。
(15) Ruser, G.; Ritter, W.; Maecke, H.R.; Bioconjugate Chem., 1990, 1, 345-349。
(16) Zhu, S. R.; Lin, H.K.; Lin, C.C.; Kou, F.P.; Chen, Y.T.; Inorg. Chim. Acta, 1995, 228, 225-232。
(18) Nicolaus, V.; Woehrle, D.; Angew. Makromol. Chem., 1992, 198, 179-190。
(19) Moran, J.K.; Greiner, D.P.; Meares, C.F.; Bioconjugate Chem., 1995, 6, 296-301。
(21) Kimura, E.; Koike, T.; Takahashi, M.; J. Chem. Soc. Chem. Commun., 1985, 385-386。
(22) Kimura, E.; Koike, T.; Uenishi, K.; Hediger, M.; Kuramoto, M.; Joko, S.; Arai, Y.; Kodama, M.; Iitaka, Y.; Inorg. Chem., 1987, 26, 2975-2983。
(24) Kimura, E.; Shinonoya, M.; Mita, T.; Iitaka, Y.; J. Chem. Soc. Chem. Commun., 1987, 1712-1714。
(25) Kimura, E.; Kotake, Y.; Koike, T.; Shionoya, M.; Shiro, M.; Inorg.Chem., 1990, 29, 4991-4996。
(27) Moi, M.K.; Meares, C.F.; DeNardo, S.J.; J. Am. Chem. Soc., 1988, 110, 6266-6267。
(28) Gansow, O.A.; Kumar, K., US 4923985, 1990。
(30) Ansari, M.H.; Ahmad, M.; Dicke, K.A.; Bioorg. Med. Chem. Lett., 1993, 3, 1067-1070。
(31) Richman, J.E.; Atkins, T.J.; J. Am .Chem. Soc., 1974, 96, 2268-2270。
(33) Parker, D.; Millican, T.A., WO 89/01476, 1989。
(34) Cox, J.P.L.; Jankowski, K.J.; Kataky, R.; Parker, D.; Beeley, N.R.A.; Boyce, B.A.; Eaton, M.A.W.; Millar, K.; Millican, A.T.; Harrison, A.; Walker, C.; J. Chem. Soc. Chem. Commun., 1989, 797-798。
(36) Schaefer, M.; Meyer, D.; Beaute, S.; Doucet, D.; Magn. Reson .Med., 1991, 22, 238-241。
(37) Muller, F.R.; Handel, H.; Tetrahedron Lett., 1982, 23, 2769-2772。
(39) Wagler, T.R.; Burrows, C.J.; J. Chem. Soc. Chem. Commun., 1987, 277-278。
(40) Marecek, J.F.; Burrows, C.J.; Tetrahedron Lett., 1986, 27, 5943-5946。
(42) Guilard, R.; Meunier, I.; Jean, C.; Boisselier-Cocolios, B., EP 427595, 1991。
(43) Comba, P.; Curtis, N.F.; Lawrance, G.A.; Sargeson, A.M.; Skelton, B.W.; White, A.H.; Inorg. Chem., 1986, 25, 4260-4267。
(45) Comba, P.; Curtis, N.F.; Lawrance, G.A.; O′Leary, M.A.; Skelton, B.W.; White, A.H.; J. Chem. Soc. Dalton Trans., 1988, 497-502。
(46) Comba, P.; Curtis, N.F.; Lawrance, G.A.; O′Leary, M.A.; Skelton, B.W.; White, A.H.; J. Chem. Soc. Dalton Trans., 1988, 2145-2152。
(48) Lawrance, G.A.; Manning, T.M.; Maeder, M.; Martinez, M.; Oleary, M.A.; Patalinghug, W.C.; Skelton, B.W.; White, A.H.; J. Chem. Soc. Dalton Trans., 1992, 1635-1641。
(49) Hambley, T.W.; Lawrance, G.A.; Maeder, M.; Wilkes, E.N.; J. Chem. Soc. Dalton Trans., 1992, 1283-1289。
(51) Comba, P.; Hilfenhaus, P.; J. Chem. Soc. Dalton Trans., 1995, 3269-3274。
(52) Bernhardt, P.V.; Sharpe, P.C.; Inorg. Chem., 2000, 39, 4123-4129。
(54) Edlin, C.D.; Faulkner, S.; Parker, D.; Wilkinson, M.P.; Woods, M.; Lin, J.; Lasri, E.; Neth, F.; Port, M.; New J. Chem., 1998, 1359-1364。
(55) Tundo, P.; Tetrahedron Lett., 1978, 47, 4693-4696。
(57) Takenouchi, K.; Tabe, M.; Watanabe, K.; Hazato, A.; Kato, Y.; Shionoya, M.; Koike, T.; Kimura, E.J.; Org. Chem., 1993, 58, 6895-6899。
(58) Mishra, A.K.; Gestin, J.F.; Benoist, E.; Faivrechauvet, A.; Chatal, J.F.; New J. Chem., 1996, 20, 585-588。
(60) Argese, M.; Ripa, G.; Scala, A.; Valle, V., WO 97/49691, 1997。
(61) Ripa, G.; Argese, M., WO 98/49151, 1998。
(63) Herve, G.; Bernard, H.; Le Bris, N.; Yaouanc, J.J.; Handel, H.; Tetrahedron Lett., 1998, 39, 6861-6864。
(64) Herve, G.; Bernard, H.; Le Bris, N.; Le Baccon, M.; Yaouanc, J.J.; Handel, H.; Tetrahedron Lett., 1999, 40, 2517-2520。
(66) Argese, M.; Manfredi, G.; Rebasti, F.; Ripa, G., WO 00/53588, 2000。
(67) Ferrari, M.; Giovenzana, G.B.; Palmisano, G.; Sisti, M.; Synth. Commun., 2000, 30, 15-21。
(69) Weisman, G.R.; Wong, E.H.; Hill, D.C.; Rogers, M.E.; Reed, D.P.; Calabrese, J.C.; Chem. Commun., 1996, 947-948。
(70) Hubin, T.J.; McCormick, J.M.; Collinson, S.R.; Alcock, N.W.; Busch, D.H.; Chem. Commun., 1998, 1675-1676。
(72) Wong, E.H.; Weisman, G.R.; Hill, D.C.; Reed, D.P.; Rogers, M.E.; Condon, J.S.; Fagan, M.A.; Calabrese, J.C.; Lam, K.C.; Guzei, I.A.; Rheingold, A.L.; J. Am. Chem. Soc., 2000, 122, 10561-10572。
(73) Hiler, G.D.; Perkins, C.M., WO 00/32601, 2000。
Claims (3)
- 式(I):
基AまたはBの一方は−(CH2)−基であり、他方の基は、−CH(R′)−CH2−基[式中、R′は、1〜12の炭素原子を含む飽和もしくは不飽和脂肪族基であるか、または−(CH2)n−O−R1′基[式中、nは0および4の間の整数であり、R1′は水素原子または1〜8の炭素原子を含む飽和もしくは不飽和脂肪族基である]であるか、または−(CH2)n−C(=O)−O−R1′基[式中、nおよびR1′は前記と同意義である]であるか、または−(CH2)n−R2′基[式中、R2′は非置換フェニル基であるか、またはアミノ、ニトロ、クロロ、ブロモ、ヨード、メトキシもしくはヒドロキシ基から選ばれる1またはそれ以上の基で置換されているフェニル基である]である]であるか、
または、−CH(R″)−基[式中、R″は、1〜12の炭素原子を含む飽和もしくは不飽和脂肪族基であるか、または−(CH2)n−O−R1″基[式中、nは0および4の間の整数であり、R1″は水素原子または1〜8の炭素原子を含む飽和もしくは不飽和脂肪族基である]であるか、または−(CH2)n−C(=O)−O−R1″基[式中、nおよびR1″は前記と同意義である]であるか、または−(CH2)n−R2″基[式中、R2″は非置換フェニル基であるか、またはアミノ、ニトロ、クロロ、ブロモ、ヨード、メトキシもしくはヒドロキシ基から選ばれる1またはそれ以上の基で置換されているフェニル基である]である]であり、
そして基Dは−(CH2)m−基[式中、mは0または1に等しい]である]で示される化合物の製造方法であって、以下の連続反応工程:工程(a):式(II):
- 前記工程(a)で使用される式(III)の化合物において、RおよびR1が、互いに独立に、水素原子またはメチル基である請求項1の方法。
- 式(VI):
基AまたはBの一方は−(CH2)−基であり、他方の基は、−CH(R′)−CH2−基[式中、R′は、1〜12の炭素原子を含む飽和もしくは不飽和脂肪族基であるか、または−(CH2)n−O−R1′基[式中、nは0および4の間の整数であり、R1′は水素原子または1〜8の炭素原子を含む飽和もしくは不飽和脂肪族基である]であるか、または−(CH2)n−C(=O)−O−R1′基[式中、nおよびR1′は前記と同意義である]であるか、または−(CH2)n−R2′基[式中、R2′は非置換フェニル基であるか、またはアミノ、ニトロ、クロロ、ブロモ、ヨード、メトキシもしくはヒドロキシ基から選ばれる1またはそれ以上の基で置換されているフェニル基である]である]であるか、
または、−CH(R″)−基[式中、R″は、1〜12の炭素原子を含む飽和もしくは不飽和脂肪族基であるか、または−(CH2)n−O−R1″基[式中、nは0および4の間の整数であり、R1″は水素原子または1〜8の炭素原子を含む飽和もしくは不飽和脂肪族基である]であるか、または−(CH2)n−C(=O)−O−R1″基[式中、nおよびR1″は前記と同意義である]であるか、または−(CH2)n−R2″基[式中、R2″は非置換フェニル基であるか、またはアミノ、ニトロ、クロロ、ブロモ、ヨード、メトキシもしくはヒドロキシ基から選ばれる1またはそれ以上の基で置換されているフェニル基である]である]であり、
そして基Dは−(CH2)m−基[式中、mは0または1に等しい]であり、そして、
RおよびR1は、同じであっても異なっていてもよく、個別に水素原子であるかまたはメチル、エチル、直線状もしくは分枝プロピルまたは直線状もしくは分枝ブチル基から選ばれる基である]で示される化合物。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR0112550A FR2830253B1 (fr) | 2001-09-28 | 2001-09-28 | Nouveau procede de preparation de macrocycles azotes c-fonctionnalises et nouveaux intermediaires obtenus |
| FR01/12550 | 2001-09-28 | ||
| PCT/FR2002/003319 WO2003029228A1 (fr) | 2001-09-28 | 2002-09-27 | Nouveau procede de preparation de macrocycles azotes c-fonctionnalises et nouveaux intermediaires obtenus |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2005508930A JP2005508930A (ja) | 2005-04-07 |
| JP2005508930A5 JP2005508930A5 (ja) | 2006-01-05 |
| JP4699693B2 true JP4699693B2 (ja) | 2011-06-15 |
Family
ID=8867751
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2003532478A Expired - Fee Related JP4699693B2 (ja) | 2001-09-28 | 2002-09-27 | C−官能基化含窒素大環式化合物製造のための新規方法および得られる新規中間体 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US7312327B2 (ja) |
| EP (1) | EP1430036A1 (ja) |
| JP (1) | JP4699693B2 (ja) |
| CA (1) | CA2461344A1 (ja) |
| FR (1) | FR2830253B1 (ja) |
| WO (1) | WO2003029228A1 (ja) |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2818254B1 (fr) * | 2000-12-15 | 2003-02-28 | Immunotech Sa | Conditionnement pour colorants photosensibles |
| FR2856063B1 (fr) * | 2003-06-13 | 2005-10-07 | Air Liquide | Procede de preparation du cis-8b-methyldecahydro-2a,4a,6a, 8a-tetraazacyclopenta[fg]acenaphthylene,ou du cis-decahydro-2a,4a,6a,8a-tetraazacyclopenta[fg] acenaphthylene, du cyclene, et de cyclenes fonctionnalises |
| AU2005280639A1 (en) * | 2004-02-17 | 2006-03-09 | Billy T. Fowler | Methods, compositions, and apparatuses for forming macrocyclic compounds |
| US7296576B2 (en) | 2004-08-18 | 2007-11-20 | Zyvex Performance Materials, Llc | Polymers for enhanced solubility of nanomaterials, compositions and methods therefor |
| FR2928144B1 (fr) * | 2008-02-28 | 2011-09-16 | Centre Nat Rech Scient | Materiaux pour l'extraction solide/liquide de cations de metaux lourds a base de polyazacycloalcanes n-fonctionnalises supportes |
| US9060847B2 (en) * | 2008-05-19 | 2015-06-23 | University Of Rochester | Optical hydrogel material with photosensitizer and method for modifying the refractive index |
| US9144491B2 (en) | 2011-06-02 | 2015-09-29 | University Of Rochester | Method for modifying the refractive index of an optical material |
| FR2982611B1 (fr) * | 2011-11-16 | 2014-01-03 | Univ Bretagne Occidentale | Procedes de preparation de composes tetraazacycloalcanes fonctionnels a l'aide d'un compose cyclique bisaminal particulier. |
Family Cites Families (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS53144593A (en) * | 1977-05-19 | 1978-12-15 | Iwao Tabuse | Alklycyclum |
| US4678667A (en) * | 1985-07-02 | 1987-07-07 | 501 Regents of the University of California | Macrocyclic bifunctional chelating agents |
| FR2614020B1 (fr) * | 1987-04-14 | 1989-07-28 | Guerbet Sa | Nouveaux ligands cycliques azotes, complexes metalliques formes par ces ligands, compositions de diagnostic contenant ces complexes et procede de preparation des ligands. |
| US5049667A (en) * | 1987-04-14 | 1991-09-17 | Guerbet S.A. | Nitrogen-containing cyclic ligands |
| DE3713842A1 (de) * | 1987-04-22 | 1988-11-17 | Schering Ag | Substituierte cyclische komplexbildner, komplexe und komplexsalze, verfahren zu deren herstellung und diese enthaltende pharmazeutische mittel |
| DE3869251D1 (de) * | 1987-07-16 | 1992-04-23 | Nycomed As | Aminopolycarbonsaeure und ihre derivate. |
| DE3728600A1 (de) * | 1987-08-27 | 1989-03-09 | Hoechst Ag | Verfahren zur markierung von substanzen mit technetium oder rhenium |
| BR8907282A (pt) * | 1988-12-23 | 1991-03-12 | Dow Chemical Co | Processo para preparar complexos de metal funcionalizados com isotiocianato |
| EP0730616B1 (en) * | 1993-11-26 | 2003-02-05 | Dow Global Technologies Inc. | Process for preparing polyazamacrocycles |
| GB9504922D0 (en) * | 1995-03-10 | 1995-04-26 | Nycomed As | Process |
| WO2000021941A1 (en) * | 1998-10-13 | 2000-04-20 | Synchem Reserach, Inc. | Biomimetic chelating agents and methods |
| EP1135392A2 (en) * | 1998-11-30 | 2001-09-26 | The Procter & Gamble Company | Process for preparing cross-bridged tetraaza macrocycles |
| DE19856481C1 (de) * | 1998-12-02 | 2000-07-06 | Schering Ag | Verfahren zur Herstellung von Cyclen |
| IT1309601B1 (it) * | 1999-03-09 | 2002-01-24 | Bracco Spa | Processo per la preparazione di 1,4,7,10 tetraazaciclododecano. |
-
2001
- 2001-09-28 FR FR0112550A patent/FR2830253B1/fr not_active Expired - Fee Related
-
2002
- 2002-09-27 JP JP2003532478A patent/JP4699693B2/ja not_active Expired - Fee Related
- 2002-09-27 US US10/490,948 patent/US7312327B2/en not_active Expired - Fee Related
- 2002-09-27 WO PCT/FR2002/003319 patent/WO2003029228A1/fr not_active Ceased
- 2002-09-27 CA CA002461344A patent/CA2461344A1/fr not_active Abandoned
- 2002-09-27 EP EP02783226A patent/EP1430036A1/fr not_active Withdrawn
Also Published As
| Publication number | Publication date |
|---|---|
| JP2005508930A (ja) | 2005-04-07 |
| FR2830253A1 (fr) | 2003-04-04 |
| US7312327B2 (en) | 2007-12-25 |
| EP1430036A1 (fr) | 2004-06-23 |
| FR2830253B1 (fr) | 2005-02-04 |
| US20040206940A1 (en) | 2004-10-21 |
| CA2461344A1 (fr) | 2003-04-10 |
| WO2003029228A1 (fr) | 2003-04-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| FI93830C (fi) | Typpipitoisia syklisiä ligandeja, menetelmä niiden valmistamiseksi sekä niiden käyttö metallikompleksien ja näitä komplekseja sisältävien diagnostisten koostumusten valmistamiseksi | |
| KR100269081B1 (ko) | N-베타-히드록시알킬-트리-n-카르복시알킬-1,4,7,10-테트라아자시클로도데칸및n-베타-히드록시알킬-트리-n-카르복시알킬-1,4,8,11-테트라아자시클로테트라데칸유도체및그들의금속착화합물의제조방법 | |
| Denat et al. | Strategies for the regioselective N-functionalization of tetraazacycloalkanes. From cyclam and cyclen towards more sophisticated molecules | |
| EP2922830B1 (en) | Synthesis of imidazo(1,2-a)pyrazin-4-ium salts for the synthesis of 1,4,7-triazacyclononane (tacn) and n- and/or c-functionalized derivatives thereof | |
| US5587451A (en) | Process for preparing polyazamacrocycles | |
| JP4699693B2 (ja) | C−官能基化含窒素大環式化合物製造のための新規方法および得られる新規中間体 | |
| JP2004509152A (ja) | 放射性金属標識分子のプロキレート剤 | |
| JPH11509516A (ja) | テトラアザシクロアルカンの製造方法 | |
| JP3119872B2 (ja) | 前もって固定された対称性をもつキレート化剤のための中間体およびそれらの製造方法 | |
| SK357392A3 (en) | Process for preparing mono-n-substituted derivatives of tetraazacyclododecane and tetraazacyclotetradecane | |
| AU679140B2 (en) | Process for preparing polyazamacrocycles | |
| US5705637A (en) | Polyazacycloalkane compounds | |
| JP5518465B2 (ja) | テトラアザ大員環誘導体の金属錯体 | |
| US7659393B2 (en) | Method of preparing cis-8b-methyldecahydro-2a,4a,6a,8a-tetraazacyclopenta[fg]acenaphthylene, cis-decahydro-2a,4a,6a,8a-tetraazacyclopenta[fg]acenaphthylene, cyclene and functionalised cyclenes | |
| Formanovsky et al. | One-Stage Monosubstitution in Cyclen-Two Novel Examples | |
| KR20190108383A (ko) | 옥사미드 및 수소화알루미늄리튬을 이용한 시클렌의 신규 합성법 | |
| CZ288287B6 (en) | Process for preparing 5-(alkoxymethyl)-2,3-pyridine dicarboximide compounds | |
| JPH10511977A (ja) | ポリアザシクロアルカン化合物 | |
| PL212714B1 (pl) | Nowe związki hybrydowe porfiryna-furoksan | |
| Formanovsky et al. | Shemyakin and Ovchinnikov Institute of Bioorganic Chemistry, 117871, Moscow, Russia |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20050907 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20050907 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20081203 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20081209 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20090306 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20090313 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20090609 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20090825 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20091225 |
|
| A911 | Transfer to examiner for re-examination before appeal (zenchi) |
Free format text: JAPANESE INTERMEDIATE CODE: A911 Effective date: 20100412 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20100817 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20101117 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20110208 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20110303 |
|
| LAPS | Cancellation because of no payment of annual fees |