JP2006505562A - 持続放出組成物の放出プロフィールの調節方法 - Google Patents
持続放出組成物の放出プロフィールの調節方法 Download PDFInfo
- Publication number
- JP2006505562A JP2006505562A JP2004544829A JP2004544829A JP2006505562A JP 2006505562 A JP2006505562 A JP 2006505562A JP 2004544829 A JP2004544829 A JP 2004544829A JP 2004544829 A JP2004544829 A JP 2004544829A JP 2006505562 A JP2006505562 A JP 2006505562A
- Authority
- JP
- Japan
- Prior art keywords
- microparticles
- triamcinolone
- sustained release
- biologically active
- epo
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images


















Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
- A61K9/0024—Solid, semi-solid or solidifying implants, which are implanted or injected in body tissue
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/56—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids
- A61K31/57—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids substituted in position 17 beta by a chain of two carbon atoms, e.g. pregnane or progesterone
- A61K31/573—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids substituted in position 17 beta by a chain of two carbon atoms, e.g. pregnane or progesterone substituted in position 21, e.g. cortisone, dexamethasone, prednisone or aldosterone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1629—Organic macromolecular compounds
- A61K9/1641—Organic macromolecular compounds obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyethylene glycol, poloxamers
- A61K9/1647—Polyesters, e.g. poly(lactide-co-glycolide)
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1682—Processes
- A61K9/1694—Processes resulting in granules or microspheres of the matrix type containing more than 5% of excipient
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Abstract
Description
本出願は、2002年10月17日に提出された米国仮出願第60/419,430号の利益を主張する。
上記出願の教示は、その全体が参照により本明細書中に援用される。
多くの病気または症状には、予防または治療の最大効果を提供するため、一定レベルまたは持続レベルの医薬または生物学的に活性な薬剤の投与が必要とされる。これは、複数投薬養生法により、または持続様式で医薬を放出する系の使用により行なわれる。
本発明は、コルチコステロイドを同時投与すると、生体適合性ポリマーおよびその内部に組み込まれた生物学的に活性な不安定な薬剤(labile agent)を含有する持続放出組成物からの該生物学的に活性な不安定な薬剤の放出プロフィールが調節(modified)され得る、という意外な知見に基づくものである。放出プロフィールの調節は、カプセル化された生物学的に活性な不安定な薬剤の生物学的利用能の増大を生じる。
本発明の好ましい態様を以下に記載する。
動物
特に記載する以外は、雄性Sprague-Dawleyラット(体重350〜500グラム)(Charles River Laboratories, Inc.)を、少なくとも6日間の標準的な動物収容における順化後に本明細書に記載の試験に使用した。本明細書に記載のほとんどの試験では、動物は、少なくとも7日間、順化させた。
マイクロ粒子の投与前に免疫抑制した動物を、持続放出組成物の投与後第0〜14日(日曜以外)すなわち投与後第0〜6日および第8〜13日に毎日、およびその後は1週間に3回、5mg/kgのシクロスポリンを腹腔内投与することにより、シクロスポリン(Sandimmune, Sandoz; CS)で処置した。
マイクロ粒子とコルチコステロイドを含んだ、生物学的に活性で不安定な薬剤の投与は具体的な研究について下記に詳細に記載されている。
組み換えヒトエリトロポエチン(EPO)を含有するマイクロ粒子を、1998年2月10日にZaleらに発行された米国特許第5,716,644号(その全内容は参照により本明細書により援用される)に記載の手順に従って作製した。一般的には、EPO含有マイクロ粒子を、Cincinnati, OhioのAlkermes, Inc.製のカタログ番号5050DL2.5Aを有するポリマー[これは50:50のラクチド/グリコリド比、およびポリマー相中の賦形剤としてMg(OH)2を含むクロロホルム中で測定したとき0.24のIVを有するポリ(ラクチド−コ−グリコリド)25KDポリマーである]を用いて調製した。指示された場合は、それぞれの表示名目上の負荷(0.25%、2%、14%のヒドロコルチゾンと2%のトリアムシノロン)がもたらされた、ヒドロコルチゾンまたはトリアムシノロンアセトニド(両方ともSigmaより購入)をポリマー相に加えると、EPOをJohnson & Johnson, New Brunswick, New Jerseyから入手し、安定化した後、マイクロ粒子中に安定化したEPOの総重量の約1.6%w/wのEPO負荷を用い、米国特許第5,716,644号に記載されるように硫酸アンモニウムでカプセル化した。
本明細書に記載されたエキセンディン含有マイクロ粒子は、下記のコアセルベーション処理によって調製した。
コアセルベーション処理は、水-油-油(W/O/O)処理としても本明細書中にふれているが、水性の薬剤と有機ポリマー溶液を用いた油中水型エマルションの形成を要する。次いで、相分離を誘導させるためとポリマーを析出させるために、油(典型的にシリコーン油)を油中の水エマルションに加えた。初期のマイクロ粒子を、油とポリマー溶媒を取り除く溶媒中で失活した。
油中水型エマルションは超音波処理を使用して作製した。エマルションの水相は溶解したエキセンディン4や様々な賦形剤を水中に含んだ。典型的には賦形剤としてスクロースと硫酸アンモニウムが存在したが、他の賦形剤や賦形剤の組み合わせを調査した。PLG相には、塩化メチレンに溶解したポリマーが含まれた。
コアセルベーションは、胚マイクロ粒子が形成される。攪拌しながらシリコーン油を一定の割合で内部エマルションに加えることによって誘導され、形成された胚マイクロ粒子は比較的軟らかく、硬化を要する。
胚マイクロ粒子を、穏やかに攪拌しながらヘプタン/エタノール混合溶媒に加えた。混合溶媒は胚マイクロ粒子を硬化した。約3℃で約1時間硬化させた後、混合溶媒をデカントし、純ヘプタンを3℃で加え、約1時間混ぜた。
硬化段階の後、マイクロ粒子を乾燥チャンバの中の微細メッシュ細孔板に移して集めた。硬化用の容器の、最終ヘプタン洗浄が行われた。温度を約3℃から約38℃に上げていきつつ、4日間、窒素ガスによりマイクロ粒子を乾燥させた。
インスリン含有マイクロ粒子およびhFSH含有マイクロ粒子を、Herbertらに付与された米国特許第5,922,253号およびGombotzらに付与された米国特許第5,019,400号(両者の全教示は、参照により本明細書に援用される)に記載の方法にしたがって調製した。
該ポリマー溶液へのタンパク質凍結乾燥物の添加によるポリマー/タンパク質混合物の形成
ポリマー/タンパク質混合物の任意のホモジネーション
超音波処理または他の液滴形成手段によるポリマー/タンパク質混合物の霧化、および液体窒素との接触による液滴の凍結
ポリマー/タンパク質液滴から抽出溶媒(例えば、−80℃エタノール)中へのポリマー溶媒の抽出、それによるポリマー/タンパク質マトリックス含有粒子の形成
ろ過による抽出溶媒からの粒子の単離
蒸発による残留溶媒の除去
注射可能製品を提供するための粒子のサイズ調整。
インスリン含有マイクロ粒子を、Cincinnati, OhioのAlkermes, Inc.から購入したポリマー(カタログ番号5050DL2.5Aを有し、50:50のラクチド/グリコリド比およびクロロホルム中で測定したとき0.24のIVを有するポリ(ラクチド−コ−グリコリド)25 kDポリマーである)を用いて調製した。インスリンは、Sigma, St. Louis, MOから購入した組換えヒトインスリンであった。インスリンの名目上負荷は10%(実負荷5.8%)であった。
使用したポリマーは、Cincinnati, OHのAlkermes, Inc.から購入した。このポリマーは、50:50のラクチド;グリコリド比を有し、10 kDの分子量およびカルボン酸末端基を有するポリ(ラクチド−コ−グリコリド)である。
トリアムシノロンアセトニド含有マイクロ粒子(2%負荷)を、以下のようにして調製した。42 mgのトリアムシノロンアセトニドをベンジルアルコールに溶解した。次いで、トリアムシノロン溶液を、約24.3 mLの6% PLG(Cincinnati, OhioのAlkermes, Inc.から購入したポリマー(カタログ番号5050DL2.5Aを有し、50:50のラクチド/グリコリド比およびクロロホルム中で測定したとき0.24のIVを有するポリ(ラクチド−コ−グリコリド)25 kDポリマーである)塩化メチレン溶液に添加した。得られた均一な溶液を、5% PVAの攪拌溶液に添加した。エマルジョンの顕微鏡検査により液滴の直径が約150〜75ミクロンであることが示されるまで、攪拌速度を上げた。次いで、エマルジョンを、攪拌冷水にゆっくりと添加した。約45分の攪拌後、懸濁液を4℃で沈降させた。マイクロ粒子をろ過により回収し、冷水で洗浄し、凍結し、凍結乾燥により乾燥させた。
プラセボマイクロ粒子は、トリアムシノロンマイクロ粒子の調製方法にしたがい、トリアムシノロン非存在で調製した。
ヒドロコルチゾン含有マイクロ粒子は、トリアムシノロンマイクロ粒子について先に詳述した手順にしたがって、2%または20%負荷のいずれかで調製した。
ブデソニド含有マイクロ粒子は、トリアムシノロンマイクロ粒子について先に詳述した手順にしたがって調製し、2.0%または2.2%負荷を有した。
デキサメタゾン含有マイクロ粒子は、トリアムシノロンマイクロ粒子について先に詳述した手順にしたがって調製し、2%負荷を有した。
同時投与後のエリスロポエチン含有マイクロからのエリスロポエチン放出に対するヒドロコルチゾンまたはトリアムシノロンの薬理学的効果
酢酸ヒドロコルチゾンまたはトリアムシノロンジアセテートをインビボで雄性Sprague-Dawleyラットに同時投与した場合の、エリスロポエチン(EPO)含有マイクロ粒子から放出されたEPOに対する薬物動態学的(PK)/薬力学的(PD)応答を測定した。使用した動物の総数は、16匹であり、平均体重は、400〜450 gmであった。動物は、試験前に少なくとも6日間、順化させた。
ラットを、第0〜14日に毎日(日曜以外)、およびその後は1週間に3回、シクロスポリン(Sandimmune, Sandoz; CS)5mg/kg ipで免疫抑制した。第0日および第1日は、動物に、シクロスポリンとともに全身ヒドロコルチゾンを与えた。
動物を5%ハロタンで麻酔した。各動物を剃毛し、背中をアルコールで拭いた。以下の表1にしたがって、酢酸ヒドロコルチゾン(Sigma Fine Chemecals, カタログ番号86H1304)またはトリアムシノロンジアセテート(Sigma Fine Chemecals, カタログ番号127F0812)とともにあらかじめバイアルに封入したEPO含有マイクロ粒子を、0.75mLビヒクル(3%カルボキシメチルセルロース、0.1% Tween20、0.9% NaCl、pH約6)を用いて再懸濁した。マイクロ粒子は、上記のようにして調製した。マイクロ粒子を、1mL注射器に取り付けた21ゲージ薄壁針を用いて肩甲骨間部位に注射した。動物には、単独(A群)、あるいは合計5 mgの酢酸ヒドロコルチゾン(B群)または1 mg(C群)もしくは5 mg(D群)のトリアムシノロンジアセテートとの組み合わせのいずれかで合計10,000 UのEPOを与えるように投薬した。埋め込み後35日間動物を追跡した。

EPO血清レベルを評価するため、血清試料(400μL)を、マイクロ粒子投与に対して以下の日に尾静脈から採取した:前採血(pre-bleed)、第1、2、4、7、10、14、17、21、24、28、31および35日。凝固後、試料を遠心分離し、-70℃で凍結した。血清EPOレベルを、ELISA(Genzyme)により製造業者の指示書(カタログ番号80-3775-00)にしたがって定量した。
EPO血清レベル
ラットにヒドロコルチゾンまたはトリアムシノロンと同時投与したEPO含有マイクロ粒子からのEPOの放出の効果の結果を図1に示す。図1は、EPO含有マイクロ粒子、酢酸ヒドロコルチゾン(5 mg)と混合したEPO含有マイクロ粒子、またはトリアムシノロンジアセテート(1 mgまたは5 mg)と混合したEPO含有マイクロ粒子を投与したラットにおける血清EPOレベル(mU/ml)の時間(日)に対するグラフである。図1に示すように、約10,000mU/mL付近またはそれ以上の初期ピーク後、血清EPOレベルは、第17日まで一定に減少した。第24日までには、処置群の明白な分類が観察された。EPO単独処置群(A群)は、39.7 mU/mL ± 32.66 mU/mLに低下した。EPO含有マイクロ粒子に加えて第2の薬剤を与えた群は、210 ± 32.66 mU/mL(B群)、127.53 ± 66.7 mU/mL(C群)および302.3 ± 90.5 mU/mL(D群)で、より高い血清レベルを示した。第35日にA群およびC群は、検出限界未満に低下したが、5 mgヒドロコルチゾン(B群)または5 mgトリアムシノロン(D群)のいずれかを与えた2群は、なお、それぞれ241.5 ± 43.9 mU/mLおよび433.18 ± 177.37 mU/mLの血清EPOレベルを有した。
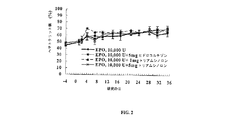
EPO含有マイクロ粒子を投与、またはEPO含有マイクロ粒子とヒドロコルチゾンもしくはトリアムシノロンを同時投与したラットのヘマトクリット試験の結果を図2に示す。図2は、EPO含有マイクロ粒子、酢酸ヒドロコルチゾン(5 mg)と混合したEPO含有マイクロ粒子、またはトリアムシノロンジアセテート(1 mgまたは5 mg)と混合したEPO含有マイクロ粒子を投与したラットにおけるヘマトクリット値(%)の時間(日)に対するグラフである。ヘマトクリット値は、すべての群で、試験初期では一定に増加し、第24日までに安定状態に達し、このとき、すべての動物は、60%を超えるヘマトクリット値を有した。ヘマトクリット値に関して群間に有意差はなかったが、値は、第36日に、EPOのみを与えた動物(A群動物)において減少したようであった。これは、EPO含有マイクロ粒子単独を投与した動物(A群)およびEPO含有マイクロ粒子+1 mg トリアムシノロンを投与した動物(C群)においてヘマトクリット値が60%半ばに減少したが、より高レベルのヒドロコルチゾン(B群)またはトリアムシノロン(D群)を与えた群は、なお70%以上であるヘマトクリット値を有したという事実により証明された。
高用量のトリアムシノロンジアセテートで処置したラット(5 mg; D群)において、第35日での検死において見られた残留ポリマーの量は、ヒドロコルチゾン(5 mg; B群)または低用量(1 mg)のトリアムシノロンジアセテート(C群)を投与した動物におけるよりも多かった。マイクロスフェアデポット(depot)を重壘(overlying)した皮膚の色は、BおよびD群のほとんどのラットにおいて青白かった。また、EPO含有マイクロ粒子とヒドロコルチゾンまたはトリアムシノロンジアセテートとの同時投与は、皮下組織におけるマイクロスフェアデポットの周囲の線維増多の量を減少させ、マイクロスフェア塊内で通常起こる肉芽腫炎症反応の強度を低下した。
共カプセル化したEPOとヒドロコルチゾンとを含有するマイクロ粒子の投与およびヒドロコルチゾンと同時投与したEPO含有マイクロ粒子
免疫不全ヌードラット(Tac: N: NIH-rnufDF, 体重範囲: 350〜450 gm)への、共カプセル化したEPOと種々のレベル(0, 0.25, 2および14%)のヒドロコルチゾンとを含有するマイクロ粒子、およびヒドロコルチゾンと同時投与したEPO含有マイクロ粒子の投与の薬力学的および薬物動態学的効果を調べた。
EPO含有マイクロ粒子を上記の手順にしたがって調製した。0.25%、2%および14%[%は、名目上ヒドロコルチゾン負荷(w/w)をいう]で共カプセル化したヒドロコルチゾンとEPOとを含有するマイクロ粒子は、上記のようにして調製した。同時投与したヒドロコルチゾンは、Sigma, St. Louis, MOから購入した。
マイクロ粒子を、実施例1に記載のようにして投与し、表2に概要を示す。動物には、合計10,000単位のEPO含有マイクロ粒子(1群)、または0.25%ヒドロコルチゾン(2群)、2%ヒドロコルチゾン(3群)、14%ヒドロコルチゾン(4群)と共カプセル化した10,000単位のEPOまたは5 mgヒドロコルチゾンと同時投与した合計10,000単位のEPOを与えるように投薬した。未処理群(6群)もまた本試験に含めた。試料回収時間点は、前採血、第1、5、8、12、15、19、22、26、29、34、41、48および55日とした。
EPO血清レベルを評価するため、血清試料(400μL)を、表2に示した日に尾静脈から採取した。凝固後、試料を約5分間約13000 rpmで遠心分離し、-70℃で凍結した。血清EPOレベルを、ELISA(Genzyme)により製造業者の指示書(カタログ番号80-3775-00)にしたがって定量した。
EPO血清レベル
ヒドロコルチゾンと共カプセル化または同時投与したEPO含有マイクロ粒子からのEPOの放出の効果の結果を図3Aに示す。図3Aは、種々のレベルのヒドロコルチゾンと共カプセル化したEPOを含有するマイクロ粒子、および酢酸ヒドロコルチゾン(5 mg)と混合したEPO含有マイクロ粒子を投与したラットにおける血清EPOレベル(mU/ml)の時間(日)に対するグラフである。図3Aに示すように、共カプセル化または同時投与のいずれかでヒドロコルチゾンを与えたすべての処置群は、循環EPO血清レベルの増加を示した。より具体的には、EPO単独処置ラットでは、血清EPOレベルは第26日に非検出可能レベルまで減少したが、0.25%のヒドロコルチゾンを与えた低用量群では第34日まで非検出可能限界に達しなかった。持続期間における用量依存的増加が見られ、このとき、それぞれ2%および14%ヒドロコルチゾンを用いた両群は、第41日に10 mU/mLの血清EPOレベルを有した。
ヒドロコルチゾンと共カプセル化または同時投与したEPO含有マイクロ粒子からのEPOの放出の効果の結果を図3Bに示す。図3Bは、表2の群について、ラットにおけるヘマトクリット値(%)の時間(日)に対するグラフである。このグラフは、試験全体を通して未処置動物ではヘマトクリットが低いまま(45〜50%)であったが、処置ラットでは、60〜70%に達するヘマトクリット値が得られたことを示す。EPOのみを与えたラットにおけるヘマトクリットのベースラインへの戻りが第38日に観察されたが、ヒドロコルチゾンと共カプセル化したEPOを与えた群はすべて、少なくとも第56日までベースラインに戻らなかった。
ヒドロコルチゾン含有マイクロ粒子と同時投与したか、またはトリアムシノロンアセトニドと混合したEPO含有マイクロ粒子
プラセボマイクロ粒子、ヒドロコルチゾン含有マイクロ粒子、またはトリアムシノロンアセトニドと混合したプラセボマイクロ粒子と混合したEPO含有マイクロ粒子のラットへの投与の薬力学的および薬物動態学的効果、ならびにかかる投与の免疫原性を調べた。
EPO含有マイクロ粒子を先に概要を示した手順にしたがって調製した。ヒドロコルチゾン含有マイクロ粒子は、上記の手順にしたがって調製した。プラセボマイクロ粒子は、先に概要を示した手順にしたがって調製した。
マイクロ粒子の投与は、実施例1に記載の通りとし、表3に概要を示す。動物に、合計100 mgのプラセボマイクロ粒子(A群)、5 mgのトリアムシノロンアセトニドと100 mgのプラセボマイクロ粒子との混合物(B群)、および100 mgの20% w/wヒドロコルチゾン含有マイクロ粒子(C群)との組み合わせで合計10,000単位EPOを与えるように投薬した。試料回収時間点は、前採血、第1、2、6、12、19、および26日とした。
EPO血清レベルを評価するため、0.4 mL試料を、表3に示した日に尾静脈から採取した(1群あたり4匹の動物)。凝固後、試料を遠心分離し、凍結した(-70℃)。血清EPOレベルを、ELISA(R&D Systems)により製造業者の指示書(カタログ番号 DEPOO)にしたがって定量し、データを用量および体重に対して正規化した。第12日から開始し、EPO抗体レベルについてもまた、ELISAを用いて毎週評価した。検出する抗体は免疫グロブリンの重鎖(γ)および軽鎖の両方と反応性であるため、このアッセイにより、すべての抗体サブクラスが検出される。ヘマトクリットアッセイ解析を、実施例1に記載のようにして行ない、マイクロ粒子投与の時間に対して以下の間隔:前採血、第1、2、6、12、19、および26日で試験した。
血清EPOレベル
プラセボマイクロ粒子、プラセボマイクロ粒子と混合したトリアムシノロンアセトニド、ヒドロコルチゾン含有マイクロ粒子と混合したEPO含有マイクロ粒子からのEPOの放出を図4に示す。図4は、上記製剤の各々を投与したラットにおける血清EPOレベル(mU/mL)の時間(日)に対するグラフである。図4に示すように、血清EPOレベルは、対照群(EPO含有マイクロ粒子およびプラセボマイクロ粒子を投与した動物;A群)において急激に低下し、第12日以降は、EPOは検出されなかった。
EPO含有マイクロ粒子+プラセボマイクロ粒子、プラセボマイクロ粒子と混合したトリアムシノロンアセトニド、およびヒドロコルチゾン含有マイクロ粒子の投与のヘマトクリット値に対する結果を図5に示す。図5は、上記製剤の各々を投与したラットにおけるヘマトクリット値(%)の時間(日)に対するグラフである。図5は、全試験での群の平均ヘマトクリットを示す。EPO含有マイクロ粒子+プラセボマイクロ粒子を与えた対照群(A群)でのヘマトクリットは、第0日〜第6日まで正常に増加した。しかしながら、第6日以降は、第6日の60.6% ± 3.11%から第33日の47.0% ± 3.56%まで、ヘマトクリット値において一定の低下があった。EPO含有マイクロ粒子+コルチコステロイドを投与した群の動物は、第12日までに対照群よりも有意に高いヘマトクリットレベルに達した。プラセボマイクロ粒子と混合したトリアムシノロンアセトニドと同時投与したEPOマイクロ粒子は、第19日に、対照(56.0% ± 6.68)よりも有意に高い(p<.05)極大の69.3% ± 3.3%までのヘマトクリット値を誘導した。20%ヒドロコルチゾンを負荷したEPO含有マイクロ粒子(C群)もまた、70.5% ± 1.91%(p<.05)の高ヘマトクリットの維持を補助した。このため、プラセボマイクロ粒子とのヒドロコルチゾンマイクロ粒子および混合されたトリアムシノロンアセトニドの投与は、同等の薬力学効果を有した。
EPO含有マイクロ粒子から放出されたEPOにより惹起された免疫応答、および第2の薬剤の抗体産生に対する影響を評価するため、血清を、抗EPO抗体の存在および力価について、ELISAにより試験した。この評価の結果を図6A、6Bおよび6C(それぞれ、第12、19および33日にアッセイ)に示す。発生のパーセント対幾何平均力価を図6A、6Bおよび6Cに示す。これらは、投与後、第12日(図6A)、第19日(図6B)および第33日(図6C)において、合計100 mgのプラセボマイクロ粒子との組み合わせで合計10,000単位のEPOを投与したラットの血清において検出されたEPOに対する抗体発生(力価)のグラフである。
デキサメタゾン含有マイクロ粒子、ブデソニド含有マイクロ粒子、およびトリアムシノロンアセトニド含有マイクロ粒子と同時投与したEPO含有マイクロ粒子
プラセボマイクロ粒子、トリアムシノロンアセトニド含有マイクロ粒子、デキサメタゾン含有マイクロ粒子およびブデソニド含有マイクロ粒子と混合したEPO含有マイクロ粒子のラットへの投与の薬力学的および薬物動態学的効果を調べた。
EPO含有マイクロ粒子を上記の手順にしたがって調製した。デキサメタゾン含有マイクロ粒子、ブデソニド含有マイクロ粒子およびトリアムシノロンアセトニド含有マイクロ粒子は、上記のようにして調製した。プラセボマイクロ粒子は、先に概要を示した手順にしたがって調製した。
ラットを、14日間、毎日(日曜以外)、およびその後3回/週で5mg/kgのシクロスポリン(Sandimmune, Sandoz; CS)のi.p.のみの投与により免疫抑制した。
マイクロ粒子の投与は、実施例1に記載の通りとし、表4に概要を示す。動物に、表4に記載のカプセル化コルチコステロイドとの組み合わせで合計10,000単位のEPOを与えるように投薬した。試料回収時間点は、前採血、第1、2、5、8、12、15、19、22、26、29、33および36日とした。
EPO血清レベルを評価するため、0.4 mL試料を、表4に示した日に尾静脈から採取した(1群あたり4匹の動物)。凝固後、試料を遠心分離し、凍結した(-80℃)。血清EPOレベルを、ELISA(R&D Systems)により製造業者の指示書(カタログ番号 DEPOO)にしたがって定量し、データを用量および体重に対して正規化した。
血清EPOレベル
プラセボマイクロ粒子、デキサメタゾン含有マイクロ粒子、ブデソニド含有マイクロ粒子、およびトリアムシノロンアセトニド含有マイクロ粒子と混合したEPO含有マイクロ粒子からのEPOの放出を図7Aに示す。図7Aは、上記製剤の各々を投与したラットにおける血清EPOレベル(mU/mL)の時間(日)に対するグラフである。図7Aに示すように、EPO含有マイクロ粒子とともにトリアムシノロンアセトニド含有マイクロ粒子、デキサメタゾン含有マイクロ粒子およびブデソニド含有マイクロ粒子を同時投与した結果としての生物学的利用能の有意な改善が認められ、放出の持続期間の顕著な延長を伴う。例えば、EPO含有マイクロ粒子と同時投与したトリアムシノロンアセトニドマイクロ粒子で処置した群は、放出の持続期間および定常状態の値に関して対照(プラセボ)と最も大きな差を有する。この試験は、第28日で終了し、その時点で、トリアムシノロン処置動物では、なお検出可能な血清EPOレベル(>12.5 mIU/mL)があった。定常状態(第7〜25日)値は、対照と比べ、この群において、60.36 mIU/mL ± 7.7 mIU/mL対(対照において)19.45 mIU/mL ± 5.28 mIU/mL(p<0.001)と、有意に多かった。また、デキサメタゾンおよびブデソニドの両方は、対照に比べ、55.2 ± 10.7 mIU/mLおよび43.7 ± 9.8 mIU/mL(p<0.01)と、有意に高い定常状態値(第7〜25日)を有した。
プラセボマイクロ粒子、デキサメタゾン含有マイクロ粒子、ブデソニド含有マイクロ粒子、およびトリアムシノロンアセトニド含有マイクロ粒子と混合した
EPO含有マイクロ粒子の投与のヘマトクリット値に対する結果を図7Bに示す。図7Bは、上記製剤の各々を投与したラットにおけるヘマトクリット値(%)の時間(日)に対するグラフである。すべての群は、最大ヘマトクリットの時点において対照よりも有意に高かった。例えば、第11日までに、プラセボにおけるヘマトクリットは、その最大67 ± 2.2%に達した。しかしながら、トリアムシノロンアセトニドは、第21日に72.5 ± 4.4の極大ヘマトクリット応答を誘導した。また、この群でデキサメタゾンは、ヘマトクリットを増加させることが認められ、この群の平均は第14日において最高の74.3 ± 2.6%であった。ブデソニドもまた、ヘマトクリットを増加させることが認められ、第11日において76.8% ± 2.5%であった。
種々の用量でブデソニド含有マイクロ粒子およびトリアムシノロンアセトニド含有マイクロ粒子と同時投与したEPO含有マイクロ粒子および共カプセル化
プラセボマイクロ粒子、トリアムシノロンアセトニド含有マイクロ粒子およびブデソニド含有マイクロ粒子と混合したEPO含有マイクロ粒子、ならびに共カプセル化したEPOおよびトリアムシノロンを有するマイクロ粒子のラットへの投与の薬力学的および薬物動態学的効果を調べた。
EPO含有マイクロ粒子を上記の手順にしたがって調製した。ブデソニド含有マイクロ粒子およびトリアムシノロン含有マイクロ粒子は、上記のようにして調製した。プラセボマイクロ粒子は、上記の手順にしたがって調製した。共カプセル化したEPOおよびトリアムシノロンを有するマイクロ粒子は、上記のようにして調製した。
ラットを、14日間、毎日(日曜以外)、およびその後3回/週で5mg/kgのシクロスポリン(Sandimmune, Sandoz; CS)をi.p.のみで投与することにより免疫抑制した。
マイクロ粒子の投与は、実施例1に記載の通りとし、表5に概要を示す。動物に、表5に記載のトリアムシノロンアセトニドと共カプセル化したか、または別個にカプセル化したコルチコステロイドとの組み合わせで、合計10,000単位EPOを与えるように投薬した。試料回収時間点は、前採血、第1、2、5、8、12、15、19、22、26、29、33および36日とした。
EPO血清レベルを評価するために、1〜7日目まで尾静脈から0.4mL試料を収集し、表5に示される残りの日に0.5mLの試料を収集した(群当たり4匹)。凝固後、試料を遠心分離し、凍結させた(-80℃)。血清EPOレベルをELISA(R&D Systems)により製造業者の指示書(Cat.No.DEP00)に従って定量し、データを投薬量および体重について標準化した。
血清EPOレベル
プラセボマイクロ粒子、トリアムシノロンアセトニド含有マイクロ粒子、およびブデソニド含有マイクロ粒子と混合したEPO含有マイクロ粒子ならびに共カプセル化されたEPOおよびトリアムシノロンアセトニドを有するマイクロ粒子からのEPOの放出を図8Aに示す。これは、時間(日)に対する上記の製剤の各々を投与したラットにおける血清EPOレベル(mU/mL)の経過のグラフである。図8Aに示されるように、ブデソニド処理およびトリアムシノロン処理した動物は両方ともEPOの放出期間の延長を示した。例えば、25mgおよび50mgのブデソニド群ならびに20mgトリアムシノロン群は両方とも、29日目の研究の終わりまで検出可能な血清レベルのEPOを有した。このとき、トリアムシノロン処理動物におけるEPOの検出可能な血清レベルは、>14.0mIU/mLであり、ブデソニド群(25mgおよび50mg)は、13.3mIU/mLおよび13.4mIU/mLそれぞれのレベルを有した。全ての処理群は、定常状態の血清レベル(5〜22日目)において有意な増大を示し、破裂後(5〜33日目)AUC(濃度曲線下面積)は有意に増大した。
プラセボマイクロ粒子、トリアムシノロン含有マイクロ粒子、およびブデソニド含有マイクロ粒子と混合したEPO含有マイクロ粒子ならびに共カプセル化されたEPOおよびトリアムシノロンを有するマイクロ粒子の投与の結果を図8Bに示す。これは時間(日)に対する上記の製剤の各々を投与したラットにおけるヘマトクリット値(%)のグラフである。図8Bは、トリアムシノロンおよびブデソニド群が両方とも類似した経過でパックされた赤血球体積を上昇させたことを示す。
小胞刺激ホルモン含有マイクロ粒子からの小胞刺激ホルモンの放出に対する第2薬剤含有マイクロ粒子の局所送達の効果
ヒドロコルチゾン含有マイクロ粒子またはトリアムシノロンアセトニド含有マイクロ粒子と共に雄Sprague-Dawleyラットにインビボで同時投与されたときのhFSH含有マイクロ粒子から放出されたヒト小胞刺激ホルモン(hFSH)に対する薬物動態学的応答が測定された。
ヒトFSH含有マイクロ粒子を上記の手順に従って調製した。ヒドロコルチゾン含有マイクロ粒子を上記の手順に従って調製した。トリアムシノロン含有マイクロ粒子を上記のように調製した。プラセボマイクロ粒子を上記の手順に従って調製した。
マイクロ粒子投与および試料収集を実施例1に記載のように行った。処置群を表6に要約する。動物を、全75mgのプラセボマイクロ粒子(群A)、10mgの2%w/wトリアムシノロンマイクロ粒子(群B)、または15mgの2%w/wヒドロコルチゾン含有マイクロ粒子(群C)と組み合わせた全15mgのhFSH含有マイクロ粒子を投与した。本研究のラットをシクロスポリン(Sandimmune, Sandoz; CS)、5mg/kgのみのipで毎日14日間、その後週に3回免疫抑制した。試料収集時点は、前採血(pre-bleed)、6時間、12時間、1、2、4、7、10、14、17、21、24、28、31、35および38日目であった。
血清HFSHレベル
血清hFSHレベルを測定するために、0.4mLの血清を表6に示された日に尾静脈から収集した(グループ当たり4匹の動物)。凝固後、試料を遠心分離し、凍結させた(-70℃)。血清hFSHレベルを製造業者の指示に従ってELISAにより定量した(American Research Products; Cat.No.P-2035)。
血清HFSHレベル
プラセボマイクロ粒子ヒドロコルチゾン含有マイクロ粒子またはトリアムシノロンアセトニド含有マイクロ粒子のいずれかと共に同時投与されたhFSH含有マイクロ粒子の投与後、血清試料を表6に示されるように収集し、製造業者の指示(American Research Products; Cat.No.P-2035)に従って血清hFSHレベルについてELISAにより試験した。図9は時間(日)に対する、全75mgのプラセボマイクロ粒子、10mgの2%w/wトリアムシノロンアセトニドマイクロ粒子、または15mgの2%w/wヒドロコルチゾン含有マイクロ粒子と組み合わせたhFSH含有粒子を投与したラットにおける血清hFSHレベル(mIU/mL)のグラフの形態で研究の経過の間の各群の薬物動態学的プロフィールを示す。図9に示されるように、hFSHの血清レベルにおける破裂相の間には有意な差異はなく、140.8±35.2mIU/mL〜200.3±35.3mIU/mLの範囲のCmax値を有した。hFSH放出プロフィールは、全ての群で二相曲線を示し、血清レベルは4日目まで減少し、10日目に再びピークに増大した。ヒドロコルチゾン含有マイクロ粒子で処理したラット(群C)におけるhFSHの10日目の血清レベルは、114.1±18.9mIU/mLで最も高かったが、このレベルは、プラセボマイクロ粒子を与えたラット(群A)におけるレベルと有意には異なっていなかった(69.0±20.1mIU/mL)。21日目までに、ヒドロコルチゾン処理動物における血清hFSHレベル、群Cは、検出限界未満に低下した。対照群Aは、21日目までに1.3±2.6mIU/mLの血清レベルを有し、また24日目までに検出可能なレベル未満になった。
小胞刺激ホルモン含有マイクロ粒子からの小胞刺激ホルモンの放出における第2薬剤含有マイクロ粒子の局所送達の効果
Sprague-Dawleyラットにトリアムシノロンアセトニド含有マイクロ粒子と共にインビボで同時投与したときのhFSH含有マイクロ粒子から放出されるヒト小胞刺激ホルモン(hFSH)に対する薬物動態応答を測定した。
ヒトFSH含有マイクロ粒子を上記の手順に従って調製した。トリアムシノロンアセトニド含有マイクロ粒子を上記のように調製した。プラセボマイクロ粒子を上記の手順に従って調製した。
マイクロ粒子投与および試料収集を実施例1に記載のように行った。処理群を表7に要約した。全100mgのプラセボマイクロ粒子(群A)、ならびに10mgの2%w/wトリアムシノロンマイクロ粒子および90mgのプラセボマイクロ粒子(群B)と組み合わせて全15mgのhFSH含有マイクロ粒子を動物に与えた。本研究のラットをシクロスポリン(Sandimmune, Sandoz; CS)で毎日5mg/kgのみのip(日曜日を除く)を14日間、その後、週当たり3回免疫抑制した。試料収集時点は、前採血、6時間、12時間、および1、2、4、7、10、14、17、21、23、27および30日であった。
血清HFSHレベル
血清hFSHレベルを測定するために、0.4mLの血清を、表7に示される日に尾静脈から収集した(群当たり4匹の動物)。凝固後、試料を遠心分離し、凍結させた(-70℃)。血清hFSHレベルを製造業者の指示(American Research Products; Cat.No.P-2035)に従ってELISAにより定量した。
血清HFSHレベル
プラセボマイクロ粒子、またはトリアムシノロンアセトニド含有マイクロ粒子+プラセボと共に同時投与したhFSH含有マイクロ粒子の投与後に血清試料を表7に示されるように収集し、製造業者の指示(American Research Products; Cat.No.P-2035)に従って血清hFSHレベルについてELISAにより試験した。図10は、時間(日)に対する、全100mgのプラセボマイクロ粒子または10mgの2%w/wトリアムシノロンアセトニドマイクロ粒子および90mgのプラセボマイクロ粒子と組み合わせてhFSH含有マイクロ粒子を投与したラットにおける血清hFSHレベル(mIU/mL)のグラフの形態で研究の過程の間の各群の薬物動態学的プロフィールを示す。図10に示されるように、トリアムシノロンアセトニド処理動物は、6時間から3日目の時点まで群A(FSHマイクロ粒子単独)と比べて血清FSHレベルにおいて有意な減少を示した。例えば、10時間の時点で、群Aの血清FSHレベルは218.3±56.6mIU/mLであり、一方、トリアムシノロンアセトニン処理群ではわずか102.2±17.6mIU/mLであった。さらに、トリアムシノロン処理群の全体の放出プロフィールは、20日目に対照群と比べて血清FSHレベルにおいて有意な増大を示した。
インスリン含有マイクロ粒子からのインスリンの放出に対する第2薬剤の局所送達の効果
雄Sprague-Dawleyラットに投与したインスリン含有マイクロ粒子の薬物動態学的プロフィールに対するヒドロコルチゾンおよびトリアムシノロンアセトニドの効果を評価した。
インスリン含有マイクロ粒子を上記のように調製した。トリアムシノロンアセトニド含有マイクロ粒子を上記のように調製した。酢酸ヒドロコルチゾン含有マイクロ粒子を上記のようにして調製した。
マイクロ粒子投与を実施例1に記載のように行い、処理群を表8に要約した。60mgのインスリン含有マイクロ粒子+75mgのプラセボ(群A)、10mgの2%w/wトリアムシノロンアセトニド含有マイクロ粒子(群B)および15mgの2%w/wヒドロコルチゾン含有マイクロ粒子(群C)をラットに投与した。本研究のラットをシクロスポリン(Sandimmune, Sandoz; CS)で毎日5mg/kgのみのipで14日間(日曜日を除く)、その後週に3回免疫抑制した。試料収集時点は、前採血、6時間、12時間、ならびに1、2、4、7、10、14、17、21、24、28、31、35および38日であった。
血清インスリンレベルを評価するために、0.4mL試料の血清を表8に示される日に尾静脈から収集した(群当たり4匹の動物)。凝固後、試料を遠心分離し、アリコートに分け(3セット、54μL各チューブ)、凍結させた(-80℃)。血清インスリンレベルを製造業者の指示(Cat.No.008-10-1132-01)に従ってELISA(ALPCO)により定量した。
製造業者により記載されたようにQiagen RNeasyキットを用いてミクロスフェアベッドからRNAを抽出した。精製RNAを用いて、製造業者により記載されるようにPromegaのReverse Transcriptaseキットを使用してcDNAを合成した。オステオポンチンcDNAをリアルタイムポリメラーゼ連鎖反応およびOligos Etc., Wilsonville, ORから入手したオステオポンチン特異的プライマーを用いて試料中で測定した。オステオポンチンmRNAコピー数をGAPDH mRNAレベルに標準化した。
血清インスリンレベル
プラセボマイクロ粒子、ヒドロコルチゾンまたはトリアムシノロンアセトニド含有マイクロ粒子と共に同時投与したインスリン含有マイクロ粒子の投与後、血清試料を表8に示されるように収集し、ELISA(ALPCO Ultrasensitive Insulin)により血清インスリンレベルについて試験した。図11は、時間(日)に対する、60mgのインスリン含有マイクロ粒子+75mgのプラセボ、10mgの2%w/wトリアムシノロンアセトニド含有マイクロ粒子または15mgの2%w/wヒドロコルチゾン含有マイクロ粒子を投与したラットにおける血清インスリンレベル(mIU/mL)のグラフの形態で研究の過程の間の各群に関する薬物動態学的プロフィールを示す。図11に示されるように、インスリンの血清レベルにおける破裂相の間には対象と比較して処理群に有意な差異はなかった。インスリン放出プロフィールは、全ての群で安定な放出曲線を示し、2日目までに血清レベルは低下し、約17日までわずかに増加した。
生物学的利用能に関して、インスリンマイクロ粒子(群B)と共に同時投与されたトリアムシノロンマイクロ粒子を与えた群は、2045.0±620.3mIU/mLで最も高い濃度曲線下面積を有し(表5)、これは表9に示されたように1021.3±396.7mIU/mLの対照動物(群A)よりも有意に高かった(p=0.05)。破裂後のAUC(2〜35日)は、対照における614.6±213.9mIU/mLと比べて1744.8±582.4mIU/mLでトリアムシノロンアセトニド処理ラットが最も高く、破裂後のAUCにおけるこの差異は有意であった(p=0.05)。さらに、2〜38日間の平均血清インスリンレベルは、対照と比べてトリアムシノロンアセトニド処理動物において高く(対照群16.9±5.9、トリアムシノロン群48.0±16.4)、対照とは有意に異なっていた(p<0.05)。これらのデータは、トリアムシノロンがマイクロ粒子からのインスリン放出の生物学的利用能を増大させることを示す。
RT-PCR
注射の14日後にミクロスフェアベッドから抽出されたオステオポンチンmRNAのレベルを、リアルタイムトランスクリプターゼPCRおよびOligs Etc. Wilsonville, ORから入手したオステオポンチン特異的マーカーにより測定した。リアルタイムリバーストランスクリプターゼ分析の結果を図12に示す。これは、60mgのインスリン含有マイクロ粒子+75mgのプラセボ(プラセボ)、10mgの2%w/wトリアムシノロンアセトニド含有マイクロ粒子(トリアムシノロン)または15mgの2%w/wヒドロコルチゾン含有マイクロ粒子(ヒドロコルチゾン)を投与したラットにおける投与の14日後のプラセボオステオポンチンmRNA発現レベル(コピー数/50ng cDNA)のヒストグラムである。図12に示されるように、トリアムシノロンアセトニド含有マイクロ粒子とインスリン含有マイクロ粒子との同時注射は、プラセボマイクロ粒子対照よりも93%低いオステオポンチンmRNAレベルに対して最も劇的な効果を有した。ヒドロコルチゾン含有マイクロ粒子は、対照と比べてオステオポンチンmRNAレベルを73%抑制した。
注射部位の免疫組織化学分析もまた行った。これらの研究は、インスリン含有マイクロ粒子と共に同時投与されたトリアムシノロンマイクロ粒子が、注射後14日目にインスリン含有マイクロ粒子へのマクロファージ、単球、およびT細胞の浸潤を劇的に減少させることを示した。ヒドロコルチゾンマイクロ粒子はまた、炎症細胞補充を減少させるが、それらの効果は、トリアムシノロンマイクロ粒子よりも低い。
プラセボマイクロ粒子、またはトリアムシノロンアセトニドまたはヒドロコルチゾン含有マイクロ粒子と共に雄Sprague-Dawleyラットに同時投与したインスリン含有粒子からのインスリンの放出、および注射部位での種々のサイトカインの発現に対する効果を測定した。
インスリン含有マイクロ粒子を上記のように調製した。トリアムシノロンアセトニド含有マイクロ粒子および酢酸ヒドロコルチゾン含有粒子を実施例8に記載されるように調製した。プラセボマイクロ粒子は実施例8で使用したものと同じであった。本研究で使用したラットは、実施例8に記載されるようにシクロスポリンを用いて免疫抑制した。
マイクロ粒子投与、試料収集および分析は、実施例8に記載したとおりであり、表10に要約した。60mgのインスリン含有マイクロ粒子+25mgのプラセボ(群A)、10mgの2%w/wトリアムシノロンアセトニド含有マイクロ粒子(群B)または15mgの2%w/wヒドロコルチゾン含有マイクロ粒子(群C)の投薬量をラットに投与した。試料収集時点は、前採血、6時間、12時間、ならびに1、2、4、7、14、21、28、および35日であった。
血清インスリンレベル
血清インスリンレベルを測定するために、マイクロ粒子投与に関して以下の日に血清試料(400μL)を尾静脈から収集した:前採血、1、2、4、7、10、14、17、21、28、31および35。凝固後、試料を実施例8に記載されるように凍結のために調製し、血清インスリンレベルを実施例8に記載されるように定量した。
RNAを製造業者により記載されるようにQiagen RNeasyキットを用いてミクロスフェアベッドから抽出した。製造業者により記載されるようにPromegaのReverse Transcriptaseキットを使用して、精製RNAを用いてcDNAを作製した。オステオポンチンcDNAを、Oligos Etc. Wilsonville, ORから入手したリアルタイムポリメラーゼ連鎖反応およびオステオポンチン特異的プライマーを用いて試料中で測定した。オステオポンチンmRNAコピー数をGAPDH mRNAレベルに標準化した。炎症前ケモカイン発現をBioSourceのChemokine Panel AおよびB PCRキットを用いて可視化した。種々のケモカインの発現をエチジウムブロミド含有2%アガロースゲル上で可視化した。
血清インスリンレベル
図13は、血清インスリンレベルに対する、プラセボマイクロ粒子、トリアムシノロンアセトニド含有マイクロ粒子またはヒドロコルチゾン含有マイクロ粒子と共に同時投与されたインスリン含有マイクロ粒子の効果の結果を示す。図13に示されるように、群A動物(インスリン含有マイクロ粒子+プラセボマイクロ粒子を投与された)は、31日後に検出可能な血清インスリンを有さない最も短い薬物動態学的プロフィールを示した。群B動物(インスリン含有マイクロ粒子+トリアムシノロンアセトニド含有マイクロ粒子)は、2日目から研究の終わり(35日目)(このとき、インスリンは血清中でまだ測定可能であった)まで最も高いインスリンのレベルを示した。第2薬剤の存在はまた、インスリン含有マイクロ粒子+トリアムシノロンアセトニドおよびヒドロコルチゾン含有マイクロ粒子、それぞれを投与した群について149.6%および38.07%でプラセボ処理群と比べて破裂後のAUCをも増大させた。
いくつかの炎症前サイトカインのmRNAレベルの分析は、オステオポンチン、RANTES、MIP-1α、MIP-1β、MCP-1、およびMIP-2を含む多数の炎症前化学走化性因子のmRNAの存在を示した。オステオポンチンmRNAレベルを定量し、注射の7日後、図14に示されるようにプラセボ群において最も高いことが見出された。これは投与後7日目および35日目に60mgのインスリン含有マイクロ粒子+25mgのプラセボ(プラセボ)、10mgの2%w/wトリアムシノロンアセトニド含有マイクロ粒子(トリアムシノロン)または15mgの2%w/wヒドロコルチゾン含有マイクロ粒子(ヒドロコルチゾン)を投与したラットにおけるオステオポンチンmRNA発現レベル(コピー数/50ng cDNA)のヒストグラムである。インスリン含有マイクロ粒子+トリアムシノロンアセトニド含有マイクロ粒子を投与した動物群は、プラセボよりも200倍低いオステオポンチンmRNA転写物を有し、インスリン含有マイクロ粒子+ヒドロコルチゾン含有マイクロ粒子を投与した群は、プラセボ群よりも約2分の1多いオステオポンチン転写物を示した。35日目に、オステオポンチンmRNAのレベルは全ての群で低かった。
プラセボマイクロ粒子、またはトリアムシノロン含有マイクロ粒子と共に雄Sprague-Dawleyラットに同時投与されたエキセンディン含有マイクロ粒子の投与後のエキセンディン放出の薬物動態学的プロフィールに対する効果を測定した。
エキセンディン含有マイクロ粒子を上記のように調製した。トリアムシノロンアセトニド含有マイクロ粒子を上記のように調製した。プラセボマイクロ粒子を上記のように調製した。
マイクロ粒子投与は実施例1に記載されるとおりであり、処理群を表11に要約した。IF-1と称する120mgのエキセンディン含有マイクロ粒子+30mgのプラセボ(群A)または10mgの2%w/wトリアムシノロン含有マイクロ粒子(群B)の投薬量をラットに投与した。SF-2と称する40mgのエキセンディン含有マイクロ粒子+30mgのプラセボ(群C)または10mgの2%w/wトリアムシノロン含有マイクロ粒子(群D)をまたラットに投与した。試料収集時点は、前採血、2時間、6時間、10時間、および1、2、4、7、10、14、17、21、24、29、32、36および39日であった。
血漿エキセンディンレベルを評価するために、0.25mL試料の血漿を0および1日目に尾静脈から収集し、0.4mL試料を表11に示された残りの日に収集した(群当たり4匹の動物)。試料を遠心分離し、血漿画分を凍結させた(-80℃)。血漿エキセンディンレベルを下記のIRMAにより定量した。
血漿中のエキセンディン4を定量する方法はサンドウィッチ免疫アッセイ、固相モノクローナル抗体EXE4:2-8.4により分析物を捕捉し、放射性ヨウ素化モノクローナル抗体GLP-1:3-3により検出した。結合カウントを標準較正曲線から定量した。このアッセイは、エキセンディン4に特異的であり、エキセンディン4(3-39)主要代謝物またはGLP-1を検出しない。典型的な標準線範囲は、トレーサー抗体の年に依存して30pg/mL〜2000pg/mLである。
血漿エキセンディン4レベル
図15は、注射後の時間(日)に対するエキセンディン血漿レベル(pg/mL)のグラフの形態で血漿エキセンディンレベルに対するプラセボマイクロ粒子およびトリアムシノロンアセトニド含有マイクロ粒子と共に同時投与したエキセンディン4含有マイクロ粒子の効果の結果を示す。図15に示されるように、群B(Lot02-002-82およびトリアムシノロン)の薬物動態学的プロフィールは対照(群A)を上回って改善した。具体的には、32日目の血漿レベルは検出可能なままであったが、これは対照群については検出可能であった最後の日であったという点でトリアムシノロンアセトニド処理群(群B)についてバイオアベイラビリティの増強が観察された。群Bについては39日目にまだ検出可能であったことは留意すべきであり、トリアムシノロンアセトニド含有マイクロ粒子と共に同時投与された場合、エキセンディンの放出期間内に実質的な増大を示す。Caveレベル、CmaxおよびAUCはまた、エキセンディン含有マイクロ粒子とのトリアムシノロンアセトニド含有マイクロ粒子の同時投与の結果として望ましく調節された。
Claims (40)
- 処置の必要な被験体に、生物学的に活性な不安定な薬剤を中に組み込んだ生体適合性ポリマーを含有してなる持続放出組成物の有効量(ここで、該不安定な薬剤が少なくとも約2週間の期間放出される)、およびコルチコステロイドを投与することを含む、生物学的に活性な不安定な薬剤のインビボ持続放出のための方法。
- コルチコステロイドが、持続放出組成物に同時に組み込まれている、請求項1記載の方法。
- コルチコステロイドが、第2の生体適合性ポリマーに別個に組み込まれている、請求項1記載の方法。
- 第2の生体適合性ポリマーが、持続放出組成物の生体適合性ポリマーと同じである、請求項3記載の方法。
- 第2の生体適合性ポリマーが、持続放出組成物の生体適合性ポリマーとは異なる、請求項4記載の方法。
- コルチコステロイドが、持続放出組成物に内包されていないが持続放出組成物と混ざり合っている、請求項1記載の方法。
- コルチコステロイドが、21−アセトキシプレグネノロン、アルクロメタゾン、アルゲストン、アムシノニド、ベクロメタゾン、ベタメタゾン、ブデソニド、クロロプレドニゾン、クロベタゾール、クロベタゾン、クロコルトロン、クロプレドノール、コルチコステロン、コルチゾン、コルチバゾール、デフラザコート、デソニド、デソキシメタゾン、デキサメタゾン、ジスフロラゾン、ジフルコルトロン、ジフルプレドナート、エノクソロン、フルアザコート、フルクロロニド、フルメタゾン、フルニソリド、フルシノロンアセトニド、フルオシノニド、フルオコルチンブチル、フルコルトロン、フルオロメトロン、酢酸フルペロロン、フルプレドニデンアセテート、フルプレドニソロン、フルランドレノリド、プロピオン酸フルチカゾン、ホルモコルタール、ハルシノニド、ハロベタゾールプロピオン酸塩、ハロメタソン、酢酸ハロプレドン、ヒドロコルタメート、ヒドロコルチゾン、ロテプレドノールエタボネート、マジプレドン、メドリゾン、メプレドニゾン、メチルプレドニゾロン、モメタゾンフロエート、パラメタゾン、プレドニカルベート、プレドニゾロン、プレドニゾロン25−ジエチルアミノアセテート、リン酸プレドニゾンナトリウム、プレドニゾン、プレドニバル、プレドニリデン、リメキゾロン、チクソコルトール、トリアムシノロンアセトニド、トリアムシノロンアセトニド21―酸メチルエステル、トリアムシノロンベネトニド、トリアムシノロンヘキサアセトニド、アセト酢酸トリアムシノロン、薬学的に許容され得るこれらの混合物、およびこれらの塩から選択される、請求項1記載の方法。
- コルチコステロイドが、トリアムシノロンアセトニド、トリアムシノロンアセトニド21―酸メチルエステル、トリアムシノロンベネトニド、トリアムシノロンヘキサアセトニド、アセト酢酸トリアムシノロン、薬学的に許容され得るこれらの混合物から選択される、請求項7記載の方法。
- 不安定な薬剤が、少なくとも約3週間の期間放出される、請求項1記載の方法。
- 不安定な薬剤が、少なくとも約4週間の期間放出される、請求項9記載の方法。
- 生体適合性ポリマーが、ポリ(ラクチド)、ポリ(グリコリド)、ポリ(ラクチド−コ−グリコリド)、ポリ(乳酸)、ポリ(グリコール酸)、ポリカーボネート、ポリエステルアミド、ポリ酸無水物、ポリ(アミノ酸)、ポリオルトエステル、ポリ(ジオキサノン)、ポリ(アルキレンアルキレート)、ポリエチレングリコールとポリオルトエステルのコポリマー、ポリウレタン、そのブレンドおよびそのコポリマーから選択される、請求項1記載の方法。
- 生体適合性ポリマーが、ポリ(ラクチド−コ−グリコリド)である、請求項11記載の方法。
- 持続放出組成物がマイクロ粒子の形態である、請求項1記載の方法。
- 生物学的に活性な不安定な薬剤がペプチドである、請求項1記載の方法。
- ペプチドがエキセンディン-4である、請求項14記載の方法。
- 生物学的に活性な不安定な薬剤がタンパク質である、請求項1記載の方法。
- タンパク質が、免疫グロブリン、抗体、サイトカイン、インターロイキン、インターフェロン、エリスロポエチン、ヌクレアーゼ、腫瘍壊死因子、コロニー刺激因子、インスリン、酵素、腫瘍サプレッサー、血液タンパク質、ホルモン、ワクチン、抗原、血液凝固因子、および成長因子から選択される、請求項16記載の方法。
- タンパク質がエリスロポエチンである請求項16記載の方法。
- タンパク質が卵胞刺激ホルモンである、請求項16記載の方法。
- タンパク質がインスリンである請求項16記載の方法。
- 有効量の生物学的に活性な不安定な薬剤を中に組み込んだ生体適合性ポリマーを含有してなる持続放出組成物(ここで、該不安定な薬剤が少なくとも約2週間の期間放出される)、およびコルチコステロイドを含んでなる医薬組成物。
- コルチコステロイドが、持続放出組成物に同時に組み込まれている、請求項21記載の医薬組成物。
- コルチコステロイドが、第2の生体適合性ポリマーに別個に組み込まれている、請求項21記載の医薬組成物。
- 第2の生体適合性ポリマーが、持続放出組成物の生体適合性ポリマーと同じである、請求項23記載の医薬組成物。
- 第2の生体適合性ポリマーが、持続放出組成物の生体適合性ポリマーとは異なる、請求項23記載の医薬組成物。
- コルチコステロイドが、持続放出組成物に内包されていないが持続放出組成物と混ざり合っている、請求項21記載の医薬組成物。
- コルチコステロイドが21−アセトキシプレグネノロン、アルクロメタゾン、アルゲストン、アムシノニド、ベクロメタゾン、ベタメタゾン、ブデソニド、クロロプレドニゾン、クロベタゾール、クロベタゾン、クロコルトロン、クロプレドノール、コルチコステロン、コルチゾン、コルチバゾール、デフラザコート、デソニド、デソキシメタゾン、デキサメタゾン、ジスフロラゾン、ジフルコルトロン、ジフルプレドナート、エノクソロン、フルアザコート、フルクロロニド、フルメタゾン、フルニソリド、フルシノロンアセトニド、フルオシノニド、フルオコルチンブチル、フルコルトロン、フルオロメトロン、酢酸フルペロロン、フルプレドニデンアセテート、フルプレドニソロン、フルランドレノリド、プロピオン酸フルチカゾン、ホルモコルタール、ハルシノニド、ハロベタゾールプロピオン酸塩、ハロメタソン、酢酸ハロプレドン、ヒドロコルタメート、ヒドロコルチゾン、ロテプレドノールエタボネート、マジプレドン、メドリゾン、メプレドニゾン、メチルプレドニゾロン、モメタゾンフロエート、パラメタゾン、プレドニカルベート、プレドニゾロン、プレドニゾロン25−ジエチルアミノアセテート、リン酸プレドニゾンナトリウム、プレドニゾン、プレドニバル、プレドニリデン、リメキゾロン、チクソコルトール、トリアムシノロンアセトニド、トリアムシノロンアセトニド21―酸メチルエステル、トリアムシノロンベネトニド、トリアムシノロンヘキサアセトニド、アセト酢酸トリアムシノロン、薬学的に許容され得るこれらの混合物、およびこれらの塩から選択される、請求項21記載の医薬組成物。
- コルチコステロイドが、トリアムシノロンアセトニド、トリアムシノロンアセトニド21―酸メチルエステル、トリアムシノロンベネトニド、トリアムシノロンヘキサアセトニド、アセト酢酸トリアムシノロン、薬学的に許容され得るこれらの混合物から選択される、請求項27記載の医薬組成物。
- 持続放出組成物が、約2週間以上の期間にわたる不安定な薬剤の目標放出期間を有する、請求項21記載の医薬組成物。
- 目標放出期間が約3週間以上である、請求項29記載の医薬組成物。
- 生体適合性ポリマーが、ポリ(ラクチド)、ポリ(グリコリド)、ポリ(ラクチド−コ−グリコリド)、ポリ(乳酸)、ポリ(グリコール酸)、ポリカーボネート、ポリエステルアミド、ポリ酸無水物、ポリ(アミノ酸)、ポリオルトエステル、ポリ(ジオキサノン)、ポリ(アルキレンアルキレート)、ポリエチレングリコールとポリオルトエステルのコポリマー、ポリウレタン、そのブレンドおよびそのコポリマーから選択される、請求項21記載の方法。
- 生体適合性ポリマーがポリ(ラクチド−コ−グリコリド)である、請求項31記載の医薬組成物。
- 持続放出組成物がマイクロ粒子の形態である、請求項21記載の医薬組成物。
- 生物学的に活性な不安定な薬剤がペプチドである、請求項21記載の医薬組成物。
- ペプチドがエキセンディン-4である、請求項34記載の医薬組成物。
- 生物学的に活性な不安定な薬剤がタンパク質である、請求項21記載の医薬組成物。
- タンパク質が、免疫グロブリン、抗体、サイトカイン、インターロイキン、インターフェロン、エリスロポエチン、ヌクレアーゼ、腫瘍壊死因子、コロニー刺激因子、インスリン、酵素、腫瘍サプレッサー、血液タンパク質、ホルモン、ワクチン、抗原、血液凝固因子、および成長因子から選択される、請求項36記載の医薬組成物。
- タンパク質がエリスロポエチンである、請求項36記載の医薬組成物。
- タンパク質が卵胞刺激ホルモンである、請求項36記載の医薬組成物。
- タンパク質がインスリンである、請求項36記載の医薬組成物。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US41943002P | 2002-10-17 | 2002-10-17 | |
| PCT/US2003/032017 WO2004034975A2 (en) | 2002-10-17 | 2003-10-08 | Sustained release profile modification |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2006505562A true JP2006505562A (ja) | 2006-02-16 |
| JP2006505562A5 JP2006505562A5 (ja) | 2006-11-16 |
Family
ID=32108081
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2004544829A Pending JP2006505562A (ja) | 2002-10-17 | 2003-10-08 | 持続放出組成物の放出プロフィールの調節方法 |
Country Status (6)
| Country | Link |
|---|---|
| US (2) | US20040121009A1 (ja) |
| EP (1) | EP1567127A4 (ja) |
| JP (1) | JP2006505562A (ja) |
| AU (1) | AU2003284028B2 (ja) |
| CA (1) | CA2501298A1 (ja) |
| WO (1) | WO2004034975A2 (ja) |
Families Citing this family (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6824822B2 (en) | 2001-08-31 | 2004-11-30 | Alkermes Controlled Therapeutics Inc. Ii | Residual solvent extraction method and microparticles produced thereby |
| US7456254B2 (en) * | 2004-04-15 | 2008-11-25 | Alkermes, Inc. | Polymer-based sustained release device |
| CN1968700A (zh) * | 2004-04-15 | 2007-05-23 | 阿尔克姆斯有限公司 | 聚合物基的持续释放装置 |
| US11246913B2 (en) | 2005-02-03 | 2022-02-15 | Intarcia Therapeutics, Inc. | Suspension formulation comprising an insulinotropic peptide |
| EP2347762B1 (en) | 2005-08-19 | 2019-05-08 | Amylin Pharmaceuticals, LLC | Exendin for treating diabetes and reducing body weight |
| KR101200728B1 (ko) | 2006-08-09 | 2012-11-13 | 인타르시아 세라퓨틱스 인코포레이티드 | 삼투성 전달 시스템 및 피스톤 조립체 |
| GR1005808B (el) * | 2006-11-27 | 2008-02-06 | Bionature E.A. Limited | Επιταση της δρασεως της ερυθροποιητινης με αγωνιστες των μεμβρανικων στεροειδων υποδοχεων |
| WO2008118712A1 (en) * | 2007-03-22 | 2008-10-02 | Alkermes, Inc. | Coacervation process |
| AU2008244523B2 (en) | 2007-04-23 | 2012-02-16 | Intarcia Therapeutics, Inc. | Suspension formulations of insulinotropic peptides and uses thereof |
| US8470360B2 (en) | 2008-04-18 | 2013-06-25 | Warsaw Orthopedic, Inc. | Drug depots having different release profiles for reducing, preventing or treating pain and inflammation |
| EP2240155B1 (en) | 2008-02-13 | 2012-06-06 | Intarcia Therapeutics, Inc | Devices, formulations, and methods for delivery of multiple beneficial agents |
| US20090263456A1 (en) * | 2008-04-18 | 2009-10-22 | Warsaw Orthopedic, Inc. | Methods and Compositions for Reducing Preventing and Treating Adhesives |
| US20110263496A1 (en) * | 2008-05-21 | 2011-10-27 | Amylin Pharmaceuticals, Inc. | Exendins to lower cholesterol and triglycerides |
| SI2462246T1 (en) | 2009-09-28 | 2018-01-31 | Intarcia Therapeutics, Inc. | Fast-setting and / or cessation of substantially unchanged delivery of the product |
| UA111162C2 (uk) | 2010-08-04 | 2016-04-11 | Флекшен Терап'Ютікс, Інк. | Ін'єкційна композиція ацетоніду триамцинолону для лікування болю |
| US20120208755A1 (en) | 2011-02-16 | 2012-08-16 | Intarcia Therapeutics, Inc. | Compositions, Devices and Methods of Use Thereof for the Treatment of Cancers |
| WO2014027253A1 (en) * | 2012-08-14 | 2014-02-20 | Wockhardt Limited | Pharmaceutical microparticulate compositions of polypeptides |
| US9889085B1 (en) | 2014-09-30 | 2018-02-13 | Intarcia Therapeutics, Inc. | Therapeutic methods for the treatment of diabetes and related conditions for patients with high baseline HbA1c |
| KR20240042548A (ko) | 2015-06-03 | 2024-04-02 | 인타르시아 세라퓨틱스 인코포레이티드 | 임플란트 배치 및 제거 시스템들 |
| MA53353A (fr) | 2016-05-16 | 2021-06-09 | Intarcia Therapeutics Inc | Polypeptides sélectifs pour le récepteur du glucagon et méthodes pour leur utilisation |
| USD860451S1 (en) | 2016-06-02 | 2019-09-17 | Intarcia Therapeutics, Inc. | Implant removal tool |
| USD840030S1 (en) | 2016-06-02 | 2019-02-05 | Intarcia Therapeutics, Inc. | Implant placement guide |
| EP3565580B1 (en) | 2017-01-03 | 2024-03-06 | i2o Therapeutics, Inc. | Continuous administration of exenatide and co-adminstration of acetaminophen, ethinylestradiol or levonorgestrel |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH08503695A (ja) * | 1992-09-10 | 1996-04-23 | チルドレンズ メディカル センター コーポレイション | 局所麻酔剤の持続性送達のための生分解性ポリマーマトリックス |
| JPH11504634A (ja) * | 1995-05-01 | 1999-04-27 | チルドレンズ メディカル センター コーポレイション | 局所麻酔薬とグルココルチコイドとの組み合わせによる延長された神経遮断 |
| JPH11506450A (ja) * | 1995-06-02 | 1999-06-08 | オキュレックス ファーマシューティカルズ,インコーポレイティド | 親水性及び疎水性薬剤の組合せによる薬剤の制御放出のための改良処方物 |
| JP2000511941A (ja) * | 1997-07-02 | 2000-09-12 | ユーロ−セルティーク エス.エイ. | 関節間隙および身体間隙における持効性麻酔 |
| WO2001043762A2 (en) * | 1999-12-16 | 2001-06-21 | Eli Lilly And Company | Polypeptide compositions with improved stability |
| WO2001087267A1 (en) * | 2000-02-28 | 2001-11-22 | Gel-Del Technologies, Inc. | Protein matrix materials, devices and methods of making and using thereof |
| JP2002520120A (ja) * | 1998-07-17 | 2002-07-09 | スカイファーマ インコーポレーテッド | 封入物質の制御放出のための生分解性組成物 |
| JP2002234833A (ja) * | 2001-02-08 | 2002-08-23 | Taiyo Yakuhin Kogyo Kk | 徐放型マイクロカプセルの製造法 |
Family Cites Families (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4542025A (en) * | 1982-07-29 | 1985-09-17 | The Stolle Research And Development Corporation | Injectable, long-acting microparticle formulation for the delivery of anti-inflammatory agents |
| US4530840A (en) * | 1982-07-29 | 1985-07-23 | The Stolle Research And Development Corporation | Injectable, long-acting microparticle formulation for the delivery of anti-inflammatory agents |
| US5811128A (en) * | 1986-10-24 | 1998-09-22 | Southern Research Institute | Method for oral or rectal delivery of microencapsulated vaccines and compositions therefor |
| US5019400A (en) * | 1989-05-01 | 1991-05-28 | Enzytech, Inc. | Very low temperature casting of controlled release microspheres |
| US5707644A (en) * | 1989-11-04 | 1998-01-13 | Danbiosyst Uk Limited | Small particle compositions for intranasal drug delivery |
| US5922340A (en) * | 1992-09-10 | 1999-07-13 | Children's Medical Center Corporation | High load formulations and methods for providing prolonged local anesthesia |
| US5424286A (en) * | 1993-05-24 | 1995-06-13 | Eng; John | Exendin-3 and exendin-4 polypeptides, and pharmaceutical compositions comprising same |
| AU677115B2 (en) * | 1994-12-19 | 1997-04-10 | University Of Miami | Biodegradable injectable drug delivery polymer |
| GB9513118D0 (en) * | 1995-06-28 | 1995-08-30 | Merck Sharp & Dohme | Therapeutic agents |
| CA2192782C (en) * | 1995-12-15 | 2008-10-14 | Nobuyuki Takechi | Production of microspheres |
| CN1126537C (zh) * | 1996-05-23 | 2003-11-05 | 株式会社三养社 | 可局部给药,可生物降解和持续释放的用于治疗牙周炎的药物组合物及其制备方法 |
| US6046187A (en) * | 1996-09-16 | 2000-04-04 | Children's Medical Center Corporation | Formulations and methods for providing prolonged local anesthesia |
| US6203813B1 (en) * | 1997-01-13 | 2001-03-20 | Lance L. Gooberman | Pharmaceutical delivery device and method of preparation therefor |
| WO2004035762A2 (en) * | 2002-10-17 | 2004-04-29 | Alkermes Controlled Therapeutics, Inc. Ii | Microencapsulation and sustained release of biologically active polypeptides |
-
2003
- 2003-10-08 AU AU2003284028A patent/AU2003284028B2/en not_active Ceased
- 2003-10-08 WO PCT/US2003/032017 patent/WO2004034975A2/en active IP Right Grant
- 2003-10-08 US US10/681,571 patent/US20040121009A1/en not_active Abandoned
- 2003-10-08 CA CA002501298A patent/CA2501298A1/en not_active Abandoned
- 2003-10-08 EP EP03776257A patent/EP1567127A4/en active Pending
- 2003-10-08 JP JP2004544829A patent/JP2006505562A/ja active Pending
-
2007
- 2007-07-17 US US11/826,694 patent/US20080057131A1/en not_active Abandoned
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH08503695A (ja) * | 1992-09-10 | 1996-04-23 | チルドレンズ メディカル センター コーポレイション | 局所麻酔剤の持続性送達のための生分解性ポリマーマトリックス |
| JPH11504634A (ja) * | 1995-05-01 | 1999-04-27 | チルドレンズ メディカル センター コーポレイション | 局所麻酔薬とグルココルチコイドとの組み合わせによる延長された神経遮断 |
| JPH11506450A (ja) * | 1995-06-02 | 1999-06-08 | オキュレックス ファーマシューティカルズ,インコーポレイティド | 親水性及び疎水性薬剤の組合せによる薬剤の制御放出のための改良処方物 |
| JP2000511941A (ja) * | 1997-07-02 | 2000-09-12 | ユーロ−セルティーク エス.エイ. | 関節間隙および身体間隙における持効性麻酔 |
| JP2002520120A (ja) * | 1998-07-17 | 2002-07-09 | スカイファーマ インコーポレーテッド | 封入物質の制御放出のための生分解性組成物 |
| WO2001043762A2 (en) * | 1999-12-16 | 2001-06-21 | Eli Lilly And Company | Polypeptide compositions with improved stability |
| WO2001087267A1 (en) * | 2000-02-28 | 2001-11-22 | Gel-Del Technologies, Inc. | Protein matrix materials, devices and methods of making and using thereof |
| JP2002234833A (ja) * | 2001-02-08 | 2002-08-23 | Taiyo Yakuhin Kogyo Kk | 徐放型マイクロカプセルの製造法 |
Also Published As
| Publication number | Publication date |
|---|---|
| US20080057131A1 (en) | 2008-03-06 |
| EP1567127A4 (en) | 2007-02-21 |
| CA2501298A1 (en) | 2004-04-29 |
| WO2004034975A3 (en) | 2005-06-02 |
| AU2003284028A1 (en) | 2004-05-04 |
| EP1567127A2 (en) | 2005-08-31 |
| US20040121009A1 (en) | 2004-06-24 |
| AU2003284028B2 (en) | 2007-05-10 |
| WO2004034975A2 (en) | 2004-04-29 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20080057131A1 (en) | Method of modifying the release profile of sustained release compositions | |
| US20200390891A1 (en) | Polymer-based sustained release device | |
| US8877252B2 (en) | Polymer-based sustained release device | |
| US20060110423A1 (en) | Polymer-based sustained release device | |
| WO2004035754A2 (en) | Microencapsulation and sustained release of biologically active polypeptides | |
| AU2006235955A1 (en) | Poly (lactide-co-glycolide)-based sustained release microcapsules comprising a polypeptide and a sugar |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20060927 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20060927 |
|
| A711 | Notification of change in applicant |
Free format text: JAPANESE INTERMEDIATE CODE: A711 Effective date: 20061113 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A821 Effective date: 20061113 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20100212 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20100428 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20100511 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20100607 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20100614 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20101025 |