JP4558835B2 - 重合体、半導体膜、電極、電極活物質、電気化学素子および蓄電デバイス - Google Patents
重合体、半導体膜、電極、電極活物質、電気化学素子および蓄電デバイス Download PDFInfo
- Publication number
- JP4558835B2 JP4558835B2 JP2009178610A JP2009178610A JP4558835B2 JP 4558835 B2 JP4558835 B2 JP 4558835B2 JP 2009178610 A JP2009178610 A JP 2009178610A JP 2009178610 A JP2009178610 A JP 2009178610A JP 4558835 B2 JP4558835 B2 JP 4558835B2
- Authority
- JP
- Japan
- Prior art keywords
- group
- atom
- polymer
- saturated hydrocarbon
- active material
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 0 CC(C)C#Cc(cc1)ccc1C#CC(*1)=C(*)*C1=C1*C(CC*)=C(*)*1 Chemical compound CC(C)C#Cc(cc1)ccc1C#CC(*1)=C(*)*C1=C1*C(CC*)=C(*)*1 0.000 description 5
- UXYULNILNJCKTO-ISLYRVAYSA-N C(S1)=C(c2ccccc2)S/C1=C1/SC(c2ccccc2)=CS1 Chemical compound C(S1)=C(c2ccccc2)S/C1=C1/SC(c2ccccc2)=CS1 UXYULNILNJCKTO-ISLYRVAYSA-N 0.000 description 1
- ZTUNWLVWZUOXLR-XQRVVYSFSA-N CCC/C=C1\[ClH]C1 Chemical compound CCC/C=C1\[ClH]C1 ZTUNWLVWZUOXLR-XQRVVYSFSA-N 0.000 description 1
- JXMGMMVUCAFFHT-ISLYRVAYSA-N IC(S1)=C(c2ccccc2)S/C1=C1\SC(I)=C(c2ccccc2)S1 Chemical compound IC(S1)=C(c2ccccc2)S/C1=C1\SC(I)=C(c2ccccc2)S1 JXMGMMVUCAFFHT-ISLYRVAYSA-N 0.000 description 1
Images







Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G61/00—Macromolecular compounds obtained by reactions forming a carbon-to-carbon link in the main chain of the macromolecule
- C08G61/12—Macromolecular compounds containing atoms other than carbon in the main chain of the macromolecule
- C08G61/122—Macromolecular compounds containing atoms other than carbon in the main chain of the macromolecule derived from five- or six-membered heterocyclic compounds, other than imides
- C08G61/123—Macromolecular compounds containing atoms other than carbon in the main chain of the macromolecule derived from five- or six-membered heterocyclic compounds, other than imides derived from five-membered heterocyclic compounds
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01G—CAPACITORS; CAPACITORS, RECTIFIERS, DETECTORS, SWITCHING DEVICES OR LIGHT-SENSITIVE DEVICES, OF THE ELECTROLYTIC TYPE
- H01G11/00—Hybrid capacitors, i.e. capacitors having different positive and negative electrodes; Electric double-layer [EDL] capacitors; Processes for the manufacture thereof or of parts thereof
- H01G11/22—Electrodes
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01G—CAPACITORS; CAPACITORS, RECTIFIERS, DETECTORS, SWITCHING DEVICES OR LIGHT-SENSITIVE DEVICES, OF THE ELECTROLYTIC TYPE
- H01G11/00—Hybrid capacitors, i.e. capacitors having different positive and negative electrodes; Electric double-layer [EDL] capacitors; Processes for the manufacture thereof or of parts thereof
- H01G11/22—Electrodes
- H01G11/30—Electrodes characterised by their material
- H01G11/48—Conductive polymers
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01G—CAPACITORS; CAPACITORS, RECTIFIERS, DETECTORS, SWITCHING DEVICES OR LIGHT-SENSITIVE DEVICES, OF THE ELECTROLYTIC TYPE
- H01G11/00—Hybrid capacitors, i.e. capacitors having different positive and negative electrodes; Electric double-layer [EDL] capacitors; Processes for the manufacture thereof or of parts thereof
- H01G11/54—Electrolytes
- H01G11/56—Solid electrolytes, e.g. gels; Additives therein
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01G—CAPACITORS; CAPACITORS, RECTIFIERS, DETECTORS, SWITCHING DEVICES OR LIGHT-SENSITIVE DEVICES, OF THE ELECTROLYTIC TYPE
- H01G11/00—Hybrid capacitors, i.e. capacitors having different positive and negative electrodes; Electric double-layer [EDL] capacitors; Processes for the manufacture thereof or of parts thereof
- H01G11/54—Electrolytes
- H01G11/58—Liquid electrolytes
- H01G11/62—Liquid electrolytes characterised by the solute, e.g. salts, anions or cations therein
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M10/00—Secondary cells; Manufacture thereof
- H01M10/05—Accumulators with non-aqueous electrolyte
- H01M10/052—Li-accumulators
- H01M10/0525—Rocking-chair batteries, i.e. batteries with lithium insertion or intercalation in both electrodes; Lithium-ion batteries
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/36—Selection of substances as active materials, active masses, active liquids
- H01M4/60—Selection of substances as active materials, active masses, active liquids of organic compounds
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/36—Selection of substances as active materials, active masses, active liquids
- H01M4/60—Selection of substances as active materials, active masses, active liquids of organic compounds
- H01M4/602—Polymers
- H01M4/606—Polymers containing aromatic main chain polymers
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K85/00—Organic materials used in the body or electrodes of devices covered by this subclass
- H10K85/10—Organic polymers or oligomers
- H10K85/111—Organic polymers or oligomers comprising aromatic, heteroaromatic, or aryl chains, e.g. polyaniline, polyphenylene or polyphenylene vinylene
- H10K85/113—Heteroaromatic compounds comprising sulfur or selene, e.g. polythiophene
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K85/00—Organic materials used in the body or electrodes of devices covered by this subclass
- H10K85/10—Organic polymers or oligomers
- H10K85/141—Organic polymers or oligomers comprising aliphatic or olefinic chains, e.g. poly N-vinylcarbazol, PVC or PTFE
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G2261/00—Macromolecular compounds obtained by reactions forming a carbon-to-carbon link in the main chain of the macromolecule
- C08G2261/10—Definition of the polymer structure
- C08G2261/14—Side-groups
- C08G2261/141—Side-chains having aliphatic units
- C08G2261/1412—Saturated aliphatic units
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G2261/00—Macromolecular compounds obtained by reactions forming a carbon-to-carbon link in the main chain of the macromolecule
- C08G2261/30—Monomer units or repeat units incorporating structural elements in the main chain
- C08G2261/33—Monomer units or repeat units incorporating structural elements in the main chain incorporating non-aromatic structural elements in the main chain
- C08G2261/334—Monomer units or repeat units incorporating structural elements in the main chain incorporating non-aromatic structural elements in the main chain containing heteroatoms
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G2261/00—Macromolecular compounds obtained by reactions forming a carbon-to-carbon link in the main chain of the macromolecule
- C08G2261/30—Monomer units or repeat units incorporating structural elements in the main chain
- C08G2261/34—Monomer units or repeat units incorporating structural elements in the main chain incorporating partially-aromatic structural elements in the main chain
- C08G2261/342—Monomer units or repeat units incorporating structural elements in the main chain incorporating partially-aromatic structural elements in the main chain containing only carbon atoms
- C08G2261/3422—Monomer units or repeat units incorporating structural elements in the main chain incorporating partially-aromatic structural elements in the main chain containing only carbon atoms conjugated, e.g. PPV-type
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G2261/00—Macromolecular compounds obtained by reactions forming a carbon-to-carbon link in the main chain of the macromolecule
- C08G2261/50—Physical properties
- C08G2261/51—Charge transport
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01G—CAPACITORS; CAPACITORS, RECTIFIERS, DETECTORS, SWITCHING DEVICES OR LIGHT-SENSITIVE DEVICES, OF THE ELECTROLYTIC TYPE
- H01G11/00—Hybrid capacitors, i.e. capacitors having different positive and negative electrodes; Electric double-layer [EDL] capacitors; Processes for the manufacture thereof or of parts thereof
- H01G11/04—Hybrid capacitors
- H01G11/06—Hybrid capacitors with one of the electrodes allowing ions to be reversibly doped thereinto, e.g. lithium ion capacitors [LIC]
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E60/00—Enabling technologies; Technologies with a potential or indirect contribution to GHG emissions mitigation
- Y02E60/10—Energy storage using batteries
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E60/00—Enabling technologies; Technologies with a potential or indirect contribution to GHG emissions mitigation
- Y02E60/13—Energy storage using capacitors
Description

本実施形態では、本発明の重合体を膜あるいは層の状態で用いる基本的な形態を説明する。まず、導電性支持体に一般式(1)で示される重合体を含む重合体膜または重合体層が支持された電極を説明する。このような電極は、次の実施形態で説明する蓄電デバイスである二次電池や二次電池以外の電気二重層キャパシタなどに用いてもよいし、生化学反応を利用するバイオチップなどの電気化学素子に好適に用いることができる。
以下、図面を参照しながら、本発明による重合体を蓄電材料として用いる蓄電デバイスの実施形態を説明する。本実施形態では、リチウム二次電池を例に挙げて本発明による蓄電デバイスおよび本発明による蓄電材料を説明する。しかし、本発明はリチウム二次電池やリチウム二次電池の正極活物質に限られず、化学反応を利用したキャパシタなどにも好適に用いられる。
一般式(3)においてXがSであり、R1およびR2がフェニル基であるポリ−(2,6−ジフェニルテトラチアフルバレン)−(1,3−ジエチニルベンゼン)共重合体(化合物7)を以下の式(R3)に示すように、前駆体化合物8の合成を行い、得られた化合物8と化合物9をカップリングすることにより合成した。以下合成法を順に示す。
化合物8を以下の式(R4)に従って合成した。
窒素雰囲気下、30mlのシュレンク管に合成した0.7g(1.15mmol)の化合物8をとり、30mlのTHFを加えた。これに、10.95mg(0.0575mmol)のヨウ化銅を加え、さらに66.4mg(0.0575mmol)のテトラキス(トリフェニルホスフィン)パラジウム(以下、Pd(PPh3)4)、1mlを加え撹拌した。この溶液に0.0145g(1.15mmol)の化合物9である1,3−ジエチニルベンゼンおよび1mlのトリエチルアミンを加え、60℃で24時間撹拌した。その後、ろ過し、1N塩酸水溶液およびエタノールで生成物を洗浄し、乾燥後赤褐色粉末を得た。収率は62%であった。
一般式(3)においてXがSであり、R1およびR2がメチル基であるポリ−(4,4’−ジメチルテトラチアフルバレン)−(1,3−ジエチニルベンゼン)共重合体(化合物10)を以下の式(R5)に示すように、前駆体化合物11の合成を行い、得られた化合物11と化合物9をカップリングすることにより合成した。以下合成法を順に示す。
化合物11を以下の式(R6)に従って合成した。
化合物11(0.12g、0.25mmol)および2mlのトリエチルアミンを10mlのN−メチルピロリドン(0.12g、0.25mmol)に溶解し、10分間窒素バブリングを行った後、Pd(PPh3)4(0.060g、0.05mml)、CuI(0.020g、0.10mmol)および1,3−ジエチニルベンゼン(0.049、0.39mmol)を加え、100℃で攪拌した。溶液の色は暗赤色から濃い赤橙色へと変化した。24時間攪拌した後、反応液を水に投入し、黒赤色の固体を得た。固体をメタノール、アセトンの順で攪拌洗浄した後、桐山ロートで単離し、自然乾燥させ、こげ茶色の生成物を得た。生成物が化合物10であることをH−NMR(CDCl3)およびIR測定(KBr法)により確認した。収量は0.07g(79%)であった。GPC測定から得られたピーク分子量はポリスチレン換算で36484であった。
一般式(2)においてXがSであり、R1およびR2がデシル基であるポリ−(4,4’−ジデシルテトラチアフルバレン)−(1,4−ジエチニルベンゼン)共重合体(化合物14)を以下の式(R7)に示すように、前駆体化合物12の合成を行い、得られた化合物12と化合物13とをカップリングすることにより合成した。以下合成法を順に示す。
1000mlのナスフラスコ中、350mlのジメチルスルホキシド(DMSO)に16.9gの1−ドデカンを溶解し、15mlのH2Oおよび54gのN−ブロモスクシンイミド(NBS)を加え、室温で4時間攪拌した。その後、エーテルで抽出し、乾燥、溶媒を減圧除去した。精製後、無色透明液体を得た。収率は59%であった。
1000mlのナスフラスコ中、110mlのアセトンに14gの化合物12a(1−ブロモ−2−ドデカノール)を溶解し、66mlの蒸留水および11mlの硫酸にあらかじめ溶解させた15gのニクロム酸ナトリウム2水和物を滴下した。室温で1.5時間攪拌後、250mlのエーテルを加え、脱水、溶媒除去後、白色固体を得た。収率は80%であった。
1000mlのナスフラスコ中、400mlのアセトンに9.2gの化合物12b(1−ブロモ−2−ドデカノン)を溶解し、50℃に加熱した。その後、5.6gのキサントゲン酸カリウムを加え、4時間還流した。還流後、蒸留水に反応液を注ぎ、エーテルにて抽出し、乾燥、溶媒除去を行い、黄色結晶を得た。収率は45%であった。
1000mlのナスフラスコ中、600mlの脱水トルエンに44gの化合物12c(O−エチル−1−キサンチルドデカン−2−オン)を溶解し、沸点近くまで過熱した。その後、120gの5硫化2リンを徐々に投入し、約20時間還流した。得られた溶液をろ過し、エーテルで抽出、乾燥し、溶媒除去後、赤色油状の目的物を得た。収率は63%であった。
窒素ガス気流下、500mlのシュレンク管に3.3gの化合物12d(4−デシル−1,3−ジチオール−2−チオン)を入れ、140mlのアセトンに溶解させた。あらかじめアセトン210mlに溶解させた48gのm−クロロ過安息香酸を滴下した。滴下後30分攪拌し、アセトンを除去後、塩化メチレン220mlに溶解し、均一になったところで20gのヘキサフルオロリン酸ナトリウムを加えた。室温で1時間攪拌し、200mlのアセトニトリルを加えた。15分攪拌し、56mlのトリエチルアミンを加え、さらに1時間攪拌した。その後、エーテルで抽出し、乾燥、溶媒除去後、精製、再結晶を行い、橙色粉末を得た。収率は22%であった。
窒素ガス気流下、100mlのシュレンク管に1.1gの化合物12e(2,6−ジデシルTTF)を25mlのTHFに溶解し、ドライアイス−メタノール浴で−78℃まで冷却した。その後4.4mlのブチルリチウム(BuLi)を滴下し、10分攪拌した。反応液に、1.5mlのパーフルオロヘキシルヨージド(PFHI)を滴下し、−78℃で1時間攪拌し、続いて室温で1時間攪拌した後、蒸留水を滴下し反応を終了させた。エーテルにて抽出、脱水、溶媒除去後、精製、再結晶を得て橙色粉末を得た。収率は35%であった。
得られた前駆体化合物12と市販の化合物13とを、化合物10の合成と同じ方法により、式(R7)に従って反応させ、茶褐色粉末を得た。収率は40%であった。
比較のため、テトラチアフルバレン骨格を主鎖ではなく、側鎖に含む化合物15、および、テトラチアフルバレン骨格間にアセチレンおよびフェニル基を含まない化合物16を合成した。
下記化学式(15)で示される重合体(化合物15)を合成した。化学式(15)の重合体(poly−TTF)は、ポリビニルアルコールとテトラチアフルバレンカルボキシル誘導体を脱水縮合により反応させて合成した。化合物15の重量平均分子量はおよそ50000であった。
下記、化学式(16)で示される重合体(化合物16)を合成した。化学式16で表される重合体は、以下に示す反応式(R9)に従って合成した。
デカン−1−エン(化合物16a、126.4g、0.09mol)を2000mlのナスフラスコに取り、DMSO(1500ml)、蒸留水(88ml)、NBS(320g、1.8mol)を加え4時間攪拌した。その後、エーテルで抽出し、乾燥、溶媒を除去し、得られた試料をシリカゲルを用いたカラムクロマトグラフィーで生成し、無色透明液体を得た。収率は98%であった。
化合物16b(210g、860mmol)を2000mlナスフラスコに取り、アセトン(900ml)に溶解させ、蒸留水(900ml)に硫酸(160ml)、2クロム酸ナトリウム2水和物(260g、880mmol)を溶解させたものを加え1.5時間攪拌した。その後、エーテルを加え、さらに1時間攪拌した。エーテルで抽出し、乾燥、溶媒を除去し、得られた試料をシリカゲルを用いたカラムクロマトグラフィーで生成し、白色固体を得た。収率は92%であった。
2000mlのナスフラスコに、アセトン(1400ml)を取り、化合物16c(150g、620mmol)を加え50℃に加熱した。キサントゲン酸カリウム(100g、620mmol)を少量ずつ加え、4時間還流した。その後、蒸留水に反応液を注ぎ、エーテルで抽出した後、乾燥、溶媒除去し、黄色透明液体を得た。収率は77%であった。
2000mlのナスフラスコに、脱水トルエン(1300ml)を取り、化合物16d(130g、450mmol)を溶解し、沸点近くまで加熱した。その後5硫化2リン(171g、770mmol)をゆっくり加え20時間還流した。得られた溶液を濾過し、5硫化2リンを除去、エーテル抽出を行った。乾燥、溶媒除去し、黄色粉末を得た。収率は82%であった。
窒素ガス気流下、500mlのシュレンク管に化合物16e(3.1g、12mmol)を取り、アセトン140mlに溶解させ、20℃に温度を保持した。そこにあらかじめアセトン(210ml)に溶解させたm−クロロ安息香酸(48g、300mmol)を滴下し、滴下後30分攪拌し、アセトン除去後塩化メチレン(220ml)に溶解させた。そこへヘキサフルオロリン酸ナトリウム(20g、120mmol)を加えた。室温で1時間攪拌後、アセトニトリル(200ml)を加え、温度を20℃に保ちながら15分間攪拌し、トリエチルアミン(56ml)を加えさらに1時間攪拌した。その後、エーテル抽出、乾燥、溶媒除去し、橙色粉末を得た。収率は23%であった。
窒素ガス気流下、100mlのシュレンク管に化合物16f(0.99g、2.3mmol)を取り、THF(25ml)に溶解させ−78℃まで冷却した。そこにブチルリチウム(4.4ml、1.53mol/Lへキサン溶液)をシリンジで滴下し、10分間攪拌した。その後、トリフルオロヘキシリヨード(PFHI、1.5ml)を滴下し、−78℃で1時間、室温で1時間攪拌し、蒸留水を加え反応を終了させた。その後、エーテルで抽出し、乾燥、溶媒除去し、ヘキサンで再結晶を行ったのち、橙色粉末を得た。収率は40%であった。
窒素ガス気流下、50mlのシュレンク管に、Ni(cod)2(0.28g、1.0mmol)、1,5−cod(0.11g、1.0mmol)を加え、DMF7mlに溶解させた。そこに2,2’−ビピリジン(0.19g、1.2mmol)を加え、溶液の色が紫色になったことを確認したのち化合物16g(0.46g、0.67mmol)を加えた。50℃で24時間攪拌後、メタノールに反応溶液を直接投入した。得られた粉末を洗浄、濾過、メタノールを用い再沈殿を行い、乾燥し、茶色粉末を得た。
1.1 蓄電デバイス用電極の作製
化合物7、7’、10および14を用いて実施例の電極を作製した。化合物7は混合前に乳鉢で粉砕してから用いた。乳鉢粉砕後の粒子径はおよそ10μm程度であった。37.5mgの化合物7と導電助剤として100mgアセチレンブラックとを均一に混合し、さらに結着剤としてポリテトラフルオロエチエン25mgを加えて混合し、ペーストを得た。得られたペーストをアルミニウム金網の上に圧着し、真空乾燥を行ない、これを直径13.5mmの円盤状に打ち抜き裁断して電極膜を作製した。重合体化合物の塗布重量は、単位面積あたり1.0mg/cm2であった。同様にして、化合物7’、10および14を用いて電極を作製した。化合物7、7’、10および14を用いた電極を以下、電極A、A’、BおよびCと呼ぶ。
電極A、A’、BおよびCと比較例の電極D、Eの電気化学的な酸化還元反応に伴う安定性評価を行った。安定性評価セルは、電極A、A’BおよびCと比較例の電極D、Eを作用極とし、対極および参照電極としてそれぞれリチウム金属を電解液に浸したビーカーセル内に配置して構成した。電解液には、炭酸プロピレン(PC)を溶媒とし、6フッ化リン酸リチウムを支持電解質塩として溶解したものを用いた。濃度は1mol/Lに調製した。
次に、蓄電デバイスを作製し評価を行った。
電極A、A’BおよびC並びに比較例の電極DおよびEをそれぞれ正極とし、負極として、金属リチウム(厚み300μm)を用いた。負極は、直径15mmの円盤状に打ち抜き、直径15mmの円盤状の集電板(ステンレス製)に貼り付けて作製した。
蓄電デバイスの容量評価及び繰り返し特性評価は、各材料の酸化還元が起こる電位領域を充放電電圧範囲として実施した。具体的には、蓄電デバイスA1、A1’は充電上限電圧を4V、放電下限電圧を3.2Vの電圧範囲とし、蓄電デバイスB1は、充電上限電圧を4.0V、放電下限電圧を3.2Vの電圧範囲、蓄電デバイスC1〜E1は、充電上限電圧を4.0V、放電下限電圧を3.0Vの電圧範囲とした。0.1mAの定電流充放電を行い、充電終了後、放電を開始するまでの休止時間はゼロとした。この充放電を50サイクル繰り返した。蓄電デバイスA1’の1〜2サイクル目の充放電カーブを図3に示す。また、蓄電デバイスA1、B1およびC1の3サイクル目の充放電カーブをそれぞれ図4、図5および図6に示す。
上述の蓄電デバイスとは異なる構造を備えた蓄電デバイスを作製し、評価を行った。
図7および図8は、作製したラミネート型リチウム二次電池の模式的な断面および上面を示している。
蓄電デバイスA2、B2、C2および比較例の蓄電デバイスE2の評価として、充放電容量評価を行い、ついで充放電サイクル特性評価を行った。本実施例では、負極としてLi金属よりもサイクル特性に優れる活性炭を用いているため、より長いサイクル回数のサイクル特性を評価することができる。
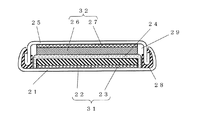
22 正極集電体
23 正極活物質層
24 セパレータ
25 封口板
26 負極活物質層
27 負極集電体
28 ガスケット
29 電解液
31 正極
32 負極
41 正極活物質粒子
42 導電剤部
Claims (24)
- 下記一般式(2)で示される構造を有し、
一般式(2)中、Xは酸素原子、硫黄原子、セレン原子、またはテルル原子、R1、R2はそれぞれ独立した鎖状飽和炭化水素基、鎖状不飽和炭化水素基、環状飽和炭化水素基、環状不飽和炭化水素基、フェニル基、水素原子、ヒドロキシル基、シアノ基、アミノ基、ニトロ基およびニトロソ基よりなる群から選ばれる少なくとも1種を含み、前記鎖状飽和炭化水素基、鎖状不飽和炭化水素基、環状飽和炭化水素基および環状不飽和炭化水素基は、それぞれ、炭素原子、酸素原子、窒素原子、硫黄原子、ケイ素原子よりなる群から選ばれる少なくとも1種を含む重合体。
- 下記一般式(3)で示される構造を有し、
一般式(3)中、Xは酸素原子、硫黄原子、セレン原子、またはテルル原子、R1、R2はそれぞれ独立した鎖状飽和炭化水素基、鎖状不飽和炭化水素基、環状飽和炭化水素基、環状不飽和炭化水素基、フェニル基、水素原子、ヒドロキシル基、シアノ基、アミノ基、ニトロ基およびニトロソ基よりなる群から選ばれる少なくとも1種を含み、前記鎖状飽和炭化水素基、鎖状不飽和炭化水素基、環状飽和炭化水素基および環状不飽和炭化水素基は、それぞれ、炭素原子、酸素原子、窒素原子、硫黄原子、ケイ素原子よりなる群から選ばれる少なくとも1種を含む重合体。
- 前記Xは硫黄原子である請求項1または2に記載の重合体。
- 前記nは4以上である請求項1から3のいずれかに記載の重合体。
- 前記Xは硫黄原子であり、前記R1およびR2は鎖状飽和炭化水素基である請求項1に記載の重合体。
- 前記Xは硫黄原子であり、前記R1およびR2はそれぞれフェニル基である請求項1に記載の重合体。
- 前記Xは硫黄原子であり、前記R1およびR2はそれぞれフェニル基である請求項2に記載の重合体。
- 前記Xは硫黄原子であり、前記R1およびR2はそれぞれメチル基である請求項2に記載の重合体。
- 請求項1から8のいずれかによって規定される重合体を含む半導体膜。
- 導電性支持体と、
前記導電性支持体上に設けられており、請求項1から8のいずれかによって規定される重合体を含む重合体膜と、
を備えた電極。 - 前記重合体膜は導電性物質を含む請求項10に記載の電極。
- 下記一般式(2)で示される構造を有し、
一般式(2)中、Xは酸素原子、硫黄原子、セレン原子、またはテルル原子、R1、R2はそれぞれ独立した鎖状飽和炭化水素基、鎖状不飽和炭化水素基、環状飽和炭化水素基、環状不飽和炭化水素基、フェニル基、水素原子、ヒドロキシル基、シアノ基、アミノ基、ニトロ基およびニトロソ基よりなる群から選ばれる少なくとも1種を含み、前記鎖状飽和炭化水素基、鎖状不飽和炭化水素基、環状飽和炭化水素基および環状不飽和炭化水素基は、それぞれ、炭素原子、酸素原子、窒素原子、硫黄原子、ケイ素原子よりなる群から選ばれる少なくとも1種を含む電極活物質。
- 下記一般式(3)で示される構造を有し、
一般式(3)中、Xは酸素原子、硫黄原子、セレン原子、またはテルル原子、R1、R2はそれぞれ独立した鎖状飽和炭化水素基、鎖状不飽和炭化水素基、環状飽和炭化水素基、環状不飽和炭化水素基、フェニル基、水素原子、ヒドロキシル基、シアノ基、アミノ基、ニトロ基およびニトロソ基よりなる群から選ばれる少なくとも1種を含み、前記鎖状飽和炭化水素基、鎖状不飽和炭化水素基、環状飽和炭化水素基および環状不飽和炭化水素基は、それぞれ、炭素原子、酸素原子、窒素原子、硫黄原子、ケイ素原子よりなる群から選ばれる少なくとも1種を含む電極活物質。
- 前記Xは硫黄原子である請求項12または13に記載の電極活物質。
- 前記nは4以上である請求項12から14のいずれかに記載の電極活物質。
- 前記Xは硫黄原子であり、前記R1およびR2は鎖状飽和炭化水素基である請求項12に記載の電極活物質。
- 前記Xは硫黄原子であり、前記R1およびR2はそれぞれフェニル基である請求項12に記載の電極活物質。
- 前記Xは硫黄原子であり、前記R1およびR2はそれぞれフェニル基である請求項13に記載の電極活物質。
- 前記Xは硫黄原子であり、前記R1およびR2はそれぞれメチル基である請求項13に記載の電極活物質。
- 正極と、負極と、前記正極および前記負極の間に配置された電解液とを備え、
前記正極および前記負極の少なくとも一方が、請求項12から19のいずれかに規定される電極活物質を含む電気化学素子。 - 前記電解液が4級アンモニウムカチオンまたはリチウムイオンとアニオンとの塩を含んでいる請求項20に記載の電気化学素子。
- 請求項12から19のいずれかに規定される電極活物質を含む正極と、
リチウムイオンを吸蔵放出可能な負極活物質を含む負極と、
前記リチウムイオンとアニオンとからなる塩を含み、前記正極および負極の間に満たされた電解液と、
を備えた蓄電デバイス。 - 請求項22に規定される蓄電デバイスを備えた携帯型電子機器。
- 請求項22に規定される蓄電デバイスを備えた車両。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2009178610A JP4558835B2 (ja) | 2008-07-31 | 2009-07-31 | 重合体、半導体膜、電極、電極活物質、電気化学素子および蓄電デバイス |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2008198501 | 2008-07-31 | ||
| JP2009178610A JP4558835B2 (ja) | 2008-07-31 | 2009-07-31 | 重合体、半導体膜、電極、電極活物質、電気化学素子および蓄電デバイス |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2010053358A JP2010053358A (ja) | 2010-03-11 |
| JP2010053358A5 JP2010053358A5 (ja) | 2010-07-08 |
| JP4558835B2 true JP4558835B2 (ja) | 2010-10-06 |
Family
ID=41610202
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2009178610A Expired - Fee Related JP4558835B2 (ja) | 2008-07-31 | 2009-07-31 | 重合体、半導体膜、電極、電極活物質、電気化学素子および蓄電デバイス |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US8481674B2 (ja) |
| EP (1) | EP2308912B1 (ja) |
| JP (1) | JP4558835B2 (ja) |
| CN (1) | CN102015821B (ja) |
| WO (1) | WO2010013492A1 (ja) |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8530086B2 (en) | 2010-07-01 | 2013-09-10 | Panasonic Corporation | Non-aqueous electrolyte secondary battery |
| JP6006789B2 (ja) * | 2011-06-03 | 2016-10-12 | ▲蘇▼州宝▲時▼得▲電▼▲動▼工具有限公司 | 電池 |
| JP2013239305A (ja) * | 2012-05-14 | 2013-11-28 | Nitto Denko Corp | 蓄電デバイス、それに用いる正極並びに多孔質シート、およびドープ率向上方法 |
| JP2014139927A (ja) * | 2012-12-20 | 2014-07-31 | Nitto Denko Corp | 蓄電デバイス、およびそれに用いる電極並びに多孔質シート |
| US9379418B2 (en) | 2013-06-20 | 2016-06-28 | Hrl Laboratories, Llc | Battery with reference electrode for voltage monitoring |
| US10593988B2 (en) | 2013-06-20 | 2020-03-17 | GM Global Technology Operations LLC | Electrochemical cell for lithium-based batteries |
| CN104558541B (zh) * | 2014-12-19 | 2017-04-05 | 华南理工大学 | 基于炔酮中间体的共轭高分子聚合物及其制备方法与应用 |
| CN110358058B (zh) * | 2019-07-16 | 2022-02-18 | 武汉轻工大学 | 一种有机光电材料及其制备方法、发光器件以及显示装置 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH01289013A (ja) * | 1988-05-16 | 1989-11-21 | Nippon Telegr & Teleph Corp <Ntt> | 有機機能膜の作製方法 |
| JP2004111374A (ja) * | 2002-08-29 | 2004-04-08 | Matsushita Electric Ind Co Ltd | 電気化学素子 |
| JP2004342605A (ja) * | 2003-04-22 | 2004-12-02 | Matsushita Electric Ind Co Ltd | 電気化学素子および電気化学素子用電極活物質 |
| JP2008159275A (ja) * | 2006-12-20 | 2008-07-10 | Matsushita Electric Ind Co Ltd | 電極活物質およびこれを用いた蓄電デバイス |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7282298B2 (en) | 2002-08-29 | 2007-10-16 | Matsushita Electric Industrial Co., Ltd. | Electrochemical device |
| US8034484B2 (en) | 2003-04-22 | 2011-10-11 | Panasonic Corporation | Electrochemical device and electrode active material for electrochemical device |
| JP4534875B2 (ja) * | 2005-06-10 | 2010-09-01 | セイコーエプソン株式会社 | 発光素子、発光素子の製造方法、電子デバイスおよび電子機器 |
-
2009
- 2009-07-31 JP JP2009178610A patent/JP4558835B2/ja not_active Expired - Fee Related
- 2009-07-31 WO PCT/JP2009/003656 patent/WO2010013492A1/ja active Application Filing
- 2009-07-31 CN CN2009801154894A patent/CN102015821B/zh not_active Expired - Fee Related
- 2009-07-31 EP EP09802737.8A patent/EP2308912B1/en not_active Not-in-force
- 2009-07-31 US US12/995,374 patent/US8481674B2/en not_active Expired - Fee Related
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH01289013A (ja) * | 1988-05-16 | 1989-11-21 | Nippon Telegr & Teleph Corp <Ntt> | 有機機能膜の作製方法 |
| JP2004111374A (ja) * | 2002-08-29 | 2004-04-08 | Matsushita Electric Ind Co Ltd | 電気化学素子 |
| JP2004342605A (ja) * | 2003-04-22 | 2004-12-02 | Matsushita Electric Ind Co Ltd | 電気化学素子および電気化学素子用電極活物質 |
| JP2008159275A (ja) * | 2006-12-20 | 2008-07-10 | Matsushita Electric Ind Co Ltd | 電極活物質およびこれを用いた蓄電デバイス |
Also Published As
| Publication number | Publication date |
|---|---|
| US20110086267A1 (en) | 2011-04-14 |
| US8481674B2 (en) | 2013-07-09 |
| CN102015821A (zh) | 2011-04-13 |
| CN102015821B (zh) | 2013-03-20 |
| JP2010053358A (ja) | 2010-03-11 |
| EP2308912A1 (en) | 2011-04-13 |
| EP2308912A4 (en) | 2013-04-03 |
| WO2010013492A1 (ja) | 2010-02-04 |
| EP2308912B1 (en) | 2016-01-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Xie et al. | Nanostructured conjugated polymers: toward high-performance organic electrodes for rechargeable batteries | |
| JP4633863B2 (ja) | 蓄電材料および蓄電デバイス | |
| JP4558835B2 (ja) | 重合体、半導体膜、電極、電極活物質、電気化学素子および蓄電デバイス | |
| US20050008934A1 (en) | Redox active reversible electrode and novel battery using the same | |
| JP4445583B2 (ja) | 蓄電デバイス用電極活物質および蓄電デバイスならびに電子機器および輸送機器 | |
| JP4633864B2 (ja) | 蓄電材料および蓄電デバイス | |
| JP5413710B2 (ja) | 電極活物質と、その製造方法及びそれを用いた電池 | |
| EP2518797B1 (en) | Electrode, method for electrode fabrication and electricity storage device | |
| JP5526399B2 (ja) | 電極活物質、その製造方法及び二次電池 | |
| JP5383913B2 (ja) | 非水電解質二次電池 | |
| JP2012219109A (ja) | ラジカル化合物、その製造方法及び二次電池 | |
| JP2012221575A (ja) | ラジカル化合物、その製造方法及び二次電池 | |
| JP2012221574A (ja) | ラジカル化合物及びその製造方法、電極活物質、並びに二次電池 | |
| JP2010044951A (ja) | 電極活物質及びこれを用いた電極 | |
| JP5333887B2 (ja) | 電極活物質及びこれを用いた電極 | |
| JP2011146346A (ja) | 二次電池用活物質及び二次電池 | |
| JP5218962B2 (ja) | 複合体電極 | |
| JP4752217B2 (ja) | 活物質、電池および重合体 | |
| JP2006073241A (ja) | 電極活物質、電池および重合体 | |
| JP2013077392A (ja) | 蓄電材料及び蓄電デバイス | |
| JP2009238492A (ja) | 複合体電極 | |
| JP2013069589A (ja) | 蓄電材料及び蓄電デバイス | |
| JP2012015377A (ja) | 蓄電デバイス |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20100526 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20100526 |
|
| A871 | Explanation of circumstances concerning accelerated examination |
Free format text: JAPANESE INTERMEDIATE CODE: A871 Effective date: 20100526 |
|
| TRDD | Decision of grant or rejection written | ||
| A975 | Report on accelerated examination |
Free format text: JAPANESE INTERMEDIATE CODE: A971005 Effective date: 20100622 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20100629 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20100721 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130730 Year of fee payment: 3 |
|
| LAPS | Cancellation because of no payment of annual fees |