JP2005296003A - 三量体Gタンパク質αサブユニットGm1のプロモーター及びその利用 - Google Patents
三量体Gタンパク質αサブユニットGm1のプロモーター及びその利用 Download PDFInfo
- Publication number
- JP2005296003A JP2005296003A JP2005070977A JP2005070977A JP2005296003A JP 2005296003 A JP2005296003 A JP 2005296003A JP 2005070977 A JP2005070977 A JP 2005070977A JP 2005070977 A JP2005070977 A JP 2005070977A JP 2005296003 A JP2005296003 A JP 2005296003A
- Authority
- JP
- Japan
- Prior art keywords
- polynucleotide
- protein
- substance
- subunit
- promoter
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images


Landscapes
- Investigating Or Analysing Biological Materials (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Peptides Or Proteins (AREA)
Abstract
【解決手段】三量体Gタンパク質αサブユニットGm1のプロモーター領域である塩基配列を有することを特徴とするポリヌクレオチド、該ポリヌクレオチドもしくはその形質転換細胞を用いたシグナル伝達調節物質のスクリーニング方法を提供する。
【選択図】なし
Description
詳述すれば、Gタンパク質はαβγのサブユニットからなる。αサブユニットにGDPが結合した不活性型のGタンパク質は、ホルモンや神経伝達物質などの受容体アゴニストがGPCRに結合すると、αサブユニットからGDPが放出され、これに代えてGTPが結合する。αサブユニットにGTPが結合した活性型G-タンパク質は、GPCRから離れるとともに、GTP結合型αサブユニットとβγサブユニットとに解離する。活性型Gタンパク質は、標的エフェクターであるアデニル酸シクラーゼ、Ca2+チャネル、K+チャネル、ホスホリパーゼCβ等を促進又は抑制することにより、多彩な細胞機能を調節する。活性型Gタンパク質のαサブユニットに結合したGTPは、αサブユニットが有するGTPaseの作用によりGDPとなり、不活性型に戻る。
これまで、哺乳動物の細胞において多種類のGタンパク質が同定されている。これらのGタンパク質はそれぞれ固有の組織分布を有し、例えば、神経細胞、嗅細胞、視細胞、味細胞、肺細胞、腎細胞又は肝細胞等に分布するGタンパク質が知られている(例えば、非特許文献2参照)。これらのGタンパク質はいずれも、GTPが結合するとともにGTPaseを活性化する部位及びαβγの3量体形成ドメインを有している(例えば、非特許文献2参照)。
このように、Gタンパク質は、固有の組織において細胞のシグナル伝達系に関与することによりホルモン受容、神経伝達、細胞の増殖・分化など重要な役割を果たす。
また、Gタンパク質αサブユニットが正常な機能を失うことにより、様々な疾患が誘発されることが明らかになってきている。例えば、Gタンパク質αサブユニットGsが定常的に活性化されることにより、下垂体腫瘍、甲状腺腫瘍又はMcCune-Albright症候群等の疾患が誘発される。またGタンパク質αサブユニットGsの機能が消失することにより、偽性副甲状腺機能低下症が誘発される。またGタンパク質αサブユニットGi2が定常的に活性化されることにより、下垂体腫瘍、副腎皮質腫瘍又は子宮腫瘍が誘発される(例えば、非特許文献3参照)。
尚、Gタンパク質の発現量の低下は、細胞外からのシグナルを細胞内に伝える機能の減衰を引き起こす。このことより、Gm1の発現量の低下が、これら神経・精神疾患(統合失調症、躁鬱病、うつ病および不安症)の症状に関わっていることが示唆され、三量体Gタンパク質αサブユニットGm1をコードする遺伝子の転写を制御する能力を有する塩基配列からなるポリヌクレオチドを同定できれば、当該Gm1の発現量の異常、又は、当該Gm1のプロモーターを介したシグナル伝達の異常に起因する疾患(特に、神経・精神疾患)の治療・改善・予防のための医薬(即ち、治療薬、改善薬又は予防薬)の有効成分としての化合物若しくはその薬学的に許容される塩を探索するための方法において利用することができる。
本発明は、
1.三量体Gタンパク質αサブユニットGm1のプロモーター領域である塩基配列を有することを特徴とするポリヌクレオチド(以下、本発明ポリヌクレオチドと記すこともある。);
2.プロモーター領域である塩基配列が、以下の(1)〜(4)のいずれかに記載される塩基配列であることを特徴とする前項1記載のポリヌクレオチド。
(1)配列番号1で示される塩基配列
(2)配列番号1で示される塩基配列における塩基番号603番から3871番までの領域で示される塩基配列
(3)上記(1)又は(2)の塩基配列において1個もしくは複数個の塩基が欠失、置換もしくは付加された塩基配列からなり、かつ三量体Gタンパク質αサブユニットGm1をコードする遺伝子の転写を制御する能力を有する塩基配列
(4)上記(1)又は(2)の塩基配列からなるポリヌクレオチドとストリンジェントな条件下にハイブリダイズするポリヌクレオチドの塩基配列に相補であり、かつ三量体Gタンパク質αサブユニットGm1をコードする遺伝子の転写を制御する能力を有する塩基配列;
3.請求項1又は2記載のポリヌクレオチドを含有することを特徴とするプラスミド(以下、本発明プラスミドと記すこともある。);
4.請求項1又は2記載のポリヌクレオチドを含有し、かつ当該ポリヌクレオチドの下流(3’側)に当該ポリヌクレオチドによって転写が制御されるポリヌクレオチドを含有することを特徴とするプラスミド;
5.請求項1又は2記載のポリヌクレオチドを含有し、かつ当該ポリヌクレオチドの下流(3’側)にレポーター遺伝子を含有することを特徴とするプラスミド;
6.前項1,2、3又は4記載のプラスミドが導入されてなる形質転換細胞(以下、本発明形質転換細胞と記すこともある。);
7.前項5記載のプラスミドが導入されてなる形質転換細胞;
8.三量体Gタンパク質αサブユニットGm1のプロモーターを介したシグナル伝達調節物質の探索方法であって、
(1)請求項7記載の形質転換細胞に被験物質を接触させる第一工程、及び、
(2)前記第一工程後に、レポーター遺伝子の発現量又はそれと相関する指標値をモニターする第二工程、
(3)前記第二工程によりモニターされた発現量又はそれと相関する指標値の変化に基づき前記物質が有する三量体Gタンパク質αサブユニットGm1のプロモーターを介したシグナル伝達調節能力を評価する第三工程、及び
(4)前記第三工程で評価されたシグナル伝達調節能力に基づき三量体Gタンパク質αサブユニットGm1のプロモーターを介したシグナル伝達調節能力を有する物質を選抜する第四工程、
を有することを特徴とする探索方法(以下、本発明探索方法と記すこともある。);
9.物質が有する三量体Gタンパク質αサブユニットGm1のプロモーターを介したシグナル伝達調節能力を評価する方法であって、
(1)請求項6記載の形質転換細胞に被験物質を接触させる第一工程、及び、
(2)前記第一工程後に、レポーター遺伝子の発現量又はそれと相関する指標値をモニターする第二工程、及び
(3)前記第二工程によりモニターされた発現量又はそれと相関する指標値の変化に基づき前記物質が有する三量体Gタンパク質αサブユニットGm1のプロモーターを介したシグナル伝達調節能力を評価する第三工程、
を有することを特徴とする評価方法(以下、本発明評価方法と記すこともある。);
10.請求項1記載のポリヌクレオチドと結合する物質の探索方法であって、
(1)請求項1記載のポリヌクレオチドと被験物質とを接触させる第一工程、及び
(2)前記第一工程後に、当該ポリヌクレオチドと被験物質との複合体生成の有無を調べる第二工程、及び
(3)前記第二工程で得られた複合体生成の有無結果に基づき当該ポリヌクレオチドと結合する物質を選抜する第三工程、
を有することを特徴とする探索方法(以下、本発明結合物質探索方法と記すこともある。);
11.請求項1記載のポリヌクレオチドと結合する物質の精製方法であって、
(1)請求項1記載のポリヌクレオチドと試料とを接触させて、当該ポリヌクレオチドと当該試料中に含有される当該ポリヌクレオチドと結合する物質との複合体を生成させる第一工程、及び、
(2)前記第一工程後、生成させた複合体から当該結合物質を単離する第二工程、
を有することを特徴とする精製方法(以下、本発明精製方法と記すこともある。);
12.請求項6記載の形質転換細胞と、レポーター遺伝子の発現量又はそれと相関する指標値の測定試薬とを含む、三量体Gタンパク質αサブユニットGm1のプロモーターを介したシグナル伝達調節物質のスクリーニングキット(以下、本発明キットと記すこともある。);
13.請求項8又は10記載の探索方法により得られる、三量体Gタンパク質αサブユニットGm1のプロモーターを介したシグナル伝達調節能力を有する化合物若しくはその薬学的に許容される塩を有効成分として含み、当該有効成分が薬学的に許容される担体中に製剤化されてなることを特徴とする神経・精神疾患用医薬(以下、本発明医薬と記すこともある。);
等を提供するものである。
本発明で用いられる遺伝子工学的技術は、たとえば、「Molecular Cloning:A Laboratory Manual 2nd edition」(1989), Cold Spring Harbor Laboratory Press及び D.,M.,Glover著、DNA クローニング(DNA Cloning)、IRL発行、1985年などに記載されている通常の方法に準じて行うことができる。
本発明ポリヌクレオチドを含有しかつ当該ポリヌクレオチドの下流(3’側)にレポーター遺伝子を含有するプラスミドが導入されてなる形質転換細胞は、当該ポリヌクレオチドを含有しておらずかつ当該ポリヌクレオチドの下流(3’側)にレポーター遺伝子を含有するプラスミドが導入されてなる形質転換細胞に比べて、レポーター遺伝子の発現量が高くなることを見出し、当該ポリヌクレオチドが三量体Gタンパク質αサブユニットGm1をコードする遺伝子の転写を制御する能力を有する塩基配列を有するポリヌクレオチドであることが確認された。
(1)配列番号1で示される塩基配列
(2)配列番号1で示される塩基配列における塩基番号603番から3871番までの領域で示される塩基配列(因みに、配列番号2で示される塩基配列に相当する塩基配列である。)
(3)上記(1)又は(2)の塩基配列において1個もしくは複数個の塩基が欠失、置換もしくは付加された塩基配列からなり、かつ三量体Gタンパク質αサブユニットGm1をコードする遺伝子の転写を制御する能力(以下、当該能力を本転写制御能力と記すこともある。)を有する塩基配列
(4)上記(1)又は(2)の塩基配列からなるポリヌクレオチドとストリンジェントな条件下にハイブリダイズするポリヌクレオチドの塩基配列に相補であり、かつ三量体Gタンパク質αサブユニットGm1をコードする遺伝子の転写を制御する能力を有する塩基配列
本発明において「転写活性を喪失しない改変」とは、例えば、既に同定されている各種の転写調節配列(即ち、転写を制御する能力を有する塩基配列)と相同性が低い部分について行うことができる改変等を意味するものである。このような改変の一つでもある「1個もしくは複数個の塩基が欠失、置換もしくは付加された塩基配列」において、三量体Gタンパク質αサブユニットGm1をコードする遺伝子の転写を制御する能力を損なうことなく、どの塩基を何個欠失、置換もしくは付加できるかの指標は、後述する方法により評価及び確認することができる。
このような改変は人為的に変異導入することによって作製してもよい。具体的には、例えば、A.Greener,M.Callahan、Strategies(1994)7,32−34等に記載される方法を用いてランダムに変異を導入することによって取得することができ、、W.Kramer,et al.、Nucleic Acids Research(1984)12,9441もしくはW.Kramer,H.J.Frits、Methods in Enzymology(1987)154,350等に記載のギャップド・デュープレックス(gapped duplex)法、またはT.A.Kunkel、Proc. of Natl. Acad. Sci. U.S.A.(1985)82,488もしくはT.A.Kunkel,et al.、Methods in Enzymology(1987)154,367等に記載のクンケル(Kunkel)法を用いて部位特異的に変異を導入することによって取得することができる。あるいは、配列番号1で示される塩基配列からなるポリヌクレオチド等のうち1ヶ所ないし数カ所の部分塩基配列を、他のプロモーターのポリヌクレオチドの一部と入れ換えたキメラDNAを作製することによって取得することができ、例えば、S.Henikoff,et al.、Gene(1984)28,351、C.Yanisch−Perron, et al.、Gene(1985)33,103等に記載された方法を用いることができる。
化学合成法を用いて調製する場合、DNA自動合成機、例えばDNA合成機モデ
ル380A (ABI社製)等を用いることができる。
次に、PCRを用いて本発明ポリヌクレオチドを調製する方法について説明する。鋳型とするゲノムライブラリーは、例えば、「Molecular Cloning:A Laboratory Manual 2nd edition」(1989)、Cold Spring Harbor Laboratory Press等に記載されている方法に準じてヒト、マウス、ラット等の哺乳動物の組織から調製することができる。また、ヒトゲノムDNA(クローンテック製)等の市販のゲノムDNAや、ヒトゲノムウォーカーキット(クローンテック製)等の市販ゲノムライブラリーを用いることができる。次いで、増幅させるプロモーターに対応したプライマー、例えば配列番号1で示される塩基配列を有する本発明ポリヌクレオチドを調製する場合であれば、例えば配列番号1で示される塩基配列における塩基番号1番から20番までの領域で示される塩基配列に対して相補的な塩基配列からなるプライマーと、配列番号1における塩基番号3851番から3871番までの領域で示される塩基配列に対して相補的な塩基配列からなるプライマーとを用いてPCRを行う。尚、前記プライマーは、配列番号1で示される塩基配列に基づいて適宜設計することができ、また、その5’末端側に、制限酵素認識配列等を付加してもよい。前記のようにして増幅されたDNAは、「Molecular Cloning:A Laboratory Manual 2nd edition」(1989), Cold Spring Harbor Laboratory Press、「Current Protocols In Molecular Biology」(1987), John Wiley & Sons,Inc.ISBN0−471−50338−X等に記載される通常の方法に準じてベクターにクローニングすることができる。具体的には例えばInvitrogen製のTAクローニングキットに含まれるプラスミドベクターやStratagene製のpBluescriptIIなどのプラスミドベクターを用いてクローニングすることができる。クローニングされたポリヌクレオチドの塩基配列は、F.Sanger,S.Nicklen,A.R.Coulson著、Proceedings of National Academy of Science U.S.A.(1977),74,5463−5467等に記載されるダイデオキシターミネーティング法などにより分析することができる。
まず、プローブに用いるDNAを標識する。プローブに用いるDNAとしては、調製しようとする本発明ポリヌクレオチドの塩基配列の少なくとも一部を有するポリヌクレオチド、例えば、配列番号1で示される塩基配列における塩基番号603番から3871番までの領域で示される塩基配列もしくはその連続した一部の塩基配列からなるポリヌクレオチドであってその鎖長が20塩基以上1000塩基以下であるポリヌクレオチド、前記ポリヌクレオチドの塩基配列において1個もしくは複数個の塩基が欠失、置換もしくは付加された塩基配列からなるポリヌクレオチド、前記ポリヌクレオチドとストリンジェントな条件下にハイブリダイズするポリヌクレオチド等を挙げることができる。
具体的には、配列番号1で示される塩基配列における塩基番号100番から 番までの領域で示される塩基配列からなるDNA、配列番号1で示される塩基配列における塩基番号200番から3871番までの領域で示される塩基配列からなるDNA、配列番号1で示される塩基配列における塩基番号300番から3871番までの領域で示される塩基配列からなるDNA、配列番号1で示される塩基配列における塩基番号400番から3871番までの領域で示される塩基配列からなるDNA、配列番号1で示される塩基配列における塩基番号500番から3871番までの領域で示される塩基配列からなるDNA等を挙げることができる。
プローブに用いる前記DNAは、例えば、化学合成法、PCR、ハイブリダイゼーション法等、「本発明ポリヌクレオチドの調製方法」として前述した通常のポリヌクレオチドの調製方法によって得ることができる。
プローブに用いる前記DNAを放射性同位元素により標識するには、例えば、ベーリンガー製、宝酒造製のRandom Labelling Kit等を用いることができ、通常のPCR反応組成中のdCTPを[α−32P]dCTPに替えて、プローブに用いる前記DNAを鋳型にしてPCR反応を行うことにより、標識を行うこともできる。また、プローブに用いるDNAを蛍光色素又は酵素で標識する場合には、例えば、ECF Direct Nucleic Acid Labelling and Ditection System(Amersham Pharmacia Biotech製)又はAlkphos Direct DNA laleling and detection kit(Amersham Pharmacia Biotech製)等を用いることができる。プローブをハイブリダイズさせるDNAライブラリーとしては、例えば、ラットなどのげっ歯類等の動物由来のゲノムDNAライブラリー等を使用することができる。当該DNAライブラリーには、市販のゲノムDNAライブラリーを用いることもできるし、また「Molecular Cloning:A Laboratory Manual 2nd edition」(1989),Cold Spring Harbor Laboratory Pressや「Current Protocols In Molecular Biology」(1987),John Wiley & Sons,Inc.ISBN0−471−50338−X等に記載される通常のライブラリー作製法に従い、例えば、Stratagene製のλ FIX II、λ EMBL3、λ EMBL4、λ DASH II等のλベクターを用い、Gigapack packaging Extracts(Stratagene製)等をin vitroパッケージングに用いてゲノムDNAライブラリーを作製し、これを用いることもできる。
本発明ポリヌクレオチド又は本発明プラスミドを導入する宿主細胞としては、大腸菌(例えばK12)、バチルス属細菌(例えばMI114)等の細菌、酵母(例えばAH22)、植物細胞、動物細胞等の細胞をあげることができ、本発明ポリヌクレオチド又は本発明プラスミドが細胞内で増幅可能な形態を保てる細胞であればよい。好ましくは動物細胞(例えばPC-12細胞、Neuro-2a細胞、IMR-32細胞、COS-7細胞、Vero細胞、CHO細胞、ES細胞等)、特に好ましくは神経細胞を挙げることができる。尚、ES細胞の場合には、本発明形質転換細胞として、本発明ポリヌクレオチド又は本発明プラスミドが導入されたES細胞を分化させた転換細胞や、ES細胞を分化した後、当該細胞に本発明ポリヌクレオチド又は本発明プラスミドが導入された転換細胞を含む。また本発明形質転換細胞は、これら転換細胞から発生・作製された動物個体(即ち、形質転換動物)内に存在する状態での細胞をも含むものである。
本発明ポリヌクレオチド又は本発明プラスミドの宿主細胞への導入法としては、細胞に応じた導入方法を適用することができ、例えば動物細胞にはリン酸カルシウム法、電気導入法、DEAEデキストラン法、ミセル形成法などを適用することができる。具体的には例えば、リン酸カルシウム法としては、Grimm,S. et al.,Proc.Natl.Acad.Sci.USA,93,10923−10927等に記載の方法を挙げることができ、電気導入法およびDEAEデキストラン法としては、Ting,A.T. et al.,EMBO J.,15,6189−6196等に記載の方法を挙げることができ、ミセル形成法としては、Hawkins,C.J. et al.,Proc.Natl.Acad.Sci.USA,93,13786−13790等に記載の方法を挙げることができる。ミセル形成法としては例えば、リポフェクトアミン(GibcoBRL製)やフュージーン(Boehringer Mannheim製)等の市販の試薬を使用することが可能である。
(1)本発明ポリヌクレオチドを含有しかつ当該ポリヌクレオチドの下流(3’側)にレポーター遺伝子を含有するプラスミドが導入されてなる形質転換細胞に被験物質を接触させる第一工程、及び、
(2)前記第一工程後に、レポーター遺伝子の発現量又はそれと相関する指標値をモニターする第二工程、
(3)前記第二工程によりモニターされた発現量又はそれと相関する指標値の変化に基づき前記物質が有する三量体Gタンパク質αサブユニットGm1のプロモーターを介したシグナル伝達調節能力を評価する第三工程、及び
(4)前記第三工程で評価されたシグナル伝達調節能力に基づき三量体Gタンパク質αサブユニットGm1のプロモーターを介したシグナル伝達調節能力を有する物質を選抜する第四工程、
を有することを特徴とする探索方法(即ち、本発明探索方法)である。当該探索方法は、いわゆるレポータージーンアッセイを用いる、基づき三量体Gタンパク質αサブユニットGm1のプロモーターを介したシグナル伝達調節能力を有する物質の探索方法である。
当該細胞と被験物質との接触は、当該細胞が成育可能な条件で培養しながら行えばよい。例えば、哺乳動物細胞を宿主とする本発明形質転換細胞の場合には、適宜ウシ胎児血清等の哺乳動物由来の血清を添加したD−MEM、OPTI−MEM、RPMI1640培地(Gibco−BRL製)等の市販の培地中で培養できる。培養は、通常約30℃〜約40℃、約2%(V/V)〜10%(V/V)二酸化炭素存在下で実施すればよく、約35℃〜約37℃、約4%(V/V)〜約6%(V/V)二酸化炭素存在下で実施するのがより好ましい。
被験物質と接触させる際の当該細胞の細胞数は、例えば96ウェルプレートを用いる場合、通常約1×104個/ウェル〜約1×105個/ウェルであればよく、約5×104個/ウェル〜約8×104個/ウェルが好ましい。
転写制御率(%)={(測定値1−測定値2)/測定値2}×100
被験物質の本転写制御能力を表わす転写制御率が、統計学的に有意な値を示す物質、具体的に好ましくは、例えば、30%以上を示す物質、より好ましくは50%以上を示す物質を、本転写制御能力を有する物質として選抜する。
このような探索方法により選抜された物質またはその薬学的に許容される塩は、それらを有効成分として含み、当該有効成分が薬学的に許容される担体中に製剤化されてなることを特徴とする転写制御剤として利用してもよい。
本発明ポリヌクレオチドと結合する物質の選抜方法は、本発明ポリヌクレオチドと被験物質とを接触させる第一工程、前記第一工程後に当該プロモーターと被験物質との複合体生成の有無を調べる第二工程及び前記第二工程で得られた複合体生成の有無結果に基づき当該ポリヌクレオチドと結合する物質を選抜する第三工程を含む。
本発明ポリヌクレオチドと被験物質とを接触させる第一工程に使用されるポリヌクレオチドは、本発明ポリヌクレオチドを、例えば市販のDNA標識キットを用い、ラジオアイソトープ若しくは蛍光色素化合物で標識して用いると、当該ポリヌクレオチドと被験物質との複合体の検出が容易になる点で好ましい。当該ポリヌクレオチドを放射性同位元素により標識するには、例えば、市販のRandom Labelling Kit等を用いることができ、通常のPCR反応組成中のdCTPを[α−32P]dCTPに替えて、当該DNAを鋳型にしてPCR反応を行うことにより、標識を行うこともできる。また、当該DNAを蛍光色素で標識する場合には例えば、ECF Direct Nucleic Acid Labelling and Detection System等を用いることができる。
当該ポリヌクレオチドと被験物質とを、約4℃〜約37℃、好ましくは約20℃〜約30℃で、約5分間〜約60分間、好ましくは約10分間〜約30分間適当なバッファー中、例えばトリス、Hepes、MES等のバッファー中、好ましくはHepesバッファー中で接触させる。被験物質の濃度は通常約0.1μM〜約1,000μMであればよく、1μM〜100μMが好ましい。当該DNAの量は通常約1fmol〜約10fmolであればよく、2fmol〜7fmolが好ましい。
本発明精製方法は、本発明ポリヌクレオチドと試料とを接触させて、当該ポリヌクレオチドと当該試料中に含有される当該ポリヌクレオチドと結合する物質との複合体を生成させる第一工程、及び、前記第一工程後、生成させた複合体から当該結合物質を単離する第二工程を含む。
本発明ポリヌクレオチドと被験物質とを接触させる際には、通常、当該ポリヌクレオチドを担体に結合させた形態で被験物質との接触を行うと、当該ポリヌクレオチドもしくは当該ポリヌクレオチドと結合物質との複合体を容易に回収できる点で好ましい。当該ポリヌクレオチドを結合させる担体の種類は特に限定されないが、例えば、市販のアフィニティークロマトグラフィー用担体、好ましくは臭化シアン活性化セファロース4B(Amersham Pharmacia Biotech製)等を使用することができる。当該ポリヌクレオチドを担体に結合させる場合には、当該ポリヌクレオチドを直接担体に結合させる方法と、スペーサーを介して結合させる方法がある。結合させる際の条件は、例えば、当該ポリヌクレオチドと臭化シアン活性化セファロース4Bを混合し、約4℃〜約10℃で1晩1000rpmで撹拌し、当該ポリヌクレオチドをセファロース上に固定する。ついで、未反応の臭化シアンの活性基を無くすために、アミノ基を持つ化合物を含んだバッファー、例えば、1Mグリシンを含む炭酸水素ナトリウム溶液中で、例えば約4℃〜約10℃で1晩放置する。得られたゲルは、通常のバッチ法により被験物質と接触させてもよく、また、そのゲルを市販のクロマトグラフ管に充填することで、本発明ポリヌクレオチドと結合する物質用アフィニティーカラムを作製し、通常のカラムクロマトグラフィー法により被験物質と接触させてもよい。
本発明精製方法によって、本発明ポリヌクレオチドの本転写制御能力を制御する物質の選抜方法によって選抜される物質又は本発明ポリヌクレオチドと結合する物質の選抜方法によって選抜される物質を精製することができる。
当該キットは、前述した本発明探索方法に使用できる。
本発明キットは、さらに、本発明ポリヌクレオチド(ここではDNA)を有しないプラスミドが導入されてなる対照細胞を含んでいてもよい。このような対照細胞はネガティブコントロールとして利用することができる。
従って、本発明転写制御剤の有効成分としての物質は、本発明ポリヌクレオチドの制御下にある遺伝子の細胞内での発現を制御することによって、当該遺伝子の翻訳産物の発現過多もしくは発現過少に起因する疾患のための医薬として有用である。また、本発明ポリヌクレオチドの制御下に連結した所望のDNA、例えば神経・精神疾患との関連が推定されるDNAや行動や記憶との関連が推定されるDNA等の神経細胞における作用や、Gm1の発現量の異常、又は、Gm1のプロモーターを介したシグナル伝達の異常に起因する疾患(特に、神経・精神疾患)や行動・記憶等への影響を検討する際の転写調節剤として利用することもできる。
マウスGm1プロモーター領域を含むポリヌクレオチド配列を以下のようにして単離した。
ファージライブラリーのスクリーニングを行なうために、以下に示す手順によってマウスGm1遺伝子の5'上流領域を含むプローブDNAを作製した。
マウスC57BL/6由来のゲノムDNA1μgを鋳型に用い、10μMのフォワードプライマーmGmg−1(5’tgttctcccgacccttcagggatcttcttt;配列番号4)、10μMのリバースプライマーmGmg−2(5’−acctatgagcagcaatggatagagtctat;配列番号5)およびTAKARATaqポリメラーゼ(TAKARALATaq with GC Buffer, 宝酒造社製)を用いてPCRを行うことにより増幅DNAを得た。
PCR条件は、95℃で30秒間、次いで60℃で30秒間、次いで72℃で2分間の保温を1サイクルとしてこれを35サイクル行った。
得られたDNAをアガロースゲル電気泳動した後、QIAquick Gel Extraction kit(QIAGEN製)を用いて精製し、回収した。この精製・回収されたDNAをプローブ用鋳型DNAとして以下の実験で用いた。
次に当該プローブ用鋳型DNA10μlを、Alkphos Direct DNA labeling and detectionキット(アマシャムバイオサイエンス社製)を用い、当該キットに添付されたプロトコールに従って、アルカリフォスフォターゼでラベルされたプローブDNAを調製した。
マウス129/sv由来ゲノムライブラリー(STRATAGENE社製)を用い、以下の手順でマウスGm1プロモーターの単離を行なった。
15cmのNZYプレート20枚に1x106のファージを播き、これを37℃で8時間培養することにより、プラークを形成させた。プラーク形成されたプレートにニトロセルロースメンブレンを接触させることにより、形成されたプラークをニトロセルロースメンブレン上に移した。このニトロセルロースメンブレンと、アルカリフォスフォターゼでラベルされたプローブDNAを、Alkphos Direct DNA labeling and detectionキット(アマシャムバイオサイエンス社製)を用い、当該キットに添付されたプロトコールに従って、ハイブリダイゼーションを行なった。またハイブリダイゼーションによるシグナルの検出も、Alkphos Direct DNA labeling and detectionキット(アマシャムバイオサイエンス社製)を用い、当該キットに添付されたプロトコールに従って行なった。
ハイブリダイゼーションによりシグナルが検出されたファージより、Wizard Lambda Prepsキット(プロメガ社製)を用い、その添付プロトコールに従って、DNAを回収しマウスGm1プロモーター領域を含むポリヌクレオチド配列をクローニングした。
マウスGm1プロモーター領域を含むポリヌクレオチド配列を有するレポーターベクターであるpGL−Gm1およびその一部のポリヌクレオチド配列を有するレポーターベクターであるpGL−Gm1/SacIを以下のようにして作製した。
実施例1より得られたマウスGm1プロモーター領域を含むファージDNAをSacIで消化した後、得られた消化物をアガロースゲル電気泳動した後、QIAquick Gel Extraction kit(QIAGEN製)を用いて精製し、回収した。この精製・回収されたDNAをインサートDNAとして以下の実験で用いた。pGL3をSacIで消化した後、アルカリフォスフォターゼ処理することにより、ベクターを調製した。次いで、調製されたベクター50ngとインサートDNA10ngとをT4ライゲースを用いて連結することにより、Gm1プロモーターレポーターベクターpGL−Gm1/SacIを作製した。当該ベクターpGL−Gm1/SacIにおいては、配列番号2で示される塩基配列(当該配列は、配列番号1における塩基番号603番から3871番で示される塩基配列に相当する。)が、pGL3のSacI部位に挿入されている。
さらに実施例1で得られたマウスGm1プロモーター領域を含むファージDNAをNheIで消化した後、得られた消化物をアガロースゲル電気泳動した後、QIAquick Gel Extraction kit(QIAGEN製)を用いて精製し、回収した。この精製・回収されたDNAをインサートDNAとして以下の実験で用いた。次いでpGL3をNheIで消化した後、アルカリフォスフォターゼ処理することにより、ベクターを調製した。調製されたベクター50ngとインサートDNA10ngとをT4ライゲースを用いて連結することにより、Gm1プロモーターレポーターベクターpGL−Gm1/NheIを作製した。次にpGL―Gm1/SacIをNdeIおよびXhoIで二重消化した後、得られた消化物をアガロースゲル電気泳動した後、2kbのDNA断片をQIAquick Gel Extraction kit(QIAGEN製)を用いて精製し、回収した。この精製・回収されたDNAをインサートDNAとして以下の実験で用いた。pGL−Gm1/NheIをNdeIおよびXhoIで二重消化した後、得られた消化物(ベクター)50ngとインサートDNA10ngとをT4ライゲースを用いて連結することにより、Gm1プロモーターレポーターベクターpGL−Gm1を作製した。当該ベクターpGL−Gm1においては、配列番号1で示される塩基配列が、pGL3のSacI部位とNheI部位との間に挿入されている。
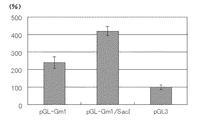
実施例1により得られた本発明Gm1プロモーターの転写活性化能を以下のようにして測定した。
PC12細胞を96穴プレートの各ウェル内に5×104cells/wellで播種し、約24時間培養した。培養された細胞に実施例2により得たGm1プロモーターレポーターベクター(pGL‐Gm1;0.2μg)又はGm1プロモーターベクター(pGL‐Gm1/SacI;0.2μg)を、リポフェクション法を用いてトランスフェクションすることにより、試験細胞を調製した。尚、上記と同じように播種された細胞に、Gm1プロモーターを有しないレポーターベクター(pGL3;0.2μg)を、リポフェクション法を用いてトランスフェクションすることにより対照細胞を調製した。
次いで、この細胞を37℃で24時間培養した後、ウェル内の培地を除き、PBS緩衝溶液で洗浄した。洗浄された細胞を細胞溶解液(ピッカジーンルシフェラーゼキット、東洋インキ製造社製)で溶解した後、ウェル内に発光基質(ピッカジーンルシフェラーゼキット、東洋インキ製造社製)を添加し、各ウェルの蛍光強度を、ルミノメーターを用いて測定した。対照として、対照細胞も同様にして蛍光強度を測定した。
対照細胞のルシフェラーゼ活性を100%として、試験細胞のルシフェラーゼ活性が130%以上を示す場合を、転写調節能を有するとした。
脳における三量体Gタンパク質αサブユニットGm1タンパク質の発現分布の解析を、抗Gm1抗体を用いて蛍光組織免疫染色法より行った。即ち、パラホルムアルデヒドを用いて固定されたマウス脳切片(Novagen社製)を脱パラフィンした後、0.01Mクエン酸溶液中でオートクレーブ処理(120℃、15分)した。
次に、脳切片をTSAキット#12(モルキュラープローブ社製)に付属されたブロッキング試薬を用いて希釈された抗Gm1抗体(1/100希釈)中で、4℃で1時間インキュベーションした。次いで、脳切片をPBS溶液で2回洗浄した後、脳切片をTSAキット#12(モルキュラープローブ社製)に付属されたHRPラベル抗ウサギIgG抗体中で、4℃で1時間インキュベーションした。次いで、脳切片をPBS溶液で2回洗浄した後、TSAキット#12(モルキュラープローブ社製)に付属されたチラミドシグナル増幅キットを用いて、当該キットに添付されたプロトコールに従って、蛍光シグナルの検出を行なった。蛍光シグナルは蛍光顕微鏡を用いて観察された。
ヒト精神疾患患者の脳における三量体Gタンパク質αサブユニットGm1タンパク質の発現の解析を、抗Gm1抗体を用いた組織免疫染色法により行った。
即ち、パラホルムアルデヒドを用いて固定されたヒト脳切片スライドLandMark Low Density Neuropsychiatric Tissue MicroArrays(Ambion社製)を脱パラフィンした後、0.01Mクエン酸溶液中でオートクレーブ処理(120℃、15分)した。
次に、脳切片を含むスライド(以下、「スライド」と略称する)を超純水で洗浄した後、3%過酸化水素水を含むメタノール中で、室温で10分間インキュベートした。
次にインキュベートされたスライドをTN溶液(0.1M Tris pH7.5, 0.15M NaCl)で3回洗浄した後、TSAビオチンシステムNEL700(PerkinElmer社製)に付属されたブロッキング試薬中で、室温で30分間インキュベートした。
次に、スライドをTSAビオチンシステムNEL700(PerkinElmer社製)に付属されたブロッキング試薬を用いて希釈された抗Gm1抗体(1/100希釈)中で、4℃で1時間インキュベーションした後、これをTN溶液(0.1M Tris pH7.5, 0.15M NaCl)で3回洗浄した。
次に、スライドをTSAビオチンシステムNEL700(PerkinElmer社製)に付属されたブロッキング試薬に希釈されたHRPラベル抗ウサギIgG抗体(1/200希釈)中で、4℃で1時間インキュベーションした後、これをTN溶液(0.1M Tris pH7.5, 0.15M NaCl)で3回洗浄した。
次に、スライドをTSAビオチンシステムNEL700(PerkinElmer社製)に付属されたビオチン化チラミド増幅試薬中で、室温で10分間インキュベーションした後、これをTN溶液(0.1M Tris pH7.5, 0.15M NaCl)で3回洗浄した。
次に、スライドをTSAビオチンシステムNEL700(PerkinElmer社製)に付属された抗HRPラベル抗アビジン抗体中で、室温で30分間インキュベーションした後、これをTN溶液(0.1M Tris pH7.5, 0.15M NaCl)で3回洗浄した。
このようにして得られたスライドを、DABタブレット1錠(シグマ社製)を15ml超純水に溶かしたものを用いて発色させた後、顕微鏡で観察した。
マウスGm1プロモーター領域の一部を含むポリヌクレオチド配列を有するレポーターベクターであるpGL-Gm1/843-3900を以下のようにして構築する。
マウスGm1のプロモーター領域の一部を含むDNAを増幅するために、マウスGm1プロモーター領域を含むファージDNA10ngを鋳型に用い10μMのフォワードプライマーprmGmg-843(5’−atcatcacaacaggaaagtaataaaa;配列番号6)、10μMのリバースプライマーprmGmg-3900(5’−attgtgcatccttcgaacgtccca;配列番号7)およびTAKARA LA Taqポリメラーゼ(TAKARA LA Taq with GC Buffer,宝酒造製)を用いてPCRを行なう。
PCR条件は、95℃で30秒間次いで60℃で30秒間次いで72℃で2分間の保温を1サイクルとしてこれを35サイクル行なう。得られたDNAをアガロースゲル電気泳動した後、QIAquick Gel Extraction kit(QIAGEN製)を用いて精製、回収する。この精製・回収されたDNAをインサートDNAとして以下の実験で用いる。
インサートDNA10ngとpDriveベクター(QIAGEN社製)50ngとをT4ライゲースを用いて連結することにより、pDrive−Gm1/843-3900を得る。
得られたマウスGm1プロモーター領域の一部を含むプラスミドpDrive−Gm1/843-3900をMluIおよびSacIで二重消化した後、得られた消化物をアガロースゲル電気泳動した後、QIAquick Gel Extraction kit(QIAGEN製)を用いて精製し、回収する。この精製・回収されたDNAをインサートDNAとして以下の実験で用いた。pGL3をMluIおよびSacIで二重消化した後、得られた消化物(ベクター)50ngとインサートDNA10ngとをT4ライゲースを用いて連結することにより、Gm1プロモーターレポーターベクターpGL-Gm1/843-3900を作製する。
マウスGm1プロモーター領域の一部を含むポリヌクレオチド配列を有するレポーターベクターであるpGL-Gm1/1232-3900を、フォワードプライマーprmGmg-1232(5’−tggtgccctcttctggtgtgtct;配列番号8)および10μMのリバースプライマーprmGmg-3900(5’−attgtgcatccttcgaacgtccca;配列番号7)を用いて、上記と同様な手順で構築する。
マウスGm1プロモーター領域の一部を含むポリヌクレオチド配列を有するレポーターベクターであるpGL-Gm1/1989-3900を、フォワードプライマーprmGmg-1989(5’−atgccatatacttgagtcacagtttgtgaa;配列番号9)および10μMのリバースプライマーprmGmg-3900(5’−attgtgcatccttcgaacgtccca;配列番号8)を用いて、上記と同様な手順で構築する。
マウスGm1プロモーター領域の一部を含むポリヌクレオチド配列を有するレポーターベクターであるpGL-Gm1/2937-3900を、フォワードプライマーprmGmg-2937(5’−ctcccgagccttcagggatcttcttt;配列番号10)および10μMのリバースプライマーprmGmg-3900(5’−attgtgcatccttcgaacgtccca;配列番号8)を用いて、上記と同様な手順で構築する。
マウスGm1プロモーター領域の一部を含むポリヌクレオチド配列を有するレポーターベクターであるpGL-Gm1/848-3776を、フォワードプライマーprmGmg-843(5’−atcatcacaacaggaaagtaataaaa;配列番号6)および10μMのリバースプライマーprmGmg-3776(5’−agactgctccggttaccgtaaatactgcct;配列番号11)を用いて、上記と同様な手順で構築する。
マウスGm1プロモーター領域の一部を含むポリヌクレオチド配列を有するレポーターベクターであるpGL-Gm1/1232-3776を、フォワードプライマーprmGmg-1232(5’−tggtgccctcttctggtgtgtct;配列番号8)および10μMのリバースプライマーprmGmg-3776(5’−agactgctccggttaccgtaaatactgcct;配列番号11)を用いて、上記と同様な手順で構築する。
マウスGm1プロモーター領域の一部を含むポリヌクレオチド配列を有するレポーターベクターであるpGL-Gm1/1989-3900を、フォワードプライマーprmGmg-1989(5’−atgccatatacttgagtcacagtttgtgaa;配列番号9)および10μMのリバースプライマーprmGmg-3776(5’−agactgctccggttaccgtaaatactgcct;配列番号11)を用いて、上記と同様な手順で構築する。
マウスGm1プロモーター領域の一部を含むポリヌクレオチド配列を有するレポーターベクターであるpGL-Gm1/2937-3900を、フォワードプライマーprmGmg-2937(5’−ctcccgagccttcagggatcttcttt;配列番号10)および10μMのリバースプライマーprmGmg-3776(5’−agactgctccggttaccgtaaatactgcct;配列番号11)を用いて、上記と同様な手順で構築する。
実施例6により得られた本発明のGm1プロモーターの一部の転写活性化能を以下のようにして測定する。
PC12細胞を96穴プレートの各ウェル内に5×104cells/wellで播種し、約24時間培養する。培養された細胞に実施例6により得られたGm1プロモーターレポーターベクター(pGL-Gm1/843-3900または、pGL-Gm1/1232-3900または、pGL-Gm1/1989-3900または、pGL-Gm1/2937-3900または、pGL-Gm1/848-3776または、pGL-Gm1/1232-3776または、pGL-Gm1/1989-3776または、pGL-Gm1/2937-3776;0.2μg)を、リポフェクション法を用いてトランスフェクションすることにより、試験細胞を調製する。尚、上記と同じように播種された細胞を用いて、Gm1プロモーターを有しないレポーターベクター(pGL3;0.2μg)を、リポフェクション法を用いてトランスフェクションすることにより、対照細胞を調製する。
次いで、この細胞を37℃で24時間培養した後、ウェル内の培地を除き、PBS緩衝溶液で洗浄した。洗浄された細胞を細胞溶解液(ピッカジーンルシフェラーゼキット、東洋インキ製造社製)で溶解した後、ウェル内に発光基質(ピッカジーンルシフェラーゼキット、東洋インキ製造社製)を添加し、各ウェルの蛍光強度を、ルミノメーターを用いて測定する。対照として、対照細胞も同様にして蛍光強度を測定する。
対照細胞のルシフェラーゼ活性を100%として、試験細胞のルシフェラーゼ活性が130%以上を示す場合を、転写調節能を有するとする。
PC12細胞を96穴プレートの各ウェル内に5×104cells/wellで播種し、約24時間培養する。培養された細胞に実施例2により得られたGm1プロモーターレポーターベクター(pGL‐Gm1;0.2μg)を、リポフェクション法を用いてトランスフェクションすることにより、試験細胞を調製する。
次いで、この細胞を約24時間培養した後、ウェル内の培地を除いた。ウェル内に、新たに0.10mlの新鮮な培地を加えた後、さらに0.1nM〜10nMの被験物質を添加し、これを37℃で24時間培養する。
次いで、ウェル内の培地を除き、PBS緩衝溶液で洗浄した。洗浄された細胞を細胞溶解液(ピッカジーンルシフェラーゼキット、東洋インキ製造社製)で溶解した後、ウェル内に発光基質(ピッカジーンルシフェラーゼキット、東洋インキ製造社製)を添加し、各ウェルの発光強度を、ルミノメーターを用いて測定する。対照として、何も接触させない細胞についても同様にして発光強度を測定する。
何も接触させない場合のルシフェラーゼ活性を100%として、被験物質とを接触させた場合のルシフェラーゼ活性のパーセンテージが50%以下になる物質又は150%以上になる物質をシグナル伝達調節物質として選択する。
実施例2により得られた本発明のGm1プロモーターの一部の転写活性化能を以下のようにして測定する。
PC12細胞を96穴プレートの各ウェル内に5×104cells/wellで播種し、約24時間培養する。培養された細胞に実施例2により得られたGm1プロモーターレポーターベクター(pGL‐Gm1又はpGL−Gm1/SacI;0.2μg)を、リポフェクション法を用いてトランスフェクションすることにより、試験細胞を調製する。また、Gm1プロモーターを有しないレポーターベクター(pGL3;0.2μg)を、リポフェクション法を用いてトランスフェクションすることにより、対照細胞を調製する。
次いで、これら細胞を約24時間培養した後、ウェル内の培地を除いた。ウェル内に、新たに0.10mlの新鮮な培地を細胞に加えた後、さらに0.1nM〜10nMの被験物質を添加し、これを37℃で24時間培養する。
次いで、ウェル内の培地を除き、PBS緩衝溶液で洗浄した。洗浄された細胞を細胞溶解液(ピッカジーンルシフェラーゼキット、東洋インキ製造社製)で溶解した後、ウェル内に発光基質(ピッカジーンルシフェラーゼキット、東洋インキ製造社製)を添加し、各ウェルの発光強度を、ルミノメーターを用いて測定する。対照として、何も接触させない細胞についても同様にして発光強度を測定する。
被験物質を接触させた場合と何も接触させてない場合のルシフェラーゼ活性の比が対照細胞に比べて、試験細胞で1.3倍以上または0.7倍以下になる物質をシグナル伝達物質として選択する。
実施例3に記載された本発明プラスミドpGL−Gm1 5μgを、XhoIおよびMluI各10Uを用いて20μLの反応液中で37℃で1時間消化する。制限酵素消化反応液をアガロースゲル電気泳動し、QIAquick Gel Extraction Kitを用いてインサートDNA断片を精製する。得られたDNA断片1μg、[α−32P]dCTP(Amersham Pharmacia Biotech製) 5μL、非標識ヌクレオチド(dATP,dTTP,dGTP)(宝酒造製)各1μL、10×klenow Buffer(宝酒造製)2μLおよびklenow酵素(宝酒造製) 1μLを加え蒸留水で合計20μLとし、37℃で1時間インキュベートすることで、標識DNA断片を得る。次に、未反応[α−32P]dCTP と標識DNA断片を分離するために反応液を10mMトリス塩酸、1mMEDTA(以後TE溶液と表記する。)で平衡化したProbeQuant G−50 Micro Columns(Amersham Pharmacia Biotech製)に供し、遠心分離(室温、1200rpm、1分間)を行い、標識DNAを溶出する。得られた溶出液の放射活性をシンチレーターで測定し、104cpm/μLになるようTE溶液で希釈して標識DNA溶液を得る。次いで、5×バインディングバッファー(50mM Hepes−水酸化カリウム(pH7.8)、250mM塩化カリウム、5mM EDTA(pH8.0)、25mM塩化マグネシウム、50%(W/V)グリセロール、25mMジチオスレイトール、3.5mM PMSF、10μg/mL Aprotinin、10μg/mL Pepstatin、10μg/mL Leupeptin、および5mM sodium orthovanadateを含有する。)5μL、2μg/μL polydIdC 1.5μL、10μM被験物質5μLおよび標識DNA溶液2μLを混合し蒸留水で合計25μLにする。該液を室温で30分間インキュベートした後、ポリアクリルアミドゲル電気泳動をおこなう。電気泳動後、ゲルをゲル板よりはがして、ワットマン3MMろ紙上にて80℃、1時間真空ポンプにて乾燥後、イメージングプレートに2時間感光後、BAS2000で画像イメージを取得する。被験物質が標識DNAと結合しDNA−被験物質複合体が形成された結果、ゲル上での移動度が遊離のDNAより小さくなり、画像イメージ上のバンドのシフトが検出された場合の被験物質を本発明ポリヌクレオチドへの結合物質として選抜する。
(1)本発明ポリヌクレオチド結合物質精製用アフィニティーカラムの作製
本発明プラスミドpGL−Gm1 200μgをXhoI、MluI 各50Uを用いて200μLの反応液中で37℃で1時間消化する。制限酵素消化反応液をアガロースゲル電気泳動し、QIAquick Gel Extraction Kitを用いて精製することにより該DNAを得る。2mLの臭化シアン活性化セファロース4Bに44μgの該DNAを加え、4℃で1晩1000rpmで撹拌し、該DNAをセファロース上に固定する。ついで、未反応の臭化シアンの活性基を無くすために、1Mグリシンを含む炭酸水素ナトリウム溶液(pH9.5) 20mLを加え4℃で1晩放置する。こうして得られたゲルを10×300mmクロマトグラフ管(イワキガラス製)に充填することで、本発明ポリヌクレオチドへの結合物質用アフィニティーカラムを作製する。
ヒト神経由来培養細胞IMR−32を15cmシャーレ(ファルコン社製)40枚にシャーレあたり1×107個の細胞を捲き込む。10%(W/V)FBSを添加したD−MEM培地(高グルコース)を用い、37℃、5%(V/V)二酸化炭素存在下で二晩培養する。二晩後、培地を除去した後、15mLのリン酸緩衝液でシャーレの器壁を1回洗浄した後、該シャーレにトリプシン−EDTA溶液(0.05%(W/V)トリプシン、0.53mM EDTAを含む。Gibco製)2mLを細胞が浸るように添加し、37℃で5分放置する。これに、FBS含有培地をトリプシン−EDTA溶液の約10倍量添加し、細胞懸濁液を得る。得られた細胞懸濁液を遠心分離(室温、1,300rpm、5分間)し、上清を除去する。細胞沈殿を15mLのリン酸緩衝液に懸濁し、再度室温で1,300rpm、5分間遠心分離し、上清を除去する。その後、細胞沈殿を氷冷した10mM Hepes−水酸化カリウム(pH7.8)、10mM 塩化カリウム、0.1mM EDTA(pH8.0)溶液(以下、バッファーAと記す。)10mLに懸濁する。10分間氷上で冷却した後、4℃、1,300rpm、5分間遠心分離する。上清を除去後、細胞沈殿を30mlのバッファーAで再懸濁し、ダウンスホモジナイザーペッスルB(Wheaton製)を用いて氷上で冷却しながら細胞を完全に破砕する。得られた細胞破砕液を遠心分離(4℃、1300rpm、5分間)し、上清を除去する。この沈殿を50mM Hepes−KOH(pH7.8)、420mM 塩化カリウム、0.1mM EDTA(pH8.0)、5mM塩化マグネシウム、2%(W/V)グリセロール溶液(以下バッファーBと記す。)2mLに懸濁する。4℃で30分間ローテーターにて穏やかに回転させた後、遠心分離(4℃、24000g、30分間)する。上清を回収し、蒸留水で5倍に希釈した希釈液を精製の試料とする。
前記した試料を(1)記載のアフィニティーカラムにロードする。さらに、5倍希釈したバッファーB 10mLをロードすることにより、複合体形成しなかった試料中成分を除去する。その後、塩化カリウム濃度を1Mまで上昇させるグラジエントを行い本発明ポリヌクレオチドへの結合物質を溶出することによって、本発明ポリヌクレオチドへの結合物質を含む画分を得る。
PCRのために設計されたプライマー
配列番号5
PCRのために設計されたプライマー
配列番号6
PCRのために設計されたプライマー
配列番号7
PCRのために設計されたプライマー
配列番号8
PCRのために設計されたプライマー
配列番号9
PCRのために設計されたプライマー
配列番号10
PCRのために設計されたプライマー
配列番号11
PCRのために設計されたプライマー
Claims (13)
- 三量体Gタンパク質αサブユニットGm1のプロモーター領域である塩基配列を有することを特徴とするポリヌクレオチド。
- プロモーター領域である塩基配列が、以下の(1)〜(4)のいずれかに記載される塩基配列であることを特徴とする請求項1記載のポリヌクレオチド。
(1)配列番号1で示される塩基配列
(2)配列番号1で示される塩基配列における塩基番号603番から3871番までの領域で示される塩基配列
(3)上記(1)又は(2)の塩基配列において1個もしくは複数個の塩基が欠失、置換もしくは付加された塩基配列からなり、かつ三量体Gタンパク質αサブユニットGm1をコードする遺伝子の転写を制御する能力を有する塩基配列
(4)上記(1)又は(2)の塩基配列からなるポリヌクレオチドとストリンジェントな条件下にハイブリダイズするポリヌクレオチドの塩基配列に相補であり、かつ三量体Gタンパク質αサブユニットGm1をコードする遺伝子の転写を制御する能力を有する塩基配列 - 請求項1又は2記載のポリヌクレオチドを含有することを特徴とするプラスミド。
- 請求項1又は2記載のポリヌクレオチドを含有し、かつ当該ポリヌクレオチドの下流(3’側)に当該ポリヌクレオチドによって転写が制御されるポリヌクレオチドを含有することを特徴とするプラスミド。
- 請求項1又は2記載のポリヌクレオチドを含有し、かつ当該ポリヌクレオチドの下流(3’側)にレポーター遺伝子を含有することを特徴とするプラスミド。
- 請求項1、2、3又は4記載のプラスミドが導入されてなる形質転換細胞。
- 請求項5記載のプラスミドが導入されてなる形質転換細胞。
- 三量体Gタンパク質αサブユニットGm1のプロモーターを介したシグナル伝達調節物質の探索方法であって、
(1)請求項7記載の形質転換細胞に被験物質を接触させる第一工程、及び、
(2)前記第一工程後に、レポーター遺伝子の発現量又はそれと相関する指標値をモニターする第二工程、
(3)前記第二工程によりモニターされた発現量又はそれと相関する指標値の変化に基づき前記物質が有する三量体Gタンパク質αサブユニットGm1のプロモーターを介したシグナル伝達調節能力を評価する第三工程、及び
(4)前記第三工程で評価されたシグナル伝達調節能力に基づき三量体Gタンパク質αサブユニットGm1のプロモーターを介したシグナル伝達調節能力を有する物質を選抜する第四工程、
を有することを特徴とする探索方法。 - 物質が有する三量体Gタンパク質αサブユニットGm1のプロモーターを介したシグナル伝達調節能力を評価する方法であって、
(1)請求項6記載の形質転換細胞に被験物質を接触させる第一工程、及び、
(2)前記第一工程後に、レポーター遺伝子の発現量又はそれと相関する指標値をモニターする第二工程、及び
(3)前記第二工程によりモニターされた発現量又はそれと相関する指標値の変化に基づき前記物質が有する三量体Gタンパク質αサブユニットGm1のプロモーターを介したシグナル伝達調節能力を評価する第三工程、
を有することを特徴とする評価方法。 - 請求項1記載のポリヌクレオチドと結合する物質の探索方法であって、
(1)請求項1記載のポリヌクレオチドと被験物質とを接触させる第一工程、及び
(2)前記第一工程後に、当該ポリヌクレオチドと被験物質との複合体生成の有無を調べる第二工程、及び
(3)前記第二工程で得られた複合体生成の有無結果に基づき当該ポリヌクレオチドと結合する物質を選抜する第三工程、
を有することを特徴とする探索方法。 - 請求項1記載のポリヌクレオチドと結合する物質の精製方法であって、
(1)請求項1記載のポリヌクレオチドと試料とを接触させて、当該ポリヌクレオチドと当該試料中に含有される当該ポリヌクレオチドと結合する物質との複合体を生成させる第一工程、及び、
(2)前記第一工程後、生成させた複合体から当該結合物質を単離する第二工程、
を有することを特徴とする精製方法。 - 請求項6記載の形質転換細胞と、レポーター遺伝子の発現量又はそれと相関する指標値の測定試薬とを含む、三量体Gタンパク質αサブユニットGm1のプロモーターを介したシグナル伝達調節物質のスクリーニングキット。
- 請求項8又は10記載の探索方法により得られる、三量体Gタンパク質αサブユニットGm1のプロモーターを介したシグナル伝達調節能力を有する化合物若しくはその薬学的に許容される塩を有効成分として含み、当該有効成分が薬学的に許容される担体中に製剤化されてなることを特徴とする神経・精神疾患用医薬。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2005070977A JP2005296003A (ja) | 2004-03-15 | 2005-03-14 | 三量体Gタンパク質αサブユニットGm1のプロモーター及びその利用 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2004072244 | 2004-03-15 | ||
| JP2005070977A JP2005296003A (ja) | 2004-03-15 | 2005-03-14 | 三量体Gタンパク質αサブユニットGm1のプロモーター及びその利用 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2005296003A true JP2005296003A (ja) | 2005-10-27 |
Family
ID=35328294
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2005070977A Pending JP2005296003A (ja) | 2004-03-15 | 2005-03-14 | 三量体Gタンパク質αサブユニットGm1のプロモーター及びその利用 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP2005296003A (ja) |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2002034575A (ja) * | 2000-07-28 | 2002-02-05 | Shiseido Co Ltd | ヒトII型5α−レダクターゼのプロモーター遺伝子およびその用途 |
| EP1382613A1 (en) * | 2002-07-16 | 2004-01-21 | Sumitomo Chemical Company, Limited | Novel G protein, polynucleotide encoding the same and utilization thereof |
-
2005
- 2005-03-14 JP JP2005070977A patent/JP2005296003A/ja active Pending
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2002034575A (ja) * | 2000-07-28 | 2002-02-05 | Shiseido Co Ltd | ヒトII型5α−レダクターゼのプロモーター遺伝子およびその用途 |
| EP1382613A1 (en) * | 2002-07-16 | 2004-01-21 | Sumitomo Chemical Company, Limited | Novel G protein, polynucleotide encoding the same and utilization thereof |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1141017B1 (en) | Characterization of the soc/crac calcium channel protein family | |
| JP4790836B2 (ja) | 副甲状腺ホルモンのレセプターとそれをコードしているdna | |
| Andriamampandry et al. | Cloning and functional characterization of a gamma‐hydroxybutyrate receptor identified in the human brain | |
| EP1270724A2 (en) | Guanosine triphosphate-binding protein coupled receptors | |
| KR20140147800A (ko) | 소형 분자로 전압-개폐된 나트륨 채널 (SCNxA)의 알파 아단위에 관련된 질환의 치료 | |
| CN106459993A (zh) | Fgfr融合体 | |
| US20040087478A1 (en) | Screening method | |
| US20030235833A1 (en) | Guanosine triphosphate-binding protein coupled receptors | |
| US20080248009A1 (en) | Regulation of acheron expression | |
| JPWO2000029571A1 (ja) | 新規膜貫通蛋白質をコードする遺伝子 | |
| KR20040010169A (ko) | 신규 g 단백질, 이를 코딩하는 폴리뉴클레오티드 및 이의이용 | |
| CA2347068A1 (en) | New transcription factor of mhc class ii genes, substances capable of inhibiting this new transcription factor and medical uses of these substances | |
| JP2005296003A (ja) | 三量体Gタンパク質αサブユニットGm1のプロモーター及びその利用 | |
| US11459613B2 (en) | Methods of characterizing resistance to modulators of Cereblon | |
| JP2004350672A (ja) | 新規g−タンパク質及びその利用 | |
| US20060115816A1 (en) | Splice variant cannabinoid receptor (cb1b) | |
| WO2006068326A1 (ja) | 新規ポリペプチドおよびその用途 | |
| US7063959B1 (en) | Compositions of the SOC/CRAC calcium channel protein family | |
| US20070178467A1 (en) | Gm1 promoter and use thereof | |
| US20060148030A1 (en) | Nuclear receptor err y 3 | |
| CA3148273A1 (en) | Compositions and methods utilizing a novel human foxo3 isoform | |
| JP2001309792A (ja) | スクリーニング方法 | |
| WO1999067369A1 (en) | Cell cycle regulatory factor | |
| WO2003018776A9 (en) | IDENTIFICATION OF THE IκBNS PROTEIN AND ITS PRODUCTS | |
| JP2002522020A (ja) | オーファンサイトカインレセプター |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| RD05 | Notification of revocation of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7425 Effective date: 20080131 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20080207 |
|
| RD05 | Notification of revocation of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7425 Effective date: 20080514 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20100928 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20110215 |