JP2004346071A - 3’窒素含有ポリヌクレオチドの合成のための固体支持体試薬 - Google Patents
3’窒素含有ポリヌクレオチドの合成のための固体支持体試薬 Download PDFInfo
- Publication number
- JP2004346071A JP2004346071A JP2004145724A JP2004145724A JP2004346071A JP 2004346071 A JP2004346071 A JP 2004346071A JP 2004145724 A JP2004145724 A JP 2004145724A JP 2004145724 A JP2004145724 A JP 2004145724A JP 2004346071 A JP2004346071 A JP 2004346071A
- Authority
- JP
- Japan
- Prior art keywords
- polynucleotide
- synthesis
- nitrogen
- linker
- solid support
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Withdrawn
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H21/00—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Biochemistry (AREA)
- Molecular Biology (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Biotechnology (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Saccharide Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
【解決手段】3’−窒素官能化ポリヌクレオチドの自動化ポリヌクレオチド合成に使用するためのポリヌクレオチド合成支持体に関するものであって、構造式:TO−Q−NR1(C=Y)OCR2R3CHR4X1−Z−W(式中、Tは酸開裂性ヒドロキシル保護基であり;Qは窒素および酸素を結合するリンカーであり;R1は窒素置換基であり;R2からR4までは別々に水素または低級アルキル基であり;Yは炭素に対して負に帯電した原子であり;X1は炭素に対して負に帯電した原子であり;Zはボンドまたはスペーサーアームであり;そしてWはZに結合できる誘導化固体合成支持体である)を有するポリヌクレオチド合成試薬。
【選択図】なし
Description
(1)3’末端で窒素基を含むポリヌクレオチドの合成のための固体支持体試薬であって、次式:
Qは窒素および酸素を結合するリンカーであり;
R1は窒素置換基であり;
R2からR4までは別々に水素または低級アルキル基であり;
Yは炭素に対して負に帯電した原子であり;
X1は炭素に対して負に帯電した原子であり;
Zはボンドまたはスペーサーアームであり;そしてWはZに結合できる誘導化固体合成支持体である)
の化合物を含有する固体支持体試薬。
(2)Tは4,4’−ジメトキシトリチル、モノメトキシトリチル、α−ナフチルジフェニルメチル、またはトリ(p−メトキシフェニル)メチルである、項目1に記載の化合物。
(3)Tが4,4’−ジメトキシトリチルである、項目1に記載の化合物。
(4)Qが低級アルキル、低級アルキレンオキシド、または、固体支持体試薬の窒素と組み合わせるとき、アミド、カルバメート、スルホンアミド、または尿素、またはそれらのいずれかの組合せである、項目1に記載の化合物。
(5)Qが低級アルキルまたは低級アルキレンオキシドである、項目1に記載の化合物。
(6)R2からR4までが水素である、項目1に記載の化合物。
(7)Yが酸素または硫黄である、項目1に記載の化合物。
(8)Yが酸素である、項目1に記載の化合物。
(9)X1がスルホニル、カルボニル、スルホキシド、パーフルオロ低級アルキル、またはスルホニル−、カルボニル−、スルホキシド−、ニトロ−、シアノ−、またはパーフルオロ低級アルキル置換アリールである、項目1に記載の化合物。
(10)X1がスルホニル、カルボニル、スルホキシドである、項目1に記載の化合物。
(11)Zが、アミノ終端リンカーで誘導化された固体合成支持体の終端窒素と組合せて、カルバメート、尿素、アミド、スルホンアミド、または次式の基である、項目1に記載の化合物。
(12)Wがアミノ終端リンカーで誘導化されたCPGである、項目1に記載の化合物。
(13)Wがアミノ終端リンカーで誘導化された有孔ポリスチレンである、項目1に記載の化合物。
(14)3’−末端で窒素基を含むポリヌクレオチドの合成のための固体支持体試薬であって、次式:
Qは窒素および酸素を結合するリンカーであり;
R1は窒素置換基であり;
R2とR3は別々に水素または低級アルキル基であり;
Yは炭素に対して負に帯電した原子であり;
X2は炭素に対して負に帯電した原子であり;
Zはボンドまたはスペーサーアームであり;そしてWはZに結合できる誘導化固体合成支持体である)
の化合物を含有する固体支持体試薬。
(15)Tが4,4’−ジメトキシトリチル、モノメトキシトリチル、α−ナフチルジフェニルメチル、またはトリ(p−メトキシフェニル)メチルである、項目14に記載の化合物。
(16)Tが4,4’−ジメトキシトリチルである、項目14に記載の化合物。
(17)Qが低級アルキル、低級アルキレンオキシド、または、固体支持体試薬の窒素と組み合わせるときアミド、カルバメート、スルホンアミド、または尿素、またはそれらのいずれかの組合せである、項目14に記載の化合物。
(18)Qが低級アルキルまたは低級アルキレンオキシドである、項目14に記載の化合物。
(19)R2とR3が水素である、項目14に記載の化合物。
(20)Yが酸素または硫黄である、項目14に記載の化合物。
(21)Yが酸素である、項目14に記載の化合物。
(22)X2がスルホニル、カルボニル、スルホキシド、シアノ、パーフルオロ低級アルキル、またはスルホニル−、カルボニル−、スルホキシド−、ニトロ−、シアノ−、またはパーフルオロ低級アルキル置換アリールである、項目14に記載の化合物。
(23)X2がスルホニル、カルボニル、またはシアノである、項目14に記載の化合物。
(24)Zが、窒素終端リンカーで誘導化された固体合成支持体の終端窒素と組合せて、カルバメート、尿素、アミド、スルホンアミド、または次式の基である、項目14に記載の化合物。
(25)Wがアミノ終端リンカーで誘導化されたCPGである、項目14に記載の化合物。
(26)Wがアミノ終端リンカーで誘導化された有孔ポリスチレンである、項目14に記載の化合物。
(27)(a)次式:
Qは窒素および酸素を結合するリンカーであり;
R1は窒素置換基であり;
R2からR4までは別々に水素または低級アルキル基であり;
Yは炭素に対して負に帯電した原子であり;
X1は炭素に対して負に帯電した原子であり;
Zはボンドまたはスペーサーアームであり;そしてWはZに結合できる誘導化固体合成支持体である)
の化合物を含む固体支持体試薬を用意し;
(b)固体支持体を酸で処理して酸開裂性ヒドロキシル保護基を除去し;
(c)保護ヌクレオシド単量体と弱酸を添加し、結合体を形成し;
(d)固体支持体上の未反応部位をキャッピングし;
(e)酸化試薬を添加し;
(f)ポリヌクレオチド鎖伸長が完了するまで工程(b)−(e)を繰り返し;
(g)ポリヌクレオチドを固体支持体から開裂し;そして(h)ポリヌクレオチドを脱保護する各工程からなる3’−末端に窒素原子を有するポリヌクレオチドを合成する方法。
(28)(a)次式:
Qは窒素および酸素を結合するリンカーであり;
R1は窒素置換基であり;
R2とR3は別々に水素または低級アルキル基であり;
Yは炭素に対して負に帯電した原子であり;
X2は炭素に対して負に帯電した原子であり;
Zはボンドまたはスペーサーアームであり;そしてWはZに結合できる誘導化固体合成支持体である)
の化合物を含む固体支持体試薬を用意し;
(b)固体支持体を酸で処理して酸開裂性ヒドロキシル保護基を除去し;
(c)保護ヌクレオシド単量体と弱酸を添加し、結合体を形成し;
(d)固体支持体上の未反応部位をキャッピングし;
(e)酸化試薬を添加し;
(f)ポリヌクレオチド鎖伸長が完了するまで工程(b)−(e)を繰り返し;
(g)ポリヌクレオチドを固体支持休から開裂し;そして(h)ポリヌクレオチドを脱保護する各工程からなる3’−末端に窒素原子を有するポリヌクレオチドを合成する方法。
Zはボンドまたはスペーサーアームであり;そしてWはZに結合できる誘導化固体合成支持体である。
いずれかの局面の他の好適例では、Qは、低級アルキル、低級アルキレンオキシド、または、固体支持体の窒素と組み合わせるとき、アミド、カルバメート、スルホンアミド、または尿素、またはそれらのいずれかの組合せである。さらに好ましくは、Qは低級アルキルまたは低級アルキレンオキシドである。
I.定義
ここで使用される「低級アルキル」の語は直鎖、分枝鎖、および1ないし8個の炭素原子を含有する環状アルキル基を示す。
本発明の好適例を以下に詳細に説明する。本発明は好適例により説明するが、これらの例によって本発明が制限されるものではない。また、本発明は本発明の請求の範囲内に含まれる代替、改変、および等価のものを含む。
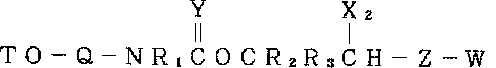
好ましくは、X2はスルホニル、カルボニル、スルホキシド、シアノ、パーフルオロ低級アルキル、またはスルホニル−、カルボニル−、スルホキシド−、ニトロ−、シアノ−、またはパーフルオロ低級アルキル置換アリールである。さらに好ましくは、X2はスルホニル、カルボニル、またはシアノである。
A.式IのX1含有固体支持体の合成:
以下は式Iの化合物のための好ましい一般の合成法である。一般に、反応機構はヒドロキシル保護アルコールアミン(Tアミン)を調製し、ジオールを当量のホスゲン、次いでTアミンと反応させてヒドロキシル保護カルバメートアルコール(T−CA)を調製することを含む。T−CAを次に処理して活性T−CAリンカーを形成し、アミノ誘導化固体基体と反応させる。
HOQNHR1
(式中の可変要素は上に定義されている)で示されるアミノアルコール(約1.0当量)を有機溶媒、例えば、メタノール、エーテル、塩化メチレン等に溶解し、塩基に不安定なアミノ保護試薬(約1.1当量)、例えば、エチルトリフルオロアセテートを−5ないし25℃の温度にてアミノアルコール溶液に一滴ずつ添加し、0ないし40℃の温度にて1〜6時間攪拌し、その後、真空下に蒸発させる。市販品として入手できるアミノアルコールの例としてはアミノエタノール、6−アミノ−1−ヘキサノール、アミノシクロヘキサノール、2−(2−アミノエトキシ)エタノール、ロイシノール等を含む。残渣を水と混合できない有機溶媒、例えば、塩化メチレン、エーテル、エチルアセテート等に溶解し、水で洗浄し、硫酸ナトリウムで乾燥させる。次いで溶媒を真空下に蒸発させて保護アミノアルコール生成物を与える。
T保護アミノアルコールを極性有機溶媒、例えばメタノールに溶解し、、塩基性水溶液、例えば、4Nの水酸化ナトリウムを0と25℃の間で添加し、反応物を10分と2時間の間、25と60℃の間で攪拌する。有機溶媒を真空下に蒸発させ、残渣を水と混合しない有機溶媒、例えば、エチルアセテート、エーテル等に溶解し、水で洗浄し、硫酸ナトリウムで乾燥させる。溶媒を真空下に蒸発させて次の式で示されるTアミンを与える。
(式中の可変要素は上に定義されている。)
T−CAを形成するために、次式の乾燥ジオールを使用する。
(式中、Z’は若干のケースではZに等しく、他のケースではZへの前駆体であり、後にT−CAを活性化するために使用される方法に依存する。他の可変要素は上に示した通りである。乾燥ジオール(2ないし10当量)および第三アミン(1.0当量)、例えばジイソプロピルエチルアミンを非プロトン性有機溶媒、例えば、ピリジンに溶解し、ホスゲン当量(1.0当量)、例えば、4−ニトロフェニルクロロホルメートを0と25℃の間で添加し、溶液を室温にて10分と2時間の間攪拌する。この溶液を次に上記で調製した(約0.25ないし1当量)Tアミンおよび第三アミン(約1.0当量)、例えばジイソプロピルエチルアミン、トリエチルアミン等を非プロトン性溶媒、例えばピリジンに溶解した溶液に添加する。反応物を10分と2時間の間、0と25℃の間で攪拌し、溶媒を真空下に蒸発させ、残渣を水と混合しない有機溶媒、例えば、エチルアセテートに溶解し、水で洗浄し、硫酸ナトリウムで乾燥させる。溶媒を真空下に蒸発させて次の式で示されるTカルバメートアルコールアミン(T−CA)を得た。
上のT−CAを次に二つの好適法のいずれか一つを使用して活性リンカーに変換する。第一の好適法では、T−CAをアミン塩基、例えば、4−ジメチルアミノピリジン、および無水物、例えば無水琥珀酸を用いて、非プロトン性溶媒、例えば、塩化メチレン中で、10と60℃の間にて10と60分の間、処理する。溶液を弱酸水溶液、例えばクエン酸で洗浄し、硫酸ナトリウムで乾燥させ、真空下に濃縮してTカルバメートアルコールリンカー(T−CAリンカー)を得る。このT−CAリンカーを次に等モル溶液の1−ビドロキシベンゾトリアゾール(HOBT)および2−(1H−ベンゾトリアゾール−1−イル)−1,1,3,3−テトラメチルウロニウム(HBTU)(N,N−ジメチルホルムアミドの0.45M溶液)を極性非プロトン性溶媒、例えば、ジメチルホルムアミド中で処理し、次に第三アミン、例えば、ジイソプロピルエチルアミンを添加して、活性化する。反応物を10と60分の間、5と35℃の間で攪拌し、活性T−CAリンカーを得る。
次は式Iの化合物のための好適な代替の一般化した合成法である。上記のようなジオール(1ないし5当量)および第三アミン(1.1当量)、例えば、トリエチルアミンを非プロトン性有機溶媒、例えば、塩化メチレンに溶解し、酸に不安定なアルコール保護試薬(1当量)、例えばジメトキシトリチルクロリドを添加し、0と25℃の間で10分から5時間攪拌する。溶液を水で洗浄し、硫酸ナトリウムで乾燥し、モノ保護ジオールを得る。
次は式IIの化合物のための好ましい一般的合成法である。出発物質は次式によって定義されるX2リンカーである。
X2リンカーがアミン、アルコール、またはチオールを含むならば、先ず塩基に不安定でなく、酸に不安定でない保護基、例えば、ベンジルまたはシリルで保護される。これらおよび他の応用できる保護基を使用するためのプロトコルはほかの場所で見出すことができる、例えば、Greene and Wuts,Protective Groups in Organic Synthesis,2nd Edition(John Wiley,New York,1991)。
乾燥極性非プロトン性溶媒中のケトンまたはアルデヒド(1当量)を−40と0℃の間で一滴ずつ添加し、反応物を−40と25℃の間で10分と2時間の間で攪拌し、反応物を水で冷却し、真空下に蒸発させて濃縮する。次に残渣を水に混合しない溶媒、例えば酢酸エチル、エーテル、塩化メチレン等に溶解し、水で洗浄し、硫酸ナトリウムで乾燥させ、真空下に濃縮し、次の式で定義されるアルコールリンカーを得る。
次にアルコールリンカーを先の方法Aの活性T−CAリンカーのための上記第二の好ましい活性化法に従って活性化し、活性化アルコールリンカーを与える。
本発明の固体支持体の好適な有用性はその3’末端に位置する窒素原子を含有するポリヌクレオチドの合成にある。ポリヌクレオチドを生成するために使用される化学の詳細な説明は他にもある、例えば、Caruthers et al.,米国特許第4,458,066号;Caruthers et al.,米国特許第4,415,732号;Caruthers et al.,Genetic Engineering,4:1−17(1982);Users Manual Model 392 and 394 Polynucleotide Synthesizers,6−1から6−22頁まで,Applied Biosystems,Part No.901237(1991)。従って、これらの文献はこれらの記述について参考文献として組み込まれる。
次の実施例は本発明の固体支持体試薬の調製と応用を示すものである。使用されるパラメーターの数値は本発明の例示であり制限されるものではない。
N−トリフルオロアセチル−6−アミノ−1−ヘキサノールの合成
6−アミノ−1−ヘキサノール(179g)(Aldrich Chemical Company,Inc.,Milwaukee,WI)をメタノール(358ml)に溶解し、エチルトリフルオロアセテート(239g)(Aldrich)を20分かけて溶液に滴下した。反応物を2.5時間攪拌した後、溶媒を真空下に除去し、残渣を塩化メチレン(250ml)に溶解し、その後、溶液を水(3×300ml)および飽和塩化ナトリウム溶液(200ml)で洗浄し、硫酸ナトリウムで乾燥させた。最後に、溶媒を真空下に除去し、白色固体を得た(299g)。
1−O−(4,4’−ジメトキシトリチル)−N−トリフルオロアセチル−6−アミノヘキサンの合成
実施例1からのN−トリフルオロアセチル−6−アミノ−1−ヘキサノール(10g)およびジイソプロピルエチルアミン(12.1g)(Aldrich)を塩化メチレン(100ml)に溶解し、5℃まで氷冷却し、ジメトキシトリチルクロライド(17.5g)(Aldrich)を冷却混合物に添加した。混合物を終夜(15時間)攪拌し、その間に温度は最初の1時間5℃に保持し、次いで室温まで上昇させた。塩化メチレン(100ml)を添加し、混合物を飽和重炭酸ナトリウム溶液(100ml)で、次に飽和塩化ナトリウム溶液(100ml)で洗浄した。次に混合物を硫酸ナトリウムで乾燥し、溶媒を真空下に除去して所望の生成物(29g)を得た。
1−O−(4,4’−ジメトキシトリチル)−6−アミノヘキサンの合成
実施例2からの1−O−(4,4’−ジメトキシトリチル)−N−トリフルオロアセチル−6−アミノヘキサン(20.9g)をメタノール(100ml)に溶解し、氷冷し、4N水酸化ナトリウム(16.6ml)を添加した。反応物を室温まで温め、ヒートガンを使用して10分間50℃まで加熱し、次に終夜(15時間)室温で攪拌した。メタノールを真空下に除去し、残渣を水(100ml)および酢酸エチル(150ml)と混合した。次に有機層を飽和塩化ナトリウム水溶液(2×100ml)で洗浄し、硫酸ナトリウムで乾燥させた。溶媒を真空下に除去し、オイル(16.9g)として生成物を得た。
1−O−(4,4’−ジメトキシトリチル)−6−アミノヘキサンおよび2.2’−スルホニルジエタノールのカルバメート付加物の合成
1−O−(4,4’−ジメトキシトリチル)−6−アミノヘキサンおよび2.2’−スルホニルジエタノールのカルバメート付加物の合成前に、2,2’−スルホニルジエタノール(Aldrich)を次の方法を用いて乾燥した。2,2’−スルホニルジエタノール(65%水溶液200g)をアセトニトリル(200ml)と混合し、混合物を蒸溜し、蒸溜物を70℃と90℃の沸点範囲で収集した(240ml)。蒸溜ポットを室温まで冷却し、さらにアセトニトリルを添加し(200ml)、蒸溜工程を繰り返し、さらに蒸溜物を収集した(270ml)。蒸溜頭部を次にスタークトラップと置き換え、トルエン(150ml)を添加し、混合物を還流させた。トラッピング3時間後、水を回収し(12ml)、溶媒を真空下に除去し、粘性オイル(105g)を得た。
1−O−(4,4’−ジメトキシトリチル)−6−アミノヘキサンと2.2’−スルホニルジエタノールのカルバメート付加物のスクシニルエステルの合成
実施例4からのカルバメート付加物(2.0g)および4−ジメチルアミノピリジン(0.49g)(Aldrich)をアルゴン下に塩化メチレン(20ml)に溶解し、無水琥珀酸(0.41g)を室温にて攪拌溶液に添加した。5分後、追加の塩化メチレン(100ml)を添加し溶液を冷クエン酸(クエン酸10%水溶液、各5×100ml)および飽和塩化ナトリウム溶液(2×各100ml)で洗浄した。洗浄した溶液を硫酸ナトリウムで乾燥させて溶媒を真空下に除去し、オイル(2.1g)として生成物を得た。
3’−アミノリンカー支持体を形成する3−アミノプロピル制御有孔ガラスへのスクシニルエステルの結合
実施例5からのスクシニルエステル(0.4g)をアルゴン下にジメチルホルムアミド(10ml)に溶解し、等モルの1−ヒドロキシベンゾトリアゾール(HOBT)および2−(1H−ベンゾトリアゾール−1−イル)−1,1,3,3−テトラメチルウロニウム(HBTU)(ジメチルホルムアミド中0.45M溶液1.14ml)(Applied Biosystems Division of the Perkin Elmer Corporation,Foster City,CA(ABI))を攪拌溶液に添加し、続いてジイソプロピルエチルアミン(0.14g)を添加した。室温で15分後、3−アミノプロピル−CPG(1グラムにつき40μモルの装填量を有する5.58gの物質)(ABI)を攪拌溶液に添加し、攪拌を停止し、反応物を時々静かに2.5時間攪拌した。スラリーを中粒度ガラス濾過器に移し、CPG支持体を塩化メチレン(5×20ml)で洗浄し、キャッピング試薬(テトラヒドロフラン中0.5MのN−メチルイミダゾール10mlおよびテトラヒドロフラン中10%無水酢酸10%2,6−ルチジン10ml)で処理し、30分間静止した。次に溶液を除去し、CPGを塩化メチレン(5×20ml)で洗浄し、真空下に乾燥させて白色固体(5.64g)を得た。
本発明の固体支持体を使用する3’−アミノヘキシルポリヌクレオチドの合成
3’−アミノポリヌクレオチドの合成は標準プロトコルと試薬(ABI)を使用するApplied Biosystems 394ポリヌクレオチド合成器を使用して行われた。394ポリヌクレオチド合成器によって使用される簡単な化学は上記IV.有用性に記載されている。合成に使用される固体支持体は実施例1−6に記載されているものであった(32mg)。ポリヌクレオチドの塩基配列は5’−AGC TAG CT−3’であった。生成物はまだ結合している末端トリチル基で合成支持体から開裂され、HPLC分析によって約80%の純度であると決定された。
3’−アミノヘキシルポリヌクレオチドへの螢光染料の結合
0.1Mトリエチルアンモニウムアセテート、pH7(100μl)中の実施例7からの粗ポリヌクレオチド(20%)を6−カルボキシフルオレセイン−N−ヒドロキシスクシンイミド(6−FAM)エステル(100μlジメチルホルムアミド中1mg)(Research Organics,Inc.,Cleveland,OH)の溶液に添加し、次に1M NaHCO3/Na2CO3 pH9.0溶液(100μl)を添加した。溶液を攪拌し、室温で30分間放置した。次に混合物をPD−10ゲルフィルターカラム(Pharmacia,Piscataway,NJ)に加え、単一の画分を収集し(2.5mlの空隙容量を溶出した後に収集した1ml画分)、5’−AGCTAGCT−3’−アミノヘキシル−6−FAMを得た。
Claims (1)
- 3’末端で窒素基を含むポリヌクレオチドの合成のための固体支持体試薬であって、次式:
Qは窒素および酸素を結合するリンカーであり;
R1は窒素置換基であり;
R2からR4までは別々に水素または低級アルキル基であり;
Yは炭素に対して負に帯電した原子であり;
X1は炭素に対して負に帯電した原子であり;
Zはボンドまたはスペーサーアームであり;そしてWはZに結合できる誘導化固体合成支持体である)
の化合物を含有する固体支持体試薬。
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US08/293,637 US5552471A (en) | 1994-08-17 | 1994-08-17 | Solid support reagents for the synthesis of 3'-Nitrogen containing polynucleotides |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP50733396A Division JP3754449B2 (ja) | 1994-08-17 | 1995-07-18 | 3’窒素含有ポリヌクレオチドの合成のための固体支持体試薬 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2004346071A true JP2004346071A (ja) | 2004-12-09 |
Family
ID=23129907
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP50733396A Expired - Fee Related JP3754449B2 (ja) | 1994-08-17 | 1995-07-18 | 3’窒素含有ポリヌクレオチドの合成のための固体支持体試薬 |
| JP2004145724A Withdrawn JP2004346071A (ja) | 1994-08-17 | 2004-05-14 | 3’窒素含有ポリヌクレオチドの合成のための固体支持体試薬 |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP50733396A Expired - Fee Related JP3754449B2 (ja) | 1994-08-17 | 1995-07-18 | 3’窒素含有ポリヌクレオチドの合成のための固体支持体試薬 |
Country Status (7)
| Country | Link |
|---|---|
| US (2) | US5552471A (ja) |
| EP (1) | EP0777675B1 (ja) |
| JP (2) | JP3754449B2 (ja) |
| AU (1) | AU684216B2 (ja) |
| CA (1) | CA2197302C (ja) |
| DE (1) | DE69507618T2 (ja) |
| WO (1) | WO1996005215A1 (ja) |
Families Citing this family (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5552471A (en) * | 1994-08-17 | 1996-09-03 | The Perkin-Elmer Corporation | Solid support reagents for the synthesis of 3'-Nitrogen containing polynucleotides |
| US5736626A (en) * | 1996-01-29 | 1998-04-07 | The Perkin-Elmer Corporation | Solid support reagents for the direct synthesis of 3'-labeled polynucleotides |
| US5789451A (en) * | 1996-07-29 | 1998-08-04 | The Dow Chemcial Company | Alkanolamine/carbon dioxide adduct and polyurethane foam therewith |
| CA2319587C (en) * | 1998-02-11 | 2004-09-21 | University Of Houston | Method and apparatus for chemical and biochemical reactions using photo-generated reagents |
| US7094943B2 (en) | 1998-04-27 | 2006-08-22 | Hubert Köster | Solution phase biopolymer synthesis |
| US6800287B2 (en) | 1998-09-25 | 2004-10-05 | Yeda Research And Development Co., Ltd. | Copolymer 1 related polypeptides for use as molecular weight markers and for therapeutic use |
| US6184347B1 (en) | 1998-11-19 | 2001-02-06 | Agilent Technologies Inc. | Minimization of blooming in high-density arrays by using reactive wash reagents |
| US6432642B1 (en) * | 1999-01-15 | 2002-08-13 | Pe Corporation (Ny) | Binary probe and clamp composition and methods for a target hybridization detection |
| US6476191B1 (en) | 1999-02-05 | 2002-11-05 | Mixture Sciences, Inc. | Volatilizable solid phase supports for compound synthesis |
| US6255476B1 (en) | 1999-02-22 | 2001-07-03 | Pe Corporation (Ny) | Methods and compositions for synthesis of labelled oligonucleotides and analogs on solid-supports |
| US7211654B2 (en) * | 2001-03-14 | 2007-05-01 | Regents Of The University Of Michigan | Linkers and co-coupling agents for optimization of oligonucleotide synthesis and purification on solid supports |
| US6855501B2 (en) * | 2001-03-26 | 2005-02-15 | Linden Technologies, Inc. | Transfer of arrayed chemical compositions |
| US20020136772A1 (en) * | 2001-03-26 | 2002-09-26 | Tai-Nang Huang | Polymer synthesis |
| DE10324063A1 (de) * | 2003-05-27 | 2004-12-23 | Robert Heinrich | Verfahren zur Herstellung von auf porösem Glas gebundenen Nucleotiden |
| WO2013024694A1 (ja) * | 2011-08-12 | 2013-02-21 | 独立行政法人産業技術総合研究所 | アミノ化オリゴヌクレオチド用固相担体 |
| JP6457999B2 (ja) | 2013-03-14 | 2019-01-23 | ザ チルドレンズ メディカル センター コーポレーション | 処置に関してがんの対象を識別するためのcd36の使用 |
| JP6727129B2 (ja) | 2014-03-26 | 2020-07-22 | ザ チルドレンズ メディカル センター コーポレーション | 環状プロサポシンペプチドおよびその使用 |
| CN109880247B (zh) * | 2019-02-18 | 2022-08-19 | 邹晗 | 一种含超分散碳纳米管的聚苯乙烯电子载带材料及其制备方法 |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4458066A (en) * | 1980-02-29 | 1984-07-03 | University Patents, Inc. | Process for preparing polynucleotides |
| US4415732A (en) * | 1981-03-27 | 1983-11-15 | University Patents, Inc. | Phosphoramidite compounds and processes |
| US5118605A (en) * | 1984-10-16 | 1992-06-02 | Chiron Corporation | Polynucleotide determination with selectable cleavage sites |
| DE3500180A1 (de) * | 1985-01-04 | 1986-07-10 | Ernst Prof. Dr. 7400 Tübingen Bayer | Pfropfcopolymerisate aus vernetzten polymeren und polyoxyethylen, verfahren zu ihrer herstellung und ihre verwendung |
| JP3022967B2 (ja) * | 1985-03-15 | 2000-03-21 | アンチバイラルズ インコーポレイテッド | 立体規則性ポリヌクレオチド結合ポリマー |
| US5141813A (en) * | 1989-08-28 | 1992-08-25 | Clontech Laboratories, Inc. | Multifunctional controlled pore glass reagent for solid phase oligonucleotide synthesis |
| US5552471A (en) * | 1994-08-17 | 1996-09-03 | The Perkin-Elmer Corporation | Solid support reagents for the synthesis of 3'-Nitrogen containing polynucleotides |
-
1994
- 1994-08-17 US US08/293,637 patent/US5552471A/en not_active Expired - Lifetime
-
1995
- 1995-07-18 JP JP50733396A patent/JP3754449B2/ja not_active Expired - Fee Related
- 1995-07-18 WO PCT/US1995/009105 patent/WO1996005215A1/en active IP Right Grant
- 1995-07-18 EP EP95927284A patent/EP0777675B1/en not_active Expired - Lifetime
- 1995-07-18 CA CA002197302A patent/CA2197302C/en not_active Expired - Fee Related
- 1995-07-18 DE DE69507618T patent/DE69507618T2/de not_active Expired - Lifetime
- 1995-07-18 AU AU31363/95A patent/AU684216B2/en not_active Ceased
- 1995-12-21 US US08/576,206 patent/US5625052A/en not_active Expired - Lifetime
-
2004
- 2004-05-14 JP JP2004145724A patent/JP2004346071A/ja not_active Withdrawn
Also Published As
| Publication number | Publication date |
|---|---|
| US5552471A (en) | 1996-09-03 |
| EP0777675B1 (en) | 1999-01-27 |
| JP3754449B2 (ja) | 2006-03-15 |
| US5625052A (en) | 1997-04-29 |
| CA2197302A1 (en) | 1996-02-22 |
| AU3136395A (en) | 1996-03-07 |
| EP0777675A1 (en) | 1997-06-11 |
| JPH10508827A (ja) | 1998-09-02 |
| DE69507618T2 (de) | 1999-09-09 |
| WO1996005215A1 (en) | 1996-02-22 |
| DE69507618D1 (de) | 1999-03-11 |
| AU684216B2 (en) | 1997-12-04 |
| CA2197302C (en) | 2003-05-20 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP3754449B2 (ja) | 3’窒素含有ポリヌクレオチドの合成のための固体支持体試薬 | |
| JP4127870B2 (ja) | 3’−標識ポリヌクレオチドの直接合成用の固体支持体試薬 | |
| Scaringe | RNA oligonucleotide synthesis via 5′-silyl-2′-orthoester chemistry | |
| US5112962A (en) | Labile anchors for solid phase polynucleotide synthesis | |
| US7227017B2 (en) | Process for the preparation of phosphorothioate oligonucleotides | |
| US6262251B1 (en) | Method for solution phase synthesis of oligonucleotides | |
| JP3161730B2 (ja) | オリゴヌクレオチド合成の方法および手段 | |
| JP2002511840A (ja) | オリゴヌクレオチド及びペプチドの液相合成方法 | |
| US6768005B2 (en) | Process | |
| KR20030032924A (ko) | 포스포로티오에이트 트리에스테르의 제조 방법 | |
| US7098326B2 (en) | Methods for the integrated synthesis and purification of oligonucleotides | |
| US8067581B2 (en) | Biomolecules having multiple attachment moieties for binding to a substrate surface | |
| US7164014B2 (en) | Protected linker compounds | |
| EP1006121B1 (en) | A universal polymer support for the synthesis of oligonucleotides | |
| Leuck | Pieken et al.(45) Date of Patent: Aug. 29, 2006 | |
| JPH1171392A (ja) | ピューロマイシンを3′−リン酸末端にもつ核酸化合物及びその合成方法 | |
| AU1357300A (en) | method for solution phase synthesis of oligonucleotides |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A132 Effective date: 20080415 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20080702 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20080707 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20081014 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20090408 |
|
| A711 | Notification of change in applicant |
Free format text: JAPANESE INTERMEDIATE CODE: A712 Effective date: 20090520 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20090707 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20090710 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20090807 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20090812 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20090907 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20090910 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20091008 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20091126 |
|
| A761 | Written withdrawal of application |
Free format text: JAPANESE INTERMEDIATE CODE: A761 Effective date: 20100325 |