JP3974557B2 - 変異解析方法及びそのためのシステム - Google Patents
変異解析方法及びそのためのシステム Download PDFInfo
- Publication number
- JP3974557B2 JP3974557B2 JP2003150853A JP2003150853A JP3974557B2 JP 3974557 B2 JP3974557 B2 JP 3974557B2 JP 2003150853 A JP2003150853 A JP 2003150853A JP 2003150853 A JP2003150853 A JP 2003150853A JP 3974557 B2 JP3974557 B2 JP 3974557B2
- Authority
- JP
- Japan
- Prior art keywords
- nucleic acid
- primer
- sequence
- pcr
- amplification
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Images






Landscapes
- Apparatus Associated With Microorganisms And Enzymes (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
- Investigating Or Analysing Biological Materials (AREA)
- Investigating Or Analysing Materials By The Use Of Chemical Reactions (AREA)
Description
【発明の属する技術分野】
本発明は、変異解析方法及びそのためのシステムに関する。より詳しくは、遺伝子の配列を個体間や集団内で比較するための一塩基多型(SNP)を解析するための方法と該方法のためのシステムに関する。
【0002】
【従来の技術】
ヒトゲノム解読後の次なる研究である「ポストゲノム研究」の重要な課題として、個体ごとの配列の微細な違いを調べる一塩基多型解析がある。ヒトにおける一塩基多型(SNP)の総体は、個体の「個性」そのものを意味する。SNPはヒトゲノム中1500塩基に一ヶ所の頻度で現れ、その中には疾病に対する罹りやすさや治癒のしやすさ、薬剤に対する感受性、あるいは環境ストレス応答に影響しているものが多数存在すると考えられている。したがってポストゲノム研究が進むと、同じ診断名や類似の症状の疾患であっても、その背景となる疾患を起こす仕組みの違いが分子レベルで明らかとなる。そして、その違いを考慮に入れた薬剤の使い分けなど、医療の個別化(オーダーメイド化)が可能になることが期待される。また、個人個人の疾患に対する罹りやすさ(疾患易罹患性)のリスク判定が可能となり、必要に応じて疾患を避けるためのライフスタイルを構築することで、疾患の予防、発症の遅延、早期発見・早期治療が実現できると考えられる。
【0003】
SNPは、減数分裂したゲノム中の遺伝子配列が他の塩基に置き換わり、それが交配により遺伝的に受け継がれたものである。そのため、多くの個体からなるグループの配列を調べると、遺伝子の特定部位の配列が一定の頻度で2種類現れることになる。SNPはその存在する位置によって5種類に分類される。翻訳領域に存在しアミノ酸配列の変化を伴わないsSNP、アミノ酸変化を伴う若しくはタンパク質が作られなくなるcSNP、調節領域に存在し遺伝子発現量へ影響する可能性のあるrSNPやiSNP、他の領域に存在し遺伝子発現量への影響はほとんどないgSNPである。
【0004】
すべてのSNPは多型マーカーとして疾患の発症や増悪に関連する遺伝子をみつけるために有用であるが、臨床分野で重要なのは疾患のリスク診断や薬剤の使い分けなどに直接関係するcSNPやrSNPである。この医学的に重要と考えられるSNPの数について見積もる。30億塩基対からなるヒトゲノムには300万〜600万箇所のSNPが存在することになる。ヒトゲノム計画の結果、ヒト遺伝子の数はおおよそ26,000〜28,000種でサイズが平均1 kb(1,000塩基)であることがわかっている。このため、意味のあるSNPの存在する領域は26〜28 Mbの範囲に絞り込まれる。遺伝子からのタンパク質翻訳では、3塩基ごとの配列組み合わせであるトリプレットコドンでアミノ酸が決定される。このうち、3番目の塩基が変化しても、同じアミノ酸に翻訳されたり類似性質のアミノ酸に置き換わったりするだけなので、タンパク質の機能に影響が出るものは全体の2/3となる。よって、タンパク質の機能に関与するSNPはおおよそ17〜19 Mbの範囲に存在することになる。SNPの出現頻度を1.5 kbに1ヶ所とすると、機能に関与するSNPは最大13,000ヶ所となる。このうち、個人のSNPとして解析の意味のあるものがどのくらい含まれるかが、医療分野でのSNP解析市場を決定づけるひとつの要因になる。つまり、cSNPとrSNPを合わせても利用価値のあるSNPは遺伝病の数と同程度の数千に絞り込まれる可能性が高い。
【0005】
このように産業上重要なSNPには限りがあり、疾病との因果関係が予測されるSNPの探索が盛んに行われている。探索にはSNPを大量解析する方法が必要であり、そのための方法や装置の開発が多数報告されている。例えば、全くの未知領域からSNP候補を探し出す方法としては、複数の試料で特定領域の配列を解析して比較する従来からのDNAシーケンス法がある。また、ある程度絞り込まれたSNP候補の解析方法としては、DNAが高次構造を形成するときの安定化エネルギーが1塩基の違いで異なることを利用したSSCP法や、変性剤濃度勾配ゲル電気泳動法(DGGE)、加熱変性HPLC法(WAVE (登録商標)システム)、SNP部位を含む試料を混合して得られるキメラ2本鎖のSNP部位を選択的に化学切断する方法(CCM)、同じくキメラ部分を酵素切断するS1ヌクレアーゼ法、公知のSNPにおけるタイピングを行うインベーダー法、TaqMan法、プローブとのハイブリダイゼーション安定性の違いを比べるDNAチップ法、SNP塩基部位のみの局所的な配列を決定するミニシーケンス法(例えば、非特許文献1参照)、ミニシーケンスと質量分析を組み合わせたマスアレー法等が開発されている。さらに、SNPの臨床検査を目的とした解析方法として、生物化学発光を利用したBAMPER(Bioluminometric Assay coupled with Modified Primer Extension Reactions)法(例えば、特許文献1及び非特許文献2参照)がある。
【0006】
BAMPER法では、標的遺伝子に対して、その3’末端がSNPアレルに一致するように設計された2種類のプローブを用いて伸長反応を行い、その際に生成するピロリン酸を、ルシフェリン・ルシフェラーゼ反応による生物化学発光によって検出する。伸長反応は、用いたプローブに対応する変異が標的遺伝子に存在する場合にのみ進行するため、発光を測定することにより、数分間で標的遺伝子のSNPの配列を判定することができる。BAMPER法は、試薬を加えるだけでSNP解析ができ、しかも簡単な光学系の発光測定装置で実施可能という点では、臨床現場での使用に適した解析方法と考えられる。
【0007】
しかしながら、BAMPER法によるSNP解析は2度の相補鎖合成反応含む4段階の反応を必要とする。以下、図1にしたがって各工程について説明する。
▲1▼検出対象となるSNP部位を含む数100塩基長からなる部分のPCR増幅:
この工程は、試料の増幅のみならず、SNP部位を含む限られた領域を増幅することで、以後のSNP検出反応の擬陽性増幅反応を低減する目的がある。結果として、SNP部位3を持つセンス鎖1とSNP部位に相補な部分4を持つアンチセンス鎖1’のPCR産物と反応残査2であるdNTPとプライマーの混合物ができる。
【0008】
▲2▼PCR反応液からのPCR産物精製:
BAMPER反応では、プローブの伸長反応で生成するピロリン酸を検出するため、PCRで使用されるプライマーやdNTPからなるPCR残査2は測定の妨害となる。すなわち、PCRプライマーはPCR産物にハイブリダイズしてDNAポリメラーゼによる相補鎖合成を起こし、ピロリン酸を産出する。また、dNTPはピロリン酸を検出のための発光反応の擬似基質となる。したがって、これらを除去するため、図1では、PCR残査2をShrimp alkaline phosphataseとExonuclease Iで分解している。なお、BAMPER法の最初の報告(非特許文献2)では、ビオチン標識したプライマーでPCR増幅を行い、増幅産物をストレプトアビジン付磁気ビーズに捕捉することで余分なPCRプライマーやdNTPを除去している。補足したPCR産物は、アルカリ変性させることにより、ビーズに固定化された鎖のみを次のBAMPER伸長反応の鋳型として用いることができる。
【0009】
▲3▼BAMPER反応前段(BAMPERプローブによる相補鎖伸長反応):
精製された鋳型PCR産物に、SNP部位にプローブ5の3’末端が相補な構造を有する、20〜30塩基長程度のBAMPERプローブをハイブリダイズさせ、DNAポリメラーゼの触媒作用により相補鎖伸長反応を行わせる。ここで、SNP配列に対応したプローブ伸長反応の忠実度を高めるために、プローブの3’末端から3乃至4番目の塩基には予め試料DNA配列とミスマッチになるような塩基配列を挿入しておく(例えば、非特許文献2及び3参照)。
【0010】
プローブがSNP部位に相補な配列を3’末端に有するときのみ、伸長反応が起こり、6の伸長鎖とピロリン酸7を生成する。1塩基伸長につき1分子のピロリン酸が生成するため、ピロリン酸の量は伸長反応に伴って増加する。3’末端が非相補なプローブは伸長反応を起こさず、ピロリン酸も生成しない。通常反応サイクルは、生成するピロリン酸量を増加させるために、94℃の変性工程(10秒間)と、50〜60℃のハイブリダイズ及び伸長反応工程(10秒間)を5回程度繰り返す。BAMPER反応におけるDNA伸長鎖長は最長でも200塩基長程度のため、短時間の反応サイクルでも十分に伸長反応が起きる。
【0011】
▲4▼BAMPER反応後段(発光反応によるピロリン酸定量検出):
最後に、BAMPERプローブの伸長反応で生じたピロリン酸7をATPに変換し、ルシフェリン−ルシフェラーゼによる発光反応で検出する。すなわち、ATPとルシフェラーゼの作用で生成した励起状態の酸化ルシフェリンが基底状態に戻るときに生じる、530nm近傍の発光を検出する。SNP配列と相補なBAMPERプローブのみが発光を生じうるため、発光を生じたBAMPERプローブの3’末端の配列により、SNPの配列を決定することができる。
【0012】
【特許文献1】
特開2002−101899号公報
【非特許文献1】
A.Jalanko, et al., Clinical Chemistry, (1992), 38, p39-43
【非特許文献2】
Guo-hua Zhou,et al., Nucleic Acid research, (2001), 29, e93
【非特許文献3】
K.Okano, et al., Electrophoresis, (1998), 19, p3071-3078
【0013】
【発明が解決しようとする課題】
上記のとおり、BAMPER法によるSNP解析は2度の相補鎖合成反応含む4段階の反応を必要とする。これがより少ない工程で実施できれば、操作はさらに簡単なものとなり、処理の自動化も容易になることが期待される。
【0014】
すなわち、本発明の課題は、BAMPER法によるSNP解析技術を改良し、より臨床現場での使用に適した、簡便で低廉なSNP解析方法やそのためのシステムを提供することにある。
【0015】
【課題を解決するための手段】
前記課題を解決するために鋭意検討した結果、発明者らはBAMPER法によるSNP解析における、前記▲1▼のPCR増幅と▲3▼のBAMPERプローブによる相補鎖伸長反応は、いずれもDNAポリメラーゼによる相補鎖合成反応であることに注目した。そして、PCRに用いるどちらかのプライマーをBAMPERプローブと同様に3’末端がSNP部位に一致するように設計すれば、PCRとBAMPERプローブによる相補鎖伸長反応を一緒に行うことができると考えた。
【0016】
しかしながら、実際にはプライマーの非特異的なハイブリダイズによる擬陽性増幅が起きるため、上記の方法のみではSNP配列を特異的に検出することは困難であった。そこで、発明者らはさらに検討を行い、プライマーやPCR条件に工夫を加えることにより、SNP近傍の短い領域を選択的に増幅し、擬陽性増幅を起こすことなく、1回のPCRでSNP配列を検出する方法を見出した。
【0017】
すなわち、本発明は、標的核酸上の特定部位を含む所望の塩基長範囲の増幅産物を得るための核酸増幅方法であって、
前記標的核酸の第一鎖の塩基配列に相補な配列を含み、かつ該第一鎖上の前記特定部位に3’末端がバイブリダイズするべく設計された第1のプライマーと、前記標的核酸の第二鎖の塩基配列に相補な配列を含み、該第二鎖上の前記特定部位と塩基対をなす部位に3’末端がバイブリダイズするべく設計された第2のプライマーとを用いて核酸増幅を行うこと、及び
前記核酸増幅における変性温度を制御することにより、前記所望の塩基長範囲の増幅産物を1本鎖に解離して、次の核酸合成反応の鋳型とすることを特徴とする、前記核酸増幅方法を提供する。
【0018】
前記方法において、核酸増幅における変性温度は、最初は通常どおり高く(例えば、94℃前後に)設定して増幅反応を進め、プライマーからの短鎖の増幅産物がある程度増えたら60〜85℃に下げるように制御する。具体的には、少なくとも5サイクル目からは60〜85℃に下げるように制御することが好ましい。これにより、プライマーからの真の増幅産物である短鎖の核酸のみが解離され、次の核酸合成反応の鋳型となるため、プライマーの非特異的ハイブリダイゼーションによる擬陽性増幅が防止される。
【0019】
本発明において、所望の塩基長範囲の増幅産物とは、前記第1のプライマーと第2のプライマーの3’末端が前記標的核酸上の特定部位にハイブリダイズして合成される核酸である。したがって、所望の塩基長範囲は、前記第1のプライマーと第2のプライマーの塩基長によって規定される値(両プライマーの塩基長の和から1を減じた塩基長)となる。
【0020】
前記第1のプライマーと第2のプライマーの塩基長は18〜50塩基長で、実質的に同一の融解温度を有することが望ましい。また、前記第1のプライマーと第2のプライマーにおけるグアニン及びシトシンの割合の和は35〜65%であることが望ましい。
【0021】
本発明はまた、標的核酸上の特定部位の配列を解析するための方法を提供する。該方法は、前記標的核酸の第一鎖の塩基配列に相補な配列を含み、かつ該第一鎖上の前記特定部位に3’末端がバイブリダイズするべく設計された第1のプライマーと、前記標的核酸の第二鎖の塩基配列に相補な配列を含み、該第二鎖上の前記特定部位と塩基対をなす部位に3’末端がバイブリダイズするべく設計された第2のプライマーとを用いて核酸増幅を行うこと、及び
前記核酸増幅における加熱温度を制御することにより、前記所望の塩基長範囲の増幅産物を1本鎖に解離して、次の核酸合成反応の鋳型とすることを特徴とする核酸増幅工程、ならびに
前記増幅産物を検出することにより、前記標的核酸上の特定部位の配列を解析する工程を含む。
【0022】
ここで、検出すべき特定部位としては、例えば一塩基多型部位を挙げることができる。
また、増幅産物の検出方法としては、電気泳動など公知のいずれの方法を用いてもよいが、核酸増幅に応じて生成されるピロリン酸を生物化学発光反応によって検出する方法が、装置や操作が簡便であるという点で好ましい。
【0023】
本発明はまた、前記解析方法のためのシステムを提供する。該システムは、標的核酸及び該標的核酸上の特定部位を含む所望の塩基長範囲の増幅産物を得るためのプライマーを含む反応溶液を収める反応槽と、
前記反応槽内の温度を制御して、前記所望の塩基長範囲の増幅産物を解離、増幅させる温度制御機構と、
前記反応槽内で生じる発光を検出するための光検出機構とを有し、かつ、
前記生物化学発光を生じたプライマーの種類を認識して前記標的核酸に含まれる特定部位の配列を自動的に解析する。
【0024】
【発明の実施の形態】
本発明のSNP解析方法は、プライマーとPCR条件を工夫することにより、BAMPER法で必要とされた▲1▼PCR増幅、▲2▼精製、▲3▼SNP特異的相補鎖伸長反応の3段階の工程を単一操作で行い、操作の簡素化と迅速化を図るものである。
【0025】
特に、上記プライマーとPCR条件の工夫によって、本発明は核酸検出時に問題となる擬陽性増幅の問題を克服している点に大きな特徴がある。以下、本発明について詳細に説明する。
【0026】
1 プライマー
1)プライマーの機能
本発明では、その3’末端がいずれも検出すべき特定部位(例えば、SNP部位)に一致するように設計したプライマー組を用いることにより、特定部位近傍の短い領域を選択的に増幅する。同時に、前記プライマーはBAMPERプローブとしても機能するため、PCR増幅のみによって、標的核酸における特定部位の配列を解析することができる。
【0027】
また、本発明ではセンス鎖とアンチセンス鎖に対応するプライマーの両方の3’末端がSNP部位に一致するように設計することで、検出すべき配列に対する選択性を向上させている。すなわち、プライマーがゲノムに非特異的にハイブリダイズしておきる擬陽性増幅が起きたとしても、PCR産物中の最小の産物が目的とする真の増幅産物となる。したがって、後述するPCR増幅時の変性温度を制御することによって、短鎖の真の増幅産物を選択的に解離させ、次の核酸合成反応の鋳型とすることで、擬陽性増幅を防止することができる。なお、本明細書中において「真の増幅産物」とは、第1のプライマーと第2のプライマーの3’末端が前記標的核酸上の特定部位にハイブリダイズして合成される増幅産物を意味する。
【0028】
2)プライマー組
プライマーは、標的核酸の第一鎖の塩基配列に相補な配列を含み、かつ該第一鎖上の特定部位に3’末端がバイブリダイズするべく設計された第1のプライマーと、前記標的核酸の第二鎖の塩基配列に相補な配列を含み、該第二鎖上の前記特定部位と塩基対をなす部位に3’末端がバイブリダイズするべく設計された第2のプライマーからなる1対のプライマー組で構成される。このプライマー組は、一塩基多型検出の場合、各アレルに対応して少なくとも2組、場合によっては3組乃至4組必要となる。すなわち、プライマーの3’末端の配列としては、A/C組、A/G組、A/T組、C/G組、C/T組、G/T組、A/C/G組、A/C/T組、A/G/T組、C/G/T組、A/C/G/Tのプライマー組が考えられる。
【0029】
3)ミスマッチの導入
上記プライマーは、少なくとも片方のプライマー、可能であれば両プライマーにおいて、その3’末端から5’側に3塩基乃至4塩基目の位置に、対応するゲノム試料の配列と異なる構造(ミスマッチ)を導入することが好ましい。これにより、PCRにおける増幅反応の変異部位(例えば、SNPアレル)配列特異性が格段に高まる。従来のBAMPER法においても、同様のミスマッチをプライマーに導入しているが、通常のPCRでは、プライマー同士が離れた位置にハイブリダイズするように設計されるため、片方のプライマーにしか人工的非相補部位(ミスマッチ)を導入することができない。本発明のプライマーは3’末端がオーバーラップした構造のため、両方のプライマーにミスマッチを入れることが可能となる。これにより、解析の精度は格段に向上する。
【0030】
このようなSNP位置に3’末端が一致するようなプライマー組でのPCRでは、PCRプライマーの設計に自由度が無く、ほぼ一義的に配列位置が決まる。このため、プライマー中にパリンドロームが出現する場合はプライマー自体が高次構造をとり、鋳型DNAに対するハイブリダイゼーションが阻害されるケースが頻繁に起きる。これを回避するため、プライマーの少なくとも一方が4塩基、6塩基、あるいは8塩基等のパリンドロームを形成するような構造の場合、該パリンドロームを形成する配列部位の5’末端側から半分のいずれか1塩基を本来の配列と異なる塩基とすることでパリンドローム構造を解消する。
【0031】
4)塩基長
本発明で用いられるプライマーは、通常のPCRプライマーと同様に、18〜50塩基長、特に18〜30塩基長程度であることが好ましい。
本発明では、両プライマーの3’末端が共に標的核酸上の特定部位(SNP部位等)にハイブリダイズするように設計されている。したがって、第1のプライマーと第2のプライマーの3’末端が前記標的核酸上の特定部位にハイブリダイズして合成される「真の増幅産物」の塩基長は、前記第1のプライマーと第2のプライマーの塩基長によって規定される。すなわち、真の増幅産物の塩基長は「第1のプライマーと第2のプライマーの塩基長の和から1を減じた塩基長」となる。例えば、18〜50塩基長のプライマー組を用いた場合、所望の塩基長範囲は、35〜99塩基となる。
【0032】
5)融解温度(Tm)
本発明において、第1のプライマーと第2のプライマーのそれぞれの融解温度(Tm)は、実質的に同じであることが好ましい。ここで、実質的に同じであるとは、計算上完全に同一ではないが、その差は5℃以下程度のわずかなものであって、実験をする上ではほぼ同一のTmとして扱えることを意味する。
【0033】
6)GC含量(%)
また、前記第1のプライマーと第2のプライマーのグアニン及びシトシンの割合(GC含量(%))の和は、35%〜65%であることが望ましい。
本発明において、真の増幅産物のGC含量(%)は、両プライマーのGC含量(%)の和にほぼ等しくなる。核酸のTmはその塩基長とGC含量(%)で変化するが、後述するよう、擬陽性増幅を起こすことなく、60〜85℃の変性温度で35〜99塩基長程度の短鎖(35〜99塩基長程度)の真の増幅産物を選択的に解離させるためには、増幅産物のGC含量(%)は35%〜65%、特に40%〜60%であることが望ましい。したがって、両プライマーのGC含量(%)の和は35%〜65%、特に40%〜60%であることが望ましい。
【0034】
2.PCR条件
本発明のもう1つの特徴は、PCR増幅時の温度制御にある。核酸増幅工程においては、通常のPCR反応サイクル同様、鋳型核酸を1本鎖に解離するための変性温度、アニーリングのための温度、伸長反応のための温度に、順次温度を変化させることが必要である。特に本発明の場合、核酸増幅における変性温度は、最初は通常どおり高く、例えば90〜100℃、好ましくは94℃前後に設定して増幅反応を進めるが、プライマーからの短鎖の真の増幅産物がある程度増えたら、変性温度を60〜85℃に下げるように制御する。具体的には、少なくとも5サイクル目から60〜85℃、好ましくは80〜85℃に制御することが好ましい。これにより、短鎖の真の増幅産物のみが解離可能となり、擬陽性増幅が起きたとしてもその産物は次の核酸合成の鋳型となることはない。
【0035】
もちろん、PCR産物の変性可能な温度は伸長産物の塩基長やGC含量(%)で変化する。例えばGC含量(%)が58%とすると、PCR産物が60塩基長の場合Tmは71℃、80塩基長では76℃、100塩基長では77℃、150塩基長では79℃、200塩基長では81℃となる(図5B参照)。実質的にPCRの変性温度はこれらのTm値よりさらに5〜10℃高くする必要がある。したがって、PCR産物の塩基長が80塩基の場合、変性温度は80〜85℃が好ましい(図5B参照)。換言すれば、GC含量(%)が58%の場合、80℃の変性温度で増幅可能なPCR産物は80塩基長が限界となる。
【0036】
一方、GC含量(%)が減少すれば、前記Tm値も低下し、好ましい変性温度も低下する(80℃の変性温度で増幅可能なPCR産物の塩基長は長くなる)。また、GC含量(%)が増加すれば、前記Tm値も上昇し、好ましい変性温度も上昇する(80℃の変性温度で増幅可能なPCR産物の塩基長は短くなる)。
【0037】
本発明の真の増幅産物は、18〜50塩基長のプライマーを用いた場合、35〜99塩基長となるが、こうした短鎖の増幅産物は、GC含量35〜65%の範囲では、60〜85℃の範囲で変性可能となる。
なお、本発明では、増幅産物のTmを調整するために、必要であればTm調整剤等を用いても良い。
【0038】
3.検出方法
本発明では、標的核酸の特定部位の配列に対応するプライマー組を用いた場合にのみ、増幅反応が進行する。すなわち、増幅産物を生成したプライマー組の3’末端の配列によって、特定部位の配列を決定することができる。
【0039】
本発明において、所望の真の増幅産物の検出は、電気泳動法、エネルギートランスファーを用いた蛍光検出など周知のいずれの検出法を用いてもよいが、以下に説明する生物化学発光を利用した検出方法が、装置と操作が簡便であると言う点で好ましい。
【0040】
PCRでは高エネルギー3リン酸結合が1つ切断されると、無機ピロリン酸が1つ遊離する。すなわち、1塩基の伸長反応により1つのピロリン酸が生成する。そこで、このピロリン酸を検出することによって、間接的に増幅産物を検出することが可能となる。
【0041】
ピロリン酸の検出は、BAMPER法と同様に、ルシフェリン−ルシフェラーゼによる生物化学発光を利用して検出する。すなわち、まずピロリン酸ジホスホキナーゼあるいはATPスルフリラーゼを用いてATPに変換する。変換したATPとルシフェリンをルシフェラーゼを酵素として酸化的に反応させると、オキシルシフェリンとピロリン酸、及びその他の一連の物質が生成する。生成した励起状態の酸化ルシフェリンが基底状態に戻るときに生じる、530nm近傍の発光を検出する。発光は、標的核酸上の特定部位(例えばSNP部位)の配列と相補なプライマーのみが生じうるため、発光を生じたプライマーの3’末端の配列により、特定部位の配列を決定することができる。
【0042】
なお、ルシフェリン−ルシフェラーゼを用いた生物化学発光では、試料中にピロリン酸やルシフェラーゼの基質となるATPやdATPが含まれていてはならないし、非特異的な相補鎖合成反応の原因となる短鎖のDNAや1本鎖部分があってはならない。したがって、試料に含まれるピロリン酸やATP、dATPは予めフォスファターゼで分解し、短鎖のDNAや1本鎖部分はエキソヌクレアーゼIで分解する。
【0043】
4. 変異解析システム
生物化学発光を利用して本発明の方法を実施する場合、電気泳動やクロマトグラフィーのような分離分析手段は不要であり、またレーザーなどによる励起の必要もないため、操作の自動化に適している。本発明は、そのような自動解析のためのシステムを提供する。
【0044】
例えば、前記システムは、標的核酸及び該標的核酸上の特定部位を含む所望の塩基長範囲の増幅産物を得るためのプライマーを含む反応溶液を収める反応槽と、前記反応槽内の温度を制御して、前記所望の塩基長範囲の増幅産物を解離、増幅させる温度制御機構と、前記反応槽内で生じる発光を検出するための光検出機構とを有し、かつ、前記生物化学発光を生じたプライマーの種類を認識して前記標的核酸に含まれる特定部位の配列を自動的に解析する。
【0045】
なお、前記所望の塩基長範囲の増幅産物を解離、増幅させる温度制御機構とは、通常のPCR反応サイクルのための温度制御機構(鋳型核酸を1本鎖に解離するための変性温度、アニーリングのための温度、伸長反応のための温度に、反応槽内の温度を順次変化させる機構)に加えて、核酸増幅における変性温度を、最初は通常どおり高く(例えば94℃前後に)設定し、プライマーからの短鎖の真の増幅産物がある程度増えたら(例えば、5サイクル目以降)、変性温度を60〜85℃に下げるような制御機構を意味する。
上記システムは、例えば、臨床現場におけるSNP解析等の変異解析に好適に利用できる。
【0046】
【実施例】
以下、実施例を用いて本発明についてさらに詳細に説明するが、これらの実施例は本発明の範囲を限定するものではない。
【0047】
実施例1:電気泳動を利用したp53エキソン8の変異解析
p53エキソン8に存在する変異をSNPに見立て、本発明の方法を用いて増幅を行い、電気泳動により解析を行った。
【0048】
1.装置及び試料
サーマルサイクラーはDNA Engine PTC200(MJ Research)を使用した。PCR産物の確認にはマイクロチップ電気泳動解析システムSV1210(日立電子エンジニアリング)を使用した。ルミノメーターとしてはWallac 1420 ARVOsx(PerkinElmer)を使用した。プライマー合成はSIGMA社に委託した。
【0049】
試料としては、ヒト由来ゲノムを用いた。ゲノムは血液2mlに9倍容量の50mM NaCl溶液を加えて溶血させ、3500Gで遠心し、これを10mlの50mM NaCl溶液に懸濁して再度遠心を行い、白血球を含む沈殿を得た。この沈澱にDNAzolを1ml加え、白血球を溶解し、不溶性成分を遠心により除去した。ここに等容量のブタノールを加えて遠心し、沈殿を水100mlに再溶解し、エタノール沈殿法で低分子不純物を除去した。最後にゲノム濃度が2.5〜25 ng/μlになるように滅菌水に溶解し、試料ゲノム溶液とした。調製した試料ゲノムは、あらかじめSNPアレルの同定をおこない、2種のホモザイゴート(AA)、(TT)、及びヘテロザイゴート(AT)を、それぞれ以下の実験に使用した。
【0050】
2.方法
図2に示すp53エキソン8の領域を用いて本実施例を説明する。21がp53エキソン8のセンス鎖を表し、21’がアンチセンス鎖である。22(相補鎖では22’)が対象となる変位部位である。この部分は正確にはSNPではなく単なる配列の変異部分であるが、本発明を説明するモデルとして使用する。従来のBAMPER法では、たとえばアンチセンス鎖21’に相補なプライマー24のみで伸長反応を行うが、本発明では、プライマー23と24を用いる。
【0051】
使用した2種のプライマーは、プライマー24がゲノム試料の2本鎖のアンチセンス鎖21’に実質的に相補で、かつ、他方のプライマー23がゲノムの他方の鎖であるセンス鎖21に実質的に相補である。また、両プライマーの3’末端25、及び26は、それぞれ検出対象であるSNPのアレル22、22’の塩基に相補である。今回、検出対象であるSNPはp53エキソン8に存在するCys275Serに対応する部分である。これはアミノ酸配列として275番目のコドンの多型であり、塩基配列のアレルとしてはA/Tとなる。したがって、上記プライマー組(23及び24)は、SNP部位がAであるアレルを検出するためのものと、SNP部位がTであるアレルを検出するためのものの2組を用意した。
【0052】
さらに、各プライマーの3’末端から3番目の塩基には、選択性を高めるためにp53エキソン8(試料DNA)とは非相補な配列27及び28を導入した。すなわち、アンチセンス鎖に相補なプライマーの3’末端から3塩基目は、p53エキソン8本来の配列に相当するTからAに改変した。センス鎖に相補なプライマーでは本来の配列に相当するAからTに改変した。これにより、プライマーの3’末端がアレルに相補なプライマー組ではプライマーの3’末端近傍3塩基のうち2塩基が完全相補となりDNAポリメラーゼによる相補鎖伸長が起きるが、3’末端がアレルに非相補なプライマー組では3’末端と3塩基目の2塩基が非相補となるためプライマー3’末端近傍が鋳型DNAにハイブリダイズできず、DNAポリメラーゼによる相補鎖伸長が起き難くなる。
【0053】
プライマー23は20塩基長でプライマー24は22塩基長で、PCR時のTmが54.5℃と55.4℃になるように設計した。なお、これらの値は実際にPCRを行うときの塩強度50mMで計算した。PCRのハイブリダイゼーション時(アニーリング時)の最適温度は47.2℃であるが、本実験は50℃で行った。
【0054】
以下に、使用した2組のプライマー組の配列を示す。
SNP部位がAのアレルを検出するためのプライマー組
sense primer:5'-aac agc ttt gag gtg cgt gAt T-3'(配列番号1、22塩基長)antisense primer:5'-tct ctc cca gga cag gcT cA-3'(配列番号2、20塩基長)
SNP部位がTのアレルを検出するためのプライマー組
sense primer:5'-aac agc ttt gag gtg cgt gAt A-3'(配列番号3、22塩基長)antisense primer:5'-tct ctc cca gga cag gcT cT-3'(配列番号4、20塩基長)。
【0055】
上記プライマーによって得られるPCR産物は41 塩基長(bp)で、アレルAに対応するものが、5'-aac agc ttt gag gtg cgt gAt TgA gcc tgt cct ggg aga ga-3'(配列番号5)とアレルTに対応するものが5'-aac agc ttt gag gtg cgt gAt AgA gcc tgt cct ggg aga ga-3'(配列番号6)となる。これらのTmは計算上65.4℃となる。
【0056】
96ウェル PCRプレートに2.5〜25 ng/μlに調製したゲノムDNA試料2 μlを加え、氷上に置いた。5 unit/μl Taq.DNA ポリメラーゼを(QIAGEN社製)0.05 μl、2.5 mM dNTPsを1 μl、25 pmol/μlの各アレルに対応するプライマー組(コントロールでは滅菌水を用いる)を1 μl混合し、さらに滅菌水、及びPCR用Bufferを加えて、最終的に各ウェルあたり23 μlとなるように調製した。実際には、Taq.DNA ポリメラーゼとdNTPsとプライマー組を含む反応液をあらかじめ必要量より1検体分多く作っておき、これを23 μlずつ各ウェルに入れるほうが効率がよく、PCRの再現性も高い。なお、各ゲノムDNA試料について、コントロールとしてプライマー組の代りに滅菌水を加えたものを同様に反応に供した。
【0057】
次に、粘着シートでPCRプレート上部をシーリングし、PCRサイクル反応を行った。PCRは、最初に94℃10秒間加熱してゲノムを変性させ、次いで、50℃10秒間と80℃10秒間のサイクルを35回繰り返した。
【0058】
3.結果
SNP配列に対応する2組のプライマーを用いてPCRを行い、電気泳動分離で解析した結果を図3に示す。41、42、43は、それぞれホモザイゴート(A/A)、ホモザイゴート(T/T)、ヘテロザイゴート(A/T)のゲノムから得られた結果である。図中44、45及び46はアレルがAに相補なプライマー組、47、48及び49はアレルがTに相補なプライマー組でPCR反応を行った場合の電気泳動分離パターンである。
【0059】
ホモザイゴート(A/A)のゲノムからは、Aに相補なプライマー組で増幅した場合にのみ、特異増幅ピーク50が得られた。また、ホモザイゴート(T/T)のゲノムからは、Tに相補なプライマー組で増幅した場合にのみ、特異増幅ピーク51が得られた。一方、ヘテロザイゴート(A/T)のゲノムからは、Aに相補なプライマー組とTに相補なプライマー組のいずれで増幅した場合においても、特異的増幅ピーク52及び53が得られた。なお、すべての電気泳動パターンで共通して検出されるピーク54はプローブに相当する。
【0060】
このPCRの系では、増幅塩基長が41塩基長と短いため、温度サイクルは50℃10秒間と80℃10秒間と短時間でよい。また、変性温度も80℃と低いため、温度の上昇下降に要する時間も少なくてすむという利点がある。実際、PCRに要する時間は温度上昇と下降率を3℃/秒として計算すると、1サイクルあたり55℃から80℃の上昇に約10秒間、80℃で10秒間、80℃から55℃への下降に約10秒間、55℃で10秒間の40秒間かかる。つまり、全行程でも20分間程度で終了することになる。本実施例の場合、マイクロチップ電気泳動の所要時間は3分間程度のため、PCRから検出まで含めても、ゲノムからのSNP判定(タイピング)が短時間で可能となることが確認された。
【0061】
実施例2:PCR条件の検討
実施例1で示したアレルに対応するプライマーとゲノムを用いて、擬陽性増幅反応の変性温度依存性について検討した。
【0062】
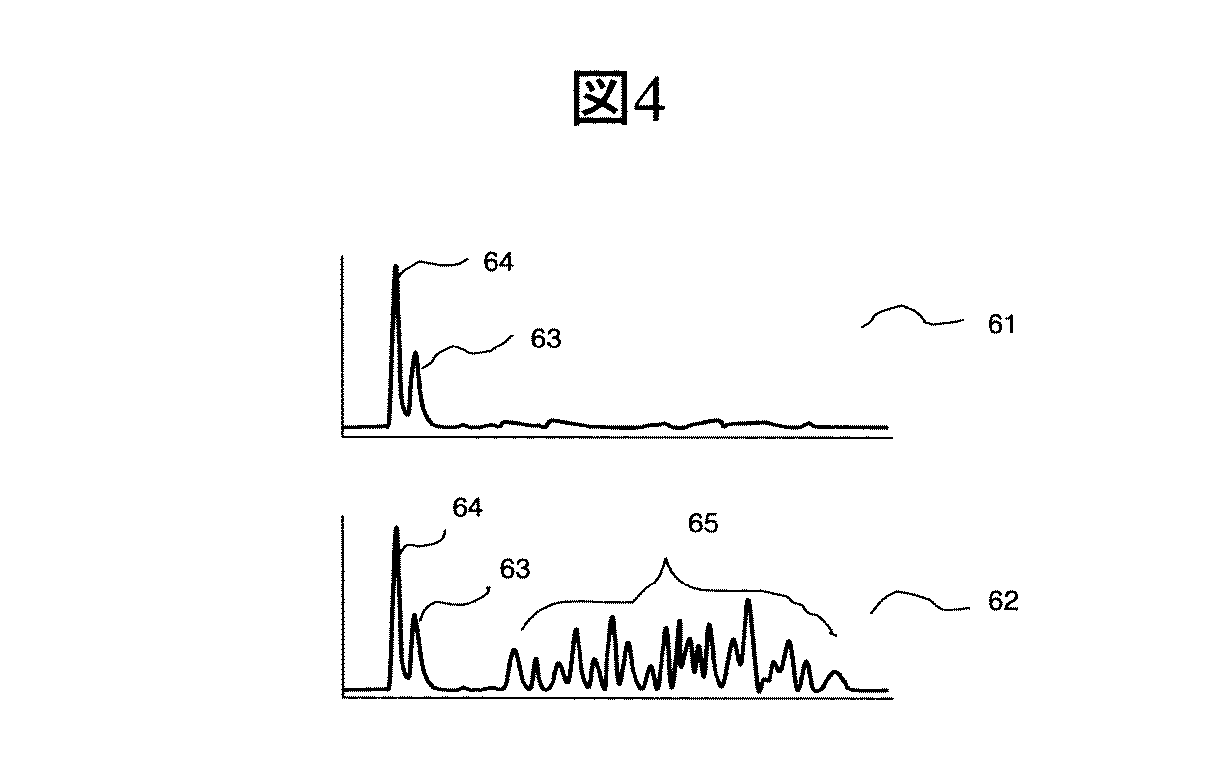
図4に、PCRサイクルにおける高い方の温度が通常のPCRと同じ94℃と本発明で用いた80℃で行ったときの増幅産物の電気泳動分離パターンを示した。63は目的のPCR増幅産物である。64はプローブ由来のピークである。パターン61に現れる一連のピーク65は、擬陽増幅に依存するものである。すなわち、PCRの高い方の温度を80℃まで下げることにより、擬陽性増幅を防止できることが確認された。これは、本発明のプライマーで生成するPCR産物が短鎖であるため、85℃程度の低温でも増幅産物の変性が十分可能なことによる。
【0063】
これをさらに検証するため、PCRの変性温度(高い方の温度)を75℃、80℃、85℃、90℃、95℃、97℃で同様に実験を行い、比較した結果を図5Aに示す。図中●で示される71のプロットは、目的のPCR産物由来の電気泳動分離ピーク63の面積の相対値を表す。図中△で示される72のプロットは、図4で観察された擬陽性増幅産物65に関わるピーク面積の相対値を表す。ここで面積の相対値とは、61と62のピーク系列でピーク面積が最大となる時の温度の面積値を1.0として計算したものである。
【0064】
図5Aより、変性温度が80〜85℃の範囲では、目的のPCR産物が得られるが、擬陽性増幅は検出限界以下になっていることがわかる。一方、90℃以上では、目的産物は得られるものの、擬陽性増幅も多くなることがわかる。なお、97℃で産物量が低下しているが、これは酵素が熱変性するためである。
【0065】
もちろん、PCR産物の変性可能な温度は伸長産物の塩基長やGC含量(%)で変化する。図5Bは伸長産物の塩基長と変性温度との関係を示したものである。本実施例ではPCR産物の塩基長が41塩基長で計算上のTm=65℃となる。GC含量(%)が58%とすると、PCR産物が60塩基長の場合Tmは71℃、80塩基長では76℃、100塩基長では77℃、150塩基長では79℃、200塩基長では81℃となる。実質的にPCRの変性温度はこれらのTm値よりさらに5〜10℃高くする必要がある。したがって、PCR産物の塩基長が80塩基の場合、変性温度は60〜85℃、好ましくは80〜85℃となる。よって、80℃の変性温度で増幅可能なPCR産物は80塩基長が限界となる。
【0066】
実施例3:生物化学発光を利用したp53エキソン8の変異解析
実施例1で調製した試料ゲノムとプライマー組を用いて、p53エキソン8に存在するCys275Serに対応する変異部分を、ルシフェリン・ルシフェラーゼによる生物化学発光を利用して解析した。検出対象となるSNP部位のアレルは実施例1と同様A/Tである。
【0067】
1.試料
シフェリン・ルシフェラーゼを用いた生物化学発光では、試料中にピロリン酸やルシフェラーゼの基質となるATPやdATPが含まれていてはならないし、非特異的な相補鎖合成反応の原因となる短鎖のDNAや1本鎖部分があってはならない。そこで、本実施例では、実施例1の方法で調製したゲノムに含まれるピロリン酸やATP、dATPを予めフォスファターゼで分解し、短鎖のDNAや1本鎖部分をエキソヌクレアーゼIで分解する必要がある。
【0068】
すなわち、384ウェルPCRプレートのウェルに、8〜56 ng/μlに調製したゲノムDNA試料を2 μl 、1 unit/μlの濃度のshrimp alkaline phosphatase (SAP)を1 μl、 10 unit/μlのexonuclease I (ExoI) を0.1 μl、10×PCR buffer(Amersham Pharmacia社製品)を0.5 μl、及び滅菌水1.4 μlを混合したものを添加した。プレートは、壁についた液体を集めるため、スピンダウンしておく。この時点の液量は5μlである。
【0069】
2.PCR反応
粘着シートでPCRプレート上部をシーリングし、サーマルサイクラーにセットした。SAPとExo Iは熱に弱いため、サーマルサイクラーのヒートリッドは使わないようにし、3 mm厚シリコンシートを被せた上に重しをのせて密閉断熱した。37℃で40分間インキュベートして酵素反応を行った後、80℃で15分間加熱して酵素を失活させ、5秒間95℃に温度上昇させてゲノムを変性させた後、氷上で急冷させた。
【0070】
上記の前処理後のゲノム試料溶液を1μlずつPCRプレートの3カ所のウェルに分注した。5 unit/μl Taq.DNA ポリメラーゼを(QIAGEN社製品)0.02 μl 、2.5 mM dNTPsを0.4 μl、25 pmol/μlの各アレルに対応するプライマー組を各0.08μl(コントロールは滅菌水を利用)、10×PCR buffer(Amersham Pharmacia社製品、15mM Mgイオンを含む)を0.8ml、10mMのMgCl2を0.45ml(PCR時の最終マグネシウム濃度は2mM)、及び滅菌水を加え、各ウェル当たりの溶液が9μlになるようにした。ここまでの操作は全て氷上で行った。なお、各ゲノムDNA試料について、コントロールとしてプライマー組の代りに滅菌水を加えたものを同様に反応に供した。
各ウェルは乾燥防止のためミネラルオイルを5 μl重層した後、PCRサイクル反応を行った。PCRは、55℃10秒間と80℃10秒間のサイクルを計35回繰り返した。
【0071】
3.生物化学発光による検出
実施例1ではPCR産物を電気泳動で分離解析したが、本実施例では生物化学発光を利用してPCR産物の検出を行った。3ウェルを一組とするPCR終了試料に対して、あらかじめ室温に戻しておいた発光試薬(キッコーマン(株))を18 μlずつ加え、5回ピペッティングして混合し、ルミノメーターで測定した。キッコーマン社製の発光試薬は「食品工業 vol.44 No.14 p25-34」記載のピロリン酸測定用の試薬で、ピルビン酸ホスホジキナーゼを用いてピロリン酸にAMPとホスホエノールピルビン酸(PEP)を作用させ、ATPを生成させるものである。この試薬では、耐熱性組換え日本産源氏ホタル由来ルシフェラーゼが用いられているため、試薬が安定で品質管理が容易という利点がある。
【0072】
アレル判定は、アレルCに相補なプライマー組から得られる信号値をIa、アレルTに相補なプライマー組から得られる信号値をIt、プライマーを含まないコントロールからの信号値をIcとして、アレルC由来の信号強度比(Ia-Ic)/(Ia+It-2×Ic)とアレルT由来の信号強度比(It-Ic)/(Ia+It-2×Ic)を計算することにより行った。判定基準は、(Ia-Ic)/(Ia+It-2×Ic)<0.2(あるいは(It-Ic)/(Ia+It-2×Ic)>0.8)の時はT/Tのホモザイゴート、(Ia-Ic)/(Ia+It-2×Ic)>0.8(あるいは(It-Ic)/(Ia+It-2×Ic)<0.2)の時はC/Cのホモザイゴート、0.4<(Ia-Ic)/(Ia+It-2×Ic)<0.6(あるいは0.4<(It-Ic)/(Ia+It-2×Ic)<0.6)の時はC/Tのヘテロザイゴートと判断することとした。
【0073】
4.結果
図6に、325検体のゲノム試料についてNCBI AF 538844のSNPを解析した結果を示した。図6の横軸は、(Ia-Ic)/(Ia+It-2×Ic)、縦軸は頻度を表す。アレルがC/Cのホモザイゴート由来のグループが81、T/Tのホモザイゴートが83、C/Tのヘテロザイゴートが82のグループを形成した。同一検体の同一SN部位をDNAシーケンシングで解析した結果、すべての検体において、本実施例の結果と一致することが判明した。この生物化学発光を利用したSNP解析は、1)ゲノム試料への不純物を分解する試薬の添加、2)PCR増幅、3)PCR産物への発光試薬の添加、といった極めて簡単な操作でSNP判定を行えるという利点がある。
【0074】
【発明の効果】
本発明によれば、3’末端が特定部位(例えば、SNP部位)に対応する一組のプライマーでPCR増幅を行うことにより、標的核酸上の特定部位の配列を正確かつ短時間で判定することができる。ゲノム試料が調製されていれば、判定は30分間以内でできる。特に、生物化学発光を利用した系は、高価で複雑な光学系機器を必要とせず、通常の簡便なルミノメーターで測定できるため、装置システムの小型化、自動化が期待できる。さらに、この系では各SNPに対応したプライマーを準備することにより、複数のゲノム試料に由来する複数のSNP判定が可能であり、汎用性に猛るという利点もある。
【0075】
【配列表】
【配列表フリーテキスト】
配列番号1−人工配列の説明:センスプライマー
配列番号2−人工配列の説明:アンチセンスプライマー
配列番号3−人工配列の説明:センスプライマー
配列番号4−人工配列の説明:アンチセンスプライマー
配列番号5−人工配列の説明:PCR増幅産物
配列番号6−人工配列の説明:PCR増幅産物
【図面の簡単な説明】
【図1】図1は、BAMPER法によるSNP解析方法の概要を示す図である。
【図2】図2は、本発明によるSNP解析方法の原理を示す図である。
【図3】図3は、本発明によるp53エキソン8のSNP解析における、電気泳動結果を示すグラフである。
【図4】図4は、p53エキソン8のSNP解析において、異なる変性温度(80℃及び94℃)でPCRを行った場合の電気泳動結果を示すグラフである。
【図5】図5は、本発明のPCR温度特性を示すグラフである。Aは、変性温度と相対増幅量の関係を示すグラフであり、Bは、塩基長と解離温度の関係を示すグラフである。
【図6】図6は、本発明を用いた検体多量解析の結果を示すグラフである。
【符号の説明】
1…PCR産物のセンス鎖、1’…PCR産物のアンチセンス鎖
2…dNTPとプライマー残査
3…センス鎖中のSNP部分
4…アンチセンス鎖中のSNP部分
5…SNP検査用プローブ
6…伸長鎖
7…ピロリン酸
21…ゲノムのセンス鎖、21’…ゲノムのアンチセンス鎖
22…ゲノムセンス鎖中のSNP部分、22’…ゲノムアンチセンス鎖中のSNP部分
23…ゲノムセンスに結合するSNP用プローブ
24…ゲノムアンチセンスに結合するSNP用プローブ
25、26…SNPに対応する識別配列
27、28…ゲノムの配列とは非相補な配列部分
29、30…ゲノムの各鎖とSNP判定用プローブのハイブリッド
31…PCR産物
41…Aホモザイゴトート検体
42…Tホモザイゴトート検体
43…ヘテロザイゴート検体
44、45、46…末端がTのSNP検査用プローブでの電気泳動結果
47、48、49…末端がAのSNP検査用プローブでの電気泳動結果
50、51、52、53…PCR産物のピーク
54…プローブのピーク
61…変性温度80℃でのPCR結果
62…変性温度94℃でのPCR結果
63…目的PCR産物
64…プローブ
65…擬陽性増幅ピーク
71…目的のPCR産物由来の電気泳動分離ピーク面積の相対値
72…擬陽性増幅産物に関わるピーク面積の合計の相対値
81…アレルAの検体グループ
82…ヘテロザイゴートの検体グループ
83…アレルTの検体グループ
Claims (7)
- 標的核酸上の一塩基多型部位を含む所望の塩基長範囲の増幅産物を得るための核酸増幅方法であって、
前記標的核酸の第一鎖の塩基配列に相補な配列を含み、かつ該第一鎖上の前記一塩基多型部位に3’末端がハイブリダイズするべく設計された第1のプライマーと、前記標的核酸の第二鎖の塩基配列に相補な配列を含み、該第二鎖上の前記一塩基多型部位と塩基対をなす部位に3’末端がハイブリダイズするべく設計された第2のプライマーとを用いて核酸増幅を行うこと、及び
前記核酸増幅における変性温度を少なくとも5サイクル目から60〜85℃に制御することにより、前記所望の塩基長範囲の増幅産物を1本鎖に解離して、次の核酸合成反応の鋳型とすることを特徴とする、前記核酸増幅方法。 - 前記所望の塩基長範囲の増幅産物が、前記第1のプライマーと第2のプライマーの3’末端が前記標的核酸上の一塩基多型部位にハイブリダイズして合成される核酸である、請求項1に記載の核酸増幅方法。
- 前記第1のプライマーと前記第2のプライマー塩基長が、いずれも18〜50であることを特徴とする、請求項1又は2に記載の核酸増幅方法。
- 前記第1のプライマーと前記第2のプライマーの融解温度が実質的に同一であることを特徴とする、請求項1〜3のいずれか1項に記載の核酸増幅方法。
- 前記第1のプライマーと前記第2のプライマーにおけるグアニン及びシトシンの割合の和が35〜65%であることを特徴とする、請求項1〜4のいずれか1項に記載の核酸増幅方法。
- 標的核酸上の一塩基多型部位の配列を解析するための方法であって、
前記標的核酸の第一鎖の塩基配列に相補な配列を含み、かつ該第一鎖上の前記一塩基多型部位に3’末端がハイブリダイズするべく設計された第1のプライマーと、前記標的核酸の第二鎖の塩基配列に相補な配列を含み、該第二鎖上の前記一塩基多型部位と塩基対をなす部位に3’末端がハイブリダイズするべく設計された第2のプライマーとを用いて核酸増幅を行うこと、及び
前記核酸増幅における変性温度を少なくとも5サイクル目から60〜85℃に制御することにより、増幅産物を1本鎖に解離して、次の核酸合成反応の鋳型とすることを特徴とする核酸増幅工程、ならびに
前記増幅産物を検出することにより、前記標的核酸上の一塩基多型部位の配列を解析する工程を含む、前記方法。 - 前記増幅産物の検出が、核酸増幅に応じて生成されるピロリン酸を利用した生物化学発光反応によって行われることを特徴とする、請求項6に記載の方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2003150853A JP3974557B2 (ja) | 2003-05-28 | 2003-05-28 | 変異解析方法及びそのためのシステム |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2003150853A JP3974557B2 (ja) | 2003-05-28 | 2003-05-28 | 変異解析方法及びそのためのシステム |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2004350563A JP2004350563A (ja) | 2004-12-16 |
| JP3974557B2 true JP3974557B2 (ja) | 2007-09-12 |
Family
ID=34046540
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2003150853A Expired - Fee Related JP3974557B2 (ja) | 2003-05-28 | 2003-05-28 | 変異解析方法及びそのためのシステム |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP3974557B2 (ja) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP7325039B2 (ja) * | 2019-07-24 | 2023-08-14 | 公立大学法人福島県立医科大学 | 一塩基の識別方法 |
| CN113832147B (zh) * | 2021-09-08 | 2024-06-14 | 华南农业大学 | 一种高效的大片段dna合成与扩增的pcr引物、方法及应用 |
-
2003
- 2003-05-28 JP JP2003150853A patent/JP3974557B2/ja not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| JP2004350563A (ja) | 2004-12-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6440707B1 (en) | Fluorescence polarization in nucleic acid analysis | |
| JP2008538496A (ja) | ウルトラディープ配列決定を用いて配列変異体を決定するための方法 | |
| JPWO2008066162A1 (ja) | Cyp2c19遺伝子増幅用プライマーセット、それを含むcyp2c19遺伝子増幅用試薬およびその用途 | |
| WO2011062258A1 (ja) | Mthfr遺伝子増幅用プライマーセット、それを含むmthfr遺伝子増幅用試薬およびその用途 | |
| WO2023241228A1 (zh) | 一种用于基因多态性分型的鉴定方法及其应用 | |
| JPWO2008066163A1 (ja) | Cyp2c9遺伝子増幅用プライマーセット、それを含むcyp2c9遺伝子増幅用試薬およびその用途 | |
| Rickert et al. | Refinement of single-nucleotide polymorphism genotyping methods on human genomic DNA: amplifluor allele-specific polymerase chain reaction versus ligation detection reaction-TaqMan | |
| JP5367365B2 (ja) | Sult1a1遺伝子増幅用プライマーセット、それを含むsult1a1遺伝子増幅用試薬およびその用途 | |
| JP4505838B2 (ja) | Nat2*6の変異の検出法ならびにそのための核酸プローブおよびキット | |
| JP3974557B2 (ja) | 変異解析方法及びそのためのシステム | |
| US20060172307A1 (en) | Method of nucleotide identification using an off-switch through proofreading 3' exonuclease-resistant modified primers by polymerases with 3' exonuclease activity | |
| JP2005328758A (ja) | 核酸増幅方法及びこれを利用した一塩基多型の解析 | |
| CN102369297B (zh) | 识别基因型的方法 | |
| JPWO2008066161A1 (ja) | Nat2遺伝子増幅用プライマーセット、それを含むnat2遺伝子増幅用試薬およびその用途 | |
| WO2004092385A1 (ja) | β3アドレナリン受容体変異遺伝子の検出法ならびにそのための核酸プローブおよびキット | |
| JP5047448B2 (ja) | Cyp2c19の変異の検出法およびそのための核酸プローブ | |
| JP4517175B2 (ja) | Nat2*7の変異の検出法ならびにそのための核酸プローブおよびキット | |
| US20250075266A1 (en) | Dna detection method and dna detection kit | |
| JP6389473B2 (ja) | 高解像能融解の較正の改善 | |
| Nagarajan et al. | Mutation Detection | |
| JP4517176B2 (ja) | Nat2*5の変異の検出法ならびにそのための核酸プローブおよびキット | |
| US20040175704A1 (en) | Compositions and methods for polynucleotide sequence detection | |
| WO2006070666A1 (ja) | 遺伝子多型の同時検出方法 | |
| TW201723189A (zh) | 用於基因型鑑定晶片之雙重等位基因特異性聚合酶鏈鎖反應的方法 | |
| JP2005110502A (ja) | Cyp2c19*3アレルの検出法およびそのための核酸プローブ |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20070227 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20070427 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20070529 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20070614 |
|
| FPAY | Renewal fee payment (prs date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20100622 Year of fee payment: 3 |
|
| LAPS | Cancellation because of no payment of annual fees |