JP2016132720A - ポリエステル樹脂ペレット及びその製造方法 - Google Patents
ポリエステル樹脂ペレット及びその製造方法 Download PDFInfo
- Publication number
- JP2016132720A JP2016132720A JP2015007949A JP2015007949A JP2016132720A JP 2016132720 A JP2016132720 A JP 2016132720A JP 2015007949 A JP2015007949 A JP 2015007949A JP 2015007949 A JP2015007949 A JP 2015007949A JP 2016132720 A JP2016132720 A JP 2016132720A
- Authority
- JP
- Japan
- Prior art keywords
- polyester resin
- acid
- ppb
- polycondensation
- pellet
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Landscapes
- Polyesters Or Polycarbonates (AREA)
Abstract
【課題】 色調とヘーズを保ったまま、副生ジエチレングリコール量を低減でき、結晶化速度を速めたポリエステル樹脂及びその製造方法を提供すること。
【解決手段】 ペレットの内部に含まれるアルカリ金属原子の含有量(PET−M)が下記式(1)を満足するポリエステル樹脂ペレット及びテレフタル酸を主成分とするジカルボン酸成分と、エチレングリコールを主成分とするジオール成分から、ポリエステル樹脂を製造し、更にそのポリエステル樹脂からポリエステル樹脂ペレットを製造する方法において、原料であるテレフタル酸中に含まれるナトリウム原子の含有量(TPA−Na)が下記式(2)を満足し、ペレットの内部に含まれるナトリウム原子の含有量(PET−Na)が下記式(3)を満足するポリエステル樹脂ペレットの製造方法。
50ppb ≦ PET−M ≦ 400ppb (1)
60ppb ≦ TPA−Na ≦ 500ppb (2)
50ppb ≦ PET−Na ≦ 400ppb (3)
【選択図】なし
【解決手段】 ペレットの内部に含まれるアルカリ金属原子の含有量(PET−M)が下記式(1)を満足するポリエステル樹脂ペレット及びテレフタル酸を主成分とするジカルボン酸成分と、エチレングリコールを主成分とするジオール成分から、ポリエステル樹脂を製造し、更にそのポリエステル樹脂からポリエステル樹脂ペレットを製造する方法において、原料であるテレフタル酸中に含まれるナトリウム原子の含有量(TPA−Na)が下記式(2)を満足し、ペレットの内部に含まれるナトリウム原子の含有量(PET−Na)が下記式(3)を満足するポリエステル樹脂ペレットの製造方法。
50ppb ≦ PET−M ≦ 400ppb (1)
60ppb ≦ TPA−Na ≦ 500ppb (2)
50ppb ≦ PET−Na ≦ 400ppb (3)
【選択図】なし
Description
本発明は、色調とヘーズを保ったまま、副生ジエチレングリコール量を低減でき、結晶化速度を速めたポリエステル樹脂及びその製造方法に関する。
ポリエチレンテレフタレートに代表されるポリエステル樹脂は、化学的、物理的性質に優れていることから、飲料ボトル等の容器、フィルム、シート、繊維等の各種用途に広範囲に使用されている。一般にポリエステル樹脂は、ジカルボン酸とジオールとをエステル化反応又はエステル交換反応、及び溶融重縮合反応を経て、更に必要に応じて、固相重縮合反応させることにより製造される。
特に、これらのポリエステル樹脂を飲料ボトル等の容器に使用する際には、ペレット状のポリエステル樹脂を射出成形し、プリフォームと呼ばれる予備成形体を得た後、延伸ブロー成形により、容器を得る手法が一般的である。
ここで、ポリエステル樹脂の重要な特性として、色調や結晶化速度、ジエチレングリコ―ル共重合量などが挙げられる。これらを制御する方法として、多くの検討がなされてきた。
ここで、ポリエステル樹脂の重要な特性として、色調や結晶化速度、ジエチレングリコ―ル共重合量などが挙げられる。これらを制御する方法として、多くの検討がなされてきた。
たとえば、結晶化速度が遅いと、特にボトルに用いた際の口栓部結晶化が遅くなるため、生産性の悪化や衛生面での問題が生じるおそれがある。そのため、結晶化を速める方法として、特許文献1では固相重縮合後にポリエチレンと接触させる方法や、特許文献2では固相重縮合後にアルカリ金属及びアルカリ土類金属を含む水と接触させる方法が知られてきた。
また、重縮合中に副生するジエチレングリコールの低減手法として、特許文献3に代表されるようなアルカリ化合物を加える方法が知られている。
前記の方法のうち、特許文献1及び2に記載される固相重縮合後に処理を行う方法は設備が必要であり、操作が煩雑で経済性の観点からも望ましいものではなかった。また、ポリエステル樹脂中のアルカリ金属量については特に記載がなく、定量的な評価がなされていない。
前記の方法のうち、特許文献1及び2に記載される固相重縮合後に処理を行う方法は設備が必要であり、操作が煩雑で経済性の観点からも望ましいものではなかった。また、ポリエステル樹脂中のアルカリ金属量については特に記載がなく、定量的な評価がなされていない。
加えて、前記文献の方法によれば、ペレット表面にポリオレフィンやアルカリ金属が付着していると考えられるため、ペレット輸送中に一部が脱落し、結晶化速度が再び遅くなってしまうことで、ペレット全体の結晶化速度が安定しないおそれがあった。
また、特許文献3に記載される方法では、アルカリ化合物の添加量が多いために、特に色調、すなわち黄色みにおいて、満足できる品質ではなかった上、異物の懸念やヘーズの悪化を招くおそれがあった。
また、特許文献3に記載される方法では、アルカリ化合物の添加量が多いために、特に色調、すなわち黄色みにおいて、満足できる品質ではなかった上、異物の懸念やヘーズの悪化を招くおそれがあった。
本発明の目的は、色調とヘーズを保ったまま、副生ジエチレングリコール量を低減でき
、結晶化速度を速めたポリエステル樹脂ペレット及びその製造方法に関する。
、結晶化速度を速めたポリエステル樹脂ペレット及びその製造方法に関する。
本発明の第一の要旨は、ポリエステル樹脂ペレットの内部に含まれるアルカリ金属原子の含有量(PET−M)が下記式(1)を満足するポリエステル樹脂ペレットである。
50ppb ≦ PET−M ≦ 400ppb (1)
本発明の第二の要旨は、テレフタル酸を主成分とするジカルボン酸成分と、エチレングリコールを主成分とするジオール成分から、ポリエステル樹脂を製造し、更にそのポリエステル樹脂からポリエステル樹脂ペレットを製造する方法において、原料であるテレフタル酸中に含まれるナトリウム原子の質量(TPA−Na)が下記式(2)を満足し、ペレットの内部に含まれるナトリウム原子の含有量(PET−Na)が下記式(3)20満足するポリエステル樹脂ペレットの製造方法である。
60ppb ≦ TPA−Na ≦ 500ppb (2)
50ppb ≦ PET−Na ≦ 400ppb (3)
50ppb ≦ PET−M ≦ 400ppb (1)
本発明の第二の要旨は、テレフタル酸を主成分とするジカルボン酸成分と、エチレングリコールを主成分とするジオール成分から、ポリエステル樹脂を製造し、更にそのポリエステル樹脂からポリエステル樹脂ペレットを製造する方法において、原料であるテレフタル酸中に含まれるナトリウム原子の質量(TPA−Na)が下記式(2)を満足し、ペレットの内部に含まれるナトリウム原子の含有量(PET−Na)が下記式(3)20満足するポリエステル樹脂ペレットの製造方法である。
60ppb ≦ TPA−Na ≦ 500ppb (2)
50ppb ≦ PET−Na ≦ 400ppb (3)
本発明のポリエステル樹脂ペレットは、色調の悪化・ヘーズの悪化を抑制したまま、副生ジエチレングリコール量を低減でき、結晶化速度を速めることができるため、品質が安定し、生産性に優れる。
以下に本発明を実施するための最良の形態を詳細に説明するが、以下に記載する構成要件の説明は、本発明の実施態様の代表例であり、本発明はこれらの内容に限定されるものではない。なお、本明細書中において、含有量を表わす「ppb」、「ppm」は、「モル」等を特記しない場合はすべて「質量ppb」、「質量ppm」を意味する。
本発明において用いられる原料は、ジカルボン酸成分に占めるテレフタル酸の割合を90モル%以上、更には96モル%以上とするのが好ましく、また、ジオール成分に占めるエチレングリコールの割合を、90モル%以上、更には95モル%以上、特には97モル%以上とするのが好ましい。テレフタル酸のジカルボン酸成分に占める割合、及びエチレングリコールのジオール成分に占める割合が前記範囲下限未満では、得られるポリエステルの成形体としての機械的強度が低下する傾向がある。
本発明において用いられる原料は、ジカルボン酸成分に占めるテレフタル酸の割合を90モル%以上、更には96モル%以上とするのが好ましく、また、ジオール成分に占めるエチレングリコールの割合を、90モル%以上、更には95モル%以上、特には97モル%以上とするのが好ましい。テレフタル酸のジカルボン酸成分に占める割合、及びエチレングリコールのジオール成分に占める割合が前記範囲下限未満では、得られるポリエステルの成形体としての機械的強度が低下する傾向がある。
本発明において用いられる原料として、テレフタル酸の代わりに、テレフタル酸のエステル形成性誘導体を使用することは可能ではあるが、アルカリ金属元素に代表されるエステル交換反応触媒を多量に添加する必要があるため、異物が発生する場合がある。
テレフタル酸以外のジカルボン酸成分としては、例えばフタル酸、イソフタル酸、フェニレンジオキシジカルボン酸、4,4’−ジフェニルジカルボン酸、4,4’−ジフェニルエーテルジカルボン酸、4,4’−ジフェニルケトンジカルボン酸、4,4’−ジフェノキシエタンジカルボン酸、4,4’−ジフェニルスルホンジカルボン酸、2,6−ナフタレンジカルボン酸等の芳香族ジカルボン酸、ヘキサヒドロテレフタル酸、ヘキサヒドロイソフタル酸等の脂環式ジカルボン酸、及び、コハク酸、グルタル酸、アジピン酸、ピメリン酸、スベリン酸、アゼライン酸、セバシン酸、ウンデカジカルボン酸、ドデカジカルボン酸等の脂肪族ジカルボン酸、これら一種又は二種以上を用いることが出来るが、特にイソフタル酸がテレフタル酸の共重合成分として好ましく、その使用量は全酸成分に対して3モル%以下であることが好ましい。
テレフタル酸以外のジカルボン酸成分としては、例えばフタル酸、イソフタル酸、フェニレンジオキシジカルボン酸、4,4’−ジフェニルジカルボン酸、4,4’−ジフェニルエーテルジカルボン酸、4,4’−ジフェニルケトンジカルボン酸、4,4’−ジフェノキシエタンジカルボン酸、4,4’−ジフェニルスルホンジカルボン酸、2,6−ナフタレンジカルボン酸等の芳香族ジカルボン酸、ヘキサヒドロテレフタル酸、ヘキサヒドロイソフタル酸等の脂環式ジカルボン酸、及び、コハク酸、グルタル酸、アジピン酸、ピメリン酸、スベリン酸、アゼライン酸、セバシン酸、ウンデカジカルボン酸、ドデカジカルボン酸等の脂肪族ジカルボン酸、これら一種又は二種以上を用いることが出来るが、特にイソフタル酸がテレフタル酸の共重合成分として好ましく、その使用量は全酸成分に対して3モル%以下であることが好ましい。
また、エチレングリコール以外のジオール成分としては、例えばジエチレングリコールが挙げられる。その他のジオール成分として、例えば、トリメチレングリコール、テトラメチレングリコール、ペンタメチレングリコール、ヘキサメチレングリコール、オクタメチレングリコール、デカメチレングリコール、ネオペンチルグリコール、2−エチル−2
−ブチル−1,3−プロパンジオール、ポリエチレングリコール、ポリテトラメチレンエーテルグリコール等の脂肪族ジオール、1,2−シクロヘキサンジオール、1,4−シクロヘキサンジオール、1,1−シクロヘキサンジメチロール、1,4−シクロヘキサンジメチロール、2,5−ノルボルナンジメチロール等の脂環式ジオール、及び、キシリレングリコール、4,4’−ジヒドロキシビフェニル、2,2−ビス(4’−ヒドロキシフェニル)プロパン、2,2−ビス(4’−β−ヒドロキシエトキシフェニル)プロパン、ビス(4−ヒドロキシフェニル)スルホン、ビス(4−β−ヒドロキシエトキシフェニル)スルホン酸等の芳香族ジオール、及び2,2−ビス(4’−ヒドロキシフェニル)プロパンのエチレンオキサイド付加物又はプロピレンオキサイド付加物、これら一種又は二種以上を共重合成分として用いることが出来る。
−ブチル−1,3−プロパンジオール、ポリエチレングリコール、ポリテトラメチレンエーテルグリコール等の脂肪族ジオール、1,2−シクロヘキサンジオール、1,4−シクロヘキサンジオール、1,1−シクロヘキサンジメチロール、1,4−シクロヘキサンジメチロール、2,5−ノルボルナンジメチロール等の脂環式ジオール、及び、キシリレングリコール、4,4’−ジヒドロキシビフェニル、2,2−ビス(4’−ヒドロキシフェニル)プロパン、2,2−ビス(4’−β−ヒドロキシエトキシフェニル)プロパン、ビス(4−ヒドロキシフェニル)スルホン、ビス(4−β−ヒドロキシエトキシフェニル)スルホン酸等の芳香族ジオール、及び2,2−ビス(4’−ヒドロキシフェニル)プロパンのエチレンオキサイド付加物又はプロピレンオキサイド付加物、これら一種又は二種以上を共重合成分として用いることが出来る。
更に、例えば、グリコール酸、p−ヒドロキシ安息香酸、p−β−ヒドロキシエトキシ安息香酸等のヒドロキシカルボン酸やアルコキシカルボン酸、及び、ステアリルアルコール、ヘネイコサノール、オクタコサノール、ベンジルアルコール、ステアリン酸、ベヘン酸、安息香酸、t−ブチル安息香酸、ベンゾイル安息香酸等の単官能成分、トリカルバリル酸、トリメリット酸、トリメシン酸、ピロメリット酸、ナフタレンテトラカルボン酸、没食子酸、トリメチロールエタン、トリメチロールプロパン、グリセロール、ペンタエリスリトール、等の三官能以上の多官能成分、等の一種又は二種以上が共重合成分として用いられてもよい。
前記の原料は、化石燃料由来であってもよいが、植物を由来とし、発酵法等を通じて得られたものであってもよい。植物由来の原料としては、エチレングリコールやテレフタル酸が知られているが、特に植物由来のエチレングリコールは入手しやすく、好適に用いることができる。なお、蒸留や活性炭ろ過等を経て、不純物を十分に除いたエチレングリコールを用いることが好ましく、純度としては90質量%以上、好ましくは95質量%以上、更に好ましくは98質量%以上、特に好ましくは99質量%以上であると、ポリエステル樹脂の色調や重合活性が良好となる。
本発明におけるポリエステル樹脂ペレットの内部に含まれるアルカリ金属原子の含有量(PET−M)は、下記式(1)を満足する必要がある。
50ppb ≦ PET−M ≦ 400ppb (1)
下限値としては、60ppb以上が好ましく、70ppb以上が特に好ましい。上限値としては、300ppb以下であることがより好ましく、250ppb以下であることが特に好ましい。
50ppb ≦ PET−M ≦ 400ppb (1)
下限値としては、60ppb以上が好ましく、70ppb以上が特に好ましい。上限値としては、300ppb以下であることがより好ましく、250ppb以下であることが特に好ましい。
前記範囲下限未満では、得られるポリエステル樹脂において、重縮合中に副生するジエチレングリコール量が増加することで、結果として共重合されるジエチレングリコールの量が多くなり、機械的強度が劣る。一方、前記範囲上限超過では、得られるポリエステル樹脂の色調が悪化する。
本発明においてポリエステル樹脂を製造するにあたっては、アルカリ金属化合物を、重縮合反応の開始までの任意の時期に添加することができる。また、アルカリ金属化合物としては、ナトリウム化合物が溶解性の点から好ましい。添加の方法に際しては、例えば、水やエチレングリコ―ルに溶解して、触媒と混合して添加してもよく、テレフタル酸とエチレングリコールのスラリー調製時に添加してもよく、エステル化反応槽に添加してもよく、重縮合反応槽に添加してもよく、又はこれらの移送配管に添加してもよいが、設備を簡素化できることから、あらかじめ、テレフタル酸とナトリウム化合物を混合しておき、スラリー調製工程に供給する方法、又はナトリウム化合物を含まないテレフタル酸とナトリウム化合物を含むテレフタル酸をあらかじめ混合して、ナトリウム含有量を上記の範囲内に調節したテレフタル酸を調製しておき、スラリー調製工程に供給する方法が好ましく用いられる。
本発明においてポリエステル樹脂を製造するにあたっては、アルカリ金属化合物を、重縮合反応の開始までの任意の時期に添加することができる。また、アルカリ金属化合物としては、ナトリウム化合物が溶解性の点から好ましい。添加の方法に際しては、例えば、水やエチレングリコ―ルに溶解して、触媒と混合して添加してもよく、テレフタル酸とエチレングリコールのスラリー調製時に添加してもよく、エステル化反応槽に添加してもよく、重縮合反応槽に添加してもよく、又はこれらの移送配管に添加してもよいが、設備を簡素化できることから、あらかじめ、テレフタル酸とナトリウム化合物を混合しておき、スラリー調製工程に供給する方法、又はナトリウム化合物を含まないテレフタル酸とナトリウム化合物を含むテレフタル酸をあらかじめ混合して、ナトリウム含有量を上記の範囲内に調節したテレフタル酸を調製しておき、スラリー調製工程に供給する方法が好ましく用いられる。
前記ナトリウム化合物としては、水酸化物、アルコキシド、酢酸塩、炭酸塩、シュウ酸塩、及びハロゲン化物、酸化物等が挙げられ、具体的には、水酸化ナトリウム、酢酸ナトリウム、臭化ナトリウム等が挙げられ、入手性の観点から、水酸化ナトリウムが特に好ましい。
テレフタル酸中に含まれるナトリウム原子の質量(TPA−Na)が下記式(2)を満足することが好ましい。
テレフタル酸中に含まれるナトリウム原子の質量(TPA−Na)が下記式(2)を満足することが好ましい。
60ppb ≦ TPA−Na ≦ 500ppb (2)
TPA−Naの下限値は、さらに好ましくは90ppb以上、特に好ましくは100ppb以上である。TPA−Naの上限値は、さらに好ましくは400ppb以下、特好ましくは300ppb以下である。
本発明において用いられるポリエステル重縮合用触媒は、一般的なポリエステル樹脂の重縮合触媒を用いてよい。具体的には、アンチモン化合物、ゲルマニウム化合物、チタン化合物、アルミニウム化合物などが挙げられる。このうち、成形体の異物低減の観点から、ゲルマニウム化合物又はチタン化合物を用いる場合が、より好ましい。
TPA−Naの下限値は、さらに好ましくは90ppb以上、特に好ましくは100ppb以上である。TPA−Naの上限値は、さらに好ましくは400ppb以下、特好ましくは300ppb以下である。
本発明において用いられるポリエステル重縮合用触媒は、一般的なポリエステル樹脂の重縮合触媒を用いてよい。具体的には、アンチモン化合物、ゲルマニウム化合物、チタン化合物、アルミニウム化合物などが挙げられる。このうち、成形体の異物低減の観点から、ゲルマニウム化合物又はチタン化合物を用いる場合が、より好ましい。
本発明において用いられるポリエステル重縮合用触媒は、そのもの単体で用いるよりも、重合速度向上・色調改良・熱安定性向上などの観点から、一般には助触媒金属化合物やリン化合物と組み合わせて使用される。特に、どの重縮合触媒を用いた場合においても、リン化合物と組み合わせることが好ましい。
本発明において重縮合触媒として具体的に用いられるアンチモン化合物としては、例えば、アンチモンの酸化物、脂肪族又は芳香族のカルボン酸の塩、ハロゲン化物、オキシハロゲン化物、アルコラート等が挙げられ、三酸化アンチモン、酢酸アンチモン、アンチモントリスエチレングリコキシドなどのグリコールに可溶性のアンチモン化合物が異物低減の観点から好ましく、三酸化アンチモンが重合活性の点で特に好ましい。
本発明において重縮合触媒として具体的に用いられるアンチモン化合物としては、例えば、アンチモンの酸化物、脂肪族又は芳香族のカルボン酸の塩、ハロゲン化物、オキシハロゲン化物、アルコラート等が挙げられ、三酸化アンチモン、酢酸アンチモン、アンチモントリスエチレングリコキシドなどのグリコールに可溶性のアンチモン化合物が異物低減の観点から好ましく、三酸化アンチモンが重合活性の点で特に好ましい。
本発明において重縮合触媒として具体的に用いられるゲルマニウム化合物としては、例えば、酢酸ゲルマニウム、二酸化ゲルマニウムが挙げられ、二酸化ゲルマニウムが入手性の点で特に好ましい。
本発明においてアンチモン化合物又はゲルマニウム化合物を重縮合触媒として用いた際に、併用される化合物としては、リン化合物が挙げられ、具体的には、例えば、正リン酸、ポリリン酸、及び、トリメチルホスフェート、トリエチルホスフェート、トリ‐n‐ブチルホスフェート、トリオクチルホスフェート、トリフェニルホスフェート、トリクレジルホスフェート、トリス(トリエチレングリコール)ホスフェート、エチルジエチルホスホノアセテート、メチルアシッドホスフェート、エチルアシッドホスフェート、イソプロピルアシッドホスフェート、ブチルアシッドホスフェート、モノブチルホスフェート、ジブチルホスフェート、ジオクチルホスフェート、トリエチレングリコールアシッドホスフェート等の5価のリン化合物、亜リン酸、次亜リン酸、ジエチルホスファイト、トリスドデシルホスファイト、トリスノニルデシルホスファイト、トリフェニルホスファイト等の3価のリン化合物等が挙げられる。その中で、正リン酸、トリス(トリエチレングリコール)ホスフェート、トリメチルアシッドホスフェート、エチルジエチルホスホノアセテート、エチルアシッドホスフェート、トリエチレングリコールアシッドホスフェート及び亜リン酸が好ましく、正リン酸、エチルアシッドホスフェートが特に好ましい。
本発明においてアンチモン化合物又はゲルマニウム化合物を重縮合触媒として用いた際に、併用される化合物としては、リン化合物が挙げられ、具体的には、例えば、正リン酸、ポリリン酸、及び、トリメチルホスフェート、トリエチルホスフェート、トリ‐n‐ブチルホスフェート、トリオクチルホスフェート、トリフェニルホスフェート、トリクレジルホスフェート、トリス(トリエチレングリコール)ホスフェート、エチルジエチルホスホノアセテート、メチルアシッドホスフェート、エチルアシッドホスフェート、イソプロピルアシッドホスフェート、ブチルアシッドホスフェート、モノブチルホスフェート、ジブチルホスフェート、ジオクチルホスフェート、トリエチレングリコールアシッドホスフェート等の5価のリン化合物、亜リン酸、次亜リン酸、ジエチルホスファイト、トリスドデシルホスファイト、トリスノニルデシルホスファイト、トリフェニルホスファイト等の3価のリン化合物等が挙げられる。その中で、正リン酸、トリス(トリエチレングリコール)ホスフェート、トリメチルアシッドホスフェート、エチルジエチルホスホノアセテート、エチルアシッドホスフェート、トリエチレングリコールアシッドホスフェート及び亜リン酸が好ましく、正リン酸、エチルアシッドホスフェートが特に好ましい。
本発明において重縮合触媒として具体的に用いられるチタン化合物としては、例えば、テトラ−n−プロピルチタネート、テトラ−i−プロピルチタネート、テトラ−n−ブチルチタネート、テトラ−n−ブチルチタネートテトラマー、テトラ−t−ブチルチタネート、テトラシクロヘキシルチタネート、テトラフェニルチタネート、テトラベンジルチタネート等のチタンアルコキシド、チタンアルコキシドの加水分解により得られるチタン酸
化物、チタンアルコキシドと珪素アルコキシド又はジルコニウムアルコキシドとの混合物の加水分解により得られるチタン−珪素又はジルコニウム複合酸化物、酢酸チタン、シュウ酸チタン、チタン酸−水酸化アルミニウム混合物、塩化チタン、塩化チタン−塩化アルミニウム混合物、臭化チタン、フッ化チタン、六フッ化チタン酸コバルト、六フッ化チタン酸マンガン、六フッ化チタン酸アンモニウム、チタンアセチルアセトナート等が挙げられ、中でも、テトラ−n−プロピルチタネート、テトラ−i−プロピルチタネート、テトラ−n−ブチルチタネート等のチタンアルコキシド、シュウ酸チタンが好ましく、テトラ−n−ブチルチタネートが重合活性及び安定性の観点から、特に好ましい。
化物、チタンアルコキシドと珪素アルコキシド又はジルコニウムアルコキシドとの混合物の加水分解により得られるチタン−珪素又はジルコニウム複合酸化物、酢酸チタン、シュウ酸チタン、チタン酸−水酸化アルミニウム混合物、塩化チタン、塩化チタン−塩化アルミニウム混合物、臭化チタン、フッ化チタン、六フッ化チタン酸コバルト、六フッ化チタン酸マンガン、六フッ化チタン酸アンモニウム、チタンアセチルアセトナート等が挙げられ、中でも、テトラ−n−プロピルチタネート、テトラ−i−プロピルチタネート、テトラ−n−ブチルチタネート等のチタンアルコキシド、シュウ酸チタンが好ましく、テトラ−n−ブチルチタネートが重合活性及び安定性の観点から、特に好ましい。
本発明においてチタン化合物を重縮合触媒として用いる際に、併用される化合物としては、リン化合物及び助触媒金属化合物が挙げられ、具体的なリン化合物としては、正リン酸、ポリリン酸、及び及び、トリメチルホスフェート、トリエチルホスフェート、トリ‐n‐ブチルホスフェート、トリオクチルホスフェート、トリフェニルホスフェート、トリクレジルホスフェート、トリス(トリエチレングリコール)ホスフェート、エチルジエチルホスホノアセテート、メチルアシッドホスフェート、エチルアシッドホスフェート、イソプロピルアシッドホスフェート、ブチルアシッドホスフェート、モノブチルホスフェート、ジブチルホスフェート、ジオクチルホスフェート、トリエチレングリコールアシッドホスフェート等の5価のリン化合物、亜リン酸、次亜リン酸、ジエチルホスファイト、トリスドデシルホスファイト、トリスノニルデシルホスファイト、トリフェニルホスファイト等の3価のリン化合物等が挙げられる。その中で、正リン酸、トリス(トリエチレングリコール)ホスフェート、トリメチルアシッドホスフェート、エチルジエチルホスホノアセテート、エチルアシッドホスフェート、トリエチレングリコールアシッドホスフェート及び亜リン酸が好ましく、トリス(トリエチレングリコール)ホスフェート、メチルアシッドホスフェート、エチルジエチルホスホノアセテート、エチルアシッドホスフェート、ブチルアシッドホスフェート及びトリエチレングリコールアシッドホスフェートが特に好ましい。
助触媒金属化合物としては、周期表第2族の元素、マンガン、鉄及びコバルトからなる群より選択される少なくとも1種を含む化合物が用いられる。周期表第2族の元素としては、マグネシウム、カルシウム等が挙げられる。具体的な助触媒金属化合物としては、これらの元素の酸化物、水酸化物、アルコキシド、酢酸塩、炭酸塩、シュウ酸塩、及びハロゲン化物等、具体的には、例えば、酸化マグネシウム、水酸化マグネシウム、マグネシウムアルコキシド、酢酸マグネシウム、炭酸マグネシウム、酸化カルシウム、水酸化カルシウム、酢酸カルシウム、炭酸カルシウム、酸化マンガン、水酸化マンガン、酢酸マンガン、酢酸第二鉄、ギ酸コバルト、酢酸コバルト、シュウ酸コバルト、炭酸コバルト、臭化コバルト、コバルトアセチルアセトナート等が挙げられる。なかでも、酢酸マグネシウム又はその水和物が、グリコールに対する溶解度が高く、液状触媒を調製しやすい点で特に好ましい。
本発明において用いるポリエステル重縮合用触媒は、得られるポリエステルの明るみ、及び触媒残渣由来の異物や重縮合活性の低下の面で、アルコール等の溶媒に不溶な固体性化合物を含まないことが好ましい。
本発明において用いるポリエステル重縮合用触媒は、溶媒、各種安定剤又は、添加剤等の成分を含んでいても良い。
本発明において用いるポリエステル重縮合用触媒は、溶媒、各種安定剤又は、添加剤等の成分を含んでいても良い。
本発明において用いるポリエステル重縮合用触媒を得る方法としては、アルコール、重縮合触媒及び併用される化合物を全て混合し、該混合物を濃縮して得ることも出来る。
この方法によって得られる触媒としては、重縮合活性・安全性・コストの点からチタン化合物を原料に含む方がより好ましい。
また、ジオールに、直接、重縮合触媒及び併用される化合物を溶解させたものを添加し
ても良い。なお、チタン化合物、助触媒金属化合物及びリン化合物を混合する際は、アルコール又はジオール中で、助触媒金属化合物、リン化合物、チタン化合物の順で混合させると、チタン化合物成分の析出が抑制され、より好ましい。助触媒金属化合物としては、重縮合活性の点からマグネシウムが特に好ましい。
この方法によって得られる触媒としては、重縮合活性・安全性・コストの点からチタン化合物を原料に含む方がより好ましい。
また、ジオールに、直接、重縮合触媒及び併用される化合物を溶解させたものを添加し
ても良い。なお、チタン化合物、助触媒金属化合物及びリン化合物を混合する際は、アルコール又はジオール中で、助触媒金属化合物、リン化合物、チタン化合物の順で混合させると、チタン化合物成分の析出が抑制され、より好ましい。助触媒金属化合物としては、重縮合活性の点からマグネシウムが特に好ましい。
本発明におけるポリエステル樹脂は、ポリエステル樹脂ペレットの内部に含まれるアルカリ金属の含有量を特定の範囲にする以外は、基本的にはポリエステル樹脂の慣用の製法を用いることが出来る。
原料調製工程では、テレフタル酸を主成分とするジカルボン酸成分とエチレングリコールを主成分とするジオール成分とを、必要に応じて用いられるその他の共重合成分等と共に、スラリー調製槽に投入し、攪拌下に混合した後、必要に応じてろ過することによって原料スラリーとする。原料スラリーにおけるテレフタル酸に対するエチレングリコールのモル比は、通常1.0〜2.0であり、好ましくは1.03〜1.7でああり、更に好ましくは1.05〜1.5である。前記モル比が1.0以上であると、エステル化反応の進行が十分となる。また、前記モル比が2.0以下であると、ポリエステル樹脂製造時に副生するジエチレングリコールの量が増加せず、耐熱性が良好となる傾向にある。
原料調製工程では、テレフタル酸を主成分とするジカルボン酸成分とエチレングリコールを主成分とするジオール成分とを、必要に応じて用いられるその他の共重合成分等と共に、スラリー調製槽に投入し、攪拌下に混合した後、必要に応じてろ過することによって原料スラリーとする。原料スラリーにおけるテレフタル酸に対するエチレングリコールのモル比は、通常1.0〜2.0であり、好ましくは1.03〜1.7でああり、更に好ましくは1.05〜1.5である。前記モル比が1.0以上であると、エステル化反応の進行が十分となる。また、前記モル比が2.0以下であると、ポリエステル樹脂製造時に副生するジエチレングリコールの量が増加せず、耐熱性が良好となる傾向にある。
テレフタル酸を主成分とするジカルボン酸成分とエチレングリコールを主成分とするジオール成分とを、エステル化反応槽で、エステル化反応させた後、得られたエステル化反応生成物であるポリエステル低分子量体を重縮合槽に移送し、前述した予め混合して得られたポリエステル重縮合用触媒を用いて、溶融重縮合反応させプレポリマーを得る。そののち、得られたプレポリマーを用いて固相重縮合反応を行う。また、これらの製造方法はいずれも連続式でも、回分式でもよく、特に制限はされない。
エステル化反応は、例えば、単一のエステル化反応槽、又は複数のエステル化反応槽を直列に接続した多段反応装置を用いて、該反応で生成する水と余剰のエチレングリコールを系外に除去しながら、エステル化反応率(原料ジカルボン酸成分の全カルボキシル基のうちジオール成分と反応してエステル化したものの割合)が、通常90%以上、好ましくは93%以上に達するまで行われる。また、得られるエステル化反応生成物としてのポリエステル低分子量体の数平均分子量は500〜5,000であるのが好ましい。
エステル化反応における反応条件としては、単一のエステル化反応槽を用いる場合、通常200〜280℃程度の温度、通常0〜400kPaG(Gは大気圧に対する相対圧力であることを表す)程度とし、攪拌下に1〜10時間程度の反応時間とする方法が一般的である。また、複数のエステル化反応槽を用いる場合は、次のような反応温度及び反応圧力にて反応を行う。第1段目のエステル化反応槽における反応温度は、通常240〜270℃、好ましくは245〜265℃、反応圧力は、通常5k〜300kPaG、好ましくは10k〜200kPaGである。更に、最終段における反応温度を、通常250〜280℃、好ましくは255〜275℃、反応圧力を通常0〜150kPaG、好ましくは0〜130kPaGとする。
なお、一般には、エステル化反応において、例えば、トリエチルアミン、トリ−n−ブチルアミン、ベンジルジメチルアミン等の第三級アミン、水酸化テトラエチルアンモニウム、水酸化テトラ−n−ブチルアンモニウム、水酸化トリメチルベンジルアンモニウム等の水酸化第四級アンモニウム、炭酸リチウム、炭酸ナトリウム、炭酸カリウム、酢酸ナトリウム等の塩基性化合物等を少量添加しておくことにより、エチレングリコールからのジエチレングリコールの副生を抑制することができるとされているが、ボトル用ポリエステル樹脂においては、特にアミン類を添加することは衛生面から望ましくない。溶融重縮合工程は、ポリエステル重縮合用触媒を用いて行われる。
溶融重縮合工程の例としては、単一の溶融重縮合槽、又は複数の溶融重縮合槽を直列に接続し、例えば、第1段目が攪拌翼を備えた完全混合型の反応器、第2段及び第3段目が攪拌翼を備えた横型プラグフロー型の反応器からなる多段反応装置を用いて、減圧下に、生成するエチレングリコールを系外に留出させながら行う方法が挙げられる。
溶融重縮合工程における反応条件の例としては、単一の重縮合槽を用いる場合、通常250〜290℃程度の温度、常圧から漸次減圧として、最終的に0.3kPa程度とし、攪拌下に1〜20時間程度の反応時間とする方法が一般的である。また、複数の重縮合槽を用いる場合の例としては、次の通りの方法が挙げられる。第1段目の重縮合槽における反応温度を、通常250〜290℃、好ましくは260〜280℃、反応圧力を、通常1〜65kPa、好ましくは2〜26kPaとする。更に、最終段における反応温度を、通常265〜300℃、好ましくは270〜295℃、反応圧力を、通常0.04〜1.0
0kPa、好ましくは0.09〜0.73kPaとする。更に、中間段を用いる場合の反応条件としては、前記第1段と最終段の条件の中間の条件が選択される。例えば、3段反応装置における第2段の反応条件の一例として、反応温度を、通常265〜295℃、好ましくは270〜285℃、反応圧力は、通常0.16〜6.53kPa、好ましくは0.30〜4.03kPaとする方法が挙げられる。
溶融重縮合工程における反応条件の例としては、単一の重縮合槽を用いる場合、通常250〜290℃程度の温度、常圧から漸次減圧として、最終的に0.3kPa程度とし、攪拌下に1〜20時間程度の反応時間とする方法が一般的である。また、複数の重縮合槽を用いる場合の例としては、次の通りの方法が挙げられる。第1段目の重縮合槽における反応温度を、通常250〜290℃、好ましくは260〜280℃、反応圧力を、通常1〜65kPa、好ましくは2〜26kPaとする。更に、最終段における反応温度を、通常265〜300℃、好ましくは270〜295℃、反応圧力を、通常0.04〜1.0
0kPa、好ましくは0.09〜0.73kPaとする。更に、中間段を用いる場合の反応条件としては、前記第1段と最終段の条件の中間の条件が選択される。例えば、3段反応装置における第2段の反応条件の一例として、反応温度を、通常265〜295℃、好ましくは270〜285℃、反応圧力は、通常0.16〜6.53kPa、好ましくは0.30〜4.03kPaとする方法が挙げられる。
本発明において、ポリエステル重縮合用触媒の反応系への添加は、ジカルボン酸成分とジオール成分の混合・調製段階、前記エステル化工程の任意の段階、又は溶融重縮合工程の初期の段階のいずれであってもよい。しかし、色調、透明性に優れたポリエステルを高反応速度で製造するためには、ポリエステル重縮合用触媒の反応系への添加を、エステル化反応率が90%以上となった段階以降に行うのが好ましく、具体的工程の例としては、多段反応装置における最終段のエステル化反応槽、又はエステル化槽から溶融重縮合工程への移送段階のポリエステル低分子量体に添加するのが好ましく、中でも、エステル化槽から溶融重縮合工程への移送段階のポリエステル低分子量体に添加するのがより好ましい。
前記溶融重縮合工程を経て得られるプレポリマーの固有粘度は、フェノール/テトラクロロエタン(質量比1/1)の混合液を溶媒として30℃で測定した値として求められる。その値は好ましくは0.50〜0.67dL/gであり、より好ましくは0.55〜0.65dL/gである。固有粘度が前記範囲下限未満では、後述する重縮合槽からの抜き出し時に、ペレット化が困難になったり、長時間の固相重縮合反応が必要となったりする傾向があるため、生産性が低下する。一方、前記範囲上限超過では、固相重縮合工程における環状三量体の低減が不十分となり、延伸ブロー成形時に金型が汚れやすくなるため、ボトルのヘーズが悪化する。
前記溶融重縮合工程により得られるプレポリマーは、通常、重縮合槽の底部に設けられた抜き出し口からストランド状に抜き出した後、該ストランド状のプレポリマーを水冷しながら、又は水冷後、カッターで切断してペレットとする。更に、該ペレット中に含まれるアセトアルデヒドや環状三量体を低減するため、固相重縮合反応に供する必要がある。固相重縮合反応は従来公知の方法、例えば、特開平2004-292803号公報の段落
[0057]から[0065]に記載されている方法等で行うことができる。本発明によって得られるポリエステル樹脂の固有粘度については、0.68〜0.90dL/gであり、より好ましくは、0.68〜0.85dL/g、更に好ましくは0.68〜0.80dL/gである。前記下限値未満では、成形体の強度が劣る。前記上限値超過では、溶融状態のポリエステル樹脂の流動性が低下し、射出成形時にポリエステル樹脂が金型へ充填される時間が長くなる。
[0057]から[0065]に記載されている方法等で行うことができる。本発明によって得られるポリエステル樹脂の固有粘度については、0.68〜0.90dL/gであり、より好ましくは、0.68〜0.85dL/g、更に好ましくは0.68〜0.80dL/gである。前記下限値未満では、成形体の強度が劣る。前記上限値超過では、溶融状態のポリエステル樹脂の流動性が低下し、射出成形時にポリエステル樹脂が金型へ充填される時間が長くなる。
本発明におけるポリエステル樹脂ペレットの固有粘度は、プレポリマーの固有粘度・プレポリマーのペレットサイズ・反応器中のガス流量又は真空度・反応温度・反応時間・反
応器中ガス組成などによって調節することができる。
なお、プレポリマーのペレットは、前記固相重縮合工程に供する前に、固相重縮合の温度よりも低温で、予備結晶化を行なってもよい。例えば、ペレットを乾燥状態で、120〜200℃、好ましくは130〜190℃、特に好ましくは150℃〜170℃で1分間〜4時間程度加熱してもよい。また、前記固相重縮合工程を経て得られたポリエステル樹脂については、熱水処理や有機溶媒による処理、リン化合物を用いた触媒失活処理などを行ってもよい。
応器中ガス組成などによって調節することができる。
なお、プレポリマーのペレットは、前記固相重縮合工程に供する前に、固相重縮合の温度よりも低温で、予備結晶化を行なってもよい。例えば、ペレットを乾燥状態で、120〜200℃、好ましくは130〜190℃、特に好ましくは150℃〜170℃で1分間〜4時間程度加熱してもよい。また、前記固相重縮合工程を経て得られたポリエステル樹脂については、熱水処理や有機溶媒による処理、リン化合物を用いた触媒失活処理などを行ってもよい。
本発明におけるポリエステル樹脂のジエチレングリコール共重合量は、ポリエステル樹脂を構成するジオールに対して、好ましくは1.5〜5モル%でありさらに好ましくは2〜4モル%であり、、特に好ましくは2〜3モル%である。前記範囲下限未満では、ヘーズが悪化する。前記範囲上限超過では、強度や熱安定性が低下する。
一般にポリエステル樹脂のジエチレングリコール共重合量は、ジエチレングリコールの添加量はもちろん、スラリーモル比、エステル化工程におけるエチレングリコール添加量、エステル化反応温度・時間・圧力によって調節できるが、本発明においては、特に、アルカリ金属化合物を、最終的に得られるポリエステル樹脂に対して、アルカリ金属原子質量換算で50ppb以上400ppb以下となるように添加することによって、色調・ヘーズに代表される、他の樹脂物性を損ねることなく、効果的に上記の範囲内に調節することが可能である。
一般にポリエステル樹脂のジエチレングリコール共重合量は、ジエチレングリコールの添加量はもちろん、スラリーモル比、エステル化工程におけるエチレングリコール添加量、エステル化反応温度・時間・圧力によって調節できるが、本発明においては、特に、アルカリ金属化合物を、最終的に得られるポリエステル樹脂に対して、アルカリ金属原子質量換算で50ppb以上400ppb以下となるように添加することによって、色調・ヘーズに代表される、他の樹脂物性を損ねることなく、効果的に上記の範囲内に調節することが可能である。
本発明におけるポリエステル樹脂の製造中に副生するジエチレングリコールの量は、ポリエステル樹脂を構成するジオールに対して、好ましくは0.5〜4モル%であり、さらに好ましくは1〜3モル%であり、特に好ましくは1〜2モル%である。前記範囲下限未満では、物性を満たすために添加されるジエチレングリコールの添加量が多くなりすぎる。一方、前記範囲上限超過では、ジエチレングリコール共重合量を低減することが出来ず、強度や熱安定性が低下するおそれがある。また、副生ジエチレングリコール量が多いということはすなわち、熱分解反応が多く生じていることを示唆しており、好ましくない。
本発明におけるポリエステル樹脂ペレットの昇温時結晶化発熱温度Tc1は、150〜200℃であることが好ましく、160〜180℃であることがより好ましく、160〜170℃であることが特に好ましい。昇温時結晶化発熱温度Tc1は、結晶化速度の指標として広く用いられるものである。前記範囲下限未満では、本発明のポリエステル樹脂ペレットをボトルに用い、口栓部の結晶化を行った際に、口栓部の寸法変化が大きくなりすぎるため、衛生性に問題が生じる。前記範囲上限超過では、結晶化速度が遅すぎ、口栓部が十分結晶化されないおそれがある。
本発明におけるポリエステル樹脂の色調b値は、4以下であり、2以下がより好ましく、1以下が特に好ましい。
本発明におけるポリエステル樹脂ペレットの色調は、反応温度、反応圧力、反応時の窒素濃度、触媒種、触媒組成、末端カルボキシル基量、色材添加量、アルカリ金属含有量などによって調節することができる。
本発明におけるポリエステル樹脂ペレットの色調は、反応温度、反応圧力、反応時の窒素濃度、触媒種、触媒組成、末端カルボキシル基量、色材添加量、アルカリ金属含有量などによって調節することができる。
本発明におけるポリエステル樹脂ペレット中のアセトアルデヒド含有量は、10ppm以下であることが好ましく、5ppm以下であることがより好ましく、3ppm以下であることが特に好ましい。アセトアルデヒド含有量が上記上限値を超えると、特に飲料用ボトルに成形した際、飲料の香味が損なわれる傾向がある。
本発明におけるポリエステル樹脂ペレット中の環状三量体の含有量は、8000ppm以下であることが好ましく、6000ppm以下であることがより好ましい。環状三量体の含有量が、上記上限値を超えると、ボトル成形時に金型表面を汚し、得られるボトルの壁面に凹凸によるヘーズを発生させる原因となる。
本発明におけるポリエステル樹脂ペレット中の環状三量体の含有量は、8000ppm以下であることが好ましく、6000ppm以下であることがより好ましい。環状三量体の含有量が、上記上限値を超えると、ボトル成形時に金型表面を汚し、得られるボトルの壁面に凹凸によるヘーズを発生させる原因となる。
本発明によって得られるポリエステル樹脂ペレットは常法によりシート、延伸フィルム、ボトル、繊維等、種々の成形体に成形することができるが、ポリエステル樹脂ペレットの製造中に副生するジエチレングリコール量が少ないため、品質のブレが少なく、それに加え、口部結晶化温度を安定的に調節できるため、特に耐圧性、耐熱性を要求される飲料ボトルに好適に用いられる。
以下に実施例により本発明を更に具体的に説明するが、本発明はこれらの実施例に限定されるものではない。なお、実施例における物性の測定は、下記により行った。
<エステル化反応率>
ポリエステル低分子量体試料を乳鉢で粉砕し、その1.0gをビーカーに精秤し、これにジメチルホルムアミド40mLを加えて攪拌しながら180℃で20分間加熱して溶解させた後、180℃のジメチルホルムアミド10mLでビーカー壁を洗浄し、室温まで冷却した。この溶液を、メトローム社製ポテンショグラフ「E−536型」自動滴定装置にて、複合pH電極「EA−120」を用い、0.1N KOHメタノール溶液で滴定した
。得られた滴定曲線の変曲点から求めた滴定量〔A(mL)〕と、JIS K8006の
方法により調製、標定した、0.1N KOHメタノール溶液のファクター〔f1〕、及
び試料質量〔W(g)〕とから、下式により、遊離の末端カルボキシル基量〔AV(meq/g)〕を求めた。
<エステル化反応率>
ポリエステル低分子量体試料を乳鉢で粉砕し、その1.0gをビーカーに精秤し、これにジメチルホルムアミド40mLを加えて攪拌しながら180℃で20分間加熱して溶解させた後、180℃のジメチルホルムアミド10mLでビーカー壁を洗浄し、室温まで冷却した。この溶液を、メトローム社製ポテンショグラフ「E−536型」自動滴定装置にて、複合pH電極「EA−120」を用い、0.1N KOHメタノール溶液で滴定した
。得られた滴定曲線の変曲点から求めた滴定量〔A(mL)〕と、JIS K8006の
方法により調製、標定した、0.1N KOHメタノール溶液のファクター〔f1〕、及
び試料質量〔W(g)〕とから、下式により、遊離の末端カルボキシル基量〔AV(meq/g)〕を求めた。
AV(meq/g)={A×f1 ×(1/10)}/W
次いで、乳鉢で粉砕した試料0.3gを三角フラスコに精秤し、これに0.5N KO
Hエタノール溶液をホールピペットで20mL加え、更に純水10mLを加えて還流冷却器をセットし、表面温度を200℃にしたプレートヒーター上で、時々攪拌しながら2時間加熱還流して試料を加水分解した。放冷後、フェノールフタレインを指示薬として0.5N 塩酸水溶液で滴定した。なお、ここで、0.5N KOHエタノール溶液と0.5N
塩酸水溶液は、JIS K8006の方法により調製、標定した。また、フェノールフタレインは、1gをエタノール90mLに溶解し、純水で100mLに定容したものを用いた。また、同一条件で試料を入れないブランクの状態においても滴定した。その際の、試料の滴定量〔Vs(mL)〕、ブランクの滴定量〔Vb(mL)〕、0.5N塩酸水溶液のファクター〔f2〕、及び試料質量〔W(g)〕とから、下式により、全カルボン酸由来のカルボキシル基量〔SV(meq/g)〕を求めた。
次いで、乳鉢で粉砕した試料0.3gを三角フラスコに精秤し、これに0.5N KO
Hエタノール溶液をホールピペットで20mL加え、更に純水10mLを加えて還流冷却器をセットし、表面温度を200℃にしたプレートヒーター上で、時々攪拌しながら2時間加熱還流して試料を加水分解した。放冷後、フェノールフタレインを指示薬として0.5N 塩酸水溶液で滴定した。なお、ここで、0.5N KOHエタノール溶液と0.5N
塩酸水溶液は、JIS K8006の方法により調製、標定した。また、フェノールフタレインは、1gをエタノール90mLに溶解し、純水で100mLに定容したものを用いた。また、同一条件で試料を入れないブランクの状態においても滴定した。その際の、試料の滴定量〔Vs(mL)〕、ブランクの滴定量〔Vb(mL)〕、0.5N塩酸水溶液のファクター〔f2〕、及び試料質量〔W(g)〕とから、下式により、全カルボン酸由来のカルボキシル基量〔SV(meq/g)〕を求めた。
SV(meq/g)={(Vb−Vs )×f2×(1/2)}/W
次いで、得られたAV(meq/g)、及びSV(meq/g)とから、下式により、エステル化反応率(%)を求めた。
エステル化反応率(%)={(SV−AV)/SV}×100
次いで、得られたAV(meq/g)、及びSV(meq/g)とから、下式により、エステル化反応率(%)を求めた。
エステル化反応率(%)={(SV−AV)/SV}×100
<固有粘度IVの測定>
ウベローデ型粘度計を使用し次の要領で求めた。すなわち、フェノール/テトラクロロエタン(質量比1/1)の混合液を溶媒として、ポリエステル試料のうち、溶融重縮合品は110℃で、固相重縮合品は120℃で、30分間溶解させた後、30℃において、濃度1.0g/dLのポリエステル樹脂溶液及び溶媒のみの落下秒数を測定し、以下の式よ
り求めた。
IV(dL/g)=((1+4KHηSP)0.5−1)/(2KHC)
(但し、ηSP=η/η0 − 1であり、ηはポリマー溶液の落下秒数、η0は溶媒の
落下秒数、Cはポリマー溶液濃度(g/dL)、KHはハギンズの定数である。KHは0.33を採用した。)
ウベローデ型粘度計を使用し次の要領で求めた。すなわち、フェノール/テトラクロロエタン(質量比1/1)の混合液を溶媒として、ポリエステル試料のうち、溶融重縮合品は110℃で、固相重縮合品は120℃で、30分間溶解させた後、30℃において、濃度1.0g/dLのポリエステル樹脂溶液及び溶媒のみの落下秒数を測定し、以下の式よ
り求めた。
IV(dL/g)=((1+4KHηSP)0.5−1)/(2KHC)
(但し、ηSP=η/η0 − 1であり、ηはポリマー溶液の落下秒数、η0は溶媒の
落下秒数、Cはポリマー溶液濃度(g/dL)、KHはハギンズの定数である。KHは0.33を採用した。)
<ジエチレングリコール(DEG)共重合量>
ウィレー型粉砕機にて、1.5mm穴の目皿を用いて粉砕した試料樹脂3gに、4N−KOH/メタノール溶液30mlを加えて還流冷却器をセットし、マグネチックスターラ付きホットプレート(表面温度200℃)上で攪拌しながら、90分間加熱還流し加水分解する。流水につけて冷却後、高純度テレフタル酸約12gを加えて、十分振とうして中和し、pHを9以下としたスラリーを、11G−4グラスフィルターを用いて濾過した後、メタノール2mlで2回洗浄して濾液と洗液を合わせ、ガスクロマトグラフィーへの供試液とする。供試液1μlをマイクロシリンジにて、(株)島津製作所ガスクロマトグラフィー(形式GC−14A)に注入し、各ジオール成分のピークの面積から、全ジオール成分に対する各ジオール成分のモル%(すなわち共重合量)を、下式に従い計算した。
ウィレー型粉砕機にて、1.5mm穴の目皿を用いて粉砕した試料樹脂3gに、4N−KOH/メタノール溶液30mlを加えて還流冷却器をセットし、マグネチックスターラ付きホットプレート(表面温度200℃)上で攪拌しながら、90分間加熱還流し加水分解する。流水につけて冷却後、高純度テレフタル酸約12gを加えて、十分振とうして中和し、pHを9以下としたスラリーを、11G−4グラスフィルターを用いて濾過した後、メタノール2mlで2回洗浄して濾液と洗液を合わせ、ガスクロマトグラフィーへの供試液とする。供試液1μlをマイクロシリンジにて、(株)島津製作所ガスクロマトグラフィー(形式GC−14A)に注入し、各ジオール成分のピークの面積から、全ジオール成分に対する各ジオール成分のモル%(すなわち共重合量)を、下式に従い計算した。
あるジオール成分のモル%=(ACO×CfCO)/(Σ(A×Cf))×100
ACO:そのジオール成分の面積(μV・秒), CfCO:そのジオール成分の補正係数
A:各ジオール成分の面積(μV・秒), Cf:各ジオール成分の補正係数
なお、ガスクロマトグラフィーの使用条件としては、
カラム :J&W社製「DB−WAX」(0.53mm×30m)
カラム温度:80℃〜160℃, 気化室温度:230℃, 検出器温度:230℃, ガス流量:キャリア(窒素):10ml/min, 水素:0.5kg/cm2, 空気:0.5k
g/cm2, 検出器:FID, 感度:102MΩ とした。
ACO:そのジオール成分の面積(μV・秒), CfCO:そのジオール成分の補正係数
A:各ジオール成分の面積(μV・秒), Cf:各ジオール成分の補正係数
なお、ガスクロマトグラフィーの使用条件としては、
カラム :J&W社製「DB−WAX」(0.53mm×30m)
カラム温度:80℃〜160℃, 気化室温度:230℃, 検出器温度:230℃, ガス流量:キャリア(窒素):10ml/min, 水素:0.5kg/cm2, 空気:0.5k
g/cm2, 検出器:FID, 感度:102MΩ とした。
<副生ジエチレングリコール(DEG)量>
重縮合中で副生するジエチレングリコール(DEG)量を、下記式によって見積もった。
副生DEG量(モル%)=ポリエステル樹脂中のDEG共重合量(モル%)−重縮合系中に添加したDEG量(モル%)
ここで定義されるDEG量(モル%)は全て、ポリエステル樹脂を構成するジオール成分に対するものである。
但し、「重縮合系中に添加したDEG量」には、スラリー調製時に添加を行う場合も含まれる。
重縮合中で副生するジエチレングリコール(DEG)量を、下記式によって見積もった。
副生DEG量(モル%)=ポリエステル樹脂中のDEG共重合量(モル%)−重縮合系中に添加したDEG量(モル%)
ここで定義されるDEG量(モル%)は全て、ポリエステル樹脂を構成するジオール成分に対するものである。
但し、「重縮合系中に添加したDEG量」には、スラリー調製時に添加を行う場合も含まれる。
<ヘーズ>
ポリエステル樹脂ペレット約5kgを、縦×横×高さが概略300×500×80mmのステンレス製バットに平坦に入れ、ヤマト科学社真空乾燥機DP63を用いて、145℃12時間の真空乾燥を実施した。得られたペレットを、射出成形機(名機製作所製M−70AIIDM)にて、シリンダー温度280℃、金型温度21℃、樹脂圧30MPaGで射出成形し、縦50mm、横100mmであり、横方向に6mmから3.5mmまで段差0.5mmの6段階の厚みを有する50.5gの段付き成形板とした。この成形板の厚み5mm部のヘーズをヘーズメーター(日本電色工業社製「NDH−300A」)にて測定した。
ポリエステル樹脂ペレット約5kgを、縦×横×高さが概略300×500×80mmのステンレス製バットに平坦に入れ、ヤマト科学社真空乾燥機DP63を用いて、145℃12時間の真空乾燥を実施した。得られたペレットを、射出成形機(名機製作所製M−70AIIDM)にて、シリンダー温度280℃、金型温度21℃、樹脂圧30MPaGで射出成形し、縦50mm、横100mmであり、横方向に6mmから3.5mmまで段差0.5mmの6段階の厚みを有する50.5gの段付き成形板とした。この成形板の厚み5mm部のヘーズをヘーズメーター(日本電色工業社製「NDH−300A」)にて測定した。
<昇温時結晶化発熱温度Tc1>
前述のポリエステル樹脂の成形板から10.0±1.0mgを切りだして精秤し、DSC(示差走査熱量計)での測定を実施した。測定条件としては、20℃から285℃まで20℃/minで昇温し、285℃で3分間保持したのち急冷した。再度20℃から285℃まで20℃/minで昇温した時の結晶化発熱ピーク温度をTc1(℃)とした。
前述のポリエステル樹脂の成形板から10.0±1.0mgを切りだして精秤し、DSC(示差走査熱量計)での測定を実施した。測定条件としては、20℃から285℃まで20℃/minで昇温し、285℃で3分間保持したのち急冷した。再度20℃から285℃まで20℃/minで昇温した時の結晶化発熱ピーク温度をTc1(℃)とした。
<色調b値>
ポリエステル樹脂ペレットを、内径36mm、深さ15mmの円柱状の粉体測色用セルに充填し、測色色差計(日本電色工業社製「ZE2000」)を用いて、JIS Z87
30の参考1に記載されるLab表色系におけるハンターの色差式の色座標b値を、反射法により測定セルを90度ずつ回転させて4箇所測定した値の単純平均値として求めた。
ポリエステル樹脂ペレットを、内径36mm、深さ15mmの円柱状の粉体測色用セルに充填し、測色色差計(日本電色工業社製「ZE2000」)を用いて、JIS Z87
30の参考1に記載されるLab表色系におけるハンターの色差式の色座標b値を、反射法により測定セルを90度ずつ回転させて4箇所測定した値の単純平均値として求めた。
<ポリエステル樹脂ペレット中のアルカリ金属量(PET−M)及びナトリウム原子の含有量(PET−Na)>
本発明において、PET−Mとは、ポリエステル樹脂ペレットの内部に含まれる、リチウム、カリウム、ナトリウム等アルカリ金属原子の質量ppbの総和であり、PET−Naとは、ポリエステル樹脂ペレットの内部に含まれる、ナトリウム原子の質量ppbのことであり、以下の方法で測定を行った。
樹脂表面の酸洗浄のため、あらかじめテフロン(登録商標)ボトルに樹脂ペレット試料を採取し、1N−HClを添加して超音波洗浄を30分間実施した後、超純水で3回洗浄した。その後、樹脂ペレット試料を石英製ケルダールフラスコに移し、硫酸と硝酸を添加して加熱分解した。分解液を超純水にて定容し、プラズマ発光分光分析装置(Agilent Technologies社製 HP4500型)を用いて定量し、ポリエステル樹脂中の質量ppbを算出した。
本発明において、PET−Mとは、ポリエステル樹脂ペレットの内部に含まれる、リチウム、カリウム、ナトリウム等アルカリ金属原子の質量ppbの総和であり、PET−Naとは、ポリエステル樹脂ペレットの内部に含まれる、ナトリウム原子の質量ppbのことであり、以下の方法で測定を行った。
樹脂表面の酸洗浄のため、あらかじめテフロン(登録商標)ボトルに樹脂ペレット試料を採取し、1N−HClを添加して超音波洗浄を30分間実施した後、超純水で3回洗浄した。その後、樹脂ペレット試料を石英製ケルダールフラスコに移し、硫酸と硝酸を添加して加熱分解した。分解液を超純水にて定容し、プラズマ発光分光分析装置(Agilent Technologies社製 HP4500型)を用いて定量し、ポリエステル樹脂中の質量ppbを算出した。
<テレフタル酸中に含まれるナトリウム原子の質量(TPA−Na)>
本発明において、TPA−Naとは、テレフタル酸中に含まれるナトリウム原子の質量ppbのことであり、以下の方法で測定を行った。
試料を白金坩堝に秤量し、硫酸を加えて加熱分解を行い、赤外炉にて灰化を行った。灰化後、硝酸を加えて加熱分解した後、分解液を純水にて定容し、原子吸光分析装置(Agilent社製 AA280Z型)を用いて定量し、テレフタル酸中の質量ppbを算出した。
本発明において、TPA−Naとは、テレフタル酸中に含まれるナトリウム原子の質量ppbのことであり、以下の方法で測定を行った。
試料を白金坩堝に秤量し、硫酸を加えて加熱分解を行い、赤外炉にて灰化を行った。灰化後、硝酸を加えて加熱分解した後、分解液を純水にて定容し、原子吸光分析装置(Agilent社製 AA280Z型)を用いて定量し、テレフタル酸中の質量ppbを算出した。
(実施例1)
スラリー調製槽、及びそれに直列に接続された2段のエステル化反応槽、及び2段目のエステル化反応槽に直列に接続された3段の溶融重縮合槽からなる連続重縮合装置を用い、スラリー調製槽に、ナトリウムを300ppbで含むテレフタル酸とエチレングリコールを、質量比で865:485の割合で連続的に供給すると共に、エチルアシッドホスフェートの0.3質量%エチレングリコール溶液を、最終的に得られるポリエステル樹脂1トン当たりリン原子としての含有量が0.194モル(6ppm)となる量で連続的に添加して、攪拌、混合することによりスラリーを調製し、このスラリーを、窒素雰囲気下で260℃、50kPaG、平均滞留時間4時間に設定された第1段目のエステル化反応槽、次いで、窒素雰囲気下で260℃、5kPaG、平均滞留時間1.5時間に設定された第2段目のエステル化反応槽に連続的に移送して、エステル化反応させた。エステル化反応率は、第1段目においては85%、第2段目においては95%であった。この際、第2段目の反応器へ、得られるポリエステル樹脂中のDEG共重合量が2.00モル%となるように、ジエチレングリコールを添加した。
スラリー調製槽、及びそれに直列に接続された2段のエステル化反応槽、及び2段目のエステル化反応槽に直列に接続された3段の溶融重縮合槽からなる連続重縮合装置を用い、スラリー調製槽に、ナトリウムを300ppbで含むテレフタル酸とエチレングリコールを、質量比で865:485の割合で連続的に供給すると共に、エチルアシッドホスフェートの0.3質量%エチレングリコール溶液を、最終的に得られるポリエステル樹脂1トン当たりリン原子としての含有量が0.194モル(6ppm)となる量で連続的に添加して、攪拌、混合することによりスラリーを調製し、このスラリーを、窒素雰囲気下で260℃、50kPaG、平均滞留時間4時間に設定された第1段目のエステル化反応槽、次いで、窒素雰囲気下で260℃、5kPaG、平均滞留時間1.5時間に設定された第2段目のエステル化反応槽に連続的に移送して、エステル化反応させた。エステル化反応率は、第1段目においては85%、第2段目においては95%であった。この際、第2段目の反応器へ、得られるポリエステル樹脂中のDEG共重合量が2.00モル%となるように、ジエチレングリコールを添加した。
その際、第2段目に設けた上部配管を通じて、酢酸マグネシウム4水和物の0.6質量%エチレングリコール溶液を、最終的に得られるポリエステル樹脂1トン当たりマグネシウム原子としての含有量が0.247モル(6ppm)となる量で連続的に添加した。
引き続いて、前記で得られたエステル化反応生成物を溶融重縮合槽に移送する際、その移送配管中のエステル化反応生成物に、テトラブチルチタネートを、チタン原子の濃度0.15質量%、水分濃度を0.5質量%としたエチレングリコール溶液として、最終的に得られるポリエステル樹脂1トン当たりチタン原子としての含有量が0.063モル(4ppm)となる量で連続的に添加しつつ、270℃、2.63kPaに設定された第1段目の溶融重縮合槽、次いで、278℃、0.53kPaに設定された第2段目の溶融重縮合槽、次いで、280℃、0.33kPaに設定された第3段目の溶融重縮合槽に連続的に移送して、得られるポリエステルプレポリマーの固有粘度が0.59dL/gとなるよ
うに各重縮合槽における滞留時間を調整して合計3.2時間で溶融重縮合させ、重縮合槽の底部に設けられた抜き出し口からストランド状に抜き出して、水冷後、カッターで切断してポリエステルプレポリマーペレットを製造した。
引き続いて、前記で得られたエステル化反応生成物を溶融重縮合槽に移送する際、その移送配管中のエステル化反応生成物に、テトラブチルチタネートを、チタン原子の濃度0.15質量%、水分濃度を0.5質量%としたエチレングリコール溶液として、最終的に得られるポリエステル樹脂1トン当たりチタン原子としての含有量が0.063モル(4ppm)となる量で連続的に添加しつつ、270℃、2.63kPaに設定された第1段目の溶融重縮合槽、次いで、278℃、0.53kPaに設定された第2段目の溶融重縮合槽、次いで、280℃、0.33kPaに設定された第3段目の溶融重縮合槽に連続的に移送して、得られるポリエステルプレポリマーの固有粘度が0.59dL/gとなるよ
うに各重縮合槽における滞留時間を調整して合計3.2時間で溶融重縮合させ、重縮合槽の底部に設けられた抜き出し口からストランド状に抜き出して、水冷後、カッターで切断してポリエステルプレポリマーペレットを製造した。
引き続いて、前記で得られたポリエステルプレポリマーペレットを、窒素雰囲気下で約160℃に保持された攪拌結晶化機内に滞留時間が約60分となるように連続的に供給して結晶化させた後、塔型の固相重縮合装置に連続的に供給し、窒素雰囲気下、205℃で、得られるポリエステル樹脂ペレットの固有粘度が0.85dL/gとなるように滞留時間を調整して24時間加熱することにより固相重縮合させた。
得られたポリエステル樹脂ペレットの評価を、前述した方法によって行った。これらの結果を表1に示す。
得られたポリエステル樹脂ペレットの評価を、前述した方法によって行った。これらの結果を表1に示す。
(比較例1)
実施例1で用いたテレフタル酸に含まれるナトリウムの量を、10ppb未満とした以外は、実施例1と同様に実施し、これらの結果を表1に示した。
実施例1と比較して、Tc1が高いことがわかる。
実施例1で用いたテレフタル酸に含まれるナトリウムの量を、10ppb未満とした以外は、実施例1と同様に実施し、これらの結果を表1に示した。
実施例1と比較して、Tc1が高いことがわかる。
(実施例2)
スラリー調製槽、及びそれに直列に接続された2段のエステル化反応槽、及び2段目のエステル化反応槽に直列に接続された3段の溶融重縮合槽からなる連続重縮合装置を用い、スラリー調製槽に、ナトリウムを300ppbで含むテレフタル酸とエチレングリコールを、質量比で166:71の割合で連続的に供給すると共に、正リン酸を、最終的に得られるポリエステル樹脂1トン当たりリン原子としての含有量が0.840モル(26ppm)、二酸化ゲルマニウムを、重縮合循環系中に留出する分も考慮し、最終的に得られるポリエステル樹脂1トンあたり0.660モル(48ppm)となるように連続的に添加して、攪拌、混合することによりスラリーを調製し、このスラリーを、窒素雰囲気下で260℃、120kPaG、平均滞留時間2.6時間に設定された第1段目のエステル化反応槽、次いで、窒素雰囲気下で260℃、0kPaG、平均滞留時間1時間に設定された第2段目のエステル化反応槽に連続的に移送して、エステル化反応させた。そのとき、エステル化反応率は、第1段目においては85%、第2段目においては97%であった。この際、第2段目の反応器へ、得られるポリエステル樹脂中のDEG共重合量が2.40モル%となるように、ジエチレングリコールを添加した。
スラリー調製槽、及びそれに直列に接続された2段のエステル化反応槽、及び2段目のエステル化反応槽に直列に接続された3段の溶融重縮合槽からなる連続重縮合装置を用い、スラリー調製槽に、ナトリウムを300ppbで含むテレフタル酸とエチレングリコールを、質量比で166:71の割合で連続的に供給すると共に、正リン酸を、最終的に得られるポリエステル樹脂1トン当たりリン原子としての含有量が0.840モル(26ppm)、二酸化ゲルマニウムを、重縮合循環系中に留出する分も考慮し、最終的に得られるポリエステル樹脂1トンあたり0.660モル(48ppm)となるように連続的に添加して、攪拌、混合することによりスラリーを調製し、このスラリーを、窒素雰囲気下で260℃、120kPaG、平均滞留時間2.6時間に設定された第1段目のエステル化反応槽、次いで、窒素雰囲気下で260℃、0kPaG、平均滞留時間1時間に設定された第2段目のエステル化反応槽に連続的に移送して、エステル化反応させた。そのとき、エステル化反応率は、第1段目においては85%、第2段目においては97%であった。この際、第2段目の反応器へ、得られるポリエステル樹脂中のDEG共重合量が2.40モル%となるように、ジエチレングリコールを添加した。
引き続いて、前記で得られたエステル化反応生成物を溶融重縮合槽に移送し、263℃、3.03kPaに設定された第1段目の溶融重縮合槽、次いで、268℃、0.33kPaに設定された第2段目の溶融重縮合槽、次いで、270℃、0.33kPaに設定された第3段目の溶融重縮合槽に連続的に移送して、得られるポリエステルプレポリマーの固有粘度が0.53dL/gとなるように各重縮合槽における滞留時間を調整して合計2.7時間で溶融重縮合させ、重縮合槽の底部に設けられた抜き出し口からストランド状に抜き出して、水冷後、カッターで切断してポリエステルプレポリマーペレットを製造した。
引き続いて、前記で得られたポリエステルプレポリマーペレットを、窒素雰囲気下で約160℃に保持された攪拌結晶化機内に滞留時間が約60分となるように連続的に供給して結晶化させた後、塔型の固相重縮合装置に連続的に供給し、窒素雰囲気下、205℃で、得られるポリエステル樹脂ペレットの固有粘度が0.74dL/gとなるように滞留時間を調整して24時間加熱することにより固相重縮合させた。
得られたポリエステル樹脂ペレットの評価を、前述した方法によって行った。これらの結果を表2に示した。この時、ヘーズは4.5%であった。
得られたポリエステル樹脂ペレットの評価を、前述した方法によって行った。これらの結果を表2に示した。この時、ヘーズは4.5%であった。
(実施例3)
実施例2で用いたテレフタル酸に含まれるナトリウムの量を、150ppbとした以外は、実施例2と同様に実施し、これらの結果を表2に示した。この時、ヘーズは3.9%であった。
実施例2で用いたテレフタル酸に含まれるナトリウムの量を、150ppbとした以外は、実施例2と同様に実施し、これらの結果を表2に示した。この時、ヘーズは3.9%であった。
(実施例4)
実施例2で用いたテレフタル酸に含まれるナトリウムの量を、100ppbとした以外は、実施例2と同様に実施し、これらの結果を表2に示した。この時、ヘーズは3.3%であった。
実施例2で用いたテレフタル酸に含まれるナトリウムの量を、100ppbとした以外は、実施例2と同様に実施し、これらの結果を表2に示した。この時、ヘーズは3.3%であった。
(比較例2)
実施例2で用いたテレフタル酸に含まれるナトリウムの量を、10ppb未満とした以外は、実施例2と同様に実施し、これらの結果を表2に示した。
実施例2と比較して、ジエチレングリコールの副生量が多く、熱安定性に劣ることが示唆される。
実施例2で用いたテレフタル酸に含まれるナトリウムの量を、10ppb未満とした以外は、実施例2と同様に実施し、これらの結果を表2に示した。
実施例2と比較して、ジエチレングリコールの副生量が多く、熱安定性に劣ることが示唆される。
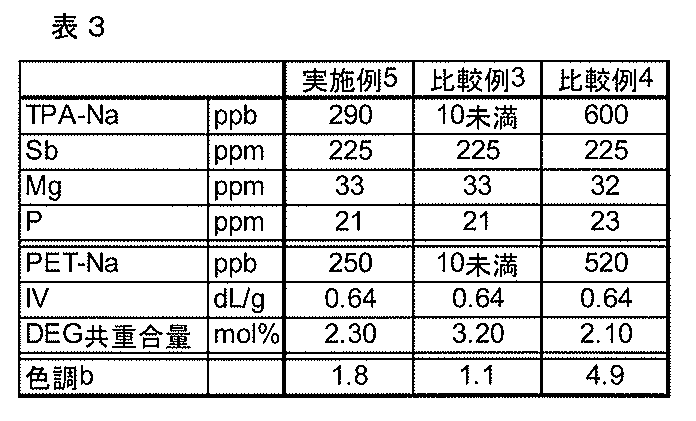
(実施例5)
スラリー調製槽、及びそれに直列に接続された2段のエステル化反応槽、及び2段目のエステル化反応槽に直列に接続された3段の溶融重縮合槽からなる連続式重縮合装置を用い、スラリー調製槽に、ナトリウムを290ppbで含むテレフタル酸とエチレングリコールを、質量比で220:123の割合で連続的に供給すると共に、エチルアシッドホスフェートのエチレングリコール溶液を、最終的に得られるポリエステル樹脂1トン当たりリン原子としての含有量が0.678モル(21ppm)となる量で連続的に添加して、攪拌、混合することによりスラリーを調製し、このスラリーを、窒素雰囲気下で260℃、相対圧力92kPaG、平均滞留時間4.5時間に設定され、反応生成物が存在する第1段目のエステル化反応槽に供給し、次いで、第1段目のエステル化反応生成物を、窒素雰囲気下で260℃、相対圧力5kPaG、平均滞留時間1.8時間に設定された第2段目のエステル化反応槽に連続的に移送して、更にエステル化反応させた。この際、第2段目の反応器へジエチレングリコールを添加しなかった。
スラリー調製槽、及びそれに直列に接続された2段のエステル化反応槽、及び2段目のエステル化反応槽に直列に接続された3段の溶融重縮合槽からなる連続式重縮合装置を用い、スラリー調製槽に、ナトリウムを290ppbで含むテレフタル酸とエチレングリコールを、質量比で220:123の割合で連続的に供給すると共に、エチルアシッドホスフェートのエチレングリコール溶液を、最終的に得られるポリエステル樹脂1トン当たりリン原子としての含有量が0.678モル(21ppm)となる量で連続的に添加して、攪拌、混合することによりスラリーを調製し、このスラリーを、窒素雰囲気下で260℃、相対圧力92kPaG、平均滞留時間4.5時間に設定され、反応生成物が存在する第1段目のエステル化反応槽に供給し、次いで、第1段目のエステル化反応生成物を、窒素雰囲気下で260℃、相対圧力5kPaG、平均滞留時間1.8時間に設定された第2段目のエステル化反応槽に連続的に移送して、更にエステル化反応させた。この際、第2段目の反応器へジエチレングリコールを添加しなかった。
引き続いて、前記で得られたエステル化反応生成物を溶融重縮合槽に移送する際、その移送配管中のエステル化反応生成物に、酢酸マグネシウム4水和物のエチレングリコール溶液と三酸化アンチモンのエチレングリコール溶液を、それぞれ最終的に得られるポリエステル樹脂に対して、マグネシウム原子としての含有量が1.36モル/トン(33ppm)、アンチモンとしての含有量が1.85モル/トン(225ppm)となる量で連続的に添加しつつ、272℃、3.5kPaに設定された第1段目の溶融重縮合槽、次いで、276℃、0.4kPaに設定された第2段目の溶融重縮合槽、次いで、277℃、0.3kPaに設定された第3段目の溶融重縮合槽に連続的に移送して、得られるポリエステル樹脂の固有粘度が0.64dL/gとなるように、各重縮合槽における滞留時間を調整して合計時間が3.3時間となるようにして溶融重縮合させ、重縮合槽の底部に設けられた抜き出し口からストランド状に抜き出して、水冷後、カッターで切断してポリエステル樹脂ペレットを製造した。得られたポリエステル樹脂ペレットの評価結果を表3に示した。
(比較例3)
実施例5で用いたテレフタル酸に含まれるナトリウムの量を、10ppb未満とした以外は、実施例5と同様に実施し、これらの結果を表3に示した。
実施例5と比較して、ジエチレングリコールの副生量が非常に多いことがわかる。
実施例5で用いたテレフタル酸に含まれるナトリウムの量を、10ppb未満とした以外は、実施例5と同様に実施し、これらの結果を表3に示した。
実施例5と比較して、ジエチレングリコールの副生量が非常に多いことがわかる。
(比較例4)
実施例5で用いたテレフタル酸に含まれるナトリウムの量を、600ppbとした以外は、実施例5と同様に実施し、これらの結果を表3に示した。
実施例5と比較して、色調b値が高く、色調の点で問題がある。
実施例5で用いたテレフタル酸に含まれるナトリウムの量を、600ppbとした以外は、実施例5と同様に実施し、これらの結果を表3に示した。
実施例5と比較して、色調b値が高く、色調の点で問題がある。
本発明におけるポリエステル樹脂ペレットは、色調及びヘーズを保ったまま、ジエチレングリコールの副生量が少なく、結晶化速度を速めることができ、各種飲料ボトル、特に耐熱圧飲料ボトルの安定製造に好適に使用できる。
Claims (3)
- ペレットの内部に含まれるアルカリ金属原子の含有量(PET−M)が下記式(1)を満足するポリエステル樹脂ペレット。
50ppb ≦ PET−M ≦ 400ppb (1) - 請求項1記載のアルカリ金属が、ナトリウムであるポリエステル樹脂ペレット。
- テレフタル酸を主成分とするジカルボン酸成分と、エチレングリコールを主成分とするジオール成分から、ポリエステル樹脂を製造し、更にそのポリエステル樹脂からポリエステル樹脂ペレットを製造する方法において、原料であるテレフタル酸中に含まれるナトリウム原子の含有量(TPA−Na)が下記式(2)を満足し、ペレットの内部に含まれるナトリウム原子の含有量(PET−Na)が下記式(3)を満足するポリエステル樹脂ペレットの製造方法。
60ppb ≦ TPA−Na ≦ 500ppb (2)
50ppb ≦ PET−Na ≦ 400ppb (3)
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015007949A JP2016132720A (ja) | 2015-01-19 | 2015-01-19 | ポリエステル樹脂ペレット及びその製造方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015007949A JP2016132720A (ja) | 2015-01-19 | 2015-01-19 | ポリエステル樹脂ペレット及びその製造方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2016132720A true JP2016132720A (ja) | 2016-07-25 |
Family
ID=56437580
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2015007949A Pending JP2016132720A (ja) | 2015-01-19 | 2015-01-19 | ポリエステル樹脂ペレット及びその製造方法 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP2016132720A (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2016132733A (ja) * | 2015-01-20 | 2016-07-25 | 三菱化学株式会社 | ポリエステル樹脂ペレット及びその製造方法 |
| JP2023167414A (ja) * | 2022-05-12 | 2023-11-24 | 三菱瓦斯化学株式会社 | 樹脂組成物及び樹脂組成物の製造方法 |
-
2015
- 2015-01-19 JP JP2015007949A patent/JP2016132720A/ja active Pending
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2016132733A (ja) * | 2015-01-20 | 2016-07-25 | 三菱化学株式会社 | ポリエステル樹脂ペレット及びその製造方法 |
| JP2023167414A (ja) * | 2022-05-12 | 2023-11-24 | 三菱瓦斯化学株式会社 | 樹脂組成物及び樹脂組成物の製造方法 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4529485B2 (ja) | ポリエステル重合触媒、その製造方法、及びそれを用いたポリエステルの製造方法 | |
| US20110301019A1 (en) | Catalyst for polyester polycondensation and method for producing polyester resin using the same | |
| JP3690255B2 (ja) | ポリエステル樹脂の製造方法及びそれにより得られるポリエステル樹脂 | |
| WO2004013203A1 (ja) | ポリエステル樹脂及びその製造方法 | |
| JP3679264B2 (ja) | ポリエステル樹脂の製造方法 | |
| TW201617379A (zh) | 製備具有少量副產物之聚(對苯二甲酸丙二酯)的方法 | |
| JP2003221437A (ja) | ポリエステル樹脂の製造方法 | |
| JP2016132720A (ja) | ポリエステル樹脂ペレット及びその製造方法 | |
| JP4529590B2 (ja) | ポリエステル樹脂及びその製造方法 | |
| KR20160047218A (ko) | 부산물의 함량이 낮은 폴리(트리메틸렌 테레프탈레이트)의 연속 제조 방법 | |
| JP5045216B2 (ja) | ポリエステル重縮合用触媒の製造方法、該触媒を用いたポリエステルの製造方法 | |
| JP2005089741A (ja) | ポリエステル樹脂及びその製造方法 | |
| JP4983043B2 (ja) | ポリエステル重縮合用触媒、その製造方法およびポリエステルの製造方法 | |
| TW202413479A (zh) | 化學回收聚對苯二甲酸乙二酯樹脂及其成形體、以及化學回收聚對苯二甲酸乙二酯樹脂之製造方法 | |
| JP2011026438A (ja) | ポリエチレンテレフタレートの製造方法 | |
| JP2006241294A (ja) | ポリエステル樹脂及び該樹脂の製造方法 | |
| JP4915296B2 (ja) | ポリエステル樹脂及びこれから得られる成型体 | |
| JP4844088B2 (ja) | ポリエステル重縮合用触媒及びそれを用いたポリエステル樹脂の製造方法 | |
| JP2014205796A (ja) | ポリエステル樹脂及びその製造方法 | |
| CN102174175A (zh) | 聚对苯二甲酸丁二醇酯及其制造方法 | |
| JP2009154888A (ja) | 炭酸飲料ボトル用ポリエステル樹脂 | |
| JP6601048B2 (ja) | フィルム用ポリエステル樹脂及びその製造方法 | |
| JP2009024088A (ja) | ゴム補強繊維用ポリエステル樹脂及びその製造方法 | |
| JP4251827B2 (ja) | ポリエステル樹脂 | |
| JP4458738B2 (ja) | ポリエステル樹脂及びこれから得られる成型体 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A711 | Notification of change in applicant |
Free format text: JAPANESE INTERMEDIATE CODE: A712 Effective date: 20170424 |