JP2008500363A - 新規化合物、該化合物を含む医薬組成物、および該化合物の使用方法 - Google Patents
新規化合物、該化合物を含む医薬組成物、および該化合物の使用方法 Download PDFInfo
- Publication number
- JP2008500363A JP2008500363A JP2007515318A JP2007515318A JP2008500363A JP 2008500363 A JP2008500363 A JP 2008500363A JP 2007515318 A JP2007515318 A JP 2007515318A JP 2007515318 A JP2007515318 A JP 2007515318A JP 2008500363 A JP2008500363 A JP 2008500363A
- Authority
- JP
- Japan
- Prior art keywords
- compound
- pharmaceutical composition
- alkyl
- fas
- cells
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 150000001875 compounds Chemical class 0.000 title claims abstract description 75
- 238000000034 method Methods 0.000 title claims description 22
- 239000008194 pharmaceutical composition Substances 0.000 title claims description 18
- 239000008024 pharmaceutical diluent Substances 0.000 claims abstract description 14
- 125000000217 alkyl group Chemical group 0.000 claims abstract description 12
- 125000003118 aryl group Chemical group 0.000 claims abstract description 11
- 125000003342 alkenyl group Chemical group 0.000 claims abstract description 10
- 125000002877 alkyl aryl group Chemical group 0.000 claims abstract description 10
- 125000003710 aryl alkyl group Chemical group 0.000 claims abstract description 10
- 125000000753 cycloalkyl group Chemical group 0.000 claims abstract description 10
- 125000004400 (C1-C12) alkyl group Chemical group 0.000 claims abstract description 7
- 125000005843 halogen group Chemical group 0.000 claims abstract description 5
- 108010039731 Fatty Acid Synthases Proteins 0.000 claims description 51
- 230000000694 effects Effects 0.000 claims description 24
- 241001465754 Metazoa Species 0.000 claims description 21
- 241000282412 Homo Species 0.000 claims description 14
- 230000004580 weight loss Effects 0.000 claims description 12
- 230000000813 microbial effect Effects 0.000 claims description 11
- 230000015572 biosynthetic process Effects 0.000 claims description 9
- 238000003786 synthesis reaction Methods 0.000 claims description 9
- LMBFAGIMSUYTBN-MPZNNTNKSA-N teixobactin Chemical compound C([C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H](CCC(N)=O)C(=O)N[C@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H]1C(N[C@@H](C)C(=O)N[C@@H](C[C@@H]2NC(=N)NC2)C(=O)N[C@H](C(=O)O[C@H]1C)[C@@H](C)CC)=O)NC)C1=CC=CC=C1 LMBFAGIMSUYTBN-MPZNNTNKSA-N 0.000 claims description 9
- 206010028980 Neoplasm Diseases 0.000 claims description 7
- 201000011510 cancer Diseases 0.000 claims description 7
- 230000002401 inhibitory effect Effects 0.000 claims description 7
- 101710151321 Melanostatin Proteins 0.000 claims description 5
- 102400000064 Neuropeptide Y Human genes 0.000 claims description 5
- 230000012010 growth Effects 0.000 claims description 5
- URPYMXQQVHTUDU-OFGSCBOVSA-N nucleopeptide y Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(N)=O)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H]1N(CCC1)C(=O)[C@@H](N)CC=1C=CC(O)=CC=1)C1=CC=C(O)C=C1 URPYMXQQVHTUDU-OFGSCBOVSA-N 0.000 claims description 5
- 230000001939 inductive effect Effects 0.000 claims description 3
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 3
- 230000005907 cancer growth Effects 0.000 claims description 2
- 230000004936 stimulating effect Effects 0.000 claims description 2
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims 1
- 101150073133 Cpt1a gene Proteins 0.000 claims 1
- 102000015303 Fatty Acid Synthases Human genes 0.000 claims 1
- 102100022089 Acyl-[acyl-carrier-protein] hydrolase Human genes 0.000 description 50
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 48
- 210000004027 cell Anatomy 0.000 description 47
- 102000004190 Enzymes Human genes 0.000 description 25
- 108090000790 Enzymes Proteins 0.000 description 25
- 239000003112 inhibitor Substances 0.000 description 25
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 20
- 235000014113 dietary fatty acids Nutrition 0.000 description 20
- 239000000194 fatty acid Substances 0.000 description 20
- 229930195729 fatty acid Natural products 0.000 description 20
- 150000004665 fatty acids Chemical class 0.000 description 18
- 230000005764 inhibitory process Effects 0.000 description 18
- 239000000243 solution Substances 0.000 description 18
- LTYOQGRJFJAKNA-KKIMTKSISA-N Malonyl CoA Natural products S(C(=O)CC(=O)O)CCNC(=O)CCNC(=O)[C@@H](O)C(CO[P@](=O)(O[P@](=O)(OC[C@H]1[C@@H](OP(=O)(O)O)[C@@H](O)[C@@H](n2c3ncnc(N)c3nc2)O1)O)O)(C)C LTYOQGRJFJAKNA-KKIMTKSISA-N 0.000 description 15
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 15
- 230000004136 fatty acid synthesis Effects 0.000 description 15
- LTYOQGRJFJAKNA-DVVLENMVSA-N malonyl-CoA Chemical compound O[C@@H]1[C@H](OP(O)(O)=O)[C@@H](COP(O)(=O)OP(O)(=O)OCC(C)(C)[C@@H](O)C(=O)NCCC(=O)NCCSC(=O)CC(O)=O)O[C@H]1N1C2=NC=NC(N)=C2N=C1 LTYOQGRJFJAKNA-DVVLENMVSA-N 0.000 description 15
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical class CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 14
- 229940079593 drug Drugs 0.000 description 13
- 239000003814 drug Substances 0.000 description 13
- ACFIXJIJDZMPPO-NNYOXOHSSA-N NADPH Chemical compound C1=CCC(C(=O)N)=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OC[C@@H]2[C@H]([C@@H](OP(O)(O)=O)[C@@H](O2)N2C3=NC=NC(N)=C3N=C2)O)O1 ACFIXJIJDZMPPO-NNYOXOHSSA-N 0.000 description 12
- 238000003556 assay Methods 0.000 description 11
- 229930027945 nicotinamide-adenine dinucleotide Natural products 0.000 description 11
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 11
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 10
- 235000019439 ethyl acetate Nutrition 0.000 description 10
- 238000007254 oxidation reaction Methods 0.000 description 9
- 238000012360 testing method Methods 0.000 description 9
- 239000003981 vehicle Substances 0.000 description 9
- GVEZIHKRYBHEFX-MNOVXSKESA-N 13C-Cerulenin Natural products CC=CCC=CCCC(=O)[C@H]1O[C@@H]1C(N)=O GVEZIHKRYBHEFX-MNOVXSKESA-N 0.000 description 8
- 241000699670 Mus sp. Species 0.000 description 8
- GVEZIHKRYBHEFX-UHFFFAOYSA-N caerulein A Natural products CC=CCC=CCCC(=O)C1OC1C(N)=O GVEZIHKRYBHEFX-UHFFFAOYSA-N 0.000 description 8
- 229950005984 cerulenin Drugs 0.000 description 8
- 238000011534 incubation Methods 0.000 description 8
- 238000012417 linear regression Methods 0.000 description 8
- 230000003647 oxidation Effects 0.000 description 8
- GVEZIHKRYBHEFX-NQQPLRFYSA-N cerulenin Chemical compound C\C=C\C\C=C\CCC(=O)[C@H]1O[C@H]1C(N)=O GVEZIHKRYBHEFX-NQQPLRFYSA-N 0.000 description 7
- 238000003818 flash chromatography Methods 0.000 description 7
- 206010006187 Breast cancer Diseases 0.000 description 6
- 208000026310 Breast neoplasm Diseases 0.000 description 6
- ZSLZBFCDCINBPY-ZSJPKINUSA-N acetyl-CoA Chemical compound O[C@@H]1[C@H](OP(O)(O)=O)[C@@H](COP(O)(=O)OP(O)(=O)OCC(C)(C)[C@@H](O)C(=O)NCCC(=O)NCCSC(=O)C)O[C@H]1N1C2=NC=NC(N)=C2N=C1 ZSLZBFCDCINBPY-ZSJPKINUSA-N 0.000 description 6
- 238000006243 chemical reaction Methods 0.000 description 6
- 231100000135 cytotoxicity Toxicity 0.000 description 6
- 230000003013 cytotoxicity Effects 0.000 description 6
- 230000001419 dependent effect Effects 0.000 description 6
- 238000002474 experimental method Methods 0.000 description 6
- 239000003188 fatty acid synthesis inhibitor Substances 0.000 description 6
- 208000015181 infectious disease Diseases 0.000 description 6
- 239000010410 layer Substances 0.000 description 6
- 238000005259 measurement Methods 0.000 description 6
- 239000002609 medium Substances 0.000 description 6
- 239000000203 mixture Substances 0.000 description 6
- 239000000047 product Substances 0.000 description 6
- UCSJYZPVAKXKNQ-HZYVHMACSA-N streptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O UCSJYZPVAKXKNQ-HZYVHMACSA-N 0.000 description 6
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 5
- 102000005870 Coenzyme A Ligases Human genes 0.000 description 5
- 108010011449 Long-chain-fatty-acid-CoA ligase Proteins 0.000 description 5
- 101000824284 Rattus norvegicus Acyl-[acyl-carrier-protein] hydrolase Proteins 0.000 description 5
- 239000003153 chemical reaction reagent Substances 0.000 description 5
- 150000002632 lipids Chemical class 0.000 description 5
- 244000005700 microbiome Species 0.000 description 5
- 230000037361 pathway Effects 0.000 description 5
- 239000013641 positive control Substances 0.000 description 5
- NKTGCVUIESDXPU-YLEPRARLSA-N triacsin C Chemical compound CCC\C=C\C\C=C\C=C\C=N\NN=O NKTGCVUIESDXPU-YLEPRARLSA-N 0.000 description 5
- 102000000452 Acetyl-CoA carboxylase Human genes 0.000 description 4
- 108010016219 Acetyl-CoA carboxylase Proteins 0.000 description 4
- 241000894006 Bacteria Species 0.000 description 4
- 108010018763 Biotin carboxylase Proteins 0.000 description 4
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 4
- QTBSBXVTEAMEQO-HQMMCQRPSA-N acetic acid Chemical compound C[14C](O)=O QTBSBXVTEAMEQO-HQMMCQRPSA-N 0.000 description 4
- 230000000844 anti-bacterial effect Effects 0.000 description 4
- 230000010261 cell growth Effects 0.000 description 4
- 239000013078 crystal Substances 0.000 description 4
- 239000002552 dosage form Substances 0.000 description 4
- 230000006870 function Effects 0.000 description 4
- 239000007788 liquid Substances 0.000 description 4
- VLTRZXGMWDSKGL-UHFFFAOYSA-N perchloric acid Chemical compound OCl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-N 0.000 description 4
- YBYRMVIVWMBXKQ-UHFFFAOYSA-N phenylmethanesulfonyl fluoride Chemical compound FS(=O)(=O)CC1=CC=CC=C1 YBYRMVIVWMBXKQ-UHFFFAOYSA-N 0.000 description 4
- 229920001184 polypeptide Polymers 0.000 description 4
- 102000004196 processed proteins & peptides Human genes 0.000 description 4
- 108090000765 processed proteins & peptides Proteins 0.000 description 4
- 230000009467 reduction Effects 0.000 description 4
- 239000000126 substance Substances 0.000 description 4
- 239000000725 suspension Substances 0.000 description 4
- 238000012546 transfer Methods 0.000 description 4
- PHIQHXFUZVPYII-ZCFIWIBFSA-N (R)-carnitine Chemical compound C[N+](C)(C)C[C@H](O)CC([O-])=O PHIQHXFUZVPYII-ZCFIWIBFSA-N 0.000 description 3
- 0 *C(*)(C(C1(CC=C)CC=C)=O)SC1=O Chemical compound *C(*)(C(C1(CC=C)CC=C)=O)SC1=O 0.000 description 3
- 108010019608 3-Oxoacyl-(Acyl-Carrier-Protein) Synthase Proteins 0.000 description 3
- 241000222120 Candida <Saccharomycetales> Species 0.000 description 3
- 108010036824 Citrate (pro-3S)-lyase Proteins 0.000 description 3
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 3
- 241000187479 Mycobacterium tuberculosis Species 0.000 description 3
- 229930182555 Penicillin Natural products 0.000 description 3
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 3
- MEFKEPWMEQBLKI-AIRLBKTGSA-O S-adenosyl-L-methionine Chemical compound O[C@@H]1[C@H](O)[C@@H](C[S+](CC[C@H]([NH3+])C([O-])=O)C)O[C@H]1N1C2=NC=NC(N)=C2N=C1 MEFKEPWMEQBLKI-AIRLBKTGSA-O 0.000 description 3
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 3
- 238000004458 analytical method Methods 0.000 description 3
- 239000012131 assay buffer Substances 0.000 description 3
- 239000000872 buffer Substances 0.000 description 3
- 229960004203 carnitine Drugs 0.000 description 3
- 201000010099 disease Diseases 0.000 description 3
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 3
- NLRZJUPPJSWAED-UHFFFAOYSA-N ethyl 2-methyl-2-pent-4-enoyl-3-sulfanyldecanoate Chemical compound CCCCCCCC(S)C(C)(C(=O)OCC)C(=O)CCC=C NLRZJUPPJSWAED-UHFFFAOYSA-N 0.000 description 3
- 239000003925 fat Substances 0.000 description 3
- 239000012091 fetal bovine serum Substances 0.000 description 3
- 238000011049 filling Methods 0.000 description 3
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 3
- 238000000338 in vitro Methods 0.000 description 3
- 238000010348 incorporation Methods 0.000 description 3
- 210000004185 liver Anatomy 0.000 description 3
- 239000012139 lysis buffer Substances 0.000 description 3
- 229940049920 malate Drugs 0.000 description 3
- BJEPYKJPYRNKOW-UHFFFAOYSA-N malic acid Chemical compound OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 3
- 239000003921 oil Substances 0.000 description 3
- 210000000056 organ Anatomy 0.000 description 3
- 239000008188 pellet Substances 0.000 description 3
- 229940049954 penicillin Drugs 0.000 description 3
- 239000002243 precursor Substances 0.000 description 3
- 230000002285 radioactive effect Effects 0.000 description 3
- 229960005322 streptomycin Drugs 0.000 description 3
- 239000006228 supernatant Substances 0.000 description 3
- PVSAPJKDKFIKHM-CQFAVKSRSA-N (3R)-3-hydroxy-3-(114C)methyl-4-(trimethylazaniumyl)butanoate Chemical compound [14CH3][C@](O)(C[N+](C)(C)C)CC([O-])=O PVSAPJKDKFIKHM-CQFAVKSRSA-N 0.000 description 2
- DJQYYYCQOZMCRC-UHFFFAOYSA-N 2-aminopropane-1,3-dithiol Chemical compound SCC(N)CS DJQYYYCQOZMCRC-UHFFFAOYSA-N 0.000 description 2
- HAZBIBXCTFPRMV-UHFFFAOYSA-N 3,3,5-trimethyl-5-octylthiolane-2,4-dione Chemical compound CCCCCCCCC1(C)SC(=O)C(C)(C)C1=O HAZBIBXCTFPRMV-UHFFFAOYSA-N 0.000 description 2
- DRYLQSIWCWNWDO-UHFFFAOYSA-N 5-decyl-3,3,5-trimethylthiolane-2,4-dione Chemical compound CCCCCCCCCCC1(C)SC(=O)C(C)(C)C1=O DRYLQSIWCWNWDO-UHFFFAOYSA-N 0.000 description 2
- JQZMUQAROJJPEK-UHFFFAOYSA-N 5-hexyl-3,3,5-trimethylthiolane-2,4-dione Chemical compound CCCCCCC1(C)SC(=O)C(C)(C)C1=O JQZMUQAROJJPEK-UHFFFAOYSA-N 0.000 description 2
- 241000220479 Acacia Species 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 2
- 241000228212 Aspergillus Species 0.000 description 2
- 241000223935 Cryptosporidium Species 0.000 description 2
- 241000194032 Enterococcus faecalis Species 0.000 description 2
- 241000588724 Escherichia coli Species 0.000 description 2
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 2
- 241000233866 Fungi Species 0.000 description 2
- 241000224467 Giardia intestinalis Species 0.000 description 2
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 2
- 101000611023 Homo sapiens Tumor necrosis factor receptor superfamily member 6 Proteins 0.000 description 2
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 2
- 102000003960 Ligases Human genes 0.000 description 2
- 108090000364 Ligases Proteins 0.000 description 2
- 241000124008 Mammalia Species 0.000 description 2
- 101001014220 Monascus pilosus Dehydrogenase mokE Proteins 0.000 description 2
- 101000755720 Neurospora crassa (strain ATCC 24698 / 74-OR23-1A / CBS 708.71 / DSM 1257 / FGSC 987) Palmitoyltransferase akr1 Proteins 0.000 description 2
- 208000008589 Obesity Diseases 0.000 description 2
- 241000315040 Omura Species 0.000 description 2
- 101000573542 Penicillium citrinum Compactin nonaketide synthase, enoyl reductase component Proteins 0.000 description 2
- 241000233872 Pneumocystis carinii Species 0.000 description 2
- 229920002594 Polyethylene Glycol 8000 Polymers 0.000 description 2
- 239000002202 Polyethylene glycol Substances 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- 241000224527 Trichomonas vaginalis Species 0.000 description 2
- 210000001789 adipocyte Anatomy 0.000 description 2
- 210000000577 adipose tissue Anatomy 0.000 description 2
- BHELZAPQIKSEDF-UHFFFAOYSA-N allyl bromide Chemical compound BrCC=C BHELZAPQIKSEDF-UHFFFAOYSA-N 0.000 description 2
- 210000004102 animal cell Anatomy 0.000 description 2
- 239000012736 aqueous medium Substances 0.000 description 2
- 210000004369 blood Anatomy 0.000 description 2
- 239000008280 blood Substances 0.000 description 2
- OSGAYBCDTDRGGQ-UHFFFAOYSA-L calcium sulfate Chemical compound [Ca+2].[O-]S([O-])(=O)=O OSGAYBCDTDRGGQ-UHFFFAOYSA-L 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 230000003915 cell function Effects 0.000 description 2
- 238000005119 centrifugation Methods 0.000 description 2
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 2
- 238000002784 cytotoxicity assay Methods 0.000 description 2
- 231100000263 cytotoxicity test Toxicity 0.000 description 2
- 150000001993 dienes Chemical class 0.000 description 2
- 238000011143 downstream manufacturing Methods 0.000 description 2
- 239000003937 drug carrier Substances 0.000 description 2
- 230000002255 enzymatic effect Effects 0.000 description 2
- 238000006911 enzymatic reaction Methods 0.000 description 2
- 239000007903 gelatin capsule Substances 0.000 description 2
- 229940085435 giardia lamblia Drugs 0.000 description 2
- 230000009036 growth inhibition Effects 0.000 description 2
- 239000001963 growth medium Substances 0.000 description 2
- IPCSVZSSVZVIGE-UHFFFAOYSA-M hexadecanoate Chemical compound CCCCCCCCCCCCCCCC([O-])=O IPCSVZSSVZVIGE-UHFFFAOYSA-M 0.000 description 2
- 102000047715 human FAS Human genes 0.000 description 2
- 230000002458 infectious effect Effects 0.000 description 2
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 2
- 150000004668 long chain fatty acids Chemical class 0.000 description 2
- 210000004072 lung Anatomy 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 230000002503 metabolic effect Effects 0.000 description 2
- 230000004060 metabolic process Effects 0.000 description 2
- 229920000609 methyl cellulose Polymers 0.000 description 2
- 239000001923 methylcellulose Substances 0.000 description 2
- 235000010981 methylcellulose Nutrition 0.000 description 2
- 235000020824 obesity Nutrition 0.000 description 2
- 235000019198 oils Nutrition 0.000 description 2
- 239000012044 organic layer Substances 0.000 description 2
- 229920001223 polyethylene glycol Polymers 0.000 description 2
- 238000002360 preparation method Methods 0.000 description 2
- 239000003755 preservative agent Substances 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- 238000012545 processing Methods 0.000 description 2
- 230000002829 reductive effect Effects 0.000 description 2
- 238000012216 screening Methods 0.000 description 2
- 239000007787 solid Substances 0.000 description 2
- 230000000638 stimulation Effects 0.000 description 2
- 239000011550 stock solution Substances 0.000 description 2
- 239000000758 substrate Substances 0.000 description 2
- 239000006188 syrup Substances 0.000 description 2
- 235000020357 syrup Nutrition 0.000 description 2
- 239000003826 tablet Substances 0.000 description 2
- 230000001225 therapeutic effect Effects 0.000 description 2
- -1 thiolate anions Chemical class 0.000 description 2
- 108010050327 trypticase-soy broth Proteins 0.000 description 2
- 210000004881 tumor cell Anatomy 0.000 description 2
- 125000004209 (C1-C8) alkyl group Chemical group 0.000 description 1
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 1
- 101710161460 3-oxoacyl-[acyl-carrier-protein] synthase Proteins 0.000 description 1
- JYCQQPHGFMYQCF-UHFFFAOYSA-N 4-tert-Octylphenol monoethoxylate Chemical compound CC(C)(C)CC(C)(C)C1=CC=C(OCCO)C=C1 JYCQQPHGFMYQCF-UHFFFAOYSA-N 0.000 description 1
- UHPMCKVQTMMPCG-UHFFFAOYSA-N 5,8-dihydroxy-2-methoxy-6-methyl-7-(2-oxopropyl)naphthalene-1,4-dione Chemical compound CC1=C(CC(C)=O)C(O)=C2C(=O)C(OC)=CC(=O)C2=C1O UHPMCKVQTMMPCG-UHFFFAOYSA-N 0.000 description 1
- 241000224422 Acanthamoeba Species 0.000 description 1
- 241000186361 Actinobacteria <class> Species 0.000 description 1
- 102000006488 Acyl-Carrier Protein S-Malonyltransferase Human genes 0.000 description 1
- 108010058912 Acyl-Carrier Protein S-Malonyltransferase Proteins 0.000 description 1
- 229920001817 Agar Polymers 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 1
- 101001074429 Bacillus subtilis (strain 168) Polyketide biosynthesis acyltransferase homolog PksD Proteins 0.000 description 1
- 101000936617 Bacillus velezensis (strain DSM 23117 / BGSC 10A6 / FZB42) Polyketide biosynthesis acyltransferase homolog BaeD Proteins 0.000 description 1
- 241000222122 Candida albicans Species 0.000 description 1
- 102000002666 Carnitine O-palmitoyltransferase Human genes 0.000 description 1
- 108010018424 Carnitine O-palmitoyltransferase Proteins 0.000 description 1
- 241001625972 Cephalosporium caerulens Species 0.000 description 1
- RGJOEKWQDUBAIZ-IBOSZNHHSA-N CoASH Chemical compound O[C@@H]1[C@H](OP(O)(O)=O)[C@@H](COP(O)(=O)OP(O)(=O)OCC(C)(C)[C@@H](O)C(=O)NCCC(=O)NCCS)O[C@H]1N1C2=NC=NC(N)=C2N=C1 RGJOEKWQDUBAIZ-IBOSZNHHSA-N 0.000 description 1
- 208000035473 Communicable disease Diseases 0.000 description 1
- 208000007163 Dermatomycoses Diseases 0.000 description 1
- 235000019739 Dicalciumphosphate Nutrition 0.000 description 1
- QRLVDLBMBULFAL-UHFFFAOYSA-N Digitonin Natural products CC1CCC2(OC1)OC3C(O)C4C5CCC6CC(OC7OC(CO)C(OC8OC(CO)C(O)C(OC9OCC(O)C(O)C9OC%10OC(CO)C(O)C(OC%11OC(CO)C(O)C(O)C%11O)C%10O)C8O)C(O)C7O)C(O)CC6(C)C5CCC4(C)C3C2C QRLVDLBMBULFAL-UHFFFAOYSA-N 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- 241000224431 Entamoeba Species 0.000 description 1
- 241001480035 Epidermophyton Species 0.000 description 1
- 241000206602 Eukaryota Species 0.000 description 1
- 229940123469 Fatty acid synthase inhibitor Drugs 0.000 description 1
- 208000014260 Fungal keratitis Diseases 0.000 description 1
- 241000223218 Fusarium Species 0.000 description 1
- 208000018522 Gastrointestinal disease Diseases 0.000 description 1
- 108010010369 HIV Protease Proteins 0.000 description 1
- 206010061598 Immunodeficiency Diseases 0.000 description 1
- 208000029462 Immunodeficiency disease Diseases 0.000 description 1
- 108090001061 Insulin Proteins 0.000 description 1
- 102000004877 Insulin Human genes 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 241000222722 Leishmania <genus> Species 0.000 description 1
- 208000019693 Lung disease Diseases 0.000 description 1
- 241001295810 Microsporidium Species 0.000 description 1
- 241001480037 Microsporum Species 0.000 description 1
- 206010028080 Mucocutaneous candidiasis Diseases 0.000 description 1
- 241000699666 Mus <mouse, genus> Species 0.000 description 1
- 241000187480 Mycobacterium smegmatis Species 0.000 description 1
- 238000010934 O-alkylation reaction Methods 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- 239000004264 Petrolatum Substances 0.000 description 1
- 241001442539 Plasmodium sp. Species 0.000 description 1
- 241000589517 Pseudomonas aeruginosa Species 0.000 description 1
- 230000006819 RNA synthesis Effects 0.000 description 1
- 239000006146 Roswell Park Memorial Institute medium Substances 0.000 description 1
- 241000235088 Saccharomyces sp. Species 0.000 description 1
- 241000242678 Schistosoma Species 0.000 description 1
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 1
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 1
- 241000295644 Staphylococcaceae Species 0.000 description 1
- 241000191967 Staphylococcus aureus Species 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 241000187180 Streptomyces sp. Species 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 102000005488 Thioesterase Human genes 0.000 description 1
- 208000002474 Tinea Diseases 0.000 description 1
- 241000223996 Toxoplasma Species 0.000 description 1
- 241000223997 Toxoplasma gondii Species 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- 241000223238 Trichophyton Species 0.000 description 1
- 241000893966 Trichophyton verrucosum Species 0.000 description 1
- 239000007983 Tris buffer Substances 0.000 description 1
- 108010059993 Vancomycin Proteins 0.000 description 1
- 241000700605 Viruses Species 0.000 description 1
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 239000008272 agar Substances 0.000 description 1
- 239000002168 alkylating agent Substances 0.000 description 1
- 229940100198 alkylating agent Drugs 0.000 description 1
- 239000003708 ampul Substances 0.000 description 1
- 238000005349 anion exchange Methods 0.000 description 1
- 238000005571 anion exchange chromatography Methods 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 230000001093 anti-cancer Effects 0.000 description 1
- 230000000843 anti-fungal effect Effects 0.000 description 1
- 230000001315 anti-hyperlipaemic effect Effects 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 229940053200 antiepileptics fatty acid derivative Drugs 0.000 description 1
- 229940121375 antifungal agent Drugs 0.000 description 1
- 239000003429 antifungal agent Substances 0.000 description 1
- 230000030741 antigen processing and presentation Effects 0.000 description 1
- 230000036528 appetite Effects 0.000 description 1
- 235000019789 appetite Nutrition 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 210000003719 b-lymphocyte Anatomy 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- ACFIXJIJDZMPPO-UHFFFAOYSA-N beta-NADPH Natural products C1=CCC(C(=O)N)=CN1C1C(O)C(O)C(COP(O)(=O)OP(O)(=O)OCC2C(C(OP(O)(O)=O)C(O2)N2C3=NC=NC(N)=C3N=C2)O)O1 ACFIXJIJDZMPPO-UHFFFAOYSA-N 0.000 description 1
- 238000012742 biochemical analysis Methods 0.000 description 1
- 238000002306 biochemical method Methods 0.000 description 1
- 230000003115 biocidal effect Effects 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 238000010170 biological method Methods 0.000 description 1
- 210000001124 body fluid Anatomy 0.000 description 1
- 239000010839 body fluid Substances 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- 229940098773 bovine serum albumin Drugs 0.000 description 1
- 238000002815 broth microdilution Methods 0.000 description 1
- 239000006172 buffering agent Substances 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 235000011132 calcium sulphate Nutrition 0.000 description 1
- 238000004364 calculation method Methods 0.000 description 1
- 235000019577 caloric intake Nutrition 0.000 description 1
- 229940095731 candida albicans Drugs 0.000 description 1
- 108020001778 catalytic domains Proteins 0.000 description 1
- 230000036978 cell physiology Effects 0.000 description 1
- 230000004663 cell proliferation Effects 0.000 description 1
- 238000001516 cell proliferation assay Methods 0.000 description 1
- 238000002737 cell proliferation kit Methods 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 239000003638 chemical reducing agent Substances 0.000 description 1
- 235000012000 cholesterol Nutrition 0.000 description 1
- RGJOEKWQDUBAIZ-UHFFFAOYSA-N coenzime A Natural products OC1C(OP(O)(O)=O)C(COP(O)(=O)OP(O)(=O)OCC(C)(C)C(O)C(=O)NCCC(=O)NCCS)OC1N1C2=NC=NC(N)=C2N=C1 RGJOEKWQDUBAIZ-UHFFFAOYSA-N 0.000 description 1
- 239000005516 coenzyme A Substances 0.000 description 1
- 229940093530 coenzyme a Drugs 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 230000005494 condensation Effects 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 238000003235 crystal violet staining Methods 0.000 description 1
- 238000012258 culturing Methods 0.000 description 1
- 238000006114 decarboxylation reaction Methods 0.000 description 1
- KDTSHFARGAKYJN-UHFFFAOYSA-N dephosphocoenzyme A Natural products OC1C(O)C(COP(O)(=O)OP(O)(=O)OCC(C)(C)C(O)C(=O)NCCC(=O)NCCS)OC1N1C2=NC=NC(N)=C2N=C1 KDTSHFARGAKYJN-UHFFFAOYSA-N 0.000 description 1
- 201000003929 dermatomycosis Diseases 0.000 description 1
- 230000001627 detrimental effect Effects 0.000 description 1
- NEFBYIFKOOEVPA-UHFFFAOYSA-K dicalcium phosphate Chemical compound [Ca+2].[Ca+2].[O-]P([O-])([O-])=O NEFBYIFKOOEVPA-UHFFFAOYSA-K 0.000 description 1
- 229940038472 dicalcium phosphate Drugs 0.000 description 1
- 229910000390 dicalcium phosphate Inorganic materials 0.000 description 1
- UVYVLBIGDKGWPX-KUAJCENISA-N digitonin Chemical compound O([C@@H]1[C@@H]([C@]2(CC[C@@H]3[C@@]4(C)C[C@@H](O)[C@H](O[C@H]5[C@@H]([C@@H](O)[C@@H](O[C@H]6[C@@H]([C@@H](O[C@H]7[C@@H]([C@@H](O)[C@H](O)CO7)O)[C@H](O)[C@@H](CO)O6)O[C@H]6[C@@H]([C@@H](O[C@H]7[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O7)O)[C@@H](O)[C@@H](CO)O6)O)[C@@H](CO)O5)O)C[C@@H]4CC[C@H]3[C@@H]2[C@@H]1O)C)[C@@H]1C)[C@]11CC[C@@H](C)CO1 UVYVLBIGDKGWPX-KUAJCENISA-N 0.000 description 1
- UVYVLBIGDKGWPX-UHFFFAOYSA-N digitonine Natural products CC1C(C2(CCC3C4(C)CC(O)C(OC5C(C(O)C(OC6C(C(OC7C(C(O)C(O)CO7)O)C(O)C(CO)O6)OC6C(C(OC7C(C(O)C(O)C(CO)O7)O)C(O)C(CO)O6)O)C(CO)O5)O)CC4CCC3C2C2O)C)C2OC11CCC(C)CO1 UVYVLBIGDKGWPX-UHFFFAOYSA-N 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 229910001873 dinitrogen Inorganic materials 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- 239000012153 distilled water Substances 0.000 description 1
- 125000002228 disulfide group Chemical group 0.000 description 1
- VHJLVAABSRFDPM-QWWZWVQMSA-N dithiothreitol Chemical compound SC[C@@H](O)[C@H](O)CS VHJLVAABSRFDPM-QWWZWVQMSA-N 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 238000005538 encapsulation Methods 0.000 description 1
- 125000004050 enoyl group Chemical group 0.000 description 1
- 229940032049 enterococcus faecalis Drugs 0.000 description 1
- 239000002532 enzyme inhibitor Substances 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- DEFVIWRASFVYLL-UHFFFAOYSA-N ethylene glycol bis(2-aminoethyl)tetraacetic acid Chemical compound OC(=O)CN(CC(O)=O)CCOCCOCCN(CC(O)=O)CC(O)=O DEFVIWRASFVYLL-UHFFFAOYSA-N 0.000 description 1
- 230000029142 excretion Effects 0.000 description 1
- 230000002550 fecal effect Effects 0.000 description 1
- 239000012894 fetal calf serum Substances 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 235000013305 food Nutrition 0.000 description 1
- 235000012631 food intake Nutrition 0.000 description 1
- 239000003205 fragrance Substances 0.000 description 1
- 239000000446 fuel Substances 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 235000011187 glycerol Nutrition 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 230000035876 healing Effects 0.000 description 1
- IPCSVZSSVZVIGE-VPMSBSONSA-N hexadecanoic acid Chemical compound CCCCCCCCCCCCCCC[14C](O)=O IPCSVZSSVZVIGE-VPMSBSONSA-N 0.000 description 1
- 229940088597 hormone Drugs 0.000 description 1
- 239000005556 hormone Substances 0.000 description 1
- 210000005260 human cell Anatomy 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 230000007813 immunodeficiency Effects 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 239000002054 inoculum Substances 0.000 description 1
- 229940125396 insulin Drugs 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 230000002427 irreversible effect Effects 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 206010023332 keratitis Diseases 0.000 description 1
- 230000003907 kidney function Effects 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 230000003902 lesion Effects 0.000 description 1
- 230000001665 lethal effect Effects 0.000 description 1
- 239000006193 liquid solution Substances 0.000 description 1
- 239000006194 liquid suspension Substances 0.000 description 1
- YNESATAKKCNGOF-UHFFFAOYSA-N lithium bis(trimethylsilyl)amide Chemical compound [Li+].C[Si](C)(C)[N-][Si](C)(C)C YNESATAKKCNGOF-UHFFFAOYSA-N 0.000 description 1
- 230000003908 liver function Effects 0.000 description 1
- 239000003589 local anesthetic agent Substances 0.000 description 1
- 229960005015 local anesthetics Drugs 0.000 description 1
- 239000008176 lyophilized powder Substances 0.000 description 1
- 239000006166 lysate Substances 0.000 description 1
- 210000002540 macrophage Anatomy 0.000 description 1
- HZZOEADXZLYIHG-UHFFFAOYSA-N magnesiomagnesium Chemical compound [Mg][Mg] HZZOEADXZLYIHG-UHFFFAOYSA-N 0.000 description 1
- 239000000391 magnesium silicate Substances 0.000 description 1
- 229910052919 magnesium silicate Inorganic materials 0.000 description 1
- 235000019792 magnesium silicate Nutrition 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 210000005075 mammary gland Anatomy 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 230000010534 mechanism of action Effects 0.000 description 1
- 238000002483 medication Methods 0.000 description 1
- 210000004379 membrane Anatomy 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 210000003470 mitochondria Anatomy 0.000 description 1
- 210000001700 mitochondrial membrane Anatomy 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 238000000329 molecular dynamics simulation Methods 0.000 description 1
- 210000000214 mouth Anatomy 0.000 description 1
- 210000004877 mucosa Anatomy 0.000 description 1
- 210000004400 mucous membrane Anatomy 0.000 description 1
- 230000007498 myristoylation Effects 0.000 description 1
- VMGAPWLDMVPYIA-HIDZBRGKSA-N n'-amino-n-iminomethanimidamide Chemical compound N\N=C\N=N VMGAPWLDMVPYIA-HIDZBRGKSA-N 0.000 description 1
- 210000003928 nasal cavity Anatomy 0.000 description 1
- 229930014626 natural product Natural products 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- 239000006186 oral dosage form Substances 0.000 description 1
- 239000007800 oxidant agent Substances 0.000 description 1
- MNBKLUUYKPBKDU-BBECNAHFSA-N palmitoyl-CoA Chemical compound O[C@@H]1[C@H](OP(O)(O)=O)[C@@H](COP(O)(=O)OP(O)(=O)OCC(C)(C)[C@@H](O)C(=O)NCCC(=O)NCCSC(=O)CCCCCCCCCCCCCCC)O[C@H]1N1C2=NC=NC(N)=C2N=C1 MNBKLUUYKPBKDU-BBECNAHFSA-N 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 239000003182 parenteral nutrition solution Substances 0.000 description 1
- 244000052769 pathogen Species 0.000 description 1
- JDKQTIKEGOOXTJ-UHFFFAOYSA-N pent-4-enoyl chloride Chemical compound ClC(=O)CCC=C JDKQTIKEGOOXTJ-UHFFFAOYSA-N 0.000 description 1
- 229940066842 petrolatum Drugs 0.000 description 1
- 235000019271 petrolatum Nutrition 0.000 description 1
- 238000004634 pharmacological analysis method Methods 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 238000011533 pre-incubation Methods 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 238000002203 pretreatment Methods 0.000 description 1
- 238000002731 protein assay Methods 0.000 description 1
- 239000012460 protein solution Substances 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- BOLDJAUMGUJJKM-LSDHHAIUSA-N renifolin D Natural products CC(=C)[C@@H]1Cc2c(O)c(O)ccc2[C@H]1CC(=O)c3ccc(O)cc3O BOLDJAUMGUJJKM-LSDHHAIUSA-N 0.000 description 1
- 150000004671 saturated fatty acids Chemical group 0.000 description 1
- 238000007789 sealing Methods 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 238000013207 serial dilution Methods 0.000 description 1
- 210000002966 serum Anatomy 0.000 description 1
- 239000012679 serum free medium Substances 0.000 description 1
- 239000002002 slurry Substances 0.000 description 1
- 239000001632 sodium acetate Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- 238000002415 sodium dodecyl sulfate polyacrylamide gel electrophoresis Methods 0.000 description 1
- QDRKDTQENPPHOJ-UHFFFAOYSA-N sodium ethoxide Chemical compound [Na+].CC[O-] QDRKDTQENPPHOJ-UHFFFAOYSA-N 0.000 description 1
- 239000008247 solid mixture Substances 0.000 description 1
- 238000005063 solubilization Methods 0.000 description 1
- 230000007928 solubilization Effects 0.000 description 1
- 239000002904 solvent Substances 0.000 description 1
- 238000010186 staining Methods 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 230000002195 synergetic effect Effects 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- 230000000542 thalamic effect Effects 0.000 description 1
- 231100001274 therapeutic index Toxicity 0.000 description 1
- 150000007970 thio esters Chemical class 0.000 description 1
- 108020002982 thioesterase Proteins 0.000 description 1
- 150000003573 thiols Chemical class 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- 229960000707 tobramycin Drugs 0.000 description 1
- NLVFBUXFDBBNBW-PBSUHMDJSA-N tobramycin Chemical compound N[C@@H]1C[C@H](O)[C@@H](CN)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](N)[C@H](O)[C@@H](CO)O2)O)[C@H](N)C[C@@H]1N NLVFBUXFDBBNBW-PBSUHMDJSA-N 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 239000000196 tragacanth Substances 0.000 description 1
- 235000010487 tragacanth Nutrition 0.000 description 1
- 229940116362 tragacanth Drugs 0.000 description 1
- 150000003626 triacylglycerols Chemical class 0.000 description 1
- AYNNSCRYTDRFCP-UHFFFAOYSA-N triazene Chemical compound NN=N AYNNSCRYTDRFCP-UHFFFAOYSA-N 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 239000001974 tryptic soy broth Substances 0.000 description 1
- 210000002700 urine Anatomy 0.000 description 1
- 229960003165 vancomycin Drugs 0.000 description 1
- MYPYJXKWCTUITO-UHFFFAOYSA-N vancomycin Natural products O1C(C(=C2)Cl)=CC=C2C(O)C(C(NC(C2=CC(O)=CC(O)=C2C=2C(O)=CC=C3C=2)C(O)=O)=O)NC(=O)C3NC(=O)C2NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(CC(C)C)NC)C(O)C(C=C3Cl)=CC=C3OC3=CC2=CC1=C3OC1OC(CO)C(O)C(O)C1OC1CC(C)(N)C(O)C(C)O1 MYPYJXKWCTUITO-UHFFFAOYSA-N 0.000 description 1
- MYPYJXKWCTUITO-LYRMYLQWSA-O vancomycin(1+) Chemical compound O([C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1OC1=C2C=C3C=C1OC1=CC=C(C=C1Cl)[C@@H](O)[C@H](C(N[C@@H](CC(N)=O)C(=O)N[C@H]3C(=O)N[C@H]1C(=O)N[C@H](C(N[C@@H](C3=CC(O)=CC(O)=C3C=3C(O)=CC=C1C=3)C([O-])=O)=O)[C@H](O)C1=CC=C(C(=C1)Cl)O2)=O)NC(=O)[C@@H](CC(C)C)[NH2+]C)[C@H]1C[C@](C)([NH3+])[C@H](O)[C@H](C)O1 MYPYJXKWCTUITO-LYRMYLQWSA-O 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 230000003612 virological effect Effects 0.000 description 1
- 239000008215 water for injection Substances 0.000 description 1
Images

Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/02—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
- C07D333/04—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom
- C07D333/26—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D333/30—Hetero atoms other than halogen
- C07D333/32—Oxygen atoms
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01N—PRESERVATION OF BODIES OF HUMANS OR ANIMALS OR PLANTS OR PARTS THEREOF; BIOCIDES, e.g. AS DISINFECTANTS, AS PESTICIDES OR AS HERBICIDES; PEST REPELLANTS OR ATTRACTANTS; PLANT GROWTH REGULATORS
- A01N43/00—Biocides, pest repellants or attractants, or plant growth regulators containing heterocyclic compounds
- A01N43/02—Biocides, pest repellants or attractants, or plant growth regulators containing heterocyclic compounds having rings with one or more oxygen or sulfur atoms as the only ring hetero atoms
- A01N43/04—Biocides, pest repellants or attractants, or plant growth regulators containing heterocyclic compounds having rings with one or more oxygen or sulfur atoms as the only ring hetero atoms with one hetero atom
- A01N43/06—Biocides, pest repellants or attractants, or plant growth regulators containing heterocyclic compounds having rings with one or more oxygen or sulfur atoms as the only ring hetero atoms with one hetero atom five-membered rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Landscapes
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Hematology (AREA)
- Diabetes (AREA)
- Obesity (AREA)
- Dentistry (AREA)
- Wood Science & Technology (AREA)
- Oncology (AREA)
- Agronomy & Crop Science (AREA)
- Pest Control & Pesticides (AREA)
- Plant Pathology (AREA)
- Child & Adolescent Psychology (AREA)
- Communicable Diseases (AREA)
- Zoology (AREA)
- Environmental Sciences (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Heterocyclic Compounds Containing Sulfur Atoms (AREA)
Abstract
医薬用希釈体および式 (II)の化合物[式中、R1およびR2は、互いに同じであっても異なっても良く、H、C1-C20 アルキル、シクロアルキル、アルケニル、アリール、アリールアルキルもしくはアルキルアリール、-CH2COR5、-CH2C(O)NR5、-C(O)R5、または-CH2OR5であり、ハロゲン原子を含有していてもよく、ここで、R5はC1-C12 アルキル基であり、R3およびR4は、互いに同じであっても異なっても良く、H、C1-C20 アルキル、シクロアルキル、アルケニル、アリール、アリールアルキルもしくはアルキルアリールである]を含む医薬組成物。
Description
脂肪酸合成酵素
脂肪酸は、細胞生理において3つの主要な役割を有する。第1に、それらは生体膜の構成単位である。第2に、脂肪酸誘導体はホルモンおよび細胞内伝達物質として機能する。第3に、本発明に特に重要なことに、脂肪酸は中性脂肪としても知られるトリアシルグリセロールとして脂肪組織に保存されうる燃料分子である。
脂肪酸は、細胞生理において3つの主要な役割を有する。第1に、それらは生体膜の構成単位である。第2に、脂肪酸誘導体はホルモンおよび細胞内伝達物質として機能する。第3に、本発明に特に重要なことに、脂肪酸は中性脂肪としても知られるトリアシルグリセロールとして脂肪組織に保存されうる燃料分子である。
4つの主要な酵素、脂肪酸合成酵素 (fatty acid synthase、FAS)、アセチル-CoA カルボキシラーゼ (ACC) 、リンゴ酸酵素およびクエン酸リアーゼが脂肪酸合成経路に関係する。主たる酵素であるFASは、前駆体であるマロニル-CoAおよびアセチル-CoAのNADPH依存的縮合を触媒し、脂肪酸を産生する。NADPHは、通常FASの反応回路の2点において必須の電子供与体として機能する還元剤である。他の3つの酵素 (すなわち、ACC、リンゴ酸酵素およびクエン酸リアーゼ)は必要な前駆体を産生する。他の酵素、例えばNADPHを産生する酵素もまた、脂肪酸合成に関係する。
FASは、Enzyme Commission(E.C.) No. 2.3.1.85 を有し、脂肪酸シンテターゼ(fatty acid synthetase)、脂肪酸リガーゼ(fatty acid ligase)、およびその系統名であるアシル-CoA:マロニル-CoA C-アシルトランスフェラーゼとしても知られる(脱炭酸、オキソアシルおよびエノイル還元、およびチオエステル加水分解)。FASが触媒する脂肪酸合成には、7つの異なる酵素または触媒ドメインが関係する: アセチルトランスアシラーゼ、マロニルトランスアシラーゼ、β-ケトアシルシンテターゼ (縮合酵素)、β-ケトアシルレダクターゼ、β-ヒドロキシアシルデヒドラーゼ、エノイルレダクターゼ、およびチオエステラーゼ. (Wakil, S.J., Biochemistry, 28: 4523-4530, 1989)。これら7つの酵素がすべて一緒になってFASを形成する。
FASが触媒する脂肪酸合成は、細菌のような下等生物と、マイコバクテリア、酵母およびヒトのような高等生物とでよく似ているが、いくつかの重要な相違がある。細菌では、7つの酵素反応は結合していない7つの別々のポリペプチドにより実行される。これはタイプ II FASに分類される。これに対して、マイコバクテリア、酵母およびヒトにおける酵素反応は多機能ポリペプチドにより実行される。例えば、酵母は2つの別々のポリペプチドから構成される複合体を有し、一方マイコバクテリアとヒトでは7つの反応がすべて1つのポリペプチドにより実行される。これらはタイプ I FASに分類される。
FAS阻害薬
様々な化合物が脂肪酸合成酵素 (FAS)を阻害することが知られている。FAS阻害薬は、化合物が精製FASの酵素活性を阻害する能力により同定することができる。FAS活性は、放射性標識前駆体 (すなわち、アセチル-CoAまたはマロニル-CoA)の脂肪酸への組み込みを測定することにより、あるいは分光光度法でNADPH酸化を測定することにより、評価することができる (Dils, et al., Methods Enzymol., 35: 74-83)。
様々な化合物が脂肪酸合成酵素 (FAS)を阻害することが知られている。FAS阻害薬は、化合物が精製FASの酵素活性を阻害する能力により同定することができる。FAS活性は、放射性標識前駆体 (すなわち、アセチル-CoAまたはマロニル-CoA)の脂肪酸への組み込みを測定することにより、あるいは分光光度法でNADPH酸化を測定することにより、評価することができる (Dils, et al., Methods Enzymol., 35: 74-83)。
以下の表1に幾つかのFAS阻害薬を挙げる。
[表1]

[表1]
脂肪酸合成経路における4つの酵素のうち、FASが脂肪酸への経路内でのみ作用するのに対して他の3つの酵素は他の細胞機能に関係するため、FASは好ましい阻害標的である。それゆえ、他の3つの酵素の1つを阻害すると正常細胞により影響すると思われる。
FASにより実行される7つの酵素工程のうち、縮合酵素(すなわち、β-ケトアシルシンテターゼ)およびエノイルレダクターゼにより触媒される工程は、脂肪合成を減少または停止させる阻害薬にとって最も一般的な候補である。FAS複合体の縮合酵素は、構造および機能に関して十分に特徴決定されている。縮合酵素の活性部位は重要なシステインチオールを含み、これは抗高脂血症薬、例えば阻害薬であるセルレニン、の標的である。
好ましい縮合酵素の阻害薬には広範な化合物が含まれ、それにはアルキル化剤、酸化剤、およびジスルフィド交換を受けうる試薬が含まれる。酵素の結合ポケットは長鎖E, E, ジエンを好む。
基本的に、側鎖ジエンおよびチオラートアニオンとの反応性を示す基を含む試薬は、縮合酵素の良好な阻害薬でありうる。セルレニン [(2S, 3R)-2,3-エポキシ-4-オキソ-7,10 ドデカジエノイルアミド]は一例である:
セルレニンは、脂肪酸合成酵素の縮合酵素の活性部位における重要なシステインチオール基に共有結合し、この鍵となる酵素工程を不活性化する(Funabashi, et al., J. Biochem., 105: 751-755, 1989)。セルレニンは他の活性を有することが指摘されているが、これらは、ヒト細胞の適当なモデルとならない微生物において生じるか(例えば、真菌におけるコレステロール合成阻害、 Omura (1976), Bacteriol. Rev., 40: 681-697; またはウイルスにおけるRNA合成低下、Perez, et al. (1991), FEBS, 280: 129-133)、非常に高い薬物濃度において生じるか(5 mg/mlでのウイルスHIVプロテアーゼの阻害、Moelling, et al. (1990), FEBS, 261:373-377)、あるいは内因性脂肪酸合成の阻害の直接的結果でありうる (Bリンパ球およびマクロファージにおける抗原処理の阻害、 Falo, et al. (1987), J. Immunol., 139: 3918-3923)。一部のデータは、セルレニンがタンパク質のミリストイル化を特異的に阻害しないことを示唆する(Simon, et al., J. Biol. Chem., 267:3922-3931, 1992)。
さらに幾つかのFAS阻害薬が米国特許出願第08/096908号およびそのCIP(1994年1月24日出願)に記載されており、その開示は引用により本明細書に含まれる。そこに含まれるのは、脂肪酸合成酵素、クエン酸リアーゼ、CoA カルボキシラーゼおよびリンゴ酸酵素の阻害薬である。
Tomodaら (Tomoda et..al., Biochim. Biophys. Act 921:595-598 1987; Omura el. al., J. Antibiotics 39:1211-1218 1986) はTriacsin C (WS-1228Aとも呼ばれる) について記載しており、これはStreptomyces sp. SK-1894の産物である天然アシル-CoAシンテターゼ阻害薬である。Triacsin Cの化学構造は、1-ヒドロキシ-3-(E, E, E-2’,4’,7’-ウンデカトリエニルイジン(undecatrienylidine)) トリアゼンである。Triacsin C は、ラット肝臓アシル-CoAシンテターゼを8.7 Mで50% 阻害する; 関連化合物のTriacsin Aは、アシル-CoAシンテターゼを長鎖脂肪酸と競合するメカニズムにより阻害する。アシル-CoAシンテターゼの阻害は動物細胞にとって有害である。Tomoda et al. (Tomoda el. al., J. Biol. Chem. 266:4214-4219, 1991) は、Triacsin CがRaji細胞において1.0μMにて成長阻害を引き起こすと教示しており、またVeroおよびHela細胞の成長を阻害することも示されている。Tomoda el. al.はさらに、アシル-CoAシンテターゼが動物細胞に必須であること、およびこの酵素の阻害は致死的な効果を有することを教示している。
米国特許第5981575号 (その開示は引用により本明細書に含まれる)において、ある化合物ファミリー (γ-置換-α-メチレン-β-カルボキシ-γ-ブチロラクトン) が脂肪酸合成を阻害すること、腫瘍細胞の増殖を阻害すること、および体重減少を誘発することが示されている。’575特許に開示される化合物は、治療適用について天然産物のセルレニンを上回るいくつかの利点を有する。: [1] それらは反応性の高いセルレニンのエポキシド基を含まない、[2] それらは水溶液中で安定かつ可溶性である、[3] それらは2段階合成反応によって製造することがき、よって容易に大量を合成可能である、[4] それらは生化学的および薬理学的分析のため容易に高比放射能にトリチウム化される。このファミリーの化合物(その多くが脂肪酸合成酵素阻害薬である)の合成は’575特許に記載されており、同様にFAS発現腫瘍細胞の処置手段としてのその用途、および体重減少の手段としてのその用途も記載されている。’575特許はまた、体重減少の手段として脂肪細胞塊 (脂肪細胞数および大きさ) を体系的に減少させるためのいずれかの脂肪酸合成酵素阻害薬の使用を開示する。
FAS阻害化合物の他の開示には、特許出願PCT/US03/20960およびPCT/US03/21700が含まれ、これらの開示は引用により本明細書に含まれる。
マウスおよびヒトにおける脂肪酸合成の主要部位は肝臓 (参照:Roncari, Can. J. Biochem., 52:221-230, 1974; Triscari et al., 1985, Metabolism, 34: 580-7; Barakat et al., 1991, Metabolism, 40: 280-5)、乳腺 (参照: Thompson, et al., Pediatr. Res., 19:139-143, 1985) および脂肪組織(Goldrick et al., 1974, Clin. Sci. Mol. Med. , 46:469-79)である。
抗菌物質としての脂肪酸合成阻害薬
セルレニンはもともと、有力な抗真菌抗生物質としてCephalosporium caerulensの培養液から単離された。構造的には、セルレニンは (2R,3S)-エポキシ-4-オキソ-7,10-トランス,トランス-ドデカン酸アミドとして特徴決定されている。その作用メカニズムは、不可逆的な結合による、脂肪酸生合成に必要な縮合酵素のβ-ケトアシル-ACP合成酵素の阻害である。セルレニンは抗真菌薬として分類されており、主にCandidaおよびSaccharomyces spに対するものである。さらに、幾つかのインビトロ活性が、ある種の細菌、放線菌およびマイコバクテリア(ただし結核菌(Mycobacterium tuberculosis)に対しては活性がない)に対して示されている。脂肪酸合成阻害薬およびセルレニンの活性は特に、原生動物、例えばToxoplasma gondii、または他の感染性真核性病原、例えばPneumocystis carinii、Giardia lamblia、Plasmodium sp.、Trichomonas vaginalis、Cryptosporidium、Tiypanosoma、LeishmaniaおよびSchistosomaについては評価されていない。
セルレニンはもともと、有力な抗真菌抗生物質としてCephalosporium caerulensの培養液から単離された。構造的には、セルレニンは (2R,3S)-エポキシ-4-オキソ-7,10-トランス,トランス-ドデカン酸アミドとして特徴決定されている。その作用メカニズムは、不可逆的な結合による、脂肪酸生合成に必要な縮合酵素のβ-ケトアシル-ACP合成酵素の阻害である。セルレニンは抗真菌薬として分類されており、主にCandidaおよびSaccharomyces spに対するものである。さらに、幾つかのインビトロ活性が、ある種の細菌、放線菌およびマイコバクテリア(ただし結核菌(Mycobacterium tuberculosis)に対しては活性がない)に対して示されている。脂肪酸合成阻害薬およびセルレニンの活性は特に、原生動物、例えばToxoplasma gondii、または他の感染性真核性病原、例えばPneumocystis carinii、Giardia lamblia、Plasmodium sp.、Trichomonas vaginalis、Cryptosporidium、Tiypanosoma、LeishmaniaおよびSchistosomaについては評価されていない。
処置に対する感受性が特に高い感染性疾患は、感染動物の外部から接近可能な表面に病変を引き起こす疾患である。外部から接近可能な表面には、非侵襲性手段により (皮膚の切断または穿刺なしに) 到達可能なすべての表面が含まれ、例えばその皮膚表面、粘膜、例えば鼻腔、口腔、胃腸管または泌尿生殖器の表面および肺表面、例えば肺胞嚢、などを覆う粘膜、である。感受性が高い疾患には以下が含まれる: (1) 皮膚真菌症または白癬、特にMicrosporum、Trichophyton、EpidermophytonまたはMucocutaneous candidiasisにより起こるもの; (2) 真菌性角膜炎、特にAspergillus、FusariumまたはCandidaにより起こるもの; (3) アメーバ性角膜炎、特にAcanthamoebaにより起こるもの; (4) 胃腸管疾患、特にGiardia lamblia、Entamoeba、Cryptosporidium、MicrosporidiumまたはCandidaにより起こるもの (通常免疫不全の動物における) ; (5) 泌尿生殖器感染、特にCandida albicansまたはTrichomonas vaginalisにより起こるもの; および(6) 肺疾患、特に結核菌、AspergillusまたはPneumocystis cariniiにより起こるもの。脂肪酸合成阻害薬による処置に対する感受性の高い感染性生物には、結核菌、特に多剤耐性株、および原生動物、例えばトキソプラズマが含まれる。
脂肪酸合成を阻害する化合物はいずれも、微生物細胞増殖阻害に使用可能である。しかしながら、患者に投与された化合物は、患者と標的微生物細胞の両方に等しく毒性であってはならない。したがって、標的微生物細胞にのみ、またはそれに主に影響する阻害薬を選択することが有益である。
自身が内部合成した脂肪酸に依存する真核性微生物細胞は、タイプ I FASを発現する。これは、FAS阻害薬が成長阻害を示すこと、および、外部から添加した脂肪酸は正常な患者細胞を保護できるがこれら微生物細胞は保護できないこと、の両方により示される。それゆえ、細胞による脂肪酸合成を妨げる物質が感染の処置に使用できる。真核生物において、脂肪酸は、基質としてアセチル CoA、マロニル CoAおよびNADPHを用いてタイプ I FASにより合成される。したがって、この経路に基質を供給しうる酵素もまた脂肪酸合成速度に影響でき、よって内部合成した脂肪酸に依存する微生物において重要である。これらいずれの酵素の発現または活性の阻害も、内部合成した脂肪酸に依存する微生物細胞の成長に影響する。
タイプ I FASの生成物は各種生物において異なる。例えば、fungus S. cerevisiaeでは、その生成物は主にコエンザイムAにエステル化されるパルミテートおよびステアレートである。Mycobacterium smegmatisでは、生成物は16〜24炭素長の範囲の飽和脂肪酸CoAエステルである。これら脂質はしばしばさらに処理され、各種脂肪成分に対する細胞要求を満たす。
下流の脂肪酸の処理および利用における重要な工程の阻害は、細胞が内因性脂肪酸に依存するか細胞外部から供給された脂肪酸を利用するかに関わらず細胞機能を阻害することが予想され、よってこれら下流の工程の阻害薬は内因性脂肪酸に依存する微生物細胞に十分に選択的でない可能性がある。しかしながら、かかる微生物にタイプ I 脂肪酸合成阻害薬を投与すると、下流の脂肪酸の処理および/または利用の阻害薬による阻害に対する感受性が高まることが発見された。この相乗効果のため、脂質の生合成および/または利用における下流の工程の阻害薬の1以上と組み合わせた脂肪酸合成阻害薬の投与は、内部合成された脂肪酸に依存する微生物細胞に選択的に影響するであろう。好ましい組合せには、FASおよびアセチルCoAカルボキシラーゼの阻害薬、またはFASおよびMASの阻害薬が含まれる。
哺乳動物がタイプ I FASを発現する生物の細胞に感染したと判断された場合、あるいはFASが患者体液から検出された場合、その哺乳動物または患者は脂肪酸合成阻害薬を投与することにより処置できる (特許第5614551号)。
食欲を抑制し体重減少を刺激するためのニューロペプチド Yの阻害は、国際特許出願PCT/US01/05316(その開示は引用により本明細書に含まれる)に記載されている。この出願は、しかしながら、本出願に開示されるいずれも化合物も記載または開示していない。
体重減少を刺激するためのカルニチンパルミトイルトランスフェラーゼ-1(CPT-1) の刺激は、米国特許出願第60/354480号(その開示は引用により本明細書に含まれる)に記載されている。この出願もまた、本出願に開示されるいずれも化合物も記載または開示していない。
癌細胞の増殖を阻害するためのFAS阻害薬の使用は、米国特許第5759837号(その開示は引用により本明細書に含まれる)に記載されている。この出願は、本出願に開示されるいずれも化合物も記載または開示していない。
本発明の目的は、様々な治療的価値のある性質、例えばFAS阻害、NPY阻害、CPT-1刺激、体重減少誘導能力、および抗癌および抗菌性、を有する式 Iの化合物の新規クラスを提供することである。
本発明のさらなる目的は、式 IIの化合物および医薬用希釈体を含む医薬組成物を提供することである。
本発明のさらなる目的は、式 IIの化合物および医薬用希釈体を含む医薬組成物を投与することにより動物およびヒトにおいて体重減少を誘導する方法を提供することである。
本発明のさらなる目的は、ヒトまたは動物に式 IIの化合物および医薬用希釈体を含む医薬組成物を投与することによりCPT-1活性を刺激する方法を提供することである。
本発明のさらなる目的は、式 IIの化合物および医薬用希釈体を含む医薬組成物を投与することによりヒトまたは動物においてニューロペプチド Yの合成を阻害する方法を提供することである。
本発明のさらなる目的は、式 IIの化合物および医薬用希釈体を含む医薬組成物を投与することによりヒトまたは動物において脂肪酸合成酵素活性を阻害する方法を提供することである。
本発明のさらなる目的は、式 IIの化合物および医薬用希釈体を含む医薬組成物を投与することにより動物およびヒトにおいて癌を処置する方法を提供することである。
本発明のまたさらなる目的は、式 IIの化合物および医薬用希釈体を含む医薬組成物を投与することにより動物およびヒトにおいて癌細胞の増殖を予防する方法を提供することである。
本発明のさらなる目的は、式 IIの化合物および医薬用希釈体を含む医薬組成物を投与することにより侵襲性微生物細胞の増殖を阻害する方法を提供することである。
本発明の化合物は、従来の方法により製造することができる。多くの化合物の合成を実施例に記載する。これら化合物は、肥満、癌または微生物感染の処置に有用でありうる。
本発明のある実施態様は、以下の一般式を有する化合物である:
[式中、
R1およびR2は、互いに同じであっても異なっても良く、H、C1-C20 アルキル、シクロアルキル、アルケニル、アリール、アリールアルキルもしくはアルキルアリール、-CH2COR5、-CH2C(O)NR5、-C(O)R5または-CH2OR5であり、ハロゲン原子を含有していてもよく、ここで、R5は、C1-C12 アルキル基である;
R3およびR4は、互いに同じであっても異なっても良く、H、C1-C20 アルキル、シクロアルキル、アルケニル、アリール、アリールアルキルまたはアルキルアリールである;
ただし、R4が-(CH2)7CH3の場合、R3はメチルであり、R1は-CH3であり、R2は-CH2-CH=CH2でない、
さらにR4が-CH3の場合、R3はHであり、R1は-CH3であり、R2は-CH3または-CH=C(CH3)CH2CH2CH=C(CH3)2でない]。
R1およびR2は、互いに同じであっても異なっても良く、H、C1-C20 アルキル、シクロアルキル、アルケニル、アリール、アリールアルキルもしくはアルキルアリール、-CH2COR5、-CH2C(O)NR5、-C(O)R5または-CH2OR5であり、ハロゲン原子を含有していてもよく、ここで、R5は、C1-C12 アルキル基である;
R3およびR4は、互いに同じであっても異なっても良く、H、C1-C20 アルキル、シクロアルキル、アルケニル、アリール、アリールアルキルまたはアルキルアリールである;
ただし、R4が-(CH2)7CH3の場合、R3はメチルであり、R1は-CH3であり、R2は-CH2-CH=CH2でない、
さらにR4が-CH3の場合、R3はHであり、R1は-CH3であり、R2は-CH3または-CH=C(CH3)CH2CH2CH=C(CH3)2でない]。
好ましくは、R1およびR2は、各々独立にC1-C12 アルキルである。好ましい実施態様において、R1およびR2は、各々-CH2-CH=CH2である。
好ましくは、R3およびR4は、各々独立にC1-C12 アルキル基である。さらに好ましくは、R4は、C1-C8 アルキル基であり、最も好ましくは-CH3である。
本発明の組成物は、ヒトおよび他の動物への投与のため、錠剤、カプセル、丸剤、粉末、顆粒、滅菌非経口溶液または懸濁液、経口溶液または懸濁液、好適な量の化合物を含む水中油滴および油中水滴エマルジョン、坐剤および液体懸濁液または溶液などの単位投与形態にて提供することができる。本明細書で使用する場合、用語「医薬用希釈体」と「医薬用担体」は同義である。経口投与のため、固体または液体の単位投与形態のいずれも調製可能である。固体組成物、例えば錠剤を調製するため、化合物は常套的な成分と混合することができ、それは例えばタルク、ステアリン酸マグネシウム、第二リン酸カルシウム、ケイ酸アルミニウムマグネシウム、硫酸カルシウム、デンプン、乳糖、アカシア、メチルセルロース、および医薬用希釈体または担体として機能的に同様の物質である。カプセルは、化合物を不活性な医薬用希釈体と混合し、この混合物を適当な大きさの硬ゼラチンカプセルに充填することによって調製する。軟ゼラチンカプセルは、化合物と許容される植物油、軽流動ワセリンまたは他の不活性油とのスラリーの機械封入によって調製する。
液体単位投与形態または経口投与形態、例えばシロップ、エリキシルおよび懸濁液が調製可能である。これら形態は、水性媒体に糖、芳香族系香料および保存剤とともに溶解して、シロップとすることができる。懸濁液は、水性媒体を用いて、懸濁剤、例えばアカシア、トラガカント、メチルセルロースなどの補助のもと調製可能である。
非経口投与のため、液体単位投与形態を、化合物と滅菌媒体とを利用して調製可能である。溶液の調製においては、化合物を注射用水に溶解し、濾過滅菌し、そして適当なバイアルまたはアンプルに充填し密閉すればよい。アジュバント、例えば局所麻酔薬、保存剤および緩衝剤を媒体に溶解することができる。組成物をバイアルに充填後凍結して、水分を吸引により除去することができる。凍結乾燥粉末は、続いてバイアル中で計量し、使用前に再構成することができる。
本発明の化合物について想定される臨床治療上の適用には以下が含まれる: (1)侵襲性微生物、例えばstaphylococciおよびenterococci、による感染; (2) その細胞が脂肪酸合成酵素を過剰発現する様々な組織において生じる癌、および(3)過剰なカロリーの摂取による肥満。治療の投与量および期間は、以下のような様々な因子に依存する: (1) 患者の年齢、体重および臓器機能(例えば、肝臓および腎臓機能); (2) 処置しようとする疾患過程の性質および程度、ならびに何らかの既存の顕著な併存疾患および使用中の併用薬、および(3) 薬物関連パラメーター、例えば投与経路、治癒に必要な投与頻度および期間、および薬物の治療インデックス。通常、投与量は、血清レベル1 ng/ml〜100 ng/mlを達成し、標的部位での有効濃度が約 1μg/ml〜10μg/mlに到達するよう選択される。
本発明を以下の実施例により説明するが、本発明はこれらに限定されない。
本発明の化合物は、以下に記載のように合成した。化合物の生物活性は、以下のように示した: 以下について試験した: (1) 精製ヒトFASの阻害、(2) 完全細胞における脂肪酸合成活性阻害、および (3)高レベルのFASおよび脂肪酸合成活性を有することが知られている培養MCF-7ヒト乳癌細胞に対する細胞毒性(クリスタルバイオレットおよびXTTアッセイによる)。細胞毒性のレベルの低い化合物を選択し、Balb/Cマウスにおいて体重減少について試験した。さらに、有意な体重減少および低レベルの細胞毒性を示した群の代表的化合物を、脂肪酸酸化およびカルニチンパルミトイルトランスフェラーゼ(CPT-1) 活性に対する効果について試験し、またノザン解析によりBalb/Cマウスにおける視床下部NPY発現に対する効果について試験した。一部の化合物はまた、グラム陽性および/または陰性細菌に対する活性についても試験した。
化合物の化学合成
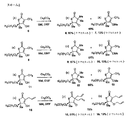
2-tert-ブチル-5-メチル-5-オクチル-[l,3]-オキサチオラン-4-オン (1(図1に示す), 2.2 g, 7.68 mmol) (EtOH (29 mL) 中)にNaOEt (2.1 M, 4.75 mL, 9.9 mmol) を添加し、その溶液を室温で撹拌した。40分後、その溶液をHCl (1 N, 30 mL) に注入し、Et2O (3 x 30 mL)で抽出した。次いで、この混合有機物をH20 (5 x 30 mL)で洗浄し、乾燥し(MgS04)、ろ過し、蒸発させて、粗製遊離チオールを得て、それをCH2Cl2 (86 mL) に溶解して0℃に冷却した。NEt3 (1.6 mL, 11.5 mmol) および4-ペンテノイルクロライド (1.10 mL, 9.98 mmol) 添加し、この溶液を0℃で1時間撹拌した。NH4Cl (飽和、150 mL) を添加し、溶液をCH2C12で抽出した。有機層を乾燥し (MgS04)、ろ過し、蒸発させた。フラッシュクロマトグラフィー 5%EtOAc/ヘキサンにより、2-(4-ペンテノイル)-スルファニル-2-メチル-デカン酸エチルエステル (2) (2.29 g, 91%)が得られた。
H1 NMR (300 MHz, CDCl3)δ 0.86 (t, J= 6.9 Hz, 3 H), 1.23 (m, 15 H), 1.60 (s, 3 H), 1.76-1.78 (m, 2 H), 2.34-2.36 (m, 2 H), 2.53-2.59(m, 2 H), 4.16 (q, J= 7.2 Hz, 2 H), 4.98 (d, J= 10.3 Hz, 1 H), 5.01 (d, J= 17.6 Hz, 1 H), 5.77 (ddt, J= 10.3, 17.6, 6.3 Hz, 1 H).
H1 NMR (300 MHz, CDCl3)δ 0.86 (t, J= 6.9 Hz, 3 H), 1.23 (m, 15 H), 1.60 (s, 3 H), 1.76-1.78 (m, 2 H), 2.34-2.36 (m, 2 H), 2.53-2.59(m, 2 H), 4.16 (q, J= 7.2 Hz, 2 H), 4.98 (d, J= 10.3 Hz, 1 H), 5.01 (d, J= 17.6 Hz, 1 H), 5.77 (ddt, J= 10.3, 17.6, 6.3 Hz, 1 H).
H1 NMR (300 MHz, CDC13)δ 0.85 (t, J= 6.9 Hz, 3 H), 1.24 (m, 12 H), 1.65 (s, 3 H), 1.81-1.86 (m, 2 H), 3.02 (d, J= 6.4 Hz, 2 H), 5.12 (dq, J=10.6, 1.5 Hz, 1 H), 5.20 (dq, J= 17.3, 1.5 Hz, 1 H), 5.84 (ddt, J= 10.6, 17.3, 6.4 Hz, IH). 13C NMR (100 MHz, CDCl3)δ 14.1, 22.6, 25.2, 26.1, 26.9, 29.1, 29.3, 29.5, 31.8, 38.5, 57.5, 111.5, 117.4, 134.4, 180.8, 195.4.
3-アリル-4-ヒドロキシ-5-メチル-5-オクチル-5-H-チオフェ-2-オン (3, 695 mg, 2.5 mmol) (DMF (14 mL)中、-40℃に冷却)にNaH (油中60%, 118 mg, 2.95 mmol)を添加し、その溶液0℃まで温め、25分撹拌した。アリルブロミド (0.34 mL, 3.94 mmol) を添加し、氷浴を取り除き、反応を室温まで温めて20時間撹拌した。HCl (1 N, 30 mL) を添加し、その溶液をEt20 (3 x 30 mL)で抽出した。この混合有機物を乾燥し (MgS04)、ろ過し、蒸発させた。フラッシュクロマトグラフィー2% EtOAc/Hex- 10%EtOAc/Hexにより純粋な4 (441 mg, 56%) およびO-アルキル化副産物 (64 mg, 8%) を得た; 全収率 (64%)。
C-アルキル化生成物 H1 NMR (300 MHz, CDCl3)δ 0.86 (t, J= 6.5 Hz, 3 H), 1.25 (m, 11 H), 1.43-1.47 (m, 1 H), 1.54 (s, 3 H), 1.79-1.84 (m, 2 H), 2.43-2.47 (m, 4 H), 5.05-5.11 (m, 4 H), 5.57-5.69 (2 H). C3 NMR (100 MHz, CDC13) δ14.1, 22.6, 25.1, 25.8, 29.1, 29.2, 29.5, 31.8, 40.2, 40.7, 41.3, 62.8, 64.8, 120.3, 120.4, 131.2, 131.2, 203.9, 213.5.
5 (以下の図2に示す;その合成はPCT出願PCT/US03/021700に記載されている, 200 mg, 0.78 mmol) (DMF (4.3 mL) に溶解)にCS2C03 (304 mg, 0.94 mmol) およびMeI (78μL, 1.25 mmol)を添加した。この溶液を室温で1時間撹拌した。次いで、その混合物をNH4Cl/1N HCl (3:1, 20 mL) に注入し、Et20 (3 x 15 mL)で抽出した。その後Et20 層をH20(3 x 15 mL)で洗浄し、乾燥し (MgS04) 、ろ過し、蒸発させて、粗製6/7を得た。フラッシュクロマトグラフィー5%EtOAc/ヘキサン-20%EtOAc/ヘキサンにより、6 (120 mg) および7 (14 mg) を得た。全収率48%。
6: H1 NMR (300 MHz, CDC13)δ 0.86 (t, J = 6.99 Hz, 3 H), 1.25 (m, 14 H), 1.29 (s, 3 H), 1.41-1.49 (m, 1 H), 1.65 (s, 3 H), 1.76-1.82 (m, 1 H), 1.96-2.01 (m, 1 H) ; C3 NMR (100 MHz, CDCl3)δ 14.0, 22.2, 22.5, 24.4, 25.6, 28.1, 29.1, 29.2, 29.4, 31.7, 40.6, 53.6, 65.1, 204.9, 215.4.
8 (以下の図2に示す, 140 mg, 0.61 mmol) およびMeI (65μL, 1.06 mmol)に反応を室温で一晩撹拌したことを除き上記の操作を行い、9 (83 mg) および10 (13 mg) を得た。フラッシュクロマトグラフィー (2%EtOAc-5%EtOAc/ヘキサン)後全収率65 %。
9: H1 NMR (400 MHz, CDCl3) δ 0.80 (t, J= 6.8 Hz, 3 H), 1.19 (m, 10 H), 1.25 (s, 3 H), 1.41-1.46 (m, 1 H), 1.65 (s, 3 H), 1.72-1.76 (m, 1 H), 1.88-1.95 (m, 1 H). 13C NMR (100 MHz, CDC13) δ13.9, 22.2, 22.4, 24.4, 25.6, 28.1, 29.1, 31.4, 40.6, 53.6, 65.1, 204.9, 215.4.
11 (以下の図2に示す, 209 mg, 0.74 mmol) およびMeI (73μL, 1.18 mmol) に一晩上記の操作を行い、フラッシュクロマトグラフィー 5%EtOAc/ヘキサン後に12 (151 mg, 69%) を得た。(ここでO-アルキル化は回収されなかったが、存在していた)。
H1 NMR (400 MHz, CDCl3)δ 0.83 (t, J= 5.1 Hz, 3 H), 1.21 (m, 18 H), 1.26 (s, 3 H), 1.42-1.46 (m, 1 H), 1.70 (s, 3 H), 1.71-1.74 (m, 1 H), 1.89-1.96 (m, 1 H). C3 NMR (100 MHz,CDCl3) δ 14.0, 22.2, 22.6, 24.4, 25.6, 28.1, 29.2, 29.2, 29.4, 29.4, 29.4, 31.8, 40.6, 53.5, 65.0, 204.8, 215.4. ±
14 (以下の図2に示す, 177 mg, 0.57 mmol) およびアリルブロミド (66μL, 0.76 mmol) に一晩上記の操作を行い、15 (126 mg) および16 (30 mg) を得た。フラッシュクロマトグラフィー 5% EtOAc/ヘキサン後全収率78%。
H1 NMR (300 MHz,CDCl3) δ 0.85 (t, J= 7.02 Hz, 3 H), 1.23 (m, 15 H), 1.40-1.50 (m, 1 H), 1.53 (s, 3 H), 1.75- 1.86 (m, 2 H), 2.37-2.50 (m, 4 H), 5.03-5.09 (m, 4 H), 5.52-5.66 (m, 2 H).
ZR-75-1 ヒト乳癌細胞からのFAS精製の生物学的および生化学的方法
ヒトFASは、American Type Culture Collectionより入手した培養ZR-75-1 ヒト乳癌細胞から精製した。出典Linn et al., 1981およびKuhajda et al., 1994の方法は、低張溶解、連続的ポリエチレングリコール (PEG) 沈澱、および陰イオン交換クロマトグラフィーを利用する。ZR-75-1 細胞は、37℃、5% C02にてRPMI培養培地(10% ウシ胎児血清、ペニシリンおよびストレプトマイシン含有)中で培養する。
ヒトFASは、American Type Culture Collectionより入手した培養ZR-75-1 ヒト乳癌細胞から精製した。出典Linn et al., 1981およびKuhajda et al., 1994の方法は、低張溶解、連続的ポリエチレングリコール (PEG) 沈澱、および陰イオン交換クロマトグラフィーを利用する。ZR-75-1 細胞は、37℃、5% C02にてRPMI培養培地(10% ウシ胎児血清、ペニシリンおよびストレプトマイシン含有)中で培養する。
集密細胞の10個のT150 フラスコを1.5 ml 溶解バッファー (20 mM トリス-HCl, pH 7.5, 1 mM EDTA, 0.1 mM フェニルメタンスルホニルフルオライド(PMSF), 0.1 % Igepal CA-630) にて溶解し、加圧型細胞破砕装置で氷上にて20打で破砕する。溶解物をJA-20ローター(Beckman) で20,000 rpmにて30分間4℃で遠心分離し、上清を溶解バッファーで42 mlとする。50% PEG 8000溶液(溶解バッファー中) を上清にゆっくりと添加し、最終濃度7.5%とする。60分間4℃で振盪した後、その溶液をJA-20ローター (Beckman) で15,000 rpm にて30分間4℃で遠心する。その後固体 PEG 8000を上清に添加し、最終濃度15%とする。上記のように振盪および遠心を繰り返した後、ペレットを一晩4℃で10 ml 緩衝液 A (20 mM K2HPO4, pH 7.4)に再懸濁する。0.45μM ろ過の後、そのタンパク質溶液をMono Q 5/5 陰イオン交換カラム (Pharmacia)にアプライする。カラムを15分間緩衝液 Aにて1 ml/分で洗浄し、結合物質を60分での1 M KClまでの直線性60-ml勾配により溶出する。FAS (MW〜270 kD) は、典型的には0.25 M KClにおいて3つの0.5 ml画分に溶出され、4-15% SDS-PAGEおよびCoomassie G250 染色 (Bio-Rad)により同定される。FASタンパク質濃度は、Coomassie Plus Protein Assay Reagent (Pierce) により製造元の説明にしたがいBSAをスタンダートとして用いて測定する。この方法により、Coomassie染色ゲルで判断して実質的に純粋なFAS調製物(>95%) が得られる。
FAS酵素活性の測定および化合物のIC50の決定
FAS活性は、マロニル-CoA依存的NADPH酸化を分光光度法でOD340にて96ウェルプレートにおいて観察することにより測定する (Dils et al およびArslanian et al, 1975)。各ウェルは、2μg 精製FAS、100 mM K2HP04、pH 6.5, 1 mM ジチオスレイトール (Sigma)および187.5 μM β-NADPH (Sigma)を含む。阻害薬のストック溶液はDMSO中に2、1および0.5 mg/mlで調製し、1μlのストックを各ウェルに添加した場合に最終濃度20、10および5 g/mlを得る。各実験につき、DMSOコントロール、阻害薬およびブランク (FAS酵素なし) とともに、セルレニン (Sigma) をポジティブコントロールとして、いずれもデュプリケートで行う。
FAS活性は、マロニル-CoA依存的NADPH酸化を分光光度法でOD340にて96ウェルプレートにおいて観察することにより測定する (Dils et al およびArslanian et al, 1975)。各ウェルは、2μg 精製FAS、100 mM K2HP04、pH 6.5, 1 mM ジチオスレイトール (Sigma)および187.5 μM β-NADPH (Sigma)を含む。阻害薬のストック溶液はDMSO中に2、1および0.5 mg/mlで調製し、1μlのストックを各ウェルに添加した場合に最終濃度20、10および5 g/mlを得る。各実験につき、DMSOコントロール、阻害薬およびブランク (FAS酵素なし) とともに、セルレニン (Sigma) をポジティブコントロールとして、いずれもデュプリケートで行う。
アッセイは、Molecular Devices SpectraMax Plus Spectrophotometerにて行う。FAS、緩衝液、阻害薬およびコントロールを含むプレートを37℃に加熱した分光光度計に設置する。速度論的プロトコールを用いて、100μlの100 mM K2HP04, pH6.5を含むデュプリケートウェルにてブランク測定し、プレートをOD340 にて10秒間隔で5分間読みとり、マロニル-CoA依存的NADPH酸化を測定する。プレートを分光光度計から取り除き、マロニル-CoA (67.4μM, 最終濃度/ウェル) およびアルキニル-CoA (61.8μM, 最終濃度/ウェル) をブランクを除く各ウェルに添加する。プレートを上記のようにして速度論的プロトコールにより再度読み取り、マロニル-CoA依存的NADPH酸化を測定する。マロニル-CoA依存的NADPH酸化およびマロニル-CoA非依存的NADPH酸化についてのΔ OD340の相違が特異的FAS活性である。FAS調製物の純度からマロニル-CoA非依存的NADPH酸化は無視できる。
化合物のFASに対するIC50は、Δ OD340を試験した各阻害薬濃度についてプロットし、直線回帰を用い、ベストフィットライン、r2値、および95%信頼区間を計算することにより決定される。FASの50% 阻害をもたらす化合物濃度がIC50である。Δ OD340 対 時間のグラフは、SOFTmax PRO ソフトウェア (Molecular Devices) により各化合物濃度についてプロットする。直線回帰、ベストフィットライン、r2および95% 信頼区間の計算は、Prism Version 3.0 (Graph Pad Software)により計算する。
クリスタルバイオレット細胞増殖アッセイ
クリスタルバイオレットアッセイでは、細胞増殖は測定されるが細胞毒性は測定されない。このアッセイは、96ウェルプレートにおける固定細胞のクリスタルバイオレット染色およびその後の可溶化、ならびに分光光度計におけるOD490の測定を用いる。OD490は、測定単位時間あたりの細胞増殖に相当する。細胞を対象化合物および媒体コントロールで処置し、各化合物についてIC50を計算する。
クリスタルバイオレットアッセイでは、細胞増殖は測定されるが細胞毒性は測定されない。このアッセイは、96ウェルプレートにおける固定細胞のクリスタルバイオレット染色およびその後の可溶化、ならびに分光光度計におけるOD490の測定を用いる。OD490は、測定単位時間あたりの細胞増殖に相当する。細胞を対象化合物および媒体コントロールで処置し、各化合物についてIC50を計算する。
具体的化合物の癌細胞に対する細胞毒性を測定するため、5 x 104 MCF-7 ヒト乳癌細胞(American Type Culture Collection より入手)を24ウェルプレートの各ウェルにDMEM培地(10% ウシ胎児血清、ペニシリンおよびストレプトマイシン含有)中にて播種する。一晩37℃、5% C02にて培養した後、DMSOに溶解した試験化合物を容量1μlにて以下の濃度でウェルに添加する: 50、40、30、20および10μg/ml(トリプリケート)。必要に応じてさらなる濃度を試験する。1μlのDMSOを媒体コントロールとしてトリプリケートのウェルに添加する。ポジティブコントロールとして、C75を10および5μg/mlにて用いる。
72時間のインキュベーションの後、細胞を0.5 mlのクリスタルバイオレット染色液 (0.5%、25% メタノール中)により各ウェルにおいて染色する。10分後、ウェルをすすぎ、風乾し、そして2時間振盪しながら0.5 mlの10% ドデシル硫酸ナトリウムで可溶化する。各ウェルから100μlを96ウェルプレートへ移した後、プレートをMolecular Devices SpectraMax Plus Spectrophotometer上でOD490にて読み取る。平均 OD490 値は、SOFTmax Pro Software (Molecular Devices) により計算し、IC50値は、直線回帰解析によりPrism version 3.02 (Graph Pad Software, San Diego)を用いて決定する。
XTT細胞毒性アッセイ
XTTアッセイは、[51Cr]放出細胞毒性アッセイの非放射性代替法である。XTTは、代謝が活性な生存細胞によってのみホルマザン色素に還元されるテトラゾリウム塩である。XTTの還元は、分光光度計にてOD490-OD650として測定する。
XTTアッセイは、[51Cr]放出細胞毒性アッセイの非放射性代替法である。XTTは、代謝が活性な生存細胞によってのみホルマザン色素に還元されるテトラゾリウム塩である。XTTの還元は、分光光度計にてOD490-OD650として測定する。
具体的化合物の癌細胞に対する細胞毒性を測定するため、9 x 103 MCF-7 ヒト乳癌細胞(American Type Culture Collectionより入手)を96ウェルプレートの各ウェルにDMEM 培地(10% ウシ胎児血清、インシュリン、ペニシリンおよびストレプトマイシン含有)中にて播種する。37℃、5% C02で一晩培養した後、DMSOに溶解した試験化合物を容量1μlにて以下の濃度でウェルに添加する:80、40、20、10、5、2.5、1.25および0.625μg/ml(トリプリケート)。必要に応じて、さらなる濃度を試験する。1μlのDMSOを媒体コントロールとしてトリプリケートのウェルに添加する。ポジティブコントロールとして、C75を40、20、10、15、12.5、10および5μg/ml(トリプリケート)にて用いる。
72時間のインキュベーション後、製造元の指示にしたがい(Cell Proliferation Kit II (XTT) Roche)、細胞をXTT 試薬と4時間インキュベートする。プレートをMolecular Devices SpectraMax Plus Spectrophotometer上でOD490およびOD650にて読み取る。3つのXTT試薬含有細胞不含ウェルがプレートブランクの役割を果たす。XTTデータは、OD490-OD650として報告される。平均および標準誤差は、SOFTmax Pro software (Molecular Dynamics)により計算する。
化合物のIC50は、コントロールと比較してOD490-OD650の50%の減少をもたらす薬物の濃度と規定される。OD490-OD650は、SOFTmax PRO software (Molecular Devices) により各化合物濃度について計算する。IC50は、コントロールに対する割合としてのFAS活性を薬物濃度に対してプロットして直線回帰により計算する。直線回帰、ベストフィットライン、r2および95% 信頼区間は、Prism Version 3.0 (Graph Pad Software)を用いて決定する。
全脂質への[14C]アセテートの取り込みの測定および化合物のIC50の決定
このアッセイは、[14C]アセテートの全脂質への取り込みを測定し、インビトロでの脂肪酸合成経路活性を測定するものである。これはインビトロにおける脂肪酸合成の阻害の測定に利用される。
このアッセイは、[14C]アセテートの全脂質への取り込みを測定し、インビトロでの脂肪酸合成経路活性を測定するものである。これはインビトロにおける脂肪酸合成の阻害の測定に利用される。
上記のように培養したMCF-7 ヒト乳癌細胞を、5 x 104 細胞で24ウェルプレートの各ウェルに播種する。一晩インキュベーションした後、DMSOに溶解した試験化合物を5、10および20 g/ml(トリプリケート)で添加し、必要であればより低い濃度も試験する。DMSOを媒体コントロールとしてトリプリケートのウェルに添加する。ポジティブコントロールとして、C75を5および10μg/ml(トリプリケート)で用いる。4時間のインキュベーションの後、0.25μCi の[14C]アセテート (容量10μl) を各ウェルに添加する。
さらに2時間インキュベーションした後、培地を各ウェルから吸引除去し、800μlのクロロホルム:メタノール(2:1) および700μlの4 mM MgC12 を各ウェルに添加する。各ウェルの内容物を1.5 Eppendorfチューブに移し、高速Eppendorf Microcentrifuge 5415Dにより最高速度で2分間遠心する。水層 (上層) を除去した後、さらに700μlのクロロホルム: メタノール (2:1)および500μlの4 mM MgCl2 を各チューブに添加し、続いて上記のように1分間遠心する。水層をパスツールピペットで除去し、廃棄する。さらに400μlのクロロホルム:メタノール(2:1)および200μlの4 mM MgC12を各チューブに添加し、次いで遠心し、そして水層を廃棄する。低層 (有機層) をシンチレーションバイアルに移し、40℃でN2ガス下乾燥する。乾燥したら、3 mlのシンチラント (APB#NBC5104) を添加し、バイアルを14Cについてカウントする。Beckman Scintillation カウンターにより平均cpm値をトリプリケートで計算する。
化合物のIC50は、コントロールと比較して[14C]アセテートの脂質への取り込みの50%の減少をもたらす薬物の濃度と規定される。これは、試験した各阻害薬濃度についての平均cpmをプロットし、直線回帰を行い、ベストフィットライン、r2値、および95% 信頼区間を計算することにより決定する。平均cpm値は、Beckman Scintillation カウンター (モデル LS6500) により各化合物濃度について計算する。直線回帰、ベストフィットライン、r2および95%信頼区間の計算は、Prism Version 3.0 (Graph Pad Software)により行う。
カルニチンパルミトイルトランスフェラーゼ-1 (CPT-1)アッセイ
CPT-1は、マロニル-CoAにより阻害されるアシル-CoAからアシル-カルニチンへの長鎖脂肪酸のATP依存的転移を触媒する。CPT-1は活性にミトコンドリア膜を必要とするので、酵素活性は透過処理された細胞またはミトコンドリアにおいて測定する。このアッセイは、透過処理された細胞を使用して、[メチル-14C] L-カルニチンの有機溶媒可溶性(organically soluble)アシル-カルニチン誘導体への転移を測定する。
CPT-1は、マロニル-CoAにより阻害されるアシル-CoAからアシル-カルニチンへの長鎖脂肪酸のATP依存的転移を触媒する。CPT-1は活性にミトコンドリア膜を必要とするので、酵素活性は透過処理された細胞またはミトコンドリアにおいて測定する。このアッセイは、透過処理された細胞を使用して、[メチル-14C] L-カルニチンの有機溶媒可溶性(organically soluble)アシル-カルニチン誘導体への転移を測定する。
MCF-7 細胞は、DMEM(10% ウシ胎児血清含有)中106細胞にて24ウェルプレートにトリプリケートでコントロール、薬物、およびマロニル-CoAについて播種する。アッセイ開始の2時間前、10 mg/ml(DMSO中)のストック溶液から調製した個々の濃度の薬物、薬物不含DMSOからなる媒体コントロールを添加する。マロニル-CoAはインタクトな細胞へは入れないので、薬物とプレインキュベートしていない細胞にのみアッセイ緩衝液において添加する。一晩37℃でインキュベーションした後、培地を除去し、700μlのアッセイ緩衝液(50 mM イミダゾール、70 mM KCl、80 mM ショ糖、1 mM EGTA、2 mM MgC12、1 mM DTT、1 mM KCN、1 mM ATP,0.1 % 脂肪酸不含ウシ血清アルブミン、70μM パルミトイル-CoA、0.25μCi [メチル-14C]L-カルニチン、40 g ジギトニンからなる、薬物、DMSO媒体コントロールまたは20μM マロニル-CoA含有)と置き換える。アッセイ緩衝液中の薬物およびDMSOの濃度は、2時間のプレインキュベーションに使用したものと同じである。6分間37℃でのインキュベーション後、反応を500μlの氷冷4 M 過塩素酸を添加して停止させる。その後細胞を回収し、13,000 x gで5分間遠心する。ペレットを500μlの氷冷2mM 過塩素酸で洗浄し、再び遠心する。得られるペレットを800μlのdH20に再懸濁し、150μlのブタノールで抽出する。ブタノール相を液体シンチレーションでカウントすると、アシルカルニチン誘導体が示される。
新規FAS阻害薬についての体重減少スクリーン
Balb/C マウス(Jackson Labs) を初期体重減少スクリーニングに利用する。動物を一定温度の12時間明暗サイクル室で飼育し、マウス餌および水を自由に摂取させる。各実験につきトリプリケートにて3匹のマウスを各試験化合物および媒体コントロールに使用する。実験のため、マウスは試験化合物ごとに別々に1ケージ3匹づつ飼育する。化合物をDMSOに10 mg/mlで希釈し、約100μlのDMSO中にて60 mg/kgを、または媒体単独を、マウスに腹腔内注射する。マウスを観察し、毎日体重を測定する; 平均体重および標準誤差はエクセル (Microsoft)で計算する。実験は、処置された動物が処置前の体重に到達するまで継続する。
Balb/C マウス(Jackson Labs) を初期体重減少スクリーニングに利用する。動物を一定温度の12時間明暗サイクル室で飼育し、マウス餌および水を自由に摂取させる。各実験につきトリプリケートにて3匹のマウスを各試験化合物および媒体コントロールに使用する。実験のため、マウスは試験化合物ごとに別々に1ケージ3匹づつ飼育する。化合物をDMSOに10 mg/mlで希釈し、約100μlのDMSO中にて60 mg/kgを、または媒体単独を、マウスに腹腔内注射する。マウスを観察し、毎日体重を測定する; 平均体重および標準誤差はエクセル (Microsoft)で計算する。実験は、処置された動物が処置前の体重に到達するまで継続する。
選択した化合物を代謝ケージ(metabolic cage)で飼育された動物で試験する。動物への投与は、1つの代謝ケージにつき、3匹の動物を用いたスクリーニング実験と同一である。動物の体重、水および食餌消費、ならびに尿および糞排泄は毎日測定する。
抗菌性
培養液微量希釈アッセイを使用して化合物の抗菌活性を評価する。化合物は2段階希釈にて試験し、可視的な成長を阻害する濃度 (コントロールの10%でのOD600) をMICとして定義する。試験した微生物にはStaphylococcus aureus (ATCC # 29213)、Enterococcus faecalis (ATCC # 29212)、Pseudomonas aeruginosa (ATCC # 27853)、およびEscherichia coli (ATCC # 25922)が含まれる。アッセイは、2種類の成長培地、Mueller Hinton BrothおよびTrypticase Soy Brothにおいて行う。
培養液微量希釈アッセイを使用して化合物の抗菌活性を評価する。化合物は2段階希釈にて試験し、可視的な成長を阻害する濃度 (コントロールの10%でのOD600) をMICとして定義する。試験した微生物にはStaphylococcus aureus (ATCC # 29213)、Enterococcus faecalis (ATCC # 29212)、Pseudomonas aeruginosa (ATCC # 27853)、およびEscherichia coli (ATCC # 25922)が含まれる。アッセイは、2種類の成長培地、Mueller Hinton BrothおよびTrypticase Soy Brothにおいて行う。
血液 (Tsoy/5% ヒツジ血液) アガープレートにT soy 培養液(10% グリセリン含有)中で維持された凍結ストックから植菌し、一晩37℃でインキュベートする。コロニーを滅菌培養液に濁度が0.5 McFarland スタンダードの濁度と一致するように懸濁する。その植菌材料を1:10にて滅菌培養液 (Mueller HintonまたはTrypticase Soy)に希釈し、195μlを96ウェルプレートの各ウェルに分注する。試験化合物(DMSOに溶解)を容量5μlにて以下の濃度でウェルに添加する: 25、12.5、6.25、3.125、1.56および0.78μg/ml(デュプリケート)。要すればさらなる濃度を試験する。デュプリケートウェルに添加した5μlのDMSOは媒体コントロールである。ポジティブコントロール化合物であるバンコマイシン (E. faecalisおよびS. aureus) およびトブラマイシン (E. coliおよびP. aeruginosa)の段階希釈を各実験に含める。
37℃24時間のインキュベーション後、プレートをOD600 にてMolecular Devices SpectraMax Plus Spectrophotometerで読み取る。平均OD600値はSOFTmax Pro Software (Molecular Devices)を用いて計算し、MIC値は直線回帰解析によりPrism version 3.02 (Graph Pad Software, San Diego)を使用して決定する。MICは、媒体コントロール測定値の10%と等しいOD600測定値を与えるのに必要な化合物の濃度と定義される。
β-酸化アッセイ-酸可溶性産物の単離
24ウェルプレート(1 ml/ウェル)に2.5 x 105 細胞/ウェルを準備した。その細胞を一晩インキュベートした。
24ウェルプレート(1 ml/ウェル)に2.5 x 105 細胞/ウェルを準備した。その細胞を一晩インキュベートした。
翌日、可溶化パルミテート溶液を準備した。50μlの(1-14C) パルミチン酸を2 ml遠心管に添加し、窒素ガス下乾燥した。2 mlのα-CD (α-シクロデキストラン)(10 mg/ml、10 mM トリス中)を添加した。この溶液を37℃水浴で30分間インキュベートした。
25μlの前記溶液を2.5μlの200 M カルニチンおよび222.5μlの血清不含培地(細胞に使用するもの)に添加して放射性混合物を準備した。
次いで、細胞を試験化合物でトリプリケートにて処理し、37℃で60分間インキュベートした。培地を除去し、250μlの放射性混合物を添加した。試験化合物を再び添加し、さらに37℃で60分間インキュベートした。反応を50μlの2.6 N HCl04にて停止させた。プレートの内容物を1.5 ml遠心管に移し、次いで50μlの4N KOHを添加し、その管を60℃水浴にて30分間インキュベートした。酢酸ナトリウム (1M, 75μl)および硫酸 (3N, 50μl) をこの溶液に添加し、ボルテックスした。その管を7分間1000 rpmで室温にて遠心した。一部(225μl) を除去し、以下を添加した (各添加後にボルテックス、最後は2回): 938μl 1:1 クロロホルム: メタノール; 468μl クロロホルム; 281μl 蒸留水。その管を1000 rpmで5分間遠心した。上相を大きなガラス製シンチレーションバイアルにとり、5 mlのBudget 溶媒 (シンチレーション液) を添加した。この管を十分にボルテックスした。最後に、C14を1分間カウントした。
生物学的試験の結果
Claims (20)
- 以下の式を有する化合物:
R1およびR2は、互いに同じであっても異なっても良く、H、C1-C20 アルキル、シクロアルキル、アルケニル、アリール、アリールアルキルもしくはアルキルアリール、-CH2COR5、-CH2C(O)NR5、-C(O)R5または-CH2OR5であって、ハロゲン原子を含有していてもよく、ここで、R5は、C1-C12 アルキル基である、
R3およびR4は、互いに同じであっても異なっても良く、H、C1-C20 アルキル、シクロアルキル、アルケニル、アリール、アリールアルキルもしくはアルキルアリールである;
ただし、R4が-(CH2)7CH3の場合、R3はメチルであり、R1は-CH3であり、R2は-CH2-CH=CH2でない、
さらに、R4が-CH3の場合、R3はHであり、R1は-CH3であり、R2は-CH3または-CH=C(CH3)CH2CH2CH=C(CH3)2でない]。 - R1およびR2が各々独立にC1-C12 アルキルである、請求項1の化合物。
- R1およびR2が各々-CH2-CH=CH2である、請求項2の化合物。
- R3およびR4が各々独立にC1-C12 アルキル基である、請求項1の化合物。
- R4がC1-C6 アルキル基である、請求項4の化合物。
- R4が-CH3である、請求項4の化合物。
- 以下の構造を有する、請求項1の化合物。
- 以下の構造を有する、請求項1の化合物。
- 以下の構造を有する、請求項1の化合物。
- 以下の構造を有する、請求項1の化合物。
- 以下の構造を有する、請求項1の化合物。
- 医薬用希釈体および以下の式 IIの化合物を含む医薬組成物:
R5およびR6は、互いに同じであっても異なっても良く、H、C1-C20 アルキル、シクロアルキル、アルケニル、アリール、アリールアルキルもしくはアルキルアリール、-CH2COR9、-CH2C(O)NR9、-C(O)R9または-CH20R9であって、ハロゲン原子を含有していてもよく、ここでR9はC1-C12 アルキル基である;
R7およびR8は、互いに同じであっても異なっても良く、H、C1-C20 アルキル、シクロアルキル、アルケニル、アリール、アリールアルキルまたはアルキルアリールである]。 - 以下の式 Iの化合物および医薬用希釈体を含む、請求項12の医薬組成物:
R1およびR2は、互いに同じであっても異なっても良く、H、C1-C20 アルキル、シクロアルキル、アルケニル、アリール、アリールアルキルもしくはアルキルアリール、-CH2COR5、-CH2C(O)NR5、-C(O)R5または-CH2OR5であって、ハロゲン原子を含有していてもよく、ここで、R5は、C1-C12 アルキル基である、
R3およびR4は、互いに同じであっても異なっても良く、H、C1-C20 アルキル、シクロアルキル、アルケニル、アリール、アリールアルキルもしくはアルキルアリールである;
ただし、R4が-(CH2)7CH3の場合、R3はメチルであり、R1は-CH3であり、R2は-CH2-CH=CH2でない、
さらに、R4が-CH3の場合、R3はHであり、R1は-CH3であり、R2は-CH3または-CH=C(CH3)CH2CH2CH=C(CH3)2でない]。 - 請求項12の医薬組成物を投与することにより、動物およびヒトにおいて体重減少を誘導する方法。
- ヒトまたは動物に請求項12の医薬組成物を投与することにより、CPT-1活性を刺激する方法。
- 請求項12の医薬組成物を投与することにより、ヒトまたは動物においてニューロペプチド Yの合成を阻害する方法。
- 請求項12の医薬組成物を投与することにより、ヒトまたは動物において脂肪酸合成酵素活性を阻害する方法。
- 請求項12の医薬組成物を投与することにより、動物およびヒトにおいて癌を処置する方法。
- 請求項12の医薬組成物を投与することにより、動物およびヒトにおいて癌細胞の増殖を予防する方法。
- 請求項12の医薬組成物を投与することにより、侵襲性微生物細胞の増殖を阻害する方法。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US57463904P | 2004-05-26 | 2004-05-26 | |
| PCT/US2005/018443 WO2005117590A2 (en) | 2004-05-26 | 2005-05-25 | Novel compounds, pharmaceutical compositions containing same, and methods of use for same |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2008500363A true JP2008500363A (ja) | 2008-01-10 |
| JP2008500363A5 JP2008500363A5 (ja) | 2008-07-17 |
Family
ID=35463277
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2007515318A Pending JP2008500363A (ja) | 2004-05-26 | 2005-05-25 | 新規化合物、該化合物を含む医薬組成物、および該化合物の使用方法 |
Country Status (13)
| Country | Link |
|---|---|
| US (1) | US20090005435A1 (ja) |
| EP (1) | EP1758577A4 (ja) |
| JP (1) | JP2008500363A (ja) |
| KR (1) | KR20070095754A (ja) |
| CN (1) | CN101022792A (ja) |
| AU (1) | AU2005249437A1 (ja) |
| BR (1) | BRPI0510397A (ja) |
| CA (1) | CA2568639A1 (ja) |
| IL (1) | IL179530A0 (ja) |
| MX (1) | MXPA06013687A (ja) |
| RU (1) | RU2006146051A (ja) |
| WO (1) | WO2005117590A2 (ja) |
| ZA (1) | ZA200700024B (ja) |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007014248A2 (en) * | 2005-07-26 | 2007-02-01 | Johns Hopkins University | Method of reducing food intake |
| EP2083831B1 (en) * | 2006-09-22 | 2013-12-25 | Merck Sharp & Dohme Corp. | Method of treatment using fatty acid synthesis inhibitors |
| JP5896601B2 (ja) * | 2007-10-05 | 2016-03-30 | ジェンザイム コーポレーション | セラミド誘導体を用いる多発性嚢胞腎疾患の治療方法 |
| WO2010118324A2 (en) | 2009-04-09 | 2010-10-14 | Nuclea Biotechnologies, LLC | Antibodies against fatty acid synthase |
| JP6285442B2 (ja) * | 2012-09-07 | 2018-02-28 | ヤンセン ファーマシューティカ エヌ.ベー. | がん治療用の脂肪酸合成酵素(fasn)阻害剤として有用なイミダゾリン−5−オン誘導体 |
| EP3220901B1 (en) | 2014-11-20 | 2020-02-19 | VIB vzw | Means and methods for treatment of early-onset parkinson's disease |
| KR20220130702A (ko) | 2020-01-23 | 2022-09-27 | 바스프 에스이 | 아민 또는 암모늄 염을 함유하는 글루포시네이트 제형 |
| CA3196217A1 (en) | 2020-10-27 | 2022-05-05 | Murat Mertoglu | Pesticide microemulsion compositions |
| MX2023009193A (es) | 2021-02-05 | 2023-08-21 | Basf Se | Composiciones herbicidas liquidas. |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004005277A1 (en) * | 2002-07-09 | 2004-01-15 | Fasgen, Inc. | Novel compunds, pharmaceutical compositions containing same, and methods of use for same |
| JP2005523270A (ja) * | 2002-02-08 | 2005-08-04 | ジョーンズ・ホプキンス・ユニバーシティ・スクール・オブ・メディシン | 体重を減少させる方法としてのcpt−1の促進 |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE3427847A1 (de) * | 1983-12-09 | 1985-06-13 | Bayer Ag, 5090 Leverkusen | Thiolan-2,4-dion-3-carboxamide |
| US5759837A (en) * | 1989-01-17 | 1998-06-02 | John Hopkins University | Chemotherapy for cancer by inhibiting the fatty acid biosynthetic pathway |
| US5614551A (en) * | 1994-01-24 | 1997-03-25 | The Johns Hopkins University | Inhibitors of fatty acid synthesis as antimicrobial agents |
| US5981575A (en) * | 1996-11-15 | 1999-11-09 | Johns Hopkins University, The | Inhibition of fatty acid synthase as a means to reduce adipocyte mass |
-
2005
- 2005-05-25 WO PCT/US2005/018443 patent/WO2005117590A2/en active Application Filing
- 2005-05-25 CA CA002568639A patent/CA2568639A1/en not_active Abandoned
- 2005-05-25 AU AU2005249437A patent/AU2005249437A1/en not_active Abandoned
- 2005-05-25 EP EP05754100A patent/EP1758577A4/en not_active Withdrawn
- 2005-05-25 JP JP2007515318A patent/JP2008500363A/ja active Pending
- 2005-05-25 BR BRPI0510397-5A patent/BRPI0510397A/pt not_active IP Right Cessation
- 2005-05-25 MX MXPA06013687A patent/MXPA06013687A/es not_active Application Discontinuation
- 2005-05-25 KR KR1020067026941A patent/KR20070095754A/ko not_active Application Discontinuation
- 2005-05-25 ZA ZA200700024A patent/ZA200700024B/en unknown
- 2005-05-25 US US11/597,538 patent/US20090005435A1/en not_active Abandoned
- 2005-05-25 RU RU2006146051/04A patent/RU2006146051A/ru not_active Application Discontinuation
- 2005-05-25 CN CNA2005800233119A patent/CN101022792A/zh active Pending
-
2006
- 2006-11-23 IL IL179530A patent/IL179530A0/en unknown
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2005523270A (ja) * | 2002-02-08 | 2005-08-04 | ジョーンズ・ホプキンス・ユニバーシティ・スクール・オブ・メディシン | 体重を減少させる方法としてのcpt−1の促進 |
| WO2004005277A1 (en) * | 2002-07-09 | 2004-01-15 | Fasgen, Inc. | Novel compunds, pharmaceutical compositions containing same, and methods of use for same |
Also Published As
| Publication number | Publication date |
|---|---|
| BRPI0510397A (pt) | 2007-11-13 |
| ZA200700024B (en) | 2008-06-25 |
| MXPA06013687A (es) | 2007-10-18 |
| EP1758577A2 (en) | 2007-03-07 |
| RU2006146051A (ru) | 2008-07-10 |
| CN101022792A (zh) | 2007-08-22 |
| WO2005117590A3 (en) | 2006-07-27 |
| IL179530A0 (en) | 2007-05-15 |
| AU2005249437A1 (en) | 2005-12-15 |
| US20090005435A1 (en) | 2009-01-01 |
| EP1758577A4 (en) | 2010-05-05 |
| KR20070095754A (ko) | 2007-10-01 |
| CA2568639A1 (en) | 2005-12-15 |
| WO2005117590A2 (en) | 2005-12-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US7649012B2 (en) | Compounds, pharmaceutical compositions containing same, and methods of use for same | |
| JP2008500363A (ja) | 新規化合物、該化合物を含む医薬組成物、および該化合物の使用方法 | |
| JP2005533107A (ja) | 新規の化合物、それを含有する医薬組成物、およびその使用方法 | |
| JP2010509335A (ja) | 新規の化合物、それを含有する医薬組成物、及びその使用方法 | |
| US20100029752A1 (en) | Novel compounds, pharmaceutical compositions containing same, and methods of use for same | |
| US20100029761A1 (en) | Novel compounds, pharmaceutical compositions containing same, and methods of use for same |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20080522 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20080522 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20110823 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20120131 |