JP2004533226A - B7−4に対するpd−1、aレセプター、およびその使用 - Google Patents
B7−4に対するpd−1、aレセプター、およびその使用 Download PDFInfo
- Publication number
- JP2004533226A JP2004533226A JP2002577908A JP2002577908A JP2004533226A JP 2004533226 A JP2004533226 A JP 2004533226A JP 2002577908 A JP2002577908 A JP 2002577908A JP 2002577908 A JP2002577908 A JP 2002577908A JP 2004533226 A JP2004533226 A JP 2004533226A
- Authority
- JP
- Japan
- Prior art keywords
- protein
- cells
- cell
- nucleic acid
- molecule
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 150000001875 compounds Chemical class 0.000 claims abstract description 110
- 230000011664 signaling Effects 0.000 claims abstract description 41
- 101710089372 Programmed cell death protein 1 Proteins 0.000 claims description 198
- 238000000034 method Methods 0.000 claims description 139
- 210000001744 T-lymphocyte Anatomy 0.000 claims description 70
- 238000012360 testing method Methods 0.000 claims description 57
- 230000026731 phosphorylation Effects 0.000 claims description 41
- 238000006366 phosphorylation reaction Methods 0.000 claims description 41
- 108010015499 Protein Kinase C-theta Proteins 0.000 claims description 28
- 102000001892 Protein Kinase C-theta Human genes 0.000 claims description 26
- 108040008097 MAP kinase activity proteins Proteins 0.000 claims description 22
- 102000019149 MAP kinase activity proteins Human genes 0.000 claims description 22
- 239000003446 ligand Substances 0.000 claims description 21
- 102100033019 Tyrosine-protein phosphatase non-receptor type 11 Human genes 0.000 claims description 16
- 238000012216 screening Methods 0.000 claims description 16
- 101710116241 Tyrosine-protein phosphatase non-receptor type 11 Proteins 0.000 claims description 12
- 101001052493 Homo sapiens Mitogen-activated protein kinase 1 Proteins 0.000 claims description 6
- 102100024193 Mitogen-activated protein kinase 1 Human genes 0.000 claims description 6
- 101000876610 Dictyostelium discoideum Extracellular signal-regulated kinase 2 Proteins 0.000 claims description 5
- 102100023990 60S ribosomal protein L17 Human genes 0.000 claims 15
- 238000003556 assay Methods 0.000 abstract description 45
- 108090000623 proteins and genes Proteins 0.000 description 247
- 210000004027 cell Anatomy 0.000 description 237
- 150000007523 nucleic acids Chemical class 0.000 description 218
- 102000039446 nucleic acids Human genes 0.000 description 204
- 108020004707 nucleic acids Proteins 0.000 description 204
- 108090000765 processed proteins & peptides Proteins 0.000 description 199
- 102100040678 Programmed cell death protein 1 Human genes 0.000 description 183
- 102000004196 processed proteins & peptides Human genes 0.000 description 151
- 230000000694 effects Effects 0.000 description 135
- 102000004169 proteins and genes Human genes 0.000 description 134
- 210000002865 immune cell Anatomy 0.000 description 127
- 235000018102 proteins Nutrition 0.000 description 126
- 229920001184 polypeptide Polymers 0.000 description 123
- 125000003729 nucleotide group Chemical group 0.000 description 116
- 239000002773 nucleotide Substances 0.000 description 113
- 230000014509 gene expression Effects 0.000 description 101
- 235000001014 amino acid Nutrition 0.000 description 79
- 150000001413 amino acids Chemical class 0.000 description 76
- 229940024606 amino acid Drugs 0.000 description 75
- 230000002401 inhibitory effect Effects 0.000 description 73
- 230000027455 binding Effects 0.000 description 72
- 230000028993 immune response Effects 0.000 description 69
- 239000013598 vector Substances 0.000 description 67
- 239000003795 chemical substances by application Substances 0.000 description 63
- 241000282414 Homo sapiens Species 0.000 description 61
- 230000000139 costimulatory effect Effects 0.000 description 60
- 241001465754 Metazoa Species 0.000 description 54
- 239000000427 antigen Substances 0.000 description 54
- 239000003153 chemical reaction reagent Substances 0.000 description 54
- 102000005962 receptors Human genes 0.000 description 54
- 108020003175 receptors Proteins 0.000 description 54
- 102000036639 antigens Human genes 0.000 description 52
- 108091007433 antigens Proteins 0.000 description 51
- 239000013604 expression vector Substances 0.000 description 49
- 230000000692 anti-sense effect Effects 0.000 description 47
- 108020004999 messenger RNA Proteins 0.000 description 47
- 230000003993 interaction Effects 0.000 description 46
- 230000004913 activation Effects 0.000 description 45
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 44
- 125000003275 alpha amino acid group Chemical group 0.000 description 40
- 102000037865 fusion proteins Human genes 0.000 description 37
- 108020001507 fusion proteins Proteins 0.000 description 37
- 108091028043 Nucleic acid sequence Proteins 0.000 description 36
- 230000005764 inhibitory process Effects 0.000 description 36
- 108090000695 Cytokines Proteins 0.000 description 35
- 239000012634 fragment Substances 0.000 description 35
- 210000001519 tissue Anatomy 0.000 description 35
- 102000004127 Cytokines Human genes 0.000 description 34
- 230000006870 function Effects 0.000 description 33
- 230000001105 regulatory effect Effects 0.000 description 32
- 102100039498 Cytotoxic T-lymphocyte protein 4 Human genes 0.000 description 31
- 101000889276 Homo sapiens Cytotoxic T-lymphocyte protein 4 Proteins 0.000 description 31
- 239000000523 sample Substances 0.000 description 30
- 239000000203 mixture Substances 0.000 description 29
- 201000010099 disease Diseases 0.000 description 28
- 239000000126 substance Substances 0.000 description 28
- 108091008874 T cell receptors Proteins 0.000 description 25
- 102000016266 T-Cell Antigen Receptors Human genes 0.000 description 25
- 230000003213 activating effect Effects 0.000 description 25
- 230000004940 costimulation Effects 0.000 description 25
- 230000009261 transgenic effect Effects 0.000 description 25
- 125000000539 amino acid group Chemical group 0.000 description 24
- 108091008034 costimulatory receptors Proteins 0.000 description 23
- 241000699666 Mus <mouse, genus> Species 0.000 description 22
- 239000012472 biological sample Substances 0.000 description 22
- 210000000349 chromosome Anatomy 0.000 description 22
- 239000002299 complementary DNA Substances 0.000 description 22
- 238000009396 hybridization Methods 0.000 description 22
- 108091008042 inhibitory receptors Proteins 0.000 description 22
- 210000000612 antigen-presenting cell Anatomy 0.000 description 21
- 230000000903 blocking effect Effects 0.000 description 21
- 230000000295 complement effect Effects 0.000 description 20
- 108091026890 Coding region Proteins 0.000 description 19
- 206010028980 Neoplasm Diseases 0.000 description 19
- 108700019146 Transgenes Proteins 0.000 description 19
- 238000004458 analytical method Methods 0.000 description 19
- 239000003814 drug Substances 0.000 description 19
- -1 I) ATA Chemical compound 0.000 description 18
- 108091034117 Oligonucleotide Proteins 0.000 description 18
- 230000004927 fusion Effects 0.000 description 18
- 238000000338 in vitro Methods 0.000 description 18
- 239000013615 primer Substances 0.000 description 18
- 101000914514 Homo sapiens T-cell-specific surface glycoprotein CD28 Proteins 0.000 description 17
- 241001529936 Murinae Species 0.000 description 17
- 102100027213 T-cell-specific surface glycoprotein CD28 Human genes 0.000 description 17
- 210000003719 b-lymphocyte Anatomy 0.000 description 17
- 230000035772 mutation Effects 0.000 description 17
- 238000002360 preparation method Methods 0.000 description 17
- 238000007423 screening assay Methods 0.000 description 17
- 102000004190 Enzymes Human genes 0.000 description 16
- 108090000790 Enzymes Proteins 0.000 description 16
- 230000000875 corresponding effect Effects 0.000 description 16
- 208000035475 disorder Diseases 0.000 description 16
- 229940088598 enzyme Drugs 0.000 description 16
- 210000004408 hybridoma Anatomy 0.000 description 16
- 238000004519 manufacturing process Methods 0.000 description 16
- 238000003259 recombinant expression Methods 0.000 description 16
- 102000053602 DNA Human genes 0.000 description 15
- 239000005557 antagonist Substances 0.000 description 15
- 230000001413 cellular effect Effects 0.000 description 15
- 239000000816 peptidomimetic Substances 0.000 description 15
- 238000013518 transcription Methods 0.000 description 15
- 230000035897 transcription Effects 0.000 description 15
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 14
- FWMNVWWHGCHHJJ-SKKKGAJSSA-N 4-amino-1-[(2r)-6-amino-2-[[(2r)-2-[[(2r)-2-[[(2r)-2-amino-3-phenylpropanoyl]amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]hexanoyl]piperidine-4-carboxylic acid Chemical compound C([C@H](C(=O)N[C@H](CC(C)C)C(=O)N[C@H](CCCCN)C(=O)N1CCC(N)(CC1)C(O)=O)NC(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 FWMNVWWHGCHHJJ-SKKKGAJSSA-N 0.000 description 14
- 108060003951 Immunoglobulin Proteins 0.000 description 14
- 230000002159 abnormal effect Effects 0.000 description 14
- 238000005516 engineering process Methods 0.000 description 14
- 102000018358 immunoglobulin Human genes 0.000 description 14
- 230000001965 increasing effect Effects 0.000 description 14
- 108020004414 DNA Proteins 0.000 description 13
- 238000001727 in vivo Methods 0.000 description 13
- 230000006698 induction Effects 0.000 description 13
- 238000003752 polymerase chain reaction Methods 0.000 description 13
- 230000004936 stimulating effect Effects 0.000 description 13
- 210000004881 tumor cell Anatomy 0.000 description 13
- 108020004511 Recombinant DNA Proteins 0.000 description 12
- 230000005540 biological transmission Effects 0.000 description 12
- 230000003828 downregulation Effects 0.000 description 12
- 229940079593 drug Drugs 0.000 description 12
- 230000002163 immunogen Effects 0.000 description 12
- 239000013612 plasmid Substances 0.000 description 12
- 230000035755 proliferation Effects 0.000 description 12
- 150000003384 small molecules Chemical class 0.000 description 12
- 238000011282 treatment Methods 0.000 description 12
- 108700018351 Major Histocompatibility Complex Proteins 0.000 description 11
- 230000000890 antigenic effect Effects 0.000 description 11
- 230000001939 inductive effect Effects 0.000 description 11
- 238000002372 labelling Methods 0.000 description 11
- 239000000463 material Substances 0.000 description 11
- 230000020382 suppression by virus of host antigen processing and presentation of peptide antigen via MHC class I Effects 0.000 description 11
- 230000001225 therapeutic effect Effects 0.000 description 11
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 description 10
- 206010020751 Hypersensitivity Diseases 0.000 description 10
- 108010002350 Interleukin-2 Proteins 0.000 description 10
- 241000124008 Mammalia Species 0.000 description 10
- 108010076504 Protein Sorting Signals Proteins 0.000 description 10
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 10
- 238000001514 detection method Methods 0.000 description 10
- 239000012636 effector Substances 0.000 description 10
- 239000011159 matrix material Substances 0.000 description 10
- 239000002609 medium Substances 0.000 description 10
- 230000037361 pathway Effects 0.000 description 10
- 230000002829 reductive effect Effects 0.000 description 10
- 241000894007 species Species 0.000 description 10
- 238000006467 substitution reaction Methods 0.000 description 10
- 230000003827 upregulation Effects 0.000 description 10
- 108091008875 B cell receptors Proteins 0.000 description 9
- 206010011968 Decreased immune responsiveness Diseases 0.000 description 9
- 108010008281 Recombinant Fusion Proteins Proteins 0.000 description 9
- 102000007056 Recombinant Fusion Proteins Human genes 0.000 description 9
- 239000011324 bead Substances 0.000 description 9
- 230000008901 benefit Effects 0.000 description 9
- 230000001086 cytosolic effect Effects 0.000 description 9
- 210000003917 human chromosome Anatomy 0.000 description 9
- 239000000047 product Substances 0.000 description 9
- 230000019491 signal transduction Effects 0.000 description 9
- 238000003786 synthesis reaction Methods 0.000 description 9
- 208000023275 Autoimmune disease Diseases 0.000 description 8
- 108090000994 Catalytic RNA Proteins 0.000 description 8
- 102000053642 Catalytic RNA Human genes 0.000 description 8
- 238000002965 ELISA Methods 0.000 description 8
- 102000009109 Fc receptors Human genes 0.000 description 8
- 108010087819 Fc receptors Proteins 0.000 description 8
- 102000000588 Interleukin-2 Human genes 0.000 description 8
- 206010035226 Plasma cell myeloma Diseases 0.000 description 8
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 8
- 239000000556 agonist Substances 0.000 description 8
- 230000004071 biological effect Effects 0.000 description 8
- 239000012707 chemical precursor Substances 0.000 description 8
- 230000002068 genetic effect Effects 0.000 description 8
- 238000002744 homologous recombination Methods 0.000 description 8
- 230000006801 homologous recombination Effects 0.000 description 8
- 230000036039 immunity Effects 0.000 description 8
- 210000004698 lymphocyte Anatomy 0.000 description 8
- 239000003550 marker Substances 0.000 description 8
- 239000012528 membrane Substances 0.000 description 8
- 201000000050 myeloid neoplasm Diseases 0.000 description 8
- 108091092562 ribozyme Proteins 0.000 description 8
- 238000010561 standard procedure Methods 0.000 description 8
- 239000003053 toxin Substances 0.000 description 8
- 231100000765 toxin Toxicity 0.000 description 8
- 108700012359 toxins Proteins 0.000 description 8
- 230000003612 virological effect Effects 0.000 description 8
- 241000588724 Escherichia coli Species 0.000 description 7
- 108010070675 Glutathione transferase Proteins 0.000 description 7
- 102100029100 Hematopoietic prostaglandin D synthase Human genes 0.000 description 7
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 7
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 7
- 230000006044 T cell activation Effects 0.000 description 7
- 230000006052 T cell proliferation Effects 0.000 description 7
- 108091023040 Transcription factor Proteins 0.000 description 7
- 102000040945 Transcription factor Human genes 0.000 description 7
- 230000001580 bacterial effect Effects 0.000 description 7
- 238000004113 cell culture Methods 0.000 description 7
- 238000003776 cleavage reaction Methods 0.000 description 7
- 238000004132 cross linking Methods 0.000 description 7
- 230000003247 decreasing effect Effects 0.000 description 7
- 230000003053 immunization Effects 0.000 description 7
- 230000001404 mediated effect Effects 0.000 description 7
- 239000002245 particle Substances 0.000 description 7
- 239000008194 pharmaceutical composition Substances 0.000 description 7
- 230000010076 replication Effects 0.000 description 7
- 230000004044 response Effects 0.000 description 7
- 230000007017 scission Effects 0.000 description 7
- 239000000243 solution Substances 0.000 description 7
- 230000000638 stimulation Effects 0.000 description 7
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 7
- 241000894006 Bacteria Species 0.000 description 6
- 239000004472 Lysine Substances 0.000 description 6
- 102000043131 MHC class II family Human genes 0.000 description 6
- 108091054438 MHC class II family Proteins 0.000 description 6
- 241000699670 Mus sp. Species 0.000 description 6
- 230000009471 action Effects 0.000 description 6
- 239000002671 adjuvant Substances 0.000 description 6
- 238000010171 animal model Methods 0.000 description 6
- 238000013459 approach Methods 0.000 description 6
- 230000015572 biosynthetic process Effects 0.000 description 6
- 230000002759 chromosomal effect Effects 0.000 description 6
- 238000007796 conventional method Methods 0.000 description 6
- 230000006378 damage Effects 0.000 description 6
- 238000012217 deletion Methods 0.000 description 6
- 230000037430 deletion Effects 0.000 description 6
- 230000005714 functional activity Effects 0.000 description 6
- 238000001415 gene therapy Methods 0.000 description 6
- 230000006450 immune cell response Effects 0.000 description 6
- 238000002347 injection Methods 0.000 description 6
- 239000007924 injection Substances 0.000 description 6
- 230000003834 intracellular effect Effects 0.000 description 6
- 244000005700 microbiome Species 0.000 description 6
- 238000010369 molecular cloning Methods 0.000 description 6
- 238000000746 purification Methods 0.000 description 6
- 230000028327 secretion Effects 0.000 description 6
- 239000002904 solvent Substances 0.000 description 6
- 230000014616 translation Effects 0.000 description 6
- MTCFGRXMJLQNBG-REOHCLBHSA-N (2S)-2-Amino-3-hydroxypropansäure Chemical compound OC[C@H](N)C(O)=O MTCFGRXMJLQNBG-REOHCLBHSA-N 0.000 description 5
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 5
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 5
- 241000282412 Homo Species 0.000 description 5
- 102000009786 Immunoglobulin Constant Regions Human genes 0.000 description 5
- 108010009817 Immunoglobulin Constant Regions Proteins 0.000 description 5
- AYFVYJQAPQTCCC-GBXIJSLDSA-N L-threonine Chemical compound C[C@@H](O)[C@H](N)C(O)=O AYFVYJQAPQTCCC-GBXIJSLDSA-N 0.000 description 5
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 5
- 229940124060 PD-1 antagonist Drugs 0.000 description 5
- 102000007079 Peptide Fragments Human genes 0.000 description 5
- 108010033276 Peptide Fragments Proteins 0.000 description 5
- 108010091086 Recombinases Proteins 0.000 description 5
- 102000018120 Recombinases Human genes 0.000 description 5
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 5
- 235000014680 Saccharomyces cerevisiae Nutrition 0.000 description 5
- 108010090804 Streptavidin Proteins 0.000 description 5
- 238000007792 addition Methods 0.000 description 5
- 230000006907 apoptotic process Effects 0.000 description 5
- 230000006470 autoimmune attack Effects 0.000 description 5
- 230000033228 biological regulation Effects 0.000 description 5
- 229960002685 biotin Drugs 0.000 description 5
- 235000020958 biotin Nutrition 0.000 description 5
- 239000011616 biotin Substances 0.000 description 5
- 210000002798 bone marrow cell Anatomy 0.000 description 5
- 230000004663 cell proliferation Effects 0.000 description 5
- 210000004978 chinese hamster ovary cell Anatomy 0.000 description 5
- 238000010367 cloning Methods 0.000 description 5
- 238000000576 coating method Methods 0.000 description 5
- 239000003184 complementary RNA Substances 0.000 description 5
- 239000003623 enhancer Substances 0.000 description 5
- 239000000499 gel Substances 0.000 description 5
- 210000004754 hybrid cell Anatomy 0.000 description 5
- 238000002649 immunization Methods 0.000 description 5
- 239000003018 immunosuppressive agent Substances 0.000 description 5
- 210000002540 macrophage Anatomy 0.000 description 5
- 210000004962 mammalian cell Anatomy 0.000 description 5
- 238000013507 mapping Methods 0.000 description 5
- 230000004048 modification Effects 0.000 description 5
- 238000012986 modification Methods 0.000 description 5
- 210000001616 monocyte Anatomy 0.000 description 5
- 238000002703 mutagenesis Methods 0.000 description 5
- 231100000350 mutagenesis Toxicity 0.000 description 5
- 210000000287 oocyte Anatomy 0.000 description 5
- 102000054765 polymorphisms of proteins Human genes 0.000 description 5
- 102000040430 polynucleotide Human genes 0.000 description 5
- 108091033319 polynucleotide Proteins 0.000 description 5
- 239000002157 polynucleotide Substances 0.000 description 5
- 230000002265 prevention Effects 0.000 description 5
- 108091008146 restriction endonucleases Proteins 0.000 description 5
- 210000003491 skin Anatomy 0.000 description 5
- 239000011780 sodium chloride Substances 0.000 description 5
- 238000001890 transfection Methods 0.000 description 5
- 238000013519 translation Methods 0.000 description 5
- 102000002260 Alkaline Phosphatase Human genes 0.000 description 4
- 108020004774 Alkaline Phosphatase Proteins 0.000 description 4
- 108020005544 Antisense RNA Proteins 0.000 description 4
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 4
- 102000017420 CD3 protein, epsilon/gamma/delta subunit Human genes 0.000 description 4
- 108050005493 CD3 protein, epsilon/gamma/delta subunit Proteins 0.000 description 4
- 102000014914 Carrier Proteins Human genes 0.000 description 4
- 108091033380 Coding strand Proteins 0.000 description 4
- 108020004705 Codon Proteins 0.000 description 4
- 239000003155 DNA primer Substances 0.000 description 4
- 108091029865 Exogenous DNA Proteins 0.000 description 4
- 238000012413 Fluorescence activated cell sorting analysis Methods 0.000 description 4
- 241000238631 Hexapoda Species 0.000 description 4
- 108091054437 MHC class I family Proteins 0.000 description 4
- 108091092724 Noncoding DNA Proteins 0.000 description 4
- 108700001237 Nucleic Acid-Based Vaccines Proteins 0.000 description 4
- 229940122544 PD-1 agonist Drugs 0.000 description 4
- 102100021566 Protein kinase C theta type Human genes 0.000 description 4
- 108700008625 Reporter Genes Proteins 0.000 description 4
- 108010083644 Ribonucleases Proteins 0.000 description 4
- 102000006382 Ribonucleases Human genes 0.000 description 4
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 4
- 230000005867 T cell response Effects 0.000 description 4
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 description 4
- 239000004473 Threonine Substances 0.000 description 4
- ISAKRJDGNUQOIC-UHFFFAOYSA-N Uracil Chemical compound O=C1C=CNC(=O)N1 ISAKRJDGNUQOIC-UHFFFAOYSA-N 0.000 description 4
- 241000700605 Viruses Species 0.000 description 4
- 238000010521 absorption reaction Methods 0.000 description 4
- 230000002776 aggregation Effects 0.000 description 4
- 238000004220 aggregation Methods 0.000 description 4
- 230000007815 allergy Effects 0.000 description 4
- 230000004075 alteration Effects 0.000 description 4
- 239000003242 anti bacterial agent Substances 0.000 description 4
- 230000001363 autoimmune Effects 0.000 description 4
- 108091008324 binding proteins Proteins 0.000 description 4
- 210000004899 c-terminal region Anatomy 0.000 description 4
- 201000011510 cancer Diseases 0.000 description 4
- 238000000423 cell based assay Methods 0.000 description 4
- 238000006243 chemical reaction Methods 0.000 description 4
- 238000003200 chromosome mapping Methods 0.000 description 4
- 239000011248 coating agent Substances 0.000 description 4
- 239000002254 cytotoxic agent Substances 0.000 description 4
- 229940127089 cytotoxic agent Drugs 0.000 description 4
- 231100000599 cytotoxic agent Toxicity 0.000 description 4
- 239000006185 dispersion Substances 0.000 description 4
- 239000003937 drug carrier Substances 0.000 description 4
- 210000003527 eukaryotic cell Anatomy 0.000 description 4
- 230000004077 genetic alteration Effects 0.000 description 4
- 231100000118 genetic alteration Toxicity 0.000 description 4
- 210000004602 germ cell Anatomy 0.000 description 4
- FDGQSTZJBFJUBT-UHFFFAOYSA-N hypoxanthine Chemical compound O=C1NC=NC2=C1NC=N2 FDGQSTZJBFJUBT-UHFFFAOYSA-N 0.000 description 4
- 230000005931 immune cell recruitment Effects 0.000 description 4
- 229960003444 immunosuppressant agent Drugs 0.000 description 4
- 230000001976 improved effect Effects 0.000 description 4
- 238000007901 in situ hybridization Methods 0.000 description 4
- 239000004615 ingredient Substances 0.000 description 4
- 235000018977 lysine Nutrition 0.000 description 4
- 230000007246 mechanism Effects 0.000 description 4
- 229940023146 nucleic acid vaccine Drugs 0.000 description 4
- 244000052769 pathogen Species 0.000 description 4
- 230000000144 pharmacologic effect Effects 0.000 description 4
- 229920001223 polyethylene glycol Polymers 0.000 description 4
- 239000002987 primer (paints) Substances 0.000 description 4
- 230000008569 process Effects 0.000 description 4
- 230000000069 prophylactic effect Effects 0.000 description 4
- 230000000717 retained effect Effects 0.000 description 4
- 210000002966 serum Anatomy 0.000 description 4
- 208000024891 symptom Diseases 0.000 description 4
- 230000009885 systemic effect Effects 0.000 description 4
- 238000011191 terminal modification Methods 0.000 description 4
- 238000002054 transplantation Methods 0.000 description 4
- 239000013603 viral vector Substances 0.000 description 4
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Chemical compound O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 4
- 208000030507 AIDS Diseases 0.000 description 3
- 206010000234 Abortion spontaneous Diseases 0.000 description 3
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 3
- 102000000844 Cell Surface Receptors Human genes 0.000 description 3
- 108010001857 Cell Surface Receptors Proteins 0.000 description 3
- 108010047041 Complementarity Determining Regions Proteins 0.000 description 3
- 230000004568 DNA-binding Effects 0.000 description 3
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 3
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 3
- 108010007457 Extracellular Signal-Regulated MAP Kinases Proteins 0.000 description 3
- 102000007665 Extracellular Signal-Regulated MAP Kinases Human genes 0.000 description 3
- 108010054218 Factor VIII Proteins 0.000 description 3
- 102000001690 Factor VIII Human genes 0.000 description 3
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 3
- 102000008949 Histocompatibility Antigens Class I Human genes 0.000 description 3
- 101001087416 Homo sapiens Tyrosine-protein phosphatase non-receptor type 11 Proteins 0.000 description 3
- 108010054477 Immunoglobulin Fab Fragments Proteins 0.000 description 3
- 102000001706 Immunoglobulin Fab Fragments Human genes 0.000 description 3
- 108010067060 Immunoglobulin Variable Region Proteins 0.000 description 3
- 102000017727 Immunoglobulin Variable Region Human genes 0.000 description 3
- 102100034343 Integrase Human genes 0.000 description 3
- 108010043610 KIR Receptors Proteins 0.000 description 3
- 102000002698 KIR Receptors Human genes 0.000 description 3
- XUJNEKJLAYXESH-REOHCLBHSA-N L-Cysteine Chemical compound SC[C@H](N)C(O)=O XUJNEKJLAYXESH-REOHCLBHSA-N 0.000 description 3
- DCXYFEDJOCDNAF-REOHCLBHSA-N L-asparagine Chemical compound OC(=O)[C@@H](N)CC(N)=O DCXYFEDJOCDNAF-REOHCLBHSA-N 0.000 description 3
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 3
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 3
- AGPKZVBTJJNPAG-WHFBIAKZSA-N L-isoleucine Chemical compound CC[C@H](C)[C@H](N)C(O)=O AGPKZVBTJJNPAG-WHFBIAKZSA-N 0.000 description 3
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 3
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 description 3
- 101710163270 Nuclease Proteins 0.000 description 3
- 238000012408 PCR amplification Methods 0.000 description 3
- 239000012270 PD-1 inhibitor Substances 0.000 description 3
- 239000012668 PD-1-inhibitor Substances 0.000 description 3
- 102000004160 Phosphoric Monoester Hydrolases Human genes 0.000 description 3
- 108090000608 Phosphoric Monoester Hydrolases Proteins 0.000 description 3
- 108091000080 Phosphotransferase Proteins 0.000 description 3
- 239000002202 Polyethylene glycol Substances 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 108010092799 RNA-directed DNA polymerase Proteins 0.000 description 3
- 241000700159 Rattus Species 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- 230000020385 T cell costimulation Effects 0.000 description 3
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 description 3
- 230000003321 amplification Effects 0.000 description 3
- 239000000074 antisense oligonucleotide Substances 0.000 description 3
- 238000012230 antisense oligonucleotides Methods 0.000 description 3
- 230000002238 attenuated effect Effects 0.000 description 3
- 210000004369 blood Anatomy 0.000 description 3
- 239000008280 blood Substances 0.000 description 3
- 239000006143 cell culture medium Substances 0.000 description 3
- 210000000170 cell membrane Anatomy 0.000 description 3
- 230000008859 change Effects 0.000 description 3
- 230000008878 coupling Effects 0.000 description 3
- 238000010168 coupling process Methods 0.000 description 3
- 238000005859 coupling reaction Methods 0.000 description 3
- 125000000151 cysteine group Chemical group N[C@@H](CS)C(=O)* 0.000 description 3
- 230000007423 decrease Effects 0.000 description 3
- 238000013461 design Methods 0.000 description 3
- 239000002612 dispersion medium Substances 0.000 description 3
- 210000002308 embryonic cell Anatomy 0.000 description 3
- 210000001671 embryonic stem cell Anatomy 0.000 description 3
- 210000003979 eosinophil Anatomy 0.000 description 3
- 239000000284 extract Substances 0.000 description 3
- 229960000301 factor viii Drugs 0.000 description 3
- 238000009472 formulation Methods 0.000 description 3
- 239000007789 gas Substances 0.000 description 3
- 238000001476 gene delivery Methods 0.000 description 3
- 210000003630 histaminocyte Anatomy 0.000 description 3
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 3
- 210000005260 human cell Anatomy 0.000 description 3
- 230000016784 immunoglobulin production Effects 0.000 description 3
- 238000001114 immunoprecipitation Methods 0.000 description 3
- 230000001506 immunosuppresive effect Effects 0.000 description 3
- 230000002452 interceptive effect Effects 0.000 description 3
- AGPKZVBTJJNPAG-UHFFFAOYSA-N isoleucine Natural products CCC(C)C(N)C(O)=O AGPKZVBTJJNPAG-UHFFFAOYSA-N 0.000 description 3
- 229960000310 isoleucine Drugs 0.000 description 3
- 230000021633 leukocyte mediated immunity Effects 0.000 description 3
- 239000007788 liquid Substances 0.000 description 3
- 230000007774 longterm Effects 0.000 description 3
- 210000001161 mammalian embryo Anatomy 0.000 description 3
- 230000031864 metaphase Effects 0.000 description 3
- 230000011987 methylation Effects 0.000 description 3
- 238000007069 methylation reaction Methods 0.000 description 3
- 230000005012 migration Effects 0.000 description 3
- 238000013508 migration Methods 0.000 description 3
- 238000012544 monitoring process Methods 0.000 description 3
- 210000000440 neutrophil Anatomy 0.000 description 3
- 238000003199 nucleic acid amplification method Methods 0.000 description 3
- 230000001717 pathogenic effect Effects 0.000 description 3
- 229940121655 pd-1 inhibitor Drugs 0.000 description 3
- 238000002823 phage display Methods 0.000 description 3
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 3
- 102000020233 phosphotransferase Human genes 0.000 description 3
- 230000003169 placental effect Effects 0.000 description 3
- 239000000843 powder Substances 0.000 description 3
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 3
- 210000001236 prokaryotic cell Anatomy 0.000 description 3
- 230000002062 proliferating effect Effects 0.000 description 3
- 238000007894 restriction fragment length polymorphism technique Methods 0.000 description 3
- 238000002741 site-directed mutagenesis Methods 0.000 description 3
- 239000007787 solid Substances 0.000 description 3
- 210000001082 somatic cell Anatomy 0.000 description 3
- 208000000995 spontaneous abortion Diseases 0.000 description 3
- 238000007910 systemic administration Methods 0.000 description 3
- 239000003826 tablet Substances 0.000 description 3
- QJJXYPPXXYFBGM-SHYZHZOCSA-N tacrolimus Natural products CO[C@H]1C[C@H](CC[C@@H]1O)C=C(C)[C@H]2OC(=O)[C@H]3CCCCN3C(=O)C(=O)[C@@]4(O)O[C@@H]([C@H](C[C@H]4C)OC)[C@@H](C[C@H](C)CC(=C[C@@H](CC=C)C(=O)C[C@H](O)[C@H]2C)C)OC QJJXYPPXXYFBGM-SHYZHZOCSA-N 0.000 description 3
- 230000008685 targeting Effects 0.000 description 3
- 230000009466 transformation Effects 0.000 description 3
- 230000005945 translocation Effects 0.000 description 3
- 230000001960 triggered effect Effects 0.000 description 3
- 241000701447 unidentified baculovirus Species 0.000 description 3
- 238000011144 upstream manufacturing Methods 0.000 description 3
- 229960005486 vaccine Drugs 0.000 description 3
- 238000001262 western blot Methods 0.000 description 3
- YSAJFXWTVFGPAX-UHFFFAOYSA-N 2-[(2,4-dioxo-1h-pyrimidin-5-yl)oxy]acetic acid Chemical compound OC(=O)COC1=CNC(=O)NC1=O YSAJFXWTVFGPAX-UHFFFAOYSA-N 0.000 description 2
- OVONXEQGWXGFJD-UHFFFAOYSA-N 4-sulfanylidene-1h-pyrimidin-2-one Chemical compound SC=1C=CNC(=O)N=1 OVONXEQGWXGFJD-UHFFFAOYSA-N 0.000 description 2
- ZLAQATDNGLKIEV-UHFFFAOYSA-N 5-methyl-2-sulfanylidene-1h-pyrimidin-4-one Chemical compound CC1=CNC(=S)NC1=O ZLAQATDNGLKIEV-UHFFFAOYSA-N 0.000 description 2
- 102100026445 A-kinase anchor protein 17A Human genes 0.000 description 2
- 108020000948 Antisense Oligonucleotides Proteins 0.000 description 2
- 239000004475 Arginine Substances 0.000 description 2
- DCXYFEDJOCDNAF-UHFFFAOYSA-N Asparagine Natural products OC(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-N 0.000 description 2
- 201000001320 Atherosclerosis Diseases 0.000 description 2
- 102100027205 B-cell antigen receptor complex-associated protein alpha chain Human genes 0.000 description 2
- 101710095183 B-cell antigen receptor complex-associated protein alpha chain Proteins 0.000 description 2
- 102100027203 B-cell antigen receptor complex-associated protein beta chain Human genes 0.000 description 2
- 101710166261 B-cell antigen receptor complex-associated protein beta chain Proteins 0.000 description 2
- 102100027314 Beta-2-microglobulin Human genes 0.000 description 2
- 108010029697 CD40 Ligand Proteins 0.000 description 2
- 101150013553 CD40 gene Proteins 0.000 description 2
- 102100032937 CD40 ligand Human genes 0.000 description 2
- 108010021064 CTLA-4 Antigen Proteins 0.000 description 2
- 102000008203 CTLA-4 Antigen Human genes 0.000 description 2
- 229940045513 CTLA4 antagonist Drugs 0.000 description 2
- 241000282472 Canis lupus familiaris Species 0.000 description 2
- 241000283707 Capra Species 0.000 description 2
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 2
- 229930105110 Cyclosporin A Natural products 0.000 description 2
- PMATZTZNYRCHOR-CGLBZJNRSA-N Cyclosporin A Chemical compound CC[C@@H]1NC(=O)[C@H]([C@H](O)[C@H](C)C\C=C\C)N(C)C(=O)[C@H](C(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)N(C)C(=O)CN(C)C1=O PMATZTZNYRCHOR-CGLBZJNRSA-N 0.000 description 2
- 108010036949 Cyclosporine Proteins 0.000 description 2
- 241000701022 Cytomegalovirus Species 0.000 description 2
- 102000004163 DNA-directed RNA polymerases Human genes 0.000 description 2
- 241000702421 Dependoparvovirus Species 0.000 description 2
- 108010010803 Gelatin Proteins 0.000 description 2
- 206010071602 Genetic polymorphism Diseases 0.000 description 2
- 239000004471 Glycine Substances 0.000 description 2
- 208000009329 Graft vs Host Disease Diseases 0.000 description 2
- 108010017213 Granulocyte-Macrophage Colony-Stimulating Factor Proteins 0.000 description 2
- 102100039620 Granulocyte-macrophage colony-stimulating factor Human genes 0.000 description 2
- 101000718019 Homo sapiens A-kinase anchor protein 17A Proteins 0.000 description 2
- 101000914484 Homo sapiens T-lymphocyte activation antigen CD80 Proteins 0.000 description 2
- 108010001336 Horseradish Peroxidase Proteins 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 2
- UGQMRVRMYYASKQ-UHFFFAOYSA-N Hypoxanthine nucleoside Natural products OC1C(O)C(CO)OC1N1C(NC=NC2=O)=C2N=C1 UGQMRVRMYYASKQ-UHFFFAOYSA-N 0.000 description 2
- 102100034980 ICOS ligand Human genes 0.000 description 2
- 108090000978 Interleukin-4 Proteins 0.000 description 2
- 102000004388 Interleukin-4 Human genes 0.000 description 2
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 description 2
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 2
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 2
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 2
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 description 2
- KZSNJWFQEVHDMF-BYPYZUCNSA-N L-valine Chemical compound CC(C)[C@H](N)C(O)=O KZSNJWFQEVHDMF-BYPYZUCNSA-N 0.000 description 2
- 108091026898 Leader sequence (mRNA) Proteins 0.000 description 2
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 2
- 206010025323 Lymphomas Diseases 0.000 description 2
- 102000043129 MHC class I family Human genes 0.000 description 2
- 208000012902 Nervous system disease Diseases 0.000 description 2
- 108010032107 Non-Receptor Type 11 Protein Tyrosine Phosphatase Proteins 0.000 description 2
- 108020004711 Nucleic Acid Probes Proteins 0.000 description 2
- 108091005461 Nucleic proteins Chemical group 0.000 description 2
- 108700026244 Open Reading Frames Proteins 0.000 description 2
- 108091093037 Peptide nucleic acid Proteins 0.000 description 2
- 206010057249 Phagocytosis Diseases 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- 102000045595 Phosphoprotein Phosphatases Human genes 0.000 description 2
- 108700019535 Phosphoprotein Phosphatases Proteins 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- ONIBWKKTOPOVIA-UHFFFAOYSA-N Proline Natural products OC(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-N 0.000 description 2
- 239000004365 Protease Substances 0.000 description 2
- 108010029485 Protein Isoforms Proteins 0.000 description 2
- 102000001708 Protein Isoforms Human genes 0.000 description 2
- 102000009516 Protein Serine-Threonine Kinases Human genes 0.000 description 2
- 108010009341 Protein Serine-Threonine Kinases Proteins 0.000 description 2
- 102000002727 Protein Tyrosine Phosphatase Human genes 0.000 description 2
- 102000004022 Protein-Tyrosine Kinases Human genes 0.000 description 2
- 108090000412 Protein-Tyrosine Kinases Proteins 0.000 description 2
- 108010039491 Ricin Proteins 0.000 description 2
- 108010084592 Saporins Proteins 0.000 description 2
- 238000012300 Sequence Analysis Methods 0.000 description 2
- 238000002105 Southern blotting Methods 0.000 description 2
- 230000017274 T cell anergy Effects 0.000 description 2
- 102100027222 T-lymphocyte activation antigen CD80 Human genes 0.000 description 2
- QJJXYPPXXYFBGM-LFZNUXCKSA-N Tacrolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1\C=C(/C)[C@@H]1[C@H](C)[C@@H](O)CC(=O)[C@H](CC=C)/C=C(C)/C[C@H](C)C[C@H](OC)[C@H]([C@H](C[C@H]2C)OC)O[C@@]2(O)C(=O)C(=O)N2CCCC[C@H]2C(=O)O1 QJJXYPPXXYFBGM-LFZNUXCKSA-N 0.000 description 2
- 239000004098 Tetracycline Substances 0.000 description 2
- 108091036066 Three prime untranslated region Proteins 0.000 description 2
- IQFYYKKMVGJFEH-XLPZGREQSA-N Thymidine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 IQFYYKKMVGJFEH-XLPZGREQSA-N 0.000 description 2
- 102100040245 Tumor necrosis factor receptor superfamily member 5 Human genes 0.000 description 2
- KZSNJWFQEVHDMF-UHFFFAOYSA-N Valine Natural products CC(C)C(N)C(O)=O KZSNJWFQEVHDMF-UHFFFAOYSA-N 0.000 description 2
- 208000027418 Wounds and injury Diseases 0.000 description 2
- 230000021736 acetylation Effects 0.000 description 2
- 238000006640 acetylation reaction Methods 0.000 description 2
- 239000002253 acid Substances 0.000 description 2
- 239000012190 activator Substances 0.000 description 2
- 239000004480 active ingredient Substances 0.000 description 2
- 238000001042 affinity chromatography Methods 0.000 description 2
- 235000004279 alanine Nutrition 0.000 description 2
- 239000013566 allergen Substances 0.000 description 2
- 208000026935 allergic disease Diseases 0.000 description 2
- 230000009435 amidation Effects 0.000 description 2
- 238000007112 amidation reaction Methods 0.000 description 2
- 230000000844 anti-bacterial effect Effects 0.000 description 2
- 239000003429 antifungal agent Substances 0.000 description 2
- 229940121375 antifungal agent Drugs 0.000 description 2
- 239000002246 antineoplastic agent Substances 0.000 description 2
- 229940041181 antineoplastic drug Drugs 0.000 description 2
- 238000003782 apoptosis assay Methods 0.000 description 2
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 2
- 229960005070 ascorbic acid Drugs 0.000 description 2
- 235000010323 ascorbic acid Nutrition 0.000 description 2
- 239000011668 ascorbic acid Substances 0.000 description 2
- 235000009582 asparagine Nutrition 0.000 description 2
- 229960001230 asparagine Drugs 0.000 description 2
- 235000003704 aspartic acid Nutrition 0.000 description 2
- 210000003651 basophil Anatomy 0.000 description 2
- 230000009286 beneficial effect Effects 0.000 description 2
- 108010081355 beta 2-Microglobulin Proteins 0.000 description 2
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 2
- 239000011230 binding agent Substances 0.000 description 2
- 239000013060 biological fluid Substances 0.000 description 2
- 239000012620 biological material Substances 0.000 description 2
- 239000002981 blocking agent Substances 0.000 description 2
- 150000001720 carbohydrates Chemical class 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- 230000003197 catalytic effect Effects 0.000 description 2
- 230000025084 cell cycle arrest Effects 0.000 description 2
- 230000003915 cell function Effects 0.000 description 2
- 230000036755 cellular response Effects 0.000 description 2
- OSASVXMJTNOKOY-UHFFFAOYSA-N chlorobutanol Chemical compound CC(C)(O)C(Cl)(Cl)Cl OSASVXMJTNOKOY-UHFFFAOYSA-N 0.000 description 2
- 229960001265 ciclosporin Drugs 0.000 description 2
- 238000011260 co-administration Methods 0.000 description 2
- 238000002742 combinatorial mutagenesis Methods 0.000 description 2
- 230000002860 competitive effect Effects 0.000 description 2
- 230000021615 conjugation Effects 0.000 description 2
- 239000012228 culture supernatant Substances 0.000 description 2
- 238000012258 culturing Methods 0.000 description 2
- 235000018417 cysteine Nutrition 0.000 description 2
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 2
- 230000016396 cytokine production Effects 0.000 description 2
- 231100000135 cytotoxicity Toxicity 0.000 description 2
- 230000003013 cytotoxicity Effects 0.000 description 2
- 230000002950 deficient Effects 0.000 description 2
- 230000001419 dependent effect Effects 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- 230000018109 developmental process Effects 0.000 description 2
- 238000002405 diagnostic procedure Methods 0.000 description 2
- 239000003085 diluting agent Substances 0.000 description 2
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 2
- 239000002552 dosage form Substances 0.000 description 2
- 230000002222 downregulating effect Effects 0.000 description 2
- 238000004520 electroporation Methods 0.000 description 2
- 230000002708 enhancing effect Effects 0.000 description 2
- 230000007613 environmental effect Effects 0.000 description 2
- 230000002255 enzymatic effect Effects 0.000 description 2
- 238000001976 enzyme digestion Methods 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- 210000003754 fetus Anatomy 0.000 description 2
- 238000011049 filling Methods 0.000 description 2
- 239000012530 fluid Substances 0.000 description 2
- GNBHRKFJIUUOQI-UHFFFAOYSA-N fluorescein Chemical compound O1C(=O)C2=CC=CC=C2C21C1=CC=C(O)C=C1OC1=CC(O)=CC=C21 GNBHRKFJIUUOQI-UHFFFAOYSA-N 0.000 description 2
- 239000008273 gelatin Substances 0.000 description 2
- 229920000159 gelatin Polymers 0.000 description 2
- 235000019322 gelatine Nutrition 0.000 description 2
- 235000011852 gelatine desserts Nutrition 0.000 description 2
- 235000013922 glutamic acid Nutrition 0.000 description 2
- 239000004220 glutamic acid Substances 0.000 description 2
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 2
- RWSXRVCMGQZWBV-WDSKDSINSA-N glutathione Chemical compound OC(=O)[C@@H](N)CCC(=O)N[C@@H](CS)C(=O)NCC(O)=O RWSXRVCMGQZWBV-WDSKDSINSA-N 0.000 description 2
- 235000011187 glycerol Nutrition 0.000 description 2
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 2
- 229910052737 gold Inorganic materials 0.000 description 2
- 239000010931 gold Substances 0.000 description 2
- 208000024908 graft versus host disease Diseases 0.000 description 2
- 208000019622 heart disease Diseases 0.000 description 2
- 239000001257 hydrogen Substances 0.000 description 2
- 229910052739 hydrogen Inorganic materials 0.000 description 2
- 230000006303 immediate early viral mRNA transcription Effects 0.000 description 2
- 238000003125 immunofluorescent labeling Methods 0.000 description 2
- 229940072221 immunoglobulins Drugs 0.000 description 2
- 229960001438 immunostimulant agent Drugs 0.000 description 2
- 239000003022 immunostimulating agent Substances 0.000 description 2
- 230000003308 immunostimulating effect Effects 0.000 description 2
- 230000001861 immunosuppressant effect Effects 0.000 description 2
- 208000015181 infectious disease Diseases 0.000 description 2
- 206010022000 influenza Diseases 0.000 description 2
- 239000003112 inhibitor Substances 0.000 description 2
- 208000014674 injury Diseases 0.000 description 2
- 230000004073 interleukin-2 production Effects 0.000 description 2
- 230000009878 intermolecular interaction Effects 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- 239000007951 isotonicity adjuster Substances 0.000 description 2
- 210000002510 keratinocyte Anatomy 0.000 description 2
- 239000002502 liposome Substances 0.000 description 2
- 239000000314 lubricant Substances 0.000 description 2
- 239000006166 lysate Substances 0.000 description 2
- 230000002101 lytic effect Effects 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 238000004949 mass spectrometry Methods 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- 229930182817 methionine Natural products 0.000 description 2
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 2
- OSWPMRLSEDHDFF-UHFFFAOYSA-N methyl salicylate Chemical compound COC(=O)C1=CC=CC=C1O OSWPMRLSEDHDFF-UHFFFAOYSA-N 0.000 description 2
- LXCFILQKKLGQFO-UHFFFAOYSA-N methylparaben Chemical compound COC(=O)C1=CC=C(O)C=C1 LXCFILQKKLGQFO-UHFFFAOYSA-N 0.000 description 2
- 238000000520 microinjection Methods 0.000 description 2
- 230000011278 mitosis Effects 0.000 description 2
- 238000000302 molecular modelling Methods 0.000 description 2
- 208000010125 myocardial infarction Diseases 0.000 description 2
- 210000000822 natural killer cell Anatomy 0.000 description 2
- 230000025020 negative regulation of T cell proliferation Effects 0.000 description 2
- 230000007302 negative regulation of cytokine production Effects 0.000 description 2
- 230000012177 negative regulation of immune response Effects 0.000 description 2
- 239000002853 nucleic acid probe Substances 0.000 description 2
- 210000004940 nucleus Anatomy 0.000 description 2
- 239000002674 ointment Substances 0.000 description 2
- 210000000056 organ Anatomy 0.000 description 2
- 238000010647 peptide synthesis reaction Methods 0.000 description 2
- 210000001986 peyer's patch Anatomy 0.000 description 2
- 230000008782 phagocytosis Effects 0.000 description 2
- 239000000546 pharmaceutical excipient Substances 0.000 description 2
- 239000012071 phase Substances 0.000 description 2
- 239000002953 phosphate buffered saline Substances 0.000 description 2
- 230000036470 plasma concentration Effects 0.000 description 2
- 230000008488 polyadenylation Effects 0.000 description 2
- 230000035935 pregnancy Effects 0.000 description 2
- 230000005522 programmed cell death Effects 0.000 description 2
- 230000001737 promoting effect Effects 0.000 description 2
- 238000000159 protein binding assay Methods 0.000 description 2
- 108020000494 protein-tyrosine phosphatase Proteins 0.000 description 2
- 239000012857 radioactive material Substances 0.000 description 2
- 230000009257 reactivity Effects 0.000 description 2
- 238000010188 recombinant method Methods 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 210000002345 respiratory system Anatomy 0.000 description 2
- 230000004043 responsiveness Effects 0.000 description 2
- 230000001177 retroviral effect Effects 0.000 description 2
- 150000003839 salts Chemical class 0.000 description 2
- 230000035939 shock Effects 0.000 description 2
- 238000002415 sodium dodecyl sulfate polyacrylamide gel electrophoresis Methods 0.000 description 2
- 230000009870 specific binding Effects 0.000 description 2
- 210000004988 splenocyte Anatomy 0.000 description 2
- 238000007920 subcutaneous administration Methods 0.000 description 2
- 235000000346 sugar Nutrition 0.000 description 2
- 239000006228 supernatant Substances 0.000 description 2
- 238000002198 surface plasmon resonance spectroscopy Methods 0.000 description 2
- 239000000725 suspension Substances 0.000 description 2
- 230000002194 synthesizing effect Effects 0.000 description 2
- 201000000596 systemic lupus erythematosus Diseases 0.000 description 2
- 229960002180 tetracycline Drugs 0.000 description 2
- 229930101283 tetracycline Natural products 0.000 description 2
- 235000019364 tetracycline Nutrition 0.000 description 2
- 150000003522 tetracyclines Chemical class 0.000 description 2
- RWQNBRDOKXIBIV-UHFFFAOYSA-N thymine Chemical compound CC1=CNC(=O)NC1=O RWQNBRDOKXIBIV-UHFFFAOYSA-N 0.000 description 2
- 230000000699 topical effect Effects 0.000 description 2
- 231100000331 toxic Toxicity 0.000 description 2
- 230000002588 toxic effect Effects 0.000 description 2
- 230000001988 toxicity Effects 0.000 description 2
- 231100000419 toxicity Toxicity 0.000 description 2
- 230000014621 translational initiation Effects 0.000 description 2
- 230000032258 transport Effects 0.000 description 2
- 241000701161 unidentified adenovirus Species 0.000 description 2
- 241001430294 unidentified retrovirus Species 0.000 description 2
- 229940035893 uracil Drugs 0.000 description 2
- 238000002255 vaccination Methods 0.000 description 2
- 239000004474 valine Substances 0.000 description 2
- 239000003981 vehicle Substances 0.000 description 2
- 230000029812 viral genome replication Effects 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- 210000005253 yeast cell Anatomy 0.000 description 2
- NNJPGOLRFBJNIW-HNNXBMFYSA-N (-)-demecolcine Chemical compound C1=C(OC)C(=O)C=C2[C@@H](NC)CCC3=CC(OC)=C(OC)C(OC)=C3C2=C1 NNJPGOLRFBJNIW-HNNXBMFYSA-N 0.000 description 1
- YMXHPSHLTSZXKH-RVBZMBCESA-N (2,5-dioxopyrrolidin-1-yl) 5-[(3as,4s,6ar)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]pentanoate Chemical compound C([C@H]1[C@H]2NC(=O)N[C@H]2CS1)CCCC(=O)ON1C(=O)CCC1=O YMXHPSHLTSZXKH-RVBZMBCESA-N 0.000 description 1
- NLEBIOOXCVAHBD-YHBSTRCHSA-N (2r,3r,4s,5s,6r)-2-[(2r,3s,4r,5r,6s)-6-dodecoxy-4,5-dihydroxy-2-(hydroxymethyl)oxan-3-yl]oxy-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound O[C@@H]1[C@@H](O)[C@@H](OCCCCCCCCCCCC)O[C@H](CO)[C@H]1O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 NLEBIOOXCVAHBD-YHBSTRCHSA-N 0.000 description 1
- RLTLDVPHEYUGFZ-QSVFAHTRSA-N (2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-1-[(2s)-2-[[(2s)-1-[(2s)-2-[[(2s)-2-amino-4-methylpentanoyl]amino]-3-hydroxypropanoyl]pyrrolidine-2-carbonyl]amino]-3-phenylpropanoyl]pyrrolidine-2-carbonyl]amino]-3-phenylpropanoyl]amino]-3-carboxypropanoyl]amino]-4-methyl Chemical compound CC(C)C[C@H](N)C(=O)N[C@@H](CO)C(=O)N1CCC[C@H]1C(=O)N[C@H](C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(O)=O)CC1=CC=CC=C1 RLTLDVPHEYUGFZ-QSVFAHTRSA-N 0.000 description 1
- MZOFCQQQCNRIBI-VMXHOPILSA-N (3s)-4-[[(2s)-1-[[(2s)-1-[[(1s)-1-carboxy-2-hydroxyethyl]amino]-4-methyl-1-oxopentan-2-yl]amino]-5-(diaminomethylideneamino)-1-oxopentan-2-yl]amino]-3-[[2-[[(2s)-2,6-diaminohexanoyl]amino]acetyl]amino]-4-oxobutanoic acid Chemical compound OC[C@@H](C(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@@H](N)CCCCN MZOFCQQQCNRIBI-VMXHOPILSA-N 0.000 description 1
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 1
- OWEGMIWEEQEYGQ-UHFFFAOYSA-N 100676-05-9 Natural products OC1C(O)C(O)C(CO)OC1OCC1C(O)C(O)C(O)C(OC2C(OC(O)C(O)C2O)CO)O1 OWEGMIWEEQEYGQ-UHFFFAOYSA-N 0.000 description 1
- RVCKCEDKBVEEHL-UHFFFAOYSA-N 2,3,4,5,6-pentachlorobenzyl alcohol Chemical compound OCC1=C(Cl)C(Cl)=C(Cl)C(Cl)=C1Cl RVCKCEDKBVEEHL-UHFFFAOYSA-N 0.000 description 1
- SGAKLDIYNFXTCK-UHFFFAOYSA-N 2-[(2,4-dioxo-1h-pyrimidin-5-yl)methylamino]acetic acid Chemical compound OC(=O)CNCC1=CNC(=O)NC1=O SGAKLDIYNFXTCK-UHFFFAOYSA-N 0.000 description 1
- IDOQDZANRZQBTP-UHFFFAOYSA-N 2-[2-(2,4,4-trimethylpentan-2-yl)phenoxy]ethanol Chemical compound CC(C)(C)CC(C)(C)C1=CC=CC=C1OCCO IDOQDZANRZQBTP-UHFFFAOYSA-N 0.000 description 1
- FBUTXZSKZCQABC-UHFFFAOYSA-N 2-amino-1-methyl-7h-purine-6-thione Chemical compound S=C1N(C)C(N)=NC2=C1NC=N2 FBUTXZSKZCQABC-UHFFFAOYSA-N 0.000 description 1
- UWAMFJWBEGHFQE-UHFFFAOYSA-N 2-amino-7-methyl-3h-purin-6-one;5-(methylaminomethyl)-1h-pyrimidine-2,4-dione Chemical compound CNCC1=CNC(=O)NC1=O.N1=C(N)NC(=O)C2=C1N=CN2C UWAMFJWBEGHFQE-UHFFFAOYSA-N 0.000 description 1
- ASJSAQIRZKANQN-CRCLSJGQSA-N 2-deoxy-D-ribose Chemical group OC[C@@H](O)[C@@H](O)CC=O ASJSAQIRZKANQN-CRCLSJGQSA-N 0.000 description 1
- GYJNVSAUBGJVLV-UHFFFAOYSA-N 3-(dimethylazaniumyl)propane-1-sulfonate Chemical compound CN(C)CCCS(O)(=O)=O GYJNVSAUBGJVLV-UHFFFAOYSA-N 0.000 description 1
- UMCMPZBLKLEWAF-BCTGSCMUSA-N 3-[(3-cholamidopropyl)dimethylammonio]propane-1-sulfonate Chemical compound C([C@H]1C[C@H]2O)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC(=O)NCCC[N+](C)(C)CCCS([O-])(=O)=O)C)[C@@]2(C)[C@@H](O)C1 UMCMPZBLKLEWAF-BCTGSCMUSA-N 0.000 description 1
- GUQQBLRVXOUDTN-XOHPMCGNSA-N 3-[dimethyl-[3-[[(4r)-4-[(3r,5s,7r,8r,9s,10s,12s,13r,14s,17r)-3,7,12-trihydroxy-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-17-yl]pentanoyl]amino]propyl]azaniumyl]-2-hydroxypropane-1-sulfonate Chemical compound C([C@H]1C[C@H]2O)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC(=O)NCCC[N+](C)(C)CC(O)CS([O-])(=O)=O)C)[C@@]2(C)[C@@H](O)C1 GUQQBLRVXOUDTN-XOHPMCGNSA-N 0.000 description 1
- KOLPWZCZXAMXKS-UHFFFAOYSA-N 3-methylcytosine Chemical compound CN1C(N)=CC=NC1=O KOLPWZCZXAMXKS-UHFFFAOYSA-N 0.000 description 1
- GJAKJCICANKRFD-UHFFFAOYSA-N 4-acetyl-4-amino-1,3-dihydropyrimidin-2-one Chemical compound CC(=O)C1(N)NC(=O)NC=C1 GJAKJCICANKRFD-UHFFFAOYSA-N 0.000 description 1
- TVZGACDUOSZQKY-LBPRGKRZSA-N 4-aminofolic acid Chemical compound C1=NC2=NC(N)=NC(N)=C2N=C1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 TVZGACDUOSZQKY-LBPRGKRZSA-N 0.000 description 1
- WPYRHVXCOQLYLY-UHFFFAOYSA-N 5-[(methoxyamino)methyl]-2-sulfanylidene-1h-pyrimidin-4-one Chemical compound CONCC1=CNC(=S)NC1=O WPYRHVXCOQLYLY-UHFFFAOYSA-N 0.000 description 1
- LQLQRFGHAALLLE-UHFFFAOYSA-N 5-bromouracil Chemical compound BrC1=CNC(=O)NC1=O LQLQRFGHAALLLE-UHFFFAOYSA-N 0.000 description 1
- VKLFQTYNHLDMDP-PNHWDRBUSA-N 5-carboxymethylaminomethyl-2-thiouridine Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C(=S)NC(=O)C(CNCC(O)=O)=C1 VKLFQTYNHLDMDP-PNHWDRBUSA-N 0.000 description 1
- ZFTBZKVVGZNMJR-UHFFFAOYSA-N 5-chlorouracil Chemical compound ClC1=CNC(=O)NC1=O ZFTBZKVVGZNMJR-UHFFFAOYSA-N 0.000 description 1
- KSNXJLQDQOIRIP-UHFFFAOYSA-N 5-iodouracil Chemical compound IC1=CNC(=O)NC1=O KSNXJLQDQOIRIP-UHFFFAOYSA-N 0.000 description 1
- KELXHQACBIUYSE-UHFFFAOYSA-N 5-methoxy-1h-pyrimidine-2,4-dione Chemical compound COC1=CNC(=O)NC1=O KELXHQACBIUYSE-UHFFFAOYSA-N 0.000 description 1
- LRSASMSXMSNRBT-UHFFFAOYSA-N 5-methylcytosine Chemical compound CC1=CNC(=O)N=C1N LRSASMSXMSNRBT-UHFFFAOYSA-N 0.000 description 1
- DCPSTSVLRXOYGS-UHFFFAOYSA-N 6-amino-1h-pyrimidine-2-thione Chemical compound NC1=CC=NC(S)=N1 DCPSTSVLRXOYGS-UHFFFAOYSA-N 0.000 description 1
- CJIJXIFQYOPWTF-UHFFFAOYSA-N 7-hydroxycoumarin Natural products O1C(=O)C=CC2=CC(O)=CC=C21 CJIJXIFQYOPWTF-UHFFFAOYSA-N 0.000 description 1
- MSSXOMSJDRHRMC-UHFFFAOYSA-N 9H-purine-2,6-diamine Chemical compound NC1=NC(N)=C2NC=NC2=N1 MSSXOMSJDRHRMC-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 108010022752 Acetylcholinesterase Proteins 0.000 description 1
- 102000012440 Acetylcholinesterase Human genes 0.000 description 1
- 206010069754 Acquired gene mutation Diseases 0.000 description 1
- 108010088751 Albumins Proteins 0.000 description 1
- 102000009027 Albumins Human genes 0.000 description 1
- 108700028369 Alleles Proteins 0.000 description 1
- 102100023635 Alpha-fetoprotein Human genes 0.000 description 1
- 101000669426 Aspergillus restrictus Ribonuclease mitogillin Proteins 0.000 description 1
- 208000032116 Autoimmune Experimental Encephalomyelitis Diseases 0.000 description 1
- 108090001008 Avidin Proteins 0.000 description 1
- 102000019260 B-Cell Antigen Receptors Human genes 0.000 description 1
- 108010012919 B-Cell Antigen Receptors Proteins 0.000 description 1
- 102100038080 B-cell receptor CD22 Human genes 0.000 description 1
- 108010045634 B7 Antigens Proteins 0.000 description 1
- 102000005738 B7 Antigens Human genes 0.000 description 1
- 231100000699 Bacterial toxin Toxicity 0.000 description 1
- DWRXFEITVBNRMK-UHFFFAOYSA-N Beta-D-1-Arabinofuranosylthymine Natural products O=C1NC(=O)C(C)=CN1C1C(O)C(O)C(CO)O1 DWRXFEITVBNRMK-UHFFFAOYSA-N 0.000 description 1
- 102100026189 Beta-galactosidase Human genes 0.000 description 1
- 108010039209 Blood Coagulation Factors Proteins 0.000 description 1
- 102000015081 Blood Coagulation Factors Human genes 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- 101100026251 Caenorhabditis elegans atf-2 gene Proteins 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- UXVMQQNJUSDDNG-UHFFFAOYSA-L Calcium chloride Chemical compound [Cl-].[Cl-].[Ca+2] UXVMQQNJUSDDNG-UHFFFAOYSA-L 0.000 description 1
- 240000001432 Calendula officinalis Species 0.000 description 1
- 235000005881 Calendula officinalis Nutrition 0.000 description 1
- 108010035563 Chloramphenicol O-acetyltransferase Proteins 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- 102100039496 Choline transporter-like protein 4 Human genes 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 102000008186 Collagen Human genes 0.000 description 1
- 108010035532 Collagen Proteins 0.000 description 1
- 108020004635 Complementary DNA Proteins 0.000 description 1
- 108020004394 Complementary RNA Proteins 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- 241000557626 Corvus corax Species 0.000 description 1
- 108010051219 Cre recombinase Proteins 0.000 description 1
- 239000004971 Cross linker Substances 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- 150000008574 D-amino acids Chemical class 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- KDXKERNSBIXSRK-RXMQYKEDSA-N D-lysine Chemical compound NCCCC[C@@H](N)C(O)=O KDXKERNSBIXSRK-RXMQYKEDSA-N 0.000 description 1
- 108020003215 DNA Probes Proteins 0.000 description 1
- 101710177611 DNA polymerase II large subunit Proteins 0.000 description 1
- 101710184669 DNA polymerase II small subunit Proteins 0.000 description 1
- 239000003298 DNA probe Substances 0.000 description 1
- 230000007018 DNA scission Effects 0.000 description 1
- 238000001712 DNA sequencing Methods 0.000 description 1
- 230000006820 DNA synthesis Effects 0.000 description 1
- 108010014303 DNA-directed DNA polymerase Proteins 0.000 description 1
- 102000016928 DNA-directed DNA polymerase Human genes 0.000 description 1
- 108090000626 DNA-directed RNA polymerases Proteins 0.000 description 1
- NNJPGOLRFBJNIW-UHFFFAOYSA-N Demecolcine Natural products C1=C(OC)C(=O)C=C2C(NC)CCC3=CC(OC)=C(OC)C(OC)=C3C2=C1 NNJPGOLRFBJNIW-UHFFFAOYSA-N 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- BWGNESOTFCXPMA-UHFFFAOYSA-N Dihydrogen disulfide Chemical compound SS BWGNESOTFCXPMA-UHFFFAOYSA-N 0.000 description 1
- 102000016607 Diphtheria Toxin Human genes 0.000 description 1
- 108010053187 Diphtheria Toxin Proteins 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- 238000008157 ELISA kit Methods 0.000 description 1
- 206010014596 Encephalitis Japanese B Diseases 0.000 description 1
- 241000792859 Enema Species 0.000 description 1
- 108010013369 Enteropeptidase Proteins 0.000 description 1
- 102100029727 Enteropeptidase Human genes 0.000 description 1
- YQYJSBFKSSDGFO-UHFFFAOYSA-N Epihygromycin Natural products OC1C(O)C(C(=O)C)OC1OC(C(=C1)O)=CC=C1C=C(C)C(=O)NC1C(O)C(O)C2OCOC2C1O YQYJSBFKSSDGFO-UHFFFAOYSA-N 0.000 description 1
- 241000702191 Escherichia virus P1 Species 0.000 description 1
- 241000206602 Eukaryota Species 0.000 description 1
- 108700024394 Exon Proteins 0.000 description 1
- 208000009386 Experimental Arthritis Diseases 0.000 description 1
- 108010046276 FLP recombinase Proteins 0.000 description 1
- 101150009958 FLT4 gene Proteins 0.000 description 1
- 108010074860 Factor Xa Proteins 0.000 description 1
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 1
- 241000233866 Fungi Species 0.000 description 1
- 108010001515 Galectin 4 Proteins 0.000 description 1
- 102100039556 Galectin-4 Human genes 0.000 description 1
- 241000287828 Gallus gallus Species 0.000 description 1
- 108700004714 Gelonium multiflorum GEL Proteins 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 108010024636 Glutathione Proteins 0.000 description 1
- 241000288105 Grus Species 0.000 description 1
- HVLSXIKZNLPZJJ-TXZCQADKSA-N HA peptide Chemical compound C([C@@H](C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](C(C)C)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](C)C(O)=O)NC(=O)[C@H]1N(CCC1)C(=O)[C@@H](N)CC=1C=CC(O)=CC=1)C1=CC=C(O)C=C1 HVLSXIKZNLPZJJ-TXZCQADKSA-N 0.000 description 1
- 208000031886 HIV Infections Diseases 0.000 description 1
- 102100029966 HLA class II histocompatibility antigen, DP alpha 1 chain Human genes 0.000 description 1
- 101710154606 Hemagglutinin Proteins 0.000 description 1
- 208000005176 Hepatitis C Diseases 0.000 description 1
- 208000007514 Herpes zoster Diseases 0.000 description 1
- 108010088652 Histocompatibility Antigens Class I Proteins 0.000 description 1
- 108700005087 Homeobox Genes Proteins 0.000 description 1
- 101000884305 Homo sapiens B-cell receptor CD22 Proteins 0.000 description 1
- 101000889282 Homo sapiens Choline transporter-like protein 4 Proteins 0.000 description 1
- 101000864089 Homo sapiens HLA class II histocompatibility antigen, DP alpha 1 chain Proteins 0.000 description 1
- 101000930802 Homo sapiens HLA class II histocompatibility antigen, DQ alpha 1 chain Proteins 0.000 description 1
- 101000968032 Homo sapiens HLA class II histocompatibility antigen, DR beta 3 chain Proteins 0.000 description 1
- 101100127397 Homo sapiens PRKCQ gene Proteins 0.000 description 1
- 101001117317 Homo sapiens Programmed cell death 1 ligand 1 Proteins 0.000 description 1
- 101000611936 Homo sapiens Programmed cell death protein 1 Proteins 0.000 description 1
- 101000934346 Homo sapiens T-cell surface antigen CD2 Proteins 0.000 description 1
- 101001050288 Homo sapiens Transcription factor Jun Proteins 0.000 description 1
- 102000003839 Human Proteins Human genes 0.000 description 1
- 108090000144 Human Proteins Proteins 0.000 description 1
- 241000701109 Human adenovirus 2 Species 0.000 description 1
- 241000701044 Human gammaherpesvirus 4 Species 0.000 description 1
- 241000713772 Human immunodeficiency virus 1 Species 0.000 description 1
- 241000713340 Human immunodeficiency virus 2 Species 0.000 description 1
- 102000004157 Hydrolases Human genes 0.000 description 1
- 108090000604 Hydrolases Proteins 0.000 description 1
- 102000009438 IgE Receptors Human genes 0.000 description 1
- 108010073816 IgE Receptors Proteins 0.000 description 1
- 108700005091 Immunoglobulin Genes Proteins 0.000 description 1
- 102000012745 Immunoglobulin Subunits Human genes 0.000 description 1
- 108010079585 Immunoglobulin Subunits Proteins 0.000 description 1
- 206010062016 Immunosuppression Diseases 0.000 description 1
- 208000026350 Inborn Genetic disease Diseases 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- 229930010555 Inosine Natural products 0.000 description 1
- UGQMRVRMYYASKQ-KQYNXXCUSA-N Inosine Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C2=NC=NC(O)=C2N=C1 UGQMRVRMYYASKQ-KQYNXXCUSA-N 0.000 description 1
- 102100022339 Integrin alpha-L Human genes 0.000 description 1
- 102100037850 Interferon gamma Human genes 0.000 description 1
- 108010074328 Interferon-gamma Proteins 0.000 description 1
- 108010002586 Interleukin-7 Proteins 0.000 description 1
- 108010002335 Interleukin-9 Proteins 0.000 description 1
- 108091092195 Intron Proteins 0.000 description 1
- 108010044467 Isoenzymes Proteins 0.000 description 1
- 201000005807 Japanese encephalitis Diseases 0.000 description 1
- 241000710842 Japanese encephalitis virus Species 0.000 description 1
- 235000019766 L-Lysine Nutrition 0.000 description 1
- ONIBWKKTOPOVIA-BYPYZUCNSA-N L-Proline Chemical compound OC(=O)[C@@H]1CCCN1 ONIBWKKTOPOVIA-BYPYZUCNSA-N 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 102100025584 Leukocyte immunoglobulin-like receptor subfamily B member 1 Human genes 0.000 description 1
- 101710145805 Leukocyte immunoglobulin-like receptor subfamily B member 3 Proteins 0.000 description 1
- 102000005482 Lipopolysaccharide Receptors Human genes 0.000 description 1
- 108010031801 Lipopolysaccharide Receptors Proteins 0.000 description 1
- 108060001084 Luciferase Proteins 0.000 description 1
- 239000005089 Luciferase Substances 0.000 description 1
- 108010064548 Lymphocyte Function-Associated Antigen-1 Proteins 0.000 description 1
- 102000043136 MAP kinase family Human genes 0.000 description 1
- 108091054455 MAP kinase family Proteins 0.000 description 1
- 241000829100 Macaca mulatta polyomavirus 1 Species 0.000 description 1
- GUBGYTABKSRVRQ-PICCSMPSSA-N Maltose Natural products O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@@H](CO)OC(O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-PICCSMPSSA-N 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 244000246386 Mentha pulegium Species 0.000 description 1
- 235000016257 Mentha pulegium Nutrition 0.000 description 1
- 235000004357 Mentha x piperita Nutrition 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- 101100519207 Mus musculus Pdcd1 gene Proteins 0.000 description 1
- 206010028372 Muscular weakness Diseases 0.000 description 1
- 102100038895 Myc proto-oncogene protein Human genes 0.000 description 1
- 101710135898 Myc proto-oncogene protein Proteins 0.000 description 1
- SGSSKEDGVONRGC-UHFFFAOYSA-N N(2)-methylguanine Chemical compound O=C1NC(NC)=NC2=C1N=CN2 SGSSKEDGVONRGC-UHFFFAOYSA-N 0.000 description 1
- NQTADLQHYWFPDB-UHFFFAOYSA-N N-Hydroxysuccinimide Chemical compound ON1C(=O)CCC1=O NQTADLQHYWFPDB-UHFFFAOYSA-N 0.000 description 1
- 230000004988 N-glycosylation Effects 0.000 description 1
- 206010029260 Neuroblastoma Diseases 0.000 description 1
- 102000008763 Neurofilament Proteins Human genes 0.000 description 1
- 108010088373 Neurofilament Proteins Proteins 0.000 description 1
- 208000025966 Neurological disease Diseases 0.000 description 1
- 102000007607 Non-Receptor Type 11 Protein Tyrosine Phosphatase Human genes 0.000 description 1
- 102100023172 Nuclear receptor subfamily 0 group B member 2 Human genes 0.000 description 1
- 108700020796 Oncogene Proteins 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 102000016979 Other receptors Human genes 0.000 description 1
- 101710093908 Outer capsid protein VP4 Proteins 0.000 description 1
- 101710135467 Outer capsid protein sigma-1 Proteins 0.000 description 1
- 101710160107 Outer membrane protein A Proteins 0.000 description 1
- 102000042846 PKC family Human genes 0.000 description 1
- 108091082203 PKC family Proteins 0.000 description 1
- 108090000526 Papain Proteins 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- 102000057297 Pepsin A Human genes 0.000 description 1
- 108090000284 Pepsin A Proteins 0.000 description 1
- 108091005804 Peptidases Proteins 0.000 description 1
- 102000035195 Peptidases Human genes 0.000 description 1
- 108010067902 Peptide Library Proteins 0.000 description 1
- 108010004729 Phycoerythrin Proteins 0.000 description 1
- 241000276498 Pollachius virens Species 0.000 description 1
- 229920002732 Polyanhydride Polymers 0.000 description 1
- 229920000954 Polyglycolide Polymers 0.000 description 1
- 229920001710 Polyorthoester Polymers 0.000 description 1
- 229920002685 Polyoxyl 35CastorOil Polymers 0.000 description 1
- 102100024216 Programmed cell death 1 ligand 1 Human genes 0.000 description 1
- 101710176177 Protein A56 Proteins 0.000 description 1
- 102000003923 Protein Kinase C Human genes 0.000 description 1
- 108090000315 Protein Kinase C Proteins 0.000 description 1
- 101710171617 Protein kinase C theta type Proteins 0.000 description 1
- 101000762949 Pseudomonas aeruginosa (strain ATCC 15692 / DSM 22644 / CIP 104116 / JCM 14847 / LMG 12228 / 1C / PRS 101 / PAO1) Exotoxin A Proteins 0.000 description 1
- 230000010799 Receptor Interactions Effects 0.000 description 1
- 206010038997 Retroviral infections Diseases 0.000 description 1
- 108091028664 Ribonucleotide Proteins 0.000 description 1
- 108090000829 Ribosome Inactivating Proteins Proteins 0.000 description 1
- 241000283984 Rodentia Species 0.000 description 1
- 102000014400 SH2 domains Human genes 0.000 description 1
- 108050003452 SH2 domains Proteins 0.000 description 1
- 101150036867 SYP gene Proteins 0.000 description 1
- 206010039491 Sarcoma Diseases 0.000 description 1
- 208000034189 Sclerosis Diseases 0.000 description 1
- 229920002684 Sepharose Polymers 0.000 description 1
- 101710150701 Serine/threonine-protein kinase C Proteins 0.000 description 1
- 101710084578 Short neurotoxin 1 Proteins 0.000 description 1
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 1
- DWAQJAXMDSEUJJ-UHFFFAOYSA-M Sodium bisulfite Chemical compound [Na+].OS([O-])=O DWAQJAXMDSEUJJ-UHFFFAOYSA-M 0.000 description 1
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 1
- 241000251131 Sphyrna Species 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 108700005078 Synthetic Genes Proteins 0.000 description 1
- 230000024932 T cell mediated immunity Effects 0.000 description 1
- 102100025237 T-cell surface antigen CD2 Human genes 0.000 description 1
- 241000223892 Tetrahymena Species 0.000 description 1
- RYYWUUFWQRZTIU-UHFFFAOYSA-N Thiophosphoric acid Chemical group OP(O)(S)=O RYYWUUFWQRZTIU-UHFFFAOYSA-N 0.000 description 1
- 108090000190 Thrombin Proteins 0.000 description 1
- 101710182532 Toxin a Proteins 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- 102100023132 Transcription factor Jun Human genes 0.000 description 1
- 101710150448 Transcriptional regulator Myc Proteins 0.000 description 1
- 206010052779 Transplant rejections Diseases 0.000 description 1
- 229920004929 Triton X-114 Polymers 0.000 description 1
- 108090000631 Trypsin Proteins 0.000 description 1
- 102000004142 Trypsin Human genes 0.000 description 1
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 1
- 102000000852 Tumor Necrosis Factor-alpha Human genes 0.000 description 1
- 102100021657 Tyrosine-protein phosphatase non-receptor type 6 Human genes 0.000 description 1
- 101710128901 Tyrosine-protein phosphatase non-receptor type 6 Proteins 0.000 description 1
- 102000016663 Vascular Endothelial Growth Factor Receptor-3 Human genes 0.000 description 1
- ZVNYJIZDIRKMBF-UHFFFAOYSA-N Vesnarinone Chemical compound C1=C(OC)C(OC)=CC=C1C(=O)N1CCN(C=2C=C3CCC(=O)NC3=CC=2)CC1 ZVNYJIZDIRKMBF-UHFFFAOYSA-N 0.000 description 1
- 108020000999 Viral RNA Proteins 0.000 description 1
- 208000036142 Viral infection Diseases 0.000 description 1
- 239000005862 Whey Substances 0.000 description 1
- 102000007544 Whey Proteins Human genes 0.000 description 1
- 108010046377 Whey Proteins Proteins 0.000 description 1
- 241000269370 Xenopus <genus> Species 0.000 description 1
- RJZZTNAWUTTWMJ-WYIOVZGUSA-N [(2r,3s,5s)-5-amino-2-[2-(4-methoxyphenyl)-2,2-diphenylethyl]-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-3-yl]oxyphosphonamidous acid Chemical compound C1=CC(OC)=CC=C1C(C=1C=CC=CC=1)(C=1C=CC=CC=1)C[C@@H]1[C@@H](OP(N)O)C[C@](N)(N2C(NC(=O)C(C)=C2)=O)O1 RJZZTNAWUTTWMJ-WYIOVZGUSA-N 0.000 description 1
- 230000005856 abnormality Effects 0.000 description 1
- 239000003070 absorption delaying agent Substances 0.000 description 1
- 229940022698 acetylcholinesterase Drugs 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 125000000641 acridinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3C=C12)* 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 230000010933 acylation Effects 0.000 description 1
- 238000005917 acylation reaction Methods 0.000 description 1
- 229960000643 adenine Drugs 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- 238000001261 affinity purification Methods 0.000 description 1
- 238000012867 alanine scanning Methods 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 230000029936 alkylation Effects 0.000 description 1
- 238000005804 alkylation reaction Methods 0.000 description 1
- 230000000172 allergic effect Effects 0.000 description 1
- 230000000735 allogeneic effect Effects 0.000 description 1
- 108010026331 alpha-Fetoproteins Proteins 0.000 description 1
- 108010001818 alpha-sarcin Proteins 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 125000003368 amide group Chemical group 0.000 description 1
- 229960003896 aminopterin Drugs 0.000 description 1
- 239000003708 ampul Substances 0.000 description 1
- 239000002269 analeptic agent Substances 0.000 description 1
- 230000033115 angiogenesis Effects 0.000 description 1
- 230000005809 anti-tumor immunity Effects 0.000 description 1
- 230000005875 antibody response Effects 0.000 description 1
- 210000000628 antibody-producing cell Anatomy 0.000 description 1
- 102000025171 antigen binding proteins Human genes 0.000 description 1
- 108091000831 antigen binding proteins Proteins 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- 230000001640 apoptogenic effect Effects 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- FIVPIPIDMRVLAY-UHFFFAOYSA-N aspergillin Natural products C1C2=CC=CC(O)C2N2C1(SS1)C(=O)N(C)C1(CO)C2=O FIVPIPIDMRVLAY-UHFFFAOYSA-N 0.000 description 1
- 238000012098 association analyses Methods 0.000 description 1
- 239000000305 astragalus gummifer gum Substances 0.000 description 1
- 210000001130 astrocyte Anatomy 0.000 description 1
- 208000010668 atopic eczema Diseases 0.000 description 1
- 230000001908 autoinhibitory effect Effects 0.000 description 1
- 239000000688 bacterial toxin Substances 0.000 description 1
- 230000003385 bacteriostatic effect Effects 0.000 description 1
- 230000004888 barrier function Effects 0.000 description 1
- DZBUGLKDJFMEHC-UHFFFAOYSA-N benzoquinolinylidene Chemical group C1=CC=CC2=CC3=CC=CC=C3N=C21 DZBUGLKDJFMEHC-UHFFFAOYSA-N 0.000 description 1
- 235000019445 benzyl alcohol Nutrition 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 108010005774 beta-Galactosidase Proteins 0.000 description 1
- IQFYYKKMVGJFEH-UHFFFAOYSA-N beta-L-thymidine Natural products O=C1NC(=O)C(C)=CN1C1OC(CO)C(O)C1 IQFYYKKMVGJFEH-UHFFFAOYSA-N 0.000 description 1
- 239000003833 bile salt Substances 0.000 description 1
- 229940093761 bile salts Drugs 0.000 description 1
- 238000004166 bioassay Methods 0.000 description 1
- 230000003115 biocidal effect Effects 0.000 description 1
- 229920002988 biodegradable polymer Polymers 0.000 description 1
- 239000004621 biodegradable polymer Substances 0.000 description 1
- 238000007413 biotinylation Methods 0.000 description 1
- 230000006287 biotinylation Effects 0.000 description 1
- 210000002459 blastocyst Anatomy 0.000 description 1
- 239000003114 blood coagulation factor Substances 0.000 description 1
- 210000004556 brain Anatomy 0.000 description 1
- 210000005013 brain tissue Anatomy 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- DQXBYHZEEUGOBF-UHFFFAOYSA-N but-3-enoic acid;ethene Chemical compound C=C.OC(=O)CC=C DQXBYHZEEUGOBF-UHFFFAOYSA-N 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 239000001110 calcium chloride Substances 0.000 description 1
- 229910001628 calcium chloride Inorganic materials 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 229910000389 calcium phosphate Inorganic materials 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 235000014633 carbohydrates Nutrition 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 229910002092 carbon dioxide Inorganic materials 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 230000020411 cell activation Effects 0.000 description 1
- 230000022131 cell cycle Effects 0.000 description 1
- 230000030833 cell death Effects 0.000 description 1
- 239000013592 cell lysate Substances 0.000 description 1
- 238000001516 cell proliferation assay Methods 0.000 description 1
- 230000033077 cellular process Effects 0.000 description 1
- 230000005754 cellular signaling Effects 0.000 description 1
- 239000002738 chelating agent Substances 0.000 description 1
- 235000013330 chicken meat Nutrition 0.000 description 1
- 229960004926 chlorobutanol Drugs 0.000 description 1
- 230000010428 chromatin condensation Effects 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 230000008711 chromosomal rearrangement Effects 0.000 description 1
- 238000013377 clone selection method Methods 0.000 description 1
- 238000012411 cloning technique Methods 0.000 description 1
- 238000000975 co-precipitation Methods 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 229920001436 collagen Polymers 0.000 description 1
- 229940075614 colloidal silicon dioxide Drugs 0.000 description 1
- 230000006957 competitive inhibition Effects 0.000 description 1
- 108010047295 complement receptors Proteins 0.000 description 1
- 102000006834 complement receptors Human genes 0.000 description 1
- 230000009918 complex formation Effects 0.000 description 1
- 238000012790 confirmation Methods 0.000 description 1
- 230000001268 conjugating effect Effects 0.000 description 1
- 238000010276 construction Methods 0.000 description 1
- 238000011109 contamination Methods 0.000 description 1
- 239000013068 control sample Substances 0.000 description 1
- 229910000366 copper(II) sulfate Inorganic materials 0.000 description 1
- JZCCFEFSEZPSOG-UHFFFAOYSA-L copper(II) sulfate pentahydrate Chemical compound O.O.O.O.O.[Cu+2].[O-]S([O-])(=O)=O JZCCFEFSEZPSOG-UHFFFAOYSA-L 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 244000038559 crop plants Species 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 108010057085 cytokine receptors Proteins 0.000 description 1
- 102000003675 cytokine receptors Human genes 0.000 description 1
- 210000000805 cytoplasm Anatomy 0.000 description 1
- 230000003436 cytoskeletal effect Effects 0.000 description 1
- 230000034994 death Effects 0.000 description 1
- 230000001934 delay Effects 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- 210000004443 dendritic cell Anatomy 0.000 description 1
- 230000008021 deposition Effects 0.000 description 1
- 210000004207 dermis Anatomy 0.000 description 1
- 239000003599 detergent Substances 0.000 description 1
- 239000008121 dextrose Substances 0.000 description 1
- 206010012601 diabetes mellitus Diseases 0.000 description 1
- 230000029087 digestion Effects 0.000 description 1
- UGMCXQCYOVCMTB-UHFFFAOYSA-K dihydroxy(stearato)aluminium Chemical compound CCCCCCCCCCCCCCCCCC(=O)O[Al](O)O UGMCXQCYOVCMTB-UHFFFAOYSA-K 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 229940000406 drug candidate Drugs 0.000 description 1
- 238000012377 drug delivery Methods 0.000 description 1
- 238000002101 electrospray ionisation tandem mass spectrometry Methods 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 206010014599 encephalitis Diseases 0.000 description 1
- 210000002889 endothelial cell Anatomy 0.000 description 1
- 239000007920 enema Substances 0.000 description 1
- 229940079360 enema for constipation Drugs 0.000 description 1
- 210000002615 epidermis Anatomy 0.000 description 1
- 210000000981 epithelium Anatomy 0.000 description 1
- 235000020776 essential amino acid Nutrition 0.000 description 1
- 239000003797 essential amino acid Substances 0.000 description 1
- RTZKZFJDLAIYFH-UHFFFAOYSA-N ether Substances CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 1
- 239000005038 ethylene vinyl acetate Substances 0.000 description 1
- 208000012997 experimental autoimmune encephalomyelitis Diseases 0.000 description 1
- 238000013401 experimental design Methods 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 230000001605 fetal effect Effects 0.000 description 1
- 210000002950 fibroblast Anatomy 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 235000019634 flavors Nutrition 0.000 description 1
- 238000000684 flow cytometry Methods 0.000 description 1
- MHMNJMPURVTYEJ-UHFFFAOYSA-N fluorescein-5-isothiocyanate Chemical compound O1C(=O)C2=CC(N=C=S)=CC=C2C21C1=CC=C(O)C=C1OC1=CC(O)=CC=C21 MHMNJMPURVTYEJ-UHFFFAOYSA-N 0.000 description 1
- 239000007850 fluorescent dye Substances 0.000 description 1
- 229960002949 fluorouracil Drugs 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 238000013467 fragmentation Methods 0.000 description 1
- 238000006062 fragmentation reaction Methods 0.000 description 1
- 238000004108 freeze drying Methods 0.000 description 1
- 238000010230 functional analysis Methods 0.000 description 1
- 230000002538 fungal effect Effects 0.000 description 1
- IECPWNUMDGFDKC-MZJAQBGESA-N fusidic acid Chemical class O[C@@H]([C@@H]12)C[C@H]3\C(=C(/CCC=C(C)C)C(O)=O)[C@@H](OC(C)=O)C[C@]3(C)[C@@]2(C)CC[C@@H]2[C@]1(C)CC[C@@H](O)[C@H]2C IECPWNUMDGFDKC-MZJAQBGESA-N 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 238000012239 gene modification Methods 0.000 description 1
- 208000016361 genetic disease Diseases 0.000 description 1
- 230000005017 genetic modification Effects 0.000 description 1
- 235000013617 genetically modified food Nutrition 0.000 description 1
- 238000003205 genotyping method Methods 0.000 description 1
- 230000000762 glandular Effects 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- FIVPIPIDMRVLAY-RBJBARPLSA-N gliotoxin Chemical compound C1C2=CC=C[C@H](O)[C@H]2N2[C@]1(SS1)C(=O)N(C)[C@@]1(CO)C2=O FIVPIPIDMRVLAY-RBJBARPLSA-N 0.000 description 1
- 229960003180 glutathione Drugs 0.000 description 1
- 125000005456 glyceride group Chemical group 0.000 description 1
- 210000000224 granular leucocyte Anatomy 0.000 description 1
- 210000003714 granulocyte Anatomy 0.000 description 1
- 230000012010 growth Effects 0.000 description 1
- 239000001963 growth medium Substances 0.000 description 1
- ZJYYHGLJYGJLLN-UHFFFAOYSA-N guanidinium thiocyanate Chemical compound SC#N.NC(N)=N ZJYYHGLJYGJLLN-UHFFFAOYSA-N 0.000 description 1
- 230000003394 haemopoietic effect Effects 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 229910001385 heavy metal Inorganic materials 0.000 description 1
- 239000000185 hemagglutinin Substances 0.000 description 1
- 210000003958 hematopoietic stem cell Anatomy 0.000 description 1
- 208000002672 hepatitis B Diseases 0.000 description 1
- 210000003494 hepatocyte Anatomy 0.000 description 1
- 238000004128 high performance liquid chromatography Methods 0.000 description 1
- 229940088597 hormone Drugs 0.000 description 1
- 239000005556 hormone Substances 0.000 description 1
- 235000001050 hortel pimenta Nutrition 0.000 description 1
- 102000048362 human PDCD1 Human genes 0.000 description 1
- 230000008348 humoral response Effects 0.000 description 1
- 230000002209 hydrophobic effect Effects 0.000 description 1
- 238000003384 imaging method Methods 0.000 description 1
- 230000003100 immobilizing effect Effects 0.000 description 1
- 230000007124 immune defense Effects 0.000 description 1
- 230000001900 immune effect Effects 0.000 description 1
- 230000036737 immune function Effects 0.000 description 1
- 230000032832 immune response to tumor cell Effects 0.000 description 1
- 208000026278 immune system disease Diseases 0.000 description 1
- 238000010166 immunofluorescence Methods 0.000 description 1
- 230000003259 immunoinhibitory effect Effects 0.000 description 1
- 239000002955 immunomodulating agent Substances 0.000 description 1
- 229940121354 immunomodulator Drugs 0.000 description 1
- 229940125721 immunosuppressive agent Drugs 0.000 description 1
- 230000002637 immunotoxin Effects 0.000 description 1
- 239000002596 immunotoxin Substances 0.000 description 1
- 229940051026 immunotoxin Drugs 0.000 description 1
- 231100000608 immunotoxin Toxicity 0.000 description 1
- 230000001771 impaired effect Effects 0.000 description 1
- 239000007943 implant Substances 0.000 description 1
- 238000000099 in vitro assay Methods 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 239000003701 inert diluent Substances 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 239000007972 injectable composition Substances 0.000 description 1
- 229940102223 injectable solution Drugs 0.000 description 1
- 229960003786 inosine Drugs 0.000 description 1
- 230000010354 integration Effects 0.000 description 1
- 238000012482 interaction analysis Methods 0.000 description 1
- 239000000138 intercalating agent Substances 0.000 description 1
- 230000031146 intracellular signal transduction Effects 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- 230000000622 irritating effect Effects 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 238000011813 knockout mouse model Methods 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 210000001821 langerhans cell Anatomy 0.000 description 1
- 239000010410 layer Substances 0.000 description 1
- 239000000787 lecithin Substances 0.000 description 1
- 235000010445 lecithin Nutrition 0.000 description 1
- 229940067606 lecithin Drugs 0.000 description 1
- 231100000636 lethal dose Toxicity 0.000 description 1
- 208000032839 leukemia Diseases 0.000 description 1
- 210000000265 leukocyte Anatomy 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- XMGQYMWWDOXHJM-UHFFFAOYSA-N limonene Chemical compound CC(=C)C1CCC(C)=CC1 XMGQYMWWDOXHJM-UHFFFAOYSA-N 0.000 description 1
- 150000002632 lipids Chemical class 0.000 description 1
- 238000001638 lipofection Methods 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 230000005923 long-lasting effect Effects 0.000 description 1
- 231100000053 low toxicity Toxicity 0.000 description 1
- HWYHZTIRURJOHG-UHFFFAOYSA-N luminol Chemical compound O=C1NNC(=O)C2=C1C(N)=CC=C2 HWYHZTIRURJOHG-UHFFFAOYSA-N 0.000 description 1
- 210000002751 lymph Anatomy 0.000 description 1
- 210000003563 lymphoid tissue Anatomy 0.000 description 1
- 229920002521 macromolecule Polymers 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- 201000004792 malaria Diseases 0.000 description 1
- 210000005075 mammary gland Anatomy 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 230000008774 maternal effect Effects 0.000 description 1
- 230000013011 mating Effects 0.000 description 1
- 230000010534 mechanism of action Effects 0.000 description 1
- 201000001441 melanoma Diseases 0.000 description 1
- 229960000485 methotrexate Drugs 0.000 description 1
- IZAGSTRIDUNNOY-UHFFFAOYSA-N methyl 2-[(2,4-dioxo-1h-pyrimidin-5-yl)oxy]acetate Chemical compound COC(=O)COC1=CNC(=O)NC1=O IZAGSTRIDUNNOY-UHFFFAOYSA-N 0.000 description 1
- 239000004292 methyl p-hydroxybenzoate Substances 0.000 description 1
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 1
- 229960001047 methyl salicylate Drugs 0.000 description 1
- 229960002216 methylparaben Drugs 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 230000003278 mimic effect Effects 0.000 description 1
- 230000000394 mitotic effect Effects 0.000 description 1
- 108091005601 modified peptides Proteins 0.000 description 1
- 230000009149 molecular binding Effects 0.000 description 1
- 238000001823 molecular biology technique Methods 0.000 description 1
- 239000000178 monomer Substances 0.000 description 1
- NKAAEMMYHLFEFN-UHFFFAOYSA-M monosodium tartrate Chemical compound [Na+].OC(=O)C(O)C(O)C([O-])=O NKAAEMMYHLFEFN-UHFFFAOYSA-M 0.000 description 1
- 210000000472 morula Anatomy 0.000 description 1
- 238000010172 mouse model Methods 0.000 description 1
- 239000002324 mouth wash Substances 0.000 description 1
- 229940051866 mouthwash Drugs 0.000 description 1
- 201000006417 multiple sclerosis Diseases 0.000 description 1
- 210000003205 muscle Anatomy 0.000 description 1
- 230000036473 myasthenia Effects 0.000 description 1
- 239000002636 mycotoxin Substances 0.000 description 1
- ZTLGJPIZUOVDMT-UHFFFAOYSA-N n,n-dichlorotriazin-4-amine Chemical compound ClN(Cl)C1=CC=NN=N1 ZTLGJPIZUOVDMT-UHFFFAOYSA-N 0.000 description 1
- MYYQSUKBWORIIV-UHFFFAOYSA-N n-(3-methylbutyl)-2-methylsulfanyl-7h-purin-6-amine Chemical compound CSC1=NC(NCCC(C)C)=C2NC=NC2=N1 MYYQSUKBWORIIV-UHFFFAOYSA-N 0.000 description 1
- 239000007922 nasal spray Substances 0.000 description 1
- 229940097496 nasal spray Drugs 0.000 description 1
- 239000006218 nasal suppository Substances 0.000 description 1
- 201000009240 nasopharyngitis Diseases 0.000 description 1
- 239000006199 nebulizer Substances 0.000 description 1
- 230000015240 negative regulation of T cell cytokine production Effects 0.000 description 1
- 210000005044 neurofilament Anatomy 0.000 description 1
- 210000002569 neuron Anatomy 0.000 description 1
- 230000004987 nonapoptotic effect Effects 0.000 description 1
- 239000002736 nonionic surfactant Substances 0.000 description 1
- 231100000956 nontoxicity Toxicity 0.000 description 1
- 239000000346 nonvolatile oil Substances 0.000 description 1
- 239000002777 nucleoside Substances 0.000 description 1
- 150000003833 nucleoside derivatives Chemical class 0.000 description 1
- 229920002113 octoxynol Polymers 0.000 description 1
- HEGSGKPQLMEBJL-RKQHYHRCSA-N octyl beta-D-glucopyranoside Chemical compound CCCCCCCCO[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O HEGSGKPQLMEBJL-RKQHYHRCSA-N 0.000 description 1
- 210000004248 oligodendroglia Anatomy 0.000 description 1
- 238000011275 oncology therapy Methods 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 239000007968 orange flavor Substances 0.000 description 1
- 230000002018 overexpression Effects 0.000 description 1
- 238000012261 overproduction Methods 0.000 description 1
- QUANRIQJNFHVEU-UHFFFAOYSA-N oxirane;propane-1,2,3-triol Chemical compound C1CO1.OCC(O)CO QUANRIQJNFHVEU-UHFFFAOYSA-N 0.000 description 1
- 229940094443 oxytocics prostaglandins Drugs 0.000 description 1
- 108010029340 p2Ca peptide Proteins 0.000 description 1
- 230000020477 pH reduction Effects 0.000 description 1
- 210000000496 pancreas Anatomy 0.000 description 1
- 229940055729 papain Drugs 0.000 description 1
- 235000019834 papain Nutrition 0.000 description 1
- 244000045947 parasite Species 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 230000032696 parturition Effects 0.000 description 1
- 230000008506 pathogenesis Effects 0.000 description 1
- 230000001575 pathological effect Effects 0.000 description 1
- 230000007170 pathology Effects 0.000 description 1
- 229940111202 pepsin Drugs 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- 230000002974 pharmacogenomic effect Effects 0.000 description 1
- 229960003742 phenol Drugs 0.000 description 1
- 150000004633 phorbol derivatives Chemical class 0.000 description 1
- 239000002644 phorbol ester Substances 0.000 description 1
- 125000002467 phosphate group Chemical group [H]OP(=O)(O[H])O[*] 0.000 description 1
- 150000008300 phosphoramidites Chemical class 0.000 description 1
- 238000003566 phosphorylation assay Methods 0.000 description 1
- 230000004962 physiological condition Effects 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 210000002381 plasma Anatomy 0.000 description 1
- 229920003023 plastic Polymers 0.000 description 1
- 239000004033 plastic Substances 0.000 description 1
- 229920001200 poly(ethylene-vinyl acetate) Polymers 0.000 description 1
- 238000002264 polyacrylamide gel electrophoresis Methods 0.000 description 1
- 239000008389 polyethoxylated castor oil Substances 0.000 description 1
- 239000004633 polyglycolic acid Substances 0.000 description 1
- 229920002704 polyhistidine Polymers 0.000 description 1
- 229930001119 polyketide Natural products 0.000 description 1
- 125000000830 polyketide group Chemical group 0.000 description 1
- 229920005862 polyol Polymers 0.000 description 1
- 150000003077 polyols Chemical class 0.000 description 1
- 229920001451 polypropylene glycol Polymers 0.000 description 1
- 230000027317 positive regulation of immune response Effects 0.000 description 1
- 230000003449 preventive effect Effects 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- 239000003380 propellant Substances 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 150000003180 prostaglandins Chemical class 0.000 description 1
- 230000004952 protein activity Effects 0.000 description 1
- 229940076376 protein agonist Drugs 0.000 description 1
- 229940076372 protein antagonist Drugs 0.000 description 1
- 230000004853 protein function Effects 0.000 description 1
- 238000001742 protein purification Methods 0.000 description 1
- 238000001243 protein synthesis Methods 0.000 description 1
- 230000004850 protein–protein interaction Effects 0.000 description 1
- 150000003212 purines Chemical class 0.000 description 1
- 150000003230 pyrimidines Chemical class 0.000 description 1
- 238000011002 quantification Methods 0.000 description 1
- 230000002285 radioactive effect Effects 0.000 description 1
- ZAHRKKWIAAJSAO-UHFFFAOYSA-N rapamycin Natural products COCC(O)C(=C/C(C)C(=O)CC(OC(=O)C1CCCCN1C(=O)C(=O)C2(O)OC(CC(OC)C(=CC=CC=CC(C)CC(C)C(=O)C)C)CCC2C)C(C)CC3CCC(O)C(C3)OC)C ZAHRKKWIAAJSAO-UHFFFAOYSA-N 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 230000007115 recruitment Effects 0.000 description 1
- 230000014493 regulation of gene expression Effects 0.000 description 1
- 230000022532 regulation of transcription, DNA-dependent Effects 0.000 description 1
- 230000003252 repetitive effect Effects 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- PYWVYCXTNDRMGF-UHFFFAOYSA-N rhodamine B Chemical compound [Cl-].C=12C=CC(=[N+](CC)CC)C=C2OC2=CC(N(CC)CC)=CC=C2C=1C1=CC=CC=C1C(O)=O PYWVYCXTNDRMGF-UHFFFAOYSA-N 0.000 description 1
- 239000002336 ribonucleotide Substances 0.000 description 1
- 238000007363 ring formation reaction Methods 0.000 description 1
- CVHZOJJKTDOEJC-UHFFFAOYSA-N saccharin Chemical compound C1=CC=C2C(=O)NS(=O)(=O)C2=C1 CVHZOJJKTDOEJC-UHFFFAOYSA-N 0.000 description 1
- 229940081974 saccharin Drugs 0.000 description 1
- 235000019204 saccharin Nutrition 0.000 description 1
- 239000000901 saccharin and its Na,K and Ca salt Substances 0.000 description 1
- 210000003296 saliva Anatomy 0.000 description 1
- 201000004409 schistosomiasis Diseases 0.000 description 1
- 238000003345 scintillation counting Methods 0.000 description 1
- 230000003248 secreting effect Effects 0.000 description 1
- 210000000582 semen Anatomy 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 238000012163 sequencing technique Methods 0.000 description 1
- 230000009131 signaling function Effects 0.000 description 1
- 229960002930 sirolimus Drugs 0.000 description 1
- QFJCIRLUMZQUOT-HPLJOQBZSA-N sirolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 QFJCIRLUMZQUOT-HPLJOQBZSA-N 0.000 description 1
- 208000017520 skin disease Diseases 0.000 description 1
- 239000001509 sodium citrate Substances 0.000 description 1
- NLJMYIDDQXHKNR-UHFFFAOYSA-K sodium citrate Chemical compound O.O.[Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O NLJMYIDDQXHKNR-UHFFFAOYSA-K 0.000 description 1
- 235000010267 sodium hydrogen sulphite Nutrition 0.000 description 1
- 239000007790 solid phase Substances 0.000 description 1
- 230000037439 somatic mutation Effects 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 125000006850 spacer group Chemical group 0.000 description 1
- 210000000278 spinal cord Anatomy 0.000 description 1
- 210000004989 spleen cell Anatomy 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 238000003153 stable transfection Methods 0.000 description 1
- 238000012409 standard PCR amplification Methods 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 210000000130 stem cell Anatomy 0.000 description 1
- 230000001954 sterilising effect Effects 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 150000005846 sugar alcohols Polymers 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 239000002511 suppository base Substances 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 230000004083 survival effect Effects 0.000 description 1
- 230000009747 swallowing Effects 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 230000009897 systematic effect Effects 0.000 description 1
- 208000001608 teratocarcinoma Diseases 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 231100001274 therapeutic index Toxicity 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- RTKIYNMVFMVABJ-UHFFFAOYSA-L thimerosal Chemical compound [Na+].CC[Hg]SC1=CC=CC=C1C([O-])=O RTKIYNMVFMVABJ-UHFFFAOYSA-L 0.000 description 1
- 229940033663 thimerosal Drugs 0.000 description 1
- 238000003161 three-hybrid assay Methods 0.000 description 1
- 229960004072 thrombin Drugs 0.000 description 1
- 230000001732 thrombotic effect Effects 0.000 description 1
- 229940104230 thymidine Drugs 0.000 description 1
- 238000011200 topical administration Methods 0.000 description 1
- 230000002103 transcriptional effect Effects 0.000 description 1
- 238000010361 transduction Methods 0.000 description 1
- 230000026683 transduction Effects 0.000 description 1
- 230000001131 transforming effect Effects 0.000 description 1
- 230000001052 transient effect Effects 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- PZYFJWVGRGEWGO-UHFFFAOYSA-N trisodium;hydrogen peroxide;trioxido(oxo)vanadium Chemical compound [Na+].[Na+].[Na+].OO.OO.OO.[O-][V]([O-])([O-])=O PZYFJWVGRGEWGO-UHFFFAOYSA-N 0.000 description 1
- 210000002993 trophoblast Anatomy 0.000 description 1
- 239000012588 trypsin Substances 0.000 description 1
- 201000008827 tuberculosis Diseases 0.000 description 1
- 238000003160 two-hybrid assay Methods 0.000 description 1
- 238000010396 two-hybrid screening Methods 0.000 description 1
- ORHBXUUXSCNDEV-UHFFFAOYSA-N umbelliferone Chemical compound C1=CC(=O)OC2=CC(O)=CC=C21 ORHBXUUXSCNDEV-UHFFFAOYSA-N 0.000 description 1
- HFTAFOQKODTIJY-UHFFFAOYSA-N umbelliferone Natural products Cc1cc2C=CC(=O)Oc2cc1OCC=CC(C)(C)O HFTAFOQKODTIJY-UHFFFAOYSA-N 0.000 description 1
- 238000001291 vacuum drying Methods 0.000 description 1
- 230000002792 vascular Effects 0.000 description 1
- 230000009385 viral infection Effects 0.000 description 1
- 239000008215 water for injection Substances 0.000 description 1
- 230000029663 wound healing Effects 0.000 description 1
Images
























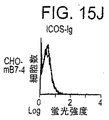

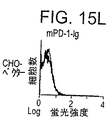










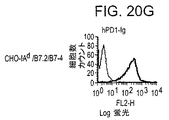







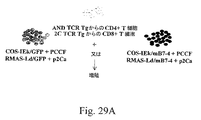







Classifications
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/5005—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells
- G01N33/5008—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells for testing or evaluating the effect of chemical or biological compounds, e.g. drugs, cosmetics
- G01N33/5044—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells for testing or evaluating the effect of chemical or biological compounds, e.g. drugs, cosmetics involving specific cell types
- G01N33/5047—Cells of the immune system
- G01N33/505—Cells of the immune system involving T-cells
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/5005—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells
- G01N33/5008—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells for testing or evaluating the effect of chemical or biological compounds, e.g. drugs, cosmetics
- G01N33/5044—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells for testing or evaluating the effect of chemical or biological compounds, e.g. drugs, cosmetics involving specific cell types
- G01N33/5047—Cells of the immune system
- G01N33/5052—Cells of the immune system involving B-cells
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/68—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving proteins, peptides or amino acids
- G01N33/6863—Cytokines, i.e. immune system proteins modifying a biological response such as cell growth proliferation or differentiation, e.g. TNF, CNF, GM-CSF, lymphotoxin, MIF or their receptors
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2333/00—Assays involving biological materials from specific organisms or of a specific nature
- G01N2333/435—Assays involving biological materials from specific organisms or of a specific nature from animals; from humans
- G01N2333/705—Assays involving receptors, cell surface antigens or cell surface determinants
- G01N2333/70503—Immunoglobulin superfamily, e.g. VCAMs, PECAM, LFA-3
- G01N2333/70532—B7 molecules, e.g. CD80, CD86
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2333/00—Assays involving biological materials from specific organisms or of a specific nature
- G01N2333/90—Enzymes; Proenzymes
- G01N2333/91—Transferases (2.)
- G01N2333/912—Transferases (2.) transferring phosphorus containing groups, e.g. kinases (2.7)
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Immunology (AREA)
- Engineering & Computer Science (AREA)
- Biomedical Technology (AREA)
- Molecular Biology (AREA)
- Cell Biology (AREA)
- Chemical & Material Sciences (AREA)
- Hematology (AREA)
- Urology & Nephrology (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Analytical Chemistry (AREA)
- Microbiology (AREA)
- Biotechnology (AREA)
- Food Science & Technology (AREA)
- Physics & Mathematics (AREA)
- Biochemistry (AREA)
- General Physics & Mathematics (AREA)
- Pathology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Tropical Medicine & Parasitology (AREA)
- Toxicology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Veterinary Medicine (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Peptides Or Proteins (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Investigating Or Analysing Biological Materials (AREA)
Abstract
Description
本明細書に開示される発明は、ナショナル インスティテュート オブ ヘルスにより授与されるAI39671、AI44690、CA84500、およびAI41584下にサポートされた。合衆国政府はそれゆえ、本発明の所定の権利を有し得る。
【0002】
本発明は、2001年4月2日に提出されたU.S.S.N.60/281,064について優先権を主張する。この出願は本明細書中に、その全体が引用により組み込まれる。
T細胞が外来タンパク質に対して反応するためには、2つのシグナルが抗原提示細胞(APC)により静止Tリンパ球に提供されなければならない(Jenkins,M.およびSchuwartz,R.(1987)J.Exp.Med.165:302−319;Mueller,D.L.ら(1990)J.Immunol.144:3701−3709)。免疫反応に対する特異性を賦与する第一シグナルは、主要組織適合複合体(MHC)との関連で提示される外来抗原性ペプチドを認識した後にT細胞レセプター(TCR)を経て変換される。副刺激と称される第二のシグナルは、T細胞を増殖させ、機能的にする(Lenschowら、(1996)Annu.Rev.Immunol.14:233)。副刺激は、抗原特異性でも、MHC制限性でもなく、APCにより発現される1以上の別個の細胞表面分子により提供させると考えられる(Jenkins,M.K.ら(1988)J.Immunol.140:3324−3330;Linsley,P.S.ら(1991)J.Exp.Med.173:721−730;Gimmi,C.D.ら1991 Proc.Natl.Acad.Sci.USA 88;6575−6579;Young,J.W.ら(1992)J.Clin.Invest.90:229−237;Koulova,L.ら(1991)J.Exp.Med.173:759−762;Reiser.H.ら(1992)Proc.Natl.Acad.Sci.USA 89:271−275;van−Seventer,G.A.ら(1990)J.Immunol.144:4579−4586;LaSalle,J.M.ら(1991)J.Immunol.147:774−80;Dustin,M.I.ら(1989)J.Exp.Med.169:503;Amitage,R.J.ら(1992)Nature357:80−82;Liu,Y.ら(1992)J.Exp.Med.175:437−445)。
【0003】
APC上で発現されるCD80(B7−1)およびCD86(B7−2)タンパク質は重要な副刺激分子である(Freemanら(1991)J.Exp.Med.174:625;Freemanら(1989)J.Immunol.143:2714;Azumaら(1993)Nature366:76;Freemanら(1993)Science262:909)。B7−2は一次免疫反応中に主な役割を果たすと考えられるが、免疫反応の過程において後の方でアップレギュレートされるB7−1は、一次T細胞反応の延長または二次T細胞反応の副刺激において重要であり得る(Bluestone(1995)Immunity2:555)。
【0004】
B7−1およびB7−2が結合する1つのレセプターであるCD28は本質的に静止T細胞上で発現され、活性化後発現が増大する。T細胞レセプターによるシグナリング後、CD28のライゲーションおよび副刺激シグナルの変換はT細胞を増大させ、IL−2を分泌させる(Linsley,P.S.ら(1991)J.Exp.Med.173:721−730;Gimmi,C.D.ら(1991)Proc.Natl.Acad.Sci.USA88:6575−6579;June,C.H.ら(1990)Immunol.Today11:211−6;Harding,F.A.ら(1992)Nature356:607−609)。CTLA4(CD152)と称する二次レセプターはCD28と相同性であるが、静止T細胞上で発現されず、T細胞活性化後に現れる(Brunet,,J.ら(1987)Nature328:267−270)。CTLA4はT細胞反応の負の制御において重要であるように思われる(Waterhouseら(1995)Science270:985)。CTLA4のブロックは、抑制シグナルを除去することが見いだされ、一方CTLA4の凝集はT細胞反応をダウンレギュレートする抑制シグナルを提供することが見いだされている(AllisonおよびKrummel(1995)Science270:932)。B7分子はCD28についてよりもCTLA4についてより高い親和性を有し(Linsley,P.S.ら(1991)J.Exp.Med.174:561−569)、B7−1およびB7−2はCTLA4分子の別個の領域に結合し、CTLA4に対する結合の異なる反応の仕組みを有することが判明している(Linsleyら(1994)Immunity1:793)。CD28およびCTLA4に関連する新規分子であるICOSが同定され、新規B7ファミリーのメンバーであるそのリガンドを有するために(Aicher,Aら(2000)J.Immunol.30:1040−7;Brodie D.ら(2000)Curr.Biol.10:333−6;Ling V.ら(2000)J.Immunol.164:1653−7;Yoshinaga S.K.ら(1999)Nature402:827−32))、IL−10の産生において重要であると考えられる(Hutloffら(1999)Nature397:263;WO98/38216)。T細胞がさらなる副刺激シグナルを受容することなくT細胞レセプターによってのみ刺激されるならば、これらは非反応性になるか、アネルギーになるか、または死に、その結果、免疫反応のダウンレギュレーションに至る。
【0005】
B7:CD28/CTLA4副刺激経路の重要性は、インビトロといくつかのインビボモデルシステムにおいて証明されている。この副刺激経路をブロックすると、ネズミおよびヒト系において抗原特異的な耐性が発生する(Harding,F.A.ら(1992)Nature356:607−609;Lenschow,D.J.ら(1992)Science257:789−792;Turka,L.A.ら(1992)Proc.Natl.Acad.Sci.USA89:11102−11105;Gimmi,C.D.ら(1993)Proc.Natl.Acad.Sci.USA90:6586−6590;Boussiotis,V.ら(1993)J.Exp.Med.178:1753−1763)。反対に、B7ネガティブなネズミ腫瘍細胞によるB7の発現は、腫瘍拒絶および腫瘍攻撃に対する長期間持続する防御を伴うT細胞により媒介される特異的免疫を誘発する(Chen,L.ら(1992)Cell71:1093−1102;Townsend,S.E.およびAllison,J.P.(1993)Science259:368−370;Baskar,S.ら(1993)Proc.Natl.Acad.Sci90:5687−5690)。従って、副刺激経路の操作は、ヒトにおける免疫反応を刺激または抑制するための非常に大きな可能性をもたらす。
【0006】
発明の概要
本発明は、少なくとも一部は、PD−1が、抗原提示細胞上に発現されるB7−4分子のレセプターであるという発見に基づく。PD−1はCTLA4と同様に負のシグナルを免疫細胞に伝達する。B4−7分子は抗原提示細胞の表面上に発現され、副刺激シグナルを免疫細胞に提供し、これらが結合する分子に応じて、免疫細胞に下方調節シグナルを伝達することができる。従って、PD−1、B7−4および/または、B7−4とPD−1間の相互作用の結果、免疫応答が調節される。
【0007】
従って、一態様において、本発明は免疫反応を調節する方法であって、免疫細胞をPD−1によるシグナリングを調節する化学物質と接触させ、これにより免疫反応を調節することを含む方法に関する。免疫細胞は、例えば、T細胞、B細胞、または骨髄細胞であってよい。一態様において、接触ステップはインビボで起こる。もう一つ別の態様において、接触ステップはインビトロで起こる。
1つの具体例では、免疫反応がダウンレギュレートされる。これは、例えば、PD−1を認識する活性化抗体、抑制性レセプターに結合するB7−4形態、およびPD−1に結合する小分子から成る群から選択される試薬を用いてPD−1を経るシグナリングを刺激することにより生じてよい。
1つの具体例において、PD−1を経るシグナリングの刺激により、免疫細胞において、免疫細胞においてアネルギーが誘導される。
該方法は、免疫細胞を、免疫反応をダウンレギュレートするさらなる試薬と接触させることをさらに含んでよい。
【0008】
さらに他の具体例においては、免疫反応がPD−1を経るシグナリングの阻害によりアップレギュレートされる。PD−1を経するシグナリングは、PD−1を認識するブロッキング抗体、B7−4の非活性化形態、B7−4を認識する抗体、およびPD−1の溶解形態から成る群から選択される試薬を用いて阻害されてよい。
【0009】
他の態様では、本発明は、免疫細胞における抑制性レセプターとのB7−4の相互作用を調節する方法であって、免疫細胞上の抑制性レセプターとのB7−4の相互作用が調節されるように、B7−4を発現する抗原提示細胞と、B7−4形態、PD−1形態、B7−4とPD−1の相互作用を調節する試薬から成る群から選択される試薬とを接触させることを含む方法を含む。免疫細胞は、例えば、T細胞、B細胞、または骨髄細胞であってよい。1つの具体例において、接触させるステップはインビトロで行う。他の具体例では、接触させるステップはインビボで行う。本方法は、免疫細胞または抗原提示細胞を、免疫反応を調節するさらなる試薬と接触させることをさらに含んでよい。
【0010】
他の態様において、本発明は、非アポトーシス機構により免疫細胞の活性化を阻害する方法であって、免疫細胞の活性化が阻害されるように、免疫細胞におけるPD−1の活性または発現を増すことを含む方法を含む。
他の態様において、本発明は、免疫細胞においてPD−1を経るシグナリングを阻害する抗原および試薬を含むワクチンを含む。
他の態様において、本発明は、免疫細胞におけるPD−1を経るシグナリングを促進する抗原および試薬を含む組成物を含む。
【0011】
他の態様において、本発明は、免疫反応のアップレギュレーションから利益を得るであろう疾患を有する対象を治療する方法であって、免疫反応のアップレギュレーションから利益を得るであろう疾患が治療されるように、対象の免疫細胞におけるPD−1を経るシグナリングを阻害する試薬を投与することを含む方法を含む。疾患は、例えば、腫瘍、神経学的疾患、または免疫抑制疾患であってよい。1つの具体例において、PD−1を経るシグナリングを阻害する試薬には、PD−1またはB7−4の溶解形態が含まれる。1つの具体例では、方法は、免疫反応をアップレギュレートする第2の試薬を対象に投与することをさらに含む。
【0012】
他の態様においては、発明は、免疫反応のダウンレギュレーションから利益を得るであろう疾患を有する対象を治療する方法であって、免疫反応のダウンレギュレーションから利益を得るであろう疾患が治療されるように、対象の免疫細胞においてPD−1を経するシグナリングを刺激する試薬を投与することを含む方法を含む。疾患は、例えば、移植、アレルギー、または自己免疫疾患であってよい。1つの具体例においては、PD−1を経るシグナリングを刺激する試薬は、PD−1を経るシグナリングを刺激する抗体、二重特異性抗体、または溶解B7−4である。方法は、免疫反応をダウンレギュレートする第2の試薬を対象に投与することをさらに含んでよい。
【0013】
他の態様では、本発明は、B7−4またはPD−1の活性を調節する化合物をスクリーニングするための細胞に基くアッセイであって、B7−4標的分子またはPD−1標的分子を発現している細胞を試験化合物と接触させて、試験化合物がB7−4またはPD−1標的分子の活性を調節する能力を決定することを含むアッセイを含む。
【0014】
さらに他の態様において、発明は、B7−4またはPD−1の標的分子に対する結合を調節する化合物をスクリーニングするためのセルフリーのアッセイであって、B7−4またはPD−1タンパク質または生物学的活性なその部分を試験化合物と接触させること、および試験化合物がB7−4またはPD−1タンパク質または生物学的に活性なその部分と結合する能力を決定することを含むアッセイを含む。
【0015】
図面の簡単な説明
図1は、ヒトの分泌B7−4、B7−4Sをエンコードするヌクレオチド配列(配列番号1)を表す。
図2は、ヒトB7−4、B7−4Mをエンコードするヌクレオチド配列(配列番号3)を表す。
図3は、ヒトB7−4Sのアミノ酸配列(配列番号2)を示し、シグナル、IgV、IgC、および疎水性末端ドメインを示す。
図4は、ヒトB7−4Mのアミノ酸配列(配列番号4)を示し、シグナル、IgV、IgC、および膜貫通および細胞質を示す。
図5A−5Bは、ネズミのB7−4のヌクレオチド配列(配列番号22)を示す。
図6は、ネズミのB7−4のアミノ酸配列(配列番号23)を示す。
図7は、ヒトB7−MおよびネズミB7−4アミノ酸配列のアライメントを示す(それぞれ、配列番号4および23)。
図8は、B7−4M−トランスフェクトCOS細胞によるCD28Ig、CTLA4−Ig、およびコントロールIgの結合のFACS分析の結果を示す。
図9は、B7−4M−トランスフェクトCOS細胞によるIgGおよびネズミICOS−ヒス融合タンパク質の結合のFACS分析の結果を示す。
図10は、B7−4M−トランスフェクトCOS細胞に対するIgM、BB1および133抗体の結合の、FACS分析の結果を示す。
図11は、B7−4M(292)でトランスフェクトしたCOS細胞がT細胞の増殖を副刺激することができることを示す。
図12は、B7−4M(292)でトランスフェクトしたCOS細胞がT細胞の増殖を副刺激することができることを示す。
図13A−13Dは、B7−4MトランスフェクトCOS細胞へのPD−1の結合を示す。
図14A−14Fは、追加されたPD−1の、およびFlt4でないの、PD−1のB7−4トランスフェクトCOS細胞への結合を完了する能力を示す。
図15A−15Lは、フローサイトメトリーにより決定される、B7−4トランスフェクトCHO細胞へ結合するPD−1の能力を示す。
図16は、BIACORE分析により決定される、B7−4トランスフェクトCHO細胞へ結合するPD−1の能力を示す。
図17は、T細胞へネガティブシグナルを伝達するB7−4Mの能力を示す。
図18A−18Cは、B7−4の存在下に刺激されたヒトT細胞におけるT細胞増殖およびサイトカイン産生の阻害を示す。
図19A−19Bは、CD28副刺激の存在下でのT細胞レセプター/B7−4の活性化がT細胞の増殖の阻害を生じることを示す。
図20A−20Iは、B7−4を発現しているCHO細胞に対するPD−1の結合を示す。
図21A−21Dは、CD28シグナルの阻害におけるB7−4の活性を示す。
図22A−22Dは、サイトカインELISAにより測定される、PD−1:B7−4経路によるサイトカイン産生の阻害を示す。
図23A−23Cは、サイトカインmRNAレベルにより測定される、PD−1:B7−4経路によるサイトカイン産生の阻害を示す。
図24A−24Cは、PD−1:B7−4経路の活性のメカニズムが細胞周期の停止であることを示す。
図25A−25Bは、B7−4およびPD−1の間の相互作用を阻害する、B7−4に対する抗体の能力を示す。
図26はPD−1に対する抗体がB7−4とPD−1間の相互作用を阻害する能力を表す。
図27は実験的自己免疫脳脊髄炎のネズミモデルにおいて溶解性B7−4Fcが疾患を悪化させる能力を表す。
図28A〜28Bは、有糸細胞分裂に関するPD−1:B7−4相互作用の影響を表す。T細胞をCSFEで標識し、ctrl.FcまたはmB7−4Fcbビーズで刺激した。表示した時点で、FACS分析を行った。Live−gated事象を図示する。図28A:CD4+T細胞。図28B:CD8+T細胞。PD−1:B7−4相互用の結果、CD4+およびCD8+T細胞の両方の有糸分裂が減少する。
図29Aから29CはPD−1:B7−4相互作用によるCD4+およびCD8+T細胞の両方の阻害を表す。図29Aは細胞系および実験デザインを概略的に表す。安定な抗原提示細胞(APC)系を、レトロウイルステクノロジーを用いてGFPまたはmB7−4/GFPを発現するように操作する。TCRトランスジェニック(Tg)マウスから得られる5×104の精製LN T細胞をAPC;ペプチドで2、2〜3、3、または4日間刺激した。CD4+T細胞を含む実験に関して、APC:T細胞比は10μM PCCFペプチドで1:10であった。CD8+T細胞を含む実験に関して、APC:T細胞比は1mMp2Caペプチドで1:1であった。図29BはPD−1:B7−4相互作用によるCD4+T細胞の増殖の阻害を表す。図29CはPD−1:B7−4相互作用によるCD8+T細胞の増殖の阻害を表す。
図30A〜30Bは、PD−1:B7−4相互作用によるCD4+(CD8+でない)T細胞増殖の阻害を克服するための副刺激の能力を表す。図30A:CD4+T細胞 図30B:CD8+T細胞。
図31A〜31BはPD−1シグナリング経路において関与するタンパク質を同定するために用いられるスクリーニングアッセイを概略的に表す。図31Aはアッセイのステップの概略図を表す。図31BはヒトPD−1のフラグメント(配列番号24)、マウスPD−1のフラグメント(配列番号25)およびアッセイにおいて用いられるペプチドの配列を表す。アッセイにおいて用いられるペプチドはITIMペプチド:PD−1_Py1(配列番号26);PD−1_Y1F(配列番号27);PD−1_Y1(配列番号28);PD−1_Py2(配列番号29);PD−1_Y2F(配列番号30);PD−1_Y2(配列番号:31);および他のペプチド:PD−1_K212_(配列番号32);PD−1_K212D(配列番号33);PD−1_K335(配列番号34);PD−1_K335D(配列番号35);PD−1_Ctail1(配列番号36);およびPD−1_Ctail2(配列番号37)であった。
図32は、PDL1.Ig融合タンパク質の存在下または不在下での、刺激されていない、または5分、30分、または90分間刺激されたヒトT細胞の分画細胞抽出物を含んでいるウェスタンブロットの写真である。ブロットをリン酸化ERK1および2に特異的な抗体でプローブした。
図33はB7−4.Ig融合タンパク質の存在(+)または不在(−)下での、刺激されていない、または5分、30分、90分、または2時間刺激されたヒトT細胞の分画細胞抽出物を含んでいるウェスタンブロットの写真である。ブロットは、リン酸化PKC−θ(抗−ホスホ−PKC−θ)を特異的に検出する、リン酸化PKC−θに特異的な抗体でプローブした。ゲルを細長く切り、リン酸化および非リン酸化PKC−θ(抗−PKC−θ)の両方を認識する抗体でプローブした。
【0016】
発明の詳細な記載
すでに特徴づけられたBリンパ球活性化抗原、たとえばB7−1およびB7−2に加えて、免疫細胞の副刺激を調節する抗原提示細胞の表面上に他の抗原が存在する。例えば、B7−4ポリペプチドは、ケラチン生成細胞および胎盤cDNAライブラリーから単離されている。B7−4は、T細胞を副刺激または阻害することも見出されている。本発明は、B7−4に対するレセプターとしてPD−1を同定する。
【0017】
免疫細胞は、活性化シグナルを伝達するレセプターを有する。たとえば、T細胞はT細胞レセプターおよびCD3複合体を有し、B細胞はB細胞レセプターを有し、脊髄細胞はFcレセプターを有する。加えて、免疫細胞は、副刺激シグナルまたはレセプター媒介性シグナリングを阻害するシグナルを伝達するレセプターを提供するシグナルを伝達するレセプターを有する。たとえば、CD28はT細胞に副刺激シグナルを伝達する。T細胞レセプターのライゲーション後、CD28のライゲーションの結果、たとえばIL−2rα、IL−2rβ、およびIL−2rγレセプターのアップレギュレーションにより特徴づけられる副刺激シグナルが生じ、IL−2メッセンジャーRNAの転写が増大し、サイトカイン遺伝子(IL−2、IFN−γ、GM−CSF、およびTNF−αを含む)の発現が増大する。副刺激シグナルの伝達により、細胞が細胞サイクルを進行し、こうしてT細胞増殖が増大する(Greenfieldら(1998)Crit.Rev.Immunol.18:389)。B7ファミリー分子、たとえばB7−4による副刺激シグナルを細胞に伝達するT細胞上のレセプターの結合(たとえば、サイトカイン分泌および/またはT細胞の増殖に至る副刺激レセプターのライゲーション)の結果、副刺激が生じる。従って、B7ファミリー分子、たとえばB7−4と免疫細胞上の副刺激シグナルを伝達するレセプター間の相互作用の阻害の結果、免疫応答および/または特異的非同調性(免疫細胞アネルギーと称する)がダウンモジュレートされる。この相互作用の阻害は、抗CD28Fabフラグメント、B7−1、B7−2および/またはB7−4に対する抗体、またはB7ファミリーメンバー分子が競合的インヒビターとして結合出来るレセプターの溶解形態を用いることにより達成できる。
【0018】
副刺激分子と結合する阻害レセプターはさらに免疫細胞上でも同定された。CTLA4の活性化は、たとえば負のシグナルをT細胞に伝達する。CTLA4の関与はIL−2産生を阻害し、細胞サイクル阻止を誘発できる(KrummelおよびAllison(1996)J.Exp.Med.183:2533)。加えて、CTLA4が欠失したマウスはリンパ組織増殖疾患を発症する(Tivolら(1995)Immunity3:541;Waterhouseら(1995)Science270:985)。CTLA4の抗体でのブロックは、阻害シグナルをブロックし、一方CTLA4の抗体との凝集は阻害シグナルを伝達する。従って、副刺激分子が結合するレセプターに応じて(すなわち、副刺激レセプター、たとえば、CD28または阻害レセプター、たとえばCTLA4)、B7−4を含むある種のB7分子はT細胞副刺激または阻害を促進できる。
PD−1は分子のイムノグロブリンファミリーのメンバーである(Ishidaら(1992)EMBO J.11:3887;Shinoharaら(1994)Genomics23:704)。PD−1はプログラムされた細胞死のモジュレーターを同定するためにデザインされたサブトラクションクローニングに基づく方法を用いてすでに同定された(Ishidaら(1992)EMBO J.11:3887−95;Woroniczら(1995)Curr.Top.Microbiol.Immunol.200:137)。PD−1はリンパ球生存において、たとえばクローン選択中に役割を果たすと考えられる(Honjo(1992)Science258:591;Agataら(1996)Int.Immunology.8:765;Nishimuraら(1996)Int.Immunology.8:773)。PD−1はB細胞応答のレギュレーターとしても関与した(Nishimura(1998)Int.Immunology.10:1563)。T細胞上にのみ見いだされるCTLA4と違って、PD−1はB細胞および骨髄細胞上においても見いだされる。
【0019】
PD−1がB7−4と結合するという事実から、PD−1はCTLA4に関する阻害レセプターのファミリーに帰属される。副刺激レセプターの関与(活性化を生じるための)の結果、免疫細胞において副刺激シグナルが生じ、一方阻害レセプター、例えばCTL4またはPD−1の関与(例えば、架橋または凝集による)は、免疫細胞における阻害シグナルの伝達に至り、その結果、免疫細胞応答および/または免疫細胞アネルギーがダウンモジュレーションされる。阻害シグナルの伝達は免疫細胞応答のダウンモジュレーションに至り(その結果として全体的な免疫応答がダウンモジュレートされる)、免疫細胞における阻害シグナルの(例えば、PD−1に対する非活性化抗体を用いることによる)防止は、免疫細胞応答のアップモジュレーションに至る(その結果、免疫応答がアップモジュレートされる)。
本発明は、PD−1の活性および/または発現;PD−1とその天然のリガンド間の相互作用の調節に有用な化学物質、およびPD−1とその天然リガンド、例えばPD−1としても知られるB7−4間の相互作用の調節により免疫応答を調節する化学物質を利用する。これらの方法において使用される調節剤の例を次にさらに記載する。
【0020】
B7−4およびPD−1核酸およびポリペプチド分子
一態様において、PD−1の活性および/または発現の調節に有用な調節剤はB7−4および/またはPD−1核酸分子、好ましくはヒトB7−4および/またはPD−1核酸分子である。
一態様において、本発明の単離された核酸分子は、真核生物B7−4またはPD−1ポリペプチドをエンコードする。分子のB7−4ファミリーは、シグナルドメイン、IgVドメインおよびIgCドメインを含む多くの保存されたドメインを共有する。IgVドメインおよびIgCドメインは当該分野で認知されるIgスーパーファミリーメンバードメインである。これらのドメインは、Igフォールドと呼ばれる独立したフォールディングパターンを有する構造単位に対応する。Igフォールドは2つのβシートのサンドイッチからなり、それぞれ、全部ではないがほとんどのドメインにおいて2シート間に保存されたジスルフィド結合がある、5〜10個のアミノ酸の平行でないβ鎖からなる。IgのIgCドメイン、およびMHC分子は同じタイプの配列パターンを共有し、Igスーパーファミリー内のC1−セットと呼ばれる。他のIgCドメインは他のセットに含まれる。IgVドメインも配列パターンを共有し、Vセットドメインと呼ばれる。IgVドメインはC−ドメインよりも長く、β鎖のさらなる対を形成する。
ヒトB7−4分子の2つの形態が同定されている。一の形態は天然に存在するB7−4可溶性ポリペプチド、すなわち短い親水性ドメインを有し、貫膜ドメインを有さず、本明細書においてはB7−4Sと称する(配列番号2に示す)。第二の形態は、細胞関連ポリペプチド、すなわち貫膜および細胞質ドメインを有し、B7−4Mと称するものである(配列番号4に示す)。
【0021】
B7−4タンパク質は、シグナル配列、ならびにIgVドメインおよびIgCドメインを含む。配列番号2のシグナル配列は、およそアミノ酸1ないしおよそアミノ酸18までで示される。配列番号4のシグナル配列は、およそアミノ酸1ないしおよそアミノ酸18までで示される。配列番号2のIgVドメインはおよそアミノ酸19ないしおよそアミノ酸134間で示され、配列番号4のIgVドメインはおよそアミノ酸19ないしおよそアミノ酸134間で示す。配列番号2のIgCドメインをおよそアミノ酸135ないしおよそアミノ酸227まで示され、配列番号4のIgCドメインはおよそアミノ酸135ないしおよそアミノ酸227まで示される。配列番号2において例示されるB7−4の親水性テールは、およそアミノ酸228からおよそアミノ酸245まで示される親水性テールを含む。配列番号4に例示されるB7−4ポリペプチドは、配列番号4のおよそアミノ酸239からおよそアミノ酸259に示す貫膜ドメインと配列番号4のおよそアミノ酸260からおよそアミノ酸290までに示す細胞質ドメインを含む。
【0022】
ネズミB7−4分子も同定された。ネズミcDNA配列を図5Aから5Bに示し、ネズミB7−4アミノ酸配列を図6に示す。本発明はさらにこれらのネズミB7−4分子に関する。
【0023】
PD−1分子は、B7−4に結合するレセプターとして本明細書中に同定される。PD−1分子は、イムノグロブリン遺伝子スーパーファミリーのメンバーである。PD−1(Ishidaら(1992)EMBO J.11:3887;Shinoharaら(1994)Genomics23:704;米国特許第5698520号)は、イムノグロブリンスーパーファミリードメイン、貫膜ドメイン、およびイムノレセプターチロシンベースの阻害モチーフ(ITIM)を含む細胞内領域を含む。これらの特性は、免疫阻害レセプターと呼ばれるさらに大きな分子のファミリーを定義し、これらはgp49B、PIR−B、およびキラー阻害レセプター(KIR)も含む(VivierおよびDaeron(1997)Immunol.Today 18:286)。これらのレセプターのチロシルリン酸化ITIMモチーフがホスファターゼを含むSH−2ドメインと相互作用し、これが阻害シグナルを生じると考えられる。これらの免疫阻害レセプターのサブセットはMHC分子、例えば、KIRと結合し、CTLA4はB7−1およびB7−2と結合する。MHCとB7遺伝子の間には系統発生論的関係があることが提案されている(Henryら(1999)Immunol.Today20(6):285−8)。
【0024】
本PD−1のヌクレオチド配列を配列番号10および11に示し、PD−1のアミノ酸配列を配列番号12に示す(Ishidaら(1992)EMBO J.11:3887;Shinoharaら(1994)Genomics23:704;米国特許第5698520号参照)。PD−1は、アポトーシス細胞死に関連するタンパク質を選択するためのサブトラクションクローニングベースの方法を用いてすでに同定されている。PD−1は本明細書において、B7−4と結合するその能力に基づいて分子のCD28/CTLA−4ファミリーのメンバーとして同定されている。CTLA4と同様に、PD−1は抗CD3に応答してT細胞の表面上で迅速に誘発される(Agataら(1996)Int.Immunol.8:765)。しかしながらCTLA4と対照的に、PD−1はB−細胞の表面上でも誘発される(抗IgMに応答して)。PD−1はさらに、胸腺細胞および骨髄細胞のサブセット上でも発現される(Agataら(1996)前出;Nishimuraら(1996)Int.Immunol.8:773)。本発明は、PD−1のリガンドとしてB7−4を同定する。
本発明の様々な態様を次のサブセクションにおいてさらに詳細に記載する。
【0025】
1.定義
本明細書において用いられる「免疫細胞」なる用語は、造血起源のものであり、免疫応答において役割を果たすものを包含する。免疫細胞は、リンパ球、例えばB細胞およびT細胞;ナチュラルキラー細胞;骨髄細胞、例えば単球、マクロファージ、好酸球、肥満細胞、好塩基球、および顆粒球を包含する。
本明細書において用いられる「T細胞」なる用語は、T細胞レセプター(TCR)を有する細胞を包含する。好ましくは、「T細胞」なる用語は、CD4+T細胞および/またはCD8+T細胞を包含する。T細胞なる用語はさらに、Tヘルパー1タイプT細胞およびTヘルパー2タイプT細胞の両方を包含する。
【0026】
「抗原提示細胞」なる用語は、プロフェッショナル抗原提示細胞(例えば、Bリンパ球、単球、樹状細胞、ランゲルハンス細胞)ならびに他の抗原提示細胞(例えば、ケラチン生成細胞、内皮細胞、星状細胞、繊維芽細胞、乏突起膠細胞)を包含する。
【0027】
本明細書において用いられる「免疫応答」なる用語は、T細胞副刺激の調節により影響を受けるT細胞性および/またはB細胞性免疫応答を包含する。免疫応答の例は、T細胞応答、例えば、サイトカイン産生、および細胞毒性を包含する。加えて、免疫応答なる用語は、T細胞活性化、例えば抗体産生(体液性応答)およびサイトカイン反応性細胞、例えばマクロファージの活性化により間接的に影響を受ける免疫応答を包含する。
【0028】
本明細書において用いられる場合、「副刺激レセプター」なる用語は、免疫細胞に副刺激シグナルを伝達するレセプター、例えばCD28を包含する。本明細書において用いる場合、「阻害レセプター」なる用語は、負のシグナルを免疫細胞に伝達するレセプター(例えば、CTLA4またはPD−1)を包含する。阻害レセプターにより変換される阻害シグナルは、副刺激レセプター(例えばCD28)が免疫細胞上に存在しなくても、従って阻害レセプターと副刺激分子の間の副刺激分子の結合に関する競合の関係でなくとも生じ得る(Fallarinoら(1998)J.Exp.Med.188:205)。阻害シグナルの免疫細胞への伝達は、免疫細胞における非反応性またはアネルギーまたはプログラムされた死になり得る。好ましくは、阻害シグナルの伝達は、アポトーシスに関与しないメカニズムにより作動する。
【0029】
本明細書において用いる場合、「アポトーシス」なる用語は、当該分野において公知の技術を用いて特徴づけることができるプログラムされた細胞死を包含する。アポトーシス細胞死は、例えば、細胞収縮、膜水疱および細胞フラグメンテーションにおいて最高に達しているクロマチン縮合により特徴づけることができる。アポトーシスを受ける細胞は、ヌクレオソーム間DNA開裂の特徴的なパターンも示す。
レセプターと結合するB7−4分子の形態に応じて、いずれかのシグナルが(例えば、レセプターの架橋を生じるB7−4分子の多価形態により)伝達または刺激され、またはシグナルが(例えば、B7−4の可溶性多価形態により)、例えばレセプターとの結合について活性化形態B7−4分子と競合することにより、阻害され得る。しかしながら、溶解性分子が刺激性であり得る例もある。様々な調節剤の効果は、本明細書に記載するような慣例的なスクリーニングアッセイを用いて容易に証明することができる。
【0030】
本明細書において用いる場合、活性化された免疫細胞に関する「副刺激」なる用語は、副刺激分子が、増殖またはエフェクター機能を誘発する第二の非活性化レセプター性シグナル(「副刺激シグナル」)を提供する能力を包含する。例えば、副刺激シグナルの結果、サイトカインが例えばT細胞レセプター性シグナルを受容したT細胞において分泌され得る。細胞−レセプター性シグナル、例えば活性化レセプターを受容した免疫細胞は、本明細書において「活性化免疫細胞」と称する。
【0031】
本明細書において使用する場合、「活性化レセプター」なる用語は、抗原、複合化抗原(例えばMHC分子に関連して)を結合するか、または抗体と結合する免疫細胞レセプターを包含する。かかる活性化レセプターは、T細胞レセプター(TCR)、B細胞レセプター(BCR)、サイトカインレセプター、LPSレセプター、補体レセプター、およびFcレセプターを包含する。
例えば、T細胞レセプターはT細胞上に存在し、CD3分子と関連する。T細胞レセプターは、MHC分子に関連する抗原により(ならびにポリクローナルT細胞活性化試薬により)刺激される。TCRを介したT細胞活性化の結果、環状ヌクレオチド変化、RNA転写変化、タンパク質合成変化、および細胞体積変化が生じる。
【0032】
B細胞レセプターは、B細胞上に提示される。B細胞抗原レセプターはメンブランIg(mIg)と他のトランスメンブランポリペプチド(例えば、IgαおよびIgβ)間の複合体である。mIgのシグナル変換機能は、オリゴマーまたはマルチマー抗原によるレセプター分子の架橋により引き起こされる。B細胞は、抗イムノグロブリン抗体によっても活性化できる。BCR活性化により、B細胞においてチロシンリン酸化を含む多くの変化が起こる。
Fcレセプターは、免疫応答に関与する多くの細胞上で見いだされる。Fcレセプター(FcR)はイムノグロブリン分子(Ig)のFc部分の細胞表面レセプターである。現在のところ同定されているヒトFcRには、IgG(FcγRと称する)、IgE(FcεR1)、IgA(Fcα)、および重合IgM/A(FcμαR)を認識するものが含まれる。FcRは次の細胞タイプにおいて見いだされる:FcεRI(肥満細胞)、FcεR.II(多くのリンパ球)、FcαR(好中球)、およびFcμαR(腺上皮、肝細胞)(Hogg,N.(1988)Immunol.Today9:185−86)。広く研究されているFcγRは細胞性免疫防御の中心であり、炎症のメディエーターおよび自己免疫疾患の発生機序に関与する加水分解酵素の放出の刺激の原因である(Unkeless,J.C.ら(1988)Annu.Rev.Immunol.6:251−81)。マクロファージ/単球、多形核白血球、およびナチュラルキラー(NK)細胞FcγRはIgGにより媒介される特異的認識の要素を賦与するので、FcγRはエフェクター細胞とIgを放出するリンパ球間の非常に重要な結合を提供する。ヒト白血球はIgG:hFcγRI(単球/マクロファージ上に見いだされる)、hFcγRII(単球、好中球、好酸球、血漿板、おそらくはB細胞、およびK562細胞系上)、およびFcγIII(NK細胞、好中球、好酸球、およびマクロファージ上)の少なくとも3つの異なるレセプターを有する。
【0033】
T細胞に関して、T細胞に対する副刺激シグナルの伝達は、シクロスポリンAにより阻害されないシグナリング経路を含む。加えて、副刺激シグナルはT細胞においてサイトカイン分泌(例えば、IL−2および/またはIL−10)および/または抗原に対する非反応性の誘発、アネルギーの誘発、T細胞における細胞死の誘発を防止することができる。
【0034】
本明細書において使用する場合、「阻害シグナル」なる用語は、免疫細胞上の分子の阻害レセプター(例えばCTLA4またはPD−1)により伝達されるシグナルを意味する。かかるシグナルは、活性化レセプター(例えば、TCR、CD3、BCR、またはFc分子による)により産生されるシグナルを拮抗し、その結果、例えば第二のメッセンジャー生成の阻害;増殖の阻害;免疫細胞におけるエフェクター機能の阻害、たとえば食作用の低下、抗体産生の低下、細胞毒性の低下、免疫細胞がメディエーター(例えば、サイトカイン(例えばIL−2)および/またはアレルギー反応のメディエーター)を産生できないこと;またはアネルギーの発生が起こる。
【0035】
本明細書において用いられる場合、「非反応性」なる用語は、刺激、例えば活性化レセプターまたはサイトカインによる刺激に対する免疫反応の不応性を包含する。非反応性は例えば、免疫抑制物質への暴露または高用量の抗原への暴露のために起こり得る。本明細書において用いられる場合、「アネルギー」または「耐性」なる用語は、活性化レセプター媒介性刺激に対する不応性を包含する。かかる不応性は、一般に抗原特異性であり、耐性化している抗原への暴露が終わった後にも存続する。例えば、T細胞におけるアネルギー(非反応性に対して)はサイトカイン、例えばIL−2がないことにより特徴づけられる。T細胞アネルギーは、T細胞が抗原に暴露され、第二シグナル(副刺激シグナル)の不在下で第一シグナル(T細胞レセプターまたはCD−3媒介性シグナル)を受容する場合に起こる。これらの条件下で、細胞を同じ抗原に再暴露すると(暴露が副刺激分子の存在下で起こる場合でも)、結果としてサイトカインが産生されず、従って増殖しない。しかしながら、アネルギーT細胞は、関連しない抗原に対する応答を起こし、サイトカイン(例えば、IL−2)とともに培養するならば増殖し得る。例えば、T細胞アネルギーは、ELISAにより測定されるようなTリンパ球によるIL−2産生がないこと、あるいはインジケーター細胞系を用いる増殖分析によりを観察できる。別法として、レポーター遺伝子構築物を用いることができる。例えば、アネルギーT細胞は、5’IL−2遺伝子エンハンサーの制御下でヘテロローガスなプロモーターによるか、またはエンハンサー内に見いだすことができるAP1配列のマルチマーにより誘発されるIL−2遺伝子転写を開始しない(Kangら、(1992)Science257:1134)。
【0036】
B7−4タンパク質および核酸分子は、ある種の保存された構造的および機能的特徴を有する分子のファミリーを含む。同様に、PD−1タンパク質および核酸分子は、保存された構造的および機能的特徴を有する分子のファミリーのメンバーである。「ファミリー」なる用語は、タンパク質および核酸分子についていう場合、共通の構造的ドメインまたはモチーフを有し、本明細書において定義するような十分なアミノ酸またはヌクレオチド配列相同性を有する2以上のタンパク質または核酸分子を意味することを意図される。かかるファミリーメンバーは、天然に存在するかまたは天然に存在せず、同じまたは異なる種のいずれかから得ることができる。例えば、ファミリーはヒト起源の第一のタンパク質、ならびに他のヒト起源の異なるタンパク質を含むことができるか、あるいは別に、ヒト以外の起源の相同物を含むことができる。ファミリーのメンバーはさらに共通の機能的特徴を有し得る。本明細書において記載するB7−4分子は分子のB7ファミリーのメンバーである。本明細書において用いられる「B7ファミリー」または「B7分子」なる用語は、B7ポリペプチド、例えばB7−1、B7−2、B7−(抗体BB−1により認識される)、B7h(Swallowら(1999)Immunity11:423)、および/またはB7−4と配列相同性を共有する副刺激分子を含む。例えば、デフォルトのパラメータを用いてNCBIのBLASTプログラム(ギャップペナルティーが存在11および拡張1に設定されたBlosurm62マトリックス(http://www.ncbi.nlm.nih.gov))を用いて比較した場合に、ヒトB7−1およびB7−2は約26%のアミノ酸配列同一性を共有する。
【0037】
好ましいB7ポリペプチドは、免疫細胞に副刺激または阻害シグナルを提供でき、これにより免疫細胞応答を促進または阻害できる。例えば、副刺激レセプターと結合した場合に、B7−4は免疫細胞の副刺激を誘発できるか、または例えば溶解形態において存在する場合に免疫細胞副刺激を阻害できる。阻害レセプターと結合する場合、B7−4分子は阻害シグナルを免疫細胞に伝達することができる。一態様において、B7ファミリーメンバーは免疫細胞上の1以上のレセプター、好ましいB7ファミリーのメンバーには、B7−4、B7−2、B7−3(抗体BB−1により認識される)、B7h、およびB7−4、および溶解性フラグメントまたはその誘導体が含まれる。例えばCTLA4、CD28、ICOS、PD−1および/または他のレセプターと結合し、レセプターに応じて、免疫細胞、好ましくはT細胞に阻害シグナルまたは副刺激シグナルを伝達する能力を有する。
【0038】
好ましいPD−1分子は免疫細胞に阻害シグナルを伝達でき、これにより免疫細胞エフェクター機能を阻害するか、あるいは例えば溶解性モノマー形態において存在する場合に免疫細胞の副刺激を(例えば、競合的阻害により)促進することができる。PD−1誘導性シグナリング(本明細書中ではPD−1を経るシグナリングとも呼ばれる)は、本明細書中、その細胞表面上でPD−1を発現する免疫細胞において、レセプターの、PD−1を活性化するリガンドまたは他の分子への結合により直接的または間接的に誘導される1またはそれ以上の細胞現象と定義される。PD−1の活性化により、細胞の変化を生じる二次的なシグナリング現象が誘発される。そのような細胞現象は、例えば、T細胞活性化アッセイにおける副刺激の阻害により、またはSHP−1分子の増加したリン酸化、ERK1/2分子の低下したリン酸化、またはPKC−θ分子の低下したリン酸化により検出することができる。適当な条件下で、PD−1誘導性のシグナリングは、免疫細胞による免疫反応のダウンレギュレーションを生じる。
【0039】
好ましいPD−1ファミリーメンバーは、1以上のレセプター、例えばB7−1、B7−2、B7−4、および/または抗原提示細胞上の他の分子と結合し、PD−1と配列同一性を共有する。
加えて、一態様において、あるタンパク質ファミリーのメンバーであるタンパク質は、1以上の他のファミリーメンバーのタンパク質に対して生じる抗体により結合される。例えば、抗BB1抗体は、B7−4分子を認識する。
【0040】
本明細書において用いられる場合、B7−4またはPD−1ポリペプチドに関する「活性」なる用語は、B7−4またはPD−1タンパク質の構造に固有の活性を包含する。B7−4に関して、「活性」なる用語は、免疫細胞副刺激を、例えば免疫細胞において副刺激シグナルを調節することにより調節することにより、または免疫細胞における阻害シグナルを、例えば免疫細胞上のナチュラルレセプターを束縛することより調節する能力を包含する。B7−4分子の活性化形態が副刺激レセプターと結合する場合、副刺激シグナルは免疫細胞において生じる。B7−4分子の活性化形態が阻害レセプターと結合する場合、阻害シグナルは免疫細胞において生じる。
【0041】
副刺激シグナルを調節すると、免疫細胞のエフェクター機能が調節される。従って、「B7−4活性」なる用語は、B7−4ポリペプチドがその天然のレセプターと結合する能力、免疫細胞副刺激または阻害シグナルを調節する能力、および免疫細胞応答を調節する能力を包含する。
PD−1に関して、「活性」なる用語は、PD−1ポリペプチドが免疫細胞において、例えば抗原提示細胞上の天然のリガンドを束縛することにより阻害シグナルを調節する能力を包含する。PD−1は阻害シグナルをCTLA4と同様の方法で免疫細胞に伝達する。免疫細胞において阻害シグナルを調節すると、免疫細胞の増殖および/または免疫細胞によるサイトカイン分泌が調節される。PD−1はさらに、B7分子の結合に関して副刺激レセプターと競合することにより副刺激シグナルを調節することができる。従って、「PD−1活性」なる用語は、PD−1ポリペプチドがその天然のリガンドと結合する能力、免疫細胞副刺激または阻害シグナルを調節する能力、および免疫応答を調節する能力を包含する。
【0042】
本明細書において用いられる「天然に存在する」核酸分子なる用語は、天然に存在するヌクレオチド配列を有する(例えば、天然のタンパク質をエンコードする)RNAまたはDNA分子を意味する。
本明細書において用いられる場合、「アンチセンス」核酸分子は、タンパク質をエンコードする「センス」核酸配列と相補性、例えば二本鎖cDNA分子のコーティング鎖と相補性、mRNA配列と相補性、または遺伝子のコーティング鎖と相補性であるヌクレオチド配列を含む。従って、アンチセンス核酸分子は、センス核酸分子と水素結合できる。
【0043】
本明細書において用いられる場合、「コーティング領域」とは、アミノ酸残基中に翻訳されるコドンを含むヌクレオチド配列の領域を意味し、一方、「非コーディング領域」なる用語は、アミノ酸中に翻訳されないヌクレオチド配列の領域(例えば、5’および3’非翻訳領域)を意味する。
本明細書において用いられる場合、「ベクター」なる用語は、これが結合しているもう一つの核酸分子を輸送できる核酸分子を意味する。ベクターの一のタイプは「プラスミド」であり、これはその中に別のDNAセグメントをライゲートできる環状二本鎖DNAループを意味する。もう一つ別のタイプのベクターはウイルスベクターであり、ここにおいては別のDNAセグメントをウイルスゲノム中にライゲートできる。ある種のベクターはこれらが導入される宿主細胞において自己複製できる(例えば、細菌性複製起源を有する細菌ベクターおよびエピソーム哺乳動物ベクター)。他のベクター(例えば、非エピソーム哺乳動物ベクター)は宿主細胞中へ導入されると宿主細胞のベクター中に組み入れられ、これにより宿主ゲノムとともに複製される。さらに、ある種のベクターは操作可能に結合される遺伝子の発現を行うことができる。さらに、かかるベクターは本明細書において「組み換え発現ベクター」または単に「発現ベクター」と称する。一般に、組み換えDNA技術において有用な発現ベクターは、プラスミドの形態であることが多い。本明細書において、プラスミドが最も一般的に用いられるベクターの形態であるので、「プラスミド」および「ベクター」は交換可能に用いられる。しかしながら、本発明はかかる他の発現ベクターの形態、例えばウイルスベクター(例えば、複製欠損レトロウイルス、アデノウイルスおよびアデノ関連ウイルス)を包含し、これらは同等の機能を果たす。
【0044】
本明細書において用いる場合、「宿主細胞」なる用語は、本発明の組み換え発現ベクターなどの本発明の核酸分子がその中に導入されている細胞を意味する。「宿主細胞」または「組み換え宿主細胞」なる用語は本明細書において交換可能に用いられる。かかる用語は特定の対象細胞のみでなく、かかる細胞の子孫または可能性のある子孫も意味すると理解されるべきである。ある種の修飾が突然変異または環境的影響のために次の世代において起こり得るので、かかる子孫は実際には親細胞と同一でないが、それでも本明細書において用いられる用語の範囲内に含まれる。
本明細書において用いられる場合、「トランスジェニック動物」なる用語は、ヒト以外の動物、好ましくは哺乳動物、さらに好ましくはマウスを意味し、該動物の1以上の細胞は「トランス遺伝子」を含む。「トランス遺伝子」なる用語は、トランスジェニック動物が発生する細胞のゲノム中に組み入れられ、成体動物のゲノム中に残存する外因性DNA、例えばトランスジェニック動物の1以上のセルタイプまたは組織においてエンコードされた遺伝子産物の発現を行うものを意味する。
【0045】
本明細書において用いられる場合、「相同性組み換え動物」なる用語は、ある種のヒト以外のトランスジェニック動物、好ましくは哺乳動物、さらに好ましくはマウスを意味し、ここにおいて外因性遺伝子は、動物の発生前の外因性遺伝子と動物の細胞中に導入された外因性DNA分子、例えば動物の胚細胞の間のホモローガスな組み換えにより変更されている。
本明細書において用いられる場合、「単離されたタンパク質」な用語は、細胞から単離されるかあるいは組み換えDNA技術により産生されるか場合は他のタンパク質、細胞物質および培地、あるいは化学的に合成される場合には化学的前駆体または他の化学物質が実質的にないタンパク質を意味する。「単離された」または「精製された」タンパク質または生物学的に活性なタンパク質それ自体は、B7−4またはPD−1タンパク質が由来する細胞または組織源から得られる細胞物質または他の汚染タンパク質の調製物を含む。「細胞物質が実質的にない」なる用語には、タンパク質が、それが単離または組換え産生される細胞の細胞成分から単離される、B7−4またはPD−1タンパク質の調製物が含まれる。「細胞物質が実質的にない」とは、約30%(乾燥重量)未満の非B7−4またはPD−1タンパク質、さらに好ましくは約20%未満の非B7−4またはPD−1タンパク質、なおさらに好ましくは約10%未満の非B7−4またはPD−1タンパク質、最も好ましくは約5%未満の非B7−4またはPD−1タンパク質を有するB7−4またはPD−1タンパク質の調製物を包含する。B7−4またはPD−1タンパク質またはその生物学的に活性なタンパク質が組み換え的に産生される場合、これは好ましくは実質的に培地がない、すなわち、培地がタンパク質調製物の体積の約20%未満であり、さらに好ましくは約10%未満、最も好ましくは約5%未満である。
【0046】
「化学前駆体または他の化学物質が実質的にない」なる言い回しは、タンパク質が、該タンパク質の合成に関与する化学前駆体または他の化学物質から分離されているB7−4またはPD−1タンパク質の調製物を包含する。一態様において、「化学前駆体または他の化学物質が実質的にない」なる言い回しは、約30%(乾燥重量)未満の化学前駆体または非B7−4またはPD−1化学物質、さらに好ましくは約20%未満の化学前駆体または非B7−4またはPD−1化学物質、さらに好ましくは約10%未満の化学前駆体または非B7−4またはPD−1化学物質、最も好ましくは約5%未満の化学前駆体または非B7−4またはPD−1化学物質を有するB7−4またはPD−1タンパク質の調製物を包含する。
【0047】
本明細書において用いられる「抗体」なる用語は、抗体の「抗原結合部分」(または単に「抗体部分」)も包含する。本明細書において用いられる「抗原結合部分」なる用語は、抗原と特異的に結合する能力を保持する抗体の1以上のフラグメントを意味する(たとえば、B7−4)。抗体の抗原結合機能は完全長抗体により実施され得ることが証明されている。抗体の「抗原結合タンパク質」なる用語に含まれる結合フラグメントの例は、(i)Fabフラグメント(VL、VH、CLおよびCH1ドメインからなる一価フラグメント);(ii)F(ab’)2フラグメント(ヒンジ領域でジスルフィドブリッジにより結合された2のFabフラグメントからなる二価フラグメント);(iii)VHおよびCH1ドメインからなるFdフラグメント;(iv)VLおよびVHドメインからなるFvフラグメント;抗体の1つの腕のVLおよびVHドメインからなるFvフラグメント;(v)VHドメインからなるdAbフラグメント(Wardら(1989)Nature341:544−546);および(vi)単離された相補性決定領域(CDR)を包含する。さらに、Fvフラグメントの2つの領域、VLおよびVHは別々の遺伝子によりコードされるが、これらは組み換え法を用いて、VLおよびVH領域対が一価分子(一本鎖Fv(scFv)とも呼ばれる;たとえば、Birdら(1988)Science242:423−426;およびHustonら(1988)Proc.Natl.Acad.Sci.USA85:5879−5883;およびOsbournら(1998)Natl.Biotechnol.16:778)を形成する一つのタンパク質鎖として調製することができる合成リンカーにより結合できる。このような一本鎖抗体はさらに抗体の「抗体結合部分」なる用語に含まれることを意図される。完全IgG分子または他のイソタイプをエンコードする発現ベクターを生じるために、特異的scFvの任意のVHおよびVL配列はヒトイムノグロブリン定常領域cDNAまたはゲノム配列と結合できる。VHおよびV1は、タンパク質化学または組み換えDNA技術のいずれかを用いてFab、Fvまたはイムノグロブリンの他のフラグメントの生成において用いることができる。一本鎖抗体の他の形態、たとえば、二重特異性抗体(daibody)も含まれる。二重特異性抗体は、二価、二重特異性抗体であり、ここにおいてVHおよびVLドメインは一本鎖ポリペプチド上に発現されるが、同じ鎖上の2つのドメイン間で対合するには短すぎ、そのために該ドメインは別の鎖の相補性ドメインと対になり、2つの抗原結合部位を形成する(たとえば、Holliger,P.ら(1993)Proc.Natl.Acad.Sci.USA90:6444−6448;Poljak,R.J.ら(1994)Structure2:1121−1123参照)。
【0048】
さらに、抗体またはその抗原結合部分は、抗体または抗体部分と1以上の他のタンパク質またはペプチドとの共有または非共有結合により形成される、より大きなイムノグロブリン分子の一部であり得る。かかる免疫接着(immunoadhesion)分子の例は、四量体scFv分子を作るためのストレプトアビジンコア領域の使用(Kipriyanov,S.M.ら(1995)Human Antibodies and Hybridomas6:93−101)および二価およびビオチニル化scFv分子を作るためのシステイン残基、マーカーペプチドおよびC−末端ポリヒスチジンタグの使用(Kipriyanov,S.Mら(1994)Mol.Immunol.31:1047−1058)を包含する。抗体部分、たとえば、FabおよびF(ab’)2フラグメントは、慣例の技術、たとえば、それぞれ完全抗体のパパインまたはペプシン消化を用いて、完全抗体から調製することができる。さらに、抗体、抗体部分および免疫接着分子は、本明細書に記載するような標準的組み換えDNA技術を用いて得ることができる。
【0049】
抗体は、ポリクローナルまたはモノクローナル;外因性、同種異系、または同系;あるいはその修飾された形態、たとえばヒト化、キメラなどであってもよい。好ましくは、本発明の抗体は、B7−4分子と特異的または実質的に特異的に結合する。本明細書において用いられる「モノクローナル抗体」および「モノクローナル抗体組成物」なる用語は、抗原の特定のエピトープと免疫反応できる抗原結合部位の1種のみを含む小唄異分子の集団を意味し、一方、「ポリクローナル抗体」および「ポリクローナル抗体組成物」なる用語は、特定の抗原と相互作用できる抗原結合部位の複数腫を含む抗体分子の手段を意味する。モノクローナル抗体組成物は、典型的には、これが免疫反応する特定の抗原に対して単一結合親和性を示す。
【0050】
本明細書において用いられる「ヒト化抗体」なる用語は、ヒト細胞により製造されるさらによく似た抗体に変更されたさまざまな可変および定常領域を有するヒト以外の細胞により製造される抗体を包含することを意図される。たとえば、非ヒト抗体アミノ酸配列を、ヒト生殖細胞系配列において見いだされるアミノ酸を組み入れるように変更することによる。本発明のヒト化抗体は、たとえばCDRにおいて、ヒト生殖細胞系イムノグロブリン配列(たとえば、インビトロでの部位特異性突然変異誘発またはインビボでの体細胞突然変異により導入される突然変異)によりによりエンコードされないアミノ酸残基を含み得る。本明細書において用いられる「ヒト化抗体」なる用語はさらに、マウスなどの別の哺乳動物種から誘導されるCDR配列がヒトフレームワーク配列上に移植されている抗体も包含する。
【0051】
本明細書において用いられる「単離された抗体」とは、異なる抗原特異性を有する他の抗体が実質的にない抗体を意味することを意図される(たとえば、B7−4と特異的に結合する単離された抗体は、B7−4以外の抗原と特異的に結合する抗体が実質的にない)。さらに、単離された抗体は他の細胞性物質および/または化学物質が実質的にない。
特定のアミノ酸配列とタンパク質をコードするヌクレオチド配列間には、遺伝子コード(後記)により決められる既知の明確な対応が存在する。同様に、特定核酸分子のヌクレオチド配列と核酸分子によりエンコードされるアミノ酸配列の間には、遺伝子コードにより決定されるような既知の明確な対応が存在する。
【0052】
遺伝子コード
アラニン (Ala, A) GCA, GCC, GCG, GCT
アルギニン (Arg, R) AGA, ACG, CGA, CGC, CGG, CGT
アスパラギン (Asn, N) AAC, AAT
アスパラギン酸 (Asp, D) GAC, GAT
システイン (Cys, C) TGC, TGT
グルタミン酸 (Glu, E) GAA, GAG
グルタミン (Gln, Q) CAA, CAG
グリシン (Gly, G) GGA, GGC, GGG, GGT
ヒスチジン (His, H) CAC, CAT
イソロイシン (Ile, I) ATA, ATC, ATT
ロイシン (Leu, L) CTA, CTC, CTG, CTT, TTA, TTG
リジン (Lys, K) AAA, AAG
メチオニン (Met, M) ATG
フェニルアラニン (Phe, F) TTC, TTT
プロリン (Pro, P) CCA, CCC, CCG, CCT
セリン (Ser, S) AGC, AGT, TCA, TCC, TCG, TCT
トレオニン (Thr, T) ACA, ACC, ACG, ACT
トリプトファン (Trp, W) TGG
チロシン (Tyr, Y) TAC, TAT
バリン (Val, V) GTA, GTC, GTG, GTT
終結シグナル (end) TAA, TAG, TGA
【0053】
遺伝子コードの重要でよく知られた特性は、その重複性(redundancy)であり、これにより、タンパク質を製造するために用いられるほとんどのアミノ酸に関して、1以上のコーディングヌクレオチドトリプレットを用いることができる(前記)。従って、多くの異なるヌクレオチド配列は所定のアミノ酸配列についてコードすることができる。かかるヌクレオチド配列は、結果として全ての生物において同じアミノ酸配列を産生するので、機能的に等価であると考えられる(ある生物はあるアミノ酸配列を他のものよりも有効に翻訳することができるが)。さらに、時折、プリンまたはピリミジンのメチル化変異体を所定のヌクレオチド配列中に見いだすことができる。かかるメチル化は、トリヌクレオチドコドンと対応するアミノ酸間のコーディング関係に影響を及ぼさない。
【0054】
前記事項を考慮すると、本発明のB7−4またはPD−1ポリペプチドをコードするDNAまたはRNA分子のヌクレオチド配列(またはその任意の部分)は、DNAまたはRNA分子をアミノ酸配列に本湯アクするために遺伝子コードを用いて、B7−4またはPD−1アミノ酸配列を誘導するために用いることができる。同様に、任意のB7−4またはPD−1アミノ酸配列に関して、B7−4またはPD−1タンパク質をエンコードできる対応するヌクレオチド配列を遺伝子コード(その重複性のために、所定のアミノ酸配列について複数の核酸配列を産生するであろう)から推測できる。かくして、B7−4またはPD−1ヌクレオチド配列の本明細書における記載および/または開示は、該ヌクレオチド配列によりエンコードされるアミノ酸配列の記載および/または開示も包含すると考えられるべきである。同様に、本明細書におけるB7−4またはPD−1アミノ酸配列の記載および/または開示も、アミノ酸配列をエンコードできるあらゆる可能なヌクレオチド配列の記載および/開示も包含すると考えるべきである。
【0055】
「小分子」なる用語は、専門用語であり、約1000分子量未満または約500分子量未満の分子を包含する。もう一つ別の具体例において、小分子はオリゴマーでない。活性についてスクリーンできる小分子化合物の例は、これらに限定されないが、ペプチド、ペプチド模倣物、核酸、炭水化物、小有機分子(たとえば、ポリケチド(polyketide))(Caneら1998 Science282:63)、および天然の生成物抽出物ライブラリーを包含する。もう一つ別の具体例において、化合物は小さな有機非ペプチド化合物である。さらにも一つの具体例において、小分子は生合成物でない。
【0056】
II.単離された核酸分子
一具体例において、特許権を要求する方法において用いられる薬剤は、B7−4またはPD−1タンパク質またはその生物学的に活性な部分をエンコードする単離された核酸分子を含む。B7−4または核酸をエンコードするPD−1エンコーディングを同定するためにハイブリダイゼーションプローブとして使用するために十分な核酸フラグメント(たとえば、B7−4またはPD−1mRNA)およびB7−4またはPD−1核酸分子の増幅または突然変異のPCRプライマーとして使用するフラグメントも提供される。本明細書において用いられる場合、「核酸分子」なる用語は、DNA分子(たとえば、cDNAまたはゲノムDNA)およびRNA分子(たとえば、mRNA)およびヌクレオチド類似体を用いて生じるDNAまたはRNAの類似体を包含することを意図される。核酸分子は一本鎖または二本鎖であり得るが、好ましくは二本鎖DNAである。
【0057】
「単離された」核酸分子は、核酸分子の天然源において存在する他の核酸から分離されるものである。たとえば、ゲノムDNAに関して、「単離された」なる用語は、ゲノムDNAが自然に結合する染色体から分離される核酸分子を包含する。好ましくは、「単離された」核酸分子は、核酸分子が由来する生物のゲノムDNAにおいて、自然に核酸分子の側面に位置する配列(すなわち、核酸の5’および3’末端に位置する配列)がない。がない。たとえば、様々な具体例において、単離されたB7−4またはPD−1核酸分子は、約5kb、4kb、3kb、2kb、1kb、0.5kbまたは0.1kb未満の、核酸が由来する細胞のゲノムDNAにおいて自然に核酸の側面に位置する核酸配列を含み得る。さらに、「単離された」核酸分子、たとえばcDNA分子は、他の細胞生物質、または組み換え技術により賛成される場合には細胞培地が実質的にないか、あるいは化学的に合成される場合には化学前駆体または他の化学物質が実質的にない。しかしながら、「単離された」B7−4またはPD−1核酸分子は、通常ゲノムDNAにおいてB7−4またはPD−1配列の側面に位置しない他のヌクレオチド配列と結合し得る(たとえば、B7−4またはPD−1ヌクレオチド配列はベクター配列と結合することができる)。ある好ましい具体例において、「単離された」核酸分子、たとえば、cDNA分子も他の物質がない。しかしながら、B7−4またはPD−1核酸分子が、「単離された」とみなされる他の細胞生物質がないものである必要はない(たとえば、他の哺乳動物DNAから分離され、細菌細胞中に挿入されたB7−4またはPD−1DNA分子も「単離された」と見なされる)。
【0058】
本発明の核酸分子、たとえば、配列番号1、3、10、または11のヌクレオチド配列を有する核酸分子またはその一部を、標準的分子生物学技術および本明細書において記載する配列情報を用いて単離することができる。たとえば、配列番号1、3、10、または11の核酸配列の全部または一部をハイブリダイゼーションプローブとして用いて、B7−4またはPD−1核酸分子を、標準的ハイブリダイゼーションおよびクローニング技術(たとえば、Sambrook,J.ら Molecular Cloning:A Laboratory Manual.第2版、Cold Spring Harbor Laboratory,Cold Spring Harbor Laboratory Press,Cold Spring Harbor,NY,1989に記載)を用いて単離することができる。
さらに、配列番号1、3、10、または11の全部または一部を含む核酸分子は、それぞれ配列番号1、3、10、または11の配列に基づいて設計された合成オリゴヌクレオチドプライマーを用いてポリメラーゼ連鎖反応(PCR)により単離することができる。
【0059】
本発明の核酸は、標準的PCR増幅技術に従って、テンプレートおよび適当なオリゴヌクレオチドプライマーとして、cDNA、mRNAまたは別法としてゲノムDNAを用いて増幅することができる。このようにして増幅された核酸は、適当なベクター中にクローンし、DNA配列分析により特徴化することができる。さらに、B7−4またはPD−1ヌクレオチド配列に対応するオリゴヌクレオチドは、標準的合成技術により、たとえば自動化DNA合成器を用いて調製することができる。
好ましい具体例において、本発明の単離された核酸分子は、配列番号1、3、10、または11に示すヌクレオチド配列を含む。
【0060】
もう一つ別の好ましい態様において、本発明の単離された核酸分子は、配列番号1、3、10、または11に示す核酸配列の補体、あるいはこれらのヌクレオチド非あれ角任意のものの一部である核酸分子を含む。配列番号1、3、10、または11に示すヌクレオチド配列と相補生である核酸分子は、それぞれ配列番号1、3、10、または11において示すヌクレオチド配列と十分相補性であるものであるので、これはそれぞれ配列番号1、3、10、または11に示すヌクレオチド配列とハイブリッド形成でき、これにより安定な二本鎖を形成する。的確な補体は規定のヌクレオチド配列と100%相同性である。さらにもう一つ別の好ましい具体例において、本発明の単離された核酸分子は、少なくとも約50%、55%、60%、65%、70%、75%、80%、85%、90%、95%、98%またはそれ以上配列番号1、3、10、または11に示すヌクレオチド配列(たとえば、ヌクレオチド配列の完全な長さ)、またはこれらのヌクレオチド配列の任意のものの一部と一致する。
【0061】
さらに、本発明の核酸分子は、配列番号1、3、10、または11の核酸配列の一部のみ、たとえば、B7−4またはPD−1タンパク質の生物学的に活性な部分をエンコードするプローブまたはプライマーまたはフラグメントとして用いることができるフラグメントを含み得る。B7−4またはPD−1遺伝子のクローニングから決定されるヌクレオチド配列は、他のB7−4またはPD−1ファミリーメンバー、ならびに他の種から得られるB7−4またはPD−1ファミリー相当物の同定および/またはクローニングにおいて用いるために設計されたプローブおよびプライマーの生成を可能にする。プローブ/プライマーは、典型的には実質的に精製されたオリゴヌクレオチドを含む。オリゴヌクレオチドは典型的には、ストリンジェントな条件下で、少なくとも約12または15、好ましくは約20または25、さらに好ましくは約30、35、40、45、50、55、60、65、または75の、配列番号1、3、10、または11のセンス配列の連続したヌクレオチドとハイブリッド形成するヌクレオチド配列、あるいは配列番号1、3、10、または11の天然に存在する対立遺伝子変異体または突然変異体の領域を含む。一例において、本発明の核酸分子は、少なくとも350、400、450、500、550、600、650、700、750、または800ヌクレオチドの長さのヌクレオチド配列を含み、配列番号1、3、10、または11の核酸分子とストリンジェントなハイブリダイゼーション条件下でハイブリッド形成する。
【0062】
もう一つ別の具体例において、第二の核酸分子は、配列番号1、3、10、または11の、少なくとも約500、600、700、800、900、または1000の連続したヌクレオチドを含む。
一具体例において、たとえばプローブとして用いられる本発明の核酸分子は、配列番号1のおよそヌクレオチド810からおよそ850または配列番号1のおよそヌクレオチド320から854の配列番号1の一部を含まない。もう一つ別の具体例において、本発明の核酸分子は、配列番号3のおよそヌクレオチド314からおよそ734、またはおよそヌクレオチド835からおよそ860、またはおよそヌクレオチド1085からおよそ1105またはおよそヌクレオチド1286からおよそ1536の配列番号3の一部を含まない。
【0063】
一具体例において、本発明の核酸分子は、配列番号1または配列番号3の少なくとも約500の連続したヌクレオチドを含む。好ましい具体例において、本発明の核酸分子は、少なくとも約600、少なくとも約800、少なくとも約900または少なくとも約950の配列番号1の連続したヌクレオチドを含む。もう一つ別の具体例において、本発明の核酸分子は、少なくとも約1500または1550の配列番号3のヌクレオチドを含む。
【0064】
好ましくは、本発明の単離された核酸分子は、配列番号1(ヌクレオチド59〜793)または配列番号3(ヌクレオチド53〜922)のコーディング領域の少なくとも一部を含む。もう一つ別の具体例において、B7−4核酸分子は、配列番号1のおよそヌクレオチド1からおよそヌクレオチド319を含む。もう一つ別の具体例において、B7−4核酸分子は、配列番号1のおよそヌクレオチド855からおよそヌクレオチド968を含む。もう一つ別の具体例において、B7−4核酸分子は、配列番号3のおよそヌクレオチド1からおよそヌクレオチド314を含む。もう一つ別の具体例において、B7−4核酸分子は、配列番号3のおよそヌクレオチド955からおよそヌクレオチド1285を含む。もう一つ別の具体例において、B7−4核酸分子は、配列番号3のおよそヌクレオチド1535からおよそヌクレオチド1552を含む。
【0065】
他の具体例において、本発明の核酸分子は:少なくとも約500、少なくとも約600、少なくとも約700、少なくとも約800、少なくとも約900または少なくとも約1000の連続した配列番号1または配列番号3のヌクレオチドを含む核酸分子と、少なくとも70%の同一性、さらに好ましくは80%の同一性、さらにいっそう好ましくは90%の同一性を有する。
【0066】
B7−4またはPD−1ヌクレオチド配列に基づくプローブは、転写物または同一または相同性のタンパク質をエンコードするゲノム配列を検出するために用いることができる。好ましい具体例において、プローブはさらにこれに結合した標識基を含む。たとえば、標識基は、放射性同位元素、蛍光化合物、酵素、または酵素補助因子であり得る。そのようなプローブを、例えば、B7−4またはPD−1−エンコーディング核酸の対象からの細胞のサンプル中でのレベルを測定すること、例えば、B7−4またはPD−1のmRNAレベルを測定することまたは、ゲノムB7−4またはPD−1遺伝子が変異したかまたは欠失したかどうかを測定することにより、B7−4またはPD−1タンパク質を誤発現する細胞または組織を同定するための診断試験キットの部分として用いることができる。
【0067】
「B7−4またはPD−1タンパク質の生物活性部分」をコードしている核酸フラグメントは、B7−4またはPD−1生物活性(B7−4またはPD−1タンパク質の生物活性は、本明細書に記載されるものである)を有するポリペプチドをコードする、配列番号1、3、10、または11のヌクレオチド配列の部分を単離すること、B7−4またはPD−1タンパク質のコードされる部分を(例えば、インビトロで組換え発現により)発現させること、およびB7−4またはPD−1タンパク質のコードされる部分の活性を評価することにより調製することができる。
【0068】
遺伝子コードの縮重のために配列番号1、3、10、または11から異なり、そして、配列番号1、3、10、または11によりコードされるものと同じB7−4またはPD−1タンパク質をコードする核酸分子が本発明に包含される。従って他の具体例において、本発明の単離核酸分子は、配列番号2、4、または12に示すアミノ酸配列を有するタンパク質をコードしているヌクレオチド配列を有する。他の具体例において、本発明の単離核酸分子は、B7−4またはPD−1タンパク質をコードしているヌクレオチド配列を有する。
【0069】
配列番号1、3、10、または11に示すB7−4またはPD−1ヌクレオチド配列に加えて、当該分野の専門家には、B7−4またはPD−1タンパク質のアミノ酸配列に変化を生じるDNA配列多型が集団内(例えば、ヒトの集団)に存在し得ることが分かるであろう。B7−4またはPD−1遺伝子におけるそのような遺伝子多型は、天然対立変異により集団内の個人の間に存在してよい。本明細書中に用いられる、「遺伝子」および「組換え遺伝子」なる用語は、B7−4またはPD−1タンパク質、好ましくは哺乳動物のB7−4またはPD−1タンパク質をコードしているオープンリーディングフレームを含む核酸分子を意味し、非コーディング調節配列、およびイントロンをさらに含むことができる。そのような天然対立変種には、機能および非機能B7−4またはPD−1タンパク質の両方が含まれ、B7−4またはPD−1遺伝子のヌクレオチド配列において1−5%の変異を典型的に生じ得る。天然対立変異の結果であり、およびB7−4またはPD−1タンパク質の機能活性を変更しないB7−4またはPD−1遺伝子におけるそのようなヌクレオチド変異および生じるアミノ酸多型は、本発明の範囲内にあるものとする。
【0070】
さらに、他のB7−4またはPD−1ファミリーのメンバーをコードしており、すなわち、配列番号1、3、10、または11のB7−4またはPD−1ファミリーとは異なるヌクレオチド配列を有する核酸分子は、本発明の範囲内にあるものとする。例えば、他のB7−4またはPD−1cDNAは、ヒトB7−4またはPD−1のヌクレオチド配列に基き同定することができる。さらに、異なる種からのB7−4またはPD−1タンパク質をコードしており、つまり配列番号1、3、10、または11のB7−4またはPD−1配列とは異なるヌクレオチド配列を有する核酸分子は、本発明の範囲内にあるものとする。例えば、マウスのB7−4またはPD−1cDNAは、ヒトのB7−4またはPD−1分子のヌクレオチド配列に基いて同定することができる。
【0071】
本発明のB7−4またはPD−1cDNAの天然対立変種およびホモログに対応する核酸分子は、本明細書中に開示するB7−4またはPD−1核酸に対するそのホモロジーに基き、本明細書中に開示するcDNAまたはその部分を基準に従いハイブリダイゼーションプローブとして用いて、単離することができる。例えば、B7−4またはPD−1DNAは、配列番号1、3、10、または11の全部または部分を用いてヒトゲノムDNAライブラリーから、標準的ハイブリダイゼーション技術(たとえば、Sambrook,J.らMolecular Cloning:A Laboratory Manual、第2版、Cold Spring Harbor Laboratory,Cold Spring Harbor,NY,1989に記載)を用いて、単離することができる。さらに、配列番号1、3、10、または11の配列に基づいて設計されたオリゴヌクレオチドプライマーを用いてポリメラーゼ連鎖反応により、B7−4またはPD−1遺伝子の全部または一部を含む核酸分子を単離することができる。たとえば、mRNAは細胞から単離することができ(たとえば、Chirgwinら(1979)Biochemistry18:5294−5299のグアニジニウム−チオシアネート抽出法による)、cDNAは逆転写酵素(たとえば、Gibco/BRL,Bethesda,MDから入手可能なMoloney MLV逆転写酵素;またはSeikagaku America,Inc.,St.Petersburg,FLから入手可能なAMV逆転写酵素)。PCR増幅の合成オリゴヌクレオチドプライマーは、配列番号1、3、10、または11に示すヌクレオチド配列に基づいて設計できる。本発明の核酸分子は、標準的pCR2.1プラスミド増幅技術に従って、テンプレートおよび適当なオリゴヌクレオチドプライマーとしてcDNA、あるいは別法としてゲノムDNAを用いて増幅することができる。このようにして増幅された核酸を適当なベクター中にクローンし、DNA配列分析により特徴づけることができる。さらに、B7−4またはPD−1ヌクレオチド配列に対応するオリゴヌクレオチドは、たとえば自動化DNA合成器を用いて、標準的合成技術により調製することができる。
【0072】
もう一つ別の具体例において、本発明の単離された核酸分子は、少なくとも15、20、25、30またはそれ以上のヌクレオチドの長さであり、ストリンジェントな条件下で、配列番号1、3、10、または11のヌクレオチド配列を含む核酸分子とハイブリッド形成する。他の具体例において、核酸分子は少なくとも30、50、100、150、200、250、300、350、400、450、500、550、または600ヌクレオチドの長さである。本明細書において用いられる場合、「ストリンジェントな条件下でハイブリッド形成する」なる用語は、互いに少なくとも30%、40%、50%、または60%相同性であるヌクレオチド配列が典型的に互いにハイブリッド形成した状態であるハイブリダイゼーションおよび洗浄の条件を説明することを意図される。好ましくは、互いに少なくとも約70%、さらに好ましくは少なくとも約80%の1以上、なおいっそう好ましくは少なくとも約85%または90%相同性である配列が互いにハイブリッド形成したままであるような条件である。このようなストリンジェントな条件は、当業者には一般的であり、Current Protocols in Molecular Biology,John Wiley & Sons,N.Y.(1989)、6.3.1−6.3.6において見いだすことができる。ストリンジェントなハイブリダイゼーション条件の好ましい非制限的な例は、6X塩化ナトリウム/クエン酸ナトリウム(SSC)中、約45度でハイブリッド形成し、続いて50〜65℃で0.2×SSC、0.1%SDS中1回以上洗浄する。好ましくは、ストリンジェントな条件下で配列番号1、3、10、または11の配列とハイブリッド形成する本発明の単離された核酸分子は、天然に存在する核酸分子に対応する。
【0073】
本明細書において用いられる場合、「天然に存在する」核酸分子とは、天然に存在する(たとえば、天然のタンパク質をエンコードする)ヌクレオチド配列を有するRNAまたはDNA分子を意味する。配列番号1、3、10、および11に示すB7−4またはPD−1ヌクレオチド配列に加えて、集団内にはB7−4またはPD−1のヌクレオチドまたはアミノ酸配列に小さな変化を起こすDNA配列多形性が存在し得ることは当業者には理解されるであろう。B7−4またはPD−1遺伝子におけるこのような遺伝子多形性は、天然の対立遺伝子バリエーションのために集団内の角固体間に存在し得る。このような天然の対立遺伝子バリエーションの結果、典型的には遺伝子の配列において1〜2%の差異が生じる。このようなヌクレオチドバリエーションと結果として得られる、天然の対立遺伝子バリエーションの結果であり、B7−4またはPD−1ポリペプチドの機能的活性を変更しないB7−4またはPD−1におけるアミノ酸多形性は本発明の範囲内に含まれる。
【0074】
集団内に存在し得るB7−4またはPD−1配列の天然に存在する対立変異体に加えて、たとえば配列番号1、3、10、または11のヌクレオチド配列中への突然変異により小さな変化を導入することができ、これにより、B7−4またはPD−1タンパク質の機能的活性を変更することなく、エンコードされたタンパク質のアミノ酸配列において変化を生じることは当業者にはさらに理解されるであろう。たとえば、「非必須」アミノ酸残基でのアミノ酸置換につながるヌクレオチド置換を配列番号1、3、10、または11の配列において行うことができる。「非必須」アミノ酸残基は、B7−4またはPD−1分子の機能的活性を変更することなく、B7−4核酸分子の野生型配列(たとえば、配列番号1、3、10、または11)から変更できる残基である。好ましくは、B7−4のレセプターとの結合またはPD−1の天然のリガンドに対する結合に必要であることが見いだされている(たとえば、アラニン走査突然変異誘発スクリーンまたは他の技術的に承認されたスクリーニング分析を用いて同定される)B7−4またはPD−1の細胞外ドメインにおける残基は変更されない。B7−4について、非必須で、従って置換を受けやすい残基の例は、B7ファミリーメンバー(またはB7−4ファミリーメンバー)のアミノ酸整列を行い、保存されない残基を決定することにより、当業者により確認され得る。このような残基は、保存されなかったので、置換をより受けやすい。
【0075】
したがって、本発明のもう一つの態様は、B7−4またはPD−1活性に必須でない核酸分子に関する。このようなB7−4またはPD−1タンパク質は、配列番号2、4、または12からのアミノ酸配列が異なり、さらに固有のB7−4活性を保持するか、PD−1の場合において、B7−4と結合する能力を有する。B7−4と結合する能力を保持する。B7−4またはPD−1タンパク質の天然でない変異体をエンコードする単離された核酸分子は、1以上のヌクレオチド置換、付加または欠失を配列番号1、3、10、または11のヌクレオチド配列中に導入することにより、1以上のアミノ酸置換、付加または欠失がエンコードされたタンパク質中に導入されるように形成することができる。標準的技術、たとえば、部位指向性突然変異誘発およびPCRによる突然変異誘発により、突然変異を配列番号1、3、10、または11中に導入することができる。好ましくは、保存的アミノ酸置換は1以上の非必須アミノ酸残基で行われる。「保存的アミノ酸置換」は、アミノ酸残基が類似した側鎖有するアミノ酸残基で置換されたものである。塩基性側鎖(たとえば、リシン、アルギニン、ヒスチジン)、酸性側鎖(たとえば、アスパラギン酸、グルタミン酸)、非荷電極性側鎖(たとえば、グリシン、アスパラギン、グルタミン、セリン、巣レオ任、チロシン、システイン)、非極性側鎖(たとえば、アラニン、バリン、ロイシン、イソロイシン、プロリン、フェニルアラニン、メチオニン、トリプトファン)、ベータ−分岐側鎖(たとえば、スレオニン、バリン、イソロイシン)および芳香族側鎖(たとえば、チロシン、フェニルアラニン、トリプトファン、ヒスチジン)を含む類似した側鎖を有するアミノ酸残基のファミリーが当該分野において定義されている。従って、B7−4またはPD−1における非必須アミノ酸残基は、好ましくは同じ側鎖ファミリーからの別のアミノ酸残基で置換される。
【0076】
別法として、もう一つ別の具体例において、突然変異は、たとえば飽和突然変異誘発により、B7−4またはPD−1コーディング配列の全部または一部と共にランダに導入することができ、結果として得られる突然変異体はDNAと結合および/または転写の活性化の能力についてスクリーンすることができ、機能的活性を保持する突然変異体が同定される。突然変異誘発後、エンコードされたB7−4またはPD−1突然変異タンパク質を宿主細胞において組み換え的に発現することができ、突然変異タンパク質の機能的活性をB7−4またはPD−1活性を評価するために当該分野において利用可能な分析法を用いて定量できる。
【0077】
したがって、本発明のもう一つの態様は、活性に必須でないアミノ酸残基において変化を含むB7−4またはPD−1タンパク質をエンコードする核酸分子に関する。
本発明のさらにもう一つの態様は、B7−4またはPD−1融合タンパク質をエンコードする単離された核酸分子に関する。少なくとも、B7−4またはPD−1タンパク質をエンコードする第一ヌクレオチド配列、非B7−4またはPD−1タンパク質をエンコードする第二のヌクレオチド配列と操作可能に結合するポリペプチドまたはペプチド、ポリペプチドまたはペプチドを含むこのような核酸分子を標準的組み換えDNA技術により調製することができる。
【0078】
好ましい具体例において、突然変異B7−4タンパク質を:1)活性化された免疫細胞の増殖および/またはエフェクター機能を副刺激する能力(または、たとえば溶解形態における副刺激を阻害する);2)抗B−7ファミリーまたは抗B7−4抗体と結合する能力;および/または3)B7−4の天然のレセプター(たとえばPD−1)と結合する能力に関して分析することができる。
好ましい具体例において、突然変異B7−4タンパク質を:1)活性化された免疫細胞の増殖および/またはエフェクター機能を副刺激する能力(または、たとえば溶解形態);2)抗PD−1抗体と結合する能力;および/または3)PD−1の天然のリガンド(たとえばB7−4)と結合する能力に関して分析することができる。
【0079】
前記のB7−4またはPD−1タンパク質をエンコードする核酸分子に加えて、それに対してアンチセンスである単離された核酸分子を調節剤として用いることができる。「アンチセンス」核酸は、タンパク質をエンコードする「センス」核酸に対して相補性である、たとえば二本鎖cDNA分子のコーディング鎖に対して相補性であるか、またはmRNA配列に対して相補性であるヌクレオチド配列を含む。従って、アンチセンス核酸はセンス核酸に水素結合できる。アンチセンス核酸は全B7−4またはPD−1コーディング鎖に対して、またはその一部に対してのみ相補性であり得る。一具体例において、アンチセンス核酸分子はB7−4またはPD−1をエンコードするヌクレオチド配列のコーディング鎖の「コーディング領域」に対してアンチセンスである。「コーディング領域」なる用語は、アミノ酸残基中に翻訳されるコドンを含むヌクレオチド配列の領域を意味する。もう一つの具体例において、アンチセンス核酸分子は、B7−4またはPD−1をエンコードするヌクレオチド配列のコーディング鎖の「非コーディング領域」に対してアンチセンスである。「非コーディング領域」なる用語は、アミノ酸中に翻訳されないコーディング領域の側面に位置する5’および3’配列(5’および3’未翻訳領域とも称する)を意味する。
【0080】
本明細書において開示されているB7−4またはPD−1をエンコードするコーディング配列が与えられると、本発明のアンチセンス核酸配列をワトソンとクリックの塩基対合則に従って設計できる。アンチセンス核酸分子は、B7−4またはPD−1mRNAの全コーディング領域に対して相補性であり得るが、さらに好ましくは、B7−4またはPD−1mRNAのコーディングまたは非コーディング領域の一部のみに対してアンチセンスであるオリゴヌクレオチドである。たとえば、アンチセンスオリゴヌクレオチドはB7−4またはPD−1mRNAの翻訳開始部位を取り巻く領域に対して相補性であり得る。アンチセンスオリゴヌクレオチドは、たとえば、約5、10、20、25、30、35、40、45または50ヌクレオチドの長さであり得る。本発明のアンチセンス核酸は、当該分野において公知の方法を用いて化学合成および控訴結紮反応を用いて構築することができる。たとえば、アンチセンス核酸分子(たとえば、アンチセンスヌクレオチド)は、天然に存在するヌクレオチド、あるいは分子の生物学的安定性を増大するため、またはアンチセンスとセンス核酸間に形成される二本鎖の物理的安定性を増大させるために設計された様々に修飾されたヌクレオチドを用いて、化学的に合成することができる。たとえばホスホロチオエート誘導体およびアクリジン置換ヌクレオチドを用いることができる。アンチセンス核酸を生成するために用いることができる修飾されたヌクレオチドの例は、5−フルオロウラシル、5−ブロモウラシル、5−クロロウラシル、5−ヨードウラシル、ヒポキサンチン、キサチン、4−アセチルシトシン、5−(カルボキシヒドロキシメチル)ウラシル、5−カルボキシメチルアミノメチル−2−チオウリジン、5−カルボキシメチルアミノメチルウラシル、ジヒドウラシル、ベータ−D−ガラクトシルケオシン、イノシン、N6−イドペンテニルアデニン、2−メチルグアニン、3−メチルシトシン、5−メチルシトシン、N6−アデニン、7−メチルグアニン5−メチルアミノメチルウラシル、5−メトキシアミノメチル−2−チオウラシル、ベータ−D−マンノシルケオシン、5’−メトキシカルボキシメチルウラシル、5−メトキシウラシル、2−メチルチオ−N6−イソペンチルアデニン、ウラシル−5−オキシ酢酸(v)、ワイブトキソシン、シュードウラシル、ケオシン、2−チオシトシン、5−メチル−2−チオウラシル、2−チオウラシル、4−チオウラシル、5−メチルウラシル、ウラシル−5−オキシ酢酸メチルエステル、ウラシル−5−オキシ酢酸(v)、5−メチル−2−チオウラシル、3−(3−アミノ−3−N−2−カルボキシプロピル)ウラシル、(acp3)w、および2,6−ジアミノプリンを包含する。別法として、その中に核酸がアンチセンス配向においてサブクローンされた発現ベクターを用いて、アンチセンス核酸を生物学的に産生することができる(すなわち、次のサブセクションにおいてさらに記載するように、挿入された核酸から転写されたRNAは、関心のある標的核酸に対してアンチセンス配向を有するものである)。
【0081】
本発明のアンチセンス核酸分子は、典型的には対照に対して投与されるか、またはB7−4またはPD−1タンパク質をエンコードする細胞性mRNAおよび/またはゲノムDNAとハイブリッド形成するかまたは結合して、たとえば、転写および/または翻訳を阻害することによりタンパク質の発現を阻害するようにその場で生成される。ハイブリダイゼーションは、慣用のヌクレオチド相補性により、安定な二本鎖を形成するか、または、たとえば、DNA二本鎖と結合するアンチセンス核酸分子の場合においては二重らせんの大きな溝における特異的な相互作用による。本発明のアンチセンス核酸分子の投与経路の例は、組織部位での直接注射を包含する。別法として、アンチセンス核酸分子は、選択された細胞を標的とするために修飾でき、次いで全身的に投与できる。たとえば、全身性投与に関して、アンチセンス分子は、たとえば、アンチセンス核酸分子をペプチドあるい細胞表面レセプターまたは抗原と結合する抗体と結合させることにより、レセプターまたは選択された細胞表面上に発現された抗原と特異的に結合するように修飾できる。アンチセンス核酸分子はさらに、本明細書に記載するベクターを用いて細胞に送達することもできる。十分なアンチセンス分子の細胞内濃度を達成するために、アンチセンス核酸分子が強力なpol IIまたはpol IIIプロモーターの制御下におかれるベクター構築物が好ましい。
【0082】
さらにもう一つの具体例において、本発明のアンチセンス核酸はα−アノマー核酸分子である。α−アノマー核酸分子は、相補性RNAと特異的二本鎖ハイブリッドを形成し、ここにおいて、通常のβ−ユニットと反対に、鎖は互いに平行である(Gaultierら(1987)Nucleic Acids.Res.15:6625−6641)。アンチセンス核酸分子は、2’−o−メチルリボヌクレオチド(Inoueら(1987)Nucleic Acids.Res.15:6131−6148)またはキメラRNA−DNA類似体(Inoueら(1987)FEBS Lett.215:327−330)も含み得る。
【0083】
さらにもう一つの具体例において、本発明のアンチセンス核酸分子はリボザイムである。リボザイムはリボヌクレアーゼ活性を有する触媒RNA分子であり、これらが相補性領域を有するmRNAなどの一本鎖核酸分子を開裂できる。したがって、リボザイム(たとえば、B7−4またはPD−1mRNA転写物を触媒により開裂させ、これによりB7−4またはPD−1mRNAの翻訳を阻害するために、ハンマーヘッド型リボザイム(HaseloffおよびGerlach(1988)Nature334:585−591))を使用できる。B7−4またはPD−1エンコーディング核酸について特異性を有するリボザイムを、本明細書において開示するB7−4またはPD−1cDNAのヌクレオチド配列(すなわち、配列番号1、3、10、または11)に基づいて設計することができる。たとえば、活性部位のヌクレオチド配列が、B7−4またはPD−1エンコーディングmRNAにおいて開裂されるヌクレオチド配列と相補性であるTetrahymena L−19IVS RNAの誘導体を構築できる。たとえば、Cechら、米国特許第4987071号;およびCechら、米国特許第5116742号参照。別法として、RNA分子のプールから特異的リボヌクレアーゼ活性を有する触媒RNAを選択するために、B7−4またはPD−1mRNAを用いることができる。たとえば、Bartel,D.およびSzostak,J.W.(1993)Science261:1411−1418参照。
【0084】
別法として、B7−4またはPD−1遺伝子発現は、B7−4またはPD−1の調節領域(たとえば、B7−4またはPD−1プロモーターおよび/;またはエンハンサー)に対して相補性のヌクレオチド配列を標的にすることにより阻害することができ、標的細胞においてB7−4またはPD−1遺伝子の転写を防止する三重らせん構造が形成される。たとえば、一般に、Helene,C.(1991)Anticancer Drug Des.6(6):569−84;Helene,C.ra(1992)Ann.N.Y.Acad.Sci.660:27−36;およびMaher,L.J.(1992)Bioessays14(12):807−15参照。
【0085】
さらにもう一つの具体例において、本発明のB7−4またはPD−1核酸分子は、塩基部分、糖部分、またはホスフェート主鎖部分で、たとえば分子の安定性、ハイブリダイゼーション、または溶解度を向上させるために修飾することができる。たとえば、ペプチド核酸を生成させるために核酸分子のデオキシリボースホスフェート主鎖を修飾することができる(Hyrup,B.およびNielsen,P.E.(1996)Bioorg.Med.Chem.4(1):5−23)。本明細書において用いられる場合、「ペプチド核酸」または「PNA」なる用語は、デオキシリボース主鎖がシュードペプチド主鎖により置換され、4の天然の核酸塩基のみが保持されている核酸模倣物、たとえばDNA模倣物を意味する。PNAの天然の主鎖は、低イオン強度の条件下で、DNAおよびRNAと特異的ハイブリダイゼーションを可能にすることが示されている。PNAオリゴマーの合成は、HyrupおよびNielsen(1996)前出およびPerry−O’Keefeら(1996)Proc.Natl.Acad.Sci.USA93:14670−675において記載されているような標準的固相ペプチド合成法を用いて実施することができる。
【0086】
B7−4またはPD−1核酸分子のPNAは、治療および診断用途において用いることができる。たとえば、たとえば、転写または翻訳阻止の誘発あるいは複製の阻害により、遺伝子発現の配列特異性調節をするために、アンチセンスまたは抗原剤(antigen agent)として、PNAを用いることができる。B7−4またはPD−1核酸分子のPNAは、遺伝子における一塩基対突然変異の分析(たとえば、PNA−指向性PCRクランピング)においても;他の酵素(たとえば、S1ヌクレアーゼ(HyrupおよびNielsen(1996)前出))との組み合わせにおいて用いられる場合、「人工の制限酵素」として;またはDNAシーケンシングまたはハイブリダイゼーションのプローブまたはプライマーとして(Hyrup B.およびNielsen(1996)前出;Perry−O’Keefeeら(1996)前出)として用いることができる。
【0087】
もう一つの具体例において、親油性または他のヘルパー基をPNAに結合させるか、PNA−DNAキメラを形成するか、または当該分野において公知のリポソームまたは他のドラッグデリバリー技術により、B7−4またはPD−1のPNAを修飾することができる(たとえば、その安定性または細胞吸収を向上させるために)。たとえば、B7−4またはPD−1核酸分子のPNA−DNAキメラを産生することができ、これはPNAとDNAの利点を兼ね備える。このようなキメラは、DNA認識酵素(たとえば、RNAseHおよびDNAポリメラーゼ)がDNAタンパク質と相互作用するのを許容し、PNAタンパク質は高い結合親和性および特異性を提供する。PNA−DNAキメラは、塩基スタッキング、核酸塩基間の結合の数、および方向の点に関して、選択された適当な長さのリンカーを用いて結合させることができる(Hyrup B.およびNielsen(1996)前出)。PNA−DNAキメラの合成は、Hyrup B.およびNielsen(1996)前出およびFinn P.J.ら(1996)Nucleic Acids Res.24(17):3357−63に記載されているようにして行うことができる。例えば、DNA鎖は、標準的ホスホルアミダイトカップリング化学反応を用いて、固体支持体上で合成することができる。修飾されたヌクレオシド類似体(例えば、5’−(4−メトキシトリチル)アミノ−5’−デオキシ−チミジンホスホルアミダイト)を、PNAおよびDNAの5’末端間のリンカーとして用いることができる(Mag,M.ら(1989)Nucleic Acids Res.17:5973−88)。PNAモノマーを次に段階的な方法でカップリングさせて、5’PNAセグメントと3’DNAセグメントを有するキメラ分子を生成させる(Finn P.J.ら(1996)前出)。別法として、5’DNAセグメントおよび3’PNAセグメントを有するキメラ分子を合成することができる(Peterser,K.H.ら(1975)Bioorganic Med.Chem.Lett.5:1119−11124)。
【0088】
他の具体例において、オリゴヌクレオチドは、他の付加された基、たとえばペプチド(例えば、インビボで宿主細胞レセプターを標的にするため)、あるいは細胞膜(例えば、Letsingerら(1989)Proc.Natl.Acad.Sci.USA86:6553−6556;Lemaitreら(1987)Proc.Natl.Acad.Sci.USA84:648−652;PCT公開番号WO88/09810参照)または血液脳関門(例えば、PCT公開番号WO89/10134)を貫通する輸送を促進する化学物質を含んでもよい。加えて、オリゴヌクレオチドは、ハイブリダイゼーション誘発性開裂剤(例えば、Krolら(1988)Biotechniques6:958−976)または挿入剤(intercalating agent)(例えば、Zon(1988)Pharm.Res.5:539−549参照)で修飾することができる。この目的のために、オリゴヌクレオチドをもう一つの分子(例えば、ペプチド、ハイブリダイゼーション誘発性架橋剤、輸送因子、またはハイブリダイゼーション誘発性開裂剤)と結合させることができる。
【0089】
III.単離されたB7−4またはPD−1タンパク質および抗B7−4またはPD−1抗体
加えて、単離されたB7−4またはPD−1タンパク質およびその生物学的に活性な部分、ならびに抗B7−4またはPD−1抗体を調節剤として用いることができる。一具体例において、自然のB7−4またはPD−1タンパク質を、標準的タンパク質精製技術を用いて、適当な精製スキームにより、細胞または組織供給源から単離することができる。もう一つ別の具体例において、B7−4またはPD−1タンパク質は、組み換えDNA技術により産生される。組み換え発現の代わりに、B7−4またはPD−1タンパク質またはポリペプチドを、標準的ペプチド合成技術を用いて化学的に合成することができる。
本発明のもう一つの態様は、単離されたB7−4またはPD−1タンパク質に関する。好ましくは、B7−4またはPD−1タンパク質は、配列番号1、3、10または11によりエンコードされるアミノ酸配列を含む。もう一つの好ましい具体例において、タンパク質は配列番号2、4、または12のアミノ酸配列を含む。他の具体例において、タンパク質は、配列番号2、4、または12に示すアミノ酸配列と、少なくとも50%、少なくとも60%のアミノ酸同一性、さらに好ましくは70%アミノ酸同一性、さらに好ましくは80%、なお一層好ましくは90%または95%のアミノ酸同一性を有する。
【0090】
他の具体例において、本発明は、B7−4またはPD−1タンパク質の単離された部分を提供する。例えば、B7−4タンパク質は、シグナル配列、ならびにIgVドメインおよびIgCドメインを含む。配列番号2のシグナル配列は、およそアミノ酸1からおよそアミノ酸18までで示される。配列番号4のシグナル配列は、およそアミノ酸1からおよそアミノ酸18までで示される。配列番号2のIgVドメインは、およそアミノ酸19からおよびアミノ酸134までで示され、配列番号4のIgVドメインは、およそアミノ酸19からおよびアミノ酸134までで示される。配列番号2のIgCドメインは、およそアミノ酸135からおよびアミノ酸227までで示され、配列番号4のIgCドメインは、およそアミノ酸135からおよびアミノ酸227までで示される。配列番号2において例示されるB7−4分子は、およそアミノ酸228からおよそアミノ酸245で示される親水性テールを含む。配列番号4に例示されるB7−4ポリペプチドは、配列番号4のおよそアミノ酸239からおよそアミノ酸259で示される貫膜ドメインと配列番号4のおよそアミノ酸260からおよそアミノ酸290で示される細胞質ドメインを含む。
【0091】
PD−1ポリペプチドは288アミノ酸の長さであり、そのドメイン構造は当該分野において公知である(Shinoharaら(1994(Genomics23:704)。予想されるタンパク質の成熟形態は約268アミノ酸を含み、細胞外ドメイン(147アミノ酸)、貫膜ドメイン(27アミノ酸)、貫膜領域(27アミノ酸)および細胞質ドメイン(94アミノ酸)を含む。4の潜在的なN−グリコシル化部位が細胞外ドメインにおいて見いだされている(米国特許代5698520号)。2つのシステイン残基(cys54とcys123)間の68アミノ酸残基はV−セット配列のジスルフィド結合したイムノグロブリンドメインと類似性を有する(米国特許第5698520号)。
【0092】
本発明はさらに、B7−4またはPD−1タンパク質の溶解形態に関する。かかる形態は、例えば配列番号2に示すように天然に存在し得るか、または処理することができ、例えばB7−4またはPD−1タンパク質の細胞外ドメインを含み得る。B7−4細胞外ドメインの例は、配列番号4のアミノ酸19〜238を含む。PD−1細胞外ドメインの例は、配列番号12のおよそアミノ酸21〜288を含む。
一具体例において、B7−4ポリペプチドの細胞外ドメインは、B7−4ポリペプチドの成熟形態、例えば、IgVおよびIgCドメインを含むが、B7−4ポリペプチドの貫膜および細胞質ドメイン(例えば、配列番号4のおよそアミノ酸19からアミノ酸238)または配列番号2のおよそアミノ酸19からアミノ酸245を含まない。
【0093】
一具体例において、PD−1ポリペプチドの細胞外ドメインは、PD−1ポリペプチドの成熟形態、例えば、イムノグロブリンスーパーファミリードメイン(例えば、V−セット配列)を含むが、PD−1ポリペプチドの貫膜および細胞質ドメイン(例えば、配列番号12およそアミノ酸21〜288)を含まない。
B7−4またはPD−1タンパク質の生物学的に活性な部分は、完全長B7−4またはPD−1タンパク質よりも少ないアミノ酸を含み、B7−4またはPD−1タンパク質の少なくとも1つの活性、好ましくは自然の結合パートナーと結合する能力を示す、B7−4またはPD−1タンパク質のアミノ酸配列と十分相同性であるかまたはこれに由来するアミノ酸配列を含むペプチドを含む。典型的には、生物学的に活性なタンパク質は、B7−4またはPD−1タンパク質の少なくとも1つの活性を有するドメインまたはモチーフを含む。B7−4またはPD−1タンパク質の生物学的に活性な部分は、例えば少なくとも10、25、50、100、150、200またはそれ以上のアミノ酸の長さであるポリペプチドであり得る。
【0094】
2つのアミノ酸または2つの核酸配列間の同一性(%)を決定するために、最適の比較の目的で配列を整列させる(たとえば、最適な整列のために、第一および第二アミノ酸または核酸配列の一方または両方にギャップを導入し、一致しない配列を比較の目的で無視することができる)。好ましい具体例において、比較のために整列された参考配列の長さは、参考配列の長さの、少なくとも30%、好ましくは少なくとも40%、さらに好ましくは少なくとも50%、なおさらに好ましくは少なくとも60%、なおいっそう好ましくは少なくとも70%、80%、または90%のものである。対応する位置での残基または核酸を次いで比較し、一方の配列の位置が他方における対応する位置と同じ残基または核酸で占められているならば、分子はこの位置で同一である。2つの配列間の同一性(%)はしたがって2つの配列により共有される同じ位置の数の関数である(すなわち、同一性(%)=(同じ位置の数)/(位置の合計数)×100)。2つの配列間の同一性(%)は、2つの配列の最適な整列のために導入する必要のあるギャップの数、および各ギャップの長さを考慮して、配列により共有される同じ位置の数の関数である。本明細書において用いられる場合、アミノ酸または核酸「同一性」とは、アミノ酸または核酸「相同性」と等しい。
【0095】
配列の比較および2つの配列間の同一性(%)の決定は、数学アルゴリズムを用いて行うことができる。好ましい具体例において、2つのアミノ酸配列間の同一性は、GCGソフトウェアパッケージのGAPプログラムを用い、Blosum62マトリックスまたはPAM250マトリックスのいずれか、および16、14、12、10、8、6、または4のギャップ重量および1、2、3、4、5、または6の長さ重量を用いて決定される。さらにもう一つの好ましい具体例において、2つのヌクレオチド配列間の同一性(%)は、GCGソフトウェアパッケージのGAPプログラムを用い、NWSgapdna.CPMマトリックス、および40、50、60、70、または80のギャップ重量および1、2、3、4、5、または6の長さ重量を用いて決定される。
【0096】
本発明の核酸およびタンパク質配列はさらに、たとえば、他のファミリーメンバーまたは関連する配列を同定するために公共のデータベースに対して検索を行うための「問い合わせ配列」として用いることができる。このような検索は、Altschulら(1990)J.Mol.Biol.215:403−10のNBLASTおよびXBLASTプログラム(バージョン2.0)を用いて行うことができる。BLASTヌクレオチド検索は、NBLASTプログラム、スコア=100、ワード長=12で行うことができ、本発明のB7−4またはPD−1核酸と相同性のヌクレオチド配列を得る。BLASTタンパク質検索は、XBLASTプログラム、スコア=50、ワード長=3で行うことができ、本発明のB7−4またはPD−1タンパク質分子と相同性のアミノ酸配列を得る。比較の目的でギャップのあるアラインメントを得るために、Altschulら(1997)Nucleic Acids.Res.25(17):3389−3402に記載されているようにしてGappoed BLASTを用いることができる。BLASTおよびGapped BLASTプログラムを用いる場合、それぞれプログラム(たとえばXBLASTおよびNBLAST)のデフォルトパラメータを用いることができる。たとえば、デフォルトBlastinマトリックス1〜3(ギャップペナルティーを:イグジステンス11およびエクステンション1に設定)を用いて本発明のヌクレオチド配列を分析した。デフォルトセッティング:Blosum62マトリックス(ギャップペナルティーを、イグジステンス11およびエクステンション1に設定)を用いて本発明のアミノ酸配列を分析した。(例えば、NCBIウェブ頁を参照)。
【0097】
本発明はさらにB7−4またはPD−1キメラまたは融合タンパク質も提供する。本明細書において用いる場合、B7−4またはPD−1「キメラタンパク質」または「融合タンパク質」は非B7−4またはPD−1ポリペプチドと操作可能に結合されたB7−4またはPD−1ポリペプチドを含む。「B7−4またはPD−1ポリペプチド」とは、B7−4またはPD−1ポリペプチドに対応するアミノ酸配列を有するポリペプチドを意味し、一方、「非B7−4またはPD−1ポリペプチド」とは、B7−4またはPD−1タンパク質、たとえばB7−4またはPD−1タンパク質と異なり、同一または異なる生物から由来するタンパク質と実質的に相同性でないタンパク質に対応するアミノ酸配列を有するポリペプチドを意味する。B7−4またはPD−1融合タンパク質内で、B7−4またはPD−1ポリペプチドはB7−4またはPD−1タンパク質の全部または一部に対応し得る。好ましい具体例において、B7−4またはPD−1融合タンパク質はB7−4またはPD−1タンパク質の少なくとも一つの生物学的に活性な部分、たとえば、B7−4またはPD−1タンパク質の細胞外ドメインを含む。融合タンパク質内で、「操作可能に結合した」なる用語は、B7−4またはPD−1ポリペプチドおよび非B7−4またはPD−1ポリペプチドは互いにフレーム内で融合することを示すことを意図される。非B7−4またはPD−1ポリペプチドはB7−4またはPD−1ポリペプチドのN−末端またはC−末端と融合させることができる。
【0098】
たとえば、一具体例において、融合タンパク質は、B7−4またはPD−1配列がGST配列のC末端と融合したGST−B7−4またはGST−PD−1融合タンパク質である。もう一つの具体例において、融合タンパク質は、B7−4またはPD−1ヌクレオチド配列がpCEP4−HAなどのベクター(Herrscher,R.F.ら(1995)Genes Dev.9:3067−3082)中に挿入されているB7−4またはPD−1−HA融合タンパク質であり、したがってB7−4またはPD−1配列はインフルエンザ赤血球凝集素エピトープタグにフレーム内で融合する。かかる融合タンパク質は、組み換えB7−4またはPD−1タンパク質の精製を促進できる。
【0099】
B7−4またはPD−1融合タンパク質は、B7−4活性を有する第一ペプチドをエンコードするヌクレオチド配列および第一ペプチドの溶解性、親和性、安定性または価を変更する部分、たとえばイムノグロブリン定常領域に対応する第二のペプチドをエンコードするヌクレオチド配列の組み換え発現により産生することができる。好ましくは、第一ペプチドは、配列番号4に示す配列のB7−4ポリペプチドの部分(たとえば、免疫細胞の副刺激または阻害を調節するために十分である配列番号4に示す配列のアミノ酸残基1〜238または19〜238(シグナル配列の開裂後))からなる。もう一つの好ましい具体例において、第一ペプチドはPD−1ポリペプチドの一部(たとえば、免疫細胞の副刺激または阻害を調節するために十分な配列番号12に示す配列のアミノ酸残基1〜288(またはシグナルペプチドの開裂後の21〜288)の部分)からなる。第二のペプチドは、イムノグロブリン定常領域、たとえば、ヒトCγ1ドメインまたはCγ4ドメイン(たとえば、ヒンジ、ヒトIgCγ1のCH2およびCH3、またはヒトIgCγ4、たとえば、Caponら、米国特許第5116964号;第5580756号;第5844095号などを参照(これらは出典明示により本発明の一部とする))を含み得る。結果として得られる融合タンパク質は変更されたB7−4またはPD−1溶解性、結合親和性、安定性および/または価(すなわち、分子あたりの利用可能な結合部位の数)を有し、タンパク質の精製効率を増大させることができる。組み換え技により産生される融合タンパク質およびペプチドは、該タンパク質またはペプチドを含む細胞および培地の混合物から分泌され、単離することができる。別法として、タンパク質またはペプチドは細胞質に保持され、細胞を収穫し、溶解させ、タンパク質を単離する。細胞培養物は典型的には、宿主細胞、培地および他の副生成物を含む。細胞培養に適した培地は当該分野において周知である。タンパク質およびペプチドを細胞培養培地、宿主細胞、または両方から、タンパク質およびペプチド精製について当該分野において公知の技術を用いて単離することができる。宿主細胞をトランスフェクトし、タンパク質およびペプチドを精製するための技術は当該分野において公知である。
【0100】
特に好ましいB7−4またはPD−1Ig融合タンパク質は、ヒトB7−4またはイムノグロブリン定常領域に結合したPD−1の細胞外ドメイン部分または可変領域様ドメインを包含する。イムノグロブリン定常領域は、イムノグロブリン構造に固有のエフェクター活性を減少または除去する遺伝子修飾を含み得る。例えば、B7−4またはPD−1ポリペプチドの細胞外部分をエンコードするDNAを、ヒンジ、例えばWO97/28267に記載されているように部位指向性突然変異誘発により修飾されたヒトIgGγ1および/またはIgGγ4のCH2およびCH3領域をエンコードするDNAと結合させることができる。例えば、B7−4の融合タンパク質およびイムノグロブリン融合タンパク質を、本明細書においては交換可能に、「B7−4Ig」または「B7−4Fc」と称する。「Ig」または「Fc」を組み入れた他のバリエーションも用いることができる。
【0101】
好ましくは、本発明のB7−4またはPD−1融合タンパク質は、標準的組み換えDNA技術により産生される。例えば、異なるポリペプチド配列をコードするDNAフラグメントを慣例の技術にしたがって、例えば、結紮のために平滑末端またはスタッガー末端、適当な末端を得るために制限酵素消化、必要に応じて付着末端のフィリングイン、望ましくない結合を回避するためのアルカリホスファターゼ処理、および酵素結紮を用いて、フレーム内で結紮される。もう一つの具体例において、自動化DNA合成器を包含する慣用の技術を用いて融合遺伝子を合成することができる。別法として、2つの連続した遺伝子貫の相補性オーバーハングをもたらすアンカープライマーを用いて、遺伝子フラグメントのPCR増幅を行うことができ、これを続いてアニールし、再増幅させて、キメラ遺伝子配列が生じる(例えば、Current Protocols in Molecular Biology,Ausubelら編、John Wiley & Sons:1992)。さらに、すでに融合部分(例えば、GSTポリペプチドまたはHAエピトープタグ)をエンコードした多くの発現ベクターが商業的に入手可能である。B7−4またはPD−1エンコーディング核酸を、かかる発現ベクター中にクローンでき、融合部分をB7−4またはPD−1タンパク質にフレーム内で結合させる。
【0102】
もう一つの具体例において、融合タンパク質は、そのN−末端でヘテロローガスなシグナル配列を含むB7−4またはPD−1タンパク質である。ある宿主細胞(例えば、哺乳動物宿主細胞)、B7−4またはPD−1の発現および/分泌はヘテロローガスなシグナル配列の使用により増大させることができる。
【0103】
本発明のB7−4またはPD−1融合タンパク質を医薬組成物中に組み入れ、対象にインビボで投与することができる。B7−4またはPD−1融合タンパク質は、例えば、自己免疫疾患などの免疫学的障害の治療において、または移植片の拒絶を阻害する場合において、免疫反応の治療的調節に有用である。さらに、本発明のB7−4またはPD−1−融合タンパク質(例えば、完全長タンパク質またはその一部)を、対象において抗B7−4またはPD−1抗体を産生するため、B7−4またはPD−1を精製するため、そしてスクリーニング分析においてはB7−4とB7−4レセプター、例えばPD−1との相互作用を阻害する分子を同定するために免疫原として用いることができる。
【0104】
好ましくは、本発明のB7−4またはPD−1キメラまたは融合タンパク質は、標準的組み換えDNA技術により産生される。例えば、異なるポリペプチド配列をコードするDNAフラグメントは、慣例の技術にしたがって、例えば結紮のための平滑化またはスタッガー末端、適当な末端を得るための制限酵素消化、必要に応じて付着末端のフィリングイン、望ましくない結合を回避するためのアルカリホスファターゼ処理、および酵素結紮により、フレーム内で結紮される。もう1つの具体例において、融合遺伝子を、自動化DNA合成器を含む慣例の技術により合成することができる。別法として、2つの連続した核酸フラグメントの相補性オーバーハングをもたらすアンカープライマーを用いて核酸(例えば、遺伝子)フラグメントのPCR増幅を行うことができ、これをその後アニールし、再増幅して、キメラ核酸配列を得る(例えば、Current Protocols in Molecular Biology、Ausubelら編、John Wiley & Sons;1992参照)。さらに、すでに融合部分(例えば、GSTポリペプチド)をエンコードした多くの発現ベクターが商業的に入手可能である。B7−4またはPD−1エンコーディング核酸をかかる発現ベクター中にクローンでき、融合部分をB7−4またはPD−1タンパク質とフレーム内で結合させる。
【0105】
本発明はさらに、B7−4またはPD−1作用物質(模倣物)またはB7−4またはPD−1拮抗物質のいずれかとして機能するB7−4またはPD−1タンパク質変異体に対に関する。B7−4またはPD−1タンパク質の変異体を、突然変異誘発、例えば不連続点突然変異またはB7−4またはPD−1タンパク質の切断により生成させることができる。B7−4またはPD−1タンパク質の作用物質は、B7−4またはPD−1タンパク質の天然に存在する形態の生物学的活性の実質的に同じもの、またはサブセットを保有する。B7−4またはPD−1タンパク質の拮抗物質は、例えばB7−4またはPD−1タンパク質の細胞活性を拮抗的に調節することにより、B7−4またはPD−1タンパク質の天然に存在する形態の1以上の活性を阻害できる。かくして、特異的生物学的効果は、制限された機能を有する変異体での処置により惹起できる。一具体例において、タンパク質の天然に存在する形態の生物学的活性のサブセットを有する変異体での対象の治療は、B7−4またはPD−1タンパク質の天然に存在する形態での治療と比べて、対象における副作用が少ない。
【0106】
一具体例において、B7−4またはPD−1作用物質(模倣物)またはB7−4またはPD−1拮抗物質のいずれかとして機能するB7−4またはPD−1タンパク質の変異体は、変異体、例えば、B7−4またはPD−1タンパク質の点変異体または切断型変異体のコンビナトリアルライブラリーを、B7−4またはPD−1タンパク質作用物質または拮抗物質活性についてスクリーニングすることにより同定することができる。一具体例において、B7−4またはPD−1変異体の多様性ライブラリーは、核酸レベルでのコンビナトリアル突然変異誘発により生成され、多様性遺伝子ライブラリーによりエンコードされる。B7−4またはPD−1変異体の多様性ライブラリーは、例えば、合成オリゴヌクレオチドの混合物を遺伝子配列中に酵素により結紮することにより産生でき、潜在的なB7−4またはPD−1配列の縮退セットが個々のポリペプチド、または別法として、B7−4またはPD−1配列を含むさらに大きな融合タンパク質(例えば、ファージディスプレー用)のセットとして発現可能である。縮退オリゴヌクレオチド配列から潜在的なB7−4またはPD−1変異体のライブラリーを産生するために用いることができる方法は数多くある。縮退遺伝子配列の化学合成は、自動DNA合成器において行うことができ、合成遺伝子を次いで適当な発現ベクター中に結紮する。1つの混合物において遺伝子の縮退セットの使用は、1つの混合物において、潜在的なB7−4またはPD01配列の所望のセットをエンコードする配列の全ての準備を可能にする。縮退オリゴヌクレオチドを合成する方法は、当該分野において公知である(例えば、Narang,S.A.(1983)Tetrahedron39:3;Itakuraら(1984)Annu.Rev.Biochem.53:323;Itakuraら(1984)Science198:1056;Ikeら(1983)Nucleic Acids Res.11:477参照)。
【0107】
加えて、スクリーニングとその後のB7−4またはPD−1タンパク質の変異体の選択のために、B7−4またはPD−1フラグメントの多様性集団を生じさせるために、B7−4またはPD−1タンパク質コーディング配列のフラグメントのライブラリーを用いることができる。一具体例において、B7−4またはPD−1コーディング配列の二本鎖PCRフラグメントを、分子当たりほぼ1回だけニッキングが起こる条件下でヌクレアーゼで処理し、二本鎖DNAを変性させ、DNAを変性させて、異なるニック生成物から得られるセンス/アンチセンスを含み得る二本鎖DNAを形成し、一本鎖タンパク質をS1ヌクレアーゼでの処理により改質された二本鎖から除去し、結果として得られるフラグメントライブラリーを発現ベクター中に結紮することにより、コーディング配列フラグメントのライブラリーを生じさせることができる。この方法により、様々なサイズのB7−4またはPD−1タンパク質のN−末端、C−末端および内部フラグメントをエンコードする発現ライブラリーを誘導することができる。
【0108】
点突然変異または切断により調製されるコンビナトリアルライブラリーの遺伝子産物をスクリーニングするための技術、および選択された性質を有する遺伝子産物についてcDNAライブラリーをスクリーニングするための技術がいくつか当該分野において知られている。かかる技術は、B7−4またはPD−1タンパク質の組み合わせ突然変異誘発により生じる遺伝子ライブラリーの急速なスクリーニングに適合させ得る。大きな遺伝子ライブラリーをスクリーニングするための高スループット分析ができる最も広く用いられている技術は、典型的には、遺伝子ライブラリーを複製可能な発現ベクター中にクローンし、適当な細胞を結果として得られるベクターのライブラリーで形質転換し、コンビナトリアルライブラリーを、所望の活性の検出が、その産物が検出された遺伝子をエンコードするベクターの単離を促進する条件下で発現することを含む。ライブラリーのける機能的変異体の頻度を向上させる新規技術である反復的アンサンブル突然変異誘発(REM)新規技術をB7−4またはPD−1変異体を同定するためのスクリーニング分析との組み合わせにおいて用いることができる(ArkinおよびYouvan(1992)Proc.Natl.Acad.Sci.USA89:7811−7815;Delagraveら(1993)Protein Eng.6(3):327−331)。
【0109】
一具体例において、多様性B7−4またはPD−1ライブラリーを分析するために細胞ベースの分析を利用することができる。例えば、発現ベクターのライブラリーを通常B7−4またはPD−1を合成し、分泌する細胞系中にトランスフェクトすることができる。トランスフェクトされた細胞を次いで培養すると、B7−4またはPD−1および特定の変異体B7−4またはPD−1が分泌され、細胞上清における変異体の発現のB7−4またはPD−1活性に対する影響を、例えば多くの機能的分析のいずれかにより検出することができる。プラスミドDNAを次いで阻害、または別法としてB7−4またはPD−1活性の増強を行う細胞から回収でき、個々のクローンをさらに特徴づけることができる。
【0110】
天然に存在するアミノ酸のみからなるB7−4またはPD−1ポリペプチドに加えて、B7−4またはPD−1ペプチド模倣物も提供される。ペプチド類似体は通常製薬業界においてテンプレートペプチドと類似した性質を有する非ペプチド薬剤として使用される。これらのタイプの非ペプチド化合物は、「ペプチド模倣物」と称し(Fauchere,J(1986)Adv.Drug Res.15:29;VeberおよびFreidinger(1985)TINS p392;およびEvansら(1987)J.Med.Chem.30:1229(出典明示により本発明の一部として参照される))、通常コンピューター化された分子モデリングの助けをかりて発展される。等しい治療的または予防的効果を生じるために、治療的に有用なペプチドと構造的に類似しているペプチド模倣物を用いることができる。一般に、ペプチド模倣物は、ヒトB7−4またはPD−1などのパラダイムポリペプチド(すなわち、生物学的または薬理学的活性を有するポリペプチド)と構造的に類似しているが、当該分野において公知の方法および次の文献においてさらに記載されている方法により、−CH2NH−、−CH2S−、−CH2−CH2−、−CH=CH−(シスおよびトランス)、−COCH2−、CH(OH)CH2−、および−CH2SO−からなる群から選択される結合により所望により置換されていてもよい1以上のペプチド結合を有する:Spatola,A.F.“Chemisty and Biochemistry of Amino Acids,Peptides,and Proteins”WEinstein,B.編、Marcel Dekker,New York、p267(1983);Spotla,A.F.、Vega Data(1983年3月)、第1巻、第3刷、“Peptide Backbone Modifications”(概論);Morley,J.S.(1980)Trends Pharm.Sci.pp463〜468(概論);Hudson,D.ら(1979)Int.J.Pept.Prot.Res.14:177〜185(−CH2NH−、CH2CH2−);Spatola,A.ら(1986)Life Sci.38:1243〜1249(−CH2S−);Hann,M.M.(1982)J.Chem.Soc.Perkin Trans.I.307〜314(−CH−CH−、シスおよびトランス);Almquist,R.G.ら(190)J.Med.Chem.23:1392〜1398(−COCH2−);Jennings−White,C.ら(1982)Tetrahedron Lett.23:2533(−COCH2−);Szelke,M.ら、European Appln.EP45665(1982)CA:97:39405(1982)(−CH(OH)CH2−);Holladay,M.W.ら(1983)Tetrahedron Lett.(1983)24:4401〜4404(−C(OH)CH2−);およびHruby,V.J.(1982)Life Sci.(1982)31:189〜199(−CH2−S−)(それぞれは出典明示により本発明の一部として参照される)。特に好ましい非ペプチド結合は−CH2NH−である。かかるペプチド模倣物は、たとえば、より経済的な製造、より高い化学的安定性、向上された薬理学的性質(半減期、吸収、効能、効力など)、変更された特異性(たとえば、広範囲の生物学的活性)、減少した抗原性などを包含する、ポリペプチドの具体例よりも著しく優れた利点を有し得る。ペプチド模倣物の標識は、通常、1以上の標識を、直接またはスペーサー(たとえばアミド基)を介して、定量的構造−活性データおよび/または分子モデリングにより予想されるペプチド模倣物上の干渉しない位置に共有結合させることを含む。このような干渉しない位置は、一般的に治療的効果を生じるためにペプチド模倣物が結合する巨大分子と直接接触しない位置である。ペプチド模倣物の誘導(たとえば標識)は、該ペプチド模倣物の所望の生物学邸または薬理学的活性を実質的に干渉すべきではない。
【0111】
さらに安定なペプチドを生じさせるために、B7−4またはPD−1アミノ酸配列の1以上のアミノ酸の同じタイプのD−アミノ酸での系統的な置換(たとえば、L−リシンの代わりにD−リシン)を用いることができる。加えて、B7−4またはPD−1アミノ酸配列または実質的に同一の配列バリエーションを含む限定されたペプチドは、当該分野において公知の方法(RizoおよびGierasch(1992)Annu.Rev.Biochem.61:387、出典明示により本発明の一部として参照される);たとえば、ペプチドを環化させる分子内ジスルフィドブリッジを形成できる内部システイン残基を添加することにより得ることができる。
【0112】
本発明において同定されたB7−4またはPD−1ポリペプチドのアミノ酸配列により、当業者らはB7−4またはPD−1ポリペプチド配列およびその配列変異体に対応するポリペプチドを産生することが可能になる。かかるポリペプチドはB7−4またはPD−1ポリペプチドをエンコードするポリヌクレオチドの発現により、原核または真核宿主細胞において、しばしばさらに大きなポリペプチドの一部として、産生することができる。別法として、かかるペプチドは化学的方法により合成することができる。組み換え宿主においてヘテロローガスなタンパク質を発現する方法、ポリペプチドの化学合成、およびインビトロの翻訳は当該分野において一般的であり、Maniatisら、Molecular Cloning:A Laboratory Manual(1989)、第2版、Cold Spring Harbor,N.Y.;BergerおよびKimmel、Methods in Enzymology、第152巻、Guide to Molecular Cloning Techniques(1987)、Academic Press,Inc.,San Diego,Calif,;Merrifield,J.(1969)J.Am.Chem.Soc.91:501;Chaiken I.M.(1981)CRC Crit.Rev.BIochem.11:255;Kaiserら(1989)Science243:187;Merrifield,B.(1986)Science232:342;Kent,S.B.H.(1988)Annu.Rev.Biochem.57:957;およびOfford,R.E.(1980)Semisynthetic Proteins,Wiley Publishing(出典明示により本発明の一部として参照される)。
【0113】
ペプチドは、典型的には直接化学合成により産生することができ、たとえば、B7−4/PD−1相互作用の作用物質または拮抗物質として用いることができる。ペプチドは、N−末端および/またはC−末端と共有結合により結合する非ペプチド部分で修飾されたペプチドとして産生することができる。ある好ましい具体例において、カルボキシ末端またはアミノ末端のいずれか、または両方は化学的に修飾される。末端アミノおよびカルボキシ基の最も一般的な修飾は、それぞれアセチル化およびアミド化である。アミノ末端修飾、たとえばアシル化(たとえば、アセチル化)またはアルキル化(たとえば、メチル化)およびカルボキシ末端修飾、たとえばアミド化、ならびに環化を含む他の末端修飾を本発明の様々な具体例に組み入れることができる。あるアミノ末端および/またはカルボキシ末端修飾および/またはコア配列に対するペプチド伸長は、有利な物理的、化学的、生物学的、および薬理学的性質、たとえば向上された安定性、増大した有効性および/または効力、血清プロテアーゼに対する耐性、望ましい薬物動態学的性質などをもたらし得る。たとえば、患者における副刺激を変更することにより、疾患を治療するためにペプチドを治療的に用いることができる。
【0114】
単離されたB7−4またはPD−1タンパク質、またはその一部またはフラグメント(またはかかるポリペプチドをエンコードする核酸分子)を、ポリクローナルおよびモノクローナル抗体調製のための標準的技術を用いてB7−4またはPD−1と結合する抗体を生じるために免疫原として用いることができる。完全長B7−4またはPD−1タンパク質を用いることができるか、あるいは本発明は免疫原として用いられるB7−4またはPD−1の抗原性ペプチドフラグメントを提供する。B7−4またはPD−1の抗原性ペプチドは少なくとも8のアミノ酸残基を含み、B7−4またはPD−1のエピトープを含み、したがって該ペプチドに対して生じる抗体は、B7−4PD−1と特異的免疫複合体を形成する。好ましくは、抗原性ペプチドは少なくとも10のアミノ酸残基、さらに好ましくは少なくとも15アミノ酸残基、さらに一層好ましくは少なくとも20アミノ酸残基、最も好ましくは少なくとも30アミノ酸残基を含む。
【0115】
別法として、B7−4またはPD−1ポリペプチドの抗原性ペプチドフラグメントを免疫原として用いることができる。B7−4またはPD−1ポリペプチドの抗原性ペプチドフラグメントは、典型的に配列番号2、4、12、または39に示すアミノ酸配列の少なくとも8アミノ酸残基を含み、B7−4またはPD−1ポリペプチドのエピトープを含むので、該ペプチドに対して生じる抗体はB7−4またはPD−1分子と免疫複合体を形成する。抗原性ペプチドにより含まれる好ましいエピトープはタンパク質の表面上に位置するB7−4またはPD−1の領域、例えば親水性領域である。一具体例において、抗体はB7−4またはPD−1分子の一部(例えば、細胞外部分)を含む分子と実質的に特異的に結合する。もう一つの具体例において、抗体はB7−4またはPD−1ポリペプチドと特異的に結合する。
【0116】
好ましくは、抗原性ペプチドは少なくとも約10のアミノ酸残基、さらに好ましくは少なくとも約15のアミノ酸残基、さらに好ましくは少なくとも約20のアミノ酸残基、最も好ましくは少なくとも約30のアミノ酸残基を含む。抗原性ペプチドにより含まれる好ましいエピトープは、タンパク質の表面上に位置し、B7−4またはPD−1ポリペプチドに独特なB7−4またはPD−1ポリペプチドの領域、例えば親水性領域である。一具体例において、かかるエピトープは1つの種、例えばマウスまたはヒトから得られるB7−4またはPD−1タンパク質に特異的であり得る(すなわち、全種にわたって保存されないB7−4またはPD−1ポリペプチドの領域におよぶ抗原性ペプチドが免疫原として用いられ;このような非保存残基を本発明において提供されるアラインメントを用いて決定することができる)。親水性領域を識別するためにB7−4またはPD−1タンパク質の標準的疎水性分析を行うことができる。
【0117】
B7−4またはPD−1免疫原は典型的には、適当な対象(たとえば、ウサギ、ヤギ、マウスまたは他の哺乳動物)を免疫源で免疫化することにより抗体を調製するために用いられる。適当な免疫原調製物は、たとえば、組み換えにより発現されたB7−4またはPD−1タンパク質またはペプチドフラグメント、または化学的に合成されたB7−4またはPD−1ペプチドフラグメントを含み得る。調製物は、さらにアジュバント、たとえばフロインドの完全または不完全アジュバントまたは類似した免疫刺激剤を含む。適当な対象を免疫原B7−4またはPD−1調製物で免疫化することにより、ポリクローナル抗B7−4またはPD−1抗体応答が誘発される。
【0118】
もう一つの具体例において、核酸ワクチンを様々な手段により、たとえば、注射(たとえば、筋肉内、皮内、または皮膚中に粒子を注入するために粒子加速器または圧縮気体を使用する遺伝子銃を用いた真皮中DNAコートされた金粒子のバイオリスティック注射(Haynesら(1996)J.Biotechnol.44:37))により投与することができる。別法として、核酸ワクチンを非侵襲性手段により投与することができる。たとえば、純粋または脂質処方されたDNAを、呼吸器系または標的とされる他の場所、たとえば、パイエル板にDNAの経口送達により送達することができる(Schubbert(1997)Proc.Natl.Acad.Sci.USA94:961)。弱毒化微生物を粘膜表面への送達に用いることができる(Sizemoreら(1995)Science270:29)。
【0119】
本発明のさらにもう一つの具体例は、抗B7−4抗体または抗PD−1抗体に関する。かかる抗体は、たとえば、動物を免疫原性B7−4またはPD−1タンパク質、あるいはB7−4またはPD−1ポリペプチドに独特のその免疫原性部分で免疫化し、次いでB7−4またはPD−1タンパク質、またはそのフラグメントと特異的に結合する抗体を動物から単離することにより得られる。
ポリクローナル抗B7−4またはPD−1抗体は、前記のように、適当な対象をB7−4またはPD−1免疫原で免疫化することにより調製することができる。免疫化された対象における抗B7−4またはPD−1抗体価を、標準的技術、たとえば固定化B7−4またはPD−1ポリペプチドを用いた酵素結合免疫吸着検定法(ELISA)を用いて、長時間にわたってモニターすることができる。必要に応じて、B7−4またはPD−1ポリペプチドに対する抗体分子を哺乳動物から(たとえば血液から)単離し、プロテインAクロマトグラフィーなどの周知の技術により精製して、IgGフラクションを得ることができる。免疫化後の適当な時点で、たとえば、抗B7−4またはPD−1抗体価が最高の時に、抗体産生細胞を対象から得、もともとKohlerおよびMilstein(1975)Nature256:495−497に記載されている標準的技術、たとえばハイブリドーマ技術(Brownら(1981)J.Immunol.127:539−46;Brownら(1980)J.Biol.Chem.255:4980−83;Yehら(1976)Proc.Natl.Acad.Sci.76:2927−31;およびYehら(1982)Int.J.Cancer29:269−75)も参照)、最近のヒトB細胞ハイブリドーマ技術(Kozborら(1983)Immunol.Today4:72)、EBV−ハイブリドーマ技術(Coleら(1985)Monoclonal Antibodies and Cancer Therapy,Alan R.Liss,Inc.、pp77−96)またはトリオーマ技術によりモノクローナル抗体を調製することができる。モノクローナル抗体ハイブリドーマを産生する技術は周知である(一般に、Kenneth R.H.in Monoclonal Antibodies:A New Dimension In Biological Analyses、Plenum Publishing Corp.,New York,New York(1997)Somatic Cell Genet.3:231−36参照)。簡単に説明すると、前記のB7−4またはPD−1免疫原で免疫化した哺乳動物から得られる重要な細胞系(典型的には、骨髄腫)をリンパ腫(典型的には脾細胞)と融合させ、結果として得られるハイブリドーマ細胞の培養上清をスクリーンして、B7−4またはPD−1ポリペプチドと好ましくは特異的に結合するハイブリドーマ産生モノクローナル抗体を同定する。
【0120】
リンパ球および永久増殖細胞系を融合させるために用いられる多くの一般的なプロトコルのいずれかを、抗B7−4またはPD−1モノクローナル抗体を生じさせる目的で適用できる(たとえば、Galfre,G.ら(1977)Nature266:55052;Gefterら(1977)前出;Lerner(1981);Kenneth(1980)前出参照)。さらに、当業者らは、有用であるこのような方法の多くのバリエーションがあることを理解するであろう。典型的には、永久増殖細胞系(たとえば、骨髄腫細胞系)は、リンパ球と同じ哺乳動物の種から得られる。たとえば、ネズミハイブリドーマは、本発明の免疫原調製物で免疫化されたマウスから得られるリンパ球を永久増殖マウス細胞系と融合することにより調製できる。好ましい永久増殖細胞系は、ヒポキサンチン、アミノプテリンおよびチミジンを含む培地(「HAT培地」)に対して感受性であるマウス骨髄腫細胞系である。多くの骨髄腫細胞系いずれか、たとえば、P3−NS1/1−Ag4−1、P3−x63−Ag8.653またはSp2/O−Ag14骨髄腫細胞系を標準的技術に従って融合パートナーとして用いることができる。これらの骨髄腫細胞系は、アメリカン・タイプ・カルチャー・コレクション(ATCC)(ROckville,Md.)から入手可能である。典型的には、HAT−感受性マウス骨髄腫細胞を、ポリエチレングリコール(「PEG」)を用いてマウス脾臓細胞と融合させる。融合の結果得られるハイブリドーマ細胞を次いでHAT培地を用いて選択し、これは未融合および非生産的に融合された骨髄腫細胞を殺す(未融合脾細胞は形質転換されないので数日後に死ぬ)。たとえば標準的ELISA分析を用いて、B7−4またはPD−1分子と結合する抗体についてハイブリドーマ培養上清をスクリーンすることにより、本発明のモノクローナル抗体を産生するハイブリドーマ細胞を検出する。
【0121】
モノクローナル抗体−分泌ハイブリドーマを調製するための代替法として、組み換え組み合わせイムノグロブリンライブラリー(たとえば、抗体ファージディスプレーライブラリー)をB7−4またはPD−1でスクリーンし、これによりB7−4またはPD−1ポリペプチドと結合するイムノグロブリンライブラリーメンバーを単離することにより、モノクローナル抗B7−4またはPD−1抗体を同定することができる。産生およびスクリーニングファージディスプレーライブラリーのキットは商業的に入手可能である(たとえば、Pharmacia Recombinant Phage Antibody System、カタログ番号27−9400−01;およびStratagene SurfZAP Phage Display Kit、カタログ番号240612)。加えて、抗体ディスプレーライブラリーを生じ、スクリーンするのに用いられる方法および試薬の例は、たとえば、Ladnerら、米国特許第5233409;Kangら、国際特許公開番号WO92/18619;Dowerら、国際特許公開番号WO91/17271;Winterら、国際特許公開番号WO92/20791;Marklandら、国際特許公開番号92/15679;Breitlingら、国際特許公開番号WO93/01288;McCaffertyら、国際特許公開番号WO92/01047;Garrardら、国際特許公開番号WO92/09690;Ladnerら、国際特許公開番号WO90/02809;Fuchsら(1991)Biotechnology(NY)9:1369−1372;Hayら(1992)Hum.Antibod.Hybridomas3:81−85;Huseら(1989)Science246:1275−1281;Griffithsら(1993)EMBO J.12:725−734;Hawkinsら(1992)J.Mol.Biol.226:889−896;Clarksonら(1991)Nature352:624−628;Gramら(1992)Proc.Natl.Acad.Sci.USA89:3576−3580;Garrardら(1991)Biotechnology(NY)9:1373−1377;Hoogenboomら(1991)Nucleic Acids Res.19:4133−4137;Barbasら(1991)Proc.Natl.Acad.Sci.USA88:7978−7982;およびMcCaffertyら(1990)Nature348:552−554において見いだすことが起きる。
【0122】
加えて、標準的組み換えDNA技術を用いて調製できる、ヒトおよび非ヒトタンパク質の両方を含む、組み換え抗B7−4またはPD−1抗体、例えばキメラおよびヒト化モノクローナル抗体は、本発明の範囲内に含まれる。このようなキメラおよびヒト化モノクローナル抗体は、当該分野において公知の組み換えDNA技術により、例えば、Robinsonら、国際特許公開番号PCT/US86/00269;Akiraら、欧州特許出願第184187号;Taniguchi,M.ら、欧州特許出願第171496号;Morrisonら、欧州特許出願第173494号;Neubergerら、PCT出願番号WO86/01533;Cabillyら、米国特許第4816567号;Cabillyら、欧州特許出願第125023号;Betterら(1988)Science240:1041−1043;Liuら(1987)Proc.Natl.Acad.Sci.USA84:3439−3443;Liuら(1987)J.Immunol.139:3521−3526;Sunら(1987)Proc.Natl.Acad.Sci.84:214−218;Nishimuraら(1987)Cancer Res.47:999−1005;Woodら(1985)Nature314:446−449;およびShawら(1988)J.Natl.Cancer Inst.80:1553−1559);Morrison,S.L.(1985)Science229:1202−1207;Oiら(1986)Biotechniques4:214;Winter、米国特許第5225539号;Jonesら(1986)Nature321:552−525;Verhoeyenら(1988)Science239:1534;およびBeidlerら(1988)J.Immunol.141:4053−4060に記載されている方法を用いて産生できる。
【0123】
加えて、米国特許第5565332号において開示されたものなどの標準的プロトコルに従ってヒト化抗体を調製することができる。もう一つの具体例において、抗体鎖または特異的結合対メンバーを、特異的結合対メンバーのポリペプチド鎖と複製可能な遺伝子ディスプレーパッケージの成分の融合物をエンコードする核酸分子を含むベクターおよび単一の結合対メンバーの第二のポリペプチド鎖をエンコードする核酸分子を含むベクター間の組み合わせにより、当該分野において公知の技術、例えば米国特許第5565332号、第5871907号、または第5733743号に記載されている技術を用いて産生することができる。細胞におけるタンパク質機能を阻害するために細胞内抗体を使用することも、当該分野において公知である(例えば、Carlson,J.R.(1988)Mol.Cell.Biol.8:2638−2646;Biocca,S.(1990)EMBO J.9:101−108;Werge,T.M.ら(1990)FEBS Lett.274:193−198;Carlson,J.R.(1993)Proc.Natl.Acad.Sci.USA90:7427−7428;Narasco,W.A.ら(1993)Proc.Natl.Acad.Sci.USA90:7889−7893;Bioccal,S.ら(1994)Biotechnology(NY)12:396−399;Chen,S−Y.ら(1994)Hum.Gene Ther.5:595−601;Duan,Lら(1994)Proc.Ntl.Acad.Sci.USA91:5075−5079;Chen,S−Y.ら(1994)Proc.Natl.Acad.Sci.USA91:5932−5936;Beerli,R.R.ら(1993)J.Biol.Chem.269:23931−23936;Beerli,R.R.ら(1994)Biochem.Biophys.Res.Commun.204:666−672;Mhashikar,A.M.ら(1995)EMBO J.14:1542−1551;Richardson,J.H.ら(1995)PRoc.Natl.Acad.Sci.USA92:3137−3141;PCT公開番号を94/02610(Marascoら);およびPCT公開番号WO95/03832(Duanら))。
【0124】
一具体例において、本発明において用いられる抗体は、二重特異性抗体である。二重特異性抗体は1つの抗体分子内に2つの異なる抗原の結合部位を有する。抗原結合は同時または連続的である。トリオーマおよびハイブリッドハイブリドーマは二重特異性抗体を分泌できる細胞系の2つの例である。ハイブリッドハイブリドーマまたはトリオーマにより産生される二重特異性抗体の例は、米国特許第4474893号に開示されている。二重特異性抗体は化学的手段(Staerzら(1985)Nature314:628、およびPerezら(1985)Nature316:354)およびハイブリドーマ技術(StaerzおよびBevan(1986)Proc.Natl.Acad.Sci.USA、83:1453、およびStaerzおよびBevan(1986)Immunol.Today7:241)により構築される。二重特異性抗体は、米国特許第5959084号にも記載されている。二重特異性抗体のフラグメントは米国特許第5798229号に記載されている。
【0125】
二重特異性剤は、ハイブリドーマまたは異なる抗体を調製する他の細胞を融合することによりヘテロハイブリドーマを調製し、続いて両抗体を産生し、コアセンブルするクローンを同定することにより、調製することができる。これらはさらに、完全イムノグロブリン鎖またはその一部、たとえばFabおよびFv配列の化学的または遺伝子結合によっても調製することができる。抗体成分はPD−1またはB7−4と結合することができる。
【0126】
B7−4またはPD−1ポリペプチドを標準的技術、例えば、アフィニティークロマトグラフィーまたは免疫沈降により単離するために、抗B7−4またはPD−1抗体(例えば、モノクローナル抗体)を用いることができる。抗B7−4またはPD−1抗体は、細胞からの天然のB7−4またはPD−1ポリペプチド、および組み換えにより産生されたB7−4または宿主細胞において発現されたPD−1ポリペプチドの精製を促進できる。さらに、B7−4またはPD−1タンパク質を検出するために(例えば、細胞溶解物または細胞上清において)、抗B7−4またはPD−1抗体を用いることができる。抗体を検出可能な物質とカップリング(すなわち、物理的結合)させることにより、検出を促進できる。従って、一具体例において、本発明の抗B7−4またはPD−1抗体は、検出可能な物質で標識される。検出可能な物質の例は、様々な酵素、補欠分子、蛍光物質、発光物質および放射性物質を包含する。適当な酵素の例は、ホースラディッシュペルオキシダーゼ、アルカリホスファターゼ、β−ガラクトシダーゼ、またはアセチルコリンエステラーゼを包含し;適当な補欠分子複合体の例は、ストレプトアビジン/ビオチンおよびアビジン/ビオチンを包含し;適当な蛍光物質の例は、ウンベリフェロン、フルオレセイン、フルオレセインイソチオシアネート、ローダミン、ジクロロトリアジニルアミンフルオレセイン、アンシルクロリドまたはフィコエリスリンを包含し;発光物質の例はルミノールを包含し;適当な放射性物質の例は、125I、131I、35S、および3Hを包含する。
【0127】
IV.組み換え発現ベクターおよび宿主細胞
B7−4またはPD−1ファミリータンパク質(またはその一部)をエンコードする核酸分子は、ベクター、好ましくは発現ベクター中に含まれ得る。本明細書において用いられる場合、「ベクター」なる用語は、それが結合しているもう一つ別の核酸を輸送できる核酸分子を意味する。ベクターの1つのタイプは「プラスミド」であり、これはその中にさあなるDNAセグメントを結紮できる環状二本鎖DNAループを意味する。もう一つ別のタイプのベクターはウイルスベクターであり、ここにおいてさらなるDNAセグメントはウイルスゲノム中に結紮できる。ある種のベクターは、これらが導入される宿主細胞において自発的に複製できる(例えば、複製の細菌起源を有する細菌ベクターおよびエピソーム哺乳動物ベクター)。他のベクター(例えば、非エピソーム哺乳動物ベクター)は、宿主細胞中に導入する際に、宿主細胞のゲノム中に組み入れられ、その結果、宿主細胞ゲノムとともに複製される。さらに、ある種のベクターはこれらが操作可能に結合される遺伝子の発現を行うことができる。かかるベクターを本発明において「発現ベクター」と称する。一般に、組み換えDNA技術において有用な発現ベクターは、しばしばプラスミドの形態である。プラスミドはベクターの最も一般的に用いられる形態であるので、本明細書において、「プラスミド」および「ベクター」は交換可能に用いることができる。しかしながら、本発明は、発現ベクターのこのような他の形態、例えば、相当する役割を果たすウイルスベクター(例えば、複製能欠損レトロウイルス、アデノウイルスおよびアデノ関連ウイルス)を包含することを意図される。
【0128】
組み換え発現ベクターは、発現、例えば、宿主細胞における核酸分子の指示細胞においてPD−1またはB7−4の構成性発現または誘導発現に適した形態の本発明の核酸分子を含み得る。このことは、組み換え発現ベクターは、発現される核酸はいれつに操作可能に結合された、発現に用いられる宿主細胞に基づいて選択される1以上の調節配列を含むことを意味する。組み換え発現ベクター内で、「操作可能に結合した」とは、興味のあるヌクレオチド配列が調節配列と、ヌクレオチド配列の発現を許容する方法で結合することを意味する(例えば、インビトロ転写/翻訳システムまたはベクターが宿主細胞中に導入される場合は宿主細胞において)。「調節配列」なる用語は、プロモーター、エンハンサーおよび他の発現調節エレメント(たとえば、ポリアデニル化シグナル)を包含する。このような調節配列は、たとえば、Goeddel(1990)Methods Enzymol.185:3−7に記載されている。調節配列は、多くのタイプの宿主細胞においてヌクレオチド配列の構成性発現を行うもの、およびある種の宿主細胞(たとえば、組織特異性調節配列)においてのみヌクレオチド配列の発現を行うものを包含する。発現ベクターのデザインは、形質転換される宿主細胞の選択、所望のタンパク質の発現レベルなどの因子に依存し得る。本発明の発現ベクターを宿主細胞中に導入することができ、結果として、本明細書において記載される核酸(たとえば、B7−4またはPD−1ファミリータンパク質、B7−4またはPD−1タンパク質の変異体、融合タンパク質など)によりエンコードされる融合タンパク質またはペプチドを包含するタンパク質またはペプチドが生じる。
【0129】
本発明の組み換え発現ベクターは、原核または真核細胞におけるB7−4またはPD−1タンパク質を発現するために設計できる。たとえば、B7−4またはPD−1タンパク質は、細菌細胞、たとえば、イー・コリ、昆虫細胞(バクロウイルス発現ベクターを使用)酵母細胞または哺乳動物細胞において発現できる。適当な宿主細胞はGoeddel(1990)前出においてさらに議論されている。別法として、組み換え発現ベクターを、インビトロで、たとえばT7プロモーター調節配列およびT7ポリメラーゼを用いて転写し、翻訳することができる。
【0130】
原核細胞におけるタンパク質の発現は、融合または非融合タンパク質のいずれかの発現を行う構成性または誘導プロモーターを含むベクターを用いてイー・コリにおいて行われることが最も多い。融合ベクターは多くのアミノ酸をその中でエンコードされるタンパク質、通常組み換えタンパク質のアミノ末端に加える。このような融合ベクターは、典型的には次の3つの目的に役立つ:1)組み換えタンパク質の発現を増大させる;2)組み換えタンパク質の溶解度を増大させる;および3)アフィニティー精製においてリガンドとして作用することにより組み換えタンパク質の精製を助ける。しばしば、融合発現ベクターにおいて、原核開裂部位を融合部分と組み換えタンパク質の接合点で導入し、融合タンパク質の精製後、組み換えタンパク質の融合部分からの除去を可能にする。このような酵素、およびその同源認識配列は、ファクターXa、トロンビンおよびエンテロキナーゼを含む。典型的な融合発現ベクターは、それぞれグルタチオン−S−トランスフェラーゼ(GST)、マルトースE結合タンパク質、またはプロテインAを標的組み換えタンパク質と融合させるpGEX(Pharmacia Biotin Inc;Smith,D.B.およびJohnson,K.S.(1988)Gene67:31−40)、pMAL(New England Biolabs,Beverly,MA)およびpRIT5(Pharmacia,Piscataway,NJ)を含む。
【0131】
精製融合タンパク質は、B7−4またはPD−1活性分析(たとえば、以下に詳細に記載する直接分析または競合アッセイ)において、あるいはたとえばB7−4またはPD−1タンパク質に対して特異的な抗体を生成するために用いることができる。
適当な誘導非融合イー・コリ発現ベクターの例は、pTrc(Amannら(1988)Gene69:301−315)およびpET11d(Studierら(1990)Methods Enzymol.185:60−89)。pTrcベクターからの標的遺伝子発現は、ハイブリッドtrp−lac融合プロモーターから宿主RNAポリメラーゼ転写に依存する。pET11dベクターからの標的遺伝子発現は、同時発現されたウイルスRNAポリメラーゼ(T7gn1)により影響を受けるT7gn10−lac融合プロモーターからの転写に依存する。このウイルスポリメラーゼは、lacUV5プロモーターの転写制御下、T7gn1遺伝子を有するレジデントプロファージから宿主下部BL21(DE3)またはHMS174(DE3)により供給される。
【0132】
イー・コリにおける組み換えタンパク発現を最大にするための一方法は、組み換えタンパク質をタンパク質分解により開裂させる能力が損なわれている宿主細菌においてタンパク質を発現することである。もう一つの方法は、各アミノ酸についての個々のコドンが位・コリにおいて優先的に利用されるものであるように、発現ベクター中に挿入される核酸の核酸配列を変更することである(Wadaら(1992)Nucleic Acids Res.20:2111−2118)。本発明の核酸は配列のこのような変更は、標準的DNA合成技術により行うことができる。
【0133】
もう一つの具体例において、B7−4またはPD−1発現ベクターは、酵母発現ベクターである。酵母エス・セレビシエにおける発現ベクターの例は、pYepSec1(Baldariら(1987)EMBO J.6:229−234)、pMFあ(KurjanおよびHerskowitz(1982)Cell30:933−943)、pJRY88(Schultzら(1987)Gene54:113−123)、pYES2(Invitrogen Corporation,San Diego,CA)、およびpicZ(Invitrogen Corp,San Diego,CA)を包含する。
【0134】
別法として、B7−4またはPD−1ポリペプチドはバクロウイルス発現ベクターを用いて昆虫細胞において発現することができる。培養された昆虫細胞(たとえば、Sf9細胞)におけるタンパク質の発現に利用できるバクロウイルスベクターは、pAcシリーズ(Smithら(1983)Mol.Cell Biol.3:2156−2165)およびpVLシリーズ(Lucklow,V.A.およびSummers,M.D.(1989)Virology170−:31−39)を包含する。
【0135】
さらにもう一つの具体例において、本発明の核酸は、哺乳動物発現ベクターを用いて哺乳動物細胞において発現される。哺乳動物発現ベクターの例は、pMex−NeoI、pCDM8(Seed,B.(1987)Nature329:840)およびpMT2PC(Kaufmanら(1987)EMBO J.6:187−195)を包含する。哺乳動物細胞において用いられる場合、発現ベクターの制御機能は、しばしばウイルス調節エレメントによりもたらされる。たとえば、通常用いられるプロモーターは、ポリオーマ、アデノウイルス2、サイトメガロウイルスおよびシミアンウイルス40から得られる。原核および真核細胞の両方の他の適当な発現系については、Sambrook,J.ら、Molecular Cloning:A Laboratory Manual.第2版、Cold Spring Harbor Laboratory,Cold Spring Harbor Laboratory Press,Cold Spring Harbor,NY、1989の第16章および第17章参照。
【0136】
もう一つの具体例において、組み換え哺乳動物発現ベクターは、特定のセルタイプにおいて優先的に核酸を発現できる(たとえば、核酸を発現するために、組織特異性調節エレメントが用いられる)。組織特異性調節エレメントは当該分野において公知である。組織特異性プロモーターの非限定的例は、アルブミンプロモーター(肝臓特異性;Pinkertら(1987)Genes Dev.1:268−277)、リンパ特異性プロモーター(CalameおよびEaton(1988)Adv.Immunol.43:235−275)、特にT細胞レセプターのプロモーター(WinotoおよびBaltimore(1989)EMZBO J.8:729−733)およびイムノグロブリン(Banerjiら(1983)Cell33:729−740;QueenおよびBaltimore(1983)Cell33:741−748)、ニューロン特異性プロモーター(たとえば、神経フィラメントプロモーター;ByneおよびRuddle(1989)Proc.Natl.Acad.Sci.USA86:5473−5477)、膵臓特異性プロモーター(Edlundら(1985)Science230:912−916)、および乳腺特異性プロモーター(たとえば、乳清プロモーター;米国特許大4873316号および欧州特許出願公開番号264166)を包含する。発展的に調節されるプロモーター、たとえば、ネズミホメオボックスプロモーター(KesselおよびGruss(1990)Science249:374−379)およびα−フェトプロテインプロモーター(CampesおよびTilghman(1989)Genes Dev.3:537−546)も含まれる。
【0137】
さらに、哺乳動物細胞において使用される誘導調節システム、たとえば、遺伝子発現が重金属イオンにより調節されるシステム(たとえば、Mayoら(1982)Cell29:99−108;Brinsterら(1982)Nature296:39−42;Searleら(1985)Mol.Cell.Biol.5:1480−1489参照)、熱ショック(たとえば、Noureら(1991)Heat Shock Response、Nouer,L.編、CRC,Boca Raton、FL、pp167−220参照)、ホルモン(たとえば、Leeら(1981)Nature294:228−232;Hynesら(1981)Proc.Natl.Acad.Sci.USA78:2038−2042;Klockら(1987)Nature329:734−736;IsraelおよびKaufman(1989)Nucleic Acids.Res.17:2589−2604;およびPCT公開番号WO93/23431参照)、FK506関連分子(たとえば、PCT公開番号WO94/18317参照)またはテトラサイクリン(Gossen,M.およびBujard,H.(1992)Proc.Natl.Acad.Sci.USA89:5547−5551;Gossen,M.ら(1995)Science268:1766−1769;PCT公開番号WO94/29442lおよびPCT公開番号WO96/01313)は当該分野において公知である。従って、もう一つの具体例において、本発明はB7−4またはPD−1DNAが誘導真核プロモーターと操作可能に結合する組み換え発現ベクターを提供し、その結果、真核細胞におけるB7−4またはPD−1タンパク質の誘導発現を許容する。
【0138】
本発明はさらに、アンチセンス配向で発現ベクター中にクローンされた本発明のDNA分子を含む組み換え発現ベクターを提供する。すなわち、DNA分子は、B7−4またはPD−1mRNAに対してアンチセンスであるRNA分子の発現(DNA分子の転写による)を許容する方法で調節配列と操作可能に結合する。さまざまなセルタイプにおいてアンチセンスRNA分子の連続した発現を行うアンチセンス配向でクローンされた核酸と操作可能に結合する調節配列を選択できる。たとえば、アンチセンスRNAの構成性、組織特異性またはセルタイプ特異性発現を行うウイルスプロモーターおよび/エンハンサー、または調節配列を選択できる。アンチセンス発現ベクターは、アンチセンス核酸が有効性の高い調節領域の制御下で賛成される組み換えプラスミド、ファージミドまたは弱毒化ウイルスの形態であり得、その活性は、ベクターが導入されるセルタイプにより決定できる。アンチセンス遺伝子の遺伝子発現の調節についての議論に関しては、Weintraub,H.ら(1986)「遺伝子分析の分子ツールとしてのアンチセンスRNA」Reviews−Trends in Genetics、第1(1)巻参照。
【0139】
本発明はさらに、本発明の組み換え発現ベクターが導入された宿主細胞に関する。このような用語は、特定の対象細胞だけでなく、かかる細胞の子孫または可能性のある子孫も意味する。突然変異または環境的影響のいずれかのためにその後の世代においてある修飾が起こりえるので、このような子孫は、実際には親細胞と同一で内科もしれないが、それでも本発明において用いられる用語の範囲内に含まれる。
宿主細胞は、任意の原核または真核細胞である。たとえば、B7−4またはPD−1タンパク質を細菌細胞、たとえば、イー・コリ、昆虫細胞、酵母または哺乳動物細胞(たとえば、チャイニーズ・ハムスター卵巣細胞(CHO)またはCOS細胞)において発現することができる。他の適当な宿主細胞は当業者には公知である。
【0140】
ベクターDNAを、慣用の形質転換またはトランスフェクション技術により原核または真核細胞中に導入することができる。本明細書において用いられる場合、「形質転換」および「トランスフェクション」なる用語は、リン酸カルシウムまたは塩化カルシウム共沈、DEAE−デキストランによるトランスフェクション、リポフェクション(lipofection)、またはエレクトロポレーションを含む、外来核酸(たとえば、DNA)を宿主細胞に導入するための様々な当該分野で承認された技術を意味する。宿主細胞を形質転換またはトランスフェクトするための適当な方法は、Sambrookら(Molecular Cloning:A Laboratory Mannual.第2版、Cold Spring Harbor Laboratory,Cold Spring Harbor Laboratory Press,Cold Spring Harbor,NY,1989)、および他の実験室マニュアルにおいて見いだすことができる。
【0141】
哺乳動物の安定なトランスフェクションに関して、使用される発現ベクターおよびトランスフェクション技術に応じて、細胞の小さなフラクションのみがそのゲノム中に外来DNAを組み入れることができることが知られている。これらの構成要素を同定し、選択するために、選択マーカー(たとえば、抗生物質に対して耐性)をエンコードする遺伝子は、一般に関心のある遺伝子と共に宿主細胞中に導入される。好ましい選択マーカーは、G418、ヒグロマイシンおよびメトトレキサートなどの薬剤に対して耐性を賦与するものを包含する。選択マーカーをエンコードする核酸は、B7−4またはPD−1タンパク質をエンコードするものと同じベクター上で宿主細胞中に導入できるか、または別のベクター上に導入できる。導入された核酸で安定にトランスフェクトされた細胞は、薬剤選択により同定できる(たとえば、選択マーカー遺伝子を組み入れた細胞は生存し、他の細胞は死ぬ)。
【0142】
本発明の宿主細胞、たとえば、培養物における原核または真核宿主細胞を、B7−4またはPD−1タンパク質を産生(すなわち、発現)するために用いることができる。したがって、本発明はさらに、本発明の宿主細胞を用いてB7−4またはPD−1タンパク質を産生するための方法を提供する。一具体例において、方法は、宿主細胞(B7−4またはPD−1タンパク質をエンコードする組み換え発現ベクターが導入されている)を適当な培地中で培養して、B7−4またはPD−1タンパク質が産生されるようにすることを含む。もう一つの具体例において、方法はさらに、培地または宿主細胞からB7−4またはPD−1タンパク質を単離することを含む。
【0143】
ある宿主細胞は、ヒト以外のトランスジェニック動物を調製するために用いることができる。たとえば、一具体例において、宿主細胞は、その中にB7−4またはPD−1コーディング配列が導入された受精卵母細胞または胚幹細胞である。かかる宿主細胞を次いで、外因性B7−4またはPD−1配列がそのゲノム中に導入されているヒト以外のトランスジェニック動物、または外因性B7−4またはPD−1配列が変更されているホモローガスな組み換え動物を創るために用いることができる。このような動物は、B7−4またはPD−1ポリペプチドの機能および/または活性を研究するため、およびB7−4またはPD−1活性の修飾物質を同定および/または評価するために有用である。本明細書において用いられる場合、「トランスジェニック動物」とは、ヒト以外の動物、好ましくは哺乳動物、さらに好ましくは齧歯類、たとえばラットまたはマウスであって、該動物の1以上の細胞がトランス遺伝子であるものである。トランスジェニック動物の他の例は、ヒト以外の霊長類、ヒツジ、イヌ、ウシ、ヤギ、ニワトリ、両生類などを包含する。トランス遺伝子は、トランスジェニック動物が発生し、成体動物のゲノムにおいて保持される細胞のゲノム中に組み入れられ、その結果、該トランスジェニック動物の1以上のセルタイプまたは組織においてエンコードされた遺伝子産物の発現が行われる外因性DNAである。本明細書において用いられる場合、「ホモローガスな組み換え動物」はヒト以外、好ましくは哺乳動物、さらに好ましくはマウスであり、ここにおいて外因性B7−4またはPD−1遺伝子は、該動物の発生前に、該動物の細胞、たとえば該動物の胚細胞中に導入された外因性遺伝子と外因性DNA分子巻のホモローガスな組み換えにより変更されている。
【0144】
トランスジェニック動物は、たとえば、マイクロインジェクション、レトロウイルス感染、および卵母細胞を偽妊娠したメスの里親動物において発生させることにより、B7−4またはPD−1エンコーディング核酸分子を受精卵母細胞のオス生殖核中に導入することにより創ることができる。配列番号1、3、10、または11のpB7−4またはPD−1cDNA配列をトランス遺伝子としてヒト以外の動物のゲノム中に導入することができる。別法として、マウスまたはラットB7−4またはPD−1遺伝子などのヒトB7−4またはPD−1遺伝子のヒト以外の相同体をトランス遺伝子として用いることができる。別法として、B7−4またはPD−1遺伝子相同体、たとえばもう一つのB7−4またはPD−1ファミリーメンバーを、配列番号1、3、10、または11のB7−4またはPD−1ファミリーcDNA配列とのハイブリダイゼーションに基づいて単離し(前記第1章においてさらに記載)、トランス遺伝子として用いることができる。イントロン配列およびポリアデニル化シグナルも、トランス遺伝子の発現の有効性を増大させるためにトランス遺伝子中に含めることができる。組織特異性調節配列は操作可能にB7−4またはPD−1トランス遺伝子と結合させて、B7−4またはPD−1タンパク質の特定の細胞への発現を行うことができる。胚操作およびマイクロインジェクションによりトランスジェニック動物、特にマウスなどの動物を調製する方法は、当該分野において一般的になりつつあり、たとえば、米国特許第4736866号および第4870009号(両方ともLederら)、米国特許第4873191号(Wagnerら)およびHogan,B.Manipulating the Mouse Embryo(Cold Spring Harbor Laboratory Press,Cold,Spring Harbor,N.Y.,1986)に記載されている。他のトランスジェニック動物を産生するために類似の方法が用いられる。トランスジェニック創始動物を、その下のっむにおけるB7−4またはPD−1トランス遺伝子の存在および/または動物の組織細胞におけるB7−4の発現に基づいて同定することができる。トランスジェニック創始動物を次にトランス遺伝子を有するさらなる動物を飼育するために用いることができる。さらに、B7−4またはPD−1タンパク質をエンコードするトランス遺伝子を有するトランスジェニック動物をさらに、他のトランス遺伝子を有する他のトランスジェニック動物を飼育するために用いることができる。
【0145】
ホモローガスな組み換え動物を創るために、これによりB7−4またはPD−1遺伝子を変更、たとえば機能的に破壊するために欠失、付加または置換が導入されているB7−4またはPD−1遺伝子の少なくとも一部を含むベクターが調製される。B7−4またはPD−1遺伝子はヒト遺伝子(たとえば、配列番号1、3、10、または11)であり得るが、より好ましくは、ヒトB7−4またはPD−1遺伝子の非ヒト相同体である(たとえば、配列番号1、3、10、または11のヌクレオチド配列でのストリンジェントなハイブリダイゼーションにより単離されるcDNA)。たとえば、マウスゲノムにおける外因性B7−4またはPD−1遺伝子を変更するために適したホモローガスな組み換えベクターを構築するために、マウスB7−4またはPD−1遺伝子を用いることができる。好ましい具体例において、ベクターは、ホモローガスな組み換えにより、外因性B7−4またはPD−1遺伝子が機能的に破壊されるように設計される(すなわち、もはや機能的タンパク質をエンコードしない;「ノックアウト」ベクターともいう)。別法として、ベクターは、ホモローガスな組み換えにより、外因性B7−4またはPD−1遺伝子が突然変異するか、または他の方法で変更されるが、それでも機能的タンパク質をエンコードするように設計できる(たとえば、外因性B7−4またはPD−1タンパク質の発現を変更するように上流調節領域を変更できる)。ホモローガスな組み換えベクターにおいて、B7−4またはPD−1遺伝子の変更された部分は、B7−4またはPD−1遺伝子の追加の核酸配列がその5’および3’末端で側面に位置して、ベクターおよび胚幹細胞における外因性B7−4またはPD−1遺伝子により運ばれる外因性B7−4またはPD−1遺伝子間にホモローガスな組み換えが起こる。追加のフランキングB7−4またはPD−1核酸配列は外因性遺伝子でのホモローガスな組み換えが成功するために十分な長さを有する。典型的には、数キロベースのフランキングDNA(5’および3’末端の両方で)はベクター中に含まれる(たとえば、ホモローガスな組み換えベクターの記載についてはThomas,K.R.およびCapecchi,M.R.(1987)Cell51:503参照)。ベクターを胚幹細胞系中に導入し(たとえば、エレクトロポレーションによる)、導入されたB7−4またはPD−1遺伝子が外因性B7−4またはPD−1遺伝子と同様に再結合されている細胞を選択する(たとえば、Li,E.ら(1992)Cell69:915)。選択された細胞を次いで動物(たとえば、マウス)の胚盤胞中に注入して、凝集キメラを形成する(たとえば、Bradley,A.Teratocarcinomas and Embryonic Stem Cells:A Practical Approach,Robertson,E.J.編(IRL,Oxford,1987)pp113−152参照)。動物の全ての細胞がトランス遺伝子の生殖細胞伝達により同様に再結合されたDNAを含む動物を飼育するために、キメラ胚を次いで適当な偽妊娠した里親動物中に移植し、胚を出産させることができる。同様に再結合されたDNAをその胚細胞中に有する子孫を用いることができる。ホモローガスな組み換えベクターおよびホモローガスな組み換え動物を構築する方法は、Bradley,A.(1991)Curr.Biotehnol.2:823−829およびPCT国際公開番号WO90/11354(Le Mouellecら;WO91/01140(Smithiesら);WO92/0968(Zijlstraら);およびWO93/04169(Bernsら)においてさらに記載されている。
【0146】
前記に加えて、当業者は、ホモローガスな組み換えについて当該分野で公知の他の方法を本発明に適用できることを理解するであろう。酵素補助部位特異性組み込みシステムは、Creレコンビナーゼ−ロックス標的システム(たとえば、Baubonis,W.およびSauer,B.(1993)Nucleic Acids Res.21:2025−2029;およびFukushige,S.およびSauer,B.(1992)Proc.Natl.Acad.Sci.USA89:7095−7909に記載)およびFLPレコンビナーゼ−FRT標的システム(たとえば、Dang,D.T.およびPerrimon,N.(1992)Dev.Genet.13:367−375;およびFiering,S.ら(1993)Proc.Natl.Acad.Sci.USA90:8469−8473に記載)を包含する。テトラサイクリン調節誘導ホモローガス組み換えシステム、たとえばPCT公開番号WO94/29442およびPCT公開番号WO96/01313に記載されているものも用いることができる。
【0147】
たとえば、もう一つの具体例において、トランス遺伝子の規制された発現を許容する選択されたシステムを含むヒト以外のトランスジェニック動物を創ることができる。このようなシステムの一例は、バクテリオファージP1のcre/loxPレコンビナーゼシステムである。cre/loxPレコンビナーゼシステムの記述に関しては、たとえば、Laksoら(1992)Proc.Natl.Acad.Sci.USA9:6232−6236参照。レコンビナーゼシステムのもう一つの例は、サッカロミセス・セレビシエ(Saccharomyces cerevisiae)のFLPレコンビナーゼシステムである(O’Gormanら(1991)Science251:1351−1355)。トランス遺伝子の発現を調節するためにcre/loxPレコンビナーゼシステムを使用する場合、Creレコンビナーゼと選択されたタンパク質の両方をエンコードするトランス遺伝子を含む動物が必要とされる。かかる動物は、たとえば2匹のトランスジェニック(一方は選択されたタンパク質をエンコードするトランス遺伝子を含み、他方はレコンビナーゼをエンコードするトランス遺伝子を含む)を交尾させることにより、「ダブル」トランスジェニック動物の構築により提供できる。
【0148】
本明細書に記載するヒト以外のトランスジェニック動物のクローンも、Wilmut,I.ら(1997)Nature385:810−813およびPCT国際公開番号WO97/07668およびWO97/07669に記載されている方法に従って創ることができる。簡単に説明すると、トランスジェニック動物から得られる細胞、たとえば、体細胞を単離し、成長周期をでて、Go期に入らせることができる。静止細胞を次いで、たとえば電気パルスの使用により、静止細胞が単離された同じ種の動物から得られる核を取り除いた卵母細胞と融合させることができる。再構成された卵母細胞を次いで培養して、桑実胚または未分化胚芽細胞を発生させ、次いで偽妊娠したメス里親動物に移す。このメス里親動物の子孫は、細胞、たとえば体細胞がたん利される動物のクローンである。
【0149】
V.医薬組成物
B7−4またはPD−1モジュレーター(たとえば、前記のB7−4またはPD−1核酸分子、タンパク質、抗体、あるいはB7−4またはPD−1活性および/または発現のモジュレーターあるいはB7−4およびPD−1の間の相互作用のモジュレーターとして確認されている化合物を含む、B7−4またはPD−1阻害または刺激剤)を投与に適した医薬組成物中に組み入れることができる。かかる組成物は、典型的には核酸分子、タンパク質、または抗体および医薬的に許容される担体を含む。本明細書において用いられる場合、「医薬的に許容される担体」とは、医薬投与に適合するありとあらゆる溶媒、分散媒体、コーティング、抗菌剤および抗真菌剤、等張剤および吸収遅延剤などを包含することを意図される。医薬的に活性な物質のこのような媒体および薬剤の使用は当該分野においては一般的である。慣用の媒体または薬剤が活性化合物と不適合性である場合を除いて、その組成物における使用も考慮される。追加の活性化合物も組成物中に組み入れることができる。
【0150】
本発明の医薬組成物は、意図される投与経路と適合するように処方される。投与経路の例は、非経口、例えば、静脈内、皮内、皮下、経口(例えば、吸入)、経皮(局所)、経粘膜、および直腸投与を包含する。非経口、皮内、または皮下投与に用いられる溶液または懸濁液は、次の成分を含むことができる:滅菌希釈剤、例えば注射用水、塩溶液、固定油、ポリエチレングリコール、グリセリン、ポリプロピレングリコールまたは他の合成溶媒;抗菌剤、例えばベンジルアルコールまたはメチルパラベン;酸化防止剤、例えば、アスコルビン酸または重亜硫酸ナトリウム;キレート化剤、例えば、エチレンジアミンテトラ酢酸;緩衝剤、例えば酢酸塩、クエン酸塩またはリン酸塩および等張性を調節するための薬剤、例えば塩化ナトリウムまたはデキストロース。pHは酸または塩基、例えば塩酸または水酸化ナトリウムで調節することができる。非経口製剤は、アンプル、使い捨て注射器またはガラスまたはプラスチック製の複数回投与用バイアル中に封入できる。
【0151】
注射可能な用途に適した医薬組成物は、滅菌水性溶液(水溶性の場合)または分散液および滅菌注射可能な溶液または分散液の即席調製用滅菌粉末を包含する。静脈内投与に関しては、適当な担体は、生理食塩水、静菌水、Cremophor EL(BASF、Parsippany,NJ)またはリン酸塩緩衝塩溶液(PBS)を包含する。あらゆる場合において、組成物は無菌でなければならず、注射針通過が容易である程度に流動性であるべきである。これは製造および貯蔵条件下で安定でなければならず、微生物、例えば細菌および真菌の汚染作用に対して保護されなければならない。担体は、例えば、水、エタノール、ポリオール(例えば、グリセロール、プロピレングリコール、および液体ポリエチレングリコールなど)、およびその適当な混合物を含む溶媒または分散液媒体であり得る。流動性は、例えば、レシチンなどのコーティングの使用、分散液の場合には必要とされる粒子サイズの維持および界面活性剤の使用により維持される。微生物の作用の防止は、様々な抗菌剤および抗真菌剤、例えば、パラベン、クロロブタノール、フェノール、アスコルビン酸、チメロサールなどにより達成できる。多くの場合において、等張剤、たとえば、糖、ポリアルコール、例えばマンニトール、ソルビトール、塩化ナトリウムを組成物中に含むのが好ましい。注射可能な組成物の長期におよぶ吸収は、組成物中に吸収を遅らせる薬剤、例えば、アルミニウムモノステアレートおよびゼラチンを含めることにより達成できる。
【0152】
無菌注射可能な溶液は、活性化合物(例えば、B7−4またはPD−1タンパク質または抗B7−4またはPD−1抗体)を必要な量において適当な溶媒中、前記の成分またはその組み合わせと共に組み入れ、必要に応じてその後濾過滅菌することにより調製できる。一般に、活性化合物を、塩基性分散媒体と前記の内の必要な他の成分を含む無菌ビヒクル中に組み入れることにより分散液を調製することができる。無菌注射溶液を調製するための無菌粉末の場合において、好ましい調製法は、真空乾燥およびそのあらかじめ無菌濾過された溶液から活性成分と任意の追加の所望の成分の粉末が得られる凍結乾燥である。
【0153】
経口組成物は、一般に、不活性希釈剤または可食性担体を含む。これらはゼラチンカプセル中に封入できるか、または圧縮されて錠剤にできる。経口治療的投与の目的に関して、活性化合物を賦形剤と共に組み入れて、錠剤、トローチまたはカプセルの形態において使用することができる。経口組成物はさらに、洗口剤として使用するために液体担体を用いて調製することもでき、ここにおいて、液体担体中の化合物は、経口投与され、くちゅくちゅし、はき出すかまたは飲み込まれる。医薬的に適合性の結合剤、および/またはアジュバント物質を組成物の一部として含めることができる。錠剤、丸薬、トローチなどは、次の成分のうちの任意のもの、または類似した性質を有する化合物を含むことができる:バインダー、例えば微結晶セルロール、トラガカントガムまたはゼラチン;賦形剤、例えばデンプンまたはラクトース、崩壊剤、例えばアルギン酸、プリモゲル、またはコーンスターチ;滑剤、例えばステアリン酸マグネシウムまたはSterotes;潤滑剤、例えばコロイド状二酸化珪素;甘味料、例えばシュークロースまたはサッカリン;あるいはフレーバー、例えばペパーミント、メチルサリチレート、またはオレンジフレーバー。
【0154】
吸入による投与に関して、化合物は、適当なプロペラント、例えば二酸化炭素などの気体を含む圧縮された容器またはディスペンサーからエアゾルスプレー、あるいはネブライザーの形態において送達される。
全身性投与は、経粘膜または経皮手段によることもできる。経粘膜または経皮投与に関して、浸透されるバリヤに対して適当な浸透剤が処方において用いられる。このような浸透剤は当該分野において一般的に公知であり、例えば、経粘膜投与については、洗剤、胆汁酸塩、およびフシジン酸誘導体を包含する。経粘膜投与は、鼻スプレーまたは坐剤伸しようにより達成できる。経皮投与については、活性化合物は当該分野において一般的に知られている軟膏、膏薬、ジェル、またはクリームに処方される。
【0155】
化合物は、直腸送達のための坐剤(例えば、慣用の坐剤基剤、例えばカカオ脂または他のグリセリドを用いる)または貯留性浣腸剤の形態においても調製できる。
一具体例において、インプランとおよびマイクロカプセル送達システムを含む放出調節型処方などの調節剤は、化合物が体から急速に排除されるのを防ぐ担体を用いて調製される。エチレンビニルアセテート、ポリアンヒドライド、ポリグリコール酸、コラーゲン、ポリオルトエステル、およびポリ酢酸などの生物分解性ポリマーを用いることができる。かかる処方の調製法は、当業者らには自明でなければならない。物質はまた、Alza CorporationおよびNova Pharmaceuticals,Inc.から商業的に入手可能である。リポソーム懸濁液(ウイルス抗原に対するモノクローナル抗体に感染した細胞を標的とするリポソームを包含する)はさらに、医薬的に許容される担体としても用いることができる。これらは当業者に公知の方法に従って、例えば、米国特許大4522811号に記載されているようにして調製できる。
【0156】
投与の簡便性および投与量の均一性のために、単位投与形態の経口または非経口組成物を処方するのが特に有利である。本発明において用いられる単位投与形態とは、治療される患者についての単一用量として適した物理的に独立した単位を意味し;各単位は、必要とされる医薬担体と組み合わせて所望の治療的効果を生じるために計算されたあらかじめ決められた量の活性成分を含む。本発明の単位投与形態の明細は、活性化合物の独自の特性および達成されるべき特定の治療効果、および個人を治療するためのかかる活性化合物を配合する分野に固有の制限に直接依存し、指示される。
【0157】
かかる化合物の毒性および治療効果は、例えば、LD50(集団の50%に対する致死量)およびED50(集団の50%において治療的に有効な用量)を決定するために、細胞培養または実験動物において標準的製薬法により決定することができる。毒性と治療的効果の用量比は治療指数であり、LD50/ED50の比として表すことができる。大きな治療指数を示す化合物が好ましい。有毒な副作用を示す化合物を用いることができるが、未感染細胞に対する潜在的なダメージを最小限に抑え、従って副作用を軽減するために、病気に冒された組織の部位に対してかかる化合物を標的とさせるデリバリーシステムを設計するために注意しなければならない。
【0158】
細胞培養分析および動物実験から得られるデータをヒトにおいて使用する一連の用量の処方において用いることができる。かかる化合物の用量は、好ましくは、毒性が低いかまたは毒性のないED50を含む循環濃度内にある。用量は、用いられる用量および用いられる投与経路に応じてこの範囲内で変化し得る。本発明の方法において用いられる化合物に関して、治療的に有効な用量は、最初は細胞培養分析から評価できる。用量は、細胞培養において決められたIC50(すなわち、症状の最大阻害の半分を達成する試験化合物の濃度)を含む循環血漿濃度を達成するために動物モデルにおいて処方することができる。このような情報は、ヒトにおいて有用な用量をさらに正確に決定するために用いることができる。血漿中のレベルは、たとえば、高性能液体クロマトグラフィーにより測定できる。
【0159】
本発明の核酸分子は、ベクター中に挿入でき、遺伝子療法ベクターとして使用できる。遺伝子療法ベクターは、たとえば静脈内注射、局所投与(米国特許第5238470)または定位注射(たとえば、Chenら(1994)Proc.Natl.Acad.Sci.USA91:3054−3057)により対象に送達できる。遺伝子療法ベクターの医薬調製物は、許容される希釈剤中遺伝子療法ベクターを含むか、または遺伝子送達ビヒクルが埋め込まれた除法性マトリックスを含み得る。別法として、完全遺伝子送達ベクターが組み換え細胞、たとえばレトロウイルスベクターから完全な状態で産生される場合、医薬組成物は。遺伝子送達系を生じる1以上の細胞を含み得る。
医薬組成物は、容器、パック、またはディスペンサー中、投与の使用説明書とともに同封できる。
【0160】
VI.本発明の使用および方法
B7−4および/またはPD−1調節剤、たとえば、本明細書に記載された核酸分子、タンパク質、タンパク質相同体、および抗体は、次の方法の1以上において用いることができる:a)たとえば、免疫反応の減少調節による治療法;b)スクリーニングアッセイ;c)予測医学(predictive medicine)(たとえば、診断分析、予知分析、臨床試験のモニタリング、および薬理遺伝学)。本発明の単離された核酸分子は、たとえば、B7−4またはPD−1タンパク質を発現するため(たとえば、遺伝子療法用途において宿主細胞における組み換え発現ベクターによる)、B7−4またはPD−1mRNA(たとえば、生物学的サンプルにおいて)またはB7−4またはPD−1遺伝子における遺伝子変更を検出するため、およびB7−4またはPD−1活性を調節するために以下にさらに記載するようにして用いることができる。B7−4またはPD−1タンパク質は、B7−4またはPD−1タンパク質の不十分または過剰の産生により特徴づけられる障害を治療するために用いることができる。加えて、B7−4またはPD−1タンパク質は、天然に存在するB7−4またはPD−1結合パートナーについてスクリーンするため、B7−4またはPD−1活性を調節する薬剤または化合物についてスクリーンするため、ならびにB7−4またはPD−1タンパク質の不十分または過剰な産生またはB7−4またはPD−1野生型タンパク質と比較して減少しているかまたは異常な活性を有するB7−4またはPD−1タンパク質形態の産生により特徴づけられる障害を治療するために用いることができる。さらに、本発明の抗B7−4またはPD−1抗体は、B7−4またはPD−1タンパク質を検出し、単離し、B7−4またはPD−1タンパク質のバイオアベイラビリティーを調節し、たとえばB7−4またはPD−1の相互作用を調節することによりB7−4またはPD−1活性を調節するために使用できる。
【0161】
A.治療法:
本発明は、異常なB7−4またはPD−1発現または活性に関連する障害にかかるおそれがある(またはかかりやすい)か、または障害にかかっている対象を治療するための予防法および治療法の両方を提供する。
【0162】
1.予防法
一態様において、本発明は、対象において、異常なB7−4またはPD−1発現または活性に関連する疾患または状態を治療する方法であって、該対象にB7−4またはPD−1ポリペプチドまたはB7−4またはPD−1ポリペプチドまたはB7−4またはPD−1活性の少なくとも一つを調節する薬剤を投与することによる方法を提供する。異常なB7−4またはPD−1発現または活性により引き起こされるか、または一因である疾患にかかるおそれのある対象は、たとえば、本明細書に記載する診断または予防法いずれかまたは組み合わせにより確認することができる。予防薬の投与は、疾患または障害が予防されるか、またはその進行が遅れるように、B7−4またはPD−1以上に特徴的な症状が現れる前に行うことができる。B7−4またはPD−1以上または状態の種類に応じて、対象を治療するために、たとえば、B7−4またはPD−1ポリペプチド、B7−4またはPD−1作用物質またはB7−4またはPD−1拮抗剤を用いることができる。適当な薬剤は、臨床指標に基づいて決定でき、たとえば本明細書に記載するスクリーニング分析を用いて確認できる。
【0163】
2.治療法
本発明のもう一つの態様は、治療目的で、B7−4またはPD−1発現または活性を調節する方法に関する。B7−4は、免疫細胞の副刺激および増殖を阻害し、PD−1により免疫細胞に阻害シグナルを伝達することが証明されている。従って、B7−4またはPD−1の活性および/または発現ならびにB7−4およびPD−1間の相互作用は、免疫反応を調節するために調節できる。B7−4が副刺激レセプターと結合する具体例において、B7−4活性の増加調節の結果、免疫反応が増加調節され、一方、B7−4活性の減少調節の結果、免疫反応が減少調節される。B7−4が阻害レセプターと結合する具体例において、B7−4活性の増加調節の結果、免疫反応が減少調節され、一方、B7−4活性の減少調節の結果、免疫反応が増加調節される。好ましい具体例において、B7−4は阻害レセプターと結合する。特に好ましい具体例において、B7−4はPD−1と結合する。
【0164】
本発明の調節法は、細胞をB7−4またはPD−1ポリペプチドのモジュレーター、たとえばB7−4および/またはPD−1の発現または活性を調節する薬剤、またはB7−4とPD−1の相互作用を調節する薬剤と接触させることを含む。
【0165】
B7−4またはPD−1タンパク質活性を調節する薬剤は、本明細書において記載される薬剤、たとえば、核酸またはタンパク質分子、B7−4またはPD−1タンパク質の天然に存在する標的分子(たとえば、B7−4の場合はPD−1またはPD−1の場合はB7−4)、B7−4またはPD−1抗体、B7−4またはPD−1作用物質または拮抗物質、B7−4またはPD−1作用物質またはPD−1拮抗物質のペプチド模倣物、または他の小分子である。
【0166】
B7−4またはPD−1の発現を調節する薬剤は、たとえば、アンチセンス核酸分子、三本鎖オリゴヌクレオチド、リボザイムあるいはB7−4またはPD−1タンパク質の発現の組み換えベクターである。たとえば、B7−4またはPD−1ポリペプチド翻訳開始部位の周りの領域に対して相補性のオリゴヌクレオチドを合成し、使用できる。1以上のアンチセンスオリゴヌクレオチドを細胞培地に、典型的には200μg/mlで添加できるか、またはB7−4またはPD−1ポリペプチドの合成を防止するために患者に投与できる。別法として、DNAの巻き戻しおよび転写を防止するために二本鎖DNAと結合して三本鎖構造を形成するオリゴヌクレオチドを使用できる。いずれかの結果として、B7−4またはPD−1ポリペプチドの合成のいずれかがブロックされる。PD−1発現が調節される場合、好ましくは、このような調節はPD−1遺伝子のノックアウト以外の手段により行う。
【0167】
発現を調節する薬剤は、これらが細胞におけるPD−1またはB7−4の量を調節するという事実に基づき、細胞におけるPD−1またはB7−4活性の合計量も調節する。
一具体例において、B7−4の阻害活性またはPD−1の阻害活性を刺激する薬剤は、B7−4またはPD−1の作用物質である。かかる薬剤の例は、活性B7−4またはPD−1タンパク質および細胞中に導入されたB7−4またはPD−1ポリペプチドをエンコードする発現可能な核酸分子を包含する。もう一つの具体例において、薬剤はB7−4の副刺激または阻害活性あるいはPD−1の阻害活性を阻害し、PD−1のリガンドまたはPD−1の拮抗物質である。かかる薬剤の例は、アンチセンスB7−4またはPD−1核酸分子、抗B7−4または抗PD−1抗体(たとえば、非活性化抗体)、B7−4またはPD−1分子の可溶性、非活性化形態、およびB7−4またはPD−1阻害剤を包含する。
【0168】
調節剤は、インビトロ(たとえば、細胞を薬剤と接触させることによる)、あるいはインビボ(たとえば、薬剤を対象に投与することによる)で投与することができる。従って、本発明は、B7−4またはPD−1タンパク質の調節から利益を得る疾患または障害、たとえば、免疫反応の減少調節から利益を得るか、またはB7−4またはPD−1タンパク質または核酸分子の異常な発現または活性により特徴づけられる障害に苦しんでいる個体を治療する方法を提供する。一具体例において、方法は、薬剤(たとえば、本明細書に記載するスクリーニングアッセイにより同定される薬剤)、またはB7−4またはPD−1発現または活性を調節する(たとえば、増加調節または減少調節する)薬剤の組み合わせを投与することを含む。もう一つの具体例において、方法は、減少しているかまたは異常なB7−4またはPD−1発現または活性を埋め合わせる治療法として、B7−4またはPD−1タンパク質または核酸分子を投与することを含む。
【0169】
B7−4またはPD−1活性の刺激は、B7−4またはPD−1が異常に減少調節される状況および/または増加したB7−4またはPD−1活性が有益な効果を有する可能性がある状況において望ましい。同様に、B7−4またはPD−1活性の阻害は、B7−4またはPD−1が異常に増加調節される状況および/または減少したB7−4またはPD−1活性が有益な効果を有する可能性がある状況において望ましい。当業者は、B7−4が副刺激レセプターと結合する例において、B7−4の副刺激およびPD−1の副刺激は、免疫細胞副刺激、したがって免疫反応に関して反対の効果を有することを認識するであろう。このような場合において、一つの分子の活性の副刺激が望ましい場合、他の分子の活性の抑制が望ましい。
【0170】
B7−4を減少調節するために用いられる薬剤(B7−4拮抗物質)の例は、(たとえば):アンチセンス分子、B7−4を認識する抗体、B7−4と免疫細胞上のその天然に存在するレセプターの一つとの相互作用をブロックする化合物(たとえば、可溶性、一価B7−4、および本明細書において記載されるスクリーニングアッセイにおいて同定されるB7−4または化合物の溶解形態)を包含する。PD−1を減少調節するために用いられる薬剤(PD−1拮抗物質)歯(たとえば):アンチセンス分子、PD−1と結合するが、免疫細胞に阻害シグナルを形質導入しない抗体(「非活性化抗体」)、およびPD−1の溶解形態を包含する。
【0171】
B7−4を増加調節するために用いられる薬剤(B7−4作用物質)の例は、(たとえば):B7−4ポリペプチドをエンコードする核酸分子、B7−4の多価分子、B7−4の発現を増大させる化合物、およびB7−4を発現する細胞などを包含する。PD−1を増加調節するために用いられる薬剤(PD−1作用物質)の例は、(たとえば):PD−1分子と結合し、活性化させる(たとえば架橋させる)ことにより、PD−1を介して阻害シグナルを伝達する抗体、PD−1の発現を向上させる化合物、PD−1をエンコードする核酸分子、およびPD−1を介してシグナルを変換するB7−4の形態(特にB7−4の二価形態)を包含する。
【0172】
3.B7−4またはPD−1の調節による免疫反応のダウンレギュレーション
B7−4ポリペプチドの阻害機能を増加調節するか、または副刺激機能を減少調節して、免疫反応を減少調節する本発明の多くの具体例がある。減少調節は、すでに進行中の免疫反応の阻害またはブロックの形態であるか、または免疫反応の誘発の防止を含み得る。
活性化された免疫細胞の機能は、免疫細胞反応の減少調節によるか、または免疫細胞における特異的アネルギーの誘発によるか、またはその両方により阻害できる。
【0173】
例えば、抗B7−4抗体またはB7−4ポリペプチド(例えば、B7−4IgなどのB7−4ポリペプチドの可溶性モノマー形態)、および/またはB7−4と副刺激レセプターとの相互作用をブロックする抗B7−4抗体を、副刺激シグナルを阻害するため、従って免疫反応を減少調節するために用いることができる。
加えて、B7−4が阻害レセプターと結合する具体例において、阻害レセプターと結合し、これを活性化するB7−4の形態、例えば、細胞表面上の多価B7−4を、免疫反応を減少調節するために用いることができる。
【0174】
同様に、PD−1経路は、免疫反応を減少調節するために薬剤を使用することにより刺激できる。これは、B7−4と免疫細胞上の刺激レセプターとの相互作用の阻害(例えば、PD−1および/またはCTLA4の溶解形態の使用による)またはPD−1の活性化(例えば、PD−1と架橋する活性化抗体の使用による)により達成され、免疫細胞に負のシグナルを提供する。
本発明の一具体例において、PD−1活性を刺激するために用いられる活性化抗体は二重特異性抗体である。例えば、かかる抗体はPD−1結合部位と免疫細胞上、例えばT細胞、B細胞、または脊髄細胞上の細胞表面レセプターを標的とするもう一つの結合部位を含み得る。一具体例において、かかる抗体は、PD−1結合部位を含むことに加えて、分子に特定の細胞集団を狙わせるために、活性化または阻害レセプター、例えば、B細胞レセプター、T細胞レセプター、またはFcレセプターに近接した分子と結合する結合部位をさらに含む。例えば、CD3抗原、T細胞レセプター鎖、LFA−1、CD2、CTLA−4、イムノグロブリン、B細胞レセプター、Igアルファ、Igベータ、CD22、またはFcレセプターを用いることができる。かかる抗体(または他の二重特異性剤)は当該分野において承認され、例えば本明細書において記載されるようにして産生することができる。二重特異性抗体についてのこの第二抗体の選択により、阻害のために標的とされる細胞集団の選択に柔軟性がもたらされる。
【0175】
もう一つの具体例において、PC−1の細胞上の活性化または阻害レセプターの同時結紮は、PD−1による負のシグナルの生成を向上させることができる。このような同時結紮は、例えば、二重特異性剤、例えば、本明細書に記載する、PD−1とレセプターと関連する分子の両方について特異性を有する二重特異性抗体の使用により達成できる。もう一つの具体例において、PD−1による負のシグナルの伝達を向上させるために、PD−1により負のシグナルを伝達する薬剤の多価形態、例えばビーズ上または表面上に提示される薬剤を使用することができる。もう一つの具体例において、このような多価薬剤は、PD−1およびレセプターとレセプター関連分子(例えば、抗CD3およびB7−4を含むビーズ)の結紮を達成するために2つの特異性を含み得る。
【0176】
B7−4の副刺激レセプターとの相互作用をブロックまたは阻害する薬剤(例えば、B7−4の溶解形態またはB7−4に対するブロッキング抗体)ならびにB7−4による阻害シグナルを促進する薬剤またはPD−1を活性化するPD−1の作用物質(例えば、PD−1活性化抗体またはPD−1活性化小分子)は、免疫細胞増殖および/またはエフェクター機能を阻害する能力またはインビトロアッセイに添加させる場合はアネルギーを誘発する能力により同定することができる。例えば、活性化レセプターによりシグナル変換を刺激する薬剤の存在下で細胞を培養することができる。細胞活性化の当該分野において承認された多くの情報を用いて、阻害、例えば、細胞増殖またはエフェクター機能(例えば、抗体産生、サイトカイン産生、食菌作用)を活性化剤の存在下で測定することができる。試験薬剤がこの活性化をブロックする能力は、該薬剤が測定される増殖またはエフェクター機能の減少に影響を及ぼす能力を測定することにより容易に決定できる。
【0177】
本発明の一具体例において、抗原をPD−1作用物質と同時に投与することにより、特異性抗原に対して耐性が生じる。例えば、耐性は特定のタンパク質に対して生じ得る。一具体例において、免疫反応が望ましくないアレルゲンまたは外来タンパク質に対する免疫反応を阻害できる。例えば、ファクターVIIIを受ける患者は、しばしばこの凝固因子に対して抗体を生じる。B7−4による副刺激シグナルをブロックする薬剤またはPD−1による阻害シグナルを刺激する薬剤を組み換えファクターVIIIとの組み合わせで(または物理的にファクターVIIIと結合させる、例えば架橋することにより)同時投与すると、結果として減少調節される。
【0178】
一具体例において、もう一つ別のBリンパ球抗原の活性を有する第二ペプチドと融合したB7−4第一ペプチドを含む融合タンパク質(例えば、B7−1またはB7−2)を、免疫細胞上の副刺激レセプターとB7−4との相互作用をブロックし、免疫反応を減少調節するために用いることができる。別法として、2つの別々のペプチド(例えば、B7−4ポリペプチドとB7−2および/またはB7−1)、またはブロッキング抗体の組み合わせ(例えば、B7−4ポリペプチドに対する抗体と抗B7−2および/または抗B7−1モノクローナル抗体)を単一の組成物として組み合わせるか、または別々に(同時または連続して)投与して、対象における免疫細胞性免疫反応を減少調節することができる。さらに、治療的に活性な量の1以上の、B7−4ポリペプチド活性と、B7−1および/またはB7−2活性を有するペプチドを、他の減少調節試薬と併せて使用して、免疫反応に影響を及ぼすことができる。他の免疫調節剤の例は、副刺激シグナルをブロックする抗体(例えば、CD28、ICOSに対する抗体)、CTLA4により阻害シグナルを活性化する抗体、および/または他の免疫細胞マーカーに対する抗体(例えば、CD40、CD40リガンド、またはサイトカインに対する抗体)、免疫抑制剤(例えば、ラパマイシン、シクロスポリンAまたはFK506)を包含する。
【0179】
B7−4および/またはPD−1ペプチドは、細胞の破壊により免疫細胞機能をブロックする治療薬の構成においても有用である。例えば、B7−4またはPD−1ポリペプチドの部分をトキシンと結合させて、これが結合している細胞の破壊を引き起こすことができる細胞毒性剤を調製することができる。
細胞毒性剤を調製するために、本発明のポリペプチドを、当該分野で公知の技術を用いて、例えば、架橋または組み換えDNA技術により、トキシンと結合、または操作可能に結合させることができる。イムノトキシンの調製は、一般に当該分野において周知である(例えば、米国特許第4340535号およびEP44167(両方とも出典明示により本発明の一部とする)参照)。トキシン部分をポリペプチドと結合させるためにうまく使用できる多くの種類のジスルフィド結合含有リンカーが公知である。一具体例において、立体的に「込み合った」ジスルフィド結合を含むリンカーは、インビボで安定性が高く、従って作用部位で結合の前にトキシン部分の放出を防止するので好ましい。
【0180】
本発明のポリペプチドまたは抗体を結合できる広範囲に及ぶトキシンが公知である。例は:多くの有用な植物、真菌または細菌由来のトキシンを包含し、これらは例として、さまざまなA鎖トキシン、特にリシンA鎖、リボソーム不活性化タンパク質、例えばサポリンまたはゲロニン、アルファ−サルシン、アスペルギリン、レストリクトシン、リボヌクレアーゼ、例えば胎盤リボヌクレアーゼ、血管由来、ジフテリア毒素、およびシュードモナスエクソトキシンを包含する。本発明に関連して使用される好ましいトキシン部分は、炭水化物残基、デグリコシル化A鎖を修飾または除去するために処理されたトキシンA鎖である(米国特許第5776427号)。
かかる細胞毒性剤の1つまたは組み合わせ(例えば、B7−4リシン(単独またはB7−2リシンまたはB7−1−リシンとの組み合わせ)を患者に注入すると、特にさらに多量のB7−4を発現する活性化免疫細胞はリガンド形成するという事実を考慮すると、免疫細胞が死ぬ。例えば、PD−1は活性化されたリンパ球の表面上で誘発されるので、Fc−R依存性メカニズムまたは細胞毒性剤(例えば、リシン、サポリン、またはカリヘアミシン)を抗体と結合させることにより、これらの特異性細胞の減少を目的とするためにPD−1に対する抗体を使用できる。一具体例において、抗体トキシンは二重特異性抗体であり得る。かかる二重特異性抗体は、たとえばある種類の細胞、例えばTCR、BCR、またはFcR分子上のみで見いだされるマーカーを用いて、特定の細胞集団を標的とするために有用である。
【0181】
B7−4ポリペプチド副刺激機能の減少調節または防止あるいはB7−4またはPD−1阻害機能の活性化(例えば、PD−1の負のシグナリング機能の刺激による)は、例えば、組織、皮膚または臓器移植、移植片対宿主病(GVHD)、または自己免疫疾患、例えば全身性エリテマトーデス、および多発性硬化症などの免疫反応の減少調節において有用である。例えば、免疫細胞機能をブロックすると、組織移植において組織の破壊が軽減される。典型的には、組織移植において、移植片の拒絶は、免疫細胞により異物として認識されることにより開始され、続いて移植片を破壊する免疫反応が起こる。B7分子の免疫細胞上の副刺激レセプターとの相互作用を阻害またはブロックする分子(例えば、PD−リガンドまたはPD−1ポリペプチドの可溶性モノマー形態)を、別の減少調節剤とともに、または組み合わせて、移植前または移植時に投与すると、副刺激シグナルの生成を阻害することができる。さらに、B7−4副刺激シグナルの阻害、またはB7−4またはPD−1阻害シグナルの促進は、免疫細胞のアネルギー化に十分であり、したがって対象において寛容性を誘発する。B7−4による副刺激シグナルのブロックによる長期寛容性の誘発は、これらのブロッキング剤を繰り返し投与する必要性を回避する。
【0182】
対象において十分な免疫抑制または寛容性を達成するために、他の分子の副刺激機能をブロックすることも望ましい。例えば、これらの抗原またはこれらの抗原に対するブロッキング抗体のそれぞれの活性を有するペプチドの組み合わせの溶解形態(別々に、または単一組成物において一緒に)を移植前または移植時に投与することにより、B7−1およびB7−4、B7−2およびB7−4、またはB7−1およびB7−2およびB7−4の機能をブロックすることが望ましい。別法として、B7−4またはPD−1の阻害活性を促進し、B7−1および/またはB7−2の副刺激活性を阻害することが望ましい。本発明の減少調節剤と組み合わせて使用できる他の減少調節剤は、例えば、CTLA4により阻害シグナルを伝達する薬剤、CTLA4の溶解形態、CTLA4により阻害シグナルを活性化する抗体、他の免疫細胞マーカーまたは他のレセプターリガンド対の溶解形態に対するブロッキング抗体(例えば、CD40とCD40リガンド間の相互作用を破壊する薬剤(例えば、抗CD40リガンド抗体))、サイトカインに対する抗体、または免疫抑制剤を包含する。もう一つの具体例において、最適のブロッキング活性を達成するために少なくとも2つの異なるB7−4抗体の組み合わせを投与することができる。
【0183】
B7−4ポリペプチド副刺激のブロッキングまたはB7−4またはPD−1阻害機能の活性化も自己免疫疾患の治療において有用である。多くの自己免疫傷害は、自己の組織に対して反応性であり、サイトカインおよび疾患の病理に関与する自己抗体の産生を促進する免疫細胞の不適切な活性化の結果である。自己反応性免疫細胞の活性化の防止または減少調節は、症状を軽減または除去する。B7分子と副刺激レセプターのレセプター:リガンド相互作用を破壊することにより免疫細胞の副刺激をブロックする試薬の投与は、免疫細胞活性化の阻害および疾患のプロセスに関与する抗体またはサイトカインの産生の防止に有用である。加えて、B7−4またはPD−1の阻害機能を促進する薬剤は、疾患からの長期的症状の軽減につながり得る自己反応性免疫細胞の抗原特異性寛容性を誘発できる。試薬の自己免疫疾患の防止または緩和における効力は、多くのヒト自己免疫疾患のよく特徴づけられた動物モデルを用いて決定できる。例は、ネズミ実験的自己免疫日本脳炎、MRL//lpr/lprマウスまたはNZBハイブリッドマウスにおける全身性エリテマトーデス、ネズミ自己免疫コラーゲン関節炎、NODマウスおよびBBラットにおける真性糖尿病、およびネズミ実験的重症性筋無力症を包含する(例えば、Paul編、Fundamental Immunology,Raven Press,New York,1989,pp840−856参照)。
【0184】
免疫細胞活性化の阻害は、例えばIgE産生の阻害によるアレルギーおよびアレルギー反応の治療において治療的に有用である。B7−4またはPD−1阻害機能を促進する薬剤は、患者における免疫細胞によるアレルギー反応を阻害するためにアレルギーの患者に投与することができる。PD−1分子の活性化はアレルギーの治療においても有用である。免疫細胞のB7−4副刺激の阻害あるいはB7−4またはPD−1阻害経路の刺激は、適当なMHC分子と組み合わせたアレルゲンへの暴露を伴う。アレルギー反応は、アレルギーの侵入経路および肥満細胞または好塩基球上のIgEの沈着パターンによって、全身性または局所性である。従って、免疫細胞によるアレルギー反応は、B7−4の副刺激レセプターあるいはB7−4またはPD−1の阻害機能を促進する薬剤との相互作用を阻害する薬剤の阻害形態の投与(例えば、それぞれ、局所または全身性のいずれか)することにより、局所性であるか、全身性である。
【0185】
B7−4副刺激活性のブロックまたはPD−1阻害活性の刺激による免疫細胞活性化の阻害は、免疫細胞のウイルス感染においても治療的に重要である。例えば、後天性免疫不全症候群(AIDS)において、ウイルス複製は免疫細胞活性化により刺激される。B7−4/副刺激レセプター相互作用またはB7−4またはPD−1阻害機能の刺激のブロックの結果、ウイルス複製が阻害され、AIDSの進行が改善される。B7−4活性またはB7−4のその天然の結合パートナー、例えばPD−1との相互作用の刺激による免疫反応の減少調節は、通常、妊娠の持続を促進するのにも有用である。B7−4は、母親と胎児の間の接合部分を形成し、胎児の母性拒絶の防止において役割を果たすと考えられる細胞の層である胎盤栄養膜において高度に発現される。自然流産の危険のある女性(例えば、「予防分析」の項に記載されているように、B7−4活性についてのスクリーニングにより確認される女性、以前に自然流産したことがある女性または妊娠が困難である女性)は、胚または胎児の免疫学的拒絶のためにB7−4またはその天然の結合パートナー、例えばPD−1との相互作用を刺激する薬剤で処理することができる。
【0186】
B7−4活性またはB7−4のその天然の結合パートナー、例えばPD−1との相互作用の刺激による免疫反応の減少調節は、自家組織の自己免疫攻撃の治療においても有用である。例えば、B7−4は通常心臓において高度に発現され、心臓を自己免疫攻撃から防御する。これは、Balb/cPD−1ノックアウトマウスが、血栓症の心臓に対して重い自己免疫攻撃を示すという事実により明らかにされる。従って、自己免疫攻撃により引き起こされるかまたは悪化する状態(例えば、この例においては、心臓病、心筋梗塞またはアテローム性動脈硬化症)は、B7−4活性またはB7−4のその天然の結合パートナー、例えばPD−1との結合を増大させることにより緩和または改善される。従って、B7−4活性またはB7−4のB7−4との相互作用を刺激することにより、自己免疫攻撃により悪化する状態、例えば自己免疫傷害(ならびに心臓病、心筋梗塞、およびアテローム性動脈硬化症)を調節することは本発明の範囲内である。
【0187】
4.免疫反応のアップレギュレーション
免疫反応をアップレギュレーションする手段としての、B7−4副刺激活性のアップレギュレーションまたはPD−1またはB7−4の抑制性の活性の阻害も治療に有用である。免疫反応のアップレギュレーションは、既存の免疫反応を高めるか、または初期免疫反応を誘発することの形態をとることができる。例えば、B7−4副刺激活性を刺激することまたはB7−4またはPD−1抑制性活性の阻害により免疫反応を高めることは、微生物、例えば、細菌、ウイルス、または寄生虫などでの感染の場合に有用である。例えば、1つの具体例において、免疫細胞において副刺激シグナルを促進するB7−4の形態(例えば、多価形態のB7−4ペプチド(例えば、溶解性の多価形態または細胞表面で発現される形態))、または抑制性レセプターとのB7−4の相互作用を阻害する試薬、またはPD−1を経る抑制性シグナルの伝達を阻害する試薬、例えばPD−1に対する非活性化抗体または小分子(例えば、ペプチドミメティクス)PD−1アンタゴニストは、微生物のより迅速または完全なクリアランスに至る抗体および細胞媒介性の反応のアップレギュレーションが有用である状況において治療的に有用である。これらの刺激には、制限されるものではなく、帯状疱疹などのウイルス性皮膚疾患、この場合、そのような試薬は皮膚へ局所送達されてよい、が含まれる。加えて、インフルエンザ、一般感冒、および脳炎などの全身性ウイルス疾患は、そのような試薬の全身投与により緩和され得る。
【0188】
所定の例において、免疫反応をアップレギュレートする他の試薬、例えば、免疫反応をさらに高めるように副刺激レセプターを経てシグナルを伝達する他のB7ファミリーのメンバーの形態をさらに投与することが望まれ得る。
【0189】
別法として、免疫反応は、エキソビボアプローチ、例えば、免疫細胞を患者から取りだし、免疫細胞をインビトロで免疫細胞における副刺激シグナルを促進するB7−4の形態または抑制性レセプターとB7−4の相互作用を阻害する試薬、またはPD−1を経る抑制性シグナルの伝達を阻害する試薬と接触させ、次いでインビとロ刺激免疫細胞を患者へ再度導入することにより、感染した患者において高めることができる。他の具体例においては、免疫反応を高める方法には、患者からの感染細胞(例えばウイルス感染細胞)を単離すること、それらを細胞がB7−4分子の全部または部分をその表面上に発現するように、副刺激レセプターに結合するB7−4の形態をコードしている核酸分子でトランスフェクトすること、およびトランスフェクトした細胞を患者へ再度導入することが含まれる。トランスフェクトされた細胞は、副刺激シグナルを免疫細胞へと送達することができ、そしてそれにより、免疫細胞をインビボで活性化することができる。
【0190】
免疫細胞において副刺激シグナルを促進するB7−4形態、またはB7−4の抑制性レセプターとの相互作用を阻害する試薬、またはPD−1を経る抑制性シグナルの伝達を阻害する試薬を種々のポリペプチド、例えば病原体由来のポリペプチドに対するワクチンにて予防的に用いることができる。病原体(例えば、ウイルスまたは細菌)に対する免疫性を、適当なアジュバント中の免疫細胞において副刺激シグナルを促進するB7−4形態または抑制性レセプターとのB7−4の相互作用を阻害する試薬、またはPD−1を経る抑制性シグナルの伝達を阻害する試薬と共に病原体のタンパク質でワクチン予防接種することにより誘導することができる。別法として、病原抗原および副刺激レセプターに結合するB7−4形態の両方をコードする遺伝子を含んでいるベクターを、ワクチン予防接種のために用いることができる。核酸ワクチンは、種々の手段、例えば、注入(例えば、DNA−コート金粒子の、粒子を皮膚へ注入するための粒子加速器または圧縮ガスを用いる遺伝子銃での表皮への、筋肉内、経皮、またはバイオリスティック注入)により投与されることができる(Haynes et al. (1996) J. Biotechnol. 44:37))。別法として、核酸ワクチンは、非侵入性の手段により投与することができる。例えば、純粋もしくは脂質調製したDNAを、例えば、DNAの経口送達によるペイヤーパッチ(Peyers patches)など、呼吸器系へと到達することができ、またはいずれかの場所へ標的することができる(Schubbert (1997) Proc. Natl. Acad. Sci. USA 94:961)。弱毒化された微生物を粘膜表面への送達のために用いることができる(Sizemore et al. (1995) Science 270:29)。
【0191】
1つの具体例において、副刺激シグナルを伝達するB7−4ポリペプチド形態を、例えば、B7−4ポリペプチドおよびMHCクラスIαタンパク質およびβ2ミクログルブリンを同時発現してT細胞を活性化し、感染からの免疫化を提供するべくトランスフェクトした細胞によりクラスI MHCタンパク質と共に投与することができる。ワクチンが有用である病原体には、制限されるものではないが、B型肝炎、C型肝炎、エプスタイン−バー−ウイルス、サイトメガロウイルス、HIV−1、HIV−2、結核、マラリア、および住血吸虫が含まれる。
【0192】
他の適用においては、B7−4副刺激機能のアップレギュレーションまたは増強が、腫瘍の免疫化の誘導に有用である。腫瘍細胞における野生型のB7−4の内因性の発現により、腫瘍細胞に対する免疫反応が抑制される。従って、腫瘍細胞におけるB7−4とPD−1の間の相互作用の阻害が腫瘍の免疫を誘導するのに有用である。1つの具体例においては、PD−1アンタゴニスト(例えば、PD−1またはB7−4に対する非−活性化抗体または小分子PD−1またはB7−4アンタゴニスト)を腫瘍を有するまたはそれに対してリスクのある対象へと投与する。
【0193】
他の具体例においては、B7−4抗原をコードしている核酸分子でトランスフェクトした腫瘍細胞(例えば、肉腫、黒色腫、リンパ腫、白血病、神経芽腫、癌)を対象に投与して、対象における腫瘍特異的耐性を克服する。所望の場合、腫瘍細胞をトランスフェクトして、B7ポリペプチドの組み合わせ(例えばB7−1、B7−2、B7−4いずれかの組み合わせ)を発現させることができる。例えば、対象から得た腫瘍細胞をエキソビボで、B7−4ポリペプチドのみ、またはB7−1活性および/またはB7−2活性を有するペプチドとの組み合わせの発現を指向する発現ベクターでトランスフェクトすることができる。トランスフェクトされた腫瘍細胞を対象へ戻し、トランスフェクト細胞の表面上でのペプチドの発現を得る。別法として、遺伝子治療法を用いて、インビボでのトランスフェクションのために腫瘍細胞を標的することができる。
【0194】
加えて、MHCクラスIまたはMHCクラスII分子を欠く、またはMHCクラスIまたはMHCクラスII分子の十分量を発現しない腫瘍細胞を、MHCクラスIα鎖タンパク質およびβ2ミクログルブリンタンパク質またはMHCクラスIIαタンパク質およびMHCクラスIIβ鎖タンパク質の全部または部分(例えば、細胞質ドメイン切断部位)をコードしている核酸でトランスフェクトし、それにより細胞表面上でMHCクラスIまたはMHCクラスIIタンパク質を発現させることができる。Bリンパ球抗原の活性を有するペプチド(例えば、B7−1、および/またはB7−2、および/またはB7−4)と組み合わせた適当なクラスIまたはクラスIIMHCの発現により、トランスフェクトした腫瘍細胞に対するT細胞媒介性の免疫反応が誘導される。任意に、MHCクラスII関連タンパク質、不変鎖などの発現を遮断するアンチセンス構築物をコードしている遺伝子を、B7−4ポリペプチドをコードしているDNAと同時トランスフェクトして、腫瘍関連抗原の提示を促進し、腫瘍特異的免疫を誘導することもできる(例えば、本明細書中にその内容を出典明治により組み込む米国特許第5,919,639号に教示されているように)。B7ネガティブネズミ腫瘍細胞によるB7−1の発現は、腫瘍の拒絶が付随するT細胞媒介性の特異的免疫およびおよびマウスにおいて腫瘍攻撃に対する長期保護を誘導することが示されている(Chen, L., et al. (1992) Cell 71:1093-1102; Townsend, S. E. and Allison, J. P. (1993) Science 259:368-370; Baskar, S. et al. (1993) Proc. Natl. Acad. Sci. 90:5687-5690)。つまり、ヒト対象における免疫細胞媒介性の免疫反応の誘導が、対象における腫瘍特異的耐性を克服するのに十分であり得る。
【0195】
他の具体例において免疫反応は、B7−4に結合する副刺激レセプターを経るシグナル伝達により、またはB7−4に結合する抑制性レセプター、例えばPD−1を経るシグナリングを阻害することにより刺激されて、先在している耐性を克服することができる。例えば、対象が有意な免疫反応を高めることができない抗原、例えば、腫瘍特異的抗原などの自己抗原に対する免疫反応は、PD−1の抑制性の活性または抑制性のリガンドに結合するB7−4の能力を阻害する試薬を投与することにより、誘導することができる。例えば、1つの具体例において、溶解性PD−1または溶解性B7−4を用いて(例えば、PD−1FcまたはB7−4Fc)、例えば腫瘍細胞に対する免疫反応を高めることができる。1つの具体例では、自己抗原、腫瘍特異的抗原などを、PD−1の抑制性の活性またはB7−4の抑制性のリガンドに結合する能力を阻害する試薬と共に同時投与することができる。他の具体例において、免疫反応を抗原(例えば自己抗原)に対して刺激して、神経疾患を治療することができる。他の具体例では、PD−1アンタゴニストをアジュバントとして用いて、活性免疫化のプロセスにおいて外来抗原への反応をブーストすることができる。
【0196】
さらに他の具体例においては、抑制性のレセプターに結合するまたは副刺激レセプターに対するB7−4の結合と競合するB7−4形態(例えば、PD−1へ結合するB7−4形態または天然に生じる溶解性分子)の細胞における産生は、例えば、免疫反応をアップレギュレートするために、アンチセンスRNAを用いて阻害することができる。例えば、1具体例において、腫瘍細胞による抑制性B7−4分子の産生を、抗腫瘍免疫を増すために阻害することができる。
【0197】
1つの具体例では、免疫細胞を対象から得、エキソビボで、副刺激分子を結合するB7−4形態の存在下、またはB7−4またはPD−1抑制性シグナルを阻害する試薬の存在下培養して、免疫細胞集団を増殖させる。さらなる具体例において、免疫細胞を次いで対象へ投与する。例えば免疫細胞へ一次活性化シグナルおよび副刺激シグナルを当該分野で公知のごとく提供することにより、免疫細胞を刺激して、インビトロで増殖させることができる。B7−4タンパク質の種々の形態または副刺激レセプターに結合するまたはPD−1を経るシグナリングを阻害する試薬を用いて、免疫細胞の増殖を副刺激することもできる。1つの具体例においては、免疫細胞をPCT出願第WO94/29436号に記載の方法に従いエキソビボで培養する。副刺激分子は溶解性であってよく、細胞膜結合へ結合する、またはビーズなどの固体表面へ結合することができる。
【0198】
B.B7−4および/またはPD−1の調節により調節されるサイトカインの同定
本明細書に開示するB7−4およびPD−1分子を用いて、B7−4および/またはPD−1の活性の調節により産生される、またはそれに反応して免疫細胞においてその産生が高められるまたは阻害されるサイトカインを同定することができる。PD−1を発現している免疫細胞は、インビトロで、一次活性化シグナルで最適下限刺激することができ、例えば、T細胞は、ホルボールエステル、抗−CD3抗体または好ましくは抗原で、MHCクラスII分子と組み合わせて刺激することができ、および副刺激シグナルの場合は、例えばB7ファミリー抗原の刺激形態により、例えばB7ポリペプチドをコードしている核酸でトランスフェクトした、およびその表面上にペプチドを発現している細胞により、またはペプチドの溶解性刺激形態により刺激することができる。培地に放出される公知のサイトカインは、ELISAにより、またはサイトカインを遮断して免疫細胞増殖またはサイトカインにより誘導される他の細胞タイプの増殖を阻害する抗体の能力により同定することができる。例えば、IL−4ELISAキットが、IL−7ブロッキング抗体同様、Genzyme(Cambridge MA)から入手可能である。IL−9およびIL−12に対するブロッキング抗体は、Genetics Institute(Cambridge, MA)より入手可能である。サイトカイン側面にてB7−4のPD−1との相互作用を刺激または遮断する効果を次いで決定することができる。
【0199】
前記のインビトロ免疫細胞副刺激アッセイを、B7−4および/またはPD−1の調節により調節され得る新規サイトカインを同定するための方法において用いることもできる。例えば、CD28/CTLA4経路の刺激がIL−2の分泌を高めると考えられる場合、ICOS経路の刺激はIL−10の分泌を高めると考えられる(Hutloff et al. 199. Nature 397:263)。副刺激に際して誘導される特定の活性、例えば免疫細胞の増殖が公知のサイトカインへのブロッキング抗体の添加により阻害され得ない場合、活性は、未知のサイトカインの活性から生じている可能性がある。副刺激後に、このサイトカインを培地から常套法により精製し、その活性を、免疫細胞の増殖を誘導するその能力により測定することができる。
【0200】
耐性の誘導において役割を果たし得るサイトカインを同定するために、前記のごとくインビトロT細胞副刺激アッセイを用いることができる。この場合、T細胞に一次活性化シグナルを与え、選択されたサイトカインと接触させるが、副刺激シグナルは与えない。免疫細胞を洗浄および停止後、細胞を一次活性化シグナルおよび副刺激シグナルの両方で再攻撃する。免疫細胞が反応しない(例えば、サイトカインを増殖または産生しない)場合、それらは耐性となっており、サイトカインは耐性の誘導を妨げてはいない。しかし、免疫細胞が反応する場合、耐性の誘導はサイトカインにより妨げられた。耐性の誘導を妨げることができるこれらのサイトカインを、Bリンパ球抗原を遮断する試薬と組み合わせて、移植片レシピエントまたは自己免疫疾患の対象において耐性を誘導するより効果的な手段として、インビボでの妨害のための標的とすることができる。例えば、B7−4またはPD−1抑制性活性を促進する試薬と共に、対象へ、サイトカインブロッキング抗体を投与することができる。
【0201】
C.B7−4またはPD−1ポリペプチドの発現を調節する分子の同定
本発明のタンパク質およびペプチドを用いて産生される抗体を、細胞上でのB7−4またはPD−1ポリペプチドの発現を調節する分子についてのスクリーニングアッセイにおいて使用できる。例えば、B7−4またはPD−1ポリペプチドの発現いおける変化に至る細胞内シグナリング経路を調節する分子は、細胞表面上での1以上のB7−4またはPD−1ポリペプチドの発現を分析することにより同定できる。分子の存在下で適当な抗体により免疫蛍光染色が減少することは、分子が細胞内シグナルを阻害することを示す。B7−4またはPD−1ポリペプチド発現を増加調節する分子は、免疫蛍光染色を増大させる。別法として、ポリペプチドの発現に対する分子の硬化は、本発明のプローブを用いて細胞mRNAレベルを検出することにより決定できる。例えば、B7−4またはPD−1ポリペプチドを発現する細胞を試験される分子と接触させ、標準的技術、例えばノザンハイブリダイゼーション分析または検出可能なマーカーで標識されたcDNAプローブを用いたmRNAまたはポリ(A+)RNAの慣例のドットブロットにより検出される細胞においてmRNAレベルを増大または減少させる。B7−4またはPD−1ポリペプチドの発現を調節する分子は、単独あるいは前記のような可溶性ブロッキングまたは刺激剤とあわせて免疫応答を増加調節または減少調節するために治療的に有用である。例えば、B7−4の発現を阻害する分子は、第二の薬剤(例えば、免疫抑制剤)と共に投与することができる。例えば、免疫抑制剤またはPD−1の発現を阻害する分子を免疫刺激物質(例えば、アジュバント)と共に投与できる。B7−4またはPD−1を調節する能力について試験できる分子の例は、IL−4、γINF、IL−10、IL−12、GM−CSFおよびプロスタグランジンなどのサイトカインを包含する。
【0202】
D.スクリーニングアッセイ
本発明はモジュレーター、すなわち、B7−4またはPD−1タンパク質と結合し、例えばB7−4またはPD−1発現またはB7−4またはPD−1活性に対して刺激または阻害硬化を有する候補または試験化合物または薬剤(例えば、ペプチド、ペプチド模倣物、小分子または他の医薬)を同定するための方法(本明細書において「スクリーニングアッセイ」とも称する)を提供する。
【0203】
B7−4またはPD−1の活性を調節する化合物を同定するための1つのそのようなアッセイは、細胞に基づくアッセイである。アッセイの1つの具体例において、PD−1を発現している細胞を試験化合物と接触させる。細胞をさらにPD−1のアクチベーター(例えば溶解形態の、または細胞上で発現されるB7−4)と接触させ、次いでPD−1の活性をアッセイする。試験化合物の不在下での細胞のPD−1の活性と比較した、試験化合物の存在下での細胞のPD−1の増加または低下した活性が、化合物がPD−1の活性を調節し、即ち、PD−1のシグナリングを調節することを示す。同様のアッセイを、B7−4活性のモジュレーターを同定するために行うことができる。例えば、B7−4の活性化形態(溶解形態または細胞上で発現された形態)を、B7−4が結合するおよびB7−4により活性化されることが知られているレセプターを発現している細胞に、試験化合物の存在下および不在下に添加して、B7−4の活性を次いでアッセイして、試験化合物が調節活性を有するかどうかを決定することができる。
【0204】
一具体例において、本発明は、B7−4またはPD−1タンパク質またはポリペプチドまたはその生物学的に活性なタンパク質と結合するか、またはその活性を調節する候補または試験化合物、例えばB7−4またはPD−1ポリペプチドのその同源結合パートナーまたは相互作用分子と相互作用する能力を調節するものをスクリーニングするための分析法を提供する。
【0205】
本発明の試験化合物は、生物学的ライブラリー;空間的にアドレス可能な平行固相または溶液相ライブラリー;デコンボリューションを必要とする合成ライブラリー法;「1ビーズ1−化合物」ライブラリー法;およびアフィニティークロマトグラフィー選択を使用する合成ライブラリー法を含む当該分野において公知のコンビナトリアルライブラリー法において多くのアプローチを用いて得ることができる。生物学的ライブラリーアプローチはペプチドライブラリーに限定されるが、他の4つのアプローチは、ペプチド、非ペプチドオリゴマーまたは化合物の小分子ライブラリーに適用可能である(Lam,K.S.(1997)Anticancer Drug Des.12:145)。
分子ライブラリーの合成法の例は、例えば:DeWittら(1993)Proc.Natl.Acad.Sci.USA90:6909;Erbら(1994)Proc.Natl.Acad.Sci.USA91:11422;Zuckermannら(1994)J.Med.Chem.37:2678;Choら(1993)Science261:1313;Carrelら(1994)Angew.Chem.Int.Ed.Engl.33:2059;Carellら(1994)Angew.Chem,Int.Ed.Engl.33:2601;およびGallopら(1994)J.Med.Chem.37:1233において見いだすことができる。
【0206】
化合物のライブラリーは、溶液中(例えば、Hougthen(1992)Biotechniques13:412−421)、またはビーズ上(Lam(1991)Nature354:82−84)、チップ(Fodor(1993)Nature364:555−556)細菌(Lander米国特許第5223409号)、胞子(Lander米国特許第5233409)、プラスミド(Cullら(1992)Proc.Natl.Acad.Sci.USA89:1865−1869)またはファージ上(ScottおよびSimith(1990)Science249:386−390);(Devlin(1990)Science249:404−406);(Cwirlaら(1990)Proc.Natl.Acad.Sci.USA87:6378−6382);(Felici(1991)J.Mol.Biol.222:301−310):(Lander前出)で提供できる。
【0207】
もう一つの具体例において、分析法は、B7−4標的分子(細胞内相互作用分子またはPD−1レセプター)またはPD−1標的分子(例えば、B7−4または細胞内相互作用分子)を発現する細胞を試験化合物と接触させ、該試験化合物がB7−4またはPD−1標的分子の活性を調節(例えば、刺激または阻害)する能力を決定することを含む細胞ベースの分析法である。試験化合物がB7−4またはPD−1標的分子の活性を調節する能力の決定は、例えば、標的分子と結合するかまたは相互作用する分子は活性をより調節しやすいので、B7−4またはPD−1タンパク質がB7−4またはPD−1標的分子と結合または相互作用する能力を決定することにより行うことができる。B7−4またはPD−1タンパク質がその結合パートナーと結合または相互作用する能力の決定は、例えば直接結合を測定することにより達成できる。
【0208】
直接結合分析において、B7−4またはPD−1タンパク質(または他の各標的分子)を放射性同位元素または酵素標識と結合させて、B7−4またはPD−1タンパク質とB7−4またはPD−1標的分子との結合が複合体において標識されたタンパク質を検出することにより決定されるようにできる。例えば、B7−4またはPD−1分子、例えば、B7−4またはPD−1タンパク質を、125I、35S、14C、または3Hで直接または間接的のいずれかで標識し、放射性同位元素を電波放射の直接計測またはシンチレーションカウンティングにより検出できる。別法として、B7−4またはPD−1分子を酵素により、例えばホースラディッシュペルオキシダーゼ、アルカリホスファターゼ、またはルシフェラーゼで標識し、酵素標識を適当な基質の生成物への変換率を定量することにより検出できる。
【0209】
化合物がB7−4またはPD−1と標的分子間の相互作用を調節する能力を、相互作用体のいずれも標識することなく定量することも本発明の範囲に含まれる。例えば、B7−4またはPD−1のその標的分子との相互作用を、B7−4またはPD−1または標的分子のいずれも標識せずに検出するために、ミクロフィジオメーターを使用できる(McConnell,H.M.ら(1992)Science257:1906−1912)。本明細書において用いられる場合、「ミクロフィジオメーター」(例えば、Cytosensor)とは、細胞がその環境を酸性化する速度を光アドレス可能な電位差センサー(LAPS)を用いて測定する分析装置である。この酸性化速度の変化を、化合物とレセプター間の相互作用のインジケーターとして用いることができる。好ましい具体例においては、B7−4またはPD−1標的分子と結合または相互作用するB7−4またはPD−1タンパク質の能力を測定することは、B7−4、PD−1、または適当な標的分子の活性を測定することにより行うことができる。例えば、B7−4、PD−1、または適当な標的分子の活性は、細胞の第2メッセンジャー(例えば、セリン/トレオニンまたはチロシンキナーゼ活性)の誘導または誘導の阻害を検出すること、適当な基質の触媒/酵素活性を検出すること、レポーター遺伝子(検出可能なマーカー、例えば、クロラムフェニコールアセチルトランスフェラーゼをコードしている核酸に作用的に結合している標的反応性調節エレメントを含んでいる)の誘導を検出すること、またはB7−4、PD−1または適当な標的分子により調節される細胞反応を検出することにより、決定することができる。例えば、B7−4またはPD−1タンパク質の、B7−4またはPD−1標的分子に結合するまたはそれらと相互作用する能力を測定することは、例えば、増殖アッセイにおける免疫細胞の副刺激または阻害を調節する化合物の能力を測定することにより、またはB7−4またはPD−1ポリペプチドの、B7−4またはPD−1ポリペプチドの部分を認識する抗体と結合する能力を妨害することにより行うことができる。
【0210】
他の例においては、(レセプターのライゲーションによる)PD−1の活性化により、SHP−2分子のリン酸化を生じる。SHP−2(Syp、SHPTP2、PTP2C、PTPN11、PTP1D、およびBPTP3としても知られる)は、非−膜チロシンホスファターゼのファミリーのメンバーである(U.S.特許第5,589,375号およびU.S.特許第5,831,009号、その内容は本明細書中に出典明示により組み込まれる。)。それは試験した全組織において至るところで発現され、心臓および脳において最もレベルが高い(Ahmad et al., Proc. Natl. Acad. Sci. U.S.A., 90: 2197-2201(1993); Bastien et al., Biochem. Biophys. Res. Commun. 196: 124-133 (1993); Freeman et al., Proc. Natl. Acad. Sci. U.S.A., 89: 11239-11243 (1992))。
【0211】
SHPタンパク質は、ホスホチロシル残基結合によるタンパク質−タンパク質相互作用を促進する、Srcタンパク質チロシンキナーゼにおいて本来的に同定される約100個のアミノ酸の保存された領域である2つのsrcホモロジー2(SH2)ドメインを含む(Neel, Semin. Cell. Biol. 4: 419-432 (1993))。これら2つのドメインは、SHP−2ホスファターゼの調節において異なる機能を示し、その結果異なるシグナリング経路に影響を及ぼすことが示されている。N−末端SHドメインは、内部触媒ホスファターゼドメインとの分子間相互作用により自己阻害効果を生じる調節および補充ドメインとして作用する。一方、C末端SH2ドメインがシグナル伝達に必要な分子間相互作用のための他のタンパク質を補充するべく作用するにすぎない(Pei et al., Proc. Natl. Acad. Sci. U.S.A. 93: 1141-1145 (1996))。SHP−2分子のリン酸化状態により、そのリン酸化活性が調節される。
【0212】
SH2−含有ホスファターゼを含むタンパク質−チロシンホスファターゼは、ヒトを含む哺乳動物などの多様な種から酵母およびツメガエル属に至る真核生物間で高度に保存されている。タンパク質−チロシンホスファターゼの変異および変化した発現が、腫瘍疾患および発生異常などの多くの病理学的症状に関連している(Tonks, N.K., et al., Cell 87:365-368 (1996); Neel, B. G., et al., Curr. Op. Cell Biol. 9:193-204 (1997); Li, J. et al., Science275: 1943-1947 (1997))。SHP−2は、異常な免疫反応において重要な役割を果たすことが示されている(例えば、アレルギー反応において(Pazdrak et al., J. Exp. Med. 186: 561-568 (1997))。
【0213】
SHP−2分子のリン酸化は、PD−1を経るシグナリングの指標である。SHP−2のリン酸化は、制限するものでなく、PAGEゲル上でのSHP−2分子の変化した移動の検出、リン酸化アッセイ、およびSHP−2分子の活性を測定するアッセイを含む当該分野で公知の方法により容易に検出され得る。SHP−2のリン酸化の検出は直接的であってよく、または別法として、例えばダウンストリームの活性または現象の検出など、間接的であってもよい。
【0214】
他の実施例において、(レセプターのライゲーションによる)PD−1の活性化により、T細胞レセプターの活性化により誘導される、細胞外シグナル調節キナーゼ1および2(ERK1および2)のリン酸化の阻害を生じる。ERK1およびERK2(MAPK1およびMAPKとしても知られる)はそれ自体がキナーゼである(Boulton, et al., Cell 65: 663-675, (1991))。ERK分子の活性化は、セリン/トレオニンリン酸化またはチロシンリン酸化による。つまり、別に活性化されたT細胞におけるPD−1シグナリングによるERK1および2のリン酸化の阻害は、PD−1によるERK1および2の活性化の阻害を示す。ERK1および2の活性化の阻害は、ERK1および2分子のアップストリームのリン酸化の阻害から生じ得、またはERK1および2を脱リン酸化して活性を減じるホスファターゼの活性化から生じ得る。
【0215】
ERKタンパク質は、二重特異性キナーゼであるMEK分子によるリン酸化により活性化されることが公知である。活性化に際して、ERK1およびERK2は核へと移動し、そこで、c−Myc、C/EBPβ、p62、TCF/Elk−1、ATF−2およびc−Junを含む種々の転写因子を直接リン酸化および活性化することができる。
【0216】
T細胞の活性化に際してのERK1および2分子のリン酸化状態/活性化状態は、PD−1を経るシグナリングの指標である。ERK1および2のリン酸化状態/活性化状態は、当該分野で容易に利用できるアッセイを用いて当業者により容易に決定され得る。例えば、ERK1および2分子のリン酸化状態は、p42/44ERK1/2タンパク質のリン酸化形態に特異的な(ホスホ−Thr202/Tyr204特異的)抗体、そのいくつかは市場入手可能である、を用いて決定することができる。別法として、ERK1および2分子のリン酸化状態は、ゲルにおけるその移動により、リン酸化状態に関わらずERK1および2を認識する抗体を同定のために用いて決定することができる。別法として、ERK1および2の活性化状態は、ERK1および2分子のキナーゼ活性をアッセイすることにより測定することができる。ERK1および2のリン酸化/活性化状態の決定は、例えばダウンストリーム活性または現象の検出など、間接的な方法によってもよい。
【0217】
他の例においては、(レセプターのライゲーションによる)PD−1の活性化により、T細胞レセプターの活性化により誘導されるタンパク質キナーゼC−θ(PKC−θ)分子のリン酸化の阻害を生じる。PKCイソ酵素は、多くの細胞シグナリング現象において重要な役割を果たす。(PKC−θ、PKCT、PRKCT、nPKC−θ、およびPRKCQとしても知られる)PKC−θは、セリン−トレオニンキナーゼのPKCファミリーのカルシウム非依存性イソ形態である。新規セリン/トレオニンタンパク質キナーゼCイソ形態(nPKC)の1つであるPKC−θは、組織において至るところで発現され、T−細胞および胸腺細胞を含む造血細胞系統において最高レベルで見出される(Baier et al., J. Biol. Chem. 268: 4997-5004 (1993); Keenan et al., Immunology 90: 557-563 (1997); Meller et al., Cell. Immunol. 193: 185-193 (1999); Wang et al., Biochem.Biophys. Res. Commun. 91: 240-246 (1993))。ネズミの胸腺細胞におけるPKC−θタンパク質の一過性の過剰発現により、インターロイキン−2−プロモーター駆動性構築物の転写の活性化を生じ(Baier et al., Eur. J. Biochem. 225: 195-203(1994))、T−細胞シグナリング経路におけるPKC−θの役割が示される。より最近では、PKC−θは、T細胞レセプター媒介性のT細胞の活性化学の間に活性化学されることが示されており、この活性化はPKC−θ分子の血漿膜へのAPC接触部位における移動と関連する(U.S.特許第6,040,152号、その内容を出典明示により本明細書中に組み込む)。PKC−θは、アポトーシス(Datta et al., J. Biol. Chem. 272: 20317-20320 (1997))、細胞骨格のアレンジメント(Pietromonaco et al., J. Biol. Chem. 273: 7594-7603 (1998) ; Simons et al., Biochem. Biophys. Res. Commun. 253: 561-565 (1998))、増殖(Passalacqua et al., Biochem. J. 337: 113-118 (1999))、および血管形成ならびに創傷治癒(Tang et al., J. Biol. Chem. 272: 28704-28711 (1997))を含む他の細胞プロセスにも関与している。
【0218】
T細胞の活性化に際してのPKC−θ分子のリン酸化/活性化状態は、PD−1を経るシグナリングの指標である。リン酸化状態は、PKC−θ分子の活性化状態を反映し、リン酸化は活性化を示す。PKC−θのリン酸化状態/活性化状態は、当該分野で容易に利用できるアッセイを用いて当業者により容易に測定され得る。例えば、PKC−θ分子のリン酸化状態は、市場入手可能な、PKC−θタンパク質のリン酸化形態に特異的な抗体(例えば、抗−ホスホT538)を用いて測定することができる。別法として、PKC−θ分子のリン酸化状態は、ゲルにおけるその移動により、PKC−θを(リン酸化状態に関わらず)認識する抗体を検出のために用いて、決定することができる。別法として、PKC−θの活性化状態は、PKC−θ分子のキナーゼ活性をアッセイすることにより(Kupfer et al., U.S. 特許No. 6,040,152号)、またはAPC接触点における膜への移動をアッセイすることにより(Kupfer et al., U.S. 特許No. 6,040,152号)決定することができる。PKC−θリン酸化/活性化状態の測定は、ダウンストリーム活性または現象の検出など、間接的な方法によってもよい。
【0219】
さらに他の具体例では、本発明のアッセイは、B7−4またはPD−1タンパク質またはその生物活性部分を試験化合物と接触させ、試験化合物がP7−4またはPD−1タンパク質またはその生物活性部分に結合する能力を測定するセルフリーアッセイである。試験化合物のB7−4またはPD−1タンパク質への結合は、前記のごとく直接的または間接的のいずれかで決定することができる。好ましい具体例では、アッセイには、B7−4またはPD−1タンパク質またはその生物活性部分を、B7−4またはPD−1を結合してアッセイ混合物形成する公知の化合物と接触させること、アッセイ混合物を試験化合物と接触させること、および試験化合物がB7−4またはPD−1タンパク質と相互作用する能力を測定すること(ここで、試験化合物がB7−4またはPD−1タンパク質と相互作用する能力を測定することは、試験化合物が公知の化合物と比較して選択的にB7−4またはPD−1ポリペプチドまたはその生物活性部分と結合する能力を決定することを含む)が含まれる。
【0220】
他の具体例では、アッセイは、B7−4またはPD−1タンパク質またはその生物活性部分を試験化合物と接触させ、試験化合物が、B7−4またはPD−1タンパク質またはその生物活性部分の活性を調節する能力を決定するセルフリーのアッセイである。試験化合物がB7−4またはPD−1タンパク質の活性を調節する能力を決定することは、例えば、B7−4またはPD−1タンパク質がB7−4またはPD−1標的分子に結合する能力を、直接結合を決定するための前記方法の1つにより決定することにより行うことができる。B7−4またはPD−1タンパク質がその結合パートナーと結合する能力の決定は、リアルタイム生体分子相互作用分析(BIA)などの技術を用いて達成できる(Sjolander,S.およびUrbaniczky,C.(1991)Anal.Chem.63:2338−2345およびSzaboら(1995)Curr.Opin.Struct.Biol.5:699−705)。本明細書において用いられる場合、「BIA」は相互作用物質のいずれも標識せずにリアルタイムで生体特異性相互作用を研究する技術である(たとえば、BIAcore)。表面プラズモン共鳴(SPR)の光学的現象における変化を、生物学的分子間のリアルタイムな反応の指標として用いることができる。
【0221】
さらにもう一つの具体例において、無細胞分析は、B7−4またはPD−1タンパク質またはその生物学的に活性な部分を、B7−4またはPD−1タンパク質と結合する公知化合物と接触させて分析混合物を形成し、分析混合物を試験化合物と接触させ、試験化合物がB7−4またはPD−1タンパク質のその結合パートナーとの相互作用を防止する能力を決定することを含む。たとえば、試験化合物がその結合部位でB7−4またはPD−1タンパク質と優先的に相互作用する能力またはB7−4またはPD−1標的分子の活性を調節する能力を試験することができる。本発明の無細胞分析は、タンパク質(たとえば、B7−4またはPD−1タンパク質またはその生物学的に活性な部分、またはB7−4またはPD−1が結合する結合パートナー)の可溶性および/または膜結合形態の両方を使用する。膜結合形態のタンパク質が使用される無細胞分析の場合において(たとえば、細胞表面B7−4またはPD−1レセプター)、膜結合形態のタンパク質が溶液中に維持されるように可溶化剤を利用することが望ましい。かかる可溶化剤の例は、非イオン性界面活性剤、たとえば、n−オクチルグルコシド、n−ドデシルグルコシド、n−ドデシルマルトシド、オクチル−N−メチルグルカミド、デカニル−N−メチルグルカミド、TritonX−100、TritonX−114、Thesit、イソトリデシポリ(エチレングリコールエーテル)n、3−[(3−コラミドプロピル)ジメチルアミニオ]−1−プロパンスルホネート(CHAPS)、3−[(3−コラミドプロピル)ジメチルアミニオ]−2−ヒドロキシ−1−プロパンスルホネート(CHAPSO)、またはN−ドデシル=N,N−ジメチル−3−アモニオ−1−プロパンスルホネートを包含する。
【0222】
本発明の前記分析法の1以上の具体例において、タンパク質の一方または両方の複合体を形成する形態または複合体を形成しない形態の分離を促進するため、ならびに分析の自動化に対応するために、B7−4またはPD−1またはその適当な標的分子のいずれかを固定化することが望ましい。試験化合物mpB7−4またはPD−1タンパク質との結合、またはB7−4またはPD−1タンパク質の標的分子との候補化合物の存在下または不在下での相互作用は、反応物質を入れるのに適した任意の容器中有で行うことができる。このような容器の例は、マイクロタイタープレート、試験管、およびミクロ遠心管を包含する。一具体例において、タンパク質の一方または両方がマトリックスに結合するのを許容するドメインを加えた融合タンパク質を提供できる。たとえば、グルタチオン−S−トランスフェラーゼ/B7−4またはPD−1融合タンパク質またはグルタチオン−S−トランスフェラーゼ/標的融合タンパク質をグルタチオンセファロースビーズ(Sigma Chemical,St.Louis,MO)またはグルタチオン誘導化マクロタイタープレート上に吸着させることができ、これを次に試験化合物、または試験化合物と未吸着標的タンパク質またはB7−4またはPD−1タンパク質のいずれかと合わせ、混合物を、複合体形成を誘導する条件下(たとえば、塩の生理学的条件およびpH)でインキュベートする。インキュベーション後、前記のような結合していない成分、ビーズの場合は固定化されていないマトリックス、直接または間接的のいずれかで測定された複合体を除去するために、ビーズまたはマイクロタイタープレートウェルを洗浄する。別法として、複合体をマトリックスから解離させることができ、B7−4またはPD−1の結合または活性のレベルを、標準的技術を用いて決定できる。
【0223】
タンパク質をマトリックス上に固定するための他の技術も本発明のスクリーニングアッセイにおいて用いることができる。たとえば、B7−4またはPD−1タンパク質またはB7−4またはPD−1標的分子のいずれかを、ビオチンおよびストレプトアビジンの結合を利用して固定化できる。ビオチニル化B7−4またはPD−1タンパク質または標的分子を、ビオチン−NHS(N−ヒドロキシ−スクシンイミド)から当業者に周知の技術(たとえば、ビオチニル化キット、Pierce Chemicals,Rockford,IL)を用いて調製し、ストレプトアビジンコートされた96ウェルプレート(Pierce Chemical)のウェル中に固定化できる。別法として、B7−4またはPD−1タンパク質または標的分子と反応性であるが、B7−4またはPD−1タンパク質のその標的分子との結合を妨害しない抗体をプレートのウェルから誘導化し、未結合標的またはB7−4またはPD−1タンパク質を抗体結合によりウェル中にトラップできる。かかる複合体の検出方法は、GST−固定化複合体についてすでに記載した者に加えて、B7−4またはPD−1タンパク質と反応性の抗体を用いた複合体の免疫検出、ならびにB7−4またはPD−1タンパク質または標的分子と結合する酵素活性の検出に依存する酵素結合分析を包含する。
【0224】
もう一つの具体例において、試験化合物がB7−4またはPD−1タンパク質の活性を調節する能力の決定は、試験化合物がB7−4を減少調節する分子、たとえば、B7−4と相互作用する分子、またはPD−1を減少調節する分子の活性を調節する能力を決定することにより、たとえばPD−1の細胞質ドメインと相互作用することにより、達成することができる。たとえば、すでに記載したように、第二のメッセンジャーのレベルを決定できるか、適当な標的上の相互作用分子の活性を決定できるか、または相互作用対の適当な標的との結合を決定できる。
【0225】
もう一つの具体例において、B7−4またはPD−1発現のモジュレーターは、細胞を候補または試験化合物と接触させ、細胞中のB7−4またはPD−1mRNAまたはタンパク質の発現を決定する方法において同定される。B7−4またはPD−1mRNAまたはタンパク質の、候補化合物の存在下での発現のレベルを、B7−4またはPD−1mRNAまたはタンパク質の候補化合物の不在下での発現のレベルと比較する。候補化合物を次いでこの比較に基づいてB7−4またはPD−1発現のモジュレーターとして同定することができる。たとえば、B7−4またはPD−1mRNAまたはタンパク質の発現が、候補化合物の存在下での方が不在下よりも大きい(たとえば、再現可能で、統計学的に有意に大きい)場合、該候補化合物は、B7−4またはPD−1mRNAまたはタンパク質発現の刺激物質として確認される。あるいは、B7−4またはPD−1mRNAまたはタンパク質の発現が、候補化合物の存在下での方が不在下よりも小さい(たとえば、再現可能で、統計学的に有意に小さい)場合、該候補化合物は、B7−4またはPD−1mRNAまたはタンパク質発現の阻害物質として確認される。細胞におけるB7−4またはPD−1mRNAまたはタンパク質発現のレベルは、B7−4またはPD−1mRNAまたはタンパク質の検出に関して本明細書において記載する方法により決定できる。
【0226】
本発明のもう一つの具体例において、PD−1シグナリング経路に関与するペプチドおよび/またはタンパク質(たとえば、PD−1:B7−4作用物質または拮抗物質として作用できるタンパク質、PD−1シグナリングに反応してリン酸化されるタンパク質、および/またはPD−1と結合するタンパク質)は、ペプチド結合/免疫沈降および質量分析により同定できる。たとえば、T細胞(たとえば、Jurkat T細胞)はB7−4の存在下または不在下、および過バナジウム酸塩の存在下または不在下で活性化できる。タンパク質を次いでT細胞の溶解物から、たとえば抗PD−1抗体、抗ホスホチロシン(Ptyr)、またはPD−1ペプチドを用いて免疫沈降させることができる。免疫沈降したタンパク質のパターンおよびリン酸化における変化を次いで、たとえばSDS−PAGE(ドデシル硫酸ナトリウムポリアクリルアミドゲル電気泳動)を用いて比較できる。タンパク質を、次いでESI MS/MS(質量分析)を用いて分析し、同定できる。前記スクリーニングアッセイを表す概略図を図31Aに示す。
【0227】
前記スクリーニングアッセイ(たとえば、Jurkat T細胞を用いた分析法において)を用いて同定されたタンパク質を、ヒトT細胞の分析において用いることができる。このようなタンパク質を次いでPD−1作用物質または拮抗物質、たとえば小分子作用物質または拮抗物質を同定するためのシグナリングベースのスクリーニングアッセイにおいて用いることができる。
【0228】
本発明のさらにもう一つの態様において、B7−4またはPD−1と結合または相互作用し、B7−4またはPD−1活性に関与する他のタンパク質(「B7−4またはPD−1結合タンパク質」または「B7−4またはPD−1bp」)を同定するために、B7−4またはPD−1タンパク質、好ましくは膜結合形態のものを、2ハイブリッドアッセイまたは3ハイブリッドアッセイにおいて「おとりタンパク(bait proteins)」として使用できる(たとえば、米国特許第5283317号;Zervosら(1993)Cell72:223−22;Maduraら(1993)J.Biol.Chem.268:12046−12054;Bartelら(1993)Biotechniques14:920−924;Iwabuchiら(1993)Oncogene8:1693−1696;およびBrent、WO94/10300)。このようなB7−4またはPD−1結合タンパク質は、たとえば、B7−4またはPD−1によるシグナリング経路の上流または下流エレメントとしてB7−4またはPD−1タンパク質またはB7−4またはPD−1標的によるシグナルの伝播に関与する可能性がある。別法として、このようなB7−4またはPD−1結合タンパク質はB7−4またはPD−1阻害物質であり得る。
【0229】
2ハイブリッドシステムは、ほとんどの転写因子のモジュレーター性質に基づき、これは分離可能なDNA結合および活性化ドメインからなる。簡単に説明すると、該アッセイは2つの異なるDNA構築物を利用する。一方の構築物においては、B7−4またはPD−1タンパク質をコードする遺伝子を、公知転写因子(たとえば、GAL−4)のDNA結合ドメインをエンコードする遺伝子と融合させる。他の構築物において、同定されていないタンパク質(「えさ(prey)」または「サンプル」)をエンコードするDNA配列のライブラリーから得られるDNA配列を、公知転写因子の活性化ドメインをコードする遺伝子と融合させる。「おとり」および「えさ」タンパク質がインビボで相互作用でき、B7−4依存性複合体を形成するならば、転写因子のDNA結合および活性化ドメインを近接させることができる。このように近接することで、転写因子と反応しやすい転写調節部位に操作可能に結合されたレポーター遺伝子(たとえば、LacZ)の転写が可能になる。レポーター遺伝子の発現を検出でき、機能的転写因子を含む細胞クローンを単離でき、B7−4またはPD−1タンパク質と相互作用するタンパク質をエンコードするクローンされた遺伝子が得られる。
【0230】
本発明はさらに、前記スクリーニングアッセイにより同定される新規薬剤に関する。従って、さらに適当な動物モデルにおいて前記のようにして同定される薬剤を使用することも本発明の範囲内に含まれる。たとえば、本明細書に記載したようにして同定される薬剤(たとえば、B7−4またはPD−1調節剤、アンチセンスB7−4またはPD−1核酸分子、B7−4またはPD−1特異性抗体、またはB7−4またはPD−1結合パートナー)を、かかる薬剤での治療の効力、毒性、または副作用を決定するために動物モデルにおいて使用できる。別法として、本明細書に記載したようにして同定される薬剤は、かかる薬剤の作用のメカニズムを決定するために動物モデルにおいて使用できる。さらに、本発明は本明細書に記載するような治療のための前記スクリーニングアッセイにより同定される新規薬剤の使用に関する。
【0231】
E.検出分析
本発明において同定されるcDNA配列の一部またはフラグメント(および対応する完全遺伝子配列)をポリヌクレオチド試薬として多くの方法において使用できる。たとえば、これらの配列は:(i)そのそれぞれの遺伝子を染色体上にマップし;従って遺伝子疾患に関連する遺伝子領域の位置を突き止める;(ii)微小な生物学的サンプル唐子湖を同定する(組織タイピング);および(iii)生物学的サンプルにおける法医学的確認を助けるために使用できる。これらの用途は、以下のサブセクションにおいて記載する。
【0232】
1.染色体マッピング
遺伝子の配列(または配列の部分)が一度単離されたら、この配列を用いて染色体上での遺伝子の位置をマップすることができる。このプロセスは、染色体マッピングと呼ばれる。従って、本明細書中に記載するB7−4ヌクレオチド配列の部分またはフラグメントを用いて、染色体上のB7−4遺伝子の位置をマップすることができる。B7−4配列の染色体へのマッピングは、これらの配列を疾患に関連する遺伝子と関連づける重要な最初のステップである。
【0233】
簡単には、B7−4遺伝子は、B7−4ヌクレオチド配列からPCRプライマー(好ましくは15−25bp長)を調製することにより、染色体へとマップすることができる。B7−4配列のコンピューター分析を用いて、ゲノムDNA中の1個以上のエキソンをスパンせず、増幅プロセスを複雑にしているプライマーを予測することができる。これらのプライマーを次いで個々のヒト染色体を含んでいる体細胞ハイブリッドのPCRスクリーニングに用いることができる。B7−4配列に対応するヒト遺伝子を含んでいるこれらのハイブリッドのみが増幅フラグメントを生じるであろう。
【0234】
体細胞ハイブリッドは、別個の哺乳動物(例えば、ヒトおよびマウス細胞)由来の体細胞を融合することにより調製する。ヒトおよびマウス細胞のハイブリッドが増殖し、分裂するにつれ、それらはランダムな順序でヒトの染色体を徐々に失うが、マウスの染色体は保持する。特定の酵素を欠くためにマウス細胞は増殖し得ないがヒト細胞は増殖できる培地を用いることにより、必要とされる酵素をコードしている遺伝子を含む1つのヒト染色体が保持される。種々の培地を用いることにより、ハイブリッド細胞系統のパネルを確立することができる。パネル中の各細胞系統は、単一のヒト染色体または少数のヒト染色体のいずれかおよびマウス染色体の完全なセットを含み、特定のヒトの染色体への個々の遺伝子の容易なマッピングを可能とする(D'Eustachio, P. et al. (1983) Science 220:919-924)。ヒト染色体のフラグメントのみを含んでいる体細胞ハイブリッドは、転座および欠失を有するヒト染色体を用いることにより作製することもできる。
【0235】
体細胞のハイブリッドのPCRマッピングは、特定の染色体へ特定の配列を指定するための迅速な方法である。3つまたはそれ以上の配列を、単一の熱サイクラーを用いて1日あたりに指定することができる。オリゴヌクレオチドプライマーを設計するためにB7−4ヌクレオチド配列を用いて、サブローカライゼーションを、特定の染色体からのフラグメントのパネルを用いて達成することができる。その染色体へ配列をマップするために同様に用いられ得る他のマッピング戦略には、インサイチュハイブリダイゼーション(Fan, Y. et al. (1990) Proc. Natl. Acad. Sci. USA, 87:6223-27に記載されている)、標識されたフローソートされた染色体でのプレ−スクリーニング、および染色体特異cDNAライブラリーへのハイブリダイゼーションによるプレ−セレクションが含まれる。
【0236】
DNA配列の中期染色体展開物へ蛍光インサイチュハイブリダイゼーション(FISH)をさらに用いて、1回のステップで正確な染色体の位置を得ることができる。染色体展開物は、有糸分裂の紡錘体を壊すコルセミドなどの化学物質により分裂が中期で遮断された細胞を用いてなすことができる。染色体は簡単にトリプシンで処理して、次いでギムザで染色することができる。明暗バンドのパターンが各染色体において発生し、その結果、染色体を個々に同定することができる。FISH法を500または600塩基もの長さのDNA配列を用いて用いることができる。しかし、1000塩基より長いクローンが簡単な検出のために十分なシグナル強度にてユニークな染色体の位置へ結合するより高い見込みを有する。好ましくは1000塩基、およびより好ましくは2000塩基が、妥当な時間で優れた結果を得るのに十分である。この方法のレビューとしては、Verma et al., ヒト染色体: 基本技術マニュアル (Pergamon Press, New York 1988)を参照されたい。
【0237】
染色体マッピングのための試薬を個々に用いて、単一の染色体またはその染色体上の単一の部位をマークすることができ、または試薬のパネルを複数の部位および/または複数の染色体をマーキングするのに用いることができる。遺伝子の非コーディング領域に対応する試薬がマッピング目的のために好ましい。コーディング配列は、遺伝子ファミリー内で保存されているようであり、従って、染色体マッピング中の交差はハイブリダイゼーションの機会が増す。
【0238】
一度配列が正確な染色体の位置へとマップされたら、染色体における配列の物理的な位置を遺伝子マップデータと関連付けることができる(そのようなデータは、例えば、ジョンス ホプキンス大学ウェルシュメディカルライブラリーよりオンラインで入手可能なMcKusick, V., Mendelian Inheritance in Manに見出される)。遺伝子と同じ染色体領域へマップされる疾患の間の関連性は、例えば、Egeland, J. et al. (1987) Nature 325:783-787に記載されている関連分析(物理的に隣接する遺伝子の共遺伝)により同定することができる。
【0239】
さらに、B7−4遺伝子に関連する疾患に罹患している個人と罹患していない個人の間のDNA配列の差を決定することができる。変異が罹患した個人の幾人かまたは全員には認められるが、罹患していない個人においてはいずれも認められない場合、変異は特定の疾患の原因であると考えられる。罹患した個人および罹患していない個人の比較は一般に、染色体展開物から認められるまたはDNA配列に基きPCRを用いて検出される欠失または転座などの染色体における構造変化をまず探すことを含む。最終的には、幾人かの個人からの遺伝子の完全な配列決定を行い、変異の存在を確認し、多型から変異を区別することができる。
【0240】
2.組織タイピング
本発明のB7−4配列をさらに用いて、少量の生物サンプルから個人を同定することもできる。合衆国軍は、例えば、そのパネルの同定のための制限フラグメント長の多型(RFLP)の使用を考慮している。この方法において、個人のゲノムDNAを1個またはそれ以上の制限酵素で消化し、サザンブロット上にプローブして、同定のためのユニークなバンドを得る。この方法は、失われ、交換され、または転移され、ポジティブな同定を困難にしている「ドッグタグ」の現在の制限を受けない。本発明の配列は、RFLPのさらなるDNAマーカーとして有用である(米国特許第5,272,057号に記載されている)。
【0241】
さらに、本発明の配列を用いて、個人のゲノムの選択された位置の実際の塩基−対−塩基DNA配列を決定する別法を提供することができる。つまり、本明細書中に記載するB7−4ヌクレオチド配列を用いて、配列の5’および3’末端から2つのPCRプライマーを調製することができる。これらのプライマーを次いで用いて、個人のDNAを増幅し、その後それを配列決定することができる。
【0242】
この方法で調製された、個人からの対応するDNA配列のパネルにより、各個人は対立変異によりそのようなDNA配列のユニークなセットを有するので、ユニークな個人の同定が提供され得る。本発明の配列を用いて、個人および組織からのそのような個人の配列を得ることができる。本発明のB7−4ヌクレオチド配列は、ヒトゲノムの部分をユニークに示す。対立変異がこれらの配列のコーディング領域にある程度まで、および非コーディング領域においてより大きな程度まで起こる。個人のヒトの間での対立変異は、各500塩基あたり約1個の頻度で生じることが見積もられている。個人からのDNAを同定目的のために比較することができる基準として、本明細書中に記載される配列のそれぞれを、ある程度まで用いることができる。より多数の多型が非コーディング領域に起こるので、より少ない配列が個人を分かつために必要である。配列番号1または3の非コーディング配列により、それぞれが100塩基の非コーディング増幅配列を生じるおそらく10〜1000プライマーのパネルを用いてポジティブな個人の同定を容易に提供することができる。予測されるコーディング配列を用いる場合、ポジティブな個人の同定のためのプライマーのより適当な数のは、500−2000であろう。
【0243】
本明細書中に記載するB7−4ヌクレオチド配列からの試薬のパネルを用いて個人に関するユニークな同定データベースを作製する場合、これら同じ試薬を後で用いてそ個人からの組織を同定することができる。ユニークな同定データベースを用いて、生きているまたは死亡した個人のポジティブな同定を、極端に少量の組織サンプルからなすことができる。
【0244】
3.法生物学における部分的B7−4配列の使用
DNA−に基く同定法は、法医学においても用いることができる。法生物学は、例えば犯罪の犯人をポジティブに同定するための手段として、犯罪現場で見出される生物学的証拠の遺伝子タイピングを用いる科学分野である。そのような同定をなすために、PCR法を用いて、犯罪現場で見出される、例えば、髪または皮膚、または体液、例えば血液、唾液、精液などのごく少量の生物学的サンプルから採取したDNA配列を増幅することができる。増幅された配列を次いで基準と比較して、それにより生物縛的サンプルの源の同定が可能となる。
【0245】
本発明の配列を用いて、例えば、他の「同定マーカー」(即ち、特定の個人にユニークな他のDNA配列)を提供することによりDNA−に基く法医学的同定の信頼性を高めることができる、ヒトゲノムにおいて特定の位置に標的される、例えばPCRプライマーなどのポリヌクレオチド試薬を提供することができる。前記のように、実際の塩基配列情報を、制限酵素により作製されたフラグメントにより形成されるパターンに対する適格な代替として、同定のために用いることができる。非コーディング領域に標的される配列は、より多数の多型が非コーディング領域に生じるために特に適当であり、この方法を用いて個人を区別することがより簡単になる。ポリヌクレオチド試薬の例には、B7−4ヌクレオチド配列または少なくとも20塩基、好ましくは少なくとも30塩基の長さを有するその部分が含まれる。
【0246】
本明細書中に記載されるB7−4ヌクレオチド配列をさらに用いて、特定の組織、例えば脳組織を同定するためのインサイチュハイブリダイゼーション法などにおいて用いることができる、標識または標識可能プローブなどのポリヌクレオチド試薬を提供することができる。これは、法医学病理学者が未知起源の組織を与えられた場合に非常に有用であり得る。そのようなB7−4プローブのパネルを用いて、種によりおよび/または器官タイプにより組織を同定することができる。
【0247】
同様の方法で、これらの試薬、例えばB7−4プライマーまたはプローブを用いて、汚染に関して培養組織をスクリーニングする(即ち、培養物中の異なるタイプの細胞の混合物の存在をスクリーニングする)ことができる。
【0248】
F.予測医薬
本発明は、診断アッセイ、予後アッセイ、および臨床試験をモニターすることを診断(予測)目的のために用いて個人を予防的に治療する予測医薬の分野にも関する。従って、本発明の1態様は、生物学的サンプル(例えば、血液、血清、細胞、組織)の内容物中でのB7−4またはPD−1タンパク質および/または核酸の発現ならびにB7−4またはPD−1の活性を測定して、それにより個人が異常なB7−4またはPD−1の発現または活性に伴う病気または疾患に罹患しているまたはそれに伴う疾患を進行させる危険にあるかどうかを決定するための診断アッセイに関する。発明によりさらに、個人がB7−4またはPD−1タンパク質、核酸の発現または活性に伴う疾患を進行させる危険にあるかどうかを決定するための診断(または予測)アッセイも提供される。例えば、B7−4またはPD−1遺伝子における変異を生物学的サンプルにおいてアッセイすることができる。そのようなアッセイを診断または予測目的のために用いて、それによりB7−4またはPD−1タンパク質、核酸発現または活性により特徴づけられる、またはそれに伴う疾患の発症前に個人を予防処置することができる。前記の診断アッセイまたは後述するアッセイなどの本明細書中に記載するアッセイを用いて、自然流産を有する傾向を検出することもできる。
本発明の他の態様は、臨床試験におけるB7−4またはPD−1の発現または活性における試薬(例えば、薬物、化合物)の影響をモニターすることを含む。 これらおよび他の試薬を以下のセクションにさらに詳細に記載する。
【0249】
1.診断分析
生物学的サンプルにおけるB7−4またはPD−1タンパク質または核酸の存在を検出するための方法の一例は、試験対象から生物学的サンプルを得、該生物学的サンプルを、B7−4またはPD−1タンパク質をエンコードし、B7−4またはPD−1タンパク質の存在を生物学的サンプルにおいて検出するB7−4またはPD−1タンパク質または核酸(たとえばmRNA、ゲノムRNA)を検出できる化合物または薬剤と接触させることを含む。B7−4またはPD−1mRNAまたはゲノムDNAを検出するための試薬は、B7−4またはPD−1mRNAまたはゲノムDNAとハイブリッド形成できる標識された核酸プローブである。核酸プローブは、たとえば、ヒトB7−4またはPD−1核酸、たとえば、配列番号1、3、10、11、または38の核酸またはその一部、たとえば、少なくとも15、30、50、100、250または500ヌクレオチドの長さで、ストリンジェントな条件下でB7−4またはPD−1mRNAまたはゲノムDNAと特異的にハイブリッド形成するために十分なオリゴヌクレオチドである。本発明の診断分析において使用される他の適当なプローブは本明細書に記載される。
【0250】
B7−4またはPD−1タンパク質を検出するための好ましい試薬は、B7−4またはPD−1タンパク質と結合できる抗体、好ましくは検出可能な標識を有する抗体である。抗体は、ポリクローナル抗体、またはさらに好ましくはモノクローナル抗体である。完全な抗体、またはそのフラグメント(たとえば、FabまたはF(ab’)2)を使用できる。プローブまたは抗体に関する「標識された」なる用語は、検出可能な物質をプローブまたは抗体とカップリング(すなわち、物理的結合)させることによるプローブまたは抗体の直接標識、ならびに直接表紙期されたもう一つの試薬との反応性によるプローブまたは抗体の間接的標識を包含することを意図される。直接標識の例は、蛍光標識された第二抗体および蛍光標識されたストレプトアビジンで検出できるようにDNAプローブのビオチンでの末端標識を用いて第一抗体を検出することを包含する。「生物学的サンプル」なる用語は、対象から単離される組織、細胞および生物学的液体、ならびに対象内に存在する組織、細胞および液体を包含することを意図される。すなわち、本発明の検出法は、インビトロならびにインビボで生物学的サンプルにおけるB7−4またはPD−1mRNA、たんぱくしつ、またはゲノムDNAを検出するために使用できる。たとえば、B7−4またはPD−1mRNAを検出するためのインビトロ技術は、ノザンハイブリダイゼーションおよびインサイチュハイブリダイゼーションを包含する。B7−4またはPD−1タンパク質を検出するためのインビトロ技術は、酵素結合免疫吸着検定法(ELISA)、ウェスタンブロット、免疫沈降および免疫蛍光を包含する。B7−4またはPD−1ゲノムDNAを検出するためのインビトロ技術は、サザンハイブリダイゼーションを包含する。さらに、B7−4またはPD−1タンパク質を検出するためのインビボ技術は、対象中に標識された抗B7−4またはPD−1抗体を導入することを含む。たとえば、抗体は、対象におけるその存在および位置が標準的イメージ化技術により検出できる放射性マーカーで標識できる。
【0251】
一具体例において、生物学的サンプルは、試験対象から得られるタンパク質分子を含む。あるいは、生物学的サンプルは、試験対象から得られるmRNA分子または試験対象から得られるゲノムDNAを含み得る。好ましい生物学的サンプルは、慣用の手段により対象から単離される血清サンプルである。
もう一つの具体例において、方法はさらに、対照から対照生物学的サンプルを得、B7−4またはPD−1タンパク質、mRNAまたはゲノムDNAの存在が生物学的サンプルにおいて検出されるように、対照サンプルを、B7−4またはPD−1タンパク質、mRNA、またはゲノムDNAを検出できる化合物または試薬と接触させ、対照サンプルにおけるB7−4またはPD−1タンパク質、mRNAまたはゲノムDNAを、試験サンプル中のB7−4またはPD−1タンパク質、mRNAまたはゲノムDNAと比較することを含む。
【0252】
本発明は、生物学的サンプルにおけるB7−4またはPD−1の存在を検出するためのキットも包含する。たとえば、キットは、生物学的サンプルにおいてB7−4またはPD−1タンパク質またはmRNAを検出できる標識された化合物または試薬;サンプル中のB7−4またはPD−1の量を定量する手段;およびサンプル中のB7−4またはPD−1の量を基準と比較するための手段を含み得る。化合物または試薬は適当な容器中に詰めることができる。キットはさらに、B7−4またはPD−1タンパク質または核酸を検出するためのキットを使用するための説明書も含み得る。
【0253】
2.診断分析
本明細書に記載する診断法はさらに、異常なB7−4またはPD−1発現または活性に関連する疾患または障害にかかっているか、または発症する危険性のある対照を同定するために使用できる。たとえば、本明細書に記載する分析、たとえば前記診断分析または後述の分析を、B7−4またはPD−1タンパク質に関連する障害にかかっているかまたは発症する危険性のある患者を確認するために使用できる。従って、本発明は、異常なB7−4またはPD−1発現または活性に関連する疾患または障害を確認するための方法であって、試験化合物が対照から得られ、B7−4またはPD−1タンパク質または核酸(たとえば、mRNA、ゲノムDNA)が検出され、B7−4またはPD−1タンパク質または核酸の存在が、異常なB7−4またはPD−1発現または活性に関連する疾患または障害にかかっているかまたは発症する危険性のある対象を診断できる者である方法を提供する。本明細書において使用する場合、「試験サンプル」とは、関心のある対象から得られる生物学的サンプルを意味する。たとえば、試験サンプルは、生物学的液体(たとえば、血清)、細胞サンプル、または組織であり得る。
【0254】
さらに、本明細書に記載する診断分析は、異常なB7−4またはPD−1発現または活性に関連する疾患または障害を治療するために、対象に薬剤(たとえば、作用物質、拮抗物質、ペプチド模倣物、タンパク質、ペプチド、核酸、小分子、または他の医薬候補)を投与できるかどうかを決定するために使用できる。従って、本発明は、B7−4またはPD−1発現または活性に関連する障害の薬剤を用いて対象を有効に治療できるかどうかを決定する方法であって、試験サンプルが得られ、B7−4またはPD−1タンパク質または核酸発現または活性が検出される(たとえば、B7−4またはPD−1タンパク質または核酸発現または活性の発生量が、異常なB7−4またはPD−1発現または活性に関連する障害を治療するために薬剤を投与できる患者を診断できる)方法を提供する。
【0255】
本発明の方法は、B7−4またはPD−1遺伝子における遺伝子変更を検出し、それにより遺伝子が変更されている対象がB7−4またはPD−1遺伝子に関連する障害の危険性がある華道かを決定することができる。好ましい具体例において、該方法は、対象から得られる細胞のサンプルにおいて、B7−4またはPD−1タンパク質をエンコードする遺伝子の統合性に影響を及ぼす変更のすくなくとも一つにより特徴づけられる遺伝子の変更の存在または不在、またはB7−4またはPD−1遺伝子の発現の誤りを検出することを含む。たとえば、このような遺伝子変更は、1)B7−4またはPD−1遺伝子からの1以上のヌクレオチドの欠失;2)B7−4またはPD−1遺伝子に対する1以上のヌクレオチドの付加;3)B7−4またはPD−1遺伝子の1以上のヌクレオチドの置換;4)B7−4またはPD−1遺伝子の染色体の再配置;5)B7−4またはPD−1遺伝子のメッセンジャーRNA転写物のレベルの変更、6)B7−4またはPD−1遺伝子、たとえばゲノムDNAのメチル化パターンの異常な修飾、7)B7−4またはPD−1遺伝子のメッセンジャーRNA転写物の非野生型スプライシングパターンの存在、8)B7−4またはPD−1タンパク質の非野生型レベル、9)B7−4またはPD−1遺伝子の対立遺伝子損失、および10)B7−4またはPD−1タンパク質の不適当な転写後修飾の少なくとも一つの存在を確認することにより検出できる。本明細書において記載されるように、B7−4またはPD−1遺伝子の変更を検出するために用いられる当該分野において公知の技術が多数ある。好ましい生物学的サンプルは、対象から慣用の手段により単離される組織または血清サンプルであり、たとえば心臓組織サンプルである。
【0256】
ある具体例において、変更の検出は、ポリメラーゼ連鎖反応(PCR)(たとえば、米国特許代4683195号および第4683202号)、たとえばアンカーPCRまたはRACE PCR、あるいは結紮連鎖反応(LCR)(たとえば、Landegranら(1988)Science241:1077−1080;およびNakazawaら(1994)Proc.Natl.Acad.Sci.USA91:360−364)におけるプローブ/プライマーの使用を含み、後者はB7−4またはPD−1遺伝子における点突然変異の検出に特に有用である(Abravayaら(1995)Nucleic Acids Res.23:675−682)。この方法は、患者からの細胞のサンプルを集める工程、サンプルの細胞から核酸(たとえば、ゲノム、mRNAまたは両方)を単離する工程、核酸サンプルを、B7−4またはPD−1遺伝子(存在するならば)のハイブリダイゼーションおよび増幅が起こるような条件下でB7−4またはPD−1遺伝子と特異的にハイブリッド形成する1以上のプライマーと接触させる工程、および増幅産物の存在または不在を検出する工程、または増幅サンプルのサイズを検出する工程および長さを対照サンプルと比較する工程を含み得る。PCRおよび/またはLCRは、本明細書に記載される突然変異を検出するために用いられる技術のいずれかと組み合わせて予備増幅工程として使用するのが望ましい。
【0257】
別の増幅法は:自立的配列複製(Guatelli,J.C.ら(1990)Proc.Natl.Acad.Sci.USA87:1874−1878)、転写増幅システム(Kwoh,D.Y.ら(1989)Proc.Natl.Acad.Sci.USA86;1173−1177)、Q−ベータレプリカーゼ(Lizardi,P.M.ら(1988)Biotecnology6:1197)、または任意の他の核酸増幅法を含み、それに続いて当業者に周知の技術を用いて増幅された分子を検出する。これらの検出スキームは、このような分子が非常に少数で存在するならば核酸分子の検出に特に有用である。
もう一つの具体例において、サンプル細胞からのB7−4またはPD−1遺伝子の突然変異は、制限酵素開裂パターンにおける変更により同定できる。例えば、サンプルおよび対照DNAが単離され、(所望により)増幅され、1以上の制限エンドヌクレアーゼで消化され、フラグメント長サイズがゲル電気泳動により決定され、比較される。サンプルと対照DNA間のフラグメント長サイズの差は、サンプルDNAにおける突然変異を示す。さらに、配列特異的リボザイム(例えば、米国特許第5498531号参照)の使用は、リボザイム開裂部位の発生または損失により特異的突然変異の存在を採点するために使用できる。
【0258】
他の具体例において、B7−4またはPD−1における遺伝子突然変異は、サンプルと対照核酸、例えばDNAまたはRNAを、数百または数千のオリゴヌクレオチドプローブを含む高密度アレイとハイブリッド形成することにより同定できる(Cronin,M.T.ら(1996)Hum.Mutat.7:244−255;Kozal,M.J.ら(1996)Nat.Med.2:753−759)。例えば、B7−4またはPD−1における遺伝子突然変異は、Cronin,M.T.ら(1996)前出に記載されているように光生成DNAを含む二次元アレイにおいて同定できる。簡単に説明すると、プローブのハイブリダイゼーションアレイは、連続した重複プローブの直線状アレイを調製することにより配列間の塩基変化を同定するために、サンプルと対照におけるDNAの長さにわたってスキャンするために使用できる。この工程により、点突然変異の同定が可能になる。この工程に続いて、第二のハイブリダイゼーションアレイを行い、これは検出される全ての変異体または突然変異に対して相補性のより小さな分化したプローブアレイを使用することにより、特的突然変異のキャラクタライゼーションを可能にする。各突然変異アレイは、平行なプローブセットからなり、1つは野生型遺伝子に相補静であり、他は突然変異遺伝子に相補性である。
【0259】
さらにもう一つの具体例において、当該分野において公知の様々な配列化反応の任意のものを使用して、B7−4またはPD−1遺伝子を直接配列させ、サンプルB7−4またはPD−1の配列を対応する野生型(対照)配列と比較することにより突然変異を検出することができる。配列化反応の例は、MaxamおよびGilbert((1977)Proc.Natl.Acad.Sci.USA74:560)またはSanger((1977)Proc.Natl.Acad.Sci.USA74:5463)により開発された技術に基づくものを包含する。質量分析による配列化(例えば、PCT国際公開番号WO94/16101;Cohenら(1996)Adv.Chromatogr.36:127−162;およびGriffinら(1993)Appl.Biochem.Biotechnol.38:147−159)を含む様々な自動化配列化法の任意のものを、診断分析((1995)Biotechniques19:448)を行う場合に利用できると考えられる。
【0260】
B7−4またはPD−1遺伝子における突然変異を検出する他の方法は、RNA/RNAまたはRNA/DNAヘテロ二本鎖におけるミスマッチした塩基を検出するために開裂剤からの保護を使用する方法(Myerら(1985)Science230:1242)を包含する。一般に、「ミスマッチ開裂」の技術は、野生型B7−4またはPD−1配列を含む(標識された)RNAまたはDNAを組織サンプルから得られる潜在的な突然変異体RNAまたはDNAとハイブリッド形成させることによりヘテロ二本鎖を提供することにより開始する。二本鎖を、二本鎖の一本鎖領域、例えば対照とサンプル鎖間の塩基対ミスマッチのために存在するものを開裂させる薬剤で処理する。例えば、RNA/DNA二本鎖をRNAseで処理し、DNA/DNAハイブリッドをS1ヌクレアーゼで処理して、ミスマッチした領域を酵素で消化する。他の具体例において、ミスマッチ領域を消化するために、DNA/DNAまたはRNA/DNA二本鎖のいずれかを、ヒドロキシルアミンまたはオスミウムテトロキシドで処理し、ピペリジンで処理できる。ミスマッチ領域の消化後、結果として得られる物質を、変性ポリアクリルアミドゲル上サイズにより分離して、突然変異の部位を決定する。例えば、Cottonら(1988)Proc.Natl.Acad.Sci.USA85:4397;Saleebaら(1992)Methods Ezymol.217:286−295参照)。好ましい具体例において、対照DNAまたはRNAを検出のために標識できる。
【0261】
さらにもう一つの具体例において、ミスマッチ開裂反応は、細胞のサンプルから得られるB7−4またはPD−1cDNAにおける点突然変異を検出し、マップするために所定のシステムにおいて二本鎖DNAにおいてミスマッチした塩基対を認識する1以上のタンパク質(いわゆる、「DNAミスマッチ修復」酵素)を利用する。例えば、イー・コリのmutY酵素は、G/AミスマッチのAを開裂させ、HeLa細胞からのチミジンDNAグリコシラーゼはG/TミスマッチのTを開裂させる(Hsuら(1994)Carcinogenesis15:1657−1662)。具体例に従って、B7−4配列に基づくプローブ、例えば、野生型B7−4またはPD−1配列を試験細胞から得られるcDNAまたは他のDNA産物とハイブリッド形成させる。二本鎖をDNAミスマッチ修復酵素で処理し、開裂産物がもし存在するならば、これを電気泳動プロトコルなどにより検出できる。例えば、米国特許第5459039号参照。
【0262】
他の具体例において、電気泳動移動度の変更をB7−4またはPD−1遺伝子における突然変異を同定するために使用できる。例えば、突然変異体と野生型核酸間の電気泳動移動度における相違を検出するために一本鎖形態(SSCP)を使用できる(Oritaら(1989)Proc.Natl.Acad.Sci.USA:86:2766、さらにCotton(1993)Mutat.Res.285:125−144;およびHayashi(1992)Genet.Anal.Tech.Appl.9:73−79)。サンプルおよび対照B7−4またはPD−1核酸の一本鎖DNAフラグメントを変性させ、復元させることができる。一本鎖核酸の第二の構造は、配列によって変化し、結果としての電気泳動移動度における変更により、一塩基の変化でも検出可能になる。分析の感度は、RNA(DNAよりも)を用いることにより向上でき、第二の構造は配列の変化にさらに敏感である。好まし具体例において、主題の方法は、電気泳動移動度における変化に基づく別の二重線状ヘテロ二本鎖分子のヘテロ二本鎖分析を利用する(Keenら(1991)Trends Genet.7:5)。
【0263】
さらにもう一つの具体例において、変性剤の勾配を含むポリアクリルアミドゲルにおける突然変異または野生型フラグメントの移動は、変性勾配ゲル電気泳動(DGGE)を用いて分析される(Myersら(1985)Nature313:495)。分析法としてDGGEを使用する場合、DNAが完全に変性しないことを確実にするために、例えば約40bpの高融点GCリッチなDNAのGCクランプをPCRにより加えることにより、これを修飾することができる。さらにもう一つの具体例において、対照とサンプルの移動度における差異を確認するために、温度勾配を変性剤の代わりに使用する(RosenbaumおよびReissner(1987)Biophys.Chem.265:12753)。
【0264】
点突然変異を検出するための他の技術の例は、これらに限定されないが、選択的オリゴヌクレオチドハイブリダイゼーション、選択的増幅、または選択的プライマーエクステンションを包含する。例えば、公知突然変異が中心におかれるオリゴヌクレオチドプライマーを調製でき、次いで、完全なマッチが見られる場合にのみハイブリダイゼーションを許容する条件下でハイブリッド形成する(Saikiら(1986)Nature324:163);Saikiら(1989)Proc.Natl.Acad.Sci.USA86:6230)。オリゴヌクレオチドがハイブリッド形成膜と結合し、標識された標的DNAとハイブリッド形成する場合、かかる対立遺伝子特異性オリゴヌクレオチドをPCR増幅された標的DNAまたは多くの異なる突然変異とハイブリッド形成させる。
【0265】
別法として、選択的PCRプラスミド増幅に依存する対立遺伝子特異性増幅技術を本発明とあわせて使用できる。特異性増幅のプライマーとして用いられるオリゴヌクレオチドは分子の中心(したがって、増幅は異なるハイブリダイゼーション依存する)(Gibbsら(1989)Nucleic Acids Res.17:2437−2448)または一つのプライマーの3’末端(適当な条件下でミスマッチが防止できるか、またはポリメラーゼエクステンションが軽減される)に興味のある突然変異を有する(Prossnerら(1993)Tibtech11:238)。加えて、開裂に基づいて検出するために、突然変異の領域中に新規制限部位を導入するのが望ましい(Gaspariniら(1992)Mol.Cell Probes6:1)。ある具体例において、増幅は増幅のためのTaqリガーゼを用いて行うことができると予想される(Barany(1991)Proc.Natl.Acad.Sci.USA88:189)。このような場合において、結紮は完全なマッチが5’配列の3’末端にある場合にのみ起こり、増幅の存在または不在を探すことにより、特異的部位で公知の突然変異の存在を検出できる。
【0266】
本明細書において記載される方法は、例えば、少なくとも1つの核酸または本明細書において記載される抗体試薬を含み、例えばB7−4またはPD−1遺伝子を含む疾患または病気の症状または家族歴を示す患者を診断するための臨床条件において都合よく使用できる、あらかじめ包装された診断キットを使用することにより行うことができる。
さらに、B7−4またはPD−1が発現される任意のセルタイプまたは組織を本明細書に記載される診断分析において利用できる。
【0267】
VII.B7−4またはPD−1の調節剤の投与
本発明のB7−4またはPD−1調節剤は、免疫細胞による免疫反応を向上させるかまたは抑制するかのいずれかのために、インビボの医薬的投与に適した生物学的に適合性の形態において患者に投与される。「インビボ投与に適した生物学的に適合性の形態」とは、任意の有毒な影響よりもタンパク質の治療効果の方が重きをなす、投与されるタンパク質の形態を意味する。対象なる用語は、免疫反応を惹起できる生きている生物、例えば、哺乳動物を包含することを意図される。患者の例は、ヒト、イヌ、ネコ、マウス、ラット、およびそのトランスジェニック種を包含する。本明細書に記載される薬剤の投与は、治療的に活性な量の薬剤単独または医薬的に許容される担体との組み合わせを包含する任意の薬理学的形態であり得る。
【0268】
治療的に活性な量の本発明の治療組成物の投与は、所望の結果を達成するために必要な用量および時間で有効な量として定義される。例えば、B7−4またはPD−1ポリペプチドの治療的に活性な量は、個人の疾患状態、年齢、性別、および体重ならびにペプチドが個人において所望の反応を惹起する能力によって変わり得る。投薬計画は、最適の治療反応を提供するために調節できる。例えば、いくつかに分割された用量を毎日投与できるか、または治療状況の緊急性により必要とされるので、比例して減少させることができる。
【0269】
B7−4またはPD−1調節剤(例えば、ペプチド、核酸分子、抗体、ペプチド模倣物、または小分子)を都合の良い方法、例えば、注射(皮下、静脈内など)、経口投与、吸入、経皮適用、または直腸投与により投与できる。投与経路に応じて、活性化合物は、化合物を酵素、酸、および化合物を不活化し得る他の自然条件の作用から化合物を保護するために物質中でコートできる。例えば、経口投与以外によりB7−4またはPD−1調節剤を投与するためには、ペプチドをその不活化を防止する物質でコートする化、またはペプチドを該物質と共に同時投与するのが望ましい。
【0270】
B7−4またはPD−1調節剤は個人に、適当な担体、希釈剤またはアジュバント中で投与できるか、酵素阻害剤と同時投与できるか、またはリポソームなどの適当な担体中で投与できる。医薬的に許容される希釈剤は、塩溶液および水性緩衝溶液を包含する。アジュバントはその最も広い意味で用いられ、任意の免疫刺激化合物、例えばインターフェロンを包含する。本発明において意図されるアジュバントは、レゾルシノール、非イオン性界面活性剤、例えば、ポリオキシエチレンオレイルエーテルおよびn−ヘキサデシルポリエチレンエーテルを包含する。酵素阻害剤は、膵トリプシン阻害剤、ジイソプロピルフルオロホスフェート(DEEP)およびトラシロールを包含する。リポソームは、水中油中水エマルジョンならびに慣用のリポソームを包含する(Sternaら(1984)J.Neuroimmunol.7:27)。
【0271】
活性化合物は、非経口または腹腔内投与できる。分散液は、グリセロール、液状ポリエチレングリコール、およびその混合物および油中でも調製できる。通常の貯蔵および使用条件下で、これらの製剤は、微生物の成長を防止するための保存料を含んでもよい。
注射可能な用途に適した医薬組成物は、滅菌水性溶液(水溶性の場合)または分散液および滅菌注射溶液または分散液の即席調製用滅菌粉末を包含する。全ての場合において、組成物は無菌でなければならず、容易に注射針通過する程度まで流動性でなければならない。製造および貯蔵条件下で安定でなければならず、細菌および真菌などの微生物の汚染作用に対して保護されなければならない。担体は、例えば、水、エタノール、ポリオール(例えば、グリセロール、ポリエチレングリコール、および液体ポリエチレングリコールなど)、およびその適当な混合物を含む溶媒または分散媒体であり得る。適当な流動性は、例えば、レシチンなどのコーティングの使用、分散液の場合には必要な粒子サイズの意地、および界面活性剤の使用により維持できる。微生物の作用の防止は、さまざまな抗菌剤および抗進近在、例えば、パラベン、クロロブタノール、フェノール、アスコルビン酸、チメロサールなどにより達成できる。多くの場合において、等張剤、例えば、糖、ポリアルコール、例えば、マンニトール、ソルビトール、塩化ナトリウムを組成物中に含めるのが好ましい。組成物中に、吸収を遅らせる薬剤、例えばアルミニウムものステアレートおよびゼラチンを含めることにより、注射可能な組成物の長期にわたる吸収をもたらすことができる。
【0272】
滅菌注射溶液は、必要に応じて、前記の成分の一つまたは組み合わせと共に必要な量の活性化合物(例えば、B7−4またはPD−1ポリペプチド)を適当な溶媒中に組み入れることにより調製でき、続いて濾過滅菌される。一般に、活性化合物を、塩基性分散媒体および前記のものから必要とされる他の成分を含む滅菌ビヒクル中に組み入れることにより、分散液を調製する。滅菌注射溶液を調製するための滅菌粉末の場合において、好ましい調製法は、真空乾燥および凍結乾燥であり、これにより活性成分(たとえば、ペプチド)の粉末とあらかじめ濾過滅菌された溶液から得られる追加の所望の成分が得られる。
【0273】
前記のように、活性化合物が適当に保護される場合、タンパク質を、例えば不活性希釈剤または同化できる可食担体と共に経口投与できる。本明細書において使用する場合、「医薬的に許容される担体」とは、ありとあらゆる溶媒、分散媒体、コーティング、抗菌剤および抗真菌剤、等張剤および吸収遅延剤などを包含する。医薬的活性な物質についてのこのような媒体および薬剤の使用は、当該分野において一般的である。任意の慣用の媒体または薬剤が活性化合物と不適合性である場合を除いては、治療組成物におけるその使用は意図される。追加の活性化合物も組成物中に組み入れることができる。
【0274】
投与の簡便性および投与量の均一性のために、単位投与形態の非経口組成物を処方するのが特に有用である。本発明において使用される単位投与形態とは、治療される哺乳動物対象の単位投与量として適した物理的に独立した単位を意味し;。各単位は所望の治療効果を生じるために計算されたあらかじめ決められた量の活性化合物を必要とされる医薬担体と組み合わせて含む。本発明の単位投与形態の明細は、(a)活性化合物の独自の特性および達成されるべき特定の治療効果、および(b)個人において敏感性を治療するためのかかる活性化合物を配合する分野に固有の制限により決定され、直接依存する。
【0275】
本発明の一具体例において、治療的に有効な量の、B7−4またはPD−1タンパク質に対する抗体を対象に投与する。本明細書において定義するとおり、抗体の治療的に有効な量(すなわち、有効量)は、約0.001〜30mg/kg(体重)、好ましくは約0.01〜25mg/kg(体重)、さらに好ましくは約0.1〜20m/kg(体重)、なおいっそう好ましくは約1〜10mg/kg、2〜9mg/kg、3〜8mg/kg、4〜7mg/kg、または5〜6mg/kg(体重)の範囲である。当業者らは、疾患または障害の重さ、以前の治療、対象の全般的健康状態および/または年齢、および存在する他の疾患を包含するが、これらに限定されないある因子が対象を有効に治療するために必要な容量に影響を及ぼし得ることを理解するであろう。さらに、治療的に活性な量の抗体での対象の治療は、一回の治療を包含するか、または好ましくは一連の治療を包含し得る。好ましい例において、対象は約0.1〜20mg/kg(体重)の範囲の抗体で、1週間に1回で約1〜10週間、好ましくは2〜8週間、さらに好ましくは約3〜7週間、なおいっそう好ましくは約4、5、または6週間投与される。治療に使用される抗体の有効量は、特定の治療の過程にわたって増加または減少し得る。
【0276】
薬剤(例えば医薬または化合物)のB7−4またはPD−1タンパク質の発現または活性に対する影響のモニターは、基礎的医薬スクリーニングのみならず、臨床試験においても適用できる。例えば、本明細書に記載するスクリーニングアッセイにより決定される薬剤のB7−4またはPD−1遺伝子発現、タンパク質レベルを増大させるか、またはB7−4またはPD−1活性を増加調節する有効性を、減少したB7−4またはPD−1遺伝子発現、タンパク質レベル、または減少調節されたB7−4またはPD−1活性を示す対象の臨床試験においてモニターすることができる。別法として、B7−4またはPD−1遺伝子発現、タンパク質レベルを減少させるか、またはB7−4またはPD−1活性を減少調節するためのスクリーニングアッセイにより決定される薬剤の有効性は、増加したB7−4またはPD−1遺伝子発現、タンパク質レベル、または増加調節されたB7−4またはPD−1活性を示す対象の臨床試験においてモニターできる。かかる臨床試験において、B7−4またはPD−1遺伝子、および好ましくは障害に関与する他の遺伝子の発現または活性は、特定の細胞の表現系の「リードアウト」またはマーカーとして使用できる。
【0277】
例えば、限定のためではないが、B7−4またはPD−1を含み、B7−4またはPD−1活性(例えば、本明細書において記載されるスクリーニングアッセイにおいて同定される)を調節する薬剤(例えば、化合物、医薬または小分子)での治療により細胞において調節される遺伝子を同定できる。従って、薬剤のB7−4またはPD−1関連障害の、例えば臨床試験において効果を研究するために、細胞を単離し、RNAを調製し、それぞれB7−4またはPD−1およびB7−4またはPD−1に関連障害に関与する他の遺伝子の発現のレベルについて分析することができる。遺伝子発現のレベル(すなわち、遺伝子発現パターン)は、本明細書に記載するようにノザンブロット分析またはRT−PCRによるか、または本明細書において記載する方法の一つにより産生されるタンパク質の量を測定するか、またはB7−4またはPD−1または他の遺伝子の活性のレベルを測定することにより定量できる。このように、遺伝子発現パターンは、マーカー、薬剤に対する細胞の生理学的反応の表示としての働きをする。従って、この反応状態は、個人の薬剤での治療前、および治療中の様々な時点で定量できる。
【0278】
好ましい具体例において、本発明は対象の薬剤(例えば、作用物質、拮抗物質、ペプチド模倣物、タンパク質、ペプチド、核酸、小分子、または本明細書に記載されるスクリーニングアッセイにより同定される他の医薬候補)での治療の有効性をモニターする方法であって、(i)薬剤の投与前に対象から投与前サンプルを得;(ii)投与前サンプルにおいてB7−4またはPD−1タンパク質、mRNA、またはゲノムDNAの発現のレベルを検出し;(iii)1以上の投与後サンプルを対象から得;(iv)投与後サンプルにおいてB7−4またはPD−1タンパク質、mRNA、またはゲノムDNAの発現または活性のレベルを検出し;(v)投与前サンプルにおけるB7−4またはPD−1タンパク質、mRNA、またはゲノムDNAの発現または活性のレベルを、投与後サンプルにおけるB7−4またはPD−1タンパク質、mRNA、またはゲノムDNAと比較し;(vi)対象に対する薬剤の投与を適宜変更する工程を含む方法を提供する。例えば、薬剤の投与を増大させることは、B7−4またはPD−1の発現または活性を検出されるよりも高いレベルまで増大させるため、すなわち、薬剤の有効性を増大させるために望ましい。別法として、薬剤の投与を減少させることは、B7−4またはPD−1の発現または活性を検出されるより低いレベルまで減少させるため、すなわち薬剤の有効性を低下させるために望ましい。このような具体例に従って、B7−4またはPD−1発現または活性を、観察できる表現系反応がない場合でさえも、薬剤の有効性のインジケーターとして使用できる。
本発明を以下の限定的と考えるべきでない実施例によりさらに説明する。奔出癌全体にわたって言及される全ての文献、特許および公開特許出願、ならびに図面および配列リストは出典明示により本発明の一部として参照される。
【0279】
実施例
実施例1:B7−4cDNA分子の単離
ホモローガスなポリペプチドをエンコードする核酸分子の公共のデータベースを検索するために、ヒトB7−2の細胞外ドメインのタンパク質配列を使用した。ESTデータベースにおける2つの重複配列、AA292201およびAA399416を同定した。これらの配列は、ヒト活性化角化細胞および胎盤cDNAライブラリーから完全長B7−4 cDNAを単離するために使用した;
これらのESTからの配列5'-CAGCTATGGTGGTGCCGACTACAA-3'(配列番号5)および5'-AGGTGCTAGGGGACAGTGTTAGACA-3'(配列番号6)を有するオリゴヌクレオチドを合成した。これらのオリゴヌクレオチドを、濾胞性リンパ腫の場合には脾臓、活性化B細胞、INF−γ活性化角化細胞、正常な脾臓、および膵臓から得られるmRNAの逆転写により調製されたPCR反応を準備するために使用した。条件は、95℃、1分;94℃、30秒、56℃、30秒、68℃、1分を35サイクル;68℃、3分、3分4℃に保持であった。全ての鋳型は389bpの予想されるサイズのバンドをもたらした。INF−γ活性化角化細胞のPCRからの389bp生成物を、アガロースゲル電気泳動により精製し、0.12ngを、0.05mMビオチン−21−dUTPおよび前記プライマーを含むPCR反応において鋳型として使用した。条件は、94℃、1分;94℃、30秒、56℃、30秒、68℃、2分を20サイクル;68℃、5分、4℃で保持であった。ビオチニル化PCR産物をNucleospinカラム(Clontech)上で精製し、ClonCapture cDNA選択法(Clontech)においてプローブとして使用した。60ngの変性ビオチニル化PCR産物を2mMCoCl2、1X RecA緩衝液、1μgのRecAタンパク質、1X ATPで最終体積30μl中でインキュベートした。反応を37℃で15分間インキュベートした。この混合物に、0.7μgの活性化角化細胞cDNAライブラリーのプラスミドDNAおよび0.4μgのヒト胎盤cDNAライブラリーを添加し、インキュベーションを20分間続けた。50ngのEcoRV消化したラムダDNAを反応に添加し、5分間インキュベートした。0.6μlの10%SDSおよび5.6μgのプロテイナーゼKを添加し、37℃で10分間インキュベートした。1μlの0.1M PMSFを添加することにより、プロテイナーゼKを不活化した。ストレプトアビジン磁気ビーズを5μgの剪断サケ精子DNAとともに10分間プレインキュベートし、ビーズを磁石で捕獲し、上清を除去し、ビーズを30μlの結合緩衝液(1mM EDTA、1M NaCl、10mM Tris−HCl、pH7.5)中に再懸濁した。ビーズを反応に添加し、反応を30分間室温で優しく混合しながらインキュベートした。ビーズを磁石で捕獲し、上清を除去した。ビーズを1mlの洗浄緩衝液(1mM EDTA、2M NaCl、10mM Tris−HCl、pH7.5)で洗浄し、ビーズを磁石で捕獲し、上清を除去した。洗浄手順を3回繰り返した。1mlの無菌H2Oを洗浄したビーズに添加し、5分間37℃でインキュベートし、ビーズを磁石上に捕獲し、上清を除去した。捕獲されたDNAを、0.1mlの溶出緩衝液(1mM EDTA、0.1N NaOH)を添加し、5分間室温でインキュベートすることにより溶出し、ビーズを磁石で捕獲し、上清を除去し、新しい試験管中に貯蔵した。22.5μlの、担体およびpH中和剤を含む沈殿ミックスを2.5体積のエタノールと共に添加した。プラスミドDNAを遠心分離により濃縮し、H2O中に再溶解させた。プラスミドDNAをエレクトロポレーションによりイー・コリDH18B/P3中に再導入し、7.5μg/mlのテトラサイクリンおよび25μg/mlのアンピシリンを含むLB−寒天プレート上で選択した。コロニーをNytranフィルター上にのせ、32P−標識されたオリゴヌクレオチドを用いて配列:
5'-CAGCTATGGTGGTGCCGACTACAA-3'(配列番号5)、
5'-AGGTGCTAGGGGACAGTGTTAGACA-3'(配列番号6)、
5'-TCGCTTGTAGTCGGCACCACCATA-3'(配列番号9)、
に関してハイブリッド形成した。全てのオリゴはAA292201配列からのものである。最終洗浄条件は、2×SSC、0.1%SDS 55℃で20分間であった。2つのハイブリッド形成コロニーを選び、cDNA挿入物の配列を決定した。
【0280】
配列決定は、B7−4分子の2つの形態を明らかにした。第一の形態、B7−4分泌(B7−4S)は、膜アンカーがない短い親水性ドメインを有するタンパク質をエンコードする。この形態のヌクレオチドおよびアミノ酸配列をそれぞれ配列番号1および2に示す。第二の形態、B7−4膜(B7−4M)は、貫膜および短い細胞質ドメインを有するタンパク質をエンコードする。この形態のヌクレオチドおよびアミノ酸配列をそれぞれ配列番号3および4に示す。図3および4に示すように、同定されたB7−4ファミリーの両メンバーは、シグナル、IgV、およびIgCドメインを有する。B7−4M形態は、デフォルトBlosum62マトリックス(ギャップペナルティーをイグジステンス11、エクステンション1に設定)(http://www/ncbi.nlm.nih.gov参照)を用いて計算すると、B7−1およびB7−2が約26%の同一性を有する条件下で、ヒトB7−1と約21%のアミノ酸同一性を有し、ヒトB7−2と約20%のアミノ酸同一性を有する。
【0281】
実施例2
B7−4mRNAの発現:ノザンブロット分析
B7−4の溶解形態のmRNAは約1.2kbであると予想されるが、他のサイズも可能である。第二の形態のmRNAは約3.8kbであり、わずかのmRNAは1.6および6.5kbである。
B7−4ポリペプチドの発現を分析した。RNAをグアニジンチオシアネート均質化および塩化セシウム遠心分離により調製した。等量のRNA(約2μgポリ(A)+RNA)をアガロースゲル上で電気泳動させ、B7−4SおよびB7−4M形態の両方に共通の32P標識されたB7−4cDNAの一部とハイブリッド形成させた。これらのB7−4mRNAは胎盤、肺、および心臓において高度に発現され、胸腺において中程度に発現される。加えて、これらのB7−4mRNAは骨格筋、腎臓、膵臓、前立腺、精巣、卵巣、小腸、結腸、および末梢血リンパ球において弱く発現される。これらはまた肝臓または脳において非常に弱く発現されることが見いだされた。B7−4mRNAは刺激されていない単球において発現されないが、IFN−γにより強力に誘発された。同様に、これらのポリペプチドの発現は、TPA/IFN−γにより角化細胞において、IFN−γにより樹状細胞において誘発されることが見いだされた。これらのB7−4mRNAは刺激されていないB細胞において発現されなかったが、Ig架橋により誘発された。
【0282】
これらのB7−4mRNAの発現を様々な細胞系においても調べた。これらはB細胞系、例えばRaji、Ramos、LBL、Nalm6、およびDHL−4においても発現されないことが見いだされた。これらはT細胞系、例えばJurkat、Rex、CEM、HPB−ALL、Pee4、およびH9またはHTLV−1形質転換T細胞系、例えばSPPおよびMT2または骨髄系U937においても発現されなかった。
【0283】
実施例3
B7−4mRNA発現のさらなるキャラクタライゼーション:ノザンブロット分析
マウスおよびヒトの複数の組織ノザンブロット(Clontech,Palo Alto,CA)を32P−dCTP放射標識されたcDNAプローブでQuikHyb(Stratagene,La jolla,CA)中で製造業者の指示に従ってプローブした。ヒトB7−4プローブは、配列番号1のコーディング領域と3’未翻訳領域に及ぶ1kgのBamHI/NotIフラグメントを含んでいた。マウスB7−4プローブは、コーディング領域からの300bp cDNAフラグメントを含んでいた。対照アクチンプローブはClontechから供給された。ブロットを2回、室温で2×SSC、0.1%SDS中で、続いて0.2×SSC、0.1%SDS、65℃で洗浄し、オートラジオグラフィーにより調べた。
B7−4mRNAは心臓、ヒト胎盤、およびヒト肝臓において高レベルで発現され、脾臓、リンパ節、胸腺、およびマウス肝臓においては低いレベルで発現された。
【0284】
B7−4mRNAを、PU5−1.8、RAW274.7、K−Balb、M−MSV−Balb/3T3、Hepa1−6、R1.1、L1210、P38D1、P815、およびNB41A3細胞を含む様々な形質転換されたマウス細胞系において発現した。
【0285】
実施例4
B7−4mRNA発現のさらなるキャラクタライゼーション:定量的PCR、遺伝子チップハイブリダイゼーション、およびRNAブロット分析
抗原提示細胞上のB7−4mRNA発現を調べ、これらの細胞上のB7−1およびB7−2の発現と比較した。定量的PCR分析に関して、細胞RNAをデオキシリボヌクレアーゼ処理し、再抽出し、第一鎖cDNAに変換した。FAM(6−カルボキシフルオレセイン)標識されたヒトB7−4、B7−1、B7−2、およびGAPDHプローブをPE Biosystems B7−4から購入した:プライマー5'-GCCGAAGTCATCTGGACAAG-3'(配列番号13)および5'-TCTCAGTGTGCTGGTCACAT-3'(配列番号14)、プローブ5'-FAM-CACCACCACCAATTCCAAGA-3'(配列番号15):プライマー5'-ACGTGACCAAGGAAGTGAAAGAA-3'(配列番号16)および5'-TGCCAGCTCTTCAACAGAAACAT-3'(配列番号17)、プローブ5'-FAM-TGGCAACGCTGTCCTGTGGTCAC-3'(配列番号18);B7−2:プライマー5'-GGGCCGCACAAGTTTTGAT-3'(配列番号19)および5'-GCCCTTGTCCTTGATCTGAAGA-3'(配列番号20)、プローブ5'-FAM-CGGACAGTTGGACCCTGAGACTTCACA-3'(配列番号21)。
【0286】
PCR反応は、Perkin Elmer TaqMan EZキットからの試薬を使用して、製造業者の指示に従って96ウェルプレート中で始めた。標準曲線を分析した4遺伝子のそれぞれについて作成した。40サイクルのPCRを、ABI Prism7700シーケンスディテクターで行い、B7−4、B7−1、およびB7−2結果を標準化するためにGAPDHを使用した。
遺伝子チップハイブリダイゼーション分析のためにAffymetrix Mu19KsubAチップを使用した。このチップ上でThe Institute for Genomic Researchの発現された配列タグTC17781によりネズミB7−4の一部の配列を再提示する。RNA単離、チップハイブリダイゼーションおよびスキャニングを、Byrne,M.C.ら(2000)Curr.Prot.Mol.Biol.Suppl.49:22.2.1−22.2.13に記載されているようにして行った。
【0287】
RNAブロットハイブリダイゼーションに関して、1.6kbのヒトおよび3.6kbのネズミB7−4cDNAを、XhaIでの消化により切り取り、γ−32P−ATPおよびDNAポリメラーゼIのKlenowフラグメントでのランダムプライミングにより標識した。RNAブロットを、Freeman,G.J.ら(1992)J.Immunol.149:3795−3801に記載されているようにしてハイブリッド形成した。
【0288】
ヒト樹状細胞は末梢血由来であった。単核細胞をFicoll勾配上での分別後に単離した。非接着細胞を除去し、残りの細胞を150ng/mlのヒトGM−CSF(R&D Systemsおよび100ng/mlのヒトIL−4(R&G Systems)中で2日間培養した。非接着樹状細胞を単離し(CD80+CD86+HLA−DR+CD54+CD58+CD1a+)、GM−CSF単独中で培養するかまたはGM−CSF、2.5μg/mlLPS(Sigma Chemicals)、および10ng/mlのヒトインターフェロン−γで活性化した。活性化後4時間および20時間で、細胞を収穫し、RNeasyキット(Qiagen)を用いてRNAを単離した。
【0289】
ネズミ骨髄単核細胞から、磁気活性化細胞ソーティングにより顆粒球、リンパ球およびIa+細胞を免疫除去し、ペトリ皿中でGM−CSFおよびIL−4とともに培養した。樹状細胞を非接着集団として7日培養後に収穫し、75〜80%CD11c+、高IA+細胞であることが示された。細胞をLPSおよびヒトインターフェロン−γで活性化した。
RNAブロットハイブリダイゼーションによるヒト血液単球における発現の分析から、B7−2は未刺激単球により発現されないが、インターフェロン−γ治療により急速に阿蔵か調節されることが示された。もう一つの炎症性サイトカイン(proinflammatory cytokine)、腫瘍壊死因子(TNF)−αでの単球の処置は、培地単独で見られるのと類似した低レベルの誘発に至り、これはおそらくプラスチックへの接着による活性化の結果である。主な4.2kb B7−4mRNAに加えて、わずかの1.8kbB7−4mRNA種もインターフェロン−γ処置された単球において観察された。細胞表面イムノグロブリン架橋により活性化されるが、Raji細胞系によっては活性化されないヒトB細胞によるB7−4の発現も観察された。同様に、B7−1は未刺激単球により発現されないが、B7−4発現と類似した反応の仕組みでインターフェロン−γに反応して増加調節される。対照的に、B7−2mRNAは単球において本質的に発現され、レベルはインターフェロン−γまたはTNF−α処置により影響を受けない。
【0290】
ヒト樹状細胞によるB7−4、B7−1、およびB7−2mRNA発現も定量的PCRにより調べた。ヒト末梢血由来の樹状細胞を顆粒球−マクロファージコロニー刺激因子(GM−CSF)単独で処理するか、またはGM−CSF、リポポリサッカライド(LPS)、およびインターフェロン−γで活性化した。LPSおよびインターフェロン−γで活性化した結果、B7−4mRNAは急速に誘発され、誘発されない細胞と比較して、4時間で16倍増加し、20時間で34倍増加した。B7−1およびB7−2mRNAも活性化により誘発された:B7−1は4時間で21倍、20時間で22倍誘発された。B7−2は4時間でわずかの誘発を示すが;発現は20時間で5倍誘発された。LPSおよびインターフェロン−γで処置されたネズミ骨髄由来の樹状細胞によるB7−4の発現を、Genechipハイブリダイゼーションを用いて調べた。これらの細胞におけるB7−4発現はヒト樹状細胞について観察されたのと類似したパターンをたどる:誘発後6および20時間で未誘発細胞と比較してB7−4mRNAの5倍誘発。これらのデータは、B7−4が抗原提示細胞およびリンパ球により発現され、B7−1およびB7−2と類似した方法で樹状細胞状で誘発されることを示す。ヒト角化細胞のホルボールエステルおよびインターフェロン−γでの処置もB7−4を誘発した。
ネズミ組織において、約3.7kb B7−4mRNA転写物をノザンブロットハイブリダイゼーションにより検出した。ネズミB7−4mRNAの分布は、ヒトB7−4と非常に類似し、心臓、胸腺および胚で高レベルであり、腎臓、脾臓および肝臓で低レベルであった。
【0291】
実施例5
B7−4の染色体位置測定
B7−4遺伝子の染色体位置を、Quantum(Toronto,Canada)から商業的に入手可能なモノクロモソーマルブロットキットを用いて決定した。ブロットを、B7−4SおよびB7−4Mの両方を認識する配列でプローブした。この方法を用いて、B7−4ポリペプチドはヒト染色体9に位置決定され、一方、B7−1およびB7−2はヒト染色体3に位置決定された。B7−4ファミリーと限定されたアミノ酸配列同一性を共有するブチロフィリンは染色体6上の主要組織適合性複合体に位置決定された。B7−4の染色体位置は、Quantum Technologies(Canada)から入手可能なモノクロモソーマル体細胞ハイブリッドDNAテンプレートのPCR増幅においてB7−4特異性プライマーを用いて確認された。
【0292】
実施例6
B7−4分子のT細胞リガンドまたは抗体との結合
COS細胞を、ベクターDNA(pcDNAI)、またはB7−4McDNAを含む発現プラスミドのいずれかでトランスフェクトした。72時間後、トランスフェクトされたCOS細胞を、0.5mM EDTAを含むPBS中で30分間、37℃でインキュベートすることにより分離した。
B7−4Mを発現するCOS細胞が様々なT細胞レセプターおよび抗体と結合する能力を試験した。B7−4トランスフェクトCOS細胞によるCD281g、CTLA4−Ig、および対照Igの結合のFACS分析は、CD281IgもCTLA4−IgもB7−4により結合しないことを示した(図8)。B7−4Mを発現するCOS細胞がIgGおよびネズミICOS−his融合タンパク質と結合する能力も試験した。ヒトB7−4のネズミICOSとの結合は検出されなかった(図9)。図10に示すように、FACS分析から、BB1(抗B7−1および抗B7−3)抗体B7−4トランスフェクトCOS細胞の結合が明らかにされたが、IgMも133(抗B7)は明らかにされなかった。
【0293】
実施例7
B7−4分子によるT細胞増殖の副刺激
B7−4ポリペプチドのヒトT細胞増殖を副刺激する能力を試験した。ヒトCD28+T細胞を、すでに記載されているようにしてB細胞に対するモノクローナル抗体、ナチュラルキラー細胞およびマクロファージを使用した免疫磁気ビーズ除去により単離した(Gimmi,C.D.ら(1993)Proc.Natl.Acad.Sci.USA90:6586−6590)。COS細胞でトランスフェクトされたB7−4およびベクターを、トランスフェクションの72時間後に収穫し、25μg/mlのマイトマイシン−Cとともに1時間インキュベートし、次いでよく洗浄した。105のナイーブT細胞をプレート結合抗CD3mAbと、インキュベートされたDNA構築物でトランスフェクトされた20000マイトマイシン−c処理COS細胞で刺激した。
細胞増殖を、72時間のインキュベーションの最後の12時間で取り込まれた3H−チミジン(1μCi)により測定した。図11および12に示すように、B7−4を発現するCOS細胞はT細胞増殖を副刺激できる。
【0294】
実施例8
B7−4に対するネズミ抗体の生成およびB7−4の細胞表面発現の検出における使用
全ネズミまたはヒトB7−4cDNAを含む哺乳動物発現ベクター(pEF6またはpcDNA3.1(Invitrogen)を調製した。cDNA/ベクター構築物を0.9%塩溶液中、1mg/mlで溶解させた(TEまたはPBSでない)。
【0295】
免疫化前、78μlの塩溶液中1mg/mlカルジオトキシン(Sigma #C−1777)を免疫化されるマウスの各後肢の前脛骨筋中に注射した。各マウスを次いで5日間放置した。
マウスを麻酔した後、50μgの1mg/mlの精製B7−4cDNA/ベクター構築物(0.9塩溶液中)を各再生前脛骨筋中有に注射した。
抗体価を、標準的方法、たとえばELISA分析を用いて免疫化後約6日に測定した。cDNA免疫化を2〜4週ごとに3サイクル繰り返した(抗体価が1:10000より大きくなるまで)。マウスを次に、B7−4でトランスフェクトしたCOS細胞で追加免疫した。
【0296】
適当な抗体価を有するマウスから単離された脾臓細胞を収穫した。脾臓細胞を融合パートナーSP2−0と融合させて、ハイブリドーマを形成した。ハイブリドーマおよび抗体を、標準的方法で処理した(たとえば、“Antiboidies:A Laboratory Manual”,Harlow,E.およびLane,D.,Cold Spring Harbor Laboratory(1988)(出典明示により本発明の一部として参照される)参照)。
抗体2A3、10D9、5A9、および11D12はスクリーニングアッセイにおいて選択された者に含まれていた。これらの抗体は、ヒトB7−4でトランスフェクトされたCOSまたはCOH細胞と結合するが、偽トランスフェクトされた細胞またはマウスB7−4でトランスフェクトされた細胞とは結合しないことが見いだされた。様々な細胞集団に関してB7−4の存在を検出するために抗体を使用した。B7−4発現は、とりわけ、心臓、腫瘍細胞(肺腫瘍細胞、卵巣腫瘍細胞、胸部腫瘍細胞、上皮腫瘍細胞、および扁平細胞癌を含む)、胎盤、および胸腺上皮上で観察された。
B7−4の細胞表面発現を調べるために、もう一つの抗体、クローン29E.2A3.C6(マウスIgG2bκ)を使用した。B7−4はヒト乳ガン細胞系MDA−231、SKBR−3、およびMCF−7上で発現される。
【0297】
実施例9
B7−4に対する完全ヒト抗体の生成
この実施例において、B7−4またはPD−1に対する完全ヒト抗体を、ヒトイムノグロブリン遺伝子についてトランスジェニックであるマウスにおいて調製する。トランスジェニックマウスを、標準的方法を用いて、たとえばHoganら、“Manipulating the Mouse Embryo:A Laboratory Manual”,Cold Spring Harbor Laboratory(出典明示により本発明の一部として参照される)にしたがって調製するか、または商業的に購入する。胚幹細胞を区お買いされた手順に従って処理する(Tetracarcinomas and embryonic stem cells:a practical approach,Robertson,E.J.編、IRL Press,Washington,D.C.,1987;Zijlstraら(1989)Nature342:435−438;およびSchwartzbergら(1989)Science246:799−803(それぞれは出典明示により本発明の一部として参照される)。DNAクローニング法は、Sambrook,J.ら、Molecular Cloning:A Laboratory Manual、第2版、1989、Cold Spring Harbor Laboratory Press,Cold Spring Harbor,N.Y.(出典明示により本発明の一部として参照される)に従って行う。オリゴヌクレオチドを、たとえば製造業者により提供される仕様書に従って、Bio Systemsオリゴヌクレオチド合成器で合成するか、または商業的に購入する。
【0298】
トランスジェニックマウスを、精製または組み換えB7−4またはPD−1またはB7−4の細胞外ドメインの少なくとも免疫原性部分を含む融合タンパク質を用いて免疫化する。100μlnリン酸塩緩衝塩溶液(PBS)中約400μgのB7−4またはPD−1を各マウスに腹腔内注射する。血清サンプルを、眼窩後洞出血により、薬6日後に集める。
B7−4またはPD−1についての抗体反応性および特異性を、間接的酵素標識免疫吸着測定法(ELISA)を用いて評価する。B7−4またはPD−1についての抗体の抗体特異性を分析するために、いくつかのイムノグロブリンスーパーファミリー分子を対照(たとえば、CTLA4およびCD28)として試験する。B7−4またはPD−1と結合するヒト可変領域を有する抗体を、マウスイムノグロブリンと交差反応性のないヒトIgMおよびヒトIgGサブクラスについて特異的な酵素接合体により検出する。簡単に説明すると、ウェルを一夜37℃で5μg/mLのPBS中B7−4でコートすることにより、PVCマイクロタイタープレートをB7−4またはPD−1でコートする。血清サンプルを、PBS、5%血清、5%Tween−20中で希釈し、ウェル中で1時間室温でインキュベートし、続いて同じ希釈剤中で抗ヒトIgG FcおよびIgGF(ab’)ホースラディッシュペルオキシダーゼまたは抗ヒトIgM Fc−ホースラディッシュペルオキシダーゼをインキュベートする。室温で1時間後、ABTS基質(Sigma,St.Louis,Mo.)の添加により酵素活性を評価し、30分後、415〜490nmで読みとる。同じマウスからの免疫化前の血清サンプルにおいて、ヒト抗体の同じ標的抗原に対する力価も試験する。
ネズミ抗体のネズミVHおよびVLドメインの相補性決定配列を、B7−4またはPD−1に対するヒト化抗体を生成するためにヒトのフレームワーク中に移植できる(Riechmannら(1988)Nature332:323;Verhoeyenら(1988)Science239:1534)。
【0299】
実施例10
B7−4またはPD−1と反応性のヒト単鎖Fvsの生成
モノクローナル抗体分泌ハイブリドーマの調製の別法として、抗B7−4または抗PD−1抗体(抗体の単鎖Fv−様部分)を確認し、Cambridge Antibody Technology Ltd.,Melbourn,UKから得られるM13バクテリオファージ上に示されるヒトイムノグロブリン配列のコンビナトリアルライブラリーのスクリーニングにより単離した(Winterら(1994)Annu.Rev.Immunol.12:433;Hoogenboomら(1998)Immunotechnology4:1)。PD−1.FcまたはB7−4.Fcを使用し、B7−4またはPD−1と結合するイムノグロブリンラブラリーメンバーを単離した。ファージディスプレーライブラリーの生成およびスクリーニング用キットは商業的に入手可能であり、scFvを生成するために標準的方法を使用した(Helfrichら(2000)J.Immunol.Methods237:131−45;Cardosoら(2000)Scand.J.Immuno.51:337−44)。PD−1.FcまたはB7−4.Fcをプラスチック上に固定化し、パンニングおよび複数回の濃縮により特異的scFvを発現するファージを選択した(Griffithsら(1993)EMBO J.12;725)。
【0300】
B7−4のレセプターの同定
ヒトイムノグロブリンガンマ1またはネズミIgガンマ2aのいずれかのヒンジ−CH2−CH3ドメインと融合したヒトPD−1の細胞外領域の融合タンパク質(FcRおよび相補性相互作用をブロックする突然変異を有する)を、PD−1と結合するリガンドを検索するために使用した。この検索の一部として、ガンマ−インターフェロンで刺激された単球の細胞表面の染色を見いだした。B7−4は、ノザンブロットハイブリダイゼーションにより観察されるように、ガンマインターフェロンで刺激した後の単球において誘発される。
PD−1−Fc(ヒトIgガンマ1)の一時的にB7−40発現ベクターでトランスフェクトされたCOS細胞の表面との結合を試験した。COS細胞を、LipofectAMINEトランスフェクト試薬を用いてB7−4MまたはB7−1のいずれかでトランスフェクトした。48時間後、細胞をヒトPD−1−Fc、ネズミPD−1−Fc、CTLA4−Fc、F1t4−Fc、またはIgGで染色し、続いてフィコエリスリン(PE)と接合した抗IgGで染色した。細胞を次いでフローサイトメトリーにより分析した。図13A〜13Dに示すように、B7−4を発現するCOS細胞はヒトPD−1−FcおよびネズミPD−1−Fcの両方を結合したが、CTLA4−Fc、F1t4−Fc、またはヒトIgGを結合しなかった。正の対照として、COS細胞を発現するB7−1がCTLA4−Fcを結合するが、PD−1−FcまたはIgGを結合しないことが示された。
【0301】
加えて、トランスフェクトしたCOS細胞単層のインサイチュ分析を行った。単層をPD−1Fc、CTLA4FcまたはヒトIgG1でプローブし、結合をFc部分に対する、アルカリホスファターゼと結合する第二抗体で検出した。結合は、発色基質5−ブロモ−4−クロロ−3−インドリルホスフェートおよびニトロブルーテトラゾリウムおよび光学顕微鏡で可視化された。平行して、B7−4でトランスフェクトした細胞はPD−1−Fcと結合するが、CTLA4−Fc(ヒトIgガンマ1)またはF1t4−Fc(ヒトIgガンマ1と結合するネズミF1t4の細胞外領域)と結合しないことが見いだされた。平行して、PD−1Fcは偽トランスフェクトされたB7−1またはB7−2トランスフェクトCOS細胞の表面と結合しなかった。
もう一つの実験において、PD−1−FcのB7−1またはB7−2の溶解形態との結合およびB7−4との結合は、BIACOREベースの分析法を用いて検出されなかった。平行して、CTLA4はB7−1と結合するが、B7−4と結合しないことが示された。PD−1−FcまたはCTLA4−Fcを固定化し、条件は本質的に、Fitzら(1997)Oncogene15:613に記載されている通りであった。完全長B7−4MまたはB7−4−Fcでトランスフェクトされた細胞からの濃縮されたCOS細胞培地を注入し、リアルタイム生体分子相互作用分析(BIA)を用いて相互作用を測定した(Sjolander,S.およびUrbaniczky,C.(1991)Anal.Chem.63:2338−2345およびSzaboら(1995)Curr.Opin.Struct.Biol.5:699−705)。ヒトB7−4はヒトおよびマウスPD−1と結合することが見いだされたが、この結合は、同時注入されたPD−1−Fc(CTLA4−FCではなく)との競合により阻害された。これらの実験は、可溶性B7−4−Fc融合タンパク質の固定化PD−1−Fcとの結合だけでなく、B7−4McDNAトランスフェクト細胞の順化培地におけるB7−4の溶解形態の存在も示し、これはおそらく脱落の結果である。
【0302】
図14A〜14Fは、PD−1がPD−1のB7−4に対する結合を競合的に阻害し、F1t4(血管内皮成長因子Cのレセプター)は阻害しないことを示す。ヒトPD−1ガンマ2a融合タンパク質のB7−4Mを発現するCOS細胞との結合をパネルAに示す。結合は、PEと結合する抗ガンマ2a特異性試薬で検出した。IgG1と結合するヒトPD−1を:50μg/ml、6.25μg/ml、100μg/ml、または25μg/mlで添加し、結合に関して競合することが見いだされた。対照として、100μg/mlのF1t4G1はPD−1のB7−4との結合に関して競合しないことが見いだされた。
【0303】
さらにもう一つの実験において、B7−4がPD−1と結合する能力をフローサイトメトリーおよびBIACORE結合分析により決定した。フローサイトメトリーで検出すると、ヒトおよびネズミPD−1.IG融合タンパク質は、CHO上で発現されたヒトおよびネズミB7−4の両方と結合した(図15A〜15F)。しかしながら、ヒトCTLAIg、ヒトCD28.Ig、またはヒトICOS.IgのいずれもB7−4発現細胞系のいずれかと結合しなかった。PD−1融合タンパク質はベクター単独でトランスフェクトされたCHO細胞と結合しなかった。さらに、PD−1:B7−4相互作用は、BIACORE装置を用いた表面プラズモン共鳴を用いて確認した。ヒトおよびネズミPD−1.Igタンパク質およびヒトCTLA−4.Igをデキストランチップの流動細胞表面上に固定化し、可溶性ヒトB7−4.Igとの結合に関して試験した。B7−4.IgはヒトおよびネズミPD−1.Igの両方と結合するが、ヒトCTLA−4.Igと結合しなかった(図16)。この結合は、同時注入された可溶性PD−1.Igとの競合によりブロックされるが、CTLA−4.Igによってはブロックされなかった。ヒトB7−1およびB7−2の溶解形態は、固定化ヒトPD−1を結合しなかった。
【0304】
BIACORE分析を、PD−1.Fc:B7−4.Fc結合速度論を分析するためにも用いた。BIA2000、CM5センサーチップを用いた。50−150RU PD−1.Fcをチップと結合させるためにNHS/EDC固定化を用いた。イオン再生緩衝液(30%1.83M MgCl2、0.46M KSCN、0.92M尿素、1.83Mグアニジン−HCl)を20回注入することにより表面を調整した。流動緩衝液(running buffer)は、3.4mM EDTA、0.005%Tween−20、および100μg/mlBSA(ウシ血清アルブミン)(25℃)を含むPBS(リン酸塩緩衝液)であった。使用した参考表面は(1)ブランク活性化(NHS/EDC)とそれに続くエタノールアミンブロック、(2)突然変異mIGG2aであった。使用したB7−4.Fcの濃度(未検出マルチマー、SECにより定量)を、ELISAoyobiBCAタンパク分析(Pierce)により定量した。使用したB7−4.mFcの範囲は、20nM〜2μMであった。流速は60μl/分であった。会合は3〜4分で、4分解離した。注射を3回行い、ランダム化した。分析は、BIAevaluationソフトウェア(1:1Langmuirおよび全般的分析)を用いて行った。分析結果から、PD−1.Fc:hB7−4.Fc結合が1:1モデルに適合することが示された。PD−1.Fc:hB7−4.Fcに関して計算された反応速度定数(KD)は186nMであった(kon=6.57±1.2×e4M−1s−1;koff=0.122±0.0007s−1)。
これらのデータは、PD−1がB7−4と結合し、この相互作用はPD−1の作用を調節することを示す。
【0305】
実施例12
B7−4は負のシグナルを免疫細胞に伝達できる
この実施例において、1ウェルあたり5×105Jurkat T細胞を抗CD3コートされたビーズ(1:1の比)および可溶性抗CD28で刺激した。B7−4を発現するCOS細胞またはEZZと称する負の対照をウェル中に滴定した。上清を48時間で収穫し、ヒトIL−2についてELISAにより分析した。図17は、COS B7−4細胞の数が増大すると(図の右側のバー)、IL−2産生が減少することを示す。
同様の分析様式、たとえばPBMCからのヒトPHA−ブラストが固定化抗CD3および可溶性抗CD28で刺激されるものを用いて、T細胞増殖における減少を、B7−4を発現するCOS細胞において滴定することにより観察した。
【0306】
実施例13
PD−1:B7−4相互作用は、CD−3によるT細胞増殖、サイトカイン増殖、およびT細胞活性化マーカーの発現を阻害する
PD−1:B7−4相互作用の機能的重要性を調べるために、B7−4のそのレセプターとの相互作用の機能的影響もヒトT細胞を用いて調べた。末梢血単核細胞をFicoll−Hypaque勾配遠心分離により単離した。モノクローナル抗体および免疫磁気ビーズ(PerspectiveBiosystems)のカクテルを用いた負の選択により、CD4+T細胞集団(85〜90%純度)を精製した。抗CD3、対照IgGおよび融合タンパク質をポリウレタンコートされたトシル活性化Dynabeads(Dynal)に製造業者の指示に従い、すでに記載されているようにして(Blarir,P.J.ら(1998)J.Immunol.160:12−15)共有結合させた。表示された濃度の抗CD3抗体(UCHT1、Pharmingen)を1×107ビーズ/mlの0.1Mリン酸塩緩衝液pH7.4に添加した。結合中5μg/mlの一定の合計Ig濃度を維持するために対照IgGをビーズ懸濁液に添加した。同様に、抗CD3/B7−4.Ig(γ2a)ビーズを表示された抗CD3抗体濃度、合計結合タンパク質(1μg/107ビーズ)のB7−4.Ig、残りの全結合タンパク質を構成する対照IgGで調製した。105T細胞を96ウェル平底プレート中で培養し、ビーズを1細胞に対して1ビーズの比で表示された濃度の抗CD28抗体(CD28.2、Pharmingen)の存在下または不在下で添加した。培養物を4日間の分析中の最後の6時間に1μCi3H−チミジン/ウェルで標識することにより、増殖を定量した。サイトカインELISAの分析のために、培養を前記のように開始し、上清を表示された時間に収穫した。インターフェロン−γ、IL−10およびIL−2濃度を商業的に入手可能なELISAキット(Genzyme)を用いて定量した。
【0307】
末梢血単核細胞(PBMC)から得られる精製されたCDR−T細胞を抗CD3mAbでコートされたビーズおよびヒトB7−4.Igまたは対照Igのいずれかで活性化した。増殖およびサイトカイン産生を刺激後36時間に評価した。抗CD3mAb/B7−4.Igでコートされたビーズで活性化された細胞は、抗CD3mAb/対照Ig活性化細胞と比較して増殖において69%の減少を示した(図18A)。増殖は、異なる濃度のB7−4.Igでコートされたビーズを使用した場合に(0.25、0.5、1、および2μgB7−4.Ig/107ビーズ)、用量に依存した方法で阻害された。
さらに、B7−4の存在下での細胞の活性化も、サイトカイン分泌を弱める。B7−4の存在下で、インターフェロン−γ(図18B)およびIL−10(図18C)分泌はそれぞれ約80%および60%減少した。IL−2産生はこれらの活性化条件下で24時間および96時間の両方で検出できなかった。しかしながら、以下の実施例14のようなビーズを用いたT細胞の活性化は、B7−4がIL−2産生を刺激の2、3、および4日に阻害できることを示した。B7−4はさらに、ビーズ:細胞比が1;1、1:2、および4:1でJurkat T細胞におけるIL−2産生を阻害した。さらに、可溶性抗CD28の形態の副刺激が提供される条件下で、B7−4の存在下での細胞の活性化も、IL−2産生における減少につながる。PD−1:B7−4相互作用も、IL−10およびIFN−γ産生の減少につながる。従って、B7−4の存在下でのネズミおよびヒトT細胞の活性化は、増殖およびサイトカイン分泌の両方の阻害につながる。
【0308】
さらに別の実験で、B7−4の存在下でのT細胞の活性化の結果、T細胞活性化マーカーの発現が減少することが示された。たとえば、T細胞がB7−4の存在下で活性化され、初期活性化マーカーCD69の発現をフローサイトメトリーで調べる場合、結果は次の通りであった(CD69発現について陽性(%))
【0309】

実施例14
PD−1:B7−4阻害速度論はPD−1発現と相関する
この実施例は、T細胞活性化中のPD−1発現の時期とT細胞増殖を阻害するPD−Laの能力の関係を説明する。T細胞活性化は、トシル−活性化ビーズ(107)でコートされた4μg抗CD3および2μgの対照Fc(ctrl.Fc)またはB7−4.Fc融合タンパク質のいずれかを用いて行った。5×104精製Balb/cLN T細胞を96ウェルプレートフォーマットにおいて1×105ビーズで刺激した。増殖に関しては、プレートを培養期間の最後の10時間にパルスをかけた。IL−2ELISAに関しては、上清を平行ウェルから収穫した。
【0311】
T細胞活性化中のPD−1発現の速度を決定するために、精製Balb/cLN T細胞を抗CD3/CTRL.Fcビーズで刺激した。刺激の0、1、2、3、4、および5日に、細胞を収穫し、洗浄し、ビオチニル化抗ネズミPD−1(Agata,Y.ら(1996)Int.Immunol.8:765−72に記載されているようにして、モノクローナル抗体J43から調製)またはビオチニル化ハムスター対照、続いてPE−ストレプトアビジンを用いて染色した。データをlive−gated細胞について計算し、陽性(%)をアイソトープ対照に対して計算した。PD−1発現はT細胞活性化期間中にわたって次のように増加した:0日(0%);1日(27%);2日(37%);3日(40%);4日(74%);および5日(83%)。
抗CD3/ctrl.Fcビーズまたは抗CD3/mB7−4.Fcのいずれかを用いて活性化されたBalb/cLN T細胞の増殖を比較する場合、T細胞増殖のB7−4阻害の反応速度は、PD−1の発現速度と相関し、B7−4がそのPD−1との相互作用によりT細胞活性化を阻害するために作用するというさらなる証拠が得られる。
【0312】
実施例15
PD−1:B7−4相互作用の結果はT細胞レセプターとCD28シグナルの強度に依存する
T細胞レセプター、CD28およびPD−1によるシグナル間の関係を調べるために、ヒトCD4+T細胞を最適以下または最適の濃度の抗CD3mAb、固定された濃度のB7−4.Igおよび濃度が増加する可溶性抗CD28mAbで刺激した。抗CD3mAbでコートされたビーズを用いて、最適以下または最適のT細胞刺激に必要な濃度を確立した。最適以下のT細胞レセプター関与条件下で(抗CD3mAb1μg/ml)、副刺激の不在下で最小の増殖が観察された(図19A)。濃度が増加する可溶性抗CD28mAbを添加すると、増殖が30倍まで増加した。これらの条件下で、B7−4の存在下でのT細胞の活性化の結果、増殖が80%減少した(図19A)。B7−4刺激による増殖の阻害を救うためには最大レベルの副刺激(抗Cd28、250ng/ml)が必要であった。対照的に、T細胞レセプター活性化(抗CD3mAb、2μg/ml)の飽和条件下では、B7−4によるT細胞増殖の阻害は、CD28副刺激が存在しない場合にのみ観察された(図19B)。
【0313】
T細胞活性化中の異なる時点でのPD−1:B7−4による増殖の阻害を立て直すための副刺激の能力を調べるために、Balb/cLN T細胞を実施例14と同様に1μg/mlの可溶性抗CD28の存在下または不在下で刺激した。増殖を2、3、および4日に測定した。抗CD28はPD−1:B7−4.Fcによる阻害をT細胞活性化の初期の時点(2日)で逆転させたが、後期(3日および4日)では逆転させなかった。阻害の逆転は用量に依存し、抗CD28濃度の範囲全体(160ng/ml、800ng/ml、1μg/ml、4μg/ml、および20μg/ml)にわたって見られた。しかしながら、強い副刺激(1μg/ml抗CD28)の同じ条件下で、B7−4.FcはIL−2産生を調べたすべての時点(2日、3日および4日)で阻害する。B7−4.Fcはまた、IL−2産生を可溶性抗CD28副刺激の広い範囲(100ng/ml、160ng/ml、500ng/ml、800ng/ml、4μg/ml、5μg/ml、10μg/ml、および20μg/ml)にわたって阻害する。
ICOSによる副刺激もPD−1:B7−4による増殖の阻害を立て直す。
【0314】
実施例16
B7−4のCD28シグナルおよびサイトカイン産生を阻害する能力
PD−1:B7−4経路の阻害効果は、TCRおよびCD28によりシグナルの強度を決定するようであり(前記実施例参照)、これにより弱いCD3/CD28による反応は容易に減少調節される。CD28シグナルとPD−1:B7−4経路の相互作用を研究するために、予備活性化されたDO11.10CD4+T細胞をCHO−IAd/B7.2またはCHO−IAd/B7.2/B7−4により提示されたOVAペプチドで活性化した。
B7−4の検出のために、5×104CHO形質転換細胞を5μg/mlのヒトPD−1Ig(hPD−1−Ig)(Genetics Institute,Cambridge,MA)と共にインキュベートし、ヤギ抗ネズミIgG2a−フィコエリスリン(PE)(Southern Biotechnology Associates Inc.,Birmingham,AL)で成長させた。加えて、細胞を5μg/mlの抗IAd−PEまたはB7.2−PE(Pharmingen,San Diego,CA)で別々に染色した。各工程後に細胞を3回PBS/1%BSA/0.02%アジ化ナトリウムで洗浄した。最終のインキュベーション後、細胞を1%パラホルムアルデヒドで固定した。100000の事象をFACSCalibar(Becton Dickinson,Mountain View,CA)で分析した。全てのイソタイプ対照をPahrmingenから入手した。
【0315】
脾臓細胞をDO11.10マウスから調製し、Tris−NH4Clで処理して、赤血球を除去した。細胞を1μg/mlのOVAペプチドと共に72時間(Analytical Biotechnology Services,Boston,MA)、10%FCS(Sigma,St.Ouis,MO)、2mMのL−グルタミン、100U/mlのペニシリン、100μg/mlのストレプトマイシン、250ng/mlのアンホテリシンB、10mMのHEPES、50μMの2−ME(すべてLife Technologiesから入手)および15mg/mlのゲンタマイシン(Bio Whittaker,Walkersville,MD)で補足されたRPMI1640(Life Technologies,Grand Island,New York)中で培養した。CD4+T細胞を、磁気活性化細胞選別分離カラム(MIltenyi Biotec,Auburn,CA)を用いた正の淘汰により精製し、結果としての純度は98%より高かった。細胞を再刺激前に一夜静置した。
CHO細胞の増殖を、50μg/mlのマイトマイシンC(Bristol Laboratories,Princeton,NJ)とともに16時間37℃でインキュベートすることにより阻害した。インキュベーション期間の最後に、細胞を10mM PBS中EDTAで収穫し、2回洗浄し、氷上に1時間放置した。細胞をその後3回洗浄し、培地中に再懸濁させた。105の予備活性化されたCD4+T細胞を、様々な濃度のOVAペプチドおよび104マイトマイシンCで処理されたCHO形質転換体とともに96ウェルプレート中で培養した。増殖を分析するために、培養物を48時間インキュベートし、1μCi/ウェルの[3H]チミジン(New England Nuclear,Boston,MA)でインキュベーション期間の最後の6時間に適用した。
【0316】
B7およびIAdの発現は、すべてのCHO形質転換体に関して類似していた(図20)。予想されるように、B7.2の導入により、すべての抗原濃度でT細胞による増殖反応が増大した(図21A〜21D)。しかしながら、B7−4は低いペプチド濃度(0.01μg/mlおよび0.001μg/ml)で反応を阻害した(それぞれ図21Cおよび21D)。
PD−1:B7−4経路のサイトカイン産生を阻害する能力を扱うために、CHO細胞形質転換体により提示されるOVAペプチドで活性化されたDO11.10CD4+T細胞からの上清を分析した。上清のアリコートを培養の開始後の様々な時点で収穫した。IL−2、IL−4、IFN−γおよびIL−10レベルを、mAbおよびPharmingenから入手した組み換えサイトカイン標準を用いて分析した。検出限界は次の通りであった:IL−2、20pg/ml、IL−4、40pg/ml、IFN0γ、100pg/mlおよびIL−10、200pg/ml。IL−2(図22A)。IL−4(図22B)、IFN−γ(図22C)、およびIL−10(図22D)の産生は、DO11.10CD4+T細胞を0.1μg/mlのペプチドおよびB7−4とともに培養した場合に著しく阻害された。この濃度で、増殖の弱い阻害しかなかった。しかしながら、B7−4はサイトカイン産生を0.01μg/mlペプチドで著しく阻害し、これは増殖の阻害と一致した(図23A〜23C)。IL−10はこれらの活性化条件下で検出されなかった。従って、B7−4によるPD−1関与は、T細胞増殖が影響を受けない場合でもサイトカイン産生を減少調節することができる。
【0317】
減少したサイトカイン産生が減少したmRNAレベルによるかどうかを確かめるために、RNA分析を使用した。CD4+T細胞を様々なCHO細胞形質転換体および0.01μg/mlのOVAペプチドで再刺激した。48時間後、細胞を収穫し、TRIzol試薬(Life Technologies)を用いてmRNAを単離した。5μgのmRNAを、RiboQUantマルチプローブキットmCK1を製造業者の指示(Pharmingen)に従ってRNase保護分析によりサイトカインレベルに関して分析した。IL−4、IL−10、IL−13、IL−2、IL−6およびIFN−γmRNAの転写レベルを、CHO−IAd/B7.2により提示された0.01μg/mlのOVAペプチドで刺激した後に予備活性化したDO11−10CD4+T細胞において検出した。しかしながら、B7−4の導入は著しくサイトカインmRNAレベルを減少させた。未刺激T細胞培養物またはCHO−IAdにより提示されたペプチドで活性化されたT細胞におけるサイトカインについてmRNAの最小の増加調節があった。この結果、少なくとも抗原性刺激が弱いかまたは限定されている場合、PD−1:B7−4経路の強力なB7/CD28シグナルを拮抗する能力、および少なくとも強力な抗原性刺激の条件におけるサイトカイン産生の阻害がさらに証明された。
【0318】
実施例17
PD−1:B7−4経路の作用メカニズム
CTLA−4の架橋は、ナイーブT細胞における細胞サイクル発達を阻害することが示されている(Krummel,M.F.およびAllison,J.P.(1996)J.Exp.Med.183:2533−2540;Walunas,T.L.ら(1996)J.Exp.Med.183:2541−1550)。PD−1はアポトーシスを受けるネズミ細胞系から単離されたので、PD−1:B7−4経路の作用の可能なメカニズムは、プログラムされた細胞の死を増大させる。この問題を取り扱うために、DO11.10CD4+T細胞を0.01μg/mlのペプチドおよび様々な濃度のCHO形質転換体で際し激し、細胞サイクル発達を分析した。CD4+T細胞を前記のように0.01μg/mlのペプチドで再刺激した。培養の36時間後、細胞を開趣旨、抗CD4−FITCで染色した。細胞をPBS中で洗浄し、70%エタノール中で1時間氷上で固定し、10μg/mlのRNase(Sigma)および50μg/mlのプロピジウムヨージド(Sigma)を含有するPBS中に再懸濁させた。染色の1時間以内に分析を行った。
【0319】
48時間後、細胞を回収し、CD4−FITCで染色した。浸透後、細胞をプロピジウムヨージドとともにインキュベートして、G0/G1、S/G2およびサブジプロイド集団を分析した。CHO−IAdにより提示されたペプチドで再刺激されたCD4+T細胞はサブジプロイド集団において高い割合の細胞を有し、このことはアポトーシスを示す(図24A)。CD4+T細胞がCHO−IAd/B7−2により提示されるペプチドにより刺激される培地においては、S/G2期において細胞の数が増大し、サブジプロイド集団において数が減少し(図24B)、このことは細胞が循環し、B7/CD28副刺激によりアポトーシスから救済されることを示す。B7−4の導入は、G0/G1期における細胞数の増大につながる(図24C)。B7−4培地におけるアポトーシスのレベルはCHO−IAd/B7培地に匹敵した。これは、アネキシン染色により確認された。PD−1:B7−4経路による細胞発達の阻害は、T細胞活性化の減少調節におけるその役割を確認する。
【0320】
B7−4.Fcを用いたさらに別の実験で、T細胞に関するPD−1:B7−4関与がアポトーシスにつながらないことを確認した。細胞を抗CD3または抗CD3/B7−4.Fcで処理するか、または照射し、プロピジウムヨージドおよびアネキシンで染色し、フローサイトメトリーにより分析した。アポトーシスの細胞を、プロピジウムヨージドおよびアネキシンの両方について陽性であったものとして確認した。平行して行った対照実験は、抗CD3/B7.4。Fc9が48時間および72時間で増殖を阻害することを示した。結果は次の通りである:
DNA含量を分析するさらに別の実験は、B7−4.Fcの存在下でのT細胞の活性化は、細胞サイクル阻止をもたらすことを証明した。T細胞をctrl.FcまたはmB7−4Fcビーズで刺激した。4日に、細胞分裂のパーセンテージをプロピジウムヨージド染色により定量した。mB7−4.Fcで刺激されたT細胞は、半数の細胞(11%対21%(対照))でDNA含量が増大し、このことは細胞分裂を示す。他の2つの実験において、パーセンテージは類似していた:14%対28%、および11%対24%。
有糸分列の分析も調べた。T細胞をCSFEで標識し、実施例14と同様にctrl.FcまたはmB7−4.Fcビーズで刺激した。1、2、3、および4日に、FACS分析を行った。live−gated事象のみを分析した。パーセンテージは非分割細胞についてである。次のデータが示すように、B7−4での処理は細胞分裂を阻害する:
【0322】
実施例18
副刺激により誘発されたかまたは外因的に添加されたIL−2は、PD−1:B7−4相互作用により誘発される増殖阻害を克服できる
精製されたBalb/cLN T細胞を実施例14と同様に1μg/mlの可溶性抗CD28および10μg/mlの抗IL−2の存在下または不在下で刺激した。増殖を2日に測定した。抗CD28による副刺激の存在下で、抗IL−2の添加は、PD−1:B7−4による阻害を回復し、このことは抗CDがIL−2産生を誘発することにより阻害効果を逆転させることを示唆する。抗IL−2の添加は、ICOSリガンドによる副刺激の存在下でPD−1:B7−4による阻害も回復する。
【0323】
もう一つの例において、Balb/cLN T細胞を実施例14と同様に外因性IL−2の添加で刺激した。増殖を3日で測定した場合、外因性IL−2はPD−1:B7−4による増殖の阻害を用量に依存した方法で逆転させる(IL−2濃度範囲:3pg/ml、10pg/ml、30pg/ml、100pg/ml、および300pg/ml)。さらに、外因性IL−2の添加(U/ml rhIL−2)はPD−1:B7−4による増殖の阻害をあらゆる時点で逆転させる(2,3、4、および5日に測定された増殖)。
【0324】
実施例19
CD8+T細胞はB7−4による阻害を受けやすい
この実施例は、B7−4に反応したCD4+とCD8+T細胞活性化間の差異を説明する。
安定な抗原提示細胞(APC)系を、レトロウイルス技術を用いてGFPまたはmB7−4/GFPを発現するように操作した。図29Aは、使用した細胞の概略図を示す。CD4+T細胞実験に関して、APC:T細胞比は10μM PCCFペプチドで1:10であった。CD8+T細胞実験に関して、APC:T細胞比は1mM p2Caペプチドで1:1であった。TCRトランスジェニックマウスから得られる5×104の精製LN T細胞を、照射APCとペプチドで2、3、または4日間刺激した。図29Bおよび29Cに示すように、CD4+およびCD8+T細胞は両方ともPD−1:B7−4相互作用により阻害された。
抗CD28の添加はPD−1:B7−4によるCD4+T細胞の増殖の阻害を克服できるが(図30A)、CD8+T細胞の増殖の阻害は克服できない(図30B)。さらに、外因性IL−2またはIL―5(それぞれ20U/mlおよび50ng/ml、0日に添加)は、PD−1:B7−4によるCD8+T細胞の阻害を克服できる(2日に測定した増殖)。CD8+T細胞は、本質的にIL−2を産生できないので阻害をより受けやすい。
【0325】
実施例20
PD−1:B7−4シグナリング経路の関与によるシグナリング分子の補充
T細胞におけるPD−1:B7−4経路の作用のメカニズムをさらに調べるために、Jurkatベースのシステムを使用した。Jurkat細胞は本質的に低レベルのPD−1を発現する。Jurkat細胞を抗CD3/ctrl.Igまたは抗CD3/hB7−4.Igでコートしたビーズで活性化した。PD−1とCD3の結紮により、抗CD3単独で刺激された細胞と比較して、Jurkat細胞によるIL−2分泌が阻害される。細胞を溶解させ、抗SHP−2免疫沈降に付し、ゲルにかけ、膜に移した。膜を4G10−HRP(ホースラディッシュペルオキシダーゼ接合抗ホスホチロシン抗体)でイムノブロットし、続いて抗SHP−2でイムノブロットして、免疫沈降におけるSHP−2発現を確認した。抗CD3およびhB7−4.Igに関して、PD−1とTCRの結紮は、TCR活性化のみと比較して、結果として急速にSHP−2をリン酸化状態に変換する。SHP−1はこれらの条件下でリン酸化されない。このデータは、SHP−2の補充がPD−1:B7−4経路によるTCRシグナリング事象を減少調節するためのメカニズムであることを示唆する。
同様の実験条件下で、抗Zap70免疫沈降の結果、Zap70結合p23pTyrがCD3およびPD−1の同時結紮(抗CD−3およびB7−4.IGを使用)により阻害される。Jurkat溶解物のCD3ζ免疫沈降および抗pTyrブロッティングの結果、CD3とPD−1の同時結紮により(抗CD3およびB7−4.Igを使用)、CD3ζリン酸化が阻害される。この系におけるCD3ζの阻害は、CD28刺激に関して存続する。
【0326】
実施例21
ビオチニル化ヒトB7−4FcのヒトPD−1Fcに対する結合の阻害
PD−1またはB7−4の細胞外領域をネズミIgγ2aのヒンジ−CH2−CH3ドメインと結合させることにより、Fc融合タンパク質が生じた。組み換えタンパク質を、一時的にLipofectAMINE(Gibco−BRL)でトランスフェクトしたCOS細胞または安定にトランスフェクトされたCHO細胞系において産生し、プロテインA−セファロースを用いて順化培地から精製した。
B7−4またはPD−1に対する抗体がヒトB7−4FcおよびヒトPD−1Fcの相互作用を阻害する能力を、標準的ELISA法を用いて試験した。簡単に説明すると、ヒトPD−1Fc分子を、96ウェルプレート中に固定化し、ブロックし、洗浄した。ビオチニル化PD−1Fc分子(100ng/ml)をウェルに約2000、700、200、70、25、8、および1.18ng/mlの濃度で添加した(図25A)。ウェルをストレプトアビジン接合ホースラディッシュペルオキシダーゼとともにインキュベートし、洗浄し、標準的方法を用いて発色させた。B7−4FcのED50は108ng/mlであることが判明した。
【0327】
ヒトB7−4(10D9および11D12)またはヒトイムノグロブリンのscFc部分(B7−4−1、B7−4−6、およびB7−4−12)に対するネズミ抗体がビオチニル化ヒトB7−4FcのヒトPD−1Fcに対する結合を阻害する能力を7種の阻害剤濃度で試験した。IC50は、0.5nMから24nMの範囲であることが判明し、データを図25に示す。
【0328】
PD−1特異性scFvも、B7−4 FcのPD−1 Fcに対する結合を阻害する能力について、前記と同じELISA法を用いて試験した。PD−1と反応性のヒトscFv(PD1−17scFv)は図26に示すように特定の結合(10−7から10−8の間のEc50)を阻害することが判明した。PD1−17scFvのVLおよびVHドメインを、完全IgGを生成するために使用した。簡単に説明すると、VHおよびVLコーディング領域を発現ベクター中のゲノムCHおよびCL遺伝子配列と結合させた。結果として得られるベクターを一時的にヒト293細胞でトランスフェクトし、IgGを順化培地から収穫した。移植された全IgG分子の効力は、scFv抗体についてよりも高かった(10−8から10−9の間のEc50)。
【0329】
実施例22
可溶性B7−4Fcの投与はネズミモデルにおいて疾患を悪化させる
B7−4/PD−1経路の調節がインビボで免疫調節活性を有するかどうかを確認するために、ヒト多発性硬化症と多くの臨床および病理学的特徴を共有する実験的自己免疫脳脊髄炎(EAE)モデルにおいて、たんぱく質をネズミモデルにおいて評価した。メスSJL/Jマウスを、完全フロインドアジュバント中100μgのプロテオリピドタンパク質(PLP)で免疫化した。10日後、脾臓を収穫し、単細胞懸濁液に処理し、次いでインビトロで5μgのPLP中に96時間再懸濁させた。細胞を3回PBS中で洗浄し、次いで15×106細胞をナイーブSJL/Jマウスに腹腔内注射により移入した。自己反応性T細胞の養子移入の結果、受容者マウスの急性麻痺に至り、これは尾の弾力性が失われ、続いて全後肢の麻痺の進行として現れた。この麻痺エピソードは、CNSにおける活性化T細胞およびマクロファージの著しい浸潤と一致する。ほとんどの条件下で、これは短期間の麻痺後に自発的に回復が起こる疾患の急性モデルである。B7−4Fcの評価に関して、マウスに100μlの滅菌塩溶液中200μgのタンパク質を、細胞移入後の0、2、4、7および11日に皮下注射した(n=10)。対照マウス(n=10)は等体積の塩溶液のみを受容した。全ての動物を定期的に疾患の臨床的徴候に関してモニターし、次のように採点した。1尾の弾力性の損失;2後肢虚弱/部分的な後肢麻痺;3完全な後肢麻痺;4後および前肢麻痺;5瀕死。
【0330】
図27に示す実験において、臨床的疾患の発生率および開始は、両群において類似していた。しかしながら、B7−4Fcで治療されたマウスは重度の疾患を発症し、動物のほとんどは急速に進行して、完全に後および前肢が麻痺した(B7−4Fcおよび対照マウスについてそれぞれ9/10および1/10)。臨床的徴候に関連する死亡率は対照群において10%であり、B7−4Fc処置マウスにおいて70%であった。加えて、臨床的疾患からの回復は、B7−4Fc処置マウスにおいて実質的に遅くなり、このマウスは治療を11日に中止したにもかかわらず生存した。
結論として、T細胞による自己免疫の養子移入モデルを使用して、可溶性B7−4Fcの投与は、疾患の臨床的徴候を悪化させ、結果として、致死率が増大し、麻痺からの回復が遅れる。これらの知見は、CNS中への炎症性細胞の活性化/浸潤の向上と一致し、インビボでB7−4Fcタンパク質についての免疫調節可能性をはっきりとに示す。
【0331】
実施例23
PD−1シグナリング分子の同定
この実施例は、PD−1シグナリング経路に関与するタンパク質を同定するためのスクリーニングアッセイを記載する。スクリーニングアッセイの概略図を図31Aに示す。1×108(実験1)または5×108(実験2)JurkatT細胞を過バナジウム酸塩の存在下および不在下で処理した。細胞を次いで溶解緩衝液(1%NP−40、150mM NaCl、Tris pH7.6、プロテアーゼ阻害剤、およびホスファターゼ阻害剤)中で溶解させた。タンパク質を次いで100μgのビオチニル化ペプチドで4時間4℃で免疫沈降させた。次のペプチドを免疫沈降に使用した:ITIMペプチドY(PO4)、F、およびY、ならびにC’(非ITIM)。ペプチドの配列を図31Bに示す。ストレプトアビジンアガロースを30分間4℃で添加し、サンプルを次いで4回1%溶解緩衝液+0.45M NaCl中で洗浄し、PBS(リン酸塩緩衝溶液)で2回洗浄した。サンプルを次いでSDS−PAGEサンプル緩衝液中で沸騰させ、4〜20%tris−グリシンNOVEX勾配ゲル(減少)上にかけた。ゲルをDaichiプロトコル(実験1)またはMannプロトコル(実験2)を用いて銀染色した。PD−1相互作用タンパク質をゲルから切り取り、質量分析により分析した。
【0332】
次のPD−1相互作用タンパク質を同定した:
実験1(ITIMペプチド)
47kD YおよびpY −過バナジウム酸塩
95kD Y、pY、およびF +過バナジウム酸塩
実験2(ITIMペプチド)
75kD YおよびpY −過バナジウム酸塩
70kD YおよびpY +過バナジウム酸塩
C−ペプチド
22kD −過バナジウム酸塩
46kD +過バナジウム酸塩
【0333】
実施例24
【0334】
PD−1;B7−4シグナリング経路のエンゲージメントによるERK1/2リン酸化ERK1/2リン酸化の阻害
ERK1および2は、T細胞レセプターの活性化に反応して活性化される。この活性化は、ERK1および2分子のリン酸化により検出可能である。PD−1媒介性のシグナリングに関与するシグナリング経路をさらに解明するために、ERK1/2活性化におけるPD−1活性化の効果を調べた。
【0335】
T細胞(他に、T細胞芽細胞とも呼ばれる)を、PHAで刺激された末梢血単核細胞から得た。T細胞を、抗−CD3(1μg/107ビーズ)、およびアイソタイプコントロールIg(IgG2a)またはPDL1.Ig融合タンパク質(4μg/107ビーズ)のいずれかでコートされたトシル−活性化ビーズと溶解性抗−CD28(5μg/ml)の存在下で、5、30、または90分間接触させることにより活性化した。刺激後、細胞およびビーズを氷冷PBS上で一度洗浄し、SDS−PAGEにより溶解およびフラクション化した。フラクション化した細胞を次いでニトロセルロースフィルターに移し、これを次いで、リン酸化ERK1および2を特異的に認識するウサギポリクローナル抗体(Cell Signaling Technologies)でプローブした。ブロットを次いで二次抗体、HRPコンジュゲート抗−ウサギIgG(Santa Cruz Biotechnologies)でプローブした。リン酸化したERK1およびERK2を示している結合抗体を、化学ルミネセンスを用いて検出した。ニトロセルロースフィルターを次いで細長く切り、リン酸化されたようとされまいとERK1およびERK2に特異的な抗体で再プローブし、トータルのERK1およびERK2の等量が細胞抽出物中に存在することを立証した。
【0336】
抗−ホスホERK特異的抗体でのプロービングの結果を図32に示す。刺激されていないT細胞は、検出可能なリン酸化ERK1/2を有しなかった。抗−CD3およびCD28で刺激されたT細胞は、刺激のわずか5分後に検出可能なリン酸化ERK1/2を有した。これに対して、抗−CD3およびCD28、およびPD−L1(PD−1を活性化するための)で刺激されたT細胞は、ERK1/2リン酸化の検出可能な増加を示さなかった。これらの結果は、PD−1シグナリングがERKの活性化を抑制することを示す。
【0337】
実施例25:PD−1;B7−4シグナリング経路のエンゲージメントによるPKC−θリン酸化の阻害
PKC−θは、T細胞レセプターの活性化に反応して活性化されることが知られる。PD−1媒介性のシグナリングに関与するシグナリング経路をさらに解明するために、PKC−θの活性化におけるPD−1の活性化の効果を調べた。
実施例24におけるように単離されたヒトのCD28+T細胞を、抗−CD3(1μg/107ビーズ)およびアイソタイプコントロールIg(IgG2a)またはPDL1.Ig融合タンパク質(4μg/107ビーズ)のいずれかでコートされたトシル−活性化ビーズと、溶解性抗−CD28(5μg/ml)の存在下に5分、30分、または90分または2時間接触させることにより活性化した。刺激後、細胞およびビーズを1度氷冷PBSで洗浄し、SDS−PAGEにより溶解およびフラクション化した。フラクション化した細胞を次いでニトロセルロースフィルターに移し、これを次いでT538でリン酸化されるPKC−θを特異的に認識するウサギポリクローナル抗体(Cess Signaling Technologiesから入手可能な抗−ホスホT538抗体)でプローブした。ニトロセルロースフィルターを次いで二次抗体、HRP−コンジュゲート抗−ネズミIgG(Transduction Laboratories)でプローブした。リン酸化したPKC−θを示す結合抗体を化学ルミネセンスを用いて検出した。ニトロセルロースフィルターを次いで細く切り、PKC−θに特異的なモノクローナル抗体(等量のPKC−θを立証するためのSanta Cruz Biotechnologiesから入手可能な抗−PKC−θ、E7)で再プローブし、等量のPKC−θが細胞抽出物中に存在することを立証した。
【0338】
抗−ホスホPKC−θ特異的抗体でプロービングした結果を図33に示す。刺激されていないT細胞は検出可能なリン酸化されたPKC−θを有しなかった。抗−ホスホPKC−θおよびCD28で刺激されたT細胞は、刺激の30分後に検出可能なリン酸化されたPKC−θを有した。このリン酸化は、抗−CD3およびCD28およびさらにPD−L1(PD−1を活性化するための)で刺激されたT細胞のものよりも有意に高かった。等量のトータルのPKC−θが全細胞抽出物中に存在する事実は、リン酸化されたPKC−θの増加が、PKC−θ分子の存在下での全体的な増加からは生じないが、そのリン酸化の調節キナーゼによる増加または調節ホスファターゼによるその脱リン酸化の低下から生じることを示す。PKC−θ分子のこのリン酸化は、PKC−θ酵素の増加した活性化コンピテント構造と関連することが報告されている(Liu et al., Biochem. J. 361: 255-265 (2002))。PKC−θ分子のPD−1のシグナリングの活性化に反応して低下したリン酸化は、PD−1シグナリングがPKC−θの活性化を抑制することを示す。
【0339】
均等
当業者らは、慣例の実験のみを用いて、本明細書に記載される本発明の具体例の多くの同等物を認識し、確認することができるであろう。このような均等物は請求の範囲に含まれることを意図される。
【図面の簡単な説明】
【0340】
【図1】図1は、ヒトの分泌B7−4、B7−4Sをエンコードするヌクレオチド配列(配列番号1)を表す。
【図2】図2は、ヒトB7−4、B7−4Mをエンコードするヌクレオチド配列(配列番号3)を表す。
【図3】図3は、ヒトB7−4Sのアミノ酸配列(配列番号2)を示し、シグナル、IgV、IgC、および疎水性末端ドメインを示す。
【図4】図4は、ヒトB7−4Mのアミノ酸配列(配列番号4)を示し、シグナル、IgV、IgC、および膜貫通および細胞質を示す。
【図5】図5A−5Bは、ネズミのB7−4のヌクレオチド配列(配列番号22)を示す。
【図6】図6は、ネズミのB7−4のアミノ酸配列(配列番号23)を示す。
【図7】図7は、ヒトB7−MおよびネズミB7−4アミノ酸配列のアライメントを示す(それぞれ、配列番号4および23)。
【図8】図8は、B7−4M−トランスフェクトCOS細胞によるCD28Ig、CTLA4−Ig、およびコントロールIgの結合のFACS分析の結果を示す。
【図9】図9は、B7−4M−トランスフェクトCOS細胞によるIgGおよびネズミICOS−ヒス融合タンパク質の結合のFACS分析の結果を示す。
【図10】図10は、B7−4M−トランスフェクトCOS細胞に対するIgM、BB1および133抗体の結合の、FACS分析の結果を示す。
【図11】図11は、B7−4M(292)でトランスフェクトしたCOS細胞がT細胞の増殖を副刺激することができることを示す。
【図12】図12は、B7−4M(292)でトランスフェクトしたCOS細胞がT細胞の増殖を副刺激することができることを示す。
【図13】図13A−13Dは、B7−4MトランスフェクトCOS細胞へのPD−1の結合を示す。
【図14】図14A−14Fは、追加されたPD−1の、およびFlt4でないの、PD−1のB7−4トランスフェクトCOS細胞への結合を完了する能力を示す。
【図15】図15A−15Lは、フローサイトメトリーにより決定される、B7−4トランスフェクトCHO細胞へ結合するPD−1の能力を示す。
【図16】図16は、BIACORE分析により決定される、B7−4トランスフェクトCHO細胞へ結合するPD−1の能力を示す。
【図17】図17は、T細胞へネガティブシグナルを伝達するB7−4Mの能力を示す。
【図18】図18A−18Cは、B7−4の存在下に刺激されたヒトT細胞におけるT細胞増殖およびサイトカイン産生の阻害を示す。
【図19】図19A−19Bは、CD28副刺激の存在下でのT細胞レセプター/B7−4の活性化がT細胞の増殖の阻害を生じることを示す。
【図20】図20A−20Iは、B7−4を発現しているCHO細胞に対するPD−1の結合を示す。
【図21】図21A−21Dは、CD28シグナルの阻害におけるB7−4の活性を示す。
【図22】図22A−22Dは、サイトカインELISAにより測定される、PD−1:B7−4経路によるサイトカイン産生の阻害を示す。
【図23】図23A−23Cは、サイトカインmRNAレベルにより測定される、PD−1:B7−4経路によるサイトカイン産生の阻害を示す。
【図24】図24A−24Cは、PD−1:B7−4経路の活性のメカニズムが細胞周期の停止であることを示す。
【図25】図25A−25Bは、B7−4およびPD−1の間の相互作用を阻害する、B7−4に対する抗体の能力を示す。
【図26】図26はPD−1に対する抗体がB7−4とPD−1間の相互作用を阻害する能力を表す。
【図27】図27は実験的自己免疫脳脊髄炎のネズミモデルにおいて溶解性B7−4Fcが疾患を悪化させる能力を表す。
【図28】図28A〜28Bは、有糸細胞分裂に関するPD−1:B7−4相互作用の影響を表す。T細胞をCSFEで標識し、ctrl.FcまたはmB7−4Fcbビーズで刺激した。表示した時点で、FACS分析を行った。Live−gated事象を図示する。図28A:CD4+T細胞。図28B:CD8+T細胞。PD−1:B7−4相互用の結果、CD4+およびCD8+T細胞の両方の有糸分裂が減少する。
【図29】図29Aから29CはPD−1:B7−4相互作用によるCD4+およびCD8+T細胞の両方の阻害を表す。図29Aは細胞系および実験デザインを概略的に表す。安定な抗原提示細胞(APC)系を、レトロウイルステクノロジーを用いてGFPまたはmB7−4/GFPを発現するように操作する。TCRトランスジェニック(Tg)マウスから得られる5×104の精製LN T細胞をAPC;ペプチドで2、2〜3、3、または4日間刺激した。CD4+T細胞を含む実験に関して、APC:T細胞比は10μM PCCFペプチドで1:10であった。CD8+T細胞を含む実験に関して、APC:T細胞比は1mMp2Caペプチドで1:1であった。図29BはPD−1:B7−4相互作用によるCD4+T細胞の増殖の阻害を表す。図29CはPD−1:B7−4相互作用によるCD8+T細胞の増殖の阻害を表す。
【図30】図30A〜30Bは、PD−1:B7−4相互作用によるCD4+(CD8+でない)T細胞増殖の阻害を克服するための副刺激の能力を表す。図30A:CD4+T細胞 図30B:CD8+T細胞。
【図31】図31A〜31BはPD−1シグナリング経路において関与するタンパク質を同定するために用いられるスクリーニングアッセイを概略的に表す。図31Aはアッセイのステップの概略図を表す。図31BはヒトPD−1のフラグメント(配列番号24)、マウスPD−1のフラグメント(配列番号25)およびアッセイにおいて用いられるペプチドの配列を表す。アッセイにおいて用いられるペプチドはITIMペプチド:PD−1_Py1(配列番号26);PD−1_Y1F(配列番号27);PD−1_Y1(配列番号28);PD−1_Py2(配列番号29);PD−1_Y2F(配列番号30);PD−1_Y2(配列番号:31);および他のペプチド:PD−1_K212_(配列番号32);PD−1_K212D(配列番号33);PD−1_K335(配列番号34);PD−1_K335D(配列番号35);PD−1_Ctail1(配列番号36);およびPD−1_Ctail2(配列番号37)であった。
【図32】図32は、PDL1.Ig融合タンパク質の存在下または不在下での、刺激されていない、または5分、30分、または90分間刺激されたヒトT細胞の分画細胞抽出物を含んでいるウェスタンブロットの写真である。ブロットをリン酸化ERK1および2に特異的な抗体でプローブした。
【図33】図33はB7−4.Ig融合タンパク質の存在(+)または不在(−)下での、刺激されていない、または5分、30分、90分、または2時間刺激されたヒトT細胞の分画細胞抽出物を含んでいるウェスタンブロットの写真である。ブロットは、リン酸化PKC−θ(抗−ホスホ−PKC−θ)を特異的に検出する、リン酸化PKC−θに特異的な抗体でプローブした。ゲルを細長く切り、リン酸化および非リン酸化PKC−θ(抗−PKC−θ)の両方を認識する抗体でプローブした。
Claims (9)
- PD−1のシグナリングを調節する化合物をスクリーニングするための方法であって、
a)PD−1リガンドの存在下に活性化されたT細胞を試験化合物と接触させること;および、
b)SHP−2のリン酸化を調節する試験化合物の能力を測定すること;
それにより、PD−1のシグナリングを調節する化合物を同定すること;
を含む方法。 - 試験化合物がPD−1シグナリングをアップモジュレートし、それによりSHP−2のリン酸化をアップモジュレートする、請求項1記載の方法。
- 試験化合物がPD−1のシグナリングをダウンモジュレートし、それによりSHP−2のリン酸化をダウンモジュレートする、請求項1記載の方法。
- PD−1のシグナリングを調節する化合物をスクリーニングするための方法であって、
a)PD−1リガンドの存在下に活性化されたT細胞を試験化合物と接触させること;および、
b)試験化合物がERK1またはERK2のリン酸化を調節する能力を決定すること;
それにより、PD−1のシグナリングを調節する化合物を同定すること、
を含む方法。 - 試験化合物がPD−1のシグナリングをアップモジュレートし、それにより、ERK1またはERK2のリン酸化をダウンモジュレートする、請求項4記載の方法。
- 試験化合物がPD−1のシグナリングをダウンモジュレートし、それによりERK1またはERK2のリン酸化をアップモジュレートする、請求項4記載の方法。
- PD−1のシグナリングを調節する化合物をスクリーニングする方法であって、
a)PD−1リガンドの存在下に活性化されたT細胞を試験化合物と接触させること;および、
b)試験化合物がPKC−θのリン酸化を調節する能力を測定すること;
それにより、PD−1のシグナリングを調節する化合物を同定すること、
を含む方法。 - 試験化合物がPD−1のシグナリングをアップモジュレートし、それによりPKC−θのリン酸化をダウンモジュレートする、請求項7記載の方法。
- 試験化合物がPD−1のシグナリングをダウンモジュレートし、それによりPKC−θのリン酸化をアップモジュレートする、請求項7記載の方法。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US28106401P | 2001-04-02 | 2001-04-02 | |
| PCT/US2002/010371 WO2002079499A1 (en) | 2001-04-02 | 2002-04-02 | Pd-1, a receptor for b7-4, and uses therefor |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2004533226A true JP2004533226A (ja) | 2004-11-04 |
| JP2004533226A5 JP2004533226A5 (ja) | 2005-12-22 |
Family
ID=23075790
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2002577908A Pending JP2004533226A (ja) | 2001-04-02 | 2002-04-02 | B7−4に対するpd−1、aレセプター、およびその使用 |
Country Status (12)
| Country | Link |
|---|---|
| US (3) | US7105328B2 (ja) |
| EP (2) | EP1383912A4 (ja) |
| JP (1) | JP2004533226A (ja) |
| AU (1) | AU2002338272B2 (ja) |
| BR (1) | BR0208637A (ja) |
| CA (1) | CA2442066C (ja) |
| MX (1) | MXPA03008959A (ja) |
| NO (1) | NO20034387L (ja) |
| NZ (1) | NZ528265A (ja) |
| PL (1) | PL367162A1 (ja) |
| WO (1) | WO2002079499A1 (ja) |
| ZA (1) | ZA200307310B (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007100098A1 (ja) | 2006-03-03 | 2007-09-07 | Kyoto University | 細胞表面機能分子の細胞外領域多量体 |
Families Citing this family (104)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA3016482A1 (en) * | 1999-11-30 | 2001-06-07 | Mayo Foundation For Medical Education And Research | B7-h1, a novel immunoregulatory molecule |
| US6803192B1 (en) * | 1999-11-30 | 2004-10-12 | Mayo Foundation For Medical Education And Research | B7-H1, a novel immunoregulatory molecule |
| US7030219B2 (en) | 2000-04-28 | 2006-04-18 | Johns Hopkins University | B7-DC, Dendritic cell co-stimulatory molecules |
| AU2002338272B2 (en) * | 2001-04-02 | 2007-10-11 | Dana-Farber Cancer Institute, Inc. | PD-1, a receptor for B7-4, and uses therefor |
| US7794710B2 (en) * | 2001-04-20 | 2010-09-14 | Mayo Foundation For Medical Education And Research | Methods of enhancing T cell responsiveness |
| US7875612B2 (en) | 2001-04-24 | 2011-01-25 | Purdue Research Foundation | Folate mimetics and folate-receptor binding conjugates thereof |
| ATE426414T1 (de) * | 2002-05-15 | 2009-04-15 | Endocyte Inc | Vitamin-mitomycin-konjugate |
| SI2206517T1 (sl) | 2002-07-03 | 2023-12-29 | Ono Pharmaceutical Co., Ltd. | Imunopotencirni sestavki,ki obsegajo protitelesa proti PD-L1 |
| US7052694B2 (en) * | 2002-07-16 | 2006-05-30 | Mayo Foundation For Medical Education And Research | Dendritic cell potentiation |
| US7432351B1 (en) | 2002-10-04 | 2008-10-07 | Mayo Foundation For Medical Education And Research | B7-H1 variants |
| US7449300B2 (en) * | 2002-11-21 | 2008-11-11 | Mayo Foundation For Medical Education And Research | Detection of antibodies specific for B7-H1 in subjects with diseases or pathological conditions mediated by activated T cells |
| EP2517730A3 (en) | 2003-01-27 | 2013-01-02 | Endocyte, Inc. | Vitamin receptor binding drug delivery conjugates |
| US7501119B2 (en) * | 2004-06-30 | 2009-03-10 | Mayo Foundation For Medical Education And Research | Methods and molecules for modulating an immune response |
| US20060099203A1 (en) | 2004-11-05 | 2006-05-11 | Pease Larry R | B7-DC binding antibody |
| CN101098854B (zh) * | 2004-07-23 | 2012-12-05 | 恩多塞特公司 | 二价连接体及其轭合物 |
| AU2005295038B2 (en) | 2004-10-06 | 2012-05-17 | Mayo Foundation For Medical Education And Research | B7-H1 and methods of diagnosis, prognosis, and treatment of cancer |
| US8044200B2 (en) * | 2005-03-16 | 2011-10-25 | Endocyte, Inc. | Synthesis and purification of pteroic acid and conjugates thereof |
| CN103059138B (zh) | 2005-05-09 | 2015-10-28 | 小野药品工业株式会社 | 程序性死亡-1(pd-1)的人单克隆抗体及使用抗pd-1抗体来治疗癌症的方法 |
| AU2011203119C1 (en) * | 2005-05-09 | 2018-06-14 | E. R. Squibb & Sons, L.L.C. | Human monoclonal antibodies to programmed death 1(PD-1) and methods for treating cancer using anti-PD-1 antibodies alone or in combination with other immunotherapeutics |
| WO2006133396A2 (en) | 2005-06-08 | 2006-12-14 | Dana-Farber Cancer Institute | Methods and compositions for the treatment of persistent infections and cancer by inhibiting the programmed cell death 1 (pd-1) pathway |
| ME02260B (me) | 2005-07-01 | 2016-02-29 | Medarex Inc | Humana monoklonska antitela za ligand programirane smrti 1 (pd-l1) |
| US8465724B2 (en) * | 2005-08-19 | 2013-06-18 | Endocyte, Inc. | Multi-drug ligand conjugates |
| US20080280937A1 (en) * | 2005-08-19 | 2008-11-13 | Christopher Paul Leamon | Ligand Conjugates of Vinca Alkaloids, Analogs, and Derivatives |
| EP2061504A4 (en) * | 2006-09-20 | 2010-01-27 | Univ Johns Hopkins | COMBINATION THERAPY FOR CANCER AND INFECTION DISEASES WITH ANTI-B7-H1 ANTIBODIES |
| AU2013200388B2 (en) * | 2006-12-27 | 2014-10-23 | Dana-Farber Cancer Institute, Inc. | Compositions and methods for the treatment of infections and tumors |
| NZ626867A (en) | 2006-12-27 | 2014-09-26 | Harvard College | Compositions and methods for the treatment of infections and tumors |
| US20100104626A1 (en) * | 2007-02-16 | 2010-04-29 | Endocyte, Inc. | Methods and compositions for treating and diagnosing kidney disease |
| CA2680535C (en) * | 2007-03-14 | 2016-09-20 | Endocyte, Inc. | Binding ligand linked drug delivery conjugates of tubulysins |
| HRP20131167T1 (hr) | 2007-06-18 | 2014-01-03 | Merck Sharp & Dohme B.V. | Antitijela za humani receptor programirane smrti pd-1 |
| US9138484B2 (en) | 2007-06-25 | 2015-09-22 | Endocyte, Inc. | Conjugates containing hydrophilic spacer linkers |
| US9877965B2 (en) | 2007-06-25 | 2018-01-30 | Endocyte, Inc. | Vitamin receptor drug delivery conjugates for treating inflammation |
| US9187521B2 (en) | 2007-10-25 | 2015-11-17 | Endocyte, Inc. | Tubulysins and processes for preparing |
| US8647822B2 (en) | 2007-11-28 | 2014-02-11 | Oregon Health & Science University | Determining whether a test compound modulates PD-1 activity in activated immune cells using gene expression profiles |
| AU2009288730B2 (en) | 2008-08-25 | 2013-06-20 | Amplimmune, Inc. | Compositions of PD-1 antagonists and methods of use |
| US8552154B2 (en) | 2008-09-26 | 2013-10-08 | Emory University | Anti-PD-L1 antibodies and uses therefor |
| DK2370593T3 (en) * | 2008-11-28 | 2016-07-04 | Univ Emory | A method for determining the effect of PD-1 Antagonists |
| MY188826A (en) | 2008-12-09 | 2022-01-06 | Genentech Inc | Anti-pd-l1 antibodies and their use to enhance t-cell function |
| WO2010104617A2 (en) | 2009-01-23 | 2010-09-16 | Salvatore Albani | Novel methods to induce a state of immune tolerance |
| CA2775761C (en) | 2009-09-30 | 2018-08-28 | Memorial Sloan-Kettering Cancer Center | Combination immunotherapy for the treatment of cancer |
| US20130202623A1 (en) * | 2010-02-16 | 2013-08-08 | Nicolas Chomont | Pd-1 modulation and uses thereof for modulating hiv replication |
| TW201134488A (en) * | 2010-03-11 | 2011-10-16 | Ucb Pharma Sa | PD-1 antibodies |
| US8993731B2 (en) | 2010-03-11 | 2015-03-31 | Ucb Biopharma Sprl | PD-1 antibody |
| US9783578B2 (en) | 2010-06-25 | 2017-10-10 | Aurigene Discovery Technologies Limited | Immunosuppression modulating compounds |
| US8907053B2 (en) | 2010-06-25 | 2014-12-09 | Aurigene Discovery Technologies Limited | Immunosuppression modulating compounds |
| GB201103631D0 (en) * | 2011-03-03 | 2011-04-13 | Univ Leeds | Identification of candidate therapeutics |
| WO2012168944A1 (en) | 2011-06-08 | 2012-12-13 | Aurigene Discovery Technologies Limited | Therapeutic compounds for immunomodulation |
| CA2850245C (en) * | 2011-10-17 | 2020-04-28 | Herlev Hospital | Pd-l1 based immunotherapy |
| US10080805B2 (en) | 2012-02-24 | 2018-09-25 | Purdue Research Foundation | Cholecystokinin B receptor targeting for imaging and therapy |
| WO2013132317A1 (en) | 2012-03-07 | 2013-09-12 | Aurigene Discovery Technologies Limited | Peptidomimetic compounds as immunomodulators |
| US20140080175A1 (en) | 2012-03-29 | 2014-03-20 | Endocyte, Inc. | Processes for preparing tubulysin derivatives and conjugates thereof |
| JP2015512910A (ja) | 2012-03-29 | 2015-04-30 | オーリジーン ディスカバリー テクノロジーズ リミテッドAurigene Discovery Technologies Limited | ヒトpd1のbcループに由来する免疫調節性環状化合物 |
| HK1203971A1 (en) | 2012-05-15 | 2015-11-06 | Bristol-Myers Squibb Company | Cancer immunotherapy by disrupting pd-1/pd-l1 signaling |
| US20150079192A1 (en) * | 2012-05-21 | 2015-03-19 | Marv Enterprises, LLC | Treatment of cancer by manipulating the immune system |
| CN111499755A (zh) * | 2012-08-03 | 2020-08-07 | 丹娜法伯癌症研究院 | 抗-pd-l1和pd-l2双结合抗体单一试剂及其使用方法 |
| US9562892B2 (en) | 2012-10-02 | 2017-02-07 | New York University | Methods and agents for modulating protocadherin-18 activity |
| WO2014059173A2 (en) * | 2012-10-10 | 2014-04-17 | Sangamo Biosciences, Inc. | T cell modifying compounds and uses thereof |
| IN2015DN04147A (ja) | 2012-10-16 | 2015-10-16 | Endocyte Inc | |
| JP6742903B2 (ja) | 2013-05-02 | 2020-08-19 | アナプティスバイオ インコーポレイティッド | プログラム死−1(pd−1)に対する抗体 |
| CA2913490A1 (en) * | 2013-06-06 | 2014-12-11 | Dana-Farber Cancer Institute, Inc. | Compositions and methods for identification, assessment, prevention, and treatment of cancer using pd-l1 isoforms |
| PT3041828T (pt) | 2013-09-06 | 2018-10-09 | Aurigene Discovery Tech Ltd | Derivados 1,3,4-0xadiazoles como imonomoduladores |
| HUE038169T2 (hu) | 2013-09-06 | 2018-09-28 | Aurigene Discovery Tech Ltd | 1,2,4-Oxadiazol származékok mint immunomodulátorok |
| HRP20181271T1 (hr) | 2013-09-06 | 2018-10-05 | Aurigene Discovery Technologies Limited | Ciklički peptidomimetički spojevi kao imunomodulatori |
| WO2015048312A1 (en) | 2013-09-26 | 2015-04-02 | Costim Pharmaceuticals Inc. | Methods for treating hematologic cancers |
| ES2969350T3 (es) | 2013-12-20 | 2024-05-17 | Intervet Int Bv | Anticuerpos murinos caninizados anti-PD-1 canino |
| JOP20200094A1 (ar) | 2014-01-24 | 2017-06-16 | Dana Farber Cancer Inst Inc | جزيئات جسم مضاد لـ pd-1 واستخداماتها |
| JOP20200096A1 (ar) | 2014-01-31 | 2017-06-16 | Children’S Medical Center Corp | جزيئات جسم مضاد لـ tim-3 واستخداماتها |
| AU2015229103C9 (en) | 2014-03-14 | 2020-11-26 | Immutep S.A.S | Antibody molecules to LAG-3 and uses thereof |
| TWI691335B (zh) | 2014-07-09 | 2020-04-21 | 英屬開曼群島商博笛生物科技有限公司 | 用於治療腫瘤的抗pd-l1組合 |
| CN112587672A (zh) | 2014-09-01 | 2021-04-02 | 博笛生物科技有限公司 | 用于治疗肿瘤的抗-pd-l1结合物 |
| WO2016040882A1 (en) | 2014-09-13 | 2016-03-17 | Novartis Ag | Combination therapies of egfr inhibitors |
| HUE058274T2 (hu) | 2015-03-10 | 2022-07-28 | Aurigene Discovery Tech Ltd | 1,2,4-oxadiazol és tiadiazol-vegyületek, mint immunmodulátorok |
| SG10201913297TA (en) | 2015-03-13 | 2020-02-27 | Cytomx Therapeutics Inc | Anti-pdl1 antibodies, activatable anti-pdl1 antibodies, and methods of use thereof |
| EP3302467A4 (en) | 2015-05-31 | 2019-01-02 | Curegenix Corporation | Combination compositions for immunotherapy |
| US10513558B2 (en) | 2015-07-13 | 2019-12-24 | Cytomx Therapeutics, Inc. | Anti-PD1 antibodies, activatable anti-PD1 antibodies, and methods of use thereof |
| CN115887671A (zh) | 2015-07-16 | 2023-04-04 | 百欧肯治疗有限公司 | 治疗癌症的组合物及方法 |
| WO2017024465A1 (en) | 2015-08-10 | 2017-02-16 | Innovent Biologics (Suzhou) Co., Ltd. | Pd-1 antibodies |
| HK1251158A1 (zh) | 2015-09-29 | 2019-01-25 | 细胞基因公司 | Pd-1結合蛋白及其使用方法 |
| WO2017102920A1 (en) | 2015-12-18 | 2017-06-22 | Intervet International B.V. | Caninized human antibodies to human and canine il-4r alpha |
| US11091556B2 (en) | 2015-12-18 | 2021-08-17 | Intervet Inc. | Caninized human antibodies to human IL-4R alpha |
| CA3018332A1 (en) | 2016-03-21 | 2017-09-28 | Dana-Farber Cancer Institute, Inc. | T-cell exhaustion state-specific gene expression regulators and uses thereof |
| WO2017220602A1 (en) | 2016-06-21 | 2017-12-28 | Herlev Hospital | Pdl1 peptides for use in cancer vaccines |
| WO2018041118A1 (en) * | 2016-08-31 | 2018-03-08 | Beijing Biocytogen Co., Ltd | Genetically modified non-human animal with human or chimeric pd-l1 |
| CN107815466B (zh) | 2016-08-31 | 2020-03-13 | 百奥赛图江苏基因生物技术有限公司 | 人源化基因改造动物模型的制备方法及应用 |
| PE20191102A1 (es) | 2016-09-14 | 2019-08-23 | Abbvie Biotherapeutics Inc | Anticuerpos anti-pd-1 y sus usos |
| US10766958B2 (en) | 2016-09-19 | 2020-09-08 | Celgene Corporation | Methods of treating vitiligo using PD-1 binding antibodies |
| KR102257154B1 (ko) | 2016-09-19 | 2021-05-28 | 셀진 코포레이션 | Pd-1 결합 단백질을 사용하는 면역 질환의 치료 방법 |
| WO2018071809A1 (en) * | 2016-10-14 | 2018-04-19 | Ariad Pharmaceuticals, Inc. | Inducible t-cell system and uses thereof |
| WO2018085468A1 (en) | 2016-11-01 | 2018-05-11 | Anaptysbio, Inc. | Antibodies directed against programmed death- 1 (pd-1) |
| CA3049440A1 (en) | 2017-01-09 | 2018-07-12 | Tesaro, Inc. | Methods of treating cancer with anti-pd-1 antibodies |
| AU2018278327B2 (en) | 2017-06-01 | 2023-03-16 | Cytomx Therapeutics, Inc. | Activatable anti-pdl1 antibodies and methods of use thereof |
| EP3634584B1 (en) | 2017-06-09 | 2024-09-18 | Providence Health & Services - Oregon | Tumor-infiltrating t-cells for use in the treatment of cancer |
| CN111133315A (zh) | 2017-08-11 | 2020-05-08 | 得克萨斯州大学系统董事会 | 用于治疗癌症转移的靶向激酶 |
| WO2019061324A1 (en) | 2017-09-29 | 2019-04-04 | Curis Inc. | CRYSTALLINE FORMS OF IMMUNOMODULATORS |
| CA3078872A1 (en) | 2017-10-11 | 2019-04-18 | Aurigene Discovery Technologies Limited | Crystalline forms of 3-substituted 1,2,4-oxadiazole |
| WO2019087087A1 (en) | 2017-11-03 | 2019-05-09 | Aurigene Discovery Technologies Limited | Dual inhibitors of tim-3 and pd-1 pathways |
| EP3706798A1 (en) | 2017-11-06 | 2020-09-16 | Aurigene Discovery Technologies Limited | Conjoint therapies for immunomodulation |
| WO2019090390A1 (en) * | 2017-11-08 | 2019-05-16 | University Of Canberra | Immunogenic compositions and uses therefor |
| CA3097369A1 (en) | 2018-04-17 | 2019-10-24 | Celldex Therapeutics, Inc. | Anti-cd27 and anti-pd-l1 antibodies and bispecific constructs |
| CN108828227B (zh) * | 2018-05-17 | 2020-06-30 | 浙江大学 | PD-L1剪接体c作为预测肿瘤预后靶标的应用 |
| MA56523A (fr) | 2019-06-18 | 2022-04-27 | Janssen Sciences Ireland Unlimited Co | Combinaison de vaccins contre le virus de l'hépatite b (vhb) et d'anticorps anti-pd-1 ou anti-pd-l1 |
| EP3986460A2 (en) | 2019-06-18 | 2022-04-27 | Janssen Sciences Ireland Unlimited Company | Combination of hepatitis b virus (hbv) vaccines and anti-pd-1 antibody |
| CN113030475B (zh) * | 2021-05-25 | 2021-08-10 | 泛肽生物科技(浙江)有限公司 | 一种基于细胞线粒体质量评估的t细胞pd-1检测方法 |
| AU2022288058A1 (en) | 2021-06-07 | 2023-11-16 | Agonox, Inc. | Cxcr5, pd-1, and icos expressing tumor reactive cd4 t cells and their use |
| NL2035804B1 (en) * | 2023-09-15 | 2025-03-25 | Umc Utrecht Holding Bv | Immune modulating inhibitory receptors |
Family Cites Families (72)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2437213A1 (fr) | 1978-09-28 | 1980-04-25 | Cm Ind | Produits cytotoxiques formes par liaison covalente de la chaine a de la ricine avec un anticorps et leur procede de preparation |
| EP0044167A3 (en) | 1980-07-14 | 1982-04-21 | The Regents Of The University Of California | Antibody targeted cytotoxic agent |
| NO812612L (no) | 1980-08-06 | 1982-02-08 | Ferring Pharma Ltd | Enzym-inhibitorer. |
| US4873191A (en) | 1981-06-12 | 1989-10-10 | Ohio University | Genetic transformation of zygotes |
| US4474893A (en) | 1981-07-01 | 1984-10-02 | The University of Texas System Cancer Center | Recombinant monoclonal antibodies |
| US4522811A (en) | 1982-07-08 | 1985-06-11 | Syntex (U.S.A.) Inc. | Serial injection of muramyldipeptides and liposomes enhances the anti-infective activity of muramyldipeptides |
| US4870009A (en) | 1982-11-22 | 1989-09-26 | The Salk Institute For Biological Studies | Method of obtaining gene product through the generation of transgenic animals |
| US4816567A (en) | 1983-04-08 | 1989-03-28 | Genentech, Inc. | Recombinant immunoglobin preparations |
| US4736866A (en) | 1984-06-22 | 1988-04-12 | President And Fellows Of Harvard College | Transgenic non-human mammals |
| JPS6147500A (ja) | 1984-08-15 | 1986-03-07 | Res Dev Corp Of Japan | キメラモノクロ−ナル抗体及びその製造法 |
| EP0173494A3 (en) | 1984-08-27 | 1987-11-25 | The Board Of Trustees Of The Leland Stanford Junior University | Chimeric receptors by dna splicing and expression |
| GB8422238D0 (en) | 1984-09-03 | 1984-10-10 | Neuberger M S | Chimeric proteins |
| JPS61134325A (ja) | 1984-12-04 | 1986-06-21 | Teijin Ltd | ハイブリツド抗体遺伝子の発現方法 |
| US4683202A (en) | 1985-03-28 | 1987-07-28 | Cetus Corporation | Process for amplifying nucleic acid sequences |
| US4683195A (en) | 1986-01-30 | 1987-07-28 | Cetus Corporation | Process for amplifying, detecting, and/or-cloning nucleic acid sequences |
| US5225539A (en) | 1986-03-27 | 1993-07-06 | Medical Research Council | Recombinant altered antibodies and methods of making altered antibodies |
| DE122007000007I2 (de) | 1986-04-09 | 2010-12-30 | Genzyme Corp | Genetisch transformierte Tiere, die ein gewünschtes Protein in Milch absondern |
| US4902505A (en) | 1986-07-30 | 1990-02-20 | Alkermes | Chimeric peptides for neuropeptide delivery through the blood-brain barrier |
| US4987071A (en) | 1986-12-03 | 1991-01-22 | University Patents, Inc. | RNA ribozyme polymerases, dephosphorylases, restriction endoribonucleases and methods |
| US5116742A (en) | 1986-12-03 | 1992-05-26 | University Patents, Inc. | RNA ribozyme restriction endoribonucleases and methods |
| US4904582A (en) | 1987-06-11 | 1990-02-27 | Synthetic Genetics | Novel amphiphilic nucleic acid conjugates |
| US4873316A (en) | 1987-06-23 | 1989-10-10 | Biogen, Inc. | Isolation of exogenous recombinant proteins from the milk of transgenic mammals |
| US5080891A (en) | 1987-08-03 | 1992-01-14 | Ddi Pharmaceuticals, Inc. | Conjugates of superoxide dismutase coupled to high molecular weight polyalkylene glycols |
| DE768377T1 (de) | 1988-09-02 | 1998-01-02 | Dyax Corp | Herstellung und Auswahl von Rekombinantproteinen mit verschiedenen Bindestellen |
| US5223409A (en) | 1988-09-02 | 1993-06-29 | Protein Engineering Corp. | Directed evolution of novel binding proteins |
| US5272057A (en) | 1988-10-14 | 1993-12-21 | Georgetown University | Method of detecting a predisposition to cancer by the use of restriction fragment length polymorphism of the gene for human poly (ADP-ribose) polymerase |
| US5116964A (en) | 1989-02-23 | 1992-05-26 | Genentech, Inc. | Hybrid immunoglobulins |
| FR2646438B1 (fr) | 1989-03-20 | 2007-11-02 | Pasteur Institut | Procede de remplacement specifique d'une copie d'un gene present dans le genome receveur par l'integration d'un gene different de celui ou se fait l'integration |
| US5328470A (en) | 1989-03-31 | 1994-07-12 | The Regents Of The University Of Michigan | Treatment of diseases by site-specific instillation of cells or site-specific transformation of cells and kits therefor |
| US5459039A (en) | 1989-05-12 | 1995-10-17 | Duke University | Methods for mapping genetic mutations |
| ATE423195T1 (de) | 1989-07-25 | 2009-03-15 | Cell Genesys Inc | Homologe rekombination für universelle donorzellen und chimerische säugetierzellen |
| US6641809B1 (en) | 1990-03-26 | 2003-11-04 | Bristol-Myers Squibb Company | Method of regulating cellular processes mediated by B7 and CD28 |
| US5427908A (en) | 1990-05-01 | 1995-06-27 | Affymax Technologies N.V. | Recombinant library screening methods |
| ATE185601T1 (de) | 1990-07-10 | 1999-10-15 | Cambridge Antibody Tech | Verfahren zur herstellung von spezifischen bindungspaargliedern |
| US5393788A (en) | 1990-07-10 | 1995-02-28 | Smithkline Beecham Corporation | Phenylalkyl oxamides |
| GB9015198D0 (en) | 1990-07-10 | 1990-08-29 | Brien Caroline J O | Binding substance |
| WO1992008802A1 (en) | 1990-10-29 | 1992-05-29 | Cetus Oncology Corporation | Bispecific antibodies, method of production, and uses thereof |
| CA2405246A1 (en) | 1990-12-03 | 1992-06-11 | Genentech, Inc. | Enrichment method for variant proteins with alterred binding properties |
| AU1545692A (en) | 1991-03-01 | 1992-10-06 | Protein Engineering Corporation | Process for the development of binding mini-proteins |
| DE69233750D1 (de) | 1991-04-10 | 2009-01-02 | Scripps Research Inst | Bibliotheken heterodimerer Rezeptoren mittels Phagemiden |
| DE69230142T2 (de) | 1991-05-15 | 2000-03-09 | Cambridge Antibody Technology Ltd. | Verfahren zur herstellung von spezifischen bindungspaargliedern |
| US5844095A (en) | 1991-06-27 | 1998-12-01 | Bristol-Myers Squibb Company | CTLA4 Ig fusion proteins |
| DE4122599C2 (de) | 1991-07-08 | 1993-11-11 | Deutsches Krebsforsch | Phagemid zum Screenen von Antikörpern |
| AU2515992A (en) | 1991-08-20 | 1993-03-16 | Genpharm International, Inc. | Gene targeting in animal cells using isogenic dna constructs |
| ES2136092T3 (es) | 1991-09-23 | 1999-11-16 | Medical Res Council | Procedimientos para la produccion de anticuerpos humanizados. |
| US5660827A (en) | 1992-03-05 | 1997-08-26 | Board Of Regents, The University Of Texas System | Antibodies that bind to endoglin |
| US5733743A (en) | 1992-03-24 | 1998-03-31 | Cambridge Antibody Technology Limited | Methods for producing members of specific binding pairs |
| DE69334150T2 (de) | 1992-05-14 | 2008-03-13 | Baylor College Of Medicine, Houston | Mutierte steroidhormonrezeptoren, methoden für ihre benutzung und molekularer schalter für gentherapie |
| WO1994002610A1 (en) | 1992-07-17 | 1994-02-03 | Dana-Farber Cancer Institute | Method of intracellular binding of target molecules |
| MX9304769A (es) | 1992-08-05 | 1994-05-31 | Max Planck Gesellschaft | Subfamilia ptp-d de fosfatasas de la proteina tirosina. |
| US5589375A (en) | 1992-10-06 | 1996-12-31 | Max-Planck-Gesellschaft Zur Forderung Der Wissenschaften E. V. | PTP 1D: a novel protein tyrosine phosphatase |
| EP0672131B1 (en) | 1992-10-30 | 2003-12-17 | The General Hospital Corporation | A novel cell cycle control protein |
| EP0679196B1 (en) | 1993-01-07 | 2004-05-26 | Sequenom, Inc. | Dna sequencing by mass spectrometry |
| PL310327A1 (en) | 1993-02-12 | 1995-12-11 | Univ Leland Stanford Junior | Adjustable transcription of target genes and other biological processes |
| ATE293686T1 (de) | 1993-06-04 | 2005-05-15 | Us Navy | Verfahren zur selektiven stimulierung der t- zellproliferation. |
| US5866755A (en) | 1993-06-14 | 1999-02-02 | Basf Aktiengellschaft | Animals transgenic for a tetracycline-regulated transcriptional inhibitor |
| EP0705334A1 (en) | 1993-06-14 | 1996-04-10 | Basf Aktiengesellschaft | Tight control of gene expression in eucaryotic cells by tetracycline-responsive promoters |
| CA2168349A1 (en) | 1993-07-30 | 1995-02-09 | Lingxun Duan | Intracellular immunization |
| UA40577C2 (uk) | 1993-08-02 | 2001-08-15 | Мерк Патент Гмбх | Біспецифічна молекула, що використовується для лізису пухлинних клітин, спосіб її одержання, моноклональне антитіло (варіанти), фармацевтичний препарат, фармацевтичний набір (варіанти), спосіб видалення пухлинних клітин |
| US5498531A (en) | 1993-09-10 | 1996-03-12 | President And Fellows Of Harvard College | Intron-mediated recombinant techniques and reagents |
| JPH07291996A (ja) | 1994-03-01 | 1995-11-07 | Yuu Honshiyo | ヒトにおけるプログラムされた細胞死に関連したポリペプチド、それをコードするdna、そのdnaからなるベクター、そのベクターで形質転換された宿主細胞、そのポリペプチドの抗体、およびそのポリペプチドまたはその抗体を含有する薬学的組成物 |
| US6576236B1 (en) * | 1994-07-01 | 2003-06-10 | Dana Farber Cancer Institute | Methods for stimulating T cell responses by manipulating a common cytokine receptor γ chain |
| GB9517780D0 (en) | 1995-08-31 | 1995-11-01 | Roslin Inst Edinburgh | Biological manipulation |
| GB9517779D0 (en) | 1995-08-31 | 1995-11-01 | Roslin Inst Edinburgh | Biological manipulation |
| US6750334B1 (en) | 1996-02-02 | 2004-06-15 | Repligen Corporation | CTLA4-immunoglobulin fusion proteins having modified effector functions and uses therefor |
| WO1997049430A1 (en) | 1996-06-26 | 1997-12-31 | Antigen Express, Inc. | Immunotherapy by modulation of antigen presentation |
| US6040152A (en) | 1996-12-31 | 2000-03-21 | National Jewish Medical And Research Center | Method and assay for regulation of T cell proliferation |
| JP3521382B2 (ja) | 1997-02-27 | 2004-04-19 | 日本たばこ産業株式会社 | 細胞間接着及びシグナル伝達を媒介する細胞表面分子 |
| MXPA02001877A (es) * | 1999-08-23 | 2002-08-20 | Dana Farber Cancer Inst Inc | Pd-1, un receptor para b7-4, y usos del mismo. |
| HUP0202882A2 (en) | 1999-08-23 | 2002-12-28 | Dana Farber Cancer Inst Inc | Novel b7-4 molecules and uses therefor |
| AU2002338272B2 (en) * | 2001-04-02 | 2007-10-11 | Dana-Farber Cancer Institute, Inc. | PD-1, a receptor for B7-4, and uses therefor |
| IT1395574B1 (it) | 2009-09-14 | 2012-10-16 | Guala Dispensing Spa | Dispositivo di erogazione |
-
2002
- 2002-04-02 AU AU2002338272A patent/AU2002338272B2/en not_active Ceased
- 2002-04-02 PL PL02367162A patent/PL367162A1/xx unknown
- 2002-04-02 CA CA002442066A patent/CA2442066C/en not_active Expired - Lifetime
- 2002-04-02 US US10/115,615 patent/US7105328B2/en not_active Expired - Lifetime
- 2002-04-02 MX MXPA03008959A patent/MXPA03008959A/es active IP Right Grant
- 2002-04-02 WO PCT/US2002/010371 patent/WO2002079499A1/en active IP Right Grant
- 2002-04-02 EP EP02757942A patent/EP1383912A4/en not_active Withdrawn
- 2002-04-02 BR BR0208637-9A patent/BR0208637A/pt not_active IP Right Cessation
- 2002-04-02 JP JP2002577908A patent/JP2004533226A/ja active Pending
- 2002-04-02 EP EP10191193A patent/EP2388590A1/en not_active Withdrawn
- 2002-04-02 NZ NZ528265A patent/NZ528265A/en unknown
-
2003
- 2003-09-18 ZA ZA200307310A patent/ZA200307310B/en unknown
- 2003-10-01 NO NO20034387A patent/NO20034387L/no not_active Application Discontinuation
-
2006
- 2006-09-11 US US11/519,186 patent/US7700301B2/en not_active Expired - Fee Related
-
2010
- 2010-04-16 US US12/761,704 patent/US20110033884A1/en not_active Abandoned
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007100098A1 (ja) | 2006-03-03 | 2007-09-07 | Kyoto University | 細胞表面機能分子の細胞外領域多量体 |
| EP2468765A1 (en) | 2006-03-03 | 2012-06-27 | ONO Pharmaceutical Co., Ltd. | Multimer of extracellular domain of cell surface functional molecule |
| EP2889309A1 (en) | 2006-03-03 | 2015-07-01 | Ono Pharmaceutical Co., Ltd. | Tetramer of extracellular domain of PD-L1 |
Also Published As
| Publication number | Publication date |
|---|---|
| US20110033884A1 (en) | 2011-02-10 |
| AU2002338272B2 (en) | 2007-10-11 |
| CA2442066A1 (en) | 2002-10-10 |
| NZ528265A (en) | 2005-10-28 |
| US20030044768A1 (en) | 2003-03-06 |
| EP2388590A1 (en) | 2011-11-23 |
| NO20034387D0 (no) | 2003-10-01 |
| ZA200307310B (en) | 2004-09-20 |
| US7105328B2 (en) | 2006-09-12 |
| PL367162A1 (en) | 2005-02-21 |
| US20080274490A1 (en) | 2008-11-06 |
| WO2002079499A1 (en) | 2002-10-10 |
| NO20034387L (no) | 2003-11-28 |
| EP1383912A1 (en) | 2004-01-28 |
| BR0208637A (pt) | 2004-12-07 |
| EP2388590A8 (en) | 2011-12-28 |
| CA2442066C (en) | 2005-11-01 |
| US7700301B2 (en) | 2010-04-20 |
| MXPA03008959A (es) | 2004-10-15 |
| EP1383912A4 (en) | 2007-11-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US7105328B2 (en) | Methods for screening for compounds that modulate pd-1 signaling | |
| JP4896327B2 (ja) | Pd−1、b7−4の受容体、およびその使用 | |
| US7029674B2 (en) | Methods for downmodulating immune cells using an antibody to PD-1 | |
| AU2002338272A1 (en) | PD-1, a receptor for B7-4, and uses therefor | |
| JP2004501631A (ja) | Pd−l2分子:新規pd−1リガンドおよびその使用 | |
| HK1161299A (en) | Assays for screening anti-pd-1 antibodies and uses thereof | |
| AU2002307063A1 (en) | Module of PD-1 interactions with its ligands |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20050323 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20050323 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20080108 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20080408 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20080415 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20080508 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20090317 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20090617 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20090624 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20091006 |