JP4228552B2 - 1,5,9−シクロドデカトリエンのエポキシ化反応混合液の精製処理方法 - Google Patents
1,5,9−シクロドデカトリエンのエポキシ化反応混合液の精製処理方法 Download PDFInfo
- Publication number
- JP4228552B2 JP4228552B2 JP2001149699A JP2001149699A JP4228552B2 JP 4228552 B2 JP4228552 B2 JP 4228552B2 JP 2001149699 A JP2001149699 A JP 2001149699A JP 2001149699 A JP2001149699 A JP 2001149699A JP 4228552 B2 JP4228552 B2 JP 4228552B2
- Authority
- JP
- Japan
- Prior art keywords
- cyclododecatriene
- oil layer
- reaction mixture
- epoxidation reaction
- epoxidation
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D301/00—Preparation of oxiranes
- C07D301/02—Synthesis of the oxirane ring
- C07D301/03—Synthesis of the oxirane ring by oxidation of unsaturated compounds, or of mixtures of unsaturated and saturated compounds
- C07D301/12—Synthesis of the oxirane ring by oxidation of unsaturated compounds, or of mixtures of unsaturated and saturated compounds with hydrogen peroxide or inorganic peroxides or peracids
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Inorganic Chemistry (AREA)
- Epoxy Compounds (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Description
【発明の属する技術分野】
本発明は、1,5,9−シクロドデカトリエンのエポキシ化反応混合液の精製処理方法に関するものであり、更に詳しく述べるならば1,5,9−シクロドデカトリエンを、触媒の存在下に過酸化水素によりエポキシ化して得られた反応混合液の精製処理方法に関するものである。本発明の精製処理方法は、ナイロン12の原料となるラウロラクタムの中間体となる1,2−エポキシ−5,9−シクロドデカジエンの製造に有用なものである。
【0002】
【従来の技術】
オレフィン化合物を過酸化水素によりエポキシ化する方法は一般的によく知られている。例えば、タングステン化合物、四級オニウム塩及び鉱酸を含む触媒存在下、オレフィン化合物を過酸化水素によりエポキシ化する方法としては、特公平1−33471号公報、特公平3−74235号公報、特開平5−213919号公報、特開昭62−230778、特開昭62−234550等に開示されている。
しかし、上記の公報には、エポキシ化反応により得られる反応混合液から、目的生成物としてエポキシ化合物を安全に収率よく単離する、工業的な処理方法については何ら開示されていない。
通常、オレフィン化合物を過酸化水素によりエポキシ化して得られた反応混合液の処理方法としては、この反応混合液中に含まれる油層部と水層部とを、分離槽を用いて互に分液し、分液された油層部を蒸留することによって目的とするエポキシ化合物を収得する方法が行われる。
しかし、タングステン化合物、四級オニウム塩及び鉱酸を含む触媒の存在下に、1,5,9−シクロドデカトリエンを過酸化水素によりエポキシ化して得られた反応混合液では、生成した1,2−エポキシ−5,9−シクロドデカジエンを含む油層部と、水層部との分液性があまり良くなく、油層部には、数%の水層部が懸濁混入した状態となる。この油層部中の水層部は、長時間静置してもこれを完全に分離する事は困難である。
この油層部中に混入した水層液粒子中には、触媒成分のタングステン酸化合物、鉱酸、及び未反応の過酸化水素が溶解しているため、エポキシ化反応液の油層部をそのまま蒸留すると、蒸留工程において、目的製品である1,2−エポキシ−5,9−シクロドデカジエンが、これらの触媒の存在下に重合するため、目的製品の蒸留取得率が低下するという問題がある。
さらに、油層部中の混入水層部にリンタングステン酸等の超強酸化合物が存在すると、得られた1,2−エポキシ−5,9−シクロドデカジエンがこれと激しく反応し、発熱するという現象も発生し得る。
また、蒸留工程では水層部と油層部に溶解している未反応過酸化水素及び副生成物の有機過酸化物が熱分解する現象も発生することがあり、エポキシ化反応により生成する反応混合液の油層部をそのまま蒸留する方法は工業的にみて安全とは言えない。
従って、油層部を蒸留する前に油層中に含有している過酸化物および触媒成分を失活させ、或は、抽出除去させる必要がある。
【0003】
【発明が解決しようとする課題】
本発明の課題は、1,5,9−シクロドデカトリエンを、タングステン化合物、四級オニウム塩及び鉱酸を含む触媒の存在下、過酸化水素によりエポキシ化して得られた反応混合液を、それに含まれる目的生成物、すなわち1,2−エポキシ−5,9−シクロドデカジエンをより安全に収率よく蒸留分離し得るように、精製処理する方法を提供することにある。
【0004】
【課題を解決するための手段】
本発明者らは、上記課題の解決手段を鋭意検討した結果、下記本発明方法を見出し、これを完成した。本発明に係る1,5,9−シクロドデカトリエンのエポキシ化反応混合液の精製処理方法は、タングステン化合物、四級オニウム塩及び鉱酸を含む触媒存在下、1,5,9−シクロドデカトリエンを過酸化水素によりエポキシ化して得られ、かつ1,2−エポキシ−5,9−シクロドデカジエンと、前記触媒成分と、並びに未反応の過酸化水素及び1,5,9−シクロドデカトリエンとを含有する反応混合液の、少なくとも油層部に対して、処理後の反応混合液の水層部のpHが8〜13になるようにアルカリ水溶液による処理のみを施して残留触媒を失活及び抽出し、未反応過酸化水素を分解することを特徴とするものである。上記本発明方法において、反応混合液からその油層部を分別してこれを前記アルカリ水溶液処理に供してもよいし、或はアルカリ水溶液処理された反応混合液を分別に供して、それに含まれる油層部を分別してもよい。このアルカリ水溶液処理された油層部分別液を蒸留に供して1,2−エポキシシクロドデカジエンを分離することができる。
【0005】
【発明の実施の形態】
本発明のエポキシ化反応混合液の精製処理方法を詳しく説明する。
前記エポキシ化反応に使用されるエポキシ化触媒用タングステン化合物としては、タングステン原子を含有する無機酸又はその塩から選ばれることが好ましく、このタングステン原子含有無機酸及びその塩は、例えばタングステン酸、タングステン酸ナトリウム、タングステン酸カリウム、タングステン酸リチウム、タングステン酸アンモニウム等のタングステン酸やその塩、十二タングステン酸ナトリウム、十二タングステン酸カリウム、十二タングステン酸アンモニウム等の十二タングステン酸塩、リンタングステン酸、リンタングステン酸ナトリウム、ケイタングステン酸、ケイタングステン酸ナトリウム、リンバナドタングステン酸、リンモリブドタングステン酸等のヘテロポリ酸及びその塩を包含し、好ましくは、タングステン酸、タングステン酸ナトリウム、タングステン酸カリウム、及びリンタングステン酸が用いられる。これらタングステン化合物は単独で用いられてもよく、或はその二種以上を混合して使用してもよい。
【0006】
本発明のエポキシ化反応において使用されるタングステン化合物の量は、タングステン原子に換算して、1,5,9−シクロドデカトリエンに対して、好ましくは0.0007〜5重量%であり、より好ましくは0.002〜3重量%である。
【0007】
エポキシ化触媒として使用される四級オニウム塩としては、例えば、トリオクチルメチルアンモニウムクロライド、トリデシルメチルアンモニウムクロライド、トリオクチルメチルアンモニウムブロマイド、ベンジルジメチルテトラデシルアンモニウムクロライド、ベンジルトリエチルアンモニウムクロライド、ジメチルジドデシルアンモニウムクロライド、ベンジルトリブチルアンモニウムクロライド、ベンジルトリブチルアンモニウムアイオダイド、フェニルトリメチルアンモニウムクロライド等の四級アンモニウムハライド、硫酸水素トリオクチルメチルアンモニウム等の四級アンモニウム硫酸水素塩、過塩素酸トリオクチルメチルアンモニウム等の四級アンモニウム過塩素酸塩、リン酸二水素トリオクチルメチルアンモニウム等の四級アンモニウムリン酸二水素塩、硝酸トリオクチルメチルアンモニウム等の四級アンモニウム硝酸塩、珪弗化水素酸トリオクチルメチルアンモニウム等の四級アンモニウム珪弗化水素酸塩、酢酸トリオクチルメチルアンモニウム等の四級アンモニウム酢酸塩等が用いられる。これらの中で、好ましくは四級アンモニウムハライドが用いられ、より好ましくはトリオクチルメチルアンモニウムクロライド、及びトリデシルメチルアンモニウムクロライドが用いられる。
【0008】
エポキシ化触媒に使用される四級オニウム塩の使用量は、1,5,9−シクロドデカトリエン重量に対して、好ましくは0.0003〜4重量%であり、より好ましくは0.003〜2.5重量%である。
【0009】
エポキシ化触媒に使用される鉱酸は、例えば、リン酸、硫酸、塩酸、過塩素酸、ヘキサフルオロ珪酸、硝酸、テトラフルオロ珪酸等が挙げられるが、好ましくはリン酸および硫酸が用いられ、より好ましくはリン酸が用いられる。これら鉱酸は、単独で用いられてもよく、或はその、二種以上を混合して使用してもよい。
【0010】
鉱酸の使用量は、1,5,9−シクロドデカトリエン重量に対して、好ましくは0.001〜5重量%であり、より好ましくは0.005〜3重量%である。
【0011】
エポキシ化反応において使用される過酸化水素は、どのような濃度のものを用いても構わないが、取り扱い上の安全性や経済性を考慮すると、10〜70重量%濃度の水溶液を用いるのが好ましい。その使用量は、1,5,9−シクロドデカトリエン重量に対して0.05〜1.2倍モルであることが好ましく、より好ましくは0.05〜1.0倍モルであり、更に好ましくは0.1〜0.8倍モルである。
【0012】
エポキシ化反応の原料として用いられる1,5,9−シクロドデカトリエンは、市販品をそのまま用いてもよく、あるいはそれを精製して用いても何ら差し支えない。また、シス体あるいはトランス体等の異性体のいかなるものを使用しても何ら問題はない。これら異性体は混合物で使用することもできる。
【0013】
エポキシ化反応において、反応溶媒として有機溶媒を使用することもできる。使用する有機溶媒は、水と均一に混ざり合わず、かつ、エポキシ化反応を阻害しないものであれば特に制限はされない。この反応有機溶媒として例えば、クロロホルム、ジクロロエタン、ジクロロメタン等の脂肪族ハロゲン化炭化水素、シクロヘキサン、n−ヘプタン等の脂肪族非ハロゲン炭化水素、ベンゼン、トルエン、キシレン等の芳香族炭化水素などを用いることができる。これらは単独でも、二種以上を混合して使用することもできる。これら有機溶媒の使用量は、1,5,9−シクロドデカトリエン重量に対し、好ましくは0〜20重量倍であり、より好ましくは0〜10重量倍である。
【0014】
前記エポキシ化反応は、1,5,9−シクロドデカトリエンと過酸化水素水溶液とが互に分離している二液相で行なうことが好ましく、例えば、窒素等の不活性ガス雰囲気下にて、1,5,9−シクロドデカトリエン、過酸化水素水溶液、並びに、タングステン化合物、四級オニウム塩および鉱酸を含む触媒を混合し、この混合物を常圧又は加圧下で加熱撹拌する等の方法によって行われる。反応温度には、特に制限はないが、好ましくは、20〜120℃であり、より好ましくは30〜120℃である。
【0015】
本発明のエポキシ化反応混合液の精製処理方法において、エポキシ化反応混合液に、直接アルカリ水溶液を加えてもよいが、エポキシ化反応混合液の二液相(以下油層部と水層部と記載する)を分液した後に、その油層部分別液をアルカリ水溶液で処理することが好ましい。前者の方法においてはアルカリ水溶液処理された反応混合液からその油層部を分別すればよい。
【0016】
本発明方法で使用するアルカリ水溶液は、アルカリ金属またはアルカリ土類金属を含むアルカリ性無機化合物、又はアンモニアを含有する水溶液であり、この水溶液のpH値は8以上であり、好ましくは10以上であり、さらに好ましくは11以上である。これらのアルカリ性無機化合物としては、アルカリ金属の水酸化物、アルカリ金属の炭酸塩、アルカリ金属の重炭酸塩、アルカリ金属の亜硫酸塩、アルカリ土類金属の水酸化物、アルカリ土類金属の炭酸塩、アルカリ土類金属の重炭酸塩、及びアルカリ土類金属の亜硫酸塩等である。好ましくは、アルカリ金属の水酸化物、アルカリ金属の炭酸塩、アルカリ金属の重炭酸塩、アルカリ金属の亜硫酸塩が用いられ、より好ましくはアルカリ金属の水酸化物が用いられる。
【0017】
アルカリ金属の水酸化物およびアルカリ土類金属の水酸化物の具体例としては、水酸化カリウム、水酸化ナトリウム、水酸化マグネシウム、水酸化バリウム、水酸化カルシウム等が挙げられる。
アルカリ金属の炭酸塩およびアルカリ土類金属の炭酸塩の具体例としては、炭酸カリウム、炭酸ナトリウム、炭酸マグネシウム、炭酸カルシウム等が挙げられる。
アルカリ金属の重炭酸塩の具体例としては、重炭酸カリウム、重炭酸ナトリウム等が挙げられる。
また、アルカリ金属の亜硫酸塩の具体例としては、亜硫酸カリウム、亜硫酸ナトリウム等が挙げられる。
好ましくは、水酸化ナトリウム、水酸化カリウム、亜硫酸ナトリウムが用いられより好ましくは水酸化ナトリウム、水酸化カリウムが用いられる。これらのアルカリ金属化合物およびアルカリ土類金属化合物は、単独で用いられてもよく、もしくはその複数種を混合して用いてもよい。
【0018】
これらのアルカリ金属化合物およびアルカリ土類金属化合物による処理方法には、特に制限はなく、固体形状化合物をそのままエポキシ化反応混合液に添加し、これに水を加える方法、もしくは予じめアルカリ水溶液に調製し、これをエポキシ化反応混合液に添加する方法等があるが、取扱いの容易さから、予じめアルカリ水溶液に調製し、これをエポキシ化反応混合液に添加する方法が好ましい。
【0019】
アルカリ水溶液のアルカリ化合物濃度としては、通常0.01〜60重量%であり、好ましくは0.1〜30重量%であり、さらに好ましくは0.5〜10重量%である。アルカリ水溶液の添加量は、処理後の水層部の pH が8〜13の範囲となるようにコントロールされる。
【0020】
通常、処理に用いるアルカリ水溶液の添加量は、油層部の合計重量に対して0.1〜20重量%であり、好ましくは0.5〜10重量%でありさらに好ましくは1.0〜5重量%である。アルカリ添加量があまりに多いと、分別された水層部の処理が必要となるという新たな問題を生ずる。
【0021】
本発明の処理方法における処理温度には、特に制限はなく、0〜120℃であることが好ましく、より好ましくは15〜80℃であり、更に好ましくは20〜60℃の範囲内である。それがあまりに高いと1,2−エポキシ−5,9−シクロドデカジエンの収率が低下する傾向が認められる。
【0022】
本発明方法に用いられる処理装置としては、エポキシ化反応混合液中の油層部とアルカリ水溶液とが、十分に混合し接触できる撹拌装置を備えたものであれば特に制限はなく、例えば、槽型装置あるいはスタティックミキサー装置を用いることが好ましい。
【0023】
本発明方法の処理時間は、処理装置の形式により異なる。槽型装置を用いる場合の処理時間は、1〜90分であることが好ましく、より好ましくは2〜60分であり、更に好ましくは5〜40分である。スタティックミキサー装置を用いる場合の処理時間は、0.01〜5秒であることが好ましく、より好ましくは0.05〜3秒であり、更に好ましくは0.1〜2秒である。
【0024】
本発明方法による処理は通常、常圧で行われるが、必要ならば加圧または減圧下で施しても差し支えない。
【0025】
本発明方法によるエポキシ化反応混合液の処理は、バッチ式あるいは連続式装置で行うことができるが、本発明の効果を工業的に十分発揮させるためには、単数又は複数の処理装置を有する連続式のものが好ましい。
【0026】
本発明による処理を施すことにより、エポキシ化反応混合液中に含まれる触媒の失活、残留触媒の抽出およびエポキシ化反応混合液中に残存する未反応の過酸化物類の分解が促進され、特に触媒成分が反応液から十分に抽出分離されるため、蒸留工程において、目的製品、すなわち1,2−エポキシ−5,9−シクロドデカジエンの重合、及び/又は熱分解による損失がなく、高収率で安全に回収することができる。
【0027】
本発明方法により処理して得られる反応混合液の油層部に含まれる1,2−エポキシ−5,9−シクロドデカジエンは、通常の蒸留で精製、分離される。蒸留装置としては、通常のスニダー型単蒸留装置や、規則充填塔型蒸留装置、多孔板塔型蒸留装置、泡鐘塔型蒸留装置等が挙げられる。
蒸留条件は、特に制限はなく、常圧下、若干の加圧下あるいは減圧下で実施できる。蒸留温度は、蒸留時の圧力に影響されるが、好ましくは200℃以下、より好ましくは180℃以下である。
【0028】
【実施例】
以下、実施例および比較例で本発明を具体的に示す。
(1)pHの測定方法
使用するアルカリ水溶液のpH測定において、試料1mol を計り取り、イオン交換水1lに溶解させ、そのpHを室温下、堀場製D−24型pHメーターによって測定した。
イオン交換水のpHは、そのまま測定した。
実施例1
1,5,9−シクロドデカトリエンのエポキシ化反応液のモデルとして5000mlガラスフラスコに1,5,9−シクロドデカトリエン4500g(27.8mol )と、四級オニウム塩としてトリオクチルメチルアンモニウムクロライド1.14g(250ppm )とを仕込み、窒素気流下、750rpm で撹拌しながら75℃まで昇温した。
反応温度が75℃に到達した後、この1,5,9−シクロドデカトリエン溶液の中に、60重量%過酸化水素393g(6.9mol )、タングステン酸ソーダ1.14g(250ppm )とリン酸1.14g(250ppm )を溶解した溶液を25分間で滴下した。その後、75℃で90分間反応を行い、冷却して1,5,9−シクロドデカトリエンのエポキシ化反応混合液のモデル液4890gを得た。
このモデル反応液の全量を5000mlの分液ロートに移し、4611gの油層部と279gの水層部に分液した。
次に、撹拌機を備えた1000mlガラスフラスコにこの分液で得た油層部の一部750gと2.0重量%水酸化ナトリウム水溶液20gとを仕込み、これらを45℃の温度で20分間撹拌混合した。
この混合処理液を常温まで冷却し、分液ロートを用いて、油層部と水層部、すなわち水酸化ナトリウム水溶液部とを互に分別した。この時、水層部のpHは9.5であった。
分別された油層部中のW(タングステン)およびP(リン)等の触媒元素濃度をプラズマ励起発光分光分析法(ICP−AES分析法)で測定し、また過酸化物の濃度をヨードメトリー滴定法で定量した。その結果、表1に示したように油層中のW濃度は10ppm であり、P濃度は1ppm 以下であり、過酸化物濃度は0.0056mmol/gであった。
さらに、水酸化ナトリウム処理後の油層部分別液500gをスニーダー型蒸留装置で蒸留した。蒸留条件は、それぞれ1,5,9−シクロドデカトリエンの留出温度が76℃/0.25kPa 、1,2−エポキシ−5,9−シクロドデカジエンの留出温度は97℃/0.25kPa で実施した。その結果、163.1gの1,2−エポキシ−5,9−シクロドデカジエンが得られた。これは、99.4%の蒸留収率に相当する。
【0029】
実施例2
実施例1における1,5,9−シクロドデカトリエンのエポキシ化モデル反応液の油層部750gと4.0重量%水酸化カリウム水溶液75gとを、撹拌機を備えた1000mlガラスフラスコ中に仕込み、25℃で10分間撹拌混合し、油層部と水層部とを分別した。分別した水層部と油層部の各々の分析を実施例1と同様に行った。
その結果、水層部のpHは10.8であった。また、油層部のW濃度は8ppm であり、P濃度は1ppm 以下であり、過酸化物濃度は0.0048mmol/gであった。
さらに、実施例1と同様に分別された油層部分別液500gを蒸留した結果、163.5gの1,2−エポキシ−5,9−シクロドデカジエンが得られた。これは、99.7%の蒸留収率に相当する。
【0030】
実施例3
実施例2において、アルカリ処理液を10wt%亜硫酸ナトリウム50gに変えたことを除き、その他は実施例2と同様に実験を行った。
その結果、処理されたエポキシ化反応混合物の水層部のpHは10.1であった。また、油層部のW濃度は15ppm であり、P濃度は1ppm 以下であり、過酸化物濃度は0.0081mmol/gであった。
さらに、実施例1と同様に分別された油層部分別液500gを蒸留した結果、162.5gの1,2−エポキシ−5,9−シクロドデカジエンが得られた。これは、99.2%の蒸留収率に相当する。
【0031】
比較例1
実施例1で調製したモデル反応液をそのまゝ油層部と水層部とに分別した後、水層部の一部分が懸濁している油層部のpHを測定したところ、そのpHは3.6であった。また油層部中のW,P元素量及び過酸化物量を実施例1と同様な方法で定量分析した。
その結果、油層部中のW量は117ppm であり、P量は4.1ppm であり、過酸化物濃度は0.0163mmol/であった。
次に、スニダー型蒸留装置で油層部分別液500gを無処理のまま実施例1と同じ蒸留条件下で蒸留した結果、160.6gの1,2−エポキシ−5,9−シクロドデカジエンが得られた。これは、97.9%の蒸留収率に相当する。
【0032】
比較例2
実施例2において、4重量%水酸化カリウム水溶液を蒸留水に変えて用いたことを除き、その他は実施例2と同様に実験を行い、定量分析を行った。
その結果、水層部のpHは6.2であった。また油層中のW濃度は102ppm であり、P濃度は3.6ppm であり、過酸化物濃度は0.0147mmol/gであった。
さらに、水処理後の油層部分別液500gを実施例1と同様の蒸留条件下で蒸留した結果、161.2gの1,2−エポキシ−5,9−シクロドデカジエンが得られた。これは、98.2%の蒸留収率に相当する。
【0033】
実施例1〜3および比較例1と2の処理条件および結果をまとめて表1に示す。
【表1】
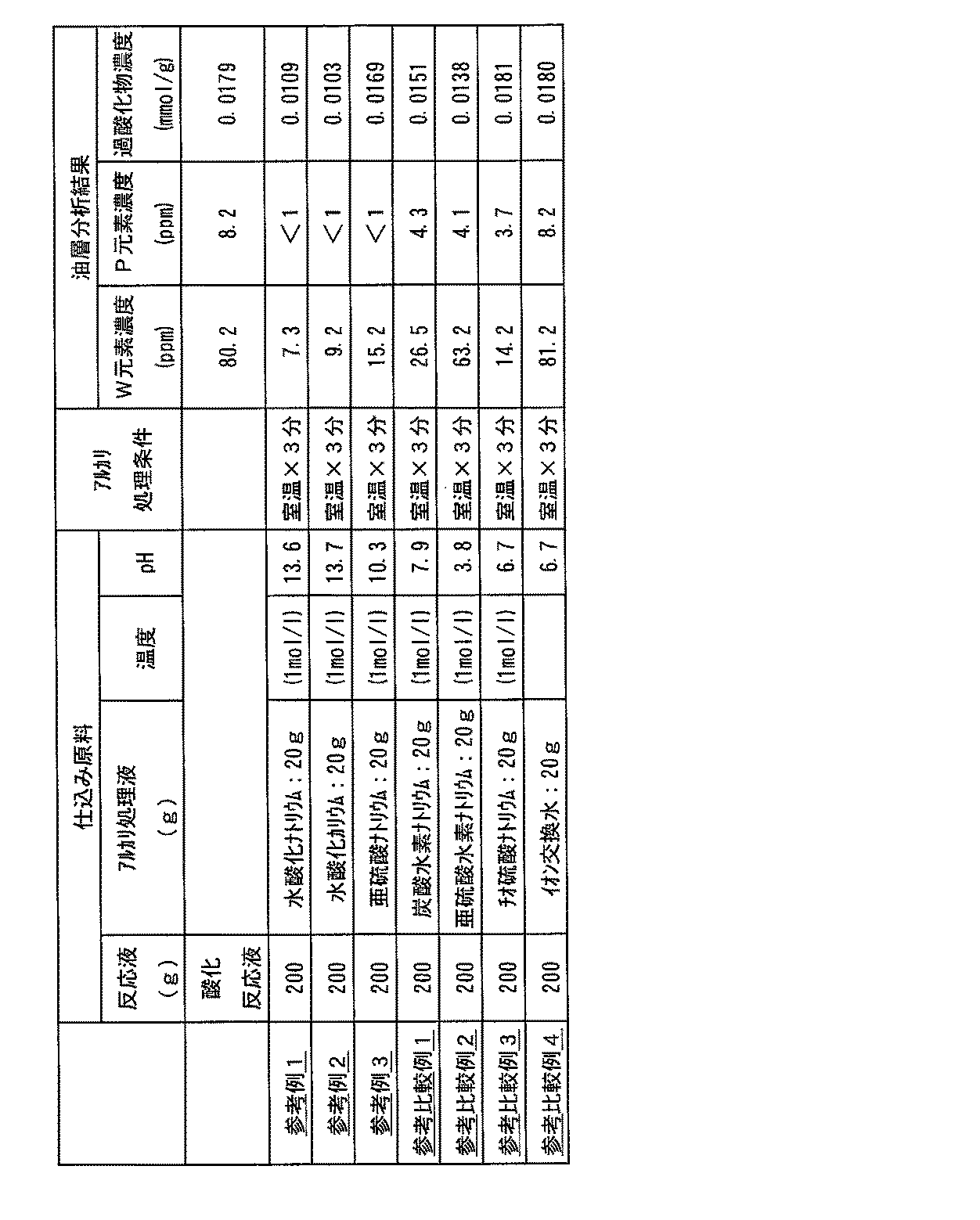
参考例1
パイロット実験で得られたエポキシ化反応混合液油層部(1,2−エポキシ−5,9−シクロドデカジエンを22.0重量%含み、さらにタングステン濃度80.2ppm 、リン濃度8.2ppm 、過酸化物濃度0.0179mmol/g、及び未反応の1,5,9−シクロドデカトリエンを反応溶媒として含む)200gに、1mol /lの水酸化ナトリウム水溶液20g(pH測定値13.6)を加え、室温下で3分間振とうし、静置分離後、分離した油層部の分析をおこなった。その結果、この油層部の過酸化物濃度は、0.0109mmol/gに低減していた。
さらに、タングステン濃度は、7.3ppm に低下しており、リンは1ppm 以下であった。この結果を表2にまとめて示す。
【0035】
参考例2
1mol /lの水酸化ナトリウム水溶液20g(pH測定値13.6)の代りに1mol /lの水酸化カリウム水溶液20g(pH測定値13.7)を使用して、参考例1と同様の処理を行った。得られた結果を表2にまとめて示す。
【0036】
参考例3
1mol /lの水酸化ナトリウム水溶液20g(pH測定値13.6)の代りに1mol /lの亜硫酸ナトリウム水溶液20g(pH測定値10.3)を使用して参考例1と同様の処理を行った。得られた結果を表2にまとめて示す。
【0037】
参考比較例1
1mol /lの水酸化ナトリウム水溶液20g(pH測定値13.6)の代りに1mol /lの炭酸水素ナトリウム水溶液20g(pH測定値7.9)を使用して参考例1と同様の処理を行った。得られた結果を表2にまとめて示す。
【0038】
参考比較例2
1mol /lの水酸化ナトリウム水溶液20g(pH測定値13.6)の代りに1mol /lの亜硫酸水素ナトリウム水溶液20g(pH測定値3.8)を使用して参考例1と同様の処理を行った。得られた結果を表2にまとめて示す。
【0039】
参考比較例3
1mol /lの水酸化ナトリウム水溶液20g(pH測定値13.6)の代りに1mol /lのチオ硫酸ナトリウム水溶液20g(pH測定値6.7)を使用して参考例1と同様の処理を行った。得られた結果を表2にまとめて示す。
【0040】
参考比較例4
1mol /lの水酸化ナトリウム水溶液20g(pH測定値13.6)の代りにイオン交換水20g(pH測定値6.7)を使用して参考例1と同様の処理を行った。得られた結果を表2にまとめて示す。
【0041】
参考例1〜3および参考比較例1〜4の処理条件および結果をまとめて表2に示す。
【表2】
【発明の効果】
本発明により、タングステン化合物、四級オニウム塩及び鉱酸を含む触媒の存在下、1,5,9−シクロドデカトリエンを過酸化水素によりエポキシ化して得られ、1,2−エポキシ−5,9−シクロドデカジエン及び該触媒成分を含有する反応混合液の少なくとも油層部にアルカリ水溶液処理のみを施し、このとき、処理後の反応混合液の水層部のpHが8〜13になるようにし、それによって残留触媒を失活及び抽出し、並びに未反応過酸化水素を分解し、目的物化合物、すなわち1,2−エポキシ−5,9−シクロドデカジエンを安全に高収率で捕集できる、精製処理された反応混合液を得ることができる。
また本発明方法の処理を施すことにより、反応液中に残存している触媒成分を高効率をもって抽出処理することができる。
Claims (5)
- タングステン化合物、四級オニウム塩及び鉱酸を含む触媒の存在下に、1,5,9−シクロドデカトリエンを過酸化水素によりエポキシ化して得られ、かつ1,2−エポキシ−5,9−シクロドデカジエンと、前記触媒成分と、並びに未反応の過酸化水素及び1,5,9−シクロドデカトリエンとを含有する反応混合液の、少なくとも油層部に対して、処理後の反応混合液の水層部のpHが8〜13になるようにアルカリ水溶液による処理のみを施して残留触媒を失活及び抽出し、並びに未反応過酸化水素を分解することを特徴とする、1,5,9−シクロドデカトリエンのエポキシ化反応混合液の精製処理方法。
- 前記エポキシ化反応において、前記1,5,9−シクロドデカトリエンが反応溶媒として用いられる請求項1に記載の1,5,9−シクロドデカトリエンのエポキシ化反応混合液の精製処理方法。
- 前記エポキシ化反応混合液から、その油層部を分別し、この分別液に前記アルカリ水溶液処理を施す、請求項1又は2に記載の1,5,9−シクロドデカトリエンのエポキシ化反応混合液の精製処理方法。
- 前記エポキシ化反応混合液に前記アルカリ水溶液処理を施し、得られた処理反応混合液から、その油層部を分別する、請求項1〜3のいずれか1項に記載の1,5,9−シクロドデカトリエンのエポキシ化反応混合液の精製処理方法。
- 請求項3又は4に記載の方法により前記分別・精製された、又は前記精製・分別された油層部分別液に、さらに蒸留を施すことを含む、1,2−エポキシ−5,9−シクロドデカジエンの分離方法。
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2001149699A JP4228552B2 (ja) | 2000-07-04 | 2001-05-18 | 1,5,9−シクロドデカトリエンのエポキシ化反応混合液の精製処理方法 |
| DE60105236T DE60105236T2 (de) | 2000-07-04 | 2001-06-29 | Reinigungsmethode für eine flüssige Reaktionsmischung aus der Epoxidierung von 1,5,9-Cyclododecatrien |
| EP01115892A EP1170291B1 (en) | 2000-07-04 | 2001-06-29 | Refining treatment method of liquid reaction mixture obtained from epoxidation reaction of 1,5,9-cyclododecatriene |
| ES01115892T ES2225359T3 (es) | 2000-07-04 | 2001-06-29 | Metodo de tratamiento por refino de una mezcla de reaccion liquida obtenida a partir de la reaccion de epoxidacion de 1,5,9-ciclododecatrieno. |
| US09/895,192 US6420575B1 (en) | 2000-07-04 | 2001-07-02 | Refining treatment method of liquid reaction mixture obtained from epoxidation reaction of 1,5,9-cyclododecatriene |
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2000202688 | 2000-07-04 | ||
| JP2000-202688 | 2000-07-04 | ||
| JP2001149699A JP4228552B2 (ja) | 2000-07-04 | 2001-05-18 | 1,5,9−シクロドデカトリエンのエポキシ化反応混合液の精製処理方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2002080469A JP2002080469A (ja) | 2002-03-19 |
| JP4228552B2 true JP4228552B2 (ja) | 2009-02-25 |
Family
ID=26595371
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2001149699A Expired - Fee Related JP4228552B2 (ja) | 2000-07-04 | 2001-05-18 | 1,5,9−シクロドデカトリエンのエポキシ化反応混合液の精製処理方法 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US6420575B1 (ja) |
| EP (1) | EP1170291B1 (ja) |
| JP (1) | JP4228552B2 (ja) |
| DE (1) | DE60105236T2 (ja) |
| ES (1) | ES2225359T3 (ja) |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR100938325B1 (ko) * | 2001-06-13 | 2010-01-22 | 오르보테크 엘티디. | 에너지 전달 시스템 |
| JP4639606B2 (ja) * | 2003-03-06 | 2011-02-23 | 住友化学株式会社 | プロピレンオキサイドの製造方法 |
| DE10344595A1 (de) * | 2003-09-25 | 2005-05-12 | Basf Ag | Verfahren zur Herstellung eines Ketons |
| JP4444642B2 (ja) | 2003-12-15 | 2010-03-31 | 高砂香料工業株式会社 | 新規な多成分系酸化触媒及びこれを用いたエポキシ化合物の製造方法 |
| BRPI0711689A2 (pt) * | 2006-06-23 | 2011-12-20 | Dow Global Technologies Inc | processo para produzir um epóxido a partir de uma olefina, processo para preparar uma composição de resina epóxi curável, composição de resina epóxi curável, resina epóxi curada, composição e revestimento com tenacidade melhorada |
| JP5517237B2 (ja) * | 2008-09-17 | 2014-06-11 | 日本化薬株式会社 | エポキシ化合物の製造方法、エポキシ化合物、硬化性樹脂組成物及びその硬化物 |
| JP6641681B2 (ja) * | 2013-10-02 | 2020-02-05 | 三菱ケミカル株式会社 | エポキシ化合物の製造方法 |
| ES2664174T3 (es) | 2014-04-24 | 2018-04-18 | Firmenich Sa | Epóxido macrocíclico insaturado como ingrediente perfumante |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5274140A (en) * | 1979-07-19 | 1993-12-28 | Instituto Guido Donegani, S.P.A. | Process for catalytically epoxidizing olefin with hydrogen peroxide |
| DE3002838B1 (de) * | 1980-01-26 | 1981-05-27 | Degussa Ag, 6000 Frankfurt | Verfahren zur Herstellung von 1,2-Epoxy-5,9-cyclododecadien |
| IT1205277B (it) | 1982-11-10 | 1989-03-15 | Montedison Spa | Nuovo composizioni perossidiche a base di tungsteno e fosforo o arsenico |
| JPS62234550A (ja) | 1985-12-24 | 1987-10-14 | San Petoro Chem:Kk | 触媒およびその使用法 |
| JPS62230778A (ja) | 1986-03-19 | 1987-10-09 | エフ エム シ− コ−ポレ−シヨン | エポキシ化方法 |
| JPH05133471A (ja) | 1991-03-12 | 1993-05-28 | Dxl Internatl Inc | ダイヤフラム・アセンブリ |
| JPH05213919A (ja) | 1992-02-04 | 1993-08-24 | Tosoh Corp | 脂環式オレフィンのエポキシ化法 |
| US6043383A (en) * | 1998-04-14 | 2000-03-28 | Ube Industries, Ltd. | Process for producing 1,2-epoxy-5,9-cyclododecadiene |
-
2001
- 2001-05-18 JP JP2001149699A patent/JP4228552B2/ja not_active Expired - Fee Related
- 2001-06-29 ES ES01115892T patent/ES2225359T3/es not_active Expired - Lifetime
- 2001-06-29 EP EP01115892A patent/EP1170291B1/en not_active Expired - Lifetime
- 2001-06-29 DE DE60105236T patent/DE60105236T2/de not_active Expired - Lifetime
- 2001-07-02 US US09/895,192 patent/US6420575B1/en not_active Expired - Lifetime
Also Published As
| Publication number | Publication date |
|---|---|
| US20020045791A1 (en) | 2002-04-18 |
| EP1170291B1 (en) | 2004-09-01 |
| DE60105236T2 (de) | 2005-09-01 |
| US6420575B1 (en) | 2002-07-16 |
| JP2002080469A (ja) | 2002-03-19 |
| DE60105236D1 (de) | 2004-10-07 |
| EP1170291A1 (en) | 2002-01-09 |
| ES2225359T3 (es) | 2005-03-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4228552B2 (ja) | 1,5,9−シクロドデカトリエンのエポキシ化反応混合液の精製処理方法 | |
| EP0958861B1 (en) | Activation method of titanium silicalite and its use in oxidation processes with hydrogen peroxide | |
| US6828459B2 (en) | Method for producing cyclohexanone oxime | |
| JP6818741B2 (ja) | 1,4−ビス(エトキシメチル)シクロヘキサンを調製する方法 | |
| TW201136907A (en) | Epoxidation of an olefin | |
| EP1700846A1 (en) | Process for producing cyclohexanone oxime | |
| EP0980348B1 (en) | Process for purifying fluoromethyl 1,1,1,3,3,3-hexafluoroisopropyl ether | |
| EP0135295B1 (en) | Purification of tertiary butyl hydroperoxide containing primary and secondary alkyl hydroperoxide contaminants | |
| KR101904568B1 (ko) | 하이드록실아민의 제조 방법 | |
| EP1674450B1 (en) | Process for producing cyclohexanone oxime | |
| JP2003192680A (ja) | 環状モノオレフィンのエポキシ化方法 | |
| EP1318141A1 (en) | Process for treating an aqueous medium containing phosphate, cyclohexanone and cyclohexanone oxime | |
| CS207291A3 (en) | Process for treating amides | |
| RU2159675C1 (ru) | Способ активирования титансодержащего силикалита, титансодержащий силикалитный катализатор и способ окисления органического субстрата | |
| JP2003160518A (ja) | シクロドデカトリエンの回収方法 | |
| EP4039672B1 (en) | Cyclododecanone and preparation method therefor | |
| JP3673600B2 (ja) | 高純度シクロヘキセンオキサイドの製造法 | |
| JP2003089662A (ja) | シクロドデカトリエンの蒸留精製方法 | |
| JP4111680B2 (ja) | ビストリフルオロメチルヒドロキシブテン−1の精製方法 | |
| JPH08193062A (ja) | ε−カプロラクタムの製造方法 | |
| JP2712675B2 (ja) | フェニル クロロチオホルメイト類の工業的製造法 | |
| JP4239288B2 (ja) | ε−カプロラクタムの製造方法 | |
| JPH0770097A (ja) | クロロスチレンオキシドの製造方法 | |
| JP3364287B2 (ja) | ヒドロキシ芳香族化合物の製造方法 | |
| JP2003012658A (ja) | エポキシシクロドデカンを高収率で製造する方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20040216 |
|
| A711 | Notification of change in applicant |
Free format text: JAPANESE INTERMEDIATE CODE: A711 Effective date: 20040209 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20040526 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20071204 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20080229 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20080708 |
|
| A711 | Notification of change in applicant |
Free format text: JAPANESE INTERMEDIATE CODE: A711 Effective date: 20080709 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A821 Effective date: 20080709 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20081008 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20081111 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20081124 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20111212 Year of fee payment: 3 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20121212 Year of fee payment: 4 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20121212 Year of fee payment: 4 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20121212 Year of fee payment: 4 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20121212 Year of fee payment: 4 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20131212 Year of fee payment: 5 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| LAPS | Cancellation because of no payment of annual fees |