JP2004509078A - Apo−2LレセプターアゴニストとCPT−11の相乗作用 - Google Patents
Apo−2LレセプターアゴニストとCPT−11の相乗作用 Download PDFInfo
- Publication number
- JP2004509078A JP2004509078A JP2002515307A JP2002515307A JP2004509078A JP 2004509078 A JP2004509078 A JP 2004509078A JP 2002515307 A JP2002515307 A JP 2002515307A JP 2002515307 A JP2002515307 A JP 2002515307A JP 2004509078 A JP2004509078 A JP 2004509078A
- Authority
- JP
- Japan
- Prior art keywords
- apo
- antibody
- cpt
- cells
- receptor
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images














Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
- A61K39/39533—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals
- A61K39/39541—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals against normal tissues, cells
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/4738—Quinolines; Isoquinolines ortho- or peri-condensed with heterocyclic ring systems
- A61K31/4745—Quinolines; Isoquinolines ortho- or peri-condensed with heterocyclic ring systems condensed with ring systems having nitrogen as a ring hetero atom, e.g. phenantrolines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- A61K38/177—Receptors; Cell surface antigens; Cell surface determinants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Abstract
Description
(発明の分野)
この発明は、一般に、哺乳動物細胞においてアポトーシスを誘発する方法に関する。特に、本発明は、哺乳動物細胞においてアポトーシスを相乗的に誘発するためのApo−2レセプターアゴニストとCPT−11の使用に関する。本発明によって考慮される種々のApo−2Lレセプターアゴニストには、Apo−2リガンド又はTRAILとして公知のリガンド、並びに一又は複数のApo−2Lレセプターに対するアゴニスト抗体が含まれる。
【0002】
(発明の背景)
様々な分子、例えば腫瘍壊死因子−α(「TNF−α」)、腫瘍壊死因子−β(「TNF−β」すなわち「リンホトキシン−α」)、リンホトキシン−β(「LT−β」)、CD30リガンド、CD27リガンド、CD40リガンド、OX−40リガンド、4−1BBリガンド、Apo−1リガンド(Fasリガンド又はCD95リガンドとも称される)、Apo−2リガンド(TRAILとも称される)、Apo−3リガンド(TWEAKとも称される)、オステオプロテゲリン(OPG)、APRIL、RANKリガンド(TRANCEとも称される)及びTALL−1(B1yS、BAFF又はTHANKとも称される)が、サイトカインの腫瘍壊死因子(「TNF」)ファミリーのメンバーとして同定されている[例えば、Gruss及びDower, Blood, 85:3378−3404(1995);Pittiら, J. Biol. Chem., 271:12687−12690(1996);Wileyら, Immunity, 3:673−682(1995);Browning等, Cell, 72:847−856 (1993);Armitage等, Nature, 357:80−82 (1992)、1997年1月16日に公開された国際公開第97/01633号;1997年7月17日に公開された国際公開第97/25428号;Marstersら, Curr. Biol., 8:525−528(1998);Simonetら, Cell, 89:309−319(1997);Chicheporticheら, Biol. Chem., 272:32401−32410(1997);Hahneら, J. Exp. Med., 188:1185−1190(1998);1998年7月2日に公開された国際公開第98/28426号;1998年10月22日に公開された国際公開第98/46751号;1998年5月7日に公開された国際公開第98/18921号;Mooreら, Science, 285:260−263(1999);Shuら, J. Leukocyte Biol., 65:680(1999);Schneiderら, J. Exp. Med., 189:1747−1756(1999);Mukhopadhyayら, J. Biol. Chem., 274:15978−15981(1999)を参照のこと]。これらの分子のなかでも、TNF−α、TNF−β、CD30リガンド、4−1BBリガンド、Apo−1リガンド、Apo−2リガンド(Apo2L/TRAIL)及びApo−3リガンド(TWEAK)は、アポトーシス性細胞死に関与していることが報告されている。TNF−αとTNF−βの両方とも、感受性腫瘍細胞におけるアポトーシス性死を誘発することが報告されている[Schmidら, Proc. Natl. Acad. Sci., 83:1881(1986);Dealtryら, Eur. J. Immunol., 17:689(1987)]。
【0003】
最近、TNFサイトカインファミリーのメンバーであると考えられる更なる分子が同定され、アポトーシスに関与することが報告されている。例えば、Pittiら, J. Biol. Chem., 271:12687−12690(1996)において、Apo−2リガンドと呼ばれる分子が記載されている。1997年7月17日公開の国際公開第97/25428号をまた参照されたい。全長ヒトApo−2リガンドは様々な哺乳動物細胞においてアポトーシスを誘発する281のアミノ酸ポリペプチドであると報告されている。他の研究者はTRAIL[Wileyら, Immunity, 3:673−682(1995);1997年1月16日公開の国際公開第97/01633号]及びAGP−1[1997年12月11日公開の国際公開第97/46686号]と呼ばれる関連したポリペプチドを開示している。
【0004】
また、TNFファミリーの様々な分子が免疫システムの発達又は機能の役割を目的としている[Grussら, Blood, 85:3378(1995)]。Zheng等は、TNF−αがCD8ポジティブT細胞の刺激後アポトーシスに関与していることを報告している[Zheng等, Nature, 377:348−351(1995)]。他の研究者は、CD30リガンドが胸腺における自己反応性T細胞の欠失に関与していることを報告している[Amakawa等, プログラム細胞死に関するコールドスプリングハーバー研究所のシンポジウム、要約集、第10巻、(1995)]。CD40リガンドは、増殖、免疫グロブリン分泌、及び生存を含むB細胞の多くの機能を活性化する[Renshaw等, J. Exp. Med.,180:1889(1994)]。他の最近同定されたTNFファミリーサイトカイン、TALL−1(BlyS)は、ある条件下において、B細胞増殖及び免疫グロブリン分泌を誘発することが報告されている[Moore等, 上掲;Schneider等, 上掲;Mackay等, J. Exp. Med., 190:1697(1999)]。
【0005】
マウスのFas/Apo−1レセプター又はリガンド遺伝子(それぞれlpr及びgldと呼称される)における変異が幾つかの自己免疫疾患に関連しており、Apo−1リガンドが末梢の自己反応性リンパ球のクローン除去を調節する役割を担っていることを示している[Krammer等, Curr. Op. Immunol.,6:279−289(1994);Nagata等, Science, 267:1449−1456(1995)]。また、Apo−1リガンドは、CD4ポジティブTリンパ球及びBリンパ球においてポスト刺激性アポトーシスを誘発することが報告されており、それらの機能がもはや必要でなくなった際の活性化リンパ球の除去に関与している[Krammer等, 上掲;Nagata等, 上掲]。Apo−1レセプターと特異的に結合するアゴニストのマウスモノクローナル抗体は、TNF−αに匹敵するか類似する細胞死滅活性を示すことが報告されている[Yonehara等, J. Exp. Med., 169:1747−1756(1989)]。
このようなTNFファミリーのサイトカインが介在する種々の細胞反応誘導は、特異的な細胞レセプターに結合することにより開始されると考えられている。以前、約55−kDa(TNFR1)と75−kDa(TNFR2)の2つの異なるTNFレセプターが同定されている[Hohman等, J. Biol. Chem., 264:14927−14934(1989);Brockhaus等, Proc. Natl. Acad. Sci., 87:3127−3131(1990);1991年3月20日に公開された欧州特許第417563号;Loetscher等, Cell, 61:351(1990);Schall等, Cell, 61:361(1990);Smith等, Science, 248:1019−1023(1990);Lewis等, Proc. Natl. Acad. Sci., 88:2830−2834(1991);Goodwin等, Mol. Cell. Biol., 11:3020−3026(1991)]。これらのTNFRは細胞外、膜貫通及び細胞内領域を含む細胞表面レセプターの典型的な構造を共有することが分かった。双方のレセプターの細胞外部分はまた可溶性TNF結合タンパク質として天然に見出される[Nophar, Y等, EMBO J., 9:3269(1990);及びKohno, T等, Proc. Natl. Acad. Sci. U.S.A., 87:8331(1990);Hale等, J. Cell. Biochem. 増補15F, 1991, p.113(P424)]。
【0006】
1型又は2型のTNFR(TNFR1及びTNFR2)の細胞外部分は、NH2−末端から出発して、1から4とされる4つのシステインに富んだドメイン(CRD)の反復アミノ酸配列パターンを含む[Schall等, 上掲;Loetscher等, 上掲;Smith等, 上掲;Nophar等, 上掲;Kohno等, 上掲;Banner等, Cell, 73:431−435(1993)]。CRDの類似の反復パターンが、p75神経成長因子レセプター(NGFR)[Johnson等, Cell, 47:545(1986);Radeke等, Nature, 325:593(1987)]、B細胞抗原CD40[Stamenkovic等, EMBO J., 8:1403(1989)]、T細胞抗原OX40[Mallet等, EMBO J., 9:1063(1990)]及びFas抗原[Yonehara等, 上掲、及びItoh等, Cell, 66:233−243(1991)]を含む幾つかの他の細胞表面タンパク質に存在している。また、CRDはショープ(Shope)及び粘液腫ポックスウィルスの可溶型TNFR(sTNFR)様のT2タンパク質にも見出されている[Upton等, Virology, 160:20−29(1987);Smith等, Biochem. Biophys. Res. Commun., 176:335(1991);Upton等, Virology, 184:370(1991)]。これらの配列の最適なアラインメントは、システイン残基の位置が良好に保存されていることを示している。これらレセプターは、しばしば集合的に、TNF/NGFレセプタースーパーファミリーのメンバーと称される。
リンホトキシン−αを除き、今日までに同定されているTNFファミリーのリガンドは、II型の膜貫通タンパク質であり、そのC末端は細胞外にある。これに対して、今日までに同定されているTNFレセプター(TNFR)ファミリーのほとんどのレセプター類はI型の膜貫通タンパク質である。しかしながら、TNFリガンド及びレセプターファミリーの双方において、ファミリーメンバー間で同定された相同性は、主として細胞外ドメイン(「ECD」)において見出されている。TNF−α、Apo−1リガンド及びCD40リガンドを含むTNFファミリーサイトカインのいくつかは、細胞表面においてタンパク分解的に切断され;各場合に得られたタンパク質は、典型的には、可溶性サイトカインとして機能するホモ三量体分子を形成する。また、TNFレセプターファミリーのタンパク質は、通常、タンパク分解的に切断され、同族のサイトカインの阻害剤として機能し得る可溶性レセプターのECDを放出する。
【0007】
より最近になって、TNFRファミリーの他のメンバーが同定されている。Bulow等によるScience, 278:138−141(1997)において、研究者は膜活性剤及びCAML−インタラクターと称される原形質膜レセプター又は「TACI」を記載している。TACIレセプターはTNFRファミリーのシステインリッチモチーフ特性を包含すると報告されている。インビトロアッセイにおいて、TACI−特異性抗体を有するトランスフェクトされたJurkat細胞の表面上でのTACIの架橋結合はNF−KBの活性化を引き起こす[また、1998年9月18日公開の国際公開第98/39361号を参照]。
Laabi等, EMBO J., 11:3897−3904(1992)は、その発現がB細胞末端成熟と一致することが見出されている「BCM」と呼ばれる新しい遺伝子の同定を報告した。BCM通常cDNAのオープンリーディングフレームは、単一膜貫通ドメインで184アミノ酸長ポリペプチドであると予測された。これらの研究者は後に、この遺伝子を「BCMA」と名付けた[Laabi等, Nucleic Acids Res., 22:1147−1154(1994)]。BCMAmRNA発現は、Bリンパ球段階前を表すヒト悪性B細胞株の非存在であり、よってリンパ球の分化の段階に関わっていると考えられることが報告された[Gras等, Int. Immunology, 7:1093−1106(1995)]。Madry等, Int. Immunology, 10:1693−1702(1998)において、マウスBCMAcDNAのクローニングが記載された。マウスBCMAcDNAは、ヒトBCMAポリペプチドに対して62%同一性を有する185アミノ酸長ポリペプチドをコードすることが報告された。マウス及びヒトBCMAタンパク質配列のアラインメントは、N末端領域で6システインの保存されたモチーフ、BCMAタンパク質はTNFRスーパーファミリーに属するという予測を示した[Madry等, 上掲]。
【0008】
Marsters等、Curr. Biol., 6:750(1996)において、研究者は、細胞外のシステインに富んだ反復についてTNFRファミリーに対して類似性を示し、細胞質死ドメイン配列を含む点でTNFR1及びCD95に似ており、Apo−3と称される、ヒトのポリペプチド天然配列の全長を開示している[Marsters等,Curr. Biol., 6:1669(1996)もまた参照されたい]。他の研究者によれば、Apo−3はDR3、wsl−1、TRAMP及びLARDとも称されている[Chinnaiyan等, Science, 274:990(1996);Kitson等, Nature, 384:372(1996);Bodmer等, Immunity, 6:79(1997);Screaton等, Proc. Natl. Acad. Sci., 94:4615−4619(1997)]。
パン(Pan)らは、「DR4」と称される他のTNFレセプターファミリーのメンバーを開示している[Pan等, Science, 276:111−113(1997);また、1998年7月30日公開の国際公開第98/32856号参照]。DR4は、細胞自殺機構に関与できる細胞質死ドメインを含むことが報告された。パン等は、DR4がApo2L/TRAILとして知られているリガンドに対するレセプターであると考えられることをさらに開示している。
【0009】
Sheridan等, Science, 277:818−821 (1997)及びパン等, Science, 277:815−818 (1997)においては、Apo2L/TRAILに対するレセプターと思われる他の分子が開示されている[1998年11月19日公開の国際公開第98/51793号;1998年9月24日公開の国際公開第98/41629号を参照のこと]。この分子は、DR5と称される(あるいは、Apo−2;TRAIL−R、TR6、Tango−63、hAPO8、TRICK2又はKILLERとも称される[Screaton等,Curr.Biol., 7:693−696(1997);Walczak等,EMBO J., 16:5386−5387(1997);Wu等,Nature Genetics, 17:141−143(1997);1998年8月20日に公開の国際公開第98/35986号;1998年10月14日に公開の欧州特許第870,827号;1998年10月22日に公開の国際公開第98/46643号;1999年1月21日に公開の国際公開第99/02653号;1999年2月25日に公開の国際公開第99/09165号;1999年3月11日に公開の国際公開第99/11791号]。DR4のように、DR5は細胞質死ドメインを含み、アポトーシスのシグナル伝達が可能であると報告されている。Apo−2L/TRAIL及びDR5の間で形成された複合体の結晶構造は、Hymowitzら,Molecular Cell, 4:563−571(1999)に開示されている。
【0010】
さらに他の死ドメインを含むレセプターであるDR6が最近同定された[Pan等, FEBS Letters, 431:351−356(1998)]。4つの推定の細胞外システインリッチドメイン及び細胞質死ドメインの他に、DR6は細胞質領域におけるプロリンに富む領域とオーバーラップするような予想されるロイシンジッパー配列を含むと考えられている。プロリンに富むモチーフはsrc相同性−3ドメインと結合する配列に類似しており、それは多くの細胞内シグナル伝達分子に見出されるものである。
最近同定されたレセプターのさらなるグループは、「デコイレセプター」と称され、シグナル伝達分子というよりはむしろ、阻害剤として機能すると考えられている。このグループには、DCR1(TRID,LIT,又はTRAIL−R3とも称される)[Pan等, Science, 276:111−113(1997);Sheridan等, Science, 277:818−821 (1997);McFarlane等, J. Biol. Chem., 272:25417−25420(1997);Schneider等, FEBS Letters, 416:329−334(1997);Degli−Esposti等, J. Exp. Med., 186:1165−1170(1997);及びMongkolsapaya等, J. Immunol., 160:3−6(1998)]及びDCR2(TRUNDD,又はTRAIL−R4とも称される)[Marsters等, Curr. Biol., 7:1003−1006(1997);Pan等, FEBS Letters, 424:41−45(1998);Degli−Esposti等, Immunity, 7:813−820(1997)]が含まれ、両者とも細胞表面分子であり、更にOPG[Simonet等, 上掲;Emery等, 下記]及びDCR3[Pitti等, Nature, 396:699−703(1998)]も含まれ、これら両者は分泌性の可溶性タンパク質である。
【0011】
最近になって、TNFRファミリーの他の同定されているメンバーには、CAR1、HVEM、GITR、ZTNFR−5、NTR−1、及びTNFL1が含まれる[Brojatsch等, Cell, 87:845−855 (1996);Montgomery等, Cell, 87:427−436 (1996);Marsters等, J.Biol.Chem., 272:14029−14032 (1997);Nocentini等, Proc. Natl. Acad. Sci. USA 94:6216−6221(1997);Emery等, J. Biol. Chem., 273:14363−14367(1998);1999年1月28日公開の国際公開第99/04001号;1999年2月18日公開の国際公開第99/07738号;1999年7月8日公開の国際公開第99/33980号]。
Tewariらにより最近概説されているように、TNFR1、TNFR2及びCD40は、転写因子、NF−KBの活性化を通して、炎症誘発性及び同時刺激性サイトカイン、サイトカインレセプター、及び細胞接着分子の発現を変調する[Tewari等, Curr. Op. Genet. Develop., 6:39−44(1996)]。NF−KBは、そのサブユニットが保存Rel領域を含有する二量体転写因子のファミリーの原型である[Verma等, Genes Develop., 9:2723−2735(1996);Baldwin, Ann. Rev. Immunol., 14:649−681(1996)]。その潜伏形態において、NF−KBはIKB阻害剤ファミリーのメンバーと複合化しており;所定の刺激に反応してIKBの不活性化の際に、放出されたNF−KBが、特異的DNA配列と結合する核に転座して遺伝子転写を活性化する。前記したように、今日までに同定されたTNFRメンバーは細胞内死ドメイン領域を含むか、又は欠いている。例えばTNFR2、CD40、HVEM、及びGITRのような、死のドメインを欠くいくつかのTNFR分子は、NF−KB活性の調節の能力がある[例えばLotz等, J. Leukocyte Biol., 60:1−7(1996)参照]。
サイトカインのTNFファミリー及びそれらのレセプター類の概説については、Ashkenazi及びDixit, Science, 281:1305−1308(1998);Golstein, Curr. Biol., 7:750−753(1997);Gruss及びDower, 上掲、及びNagata, Cell, 88:355−365(1997)を参照されたい。
【0012】
(発明の概要)
出願人は、Apo−2リガンド又は他のApo−2LレセプターアゴニストとCPT−11が、哺乳動物細胞、特に哺乳動物癌細胞においてアポトーシスの誘発に相乗的に作用することができることを見出した。
本発明は、哺乳動物細胞においてアポトーシスを誘発するのためのApo−2リガンド及びCPT−11の様々な使用方法を提供する。例えば、本発明は、哺乳動物細胞、例えば癌細胞を、CPT−11及び一又は複数のApo−2リガンドレセプターアゴニストにさらすことを含んでなるアポトーシスを誘発する方法を提供し、CPT−11はApo−2リガンドレセプターアゴニストの前に投与されて、細胞を前処理する。
細胞は、細胞培地中又は哺乳動物、例えば細胞のアポトーシスの誘発が所望される症状又は癌に罹っている哺乳動物中にあってもよい。従って、本発明はここで開示されるように、Apo−2リガンド及びCPT−11の有効量を投与することを含んでなる癌を罹っている哺乳動物を治療する方法を含む。
【0013】
場合によっては、本方法はApo−2リガンドのアポトーシス活性に似たアゴニストの抗−Apo−2リガンドレセプター抗体を使用しうる。よって、本発明は哺乳動物細胞のアポトーシスを誘発するためのApo−2リガンドレセプターアゴニスト抗体及びCPT−11の様々な使用方法を提供する。好ましい実施態様では、アゴニスト抗体は、DR4又はDR5レセプターに対するモノクローナル抗体を含むであろう。
また任意の実施態様では、哺乳動物癌細胞を、有効量のCPT−11及びApo−2リガンドレセプターアゴニストにさらすことを含む、哺乳動物癌細胞のアポトーシスを高める方法を提供することにあり、ここで、該哺乳動物癌細胞を、該Apo−2リガンドレセプターアゴニストにさらす前に、約6〜72時間CPT−11にさらす。本方法は、有効量のCPT−11に前記哺乳動物癌細胞を暴露させることを含み、前記細胞においてDR4レセプター又はDR5レセプターのアップレギュレーションを誘導する。場合によっては、哺乳動物癌細胞を、前記Apo−2リガンドレセプターアゴニストにさらす前に、約24〜48時間CPT−11にさらす。Apo−2リガンドレセプターアゴニストは、Apo2Lポリペプチド又は抗−DR4レセプター抗体又は抗−DR5レセプター抗体を含んでいてもよい。
【0014】
さらに、任意の実施態様では、有効量のCPT−11及びApo−2リガンドレセプターアゴニストを癌を患っている哺乳動物に投与することを含んでなる、哺乳動物の癌を治療する方法を提供し、ここで該CPT−11は、Apo−2リガンドレセプターアゴニストを投与する前の約6〜72時間に投与される。場合によっては、Apo−2リガンドレセプターアゴニストは、Apo2Lポリペプチド、抗−DR4レセプター抗体又は抗−DR5レセプター抗体を含む。
本発明はまた、Apo−2リガンド又はApo−2Lレセプターアゴニスト抗体及び/又はCPT−11を含んでなる組成物を提供する。場合によっては、本発明の組成物は、製薬的に許容可能な担体又は希釈剤を含みうる。好ましくは、該組成物は、哺乳動物細胞のアポトーシスを相乗的に誘発する効果的な量のApo−2リガンド又はアゴニスト抗体及び/又はCRT−11を含みうる。
本発明はまた、Apo−2リガンド又はApo−2Lレセプターアゴニスト抗体及び/又はCPT−11を含む製造品又はキットを提供する。
【0015】
(発明の詳細な説明)
I.定義
「アポトーシス」及び「アポトーシス活性」という用語は広義に使用され、典型的には、細胞質の凝集、原形質膜の微絨毛の喪失、核の分節化、染色体DNAの分解又はミトコンドリア機能の喪失を含む一又は複数の特徴的な細胞変化を伴う、哺乳動物における細胞死の規則的又はコントロールされた形態を称す。この活性は、当該技術で公知の技術、例えば細胞生死判別アッセイ、FACS分析又はDNA電気泳動法、そしてより詳細にはアネキシンVの結合、DNAの断片化、PARP切断、細胞収縮、小胞体の拡大、細胞断片化、及び/又は膜ベジクル(アポトーシス体と呼ばれる)の生成により、決定し測定することができる。これらの技術及びアッセイは、当該分野において、例えば国際公開第97/25428号及び国際公開第97/01633号に記載されている。場合によっては、アポトーシス活性は実施例に記載されているアッセイを使用して測定することもできる。
ここで使用される場合、「相乗」又は「相乗効果」又は「相乗的に」という用語は、組み合わせた効果がその個々の効果の合計よりもより大きいような2又はそれ以上の薬剤の相互作用を称する。
【0016】
「Apo−2リガンド」、「Apo−2L」又は「TRAIL」という用語は、Pitti等, J. Biol. Chem., 271:12687−12690 (1996)の図1Aに示されたアミノ酸配列のアミノ酸残基95−281、114−281、残基91−281、残基92−281、残基41−281、残基15−281、又は残基1−281(ここでは、配列リストで配列番号:1として提供されている)、並びに上記配列の生物活性な(例えばアポトーシス活性)断片、欠失、挿入又は置換変異体を含むポリペプチドを称する。一実施態様において、ポリペプチド配列は、配列番号:1の残基114−281を含む。場合によっては、ポリペプチド配列は、配列番号:1の少なくとも残基91−281又は残基92−281を有する。他の好適な実施態様では、生物活性な断片又は変異体は、上記配列の何れかと少なくとも約80%のアミノ酸配列同一性、より好ましくは少なくとも90%のアミノ酸配列同一性、そして更により好ましくは少なくとも約95%、96%、97%、98%、又は99%のアミノ酸配列同一性を有する。本定義は、(Pitti等, 上掲に提供される配列(配列番号:1)の番号を用いて)位置203、218又は269で少なくとも1のアミノ酸がアラニン残基によって置換されて、Pitti等, J. Biol. Chem., 271:12687−12690(1996)の図1Aのアミノ酸91−281を含んでなるApo−2リガンドの置換変異体を包含する。本定義は、例えばヒト組織型又は他の供給源のようなApo−2リガンド供給源から単離されるか、組み換え又は合成法により調製されたApo−2リガンドを包含する。Apo−2リガンドという用語はまた上掲の国際公開第97/25428号及び上掲の国際公開第97/01633号に記載されたポリペプチドを称する。Apo−2リガンドポリペプチドはポリエチレングリコールのような一又は複数のポリマー分子に結合しうるものと考えられる。
【0017】
「CPT−11」という用語は一般的な意味で使用され、トポイソメラーゼI阻害剤クラスの化学療法又は化学療法剤を称する。ここで使用される「CPT−11」という用語は、化学名(4S)−4、11−ジエチル−4−ヒドロキシ−9−[(4−ピペリジノ−ピペリジノ)カルボニルオキシル]−1H−ピラノ[3’,4’:6,7]インドリジノ[1,2−b]キノリン−3,14(4H,12H)ジオン塩酸塩三水和物、及び名称イリノテカン、カンプトセシン、トポテカン、又はCamptosar(登録商標)(商品名)、同様にその水溶性誘導体又はこのような薬剤の製薬的に許容可能な塩を有する化学療法剤を含む。イリノテカン塩酸塩は、実験式C33H38N4O6 *HCI*3H2O及び分子量約677.19を有する。このような化学名及び化学式は、当業者によく知られている。Camptosar(商品名)は、Pharmacia & Upjohnより商業的に入手可能で、FDAにより米国での販売が許可されている。該分子を表示するCamptosar(商品名)の製品挿入物は、5−FUに基づく治療に続いて、病気が再発又は進行した転移性直腸結腸癌を有するヒト患者の治療に使用することができる。CPT−11は、ポリエチレングリコール等の一又は複数のポリマー分子に結合可能であると考えられている。
【0018】
ここで同定されているApo−2Lポリペプチド配列に対する「パーセント(%)アミノ酸配列同一性」は、配列を整列させ、最大のパーセント配列同一性を得るために必要ならば間隙を導入し、如何なる保存的置換も配列同一性の一部と考えないとした、Apo−2L配列のアミノ酸残基と同一である候補配列中のアミノ酸残基のパーセントとして定義される。パーセント核酸配列同一性を決定する目的のためのアラインメントは、当業者の知る範囲にある種々の方法、例えばBLAST、BLAST−2、ALIGN、ALIGN−2又はMegalign(DNASTAR)ソフトウエアのような公に入手可能なコンピュータソフトウエアを使用することにより達成可能である。当業者であれば、比較される配列の全長に対して最大のアラインメントを達成するために必要な任意のアルゴリズムを含む、アラインメントを測定するための適切なパラメータを決定することができる。場合によっては、%アミノ酸配列同一性値は、配列比較コンピュータプログラムALIGN−2を用いて得られる。ALIGN−2配列比較コンピュータプログラムはジェネンテク社によって作成され、ソースコードは米国著作権庁, Washington D.C., 20559に使用者用書類とともに提出され、米国著作権登録番号TXU510087の下で登録されている。ALIGN−2プログラムはジェネンテク社、South San Francisco, Californiaを通して公的に入手可能である。ALIGN−2プログラムは、UNIXオペレーティングシステム、好ましくはデジタルUNIX V4.0Dでの使用のためにコンパイルされる。全ての配列比較パラメータは、ALIGN−2プログラムによって設定され変動しない。しかしながら、%アミノ酸配列同一性は、配列比較プログラムNCBI−BLAST2(Altschul等, Nucleic Acids Res. 25:3389−3402 (1997))を用いて決定してもよい。NCBI−BLAST2配列比較プログラムは、http://ww.ncbi.nlm.nih.govからダウンロードできる。NCBI−BLAST2は幾つかの検索パラメータを使用し、それら検索パラメータの全てはデフォルト値に設定され、例えば、unmask=可、鎖=全て、予測される発生=10、最小低複合長=15/5、マルチパスe−値=0.01、マルチパスの定数=25、最終ギャップアラインメントのドロップオフ=25、及びスコアリングマトリクス=BLOSUM62を含む。
【0019】
「アゴニストの抗−Apo−2リガンドレセプター抗体」に関して使用される「抗体」という用語は広義に使用され、特に無傷のモノクローナル抗体、ポリクローナル抗体、少なくとも2つの無傷の抗体から形成される多重特異性抗体(例えば二重特異性抗体)、及びそれらが一又は複数のApo−2リガンドと結合し、及び/又は一又は複数のApo−2リガンドレセプターを発現している哺乳動物細胞のアポトーシスシグナル伝達経路を活性化させるか、又はApo−2リガンドのアポトーシス活性を模倣する(例えば、それに匹敵するもしくは少なくとも等しい)か、又はApo−2よりも大きなアポトーシス活性を有する限り抗体断片をカバーする。
【0020】
「Apo−2リガンドレセプター」は、当該分野において「DR4」及び「DR5」と称されるレセプターを含む。Pan等は、「DR4」と称される他のTNFレセプターファミリーのメンバーを開示している[Pan等, Science, 276:111−113(1997);また1998年7月30日公開の国際公開第98/32856号を参照]。DR4レセプターは、細胞自殺機構に関わることが可能な細胞質死ドメインを含むことが報告されている。Pan等は、DR4がApo2L/TRAILとして知られるリガンドのレセプターであると考えられると開示している。全長DR4レセプターのアミノ酸配列は、ここでは配列番号:2として提供されている。Sheridan等, Science, 277:818−821 (1997)及びPan等, Science, 277:815−818 (1997)においては、Apo−2L/TRAILの他のレセプターが記載されている。[1998年11月19日発行の国際公開第98/51793号;1998年9月24日発行の国際公開第98/41629号を参照のこと]。このレセプターはDR5とも呼ばれる。(該レセプターはまた、Apo−2;TRAIL−R、TR6、Tango−63、hAPO8、TRICK2又はKILLERとも呼ばれる;Screaton等,Curr.Biol., 7:693−696(1997);Walczak等,EMBO J., 16:5386−5387(1997);Wu等,Nature Genetics, 17:141−143(1997);1998年8月20日に公開の国際公開第98/35986号(米国特許第6,072,047号に対応);1998年10月14日に公開の欧州特許第870,827号;1998年10月22日に公開の国際公開第98/46643号;1999年1月21日に公開の国際公開第99/02653号;1999年2月25日に公開の国際公開第99/09165号;1999年3月11日に公開の国際公開第99/11791号]。DR4のように、DR5は細胞質死ドメインを含み、アポトーシスのシグナル伝達が可能であると報告されている。国際公開第98/35986号(米国特許第6,072,047号に対応)の全長DR5レセプター配列は、440のアミノ酸のポリペプチドであることが報告されており、そのアミノ酸配列は配列番号:3で提供されている。国際公開第98/51793号の全長DR5レセプター配列は、441のアミノ酸のポリペプチドであることが報告されており、そのアミノ酸配列は配列番号:4で提供されている。前記されるように、Apo−2Lに対する他のレセプターはDcR1、DcR2、及びOPGを含む[Sheridan等、上掲;Marsters等、上掲;及びSimonet等、上掲を参照のこと]。ここで使用される場合の「Apo−2Lレセプター」という用語は天然配列レセプター及びレセプター変異体を含む。これらの用語は、ヒトを含む様々な動物で発現されるApo−2Lレセプターを含む。Apo−2Lレセプターは、様々なヒト組織系統で自然に発生して内因的に発現されても、あるいは組換え又は合成方法によって発現されてもよい。「天然配列Apo−2Lレセプター」は、自然から誘導したApo−2Lレセプターと同じアミノ酸配列を有するポリペプチドを含む。よって、天然配列Apo−2Lレセプターは、任意の動物から自然に発生したApo−2Lレセプターのアミノ酸配列を有することができる。このような天然配列Apo−2Lレセプターは、自然から単離することができ、あるいは組換え又は合成法によって作成することもできる。「天然配列Apo−2Lレセプター」なる用語は、特にレセプターの自然に発生する切断又は分泌形態(例えば、細胞外ドメイン配列を含む可溶化形態など)、自然に発生する変異形態(例えば選択的にスプライシングされた形態)及び自然に発生する対立遺伝子変異体を含む。レセプター変異体は天然配列Apo−2Lレセプターの断片又は欠失突然変異を含みうる。
【0021】
ここで使用される「モノクローナル抗体」という用語は、実質的に均一な抗体の集団から得られる抗体を称する、すなわち、集団に含まれる個々の抗体が、少量存在しうる自然に生じる可能な突然変異を除いて同一である。モノクローナル抗体は高度に特異的であり、一つの抗原部位に指向する。さらに、異なる決定基(エピトープ)に対する異なる抗体を典型的に含む従来の(ポリクローナル)抗体調製物とは反対に、各モノクローナル抗体は抗原の単一の決定基に指向する。それらの特性に加えて、モノクローナル抗体は、ハイブリドーマ培地で合成され、他の免疫グロブリンで汚染されていないという点で有利である。「モノクローナル」との修飾は、実質的に均一な抗体集団から得られたという抗体の特徴を示し、抗体を何か特定の方法で生産しなければならないと解釈されるものではない。例えば、本発明に従って使用されるモノクローナル抗体は、最初にKohler等, Nature, 256:495 (1975)に記載されたハイブリドーマ法によって作ることができ、あるいは組換えDNA法によって作ることができる(例えば米国特許第4,816,567号参照)。「モノクローナル抗体」は、また、Clackson等, Nature, 352:624−628(1991)及びMarks等, J. Mol.Biol., 222:581−597(1991)に記載された技術を用いてファージ抗体ライブラリから単離することができる。
ここで、モノクローナル抗体は特に、「キメラ」抗体(免疫グロブリン)を含み、それは、重鎖又は軽鎖の一部が特定の種から誘導された又は特定の抗体クラス又はサブクラスに属する抗体の対応する配列と同一又は相同であるが、鎖の残りの部分は他の種から誘導された又は他の抗体クラス又はサブクラスに属する抗体の対応する配列と同一又は相同である抗体、並びにそれらが所望の生物学的活性を示す限りにおいてそれらの抗体の断片である(米国特許第4,816,567号;Morrison等, Proc. Natl. Acad. Sci. USA, 81:6851−6855 (1984))。
【0022】
非ヒト(例えばマウス)抗体の「ヒト化」形とは、キメラ免疫グロブリン、免疫グロブリン鎖、あるいはそれらの断片(例えばFv、Fab、Fab’、F(ab’)2あるいは抗体の他の抗原結合サブ配列)であって、非ヒト免疫グロブリンに由来する最小配列を含むものである。大部分において、ヒト化抗体はレシピエントの相補性決定領域(CDR)の残基が、マウス、ラット又はウサギのような所望の特異性、親和性及び能力を有する非ヒト(ドナー抗体)のCDRの残基によって置換されたヒト免疫グロブリン(レシピエント抗体)である。ある場合には、ヒト免疫グロブリンのFvフレームワーク領域(FR)残基は、対応する非ヒト残基によって置換されている。更に、ヒト化抗体は、レシピエント抗体にも、移入されたCDRもしくはフレームワーク配列にも見出されない残基を含んでもよい。これらの修飾は抗体の特性を更に洗練し、最適化するために行われる。一般に、ヒト化抗体は、全てあるいはほとんど全てのCDR領域が非ヒト免疫グロブリンのものに対応し、全てあるいはほとんど全てのFR領域がヒト免疫グロブリン配列のものである、少なくとも1つ、典型的には2つの可変ドメインの実質的に全てを含む。ヒト化抗体は、最適には免疫グロブリン定常領域(Fc)、典型的にはヒトの免疫グロブリンの定常領域の少なくとも一部を含んでなる。更なる詳細については、Jones等, Nature, 321:522−525(1986);Reichmann等, Nature, 332:323−329(1988);及びPresta, Curr. Op. Struct. Biol., 2:593−596(1992)参照。ヒト化抗体はPRIMATIZEDTMを含み、抗体の抗原結合領域が、関心ある抗原でマカク属サルに免疫性を与えることで生成される抗体から得られる。
【0023】
抗体は、典型的には、特定の抗原に結合特異性を示すタンパク質又はポリペプチドである。天然抗体は、通常はヘテロ四量体糖タンパク質であり、2つの同一な軽(L)鎖と2つの同一な重(H)鎖とからなる。典型的には、各軽鎖は共有ジスルフィド結合によって重鎖に結合しているが、ジスルフィド結合の数は異なる免疫グロブリンアイソタイプの重鎖の間で変化する。また各重鎖及び軽鎖は、規則的に離間した鎖間ジスルフィド架橋を有する。各重鎖は、一端に多数の定常ドメインに続く可変ドメイン(VH)を持つ。各軽鎖は、一端に可変ドメイン(VL)を、他端に定常ドメインを有し;軽鎖の定常ドメインは重鎖の第1の定常ドメインと整列し、軽鎖の可変ドメインは重鎖の可変ドメインと整列している。特定のアミノ酸残基が軽鎖と重鎖の可変ドメイン間の界面を形成すると考えられている[Chothia等, J. Mol. Biol., 186:651−663 (1985);Novotny及びHaber, Proc. Natl. Acad. Sci. USA. 82:4592−4596 (1985)]。任意の脊髄動物種からの抗体の軽鎖は、それらの定常ドメインのアミノ酸配列に基づいて、カッパ及びラムダと呼ばれる2つの明確に区別される型に割り当てることができる。それらの重鎖の定常ドメインのアミノ酸配列に応じて、免疫グロブリンは異なるクラスに割り当てられる。免疫グロブリンには5つの大きなクラス:IgA、IgD、IgE、IgG及びIgMがあり、そのうちの幾つかは、サブクラス(アイソタイプ)、例えば、IgG−1、IgG−2、IgG−3、及びIgG−4;IgA−1及びIgA−2に更に分けることができる。免疫グロブリンの異なるクラスに対応する重鎖定常ドメインは、各々、アルファ、デルタ、イプシロン、ガンマ、及びミューと呼ばれる。
【0024】
「抗体断片」は無傷の抗体の一部、一般的には無傷の抗体の抗原結合性又は可変領域を含む。抗体断片の例は、Fab、Fab’、F(ab’)2及びFv断片、ダイアボディ(diabodies)、一本鎖抗体分子、及び抗体断片から形成された多重特異性抗体を含む。
ここで「可変」という用語は、抗体間で配列が異なり各特定の抗体のその抗原に対する結合性及び特異性の使用される可変ドメインの或る一部を記述するのに用いられる。しかしながら、可変性は抗体の可変ドメインを通して常に均一に分布しているわけではない。典型的には、軽鎖と重鎖の両方の可変ドメインにおいて相補性決定領域(CDR)又は高頻度可変領域と呼ばれる3つのセグメントに集中している。可変ドメインのより高度に保存される部分はフレームワーク(FR)と呼ばれる。天然重鎖及び軽鎖の可変ドメインは各々4つのFR領域を含み、これは大きくβ−シート配置をとり、3つのCDRに結合してループ状結合を形成するが、β−シート構造の一部を形成する場合もある。各鎖のCDRは、FR領域の直近に保持され、他の鎖からのCDRとともに抗体の抗原結合部位の形成に寄与する[Kabat, E.A.等, Sequences of Proteins of Immunological Interest, National Institutes of Health, Bethesda, MD (1987)参照]。定常ドメインは抗体の抗原への結合には直接含まれないが、抗体依存的細胞毒性における抗体の寄与といった様々なエフェクター機能を示す。
ここで、モノクローナル抗体は、起源の種又は免疫グロブリンクラス又はサブクラスの命名にかかわらず、キメラ、定常ドメインを有する抗−Apo−2Lレセプター抗体の可変(高頻度可変を含む)ドメインをスプライシングすることによって生産されるハイブリッド及び組換え抗体(例えば「ヒト化」抗体)、又は重鎖を有する軽鎖、又は他の種からの鎖を有するある種からの鎖、又は異種タンパク質との融合体、並びにそれが所望の生物的活性を有する限り抗体断片(例えばFab、F(ab’)2及びFv)を特に含む。例えば、米国特許第4,816,567号、及びMage等, Monoclonal Antibody Production Techniques and Applications, pp.79−97(Marcel Dekker, Inc.:ニューヨーク, 1987)を参照。
【0025】
「ヒト抗体」は、ヒトによって生産される抗体のアミノ酸配列に一致するアミノ酸配列を有するもの、及び/又はここにおいて開示されたヒト抗体を製造するいずれかの技術を使用して製造されたものである。ヒト抗体この定義は、特に非ヒト抗原結合残基を含んでなるヒト化抗体を除く。ヒト抗体は、この分野で知られている種々の技術を使用することによって生産することが可能である。一実施態様では、ヒト抗体は、ヒト抗体を発現するファージライブラリーから選択される(Vaughan等, Nature Biotechnology 14:309−314 (1996):Sheetsら. PNAS, (USA)95:6157−6162(1998);Hoogenboom及びWinter, J. Mol. Biol., 227:381 (1991);Marks等, J. Mol. Biol., 222:581 (1991))。また、ヒト抗体は、ヒト免疫グロブリン座位をトランスジェニック動物、例えば内在性免疫グロブリン遺伝子が部分的又は完全に不活性化されたマウスに導入することにより産生することができる。この試みでは、遺伝子再構成、構築及び抗体レパートリーを含む、あらゆる点でヒトに見られるものと密接に類似しているヒト抗体の産生が観察される。このアプローチ法は、例えば米国特許第5,545,807号;第5,545,806号;第5,569,825号;第5,625,126号;第5,633,425号;第5,661,016号、及び次の科学文献:Marks等, Bio/Technology 10:779−783 (1992);Lonberg等, Nature 368:856−859 (1994);Morrison等, Nature 368:812−13 (1994);Fishwild等, Nature Biotechnology 14:845−51 (1996);Neuberger, Nature Biotechnology 14:826 (1996);Lonberg及びHuszar, Intern. Rev. Immunol. 13:65−93 (1995)。あるいは、ヒト抗体は、標的抗原に対する抗体を生産するヒトBリンパ球の固定化によって調製されてもよい(そのようなBリンパ球は、個々から回収されてもよいし、インビトロで免疫化してもよい)。例えば、Cole等, Monoclonal Antibodies and Cancer Therapy, Alan R. Liss. p.77(1985)及びBoerner等, J. Immunol., 147(1):86−95(1991);及び米国特許第5,750,373号を参照のこと。
【0026】
「Fc領域」という用語は、無傷の抗体のパパイン消化で生じ得る免疫グロブリン重鎖のC末端領域を定義するために使用される。Fc領域は、天然配列Fc領域又は変異体Fc領域である。免疫グロブリン重鎖のFc領域の境界は変化するかも知れないが、ヒトIgG重鎖のFc領域は、通常、Cys226の位置のアミノ酸残基、又は約位置Pro230からFc領域のカルボキシル末端まで伸展すると定義される(ここにおいては、Kabatら,上掲の番号方式を使用)。免疫グロブリンのFc領域は、一般的に、二つの定常ドメイン、CH2ドメイン及びCH3ドメインを含み、場合によってはCH4ドメインを含む。
ここで「Fc領域鎖」とは、Fc領域の二つのポリペプチド鎖のうちの一つを意味する。
ヒトIgGFc領域の「CH2ドメイン」(「Cγ2」ドメインとも呼ばれる)は通常アミノ酸残基で約位置231からアミノ酸残基で約位置340まで伸展する。CH2ドメインは、他のドメインと密接には対をなさないという点で独特である。むしろ、2つのN−結合分岐炭水化物鎖が未変性の天然IgG分子の2つのCH2ドメインの間に介在されている。炭水化物はドメイン−ドメイン対形成に対する代替物を提供し、CH2ドメインを安定化させるのに役立つと推測されてきた。Burton, Molec. Immunol. 22:161−206 (1985)。ここにおけるCH2ドメインは、天然配列CH2ドメイン又は変異体CH2ドメインであってもよい。
「CH3ドメイン」は、残基C末端からFc領域のCH2ドメインへの伸長を含む(すなわち、IgGのアミノ酸残基で約位置341からアミノ酸残基で約位置447)。ここにおけるCH3領域は、天然配列CH3ドメイン又は変異体CH3ドメインでもよい(例えば、その一つの鎖の導入された「突出部(protroberance)」を伴うCH3ドメイン、及びその他の鎖の相当する導入された「空洞」;米国特許第5,821,333号を参照のこと)。
【0027】
「ヒンジ領域」は、ヒトIgG1の約Glu216、又は約Cys226から約Pro230の伸展として一般に定義されている(Burton, Molec. Immunol. 22:161−206 (1985))。他のIgGアイソタイプのヒンジ領域は、重鎖間S−S結合を形成する最初と最後のシステイン残基を同じ位置に配することにより、IgG1と整列させられうる。ここにおけるヒンジ領域は、天然配列ヒンジ領域又は変異体ヒンジ領域であってもよい。変異体ヒンジ領域の二つのポリペプチド鎖は、一般的に、ポリペプチド鎖当たり少なくとも一つのシステイン残基を保持し、変異体ヒンジ領域の二つのポリペプチド鎖は、二つの鎖の間でジスフィルド結合を形成できる。ここにおける好ましいヒンジ領域は、天然配列ヒトヒンジ領域で、例えば天然配列ヒトIgG1ヒンジ領域である。
「機能的Fc領域」は、天然配列Fc領域の少なくとも一つの「エフェクター機能」を有する。模範的な「エフェクター機能」は、C1q結合を含む;補体依存性細胞障害性(CDC);Fcレセプター結合;抗体依存性細胞媒介障害活性(ADCC);ファゴサイトーシス;細胞表層レセプターの下方制御(例えば、B細胞レセプター;BCR)等。このようなエフェクター機能は、一般的に、結合ドメインと結合するFc領域を必要とし(例えば、抗体可変ドメイン)、そのような抗体エフェクター機能を評価するための分野で知られた種々のアッセイを使用して評価できる。
「天然配列Fc領域」は、天然に見出されるFc領域のアミノ酸配列と同一のアミノ酸配列を含む。「変異体Fc領域」は、少なくとも一つのアミノ酸修飾によって天然配列Fc領域のアミノ酸配列とは異なるアミノ酸配列を含む。好ましくは、変異体Fc領域は、天然配列Fc領域又は親ポリペプチドのFc領域と比較し、少なくとも一つの一アミノ酸置換を有する、例えば、天然配列Fc領域又は親ポリペプチドのFc領域において、約1から約10アミノ酸置換、及び好ましくは約1から約5アミノ酸置換である。ここにおける変異体Fc領域は、好ましくは、天然配列Fc領域及び/又は親ポリペプチドのFc領域と少なくとも約80%配列同一性、最も好ましくは少なくとも約90%配列同一性、さらに好ましくは少なくとも約95%配列同一性を有する。
【0028】
「Fcレセプター」又は「FcR」という用語は、抗体のFc領域に結合するレセプターを記述するために使用される。好適なFcRは、天然配列ヒトFcRである。さらに好適なFcRは、IgG抗体(ガンマレセプター)に結合し、FcγRI、FcγRII及びFcγRIIIサブクラスのレセプターを含むものであり、これらのレセプターの対立遺伝子変異体及び選択的スプライシング型を含む。FcγRIIレセプターは、FcγRIIA(「活性化レセプター」)及びFcγRIIB(「阻害レセプター」)を含み、それらは、主としてその細胞質ドメインにおいて異なる類似のアミノ酸配列を有する。活性化レセプターFcγRIIAは、その細胞質ドメインに、免疫レセプターチロシン−ベース活性化モチーフ(ITIM)を有する。阻害レセプターFcγRIIBは、その細胞質ドメインに、免疫レセプターチロシン−ベース活性化モチーフ(ITAM)を有する(Daeron, Annu. Rev. Immunol., 15:203−234(1997)に概説されている)。FcRsはRavetch及びKinet, Annu. Rev. Immunol 9:457−92 (1991);Capel等, Immunomethods 4:25−34 (1994);及びde Haas等, J. Lab. Clin. Med. 126:330−41 (1995)において概説されている。将来同定されるものも含む他のFcRsが、ここにおける「FcR」なる用語によって包含される。この用語は胎児への母性IgGsの移動の原因である新生児レセプター、FcRnもまた含む(Guyer等, J. Immumol. 117:587 (1976)及びKim等, J. Immunol. 24:249 (1994))。
「親和性成熟」抗体とは、変化を有しない親抗体と比較し、抗原に対する抗体の親和性に改良を生じせしめる、抗体の一又は複数のCDRにおける一又は複数の変化を伴うものである。好ましい親和性成熟抗体は、標的抗原に対してナノモル又はピコモルの親和性を有する。親和性成熟抗体は、当該分野において知られる手順によって生産される。Marksら. Bio/Technology, 10:779−783(1992)は、VH及びVLドメイン混合による親和性成熟について記載している。CDR及び/又はフレームワーク残基のランダム突然変異誘発は、Barbas等, Proc Nat Acad. Sci, USA 91:3809−3813(1994);Schier 等, Gene, 169:147−155(1995);Yelton等, J. Immunol., 155:1994−2004(1995);Jackson等, J. Immunol., 154(7):3310−9(1995);及びHawkins等, J. Mol. Biol., 226:889−896(1992)に記載されている。
【0029】
ここで用いられる場合の「アゴニスト」及び「アゴニスト性」という用語は、直接的又は間接的に、実質的にApo−2リガンドに対するレセプターの生物学的活性又は活性化を含む、促進する又は向上させることのできる分子を称するか、又は記述する。場合によっては、「アゴニストApo−2Lレセプター抗体」は、Apo−2リガンドに類似する又は匹敵する活性を有する抗体である。好ましくは、アゴニストは哺乳動物細胞、好ましくは哺乳動物癌細胞のアポトーシスを含むことのできる分子である。さらに好ましくは、アゴニストはApo−2Lレセプターに対する抗体であり、該抗体は実施例1に記載されるApo−2Lポリペプチドよりも大きいか又は等しいアポトーシス活性を有する。場合によっては、このような分子のアゴニスト活性は、9D細胞あるいはDR4又はDR5のようなApo−2Lに対するレセプターを発現する他の細胞のアポトーシスを検査するための実施例2に記載されたアッセイにおいて、Fc免疫グロブリン又は補体(以下に記載)を用いて、分子、単独又は架橋結合した形態でアッセイすることにより決定される。アゴニストは一又は複数のポリマー分子、例えばポリエチレングリコールに結合すると考えられる。
【0030】
「単離された」とは、ここで開示された種々のタンパク質を記述するために使用するときは、その自然環境の成分から同定され分離され及び/又は回収されたタンパク質を意味する。その自然環境の汚染成分とは、タンパク質の診断又は治療への使用を典型的には妨害する物質であり、酵素、ホルモン、及び他のタンパク質様又は非タンパク質様溶質が含まれる。好ましい実施態様において、タンパク質は、(1)スピニングカップシークエネーターを使用することにより、N末端あるいは内部アミノ酸配列の少なくとも15残基を得るのに充分なほど、あるいは、(2)クーマシーブルーあるいは好ましくは銀染色を用いた非還元あるいは還元条件下でのSDS−PAGEによる均一性が得られるように充分なほど精製される。タンパク質の自然環境の少なくとも1つの成分が存在しないため、単離されたタンパク質には、組換え細胞内のインサイツのタンパク質が含まれる。しかしながら、通常は、単離されたタンパク質は少なくとも1つの精製工程により調製される。
ここでの目的における「生物学的に活性な」又は「生物学的活性」とは、(a)インビボ及び/又はエキソビボで、少なくとも一種類の哺乳動物細胞(例えば癌細胞)又はウイルス感染した細胞においてアポトーシスを誘発又は刺激する能力を有する;(b)抗体、例えば免疫原を産生する能力がある;又は(c)天然又は天然発生Apo−2リガンドポリペプチドの活性を保持していることを意味する。
【0031】
ここで使用される場合の「成長阻害剤」とは、インビトロ及び/又はインビボにおいて細胞の成長を阻害する化合物又は組成物のことを称する。従って、成長阻害剤には、S期の細胞の割合を著しく減少させるようなものでありうる。成長阻害剤の例には、細胞周期の進行をブロックする薬剤(S期以外の時点において)、例えばG1停止及びM期停止を誘発する薬剤が含まれる。典型的なM期ブロッカーには、ビンカス(ビンクリスチン及びビンブラスチン)、タキソール(登録商標)、トポIIインヒビター、例えばドキソルビシン、エピルビシン、ダウノルビシン、エトポシド、及びブレオマイシンが含まれる。また、G1を停止させるこれらの薬剤、例えばDNAアルキル化剤、例えばタモキシフェン、プレドニソン、ダカーバジン、メクロレタミン、シスプラチン、メトトレキセート、5−フルオロウラシル、及びara−CがS期停止に溢流する。更なる情報は、Murakamiらにより「細胞周期の調節、オンコジーン、及び抗新生物薬」と題された、癌の分子的基礎、Mendelsohn及びIsrael編、第1章(WB Saunders;Philadelphia, 1995)、特に13頁に見出すことができる。
【0032】
この出願で使用される「プロドラッグ」という用語は、親薬物に比べて、癌細胞に対する細胞毒性が低く、より活性な親形態に、酵素的に活性化又は転換され得る製薬的に活性な物質の先駆体又は誘導体形態を称する。例えば、Wilman, 「Prodrugs in Cancer Chemotherapy」, Biochemical Society Transactions, 14, pp.375−382, 615th Meeting Belfast(1986)及びStella等,「Prodrugs:A Chemical Approach to Targeted Drug Delivery」, Directed Drug Delivery, Borchardt等,(編), pp.247−267, Humana Press(1985)を参照。 限定するものではないが、本発明のプロドラッグには、ホスファート含有プロドラッグ、チオホスファート含有プロドラッグ、スルファート含有プロドラッグ、ペプチド含有プロドラッグ、D−アミノ酸変性プロドラッグ、グリコシル化プロドラッグ、βラクタム含有プロドラッグ、任意に置換されたフェノキシアセトアミド含有プロドラッグ又は任意に置換されたフェニルアセトアミド含有プロドラッグ、より活性のある細胞毒のない薬剤に転換可能な5−フルオロシトシン及び他の5−フルオロウリジンプロドラッグが含まれる。限定するものではないが、本発明で使用されるプロドラッグ形態に誘導体化可能な細胞障害剤の例には、下記の化学療法剤が含まれる。
ここで用いられる「細胞障害剤」という用語は、細胞の機能を阻害し又は妨害し、及び/又は細胞の破壊を引き起こす物質のことをいう。この用語は放射活性アイソトープ(例えばAt211,I131,I125,Y90,Re186,Re188,Sm153,Bi212,P32,及びLuの放射性アイソトープ)、化学療法剤、及び断片及び/又はその変異体を含む細菌性、真菌性、植物又は動物起源の酵素的に活性な毒素又は小分子毒素等の毒素を含むことが意図されている。
【0033】
「化学療法剤」は、癌のような症状の治療に有用な化学的化合物である。化学療法剤の例には、チオテパ及びシクロホスファミド(CYTOXAN(登録商標))のようなアルキル化剤;ブスルファン、インプロスルファン及びピポスルファンのようなスルホン酸アルキル類、;ベンゾドーパ(benzodopa)、カルボコン、メツレドーパ(meturedopa)、及びウレドーパ(uredopa)のようなアジリジン類;アルトレートアミン(altretamine)、トリエチレンメラミン、トリエチレンホスホラミド、トリエチレンチオホスホラミド(triethylenethiophosphaoramide)及びトリメチローロメラミン(trimethylolomelamine)を含むエチレンイミン類及びメチラメラミン類;アセトゲニン(acetogenins)(特にブラタシン(bullatacin)及びブラタシノン(bullatacinone));カンプトセシン(合成類似体トポテカン(topotecan)を含む);ブリオスタチン;カリスタチン(callystatin);CC−1065(そのアドゼレシン(adozelesin)、カルゼレシン(carzelesin)及びバイゼレシン(bizelesin)合成類似体を含む);クリプトフィシン(cryptophycin)(特にクリプトフィシン1及びクリプトフィシン8);ドラスタチン(dolastatin );デュカロマイシン(duocarmycin )(合成類似体、KW−2189及びCBI−TMIを含む); エレトロビン(eleutherobin);パンクラチスタチン(pancratistatin );サルコデイチン(sarcodictyin);スポンジスタチン(spongistatin );クロランブシル、クロロナファジン(chlornaphazine)、チョロホスファミド(cholophosphamide)、エストラムスチン、イホスファミド、メクロレタミン、メクロレタミンオキシドヒドロクロリド、メルファラン、ノベンビチン(novembichin)、フェネステリン(phenesterine)、プレドニムスチン(prednimustine)、トロフォスファミド(trofosfamide)、ウラシルマスタードなどのナイトロジェンマスタード;ニトロスレアス(nitrosureas)、例えばカルムスチン(carmustine)、クロロゾトシン(chlorozotocin)、フォテムスチン(fotemustine)、ロムスチン(lomustine)、ニムスチン、ラニムスチン;エネジイン(enediyne) 抗生物質等の抗生物質(例えば、カリケアマイシン(calicheamicin)、特にカリケアマイシンγ1 I及びカリケアマイシンθI 1、例えば、Agnew Chem Intl. Ed. Engl., 33:183−186(1994)を参照のこと;ダイネミシンA(dynemicinA)を含むダイネミシン(dynemicin);エスペラマイシン(esperamicin); 同様にネオカルチノスタチン発光団及び関連色素蛋白エネジイン(enediyne) 抗生物質発光団)、アクラシノマイシン(aclacinomysins)、アクチノマイシン、オースラマイシン(authramycin)、アザセリン、ブレオマイシン(bleomycins)、カクチノマイシン(cactinomycin)、カラビシン(carabicin)、カリミノマイシン(carminomycin)、カルジノフィリン(carzinophilin)、クロモマイシン、ダクチノマイシン、ダウノルビシン、デトロビシン(detorubicin)、6−ジアゾ−5−オキソ−L−ノルロイシン、ドキソルビシン(モルフォリノ−ドキソルビシン、シアノモルフォリノ−ドキソルビシン、2−ピロリノ−ドキソルビシン及びデオキシドキソルビシンを含む)、エピルビシン、エソルビシン(esorubicin)、イダルビシン、マセロマイシン(marcellomycin)、マイトマイシン(mitomycins)、マイコフェノール酸(mycophenolic acid)、ノガラマイシン(nogalamycin)、オリボマイシン(olivomycins)、ペプロマイシン、ポトフィロマイシン(potfiromycin)、ピューロマイシン、クエラマイシン(quelamycin)、ロドルビシン(rodorubicin)、ストレプトニグリン、ストレプトゾシン、ツベルシジン(tubercidin)、ウベニメクス、ジノスタチン(zinostatin)、ゾルビシン(zorubicin);メトトレキセート及び5−フルオロウラシル(5−FU)のような抗−代謝産物;デノプテリン(denopterin)、メトトレキセート、プテロプテリン(pteropterin)、トリメトレキセート(trimetrexate)のような葉酸類似体;フルダラビン(fludarabine)、6−メルカプトプリン、チアミプリン、チオグアニンのようなプリン類似体;アンシタビン、アザシチジン(azacitidine)、6−アザウリジン(azauridine)、カルモフール、シタラビン、ジデオキシウリジン、ドキシフルリジン、エノシタビン(enocitabine)、フロキシウリジン(floxuridine)、5−FUのようなピリミジン類似体;カルステロン(calusterone)、プロピオン酸ドロモスタノロン、エピチオスタノール、メピチオスタン、テストラクトン(testolactone)のようなアンドロゲン類;アミノグルテチミド、ミトタン、トリロスタンのような抗副腎剤;フロリン酸(frolinic acid)のような葉酸リプレニッシャー(replenisher);アセグラトン;アルドホスファミドグリコシド;アミノレブリン酸;アムサクリン(amsacrine);ベストラブシル(bestrabucil);ビサントレン(bisantrene);エダトラキセート(edatraxate);デフォファミン(defofamine);デメコルシン(demecolcine);ジアジコン(diaziquone);エルフォルニチン(elfornithine);酢酸エリプチニウム(elliptinium acetate);エポチロン(epothilone);エトグルシド(etoglucid);硝酸ガリウム;ヒドロキシ尿素;レンチナン;ロニダミン(lonidamine);メイタンシン(maytansine)及びアンサマイトシン(ansamitocin )のようなメイタンシノイド(maytansinoid);ミトグアゾン(mitoguazone);ミトキサントロン;モピダモール(mopidamol);ニトラクリン(nitracrine);ペントスタチン;フェナメット(phenamet);ピラルビシン;ポドフィリン酸(podophyllinic acid);2−エチルヒドラジド;プロカルバジン;PSK(登録商標);ラゾキサン(razoxane);リゾキシン(rhizoxin);シゾフィラン;スピロゲルマニウム(spirogermanium);テニュアゾン酸(tenuazonic acid);トリアジコン(triaziquone);2,2’,2’’−トリクロロトリエチルアミン;トリコテセン(trichothecenes)(特に、T−2トキシン、ベラキュリンA(verracurin A)、ロリデンA(roridin A)及びアングイデン(anguidine));ウレタン;ビンデシン;ダカルバジン;マンノムスチン(mannomustine);ミトブロニトール;ミトラクトール(mitolactol);ピポブロマン(pipobroman);ガシトシン(gacytosine);アラビノシド(「Ara−C」);シクロホスファミド;チオテパ;タキソイド、例えばパクリタキセル(タキソール(登録商標)、Bristol−Myers Squibb Oncology, Princeton, NJ)、及びドキセタキセル(タキソテア(登録商標)、Rhone−Poulenc Rorer, Antony, France);クロランブシル;ゲンシタビン(gemcitabine);6−チオグアニン;メルカプトプリン;メトトレキセート;シスプラチン及びカルボプラチンのようなプラチナ類似体;ビンブラスチン;プラチナ;エトポシド(VP−16);イフォスファミド;マイトマイシンC;ミトキサントン;ビンクリスチン;ビノレルビン;ナベルビン(navelbine);ノバントロン(novantrone);テニポシド;ダウノマイシン;アミノプテリン;キセローダ(xeloda);イバンドロナート(ibandronate);CPT−11;トポイソメラーゼインヒビターRFS2000;ジフルオロメチロールニチン(DMFO);レチノイン酸;カペシタビン(capecitabine);並びに上述したものの製薬的に許容可能な塩類、酸類又は誘導体が含まれる。また、この定義には、腫瘍に対するホルモン作用を調節又は阻害するように働く抗ホルモン剤、例えばタモキシフェン、ラロキシフェン(raloxifene)、4(5)−イミダゾール類を阻害するアロマターゼ、4−ヒドロキシタモキシフェン、トリオキシフェン(trioxifene)、ケオキシフェン(keoxifene)、LY117018、オナプリストーン(onapristone)、及びトレミフェン(Fareston)を含む抗エストロゲン;及び抗アンドロゲン、例えばフルタミド(flutamide)、ニルタミド(nilutamide)、ビカルタミド、ロイプロリド、及びゴセレリン;並びに上記のものの製薬的に許容可能な塩類、酸類又は誘導体が含まれる。
【0034】
「サイトカイン」という用語は、一つの細胞集団から放出されるタンパク質であって、他の細胞に対して細胞間メディエータとして作用するものの包括的な用語である。そのようなサイトカインの例は、リンフォカイン、モノカイン、及び伝統的なポリペプチドホルモンである。サイトカインには、成長ホルモン、例えばヒト成長ホルモン、N−メチオニルヒト成長ホルモン、及びウシ成長ホルモン;副甲状腺ホルモン;チロキシン;インスリン;プロインスリン;リラクシン;プロリラクシン;卵胞刺激ホルモン(FSH)、副甲状腺刺激ホルモン(TSH)、及び黄体形成ホルモン(LH)のような糖タンパク質ホルモン;肝臓成長因子;繊維芽細胞成長因子;プロラクチン;胎盤ラクトゲン;腫瘍壊死因子−α及び−β;マレリアン(mullerian)阻害物質;マウス性腺刺激ホルモン関連ペプチド;インヒビン;アクチビン;血管内皮成長因子;インテグリン;トロンボポエチン(TPO);NGF−αのような神経成長因子;血小板成長因子;TGF−α及びTGF−βのようなトランスフォーミング成長因子(TGF);インスリン様成長因子I及びII;エリスロポイエチン(EPO);オステオインダクティブ因子;インターフェロン、例えばインターフェロンα、β、γコロニー刺激因子(CSF)、例えばマクロファージ−CSF(M−CSF);顆粒球−マクロファージ−CSF(GM−CSF)及び顆粒球−CSF(G−CSF);IL−1、IL−1α、IL−2、IL−3、 IL−4、IL−5、IL−6、IL−7、 IL−8、IL−9、IL−10、IL−11、IL−12等のインターロイキン(IL);TNF−α又はTNF−βのような腫瘍壊死因子;及びLIF及びキットリガンド(KL)を含む他のポリペプチド因子が含まれる。ここで使用される場合は、サイトカインなる用語は天然源由来あるいは組換え細胞培養由来のタンパク質及び天然配列サイトカインの生物的に活性な等価物を含む。
【0035】
ここで使用される場合の「前処理」又は「前処理する」なる用語は、哺乳動物細胞をApo−2レセプターアゴニストにさらす前に、CPT−11(又は他の化学療法剤)に暴露させることを称する。哺乳動物細胞、特に癌細胞をCPT−11で前処理すると、該癌細胞内又は細胞上でDR4又はDR5レセプター発現が高められるかアップレギュレーションされて、Apo−2リガンドレセプターアゴニストに対して敏感になると考えられる。好ましくは、細胞の前処理に使用されるCPT−11の量は、前記哺乳動物細胞内又は細胞上において、同じ条件下でCPT−11にさらされなかった同様の哺乳動物細胞と比較して、約0.5〜約5倍、好ましくは約1〜約4倍、より好ましくは約2〜約4倍になるまで、DR4又はDR5レセプター発現が高められるかアップレギュレーションされるのに十分な量とされる。
「処理」又は「治療」とは、双方とも、治癒的処理、及び予防的又は防護的処置を称する。
「有効量」という用語は、哺乳類の疾病や疾患の治療のために有効な薬剤の量に相当する。癌の場合は、治療的有効量の薬剤により、癌細胞の数を減少させ;腫瘍の大きさを小さくし;癌細胞の周辺器官への浸潤を阻害(すなわち、ある程度に遅く、好ましくは止める)し;腫瘍の転移を阻害(すなわち、ある程度に遅く、好ましくは止める)し;腫瘍の成長をある程度阻害し;及び/又は疾患に関連する一又は複数の症状をある程度和らげることが可能である。ある程度、薬剤は、成長を妨げ及び/又は現存の癌細胞を殺すことが可能で、細胞分裂停止性及び/又は細胞障害性である。癌治療に対しては、インビボにおける効力は、腫瘍の負荷又は体積の評価、例えば病状の進行時間(TTP)の評価、及び/又は応答速度(RR)を決定することにより測定される。
【0036】
処置又は治療を目的とした「哺乳動物」とは、ヒト、家畜及び農場飼育動物、及び動物園、スポーツ、又はペット動物、例えば犬、ウマ、ネコ、ウシなどを含む哺乳類に分類される任意の動物を称する。好ましくは、哺乳動物はヒトである。
「癌」、「癌性」又は「悪性」という用語は、典型的には調節されない細胞成長を特徴とする、哺乳動物における生理学的状態を称するか記述する。癌の例には、これらに限定されるものではないが、癌腫、リンパ腫、芽細胞腫、肉腫、及び白血病が含まれる。このような癌のより特定の例には、大腸癌、結腸直腸癌、直腸癌、扁平上皮細胞癌、小細胞肺癌、非小細胞肺癌、ホジキン及び非ホジキンリンパ腫、睾丸癌、骨髄腫、食道癌、胃腸癌、腎臓癌、膵臓癌、グリア芽細胞腫、子宮頸癌、卵巣癌、神経膠腫、肝臓癌、膀胱癌、肝細胞腫(hepatoma)、乳癌、子宮体癌、唾液腺癌、腎臓癌、肝臓癌、前立腺癌、産卵口癌、甲状腺癌、肝癌(hepatic carcinoma)及び様々な種類の頭部及び頸部の癌が含まれる。
【0037】
II. 方法と材料
A.方法
一般に、哺乳動物細胞においてアポトーシスを誘発する本発明の方法は、有効量のApo−2リガンドとCPT−11又は有効量のApo−2Lレセプターアゴニスト抗体とCPT−11に細胞をさらすことを含んでなり、ここで、該細胞は該Apo−2L又はApo−2Lレセプターアゴニスト抗体に暴露される前にCPT−11に暴露される。通常、用いられるApo−2L(又はアゴニスト抗体)の量はアポトーシスを誘発するのに効果的な量である。また、通常用いられるCPT−11の量は、前記細胞内又は細胞上におけるDR4又はDR5レセプターの発現を高めるのに有効な量とされる。これは、例えば、以下と実施例に記載する方法に従ってインビボ又はエキゾビボで達成することができる。Apo−2リガンド又はアゴニスト抗体とCPT−11で治療される典型的な症状又は疾患には良性又は悪性癌が含まれる。
【0038】
1.アポトーシス機構のエレメント
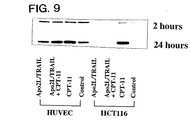
殺傷活性の増加に相関したアポトーシス機構のあるエレメントをさらに理解することで、哺乳動物細胞のアポトーシスを誘導するための方法を容易に実行することができるようになる。これに関連して、実施例3に提供されたデータにより、Apo2L/TRAIL+CPT−11処理で増加した殺傷活性と相関したアポトーシス機構のエレメントが同定される。
以下に詳細に論議するように、反応性の腫瘍細胞であって正常細胞ではないものをApo2L/TRAILで処理すると、DR5レセプターの一過性のアップレギュレーションを誘発することができる。同様に、CPT−11にさらすことで、DR5レセプター及び/又はDR4レセプターがアップレギュレーションする結果となる。約24時間にわたるApo2L/TRAIL+CPT−11の組合せ処理により、対照と比較してDR4及びDR5の発現が増強される結果となった。さらに、種々の細胞を20−22時間、CPT−11で前処理し、続いて2時間、Apo2L/TRAILで処理することで、DR5及びDR4 mRNA、並びにカスパーゼ−3様切断/活性化、及びアポトーシスが最も高く誘発された。カスパーゼインヒビターZ−VADの添加により、DR5及びDR4 mRNAの発現レベルがさらに増強されることが見出された。
【0039】
実施例3に提供されたデータは、CPT−11及びApo2L/TRAILが、p53依存性及びp53独立性経路でそれぞれ別個にアポトーシスを誘発する証拠を提供するものである。特に、HCT−116細胞をApo2L/TRAIL単独で処理するとp53発現を誘発しなかったが、CPT−11及びApo2L/TRAILとCPT−11を組合せると、p53タンパク質が強く誘発される結果となった。さらに、Apo2L/TRAILはDR5 mRNA発現の一過性のアップレギュレーションを媒介したが、CPT−11はDR5及びDR4 mRNA発現の双方を増加した。CPT−11は単独で、細胞周期停止に関与しているp21タンパク質、p53誘発性、サイクリン依存性キナーゼインヒビターの実質的なアップレギュレーションを誘発した。p21のCPT−11誘発性蓄積は、Apo2L/TRAILで同時処理(co−treatment)することにより、カスパーゼ依存的に妨げられた。さらに、G2−M期での蓄積よりはむしろ、Apo2L/TRAILで同時処理された細胞はアポトーシスを受ける。よって、Apo2L/TRAIL及びCPT−11の組合せ処理により、p21の分解、DR4及びDR5のアップレギュレーションが誘導され、細胞周期停止及びおそらくDNA修復よりもむしろアポトーシス経路へ癌細胞を至らしめる。このことは、組合せ処理を用い、インビボで示された抗−腫瘍活性を増強させることと明確に一致しており(Ashkenaziら, J. Clinical Investigation. 104:155−162(1999);Gliniakら, Cancer Research. 59:6153−6158(1999))、これらのデータは、Apo2L/TRAIL及びCPT−11処理が抗−腫瘍活性の増強を媒介する、潜在的メカニズムを提供するものである。
【0040】
実施例3のデータは、アポトーシスを被ったHCT116細胞におけるFLICE阻害タンパク質(FLIP)のタンパク質発現に何の変化もなかったことを示しており、この実験系におけるFLIPの実質的な抗−アポトーシス関与を除外するものであり、該データはメラノーマ腫瘍における研究と一致する(Griffithら, Current Opinion in Immunology. 10:559−563(1998);Leverkusら, Cancer Research, 60:553−559(2000);Zhangら, Cancer Research, 59:2747−2753(1999))。さらに、一般的なカスパーゼインヒビター、ZVADの存在により、Apo2L/TRAIL媒介性アポトーシス、p21の分解、及びCPT−11により媒介されるG2−M相細胞停止の破壊が効果的にブロックされ、これはp21がApo2L/TRAIL媒介性アポトーシスにおいて制御的な役割を担っていることの証拠を提供するものである(また、Xuら, Biochem. Biophys. Res. Comm., 269:179−190(2000)を参照)。しかしながら、これらの条件でp21の過剰発現を誘発すると、近位カスパーゼ活性が阻害されてアポトーシスが防止される。
ここでのデータは、異なる経路、それぞれDNAダメージ及び死亡シグナル伝達レセプターを通してアポトーシスを媒介するCPT−11及びApo2L/TRAILという2つの薬剤が、互いに共同して作用可能であるといった、新規のメカニズムを記載するものである。すなわち、CPT−11によるp21誘発のApo2L/TRAIL阻害により、G2/M細胞周期停止における細胞の蓄積が予め除外され、よってアポトーシスの増強が促進される。さらに、これらの薬剤の組合せによる死亡レセプターのアップレギュレーションは、観察され増強したアポトーシス活性にも寄与する。
【0041】
2.Apo−2Lレセプターアゴニストに誘発されるアポトーシスの調節
ここに開示したようにして、哺乳動物癌細胞におけるアポトーシスを調節及び増強することが可能であり、これは、細胞を、有効量のCPT−11及びApo−2Lレセプターアゴニストに、Apo−2リガンドレセプターアゴニストの投与の前に、CPT−11を投与することによってさらした際に生じる。特に、実施例3及び図5に示すように、細胞を20−22時間、CPT−11で前処理し、続いて2時間、Apo2L/TRAILで処理することで、DR5及びDR4 mRNA、並びにカスパーゼ−3様切断/活性化、及びアポトーシスが最も高く誘発された。よって本発明の重要な側面は、Apo−2Lレセプターアゴニスト及び化学療法剤、例えばCPT−11を使用し、哺乳動物細胞においてアポトーシスを誘発される改善された方法にあり、ここで、該方法は、Apo−2Lレセプターアゴニストで処理する前に、化学療法剤で細胞を前処理することを含む。
【0042】
Apo−2Lレセプターアゴニストで処理する前に、哺乳動物細胞をCPT−11等の化学療法剤で前処理する方法は、これらの薬剤の同時投与よりもいくつかの利点を有し得る。特に上述したように、これらの方法は、これらの薬剤の組合せ投与にとって最適な条件を同定することによって、処理様式を促進することができる。従って、アポトーシス反応を最適化する方法を同定することによって、医者は、より簡便で患者に親切な方式でこれらの薬剤を付与することができるであろう。特に、アポトーシス反応を最適化する方法を使用することで、医者は複数回の注射というよりはむしろ単一のボーラスでこれらの薬剤を投与し、またこれらの薬剤をより低濃度で投与し、又はこれらの薬剤をより短い期間で投与することができるようになる。
【0043】
CPT−11に類似した生理学的効果を有する付加的な化学療法剤を、ここで開示した方法に使用することができる。特に、異なる抗癌遺伝毒性ストレス化学物質、例えばドキソルビシン、エトポシド、CDDP及びガンマ放射線処理にさらすことで、いくつかの腫瘍株化細胞におけるApo2L/TRAIL死亡レセプターDR5の選択的p53依存性アップレギュレーションがなされる結果となる(例えば、Kimら, Clin. Cancer Res. 6(2):335−346(2000);Gibsonら, Mol. Cell Biol. 20(1):205−212(2000);Keaneら, Cancer Res. 59(3):734−741(1999);Naganeら, Cancer Res. 60(4):847−853(2000);Wuら, Nature Genetics. 17:141−3(1997)及びWuら, Oncogene. 18:6411−6418(1999)を参照)。p53独立性様式におけるDR5のアップレギュレーションは、TNF−αで腫瘍細胞を処理することにより(Sheikhら, Cancer Research 58:1593−1598(1998))、あるいは異なる神経膠腫株化細胞におけるいくつかの化学療法剤により(Naganeら, Cancer Research 60:847−853(2000))示されている。さらに、DR5のアップレギュレーションは、カスパーゼ依存性Apo2L/TRAIL媒介性アポトーシスに対する反応性が増加することと、最も多くのケースで相関している(Chinnaiyanら, Proc. Nat. Acad. Sci., 97:1754−1759(2000))。
【0044】
ここで開示したように、一つは殺傷活性を増加させることに関連した細胞アポトーシス機構を調節する薬剤で細胞を前処理することにより、哺乳動物癌細胞におけるApo−2Lレセプターアゴニスト媒介性アポトーシスを増強することができる。ここに開示する本発明の典型的な実施態様には、DR4のアップレギュレーション、DR5のアップレギュレーション及び/又はp53タンパク質の誘発を含む、一又は複数の生理学的事象に効果を及ぼす薬剤で細胞を前処理することにより、Apo−2Lレセプターアゴニスト媒介性アポトーシスに細胞を感作させるための方法が含まれる。好ましくは、薬剤は、CPT−11、ドキソルビシン、5−フルオロウラシル、インターフェロン(例えば、インターフェロンアルファ又はインターフェロンガンマ)、エトポシド、シスジアミンジクロロプラチナム(II)(CDDP)、TNF−α及びガンマインターフェロンからなる群から選択される。より好ましい実施態様では、薬剤はCPT−11である。
【0045】
本発明の一実施態様においては、有効量のCPT−11及びApo−2リガンドレセプターアゴニストに細胞を暴露させることを含む、哺乳動物癌細胞でアポトーシスを誘導する方法を提供するものであり、ここで細胞はApo−2リガンドレセプターアゴニストの前にCPT−11に暴露される。好ましくは、これらの方法において、所定量のCPT−11の投与により、該細胞中又は細胞上でDR4及び/又はDR5がアップレギュレーションされる結果となる。DR4及び/又はDR5のアップレギュレーション又は増強した発現度合いは、実施例に記載されている技術を含む、例えばDR4又はDR5mRNAの発現を測定する公知の技術を使用し、CPT−11に曝さない対照細胞と比較することによってアッセイ及び測定される。このようなアッセイは、DR4又はDR5の所望の又は最適なアップレギュレーションを誘発させ得る、前処理の最適時間を決定するために、CPT−11に細胞をさらした後の選択した時間点で実施する。インビトロアッセイ法を使用することで、本出願人は、CPT−11によるDR5発現の誘発が、2時間の暴露又はインキュベート後に観察され、特にDR5発現は、50マイクログラム/mlのCPT−11に6時間細胞をさらした後にインビトロで誘発され得ることを見出した。任意に、Apo−2レセプターアゴニストにさらす前に、約1時間〜約5日、好ましくは約2時間〜約24、48又は72時間、より好ましくは約6時間〜24時間又は48時間、細胞をCPT−11にさらす。該方法において、Apo−2リガンドレセプターアゴニストは、典型的にはApo2L/TRAIL又は抗−DR4レセプター抗体を含む。本発明のさらなる実施態様にはこれらの方法の変形例が含まれ、例えば、付加的な治療様式、例えば一又は複数の成長阻害剤又は放射線に癌細胞をさらすといった様式を使用する。該方法の好ましい実施態様では、癌細胞には、直腸結腸癌細胞が含まれる。
【0046】
典型的には、CPT−11の有効量は、CPT−11に暴露された哺乳動物細胞内又は細胞上において、DR4又はDR5、もしくはDR4とDR5の双方がアップレギュレーションされるのに十分な量である。さらに典型的には、有効量のCPT−11はp53タンパク質を誘発する。使用されるCPT−11の典型的な用量は、Physician’s Desk Reference(PDR)に従った標準的な臨床量を含み、約1マイクログラム/ml〜約100マイクログラム/ml、場合によっては約2マイクログラム/ml〜約50マイクログラム/mlの範囲が含まれ、臨床的使用用として、さらに好ましくは約0.05mg/kg〜約2.5mg/kgの範囲が含まれ、一方、Apo−2リガンドの典型的な用量は0.1mg/kg〜約12.0mg/kgの範囲を含む。
【0047】
B. 材料
本方法において使用することができるApo−2Lには、上掲のPitti等、上掲の国際公開第97/25428号、及び上掲の国際公開第97/0163号に記載されたApo−2Lポリペプチド(TRAILと称されるポリペプチド)を含む。全長ポリペプチド並びに細胞外ドメイン(ECD)配列を含んでなるApo−2Lの可溶型のような、Apo−2Lの様々な形態を使用することができると考えられる。そのような可溶型ECD配列の例には、ここで配列番号:1であり、Pitti等, J. Biol. Chem., 271:12687−12690 (1996)の図1Aに示されたApo−2L配列のアミノ酸114−281、95−281、91−281又は92−281を含んでなるポリペプチドが含まれる。アミノ酸92−281を含んでなるポリペプチはApo−2Lの天然に切断した型であると現在は信じられている。本出願人は、CHO細胞においてヒトApo−2Lを発現させ、92−281ポリペプチドがApo−2Lの発現型であることを見出した。国際公開第97/25428号に記載された共有結合的に修飾された型のようなApo−2Lの修飾型が含まれる。特に、ポリエチレングリコールのような非タンパク性ポリマーに結合したApo−2Lが本方法に使用されるものに含まれる。Apo−2Lポリペプチドは国際公開第97/25428号に記載された方法の何れかにより調製することができる。
該方法で使用されうるアポトーシス活性を有するApo−2リガンドの変異体は、例えばアラニンスキャン技術により同定されたものを含む。特に置換変異体は、位置203、218又は269でアミノ酸の少なくとも1種がアラニン残基で置換された、Pitti等, J. Biol. Chem., 271:12687−12690(1996)の図1Aのアミノ酸91−281を含んでなる。場合によっては、Apo−2リガンド変異体は1又は複数のこれら3つの異なった部位の置換を含みうる。
【0048】
Apo−2Lのアポトーシス活性を模倣する分子がまた現在開示した方法において使用しうると考えられる。そのような分子の例には、Apo−2Lに少なくとも匹敵するか、もしくは同様な方法でアポトーシスを誘発しうるアゴニスト抗体が含まれる。特に、これらのアゴニスト抗体はApo−2Lに対するレセプターの一又は複数に結合する抗体を含む。好ましくは、アゴニスト抗体は、DR4又はDR5のような細胞質死ドメインを含むApo−2Lレセプターに対する。さらに好ましくは、アゴニスト抗体は、レセプターの様なものに結合し、その結合は実施例2に記載されているように、例えばFACS分析又はELISAを用いて決定することができる。DR5(又はApo−2)と呼ばれるレセプターに対するアゴニスト抗体は、以下に記載するような融合技術を使用して調製されている。DR5又はApo−2レセプターアゴニスト抗体の一つは3F11.39.7と呼ばれ、1998年1月13日に寄託番号HB−12456としてATCCに寄託されている。他のDR5レセプター抗体は3H3.14.5を含み、ここで示されるようにATCCに寄託された。Apo−2Lレセプター抗体のアゴニスト活性は、アポトーシス活性の様々な検査方法を使用して測定することができ、さらに場合によっては、このような抗体のアポトーシス活性は、9D細胞又はDR4又はDR5のようなApo−2Lレセプターを発現する他の細胞のアポトーシスを検査するための実施例2に記載されたアッセイにおいて、Fc免疫グロブリン又は補体(下記)を用いて、抗体、単独又は架橋結合した形態でアッセイすることにより決定される。
さらに、DR4と呼ばれる他のApo−2Lレセプターに対するアゴニスト抗体もまた、調製される。DR4アゴニスト抗体の1つは、4H6.17.8と称され、1998年1月13日に寄託番号HB−12455としてATCCに寄託されている。またさらにアゴニストDR4抗体は、以下に記載されるようにATCCに寄託されている抗体4E7.24.3、1H5.25.9、4G7.18.8、及び5G11.17.1を含む。Apo−2Lレセプター抗体のアゴニスト活性は、アポトーシス活性の様々な検査方法を使用して測定することができ、さらに場合によっては、このような抗体のアポトーシス活性は、9D細胞又はDR4又はDR5のようなApo−2Lレセプターを発現する他の細胞のアポトーシスを検査するための実施例2に記載されたアッセイにおいて、Fc免疫グロブリン又は補体(下記)を用いて、抗体、単独又は架橋結合した形態でアッセイすることにより決定される。
【0049】
本発明において考察されているアゴニスト抗体には、1つのApo−2Lレセプター又は1以上のApo−2Lレセプターと結合する抗体が含まれる。1以上のApo−2Lレセプターを結合する抗体は、以下の実施例のように例えばELISA又はFACSにより決定される、2又はそれ以上のそれぞれ異なる抗体と結合「交差反応し」、それぞれ異なる抗体と結合する能力のある抗体として特徴づけられうる。場合によっては、2又はそれ以上の異なる抗原と「特異的に交差反応する」抗体は、第1の抗原と結合するもので、さらに第2の異なる抗原と結合し、約10μg/mLの抗体濃度で第2の抗原に対する抗体の結合能力は、(以下の実施例のように)捕獲ELISAで決定されるように第1の抗原の結合能力の約50%〜約100%(好ましくは約75%〜約100%)である。例えば抗体は、DR5(「第1抗原」)と特異的に結合しても、更にDR4のような他のApo−2レセプター(「第2抗原」)と特異的に交差反応してもよく、ここで、約10μg/mLの抗体のDR4との結合の範囲は、ここでの捕獲ELISAにおいてDR5に対する抗体の結合能力の約50%〜約100%である。様々なApo−2Lレセプターとの抗体の交差反応は、国際特許出願番号PCT/US99/13197の更なる詳細に記載される。
以下に記載されるように、このような抗体の例示的な形態としては、ポリクローナル、モノクローナル、ヒト化、二重特異性及びヘテロ抱合体抗体が含まれる。
【0050】
1.ポリクローナル抗体
本発明の抗体はポリクローナル抗体を含んでよい。ポリクローナル抗体の調製方法は当業者に知られている。ポリクローナル抗体は、哺乳動物において、例えば免疫化剤、及び所望するのであればアジュバントを、一又は複数回注射することで発生させることができる。典型的には、免疫化剤及び/又はアジュバントを複数回皮下又は腹腔内注射することにより哺乳動物に注射する。免疫化剤は、DR4又はDR5ポリペプチド(又はDR4又はDR5ECD)又はその融合タンパク質を含みうる。免疫化剤を、免疫化される哺乳動物で免疫原性が知られたタンパク質に抱合させるのが有用である。そのような免疫原性タンパク質の例は、限定するものではないが、キーホール・リンペット(keyhole limpet)ヘモシアニン、血清アルブミン、ウシサイログロブリン及び大豆トリプシンインヒビターが含まれる。使用され得るアジュバントの例には、フロイント完全アジュバント及びMPL−TDMアジュバント(モノホスホリル脂質A、合成トレハロースジコリノミコラート)が含まれる。免疫化プロトコールは、過度の実験なく当業者により選択されるであろう。哺乳動物から採血し、血清を検定して抗体価を求める。望まれるならば、抗体価が増加又は平坦化するまで哺乳動物に追加免疫を施す。
【0051】
2.モノクローナル抗体
本発明の抗体は、あるいは、モノクローナル抗体であってもよい。モノクローナル抗体は、Kohler及びMilstein, Nature, 256:495 (1975)に記載されているようなハイブリドーマ法を使用することで調製することができる。ハイブリドーマ法では、マウス、ハムスター又は他の適切な宿主動物を典型的には免疫化剤により免疫化することで、免疫化剤に特異的に結合する抗体を産生するかあるいは産生可能なリンパ球を誘発する。あるいは、リンパ球をインビトロで免疫化することもできる。
免疫化剤は、典型的にはDR4又はDR5ポリペプチド又はその融合タンパク質、例えばDR4又はDR5ECD−IgG融合タンパク質を含む。
一般にヒト由来の細胞が望まれる場合には末梢血リンパ球(「PBL」)が使用され、あるいは非ヒト哺乳動物源が望まれている場合は、脾臓細胞又はリンパ節細胞が使用される。次いで、ポリエチレングリコール等の適当な融合剤を用いてリンパ球を不死化株化細胞と融合させ、ハイブリドーマ細胞を形成する[Goding, Monoclonal Antibodies:Principles and Practice, Academic Press, (1986) pp. 59−103]。不死化株化細胞は、通常は、形質転換した哺乳動物細胞、特に齧歯動物、ウシ、及びヒト由来の骨髄腫細胞である。通常、ラット又はマウスの骨髄腫細胞株が使用される。ハイブリドーマ細胞は、好ましくは、未融合の不死化細胞の生存又は成長を阻害する一又は複数の物質を含有する適切な培地で培養される。例えば、親の細胞が、酵素のヒポキサンチングアニンホスホリボシルトランスフェラーゼ(HGPRT又はHPRT)を欠いていると、ハイブリドーマの培地は、典型的には、ヒポキサチン、アミノプチリン及びチミジンを含み(「HAT培地」)、この物質がHGPRT欠乏性細胞の増殖を阻止する。
【0052】
好ましい不死化株化細胞は、効率的に融合し、選択された抗体生成細胞による安定した高レベルの抗体発現を支援し、HAT培地のような培地に対して感受性である。より好ましい不死化株化細胞はマウス骨髄腫株であり、これはカリフォルニア州サンディエゴのSalk Institute Cell Distribution Centerやバージニア州マナッサスのアメリカン・タイプ・カルチャー・コレクションより入手可能である。そのようなマウス骨髄腫細胞株の例は、以下の実施例2に記載されたP3X63AgU.1である。ヒトモノクローナル抗体産生ためのヒト骨髄腫及びマウス−ヒト異種骨髄腫株化細胞も記載されている[Kozbor, J. Immunol., 133:3001 (1984);Brodeur等, Monoclonal Antibody Production Techniques and Applications, Marcel Dekker, Inc., New York, (1987) pp. 51−63]。
ハイブリドーマ細胞が培養される培地は、次いでApo−2Lレセプターに対するモノクローナル抗体の存在について検定することができる。好ましくは、ハイブリドーマ細胞によって産生されたモノクローナル抗体の結合特異性は免疫沈降又はラジオイムノアッセイ(RIA)又は酵素結合免疫測定法(ELISA)等のインビトロ結合検定法によって測定する。このような技術及びアッセイは、当該分野において公知である。モノクローナル抗体の結合親和性は、例えばMunson及びPollard, Anal. Biochem., 107:220 (1980)によるスキャッチャード分析法によって測定することができる。
【0053】
所望のハイブリドーマ細胞が同定された後、クローンを制限希釈工程を経てサブクローニングし、標準的な方法で増殖させることができる[Goding, 上掲]。この目的のための適当な培地には、例えば、ダルベッコの改変イーグル培地又はRPMI−1640倍地が含まれる。あるいは、ハイブリドーマ細胞は哺乳動物においてインビボで腹水として成長させることもできる。
サブクローンによって分泌されるモノクローナル抗体は、例えばプロテインAセファロース法、ヒドロキシルアパタイトクロマトグラフィー法、ゲル電気泳動法、透析法又はアフィニティークロマトグラフィー等の従来の免疫グロブリン精製方法によって培地又は腹水液から分離又は精製される。
また、モノクローナル抗体は、組換えDNA法、例えば米国特許第4,816,567号に記載された方法により作成することができる。本発明のモノクローナル抗体をコードするDNAは、従来の手順によって(例えば、マウス抗体の重鎖及び軽鎖をコードする遺伝子に特異的に結合可能なオリゴヌクレオチドプローブを使用して)、容易に単離し配列決定することができる。本発明のハイブリドーマ細胞はそのようなDNAの好ましい供給源となる。ひとたび単離されたら、DNAは発現ベクター内に配することができ、これが宿主細胞、例えばサルCOS細胞、チャイニーズハムスター卵巣(CHO)細胞、あるいは免疫グロブリンタンパク質を生成しない骨髄腫細胞内にトランスフェクトされ、組換え宿主細胞内でモノクローナル抗体の合成をすることができる。また、DNAは、例えば相同マウス配列に換えてヒト重鎖及び軽鎖定常ドメインのコード配列を置換することにより[米国特許第4,816,567号;Morrison等, 上掲]、又は免疫グロブリンコード配列に非免疫グロブリンポリペプチドのコード配列の一部又は全部を共有結合することにより修飾することができる。このような非免疫グロブリンポリペプチドは、本発明の抗体の定常ドメインの代わりに置換するか、本発明の抗体の一つの抗原結合部位の可変ドメインの代わりに置換し、キメラ性二価抗体を産生することができる。場合によっては、キメラ抗体は、ここで開示される4H6.17.8、3F11.39.7、4E7.24.3、1H5.25.9、4G7.18.8、5G11.17.1、及び3H3.14.5抗体から選択される抗Apo−2Lレセプター抗体の少なくとも1つの可変又は高頻度可変ドメインを含んで構築されうる。
場合によっては、本発明のアゴニスト抗体は、ここに開示した4H6.17.8、3F11.39.7、4E7.24.3、1H5.25.9、4G7.18.8、5G11.17.1、及び3H3.14.5抗体と同じエピトープに結合する。このことは、ここに記載するような種々のアッセイを実施することにより決定できる。例えば、モノクローナル抗体が、ここにおいて特別に言及したDR4又はDR5抗体と同じ特異性を有するか否かを決定するために、阻止アッセイ又はアポトーシス誘発アッセイにおいて、その活性を比較することができる。
【0054】
本発明の抗体は、「架橋」抗体を含む。ここにおいて使用されている「架橋」という用語は、一(又は単)分子を形成するための、少なくとも二つのIgG分子が互いに結合することを称する。Apo−2Lレセプター抗体は種々のリンカー分子を使用して架橋されてもよく、場合によっては、DR4抗体は、抗IgG分子、補体、化学修飾又は分子工学を使用して架橋される。一度抗体が細胞表層膜へ結合すると、補体が抗体分子に対して比較的に高い親和性を有することは、当業者とっては高く評価されることである。従って、細胞表層膜へ結合している二又はそれ以上の抗体を連結するための架橋分子として、補体を使用してもよいと考えられる。種々のマウスIgアイソタイプの中で、IgM、IgG2a及びIgG2bは、補体を固定することが知られている。
本発明の抗体は、抗体の多価型と同様に、二量体抗体を任意に含むことができる。当業者は、当該分野で知られている技術及びここにおける抗Apo−2Lレセプター抗体を使用して、そのような二量体又は多価型を組み立てることができる。
【0055】
本発明の抗体は一価抗体を含み得る。一価抗体の調製方法は当該分野においてよく知られてる。例えば、一つの方法は免疫グロブリン軽鎖と修飾重鎖の組換え発現を含む。重鎖は一般的に、重鎖の架橋を防止するようにFc領域の任意のポイントで切断される。あるいは、関連したシステイン残基を他のアミノ酸残基で置換するか欠失させて架橋を防止する。
一価抗体の調製にはインビトロ法がまた適している。抗体の消化による、その断片、特にFab断片の調製は、当該分野において知られている慣用的技術を使用して達成できる。例えば、消化はパパインの使用により行うことができる。パパイン消化の例は、94/12/22に公開された国際公開第94/29348号、及び米国特許第4,342,566号に記載されている。抗体のパパイン消化は、典型的には、Fab断片と呼ばれ、各々が単一の抗原結合部位を有する2つの同一の抗原結合断片と、残りのFc断片を生成する。ペプシン処理により、2つの抗原結合部位を有し、抗原の架橋が尚も可能なF(ab’)2断片が得られる。
また、抗体の消化により生産されたFab断片は、軽鎖の定常ドメインと重鎖の第1定常ドメイン(CH1)を含む。Fab’断片は、抗体のヒンジ領域から一又は複数のシステインを含む重鎖CH1ドメインのカルボキシ末端に幾つかの残基が付加されているということで、Fab断片とは異なっている。Fab’−SHとは、定常ドメインのシステイン残基が遊離のチオール基を担持しているFab’に対するここでの命名である。F(ab’)2抗体断片は、本来は、それらの間にヒンジシステインを有するFab’断片の対として生産された。抗体断片の他の化学的結合もまた知られている。
また、Iliades等, FEBS Letters, 409:437−441 (1997)に記載されているように、一本鎖Fv断片も産生されうる。このような一本鎖断片の種々のリンカーを用いた結合は、Kortt等, Protein Engineering, 10:423−433 (1997)に記載されている。
【0056】
上記に記載の抗体に加えて、キメラ又はハイブリッド抗体が、架橋剤を含む合成タンパク質化学における周知の方法を用いてインビトロで調製されうると考えられる。例えば、免疫毒素はジスルフィド交換反応を用いて、又はチオエーテル結合の形成により作成することができる。この目的に好適な試薬の例には、イミノチオレート及びメチル−4−メルカプトブチルイミデートが含まれる。
本発明のApo−2Lレセプター抗体は、さらにヒト化抗体又はヒト抗体を含む。非ヒト(例えばマウス)抗体のヒト化型とは、キメラ免疫グロブリン、免疫グロブリン鎖あるいはその断片(例えばFv、Fab、Fab’、F(ab’)2あるいは抗体の他の抗原結合性サブ配列)であって、非ヒト免疫グロブリンに由来する最小配列を含むものである。ヒト化抗体はレシピエントの相補性決定領域(CDR)の残基が、マウス、ラット又はウサギのような所望の特異性、親和性及び能力を有する非ヒト種(ドナー抗体)のCDRの残基によって置換されたヒト免疫グロブリン(レシピエント抗体)を含む。ある場合には、ヒト免疫グロブリンのFvフレームワーク残基は、対応する非ヒト残基によって置換されている。また、ヒト化抗体は、レシピエント抗体にも、移入されたCDRもしくはフレームワーク配列にも見出されない残基を含んでいてもよい。一般に、ヒト化抗体は、全てあるいはほとんど全てのCDR領域が非ヒト免疫グロブリンのものに対応し、全てあるいはほとんど全てのFR領域がヒト免疫グロブリン共通配列のものである、少なくとも1つ、典型的には2つの可変ドメインの実質的に全てを含む。ヒト化抗体は、最適には免疫グロブリン定常領域(Fc)、典型的にはヒトの免疫グロブリンの定常領域の少なくとも一部を含んでなる[Jones等, Nature, 321:522−525 (1986);Riechmann 等, Nature, 332:323−329 (1988);及びPresta, Curr. Op Struct. Biol., 2:593−596 (1992)]。
【0057】
非ヒト抗体をヒト化する方法はこの分野で良く知られている。一般的に、ヒト化抗体には非ヒト由来の一又は複数のアミノ酸残基が導入されている。これら非ヒトアミノ酸残基は、しばしば、典型的には「移入」可変ドメインから得られる「移入」残基と称される。ヒト化は基本的にウィンター(Winter)及び共同研究者の方法[Jones等, Nature, 321:522−525 (1986);Riechmann等, Nature, 332:323−327 (1988);Verhoeyen等, Science, 239:1534−1536 (1988)]に従って、齧歯動物のCDR又はCDR配列でヒト抗体の対応する配列を置換することにより実施される。よって、このような「ヒト化」抗体は、無傷のヒト可変ドメインより実質的に少ない分が非ヒト種由来の対応する配列で置換されたキメラ抗体である(米国特許第4,816,567号)。実際には、ヒト化抗体は典型的には幾つかのCDR残基及び場合によっては幾つかのFR残基が齧歯類抗体の類似部位からの残基によって置換されたヒト抗体である。そのような移入残基又は移入可変ドメイン(又はCDR)の元になるものは、ここで開示される寄託された抗Apo−2Lレセプター抗体4H6.17.8、3F11.39.7、4E7.24.3、1H5.25.9、4G7.18.8、5G11.17.1、及び3H3.14.5を含む。
抗原性の軽減のためには、ヒト化抗体を作成するために使用するヒトの可変ドメイン、軽鎖及び重鎖両方の選択が非常に重要である。「ベストフィット法」に従うと、齧歯動物抗体の可変ドメインの配列を既知のヒト可変ドメイン配列のライブラリ全体に対してスクリーニングする。齧歯動物のものと最も近いヒトの配列を次にヒト化抗体のヒトフレームワーク(FR)として受け入れる[Sims等, J. Immunol., 151:2296−2308 (1993);Chothia及びLesk, J. Mol. Biol., 196:901−917 (1987)]。他の方法では、軽鎖又は重鎖の特定のサブグループのヒト抗体全てのコンセンサス配列から誘導される特定のフレームワークを使用する。同じフレームワークを幾つかの異なるヒト化抗体に使用できる[Carter等, Proc. Natl. Acad. Sci. USA, 89:4285−4289 (1992);Presta等, J. Immunol., 151:2623−2632 (1993)]。
【0058】
さらに、抗体は、抗原に対する高親和性や他の好ましい生物学的性質を保持してヒト化することが重要である。この目標を達成するべく、好ましい方法では、親及びヒト化配列の三次元モデルを使用して、親配列及び様々な概念的ヒト化産物の分析工程を経てヒト化抗体を調製する。三次元免疫グロブリンモデルは一般的に入手可能であり、当業者にはよく知られている。選択された候補免疫グロブリン配列の推測三次元立体配座構造を図解し、表示するコンピュータプログラムは入手可能である。これら表示を見ることで、候補免疫グロブリン配列の機能における残基の役割の分析、すなわち候補免疫グログリンの抗原と結合する能力に影響を及ぼす残基の分析が可能となる。このようにして、例えば標的抗原に対する親和性を高めるといった、望ましい抗体特徴が得られるように、FR残基を共通及び移入配列から選択し、組み合わせることができる。一般的に、CDR残基は、直接かつ最も実質的に抗原結合性に影響を及ぼしている[1994年、3月3日に公開された国際公開第94/04679号を参照]。
【0059】
ヒトモノクローナル抗体は、ヒトBリンパ球を融合パートナーとして使用する、Kohler及びMilsteinによって最初に記載されたハイブリドーマ法を適応することによって作製することができる。関心ある抗体を生産するヒトBリンパ球は、例えば、インフォームド・コンセントを得た後に、ヒト個人から単離される。例えば、個人は、全身性紅斑性狼瘡(Shoenfeldら,J.Clin. Invest., 70:205(1982))、免疫媒介血小板新生紫斑病(ITP)(Nugentら,Blood, 70(1):16−22(1987))、又は癌のようなある疾患とともに生じる自己抗原に対する抗体を生産している。或いは、又は付加的に、リンパ球はインビトロで免疫化される。例えば、単離されたヒト末梢血リンパ球をlysomotrophic agent(例えばL−ロイシン−O−メチルエステル、L−グルタミン酸ジメチルエステル又はL−ロイシル−L−ロイシン−O−メチル エステル)(米国特許第5,567,610号、Borrebaeck等,)へ暴露させる;及び/又はインビトロでT細胞欠損ヒト末梢血リンパ球を8−メルカプトグアノシン及びサイトカインのようなアジュバントで処理(米国特許第5,229,275号、Goroff等.)してもよい。
被検者より回収されたかインビトロで免疫化されたBリンパ球は、次には通常、ヒトモノクローナル抗体を生成するために不死化される。限定されるものではないが、Bリンパ球を不死化する技術は、(a)ヒトBリンパ球とヒト、マウスミエローマ又はマウス−ヒトヘテロミエローマ細胞との融合;(b)ウイルスによる形質転換(例えば、エプスタイン・バーウイルス;例えば、Nugentら,上掲を参照のこと);(c)リンパ芽球腫細胞株との融合;又は(d)リンパ腫細胞との融合、を含む。
【0060】
リンパ球を、ポリエチレングリコールのような適当な融合剤を用いて骨髄腫細胞と融合させ、ハイブリドーマ細胞を形成する(Goding, Monoclonal Antibodies:Principles and Practice, pp.590−103(Academic Press, 1986))。このようにして調製されたハイブリドーマ細胞を、融合していない親の骨髄腫細胞の成長又は生存を阻害する一又は複数の物質を好ましくは含む適当な培地に蒔き、成長させる。例えば、親の骨髄腫細胞が酵素ヒポキサンチングアニジンホスホリボシルトランスフェラーゼ(HGPRT又はHPRT)を欠失するならば、ハイブリドーマのための培地は、典型的には、HGPRT欠失細胞の成長を妨げる物質であるヒポキサンチン、アミノプテリン及びチミジンを含有するであろう(HAT培地)。適切なヒトミエローマ及びマウス−ヒトヘテロミエローマ細胞株が記載されている(Kozbor, J. Immunol., 133:3001 (1984);Brodeur等, Monoclonal Antibody Production Techniques and Applications, pp. 51−63 (Marcel Dekker, Inc., New York, 1987))。ハイブリドーマ細胞が生育している培地を、抗原に対するモノクローナル抗体の産生について検定する。好ましくは、ハイブリドーマ細胞により産生されるモノクローナル抗体の結合特異性は、免疫沈降又はインビトロ結合検定、例えばラジオイムノアッセイ(RIA)又は酵素結合免疫吸着検定(ELISA)によって測定する。
所望の特異性、親和性、及び/又は活性の抗体を産生するハイブリドーマ細胞が同定された後、クローンを限界希釈法によりサブクローニングし、標準的な方法により成長させることができる(Goding, Monoclonal Antibodies:Principles and Practice, pp.59−103(Academic Press, 1986))。この目的に対して好適な培地は、例えば、D−MEM又はRPMI−1640培地を包含する。サブクローンにより分泌されたモノクローナル抗体は、例えばプロテインA−クロマトグラフィー、ゲル電気泳動、透析、又はアフィニティークロマトグラフィーのような常套的な免疫グロブリン精製法により、培地、腹水、又は血清から好適に分離される。
【0061】
また、ヒト抗体は、ヒト抗体を生産することのできるマウスのような非ヒト宿主を使用して生成してもよい。上記のように、免疫化することで、内在性免疫グロブリンが生成されない状態でもヒト抗体の完全リパートリを生成することができるトランスジェニックマウスを利用することが可能である。例えば、キメラ及び生殖系列変異マウスにおいて抗体重鎖結合領域(JH)遺伝子をホモ接合的に欠失させると内在性抗体生産の完全な阻害が生じることが記述されている。このような生殖系列変異マウスにヒト生殖系列免疫グロブリン遺伝子配列を移すと、抗原投与時にヒト抗体の生成が生じる。例えば、Jakobovits等, Proc. Natl. Acad. Sci. USA, 90:2551(1993);Jakobovits等, Nature, 362:255−258 (1993);Bruggermann等, Year in Immuno., 7:33 (1993);米国特許第5,591,669号;米国特許第5,589,369号;及び米国特許第5,545,807号を参照のこと。また、ヒト抗体は、SCID−huマウス(Duchosalら,Nature 355:258−262(1992))を使用して作製してもよい。
その他の実施態様では、ヒト抗体は、ヒト抗体ファージディスプレイライブラリより選択してもよい。抗体又はその断片のライブラリの調製は当該分野で良く知られており、宿主細胞へ導入可能な形質転換ベクターのファミリーの構築のためには、いずれの既知の方法を使用してもよい。ファージ中の抗体軽鎖及び重鎖(Huseら,Science, 246:1275(1989))、又はファージ或いはファージミド中の融合タンパク質のライブラリは、既知の方法によって調製が可能である。例えば、Vaughan等, Nature Biotechnology 14:309 −314(1996);Barbasら,Proc. Nalt. Acad. Sci., USA, 88:7978−7982(1991);Marks等, J.Mol.Biol., 222:581−597(1991);Hoogenboom及びWinter, J. Mol. Biol., 227:381−388 (1992);Barbasら,Proc. Nalt. Acad. Sci., USA, 89:4457−4461(1992);Griffiths等, EMBO Journal, 13:3245−3260(1994);de Kruifら,J. Mol. Biol., 248:97−105(1995);国際公開第98/05344号;国際公開第98/15833号;国際公開第97/47314;国際公開第97/44491号;国際公開第97/35196号;国際公開第95/34648号;米国特許第5,712,089号;米国特許第5,702,892号;米国特許第5,427,908号;米国特許第5,403,484号;米国特許第5,432,018号;米国特許第5,270,170号;国際公開第92/06176号;国際公開第99/06587号;米国特許第5,514,548号;国際公開第97/08320号;及び米国特許第5,702,892号を参照のこと。関心ある抗原は、標的抗原と結合するファージ抗体を選択するための分野において既知の方法を使用し、ファージライブラリに対して選別される。
【0062】
ここに記載のApo−2Lレセプター抗体は、場合によっては一又は複数の所望する生物活性又は特性を有する。そのような抗体は、限定されないが、キメラ、ヒト化、ヒト、及び親和性成熟抗体を包含してもよい。記載されているように、抗体は、これらの所望する活性又は特性を達成するための種々の技術を使用して構築又は設計される。一実施態様では、Apo−2Lレセプター抗体は、少なくとも105M−1、好ましくは106M−1から107M−1の範囲、さらに好ましくは少なくとも108M−1から1012M−1の範囲、及びよりさらに好ましくは少なくとも109M−1から1012M−1の範囲のDR4又はDR5レセプター結合親和性を有する。抗体の結合親和性は、スキャッチャード分析法(Munsonら、上掲を参照のこと)を包含する当該分野において既知の技術に従った抗体の試験による過度の実験なしに測定することができる。例えば、実施例2に記載のように、DR4−IgGレセプター構成物への結合親和性のために、DR4抗体をアッセイすることができる。
【0063】
その他の実施態様では、本発明のApo−2Lレセプター抗体は、Apo−2Lが結合するDR4又はDR5上の同じエピトープと結合するか、又はApo−2Lが結合するDR4又はDR5上のエピトープと一致又は重複するDR4又はDR5上のエピトープと結合する。また、Apo−2リガンドがDR4又はDR5へ結合することを妨げる立体構造を作り出すように、抗体は相互作用する。本発明の抗体のエピトープ結合特性は、当該分野において既知の技術を用いて決定されてもよい。例えば、抗体は、競争阻害アッセイのような、DR4又はDR5へのApo−2Lの結合をブロック又は阻害する抗体の能力を測定するためのインビトロアッセイにおいて試験されてもよい。場合によっては、例えばDR4−IgG構成物(実施例2に記載のような)又は細胞発現DR4へApo−2Lポリペプチド(実施例1に記載のような)の結合を阻害するDR4抗体の能力を測定するために、抗体は競争阻害アッセイにおいて試験されてもよい。場合によっては、抗体は、少なくとも50%、好ましくは少なくとも75%、及びなおさらに好ましくは少なくとも90%、レセプターへのApo−2Lの結合を遮断又は阻害することができ、これは一例として、Apo−2リガンド(TRAIL)の可溶型及びDR4 ECD−IgG(実施例2に記載のような)を使用するインビトロ競争阻害アッセイにおいて測定されてもよい。
好ましい実施態様では、抗体は、Apo−2リガンド(TRAIL)に匹敵する、又はこれに類する活性を有するアゴニスト抗体を含む。好ましくは、そのようなアゴニストDR4又はDR5抗体は、少なくとも一種類の癌又は腫瘍細胞株或いは原発腫瘍においてアポトーシスを誘導する。アゴニストDR4又はDR5抗体のアポトーシス活性は、既知のインビトロ又はインビボアッセイを使用して測定してもよい。そのようなインビトロ又はインビボアッセイの例は、下記の実施例部分に詳細に記載されている。インビトロでは、アポトーシス活性は、アネキシンV結合(AnnexinV binding)のような既知の技術を使用して測定することが可能である。インビボでは、アポトーシス活性は、例えば、腫瘍の負担又は容積の減少を測定することで決定してもよい。
【0064】
3.二重特異性抗体
二重特異性抗体は、少なくとも2つの異なる抗原に対して結合特異性を有するモノクローナル抗体、好ましくはヒトもしくはヒト化抗体である。本ケースにおいて、結合特異性の一方はApo−2Lレセプターに対してであり、他方は任意の他の抗原、好ましくは細胞表面タンパク質又はレセプター又はレセプターサブユニットに対してである。
二重特異性抗体を生成する方法は当該技術分野において周知である。伝統的には、二重特異性抗体の組換え生成方法は、二つの重鎖が異なる特異性を持つ二つの免疫グロブリン重鎖/軽鎖対の同時発現に基づく[Milstein及びCuello, Nature, 305:537−539 (1983)]。免疫グロブリンの重鎖と軽鎖を無作為に取り揃えたため、これらハイブリドーマ(クアドローマ)は10種の異なる抗体分子の潜在的混合物を作成でき、その内一種のみが正しい二重特異性構造を有する。正しい分子の精製は、アフィニティークロマトグラフィー工程によって通常達成される。同様の手順が1993年5月13日公開の国際公開第93/08829号、及びTraunecker等, EMBO J.,10:3655−3659 (1991)に開示されている。
所望の結合特異性(抗体−抗原結合部位)を有する抗体可変ドメインを免疫グロブリン定常ドメイン配列に融合することができる。融合は、好ましくは少なくともヒンジ部、CH2及びCH3領域の一部を含む免疫グロブリン重鎖定常ドメインとのものである。少なくとも一つの融合には軽鎖結合に必要な部位を含む第一の重鎖定常領域(CH1)が存在することが望ましい。免疫グロブリン重鎖融合をコードするDNA、及び望むのであれば免疫グロブリン軽鎖を、別々の発現ベクターに挿入し、適当な宿主生物に同時形質移入する。二重特異性抗体を生成するための更なる詳細については、例えばSuresh等, Methods in Enzymology, 121:210(1986)を参照されたい。
【0065】
4.ヘテロ抱合体抗体
ヘテロ抱合抗体もまた本発明の範囲に入る。ヘテロ抱合抗体は、2つの共有的に結合した抗体からなる。このような抗体は、例えば、免疫系細胞を不要な細胞に対して標的化させるため[米国特許第4,676,980号]及びHIV感染の治療のために[国際公開第91/00360号;国際公開号92/200373号;欧州特許第03089号]提案された。本抗体は、架橋剤に関連したものを含む合成タンパク質化学における既知の方法を使用してインビトロで調製することができると考えられる。例えば、ジスルフィド交換反応を使用するか又はチオエーテル結合を形成することにより免疫毒素を作成することができる。この目的に対して好適な試薬の例には、イミノチオレート及びメチル−4−メルカプトブチリミデート、及び例えば米国特許第4,676,980号に開示されているものが含まれる。
【0066】
5.トリアボディ(triabodies)
トリアボディも本発明の範囲内である。このような抗体は、例えば上掲のIliades等及び上掲のKortt等に記載されている。
【0067】
6.他の修飾
Apo−2Lレセプター抗体の他の修飾はここにおいて考慮されている。また、本発明の抗体は、プロドラッグ(例えばペプチジル化学療法剤、国際公開第81/01145号を参照)を活性な抗癌剤に転化させるプロドラッグ活性化酵素、又は細胞障害剤(毒素分子等)に抗体をコンジュゲートさせることにより修飾することができる。例えば国際公開第88/07378及び米国特許第4,975,278号を参照されたい。また、この技術は、「抗体依存性酵素媒介性プロドラッグ治療法」(ADEPT)と呼ばれている。
ADEPTに有用な免疫コンジュゲートの酵素成分には、より活性な細胞毒形態に転化するように、プロドラッグに作用し得る任意の酵素が含まれる。限定するものではないが、この発明の方法に有用な酵素には、ホスファート含有プロドラッグを遊離の薬剤に転化するのに有用なアルカリ性ホスファターゼ;スルファート含有プロドラッグを遊離の薬剤に転化するのに有用なアリールスルファターゼ;非毒性5−フルオロシトシンを抗癌剤5−フルオロウラシルに転化するのに有用なシトシンデアミナーゼ;プロテアーゼ、例えばセラチアプロテアーゼ、サーモリシン、サブチリシン、カルボキシペプチダーゼ及びカテプシン(例えば、カテプシンB及びL)で、ペプチド含有プロドラッグを遊離の薬剤に転化するのに有用なもの;カスパーゼ、例えばカスパーゼ−3;D−アミノ酸置換基を含有するプロドラッグの転化に有用なD−アラニルカルボキシペプチダーゼ;炭水化物切断酵素、例えばグリコシル化プロドラッグを遊離の薬剤に転化するのに有用なノイラミニダーゼ及びβガラクトシダーゼ;βラクタムで誘導体化された薬剤を遊離の薬剤に転化させるのに有用なβ−ラクタマーゼ;及びペニシリンアミダーゼ、例えばそれぞれフェノキシアセチル又はフェニルアセチル基で、それらのアミン性窒素において誘導体化された薬剤を遊離の薬剤に転化するのに有用なペニシリンVアミダーゼ又はペニシリンGアミダーゼが含まれる。あるいは、当該技術において「アブザイム」としてもまた公知の酵素活性を有する抗体を、遊離の活性薬剤に本発明のプロドラッグを転化させるために使用することもできる(例えば、Massey, Nature 328:457−458(1987)を参照)。抗体−アブザイムコンジュゲートは、ここで記載されているようにして、腫瘍細胞個体群にアブザイムを送達するために調製することができる。
【0068】
この酵素は、当該分野においてよく知られている技術、例えばヘテロ二官能性架橋試薬を使用することにより、抗体に共有的に結合させることができる。あるいは、本発明の抗体の少なくとも抗原結合領域を本発明の酵素の少なくとも機能的に活性な部位に結合せしめてなる融合タンパク質を、当該技術においてよく知られている組換えDNA技術を使用して作成することができる(Neuberger等, Nature 312:604−608[1984])。
さらなる抗体の修飾が考察されている。例えば、抗体は種々の非タンパク質様ポリマー、例えばポリエチレングリコール、ポリプロピレングリコール、ポリオキシアルキレン類、又はポリエチレングリコールとポリプロピレングリコールのコポリマーに結合してもよい。また抗体は、例えばコアセルベーション技術又は界面重合により調製されたマイクロカプセル(例えば、それぞれヒドロキシメチルセルロース又はゼラチン−マイクロカプセル及びポリ−(メチルメタクリレート)マイクロカプセル)にコロイド状薬物送達系(例えば、リポソーム、アルブミンミクロスフィア、マイクロエマルション、ナノ粒子及びナノカプセル)又はマクロエマルションに捕捉することができる。このような技術はRemington’s Pharmaceutical Sciences, 16th edition, A. Oslo編(1980)に開示されている。抗体の血清半減期を増大させるために、米国特許第5,739,277号に記載されているように抗体(特に抗体断片)へサルベージレセプター結合エピトープを組み込んでもよい。ここで使用される「サルベージレセプター結合エピトープ」という用語は、IgG分子のインビボでの血清半減期を増加させる原因となるIgG分子(例えばIgG1、IgG2、IgG3又はIgG4)のFc領域のエピトープを称する。
【0069】
7.組み換え体方法
本発明はまたここに開示した抗体をコードしている単離された核酸、該核酸を含むベクター及び宿主細胞、及び抗体の生産に対する組換え技術を提供する。
抗体の組換え生産のために、それをコードする核酸が単離され、さらなるクローニング(DNAの増幅)又は発現のために、複製可能なベクター内に挿入される。抗体をコードするDNAは直ぐに単離され、従来の手法(例えば、抗体をコードする遺伝子に特異的に結合可能なオリゴヌクレオチドを使用するもの)を用いて配列決定される。多くのベクターが入手可能である。ベクター成分としては、一般に、これらに制限されるものではないが、次のものの一又は複数が含まれる:シグナル配列、複製開始点、一又は複数のマーカー遺伝子、エンハンサーエレメント、プロモーター、及び転写終結配列である。
ここにおける方法は、抗Apo−2Lレセプター抗体軽鎖又は重鎖(又は両軽鎖及び重鎖の両方)をコードするDNA配列を含んでなるベクターを提供する、宿主細胞をベクターでトランスフェクト又は形質転換する、及び組み換え体抗Apo−2Lレセプター抗体産物を産生するのに十分な条件下で宿主細胞を培養する段階を含む、キメラ又は組み換え体抗Apo−2Lレセプター抗体の生産のための方法を包含する。
【0070】
(i)シグナル配列成分
本発明の抗Apo−2Lレセプター抗体は直接的に組換え手法によって生産されるだけではなく、好ましくはシグナル配列あるいは成熟タンパク質あるいはポリペプチドのN末端に特異的切断部位を有する他のポリペプチドである異種性ポリペプチドとの融合ペプチドとしても産生される。好ましく選択された異種シグナル配列は宿主細胞によって認識され加工される(すなわち、シグナルペプチダーゼによって切断される)ものである。天然抗体シグナル配列を認識せずプロセシングしない原核生物宿主細胞に対しては、シグナル配列は、例えばアルカリホスファターゼ、ペニシリナーゼ、lppあるいは熱安定なエンテロトキシンIIリーダーの群から選択される原核生物シグナル配列により置換される。酵母の分泌に関しては、天然シグナル配列は、酵母インベルターゼリーダー、α因子リーダー(酵母菌属(Saccharomyces)及びクルイベロマイシス(Kluyveromyces)α因子リーダーを含む)、又は酸ホスフォターゼリーダー、白体(C.albicans)グルコアミラーゼリーダー、又は国際公開第90/13646号に記載されているシグナルにより置換されうる。哺乳動物細胞の発現においては、哺乳動物のシグナル配列並びにウイルス分泌リーダー、例えば単純ヘルペスgDシグナルが利用できる。このような前駆体領域のDNAは、抗体をコードするDNAにリーディングフレームが結合される。
【0071】
(ii)複製開始点成分
発現及びクローニングベクターは共に一又は複数の選択された宿主細胞においてベクターの複製を可能にする核酸配列を含む。一般に、この配列はクローニングベクターにおいて、宿主染色体DNAとは独立にベクターが複製することを可能にするものであり、複製開始点又は自律的複製配列を含む。そのような配列は様々な細菌、酵母及びウイルスに対してよく知られている。プラスミドpBR322に由来する複製開始点は大部分のグラム陰性細菌に好適であり、2μプラスミド開始点は酵母に適しており、様々なウイルス開始点(SV40、ポリオーマ、アデノウイルス、VSV又はBPV)は哺乳動物細胞におけるクローニングベクターに有用である。一般には、哺乳動物の発現ベクターには複製開始点成分は不要である(SV40開始点が典型的には初期プロモーターを有しているため用いられる)。
【0072】
(iii)選択遺伝子成分
発現及びクローニングベクターは、選択可能マーカーとも称される選択遺伝子を含む。典型的な選択遺伝子は、(a)アンピシリン、ネオマイシン、メトトレキセートあるいはテトラサイクリンのような抗生物質あるいは他の毒素に耐性を与え、(b)栄養要求性欠陥を補い、又は(c)例えばバシラス菌に対する遺伝子コードD−アラニンラセマーゼのような、複合培地から得られない重要な栄養素を供給する、タンパク質をコードする。
選択技術の一例においては、宿主細胞の成長を抑止する薬物が用いられる。異種性遺伝子で首尾よく形質転換したこれらの細胞は、抗薬物性を付与し、選択療法を生存するタンパク質を生産する。このような優性選択の例としては、薬物ネオマイシン、ミコフェノール酸又はハイグロマイシンが使用される。
哺乳動物細胞に適切な選択可能なマーカーの他の例は、抗体核酸を捕捉することのできる細胞成分を同定することのできるもの、例えばDHFR、チミジンキナーゼ、メタロチオネインI及びII、好ましくは、霊長類メタロチオネイン遺伝子、アデノシンデアミナーゼ、オルニチンデカルボキシラーゼ等である。
例えば、DHFR選択遺伝子によって形質転換された細胞は、先ず、DHFRの競合的アンタゴニストであるメトトリキセート(Mtx)を含む培地において形質転換物の全てを培養することで同定される。野生型DHFRを用いた場合の好適な宿主細胞は、DHFR活性に欠陥のあるチャイニーズハムスター卵巣(CHO)株化細胞である。
あるいは、抗Apo−2Lレセプター抗体をコードするDNA配列、野生型DHFRタンパク質、及びアミノグリコシド3’−ホスホトランスフェラーゼ(APH)のような他の選択可能マーカーで形質転換あるいは同時形質転換した宿主細胞(特に、内在性DHFRを含む野生型宿主)は、カナマイシン、ネオマイシンあるいはG418のようなアミノグリコシド抗生物質のような選択可能マーカーの選択剤を含む培地における細胞増殖により選択することができる。米国特許第4,965,199号を参照。
【0073】
酵母中での使用に好適な選択遺伝子は酵母プラスミドYRp7に存在するtrp1遺伝子である(Stinchcomb等, Nature, 282:39(1979))。trp1遺伝子は、例えば、ATCC第44076号あるいはPEP4−1のようなトリプトファン内で成長する能力に欠ける酵母の突然変異株に対する選択マーカーを提供する。Jones, Genetics, 85:12 (1977)。酵母宿主細胞ゲノムにtrp1破壊が存在することは、トリプトファンの不存在下における成長による形質転換を検出するための有効な環境を提供する。同様に、Leu2欠陥酵母株(ATCC 20,622あるいは 38,626)は、Leu2遺伝子を有する既知のプラスミドによって補完される。
さらに、1.6μmの円形プラスミドpKD1由来のベクターは、クルイヴェロマイシス(Kluyveromyces)酵母の形質転換に用いることができる。あるいは、組換え子ウシのキモシンの大量生産のための発現系がK.ラクティス(lactis)に対して報告されている。Van den Berg, Bio/Technology, 8:135 (1990)。クルイヴェロマイシスの工業的な菌株からの、組換えによる成熟したヒト血清アルブミンを分泌する安定した複数コピー発現ベクターも開示されている。Fleer 等, Bio/Technology,9:968−975 (1991)。
【0074】
(iv)プロモーター成分
通常、発現及びクローニングベクターは、宿主生体により認識され、抗体核酸に作用可能に結合するプロモーターを含む。原核生物宿主での使用に好適なプロモーターは、phoAプロモーター、βラクタマーゼ及びラクトースプロモーター系、アルカリホスファターゼ、トリプトファン(trp)プロモーター系、及びハイブリッドプロモーター、例えばtacプロモーターを含む。しかし、他の既知の細菌プロモーターも好適である。細菌系で使用するプロモータもまた抗Apo−2Lレセプター抗体をコードするDNAと作用可能に結合したシャイン・ダルガーノ(S.D.)配列を有する。
真核生物に対してもプロモーター配列が知られている。事実上、全ての真核生物の遺伝子は、転写開始部位からおよそ25ないし30塩基上流に位置するAT富化領域を有する。多数の遺伝子の転写開始位置から70ないし80塩基上流に見出される他の配列は、Nが任意のヌクレオチドであるCNCAAT領域である。大部分の真核生物遺伝子の3’末端には、コード配列の3’末端へのポリA尾部の付加に対するシグナルであるAATAAA配列がある。これらの配列は全て真核生物の発現ベクターに適切に挿入される。
【0075】
酵母宿主と共に用いて好適なプロモーター配列の例としては、3−ホスホグリセラートキナーゼ又は他の糖分解酵素、例えばエノラーゼ、グリセルアルデヒド−3−リン酸デヒドロゲナーゼ、ヘキソキナーゼ、ピルビン酸デカルボキシラーゼ、ホスホフルクトキナーゼ、グルコース−6−リン酸イソメラーゼ、3−ホスホグリセレートムターゼ、ピルビン酸キナーゼ、トリオセリン酸イソメラーゼ、ホスホグルコースイソメラーゼ、及びグルコキナーゼが含まれる。
他の酵母プロモーターとしては、成長条件によって転写が制御される付加的利点を有する誘発的プロモーターであり、アルコールデヒドロゲナーゼ2、イソチトクロムC、酸ホスファターゼ、窒素代謝と関連する分解性酵素、メタロチオネイン、グリセルアルデヒド−3−リン酸デヒドロゲナーゼ、及びマルトース及びガラクトースの利用を支配する酵素のプロモーター領域がある。酵母の発現に好適に用いられるベクターとプロモータは欧州特許第73657号に更に記載されている。また酵母エンハンサーも酵母プロモーターと共に好適に用いられる。
【0076】
哺乳動物の宿主細胞におけるベクターからの抗Apo−2Lレセプター抗体転写は、例えば、ポリオーマウィルス、伝染性上皮腫ウィルス、アデノウィルス(例えばアデノウィルス2)、ウシ乳頭腫ウィルス、トリ肉腫ウィルス、サイトメガロウィルス、レトロウィルス、B型肝炎ウィルス及び最も好ましくはサルウィルス40(SV40)のようなウィルスのゲノムから得られるプロモーター、異種性哺乳動物プロモーター、例えばアクチンプロモーター又は免疫グロブリンプロモーター、ヒートショックプロモーターによって、提供されるこのようなプロモーターが宿主細胞系に適合し得る限り、調節される。
SV40ウィルスの初期及び後期プロモーターは、SV40ウイルスの複製起点をさらに含むSV40制限断片として簡便に得られる。ヒトサイトメガロウィルスの最初期プロモーターは、HindIIIE制限断片として簡便に得られる。ベクターとしてウシ乳頭腫ウィルスを用いて哺乳動物宿主でDNAを発現する系が、米国特許第4,419,446号に開示されている。この系の変更例は米国特許第4,601,978号に開示されている。また、単純ヘルペスウイルス由来のチミジンキナーゼプロモーターの調節下でのマウス細胞におけるヒトβインターフェロンcDNAの発現について、Reyes等, Nature, 297:598−601(1982)を参照されたい。あるいは、ラウス肉腫ウィルス長末端反復をプロモーターとして使用することができる。
【0077】
(v)エンハンサーエレメント成分
より高等の真核生物による本発明の抗Apo−2Lレセプター抗体をコードしているDNAの転写は、ベクター中にエンハンサー配列を挿入することによって増強され得る。哺乳動物の遺伝子由来の多くのエンハンサー配列が現在知られている(グロビン、エラスターゼ、アルブミン、α−フェトプロテイン及びインスリン)。しかしながら、典型的には、真核細胞ウィルス由来のエンハンサーが用いられるであろう。例としては、複製開始点の後期側のSV40エンハンサー(100−270塩基対)、サイトメガロウィルス初期プロモーターエンハンサー、複製開始点の後期側のポリオーマエンハンサー及びアデノウィルスエンハンサーが含まれる。真核生物のプロモーターの活性化のための増強要素については、Yaniv, Nature, 297:17−18 (1982)もまた参照のこと。エンハンサーは、抗体コード配列の5’又は3’位でベクター中にスプライシングされ得るが、好ましくはプロモーターから5’位に位置している。
【0078】
(vi)転写終結成分
真核生物宿主細胞(酵母、真菌、昆虫、植物、動物、ヒト、又は他の多細胞生物由来の有核細胞)に用いられる発現ベクターは、また転写の終結及びmRNAの安定化に必要な配列を含む。このような配列は、真核生物又はウィルスのDNA又はcDNAの5’、時には3’の非翻訳領域から一般に取得できる。これらの領域は、多価抗体をコードしているmRNAの非翻訳部分にポリアデニル化断片として転写されるヌクレオチドセグメントを含む。一つの有用な転写終結成分はウシ成長ホルモンポリアデニル化領域である。国際公開第94/11026号とそこに開示した発現ベクターを参照されたい。
【0079】
(vii)宿主細胞の選択及び形質転換
ここに記載のベクターにDNAをクローニングあるいは発現するために適切な宿主細胞は、上述の原核生物、酵母、又は高等真核生物細胞である。この目的にとって適切な原核生物は、真正細菌、例えばグラム陰性又はグラム陽性生物体、例えばエシェリチアのような腸内菌科、例えば大腸菌、エンテロバクター、エルウィニア(Erwinia)、クレブシエラ、プロテウス、サルモネラ、例えばネズミチフス菌、セラチア属、例えばセラチア・マルセスキャンス及び赤痢菌属、並びに桿菌、例えば枯草菌及びB.リチェフォルミス(licheniformis)(例えば、1989年4月12日に公開された DD 266,710に開示されたバシリ・リチェニフォルミス41P)、シュードモナス属、例えば緑膿菌及びストレプトマイセス属を含む。一つの好適な大腸菌クローニング宿主は大腸菌294(ATCC31446)であるが、他の大腸菌B、大腸菌X1776(ATCC31537)及び大腸菌W3110(ATCC27325)のような株も好適である。これらの例は限定するものではなく例示的なものである。
原核生物に加えて、糸状菌又は酵母菌のような真核微生物は、Apo−2Lレセプター抗体をコードするベクターのための適切なクローニング又は発現宿主である。サッカロミセス・セレヴィシア、又は一般的なパン酵母は下等真核生物宿主微生物のなかで最も一般的に用いられる。しかしながら、多数の他の属、種及び菌株も、一般的に入手可能でここで使用でき、例えば、シゾサッカロマイセスポンベ;クルイベロマイセス宿主、例えばK.ラクティス、K.フラギリス(ATCC12424)、K.ブルガリカス(ATCC16045)、K.ウィッケラミイ(ATCC24178)、K.ワルチイ(ATCC56500)、K.ドロソフィラルム(ATCC36906)、K.サーモトレランス、及びK.マルキシアナス;ヤローウィア(欧州特許第402,226号);ピチア・パストリス(欧州特許第183,070号);カンジダ;トリコデルマ・リーシア(欧州特許第244,234号);アカパンカビ;シュワニオマイセス、例えばシュワニオマイセス・オクシデンタリス;及び糸状真菌、例えばパンカビ属、アオカビ属、トリポクラジウム、及びコウジカビ属(Aspergillus)宿主、例えば偽巣性コウジ菌(A. nidulans)及びクロカビ(A. niger)が使用できる。
【0080】
グリコシル化抗体の発現に適切な宿主細胞は、多細胞生物から誘導される。無脊椎動物細胞の例には、植物及び昆虫細胞が含まれる。多数のバキュロウィルス株及び変異体及び対応する許容可能な昆虫宿主細胞、例えばスポドプテラ・フルギペルダ(毛虫)、アエデス・アエジプティ(蚊)、アエデス・アルボピクトゥス(蚊)、ドゥロソフィラ・メラノガスター(ショウジョウバエ)、及びボンビクス・モリが同定されている。トランスフェクションのための種々のウィルス株、例えば、オートグラファ・カリフォルニカNPVのL−1変異体とボンビクス・モリ NPVのBm−5株が公に利用でき、このようなウィルスは本発明においてここに記載したウィルスとして使用でき、特にスポドプテラ・フルギペルダ(Spodoptera frugiperda)細胞のトランスフェクションに使用できる。
綿花、コーン、ジャガイモ、大豆、ペチュニア、トマト、及びタバコのような植物細胞培養を宿主として利用することができる。
しかしながら、脊椎動物細胞におけるものが最も興味深く、培養(組織培養)中での脊椎動物細胞の増殖は常套的な手順になった。有用な哺乳動物宿主株化細胞の例は、SV40によって形質転換されたサル腎臓CV1株 (COS−7, ATCC CRL 1651);ヒト胚腎臓株[293又は懸濁培養での成長のためにサブクローン化された293細胞、Grahamほか, J. Gen Virol., 36:59 (1977)];ハムスター乳児腎細胞(BHK, ATCC CCL 10);チャイニーズハムスター卵巣細胞/−DHFR(CHO, Urlaub, Proc. Natl. Acad. Sci. USA, 77:4216 (1980));マウスのセルトリ細胞[TM4, Mather, Biol. Reprod., 23:243−251 (1980)];サルの腎細胞 (CVI ATCC CCL 70);アフリカミドリザルの腎細胞(VERO−76, ATCC CRL−1587);ヒト子宮頸癌細胞 (HELA, ATCC CCL 2);イヌ腎細胞 (MDCK, ATCC CCL 34); バッファローラット肝細胞 (BRL 3A, ATCC CRL 1442);ヒト肺細胞 (W138, ATCC CCL 75);ヒト肝細胞 (Hep G2, HB 8065);マウス乳房腫瘍(MMT 060562, ATTC CCL51);TRI細胞[Motherほか, Annals N.Y. Acad. Sci., 383:44−68 (1982)];MRC5細胞;FS4細胞;ヒト肝癌株(HepG2);及びミエローマ又はリンパ腫細胞(例えば、Y0, J558L, P3及びNS0細胞)(米国特許第5,807,715号を参照のこと)である。
宿主細胞は、抗体生成のための上述した発現又はクローニングベクターで形質転換し、プロモーターを誘導し、形質転換体を選択し、又は所望の配列をコードしている遺伝子を増幅するために適当に修飾された常套的栄養培地で培養する。
【0081】
(viii)宿主細胞の培養
本発明の抗体を生成するために用いられる宿主細胞は種々の培地において培養することができる。市販培地、例えばハム(Ham)のF10(シグマ)、最小必須培地((MEM),シグマ)、RPMI−1640(シグマ)及びダルベッコの改良イーグル培地((DMEM),シグマ)が宿主細胞の培養に好適である。また、Ham等, Meth. Enz. 58:44 (1979), Barnes等, Anal. Biochem. 102:255 (1980), 米国特許第4,767,704号;同4,657,866号;同4,927,762号;同4,560,655号;又は同5,122,469号;国際公開第90/03430;国際公開第87/00195;又は米国特許再発行第30,985号に記載された任意の培地も宿主細胞に対する培地として使用できる。これらの培地はいずれも、ホルモン及び/又は他の成長因子(例えばインスリン、トランスフェリン、又は表皮成長因子)、塩類(例えば、塩化ナトリウム、カルシウム、マグネシウム及びリン酸塩)、バッファー(例えばHEPES)、ヌクレオチド(例えばアデノシン及びチミジン)、抗生物質(例えば、ゲンタマイシン(商品名)薬)、微量元素(最終濃度がマイクロモル範囲で通常存在する無機化合物として定義される)及びグルコース又は同等のエネルギー源を必要に応じて補充することができる。任意の他の必要な補充物質もまた当業者に知られている適当な濃度で含むことができる。培養条件、例えば温度、pH等々は、発現のために選ばれた宿主細胞について以前から用いられているものであり、当業者には明らかであろう。
【0082】
(ix)精製
組換え技術を使用する場合、抗体は細胞内、細胞膜周辺腔内に生成されるか、又は培地に直接分泌され得る。抗体が細胞内に生成される場合、第1段階として、粒状屑、宿主細胞又は溶菌断片を、例えば遠心分離又は限外濾過にかけて取り除く。Carter等, Bio/Technology 10:163−167(1992)は、大腸菌の細胞膜周辺腔に分泌されるタンパク質を単離するための手順について記載している。簡単に述べると、細胞ペーストを酢酸ナトリウム(pH3.5)、EDTA、及びフェニルメチルスルホニルフロリド(PMSF)の存在下で、30分以上かけて解凍する。細胞屑は遠心分離により除去することができる。抗体が培地へ分泌されている場合、そのような発現系からの上清は、一般的には第1に市販のタンパク質濃縮フィルター、例えばAmicon又はMillipore Pelliconの限外濾過ユニットを用いて濃縮する。PMSFなどのプロテアーゼ阻害剤を上記の任意の工程に含めてタンパク質分解を阻害してもよく、抗生物質を含めて外来性の汚染物の成長を防止してもよい。
【0083】
細胞から調製した抗体組成物は、例えば、ヒドロキシアパタイトクロマトグラフィー、ゲル電気泳動、透析、及びアフィニティクロマトグラフィを用いて精製でき、アフィニティクロマトグラフィが好ましい精製技術である。アフィニティリガンドとしてのプロテインAの適合性は、抗体に存在する任意の免疫グロブリンFc領域の種及びアイソタイプに依存する。プロテインAは、ヒトγ1、γ2、又はγ4重鎖に基づく抗体の精製に用いることができる(Lindmark等, J. Immunol. Meth. 62:1−13 (1983))。プロテインGは、全てのマウスアイソタイプ及びヒトγ3に推奨されている(Guss等, EMBO J. 5:15671575 (1986))。アフィニティリガンドが結合されるマトリクスはアガロースであることが最も多いが、他のマトリクスも使用可能である。孔制御ガラスやポリ(スチレンジビニル)ベンゼン等の機械的に安定なマトリクスは、アガロースで達成できるものより早い流速及び短い処理時間を可能にする。抗体がCH3ドメインを含む場合、Bakerbond ABX(商品名)樹脂(J.T. Baker, Phillipsburg, NJ)が精製に有用である。イオン交換カラムでの分画、エタノール沈殿、逆相HPLC、シリカ上のクロマトグラフィー、アニオン又はカチオン交換樹脂(例えばポリアスパラギン酸カラム)上でのヘパリンSEPHAROSE(商品名)クロマトグラフィー、クロマトフォーカシング、SDS−PAGE、及び硫酸アンモニウム沈殿などの他の技術も、回収される抗体に応じて利用可能である。
【0084】
C.製剤
Apo−2リガンド又はApo−2Lレセプターアゴニスト抗体とCPT−11は好ましくは担体で投与される。分子は、単一の担体で投与することができ、あるいは別個の担体に含めることができる。適した担体とその製剤は、Osloらにより編集されたRemington’s Pharmaceutical Sciences, 16th ed.., 1980, Mack Publishing Co.に記載されている。典型的には、適当量の製薬的に許容可能な塩が製剤を等張にするために担体中に使用される。担体の例には、生理食塩水、リンゲル液及びブドウ糖液が含まれる。溶液のpHは好ましくは約5から約8、より好ましくは約7.4から約7.8である。例えば投与経路及び投与される薬剤の濃度に応じてある種の担体がより好ましいことは当業者には明らかであろう。担体は凍結乾燥された製剤又は水性溶液の形態をとりうる。
許容できる担体、賦形剤又は安定剤は、好ましくは、用いる投与量及び濃度では細胞及び/又はレシピエントに対して無毒性であり、リン酸、クエン酸及び他の有機酸等の緩衝液;アスコルビン酸及びメチオニンを含む抗酸化剤;防腐剤(例えば、オクタデシルジメチルベンジルアンモニウムクロリド;塩化ヘキサメトニウム;塩化ベンズアルコニウム;塩化ベンゼトニウム;フェノール、ブチル又はベンジルアルコール;アルキルパラベン類、例えばメチル又はプロピルパラベン;カテコール;レゾルシノール;シクロヘキサノール;3−ペンタノール;及びm−クレゾール);低分子量(残基数10個未満)ポリペプチド;血清アルブミン、ゼラチン又は免疫グロブリン等のタンパク質;ポリビニルピロリドン等の親水性重合体;グリシン、グルタミン、アスパラギン、ヒスチジン、アルギニン、又はリシン等のアミノ酸;グルコース、マンノース又はデキストリン等の単糖類、二糖類及び他の炭水化物;EDTA等のキレート剤;スクロース、マンニトール、トレハロース又はソルビトール等の糖類、ナトリウム等の塩形成対イオン;及び/又はTWEENTM、PLURONICSTM又はポリエチレングリコール(PEG)等の非イオン性界面活性剤を含む。
【0085】
また、製剤は、治療される特定の効能に必要な一以上の活性化合物、好ましくは互いに悪影響を及ぼさない相補的活性を有するものを含有し得る。あるいは、又は加えて、組成物は、細胞障害剤、サイトカイン又は成長阻害剤を含有してもよい。このような分子は、好適には、意図した目的に有効な量で組合せて存在する。
またApo−2L又はアゴニスト抗体とCPT−11は、例えばコアセルベーション法又は界面重合により調製されたマイクロカプセル、例えばそれぞれヒドロキシメチルセルロース又はゼラチン−マイクロカプセル及びポリ−(メチルメタクリレート)マイクロカプセルに、コロイド状薬物送達系(例えば、リポソーム、アルブミンミクロスフィア、マイクロエマルション、ナノ粒子及びナノカプセル)又はマクロエマルションに捕捉することができる。このような技術はRemington’s Pharmaceutical Sciences, 16th edition, Oslo,A.編(1980)に開示されている。
インビボ投与に使用される製剤は滅菌されていなくてはならない。これは、滅菌濾過膜を通す濾過により容易に達成される。
徐放性調製物を調製してもよい。徐放性調製物の適切な例には、抗体を含む固体疎水性重合体の半透性マトリックスが含まれ、このマトリックスはフィルム又はマイクロカプセル等の成形品の形態である。徐放性マトリックスの例には、ポリエステル、ヒドロゲル(例えばポリ(2−ヒドロキシエチル−メタクリレート)、又はポリ(ビニルアルコール))、ポリラクチド(米国特許第3,773,919号)、L−グルタミン酸とγ−エチル−L−グルタマートのコポリマー、非分解性エチレン−酢酸ビニル、分解性の乳酸−グリコール酸コポリマー、例えばLUPRON DEPOTTM(乳酸−グリコール酸コポリマー及び酢酸ロイプロリドからなる注入可能なミクロスフィア)、及びポリ−D−(−)−3−ヒドロキシ酪酸が含まれる。エチレン−酢酸ビニルや乳酸−グリコール酸等のポリマーは100日以上分子を放出できるが、特定のヒドロゲルはより短い時間タンパク質を放出する。
【0086】
D.投与方法
Apo−2L又はApo−2Lレセプターアゴニスト抗体とCPT−11は、公知の方法、例えばボーラスとしてもしくは一定時間にわたる連続注入による静脈内投与、筋肉内、腹腔内、脳脊髄内、皮下、関節内、滑液包内、くも膜下腔内、経口、局所的、又は吸入経路により投与することができる。場合によっては、投与は、様々な市販の装置を使用するミニポンプ流入によって実施することもできる。
Apo−2リガンド又はアゴニスト抗体とCPT−11の投与の有効な用量は経験的に決定することができ、そのような決定を行うことは当業者の技量の範囲にある。単独に使用されるApo−2リガンドの効果的な用量又は量は一日当り約1μg/kgから約100mg/kg体重あるいはそれ以上までの範囲であると現在は考えられる。単独に使用されるCPT−11の効果的な用量又は量は約1mg/m2から約150mg/m2の範囲である。用量の種間スケーリングは、例えば、Mordenti等, Pharmaceut. Res., 8:1351(1991)に開示されているような当該分野で知られている方法で実施することができる。当業者であれば、投与されなければならないApo−2リガンド又はアゴニスト抗体とCPT−11の用量は、例えばApo−2リガンド又はアゴニスト抗体とCPT−11を受ける哺乳動物、投与経路、及び哺乳動物に投与されている他の薬物又は治療法に応じて変化することを理解するであろう。
細胞の型及び/又は病気の重症度に応じて、例えば一又は複数の別個の投与又は連続注入の何れであれ、約1μg/kgないし15mg/kg(例えば0.1−20mg/kg)のアゴニスト抗体が、投与の最初の候補用量である。上述した要因に応じて、典型的な一日の投与量は約1μg/kgから100mg/kgあるいはそれ以上の範囲である。数日間又はそれ以上の繰り返し投与の場合、状態によっては、病気の徴候の望ましい抑制が生じるまで処置を維持する。しかしながら、他の投薬計画も有効であろう。
【0087】
CPT−11で細胞を前処理すると、選択された集団の細胞中でのアポトーシスを誘発するのに必要なApo−2Lレセプターアゴニストの量が低減すると考えられている。例えば、CPT−11で細胞を前処理すると、少なくとも25%、好ましくは少なくとも50%まで、哺乳動物細胞におけるアポトーシスの誘発(等しい量又は程度)に必要なApo−2Lレセプターアゴニストの量が低減する。
一又は複数のApo−2Lレセプターアゴニストが本方法において使用されると考えられている。例えば、当業者であれば、Apo−2リガンド、DR4アゴニスト抗体、DR5アゴニスト抗体、又はそれらの組合せを使用するであろう。場合によっては、Apo−2Lレセプターアゴニスト抗体は、DR4及びDR5の双方に結合する交差反応抗体を含むであろう。
【0088】
さらなる治療法を本方法において使用することも考えられる。一又は複数の他の治療法には、これらに限定されるものではないが、他の化学療法(又は化学療法剤)及び/又は放射線療法、免疫アジュバント、成長阻害因子、サイトカイン、及び他の非Her−2抗体をベースとした治療法が含まれる。例としては、インターロイキン(例えば、IL−1、IL−2、IL−3、IL−6)、白血病抑制因子、インターフェロン、TGF−β、エリスロポイエチン、トロンボポイエチン、及び抗VEGF抗体が含まれる。哺乳動物においてアポトーシスを誘発することが知られている他の薬剤もまた使用することができ、それら薬剤には、TNF−α、TNF−β(リンホトキシン−α)、CD30リガンド、4−1BBリガンド、及びApo−1リガンドが含まれる。
本発明により考えられる化学療法には、当該分野で知られ、商業的に入手可能な化学物質又は薬物、例えばアドリアマイシン、ドキソルビシン、5−フルオロウラシル、シトシンアラビノシド(「Ara−C」)、シクロホスファミド、ロイコボリン、チオテパ、ブスルファン、サイトキシン、タキソール、トキソテレ(Toxotere)、メトトレキサート、シスプラチン、メルファラン、ビンブラスチン、ブレオマイシン、エトポシド、イフォスファミド、マイトマイシンC、マイトキサントロン、ビンクレイスチン、ビノレルビン、カルボプラチン、テニポシド、ダウノマイシン、カルミノマイシン、アミノプテリン、ダクチノマイシン、マイトマイシン、エスペラマイシン(米国特許第4,675,187号参照)、メルファラン及び他の関連ナイトロジェン・マスタードが含まれる。また含まれるものは、タモキシフェン及びオナプリストーンのような腫瘍のホルモン作用を調節又は阻害するように作用する薬剤である。
【0089】
このような化学療法の調製と用量スケジュールは、製造者の指示書に従って使用されるか、有能な実務者により経験的に決定されて使用される。このような化学療法の調製と用量スケジュールは、Chemotherapy Service Ed., M.C. Perry, Williams & Wilkins, Baltimore, MD (1992)にも記載されている。化学療法剤は、Apo−2L又はアゴニスト抗体及び/又はCPT−11の投与の前でも後でもよく、またはそれと同時に与えてもよい。
化学療法は、好ましくは、上述したもののような担体で投与される。化学療法の投与方法は、Apo−2リガンド又はアゴニスト抗体又はCPT−11に対して使用するものと同じものとでき、あるいは異なった方法で投与することもできる。
【0090】
放射線療法は、当該分野で一般的に使用され、当業者に知られているプロトコルに従って施すことができる。このような療法には、セシウム、イリジウム、ヨウ素又はコバルト照射が含まれる。放射線療法は、体全体の照射でも、体の中又は上の特定の部位又は組織に局所的に向けられるものでもよい。典型的には、放射線療法は、約1週から約2週の期間にわたって、パルスで施される。しかしながら、放射線療法をより長い時間、施してもよい。場合によっては、放射線療法は、単一用量又は複数の連続用量として施されうる。
Apo−2リガンド又はアゴニスト抗体とCPT−11の投与に続いて、インビトロの治療細胞を分析することができる。インビボ治療があった場合、治療された哺乳動物は熟練した実務者によく知られた様々な方法でモニターすることができる。例えば、身体的に、バイオプシーにより、あるいは標準的なX線画像法により腫瘍を観察することができる。
【0091】
III. 製造品
本発明の他の実施態様では、前述した疾患の治療に有用な物質を含有する製造品が提供される。製造品は容器及びラベルを含んでなる。適切な容器には、例えばボトル、バイアル、シリンジ、及び試験管が含まれる。容器はガラス又はプラスチックのような様々な材料で形成することができる。容器は病状の治療に有効な組成物を収容しており、滅菌したアクセスポートを有している(例えば、容器は皮下注射針により貫通可能なストッパーを具備する静脈溶液用のバック又はバイアルであってよい)。組成物中の活性剤はApo−2リガンド又はアゴニスト抗体とCPT−11である。容器上の又は容器に伴うラベルには、組成物が、選択された病状の治療に使用されることが示されている。製造品は、製薬的に許容可能なバッファー、例えばリン酸緩衝生理食塩水、リンガル液及びブドウ糖液を収容する第2の容器をさらに具備する。さらに、他のバッファー、希釈剤、フィルター、針、シリンジ、及び使用指示書を含むパッケージ挿入物を含む、市販及び使用者の観点から望ましい他の材料をさらに含んでいてもよい。
【0092】
以下の実施例は例示するためにのみ提供されるものであって、限定するものではない。本明細書で引用した全ての文献を、出典明示によりここに取り込む。
実施例1
この実施例は、インビボのApo−2リガンド及びCPT−11による腫瘍成長の相乗的抑制について例証するものである。
大腸癌細胞株COLO205(NCIから入手可能)が育成され、サプライヤー法によって維持された。簡単に述べると、COLO205細胞が10%胎児ウシ血清と2.0mML−グルタミンを含有する高グルコースDMEM/F12(50:50)培地で培養された。アミノ酸114−281(配列番号:1)を含んでなるApo−2リガンドが大腸菌で調製された。ヒトApo−2Lの細胞外タンパク質(アミノ酸114−281;上掲のPitti等参照)は、加えられた開始メチオニンコドンを有するpS1346発現プラスミド(Scholtissek等, Gene, 62:55−64(1988))中にサブクローン化され、10L又は100Lの発酵器でtrpプロモーターのコントロールの下、大腸菌株W3110中に発現された。組換えヒト可溶性Apo−2Lを含有する細胞ペーストが、0.1M トリス/0.2M NaCL/50mM EDTAを含有する緩衝液、pH8.0で抽出された。抽出物は、40%硝酸アンモニウムにより沈殿させた。ヒドロキシアパタイト及びNi−NTAアガロースカラムでの2つの連続したクロマトグラフィー分離段階により、精製度>98%均一性を達成した。(しかし、それはポリヒスチジンタグが欠け、組換え可溶性114−281アミノ酸Apo−2L断片が内因性のヒスチジン残基を通じてNi−NTAカラムに結合したと考えられる)。純度は、ドデシル硫酸ナトリウム(SDS)−ポリアクリルアミドゲル電気泳動及び硝酸銀又はカーマシーブルー染色により、アミノ酸配列分析、及び高速液体クロマトグラフィ−(HPLC)によるサイズ除外にて決定される。CPT−11(Camptosar(登録商標))はPharmacia&Upjohnから入手した。
【0093】
胸腺欠損ヌードマウス(Jackson Laboratories)に500万のCOLO205大腸癌細胞を皮下注射し、腫瘍を約120mm3まで成長させた。腫瘍を持つマウスは、1グループにつき9匹の4グループ中にランダム化され、ビヒクル(20mMトリス、8%トレハロース、0.01Tween−20、pH7.5)、Apo−2L(0−4及び7−11日目に30mg/kg/日)、又はCPT−11(0、4及び8日目に80mg/kg/日)、又はApo−2L(0−4及び7−11日目に30mg/kg/日)+CPT−11(0、4及び8日目に80mg/kg/日)の何れかで治療した。腫瘍の用量は、34日までの表示された日に測定された。
図1に示されるように、Apo−2L(白抜き三角)又はCPT−11(白抜き四角)は、それぞれ、治療期間の腫瘍成長を抑制したが、それぞれのグループの9動物全てで数日後、腫瘍成長が再開した。これに対して、Apo−2LとCPT−11の組み合わせ(塗りつぶされた三角)では、かなりの腫瘍減縮が起り、その結果、組み合わせ治療グループの9のうち8の動物で完全に腫瘍が消失した。
この実験の結果は、Apo−2LリガンドとCPT−11治療がインビボの癌細胞の成長を相乗的に抑制することを示している。
【0094】
実施例2
この実施例はインビボのDR4レセプターアゴニスト抗体、4H6.17.8(「4H6」)及びCPT−11による腫瘍成長の相乗的抑制について例示するものである。
アゴニスト抗体が、次のようにして調製された。可溶性のDR4ECDイムノアドヘシン作成物を調製した。成熟DR4 ECD配列(Panらにより示されたアミノ酸1−218、上掲)を、pCMV−1Flagベクター(Kodak)のFlagシグナル配列の下流にクローニングし、既に記載されているようにして[Aruffo等,Cell, 61:1303−1313(1990)]ヒト免疫グロブリンG1重鎖のCH1、ヒンジ及びFc領域に融合した。イムノアドヘシンは、ヒト293細胞に一過性形質移入して発現させ、Ashkenazi等, Proc. Natl. Acad. Sci., 88:10535−10539(1991)により記載されたプロテインAアフィニティークロマトグラフィーにより細胞上清から精製した。
0.5μg/50μlのDR4ECDイムノアドヘシンタンパク質(上記)(Ribi Immunochemical Research Inc., Hamilton, MTから購入したMPL−TDMアジュバントで希釈)を、各々の後足の裏の柔らかい部分に、3−4日間隔で11回注射することで、Balb/cマウス(チャールズリヴァー研究所から得たもの)を免疫化した。
最終の追加免疫から3日後に、膝窩リンパ節をマウスから取り出し、単細胞懸濁液を、1%のペニシリン−ストレプトマイシンが補われたDMEM培地(Biowhitakker Corp.から得たもの)中で調製した。次いで、リンパ節細胞を35%のポリエチレングリコールを使用してマウス骨髄腫細胞P3X63AgU.1(ATCC CRL 1597)と融合させ、96−ウェル培養プレートで培養した。融合の結果得られたハイブリドーマをHAT培地において選択した。融合から10日後に、ハイブリドーマ培養の上清をELISAでスクリーニングし、DR4ECDイムノアドヘシンタンパク質(上記)に結合するモノクローナル抗体の存在を試験した。
【0095】
ELISAでは、各々のウェルに、PBS中に2μg/mlのヤギ抗ヒトIgGFc(Cappel Laboratoriesから購入)が入ったものを50μl添加して、96−ウェルマイクロタイタープレート(Maxisorp; Nunc, Kamstrup, Denmark)をコートし、4℃で一晩インキュベートした。次いでプレートを洗浄用バッファー(0.05%のトゥイーン20を含有するPBS)で3回洗浄した。次いで、マイクロタイタープレートのウェルを、PBSに2.0%のウシ血清アルブミンが入ったもの200μlでブロックし、室温で1時間インキュベートした。次いで、プレートを洗浄用バッファーで再度3回洗浄した。
洗浄工程後、アッセイ用バッファー中の0.4μg/mlのDR4ECDイムノアドヘシンタンパク質50μlを各ウェルに添加した。プレートを振盪装置上で、室温で1時間インキュベートし、続いて洗浄用バッファーで3回洗浄した。
洗浄工程に続いて、100μlのハイブリドーマ上清又はプロテインG−セファロースカラム精製抗体(10μg/ml)を指定ウェルに添加した。100μlのP3X63AgU.1骨髄腫細胞条件培地を他の指定ウェルに対照として添加した。プレートを振盪装置上で、室温で1時間インキュベートし、続いて洗浄用バッファーで3回洗浄した。
【0096】
次に、アッセイ用バッファー(0.5%のウシ血清アルブミン、0.05%のトゥイーン20がPBS中に入ったもの)で、1:1000に希釈されたHRP−抱合ヤギ抗マウスIgG Fc(Cappel Laboratoriesから購入)の50μlを各々のウェルに添加し、プレートを振盪装置上で、室温で1時間インキュベートした。プレートを洗浄用バッファーで3回洗浄し、次いで各々のウェルに50μlの基質(TMBマイクロウェルペルオキシダーゼ基質;Kirkegaard & Perry, Gaithersburg, MD)を添加し、室温で10分間インキュベートした。50μlのTMB1−成分終止溶液(ジエチルグリコール;Kirkegaard & Perry)を各々のウェルに添加して反応を終了させ、450nmでの吸光度を自動マイクロタイタープレート読取装置で読み取った。
最初にELISAでスクリーニングしたハイブリドーマ上清は、CD4−IgGではなくDR4−IgGに結合するそれらの能力について考慮した。ELISAで陽性とされた上清は、9D細胞(DR4を発現するヒトBリンパ細胞系;Genentech, Inc.)及びFITC抱合ヤギ抗マウスIgGを用いたFACS分析でさらに分析した。この分析のために、セルソーター用バッファー(1%のFCS及び0.02%のNaN3を含むPBS)中の細胞懸濁物(4x106細胞/ml)の25μlを、U字底のミクロタイターウェルに添加し、100μl培養上清又はセルソーター用バッファー中の精製抗体(10μg/ml)と混合し、氷上で30分間インキュベートした。次いで、細胞を洗浄し、100μlのFITC抱合ヤギ抗マウスIgGとともに4℃で30分間インキュベートした。次いで、細胞を2回洗浄し、150μlのセルソーター用バッファーに再懸濁し、FACScan(Becton Dickinson, Mountain View, CA)によって分析した。
【0097】
9D細胞のFACS染色は、抗体、4E7.24.3及び4H6.17.8が、9Dが細胞上のDR4レセプターを認識したことを明らかにした。ハイブリドーマ上清及び精製抗体を、DR4に媒介された9D細胞のアポトーシスを誘発する能力について試験した。9D細胞(5x105細胞/0.5ml)を5μgのDR4mAbs(4E7.24.3又は4H6.17.8)又はIgG対照抗体とともに200μl完全RPMI培地中、4℃で15分間インキュベートした。次いで、細胞を、10μgのヤギ抗マウスIgGFc抗体(ICN Pharmaceuticals)を共なう又は共なわない、300μlの完全RPMI中、37℃で5分間インキュベートした。この時点で、細胞を7%のCO2存在下、37℃で一晩インキュベートした。次いで細胞を回収し、PBSで1回洗浄した。細胞のアポトーシスを、製造者の推奨(Clontech)に従ってホスファチジルセリンに結合するFITC−アネキシンVの染色によって決定した。細胞をPBS中で洗浄し、200μlの結合バッファー中に再懸濁させた。10μlのアネキシン−V−FITC(1μg/ml)及び10μlのヨウ化プロピジウムを細胞に添加した。暗中で15分間インキュベートした後、9D細胞をFACSで分析した。
どちらのDR4抗体も(ヤギ抗マウスIgGFc無しで)対照抗体に比較して9D細胞のアポトーシスを誘発した。しかし、両方のDR4抗体のアゴニスト活性は、ヤギ抗マウスIgGFc存在下でのDR4レセプター交差結合によって高められた。両方のDR4抗体によるこの高められたアポトーシスは、9D細胞におけるApo−2Lのアポトーシス活性に匹敵した。
【0098】
4H6.17.8モノクローナル抗体+CPT−11の効果(図2に示される他の治療グループと比較して)を試験したインビボ研究は、抗体治療グループにおいて抗DR4抗体4H6(5mg/kg;上述のようにして調製)が研究期間に毎週2回のマウスへのi.p.注射により投与されたこと以外は、原則的に前記実施例1に記載されたようにして実施された。Apo−2L治療グループにおいては、60mg/kg/日で0−4日目にi.p.注射によりApo−2Lを投与した。CPT−11おいては、80mg/kgで0、4及び8日目にi.v.注射によりCPT−11を投与した。
結果は図2に示される。それぞれの薬剤単独では、腫瘍の進行をかなり遅らせた。Apo−2L又は抗DR4モノクローナル抗体とCPT−11の組み合わせでは、単一薬剤での治療と比較して腫瘍進行がより大きく遅れ、腫瘍退行を引き起こした。抗DR4モノクローナル抗体は、Apo−2L単独薬剤として、及びCPT−11との組み合わせの両方よりも効果的であった。有効(当初の50%よりも多く減少した腫瘍の容量)は、抗DR4抗体+CPT−11で治療した10のマウス全てにおいて引き起こされたが、Apo−2L+CPT−11での治療では10のうち6のマウスのみであった。これらの結果から、Apo−2Lレセプターアゴニストは、CPT−11と協同して、それぞれ単一薬剤での効果の加法合計を超越して、腫瘍進行の相乗的に抑制することを示す。
【0099】
実施例3
この実施例はCPT−11とApo2L/TRAILの生理学的効果を記載し、Apo2l/TRAILにさらす前にCPT−11で細胞を前処理することで、DR5及びDR4mRNA、並びにカスパーゼ(caspase)−3−様切断/活性化、及びアポトーシスが最も誘発されることを示すものである。
ここで使用される略語には:Apo2L/TRAIL、Apo2リガンド/腫瘍壊死因子関連アポトーシス誘発性リガンド(実施例1及び2に記載されたようにして調製);DR、死亡レセプター;DcR、デコイレセプター;FADD、死亡ドメインを有するFas−結合性タンパク質;CPT、カンプトテシン;CPT−11、イリノテカン;HUVEC、ヒト臍静脈内皮細胞;TNF、腫瘍壊死因子;FLIP、flice−抑制タンパク質;CDDP、シスジアミンジクロロプラチナム(II);CDK、サイクリン依存性キナーゼが含まれる。
細胞培養のために、ヒト腫瘍大腸癌株化細胞HCT116をアメリカン・タイプ・カルチャー・コレクション(Manassas, VA)から得た。細胞を、10%の胎児ウシ血清、1mMのグルタミン、100単位/mlのペニシリン及び100μg/mlのストレプトマイシンを含有するRPMI1640培地で培養した。薬剤処理する前に、150cmのプレートで24時間、細胞を継代培養した。ヒト臍静脈内皮細胞(HUVEC)をセル・システム(Cell Systems)(#2V0−C75;Kirkland, WA)から得て、CS−CTM完全培地(#4Z0−500, セル・システム)においてインキュベートした。
【0100】
細胞生存度を測定するために、AlamarBlueTMアッセイを使用した。96ウェル組織培養プレートにおいて、HCT116大腸癌細胞(〜10000細胞/ウェル)を10%FBSのRPMI1640培地にて一晩インキュベートした。翌日培地を取り除き、Apo2L/TRAIL単独(1μg/mL)、CPT−11(50μg/mL)又はApo2L/TRAIL+CPT11を含有する血清フリーの培地で、細胞を24時間インキュベートした。24時間のインキュベート時間の最後の6時間に、AlamarBlueTMをウェルに添加した。励起530nm及び発光590nmにて、96ウェル蛍光光度プレート読取機(CytoFlour multi−well plate reader series 4000, PerSeptive Biosystems;Framingham, MA)を使用して、蛍光を読み取った。さらに、分子プローブであるLive/Dead(登録商標)生存度/細胞障害性キット(Eugene, OR)を使用し、生存又は死亡した細胞の存在を評価した。これらのアッセイにおいては、フルオレセイン(カルセイン)とローダミン(エチジウムホモダイマー−1)フィルターを具備するAxiovert25(Zeiss;Thornwood, NY)蛍光顕微鏡下で培養物を観察する15分前に、カルセイン(Calcein)−Am(4μM)とエチジウムホモダイマー−1(2μM)を添加して細胞を処理した。
【0101】
また、クリスタルバイオレットアッセイも使用した。96ウェル組織培養プレートにおいて、HCT116大腸癌細胞(約20000細胞/ウェル)を10%胎児ウシ血清(FBS)のRPMI1640培地にて一晩インキュベートした。翌日培地を取り除き、上述した種々の薬剤を含有する新鮮な培地で、細胞を24又は48時間インキュベートした。各処理の終わりに培地を取り除き、0.5%のクリスタルバイオレット溶液を各ウェルに100μl添加し、水で洗浄する前に10分間、室温でインキュベートした。ウェルを乾燥させた後、0.5NのHClを含有するエタノールを各ウェルに100μl添加した。ついで、プレートを96ウェルプレート読取機(Spectra Max 340pc, Molecular Devices Corporation, Sunnyvale, CA)を使用し、540nmで読み取った。
【0102】
カスパーゼ活性をカスパーゼ−3アッセイキット(Clontech;Palo Alto, CA)により測定した。該アッセイを製造者の指示書に従い行った。HCT116細胞を、10%のFCS、1mMのグルタミン、100単位/mlのペニシリン及び100μg/mlのストレプトマイシンを含有するRPMI培地で培養し、図の説明文に記載しているような種々の処理を施した。処理後、細胞を収集し、冷PBSで1回洗浄し、アッセイ時間まで−20℃で凍結した。細胞ペレットを解凍し、キットにて提供される細胞溶解バッファーにより10分間、氷上で溶解させた。溶菌液を、反応バッファー中にて37℃で1時間、蛍光発生的カスパーゼ基質(Z−DEVD−AFE, 100μM)と共にインキュベートした。サンプルを、400/30nmの励起フィルター及び508/20nmの発光フィルターを具備するCytoFluor multi−well plate reader(PerSeptive Biosystems)にて分析した。相対蛍光レベルを各サンプルのタンパク質濃度に対して規準化した。
【0103】
全RNAを、製造者の指示書に従い、RNA Stat−60TM溶液(Tel−Test, Inc (Friendsweed, TX)を使用して単離した。Quantigene bDNATMシグナル増幅キット(Chiron diagnostics;East Walpole, MA)を使用し、mRNAレベルを評価した。GAPDHプローブ対の配列は製造者の推奨に従って。DR4プローブ対は、核酸残基9−582の領域に配列させた。5捕獲プローブ、16標識プローブ及び8ブロックプローブが存在した。DR5プローブ対を核酸残基13−591の領域に配列した。DR5に対する5捕獲プローブ、17レベルプローブ及び5ブロックプローブが存在する。組換えDcR1、DcR2、DR4及びDR5構築物を使用し、インビトロ転写からのRNAを使用し、プローブの特異性をテストした。DR4及びDR5プローブ対の両方ともそれら自身のRNA転写に対して高い特異性があった。各プローブ対からのシグナルは、テストされた濃度では直線状であった。約2μgの全RNA/ウェルを使用して、製造者の指示書に従いbDNAアッセイを行った。
【0104】
ウエスタンブロット法のために、10%のグリセロール、1%のトリトン−x100、150mMのNaCl及びプロテアーゼインヒビターを含有する20mMのトリス−HCl、pH7.4中で細胞を溶解した。(1mMのPMSF、10μg/mlのアプロチニン、10μg/mlのロイペプチン、シグマ)。ウェル当たり全タンパク質の50μgのアリコットを、NuPAGE MES SDSラニングバッファー(Novex, San Diego, CA)を有するNuPAGE 4−12%ビス−トリスゲル中で分離した。ゲルを、NuPAGEトランスファーバッファーと共に、セミドライトランスファー装置(Bio−dad;Hercules, CA)により、0.2μMの純粋ニトロセルロース膜(Bio7−Rad)にトランスファーさせた。5%のノンファットドライミルクを含有するPBSで膜をブロックし、興味あるタンパク質の第1抗体、続いて西洋ワサビペルオキシダーゼ結合第2抗体(Amersham;Braunschweig, Germany)と共にインキュベートした。増強された化学発光系により免疫反応バンドを可視化した(Amersham;Braunschweig, Germany)。この研究で使用した抗体は:抗−カスパーゼ−3(Stratagene, #200021;La Jolla, CA)、抗−p53及び抗−p21(Oncogene, #OP43及び#OP64;Cambridge, MA)、及び抗−FLIP(#343002, Calbiochem, San Diego, CA)である。抗体は、製造者に推奨される濃度で使用した。
【0105】
細胞周期分析をFACS分析を介して行った。HCT116及びHUVEC細胞をインキュベートし、図の説明文に記載したようにして処理した。処理後、培地中に浮遊する細胞とプレートで生存している細胞の両方を収集した。周期テストプラスDNATM試薬キット(Beckon Dickinson;San Jose, CA)を使用し、製造者の指示書に従って細胞を染色した。染色した細胞をFACSソーター(Becton Dickinson)において分析した。sub−G1 DNA内容物を含有するアポトーシス細胞のパーセンテージをCellQuestプログラムを使用して定量した。細胞周期における各相で生存している細胞のパーセンテージをModFit’ LTプログラムを使用して定量した。
【0106】
図5は、インビトロにおけるHCT116細胞のApo2L/TRAIL媒介性アポトーシスのCPT−11感作が時間依存していることを示すアポトーシスの時間特性を提供するものである。腫瘍細胞をカルセイン−AMとエチジウムホモダイマー−1(それぞれ、緑及び赤の蛍光)で染色し、続いてApo2L/TRAIL又はCPT−11を単独で、又は組み合わせて、これと共にインキュべートした。これらの条件下では、緑の蛍光は生存している細胞を表し;赤い染色は死亡している細胞を示す。処理2時間後、Apo2L/TRAIL及びApo2L/TRAIL+CPT−11の組み合わせにより、細胞収縮及び単層からの細胞剥離を含む、アポトーシスに特徴的な細胞変化が誘発された。しかしながら、この時点においては、Apo2L/TRAILとApo2L/TRAIL+CPT−11との間の細胞殺傷性において、あまり差はなかった。これに対し、Apo2L/TRAIL+CPT−11の組み合わせにより、エチジウムホモダイマーの取り込みに示されるように、24時間までの細胞死が明確に増加し、全細胞密度がはっきりと減少する結果となった。CPT−11単独でインキュベートしても、2時間後に何の形態的変化も見られなかった。しかしながら、処理24時間後には、いくつかの死亡した細胞がはっきりと存在していた。細胞生存度の定量的分析の結果(図4)は、形態学的蛍光データと一致した。処理24時間後、Apo2L/TRAIL+CPT−11の組み合わせにより、Apo2L単独のものと比較して、26%アポトーシスが増加する結果となった。
【0107】
クリスタルバイオレットアッセイを行い、いくつかのHCT116細胞をApo2L/TRAIL(100ng/m)とCPT−11(50マクログラム/ml)の組合せ処理に全体で24時間さらし、続いて、培地の単独存在においてさらなる24時間インキュベートした。連続処理された細胞グループにおいて、HCT116細胞を、最初の24時間CPT−11にさらし、ついで培地を変えて、さらなる24時間、Apo2L/TRAIL含有培地にさらした。これらの条件下では、連続処理された細胞における全細胞殺傷性は、組合せ処理で処理された細胞サンプルよりも約6%増強されていた(p<0.001、t−Test)。さらに、連続及び組合せ処理を比較する相対生存活性は、54%と同程度まで低減した。この効果は異なる濃度のCPT−11及びApo2L/TRAILでも観察された(データは図示せず)。
【0108】
Apo2L/TRAIL誘発性アポトーシスはカスパーゼ−3活性に関与していることが以前に報告されている(例えば、Muhlenbeckら, J.B.C. 273:33091−8(1998))ので、キャズパーゼ−3活性化のレベルをこれらの条件下で測定した。特に、経時的なカスパーゼ−3活性の評価を、上述したような蛍光定量的及びウエスタンブロット分析によりモニターした。カスパーゼ−3活性化のウエスタンブロット分析では、処理2時間後に、Apo2L/TRAILによりp24,p20及びp17形でカスパーゼ−3の切断がかなり誘発されたことが示された(図5)。カスパーゼ−3活性化は、蛍光定量的アッセイにより別途、確認された(図5)。興味深いことに、処理2時間後に、Apo2L/TRAIL+CPT−11の組合せにより同程度のカスパーゼ切断及び活性が誘発された。CPT−11単独では、カスパーゼ−3様活性は弱くはあるが確認可能で顕著であったが、切断はウエスタンブロットでは検出されなかった。24時間後、Apo2L/TRAIL+CPT−11の組合せインキュベートにより、Apo2L/TRAIL又はCPT−11単独のものと比較して、カスパーゼ−3プロセシング及び活性化がはっきりと増加した。さらに、細胞をCPT−11単独で一晩インキュベートし、続いてCPT−11に加えてApo2L/TRAILと共に2時間処理する組合せ処理の変形により、最も高い程度でカスパーゼ−3切断及び活性化がなされる結果となった。特に、20−22時間CPT−11で、続いて2時間Apo2L/TRAILで細胞を前処理することで、DR5及びDR4 mRNA、並びにカスパーゼ−3様切断/活性化、及びアポトーシスが最も高く誘発された。以上のことから、これらの結果には、Apo2L/TRAIL+CPT−11の組合せ処理後にカスパーゼ−3活性化が増強され、腫瘍アポトーシスの増加に至ることが示されている。
【0109】
Apo2L/TRAILレセプターDR5及びDR4の発現におけるApo2L/TRAIL及びCPT−11の効果を調査するために、bDNAアッセイを使用した。HCT116細胞を、Apo2L/TRAIL又はCPT−11を単独で、さらには組合せでの処理前及び処理後に分析した。Apo2L/TRAILは、処理2時間後に、DR5mRNA発現において、対照と比較して2倍の一過性増加を誘発した(図6)。DR5発現におけるApo2L/TRAIL誘発性変化は、処理24時間後に、対照のレベルまで戻った。これに対して、CPT−11単独では、処理24時間後に、DR4及びDR5mRNAの双方において2.5倍増加したが、2時間後では増加がない結果となった(図6)。22時間CPT−11単独で処理し、続いて2時間Apo2L/TRAIL+CPT−11で処理すると、DR5の発現が最も高くアップレギュレーションされる結果となった(3ないし4倍)。30分間の全ての処理によっては、レセプター発現に何の変化も見られなかった。カスパーゼ−3の切断/活性化レベルの時間特性は、それぞれ2及び24時間後にDR5及び/又はDR4のアップレギュレーションに続くものであった(図5)。これに対して、HUVEC細胞におけるDR5及びDR4発現は、これらの処理のいずれによっても何の影響も受けなかった。これらの結果により、Apo2L/TRAILにより腫瘍細胞中でDR5がアップレギュレーションされ、それ自身のアポトーシス活性が増強されることが示唆される。Apo2L/TRAIL+CPT−11の組合せ処理により、DR5及びDR4がさらにアップレギュレーションされることは、この観察を支持するものである。さらに、一般的なカスパーゼインヒビター、Z−VADの存在下で同様の実験を行い、生存している細胞よりはむしろ全細胞集団を、24時間後に分析した。Apo2L/TRAIL+CPT−11とZ−VADを組合せることで、Z−VADのないそれぞれの対照と比較して、さらに増加(DR5に対しては1.7対2.7倍、DR4に対しては1.9対3.0倍の増加)する結果となった(図7)。これらの結果は、これらの死亡レセプターのアップレギュレーションが細胞死の増加に寄与するという見解に一致した。
【0110】
Apo2L/TRAIL及びCPT−11による、DR5アップレギュレーションにおけるp53の関与を決定するために、p53タンパク質の発現レベルをウエスタンブロット分析により測定した。HCT116腫瘍及び正常なHUVEC細胞をApo2L/TRAIL又はCPT−11を単独で、及び組合せで処理した後、アリコットを分析した。Apo2L/TRAIL媒介性アポトーシスがp53に非依存的である(例えば、Ashkenaziら, Current Opinion in Cell Biology 11:255−260(1999)及びRiegerら, FEBS Letters 427:124−128(1998))ことを示した以前の報告と一致して、Apo2L/TRAILは、分析した任意の時間点においてp53タンパク質レベルを増加させなかった(図8)。これに対して、以前に報告されたように(McDonaldら, British Journal of Cancer 78:745−51(1998))、CPT−11と共にインキュベートすると、腫瘍及び正常細胞の双方において、処理2時間後と同様に早く、p53発現の強くて持続性のある誘発が生じる結果となった。少なくとも24時間、p53のCPT−11誘発は持続し、この誘発はApo2L/TRAILの添加による影響を受けなかった。
【0111】
アポトーシス対細胞停止におけるp21の役割も調べた。ウエスタンブロットにより、p21及びp53タンパク質レベルについて並行して、アリコットを分析した。以前に示したように、CPT−11単独では、双方の細胞型において強くp53を誘発した(図8)。また、CPT−11は、腫瘍及び正常細胞の双方において、24時間後に、p21タンパク質のかなりの誘発を媒介した(図9)。Apo2L/TRAIL単独ではp53又はp21の発現に影響はなかった。またApo2L/TRAIL+CPT−11による組合せ処理により、正常細胞における処理24時間後に、p53及びp21の強い発現が誘発された。驚くべきことに、HCT116腫瘍細胞の組合せ処理におけるp21タンパク質レベルは、CPT−11単独と類似した規模でのp53発現の増加にかかわらず、基底レベルのままであった。これらのデータにより、Apo2L/TRAILは、CPT−11処理後のp53における増加と関連して、p21の蓄積を抑制することの証拠を提供するものである。
【0112】
さらに、腫瘍アポトーシスのインビトロ実験モデルにおけるFLIPの関連可能性を研究した。HCT116細胞を先に記載したようにして、2及び24時間処理した。細胞溶解物を得、抗−FLIP抗体を使用するウエスタンブロット分析用に処理した。図10には、FLIPのタンパク質レベルは如何なる実験処理によっても影響を受けないことが示されている。(これは、FLIPがこの大腸癌株化細胞を使用するアポトーシス調節の因子ではないことを示している)。
【0113】
処理された細胞の細胞周期特性、特に細胞周期停止の出現におけるp21誘発と変化との間の正の相関性を測定するために、HCT116細胞を、Apo2L/TRAIL又はCPT−11を単独で、さらに組合せで処理した2、6及び24時間後に細胞周期分析にかけた。6時間後(図11)、CPT−11単独では、細胞周期特性においてかなりのシフトが誘発され、結果としてGo−G1細胞周期停止が生じた(76%)。また、この変化は、Apo2L/TRAIL+CPT−11の組合せ処理においても、より少ない程度ではあるが(55%)、認めることができ、Apo2L/TRAIL単独では誘発されなかった(43%vs.30%対照)。24時間までに、CPT−11処理により、G2−M期停止(43%vs. 19%対照)の出現、及びGo−G1期の明確な低減により特徴付けられる全く異なった結果を示した。より重要なことに、Apo2L/TRAIL+CPT−11の組合せは、処理24時間後において、このG2−M停止の出現(17%vs.19%対照)を完全に妨げた。これらのデータは、p21のCPT−11誘発におけるApo2L/TRAIL媒介性抑制と一致した。
【0114】
同様の実験条件下で24時間、正常細胞の細胞周期分析したところ、処理の間では何の差異も示されなかった。また、これらの細胞周期分析により、Apo2L/TRAIL+CPT−11の組合せ処理のアポトーシス活性の増加が確認された。対照細胞は1%であるのに対し、Apo2L/TRAIL+CPT−11(24時間)は42%アポトーシス、Apo2L/TRAIL単独では19%、CPT−11処理では9%という結果となった(図11)。これらの結果は、細胞周期停止経路よりも、むしろApo2L/TRAIL+CPT−11の組合せ処理が、p21誘発を防止し、癌細胞をアポトーシスに向かわせることにより腫瘍抑制を媒介するといった概念をさらに支持するものである。CPT−11インキュベートによる、p21タンパク質レベルにおけるさらに詳細な時間特性の変化では、処理18時間までの増加が処理4時間と同様に早く増加したことを示した。
【0115】
近年の研究では、カスパーゼ基質(例えば、Zhangら, Oncogene 18:1131−1138(1999);Levkauら, Molecular Cell 1:553−563(1998);Gervaisら, Journal of Biological Chemistry 273:19207−19212(1998)を参照)として、又はカスパーゼ活性化のインヒビター(例えば、Suzukiら, Oncogene 17:931−939(1998);Suzukiら, Oncogene 18:1239−44(1999);Suzukiら, Molecular & Cellular Biology 19:3842−3847(1999);Suzukiら, Oncogene 19:1346−1353(2000)を参照)として、アポトーシスプロセスにおけるp21に対する反対の役割が示されている。p21のタンパク質レベルにおけるカスパーゼApo2L/TRAILの役割を決定するために、一般的なカスパーゼインヒビター、Z−VADが存在するが、先に記載したようにして、HCT116細胞を処理した。カスパーゼ阻害により、Z−VADなしの同様の処理のものと比較して、Apo2L/TRAIL+CPT−11の組合せグループにおけるp21が、細胞内レベルにおいて顕著に増加する結果となった(図12)。大きな違いは、残存処理グループで観察されなかった。興味あることに、腫瘍細胞をCPT−11で一晩プレインキュベートし、次いで分析前にApo2L/TRAILで2時間処理した場合、24時間の規則的な組合せ処理において検出されたようにp21レベルにおいて同様の減少があった。p21の分解は、これらの条件下で約15Kdに切断された断片が存在することで確認された。この実験により、カスパーゼ活性を阻害することで、Apo2L/TRAILで誘発されるp21の他の強い分解が防止されることが示された。さらに、カスパーゼ活性化、p21レベル及び細胞周期停止の間の相関性を示すために、細胞周期分析を実施し、Apo2L/TRAILがカスパーゼインヒビター、Z−VADの存在下においてCPT−11誘発性G2−M停止を妨げないことを示した(図13)。
【0116】
Apo2L/TRAILとCPT−11の組合せ療法を検査するためにさらなる実験を行い、2つの異なる条件の組合せ治療を比較した。最初の条件(組合せ)において、腫瘍細胞を、インビトロで24時間、Apo2L/TRAIL及びCPT−11にさらし、続いてさらなる24時間、培地単独の存在下でインキュベートした。第2グループ(連続)においては、最初の24時間CPT−11にさらし、ついで、さらなる24時間、Apo2L/TRAILを単独で含有する培地に変えた。図14Aには、連続処理における全細胞殺傷性が、組合せグループよりも約6%増強されていた(p<0.001、t−Test)。さらに、連続及び組合せ処理を比較する相対殺傷活性は、54%と同程度まで増加した。この効果は異なる濃度のApo2L/TRAILでも見られた(図14A)。さらに、組合せ処理を超えた連続治療の増加したアポトーシス効果は、細胞が薬剤フリーの培地でさらに4日間インキュベートし、続いて2日間の薬剤暴露の場合、さらに68%まで増強された(図14B)。
また、CPT−11、SN38の活性な代謝産物が代わりに用いられた場合、増大した腫瘍細胞死が組合わせ及び連続的処理において認められたが(図15)、これは、腫瘍のアポトーシスの増大がCPT−11化合物の代謝における変化を反映するものではないことを示している。
【0117】
材料の寄託
次の材料をアメリカン・タイプ・カルチャー・コレクション,10801 University Boulevard., Manassas, Virginia, USA(ATCC)に寄託した:
材料 ATCC寄託番号 寄託日
4E7.24.3 HB−12454 1998年1月13日
4H6.17.8 HB−12455 1998年1月13日
1H5.25.9 HB−12695 1999年4月 1日
4G7.18.8 PTA−99 1999年5月21日
5G11.17.1 HB−12694 1999年4月 1日
3F11.39.7 HB−12456 1998年1月13日
3H3.14.5 HB−12534 1998年6月 2日
【0118】
この寄託は、特許手続き上の微生物の寄託の国際的承認に関するブダペスト条約及びその規則(ブダペスト条約)の規定に従って行われた。これは、寄託の日付から30年間、寄託の生存可能な培養が維持されることを保証するものである。寄託物はブダペスト条約の条項に従い、またジェネンテク社とATCCとの間の合意に従い、ATCCから入手することができ、これは、どれが最初であろうとも、関連した米国特許の発行時又は任意の米国又は外国特許出願の公開時に、寄託培養物の後代を永久かつ非制限的に入手可能とすることを保証し、35USC122条及びそれに従う特許庁長官規則(特に参照番号886OG638の37CFR第1.14条を含む)に従って権利を有すると米国特許庁長官が決定した者に後代を入手可能とすることを保証するものである。
本出願の譲受人は、寄託した培養物が、適切な条件下で培養されていた場合に死亡もしくは損失又は破壊されたならば、材料は通知時に同一の他のもの即座に取り替えることに同意する。寄託物質の入手可能性は、特許法に従いあらゆる政府の権限下で認められた権利に違反して、本発明を実施するライセンスであるとみなされるものではない。
上記の文書による記載は、当業者に本発明を実施できるようにするために十分であると考えられる。本発明は、ここに呈された実施例によりその範囲が限定されるものではない。実際、ここに示し記載したものに加えて、本発明を様々に改変することは、前記の記載から当業者にとっては明らかなものであり、添付の請求の範囲内に入るものである。
【図面の簡単な説明】
【図1】胸腺欠損ヌードマウスに皮下注射された、Apo−2L(白抜き三角)、CPT−11(白抜き四角)、Apo−2L+CPT−11(塗りつぶした三角)、又はビヒクル単独(白抜き丸)のヒト大腸癌細胞の成長に対する効果を示す。
【図2】胸腺欠損ヌードマウスに皮下注射された、Apo−2L(60mg/kg)(白抜き四角)、CPT−11(80mg/kg)(塗りつぶした三角)、Apo−2L(「Apo2L.0」)+CPT−11(塗りつぶした四角)、抗−DR4 mAb 4H6(白抜き三角)、抗−DR4 mAb+CPT−11(塗りつぶした三角)又はビヒクル単独(塗りつぶした丸)のヒト大腸癌細胞の成長に対する効果を示す。
【図3】図3A−3Hは、死亡した又は生存している腫瘍細胞の蛍光特性を示す。HCT116培養体を、それぞれ2及び24時間、Apo2L/TRAIL、CPT−11及びApo2L/TRAIL+CPT−11で処理した。次に、蛍光染料を使用して室温で30分間インキュベートし、細胞を蛍光顕微鏡で検査した。カルセイン−ポジティブ細胞(緑の標識)は生存している細胞を示すのに対し、エチジウムホモダイマー−1でポジティブ染色(赤の標識)されたものは、死亡しているか、かなりの損傷がある細胞を表し、これを図3E、3F及び3Gに示した。
【図4】CPT−11がインビトロでApo2L/TRAIL媒介性アポトーシスを増強させることを示す。HCT116培養体を、CPT−11(50μg/ml)、Apo2L/TRAIL(1μg/ml)、及びApo2L/TRAIL+CPT−11と共に24時間インキュベートした。生存している細胞の数をalamarBlueアッセイ(平均+SD、n=2)により測定した。サンプル中で生存している細胞のパーセンテージを対照処理に対して規準化した。
【図5】図5A−5Dは、Apo2L/TRAIL誘発性カスパーゼ−3活性のCPT−11感作がどのように時間依存しているかを示す。HCT116細胞を、Apo2L/TRAIL(1μg/ml)、CPT−11(50μg/ml)、及びApo2L/TRAIL+CPT−11で、2及び24時間処理した。細胞溶解物の等量のアリコットを、ウエスタンブロット分析によるプロ−カスパーゼ−3プロセシング(5A及び5B)、及び蛍光定量的アッセイによるカスパーゼ−3活性(5C及び5D)で評価した。
【図6】図6A−6Bは、Apo2L/TRAIL及びCPT−11単独で、又は組合せでの処理後の、DR5(6A)及びDR4(6B)遺伝子発現におけるHCT116の変化を示す。DR5及びDR4 mRNAレベルを、それぞれ2、6及び24時間、Apo2L/TRAIL(1μg/ml)、CPT−11(50μg/ml)、Apo2L/TRAIL+CPT−1とインキュベートした後、bDNAアッセイにより測定した。相対値はGAPDH対する比率として算出され、未処理の対照培養に対して規準化した。
【図7】図7A−7Bは、Apo2L/TRAIL及びCPT−11単独で、又は組合せでの処理後の、HCT116細胞におけるDR5(7A)及びDR4(7B)誘発を、Z−VADがブロックしないことを示す。DR5及びDR4 mRNAレベルを、それぞれ2、6及び24時間、Apo2L/TRAIL(1μg/ml)、CPT−11(50μg/ml)、及びApo2L/TRAIL+CPT−11とインキュベートした後、bDNAアッセイにより測定した。相対値はGAPDHに対する比率として算出され、未処理の対照培養体に対して規準化した。
【図8】CPT−11(Apo2L/TRAIL処理なし)により、HCT116及びHUVEC細胞におけるp53タンパク質レベルが増加するといった結果を示す。p53タンパク質レベルは、2及び24時間処理されたHCT116及びHUVEC細胞培養体におけるウエスタンブロット分析により特徴付けられた。
【図9】Apo2L/TRAIL処理により、p21タンパク質レベルのCPT−11媒介性誘発が、腫瘍細胞では抑制されて、HUVEC細胞では抑制されないことを示す。ヒトHCT116大腸腫瘍及び正常なHUVEC細胞を、2及び24時間、Apo2L/TRAIL(1μg/ml)、CPT−11(50μg/ml)、及びApo2L/TRAIL+CPT−1で処理した。細胞溶解物の等量のアリコット(50μg/レーン)を、ウエスタンブロット分析によるp21タンパク質発現についてテストした。
【図10】カスパーゼ−8インヒビター、FLIPタンパク質レベルが処理後に変化しないことを示す。ヒトHCT116大腸腫瘍細胞を、2及び24時間、Apo2L/TRAIL(1μg/ml)、CPT−11(50μg/ml)、及びApo2L/TRAIL+CPT−11(1μg/ml)で処理した。処理後、細胞培養体をフローサイトメトリー細胞周期分析にかけた。
【図11】Apo2L/TRAILによりCPT−11誘発性のG2/M細胞周期停止が抑制されることを示す。HCT116腫瘍細胞を、2、6及び24時間、Apo2L/TRAIL(1μg/ml)、CPT−11(50μg/ml)、及びApo2L/TRAIL+CPT−11で処理した。処理後、細胞培養体をフローサイトメトリー細胞周期分析にかけた。
【図12】カスパーゼインヒビター、Z−VAD(I)がp53及びp21のレベルにおいて異なる効果を有することを示す。細胞培養体を、24時間、20μMのZ−VADで、又はこれを用いないで処理した。細胞溶解物を、p53及びp21のタンパク質レベルのためのウエスタンブロット分析にかけた。
【図13】カスパーゼインヒビターZVADの存在下でApo2L/TRAIL処理をすると、CPT−11誘発性G2−M停止の防止に失敗することを示す。20μMのZ−VADの存在下で、HCT116腫瘍細胞を24時間、Apo2L/TRAIL(1μg/ml)、CPT−11(50μg/ml)、及びApo2L/TRAIL+CPT−11で処理した。処理後、細胞培養体をフローサイトメトリー細胞周期分析にかけた。
【図14】図14A−14Bは、連続処理により、腫瘍細胞における全細胞殺傷性が増強されることを示す。A.組合せグループにおいて、HCT116細胞を、CPT−11(10マイクログラム/ml)及び異なる濃度のApo2L/TRAIL(図に示す)に24時間さらし、さらなる24時間、培地単独の存在下でインキュベートした。連続グループにおいては、細胞を、最初の24時間CPT−11にさらし、ついで、さらなる24時間、Apo2L/TRAIL含有培地に変えた。細胞生存性を実施例に記載したようなクリスタルバイオレットアッセイにより測定した(平均±SD、n=4)。B.HCT116細胞(組合せ)を、CPT−11(10マイクログラム/ml)及び異なる濃度のApo2L/TRAILに、全部で24時間さらし、さらなる120時間、培地単独の存在下でインキュベートした。連続グループにおいては、細胞を、最初の24時間CPT−11にさらし、ついで、さらなる24時間、Apo2L/TRAIL含有培地に変え、薬剤フリーの培地で96時間インキュベートし、上述した様にして細胞生存性をテストした。
【図15】組合せ及び連続処理にCPT−11の代わりにSN−38で置き換えると、同様に増強された腫瘍細胞殺傷性となる結果を示す。組合せグループにおいて、HCT116細胞を、SN−38(0.05マイクログラム/ml)及び2つの濃度のApo2L/TRAIL(図に示す)に、全体で24時間さらし、さらなる24時間、培地単独の存在下でインキュベートした。連続グループにおいては、細胞を、最初の24時間SN−38にさらし、ついで、さらなる24時間、培地のみを含有するApo2L/TRAILに変えた。細胞生存性を実施例に記載したようなクリスタルバイオレットアッセイにより測定した(平均±SD、n=4)。
Claims (27)
- 有効量のCPT−11とApo−2リガンドレセプターアゴニストに哺乳動物細胞をさらすことを含む、哺乳動物細胞においてアポトーシスを増強させる方法であって、前記哺乳動物細胞を、Apo−2リガンドレセプターアゴニストにさらす前に、約6時間から約72時間、CPT−11にさらす方法。
- 前記哺乳動物細胞をCPT−11にさらし、該細胞においてDR4レセプターのアップレギュレーションを誘導する、請求項1に記載の方法。
- 前記哺乳動物細胞をCPT−11にさらし、該細胞においてDR5レセプターのアップレギュレーションを誘導する、請求項1に記載の方法。
- 前記哺乳動物細胞を、Apo−2リガンドレセプターアゴニストにさらす前に、約24又は48時間、CPT−11にさらす、請求項1に記載の方法。
- 前記Apo−2リガンドレセプターアゴニストがApo2Lポリペプチドを含んでなる、請求項1に記載の方法。
- 前記Apo−2リガンドレセプターアゴニストが抗−DR4レセプター抗体を含んでなる、請求項1に記載の方法。
- 前記抗−DR4レセプター抗体がモノクローナル抗体である、請求項6に記載の方法。
- 前記抗−DR4レセプターモノクローナル抗体がキメラ抗体を含んでなる、請求項7に記載の方法。
- 前記抗−DR4レセプターモノクローナル抗体がヒト抗体を含んでなる、請求項7に記載の方法。
- 前記Apo−2リガンドレセプターアゴニストが抗−DR5レセプター抗体を含んでなる、請求項1に記載の方法。
- 前記抗−DR5レセプター抗体がモノクローナル抗体である、請求項10に記載の方法。
- 前記抗−DR5レセプターモノクローナル抗体がキメラ抗体を含んでなる、請求項11に記載の方法。
- 前記抗−DR5レセプターモノクローナル抗体がヒト抗体を含んでなる、請求項11に記載の方法。
- 前記Apo−2リガンドレセプターアゴニストが、一以上のApo−2リガンドレセプターと交差反応する抗−Apo−2リガンドレセプター抗体である、請求項1に記載の方法。
- 哺乳動物細胞を一又は複数の成長阻害剤にさらすことをさらに含む、請求項1に記載の方法。
- 哺乳動物細胞を放射線にさらすことをさらに含む、請求項1に記載の方法。
- 哺乳動物細胞が結腸直腸癌細胞である、請求項1に記載の方法。
- 有効量のCPT−11及びApo−2リガンドレセプターアゴニストに哺乳動物細胞をさらすことを含む、哺乳動物癌細胞におけるアポトーシスを増強する方法であって、(a)該哺乳動物癌細胞を、Apo−2リガンドレセプターアゴニストにさらす前に、約6時間〜約72時間、CPT−11にさらし、(b)前記Apo−2リガンドレセプターアゴニストが、配列番号:1のアミノ酸残基114−281を含んでなるApo−2リガンドポリペプチド、抗−DR4レセプター抗体及び抗−DR5レセプター抗体からなる群から選択される方法。
- 前記哺乳動物癌細胞をCPT−11にさらすことにより、該細胞においてDR4レセプターのアップレギュレーションを誘導する、請求項18に記載の方法。
- 前記哺乳動物癌細胞をCPT−11にさらすことにより、該細胞においてDR5レセプターのアップレギュレーションを誘導する、請求項18に記載の方法。
- 前記抗−DR4レセプター抗体又は抗−DR5レセプター抗体が、キメラ、ヒト化又はヒト抗体である、請求項18に記載の方法。
- 前記哺乳動物癌細胞が結腸直腸癌細胞である、請求項18に記載の方法。
- 前記Apo−2リガンドポリペプチドが配列番号:1のアミノ酸残基114−281からなる、請求項18に記載の方法。
- 有効量のCPT−11及びApo−2リガンドレセプターアゴニストを、癌のある哺乳動物に投与することを含む哺乳動物の癌の治療方法であって、前記CPT−11を、Apo−2リガンドレセプターアゴニストを投与するより約6時間〜72時間前に投与する方法。
- 前記Apo−2リガンドレセプターアゴニストがApo2Lポリペプチドを含んでなる、請求項24に記載の方法。
- 前記Apo−2リガンドレセプターアゴニストが抗−DR4レセプター抗体を含んでなる、請求項24に記載の方法。
- 前記Apo−2リガンドレセプターアゴニストが抗−DR5レセプター抗体を含んでなる、請求項24に記載の方法。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US22125600P | 2000-07-27 | 2000-07-27 | |
| PCT/US2001/023691 WO2002009755A2 (en) | 2000-07-27 | 2001-07-27 | Apo-2l receptor agonist and cpt-11 synergism |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2004509078A true JP2004509078A (ja) | 2004-03-25 |
| JP2004509078A5 JP2004509078A5 (ja) | 2008-09-04 |
Family
ID=22827042
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2002515307A Pending JP2004509078A (ja) | 2000-07-27 | 2001-07-27 | Apo−2LレセプターアゴニストとCPT−11の相乗作用 |
Country Status (12)
| Country | Link |
|---|---|
| EP (2) | EP1303293B1 (ja) |
| JP (1) | JP2004509078A (ja) |
| AT (1) | ATE415978T1 (ja) |
| AU (1) | AU2001279055A1 (ja) |
| CA (1) | CA2415473A1 (ja) |
| CY (1) | CY1108815T1 (ja) |
| DE (1) | DE60136816D1 (ja) |
| DK (1) | DK1303293T3 (ja) |
| ES (1) | ES2317924T3 (ja) |
| IL (1) | IL153948A0 (ja) |
| PT (1) | PT1303293E (ja) |
| WO (1) | WO2002009755A2 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2009504186A (ja) * | 2005-08-16 | 2009-02-05 | ジェネンテック・インコーポレーテッド | 細胞/組織におけるgalnac−t14発現を試験することによるap02l/trailへのアポトーシス感受性 |
Families Citing this family (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7842668B1 (en) | 2001-11-13 | 2010-11-30 | Genentech, Inc. | Apo-2 ligand/trail formulations |
| US7741285B2 (en) | 2001-11-13 | 2010-06-22 | Genentech, Inc. | APO-2 ligand/trail formulations |
| DK1450847T3 (da) | 2001-11-13 | 2010-12-13 | Genentech Inc | Apo2-ligand/TRAIL-formuleringer og anvendelser deraf |
| EP2500032A1 (en) | 2002-06-24 | 2012-09-19 | Genentech, Inc. | APO-2 ligand/trail variants and uses thereof |
| EP1534336A4 (en) * | 2002-08-15 | 2005-12-14 | Human Genome Sciences Inc | ANTIBODIES IMMUNOSPECIFICALLY FIXING TO TRAIL RECEPTORS |
| KR20060132006A (ko) | 2004-03-23 | 2006-12-20 | 비오겐 아이덱 엠에이 아이엔씨. | 수용체 커플링제 및 이의 치료적 용도 |
| JO3000B1 (ar) | 2004-10-20 | 2016-09-05 | Genentech Inc | مركبات أجسام مضادة . |
| US8029783B2 (en) | 2005-02-02 | 2011-10-04 | Genentech, Inc. | DR5 antibodies and articles of manufacture containing same |
| US8460256B2 (en) | 2009-07-15 | 2013-06-11 | Allegiance Corporation | Collapsible fluid collection and disposal system and related methods |
| WO2012151317A1 (en) | 2011-05-03 | 2012-11-08 | Genentech, Inc. | Vascular disruption agents and uses thereof |
| WO2021058503A1 (en) * | 2019-09-24 | 2021-04-01 | Centre National De La Recherche Scientifique - Cnrs - | Method for identifying gene products involved in incomplete response of drug sensitive cell populations |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1997025428A1 (en) * | 1996-01-09 | 1997-07-17 | Genentech, Inc. | Apo-2 ligand |
| WO1998032856A1 (en) * | 1997-01-28 | 1998-07-30 | Human Genome Sciences, Inc. | Death domain containing receptor 4 (dr4: death receptor 4), member of the tnf-receptor superfamily and binding to trail (ap02-l) |
| WO1999009165A1 (en) * | 1997-08-15 | 1999-02-25 | Idun Pharmaceuticals, Inc. | Trail receptors, nucleic acids encoding the same, and methods of use thereof |
| WO1999048527A1 (en) * | 1998-03-27 | 1999-09-30 | Genentech, Inc. | Apo-2 ligand-anti-her-2 antibody synergism |
Family Cites Families (82)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3773919A (en) | 1969-10-23 | 1973-11-20 | Du Pont | Polylactide-drug mixtures |
| USRE30985E (en) | 1978-01-01 | 1982-06-29 | Serum-free cell culture media | |
| FR2413974A1 (fr) | 1978-01-06 | 1979-08-03 | David Bernard | Sechoir pour feuilles imprimees par serigraphie |
| WO1981001145A1 (en) | 1979-10-18 | 1981-04-30 | Univ Illinois | Hydrolytic enzyme-activatible pro-drugs |
| US4342566A (en) | 1980-02-22 | 1982-08-03 | Scripps Clinic & Research Foundation | Solid phase anti-C3 assay for detection of immune complexes |
| US4419446A (en) | 1980-12-31 | 1983-12-06 | The United States Of America As Represented By The Department Of Health And Human Services | Recombinant DNA process utilizing a papilloma virus DNA as a vector |
| NZ201705A (en) | 1981-08-31 | 1986-03-14 | Genentech Inc | Recombinant dna method for production of hepatitis b surface antigen in yeast |
| US4601978A (en) | 1982-11-24 | 1986-07-22 | The Regents Of The University Of California | Mammalian metallothionein promoter system |
| US4560655A (en) | 1982-12-16 | 1985-12-24 | Immunex Corporation | Serum-free cell culture medium and process for making same |
| US4657866A (en) | 1982-12-21 | 1987-04-14 | Sudhir Kumar | Serum-free, synthetic, completely chemically defined tissue culture media |
| US4816567A (en) | 1983-04-08 | 1989-03-28 | Genentech, Inc. | Recombinant immunoglobin preparations |
| US4675187A (en) | 1983-05-16 | 1987-06-23 | Bristol-Myers Company | BBM-1675, a new antibiotic complex |
| DD266710A3 (de) | 1983-06-06 | 1989-04-12 | Ve Forschungszentrum Biotechnologie | Verfahren zur biotechnischen Herstellung van alkalischer Phosphatase |
| US4767704A (en) | 1983-10-07 | 1988-08-30 | Columbia University In The City Of New York | Protein-free culture medium |
| US4965199A (en) | 1984-04-20 | 1990-10-23 | Genentech, Inc. | Preparation of functional human factor VIII in mammalian cells using methotrexate based selection |
| US5807715A (en) | 1984-08-27 | 1998-09-15 | The Board Of Trustees Of The Leland Stanford Junior University | Methods and transformed mammalian lymphocyte cells for producing functional antigen-binding protein including chimeric immunoglobulin |
| US4879231A (en) | 1984-10-30 | 1989-11-07 | Phillips Petroleum Company | Transformation of yeasts of the genus pichia |
| GB8516415D0 (en) | 1985-06-28 | 1985-07-31 | Celltech Ltd | Culture of animal cells |
| US4676980A (en) | 1985-09-23 | 1987-06-30 | The United States Of America As Represented By The Secretary Of The Department Of Health And Human Services | Target specific cross-linked heteroantibodies |
| US4927762A (en) | 1986-04-01 | 1990-05-22 | Cell Enterprises, Inc. | Cell culture medium with antioxidant |
| GB8610600D0 (en) | 1986-04-30 | 1986-06-04 | Novo Industri As | Transformation of trichoderma |
| US5567610A (en) | 1986-09-04 | 1996-10-22 | Bioinvent International Ab | Method of producing human monoclonal antibodies and kit therefor |
| GB8705477D0 (en) | 1987-03-09 | 1987-04-15 | Carlton Med Prod | Drug delivery systems |
| US4975278A (en) | 1988-02-26 | 1990-12-04 | Bristol-Myers Company | Antibody-enzyme conjugates in combination with prodrugs for the delivery of cytotoxic agents to tumor cells |
| US5223409A (en) | 1988-09-02 | 1993-06-29 | Protein Engineering Corp. | Directed evolution of novel binding proteins |
| EP0435911B1 (en) | 1988-09-23 | 1996-03-13 | Cetus Oncology Corporation | Cell culture medium for enhanced cell growth, culture longevity and product expression |
| GB8823869D0 (en) | 1988-10-12 | 1988-11-16 | Medical Res Council | Production of antibodies |
| US5175384A (en) | 1988-12-05 | 1992-12-29 | Genpharm International | Transgenic mice depleted in mature t-cells and methods for making transgenic mice |
| FR2646437B1 (fr) | 1989-04-28 | 1991-08-30 | Transgene Sa | Nouvelles sequences d'adn, leur application en tant que sequence codant pour un peptide signal pour la secretion de proteines matures par des levures recombinantes, cassettes d'expression, levures transformees et procede de preparation de proteines correspondant |
| EP0402226A1 (en) | 1989-06-06 | 1990-12-12 | Institut National De La Recherche Agronomique | Transformation vectors for yeast yarrowia |
| EP0739904A1 (en) | 1989-06-29 | 1996-10-30 | Medarex, Inc. | Bispecific reagents for aids therapy |
| DE10399023I2 (de) | 1989-09-12 | 2006-11-23 | Ahp Mfg B V | TFN-bindende Proteine |
| US5229275A (en) | 1990-04-26 | 1993-07-20 | Akzo N.V. | In-vitro method for producing antigen-specific human monoclonal antibodies |
| US5427908A (en) | 1990-05-01 | 1995-06-27 | Affymax Technologies N.V. | Recombinant library screening methods |
| US5723286A (en) | 1990-06-20 | 1998-03-03 | Affymax Technologies N.V. | Peptide library and screening systems |
| US5633425A (en) | 1990-08-29 | 1997-05-27 | Genpharm International, Inc. | Transgenic non-human animals capable of producing heterologous antibodies |
| US5625126A (en) | 1990-08-29 | 1997-04-29 | Genpharm International, Inc. | Transgenic non-human animals for producing heterologous antibodies |
| DK0814159T3 (da) | 1990-08-29 | 2005-10-24 | Genpharm Int | Transgene, ikke-humane dyr, der er i stand til at danne heterologe antistoffer |
| US5545806A (en) | 1990-08-29 | 1996-08-13 | Genpharm International, Inc. | Ransgenic non-human animals for producing heterologous antibodies |
| US5661016A (en) | 1990-08-29 | 1997-08-26 | Genpharm International Inc. | Transgenic non-human animals capable of producing heterologous antibodies of various isotypes |
| IL99553A0 (en) | 1990-09-28 | 1992-08-18 | Ixsys Inc | Compositions containing oligonucleotides linked to expression elements,a kit for the preparation of vectors useful for the expression of a diverse population of random peptides and methods utilizing the same |
| US5122469A (en) | 1990-10-03 | 1992-06-16 | Genentech, Inc. | Method for culturing Chinese hamster ovary cells to improve production of recombinant proteins |
| WO1992009690A2 (en) | 1990-12-03 | 1992-06-11 | Genentech, Inc. | Enrichment method for variant proteins with altered binding properties |
| WO1992020373A1 (en) | 1991-05-14 | 1992-11-26 | Repligen Corporation | Heteroconjugate antibodies for treatment of hiv infection |
| WO1994004679A1 (en) | 1991-06-14 | 1994-03-03 | Genentech, Inc. | Method for making humanized antibodies |
| US5270170A (en) | 1991-10-16 | 1993-12-14 | Affymax Technologies N.V. | Peptide library and screening method |
| WO1993008829A1 (en) | 1991-11-04 | 1993-05-13 | The Regents Of The University Of California | Compositions that mediate killing of hiv-infected cells |
| DE69333082T2 (de) | 1992-02-11 | 2004-05-06 | Cell Genesys, Inc., Foster City | Erzielen von homozygotem durch zielgerichtete genetische ereignisse |
| PT752248E (pt) | 1992-11-13 | 2001-01-31 | Idec Pharma Corp | Aplicacao terapeutica de anticorpos quimericos e marcados radioactivamente contra antigenios de diferenciacao restrita de linfocitos b humanos para o tratamento do linfoma de celulas b |
| DE614989T1 (de) | 1993-02-17 | 1995-09-28 | Morphosys Proteinoptimierung | Verfahren für in vivo Selektion von Ligandenbindende Proteine. |
| DE69405102T2 (de) | 1993-06-03 | 1998-01-15 | Therapeutic Antibodies Inc | Antikoerperfragmente in therapie |
| SE9304060D0 (sv) | 1993-12-06 | 1993-12-06 | Bioinvent Int Ab | Sätt att selektera specifika bakteriofager |
| US5516637A (en) | 1994-06-10 | 1996-05-14 | Dade International Inc. | Method involving display of protein binding pairs on the surface of bacterial pili and bacteriophage |
| US5731168A (en) | 1995-03-01 | 1998-03-24 | Genentech, Inc. | Method for making heteromultimeric polypeptides |
| US5739277A (en) | 1995-04-14 | 1998-04-14 | Genentech Inc. | Altered polypeptides with increased half-life |
| US5702892A (en) | 1995-05-09 | 1997-12-30 | The United States Of America As Represented By The Department Of Health And Human Services | Phage-display of immunoglobulin heavy chain libraries |
| IL122408A0 (en) | 1995-06-29 | 1998-06-15 | Immunex Corp | Dna encoding a trail polypeptide and its preparation |
| AU725609C (en) | 1995-08-18 | 2002-01-03 | Morphosys Ag | Protein/(poly)peptide libraries |
| CN1198837C (zh) | 1996-03-20 | 2005-04-27 | 戴克斯公司 | 组织纤溶酶原激活物(tPA)的纯化 |
| AU3135097A (en) | 1996-05-22 | 1997-12-09 | Johns Hopkins University, The | Methods of detection utilizing modified bacteriophage |
| AU3381097A (en) | 1996-06-07 | 1998-01-05 | Amgen, Inc. | Tumor necrosis factor-related polypeptide |
| WO1997047314A1 (en) | 1996-06-10 | 1997-12-18 | The Scripps Research Institute | Use of substrate subtraction libraries to distinguish enzyme specificities |
| AU3737297A (en) | 1996-08-05 | 1998-02-25 | Brigham And Women's Hospital | Bacteriophage-mediated gene therapy |
| JP2001512560A (ja) | 1996-10-08 | 2001-08-21 | ユー―ビスイス ベスローテン フェンノートシャップ | 標的に対し特異的な親和性を有するペプチドおよびタンパク質の選択のための方法および手段 |
| DK0939804T4 (da) | 1996-10-25 | 2011-07-18 | Human Genome Sciences Inc | Neutrokin-alpha |
| EP2003203A1 (en) | 1996-12-23 | 2008-12-17 | Immunex Corporation | Ligand for receptor activator of nf-kappa b, ligand is member of TNF superfamily |
| US6072047A (en) | 1997-02-13 | 2000-06-06 | Immunex Corporation | Receptor that binds trail |
| US5969102A (en) | 1997-03-03 | 1999-10-19 | St. Jude Children's Research Hospital | Lymphocyte surface receptor that binds CAML, nucleic acids encoding the same and methods of use thereof |
| US20010010924A1 (en) | 1997-03-14 | 2001-08-02 | Keith Charles Deen | Tumor necrosis factor related receptor, tr6 polynecleotides |
| CA2644454A1 (en) | 1997-03-17 | 1998-09-24 | Human Genome Sciences, Inc. | Death domain containing receptor 5 |
| EP1007562A4 (en) | 1997-04-16 | 2000-09-27 | Millennium Pharm Inc | TANGO-63d AND TANGO-63e PROTEINS RELATED TO THE TUMOR NECROSIS FACTOR RECEPTOR |
| ID23855A (id) | 1997-04-16 | 2000-05-25 | Amgen Inc | Protein pengikat osteoprotegerin dan reseptor |
| EP0981618B2 (en) | 1997-05-15 | 2011-08-24 | Genentech, Inc. | Anti-apo-2 antibody |
| AU8400398A (en) | 1997-07-11 | 1999-02-08 | Trustees Of The University Of Pennsylvania, The | Nucleic acid encoding a novel chemotherapy-induced protein, and methods of use |
| WO1999004001A1 (en) | 1997-07-21 | 1999-01-28 | Zymogenetics, Inc. | Tumor necrosis factor receptor ztnfr-5 |
| WO1999006587A2 (en) | 1997-08-01 | 1999-02-11 | Morphosys Ag | Novel method and phage for the identification of nucleic acid sequences encoding members of a multimeric (poly)peptide complex |
| EP1019502A2 (en) | 1997-08-06 | 2000-07-19 | Regeneron Pharmaceuticals, Inc. | Human orphan receptor ntr-1 |
| WO1999011791A2 (en) | 1997-09-05 | 1999-03-11 | University Of Washington | Tumor necrosis factor family receptors and ligands, encoding nucleic acids and related binding agents |
| AU2093499A (en) | 1997-12-30 | 1999-07-19 | Chiron Corporation | Members of tnf and tnfr families |
| EP1053256B9 (en) * | 1998-01-26 | 2012-01-18 | Genentech, Inc. | Antibodies to death receptor 4 (dr4) and uses thereof |
| ATE330972T1 (de) * | 1999-06-09 | 2006-07-15 | Genentech Inc | Apo-2l rezeptor-agonist und cpt-11 synergie- effekt |
| KR102213110B1 (ko) | 2014-03-21 | 2021-02-09 | 에스케이플래닛 주식회사 | 끊김 없는 서비스를 위한 비콘 장치 및 그의 제어 방법 |
-
2001
- 2001-07-27 EP EP01957295A patent/EP1303293B1/en not_active Expired - Lifetime
- 2001-07-27 IL IL15394801A patent/IL153948A0/xx unknown
- 2001-07-27 EP EP08017358A patent/EP2014303A2/en not_active Withdrawn
- 2001-07-27 AU AU2001279055A patent/AU2001279055A1/en not_active Abandoned
- 2001-07-27 CA CA002415473A patent/CA2415473A1/en not_active Abandoned
- 2001-07-27 DE DE60136816T patent/DE60136816D1/de not_active Expired - Lifetime
- 2001-07-27 ES ES01957295T patent/ES2317924T3/es not_active Expired - Lifetime
- 2001-07-27 JP JP2002515307A patent/JP2004509078A/ja active Pending
- 2001-07-27 DK DK01957295T patent/DK1303293T3/da active
- 2001-07-27 WO PCT/US2001/023691 patent/WO2002009755A2/en active Application Filing
- 2001-07-27 AT AT01957295T patent/ATE415978T1/de not_active IP Right Cessation
- 2001-07-27 PT PT01957295T patent/PT1303293E/pt unknown
-
2009
- 2009-02-18 CY CY20091100184T patent/CY1108815T1/el unknown
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1997025428A1 (en) * | 1996-01-09 | 1997-07-17 | Genentech, Inc. | Apo-2 ligand |
| WO1998032856A1 (en) * | 1997-01-28 | 1998-07-30 | Human Genome Sciences, Inc. | Death domain containing receptor 4 (dr4: death receptor 4), member of the tnf-receptor superfamily and binding to trail (ap02-l) |
| WO1999009165A1 (en) * | 1997-08-15 | 1999-02-25 | Idun Pharmaceuticals, Inc. | Trail receptors, nucleic acids encoding the same, and methods of use thereof |
| WO1999048527A1 (en) * | 1998-03-27 | 1999-09-30 | Genentech, Inc. | Apo-2 ligand-anti-her-2 antibody synergism |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2009504186A (ja) * | 2005-08-16 | 2009-02-05 | ジェネンテック・インコーポレーテッド | 細胞/組織におけるgalnac−t14発現を試験することによるap02l/trailへのアポトーシス感受性 |
| JP2011250788A (ja) * | 2005-08-16 | 2011-12-15 | Genentech Inc | 細胞/組織におけるgalnac−t14発現を試験することによるapo2l/trailへのアポトーシス感受性 |
Also Published As
| Publication number | Publication date |
|---|---|
| AU2001279055A1 (en) | 2002-02-13 |
| ATE415978T1 (de) | 2008-12-15 |
| DE60136816D1 (de) | 2009-01-15 |
| WO2002009755A3 (en) | 2003-02-13 |
| CY1108815T1 (el) | 2014-04-09 |
| DK1303293T3 (da) | 2009-03-30 |
| ES2317924T3 (es) | 2009-05-01 |
| CA2415473A1 (en) | 2002-02-07 |
| IL153948A0 (en) | 2003-07-31 |
| EP2014303A2 (en) | 2009-01-14 |
| PT1303293E (pt) | 2009-03-11 |
| EP1303293A2 (en) | 2003-04-23 |
| WO2002009755A2 (en) | 2002-02-07 |
| EP1303293B1 (en) | 2008-12-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1192185B1 (en) | Apo-2L RECEPTOR AGONIST AND CPT-11 SYNERGISM | |
| US20110059104A1 (en) | Apo-2L receptor agonist and CPT-11 synergism | |
| AU2002250352C1 (en) | Combination therapy | |
| CA2594918C (en) | Dr5 antibodies and uses thereof | |
| JP4589586B2 (ja) | Dr4抗体とその利用 | |
| JP2008513367A (ja) | デスレセプターリガンド及びcd20抗体の使用方法 | |
| JP2008512479A (ja) | デスレセプターリガンド及びcd20抗体の使用方法 | |
| TW200950778A (en) | Methods of using Apo2L/TRAIL to treat cancer | |
| RU2395294C2 (ru) | Способы применения агонистов аро-2l-рецепторов и активаторов nk-клеток | |
| EP1303293B1 (en) | Sequential administration of cpt-11 and apo-2l polypeptide | |
| EP1658859A1 (en) | APO-2L receptor agonist and CPT-11 synergism | |
| AU2010202871A1 (en) | APO-2L receptor agonist and CPT-11 synergism | |
| AU2005202006A1 (en) | APO-21 receptor agonist and CPT-11 synergism | |
| TWI419903B (zh) | Dr5抗體及其用途 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20040128 |
|
| A072 | Dismissal of procedure [no reply to invitation to correct request for examination] |
Free format text: JAPANESE INTERMEDIATE CODE: A072 Effective date: 20040817 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20080718 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20080718 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20110517 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20111115 |