FR2486071A2 - Procede de preparation de composes aryloxyalcanoiques optiquement actifs - Google Patents
Procede de preparation de composes aryloxyalcanoiques optiquement actifs Download PDFInfo
- Publication number
- FR2486071A2 FR2486071A2 FR8015103A FR8015103A FR2486071A2 FR 2486071 A2 FR2486071 A2 FR 2486071A2 FR 8015103 A FR8015103 A FR 8015103A FR 8015103 A FR8015103 A FR 8015103A FR 2486071 A2 FR2486071 A2 FR 2486071A2
- Authority
- FR
- France
- Prior art keywords
- optically active
- alkali metal
- salt
- acid
- carbon atoms
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/347—Preparation of carboxylic acids or their salts, halides or anhydrides by reactions not involving formation of carboxyl groups
- C07C51/367—Preparation of carboxylic acids or their salts, halides or anhydrides by reactions not involving formation of carboxyl groups by introduction of functional groups containing oxygen only in singly bound form
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/347—Preparation of carboxylic acids or their salts, halides or anhydrides by reactions not involving formation of carboxyl groups
- C07C51/353—Preparation of carboxylic acids or their salts, halides or anhydrides by reactions not involving formation of carboxyl groups by isomerisation; by change of size of the carbon skeleton
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
L'INVENTION CONCERNE LA PREPARATION DE DERIVES ARYLOXYALCANOIQUES OPTIQUEMENT ACTIFS, DE FORMULE: (CF DESSIN DANS BOPI) DANS LAQUELLE AR REPRESENTE UN RADICAL HYDROCARBONE AROMATIQUE MONO OU BICYCLIQUE EVENTUELLEMENT SUBSTITUE, R UN RADICAL ALCOYL (C-C)
Description
La demande de brevet princ7oal revendique un procédé de préparation d'acides (D) phénoxy-2 propioniques, optiquement actifs à partir d'une solution aqueuse ou hydroalcoolique du sel alcalin de l'acide chloro-2 propionique et d'une solution aqueuse du sel alcalin d'un phénol.
La présente demande de certificat d'addition concerne un procédé, similaire à celui revendiqué dans la demande principale et destiné à la préparation d'acides et dérivés d'acides aryloxyalcanoiques optiquement actifs, utilisables comme herbicides, et plus particulièrement d'acides et dérivés d'acides aryloxyalcanoïques présentant une teneur élevée en isomère de configuration absolue n.
Elle concerne de plus les composés optiquement actifs ainsi obtenus.
Les composés optiquement actifs susceptibles d'être préparés selon le procédé de l'invention répondent è la formule générale

(formule I) dans laquelle
Ar représente un radical hydrocarboné aromatique, mono
ou bicyclique, contenant de 6 à 10 atomes de car
bone (tel que phényle,- naphtyle-l ou. naphtyle-2),
ledit radical étant éventuellement substitué
par un à trois substituants, identiques ou diffé
rents, choisis parmi
-les atomes d'halogène (de préférence chlore ou
brome),
-les radicaux alcoyle contenant de 1 à 4 atomes de
carbone,
-le radical phénoxy lui-même éventuellement substi
tué par 1 à 3 substituants, identiques ou
différents, choisis parmi les atomes d'halogènes
(de préférence chlore), les radicaux alcoyle conte
nant de 1 à 4 atomes de carbone (de préférence
méthyle), le radical trifluorométhyle et le radical
nitro cet le radical pyridyloxy lui-même éventuellement
substitué par 1 à 3 substituants choisis parmi les
atomes dshalogènes (de préférence chlore), les
radicaux alcoyle contenant de 1 à 4 atomes de car
bone (de préférence méthyle) et le radical trifluo-
méthyle, ces substituants pouvant être identiques
ou différents.
(formule I) dans laquelle
Ar représente un radical hydrocarboné aromatique, mono
ou bicyclique, contenant de 6 à 10 atomes de car
bone (tel que phényle,- naphtyle-l ou. naphtyle-2),
ledit radical étant éventuellement substitué
par un à trois substituants, identiques ou diffé
rents, choisis parmi
-les atomes d'halogène (de préférence chlore ou
brome),
-les radicaux alcoyle contenant de 1 à 4 atomes de
carbone,
-le radical phénoxy lui-même éventuellement substi
tué par 1 à 3 substituants, identiques ou
différents, choisis parmi les atomes d'halogènes
(de préférence chlore), les radicaux alcoyle conte
nant de 1 à 4 atomes de carbone (de préférence
méthyle), le radical trifluorométhyle et le radical
nitro cet le radical pyridyloxy lui-même éventuellement
substitué par 1 à 3 substituants choisis parmi les
atomes dshalogènes (de préférence chlore), les
radicaux alcoyle contenant de 1 à 4 atomes de car
bone (de préférence méthyle) et le radical trifluo-
méthyle, ces substituants pouvant être identiques
ou différents.
A représente un radical
dans lesquels:
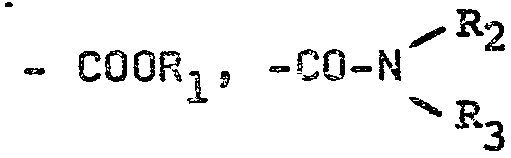
et CO z
R1 représente un atome d'hydrogène, u. équivalent d'un
cation dune base organique ou minérale (par exem
ple un équivalent d'un cation d'un métal alcalin ou
alcalino-terreux ou d'un cation ammonium éventuel
lement substitué), ou un radical alcoyle contenant
de 1 à 12 atomes de carbone, éventuellement substi
tué par 1 ou plusieurs atomes d'halogènes ou radi
caux hydroxy,
R2 et R3, identiques ou différents, représentent
chacun un atome d'hydrogène, ou un radical alcoyle
contenant de 1 à 6 atomes de carbone, éventuellement
substitué par un ou plusieurs OH, NH2 ou alcoxy (C1-c4),
ou un radical phényle éventuellement substitué par
un ou plusieurs halogènes, alcoyles (C1-C4) ou
alkcxy (C1-C4) ou (R2 et/ou Ra représen
tent) un radical alcényle contenant de 2 à 4 atomes
de carbone,
Z représente un atome d'halogène (de préférence
chlore),
R représente un radical alcoyle contenant de 1 à 4
atomes de carbone (de préférence méthyle),...
dans lesquels:
et CO z
R1 représente un atome d'hydrogène, u. équivalent d'un
cation dune base organique ou minérale (par exem
ple un équivalent d'un cation d'un métal alcalin ou
alcalino-terreux ou d'un cation ammonium éventuel
lement substitué), ou un radical alcoyle contenant
de 1 à 12 atomes de carbone, éventuellement substi
tué par 1 ou plusieurs atomes d'halogènes ou radi
caux hydroxy,
R2 et R3, identiques ou différents, représentent
chacun un atome d'hydrogène, ou un radical alcoyle
contenant de 1 à 6 atomes de carbone, éventuellement
substitué par un ou plusieurs OH, NH2 ou alcoxy (C1-c4),
ou un radical phényle éventuellement substitué par
un ou plusieurs halogènes, alcoyles (C1-C4) ou
alkcxy (C1-C4) ou (R2 et/ou Ra représen
tent) un radical alcényle contenant de 2 à 4 atomes
de carbone,
Z représente un atome d'halogène (de préférence
chlore),
R représente un radical alcoyle contenant de 1 à 4
atomes de carbone (de préférence méthyle),...
L1invention concerne plus particulièrement la préparation des acides et sels d'acides aryloxyalcanoïques, optiquement actifs, de formule

(formule -Il) dans laquelle Ar et R ont la même signification que dans la formule I et M représente un cation d'un métal alcalin (par exemple sodium ou potassium) ou un atome d'hydrogène.
(formule -Il) dans laquelle Ar et R ont la même signification que dans la formule I et M représente un cation d'un métal alcalin (par exemple sodium ou potassium) ou un atome d'hydrogène.
Le composé de formule I et celui de formule Il présentent un atome de carbone substitué de façon asymétrique.
Chacun de ces composés peut donc exister sous deux formes énantiomères dont l'une présente la configuration absolue 0 et l'autre la configuration absolue L. (Selon une autre nomenclature équivalente, les lettres R et'S sont quelquefois utilisées respectivement au lieu de D et de L pour repérer les configurations absolues. Dans ce qui suit; nous utiliserons systématiquement la première de ces nomenclatures, c'est-à-dire les lettres D et L).
Le composé de formule I (ou de formule Il) comprenant en proportions pondérales égales l'isomère de configuration absolue et l'isomare de configuration absolue L est le composé racémique, optiquement inactif.
Dans ce qui suit, on entendra par composé optiquement actif un composé constitué en majeure partie (en poids) ou dans sa totalité par l'un des isomères de ce composé. Par pureté optique d'un composé optiquement actif1 c'est-à-dire constitué de façon prépondérante par l'un des deux isomères, on entendra le pourcentage pondéral de l'isomère prépondérant contenu dans ce composé. Par composé optique ment actif de pureté optique élevée, on entendra un composé dans lequel le pourcentage pondéral de l'isomère prépondérant est d-'au moins 90 %.
Il est connu que de nombreux composés répondant aux formules I et II présentent d'excellentes propriétés herbicides et plusieurs d'entre eux sont actuellement commercialisés pour cette utilisation tels que notamment - l'acide (méthyl-2 chloro-4@phénoxy)-2 propionique (ou
Mecoprop), - l'acide (trichloro-2,4,5-phénoxy)-2 propionique (ou
Fenoprop), - l'acide (méthyl-2 chloro-4mphénoxy)-2 propionique (ou
Dichlorprop), n c - l'acide (méthyl-2 phénoxy)-2 propionique, ces acides étant généralement commercialisés sous forme de leurs sels de sodium ou despotassium ou de leurs sels d'amine, ou de~leurs esters avec des alcanols inférieurs ou :: r- esent - le (α-naphtoxy)-2 - NçN diéthylpropionamide (ou
Napropamide), les I - le [(dichloro-2,4 phénoxy)-4 phénoxy ]-2 propionate de méthyle (ou Diclofop). 3 assez
Ces composés ont dte Jusqu'à présent généralement commercialisés-sous leur forme@racémique. Il a été toutefois observé@que, pour plusieurs des composés mentionnés ci-dessus, l'un-des isomères@présente une activité herbicide très supérieure à desèrégale à celle du composé racémique correspondant. I1 appas-tit donc très désirable de disposer d'un procédé permettant de préparer ces composés herbicides sous leur forme@optiquement active, composée en majeure partie, ou en totalité, par l'isomère présentant la meilleure activité herbicide Dans le cas des composés herbicides cités plus hautfail a été observé que l'isomère de configuration D présentelarpresque totalité de l'activité herbicide alors que isomère de configuration L ne présente pratiquement aucunecactivité herbicide.
Mecoprop), - l'acide (trichloro-2,4,5-phénoxy)-2 propionique (ou
Fenoprop), - l'acide (méthyl-2 chloro-4mphénoxy)-2 propionique (ou
Dichlorprop), n c - l'acide (méthyl-2 phénoxy)-2 propionique, ces acides étant généralement commercialisés sous forme de leurs sels de sodium ou despotassium ou de leurs sels d'amine, ou de~leurs esters avec des alcanols inférieurs ou :: r- esent - le (α-naphtoxy)-2 - NçN diéthylpropionamide (ou
Napropamide), les I - le [(dichloro-2,4 phénoxy)-4 phénoxy ]-2 propionate de méthyle (ou Diclofop). 3 assez
Ces composés ont dte Jusqu'à présent généralement commercialisés-sous leur forme@racémique. Il a été toutefois observé@que, pour plusieurs des composés mentionnés ci-dessus, l'un-des isomères@présente une activité herbicide très supérieure à desèrégale à celle du composé racémique correspondant. I1 appas-tit donc très désirable de disposer d'un procédé permettant de préparer ces composés herbicides sous leur forme@optiquement active, composée en majeure partie, ou en totalité, par l'isomère présentant la meilleure activité herbicide Dans le cas des composés herbicides cités plus hautfail a été observé que l'isomère de configuration D présentelarpresque totalité de l'activité herbicide alors que isomère de configuration L ne présente pratiquement aucunecactivité herbicide.
L'invention concerneedonc plus particulièrement la préparation des dérivés optiquement actifs des composés selon les formules I et Il,,- lesdits dérivés étant constitués en totalité ou en majeure partie par l'isomère de configuration D.
I1 a été proposé (brevet français 1 479 271) de préparer l'isomère de configuration D de certains acides phénoxy-2 propioniques, en faisant réagir un sel alcalin, dextrogyre dans l'eau, de l'acide chloro-2 propionique, sur un seul alcalin d'un phénol, en présence d'un solvant organique inerte de point d'ébullition élevé, tel qu'un hydrocarbure aromatique, à la température d'ébullition du solvant, puis en acidifiant le sel alcalin ainsi obtenu, par traitement au moyen d'un acide fort, pour donner enfin l'acide D-phénoxy-2 propionique.
Selon ce procédé, le passage du chloro-2 propionate de métal alcalin au (D)-phenoxy-2 propionate de métal alcalin s'effectue avec inversion de configuration de type inversion de Walden.
Le procédé revendiqué par ce brevet français permet d'obtenir avec un bon rendement des acides phénoxy-2 propioniques dont la teneur en isomère D est très élevée. Sur un plan pratique, selon la technique décrite dans ce brevet, il est préferé d'utiliser des réactifs anhydres et/ou d'éliminer l'eau présente dans le mélange réactionnel, avant le début de la réaction, par entrainement 'azéotropique.
Industriellement, l'utilisation de réactifs anhydres présente de sérieux inconvénients. Elle est onéreuse puisqu'elle implique une déshydratation préalable des réactifs. De plus certains réactifs, et en particulier le chloro-2 propionate de sodium, sont difficiles à préparer à l'état anhydre.
Par ailleurs, plusieurs de ces réactifs anhydres sont des solides dont la mise en oeuvre est difficile et qui conduisent à des mélanges réactionnels hétérogènes, difficiles à agiter et de ce fait peu réactifs.
Par ailleurs enfin, l'élimination de l'eau par entrainement azéotropique nécessite l'utilisation d'un solvant auxiliaire qu'il est ensuite nécessaire de récupérer et recycler.
La présente invention se propose de remédier à ces inconvénients.
Un but de la présente Invention est de fournir un procédé amélioré de préparation des acides et dérivés d'acides aryluxyalcanoïques optiquement actifs selon les formules I et 11.
Un autre but de l'invention est de permettre la préparation de ces composés a partir de solutions aqueuses ou hydroorganiques d'halogéno-2 alcanoate alcalin optiquement actif, éventuellement préparées in situ
Un autre but de l'invention est de permettre la préparation de ces composés avec un excellent rendement à partir d'un helogéno-2 alcanoate d'alcoyle ou d'un halogéno-2 alcanoate alcalin, optiquement actifs
Un autre but de l'invention est de Dermettre l'obtention de composés présentant un degré de pureté optique élevé.
Un autre but de l'invention est de permettre la préparation de ces composés avec un excellent rendement à partir d'un helogéno-2 alcanoate d'alcoyle ou d'un halogéno-2 alcanoate alcalin, optiquement actifs
Un autre but de l'invention est de Dermettre l'obtention de composés présentant un degré de pureté optique élevé.
I1 a maintenant été trouvé que ces buts pouvaient être atteints selon un nouveau procédé qui fait l'objet de la présente invention.
Ce procédé, distinct de celui revendiqué par la demande de brevet principal, est un procédé de préparation des composés aryloxyalcanoïques optiquement actifs répondant aux formules I et 11, par action d'un sel de métal alcalin, optiquement actif, d'un acide halogéno-2 alca noïque contenant de 3 à 6 atomes de carbone, sur un sel alcalin d'un phénol de formule::
Ar - OH (formule III) dans laquelle Ar a la même signification que dans la formule I, pour donner le sel de métal alcalin répondant à la formule II pour laquelle M représente un cation de métal alcalin, acidification de ce sel pour donner l'acide correspondant (composé selon la formule II pour lequel M représente un atome d'hydrogène), puis, éventuellement transformation de cet acide selon des méthodes en soi connues, pour donner l'un quelconque-des composés selon la formule I. I1 est caractérisé en ce que le sel de métal alcalin répondant à la formule Il est préparé en faisant réagir la solution aqueuse ou hydroorganique du sel alcalin de l'acide halogéno-2 alcanoïque sur une solution aqueuse du sel alcalin du phénol, à une température élevée,-sous une pression inférieure à la pression de vapeur de l'eau à la température de la réaction. (Les pressions mentionnées dans la présente demande, sont des pressions absolues, la valeur zéro correspondant au vide absolu).
Ar - OH (formule III) dans laquelle Ar a la même signification que dans la formule I, pour donner le sel de métal alcalin répondant à la formule II pour laquelle M représente un cation de métal alcalin, acidification de ce sel pour donner l'acide correspondant (composé selon la formule II pour lequel M représente un atome d'hydrogène), puis, éventuellement transformation de cet acide selon des méthodes en soi connues, pour donner l'un quelconque-des composés selon la formule I. I1 est caractérisé en ce que le sel de métal alcalin répondant à la formule Il est préparé en faisant réagir la solution aqueuse ou hydroorganique du sel alcalin de l'acide halogéno-2 alcanoïque sur une solution aqueuse du sel alcalin du phénol, à une température élevée,-sous une pression inférieure à la pression de vapeur de l'eau à la température de la réaction. (Les pressions mentionnées dans la présente demande, sont des pressions absolues, la valeur zéro correspondant au vide absolu).
Comme indiqué précédemment, le procédé revendiqué par la présente demande de certificat d'addition est distinct de celui revendiqué dans la demande de brevet principal, c'est-à-dire qu'il n'inclut pas la préparation d'.acides ou sels d'acides (D) phénoxy-2 propioniques, par action d'une solution aqueuse ou hydroalcoolique d'un sel alcalin de l'acide chloro-2 propionique optiquement actif, sur une solution aqueuse d'un sel alcalin de phénols de formule

dans laquelle X représente un atome d'halogène ou le radical méthyle et n est un nombre entier de 1 à 5 (étant entendu que lorsque n est différent de 1, les substituants
X peuvent etre identiques ou différents)
La température à utiliser pour la mise en oeuvre du procédé revendiqué peut être choisie selon la réactivité du sel alcalin du phénol de formule III et celle du sel alcalin de l'halogéno-2 alcanoate alcalin utilisé. On conçoit par- exemple que cette température ne sera pas forcément la même dans le cas d'un chloro-2 propionate de métal alcalin que dans le cas d'un bromo-2 propionate alcalin puisque ces composés ont des réactivités assez différentes.
dans laquelle X représente un atome d'halogène ou le radical méthyle et n est un nombre entier de 1 à 5 (étant entendu que lorsque n est différent de 1, les substituants
X peuvent etre identiques ou différents)
La température à utiliser pour la mise en oeuvre du procédé revendiqué peut être choisie selon la réactivité du sel alcalin du phénol de formule III et celle du sel alcalin de l'halogéno-2 alcanoate alcalin utilisé. On conçoit par- exemple que cette température ne sera pas forcément la même dans le cas d'un chloro-2 propionate de métal alcalin que dans le cas d'un bromo-2 propionate alcalin puisque ces composés ont des réactivités assez différentes.
Cette température sera choisie assez élevée pour permettre à la réaction de s'effectuer dans des conditions satisfaisantes, elle devra toutefois rester inférieure aux températures pour lesquelles la dégradation thermique des produits ou réactifs mis en jeu dans la réaction et éventuellement l'autoracémisation des composés optiquement actifs.
Lorsque cette réaction est effectuée en utilisant comme halogéno-2 alcanoate alcalin, le chloro-2 propionate de sodium, de bons résultats sont obtenus en opérant entre 70 et 1600C.
De préférence, les conditions utilisées pour la mise en oeuvre du procédé selon l'invention sont celles indiquées ci-après, en a, b, G, d, e et f.
a - l'acide halogéno-2 alcanoique est l'acide chloro-2
propionique,
b- comme sels de métal alcalin des réactifs de départ,
on utilise respectivement le sel de sodium de
l'acide chloro-2 propionique et le sel de sodium du
phénol de formule III et on obtient ainsi le compo
sé selon la formule II pour lequel R représente le
radical méthyle et M un atome de sodium,
c- la réaction est effectuée sous une pression infé-
rieure ou au plus égale à la pression atmosphéri
que, et de préférence, comprise entre 0,1 et 1 bar.
propionique,
b- comme sels de métal alcalin des réactifs de départ,
on utilise respectivement le sel de sodium de
l'acide chloro-2 propionique et le sel de sodium du
phénol de formule III et on obtient ainsi le compo
sé selon la formule II pour lequel R représente le
radical méthyle et M un atome de sodium,
c- la réaction est effectuée sous une pression infé-
rieure ou au plus égale à la pression atmosphéri
que, et de préférence, comprise entre 0,1 et 1 bar.
Elle doit être choisie d'autant plus basse que la
température de la réaction a été choisie elle-même
plus basse. A titre d'exemple, lorsqu'on opère à
80-900C, cette pression doit avantageusement être
de l'ordre de 0,1 à 0,7 bars,
d- la solution aqueuse ou hydroorganique du selalca-
lin de l'acide halogéno-2 alcanoïque est une solu
tion de concentration élevée, proche de la limite
de saturation. Lorsqu'on utilise une solution
hydroorganique, cette solution contient un ou
plusieurs solvants organiques. Ces solvants doivent
présenter un bon pouvoir dissolvant vis-à-vis du
sel alcalin de l'acide halogéno-2 alcanoïque, être
inertes, miscibles à l'eau et présenter un point
d'ébullition inférieur à celui de l'eau, dans les
conditions de la réaction.Ces solvants organiques sont avantageusement choisis parmi les alcools, et.
température de la réaction a été choisie elle-même
plus basse. A titre d'exemple, lorsqu'on opère à
80-900C, cette pression doit avantageusement être
de l'ordre de 0,1 à 0,7 bars,
d- la solution aqueuse ou hydroorganique du selalca-
lin de l'acide halogéno-2 alcanoïque est une solu
tion de concentration élevée, proche de la limite
de saturation. Lorsqu'on utilise une solution
hydroorganique, cette solution contient un ou
plusieurs solvants organiques. Ces solvants doivent
présenter un bon pouvoir dissolvant vis-à-vis du
sel alcalin de l'acide halogéno-2 alcanoïque, être
inertes, miscibles à l'eau et présenter un point
d'ébullition inférieur à celui de l'eau, dans les
conditions de la réaction.Ces solvants organiques sont avantageusement choisis parmi les alcools, et.
plus particulièrement parmi les alcanols inférieurs (c'est-à-dire comprenant de 1 à 4 atomes de carbone).
Lorsque l'acide halogéno-2 alcanoïque est l'acide chloro-2 propionique, on utilise avantageusement une solution aqueuse ou hydroorganique comprenant au moins 20 % en poids de chloro-2 propionate de sodium, le complément étant constitué par de l'eau et éventuellement par un solvant organique tel qu'un alcanol inférieur (par exemple méthanol ou éthanol).
La limite supérieure de la quantité de chloro-2 propionate de sodium présente dans la solution correspond a la limite de saturatic en chloro-2 propionate de sodium de la solution dans les conditions de température et pression utilisées.
Avantageusement, cette solution comprend en poids de 20 à 50 % de chlore-2 propionate de sodium.
e - la solution agueuse du sel alcalin du phénol de formule IIIest une solution de concentration élevée proche de la limite de saturation, Elle comprend avantageusement au moins 50 a en poids du sel alcalin du phénol de formule III. La limite supérieure de la quantité de ce sel utilise correspond à la limite de saturation de la solution, dans les conditions de teinpérature et pression utilisées.
De préférence, on utilise uno solution contenant de 50 à 30 % en poids de sel de sodium du phénol selon la formule III. Elle peut etre préparée par saponi- fication du phénol selon la formule III par une solution aqueuse de soude industrielle. Cette solution est de pré- férence concentrée jusqu'au voisinage de sa limite de saturation, par un chauffage effectué a une température comprise entre 70 et 160 C, sous une pression inférieure à la pression de vapeur de l'eau a cette température.
Avantageusement, ce chauffage est effectué en utilisant les mêmes conditions de température et pression que pour la réaction selon l'invention.
f- le sel alcalin de l'acide halogéno-2 alcanoïque est composé en majeure partie par 1 isomère re de configuration L (avantageusement, il comorend au moins 90 9 d'isomère L). Du fait que la réaction selon l'invention s'effectue avec inversion de Walden, avec une très faible diminution de la pureté optique, on obtient ainsi des composés selon les formules I et II composés en majeure partie de l'isomère de configuration D.
(I1 serait bien sur possible si on le désirait, de préparer des composés I et II composés en majeure partie de l'énantiomère de configuration L en partant d'un sel alcalin de l'acide halogéno-2 alcanoique en majeure par- tie par l'isomère D).
Le sel de métal alcalin optiquement actif, de l'acide halogéno-2 alcanorque utilisé comme matière.de départ peut être préparé selon une méthode en soi connue, en une opération séparée, par saponification par une base minérale telle que la soude, d'un halogéno-2 alcanoate d'alcoyle optiquement actif. Ce sel alcalin peut également être préparé in situ, selon un procédé en soi connu (cf brevet français 1 479 271) selon lequel un chloro-2 propionate d'alcoylc est could dans le m!- lange léactionnel contenant le phenol et une base minerale telle que la soude en maintenant la température entre,5 et 35 C.Lorsqu'on opère selon ces conditions, il a été observé qu'une élévation de température au dessus des valeurs indiquées précedemment entraînait à la fois une très importante baisse du rendement en chloro-2 propionate alcalin, due à la formation de lactate de métal alcalin et également une baisse rapide de la pureté optique du chloro-2 propionate alcalin formé. La réaction de saponification étant exothermique, il est alors nécessaire de refroidir le mélange réactionnel, pour maintenir sa température entre 5 et 350C pendant toute la durée de cette réaction, ce qui constitue un inconvénient sérieux dans le cas d'une fabrication industrielle.
I1 a été trouvé que, selon une veriante du procédé selon l'invention, il était possible d'effectuer cette préparation in situ du sel alcalin optiquement actif de l'acide halogénoalcanoique, à une température élevée (supérieure à 40 C et de préférence comprise entre 70 et 160 C) en évitant complètement les inconvéni,ents mentionnés précédemment. Cette variante du procédé selon l'invention concerne à la fois le procédé revendiqué dans la demande de brevet principal et celui revendiqué dans la présente demande de certificat d'addition ; et de ce fait, la restriction mentionnée plus haut ne concerne pas cette variante du procédé de l'invention.
Selon cette variante du procédé de l'invention, (variante faisant également partie de l'invention) le sel alcalin optiquement actif de l'acide halogéno-2 alcanoïque est préparé in situ, dans le réacteur contenant la solution aqueuse du sel de métal alcalin du phénol de formule III, à température élevée (de préférence comprise entre 70 et 160 C), en introduisant, dans ce réacteur, simultanément et en a,uantités sensiblement stoéchiométriques, d'une part une solution aqueuse d'un hydroxyde alcalin (de préférence soude) et d'autre part un halogéno-2 alcanoate d'alcoyle optiquement actif de formule ::

(formule @v) dans laquelle hal représente un atome d'halogène, de préférence chlore, R a la-même signification que dans la formule I et R4 représente un radical alcoyle (Cl-C6),sous- une pression a la fois supérieure à a pression de vaporisation (ou d'ébullition) de l'halogeno-2 alcanoate d'alcoyle selon la formule IV , et inférieure à la pression de vapeur de l'eau à cette température. Par quantités sensiblement stoéchiométriques, on entend quantités telles que le rapport molaire des deux réactifs introduits (soude et halogéno-2 alcanoate d'alcoyle) reste compris entre 0,8 et 1,2 et de préférence voisin de 1. Dans ces conditions, on observe que,bien que la température soit élevée, il n'y a pratiquement ni formation de lactate alcalin ni baisse de la pureté optique des composés optiquement actifs(tels que le balogéno propionate d'alcoyle et son sel alcalin) présents dans le mélange réactionnel et que l'on obtient le produit résultant avec une excellente pureté optique et un excellent rendement.
(formule @v) dans laquelle hal représente un atome d'halogène, de préférence chlore, R a la-même signification que dans la formule I et R4 représente un radical alcoyle (Cl-C6),sous- une pression a la fois supérieure à a pression de vaporisation (ou d'ébullition) de l'halogeno-2 alcanoate d'alcoyle selon la formule IV , et inférieure à la pression de vapeur de l'eau à cette température. Par quantités sensiblement stoéchiométriques, on entend quantités telles que le rapport molaire des deux réactifs introduits (soude et halogéno-2 alcanoate d'alcoyle) reste compris entre 0,8 et 1,2 et de préférence voisin de 1. Dans ces conditions, on observe que,bien que la température soit élevée, il n'y a pratiquement ni formation de lactate alcalin ni baisse de la pureté optique des composés optiquement actifs(tels que le balogéno propionate d'alcoyle et son sel alcalin) présents dans le mélange réactionnel et que l'on obtient le produit résultant avec une excellente pureté optique et un excellent rendement.
Pour cette variante, on utilise avanta- geusement comme réactifs de départ, d'une part une solution aqueuse de soude, proche de la limite de saturation et d'autre part un halogéno-2 alcanoate d'alcoyle selon la formule IV composé en majeure partie par l'isomère de configuration L, présentant an pouvoir rotatoire levo- gyre (mesure effectuée sans solvant) et comprenant de préférence au moins 90 % en poids d'isomère t. On obtient ainsi in situ une solution hydroorganique du sel alcalin optiquement actif de l'acide halogénoalcanoique et ledit sel réagit au fur et à mesure de sa formation sur le sel alcalin du phénol selon les conditions indiquées précé- demment.
Pour effectuer cette préparation in situ des sels de métaux alcalins des acides halogéno-2 alcanoiques, on peut opérer selon les mêmes conditions de température que selon le procédé décrit précédemment par exemple à des températures comprises entre 70 et 160 C.
La pression à utiliser pour cette varian- te du procédé selon l'invention varie dans des limites qui peuvent être. déterminées à partir des diagrammes de vaporisation de l'eau et de l'halogéno-2 alcanoate d'alcoyle optiquement actif utilisé.
A A titre indicatif, lorsqu'on utilise le.
chloro-propionate de méthyle, en opérant à 90 C, cette pression doit être comprise entre 0,24 bar (pression de vapeur du chloro-2 propionate de méthyle à 90 C) et 0,69 bar (pression de vapeur de l'eau à 90 C).
Lorsqu'on désire.opérer sous une pression plus faible, il peut ,être avantageux de remplacer le chloro-2 propionate de méthyle par un ester moins ns vo- latil tel que par exemple un chloro-2 propionate d d'éthyle, propyle ou butyle
Le procédé selon l'invention peut être mis en oeuvre de façon discontinue, comme indiqué dans les exemples. Il peut également être mis en oeuvre en continu en coulant graduellement une solution aqueuse ou hydroorganique d'halogéno-2 alcanoate de métal alcalin (ou selon la variante l'halogéno-2 alcanoate d'alcoyle et la soude) dans la solution aqueuse du sel alcalin du phénol de formule III, selon les conditions indiquées précédemment et en éliminant le produit de la réaction.
Le procédé selon l'invention peut être mis en oeuvre de façon discontinue, comme indiqué dans les exemples. Il peut également être mis en oeuvre en continu en coulant graduellement une solution aqueuse ou hydroorganique d'halogéno-2 alcanoate de métal alcalin (ou selon la variante l'halogéno-2 alcanoate d'alcoyle et la soude) dans la solution aqueuse du sel alcalin du phénol de formule III, selon les conditions indiquées précédemment et en éliminant le produit de la réaction.
Le procédé selon l'invention fournit en un premier temps les sels de métaux alcalins selon la formule II avec M xeprdsentant un atome de métal alcalin, puis par acidification de ces sels, les acides libres correspondants (composés selon la formule II avec M représentant un atome d'hydrogène).
A partir de ces acides libres et de leurs sels, les divers composés selon la formule I peu 'rent entre obtenus selon des procédés en soi connus ::
des halogénures d'acides selon la formule I pour laquelle A représente un radical COZ peuvent être obtenus par réaction de ces acides libres avec un agent d'halogénation tel que SOCl2, PC15 et PCl3,
- à. partir de ces halogénures d'acides, on peut, préparer les amides correspondants (composés selon la formule I pour lesquels A représente le radical

en faisant réagir ces halogénures d'acides sur
une. amine de formule HN R2R3, à a partir de ces acides libres ou des halogénures d'acides, on peut obtenir des esters (compo sés selon la formule I pour lesquels A représente COOR1 et R1 représente un radical alcoyle éventuellement substi tué), par estérification au moyen d'un alcanol approprié.
des halogénures d'acides selon la formule I pour laquelle A représente un radical COZ peuvent être obtenus par réaction de ces acides libres avec un agent d'halogénation tel que SOCl2, PC15 et PCl3,
- à. partir de ces halogénures d'acides, on peut, préparer les amides correspondants (composés selon la formule I pour lesquels A représente le radical
en faisant réagir ces halogénures d'acides sur
une. amine de formule HN R2R3, à a partir de ces acides libres ou des halogénures d'acides, on peut obtenir des esters (compo sés selon la formule I pour lesquels A représente COOR1 et R1 représente un radical alcoyle éventuellement substi tué), par estérification au moyen d'un alcanol approprié.
D'autres esters peuvent également être obtenus par transestérification directe au moyen d'un alcanol approprié.
Certains sels des acides libres selon la formule II sont obtenus au ours du procédé de l'in Invention (sels de.métaux alcalins et principalement sels de sodium et potassium). D'autres sels peuvent être obtenus à partir des acides libres selon la formule II, par saponification par une base minérale ou organique appropriée.
Le procédé selon l'invention peut être utilisé pour la préparation des différents composés optiquement actifs répondant à la formule I. I1 est tout particulièrement adapté à la préparation de composés suivants obtenus sous une forme optiquement active à tencur en isomère D très elevee acide (méthyl-2 chloro-4 phénoxy)-2 propionique acide naphtoxy-l propionique acide [(dichloro-2,4 phénoxy)-4 phénoxy] -2 propionique.
Les exemples ci-après sont décrits pour illustrer l'invention sans toutefois la limiter.
On observera d'après ces exemples que, le procédé selon l'invention permet d'obtenir, à partir de chloro-2 propionates de métaux alcalins ou d'alcoyles comprenant 94 à 98 % d'isomère L, des acides phénoxy-2 propioniques dont la teneur en isomère D est généralement comprise entre 91,5 et 95,9 %.
On observera également d'après ces exemples que le rendement en acide ph6noxypropionique reste, généralement compris entre 96 et 98 % lorsque le produit de départ est le chloro-2 propionate de sodium, et entre 91 et 95 % lorsque le produit de départ est le chloro-2 propionate de méthyle.
Dans les exemples ci-après, donnés à titre non limitatif, les concentrations sont calculées en g/ml.
Exemple 1
On utilise un réacteur de 500 ml équipé d'une agitation et d'un tube de dégagement prolongé par un réfrigérant (et permettant ainsi de condenser les Ywa peurs dégagées), l'ensemble étant relié à un système permettant d'opérer sous pression réduite.
On utilise un réacteur de 500 ml équipé d'une agitation et d'un tube de dégagement prolongé par un réfrigérant (et permettant ainsi de condenser les Ywa peurs dégagées), l'ensemble étant relié à un système permettant d'opérer sous pression réduite.
Dans ce réacteur, on introduit 171 g (1,2 mole) de méthyl-2-chloro-4 phénol et 92 g d'une solution aqueuse de soude comprenant 44 g de soude et 44 g d'eau et on chauffe le mélange, avec agitation, à 900C, sous 0,25 bar (188 mm Hg) pendant 15 minutes.
On coule alors dans ce mélange, en 2 heures 15 minutes, avec un débit constant, 200 ml d'une solution aqueuse comprenant 130,5 g (1 mole) de chloro-2 propionate de sodium de pouvoir rotatoire [α] 20 = + 30,90 (C = 0,1 eau) et 125 g d'eau, tout en maintenant la température à 900C et la pression à 0,25 bar. Une fois la coulée terminée, le mélange réactionnel est maintenu pen- dant 15 minutes, sous agitation, à 900C et sous 0,25 bar.
On coule alors ce mélange dans 550 ml d'eau, puis on acidifie par HCl ION de façon a l'amener à pH = 6. Il est ensuite lavé par 4 x 200 ml de perchloréthylène puis décanté. La phase aqueuse est amenée à pH = 0,5 et l'acide (méthyl-2 chloro-4 phénoxy > 2 propionique (désigné dans ce qui suit par acide MCPP)est repréci pité puis extrait par 2 x 300 ml de perchloréthylène. La phase organique est recueillie puis distille sous pres- sion reduite et on obtient 199,5 g d'acide MCPP (0,93 mole), de titre alcalimétrique = 100 %, soit un rendement de 93 % par rapport au chloro-2 propionate de sodium.
Dans la première phase organique séparée, on récupère 9,6 g d'acide, ce qui porte le rendement total à 97 %.
L'acide MCPP obtenu présente un pouvoir rotatoire [α]D20 = + 25,86 (C = 0,1 acétone). Compte-tenu du pouvoir rotatoire théorique de l'acide (D) MCPP opti- quernent pur, mesuré selon les mêmes conditions[α]D20 = + 28,150, l'acide MCPP obtenu est composé pour 95,9 % par de l'isomère D, et pour le complément par de l'isomère t et sa pureté optique est donc de 95,9 e.
Compte-tenu du pouvoir rotatoire théorique du L chloro-2 propionate de sodium optiquement pur [α] D20 = + 40 (C = 0,1 eau), le chloro-2 propionate de sodium utilise comme matiere de depart etait constitue pour 98 % par de l'isomère L (et pour le complément par l'isomère D) et sa pureté optique était doc de 98 %.
Essai comparatif
On effectue l'essai décrit dans l'exem- ple 1, en utilisant les mêmes quantités des mêmes réactifs et en opérant selon les mêmes conditions, si ce n'est-qu'au lieu d'opérer sous 0,25 bar, on opère sous pression atmosphérique (1,013 bar), supérieure à la pression de vapeur de l'eau à 900C (0,69 bar). Dans ces conditions, le rendement total en acide MCPP est de 92 % et son pouvoir rotatoire est [α]D20 = + 20,8 (C = 0,1 acétone), soit une pureté optique de 87 %.
On effectue l'essai décrit dans l'exem- ple 1, en utilisant les mêmes quantités des mêmes réactifs et en opérant selon les mêmes conditions, si ce n'est-qu'au lieu d'opérer sous 0,25 bar, on opère sous pression atmosphérique (1,013 bar), supérieure à la pression de vapeur de l'eau à 900C (0,69 bar). Dans ces conditions, le rendement total en acide MCPP est de 92 % et son pouvoir rotatoire est [α]D20 = + 20,8 (C = 0,1 acétone), soit une pureté optique de 87 %.
Exemple 2
On opère selon les conditions décrites à l'exemple 1, avec toutefois les seules différences suivantes : la température est de 150 C (au lieu de 90 C) et la pression de 1,013 bar (760 mm Hg) au lieu de c,25 bar.
On opère selon les conditions décrites à l'exemple 1, avec toutefois les seules différences suivantes : la température est de 150 C (au lieu de 90 C) et la pression de 1,013 bar (760 mm Hg) au lieu de c,25 bar.
On obtient ainsi 205 g d'acide MCPP de titre alcalimétrique 100 % et on récupère de plus 2 g d'acide MCPP. dans la première phase organique séparée, soit un rendement total de 96,5 %. Pouvoir rotatoire[α]D20 = + 24,8 (C = 0,1 acétone), soit une purete optique de 94 %.
Exemple 3
On opère comme selon les conditions décrites dans l'exemple 2, avec toutefois la seule différence suivante : la température est de 130 C, au lieu de 150 C. On obtient ainsi 200 g d'acide MCPP et on récupère de plus 9,5 g de cet acide dans la première phase organi que séparée, soit un rendement total de 97,6 %, Pouvoir rotatoire [α]D20 = + 24,52 (C = 0,1 acétone), soit une pureté optique de 93,5 %.
On opère comme selon les conditions décrites dans l'exemple 2, avec toutefois la seule différence suivante : la température est de 130 C, au lieu de 150 C. On obtient ainsi 200 g d'acide MCPP et on récupère de plus 9,5 g de cet acide dans la première phase organi que séparée, soit un rendement total de 97,6 %, Pouvoir rotatoire [α]D20 = + 24,52 (C = 0,1 acétone), soit une pureté optique de 93,5 %.
Exemple 4
On opère selon les conditions déorites à l'exemple 1 avec toutefois les seules différences suivantes : - la pression est de 0,26 bar (au lieu de 0,25 bar); - la solution aqueuse de chloro-2 propionate de sodium est remplacée par une solution hydroxyméthanollque cons tituée par :
. 130,5 g de chloro-2 propionate de sodium (1 mole),
. 42,0 g de méthanol,
. 93,0 g d'eau.
On opère selon les conditions déorites à l'exemple 1 avec toutefois les seules différences suivantes : - la pression est de 0,26 bar (au lieu de 0,25 bar); - la solution aqueuse de chloro-2 propionate de sodium est remplacée par une solution hydroxyméthanollque cons tituée par :
. 130,5 g de chloro-2 propionate de sodium (1 mole),
. 42,0 g de méthanol,
. 93,0 g d'eau.
On obtient ainsi 206 g d'acide MCPP et on récupère de plus 2,4 g de cet acide dans la premiè- re phase organique séparée, soit un rendement total de 97,2 %. [α]D20 = + 24,63 (C = 0,1 acétone), soit une pureté optique de 93,7 %
La solution hydroxyméthanolique de dé part a été préparée dans une opération séparée en traitant 122,5 g de chloro-2 propionate de méthyle [α]D20 = pureté:99,9 % - 24,85 (sans solvant)/ par 133 g d'une solution aqueuse de soude à 30 % (1 mole) et 10 g de méthanol, à une température inférieure à 35 C, sous pression normale.
La solution hydroxyméthanolique de dé part a été préparée dans une opération séparée en traitant 122,5 g de chloro-2 propionate de méthyle [α]D20 = pureté:99,9 % - 24,85 (sans solvant)/ par 133 g d'une solution aqueuse de soude à 30 % (1 mole) et 10 g de méthanol, à une température inférieure à 35 C, sous pression normale.
Compte-tenu du pouvoir rotatoire 'théo- rique du L chloro-2 propionate de méthyle optiquement pur D20 = - 27,80 (sans solvant), le chloro-2 oropionate de méthyle utilisé comme matière de départ était constitué pour 94,7 % par de l'isomère L (pureté optique 94,7 %)@
Exemple 5
On opère selon les conditions décrites à l'exemple 4,avec toutefois les différences suivantes : - la température est de 135 C (au lieu de 900C), - la pression est de 1,013 bar (au lieu de 0,26 bar).
Exemple 5
On opère selon les conditions décrites à l'exemple 4,avec toutefois les différences suivantes : - la température est de 135 C (au lieu de 900C), - la pression est de 1,013 bar (au lieu de 0,26 bar).
On obtient au total 208,4 g d'acide
MCPP (rendement total 97,1 %), de pouvoir rotatoire [α]D20 = + 24,0 (C = 0,1 acétone), soit une pureté optique de 92,7 %.
MCPP (rendement total 97,1 %), de pouvoir rotatoire [α]D20 = + 24,0 (C = 0,1 acétone), soit une pureté optique de 92,7 %.
Exemple 6
On utilise le même dispositif qu'à l'exemple 1. Dans le réacteur, on introduit 171,5 g de méthyl2 chloro-4 phénol, soit 1,2 mole compte-tenu de la pureté (99,8 %) et 88 g d'une solution aqueuse à 50 % de soude (soit 1,1 mole) et on chauffe ce mélange à 900C sous une pression de 0,4 bar, et on maintient à cette température et pression pendant 20 minutes, sous agitation.
On utilise le même dispositif qu'à l'exemple 1. Dans le réacteur, on introduit 171,5 g de méthyl2 chloro-4 phénol, soit 1,2 mole compte-tenu de la pureté (99,8 %) et 88 g d'une solution aqueuse à 50 % de soude (soit 1,1 mole) et on chauffe ce mélange à 900C sous une pression de 0,4 bar, et on maintient à cette température et pression pendant 20 minutes, sous agitation.
Dans ce mélange, on coule simultanément, en quantites stoéchiométriques, d'une part 124 g de chloro-2 propionate de méthyle, de pureté 98,5 % (soit 1 mole) et de pouvoir rotatoire [α]D20 = - 25 ,3 (sans solvant) et d'autre part 80 g d'une solution aqueuse à 50 z de soude (1 mole). La durée de la coulée des deux réactifs est de 1 heure. Pendant toute cette durée, la tempéra- ture est maintenue à 900C et la pression à 0,4 bar et on maintient le mélange réactionnel, encore pendant 30 minutes sous cette température et cette pression, sous agita tion.
On coule alors ce mélange dans 550 ml d'eau; puis on acidifie par HCl 10 N, de façon à l'amener à Ph = 5,5 et on lave par 4 x 200 ml de perchloréthylène.
La phase organique qui contient le méthyl-2 chloro-4 ph- nol en excès et une faible quantité d'acide MCPP est sé- parée. La phase aqueuse est acidifiée par HCl 10 N jusqu'à pH= 0,5 et l'acide (méthyl-2-chloro-4 phénoxy)-2 propionique est reprécipité puis extrait par 2 x 300 mi de perchloréthylêne. La phase organique est recueillie puis distillée sous pression réduite et on obtient 200 g d'a- cide MCPP, de titre alcalimétrique = 99,6 % et de pouvoir rotatoire [α]D20 = + 24,2 (C = 0,1 acétone). Dans la première phase organique séparée, on récupère encore 1,7g d'acide MCPP.
- pureté optique (ou % d'isomère D) de l'acide MCPP obtenu : 92,8 %, - pureté optique (ou . d'isomère t.) du chloro-2 proplona te de méthyle de départ : 94,7 %, rendement total en acide MCPP par rapport au chloro-2 propionate de méthyle : 94 t.
Exemples 7 à 9
On opère comme à l'exemple 6, à partir des mêmes quantités des mêmes réactifs, mais en faisant varier la température et la pression maintenues pendant la coulée de chloro-2 propionate de méthyle, et de la solution aqueuse de soude, ainsi que la durée de cette 'coulée. Les résultats obtenus sont consignés dans le tableau ci-après ::

On opère comme à l'exemple 6, à partir des mêmes quantités des mêmes réactifs, mais en faisant varier la température et la pression maintenues pendant la coulée de chloro-2 propionate de méthyle, et de la solution aqueuse de soude, ainsi que la durée de cette 'coulée. Les résultats obtenus sont consignés dans le tableau ci-après ::
Exemple <SEP> COULEE <SEP> DES <SEP> REACTIFS <SEP> ACIDE <SEP> MCPP
<tb> <SEP> No <SEP> Tempéra- <SEP> Pression <SEP> Durée <SEP> Rendements <SEP> % <SEP> isomères
<tb> <SEP> ture <SEP> (bars) <SEP> total <SEP> %
<tb> <SEP> ( C)
<tb> <SEP> 7 <SEP> 80 <SEP> 0,4 <SEP> 2h <SEP> 92,5 <SEP> 92,7
<tb> <SEP> 8 <SEP> 90 <SEP> 0,4 <SEP> 1h <SEP> 45 <SEP> 95,6 <SEP> 93,8
<tb> <SEP> 9 <SEP> 130 <SEP> 1,013 <SEP> 1h <SEP> 20 <SEP> 93,0 <SEP> 91,5
<tb>
Exemple 10
On utilise le même dispositif qu'à l'exemple 1.Dans le réacteur, on introduit 172,8 g d'α- naphtol (soit 1,2 mole) et 88 g d'une solution aqueuse à 50 % de soude (soit 1,1 mole) et on chauffe ce à 100 C, sous 0,3 bars, et on maintient sous cette température et cette pression, pendant 15 minutes, sous agitation
On coule alors, en deux heures, dans ce mélange 265,5 g d'une solution hydroxyméthanolique de chloro-2 propionate de sodium ayant la même composition qu'à l'exemple 4 et préparée comme décrit dans cet exemple à partir du même chioro-2 propionate de méthyle.
<tb> <SEP> No <SEP> Tempéra- <SEP> Pression <SEP> Durée <SEP> Rendements <SEP> % <SEP> isomères
<tb> <SEP> ture <SEP> (bars) <SEP> total <SEP> %
<tb> <SEP> ( C)
<tb> <SEP> 7 <SEP> 80 <SEP> 0,4 <SEP> 2h <SEP> 92,5 <SEP> 92,7
<tb> <SEP> 8 <SEP> 90 <SEP> 0,4 <SEP> 1h <SEP> 45 <SEP> 95,6 <SEP> 93,8
<tb> <SEP> 9 <SEP> 130 <SEP> 1,013 <SEP> 1h <SEP> 20 <SEP> 93,0 <SEP> 91,5
<tb>
Exemple 10
On utilise le même dispositif qu'à l'exemple 1.Dans le réacteur, on introduit 172,8 g d'α- naphtol (soit 1,2 mole) et 88 g d'une solution aqueuse à 50 % de soude (soit 1,1 mole) et on chauffe ce à 100 C, sous 0,3 bars, et on maintient sous cette température et cette pression, pendant 15 minutes, sous agitation
On coule alors, en deux heures, dans ce mélange 265,5 g d'une solution hydroxyméthanolique de chloro-2 propionate de sodium ayant la même composition qu'à l'exemple 4 et préparée comme décrit dans cet exemple à partir du même chioro-2 propionate de méthyle.
Après la fin de la coulée, on maintient à 100 C pendant 30 minutes.
On ajoute au mélange 600 mi d'eau et on amène le pH à 5,5 par addition d'HCl ION. On extrait le naphtol libre par 6 fois 100 ml de perchloréthylène à 850C.
A la phase aqueuse maintenue à 85 C, on ajoute 500 mi de perchloréthylène, avant d'amener à pH = 0,5 par addition d'HCl ION. On sépare la phase organique qui est lavée avec deux fois 100 ml d'eau chaude, on élimine ensuite le solvant par évaporation sous vide à 135 C jusqu'à poids constant.
On obtient ainsi 202 g d'acide naphtoxy1 propionique fondant à 125 C, de titre alcalimétrique 99,8 %, de pouvoir rotatoire[α]D20= -39,2 (C=0,01acétone).
Rendement 93,5 %.
Par deux recristallisations successives dans le toluène, on augmente la pureté optique du composé obtenu et on obtient ainsi un acide naphtoxy-1 propionique de pouvoir rotatoire[α]D20 = -45,6 (C= 0,01 acétone), fondant à 128 C. Une troisième recristallisation n'augmente pas le pouvoir rotatoire et il est vraisemblable que le composé ainsi obtenu est constitué dans sa totalité par l'isomère de configuration absolue D.
Exemple 11
On utilise le même dispositif qu'à l'exemple 1. Dans le réacteur on introduit 306 g de (dichloro-2,4 phénoxy)-4 phénol (soit 1,2 mole) et 88 g d'une solution aqueuse à 50 % de soude (soit 1,1 mole) et on chauffe ce mélange à 90 C sous 0,3 bar pendant 1 heure.
On utilise le même dispositif qu'à l'exemple 1. Dans le réacteur on introduit 306 g de (dichloro-2,4 phénoxy)-4 phénol (soit 1,2 mole) et 88 g d'une solution aqueuse à 50 % de soude (soit 1,1 mole) et on chauffe ce mélange à 90 C sous 0,3 bar pendant 1 heure.
On coule dans ce mélange en 1 h 30 265,5 g de la même slution hydroxyméthanolique de chloro-2 propionate de sodium qu'à l'exemple 4. Une fois la coulée terminée, on maintient pendant 30 minutes à 90 C.
On coule alors ce mélange dans 2500 ml d'eau puis amene à pH = 6 par addition d'HCl ION. Le mélange est lavé par 6 x 300 mi de, xylène, puis décanté.
La phase aqueuse est amenée à pH = ,5 et l'acide [ (dichloro-2,4 phénoxy)-4 phénoxy] -2 propionique précipite. On l'extrait par 2 x 300 ml de xylène. La phase organiques est recueillie puis distillée sous pression réduite et on obtient 293 g (Q,896 mole) d'acide [(dichloro-2,4 phénoxy)-4 phénoxy] -2 propionique, de titre alcalimétrique = 99,7 %, soit un rendement de 89,6 % par repport au chloro-2 propionate de méthyle de départ. On récupère encore 14 g d'acide dans la première phase organique séparé (rendement total : 93,9 %).
L'acide obtenu présente un pouvoir rotatoire est D20 = + 15,60 .(C-0,l chloroforme).
On dissout 30 g de cet acide dans 250 ml de méthanol, et après avoir ajouté 0,5 ml d'H2S04, on chauffe à l'ébullition Pendant 2 heures en éliminant l'eau formée et une tartie du méthanol par distillation. On ajoute alors 200 ml de xylène et sépare le méthanol par distillation. La solution xylénique de l'ester formé est lavée par une solution aqueuse de bicarbonate de soude pour neutraliser H2SO4 et l'acide non estérifié. Après élimination du xylene par distillation sous pression réduite, on obtient 29 g de
t(dichloro-2,4 phénoxy)-4 phénoxy3 -2 propionate de méthyle soit un rendement de 92,7 %, de pouvoir rotatoire 20 20 = + 260 (C=O,1 chloroforme).
t(dichloro-2,4 phénoxy)-4 phénoxy3 -2 propionate de méthyle soit un rendement de 92,7 %, de pouvoir rotatoire 20 20 = + 260 (C=O,1 chloroforme).
Claims (3)
- 2 représente un atome d'halogèneR représente un radical alcoyle contenant de 1 à 4atomes de carbone,Par action d'un sel de métal alcalin, optiquement actif d'un acide halogéno-2 alcanoique contenant de 3à 6 atomes de carbone, sur un sel de métal alcalin du phénol de formuleAr - OHdans laquelle Ar a la même signification que précédemment, et, obtention d'un sel alcalin, optiquement actif, répondant à là formule :
 dans laquelle Ar et R ont la mme signification que précédemment et M représente un cation de métal alcalin, puis, éventuellement, transformation de ce sel alcalin selon des méthodes en sol connues pour donner le composé aryloxyalcanoïque optiquement actif, de formule générale::
dans laquelle Ar et R ont la mme signification que précédemment et M représente un cation de métal alcalin, puis, éventuellement, transformation de ce sel alcalin selon des méthodes en sol connues pour donner le composé aryloxyalcanoïque optiquement actif, de formule générale:: dans laquelle Ar, R et A ont la même signification que précédemment,Ledit procédé étant caractérisé en ce que le sel de métal alcalin d'acide aryloxyalcanoïque est prépare en faisant réagir une solution aqueuse ou hydroorganique du sel de métal alcalin, optiquement actif de l'acide halogéno-2 alcanoïque sur une solution aqueuse du sel de métal alcalin du phénol, à une température élevée1 sous une pression inférieure à la pression de vapeur de l'eau à la température de la réaction, sous réserve que ce procédé ne concerne pas la préparation d'acides (D) phénoxy-2 propioniques ou de leurs sels alcalins, par action d'une solution aqueuse ou hydroalcoolique d'un sel alcalin de l'acide chloro-2 propionique, optiquement actif, sur une solution aqueuse d'un sel alcalin de phénols de formule
dans laquelle Ar, R et A ont la même signification que précédemment,Ledit procédé étant caractérisé en ce que le sel de métal alcalin d'acide aryloxyalcanoïque est prépare en faisant réagir une solution aqueuse ou hydroorganique du sel de métal alcalin, optiquement actif de l'acide halogéno-2 alcanoïque sur une solution aqueuse du sel de métal alcalin du phénol, à une température élevée1 sous une pression inférieure à la pression de vapeur de l'eau à la température de la réaction, sous réserve que ce procédé ne concerne pas la préparation d'acides (D) phénoxy-2 propioniques ou de leurs sels alcalins, par action d'une solution aqueuse ou hydroalcoolique d'un sel alcalin de l'acide chloro-2 propionique, optiquement actif, sur une solution aqueuse d'un sel alcalin de phénols de formule dans laquelle X représente un atome d'halogène ou leradical méthyle et n est un nombre entier de 1 à 3.2) Procédé de préparation d'un sel de métal alcalin,optiquement actif, de formule
dans laquelle X représente un atome d'halogène ou leradical méthyle et n est un nombre entier de 1 à 3.2) Procédé de préparation d'un sel de métal alcalin,optiquement actif, de formule dans laquelle Ar, R et M ont la même significationque dans la revendication l, par action d'un sel demétal alcalin, optiquement actif d'un acide halogéno
dans laquelle Ar, R et M ont la même significationque dans la revendication l, par action d'un sel demétal alcalin, optiquement actif d'un acide halogéno - 2 alcanoique contenant de 3 à 6 atomes de carbone,sur un sel de métal alcalin d'un phénol de formuleAr - OH, dans laquelle Ar a la même significationque précédemment, selon les conditions de pression,température et milieu indiquées dans la revendication1,ledit procédé étant caractérisé en ce que le sel demétal alcalin, optiquement actif de l'acide halogêno- 2 2 alcanolque est préparé in situ dans le réacteurcontenant la solution aqueuse du sel de métal alcalindu phénol, en introduisant dansce réacteur, simultanément, et en quantités sensiblenient stoechiométriques, d'une part une solution @@aqueuse d'hydroxyde de métal alcalin et d'autre partun halogéno-2 alcanoate d'alcoyle optiquement actifde formule
 dans laquelle hal représente un atome d'halogène,R a -la même signification que dans la revendication 1et R4 représente un radical alcoyle contenant de l à
dans laquelle hal représente un atome d'halogène,R a -la même signification que dans la revendication 1et R4 représente un radical alcoyle contenant de l à - 6 atomes de carbone, à une température élevée, sousune pression inférieure à la pression de vapeur del'eau à cette température et supérieure à la pressionde vapeur de l'hal-ogéno-2 propionate d'alcoyle àcette température, étant entendu que la réserve figurantdans la revendication l ne concerne pas cette revendication 2.3) Procédé selon l'une des revendications l ou 2, caracteri-se en ce que l'acide halogéno-2 alcanoïque estl'acide chloro-2 propionique.4 > Procédé selon la revendication 3), caractérisé ence que le sel de métal alcalin optiquement actif del'acide chloro-2 propionique et le sel de métalalcalin du phénol sont des sels de sodium..5) Procédé selon la revendication 4), caractérisé ence que la réaction entre le sel sodium du phénolet le sel de sodium optiquement actif de l'acidechloro-propionique est effectuee à une températurecomprise entre 70 et 1600C sous une pression inférieure ou au plus égale à la pression atmosphérique... 6) Procédé selon la revendication 5), caractérisé ence que la pression est comprise entre 0,1 et 1 bar.7) Procédé selon l'une des revendications 5 ou 6, caractériséen ce que la solution aqueuse ou hydroorganique dechloropropionate de sodium optiquement actif contientau moins 20 % en poids de chloro-2 propionate desodium.8) Procédé selon la revendication 5, 6 ou 7, caractérisé ence que la solution aqueuse du sel de métal alcalin duphénol contient au moins 50 % en poids de ce sel.9) Procédé selon la revendication 8), caractérise en ceque le chloro-2 propionate de sodium optiquement : actifest constitué pour au moins 90 % en poids par l'isomèrede configuration L.10) Composé optiquement actif répondant à la formule générale
 dans laquelle Ar, R et A ont la même signification quedans la revendication 1, ledit composé étant obtenuau moyen d'un procédé selon l'une des revendications 1à 9.11) Composé selon la revendication 10 caractérisé en ce qu'ilcomprend au moins 90 % en poids d'isomère de configurationabsolue D.
dans laquelle Ar, R et A ont la même signification quedans la revendication 1, ledit composé étant obtenuau moyen d'un procédé selon l'une des revendications 1à 9.11) Composé selon la revendication 10 caractérisé en ce qu'ilcomprend au moins 90 % en poids d'isomère de configurationabsolue D.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR8015103A FR2486071A2 (fr) | 1979-06-29 | 1980-07-03 | Procede de preparation de composes aryloxyalcanoiques optiquement actifs |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR7917423A FR2460286A1 (fr) | 1979-06-29 | 1979-06-29 | Procede de preparation d'acide phenoxy-2 propioniques optiquement actifs |
| FR8015103A FR2486071A2 (fr) | 1979-06-29 | 1980-07-03 | Procede de preparation de composes aryloxyalcanoiques optiquement actifs |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| FR2486071A2 true FR2486071A2 (fr) | 1982-01-08 |
| FR2486071B2 FR2486071B2 (fr) | 1984-02-10 |
Family
ID=26221241
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| FR8015103A Granted FR2486071A2 (fr) | 1979-06-29 | 1980-07-03 | Procede de preparation de composes aryloxyalcanoiques optiquement actifs |
Country Status (1)
| Country | Link |
|---|---|
| FR (1) | FR2486071A2 (fr) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4757155A (en) * | 1986-12-22 | 1988-07-12 | The Dow Chemical Company | Process for the preparation of optically active 2-[4-(2',4'-dihalophenoxy)phenoxy]propionic acids |
| US4760172A (en) * | 1986-12-22 | 1988-07-26 | The Dow Chemical Company | Process for the preparation of optically active 2-(4-methoxyphenoxy) propionic acid |
| FR2719042A1 (fr) * | 1994-04-25 | 1995-10-27 | Rhone Poulenc Chimie | Procédé de préparation d'un acide alpha-(hydroxyphénoxy)- alcanecarboxylique optiquement actif. |
Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DD64972A (fr) * | ||||
| DD64723A (fr) * | ||||
| GB679676A (en) * | 1950-04-03 | 1952-09-24 | Dow Chemical Co | Improved method for the production of salts of aromatic-oxy-aliphatic carboxylic acids |
| FR504M (fr) * | 1959-01-07 | 1961-05-15 | ||
| FR1479271A (fr) * | 1965-12-17 | 1967-05-05 | Rhone Poulenc Sa | Nouveau procédé de préparation d'acides phénoxy-2 propioniques optiquement actifs |
| DE2251556A1 (de) * | 1971-10-21 | 1973-04-26 | Menarini Sas | Derivate der 1-halogen-2-naphthyloxyessigsaeure sowie verfahren zu ihrer herstellung |
| US4035416A (en) * | 1972-11-02 | 1977-07-12 | The Dow Chemical Company | Process for making phenoxyalkanoic acids |
| EP0009285A1 (fr) * | 1978-09-19 | 1980-04-02 | Akzo N.V. | Procédé de préparation d'acides 2-phénoxy-propioniques optiquement actifs et compositions herbicides les contenant |
| DE2854542A1 (de) * | 1978-12-16 | 1980-06-19 | Hoechst Ag | Verfahren zur herstellung von optisch aktiven d-(phenoxi- bzw. benzyl)- phenoxipropionsaeuren und ihren alkalisalzen |
-
1980
- 1980-07-03 FR FR8015103A patent/FR2486071A2/fr active Granted
Patent Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DD64972A (fr) * | ||||
| DD64723A (fr) * | ||||
| GB679676A (en) * | 1950-04-03 | 1952-09-24 | Dow Chemical Co | Improved method for the production of salts of aromatic-oxy-aliphatic carboxylic acids |
| FR504M (fr) * | 1959-01-07 | 1961-05-15 | ||
| FR1479271A (fr) * | 1965-12-17 | 1967-05-05 | Rhone Poulenc Sa | Nouveau procédé de préparation d'acides phénoxy-2 propioniques optiquement actifs |
| DE2251556A1 (de) * | 1971-10-21 | 1973-04-26 | Menarini Sas | Derivate der 1-halogen-2-naphthyloxyessigsaeure sowie verfahren zu ihrer herstellung |
| US4035416A (en) * | 1972-11-02 | 1977-07-12 | The Dow Chemical Company | Process for making phenoxyalkanoic acids |
| EP0009285A1 (fr) * | 1978-09-19 | 1980-04-02 | Akzo N.V. | Procédé de préparation d'acides 2-phénoxy-propioniques optiquement actifs et compositions herbicides les contenant |
| DE2854542A1 (de) * | 1978-12-16 | 1980-06-19 | Hoechst Ag | Verfahren zur herstellung von optisch aktiven d-(phenoxi- bzw. benzyl)- phenoxipropionsaeuren und ihren alkalisalzen |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4757155A (en) * | 1986-12-22 | 1988-07-12 | The Dow Chemical Company | Process for the preparation of optically active 2-[4-(2',4'-dihalophenoxy)phenoxy]propionic acids |
| US4760172A (en) * | 1986-12-22 | 1988-07-26 | The Dow Chemical Company | Process for the preparation of optically active 2-(4-methoxyphenoxy) propionic acid |
| FR2719042A1 (fr) * | 1994-04-25 | 1995-10-27 | Rhone Poulenc Chimie | Procédé de préparation d'un acide alpha-(hydroxyphénoxy)- alcanecarboxylique optiquement actif. |
| EP0679629A1 (fr) * | 1994-04-25 | 1995-11-02 | Rhone-Poulenc Chimie | Procédé de préparation d'un acide alpha-(hydroxyphenoxy)-alcanecarboxylique optiquement actif et de ses dérivés |
| US5654338A (en) * | 1994-04-25 | 1997-08-05 | Rhone-Poulenc Chimie | Preparation of optically active α-(hydroxyphenoxy)alkanecarboxylic acids and derivatives thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| FR2486071B2 (fr) | 1984-02-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| BE1006426A6 (fr) | Procede de preparation de la n-methyl-3-phenyl-3-[4-(trifluoromethyl)-phenoxy]-propylamine et de ses sels d'addition d'acide. | |
| FR3012290A1 (fr) | Procede de preparation de sels d'acides carboxyliques herbicides | |
| FR2832146A1 (fr) | Base libre de tamsulosine racemique et ses procedes de preparation | |
| FR2459221A1 (fr) | Procede de preparation de chloro-2 propionate d'alcoyle par chloration de lactate d'alcoyle | |
| EP2170801B1 (fr) | Nouveau procede de synthese du fenofibrate | |
| CH653981A5 (fr) | Procede de preparation de composes aryloxyalcanoiques optiquement actifs. | |
| CH621764A5 (fr) | ||
| FR2486071A2 (fr) | Procede de preparation de composes aryloxyalcanoiques optiquement actifs | |
| FR2793490A1 (fr) | Procede de preparation du (1r,2s,4r)-(-)-2-[(2'-{n,n- dimethylamino}-ethoxy)]-2-[phenyl]-1,7,7-tri-[methyl]- bicyclo[2.2.1]-heptane et ses sels d'addition d'acide pharmaceutiquement acceptables | |
| EP0679629B1 (fr) | Procédé de préparation d'un acide alpha-(hydroxyphenoxy)-alcanecarboxylique optiquement actif et de ses dérivés | |
| EP0023459B1 (fr) | Procédé de préparation d'acides hydroxyarylglycoliques racémiques | |
| EP0007882B1 (fr) | Procédé de préparation d'alcools allyliques à partir d'halogénures d'allyle | |
| EP0544740B1 (fr) | Procede de transformation de l'acide (benzoyl-3 phenyl)-2 propionique-r(-) en isomere s(+) | |
| EP0183591B1 (fr) | Procédé de préparation des carbonates de bromo-1 éthyle et d'hydrocarbyle et nouveaux carbonates de bromo-1 éthyle et d'hydrocarbyle | |
| EP2358665B1 (fr) | Procede de preparation de l'hemifumarate d'eplivanserine | |
| FR2769911A1 (fr) | Procede pour preparer des derives de 5-perfluoroalkyluracile | |
| US4760172A (en) | Process for the preparation of optically active 2-(4-methoxyphenoxy) propionic acid | |
| FR2599737A1 (fr) | Procede pour la fixation de groupes alkyles, alkenyles, cycloalkyles ou aralkyles sur une chaine carbonee portant un groupement fonctionnel | |
| FR2853901A1 (fr) | Procede de preparation de derives de l'acide hexahydropyridazine-3-carboxylique | |
| CA1111447A (fr) | Composes halogenes, procede de preparation et application a la preparation du metaphenoxy benzaldehyde | |
| CH627148A5 (fr) | ||
| EP0260262A1 (fr) | Procede de preparation d'aldehydes par formylation | |
| CH618670A5 (en) | Process for preparing pyrogallol and certain of its derivatives | |
| EP0575303A1 (fr) | Un procédé pour la préparation de l'acide 5-(2,5-diméthylphénoxy)-2,2-diméthyl pentanoique | |
| FR2530242A1 (en) | Antiinflammatory 2-phenyl:benzoxazolyl-propionic acid prepn. |