JP2019504892A - 癌治療のための改変哺乳類細胞 - Google Patents
癌治療のための改変哺乳類細胞 Download PDFInfo
- Publication number
- JP2019504892A JP2019504892A JP2018560705A JP2018560705A JP2019504892A JP 2019504892 A JP2019504892 A JP 2019504892A JP 2018560705 A JP2018560705 A JP 2018560705A JP 2018560705 A JP2018560705 A JP 2018560705A JP 2019504892 A JP2019504892 A JP 2019504892A
- Authority
- JP
- Japan
- Prior art keywords
- cell
- promoter
- pharmaceutical composition
- nucleic acid
- modified mammalian
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 210000004962 mammalian cell Anatomy 0.000 title claims abstract description 254
- 206010028980 Neoplasm Diseases 0.000 title claims abstract description 162
- 201000011510 cancer Diseases 0.000 title claims abstract description 43
- 238000011282 treatment Methods 0.000 title abstract description 26
- 150000007523 nucleic acids Chemical class 0.000 claims abstract description 277
- 102000039446 nucleic acids Human genes 0.000 claims abstract description 275
- 108020004707 nucleic acids Proteins 0.000 claims abstract description 275
- 239000002955 immunomodulating agent Substances 0.000 claims abstract description 259
- 229940121354 immunomodulator Drugs 0.000 claims abstract description 248
- 210000004027 cell Anatomy 0.000 claims abstract description 236
- 108090000623 proteins and genes Proteins 0.000 claims abstract description 220
- 108010019670 Chimeric Antigen Receptors Proteins 0.000 claims abstract description 215
- 230000002584 immunomodulator Effects 0.000 claims abstract description 205
- 102000004169 proteins and genes Human genes 0.000 claims abstract description 193
- 239000008194 pharmaceutical composition Substances 0.000 claims abstract description 159
- 230000001225 therapeutic effect Effects 0.000 claims abstract description 135
- 210000001744 T-lymphocyte Anatomy 0.000 claims abstract description 111
- 108091008874 T cell receptors Proteins 0.000 claims abstract description 106
- 102000016266 T-Cell Antigen Receptors Human genes 0.000 claims abstract description 102
- 102000037984 Inhibitory immune checkpoint proteins Human genes 0.000 claims abstract description 95
- 108091008026 Inhibitory immune checkpoint proteins Proteins 0.000 claims abstract description 95
- 229940076838 Immune checkpoint inhibitor Drugs 0.000 claims abstract description 94
- 239000012274 immune-checkpoint protein inhibitor Substances 0.000 claims abstract description 94
- 238000000034 method Methods 0.000 claims abstract description 66
- 239000000427 antigen Substances 0.000 claims description 133
- 108091007433 antigens Proteins 0.000 claims description 133
- 102000036639 antigens Human genes 0.000 claims description 133
- 210000002865 immune cell Anatomy 0.000 claims description 104
- 239000003795 chemical substances by application Substances 0.000 claims description 79
- 230000001939 inductive effect Effects 0.000 claims description 79
- 230000014509 gene expression Effects 0.000 claims description 71
- 230000004068 intracellular signaling Effects 0.000 claims description 68
- -1 BLTA Proteins 0.000 claims description 60
- 229940045513 CTLA4 antagonist Drugs 0.000 claims description 53
- 230000003308 immunostimulating effect Effects 0.000 claims description 49
- 108090000765 processed proteins & peptides Proteins 0.000 claims description 48
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 46
- 239000003112 inhibitor Substances 0.000 claims description 44
- 239000013598 vector Substances 0.000 claims description 44
- 102000004196 processed proteins & peptides Human genes 0.000 claims description 41
- 230000008685 targeting Effects 0.000 claims description 40
- 229920001184 polypeptide Polymers 0.000 claims description 38
- 108010002350 Interleukin-2 Proteins 0.000 claims description 36
- 108010003723 Single-Domain Antibodies Proteins 0.000 claims description 32
- 210000000130 stem cell Anatomy 0.000 claims description 30
- 239000000411 inducer Substances 0.000 claims description 29
- 210000003819 peripheral blood mononuclear cell Anatomy 0.000 claims description 27
- 230000002238 attenuated effect Effects 0.000 claims description 25
- 108010074108 interleukin-21 Proteins 0.000 claims description 23
- 102000003812 Interleukin-15 Human genes 0.000 claims description 22
- 108090000172 Interleukin-15 Proteins 0.000 claims description 22
- 239000012642 immune effector Substances 0.000 claims description 22
- 108010002586 Interleukin-7 Proteins 0.000 claims description 21
- 230000006044 T cell activation Effects 0.000 claims description 21
- 230000006698 induction Effects 0.000 claims description 21
- 102100021569 Apoptosis regulator Bcl-2 Human genes 0.000 claims description 20
- 230000001419 dependent effect Effects 0.000 claims description 20
- 101710149863 C-C chemokine receptor type 4 Proteins 0.000 claims description 19
- 102100032976 CCR4-NOT transcription complex subunit 6 Human genes 0.000 claims description 19
- 102100032101 Tumor necrosis factor ligand superfamily member 9 Human genes 0.000 claims description 19
- 210000000822 natural killer cell Anatomy 0.000 claims description 19
- 150000003384 small molecules Chemical group 0.000 claims description 19
- 108010065805 Interleukin-12 Proteins 0.000 claims description 18
- 102000013462 Interleukin-12 Human genes 0.000 claims description 18
- 108010074708 B7-H1 Antigen Proteins 0.000 claims description 17
- 102000008096 B7-H1 Antigen Human genes 0.000 claims description 17
- 102100024025 Heparanase Human genes 0.000 claims description 17
- 101000971171 Homo sapiens Apoptosis regulator Bcl-2 Proteins 0.000 claims description 17
- 102100030983 Lymphocyte expansion molecule Human genes 0.000 claims description 17
- 108010037536 heparanase Proteins 0.000 claims description 17
- 101001063413 Homo sapiens Lymphocyte expansion molecule Proteins 0.000 claims description 16
- 101000638251 Homo sapiens Tumor necrosis factor ligand superfamily member 9 Proteins 0.000 claims description 16
- 239000000203 mixture Substances 0.000 claims description 11
- 101100407308 Mus musculus Pdcd1lg2 gene Proteins 0.000 claims description 10
- 108700030875 Programmed Cell Death 1 Ligand 2 Proteins 0.000 claims description 10
- 102100024213 Programmed cell death 1 ligand 2 Human genes 0.000 claims description 10
- 239000013603 viral vector Substances 0.000 claims description 9
- 210000003719 b-lymphocyte Anatomy 0.000 claims description 8
- 238000002347 injection Methods 0.000 claims description 8
- 239000007924 injection Substances 0.000 claims description 8
- 230000000735 allogeneic effect Effects 0.000 claims description 5
- 230000005966 endogenous activation Effects 0.000 claims description 5
- 241000700584 Simplexvirus Species 0.000 claims description 4
- 238000001802 infusion Methods 0.000 claims description 4
- 230000001177 retroviral effect Effects 0.000 claims description 4
- 238000004520 electroporation Methods 0.000 claims description 3
- 102100034458 Hepatitis A virus cellular receptor 2 Human genes 0.000 claims description 2
- 229940126547 T-cell immunoglobulin mucin-3 Drugs 0.000 claims description 2
- 102100023990 60S ribosomal protein L17 Human genes 0.000 claims 1
- 108010021064 CTLA-4 Antigen Proteins 0.000 claims 1
- 102000008203 CTLA-4 Antigen Human genes 0.000 claims 1
- 101710083479 Hepatitis A virus cellular receptor 2 homolog Proteins 0.000 claims 1
- 102000017578 LAG3 Human genes 0.000 claims 1
- 101150030213 Lag3 gene Proteins 0.000 claims 1
- 101710089372 Programmed cell death protein 1 Proteins 0.000 claims 1
- 238000009169 immunotherapy Methods 0.000 abstract description 7
- 238000002619 cancer immunotherapy Methods 0.000 abstract description 4
- 235000018102 proteins Nutrition 0.000 description 186
- 210000004881 tumor cell Anatomy 0.000 description 55
- 230000027455 binding Effects 0.000 description 53
- 230000002519 immonomodulatory effect Effects 0.000 description 42
- 230000006870 function Effects 0.000 description 40
- 102000000588 Interleukin-2 Human genes 0.000 description 35
- 102100039498 Cytotoxic T-lymphocyte protein 4 Human genes 0.000 description 32
- 101000889276 Homo sapiens Cytotoxic T-lymphocyte protein 4 Proteins 0.000 description 32
- 101100519207 Mus musculus Pdcd1 gene Proteins 0.000 description 32
- 230000011664 signaling Effects 0.000 description 28
- 239000003446 ligand Substances 0.000 description 27
- 230000028327 secretion Effects 0.000 description 26
- 102000001301 EGF receptor Human genes 0.000 description 25
- 108060006698 EGF receptor Proteins 0.000 description 25
- 102000037982 Immune checkpoint proteins Human genes 0.000 description 25
- 108091008036 Immune checkpoint proteins Proteins 0.000 description 25
- 230000000139 costimulatory effect Effects 0.000 description 23
- 108060003951 Immunoglobulin Proteins 0.000 description 22
- 102000018358 immunoglobulin Human genes 0.000 description 22
- 102100021592 Interleukin-7 Human genes 0.000 description 20
- BGFTWECWAICPDG-UHFFFAOYSA-N 2-[bis(4-chlorophenyl)methyl]-4-n-[3-[bis(4-chlorophenyl)methyl]-4-(dimethylamino)phenyl]-1-n,1-n-dimethylbenzene-1,4-diamine Chemical compound C1=C(C(C=2C=CC(Cl)=CC=2)C=2C=CC(Cl)=CC=2)C(N(C)C)=CC=C1NC(C=1)=CC=C(N(C)C)C=1C(C=1C=CC(Cl)=CC=1)C1=CC=C(Cl)C=C1 BGFTWECWAICPDG-UHFFFAOYSA-N 0.000 description 18
- 108010008014 B-Cell Maturation Antigen Proteins 0.000 description 18
- 102000006942 B-Cell Maturation Antigen Human genes 0.000 description 18
- 102100025570 Cancer/testis antigen 1 Human genes 0.000 description 18
- 101000856237 Homo sapiens Cancer/testis antigen 1 Proteins 0.000 description 18
- 101001012157 Homo sapiens Receptor tyrosine-protein kinase erbB-2 Proteins 0.000 description 18
- 108010021625 Immunoglobulin Fragments Proteins 0.000 description 18
- 102000008394 Immunoglobulin Fragments Human genes 0.000 description 18
- 102100030086 Receptor tyrosine-protein kinase erbB-2 Human genes 0.000 description 18
- 239000004098 Tetracycline Substances 0.000 description 18
- 229960002180 tetracycline Drugs 0.000 description 18
- 229930101283 tetracycline Natural products 0.000 description 18
- 235000019364 tetracycline Nutrition 0.000 description 18
- 150000003522 tetracyclines Chemical class 0.000 description 18
- 102100030703 Interleukin-22 Human genes 0.000 description 17
- 229960001438 immunostimulant agent Drugs 0.000 description 17
- 239000003022 immunostimulating agent Substances 0.000 description 17
- SGKRLCUYIXIAHR-AKNGSSGZSA-N (4s,4ar,5s,5ar,6r,12ar)-4-(dimethylamino)-1,5,10,11,12a-pentahydroxy-6-methyl-3,12-dioxo-4a,5,5a,6-tetrahydro-4h-tetracene-2-carboxamide Chemical compound C1=CC=C2[C@H](C)[C@@H]([C@H](O)[C@@H]3[C@](C(O)=C(C(N)=O)C(=O)[C@H]3N(C)C)(O)C3=O)C3=C(O)C2=C1O SGKRLCUYIXIAHR-AKNGSSGZSA-N 0.000 description 16
- 230000004913 activation Effects 0.000 description 16
- 229960003722 doxycycline Drugs 0.000 description 16
- 229940126546 immune checkpoint molecule Drugs 0.000 description 16
- 102100024222 B-lymphocyte antigen CD19 Human genes 0.000 description 15
- 102000017420 CD3 protein, epsilon/gamma/delta subunit Human genes 0.000 description 15
- 108050005493 CD3 protein, epsilon/gamma/delta subunit Proteins 0.000 description 15
- 102000004127 Cytokines Human genes 0.000 description 15
- 108090000695 Cytokines Proteins 0.000 description 15
- 101000980825 Homo sapiens B-lymphocyte antigen CD19 Proteins 0.000 description 15
- 230000000694 effects Effects 0.000 description 15
- 108010022366 Carcinoembryonic Antigen Proteins 0.000 description 14
- 230000000973 chemotherapeutic effect Effects 0.000 description 14
- 230000003013 cytotoxicity Effects 0.000 description 14
- 231100000135 cytotoxicity Toxicity 0.000 description 14
- 102100025475 Carcinoembryonic antigen-related cell adhesion molecule 5 Human genes 0.000 description 13
- 201000010099 disease Diseases 0.000 description 13
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 13
- 230000002401 inhibitory effect Effects 0.000 description 13
- 102100038080 B-cell receptor CD22 Human genes 0.000 description 12
- 102100022005 B-lymphocyte antigen CD20 Human genes 0.000 description 12
- 101000884305 Homo sapiens B-cell receptor CD22 Proteins 0.000 description 12
- 101000897405 Homo sapiens B-lymphocyte antigen CD20 Proteins 0.000 description 12
- 101000934338 Homo sapiens Myeloid cell surface antigen CD33 Proteins 0.000 description 12
- 102100025243 Myeloid cell surface antigen CD33 Human genes 0.000 description 12
- 239000012634 fragment Substances 0.000 description 12
- 230000035755 proliferation Effects 0.000 description 12
- 102100041003 Glutamate carboxypeptidase 2 Human genes 0.000 description 11
- 101000892862 Homo sapiens Glutamate carboxypeptidase 2 Proteins 0.000 description 11
- 101001137987 Homo sapiens Lymphocyte activation gene 3 protein Proteins 0.000 description 11
- 101000946843 Homo sapiens T-cell surface glycoprotein CD8 alpha chain Proteins 0.000 description 11
- 102100039688 Insulin-like growth factor 1 receptor Human genes 0.000 description 11
- 102100022339 Integrin alpha-L Human genes 0.000 description 11
- 102100020862 Lymphocyte activation gene 3 protein Human genes 0.000 description 11
- 102000003735 Mesothelin Human genes 0.000 description 11
- 108090000015 Mesothelin Proteins 0.000 description 11
- 102100029215 Signaling lymphocytic activation molecule Human genes 0.000 description 11
- 102100034922 T-cell surface glycoprotein CD8 alpha chain Human genes 0.000 description 11
- 108700019146 Transgenes Proteins 0.000 description 11
- 108700020467 WT1 Proteins 0.000 description 11
- 230000001461 cytolytic effect Effects 0.000 description 11
- 229920001481 poly(stearyl methacrylate) Polymers 0.000 description 11
- 102100031585 ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase 1 Human genes 0.000 description 10
- 101000777636 Homo sapiens ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase 1 Proteins 0.000 description 10
- 101001103039 Homo sapiens Inactive tyrosine-protein kinase transmembrane receptor ROR1 Proteins 0.000 description 10
- 101001034652 Homo sapiens Insulin-like growth factor 1 receptor Proteins 0.000 description 10
- 101001103036 Homo sapiens Nuclear receptor ROR-alpha Proteins 0.000 description 10
- 102100039615 Inactive tyrosine-protein kinase transmembrane receptor ROR1 Human genes 0.000 description 10
- 230000006037 cell lysis Effects 0.000 description 10
- 239000003814 drug Substances 0.000 description 10
- 210000000987 immune system Anatomy 0.000 description 10
- 229960002621 pembrolizumab Drugs 0.000 description 10
- SSOORFWOBGFTHL-OTEJMHTDSA-N (4S)-5-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-6-amino-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[2-[(2S)-2-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-6-amino-1-[[(2S)-1-[[(2S)-1-[[(2S,3S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-6-amino-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-5-amino-1-[[(2S)-1-[[(2S)-1-[[(2S)-6-amino-1-[[(2S)-6-amino-1-[[(2S)-1-[[(2S)-1-[[(2S)-5-amino-1-[[(2S)-5-carbamimidamido-1-[[(2S)-5-carbamimidamido-1-[[(1S)-4-carbamimidamido-1-carboxybutyl]amino]-1-oxopentan-2-yl]amino]-1-oxopentan-2-yl]amino]-1,5-dioxopentan-2-yl]amino]-5-carbamimidamido-1-oxopentan-2-yl]amino]-5-carbamimidamido-1-oxopentan-2-yl]amino]-1-oxohexan-2-yl]amino]-1-oxohexan-2-yl]amino]-5-carbamimidamido-1-oxopentan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-1,5-dioxopentan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-3-hydroxy-1-oxopropan-2-yl]amino]-3-hydroxy-1-oxopropan-2-yl]amino]-3-hydroxy-1-oxopropan-2-yl]amino]-1-oxopropan-2-yl]amino]-1-oxohexan-2-yl]amino]-3-hydroxy-1-oxopropan-2-yl]amino]-1-oxo-3-phenylpropan-2-yl]amino]-3-methyl-1-oxopentan-2-yl]amino]-3-methyl-1-oxobutan-2-yl]amino]-5-carbamimidamido-1-oxopentan-2-yl]amino]-1-oxohexan-2-yl]amino]-3-methyl-1-oxobutan-2-yl]amino]-5-carbamimidamido-1-oxopentan-2-yl]amino]-3-methyl-1-oxobutan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-1-oxopropan-2-yl]amino]-5-carbamimidamido-1-oxopentan-2-yl]carbamoyl]pyrrolidin-1-yl]-2-oxoethyl]amino]-3-(1H-indol-3-yl)-1-oxopropan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-1-oxo-3-phenylpropan-2-yl]amino]-5-carbamimidamido-1-oxopentan-2-yl]amino]-1-oxohexan-2-yl]amino]-3-methyl-1-oxobutan-2-yl]amino]-5-carbamimidamido-1-oxopentan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-1-oxo-3-phenylpropan-2-yl]amino]-3-(1H-imidazol-4-yl)-1-oxopropan-2-yl]amino]-3-methyl-1-oxobutan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-4-[[(2S)-2-[[(2S)-2-[[(2S)-2,6-diaminohexanoyl]amino]-3-methylbutanoyl]amino]propanoyl]amino]-5-oxopentanoic acid Chemical compound CC[C@H](C)[C@H](NC(=O)[C@@H](NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCCN)NC(=O)[C@@H](NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@H](C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@@H]1CCCN1C(=O)CNC(=O)[C@H](Cc1c[nH]c2ccccc12)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](Cc1ccccc1)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCCN)NC(=O)[C@@H](NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](Cc1ccccc1)NC(=O)[C@H](Cc1c[nH]cn1)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](NC(=O)[C@@H](N)CCCCN)C(C)C)C(C)C)C(C)C)C(C)C)C(C)C)C(C)C)C(=O)N[C@@H](Cc1ccccc1)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C)C(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O SSOORFWOBGFTHL-OTEJMHTDSA-N 0.000 description 9
- 102100024263 CD160 antigen Human genes 0.000 description 9
- 101000761938 Homo sapiens CD160 antigen Proteins 0.000 description 9
- 101000979342 Homo sapiens Nuclear factor NF-kappa-B p105 subunit Proteins 0.000 description 9
- 101000851007 Homo sapiens Vascular endothelial growth factor receptor 2 Proteins 0.000 description 9
- 102100023050 Nuclear factor NF-kappa-B p105 subunit Human genes 0.000 description 9
- 108010076504 Protein Sorting Signals Proteins 0.000 description 9
- 102100033177 Vascular endothelial growth factor receptor 2 Human genes 0.000 description 9
- 102000040856 WT1 Human genes 0.000 description 9
- 101150084041 WT1 gene Proteins 0.000 description 9
- 229940079593 drug Drugs 0.000 description 9
- 239000012636 effector Substances 0.000 description 9
- 230000028993 immune response Effects 0.000 description 9
- 241000894007 species Species 0.000 description 9
- 230000004083 survival effect Effects 0.000 description 9
- LAMIXXKAWNLXOC-INIZCTEOSA-N (S)-HDAC-42 Chemical compound O=C([C@@H](C(C)C)C=1C=CC=CC=1)NC1=CC=C(C(=O)NO)C=C1 LAMIXXKAWNLXOC-INIZCTEOSA-N 0.000 description 8
- 101000994375 Homo sapiens Integrin alpha-4 Proteins 0.000 description 8
- 101000935040 Homo sapiens Integrin beta-2 Proteins 0.000 description 8
- 101000716102 Homo sapiens T-cell surface glycoprotein CD4 Proteins 0.000 description 8
- 102100032818 Integrin alpha-4 Human genes 0.000 description 8
- 102100032816 Integrin alpha-6 Human genes 0.000 description 8
- 102100025390 Integrin beta-2 Human genes 0.000 description 8
- 108010064548 Lymphocyte Function-Associated Antigen-1 Proteins 0.000 description 8
- 241001465754 Metazoa Species 0.000 description 8
- 102100036011 T-cell surface glycoprotein CD4 Human genes 0.000 description 8
- 102100022153 Tumor necrosis factor receptor superfamily member 4 Human genes 0.000 description 8
- 241000700605 Viruses Species 0.000 description 8
- 230000000903 blocking effect Effects 0.000 description 8
- 229960005386 ipilimumab Drugs 0.000 description 8
- 102000005962 receptors Human genes 0.000 description 8
- 108020003175 receptors Proteins 0.000 description 8
- 230000004044 response Effects 0.000 description 8
- 102100022297 Integrin alpha-X Human genes 0.000 description 7
- 108010074687 Signaling Lymphocytic Activation Molecule Family Member 1 Proteins 0.000 description 7
- 101710165473 Tumor necrosis factor receptor superfamily member 4 Proteins 0.000 description 7
- 239000012190 activator Substances 0.000 description 7
- 230000001086 cytosolic effect Effects 0.000 description 7
- 238000005516 engineering process Methods 0.000 description 7
- 208000005017 glioblastoma Diseases 0.000 description 7
- 230000003834 intracellular effect Effects 0.000 description 7
- 230000001105 regulatory effect Effects 0.000 description 7
- 102100027207 CD27 antigen Human genes 0.000 description 6
- 101150013553 CD40 gene Proteins 0.000 description 6
- 101000914511 Homo sapiens CD27 antigen Proteins 0.000 description 6
- 101001078158 Homo sapiens Integrin alpha-1 Proteins 0.000 description 6
- 101000994365 Homo sapiens Integrin alpha-6 Proteins 0.000 description 6
- 101001035237 Homo sapiens Integrin alpha-D Proteins 0.000 description 6
- 101001046687 Homo sapiens Integrin alpha-E Proteins 0.000 description 6
- 101000935043 Homo sapiens Integrin beta-1 Proteins 0.000 description 6
- 101000971538 Homo sapiens Killer cell lectin-like receptor subfamily F member 1 Proteins 0.000 description 6
- 101000633786 Homo sapiens SLAM family member 6 Proteins 0.000 description 6
- 101000801234 Homo sapiens Tumor necrosis factor receptor superfamily member 18 Proteins 0.000 description 6
- 102100025323 Integrin alpha-1 Human genes 0.000 description 6
- 102100039904 Integrin alpha-D Human genes 0.000 description 6
- 102100022341 Integrin alpha-E Human genes 0.000 description 6
- 102100022338 Integrin alpha-M Human genes 0.000 description 6
- 102100025304 Integrin beta-1 Human genes 0.000 description 6
- 102100021458 Killer cell lectin-like receptor subfamily F member 1 Human genes 0.000 description 6
- 102100029197 SLAM family member 6 Human genes 0.000 description 6
- 102100033728 Tumor necrosis factor receptor superfamily member 18 Human genes 0.000 description 6
- 102100040245 Tumor necrosis factor receptor superfamily member 5 Human genes 0.000 description 6
- 239000000556 agonist Substances 0.000 description 6
- 150000001413 amino acids Chemical group 0.000 description 6
- 150000001875 compounds Chemical class 0.000 description 6
- 238000010586 diagram Methods 0.000 description 6
- 108040006849 interleukin-2 receptor activity proteins Proteins 0.000 description 6
- 210000004698 lymphocyte Anatomy 0.000 description 6
- 102000040430 polynucleotide Human genes 0.000 description 6
- 108091033319 polynucleotide Proteins 0.000 description 6
- 239000002157 polynucleotide Substances 0.000 description 6
- 239000000126 substance Substances 0.000 description 6
- 229960000575 trastuzumab Drugs 0.000 description 6
- 102100029822 B- and T-lymphocyte attenuator Human genes 0.000 description 5
- 101710144268 B- and T-lymphocyte attenuator Proteins 0.000 description 5
- 101000633780 Homo sapiens Signaling lymphocytic activation molecule Proteins 0.000 description 5
- 101000914484 Homo sapiens T-lymphocyte activation antigen CD80 Proteins 0.000 description 5
- 108010061593 Member 14 Tumor Necrosis Factor Receptors Proteins 0.000 description 5
- 108010072866 Prostate-Specific Antigen Proteins 0.000 description 5
- 102100038358 Prostate-specific antigen Human genes 0.000 description 5
- 102100027744 Semaphorin-4D Human genes 0.000 description 5
- 102100027222 T-lymphocyte activation antigen CD80 Human genes 0.000 description 5
- 102100028785 Tumor necrosis factor receptor superfamily member 14 Human genes 0.000 description 5
- 239000005557 antagonist Substances 0.000 description 5
- 230000010056 antibody-dependent cellular cytotoxicity Effects 0.000 description 5
- 238000002784 cytotoxicity assay Methods 0.000 description 5
- 231100000263 cytotoxicity test Toxicity 0.000 description 5
- 210000004408 hybridoma Anatomy 0.000 description 5
- 229940072221 immunoglobulins Drugs 0.000 description 5
- 230000004957 immunoregulator effect Effects 0.000 description 5
- 238000000338 in vitro Methods 0.000 description 5
- 238000004519 manufacturing process Methods 0.000 description 5
- 230000007246 mechanism Effects 0.000 description 5
- 230000001404 mediated effect Effects 0.000 description 5
- 201000001441 melanoma Diseases 0.000 description 5
- 230000003612 virological effect Effects 0.000 description 5
- 102100032937 CD40 ligand Human genes 0.000 description 4
- 108091033409 CRISPR Proteins 0.000 description 4
- 108091026890 Coding region Proteins 0.000 description 4
- 241000701022 Cytomegalovirus Species 0.000 description 4
- 108010017213 Granulocyte-Macrophage Colony-Stimulating Factor Proteins 0.000 description 4
- 108010004889 Heat-Shock Proteins Proteins 0.000 description 4
- 102000002812 Heat-Shock Proteins Human genes 0.000 description 4
- 102000002698 KIR Receptors Human genes 0.000 description 4
- 108010043610 KIR Receptors Proteins 0.000 description 4
- 102000043129 MHC class I family Human genes 0.000 description 4
- 241000282567 Macaca fascicularis Species 0.000 description 4
- 102000014128 RANK Ligand Human genes 0.000 description 4
- 108010025832 RANK Ligand Proteins 0.000 description 4
- 102000040945 Transcription factor Human genes 0.000 description 4
- 108091023040 Transcription factor Proteins 0.000 description 4
- 210000004369 blood Anatomy 0.000 description 4
- 239000008280 blood Substances 0.000 description 4
- 238000002659 cell therapy Methods 0.000 description 4
- 210000004978 chinese hamster ovary cell Anatomy 0.000 description 4
- 230000004186 co-expression Effects 0.000 description 4
- 231100000433 cytotoxic Toxicity 0.000 description 4
- 230000001472 cytotoxic effect Effects 0.000 description 4
- 238000006471 dimerization reaction Methods 0.000 description 4
- 239000003623 enhancer Substances 0.000 description 4
- 238000001415 gene therapy Methods 0.000 description 4
- 230000012010 growth Effects 0.000 description 4
- 230000036039 immunity Effects 0.000 description 4
- 230000006872 improvement Effects 0.000 description 4
- 210000004263 induced pluripotent stem cell Anatomy 0.000 description 4
- 230000010354 integration Effects 0.000 description 4
- 208000032839 leukemia Diseases 0.000 description 4
- 208000002154 non-small cell lung carcinoma Diseases 0.000 description 4
- 229960002087 pertuzumab Drugs 0.000 description 4
- 238000002360 preparation method Methods 0.000 description 4
- 230000008569 process Effects 0.000 description 4
- 230000009467 reduction Effects 0.000 description 4
- 230000004936 stimulating effect Effects 0.000 description 4
- 208000024891 symptom Diseases 0.000 description 4
- 210000001519 tissue Anatomy 0.000 description 4
- 230000009258 tissue cross reactivity Effects 0.000 description 4
- 238000013518 transcription Methods 0.000 description 4
- 230000035897 transcription Effects 0.000 description 4
- 238000001890 transfection Methods 0.000 description 4
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 description 4
- 108010082808 4-1BB Ligand Proteins 0.000 description 3
- MJZJYWCQPMNPRM-UHFFFAOYSA-N 6,6-dimethyl-1-[3-(2,4,5-trichlorophenoxy)propoxy]-1,6-dihydro-1,3,5-triazine-2,4-diamine Chemical compound CC1(C)N=C(N)N=C(N)N1OCCCOC1=CC(Cl)=C(Cl)C=C1Cl MJZJYWCQPMNPRM-UHFFFAOYSA-N 0.000 description 3
- 101150051188 Adora2a gene Proteins 0.000 description 3
- 101710145634 Antigen 1 Proteins 0.000 description 3
- 101150017888 Bcl2 gene Proteins 0.000 description 3
- 108010056102 CD100 antigen Proteins 0.000 description 3
- 108010017009 CD11b Antigen Proteins 0.000 description 3
- 102100038078 CD276 antigen Human genes 0.000 description 3
- 101710185679 CD276 antigen Proteins 0.000 description 3
- 108010029697 CD40 Ligand Proteins 0.000 description 3
- 108010062802 CD66 antigens Proteins 0.000 description 3
- 102100027217 CD82 antigen Human genes 0.000 description 3
- 101710139831 CD82 antigen Proteins 0.000 description 3
- 102100024533 Carcinoembryonic antigen-related cell adhesion molecule 1 Human genes 0.000 description 3
- 206010009944 Colon cancer Diseases 0.000 description 3
- 206010061818 Disease progression Diseases 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- 102100039620 Granulocyte-macrophage colony-stimulating factor Human genes 0.000 description 3
- 102100029360 Hematopoietic cell signal transducer Human genes 0.000 description 3
- 241000282412 Homo Species 0.000 description 3
- 101000990188 Homo sapiens Hematopoietic cell signal transducer Proteins 0.000 description 3
- 101001046683 Homo sapiens Integrin alpha-L Proteins 0.000 description 3
- 101001046686 Homo sapiens Integrin alpha-M Proteins 0.000 description 3
- 101001046668 Homo sapiens Integrin alpha-X Proteins 0.000 description 3
- 101001015037 Homo sapiens Integrin beta-7 Proteins 0.000 description 3
- 101001109503 Homo sapiens NKG2-C type II integral membrane protein Proteins 0.000 description 3
- 101001109501 Homo sapiens NKG2-D type II integral membrane protein Proteins 0.000 description 3
- 101000589305 Homo sapiens Natural cytotoxicity triggering receptor 2 Proteins 0.000 description 3
- 101000873418 Homo sapiens P-selectin glycoprotein ligand 1 Proteins 0.000 description 3
- 101000692259 Homo sapiens Phosphoprotein associated with glycosphingolipid-enriched microdomains 1 Proteins 0.000 description 3
- 101000633778 Homo sapiens SLAM family member 5 Proteins 0.000 description 3
- 101000633784 Homo sapiens SLAM family member 7 Proteins 0.000 description 3
- 101000596234 Homo sapiens T-cell surface protein tactile Proteins 0.000 description 3
- 101000795169 Homo sapiens Tumor necrosis factor receptor superfamily member 13C Proteins 0.000 description 3
- 101000679857 Homo sapiens Tumor necrosis factor receptor superfamily member 3 Proteins 0.000 description 3
- 101000666896 Homo sapiens V-type immunoglobulin domain-containing suppressor of T-cell activation Proteins 0.000 description 3
- 108700002232 Immediate-Early Genes Proteins 0.000 description 3
- 102100033016 Integrin beta-7 Human genes 0.000 description 3
- 108010064593 Intercellular Adhesion Molecule-1 Proteins 0.000 description 3
- 102100037877 Intercellular adhesion molecule 1 Human genes 0.000 description 3
- 108091054437 MHC class I family Proteins 0.000 description 3
- 241000124008 Mammalia Species 0.000 description 3
- 102100022430 Melanocyte protein PMEL Human genes 0.000 description 3
- 206010027476 Metastases Diseases 0.000 description 3
- 241000699666 Mus <mouse, genus> Species 0.000 description 3
- 102100022683 NKG2-C type II integral membrane protein Human genes 0.000 description 3
- 102100022680 NKG2-D type II integral membrane protein Human genes 0.000 description 3
- 108010004222 Natural Cytotoxicity Triggering Receptor 3 Proteins 0.000 description 3
- 102100032851 Natural cytotoxicity triggering receptor 2 Human genes 0.000 description 3
- 102100032852 Natural cytotoxicity triggering receptor 3 Human genes 0.000 description 3
- 102100038082 Natural killer cell receptor 2B4 Human genes 0.000 description 3
- 101710141230 Natural killer cell receptor 2B4 Proteins 0.000 description 3
- 102100034925 P-selectin glycoprotein ligand 1 Human genes 0.000 description 3
- 108700008625 Reporter Genes Proteins 0.000 description 3
- 102100029216 SLAM family member 5 Human genes 0.000 description 3
- 102100029198 SLAM family member 7 Human genes 0.000 description 3
- 101800001271 Surface protein Proteins 0.000 description 3
- 102100035268 T-cell surface protein tactile Human genes 0.000 description 3
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 3
- 102000000852 Tumor Necrosis Factor-alpha Human genes 0.000 description 3
- 102100029690 Tumor necrosis factor receptor superfamily member 13C Human genes 0.000 description 3
- 102100033733 Tumor necrosis factor receptor superfamily member 1B Human genes 0.000 description 3
- 101710187830 Tumor necrosis factor receptor superfamily member 1B Proteins 0.000 description 3
- 102100022156 Tumor necrosis factor receptor superfamily member 3 Human genes 0.000 description 3
- 108010079206 V-Set Domain-Containing T-Cell Activation Inhibitor 1 Proteins 0.000 description 3
- 102100038929 V-set domain-containing T-cell activation inhibitor 1 Human genes 0.000 description 3
- 102100038282 V-type immunoglobulin domain-containing suppressor of T-cell activation Human genes 0.000 description 3
- 102000013529 alpha-Fetoproteins Human genes 0.000 description 3
- 108010026331 alpha-Fetoproteins Proteins 0.000 description 3
- 230000000259 anti-tumor effect Effects 0.000 description 3
- 239000002246 antineoplastic agent Substances 0.000 description 3
- 230000008901 benefit Effects 0.000 description 3
- 239000011575 calcium Substances 0.000 description 3
- 238000004113 cell culture Methods 0.000 description 3
- 230000003915 cell function Effects 0.000 description 3
- 238000003501 co-culture Methods 0.000 description 3
- 125000000151 cysteine group Chemical class N[C@@H](CS)C(=O)* 0.000 description 3
- 210000001151 cytotoxic T lymphocyte Anatomy 0.000 description 3
- 230000007423 decrease Effects 0.000 description 3
- 238000012217 deletion Methods 0.000 description 3
- 230000037430 deletion Effects 0.000 description 3
- 210000004443 dendritic cell Anatomy 0.000 description 3
- 230000005750 disease progression Effects 0.000 description 3
- 210000003958 hematopoietic stem cell Anatomy 0.000 description 3
- 210000005260 human cell Anatomy 0.000 description 3
- 238000001727 in vivo Methods 0.000 description 3
- 230000001965 increasing effect Effects 0.000 description 3
- 230000005865 ionizing radiation Effects 0.000 description 3
- 238000012423 maintenance Methods 0.000 description 3
- 210000002901 mesenchymal stem cell Anatomy 0.000 description 3
- 230000035772 mutation Effects 0.000 description 3
- 238000007911 parenteral administration Methods 0.000 description 3
- 239000002245 particle Substances 0.000 description 3
- 229950011485 pascolizumab Drugs 0.000 description 3
- 239000003755 preservative agent Substances 0.000 description 3
- 230000005855 radiation Effects 0.000 description 3
- 210000003289 regulatory T cell Anatomy 0.000 description 3
- 238000012552 review Methods 0.000 description 3
- 229960004641 rituximab Drugs 0.000 description 3
- 210000002966 serum Anatomy 0.000 description 3
- 230000001988 toxicity Effects 0.000 description 3
- 231100000419 toxicity Toxicity 0.000 description 3
- 238000012546 transfer Methods 0.000 description 3
- 230000004614 tumor growth Effects 0.000 description 3
- 210000003171 tumor-infiltrating lymphocyte Anatomy 0.000 description 3
- 229960003824 ustekinumab Drugs 0.000 description 3
- 208000003950 B-cell lymphoma Diseases 0.000 description 2
- 206010006187 Breast cancer Diseases 0.000 description 2
- 208000026310 Breast neoplasm Diseases 0.000 description 2
- 102100038077 CD226 antigen Human genes 0.000 description 2
- 102100025221 CD70 antigen Human genes 0.000 description 2
- 102100035793 CD83 antigen Human genes 0.000 description 2
- 238000010354 CRISPR gene editing Methods 0.000 description 2
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 2
- 241000282472 Canis lupus familiaris Species 0.000 description 2
- 102000000844 Cell Surface Receptors Human genes 0.000 description 2
- 108010001857 Cell Surface Receptors Proteins 0.000 description 2
- 102100025064 Cellular tumor antigen p53 Human genes 0.000 description 2
- 102000019034 Chemokines Human genes 0.000 description 2
- 108010012236 Chemokines Proteins 0.000 description 2
- 102100027816 Cytotoxic and regulatory T-cell molecule Human genes 0.000 description 2
- 108020004414 DNA Proteins 0.000 description 2
- 229920002307 Dextran Polymers 0.000 description 2
- 102100025137 Early activation antigen CD69 Human genes 0.000 description 2
- 101000585551 Equus caballus Pregnancy-associated glycoprotein Proteins 0.000 description 2
- 108010008165 Etanercept Proteins 0.000 description 2
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 2
- 108010021468 Fc gamma receptor IIA Proteins 0.000 description 2
- 208000007212 Foot-and-Mouth Disease Diseases 0.000 description 2
- 241000710198 Foot-and-mouth disease virus Species 0.000 description 2
- 102100022086 GRB2-related adapter protein 2 Human genes 0.000 description 2
- 102100022132 High affinity immunoglobulin epsilon receptor subunit gamma Human genes 0.000 description 2
- 102000018713 Histocompatibility Antigens Class II Human genes 0.000 description 2
- 101000884298 Homo sapiens CD226 antigen Proteins 0.000 description 2
- 101000946856 Homo sapiens CD83 antigen Proteins 0.000 description 2
- 101000721661 Homo sapiens Cellular tumor antigen p53 Proteins 0.000 description 2
- 101000934374 Homo sapiens Early activation antigen CD69 Proteins 0.000 description 2
- 101000900690 Homo sapiens GRB2-related adapter protein 2 Proteins 0.000 description 2
- 101000824104 Homo sapiens High affinity immunoglobulin epsilon receptor subunit gamma Proteins 0.000 description 2
- 101000777628 Homo sapiens Leukocyte antigen CD37 Proteins 0.000 description 2
- 101001047640 Homo sapiens Linker for activation of T-cells family member 1 Proteins 0.000 description 2
- 101001090688 Homo sapiens Lymphocyte cytosolic protein 2 Proteins 0.000 description 2
- 101000578784 Homo sapiens Melanoma antigen recognized by T-cells 1 Proteins 0.000 description 2
- 101001124867 Homo sapiens Peroxiredoxin-1 Proteins 0.000 description 2
- 101000702132 Homo sapiens Protein spinster homolog 1 Proteins 0.000 description 2
- 241000700588 Human alphaherpesvirus 1 Species 0.000 description 2
- 241000701806 Human papillomavirus Species 0.000 description 2
- 206010021143 Hypoxia Diseases 0.000 description 2
- 102100034980 ICOS ligand Human genes 0.000 description 2
- 108010054477 Immunoglobulin Fab Fragments Proteins 0.000 description 2
- 102000001706 Immunoglobulin Fab Fragments Human genes 0.000 description 2
- 108010091135 Immunoglobulin Fc Fragments Proteins 0.000 description 2
- 102000018071 Immunoglobulin Fc Fragments Human genes 0.000 description 2
- 108010067060 Immunoglobulin Variable Region Proteins 0.000 description 2
- 102000017727 Immunoglobulin Variable Region Human genes 0.000 description 2
- 108010041100 Integrin alpha6 Proteins 0.000 description 2
- 108010030465 Integrin alpha6beta1 Proteins 0.000 description 2
- 102100037850 Interferon gamma Human genes 0.000 description 2
- 108010074328 Interferon-gamma Proteins 0.000 description 2
- 102100031586 Leukocyte antigen CD37 Human genes 0.000 description 2
- 102100024032 Linker for activation of T-cells family member 1 Human genes 0.000 description 2
- 102100029204 Low affinity immunoglobulin gamma Fc region receptor II-a Human genes 0.000 description 2
- 102100034709 Lymphocyte cytosolic protein 2 Human genes 0.000 description 2
- 241000829100 Macaca mulatta polyomavirus 1 Species 0.000 description 2
- 102100028389 Melanoma antigen recognized by T-cells 1 Human genes 0.000 description 2
- 102000000440 Melanoma-associated antigen Human genes 0.000 description 2
- 108050008953 Melanoma-associated antigen Proteins 0.000 description 2
- 101100236305 Mus musculus Ly9 gene Proteins 0.000 description 2
- 241000699670 Mus sp. Species 0.000 description 2
- 241000204031 Mycoplasma Species 0.000 description 2
- 108010057466 NF-kappa B Proteins 0.000 description 2
- 102000003945 NF-kappa B Human genes 0.000 description 2
- 108010004217 Natural Cytotoxicity Triggering Receptor 1 Proteins 0.000 description 2
- 102100032870 Natural cytotoxicity triggering receptor 1 Human genes 0.000 description 2
- 101100445499 Neurospora crassa (strain ATCC 24698 / 74-OR23-1A / CBS 708.71 / DSM 1257 / FGSC 987) erg-1 gene Proteins 0.000 description 2
- 241000283973 Oryctolagus cuniculus Species 0.000 description 2
- 102100026066 Phosphoprotein associated with glycosphingolipid-enriched microdomains 1 Human genes 0.000 description 2
- 108010067965 Phytochrome B Proteins 0.000 description 2
- 108010047620 Phytohemagglutinins Proteins 0.000 description 2
- 206010060862 Prostate cancer Diseases 0.000 description 2
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 2
- 241000700159 Rattus Species 0.000 description 2
- 108020004511 Recombinant DNA Proteins 0.000 description 2
- 108700025832 Serum Response Element Proteins 0.000 description 2
- 206010041067 Small cell lung cancer Diseases 0.000 description 2
- 230000005867 T cell response Effects 0.000 description 2
- 102100025237 T-cell surface antigen CD2 Human genes 0.000 description 2
- 238000010459 TALEN Methods 0.000 description 2
- 102000018679 Tacrolimus Binding Proteins Human genes 0.000 description 2
- 108010027179 Tacrolimus Binding Proteins Proteins 0.000 description 2
- 108020000411 Toll-like receptor Proteins 0.000 description 2
- 102000002689 Toll-like receptor Human genes 0.000 description 2
- 102000003425 Tyrosinase Human genes 0.000 description 2
- 108060008724 Tyrosinase Proteins 0.000 description 2
- 238000012452 Xenomouse strains Methods 0.000 description 2
- 101001038499 Yarrowia lipolytica (strain CLIB 122 / E 150) Lysine acetyltransferase Proteins 0.000 description 2
- 230000003213 activating effect Effects 0.000 description 2
- OIRDTQYFTABQOQ-KQYNXXCUSA-N adenosine Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O OIRDTQYFTABQOQ-KQYNXXCUSA-N 0.000 description 2
- 229960000548 alemtuzumab Drugs 0.000 description 2
- 235000001014 amino acid Nutrition 0.000 description 2
- 230000000890 antigenic effect Effects 0.000 description 2
- 239000003963 antioxidant agent Substances 0.000 description 2
- 229950003145 apolizumab Drugs 0.000 description 2
- 230000009286 beneficial effect Effects 0.000 description 2
- 229960000397 bevacizumab Drugs 0.000 description 2
- 230000004071 biological effect Effects 0.000 description 2
- 229960000074 biopharmaceutical Drugs 0.000 description 2
- 230000037396 body weight Effects 0.000 description 2
- 239000000872 buffer Substances 0.000 description 2
- 229910052791 calcium Inorganic materials 0.000 description 2
- 229940022399 cancer vaccine Drugs 0.000 description 2
- 238000009566 cancer vaccine Methods 0.000 description 2
- 230000024245 cell differentiation Effects 0.000 description 2
- 230000010001 cellular homeostasis Effects 0.000 description 2
- 108010072917 class-I restricted T cell-associated molecule Proteins 0.000 description 2
- 238000011109 contamination Methods 0.000 description 2
- 235000018417 cysteine Nutrition 0.000 description 2
- 229940127089 cytotoxic agent Drugs 0.000 description 2
- 230000003111 delayed effect Effects 0.000 description 2
- 230000004069 differentiation Effects 0.000 description 2
- 239000000539 dimer Substances 0.000 description 2
- 239000002158 endotoxin Substances 0.000 description 2
- 230000002708 enhancing effect Effects 0.000 description 2
- 210000003527 eukaryotic cell Anatomy 0.000 description 2
- IJJVMEJXYNJXOJ-UHFFFAOYSA-N fluquinconazole Chemical compound C=1C=C(Cl)C=C(Cl)C=1N1C(=O)C2=CC(F)=CC=C2N=C1N1C=NC=N1 IJJVMEJXYNJXOJ-UHFFFAOYSA-N 0.000 description 2
- 229950004923 fontolizumab Drugs 0.000 description 2
- 238000009472 formulation Methods 0.000 description 2
- 230000004927 fusion Effects 0.000 description 2
- 108020001507 fusion proteins Proteins 0.000 description 2
- 102000037865 fusion proteins Human genes 0.000 description 2
- 229960003297 gemtuzumab ozogamicin Drugs 0.000 description 2
- 239000003102 growth factor Substances 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- 210000002443 helper t lymphocyte Anatomy 0.000 description 2
- 230000008004 immune attack Effects 0.000 description 2
- 230000005847 immunogenicity Effects 0.000 description 2
- 230000001506 immunosuppresive effect Effects 0.000 description 2
- 230000001976 improved effect Effects 0.000 description 2
- 239000012678 infectious agent Substances 0.000 description 2
- 230000005764 inhibitory process Effects 0.000 description 2
- 230000000977 initiatory effect Effects 0.000 description 2
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- 230000009545 invasion Effects 0.000 description 2
- PGHMRUGBZOYCAA-UHFFFAOYSA-N ionomycin Natural products O1C(CC(O)C(C)C(O)C(C)C=CCC(C)CC(C)C(O)=CC(=O)C(C)CC(C)CC(CCC(O)=O)C)CCC1(C)C1OC(C)(C(C)O)CC1 PGHMRUGBZOYCAA-UHFFFAOYSA-N 0.000 description 2
- PGHMRUGBZOYCAA-ADZNBVRBSA-N ionomycin Chemical compound O1[C@H](C[C@H](O)[C@H](C)[C@H](O)[C@H](C)/C=C/C[C@@H](C)C[C@@H](C)C(/O)=C/C(=O)[C@@H](C)C[C@@H](C)C[C@@H](CCC(O)=O)C)CC[C@@]1(C)[C@@H]1O[C@](C)([C@@H](C)O)CC1 PGHMRUGBZOYCAA-ADZNBVRBSA-N 0.000 description 2
- 239000002502 liposome Substances 0.000 description 2
- 210000001165 lymph node Anatomy 0.000 description 2
- 210000003071 memory t lymphocyte Anatomy 0.000 description 2
- 229910052751 metal Inorganic materials 0.000 description 2
- 239000002184 metal Substances 0.000 description 2
- 230000001394 metastastic effect Effects 0.000 description 2
- 206010061289 metastatic neoplasm Diseases 0.000 description 2
- 238000012737 microarray-based gene expression Methods 0.000 description 2
- 230000000813 microbial effect Effects 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- 239000003607 modifier Substances 0.000 description 2
- 238000012243 multiplex automated genomic engineering Methods 0.000 description 2
- 230000037361 pathway Effects 0.000 description 2
- 238000002823 phage display Methods 0.000 description 2
- 230000001885 phytohemagglutinin Effects 0.000 description 2
- 108010086652 phytohemagglutinin-P Proteins 0.000 description 2
- 239000013612 plasmid Substances 0.000 description 2
- 239000000047 product Substances 0.000 description 2
- 238000003127 radioimmunoassay Methods 0.000 description 2
- 230000002829 reductive effect Effects 0.000 description 2
- 230000010076 replication Effects 0.000 description 2
- 230000003248 secreting effect Effects 0.000 description 2
- 208000000587 small cell lung carcinoma Diseases 0.000 description 2
- 230000009870 specific binding Effects 0.000 description 2
- 239000003381 stabilizer Substances 0.000 description 2
- 150000003431 steroids Chemical class 0.000 description 2
- 230000000638 stimulation Effects 0.000 description 2
- 229950001072 tadocizumab Drugs 0.000 description 2
- 101150047061 tag-72 gene Proteins 0.000 description 2
- 108010078373 tisagenlecleucel Proteins 0.000 description 2
- 230000009261 transgenic effect Effects 0.000 description 2
- 241001430294 unidentified retrovirus Species 0.000 description 2
- QGVLYPPODPLXMB-UBTYZVCOSA-N (1aR,1bS,4aR,7aS,7bS,8R,9R,9aS)-4a,7b,9,9a-tetrahydroxy-3-(hydroxymethyl)-1,1,6,8-tetramethyl-1,1a,1b,4,4a,7a,7b,8,9,9a-decahydro-5H-cyclopropa[3,4]benzo[1,2-e]azulen-5-one Chemical compound C1=C(CO)C[C@]2(O)C(=O)C(C)=C[C@H]2[C@@]2(O)[C@H](C)[C@@H](O)[C@@]3(O)C(C)(C)[C@H]3[C@@H]21 QGVLYPPODPLXMB-UBTYZVCOSA-N 0.000 description 1
- AFWRJOYNLMVZQO-GMFATLNBSA-N (1r,2r,4as,8as)-1-[(1e,3e)-5-hydroxy-3-methylpenta-1,3-dienyl]-2,5,5,8a-tetramethyl-3,4,4a,6,7,8-hexahydro-1h-naphthalen-2-ol Chemical compound CC1(C)CCC[C@]2(C)[C@@H](/C=C/C(=C/CO)/C)[C@](C)(O)CC[C@H]21 AFWRJOYNLMVZQO-GMFATLNBSA-N 0.000 description 1
- ZADWXFSZEAPBJS-SNVBAGLBSA-N (2r)-2-amino-3-(1-methylindol-3-yl)propanoic acid Chemical compound C1=CC=C2N(C)C=C(C[C@@H](N)C(O)=O)C2=C1 ZADWXFSZEAPBJS-SNVBAGLBSA-N 0.000 description 1
- YQYGGOPUTPQHAY-KIQLFZLRSA-N (4S)-4-[[(2S)-2-[[(2S)-2-[2-[6-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S,3S)-1-[[(2S)-5-amino-1-[[(4S,7R)-7-[[(2S)-1-[(2S)-6-amino-2-[[(2R)-2-[[(2S)-5-amino-2-[[(2S,3R)-2-[[(2S)-6-amino-2-[[(2S)-4-carboxy-2-hydrazinylbutanoyl]amino]hexanoyl]amino]-3-methylpentanoyl]amino]-5-oxopentanoyl]amino]propanoyl]amino]hexanoyl]pyrrolidine-2-carbonyl]amino]-2-methyl-5,6-dioxooctan-4-yl]amino]-1,5-dioxopentan-2-yl]amino]-3-hydroxy-1-oxobutan-2-yl]amino]-3-methyl-1-oxobutan-2-yl]amino]-5-carbamimidamido-1-oxopentan-2-yl]amino]-1-oxo-3-phenylpropan-2-yl]amino]-5-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S,3S)-2-[[(2S)-4-amino-2-[[(2S)-2-amino-3-hydroxypropanoyl]amino]-4-oxobutanoyl]amino]-3-hydroxybutanoyl]amino]-3-hydroxypropanoyl]amino]-4-carboxybutanoyl]amino]-3-hydroxypropanoyl]amino]-3-phenylpropanoyl]amino]-6-oxohexyl]hydrazinyl]-3-phenylpropanoyl]amino]-3-hydroxypropanoyl]amino]-5-[[(2S)-1-[[(2S,3S)-1-[[(2S)-4-amino-1-[[(2S)-1-hydroxy-3-oxopropan-2-yl]amino]-1,4-dioxobutan-2-yl]amino]-3-hydroxy-1-oxobutan-2-yl]amino]-3-hydroxy-1-oxopropan-2-yl]amino]-5-oxopentanoic acid Chemical compound CC[C@@H](C)[C@H](NC(=O)[C@H](CCCCN)NC(=O)[C@H](CCC(O)=O)NN)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@H](C)C(=O)N[C@@H](CCCCN)C(=O)N1CCC[C@H]1C(=O)N[C@H](C)C(=O)C(=O)[C@H](CC(C)C)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](NC(=O)[C@@H](NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](Cc1ccccc1)NC(=O)C(CCCCNN[C@@H](Cc1ccccc1)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H]([C@H](C)O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CO)C=O)NC(=O)[C@H](Cc1ccccc1)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC(N)=O)NC(=O)[C@@H](N)CO)[C@H](C)O)C(C)C)[C@H](C)O YQYGGOPUTPQHAY-KIQLFZLRSA-N 0.000 description 1
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 1
- GZCWLCBFPRFLKL-UHFFFAOYSA-N 1-prop-2-ynoxypropan-2-ol Chemical compound CC(O)COCC#C GZCWLCBFPRFLKL-UHFFFAOYSA-N 0.000 description 1
- 102100024341 10 kDa heat shock protein, mitochondrial Human genes 0.000 description 1
- 101710122378 10 kDa heat shock protein, mitochondrial Proteins 0.000 description 1
- IHWDSEPNZDYMNF-UHFFFAOYSA-N 1H-indol-2-amine Chemical compound C1=CC=C2NC(N)=CC2=C1 IHWDSEPNZDYMNF-UHFFFAOYSA-N 0.000 description 1
- FSPQCTGGIANIJZ-UHFFFAOYSA-N 2-[[(3,4-dimethoxyphenyl)-oxomethyl]amino]-4,5,6,7-tetrahydro-1-benzothiophene-3-carboxamide Chemical compound C1=C(OC)C(OC)=CC=C1C(=O)NC1=C(C(N)=O)C(CCCC2)=C2S1 FSPQCTGGIANIJZ-UHFFFAOYSA-N 0.000 description 1
- UQSASSBWRKBREL-UHFFFAOYSA-K 2-hydroxyethyl(trimethyl)azanium;iron(3+);2-oxidopropane-1,2,3-tricarboxylate;trihydrate Chemical compound O.O.O.[Fe+3].C[N+](C)(C)CCO.[O-]C(=O)CC([O-])(C([O-])=O)CC([O-])=O UQSASSBWRKBREL-UHFFFAOYSA-K 0.000 description 1
- 108020005345 3' Untranslated Regions Proteins 0.000 description 1
- LKKMLIBUAXYLOY-UHFFFAOYSA-N 3-Amino-1-methyl-5H-pyrido[4,3-b]indole Chemical compound N1C2=CC=CC=C2C2=C1C=C(N)N=C2C LKKMLIBUAXYLOY-UHFFFAOYSA-N 0.000 description 1
- WEVYNIUIFUYDGI-UHFFFAOYSA-N 3-[6-[4-(trifluoromethoxy)anilino]-4-pyrimidinyl]benzamide Chemical compound NC(=O)C1=CC=CC(C=2N=CN=C(NC=3C=CC(OC(F)(F)F)=CC=3)C=2)=C1 WEVYNIUIFUYDGI-UHFFFAOYSA-N 0.000 description 1
- 108020003589 5' Untranslated Regions Proteins 0.000 description 1
- 102100030310 5,6-dihydroxyindole-2-carboxylic acid oxidase Human genes 0.000 description 1
- 101710163881 5,6-dihydroxyindole-2-carboxylic acid oxidase Proteins 0.000 description 1
- 101710163573 5-hydroxyisourate hydrolase Proteins 0.000 description 1
- 102100038222 60 kDa heat shock protein, mitochondrial Human genes 0.000 description 1
- 101710154868 60 kDa heat shock protein, mitochondrial Proteins 0.000 description 1
- 102100030840 AT-rich interactive domain-containing protein 4B Human genes 0.000 description 1
- 102000013563 Acid Phosphatase Human genes 0.000 description 1
- 108010051457 Acid Phosphatase Proteins 0.000 description 1
- 102000007469 Actins Human genes 0.000 description 1
- 108010085238 Actins Proteins 0.000 description 1
- 235000002198 Annona diversifolia Nutrition 0.000 description 1
- 102000006306 Antigen Receptors Human genes 0.000 description 1
- 108010083359 Antigen Receptors Proteins 0.000 description 1
- 241000219195 Arabidopsis thaliana Species 0.000 description 1
- 102100035526 B melanoma antigen 1 Human genes 0.000 description 1
- 229940125565 BMS-986016 Drugs 0.000 description 1
- 102100021663 Baculoviral IAP repeat-containing protein 5 Human genes 0.000 description 1
- 102100026596 Bcl-2-like protein 1 Human genes 0.000 description 1
- 108060000903 Beta-catenin Proteins 0.000 description 1
- 102000015735 Beta-catenin Human genes 0.000 description 1
- 206010005003 Bladder cancer Diseases 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- 208000003174 Brain Neoplasms Diseases 0.000 description 1
- 239000002126 C01EB10 - Adenosine Substances 0.000 description 1
- 108010046080 CD27 Ligand Proteins 0.000 description 1
- 102100037904 CD9 antigen Human genes 0.000 description 1
- AOPORIRPXVMWSL-DEOSSOPVSA-N CN(C(=S)[C@H]1N(CCC1)C(=O)NCC1=C(C=C(C=C1)C(=O)N1C2=C(NC3=C(C1)C=NN3C)C=CC=C2)C)C Chemical compound CN(C(=S)[C@H]1N(CCC1)C(=O)NCC1=C(C=C(C=C1)C(=O)N1C2=C(NC3=C(C1)C=NN3C)C=CC=C2)C)C AOPORIRPXVMWSL-DEOSSOPVSA-N 0.000 description 1
- 102000004631 Calcineurin Human genes 0.000 description 1
- 108010042955 Calcineurin Proteins 0.000 description 1
- 241000282832 Camelidae Species 0.000 description 1
- 101100507655 Canis lupus familiaris HSPA1 gene Proteins 0.000 description 1
- 102100024423 Carbonic anhydrase 9 Human genes 0.000 description 1
- 102000013392 Carboxylesterase Human genes 0.000 description 1
- 108010051152 Carboxylesterase Proteins 0.000 description 1
- 102000012406 Carcinoembryonic Antigen Human genes 0.000 description 1
- 102000053642 Catalytic RNA Human genes 0.000 description 1
- 108090000994 Catalytic RNA Proteins 0.000 description 1
- 241000700198 Cavia Species 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- 102000009410 Chemokine receptor Human genes 0.000 description 1
- 108050000299 Chemokine receptor Proteins 0.000 description 1
- 108091035707 Consensus sequence Proteins 0.000 description 1
- 241000699802 Cricetulus griseus Species 0.000 description 1
- 108010025464 Cyclin-Dependent Kinase 4 Proteins 0.000 description 1
- 102100036252 Cyclin-dependent kinase 4 Human genes 0.000 description 1
- 108010072210 Cyclophilin C Proteins 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- WQZGKKKJIJFFOK-QTVWNMPRSA-N D-mannopyranose Chemical compound OC[C@H]1OC(O)[C@@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-QTVWNMPRSA-N 0.000 description 1
- 230000005778 DNA damage Effects 0.000 description 1
- 231100000277 DNA damage Toxicity 0.000 description 1
- 102000052510 DNA-Binding Proteins Human genes 0.000 description 1
- 108700020911 DNA-Binding Proteins Proteins 0.000 description 1
- 230000004568 DNA-binding Effects 0.000 description 1
- 241000252212 Danio rerio Species 0.000 description 1
- 241000702421 Dependoparvovirus Species 0.000 description 1
- 102100029721 DnaJ homolog subfamily B member 1 Human genes 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- UPEZCKBFRMILAV-JNEQICEOSA-N Ecdysone Natural products O=C1[C@H]2[C@@](C)([C@@H]3C([C@@]4(O)[C@@](C)([C@H]([C@H]([C@@H](O)CCC(O)(C)C)C)CC4)CC3)=C1)C[C@H](O)[C@H](O)C2 UPEZCKBFRMILAV-JNEQICEOSA-N 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- 108010066687 Epithelial Cell Adhesion Molecule Proteins 0.000 description 1
- 102000018651 Epithelial Cell Adhesion Molecule Human genes 0.000 description 1
- 241000283086 Equidae Species 0.000 description 1
- 241000214054 Equine rhinitis A virus Species 0.000 description 1
- 241000588724 Escherichia coli Species 0.000 description 1
- 102100038595 Estrogen receptor Human genes 0.000 description 1
- 108700024394 Exon Proteins 0.000 description 1
- 102000010834 Extracellular Matrix Proteins Human genes 0.000 description 1
- 108010037362 Extracellular Matrix Proteins Proteins 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- 102000004641 Fetal Proteins Human genes 0.000 description 1
- 108010003471 Fetal Proteins Proteins 0.000 description 1
- 102100028073 Fibroblast growth factor 5 Human genes 0.000 description 1
- 108090000380 Fibroblast growth factor 5 Proteins 0.000 description 1
- 102000054184 GADD45 Human genes 0.000 description 1
- 101710113436 GTPase KRas Proteins 0.000 description 1
- 102100040510 Galectin-3-binding protein Human genes 0.000 description 1
- 101710197901 Galectin-3-binding protein Proteins 0.000 description 1
- 102100031351 Galectin-9 Human genes 0.000 description 1
- 101710121810 Galectin-9 Proteins 0.000 description 1
- 101000834253 Gallus gallus Actin, cytoplasmic 1 Proteins 0.000 description 1
- 108700028146 Genetic Enhancer Elements Proteins 0.000 description 1
- 108700039691 Genetic Promoter Regions Proteins 0.000 description 1
- 208000034951 Genetic Translocation Diseases 0.000 description 1
- MXCOJKLBLFWFNI-VBFYFJBNSA-N Gibberellin A15 Natural products O=C(O)[C@H]1[C@@H]2[C@@]3(C)C(=O)OC[C@]2([C@H]2[C@@]41CC(=C)[C@H](C4)CC2)CCC3 MXCOJKLBLFWFNI-VBFYFJBNSA-N 0.000 description 1
- BKBYHSYZKIAJDA-UHFFFAOYSA-N Gibberellin A29 Natural products C12CCC(C3)(O)C(=C)CC23C(C(O)=O)C2C3(C)C(=O)OC21CC(O)C3 BKBYHSYZKIAJDA-UHFFFAOYSA-N 0.000 description 1
- NYLKJADFYRJQOI-DMQYUYNFSA-N Gibberellin A35 Natural products C([C@@H]1C[C@]2(CC1=C)[C@H]1C(O)=O)[C@H](O)[C@H]2[C@@]2(OC3=O)[C@H]1[C@@]3(C)[C@@H](O)CC2 NYLKJADFYRJQOI-DMQYUYNFSA-N 0.000 description 1
- KSBJAONOPKRVRR-UHFFFAOYSA-N Gibberellin A44 Natural products C12CCC(C3)(O)C(=C)CC23C(C(O)=O)C2C3(C)C(=O)OCC21CCC3 KSBJAONOPKRVRR-UHFFFAOYSA-N 0.000 description 1
- FKMJLJSSQHSAEF-WQODXWHISA-N Gibberellin A50 Natural products O=C(O)[C@H]1[C@@H]2[C@@]3(C)[C@@H](O)[C@@H](O)C[C@@]2(OC3=O)[C@@H]2[C@@H](O)C[C@H]3C(=C)C[C@@]12C3 FKMJLJSSQHSAEF-WQODXWHISA-N 0.000 description 1
- JKQCGVABHHQYKQ-YZVDRFENSA-N Gibberellin A61 Natural products O=C(O)[C@H]1[C@@H]2[C@]3(C)C(=O)O[C@]2([C@H](O)CC3)[C@H]2[C@]31CC(=C)[C@@H](C3)CC2 JKQCGVABHHQYKQ-YZVDRFENSA-N 0.000 description 1
- 208000032612 Glial tumor Diseases 0.000 description 1
- 206010018338 Glioma Diseases 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Natural products NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- 102000003886 Glycoproteins Human genes 0.000 description 1
- 108090000288 Glycoproteins Proteins 0.000 description 1
- 108010042283 HSP40 Heat-Shock Proteins Proteins 0.000 description 1
- 102100034051 Heat shock protein HSP 90-alpha Human genes 0.000 description 1
- 102100031573 Hematopoietic progenitor cell antigen CD34 Human genes 0.000 description 1
- 241000238631 Hexapoda Species 0.000 description 1
- 102100026122 High affinity immunoglobulin gamma Fc receptor I Human genes 0.000 description 1
- 108010088652 Histocompatibility Antigens Class I Proteins 0.000 description 1
- 108010027412 Histocompatibility Antigens Class II Proteins 0.000 description 1
- 101000792935 Homo sapiens AT-rich interactive domain-containing protein 4B Proteins 0.000 description 1
- 101000874316 Homo sapiens B melanoma antigen 1 Proteins 0.000 description 1
- 101000934356 Homo sapiens CD70 antigen Proteins 0.000 description 1
- 101000738354 Homo sapiens CD9 antigen Proteins 0.000 description 1
- 101001066158 Homo sapiens Growth arrest and DNA damage-inducible protein GADD45 alpha Proteins 0.000 description 1
- 101001016865 Homo sapiens Heat shock protein HSP 90-alpha Proteins 0.000 description 1
- 101000777663 Homo sapiens Hematopoietic progenitor cell antigen CD34 Proteins 0.000 description 1
- 101001068133 Homo sapiens Hepatitis A virus cellular receptor 2 Proteins 0.000 description 1
- 101000913074 Homo sapiens High affinity immunoglobulin gamma Fc receptor I Proteins 0.000 description 1
- 101001019455 Homo sapiens ICOS ligand Proteins 0.000 description 1
- 101000998120 Homo sapiens Interleukin-3 receptor subunit alpha Proteins 0.000 description 1
- 101000868279 Homo sapiens Leukocyte surface antigen CD47 Proteins 0.000 description 1
- 101000917858 Homo sapiens Low affinity immunoglobulin gamma Fc region receptor III-A Proteins 0.000 description 1
- 101000917839 Homo sapiens Low affinity immunoglobulin gamma Fc region receptor III-B Proteins 0.000 description 1
- 101001014223 Homo sapiens MAPK/MAK/MRK overlapping kinase Proteins 0.000 description 1
- 101000934372 Homo sapiens Macrosialin Proteins 0.000 description 1
- 101000946889 Homo sapiens Monocyte differentiation antigen CD14 Proteins 0.000 description 1
- 101000581981 Homo sapiens Neural cell adhesion molecule 1 Proteins 0.000 description 1
- 101001062222 Homo sapiens Receptor-binding cancer antigen expressed on SiSo cells Proteins 0.000 description 1
- 101000738771 Homo sapiens Receptor-type tyrosine-protein phosphatase C Proteins 0.000 description 1
- 101000973629 Homo sapiens Ribosome quality control complex subunit NEMF Proteins 0.000 description 1
- 101000934346 Homo sapiens T-cell surface antigen CD2 Proteins 0.000 description 1
- 101000934341 Homo sapiens T-cell surface glycoprotein CD5 Proteins 0.000 description 1
- 101000809875 Homo sapiens TYRO protein tyrosine kinase-binding protein Proteins 0.000 description 1
- 101000655352 Homo sapiens Telomerase reverse transcriptase Proteins 0.000 description 1
- 101001050288 Homo sapiens Transcription factor Jun Proteins 0.000 description 1
- 101000851376 Homo sapiens Tumor necrosis factor receptor superfamily member 8 Proteins 0.000 description 1
- 101000671653 Homo sapiens U3 small nucleolar RNA-associated protein 14 homolog A Proteins 0.000 description 1
- 206010062904 Hormone-refractory prostate cancer Diseases 0.000 description 1
- 241000701044 Human gammaherpesvirus 4 Species 0.000 description 1
- 206010020751 Hypersensitivity Diseases 0.000 description 1
- 101710093458 ICOS ligand Proteins 0.000 description 1
- 108010031794 IGF Type 1 Receptor Proteins 0.000 description 1
- 108010009817 Immunoglobulin Constant Regions Proteins 0.000 description 1
- 102000009786 Immunoglobulin Constant Regions Human genes 0.000 description 1
- 102100023915 Insulin Human genes 0.000 description 1
- 108090001061 Insulin Proteins 0.000 description 1
- 102000048143 Insulin-Like Growth Factor II Human genes 0.000 description 1
- 108090001117 Insulin-Like Growth Factor II Proteins 0.000 description 1
- 102100034343 Integrase Human genes 0.000 description 1
- 108010061833 Integrases Proteins 0.000 description 1
- 108010030506 Integrin alpha6beta4 Proteins 0.000 description 1
- 102000018682 Interleukin Receptor Common gamma Subunit Human genes 0.000 description 1
- 108010066719 Interleukin Receptor Common gamma Subunit Proteins 0.000 description 1
- 108010002352 Interleukin-1 Proteins 0.000 description 1
- 102000000589 Interleukin-1 Human genes 0.000 description 1
- 102100033493 Interleukin-3 receptor subunit alpha Human genes 0.000 description 1
- 108090000978 Interleukin-4 Proteins 0.000 description 1
- 102000004388 Interleukin-4 Human genes 0.000 description 1
- 102100034872 Kallikrein-4 Human genes 0.000 description 1
- 102100031413 L-dopachrome tautomerase Human genes 0.000 description 1
- 101710093778 L-dopachrome tautomerase Proteins 0.000 description 1
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 1
- 108700003970 LIT-001 Proteins 0.000 description 1
- 241000282838 Lama Species 0.000 description 1
- 108090001090 Lectins Proteins 0.000 description 1
- 102000004856 Lectins Human genes 0.000 description 1
- 241000713666 Lentivirus Species 0.000 description 1
- 108010028275 Leukocyte Elastase Proteins 0.000 description 1
- 102100032913 Leukocyte surface antigen CD47 Human genes 0.000 description 1
- 102100029185 Low affinity immunoglobulin gamma Fc region receptor III-B Human genes 0.000 description 1
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 1
- 206010025323 Lymphomas Diseases 0.000 description 1
- 102100031520 MAPK/MAK/MRK overlapping kinase Human genes 0.000 description 1
- 108010010995 MART-1 Antigen Proteins 0.000 description 1
- 102000016200 MART-1 Antigen Human genes 0.000 description 1
- 108091054438 MHC class II family Proteins 0.000 description 1
- 241000282553 Macaca Species 0.000 description 1
- 102000007651 Macrophage Colony-Stimulating Factor Human genes 0.000 description 1
- 108010046938 Macrophage Colony-Stimulating Factor Proteins 0.000 description 1
- 102100025136 Macrosialin Human genes 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 102100025912 Melanopsin Human genes 0.000 description 1
- 206010027480 Metastatic malignant melanoma Diseases 0.000 description 1
- 102100035877 Monocyte differentiation antigen CD14 Human genes 0.000 description 1
- 241001529936 Murinae Species 0.000 description 1
- 241000699660 Mus musculus Species 0.000 description 1
- 101100341510 Mus musculus Itgal gene Proteins 0.000 description 1
- 102000027581 NK cell receptors Human genes 0.000 description 1
- 108091008877 NK cell receptors Proteins 0.000 description 1
- 102100027347 Neural cell adhesion molecule 1 Human genes 0.000 description 1
- 206010029260 Neuroblastoma Diseases 0.000 description 1
- 102100033174 Neutrophil elastase Human genes 0.000 description 1
- 102000004473 OX40 Ligand Human genes 0.000 description 1
- 108010042215 OX40 Ligand Proteins 0.000 description 1
- YGACXVRLDHEXKY-WXRXAMBDSA-N O[C@H](C[C@H]1c2c(cccc2F)-c2cncn12)[C@H]1CC[C@H](O)CC1 Chemical compound O[C@H](C[C@H]1c2c(cccc2F)-c2cncn12)[C@H]1CC[C@H](O)CC1 YGACXVRLDHEXKY-WXRXAMBDSA-N 0.000 description 1
- 108091034117 Oligonucleotide Proteins 0.000 description 1
- 102000043276 Oncogene Human genes 0.000 description 1
- 108700020796 Oncogene Proteins 0.000 description 1
- 102000016979 Other receptors Human genes 0.000 description 1
- 102000004020 Oxygenases Human genes 0.000 description 1
- 108090000417 Oxygenases Proteins 0.000 description 1
- 108090000526 Papain Proteins 0.000 description 1
- 102000057297 Pepsin A Human genes 0.000 description 1
- 108090000284 Pepsin A Proteins 0.000 description 1
- 102000010292 Peptide Elongation Factor 1 Human genes 0.000 description 1
- 108010077524 Peptide Elongation Factor 1 Proteins 0.000 description 1
- 102100024968 Peptidyl-prolyl cis-trans isomerase C Human genes 0.000 description 1
- 206010034960 Photophobia Diseases 0.000 description 1
- 108090000679 Phytochrome Proteins 0.000 description 1
- 241000276498 Pollachius virens Species 0.000 description 1
- 229920002873 Polyethylenimine Polymers 0.000 description 1
- 108010039918 Polylysine Proteins 0.000 description 1
- 102100037935 Polyubiquitin-C Human genes 0.000 description 1
- 102100025803 Progesterone receptor Human genes 0.000 description 1
- 239000004365 Protease Substances 0.000 description 1
- 108010071563 Proto-Oncogene Proteins c-fos Proteins 0.000 description 1
- 102000007568 Proto-Oncogene Proteins c-fos Human genes 0.000 description 1
- 101710100968 Receptor tyrosine-protein kinase erbB-2 Proteins 0.000 description 1
- 102100029165 Receptor-binding cancer antigen expressed on SiSo cells Human genes 0.000 description 1
- 102100020718 Receptor-type tyrosine-protein kinase FLT3 Human genes 0.000 description 1
- 101710151245 Receptor-type tyrosine-protein kinase FLT3 Proteins 0.000 description 1
- 102100037422 Receptor-type tyrosine-protein phosphatase C Human genes 0.000 description 1
- 108010008281 Recombinant Fusion Proteins Proteins 0.000 description 1
- 102000007056 Recombinant Fusion Proteins Human genes 0.000 description 1
- 102100022213 Ribosome quality control complex subunit NEMF Human genes 0.000 description 1
- 101150036449 SIRPA gene Proteins 0.000 description 1
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 1
- 101710173693 Short transient receptor potential channel 1 Proteins 0.000 description 1
- 101710173694 Short transient receptor potential channel 2 Proteins 0.000 description 1
- 102000010841 Signaling Lymphocytic Activation Molecule Family Human genes 0.000 description 1
- 108010062314 Signaling Lymphocytic Activation Molecule Family Proteins 0.000 description 1
- 102000008115 Signaling Lymphocytic Activation Molecule Family Member 1 Human genes 0.000 description 1
- 108010052160 Site-specific recombinase Proteins 0.000 description 1
- 102100037253 Solute carrier family 45 member 3 Human genes 0.000 description 1
- 102000007451 Steroid Receptors Human genes 0.000 description 1
- 108010085012 Steroid Receptors Proteins 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 108010002687 Survivin Proteins 0.000 description 1
- 230000024932 T cell mediated immunity Effects 0.000 description 1
- 102100025244 T-cell surface glycoprotein CD5 Human genes 0.000 description 1
- 101150031162 TM4SF1 gene Proteins 0.000 description 1
- 229940126302 TTI-621 Drugs 0.000 description 1
- 102100038717 TYRO protein tyrosine kinase-binding protein Human genes 0.000 description 1
- 108010017842 Telomerase Proteins 0.000 description 1
- 102100032802 Tetraspanin-8 Human genes 0.000 description 1
- WDLRUFUQRNWCPK-UHFFFAOYSA-N Tetraxetan Chemical compound OC(=O)CN1CCN(CC(O)=O)CCN(CC(O)=O)CCN(CC(O)=O)CC1 WDLRUFUQRNWCPK-UHFFFAOYSA-N 0.000 description 1
- 108010034949 Thyroglobulin Proteins 0.000 description 1
- 102000009843 Thyroglobulin Human genes 0.000 description 1
- 102100033571 Tissue-type plasminogen activator Human genes 0.000 description 1
- 108050006955 Tissue-type plasminogen activator Proteins 0.000 description 1
- 108010043645 Transcription Activator-Like Effector Nucleases Proteins 0.000 description 1
- 102100023132 Transcription factor Jun Human genes 0.000 description 1
- 102000004887 Transforming Growth Factor beta Human genes 0.000 description 1
- 108090001012 Transforming Growth Factor beta Proteins 0.000 description 1
- 102100034902 Transmembrane 4 L6 family member 1 Human genes 0.000 description 1
- 102000008579 Transposases Human genes 0.000 description 1
- 108010020764 Transposases Proteins 0.000 description 1
- LVTKHGUGBGNBPL-UHFFFAOYSA-N Trp-P-1 Chemical compound N1C2=CC=CC=C2C2=C1C(C)=C(N)N=C2C LVTKHGUGBGNBPL-UHFFFAOYSA-N 0.000 description 1
- 108060008683 Tumor Necrosis Factor Receptor Proteins 0.000 description 1
- 102000044209 Tumor Suppressor Genes Human genes 0.000 description 1
- 108700025716 Tumor Suppressor Genes Proteins 0.000 description 1
- 102000015098 Tumor Suppressor Protein p53 Human genes 0.000 description 1
- 108010078814 Tumor Suppressor Protein p53 Proteins 0.000 description 1
- 102100036857 Tumor necrosis factor receptor superfamily member 8 Human genes 0.000 description 1
- 102100033254 Tumor suppressor ARF Human genes 0.000 description 1
- 102100040099 U3 small nucleolar RNA-associated protein 14 homolog A Human genes 0.000 description 1
- 108010056354 Ubiquitin C Proteins 0.000 description 1
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 description 1
- 241000700618 Vaccinia virus Species 0.000 description 1
- 241001416177 Vicugna pacos Species 0.000 description 1
- 108010017070 Zinc Finger Nucleases Proteins 0.000 description 1
- WTIJXIZOODAMJT-WBACWINTSA-N [(3r,4s,5r,6s)-5-hydroxy-6-[4-hydroxy-3-[[5-[[4-hydroxy-7-[(2s,3r,4s,5r)-3-hydroxy-5-methoxy-6,6-dimethyl-4-(5-methyl-1h-pyrrole-2-carbonyl)oxyoxan-2-yl]oxy-8-methyl-2-oxochromen-3-yl]carbamoyl]-4-methyl-1h-pyrrole-3-carbonyl]amino]-8-methyl-2-oxochromen- Chemical compound O([C@@H]1[C@H](C(O[C@H](OC=2C(=C3OC(=O)C(NC(=O)C=4C(=C(C(=O)NC=5C(OC6=C(C)C(O[C@@H]7[C@@H]([C@H](OC(=O)C=8NC(C)=CC=8)[C@@H](OC)C(C)(C)O7)O)=CC=C6C=5O)=O)NC=4)C)=C(O)C3=CC=2)C)[C@@H]1O)(C)C)OC)C(=O)C1=CC=C(C)N1 WTIJXIZOODAMJT-WBACWINTSA-N 0.000 description 1
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 230000006786 activation induced cell death Effects 0.000 description 1
- 229960002964 adalimumab Drugs 0.000 description 1
- 229960005305 adenosine Drugs 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 230000002776 aggregation Effects 0.000 description 1
- 238000004220 aggregation Methods 0.000 description 1
- 239000013566 allergen Substances 0.000 description 1
- 208000026935 allergic disease Diseases 0.000 description 1
- 230000000172 allergic effect Effects 0.000 description 1
- 230000007815 allergy Effects 0.000 description 1
- 102000002287 alpha Subunit Glycoprotein Hormones Human genes 0.000 description 1
- 108010000732 alpha Subunit Glycoprotein Hormones Proteins 0.000 description 1
- UPEZCKBFRMILAV-UHFFFAOYSA-N alpha-Ecdysone Natural products C1C(O)C(O)CC2(C)C(CCC3(C(C(C(O)CCC(C)(C)O)C)CCC33O)C)C3=CC(=O)C21 UPEZCKBFRMILAV-UHFFFAOYSA-N 0.000 description 1
- 230000004075 alteration Effects 0.000 description 1
- WNROFYMDJYEPJX-UHFFFAOYSA-K aluminium hydroxide Chemical compound [OH-].[OH-].[OH-].[Al+3] WNROFYMDJYEPJX-UHFFFAOYSA-K 0.000 description 1
- 125000000539 amino acid group Chemical group 0.000 description 1
- 210000000612 antigen-presenting cell Anatomy 0.000 description 1
- 230000005975 antitumor immune response Effects 0.000 description 1
- 229950002882 aselizumab Drugs 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 208000006673 asthma Diseases 0.000 description 1
- 229960003852 atezolizumab Drugs 0.000 description 1
- 239000012752 auxiliary agent Substances 0.000 description 1
- 229940120638 avastin Drugs 0.000 description 1
- 239000000022 bacteriostatic agent Substances 0.000 description 1
- IXORZMNAPKEEDV-OHSBXDGMSA-N berelex Chemical compound C([C@@]1(O)C(=C)C[C@@]2(C1)[C@H]1C(O)=O)C[C@H]2[C@]2(C=C[C@@H]3O)C1[C@@]3(C)C(=O)O2 IXORZMNAPKEEDV-OHSBXDGMSA-N 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 230000003115 biocidal effect Effects 0.000 description 1
- 230000005540 biological transmission Effects 0.000 description 1
- 239000000090 biomarker Substances 0.000 description 1
- 238000001574 biopsy Methods 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 229960003008 blinatumomab Drugs 0.000 description 1
- 238000004820 blood count Methods 0.000 description 1
- 230000036760 body temperature Effects 0.000 description 1
- 210000001185 bone marrow Anatomy 0.000 description 1
- 208000026555 breast adenosis Diseases 0.000 description 1
- 239000007975 buffered saline Substances 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 229910000389 calcium phosphate Inorganic materials 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- 235000014633 carbohydrates Nutrition 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 229920006317 cationic polymer Polymers 0.000 description 1
- 239000006143 cell culture medium Substances 0.000 description 1
- 230000030833 cell death Effects 0.000 description 1
- 230000011712 cell development Effects 0.000 description 1
- 230000010261 cell growth Effects 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 230000012292 cell migration Effects 0.000 description 1
- 230000004663 cell proliferation Effects 0.000 description 1
- 239000002458 cell surface marker Substances 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 230000007969 cellular immunity Effects 0.000 description 1
- 229960005395 cetuximab Drugs 0.000 description 1
- 239000002738 chelating agent Substances 0.000 description 1
- 238000012412 chemical coupling Methods 0.000 description 1
- 238000002512 chemotherapy Methods 0.000 description 1
- ZYVSOIYQKUDENJ-WKSBCEQHSA-N chromomycin A3 Chemical compound O([C@@H]1C[C@@H](O[C@H](C)[C@@H]1OC(C)=O)OC=1C=C2C=C3C[C@H]([C@@H](C(=O)C3=C(O)C2=C(O)C=1C)O[C@@H]1O[C@H](C)[C@@H](O)[C@H](O[C@@H]2O[C@H](C)[C@@H](O)[C@H](O[C@@H]3O[C@@H](C)[C@H](OC(C)=O)[C@@](C)(O)C3)C2)C1)[C@H](OC)C(=O)[C@@H](O)[C@@H](C)O)[C@@H]1C[C@@H](O)[C@@H](OC)[C@@H](C)O1 ZYVSOIYQKUDENJ-WKSBCEQHSA-N 0.000 description 1
- 230000000295 complement effect Effects 0.000 description 1
- 210000002808 connective tissue Anatomy 0.000 description 1
- 108091008034 costimulatory receptors Proteins 0.000 description 1
- 238000004132 cross linking Methods 0.000 description 1
- 108040004564 crotonyl-CoA reductase activity proteins Proteins 0.000 description 1
- 102000003675 cytokine receptors Human genes 0.000 description 1
- 108010057085 cytokine receptors Proteins 0.000 description 1
- 210000000805 cytoplasm Anatomy 0.000 description 1
- 229960002806 daclizumab Drugs 0.000 description 1
- 230000006378 damage Effects 0.000 description 1
- 108010048522 dapirolizumab pegol Proteins 0.000 description 1
- 229950005026 dapirolizumab pegol Drugs 0.000 description 1
- 230000002498 deadly effect Effects 0.000 description 1
- 230000002950 deficient Effects 0.000 description 1
- 230000002939 deleterious effect Effects 0.000 description 1
- 239000000412 dendrimer Substances 0.000 description 1
- 229920000736 dendritic polymer Polymers 0.000 description 1
- 230000029087 digestion Effects 0.000 description 1
- 230000000447 dimerizing effect Effects 0.000 description 1
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 1
- 238000010494 dissociation reaction Methods 0.000 description 1
- 230000005593 dissociations Effects 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 230000003828 downregulation Effects 0.000 description 1
- 229950009791 durvalumab Drugs 0.000 description 1
- UPEZCKBFRMILAV-JMZLNJERSA-N ecdysone Chemical compound C1[C@@H](O)[C@@H](O)C[C@]2(C)[C@@H](CC[C@@]3([C@@H]([C@@H]([C@H](O)CCC(C)(C)O)C)CC[C@]33O)C)C3=CC(=O)[C@@H]21 UPEZCKBFRMILAV-JMZLNJERSA-N 0.000 description 1
- 108010057988 ecdysone receptor Proteins 0.000 description 1
- 229960002224 eculizumab Drugs 0.000 description 1
- 229960000284 efalizumab Drugs 0.000 description 1
- 210000003162 effector t lymphocyte Anatomy 0.000 description 1
- 229940056913 eftilagimod alfa Drugs 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 230000013020 embryo development Effects 0.000 description 1
- 210000001671 embryonic stem cell Anatomy 0.000 description 1
- 210000002257 embryonic structure Anatomy 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 229940073621 enbrel Drugs 0.000 description 1
- 229940088598 enzyme Drugs 0.000 description 1
- 210000000981 epithelium Anatomy 0.000 description 1
- 229950009760 epratuzumab Drugs 0.000 description 1
- 229950004292 erlizumab Drugs 0.000 description 1
- 108010038795 estrogen receptors Proteins 0.000 description 1
- 229960000403 etanercept Drugs 0.000 description 1
- 239000013604 expression vector Substances 0.000 description 1
- 210000002744 extracellular matrix Anatomy 0.000 description 1
- 229950001563 felvizumab Drugs 0.000 description 1
- 230000001605 fetal effect Effects 0.000 description 1
- 238000001476 gene delivery Methods 0.000 description 1
- 102000034356 gene-regulatory proteins Human genes 0.000 description 1
- 108091006104 gene-regulatory proteins Proteins 0.000 description 1
- GFEIVLLXVCBXOQ-UHFFFAOYSA-N gibberelline Natural products CC12CCCC3(C(O)OC1=O)C4CCC5(O)CC4(CC5=C)C(C23)C(=O)O GFEIVLLXVCBXOQ-UHFFFAOYSA-N 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- RWSXRVCMGQZWBV-WDSKDSINSA-N glutathione Chemical compound OC(=O)[C@@H](N)CCC(=O)N[C@@H](CS)C(=O)NCC(O)=O RWSXRVCMGQZWBV-WDSKDSINSA-N 0.000 description 1
- 210000003714 granulocyte Anatomy 0.000 description 1
- 239000000122 growth hormone Substances 0.000 description 1
- 230000003394 haemopoietic effect Effects 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 229940022353 herceptin Drugs 0.000 description 1
- 230000006801 homologous recombination Effects 0.000 description 1
- 238000002744 homologous recombination Methods 0.000 description 1
- 229940088597 hormone Drugs 0.000 description 1
- 230000009215 host defense mechanism Effects 0.000 description 1
- 229940084986 human chorionic gonadotropin Drugs 0.000 description 1
- 238000011577 humanized mouse model Methods 0.000 description 1
- 229940048921 humira Drugs 0.000 description 1
- 230000007954 hypoxia Effects 0.000 description 1
- 229940091204 imlygic Drugs 0.000 description 1
- 230000005746 immune checkpoint blockade Effects 0.000 description 1
- 230000001900 immune effect Effects 0.000 description 1
- 230000036737 immune function Effects 0.000 description 1
- 102000027596 immune receptors Human genes 0.000 description 1
- 108091008915 immune receptors Proteins 0.000 description 1
- 230000037451 immune surveillance Effects 0.000 description 1
- 230000006058 immune tolerance Effects 0.000 description 1
- 230000003053 immunization Effects 0.000 description 1
- 230000002163 immunogen Effects 0.000 description 1
- 230000000957 immunotolerogenic effect Effects 0.000 description 1
- 238000000530 impalefection Methods 0.000 description 1
- 102000006639 indoleamine 2,3-dioxygenase Human genes 0.000 description 1
- 108020004201 indoleamine 2,3-dioxygenase Proteins 0.000 description 1
- 229950009034 indoximod Drugs 0.000 description 1
- 239000007972 injectable composition Substances 0.000 description 1
- 238000011081 inoculation Methods 0.000 description 1
- 229950004101 inotuzumab ozogamicin Drugs 0.000 description 1
- 238000003780 insertion Methods 0.000 description 1
- 230000037431 insertion Effects 0.000 description 1
- 229940125396 insulin Drugs 0.000 description 1
- 102000006495 integrins Human genes 0.000 description 1
- 108010044426 integrins Proteins 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 230000000968 intestinal effect Effects 0.000 description 1
- 230000002601 intratumoral effect Effects 0.000 description 1
- 238000011835 investigation Methods 0.000 description 1
- FZWBNHMXJMCXLU-BLAUPYHCSA-N isomaltotriose Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1OC[C@@H]1[C@@H](O)[C@H](O)[C@@H](O)[C@@H](OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C=O)O1 FZWBNHMXJMCXLU-BLAUPYHCSA-N 0.000 description 1
- BPHPUYQFMNQIOC-NXRLNHOXSA-N isopropyl beta-D-thiogalactopyranoside Chemical compound CC(C)S[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1O BPHPUYQFMNQIOC-NXRLNHOXSA-N 0.000 description 1
- 108010024383 kallikrein 4 Proteins 0.000 description 1
- 239000002523 lectin Substances 0.000 description 1
- 231100000518 lethal Toxicity 0.000 description 1
- 230000001665 lethal effect Effects 0.000 description 1
- 230000021633 leukocyte mediated immunity Effects 0.000 description 1
- 208000013469 light sensitivity Diseases 0.000 description 1
- 229950002950 lintuzumab Drugs 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 229950011263 lirilumab Drugs 0.000 description 1
- 230000007108 local immune response Effects 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 201000005202 lung cancer Diseases 0.000 description 1
- 208000020816 lung neoplasm Diseases 0.000 description 1
- 210000002751 lymph Anatomy 0.000 description 1
- 210000003810 lymphokine-activated killer cell Anatomy 0.000 description 1
- 210000002540 macrophage Anatomy 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 229950008001 matuzumab Drugs 0.000 description 1
- 239000002609 medium Substances 0.000 description 1
- 108010005417 melanopsin Proteins 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 210000004379 membrane Anatomy 0.000 description 1
- 229960005558 mertansine Drugs 0.000 description 1
- ANZJBCHSOXCCRQ-FKUXLPTCSA-N mertansine Chemical compound CO[C@@H]([C@@]1(O)C[C@H](OC(=O)N1)[C@@H](C)[C@@H]1O[C@@]1(C)[C@@H](OC(=O)[C@H](C)N(C)C(=O)CCS)CC(=O)N1C)\C=C\C=C(C)\CC2=CC(OC)=C(Cl)C1=C2 ANZJBCHSOXCCRQ-FKUXLPTCSA-N 0.000 description 1
- 108020004999 messenger RNA Proteins 0.000 description 1
- 150000002739 metals Chemical class 0.000 description 1
- 230000009401 metastasis Effects 0.000 description 1
- 208000021039 metastatic melanoma Diseases 0.000 description 1
- 230000006540 mitochondrial respiration Effects 0.000 description 1
- 102000035118 modified proteins Human genes 0.000 description 1
- 108091005573 modified proteins Proteins 0.000 description 1
- 238000010369 molecular cloning Methods 0.000 description 1
- 238000009126 molecular therapy Methods 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- 210000001616 monocyte Anatomy 0.000 description 1
- 230000001459 mortal effect Effects 0.000 description 1
- 210000003205 muscle Anatomy 0.000 description 1
- 210000000581 natural killer T-cell Anatomy 0.000 description 1
- 230000017066 negative regulation of growth Effects 0.000 description 1
- 210000000944 nerve tissue Anatomy 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 210000000440 neutrophil Anatomy 0.000 description 1
- 229950010203 nimotuzumab Drugs 0.000 description 1
- 229960003301 nivolumab Drugs 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 238000011580 nude mouse model Methods 0.000 description 1
- 229950005751 ocrelizumab Drugs 0.000 description 1
- 238000011275 oncology therapy Methods 0.000 description 1
- 244000309459 oncolytic virus Species 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 230000002018 overexpression Effects 0.000 description 1
- 229960000402 palivizumab Drugs 0.000 description 1
- 229960001972 panitumumab Drugs 0.000 description 1
- 229940055729 papain Drugs 0.000 description 1
- 235000019834 papain Nutrition 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 230000007170 pathology Effects 0.000 description 1
- 229940111202 pepsin Drugs 0.000 description 1
- 210000005259 peripheral blood Anatomy 0.000 description 1
- 239000011886 peripheral blood Substances 0.000 description 1
- 230000002688 persistence Effects 0.000 description 1
- QGVLYPPODPLXMB-QXYKVGAMSA-N phorbol Natural products C[C@@H]1[C@@H](O)[C@]2(O)[C@H]([C@H]3C=C(CO)C[C@@]4(O)[C@H](C=C(C)C4=O)[C@@]13O)C2(C)C QGVLYPPODPLXMB-QXYKVGAMSA-N 0.000 description 1
- 239000002953 phosphate buffered saline Substances 0.000 description 1
- 108091008695 photoreceptors Proteins 0.000 description 1
- 230000004962 physiological condition Effects 0.000 description 1
- 230000035790 physiological processes and functions Effects 0.000 description 1
- 229950010773 pidilizumab Drugs 0.000 description 1
- 210000001778 pluripotent stem cell Anatomy 0.000 description 1
- 229920000656 polylysine Polymers 0.000 description 1
- 230000029279 positive regulation of transcription, DNA-dependent Effects 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 210000004986 primary T-cell Anatomy 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 230000000861 pro-apoptotic effect Effects 0.000 description 1
- 108090000468 progesterone receptors Proteins 0.000 description 1
- 238000004393 prognosis Methods 0.000 description 1
- 210000001236 prokaryotic cell Anatomy 0.000 description 1
- 230000000644 propagated effect Effects 0.000 description 1
- 210000002307 prostate Anatomy 0.000 description 1
- 108010079891 prostein Proteins 0.000 description 1
- 210000001938 protoplast Anatomy 0.000 description 1
- 238000001959 radiotherapy Methods 0.000 description 1
- 229960003876 ranibizumab Drugs 0.000 description 1
- 108010014186 ras Proteins Proteins 0.000 description 1
- 102000016914 ras Proteins Human genes 0.000 description 1
- 230000006798 recombination Effects 0.000 description 1
- 238000005215 recombination Methods 0.000 description 1
- 230000007115 recruitment Effects 0.000 description 1
- 230000014493 regulation of gene expression Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 238000002271 resection Methods 0.000 description 1
- 229960003254 reslizumab Drugs 0.000 description 1
- 230000032554 response to blue light Effects 0.000 description 1
- 108091008146 restriction endonucleases Proteins 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 108091092562 ribozyme Proteins 0.000 description 1
- 102220278926 rs759871071 Human genes 0.000 description 1
- 229950005374 ruplizumab Drugs 0.000 description 1
- 229950008684 sibrotuzumab Drugs 0.000 description 1
- 108091006024 signal transducing proteins Proteins 0.000 description 1
- 102000034285 signal transducing proteins Human genes 0.000 description 1
- 210000003491 skin Anatomy 0.000 description 1
- 239000000243 solution Substances 0.000 description 1
- 239000002904 solvent Substances 0.000 description 1
- 229950006551 sontuzumab Drugs 0.000 description 1
- 210000000952 spleen Anatomy 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 239000011885 synergistic combination Substances 0.000 description 1
- 229940126625 tavolimab Drugs 0.000 description 1
- 238000010998 test method Methods 0.000 description 1
- 210000001550 testis Anatomy 0.000 description 1
- ZRKFYGHZFMAOKI-QMGMOQQFSA-N tgfbeta Chemical compound C([C@H](NC(=O)[C@H](C(C)C)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CC(C)C)NC(=O)CNC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](N)CCSC)C(C)C)[C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(O)=O)C1=CC=C(O)C=C1 ZRKFYGHZFMAOKI-QMGMOQQFSA-N 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- 125000003396 thiol group Chemical group [H]S* 0.000 description 1
- 210000001541 thymus gland Anatomy 0.000 description 1
- 229960002175 thyroglobulin Drugs 0.000 description 1
- 229960003989 tocilizumab Drugs 0.000 description 1
- 239000008181 tonicity modifier Substances 0.000 description 1
- 238000011200 topical administration Methods 0.000 description 1
- 229960005267 tositumomab Drugs 0.000 description 1
- 210000003014 totipotent stem cell Anatomy 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 230000005026 transcription initiation Effects 0.000 description 1
- 230000005030 transcription termination Effects 0.000 description 1
- 230000002103 transcriptional effect Effects 0.000 description 1
- 230000002463 transducing effect Effects 0.000 description 1
- 238000010361 transduction Methods 0.000 description 1
- 230000026683 transduction Effects 0.000 description 1
- 238000003151 transfection method Methods 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 238000013519 translation Methods 0.000 description 1
- 230000032258 transport Effects 0.000 description 1
- 229950007217 tremelimumab Drugs 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- 229950003364 tucotuzumab celmoleukin Drugs 0.000 description 1
- 108700008509 tucotuzumab celmoleukin Proteins 0.000 description 1
- 102000003298 tumor necrosis factor receptor Human genes 0.000 description 1
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 1
- 241000701161 unidentified adenovirus Species 0.000 description 1
- 210000002444 unipotent stem cell Anatomy 0.000 description 1
- 230000003827 upregulation Effects 0.000 description 1
- 229950005972 urelumab Drugs 0.000 description 1
- 201000005112 urinary bladder cancer Diseases 0.000 description 1
- 229950004362 urtoxazumab Drugs 0.000 description 1
- 229950001067 varlilumab Drugs 0.000 description 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- 229940055760 yervoy Drugs 0.000 description 1
Images



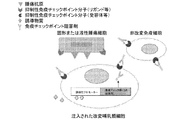











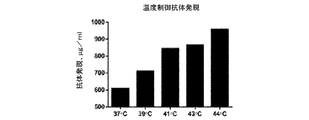



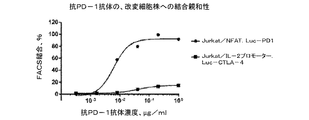
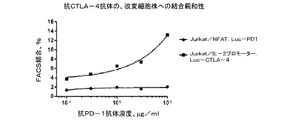
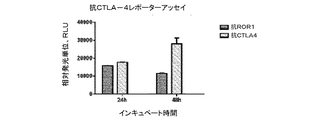


























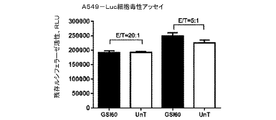

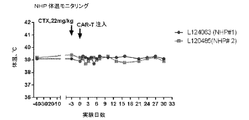




Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K35/00—Medicinal preparations containing materials or reaction products thereof with undetermined constitution
- A61K35/12—Materials from mammals; Compositions comprising non-specified tissues or cells; Compositions comprising non-embryonic stem cells; Genetically modified cells
- A61K35/14—Blood; Artificial blood
- A61K35/17—Lymphocytes; B-cells; T-cells; Natural killer cells; Interferon-activated or cytokine-activated lymphocytes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/461—Cellular immunotherapy characterised by the cell type used
- A61K39/4611—T-cells, e.g. tumor infiltrating lymphocytes [TIL], lymphokine-activated killer cells [LAK] or regulatory T cells [Treg]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/463—Cellular immunotherapy characterised by recombinant expression
- A61K39/4631—Chimeric Antigen Receptors [CAR]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/463—Cellular immunotherapy characterised by recombinant expression
- A61K39/4632—T-cell receptors [TCR]; antibody T-cell receptor constructs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/463—Cellular immunotherapy characterised by recombinant expression
- A61K39/4633—Antibodies or T cell engagers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/463—Cellular immunotherapy characterised by recombinant expression
- A61K39/4636—Immune checkpoint inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/464—Cellular immunotherapy characterised by the antigen targeted or presented
- A61K39/4643—Vertebrate antigens
- A61K39/4644—Cancer antigens
- A61K39/464402—Receptors, cell surface antigens or cell surface determinants
- A61K39/464403—Receptors for growth factors
- A61K39/464404—Epidermal growth factor receptors [EGFR]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/464—Cellular immunotherapy characterised by the antigen targeted or presented
- A61K39/4643—Vertebrate antigens
- A61K39/4644—Cancer antigens
- A61K39/464402—Receptors, cell surface antigens or cell surface determinants
- A61K39/464416—Receptors for cytokines
- A61K39/464417—Receptors for tumor necrosis factors [TNF], e.g. lymphotoxin receptor [LTR], CD30
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/464—Cellular immunotherapy characterised by the antigen targeted or presented
- A61K39/4643—Vertebrate antigens
- A61K39/4644—Cancer antigens
- A61K39/464484—Cancer testis antigens, e.g. SSX, BAGE, GAGE or SAGE
- A61K39/464488—NY-ESO
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2803—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily
- C07K16/2809—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily against the T-cell receptor (TcR)-CD3 complex
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2803—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily
- C07K16/2818—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily against CD28 or CD152
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2863—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against receptors for growth factors, growth regulators
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/32—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against translation products of oncogenes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0634—Cells from the blood or the immune system
- C12N5/0636—T lymphocytes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/10—Cells modified by introduction of foreign genetic material
- C12N5/12—Fused cells, e.g. hybridomas
- C12N5/16—Animal cells
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/505—Medicinal preparations containing antigens or antibodies comprising antibodies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/505—Medicinal preparations containing antigens or antibodies comprising antibodies
- A61K2039/507—Comprising a combination of two or more separate antibodies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/51—Medicinal preparations containing antigens or antibodies comprising whole cells, viruses or DNA/RNA
- A61K2039/515—Animal cells
- A61K2039/5156—Animal cells expressing foreign proteins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/51—Medicinal preparations containing antigens or antibodies comprising whole cells, viruses or DNA/RNA
- A61K2039/515—Animal cells
- A61K2039/5158—Antigen-pulsed cells, e.g. T-cells
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/57—Medicinal preparations containing antigens or antibodies characterised by the type of response, e.g. Th1, Th2
- A61K2039/572—Medicinal preparations containing antigens or antibodies characterised by the type of response, e.g. Th1, Th2 cytotoxic response
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2239/00—Indexing codes associated with cellular immunotherapy of group A61K39/46
- A61K2239/46—Indexing codes associated with cellular immunotherapy of group A61K39/46 characterised by the cancer treated
- A61K2239/47—Brain; Nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2239/00—Indexing codes associated with cellular immunotherapy of group A61K39/46
- A61K2239/46—Indexing codes associated with cellular immunotherapy of group A61K39/46 characterised by the cancer treated
- A61K2239/48—Blood cells, e.g. leukemia or lymphoma
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2239/00—Indexing codes associated with cellular immunotherapy of group A61K39/46
- A61K2239/46—Indexing codes associated with cellular immunotherapy of group A61K39/46 characterised by the cancer treated
- A61K2239/55—Lung
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/73—Inducing cell death, e.g. apoptosis, necrosis or inhibition of cell proliferation
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/40—Regulators of development
- C12N2501/48—Regulators of apoptosis
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/50—Cell markers; Cell surface determinants
- C12N2501/51—B7 molecules, e.g. CD80, CD86, CD28 (ligand), CD152 (ligand)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2510/00—Genetically modified cells
- C12N2510/02—Cells for production
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2740/00—Reverse transcribing RNA viruses
- C12N2740/00011—Details
- C12N2740/10011—Retroviridae
- C12N2740/16011—Human Immunodeficiency Virus, HIV
- C12N2740/16041—Use of virus, viral particle or viral elements as a vector
- C12N2740/16043—Use of virus, viral particle or viral elements as a vector viral genome or elements thereof as genetic vector
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Immunology (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Genetics & Genomics (AREA)
- Engineering & Computer Science (AREA)
- Cell Biology (AREA)
- Biochemistry (AREA)
- Microbiology (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- Biomedical Technology (AREA)
- Epidemiology (AREA)
- Biotechnology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Molecular Biology (AREA)
- Zoology (AREA)
- Biophysics (AREA)
- Mycology (AREA)
- Wood Science & Technology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Oncology (AREA)
- General Engineering & Computer Science (AREA)
- Hematology (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Virology (AREA)
- Developmental Biology & Embryology (AREA)
- Dermatology (AREA)
- Medicines Containing Material From Animals Or Micro-Organisms (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
Abstract
Description
本出願は、2016年2月4日に出願された国際特許出願第PCT/CN2016/073489号および2016年6月30日に出願された国際特許出願第PCT/CN2016/087855号の優先権の利益を主張するものであり、これらそれぞれの内容は、参照によりその全体が本明細書に組み込まれるものとする。
Tを使用することが報告されている。
前記組換えT細胞受容体の細胞内シグナル伝達ドメインによって誘導可能である。
をコードする第2の異種核酸を含む。一部の実施形態において、前記免疫調節剤をコードする異種核酸と前記治療用タンパク質をコードする前記第2の異種核酸が、同一のプロモーターに機能的に連結される。一部の実施形態において、前記免疫調節剤をコードする異種核酸と前記治療用タンパク質をコードする前記第2の異種核酸が、異なるプロモーターに機能的に連結される。一部の実施形態において、前記改変哺乳類細胞は、免疫調節剤と2種以上の治療用タンパク質と発現する。
本明細書で使用される「治療」という語は、臨床病理学の過程で、治療対象の個体または細胞の自然経過を改変するように設計された臨床的介入のことを指す。治療の望ましい効果としては、疾患の進行速度の低下、病状の改善または緩和、および寛解または予後の改善等が含まれる。例えば、癌と関連した1つまたは複数の症状が軽減または解消された場合に個体の癌に対する「治療」が成功したこととなり、その例は、癌性細胞の増殖の低下(または該細胞の破壊)、前記疾患に由来する症状の減少、前記疾患に苦しむ患者の生活の質の改善、前記疾患の治療に必要な他の薬物の投与量の減少、および/または個体の生存期間の延長等を含むが、これらに限定されない。
る。この遅延は、前記疾患の過程および/または治療を受ける個体によってその期間の長さが異なりうる。当業者にとっては明らかなことであるが、十分な、または有意な遅延は、事実上、個体において前記疾患が発症しない予防をも包含しうる。例えば、転移の発生を含む末期癌の遅延であってもよい。
位を有する、2個の同一の抗原結合断片と、残りの部分である、その名前が容易に結晶化する能力を反映した、「Fc」断片とが生成する。ペプシン処理により、2個の抗原結合部位を有し、抗原を架橋することが依然可能な、F(ab’)2断片が生成する。
nberg et al., Nature 374:168-73 (1995);Hassanzadeh-Ghassabeh et al., Nanomedicine (Lond), 8:1013-26 (2013)を参照のこと)。
et al., Hybridoma 14 (3): 253-260 (1995);Harlow et al., Antibodies: A Laboratory Manual, (Cold Spring Harbor Laboratory Press, 2nd ed. 1988);Hammerling et al., Monoclonal Antibodies and T-Cell Hybridomas 563-681 (Elsevier, N.Y., 1981))、組換えDNA法(例えば米国特許第4,816,567号を参照のこと)、ファージディスプレイ技術(例えば、Clackson et al., Nature 352: 624-628 (1991);Marks et al., J. Mol. Biol. 222: 581-597 (1992);Sidhu et al., J. Mol. Biol. 338(2): 299-310 (2004);Lee et al., J. Mol. Biol. 340(5): 1073-1093 (2004);Fellouse, Proc. Natl. Acad. Sci. USA 101(34): 12467-12472 (2004);およびLee et al., J. Immunol. Methods 284(1-2): 119-132 (2004)を参照のこと)、ならびに、ヒト免疫グロブリン遺伝子座またはヒト免疫グロブリン配列をコードする遺伝子の一部または全体を有する動物において、ヒトまたはヒト様抗体を製造するための技術(例えば、WO1998/24893;WO1996/34096;WO1996/33735;WO1991/10741;Jakobovits et al., Proc. Natl. Acad. Sci. USA 90: 2551 (1993);Jakobovits et al., Nature 362: 255-258 (1993);Bruggemann et al., Year in Immunol. 7:33 (1993);米国特許第5,545,807号、第5,545,806号、第5,569,825号、第5,625,126号、第5,633,425号、および第5,661,016号;Marks et al., Bio/Technology 10: 779-783 (1992);Lonberg et al., Nature 368: 856-859 (1994);Morrison, Nature 368: 812-813 (1994);Fishwild et al., Nature Biotechnol. 14: 845-851 (1996);Neuberger, Nature Biotechnol. 14: 826 (1996);ならびにLonberg and Huszar, Intern. Rev. Immunol. 13: 65-93 (1995)を参照のこと)等を含む各種技術によって作製されたものであってよい。
または軽鎖の一部が、特定の種に由来する抗体中の、または特定の抗体クラスもしくはサブクラスに属する抗体中の対応する配列と同一または相同であり、一方、鎖の残りの部分は、他の種に由来する抗体中の、または他の抗体クラスもしくはサブクラスに属する抗体中の、対応する配列と同一または相同である(例えば米国特許第4,816,567号;およびMorrison et al., Proc. Natl. Acad. Sci. USA 81:6851-6855 (1984)を参照のこと)。キメラ抗体としては、PRIMATTZED(登録商標)抗体などが挙げられ、該抗体においては抗原結合領域が、例えば、目的の抗原でマカクザルを免疫すること等によって産生された抗体に由来する。
Op. Biotech. 5:428-433 (1994);ならびに米国特許第6,982,321号および第7,087,409号を参照のこと。
and Winter, J. Mol. Biol. 227:381 (1991);Marks et al., J. Mol. Biol. 222:581 (1991)、Cole et al., Monoclonal Antibodies and Cancer Therapy, Alan R. Liss, 77 (1985);Boerner et al., J. Immunol. 147(1):86-95 (1991)に記載の方法も、ヒトモノクローナル抗体の調製に利用可能である。van Dijk and van de Winkel, Curr. Opin. Pharmacol. 5: 368-74 (2001)も参照のこと。ヒト抗体の作製は、前記抗原を、抗原チャレンジに応答してこのような抗体を産生するが、内在性遺伝子座は機能しないように改変したトランスジェニック動物に投与することによって行うことができ、該トランスジェニック動物の例は免疫化ゼノマウス(xenomouse)(例えば、XENOMOUSE(登録商標)技術に関して米国特許第6,075,181号および第6,150,584号を参照のこと)を含む。また、例えば、ヒトB細胞ハイブリドーマ技術によって生成されたヒト抗体に関してLi et al., Proc. Natl. Acad. Sci. USA 103:3557-3562 (2006)を参照のこと。
の標的に対して結合する場合と比べ、この標的に対して、より高い親和性と結合活性で、より容易に、および/またはより長い持続時間で、結合する抗体である。一実施形態においては、抗体の無関係な標的への結合の程度は、例えばラジオイムノアッセイ(RIA)による測定において、該抗体の標的への結合の約10%未満である。ある実施形態においては、標的に特異的に結合する抗体は、≦1μM、≦100nM、≦10nM、≦1nM、または≦0.1nMの解離定数(Kd)を有する。ある実施形態においては、抗体は、異なる種に由来するタンパク質間で保存されている該タンパク質上のエピトープに特異的に結合する。他の一実施形態において、特異的な結合は排他的な結合を含みうるが、必ずしもそれが必要というわけではない。
本発明の一側面により、a)免疫調節剤をコードする異種核酸を含む改変哺乳類細胞であって、前記異種核酸がプロモーターに機能的に連結した、改変哺乳類細胞;およびb)薬理学的に許容される賦形剤、を含む医薬組成物が提供される。一部の実施形態において、前記改変哺乳類細胞は免疫細胞である。一部の実施形態において、前記改変哺乳類細胞は幹細胞である。一部の実施形態において、前記プロモーターは誘導性である。一部の実施形態において、前記免疫調節剤は免疫チェックポイント阻害剤である。一部の実施形態において、前記免疫調節剤は免疫賦活剤である。一部の実施形態において、前記免疫調節剤は分泌タンパク質である。一部の実施形態において、前記免疫調節剤は抗体である。一部の実施形態において、前記改変哺乳類細胞は、さらに、腫瘍抗原を認識するターゲティング分子をその表面上に発現する。一部の実施形態において、前記改変哺乳類細胞は、さらに、治療用タンパク質(例えば第2の免疫調節剤、または免疫調節剤ではない治療用タンパク質)をコードする第2の異種核酸を含む。
異なる。例えば、本発明の改変哺乳類細胞が発現する免疫調節剤は、該改変哺乳類細胞を、それを必要とする個体に直接投与することによってその個体に送達することができ、その際に該免疫調節剤を該改変哺乳類細胞から単離または精製する必要がない。一部の実施形態において、前記医薬組成物はヒト個体等の個体に投与するのに適したものである。一部の実施形態において、前記医薬組成物は注射に適したものである。一部の実施形態において、前記医薬組成物は輸液に適したものである。一部の実施形態において、前記医薬組成物は細胞培養培地を実質的に含まない。一部の実施形態において、前記医薬組成物はエンドトキシンまたはアレルゲンタンパク質を実質的に含まない。一部の実施形態において、「実質的に含まない」とは、医薬組成物の総容量または重量に対し、約10%、5%、1%、0.1%、0.01%、0.001%、1ppm、またはそれ未満、のいずれかよりも少ないことを意味する。一部の実施形態において、前記医薬組成物はマイコプラズマ、微生物因子、および/または感染症因子を含まない。
本出願人の医薬組成物は、前記改変哺乳類細胞を任意の数含んでいてよい。一部の実施形態において、前記医薬組成物は、1コピーの前記改変哺乳類細胞を含む。一部の実施形態において、前記医薬組成物は、前記改変哺乳類細胞を、少なくとも約1、10、100、1000、104、105、106、107、108、またはそれよりも多い、いずれかのコピー数で含む。一部の実施形態において、前記医薬組成物は、1種類の改変哺乳類細胞を含む。一部の実施形態において、前記医薬組成物は少なくとも2種類の改変哺乳類細胞を含み、これらの異なる種類の改変哺乳類細胞では、細胞源、細胞型、発現する治療用タンパク質、免疫調節剤、および/またはプロモーター等が異なる。
ト等)T細胞であって、前記異種核酸がプロモーターに機能的に連結した、改変哺乳類T細胞;およびb)薬理学的に許容される賦形剤、を含む医薬組成物が提供される。一部の実施形態において、前記改変哺乳類T細胞は細胞傷害性T細胞、ヘルパーT細胞、TIL、LAK細胞、CAR−T、またはTCR−Tから選択される。一部の実施形態において、前記プロモーターは誘導性である。一部の実施形態において、前記プロモーターはT細胞活性化依存プロモーターである。一部の実施形態において、前記免疫調節剤は免疫チェックポイント阻害剤である。一部の実施形態において、前記免疫調節剤は免疫賦活剤である。一部の実施形態において、前記免疫調節剤は分泌タンパク質である。一部の実施形態において、前記免疫調節剤は抗体である。一部の実施形態において、前記改変哺乳類T細胞は、さらに、腫瘍抗原(CARまたはTCR等)を認識するターゲティング分子をその表面上に発現する。一部の実施形態において、前記改変哺乳類T細胞は、さらに、治療用タンパク質(例えば第2の免疫調節剤、または免疫調節剤ではない治療用タンパク質)をコードする第2の異種核酸を含む。
する。一部の実施形態において、前記改変哺乳類細胞は複数コピーの前記異種核酸を有する。一部の実施形態において、前記改変哺乳類細胞は少なくとも一種のさらなる異種核酸を含み、その例は、第2の免疫調節剤もしくは免疫調節剤ではない治療用タンパク質をコードする第2の異種核酸;または、細胞内のバイオマーカーの発現に対するレポーターをコードする第2の異種核酸を含む。一部の実施形態において、前記改変哺乳類細胞は2種類以上の異種核酸を含み、これらはそれぞれ異なる治療用タンパク質(例えば免疫調節剤または非免疫調節剤)をコードする。
まれていてよい。
前記改変哺乳類細胞は、免疫調節剤を任意の数(例えば1個、2個、3個、4個、5個、もしくは6個のいずれか、またはそれ以上)発現してよい。一部の実施形態において、前記改変哺乳類細胞は、単一の免疫調節剤をコードする異種核酸を含む。一部の実施形態において、前記改変哺乳類細胞は、少なくとも2つの免疫調節剤をコードする1つまたは複数の異種核酸を含む。一部の実施形態において、前記少なくとも2つの免疫調節剤をコードする異種核酸は、同一のプロモーターに機能的に連結している。一部の実施形態において、前記少なくとも2つの免疫調節剤をコードする異種核酸は、異なるプロモーターに機能的に連結している。
おいて、前記免疫調節剤は抗体である。モノクローナル抗体等の天然抗体は、特定の抗原と免疫学的な反応性を有する免疫グロブリン分子である。本明細書で使用される「抗体」という語は遺伝子操作された形態のものを含み、その例はキメラ抗体(例えばヒト化マウス抗体)、ヘテロ結合抗体(heteroconjugate antibody)(例えば二重特異性抗体)、組換え一本鎖Fv断片(scFv)、単一ドメイン抗体、および重鎖のみ抗体などを含む。「抗体」という語はまた、Fab’、F(ab’)2、scFv、およびVHH等の抗原結合型の抗体も含む。一部の実施形態において、前記抗体はアゴニスト抗体である。一部の実施形態において、前記抗体はアンタゴニスト抗体である。一部の実施形態において、前記抗体はモノクローナル抗体である。一部の実施形態において、前記抗体は全長抗体である。一部の実施形態において、前記抗体は、VH、VL、VNAR、VHH、Fab、Fab’、F(ab’)2、Fv、ミニボディ(minibody)、scFv、sc(Fv)2、トリボディ(tribody)、テトラボディ(tetrabody)、BiTE、ミニボディ(minibody)、scFv−Fc、トリアボディ(triabody)、および全長抗体の他の抗原結合サブ配列、またはこれらの人為的な組み合わせ、からなる群より選択される抗原結合断片である。一部の実施形態において、前記抗体はヒト抗体、ヒト化抗体、またはキメラ抗体である。一部の実施形態において、前記抗体は一価抗体である。一部の実施形態において、前記抗体は二価抗体または四価抗体等の多価抗体である。一部の実施形態において、前記抗体は二重特異性抗体である。一部の実施形態において、前記抗体は多重特異性抗体である。一部の実施形態において、前記抗体は単一ドメイン抗体である。一部の実施形態において、前記抗体は重鎖のみ抗体である。一部の実施形態において、前記抗体は、抗体断片(Fc含有融合タンパク質等)、または全長抗体の任意の他の機能的バリアントもしくは誘導体を含む、融合タンパク質である。
おいて、前記免疫調節抗体は重鎖と軽鎖とを含む。一部の実施形態において、前記改変哺乳類細胞は、さらに、腫瘍抗原を認識するターゲティング分子をその表面上に発現する。一部の実施形態において、前記改変哺乳類細胞は、さらに、治療用タンパク質(例えば第2の免疫調節剤、または免疫調節剤ではない治療用タンパク質)をコードする第2の異種核酸を含む。
e Reviews Cancer 12: 252-264 (2012)を参照のこと)。前記免疫調節剤は、前記免疫チェックポイント分子に特異的に結合する抗体、天然リガンド、または改変タンパク質でありうる。
体等)を含む。一部の実施形態において、前記免疫チェックポイント阻害剤は分泌性である。一部の実施形態において、前記免疫チェックポイント阻害剤は、抗CTLA−4(イピリムマブ(Ipilimumab)、トレメリムマブ(Tremelimumab)、KAHR−102等)、抗TIM−3(F38−2E2、ENUM005等)、抗LAG−3(BMS−986016、IMP701、IMP321、C9B7W等)、抗KIR(リリルマブ(Lirilumab)およびIPH2101等)、抗PD−1(ニボルマブ(Nivolumab)、ピディリズマブ(Pidilizumab)、ペムブロリズマブ(Pembrolizumab)、BMS−936559、アテゾリズマブ(atezolizumab)、ラムブロリズマブ(Lambrolizumab)、MK−3475、AMP−224、AMP−514、STI−A1110、TSR−042等)、抗PD−L1(KY−1003(EP20120194977)、MCLA−145、RG7446、BMS−936559、MEDI−4736、MSB0010718C、AUR−012、STI−A1010、PCT/US2001/020964、MPDL3280A、AMP−224、ダピロリズマブ・ペゴル(Dapirolizumab pegol)(CDP−7657)、MEDI−4920等)、抗CD73(AR−42(OSU−HDAC42、HDAC−42、AR42、AR 42、OSU−HDAC 42、OSU−HDAC−42、NSC D736012、HDAC−42、HDAC 42、HDAC42、NSCD736012、NSC−D736012)、MEDI−9447等)、抗B7−H3(MGA271、DS−5573a、8H9等)、抗CD47(CC−90002、TTI−621、VLST−007等)、抗BTLA、抗VISTA、抗A2aR、抗B7−1、抗B7−H4、抗CD52(アレムツズマブ(alemtuzumab)等)、抗IL−10、抗IL−35、および抗TGF−β(フレソルミマブ(Fresolumimab)等)からなる群より選択される、抑制性免疫チェックポイントタンパク質を標的とする抗体(例えばアンタゴニスト抗体)である。一部の実施形態において、前記免疫チェックポイント阻害剤は、PD−1、PD−L1、PD−L2、CTLA−4、BLTA、TIM−3、およびLAG−3からなる群より選択される抑制性チェックポイント分子の阻害剤である。
形態において、前記抗PD−1抗体はラムブロリズマブ(Lambrolizumab)である。ラムブロリズマブ(ペムブロリズマブ(Pembrolizumab)またはMK−3475とも呼ばれる;商品名KEYTRUDA(登録商標))は、2014年9月4日付けで米国FDAに認可されたヒト化抗PD−1 IgG4 mAbである。この薬剤は当初、転移性メラノーマの治療に使用された。ペムブロリズマブは、PD−L1を発現する腫瘍を有し、他の化学療法剤による治療に失敗した患者における転移性非小細胞肺癌の治療に対して、2015年10月2日付けで米国FDAに認可された。
一部の実施形態において、前記改変哺乳類細胞は、さらに、治療用タンパク質をコードする第2の異種核酸を含む。本明細書が企図する治療用タンパク質の例は、治療効果を有する、任意のタンパク質またはポリペプチドに基づいた薬剤を含む。従って、免疫調節剤は治療用タンパク質の1種であると考えられる。前記改変哺乳類細胞は、前記免疫調節剤に加え、治療用タンパク質を任意の数(例えば1個、2個、3個、4個、5個、もしくは6個のいずれか、またはそれ以上)発現してよい。一部の実施形態において、前記治療用タンパク質は免疫調節剤である。一部の実施形態において、前記治療用タンパク質は免疫調節剤ではない。一部の実施形態において、前記改変哺乳類細胞は、前記免疫調節剤に加え、2つ以上のさらなる免疫調節剤、2つ以上の免疫調節剤ではない治療用タンパク質、またはさらなる免疫調節剤と免疫調節剤ではない治療用タンパク質との組み合わせなどを含む、2つ以上の治療用タンパク質を発現する。
に導入される。一部の実施形態において、前記免疫調節剤をコードする異種核酸と、前記治療用タンパク質をコードする第2の異種核酸とが、別々の異種遺伝子発現カセットを介して前記改変哺乳類細胞に導入される。
は細胞表面分子のリガンドである。一部の実施形態において、前記治療用タンパク質は細胞表面分子の阻害剤である。一部の実施形態において、前記治療用タンパク質は細胞表面分子の賦活剤である。一部の実施形態において、前記治療用タンパク質は細胞表面分子である。一部の実施形態において、前記治療用タンパク質は化学療法剤である。一部の実施形態において、前記治療用タンパク質は腫瘍抗原に特異的に結合する。
ozogamicin)、イノツズマブ・オゾガマイシン(inotuzumab ozogamicin)、イピリムマブ(ipilimumab)、ラベツズマブ(labetuzumab)、リンツズマブ(lintuzumab)、マツズマブ(matuzumab)、メポリズマブ(mepolizumab)、モタビズマブ(motavizumab)、モトビズマブ(motovizumab)、ナタリズマブ(natalizumab)、ニモツズマブ(nimotuzumab)、ノロビズマブ(nolovizumab)、ヌマビズマブ(numavizumab)、オクレリズマブ(ocrelizumab)、オマリズマブ(omalizumab)、パリビズマブ(palivizumab)、パスコリズマブ(pascolizumab)、ペクフシツズマブ(pecfusituzumab)、ペクツズマブ(pectuzumab)、ペキセリズマブ(pexelizumab)、ラリビズマブ(ralivizumab)、ラニビズマブ(ranibizumab)、レスリビズマブ(reslivizumab)、レスリズマブ(reslizumab)、レシビズマブ(resyvizumab)、ロベリズマブ(rovelizumab)、ルプリズマブ(ruplizumab)、シブロツズマブ(sibrotuzumab)、シプリズマブ(siplizumab)、ソンツズマブ(sontuzumab)、タカツズマブ・テトラキセタン(tacatuzumab tetraxetan)、タドシズマブ(tadocizumab)、タリズマブ(talizumab)、テフィバズマブ(tefibazumab)、トシリズマブ(tocilizumab)、トラリズマブ(toralizumab)、ツコツズマブ・セルモロイキン(tucotuzumab celmoleukin)、ツクシツズマブ(tucusituzumab)、ウマビズマブ(umavizumab)、ウルトキサズマブ(urtoxazumab)、ウステキヌマブ(ustekinumab)、ビシリズマブ(visilizumab)、および抗インターロイキン−12(ABT−874/J695、Wyeth Research and Abbott Laboratories)を含むが、これらに限定されない。一部の実施形態において、前記治療用タンパク質は抗HER2抗体である。一部の実施形態において、前記抗HER2抗体はHER2に結合し、HER2+癌細胞の細胞増殖または成長を阻害する。一部の実施形態において、前記抗HER2抗体はHER2に結合し、HER2が他のHER受容体と二量体化することを阻害する。一部の実施形態において、前記抗HER2抗体はトラスツズマブ(trastuzumab)またはペルツズマブ(pertuzumab)である。一部の実施形態において、前記抗HER2抗体はトラスツズマブまたはペルツズマブではない。
、該治療用タンパク質のためのプロモーターは、本明細書に記載した免疫調節剤のそれらのための任意の特性を有していてよい。
前記免疫調節剤をコードする異種核酸、前記細胞表面分子(CARもしくはTCR)、または本明細書に記載される任意の他の治療用タンパク質は、プロモーターに機能的に連結していてよい。一部の実施形態において、前記免疫調節剤、細胞表面分子(CARまたはTCR等)、および他の治療用タンパク質のそれぞれは、異なるプロモーターにより駆動されるものであってよい。一部の実施形態において、前記免疫調節剤、細胞表面分子(CARまたはTCR等)、および他の治療用タンパク質は、同一のプロモーターによって駆動されるものであってよい。
とCMV初期エンハンサーとの組み合わせ(CAGG)を含むが、これらに限定されない。導入遺伝子の発現の駆動に対するこのような構成的プロモーターの有効性は、極めて多数の研究によって広範に比較されてきた。例えば、Michael C. Miloneらは、初代ヒトT細胞におけるキメラ抗原受容体発現の駆動に対するCMV、hEF1α、UbiC、およびPGKの有効性を比較し、その結果、hEF1αプロモーターが導入遺伝子の最高レベルの発現を誘導しただけではなく、CD4およびCD8ヒトT細胞において最適に維持されたと結論づけた(Molecular Therapy, 17(8): 1453-1464 (2009))。一部の実施形態において、前記異種核酸中のプロモーターはhEF1αプロモーターである。構成的プロモーターに機能的に連結した免疫チェックポイント阻害剤をコードする異種核酸を含む改変哺乳類細胞であって、該免疫チェックポイント阻害剤が、腫瘍細胞上に発現する抑制性免疫チェックポイント分子を遮断するものである例を、図1に示す。構成的プロモーターに機能的に連結した免疫チェックポイント阻害剤をコードする異種核酸を含む改変哺乳類細胞であって、該免疫チェックポイント阻害剤が、該改変哺乳類細胞および非改変免疫細胞上に発現する抑制性免疫チェックポイント分子を遮断するものである例を、図2に示す。
の実施形態において、前記誘導物質は化合物等の小分子である。一部の実施形態において、前記小分子は、ドキシサイクリン、テトラサイクリン、アルコール、金属、またはステロイド類からなる群より選択される。化学的に誘導されるプロモーターが最も広範に研究されてきた。このようなプロモーターの例は、ドキシサイクリン、テトラサイクリン、アルコール、ステロイド類、金属、および他の化合物等の小分子化学物質の有無によって転写活性が調節されるプロモーターを含む。リバーステトラサイクリン制御性トランス活性化因子(reverse tetracycline-controlled transactivator)(rtTA)およびテトラサイクリン応答エレメントプロモーター(tetracycline-responsive element promoter)(TRE)を用いたドキシサイクリン誘導系が、現在のところ最も成熟した系である。WO9429442は、テトラサイクリン応答性プロモーターによる真核細胞中の遺伝子発現の厳格な制御について記載している。WO9601313は、テトラサイクリンによって制御される転写モジュレーターを開示している。さらに、Tet−on系などのTet技術が、例えばTetSystems.comのウェブサイト上に記載されている。公知の化学的に制御される任意のプロモーターを用いて本出願の治療用タンパク質の発現を駆動してよい。
(2012);Nature 500: 472-476 (2013);Nature Neuroscience 18:1202-1212 (2015)を参照のこと)。このような遺伝子調節系は、(1)DNA結合、または(2)転写活性化ドメインのDNA結合タンパク質へのリクルート、の制御に基づき大きく2つのカテゴリーに分けることができる。例えば、青色光(480nm)に応答して細胞内カルシウムの増加を引き起こし、その結果NFATのカルシニューリン媒介動員が起こる、メラノプシンに基づいた合成哺乳類青色光制御転写系が開発され、哺乳類細胞において試験された。より最近、Motta-Menaらは、ヒト細胞株およびゼブラフィッシュ胚において転写開始の高レベルの青色光感受性制御を可能にする、天然EL222転写因子から開発された新規誘導
性遺伝子発現系を記載した(Nat. Chem. Biol. 10(3):196-202 (2014))。さらに、Arabidopsis thalianaの光受容体フィトクロムB(PhyB)とフィトクロム相互作用因子6(PIF6)の赤色光誘導相互作用が、赤色光誘導型の遺伝子発現調節のために利用された。さらに、紫外線B(UVB)誘導性遺伝子発現系も開発され、哺乳類細胞における標的遺伝子転写において有効であることが示された(Gene and Cell Therapy: Therapeutic Mechanisms and Strategies, Fourth Edition CRC Press, Jan. 20th, 2015の第25章)。本明細書に記載される任意の光誘導性プロモーターを用いて本発明の治療用タンパク質の発現を駆動してよい。
自殺遺伝子をhsp70Bプロモーターに連結し、マウス乳癌を有するヌードマウスでこの系を試験した。hsp70B−HSVtkコード配列を腫瘍に投与し、熱処理したマウスは、熱処理を行わなかった対照と比較して、腫瘍の退縮と有意な生存率の向上を示した(Hum. Gene Ther. 11:2453 (2000))。当該分野で公知のさらなる熱誘導性プロモーターを、例えばGene and Cell Therapy: Therapeutic Mechanisms and Strategies, Fourth Edition CRC Press, Jan. 20th, 2015の第25章に見出すことができる。本明細書に議論される任意の熱誘導性プロモーターを用いて本発明の治療用タンパク質の発現を駆動してよい。
5:472-484 (2005))。本願発明者らは、ヒトT細胞株において、NFATプロモーターが、PMA/PHA−P活性化の存在下でレポーター遺伝子の発現を開始するのに有効であることを見出した。核内因子カッパB(NFκB)を含む他の経路も、T細胞活性化を介した導入遺伝子発現の制御に用いることができる。
上記改変哺乳類細胞のいずれも、細胞表面分子をさらに発現してよい。該細胞表面分子は細胞外ドメインと膜貫通ドメインとを有する。一部の実施形態において、前記細胞表面分子は、さらに、一次細胞内シグナル伝達ドメインおよび/または共刺激シグナル伝達ドメイン等の、細胞内エフェクタードメインを有する。一部の実施形態において、前記細胞表面分子は内在性分子である。一部の実施形態において、前記細胞表面分子は異種分子である。一部の実施形態において、前記細胞表面分子は改変分子である。一部の実施形態において、前記細胞表面分子は前記改変哺乳類細胞の異種核酸によってコードされる。一部の実施形態において、前記細胞表面分子は、プロモーター(構成的プロモーターまたは誘導性プロモーター等)に機能的に連結した第2の異種核酸によってコードされる。一部の実施形態において、前記細胞表面分子は、細胞をCELL SQUEEZE(登録商標)等のマイクロ流体システムに通過させながらタンパク質を細胞膜に挿入することによって、前記改変哺乳類細胞に導入される(例えば、米国特許出願公開第20140287509号を参照のこと)。前記細胞表面分子は、例えば該改変哺乳類細胞をターゲティングすることにより、シグナルを変換することにより、および/または該改変哺乳類細胞の細胞毒性を増強することにより、前記改変哺乳類細胞の機能を増強するものであってよい。一部の実施形態において、前記改変哺乳類細胞は、CARまたはTCR等の細胞表面分子を発現しない。
よび任意に1つまたは複数の共刺激シグナル伝達ドメインを有するCARと比べ、約90%、80%、70%、60%、50%、40%、30%、20%、10%、またはそれ未満、のいずれかの免疫エフェクター機能(標的細胞に対する細胞溶解機能等)しか有しない細胞内シグナル伝達ドメインを有する。一部の実施形態において、前記CAR単独では標的細胞の細胞溶解を誘発しない。一部の実施形態において、前記細胞内シグナル伝達ドメインは、CAR含有細胞の増殖および/または生存を促進するシグナルを生成する。一部の実施形態において、前記CARは、CD−28、CD137、CD3、CD27、CD40、ICOS、GITR、およびOX40のシグナル伝達ドメインから選択される1つまたは複数の細胞内シグナル伝達ドメインを含む。天然に存在する分子のシグナル伝達ドメインは、該分子の細胞内(すなわち細胞質)部分全体もしくは天然の細胞内シグナル伝達ドメイン全体、またはその断片もしくは誘導体を含みうる。
、IL−2Rガンマ、IL−7Rアルファ、ITGA4、VLAl、CD49a、ITGA4、IA4、CD49D、ITGA6、VLA−6、CD49f、ITGAD、CD11d、ITGAE、CD103、ITGAL、CD11a、LFA−1、ITGAM、CD11b、ITGAX、CD11c、ITGB1、CD29、ITGB2、CD18、LFA−1、ITGB7、NKG2D、NKG2C、TNFR2、TRANCE/RANKL、DNAM1(CD226)、SLAMF4(CD244、2B4)、CD84、CD96(Tactile)、CEACAM1、CRTAM、Ly9(CD229)、CD160(BY55)、PSGLl、CD100(SEMA4D)、CD69、SLAMF6(NTB−A、Lyl08)、SLAM(SLAMFl、CD150、IPO−3)、BLAME(SLAMF8)、SELPLG(CD162)、LTBR、LAT、GADS、SLP−76、PAG/Cbp、およびCD19a;ならびに、CD83に特異的に結合するリガンドを含む。
、CD137の機能的シグナル伝達ドメインとを含む。一部の実施形態において、前記細胞内シグナル伝達ドメインは、CD3ゼータの非機能的または減弱化一次シグナル伝達ドメインと、CD137の機能的シグナル伝達ドメインとを含む。一部の実施形態において、前記細胞内シグナル伝達ドメインは、CD137の機能的シグナル伝達ドメインからなる、または本質的になる。
EA、EGFR(EGFRvIII等)、GD2、HER2、IGF1R、メソテリン、PSMA、ROR1、およびWT1からなる群より選択される腫瘍抗原を標的とする。一部の実施形態において、前記CARは、前記改変哺乳類免疫細胞が腫瘍細胞に結合した際に、および該改変哺乳類免疫細胞による免疫調節剤の分泌の際に、免疫細胞(該改変哺乳類免疫細胞を含む)の細胞溶解機能、サイトカイン分泌、および/または増殖を引き起こす。一部の実施形態において、前記CARは、免疫エフェクター機能が無効化または減弱化した細胞内シグナル伝達ドメインを含む。一部の実施形態において、前記CARは、短縮型CARである。一部の実施形態において、前記CARは、一次細胞内シグナル伝達ドメイン(CD3ζ等)を含まない。一部の実施形態において、前記CARは、非機能的な、または減弱化した一次細胞内シグナル伝達ドメイン(変異CD3ζ等)を含む。一部の実施形態において、前記CAR単独では標的細胞の細胞溶解を誘発しない。
おいて、前記改変哺乳類細胞がT細胞である場合、該T細胞受容体は内在性T細胞受容体である。一部の実施形態において、前記TCRを有する改変哺乳類細胞は前選択される(pre-selected)。一部の実施形態において、前記T細胞受容体は組換えTCRである。一部の実施形態において、前記TCRは腫瘍抗原に対して特異的である。一部の実施形態において、前記腫瘍抗原は、CD19、BCMA、NY−ESO−1、VEGFR2、MAGE−A3、VEGFR2、MAGE−A3、CD20、CD22、CD33、CD38、CEA、EGFR(EGFRvIII等)、GD2、HER2、IGF1R、メソテリン、PSMA、ROR1、WT1、および臨床的に重要な他の腫瘍抗原からなる群より選択される。一部の実施形態において、前記腫瘍抗原は腫瘍細胞の細胞内タンパク質に由来する。腫瘍抗原(腫瘍関連抗原を含む)に対して特異的なTCRは多数記載されており、それらの例は、NY−ESO−1癌精巣抗原、p53腫瘍抑制抗原、ならびに、メラノーマ(MARTI、gp100等)、白血病(WT1、副組織適合抗原等)、および乳癌(HER2、NY−BR1等)の腫瘍抗原に対するTCRを含む。本出願においては、当該分野で公知の任意のTCRを用いてよい。一部の実施形態において、前記TCRは腫瘍抗原に対して増強された親和性を有する。TCRの例、および該TCRを哺乳類細胞に導入する方法は、例えば、米国特許第5830755号およびKessels et al. Immunotherapy
through TCR gene transfer. Nat. Immunol. 2, 957-961 (2001)に記載されている。一部の実施形態において、前記改変哺乳類細胞はTCR−T細胞である。
ロモーターに機能的に連結した免疫チェックポイント阻害剤をコードする異種核酸と、構成的プロモーターに機能的に連結したCARをコードする第2の異種核酸と、を含む改変哺乳類細胞であって、該免疫チェックポイント阻害剤が、腫瘍細胞上に発現した抑制性免疫チェックポイント分子を遮断する、改変哺乳類細胞の例を、図5に示す。CAR誘導性プロモーターに機能的に連結した免疫チェックポイント阻害剤をコードする異種核酸と、構成的プロモーターに機能的に連結したCARをコードする第2の異種核酸を含む改変哺乳類細胞であって、該免疫チェックポイント阻害剤が、該改変哺乳類細胞および非改変免疫細胞上に発現した抑制性免疫チェックポイント分子を遮断する、改変哺乳類細胞の例を、図6に示す。
ーゼ、CD137L、LEM、およびBcl−2からなる群より選択される。一部の実施形態において、前記TCRは、第2のプロモーターに機能的に連結した第3の異種核酸によってコードされる。一部の実施形態において、前記第2のプロモーターは構成的プロモーターである。一部の実施形態において、前記第2のプロモーターは、例えば、誘導物質(例えばテトラサイクリンまたはドキシサイクリンなどの小分子等)、照射、温度、酸化還元状態、腫瘍環境、および前記改変哺乳類TCR−T細胞の活性化状態、から選択される誘導条件によって誘導可能である。一部の実施形態において、前記TCRは、CD19、BCMA、NY−ESO−1、VEGFR2、MAGE−A3、CD20、CD22、CD33、CD38、CEA、EGFR(EGFRvIII等)、GD2、HER2、IGF1R、メソテリン、PSMA、ROR1、およびWT1からなる群より選択される腫瘍抗原を標的とする。
1、GAGE−2、p15等の腫瘍特異的多系列抗原(tumor-specific multilineage antigens);CEA等の過剰発現胎児性抗原;p53、Ras、HER2/neu等の過剰発現腫瘍遺伝子および変異腫瘍抑制遺伝子;染色体転座の結果生ずる固有の腫瘍抗原;BCR−ABL、E2A−PRL、H4−RET、IGH−IGK、MYL−RARなど;ならびに、エプスタイン・バーウイルス抗原EBVAならびにヒトパピローマウイルス(HPV)抗原E6およびE7等のウイルス抗原。他の大型の、タンパク質に基づいた抗原の例は、TSP−180、MAGE−4、MAGE−5、MAGE−6、RAGE、NY−ESO、p185erbB2、p180erbB−3、c−met、nm−23HI、PSA、TAG−72、CA19−9、CA72−4、CAM17.1、NuMa、K−ras、ベータカテニン、CDK4、Mum−1、p15、p16、43−9F、5T4、791Tgp72、アルファ−フェトプロテイン、ベータ−HCG、BCA225、BTAA、CA125、CA15−3\CA27.29\BCAA、CA195、CA242、CA−50、CAM43、CD68\P1、CO−029、FGF−5、G250、Ga733\EpCAM、HTgp−175、M344、MA−50、MG7−Ag、MOV18、NB/70K、NY−CO−1、RCAS1、SDCCAG16、TA−90\Mac−2結合タンパク質\シクロフィリンC関連タンパク質、TAAL6、TAG72、TLP、およびTPSを含む。
胞内シグナル伝達ドメイン(変異CD3ζ等)を含む。一部の実施形態において、前記CAR単独では標的細胞の細胞溶解を誘発しない。一部の実施形態において、前記医薬組成物はNSCLC等の肺癌を治療するのに有用である。
、前記CAR単独では標的細胞の細胞溶解を誘発しない。一部の実施形態において、前記医薬組成物は膠芽腫の治療に有用である。

−12およびIL−15と共に培養したUCB由来T細胞が、固有のセントラルメモリー/エフェクター表現型で150倍を超える増殖を引き起こしたことを示す報告もある(Leukemia (2015) 29: 415-422)。さらに、LEMはミトコンドリア呼吸に対する効果を介してCD8+ T細胞免疫を促進し(Science (2015) 348(6238): 995-1001)、ヘパラナーゼはCARリダイレクトTリンパ球(CAR-redirected T-lymphocytes)の腫瘍浸潤と抗腫瘍活性を促進する(Nat. Med. (2015) 21(5): 524-529)。
R−T細胞であって、前記異種核酸がプロモーターに機能的に連結し、前記改変CAR−T細胞がCARを発現する、改変哺乳類CAR−T細胞;およびb)薬理学的に許容される賦形剤、を含む医薬組成物が提供される。一部の実施形態において、前記異種核酸は免疫チェックポイント阻害剤と免疫賦活剤の両者をコードする。一部の実施形態において、前記異種核酸は少なくとも2個の免疫賦活剤をコードする。一部の実施形態において、前記免疫チェックポイント阻害剤は、PD−1、PD−L1、PD−L2、CTLA−4、BLTA、TIM−3、またはLAG−3からなる群より選択される免疫チェックポイント分子の阻害剤である。一部の実施形態において、前記免疫チェックポイント阻害剤は抗体(全長抗体、scFv、単一ドメイン抗体、重鎖のみ抗体、またはFab等)である。一部の実施形態において、前記免疫賦活剤はIL−2、IL−7、IL−15、IL−21、IL−12、CCR4、CCR2b、ヘパラナーゼ、CD137L、LEM、およびBcl−2からなる群より選択される。一部の実施形態において、前記CARは、EGFRvIII等のEGFR、BCMA、またはNY−ESO−1等の腫瘍抗原を標的とする。一部の実施形態において、前記改変CAR−T細胞は、さらに、治療用タンパク質(第2の免疫調節剤;または、免疫調節剤ではない治療用タンパク質、例えば化学療法抗体等)をコードする第2の異種核酸を含む。一部の実施形態において、前記CARは、前記プロモーターに機能的に連結した異種核酸によってコードされる。一部の実施形態において、前記CARは、第2のプロモーターに機能的に連結した第2の異種核酸によってコードされる。一部の実施形態において、前記プロモーターおよび/または前記第2のプロモーターは、hEF1αプロモーター等の構成的プロモーターである。一部の実施形態において、前記プロモーターおよび/または第2のプロモーターは、例えば、誘導物質(例えばテトラサイクリンまたはドキシサイクリンなどの小分子等)、照射、温度、酸化還元状態、腫瘍環境、および前記改変哺乳類CAR−T細胞の活性化状態、から選択される誘導条件によって誘導可能である。一部の実施形態において、前記CARは、前記改変哺乳類CAR−T細胞が腫瘍細胞に結合した際に、および該改変CAR−T細胞による免疫調節剤の分泌の際に、該改変CAR−T細胞を含むT細胞の細胞溶解機能、サイトカイン分泌、および/または増殖を引き起こす。一部の実施形態において、前記CARは免疫エフェクター機能が無効化または減弱化した細胞内シグナル伝達ドメインを含む。一部の実施形態において、前記CARは短縮型CARである。一部の実施形態において、前記CARは一次細胞内シグナル伝達ドメイン(CD3ζ等)を含まない。一部の実施形態において、前記CARは、非機能的な、または減弱化した一次細胞内シグナル伝達ドメイン(変異CD3ζ等)を含む。一部の実施形態において、前記CAR単独では標的細胞の細胞溶解を誘発しない。一部の実施形態において、前記CARは、CD8α SP、腫瘍抗原(EGFRvIIIなどのEGFR、BCMA、またはNY−ESO−1等)に特異的に結合するターゲティングドメイン、CD8αヒンジおよび膜貫通ドメイン、ならびにCD137細胞質ドメインを含む。
おいて、前記プロモーターは誘導性である。一部の実施形態において、前記プロモーターは、IL−2プロモーター、NFATプロモーター、またはNFκBプロモーター等の、T細胞活性化依存プロモーターである。一部の実施形態において、前記免疫チェックポイント阻害剤は、PD−1、PD−L1、PD−L2、CTLA−4、BLTA、TIM−3、またはLAG−3からなる群より選択される免疫チェックポイント分子の阻害剤である。一部の実施形態において、前記免疫チェックポイント阻害剤は抗体(全長抗体、scFv、単一ドメイン抗体、重鎖のみ抗体、またはFab等)である。一部の実施形態において、前記免疫賦活剤はIL−2、IL−7、IL−15、IL−21、IL−12、CCR4、CCR2b、ヘパラナーゼ、CD137L、LEM、およびBcl−2からなる群より選択される。一部の実施形態において、前記CARは、EGFRvIII等のEGFR、BCMA、またはNY−ESO−1等の腫瘍抗原を標的とする。
IL−7、IL−15、IL−21、IL−12、CCR4、CCR2b、ヘパラナーゼ、CD137L、LEM、およびBcl−2からなる群より選択される。一部の実施形態において、前記CARは、EGFRvIII等のEGFR、BCMA、またはNY−ESO−1等の腫瘍抗原を標的とする。一部の実施形態において、前記CARは短縮型CARである。一部の実施形態において、前記CARは一次細胞内シグナル伝達ドメイン(CD3ζ等)を含まない。一部の実施形態において、前記CARは、非機能的な、または減弱化した一次細胞内シグナル伝達ドメイン(変異CD3ζ等)を含む。一部の実施形態において、前記CAR単独では標的細胞の細胞溶解を誘発しない。一部の実施形態において、前記CARは、CD8α SP、腫瘍抗原(EGFRvIIIなどのEGFR;NY−ESO−1;またはBCMA等)に特異的に結合するターゲティングドメイン、CD8αヒンジおよび膜貫通ドメイン、ならびにCD137細胞質ドメインを含む。
一部の実施形態において、前記医薬組成物はさらに第2の細胞を含み、該第2の細胞は、CARまたはTCRを発現する哺乳類免疫細胞(T細胞等)である。上節に記載のCARまたはTCRのうち任意のものが該第2の細胞によって発現されてよく、その場合に前記改変哺乳類細胞は前記免疫調節剤および任意の1つまたは複数のさらなる治療用タンパク質(他の免疫調節剤または非免疫調節剤等)のみを発現するものであってよい。腫瘍部位において、前記医薬組成物の改変哺乳類細胞は、免疫調節剤を分泌することによって抑制性免疫チェックポイントを遮断するか、あるいは刺激性免疫チェックポイントを活性化することが可能である一方で、CARまたはTCRを発現する前記第2の哺乳類免疫細胞は、腫瘍細胞へのリクルートが可能である。前記免疫調節剤および前記CARまたはTCRからのシグナルの組み合わせによって、前記第2の哺乳類免疫細胞の活性化が可能となり、腫瘍細胞に対する強固な免疫応答が引き起こされうる。これら二成分の医薬組成物によって、前記改変哺乳類細胞による前記免疫調節剤およびさらなる治療用タンパク質の分泌と、CARまたはTCRを発現する前記第2の哺乳類免疫細胞の活性化と、を独立に制御(タイミングや量など)することが可能となる。前記2種の細胞の正確な制御は、前記免疫調節剤またはCARもしくはTCRによって引き起こされる望ましくない副作用を軽減することにおいて有用であろう。
第2の哺乳類免疫細胞はPBMC、T細胞、またはNK細胞である。一部の実施形態において、前記CARは、EGFRvIII等の腫瘍抗原を標的とする。
本発明の医薬組成物は治療目的に対して有用である。従って、免疫調節剤または他の治療用タンパク質を発現する産生細胞等の改変哺乳類細胞を含む他の組成物とは異なり、本発明の医薬組成物は、個体への投与に適した薬理学的に許容される賦形剤を含む。
る方法が当該分野で公知である。一部の実施形態において、前記医薬組成物は、細胞培養において使用される前記改変哺乳類免疫細胞以外の動物源のタンパク質など、アレルギー性の効果をもたらしうる外来タンパク質を実質的に含まない。一部の実施形態において、「実質的に含まない」とは、医薬組成物の総容量または重量に対し約10%、5%、1%、0.1%、0.01%、0.001%、1ppm、またはそれ未満、のいずれかよりも少ないことを意味する。一部の実施形態において、前記医薬組成物はGMPレベルの工場で調製される。一部の実施形態において、非経口投与用の前記医薬組成物に含まれるエンドトキシンは、約5EU/kg体重/時間(EU/kg body weight/hr)未満である。一部の実施形態において、静脈内投与用の前記医薬組成物中の改変哺乳類細胞の少なくとも約70%は生存している。一部の実施形態において、前記医薬組成物は、米国薬局方(USP)に記載の14日間直接接種試験法(14-day direct inoculation test method)を用いた評価において、「増殖なし(no growth)」の結果となる。一部の実施形態において、前記医薬組成物の投与に先立って、前記改変哺乳類細胞および前記薬理学的に許容される賦形剤の両者を含んだ試料を、最終採取の約48〜72時間前に(または培養物の最後の再供給と同時に)無菌性試験のために採取すべきである。一部の実施形態において、前記医薬組成物にはマイコプラズマの混入がない。一部の実施形態において、前記医薬組成物は検出可能な微生物因子を含まない。一部の実施形態において、前記医薬組成物は、HIV−I型、HIV−II型、HBV、HCV、ヒトTリンパ好性ウイルスI型、およびヒトTリンパ好性ウイルスII型等の感染症因子を含まない。
本明細書に記載される医薬組成物のいずれかを調製する方法がさらに提供され、該方法は、前記異種核酸を含むベクターを哺乳類細胞に導入することを含む。
ト阻害剤等)をコードする配列を有する自己不活性化レンチウイルスベクター、および/またはキメラ抗原受容体を有する自己不活性化レンチウイルスベクターを、当該分野で公知のプロトコルによりパッケージングすることができる。得られたレンチウイルスベクターを、当該分野で公知の手法を用いて哺乳類細胞(初代ヒトT細胞等)の形質導入に使用することができる。
化学的、または生物学的方法で宿主細胞に導入することができる。
なレポーターアッセイは、自家開発の安定なレポーター腫瘍細胞に対して行うことができる。改変T細胞の場合、サイトカイン放出アッセイを、該改変T細胞による免疫チェックポイント阻害剤の分泌に応じたT細胞回復レベルの検出のために行うことができる。改変CAR−T細胞の場合、腫瘍細胞へのCAR−T細胞毒性の増強に対する免疫調節剤(免疫チェックポイント阻害剤等)分泌の能力をin vitro共培養アッセイによりアッセイすることができ、該アッセイにおいては、T細胞と腫瘍細胞の共培養をいくつかの比率で一定時間行う。
本出願の一側面は、上記医薬組成物のいずれかを使用した癌の治療方法に関する。
、前記改変哺乳類細胞は、さらに、治療用タンパク質(第2の免疫調節剤、例えば免疫賦活剤;または、免疫調節剤ではない治療用タンパク質、例えば抗HER2抗体などの化学療法抗体等)をコードする第2の異種核酸を含む。一部の実施形態において、前記免疫賦活剤はIL−2、IL−7、IL−15、IL−21、IL−12、CCR4、CCR2b、ヘパラナーゼ、CD137L、LEM、およびBcl−2からなる群より選択される。一部の実施形態において、前記CARは、第2のプロモーターに機能的に連結した第3の異種核酸によってコードされる。一部の実施形態において、前記第2のプロモーターは構成的プロモーターである。一部の実施形態において、前記第2のプロモーターは、例えば、誘導物質(例えばテトラサイクリンまたはドキシサイクリンなどの小分子等)、照射、温度、酸化還元状態、腫瘍環境、および前記改変哺乳類免疫細胞の活性化状態、から選択される誘導条件によって誘導可能である。一部の実施形態において、前記CARまたはTCRは、EGFRvIII等のEGFR、BCMA、またはNY−ESO−1等の腫瘍抗原を標的とする。一部の実施形態において、前記改変哺乳類免疫細胞は前記個体から得られる。一部の実施形態において、前記改変哺乳類免疫細胞は前記個体に対し同種異系である。
において、前記改変哺乳類細胞は免疫細胞(PBMC、NK細胞、またはT細胞等)である。一部の実施形態において、前記改変哺乳類細胞は幹細胞である。一部の実施形態において、前記プロモーターは、誘導物質(例えばテトラサイクリンまたはドキシサイクリンなどの小分子等)、照射、温度、酸化還元状態、腫瘍環境、および前記改変哺乳類細胞の活性化状態、から選択される誘導条件によって誘導可能である。一部の実施形態において、前記免疫調節剤は免疫チェックポイント阻害剤(CTLA−4の阻害剤またはPD−1の阻害剤等)である。一部の実施形態において、前記免疫調節剤は免疫賦活剤である。一部の実施形態において、前記免疫調節剤は分泌タンパク質である。一部の実施形態において、前記免疫調節剤は抗体(全長抗体、scFv、単一ドメイン抗体、重鎖のみ抗体、またはFab等)である。一部の実施形態において、前記改変哺乳類細胞は、さらに、腫瘍抗原を認識するターゲティング分子をその表面上に発現する。一部の実施形態において、前記改変哺乳類細胞は、さらに、治療用タンパク質(第2の免疫調節剤、例えば免疫賦活剤;または、免疫調節剤ではない治療用タンパク質、例えば化学療法抗体;等)をコードする第2の異種核酸を含む。一部の実施形態において、前記免疫賦活剤はIL−2、IL−7、IL−15、IL−21、IL−12、CCR4、CCR2b、ヘパラナーゼ、CD137L、LEM、およびBcl−2からなる群より選択される。一部の実施形態において、前記第2の哺乳類免疫細胞はPBMC、T細胞、またはNK細胞である。一部の実施形態において、前記CARまたはTCRは、EGFRvIII等のEGFR、BCMA、またはNY−ESO−1等の腫瘍抗原を標的とする。一部の実施形態において、前記改変哺乳類細胞および/または前記第2の哺乳類免疫細胞は、前記個体から得られる。一部の実施形態において、前記改変哺乳類細胞および/または前記第2の哺乳類免疫細胞は、前記個体に対し同種異系である。
原を標的とする。一部の実施形態において、前記改変哺乳類細胞および/または前記第2の哺乳類免疫細胞は、前記個体から得られる。一部の実施形態において、前記改変哺乳類細胞および/または前記第2の哺乳類免疫細胞は、前記個体に対し同種異系である。
cancer)、肝臓癌、肺癌(小細胞肺癌、非小細胞肺癌、肺腺癌、および肺扁平上皮癌等)、リンパ腫(T細胞リンパ腫を除く)、髄芽腫、メラノーマ、中皮腫、転移性扁平上皮頸部癌、口腔癌(mouth cancer)、多発性内分泌腫瘍症候群、骨髄異形成症候群、骨髄異形成性/骨髄増殖性疾患、鼻腔および副鼻腔癌、上咽頭癌、神経芽細胞腫、神経内分泌癌、口腔咽頭癌、卵巣癌(卵巣上皮癌、卵巣胚細胞腫瘍、卵巣低悪性度腫瘍等)、膵臓癌、副甲状腺癌、陰茎癌、腹膜癌、咽頭癌、褐色細胞腫、松果体芽腫およびテント上未分化神経外胚葉腫瘍、下垂体腫瘍、胸膜肺芽腫、原発性中枢神経系リンパ腫(小膠細胞腫)、肺リンパ管筋腫症、直腸癌、腎臓癌、腎盂尿管癌(移行細胞癌)、横紋筋肉腫、唾液腺癌、皮膚癌(非メラノーマ(扁平上皮癌(squamous cell carcinoma)等)、メラノーマ、およびメルケル細胞癌等)、小腸癌、扁平上皮癌(squamous cell cancer)、精巣癌、咽喉癌、甲状腺癌、結節性硬化症、尿道癌、腟癌、外陰癌、ウィルムス腫瘍、母斑症関連異常血管増殖、浮腫(脳腫瘍関連等)、ならびにメーグス症候群を含むが、これらに限定されない。
に直接注射される。一部の実施形態において、前記医薬組成物は腫瘍部位に局所的に、例えば直接腫瘍細胞に、または腫瘍細胞を有する組織に投与される。
EM、およびBcl−2からなる群より選択される。一部の実施形態において、前記CARは、第2のプロモーターに機能的に連結した第3の異種核酸によってコードされる。一部の実施形態において、前記第2のプロモーターは構成的プロモーターである。一部の実施形態において、前記第2のプロモーターは、例えば、誘導物質(例えばテトラサイクリンまたはドキシサイクリンなどの小分子等)、照射、温度、酸化還元状態、腫瘍環境、および前記改変哺乳類免疫細胞の活性化状態、から選択される誘導条件によって誘導可能である。一部の実施形態において、前記CARまたはTCRは、EGFRvIII等の腫瘍抗原を標的とする。一部の実施形態において、前記改変哺乳類免疫細胞は前記個体から得られる。一部の実施形態において、前記改変哺乳類免疫細胞は前記個体に対し同種異系である。一部の実施形態において、前記固形癌は、メラノーマ、乳癌、肺癌、肝臓癌、白血病、リンパ腫、胃癌、結腸癌、骨癌、脳腫瘍、膵臓癌、および卵巣癌からなる群より選択される。一部の実施形態において、前記医薬組成物は輸液により投与される。一部の実施形態において、前記医薬組成物は腫瘍内注射により投与される。
提供される。一部の実施形態において、前記改変哺乳類細胞を含む医薬組成物は、前記第2の改変哺乳類免疫細胞を含む医薬組成物の投与よりも先に投与される。一部の実施形態において、前記改変哺乳類細胞を含む医薬組成物は、前記第2の改変哺乳類免疫細胞を含む医薬組成物の投与よりも後に投与される。一部の実施形態において、前記改変哺乳類細胞は免疫細胞(PBMC、NK細胞、またはT細胞等)である。一部の実施形態において、前記改変哺乳類細胞は幹細胞である。一部の実施形態において、前記プロモーターは、誘導物質(例えばテトラサイクリンまたはドキシサイクリンなどの小分子等)、照射、温度、酸化還元状態、腫瘍環境、および前記改変哺乳類細胞の活性化状態、から選択される誘導条件によって誘導可能である。一部の実施形態において、前記免疫調節剤は免疫チェックポイント阻害剤(CTLA−4の阻害剤またはPD−1の阻害剤等)である。一部の実施形態において、前記免疫調節剤は免疫賦活剤である。一部の実施形態において、前記免疫調節剤は分泌タンパク質である。一部の実施形態において、前記免疫調節剤は抗体(全長抗体、scFv、単一ドメイン抗体、重鎖のみ抗体、またはFab等)である。一部の実施形態において、前記改変哺乳類細胞は、さらに、腫瘍抗原を認識するターゲティング分子をその表面上に発現する。一部の実施形態において、前記改変哺乳類細胞は、さらに、治療用タンパク質(第2の免疫調節剤、例えば免疫賦活剤;または、免疫調節剤ではない治療用タンパク質、例えば化学療法抗体等)をコードする第2の異種核酸を含む。一部の実施形態において、前記免疫賦活剤はIL−2、IL−7、IL−15、IL−21、IL−12、CCR4、CCR2b、ヘパラナーゼ、CD137L、LEM、およびBcl−2からなる群より選択される。一部の実施形態において、前記第2の哺乳類免疫細胞はPBMC、T細胞、またはNK細胞である。一部の実施形態において、前記CARまたはTCRは、EGFRvIII等の腫瘍抗原を標的とする。一部の実施形態において、前記改変哺乳類細胞および/または前記第2の哺乳類免疫細胞は、前記個体から得られる。一部の実施形態において、前記改変哺乳類細胞および/または前記第2の哺乳類免疫細胞は、前記個体に対し同種異系である。一部の実施形態において、前記固形癌は、メラノーマ、乳癌、肺癌、肝臓癌、白血病、リンパ腫、胃癌、結腸癌、骨癌、脳腫瘍、膵臓癌、および卵巣癌からなる群より選択される。一部の実施形態において、前記医薬組成物は輸液により投与される。一部の実施形態において、前記医薬組成物は腫瘍内注射により投与される。
2b、ヘパラナーゼ、CD137L、LEM、およびBcl−2からなる群より選択される。一部の実施形態において、前記改変哺乳類細胞は前記個体から得られる。一部の実施形態において、前記改変哺乳類細胞は前記個体に対し同種異系である。一部の実施形態において、前記液性癌は白血病またはリンパ腫である。一部の実施形態において、前記医薬組成物は輸液により投与される。
態において、前記免疫調節剤は抗体(全長抗体、scFv、単一ドメイン抗体、重鎖のみ抗体、またはFab等)である。一部の実施形態において、前記改変哺乳類細胞は、さらに、腫瘍抗原を認識するターゲティング分子をその表面上に発現する。一部の実施形態において、前記改変哺乳類細胞は、さらに、治療用タンパク質(第2の免疫調節剤、例えば免疫賦活剤;または、免疫調節剤ではない治療用タンパク質、例えば化学療法抗体等)をコードする第2の異種核酸を含む。一部の実施形態において、前記免疫賦活剤はIL−2、IL−7、IL−15、IL−21、IL−12、CCR4、CCR2b、ヘパラナーゼ、CD137L、LEM、およびBcl−2からなる群より選択される。一部の実施形態において、前記第2の哺乳類免疫細胞はPBMC、T細胞、またはNK細胞である。一部の実施形態において、前記CARまたはTCRは、BCMAまたはNY−ESO−1等の腫瘍抗原を標的とする。一部の実施形態において、前記改変哺乳類細胞および/または前記第2の哺乳類免疫細胞は、前記個体から得られる。一部の実施形態において、前記改変哺乳類細胞および/または前記第2の哺乳類免疫細胞は、前記個体に対し同種異系である。一部の実施形態において、前記液性癌は白血病またはリンパ腫である。一部の実施形態において、前記医薬組成物は輸液により投与される。
節剤および/または他の治療用タンパク質の発現を誘導することを含む。前記改変哺乳類細胞および/または前記第2の哺乳類免疫細胞の誘導は、個体への投与前に、または個体への投与後に行われうる。一部の実施形態において、前記プロモーターは誘導条件によって誘導可能であり、前記方法はさらに該誘導条件を個体に適用することを含む。一部の実施形態において、前記プロモーターは誘導物質(例えばテトラサイクリンまたはドキシサイクリンなどの小分子等)によって誘導可能であり、前記方法はさらに、該誘導物質の有効量を個体に投与して免疫調節剤および/または他の治療用タンパク質の発現を誘導することを含む。一部の実施形態において、前記誘導物質は全身投与される。一部の実施形態において、前記誘導物質は腫瘍部位に局所的に、例えば直接腫瘍細胞に、または腫瘍細胞を有する組織に、投与される。一部の実施形態において、前記プロモーターは照射(光または電離放射線等)によって誘導可能であり、前記方法はさらに、個体に、例えば全身に、または腫瘍部位に局所的に、照射を適用することを含む。一部の実施形態において、前記プロモーターは熱によって誘導可能であり、前記方法はさらに、個体に、例えば腫瘍部位に局所的に、熱を加えることを含む。
の治療法が提供される。一部の実施形態において、前記改変哺乳類免疫細胞はPBMC、T細胞、またはNK細胞である。一部の実施形態において、前記プロモーターは、例えばCARまたはTCRの細胞内シグナル伝達ドメイン等によって、誘導可能である。一部の実施形態において、前記プロモーターは、IL−2プロモーター、NFATプロモーター、またはNFκBプロモーター等の、T細胞活性化依存プロモーターである。一部の実施形態において、前記免疫調節剤は免疫チェックポイント阻害剤(CTLA−4の阻害剤またはPD−1の阻害剤等)である。一部の実施形態において、前記免疫調節剤は免疫賦活剤である。一部の実施形態において、前記免疫調節剤は分泌タンパク質である。一部の実施形態において、前記免疫調節剤は抗体(全長抗体、scFv、単一ドメイン抗体、重鎖のみ抗体、またはFab等)である。一部の実施形態において、前記改変哺乳類細胞は、さらに、治療用タンパク質(第2の免疫調節剤、例えば免疫賦活剤;または、免疫調節剤ではない治療用タンパク質、例えば抗HER2抗体などの化学療法抗体等)をコードする第2の異種核酸を含む。一部の実施形態において、前記免疫賦活剤はIL−2、IL−7、IL−15、IL−21、IL−12、CCR4、CCR2b、ヘパラナーゼ、CD137L、LEM、およびBcl−2からなる群より選択される。一部の実施形態において、前記CARは、第2のプロモーターに機能的に連結した第3の異種核酸によってコードされる。一部の実施形態において、前記第2のプロモーターは構成的プロモーターである。一部の実施形態において、前記第2のプロモーターは、例えば、誘導物質(例えばテトラサイクリンまたはドキシサイクリンなどの小分子等)、照射、温度、酸化還元状態、腫瘍環境、および前記改変哺乳類免疫細胞の活性化状態、から選択される誘導条件によって誘導可能である。一部の実施形態において、前記CARまたはTCRは、EGFRvIII等のEGFR、BCMA、またはNY−ESO−1等の腫瘍抗原を標的とする。一部の実施形態において、前記改変哺乳類免疫細胞は前記個体から得られる。一部の実施形態において、前記改変哺乳類免疫細胞は前記個体に対し同種異系である。一部の実施形態において、前記癌は、白血病、リンパ腫、メラノーマ、乳癌、肺癌、肝臓癌、白血病、リンパ腫、胃癌、結腸癌、骨癌、脳腫瘍、膵臓癌、および卵巣癌からなる群より選択される。一部の実施形態において、前記医薬組成物は(輸液等により)全身投与され、前記プロモーターは腫瘍部位で(例えば誘導物質の局所投与により、または局所的な加熱もしくは照射により)局所的に誘導される。
1、IL−12、CCR4、CCR2b、ヘパラナーゼ、CD137L、LEM、およびBcl−2からなる群より選択される。一部の実施形態において、前記第2の哺乳類免疫細胞はPBMC、T細胞、またはNK細胞である。一部の実施形態において、前記CARまたはTCRは、EGFRvIII等のEGFR、BCMA、またはNY−ESO−1等の腫瘍抗原を標的とする。一部の実施形態において、前記改変哺乳類細胞および/または前記第2の哺乳類免疫細胞は、前記個体から得られる。一部の実施形態において、前記改変哺乳類細胞および/または前記第2の哺乳類免疫細胞は、前記個体に対し同種異系である。一部の実施形態において、前記癌は、白血病、リンパ腫、メラノーマ、乳癌、肺癌、肝臓癌、白血病、リンパ腫、胃癌、結腸癌、骨癌、脳腫瘍、膵臓癌、および卵巣癌からなる群より選択される。一部の実施形態において、前記医薬組成物は(輸液等により)全身投与され、前記プロモーターは腫瘍部位で(例えば誘導物質の局所投与により、または局所的な加熱もしくは照射により)局所的に誘導される。
本明細書に記載される医薬組成物のいずれかを含有するキット、単位用量(unit dosages)、および製品がさらに提供される。
変哺乳類免疫細胞がさらにCARまたはTCRを発現する、改変哺乳類免疫細胞;およびb)薬理学的に許容される賦形剤、を含む医薬組成物と、(2)該医薬組成物を使用するための説明書と、を含むキットが提供される。一部の実施形態において、前記改変哺乳類免疫細胞はPBMC、T細胞、またはNK細胞である。一部の実施形態において、前記プロモーターは、例えばCARまたはTCRの細胞内シグナル伝達ドメイン等によって、誘導可能である。一部の実施形態において、前記プロモーターは、IL−2プロモーター、NFATプロモーター、またはNFκBプロモーター等の、T細胞活性化依存プロモーターである。一部の実施形態において、前記免疫調節剤は免疫チェックポイント阻害剤(CTLA−4の阻害剤またはPD−1の阻害剤等)である。一部の実施形態において、前記免疫調節剤は免疫賦活剤である。一部の実施形態において、前記免疫調節剤は分泌タンパク質である。一部の実施形態において、前記免疫調節剤は抗体(全長抗体、scFv、単一ドメイン抗体、重鎖のみ抗体、またはFab等)である。一部の実施形態において、前記改変哺乳類細胞は、さらに、治療用タンパク質(第2の免疫調節剤、例えば免疫賦活剤;または、免疫調節剤ではない治療用タンパク質、例えば抗HER2抗体などの化学療法抗体等)をコードする第2の異種核酸を含む。一部の実施形態において、前記免疫賦活剤はIL−2、IL−7、IL−15、IL−21、IL−12、CCR4、CCR2b、ヘパラナーゼ、CD137L、LEM、およびBcl−2からなる群より選択される。一部の実施形態において、前記CARは、第2のプロモーターに機能的に連結した第3の異種核酸によってコードされる。一部の実施形態において、前記第2のプロモーターは構成的プロモーターである。一部の実施形態において、前記第2のプロモーターは、例えば、誘導物質(例えばテトラサイクリンまたはドキシサイクリンなどの小分子等)、照射、温度、酸化還元状態、腫瘍環境、および前記改変哺乳類免疫細胞の活性化状態、から選択される誘導条件によって誘導可能である。一部の実施形態において、前記CARまたはTCRは、EGFRvIII等のEGFR、BCMA、またはNY−ESO−1等の腫瘍抗原を標的とする。一部の実施形態において、前記キットはさらに誘導物質を含む。
機能的な、または減弱化した一次細胞内シグナル伝達ドメイン(変異CD3ζ等)を含む。
はPBMC、T細胞、またはNK細胞である。一部の実施形態において、前記CARまたはTCRは、EGFRvIII等のEGFR、BCMA、またはNY−ESO−1等の腫瘍抗原を標的とする。一部の実施形態において、前記医薬組成物と、前記第2の哺乳類免疫細胞を含む組成物とが、投与の前に混合される。一部の実施形態において、前記キットはさらに誘導物質を含む。
載の併用療法を含む製品およびキットも企図される。
構成的プロモーターhEF1α、ドキシサイクリン誘導性プロモーター(TETON(登録商標)等)、NFAT依存誘導性プロモーター、または熱誘導性プロモーター(ヒト熱ショックタンパク質70プロモーターHSP70p等)によって駆動される抗体遺伝子を有する自己不活性化レンチウイルスベクターを設計および調製した。各抗体遺伝子は、PD−1、CTLA−4、および任意の他の目的とする標的から選択される、固有の抗原に対して特異的に抗体を発現することができる。初代ヒト末梢血単核球(PBMC)を、健常ドナー由来の末梢血を密度勾配遠心することで調製した。磁気ビーズ分離を用いてPBMCからヒト初代T細胞を精製し、プレ活性化した。次いで、プレ活性化されたT細胞に前記レンチウイルスベクターを形質導入し、ex vivoで数日間増殖させた。あるいは、293−6E細胞、Jurkat細胞、間葉系幹細胞、精製ヒトB細胞、および他のPBMC細胞等の他の宿主細胞に前記レンチウイルスベクターを形質導入し、抗体の発現に使用することができる。抗体の分泌を、均一性時間分解蛍光(homogenous time-resolved fluorescence)(HTFR)法を用いて検出した。あるいは、分泌された抗体の検出を、組換え抗原タグタンパク質により、酵素結合イムノソルベントアッセイ(ELISA)で行うことができる。分泌された抗体の生物活性を、in vitroレポーターアッセイを用いて評価した。
pMDLg/pRRE(Addgene #12251)、pRSV−Rev(Addgene #12253)、およびpMD2.G(Addgene #12259)を含むレンチウイルスパッケージングプラスミド混合物を、抗体発現プラスミドpLLV−プロモーター−抗PD−1ベクター(すなわち、pLLV−hEF1α−抗PD−1、pLLV−TetOn−抗PD−1、pLLV−NFAT−抗PD−1、もしくはpLLV−HSP70p−抗PD−1)、またはpLLV−プロモーター−抗CTLA−4ベクター(すなわち、pLLV−hEF1α−抗CTLA−4、pLLV−TetOn−抗CTLA−4、pLLV−NFAT−抗CTLA−4、もしくはpLLV−HSP70p−抗CTLA−4)と、ポリエーテルイミド(PEI)と共に、あらかじめ最適化された比率で予備混合した後、適切に混合して室温で5分間インキュベートした。次いで、トランスフェクション混合物をHEK293細胞に滴下し、おだやかに混合した。その後、細胞を37℃の5%CO2細胞インキュベーター内で一晩インキュベートした。4℃にて500g
で10分間遠心分離した後、上清を回収した。
白血球を回収し、細胞濃度をR10培地内で5×106細胞/mLに調整した。その後、白血球を0.9%NaCl溶液と1:1(v/v)の比率で混合した。3mLのlymphoprep培地を15mLの遠心管に加え、lymphoprep上に6mLの希釈リンパ球混合物をゆっくりと重層した。リンパ球混合物を800gで30分間、20℃にてブレーキ無しで遠心分離した。その後、リンパ球のバフィーコートを200μLのピペットで回収した。回収した画分を、少なくとも6倍の0.9%NaClまたはR10で希釈し、溶液の密度を低下させた。その後、回収した画分を250gで10分間、20℃にて遠心分離した。上清を完全に吸引し、10mLのR10を細胞ペレットに加えた。該混合物をさらに250gで10分間、20℃にて遠心分離した。その後、上清を吸引した。37℃にあらかじめ温めた2mLのR10と共に100IU/mLのIL−2を細胞ペレットに加え、該細胞ペレットを静かに再懸濁した。細胞数を計数し、後の実験で使用可能なPBMC試料とした。
Miltenyi Pan T cell isolation kit (Cat#130−096−535)を用いて、製造者が提供するプロトコルに従い、次のようにPBMCからヒトT細胞を精製した。細胞数を最初に測定した。細胞懸濁物を300gで10分間遠心分離した。上清を完全に吸引し、細胞ペレットを107個の全細胞あたり40μLの緩衝液に再懸濁した。107個の全細胞あたり10μLのPan T Cell Biotin−Antibody Cocktailを添加し、十分に混合し、約5分間冷蔵庫(2〜8℃)内でインキュベートした。その後、107個の細胞あたり30μLの緩衝液を添加した。107個の細胞あたり20μLのPan T Cell MicroBead Cocktailを添加した。該混合物をよく混合し、冷蔵庫(2〜8℃)内でさらに10分間インキュベートした。最低でも500μLが磁気分離に必要であった。LSカラムを適したMACS分離装置の磁場に設置した。3mLの緩衝液でリンスすることでカラムの準備を行った。その後、細胞懸濁物を該カラム上にアプライし、濃縮されたT細胞画分に相当する非標識細胞を含むフロースルーを回収した。次いで、3mLの緩衝液でカラムを洗浄することによりT細胞を回収し、その際、濃縮されたT細胞に相当する通過分の非標識細胞を回収して、先の工程のフロースルーと合わせた。その後、T細胞をR10+100IU/mL IL−2中に再懸濁した。human T Cell Activation/Expansion Kit(Miltenyi #130−091−441)を用いて、該初代T細胞を形質導入に先立って3日間プレ活性化した。
初代ヒトB細胞も磁気ビーズ分離法により調製した。PBMCを上記の通り密度勾配遠心分離によって調製した。Miltenyi Human B cell isolation kit(Cat#130−091−151)を用いて、上記ヒトT細胞の調製と類似のプロトコルに従い、PBMCからヒトB細胞を精製した。単離されたヒトB細胞を、ex vivoで、組換えCD40とIL4タンパク質を添加したRPMI1640培地(Martina et al. PLoS ONE 3(1): e1464 (2008))中で培養した。
初代ヒトNK細胞を磁気ビーズ分離法により調製した。PBMCを上記の通り密度勾配遠心によって調製した。Miltenyi Human NK cell isolation kit (Cat#130−092−657)を用いて、上記ヒトT細胞の調製と類似のプロトコルに従い、PBMCからヒトNK細胞を精製した。単離されたヒトNK細胞を、ex vivoで、200IU/mLの組換えIL2タンパク質を添加したα−MEM培地中で培養した。
ヒトドナーの骨髄を局所麻酔下で腸骨稜から吸引によって採取し、次いで単核球をFicoll分離法により単離する。その後、細胞を洗浄し、MSC培地(ダルベッコ改変イーグル培地−低グルコース/ペニシリン/ストレプトマイシン/10%ウシ胎児血清)中に再懸濁し、組織培養フラスコに播種して、37℃、5%CO2でインキュベートする。MSCを、MSC増殖のための標準化LUMCプロトコルに従い増殖させる。週に2回、培養物を顕微鏡観察し、培地を入れ替える。>70%コンフルエンスに達したら細胞をトリプシン処理し、各種サイズのMSCの半製品(half products)(継代1)を10%ジメチルスルホキシドと共に凍結保存する。
初代ヒトT細胞、精製ヒトB細胞、および精製ヒトNK細胞を含む宿主細胞への、段階希釈したウイルスストックによる形質導入を、7μg/mLのポリブレンの存在下で、1200g、32℃、1.5時間の遠心分離によって行った。その後、トランスフェクトされた細胞を細胞培養インキュベーターに移し、適した誘導条件下で導入遺伝子の発現を行う。具体的には、pLLV−hEF1α−抗PD−1またはpLLV−hEF1α−抗CTLA−4を形質導入した宿主細胞を、誘導物質または誘導条件無しで48時間インキュベートした。pLLV−TetOn−抗PD−1またはpLLV−TetOn−抗CTLA−4を形質導入した宿主細胞を、各種濃度(0〜12μg/mL)のドキシサイクリンと共に48時間インキュベートした。pLLV−NFAT−抗PD−1またはpLLV−NFAT−抗CTLA−4を形質導入した宿主細胞を、各種濃度(0〜50ng/mL PMA・1000ng/mL PHA−P)のT細胞活性化組成物PMA/PHA−Pと共に48時間インキュベートした。pLLV−HSP70p−抗PD−1またはpLLV−HSP70p−抗CTLA−4を形質導入した宿主細胞に、形質導入直後、温度を制御したウォーターバス中で、37℃、39℃、41℃、43℃、または44℃の20分間の熱ショックを加えた。熱ショック後、細胞を6ウェルプレートに播種し直し、37℃、5%CO2の細胞培養インキュベーターでさらに3日間増殖させた。温度誘導性プロモーターを有する他の構築物については、宿主細胞を24〜72時間インキュベートした後、最適温度(37℃〜45℃等)において一定時間(例えば10分間、20分間または30分間等)誘導することができる。
℃で608.4ng/mL)と比べ、有意に高い濃度で分泌された(39℃で709.5ng/mL、41℃で844.2ng/mL、43℃で866.8ng/mL、44℃で957.8ng/mL)。
レポーター細胞株の作製
安定的にPD−1を発現するレポーター細胞株を確立した。簡単に記載すると、レンチウイルスベクターの改変を、pLVX−Puro(Clontech #632164)を用いて、GenScriptにより、本来のプロモーターをヒト伸長因子1αプロモーター(hEF1α)で置き換え、ピューロマイシン耐性遺伝子をEcoRIおよびXbaIを有するT2A連結G418耐性遺伝子で置き換えることによって行った。該ベクターは、pLLV−hEF1α−T2A−G418Rと命名され、分子クローニングの際にいくつかの新たな制限部位MluI、HpaI、およびBamHIが該ベクターに加えられた。ヒトPD−1遺伝子(NCBI参照配列番号:NM_005018.2)配列を、EcoRI/HpaIを介して、pLLV−hEF1α−T2A−G418Rベクターにクローニングし、pLLV−hEF1α−PD−1−T2A−G418Rを提供した。これをさらに、上記実施例中に記載されるようにレンチウイルスパッケージング手順で処理した。研究室内で以前に確立した宿主細胞株Jurkat/NFAT.Luc(ピューロマイシン耐性)への、PD−1遺伝子を有するレンチウイルスによる形質導入を、7μg/mLのポリブレンの存在下で、1200g、32℃、1.5時間の遠心分離によって行った。その後、形質導入された細胞を細胞培養インキュベーターに移し、適した誘導条件下で導入遺伝子の発現を行った。陽性細胞をネオマイシン(G418)により選択し、単一クローンを限界希釈法によって選択した。最良のクローンを、抗PD−1抗体を用いてFACSにより選別した。図12Aに示す通り、Jurkat/NFAT.Luc−PD−1と命名された陽性クローン(Jurkat.NFAT.Luc.PD1としても知られる)の例では95.2%がPD−1タンパク質を発現した。
A.Luc.CTLA4としても知られる)の例では30.6%がCTLA−4タンパク質を発現した。
分泌された抗PD−1抗体の結合親和性を、安定細胞株Jurkat/NFAT.Luc−PD−1上で発現するPD−1タンパク質への結合によって測定した。簡単に記載すると、5×105個のJurkat/NFAT.Luc−PD−1細胞を、実施例1の改変宿主細胞から分泌された抗PD−1抗体(0、0.1、0.3、1、3、10μg/mL)を含む段階希釈した上清と共に、室温で2時間インキュベートした。細胞洗浄を数サイクル行った後、ヒトIgGに対するフルオロフォア標識二次抗体を加え、細胞に結合した抗PD−1抗体をFACSにより検出した。Jurkat/CTLA−4細胞を陰性対照として使用した。
T細胞活性化の回復に対する抗PD−1抗体の効果を評価するため、1〜5×105個のJurkat/NFAT.Luc−PD−1レポーター細胞を、CHO/PD−L1細胞と共に、異なるE/T比(1:1、10:1、20:1、1:10、1:20等)で、実施例1の改変宿主細胞から分泌された抗PD−1抗体(例えば、world wide web.Drugbank.ca上のアクセッション番号DB09037の配列を有するペムブロリズマブ等)の存在下で、一定時間(4時間から72時間等)インキュベートする。並行して、無関係な遺伝子を有するレンチウイルスベクターを同一の細胞株に形質導入し、陰性対照とする。
ONE−GLO(商標)ルシフェラーゼアッセイ試薬を、共培養した細胞に加える。相対発光単位(relative light unit)(RLU)で測定したアッセイウェルからのルシフェラーゼ活性を、各レポーター細胞の活性化の度合いとする。
ヒトリンパ腫細胞株Raji(ATCC、#CCL86)を、使用説明書に従い、10%FBSを添加したRPMI1640培地で培養した。Raji細胞は、CTLA−4リガンドであるCD80およびCD86を高レベルで発現することが示されている(International Immunology 10(4):499-506. May 1998)。抗CTLA−4抗体を、ヒトHEK293T細胞への、イピリムマブ全長IgGコード配列(world wide web.Drugbank.ca、アクセッション番号:Ipilimumab DB06186)を有するレンチウイルスベクターの形質導入によって得た。該形質導入細胞を抗CTLA4の分泌に適した条件下で培養し、上清を回収して、細胞に基づいたアッセイに用いた。1×105個のJurkat/IL−2プロモーター.Luc.CTLA−4細胞を96ウェルアッセイプレート(Corning #3610)に播種し、次いで最終濃度20μg/mLで抗ROR1または抗CTLA−4を加え、37℃の細胞培養インキュベーター内で1時間インキュベートした。その後、3.2×105個のRaji細胞を各ウェルに加え、共培養アッセイをさらに24または48時間継続した。共培養完了後、各ウェル内のルシフェラーゼ活性を、ONE−GLO(商標)ルシフェラーゼ活性アッセイキット(Promega #E6110)を用いて、使用説明書に従い測定した。プレートをPHERSTAR(商標) Plusマイクロプレートリーダー上で読み取った。図14に示すように、前記共培養アッセイを48時間行った場合、抗CTLA−4(イピリムマブ)は、関連の無い抗体(抗ROR1)との比較において、約2.4倍高いRLUシグナルを示した(27,914.00±3431.00RLU対11,585.00±303.00、平均値±標準誤差)。このデータは、上記CTLA−4レポーターアッセイにおいて、CTLA−4遮断がIL−2プロモーター駆動ルシフェラーゼ遺伝子発現を強力に回復させることができたことを示唆する。
PD−L1遺伝子および1個の標的遺伝子(EGFRvIII)、ならびにルシフェラーゼ遺伝子を、レンチウイルスベクターを用いて、ヒト膠芽腫腫瘍細胞株(U87MG)に導入した。簡単に記載すると、ヒトPD−L1遺伝子(NCBI参照配列番号:NM_014143.3)配列を、EcoRI/HpaIを介して、pLLV−hEF1α−T2A−G418Rベクターにクローニングし、ベクターpLLV−hEF1α−PD−L1−T2A−G418Rを提供した。これをさらに、上記のようにレンチウイルスパッケージング手順で処理した。ヒト上皮成長因子受容体バリアントIII(EGFR vIII)ヌクレオチド配列を、野生型EGFR(NM_005228)からエクソン2〜7を欠失させ、それによってコード配列の801塩基対をインフレーム欠失させ、繋ぎ目で新規グリシン残基を生成することによって得た(Endocrine-Related Cancer(2001)883-96)。EGFR vIIIヌクレオチド配列を、XhoI/XbaI制限酵素でpLVX−P
uroベクターにクローニングし、pLVX−EGFRvIIIベクターを提供した。これをさらに、上記のようにレンチウイルスパッケージング処理した。陽性細胞をG418とピューロマイシンによりスクリーニングし、単一クローンを限界希釈法によって得た。
示されるように、また、陰性対照との比較におけるIL−2またはIFN−ガンマの分泌の増加によって示されるように、T細胞活性化と細胞毒性の回復をもたらす可能性がある。このような効果はエフェクター/標的細胞比に対して用量依存的である可能性がある。
抗EGFRvIII−CAR構築物(GSI026)を設計し、レンチウイルスベクターに導入した。該抗EGFRvIII−CAR遺伝子は、N末端からC末端に向かって、CD8αシグナルペプチド、ヒト化抗EGFRvIII scFv、CD8αヒンジおよび膜貫通(TM)領域、CD137細胞質ドメイン(CD137 cyto)、およびCD3ζを含んだ全長抗EGFRvIII CARを含む。中国特許出願第CN201611039855.0号を参照のこと。次のT細胞の実験群を作製した:(1)T/GSI026:抗EGFRvIII−CAR遺伝子(GSI026)を有するレンチウイルスベクターを形質導入した改変ヒト初代T細胞;(2)T/抗PD−1:NFATプロモーターの制御下の抗PD−1抗体(ペムブロリズマブ)遺伝子を有するレンチウイルスベクターを形質導入した改変ヒト初代T細胞;(3)T/GSI026・抗PD−1:抗EGFRvIII−CAR遺伝子(GSI026)とNFATプロモーターの制御下の抗PD−1抗体(ペムブロリズマブ)遺伝子との両者を有するレンチウイルスベクターを形質導入した改変ヒト初代T細胞;および(4)UnT:陰性対照としての無関係の遺伝子を形質導入した改変ヒト初代T細胞。さらに、抗EGFRvIII−CAR遺伝子を有するレンチウイルスベクターを形質導入した改変ヒト初代T細胞と、NFATプロモーターの制御下の抗PD−1抗体遺伝子を有するレンチウイルスベクターを形質導入した改変ヒト初代T細胞との混合物を作製し、腫瘍細胞に対する細胞毒性について評価することができる。
D−1抗体の両者を発現するCAR−T細胞は、抗PD−1のみを発現するT細胞(0.40±0.04μg/mL)または標的細胞と共培養しなかったCAR−T細胞(0.42±0.01μg/mL)と比べて、より多くの抗PD−1抗体(0.50±0.02μg/mL)を分泌した。この結果は、前記標的細胞との共培養がCAR−T細胞におけるNFATプロモーターを誘導し、これによって該NFATプロモーターにより駆動される抗PD−1抗体遺伝子の発現が強化され、ひいては前記標的細胞に対する改変T細胞の細胞毒性が強化されることを示唆している。
次のT細胞の実験群を作製した:(1)T/GSI026:抗EGFRvIII−CAR遺伝子(GSI026)を有するレンチウイルスベクターを形質導入した改変ヒト初代T細胞;(2)T/抗CTLA−4:NFATプロモーターの制御下の抗CTLA−4抗体(イピリムマブ)遺伝子を有するレンチウイルスベクターを形質導入した改変ヒト初代T細胞;(3)T/GSI026 抗CTLA−4:抗EGFRvIII−CAR遺伝子(GSI026)とNFATプロモーターの制御下の抗CTLA−4抗体(イピリムマブ)遺伝子との両者を有するレンチウイルスベクターを形質導入した改変ヒト初代T細胞;および(4)unT:陰性対照としての無関係の遺伝子を形質導入した改変ヒト初代T細胞。さらに、抗EGFRvIII−CAR遺伝子を有するレンチウイルスベクターを形質導入した改変ヒト初代T細胞と、NFATプロモーターの制御下の抗CTLA−4抗体遺伝子を有するレンチウイルスベクターを形質導入した改変ヒト初代T細胞との混合物を作製し、腫瘍細胞に対する細胞毒性について評価することができる。
抗PD−1抗体のみ、または該抗体とキメラ抗原受容体(CAR)との組み合わせを発現する改変ヒト初代T細胞のin vivoでの有効性を、ヒト腫瘍細胞を移植したマウス異種移植モデル内で評価することができる。例えば、U87MG/VIII−Luc−PD−L1腫瘍細胞を一群のNSGマウスに移植し、ヒト膠芽腫のマウス異種移植モデルが提供される。
ターを形質導入した改変ヒト初代T細胞;ならびに(5)陰性対照としての無関係の遺伝子を形質導入した改変ヒト初代T細胞。前記抗PD−1抗体遺伝子はドキシサイクリン誘導性プロモーター(TETON(登録商標)等)またはNFATプロモーターの転写制御下にある。各処理条件における抗PD−1抗体の分泌は、マウスへの投与前に、またはマウスへの投与後に、適した条件で誘導される。
抗CTLA−4抗体のみ、または該抗体とキメラ抗原受容体(CAR)との組み合わせを発現する改変ヒト初代T細胞のin vivoでの有効性を、ヒト腫瘍細胞を移植したマウス異種移植モデル内で評価することができる。例えば、U87MG/VIII−Luc−CD80/CD86腫瘍細胞を一群のNSGマウスに移植し、ヒト膠芽腫のマウス異種移植モデルが提供される。
瘍細胞を死滅させることにおいて、より強力である可能性がある。このような効果は、形質導入された抗CTLA−4遺伝子がTet−on系の制御下である場合、ドキシサイクリン対して用量依存的である可能性がある。このような効果は、形質導入された抗CTLA−4遺伝子の発現がNFAT依存誘導性プロモーターの制御下である場合、EGFRvIII抗原特異的CAR−Tの存在に依存する可能性がある。このような効果は、形質導入された抗CTLA−4遺伝子がHSP70pの制御下である場合、熱ショックの温度または時間に依存する可能性がある。
抗PD−1抗体のみ、または該抗体とCARとの組み合わせを発現する改変ヒト初代T細胞のin vivoでの有効性を、ヒト腫瘍細胞を移植したマウス異種移植モデル内で評価することができる。例えば、ルシフェラーゼ導入遺伝子を発現するように改変されたヒト多発性骨髄腫細胞であるRPMI−8226細胞を一群のNSGマウスに移植し、ヒト多発性骨髄腫のマウス異種移植モデルが提供される。
抗CTLA−4抗体のみ、または該抗体とCARとの組み合わせを発現する改変ヒト初代T細胞のin vivoでの有効性を、ヒト腫瘍細胞を移植したマウス異種移植モデル内で評価することができる。例えば、ルシフェラーゼ導入遺伝子を発現するように改変されたヒト多発性骨髄腫細胞であるRPMI−8226細胞を一群のNSGマウスに移植し
、ヒト多発性骨髄腫のマウス異種移植モデルが提供される。
NY−ESO−1は予後不良な多発性骨髄腫において高度に発現されている(例えば、Blood 105:3939-3944 (2005)を参照のこと)。NY−ESO−1に対するTCRを用いた細胞療法が、例えばWO/2005/113595に記載されている。HLA−A*0201拘束性NY−ESO−1に対する高親和性TCRを形質導入した自己由来PBMCの養子移入が、転移性滑膜細胞肉腫および転移性メラノーマの患者に対し、臨床現場で試験されている(例えば、J. Clin. Oncol. 29:917-24 (2011)およびClinical Cancer Research 21:5 (2014)を参照のこと)。また、Rapoport APら(Nat. Med. 21 (8): 914-21 (2015))は、多発性骨髄腫に対する第I/II相臨床試験において有望な臨床結果を報告しており、該臨床試験においては、癌精巣抗原NY−ESO−1およびLAGE−1によって共有される天然に加工されたペプチド(naturally processed peptide)を認識する、親和性が強化されたT細胞受容体(TCR)を発現するように改変された自己T細胞を利用している。他の進行中のTCR−T免疫療法の臨床試験は、HLA−DP0401陽性の転移性癌患者に対する、MAGE−A3を標的としたTCR免疫療法(NCT02111850)などを含む。
ドする、6個のレンチウイルスベクターを設計した。VアルファおよびVベータ配列(野生型バリアント1G4および親和性成熟バリアント113−1G4)を、Li Y, Nature biotechnology. 2005;23:349-354およびRobbins P.F. et al, J Immunol. 2008 May 1;180(9): 6116-6131に従って設計した。両鎖の定常領域の配列を、UniProt: TCA:アクセッション番号P01848;TRBC1:アクセッション番号P01850;およびTRBC2:アクセッション番号A0A5B9からの配列に従って設計した。TCRアルファ鎖およびTCRベータ鎖配列を、T2Aペプチド(PLoS ONE 6(4): e18556)により連結した。1G4天然シグナルペプチドまたはCD8αシグナルペプチド配列を両鎖に先行させた。さらに、設計された前記TCR配列のコドン最適化を行い、ヒト細胞上での最適な発現を可能にした。前記ヌクレオチド配列の合成後、それを、PLVX−Puro(Clontech #632164)の本来のプロモーターをヒト伸長因子1αプロモーター(hEF1α)と置き換え、ピューロマイシン耐性遺伝子を欠失させるように改変したレンチウイルスベクターにクローニングした。前記TCRをコードするベクター(pLLV−LIT001〜pLLV−LIT006)は、上述の実施例に記載のように、293T細胞におけるレンチウイルスパッケージング系で生成した。
#130−091−441)を用いて2日間プレ活性化した。その後、プレ活性化されたT細胞に、上記TCRをコードするレンチウイルスベクターを形質導入し、TCRであるLIT−001〜LIT−006を発現するTCR−T細胞を作製した。
2±7.68pg/mL)または抗PD−1抗体のみを発現するT細胞(73.19±1.88pg/mL)と比べて、より高濃度のIFN−ガンマを分泌した(200.77±1.13pg/mL)。
HLA−A*0201クラスI拘束エレメントとの関連において、NY−ESO−1の残基157〜165(NY−ESO−1:157−165)に相当するペプチドSLLMWITQCを認識するTCR(LIT−006)をコードするレンチウイルスベクターを、293−6E細胞におけるレンチウイルスパッケージング系で生成した。また、hEF1αプロモーターの転写制御下にある抗CTLA−4遺伝子を有するレンチウイルスベクター(すなわちpLLV−hEF1α−抗CTLA−4)を、実施例1に記載のようにレンチウイルスパッケージング系で生成した。
を形質導入した改変ヒト初代T細胞との混合物を作製し、腫瘍細胞に対する細胞毒性について評価することができる。
初代ヒト末梢血単核球(PBMC)を、健常ドナー由来の末梢血を密度勾配遠心することで調製した。磁気ビーズ分離を用いてPBMCからヒト初代T細胞を精製した。抗HER2抗体の軽鎖および重鎖をコードする配列を設計し、2個のpcDNA3.1プラスミドベクターにそれぞれクローニングした。4μgの各プラスミドを、43msのパルス時間を用いた665Vのエレクトロポレーションにより、1×107個のヒト初代T細胞にトランスフェクトした。トランスフェクトされたT細胞をex vivoで3日間増殖させた。その後、トランスフェクトされたT細胞の一部に抗PD−1抗体(または抗CTLA−4抗体)遺伝子を有するレンチウイルスベクターを形質導入し、抗HER2および抗PD−1(または抗CTLA−4)抗体を発現させた。抗体の分泌は、LANPOWER(商標) Human Fc Detection kit(GenScript #L00656−1000)を用いたHTRF法で検出することができる。
、「SK−BR−3/Luc細胞」と呼ぶ)、これによってルシフェラーゼ活性のアッセイにより生腫瘍細胞の定量が可能となる。ハーセプチン(トラスツズマブ)として知られる抗HER2抗体は、SK−BR−3/Luc細胞に対する強力な細胞毒性を有する。
序論
肺癌は世界的に最も多い癌死の原因であり、癌関連死の5件あたり約1件、または推定160万人が関与している。米国および中国の両者で、肺癌は、男性と女性の両者における癌関連死の原因として、群を抜いて第1位となっている。米国癌協会によると、221,000人より多くの米国人が2015年中に肺癌と診断され、NSCLCが全ての肺癌のうち85%を占めている。肺癌と診断された米国内の患者のうち、全体で17.4%が診断後に5年間生存するが、発展途上国においては平均的な臨床成績は低下する。
体チロシンキナーゼ(RTK):EGFR(ErbB−1)、HER2/c−neu(ErbB−2)、Her3(ErbB−3)、およびHer4(ErbB−4)からなるサブファミリーである上皮成長因子受容体における、受容体の1つである。EGFRの主要な天然リガンドの一つはEGFである。
本実施例では、単一のベクターにコードされる、EGFRを標的とするCARと、免疫チェックポイント阻害剤を共発現するように、初代T細胞を改変した(図7A)。前記抗EGFR CARは、EGFRを過剰発現する肺癌部位にT細胞を誘導し、その肺癌部位で前記免疫チェックポイント阻害剤の部位特異的発現を引き起こす。前記抗EGFR CARが腫瘍細胞上のEGFRへ結合することにより、該CARの短縮型または変異型細胞内シグナル伝達ドメインが活性化され、それによって前記改変T細胞による減弱化された下流の免疫応答が引き起こされ、宿主内の非改変免疫細胞のリクルートによって腫瘍細胞が死滅するが、低濃度のEGFRを発現する正常細胞は死滅しない。この免疫応答はさらに、例えばT細胞メモリーや組織ホーミングを強化し、またT細胞の増殖および生存を促進する、1つまたは複数の免疫賦活剤(図7B)を過剰発現するように前記CAR−Tを改変することで増強されうる。
scFv、CD8αヒンジおよび膜貫通(TM)領域、CD137細胞質ドメイン(CD137 cyto)、およびCD3ζを含んだ全長抗EGFR CARをコードする。
る細胞株であるヒトA431細胞で免疫することにより作製した。mAb425をさらにヒト化し、マツズマブ(matuzumab)を提供した(米国特許第5,558,864号を参照のこと)。mAb425は高い親和性でEGFRに結合する。前臨床試験において、mAb425がEGFR依存性腫瘍の増殖を阻害し、VEGFの発現を阻害し、ADCCを誘導することが示された。マツズマブは、2000年代の初頭に、結腸直腸、肺、食道、および胃癌の治療に対する第II相臨床試験が行われた。しかし、さらなる臨床試験は2007年の第I相試験以降は行われなかった。2008年2月18日、タケダとメルクはマツズマブの開発をもはや続行しないことを発表した。
CARをコードする配列の隣にクローニングした。例えば、構築物GSI054においては、抗PD−1コード配列をCD137細胞質ドメインの隣にクローニングした。従って、GSI054は、N末端からC末端に向かって、CD8αシグナルペプチド、mAb425 scFv、CD8αヒンジおよびTM、ならびにCD137細胞質ドメインと、抗PD−1とを含む。
CARは、EGFRを過剰発現する腫瘍細胞の増殖を阻害する潜在的な能力を有する。
およびT2A等の自己切断可能なリンカーで置換し、高効率の複数遺伝子共発現を可能にした。
A549(ATCC# CCL−185)は周知のヒト肺癌細胞株であり、EGFRを過剰発現する。in vitroおよびin vivoアッセイを容易にするため、ホタルルシフェラーゼ遺伝子を親A549細胞に導入し、得られた細胞株をA549−Lucと命名した。
)を発現するCAR−T細胞では大きく減弱化された。短縮型CAR(GSI053)を発現するT細胞はEGFRを過剰発現する細胞に対して有意な細胞毒性を引き出すことができなかったため、低濃度でEGFRを発現する正常細胞は前記CAR−T細胞によって攻撃されず、改善されたin vivo安全性が提供される可能性がある。GSI057(短縮型CAR+抗PD−1+IL−7+CCR2b+Bcl2)を形質導入したCAR−T細胞は、A549−luc細胞と7日間共培養した際、有意な細胞毒性を示さなかった。これは予期していなかったことである。IL−7はA549細胞の増殖を促進しうる(Int J Clin Exp Pathol. 2014 Feb 15;7(3):870)ため、GSI055〜GSI057(IL−7を発現する)導入T細胞は次の実験に含めなかった。GSI060(短縮型CAR+抗PD−1+IL−21+CCR4+Bcl2)を形質導入したT細胞は、GSI053導入T細胞または抗PD−1抗体のみを発現するT細胞(T/抗PD−1)と比べ、強化された細胞毒性を示した。
ヒト初代T細胞を調製し、プレ活性化し、構築物GSI053、GSI054、GSI058〜GSI060をそれぞれ形質導入した。A549−luc細胞と3日間共培養した後、形質導入された各群の初代T細胞の上清を回収し、抗PD−1抗体の発現をLANPOWER(商標) Human Fc Detection kit(GenScript)によって検出した。簡単に記載すると、5μLのヒトIgG−GS665、5μLの抗ヒトFc抗体−Eu、および10μLの抗体試料または対照をアッセイプレート中で混合し、室温で1.5時間インキュベートした。プレートをHTRF対応機器(PHERSTAR(商標) Plus microplate reader、Ex:320〜340nm、Em:620nmおよび665nm)上で読み取った。
#433804)を用いて、使用説明書に従い検出した。非形質導入T細胞(UnT)を陰性対照として用いた。図26Bに示すように、GSI058 CAR−T細胞は、A549−luc細胞と3日間共培養した際に、135.05±1.68pg/mLのIL−21を分泌した。一方、GSI059およびGSI060 CAR−T細胞は、それぞれ100.58±0.80pg/mLおよび18.69±4.58pg/mLのIL−21を分泌した。
短縮型抗EGFR CAR(例えばGSI053〜GSI060)によって誘導される、抗PD−1を発現する改変ヒト初代T細胞のin vivoでの有効性を、ヒト腫瘍細胞を移植したマウス異種移植モデル内で評価することができる。例えば、NSGマウスの一群に肺癌細胞A549−Lucを移植し、ヒト肺癌のマウス異種移植モデルを提供する。処理群において、前記モデル化されたマウスに、レンチウイルス構築物GSI052〜GSI060を形質導入したヒト初代T細胞の各群を注入する。対照群において、前記モデル化されたマウスに、非形質導入T細胞を注入する。
。処理前後に、in vivo生物発光イメージングによって腫瘍サイズをモニタリングしてもよい。
短縮型抗EGFR CARと抗PD−1とを発現するCAR−T細胞のin vivoでの安全性を、カニクイザルのモデルにおいて評価した。
を、投与後1日目、2日目、3日目、4週間目まで、検出することができる。
LPD−1−16と命名された抗PD−1単一ドメイン抗体(sdAb)と、前記抗EGFRvIII CAR(GSI026、実施例3に記載)との両者をコードするレンチウイルスベクター(「pLIC−1042」)を設計した。pLIC−1042は、上記実施例に記載のように、293T細胞におけるレンチウイルスパッケージング系を用いて生成した。Pan T cell isolation kit(Miltenyi、Cat#130−096−535)を使用し、ヒト初代T細胞をPBMCから作製した。単離したT細胞を、T cell activation and expansion kitを用いて3日間プレ活性化した。T細胞の次の実験群を作製した:(1)T/pLIC−1042:抗EGFRvIII−CAR遺伝子(GSI026)とNFATプロモーターの制御下の抗PD−1 sdAb(LPD−1−16)遺伝子との両者を有するレンチウイルスベクターを形質導入した改変ヒト初代T細胞;(2)T/GSI026・抗PD−1:抗EGFRvIII−CAR遺伝子(GSI026)とNFATプロモーターの制御下のIgG抗PD−1抗体(ペムブロリズマブ)遺伝子との両者を有するレンチウイルスベクターを形質導入した改変ヒト初代T細胞(実施例3に記載の通り);(3)T/GSI026:抗EGFRvIII−CAR遺伝子(GSI026)を有するレンチウイルスベクターを形質導入した改変ヒト初代T細胞;および(4) UnT:陰性対照としての無関係の遺伝子を形質導入した改変ヒト初代T細胞。
した。単離したT細胞を、T cell activation and expansion kitを用いて3日間プレ活性化した。T細胞の次の実験群を作製した:(1)T/pLIC−1043:抗EGFRvIII−CAR遺伝子(GSI026)とNFATプロモーターの制御下の抗CTLA−4 sdAb(LCA−16)遺伝子との両者を有するレンチウイルスベクターを形質導入した改変ヒト初代T細胞;(2)T/GSI026・CTLA−4:抗EGFRvIII−CAR遺伝子(GSI026)とNFATプロモーターの制御下のIgG抗CTLA−4抗体(イピリムマブ)遺伝子との両者を有するレンチウイルスベクターを形質導入した改変ヒト初代T細胞(実施例4に記載の通り);(3)T/GSI026:抗EGFRvIII−CAR遺伝子(GSI026)を有するレンチウイルスベクターを形質導入した改変ヒト初代T細胞;および(4)UnT:陰性対照としての無関係の遺伝子を形質導入した改変ヒト初代T細胞。
Claims (60)
- a)免疫調節剤をコードする異種核酸を含む改変哺乳類細胞であって、前記異種核酸がプロモーターに機能的に連結した、改変哺乳類細胞;およびb)薬理学的に許容される賦形剤、を含む医薬組成物。
- 前記異種核酸が前記改変哺乳類細胞のゲノム中に存在する、請求項1に記載の医薬組成物。
- 前記改変哺乳類細胞が初代細胞である、請求項1または請求項2に記載の医薬組成物。
- 前記改変哺乳類細胞が細胞株由来である、請求項1または請求項2に記載の医薬組成物。
- 前記改変哺乳類細胞が免疫細胞である、請求項1〜4のいずれか1項に記載の医薬組成物。
- 前記免疫細胞が末梢血単核球(PBMC)、T細胞、B細胞、またはNK細胞である、請求項5に記載の医薬組成物。
- 前記改変哺乳類細胞が幹細胞である、請求項1〜4のいずれか1項に記載の医薬組成物。
- 前記改変哺乳類細胞がさらにキメラ抗原受容体(CAR)または組換えT細胞受容体(TCR)を発現する、請求項5〜7のいずれか1項に記載の医薬組成物。
- 前記改変哺乳類細胞が、前記免疫調節剤をコードする前記異種核酸と、前記CARまたは前記TCRをコードする第2の異種核酸を含むベクターを含む、請求項8に記載の医薬組成物。
- 前記CARまたは前記TCRをコードする前記第2の異種核酸が前記プロモーターに機能的に連結した、請求項9に記載の医薬組成物。
- 前記プロモーターが前記キメラ抗原受容体または前記組換えT細胞受容体の細胞内シグナル伝達ドメインによって誘導可能である、請求項8〜10のいずれか1項に記載の医薬組成物。
- 前記CARまたは前記TCRが、免疫エフェクター機能が無効化または減弱化した細胞内シグナル伝達ドメインを含む、請求項8〜11のいずれか1項に記載の医薬組成物。
- 前記医薬組成物がさらに第2の細胞を含み、前記第2の細胞はキメラ抗原受容体または組換えT細胞受容体を発現する哺乳類免疫細胞である、請求項1〜12のいずれか1項に記載の医薬組成物。
- 前記プロモーターが内在性プロモーターである、請求項1〜13のいずれか1項に記載の医薬組成物。
- 前記プロモーターが異種プロモーターである、請求項1〜13のいずれか1項に記載の医薬組成物。
- 前記プロモーターが誘導条件によって誘導可能なプロモーターである、請求項1〜15のいずれか1項に記載の医薬組成物。
- 前記誘導条件が、誘導物質、照射、温度、酸化還元状態、腫瘍環境、および前記改変哺乳類細胞の活性化状態からなる群より選択される、請求項16に記載の医薬組成物。
- 前記プロモーターが前記改変哺乳類細胞の内在性活性化シグナルによって誘導可能である、請求項17に記載の医薬組成物。
- 前記プロモーターがT細胞活性化依存プロモーターである、請求項18に記載の医薬組成物。
- 前記プロモーターが誘導物質によって誘導可能である、請求項17に記載の医薬組成物。
- 前記誘導物質が小分子である、請求項20に記載の医薬組成物。
- 前記誘導物質がポリペプチドである、請求項20に記載の医薬組成物。
- 前記ポリペプチドが前記改変哺乳類細胞によって発現される、請求項22に記載の医薬組成物。
- 前記改変哺乳類細胞がさらに、腫瘍抗原を認識するターゲティング分子をその表面上に発現する、請求項1〜23のいずれか1項に記載の医薬組成物。
- 前記免疫調節剤が免疫チェックポイント阻害剤である、請求項1〜24のいずれか1項に記載の医薬組成物。
- 前記免疫チェックポイント阻害剤がPD−1、PD−L1、PD−L2、CTLA−4、BLTA、TIM−3、またはLAG−3の阻害剤である、請求項25に記載の医薬組成物。
- 前記免疫調節剤が免疫賦活剤である、請求項1〜24のいずれか1項に記載の方法。
- 前記免疫賦活剤がIL−2、IL−7、IL−15、IL−21、IL−12、CCR4、CCR2b、ヘパラナーゼ、CD137L、LEM、およびBcl−2からなる群より選択される、請求項27に記載の方法。
- 前記免疫調節剤が抗体である、請求項1〜27のいずれか1項に記載の医薬組成物。
- 前記抗体が一本鎖抗体である、請求項29に記載の医薬組成物。
- 前記抗体が単一ドメイン抗体である、請求項29に記載の医薬組成物。
- 前記抗体が重鎖と軽鎖とを含む、請求項29または請求項30に記載の医薬組成物。
- 前記重鎖をコードする核酸と前記軽鎖をコードする核酸が、同一のプロモーターに機能的に連結される、請求項32に記載の医薬組成物。
- 前記重鎖をコードする核酸と前記軽鎖をコードする核酸が、異なるプロモーターに機能
的に連結される、請求項32に記載の医薬組成物。 - 前記重鎖をコードする核酸のためのプロモーターと前記軽鎖をコードする核酸のためのプロモーターを同時に誘導することができる、請求項34に記載の医薬組成物。
- 前記重鎖をコードする核酸のためのプロモーターと前記軽鎖をコードする核酸のためのプロモーターを順次誘導することができる、請求項34に記載の医薬組成物。
- 前記重鎖をコードする核酸のためのプロモーターと前記軽鎖をコードする核酸のためのプロモーターが、約10:1〜約1:10の強度比を有する、請求項34〜36のいずれか1項に記載の医薬組成物。
- 前記改変哺乳類細胞がさらに、治療用タンパク質をコードする第2の異種核酸を含む、請求項1〜37のいずれか1項に記載の医薬組成物。
- 前記免疫調節剤をコードする異種核酸と、前記治療用タンパク質をコードする第2の異種核酸が、同一のプロモーターに機能的に連結される、請求項38に記載の医薬組成物。
- 前記免疫調節剤をコードする異種核酸と、前記治療用タンパク質をコードする第2の異種核酸が、異なるプロモーターに機能的に連結される、請求項38に記載の医薬組成物。
- 前記治療用タンパク質が免疫調節剤ではない、請求項38〜40のいずれか1項に記載の医薬組成物。
- 前記治療用タンパク質が免疫調節剤である、請求項38〜40のいずれか1項に記載の医薬組成物。
- 前記改変哺乳類細胞が前記免疫調節剤と2種以上の治療用タンパク質を発現する、請求項41または請求項42に記載の医薬組成物。
- 個体における癌の治療法であって、請求項1〜43のいずれか1項に記載の医薬組成物の有効量を前記個体に投与することを含む、方法。
- 前記医薬組成物が全身投与される、請求項44に記載の方法。
- 前記医薬組成物が輸液により投与される、請求項45に記載の方法。
- 前記医薬組成物が腫瘍部位に局所投与される、請求項44に記載の方法。
- 前記医薬組成物が注射により投与される、請求項47に記載の方法。
- 前記免疫調節剤の発現を前記改変哺乳類細胞内で誘導することをさらに含む、請求項44〜48のいずれか1項に記載の方法。
- 前記癌が固形腫瘍である、請求項44〜49のいずれか1項に記載の方法。
- 前記癌が液性腫瘍である、請求項44〜49のいずれか1項に記載の方法。
- 前記改変哺乳類細胞が前記個体から得られる、請求項44〜51のいずれか1項に記載の方法。
- 前記改変哺乳類細胞が前記個体に対し同種異系である、請求項44〜51のいずれか1項に記載の方法。
- 前記個体がヒト個体である、請求項44〜53のいずれか1項に記載の方法。
- 前記免疫調節剤をコードする前記異種核酸を含んだベクターを哺乳類細胞に導入することを含む、請求項1〜43のいずれか1項に記載の医薬組成物を調製する方法。
- 前記ベクターがウイルスベクターである、請求項55に記載の方法。
- 前記ウイルスベクターが、レンチウイルスベクター、レトロウイルスベクター、アデノウイルスベクター、アデノ関連ウイルスベクター、単純ヘルペスウイルスベクター、およびそれらの誘導体からなる群より選択される、請求項56に記載の方法。
- 前記ベクターがエレクトロポレーションによって前記細胞に導入される、請求項55に記載の方法。
- a)請求項1〜43のいずれ1項に記載の医薬組成物;およびb)前記医薬組成物の使用のための説明書、を含むキット。
- c)キメラ抗原受容体または組換えT細胞受容体を発現する第2の哺乳類免疫細胞を含む組成物、をさらに含む、請求項59に記載のキット。
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN2016073489 | 2016-02-04 | ||
| CNPCT/CN2016/073489 | 2016-02-04 | ||
| PCT/CN2016/087855 WO2017133175A1 (en) | 2016-02-04 | 2016-06-30 | Engineered mammalian cells for cancer therapy |
| CNPCT/CN2016/087855 | 2016-06-30 | ||
| PCT/CN2017/072723 WO2017133633A1 (en) | 2016-02-04 | 2017-01-26 | Engineered mammalian cells for cancer therapy |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2019504892A true JP2019504892A (ja) | 2019-02-21 |
| JP2019504892A5 JP2019504892A5 (ja) | 2020-03-05 |
Family
ID=59499323
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2018560705A Pending JP2019504892A (ja) | 2016-02-04 | 2017-01-26 | 癌治療のための改変哺乳類細胞 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US20190038671A1 (ja) |
| EP (1) | EP3411079A4 (ja) |
| JP (1) | JP2019504892A (ja) |
| CN (1) | CN108883202A (ja) |
| WO (2) | WO2017133175A1 (ja) |
Families Citing this family (50)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10704021B2 (en) | 2012-03-15 | 2020-07-07 | Flodesign Sonics, Inc. | Acoustic perfusion devices |
| WO2015105955A1 (en) | 2014-01-08 | 2015-07-16 | Flodesign Sonics, Inc. | Acoustophoresis device with dual acoustophoretic chamber |
| CA2974651A1 (en) | 2014-01-24 | 2015-07-30 | Children's Hospital Of Eastern Ontario Research Institute Inc. | Smc combination therapy for the treatment of cancer |
| US20170151281A1 (en) | 2015-02-19 | 2017-06-01 | Batu Biologics, Inc. | Chimeric antigen receptor dendritic cell (car-dc) for treatment of cancer |
| US11708572B2 (en) | 2015-04-29 | 2023-07-25 | Flodesign Sonics, Inc. | Acoustic cell separation techniques and processes |
| US11377651B2 (en) | 2016-10-19 | 2022-07-05 | Flodesign Sonics, Inc. | Cell therapy processes utilizing acoustophoresis |
| US11214789B2 (en) | 2016-05-03 | 2022-01-04 | Flodesign Sonics, Inc. | Concentration and washing of particles with acoustics |
| WO2018071500A1 (en) | 2016-10-11 | 2018-04-19 | Agenus Inc. | Anti-lag-3 antibodies and methods of use thereof |
| US11185586B2 (en) | 2016-11-22 | 2021-11-30 | Alloplex Biotherapeutics, Inc. | Allogeneic tumor cell vaccine |
| AU2018223211A1 (en) * | 2017-02-21 | 2019-10-10 | The University Of Adelaide | T cells expressing chemokine receptors for treating cancer |
| CA3059634A1 (en) | 2017-04-13 | 2018-10-18 | Senti Biosciences, Inc. | Combinatorial cancer immunotherapy |
| JP2020517263A (ja) * | 2017-04-19 | 2020-06-18 | ユニバーシティ オブ サザン カリフォルニア | がんを治療するための組成物及び方法 |
| WO2019000223A1 (en) | 2017-06-27 | 2019-01-03 | Nanjing Legend Biotech Co., Ltd. | ENABLERS OF IMMUNE EFFECTOR CELLS OF CHIMERIC ANTIBODIES AND METHODS OF USE THEREOF |
| EP3662055A1 (en) * | 2017-08-02 | 2020-06-10 | Autolus Limited | Cells expressing a chimeric antigen receptor or engineered tcr and comprising a nucleotide sequence which is selectively expressed |
| GB201713078D0 (en) * | 2017-08-15 | 2017-09-27 | Adaptimmune Ltd | T Cell Modification |
| EP3679145A2 (en) * | 2017-09-08 | 2020-07-15 | Poseida Therapeutics, Inc. | Compositions and methods for chimeric ligand receptor (clr)-mediated conditional gene expression |
| CA3079076A1 (en) * | 2017-10-18 | 2019-04-25 | Chemotherapeutisches Forschungsinstitut Georg-Speyer-Haus | Methods and compounds for improved immune cell therapy |
| CA3085784A1 (en) | 2017-12-14 | 2019-06-20 | Flodesign Sonics, Inc. | Acoustic transducer driver and controller |
| GB201721802D0 (en) * | 2017-12-22 | 2018-02-07 | Almac Discovery Ltd | Ror1-specific antigen binding molecules |
| WO2019177011A1 (ja) * | 2018-03-13 | 2019-09-19 | 国立大学法人大阪大学 | 腫瘍免疫賦活剤 |
| US20210032661A1 (en) * | 2018-04-09 | 2021-02-04 | The Trustees Of The University Of Pennsylvania | Methods And Compositions Comprising A Viral Vector For Expression Of A Transgene And An Effector |
| CN110615842B (zh) * | 2018-06-20 | 2023-05-09 | 上海隆耀生物科技有限公司 | 一种包含共刺激受体的嵌合抗原受体及应用 |
| CN108913718A (zh) * | 2018-07-20 | 2018-11-30 | 苏州茂行生物科技有限公司 | 一种靶向EGFR vⅢ的CAR-T细胞的制备方法及应用 |
| CN108949825A (zh) * | 2018-07-30 | 2018-12-07 | 苏州茂行生物科技有限公司 | 一种靶向her2的car-t细胞的制备方法及应用 |
| US11866494B2 (en) * | 2018-08-31 | 2024-01-09 | Innovative Cellular Therapeutics Holdings, Ltd. | CAR T therapy through uses of co-stimulation |
| US20210317184A1 (en) * | 2018-09-05 | 2021-10-14 | GlaxoSmithKline Intellectual Property Devolopment Limited | T cell modification |
| EP3866813A4 (en) | 2018-10-17 | 2022-08-03 | Senti Biosciences, Inc. | COMBINATORY CANCER IMMUNOTHERAPY |
| US11419898B2 (en) | 2018-10-17 | 2022-08-23 | Senti Biosciences, Inc. | Combinatorial cancer immunotherapy |
| EP3876977A1 (en) | 2018-11-06 | 2021-09-15 | The Regents Of The University Of California | Chimeric antigen receptors for phagocytosis |
| JP2022512326A (ja) * | 2018-12-06 | 2022-02-03 | グアンドン ティーシーアールキュア バイオファーマ テクノロジー カンパニー リミテッド | 腫瘍抗原、TGF-β、および免疫チェックポイントを標的とする組み合わせTCR-T細胞療法 |
| WO2020172553A1 (en) * | 2019-02-22 | 2020-08-27 | Novartis Ag | Combination therapies of egfrviii chimeric antigen receptors and pd-1 inhibitors |
| US20220175833A1 (en) * | 2019-03-07 | 2022-06-09 | Children's Medical Center Corporation | Targeted delivery of immune-modulating vhh and vhh-fusion protein |
| US20220204575A1 (en) * | 2019-04-23 | 2022-06-30 | The Regents Of The University Of California | Modulating survival of therapeutic cells and methods, cells and nucleic acids related thereto |
| US11026973B2 (en) | 2019-04-30 | 2021-06-08 | Myeloid Therapeutics, Inc. | Engineered phagocytic receptor compositions and methods of use thereof |
| US20220291203A1 (en) * | 2019-07-25 | 2022-09-15 | Immunowake Inc. | Methods of measuring cell-mediated killing by effectors |
| CN114450397A (zh) * | 2019-08-01 | 2022-05-06 | 纪念斯隆-凯特琳癌症中心 | 用于改善免疫疗法的细胞及其用途 |
| BR112022003970A2 (pt) | 2019-09-03 | 2022-06-21 | Myeloid Therapeutics Inc | Métodos e composições para integração genômica |
| IL292355A (en) * | 2019-10-22 | 2022-06-01 | Alloplex Biotherapeutics | Compositions and methods for in vitro activation and expansion of serial killer t-cell populations and passive vaccination of a cancer patient with tumor cell-killing cells |
| CA3160443A1 (en) * | 2019-11-08 | 2021-05-14 | Mayo Foundation For Medical Education And Research | Methods and materials for using engineered mesenchymal stem cells to treat inflammatory conditions and degenerative diseases |
| CN112941028A (zh) * | 2019-12-09 | 2021-06-11 | 上海细胞治疗集团有限公司 | 纳米抗体基因修饰的间充质干细胞及其制备方法与应用 |
| US10980836B1 (en) | 2019-12-11 | 2021-04-20 | Myeloid Therapeutics, Inc. | Therapeutic cell compositions and methods of manufacturing and use thereof |
| EP4090360A1 (en) * | 2020-01-14 | 2022-11-23 | Adaptimmune Limited | Method of treatment of cancer or tumour |
| CN115515980A (zh) * | 2020-02-26 | 2022-12-23 | 拜格拉夫55公司 | C19 c38双特异性抗体 |
| US20230149461A1 (en) * | 2020-04-01 | 2023-05-18 | Nanjing Legend Biotech Co., Ltd. | Compositions and methods for reducing graft rejection in allogeneic cell therapy |
| US20230348853A1 (en) * | 2020-06-29 | 2023-11-02 | Memorial Sloan Kettering Cancer Center | C825 expressing immune cells and diagnostic uses thereof |
| WO2022093884A1 (en) * | 2020-10-27 | 2022-05-05 | Pact Pharma, Inc. | Compositions and methods for the treatment of cancer using next generation engineered t cell therapy |
| CA3197423A1 (en) | 2020-11-04 | 2022-05-12 | Daniel Getts | Engineered chimeric fusion protein compositions and methods of use thereof |
| WO2022272102A2 (en) * | 2021-06-24 | 2022-12-29 | Georgia Tech Research Corporation | Methods and compositions for remote control of t cell therapies by thermal targeting |
| CN113789299B (zh) * | 2021-10-18 | 2023-03-24 | 新鲁细胞生物科技(海南)有限公司 | 一种脐血nk细胞的体外增殖培养基、体外培养试剂盒和体外培养方法 |
| WO2024077270A1 (en) * | 2022-10-07 | 2024-04-11 | Senti Biosciences, Inc. | Armed chimeric receptors and methods of use thereof |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013040371A2 (en) * | 2011-09-16 | 2013-03-21 | Baylor College Of Medicine | Targeting the tumor microenvironment using manipulated nkt cells |
| WO2014134165A1 (en) * | 2013-02-26 | 2014-09-04 | Memorial Sloan-Kettering Cancer Center | Compositions and methods for immunotherapy |
| WO2015142661A1 (en) * | 2014-03-15 | 2015-09-24 | Novartis Ag | Regulatable chimeric antigen receptor |
| WO2015188119A1 (en) * | 2014-06-06 | 2015-12-10 | Bluebird Bio, Inc. | Improved t cell compositions |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5215964A (en) * | 1991-06-03 | 1993-06-01 | Immunobiology Research Institute, Inc. | Peptides useful in regulating the immune and nervous systems |
| IL135943A0 (en) * | 1997-11-03 | 2001-05-20 | Univ Arizona | Hyperthermic inducible expression vectors for gene theraphy and methods of use thereof |
| CA2508660C (en) * | 2002-12-23 | 2013-08-20 | Wyeth | Antibodies against pd-1 and uses therefor |
| NZ569541A (en) * | 2006-01-13 | 2012-05-25 | Us Gov Health & Human Serv | Codon optimized IL-15 and IL-15R-alpha genes for expression in mammalian cells |
| DK2958943T3 (da) * | 2013-02-20 | 2019-12-09 | Univ Pennsylvania | Behandling af cancer ved anvendelse af humaniseret anti-EGFRvIII kimær antigenreceptor |
| JP2017528433A (ja) * | 2014-07-21 | 2017-09-28 | ノバルティス アーゲー | 低い免疫増強用量のmTOR阻害剤とCARの組み合わせ |
| EP3313883B1 (en) * | 2015-06-23 | 2023-12-06 | Memorial Sloan Kettering Cancer Center | Novel pd-1 immune modulating agents |
-
2016
- 2016-06-30 WO PCT/CN2016/087855 patent/WO2017133175A1/en active Application Filing
-
2017
- 2017-01-26 WO PCT/CN2017/072723 patent/WO2017133633A1/en active Application Filing
- 2017-01-26 US US16/075,220 patent/US20190038671A1/en not_active Abandoned
- 2017-01-26 CN CN201780010175.2A patent/CN108883202A/zh active Pending
- 2017-01-26 JP JP2018560705A patent/JP2019504892A/ja active Pending
- 2017-01-26 EP EP17746953.3A patent/EP3411079A4/en not_active Withdrawn
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013040371A2 (en) * | 2011-09-16 | 2013-03-21 | Baylor College Of Medicine | Targeting the tumor microenvironment using manipulated nkt cells |
| WO2014134165A1 (en) * | 2013-02-26 | 2014-09-04 | Memorial Sloan-Kettering Cancer Center | Compositions and methods for immunotherapy |
| WO2015142661A1 (en) * | 2014-03-15 | 2015-09-24 | Novartis Ag | Regulatable chimeric antigen receptor |
| WO2015188119A1 (en) * | 2014-06-06 | 2015-12-10 | Bluebird Bio, Inc. | Improved t cell compositions |
Non-Patent Citations (2)
| Title |
|---|
| CLINICAL CANCER RESEARCH,2013年,VOL.19, NO.20,PP.5636-5646, JPN6021006274, ISSN: 0004608014 * |
| 血液フロンティア, 2016 NOV, VOL.26 NO.11, P.86-89, JPN6021006276, ISSN: 0004450935 * |
Also Published As
| Publication number | Publication date |
|---|---|
| US20190038671A1 (en) | 2019-02-07 |
| EP3411079A1 (en) | 2018-12-12 |
| WO2017133175A1 (en) | 2017-08-10 |
| EP3411079A4 (en) | 2019-10-30 |
| CN108883202A (zh) | 2018-11-23 |
| WO2017133633A1 (en) | 2017-08-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20190038671A1 (en) | Engineered mammalian cells for cancer therapy | |
| JP7217970B2 (ja) | 融合タンパク質を用いてt細胞受容体をリプログラミングするための組成物及び方法 | |
| US10829735B2 (en) | Methods for improving the efficacy and expansion of immune cells | |
| US20190375815A1 (en) | Treatment of cancer using chimeric t cell receptor proteins having multiple specificities | |
| JP2022002525A (ja) | キメラ抗原受容体発現細胞の有効性および増殖を改善するための方法 | |
| WO2020147708A1 (en) | Chimeric receptor polypeptides and uses thereof | |
| WO2020228825A1 (en) | Engineered immune cells comprsing a recognition molecule | |
| JP2020534871A (ja) | キメラエンガルフメント受容体分子および使用方法 | |
| US20210338733A1 (en) | Tn-MUC1 Chimeric Antigen Receptor (CAR) T Cell Therapy | |
| US20210038659A1 (en) | Combination therapy using a chimeric antigen receptor | |
| WO2022214089A1 (zh) | 细胞免疫治疗的应用 | |
| US20230183296A1 (en) | Compositions and methods for reducing host rejection of allogeneic cells using simian icp47 and variants thereof | |
| WO2020102770A1 (en) | Methods of dosing engineered t cells for the treatment of b cell malignancies | |
| TW202321287A (zh) | 特異性靶向間皮素之經工程改造之免疫細胞及其用途 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20200127 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20200127 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20210224 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20210519 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20211005 |