JP2009514988A - イマチニブ塩基及びイマチニブメシレート、及びそれらの調製方法 - Google Patents
イマチニブ塩基及びイマチニブメシレート、及びそれらの調製方法 Download PDFInfo
- Publication number
- JP2009514988A JP2009514988A JP2008543599A JP2008543599A JP2009514988A JP 2009514988 A JP2009514988 A JP 2009514988A JP 2008543599 A JP2008543599 A JP 2008543599A JP 2008543599 A JP2008543599 A JP 2008543599A JP 2009514988 A JP2009514988 A JP 2009514988A
- Authority
- JP
- Japan
- Prior art keywords
- imatinib
- formula
- base
- desmethyl
- compound
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 0 Cc(ccc(*)c1)c1Nc1nccc(-c2cccnc2)n1 Chemical compound Cc(ccc(*)c1)c1Nc1nccc(-c2cccnc2)n1 0.000 description 2
- BQQYXPHRXIZMDM-UHFFFAOYSA-N Cc(ccc(NC(c1ccc(CN2CCNCC2)cc1)=O)c1)c1Nc1nccc(-c2cccnc2)n1 Chemical compound Cc(ccc(NC(c1ccc(CN2CCNCC2)cc1)=O)c1)c1Nc1nccc(-c2cccnc2)n1 BQQYXPHRXIZMDM-UHFFFAOYSA-N 0.000 description 1
Images








Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/04—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms
- C07D295/14—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D295/155—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals with the ring nitrogen atoms and the carbon atoms with three bonds to hetero atoms separated by carbocyclic rings or by carbon chains interrupted by carbocyclic rings
Abstract
Description
本発明は、結晶性イマチニブ(Imatinib)塩基、デスメチルイマチニブを有さないイマチニブ及びデスメチルイマチニブメシレートを有さないイマチニブメシレート、それらの調製方法及びそれらの医薬組成物に向けられる。
イマチニブメシレート(4−(4−メチルピペラジン−1−イルメチル)−N−[4−メチル−3−[(4−ピリニン−3−イル)ピリミジン−2−イルアミノ]フェニル]べンズアミドメシレート)、すなわち次の化学構造:

イマチニブメシレートにおける不純物、又はいずれかの活性医薬成分(“API”)は所望されるず、そして極端な場合、APTを含む投与形により処理される患者に有害である。
イマチニブ塩基の新規形成現象形の発見は、改良された特徴、例えば流動性及び溶解性を有する結晶形のイマチニブ塩基を生成することにより、活性医薬成分(AP)、すなわちイマチニブメシレートの合成性能を改良する新規機会を提供する。従って多形現象形のイマチニブ塩基についての必要性が当業界において存在する。さらに、デスメチルイマチニブを有さないイマチニブ、及びデスメチルイマチニブメシレートを有さないイマチニブメシレート、及びその分離手段の提供が有益である。
1つの態様においては、本発明は、約6.4、8.1、10.2、12.8、16.1、19.4、20.4、21.7、22.1、25.8及び26.7±0.2°2-θでピークから成る列挙から選択されたいづれかの5個のピークを有する粉末XRDパターン;約159.6、146.7、136.8及び132.4±0.2ppmでシグナルを有する固体状態13C NMR スペクトル;100〜180ppmの化学シフト範囲における最低の化学シフトを示すシグナルと、もう1つのシグナルとの間で約51.2、38.3、28.4及び24.0±0.1ppmの化学シフト差異を有する固体状態13C NMRスペクトル;及びそれらの組合せから成る群から選択されたデータにより特徴づけられる結晶性イマチニブ塩基を包含する。
さらにもう1つの態様においては、本発明は、ピリジンを含む混合物からイマチニブ塩基を結晶化することを含んで成る、上記結晶性イマチニブ塩基の調製方法を包含する。
もう1つの態様においては、本発明は、1,4−ジオキサン中、イマチニブ塩基の溶液を凍結乾燥することを含んで成る非晶性イマチニブ塩基の調製方法を包含する。
1つの態様においては、本発明は、イマチニブ塩の調製のためへの本発明のいずれか1つの形のイマチニブ塩基の使用を包含する。
もう1つの態様においては、本発明は、HPLC面積%単位によれば、約0.09%以下の下記式:
下記式I:
一般式Iの化合物の選択されたバッチによりイマチニブ塩基を調製することを含んで成る。
c)イマチニブ塩基を結晶化し、HPLC面積%単位によれば、約0.09%以下のデスメチルイマチニブを有する結晶性イマチニブを得ることを含んで成る。
さらにもう1つの態様においては、本発明は、HPLC面積%単位によれば、約0.09%以下のデスメチル−イマチニブメシレートを有するイマチニブメシレート、及び医薬的に許容できる賦形剤を組合すことを含んで成る、医薬組成物の調製方法を包含する。
で表される化合物とを反応することを含んで成る、式IVのデスメチル化合物の調製方法を包含する。
1つの態様においては、本発明は、参照マーカーとして式IVのデスメチル化合物を用いてHPLCを実施することを含んで成る方法により、式IVのデスメチル化合物の存在を決定する方法を包含する。
本明細書において使用される場合、用語“イマチニブ”とは、下記式:
前記結晶性イマチニブ塩基は、PXRD又は固体状態C13NMRにより測定される場合、約20%以下の結晶フォームIのイマチニブ塩基、好ましくは約10%以下の結晶フォームI、最も好ましくは約5%以下の結晶フォームIのイマチニブ塩基を有する。典型的には、上記フォームにおける結晶形Iのイマチニブ塩基の含有率は、重量%により測定される。
前記結晶性イマチニブ塩基は、引用により本明細書に組み込まれる、2007年10月26日に出願された同時出願番号11/・・・・・(事件整理番号13150/A400US1)に開示される方法に従って調製され得る。この方法は、下記式III :
で表されるアミンと、下記式V:
で表される4−[(4−メチル−1−ピペラジニル)メチル]ベンゾイル誘導体、及び式III の化合物1g当たり約2〜約10、好ましくは約4〜約7及び最も好ましくは約5〜約6体積の量でのピリジンとを反応し、そしてイマチニブ塩基を回収することを含んで成る。イマチニブ塩基の回収は、本発明の結晶イマチニブ塩基を提供する。アルキル基は好ましくは、C1-6アルキル基である。
ピリジンを含む混合物は、溶媒、抗溶媒、及びピリジンを包含する。結晶化は、イマチニブ塩基又はその塩、ピリジン及び前記溶媒を含む溶液を供給し、そして抗溶媒を添加し、前記結晶性イマチニブ塩基の沈殿物を得ることを含んで成る方法により実施され;ここでイマチニブ塩が使用される場合、混合物はまた、追加の塩基も含んで成る。
本発明の方法においては、溶液への抗溶媒の添加は、前記混合物を提供する。好ましくは、混合物はスラリーである。抗溶媒は、溶媒と同じか又は異なった溶媒であり得る。好ましくは、抗溶媒は水である。
結晶化工程はさらに、スラリーを冷却し、そして冷却されたスラリーを維持し、前記結晶性イマチニブの収率を高めることを含んで成る。好ましくは、冷却は、約30℃〜約0℃、より好ましくは約20℃〜約15℃の温度に実施され得る。好ましくは、冷却されたスラリーは、約1〜約24時間、より好ましくは約14〜約16時間、維持される。
本発明はまた、非晶性イマチニブ塩基を包含する。非晶性イマチニブ塩基は、図5に示されるX−線粉末回折パターンにより特徴づけられ得る。非晶性イマチニブ塩基は、X−線粉末回折パターンでのいずれかの有意な回析ピークの不在により同定され得る。
非晶性イマチニブ塩基は、1,4−ジオキサン中、イマチニブ塩基の溶液を凍結乾燥することを含んで成る方法により調製され得る。
好ましくは、溶液は、イマチニブ塩基及び1,4−ジオキサンを組合し、そしてその組合せを加熱することにより供給される。好ましくは、加熱は、約50〜約110℃、より好ましくは約90℃〜約101℃の温度へ行われる。
凍結乾燥工程は、ジオキサンの凍結温度以下の温度で行われ得る。好ましくは、凍結乾燥は、約12℃〜約0℃の温度で行われる。凍結乾燥は好ましくは、減圧で、好ましくは約0.01〜約100mバール、より好ましくは約0.1〜約15mバール、最も好ましくは約1mバールの圧力で行われる。
典型的には、デスメチルイマチニブメシレートの含有率の測定は、面積%単位によってであり、そして
b)シリカに基づくC18逆相HPLCカラムに、前記溶液を注入し;
c)1−ブタンスルホン酸ナトリウム塩、KH2PO4及びH3PO4の混合物(移動相Aとして言及される)、及びアセト二トリル、メタノール及びテトラヒドロフランの混合物(移動相Bとして言及される)のグラジエント溶離剤を用いて、前記カラムからサンプルを溶出し;そして
d)UV検出器を用いて、デスメチルイマチニブメシレートの含有率を測定することを含んで成るを含んで成るHPLC方法により行われ得る。
典型的には、デスメチル−イマチニブの含有率の測定は、面積%単位によってであり、そして上記のようなHPLC方法により行われ得る。
a)下記式I:
c)一般式I(式中、nは0,1又は2である)の化合物の選択されたバッチによりイマチニブを調製することを含んで成る方法を含んで成る方法により調製され得る。
a)式Iの化合物及び式IIのデスメチル不純物を含んで成るサンプルと、水と組合し、溶液を得;
b)シリカに基づくC18逆相HPLCカラムに、前記溶液を注入し;
c)1−ブタンスルホン酸ナトリウム塩、KH2PO4及びH3PO4の混合物(移動相Aとして言及される)、及びアセト二トリルの混合物(移動相Bとして言及される)のグラジエント溶離剤を用いて、前記カラムからサンプルを溶出し;そして
d)UV検出器を用いて、式IIのデスメチル不純物の含有率を測定すること含んで成るHPLC方法により実施され得る。
結晶化が、水及びC1-3アルコールの混合物中、式Iの化合物の溶液を供給し、そして式Iの化合物を沈殿することを含んで成る。
前記溶液が、式Iの化合物と、水及びC1-3アルコールの混合物とを組合し、そしてその組合せを、約55℃〜約80℃の温度に加熱することにより供給される。好ましくは、加熱は、約55℃〜約80℃、より好ましくは約60℃〜約75℃、最も好ましくは約75℃の温度へである。
で表される4−安息香酸誘導体と、下記式:
次に、約0.15%以下の式IIのデスメチル不純物を有する供給される式Iの化合物が、下記式III :
式Iの化合物と式III のアミンとの反応は例えば2007年10月26日に出願された同時出願番号11/・・・・・(事件整理番号13150/A400US1)に開示される方法により行われ得る。
で表される4−[(4−メチル−1−ピペラジニル)メチル]ベンゾイル誘導体、及び式III の化合物1g当たり約2〜約10(7〜35当量)、より好ましくは約4〜約7及び最も好ましくは約5〜約6体積の量でのピリジンとを反応し、そしてイマチニブを回収することを含んで成る。前記方法はさらに、式Vの化合物を含んで成る反応混合物を得るために、塩基又は吸収剤の存在下でカルボン酸活性化剤と、下記式I:
で表される化合物とを反応することを含んで成る方法により調製され得る。
好ましくは、XはCl、Br又はIであり、より好ましくはXはClである。好ましくは、HBはHCl、HI又はHBrであり、より好ましくはHBはHClでアル。
好ましくは、nは0又は2のいずれかである。
好ましくは、反応混合物は、約15℃〜約25℃に暖められ、続いて、低い粘度を製造するために必要とされる場合、追加の量の溶媒が添加される。
次に、得られる式IVのデスメチル化合物は、当業界において知られているいずれかの方法、例えば抽出及び乾燥により回収され得る。
イマチニブは、イマチニブメシレートに転換される場合、式IVのデスメチル化合物から精製され得る。精製は、例えば引用により本明細書に組み込まれる譲渡されたアメリカ出願番号11/796,573号に開示される方法に従って行われ得る。
得られるイマチニブメシレートは、十分に低い量の式IVのデスメチル化合物、好ましくは約0.15%以下のデスメチル不純物IV、より好ましくは約0.10%以下のデスメチル不純物IVを有する。典型的には、イマチニブメシレートにおける式IVのデスメチルの含有率の測定は、面積%単位、好ましくはHPLCによってである。
1つの態様においては、HPLC面積%単位によれば、約0.09%以下のデスメチル−イマチニブメシレートを有するイマチニブメシレート、及び少なくとも1つの医薬的に許容できる賦形剤を含んで成る医薬組成物を包含する。
もう1つの態様においては、本発明は、HPLC面積%単位によれば、約0.09%以下のデスメチル−イマチニブメシレートを有するイマチニブメシレート、及び医薬的に許容できる賦形剤を組合すことを含んで成る、医薬組成物の調製方法を包含する。
固体及び液体組成物はまた、それらの外観を改良し、そして/又は生成物及び/又は生成物及び単位用量レベルの患者による同定を促進するために、いずれか医薬的に許容できる着色剤を用いて着色され得る。
保存剤及びキレート化剤、例えばアルコール、安息香酸ナトリウム、ブチル化されたヒドロキシルトルエン、ブチル化されたヒドロキシアニソール、及びエチレンジアミン四酢酸が、貯蔵安定性を改良するために摂取のための安全レベルで添加され得る。
その用量は、単位用量形で便利には提供され、そして医薬業界において良く知られているいずれかの方法により調製され得る。
本発明の経口投与形は好ましくは、約10mg〜約160mg、より好ましくは約20mg〜約80mg、及び最も好ましくは20, 40, 60及び80mgの投与量を有する経口カプセルの形で存在する。毎日の投与量は、1日当たり1,2又はそれ以上のカプセルを包含する。
乾燥顆粒化に変わるものとして、ブレンドされた組成物は、直接的圧縮技法を用いて、圧縮された用量形に直接的に圧縮され得る。直接的な圧縮は、顆粒を有さないより均等な錠剤を生成する。
活性成分及び賦形剤は、当業界において知られている方法に従って、組成物及び用量形に配合され得る。
式Iの化合物における式IIのデスメチル不純物の決定のための分析方法:
カラム&充填:YMC-Pack ODS-AQ;5μm、250×4.6mm(C. P. S. Analytica Part. No. AQ12SO5-2546WT)又は同等物。
移動相A :20mMの1−ブタンスルホン酸ナトリウム塩+10mMのKH2PO4、85%H3PO4によりpH2.5にする。
移動相B :アセトニトリル。
後の時間 :10分
流速 :1.0ml/分
検出器 :λ=230nm
カラム温度 :60℃
注入体積 :5μl
希釈剤 :水
典型的な保持時間は次の通りである:
カラム&充填:YMC-Pack ODS-AQ;5μm、250×4.6mm(Part. No. AQ12SO5-2546WT)又は同等物。
移動相A :水中、25mMの1−ブタンスルホン酸ナトリウム塩+25mMのKH2PO4、85%H3PO4によりpH2.3にする。
移動相B :アセトニトリル/メタノール/テトラヒドロフラン 70:10:20(v/v/v)。
後の時間 :15分
集積時間 :55分
流速 :0.9ml/分
検出器 :λ=235nm
カラム温度 :60℃
注入体積 :5μl
希釈剤 :移動相A/移動相B 8:2(v/v)
典型的な保持時間は次の通りである:
PXRD回折を、X−線粉末回折器上で実施した:Philips X’pert Pro粉末回折器、CuKα線、λ=1.5418Å。単一点検出器;走査速度3°/分及び多チャネル X’Celerator検出器活性長(2θ)=2.122°;走査速度6°/分、実験室温度:22−25℃。
DSC測定を、示差走査熱量計DSC823e (Mettler Taledo)上で実施した。PINを有する40μlのAlるつぼを、サンプル分離のために使用した。サンプルの通常の重量は1〜5mgであった。パージガスとしての窒素;50ml/分。
プログラム1:温度は50℃〜100℃、10℃/分、次に100℃〜250℃、40℃/分。
プログラム2:50℃〜250℃、10℃/分。
0℃でのピリジン(400g)中、N−(5−アミノ−2−メチルフェニル)−4−(3−ピリジル)−2−ピリジンアミン(80g)の溶液に、4−[(4−メチル−1−ピペラジニル)メチル]塩化ベンゾイル二塩酸塩(1.1当量)を添加する。反応を撹拌下で15〜20℃で1時間、維持し、次に水(400ml)を添加する。その混合物を40℃まで加熱し、次に26%NH4OH(200g)及び水(900g)を添加する。その反応混合物を、室温で一晩、撹拌下で維持する。固形物を濾過し、水により洗浄し、そして真空下で45℃で3〜4時間、乾燥する。イマチニブを、黄色の粉末として得る(135g、95%の収率、98%以上の純度)。
0℃でのピリジン(400g)中、4−[(4−メチル−1−ピペラジニル)メチル]安息香(84g)の懸濁液に、SOCl2(44.8g、1.05当量)を添加し、そしてその混合物を、30〜50℃で1〜2時間、撹拌下で維持する。0℃で冷却した後、N−(5−アミノ−2−メチルフェニル)−4−(3−ピリジル)−2−ピリジンアミン(80g)を添加する。その反応を15〜20℃で1時間、撹拌下で維持し、次に水(400ml)を添加する。その混合物を40℃まで加熱し、次に、26%NH4OH(200g)及び水(900ml)を添加する。その反応混合物を、室温で一晩、撹拌下で維持する。固形物を濾過し、水により洗浄し、そして真空下で一晩、45℃で乾燥する。本発明のイマチニブ塩基を、黄色の粉末として得る(125g、88%の収率、98%以上の純度)。
20℃でのピリジン(100g)中、4−[(4−メチル−1−ピペラジニル)メチル]安息香二塩酸塩(30g)の懸濁液に、SOCl2(11.5g、1.05当量)を添加し、そしてその混合物を、45〜50℃で1〜2時間、撹拌下で維持する。0℃で冷却した後、N−(5−アミノ−2−メチルフェニル)−4−(3−ピリジル)−2−ピリジンアミン(80g)を添加する。その反応を15〜20℃で1時間、撹拌下で維持し、次に水(100ml)を添加する。その混合物を40℃まで加熱し、次に、26%NH4OH(50g)及び水(225ml)を添加する。その反応混合物を、室温で一晩、撹拌下で維持する。固形物を濾過し、水により洗浄し、そして真空下で一晩、45℃で乾燥する。本発明のイマチニブ塩基を、黄色の粉末として得る(32g、90%の収率、98%以上の純度)。
28%のNH3(30ml)を、ピリジン(140ml)及び水(70ml)の混合物中、イマチニブメシレート(60g)の溶液に、40℃で添加した。前記溶液を、イマチニブ塩基の沈殿が生じるまで、40℃で撹拌下で維持した。追加の量の水(490ml)を40℃で添加し、次にその混合物を室温で自発的に反応せしめ、そしてそれを撹拌下で15時間、維持した。固形物を濾過し、水により洗浄し、そして真空下で40℃で一晩、乾燥した。本発明の結晶性イマチニブ塩基を、黄色の固形物として得た(50g、90%の収率)。
40〜50℃でのピリジン(376g)及び水(188g)中、イマチニブ塩基(94g)の溶液に、水(1300g)を添加した。その混合物を、撹拌下で15〜20℃で一晩、維持し、次に固形物を濾過し、水により洗浄し、そして真空下で40℃で16時間、乾燥した。イマチニブ塩基を、黄色の固形物として得た(99.6g、99%以上の純度)。
37%HCl(9ml)を、ピリジン(140ml)及び水(70ml)の混合物中、イマチニブ塩基(50g)の懸濁液に添加した。その溶液を40℃まで加熱し、木炭により処理し、そして濾過した。28%NH3(30ml)を、40℃で濾液(pH=9.3)添加し、そして溶液を、イマチニブ塩基の沈殿が生じるまで、40℃で撹拌下で維持した。追加の量の水(490ml)を40℃で添加し、次にその混合物を室温で自発的に反応せしめ、そしてそれを撹拌下で15時間、維持した。固形物を濾過し、水により洗浄し、そして真空下で40℃で一晩、乾燥した。本発明の結晶性イマチニブ塩基を、黄色の固形物として得た(50g、90%の収率)。
4−[(4−メチル−1−ピペラジニル)メチル]−N−[4−メチル−3−[[4−(3−ピリジニル)−2−ピリミジニル]アミノフェニル]ベンズアミド(98.2g)を、エタノール(1.4L)に添加する。その懸濁液に、メタンスルホン酸(19.2g)を滴下する。その溶液を、還流下で20分間、加熱し、そして次に65℃で濾過した。濾液を50%まで蒸発し、そして残渣を25℃で濾過した(濾過された材料A)。母液を乾燥まで蒸発した。この残渣及び濾過された材料Aを、エタノール(2.2L)に懸濁し、そして還流下で(30ml)の添加により溶解した。その溶液を冷却し、そして25℃で一晩、維持した。固形物を濾過し、そして65℃で乾燥した。
イマチニブ塩基(500mg)を、90℃で1,4−ジオキサン(10ml)に溶解した。その溶液を25℃に冷却し、そして-30℃でのフリーザーに置き、ここで溶液を凍結した。凍結された溶液を、凍結乾燥機に移し、そして1mバールの真空を適用し、1,4−ジオキサンの凍結乾燥を提供し、非晶性イマチニブ塩基を得た。
水(320ml)及びIPA(420ml)の混合物中、化合物Iの懸濁液を、透明な溶液を得るために75℃で加熱した。激しい撹拌下で、その反応混合物を1時間で20〜25℃まで冷却し、次に、1.5時間で0〜3℃まで冷却し、そしてこの温度で1〜2時間、撹拌下で維持した。固形物を濾過し、そして固形物のケークをIPA(180ml)により洗浄した。生成物を真空下で15時間、60〜65℃で乾燥した。純粋な化合物(I)を白色固形物(131g)として得た。
n−ブタノール(100g)中、4−クロロメチル安息香酸(10g、58.6mモル)及びピペラジン(20g、232mモル)の混合物を50℃で3時間、加熱し、次に室温で一晩、維持した。固形物を濾過し、n−ブタノールにより洗浄し、そして70℃で一晩、乾燥した。デスメチル不純物(II)を、白色固形物として得た(17.5g、86.6%の純度)。
4−クロロメチル−N−[メチル−3−(4−ピリジン−3−イル−ピリミジン−2−イルアミノ)−フェニル]−ベンズアミドの合成:
4−クロロメチルベンゾイルクロリド(15g)を、2〜3℃で、THF(370g)中、N−(5−アミノ−2−メチルフェニル)−4−(3−ピリジル)−2−ピリジンアミン(18.5g)、K2CO3(20g)の混合物に添加した。その混合物を、2〜7℃で1時間、次に15〜20℃で、さらに3時間、撹拌下で維持した。水(700g)を添加し、そしてその懸濁液を15〜20℃で1時間、撹拌下で維持した。固形物を濾過し、水により洗浄し、そして65℃で真空下で15時間、乾燥し、標記生成物を得た(27g、94%の収率)。
37%HCl(9ml)を、ピリジン(140ml)及び水(70ml)の混合物中、イマチニブ塩基(50g、0.12%のデスメチル)の懸濁液に添加した。その溶液を40℃まで加熱し、木炭により処理し、そして濾過した。28%NH3(30ml)を、40℃で濾液(pH=9.3)添加し、そして溶液を、イマチニブ塩基の沈殿が生じるまで、40℃で撹拌下で維持した。追加の量の水(490ml)を40℃で添加し、次にその混合物を室温で自発的に反応せしめ、そしてそれを撹拌下で15時間、維持した。固形物を濾過し、水により洗浄し、そして真空下で40℃で一晩、乾燥した。イマチニブ塩基を、黄色の固形物として得た(50g、90%の収率、0.08%のデスメチル含有率)。
60℃でN2下でのトルエン(35ml)及びDMF(1ml)中、化合物I(n=2、X=Cl)(20g、0.13%のデスメチル不純物IIを含む)の懸濁液に、SOCl2(20g)を1時間にわたって添加する。その混合物を、62℃で撹拌下で20時間、維持する。20℃での冷却の後、トルエン(20ml)を添加し、そしてその混合物を0.5時間、撹拌する。固形物を濾過し、トルエン(50ml)により洗浄し、そして65℃で真空下で15時間、乾燥する。生成物を白色粉末(21g)として得る。0℃でのピリジン(8.25L)中、式III (R=H)のアミン(1.5kg)の溶液に、N2下で固形物としての化合物I(n=2;X=Cl)(2.69kg)を添加した。温度は自発的に5〜10℃に上昇した。
MeSO3H(2.4g)を、60℃でのメタノール(240g)中、デスメチルイマチニブ(12g)の溶液に添加した。溶媒を真空下で蒸発し、そして残渣をエタノール(72g)及びAcOEt(360g)により採取した。その混合物を室温で一晩、撹拌し、次に固形物を濾過し、AcOEtにより洗浄し、そして真空下で75℃で乾燥し、標記生成物(13.9g)を得た。
4−[(4−メチル−1−ピペラジニル)メチル]塩化ベンゾイル二塩酸塩(4g)を、3℃でのピリジン(25ml)中、デスメチルイマチニブ(III )(5g)の溶液に添加した。その混合物を室温にし、次に追加のピリジン(25ml)を添加し、そしてその混合物を室温で6時間、撹拌した。水(50ml)及び28%NH3を添加し、そしてその混合物を真空下で蒸発乾燥した。残渣をDCM及び水により採取した。有機相を分離し、そして蒸発乾燥し、標記化合物を黄色の粉末として得た(4g)。
被膜を5個の錠剤から剥離し、そして残留物を乳鉢において微粉砕した。20mgの粉末を4mlの移動相B及び16mlの移動相Aにより採取した。その混合物を5時間、音波処理し、そして次に濾過した。濾液をHPLCに注入した。
イマチニブメシレート(4.2g、0.08%のデスメチルイマチニブを含む)を、メタノール(10.5ml)に添加し、そしてその混合物を60℃で加熱した。その溶液を20℃に撹拌下で冷却した。20℃での30分間の撹拌の後、固形物を濾過し、そして100℃の真空下で乾燥し、イマチニブメシレート(3.0g、0.08%のデスメチルイマチニブを含む)を得た。
イマチニブメシレート(4.2g、0.39%のデスメチルイマチニブを含む)を、メタノール(10.5ml)に添加し、そしてその混合物を60℃で加熱した。その溶液を20℃に撹拌下で冷却した。20℃での30分間の撹拌の後、固形物を濾過し、そして100℃の真空下で乾燥し、イマチニブメシレート(3.8g、0.38%のデスメチルイマチニブを含む)を得た。
イマチニブ塩基(60g;0.1216モル)を、1200mlのエタノールに懸濁し、そして撹拌した。反応器を、すべての実験の間、窒素流下で維持した(6L/時間)。次に、24mlの水を、前記懸濁液に添加し、そして温度を−15℃で調節した。メタンスルホン酸のエタノール溶液(79.8ml、10%v/v;0.1213モル)を、反応混合物に2分間にわたって添加した。溶液の温度を、10分間、−10℃で設定し、イマチニブ塩基を溶解し、そして播種材料(2g)を添加した。結晶化工程を、撹拌下で190分間、続け、そして温度を−5℃に連続的に高めた。懸濁液を、約−27℃でフリーザーに一晩、貯蔵した。次に、懸濁液を、1000mlのTBMEにより希釈し、窒素圧により濾過し、そして得られる結晶部分を400mlのTBMEにより洗浄した。得られる結晶フォームを、フィルターを通して窒素流により乾燥し、遊離エタノールを除去した。エタノール含有率は約7.5%であった(収量は67.95g(85%)であった)。
Claims (62)
- 約6.4、8.1、10.2、12.8、16.1、19.4、20.4、21.7、22.1、25.8及び26.7±0.2°2-θでピークから成るリストから選択されたいづれかの5個のピークを有する粉末XRDパターン;約159.6、146.7、136.8及び132.4±0.2ppmでシグナルを有する固体状態13C NMR スペクトル;100〜180ppmの化学シフト範囲における最低の化学シフトを示すシグナルと、もう1つのシグナルとの間で約51.2、38.3、28.4及び24.0±0.1ppmの化学シフト差異を有する固体状態13C NMRスペクトルから成る群から選択されたデータにより特徴づけられる結晶性イマチニブ(Imatinib)塩基。
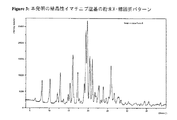
- 前記結晶性イマチニブ塩基が、図3に示される粉末XRDパターン、図1に示される固体状態13C NMR スペクトル、及び図2に示される固体状態13C NMR スペクトルから成る群から選択されたデータにより特徴づけられる請求項1記載の結晶性イマチニブ塩基。
- 約8.1、10.2、12.8、16.1及び19.4±0.2°2−θでピークを有する粉末XRDパターン;約6.4、8.1, 10.2, 19.4、20.4及び25.8±0.2°2−θでピークを有する粉末XRDパターン;約17.3、20.4、21.1及び25.8±0.2°2−θでピークを有する粉末XRDパターン;約6.4、21.7、22.1及び26.7±0.2°2−θでピークを有する粉末XRDパターン;約125.8及び108.4±0.2ppmでシグナルを有する固体状態13C NMR スペクトル;2種のピークを有するDSC曲線(第1ピークは97.6℃で吸熱ピークであり、そして第2ピークは210.5℃で吸熱ピークである);及び図4に示されるようなDSC曲線から成る群から選択されたデータによりさらに特徴づけられる請求項1又は2記載の結晶性イマチニブ塩基。
- 前記結晶性イマチニブ塩基が、イマチニブ塩基のピリジン溶媒化合物である請求項1〜3のいずれか1項記載の結晶性イマチニブ塩基。
- 前記ピリジン溶媒化合物が、半−ピリジン溶媒化合物である請求項4記載の結晶性イマチニブ塩基。
- 前記ピリジン含有率が、GCにより測定される場合、約7%(w/w)である請求項4〜5のいずれか1項記載の結晶性イマチニブ塩基。
- 前記イマチニブ塩基が、PXRD又は固体状態C13NMRにより測定される場合、約20%以下の結晶フォームIのイマチニブ塩基を有する請求項1〜6のいずれか1項記載の結晶性イマチニブ塩基。
- 請求項1〜7のいずれか1項記載の結晶性イマチニブ塩基の調製方法であって、
下記式III :
で表されるアミンと、下記式V:
で表される4−[(4−メチル−1−ピペラジニル)メチル]ベンゾイル誘導体、及び式III の化合物1g当たり約2〜約10体積の量でのピリジンとを反応せしめ、そしてイマチニブ塩基を回収する;
ことを含んで成る方法。 - XがClであり、そしてRがHである請求項8記載の方法。
- イマチニブ塩基又はその塩と、ピリジンとを組合し、混合物を得、そして前記混合物からイマチニブ塩基を結晶化することを含んで成る請求項1〜9のいずれか1項記載の方法。
- 結晶化が、イマチニブ塩基、ピリジン及び溶媒を含む溶液を調製し、そして抗溶媒を添加し、前記結晶性イマチニブ塩基の沈殿物を得ることを含んで成る請求項10記載の方法。
- 前記溶媒が、水、水相溶性有機溶媒又はそれらの混合物である請求項10〜11のいずれか1項記載の方法。
- 前記水相溶性有機溶媒が、ジメチルホルムアミド、ジメチルアセトアミド、テトラヒドロフラン、アルコール、アセトン、アセトニトリル、ジオキサン、ジメチルスルホキシド及びそれらの混合物から成る群から選択される請求項12記載の方法。
- 前記溶媒が、ジメチルホルムアミド、ジメチルアセトアミド、テトラヒドロフラン又は水である請求項12記載の方法。
- 前記溶液が、イマチニブ塩及びイマチニブ塩基を得るための追加の塩基を組合すことにより調製される請求項10〜14のいずれか1項記載の方法。
- 前記追加の塩基が、第三アミンから成る群から選択された有機塩基、及び水酸化ナトリウム又はカリウム、炭酸水素ナトリウム又はカリウム、又はアンモニウムから成る群から選択されたアルカリ塩である無機塩基である請求項15記載の方法。
- 前記溶液が、イマチニブ塩基又は塩、溶媒、ピリジン及び任意には追加の塩基の組合せを、約5℃〜約70℃の温度に加熱することにより供給される請求項10〜16のいずれか1項記載の方法。
- 前記加熱が、約40℃〜約50℃の温度へである請求項17記載の方法。
- 前記ピリジンが、イマチニブ塩基又は塩1g当たり約2〜約10体積の量で、前記溶液に存在する請求項10〜18のいずれか1項記載の方法。
- 前記スラリーを、約30℃〜約0℃の温度に、約1〜約24時間にわたって冷却することをさらに含んで成る請求項10〜19のいずれか1項記載の方法。
- 請求項1〜7のいずれか1項記載の結晶性イマチニブ塩基を、イマチニブ塩に転換することを含んで成る、イマチニブ塩の調製方法。
- 非晶性イマチニブ塩基。
- 前記非晶性イマチニブ塩基が、図5に示されるX−線粉末回折パターンにより特徴づけられ得る請求項22記載の非晶性イマチニブ塩基。
- 1,4−ジオキサン中、イマチニブ塩基の溶液を凍結乾燥することを含んで成る請求項22又は23記載の非晶性イマチニブ塩基の調製方法。
- 前記溶液が、イマチニブ塩基及び1,4−ジオキサンを組合し、そしてその組合せを、約50℃〜約110℃の温度に加熱することにより供給される請求項24記載の方法。
- 前記凍結乾燥工程が、約12℃〜約0℃の温度で実施される請求項24〜25のいずれか1項記載の方法。
- 凍結乾燥が、約0.01〜約100mバールで実施される請求項26記載の方法。
- 非晶性イマチニブ塩基をイマチニブ塩に転換することを含んで成るイマチニブ塩の調製方法。
- HPLC面積%単位によれば、約0.09%以下の下記式:
- HPLC面積%単位によれば、約0.07%以下のデスメチル−イマチニブメシレートを有する請求項29記載のイマチニブメシレート。
- HPLC面積%単位によれば、約0.09%以下の下記式:
- HPLC面積%単位によれば、0.07%以下のデスメチル−イマチニブ塩基を有する請求項31記載のイマチニブ塩基。
- HPLC面積%単位によれば、約0.09%以下のデスメチル−イマチニブを有するイマチニブの調製方法であって、
a)下記式I:
b)約0.15%以下の式IIのデスメチル不純物を有する、式Iの化合物のバッチを選択し;そして
c)一般式I(式中、nは0,1又は2である)の化合物の選択されたバッチによりイマチニブを調製することを含んで成る方法。 - 面積HPLC%単位によれば、式IIのデスメチル不純物の含有率の測定が、
a)式Iの化合物及び式IIのデスメチル不純物を含んで成るサンプルと、水と一緒にして、溶液を得;
b)シリカに基づくC18逆相HPLCカラムに、前記溶液を注入し;
c)1−ブタンスルホン酸ナトリウム塩、KH2PO4及びH3PO4の混合物(移動相Aとして言及される)、及びアセト二トリルの混合物(移動相Bとして言及される)のグラジエント溶離剤を用いて、前記カラムからサンプルを溶出し;そして
d)UV検出器を用いて、式IIのデスメチル不純物の含有率を測定する;
ことを含んで成る請求項33記載の方法。 - HPLC面積%単位によれば、約0.15%以下の式IIのデスメチル不純物を有する式Iの化合物が、水及びC1-3アルコールの混合物から式Iの化合物を結晶化することを含んで成る工程により供給される請求項33又は34記載の方法。
- 結晶化が、水及びC1-3アルコールの混合物中、式Iの化合物の溶液を供給し、そして式Iの化合物を沈殿することを含んで成る請求項35記載の方法。
- 前記溶液が、式Iの化合物と、水及びC1-3アルコールの混合物とを組合し、そしてその組合せを、約55℃〜約80℃の温度に加熱することにより供給される請求項35又は36記載の方法。
- 前記温度が、約65〜約75℃である請求項37記載の方法。
- 前記C1-3アルコールが、イソプロパノール(“IPA”)である請求項36〜38のいずれか1項記載の方法。
- 前記混合物におけるC1-3アルコール:水の比が、約80:20である請求項36〜39のいずれか1項記載の方法。
- 前記溶液が、約50℃〜約-5℃の温度に冷却される請求項36〜40のいずれか1項記載の方法。
- 冷却が、前記溶液の約35℃〜約15℃の温度への第1の冷却、及び約5℃〜約-5℃の温度への第2の冷却段階により段階的に実施される請求項41記載の方法。
- 前記第1の冷却段階が約0.5〜約3時間にわたって行なわれ、そして前記第2の冷却が約0.5〜約5時間にわたって行なわれる請求項42記載の方法。
- 前記冷却された溶液が、そのような温度で約1〜約5時間、維持される請求項41〜43のいずれか1項記載の方法。
- 段階(c)におけるようなイマチニブの調製が、HPLC面積%単位によれば、約0.15%以下の式IIのデスメチル不純物を有する式Iの化合物と、下記式III :
- 前記得られるイマチニブを結晶化することをさらに含んで成る請求項45記載の方法。
- イマチニブの少なくとも1つのバッチにおけるデスメチルイマチニブのレベルを測定し、HPLC面積%単位によれば、約0.09%以下のイマチニブのバッチを選択し、そしてHPLC面積%単位によれば、約0.09%以下のイマチニブによりイマチニブメシレートを調製することを含んで成る方法により、HPLC面積%単位によれば、約0.09%以下のデスメチルイマチニブを有するイマチニブメシレートを調製することをさらに含んで成る請求項45記載の方法。
- 下記式IV:
- HPLC面積%単位によれば、約0.15%以下のデスメチルイマチニブを有する式IVのデスメチル化合物。
- 約2.141、 2.329、 2.305、2.399、 3.463、 3.583、 7.322、 7.325、 7.923及び10.159ppmでピークを有する1H NMR (DMSO-d6)スペクトル;図6に示されるような1H NMRスペクトル;約45.71、 52.54、 54.73、 61.63、 61.42、126.67、126.69、126.93、128.76、 133.95、 134.48、137.85、139.94、141.59、165.24及び168.97ppmでピークを有する13C NMR (DMSO-d6)スペクトル;図7に示されるような13C NMRスペクトル;約1452、1528、1680及び2937cm-1で主要ピークを有するIRスペクトル;図8に示されるようなIRスペクトル;約696g/モルで[MH]+ピークを有するMSスペクトル;及び図9に示されるようなMSスペクトルから成る群から選択されたデータの少なくとも1つにより特徴づけられる式IVのデスメチル化合物。
- 請求項48〜50のいずれか1項記載の式IVのデスメチル化合物の調製方法であって、
下記式:
で表される化合物とを反応することを含んで成る方法。 - XがClであり、HBがHClであり、そしてnが0又は2のいずれかである請求項51記載の方法。
- 塩基が前記反応に添加される請求項51〜52のいずれか1項記載の方法。
- 前記塩基が、アミン及びアルカリ金属塩基から成る群から選択される請求項53記載の方法。
- 前記塩基が、トリエチルアミン(“TEA”)、ジイソプロピルアミン(“DIPEA”)、N−メチルモルホリン、それらの混合物、K2CO3、 Na2CO3、NaHCO3、KHCO3及びそれらの混合物から成る群から選択される請求項53〜54のいずれか1項記載の方法。
- 前記塩基の量が、化合物Vの1モルに当たり少なくとも1モル当量である請求項53〜55のいずれか1項記載の方法。
- 前記反応が、テトラヒドロフラン(“THF”)、メチルテトラヒドロフラン(“MeTHF”)、ジオキソラン、ジクロロメタン(“DCM”)、ジメチルホルムアミド(“DMF”)、ジメチルアセトアミド(“DMA”)、ジメチルスルホキシド(“DMSO”)、トルエン及びそれらの混合物から成る群から選択された溶媒である請求項51〜56のいずれか1項記載の方法。
- 前記反応混合物が、約15℃〜約25℃の温度に暖められ、続いて追加の量の溶媒が添加される請求項51〜57のいずれか1項記載の方法。
- 式IVのデスメチル化合物が参照マーカーである請求項48〜50のいずれか1項記載の式IVのイマチニブ化合物。
- サンプルに存在する式IVのデスメチル化合物の量の決定方法であって、(a)既知量の式IVのデスメチル化合物を含んで成る参照標準における式IVのデスメチル化合物に対応するピーク下の面積をHPLCにより測定し;(b)式IVのデスメチル化合物及びイマチニブを含んで成るサンプルにおける式IVのデスメチル化合物に対応するピーク下の面積をHPLCにより測定し;そして(c)段階(b)の面積に対して段階(a)の面積を比較することにより、サンプルにおける式IVのデスメチル化合物の量を決定することを含んで成る方法。
- HPLC面積%単位によれば、約0.09%以下のデスメチル−イマチニブメシレートを有するイマチニブメシレート、及び少なくとも1つの医薬的に許容できる賦形剤を含んで成る医薬組成物。
- HPLC面積%単位によれば、約0.09%以下のデスメチル−イマチニブメシレートを有するイマチニブメシレート、及び医薬的に許容できる賦形剤を組合すことを含んで成る、医薬組成物の調製方法。
Applications Claiming Priority (11)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US85477406P | 2006-10-26 | 2006-10-26 | |
| US86062406P | 2006-11-22 | 2006-11-22 | |
| US87442006P | 2006-12-11 | 2006-12-11 | |
| US93491107P | 2007-06-14 | 2007-06-14 | |
| US95836707P | 2007-07-05 | 2007-07-05 | |
| US96323807P | 2007-08-02 | 2007-08-02 | |
| US96761707P | 2007-09-05 | 2007-09-05 | |
| US99533207P | 2007-09-25 | 2007-09-25 | |
| US99784907P | 2007-10-05 | 2007-10-05 | |
| US97925607P | 2007-10-11 | 2007-10-11 | |
| PCT/US2007/022747 WO2008057291A2 (en) | 2006-10-26 | 2007-10-26 | Crystalline and amorphous imatinib base, imatinib mesylate- and processes for preparation thereof |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2009514988A true JP2009514988A (ja) | 2009-04-09 |
| JP2009514988A5 JP2009514988A5 (ja) | 2011-08-18 |
Family
ID=39027276
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2008541513A Pending JP2009503120A (ja) | 2006-10-26 | 2007-10-26 | イマチニブの調製方法 |
| JP2008543599A Pending JP2009514988A (ja) | 2006-10-26 | 2007-10-26 | イマチニブ塩基及びイマチニブメシレート、及びそれらの調製方法 |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2008541513A Pending JP2009503120A (ja) | 2006-10-26 | 2007-10-26 | イマチニブの調製方法 |
Country Status (6)
| Country | Link |
|---|---|
| US (2) | US20080207904A1 (ja) |
| EP (2) | EP2076507A2 (ja) |
| JP (2) | JP2009503120A (ja) |
| KR (2) | KR20090061055A (ja) |
| MX (1) | MX2008008447A (ja) |
| WO (2) | WO2008051597A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2010540465A (ja) * | 2007-09-25 | 2010-12-24 | テバ ファーマシューティカル インダストリーズ リミティド | 安定なイマチニブ組成物 |
Families Citing this family (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20100330130A1 (en) | 2009-05-22 | 2010-12-30 | Actavis Group Ptc Ehf | Substantially pure imatinib or a pharmaceutically acceptable salt thereof |
| US20110306763A1 (en) | 2009-12-10 | 2011-12-15 | Shanghai Parling Pharmatech Co., Ltd. | Process for the preparation of imatinib and salts thereof |
| WO2011095835A1 (en) | 2010-02-02 | 2011-08-11 | Actavis Group Ptc Ehf | Highly pure imatinib or a pharmaceutically acceptable salt thereof |
| EA024088B1 (ru) | 2010-06-18 | 2016-08-31 | КРКА, д.д., НОВО МЕСТО | α-ФОРМА МЕЗИЛАТА ИМАТИНИБА, СПОСОБЫ ЕЕ ПОЛУЧЕНИЯ И СОДЕРЖАЩАЯ ЕЁ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ |
| WO2012015999A2 (en) * | 2010-07-29 | 2012-02-02 | Dr. Reddy's Laboratories Ltd. | Process for the preparation of imatinib mesylate |
| CN101899035B (zh) * | 2010-09-03 | 2012-09-05 | 天津市炜杰科技有限公司 | 一种高纯度伊马替尼的制备方法 |
| GB2488788B (en) * | 2011-03-07 | 2013-07-10 | Natco Pharma Ltd | Oral formulation of phenylaminopyrymidine compound with enhanced bioavailability and pharmacological response |
| EP2691385A4 (en) | 2011-03-31 | 2014-08-13 | Ind Swift Lab Ltd | IMPROVED METHOD FOR THE PRODUCTION OF IMATINIB AND ITS MESYLATE SALT |
| KR101139431B1 (ko) | 2011-05-30 | 2012-04-27 | (주)비씨월드제약 | 이매티닙 염기의 신규한 제조방법 |
| WO2013008242A1 (en) * | 2011-07-12 | 2013-01-17 | Natco Pharma Limited | A process for the preparation of highly pure 4-(4-methyl piperazinomethyl) benzoic acid dihydrochloride |
| CN102850297B (zh) * | 2012-10-10 | 2014-07-23 | 山东金城医药化工股份有限公司 | 伊马酸的制备方法 |
| KR101558960B1 (ko) | 2013-07-18 | 2015-10-08 | 하나제약 주식회사 | Ν-5-(4-[4-메틸-피페라지노-메틸]-벤조일아미도)-2-메틸페닐-4-[3-피리딜]-2-피리미딘-아민의 신규한 제조방법 |
| CN103483314B (zh) * | 2013-09-16 | 2015-02-18 | 南京优科生物医药研究有限公司 | 一种便捷的制备甲磺酸伊马替尼α晶型的方法 |
| SE539450C2 (en) * | 2016-02-29 | 2017-09-26 | Imatinib for use in the treatment of stroke | |
| US11298355B2 (en) | 2019-05-16 | 2022-04-12 | Aerovate Therapeutics, Inc. | Inhalable imatinib formulations, manufacture, and uses thereof |
| US11464776B2 (en) | 2019-05-16 | 2022-10-11 | Aerovate Therapeutics, Inc. | Inhalable imatinib formulations, manufacture, and uses thereof |
| CN115850258A (zh) * | 2022-12-27 | 2023-03-28 | 东北林业大学 | 一种马赛替尼的合成方法 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0687834A (ja) * | 1992-04-03 | 1994-03-29 | Ciba Geigy Ag | ピリミジン誘導体及びその製法 |
| JP2005502861A (ja) * | 2001-08-10 | 2005-01-27 | サイミックス テクノロジーズ, インコーポレイテッド | 事前処方物を作製および試験するための装置および方法ならびにそのためのシステム |
| JP2005528340A (ja) * | 2002-02-07 | 2005-09-22 | ノバルティス アクチエンゲゼルシャフト | N−フェニル−2−ピリミジン−アミン誘導体 |
Family Cites Families (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4623486A (en) * | 1985-05-29 | 1986-11-18 | Pfizer Inc. | [4-substituted benzoyloxy]-N-substituted-2H-1,2-benzothiazine-3-carboxamide 1,1-dioxides having anti-arthritic activity |
| US5521184A (en) * | 1992-04-03 | 1996-05-28 | Ciba-Geigy Corporation | Pyrimidine derivatives and processes for the preparation thereof |
| CO4940418A1 (es) * | 1997-07-18 | 2000-07-24 | Novartis Ag | Modificacion de cristal de un derivado de n-fenil-2- pirimidinamina, procesos para su fabricacion y su uso |
| GB0022438D0 (en) * | 2000-09-13 | 2000-11-01 | Novartis Ag | Organic Compounds |
| JP2003119184A (ja) * | 2001-10-11 | 2003-04-23 | Toray Ind Inc | 置換ピペラジニルメチル芳香族酸誘導体の製造方法 |
| GB0201508D0 (en) * | 2002-01-23 | 2002-03-13 | Novartis Ag | Organic compounds |
| GB2398565A (en) * | 2003-02-18 | 2004-08-25 | Cipla Ltd | Imatinib preparation and salts |
| WO2004099186A1 (en) * | 2003-05-06 | 2004-11-18 | Il Yang Pharm Co., Ltd. | N-phenyl-2-pyrimidine-amine derivatives and process for the preparation thereof |
| US7300938B2 (en) * | 2003-06-02 | 2007-11-27 | Hetero Drugs Limited | Polymorphs of imatinib mesylate |
| US7507821B2 (en) * | 2004-12-30 | 2009-03-24 | Chemagis Ltd. | Process for preparing Imatinib |
| US20060223816A1 (en) * | 2006-05-08 | 2006-10-05 | Chemagis Ltd. | Imatinib mesylate alpha form and production process therefor |
| US20060223817A1 (en) * | 2006-05-15 | 2006-10-05 | Chemagis Ltd. | Crystalline imatinib base and production process therefor |
| US7550591B2 (en) * | 2007-05-02 | 2009-06-23 | Chemagis Ltd. | Imatinib production process |
-
2007
- 2007-10-26 EP EP07839811A patent/EP2076507A2/en not_active Withdrawn
- 2007-10-26 WO PCT/US2007/022637 patent/WO2008051597A1/en active Application Filing
- 2007-10-26 KR KR1020097008048A patent/KR20090061055A/ko not_active Application Discontinuation
- 2007-10-26 JP JP2008541513A patent/JP2009503120A/ja active Pending
- 2007-10-26 MX MX2008008447A patent/MX2008008447A/es not_active Application Discontinuation
- 2007-10-26 US US11/978,170 patent/US20080207904A1/en not_active Abandoned
- 2007-10-26 WO PCT/US2007/022747 patent/WO2008057291A2/en active Application Filing
- 2007-10-26 JP JP2008543599A patent/JP2009514988A/ja active Pending
- 2007-10-26 US US11/978,227 patent/US20080103305A1/en not_active Abandoned
- 2007-10-26 KR KR1020097008519A patent/KR20090061068A/ko not_active Application Discontinuation
- 2007-10-26 EP EP07839783A patent/EP1966186A1/en not_active Withdrawn
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0687834A (ja) * | 1992-04-03 | 1994-03-29 | Ciba Geigy Ag | ピリミジン誘導体及びその製法 |
| JP2005502861A (ja) * | 2001-08-10 | 2005-01-27 | サイミックス テクノロジーズ, インコーポレイテッド | 事前処方物を作製および試験するための装置および方法ならびにそのためのシステム |
| JP2005528340A (ja) * | 2002-02-07 | 2005-09-22 | ノバルティス アクチエンゲゼルシャフト | N−フェニル−2−ピリミジン−アミン誘導体 |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2010540465A (ja) * | 2007-09-25 | 2010-12-24 | テバ ファーマシューティカル インダストリーズ リミティド | 安定なイマチニブ組成物 |
Also Published As
| Publication number | Publication date |
|---|---|
| KR20090061068A (ko) | 2009-06-15 |
| JP2009503120A (ja) | 2009-01-29 |
| WO2008057291A3 (en) | 2008-07-03 |
| KR20090061055A (ko) | 2009-06-15 |
| WO2008057291A2 (en) | 2008-05-15 |
| WO2008051597A1 (en) | 2008-05-02 |
| EP1966186A1 (en) | 2008-09-10 |
| US20080207904A1 (en) | 2008-08-28 |
| EP2076507A2 (en) | 2009-07-08 |
| MX2008008447A (es) | 2008-09-15 |
| WO2008057291B1 (en) | 2008-08-21 |
| US20080103305A1 (en) | 2008-05-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP2009514988A (ja) | イマチニブ塩基及びイマチニブメシレート、及びそれらの調製方法 | |
| JP2007302658A (ja) | イマチニブメシレートの多形フォーム及び新規結晶フォーム及び非晶フォーム並びにフォームαの調製方法 | |
| TW202015698A (zh) | Tlr7/tlr8抑制劑之晶型 | |
| US20210387952A1 (en) | Solid state forms of daprodustat and process for preparation thereof | |
| CN101641345A (zh) | 晶体和无定形伊马替尼碱、甲磺酸伊马替尼及其制备方法 | |
| US20210292479A1 (en) | Solid state forms of sugammadex sodium | |
| JP2008539278A (ja) | 結晶性ロスバスタチンカルシウム | |
| JP2015522037A (ja) | ベムラフェニブコリン塩の固体形態 | |
| EP1988089A1 (en) | Imatinib base, and imatinib mesylate and processes for preparation thereof | |
| US20220135548A1 (en) | Solid state forms of n-[2-(2-{4-[2-(6,7-dimethoxy-3,4-dihydro-2(1h)-isoquinolinyl)ethyl]phenyl}-2h-tetrazol-5-yl)-4,5- dimethoxyphenyl]-4-oxo-4h-chromene-2-carboxamide and of its mesylate salt | |
| CA2518999A1 (en) | Crystalline and amorphous solids of pantoprazole and processes for their preparation | |
| WO2020051014A1 (en) | Processes for the preparation of tenapanor and intermediates thereof | |
| US20230339962A1 (en) | Solid state forms of sep-363856 and process for preparation thereof | |
| US20220153744A1 (en) | Solid state forms of acalabrutinib | |
| US20240010632A1 (en) | Solid state forms of erdafitinib salts and processes for preparation of erdafitinib | |
| WO2022177927A1 (en) | Unhydrous crystalline form of omecamtiv mecarbil dihydrobromide salt | |
| WO2022217008A1 (en) | Solid state forms of zavegepant and process for preparation thereof | |
| EP3990113A1 (en) | Solid state forms of roluperidone and salts thereof | |
| US20220289764A1 (en) | Crystalline lorlatinib : fumaric acid and solid state form thereof | |
| US20230322786A1 (en) | Solid state forms of at-001 and process for preparation thereof | |
| US20220380288A1 (en) | Solid state forms of fezagepras and process for preparation thereof | |
| US20220135566A1 (en) | Crystalline solid forms of baricitinib | |
| US20230357163A1 (en) | Solid state forms of gefapixant and process for preparation thereof | |
| US20230373998A1 (en) | Solid state forms of lorecivivint | |
| US20230278990A1 (en) | Solid state forms of n-[2-(2-{4-[2-(6,7-dimethoxy-3,4-dihydro-2(1h)-isoquinolinyl)ethyl]phenyl}-2h-tetrazol-5-yl)-4,5-dimethoxyphenyl]-4-oxo-4h-chromene-2-carboxamide mesylate salt |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20090224 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20090410 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20110104 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20110401 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20110408 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20110704 |
|
| A524 | Written submission of copy of amendment under section 19 (pct) |
Free format text: JAPANESE INTERMEDIATE CODE: A524 Effective date: 20110704 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20120110 |