JP2020007516A - セルロースアセテート及びセルロースアセテートの製造方法 - Google Patents
セルロースアセテート及びセルロースアセテートの製造方法 Download PDFInfo
- Publication number
- JP2020007516A JP2020007516A JP2018132667A JP2018132667A JP2020007516A JP 2020007516 A JP2020007516 A JP 2020007516A JP 2018132667 A JP2018132667 A JP 2018132667A JP 2018132667 A JP2018132667 A JP 2018132667A JP 2020007516 A JP2020007516 A JP 2020007516A
- Authority
- JP
- Japan
- Prior art keywords
- cellulose acetate
- pulp
- weight
- cellulose
- acetylation
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08B—POLYSACCHARIDES; DERIVATIVES THEREOF
- C08B3/00—Preparation of cellulose esters of organic acids
- C08B3/06—Cellulose acetate, e.g. mono-acetate, di-acetate or tri-acetate
Landscapes
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Biochemistry (AREA)
- Materials Engineering (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Polysaccharides And Polysaccharide Derivatives (AREA)
Abstract
Description
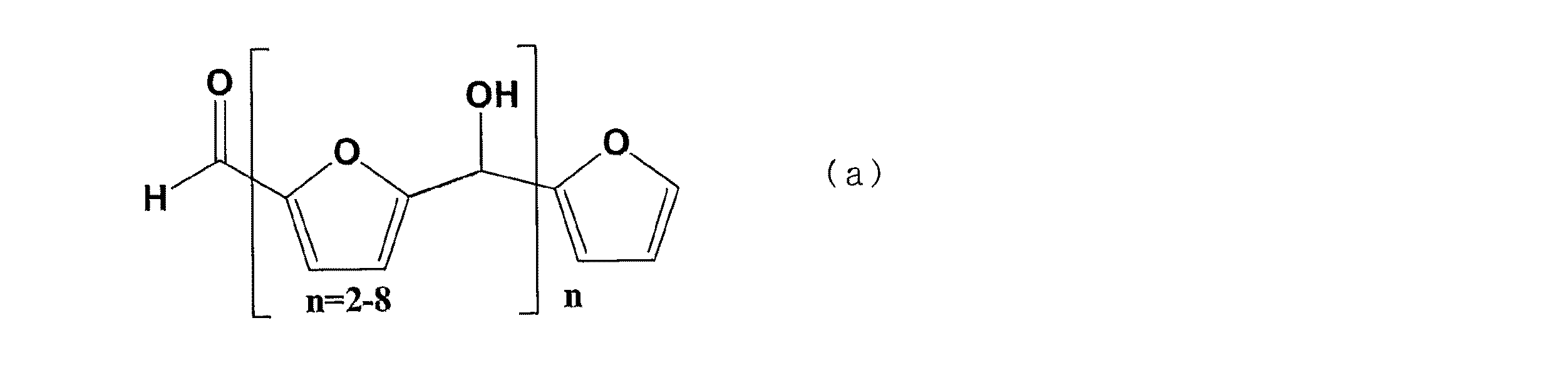
本開示のセルロースアセテートは、下記一般式(a)で表される化合物の含有量が0ppb以上100ppb以下であり、アセチル総置換度が2.3以上2.6以下である。
分析装置:Corona CAD(Thermo社製)
カラム:ULTRON AF−HILIC−CD(4.6φ×100mm,5μm)(信和化工株式会社製)
カラム温度:30℃
溶離液:A液:10mM 酢酸アンモニウム(pH6.8)、B液:アセトニトリル(HPLC用)
グラジエント:A液/B液=10/90(25min)→A液/B液=50/50(0.1min)→A液/B液=10/90(29.9min) 合計55min
流量:0.4mL/min
検出:CADPDA(Thermo社製)
A:シグナル220nm、バンド幅10nm、リファレンスOFF、スリット1nm
B:シグナル285nm、バンド幅10nm、リファレンスOFF、スリット1nm
C:シグナル380nm、バンド幅10nm、リファレンスOFF、スリット1nm
D:シグナル430nm、バンド幅10nm、リファレンスOFF、スリット1nm
スペクトル保存全てシグナル190〜600nm
注入量:15μL
本開示のセルロースアセテートのアセチル総置換度は、2.3以上2.6以下であるところ、2.4以上2.6以下であってよく、2.4以上2.5以下であってよい。アセチル総置換度をより低くしようとすれば、加水分解を行って、脱アセチル化を進行する必要があるが、脱アセチル化の進行に伴い、上記一般式(a)で表される化合物も発生しやすくなる。しかしながら、本開示のセルロースアセテートは、アセチル総置換度が2.3以上2.6以下と低いにもかかわらず、上記一般式(a)で表される化合物の含有量が十分に低く、優れた色相を有する。
DS=162.14×AV×0.01/(60.052−42.037×AV×0.01)
DS:アセチル総置換度
AV:酢化度(%)
AV(%)=(A−B)×F×1.201/試料重量(g)
A:0.2N−水酸化ナトリウム規定液の滴定量(ml)
B:ブランクテストにおける0.2N−水酸化ナトリウム規定液の滴定量(ml)
F:0.2N−水酸化ナトリウム規定液のファクター
本開示のセルロースアセテートの波長430nmにおける吸光度法色相の値は、0.65cm−1以下が好ましく、0.50cm−1以下がより好ましく、0.45cm−1以下がさらに好ましい。波長430nmにおける吸光度法色相の値は、より小さい方が色相に優れるため好ましく、下限は特に限定されない。
吸光度:分光光度計 島津製作所社製UV−1700
A:430nmの吸光度(液の黄色味を測定)
B:740nmの吸光度(液の濁りを測定:ベースライン)
セルロースアセテート濃度(重量%):絶乾セルロースアセテート重量(g)/セルロースアセテート溶液全体重量(g)×100
絶乾セルロースアセテート重量(g):セルロースアセテートの重量(g)×(1−含水率(%)/100)
含水率(%):赤外線水分計 METTLER TOLEDO HB43
本開示のセルロースアセテートは、6%粘度が60mPa・s以上が好ましく、80mPa・s以上がより好ましく、90mPa・s以上がさらに好ましく、100mPa・s以上が最も好ましい。また、160mPa・s以下、130mPa・s以下、110mPa・s以下であってよい。
本開示のセルロースアセテートのろ過度KWとしては、400ml−1以下が好ましく、200ml−1以下がより好ましく、100ml−1以下がさらに好ましく、95ml−1以下が最も好ましい。
KW=〔(2−P2/P1)/(P1+P2)〕×104
本開示のセルロースアセテートの糖鎖成分の構成比としては、マンノース単位(Man)、キシロース単位(Xyl)、及びグルコース単位(Glc)の3成分の合計を100として、マンノース単位(Man)及びキシロース単位(Xyl)の合計含量(モル%)は、5.0以下が好ましく、4.0以下がより好ましい。また、原料となるパルプ製造における環境負荷、ならびに製造コストの観点からは、マンノース単位(Man)及びキシロース単位(Xyl)の合計含量(モル%)は、1.0以上であってよい。
本開示のセルロースアセテートの製造方法は、パルプと酢酸とを接触させて前処理する工程(1)、前記前処理をした後、前記パルプに含まれるセルロースを無水酢酸と反応させてアセチル化する工程(2)、前記アセチル化により得られたセルロースアセテートを加水分解する工程(3)、及び前記加水分解によりアセチル置換度が調整されたセルロースアセテートを沈殿する工程(4)を有し、前記加水分解工程(3)までにジアセトアミドを添加するものである。ジアセトアミドは、加水分解工程(3)までの何れかの工程において添加されていればよい。
本開示のセルロースアセテートの原料となるセルロース源として、パルプを用いることができる。パルプとしては、例えば、木材パルプ、及びリンターパルプが挙げられる。特には、木材パルプを用いることができる。
本開示のセルロースアセテートの製造方法においては、予めパルプを解砕する工程を有してもよい。これにより、以降の工程で反応が効率的に均一に進み、取扱いも容易になる。解砕工程は、特に、パルプがシート状の形態で供給されるような場合に有効である。
パルプと酢酸とを接触させて前処理する工程(1)において、酢酸は、例えば、パルプに含まれるセルロース100重量部に対して、好ましくは10重量部以上500重量部以下を添加することにより接触させることができる。この時、酢酸は、99重量%以上の濃度のものを用いることができる。
前記パルプに含まれるセルロースを無水酢酸と反応させてアセチル化する工程(2)において、アセチル化は、具体的には、例えば、酢酸、無水酢酸及び硫酸からなる混合物に、前処理により活性化したパルプに含まれるセルロースを添加すること、または前処理により活性化したパルプに含まれるセルロースに、酢酸と無水酢酸との混合物及び硫酸を添加すること等により開始することができる。ここで、酢酸は、99重量%以上の濃度のものを用いることができる。硫酸は、98重量%以上の濃度のものを用いることが好ましい。
減圧条件下のアセチル化工程(2)においては、少なくともアセチル化反応開始時にアセチル化反応系内の真空度を70Torr(9.3kPa)以下とすることが好ましい。下限は特に限定されないが、例えば60Torr(8.0kPa)以上である。これにより、アセチル化反応のため無水酢酸の溶媒として酢酸を用いた場合に、酢酸の蒸発を促してアセチル化反応で消費された無水酢酸の濃度を高め、アセチル化反応を促すことができ、無水酢酸及び溶媒の使用率を低減することができ、セルロースアセテートの生産性を向上することができる。この方法では、アセチル化反応に伴う温度上昇を抑えるための酢酸及び無水酢酸の混合溶液の冷却が不要になる利点がある。
常圧条件下のセルロースアセテートの製造方法について述べる。常圧条件でアセチル化する場合には、反応温度を制御するために、添加する無水酢酸、酢酸を低温にする必要がある。
前記アセチル化により得られたセルロースアセテートは、ほぼ全ての水酸基がアセチル基に置換されている状態であり、これを所望の置換度に調整するために加水分解を行う必要がある。前記アセチル化反応の触媒として硫酸を用いた場合、当該硫酸は、硫酸エステルとしてセルロースに結合しているため、加水分解する工程(3)においては、前記アセチル化反応終了後、熱安定性向上のためこの硫酸エステルを加水分解して除去する目的もある。
前記加水分解によりアセチル置換度が調整されたセルロースアセテートを沈殿する工程(4)においては、セルロースアセテートを含む混合物と水又は希酢酸等の沈殿剤とを混合し、生成したセルロースアセテート(沈殿物)を分離して沈殿物を得ることができる。また、沈殿剤としては、希酢酸が好ましい。
重量既知のパルプを25℃で17.5%と9.45%の水酸化ナトリウム水溶液で連続的に抽出し、その抽出液の可溶部分に対して重クロム酸カリウムで酸化し、酸化に要した重クロム酸カリウムの容量からβ,γ−セルロースの重量を決定した。初期のパルプの重量からβ,γ−セルロース重量を引いた値を、パルプの不溶部分の重量、つまりα−セルロースの重量とした(TAPPI T203)。初期のパルプの重量に対する、パルプの不溶部分の重量の割合が、α−セルロース含有率(重量%)である。
アセチル総置換度は、上記の手塚の方法に従い、1H−NMRを用いた方法により測定した。
(1)セルロースアセテートの含水率測定
赤外線水分計(METTLER TOLEDO HB43)を用いて、セルロースアセテートの含水率を測定し、記録用紙に記録した。
まずサンプル調製を行った。1)三角フラスコにDMSO95.00gを計量した。2)三角フラスコにスターラー回転子を入れ、セロファン、シリコン栓をして攪拌した。3)セルロースアセテートサンプル5.00gを薬包紙等に計量し攪拌している三角フラスコ内に添加した。4)セロファン、シリコン栓をしてスターラーで1hr攪拌した。5)回転振盪機(高速)で2hr振盪した。6)回転振盪機から取り外した後30分間静置し脱泡し、サンプルを調製した。
以下の計算式で得られた数値をセルロースアセテートのその溶媒における「吸光度法色相」値とした。
吸光度法色相(cm−1)=吸光度(A−B)/セル厚(cm)/セルロースアセテート濃度(重量%)×100
吸光度:分光光度計 島津製作所社製UV−1700
A:430nmの吸光度(液の黄色味を測定)
B:740nmの吸光度(液の濁りを測定:ベースライン)
セルロースアセテート濃度(重量%):絶乾セルロースアセテート重量(g)/セルロースアセテート溶液全体重量(g)×100
絶乾セルロースアセテート重量(g):セルロースアセテートの重量(g)×(1−含水率(%)/100)
含水率(%):上記赤外線水分計で測定した値
三角フラスコに乾燥試料3.00g、95%アセトン水溶液39.90gを採取し、密栓して約1時間攪拌した。その後、回転振盪機で約1.5時間振盪して完溶させた。得られた6wt/vol%の溶液を所定のオストワルド粘度計の標線まで移し、25±1℃で約30分間整温した。計時標線間の流下時間を測定し、次式により6%粘度(mPa・s)を算出した。
6%粘度(mPa・s)=流下時間(s)×粘度計係数
粘度計係数={標準液絶対粘度(mPa・s)×溶液の密度(0.827g/cm3)}/{標準液の密度(g/cm3)×標準液の流下秒数(s)}
ろ過度は、以下の方法により測定した。95%アセトン水溶液に、20重量%濃度となるように溶解したセルロースアセテート溶液を30℃、一定圧力(2kgf/cm2)下で、所定のろ布金巾(直径15mm、ろ過面積1.77cm2)に通し、ろ過した。この時、ろ過開始後20分までのろ過量をP1(ml)、20分より60分までのろ過量をP2(ml)として測定し、下記式によりろ過度KW(ml−1)を算出した。
KW=〔(2−P2/P1)/(P1+P2)〕×104
充分に乾燥した試料200mgを精秤し、72%硫酸3mLを加え、氷水で冷却しながら超音波を用いて撹拌し、2時間以上かけて試料を完全に溶解させた。得られた溶液に蒸留水39mLを加えて十分に振盪し、窒素気流下、110℃で3時間還流した後、30分間放冷した。次いで、炭酸バリウム14gを加え、氷水で冷却しつつ超音波を用いて撹拌し、硫酸を中和した。30分後、さらに炭酸バリウム10gを加え、pH5.5〜6.5程度になるまで硫酸を中和し、ろ過した。得られた濾液を超純水で100重量倍に希釈し、分析用の試料を調製した。
高速液体クロマトグラフィ(HPLC:アジレント・テクノロジー社製Agilent 1200シリーズシステム)
検出器: CoronaPlus CAD検出器
カラム: Shodex社製、Asahipak NH2P-50 4E(250×4.6mm)
ガードカラム: Shodex社製、Asahipak NH2P-50G 4A
溶離液: 超純水/アセトニトリル(HPLC用)= 25/75(v/ v)
溶離液流量:1.0mL/分
カラム温度:20℃
上記一般式(a)で表される化合物の含有量は、次のとおり、高分子量不純物測定法にて測定した。50℃のアセトン−水溶液(50重量%−50重量%)130gで、セルロースアセテート10gを1時間洗浄した後、抽出して得た抽出液を濃縮し、上記測定条件のHPLCで分析し定量した。
セルロース原料として木材パルプ、レイアセタ−HJ(含水率約3.0%、α−セルロース含有率98重量%)を解砕機でフラッフ状に解砕した。フラッフ状のセルロースパルプ100重量部を前処理機に入れ、氷酢酸38重量部を散布して、前処理により活性化(25℃、60分)した。また、無水酢酸に対してジアセトアミドを200ppmとなるように添加して溶解させ、無水酢酸溶液を調製した。前処理によって酢酸を含浸させたセルロースパルプを、酢化反応器に撹拌しながら入れ、酢化反応器に仕込んだ。さらに酢化反応器に氷酢酸200重量部、無水酢酸溶液270重量部、硫酸1.1重量部を添加し、12℃に冷却し、10分かけて60℃に到達させた後、80分間保持して、アセチル化反応を行った。
無水酢酸に対してジアセトアミドを添加しなかった以外は、実施例1と同様にしてセルロースアセテートを得た。得られたセルロースアセテート及び各原料の物性を評価した結果は、表1に示す。
Claims (8)
- 下記一般式(a)で表される化合物の含有量が0ppb以上100ppb以下であり、
アセチル総置換度が2.3以上2.6以下である、セルロースアセテート。
- パルプと酢酸とを接触させて前処理する工程(1)、
前記前処理をした後、前記パルプに含まれるセルロースを無水酢酸と反応させてアセチル化する工程(2)、
前記アセチル化により得られたセルロースアセテートを加水分解する工程(3)、及び
前記加水分解によりアセチル置換度が調整されたセルロースアセテートを沈殿する工程(4)を有し、
前記加水分解工程(3)までにジアセトアミドを添加する、セルロースアセテートの製造方法。 - 前記アセチル化工程(2)において、前記無水酢酸にジアセトアミドを添加する、請求項2に記載のセルロースアセテートの製造方法。
- 前記無水酢酸の重量に対して50ppm以上200ppm以下のジアセトアミドを添加する、請求項3に記載のセルロースアセテートの製造方法。
- 前記前処理工程(1)において、前記酢酸にジアセトアミドを添加する、請求項2〜4のいずれか一項に記載のセルロースアセテートの製造方法。
- 前記酢酸の重量に対して350ppm以上1500ppm以下のジアセトアミドを添加する、請求項5に記載のセルロースアセテートの製造方法。
- 前記アセチル化工程(2)において、アセチル化反応開始時にアセチル化反応系内の真空度を9.3kPa以下とする、請求項2〜6の何れか一項に記載のセルロースアセテートの製造方法。
- 前記加水分解工程(3)において、反応系内の最高到達温度を100℃以上200℃以下とする、請求項2〜7の何れか一項に記載のセルロースアセテートの製造方法。
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2018132667A JP7093256B2 (ja) | 2018-07-12 | 2018-07-12 | セルロースアセテートの製造方法 |
| PCT/JP2019/027678 WO2020013312A1 (ja) | 2018-07-12 | 2019-07-12 | セルロースアセテート及びセルロースアセテートの製造方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2018132667A JP7093256B2 (ja) | 2018-07-12 | 2018-07-12 | セルロースアセテートの製造方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2020007516A true JP2020007516A (ja) | 2020-01-16 |
| JP7093256B2 JP7093256B2 (ja) | 2022-06-29 |
Family
ID=69143018
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2018132667A Active JP7093256B2 (ja) | 2018-07-12 | 2018-07-12 | セルロースアセテートの製造方法 |
Country Status (2)
| Country | Link |
|---|---|
| JP (1) | JP7093256B2 (ja) |
| WO (1) | WO2020013312A1 (ja) |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS58176201A (ja) * | 1982-04-12 | 1983-10-15 | Daicel Chem Ind Ltd | セルロ−ス・カルボン酸エステルの新規な製造方法 |
| WO2017061474A1 (ja) * | 2015-10-08 | 2017-04-13 | 株式会社ダイセル | セルロースアセテート、セルロースアセテートの製造方法および製造装置 |
| WO2018066477A1 (ja) * | 2016-10-03 | 2018-04-12 | 株式会社ダイセル | セルロースアセテートおよびセルロースアセテートの製造方法 |
| JP2018119051A (ja) * | 2017-01-25 | 2018-08-02 | 株式会社ダイセル | セルロースアセテートおよび成形体 |
| WO2018139319A1 (ja) * | 2017-01-25 | 2018-08-02 | 株式会社ダイセル | セルロースアセテートフレークの製造方法 |
-
2018
- 2018-07-12 JP JP2018132667A patent/JP7093256B2/ja active Active
-
2019
- 2019-07-12 WO PCT/JP2019/027678 patent/WO2020013312A1/ja active Application Filing
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS58176201A (ja) * | 1982-04-12 | 1983-10-15 | Daicel Chem Ind Ltd | セルロ−ス・カルボン酸エステルの新規な製造方法 |
| WO2017061474A1 (ja) * | 2015-10-08 | 2017-04-13 | 株式会社ダイセル | セルロースアセテート、セルロースアセテートの製造方法および製造装置 |
| WO2018066477A1 (ja) * | 2016-10-03 | 2018-04-12 | 株式会社ダイセル | セルロースアセテートおよびセルロースアセテートの製造方法 |
| JP2018119051A (ja) * | 2017-01-25 | 2018-08-02 | 株式会社ダイセル | セルロースアセテートおよび成形体 |
| WO2018139319A1 (ja) * | 2017-01-25 | 2018-08-02 | 株式会社ダイセル | セルロースアセテートフレークの製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP7093256B2 (ja) | 2022-06-29 |
| WO2020013312A1 (ja) | 2020-01-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR101057509B1 (ko) | 개선된 습열 안정성을 갖는 셀룰로오스 에스테르 | |
| JP6820858B2 (ja) | セルロースアセテート、セルロースアセテートの製造方法および製造装置 | |
| JP2018058941A (ja) | セルロースアセテートおよびセルロースアセテートの製造方法 | |
| WO2018139319A1 (ja) | セルロースアセテートフレークの製造方法 | |
| US11773239B2 (en) | Cellulose acetate and molded article | |
| JP7093256B2 (ja) | セルロースアセテートの製造方法 | |
| JP7526419B2 (ja) | セルロースアセテートの製造方法 | |
| JP6663979B2 (ja) | セルロースアセテート | |
| US11066484B2 (en) | Cellulose acetate and method for producing cellulose acetate | |
| CN106573990B (zh) | 乙酸纤维素薄片及其制造方法 | |
| JP2024095757A (ja) | 酢酸セルロース及び酢酸セルロースの製造方法 | |
| Egüés Artola et al. | Effect of different hemicelluloses characteristics on film forming properties |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20210514 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20220524 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20220530 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20220614 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20220617 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 7093256 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |