本発明は、バイオテクノロジー医学、生物学及び生化学の分野に関する。その適用は、ヒトの健康、動物及び植物のケアーを目的とする。更に特定的には、本発明は、治療的関心のある分子を同定するための新規なスクリーニング方法及び新規な遺伝子治療ツールを開発することができる、核酸配列を同定することを可能とし、そして更に本発明は分子の毒性及び効能並びに薬理ゲノムデータに関する情報を提供する。
本発明は、比較されるべき2つの異なる状態に由来するRNA、特に病気の器官又は組織とその健康な同等物に由来するRNA間の定性的な差(qualitative differences)を証明することに基づく核酸配列を同定するための独創的な一連の方法を主として説明する。更に特定的には、これらの方法は、病理条件及び健康な条件に関して又は比較することが望まれる2つの生理学的条件に関して差異的にスプライシングされる(differentially spliced)オルタナティブエキソン及びイントロン(alternative exons and introns)を特異的にクローニングすることを意図する。RNAにおけるこれらの定性的な差は、RNAに転写されるべき領域における挿入又は欠失の如きゲノム変化(genome alterations)によることもありうる。この一連の方法は略称DATAS:選択的スプライシングによる転写物の差異分析(Differential Analysis of Transcripts with Alternative Splicing)として同定される。
与えれた疾患の根底となる又は疾患に結び付けられる遺伝子発現変化の特徴付けは、新規な治療標的及び独創的な診断ツールの発見に関する実質的な希望を生じさせる。しかしながら、ゲノム又は相補的DNA配列の同定によって得られる情報は、ポジショナルクローニングによろうが定量的差異スクリーニング(quantitative differential screening)技術によろうが、調べている疾患に関する調節の欠陥に関与する機能に関するものは、もしあったとしても、僅かであり、そして関与する機能的ドメインに関して得られる情報は更に少ない。本発明は、2つの異なる病態生理学的条件の間で発生するRNAスプライシングの差を同定することを目的とする一連の独創的方法を記載する。このような差の同定は、定性的な差に関する情報を与えるが、これまでに記載された技術の場合のように定量的な差に関しては情報を与えない。ゆえに、本発明で開示される技術は、「定性的差異スクリーニング("qualitarive differential screening")」又はDATASという用語の下ですべて包含される。本発明の方法は、新規な標的又は治療的産物を同定するため、学的研究及び/又は診断用ツールを考案するため、核酸ライブラリーを構築するため及び例えば化合物の毒物学的プロフィル又は効能を決定するための方法を開発するために使用することができる。
本発明の第1の目的は、更に特定的には、2つの生物学的試料間で発生する定性的な遺伝的差異に相当する核酸領域を同定及び/又はクローニングする方法であって、第1の生物学的試料に由来する二本鎖cDNA又はRNAの集団を第2の生物学的試料に由来するcDNAの集団とハイブリッド形成させることを含む方法に基づいている(図1A)。
上記したとおり、定性的な遺伝的差異は、RNAスプライシングの変化又はRNAに転写されるゲノムの領域における又は欠失及び/又は挿入によることがありうる。
第1の態様では、ハイブリッド形成は、第1の生物学的試料に由来するRNAと第2の生物学的試料に由来するcDNA(一本鎖又は二本鎖の)との間で行われる。
他の態様では、ハイブリッド形成は、第1の生物学的試料に由来する二本鎖cDNAと第2の生物学的試料に由来するcDNA(二本鎖又は好ましくは一本鎖の)との間で行われる。
本発明の更に特定の目的は、2つの生理学的条件間で発生する差異的にスプライシングされた(differentially spliced)核酸領域を同定するための方法であって、試験条件に由来するRNA又は二本鎖cDNAの集団を基準条件から生じるcDNAの集団とハイブリッド形成させそして差異スプライシングイベント(differential splicing events)に相当する核酸を同定することを含む方法を提供することである。
本発明の他の特定の目的は、2つの生理学的条件間で発生する差異スプライシングされた核酸領域を同定するための方法であって、試験条件からのcDNAの第1集団を第2の(例えば、基準)条件からのcDNAの第2集団とハイブリッド形成させ、そして形成されたハイブリッドから、差異的なスプライシングイベントに相当する核酸を同定することを含む方法を提供することである。更に特定の態様では、cDNAの第1集団は一本鎖でありそして第2集団は二本鎖又は一本鎖である。該集団は典型的には、その組成又は配列が少なくとも部分的に未知である複数の異なるポリヌクレオチド配列を含む。しかしながら、特定の態様では、第1集団は選ばれたcDNA、即ち、関心のある1つ又はそれより多くの選ばれた遺伝子又はRNAに相当する1つ又はそれより多くのcDNAを含む。この特定の態様では、種々の病態生理学的状況からの選ばれた遺伝子の生物学的に関係のあるスプライシング形態を同定することができる。
本発明の他の目的は、2つの生理学的条件間で発生する差異スプライシングされた核酸をクローニングするための方法であって、試験条件から誘導されるRNA又は二本鎖cDNAの集団を基準条件から生じるcDNAの集団とハイブリッド形成させそして差異スプライシングイベントに相当する核酸をクローニングすることを含む方法を提供することである。
本発明の他の目的は、2つの生理学的条件間で発生する差異スプライシングされた核酸をクローニングするための方法であって、試験条件から誘導されるcDNAの集団、該集団は複数の異なるDNA配列を含む、を、基準条件から生じるcDNAの集団、該集団は複数の異なるDNA配列を含む、とハイブリッド形成させ、そして形成されたハイブリッドから対を形成していない領域(unpaired region)を含む核酸、該核酸は差異スプライシングされたドメインに相当する、をクローニングすることを含む方法を提供することである。
特定の態様では、本発明に従う核酸同定及び/又はクローニングの方法は、
(a)第1の試料(試験条件)に由来するRNAを第2の試料(基準条件)に由来するcDNAとハイブリッド形成させること、
(b)第2の試料(基準条件)に由来するRNAを第1の試料(試験条件)に由来するcDNAとハイブリッド形成させること、及び
(c)段階(a)及び(b)で形成されたハイブリッドから、定性的な遺伝的差異(qualitative genetic differences)に相当するこれらの核酸を同定及び/又はクローニングすること、
からなる2つのハイブリッド形成を平行して行うことを含む。
本発明は、核酸ライブラリーの作成、このようにして作成された核酸及び核酸ライブラリー並びに後記する生物学/バイオテクノロジーのすべての分野におけるこのような物質の使用も指向する。
この点で、本発明は、2つの生物学的試料間で発生する定性的な差を表すプロフィル化された核酸組成物又はライブラリーを作成する方法であって、第1の生物学的試料に由来するRNAを第2の生物学的試料から生じるcDNAとハイブリッド形成させることを含む方法も指向する。
本発明は、更に、cDNA組成物をRNAとハイブリッド形成させること又はその逆、を含むcDNA組成物をプロフィル化するための方法にも関する。
上記したとおり、本発明は、特に、生理学的状態を表す核酸を同定及びクローニングするための方法に関する。更に、同定及び/又はクローニングされた核酸は、これらの核酸が観察される生理学的状態に強い程度に一般に関与しているという点で生理学的状態の定性的特徴を表す。ゆえに、本発明の定性的方法は、病態生理学的状態の出現に機能的役割を果たす遺伝子エレメント又はそのタンパク質産生物の直接の探求を与える。
本発明の方法は、異なる生理学的状態に属する、一方ではRNA又はcDNAと他方ではcDNAの間のクロスハイブリッド形成からなる最初の段階に部分的に基づいている。この又はこれらのクロスハイブリッド形成法は、有利には、形成されたハイブリッドにおいて、対を形成していない領域、即ち、与えられた生理学的条件なおけるRNAには存在するが他の生理学的条件からのRNAには存在しない領域を証明することを可能とする。このような領域は、生理学的状態に典型的な選択的形態のスプライシングに本質的に相当するが、挿入又は欠失のような遺伝子変化の反映でもありえ、したがって下記するとおりの治療学及び診断学の分野で特に有用な遺伝子エレメントを形成する。ゆえに、本発明は、特に、クロスハイブリッド形成(1つ又は複数)の後に形成された複合体を保持してそれから定性的な差に相当する領域を推定することからなる。この方法は、ハイブリッド形成(1つ又は複数)の後形成されたハイブリッドを捨ててハイブリッド形成しなかった核酸のみを保存する、当業者に知られている定量的サブトラクション技術(Sargent and Dawid(1983), Science, 222: 135-139; Davis et al.(1984), PNAS, 81: 2194-2198; Duguid and Dinauer(1990), Nucl.Acid.Res., 18: 2789-2792; Diatchenko et al., (1996), PNAS, 93: 6025-6030)とは区別することができる。
第1の態様では、本発明は、試験試料のRNAを基準試料のcDNAとハイブリッド形成させることを含む関心のある核酸を同定するための方法に関する。このハイブリッド形成法は、形成された複合体において、調べている条件間の定性的な遺伝的差異を同定すること及びかくして例えば試験条件に特徴的なスプライシングを同定及び/又はクローニングすることを可能とする。
ゆえに、本発明の第1の変法に従えば、該方法は、基準条件に比べて生理学的試験条件において発生するスプライシングイベントに特徴的な核酸集団を発生させること(図1A、1B)を可能とする。後に説明するとおり、この集団は、核酸のクローニング及び特徴付け、診断、スクリーニング、治療及び抗体産生におけるそれらの使用、又は完全タンパク質もしくはタンパク質の断片の合成のために使用することができる。この集団は、後に示されるとおり種々の適用分野で使用することができるライブラリーを発生させるため及び標識されたプローブを発生させるために使用することもできる(図1D)。
本発明の他の変法に従えば、本方法は、基準条件に由来するRNAと試験条件に由来するcDNA間で平行して行われる前記した第1のハイブリッド形成及び第2のハイブリッド形成を含む。この変法は、1つは基準条件に対する試験条件の定性的特徴を表しそして他方が試験条件に対する基準条件の定性的特徴を表す2つの核酸集団を発生させることを可能とする(図1C)ので特に有利である。これらの2つの集団は、以下にさらに完全に説明されるとおり特定の生理学的条件の遺伝学的フィンガープリント(genetic fingerprints)として役立つ核酸ソースとして又はライブラリーとして利用することもできる(図1D)。
更なる態様では、本発明は、試験試料からのDNAsを基準試料の二本鎖cDNAとハイブリッド形成させることを含む関心のある核酸を同定するための方法に関する。このハイブリッド形成法は、形成された複合体において、調べている条件間の定性的な遺伝的差異を同定すること及びかくして例えば試験条件に特徴的なスプライシングを同定及び/又はクローニングすることを可能とする。後に開示されるとおり、この態様は、それがオルタナティブイントロン及びエキソン(alternative introns and exons)のみならず、同じ核酸ライブラリー内で、エキソン又はイントロンの欠失により形成される特異的連結部(specific junctions)も示すという点で有利である。更に、得られる配列は、オルタナティブイントロン及びエキソンのフランキング配列(flanking sequences)に関する情報も提供する。かくして、本発明は、スプライシングされなかった領域に関する情報なしに、選択的スプライシングが破壊されそしてスプライシングされた遺伝子の一部のみが保持される、US5,929,535に開示された技術の如き先行技術とは明らかに区別される。かくして、US5,929,535に開示された方法は適当なスプライスオリゴヌクレオチドのデザインを可能とすることができない。
本発明は、すべてのタイプの生物学的試料に適用することができる。特に、生物学的試料は、核酸を含有するいかなる細胞、器官、組織試料又は生検材料等であってもよい。器官、組織又は生検材料の場合には、試料は成分細胞へのアクセスを容易にするために培養させることができる。試料は、哺乳動物(特にヒト)、植物、バクテリア及び下等真核生成物(酵母、カビ細胞(fungal cells)等)から誘導されることができる。関係のある材料は、特に、腫瘍生検(tumor biopsy)、神経変性兆候を示す神経変性プラーク(neurodegenerative plaque)又は大脳ゾーン生検(cerebral zone biopsy)、皮膚試料、採血により得られる血液試料、結腸生検(colorectal biopsy)、気管支肺胞洗浄に由来する生検材料等により例示される。細胞の例は、注目すべきことに、筋肉細胞、肝細胞、繊維芽細胞、神経細胞、表皮及び皮膚細胞、血液細胞、例えばB及びTリンパ球、マスト細胞、単球、顆粒球及びマクロファージを含む。
上記したとおり、本発明に従う定性的差異スクリーニングは、他の用途のためにクローニング又は使用されるべき、基準生理学的条件(条件A)に対する与えられた生理学的条件(条件B)に特徴的な核酸の同定を可能とする。例示として、調査されるべき生理学的条件A及びBは下記の中から選ぶことができる:
A−RNA集団
本発明は、全RNA又はメッセンジャーRNAを使用することにより行うことができる。これらのRNAは、当業者に熟知されているいかなる慣用の分子生物学法によっても調製することができる。このような方法は、一般に、細胞、組織又は試料溶解(lysis)及び抽出法によるRNA回収を含む。これは、特に、カオトロピック剤、例えばチオシアン酸グアニジウム(RNAに影響を与えないで細胞を破壊する)による処理、次いで溶媒(例えば、フェノール、クロロホルム)によるRNA抽出により行うことができる。このような方法は当該技術分野で周知である(Maniatis et al., Chomczynski et al. (1987), Anal.Biochem., 162: 156参照)。これらの方法は、商業的に入手可能なキット、例えば、全RNA用のUS73750キット(Amersham)又はRneasy kit(Qujagen)を使用することにより容易に実施することができる。RNAは完全に純粋な状態にあることは必要ではなく、そして特に調製物中に残っている微量のゲノムDNA又は他の細胞成分(タンパク質等)は、それらがRNA安定性に有意に影響を与えない限りそして比較下の種々の試料の調製方法が同じである限り、妨害しないであろう。場合により、全RNA調製物の代わりにメッセンジャーRNAを使用することが更に可能である。これらは、標準方法に従い、ポリT配列によって、生物学的試料から直接又は全RNAから単離することができる。この点で、メッセンジャーRNAの調製は、例えば、US72700キット(Amersham)又はオリゴ(dT)ビーズ(Dynal)の使用を伴うキットの如き商業的に入手可能なキットを使用して行うことができる。RNA調製の有利な方法は、細胞質ゾルRNAを抽出し、次いで細胞質ゾルポリA+RNAを抽出することからなる。スプライシングされていないエキソン及びイントロンを有するプレメッセンジャーRNAにより汚染されていない細胞質ゾルRNAの選択的調製を可能とするキットは、商業的に入手可能である。これは、特にQuiagenにより市販されているRneasyキット(カタログ番号の例:74103)についてそうである。RNAは、前もって調製されたライブラリー又は他の試料から直接得ることもでき及び/又は適当な条件下に貯蔵された収集物から入手可能である。
一般に、使用されるRNA調製物は、有利には少なくとも0.1μgのRNA、好ましくは少なくとも0.5μgのRNAを含む。量は、本発明の実施を変化させないように保ちながら、使用される特定の細胞及び方法に依存して変わることができる。十分な量のRNA(好ましくは少なくとも0.1μg)を得るために、一般に少なくとも105個の細胞を含む生物学的試料を使用することが推奨される。この点で、典型的な生検試料は一般に105〜108個の細胞を含みそして典型的なペトリ皿(直径6〜10cm)上での細胞培養物は、十分な量のRNAが容易に得られうるように、約106個の細胞を含有する。
RNA調製物は、その場で使用することができ、又は好ましくは、後に使用するために溶液として又は冷凍された状態において冷所に貯蔵することができる。
B−cDNA集団
本発明の範囲内で使用されるcDNAは、慣用の分子生物学技術に従う逆転写により得ることができる。特にManiatis et alを参照されたい。逆転写は、一般に酵素、逆転写酵素及びプライマーを使用して行われる。
この点で、多くの逆転写酵素が文献に記載されておりそして商業的に入手可能である(1483188キット、Boehringer)。最も普通に使用される逆転写酵素の例は、鳥類ウイルスAMV(ニワトリ骨髄芽球症ウイルス(Avian Myeloblastosis Virus))に由来する逆転写酵素及びマウス白血病ウイルスMMLV(モロニーマウス白血病ウイルス(Moloney Murine Leukemia Virus))からの逆転写酵素を含む。逆転写活性を有するある種の熱安定性DNAポリメラーゼ、例えば、Thermus flavus及びThermus thermophilus HB-8(商業的に入手可能;Promegaカタログ番号 M1941及びM2101)から単離された熱安定性DNAポリメラーゼに言及することも価値がある。有利な変法に従えば、本発明は、AMV逆転写酵素を使用して行われる。何故ならば、42℃で活性な(37℃で活性なMMLVの逆転写酵素と対照的に)この酵素は、伸長を停止させる可能性のあるRNA二次構造を脱安定化させ、そしてそれゆえより大きい長さのRNAの逆転写を可能としそしてRNAのはるかにより忠実なコピーであるcDNA調製物を高収率で与えるからである。
本発明の更なる有利な変法に従えば、RNアーゼH活性を欠いた逆転写酵素が使用される。このタイプの酵素の使用は、いくつかの利点、特にcDNA合成の収率を増加させ、そして新しく合成されたcDNAとのヘテロ二本鎖の形成にその後参加させられるRNAのいかなる劣化も回避するという利点を有し、それによって場合によりcDNAのフェノール抽出を省くことを可能とする。RNアーゼH活性に欠けた逆転写酵素は、欠失(1つ又は複数)及び/又は突然変異誘発によりいかなる逆転写酵素からも調製されうる。更に、このような酵素も商業的に入手可能である(例えば、Life Technologies,カタログ番号 18053-017)。
逆転写酵素に適用される操作条件((濃度及び温度)は当業者には周知である。特に、10mMの最適なMg2+濃度の存在下に、一般に10〜30単位の酵素が1つの反応で使用される。
逆転写のために使用されるプライマー(1つ又は複数)は種々のタイプであることができる。それは、特に、好ましくは4〜10ヌクレオチドを含むランダムオリゴヌクレオチド、有利にはヘキサヌクレオチドであることができる。このタイプのランダムプライマーの使用は、文献に記載されておりそしてRNA分子内の種々の部位での逆転写のランダムな開始を可能とする。この技術は、全RNA(即ち、特にmRNA、tRNA及びrRNAを含む)を逆転写するために特に使用される。mRNAのみの逆転写を行うことが所望される場合には、プライマーとして、メッセンジャーRNAに対して特異的なポリAテイルから出発する逆転写の開始を可能とするオリゴdTオリゴヌクレオチドを使用することが有利である。オリゴdTオリゴヌクレオチドは、4〜20mer、有利には、約15merを含むことができる。このようなプライマーの使用は本発明の好ましい態様を表す。更に、逆転写のための標識されたプライマーを使用することが有利でありうる。実際問題として、これは、RNAの認識及び/又は選択及び/又はその後のcDNAからの選別を可能とする。これは、RNA/DNAヘテロ二本鎖を単離することも可能とすることもでき、RNA/DNAヘテロ二本鎖の形成は本発明の実施において重要な段階を表す。プライマーの標識化は、いかなるリガンド−受容体をベースとするシステムによっても行うことができ、即ち、プライマーを有する分子の親和性を仲立ちとした分離を提供するシステムによってなされうる。それは、予めストレプトアビジンでコーティングされたいかなる支持体(ビーズ、カラム、プレート等)上にも捕捉されうる、例えば、ビオチン標識からなることができる。プライマーの性質に影響を与えることなく分離を可能とするいかなる他の標識化システムも同様に使用することができる。
典型的操作条件において、この逆転写は一本鎖相補的DNA(cDNA)を発生させる。これは、本発明の第1の有利な態様を表す。
本発明を実施する第2の変法では、逆転写は二本鎖cDNAが調製されるように達成される。この結果は、第1cDNA鎖の転写の後、DNA修飾することができる酵素、例えばファージT4DNAリガーゼ、DNAポリメラーゼI及びファージT4DNAポリメラーゼを伴う慣用の分子生物学技術を使用して第2鎖(second strand)を発生させることにより達成される。
cDNA調製物はその場で使用することができ又は好ましくは後の使用のために溶液として又は冷凍された状態で、好ましくは冷所に貯蔵することができる。
上記したとおり、本発明は、典型的には、複合核酸集団(即ち、少なくとも部分的に未知であるか又は特徴付けされていない複数の異なる核酸配列、典型的には20、50又は100より多くの異なる核酸配列を含む集団)を使用して行われる。しかしながら、特定の態様では、本発明は選ばれた核酸集団を使用して行われうる。このような選ばれた核酸集団は、例えば、選ばれた遺伝子又はRNA(又はいくつかの既知のそして選ばれた遺伝子又はRNA)の配列を含むことができる。選ばれた核酸集団を使用することによって、本発明は、いかなる特定の病態生理学的条件にある選ばれた遺伝子の生物学的に関係のあるスプライシング形態(biologically relevant splicing forms)も同定するために使用することができる。かくして、本発明は、以後説明されるとおり選ばれた遺伝子又はRNAのスプライシング形態をクローニング又は同定するためにも適当である。
C−ハイブリッド形成
上記したとおり、本発明に従う方法は、異なる生理学的条件における生物学的試料に由来する又は異なる起源に由来する、一方ではRNA又はcDNAと他方ではcDNAとの独創的クロスハイブリッド形成段階に部分的に基づいている。好ましい態様では、本発明に従うハイブリッド形成は、液相で有利に行われる。更に、それは、いかなる適当な装置においても、例えば、チューブ(例えば、Eppendorffチューブ)、プレート又は分子生物学で普通に使用されるいかなる他の適当な支持体においても行うことができる。ハイブリッド形成は、10〜1000μl、例えば、10〜500μlの範囲の容積において有利に行われる。使用される特定の装置及び容積は当業者により容易に適合させられうることは理解されるべきである。使用される核酸の量は同様に当該技術分野で周知である。一般に、数μgの核酸、例えば、0.1〜100μgの範囲の核酸を使用すれば十分である。
ハイブリッド形成を行う際に考慮されるべき重要なファクターは、使用される核酸のそれぞれの量である。かくして、2つの試料からの核酸を約50〜0.02、好ましくは40〜0.1の範囲の割合で使用することが可能である。更に特に有利な方法では、cDNA/RNA比は好ましくは1に近いか又は1より大きい。実際、このような実験では、RNAはテスター化合物を形成しそしてcDNAはドライバーを形成し、そして方法の特異性を改良するために、ドライバーがテスターに対して過剰である操作条件を選ぶことが好ましい。他の特に有利な方法では、ss−cDNA/ds−cDNA比は好ましくは1に近いか又は1より大きく、更に好ましくは5より大きい。このような実験では、ss−cDNAはテスターでありそして好ましくは、ドライバー試料からのds−cDNAを追い出すように過剰に使用されるべきである。このような条件では、核酸間の協同性効果が起こりそしてミスマッチは強く退けられる。結果として、観察される唯一のミスマッチは、一般に、ドライバーcDNAには存在せずそしてそれゆえ特異的であると考えられうるテスターRNA又はss−cDNAにおける領域の存在によるものである。ゆえに、本方法の特異性を高めるために、ハイブリッド形成は、約1と約10との間に含まれるcDNA/RNA又はss−cDNA/ds−cDNA比を使用して有利に行われる。この比は操作条件(入手可能な核酸の量、生理学的条件、必要な結果等)に依存して当業者により適合させられうることは理解される。他のハイブリッド形成パラメーター(時間、温度、イオン強度)も当業者により適合させられうる。一般的に言えば、テスター及びドライバーの変性(例えば、加熱により)の後、約37℃の温度で(そして場合により下記する如き温度シフトを行うことにより)そして標準イオン強度条件下に(例えば、0.1M〜5M の範囲のNaCl)約2〜24時間ハイブリッド形成を行う。イオン強度は、特に固体支持体上のハイブリッド形成の場合に、ハイブリッド形成ストリンジェンシーを規定するファクターの1つであることが知られている。
本発明の特定の態様に従えば、ハイブリッド形成は、例えば、Kohno D.E.et al.(Biochemistry.(1977), 16(24): 5329-5341)により記載されたPERT技術(Phenol Emulsion DNA Reassociation Technique)に従って、フェノールエマルションにおいて行われる。有利には、Miller及びRibletの技術(NAR, (1995), 23: 2339)に従い、攪拌の代わりに温度サイクリング(約37℃から約60/65℃への温度シフト)下のフェノールエマルションハイブリッド形成が本発明の範囲内で使用される。特にエマルション相における、いかなる他の液相ハイブリッド形成技術も、本発明の範囲内で使用することができる。かくして、他の特に有利な態様では、ハイブリッド形成は、ホルムアミド80%を含有する溶液中で例えば40℃の温度で行われる。
支持体に固定された相手の1つとハイブリッド形成を行うこともできる。有利には、cDNAが固定化される。これは、特にビオチニル化されたプライマーを使用することによりcDNA標識化(上記参照)を利用することにより行うことができる。ビオチン部分をストレプトアビジン分子でコーティングされた磁性ビーズと接触させる。ついでcDNAは、磁場を加えることによりフィルター又はマイクロタイターディッシュウエルと接触させて保持することができる。次いで、適当なイオン強度条件の下に、RNAをcDNAと接触させる。対を形成していないRNAは洗浄により除去される。ハイブリッド形成されたRNA及びcDNAは磁界を除去すると回収される。
cDNAが二本鎖である場合には、使用されるハイブリッド形成条件は本質的に前記したハイブリッド形成条件と同様であり、そして当業者により適合させられうる。RNAと二本鎖cDNA間でヘテロ三本鎖が形成される場合には、ハイブリッド形成はホルムアミドの存在下に行うことができ、そして複合体は、例えば、60℃〜40℃、好ましくは56℃〜44℃で変わる範囲の温度にさらされてRループ複合体の形成を促進することができる。更に、ハイブリッド形成の後、ホルムアミドが媒体から除去されると、形成された三本鎖構造(triplex structures)を安定化させるための安定剤、例えばグリオキサールを加えることが望ましい(Kaback et al., (1979), Nuc. Acid Res., 6: 2499-2517)。
かくして、本発明に従うこれらのクロスハイブリッド形成は、試験されるべき各生理学的条件の定性的性質を表すcDNA/cDNAホモ二本鎖又はcDNA/RNAヘテロ二本鎖又はヘテロ三本鎖を含む組成物を発生させる。既に言及したとおり、本発明の組成物の各々において、各生理学的条件に特異的な、差異選択的スプライシング(differential alternative splicing)又は他の遺伝子変化に本質的に相当する核酸を同定及び/又はクローニングすることができる。
ゆえに、本発明は、有利には2つの生理学的条件間に発生する遺伝子差を表す核酸を同定及び/又はクローニングするための方法であって、第1生理学的条件の生物学的試料に由来するRNAを第2生理学的条件の生物学的試料に由来する一本鎖cDNAとハイブリッド形成させ、そしてこのようにして形成されたハイブリッドから、対を形成していないRNA領域を同定及び/又はクローニングすることを含む方法に関する。
この第1変法は、更に特定的には、RNAと一本鎖cDNA間のヘテロ二本鎖構造体の形成に基づいている(図2〜4参照)。この変法は、メッセンジャーRNA又は本質的にメッセンジャーRNAの逆転写により、即ち、オリゴdTプライマーの存在下に産生されたcDNAを使用して有利に実施される。
特定の態様では、本発明に従う核酸を同定及び/又はクローニングする方法は、
(a)試験条件に由来するRNAを基準条件に由来する一本鎖cDNAとハイブリッド形成させ、
(b)基準条件に由来するRNAを試験条件に由来する一本鎖cDNAとハイブリッド形成させ、そして
(c)段階(a)及び(b)で形成されたハイブリッドから、対を形成していないRNA領域を同定及び/又はクローニングする、
ことを含む。
特定の別の実施方式においては、本発明の方法は、下記の段階:
(a)生理学的条件A(rA)の生物学的試料からRNAを得、
(b)生理学的条件B(rB)の同じ生物学的試料からのRNAを得、
(c)段階(a)で与えられたrARNAの一部からcDNA(cAcDNA)を及び段階Bで与えられたrBRNAの一部からcDNA(cBcDNA)をポリTプライマーによって調製し、
(d)液相においてrARNAの一部をcBDNAの一部とハイブリッド形成させ(rA/cBヘテロ二本鎖を発生させるために)、
(e)液相においてrBRNAの一部をcAcDNAの一部とハイブリッド形成させ(rB/cAヘテロ二本鎖を発生させるために)、
(f)段階(d)及び(e)で得られたrA/cB及びrB/cAヘテロ二本鎖内の対を形成していないRNA領域を同定及び/又はクローニングする、
段階を含む。
本発明を実施する別の方式に従えば、本発明の方法は、試験条件に由来するRNAを基準条件に由来するニ本鎖cDNAとハイブリッド形成させ、そして得られる二本鎖DNA領域を同定及び/又はクローニングすることを含む。この第2の変法は、更に特定的には、Rループ型構造(図5参照)に由来する、RNAと二本鎖cDNAとのヘテロ三本鎖構造の形成に基づいている。この変法は、本質的にメッセンジャーRNA、又はメッセンジャーRNAの逆転写により、即ち、ポリTプライマーの存在下に産生されたcDNAを使用することによっても好ましく実施される。やはりこの変異態様においは、特定の態様は、本発明に従う2つの核酸集団を発生させる、2つのハイブリッド形成を平行して行うことを含む。この変法では、選択的スプライシングイベントに特異的な所望の領域は、対を形成していないRNA領域ではなくて、その代わりに相同性RNA配列により追い出されなかった二本鎖DNAである(図5参照)。
本発明の他の変法では、2つの試料間で発生する定性的な遺伝的差異(例えば、選択的スプライシングイベンド)を検出するための方法は、第1生物学的試料に由来する二本鎖cDNAを第2生物学的試料に由来するcDNA(二本鎖又は好ましくは一本鎖)とハイブリッド形成させることを含む(図6)。
前記した変法と異なり、この変法は、DNA/RNAヘテロ二本鎖又はヘテロ三本鎖構造を使用しないで、その代わりにDNA/DNA相同性ホモ二本鎖を使用する。この変法は、それがオルタナティブイントロン及びエキソンを示すのみならず、同じ核酸ライブラリー内でエキソン又はイントロンの欠失により形成された特異的連結部(specific junctions)も示すという点で有利である。更に、このようなライブラリーにおける配列はオルタナティブイントロン及びエキソンのフランキング配列に関する情報を与える。
第1の態様に従えば、この方法は、一本鎖cDNAの第1複合集団を二本鎖cDNAの第2複合集団とハイブリッド形成させることを含む。この態様は、基準条件と比較して生理学的試験条件において発生するスプライシングイベントに特徴的な核酸集団を発生することを可能とする(図1A、変法#3、図6A、図26)。後に示されるとおり、この集団は、核酸のクローニング及び特徴付け、診断、スクリーニング、治療及び抗体産生又は全タンパク質又はタンパク質断片の合成のために使用することができる。この集団は、後に示される種々の使用分野に使用されうるライブラリーを発生させるため及び標識されたプローブを発生させるために使用することもできる(図1D)。
他の態様に従えば、この方法は、一本鎖cDNAの第1集団を一本鎖cDNAの第2集団とハイブリッド形成させることを含む。この態様では、試験試料及び基準試料の両方は一本鎖cDNAの形態にある。この態様は、基準試料からの二本鎖cDNAの再アニーリングを回避し、かくして、一方の鎖が試験試料から生じそして他方が基準試料から生じるDNA/DNAホモ二本鎖のみが形成されうる。形成されるハイブリッドは、診断、スクリーニング、治療及び抗体産生又は全タンパク質又はタンパク質断片の合成において使用することができる、2つの試料間で発生する差異スプライシングイベントを表す核酸のクローニング及び特徴付けを可能とする(図1D)。
特定の態様では、本発明に従う核酸を同定及び/又はクローニングする方法は、
(a)試験条件に由来する複数の異なる一本鎖cDNAを含む核酸集団を、基準条件に由来する複数の異なる二本鎖cDNAを含む核酸集団とハイブリッド形成させ、
(b)段階(a)で形成されたハイブリッドから、対を形成していないDNA領域を同定及び/又はクローニングする、
ことを含む。
他の特定の態様では、本発明に従う核酸を同定及び/又はクローニングするための方法は、
(a)試験条件に由来する複数の異なる一本鎖cDNAを含む核酸集団を、基準条件に由来する複数の異なる一本鎖cDNAを含む核酸集団とハイブリッド形成させ、そして
(b)段階(a)で形成されたハイブリッドから、対を形成していないDNA領域を同定及び/又はクローニングする、
ことを含む。
特定の別の実施方式では、本発明の方法は、下記の段階:
(a)生理学的条件A(rA)の生物学的試料からRNAを得(rA)、
(b)生理学的条件B(rB)の同じ生物学的試料からRNAを得(rB)、
(c)段階(a)で与えられたrARNAからcDNA(cAcDNA)を及び段階Bで与えられたrBRNAからcDNA(cBcDNA)を標識された(例えばビオチニル化された)ポリTプライマーによって調製し、
(d)cBcDNAから二本鎖cDNAを調製してdcBcDNAを産生し、
(e)cAcDNAの一部をdcBcDNAの一部とハイブリッド形成させ(例えば、液相で)(dcB/cAcDNAホモ二本鎖を発生させ)、
(f)段階(e)で得られたホモ二本鎖内の対を形成していないDNA領域を同定及び/又はクローニングする、
ことを含む。
他の特定の態様では、本発明に従う核酸を同定及び/又はクローニングする方法は、
(a)1つ又はそれより多くの選ばれた遺伝子又はRNAに由来する一本鎖cDNAを含む核酸集団を、生物学的試料に由来する複数の異なる一本鎖又は二本鎖cDNAを含む核酸集団とハイブリッド形成させ、
(b)段階(a)で形成されたハイブリッドから、対を形成していないDNA領域を同定及び/又はクローニングする、
ことを含む。
特定の別の実施方式では、本発明の方法は、下記の段階:
(a)生理学的条件Aの生物学的試料からRNAを得(rA)、
(b)段階(a)のrAからcDNAを、標識された(例えば、ビオチニル化された)ポリTプライマーによって調製し(cAcDNA)、
(c)場合によりcAcDNAから二本鎖cDNAを調製してdcAcDNAを産生させ、
(d)段階(b)又は(c)の該cAcDNA又はdcAcDNAを1つ又はそれより多くの選ばれた遺伝子又はRNAに由来する一本鎖cDNAとハイブリッド形成させ(例えば、液相で)、そして
(e)段階(e)で得られたハイブリッド以内の対を形成していないDNA領域を同定及び/又はクローニングする、
ことを含む。
この最後の態様に従えば、特定の病態生理学的状況において発生するいかなる選ばれた遺伝子又はRNAの生物学的に関係のあるスプライシング形態を同定することが可能である。特に、与えられた遺伝子のss−cDNA配列を産生しそして上記の方法を行うことにより、特定の組織又は条件において発生する与えられた遺伝子のスプライシング形態の存在、性質及び/又は配列を決定することが可能である。このようにして同定された対を形成していない領域は、該特異的組織又は条件における該遺伝子の生物学的に関係のあるスプライシング形態に相当するであろう。選ばれた遺伝子又はRNAは、関心のあるいかなる遺伝子又は遺伝子のファミリーであってもよく、例えば、ホルモン、サイトカイン、成長因子、腫瘍抑制剤、受容体、イオンチャンネル、転写因子、栄養因子、凝血因子(clotting factors)、リポタンパク質等の遺伝子又は遺伝子のファミリーであることができる。それらは、哺乳動物起源のもの又はいかなる他の起源のもの、例えば、植物、ウイルス等の起源のものであってもよい。特定の態様では、選ばれた遺伝子は、受容体、例えばGタンパク質結合受容体(G-Protein-Coupled Receptor)のすべて又は一部を含む核酸分子である。
調査中の両試料(即ち、病態生理学的条件)について、細胞質ゾルポリA+RNAは、当該技術分野で知られているこれまでに記載された技術により抽出されうる。これらのRNAは、上記したとおり固有のRNアーゼH活性のある又はない逆転写酵素の作用によりcDNAに転換される。次いで、これらの一本鎖cDNAの1つは、ランダムヘキサマーによるプライミング及び当業者により知られている技術に従って二本鎖cDNAに転換される。ゆえに、調査中の条件の1つについて、一本鎖cDNA(「ドライバー」と呼ばれる)がありそして他方の条件について二本鎖cDNA(「テスター」と呼ばれる)がある。これらのcDNAは加熱により変性され、次いでドライバーがテスターに対して過剰であるように混合される。この過剰は1〜50倍、有利には10倍の間で選ばれる。2つの病態生理学的条件で出発して行われる与えられた実験では、ドライバーを発生する条件の選択は任意でありそして収集されたデータの性質に影響を与えてはいけない。実際問題として、上記したアプローチの場合におけると同じく、2つのmRNA集団間で発生する定性的な差を同定するためのストラテジーは、共通のメッセンジャーに存在するこれらの差をクローニングすることに基づいている:このストラテジーは、調査中の条件の1つにおいて独特の配列又は過剰の配列に相当する一本鎖の代わりに二本鎖内に存在する配列をクローニングすることに基づいている。cDNAの混合物を沈殿させ、次いでホルムアミド(例えば、80%)を含有する溶液中にとり取り込まれる(taken up)。ハイブリッド形成は16時間〜48時間、有利には24時間行われる。
特定の態様では、cDNAの1つの集団は試料に由来する一本鎖cDNA集団であり、該集団は、ビオチニル化プライマー(例えば、ビオチニル化オリゴdTプライマー)の存在下に逆転写酵素により得られ、かくしてビオチニル化一本鎖cDNAの発生をもたらす。
特定の態様では、一本鎖cDNA集団と二本鎖cDNA集団とのハイブリッド形成は、95℃でのDNAsの熱変性、続いて相補的配列のハイブリッド形成のために適当なイオン及び温度条件下のインキュベーションにより行われる。このハイブリッド形成から4つの主な分子種:即ち、
−3′−ビオチニル化されている、第1の試料からの一本鎖DNA、
−再アニーリングされた、第2の試料からの二本鎖DNA、
−第2の試料からの変性された一本鎖DNA、及び
−3′−ビオチニル化されている第1の試料からの一本鎖DNAと第2の試料からの変性された一本鎖DNAとのハイブリッド形成により形成されたDNA/DNAホモ二本鎖、が得られる。これらのホモ二本鎖は、2つの試料を区別する遺伝子の差異スプライシングに相当する、一本鎖DNAループの形態の対を形成していない領域(1つ又は複数)を含有する。
後にD節に開示されるように、これらのスプライシングイベント(スプライシングされた形態及びスプライシングされていない形態)に相当する配列を単離し、そして特異的核酸プローブをデザインするのに使用することができる。ハイブリッド形成産生物を沈殿させ、次いで二本鎖DNAのための4塩基認識部位(4-base recognition site)を有する制限エンドヌクレアーゼの作用に付す。ゆえに、このような制限酵素は、ハイブリッド形成期間中に形成された二本鎖cDNAを平均して256塩基毎に開裂するであろう。この酵素は、有利には付着末端を発生するように選ばれる。このような酵素としては、Sau3AI、HpaII、TaqI及びMseIの如き制限酵素が例示される。ゆえに、これらの酵素により消化された二本鎖断片は、開裂された制限部位を使用するクローニングストラテジーに利用可能である。このような断片には2つのタイプがあり:即ち、その2つの鎖が完全に相補的である完全にハイブリッド形成された断片、及び部分的にハイブリッド形成された断片、即ち、二本鎖領域により隣接された(flanked)一本鎖ループを含む断片(図6A)である。少数であるこれらの後者の断片は関心のある情報を含む。完全にハイブリッド形成された断片、これらはcDNA長の大部分から誘導されるので多数である、からそれらを分離するために、ゲル又はいかなる他の適当なマトリックスも使用される。これらの方法は、特に一本鎖DNAループを含有するDNA断片の、電気泳動又はゲルろ過期間中の、より遅い移動を利用する。この方法においては、所望の情報を含む該少数断片は、両集団における同じDNA領域に相当する多数断片から分取的に(preparatively)分離されうる。定性的な差に結び付いたポジティブ及びネガティブフィンガープリントを、同じ集団から単離することを可能とするこの変法は、RNA/DNAヘテロ二本鎖構造体で実施することもできる。この点で、すべての配列が対を形成している相同性ヘテロ二本鎖に比べて、RNAの一部が対を形成していないRNA/DNA二本鎖のより遅い移動の例は、実施例で記載されたgrb2/grb33モデルで説明される(特に図8、レーン2及び3参照)。
D−同定及び/又はクローニング
ハイブリッド形成により発生させた核酸集団から出発して、定性的な差(例えば、差異選択的スプライシングイベント)を特徴付ける領域は、当業者により知られているいかなる技術によっても同定することができる。
D1.RNA/DNAヘテロ二本鎖で出発する同定及び/又はクローニング
かくして、RNA/DNAヘテロ二本鎖の場合に(この方法の第1の変法)、これらの領域は、図3に示されたとおり、対を形成していないRNA領域(RNAループ)として本質的に現れる。ゆえにこれらの領域は、ヘテロ二本鎖及び一本鎖核酸(DNA、RNA)(過剰の未反応の核酸)を分離し、二本鎖RNA(ヘテロ二本鎖構造に参加した部分)を選択的に消化しそして最後に得られる一本鎖RNAを一本鎖DNAから分離することにより同定及びクローニングされうる。
この点で、図3に示された第1アプローチに従えば、対を形成していないRNA領域は、RNA/DNAヘテロ二本鎖に参加したRNAドメインを選択的に消化することができる酵素によるヘテロ二本鎖の処理によって同定される。このような活性を有する酵素は先行技術から知られておりそして商業的に入手可能である。それは、RNアーゼH、例えば、特に組換え技術によりE.coliに由来する商業的に入手可能なRNアーゼHを挙げることができる(Promega カタログ番号 M4281; Life Technology カタログ番号 18021)。かくしてこの第1処理は対を形成していない一本鎖RNA領域と一本鎖cDNAを含む混合物を発生させる。RNAは、当該技術分野で知られたいかなる技術によっても、特にcDNAを産生するのに使用されるこれらのプライマーの標識化に基づいてcDNAから分離することができる(上記参照)。これらのRNAは標的、関心のある遺伝子産生物を同定するため又はいかなる他の用途のための材料のソースとしても使用することができる。これらのRNAは、後に記載するとおり、同じくcDNAに転換されることができ、次いでベクターにクローニングされうる。
この点で、RNAのクローニングは種々の方法で行うことができる。1つの方法は、各RNA端部に適合性プライマーの存在下に逆転写反応のための鋳型として作用するオリゴヌクレオチドを挿入することである。プライマーは、当業者により知られている技術に従って酵素、例えば、ファージT4由来のそして供与体分子の5′リン酸基と受容体分子の3′ヒドロキシル基間の分子間ホスホジエステル結合形成を触媒するRNAリガーゼによって付加することができる。このようなRNAリガーゼは商業的に入手可能である(例えば、Life Rechnologies-GIBCO BRL カタログ番号18003)。このようにして得られたcDNAを、次いで、図3に示されたとおり、適当なプライマーを使用して慣用の技術(例えばPCR)により増幅させることができる。この技術は短いRNA分子(1000塩基より小さい)をクローニングするのに特に適している。
特異的なRNA領域をクローニング及び/又は同定するための他のアプローチは、例えは、RNAに沿ってランダムに転写を開始するランダムプライマーを使用して、RNアーゼHの如き二本鎖RNAに対して特異的に作用する酵素の消化物に対して行われる逆転写反応を含む。次いでこのようにして得られたcDNAを慣用の分子生物学技術に従って、例えば、T4ファージDNAリガーゼ(商業的に入手可能;例えば、Life Technologies-GIBCO BRL カタログ番号18003)によってcDNA端部にオリゴヌクレオチドを付加することにより形成されるプライマーを使用するPCRにより増幅される。この第2の技術は、図4と実施例に説明されている。この技術は、特に長いRNAに適合し、そして後に全イニシャル配列(entire initial sequence)を構築するのに十分な配列データの部分を与える。
特異的RNA領域をクローニング及び/又は同定するための更なるアプローチは、同じくランダムプライマーを使用する逆転写反応に基づいている(図4)。しかしながら、この変法に従えば、使用されるプライマーは少なくとも部分的にセミランダムプライマー、即ち、
−ランダム(縮重した(degenerated))領域、
−規定された程度の制限を有する最小プライミング領域、及び
−安定化領域、
を含むオリゴヌクレオチドである。
好ましくは、これらは、5′→3′方向に、
−8〜24個の規定されたヌクレオチド、好ましくは10〜18個のヌクレオチドを含む安定化領域、
この安定化領域は、それ自体、本発明のセミランダムプライマーによって行われる初期増幅に由来する断片を再増幅するのに使用されるオリゴヌクレオチドの配列に相当することができる。更に、安定化領域は、制限酵素に対応する1つ又はそれより多くの部位、好ましくは非パリンドロームの部位の配列を含むことができる。これは、例えば、このようにして増幅された断片のクローニングを簡単化することを可能とする。安定化領域の特定の例は配列GAGAAGCGTTAT(配列番号1の残基1〜12)により与えられる;
−3〜8ヌクレオチド、更に特定的には5〜7個のヌクレオチドを有するランダム領域、及び
−オリゴヌクレオチドが平均して少なくとも約60塩基対毎に、好ましくは約250塩基対毎にハイブリッド形成するように規定された最小プライミング領域、
更に好ましくは、プライミング領域は、2〜4個の規定されたヌクレオチド、好ましくは3又は4個の、例えばAGGX、式中Xは4つの塩基A、C、G又はTの1つである、の如きヌクレオチドを含む。このようなプライミング領域の存在はオリゴヌクレオチドに平均して約256塩基対毎にハイブリッド形成する能力を与える;
を含むオリゴヌクレオチドである。
特に好ましい方式では、オリゴヌクレオチドは、式:
式中、不変の塩基はPCR実験において自己塩基対形成によるバツクグラウンドを最小にするように配置されており(ordered)、そしてNは4つの塩基が示された位置においてランダム様式で存在することができることを示し、そしてXは4種の塩基A、C、G又はTの1つである、
を有する。このようなオリゴヌクレオチドは同じく本発明の目的を構成する。
この点で、クローニングされるべきRNAに対するプライミングイベントを増加させるように、反応は、
の如きオリゴヌクレオチド、各オリゴヌクレオチド集団(A、B、C、D)は単独で又は他との組み合わせにおいて使用することができる、により平行して行うことができる。
逆転写反応の後、cDNAはオリゴヌクレオチドA又はB又はC又はDを使用するPCRにより増幅される。
上記に示されたとおり、所望のオリゴヌクレオチド集団の複雑性及び特異性に依存して、縮重した位置(degenerated positions)の数は3〜8、好ましくは5〜7の範囲にあることができる。3より少ないと、ハイブリッド形成は限定されそして8より大きいとオリゴヌクレオチド集団は特異的バンドの良好な増幅を確保するのにはあまりにも複雑すぎる。
更に、これらのオリゴヌクレオチドの普遍の3′端部(制限されたプライミング領域)の長さは修正することもできる:4個の不変の塩基を有する上記したプライマーは平均して256塩基対断片の増幅を可能とするが、3個の不変の塩基を有するプライマーはより短い断片(平均して64塩基対)の増幅を可能とする。本発明の好ましい第1の態様では、プライミング領域が4個の不変の塩基を含むオリゴヌクレオチドを使用する。本発明の他の好ましい態様では、3個の不変の塩基のプライミング領域を有するオリゴヌクレオチドを使用する。実際、エキソンは137塩基の平均サイズを有するので、それらは、このようなオリゴヌクレオチドにより有利に増幅される。この点で、例えば、配列番号2,3、及び4を有するオリゴヌクレオチドも参照されたい。
最後に、一般に、RNAの同定及び/又はクローニング段階は、できるだけ多くの情報を発生するようにPCRとクローニングの種々の方法に基づく。
D2.ヘテロ二本鎖で出発する同定及び/又はクローニング
ヘテロ二本鎖構造の場合に(本方法の別の変法)、定性的に異なる領域(挿入、欠失、差異スプライシング)は本質的に、図5に示されるとおり、二本鎖DNA領域の形態で現れる。かくして、このような領域はRNAを消化することができる酵素の如き適当な酵素の存在下にそれらを処理し、次いで一本鎖DNAを消化することができる酵素により処理することによって同定及びクローニングすることができる。かくして、核酸は直接二本鎖DNAの形態で得られ、そして例えば、ベクターpMos−Blue(Amerisham, RPN5110)の如きいかなる適当なベクターにもクローニングされうる。この方法は、ヌクレアーゼ活性を有するように修飾された、所定の配列のRNA又はオリゴヌクレオチドを使用する従来述べられた方法(Landigraf et al., (1994), Biochemistry, 33: 10607-10615)とは区別されるべきである。
D3.DNA/DNAホモ二本鎖で開始する同定及び/又はクローニング(図6)
この態様では、関心のある配列(差異スプライシング)は、図6に示されたとおり、本質的に対を形成しないDNA領域の形態で現れる。このような領域は、適当な酵素の使用及び核酸精製段階を含む、本願に開示された種々の技術に従って同定及びクローニングされうる。
特定の好ましい態様では、対を形成しない領域を含む核酸の集団は、
−二本鎖DNAに特異的な制限酵素により形成されたハイブリッドを消化し、
−対を形成していない領域を含む制限断片を単離し、そして
−単離された断片を増幅する、
ことにより同定又はクローニングされる。
ハイブリッドの消化の前に、分離段階を行って汚染しているハイブリッド(例えば、同じ試料からの2つのDNA鎖間で形成された)を除去することができる。この分離は、ハイブリッド形成の前に1つの試料中のcDNAを標識しそしてハイブリッド形成の後標識されなかったcDNAを除去することにより有利に行われる。特定の態様では、1つの試料に由来するcDNAはビオチニル化され、そして分離段階は、ハイブリッド形成産生物をストレプトアビジンでコーティンされた支持体、例えば、ビーズ(例えば、磁性ビーズ)と接触させることを含む。他の標識、例えば、親和性結合による選択的分離を可能とするアフイニティー対のいかなる他方のパートナーも使用することができることは理解されるべきである。アフイニティー精製されると、2つの分子種が得られる:関心のあるホモ二本鎖及び第1の試料からの出発標識化一本鎖cDNA(starting labeled single-stranded cDNA)。
関心のある対を形成していない領域を単離するために、産生物を二本鎖DNAsに特異的な制限酵素を使用して酵素消化に付す。したがって、ホモ二本鎖のみが消化されそして第1の試料からのいかなる汚染性の標識された一本鎖cDNAも元のままである(intact)。
酵素は、好ましくは、小さなds制限断片を発生するように、ds−DNAsを頻繁に切断する酵素から選ばれる。好ましい態様では、制限酵素は4塩基開裂部位を認識する。このような開裂部位は平均して約250塩基毎に存在する。更なる好ましい態様では、付着末端を形成する。このような酵素の例は、例えば、Sau3A1、HpaII、TaqI、MseIを含む。
この処理の結果として、混合物は3つのタイプの分子種:
−その2つの鎖が完全に相補的である完全にハイブリッド形成したds断片、 −部分的にハイブリッド形成したds断片、即ち、二本鎖領域により隣接された(flanked)された1つ又はそれより多くの対を形成していない領域(即ち、一本鎖ループ)(図6A)。少数であるこれらのds断片は関心のある情報を含む。及び
−第1の試料からの標識された、消化されなかったsscDNA、
を含む。
これらの種を分離しそして部分的にハイブリッド形成されたds断片を単離するために、混合物をゲル又はいかなる他の適当なマトリックスにもよる分離方法に付すことができる。これらの方法は、特に一本鎖DNAループを含有するDNA断片の電気泳動又はゲルろ過期間中のより遅い移動を利用する。この方法において、所望の情報を含む少量断片は両集団における同一のDNA領域に相当する多量断片から分取的に(preparatively)分離されうる。
最も好ましい態様では、部分的にハイブリッド形成されたds断片は、第1の試料から生じる標識された、対を形成していないsscDNAを除去するために、混合物を上記したとおりのストレプトアビジンでコーティングされた支持体で最初に処理することにより単離する。次いで、対を形成していない領域を含むds断片を単離するために、混合物を標識された縮重したオリゴヌクレオチド(degenerated oligonuclotides)と接触させることができる(オリゴヌクレオチド捕捉)。これらの縮重したオリゴヌクレオチドは、すべての可能な配列の組み合わせを表し、かくしていかなるss配列ともハイブリッド形成することができる。これらの縮重したオリゴヌクレオチドは、更に好ましくは、長さが10〜30ヌクレオチド、更に好ましくは約24ヌクレオチドを含む。それらを特異的ハイブリッド形成が起こることを可能とする条件下に混合物と接触させ、それにより対を形成していない領域を含むds断片と特異的に反応させる。このようなハイブリッド形成は標識を使用する分離により該ds断片の捕捉及び単離を可能とする。例えば、標識はビオチンであることができそして対を形成していない領域を含むds断片はストレプトアビジンでコーティングされた支持体(例えば、磁性ビーズ)との接触により単離することができる。
次いで対を形成していない領域を含むds断片は、媒体のイオン強度を低下することにより標識されたオリゴヌクレオチドから分離させる。
単離された断片を、次いで、それらの端部の各々で、アダプター又はリンカー、それらの端部の1つに開裂された制限部位を有する、にライゲーションする。この段階は、当業者に知られている技術に従って、例えば、ファージT4DNAリガーゼによるライゲーションにより行うことができる。このようにして導入された制限部位はcDNA断片の部位と適合性であるように選ばれる。導入されたリンカーは、酵素的増幅(PCR)のためのプライマーを発生させることを可能とする、既知の配列の二本鎖cDNA配列である。
次の段階で、各々同定されるべき定性的な差を有する二本鎖を増幅する。そのために、リンカーと付加した二本鎖cDNAの熱変性の後、これらのcDNA端部の各々を特異的プライミング配列に共有結合させる。適当な特異的プライマーによるPCRに続いて、2つの種類の二本鎖cDNA:即ち、2つの病態生理学的条件を区別する定性的な差に特異的な配列を含有する断片及びこれらのスプライシングイベントのネガティブフィンガープリントを含む断片が得られる。これらの断片をクローニングすると、各スプライシングイベントについてポジティブ及びネガティブフィンガープリントが存在する選択的スプライシングライブラリーを発生する。ゆえに、このライブラリーは、オルタナテイブエキソン及びイントロンへのアクセスのみならず、これらのスプライシングされた配列の切除により形成された特異的連結部へのアクセスも与える。同じライブラリーにおいて、この差異遺伝子情報は、無差別に2つの病態生理学的条件から誘導されうる。更に、同定されたスプライシングイベントの差異な性質をチェックするようにそしてそれらが特異的に引き出される条件を決定するように、ライブラリーにおけるクローンを全mRNA集団の各々に由来するプローブとハイブリッド形成させることができる。
次いで、この方法は、増幅された断片の配列決定、配列をデータベースに記憶させること、配列をデータベースにおいて分析してスプライスドメイン及び相当する連結領域を同定し、該スプライスドメイン又は連結領域に特異的なオリゴヌクレオチドを合成し及び/又は支持体上に該オリゴヌクレオチドを付着させることを更に含むことができる。これらの種々の段階は、cDNAの産生からスプライスオリゴヌクレオチドの付着までコンピューター支援又はコンピューター操作させることができる。
そのように同定された定性的な差に由来するcDNA断片は2つの主な使用:
−適当なベクターにクローニングして調査中の2つの病態生理学的条件間で発生する定性的な差を表すライブラリーを構築すること、
−差異スプライシングイベントの同定を可能とするDNAライブラリーをスクリーニングするためのプローブとしての使用、
を有する。
本発明で使用されるベクターは、特にプラスミド、コスミド、ファージ、YAC、HAC等であることができる。かくして、これらの核酸はそのまま貯蔵することができ、又は複製のために使用されるクローニングベクターと適合性の微生物に導入することができ及び/又は培養物の形態で貯蔵することができる。
各試料について本明細書に記載された方法を行うために必要な期間は、一般に2か月より少なく、特に6週間より少ない。更に、これらの種々の方法は、全体の時間の長さを減少させそして多数の試料の処理を簡単にするように自動化することができる。
この点で、本発明の他の目的は、本発明の方法により同定及び/又はクローニングされた核酸に関する。既に述べたとおり、これらの核酸はRNA又はcDNAであることができる。更に一般的には、2つの生理学的条件に特有の選択的スプライシングに相当する核酸を本質的に含む核酸組成物に関する。更に特定的には、これらの核酸は生物学的試験試料において同定されそして基準条件下の同じ生理学的試料に存在しない選択的スプライシングに相当する。本発明は、同じく、このようにしてクローニングされた核酸の、治療的又は診断生成物として又は後に記載するとおり活性分子を同定するためのスクリーニング手段としての使用に関する。
ゆえに上記に開示された種々の方法はすべて2つの病態生理学的条件間の差異スプライシングされた遺伝子情報を表すcDNA配列のクローニングをもたらす。かくしてこれらの方法の1つに由来するクローンの全体の組は関心のある2つの条件間に発生する定性的な差を表すライブラリーを構築することを可能とする。
E−定性的ライブラリーの発生
この点で、本発明は更に生物学的試料の与えられた生理学的状態を表す核酸ライブラリーを作成する方法を指向する。この方法は、有利には、該生理学的状態の遺伝子発現(例えば、選択的スプライシング)の定性的マーカーを表すが基準状態においては存在しない核酸をクローニングして、調査されるべき2つの状態間で発生する定性的な差に特異的なライブラリーを発生させることを含む。
これらのライブラリーは、プラスミド又はファージベクターに挿入されたcDNAにより構築される。このようなライブラリーはニトロセルロースフィルター又は当業者により知られているいかなる他の支持体、例えばチップ又はバイオチップ上に付着させることができる。
定性的差異スクリーニングの特徴の1つ又は独創的特徴の1つは、この技術が、1つではなくて、有利には、2つの与えられた条件間で発生する定性的な差の全体の組を表す2つの差異ライブラリー:ライブラリー対(図1D参照)をもたらすことである。
かくして、本発明は、好ましくは、第1の生物学的試料に由来するRNAを第2の生物学的試料に由来するcDNAとハイブリッド形成することにより得られうるいかなる核酸組成物又はライブラリーにも関する。更に好ましくは、本発明のライブラリー又は組成物は、2つの生物学的試料間の発現の定性的な差を表す核酸を含みそして(i)第1の生物学的試料に由来するRNAと第2の生物学的試料に由来するcDNAとの少なくとも1つのハイブリッド形成段階、(ii)発現の定性的な差を表すこれらの核酸を選び、そして場合により(iii)該核酸をクローニングすることを含む方法により発生される。
更に、このようなライブラリーが構築されと、得られるライブラリーの特異性を改良するために、クローン選択の段階を進行させることが可能である。実際に、観察されるあるミスマッチは単に定性的な差(例えば、差異選択的スプライシング)のみによるのではなくて、例えば、逆転写欠陥から生じうるということがありうる。このようなイベントは一般に有意ではないけれども、核酸クローニングの前にそれらを防止するか又はそれらの発生率を減少させることが好ましい。これを達成するために、ライブラリークローンを調査されるべき両生理学的条件において発生するcDNA集団とハイブリッド形成させることができる(上記段階c)。非差異方式で両集団とハイブリッド形成するクローンは、非特異的であると考えられそして場合により捨てられるか又は第2プライオリティとして処理されるであろう(実際、試験試料における新しいアイソフォームの出現は、基準試料に存在する最初のアイソフォームがこの試験試料から消失したことを必ずしも示さない)。いずれかの集団の1つのみとハイブリッド形成するか又は集団の1つと優先的にハイブリッド形成するクローンは特異的であると考えられそして濃縮又は精製されたライブラリーを構築するのに優先して選ばれうる。
精製段階は、ハイブリッド形成すること又及び統計的に関係がある数(statistically relevant number)の病理試料に由来するプローブによってクローンの同一性をチェックすることにより行うこともできる。
ゆえに、本願は、同じく、生理学的条件の典型的な選択的スプライシングに特異的な核酸を含むいかなる核酸ライブラリーも指向する。これらのライブラリーは有利には、選択的スプライシングの特異的RNA領域に相当する一般に二本鎖のcDNAを含む。このようなライブラリーは、一般にクローニングベクター内に組み込まれた核酸、又は該核酸を含有する細胞培養物からなることができる。
最初のRNAの選択は、得られるライブラリーの特徴を部分的に決定する:
−両条件A及びBのRNAは当業者に知られている技術に従って単離されたmRNA又は全成熟RNA(total mature RNAs)である。かくしてライブラリーはいわゆる制限された定性的差異スクリーニングライブラリーである。何故ならば、それらは両病態生理学的条件の成熟RNAを特徴付ける定性的な差に制限されるからである。
−両条件の一方のRNAはmRNA又は成熟した全RNAであるが、これに対して
他方の条件のRNAは、細胞核から当業者に知られている技術に従って単離された、スプライシングによりプロセッシングされていないプレメッセンジャーRNAである。この状況においては、得られるライブラリーはいわゆる複合差異スクリーニングライブラリーである。何故ならば、成熟RNA間の差に制限されるのではなくて、すべてのイントロンを含む、他方には欠けている与えられた条件におけるスプライシングされた転写物の全体の組を含むからである。
−最後に、RNAは単一の病態生理学的条件から生じることができ、そしてこの場合に差異スクリーニングは同じ試料からの成熟RNA及びプレメッセンジャーRNAを含む。このような場合に、得られるライブラリーは自己由来の定性的差異スクリーニングライブラリーである。このようなライブラリーの有用性は、それらが与えられた条件において転写されたイントロンの全範囲をもっぱら含むという点にある。それらが異なる条件の成熟RNAに由来するプローブとハイブリッド形成するかどうかは、調査中の条件がイントロンを固持すると共にそれらの容易な同定を与えることにより特徴付けられるならば、迅速に確かめることを可能とする。
一般的にいえば、ライブラリーは、クローニングされた核酸により形質転換された細胞培養物の、固体培地上(特に、寒天培地上)へのスプレッディングにより発生される。形質転換は、当業者に知られているいかなる技術(トランスフェクション、リン酸カルシウム沈殿、エレクトロポレーション、バクテリオファージによる感染等)によっても行われる。細胞培養物は、一般にバクテリア培養物、例えばE.coliである。それは、真核細胞培養物、特に下等真核細胞(例えば酵母)であることもできる。このスプレッディング段階は皿又はいかなる他の適当な支持体上でも無菌条件で行うことができる。更に、寒天培地上の広がった培養物は例えば冷凍した状態で(グリセロール又はいかなる他の適当な作用物質中でも)貯蔵することができる。当然、これらのライブラリーは、「デュプリケート(duplicate)」、即ち、後に更に完全に記載されるよく知られた技術に従って作られたコピーを産生するために使用することができる。更に、このようなライブラリーは、一般に、増幅されたライブラリー、即ち、各クローンを増幅された状態で含むライブラリーを作成するのに使用される。増幅されたライブラリーは、下記の如く作成される:即ち、広がった培養物から出発して、すべての細胞クローンを回収し、そしていかなる適合性の培地でも使用して冷凍された状態で又は冷所に貯蔵するためにパッケージする。この増幅されたライブラリーは、有利にはE.coliバクテリア培養物から作成されそして無菌条件で4℃で貯蔵される。この増幅されたライブラリーは、種々の用途のために種々の支持体上にこのようなクローンを含有するいかなるその後に作成されるライブラリーの作成及び制限のない複製も可能とする。このようなライブラリーは、更に、関心のあるいかなるクローンの単離及び特徴付けも可能とする。本発明のライブラリーを構成する各クローンは、実際に生理学的条件の特徴的エレメントであり、そしてそれゆえマーカー、抗体産生、診断、遺伝子導入治療等のためのサーチの如き種々の調査のための特に興味のある標的を構成する。これらの種々の用途は以下に更に詳細に論議される。ライブラリーは、一般に、適当な支持体(例えばペトリ皿)上の寒天培地に培養物をスプレッディングすることにより上記したようにして作成される。寒天培地を使用することの利点は各コロニーを分離しそして別個に認識することができるということである。この培養物から出発して、同じデュプリケートを、当該技術分野で知られている技術に従ういかなる適当な支持体上でもレプリカ平板培養(replica-plating)により簡単に実質的量において作成することができる。かくして、デュプリケートは、フィルター、その上に細胞付着が可能である膜(ナイロン、ニトロセルロース等)によって得られうる。次いでフィルターは、核酸を変化させないいかなるパッケージング培地中にも、乾燥された状態で例えば4℃でそのまま貯蔵されうる。フィルターは、同じく細胞、タンパク質等を捨てそしてこのような成分のみを核酸として保持することのような方式で処理することができる。これらの処理法は、特に、プロテアーゼ、洗剤等の使用を含むことができる。処理されたフィルターは、いかなる装置においても又は核酸のために許容できるいかなる条件下にでも同じく貯蔵することができる。
核酸ライブラリーは、同じくバイオチップ又はいかなる他の適当な装置へのトランスファーにより核酸から直接作成することができる。
本発明は、同じく2つの生理学的条件を区別する選択的スプライシングイベントの特異的なオリゴヌクレオチドを含むいかなるライブラリーも指向する。これらは、有利には、5〜100mer、好ましくは、50mer未満、例えば、25merの範囲の一本鎖オリゴヌクレオチドである。
これらのオリゴヌクレオチドは、与えられた条件又はタイプの生理学的条件を表す選択的スプライシングに特異的である。かくして、このようなオリゴヌクレオチドは、例えば、試験核酸集団及び基準核酸集団に特徴的な選択的スプライシングイベントを表すオリゴヌクレオチドであることができる。これらのオリゴヌクレオチドは、好ましくは、例えば、特異的イントロン又はエキソンから好ましくは調査中の2つの状況の1つにおいて発現された配列から誘導されうるか又はそれらはエキソン又はイントロンの保持又は欠失により形成された連結部に相当することができる。
ある選択的スプライシングイベントがアポトーシス条件において観察されることが文献に報告されている。これは、例えば、Bclx、Bax、Fas又はGrb2遺伝子内のスプライシングについて特に当てはまる。文献及び/又はデータベースにおいて入手可能な公表されたデータ又は配列を参照することにより、スプライシングされた形態又はスプライシングされていない形態に特異的なオリゴヌクレオチドを発生させることが可能である。これらのオリゴヌクレオチドは、例えば、下記のストラテジーに従って発生させることができる:即ち、
(a)アポトーシス条件に特徴的なタンパク質又はスプライシングイベント及びスプライシングされたドメインの配列を同定すること。この同定法は公表されたデータ又はデータベースにおいて入手可能な配列の編集(compilation)に基づくことができる。
(b)このドメインの1つ又はそれより多くの領域に相当する1つ又はそれより多くのオリゴヌクレオチドを人工的に合成すること。これはゆえにハイブリッド形成により試験試料のRNAにおけるスプライシングされていない形態の同定を可能とする。
(c)スプライシングされたドメインにより分離された2つのドメイン間の連結領域に相当する1種又はそれより多くのオリゴヌクレオチドを人工的に合成すること。これらのオリゴヌクレオチドはそれゆえハイブリッド形成により試験試料のRNAにおけるスプライシングされた形態の同定を可能とする。
(d)アポトーシス条件に特徴的な他のタンパク質又はスプライシングイベントに関して上記した段階(a)〜(c)を繰り返すこと。
(e)上記に同定されたメッセンジャーのアポトーシス形態に特異的な1種又は複数種のオリゴヌクレオチドを第1の適当な支持体上に移しそして非アポトーシス形態に特異的な1種又はそれより多くのオリゴヌクレオチドを他の適当な支持体上に移すこと。
このようにして得られた2つの支持体は、細胞又は試験試料の生理学的状態及び特にそれらのアポトーシス状態を、このような細胞又は試料に由来する核酸調製物のハイブリッド形成により評価するために使用することができる。
異なる病態生理学的状態(神経変性、毒性、増殖等)に特異的なオリゴヌクレオチドを使用して、他の同様なライブラリーを発生させることができ、かくして用途の範囲を広げることができる。
オルタナティブイントロン又はエキソンライブラリーは、個々の生物、組織又は細胞培養物のゲノムに関する情報が記録されているデータベースをシステマティックに分析することにより編集されたコンピューター化されたデータベースシステムの形態にあることもてきる。このような場合に、このようなバーチャルなデータベースの精巧化により得られたデータは、2つの病態生理学的条件を平行して試験するのに役立つオリゴヌクレオチドプライマーを発生させるのに使用することができる。
コンピューター化されたデータベースは、与えられたクラスのタンパク質を表す又は特定の配列に特異的な多様なヌクレオチドプローブを誘導するのに更に使用することができる。次いで、これらのプローブは、種々のオルタナティブイントロン及びエキソンクローニング技術に由来するクローンライブラリー上に付着させて、これらの分子ライブラリーの複雑度を認識しそして与えられたクラスのタンパク質又は与えられた規定された配列が、2つの異なる病態生理学的状態を比較するとき、差異的にスプライシングされるかどうかを迅速に決定することができる。
本発明に従う更なる核酸組成物又はライブラリーは、本発明の方法(DATAS)に従って同定された配列から発生させたアンチセンスライブラリーである。このタイプのライブラリーを発生させるために、このような配列はクローニングされて、DATASのために使用されたメッセンジャーRNAに対してアンチセンスオリエンテーション(antisense orientation)に相当するRNA断片として発現される。これはいわゆるアンチセンスライブラリーをもたらす。このアプローチは、クローニングされた断片のオリエンテーションを可能とするクローニング変異体を優先的に利用する。このようなアンチセンスライブラリーの有用性は、それが細胞系のトランスフェクションを可能としそして形態的であろうが酵素的であろうが又はレポーター遺伝子又は選択剤に対する抵抗を与える遺伝子の使用により示されるすべての表現型の変化(phenotypic alterations)の監視を可能とするということである。アンチセンス発現ベクターの導入の後の表現型変化の分析は、一般に、いわゆる安定なクローン、即ち、発現ベクター及び宿主ゲノムの協同した複製を可能とするクローンの選択の後行われる。この協同は、細胞ゲノムへの発現ベクターの組込みにより可能とされるか又は発現ベクターがエピソームであるとき選択的圧力により可能とされる。このような選択的圧力は、トランスフェクションされた細胞培養物を、発現ベクターにより担われた遺伝子の産物が細胞内で発現されるときにのみ脱毒性化されうる毒性剤で処理することにより加えられる。これは宿主及び導入遺伝子複製間の同期化をもたらす。有利には、与えられた細胞内で50〜100ベクターコピーの発現を可能とするエプスタインバーウイルスに由来するエピソームベクターが使用される(Deiss et al., (1996), EMBO J., 15: 3861-3870; Kissil et al., (1995), J.Biol.Chem, 270: 27932-27936)。
これらのアンチセンスライブラリーが含有するDATAS配列に関係するこれらのアンチセンスライブラリーの利点は、その発現が抑制されて選ばれた表現型を産生する遺伝子の同定を可能とするのみならず、この遺伝子のどのスプライシングアイソフォームが影響を受けたかの同定も可能とするということである。アンチセンス断片が与えられたエキソンを標的とする場合に、タンパク質ドメイン及び従ってこのドメインが関与する機能は観察された表現型に反対の作用をすることはそれから推定できる。この点で、DATASとアンチセンスアプローチとのカップリングは機能的ゲノミクスに対する近道を表す。
本発明は、上記したスプライスオリゴヌクレオチドをデサインするための配列情報を得るための顕著な利点を与える。特に、本発明は、ポジティブ及びネガティブイベント(即ち、スプライスされたドメイン及びスプライスされていないドメイン)の両方を得ることを可能とする。かくして本発明は、2つの病態生理学的状態又は与えられた状況を区別するスプライシングによりリクルートされたエキソン−エキソン及びエキソン−イントロン連結部を特徴付ける配列のすべてに対するアクセスを与えるライブラリーの作成を可能とする。
F−DNAチップ
本発明は、更に上記した核酸組成物又はライブラリーを含むいかなる支持体材料(膜、フィルター、バイオチップ、チップ等)も指向する。これは更に特定的には細胞ライブラリー又は核酸ライブラリーであることができる。本発明は、本発明に従ういくつかのライブラリーを含むいかなるキット又は支持体にも関する。特に、基準生理学的条件に対する試験生理学的条件の定性的特徴を表すライブラリーと、コントロールとして、試験生理学的条件に対する基準生理学的条件の特徴を表すライブラリー(「ライブラリー対」」を平行して使用することが有利でありうる。かくして、本発明に従う有利なキットは、2つの生理学的条件に属する2つの差異定性的ライブラリー(differential qualitative libraries)(「ライブラリー対」)を含む。1つの特定の態様に従えば、本発明に従うキットは、例えば、異なる生理学的状態又は異なる生物学的試料に相当する上記したいくつかのライブラリー対を含む。キットは、例えば、共通の支持体上に順次に配列されたこれらの異なるライブラリー対を含むことができる。
上記したこの発明の特定の態様は、スプライスオリゴヌクレオチドアレイ、即ち、エキソンとイントロンを識別することができるオリゴヌクレオチドでコーティングされた支持体材料である。オリゴヌクレオチドはエキソン又はイントロン配列及び/又はエキソン−エキソン又はイントロン−エキソン(いかなるオリエンテーションにおいても)連結領域に対して特異的であることができる。かくして本発明の特定の目的は、支持体材料に固定化された複数のオリゴヌクレオチドを含む産生物であって、(i)該オリゴヌクレオチドは遺伝子又はRNAのエキソン−エキソン又はエキソン−イントロン連結領域に対して相補的及び特異的な配列を含み、(ii)該オリゴヌクレオチドは5〜100ヌクレオチドの長さを有し、そして(iii)該産生物は同じ遺伝子又はRNAの異なるエキソン−エキソン又はエキソン−イントロン連結領域に対して相補的及び特異的な少なくとも2組のオリゴヌクレオチドを含み、
該産生物はハイブリッド形成が起こることを可能とする条件下に核酸を含有する試料と接触させると、該試料中の連結領域の存在又は不存在の決定を可能とする、産生物も含む。
上記したとおり、支持体上の核酸は好ましくは配置されており、即ち、支持体の既知の分離した区域又は「セル」に位置している。2〜1000組の異なるオリゴヌクレオチド又はそれより多くを含む、支持体に結合した複数の(組の)オリゴヌクレオチドがあることができる。それらは、支持体材料の表面に高い又は低い密度で付着させることができる。オリゴヌクレオチドは、好ましくは、予め決定された幾何学的配列において支持体の表面に付着される。特に、支持体上の特定の「セル」の形状、サイズ及び位置は標準化して、自動的評価を可能とするか又は容易にすることができる。従って、オリゴヌクレオチドの各組は、担体材料の表面の規定された位置を有する「セル」に対応する。セルの数は状況に依存して少数〜数百まで変わることができる。
試料における連結領域の存在又は不存在の決定のための産生物の有効性を増加させるために、下記の特徴の少なくとも1つを有するオリゴヌクレオチドを使用することが特に好ましい。
−長さが10〜60ヌクレオチド、更に好ましくは長さが10〜50ヌクレオチド、なお更に好ましくは長さが10〜40ヌクレオチドであるオリゴヌクレオチド。オリゴヌクレオチド配列は、有利には標的スプライスドメイン又はスプライス連結部に集中するが、オルタナティブ配置(alternative configuration)も使用することができる。最も好ましい態様では、オリゴヌクレオチドは長さが18〜30ヌクレオチド、更に特定的には長さが約24ヌクレオチドを含有しそして本質的に標的スプライスドメインに中心を置く(即ち、オリゴヌクレオチド配列の少なくとも40%、好ましくは少なくとも45%が標的スプライス連結部の各部位から延びている)。特定の態様では、オリゴヌクレオチドはスプライス連結部に完全に中心を置いた24mer(即ち、オリゴの配列の12ヌクレオチドがスプライス連結部の各側にハイブリッド形成する)。
−25〜65%、好ましくは30〜60%のGC含有率を有するオリゴヌクレオチド。GC含有率はオリゴヌクレオチドの長さに依存して当業者により調節されうる。40merでは、30%〜60%のGC含有率を有することが好ましい。24merでは、40%〜60%のGC含有率を有することが好ましい。
−60〜80℃の溶融温度を有するオリゴヌクレオチド。溶融温度は、オリゴヌクレオチドの長さに依存して当業者により調節されうる。例えば、40merでは、65〜75℃の溶融温度を有することが好ましい。24merでは、65〜70℃の溶融温度を有することが好ましい。
−ヘアピン傾向及び/又は自己ニ量体化傾向を本質的に欠いているオリゴヌクレオチド。
上記した1つの単一産生物においては、お互いに対して均質なオリゴヌクレオチド、即ち、上記したとおりの同様な特徴を有するオリゴヌクレオチドを使用することが好ましい。
本発明の更なる目的は、核酸のアレイを作成する方法であって、
a)第1の試料に由来する複数の異なるcDNAを第2の試料に由来する複数の異なるcDNAとハイブリッド形成させ、該生物学的試料の少なくとも1つにおけるcDNAの組成又は配列は少なくとも部分的に未知であり、
b)a)で形成されたハイブリッドから、対を形成していない領域を含む核酸の集団を同定又はクローニングし、該クローニング又は同定された核酸は該試料間で差異的にスプライシングされる遺伝子の部分に相当する対を形成していない領域を含み、
c)b)でクローニング又は同定された核酸に特異的な核酸プローブ、好ましくはオリゴヌクレオチドプローブを合成し、そして
d)支持体上に該核酸を付着させて核酸のアレイを作成する、
ことを含む方法である。
本発明は、スプライスオリゴヌクレオチドのアレイを作成する方法であって、
−スプライシングされた形態及びスプライシングされていない形態の1つ又は複数の遺伝子の配列を含む核酸配列のライブラリーを提供し、
−該形態の該遺伝子におけるスプライシングにより創られた連結部の配列を決定し、該連結部は該形態の該遺伝子に特異的であり、
−該連結部配列に相補的及び特異的なオリゴヌクレオチドを合成し、該オリゴヌクレオチドは10〜100ヌクレオチド、好ましくは10〜60ヌクレオチドの長さを有し、そして
−支持体上に該オリゴヌクレオチドを付着させてスプライスオリゴヌクレオチドのアレイを作成する、
ことを含む方法に関する。
配列のライブラリーは、上記した方法により作成することができる。連結部の配列は当該技術分野で知られている種々の方法により決定することができる。典型的には、ライブラリーにおける配列を互いに比較して相補的部分を同定する。このような相補的部分は連結領域を規定する欠失した又は挿入された配列も同定する。このような連結領域に特異的なオリゴヌクレオチドは当該技術分野で知られた技術を使用して、典型的には化学的合成によりデザイン及び合成することができる。有利には、オリゴヌクレオチドは上記した特徴の少なくとも1つを示す。支持体上のこれらのオリゴの付着は、多様な技術により達成することができる(活性化された支持体との直接の結合、スペーサー基による間接の結合、化学的カップリング、非共有結合又は共有結合カップリング、電気的カップリング等)。担体材料上にポリヌクレオチドを固定化するための種々の方法が当該技術分野において、例えば、GB2,197,720、FR2,726,286及びWO97/18226に記載されている。これらの特許は参照により本明細書に組み込まれる。受動的吸着による固定化は、例えば、Inouye(J.Clin.Microbiol.28(1990)1469)に記載されている。共有結合又は紫外線による固定化は、例えばMorrissey et al.(Mol.Cell.Probe 3(1989)189)に記載されている。上記したとおり、支持体は固体又は半固体であることができそしてガラス、ポリマー、プラスチック、シリカ、金属、ゲル、ポリスチレン、テフロン(登録商標)又はナイロン又は例えばEP373203及びWO90/15070に記載されたいかなる他の材料も含むことができる。担体材料の典型的な例は3Dリンク活性化スライド(3D-link activated slides)(Motorola)を含む。核酸は好ましくは支持体の表面に規則的に位置している(ordered)。それらの密度は当業者により調節されうる。
上記方法の特定の態様では、オリゴヌクレオチド合成及び付着は、例えば光リソグラフィー又は圧電方法を使用して、同時に、即ち、チップ上のオリゴヌクレオチドのin situ合成により達成される。in situ合成方法の例はUS50,510,270及びUS5,700,637に開示されている。これらの特許は参照により本明細書に組み込まれる。
上記の方法は、有利にはコンピューター支援又はコンピューター操作される。特に、オリゴヌクレオチドのデザインは、種々のソフトウエア、例えば、ArrayDesigner2、Featurama及びPrimerFinderにより操作されうる。支持体上のオリゴヌクレオチドのスポッティングはロボット装置、例えば、MicroGridll (BioRobotics)により操作されうる。
G−プローブの発生
2つの病態生理学的状態間に発生する定性的な差を表す本発明に従うcDNA組成物の他の使用は、そのプローブを誘導することからなる。このようなプローブは実際に使用して2つの病態生理学的条件間の差異スプライシングイベントをスクリーニングすることができる。
これらのプローブ(図1D参照)は、当該技術分野で知られた慣用の技術に従って核酸ライブラリー又は集団を標識することにより調製することができる。かくして、標識化は、好ましくは、酵素的、放射性、蛍光、免疫学的手段等により行うことができる。標識化は好ましくは放射性又は蛍光性である。このタイプの標識化は、例えば、標識されたヌクレオチドを核酸集団(合成の後又は合成期間中)に導入して、慣用の方法によるそれらの可視化を可能とすることにより達成されうる。
ゆえに、1つの用途は、慣用のゲノムライブラリーをスクリーニングすることである。このようなライブラリーは、ベクターがファージ又はコスミドから誘導されるかどうかに依存して、10kb〜40kbのDNA断片を含むことができる。かくして、DATASにより発生されたプローブとハイブリッド形成しそして2つの条件間で発生する差異スプライシングイベントを表すクローンの数は、かくして選択的スプライシングにより影響を受けた遺伝子の数を、それらが調査されている一方又は他方の条件で発現されているかどうかに従って、ほぼ反映する。
好ましくは、本発明のプローブは、スプライシングイベントを同定するのに適合したゲノムDNAライブラリー(一般にヒト起源の)をスクリーニングするのに使用される。このようなゲノムライブラリーは、好ましくは、統計的に単一の差異スプライシング可能なエレメントのみ、即ち、単一のエキソン又は単一のエキソンのみを生じさせるように、制限されたサイズのDNA断片(一般にベクターにクローニングされた)からなる。ゆえに、ゲノムDNAライブラリーは、ゲノムDNAを4塩基により制限された認識部位を有する酵素で消化し、かくして制御された消化により1kbの平均サイズを有するDNA断片を得る可能性を与えることにより作成される。このような断片は、高等真核生物ゲノムを表すDNAライブラリーを構成するのに107クローンの発生を必要とする。このようなライブラリーは同じく本願の目的である。ついでこのライブラリーを定性的差異スクリーニングに由来するプローブとハイブリッド形成させる。実際、調査されそして2つの病態生理学的条件A及びBを比較する各実験では、2つのプローブ(プローブ対)が得られる。1つのプローブは条件Aの特徴的なスプライシングイベントに富んでおりそして他方のプローブはBの特徴的スプライシングマーカーに富んでいる。いずれかのプローブの1つと優先的にハイブリッド形成するゲノムライブラリーにおけるクローンは対応する病態生理学的条件において優先的にスプライシングされる配列を有する。
本発明の方法は、かくして遺伝子発現における定性的な差のシステマチックな同定を与える。これらの方法は、例えば、毒物学、薬理学又は更に薬理ゲノミックスの分野において関心のある分子の同定及び/又はクローニングに関係する多くの用途を有する。
H−用途
ゆえに、本発明は、更に、治療又は診断的価値のある分子を同定するための前記した方法、核酸又はライブラリーの使用に関する。本発明は更に特定的には病理学において変化させられるタンパク質又はタンパク質ドメインを同定するための上記した方法、核酸又はライブラリーの使用に関する。
これらの技術の主要な強みの1つは、実際、メッセンジャー内の、結果として対応するタンパク質内の与えられた障害において影響を受ける機能的ドメインの同定である。これは、病理学的状態の進展及び持続における与えられたドメインの重要性を評価することを可能とする。病理学的障害の影響を与えられたタンパク質ドメインに制限することの直接の利点は、該タンパク質ドメインが治療目的の小さな分子をスクリーニングするための関係のある標的と考えることができるということにある。この情報は更に遺伝子治療により送達されうる治療的に活性なポリペプチドをデザインするためのキーを構成する:即ち、このようなポリペプチドは特に本明細書に記載された技術により同定されるドメインに対する中和抗体に由来する一本鎖抗体であることができる。
更に特定的には、本発明に従う方法は、
−オルタナティブエキソン(alternative exons)に由来するコード配列であることができ、
−2つの病態生理学的状態間の差異スプライシングされたイントロンにより生じた非コード配列に相当することができる、
分子を提供する。これらの2つの点から種々の情報を得ることができる。
2つの病態生理学的状態を区別するエキソンの選択的スプライシングは、特定のタンパク質の1つ又はそれより多くの機能をモジュレートする(より正確な用語では、抑制又は回復する)ことができる遺伝子発現の調節機構を反映する。ゆえに、構造的及び機能的ドメインの大部分(SH2、SH3、PTB、PDZ及び種々の酵素の触媒ドメイン)はいくつかの連続的エキソン(contiguous exons)によりコードされているので、2つの形状(configurations)を考慮することができる:
i)ドメインは病理的条件においてトランケーションされる(Zhu, Q. et al., (1994), J.Exp.Med., 180(2): 461-470);これは、このようなドメインが関与するシグナル伝達経路(signaling pathways)は治療目的で回復されなければならないということを示す。
ii)ドメインは病理的障害の過程で保持されるが、これに対してそれらは健康な状態では存在しない;これらのドメインはこのようなドメインにより媒介されるシグナル伝達に拮抗することを意図した低分子量化合物に対する標的をスクリーニングすると考えられうる。
差異スプライシングされた配列は、コード配列の5′又は3′に位置した非コード領域に相当するか又は2つのコードエキソン間に存在するイントロンに相当することができる。非コード領域においては、これらの差異スプライシングは、メッセンジャー安定性又は翻訳性の変更(modification)を反映することができる(Bloom, T.J. and Beavo, J.A., (1995), Proc.Natl.Acad.Sci.USA, 93(24): 14188-14192; Ambartsumian, N.et al., (1995), Gene, 159(1): 125-130)。これらの現象の調査はこのような情報に基づいて行われるべきであり、そして相当するタンパク質をその蓄積又は消失を考慮して候補標的とみなす(qualify)かも知れない。コード配列におけるイントロンの保持は、リーディングフレーム内に停止コドンを導入することによりネイティブタンパク質のトランケーションをもたらす(Varesco., L., et al., (1994), Hum.Genet., 93(3): 281-286; Canton, H., et al., (1996), Mol.Pharmacol., 50(4), 799-807; Ion, A., et al., (1996), Am.J.Hum.Genet., 58(6): 1185-1191)。このような停止コドンが読みとられる前に、一般に多数の追加のコドンの翻訳が起こり、それにより選択的スプライシングのタンパク質マーカーとして挙動する特定の配列が翻訳されたタンパク質に付加される。これらの追加のアミノ酸は、病理学的条件に固有の選択的形態に特異的な抗体を産生させるのに使用することができる。これらの抗体は次いで診断ツールとして使用することができる。トランケーションされたタンパク質は、性質の変更(change)又は変化(alteration)すら受ける。かくして、酵素はそれらの触媒ドメイン又は調節ドメインをあいまいにする(loose)ことができ、不活性になるか又は構成的に活性化されうる。アダプターは、それらのシグナル伝達カスケードの種々のパートナーを連結する能力を失うことがある(Watanabe, K.et al., (1995), J.Biol.Chem., 270(23): 13733-13739)。受容体のスプライシング産生物は、それらの対応するリガンドに結合する能力を失った受容体の形成をもたらすことがあり(Nakajima, et.al., (1996), life Sci., 58(9): 761-768)そしてそれらの細胞外ドメインの放出により受容体の可溶性形態を発生させることもできる(Cheng J., (1994), Science, 263(5154): 1759-1762)。この場合に、診断試験は、種々の生理学的流体中の与えられたリガンドに結合する受容体の循環する可溶性形態の存在に基づいてデザインされうる。
本発明は、更に特定的には、病理学に関与するタンパク質に対して特異的な抗原性ドメインを同定するための前記した方法、核酸又はライブラリーの使用に関する。本発明は病理学的状態を診断するための上記した核酸、タンパク質又はペプチドの使用にも関する。
本発明は、病理に関与するタンパク質又はタンパク質ドメインを同定及び/又は製造するための方法であって、
(a)病理試料のメッセンジャーRNAを健康な試料のcDNAとハイブリッド形成させるか又はその逆、又は両方を平行して行うこと、
(b)形成されたハイブリッド内で、健康な状態に対して病理状態に特異的な定性的な差(対を形成しない(RNA)又は対を形成した(二本鎖DNA))に相当する領域を同定すること、
(c)段階(b)で同定された1つ又はそれより多くの領域に相当するタンパク質又はタンパク質ドメインを同定及び/又は産生させること、
を含む方法にも関する。
そのように同定された領域は、一般に、差異スプライシングに相当するが、それらは例えば挿入(1つ又は複数)又は欠失(1つ又は複数)の如き他の遺伝子変化に相当することもできる。
タンパク質(1つ又は複数)又はタンパク質ドメインを単離し、配列決定しそして治療又は診断用途において、特に他の抗体産生のために使用することができる。
この点をよりよく説明するために、本発明の定性的差異スクリーニングはがん抑制遺伝子を有利に同定することを可能とする。実際、例は、抑制遺伝子が腫瘍の進行中に不活性化される1つの方法は選択的形態(alternative forms)のスプライシングのモジュレーションによる不活性化であることを実施例は示すことができる。
このゆえに、小細胞肺癌において、RBファミリーに属するタンパク質p130(網膜芽細胞腫タンパク質)の遺伝子は、コンセンサススプライシング部位で突然変異させられる。この突然変異は、エキソン2の除去及び未成熟停止コドン(premature stop codon)の存在によるタンパク質の合成の欠如を生じさせる。この観察は、腫瘍形成におけるRBファミリーメンバーの重要性を強調するその種の最初のものであった。同様に、ある種の非小細胞肺癌において、タンパク質p161NK4A、サイクリン依存性キナーゼcdk4及びcdk6の阻害剤であるタンパク質、の遺伝子は、供与スプライシング部位(donor splicing site)で突然変異される。この突然変異は、短い半減期を有するトランケーションされたタンパク質の産生をもたらし、これはRBの不活性なリン酸化形態の蓄積をもたらす。更に、WT1、Wilmのがん抑制遺伝子は、選択的スプライシングにより発生したいくつかのメッセンジャーRNAに転写される。乳癌では、種々の変異体の相対的割合は健康な組織に比べて変更されており(modified)、それにより腫瘍の進行におけるWT1の種々の機能的ドメインの重要性を理解することへの診断ツール又は手がかりを与える。細胞形質転換期間中の種々のメッセンジャーRNA形態及びタンパク質アイソフォーム間の割合に影響を与える同じ変化プロセスがニューロフイブリンNF1の場合にも見出される。更に、スプライシング現象のモジュレーションが腫瘍進行のマーカーとして挙動するという概念はHDM2の例によりさらに支持され、その場合5つの選択的スプライシングイベントが卵巣及び膵臓癌において検出され、その発現は腫瘍進展の段階に依存して増加する。更に、頭部及び頚部癌では、p53が不活性化される機構の1つはコンセンサススプライシング部位での突然変異を伴う。
これらの少数の例は、与えられた腫瘍と隣接した健康な組織とを識別する選択的スプライシングパターンのためのシステマチックスクリーニングに基づく本発明の方法の利益を説明する。このようにして得られた結果は、既知のがん抑制遺伝子の特徴付けのみならず、定性的差異スクリーニング方法の独創的且つシステマチックな面に鑑み、新しいがん抑制遺伝子に影響を与えそうな腫瘍に対して特異的な新規な選択的スプライシングの同定も可能とする。
ゆえに、本発明は、前記した、がん抑制遺伝子又はこれらのがん抑制遺伝子内の遺伝子変化(例えば、スプライシングイベント)を同定及び/又はクローニングすることを更に指向する。この方法は、有利には、下記の段階:
(a)腫瘍試料のメッセンジャーRNAを健康な試料のcDNAとハイブリッド形成させるか又はその逆、又は両方を平行して行うこと、
(b)形成されたハイブリッド内で、健康な試料に対する腫瘍試料に特異的な領域を同定すること、
(c)段階(b)で同定された1つ又はそれより多くの領域に相当するタンパク質又はタンパク質ドメインを同定すること、
を含むことができる。
次いで、同定されたタンパク質又はタンパク質ドメインのがん抑制性を、種々の既知のモデルで試験することができる。次いで、これらのタンパク質又はそれらのネイティブ形態(健康な組織で観察されるスプライシングパターンを示す)を、種々の治療的又は診断用途に、特に抗腫瘍遺伝子治療のために使用することができる。
ゆえに、本願は、本技術を具体化する種々の観点のみならず、研究において得られる情報の利用、低分子量の化合物についてのスクリーニングアッセイの開発及び遺伝子治療又は診断ツールの開発にも関する。
これに関して、本発明は更に、遺伝毒物学における上記した方法、核酸又はライブラリーの使用、即ち、試験化合物の毒性を予言することに関する。
毒性作用物質による細胞又は組織の処理期間中に開始される遺伝子プログラムは、主としてアポトーシスプロセス又はプログラムされた細胞死と顕著に相関している。このようなアポトーシス機構を調節することにおいて選択的スプライシングプロセスの重要性は文献に記載されている。しかしながら、今日までに述べられた単一の遺伝子工学技術は、2つの与えられた病態生理学的条件に特有の選択的スプライシングによる配列変化の完全なスクリーニング及び単離を可能としない。本発明により開発された定性的差異スプライシングスクリーニング方法は、cDNAライブラリー内の2つの条件間で発生するすべてのスプライシング差を集めることを可能とする。標準毒性化合物で処理された又は処理されていない組織(又は細胞培養物)RNA配列(例えばメッセンジャーRNA)を比較することは、調べられるべき毒性作用を特徴付ける遺伝子発現の定性的な差を含むcDNAライブラリーの発生を可能とする。次いでこれらのcDNAライブラリーを、毒性について評価されるべき化学品で処理した同じ組織又は細胞から生じるRNAに由来するプローブとハイブリッド形成させることができる。これらのプローブの与えられた標準毒性条件に特異的な遺伝子配列とハイブリッド形成する相対的能力は、化合物の毒性を決定することを可能とする。更に、毒性作用物質により誘発された定性的差異ライブラリーの発生及び利用のためのDATASの使用に加えて、本発明の一部は、あるメッセンジャーRNAのスプライシングにおける調節欠陥が、当業者に知られている細胞毒性及びアポトーシス試験で決定されたIC50より低い用量で、ある毒性作用物質により誘発されうることを証明することからもなる。このような調節欠陥(又は脱調節(deregulations)をマーカーとして使用して分子(化学的又は遺伝子)の毒性及び/又は効能を評価することができる。
ゆえに、本発明は、また、生物学的試料に対して化合物により誘発されるスプライシング形態及び/又はパターンの検出に基づく、化合物の毒性及び/又は治療潜在力を検出又は監視するためのいかなる方法にも関する。本発明は更に、マーカーとしてのスプライシング形態及び/又はパターンのいかなる変更(modification)も使用して分子の毒性及び/又は効能を評価することに関する。
毒性評価又は監視は更に特定的には2つのアプローチに従って行うことができる:
第1のアプローチに従えば、一方では処理に付されなかった基準組織又は細胞培養物と他方ではその毒性が評価されるべき産生物により処理されたものとの間で、定性的差異スクリーニングを行うことができる。この産生物により特定的に誘発された定性的な差を表すクローンの分析は、次いでアポトーシスの如き毒性反応に関与するcDNAに密接に関係するイベントのこれらのクローン内の最終的検出を与える。
このようなマーカーは監視される。何故ならば、それらはが問題の産生物の用量及び問題の産生物による処理の期間の関数として生じ、それによりその毒物学的プロフィルを確立することができるからである。
ゆえに、本願は、その毒性が測定されるべき化合物によりモデル生物学的システムにおいて誘発された毒性マーカーを、上記した方法に従う定性的差異スクリーニングによって同定するための方法も指向する。これについて、本発明は、特に、試験毒性化合物による処理を受けたか又は受けていない試料のcDNA及びRNA間の定性的差異ライブラリーを作成し、そして処理後の試料の性質に特異的な毒性マーカーを調べることを含む、与えられた生物学的試料の毒性状態に特異的な核酸を同定及び/又はクローニングするための方法に関する。
第2のアプローチに従えば、与えられた基準組織又は細胞モデルについて投与量及び処理期間の関数として毒性プロフィルを完全に表すアバクス(abacus)が種々の種類の毒性産生物について作成される。各アバクスドットについて、定性的な遺伝的差異を表すcDNAライブラリーを発生させることができる。後者は定性的差異ライブラリーを表す、即ち、それらはアバクスダイアグラムにおいて選ばれたドットからの遺伝子情報及びコントロール組織又は細胞モデルにおける対応するドットからの遺伝子情報を抽出することにより得られる。実施例に記載したとおり、定性的差異スクリーニングは、1つの条件に由来するmRNAを他の条件に由来するcDNAとハイブリッド形成させることに基づいている。上記したとおり、定性的差異スクリーニングは、プレメッセンジャー種を含有する全RNA又は核RNAを使用して行うこともできる。
この点で、本発明は、与えられた生物学的試料に対する試験化合物の毒性を決定又は評価するための方法であって、
−健康な状態からの該生物学的試料及び基準毒性化合物による該試料の処理から生じる毒性の種々の段階における該生物学的試料のcDNA及びRNA間の差異ライブラリーを、
−該試験化合物により処理された生物学的試料の核酸調製物とハイブリッド形成させ、そして
−種々のライブラリーとのハイブリッド形成の程度を決定することにより試験化合物の毒性を評価する、
ことを含む方法に関する。
この方法に従えば、
−条件A(試験)からのRNAと条件B(基準)からのcDNA(rA/cB)
−条件B(基準)からのRNAと条件A(試験)からのcDNA(rB/cA)
間の、各条件(化合物投与量及び/又はインキュベーション時間)について2つのクロスハイブリッド形成を進行させることが有利である。
かくして、各アバクスドットにおける各基準毒性条件は、2つの定性的差異スクリーニングライブラリーに対応する。このようなライブラリーの1つは、定性的な差、即ち、特に正常な基準条件に特異的な選択的スプライシングイベントのフルコレクションであり、これに対して他方のライブラリーは毒性状況に特異的なスプライシングイベントのフルコレクションである。これらのライブラリーは固体支持体材料、例えば、ナイロン又はニトロセルロースフィルター又は有利にはチップ上でレプリカ平板培養される(replica-plated)。可変長(考慮されるスプライシングイベントに従って)のcDNA断片から最初に形成されたこれらのライブラリーは、予め単離された配列に由来するオリゴヌクレオチドを使用することにより最適化されうる。
化合物が医薬開発のための候補者である場合には、これは毒性アバクスダイアグラムに記録されたのと同じ組織又は細胞モデルで試験することができる。次いで分子プローブを、関心のある化合物で処理された生物学的試料から抽出されたmRNAから合成することができる。次いでこれらのプローブを、rA/cB及びrB/cAライブラリーを有するフィルター上でハイブリッド形成させる。例えば、rA/cBライブラリーは正常な条件に特異的な配列を含有することができそしてrB/cAライブラリーは毒性条件に特異的な選択的スプライシングされた種を含有することができる。次いで化合物の無害又は毒性は、試験化合物により処理された基準組織又は細胞モデルに属するmRNA由来のプローブのハイブリッド形成プロフィルを検査することにより容易に評価される:
−rA/cBライブラリーとの有効なハイブリッド形成及びrB/cAライブラリーにおけるシグナルがないことは、化合物が調べているモデルにおける毒性を持たないことを証明する
−プローブとrB/cAライブラリークローンとのポジティブハイブリッド形成は、試験化合物で誘発された毒性の証拠である。
このようなライブラリーに関係する実際の用途は、エタノール、カンプトテシン又はPMAの如き毒性作用物質による処理の後の肝細胞培養モデル、例えばHepG2系、腎上皮細胞、例えば、Hk−2系又は内皮細胞、例えばECV304系により与えられうる。
好ましい例は、毒性作用物質又は刺激剤による処理を受けているか又は受けていない皮膚培養物モデルの化粧品試験における使用により与えられうる。
ゆえに、本願の更なる目的は、文献に開示されたアバクスチャートに従う広いクラスの毒性作用物質を表す化合物により処理された基準器官、組織又は細胞培養物から造られた差異スクリーニングライブラリー(cDNAとRNA間の)である。本発明は更に当業者に知られたフィルター又は支持体材料(ニトロセルロース、ナイロン...)上へのこれらのライブラリーのスプレッディングを包含する。有利には、これらの支持体材料は、このゆえに遺伝毒性チップを規定するチップであることができる。本発明は更に種々の毒性作用物質の作用の基礎をなす機構を理解するためにこれらのライブラリーを構成する種々のクローンに関する配列決定データの利用可能性(potential exploitation)に関するものであり、そしてその毒性が決定されるべき化合物又は医薬品により処理された細胞又は組織に由来するプローブとのハイブリッド形成におけるこれらのライブラリーの使用に関する。有利には、本発明は、種々の毒性条件下に処理された皮膚細胞から作成された、上記したタイプの如き核酸ライブラリーに関する。本発明は更にこれらの個々の皮膚差異ライブラリーを含むキットに関する。
本発明は更に、前記した方法、核酸又はライブラリーを使用して、試験化合物の治療的有効性を評価する(予測する)こと又は高めること(遺伝薬理学)を指向する。
この特定の使用では、基礎となる原理は、前記した原理にきわめて似ている。基準差異ライブラリーは、器官のコントロール細胞培養物からのcDNA及びRNA及び病理学的モデルを刺激するそのカウンターパート(counterparts)からのcDNA及びRNA間で確立される。次いで産生物の治療的有効性を、病理モデルの特異的な遺伝子発現の定性的変動に拮抗するその潜在力を監視することにより評価することができる。これは、基準ライブラリーと病理モデルに由来するプローブのハイブリッド形成プロフィルの変化により証明される:即ち、処理がない場合には、プローブは疾患の特異的マーカーを含有するライブラリーとのみハイブリッド形成する。有効な産生物による処理の後、プローブは、それが病理モデルから誘導されるとしても、健康なモデル同等物のマーカーを有する他のライブラリーと優先的にハイブリッド形成する。
この点で、モデルは、更に、与えられた生物学的試料に対する試験化合物の治療的有効性を決定又は評価する方法であって、
−健康な状態及び病理的状態(種々の進展状態の)における該生物学的試料からのcDNA及びRNA間の差異ライブラリーを、
−該試験化合物により処理される生物学的試料に由来する核酸の調製物とハイブリッド形成させ、そして
−該ディファレントライブラリーとのハイブリッド形成の程度を決定することにより試験化合物の治療的潜在力を評価する、
ことを含む方法を指向する。
このような用途は、標準栄養因子により拮抗される神経変性のある面を刺激するアポトーシスモデルにより例示される。かくして、NGFの存在下に神経突起に分化するPC12褐色細胞腫系に由来する細胞は、この成長因子の除去によりアポトーシスに入る。このアポトーシスプロセスは、多くのプログラムされた細胞死マーカーの発現により伴われ、そのいくつかは選択的スプライシングにより調節されそしてIGF1によりダウンレギュレーションされる。定性的差異スクリーニングに由来する2つのライブラリーは、一方では、NGF除去に続くアポトーシスのプロセスにおける分化したPC12細胞のmRNA抽出物から発生されそして他方ではIGF−1を補充することによりアポトーシスを受けるのを阻止された分化したPC12細胞から発生される。これらのライブラリーに、アポトーシスのプロセスにおける分化したPC12に由来するmRNAから調製されたプローブをハイブリッド形成させることができ、そしてその生存は試験されるべき神経保護産生物による処理により高められる。かくして、定性的特徴を逆にするための試験化合物の有効性は、該プローブの、より良好な生存率を有する細胞を表すこれらの特異的ライブラリークローンに選択的にハイブリッド形成する能力を監視することにより認識されうる。この試験は次いでこのような化合物の誘導体の有効性又は神経保護化合物のいかなる他の新規なファミリーの有効性も試験しそしてその薬理学的プロフィルを改良するのに使用することができる。
特定の態様では、本発明の方法は、健康な神経細胞及びこの神経変性モデル細胞に由来する本発明に従う差異ライブラリーとのハイブリッド形成を行うことにより神経保護試験化合物の有効性を評価することを可能とする。
他の態様では、腫瘍及び健康な細胞試料から確立された差異ライブラリーを使用して抗腫瘍化合物を試験することに興味がある。
既に注目したとおり、本発明の方法は、化合物の種々の誘導体の、健康な試料を表すライブラリーのハイブリッド形成プロフィルと同様なハイブリッド形成プロフィルを誘発する能力を試験することによって、化合物の性質を改良するのに使用することができる。
本発明は更に薬理ゲノミクスにおいて前記した方法、核酸又はライブラリーの使用、即ち、試験化合物又は処理に対する患者の応答を評価する(予言する)ことを指向する。
薬理ゲノミクスは、どの処理が与えられた病理に対して合理的に成功するかを決定する観点で患者の遺伝子プロフィルを確立することを目標とする。本発明に記載された技術は、この点で与えられた処理に応答する病理的な条件と処理に対して応答しないか又は不十分に応答する他の条件間で発生する定性的な差を表すcDNAライブラリーを確立することを可能とし、かくして種々の治療ストラテジーを適格とすることができる。一旦これらの標準ライブラリーが確立されると、それらは患者のメッセンジャーRNAから調製されたプローブとハイブリッド形成させることができる。ハイブリッド形成の結果は、どの患者が応答性又は非応答性条件に相当するハイブリッド形成プロフィルを有するかを決定すること、かくして患者管理における処理選択を精密にすることを可能とする。
本願では、その目的は、一方では、成功しそうな最も適切な処理方式を患者の経歴に依存して示唆し、そして他方では、これらの患者が最も利益を得られそうな与えられた処理方式を登録することである。他の用途と同様に、2つの定性的差異スクリーニングライブラリーを作成する:1つは与えられた処理に応答することが知られている病理モデル又は試料に基づいており、そして他方は治療よに不十分に応答性であるか又は応答しない更なる病理モデル又は試料に基づいている。これらの2つのライブラリーを、次いで個々の患者の生検組織から抽出されたmRNAに由来するプローブとハイブリッド形成させる。このようなプローブが1つの特定の条件に特異的な選択的にスプライシングされた形態と優先的にハイブリッド形成するどうかに依存して、患者は、最初にモデルを規定するのに役立った標準処理に応答性の対象及び非応答性対象に分けられうる。
この点で、本発明は試験化合物又は処置に対する患者の応答を決定又は評価するための方法であって、
−該化合物/処理に応答性の生物学的試料からのcDNA及びRNAと該化合物/処理に不十分に応答性であるか又は非応答性である生物学的試料からのcDNA及びRNA間の差異ライブラリーを、
−患者の病理的生物学的試料に由来する核酸調製物とハイブリッド形成させ、そして
種々のライブラリーとのハイブリッド形成の程度を決定することにより患者の応答性を評価する、
ことを含む方法も指向する。
薬理ゲノミクスにおける定性的差異スクリーニングの有用性の好ましい例は、同じ組織学的起源の2つの腫瘍であって、その1つは抗腫瘍性化合物で処理されると(例えば、遺伝子治療により野生型p53タンパク質をコードするcDNAの導入)退行を示し、他方はこのような処理に対して非応答性である2つの腫瘍間の定性的差異スクリーニングにより説明される。これらの2つの条件間の定性的差異ライブラリーを構築することに由来する第1の利益は、これらのライブラリーを構成するクローンを分析することによって、第1モデルでは観察されるが第2では存在しない退行の期間中どの分子機構が引き出されるかを決定することができることである。
次いで、これらのライブラリーに由来するcDNAを有するフィルターの使用又はいかなる他の支持体材料の使用も、該処理に対するその応答が予測されるべき腫瘍生検のmRNAに由来するプローブとのハイブリッド形成を行うことを可能とする。結果を見て患者に最適化された処理方式を割り当てることが可能である。
この方法の1つの特定の例は、p53がん抑制遺伝子治療に対する腫瘍応答を決定することからなる。あるタンパク質及びある腫瘍はこの型の処置に多かれ少なかれ応答することが実際に報告されている(Rothet al., (1995)Nature Medicine, 2: 958)。ゆえに、処置を最適化するため及び受けている臨床試験における患者の登録に関する最善の選択をするために、どのタイプの腫瘍及び/又はどの患者が野生型p53遺伝子治療に感受性であるかを決定することができることが必須である。有利には、本発明の方法は、p53応答性細胞及び非応答性細胞の定性的特徴に特異的なライブラリーを提供することによりこの方法を簡単にすることを可能とする。p53に感受性又は抵抗性の細胞モデルの例は、例えば、Sabbatini et al.により記載されており(Genes Dev., (1995), 9: 2184)又はRoemer et al.により記載されている(Oncogene, (1996), 12: 2069)。患者の生検試料に由来するプローブとこれらのライブラリーのハイブリッド形成は、患者応答性の評価をより容易にするであろう。更に、特異的ライブラリーはp53応答性に関与する核酸の同定を可能とするであろう。
ゆえに、本願は、少なくとも1種の薬理学的作用物質に対する応答性が変わる病理試料又は病理モデルからの差異スクリーニングライブラリーの確立も指向する。これらのライブラリーは上記したとおりの制限されたライブラリー、複合ライブラリー又は自己由来のライブラリー(autologous libraries)であることができる。それは、当業者に知られたフィルター又は支持体材料(ニトロセルロース、ナイロン)上のこれらのライブラリーのスプレッディングにも関する。有利な方式では、これらの支持体材料はかくして薬理ゲノムチップを規定するチップであることができる。更に本発明は、種々の処理に異なって応答する病理試料をもたらした機構を解明する観点でこのようなライブラリーを形成する種々のクローンの配列決定データの利用可能性(potential exploitation)に関し、そしてこれらのライブラリーを規定するのに最初に使用された標準処理に対する応答を予言することが望まれる病理的条件から生じる生検組織に由来するプローブとのハイブリッド形成を行うためのこのようなライブラリーの使用に関する。
かくして、本発明は、スプライシング形態及び/又はパターンの変動が、薬理ゲノムマーカーのソース、即ち、患者の処理に応答する能力又は患者が処理に応答する方式を決定するためのマーカーのソースを表すことを記載する。したがって、この点で、本発明は、更に薬理ゲノムマーカー(pharmacogenomic markers)のソースとして選択的スプライシングにより発生したアイソフォームにおける個々の間の変動性の使用を指向する(スプライソソーム分析)。本発明は、薬理ゲノムマーカーのソースとして処理により誘発されたスプライシング修飾(splicing modifications)の使用にも関する。かくして、上記に説明されたとおり、本発明のDATAS法は2つの生物学的試料間で発生する定性的な差を表す核酸を発生させることを可能とする。このような核酸又はその誘導体(プローブ、プライマー、相補的酸等)は、対象の処理に応答する能力及び方式又は与えられた処理/病理に対する対象の素質等を証明するという観点で対象のスプライソソームを分析するのに使用することができる。
これらの種々の一般的例は、遺伝毒性、遺伝薬理学及び薬理ゲノミクスの研究及び潜在的診断又は治療標的に関する調査における定性的差異スクリーニングライブラリーの有用性を説明する。このようなライブラリーは、2つの病態生理学的状況間で発生する定性的な差をクローニングすることから誘導される。これらの定性的な差を表すcDNAの他の使用は、上記した特徴を有するゲノムDNAライブラリーをスクリーニングするようにデザインされたプローブを発生させることであるので、このようなアプローチは、遺伝毒性、遺伝薬理学及び薬理ゲノミクスのいかなる研究のためにも実施することができ、そして遺伝子同定のために実施することもできる。例えば、遺伝毒性の研究では、単一イントロン又は単一エキソンへのそれらのインサート(inserts)のサイズにより統計的に制限されたゲノムクローンは、基準細胞又は組織試料と基準毒性化合物により処理された同じ細胞又は組織間の定性的差異分析に由来するDATASプローブとのそれらのハイブリッド形成に従ってフィルター上に配列される。種々のクラスの毒性を表すこのようなクローンが選ばれると、それらは次いで、その毒性が評価されるべき化合物により処理された同じ細胞集団又は同じ組織試料の全メッセンジャーRNAに由来するプローブとハイブリッド形成させることができる。
本発明の他の利点及び実際の用途は限定としてではなく説明の目的で与えられる下記の実施例から明らかになるであろう。本発明の使用分野は図7に示される。
実施例
1.一本鎖cDNAを使用するRNAにおける選択的スプライシング及び他の定性的修飾の差異クローニング
一方は正常であり(mN)そして他方は病理起源(mP)である2つの条件に相当するメッセンジャーRNAを生検試料又は培養された細胞から単離する。これらのメッセンジャーRNAを逆転写(RT)により相補的なDNAs(cN)及び(cP)に転化する。次いでmN/cP及びcN/mPハイブリッドを液相において調製する(cN/mPの形成をもたらすいずれかの場合の1つを説明する図2の略図参照)。
これらのハイブリッドは、連続的に熱サイクリングに付されたフェノールエマルション(PERT技術又はPhenol Emulsion DNA Reassociation Technique)において有利に調製される(Miller, R., D. and Riblet, R., (1995), Nucleic Acids Research, 23(12): 2339-2340)。典型的には、このハイブリッド形成は、水性相(120mMリン酸ナトリウム緩衝液、2.5M NaCl、10mMEDTA)及び水性相の8%に相当しそして二回蒸留されたフェノールから形成された有機相から形成されたエマルション中で0.1〜1μgのポリA+RNAと0.1〜2μgの相補的DNAを使用して行われる。
ヘテロ二本鎖を得るために他の方法も有利に使用される:逆転写反応の後、新しく合成されたcDNAを排除クロマトグラフィーによりビオチニル化オリゴdTプライマーから分離する。このcDNA0.1〜2μgを0.3M酢酸ナトリウム及び2容積のエタノールの存在下に0.1〜1μgのポリA+RNAと共沈させる。これらの共沈した核酸を、80%ホルムアミド、40mMPIPES(ピペラジンビス(2−エタンスルホン酸))pH6.4、0.4MNaCl及び1mMEDTAからなるハイブリッド形成緩衝液30lμl中に取り込ませる。
溶液中の核酸を85℃で10分間熱変性し、次いで40℃で少なくとも16時間〜48時間ハイブリッド形成を行う。
ホルムアミドハイブリッド形成方法の利点は、それがcDNA及びRNA鎖対形成のためのより高度に選択的な条件を与えるということである。
これらの2つのハイブリッド形成技術の結果として、RNA/DNAへテロ二本鎖が得られ、その塩基対形成の程度はRTの全体のcDNA(entire cDNA)を合成する能力に依存する。観察される他の一本鎖構造は、調べている2つの病態生理学的状態を区別する選択的スプライシングに相当するRNA(及びDNA)領域である。
次いで、この方法は、このようなスプライスループにより生じる遺伝情報を特徴付けることを目的とする。
このために、ストレプトアビジンコーテッドビーズによるcDNA(ビオチニル化オリゴdTによりプライミングされた)の捕捉によりヘテロ二本鎖を精製する。有利には、これらのビーズは、磁性を有するビーズであり、これは、ヘテロ二本鎖を、ヘテロ二本鎖構造に参加していないRNAから磁性分離器の作用により分離することを可能とする。このようなビーズ及びこのような分離器は商業的に入手可能である。
この方法のこの段階で、ヘテロ二本鎖及びRNAとのハイブリッド形成に参加しなかったcDNAを単離する。この物質を次いで、cDNAとハイブリッド形成したRNAの領域を選択的に加水分解するRNアーゼHの作用に付す。この加水分解の生成物は一方ではcDNAでありそして、他方では、不完全な逆転写反応の結果としてスプライスループ又はハイブリッド形成しなかった領域に相当するRNA断片である。このRNA断片は、上記した同じ実験方法に従う磁性分離、及び汚染性RNアーゼ活性のないDNアーゼによる消化によりDNAから分離される。
1.1Grb2遺伝子のスプライシング変異体に関するDATAS方法の確認
このアプローチの実行可能性は、一方ではGrb2のコード領域に相当するRNA、及びGrb3.3のコード領域に相補的な一本鎖cDNAを使用するin vitroシステムにおいて証明された。Grb2遺伝子は、651塩基対のオープンリーディングフレームを有する。Grb33は、選択的スプライシングにより発生されそしてGrb2のSH2機能的ドメインにおける121塩基対の欠失を含むgrb2のアイソフォームである(Fath et al., (1994), Science 264; 971-4)。Grb2及びGrb33RNAは、RiboMaxキット(Promega)によってT7プロモーターにより駆動されるGrb2又はGrb33コード配列を有するプラスミドから当業者により知られている方法により合成される。産生物の分析は、合成が均質であることを示す(図8A)。可視化の目的で、Grb2RNAは、RiboProbeキット(Promega)によってin vitro転写期間中標識された塩基の組込みにより放射能標識もされた。Grb2及びGrb33cDNAは、SuperscriptIIキット(Life Technologies)及び、Grb2配列の相補体(complement)(618-639)に相当するGrb2及びGrb33に共通のビオチニル化オリゴヌクレオチドプライマーを使用して、上で得られた合成RNA生成物からの逆転写により合成された。RNA及びcDNAを供給者の教示(Promega, Life Technologies)に従って処理し、排除カラム(RNアーゼのないセファデックスG25又はG50、5Prime、3Prime)で精製しそして分光法により定量した。
DATASの第1段階は、80%ホルムアミド、40mMPIPES(pH6.4)、0.4MNaCl、1mMEDTAを含有するハイブリッド形成緩衝液30μl中で、標識されたGrb2RNA10ngを、
1.ビオチニル化grb33cDNA100ng、
2.ビオチニル化grb2cDNA100ng、
3.水
と懸濁させて一緒にすることにより行った。核酸を85℃で10分間加熱することにより変性し、その後40℃で16時間ハイブリッド形成を行う。ストレプトアビジンビーズ上に捕捉した後、試料を前記したとおりRNアーゼHで処理する。
これらの段階は、6%アクリルアミドゲルでの電気泳動、続いて標識されたgrb2RNAに由来する種の定性及び定量を可能とするInstant Imager (Packard Instruments)によるゲルの処理により分析される(図8B)。かくして、レーン2, 3及び4は、grb2/grb33及びgrb2/grb2二本鎖が定量的に形成されることを示す。grb2/grb33複合体の移動はgrb2RNAの移動より相対的に遅く(レーン2)、grb2/grb2複合体の移動はより速い(レーン3)。レーン5, 6及び7は、grb2/grb33及びgrb2/grb2複合体の80%はビーズにより捕捉され、これに対してビオチニル化されていないgrb2RNAだけがビーズ上澄液において見いだされたことを示す、ストレプトアビジンビーズにより保持されなかった試料に相当する。RNアーゼHによる処理は、ブロモフェノールブルー(BPB)より速く移動する遊離ヌクレオチドに加えて、キシレンシアノールブルー(XC(図において矢印により示された)より遅く移動する種、及び特に、grb2/grb2 複合体及びgrb2RNAに相当するレーン9及び10に対してgrb2/grb33複合体に相当するレーン8における種を放出する。レーン11、12及び13は、試料を排除カラムに通して遊離ヌクレオチドを除去した後のレーン8、9及び10に相当する。レーン8及び11に観察される移動は、grb33からgrb2を区別する121ヌクレオチドの欠失に相当するRNA分子について予測される移動である。
この結果は、2つのスプライシングアイソフォームに由来する2つの配列間で二本鎖の形成により発生したRNAループを得ることが可能であることを明らかに示す。
1.2.健康な状態及び毒性状態における肝細胞の定性的ライブラリーを発生させるためのDATAS方法の適用
より複雑な状況を検査した。分子の毒性を予言するためのツールとしてDATAS技術の適用の範囲内で、ヒト肝細胞細胞系HepG2を0.1Mエタノールで18時間処理した。処理を受けた細胞又は受けなかった細胞からRNAを抽出した。上記したDATAS変法(ビオチニル化sscDNAの調製、液相でのクロスハイブリッド形成、磁場を加えて種を分離すること、RNアーゼH消化)を、基準条件(又は条件A)における未処理細胞及び試験条件(又は条件B)における処理された細胞で行った(図9)。抽出されたRNAは放射能標識されていないので、RNアーゼH消化により発生したRNAを、T4ポリヌクレオチドキナーゼ及びガンマ−P32ATPによってRNA5′リン酸を標識されたリン酸で置換するための交換反応を行うことにより可視化した。次いで、これらの標識された産生物をアクリルアミド/尿素ゲルにローディングしそしてInstant Imager(Packaed Instruments)を使用する露光により分析した。次いで、A/B及びB/Aハイブリッド形成に由来する複合シグネーチャーを、ゲルにおいてより遅く移動しそして大きい核酸配列に相当する第1の群のシグナル及び25〜500ヌクレオチド間の移動する第2の群のシグナルで可視化することができた。これらのシグネーチャーは条件A/Aでははるかに低い強度であり、これは、エタノールがA/B及びB/Aシグナルの存在として明らかに示されたRNAスプライシングイベントのリプログラミングを誘発することができることを示唆する。
1.3..同定された核酸からのライブラリーのクローニング及び作成
次いで、RNアーゼHの作用に対して抵抗性のこれらのDNA断片をクローニングするためにいくつかの実験的な選択が考えられうる:
A.第1のアプローチはこのようなループを単離及びクローニングすることからなる(図3)。
このアプローチに従えば、当該技術分野で知られた条件に従ってRNAリガーゼにより各端部へのオリゴヌクレオチドのライゲーションを進める。次いでこれらのオリゴヌクレオチドをプライマーとして使用してRT PCRを行う。PCR産生物をクローニングしそして関心のある2つの病態生理学的条件に相当する全相補的DNAプローブでスクリーニングする。いずれかのプローブの1つと優先的にハイブリッド形成するこれらのクローンのみがスプライスループを含有し、スプライスループは次いで配列決定され及び/又はライブラリーを発生させるのに使用される。
B.第2のアプローチ(図4)は、少なくとも部分的にランダムプライマーにより開始された、RNアーゼH消化によりヘテロ二本鎖構造から開放された一本鎖RNAに対する逆転写反応を行うことからなる。かくして、これらはランダム3′及び5′配列を有するプライマー、ランダム3′端部及び規定された5′配列を有するプライマー、又はまだセミランダムオリゴヌクレオチド、即ち、縮重(degeneration)の領域及び規定された領域を含むプライマーであることができる。
ゆえに、このストラテジーに従えば、プライマーを一本鎖RNAに沿ってどこででも又はセミランダムプライマーの選択により決定される各一連の塩基においてハイブリッド形成させることができる。次いで上記したオリゴヌクレオチドに相当するプライマーを使用してPCRを行って、スプライスループ誘導配列(splice loop-derived sequences)を得る。
図10(レーン1〜12)は、いくつかのDATAS実験で得られそして下記のセミランダムオリゴヌクレオチド:
の使用にカップリングしたPCR断片のアクリルアミドゲル分析を示す。
これらの結果を同じオリゴヌクレオチドを使用してしかし鋳型として全cDNAにより得られたシグナル(レーン13)の複雑性と比較すると、DATASは定性的な差に相当する情報をフィルターする(プロフィル化する)ことを可能とすることが証明される。
この変法を、上記したプロトコル(実施例1.1)に従うgrb2RNA/grb33一本鎖cDNA二本鎖のRNアーゼH消化により発生したgrb2RNAドメインに相当するイベントをクローニングするのに使用した。そうするために、モデル
(式中、Nは上記の如く定義され、W、X及びYは各々定義された不変の塩基を表し、そしてZは定義された塩基又は3′−OH基を示す、配列番号3)から選ばれそしてgrb2欠失における断片を増幅するように選ばれた配列:
を有するオリゴヌクレオチドを使用してPCR断片の発生を可能とし、該PCR断片は、クローニング及び配列決定の後、実際grb2の欠失したドメイン(grb2 deleted domain)(grb2における194−281)から誘導されることが示された。
ゆえに、これらの2つのアプローチは、プローブとして使用することができる、試験されるべき両条件において差異スプライシングを表す核酸組成物の産生を可能とするか、又は定性的差異cDNAライブラリーを構築することを可能とする。DATAS技術の定性的な差を表すプロフィル化されたcDNAライブラリーを発生する能力を下記の実施例1.4で更に説明する。
1.4.ヒト内皮細胞を表すプロフィル化ライブラリーの作成
この実施例はヒト内皮細胞系(ECV304)を使用して行われた。遺伝子発現の定性的分析は、一方では、増殖している細胞から、そして他方では、アノイキス(anoikis)(接着支持体を除去することにより誘発されるアポトーシス)のプロセスにおける細胞から抽出されたcystolicRNAを使用することにより達成された。
ECV細胞をEarle塩(Life Sciences)を補充された199培地中で増殖させた。アノイキスは、ポリHEMA処理された培養皿(polyHEMA-treated culture dishes)上での4時間の通過により誘発された。RNA調製のために、細胞をNonidetP−40を含有する緩衝液中に溶解させた。次いで、核酸を遠心により除去する。次いで細胞質溶液をQuiagen社の教示に従ってRneasyシリカマトリックスにRNAを特異的に固定するように調節した。洗浄の後、全RNAをDEPC処理された水中に溶離する。Dynabeadsオリゴ(dT)25磁性ビーズ(Dynal)での分離により全RNAからメッセンジャーRNAを調製する。ビーズを固定緩衝液中に懸濁させた後、全RNAを室温で5分間インキュベーションする。磁性分離及び洗浄の後、ビーズを溶離緩衝液中に取り込みそして65℃でインキュベーションしてメッセンジャーRNAを放出する。
SuperScriptII又はThermoScript逆転写酵素(Life Technologies)及びオリゴ(dT)プライマーによってメッセンジャーRNAから第1DNA鎖を合成する。RNアーゼH消化の後、セファデックスG50(5Prime−3Prime)カラムに通すことにより遊離ヌクレオチドを除去する。 フェノール/エタノール抽出及びエタノール沈殿の後、試料を紫外線吸光度により定量する。
必要な量のRNA及びcDNA(この場合には各々200ng)をプールしそしてエタノール沈殿させる。試料を脱イオンされたホルムアミド(特記しないかぎり、80%(V/V))を補充されたハイブリッド形成緩衝液(40mMHepes(pH7.2)、400mMNaCl,1mMEDTA)中に30μlの容積において取り込む。70℃で5分変性した後、試料を40℃で一夜インキュベーションする。
ストレプトアビジンビーズ(Dynal)を次いで固定化緩衝液(2X=10mMトリスHCl(pH7.5)、2MNaCl、1mMEDTA)中で洗浄し次いで再調整する(reconditioned)。ハイブリッド形成試料を水で200μlの容積に希釈し、ビーズ200μlに調節しそして30℃で60分間インキュベーションする。ビーズの磁性捕捉及び洗浄の後、ビーズをRNアーゼH緩衝液150μl中に懸濁させ、次いで37℃で20分間インキュベーションする。磁性捕捉の後、ハイブリッド形成しなかった領域を上澄液中に放出し、上澄液をDエヌアーゼで処理し、次いで酸性フェノール/クロロホルムで抽出しそしてエタノール沈殿させる。少量の核酸のエタノール沈殿を市販のポリマーSeeDNA(Amersham Pharmacia Biotech)を使用して行い、非常に希薄な溶液(ng/ml範囲の)からの核酸の定量的回収を可能とする。
RNアーゼH消化に由来するRNA試料からのcDNAの合成を、ランダムヘキサヌクレオチド及びSuperscriptII逆転写酵素によって行う。次いでRNAをRNアーゼH及びRNアーゼT1の混合物で消化する。プライマー、組み込まれなかったヌクレオチド及び酵素をGlassMAX SpinカートリッジによってcDNAから分離する。スプライスループに相当するcDNAを次いで本発明において前記したセミランダムオリゴヌクレオチドを使用するPCRに付す。この場合に、選ばれたオリゴヌクレオチドは下記のとおりである:
PCR反応は、Taqポリメラーゼを使用して30サイクル行う:
・最初の変性:94℃で1分
・94℃で30秒
・55℃で30秒
・72℃で30秒
・最終伸長:72℃で5分
PCR産生物をTaqポリメラーゼの活性に由来する断片のクローニングを簡単にするように、3′端部にフローティングT(floating T)を有するpGEM−Tベクター(Promega)にクローニングする。コンピテントJM109バクテリア(Promega)における形質転換の後、得られるコロニーをニトロセルロースフィルターに移し、そして一方では増殖している細胞からの及び他方ではアノイキスにおける全cDNAに関して行われたPCRの産生物に由来するプローブと優先的にハイブリッド形成させる。同じオリゴヌクレオチド
をこれらのPCR反応のために使用する。第1の実験態様では、アポトーシスにおける細胞からのプローブと優先的にハイブリッド形成する34のクローン及び増殖している細胞からのプローブとハイブリッド形成する13クローンを単離した。
これらの13クローンのうち、3クローンはSHCタンパク質のSH2ドメインに由来する同じcDNA断片を含有する。
SHC SH2ドメインに隣接する(flanking)PCRプライマー
の使用は、アノイキスにおけるECV細胞で特異的に観察されるSHC SH2ドメイン欠失の特徴付けを可能とした。このプライマー対によって、もとのままのSH2ドメインを含有する382塩基対cDNA断片に相当する一つの増幅産生物が指数関数的に増殖しているECV細胞からのRNAから得られる。PCRをアノイキスにおける細胞からのRNAで行う場合には、更なる287塩基対の断片が観察される。この追加の断片はSCHメッセンジャーに由来するが欠失を有するメッセンジャーRNA由来のものである。
この欠失は、SHCタンパク質の52kDa及び46kDa形態をコードするメッセンジャーオープンリーディングフレームの塩基1198〜1293に相当する(Pelicci, G.et al., (1992), Cell, 70: 93-104)。
文献と共にSH2ドメインに関する構造データは、このような欠失がホスホチロシンに対するアフイニティーの損失をもたらすことを示す。何故ならば、それはリン酸化チロシンとの相互作用に関与するアミノ酸を含むからである(Waksman, G.et al., (1992), Nature, 358: 646-653)。SHCタンパク質は、それらのSH2及びPTBドメイン(ホスホチロシン結合ドメイン)を介して種々のパートナーを連結するアダプターであるので、この欠失は我々が△SHCと呼ぶSHCのネイティブな負の優性形態(native negative dominant form)を発生させる。遺伝子が配列決定されているタンパク質のSH2ドメインは2つのエキソンに担われているので、DATASにより同定される欠失はSHC遺伝子のオルタナティブエキソン(alternative eon)に相当するようである。
△SHCのタンパク質及び核酸配列は図17に示される(配列番号9及び10)。
SHC SH2ドメインは、細胞増殖及び生存力に関与する多数のシグナルの伝達に関与しているので、△SHC配列の検査は、SHCタンパク質に対するその負の優性な性質及びその種々の細胞シグナルを妨害する能力を予言することを可能とする。
本発明は、SHCのこの新規なスプライシングされた形態、このスプライシングに相当するタンパク質ドメイン、生物学的試料におけるその検出を可能とするいかなる抗体又は核酸プローブ、及び例えば、診断又は治療目的のそれらの使用にも関する。
本発明は、特に塩基1198〜1293に相当する少なくとも1つの欠失、更に特定的には、配列番号8の配列の欠失を含むいかなるSHC変異体にも関する。本発明は、更に特定的には、配列番号10の配列によりコードされた配列番号9の配列を有する△SHC変異体に関する。
ゆえに、本発明は、上記△SHC変異体を同定するため及び/又は生物学的試料におけるSHC/△SHC比のいかなる変化も同定するための、いかなる核酸プローブ、オリゴヌクレオチド又は抗体にも関する。これは特に配列番号8の配列のすべて又は一部に相補的なプローブ又はオリゴヌクレオチド、あるいはこの配列によりコードされたタンパク質ドメインに対する抗体であることができる。このようなプローブ、オリゴヌクレオチド又は抗体は、生物学的試料におけるスプライシングされていない形態(例えば、SHC)の存在を検出することを可能とする。
これらの物質は、更にスプライシングされた形態(例えば△SHC)、即ち、例えばスプライシングから生じる連結領域(配列番号10の配列においてヌクレオチド1198のまわりに位置した)に相当する、に特異的なプローブ、オリゴヌクレオチド及び/又は抗体と平行して使用することができる。
このような物質は、免疫抑制に関する疾患(がん、免疫抑制治療、AIDS等)の診断のために使用することができる。
本発明は、i)SHCタンパク質におけるスプライシングされたドメイン(特に、例えば、自己免疫疾患又は移植片拒絶及びがんにおける免疫寛容の状態を誘発するために)又はii)△SHCタンパク質により得られた追加された機能をブロックすることに基づく分子のいかなるスクリーニング法にも関する。
本発明は更に、△SHCの治療的使用及び特に、例えば、SHCタンパク質ハイパーリン酸化が証明されうるがん性細胞又はがん(ex vivo又はin vivo)の処理を指向する。ゆえに、この点で、本発明は△SHCをコードする配列を含むいかなるベクターにも関し、特にウイルスベクターに関する。このベクターは、好ましくは、哺乳動物、特にヒト起源のがん性細胞又は増殖している細胞、例えば平滑筋細胞、内皮細胞(再発狭窄症)、繊維芽細胞(繊維症)をトランスフェクションすることができる。ウイルスベクターは特に、アデルウイルス、レトロウイルス、AAV、ヘルペスベクター等により例示されうる。
2.二本鎖cDNAを使用するRNAの選択的スプライシング及び他の定性的修飾の差異クローニング(図5)
標準分子生物学法により正常な(mN)条件及び病理(mP)条件に相当するメッセンジャーRNAを産生させ、そして対応する二本鎖相補的DNAs(dsN及びdsP)を産生させる。次いで70%ホルムアミドを含有する溶液中でmNをdsPと及びmPをdsNとハイブリッド形成させることによりRループ構造を得る。条件NとP間の差異スプライシングされた核酸ドメインは二本鎖DNAの形態で残っているであろう。次いで追い出された(displaced)一本鎖DNAsをグリオキサールで処理してホルムアミドの除去時のRNA鎖の更なる追い出しを回避する。ホルムアミド及びグリオキサールの除去及びRNアーゼHによる処理の後、ミツバチ型構造が得られ、対を形成していない一本鎖DNAsはミツバチの羽を表し、そして関心のある対を形成した二本鎖ドメインはミツバチの身体を思い起こさせる。一本鎖DNAを特異的に消化する酵素、例えば、ヌクレアーゼS1又はヤエナリヌクレアーゼ(mung bean nuclease)の使用は、二本鎖形態で残ったDNAの単離を可能とし、これを次にクローニングしそして配列決定する。この第2の技術は、関心のあるドメインのRNAフィンガープリントを生じさせる第1の方法と比べて、関心のあるドメインの二本鎖DNAフィンガープリントの直接の形成を可能とする。
このアプローチは、上記したgrb2/grb33モデルに関して行われた。grb2二本鎖DNAは、grb2の配列(1〜22)及びgrb2の相補的配列(618〜639)に相当する2つのヌクレオチドプライマーを使用してgrb2一本鎖cDNAのPCR増幅により産生された。このPCR断片をアガロースゲルで精製し、アフイニティーカラム(JetQuick, Genomed)で洗浄しそして分光法により定量した。同時に、grb2及びgrb33リーディングフレームに相当する2つの合成RNAを、RiboMaxキット(Promega)によってT7プロモーターの制御下にgrb2又はgrb33cDNAを有するプラスミドベクターから産生させた。RNAを供給者により教示されたとおりに精製しそして排除カラム(Sephadex G50、5prime−3prime)で洗浄した。二本鎖grb2DNA(1〜639)600ngを、
下記の緩衝液:100mMPIPES(pH7.2)、35mMNaCl、10mMEDTA、70%脱イオン化ホルムアミド(Sigma)、中で3つの別々の反応において、
1.grb33RNA3μg
2.grb2RNA3μg
3.水
と一緒にした。
試料を56℃に加熱し、次いで10分毎に−0.2℃ずつ44℃に冷却した。次いでそれらを4℃で貯蔵した。アガロースゲルの分析は、コントロールレーン3(図11A)と比較してレーン1及び2の変化した移動パターンを示し、これは新しい複合体が形成されたことを示す。次いで試料を脱イオン化グリオキサール(Sigma)(5%v/v又は1M)で12℃で2時間処理した。次いで複合体をエタノール(0.1MNaCl、エタノール2容積)で沈殿させ、70%エタノールで洗浄し、乾燥し、次いで水中に再懸濁させる。次にそれらをRNアーゼH(Life Technology)により処理し、次いで一本鎖DNAに特異的な酵素により処理する。ヌクレアーゼS1及びヤエナリヌクレアーゼはこのような性質を有しそして商業的に入手可能である(Life Technologies, Amersham)。このような消化物(酵素を供給された緩衝液中で5分間インキュベーション)をアガロースゲル上で分析した(図11B)。有意な消化産生物が反応1に由来する複合体(grb2/grb33)からのみ得られた(図11B、レーン7及び10)。これらの消化はヤエナリヌクレアーゼ(レーン10)の場合よりヌクレアーゼS1(レーン7)によるのがより完全であると思われる。かくして、100塩基対よりわずかに大きいサイズに相当するバンド(レーン7で矢印により示された)を精製し、pMos−Blueベクター(Amersham)にクローニングしそして配列決定した。この断片はgrb33において欠失しているgrb2の120塩基対ドメインに相当する。
このアプローチは、当業者により知られた方法に従って産生された全メッセンジャーRNA集団及び全二本鎖cDNA集団で出発して今や実行することができる。基準条件に相当するRNAを試験条件に由来する二本鎖cDNAとハイブリッド形成させそしてその逆をした。上記したプロトコルの適用の後、消化物をアガロースゲル上にローディングして50〜300塩基対の範囲のサイズに相当するバンドを単離及び精製する。
このようなバンドを次いでベクター(pMos-Blue, Amersham)にクローニングして定性的差異イベントに富んだインサートのライブラリーを発生させる。
3.定性的差異スクリーニングに由来するライブラリーの構築
上記した2つの実施例は、2つの与えられた病理条件間で発生する差異スプライシングされた配列のすべて又は一部を表すcDNAのクローニングをもたらす。これらのcDNAは、このようなcDNAのプラスミド又はファージベクターへの挿入によりライブラリーの構築を可能とする。これらのライブラリーは、ニトロセルロースフィルター又は当該技術分野で知られたいかなる他の支持体材料にも付着させることができる、例えば、チツプ又はバイオチップ又は膜上に付着させることができる。前記したライブラリーを光から離して冷所に貯蔵することができる。これらのライブラリーは、慣用の技術により支持体材料上に付着及び固定されると、化合物により処理して、プラスミド又はファージの複製を可能とした宿主バクテリアを除去することができる。これらのライブラリーは、有利には、クローニングされたcDNAに相当するcDNA断片からなるが、選択的スプライシングイベントに由来するこれらの配列のみをフィルター上に付着させるようにPCRにより作成されうる。
定性的差異スクリーニングの特徴の1つ及び独創的な特徴の1つは、この方法は有利には、2つの与えられた条件間で発生する定性的な差の全体のアレイを表す1つのみならず2つの差異ライブラリー(「ライブラリー対」)をもたらすということである。特に、本発明の差異スプライシングライブラリーの1つは、基準生理学的条件と比べて試験生理学的条件の独特の定性的マーカーを表し、他方のライブラリーは、試験生理学的条件に対して基準生理学的条件の独特の定性的マーカーを表す。このライブラリーのカップルは、ライブラリー対または「差異スプライシングライブラリー」とも呼ばれる。
定性的差異スクリーニングの利益の1つは、次の節に記載のとおり、化合物の毒性を評価することを可能とするということであるので、この技術の実行の良好な実施例は、一方では、未処理HepG2細胞、そして他方ではエタノール処理された細胞、 に特異的な配列に相当するcDNAクローンを得るためのDATASの使用である。後者の細胞は、細胞毒性の兆候及び1Mエタノールに18時間さらすことから出発するヌクレオソーム間の断片化(internucleosomal fragmentation)を介したDNA分解を示す。エタノール毒性の初期マーカーを得るために、メッセンジャーRNAを、未処理細胞及び0.1Mエタノールで18時間処理された細胞から調製する。一本鎖cDNA及びRNアーゼHを使用するDATAS変法の実行の後、得られるクローニングされたcDNAをPCRにより増幅し、アガロースゲル上で電気泳動させ、次いで当業者により知られた技術にしたがってナイロンフィルターに移す。一方では未処理状態の特異的定性的な差に特異的なクローン及び他方ではエタノール処理された細胞に特異的な配列に特異的なクローンの各組について、2つの同じフィルターデュプリケート(duplicate)を作成する。かくして、クローンの各組のフィンガープリントを、一方では未処理細胞に特異的なプローブとハイブリッド形成させそして他方では0.1Mエタノールで18時間処理された細胞に特異的なプローブとハイブリッド形成させる。
図12において得られそして示された差異ハイブリッド形成プロフィルは、DATAS技術により与えられたサブトラクションの品質を認識することを可能とする。かくして、未処理細胞(NT)からのmRNAと処理された細胞(Tr)からのcDNAとのハイブリッド形成から誘導されそして未処理条件に特異的な定性的な差に相当するであろうクローンは、未処理細胞の全メッセンジャーRNA集団を表すプローブと優先的にハイブリッド形成する。反対に、RNA(Tr)/cDNA(NT)ヘテロ二本鎖に対するRNアーゼHの作用に抵抗性の産物に由来するクローンは、処理された細胞からの全メッセンジャーRNAに由来するプローブと優先的にハイブリッド形成する。
一方では処理された条件に特異的なクローンと、他方では未処理条件に特異的なクローンの2組は、2つの異なる細胞状態に特徴的な定性的差異ライブラリーの例を表す。
4.定性的差異ライブラリーの使用及び利益
本発明の差異スプライシングライブラリーの可能性のある用途を特に図13〜15に示す。かくして、これらのライブラリーは以下のために有用である:
4.1,化合物の毒性の評価(図13)
この実施例では、基準条件はAと名づけられそして毒性条件はBと名づけられる。毒性アバクスチャートは、種々の期間の時間、種々の濃度の基準毒性化合物の存在下に条件Aを処理することにより得られる。毒性アバクスチャートの種々のドットについて、定性的差異ライブラリー、即ち、この例では制限されたライブラリーrA/cB及びrB/cAを構築する(ライブラリー対)。ライブラリー対を有利には支持体上に付着させる。次いで支持体を種々の用量の試験化合物で処理された元の生物学的試料に由来するプローブとハイブリッド形成させる:産生物X、Y及びZ。試験産生物の毒性潜在力を決定するためにハイブリッド形成反応を展開する(developed):この例では、産生物Zは高度に毒性でありそして産生物Yは中間的なプロフィルを示す。毒性アバクスチャートを構築することの実行可能性はエタノール処理及びHepG2が関与する定性的差異スクリーニングライブラリーの構築に関して前記した例で明白に説明される。
4.2.医薬組成物の効能の評価(図14):
この実施例では、本発明に従う制限されたライブラリー対は、病理モデルB及び健康なモデルA(又は基準活性産生物で処理された病理モデル)で出発して構築される。差異ライブラリーrA/cB及びrB/cAを場合により支持体上に付着させる。このライブラリー対は両条件間で発生するスプライシングにおける差を完全に表す。このライブラリー対は試験化合物の効能を評価することを可能とし、即ち、病理型プロフィル(rB/cA)から出発して「健康様("healthy-like")」プロフィル(rC/cB)を発生させるその能力を決定することを可能とする。この実施例では、ライブラリー対を、試験化合物により処理されているか又はされていない条件A及びBから調製されたプローブとハイブリッド形成させる。得られうるハイブリッド形成プロフィルは図14に示される。この用途の実行可能性は健康な条件と毒性条件に特徴的な定性的差異ライブラリーの前記した構築の実行可能性と同一である。毒性条件は、病理条件により置き換えられ、そして試験化合物の多少優先的に基準条件又は病理条件とハイブリッド形成するプローブを産生する能力が評価される。
4.3.処理に対する病理試料の応答を予言すること(図15)
この実施例では、本発明に従う制限されたライブラリー対は、2つの病理モデル、その1つは与えられた産生物(例えば、野生型p53遺伝子)による処理に応答性であり:条件A;他方は非応答性である:条件B、で出発して構築される。このライブラリー対(rA/cB;rB/cA)を支持体上に付着させる。
次いでこのライブラリー対を使用して同じ産生物に対する病理試験試料の感受性を決定する。その目的で、このライブラリー対を、基準処理に対する応答を評価することが望まれる患者の生検組織に由来するプローブとハイブリッド形成させる。応答性生検試料及び非応答性生検試料のハイブリッド形成プロフィルを図15に示す。
4.4.みなし子受容体(orphan receptors)に対するリガンドの同定
膜又は核内受容体のそれらのリガンドによる活性化は、あるRNAのスプライシングにおける調節欠陥を特異的に誘発することができる。本発明のDATAS法によるこれらのイベントの同定は、受容体、特にみなし子受容体に対する天然又は合成リガンドを調べるのに使用することができる、受容体活性化を監視するためのツール(マーカー、ライブラリー、キット等)を提供する。この用途に従えば、調節欠陥と関連したマーカーを同定しそして支持体上に付着させる。種々の組成物及び/又は試験化合物により処理された又は処理されていない、調査下の受容体を(過剰)発現する全細胞RNAを抽出しそして支持体とのハイブリッド形成反応におけるプローブとして使用する。支持体上に付着されたマーカーのいくらか又はすべてとのハイブリッド形成の検出は、関心のある受容体が活性化されることそしてそれゆえ対応する組成物/化合物が該受容体のリガンドを構成し又は含有することを示す。
4.5.治療的関心のある標的の同定
これは、そのスプライシングが病理又は病理モデルにおいて変化させられる遺伝子を同定すること、及び更に特定的には、修飾されたエキソン又はイントロンを同定することにより達成される。このアプローチは、例えば、増殖、分化又はアポトーシスの現象に関与する病理又はいかなる病態生理学的プロセスにおいても変化させられる機能的ドメインをコードする配列を決定することを可能とするであろう。
差異スプライシングされた遺伝子を同定するための定性的差異スクリーニングの利益の例は、野生型p53発現の誘導を介するアポトーシス誘導のモデルへのDATASの適用により与えられる。この細胞モデルは、誘導性p53がん抑制遺伝子発現システムをトランスフェクションすることにより確立された。p53で誘導されたアポトーシスと特異的に関連する定性的な差を同定するために、誘導された細胞及び誘導されなかった細胞に由来するメッセンジャーRNAで出発してDATASを 実施した。これらの実験では、200ngのpoly+RNA及び200ngのcDNAを使用してヘテロ二本鎖を形成させた。各クロスハイブリッド形成から約100クローンが得られた。もとの条件からの全メッセンジャーRNAを表すプローブとこれらのバクテリアクローン、したがってそれらが含有するcDNA断片のハイブリッド形成は、細胞死をもたらす効力のあるp53誘導の期間中に特異的に発現された配列の同定を可能とした(図16)。
これらの断片は、存在するメッセージの品質を変えるエキソン又はイントロン配列に由来し、そしてそれらが参加するか又はそれらが妨害する機能的ドメインを、細胞死を誘発又は抑制するための処理の標的として適格とする(qualify)。
このようなアプローチは、非アポトーシス条件とアポトーシス条件間のすべての差異スプライシングイベントを含むライブラリー対の構築ももたらす。このライブラリー対は、他の病態生理学的条件又は与えられた処理に由来するプローブのハイブリッド形成する能力を試験するのに使用することができる。このようなハイブリッド形成の結果は、アポトーシスに向かう試験条件の遺伝子発現プログラムの潜在的コミットメントに関する指示を与えるであろう。
上記説明から明らかなとおり、本発明は更に:
−本願に記載の方法により同定されそしてそれらが病理条件に特徴的であることを特徴とする配列を認識するいかなる核酸プローブ、いかなるヌクレオチド、いかなる抗体にも関し、
−スクリーニングアッセイを考案することにより治療目的の有機分子の調査のための本明細書に開示された技術を適用することに由来する情報の使用であって、有機分子は健康な条件と病理学的条件間で発生する差異スプライシングされたドメインを標的とすることを特徴とするか、又はそれらは差異スプライシングの結果としてタンパク質により獲得された機能の抑制に基づいていることを特徴とする使用に関し、
−遺伝子治療用途のための本願に記載された方法に由来する情報の利用に関し、
−遺伝子治療により送達されるcDNAの使用であって、該cDNAが規定された細胞シグナル伝達経路のアンタゴニスト又はアゴニストとして挙動する、cDNAの使用に関し、
.調査の目的での診断手段又は試薬の商業的産生、
.治療用途のための分子、ポリペプチド、核酸の発生又は調査、
を目的とするオルタナティブエキソン又はイントロンの分子ライブラリーのいかなる構築又はいかなる使用にも関し、
−オルタナティブエキソン又はイントロンのアレイを含有するすべてのコンピューター化されたバーチャルライブラリーであって、該ライブラリーは2つの異なる病態生理学的条件を区別する選択的スプライシング形態を特徴付けるために核酸プローブ又はオリゴヌクレオチドのデザインを可能とすることを特徴とするライブラリーのいかなる構築又はいかなる使用にも関し、
−本発明の方法により同定又はクローニングされた選択的スプライシング産物を妨害することができるポリペプチド、センスもしくはアンチセンス核酸又は化合物を含むいかなる医薬組成物又は診断組成物にも関し、
−病理条件に固有の選択的スプライシングイベントと対照的に正常な条件を表すスプライシングパターンを回復することができるポリペプチド、センスもしくはアンチセンス核酸又は化合物を含むいかなる医薬組成物又は診断組成物にも関する。
5.毒性化合物によるRNAスプライシング機構の脱調節
この実施例は、差異スプライシング形態及び/又はプロフィルを化合物の毒性及び/又は効能を監視及び/又は決定するためのマーカーとして使用することができることを示す。
RNAスプライシング調節欠陥に対する毒性化合物の効果を下記のとおり試験した。HepG2肝細胞を種々の用量の3つの毒性化合物(エタノール、カンプトテシン、PMA(ホルボール12−ミリステート13−アセテート)で処理した。2つの細胞毒性試験(トリパンブルー、MTT)を種々の時点で行った:エタノールでは4時間及び18時間;カンプトテシンでは4時間及び18時間;PMAでは18時間及び40時間。
トリパンブルーは、生きている細胞により取り込まれうる染料である。顕微鏡下の[青]及び[白]細胞の簡単な計数は、処理後の生存細胞の百分率又は生存百分率を与える。実験ポイントは三回反復で(in triplicate)決定される。
MTT試験は、生きている細胞が可溶性テトラゾリウム塩(MTT)を不溶性ホルマザン沈殿に転化する能力を測定する比色試験である。これらの暗青色ホルマザン結晶を溶解させそしてそれらの濃度を550nmにおける吸光度を測定することにより決定することができる。かくして24ウエルの皿に150,000個の細胞を一夜播種し、次いで毒性化合物で細胞を処理した後、MTT(Sigma)50μlを加える(PBS中の5mg/mlの濃度で)。ホルマザン結晶形成反応をCO2インキュベーター中で5時間行う(37℃、5%CO2、95%湿度)。可溶化溶液(イソプロパノール−トリトンX−100(10%)中の0.1NHCl)500μlの添加の後、結晶を攪拌しながら溶解しそしてそれらの吸光度を550〜660nmで測定する。決定は、適当なコントロール(生存率、細胞死、ブランク)を使用して3回反復してなされる。
アポトーシス又はプログラムされた細胞死の試験も抗ヒストン抗体及びELISAによりDNA断片化を測定することによって行った。RocheからのCell Death ELISA Plusを使用した。
これらの3つの試験の結果(図18 A、B、C)は、下記の濃度:
・エタノール:0.1M
・カンプトテシン:1μg/ml
・PMA:50ng/ml
は、測定されたIC50値より十分に低かったことを示す。
かくして、HepG2細胞を、これらの3つの濃度のこれらの3種の化合物で、エタノール及びカンプトテシンの場合には4時間そしてPMAの場合には18時間処理した。メッセンジャーRNAをRneasyキット(Quiagen)で精製された全RNAから出発してDynal−Oligo−(dT)ビーズで精製した。Superscript逆転写酵素(Life Technologies)及びプライマーとしてランダムヘキサマーを使用してこれらのメッセンジャーRNAからcDNAを合成した。
これらの最初の鎖は、下記のオリゴヌクレオチドプライマーによるPCR増幅反応(94℃1分、55℃1分、72℃1分、30サイクル)のための鋳型として役立った:
MACH−α:
これらのプライマーはMACH−αの種々の記載されたアイソフォーム(1、2及び3、それぞれ595、550及び343塩基対を増幅する)に共通の領域に相当する。MACH−α(Caspase−8)は、プログラムされた細胞死に関与するプロテアーゼである(Boldin et al., (1996), Cell, 85: 803-815)。
BCL−X:
これらのプライマーは、bcl−Xの種々の記載されたアイソフォーム(bcl−XI、bcl−Xs、BCL−Xβ)に共通の領域に相当し(Boise et al., (1993), Cell 74: 597-608; U72398(Genbank)そしてこれらの3つのアイソフォームのための単一の204塩基対断片を増幅するであろう。
FASR:
これらのプライマーは、あるFASRアイソフォームに共通の領域に相当し、そして野生型形態FasRでは478塩基対断片を、アイソフォーム△8では452塩基対及びアイソフォーム△TMでは415塩基対を増幅するであろう。
図19に示された結果は、
・カンプトテシンはアイソフォームMACH−α1の発現の減少及びアイソフォームMACH−α3の増加を誘導する
・カンプトテシンは、新規なbcl−Xアイソフォームの出現(200塩基対付近のダブレットにおける上部バンド)を誘導する
・カンプトテシンは、Fas△TMに相当することができるより短いアイソフォームの発現により置き換えられた、野生型形態のfas受容体の減少を誘導する
・エタノ−ルはより短いアイソフォームにより置き換えられるbcl−xの消失を誘導する
・エタノールはより短いアイソフォームを犠牲にしてfas受容体の長い野生型形態の増加を誘導する、
ことを示す。
これらの結果は、低い濃度の毒性化合物による処理はあるRNAの選択的スプライシングにおける調節欠陥を誘導することができ、そしてこれを特異的な方式で誘導することができることを証明する。転写後のレベルにおけるこれらの調節欠陥の同定、特にDATAS技術の適用による同定は、かくして、分子の毒性を予言するためのツールを構成する。
6.スプライスオリゴヌクレオチドアレイ
特定の遺伝子から生じるRNAアイソフォームは、それらのスプライス連結部配列の点で異なる。本発明は、これらの配列差を利用して連結部オリゴヌクレオチドプライマーを使用することにより特定のアイソフォームの発現を分析することを今や提唱する。これらのプライマーは成熟メッセンジャーRNAのスプライス連結部を横切って特異的にハイブリッド形成するようにデザインされ、そしてそれ故アイソフォーム特異的である。このようなプライマーは、汚染ゲノムDNAにハイブリッド形成しない、従って実験の再現性を増加させる、という追加の利点を与える。
DATAS技術により同定された選択的にスプライシングされた遺伝子がスプライシング連結部分析のために選ばれた。図20に示された5つの位置に関係するこれらの遺伝子の各々についてオリゴヌクレオチドプローブを発生させた(3つの連結部プライマー及び2つのエキソンプライマー)。
exon1オリゴヌクレオチドは野生型及びより短いアイソフォームの両方を監視するであろう。
exon2オリゴヌクレオチドは野生型アイソフォームのみを監視するであろう。
jct 1−2オリゴヌクレオチドは野生型アイソフォームのみを監視するであろう。
jct 2−3オリゴヌクレオチドは野生型アイソフォームのみを監視するであろう。
jct 1−3オリゴヌクレオチドは短いアイソフォームのみを監視するであろう。
スプライシングアイソフォームの存在は、試料中の連結部1−3オリゴヌクレオチドとのハイブリッドの存在を検出することにより、又は更に好ましくは、1つの生物学的試料内の野生型(長い)アイソフォームと短いアイソフォームとの比を測定することにより決定することができる。このような測定は、中立(neutral)(例えば、もし哺乳動物アイソフォームが監視されるならば、非哺乳動物)複合RNA混合物中にスパイクされた(spiked)合成RNAを使用してこれらのオリゴヌクレオチドの各々のハイブリッド形成有効性を決定することにより行うことができる。次いで正規化係数を使用して、
(野生型/短い)=exon2/Jct1−3=Jct1−2/Jct1−3
=Jct2−3/Jct1−3
を監視することができる。
例えば、2つの生物学的試料A及びB間の野生型と短いアイソフォームとの比の変化は:
(野生型/短い)A/(野生型/短い)B=[(野生型)A/(野生型)B]×[(短い)B/(短い)A]
により計算することができる。
[(野生型)A/(野生型)B]は、エキソン2(共通エキソン)、Jct1−2又はjct2−3オリゴヌクレオチドにより得られる結果を使用することにより測定することができる。
[(短い)B/(短い)A]は、Jct1−3オリゴヌクレオチドにより得られた結果を使用して測定することができる。
これらのプライマーの各々を3つの異なる長さ(24、30及び40塩基)として発生させた。これらのプライマーを3D−link(登録商標)(Motorola)活性化スライド上に配置してマイクロアレイ分析のための三次元マトリックスを創る。確認の目的で、これらの分析は、3つの選ばれた遺伝子の各アイソフォームに相当するin vitro転写されたRNAを使用して行うことができる。
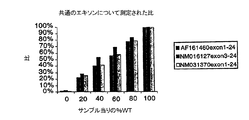
低酸素症関連モデルから単離された3つのDATASクローンは課役のmRNAに相当する:
・GenbankリファレンスAF161460.1:ホモサピエンスHSPC111mRNA
・RefseqリファレンスNM_031370.1:ホモサピエンスヘテロ核リボ核タンパク質D(Homo sapiens heterogenous nuclear ribonucleoprotein D)
・RefseqリファレンスNM_016127:ホモサピエンス仮想的タンパク質(hypothetical protein)MGC8721。
各遺伝子について、プライマーオリゴヌクレオチドの対を同定されたDATAS断片のまわりにデザインした。PCR増幅は、野生型の長いアイソフォーム及びエキソン配列に相当するいくらかの核酸配列を欠失しているより短いアイソフォームを発生させるであろう。
これらのプライマーオリゴヌクレオチドは、配列番号17及び18(AF161460.1について)、配列番号19及び20(NM_031370.1について)、配列番号21及び22(NM_016127について)である。
野生型形態及びより短い形態は同定されておりそして、
に相当する。
6.1.オリゴヌクレオチドデザイン
我々は、検出を改良するために、発生されるべきすべてのオリゴヌクレオチドのための共通の熱力学的プロフィルを作成することを決定した。ゆえに、我々は、均一な溶融温度を選びそして一定の長さを有するオリゴヌクレオチドをデザインした。我々は、24マー、30マー及び40マーを評価することを決定した。また、標的スプライス連結部(target splice junction)に対するオリゴヌクレオチドの種々の位置を、中心のあるオリゴヌクレオチドから非対称オリゴヌクレオチドまで考慮した。オリゴヌクレオチドのデザインは、高いスループット特徴を与えるソフトウエアー、例えば、Array Designer2又はFeaturamaにより支援することができる。この実施例では、Primer Finderを使用しそして下記の基準が定義されそして適用された:
−24マー及び30マーでは40〜60%の%GC及び40マーでは30%〜60%の%GC 、
−溶融温度:24マー及び30マーでは65℃〜70℃及び40マープライマーでは65〜75℃、
−プライマー濃度:50nM
−塩濃度:50mM。
有意なヘアピン傾向及び自己二量体化傾向を有するプライマーは隠れている(hidden)。
配列番号29〜79のオリゴヌクレオチドをデザインしそして合成した。それらは25μMの濃度で1Xプリンティングバッファーに取り込ませた。BioroboticsからのMicroGridIIスポッターを使用して製造者の教示(Motorola)に従ってスライドを調製してスプライスオリゴアレイを作成した。
6.2.50/50比で混合された合成プローブとのハイブリッド形成
これらのバイオチップを、短いアイソフォーム及び野生型アイソフォームの50/50混合物とハイブリッド形成させた(全体で6)。プローブ調製及びハイブリッド形成条件を下記に詳述する。
試験核酸を95℃で3分間変性し、そして遠心により冷却した。試験核酸をスライド(3D−Link(登録商標)、Motorola)上に置きそしてカバースリップを注意深く所定の位置に置いた。ハイブリッド形成温度は、与えられた長さを有するオリゴヌクレオチドのすべてについて均一である溶融温度より15℃低いと規定された。
これらのハイブリッド形成温度は:
・50℃(24マー)
・55℃(30マー)
・60℃及び50℃(40マー)
である。
ハイブリッド形成あたり下記の条件を使用した:
・断片化されたcRNAプローブ20ng
・ハイブリッド形成緩衝液(5×SSC/0.1%SDS)、14μlとするのに十分な量
・1.5μl鮭精子DNA1μg/μl。
加湿されたハイブリッド形成チャンバ中で8〜16時間インキュベーションを行った。スライドを、ハイブリッド形成のために使用された温度で2×SSC/0.1%SDSの低ストリンジェンシー溶液で洗浄した。室温で0.2×SSC及び0.1×SSC緩衝液を使用する追加の洗浄によりストリンジェンシーを増加させた。次いでスライドをスピン乾燥しそしてScan Array4000(Packard Instruments)及びScanArrayソフトウエアを使用して走査した。次いでスポット当たりの蛍光強度をQuant Arrayにより決定した。
スライドの分析は、ブロッキング緩衝液及びハイブリッド形成条件及びハイブリッド形成後の条件の適切な選択を示す低いバックグラウンド強度値を示す。更に、スポットは、ガラススライドの品質、プリントされるべき標的の適当な濃度及び使用されるプリンティング緩衝液により均一でありそして同じ形態である。予想されるとおり、赤いスポットは共通のエキソン、スキップドエキソン及び連結部1−2及び2−3に特異的なオリゴについて得られ、そして緑色のスポットは共通のエキソン及び連結部1−3に特異的なオリゴについて得られた。
両方の像を重ね合わせると、スポットは共通のエキソンに特異的なオリゴではオレンジで現れ、これは等量のCy5短形態cRNA及びCy3長形態cRNAのハイブリッド形成を示す。
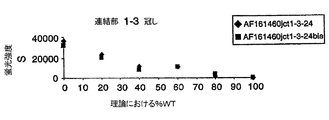
スプライス連結部に中心のあるオリゴヌクレオチドで最善の結果が得られた。しかしながら、他の可能性も考慮されそして再現性のある結果を生じた。観察から、ハイブリッド形成は連結部に中心のあるオリゴ(12/12)で高度に特異的でありそして特異性はオリゴが連結部におけるより高度に非対称的な位置に向かうと減少することが明らかである。しかしながら、僅かな非対称性はハイブリッド形成の品質に影響を与えない[(NM016127jct1−2(13/11)、jct1−2bis(12/12)、jct1−3(13/11)、jct1−3bis(12/12))。
同様な結果が30マー及び40マーを使用して得られたが、最善の特異性は30マーにより達成される。
6.3.種々の比で混合された合成プローブとのハイブリッド形成
本方法が長いアイソフォーム対短いアイソフォームの比を決定することを可能とすることを確証するために、これらの比を0、20、40、60、80,100%に調節した。次いでこれらのプローブをスライドにハイブリッド形成させた。長いアイソフォーム(Cyanine3)500ng及び短いアイソフォーム500ng(500ng)を調製した。両方の試料を断片化しそして脱塩した。次いでプローブをハイブリッド形成緩衝液中で10ng/μlの濃度に希釈しそして断片化した長い形態及び短い形態を下表に記載の如く一緒にプールした。
各試料4μlをハイブリッド形成緩衝液(5×SSC/0.1%SDS)で15μlとしそして鮭の精子DNA(1μg/μl)1.5μlを加えて後変性した(95℃で2分)。次いで試料をスライド(24マーオリゴアレイ)上に加えそしてカバースリップを注意深く置いた。加湿されたハイブリッド形成チャンバ中で50℃で16時間にわたりインキュベーションを行った。スライドを2×SSC/0.1%SDS50℃の低ストリンジェンシー溶液で洗浄した。次いで室温で0.2×SSC及び0.1×SSC緩衝液を使用する追加の洗浄によりストリンジェンシーを増加させた。次いでスライドをスピン乾燥しそしてScan Array4000(Packard Instruments)及びScanArrayソフトウエアを使用して走査した。次いでスポット当たりの蛍光強度をQuantArrayにより決定した。
野生型と短いアイソフォームとの共通のエキソンに相当する強度の比は、
(強度Cy5−バックグラウンド強度Cy5)/(強度Cy3−バックグラウンド強度Cy3)
に従って計算した。
図21Aは、3つの試験された遺伝子について計算された比は予想値と良く一致していることを示す。
比の変化を連結部1−3に関しても監視した。図21Bに示されたとおり、長いアイソフォームの量は増加すると共に蛍光強度は連結部1−3に関して減少する(AF16140のための2つの連結部オリゴヌクレオチドに関する図21B参照)。
これらの結果は、長いアイソフォーム対短いアイソフォームの比を決定することができることを確証する。
6.4.複合RNA集団とのハイブリッド形成
この実施例では、我々はcRNAの複合混合物にスパイクされた標識されたアイソフォームをハイブリッド形成させることができることを証明しそして蛍光強度の有意な値を検出するのに必要なプローブの量として感受性を評価した。
減少していく量の6つのアイソフォームの全量(20ng、5ng、1.25ng、0.32ng、0.16ng、0.08ng、0.04ng)をショウジョウバエ(Drosophila)RNA300ngにスパイクした。得られる断片化アイソフォームプローブを20ng/μl、1ng/μl、及び0.1ng/μlの濃度とした。ショウジョウバエの全RNAを線状増幅に付しそしてcRNAを1μg/μlの最終濃度にした。
下記の表はハイブリッド形成されるべき試料の組成物を記載する。
試料を95℃で3分間変性しそして遠心により冷却した。試料をガラススライド上に加えそしてカバースリップを置いた。加湿されたハイブリッド形成チャンバ中で50℃で8〜16時間にわたりインキュベーションを行った。スライドを2×SSC/0.1%SDS50℃の低ストリンジェンシー溶液で洗浄した。室温で0.2×SSC及び0.1×SSC緩衝液を使用する追加の洗浄によりストリンジェンシーは増加する。次いでスライドをスピン乾燥しそしてScan Array 4000(Packard Instruments)及びScan Arrayソフトウエアを使用して走査する。次いでスポット当たりの蛍光強度をQuant Arrayにより決定する。
図22はNM_016127について得られた像を示す(同様な結果が他の2つの遺伝子で得られた)。検出可能な最も低い量はショウジョウバエの断片化された標識されたcRNAにスパイクされた断片化された標識されたアイソフォーム0.16ngのあたりである。この結果は、全ショウジョウバエ(Drosophila)RNA3000ngを使用する場合にも観察された。
図23及び24は、長いアイソフォームと短いアイソフォームとの50/50の比は全量で0.16ngに材料の量を減少させるときでも依然として計算することができることを更に証明する。
要するに、感受性の調査は、全RNA3000ngにおけるアイソフォーム当たり26pg(6で割った0.16ng)までの蛍光強度の検出をもたらす(即ち、0.001%で存在するmRNAの検出又は総計107個の細胞について細胞当たり3コピーのmRNAの検出)。
6.5.複合ヒトRNA集団とのハイブリッド形成
ヘパトーマ細胞系HepG2に由来するRNA3mgをスプライスオリゴヌクレオチドスライドとのハイブリッド形成のためのプローブとして使用した。
像及び蛍光値はNM_016127及びNM_031370について図25に示される。Jct1−3又はJct1−3bis蛍光値は、NM_031370はその野生型カウンターパートと比較したとき有意水準で発現され、NM_016127についてそうではないことを示す。これらの結果は、ヒト試料からを含めて試料におけるスプライシング変動を検出するための本発明の方法及び生成物の特異性及び感受性を確証する。
6.6前立腺腫瘍監視
スプライスイベントのより大きいパネルで更なる確認を行った。PSA(前立腺特異的抗原又はKallikren−3(KLK3)及びのKallikrein−2(KLK2)遺伝子に重点を置いた。オリゴヌクレオチドを発生させて野生型RNAの発現及び以前に特徴付けられたPSA及びKLK2のいくつかのスプライスイベント(patent application WO03/076610参照)の発現を監視した。
PSA及びKLK2は同じ又は同様な長さの5エキソン及び4イントロンからなる同様な構造を有する極めて相同性の遺伝子である。157オリゴヌクレオチド(配列番号80〜236)が、この節で以前に述べた基準(簡単に言えば、24ヌクレオチドの長さ(7オリゴヌクレオチドはTm、GC百分率又は二次構造制限に適合するように25ヌクレオチド長さであるけれども)、65〜70℃のTm、40〜60%の%GC、自己二量体化及びヘアピン形成のないこと)に従ってデザインされた。スプライス連結部が標的化されるとき、オリゴヌクレオチドはそれらの中心に又はそれらの中心近くに連結部を位置づけるようにデザインされる。
これらのオリゴヌクレオチドは、
−PSAの5つのエキソン(配列番号80〜85)及びKLK2の5つのエキソン(配列番号175〜180)、
−PSAの4つのイントロン(配列番号98〜101)及びKLK2の4つのイントロン(配列番号185〜188)、
−PSAの4つのエキソン−エキソン連結部(配列番号86〜89)及びKLK2の4つのエキソン−エキソン連結部(配列番号181〜184)、
−PSAの8つのエキソン−イントロン連結部(配列番号90〜97)及びKLK2の8つのエキソン−イントロン連結部(配列番号189〜195)、
を標的とする。
これらのオリゴヌクレオチドの残りはPSA内のスプライスイベントを特異的に監視するようにデザインされた。
これらのオリゴヌクレオチドの大部分はスプライスイベントを識別することができる。何故ならば、それらはスプライスイベントに特徴的な特異的連結部内にあるからである。
オリゴヌクレオチド(配列番号80〜236)は方形プレート(quadriplates)としてスポットされるべき25マイクロモルの濃度の1Xプリンティング緩衝液中に取り込まれた5′端部(C6−NH2)における修飾をともなって合成された。BioRoboticsからのMicroGridllスポッターを使用して製造者の教示に従ってスライド(codelink(登録商標)、Amersham)を調製してスプライスオリゴアレイ又はスプライスチップを作成した。Arabidopsis thaliana 光化学系IクロロフィルA/B結合タンパク質遺伝子に相当するコントロールオリゴヌクレオチドもチップ上に加えた。
オリゴヌクレオチドの特異性を確認するために、野生型及び各アイソフォームに相当する個々の標識されたプローブとのハイブリッド形成を行った。これらのプローブは低酸素症関連モデルからの長いアイソフォーム及び短いアイソフォームについて前記した如くして調製する。55℃の温度で前記のようにハイブリッド形成を行った。スライドを加工しそして前記のように分析した。
Cy3及びCy5シグナルを区別することができる共焦点レーザースキャナー(Scan Array 4000、Packard Instrument)によりハイブリッド形成したアレイを読み取りそしてScan Arrayソフトウエア(Packard Instrument)を使用して各チャンネルについて別々のTIFF像を作ることにより遺伝子発現を評価する。Quant Array(Packard Instrument)はこれらの像をインポートし、スポットを横切ってグリッドを置きそして生のスポット強度からローカルバックグラウンドを差し引くことができる。Quant Arrayは各ハイブリッド形成についてエクスポートテキストファイルを発生させる。
次にGeneTrafficソフトウエア(Iobion Informatics)を使用して、試料調製並びにマイクロアレイ実験のアノテーション(MIAME)、アドレスデータ分析に必要なプロトコルを追跡しそして広範な統計的分析を可能とする。
バックグラウンドの2倍を超える蛍光シグナルを示すすべてのオリゴヌクレオチドを記録した。
図28は、野生型KLK2とハイブリッド形成したスライドの像及びポジティブオリゴヌクレオチド及びそれらのシグナル強度(バックグラウンドを超える倍比(fold)として)を示す分析を示す。23オリゴヌクレオチドがバックグラウンドの2倍を超えるシグナルを生じそしてすべて野生型KLK2とハイブリッド形成すると予測された。重要なことに、連結部と関連したアイソフォーム特異的識別性オリゴヌクレオチドはいかなるシグナルも生じなかった。
図29は、PSA−016アイソフォームとハイブリッド形成したスライドの像及びポジティブオリゴヌクレオチド及びそれらのシグナル強度(バックグラウンドを超える倍比(fold)として)を示す分析を示す。4つのオリゴヌクレオチドがバックグラウンドの2倍を超えるシグナルを生じそしてすべてPSA−016とハイブリッド形成すると予測された。重要なことであるが、識別性オリゴヌクレオチドPSA−016−ex1−ex2(配列番号122)はその対応するアイソフォームとハイブリッド形成する。他の3つのオリゴヌクレオチドはすべてPSA−016内に存在し、かくしてハイブリッド形成すると予測された(それらはPSA−016に対して独特ではないけれども)。
このようなアッセイは、スプライス変異体の発現を十分に監視するためのオリゴヌクレオチドのデザインの正当性を立証する。
次にPSA/KLK2スプライスチップを、前立腺がん患者からの正常な組織及びがん前立腺組織に由来する複合RNAから作られたプローブとハイブリッド形成させた。これらのRNAはGenomics Collaborative、USAから得られた。これらのRNAを発生させるために使用された組織は、公認の病理学者によりコントロールされた。腫瘍組織は腫瘍細胞を少なくも70%含有すると評価された。RNAはAgilent Bioanalyserでのゲル電気泳動を行うことにより品質制御されそしていかなる劣化の兆候も示さなかった。簡単に言えば、全RNA5ugを線状増幅(T7増幅法)のための出発物質として使用する。次に、得られるcRNAを50%DMSOの存在下にCy3又はCy5蛍光染料で間接標識した。標識されたプローブ(良性及び腫瘍試料)5〜6ugを化学的に断片化しそしてチップ上で共ハイブリッド形成させた。図30は一人の前立腺がん患者からの良性及び腫瘍RNAに由来するプローブと共ハイブリッド形成したPSA/KLK2スプライスチップの像を示す。
3つの追加のマッチした対をハイブリッド形成させそして分析した。表Aは、4人の患者で得られた結果を示す。患者の少なくとも一人においてバックグラウンドの2倍より高いシグナルを示すオリゴヌクレオチドのみが選ばれた。106のオリゴヌクレオチドがこの基準に合格し、その中でも63は識別性オリゴヌクレオチド、即ち、連結部(野生型形態又はスプライス変異体からのいずれかの)を標的とするオリゴヌクレオチドであった。43のオリゴヌクレオチドは少なくとも一人の患者において正常な試料と腫瘍試料間の調節異常(dysregulations)を示した(1.5倍のアツプレギュレーション又はダウンレギュレーションの閾値)。これらの43のオリゴヌクレオチドの内の6つは少なくとも3人の患者について一致していた。
他の重要な分析は、野生型形態に対するスプライス変異体の相対的豊富さを評価することからなる。これは、野生型基準に対する各オリゴヌクレオチドの強度の比を測定することで評価することができる。表BはPSA及びKLK2のエキソン1及びエキソン4から取られた2つの異なる基準を使用しそして4人の患者からの良性組織で評価された結果を示す。両基準とも同じ結果を生じるが、該結果はアイソフォームの相対的発現において患者間で大きな変動性がある。各オリゴヌクレオチドは異なる効力でその標的にハイブリッド形成するので、野生型に対する各アイソフォームの絶対百分率を誘導することは困難である。しかしながら、4人の患者にわたって与えられたオリゴヌクレオチドについてこれらの比を比較することは可能である。次いで2倍より大きい差が少なくとも二人の患者において56のオリゴヌクレオチドで観察されうる。
このタイプの分析は、大規模でのスプライス変異体の発現を監視するための実行可能性を確認する。PSA/KLK2チップは、特に連結部オリゴヌクレオチドはスプライス変異体に含まれるスプライス連結部に関する価値ある情報を与えるように特別にデサザインされうることを示す。PSA/KLK2レパートリーにおける腫瘍脱調製及び患者間の変動性の同定は、前立腺がんの検出、診断又は予後を改善するのに価値あることを証明することができる。
7.スプライス連結部同定
7.1.この実施例は、ss−cDNAの第1集団及びds−cDNAの第2集団からのスプライスドメインの同定又はクローニングを説明する。この方法は、不均一RNA集団からなる複合生物学的試料を使用して行われた。更に特定的には、下記の2つのRNA試料を使用した:
−試料1:特にhnRNPA1を発現するEC293細胞に由来するRNA、及び
−試料2:ポナステロン(ponasterone)により誘導されると、hnRNPA1bと名付けられたhnRNPA1のスプライシング変異体を発現するプラスミドpIND−マウスA1bでトランスフェクションされたEC293細胞に由来するRNA。
かくして、これらの2つの複合RNA集団は、種々の遺伝子の種々のアイソフォーム、特に、hnRNPA1コードするRNAの2つのアイソフォーム、追加のエキソンを含むhnRNPA1bアイソフォーム、を含有する。
該試料の各々からのmRNA1μgを逆転写反応において使用して、オリゴdTプライマーの存在下にss−cDNAを産生させた。これらの試料の1つの逆転写のために、ビオチニル化オリゴdTプライマーを使用して、標識されたss−cDNAの1つの集団を産生させた。次いで試料1のss−cDNAからds−cDNAを産生させた。これらの複合cDNA集団を、次いで1/5の割合のss−cDNAとds−cDNAを使用してハイブリッド形成させた。平行して、コントロール状況(試料1、「C」)に由来するビオチニル化ss−cDNA及び誘導された状況(試料2、「I」)に由来するds−cDNAを同じ割合で使用してハイブリッド形成を行った。cDNAをハイブリッド形成緩衝液(80%ホルムアミド、20%SDS)中に懸濁させ、熱変性し、そして40℃で一夜冷却することによりハイブリッド形成を行った。反応混合物中の標識された分子種をストレプトアビジンコーテッドビーズを使用して回収した。ハイブリッドをSau3AIにより消化した。対を形成していない領域を含む得られる断片をビオチニル化(半)ランダムオリゴヌクレオチド(N25又はN25GGC)とインキュベーションして、存在するすべての一本鎖配列とのハイブリッドの形成を引起し、それにより対を形成していない領域を含む断片を捕捉した。このようなハイブリッドをストレプトアビジンコーテッドビーズを使用して回収し、断片を溶離しそしてアダプターを各末端でライゲーションして、選ばれたds−cDNA断片のすべての両方の鎖の増幅反応のための鋳型配列を与えた。両鎖の増幅が行われるので、この方法は、RNAのスプライシングされた配列と対応するスプライシングされていない配列の特徴を示す核酸のライブラリーを発生させる。次いでこれらの断片をTAベクターにクローニングして、コンピュータソフトウエアを使用して配列決定及び/又は分析する。
結果を図27に示す。これらの結果は、PCR増幅すると、スプライシングされたドメインの特徴を示す核酸の集団が得られることを示す。実際に、ハイブリッド形成産生物(I/C又はC/I)を分析するときスメアー(smear)が得られ、一方このようなスメアーはコントロール実験では現れない。更に、ハイブリッド形成産生物I/C及びC/IにおいてhnRNPA1及びA1bに特異的なプライマーによりこれらの核酸を増幅すると、特異的バンドが観察され、それによりこの方法は生物学的試料を識別する生物学的に関係のあるスプライシング(biologically relevant splicings)の選別を可能とすることを証明する。
7.2.この別の実施例は、ss−cDNAの第1集団及びds−cDNAの第2集団からのスプライスイベント及びスプライス連結部の同定又はクローニングを説明する。この方法を1つ又はそれより多くの特異的RNAに富んだ不均一RNA集団からなる複合生物学的試料を使用して行った。更に特定的には、下記の2つのRNA試料を使用した:
試料1:特にPSA(Prostate Specific Antigen又はKLK3)を発現しそして潜在的にいくらかのスプライスアイソフォームを発現するMDA−2B前立腺がん細胞からの増幅されたRNA、及び
試料2:明白にPSAを発現しそして潜在的にいくらかのスプライスアイソフォームを発現するLnCap前立腺がん細胞からの増幅されたRNA。
該試料の各々からのmRNA1マイクログラムを逆転写反応において使用してPSA様cDNAに富んだss−cDNASを産生させた。このために、T7プロモーターテイルと関連したPSA遺伝子の第5エキソンにおける配列に相補的なofa配列からなるPSA特異的オリゴヌクレオチドプライマーを使用した:
次いで二本鎖cDNAをこれらのss−cDNAから産生させた。これらのds−cDNAは機能的T7プロモーターを示し、かくしてT7RNAポリメラーゼを使用して相補的RNA(cRNA)を産生させるのに役立つことができる。該試料の各々からのcRNAを逆転写反応において使用して、エキソン1の5′端部に位置したPSA特異的プライマー
の存在下にss−cDNAを産生させた。これらの試料の1つの逆転写のために、ビオチニル化プライマーを使用して標識されたss−cDNAの1つの集団を産生させた。次いでds−cDNAを試料2のss−cDNAから産生させた。これらの複合cDNA集団を、次いで1/5の割合のss−cDNAとds−cDNAを使用してハイブリッド形成させた。平行して、同じ割合で、試料1に由来するビオチニル化ss−cDNA「コントロール1」)及び試料2に由来するds−cDNA「コントロール2」のみを使用してハイブリッド形成を行った。cDNAをハイブリッド形成緩衝液(80%ホルムアミド、20%SDS)中に懸濁させ、熱変性し、そして40℃で一夜冷却することによりハイブリッド形成を行った。反応混合物中の標識された分子種をストレプトアビジンコーテッドビーズを使用して回収した。ハイブリッドをBssSI及びStyI、それぞれPSAのエキソン2及びエキソン5に位置した2つの制限酵素、により消化した。対を形成していない領域を含む得られる断片をビオチニル化(半)ランダムオリゴヌクレオチド(N25又はN25GGC)とインキュベーションして、存在するすべての一本鎖配列とのハイブリッドの形成を引起し、それにより対を形成していない領域を含む断片を捕捉した。このようなハイブリッドを、ストレプトアビジンコーテッドビーズを使用して回収し、断片を溶離しそしてアダプターを各末端でライゲーションして、選ばれたds−cDNA断片のすべての両方の鎖の増幅反応のための鋳型配列を与えた。ここで、アダプターは、BssSI及びStyI酵素の付着性末端に相当する(下記の2つのオリゴヌクレオチド:5′−GCATTAACCCTCACTAAAGGGAC−3′及び
から作成されたBssIアダプター;下記の2つのオリゴヌクレオチド:5′−GCATTAACCCTCACTAAAGGGAC−3′ 及び
から作成されたStyIアダプター。両鎖の増幅はアダプター配列に由来するプライマーにより行われるので、この方法は、RNAのスプライシングされた配列と対応するスプライシングされていない配列の両方に特徴的な核酸のライブラリーを発生させる。次いでこれらの断片をアガロース電気泳動でスメアーとして可視化し、一方2つのコントロールから生じた増幅はいかなる可視シグナルも生成しなかった。次いでそれらをTAベクターにクローニングして、コンピュータソフトウエアを使用して配列決定及び/又は分析する。この実施例のプロセスは図31に示される。
配列分析は、予想されるBssI及びStyI部位に2つのアダプター配列を示しそしてこれらの2つの部位の間にPSA遺伝子に由来する配列を示すクローニングされた断片を示した。野生型PSA配列に相当するクローンは回収されそして下記のイベントに相当するクローンも回収された:
・エキソン3の5′端部における129ヌクレオチドの欠失。このイベントは以前に記載された(Tanaka T.et al, Cancer Res.2000; 60(1): 56-9)。
・エキソン3内の119ヌクレオチドの欠失。このイベントは以前にGenbankに寄託されておりそしてその後公表された(AJ459782; Heuze Vourc'h N.et al, Eur J Biochem.2003, 270(4): 706-14
・エキソン3の3′端部の18ヌクレオチドの伸長部(イントロン3の5′端部の18ヌクレオチドに相当する)。このイベントはPCT patent application noWO03/076610に以前に記載されている。
コンセンサススプライスイベントに相当する新規なイベントも特徴付けられた:
・エキソン3の5′端部における新規な欠失。
・エキソン4及びエキソン3の大部分を包含する新規な欠失。
要するに、初めて記載された2つを含む5つのPSAアイソフォームが、関心のある遺伝子(ここではPSA)に無関係の遍在するプライマーによるPCR増幅の後それらの配列の先見的知識なしに特徴付けられた。
表A。4人の前立腺がん患者に由来するプローブとのPSA/KLK2スプライスチップのハイブリッド形成分析。強調されたオリゴヌクレオチドは連結部オリゴヌクレオチドに相当する。各患者について、正規化された強度が示される。少なくとも一人の患者においてバックグラウンドの2倍より高い蛍光シグナルを生じるオリゴヌクレオチドのみが選ばれた。ブランクは、シグナルがバックグラウンドの2倍より低いことを示す。1.5より高いアップ又はダウンレギュレーションの倍比変化(fold change)を示すオリゴヌクレオチドは黄色で強調されておりそして倍比変化の欄において青で強調される。
表B。4人の前立腺がん患者に由来するプローブによるPSA/KLK2スプライ スチップの追跡分析。少なくとも一人の患者においてバックグラウンドの2倍より高い蛍光シグナルを生じるオリゴヌクレオチドのみ(良性組織のみ)が選ばれた。比は、各オリゴヌクレオチドのシグナル強度を、2つの基準、即ち、PSA及びKLK2についてのエキソン1又はエキソン4のシグナル強度で割ることにより計算される。ブランクはシグナルがその患者でバックグラウンドの2倍より低いことを示す。与えられたオリゴヌクレオチドについては、比は黄色で強調されそして、2つの差が少なくとも2倍差異なるとき青で強調された。