JP2010148502A - β−ホスホグルコムターゼとその製造方法並びに用途 - Google Patents
β−ホスホグルコムターゼとその製造方法並びに用途 Download PDFInfo
- Publication number
- JP2010148502A JP2010148502A JP2009268783A JP2009268783A JP2010148502A JP 2010148502 A JP2010148502 A JP 2010148502A JP 2009268783 A JP2009268783 A JP 2009268783A JP 2009268783 A JP2009268783 A JP 2009268783A JP 2010148502 A JP2010148502 A JP 2010148502A
- Authority
- JP
- Japan
- Prior art keywords
- enzyme
- pgm
- dna
- glucose
- phosphoglucomutase
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Withdrawn
Links
Images





Classifications
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/52—Improvements relating to the production of bulk chemicals using catalysts, e.g. selective catalysts
Landscapes
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Enzymes And Modification Thereof (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
Abstract
【解決手段】耐熱性に優れる嫌気性好熱菌であるサーモアナエロバクター・ブロッキイ(Thermoanaerobacter brockii)ATCC35047に由来するβ−ホスホグルコムターゼ組換え酵素を精製し性質を調べたところ、65℃においても酵素反応が可能な優れた耐熱性を有することを見出した。従来の酵素と比べて、より高い温度で利用できるので、少ない酵素量で長時間の反応が可能となり、反応液の雑菌汚染の懸念も少ない。当該酵素をコードするDNA、当該酵素の製造方法とこれを用いたオリゴ糖の製造方法を提供。
【選択図】なし
Description
(1)作用
G6Pをβ−G1Pに変換し、β−G1PをG6Pに変換する;
(2)分子量
SDS−ゲル電気泳動法において、25,000±5,000ダルトン;
(3)至適温度
10mMのMg2+イオン存在下、pH6.5、30分間反応の条件下で65℃;
(4)至適pH
10mMのMg2+イオン存在下、55℃、30分間反応の条件下でpH6.5;
(4)温度安定性
10mMのMg2+イオン存在下、pH6.5、30分間保持する条件下で60℃まで安定;
(5)pH安定性
10mMのMg2+イオン存在下、37℃、24時間保持する条件下でpH4.5乃至9.0の範囲で安定;
(6)金属イオンによる活性化
Mg2+、Mn2+、Co2+イオンにより活性化される;
β−PGMをコードするDNAをサーモアナエロバクター・ブロッキイ ATCC35047からクローニングし、自律複製可能な組換えDNAの作製、酵素をコードするDNAの塩基配列の決定及び形質転換体の調製を行った。
本願と同じ出願人による特開平10−304882号公報の実施例A−2に記載の方法に従い、サーモアナエロバクター・ブロッキイ ATCC35047株のゲノムDNAを調製した。
非特許文献1記載のラクトコッカス・ラクチス由来β−PGMのアミノ酸配列に基づき、サーモアナエロバクター属微生物に由来し、且つ、同アミノ酸配列と相同性を示すアミノ酸配列を有する蛋白質を、データベース『GenBank』を対象に検索したところ、サーモアナエロバクター・テングコンジェンシス(Thermoanaerobacter tengcongensis) MB4のゲノムDNA(Accession NC 003869)において、推定ホスホヘキソムターゼをコードするDNAが認められた。なお、当該推定ホスホヘキソムターゼは配列表における配列番号3で示されるアミノ酸配列を有しており、配列表における配列番号4で示される塩基配列を有するDNAにコードされていた。サーモアナエロバクター・ブロッキイATCC35047のβ−PGMのアミノ酸配列は、上記サーモアナエロバクター・テングコンジェンシス MB4の推定ホスホヘキソムターゼのアミノ酸配列と全域にわたって相同性が高いと仮定し、サーモアナエロバクター・ブロッキイATCC35047由来のβ−PGMをコードするDNAのクローニングを試みた。
実験1−2で決定したβ−PGMをコードするDNAの塩基配列を確定させる目的で、当該塩基配列における翻訳開始コドン及び終始コドンを含むプライマー、すなわち、配列表における配列番号9及び10で示される塩基配列を有するオリゴヌクレオチドを合成し、それぞれをセンスプライマー及びアンチセンスプライマーとして用いて、実験1−1で得たゲノムDNAを鋳型としてPCRを行い、β−PGMの全域をコードするDNAの再クローニングを行い、組換えDNA、pCR−PGMを得た。pCR−PGMに含まれるDNAの塩基配列を常法のジデオキシ法により決定したところ、実験1−2で決定した配列表における配列番号2で示される塩基配列と全く同一の塩基配列を含んでいた。
組換えDNA、pCR−TbPGM中のβ−PGMをコードするDNAを発現用ベクターに挿入し、得られた発現用組換えDNAを大腸菌に導入し形質転換体を調製した。
実験2で得た形質転換体RSET−TbPGM−2を、500ml容の三角フラスコに入った100mlのLB培地(トリプトン 1%、酵母エキス 0.5%、NaCl 0.2%、pH7.5、アンピシリン 70μg/ml含有)に植菌し、27℃で24時間培養した。合計3Lの培養液から遠心分離により、湿菌体35.9gを回収し、この菌体を300mlの20mM 酢酸緩衝液(pH6.5、NaClを20mM、DTTを1mM含有)に懸濁し、超音波処理して菌体を破砕し、さらに遠心分離により不溶物を除去し、菌体破砕抽出液340mlを採取した。この菌体破砕抽出液の33%〜80%硫安飽和塩析画分を回収し、20mM 酢酸緩衝液(pH6.5)に対して透析した後、60℃で15分間熱処理を行い、生じた変性蛋白質を遠心分離で除き、熱処理液上清を回収した。

実験3の方法で得た単量体酵素及び二量体酵素と考えられるβ−PGM精製標品をそれぞれ5乃至20%(w/v)グラジエントゲルを用いたSDS−ポリアクリルアミドゲル電気泳動法に供し、同時に泳動した分子量マーカー(日本バイオ・ラッド・ラボラトリーズ株式会社製)と比較して分子量を測定したところ、いずれも分子量は25,000±5,000ダルトンであった。この値は当該酵素のアミノ酸配列(配列表における配列番号1で示されるアミノ酸配列)から算出される分子量24,363とよく一致するものであった。
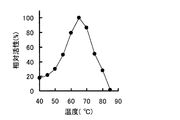
実験3の方法で得たβ−PGM精製標品のうち、単量体酵素を用いて、β−PGM活性に及ぼす温度及びpHの影響を活性測定の方法に準じて調べた。これらの結果を図3(至適温度)及び図4(至適pH)に示した。β−PGMの至適温度はpH6.5、30分間反応、10mM Mg2+イオン存在下で65℃であった。至適pHは、45℃、30分間反応、10mM Mg2+イオン存在下でpH6.5であった。なお、二量体酵素についても同様に調べたところ、単量体酵素と同じであった。
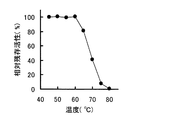
実験3の方法で得たβ−PGM精製標品のうち、単量体酵素を用いて、β−PGMの温度安定性及びpH安定性を調べた。温度安定性は、10mM MgCl2を含有する30mM MOPS緩衝液(pH6.5)を用い、酵素溶液を各温度に30分間保持し、水冷した後、残存する酵素活性を測定することにより求めた。pH安定性は、酵素を10mM MgCl2を含有する各pHの25mM緩衝液中で37℃、24時間保持した後、pHを6.5に調整し、残存する酵素活性を測定することにより求めた。これらの結果を図5(温度安定性)及び図6(pH安定性)に示した。図5から明らかなように、本酵素は60℃まで安定であった。また、図6から明らかなように、本酵素は、少なくともpH4.5乃至9.0の範囲で安定であった。なお、二量体酵素についても同様に調べたところ、単量体酵素と同じであった。
実験3の方法で得たβ−PGM精製標品のうち、単量体酵素を用いて、酵素活性に及ぼす金属イオンの影響を調べた。活性測定に用いる基質溶液から10mMのMgCl2を除いた基質溶液を用い、濃度を変えた各種金属塩存在下で活性測定した結果を表2に示す。なお、β−PGMの活性は活性測定条件と同じ10mM Mg2+イオン存在下での活性を100とした場合の相対活性で表した。
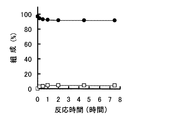
β−PGMが触媒するβ−G1PとG6Pの相互変換反応の平衡点を調べる実験を以下のとおり行った。5.3mM β−G1P(オリエンタル酵母社販売)、26.3mM MOPS緩衝液(pH6.5)、4.27mM MgCl2を含む基質溶液950μlに実験3の方法で得たβ−PGMの単量体酵素50μl(1.95単位)を加えて55℃で反応を行い、反応液を経時的にサンプリングし、沸騰湯浴中で10分間加熱し反応停止した後、分析に供した。また、β−G1Pに変えてG6P(オリエンタル酵母社販売)を用いた以外は同様に処理して分析を行った。なお、反応液中のβ−G1P及びG6Pの定量は、HPAE−PAD(High Performance Anion Exchange−Pulsed Amperometric Detection)法により下記の測定条件にて行った。
<HPAE−PAD条件>
カラム:CarboPac PA1(4x250mm、日本ダイオネクス社販売)
カラム温度:40℃
溶離液A:100mM 水酸化ナトリウム水溶液(pH11.6)
溶離液B:1M 酢酸ナトリウムを含有する100mM 水酸化ナトリウム水溶液
溶離液の容量比:溶離液A 9:溶離液B 1(0〜20分)
溶離液A 8:溶離液B 2(20〜30分)
溶離液A 5:溶離液B 5(30〜40分)
流 速:1ml/分
検 出:パルスドアンペロメトリー検出器 ED40(日本ダイオネクス社製)
β−PGMと、同じくサーモアナエロバクター・ブロッキイ ATCC35047由来のトレハロースホスホリラーゼとを組み合わせて、グルコースとG6Pとを基質としてトレハロースを酵素合成する系の構築を試みた。なお、β−PGMは実験3の方法で得た単量体酵素を、サーモアナエロバクター・ブロッキイ ATCC35047由来のトレハロースホスホリラーゼは、本願と同じ出願人による特開平10−304881号公報の実施例A−4に記載された方法で調製し、同公報記載の方法で活性測定した組換え型トレハロースホスホリラーゼの精製標品を用いた。
β−PGMと、同じくサーモアナエロバクター・ブロッキイ ATCC35047由来のコージビオースホスホリラーゼとを組み合わせて、グルコースとG6Pとを基質としてコージオリゴ糖を酵素合成する系の構築を試みた。なお、β−PGMは実験3で得た単量体酵素を、サーモアナエロバクター・ブロッキイ ATCC35047由来のコージビオースホスホリラーゼは本願と同じ出願人による特開平10−304882号公報の実施例A−4に記載された方法で調製し、同公報記載の方法で活性測定した組換え型コージビオースホスホリラーゼの精製標品を用いた。
<HPLC条件>
カラム:MCIgel CK04SS(株式会社三菱化学製造) 2本
(カラム2本を直列に連結)
溶離液:精製水
流速:0.4ml/分
カラム温度:80℃
検出:示差屈折計
サーモアナエロバクター・ブロッキイ ATCC35047を、特開平10−304882号公報の実験1に記載された培地に接種し、嫌気ファーメンターにて温度65℃で約50時間培養した。培養液を遠心分離して得た菌体を超音波破砕し、破砕液上清のβ−PGM活性を測定した。この活性を培養液1ml当たりに換算すると、0.03単位であった。破砕液上清を限外濾過膜にて濃縮し、得られた濃縮液を透析してβ−PGM活性を約4単位/ml有する粗酵素液をもとの培養液の総活性に対して約65%の収率で得た。
実験2で得た形質転換体RSET−TbPGM−2を3LのT培地(培地1L当たり、バクト−トリプトン12g、バクト−イーストエキストラクト24g、グリセロール4ml、17mM リン酸一カリウム、72mM リン酸二カリウムを含有)を用いて37℃で24時間好気的に培養した。培養液を遠心分離して得た菌体を超音波破砕し、破砕液上清を55℃で30分間熱処理し、宿主由来の非耐熱性蛋白質を変性、失活させた。熱処理液をさらに遠心分離して得た部分精製酵素液のβ−PGM活性を測定し、培養液1ml当たりに換算すると約38単位であった。
実験3で得た単量体酵素をβ−PGMとして用い、本願と同じ出願人による特開平10−304881号公報の実施例A−4に記載された方法で調製した組換え型トレハロースホスホリラーゼの精製標品を用いてグルコースとG6Pを基質としてトレハロースを調製した。反応液の終濃度でグルコースを12mM、G6P(オリエンタル酵母社販売)を10mM、酢酸ナトリウム緩衝液(pH5.5)を50mM、MgCl2を10mMとなるように調製した基質溶液に、1グラムのG6P当たり1,650単位のβ−PGMと50単位のトレハロースホスホリラーゼを加えて55℃で25時間反応させたところ、5.8mMのトレハロースを含む反応液が得られた。反応停止後、反応液に終濃度0.1mMのCaCl2を添加し、反応液中のリン酸をリン酸カルシウムとして沈澱させ遠心分離により除去した。得られたリン酸除去液に再度β−PGMとトレハロースホスホリラーゼを添加し、55℃で13時間反応させたところ反応液中のトレハロース濃度は7.7mMに達した。反応停止の後、反応液を電気透析し、反応液中に含まれるG6Pとβ−G1Pを除去したところ、固形物換算でトレハロースを62質量%、グルコースを38質量%含有する糖液が得られた。
○:β−PGM活性
図2において、
a:分子量51,000ダルトンを示すβ−PGM
b:分子量25,000ダルトンを示すβ−PGM
図7及び8において、
□:β−G1P
●:G6P
Claims (10)
- 下記の理化学的性質を有するβ−ホスホグルコムターゼ。
(1)作用
D−グルコース−6−リン酸をβ−D−グルコース−1−リン酸に変換し、β−D−グルコース−1−リン酸をD−グルコース−6−リン酸に変換する;
(2)分子量
SDS−ゲル電気泳動法において、25,000±5,000ダルトン;
(3)至適温度
10mMのMg2+イオン存在下、pH6.5、30分間反応の条件下で65℃;
(4)至適pH
10mMのMg2+イオン存在下、55℃、30分間反応の条件下でpH6.5;
(4)温度安定性
10mMのMg2+イオン存在下、pH6.5、30分間保持する条件下で60℃まで安定;
(5)pH安定性
10mMのMg2+イオン存在下、37℃、24時間保持する条件下でpH4.5乃至9.0の範囲で安定;及び
(6)金属イオンによる活性化
Mg2+、Mn2+、Co2+イオンにより活性化される; - 配列表における配列番号1に記載のアミノ酸配列か、当該アミノ酸配列において1個以上10個未満のアミノ酸残基が置換、欠失又は付加したアミノ酸配列を有する請求項1記載のβ−ホスホグルコムターゼ。
- 請求項2記載のβ−ホスホグルコムターゼをコードするDNA。
- DNAが、配列表における配列番号2に記載の塩基配列か、遺伝暗号の縮重に基づきコードするアミノ酸配列を変えることなく配列表における配列番号2に記載の塩基配列における塩基の1個又は2個以上が他の塩基に置換した塩基配列、又は、それらの塩基配列に相補的な塩基配列を有するDNAである請求項3記載のDNA。
- 請求項3又は4記載のDNAと自律複製可能なベクターDNAとを含んでなる組換えDNA。
- 請求項5記載の組換えDNAを適宜の宿主に導入して得られる形質転換体。
- 請求項1又は2記載のβ−ホスホグルコムターゼ産生能を有する微生物を栄養培地に培養し、培養物から産生したβ−ホスホグルコムターゼを採取することを特徴とするβ−ホスホグルコムターゼの製造方法。
- 微生物が、請求項6記載の形質転換体である請求項7記載のβ−ホスホグルコムターゼの製造方法。
- 下記の工程を含んでなるオリゴ糖の製造方法。
(1)請求項1又は2記載のβ−ホスホグルコムターゼをD−グルコース−6−リン酸に作用させてβ−D−グルコース−1−リン酸に変換する工程;
(2)得られるβ−D−グルコース−1−リン酸を二糖類ホスホリラーゼの共存下でグルコースと反応させオリゴ糖を生成させる工程;及び
(3)得られるオリゴ糖を採取する工程; - 二糖類ホスホリラーゼが、トレハロースホスホリラーゼ又はコージビオースホスホリラーゼである請求項9記載のオリゴ糖の製造方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2009268783A JP2010148502A (ja) | 2008-11-28 | 2009-11-26 | β−ホスホグルコムターゼとその製造方法並びに用途 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2008303324 | 2008-11-28 | ||
| JP2009268783A JP2010148502A (ja) | 2008-11-28 | 2009-11-26 | β−ホスホグルコムターゼとその製造方法並びに用途 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2010148502A true JP2010148502A (ja) | 2010-07-08 |
| JP2010148502A5 JP2010148502A5 (ja) | 2013-01-10 |
Family
ID=42568305
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2009268783A Withdrawn JP2010148502A (ja) | 2008-11-28 | 2009-11-26 | β−ホスホグルコムターゼとその製造方法並びに用途 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP2010148502A (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015019939A1 (ja) * | 2013-08-07 | 2015-02-12 | 国立大学法人新潟大学 | α-グルコシドの製造方法 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH03244398A (ja) * | 1990-02-20 | 1991-10-31 | Internatl Reagents Corp | 試料中の物質の測定法 |
| JPH10304882A (ja) * | 1996-11-08 | 1998-11-17 | Hayashibara Biochem Lab Inc | コージビオースホスホリラーゼとその製造方法並びに用途 |
| JPH10304881A (ja) * | 1996-11-08 | 1998-11-17 | Hayashibara Biochem Lab Inc | トレハロースホスホリラーゼとその製造方法並びに用途 |
| JP2000083674A (ja) * | 1998-09-17 | 2000-03-28 | Kikkoman Corp | β−ホスホグルコムターゼ遺伝子、新規な組み換え体DNA及びβ−ホスホグルコムターゼの製造法 |
-
2009
- 2009-11-26 JP JP2009268783A patent/JP2010148502A/ja not_active Withdrawn
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH03244398A (ja) * | 1990-02-20 | 1991-10-31 | Internatl Reagents Corp | 試料中の物質の測定法 |
| JPH10304882A (ja) * | 1996-11-08 | 1998-11-17 | Hayashibara Biochem Lab Inc | コージビオースホスホリラーゼとその製造方法並びに用途 |
| JPH10304881A (ja) * | 1996-11-08 | 1998-11-17 | Hayashibara Biochem Lab Inc | トレハロースホスホリラーゼとその製造方法並びに用途 |
| JP2000083674A (ja) * | 1998-09-17 | 2000-03-28 | Kikkoman Corp | β−ホスホグルコムターゼ遺伝子、新規な組み換え体DNA及びβ−ホスホグルコムターゼの製造法 |
Non-Patent Citations (3)
| Title |
|---|
| CSNC200800951004; 'ホ' 化学大辞典8 フリヘホマ ENCYCLOPAEDIA CHIMICA 第1版, 19620228, 第672頁, 左欄上段, 南條 正男 共立出版株式会社 * |
| JPN6014018090; B0K4J7 , 20080318, UniProt * |
| JPN6014047169; 'ホ' 化学大辞典8 フリヘホマ ENCYCLOPAEDIA CHIMICA 第1版, 19620228, 第672頁, 左欄上段, 南條 正男 共立出版株式会社 * |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015019939A1 (ja) * | 2013-08-07 | 2015-02-12 | 国立大学法人新潟大学 | α-グルコシドの製造方法 |
| JPWO2015019939A1 (ja) * | 2013-08-07 | 2017-03-02 | 国立大学法人 新潟大学 | α−グルコシドの製造方法 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5098086B2 (ja) | ケトース3−エピメラーゼとその製造方法並びに用途 | |
| JP5274476B2 (ja) | コリネバクテリウム属菌株から発現されたアラビノース異性化酵素及びそれを用いたタガトースの製造方法 | |
| EP1756291B1 (en) | Enzymatic decarboxylation of 2-keto-l-gulonic acid to produce xylose | |
| JP5848834B2 (ja) | アルスロバクターグロビホルミスの生産するケトース3−エピメラーゼ | |
| JP2017510302A5 (ja) | ||
| JP5224572B2 (ja) | デキストラン生成酵素遺伝子、デキストラン生成酵素およびその製造方法、デキストランの製造方法 | |
| JP5677097B2 (ja) | セロビオース2−エピメラーゼとその製造方法並びに用途 | |
| CA2582686C (en) | Method of producing uridine 5'-diphospho-n-acetylgalactosamine | |
| Poonperm et al. | Cloning, sequencing, overexpression and characterization of L-rhamnose isomerase from Bacillus pallidus Y25 for rare sugar production | |
| JP5933321B2 (ja) | 新規なα−グルコシダーゼとその製造法並びに用途 | |
| Yi et al. | Cloning of dextransucrase gene from Leuconostoc citreum HJ-P4 and its high-level expression in E. coli by low temperature induction | |
| de Pascale et al. | Recombinant thermophilic enzymes for trehalose and trehalosyl dextrins production | |
| Li et al. | Cloning, protein sequence clarification, and substrate specificity of a leucine dehydrogenase from Bacillus sphaericus ATCC4525 | |
| JP2010148502A (ja) | β−ホスホグルコムターゼとその製造方法並びに用途 | |
| CA2392463C (en) | Novel use of uridine diphosphate glucose 4-epimerase | |
| WO2019035482A1 (ja) | エピメリ化活性を有するタンパク質 | |
| JP5714241B2 (ja) | α−グルコシダーゼとその製造方法並びに用途 | |
| JP3766040B2 (ja) | シトシンヌクレオシド化合物の製造方法 | |
| JP3557271B2 (ja) | 酵素をコードするdnaとそれを含む組換えdna並びに形質転換体 | |
| JP2023554112A (ja) | 熱安定性に優れたアルロースエピマー化酵素変異体、その製造方法およびこれを用いたアルロースの製造方法 | |
| KR101479135B1 (ko) | 아스페르기루스 플라버스 유래 솔비톨 탈수소화효소와 nadh 산화효소와의 커플링에 의한 l-자일룰로스의 생산 | |
| JP2005304306A (ja) | 新規な糖質酸化酵素 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A711 | Notification of change in applicant |
Free format text: JAPANESE INTERMEDIATE CODE: A712 Effective date: 20121015 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20121120 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20121120 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20140513 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20140711 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20141111 |
|
| A761 | Written withdrawal of application |
Free format text: JAPANESE INTERMEDIATE CODE: A761 Effective date: 20150123 |