JP4248242B2 - 製油所の輸送機関用燃料のための混合成分の一体化調製 - Google Patents
製油所の輸送機関用燃料のための混合成分の一体化調製 Download PDFInfo
- Publication number
- JP4248242B2 JP4248242B2 JP2002563263A JP2002563263A JP4248242B2 JP 4248242 B2 JP4248242 B2 JP 4248242B2 JP 2002563263 A JP2002563263 A JP 2002563263A JP 2002563263 A JP2002563263 A JP 2002563263A JP 4248242 B2 JP4248242 B2 JP 4248242B2
- Authority
- JP
- Japan
- Prior art keywords
- sulfur
- organic
- liquid
- boiling
- group
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Images

Classifications
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G27/00—Refining of hydrocarbon oils in the absence of hydrogen, by oxidation
- C10G27/04—Refining of hydrocarbon oils in the absence of hydrogen, by oxidation with oxygen or compounds generating oxygen
Landscapes
- Chemical & Material Sciences (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Production Of Liquid Hydrocarbon Mixture For Refining Petroleum (AREA)
- Catalysts (AREA)
Description
適切な石油留出油、好ましくは水素化処理された留出油中の有機化合物の選択的酸素化を含む一体化方法による、輸送機関用燃料の製油所における混合のための成分の製造のための経済的方法が提供される。本発明の一体化方法は、好都合には更に酸素化の供給原料のそれ自体の供給源を、水素化処理された留出油の低沸点留分として与える。有益には、一体化方法は、高沸点留分の選択的酸化を含み、これにより炭化水素、硫黄含有有機及び/又は窒素含有有機化合物への酸素の組み込みは、酸化によって硫黄及び/又は窒素の除去を援助する。
本発明の一つの側面は、製油所の輸送機関用燃料又は製油所の輸送機関用燃料の混合成分の製造のための方法を提供し、この方法は:天然の石油から誘導される有機化合物の混合物を含んでなる有機供給原料を用意し、前記混合物は、約10°API(約1.00)ないし約100°API(約0.61)の範囲の比重を有し;分子状酸素(二原子酸素)のガス状供給源を、少なくとも一つの多価及び/又は重金属を含んでなる可溶性触媒系を含有する液体反応媒体中で、液体反応媒体をハロゲン及び/又はハロゲン含有化合物を実質的に含まないように維持しながら前記有機供給原料と接触させて、炭化水素類、酸素化された有機化合物、反応水、及び酸性副産物を含んでなる不混和性相の混合物を形成し;そして不混和性相の混合物から、炭化水素類、酸素化された有機化合物及び酸性副産物を含んでなる少なくとも一つの低密度の第1の有機液体並びに少なくとも触媒金属の一部、反応水、及び酸性副産物を含有する高密度の第2の液体を分離することを含んでなる。
本発明の方法は、好都合には酸化の供給原料の接触水素化処理を含んで、硫化水素を形成し、これはガスとして、固体収着剤上に収集され、及び/又は水性液体で洗浄して液体供給原料から分離することができる。本発明の好ましい側面において、有機供給原料の全て又は少なくとも一部は、約50℃ないし約425℃間で沸騰する物質から本質的になる石油留出油のための水素化処理法の生成物であり、この水素化処理法は、石油留出油を水素の供給源と水素化条件で、水素化処理された石油留出油からの硫黄及び/又は窒素の除去を水素化によって援助する水素添加触媒の存在中で反応させることを含む。
M[RCOCH=C(O−)R’]x
によって表される化合物からなる群から選択される触媒金属の供給源を含んでなり、ここでxは2又は3である。Mは、マンガン、コバルト、ニッケル、クロム、バナジウム、モリブデン、タングステン、スズ及びセリウムからなる群の一つ又はそれより多い一員であり、そして更に好ましくは、群は、マンガン、コバルト、又はセリウムからなる。R及びR’は、水素原子並びにメチル基、約20個までの炭素原子、そして更に好ましくは約10個までの炭素原子を有するアルキル基、アリール基、アルケニル基及びアルキニル基からなる群の同一又は異なった一員である。
Mn[RCOCH=C(O−)R’]2
Co[RCOCH=C(O−)R’]2及び/又は
Ce[RCOCH=C(O−)R’]3
によって表される化合物からなる群から選択される触媒金属の供給源を含んでなり、ここでR及びR’は、水素原子並びにメチル基、約20個までの炭素原子、そして更に好ましくは8個までの炭素原子を有するアルキル基、アリール基、アルケニル基及びアルキニル基からなる群の同一又は異なった一員である。最も好ましいものは、以下の式:
Mn[CH3COCH=C(O−)CH3]2
Co[CH3COCH=C(O−)CH3]2及び/又は
Ce[CH3COCH=C(O−)CH3]3
によって表される化合物の群から選択される金属触媒の供給源である。
酸化の供給原料が製油所ストリームの水素化から誘導される高沸点流出油留分である場合、製油所ストリームは、約200℃ないし約425℃間で沸騰する物質から本質的になる。好ましくは製油所ストリームは、約250℃ないし約400℃間で沸騰する物質、そして更に好ましくは約275℃ないし約375℃間で沸騰する物質から本質的になる。
本発明のもう一つの側面において、回収された有機相の処理は、酸化された硫黄含有及び/又は窒素含有有機化合物を選択的に抽出するために適した誘電率を有する溶媒を含んでなる少なくとも一つの不混和性液体の使用を含む。好都合には、溶媒は、約24ないし約80の範囲の誘電率を有する。有用な溶媒は、2ないし約6個の炭素原子の一価及び二価のアルコール、好ましくはメタノール、エタノール、プロパノール、エチレングリコール、プロピレングルコール、ブチレングリコール及びこれらの水溶液を含む。特に有用なものは、溶媒が水、メタノール、エタノール及びこれらの混合物からなる群から選択される化合物を含んでなる不混和性液体である。
本発明の一つの側面において、回収された反応混合物の有機相は、その後(i)イオン交換樹脂及び(ii)不均質収着剤と接触させられて、適した全酸価を有する生成物が得られる。
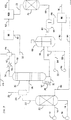
図面は、周囲条件で液体である輸送機関用燃料の混合のための成分の連続的製造のための本発明の好ましい側面を描写した略系統線図である。図1の略系統線図中の本発明の要素は、有機供給原料を、二原子酸素で、少なくとも一つの多価及び/又は重金属を含んでなる可溶性触媒系を含有する液体反応媒体中で、液体反応媒体をハロゲン及び/又はハロゲン含有化合物を実質的に含まないように維持しながら酸素化して、炭化水素類、酸素化された有機化合物、反応水、及び酸性副産物を含んでなる不混和性相の混合物を形成することを含む。不混和性相の混合物は、重力分離されて、少なくとも一つの低密度の第1の有機液体並びに少なくとも触媒金属の一部、反応水、及び酸性副産物を含有する高密度の第2の液体が回収される。有機液体は、重炭酸ナトリウム水溶液又は中和及び/又は酸化の酸性副産物の除去が可能な他の可溶性化学塩基の水溶液で洗浄され、そして酸素化された生成物が回収される。
[一般的説明]
好都合には、本発明の触媒系は、元素、組み合わせ、又はイオンの形態の、マンガン、コバルト、ニッケル、クロム、バナジウム、モリブデン、タングステン、スズ、セリウム、及びこれらの混合物からなる群から選択される触媒金属の供給源を含んでなる。触媒金属は、好ましくはマンガン、コバルト及びこれらの混合物からなる群から選択され、そして金属を使用することができる。
本発明の方法は、好都合には酸化の供給原料の接触水素化脱硫を含んで、硫化水素を形成し、これを、ガスとして、固体収着剤上に収集して、及び/又は水性液体で洗浄することによって液体供給原料から分離することができる。酸化の供給原料が、水素化処理された石油留出油からの硫黄及び/又は窒素の除去を促進するための石油留出油の水素化法の生成物である場合、本発明のために必要な過酸の量は、本発明により処理されている水素化処理された流れ中に含有された緊密に置換された硫化物を酸化するために必要な化学量論的な量である。好ましくは緊密に置換された硫化物の全てを酸化するものである量が使用されるものである。
本発明をよりよく理解するために、本発明の好ましい側面が、図1に略図的に描写される。そこで、図1において、典型的には約10°API(約1.00)ないし約100°API(約0.61)の範囲の比重を有する、天然の石油から誘導された有機化合物の混合物を含んでなる有機供給原料は、実質的に液体の形態で導管32を経由して、そして撹拌機付酸化反応器110に、空気又は窒素富化空気のような二原子酸素(分子状酸素)のガス状供給源による液相の接触酸素化のために流入する。二原子酸素のガス状供給源は、少なくとも一つの多価及び/又は重金属を含んでなる可溶性触媒系を含有する液体反応媒体中で有機供給原料と接触される。液体反応媒体は、ハロゲン及び/又はハロゲン含有化合物を実質的に含まないように維持される。酸素化反応は、約170℃ないし約210℃の範囲の温度で、そして約150psi(約10.5kg/cm2)ないし約400psi(約28kg/cm2)、好ましくは約150psi(約10.5kg/cm2)ないし約300psi(約21kg/cm2)の範囲の圧力で行われる。
塔30の塔底から、多量の硫黄含有有機化合物を有する高沸点液体留分のもう一つの部分は、酸化の供給原料として酸化反応器60に導管38を経由して供給される。
硫黄原子が、むしろ芳香族炭化水素よりも立体障害されている化合物を、従来使用された既知の脱硫系と比較して選択的に酸化するために、触媒的活性金属及び/又は活性金属含有化合物を実質的に含まないように維持された液相反応混合物中の選択された有機過酸を使用した、本発明による方法の特徴及び利益の観点から、以下の実施例が与えられる。以下の実施例は、例示であり、そして制約することを意味するものではない。
炭化水素生成物の酸素化は、原料油及び生成物の高精度炭素及び水素分析間の差によって決定した。

この実施例において、約500ppmの水準の硫黄を含有する製油所の留出油を、硫黄を約130ppmの水準で含有する水素化脱硫留出油を製造するために適した条件下で水素化処理し、これを水素化処理留出油150と規定した。水素化処理留出油150を、以下の表による温度で収集した4種の画分にカットした。
この実施例において、約500ppmの水準の硫黄を含有する製油所の留出油を、硫黄を約15ppmの水準で含有する水素化脱硫留出油を製造するために適した条件下で水素化処理し、これを水素化処理留出油15と規定した。
この実施例は、S−25と規定された水素化処理された製油所の留出油の、本発明による接触酸化を記載する。公称容積5ガロン(約19リットル)を有し、そしてチタンで製造された撹拌された反応器に、18lbs(約8.2kg)のS−25及び18.81グラムのコバルト(II)アセチルアセトナート水和物(Aldrichカタログ番号34,461−5、これは22.92重量パーセントのコバルトを含有する)を送入した。これは、水素化処理された留出油中の0.23重量パーセントのコバルト(II)アセチルアセトナート水和物濃度、又は留出油中の527ppmのコバルトを与える。
この実施例は、GS−25の重炭酸ナトリウム水溶液を使用した酸素化後の処理を記載し、これはセタン価を上昇した。実施例3のGS−25の一部を、重炭酸ナトリウム水溶液で処理し、水で洗浄し、無水の3Aモレキュラーシーブで乾燥し、そして濾過した。濾過した物質を、セタン価測定及び他の分析にかけた。大量層の処理された部分の分析は、1.67パーセントの酸素化水準、7ppmの硫黄の水準、9ppmの窒素の水準、及び2.1mgKOH/gの全酸価を決定した。この処理後の大量層のセタン価測定は、62.9と決定されたが、しかしセタン価測定エンジンはこの場合非常に滑らかに作動した。
水素化処理された製油所の留出油S−25を、蒸留によって分割して、コバルト(II)塩を含有する可溶性有機化合物を使用した接触酸素化のための供給原料を得た。約288℃より低い温度で収集された留分は、S−25−B288と規定された硫黄に貧であり且つ単環芳香族に富む留分であった。S−25−B288の分析は、10ppmの硫黄含有率、5ppmの窒素含有率、及び87.01パーセントの炭素、12.98パーセントの水素を決定し、芳香族炭素は16.5パーセントであった。
この実験のために、5ガロン(約19リットル)の耐圧反応器に、S−25−B288のもう一つの部分及びミネラルスピリット中のオクタン酸コバルト(II)を送入して、750ppmのコバルト濃度を得た。酸素化は、反応時間を39分に延長した以外は、実施例3のように行った。GS−25−B288−1と規定された、酸化されたS−25−B288−1の分析は、4.18パーセントの酸素化、及び11.8mgKOH/gの全酸価を決定した。
この実施例のために、300mLのParr耐圧反応器の底部に、S−25及びコバルト(II)ビス−アセチルアセトナート水和物を送入して、543ppmのコバルト濃度を得た。S−25の酸素化を、反応時間が33分であった以外は、実施例5のように行った。この実施例の酸素化されたS−25の分析は、11ppmの硫黄含有率、即ち45パーセントの硫黄の減少、9ppmの窒素含有率、即ち50パーセントの窒素の減少、3.61パーセントの酸素化、及び7.1mgKOH/mgの全酸価を決定した。
水素化処理された製油所の留出油S−25を、蒸留によって分割して、過酸化水素及び酢酸を使用した酸化のための供給原料を得た。約300℃より低い温度で収集された留分は、S−25−B300と規定された硫黄に貧であり且つ単環芳香族に富む留分であった。S−25−B300の分析は、3ppmの硫黄含有率、2ppmの窒素含有率、及び合計37.9パーセントの芳香族に対して、36.2パーセントの単環芳香族、1.8パーセントの二環芳香族を決定した。300℃より高い温度で収集された留分は、S−25−A300と規定された硫黄に富み且つ単環芳香族に乏しい留分であった。S−25−A300の分析は、35ppmの硫黄含有率、31ppmの窒素含有率を決定し、そして芳香族含有率は、合計22.9パーセントの芳香族に対して、15.7パーセントの単環芳香族、5.8パーセントの二環芳香族、及び1.4パーセントの三環芳香族であった。
水素化処理された製油所の留出油S−25を、蒸留によって分割して、過酸化水素及び酢酸を含有する不混和性水溶液相を使用した酸化のための供給原料を得た。約316℃より高い温度で収集されたS−25の留分は、S−25−A316と規定された硫黄に富み且つ単環芳香族に乏しい留分であった。S−25−A316の分析は、80ppmの硫黄含有率、及び102ppmの窒素含有率を決定した。
水素化処理された製油所の留出油S−25−A316の第2の酸化を、100mLの氷酢酸を送入し、しかし水を送入しないで、実施例12に記載したように行った。有機層は、27ppmの硫黄及び3ppmの窒素を含有することが見出された。水性層は81ppmの硫黄を含有していた。
実施例12及び実施例12aの両方からのフラスコの全体の内容物を混合した。次いで底部層を除去し、両方の実験からの混合された有機層を残した。有機層を無水の硫酸ナトリウムで乾燥して、工程からのいずれもの残留水を除去した。真空濾過によって廃硫酸ナトリウムを除去した後、濾液とアルミナの比が7:1ないし10:1の範囲であるような充分なアルミナを通して、濾液をパーコレートした。アルミナから出た有機層の分析は、32ppmの全硫黄及び5ppmの全窒素であった。
S−150と規定された水素化処理された製油所の留出油を、蒸留によって分割して、過酸化水素及び酢酸により形成された過酸を使用した酸化のための供給原料を得た。S−150の分析は、113ppmの硫黄含有率及び36ppmの窒素含有率を決定した。約316℃より高い温度で収集されたS−98の留分は、S−150−A316と規定される硫黄に富み且つ単環芳香族に乏しい留分であった。S−150−A316の分析は、580ppmの硫黄含有率及び147ppmの窒素含有率を決定した。
500mLの分液漏斗に、150mLのPS−150−A316及び150mLのメタノールを送入した。漏斗を振盪し、そして次いで混合物を分離させた。底部メタノール層を収集し、そして分析試験に供した。次いで50mLの生成物の部分を分析試験のために収集し、そして試料ME−14−1と規定した。
分液漏斗に50mLのPS−150−A316及び50mLの水を送入した。漏斗を振盪し、そして層を分離させた。底部の水層を収集し、そして分析試験に供した。炭化水素層を分析試験のために収集し、そしてE15−1Wと規定した。Table IIは、これらの結果を与える。
500グラムのPS−150−A316を、50グラムの無水の酸性アルミナを通してパーコレートした。収集した生成物をE16−1Aと規定し、そして分析した。データをTable IIに与える。
分析は、アルミナで処理された物質E16−1Aに対して行われ、そしてPS−150−A316と比較された。分析は、生成物中にいかなるジベンゾチオフェンも存在しないことを示し、一方原料は約3,000ppmのこの不純物を含有していた。
水素化処理された製油所の留出油S−25を、蒸留によって分割して、過酸化水素及び酢酸により形成された過酸を使用した酸化のための供給原料を得た。約288℃より低い温度で収集されたS−25の留分は、S−DF−B288と規定された硫黄に貧であり且つ単環芳香族に富む留分であった。約288℃より高い温度で収集されたS−25の留分は、S−DF−A288と規定される硫黄に富み且つ単環芳香族に乏しい留分であった。S−DF−A288の分析は、30ppmの硫黄含有率を決定した。
実施例17からのアルミナで処理された物質BA−DF−A288及び実施例6からの酸素化された物質E6−Fを混合して、DF−GPを製造した。燃料DF−GPの試験及び分析の結果をTable VIIIに与える。
Claims (19)
- 製油所の輸送機関用燃料又は製油所の輸送機関用燃料の混合成分の製造のための方法であって:
天然の石油から誘導される有機化合物の混合物を含んでなる有機供給原料を用意し、前記混合物は、約10°API(約1.00)ないし約100°API(約0.61)の範囲の比重を有し;
二原子酸素のガス状供給源を、以下の式:
M[RCOCH=C(O−)R’]x
(ここでMは、マンガン、コバルト、ニッケル、スズ及びセリウムからなる群の一つ又はそれより多い一員であり、R及びR’は、水素原子並びにメチル、約20個までの炭素原子を有するアルキル基、アリール基、アルケニル基及びアルキニル基からなる群から選択される同一又は異なった一員であり、そしてxは2又は3である)
によって表される化合物からなる群から選択される少なくとも1の触媒金属源を含んでなる可溶性触媒系を含有する液体反応媒体中で、液体反応媒体をハロゲン及び/又はハロゲン含有化合物を実質的に含まないように維持しながら、前記有機供給原料と接触させて、炭化水素類、酸素化された有機化合物、反応水、及び酸性副産物を含んでなる不混和性相の混合物を形成し;そして
不混和性相の混合物から、炭化水素類、酸素化された有機化合物及び酸性副産物を含んでなる少なくとも一つの低密度の第1の有機液体並びに触媒金属の少なくとも一部、反応水及び酸性副産物を含有する高密度の第2の液体を分離すること;
を含んでなる、前記方法。 - 前記有機供給原料が、硫黄含有及び/又は窒素含有有機化合物を含んでなり、これらの一つ又はそれより多くは液体反応媒体中で酸化され、そして前記第2の分離された液体が、酸化された硫黄含有及び/又は窒素含有有機化合物の少なくとも一部を含有する水溶液である、請求項1に記載の方法。
- 更に前記分離された有機液体を中和剤と接触させ、そして低い含有率の酸性副産物を有する生成物を回収することを含んでなる、請求項2に記載の方法。
- 更に、前記分離された第2の液体から触媒系の少なくとも一部を回収し;そして回収された触媒系の全て又は一部を前記液体反応媒体中に注入することを含んでなる、請求項1に記載の方法。
- 前記有機供給原料の全て又は一部が、約50℃ないし約425℃まで沸騰する物質から本質的になる石油留出油のための水素化処理法の生成物であり、この水素化処理法が、石油留出油を水素の供給源と水素化条件で、水素化処理された石油留出油からの硫黄及び/又は窒素の除去を水素化によって援助する水素添加触媒の存在中で反応させることを含む、請求項1に記載の方法。
- 前記水素添加触媒が、周期表のd−遷移元素からなる群から選択され、それぞれが不活性担体上に全触媒の約0.1パーセントないし約20重量パーセントの量で組み込まれた、少なくとも一つの活性金属を含んでなる、請求項5に記載の方法。
- 前記水素化処理法が、更に、硫黄に貧であり且つ単環芳香族に富む留分からなる少なくとも一つの低沸点液体、及び硫黄に富み且つ単環芳香族に乏しい留分からなる高沸点液体を得るための、蒸留による水素化処理された石油留出油の分割を含んでなり、そして前記有機供給原料が約50%よりも多い低沸点の液体を含む、請求項5に記載の方法。
- 前記触媒金属が、以下の式:
Mn[RCOCH=C(O−)R’]2
Co[RCOCH=C(O−)R’]2及び/又は
Ce[RCOCH=C(O−)R’]3
(ここでR及びR’は、水素原子並びにメチル、約20個までの炭素原子を有するアルキル基、アリール基、アルケニル基及びアルキニル基からなる群から選択される同一又は異なった一員である)
によって表される化合物からなる群から選択される少なくとも1の触媒金属源を含んでなる請求項1に記載の方法。 - 製油所の輸送機関用燃料又は製油所の輸送機関用燃料の混合成分を製造するための方法であって:
天然の石油から誘導される有機化合物の混合物で、約10°API(約1.00)ないし約100°API(約0.61)の範囲の比重を有する混合物を含んでなる有機供給原料を蒸留によって分割して、硫黄に貧であり且つ単環芳香族に富む留分からなる少なくとも一つの低沸点有機部分、及び硫黄に富み且つ単環芳香族に乏しい留分からなる高沸点有機部分を得て;
二原子酸素のガス状供給源を低沸点有機部分の少なくとも一部と、以下の式:
M[RCOCH=C(O−)R’]x
(ここでMは、マンガン、コバルト、ニッケル、スズ及びセリウムからなる群の一つ又はそれより多い一員であり、R及びR’は、水素原子並びにメチル、約20個までの炭素原子を有するアルキル基、アリール基、アルケニル基及びアルキニル基からなる群から選択される同一又は異なった一員であり、そしてxは2又は3である)
によって表される化合物からなる群から選択される触媒金属源を含んでなる可溶性触媒系を含有する液体反応媒体中で、液体反応媒体をハロゲン及び/又はハロゲン含有化合物を実質的に含まないように維持しながら接触させて、炭化水素類、酸素化された有機化合物、反応水、及び酸性副産物を含んでなる不混和性相の混合物を形成し;
不混和性相の混合物から、炭化水素類、酸素化された有機化合物及び酸性副産物を含んでなる少なくとも一つの低密度の第1の有機液体並びに少なくとも触媒金属の一部、反応水、及び酸性副産物を含有する高密度の第2の液体を分離し;そして
分離された有機液体の全て又は一部を中和剤と接触させ、これによって低い含有率の酸性副産物を有する低沸点の酸素化された生成物を回収すること;
を含んでなる、前記方法。 - 前記触媒金属が、以下の式:
Mn[RCOCH=C(O−)R’]2
Co[RCOCH=C(O−)R’]2及び/又は
Ce[RCOCH=C(O−)R’]3
(ここでR及びR’は、水素原子並びにメチル、約20個までの炭素原子を有するアルキル基、アリール基、アルケニル基及びアルキニル基からなる群から選択される同一又は異なった一員である)
によって表される化合物からなる群から選択される少なくとも1の触媒金属源を含んでなる請求項9に記載の方法。 - 前記中和剤が化学塩基の水溶液であり、前記分離された有機液体の少なくとも一部が該化学塩基の水溶液と接触させられ、そして回収された酸素化された生成物が約20mgKOH/gより少ない全酸価を示す、請求項9に記載の方法。
- 前記化学塩基が、水酸化物、炭酸塩又は重炭酸塩の形態のナトリウム、カリウム、バリウム、カルシウム及びマグネシウムからなる群から選択される化合物である、請求項11に記載の方法。
- 前記有機供給原料の全て又は一部が、約50℃ないし約425℃間で沸騰する物質から本質的になる石油留出油のための水素化法の生成物であり、この水素化法が、石油留出油を水素の供給源と水素化条件で、水素化処理された石油留出油からの硫黄及び/又は窒素の除去を水素化によって援助する水素添加触媒の存在中で反応させることを含む、請求項9に記載の方法。
- 製油所の輸送機関用燃料又は製油所の輸送機関用燃料の混合成分を製造するための方法であって:
天然の石油から誘導される有機化合物の混合物であって約75℃ないし約425℃間で沸騰する物質から本質的になる混合物を含んでなる有機供給原料を蒸留によって分割し、硫黄に貧であり且つ単環芳香族に富む留分からなる少なくとも一つの低沸点有機部分、及び硫黄に富み且つ単環芳香族に乏しい留分からなる高沸点有機部分を得て;
二原子酸素のガス状供給源を低沸点有機部分の少なくとも一部と、マンガン、コバルト、ニッケル、クロム、バナジウム、モリブデン、タングステン、スズ、セリウム、及びこれらの混合物からなる群から選択される少なくとも一つの触媒金属の供給源を含んでなる可溶性触媒系を含有する液体反応媒体中で、液体反応媒体をハロゲン及び/又はハロゲン含有化合物を実質的に含まないように維持しながら接触させて、炭化水素類、酸素化された有機化合物、反応水、及び酸性副産物を含んでなる不混和性相の混合物を形成し;
不混和性相の混合物から、炭化水素類、酸素化された有機化合物及び酸性副産物を含んでなる少なくとも一つの低密度の第1の有機液体並びに少なくとも触媒金属の一部、反応水、及び酸性副産物を含有する高密度の第2の液体を分離し;そして
分離された有機液体の全て又は一部を中和剤と接触させ、これによって低い含有率の酸性副産物を有する低沸点の酸素化された生成物を回収し;そして
高沸点有機部分を、少なくとも一つの有機過酸又は有機過酸の前駆体を含んでなる不混和性過酸含有相と、触媒的活性金属及び/又は活性金属含有化合物を実質的に含まないように維持された液体反応混合物中で、そして一つ又はそれより多い硫黄含有及び/又は窒素含有有機化合物の酸化のために適した条件下で接触させ;
不混和性過酸含有相の少なくとも一部を反応混合物の酸化された相から分離し;そして
反応混合物の酸化された相を、固体の収着剤、イオン交換樹脂、及び/又は溶媒或いは可溶性の塩基性化学化合物を含有する適切な不混和性液体と接触させて、高沸点留分より少ない硫黄及び/又はより少ない窒素を含有する高沸点生成物を得ること;
を含んでなる、前記方法。 - 前記不混和性過酸含有相が、過酸化水素及び/又はアルキルヒドロペルオキシドの供給源、2ないし約6個の炭素原子の脂肪族モノカルボン酸及び水を混合することによって形成される、請求項14に記載の方法。
- 前記不混和性過酸含有相が、過酸化水素、酢酸、及び水を混合することによって形成される、請求項14に記載の方法。
- 前記分離された過酸含有相の少なくとも一部が、反応混合物に循環される、請求項14に記載の方法。
- 更に、低沸点の酸素化された生成物の少なくとも一部を、高沸点生成物の少なくとも一部と混合して、輸送機関用燃料の製油所の混合のための成分を得ることを含んでなる、請求項14に記載の方法。
- 前記有機供給原料が、本質的に製油所ストリームの水素化処理から誘導される、約200℃ないし約425℃まで沸騰する物質からなる高沸点留出油留分である、請求項14に記載の方法。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US09/779,283 US20020148754A1 (en) | 2001-02-08 | 2001-02-08 | Integrated preparation of blending components for refinery transportation fuels |
| PCT/US2002/001394 WO2002062925A2 (en) | 2001-02-08 | 2002-01-16 | Integrated preparation of blending components for refinery transportation fuels |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2004536159A JP2004536159A (ja) | 2004-12-02 |
| JP2004536159A5 JP2004536159A5 (ja) | 2007-05-17 |
| JP4248242B2 true JP4248242B2 (ja) | 2009-04-02 |
Family
ID=25115898
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2002563263A Expired - Fee Related JP4248242B2 (ja) | 2001-02-08 | 2002-01-16 | 製油所の輸送機関用燃料のための混合成分の一体化調製 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US20020148754A1 (ja) |
| EP (1) | EP1385922A2 (ja) |
| JP (1) | JP4248242B2 (ja) |
| AU (1) | AU2002251783B2 (ja) |
| WO (1) | WO2002062925A2 (ja) |
Families Citing this family (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20050109678A1 (en) * | 2003-11-21 | 2005-05-26 | Ketley Graham W. | Preparation of components for refinery blending of transportation fuels |
| US7674368B2 (en) * | 2003-12-19 | 2010-03-09 | Shell Oil Company | Systems, methods, and catalysts for producing a crude product |
| BRPI0519500A2 (pt) * | 2004-12-29 | 2009-02-03 | Bp Corp North America Inc | processo para a dessulfurizaÇço de uma carga de alimentaÇço de destilado para produzir combustÍvel de transporte de refinaria ou componentes de mistura para combustÍvel de transporte de refinaria |
| US7744749B2 (en) * | 2005-09-08 | 2010-06-29 | Saudi Arabian Oil Company | Diesel oil desulfurization by oxidation and extraction |
| US8715489B2 (en) * | 2005-09-08 | 2014-05-06 | Saudi Arabian Oil Company | Process for oxidative conversion of organosulfur compounds in liquid hydrocarbon mixtures |
| US20070138054A1 (en) * | 2005-12-16 | 2007-06-21 | Palmer Thomas R | Selective ring opening by oxidation process |
| US20070140950A1 (en) * | 2005-12-16 | 2007-06-21 | Palmer Thomas R | In-situ generation of peroxides in petroleum streams |
| CA2647760C (en) * | 2006-04-04 | 2013-12-10 | Shell Internationale Research Maatschappij B.V. | A process for reducing the total acid number (tan) of a liquid hydrocarbonaceous feedstock |
| US20090001028A1 (en) * | 2007-06-27 | 2009-01-01 | Samuel Frisch | Two-stage oxygenation system for use with a fluidized bed reactor |
| US8926825B2 (en) * | 2010-03-19 | 2015-01-06 | Mark Cullen | Process for removing sulfur from hydrocarbon streams using hydrotreatment, fractionation and oxidation |
| US8658027B2 (en) | 2010-03-29 | 2014-02-25 | Saudi Arabian Oil Company | Integrated hydrotreating and oxidative desulfurization process |
| WO2011140572A1 (en) * | 2010-05-06 | 2011-11-10 | Sasol Technology (Pty) Ltd | Diesel engine injector fouling improvements with a highly paraffinic distillate fuel |
| US9138658B2 (en) * | 2011-01-31 | 2015-09-22 | Exxonmobil Chemical Patents Inc. | Solvent quality control in extraction processes |
| US9005433B2 (en) | 2011-07-27 | 2015-04-14 | Saudi Arabian Oil Company | Integrated process for in-situ organic peroxide production and oxidative heteroatom conversion |
| US10441944B2 (en) * | 2015-06-30 | 2019-10-15 | Hindustan Petroleum Corporation Ltd. | Catalyst composition for isomerization of paraffins |
Family Cites Families (27)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US289561A (en) * | 1883-12-04 | John f | ||
| GB289561A (ja) * | 1927-01-18 | |||
| US1972102A (en) * | 1930-12-12 | 1934-09-04 | Atlantic Refining Co | Hydrocarbon oil treatment |
| DE1105087B (de) * | 1958-07-03 | 1961-04-20 | Basf Ag | Verfahren zur Raffination von technischen Kohlenwasserstoff-gemischen |
| US3413307A (en) * | 1965-05-10 | 1968-11-26 | Exxon Research Engineering Co | Desulfurization process |
| US3551328A (en) * | 1968-11-26 | 1970-12-29 | Texaco Inc | Desulfurization of a heavy hydrocarbon fraction |
| US3595778A (en) * | 1968-12-16 | 1971-07-27 | Texaco Inc | Desulfurization process including an oxidation step with ozone and a vanadium catalyst |
| US3565793A (en) * | 1968-12-27 | 1971-02-23 | Texaco Inc | Desulfurization with a catalytic oxidation step |
| US3847798A (en) * | 1972-06-05 | 1974-11-12 | Atlantic Richfield Co | Oxidation and desulfurization of a hydrocarbon material |
| US3816301A (en) * | 1972-06-30 | 1974-06-11 | Atlantic Richfield Co | Process for the desulfurization of hydrocarbons |
| US3970545A (en) * | 1972-11-10 | 1976-07-20 | Atlantic Richfield Company | Hydrocarbon desulfurization utilizing a non-catalytic hydrogen donor step and an oxidation step |
| US3847800A (en) * | 1973-08-06 | 1974-11-12 | Kvb Eng Inc | Method for removing sulfur and nitrogen in petroleum oils |
| US4494961A (en) * | 1983-06-14 | 1985-01-22 | Mobil Oil Corporation | Increasing the cetane number of diesel fuel by partial oxidation _ |
| US4723963A (en) * | 1984-12-18 | 1988-02-09 | Exxon Research And Engineering Company | Fuel having improved cetane |
| US4830733A (en) * | 1987-10-05 | 1989-05-16 | Uop | Integrated process for the removal of sulfur compounds from fluid streams |
| GB9023257D0 (en) * | 1990-10-25 | 1990-12-05 | British Petroleum Co Plc | Desulphurisation of oil |
| US5320742A (en) * | 1991-08-15 | 1994-06-14 | Mobil Oil Corporation | Gasoline upgrading process |
| US5147526A (en) * | 1991-10-01 | 1992-09-15 | Amoco Corporation | Distillate hydrogenation |
| US5310479A (en) * | 1991-12-04 | 1994-05-10 | Mobil Oil Corporation | Process for reducing the sulfur content of a crude |
| US5288390A (en) * | 1992-03-30 | 1994-02-22 | Sun Company, Inc. (R&M) | Polycyclic aromatic ring cleavage (PARC) process |
| JP3227521B2 (ja) * | 1992-04-06 | 2001-11-12 | 舟越 泉 | 液状油中から有機硫黄化合物を回収する方法 |
| US5720901A (en) * | 1993-12-27 | 1998-02-24 | Shell Oil Company | Process for the catalytic partial oxidation of hydrocarbons |
| FR2753717B1 (fr) * | 1996-09-24 | 1998-10-30 | Procede et installation pour la production d'essences de craquage catalytique a faible teneur en soufre | |
| US5814109A (en) * | 1997-02-07 | 1998-09-29 | Exxon Research And Engineering Company | Diesel additive for improving cetane, lubricity, and stability |
| US6087544A (en) * | 1998-05-07 | 2000-07-11 | Exxon Research And Engineering Co. | Process for the production of high lubricity low sulfur distillate fuels |
| US6171478B1 (en) * | 1998-07-15 | 2001-01-09 | Uop Llc | Process for the desulfurization of a hydrocarbonaceous oil |
| US5958224A (en) * | 1998-08-14 | 1999-09-28 | Exxon Research And Engineering Co | Process for deep desulfurization using combined hydrotreating-oxidation |
-
2001
- 2001-02-08 US US09/779,283 patent/US20020148754A1/en not_active Abandoned
-
2002
- 2002-01-16 AU AU2002251783A patent/AU2002251783B2/en not_active Ceased
- 2002-01-16 JP JP2002563263A patent/JP4248242B2/ja not_active Expired - Fee Related
- 2002-01-16 WO PCT/US2002/001394 patent/WO2002062925A2/en active Application Filing
- 2002-01-16 EP EP02720809A patent/EP1385922A2/en not_active Ceased
Also Published As
| Publication number | Publication date |
|---|---|
| JP2004536159A (ja) | 2004-12-02 |
| AU2002251783B2 (en) | 2006-11-23 |
| EP1385922A2 (en) | 2004-02-04 |
| WO2002062925A2 (en) | 2002-08-15 |
| US20020148754A1 (en) | 2002-10-17 |
| WO2002062925A3 (en) | 2003-11-20 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4290547B2 (ja) | 輸送機関用燃料の製油所ブレンド用成分の酸素化プロセス | |
| US6827845B2 (en) | Preparation of components for refinery blending of transportation fuels | |
| US6881325B2 (en) | Preparation of components for transportation fuels | |
| JP4248242B2 (ja) | 製油所の輸送機関用燃料のための混合成分の一体化調製 | |
| KR20070091297A (ko) | 산화적 탈황 방법 | |
| AU2002321984A1 (en) | Process for oxygenation of components for refinery blending of transportation fuels | |
| JP2006511658A (ja) | 輸送用燃料の製油所ブレンド用成分の調製 | |
| JP2007297639A (ja) | 輸送機関用燃料 | |
| AU2002251783A1 (en) | Integrated preparation of blending components for refinery transportation fuels | |
| RU2341549C2 (ru) | Способ уменьшения содержания серы и/или азота в дистиллятном сырье | |
| AU2002245281A1 (en) | Transportation fuels | |
| KR100885497B1 (ko) | 탈질, 탈황, 함산소 화합물 제조를 위한 탄화수소 기질의선택산화 방법 | |
| WO2003002692A2 (en) | Hydrotreating of components for refinery blending of transportation fuels | |
| AU2002241897B2 (en) | Preparation of components for transportation fuels | |
| AU2007201847B2 (en) | Process for oxygenation of components for refinery blending of transportation fuels | |
| AU2002241897A1 (en) | Preparation of components for transportation fuels |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20041216 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20060817 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20060919 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20061218 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20061225 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20070315 |
|
| A524 | Written submission of copy of amendment under section 19 (pct) |
Free format text: JAPANESE INTERMEDIATE CODE: A524 Effective date: 20070315 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20081009 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20081208 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20090108 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20090113 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20120123 Year of fee payment: 3 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| LAPS | Cancellation because of no payment of annual fees |