JP2010037340A - オルガノシランの製造法 - Google Patents
オルガノシランの製造法 Download PDFInfo
- Publication number
- JP2010037340A JP2010037340A JP2009177297A JP2009177297A JP2010037340A JP 2010037340 A JP2010037340 A JP 2010037340A JP 2009177297 A JP2009177297 A JP 2009177297A JP 2009177297 A JP2009177297 A JP 2009177297A JP 2010037340 A JP2010037340 A JP 2010037340A
- Authority
- JP
- Japan
- Prior art keywords
- alkali metal
- particularly preferably
- weight
- alcohol
- formula
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- 150000001282 organosilanes Chemical class 0.000 title claims abstract description 31
- 238000004519 manufacturing process Methods 0.000 title claims abstract description 11
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims abstract description 59
- 229910052783 alkali metal Inorganic materials 0.000 claims abstract description 59
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims abstract description 54
- 238000006243 chemical reaction Methods 0.000 claims abstract description 53
- -1 alkali metal hydrogen sulfide Chemical class 0.000 claims abstract description 52
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 claims abstract description 44
- 229910000037 hydrogen sulfide Inorganic materials 0.000 claims abstract description 42
- 229910052717 sulfur Inorganic materials 0.000 claims abstract description 39
- 239000011593 sulfur Substances 0.000 claims abstract description 35
- 125000001190 organyl group Chemical group 0.000 claims abstract description 28
- 229910000288 alkali metal carbonate Inorganic materials 0.000 claims abstract description 25
- 150000008041 alkali metal carbonates Chemical class 0.000 claims abstract description 25
- 238000000034 method Methods 0.000 claims abstract description 25
- 239000007787 solid Substances 0.000 claims abstract description 21
- 239000000203 mixture Substances 0.000 claims description 37
- 239000012043 crude product Substances 0.000 claims description 24
- 239000000047 product Substances 0.000 claims description 24
- 239000000725 suspension Substances 0.000 claims description 24
- 239000000654 additive Substances 0.000 claims description 19
- 230000008569 process Effects 0.000 claims description 17
- 239000002904 solvent Substances 0.000 claims description 14
- 230000000996 additive effect Effects 0.000 claims description 11
- 125000000217 alkyl group Chemical group 0.000 claims description 11
- 238000004821 distillation Methods 0.000 claims description 11
- 125000003118 aryl group Chemical group 0.000 claims description 10
- BLRPTPMANUNPDV-UHFFFAOYSA-N Silane Chemical compound [SiH4] BLRPTPMANUNPDV-UHFFFAOYSA-N 0.000 claims description 8
- 125000001931 aliphatic group Chemical group 0.000 claims description 8
- 229910000077 silane Inorganic materials 0.000 claims description 8
- 229910001508 alkali metal halide Inorganic materials 0.000 claims description 6
- 150000008045 alkali metal halides Chemical class 0.000 claims description 6
- 229920000570 polyether Polymers 0.000 claims description 5
- 239000004721 Polyphenylene oxide Substances 0.000 claims description 4
- 125000003710 aryl alkyl group Chemical group 0.000 claims description 4
- 229910052740 iodine Inorganic materials 0.000 claims description 4
- 239000011630 iodine Substances 0.000 claims description 4
- 238000005191 phase separation Methods 0.000 claims description 4
- 229920006395 saturated elastomer Polymers 0.000 claims description 4
- 230000002378 acidificating effect Effects 0.000 claims description 3
- 230000001476 alcoholic effect Effects 0.000 claims description 3
- 229910052739 hydrogen Inorganic materials 0.000 claims description 3
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 claims description 2
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 claims description 2
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 claims description 2
- 125000003342 alkenyl group Chemical group 0.000 claims description 2
- 125000005011 alkyl ether group Chemical group 0.000 claims description 2
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 claims description 2
- 229910052794 bromium Inorganic materials 0.000 claims description 2
- 229910052801 chlorine Inorganic materials 0.000 claims description 2
- 239000000460 chlorine Substances 0.000 claims description 2
- 229910052731 fluorine Inorganic materials 0.000 claims description 2
- 239000011737 fluorine Substances 0.000 claims description 2
- 239000001257 hydrogen Substances 0.000 claims description 2
- 238000002360 preparation method Methods 0.000 claims description 2
- 239000004215 Carbon black (E152) Substances 0.000 claims 1
- 229930195733 hydrocarbon Natural products 0.000 claims 1
- 150000002431 hydrogen Chemical class 0.000 claims 1
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 claims 1
- 150000003961 organosilicon compounds Chemical class 0.000 claims 1
- 150000008044 alkali metal hydroxides Chemical class 0.000 abstract description 8
- 239000000463 material Substances 0.000 abstract description 8
- 229910052977 alkali metal sulfide Inorganic materials 0.000 abstract description 7
- 239000005077 polysulfide Substances 0.000 abstract description 3
- 229920001021 polysulfide Polymers 0.000 abstract description 3
- 150000008117 polysulfides Polymers 0.000 abstract description 3
- 238000005987 sulfurization reaction Methods 0.000 abstract description 3
- 239000002245 particle Substances 0.000 description 22
- 150000001875 compounds Chemical class 0.000 description 19
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 15
- 150000003839 salts Chemical class 0.000 description 14
- 150000001340 alkali metals Chemical class 0.000 description 13
- 238000004817 gas chromatography Methods 0.000 description 12
- 238000010626 work up procedure Methods 0.000 description 12
- 238000007873 sieving Methods 0.000 description 11
- 238000009826 distribution Methods 0.000 description 10
- 238000005259 measurement Methods 0.000 description 9
- 239000002994 raw material Substances 0.000 description 9
- 239000011541 reaction mixture Substances 0.000 description 9
- 150000004756 silanes Chemical class 0.000 description 9
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 8
- 238000001914 filtration Methods 0.000 description 8
- 239000007858 starting material Substances 0.000 description 8
- 239000007789 gas Substances 0.000 description 7
- 239000007788 liquid Substances 0.000 description 7
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 6
- SNRUBQQJIBEYMU-UHFFFAOYSA-N dodecane Chemical compound CCCCCCCCCCCC SNRUBQQJIBEYMU-UHFFFAOYSA-N 0.000 description 6
- 238000000926 separation method Methods 0.000 description 6
- 238000004448 titration Methods 0.000 description 6
- KSCAZPYHLGGNPZ-UHFFFAOYSA-N 3-chloropropyl(triethoxy)silane Chemical compound CCO[Si](OCC)(OCC)CCCCl KSCAZPYHLGGNPZ-UHFFFAOYSA-N 0.000 description 5
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 5
- 238000002050 diffraction method Methods 0.000 description 5
- 150000004820 halides Chemical class 0.000 description 5
- 238000005481 NMR spectroscopy Methods 0.000 description 4
- 239000003153 chemical reaction reagent Substances 0.000 description 4
- 239000012071 phase Substances 0.000 description 4
- 239000011734 sodium Substances 0.000 description 4
- 125000004434 sulfur atom Chemical group 0.000 description 4
- 238000005303 weighing Methods 0.000 description 4
- 238000005160 1H NMR spectroscopy Methods 0.000 description 3
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 3
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 3
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 3
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 229910000831 Steel Inorganic materials 0.000 description 3
- UCKMPCXJQFINFW-UHFFFAOYSA-N Sulphide Chemical compound [S-2] UCKMPCXJQFINFW-UHFFFAOYSA-N 0.000 description 3
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 3
- 150000001298 alcohols Chemical class 0.000 description 3
- 239000000872 buffer Substances 0.000 description 3
- 238000011049 filling Methods 0.000 description 3
- 150000007529 inorganic bases Chemical class 0.000 description 3
- 150000007522 mineralic acids Chemical class 0.000 description 3
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 3
- 239000012074 organic phase Substances 0.000 description 3
- 239000008188 pellet Substances 0.000 description 3
- 150000003138 primary alcohols Chemical class 0.000 description 3
- 239000011780 sodium chloride Substances 0.000 description 3
- 239000010959 steel Substances 0.000 description 3
- 238000005406 washing Methods 0.000 description 3
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 2
- 239000005046 Chlorosilane Substances 0.000 description 2
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 2
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 2
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical group CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 2
- URLKBWYHVLBVBO-UHFFFAOYSA-N Para-Xylene Chemical group CC1=CC=C(C)C=C1 URLKBWYHVLBVBO-UHFFFAOYSA-N 0.000 description 2
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 2
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 2
- 239000008346 aqueous phase Substances 0.000 description 2
- WDIHJSXYQDMJHN-UHFFFAOYSA-L barium chloride Chemical compound [Cl-].[Cl-].[Ba+2] WDIHJSXYQDMJHN-UHFFFAOYSA-L 0.000 description 2
- 229910001626 barium chloride Inorganic materials 0.000 description 2
- 239000006227 byproduct Substances 0.000 description 2
- 238000012512 characterization method Methods 0.000 description 2
- 239000003795 chemical substances by application Substances 0.000 description 2
- KOPOQZFJUQMUML-UHFFFAOYSA-N chlorosilane Chemical compound Cl[SiH3] KOPOQZFJUQMUML-UHFFFAOYSA-N 0.000 description 2
- 238000009833 condensation Methods 0.000 description 2
- 230000005494 condensation Effects 0.000 description 2
- LQZZUXJYWNFBMV-UHFFFAOYSA-N dodecan-1-ol Chemical compound CCCCCCCCCCCCO LQZZUXJYWNFBMV-UHFFFAOYSA-N 0.000 description 2
- 230000000694 effects Effects 0.000 description 2
- 239000000706 filtrate Substances 0.000 description 2
- 239000012467 final product Substances 0.000 description 2
- 238000004128 high performance liquid chromatography Methods 0.000 description 2
- 150000004677 hydrates Chemical class 0.000 description 2
- ZXEKIIBDNHEJCQ-UHFFFAOYSA-N isobutanol Chemical compound CC(C)CO ZXEKIIBDNHEJCQ-UHFFFAOYSA-N 0.000 description 2
- 239000007791 liquid phase Substances 0.000 description 2
- 239000012263 liquid product Substances 0.000 description 2
- IVSZLXZYQVIEFR-UHFFFAOYSA-N m-xylene Chemical group CC1=CC=CC(C)=C1 IVSZLXZYQVIEFR-UHFFFAOYSA-N 0.000 description 2
- GLDOVTGHNKAZLK-UHFFFAOYSA-N octadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCCCO GLDOVTGHNKAZLK-UHFFFAOYSA-N 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 150000007530 organic bases Chemical class 0.000 description 2
- 238000012545 processing Methods 0.000 description 2
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 2
- 230000035484 reaction time Effects 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 238000010058 rubber compounding Methods 0.000 description 2
- 150000003333 secondary alcohols Chemical class 0.000 description 2
- SQGYOTSLMSWVJD-UHFFFAOYSA-N silver(1+) nitrate Chemical compound [Ag+].[O-]N(=O)=O SQGYOTSLMSWVJD-UHFFFAOYSA-N 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L sodium carbonate Substances [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- HYHCSLBZRBJJCH-UHFFFAOYSA-M sodium hydrosulfide Chemical compound [Na+].[SH-] HYHCSLBZRBJJCH-UHFFFAOYSA-M 0.000 description 2
- 238000001228 spectrum Methods 0.000 description 2
- 238000007655 standard test method Methods 0.000 description 2
- 150000003509 tertiary alcohols Chemical class 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- HLZKNKRTKFSKGZ-UHFFFAOYSA-N tetradecan-1-ol Chemical compound CCCCCCCCCCCCCCO HLZKNKRTKFSKGZ-UHFFFAOYSA-N 0.000 description 2
- CZDYPVPMEAXLPK-UHFFFAOYSA-N tetramethylsilane Chemical compound C[Si](C)(C)C CZDYPVPMEAXLPK-UHFFFAOYSA-N 0.000 description 2
- 239000010409 thin film Substances 0.000 description 2
- ZRKGYQLXOAHRRN-UHFFFAOYSA-N triethoxy-[3-(3-triethoxysilylpropylsulfanyl)propyl]silane Chemical class CCO[Si](OCC)(OCC)CCCSCCC[Si](OCC)(OCC)OCC ZRKGYQLXOAHRRN-UHFFFAOYSA-N 0.000 description 2
- 125000004209 (C1-C8) alkyl group Chemical group 0.000 description 1
- WNXJIVFYUVYPPR-UHFFFAOYSA-N 1,3-dioxolane Chemical compound C1COCO1 WNXJIVFYUVYPPR-UHFFFAOYSA-N 0.000 description 1
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- 101710134784 Agnoprotein Proteins 0.000 description 1
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 1
- IAYPIBMASNFSPL-UHFFFAOYSA-N Ethylene oxide Chemical group C1CO1 IAYPIBMASNFSPL-UHFFFAOYSA-N 0.000 description 1
- 238000003109 Karl Fischer titration Methods 0.000 description 1
- 229910012525 LiSH Inorganic materials 0.000 description 1
- GOOHAUXETOMSMM-UHFFFAOYSA-N Propylene oxide Chemical group CC1CO1 GOOHAUXETOMSMM-UHFFFAOYSA-N 0.000 description 1
- 229910020175 SiOH Inorganic materials 0.000 description 1
- 239000004809 Teflon Substances 0.000 description 1
- 229920006362 Teflon® Polymers 0.000 description 1
- DJRSJHZEOJHZER-UHFFFAOYSA-N [SiH4].SS Chemical compound [SiH4].SS DJRSJHZEOJHZER-UHFFFAOYSA-N 0.000 description 1
- 150000001335 aliphatic alkanes Chemical class 0.000 description 1
- 229910052936 alkali metal sulfate Inorganic materials 0.000 description 1
- 125000003545 alkoxy group Chemical group 0.000 description 1
- 150000005215 alkyl ethers Chemical class 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 239000003849 aromatic solvent Substances 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 125000004432 carbon atom Chemical group C* 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- XEHUIDSUOAGHBW-UHFFFAOYSA-N chromium;pentane-2,4-dione Chemical compound [Cr].CC(=O)CC(C)=O.CC(=O)CC(C)=O.CC(=O)CC(C)=O XEHUIDSUOAGHBW-UHFFFAOYSA-N 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 229910000365 copper sulfate Inorganic materials 0.000 description 1
- ARUVKPQLZAKDPS-UHFFFAOYSA-L copper(II) sulfate Chemical compound [Cu+2].[O-][S+2]([O-])([O-])[O-] ARUVKPQLZAKDPS-UHFFFAOYSA-L 0.000 description 1
- 238000005260 corrosion Methods 0.000 description 1
- 230000007797 corrosion Effects 0.000 description 1
- 230000007423 decrease Effects 0.000 description 1
- 239000008367 deionised water Substances 0.000 description 1
- 229910021641 deionized water Inorganic materials 0.000 description 1
- KPUWHANPEXNPJT-UHFFFAOYSA-N disiloxane Chemical class [SiH3]O[SiH3] KPUWHANPEXNPJT-UHFFFAOYSA-N 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 239000010408 film Substances 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 238000000227 grinding Methods 0.000 description 1
- 229910000856 hastalloy Inorganic materials 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- BXWNKGSJHAJOGX-UHFFFAOYSA-N hexadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCO BXWNKGSJHAJOGX-UHFFFAOYSA-N 0.000 description 1
- 150000002430 hydrocarbons Chemical group 0.000 description 1
- 125000004435 hydrogen atom Chemical class [H]* 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 230000003301 hydrolyzing effect Effects 0.000 description 1
- 229940046892 lead acetate Drugs 0.000 description 1
- HXQGSILMFTUKHI-UHFFFAOYSA-M lithium;sulfanide Chemical compound S[Li] HXQGSILMFTUKHI-UHFFFAOYSA-M 0.000 description 1
- 239000000178 monomer Substances 0.000 description 1
- 238000001208 nuclear magnetic resonance pulse sequence Methods 0.000 description 1
- 229940078552 o-xylene Drugs 0.000 description 1
- TVMXDCGIABBOFY-UHFFFAOYSA-N octane Chemical compound CCCCCCCC TVMXDCGIABBOFY-UHFFFAOYSA-N 0.000 description 1
- 239000003444 phase transfer catalyst Substances 0.000 description 1
- 239000004033 plastic Substances 0.000 description 1
- 239000003495 polar organic solvent Substances 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 230000009257 reactivity Effects 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 229910052710 silicon Inorganic materials 0.000 description 1
- 239000010703 silicon Substances 0.000 description 1
- 229910001961 silver nitrate Inorganic materials 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- 239000011877 solvent mixture Substances 0.000 description 1
- 238000000527 sonication Methods 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 229910052715 tantalum Inorganic materials 0.000 description 1
- GUVRBAGPIYLISA-UHFFFAOYSA-N tantalum atom Chemical compound [Ta] GUVRBAGPIYLISA-UHFFFAOYSA-N 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- IKRMQEUTISXXQP-UHFFFAOYSA-N tetrasulfane Chemical compound SSSS IKRMQEUTISXXQP-UHFFFAOYSA-N 0.000 description 1
- 150000003568 thioethers Chemical class 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic Table
- C07F7/02—Silicon compounds
- C07F7/08—Compounds having one or more C—Si linkages
- C07F7/18—Compounds having one or more C—Si linkages as well as one or more C—O—Si linkages
- C07F7/1804—Compounds having Si-O-C linkages
- C07F7/1872—Preparation; Treatments not provided for in C07F7/20
- C07F7/1892—Preparation; Treatments not provided for in C07F7/20 by reactions not provided for in C07F7/1876 - C07F7/1888
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Silicon Polymers (AREA)
Abstract
【解決手段】一般式Iのオルガノシランを、式IIの(ハロゲンオルガニル)アルコキシシランと含水アルカリ金属硫化水素、硫黄およびアルカリ金属炭酸塩とのアルコール中での反応によって製造するための方法において、この方法は、式IIの(ハロゲン化オルガニル)アルコキシシランとアルカリ金属硫化水素とのモル比が1:0.40〜1:0.75であり、アルカリ金属硫化水素とアルカリ金属炭酸塩とのモル比が1:0.5〜1:1.5であることによって特徴付けられる。
【選択図】なし
Description
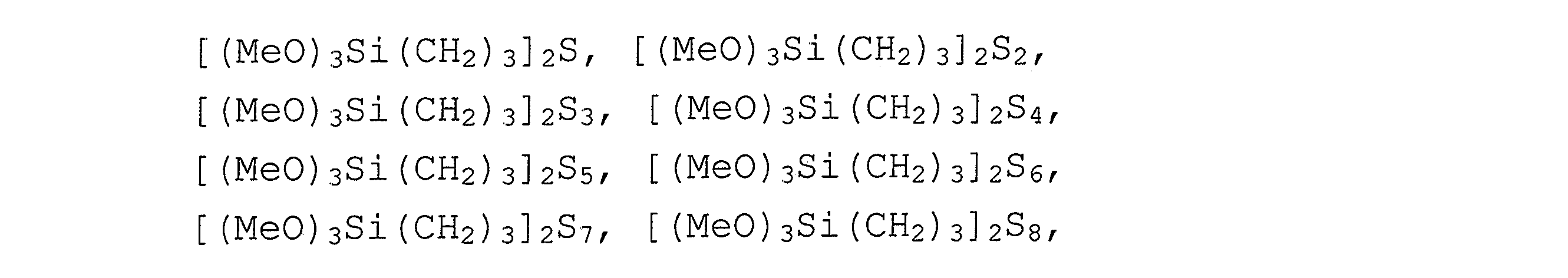
Rは、同一かまたは異なり、C1〜C8−アルキル基、特にCH3またはCH2CH3、C1〜C8−アルケニル基、C1〜C8−アリール基、C1〜C8−アラルキル基またはOR'基であり、
R'は、同一かまたは異なり、C1〜C24−、特にC1〜C4−またはC12〜C18−、特に有利にCH2CH3、分枝鎖状または非分枝鎖状の一価のアルキル基またはアルケニル基、アリール基、アラルキル基、水素(−H)、アルキルエーテル基−(CRIII 2)y'−O−Alk、但し、y'は、1〜20、有利に2〜10、特に有利に2〜5であるかまたはアルキルポリエーテル基−(CRIII 2O)y−Alkまたは−(CRIII 2−CRIII 2−O)y−Alk、但し、yは、2〜20、有利に2〜10、特に有利に2〜5であり、
RIIIは、互いに無関係にHまたはアルキル基、特にCH3基であり、Alkは、分枝鎖状または非分枝鎖状で飽和または不飽和の脂肪族、芳香族または混合された脂肪族/芳香族の一価のC1〜C30−、有利にC2〜C20−、特に有利にC6〜C18−、殊に特に有利にC10〜C18−、炭化水素基であり、
R''は、場合によってはF−、Cl−、Br−、I−、HS−、NH2−またはNHR’で置換されている、分枝鎖状または非分枝鎖状で飽和または不飽和の脂肪族、芳香族または混合された脂肪族/芳香族の2価のC1〜C30−、有利にC1〜C20−、特に有利にC1〜C10−、殊に有利にC1〜C7−炭化水素基であり、
mは、1.5〜4.5、有利に2〜2.6および3.5〜3.9、特に有利に2.1〜2.3および3.6〜3.8の平均硫黄鎖長である〕示されるオルガノシランを、式II
式IIの(ハロゲン化オルガニル)アルコキシシランとアルカリ金属硫化水素とのモル比が1:0.40〜1:0.75、特に1:0.45〜1:0.65、特に有利に1:0.5〜1:0.6、殊に有利に1:0.5〜1:0.55であり、アルカリ金属硫化水素とアルカリ金属炭酸塩とのモル比が1:0.5〜1:1.5、特に1:0.65〜1:1.3、特に有利に1:0.85〜1:1.2、殊に有利に1:0.95〜1:1.1であることによって特徴付けられる。
または
試料(硫化水素ナトリウム)約3gは、100mlの測定フラスコ中に計量供給され(計量供給量(読取り可能性1mgまたはそれ以上)、最も純粋な水約75ml中に溶解される。
Na2S + H2O → NaOH + NaSH
pH4〜pH5の間の第2の転換点の場合、溶液中に存在するNaSHは、次の方程式に従って反応される:
NaSH +HCl → NaCl + H2S↑
計算式:
MNaOH=NaOHの分子量(39.99g/mol)、
mHCl=塩酸溶液のモル濃度、
E=mgでの計量供給量、
F=塩酸の滴定量、
VP=ピペットの容量(ml)、
VMK=計量フラスコの容量(ml)。
Vi=第1の転換点(ml)、
MNaSH=NaSHの分子量(56.064g/mol)、
mHCl=塩酸溶液のモル濃度、
E=mgでの計量供給量、
F=塩酸の滴定量、
VP=ピペットの容量(ml)、
VMK=計量フラスコの容量(ml)。
一定量の試料は、異なる標準の目開きのスクリーンのスタックで分離される。
機械的篩別機(Ro-tap);正確な秤量:
精度±0.01g(Mettler社)。
(RIV)2(Hal)Si−R''−Hal III
〔式中、Hal、RおよびR''は、上記の意味を有し、RIVは、互いに無関係にRまたはHalである〕で示される化合物が使用されてよい。
単離された粗製生成物のGC分析は、内部標準としてのドデカンを用いてのガスクロマトグラフィー(FID)で実施される。
HPLC測定のための方法は、"Luginsland, H-D, Reactivity of the Sulfur Functions of the Disulfane Silane TESPD and Tetrasulfane Silane TESPT, paper presented at the ACS Meeting, April 1999, Chicago"中に記載されている。
i=シラン成分中の硫黄原子の数、
Mi=硫黄原子iを有するシラン成分のモル質量、
Ai=硫黄原子iを有するシラン成分の信号の面積、
Ri=硫黄原子iを有する硫化シラン成分の応答ファクター。
Siスペクトルは、99.35MHz(H−NMR500.13MHz)のSiに対する測定周波数を有するBrucker Advance 500-NMRスペクトロメーターで記録される。スペクトルは、内部でテトラメチルシラン(TMS=0ppm)を基準とする。試料は、CDCl3中の約30%の溶液として、緩和促進剤としてのクロムアセチルアセトネート(約0.05〜0.1モルの溶液)の添加下に測定される。パルスシーケンスとして、プロトン脱カップリングを有する逆ゲートシーケンスは、収集時間中ならびに5秒間の緩和遅延でのみ使用される。
耐圧性の反応器中で、6.9+/−1質量%の純度(固体中)、NaOH含量0.67質量%(溶液中)および含水量25質量%を有するICS-Chemie Wolfen社のNaSH水和物50kg、384μmのレーザー回折による粒子分布(中央値)を有するSolvay社のNa2CO374.8kg、17μmのレーザー回折による粒子分布(中央値)を有するCS Additive社の硫黄粉末20kg(粉砕硫黄)、エタノール193kgおよび水34kgを互いに混合する。
耐圧性の反応器中で、45.5質量%のNaSH含量を有するAkzo Nobel社のNaSH溶液85.6g、360μmのレーザー回折による粒度分布(中央値)を有するMerck社のNa2CO375kg、1mm超の粒度分布(中央値)を有するKemmax社の硫黄ペレット57.1kg、エタノール153gおよび水17gを互いに混合する。
耐圧性の反応器中で、45.5質量%のNaSH含量を有するAkzo Nobel社のNaSH溶液96g、360μmのレーザー回折による粒度分布(中央値)を有するMerck社のNa2CO375kg、1mm超の粒度分布(中央値)を有するKemmax社の硫黄ペレット29.1g、エタノール150gおよび水20gを互いに混合する。
耐圧性の反応器中で、71質量%のNaSH含量を有するICS社のNaSH55g、41μmのレーザー回折による粒度分布(中央値)を有する硫黄粉末21.9g(粉砕硫黄)、エタノール128gおよび水32gを互いに混合する。
耐圧性の反応器中で、45.5質量%のNaSH含量を有するAkzo Nobel社のNaSH溶液85.6g、BASF社のNa2CO375g、1mm超の粒度分布(中央値)を有するKemmax社の硫黄ペレット57.1g、エタノール146.2gおよび水23.8gを互いに混合する。
Claims (8)
- 一般式I
Rは、同一かまたは異なり、C1〜C8−アルキル基、C1〜C8−アルケニル基、C1〜C8−アリール基、C1〜C8−アラルキル基またはOR'基であり、
R'は、同一かまたは異なり、C1〜C24−分枝鎖状または非分枝鎖状の一価のアルキル基またはアルケニル基、アリール基、アラルキル基、水素、アルキルエーテル基−(CRIII 2)y'−O−Alk、但し、y'は、1〜20であるかまたはアルキルポリエーテル基−(CRIII 2O)y−Alkまたは−(CRIII 2−CRIII 2−O)y−Alk、但し、yは、2〜20であり、
RIIIは、互いに無関係にHまたはアルキル基であり、Alkは、分枝鎖状または非分枝鎖状で飽和または不飽和の脂肪族、芳香族または混合された脂肪族/芳香族の一価のC1〜C30−炭化水素基であり、
R''は、場合によってはF−、Cl−、Br−、I−、HS−、NH2−またはNHR’で置換されている、分枝鎖状または非分枝鎖状で飽和または不飽和の脂肪族、芳香族または混合された脂肪族/芳香族の2価のC1〜C30−炭化水素基であり、
mは、1.5〜4.5の平均硫黄鎖長である〕で示されるオルガノシランを、
式II
- 反応前、反応中または反応後に添加剤を添加する、請求項1記載のオルガノシランの製造法。
- 添加剤は、非アルコール性溶剤であるかまたは極性、プロトン性、非プロトン性、塩基性または酸性の添加剤である、請求項2記載のオルガノシランの製造法。
- 粗製生成物懸濁液からアルコール/水混合物を除去し、引続き一般式Iの形成された生成物を、固体から分離する、請求項1から3までのいずれか1項に記載のオルガノシランの製造法。
- 粗製生成物懸濁液から固体を分離し、引続きアルコール/水混合物を一般式Iの形成された生成物から分離する、請求項1から3までのいずれか1項に記載のオルガノシランの製造法。
- アルコール/水混合物を一般式Iの形成された生成物から蒸留によって分離する、請求項5記載のオルガノシランの製造法。
- アルコール/水混合物を一般式Iの形成された生成物から相分離によって分離する、請求項5記載のオルガノシランの製造法。
- 粗製生成物懸濁液からアルコール/水混合物を除去し、一般式(I)の有機珪素化合物およびアルカリ金属ハロゲン化物を含有する、残存する粗製生成物懸濁液を水と混合し、形成する相を分離する、請求項1から3までのいずれか1項に記載のオルガノシランの製造法。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE102008035623.9 | 2008-07-31 | ||
| DE102008035623A DE102008035623A1 (de) | 2008-07-31 | 2008-07-31 | Verfahren zur Herstellung von Organosilanen |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2010037340A true JP2010037340A (ja) | 2010-02-18 |
| JP5478973B2 JP5478973B2 (ja) | 2014-04-23 |
Family
ID=41066706
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2009177297A Expired - Fee Related JP5478973B2 (ja) | 2008-07-31 | 2009-07-30 | オルガノシランの製造法 |
Country Status (12)
| Country | Link |
|---|---|
| US (1) | US8129556B2 (ja) |
| EP (1) | EP2149579B1 (ja) |
| JP (1) | JP5478973B2 (ja) |
| KR (1) | KR101627374B1 (ja) |
| CN (1) | CN101638416B (ja) |
| AT (1) | ATE523516T1 (ja) |
| BR (1) | BRPI0902472A2 (ja) |
| CA (1) | CA2674253A1 (ja) |
| DE (1) | DE102008035623A1 (ja) |
| RU (1) | RU2009128886A (ja) |
| TW (1) | TW201016711A (ja) |
| ZA (1) | ZA200905339B (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2013181029A (ja) * | 2012-03-05 | 2013-09-12 | Daiso Co Ltd | 高純度のメルカプトシラン化合物の製造法。 |
| JP2021123599A (ja) * | 2020-02-06 | 2021-08-30 | エボニック オペレーションズ ゲーエムベーハー | ポリスルファンシランの製造方法 |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101885734B (zh) * | 2010-06-26 | 2012-05-23 | 丁爱梅 | 一种双-[丙基三乙氧基硅烷]-四硫化物的合成方法 |
| DE102011083109A1 (de) * | 2011-09-21 | 2013-03-21 | Wacker Chemie Ag | Verfahren zur Herstellung von Pulvern aus Alkalisalzen von Silanolen |
| US20140049467A1 (en) * | 2012-08-14 | 2014-02-20 | Pierre-Yves Laligand | Input device using input mode data from a controlled device |
| EP3838905A1 (de) * | 2019-12-18 | 2021-06-23 | Evonik Operations GmbH | Verfahren zur herstellung von polysulfansilanen mittels phasentransferkatalyse |
| EP3862358A1 (en) * | 2020-02-06 | 2021-08-11 | Evonik Operations GmbH | A process for the production of sulfur containing silanes by utilization of phase transfer catalysis |
| CN116478204B (zh) * | 2023-04-27 | 2024-10-15 | 唐山三孚新材料有限公司 | 双-[三乙氧基硅基丙基]-四硫化物的制备方法 |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2006249086A (ja) * | 2005-03-07 | 2006-09-21 | Degussa Ag | オルガノシランの製造方法 |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE10009790C1 (de) | 2000-03-01 | 2001-09-20 | Degussa | Verfahren zur Herstellung von Organosilylalkylpolysulfanen |
| US6680398B1 (en) * | 2002-08-16 | 2004-01-20 | Dow Corning Corporation | Method of making mercaptoalkylalkoxysilanes |
| ZA200601910B (en) * | 2005-03-07 | 2006-11-29 | Degussa | Process for the preparation of organosilances |
| DE102006041356A1 (de) * | 2006-09-01 | 2008-03-20 | Evonik Degussa Gmbh | Verfahren zur Herstellung von Organosilanen |
| KR20110130433A (ko) | 2009-03-20 | 2011-12-05 | 에보니크 데구사 게엠베하 | 오르가노실란의 제조 방법 |
-
2008
- 2008-07-31 DE DE102008035623A patent/DE102008035623A1/de not_active Ceased
-
2009
- 2009-07-22 BR BRPI0902472-7A patent/BRPI0902472A2/pt not_active IP Right Cessation
- 2009-07-23 EP EP09166242A patent/EP2149579B1/de not_active Not-in-force
- 2009-07-23 AT AT09166242T patent/ATE523516T1/de active
- 2009-07-24 US US12/509,089 patent/US8129556B2/en not_active Expired - Fee Related
- 2009-07-28 RU RU2009128886/04A patent/RU2009128886A/ru not_active Application Discontinuation
- 2009-07-28 TW TW098125416A patent/TW201016711A/zh unknown
- 2009-07-30 CA CA002674253A patent/CA2674253A1/en not_active Abandoned
- 2009-07-30 JP JP2009177297A patent/JP5478973B2/ja not_active Expired - Fee Related
- 2009-07-30 KR KR1020090069870A patent/KR101627374B1/ko not_active Expired - Fee Related
- 2009-07-30 ZA ZA200905339A patent/ZA200905339B/en unknown
- 2009-07-31 CN CN200910161108.8A patent/CN101638416B/zh active Active
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2006249086A (ja) * | 2005-03-07 | 2006-09-21 | Degussa Ag | オルガノシランの製造方法 |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2013181029A (ja) * | 2012-03-05 | 2013-09-12 | Daiso Co Ltd | 高純度のメルカプトシラン化合物の製造法。 |
| JP2021123599A (ja) * | 2020-02-06 | 2021-08-30 | エボニック オペレーションズ ゲーエムベーハー | ポリスルファンシランの製造方法 |
| JP7668121B2 (ja) | 2020-02-06 | 2025-04-24 | エボニック オペレーションズ ゲーエムベーハー | ポリスルファンシランの製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| TW201016711A (en) | 2010-05-01 |
| CN101638416B (zh) | 2014-07-09 |
| US20100029971A1 (en) | 2010-02-04 |
| US8129556B2 (en) | 2012-03-06 |
| CN101638416A (zh) | 2010-02-03 |
| BRPI0902472A2 (pt) | 2010-04-20 |
| KR20100014150A (ko) | 2010-02-10 |
| EP2149579B1 (de) | 2011-09-07 |
| KR101627374B1 (ko) | 2016-06-03 |
| JP5478973B2 (ja) | 2014-04-23 |
| ZA200905339B (en) | 2010-04-28 |
| EP2149579A1 (de) | 2010-02-03 |
| CA2674253A1 (en) | 2010-01-31 |
| RU2009128886A (ru) | 2011-02-10 |
| ATE523516T1 (de) | 2011-09-15 |
| DE102008035623A1 (de) | 2010-02-04 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5478973B2 (ja) | オルガノシランの製造法 | |
| JP4391607B2 (ja) | 高純度オルガノシリコンジスルファンの製造方法 | |
| KR101260517B1 (ko) | 유기실란의 제조 방법 | |
| KR101588914B1 (ko) | 아미노-관능성 유기실란의 제조 동안 생성된 염을 함유하는 잔류물을 처리하는 방법 | |
| KR101588913B1 (ko) | 암모늄 할라이드 및/또는 유기 아민 히드로할라이드를 함유하는 아미노-관능성 유기실란의 수성 처리 방법 | |
| RU2360917C2 (ru) | Способ получения (меркаптоорганил)алкоксисиланов | |
| KR20050043693A (ko) | (메르캅토오르가닐)알콕시실란의 제조 방법 | |
| JP5349307B2 (ja) | オルガノシランの製造方法 | |
| JP5044163B2 (ja) | 有機珪素化合物の製造法 | |
| RU2391291C2 (ru) | Способ получения органосиланов | |
| KR20230061436A (ko) | 상 이동 촉매작용에 의해 폴리술판 실란을 제조하는 방법 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| RD04 | Notification of resignation of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7424 Effective date: 20101227 |
|
| RD04 | Notification of resignation of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7424 Effective date: 20101228 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20120515 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20130913 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20130924 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20131216 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20140114 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20140212 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 5478973 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| S533 | Written request for registration of change of name |
Free format text: JAPANESE INTERMEDIATE CODE: R313533 |
|
| R350 | Written notification of registration of transfer |
Free format text: JAPANESE INTERMEDIATE CODE: R350 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| LAPS | Cancellation because of no payment of annual fees |