ES2953441T3 - Combinación de anticuerpos anti-hla-dr o anti-Trop-2 con inhibidores de microtúbulos, inhibidores de parp, inhibidores de la cinasa de bruton o inhibidores de la fosfoinositida 3-cinasa mejora significativamente el resultado terapéutico en el cáncer - Google Patents
Combinación de anticuerpos anti-hla-dr o anti-Trop-2 con inhibidores de microtúbulos, inhibidores de parp, inhibidores de la cinasa de bruton o inhibidores de la fosfoinositida 3-cinasa mejora significativamente el resultado terapéutico en el cáncer Download PDFInfo
- Publication number
- ES2953441T3 ES2953441T3 ES16815285T ES16815285T ES2953441T3 ES 2953441 T3 ES2953441 T3 ES 2953441T3 ES 16815285 T ES16815285 T ES 16815285T ES 16815285 T ES16815285 T ES 16815285T ES 2953441 T3 ES2953441 T3 ES 2953441T3
- Authority
- ES
- Spain
- Prior art keywords
- cancer
- antibody
- antibodies
- immunoconjugate
- seq
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 206010028980 Neoplasm Diseases 0.000 title claims abstract description 189
- 231100000782 microtubule inhibitor Toxicity 0.000 title claims abstract description 46
- 239000012661 PARP inhibitor Substances 0.000 title claims abstract description 33
- 229940121906 Poly ADP ribose polymerase inhibitor Drugs 0.000 title claims abstract description 33
- 201000011510 cancer Diseases 0.000 title claims description 67
- 230000001225 therapeutic effect Effects 0.000 title claims description 37
- 229940043355 kinase inhibitor Drugs 0.000 title abstract description 19
- 239000003757 phosphotransferase inhibitor Substances 0.000 title abstract description 19
- 229940043441 phosphoinositide 3-kinase inhibitor Drugs 0.000 title abstract description 18
- 239000002935 phosphatidylinositol 3 kinase inhibitor Substances 0.000 title description 2
- 229940127121 immunoconjugate Drugs 0.000 claims abstract description 126
- 239000003814 drug Substances 0.000 claims abstract description 80
- FJHBVJOVLFPMQE-QFIPXVFZSA-N 7-Ethyl-10-Hydroxy-Camptothecin Chemical compound C1=C(O)C=C2C(CC)=C(CN3C(C4=C([C@@](C(=O)OC4)(O)CC)C=C33)=O)C3=NC2=C1 FJHBVJOVLFPMQE-QFIPXVFZSA-N 0.000 claims abstract description 79
- 229940079593 drug Drugs 0.000 claims abstract description 34
- 230000004614 tumor growth Effects 0.000 claims abstract description 17
- 230000002195 synergetic effect Effects 0.000 claims abstract description 11
- 206010027476 Metastases Diseases 0.000 claims abstract description 7
- 238000002512 chemotherapy Methods 0.000 claims abstract description 6
- 238000001959 radiotherapy Methods 0.000 claims abstract description 5
- 241000282414 Homo sapiens Species 0.000 claims description 116
- 238000000034 method Methods 0.000 claims description 104
- -1 combrestatin Chemical compound 0.000 claims description 99
- 239000000562 conjugate Substances 0.000 claims description 72
- 238000002560 therapeutic procedure Methods 0.000 claims description 59
- 229940049595 antibody-drug conjugate Drugs 0.000 claims description 54
- 239000000611 antibody drug conjugate Substances 0.000 claims description 48
- 229960000572 olaparib Drugs 0.000 claims description 47
- FAQDUNYVKQKNLD-UHFFFAOYSA-N olaparib Chemical group FC1=CC=C(CC2=C3[CH]C=CC=C3C(=O)N=N2)C=C1C(=O)N(CC1)CCN1C(=O)C1CC1 FAQDUNYVKQKNLD-UHFFFAOYSA-N 0.000 claims description 47
- 108090000765 processed proteins & peptides Proteins 0.000 claims description 46
- 229940124597 therapeutic agent Drugs 0.000 claims description 45
- 238000011282 treatment Methods 0.000 claims description 38
- 208000003721 Triple Negative Breast Neoplasms Diseases 0.000 claims description 36
- 208000022679 triple-negative breast carcinoma Diseases 0.000 claims description 36
- UWKQSNNFCGGAFS-XIFFEERXSA-N irinotecan Chemical compound C1=C2C(CC)=C3CN(C(C4=C([C@@](C(=O)OC4)(O)CC)C=4)=O)C=4C3=NC2=CC=C1OC(=O)N(CC1)CCC1N1CCCCC1 UWKQSNNFCGGAFS-XIFFEERXSA-N 0.000 claims description 35
- 229960004768 irinotecan Drugs 0.000 claims description 33
- 229930012538 Paclitaxel Natural products 0.000 claims description 32
- 229960001592 paclitaxel Drugs 0.000 claims description 32
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 claims description 32
- XYFPWWZEPKGCCK-GOSISDBHSA-N ibrutinib Chemical compound C1=2C(N)=NC=NC=2N([C@H]2CN(CCC2)C(=O)C=C)N=C1C(C=C1)=CC=C1OC1=CC=CC=C1 XYFPWWZEPKGCCK-GOSISDBHSA-N 0.000 claims description 26
- 239000003795 chemical substances by application Substances 0.000 claims description 25
- 239000002177 L01XE27 - Ibrutinib Substances 0.000 claims description 22
- 229960001507 ibrutinib Drugs 0.000 claims description 22
- 229940122255 Microtubule inhibitor Drugs 0.000 claims description 21
- VSJKWCGYPAHWDS-FQEVSTJZSA-N camptothecin Chemical compound C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-FQEVSTJZSA-N 0.000 claims description 21
- 102000004196 processed proteins & peptides Human genes 0.000 claims description 21
- VSJKWCGYPAHWDS-UHFFFAOYSA-N dl-camptothecin Natural products C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)C5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-UHFFFAOYSA-N 0.000 claims description 19
- 229960000439 eribulin mesylate Drugs 0.000 claims description 19
- 229960003445 idelalisib Drugs 0.000 claims description 19
- KLWPJMFMVPTNCC-UHFFFAOYSA-N Camptothecin Natural products CCC1(O)C(=O)OCC2=C1C=C3C4Nc5ccccc5C=C4CN3C2=O KLWPJMFMVPTNCC-UHFFFAOYSA-N 0.000 claims description 16
- 229940127093 camptothecin Drugs 0.000 claims description 16
- 102000004127 Cytokines Human genes 0.000 claims description 14
- 108090000695 Cytokines Proteins 0.000 claims description 14
- 206010009944 Colon cancer Diseases 0.000 claims description 11
- HMABYWSNWIZPAG-UHFFFAOYSA-N rucaparib Chemical compound C1=CC(CNC)=CC=C1C(N1)=C2CCNC(=O)C3=C2C1=CC(F)=C3 HMABYWSNWIZPAG-UHFFFAOYSA-N 0.000 claims description 11
- 229950004707 rucaparib Drugs 0.000 claims description 11
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 claims description 10
- NOPNWHSMQOXAEI-PUCKCBAPSA-N (7s,9s)-7-[(2r,4s,5s,6s)-4-(2,3-dihydropyrrol-1-yl)-5-hydroxy-6-methyloxan-2-yl]oxy-6,9,11-trihydroxy-9-(2-hydroxyacetyl)-4-methoxy-8,10-dihydro-7h-tetracene-5,12-dione Chemical compound N1([C@H]2C[C@@H](O[C@@H](C)[C@H]2O)O[C@H]2C[C@@](O)(CC=3C(O)=C4C(=O)C=5C=CC=C(C=5C(=O)C4=C(O)C=32)OC)C(=O)CO)CCC=C1 NOPNWHSMQOXAEI-PUCKCBAPSA-N 0.000 claims description 9
- 229940122803 Vinca alkaloid Drugs 0.000 claims description 9
- 239000003053 toxin Substances 0.000 claims description 9
- 231100000765 toxin Toxicity 0.000 claims description 9
- 206010006187 Breast cancer Diseases 0.000 claims description 8
- 208000026310 Breast neoplasm Diseases 0.000 claims description 8
- 206010061535 Ovarian neoplasm Diseases 0.000 claims description 8
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 claims description 8
- GLVAUDGFNGKCSF-UHFFFAOYSA-N mercaptopurine Chemical compound S=C1NC=NC2=C1NC=N2 GLVAUDGFNGKCSF-UHFFFAOYSA-N 0.000 claims description 8
- 229960003048 vinblastine Drugs 0.000 claims description 8
- JXLYSJRDGCGARV-XQKSVPLYSA-N vincaleukoblastine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-XQKSVPLYSA-N 0.000 claims description 8
- 229960004528 vincristine Drugs 0.000 claims description 8
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 claims description 8
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 claims description 8
- GBABOYUKABKIAF-IELIFDKJSA-N vinorelbine Chemical compound C1N(CC=2C3=CC=CC=C3NC=22)CC(CC)=C[C@H]1C[C@]2(C(=O)OC)C1=CC([C@]23[C@H]([C@@]([C@H](OC(C)=O)[C@]4(CC)C=CCN([C@H]34)CC2)(O)C(=O)OC)N2C)=C2C=C1OC GBABOYUKABKIAF-IELIFDKJSA-N 0.000 claims description 8
- 229960002066 vinorelbine Drugs 0.000 claims description 8
- ZDZOTLJHXYCWBA-VCVYQWHSSA-N N-debenzoyl-N-(tert-butoxycarbonyl)-10-deacetyltaxol Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 ZDZOTLJHXYCWBA-VCVYQWHSSA-N 0.000 claims description 7
- 108091034117 Oligonucleotide Proteins 0.000 claims description 7
- 206010033128 Ovarian cancer Diseases 0.000 claims description 7
- 208000005718 Stomach Neoplasms Diseases 0.000 claims description 7
- 229940123237 Taxane Drugs 0.000 claims description 7
- 229960003668 docetaxel Drugs 0.000 claims description 7
- 206010017758 gastric cancer Diseases 0.000 claims description 7
- CDOOFZZILLRUQH-GDLZYMKVSA-N n-[3-[6-[4-[(2r)-1,4-dimethyl-3-oxopiperazin-2-yl]anilino]-4-methyl-5-oxopyrazin-2-yl]-2-methylphenyl]-4,5,6,7-tetrahydro-1-benzothiophene-2-carboxamide Chemical compound CN1CCN(C)C(=O)[C@H]1C(C=C1)=CC=C1NC1=NC(C=2C(=C(NC(=O)C=3SC=4CCCCC=4C=3)C=CC=2)C)=CN(C)C1=O CDOOFZZILLRUQH-GDLZYMKVSA-N 0.000 claims description 7
- 208000002154 non-small cell lung carcinoma Diseases 0.000 claims description 7
- 229960000303 topotecan Drugs 0.000 claims description 7
- UCFGDBYHRUNTLO-QHCPKHFHSA-N topotecan Chemical compound C1=C(O)C(CN(C)C)=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 UCFGDBYHRUNTLO-QHCPKHFHSA-N 0.000 claims description 7
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 claims description 7
- 229950011257 veliparib Drugs 0.000 claims description 7
- JNAHVYVRKWKWKQ-CYBMUJFWSA-N veliparib Chemical compound N=1C2=CC=CC(C(N)=O)=C2NC=1[C@@]1(C)CCCN1 JNAHVYVRKWKWKQ-CYBMUJFWSA-N 0.000 claims description 7
- IAKHMKGGTNLKSZ-INIZCTEOSA-N (S)-colchicine Chemical compound C1([C@@H](NC(C)=O)CC2)=CC(=O)C(OC)=CC=C1C1=C2C=C(OC)C(OC)=C1OC IAKHMKGGTNLKSZ-INIZCTEOSA-N 0.000 claims description 6
- MDOJTZQKHMAPBK-UHFFFAOYSA-N 4-iodo-3-nitrobenzamide Chemical compound NC(=O)C1=CC=C(I)C([N+]([O-])=O)=C1 MDOJTZQKHMAPBK-UHFFFAOYSA-N 0.000 claims description 6
- 208000001333 Colorectal Neoplasms Diseases 0.000 claims description 6
- 206010014733 Endometrial cancer Diseases 0.000 claims description 6
- 206010014759 Endometrial neoplasm Diseases 0.000 claims description 6
- 239000005551 L01XE03 - Erlotinib Substances 0.000 claims description 6
- UVSVTDVJQAJIFG-VURMDHGXSA-N LFM-A13 Chemical compound C\C(O)=C(/C#N)C(=O)NC1=CC(Br)=CC=C1Br UVSVTDVJQAJIFG-VURMDHGXSA-N 0.000 claims description 6
- 206010060862 Prostate cancer Diseases 0.000 claims description 6
- 208000000236 Prostatic Neoplasms Diseases 0.000 claims description 6
- NKANXQFJJICGDU-QPLCGJKRSA-N Tamoxifen Chemical compound C=1C=CC=CC=1C(/CC)=C(C=1C=CC(OCCN(C)C)=CC=1)/C1=CC=CC=C1 NKANXQFJJICGDU-QPLCGJKRSA-N 0.000 claims description 6
- IEDXPSOJFSVCKU-HOKPPMCLSA-N [4-[[(2S)-5-(carbamoylamino)-2-[[(2S)-2-[6-(2,5-dioxopyrrolidin-1-yl)hexanoylamino]-3-methylbutanoyl]amino]pentanoyl]amino]phenyl]methyl N-[(2S)-1-[[(2S)-1-[[(3R,4S,5S)-1-[(2S)-2-[(1R,2R)-3-[[(1S,2R)-1-hydroxy-1-phenylpropan-2-yl]amino]-1-methoxy-2-methyl-3-oxopropyl]pyrrolidin-1-yl]-3-methoxy-5-methyl-1-oxoheptan-4-yl]-methylamino]-3-methyl-1-oxobutan-2-yl]amino]-3-methyl-1-oxobutan-2-yl]-N-methylcarbamate Chemical compound CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)N[C@H](C)[C@@H](O)c1ccccc1)OC)N(C)C(=O)[C@@H](NC(=O)[C@H](C(C)C)N(C)C(=O)OCc1ccc(NC(=O)[C@H](CCCNC(N)=O)NC(=O)[C@@H](NC(=O)CCCCCN2C(=O)CCC2=O)C(C)C)cc1)C(C)C IEDXPSOJFSVCKU-HOKPPMCLSA-N 0.000 claims description 6
- 229940045799 anthracyclines and related substance Drugs 0.000 claims description 6
- 229960004562 carboplatin Drugs 0.000 claims description 6
- 229930013356 epothilone Natural products 0.000 claims description 6
- AAKJLRGGTJKAMG-UHFFFAOYSA-N erlotinib Chemical compound C=12C=C(OCCOC)C(OCCOC)=CC2=NC=NC=1NC1=CC=CC(C#C)=C1 AAKJLRGGTJKAMG-UHFFFAOYSA-N 0.000 claims description 6
- 229960001433 erlotinib Drugs 0.000 claims description 6
- ODKNJVUHOIMIIZ-RRKCRQDMSA-N floxuridine Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(F)=C1 ODKNJVUHOIMIIZ-RRKCRQDMSA-N 0.000 claims description 6
- 229960002584 gefitinib Drugs 0.000 claims description 6
- XGALLCVXEZPNRQ-UHFFFAOYSA-N gefitinib Chemical compound C=12C=C(OCCCN3CCOCC3)C(OC)=CC2=NC=NC=1NC1=CC=C(F)C(Cl)=C1 XGALLCVXEZPNRQ-UHFFFAOYSA-N 0.000 claims description 6
- 230000002401 inhibitory effect Effects 0.000 claims description 6
- PCHKPVIQAHNQLW-CQSZACIVSA-N niraparib Chemical compound N1=C2C(C(=O)N)=CC=CC2=CN1C(C=C1)=CC=C1[C@@H]1CCCNC1 PCHKPVIQAHNQLW-CQSZACIVSA-N 0.000 claims description 6
- 230000009467 reduction Effects 0.000 claims description 6
- 208000000587 small cell lung carcinoma Diseases 0.000 claims description 6
- 201000011549 stomach cancer Diseases 0.000 claims description 6
- WINHZLLDWRZWRT-ATVHPVEESA-N sunitinib Chemical compound CCN(CC)CCNC(=O)C1=C(C)NC(\C=C/2C3=CC(F)=CC=C3NC\2=O)=C1C WINHZLLDWRZWRT-ATVHPVEESA-N 0.000 claims description 6
- NRUKOCRGYNPUPR-QBPJDGROSA-N teniposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@@H](OC[C@H]4O3)C=3SC=CC=3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 NRUKOCRGYNPUPR-QBPJDGROSA-N 0.000 claims description 6
- DENYZIUJOTUUNY-MRXNPFEDSA-N (2R)-14-fluoro-2-methyl-6,9,10,19-tetrazapentacyclo[14.2.1.02,6.08,18.012,17]nonadeca-1(18),8,12(17),13,15-pentaen-11-one Chemical compound FC=1C=C2C=3C=4C(CN5[C@@](C4NC3C1)(CCC5)C)=NNC2=O DENYZIUJOTUUNY-MRXNPFEDSA-N 0.000 claims description 5
- QMCOCIWNMHBIIA-LROMGURASA-N (2s)-n-tert-butyl-1-[(2s)-1-[(2s)-2-[[(2s)-2-[[(2s)-2-(dimethylamino)-3-methylbutanoyl]amino]-3-methylbutanoyl]-methylamino]-3-methylbutanoyl]pyrrolidine-2-carbonyl]pyrrolidine-2-carboxamide Chemical compound CC(C)[C@H](N(C)C)C(=O)N[C@@H](C(C)C)C(=O)N(C)[C@@H](C(C)C)C(=O)N1CCC[C@H]1C(=O)N1[C@H](C(=O)NC(C)(C)C)CCC1 QMCOCIWNMHBIIA-LROMGURASA-N 0.000 claims description 5
- LSXOBYNBRKOTIQ-RQUBOUMQSA-N (3s,10r,13e,16s)-10-[(3-chloro-4-methoxyphenyl)methyl]-6,6-dimethyl-3-(2-methylpropyl)-16-[(1s)-1-[(2r,3r)-3-phenyloxiran-2-yl]ethyl]-1,4-dioxa-8,11-diazacyclohexadec-13-ene-2,5,9,12-tetrone Chemical compound C1=C(Cl)C(OC)=CC=C1C[C@@H]1C(=O)NCC(C)(C)C(=O)O[C@@H](CC(C)C)C(=O)O[C@H]([C@H](C)[C@@H]2[C@H](O2)C=2C=CC=CC=2)C/C=C/C(=O)N1 LSXOBYNBRKOTIQ-RQUBOUMQSA-N 0.000 claims description 5
- FDKXTQMXEQVLRF-ZHACJKMWSA-N (E)-dacarbazine Chemical compound CN(C)\N=N\c1[nH]cnc1C(N)=O FDKXTQMXEQVLRF-ZHACJKMWSA-N 0.000 claims description 5
- HAWSQZCWOQZXHI-FQEVSTJZSA-N 10-Hydroxycamptothecin Chemical compound C1=C(O)C=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 HAWSQZCWOQZXHI-FQEVSTJZSA-N 0.000 claims description 5
- CQOQDQWUFQDJMK-SSTWWWIQSA-N 2-methoxy-17beta-estradiol Chemical compound C([C@@H]12)C[C@]3(C)[C@@H](O)CC[C@H]3[C@@H]1CCC1=C2C=C(OC)C(O)=C1 CQOQDQWUFQDJMK-SSTWWWIQSA-N 0.000 claims description 5
- NDMPLJNOPCLANR-UHFFFAOYSA-N 3,4-dihydroxy-15-(4-hydroxy-18-methoxycarbonyl-5,18-seco-ibogamin-18-yl)-16-methoxy-1-methyl-6,7-didehydro-aspidospermidine-3-carboxylic acid methyl ester Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 NDMPLJNOPCLANR-UHFFFAOYSA-N 0.000 claims description 5
- GSCPDZHWVNUUFI-UHFFFAOYSA-N 3-aminobenzamide Chemical compound NC(=O)C1=CC=CC(N)=C1 GSCPDZHWVNUUFI-UHFFFAOYSA-N 0.000 claims description 5
- WYWHKKSPHMUBEB-UHFFFAOYSA-N 6-Mercaptoguanine Natural products N1C(N)=NC(=S)C2=C1N=CN2 WYWHKKSPHMUBEB-UHFFFAOYSA-N 0.000 claims description 5
- HAWSQZCWOQZXHI-UHFFFAOYSA-N CPT-OH Natural products C1=C(O)C=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)C5(O)CC)C4=NC2=C1 HAWSQZCWOQZXHI-UHFFFAOYSA-N 0.000 claims description 5
- 229930195695 Halichondrin Natural products 0.000 claims description 5
- 208000008839 Kidney Neoplasms Diseases 0.000 claims description 5
- OWPCHSCAPHNHAV-UHFFFAOYSA-N Rhizoxin Natural products C1C(O)C2(C)OC2C=CC(C)C(OC(=O)C2)CC2CC2OC2C(=O)OC1C(C)C(OC)C(C)=CC=CC(C)=CC1=COC(C)=N1 OWPCHSCAPHNHAV-UHFFFAOYSA-N 0.000 claims description 5
- 206010041067 Small cell lung cancer Diseases 0.000 claims description 5
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 claims description 5
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 claims description 5
- HWGQMRYQVZSGDQ-HZPDHXFCSA-N chembl3137320 Chemical compound CN1N=CN=C1[C@H]([C@H](N1)C=2C=CC(F)=CC=2)C2=NNC(=O)C3=C2C1=CC(F)=C3 HWGQMRYQVZSGDQ-HZPDHXFCSA-N 0.000 claims description 5
- 108010083340 cryptophycin 52 Proteins 0.000 claims description 5
- YFGZFQNBPSCWPN-UHFFFAOYSA-N cryptophycin 52 Natural products C1=CC(OC)=CC=C1CC1C(=O)NCC(C)C(=O)OC(CC(C)C)C(=O)OC(C(C)C2C(O2)C=2C=CC=CC=2)CC=CC(=O)N1 YFGZFQNBPSCWPN-UHFFFAOYSA-N 0.000 claims description 5
- 229930194832 curacin Natural products 0.000 claims description 5
- 229930188854 dolastatin Natural products 0.000 claims description 5
- 229960004679 doxorubicin Drugs 0.000 claims description 5
- YJGVMLPVUAXIQN-UHFFFAOYSA-N epipodophyllotoxin Natural products COC1=C(OC)C(OC)=CC(C2C3=CC=4OCOC=4C=C3C(O)C3C2C(OC3)=O)=C1 YJGVMLPVUAXIQN-UHFFFAOYSA-N 0.000 claims description 5
- 150000003883 epothilone derivatives Chemical class 0.000 claims description 5
- 230000009368 gene silencing by RNA Effects 0.000 claims description 5
- 229930187626 hemiasterlin Natural products 0.000 claims description 5
- 229960001428 mercaptopurine Drugs 0.000 claims description 5
- 229960005558 mertansine Drugs 0.000 claims description 5
- ANZJBCHSOXCCRQ-FKUXLPTCSA-N mertansine Chemical compound CO[C@@H]([C@@]1(O)C[C@H](OC(=O)N1)[C@@H](C)[C@@H]1O[C@@]1(C)[C@@H](OC(=O)[C@H](C)N(C)C(=O)CCS)CC(=O)N1C)\C=C\C=C(C)\CC2=CC(OC)=C(Cl)C1=C2 ANZJBCHSOXCCRQ-FKUXLPTCSA-N 0.000 claims description 5
- 206010061289 metastatic neoplasm Diseases 0.000 claims description 5
- YJGVMLPVUAXIQN-XVVDYKMHSA-N podophyllotoxin Chemical compound COC1=C(OC)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@H](O)[C@@H]3[C@@H]2C(OC3)=O)=C1 YJGVMLPVUAXIQN-XVVDYKMHSA-N 0.000 claims description 5
- 229960001237 podophyllotoxin Drugs 0.000 claims description 5
- YVCVYCSAAZQOJI-UHFFFAOYSA-N podophyllotoxin Natural products COC1=C(O)C(OC)=CC(C2C3=CC=4OCOC=4C=C3C(O)C3C2C(OC3)=O)=C1 YVCVYCSAAZQOJI-UHFFFAOYSA-N 0.000 claims description 5
- OWPCHSCAPHNHAV-LMONGJCWSA-N rhizoxin Chemical compound C/C([C@H](OC)[C@@H](C)[C@@H]1C[C@H](O)[C@]2(C)O[C@@H]2/C=C/[C@@H](C)[C@]2([H])OC(=O)C[C@@](C2)(C[C@@H]2O[C@H]2C(=O)O1)[H])=C\C=C\C(\C)=C\C1=COC(C)=N1 OWPCHSCAPHNHAV-LMONGJCWSA-N 0.000 claims description 5
- ICXJVZHDZFXYQC-UHFFFAOYSA-N spongistatin 1 Natural products OC1C(O2)(O)CC(O)C(C)C2CCCC=CC(O2)CC(O)CC2(O2)CC(OC)CC2CC(=O)C(C)C(OC(C)=O)C(C)C(=C)CC(O2)CC(C)(O)CC2(O2)CC(OC(C)=O)CC2CC(=O)OC2C(O)C(CC(=C)CC(O)C=CC(Cl)=C)OC1C2C ICXJVZHDZFXYQC-UHFFFAOYSA-N 0.000 claims description 5
- 108010029464 tasidotin Proteins 0.000 claims description 5
- 229950010740 tasidotin Drugs 0.000 claims description 5
- 201000005112 urinary bladder cancer Diseases 0.000 claims description 5
- 229960004355 vindesine Drugs 0.000 claims description 5
- UGGWPQSBPIFKDZ-KOTLKJBCSA-N vindesine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(N)=O)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1N=C1[C]2C=CC=C1 UGGWPQSBPIFKDZ-KOTLKJBCSA-N 0.000 claims description 5
- 229960000922 vinflunine Drugs 0.000 claims description 5
- NMDYYWFGPIMTKO-HBVLKOHWSA-N vinflunine Chemical compound C([C@@](C1=C(C2=CC=CC=C2N1)C1)(C2=C(OC)C=C3N(C)[C@@H]4[C@@]5(C3=C2)CCN2CC=C[C@]([C@@H]52)([C@H]([C@]4(O)C(=O)OC)OC(C)=O)CC)C(=O)OC)[C@H]2C[C@@H](C(C)(F)F)CN1C2 NMDYYWFGPIMTKO-HBVLKOHWSA-N 0.000 claims description 5
- CTLOSZHDGZLOQE-UHFFFAOYSA-N 14-methoxy-9-[(4-methylpiperazin-1-yl)methyl]-9,19-diazapentacyclo[10.7.0.02,6.07,11.013,18]nonadeca-1(12),2(6),7(11),13(18),14,16-hexaene-8,10-dione Chemical compound O=C1C2=C3C=4C(OC)=CC=CC=4NC3=C3CCCC3=C2C(=O)N1CN1CCN(C)CC1 CTLOSZHDGZLOQE-UHFFFAOYSA-N 0.000 claims description 4
- VVIAGPKUTFNRDU-UHFFFAOYSA-N 6S-folinic acid Natural products C1NC=2NC(N)=NC(=O)C=2N(C=O)C1CNC1=CC=C(C(=O)NC(CCC(O)=O)C(O)=O)C=C1 VVIAGPKUTFNRDU-UHFFFAOYSA-N 0.000 claims description 4
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 claims description 4
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 claims description 4
- 206010038389 Renal cancer Diseases 0.000 claims description 4
- RJURFGZVJUQBHK-UHFFFAOYSA-N actinomycin D Natural products CC1OC(=O)C(C(C)C)N(C)C(=O)CN(C)C(=O)C2CCCN2C(=O)C(C(C)C)NC(=O)C1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)NC4C(=O)NC(C(N5CCCC5C(=O)N(C)CC(=O)N(C)C(C(C)C)C(=O)OC4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-UHFFFAOYSA-N 0.000 claims description 4
- 229930195731 calicheamicin Natural products 0.000 claims description 4
- BWVHYDYUKQEFHG-UHFFFAOYSA-N cep-8983 Chemical compound COC1=CC=CC2=C1C1=C3C(=O)NC(=O)C3=C3CCCC3=C1N2 BWVHYDYUKQEFHG-UHFFFAOYSA-N 0.000 claims description 4
- 229960004316 cisplatin Drugs 0.000 claims description 4
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 claims description 4
- 229940111134 coxibs Drugs 0.000 claims description 4
- 239000003255 cyclooxygenase 2 inhibitor Substances 0.000 claims description 4
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 claims description 4
- 229960000390 fludarabine Drugs 0.000 claims description 4
- GIUYCYHIANZCFB-FJFJXFQQSA-N fludarabine phosphate Chemical compound C1=NC=2C(N)=NC(F)=NC=2N1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)[C@@H]1O GIUYCYHIANZCFB-FJFJXFQQSA-N 0.000 claims description 4
- 229960002949 fluorouracil Drugs 0.000 claims description 4
- 235000008191 folinic acid Nutrition 0.000 claims description 4
- 239000011672 folinic acid Substances 0.000 claims description 4
- VVIAGPKUTFNRDU-ABLWVSNPSA-N folinic acid Chemical compound C1NC=2NC(N)=NC(=O)C=2N(C=O)C1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 VVIAGPKUTFNRDU-ABLWVSNPSA-N 0.000 claims description 4
- 201000010982 kidney cancer Diseases 0.000 claims description 4
- 229960001691 leucovorin Drugs 0.000 claims description 4
- 230000001394 metastastic effect Effects 0.000 claims description 4
- 229960000485 methotrexate Drugs 0.000 claims description 4
- 238000001356 surgical procedure Methods 0.000 claims description 4
- NNJPGOLRFBJNIW-HNNXBMFYSA-N (-)-demecolcine Chemical compound C1=C(OC)C(=O)C=C2[C@@H](NC)CCC3=CC(OC)=C(OC)C(OC)=C3C2=C1 NNJPGOLRFBJNIW-HNNXBMFYSA-N 0.000 claims description 3
- URCVASXWNJQAEH-HDWVWLDDSA-N (2s,3s,4s,5r,6s)-6-[4-[(5s,5ar,8ar,9r)-5-[[(2r,4ar,6r,7r,8r,8as)-7,8-dihydroxy-2-methyl-4,4a,6,7,8,8a-hexahydropyrano[3,2-d][1,3]dioxin-6-yl]oxy]-8-oxo-5a,6,8a,9-tetrahydro-5h-[2]benzofuro[5,6-f][1,3]benzodioxol-9-yl]-2,6-dimethoxyphenoxy]-3,4,5-trihydrox Chemical compound COC1=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=CC(OC)=C1O[C@@H]1O[C@H](C(O)=O)[C@@H](O)[C@H](O)[C@H]1O URCVASXWNJQAEH-HDWVWLDDSA-N 0.000 claims description 3
- 102100025573 1-alkyl-2-acetylglycerophosphocholine esterase Human genes 0.000 claims description 3
- VSNHCAURESNICA-NJFSPNSNSA-N 1-oxidanylurea Chemical compound N[14C](=O)NO VSNHCAURESNICA-NJFSPNSNSA-N 0.000 claims description 3
- UEJJHQNACJXSKW-UHFFFAOYSA-N 2-(2,6-dioxopiperidin-3-yl)-1H-isoindole-1,3(2H)-dione Chemical compound O=C1C2=CC=CC=C2C(=O)N1C1CCC(=O)NC1=O UEJJHQNACJXSKW-UHFFFAOYSA-N 0.000 claims description 3
- YIMDLWDNDGKDTJ-QLKYHASDSA-N 3'-deamino-3'-(3-cyanomorpholin-4-yl)doxorubicin Chemical compound N1([C@H]2C[C@@H](O[C@@H](C)[C@H]2O)O[C@H]2C[C@@](O)(CC=3C(O)=C4C(=O)C=5C=CC=C(C=5C(=O)C4=C(O)C=32)OC)C(=O)CO)CCOCC1C#N YIMDLWDNDGKDTJ-QLKYHASDSA-N 0.000 claims description 3
- IDPUKCWIGUEADI-UHFFFAOYSA-N 5-[bis(2-chloroethyl)amino]uracil Chemical compound ClCCN(CCCl)C1=CNC(=O)NC1=O IDPUKCWIGUEADI-UHFFFAOYSA-N 0.000 claims description 3
- BFYIZQONLCFLEV-DAELLWKTSA-N Aromasine Chemical compound O=C1C=C[C@]2(C)[C@H]3CC[C@](C)(C(CC4)=O)[C@@H]4[C@@H]3CC(=C)C2=C1 BFYIZQONLCFLEV-DAELLWKTSA-N 0.000 claims description 3
- 108010024976 Asparaginase Proteins 0.000 claims description 3
- MLDQJTXFUGDVEO-UHFFFAOYSA-N BAY-43-9006 Chemical compound C1=NC(C(=O)NC)=CC(OC=2C=CC(NC(=O)NC=3C=C(C(Cl)=CC=3)C(F)(F)F)=CC=2)=C1 MLDQJTXFUGDVEO-UHFFFAOYSA-N 0.000 claims description 3
- 108010006654 Bleomycin Proteins 0.000 claims description 3
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 claims description 3
- DLGOEMSEDOSKAD-UHFFFAOYSA-N Carmustine Chemical compound ClCCNC(=O)N(N=O)CCCl DLGOEMSEDOSKAD-UHFFFAOYSA-N 0.000 claims description 3
- JWBOIMRXGHLCPP-UHFFFAOYSA-N Chloditan Chemical compound C=1C=CC=C(Cl)C=1C(C(Cl)Cl)C1=CC=C(Cl)C=C1 JWBOIMRXGHLCPP-UHFFFAOYSA-N 0.000 claims description 3
- NNJPGOLRFBJNIW-UHFFFAOYSA-N Demecolcine Natural products C1=C(OC)C(=O)C=C2C(NC)CCC3=CC(OC)=C(OC)C(OC)=C3C2=C1 NNJPGOLRFBJNIW-UHFFFAOYSA-N 0.000 claims description 3
- XDXDZDZNSLXDNA-TZNDIEGXSA-N Idarubicin Chemical compound C1[C@H](N)[C@H](O)[C@H](C)O[C@H]1O[C@@H]1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2C[C@@](O)(C(C)=O)C1 XDXDZDZNSLXDNA-TZNDIEGXSA-N 0.000 claims description 3
- XDXDZDZNSLXDNA-UHFFFAOYSA-N Idarubicin Natural products C1C(N)C(O)C(C)OC1OC1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2CC(O)(C(C)=O)C1 XDXDZDZNSLXDNA-UHFFFAOYSA-N 0.000 claims description 3
- 239000005517 L01XE01 - Imatinib Substances 0.000 claims description 3
- 239000005411 L01XE02 - Gefitinib Substances 0.000 claims description 3
- 239000002147 L01XE04 - Sunitinib Substances 0.000 claims description 3
- 239000005511 L01XE05 - Sorafenib Substances 0.000 claims description 3
- 239000002136 L01XE07 - Lapatinib Substances 0.000 claims description 3
- 239000005536 L01XE08 - Nilotinib Substances 0.000 claims description 3
- 239000002145 L01XE14 - Bosutinib Substances 0.000 claims description 3
- GQYIWUVLTXOXAJ-UHFFFAOYSA-N Lomustine Chemical compound ClCCN(N=O)C(=O)NC1CCCCC1 GQYIWUVLTXOXAJ-UHFFFAOYSA-N 0.000 claims description 3
- 229930192392 Mitomycin Natural products 0.000 claims description 3
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 claims description 3
- KYRVNWMVYQXFEU-UHFFFAOYSA-N Nocodazole Chemical compound C1=C2NC(NC(=O)OC)=NC2=CC=C1C(=O)C1=CC=CS1 KYRVNWMVYQXFEU-UHFFFAOYSA-N 0.000 claims description 3
- 238000012228 RNA interference-mediated gene silencing Methods 0.000 claims description 3
- FOCVUCIESVLUNU-UHFFFAOYSA-N Thiotepa Chemical compound C1CN1P(N1CC1)(=S)N1CC1 FOCVUCIESVLUNU-UHFFFAOYSA-N 0.000 claims description 3
- 208000002495 Uterine Neoplasms Diseases 0.000 claims description 3
- 229960001686 afatinib Drugs 0.000 claims description 3
- ULXXDDBFHOBEHA-CWDCEQMOSA-N afatinib Chemical compound N1=CN=C2C=C(O[C@@H]3COCC3)C(NC(=O)/C=C/CN(C)C)=CC2=C1NC1=CC=C(F)C(Cl)=C1 ULXXDDBFHOBEHA-CWDCEQMOSA-N 0.000 claims description 3
- 229950010817 alvocidib Drugs 0.000 claims description 3
- BIIVYFLTOXDAOV-YVEFUNNKSA-N alvocidib Chemical compound O[C@@H]1CN(C)CC[C@@H]1C1=C(O)C=C(O)C2=C1OC(C=1C(=CC=CC=1)Cl)=CC2=O BIIVYFLTOXDAOV-YVEFUNNKSA-N 0.000 claims description 3
- 229960002932 anastrozole Drugs 0.000 claims description 3
- YBBLVLTVTVSKRW-UHFFFAOYSA-N anastrozole Chemical compound N#CC(C)(C)C1=CC(C(C)(C#N)C)=CC(CN2N=CN=C2)=C1 YBBLVLTVTVSKRW-UHFFFAOYSA-N 0.000 claims description 3
- 108010044540 auristatin Proteins 0.000 claims description 3
- 229960003005 axitinib Drugs 0.000 claims description 3
- RITAVMQDGBJQJZ-FMIVXFBMSA-N axitinib Chemical compound CNC(=O)C1=CC=CC=C1SC1=CC=C(C(\C=C\C=2N=CC=CC=2)=NN2)C2=C1 RITAVMQDGBJQJZ-FMIVXFBMSA-N 0.000 claims description 3
- 229950010054 azaribine Drugs 0.000 claims description 3
- QQOBRRFOVWGIMD-OJAKKHQRSA-N azaribine Chemical compound CC(=O)O[C@@H]1[C@H](OC(C)=O)[C@@H](COC(=O)C)O[C@H]1N1C(=O)NC(=O)C=N1 QQOBRRFOVWGIMD-OJAKKHQRSA-N 0.000 claims description 3
- 229960002707 bendamustine Drugs 0.000 claims description 3
- YTKUWDBFDASYHO-UHFFFAOYSA-N bendamustine Chemical compound ClCCN(CCCl)C1=CC=C2N(C)C(CCCC(O)=O)=NC2=C1 YTKUWDBFDASYHO-UHFFFAOYSA-N 0.000 claims description 3
- 239000011230 binding agent Substances 0.000 claims description 3
- 229960001561 bleomycin Drugs 0.000 claims description 3
- OYVAGSVQBOHSSS-UAPAGMARSA-O bleomycin A2 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C OYVAGSVQBOHSSS-UAPAGMARSA-O 0.000 claims description 3
- GXJABQQUPOEUTA-RDJZCZTQSA-N bortezomib Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)B(O)O)NC(=O)C=1N=CC=NC=1)C1=CC=CC=C1 GXJABQQUPOEUTA-RDJZCZTQSA-N 0.000 claims description 3
- 229960001467 bortezomib Drugs 0.000 claims description 3
- UBPYILGKFZZVDX-UHFFFAOYSA-N bosutinib Chemical compound C1=C(Cl)C(OC)=CC(NC=2C3=CC(OC)=C(OCCCN4CCN(C)CC4)C=C3N=CC=2C#N)=C1Cl UBPYILGKFZZVDX-UHFFFAOYSA-N 0.000 claims description 3
- 229960003736 bosutinib Drugs 0.000 claims description 3
- 229960005539 bryostatin 1 Drugs 0.000 claims description 3
- MJQUEDHRCUIRLF-TVIXENOKSA-N bryostatin 1 Chemical compound C([C@@H]1CC(/[C@@H]([C@@](C(C)(C)/C=C/2)(O)O1)OC(=O)/C=C/C=C/CCC)=C\C(=O)OC)[C@H]([C@@H](C)O)OC(=O)C[C@H](O)C[C@@H](O1)C[C@H](OC(C)=O)C(C)(C)[C@]1(O)C[C@@H]1C\C(=C\C(=O)OC)C[C@H]\2O1 MJQUEDHRCUIRLF-TVIXENOKSA-N 0.000 claims description 3
- 229960002092 busulfan Drugs 0.000 claims description 3
- HXCHCVDVKSCDHU-LULTVBGHSA-N calicheamicin Chemical compound C1[C@H](OC)[C@@H](NCC)CO[C@H]1O[C@H]1[C@H](O[C@@H]2C\3=C(NC(=O)OC)C(=O)C[C@](C/3=C/CSSSC)(O)C#C\C=C/C#C2)O[C@H](C)[C@@H](NO[C@@H]2O[C@H](C)[C@@H](SC(=O)C=3C(=C(OC)C(O[C@H]4[C@@H]([C@H](OC)[C@@H](O)[C@H](C)O4)O)=C(I)C=3C)OC)[C@@H](O)C2)[C@@H]1O HXCHCVDVKSCDHU-LULTVBGHSA-N 0.000 claims description 3
- 229960005243 carmustine Drugs 0.000 claims description 3
- 229940047495 celebrex Drugs 0.000 claims description 3
- RZEKVGVHFLEQIL-UHFFFAOYSA-N celecoxib Chemical compound C1=CC(C)=CC=C1C1=CC(C(F)(F)F)=NN1C1=CC=C(S(N)(=O)=O)C=C1 RZEKVGVHFLEQIL-UHFFFAOYSA-N 0.000 claims description 3
- 229960004630 chlorambucil Drugs 0.000 claims description 3
- JCKYGMPEJWAADB-UHFFFAOYSA-N chlorambucil Chemical compound OC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 JCKYGMPEJWAADB-UHFFFAOYSA-N 0.000 claims description 3
- 229960001338 colchicine Drugs 0.000 claims description 3
- 229960005052 demecolcine Drugs 0.000 claims description 3
- CFCUWKMKBJTWLW-UHFFFAOYSA-N deoliosyl-3C-alpha-L-digitoxosyl-MTM Natural products CC=1C(O)=C2C(O)=C3C(=O)C(OC4OC(C)C(O)C(OC5OC(C)C(O)C(OC6OC(C)C(O)C(C)(O)C6)C5)C4)C(C(OC)C(=O)C(O)C(C)O)CC3=CC2=CC=1OC(OC(C)C1O)CC1OC1CC(O)C(O)C(C)O1 CFCUWKMKBJTWLW-UHFFFAOYSA-N 0.000 claims description 3
- INVTYAOGFAGBOE-UHFFFAOYSA-N entinostat Chemical compound NC1=CC=CC=C1NC(=O)C(C=C1)=CC=C1CNC(=O)OCC1=CC=CN=C1 INVTYAOGFAGBOE-UHFFFAOYSA-N 0.000 claims description 3
- 229950005837 entinostat Drugs 0.000 claims description 3
- FRPJXPJMRWBBIH-RBRWEJTLSA-N estramustine Chemical compound ClCCN(CCCl)C(=O)OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 FRPJXPJMRWBBIH-RBRWEJTLSA-N 0.000 claims description 3
- 229960001842 estramustine Drugs 0.000 claims description 3
- 108010038795 estrogen receptors Proteins 0.000 claims description 3
- LIQODXNTTZAGID-OCBXBXKTSA-N etoposide phosphate Chemical compound COC1=C(OP(O)(O)=O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 LIQODXNTTZAGID-OCBXBXKTSA-N 0.000 claims description 3
- 229960000752 etoposide phosphate Drugs 0.000 claims description 3
- 229960000255 exemestane Drugs 0.000 claims description 3
- 229960000556 fingolimod Drugs 0.000 claims description 3
- KKGQTZUTZRNORY-UHFFFAOYSA-N fingolimod Chemical compound CCCCCCCCC1=CC=C(CCC(N)(CO)CO)C=C1 KKGQTZUTZRNORY-UHFFFAOYSA-N 0.000 claims description 3
- 229960000961 floxuridine Drugs 0.000 claims description 3
- 229960002074 flutamide Drugs 0.000 claims description 3
- MKXKFYHWDHIYRV-UHFFFAOYSA-N flutamide Chemical compound CC(C)C(=O)NC1=CC=C([N+]([O-])=O)C(C(F)(F)F)=C1 MKXKFYHWDHIYRV-UHFFFAOYSA-N 0.000 claims description 3
- 229950005309 fostamatinib Drugs 0.000 claims description 3
- GKDRMWXFWHEQQT-UHFFFAOYSA-N fostamatinib Chemical compound COC1=C(OC)C(OC)=CC(NC=2N=C(NC=3N=C4N(COP(O)(O)=O)C(=O)C(C)(C)OC4=CC=3)C(F)=CN=2)=C1 GKDRMWXFWHEQQT-UHFFFAOYSA-N 0.000 claims description 3
- 229960005277 gemcitabine Drugs 0.000 claims description 3
- SDUQYLNIPVEERB-QPPQHZFASA-N gemcitabine Chemical compound O=C1N=C(N)C=CN1[C@H]1C(F)(F)[C@H](O)[C@@H](CO)O1 SDUQYLNIPVEERB-QPPQHZFASA-N 0.000 claims description 3
- 238000001415 gene therapy Methods 0.000 claims description 3
- 229960000908 idarubicin Drugs 0.000 claims description 3
- 229960001101 ifosfamide Drugs 0.000 claims description 3
- HOMGKSMUEGBAAB-UHFFFAOYSA-N ifosfamide Chemical compound ClCCNP1(=O)OCCCN1CCCl HOMGKSMUEGBAAB-UHFFFAOYSA-N 0.000 claims description 3
- 229960002411 imatinib Drugs 0.000 claims description 3
- KTUFNOKKBVMGRW-UHFFFAOYSA-N imatinib Chemical compound C1CN(C)CCN1CC1=CC=C(C(=O)NC=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)C=C1 KTUFNOKKBVMGRW-UHFFFAOYSA-N 0.000 claims description 3
- 229960004891 lapatinib Drugs 0.000 claims description 3
- BCFGMOOMADDAQU-UHFFFAOYSA-N lapatinib Chemical compound O1C(CNCCS(=O)(=O)C)=CC=C1C1=CC=C(N=CN=C2NC=3C=C(Cl)C(OCC=4C=C(F)C=CC=4)=CC=3)C2=C1 BCFGMOOMADDAQU-UHFFFAOYSA-N 0.000 claims description 3
- 229960002247 lomustine Drugs 0.000 claims description 3
- HAWPXGHAZFHHAD-UHFFFAOYSA-N mechlorethamine Chemical compound ClCCN(C)CCCl HAWPXGHAZFHHAD-UHFFFAOYSA-N 0.000 claims description 3
- 229960004961 mechlorethamine Drugs 0.000 claims description 3
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 claims description 3
- 229960001924 melphalan Drugs 0.000 claims description 3
- CFCUWKMKBJTWLW-BKHRDMLASA-N mithramycin Chemical compound O([C@@H]1C[C@@H](O[C@H](C)[C@H]1O)OC=1C=C2C=C3C[C@H]([C@@H](C(=O)C3=C(O)C2=C(O)C=1C)O[C@@H]1O[C@H](C)[C@@H](O)[C@H](O[C@@H]2O[C@H](C)[C@H](O)[C@H](O[C@@H]3O[C@H](C)[C@@H](O)[C@@](C)(O)C3)C2)C1)[C@H](OC)C(=O)[C@@H](O)[C@@H](C)O)[C@H]1C[C@@H](O)[C@H](O)[C@@H](C)O1 CFCUWKMKBJTWLW-BKHRDMLASA-N 0.000 claims description 3
- 229960004857 mitomycin Drugs 0.000 claims description 3
- 229960001156 mitoxantrone Drugs 0.000 claims description 3
- KKZJGLLVHKMTCM-UHFFFAOYSA-N mitoxantrone Chemical compound O=C1C2=C(O)C=CC(O)=C2C(=O)C2=C1C(NCCNCCO)=CC=C2NCCNCCO KKZJGLLVHKMTCM-UHFFFAOYSA-N 0.000 claims description 3
- 229950008835 neratinib Drugs 0.000 claims description 3
- ZNHPZUKZSNBOSQ-BQYQJAHWSA-N neratinib Chemical compound C=12C=C(NC\C=C\CN(C)C)C(OCC)=CC2=NC=C(C#N)C=1NC(C=C1Cl)=CC=C1OCC1=CC=CC=N1 ZNHPZUKZSNBOSQ-BQYQJAHWSA-N 0.000 claims description 3
- HHZIURLSWUIHRB-UHFFFAOYSA-N nilotinib Chemical compound C1=NC(C)=CN1C1=CC(NC(=O)C=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)=CC(C(F)(F)F)=C1 HHZIURLSWUIHRB-UHFFFAOYSA-N 0.000 claims description 3
- 229960001346 nilotinib Drugs 0.000 claims description 3
- 229950006344 nocodazole Drugs 0.000 claims description 3
- 229960003171 plicamycin Drugs 0.000 claims description 3
- 229950008499 plitidepsin Drugs 0.000 claims description 3
- 108010049948 plitidepsin Proteins 0.000 claims description 3
- UUSZLLQJYRSZIS-LXNNNBEUSA-N plitidepsin Chemical compound CN([C@H](CC(C)C)C(=O)N[C@@H]1C(=O)N[C@@H]([C@H](CC(=O)O[C@H](C(=O)[C@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N2CCC[C@H]2C(=O)N(C)[C@@H](CC=2C=CC(OC)=CC=2)C(=O)O[C@@H]1C)C(C)C)O)[C@@H](C)CC)C(=O)[C@@H]1CCCN1C(=O)C(C)=O UUSZLLQJYRSZIS-LXNNNBEUSA-N 0.000 claims description 3
- CPTBDICYNRMXFX-UHFFFAOYSA-N procarbazine Chemical compound CNNCC1=CC=C(C(=O)NC(C)C)C=C1 CPTBDICYNRMXFX-UHFFFAOYSA-N 0.000 claims description 3
- 229960000624 procarbazine Drugs 0.000 claims description 3
- 239000003528 protein farnesyltransferase inhibitor Substances 0.000 claims description 3
- GZUITABIAKMVPG-UHFFFAOYSA-N raloxifene Chemical compound C1=CC(O)=CC=C1C1=C(C(=O)C=2C=CC(OCCN3CCCCC3)=CC=2)C2=CC=C(O)C=C2S1 GZUITABIAKMVPG-UHFFFAOYSA-N 0.000 claims description 3
- 229960004622 raloxifene Drugs 0.000 claims description 3
- 229960003787 sorafenib Drugs 0.000 claims description 3
- 229960001052 streptozocin Drugs 0.000 claims description 3
- ZSJLQEPLLKMAKR-GKHCUFPYSA-N streptozocin Chemical compound O=NN(C)C(=O)N[C@H]1[C@@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O ZSJLQEPLLKMAKR-GKHCUFPYSA-N 0.000 claims description 3
- 229960001796 sunitinib Drugs 0.000 claims description 3
- 229960001603 tamoxifen Drugs 0.000 claims description 3
- DKPFODGZWDEEBT-QFIAKTPHSA-N taxane Chemical class C([C@]1(C)CCC[C@@H](C)[C@H]1C1)C[C@H]2[C@H](C)CC[C@@H]1C2(C)C DKPFODGZWDEEBT-QFIAKTPHSA-N 0.000 claims description 3
- 229960001278 teniposide Drugs 0.000 claims description 3
- 229960003433 thalidomide Drugs 0.000 claims description 3
- 229960001196 thiotepa Drugs 0.000 claims description 3
- 229960003087 tioguanine Drugs 0.000 claims description 3
- 206010044412 transitional cell carcinoma Diseases 0.000 claims description 3
- 229960001055 uracil mustard Drugs 0.000 claims description 3
- 206010046766 uterine cancer Diseases 0.000 claims description 3
- 229950000578 vatalanib Drugs 0.000 claims description 3
- YCOYDOIWSSHVCK-UHFFFAOYSA-N vatalanib Chemical compound C1=CC(Cl)=CC=C1NC(C1=CC=CC=C11)=NN=C1CC1=CC=NC=C1 YCOYDOIWSSHVCK-UHFFFAOYSA-N 0.000 claims description 3
- PXOMSWXCVZBBIV-PQKSKRJKSA-N (2S,3S,4S,5R,6R)-6-[(2S,3R,4S,6R)-4-amino-2-methyl-6-[[(1S,3S)-3,5,12-trihydroxy-3-(2-hydroxyacetyl)-10-methoxy-6,11-dioxo-2,4-dihydro-1H-tetracen-1-yl]oxy]oxan-3-yl]oxy-3,4,5-trihydroxyoxane-2-carboxylic acid Chemical compound C[C@H]1[C@@H]([C@H](C[C@@H](O1)O[C@H]2C[C@@](CC3=C2C(=C4C(=C3O)C(=O)C5=C(C4=O)C(=CC=C5)OC)O)(C(=O)CO)O)N)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)C(=O)O)O)O)O PXOMSWXCVZBBIV-PQKSKRJKSA-N 0.000 claims description 2
- APOKYMYZOKIMLM-LUMVZWMBSA-N (2s,3s,4s,5r,6s)-3,4,5-trihydroxy-6-[4-[[(2s,3s,4s,6r)-3-hydroxy-2-methyl-6-[[(1s,3s)-3,5,12-trihydroxy-3-(2-hydroxyacetyl)-10-methoxy-6,11-dioxo-2,4-dihydro-1h-tetracen-1-yl]oxy]oxan-4-yl]carbamoyloxymethyl]-2-nitrophenoxy]oxane-2-carboxylic acid Chemical compound N([C@H]1C[C@@H](O[C@@H](C)[C@H]1O)O[C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)C(=O)OCC(C=C1[N+]([O-])=O)=CC=C1O[C@@H]1O[C@H](C(O)=O)[C@@H](O)[C@H](O)[C@H]1O APOKYMYZOKIMLM-LUMVZWMBSA-N 0.000 claims description 2
- FPVKHBSQESCIEP-UHFFFAOYSA-N (8S)-3-(2-deoxy-beta-D-erythro-pentofuranosyl)-3,6,7,8-tetrahydroimidazo[4,5-d][1,3]diazepin-8-ol Natural products C1C(O)C(CO)OC1N1C(NC=NCC2O)=C2N=C1 FPVKHBSQESCIEP-UHFFFAOYSA-N 0.000 claims description 2
- PIMQWRZWLQKKBJ-SFHVURJKSA-N 2-[(2S)-1-[3-ethyl-7-[(1-oxido-3-pyridin-1-iumyl)methylamino]-5-pyrazolo[1,5-a]pyrimidinyl]-2-piperidinyl]ethanol Chemical compound C=1C(N2[C@@H](CCCC2)CCO)=NC2=C(CC)C=NN2C=1NCC1=CC=C[N+]([O-])=C1 PIMQWRZWLQKKBJ-SFHVURJKSA-N 0.000 claims description 2
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 claims description 2
- FVLVBPDQNARYJU-XAHDHGMMSA-N C[C@H]1CCC(CC1)NC(=O)N(CCCl)N=O Chemical compound C[C@H]1CCC(CC1)NC(=O)N(CCCl)N=O FVLVBPDQNARYJU-XAHDHGMMSA-N 0.000 claims description 2
- 241000759909 Camptotheca Species 0.000 claims description 2
- PTOAARAWEBMLNO-KVQBGUIXSA-N Cladribine Chemical compound C1=NC=2C(N)=NC(Cl)=NC=2N1[C@H]1C[C@H](O)[C@@H](CO)O1 PTOAARAWEBMLNO-KVQBGUIXSA-N 0.000 claims description 2
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 claims description 2
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 claims description 2
- 108010092160 Dactinomycin Proteins 0.000 claims description 2
- ZBNZXTGUTAYRHI-UHFFFAOYSA-N Dasatinib Chemical compound C=1C(N2CCN(CCO)CC2)=NC(C)=NC=1NC(S1)=NC=C1C(=O)NC1=C(C)C=CC=C1Cl ZBNZXTGUTAYRHI-UHFFFAOYSA-N 0.000 claims description 2
- 239000002067 L01XE06 - Dasatinib Substances 0.000 claims description 2
- 239000002146 L01XE16 - Crizotinib Substances 0.000 claims description 2
- RJURFGZVJUQBHK-IIXSONLDSA-N actinomycin D Chemical compound C[C@H]1OC(=O)[C@H](C(C)C)N(C)C(=O)CN(C)C(=O)[C@@H]2CCCN2C(=O)[C@@H](C(C)C)NC(=O)[C@H]1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)N[C@@H]4C(=O)N[C@@H](C(N5CCC[C@H]5C(=O)N(C)CC(=O)N(C)[C@@H](C(C)C)C(=O)O[C@@H]4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-IIXSONLDSA-N 0.000 claims description 2
- 229960002436 cladribine Drugs 0.000 claims description 2
- 229960005061 crizotinib Drugs 0.000 claims description 2
- KTEIFNKAUNYNJU-GFCCVEGCSA-N crizotinib Chemical compound O([C@H](C)C=1C(=C(F)C=CC=1Cl)Cl)C(C(=NC=1)N)=CC=1C(=C1)C=NN1C1CCNCC1 KTEIFNKAUNYNJU-GFCCVEGCSA-N 0.000 claims description 2
- 229960004397 cyclophosphamide Drugs 0.000 claims description 2
- 229960000684 cytarabine Drugs 0.000 claims description 2
- 229960003901 dacarbazine Drugs 0.000 claims description 2
- 229960000640 dactinomycin Drugs 0.000 claims description 2
- 229960002448 dasatinib Drugs 0.000 claims description 2
- 229960000975 daunorubicin Drugs 0.000 claims description 2
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 claims description 2
- 229950009859 dinaciclib Drugs 0.000 claims description 2
- 229960000350 mitotane Drugs 0.000 claims description 2
- 229940086322 navelbine Drugs 0.000 claims description 2
- 229960002340 pentostatin Drugs 0.000 claims description 2
- FPVKHBSQESCIEP-JQCXWYLXSA-N pentostatin Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(N=CNC[C@H]2O)=C2N=C1 FPVKHBSQESCIEP-JQCXWYLXSA-N 0.000 claims description 2
- 229960003440 semustine Drugs 0.000 claims description 2
- CILBMBUYJCWATM-PYGJLNRPSA-N vinorelbine ditartrate Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O.OC(=O)[C@H](O)[C@@H](O)C(O)=O.C1N(CC=2C3=CC=CC=C3NC=22)CC(CC)=C[C@H]1C[C@]2(C(=O)OC)C1=CC([C@]23[C@H]([C@@]([C@H](OC(C)=O)[C@]4(CC)C=CCN([C@H]34)CC2)(O)C(=O)OC)N2C)=C2C=C1OC CILBMBUYJCWATM-PYGJLNRPSA-N 0.000 claims description 2
- 190000008236 carboplatin Chemical compound 0.000 claims 2
- QAMYWGZHLCQOOJ-PWIVHLLHSA-N eribulin mesylate Chemical compound CS(O)(=O)=O.C([C@H]1CC[C@@H]2O[C@@H]3[C@H]4O[C@H]5C[C@](O[C@H]4[C@H]2O1)(O[C@@H]53)CC[C@@H]1O[C@H](C(C1)=C)CC1)C(=O)C[C@@H]2[C@@H](OC)[C@@H](C[C@H](O)CN)O[C@H]2C[C@@H]2C(=C)[C@H](C)C[C@H]1O2 QAMYWGZHLCQOOJ-PWIVHLLHSA-N 0.000 claims 2
- YKLIKGKUANLGSB-HNNXBMFYSA-N idelalisib Chemical compound C1([C@@H](NC=2[C]3N=CN=C3N=CN=2)CC)=NC2=CC=CC(F)=C2C(=O)N1C1=CC=CC=C1 YKLIKGKUANLGSB-HNNXBMFYSA-N 0.000 claims 2
- BSJGASKRWFKGMV-UHFFFAOYSA-L ammonia dichloroplatinum(2+) Chemical compound N.N.Cl[Pt+2]Cl BSJGASKRWFKGMV-UHFFFAOYSA-L 0.000 claims 1
- 102000015694 estrogen receptors Human genes 0.000 claims 1
- MNRILEROXIRVNJ-UHFFFAOYSA-N tioguanine Chemical compound N1C(N)=NC(=S)C2=NC=N[C]21 MNRILEROXIRVNJ-UHFFFAOYSA-N 0.000 claims 1
- 238000002648 combination therapy Methods 0.000 abstract description 17
- 239000012828 PI3K inhibitor Substances 0.000 abstract description 16
- 102000006354 HLA-DR Antigens Human genes 0.000 abstract description 13
- 108010058597 HLA-DR Antigens Proteins 0.000 abstract description 13
- 230000005764 inhibitory process Effects 0.000 abstract description 13
- 230000000996 additive effect Effects 0.000 abstract description 10
- 238000009169 immunotherapy Methods 0.000 abstract description 4
- 238000011301 standard therapy Methods 0.000 abstract description 2
- 229950000143 sacituzumab govitecan Drugs 0.000 description 122
- ULRUOUDIQPERIJ-PQURJYPBSA-N sacituzumab govitecan Chemical compound N([C@@H](CCCCN)C(=O)NC1=CC=C(C=C1)COC(=O)O[C@]1(CC)C(=O)OCC2=C1C=C1N(C2=O)CC2=C(C3=CC(O)=CC=C3N=C21)CC)C(=O)COCC(=O)NCCOCCOCCOCCOCCOCCOCCOCCOCCN(N=N1)C=C1CNC(=O)C(CC1)CCC1CN1C(=O)CC(SC[C@H](N)C(O)=O)C1=O ULRUOUDIQPERIJ-PQURJYPBSA-N 0.000 description 122
- 210000004027 cell Anatomy 0.000 description 87
- 239000000427 antigen Substances 0.000 description 71
- 108091007433 antigens Proteins 0.000 description 70
- 102000036639 antigens Human genes 0.000 description 70
- 230000027455 binding Effects 0.000 description 54
- 108090000623 proteins and genes Proteins 0.000 description 49
- 235000001014 amino acid Nutrition 0.000 description 45
- 241000699670 Mus sp. Species 0.000 description 41
- 238000006467 substitution reaction Methods 0.000 description 39
- 102000004169 proteins and genes Human genes 0.000 description 35
- 108010047041 Complementarity Determining Regions Proteins 0.000 description 34
- 125000003275 alpha amino acid group Chemical group 0.000 description 33
- 235000018102 proteins Nutrition 0.000 description 32
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 31
- 230000004044 response Effects 0.000 description 30
- NFGXHKASABOEEW-UHFFFAOYSA-N 1-methylethyl 11-methoxy-3,7,11-trimethyl-2,4-dodecadienoate Chemical compound COC(C)(C)CCCC(C)CC=CC(C)=CC(=O)OC(C)C NFGXHKASABOEEW-UHFFFAOYSA-N 0.000 description 28
- 101150044980 Akap1 gene Proteins 0.000 description 28
- 108010021625 Immunoglobulin Fragments Proteins 0.000 description 28
- 102000008394 Immunoglobulin Fragments Human genes 0.000 description 28
- 230000000694 effects Effects 0.000 description 26
- 239000000203 mixture Substances 0.000 description 25
- 201000010099 disease Diseases 0.000 description 23
- 102000001253 Protein Kinase Human genes 0.000 description 22
- 108060006633 protein kinase Proteins 0.000 description 22
- 230000004083 survival effect Effects 0.000 description 22
- 102100035360 Cerebellar degeneration-related antigen 1 Human genes 0.000 description 21
- 241001465754 Metazoa Species 0.000 description 21
- 208000031422 Lymphocytic Chronic B-Cell Leukemia Diseases 0.000 description 20
- 229940024606 amino acid Drugs 0.000 description 20
- 239000012634 fragment Substances 0.000 description 20
- 208000010839 B-cell chronic lymphocytic leukemia Diseases 0.000 description 19
- 101000737793 Homo sapiens Cerebellar degeneration-related antigen 1 Proteins 0.000 description 19
- 150000001413 amino acids Chemical class 0.000 description 19
- UFNVPOGXISZXJD-JBQZKEIOSA-N eribulin Chemical compound C([C@H]1CC[C@@H]2O[C@@H]3[C@H]4O[C@@H]5C[C@](O[C@H]4[C@H]2O1)(O[C@@H]53)CC[C@@H]1O[C@H](C(C1)=C)CC1)C(=O)C[C@@H]2[C@@H](OC)[C@@H](C[C@H](O)CN)O[C@H]2C[C@@H]2C(=C)[C@H](C)C[C@H]1O2 UFNVPOGXISZXJD-JBQZKEIOSA-N 0.000 description 19
- 206010058314 Dysplasia Diseases 0.000 description 18
- 239000007924 injection Substances 0.000 description 18
- 238000002347 injection Methods 0.000 description 18
- 229950000815 veltuzumab Drugs 0.000 description 18
- 102100025475 Carcinoembryonic antigen-related cell adhesion molecule 5 Human genes 0.000 description 17
- 210000003719 b-lymphocyte Anatomy 0.000 description 17
- 230000021615 conjugation Effects 0.000 description 17
- IFSDAJWBUCMOAH-HNNXBMFYSA-N idelalisib Chemical compound C1([C@@H](NC=2C=3N=CNC=3N=CN=2)CC)=NC2=CC=CC(F)=C2C(=O)N1C1=CC=CC=C1 IFSDAJWBUCMOAH-HNNXBMFYSA-N 0.000 description 17
- 125000005647 linker group Chemical group 0.000 description 17
- 238000002823 phage display Methods 0.000 description 17
- 230000003442 weekly effect Effects 0.000 description 17
- 208000015914 Non-Hodgkin lymphomas Diseases 0.000 description 16
- 208000032852 chronic lymphocytic leukemia Diseases 0.000 description 16
- 230000002829 reductive effect Effects 0.000 description 16
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 15
- 206010061902 Pancreatic neoplasm Diseases 0.000 description 15
- 239000002246 antineoplastic agent Substances 0.000 description 15
- 210000001519 tissue Anatomy 0.000 description 15
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Natural products NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 14
- 102000012338 Poly(ADP-ribose) Polymerases Human genes 0.000 description 14
- 108010061844 Poly(ADP-ribose) Polymerases Proteins 0.000 description 14
- 229920000776 Poly(Adenosine diphosphate-ribose) polymerase Polymers 0.000 description 14
- KZSNJWFQEVHDMF-UHFFFAOYSA-N Valine Natural products CC(C)C(N)C(O)=O KZSNJWFQEVHDMF-UHFFFAOYSA-N 0.000 description 14
- 230000000259 anti-tumor effect Effects 0.000 description 14
- 210000004408 hybridoma Anatomy 0.000 description 14
- 229940027941 immunoglobulin g Drugs 0.000 description 14
- 239000011159 matrix material Substances 0.000 description 14
- 208000008443 pancreatic carcinoma Diseases 0.000 description 14
- 102100030595 HLA class II histocompatibility antigen gamma chain Human genes 0.000 description 13
- 101001082627 Homo sapiens HLA class II histocompatibility antigen gamma chain Proteins 0.000 description 13
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 13
- 241000699660 Mus musculus Species 0.000 description 13
- 238000001802 infusion Methods 0.000 description 13
- 210000004072 lung Anatomy 0.000 description 13
- 201000002528 pancreatic cancer Diseases 0.000 description 13
- 208000023275 Autoimmune disease Diseases 0.000 description 12
- DCXYFEDJOCDNAF-REOHCLBHSA-N L-asparagine Chemical compound OC(=O)[C@@H](N)CC(N)=O DCXYFEDJOCDNAF-REOHCLBHSA-N 0.000 description 12
- 206010035226 Plasma cell myeloma Diseases 0.000 description 12
- 125000003295 alanine group Chemical group N[C@@H](C)C(=O)* 0.000 description 12
- 102000005962 receptors Human genes 0.000 description 12
- 108020003175 receptors Proteins 0.000 description 12
- 102100038080 B-cell receptor CD22 Human genes 0.000 description 11
- 201000009030 Carcinoma Diseases 0.000 description 11
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 11
- 101000884305 Homo sapiens B-cell receptor CD22 Proteins 0.000 description 11
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 11
- AGPKZVBTJJNPAG-WHFBIAKZSA-N L-isoleucine Chemical compound CC[C@H](C)[C@H](N)C(O)=O AGPKZVBTJJNPAG-WHFBIAKZSA-N 0.000 description 11
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 11
- 238000003776 cleavage reaction Methods 0.000 description 11
- 230000003993 interaction Effects 0.000 description 11
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 description 11
- 238000004519 manufacturing process Methods 0.000 description 11
- 229960004641 rituximab Drugs 0.000 description 11
- 230000007017 scission Effects 0.000 description 11
- 230000008685 targeting Effects 0.000 description 11
- 108091023037 Aptamer Proteins 0.000 description 10
- 102100022005 B-lymphocyte antigen CD20 Human genes 0.000 description 10
- 208000017604 Hodgkin disease Diseases 0.000 description 10
- 101000897405 Homo sapiens B-lymphocyte antigen CD20 Proteins 0.000 description 10
- 101000914324 Homo sapiens Carcinoembryonic antigen-related cell adhesion molecule 5 Proteins 0.000 description 10
- 102000015696 Interleukins Human genes 0.000 description 10
- 108010063738 Interleukins Proteins 0.000 description 10
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 10
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 10
- 208000034578 Multiple myelomas Diseases 0.000 description 10
- 241000699666 Mus <mouse, genus> Species 0.000 description 10
- 206010039491 Sarcoma Diseases 0.000 description 10
- 230000008901 benefit Effects 0.000 description 10
- 229950009760 epratuzumab Drugs 0.000 description 10
- 239000002955 immunomodulating agent Substances 0.000 description 10
- 229940121354 immunomodulator Drugs 0.000 description 10
- 238000001727 in vivo Methods 0.000 description 10
- 229950003734 milatuzumab Drugs 0.000 description 10
- 210000000056 organ Anatomy 0.000 description 10
- 239000000047 product Substances 0.000 description 10
- 241000894007 species Species 0.000 description 10
- 238000007920 subcutaneous administration Methods 0.000 description 10
- 208000014697 Acute lymphocytic leukaemia Diseases 0.000 description 9
- 108091007743 BRCA1/2 Proteins 0.000 description 9
- 102100025473 Carcinoembryonic antigen-related cell adhesion molecule 6 Human genes 0.000 description 9
- 208000010747 Hodgkins lymphoma Diseases 0.000 description 9
- 101000914326 Homo sapiens Carcinoembryonic antigen-related cell adhesion molecule 6 Proteins 0.000 description 9
- 102000014150 Interferons Human genes 0.000 description 9
- 108010050904 Interferons Proteins 0.000 description 9
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 9
- AYFVYJQAPQTCCC-GBXIJSLDSA-N L-threonine Chemical compound C[C@@H](O)[C@H](N)C(O)=O AYFVYJQAPQTCCC-GBXIJSLDSA-N 0.000 description 9
- 208000006664 Precursor Cell Lymphoblastic Leukemia-Lymphoma Diseases 0.000 description 9
- 125000000539 amino acid group Chemical group 0.000 description 9
- 210000001072 colon Anatomy 0.000 description 9
- 206010012601 diabetes mellitus Diseases 0.000 description 9
- 239000000539 dimer Substances 0.000 description 9
- 239000012636 effector Substances 0.000 description 9
- 108020001507 fusion proteins Proteins 0.000 description 9
- 102000037865 fusion proteins Human genes 0.000 description 9
- 230000003211 malignant effect Effects 0.000 description 9
- 238000009097 single-agent therapy Methods 0.000 description 9
- SJVQHLPISAIATJ-ZDUSSCGKSA-N 8-chloro-2-phenyl-3-[(1S)-1-(7H-purin-6-ylamino)ethyl]-1-isoquinolinone Chemical compound C1([C@@H](NC=2C=3N=CNC=3N=CN=2)C)=CC2=CC=CC(Cl)=C2C(=O)N1C1=CC=CC=C1 SJVQHLPISAIATJ-ZDUSSCGKSA-N 0.000 description 8
- 208000024893 Acute lymphoblastic leukemia Diseases 0.000 description 8
- 108020004414 DNA Proteins 0.000 description 8
- 241000282412 Homo Species 0.000 description 8
- 241001529936 Murinae Species 0.000 description 8
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 8
- 102000000852 Tumor Necrosis Factor-alpha Human genes 0.000 description 8
- 108010026331 alpha-Fetoproteins Proteins 0.000 description 8
- 102000013529 alpha-Fetoproteins Human genes 0.000 description 8
- 210000000481 breast Anatomy 0.000 description 8
- 229940044683 chemotherapy drug Drugs 0.000 description 8
- 230000001684 chronic effect Effects 0.000 description 8
- 150000001875 compounds Chemical class 0.000 description 8
- 201000001981 dermatomyositis Diseases 0.000 description 8
- 230000005847 immunogenicity Effects 0.000 description 8
- 230000001976 improved effect Effects 0.000 description 8
- 208000032839 leukemia Diseases 0.000 description 8
- 208000020816 lung neoplasm Diseases 0.000 description 8
- 201000001441 melanoma Diseases 0.000 description 8
- 238000011580 nude mouse model Methods 0.000 description 8
- 239000002245 particle Substances 0.000 description 8
- 210000002966 serum Anatomy 0.000 description 8
- 239000000126 substance Substances 0.000 description 8
- 108700012359 toxins Proteins 0.000 description 8
- MTCFGRXMJLQNBG-REOHCLBHSA-N (2S)-2-Amino-3-hydroxypropansäure Chemical compound OC[C@H](N)C(O)=O MTCFGRXMJLQNBG-REOHCLBHSA-N 0.000 description 7
- 102100024222 B-lymphocyte antigen CD19 Human genes 0.000 description 7
- 102100032937 CD40 ligand Human genes 0.000 description 7
- 108010022366 Carcinoembryonic Antigen Proteins 0.000 description 7
- 108010071942 Colony-Stimulating Factors Proteins 0.000 description 7
- 102100039620 Granulocyte-macrophage colony-stimulating factor Human genes 0.000 description 7
- 101000980825 Homo sapiens B-lymphocyte antigen CD19 Proteins 0.000 description 7
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 7
- 206010025323 Lymphomas Diseases 0.000 description 7
- 206010034277 Pemphigoid Diseases 0.000 description 7
- 208000031981 Thrombocytopenic Idiopathic Purpura Diseases 0.000 description 7
- 230000001154 acute effect Effects 0.000 description 7
- 235000004279 alanine Nutrition 0.000 description 7
- 230000015572 biosynthetic process Effects 0.000 description 7
- 208000000594 bullous pemphigoid Diseases 0.000 description 7
- 238000006243 chemical reaction Methods 0.000 description 7
- 229940127089 cytotoxic agent Drugs 0.000 description 7
- 208000005017 glioblastoma Diseases 0.000 description 7
- 206010073071 hepatocellular carcinoma Diseases 0.000 description 7
- 230000028993 immune response Effects 0.000 description 7
- 239000003112 inhibitor Substances 0.000 description 7
- 201000007270 liver cancer Diseases 0.000 description 7
- 208000014018 liver neoplasm Diseases 0.000 description 7
- 229960003347 obinutuzumab Drugs 0.000 description 7
- 210000000496 pancreas Anatomy 0.000 description 7
- 239000002243 precursor Substances 0.000 description 7
- 230000001105 regulatory effect Effects 0.000 description 7
- 210000004881 tumor cell Anatomy 0.000 description 7
- 210000003932 urinary bladder Anatomy 0.000 description 7
- 239000004475 Arginine Substances 0.000 description 6
- 101150013553 CD40 gene Proteins 0.000 description 6
- 102000014914 Carrier Proteins Human genes 0.000 description 6
- 102100031940 Epithelial cell adhesion molecule Human genes 0.000 description 6
- 208000021519 Hodgkin lymphoma Diseases 0.000 description 6
- 101001012157 Homo sapiens Receptor tyrosine-protein kinase erbB-2 Proteins 0.000 description 6
- 108060003951 Immunoglobulin Proteins 0.000 description 6
- 108090000723 Insulin-Like Growth Factor I Proteins 0.000 description 6
- 108010029485 Protein Isoforms Proteins 0.000 description 6
- 102000001708 Protein Isoforms Human genes 0.000 description 6
- 102100030086 Receptor tyrosine-protein kinase erbB-2 Human genes 0.000 description 6
- SSJQVDUAKDRWTA-CAYKMONMSA-N SN38 glucuronide Chemical compound C1=C2C(CC)=C3CN(C(C4=C([C@@](C(=O)OC4)(O)CC)C=4)=O)C=4C3=NC2=CC=C1O[C@@H]1O[C@H](C(O)=O)[C@@H](O)[C@H](O)[C@H]1O SSJQVDUAKDRWTA-CAYKMONMSA-N 0.000 description 6
- 210000001744 T-lymphocyte Anatomy 0.000 description 6
- 102100040245 Tumor necrosis factor receptor superfamily member 5 Human genes 0.000 description 6
- 230000000890 antigenic effect Effects 0.000 description 6
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 6
- 210000004369 blood Anatomy 0.000 description 6
- 239000008280 blood Substances 0.000 description 6
- 239000000872 buffer Substances 0.000 description 6
- 208000029742 colonic neoplasm Diseases 0.000 description 6
- 238000006471 dimerization reaction Methods 0.000 description 6
- 208000035475 disorder Diseases 0.000 description 6
- 238000009472 formulation Methods 0.000 description 6
- 230000006870 function Effects 0.000 description 6
- 229940088597 hormone Drugs 0.000 description 6
- 239000005556 hormone Substances 0.000 description 6
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 6
- 208000026278 immune system disease Diseases 0.000 description 6
- 102000018358 immunoglobulin Human genes 0.000 description 6
- 238000000338 in vitro Methods 0.000 description 6
- 230000001965 increasing effect Effects 0.000 description 6
- 238000001990 intravenous administration Methods 0.000 description 6
- 230000001404 mediated effect Effects 0.000 description 6
- 229930182817 methionine Natural products 0.000 description 6
- 125000001360 methionine group Chemical group N[C@@H](CCSC)C(=O)* 0.000 description 6
- 210000001672 ovary Anatomy 0.000 description 6
- 210000002307 prostate Anatomy 0.000 description 6
- 206010039073 rheumatoid arthritis Diseases 0.000 description 6
- 210000002784 stomach Anatomy 0.000 description 6
- 201000000596 systemic lupus erythematosus Diseases 0.000 description 6
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 5
- 102100031585 ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase 1 Human genes 0.000 description 5
- 208000003950 B-cell lymphoma Diseases 0.000 description 5
- 102000017420 CD3 protein, epsilon/gamma/delta subunit Human genes 0.000 description 5
- 108050005493 CD3 protein, epsilon/gamma/delta subunit Proteins 0.000 description 5
- 108010029697 CD40 Ligand Proteins 0.000 description 5
- 206010008342 Cervix carcinoma Diseases 0.000 description 5
- 102100032768 Complement receptor type 2 Human genes 0.000 description 5
- 231100001074 DNA strand break Toxicity 0.000 description 5
- 206010018338 Glioma Diseases 0.000 description 5
- 239000004471 Glycine Substances 0.000 description 5
- 101000777636 Homo sapiens ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase 1 Proteins 0.000 description 5
- 101000941929 Homo sapiens Complement receptor type 2 Proteins 0.000 description 5
- 101001057504 Homo sapiens Interferon-stimulated gene 20 kDa protein Proteins 0.000 description 5
- 101001055144 Homo sapiens Interleukin-2 receptor subunit alpha Proteins 0.000 description 5
- 101000878605 Homo sapiens Low affinity immunoglobulin epsilon Fc receptor Proteins 0.000 description 5
- 102100027268 Interferon-stimulated gene 20 kDa protein Human genes 0.000 description 5
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 description 5
- 102100038007 Low affinity immunoglobulin epsilon Fc receptor Human genes 0.000 description 5
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 5
- 201000011152 Pemphigus Diseases 0.000 description 5
- 208000021386 Sjogren Syndrome Diseases 0.000 description 5
- 102000013275 Somatomedins Human genes 0.000 description 5
- 206010052779 Transplant rejections Diseases 0.000 description 5
- 102100027212 Tumor-associated calcium signal transducer 2 Human genes 0.000 description 5
- 208000006105 Uterine Cervical Neoplasms Diseases 0.000 description 5
- 230000035508 accumulation Effects 0.000 description 5
- 238000009825 accumulation Methods 0.000 description 5
- 201000003710 autoimmune thrombocytopenic purpura Diseases 0.000 description 5
- 108091008324 binding proteins Proteins 0.000 description 5
- 210000004899 c-terminal region Anatomy 0.000 description 5
- 201000010881 cervical cancer Diseases 0.000 description 5
- 230000000295 complement effect Effects 0.000 description 5
- 238000002591 computed tomography Methods 0.000 description 5
- 230000000875 corresponding effect Effects 0.000 description 5
- 210000003238 esophagus Anatomy 0.000 description 5
- 230000002584 immunomodulator Effects 0.000 description 5
- 229940047124 interferons Drugs 0.000 description 5
- 210000003734 kidney Anatomy 0.000 description 5
- CBNAAKBWBABMBY-LQCKLLCCSA-N labetuzumab-sn38 Chemical compound N([C@@H](CCCN)C(=O)NC1=CC=C(C=C1)COC(=O)O[C@]1(CC)C(=O)OCC2=C1C=C1N(C2=O)CC2=C(C3=CC(O)=CC=C3N=C21)CC)C(=O)COCC(=O)NCCOCCOCCOCCOCCOCCOCCOCCOCCN(N=N1)C=C1CNC(=O)C(CC1)CCC1CN1C(=O)CC(SC[C@H](N)C(O)=O)C1=O CBNAAKBWBABMBY-LQCKLLCCSA-N 0.000 description 5
- 230000000670 limiting effect Effects 0.000 description 5
- 201000006417 multiple sclerosis Diseases 0.000 description 5
- 208000025113 myeloid leukemia Diseases 0.000 description 5
- 239000002773 nucleotide Substances 0.000 description 5
- 125000003729 nucleotide group Chemical group 0.000 description 5
- 201000001976 pemphigus vulgaris Diseases 0.000 description 5
- 239000000546 pharmaceutical excipient Substances 0.000 description 5
- 238000000746 purification Methods 0.000 description 5
- 210000000664 rectum Anatomy 0.000 description 5
- 201000000306 sarcoidosis Diseases 0.000 description 5
- 239000007787 solid Substances 0.000 description 5
- 208000011580 syndromic disease Diseases 0.000 description 5
- 238000012360 testing method Methods 0.000 description 5
- 230000001988 toxicity Effects 0.000 description 5
- 231100000419 toxicity Toxicity 0.000 description 5
- 238000011830 transgenic mouse model Methods 0.000 description 5
- 239000004474 valine Substances 0.000 description 5
- SVNJBEMPMKWDCO-KCHLEUMXSA-N (2s)-2-[[(2s)-3-carboxy-2-[[2-[[(2s)-5-(diaminomethylideneamino)-2-[[4-oxo-4-[[4-(4-oxo-8-phenylchromen-2-yl)morpholin-4-ium-4-yl]methoxy]butanoyl]amino]pentanoyl]amino]acetyl]amino]propanoyl]amino]-3-hydroxypropanoate Chemical compound C=1C(=O)C2=CC=CC(C=3C=CC=CC=3)=C2OC=1[N+]1(COC(=O)CCC(=O)N[C@@H](CCCNC(=N)N)C(=O)NCC(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CO)C([O-])=O)CCOCC1 SVNJBEMPMKWDCO-KCHLEUMXSA-N 0.000 description 4
- MZOFCQQQCNRIBI-VMXHOPILSA-N (3s)-4-[[(2s)-1-[[(2s)-1-[[(1s)-1-carboxy-2-hydroxyethyl]amino]-4-methyl-1-oxopentan-2-yl]amino]-5-(diaminomethylideneamino)-1-oxopentan-2-yl]amino]-3-[[2-[[(2s)-2,6-diaminohexanoyl]amino]acetyl]amino]-4-oxobutanoic acid Chemical compound OC[C@@H](C(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@@H](N)CCCCN MZOFCQQQCNRIBI-VMXHOPILSA-N 0.000 description 4
- QDITZBLZQQZVEE-YBEGLDIGSA-N (5z)-5-[(4-pyridin-4-ylquinolin-6-yl)methylidene]-1,3-thiazolidine-2,4-dione Chemical compound S1C(=O)NC(=O)\C1=C\C1=CC=C(N=CC=C2C=3C=CN=CC=3)C2=C1 QDITZBLZQQZVEE-YBEGLDIGSA-N 0.000 description 4
- IUVCFHHAEHNCFT-INIZCTEOSA-N 2-[(1s)-1-[4-amino-3-(3-fluoro-4-propan-2-yloxyphenyl)pyrazolo[3,4-d]pyrimidin-1-yl]ethyl]-6-fluoro-3-(3-fluorophenyl)chromen-4-one Chemical compound C1=C(F)C(OC(C)C)=CC=C1C(C1=C(N)N=CN=C11)=NN1[C@@H](C)C1=C(C=2C=C(F)C=CC=2)C(=O)C2=CC(F)=CC=C2O1 IUVCFHHAEHNCFT-INIZCTEOSA-N 0.000 description 4
- RGHYDLZMTYDBDT-UHFFFAOYSA-N 2-amino-8-ethyl-4-methyl-6-(1H-pyrazol-5-yl)-7-pyrido[2,3-d]pyrimidinone Chemical compound O=C1N(CC)C2=NC(N)=NC(C)=C2C=C1C=1C=CNN=1 RGHYDLZMTYDBDT-UHFFFAOYSA-N 0.000 description 4
- QINPEPAQOBZPOF-UHFFFAOYSA-N 2-amino-n-[3-[[3-(2-chloro-5-methoxyanilino)quinoxalin-2-yl]sulfamoyl]phenyl]-2-methylpropanamide Chemical compound COC1=CC=C(Cl)C(NC=2C(=NC3=CC=CC=C3N=2)NS(=O)(=O)C=2C=C(NC(=O)C(C)(C)N)C=CC=2)=C1 QINPEPAQOBZPOF-UHFFFAOYSA-N 0.000 description 4
- HDXDQPRPFRKGKZ-INIZCTEOSA-N 3-(3-fluorophenyl)-2-[(1s)-1-(7h-purin-6-ylamino)propyl]chromen-4-one Chemical compound C=1([C@@H](NC=2C=3NC=NC=3N=CN=2)CC)OC2=CC=CC=C2C(=O)C=1C1=CC=CC(F)=C1 HDXDQPRPFRKGKZ-INIZCTEOSA-N 0.000 description 4
- UJIAQDJKSXQLIT-UHFFFAOYSA-N 3-[2,4-diamino-7-(3-hydroxyphenyl)-6-pteridinyl]phenol Chemical compound C=1C=CC(O)=CC=1C1=NC2=NC(N)=NC(N)=C2N=C1C1=CC=CC(O)=C1 UJIAQDJKSXQLIT-UHFFFAOYSA-N 0.000 description 4
- SEJLPXCPMNSRAM-GOSISDBHSA-N 6-amino-9-[(3r)-1-but-2-ynoylpyrrolidin-3-yl]-7-(4-phenoxyphenyl)purin-8-one Chemical compound C1N(C(=O)C#CC)CC[C@H]1N1C(=O)N(C=2C=CC(OC=3C=CC=CC=3)=CC=2)C2=C(N)N=CN=C21 SEJLPXCPMNSRAM-GOSISDBHSA-N 0.000 description 4
- ZTUJNJAKTLHBEX-UHFFFAOYSA-N 6-cyclopropyl-8-fluoro-2-[2-(hydroxymethyl)-3-[1-methyl-5-[[5-(4-methylpiperazin-1-yl)pyridin-2-yl]amino]-6-oxopyridin-3-yl]phenyl]isoquinolin-1-one Chemical compound C1CN(C)CCN1C(C=N1)=CC=C1NC1=CC(C=2C(=C(C=CC=2)N2C(C3=C(F)C=C(C=C3C=C2)C2CC2)=O)CO)=CN(C)C1=O ZTUJNJAKTLHBEX-UHFFFAOYSA-N 0.000 description 4
- YEAHTLOYHVWAKW-UHFFFAOYSA-N 8-(1-hydroxyethyl)-2-methoxy-3-[(4-methoxyphenyl)methoxy]benzo[c]chromen-6-one Chemical compound C1=CC(OC)=CC=C1COC(C(=C1)OC)=CC2=C1C1=CC=C(C(C)O)C=C1C(=O)O2 YEAHTLOYHVWAKW-UHFFFAOYSA-N 0.000 description 4
- 208000026872 Addison Disease Diseases 0.000 description 4
- 102100035248 Alpha-(1,3)-fucosyltransferase 4 Human genes 0.000 description 4
- 206010002556 Ankylosing Spondylitis Diseases 0.000 description 4
- 238000012935 Averaging Methods 0.000 description 4
- CWHUFRVAEUJCEF-UHFFFAOYSA-N BKM120 Chemical compound C1=NC(N)=CC(C(F)(F)F)=C1C1=CC(N2CCOCC2)=NC(N2CCOCC2)=N1 CWHUFRVAEUJCEF-UHFFFAOYSA-N 0.000 description 4
- 241000894006 Bacteria Species 0.000 description 4
- 208000008439 Biliary Liver Cirrhosis Diseases 0.000 description 4
- 208000033222 Biliary cirrhosis primary Diseases 0.000 description 4
- 208000011691 Burkitt lymphomas Diseases 0.000 description 4
- 102100024423 Carbonic anhydrase 9 Human genes 0.000 description 4
- 108010012236 Chemokines Proteins 0.000 description 4
- 102000019034 Chemokines Human genes 0.000 description 4
- 206010008748 Chorea Diseases 0.000 description 4
- 206010009900 Colitis ulcerative Diseases 0.000 description 4
- 102100025680 Complement decay-accelerating factor Human genes 0.000 description 4
- 102100033270 Cyclin-dependent kinase inhibitor 1 Human genes 0.000 description 4
- 102000004190 Enzymes Human genes 0.000 description 4
- 108090000790 Enzymes Proteins 0.000 description 4
- 206010015218 Erythema multiforme Diseases 0.000 description 4
- 206010015226 Erythema nodosum Diseases 0.000 description 4
- 208000006168 Ewing Sarcoma Diseases 0.000 description 4
- 206010018372 Glomerulonephritis membranous Diseases 0.000 description 4
- 206010072579 Granulomatosis with polyangiitis Diseases 0.000 description 4
- 208000030836 Hashimoto thyroiditis Diseases 0.000 description 4
- 101001022185 Homo sapiens Alpha-(1,3)-fucosyltransferase 4 Proteins 0.000 description 4
- 101000856022 Homo sapiens Complement decay-accelerating factor Proteins 0.000 description 4
- 101000944380 Homo sapiens Cyclin-dependent kinase inhibitor 1 Proteins 0.000 description 4
- 101000920667 Homo sapiens Epithelial cell adhesion molecule Proteins 0.000 description 4
- 101000853002 Homo sapiens Interleukin-25 Proteins 0.000 description 4
- 101000917858 Homo sapiens Low affinity immunoglobulin gamma Fc region receptor III-A Proteins 0.000 description 4
- 101000917839 Homo sapiens Low affinity immunoglobulin gamma Fc region receptor III-B Proteins 0.000 description 4
- 101000946889 Homo sapiens Monocyte differentiation antigen CD14 Proteins 0.000 description 4
- 101000934338 Homo sapiens Myeloid cell surface antigen CD33 Proteins 0.000 description 4
- 101001128431 Homo sapiens Myeloid-derived growth factor Proteins 0.000 description 4
- 101000738771 Homo sapiens Receptor-type tyrosine-protein phosphatase C Proteins 0.000 description 4
- 101000874179 Homo sapiens Syndecan-1 Proteins 0.000 description 4
- 101000934346 Homo sapiens T-cell surface antigen CD2 Proteins 0.000 description 4
- 101000716102 Homo sapiens T-cell surface glycoprotein CD4 Proteins 0.000 description 4
- 101000934341 Homo sapiens T-cell surface glycoprotein CD5 Proteins 0.000 description 4
- 101000946843 Homo sapiens T-cell surface glycoprotein CD8 alpha chain Proteins 0.000 description 4
- 206010020850 Hyperthyroidism Diseases 0.000 description 4
- GNWHRHGTIBRNSM-UHFFFAOYSA-N IC-87114 Chemical compound CC1=CC=CC=C1N1C(=O)C2=C(C)C=CC=C2N=C1CN1C2=NC=NC(N)=C2N=C1 GNWHRHGTIBRNSM-UHFFFAOYSA-N 0.000 description 4
- 201000009794 Idiopathic Pulmonary Fibrosis Diseases 0.000 description 4
- 206010021245 Idiopathic thrombocytopenic purpura Diseases 0.000 description 4
- 208000010159 IgA glomerulonephritis Diseases 0.000 description 4
- 206010021263 IgA nephropathy Diseases 0.000 description 4
- 108090001005 Interleukin-6 Proteins 0.000 description 4
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 description 4
- KZSNJWFQEVHDMF-BYPYZUCNSA-N L-valine Chemical compound CC(C)[C@H](N)C(O)=O KZSNJWFQEVHDMF-BYPYZUCNSA-N 0.000 description 4
- 102100029185 Low affinity immunoglobulin gamma Fc region receptor III-B Human genes 0.000 description 4
- 208000005777 Lupus Nephritis Diseases 0.000 description 4
- 239000004472 Lysine Substances 0.000 description 4
- 102100035877 Monocyte differentiation antigen CD14 Human genes 0.000 description 4
- 208000000112 Myalgia Diseases 0.000 description 4
- 102100025243 Myeloid cell surface antigen CD33 Human genes 0.000 description 4
- 208000034176 Neoplasms, Germ Cell and Embryonal Diseases 0.000 description 4
- 208000031845 Pernicious anaemia Diseases 0.000 description 4
- 206010065159 Polychondritis Diseases 0.000 description 4
- 208000012654 Primary biliary cholangitis Diseases 0.000 description 4
- ONIBWKKTOPOVIA-UHFFFAOYSA-N Proline Natural products OC(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-N 0.000 description 4
- 108010072866 Prostate-Specific Antigen Proteins 0.000 description 4
- 102000007066 Prostate-Specific Antigen Human genes 0.000 description 4
- 201000004681 Psoriasis Diseases 0.000 description 4
- 102100037422 Receptor-type tyrosine-protein phosphatase C Human genes 0.000 description 4
- 206010039710 Scleroderma Diseases 0.000 description 4
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 4
- 208000027522 Sydenham chorea Diseases 0.000 description 4
- 102100035721 Syndecan-1 Human genes 0.000 description 4
- 102100025237 T-cell surface antigen CD2 Human genes 0.000 description 4
- 102100036011 T-cell surface glycoprotein CD4 Human genes 0.000 description 4
- 102100025244 T-cell surface glycoprotein CD5 Human genes 0.000 description 4
- 102100034922 T-cell surface glycoprotein CD8 alpha chain Human genes 0.000 description 4
- 208000001106 Takayasu Arteritis Diseases 0.000 description 4
- 201000006704 Ulcerative Colitis Diseases 0.000 description 4
- 208000033559 Waldenström macroglobulinemia Diseases 0.000 description 4
- 238000012452 Xenomouse strains Methods 0.000 description 4
- HGVNLRPZOWWDKD-UHFFFAOYSA-N ZSTK-474 Chemical compound FC(F)C1=NC2=CC=CC=C2N1C(N=1)=NC(N2CCOCC2)=NC=1N1CCOCC1 HGVNLRPZOWWDKD-UHFFFAOYSA-N 0.000 description 4
- 206010002026 amyotrophic lateral sclerosis Diseases 0.000 description 4
- 230000003042 antagnostic effect Effects 0.000 description 4
- 238000011319 anticancer therapy Methods 0.000 description 4
- 238000003556 assay Methods 0.000 description 4
- VSRXQHXAPYXROS-UHFFFAOYSA-N azanide;cyclobutane-1,1-dicarboxylic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OC(=O)C1(C(O)=O)CCC1 VSRXQHXAPYXROS-UHFFFAOYSA-N 0.000 description 4
- 229960000397 bevacizumab Drugs 0.000 description 4
- 229950003628 buparlisib Drugs 0.000 description 4
- 229960005395 cetuximab Drugs 0.000 description 4
- 208000025302 chronic primary adrenal insufficiency Diseases 0.000 description 4
- 230000000052 comparative effect Effects 0.000 description 4
- PZBCKZWLPGJMAO-UHFFFAOYSA-N copanlisib Chemical compound C1=CC=2C3=NCCN3C(NC(=O)C=3C=NC(N)=NC=3)=NC=2C(OC)=C1OCCCN1CCOCC1 PZBCKZWLPGJMAO-UHFFFAOYSA-N 0.000 description 4
- 125000000151 cysteine group Chemical group N[C@@H](CS)C(=O)* 0.000 description 4
- 229950006418 dactolisib Drugs 0.000 description 4
- JOGKUKXHTYWRGZ-UHFFFAOYSA-N dactolisib Chemical compound O=C1N(C)C2=CN=C3C=CC(C=4C=C5C=CC=CC5=NC=4)=CC3=C2N1C1=CC=C(C(C)(C)C#N)C=C1 JOGKUKXHTYWRGZ-UHFFFAOYSA-N 0.000 description 4
- 230000007547 defect Effects 0.000 description 4
- 229950004949 duvelisib Drugs 0.000 description 4
- 208000002169 ectodermal dysplasia Diseases 0.000 description 4
- 238000005516 engineering process Methods 0.000 description 4
- 229940088598 enzyme Drugs 0.000 description 4
- 239000013604 expression vector Substances 0.000 description 4
- 235000013922 glutamic acid Nutrition 0.000 description 4
- 239000004220 glutamic acid Substances 0.000 description 4
- 208000024908 graft versus host disease Diseases 0.000 description 4
- 230000012010 growth Effects 0.000 description 4
- 230000009036 growth inhibition Effects 0.000 description 4
- 238000003384 imaging method Methods 0.000 description 4
- 210000000987 immune system Anatomy 0.000 description 4
- 230000002163 immunogen Effects 0.000 description 4
- 229940079322 interferon Drugs 0.000 description 4
- 229940047122 interleukins Drugs 0.000 description 4
- 239000007928 intraperitoneal injection Substances 0.000 description 4
- 210000000244 kidney pelvis Anatomy 0.000 description 4
- 229950000518 labetuzumab Drugs 0.000 description 4
- 210000004185 liver Anatomy 0.000 description 4
- 201000005202 lung cancer Diseases 0.000 description 4
- 208000003747 lymphoid leukemia Diseases 0.000 description 4
- 210000002540 macrophage Anatomy 0.000 description 4
- 230000036210 malignancy Effects 0.000 description 4
- 208000020984 malignant renal pelvis neoplasm Diseases 0.000 description 4
- 201000008350 membranous glomerulonephritis Diseases 0.000 description 4
- 231100000855 membranous nephropathy Toxicity 0.000 description 4
- 229910052751 metal Inorganic materials 0.000 description 4
- 239000002184 metal Substances 0.000 description 4
- 230000009401 metastasis Effects 0.000 description 4
- 238000003032 molecular docking Methods 0.000 description 4
- 206010028417 myasthenia gravis Diseases 0.000 description 4
- KXBDTLQSDKGAEB-UHFFFAOYSA-N n-[3-[[5-fluoro-2-[4-(2-methoxyethoxy)anilino]pyrimidin-4-yl]amino]phenyl]prop-2-enamide Chemical compound C1=CC(OCCOC)=CC=C1NC1=NC=C(F)C(NC=2C=C(NC(=O)C=C)C=CC=2)=N1 KXBDTLQSDKGAEB-UHFFFAOYSA-N 0.000 description 4
- GDCJHDUWWAKBIW-UHFFFAOYSA-N n-[4-[4-[2-(difluoromethyl)-4-methoxybenzimidazol-1-yl]-6-morpholin-4-yl-1,3,5-triazin-2-yl]phenyl]-2-(dimethylamino)ethanesulfonamide Chemical compound FC(F)C1=NC=2C(OC)=CC=CC=2N1C(N=1)=NC(N2CCOCC2)=NC=1C1=CC=C(NS(=O)(=O)CCN(C)C)C=C1 GDCJHDUWWAKBIW-UHFFFAOYSA-N 0.000 description 4
- JOWXJLIFIIOYMS-UHFFFAOYSA-N n-hydroxy-2-[[2-(6-methoxypyridin-3-yl)-4-morpholin-4-ylthieno[3,2-d]pyrimidin-6-yl]methyl-methylamino]pyrimidine-5-carboxamide Chemical compound C1=NC(OC)=CC=C1C1=NC(N2CCOCC2)=C(SC(CN(C)C=2N=CC(=CN=2)C(=O)NO)=C2)C2=N1 JOWXJLIFIIOYMS-UHFFFAOYSA-N 0.000 description 4
- 230000009826 neoplastic cell growth Effects 0.000 description 4
- 201000008383 nephritis Diseases 0.000 description 4
- 208000002040 neurosyphilis Diseases 0.000 description 4
- 208000004235 neutropenia Diseases 0.000 description 4
- 201000008968 osteosarcoma Diseases 0.000 description 4
- 230000036961 partial effect Effects 0.000 description 4
- SZFPYBIJACMNJV-UHFFFAOYSA-N perifosine Chemical compound CCCCCCCCCCCCCCCCCCOP([O-])(=O)OC1CC[N+](C)(C)CC1 SZFPYBIJACMNJV-UHFFFAOYSA-N 0.000 description 4
- 229950010632 perifosine Drugs 0.000 description 4
- LHNIIDJUOCFXAP-UHFFFAOYSA-N pictrelisib Chemical compound C1CN(S(=O)(=O)C)CCN1CC1=CC2=NC(C=3C=4C=NNC=4C=CC=3)=NC(N3CCOCC3)=C2S1 LHNIIDJUOCFXAP-UHFFFAOYSA-N 0.000 description 4
- 201000006292 polyarteritis nodosa Diseases 0.000 description 4
- 208000005987 polymyositis Diseases 0.000 description 4
- 230000003389 potentiating effect Effects 0.000 description 4
- 208000005069 pulmonary fibrosis Diseases 0.000 description 4
- 201000003068 rheumatic fever Diseases 0.000 description 4
- 238000012216 screening Methods 0.000 description 4
- BLGWHBSBBJNKJO-UHFFFAOYSA-N serabelisib Chemical compound C=1C=C2OC(N)=NC2=CC=1C(=CN12)C=CC1=NC=C2C(=O)N1CCOCC1 BLGWHBSBBJNKJO-UHFFFAOYSA-N 0.000 description 4
- 208000037969 squamous neck cancer Diseases 0.000 description 4
- 238000010561 standard procedure Methods 0.000 description 4
- 208000002025 tabes dorsalis Diseases 0.000 description 4
- 208000005057 thyrotoxicosis Diseases 0.000 description 4
- 239000013638 trimer Substances 0.000 description 4
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 4
- 229960004441 tyrosine Drugs 0.000 description 4
- QDLHCMPXEPAAMD-QAIWCSMKSA-N wortmannin Chemical compound C1([C@]2(C)C3=C(C4=O)OC=C3C(=O)O[C@@H]2COC)=C4[C@@H]2CCC(=O)[C@@]2(C)C[C@H]1OC(C)=O QDLHCMPXEPAAMD-QAIWCSMKSA-N 0.000 description 4
- QDLHCMPXEPAAMD-UHFFFAOYSA-N wortmannin Natural products COCC1OC(=O)C2=COC(C3=O)=C2C1(C)C1=C3C2CCC(=O)C2(C)CC1OC(C)=O QDLHCMPXEPAAMD-UHFFFAOYSA-N 0.000 description 4
- AADVCYNFEREWOS-UHFFFAOYSA-N (+)-DDM Natural products C=CC=CC(C)C(OC(N)=O)C(C)C(O)C(C)CC(C)=CC(C)C(O)C(C)C=CC(O)CC1OC(=O)C(C)C(O)C1C AADVCYNFEREWOS-UHFFFAOYSA-N 0.000 description 3
- DVLFYONBTKHTER-UHFFFAOYSA-N 3-(N-morpholino)propanesulfonic acid Chemical compound OS(=O)(=O)CCCN1CCOCC1 DVLFYONBTKHTER-UHFFFAOYSA-N 0.000 description 3
- 102100024049 A-kinase anchor protein 13 Human genes 0.000 description 3
- 208000031261 Acute myeloid leukaemia Diseases 0.000 description 3
- 208000002267 Anti-neutrophil cytoplasmic antibody-associated vasculitis Diseases 0.000 description 3
- 102100021569 Apoptosis regulator Bcl-2 Human genes 0.000 description 3
- DCXYFEDJOCDNAF-UHFFFAOYSA-N Asparagine Natural products OC(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-N 0.000 description 3
- 206010003571 Astrocytoma Diseases 0.000 description 3
- 206010004593 Bile duct cancer Diseases 0.000 description 3
- 206010005003 Bladder cancer Diseases 0.000 description 3
- 102100037086 Bone marrow stromal antigen 2 Human genes 0.000 description 3
- 208000003174 Brain Neoplasms Diseases 0.000 description 3
- 102100021943 C-C motif chemokine 2 Human genes 0.000 description 3
- 102100032367 C-C motif chemokine 5 Human genes 0.000 description 3
- 102100024217 CAMPATH-1 antigen Human genes 0.000 description 3
- 102100032912 CD44 antigen Human genes 0.000 description 3
- 108010065524 CD52 Antigen Proteins 0.000 description 3
- 102100022002 CD59 glycoprotein Human genes 0.000 description 3
- 108010062802 CD66 antigens Proteins 0.000 description 3
- 206010007953 Central nervous system lymphoma Diseases 0.000 description 3
- 108010055166 Chemokine CCL5 Proteins 0.000 description 3
- 102000011022 Chorionic Gonadotropin Human genes 0.000 description 3
- 108010062540 Chorionic Gonadotropin Proteins 0.000 description 3
- 206010008909 Chronic Hepatitis Diseases 0.000 description 3
- 108091035707 Consensus sequence Proteins 0.000 description 3
- 102000008130 Cyclic AMP-Dependent Protein Kinases Human genes 0.000 description 3
- 108010049894 Cyclic AMP-Dependent Protein Kinases Proteins 0.000 description 3
- 206010012735 Diarrhoea Diseases 0.000 description 3
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 3
- AADVCYNFEREWOS-OBRABYBLSA-N Discodermolide Chemical compound C=C\C=C/[C@H](C)[C@H](OC(N)=O)[C@@H](C)[C@H](O)[C@@H](C)C\C(C)=C/[C@H](C)[C@@H](O)[C@@H](C)\C=C/[C@@H](O)C[C@@H]1OC(=O)[C@H](C)[C@@H](O)[C@H]1C AADVCYNFEREWOS-OBRABYBLSA-N 0.000 description 3
- 102000003951 Erythropoietin Human genes 0.000 description 3
- 108090000394 Erythropoietin Proteins 0.000 description 3
- 102000018233 Fibroblast Growth Factor Human genes 0.000 description 3
- 108050007372 Fibroblast Growth Factor Proteins 0.000 description 3
- 102100037362 Fibronectin Human genes 0.000 description 3
- 108010067306 Fibronectins Proteins 0.000 description 3
- 208000007465 Giant cell arteritis Diseases 0.000 description 3
- 208000032612 Glial tumor Diseases 0.000 description 3
- 102100041003 Glutamate carboxypeptidase 2 Human genes 0.000 description 3
- 208000009329 Graft vs Host Disease Diseases 0.000 description 3
- 206010019617 Henoch-Schonlein purpura Diseases 0.000 description 3
- 206010019755 Hepatitis chronic active Diseases 0.000 description 3
- 102100026122 High affinity immunoglobulin gamma Fc receptor I Human genes 0.000 description 3
- 101000971171 Homo sapiens Apoptosis regulator Bcl-2 Proteins 0.000 description 3
- 101000740785 Homo sapiens Bone marrow stromal antigen 2 Proteins 0.000 description 3
- 101000868273 Homo sapiens CD44 antigen Proteins 0.000 description 3
- 101000897400 Homo sapiens CD59 glycoprotein Proteins 0.000 description 3
- 101000892862 Homo sapiens Glutamate carboxypeptidase 2 Proteins 0.000 description 3
- 101000913074 Homo sapiens High affinity immunoglobulin gamma Fc receptor I Proteins 0.000 description 3
- 101000777628 Homo sapiens Leukocyte antigen CD37 Proteins 0.000 description 3
- 101000595923 Homo sapiens Placenta growth factor Proteins 0.000 description 3
- 101000914484 Homo sapiens T-lymphocyte activation antigen CD80 Proteins 0.000 description 3
- 208000031814 IgA Vasculitis Diseases 0.000 description 3
- 102100037852 Insulin-like growth factor I Human genes 0.000 description 3
- 108010065805 Interleukin-12 Proteins 0.000 description 3
- 102000013462 Interleukin-12 Human genes 0.000 description 3
- 108090000172 Interleukin-15 Proteins 0.000 description 3
- 102000003812 Interleukin-15 Human genes 0.000 description 3
- 108050003558 Interleukin-17 Proteins 0.000 description 3
- 102000013691 Interleukin-17 Human genes 0.000 description 3
- 108010002350 Interleukin-2 Proteins 0.000 description 3
- 102000000588 Interleukin-2 Human genes 0.000 description 3
- 102000004889 Interleukin-6 Human genes 0.000 description 3
- 102100037792 Interleukin-6 receptor subunit alpha Human genes 0.000 description 3
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 description 3
- 102100031586 Leukocyte antigen CD37 Human genes 0.000 description 3
- 102000004083 Lymphotoxin-alpha Human genes 0.000 description 3
- 108090000542 Lymphotoxin-alpha Proteins 0.000 description 3
- 231100000070 MTS assay Toxicity 0.000 description 3
- 238000000719 MTS assay Methods 0.000 description 3
- 208000000172 Medulloblastoma Diseases 0.000 description 3
- 101100034151 Mus musculus Rilp gene Proteins 0.000 description 3
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 description 3
- CMVRBCDBISKHME-URBSQPMJSA-N N([C@@H](CCCCN)C(=O)NC1=CC=C(C=C1)COC(=O)O[C@]1(CC)C(=O)OCC2=C1C=C1N(C2=O)CC2=C(C3=CC(O)=CC=C3N=C21)CC)C(=O)COCC(=O)NCCOCCOCCOCCOCCOCCOCCOCCOCCN(N=N1)C=C1CNC(=O)C(CC1)CCC1CN1C(=O)C=CC1=O Chemical compound N([C@@H](CCCCN)C(=O)NC1=CC=C(C=C1)COC(=O)O[C@]1(CC)C(=O)OCC2=C1C=C1N(C2=O)CC2=C(C3=CC(O)=CC=C3N=C21)CC)C(=O)COCC(=O)NCCOCCOCCOCCOCCOCCOCCOCCOCCN(N=N1)C=C1CNC(=O)C(CC1)CCC1CN1C(=O)C=CC1=O CMVRBCDBISKHME-URBSQPMJSA-N 0.000 description 3
- 208000004286 Osteochondrodysplasias Diseases 0.000 description 3
- 108010011536 PTEN Phosphohydrolase Proteins 0.000 description 3
- 102000014160 PTEN Phosphohydrolase Human genes 0.000 description 3
- QIUASFSNWYMDFS-NILGECQDSA-N PX-866 Chemical compound CC(=O)O[C@@H]1C[C@]2(C)C(=O)CC[C@H]2C2=C1[C@@]1(C)[C@@H](COC)OC(=O)\C(=C\N(CC=C)CC=C)C1=C(O)C2=O QIUASFSNWYMDFS-NILGECQDSA-N 0.000 description 3
- 102000057297 Pepsin A Human genes 0.000 description 3
- 108090000284 Pepsin A Proteins 0.000 description 3
- 102100035194 Placenta growth factor Human genes 0.000 description 3
- 101100465037 Rattus norvegicus Prickle1 gene Proteins 0.000 description 3
- 102000007056 Recombinant Fusion Proteins Human genes 0.000 description 3
- 108010008281 Recombinant Fusion Proteins Proteins 0.000 description 3
- 208000015634 Rectal Neoplasms Diseases 0.000 description 3
- 108010083644 Ribonucleases Proteins 0.000 description 3
- 102000006382 Ribonucleases Human genes 0.000 description 3
- 108010003723 Single-Domain Antibodies Proteins 0.000 description 3
- 108020004459 Small interfering RNA Proteins 0.000 description 3
- 208000021712 Soft tissue sarcoma Diseases 0.000 description 3
- 206010042971 T-cell lymphoma Diseases 0.000 description 3
- 102100027222 T-lymphocyte activation antigen CD80 Human genes 0.000 description 3
- 208000024313 Testicular Neoplasms Diseases 0.000 description 3
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 description 3
- 239000004473 Threonine Substances 0.000 description 3
- 206010043540 Thromboangiitis obliterans Diseases 0.000 description 3
- 102000036693 Thrombopoietin Human genes 0.000 description 3
- 108010041111 Thrombopoietin Proteins 0.000 description 3
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 description 3
- 206010054094 Tumour necrosis Diseases 0.000 description 3
- 208000023915 Ureteral Neoplasms Diseases 0.000 description 3
- 206010046392 Ureteric cancer Diseases 0.000 description 3
- 208000016025 Waldenstroem macroglobulinemia Diseases 0.000 description 3
- 208000008383 Wilms tumor Diseases 0.000 description 3
- 238000002835 absorbance Methods 0.000 description 3
- 229940125666 actinium-225 Drugs 0.000 description 3
- 208000009956 adenocarcinoma Diseases 0.000 description 3
- 238000004458 analytical method Methods 0.000 description 3
- 229940124650 anti-cancer therapies Drugs 0.000 description 3
- 238000013459 approach Methods 0.000 description 3
- 235000009582 asparagine Nutrition 0.000 description 3
- 229960001230 asparagine Drugs 0.000 description 3
- 229940009098 aspartate Drugs 0.000 description 3
- 238000011717 athymic nude mouse Methods 0.000 description 3
- 230000001363 autoimmune Effects 0.000 description 3
- 210000000988 bone and bone Anatomy 0.000 description 3
- 210000004556 brain Anatomy 0.000 description 3
- 230000003833 cell viability Effects 0.000 description 3
- 210000003679 cervix uteri Anatomy 0.000 description 3
- 238000010276 construction Methods 0.000 description 3
- 235000018417 cysteine Nutrition 0.000 description 3
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 3
- 230000007423 decrease Effects 0.000 description 3
- 230000003247 decreasing effect Effects 0.000 description 3
- 239000002552 dosage form Substances 0.000 description 3
- 210000004696 endometrium Anatomy 0.000 description 3
- 230000002255 enzymatic effect Effects 0.000 description 3
- 210000000981 epithelium Anatomy 0.000 description 3
- 229940105423 erythropoietin Drugs 0.000 description 3
- 208000021045 exocrine pancreatic carcinoma Diseases 0.000 description 3
- 238000002474 experimental method Methods 0.000 description 3
- 229940126864 fibroblast growth factor Drugs 0.000 description 3
- 201000003444 follicular lymphoma Diseases 0.000 description 3
- 230000002496 gastric effect Effects 0.000 description 3
- 230000014509 gene expression Effects 0.000 description 3
- 230000002068 genetic effect Effects 0.000 description 3
- 229930195712 glutamate Natural products 0.000 description 3
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 3
- 230000003394 haemopoietic effect Effects 0.000 description 3
- 201000009277 hairy cell leukemia Diseases 0.000 description 3
- 201000010536 head and neck cancer Diseases 0.000 description 3
- 208000014829 head and neck neoplasm Diseases 0.000 description 3
- 229940084986 human chorionic gonadotropin Drugs 0.000 description 3
- 230000002267 hypothalamic effect Effects 0.000 description 3
- 210000002865 immune cell Anatomy 0.000 description 3
- 238000011534 incubation Methods 0.000 description 3
- 229960000598 infliximab Drugs 0.000 description 3
- 108010028930 invariant chain Proteins 0.000 description 3
- 229960000310 isoleucine Drugs 0.000 description 3
- AGPKZVBTJJNPAG-UHFFFAOYSA-N isoleucine Natural products CCC(C)C(N)C(O)=O AGPKZVBTJJNPAG-UHFFFAOYSA-N 0.000 description 3
- 231100000225 lethality Toxicity 0.000 description 3
- 125000001909 leucine group Chemical group [H]N(*)C(C(*)=O)C([H])([H])C(C([H])([H])[H])C([H])([H])[H] 0.000 description 3
- 238000002595 magnetic resonance imaging Methods 0.000 description 3
- 208000023356 medullary thyroid gland carcinoma Diseases 0.000 description 3
- 231100000590 oncogenic Toxicity 0.000 description 3
- 230000002246 oncogenic effect Effects 0.000 description 3
- 229940111202 pepsin Drugs 0.000 description 3
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Substances [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 3
- 229920001184 polypeptide Polymers 0.000 description 3
- OXCMYAYHXIHQOA-UHFFFAOYSA-N potassium;[2-butyl-5-chloro-3-[[4-[2-(1,2,4-triaza-3-azanidacyclopenta-1,4-dien-5-yl)phenyl]phenyl]methyl]imidazol-4-yl]methanol Chemical compound [K+].CCCCC1=NC(Cl)=C(CO)N1CC1=CC=C(C=2C(=CC=CC=2)C2=N[N-]N=N2)C=C1 OXCMYAYHXIHQOA-UHFFFAOYSA-N 0.000 description 3
- 208000016800 primary central nervous system lymphoma Diseases 0.000 description 3
- 229960005562 radium-223 Drugs 0.000 description 3
- 201000008158 rapidly progressive glomerulonephritis Diseases 0.000 description 3
- 201000007444 renal pelvis carcinoma Diseases 0.000 description 3
- 230000002441 reversible effect Effects 0.000 description 3
- 238000012552 review Methods 0.000 description 3
- 201000009410 rhabdomyosarcoma Diseases 0.000 description 3
- 230000003248 secreting effect Effects 0.000 description 3
- 210000003491 skin Anatomy 0.000 description 3
- 206010041823 squamous cell carcinoma Diseases 0.000 description 3
- 206010043207 temporal arteritis Diseases 0.000 description 3
- 201000003120 testicular cancer Diseases 0.000 description 3
- 210000001550 testis Anatomy 0.000 description 3
- 125000003396 thiol group Chemical group [H]S* 0.000 description 3
- 229960003989 tocilizumab Drugs 0.000 description 3
- 231100000167 toxic agent Toxicity 0.000 description 3
- 239000003440 toxic substance Substances 0.000 description 3
- 230000009261 transgenic effect Effects 0.000 description 3
- 201000011294 ureter cancer Diseases 0.000 description 3
- FBDOJYYTMIHHDH-OZBJMMHXSA-N (19S)-19-ethyl-19-hydroxy-17-oxa-3,13-diazapentacyclo[11.8.0.02,11.04,9.015,20]henicosa-2,4,6,8,10,14,20-heptaen-18-one Chemical compound CC[C@@]1(O)C(=O)OCC2=CN3Cc4cc5ccccc5nc4C3C=C12 FBDOJYYTMIHHDH-OZBJMMHXSA-N 0.000 description 2
- INAUWOVKEZHHDM-PEDBPRJASA-N (7s,9s)-6,9,11-trihydroxy-9-(2-hydroxyacetyl)-7-[(2r,4s,5s,6s)-5-hydroxy-6-methyl-4-morpholin-4-yloxan-2-yl]oxy-4-methoxy-8,10-dihydro-7h-tetracene-5,12-dione;hydrochloride Chemical compound Cl.N1([C@H]2C[C@@H](O[C@@H](C)[C@H]2O)O[C@H]2C[C@@](O)(CC=3C(O)=C4C(=O)C=5C=CC=C(C=5C(=O)C4=C(O)C=32)OC)C(=O)CO)CCOCC1 INAUWOVKEZHHDM-PEDBPRJASA-N 0.000 description 2
- QZTKDVCDBIDYMD-UHFFFAOYSA-N 2,2'-[(2-amino-2-oxoethyl)imino]diacetic acid Chemical compound NC(=O)CN(CC(O)=O)CC(O)=O QZTKDVCDBIDYMD-UHFFFAOYSA-N 0.000 description 2
- SXGZJKUKBWWHRA-UHFFFAOYSA-N 2-(N-morpholiniumyl)ethanesulfonate Chemical compound [O-]S(=O)(=O)CC[NH+]1CCOCC1 SXGZJKUKBWWHRA-UHFFFAOYSA-N 0.000 description 2
- JHALWMSZGCVVEM-UHFFFAOYSA-N 2-[4,7-bis(carboxymethyl)-1,4,7-triazonan-1-yl]acetic acid Chemical compound OC(=O)CN1CCN(CC(O)=O)CCN(CC(O)=O)CC1 JHALWMSZGCVVEM-UHFFFAOYSA-N 0.000 description 2
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 2
- AJTVSSFTXWNIRG-UHFFFAOYSA-N 2-[bis(2-hydroxyethyl)amino]ethanesulfonic acid Chemical compound OCC[NH+](CCO)CCS([O-])(=O)=O AJTVSSFTXWNIRG-UHFFFAOYSA-N 0.000 description 2
- NUFBIAUZAMHTSP-UHFFFAOYSA-N 3-(n-morpholino)-2-hydroxypropanesulfonic acid Chemical compound OS(=O)(=O)CC(O)CN1CCOCC1 NUFBIAUZAMHTSP-UHFFFAOYSA-N 0.000 description 2
- AOJJSUZBOXZQNB-VTZDEGQISA-N 4'-epidoxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-VTZDEGQISA-N 0.000 description 2
- OVMSOCFBDVBLFW-VHLOTGQHSA-N 5beta,20-epoxy-1,7beta,13alpha-trihydroxy-9-oxotax-11-ene-2alpha,4alpha,10beta-triyl 4,10-diacetate 2-benzoate Chemical compound O([C@@H]1[C@@]2(C[C@H](O)C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)O)C(=O)C1=CC=CC=C1 OVMSOCFBDVBLFW-VHLOTGQHSA-N 0.000 description 2
- 208000030507 AIDS Diseases 0.000 description 2
- 108010088751 Albumins Proteins 0.000 description 2
- 102000009027 Albumins Human genes 0.000 description 2
- 102100022749 Aminopeptidase N Human genes 0.000 description 2
- 206010061424 Anal cancer Diseases 0.000 description 2
- 241000024188 Andala Species 0.000 description 2
- 102100029470 Apolipoprotein E Human genes 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- 102100021631 B-cell lymphoma 6 protein Human genes 0.000 description 2
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 description 2
- 231100000699 Bacterial toxin Toxicity 0.000 description 2
- 102100032412 Basigin Human genes 0.000 description 2
- 206010005949 Bone cancer Diseases 0.000 description 2
- 208000018084 Bone neoplasm Diseases 0.000 description 2
- 102100036842 C-C motif chemokine 19 Human genes 0.000 description 2
- 101710155857 C-C motif chemokine 2 Proteins 0.000 description 2
- 102100036846 C-C motif chemokine 21 Human genes 0.000 description 2
- 101710098275 C-X-C motif chemokine 10 Proteins 0.000 description 2
- 102100032528 C-type lectin domain family 11 member A Human genes 0.000 description 2
- 101710167766 C-type lectin domain family 11 member A Proteins 0.000 description 2
- 108700012439 CA9 Proteins 0.000 description 2
- 102100025221 CD70 antigen Human genes 0.000 description 2
- 102100035793 CD83 antigen Human genes 0.000 description 2
- 108010021064 CTLA-4 Antigen Proteins 0.000 description 2
- 229940045513 CTLA4 antagonist Drugs 0.000 description 2
- 102100025570 Cancer/testis antigen 1 Human genes 0.000 description 2
- 102000003952 Caspase 3 Human genes 0.000 description 2
- 108090000397 Caspase 3 Proteins 0.000 description 2
- 208000010833 Chronic myeloid leukaemia Diseases 0.000 description 2
- 208000035473 Communicable disease Diseases 0.000 description 2
- 206010010452 Congenital ectodermal dysplasia Diseases 0.000 description 2
- 102100039498 Cytotoxic T-lymphocyte protein 4 Human genes 0.000 description 2
- 102000003915 DNA Topoisomerases Human genes 0.000 description 2
- 108090000323 DNA Topoisomerases Proteins 0.000 description 2
- 230000033616 DNA repair Effects 0.000 description 2
- 230000008265 DNA repair mechanism Effects 0.000 description 2
- ZINBFGBAIFRYSH-UHFFFAOYSA-N Demethoxyviridin Natural products CC12C(O)C(O)C(=O)c3coc(C(=O)c4c5CCC(=O)c5ccc14)c23 ZINBFGBAIFRYSH-UHFFFAOYSA-N 0.000 description 2
- 206010061818 Disease progression Diseases 0.000 description 2
- 101150029707 ERBB2 gene Proteins 0.000 description 2
- 206010014967 Ependymoma Diseases 0.000 description 2
- HTIJFSOGRVMCQR-UHFFFAOYSA-N Epirubicin Natural products COc1cccc2C(=O)c3c(O)c4CC(O)(CC(OC5CC(N)C(=O)C(C)O5)c4c(O)c3C(=O)c12)C(=O)CO HTIJFSOGRVMCQR-UHFFFAOYSA-N 0.000 description 2
- 108010066687 Epithelial Cell Adhesion Molecule Proteins 0.000 description 2
- 208000000461 Esophageal Neoplasms Diseases 0.000 description 2
- 102100038595 Estrogen receptor Human genes 0.000 description 2
- 102000012673 Follicle Stimulating Hormone Human genes 0.000 description 2
- 108010079345 Follicle Stimulating Hormone Proteins 0.000 description 2
- 102100024165 G1/S-specific cyclin-D1 Human genes 0.000 description 2
- 201000010915 Glioblastoma multiforme Diseases 0.000 description 2
- SXRSQZLOMIGNAQ-UHFFFAOYSA-N Glutaraldehyde Chemical compound O=CCCCC=O SXRSQZLOMIGNAQ-UHFFFAOYSA-N 0.000 description 2
- 102000003886 Glycoproteins Human genes 0.000 description 2
- 108090000288 Glycoproteins Proteins 0.000 description 2
- 208000024869 Goodpasture syndrome Diseases 0.000 description 2
- 108010017080 Granulocyte Colony-Stimulating Factor Proteins 0.000 description 2
- 102100039619 Granulocyte colony-stimulating factor Human genes 0.000 description 2
- 102100031573 Hematopoietic progenitor cell antigen CD34 Human genes 0.000 description 2
- 201000004331 Henoch-Schoenlein purpura Diseases 0.000 description 2
- 101000833679 Homo sapiens A-kinase anchor protein 13 Proteins 0.000 description 2
- 101000757160 Homo sapiens Aminopeptidase N Proteins 0.000 description 2
- 101000771674 Homo sapiens Apolipoprotein E Proteins 0.000 description 2
- 101000971234 Homo sapiens B-cell lymphoma 6 protein Proteins 0.000 description 2
- 101000798441 Homo sapiens Basigin Proteins 0.000 description 2
- 101000713106 Homo sapiens C-C motif chemokine 19 Proteins 0.000 description 2
- 101000713085 Homo sapiens C-C motif chemokine 21 Proteins 0.000 description 2
- 101100165850 Homo sapiens CA9 gene Proteins 0.000 description 2
- 101000868215 Homo sapiens CD40 ligand Proteins 0.000 description 2
- 101000934356 Homo sapiens CD70 antigen Proteins 0.000 description 2
- 101000946856 Homo sapiens CD83 antigen Proteins 0.000 description 2
- 101000856237 Homo sapiens Cancer/testis antigen 1 Proteins 0.000 description 2
- 101000935587 Homo sapiens Flavin reductase (NADPH) Proteins 0.000 description 2
- 101000980756 Homo sapiens G1/S-specific cyclin-D1 Proteins 0.000 description 2
- 101000777663 Homo sapiens Hematopoietic progenitor cell antigen CD34 Proteins 0.000 description 2
- 101000599951 Homo sapiens Insulin-like growth factor I Proteins 0.000 description 2
- 101001046683 Homo sapiens Integrin alpha-L Proteins 0.000 description 2
- 101001046686 Homo sapiens Integrin alpha-M Proteins 0.000 description 2
- 101000935043 Homo sapiens Integrin beta-1 Proteins 0.000 description 2
- 101000935040 Homo sapiens Integrin beta-2 Proteins 0.000 description 2
- 101000599852 Homo sapiens Intercellular adhesion molecule 1 Proteins 0.000 description 2
- 101000599048 Homo sapiens Interleukin-6 receptor subunit alpha Proteins 0.000 description 2
- 101000608935 Homo sapiens Leukosialin Proteins 0.000 description 2
- 101000961414 Homo sapiens Membrane cofactor protein Proteins 0.000 description 2
- 101001133056 Homo sapiens Mucin-1 Proteins 0.000 description 2
- 101000623901 Homo sapiens Mucin-16 Proteins 0.000 description 2
- 101001133081 Homo sapiens Mucin-2 Proteins 0.000 description 2
- 101000972284 Homo sapiens Mucin-3A Proteins 0.000 description 2
- 101000972286 Homo sapiens Mucin-4 Proteins 0.000 description 2
- 101000610551 Homo sapiens Prominin-1 Proteins 0.000 description 2
- 101000716124 Homo sapiens T-cell surface glycoprotein CD1c Proteins 0.000 description 2
- 101000611023 Homo sapiens Tumor necrosis factor receptor superfamily member 6 Proteins 0.000 description 2
- 101000851376 Homo sapiens Tumor necrosis factor receptor superfamily member 8 Proteins 0.000 description 2
- 108010000521 Human Growth Hormone Proteins 0.000 description 2
- 102000002265 Human Growth Hormone Human genes 0.000 description 2
- 239000000854 Human Growth Hormone Substances 0.000 description 2
- 206010021143 Hypoxia Diseases 0.000 description 2
- 208000028622 Immune thrombocytopenia Diseases 0.000 description 2
- 108010054477 Immunoglobulin Fab Fragments Proteins 0.000 description 2
- 102000001706 Immunoglobulin Fab Fragments Human genes 0.000 description 2
- 108010067060 Immunoglobulin Variable Region Proteins 0.000 description 2
- 102000017727 Immunoglobulin Variable Region Human genes 0.000 description 2
- 102100022339 Integrin alpha-L Human genes 0.000 description 2
- 102100022338 Integrin alpha-M Human genes 0.000 description 2
- 102100022297 Integrin alpha-X Human genes 0.000 description 2
- 102100025304 Integrin beta-1 Human genes 0.000 description 2
- 102100025390 Integrin beta-2 Human genes 0.000 description 2
- 102100037877 Intercellular adhesion molecule 1 Human genes 0.000 description 2
- 102000000589 Interleukin-1 Human genes 0.000 description 2
- 108010002352 Interleukin-1 Proteins 0.000 description 2
- 102100020793 Interleukin-13 receptor subunit alpha-2 Human genes 0.000 description 2
- 108090000171 Interleukin-18 Proteins 0.000 description 2
- 102100039564 Leukosialin Human genes 0.000 description 2
- 102000009151 Luteinizing Hormone Human genes 0.000 description 2
- 108010073521 Luteinizing Hormone Proteins 0.000 description 2
- 208000028018 Lymphocytic leukaemia Diseases 0.000 description 2
- 239000007993 MOPS buffer Substances 0.000 description 2
- 108010048043 Macrophage Migration-Inhibitory Factors Proteins 0.000 description 2
- 102100037791 Macrophage migration inhibitory factor Human genes 0.000 description 2
- 241000124008 Mammalia Species 0.000 description 2
- 208000009018 Medullary thyroid cancer Diseases 0.000 description 2
- 102100039373 Membrane cofactor protein Human genes 0.000 description 2
- 206010027406 Mesothelioma Diseases 0.000 description 2
- 108091022875 Microtubule Proteins 0.000 description 2
- 102000029749 Microtubule Human genes 0.000 description 2
- 102100034256 Mucin-1 Human genes 0.000 description 2
- 102100023123 Mucin-16 Human genes 0.000 description 2
- 102100034263 Mucin-2 Human genes 0.000 description 2
- 102100022497 Mucin-3A Human genes 0.000 description 2
- 102100022693 Mucin-4 Human genes 0.000 description 2
- 101100335081 Mus musculus Flt3 gene Proteins 0.000 description 2
- 208000033761 Myelogenous Chronic BCR-ABL Positive Leukemia Diseases 0.000 description 2
- DBXNUXBLKRLWFA-UHFFFAOYSA-N N-(2-acetamido)-2-aminoethanesulfonic acid Chemical compound NC(=O)CNCCS(O)(=O)=O DBXNUXBLKRLWFA-UHFFFAOYSA-N 0.000 description 2
- 125000000729 N-terminal amino-acid group Chemical group 0.000 description 2
- 102000003729 Neprilysin Human genes 0.000 description 2
- 108090000028 Neprilysin Proteins 0.000 description 2
- 206010029260 Neuroblastoma Diseases 0.000 description 2
- KUIFHYPNNRVEKZ-VIJRYAKMSA-N O-(N-acetyl-alpha-D-galactosaminyl)-L-threonine Chemical compound OC(=O)[C@@H](N)[C@@H](C)O[C@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1NC(C)=O KUIFHYPNNRVEKZ-VIJRYAKMSA-N 0.000 description 2
- 206010030155 Oesophageal carcinoma Diseases 0.000 description 2
- 208000007641 Pinealoma Diseases 0.000 description 2
- 231100000742 Plant toxin Toxicity 0.000 description 2
- 239000002202 Polyethylene glycol Substances 0.000 description 2
- 241000288906 Primates Species 0.000 description 2
- 101710089372 Programmed cell death protein 1 Proteins 0.000 description 2
- 102100040678 Programmed cell death protein 1 Human genes 0.000 description 2
- 102100040120 Prominin-1 Human genes 0.000 description 2
- 108091030071 RNAI Proteins 0.000 description 2
- 208000006265 Renal cell carcinoma Diseases 0.000 description 2
- 208000004453 Retinal Dysplasia Diseases 0.000 description 2
- 201000000582 Retinoblastoma Diseases 0.000 description 2
- 241000283984 Rodentia Species 0.000 description 2
- 102000000395 SH3 domains Human genes 0.000 description 2
- 108050008861 SH3 domains Proteins 0.000 description 2
- 206010061934 Salivary gland cancer Diseases 0.000 description 2
- 208000000453 Skin Neoplasms Diseases 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- 102100027208 T-cell antigen CD7 Human genes 0.000 description 2
- PZBFGYYEXUXCOF-UHFFFAOYSA-N TCEP Chemical compound OC(=O)CCP(CCC(O)=O)CCC(O)=O PZBFGYYEXUXCOF-UHFFFAOYSA-N 0.000 description 2
- 108010000449 TNF-Related Apoptosis-Inducing Ligand Receptors Proteins 0.000 description 2
- 102000002259 TNF-Related Apoptosis-Inducing Ligand Receptors Human genes 0.000 description 2
- 108010008125 Tenascin Proteins 0.000 description 2
- 102100038126 Tenascin Human genes 0.000 description 2
- 206010057644 Testis cancer Diseases 0.000 description 2
- 102000011923 Thyrotropin Human genes 0.000 description 2
- 108010061174 Thyrotropin Proteins 0.000 description 2
- 108010009583 Transforming Growth Factors Proteins 0.000 description 2
- 102000009618 Transforming Growth Factors Human genes 0.000 description 2
- 102100040403 Tumor necrosis factor receptor superfamily member 6 Human genes 0.000 description 2
- 102100036857 Tumor necrosis factor receptor superfamily member 8 Human genes 0.000 description 2
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 description 2
- 108091008605 VEGF receptors Proteins 0.000 description 2
- 108010019530 Vascular Endothelial Growth Factors Proteins 0.000 description 2
- 102000005789 Vascular Endothelial Growth Factors Human genes 0.000 description 2
- 102100033177 Vascular endothelial growth factor receptor 2 Human genes 0.000 description 2
- 206010047700 Vomiting Diseases 0.000 description 2
- 206010047741 Vulval cancer Diseases 0.000 description 2
- 208000004354 Vulvar Neoplasms Diseases 0.000 description 2
- 208000017733 acquired polycythemia vera Diseases 0.000 description 2
- 229960002964 adalimumab Drugs 0.000 description 2
- 239000000654 additive Substances 0.000 description 2
- 229960000548 alemtuzumab Drugs 0.000 description 2
- 125000000217 alkyl group Chemical group 0.000 description 2
- 229940100198 alkylating agent Drugs 0.000 description 2
- 239000002168 alkylating agent Substances 0.000 description 2
- 238000004873 anchoring Methods 0.000 description 2
- 239000004037 angiogenesis inhibitor Substances 0.000 description 2
- 238000010171 animal model Methods 0.000 description 2
- 239000005557 antagonist Substances 0.000 description 2
- SARKSIVGGPWEIG-UHFFFAOYSA-N anthra[2,3-b]benzo[d]thiophene Chemical compound C1=CC=C2C=C(C=C3C4=CC=CC=C4SC3=C3)C3=CC2=C1 SARKSIVGGPWEIG-UHFFFAOYSA-N 0.000 description 2
- 230000001093 anti-cancer Effects 0.000 description 2
- 230000036436 anti-hiv Effects 0.000 description 2
- 230000006023 anti-tumor response Effects 0.000 description 2
- 230000006907 apoptotic process Effects 0.000 description 2
- 206010003230 arteritis Diseases 0.000 description 2
- 235000003704 aspartic acid Nutrition 0.000 description 2
- 230000001580 bacterial effect Effects 0.000 description 2
- 239000000688 bacterial toxin Substances 0.000 description 2
- 229960004669 basiliximab Drugs 0.000 description 2
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 2
- 230000001588 bifunctional effect Effects 0.000 description 2
- 230000036765 blood level Effects 0.000 description 2
- 210000001185 bone marrow Anatomy 0.000 description 2
- 208000030224 brain astrocytoma Diseases 0.000 description 2
- 101150061829 bre-3 gene Proteins 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- 230000003197 catalytic effect Effects 0.000 description 2
- 238000004113 cell culture Methods 0.000 description 2
- 229960003115 certolizumab pegol Drugs 0.000 description 2
- 230000000973 chemotherapeutic effect Effects 0.000 description 2
- 208000018805 childhood acute lymphoblastic leukemia Diseases 0.000 description 2
- 201000011633 childhood acute lymphocytic leukemia Diseases 0.000 description 2
- 208000024207 chronic leukemia Diseases 0.000 description 2
- 239000002299 complementary DNA Substances 0.000 description 2
- 239000013078 crystal Substances 0.000 description 2
- 238000012258 culturing Methods 0.000 description 2
- 239000002254 cytotoxic agent Substances 0.000 description 2
- 231100000599 cytotoxic agent Toxicity 0.000 description 2
- 229960002806 daclizumab Drugs 0.000 description 2
- 230000002950 deficient Effects 0.000 description 2
- SWJBYJJNDIXFSA-KUHUBIRLSA-N demethoxyviridin Chemical compound O=C1C2=C3CCC(=O)C3=CC=C2[C@]2(C)C3=C1OC=C3C(=O)C[C@H]2O SWJBYJJNDIXFSA-KUHUBIRLSA-N 0.000 description 2
- 230000001419 dependent effect Effects 0.000 description 2
- 238000013461 design Methods 0.000 description 2
- 230000005750 disease progression Effects 0.000 description 2
- VHJLVAABSRFDPM-QWWZWVQMSA-N dithiothreitol Chemical compound SC[C@@H](O)[C@H](O)CS VHJLVAABSRFDPM-QWWZWVQMSA-N 0.000 description 2
- 208000036552 dowling-degos disease 3 Diseases 0.000 description 2
- 238000012377 drug delivery Methods 0.000 description 2
- 239000003640 drug residue Substances 0.000 description 2
- 230000004064 dysfunction Effects 0.000 description 2
- 229960000284 efalizumab Drugs 0.000 description 2
- 102000052116 epidermal growth factor receptor activity proteins Human genes 0.000 description 2
- 108700015053 epidermal growth factor receptor activity proteins Proteins 0.000 description 2
- 229960001904 epirubicin Drugs 0.000 description 2
- 229960003649 eribulin Drugs 0.000 description 2
- 150000002148 esters Chemical class 0.000 description 2
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 2
- 238000013401 experimental design Methods 0.000 description 2
- 102000006815 folate receptor Human genes 0.000 description 2
- 108020005243 folate receptor Proteins 0.000 description 2
- PJZDLZXMGBOJRF-CXOZILEQSA-L folfirinox Chemical compound [Pt+4].[O-]C(=O)C([O-])=O.[NH-][C@H]1CCCC[C@@H]1[NH-].FC1=CNC(=O)NC1=O.C1NC=2NC(N)=NC(=O)C=2N(C=O)C1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1.C1=C2C(CC)=C3CN(C(C4=C([C@@](C(=O)OC4)(O)CC)C=4)=O)C=4C3=NC2=CC=C1OC(=O)N(CC1)CCC1N1CCCCC1 PJZDLZXMGBOJRF-CXOZILEQSA-L 0.000 description 2
- 229940028334 follicle stimulating hormone Drugs 0.000 description 2
- 210000001035 gastrointestinal tract Anatomy 0.000 description 2
- 229960000578 gemtuzumab Drugs 0.000 description 2
- 125000000291 glutamic acid group Chemical group N[C@@H](CCC(O)=O)C(=O)* 0.000 description 2
- 125000003630 glycyl group Chemical group [H]N([H])C([H])([H])C(*)=O 0.000 description 2
- 210000003714 granulocyte Anatomy 0.000 description 2
- 230000036541 health Effects 0.000 description 2
- 201000005787 hematologic cancer Diseases 0.000 description 2
- 230000002489 hematologic effect Effects 0.000 description 2
- 208000019691 hematopoietic and lymphoid cell neoplasm Diseases 0.000 description 2
- 231100000844 hepatocellular carcinoma Toxicity 0.000 description 2
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 2
- 230000002209 hydrophobic effect Effects 0.000 description 2
- 230000003463 hyperproliferative effect Effects 0.000 description 2
- 230000036039 immunity Effects 0.000 description 2
- 208000015446 immunoglobulin a vasculitis Diseases 0.000 description 2
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 2
- 102000006495 integrins Human genes 0.000 description 2
- 108010044426 integrins Proteins 0.000 description 2
- 108040003607 interleukin-13 receptor activity proteins Proteins 0.000 description 2
- 102000008616 interleukin-15 receptor activity proteins Human genes 0.000 description 2
- 108040002039 interleukin-15 receptor activity proteins Proteins 0.000 description 2
- 102000053460 interleukin-17 receptor activity proteins Human genes 0.000 description 2
- 108040001304 interleukin-17 receptor activity proteins Proteins 0.000 description 2
- 102000008625 interleukin-18 receptor activity proteins Human genes 0.000 description 2
- 108040002014 interleukin-18 receptor activity proteins Proteins 0.000 description 2
- 108040006852 interleukin-4 receptor activity proteins Proteins 0.000 description 2
- 230000000968 intestinal effect Effects 0.000 description 2
- 238000007912 intraperitoneal administration Methods 0.000 description 2
- 229960005386 ipilimumab Drugs 0.000 description 2
- 238000005304 joining Methods 0.000 description 2
- 229950004881 labetuzumab govitecan Drugs 0.000 description 2
- 239000003446 ligand Substances 0.000 description 2
- 201000005243 lung squamous cell carcinoma Diseases 0.000 description 2
- 229940040129 luteinizing hormone Drugs 0.000 description 2
- 208000025036 lymphosarcoma Diseases 0.000 description 2
- 231100001142 manageable toxicity Toxicity 0.000 description 2
- 239000003550 marker Substances 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- 230000010534 mechanism of action Effects 0.000 description 2
- 150000002739 metals Chemical class 0.000 description 2
- 238000012737 microarray-based gene expression Methods 0.000 description 2
- 210000004688 microtubule Anatomy 0.000 description 2
- 230000005012 migration Effects 0.000 description 2
- 238000013508 migration Methods 0.000 description 2
- 238000010369 molecular cloning Methods 0.000 description 2
- 210000000214 mouth Anatomy 0.000 description 2
- 206010028197 multiple epiphyseal dysplasia Diseases 0.000 description 2
- 238000012243 multiplex automated genomic engineering Methods 0.000 description 2
- 229960003816 muromonab-cd3 Drugs 0.000 description 2
- 201000000050 myeloid neoplasm Diseases 0.000 description 2
- YOHYSYJDKVYCJI-UHFFFAOYSA-N n-[3-[[6-[3-(trifluoromethyl)anilino]pyrimidin-4-yl]amino]phenyl]cyclopropanecarboxamide Chemical compound FC(F)(F)C1=CC=CC(NC=2N=CN=C(NC=3C=C(NC(=O)C4CC4)C=CC=3)C=2)=C1 YOHYSYJDKVYCJI-UHFFFAOYSA-N 0.000 description 2
- 229960005027 natalizumab Drugs 0.000 description 2
- 201000008026 nephroblastoma Diseases 0.000 description 2
- 229960003301 nivolumab Drugs 0.000 description 2
- 102000039446 nucleic acids Human genes 0.000 description 2
- 108020004707 nucleic acids Proteins 0.000 description 2
- 150000007523 nucleic acids Chemical class 0.000 description 2
- 229960000470 omalizumab Drugs 0.000 description 2
- 210000004789 organ system Anatomy 0.000 description 2
- 229960001756 oxaliplatin Drugs 0.000 description 2
- DWAFYCQODLXJNR-BNTLRKBRSA-L oxaliplatin Chemical compound O1C(=O)C(=O)O[Pt]11N[C@@H]2CCCC[C@H]2N1 DWAFYCQODLXJNR-BNTLRKBRSA-L 0.000 description 2
- 238000007427 paired t-test Methods 0.000 description 2
- 229960001972 panitumumab Drugs 0.000 description 2
- 230000037361 pathway Effects 0.000 description 2
- 229960002621 pembrolizumab Drugs 0.000 description 2
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 2
- 239000002953 phosphate buffered saline Substances 0.000 description 2
- 239000003123 plant toxin Substances 0.000 description 2
- 229920001481 poly(stearyl methacrylate) Polymers 0.000 description 2
- 208000037244 polycythemia vera Diseases 0.000 description 2
- 229920001223 polyethylene glycol Polymers 0.000 description 2
- 229920000642 polymer Polymers 0.000 description 2
- 238000002360 preparation method Methods 0.000 description 2
- 230000000861 pro-apoptotic effect Effects 0.000 description 2
- 229940002612 prodrug Drugs 0.000 description 2
- 239000000651 prodrug Substances 0.000 description 2
- 230000035755 proliferation Effects 0.000 description 2
- 108020001580 protein domains Proteins 0.000 description 2
- 230000005855 radiation Effects 0.000 description 2
- 238000011363 radioimmunotherapy Methods 0.000 description 2
- 206010038038 rectal cancer Diseases 0.000 description 2
- 201000001275 rectum cancer Diseases 0.000 description 2
- CXMXRPHRNRROMY-UHFFFAOYSA-N sebacic acid Chemical compound OC(=O)CCCCCCCCC(O)=O CXMXRPHRNRROMY-UHFFFAOYSA-N 0.000 description 2
- 230000019491 signal transduction Effects 0.000 description 2
- 238000002741 site-directed mutagenesis Methods 0.000 description 2
- 238000001542 size-exclusion chromatography Methods 0.000 description 2
- 239000002904 solvent Substances 0.000 description 2
- 230000009870 specific binding Effects 0.000 description 2
- 208000017572 squamous cell neoplasm Diseases 0.000 description 2
- 230000000087 stabilizing effect Effects 0.000 description 2
- 210000000130 stem cell Anatomy 0.000 description 2
- 238000003860 storage Methods 0.000 description 2
- 229910052717 sulfur Inorganic materials 0.000 description 2
- 230000009044 synergistic interaction Effects 0.000 description 2
- 101150047061 tag-72 gene Proteins 0.000 description 2
- 150000003573 thiols Chemical class 0.000 description 2
- 210000001685 thyroid gland Anatomy 0.000 description 2
- 229960005267 tositumomab Drugs 0.000 description 2
- 230000001052 transient effect Effects 0.000 description 2
- 229960000575 trastuzumab Drugs 0.000 description 2
- 241001515965 unidentified phage Species 0.000 description 2
- 230000003827 upregulation Effects 0.000 description 2
- 210000004291 uterus Anatomy 0.000 description 2
- 230000004400 visual pathway Effects 0.000 description 2
- 210000000239 visual pathway Anatomy 0.000 description 2
- 201000005102 vulva cancer Diseases 0.000 description 2
- HDTRYLNUVZCQOY-UHFFFAOYSA-N α-D-glucopyranosyl-α-D-glucopyranoside Natural products OC1C(O)C(O)C(CO)OC1OC1C(O)C(O)C(O)C(CO)O1 HDTRYLNUVZCQOY-UHFFFAOYSA-N 0.000 description 1
- 102000040650 (ribonucleotides)n+m Human genes 0.000 description 1
- WEYNBWVKOYCCQT-UHFFFAOYSA-N 1-(3-chloro-4-methylphenyl)-3-{2-[({5-[(dimethylamino)methyl]-2-furyl}methyl)thio]ethyl}urea Chemical compound O1C(CN(C)C)=CC=C1CSCCNC(=O)NC1=CC=C(C)C(Cl)=C1 WEYNBWVKOYCCQT-UHFFFAOYSA-N 0.000 description 1
- IHPYMWDTONKSCO-UHFFFAOYSA-N 2,2'-piperazine-1,4-diylbisethanesulfonic acid Chemical compound OS(=O)(=O)CCN1CCN(CCS(O)(=O)=O)CC1 IHPYMWDTONKSCO-UHFFFAOYSA-N 0.000 description 1
- VILCJCGEZXAXTO-UHFFFAOYSA-N 2,2,2-tetramine Chemical compound NCCNCCNCCN VILCJCGEZXAXTO-UHFFFAOYSA-N 0.000 description 1
- RTQWWZBSTRGEAV-PKHIMPSTSA-N 2-[[(2s)-2-[bis(carboxymethyl)amino]-3-[4-(methylcarbamoylamino)phenyl]propyl]-[2-[bis(carboxymethyl)amino]propyl]amino]acetic acid Chemical compound CNC(=O)NC1=CC=C(C[C@@H](CN(CC(C)N(CC(O)=O)CC(O)=O)CC(O)=O)N(CC(O)=O)CC(O)=O)C=C1 RTQWWZBSTRGEAV-PKHIMPSTSA-N 0.000 description 1
- FSPQCTGGIANIJZ-UHFFFAOYSA-N 2-[[(3,4-dimethoxyphenyl)-oxomethyl]amino]-4,5,6,7-tetrahydro-1-benzothiophene-3-carboxamide Chemical compound C1=C(OC)C(OC)=CC=C1C(=O)NC1=C(C(N)=O)C(CCCC2)=C2S1 FSPQCTGGIANIJZ-UHFFFAOYSA-N 0.000 description 1
- GOJUJUVQIVIZAV-UHFFFAOYSA-N 2-amino-4,6-dichloropyrimidine-5-carbaldehyde Chemical group NC1=NC(Cl)=C(C=O)C(Cl)=N1 GOJUJUVQIVIZAV-UHFFFAOYSA-N 0.000 description 1
- AUDYZXNUHIIGRB-UHFFFAOYSA-N 3-thiophen-2-ylpyrrole-2,5-dione Chemical compound O=C1NC(=O)C(C=2SC=CC=2)=C1 AUDYZXNUHIIGRB-UHFFFAOYSA-N 0.000 description 1
- FWMNVWWHGCHHJJ-SKKKGAJSSA-N 4-amino-1-[(2r)-6-amino-2-[[(2r)-2-[[(2r)-2-[[(2r)-2-amino-3-phenylpropanoyl]amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]hexanoyl]piperidine-4-carboxylic acid Chemical compound C([C@H](C(=O)N[C@H](CC(C)C)C(=O)N[C@H](CCCCN)C(=O)N1CCC(N)(CC1)C(O)=O)NC(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 FWMNVWWHGCHHJJ-SKKKGAJSSA-N 0.000 description 1
- 101710170217 A-kinase anchor protein 13 Proteins 0.000 description 1
- 102100031901 A-kinase anchor protein 2 Human genes 0.000 description 1
- 101710109888 A-kinase anchor protein 2 Proteins 0.000 description 1
- 102100040078 A-kinase anchor protein 5 Human genes 0.000 description 1
- 102100031906 A-kinase anchor protein 7 isoforms alpha and beta Human genes 0.000 description 1
- 239000007991 ACES buffer Substances 0.000 description 1
- 208000002008 AIDS-Related Lymphoma Diseases 0.000 description 1
- 108010066676 Abrin Proteins 0.000 description 1
- 102000007469 Actins Human genes 0.000 description 1
- 108010085238 Actins Proteins 0.000 description 1
- 108010059616 Activins Proteins 0.000 description 1
- 206010000830 Acute leukaemia Diseases 0.000 description 1
- 101710137115 Adenylyl cyclase-associated protein 1 Proteins 0.000 description 1
- 102100032959 Alpha-actinin-4 Human genes 0.000 description 1
- 101710115256 Alpha-actinin-4 Proteins 0.000 description 1
- 208000024827 Alzheimer disease Diseases 0.000 description 1
- 102000052587 Anaphase-Promoting Complex-Cyclosome Apc3 Subunit Human genes 0.000 description 1
- 108700004606 Anaphase-Promoting Complex-Cyclosome Apc3 Subunit Proteins 0.000 description 1
- 201000003076 Angiosarcoma Diseases 0.000 description 1
- 102400000068 Angiostatin Human genes 0.000 description 1
- 108010079709 Angiostatins Proteins 0.000 description 1
- 208000001454 Anhidrotic Ectodermal Dysplasia 1 Diseases 0.000 description 1
- 208000007860 Anus Neoplasms Diseases 0.000 description 1
- 101100524547 Arabidopsis thaliana RFS5 gene Proteins 0.000 description 1
- 206010060971 Astrocytoma malignant Diseases 0.000 description 1
- 101710192393 Attachment protein G3P Proteins 0.000 description 1
- 102100022716 Atypical chemokine receptor 3 Human genes 0.000 description 1
- 102100035526 B melanoma antigen 1 Human genes 0.000 description 1
- 208000037914 B-cell disorder Diseases 0.000 description 1
- 108010074708 B7-H1 Antigen Proteins 0.000 description 1
- 108010077805 Bacterial Proteins Proteins 0.000 description 1
- 102100021663 Baculoviral IAP repeat-containing protein 5 Human genes 0.000 description 1
- 206010004146 Basal cell carcinoma Diseases 0.000 description 1
- 208000013165 Bowen disease Diseases 0.000 description 1
- 208000019337 Bowen disease of the skin Diseases 0.000 description 1
- 206010006143 Brain stem glioma Diseases 0.000 description 1
- 102100031650 C-X-C chemokine receptor type 4 Human genes 0.000 description 1
- 108010008629 CA-125 Antigen Proteins 0.000 description 1
- 102000007269 CA-125 Antigen Human genes 0.000 description 1
- 101150108242 CDC27 gene Proteins 0.000 description 1
- 101100381481 Caenorhabditis elegans baz-2 gene Proteins 0.000 description 1
- 101100005789 Caenorhabditis elegans cdk-4 gene Proteins 0.000 description 1
- 241000282836 Camelus dromedarius Species 0.000 description 1
- 208000013627 Camurati-Engelmann disease Diseases 0.000 description 1
- 102100039510 Cancer/testis antigen 2 Human genes 0.000 description 1
- 241000282472 Canis lupus familiaris Species 0.000 description 1
- 101710169873 Capsid protein G8P Proteins 0.000 description 1
- 102100025466 Carcinoembryonic antigen-related cell adhesion molecule 3 Human genes 0.000 description 1
- 102100025470 Carcinoembryonic antigen-related cell adhesion molecule 8 Human genes 0.000 description 1
- 206010007279 Carcinoid tumour of the gastrointestinal tract Diseases 0.000 description 1
- 208000017897 Carcinoma of esophagus Diseases 0.000 description 1
- 108010078791 Carrier Proteins Proteins 0.000 description 1
- 102100026548 Caspase-8 Human genes 0.000 description 1
- 108090000538 Caspase-8 Proteins 0.000 description 1
- 101150015280 Cel gene Proteins 0.000 description 1
- 102000000844 Cell Surface Receptors Human genes 0.000 description 1
- 108010001857 Cell Surface Receptors Proteins 0.000 description 1
- 102100025064 Cellular tumor antigen p53 Human genes 0.000 description 1
- 206010008263 Cervical dysplasia Diseases 0.000 description 1
- 208000001139 Cherubism Diseases 0.000 description 1
- 206010008724 Chondroectodermal dysplasia Diseases 0.000 description 1
- 208000005243 Chondrosarcoma Diseases 0.000 description 1
- 102100039361 Chondrosarcoma-associated gene 2/3 protein Human genes 0.000 description 1
- 201000009047 Chordoma Diseases 0.000 description 1
- 208000006332 Choriocarcinoma Diseases 0.000 description 1
- 102100021809 Chorionic somatomammotropin hormone 1 Human genes 0.000 description 1
- 201000000304 Cleidocranial dysplasia Diseases 0.000 description 1
- 102100031162 Collagen alpha-1(XVIII) chain Human genes 0.000 description 1
- 206010055114 Colon cancer metastatic Diseases 0.000 description 1
- 102000007644 Colony-Stimulating Factors Human genes 0.000 description 1
- 206010052358 Colorectal cancer metastatic Diseases 0.000 description 1
- 108010053085 Complement Factor H Proteins 0.000 description 1
- 102000016550 Complement Factor H Human genes 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- 208000009798 Craniopharyngioma Diseases 0.000 description 1
- 239000004971 Cross linker Substances 0.000 description 1
- 102100026234 Cytokine receptor common subunit gamma Human genes 0.000 description 1
- XUIIKFGFIJCVMT-GFCCVEGCSA-N D-thyroxine Chemical compound IC1=CC(C[C@@H](N)C(O)=O)=CC(I)=C1OC1=CC(I)=C(O)C(I)=C1 XUIIKFGFIJCVMT-GFCCVEGCSA-N 0.000 description 1
- 102000053602 DNA Human genes 0.000 description 1
- 229940123780 DNA topoisomerase I inhibitor Drugs 0.000 description 1
- 208000005335 Dentin Dysplasia Diseases 0.000 description 1
- 102000007260 Deoxyribonuclease I Human genes 0.000 description 1
- 108010008532 Deoxyribonuclease I Proteins 0.000 description 1
- 208000013558 Developmental Bone disease Diseases 0.000 description 1
- 101100216227 Dictyostelium discoideum anapc3 gene Proteins 0.000 description 1
- 208000002699 Digestive System Neoplasms Diseases 0.000 description 1
- 102000016607 Diphtheria Toxin Human genes 0.000 description 1
- 108010053187 Diphtheria Toxin Proteins 0.000 description 1
- 206010061819 Disease recurrence Diseases 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- 102000001301 EGF receptor Human genes 0.000 description 1
- 108060006698 EGF receptor Proteins 0.000 description 1
- 238000002965 ELISA Methods 0.000 description 1
- 208000006586 Ectromelia Diseases 0.000 description 1
- 201000002650 Ellis-van Creveld syndrome Diseases 0.000 description 1
- 201000009051 Embryonal Carcinoma Diseases 0.000 description 1
- 108010079505 Endostatins Proteins 0.000 description 1
- 208000031637 Erythroblastic Acute Leukemia Diseases 0.000 description 1
- 208000036566 Erythroleukaemia Diseases 0.000 description 1
- 241000588724 Escherichia coli Species 0.000 description 1
- 208000004332 Evans syndrome Diseases 0.000 description 1
- 208000017259 Extragonadal germ cell tumor Diseases 0.000 description 1
- 206010067141 Faciodigitogenital dysplasia Diseases 0.000 description 1
- 208000002633 Febrile Neutropenia Diseases 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- 102000004641 Fetal Proteins Human genes 0.000 description 1
- 108010003471 Fetal Proteins Proteins 0.000 description 1
- 108090000382 Fibroblast growth factor 6 Proteins 0.000 description 1
- 102100028075 Fibroblast growth factor 6 Human genes 0.000 description 1
- 208000000571 Fibrocystic breast disease Diseases 0.000 description 1
- 201000008808 Fibrosarcoma Diseases 0.000 description 1
- 208000008961 Fibrous Dysplasia of Bone Diseases 0.000 description 1
- 241000724791 Filamentous phage Species 0.000 description 1
- 102100038651 Four and a half LIM domains protein 1 Human genes 0.000 description 1
- 101710127220 Four and a half LIM domains protein 1 Proteins 0.000 description 1
- 206010073655 Freeman-Sheldon syndrome Diseases 0.000 description 1
- 208000022072 Gallbladder Neoplasms Diseases 0.000 description 1
- GYHNNYVSQQEPJS-UHFFFAOYSA-N Gallium Chemical compound [Ga] GYHNNYVSQQEPJS-UHFFFAOYSA-N 0.000 description 1
- 206010061968 Gastric neoplasm Diseases 0.000 description 1
- 206010017993 Gastrointestinal neoplasms Diseases 0.000 description 1
- 206010051635 Gastrointestinal tract adenoma Diseases 0.000 description 1
- 208000015872 Gaucher disease Diseases 0.000 description 1
- 108700004714 Gelonium multiflorum GEL Proteins 0.000 description 1
- 208000021309 Germ cell tumor Diseases 0.000 description 1
- 241000026944 Giovanella Species 0.000 description 1
- 206010018378 Glomerulonephritis rapidly progressive Diseases 0.000 description 1
- 201000003200 Goldenhar Syndrome Diseases 0.000 description 1
- 102000006771 Gonadotropins Human genes 0.000 description 1
- 108010086677 Gonadotropins Proteins 0.000 description 1
- 239000007995 HEPES buffer Substances 0.000 description 1
- 208000001258 Hemangiosarcoma Diseases 0.000 description 1
- 208000002250 Hematologic Neoplasms Diseases 0.000 description 1
- 102000001554 Hemoglobins Human genes 0.000 description 1
- 108010054147 Hemoglobins Proteins 0.000 description 1
- 238000012752 Hepatectomy Methods 0.000 description 1
- 241000545744 Hirudinea Species 0.000 description 1
- 108010025076 Holoenzymes Proteins 0.000 description 1
- 206010050469 Holt-Oram syndrome Diseases 0.000 description 1
- 101000890614 Homo sapiens A-kinase anchor protein 5 Proteins 0.000 description 1
- 101000774725 Homo sapiens A-kinase anchor protein 7 isoform gamma Proteins 0.000 description 1
- 101000774727 Homo sapiens A-kinase anchor protein 7 isoforms alpha and beta Proteins 0.000 description 1
- 101000678890 Homo sapiens Atypical chemokine receptor 3 Proteins 0.000 description 1
- 101000874316 Homo sapiens B melanoma antigen 1 Proteins 0.000 description 1
- 101000897480 Homo sapiens C-C motif chemokine 2 Proteins 0.000 description 1
- 101000922348 Homo sapiens C-X-C chemokine receptor type 4 Proteins 0.000 description 1
- 101000889345 Homo sapiens Cancer/testis antigen 2 Proteins 0.000 description 1
- 101000914337 Homo sapiens Carcinoembryonic antigen-related cell adhesion molecule 3 Proteins 0.000 description 1
- 101000914320 Homo sapiens Carcinoembryonic antigen-related cell adhesion molecule 8 Proteins 0.000 description 1
- 101000721661 Homo sapiens Cellular tumor antigen p53 Proteins 0.000 description 1
- 101000745414 Homo sapiens Chondrosarcoma-associated gene 2/3 protein Proteins 0.000 description 1
- 101001055227 Homo sapiens Cytokine receptor common subunit gamma Proteins 0.000 description 1
- 101000746373 Homo sapiens Granulocyte-macrophage colony-stimulating factor Proteins 0.000 description 1
- 101000578784 Homo sapiens Melanoma antigen recognized by T-cells 1 Proteins 0.000 description 1
- 101000623900 Homo sapiens Mucin-13 Proteins 0.000 description 1
- 101000581981 Homo sapiens Neural cell adhesion molecule 1 Proteins 0.000 description 1
- 101001001487 Homo sapiens Phosphatidylinositol-glycan biosynthesis class F protein Proteins 0.000 description 1
- 101000842302 Homo sapiens Protein-cysteine N-palmitoyltransferase HHAT Proteins 0.000 description 1
- 101001109419 Homo sapiens RNA-binding protein NOB1 Proteins 0.000 description 1
- 101000936922 Homo sapiens Sarcoplasmic/endoplasmic reticulum calcium ATPase 2 Proteins 0.000 description 1
- 101001059454 Homo sapiens Serine/threonine-protein kinase MARK2 Proteins 0.000 description 1
- 101000617130 Homo sapiens Stromal cell-derived factor 1 Proteins 0.000 description 1
- 101000980827 Homo sapiens T-cell surface glycoprotein CD1a Proteins 0.000 description 1
- 101000716149 Homo sapiens T-cell surface glycoprotein CD1b Proteins 0.000 description 1
- 101000835093 Homo sapiens Transferrin receptor protein 1 Proteins 0.000 description 1
- 101000730644 Homo sapiens Zinc finger protein PLAGL2 Proteins 0.000 description 1
- 108090000144 Human Proteins Proteins 0.000 description 1
- 102000003839 Human Proteins Human genes 0.000 description 1
- 208000019758 Hypergammaglobulinemia Diseases 0.000 description 1
- 206010020649 Hyperkeratosis Diseases 0.000 description 1
- 206010020880 Hypertrophy Diseases 0.000 description 1
- 206010021042 Hypopharyngeal cancer Diseases 0.000 description 1
- 206010056305 Hypopharyngeal neoplasm Diseases 0.000 description 1
- 102000026633 IL6 Human genes 0.000 description 1
- 229940076838 Immune checkpoint inhibitor Drugs 0.000 description 1
- 206010061598 Immunodeficiency Diseases 0.000 description 1
- 102000018071 Immunoglobulin Fc Fragments Human genes 0.000 description 1
- 108010091135 Immunoglobulin Fc Fragments Proteins 0.000 description 1
- 108700005091 Immunoglobulin Genes Proteins 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- 102100026818 Inhibin beta E chain Human genes 0.000 description 1
- 102000004877 Insulin Human genes 0.000 description 1
- 108090001061 Insulin Proteins 0.000 description 1
- 108090001117 Insulin-Like Growth Factor II Proteins 0.000 description 1
- 102100025947 Insulin-like growth factor II Human genes 0.000 description 1
- 102100026720 Interferon beta Human genes 0.000 description 1
- 108010047761 Interferon-alpha Proteins 0.000 description 1
- 102000006992 Interferon-alpha Human genes 0.000 description 1
- 108090000467 Interferon-beta Proteins 0.000 description 1
- 102000003814 Interleukin-10 Human genes 0.000 description 1
- 108090000174 Interleukin-10 Proteins 0.000 description 1
- 108090000177 Interleukin-11 Proteins 0.000 description 1
- 102000003815 Interleukin-11 Human genes 0.000 description 1
- 108090000176 Interleukin-13 Proteins 0.000 description 1
- 101800003050 Interleukin-16 Proteins 0.000 description 1
- 102000049772 Interleukin-16 Human genes 0.000 description 1
- 102100030703 Interleukin-22 Human genes 0.000 description 1
- 108010065637 Interleukin-23 Proteins 0.000 description 1
- 108010002386 Interleukin-3 Proteins 0.000 description 1
- 108090000978 Interleukin-4 Proteins 0.000 description 1
- 108010002616 Interleukin-5 Proteins 0.000 description 1
- 108010002586 Interleukin-7 Proteins 0.000 description 1
- 108090001007 Interleukin-8 Proteins 0.000 description 1
- 108010002335 Interleukin-9 Proteins 0.000 description 1
- 102000000585 Interleukin-9 Human genes 0.000 description 1
- 208000005016 Intestinal Neoplasms Diseases 0.000 description 1
- 208000032177 Intestinal Polyps Diseases 0.000 description 1
- 206010061252 Intraocular melanoma Diseases 0.000 description 1
- 108010044467 Isoenzymes Proteins 0.000 description 1
- 208000007766 Kaposi sarcoma Diseases 0.000 description 1
- 208000001126 Keratosis Diseases 0.000 description 1
- 102100020880 Kit ligand Human genes 0.000 description 1
- 101150105104 Kras gene Proteins 0.000 description 1
- 150000008575 L-amino acids Chemical class 0.000 description 1
- LEVWYRKDKASIDU-IMJSIDKUSA-N L-cystine Chemical compound [O-]C(=O)[C@@H]([NH3+])CSSC[C@H]([NH3+])C([O-])=O LEVWYRKDKASIDU-IMJSIDKUSA-N 0.000 description 1
- 125000000510 L-tryptophano group Chemical group [H]C1=C([H])C([H])=C2N([H])C([H])=C(C([H])([H])[C@@]([H])(C(O[H])=O)N([H])[*])C2=C1[H] 0.000 description 1
- 206010023825 Laryngeal cancer Diseases 0.000 description 1
- 208000018142 Leiomyosarcoma Diseases 0.000 description 1
- 102000016267 Leptin Human genes 0.000 description 1
- 108010092277 Leptin Proteins 0.000 description 1
- 206010024305 Leukaemia monocytic Diseases 0.000 description 1
- 102100032352 Leukemia inhibitory factor Human genes 0.000 description 1
- 108090000581 Leukemia inhibitory factor Proteins 0.000 description 1
- 206010024503 Limb reduction defect Diseases 0.000 description 1
- 108010074338 Lymphokines Proteins 0.000 description 1
- 102000008072 Lymphokines Human genes 0.000 description 1
- 206010025312 Lymphoma AIDS related Diseases 0.000 description 1
- 208000030289 Lymphoproliferative disease Diseases 0.000 description 1
- 101710125418 Major capsid protein Proteins 0.000 description 1
- 101710156564 Major tail protein Gp23 Proteins 0.000 description 1
- 208000004059 Male Breast Neoplasms Diseases 0.000 description 1
- PEEHTFAAVSWFBL-UHFFFAOYSA-N Maleimide Chemical compound O=C1NC(=O)C=C1 PEEHTFAAVSWFBL-UHFFFAOYSA-N 0.000 description 1
- 208000006644 Malignant Fibrous Histiocytoma Diseases 0.000 description 1
- 206010025557 Malignant fibrous histiocytoma of bone Diseases 0.000 description 1
- 206010025997 Malignant neoplasm of islets of Langerhans Diseases 0.000 description 1
- 208000032271 Malignant tumor of penis Diseases 0.000 description 1
- 208000025205 Mantle-Cell Lymphoma Diseases 0.000 description 1
- 102100027754 Mast/stem cell growth factor receptor Kit Human genes 0.000 description 1
- 201000001853 McCune-Albright syndrome Diseases 0.000 description 1
- 208000007054 Medullary Carcinoma Diseases 0.000 description 1
- 102100028389 Melanoma antigen recognized by T-cells 1 Human genes 0.000 description 1
- 102000003735 Mesothelin Human genes 0.000 description 1
- 108090000015 Mesothelin Proteins 0.000 description 1
- 206010054949 Metaplasia Diseases 0.000 description 1
- 206010027457 Metastases to liver Diseases 0.000 description 1
- 206010027458 Metastases to lung Diseases 0.000 description 1
- 108010020004 Microtubule-Associated Proteins Proteins 0.000 description 1
- 102000009664 Microtubule-Associated Proteins Human genes 0.000 description 1
- 108010006519 Molecular Chaperones Proteins 0.000 description 1
- 102000013967 Monokines Human genes 0.000 description 1
- 108010050619 Monokines Proteins 0.000 description 1
- 208000003445 Mouth Neoplasms Diseases 0.000 description 1
- 108010008707 Mucin-1 Proteins 0.000 description 1
- 102000007298 Mucin-1 Human genes 0.000 description 1
- 102100023124 Mucin-13 Human genes 0.000 description 1
- 102000015728 Mucins Human genes 0.000 description 1
- 108010063954 Mucins Proteins 0.000 description 1
- 101100381525 Mus musculus Bcl6 gene Proteins 0.000 description 1
- 108010021466 Mutant Proteins Proteins 0.000 description 1
- 102000008300 Mutant Proteins Human genes 0.000 description 1
- 201000003793 Myelodysplastic syndrome Diseases 0.000 description 1
- 102100031789 Myeloid-derived growth factor Human genes 0.000 description 1
- 208000014767 Myeloproliferative disease Diseases 0.000 description 1
- GHAZCVNUKKZTLG-UHFFFAOYSA-N N-ethyl-succinimide Natural products CCN1C(=O)CCC1=O GHAZCVNUKKZTLG-UHFFFAOYSA-N 0.000 description 1
- HDFGOPSGAURCEO-UHFFFAOYSA-N N-ethylmaleimide Chemical compound CCN1C(=O)C=CC1=O HDFGOPSGAURCEO-UHFFFAOYSA-N 0.000 description 1
- 208000001894 Nasopharyngeal Neoplasms Diseases 0.000 description 1
- 206010061306 Nasopharyngeal cancer Diseases 0.000 description 1
- 206010028813 Nausea Diseases 0.000 description 1
- 206010028851 Necrosis Diseases 0.000 description 1
- 108010025020 Nerve Growth Factor Proteins 0.000 description 1
- 102000007072 Nerve Growth Factors Human genes 0.000 description 1
- 102100027347 Neural cell adhesion molecule 1 Human genes 0.000 description 1
- 208000009277 Neuroectodermal Tumors Diseases 0.000 description 1
- 101710163270 Nuclease Proteins 0.000 description 1
- 108091028043 Nucleic acid sequence Proteins 0.000 description 1
- 206010051934 Oculoauriculovertebral dysplasia Diseases 0.000 description 1
- 208000008909 Oculodentodigital dysplasia Diseases 0.000 description 1
- 208000004910 Odontodysplasia Diseases 0.000 description 1
- 206010050171 Oesophageal dysplasia Diseases 0.000 description 1
- 201000010133 Oligodendroglioma Diseases 0.000 description 1
- 208000010335 Ophthalmomandibulomelic dysplasia Diseases 0.000 description 1
- 206010031096 Oropharyngeal cancer Diseases 0.000 description 1
- 206010057444 Oropharyngeal neoplasm Diseases 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 101710160107 Outer membrane protein A Proteins 0.000 description 1
- 208000007571 Ovarian Epithelial Carcinoma Diseases 0.000 description 1
- 108060006580 PRAME Proteins 0.000 description 1
- 102000036673 PRAME Human genes 0.000 description 1
- 102100034640 PWWP domain-containing DNA repair factor 3A Human genes 0.000 description 1
- 108050007154 PWWP domain-containing DNA repair factor 3A Proteins 0.000 description 1
- 108090000526 Papain Proteins 0.000 description 1
- 241000282520 Papio Species 0.000 description 1
- 208000002774 Paraproteinemias Diseases 0.000 description 1
- 208000000821 Parathyroid Neoplasms Diseases 0.000 description 1
- 102000003982 Parathyroid hormone Human genes 0.000 description 1
- 108090000445 Parathyroid hormone Proteins 0.000 description 1
- 208000002471 Penile Neoplasms Diseases 0.000 description 1
- 206010034299 Penile cancer Diseases 0.000 description 1
- 229940083963 Peptide antagonist Drugs 0.000 description 1
- ABLZXFCXXLZCGV-UHFFFAOYSA-N Phosphorous acid Chemical group OP(O)=O ABLZXFCXXLZCGV-UHFFFAOYSA-N 0.000 description 1
- 108091000080 Phosphotransferase Proteins 0.000 description 1
- 241001425800 Pipa Species 0.000 description 1
- 208000007913 Pituitary Neoplasms Diseases 0.000 description 1
- 108010003044 Placental Lactogen Proteins 0.000 description 1
- 239000000381 Placental Lactogen Substances 0.000 description 1
- 241000276498 Pollachius virens Species 0.000 description 1
- 229920002732 Polyanhydride Polymers 0.000 description 1
- 208000006994 Precancerous Conditions Diseases 0.000 description 1
- 102100024216 Programmed cell death 1 ligand 1 Human genes 0.000 description 1
- 108010076181 Proinsulin Proteins 0.000 description 1
- 102000003946 Prolactin Human genes 0.000 description 1
- 108010057464 Prolactin Proteins 0.000 description 1
- 102100035703 Prostatic acid phosphatase Human genes 0.000 description 1
- 239000004365 Protease Substances 0.000 description 1
- 102100030616 Protein-cysteine N-palmitoyltransferase HHAT Human genes 0.000 description 1
- 241000589516 Pseudomonas Species 0.000 description 1
- 101000762949 Pseudomonas aeruginosa (strain ATCC 15692 / DSM 22644 / CIP 104116 / JCM 14847 / LMG 12228 / 1C / PRS 101 / PAO1) Exotoxin A Proteins 0.000 description 1
- 206010037549 Purpura Diseases 0.000 description 1
- 241001672981 Purpura Species 0.000 description 1
- 206010037660 Pyrexia Diseases 0.000 description 1
- 102100022491 RNA-binding protein NOB1 Human genes 0.000 description 1
- 101100372762 Rattus norvegicus Flt1 gene Proteins 0.000 description 1
- 102100020718 Receptor-type tyrosine-protein kinase FLT3 Human genes 0.000 description 1
- 101710151245 Receptor-type tyrosine-protein kinase FLT3 Proteins 0.000 description 1
- 108090000103 Relaxin Proteins 0.000 description 1
- 102000003743 Relaxin Human genes 0.000 description 1
- 108010039491 Ricin Proteins 0.000 description 1
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 1
- 208000004337 Salivary Gland Neoplasms Diseases 0.000 description 1
- 102100027732 Sarcoplasmic/endoplasmic reticulum calcium ATPase 2 Human genes 0.000 description 1
- 201000010208 Seminoma Diseases 0.000 description 1
- 229920002684 Sepharose Polymers 0.000 description 1
- 102100028904 Serine/threonine-protein kinase MARK2 Human genes 0.000 description 1
- 108020004682 Single-Stranded DNA Proteins 0.000 description 1
- 206010072610 Skeletal dysplasia Diseases 0.000 description 1
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- 108010039445 Stem Cell Factor Proteins 0.000 description 1
- 102100021669 Stromal cell-derived factor 1 Human genes 0.000 description 1
- 108010002687 Survivin Proteins 0.000 description 1
- 208000031673 T-Cell Cutaneous Lymphoma Diseases 0.000 description 1
- 208000000389 T-cell leukemia Diseases 0.000 description 1
- 208000027585 T-cell non-Hodgkin lymphoma Diseases 0.000 description 1
- 102100024219 T-cell surface glycoprotein CD1a Human genes 0.000 description 1
- 102100033082 TNF receptor-associated factor 3 Human genes 0.000 description 1
- WDLRUFUQRNWCPK-UHFFFAOYSA-N Tetraxetan Chemical compound OC(=O)CN1CCN(CC(O)=O)CCN(CC(O)=O)CCN(CC(O)=O)CC1 WDLRUFUQRNWCPK-UHFFFAOYSA-N 0.000 description 1
- 108060008245 Thrombospondin Proteins 0.000 description 1
- 102000002938 Thrombospondin Human genes 0.000 description 1
- 201000009365 Thymic carcinoma Diseases 0.000 description 1
- 208000024770 Thyroid neoplasm Diseases 0.000 description 1
- 101710183280 Topoisomerase Proteins 0.000 description 1
- 239000000365 Topoisomerase I Inhibitor Substances 0.000 description 1
- 101710120037 Toxin CcdB Proteins 0.000 description 1
- 102100026144 Transferrin receptor protein 1 Human genes 0.000 description 1
- 206010044407 Transitional cell cancer of the renal pelvis and ureter Diseases 0.000 description 1
- HDTRYLNUVZCQOY-WSWWMNSNSA-N Trehalose Natural products O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@H](O)[C@@H](O)[C@@H](O)[C@@H](CO)O1 HDTRYLNUVZCQOY-WSWWMNSNSA-N 0.000 description 1
- 208000015778 Undifferentiated pleomorphic sarcoma Diseases 0.000 description 1
- 206010046431 Urethral cancer Diseases 0.000 description 1
- 206010046458 Urethral neoplasms Diseases 0.000 description 1
- 208000008385 Urogenital Neoplasms Diseases 0.000 description 1
- 201000005969 Uveal melanoma Diseases 0.000 description 1
- 108010073929 Vascular Endothelial Growth Factor A Proteins 0.000 description 1
- 241000251539 Vertebrata <Metazoa> Species 0.000 description 1
- 208000014070 Vestibular schwannoma Diseases 0.000 description 1
- 102100032571 Zinc finger protein PLAGL2 Human genes 0.000 description 1
- 229950005186 abagovomab Drugs 0.000 description 1
- 229960000446 abciximab Drugs 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 208000004064 acoustic neuroma Diseases 0.000 description 1
- 208000009621 actinic keratosis Diseases 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 239000004480 active ingredient Substances 0.000 description 1
- 239000000488 activin Substances 0.000 description 1
- 208000021841 acute erythroid leukemia Diseases 0.000 description 1
- 230000004721 adaptive immunity Effects 0.000 description 1
- 229950009084 adecatumumab Drugs 0.000 description 1
- 208000020990 adrenal cortex carcinoma Diseases 0.000 description 1
- 208000007128 adrenocortical carcinoma Diseases 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 238000001042 affinity chromatography Methods 0.000 description 1
- 108010035879 albumin-bilirubin complex Proteins 0.000 description 1
- 229940045714 alkyl sulfonate alkylating agent Drugs 0.000 description 1
- 150000008052 alkyl sulfonates Chemical class 0.000 description 1
- HDTRYLNUVZCQOY-LIZSDCNHSA-N alpha,alpha-trehalose Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 HDTRYLNUVZCQOY-LIZSDCNHSA-N 0.000 description 1
- SRHNADOZAAWYLV-XLMUYGLTSA-N alpha-L-Fucp-(1->2)-beta-D-Galp-(1->4)-[alpha-L-Fucp-(1->3)]-beta-D-GlcpNAc Chemical compound O[C@H]1[C@H](O)[C@H](O)[C@H](C)O[C@H]1O[C@H]1[C@H](O[C@H]2[C@@H]([C@@H](NC(C)=O)[C@H](O)O[C@@H]2CO)O[C@H]2[C@H]([C@H](O)[C@H](O)[C@H](C)O2)O)O[C@H](CO)[C@H](O)[C@@H]1O SRHNADOZAAWYLV-XLMUYGLTSA-N 0.000 description 1
- 230000003321 amplification Effects 0.000 description 1
- 201000007538 anal carcinoma Diseases 0.000 description 1
- 238000013103 analytical ultracentrifugation Methods 0.000 description 1
- 208000007502 anemia Diseases 0.000 description 1
- 230000033115 angiogenesis Effects 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 230000000181 anti-adherent effect Effects 0.000 description 1
- 230000003527 anti-angiogenesis Effects 0.000 description 1
- 230000001772 anti-angiogenic effect Effects 0.000 description 1
- 230000003466 anti-cipated effect Effects 0.000 description 1
- 230000000340 anti-metabolite Effects 0.000 description 1
- 230000002001 anti-metastasis Effects 0.000 description 1
- 230000002927 anti-mitotic effect Effects 0.000 description 1
- 230000000692 anti-sense effect Effects 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 229940125644 antibody drug Drugs 0.000 description 1
- 230000010056 antibody-dependent cellular cytotoxicity Effects 0.000 description 1
- 210000000628 antibody-producing cell Anatomy 0.000 description 1
- 102000025171 antigen binding proteins Human genes 0.000 description 1
- 108091000831 antigen binding proteins Proteins 0.000 description 1
- 229940100197 antimetabolite Drugs 0.000 description 1
- 239000002256 antimetabolite Substances 0.000 description 1
- 229940045687 antimetabolites folic acid analogs Drugs 0.000 description 1
- 229940045719 antineoplastic alkylating agent nitrosoureas Drugs 0.000 description 1
- 201000011165 anus cancer Diseases 0.000 description 1
- 230000005775 apoptotic pathway Effects 0.000 description 1
- 239000012062 aqueous buffer Substances 0.000 description 1
- 239000008135 aqueous vehicle Substances 0.000 description 1
- 125000000637 arginyl group Chemical group N[C@@H](CCCNC(N)=N)C(=O)* 0.000 description 1
- 206010003246 arthritis Diseases 0.000 description 1
- FZCSTZYAHCUGEM-UHFFFAOYSA-N aspergillomarasmine B Natural products OC(=O)CNC(C(O)=O)CNC(C(O)=O)CC(O)=O FZCSTZYAHCUGEM-UHFFFAOYSA-N 0.000 description 1
- 229960003852 atezolizumab Drugs 0.000 description 1
- 229940120638 avastin Drugs 0.000 description 1
- 229930014667 baccatin III Natural products 0.000 description 1
- 230000006399 behavior Effects 0.000 description 1
- 229940022836 benlysta Drugs 0.000 description 1
- 229950000321 benralizumab Drugs 0.000 description 1
- 102000015736 beta 2-Microglobulin Human genes 0.000 description 1
- 108010081355 beta 2-Microglobulin Proteins 0.000 description 1
- 201000007180 bile duct carcinoma Diseases 0.000 description 1
- 208000026900 bile duct neoplasm Diseases 0.000 description 1
- 229920000249 biocompatible polymer Polymers 0.000 description 1
- 238000007622 bioinformatic analysis Methods 0.000 description 1
- 239000006177 biological buffer Substances 0.000 description 1
- 201000001531 bladder carcinoma Diseases 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- 238000004820 blood count Methods 0.000 description 1
- 201000008873 bone osteosarcoma Diseases 0.000 description 1
- 208000012172 borderline epithelial tumor of ovary Diseases 0.000 description 1
- 150000001639 boron compounds Chemical class 0.000 description 1
- 108010006025 bovine growth hormone Proteins 0.000 description 1
- DYODAJAEQDVYFX-UHFFFAOYSA-N brallobarbital Chemical compound BrC(=C)CC1(CC=C)C(=O)NC(=O)NC1=O DYODAJAEQDVYFX-UHFFFAOYSA-N 0.000 description 1
- 208000011803 breast fibrocystic disease Diseases 0.000 description 1
- 208000003362 bronchogenic carcinoma Diseases 0.000 description 1
- 206010006475 bronchopulmonary dysplasia Diseases 0.000 description 1
- 239000012830 cancer therapeutic Substances 0.000 description 1
- 229910002091 carbon monoxide Inorganic materials 0.000 description 1
- 230000000747 cardiac effect Effects 0.000 description 1
- 230000032823 cell division Effects 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 210000003169 central nervous system Anatomy 0.000 description 1
- 201000007335 cerebellar astrocytoma Diseases 0.000 description 1
- 230000002490 cerebral effect Effects 0.000 description 1
- 208000007287 cheilitis Diseases 0.000 description 1
- 239000013522 chelant Substances 0.000 description 1
- 239000002738 chelating agent Substances 0.000 description 1
- 238000010382 chemical cross-linking Methods 0.000 description 1
- 238000001311 chemical methods and process Methods 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 239000003638 chemical reducing agent Substances 0.000 description 1
- 201000002687 childhood acute myeloid leukemia Diseases 0.000 description 1
- 201000004018 childhood brain stem glioma Diseases 0.000 description 1
- 201000004677 childhood cerebellar astrocytic neoplasm Diseases 0.000 description 1
- 201000005793 childhood medulloblastoma Diseases 0.000 description 1
- 208000006990 cholangiocarcinoma Diseases 0.000 description 1
- 230000002759 chromosomal effect Effects 0.000 description 1
- 238000010367 cloning Methods 0.000 description 1
- 231100000096 clonogenic assay Toxicity 0.000 description 1
- 238000009643 clonogenic assay Methods 0.000 description 1
- 238000012321 colectomy Methods 0.000 description 1
- 229940047120 colony stimulating factors Drugs 0.000 description 1
- 238000011284 combination treatment Methods 0.000 description 1
- 230000002301 combined effect Effects 0.000 description 1
- 230000009918 complex formation Effects 0.000 description 1
- 239000002131 composite material Substances 0.000 description 1
- 210000002808 connective tissue Anatomy 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- 239000006184 cosolvent Substances 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 201000010100 craniodiaphyseal dysplasia Diseases 0.000 description 1
- 201000010095 craniometaphyseal dysplasia Diseases 0.000 description 1
- 239000003431 cross linking reagent Substances 0.000 description 1
- 201000007241 cutaneous T cell lymphoma Diseases 0.000 description 1
- IMBXRZKCLVBLBH-OGYJWPHRSA-N cvp protocol Chemical compound ClCCN(CCCl)P1(=O)NCCCO1.O=C1C=C[C@]2(C)[C@H]3C(=O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1.C([C@H](C[C@]1(C(=O)OC)C=2C(=C3C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C=O)=CC=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 IMBXRZKCLVBLBH-OGYJWPHRSA-N 0.000 description 1
- 208000002445 cystadenocarcinoma Diseases 0.000 description 1
- 229960003067 cystine Drugs 0.000 description 1
- 230000009089 cytolysis Effects 0.000 description 1
- 210000000805 cytoplasm Anatomy 0.000 description 1
- 210000004292 cytoskeleton Anatomy 0.000 description 1
- 231100000433 cytotoxic Toxicity 0.000 description 1
- 230000001472 cytotoxic effect Effects 0.000 description 1
- 229960002204 daratumumab Drugs 0.000 description 1
- 230000007812 deficiency Effects 0.000 description 1
- 239000003405 delayed action preparation Substances 0.000 description 1
- 229940124447 delivery agent Drugs 0.000 description 1
- 210000004443 dendritic cell Anatomy 0.000 description 1
- 210000003298 dental enamel Anatomy 0.000 description 1
- 238000001212 derivatisation Methods 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 230000018109 developmental process Effects 0.000 description 1
- 229960003957 dexamethasone Drugs 0.000 description 1
- UREBDLICKHMUKA-CXSFZGCWSA-N dexamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-CXSFZGCWSA-N 0.000 description 1
- 238000011026 diafiltration Methods 0.000 description 1
- 208000015799 differentiated thyroid carcinoma Diseases 0.000 description 1
- 238000002050 diffraction method Methods 0.000 description 1
- 230000029087 digestion Effects 0.000 description 1
- 210000002249 digestive system Anatomy 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 229940000406 drug candidate Drugs 0.000 description 1
- 238000002651 drug therapy Methods 0.000 description 1
- 230000009977 dual effect Effects 0.000 description 1
- 229950009791 durvalumab Drugs 0.000 description 1
- 239000000975 dye Substances 0.000 description 1
- 208000031068 ectodermal dysplasia syndrome Diseases 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 210000001671 embryonic stem cell Anatomy 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 230000002124 endocrine Effects 0.000 description 1
- 210000001900 endoderm Anatomy 0.000 description 1
- 201000003914 endometrial carcinoma Diseases 0.000 description 1
- 230000002357 endometrial effect Effects 0.000 description 1
- 210000002472 endoplasmic reticulum Anatomy 0.000 description 1
- 210000001163 endosome Anatomy 0.000 description 1
- 239000002158 endotoxin Substances 0.000 description 1
- 231100000655 enterotoxin Toxicity 0.000 description 1
- 108010087914 epidermal growth factor receptor VIII Proteins 0.000 description 1
- 208000037828 epithelial carcinoma Diseases 0.000 description 1
- 210000002919 epithelial cell Anatomy 0.000 description 1
- HESCAJZNRMSMJG-KKQRBIROSA-N epothilone A Chemical class C/C([C@@H]1C[C@@H]2O[C@@H]2CCC[C@@H]([C@@H]([C@@H](C)C(=O)C(C)(C)[C@@H](O)CC(=O)O1)O)C)=C\C1=CSC(C)=N1 HESCAJZNRMSMJG-KKQRBIROSA-N 0.000 description 1
- 229940082789 erbitux Drugs 0.000 description 1
- 201000004101 esophageal cancer Diseases 0.000 description 1
- 201000005619 esophageal carcinoma Diseases 0.000 description 1
- 229960005420 etoposide Drugs 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 239000003777 experimental drug Substances 0.000 description 1
- 239000013613 expression plasmid Substances 0.000 description 1
- 201000008819 extrahepatic bile duct carcinoma Diseases 0.000 description 1
- 208000024519 eye neoplasm Diseases 0.000 description 1
- 201000007741 female breast cancer Diseases 0.000 description 1
- 201000002276 female breast carcinoma Diseases 0.000 description 1
- 210000003754 fetus Anatomy 0.000 description 1
- 229950003499 fibrin Drugs 0.000 description 1
- 208000008487 fibromuscular dysplasia Diseases 0.000 description 1
- 201000010103 fibrous dysplasia Diseases 0.000 description 1
- 238000000684 flow cytometry Methods 0.000 description 1
- 150000002224 folic acids Chemical class 0.000 description 1
- 230000003325 follicular Effects 0.000 description 1
- 239000012737 fresh medium Substances 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 230000004927 fusion Effects 0.000 description 1
- 210000000232 gallbladder Anatomy 0.000 description 1
- 201000010175 gallbladder cancer Diseases 0.000 description 1
- 229910052733 gallium Inorganic materials 0.000 description 1
- 150000002270 gangliosides Chemical class 0.000 description 1
- 229950002508 gantenerumab Drugs 0.000 description 1
- 208000010749 gastric carcinoma Diseases 0.000 description 1
- 238000010353 genetic engineering Methods 0.000 description 1
- 201000007116 gestational trophoblastic neoplasm Diseases 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 230000013595 glycosylation Effects 0.000 description 1
- 238000006206 glycosylation reaction Methods 0.000 description 1
- 239000002622 gonadotropin Substances 0.000 description 1
- 239000003102 growth factor Substances 0.000 description 1
- 239000000122 growth hormone Substances 0.000 description 1
- 210000003128 head Anatomy 0.000 description 1
- 208000029427 heart-hand syndrome Diseases 0.000 description 1
- 208000025750 heavy chain disease Diseases 0.000 description 1
- 208000024200 hematopoietic and lymphoid system neoplasm Diseases 0.000 description 1
- 208000014845 hemimelia Diseases 0.000 description 1
- 208000006454 hepatitis Diseases 0.000 description 1
- 231100000283 hepatitis Toxicity 0.000 description 1
- 238000004128 high performance liquid chromatography Methods 0.000 description 1
- 201000000284 histiocytoma Diseases 0.000 description 1
- 238000002744 homologous recombination Methods 0.000 description 1
- 230000006801 homologous recombination Effects 0.000 description 1
- 238000013415 human tumor xenograft model Methods 0.000 description 1
- 230000005661 hydrophobic surface Effects 0.000 description 1
- 206010020718 hyperplasia Diseases 0.000 description 1
- 201000003535 hypohidrotic ectodermal dysplasia Diseases 0.000 description 1
- 208000035128 hypohidrotic/hair/tooth type autosomal recessive ectodermal dysplasia 10B Diseases 0.000 description 1
- 208000032771 hypohidrotic/hair/tooth type autosomal recessive ectodermal dysplasia 11B Diseases 0.000 description 1
- 201000006866 hypopharynx cancer Diseases 0.000 description 1
- 230000007954 hypoxia Effects 0.000 description 1
- 229960001001 ibritumomab tiuxetan Drugs 0.000 description 1
- 230000008938 immune dysregulation Effects 0.000 description 1
- 239000012274 immune-checkpoint protein inhibitor Substances 0.000 description 1
- 238000002649 immunization Methods 0.000 description 1
- 230000003053 immunization Effects 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 230000002779 inactivation Effects 0.000 description 1
- 230000001939 inductive effect Effects 0.000 description 1
- 230000002458 infectious effect Effects 0.000 description 1
- 208000027866 inflammatory disease Diseases 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 239000000893 inhibin Substances 0.000 description 1
- 229950002133 iniparib Drugs 0.000 description 1
- ZPNFWUPYTFPOJU-LPYSRVMUSA-N iniprol Chemical compound C([C@H]1C(=O)NCC(=O)NCC(=O)N[C@H]2CSSC[C@H]3C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@H](C(N[C@H](C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=4C=CC(O)=CC=4)C(=O)N[C@@H](CC=4C=CC=CC=4)C(=O)N[C@@H](CC=4C=CC(O)=CC=4)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C)C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CSSC[C@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CC=4C=CC=CC=4)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCCN)NC(=O)[C@H](C)NC(=O)[C@H](CCCNC(N)=N)NC2=O)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CSSC[C@H](NC(=O)[C@H](CC=2C=CC=CC=2)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H]2N(CCC2)C(=O)[C@@H](N)CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N2[C@@H](CCC2)C(=O)N2[C@@H](CCC2)C(=O)N[C@@H](CC=2C=CC(O)=CC=2)C(=O)N[C@@H]([C@@H](C)O)C(=O)NCC(=O)N2[C@@H](CCC2)C(=O)N3)C(=O)NCC(=O)NCC(=O)N[C@@H](C)C(O)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@H](C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@H](C(=O)N1)C(C)C)[C@@H](C)O)[C@@H](C)CC)=O)[C@@H](C)CC)C1=CC=C(O)C=C1 ZPNFWUPYTFPOJU-LPYSRVMUSA-N 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- 230000015788 innate immune response Effects 0.000 description 1
- 238000011081 inoculation Methods 0.000 description 1
- 229940125396 insulin Drugs 0.000 description 1
- 108040006849 interleukin-2 receptor activity proteins Proteins 0.000 description 1
- 108010074108 interleukin-21 Proteins 0.000 description 1
- 108040006858 interleukin-6 receptor activity proteins Proteins 0.000 description 1
- 230000006662 intracellular pathway Effects 0.000 description 1
- 208000020082 intraepithelial neoplasia Diseases 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000007913 intrathecal administration Methods 0.000 description 1
- 238000007914 intraventricular administration Methods 0.000 description 1
- 238000011835 investigation Methods 0.000 description 1
- 238000004255 ion exchange chromatography Methods 0.000 description 1
- 230000002427 irreversible effect Effects 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- 210000004153 islets of langerhan Anatomy 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- YWXYYJSYQOXTPL-SLPGGIOYSA-N isosorbide mononitrate Chemical compound [O-][N+](=O)O[C@@H]1CO[C@@H]2[C@@H](O)CO[C@@H]21 YWXYYJSYQOXTPL-SLPGGIOYSA-N 0.000 description 1
- 208000017169 kidney disease Diseases 0.000 description 1
- 150000002596 lactones Chemical group 0.000 description 1
- 206010023841 laryngeal neoplasm Diseases 0.000 description 1
- 201000004962 larynx cancer Diseases 0.000 description 1
- 230000003902 lesion Effects 0.000 description 1
- 210000000265 leukocyte Anatomy 0.000 description 1
- 208000002741 leukoplakia Diseases 0.000 description 1
- 208000012987 lip and oral cavity carcinoma Diseases 0.000 description 1
- 206010024627 liposarcoma Diseases 0.000 description 1
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 1
- 231100000053 low toxicity Toxicity 0.000 description 1
- 201000005249 lung adenocarcinoma Diseases 0.000 description 1
- 208000037841 lung tumor Diseases 0.000 description 1
- 210000001165 lymph node Anatomy 0.000 description 1
- 208000037829 lymphangioendotheliosarcoma Diseases 0.000 description 1
- 208000012804 lymphangiosarcoma Diseases 0.000 description 1
- 230000001926 lymphatic effect Effects 0.000 description 1
- 210000004698 lymphocyte Anatomy 0.000 description 1
- 230000000329 lymphopenic effect Effects 0.000 description 1
- 239000012931 lyophilized formulation Substances 0.000 description 1
- 125000003588 lysine group Chemical group [H]N([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 1
- 210000003712 lysosome Anatomy 0.000 description 1
- 230000001868 lysosomic effect Effects 0.000 description 1
- 201000000564 macroglobulinemia Diseases 0.000 description 1
- 229920002521 macromolecule Polymers 0.000 description 1
- 238000009115 maintenance therapy Methods 0.000 description 1
- 201000003175 male breast cancer Diseases 0.000 description 1
- 208000010907 male breast carcinoma Diseases 0.000 description 1
- 208000006178 malignant mesothelioma Diseases 0.000 description 1
- 208000026045 malignant tumor of parathyroid gland Diseases 0.000 description 1
- 201000007924 marginal zone B-cell lymphoma Diseases 0.000 description 1
- 208000021937 marginal zone lymphoma Diseases 0.000 description 1
- 231100000682 maximum tolerated dose Toxicity 0.000 description 1
- KJLLKLRVCJAFRY-UHFFFAOYSA-N mebutizide Chemical compound ClC1=C(S(N)(=O)=O)C=C2S(=O)(=O)NC(C(C)C(C)CC)NC2=C1 KJLLKLRVCJAFRY-UHFFFAOYSA-N 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 206010027191 meningioma Diseases 0.000 description 1
- 239000002207 metabolite Substances 0.000 description 1
- 201000010828 metaphyseal dysplasia Diseases 0.000 description 1
- 230000015689 metaplastic ossification Effects 0.000 description 1
- 208000037819 metastatic cancer Diseases 0.000 description 1
- 208000011575 metastatic malignant neoplasm Diseases 0.000 description 1
- 208000037970 metastatic squamous neck cancer Diseases 0.000 description 1
- MYWUZJCMWCOHBA-VIFPVBQESA-N methamphetamine Chemical compound CN[C@@H](C)CC1=CC=CC=C1 MYWUZJCMWCOHBA-VIFPVBQESA-N 0.000 description 1
- 210000003470 mitochondria Anatomy 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 102000035118 modified proteins Human genes 0.000 description 1
- 108091005573 modified proteins Proteins 0.000 description 1
- 238000001823 molecular biology technique Methods 0.000 description 1
- 201000006894 monocytic leukemia Diseases 0.000 description 1
- 208000008084 monostotic fibrous dysplasia Diseases 0.000 description 1
- 108091005763 multidomain proteins Proteins 0.000 description 1
- 230000035772 mutation Effects 0.000 description 1
- 201000005962 mycosis fungoides Diseases 0.000 description 1
- 208000001611 myxosarcoma Diseases 0.000 description 1
- OHDXDNUPVVYWOV-UHFFFAOYSA-N n-methyl-1-(2-naphthalen-1-ylsulfanylphenyl)methanamine Chemical compound CNCC1=CC=CC=C1SC1=CC=CC2=CC=CC=C12 OHDXDNUPVVYWOV-UHFFFAOYSA-N 0.000 description 1
- 210000003928 nasal cavity Anatomy 0.000 description 1
- 230000008693 nausea Effects 0.000 description 1
- 230000017074 necrotic cell death Effects 0.000 description 1
- 208000025189 neoplasm of testis Diseases 0.000 description 1
- 230000001613 neoplastic effect Effects 0.000 description 1
- 210000000885 nephron Anatomy 0.000 description 1
- 231100000417 nephrotoxicity Toxicity 0.000 description 1
- 201000011519 neuroendocrine tumor Diseases 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 206010029410 night sweats Diseases 0.000 description 1
- 230000036565 night sweats Effects 0.000 description 1
- 229950011068 niraparib Drugs 0.000 description 1
- 229910052757 nitrogen Inorganic materials 0.000 description 1
- 238000003199 nucleic acid amplification method Methods 0.000 description 1
- 238000007899 nucleic acid hybridization Methods 0.000 description 1
- 238000001668 nucleic acid synthesis Methods 0.000 description 1
- 210000004940 nucleus Anatomy 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 201000008106 ocular cancer Diseases 0.000 description 1
- 201000002575 ocular melanoma Diseases 0.000 description 1
- 208000017920 oculo-auriculo-vertebral spectrum Diseases 0.000 description 1
- 238000006384 oligomerization reaction Methods 0.000 description 1
- 238000011275 oncology therapy Methods 0.000 description 1
- 208000022982 optic pathway glioma Diseases 0.000 description 1
- 238000011369 optimal treatment Methods 0.000 description 1
- 238000005457 optimization Methods 0.000 description 1
- 230000008520 organization Effects 0.000 description 1
- 201000006958 oropharynx cancer Diseases 0.000 description 1
- 230000002138 osteoinductive effect Effects 0.000 description 1
- 229940026778 other chemotherapeutics in atc Drugs 0.000 description 1
- 230000002611 ovarian Effects 0.000 description 1
- 208000021284 ovarian germ cell tumor Diseases 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 201000002530 pancreatic endocrine carcinoma Diseases 0.000 description 1
- 238000004091 panning Methods 0.000 description 1
- 229940055729 papain Drugs 0.000 description 1
- 235000019834 papain Nutrition 0.000 description 1
- 208000004019 papillary adenocarcinoma Diseases 0.000 description 1
- 201000010198 papillary carcinoma Diseases 0.000 description 1
- 210000003695 paranasal sinus Anatomy 0.000 description 1
- 239000000199 parathyroid hormone Substances 0.000 description 1
- 229960001319 parathyroid hormone Drugs 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 230000007170 pathology Effects 0.000 description 1
- 208000030940 penile carcinoma Diseases 0.000 description 1
- 201000008174 penis carcinoma Diseases 0.000 description 1
- 230000000737 periodic effect Effects 0.000 description 1
- 201000002628 peritoneum cancer Diseases 0.000 description 1
- 208000028591 pheochromocytoma Diseases 0.000 description 1
- 239000008363 phosphate buffer Substances 0.000 description 1
- 125000002467 phosphate group Chemical group [H]OP(=O)(O[H])O[*] 0.000 description 1
- 229930004090 phosphatidylinositide Natural products 0.000 description 1
- 102000020233 phosphotransferase Human genes 0.000 description 1
- 239000000906 photoactive agent Substances 0.000 description 1
- 230000004962 physiological condition Effects 0.000 description 1
- 230000001766 physiological effect Effects 0.000 description 1
- 208000024724 pineal body neoplasm Diseases 0.000 description 1
- 201000004123 pineal gland cancer Diseases 0.000 description 1
- 201000003113 pineoblastoma Diseases 0.000 description 1
- 208000010916 pituitary tumor Diseases 0.000 description 1
- 208000010626 plasma cell neoplasm Diseases 0.000 description 1
- 238000007747 plating Methods 0.000 description 1
- 229910052697 platinum Inorganic materials 0.000 description 1
- 231100000614 poison Toxicity 0.000 description 1
- 108700028325 pokeweed antiviral Proteins 0.000 description 1
- 229920000570 polyether Polymers 0.000 description 1
- 238000003752 polymerase chain reaction Methods 0.000 description 1
- 208000001061 polyostotic fibrous dysplasia Diseases 0.000 description 1
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 1
- 239000000244 polyoxyethylene sorbitan monooleate Substances 0.000 description 1
- 229920000053 polysorbate 80 Polymers 0.000 description 1
- 229940068968 polysorbate 80 Drugs 0.000 description 1
- 235000020004 porter Nutrition 0.000 description 1
- 238000002600 positron emission tomography Methods 0.000 description 1
- 230000036515 potency Effects 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 230000001855 preneoplastic effect Effects 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 208000025638 primary cutaneous T-cell non-Hodgkin lymphoma Diseases 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- 230000002250 progressing effect Effects 0.000 description 1
- 208000037821 progressive disease Diseases 0.000 description 1
- 229940097325 prolactin Drugs 0.000 description 1
- 108010087851 prorelaxin Proteins 0.000 description 1
- 150000003180 prostaglandins Chemical class 0.000 description 1
- 108010043671 prostatic acid phosphatase Proteins 0.000 description 1
- 125000006239 protecting group Chemical group 0.000 description 1
- 238000000159 protein binding assay Methods 0.000 description 1
- 238000001814 protein method Methods 0.000 description 1
- 230000002685 pulmonary effect Effects 0.000 description 1
- 150000003212 purines Chemical class 0.000 description 1
- 150000003230 pyrimidines Chemical class 0.000 description 1
- 230000002285 radioactive effect Effects 0.000 description 1
- 238000007674 radiofrequency ablation Methods 0.000 description 1
- 229940051022 radioimmunoconjugate Drugs 0.000 description 1
- 108010061338 ranpirnase Proteins 0.000 description 1
- 238000010188 recombinant method Methods 0.000 description 1
- 230000006798 recombination Effects 0.000 description 1
- 238000005215 recombination Methods 0.000 description 1
- 208000016691 refractory malignant neoplasm Diseases 0.000 description 1
- 208000015347 renal cell adenocarcinoma Diseases 0.000 description 1
- 208000030859 renal pelvis/ureter urothelial carcinoma Diseases 0.000 description 1
- 230000008263 repair mechanism Effects 0.000 description 1
- 238000002271 resection Methods 0.000 description 1
- 230000000241 respiratory effect Effects 0.000 description 1
- 108091008146 restriction endonucleases Proteins 0.000 description 1
- 238000007142 ring opening reaction Methods 0.000 description 1
- 235000002020 sage Nutrition 0.000 description 1
- 201000003804 salivary gland carcinoma Diseases 0.000 description 1
- 201000008407 sebaceous adenocarcinoma Diseases 0.000 description 1
- 208000002477 septooptic dysplasia Diseases 0.000 description 1
- 238000012163 sequencing technique Methods 0.000 description 1
- 239000013605 shuttle vector Substances 0.000 description 1
- 230000011664 signaling Effects 0.000 description 1
- 238000012868 site-directed mutagenesis technique Methods 0.000 description 1
- 210000002027 skeletal muscle Anatomy 0.000 description 1
- 201000000849 skin cancer Diseases 0.000 description 1
- 201000008261 skin carcinoma Diseases 0.000 description 1
- 201000002314 small intestine cancer Diseases 0.000 description 1
- 150000003384 small molecules Chemical class 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 229950007874 solanezumab Drugs 0.000 description 1
- 239000007909 solid dosage form Substances 0.000 description 1
- 239000000243 solution Substances 0.000 description 1
- 125000006850 spacer group Chemical group 0.000 description 1
- 210000000952 spleen Anatomy 0.000 description 1
- 206010062920 spondyloepiphyseal dysplasia Diseases 0.000 description 1
- 201000002962 spondyloepiphyseal dysplasia with congenital joint dislocations Diseases 0.000 description 1
- 230000002269 spontaneous effect Effects 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 201000000498 stomach carcinoma Diseases 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 230000008093 supporting effect Effects 0.000 description 1
- 201000008205 supratentorial primitive neuroectodermal tumor Diseases 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 201000010965 sweat gland carcinoma Diseases 0.000 description 1
- 206010042863 synovial sarcoma Diseases 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- 229950004550 talazoparib Drugs 0.000 description 1
- 231100001274 therapeutic index Toxicity 0.000 description 1
- 230000004797 therapeutic response Effects 0.000 description 1
- 210000000115 thoracic cavity Anatomy 0.000 description 1
- 201000003067 thrombocytopenia due to platelet alloimmunization Diseases 0.000 description 1
- 201000005990 thymic dysplasia Diseases 0.000 description 1
- 208000008732 thymoma Diseases 0.000 description 1
- 201000002510 thyroid cancer Diseases 0.000 description 1
- 229940034208 thyroxine Drugs 0.000 description 1
- XUIIKFGFIJCVMT-UHFFFAOYSA-N thyroxine-binding globulin Natural products IC1=CC(CC([NH3+])C([O-])=O)=CC(I)=C1OC1=CC(I)=C(O)C(I)=C1 XUIIKFGFIJCVMT-UHFFFAOYSA-N 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 238000001890 transfection Methods 0.000 description 1
- 238000003151 transfection method Methods 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 230000014616 translation Effects 0.000 description 1
- 230000001960 triggered effect Effects 0.000 description 1
- 208000029387 trophoblastic neoplasm Diseases 0.000 description 1
- 230000005747 tumor angiogenesis Effects 0.000 description 1
- 208000001072 type 2 diabetes mellitus Diseases 0.000 description 1
- 229940121358 tyrosine kinase inhibitor Drugs 0.000 description 1
- 239000005483 tyrosine kinase inhibitor Substances 0.000 description 1
- 238000002604 ultrasonography Methods 0.000 description 1
- 208000018417 undifferentiated high grade pleomorphic sarcoma of bone Diseases 0.000 description 1
- 241001430294 unidentified retrovirus Species 0.000 description 1
- 210000000626 ureter Anatomy 0.000 description 1
- 208000010570 urinary bladder carcinoma Diseases 0.000 description 1
- 208000037965 uterine sarcoma Diseases 0.000 description 1
- 229960005486 vaccine Drugs 0.000 description 1
- 206010046885 vaginal cancer Diseases 0.000 description 1
- 208000013139 vaginal neoplasm Diseases 0.000 description 1
- 125000002987 valine group Chemical group [H]N([H])C([H])(C(*)=O)C([H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 239000013598 vector Substances 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 238000012800 visualization Methods 0.000 description 1
- 230000008673 vomiting Effects 0.000 description 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 1
- 238000001262 western blot Methods 0.000 description 1
- 229910052727 yttrium Inorganic materials 0.000 description 1
- VWQVUPCCIRVNHF-UHFFFAOYSA-N yttrium atom Chemical compound [Y] VWQVUPCCIRVNHF-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/30—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants from tumour cells
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/337—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having four-membered rings, e.g. taxol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/357—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having two or more oxygen atoms in the same ring, e.g. crown ethers, guanadrel
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/4738—Quinolines; Isoquinolines ortho- or peri-condensed with heterocyclic ring systems
- A61K31/4745—Quinolines; Isoquinolines ortho- or peri-condensed with heterocyclic ring systems condensed with ring systems having nitrogen as a ring hetero atom, e.g. phenantrolines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/50—Pyridazines; Hydrogenated pyridazines
- A61K31/502—Pyridazines; Hydrogenated pyridazines ortho- or peri-condensed with carbocyclic ring systems, e.g. cinnoline, phthalazine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
- A61K31/52—Purines, e.g. adenine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
- A61K39/39533—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals
- A61K39/3955—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals against proteinaceous materials, e.g. enzymes, hormones, lymphokines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
- A61K39/39533—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals
- A61K39/39558—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals against tumor tissues, cells, antigens
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6849—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a receptor, a cell surface antigen or a cell surface determinant
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6851—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6851—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell
- A61K47/6853—Carcino-embryonic antigens
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6889—Conjugates wherein the antibody being the modifying agent and wherein the linker, binder or spacer confers particular properties to the conjugates, e.g. peptidic enzyme-labile linkers or acid-labile linkers, providing for an acid-labile immuno conjugate wherein the drug may be released from its antibody conjugated part in an acidic, e.g. tumoural or environment
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/04—Antineoplastic agents specific for metastasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2803—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2887—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against CD20
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/30—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants from tumour cells
- C07K16/3007—Carcino-embryonic Antigens
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/505—Medicinal preparations containing antigens or antibodies comprising antibodies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/545—Medicinal preparations containing antigens or antibodies characterised by the dose, timing or administration schedule
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/20—Immunoglobulins specific features characterized by taxonomic origin
- C07K2317/24—Immunoglobulins specific features characterized by taxonomic origin containing regions, domains or residues from different species, e.g. chimeric, humanized or veneered
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/73—Inducing cell death, e.g. apoptosis, necrosis or inhibition of cell proliferation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/73—Inducing cell death, e.g. apoptosis, necrosis or inhibition of cell proliferation
- C07K2317/732—Antibody-dependent cellular cytotoxicity [ADCC]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/90—Immunoglobulins specific features characterized by (pharmaco)kinetic aspects or by stability of the immunoglobulin
- C07K2317/94—Stability, e.g. half-life, pH, temperature or enzyme-resistance
Abstract
La presente invención se refiere a la terapia combinada con fármacos, tales como inhibidores de microtúbulos, inhibidores de PARP, inhibidores de la quinasa de Bruton o inhibidores de PI3K, con anticuerpos o inmunoconjugados contra HLA-DR o Trop-2. Cuando se utilizan inmunoconjugados, preferiblemente incorporan SN-38 o pro-2PDOX. El inmunoconjugado se puede administrar a una dosis de entre 1 mg/kg y 18 mg/kg, preferiblemente 4, 6, 8, 9, 10, 12, 16 o 18 mg/kg, más preferiblemente 8 o 10 mg/kg. La terapia combinada puede reducir el tamaño de los tumores sólidos, reducir o eliminar las metástasis y es eficaz para tratar cánceres resistentes a las terapias estándar, como la radioterapia, la quimioterapia o la inmunoterapia. Preferiblemente, la terapia de combinación tiene un efecto aditivo sobre la inhibición del crecimiento tumoral. Lo más preferiblemente, la terapia combinada tiene un efecto sinérgico sobre la inhibición del crecimiento tumoral. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Combinación de anticuerpos anti-hla-dr o anti-Trop-2 con inhibidores de microtúbulos, inhibidores de parp, inhibidores de la cinasa de bruton o inhibidores de la fosfoinositida 3-cinasa mejora significativamente el resultado terapéutico en el cáncer
ANTECEDENTES DE LA INVENCIÓN
CAMPO DE LA INVENCIÓN
[0001] La presente invención se refiere a un inmunoconjugado que se une a Trop-2 para su uso en un método para tratar el cáncer que expresa Trop-2; y al menos un agente terapéutico seleccionado del grupo que consiste en un inhibidor de PARP y un inhibidor de microtúbulos, para usar en un método para tratar el cáncer que expresa Trop-2. Dicha terapia de combinación es más eficaz que el inmunoconjugado solo, el fármaco solo o los efectos combinados del fármaco y el inmunoconjugado solos. Preferiblemente, la combinación presenta efectos sinérgicos. Preferiblemente, los inmunoconjugados se administran en dosis específicas y/o programas de administración específicos que optimizan su efecto terapéutico. Las dosis optimizadas y los programas de administración de conjugados de anticuerpo y fármaco (ADC) para uso terapéutico en humanos que se describen en el presente documento muestran una eficacia superior inesperada que no podría haberse predicho a partir de estudios en modelos animales, lo que permite un tratamiento eficaz de los cánceres que son resistentes a las terapias anticancerígenas estándar., incluido el irinotecán (CPT-11), el paclitaxel u otros compuestos que inducen roturas de la cadena de ADN. Sorprendentemente, la terapia de combinación con inmunoconjugados de anticuerpo-SN38 e inhibidores de microtúbulos o inhibidores de PARP muestra efectos sinérgicos inesperados. En una forma de realización particularmente preferida, un anticuerpo conjugado con SN-38 de uso es un anticuerpo anti-Trop-2, tal como IMMU-132 (sacitizumab govitecan, también conocido como hRS7-CL2A-SN-38 o IMMU-132). Más preferiblemente, los métodos son útiles en el tratamiento del cáncer de mama triple negativo (TNBC), cáncer colorrectal metastásico, SCLC o NSCLC, así como otros cánceres que expresan Trop-2.
ANTECEDENTES DE LA INVENCIÓN
[0002] Durante muchos años, el objetivo de los científicos en el campo de la terapia farmacológica específicamente dirigida ha sido utilizar anticuerpos monoclonales (MAbs) para la administración específica de agentes tóxicos a los cánceres humanos. Se han desarrollado conjugados de MAbs que se dirigen a antígenos asociados a tumores (TAA) y agentes tóxicos adecuados, pero han tenido un éxito mixto en la terapia del cáncer en humanos y prácticamente ninguna aplicación en otras enfermedades, como enfermedades infecciosas y autoinmunes. El agente tóxico suele ser un fármaco quimioterapéutico, aunque también se han conjugado radionúclidos emisores de partículas o toxinas bacterianas o vegetales con MAb, especialmente para el tratamiento del cáncer (Sharkey y Goldenberg, CA Cancer J Clin. 2006 Jul-Aug; 56(4):226-243) y, más recientemente, con radioinmunoconjugados para la terapia preclínica de ciertas enfermedades infecciosas (Dadachova y Casadevall, QJ Nucl Med Mol Imaging 2006;50(3): 193-204).
[0003] Las ventajas de usar conjugados de MAb-fármaco quimioterapéutico son que (a) el propio fármaco quimioterapéutico está estructuralmente bien definido; (b) el fármaco quimioterapéutico se une a la proteína MAb usando químicas de conjugación muy bien definidas, a menudo en sitios específicos alejados de las regiones de unión al antígeno de los MAb; (c) los conjugados de MAb-fármaco quimioterapéutico pueden fabricarse de manera más reproducible y, por lo general, con menos inmunogenicidad que los conjugados químicos que involucran MAb y toxinas bacterianas o vegetales y, como tales, son más susceptibles de desarrollo comercial y aprobación regulatoria; y (d) los conjugados MAb-fármaco quimioterapéutico son de orden de magnitud menos tóxicos desde el punto de vista sistémico que los conjugados de MAb con radionúclido, particularmente para la médula ósea sensible a la radiación.
[0004] La camptotecina (CPT) y sus derivados son una clase de potentes agentes antitumorales. El irinotecán (también denominado CPT-11) y el topotecán son análogos de CPT que son agentes terapéuticos contra el cáncer aprobados (Iyer y Ratain, Cancer Chemother. Phamacol. 42: S31-S43 (1998)). Los CPT actúan inhibiendo la enzima topoisomerasa I al estabilizar el complejo topoisomerasa I-ADN (Liu, et al. en The Camptothecins: Unfolding Their Anticancer Potential, Liehr JG, Giovanella, b C and Verschraegen (eds), NY Acad Sci., NY 922:1 -10 (2000)). Los CPT presentan problemas específicos en la preparación de conjugados. Un problema es la insolubilidad de la mayoría de los derivados de CPT en tampones acuosos. En segundo lugar, los CPT brindan desafíos específicos para la modificación estructural para conjugar macromoléculas. Por ejemplo, el propio CPT contiene solo un grupo hidroxilo terciario en el anillo-E. El grupo funcional hidroxilo en el caso de CPT debe acoplarse a un enlazador adecuado para la posterior conjugación de proteínas; y en derivados CPT potentes, como SN-38, el metabolito activo del quimioterapéutico CPT-11, y otros derivados que contienen hidroxilo C-10, como topotecán y 10-hidroxi-CPT, la presencia de un hidroxilo fenólico en la posición C-10 complica la necesaria derivatización C-20-hidroxilo. En tercer lugar, la labilidad en condiciones fisiológicas del resto ó -lactona del anillo E de las camptotecinas da como resultado una potencia antitumoral muy reducida. Por lo tanto, el protocolo de conjugación se lleva a cabo a un pH de 7 o inferior para evitar la apertura del anillo de lactona. Sin embargo, la conjugación de un CPT bifuncional que posea un grupo reactivo con amina tal como un éster activo típicamente requeriría un pH de 8 o mayor. En cuarto lugar, preferiblemente
se incorpora un resto escindible intracelularmente en el enlazador/espaciador que conecta los CPT y los anticuerpos u otros restos de unión.
[0005] El documento WO 2015/047510 A1 está relacionado con conjugaciones anti-Trop-2 anticuerpo-fármaco y sus usos.
[0006] Existe la necesidad de métodos más efectivos para preparar y administrar conjugados de anticuerpo-fármaco, como conjugados de anticuerpo-SN-38, y una terapia de combinación más efectiva con anticuerpos o inmunoconjugados y fármacos, como inhibidores de microtúbulos, inhibidores de PARP, inhibidores de quinasa de Bruton o inhibidores de PI3K.
RESUMEN DE LA INVENCIÓN
[0007] El problema que subyace a la presente invención se resuelve mediante el objeto de las reivindicaciones independientes adjuntas. Las formas de realización preferidas pueden tomarse de las reivindicaciones dependientes adjuntas.
[0008] Más específicamente, el problema que subyace a la presente invención se resuelve en un primer aspecto mediante un inmunoconjugado que se une a Trop-2 para su uso en un método para tratar el cáncer que expresa Trop-2, en el que el método comprende:
a) administrar a un sujeto humano con el cáncer el inmunoconjugado; y
b) administrar al sujeto al menos un agente terapéutico seleccionado del grupo que consiste en un inhibidor de PARP y un inhibidor de microtúbulos
en el que el inmunoconjugado comprende un anticuerpo anti-Trop-2 conjugado con SN-38.
[0009] Más específicamente, el problema que subyace a la presente invención se resuelve en un segundo aspecto mediante al menos un agente terapéutico seleccionado del grupo que consiste en un inhibidor de PARP y un inhibidor de microtúbulos, para usar en un método para tratar el cáncer que expresa Trop-2, en el que el el método comprende:
a) administrar a un sujeto humano con cáncer el inmunoconjugado; y
b) administrar al sujeto al menos un agente terapéutico seleccionado del grupo que consiste en un inhibidor de PARP y un inhibidor de microtúbulos, en el que el inmunoconjugado comprende un anticuerpo anti-Trop-2 conjugado con SN-38.
[0010] En una forma de realización del primer y segundo aspecto, la combinación de anticuerpo o inmunoconjugado y agente terapéutico tiene un efecto sinérgico en la inhibición del crecimiento tumoral.
[0011] En una forma de realización del primer y segundo aspecto, el anticuerpo anti-Trop-2 es hRS7.
[0012] En una forma de realización del primer y segundo aspecto, el inhibidor de PARP se selecciona del grupo que consiste en olaparib, talazoparib (Bm N-673), rucaparib, veliparib, CEP 9722, MK 4827, BGB-290, ABT-888, AG014699, BSI-201, CEP-8983 y 3-aminobenzamida; o el inhibidor de microtúbulos se selecciona del grupo que consiste en un alcaloide de la vinca, un taxano, un maitansinoide, una auristatina, vincristina, vinblastina, paclitaxel, mertansina, demecolcina, nocodazol, epotilona, docetaxel, disodermolida, colchicina, combrestatina, podofilotoxina, CI-980, fenilahistinas, esteganacinas, curacinas, 2-metoxiestradiol, E7010, metoxibencenosuflonamidas, vinorelbina, vinflunina, vindesina, dolastatinas, espongistatina, rizoxina, tasidotina, halicondrinas, hemiasterlinas, criptoficina 52, MMAE y mesilato de eribulina.
[0013] En una forma de realización del primer y segundo aspecto, el inhibidor de PARP es olaparib; o el inhibidor de microtúbulos es paclitaxel o mesilato de eribulina.
[0014] En una forma de realización del primer y segundo aspecto, el cáncer es un tumor sólido y el tratamiento da como resultado una reducción del tamaño del tumor de al menos un 15 %, al menos un 20 %, al menos un 30 % o al menos un 40 %.
[0015] En una forma de realización del primer y segundo aspecto, el cáncer es metastásico y el método comprende además reducir el tamaño o eliminar las metástasis.
[0016] En una forma de realización del primer y segundo aspecto, el cáncer es refractario a otras terapias pero responde a la combinación de conjugado de anticuerpo-fármaco y agente terapéutico.
[0017] En una forma de realización del primer y segundo aspecto, el paciente no ha respondido a la terapia con irinotecan, antes del tratamiento con el conjugado de anticuerpo-fármaco.
[0018] En una forma de realización del primer y segundo aspecto, el método comprende además administrar al sujeto una o más modalidades terapéuticas adicionales seleccionadas del grupo que consiste en anticuerpos no conjugados, anticuerpos radiomarcados, anticuerpos conjugados con fármacos, anticuerpos conjugados con toxinas, terapia génica, quimioterapia, péptidos terapéuticos, terapia con citocinas, oligonucleótidos, radioterapia localizada, cirugía y terapia con ARN de interferencia.
[0019] En una forma de realización del primer y segundo aspecto, la modalidad terapéutica comprende el tratamiento con un agente seleccionado del grupo que consiste en 5-fluorouracilo, afatinib, aplidina, azaribina, anastrozol, antraciclinas, axitinib, AVL-101, AVL-291, bendamustina, bleomicina, bortezomib, bosutinib, briostatina-1, busulfán, caliqueamicina, camptotecina, carboplatino, 10-hidroxicamptotecina, carmustina, celebrex, clorambucilo, cisplatino (CDDP), inhibidores de la Cox-2, irinotecán (CPT-11), SN-38, carboplatino, cladribina, camptotecanos, ciclofosfamida, crizotinib, citarabina, dacarbazina, dasatinib, dinaciclib, docetaxel, dactinomicina, daunorrubicina, doxorrubicina, 2-pirrolinodoxorrubicina (2P-DOX), cianomorfolino doxorrubicina, glucurónido de doxorrubicina, glucurónido de epirrubicina, erlotinib, estramustina, epidofilotoxina, erlotinib, entinostat, agentes de unión al receptor de estrógeno, etopósido (VP16), glucurónido de etopósido, fosfato de etopósido, exemestano, fingolimod, flavopiridol, floxuridina (FUdR), 3',5'-O-dioleoil-FudR (FUdR-dO), fludarabina, flutamida, inhibidores de la farnesil-proteína transferasa, fostamatinib, ganetespib, GDC-0834, GS-1101, gefitinib, gemcitabina, hidroxiurea, ibrutinib, idarrubicina, idelalisib, ifosfamida, imatinib, L-asparaginasa, lapatinib, lenolidamida, leucovorina, LFM- A13, lomustina, mecloretamina, melfalán, mercaptopurina, 6-mercaptopurina, metotrexato, mitoxantrona, mitramicina, mitomicina, Mitotano, Navelbina, Neratinib, Nilotinib, Nitrosurea, Olaaparib, Plicomicina, Procarbazina, Paclitaxel, PCI-32765, Postostatina, Psi-341, Raloxifeno, Semustineine, sorafenib, estreptozocina, SU11248, sunitinib, tamoxifeno, temazolomida (una forma acuosa de DTIC), transplatino, talidomida, tioguanina, tiotepa, tenipósido, topotecán, uracilo mostaza, vatalanib, vinorelbina, vinblastina, vincristina, alcaloides de la vinca y ZD1839.
[0020] En una forma de realización del primer y segundo aspecto, el cáncer se selecciona del grupo que consiste en cáncer de mama triple negativo, cáncer de mama, cáncer de ovario, cáncer de endometrio, cáncer urotelial, cáncer de útero, cáncer de pulmón de células no pequeñas, cáncer de pulmón de células pequeñas, cáncer de estómago, cáncer de vejiga urinaria, cáncer renal, cáncer de próstata y cáncer colorrectal.
[0021] Preferiblemente, la terapia de combinación según la presente invención como se define en las reivindicaciones es más eficaz que el anticuerpo o el inmunoconjugado solos, el fármaco solo o la suma de los efectos del anticuerpo o inmunoconjugado y el fármaco. Lo más preferiblemente, la combinación exhibe efectos sinérgicos en sujetos humanos.
[0022] Como se usa en este documento, la abreviatura "CPT" puede referirse a camptotecina o cualquiera de sus derivados, como SN-38, a menos que se indique expresamente lo contrario. La presente invención, tal como se define en las reivindicaciones, proporciona métodos y composiciones mejorados para preparar y administrar inmunoconjugados de anticuerpo CPT. Ejemplos de enfermedades o condiciones que pueden tratarse con los anticuerpos o inmunoconjugados incluyen, por ejemplo, cáncer o enfermedad autoinmune.
[0023] El anticuerpo usado de acuerdo con la presente invención como se define en las reivindicaciones puede ser de varios isotipos, preferiblemente IgG1, IgG2, IgG3 o IgG4 humana, más preferiblemente comprende secuencias de región constante y bisagra de IgG1 humana. El anticuerpo o fragmento del mismo puede ser un humano-ratón quimérico, un humano-primate quimérico, un humanizado (estructura humana y regiones hipervariables murinas (CDR)), o un anticuerpo completamente humano, así como variaciones de los mismos, tales como anticuerpos IgG4. (denominados "unicuerpos"), como describen van der Neut Kolfschoten et al. (Ciencia 2007; 317:1554-1557). Más preferiblemente, el anticuerpo o fragmento del mismo puede diseñarse o seleccionarse para que comprenda secuencias de regiones constantes humanas que pertenecen a alotipos específicos, lo que puede dar como resultado una inmunogenicidad reducida cuando el anticuerpo o inmunoconjugado se administra a un sujeto humano. Los alotipos preferidos para la administración incluyen un alotipo no G1m1 (nG1m1), como G1m3, G1m3,1, G1m3,2 o G1m3,1,2. Más preferiblemente, el alotipo se selecciona del grupo que consiste en los alotipos nG1m1, G1m3, nG1m1, 2 y Km3.
[0024] Los anticuerpos de uso pueden unirse a cualquier antígeno asociado a una enfermedad conocido en la técnica. Cuando el estado de la enfermedad es cáncer, por ejemplo, se conocen en la técnica muchos antígenos expresados por o asociados con células tumorales, que incluyen, entre otros, anhidrasa carbónica IX, alfa-fetoproteína (AFP), a-actinina-4, A3, antígeno específico para el anticuerpo A33, ART-4, B7, Ba 733, BAGE, antígeno BrE3, CA125, CAMEL, CAP-1, CASP-8/m,, CCL19, CCL21, CD1, CD1a, CD2, CD3, CD4, CD5, CD8, CD11A, CD14, CD15, CD16, CD18, CD19, CD20, CD21, CD22, CD23, CD25, CD29, CD30, CD32b, CD33, CD37, CD38, CD40, CD40L, CD44, CD45, CD46, CD52, CD54, CD55, CD59, CD64, CD66a-e, CD67, CD70, CD70L, CD74, CD79a, CD80, CD83, CD95, CD126, CD132, CD133, CD138, CD147, CD154, CDC27, CDK-4/m, CDKN2A, CTLA-4, CXCR4, CXCR7, CXCL12, HIF-1a, antígeno p específico de colon (CSAp), CEA (CEACAM5), CEACAM6, c-Met, DAM, EGFR, EGFRvIII, EGP-1
(Trop-2), EGP-2, ELF2-M, Ep-CAM, factor de crecimiento de fibroblastos (FGF), Flt-1, Flt-3, receptor de folato, antígeno G250, GAGE, gp100, GRO-p, HLA-DR, HM1.24, gonadotropina coriónica humana (HCG) y sus subunidades, HER2/neu, HMGB-1, factor inducible por hipoxia (HIF-1), HSP70-2M, HST-2, Ia, IGF-1R, IFN-y, IFN-a, IFN-p, IFN-A, IL-4R, IL-6R, IL-13R, IL-15R, IL-17R, IL-18R, IL-2, IL-6, IL-8, IL-12, IL-15, IL-17, IL-18, IL-23, IL-25, factor de crecimiento similar a la insulina-1 (IGF-1), IGF-1R, KS1-4, Le-Y, LDR/FUT, inhibidor de la migración de macrófagos factor (MIF), MAGE, MAGE-3, MART-1, MART-2, NY-ESO-1, TRAG-3, mCRP, MCP-1, MIP-1A, MIP-1B, MIF, MUC1, MUC2, MUC3, MUC4, MUC5ac, MUC13, MUC16, MUM-1/2, MUM-3, NCA66, NCA95, NCA90, mucina de cáncer de páncreas, receptor PD-1, receptor PD-L1, factor de crecimiento placentario, p53, PLAGL2, fosfatasa ácida prostática, PSA, PRAME, PSMA, P1GF, ILGF, ILGF-1R, IL-6, IL-25, RS5, RANTES, T101, SAGE, S100, survivin, survivin-2B, TAC, TAG-72, tenascina, receptores TRAIL, TNF-α, antígeno Tn, antígenos de Thomson-Friedenreich, antígenos de necrosis tumoral, VEGFR, fibronectina ED-B, WT-1, antígeno 17-1A, factores del complemento C3, C3a, C3b, C5a, C5, un marcador de angiogénesis, bcl-2, bcl-6, Kras, un marcador oncogénico y un producto oncogénico (véase, por ejemplo, Sensi et al., Clin Cancer Res 2006, 12:5023-32; Parmiani et al., J Immunol 2007, 178:1975-79; Novellino et al. Cancer Immunol Immunother 2005, 54:187-207). Preferiblemente, el anticuerpo se une a CEACAM5, CEACAM6, EGP-1 (Trop-2), MUC-16, AFP, MUC5a,c, CD74, CD19, CD20, CD22 o HLA-DR. Más preferiblemente, el anticuerpo se une a Trop-2 o HLA-DR.
[0025] Los ejemplos de anticuerpos que se pueden utilizar incluyen, entre otros, hR1 (anti-IGF-1R, solicitud de patente de EE. UU. número de serie 13/688,812, presentada el 29/11/12), hPAM4 (anti-mucina, patente de EE. UU.
7,282,567), hA20 (anti-CD20, patente de EE. UU. n.° 7, 151,164), hA19 (anti-CD19, patente de EE. UU. n.° 7,109,304), hIMMU31 (anti-AFP, patente de EE. UU. n.° Patente de EE. UU. n.° 7,312,318), hLL2 (anti-CD22, Patente de EE. UU. n.° 5,789,554), hMu-9 (anti-CSAp, Patente de EE. UU. n.° 7,387,772), hL243 (anti-HLA-DR, Patente de EE. UU. n.° 7,612,180), hMN-14 (anti-CEACAM5, patente de EE. UU. n.° 6.676.924), hMN-15 (anti-CEACAM6, patente de EE. UU. n.° 8.287.865), hRS7 (anti-EGP-1, patente de EE. UU. 7,238,785), hMN- 3 (anti-CEACAM6, patente de EE. UU. n.° 7.541.440), Ab124 y Ab125 (anti-CXCR4, patente de EE. UU. n.° 7.138.496). Más preferiblemente, el anticuerpo es IMMU-31 (anti-AFP), hRS7 (anti-Trop-2), hMN-14 (anti-CEACAM5), hMN-3 (anti-CEACAM6), hMN-15 (anti-CEACAM6), hLL1 (anti-CD74), hLL2 (anti-CD22), hL243 o IMMU-114 (anti-HLA-DR), hA19 (anti-CD19) o hA20 (anti-CD20). Como se usa en este documento, los términos epratuzumab y hLL2 son intercambiables, al igual que los términos veltuzumab y hA20, hL243g4P, hL243gamma4P e IMMU-114. En una forma de realización más preferida, el anticuerpo es un anticuerpo anti-Trop-2, como hRS7, o un anticuerpo anti-HLA-DR, como hL243.
[0026] Los anticuerpos alternativos de uso incluyen, entre otros, abciximab (anti-glucoproteína IIb/IIIa), alemtuzumab (anti-CD52), bevacizumab (anti-VEGF), cetuximab (anti-EGFR), gemtuzumab (anti-CD33), ibritumomab (anti-CD20), panitumumab (anti-EGFR), rituximab (anti-CD20), tositumomab (anti-CD20), trastuzumab (anti-ErbB2), lambrolizumab (anti-receptor Pd -1), atezolizumab (anti-PD -L1), MEDI4736 (anti-PD-L1), nivolumab (anti-receptor PD-1), ipilimumab (anti-CTLA-4), abagovomab (anti-CA-125), adecatumumab (anti-EpCAM), atlizumab (anti-receptor de IL-6), benralizumab (anti-CD125), obinutuzumab (GA101, anti-CD20), CC49 (anti-TAG-72), AB-PG1-XG1-026 (anti-PSMA, solicitud de patente de EE. UU. 11/ 983,372, depositado como ATCC PTA-4405 y PTA-4406), D2/B (anti-PSMA, WO 2009/130575), tocilizumab (anti-receptor de IL-6), basiliximab (anti-CD25), daclizumab (anti-CD25), efalizumab (anti-CD11a), GA101 (anti-CD20; Glycart Roche), muromonab-CD3 (anti-receptor CD3), natalizumab (anti-integrina a 4), omalizumab (anti-IgE); anticuerpos anti-TNF-α como CDP571 (Ofei et al., 2011, Diabetes 45:881-85), MTNFAI, M2TNFAI, M3TNFAI, M3TNFABI, M302B, M303 (Thermo Scientific, Rockford, IL), infliximab (Centocor, Malvern, PA), certolizumab pegol (UCB, Bruselas, Bélgica), anti-CD40L (UCB, Bruselas, Bélgica), adalimumab (Abbott, Abbott Park, IL), Benlysta (Human Genome Sciences); anticuerpos para la terapia de la enfermedad de Alzheimer tales como Alz 50 (Ksiezak-Reding et al., 1987, J Biol Chem 263:7943-47), gantenerumab, solanezumab e infliximab; anticuerpos anti-fibrina como 59D8, T2G1s, MH1; anticuerpos anti-CD38 como MOR03087 (MorphoSys AG), MOR202 (Celgene), HuMax-CD38 (Genmab) o daratumumab (Johnson & Johnson); (anticuerpos anti-VIH tales como P4/D10 (patente de EE. UU. 8.333.971), Ab 75, Ab 76, Ab 77 (Paulik et al., 1999, Biochem Pharmacol 58:1781-90), así como los anticuerpos anti-VIH descritos y vendido por Polymun (Viena, Austria), también descrito en las patentes de EE. UU.
5.831.034, patente de EE. UU. 5.911.989 y Vcelar et al., AIDS 2007; 21 (16):2161 -2170 y Joos et al., Antimicrob. Agents Chemother. 200650(5):1773-9.
[0027] De acuerdo con la presente invención como se define en las reivindicaciones, un resto de fármaco conjugado con un anticuerpo se selecciona de camptotecina (CPT) y sus análogos y derivados y es más preferiblemente SN-38. Sin embargo, otras fracciones de fármacos que pueden utilizarse incluyen taxanos (p. ej., bacatina III, taxol), auristatinas (p. ej., MMAE), caliqueamicinas, epotilonas, antraciclinas (p. ej., doxorrubicina (DOX), epirrubicina, morfolinodoxorrubicina (morfolino-DOX), cianomorfolinodoxorrubicina (cianomorfolino-DOX), 2-pirrolinodoxorrubicina (2-PDOX), una forma de profármaco de 2-PDOX (pro-2-PDOX), topotecán, etopósido, cisplatino, oxaliplatino o carboplatino; véase, por ejemplo, Priebe W (ed j, serie de simposios ACS 574, publicado por American Chemical Society, Washington Dc , 1995 (332 páginas) y Nagy y col., Proc. Natl. Acad. Sci. e E. UU. 93:2464-2469, 1996). Como alternativa, el fármaco puede ser un inhibidor de la cinasa de Bruton, como ibrutinib (PCI-32765), PCI-45292, CC-292 (AVL-292), ONO-4059, GDC-0834, LFM-A13 o RN486, o un PI3K inhibidor, como idelalisib, Wortmannin, demethoxyviridin, perifosine, PX-866, IPI-145 (duvelisib), BAY 80-6946, BEZ235, RP6530, TGR1202, SF1126, INK1117, GDC-0941, BKM120, XL147, XL765, Palomid 529, GSK1059615, ZSTK474, PWT33597, IC87114, TG100-115, CAL263, PI-103, GNE477, CUDC-907, AEZS-136 o LY294002. Preferiblemente, el anticuerpo o fragmento del
mismo se une a al menos un resto quimioterapéutico; preferiblemente de 1 a aproximadamente 5 restos de fármaco; más preferiblemente de 6 a 8 fracciones de fármaco, lo más preferiblemente de aproximadamente 6 a aproximadamente 12 fracciones de fármaco.
[0028] Un ejemplo de un derivado de CPT soluble en agua es CPT-11. Hay disponible una gran cantidad de datos clínicos sobre la farmacología de CPT-11 y su conversión in vivo en el SN-38 activo (Iyer y Ratain, Cancer Chemother Pharmacol. 42:S31-43 (1998); Mathijssen et al., Clin Cancer Res. 7: 2182-2194 (2002); Rivory, Ann NY Acad Sci.
922:205-215, 2000)). La forma activa SN-38 es aproximadamente 2 a 3 órdenes de magnitud más potente que CPT-11. En formas de realización preferidas específicas, el inmunoconjugado puede ser un hMN-14-SN-38, hMN-3-SN-38, hMN- Conjugado 15-SN-38, IMMU-31-SN-38, hRS7- SN-38, hA20-SN-38, hL243-SN-38, hLL1-SN-38 o hLL2-SN-38.
[0029] De acuerdo con la presente invención como se define en las reivindicaciones, el cáncer incluye, entre otros, linfomas no Hodgkin, leucemias linfoides crónicas y agudas de células B, linfoma de Burkitt, linfoma de Hodgkin, linfoma agudo de células B grandes, leucemia de células pilosas, leucemia mieloide aguda leucemia, leucemia mieloide crónica, leucemia linfocítica aguda, leucemia linfocítica crónica, linfomas y leucemias de células T, mieloma múltiple, macroglobulinemia de Waldenstrom, carcinomas, melanomas, sarcomas, gliomas, cánceres de huesos y piel. Los carcinomas pueden incluir carcinomas de la cavidad oral, esófago, tracto gastrointestinal, tracto pulmonar, pulmón, estómago, colon, recto, mama, ovario, próstata, útero, endometrio, cuello uterino, vejiga urinaria, páncreas, hueso, cerebro, tejido conectivo, tiroides, hígado, vesícula biliar, vejiga urinaria (urotelial), riñón, piel, sistema nervioso central y testículos.
[0030] En ciertas formas de realización de la presente invención como se define en las reivindicaciones, el inmunoconjugado puede usarse en combinación con cirugía, radioterapia, quimioterapia, inmunoterapia con anticuerpos desnudos, incluidos anticuerpos inhibidores de puntos de control, radioinmunoterapia, inmunomoduladores, vacunas y similares. Estas terapias de combinación pueden permitir que se administren dosis más bajas de cada agente terapéutico en dichas combinaciones, reduciendo así ciertos efectos secundarios graves y reduciendo potencialmente los ciclos de terapia necesarios. Cuando la toxicidad superpuesta es nula o mínima, también se pueden administrar dosis completas de cada uno.
[0031] La dosificación óptima preferida de inmunoconjugados puede incluir una dosificación de entre 1 mg/kg y 20 mg/kg, más preferiblemente de 4 a 16 mg/kg, más preferiblemente de 8 a 10 mg/kg, preferiblemente administrada semanalmente, dos veces por semana, cada dos semanas., o cada tercera semana. El programa de dosificación óptimo puede incluir ciclos de tratamiento de dos semanas consecutivas de terapia seguido de una, dos, tres o cuatro semanas de descanso, o alternando semanas de terapia y descanso, o una semana de terapia seguida de dos, tres o cuatro semanas de descanso, o tres semanas de terapia seguidas de una, dos, tres o cuatro semanas de descanso, o cuatro semanas de terapia seguidas de una, dos, tres o cuatro semanas de descanso, o cinco semanas de terapia seguidas de una, dos, tres, cuatro o cinco semanas de descanso, o administración una vez cada dos semanas, una vez cada tres semanas o una vez al mes. El tratamiento puede extenderse por cualquier número de ciclos, preferiblemente al menos 2, al menos 4, al menos 6, al menos 8, al menos 10, al menos 12, al menos 14 o al menos 16 ciclos. Las dosis de uso ejemplares pueden incluir 1 mg/kg, 2 mg/kg, 3 mg/kg, 4 mg/kg, 5 mg/kg, 6 mg/kg, 7 mg/kg, 8 mg/kg, 9 mg/kg, 10 mg/kg, 11 mg/kg, 12 mg/kg, 13 mg/kg, 14 mg/kg, 15 mg/kg, 16 mg/kg, 17 mg/kg y 18 mg/kg. La persona con conocimientos ordinarios se dará cuenta de que una variedad de factores, como la edad, la salud general, la función o el peso de órganos específicos, así como los efectos de la terapia previa en sistemas de órganos específicos (p. ej., médula ósea) se pueden considerar para seleccionar un tratamiento óptimo. dosis de inmunoconjugado, y que la dosis y/o la frecuencia de administración pueden aumentar o disminuir durante el curso de la terapia. La dosis puede repetirse según sea necesario, y se observa evidencia de reducción del tumor después de tan solo 4 a 8 dosis. Las dosis optimizadas y los programas de administración descritos en este documento muestran una eficacia superior inesperada y una toxicidad reducida en sujetos humanos, lo que no podría haberse previsto a partir de estudios en modelos animales. Sorprendentemente, la eficacia superior permite el tratamiento de tumores que previamente se encontró que eran resistentes a una o más terapias anticancerígenas estándar, incluido el compuesto original, CPT-11, del cual se deriva in vivo SN-38.
[0032] La presente invención, tal como se define en las reivindicaciones, puede incluir el uso de CT y/o PET/CT, o MRI, para medir la respuesta del tumor a intervalos regulares. También se pueden controlar los niveles en sangre de marcadores tumorales, como CEA (antígeno carcinoembrionario), CA19-9, AFP, CA 15.3 o PSA. Las dosis y/o los programas de administración se pueden ajustar según sea necesario, de acuerdo con los resultados de las imágenes y/o los niveles sanguíneos de los marcadores.
[0033] Un resultado sorprendente con la presente invención como se define en las reivindicaciones es la tolerabilidad inesperada de altas dosis de conjugado de anticuerpo-fármaco cuando se administran a pacientes, incluso con infusiones repetidas, observándose solo toxicidades de grado relativamente bajo de náuseas y vómitos, o neutropenia manejable. Preferiblemente, la incidencia de efectos secundarios de grado 3 o superior, tales como anemia, diarrea, náuseas, vómitos o neutropenia, se limita al 26% o menos de la población tratada. Más preferiblemente, la incidencia de diarrea y neutropenia de grado 3 o superior se produce en el 26% o menos de la población tratada. Otro resultado sorprendente es la falta de acumulación del conjugado anticuerpo-fármaco, a diferencia de otros productos que han
conjugado SN-38 con albúmina, PEG u otros transportadores, en tejidos no objetivo o tejidos objetivo donde el MAb no es accesible. La falta de acumulación se asocia con una mejor tolerabilidad y ausencia de toxicidad grave incluso después de dosis repetidas o aumentadas. Estos resultados sorprendentes permiten la optimización de la dosificación y el programa de administración, con eficacias inesperadamente altas y toxicidades bajas; es decir, un índice terapéutico más alto que cuando el fármaco original de SN-38, el irinotecán, se administra solo o en combinación con otros fármacos. Esto está subrayado por la advertencia de recuadro negro en la etiqueta del producto de irinotecán, requerida por la FDA, que indica la diarrea temprana y tardía altamente severa que resulta de la terapia con irinotecán. Esto no se experimenta en la misma medida y frecuencia con estos ADC que contienen SN-38. Incluso la neutropenia febril es mucho menor con los SN-38-ADC descritos en este documento que con la terapia con irinotecán.
[0034] La presente invención, tal como se define en las reivindicaciones, prevé la reducción de tumores sólidos, en individuos con cánceres previamente resistentes, del 15 % o más, preferentemente del 20 % o más, preferentemente del 30 % o más, más preferentemente del 40 % o más de tamaño (como medido sumando el diámetro más largo de las lesiones diana, según RECIST o RECIST 1.1). El experto en la materia se dará cuenta de que el tamaño del tumor se puede medir mediante una variedad de técnicas diferentes, como el volumen total del tumor, el tamaño máximo del tumor en cualquier dimensión o una combinación de medidas de tamaño en varias dimensiones. Esto puede ser con procedimientos radiológicos estándar, como tomografía computarizada, imágenes por resonancia magnética, ultrasonografía y/o tomografía por emisión de positrones. Los medios para medir el tamaño son menos importantes que observar una tendencia de disminución del tamaño del tumor con el tratamiento con anticuerpos o inmunoconjugados, que preferiblemente da como resultado la eliminación del tumor. Sin embargo, para cumplir con las pautas RECIST, se prefiere la TC o la RM en serie y se deben repetir para confirmar las mediciones.
[0035] Mientras que el anticuerpo o inmunoconjugado puede administrarse como una inyección de bolo periódica, en formas de realización alternativas de la invención como se define en las reivindicaciones, el anticuerpo o inmunoconjugado puede administrarse mediante infusión continua. Para aumentar la Cmax y extender la p K del anticuerpo o inmunoconjugado en la sangre, se puede administrar una infusión continua, por ejemplo, mediante un catéter permanente. Dichos dispositivos son conocidos en la técnica, como los catéteres HICKMAN®, BROVIAC® o PORT-A-CATH® (ver, por ejemplo, Skolnik et al., Ther Drug Monit 32:741-48, 2010) y cualquier catéter permanente conocido. puede ser usado. También se conocen en la técnica una variedad de bombas de infusión continua y se puede usar cualquier bomba de infusión conocida. El rango de dosificación para infusión continua puede estar entre 0,1 y 3,0 mg/kg por día. Más preferiblemente, estos inmunoconjugados pueden administrarse mediante infusiones intravenosas durante períodos relativamente cortos de 2 a 5 horas, más preferiblemente de 2 a 3 horas.
[0036] En formas de realización particularmente preferidas de la presente invención como se define en las reivindicaciones, los anticuerpos o inmunoconjugados y los programas de dosificación pueden ser eficaces en pacientes resistentes a las terapias estándar. Por ejemplo, se puede administrar un inmunoconjugado hRS7-SN-38 a un paciente que no ha respondido a una terapia previa con irinotecan, el agente original de SN-38. Sorprendentemente, el paciente resistente al irinotecán puede mostrar una respuesta parcial o incluso completa a hRS7-SN-38. La capacidad del inmunoconjugado para dirigirse específicamente al tejido tumoral puede superar la resistencia tumoral mediante una orientación mejorada y una administración mejorada del agente terapéutico. Un sujeto preferido específico puede ser un paciente con Trop-2 positivo de mama, ovario, cuello uterino, endometrio, pulmón, próstata, colon, recto, estómago, esófago, vejiga (urotelial), riñón, páncreas, cerebro, tiroides, epitelio o cáncer de cabeza y cuello. Preferiblemente, el cáncer es un cáncer metastásico. Más preferiblemente, el paciente ha fracasado previamente en el tratamiento con al menos una terapia estándar contra el cáncer. Lo más preferiblemente, el paciente ha fracasado previamente en la terapia con irinotecan (CPT-11), el compuesto original de SN-38. En formas de realización preferidas alternativas, el cáncer es TNBC, no TNBC, cáncer de endometrio, pulmón u ovario.
[0037] En ciertas formas de realización preferidas de la presente invención, tal como se define en las reivindicaciones, se puede usar un inmunoconjugado, como sacituzumab govitecan, en terapia de combinación con al menos un inhibidor de microtúbulos. Se conocen en la técnica varios inhibidores de microtúbulos, tales como alcaloides de la vinca (p. ej., vincristina, vinblastina), taxanos (p. ej., paclitaxel), maitansinoides (p. ej., mertansina) y auristatinas. Otros inhibidores de microtúbulos conocidos incluyen demecolcina, nocodazol, epotilona, docetaxel, discodermolida, colchicina, combrestatina, podofilotoxina, CI-980, fenilahistinas, esteganacinas, curacinas, 2-metoxiestradiol, E7010, metoxibencenosuflonamidas, vinorelbina, vinflunina, vindesina, dolastatinas, s pongistatina, rizoxina, tasidotina, halicondrinas, hemiasterlinas, criptoficina 52, MMAE y mesilato de eribulina (véase, por ejemplo, Dumontet & Jordan, 2010, Nat Rev Drug Discov 9:790-803). Cualquier inhibidor de microtúbulos conocido puede usarse en combinación con un anticuerpo o un conjugado de anticuerpo-fármaco (ADC). Preferiblemente, el inhibidor de microtúbulos es uno que exhibe efectos sinérgicos cuando se usa en combinación con un anticuerpo o ADC. Un ejemplo potente es el anticuerpo conjugado con SN-38, como sacituzumab govitecan o labetuzumab govitecan (dirigido a CEACAMS) expresado por muchos cánceres sólidos. Lo más preferiblemente, el inhibidor de microtúbulos es paclitaxel o mesilato de eribulina.
[0038] En otras formas de realización preferidas de la presente invención como se define en las reivindicaciones, el ADC se puede usar en terapia de combinación con al menos un inhibidor de PARP. Uno de estos ADC está compuesto por SN-38 conjugado con un anticuerpo dirigido a tumores, como sacituzumab govitecan o labetuzumab govitecan.
Se conocen en la técnica varios inhibidores de PARP, como olaparib, talazoparib (BMN-673), rucaparib, veliparib, niraparib, iniparib, CEP 9722, MK 4827, BGB-290, ABT-888, AG014699, BSI-201, CEP-8983 y 3-aminobenzamida (ver, por ejemplo, Rouleau et al., 2010, Nat Rev Cancer 10:293-301, Bao et al., 2015, Oncotarget [Epub antes de la impresión, 22 de septiembre de 2015]). Cualquier inhibidor de PARP conocido puede usarse en combinación con un anticuerpo o ADC, tal como, por ejemplo, un conjugado de anticuerpo SN-38 o un conjugado de anticuerpo P2PDox. Preferiblemente, el inhibidor de PARP es uno que exhibe efectos sinérgicos cuando se usa en combinación con el anticuerpo o ADC. Esto se ha validado al usar un anticuerpo conjugado con SN-38, como sacituzumab govitecan. Lo más preferiblemente, el inhibidor de PARP es olaparib o rucaparib.
[0039] De acuerdo con la presente descripción, se puede usar un anticuerpo o inmunoconjugado en combinación con un inhibidor de quinasa de Bruton o un inhibidor de PI3K. Los inhibidores de la quinasa Bruton ejemplares incluyen, pero no se limitan a, ibrutinib (PCI-32765), PCI-45292, CC-292 (AVL-292), ONO-4059, GDC-0834, LFM-A13 o RN486. Los inhibidores de PI3K ejemplares incluyen, entre otros, idelalisib, Wortmannin, demetoxiviridina, perifosina, PX-866, IPI-145 (duvelisib), BAY 80-6946, BEZ235, RP6530, TGR1202, SF1126, INK1117, GDC-0941, BKM120, XL147, XL765, Palomid 529, GSK1059615, ZSTK474, PWT33597, IC87114, TG100-115, CAL263, PI-103, GNE477, CUDC-907, AEZS-136 o LY294002. Cualquier inhibidor de quinasa Bruton o PI3K conocido en la técnica se puede utilizar en la terapia de combinación.
BREVE DESCRIPCIÓN DE LAS FIGURAS
[0040]
FIG. 1A. Sinergia de IMMU-132 más Olaparib en BRCA1/2 w.t. TNBC (MDA-MB-468). Las curvas de dosis/respuesta para cada agente solo se probaron primero para determinar los valores CI10, CI20 o CI30 del agente único después de una incubación de 96 horas. En ensayos combinados, se probó un agente (p. ej., IMMU-132) en una línea celular dada en un rango de concentraciones (es decir, curvas de dosis/respuesta). Un conjunto de pozos solo recibió IMMU-132. Otro conjunto recibió IMMU-132 como dosis/respuesta con una cantidad constante de olaparib (p. ej., CI10-concentración). Otros dos conjuntos utilizaron olaparib en concentraciones CI20 o CI30. Para cada curva de dosis/respuesta de IMMU-132, se determinó el valor Cl50 a partir de estos datos. FIG. 1A muestra los cambios en los valores de Cl50 de olaparib cuando se combina con cantidades constantes de IMMU-132 (0,6, 1,1 o 1,8 nM de equivalentes de SN-38).
FIG. 1B. Sinergia de IMMU-132 más Olaparib en BRCA1/2 w.t. TNBC (MdA-MB-468). Las curvas de dosis/respuesta para cada agente solo se probaron primero para determinar los valores CI10, CI20 o Cl50 del agente único después de una incubación de 96 horas. En ensayos combinados, se probó un agente (p. ej., IMMU-132) en una línea celular dada en un rango de concentraciones (es decir, curvas de dosis/respuesta). Un conjunto de pozos solo recibió IMMU-132. Otro conjunto recibió IMMU-132 como dosis/respuesta con una cantidad constante de olaparib (p. ej., CI10-concentración). Otros dos conjuntos utilizaron olaparib en concentraciones CI20 o CI30. Para cada curva de dosis/respuesta de IMMU-132, se determinó el valor Cl50 a partir de estos datos. FIG. 1B muestra la misma línea celular que muestra cambios en los valores de IMMU-132 Cl50 cuando se combina con cantidades constantes de olaparib (1,2 o 3 mM).
FIG. 1C. Sinergia de IMMU-132 más Olaparib en BRCA1/2 w.t. TNBC (MDA-MB-468). Las curvas de dosis/respuesta para cada agente solo se probaron primero para determinar los valores CI10, CI20 o CI30 del agente único después de una incubación de 96 horas. En ensayos de combinación, se probó un agente (p. ej., IMMU-132) en una línea celular determinada en un rango de concentraciones (es decir, curvas de dosis/respuesta). Un conjunto de pozos solo recibió IMMU-132. Otro conjunto recibió IMMU-132 como dosis/respuesta con una cantidad constante de olaparib (p. ej., CI10-concentración). Otros dos conjuntos utilizaron olaparib en concentraciones CI20 o CI30. Para cada curva de dosis/respuesta de IMMU-132, se determinó el valor Cl50 a partir de estos datos. FIG. 1C muestra un isobolograma de valores Cl50 normalizados de tres experimentos separados que prueban la interacción de IMMU-132 y olaparib, mostrando claramente una interacción sinérgica.
FIG. 2A. Inhibición del crecimiento tumoral de IMMU-132 y Olaparib combinados en TNBC: tumores defectuosos para BRCA1/2 y PTEN. Los ratones con tumor (TV~0,3 cm3) se trataron con olaparib (1 mg; ~50 mg/kg, i.p. en un programa MF; flechas rojas) o IMMU-132 (iv semanalmente, flechas negras). Se usó como control un anti-CD20 SN-38-ADC no dirigido a tumores. HCC1806 es una línea tumoral TNBC defectuosa en BRCAI/2. Olaparib solo no tuvo efectos antitumorales significativos. IMMU-132 solo inhibió significativamente el crecimiento tumoral en comparación con todos los grupos de control (P<0,0106, AUC). IMMU-132 más olaparib mejoraron aún más las respuestas antitumorales significativamente en comparación con todos los grupos (P<0,0019; AUC). Los ratones del grupo de combinación aún no han alcanzado la supervivencia media (>80,5 días), que es más de 2 y 4 veces más larga que la monoterapia con IMMU-132 o olaparib, respectivamente (P<0,0083).
FIG. 2B. Inhibición del crecimiento tumoral de IMMU-132 y Olaparib combinados en TNBC: tumores defectuosos para BRCA1/2 y PTEN. Los ratones portadores de tumores (TV ~ 0,3 cm3) se trataron con olaparib (1 mg; ~50 mg/kg, i.p. en un programa de MF; flechas claras) o IMMU-132 (iv semanalmente, flechas oscuras). Se usó como control un anti-CD20 SN-38-ADC no dirigido a tumores. En los tumores BRCA1/2 wt, MDA-MB-468 defectuosos para PTEN, IMMU-132 solo tuvo efectos antitumorales significativos en
comparación con todos los grupos de control (P<0,0098; AUC). Sin embargo, la combinación de IMMU-132 más olaparib inhibió el crecimiento tumoral significativamente mejor que IMMU-132 u olaparib solo (P = 0,004; AUC). Esto se traduce en un beneficio de supervivencia significativo en comparación con todos los demás grupos (P <0,045).
FIG. 3A. Inhibición del crecimiento tumoral de IMMU-132 combinado e inhibidores de microtúbulos en xenoinjertos tumorales TNBC humanos. Los ratones portadores de tumores (TV~0,3 cm3) se trataron con paclitaxel o mesilato de eribulina, como se indica, solos o con IMMU-132. La terapia se administró como inyecciones semanales durante dos semanas con una semana de descanso antes de repetir. Se usó un ADC anti-CD20 SN-38-ADC no dirigido a tumores como ADC de control. Los ratones portadores de tumores HCC1806 tratados con la combinación de IMMU-132 más paclitaxel inhibieron significativamente el crecimiento tumoral en comparación con IMMU-132 solo (P<0,0195, AUC).
FIG. 3B. Inhibición del crecimiento tumoral de IMMU-132 combinado e inhibidores de microtúbulos en xenoinjertos tumorales TNBC humanos. Los ratones portadores de tumores (TV~0,3 cm3) se trataron con paclitaxel o mesilato de eribulina, como se indica, solos o con IMMU-132. La terapia se administró como inyecciones semanales durante dos semanas con una semana de descanso antes de repetir. Se usó un ADC anti-CD20 SN-38-ADC no dirigido a tumores como ADC de control. Los ratones portadores de tumores HCC1806 tratados con la combinación de IMMU-132 más paclitaxel mostraron un beneficio de supervivencia significativo en comparación con todos los demás tratamientos (P<0,006, rango logarítmico).
FIG. 3C. Inhibición del crecimiento tumoral de IMMU-132 combinado e inhibidores de microtúbulos en xenoinjertos tumorales TNBC humanos. Los ratones con tumores MDA-MB-468 demostraron efectos antitumorales significativos cuando se combinó IMMU-132 (100 o 200 mg) con paclitaxel en comparación con los ratones que recibieron monoterapia (P<0,0328, AUC).
FIG. 3D. Inhibición del crecimiento tumoral de IMMU-132 combinado e inhibidores de microtúbulos en xenoinjertos tumorales TNBC humanos. El mesilato de eribulina solo inhibe significativamente el crecimiento del tumor MDA-MB-468 (P = 0,0019, AUC). Es importante destacar que la combinación de IMMU-132 más mesilato de eribulina resultó en regresiones tumorales significativas con volúmenes tumorales medios que retrocedieron a 0,026 ± 0,033 cm3 versus 0,177 ± 0,087 cm3 y 0,3446302 cm3 para los grupos de IMMU-132 y monoterapia con eribulina, respectivamente (P <0,0009 combinado frente a todos los demás grupos de terapia, AUC).
FIG. 3E. Inhibición del crecimiento tumoral de IMMU-132 combinado e inhibidores de microtúbulos en xenoinjertos tumorales TNBC humanos. IMMU-132 más mesilato de eribulina produjo efectos antitumorales significativos en comparación con todos los demás grupos de tratamiento (P <0,0432; prueba t pareada de dos colas).
FIG. 3F. Inhibición del crecimiento tumoral de IMMU-132 combinado e inhibidores de microtúbulos en xenoinjertos tumorales TNBC humanos. IMMU-132 más mesilato de eribulina proporcionó un beneficio de supervivencia significativo en comparación con todos los grupos, excepto la combinación de mesilato de eribulina más ADC de control (P <0,0284).
FIG. 4. Actividad sinérgica de IMMU-132 más Olaparib en varias líneas celulares de TNBC humano. Se muestra un resumen de los valores del índice de combinación (CI) calculados para varias líneas celulares. Los números de CI se calculan usando la siguiente fórmula: CI=Da/Dxa Db/Dxb, donde
Da=CI50-dosis de IMMU-132 cuando se usa en combinación con una cantidad constante de Olaparib Db=CI50-dosis de Olaparib cuando se usa en combinación con una cantidad constante de IMMU-132 Dxa=CI50-dosis de IMMU-132 cuando se usa solo
Dxb=CI50 -dosis de Olaparib cuando se usa solo
aIMMU-132 valor de Cl50 (nM) cuando se combina con olaparib.
bOlaparib valor de Cl50 (nM) cuando se combina con IMMU-132.
cNúmero de índice de combinación de IC (IC<1,0 es indicativo de sinergia).
FIG. 5A. Efecto de IMMU-114 sobre Cl50 de ibrutinib.
FIG. 5B. Efecto de IMMU-114 sobre Cl50 de idelalisib.
FIG. 6A. Isobolograma que demuestra un efecto aditivo de IMMU-114 combinado con un inhibidor de la quinasa de Bruton (ibrutinib) en una línea celular de LLC humana. Se incubaron células de leucemia de células B crónicas humanas (JVM-3) en placas de 96 pocillos con un inhibidor de la cinasa de Bruton (ibrutinib) en dosis que oscilaban entre 1 x 10'5 y 3,9 x 10'9 M. Un conjunto de células recibió solo el inhibidor mientras que otros conjuntos recibieron el inhibidor más una cantidad constante de IMMU-114 (0,25, 0,5, 0,75 o 1 nM). Las placas se incubaron durante 96 h. La viabilidad celular se evaluó mediante ensayo MTS y se generaron curvas de dosis/respuesta para cada condición. Los valores de Cl50 se determinaron usando Prism Graph-Pad. Los datos se normalizaron y se generó un isobolograma, lo que indica un efecto aditivo para ibrutinib cuando se combina con IMMU-114 FIG. 6B. Isobolograma que demuestra un efecto aditivo de IMMU-114 combinado con un inhibidor de PI3K (idelalisib) en una línea celular de LLC humana. Los estudios se realizaron como se describe en la leyenda de la FIG. 6A. El isobolograma indicó un efecto aditivo para idelalisib cuando se combina con IMMU-114.
FIG. 7. Terapia in vivo de ratones desnudos atímicos, portadores de carcinoma pancreático humano Capan 1, con conjugados MAb-CL2A-SN-38.
FIG. 8. Terapia in vivo de ratones desnudos atímicos, portadores de carcinoma pancreático humano BxPC3, con conjugados MAb-CL2A-SN-38.
FIG. 9. Terapia in vivo de ratones desnudos atímicos, portadores de carcinoma de colon humano LS174T, con conjugado hMN-14-CL2A-SN-38.
FIG. 10. Curvas de supervivencia de ratones tratados con hMN14-CL-SN-38 que portan enfermedad metastásica pulmonar GW-39.
FIG. 11A. Eficacia terapéutica del ADC hRS7-SN-38 en ratones portadores de xenoinjertos de tumor de pulmón de células no pequeñas humanas. Se inyectaron ratones con tumores Calu-3 (N = 5-7) con hRS7-CL2-SN-38 cada 4 días para un total de 4 inyecciones (q4dx4). Todos los ADC y controles se administraron en las cantidades indicadas (expresadas como cantidad de SN-38 por dosis; flechas largas = inyecciones de conjugado, flechas cortas = inyecciones de irinotecán).
FIG. 11B. Eficacia terapéutica del ADC hRS7-SN-38 en ratones portadores de xenoinjertos de tumor colorrectal humano. A ratones COLO 205 portadores de tumores 5) se les inyectó 8 veces (q4dx8) el ADC o cada 2 días para un total de 5 inyecciones (q2dx5) con el MTD de irinotecán. Todos los ADC y controles se administraron en las cantidades indicadas (expresadas como cantidad de SN-38 por dosis; flechas largas = inyecciones de conjugado, flechas cortas = inyecciones de irinotecán).
FIG. 11C. Eficacia terapéutica de hRS7-SN-38 ADC en ratones portadores de xenoinjertos de cáncer de páncreas humano. Se trataron ratones con tumor Capan-1 (N = 10) (N = 10) dos veces por semana durante 4 semanas con los agentes indicados. Todos los ADC y controles se administraron en las cantidades indicadas (expresadas como cantidad de SN-38 por dosis; flechas largas = inyecciones de conjugado, flechas cortas = inyecciones de irinotecán).
FIG. 11D. Eficacia terapéutica de hRS7-SN-38 ADC en ratones portadores de xenoinjertos de cáncer de páncreas humano. Se trataron ratones portadores de tumores BxPC-3 (N = 10) dos veces por semana durante 4 semanas con los agentes indicados. Todos los ADC y controles se administraron en las cantidades indicadas (expresadas como cantidad de SN-38 por dosis; flechas largas = inyecciones de conjugado, flechas cortas = inyecciones de irinotecán).
FIG. 11E. Eficacia terapéutica de hRS7-SN-38 ADC en ratones portadores de xenoinjertos de carcinoma de pulmón de células escamosas humanas. Además del ADC administrado dos veces por semana durante 4 semanas, los ratones portadores de tumores SK-MES-1 (N = 8) recibieron la MTD de CPT-11 (q2dx5). Todos los ADC y controles se administraron en las cantidades indicadas (expresadas como cantidad de SN-38 por dosis; flechas largas = inyecciones de conjugado, flechas cortas = inyecciones de irinotecán).
FIG. 12A. Eficacia comparativa de los conjugados epratuzumab (Emab)-SN-38 y veltuzumab (Vmab)-SN-38 en el modelo de Ramos subcutáneo. A ratones desnudos (N = 10 por grupo) con tumores que promediaban aproximadamente 0,35 cm3 (0,20-0,55 cm3) se les administró 0,25 mg de Emab-SN-38 dos veces por semana durante 4 semanas.
FIG. 12B. Eficacia comparativa de los conjugados epratuzumab (Emab)-SN-38 y veltuzumab (Vmab)-SN-38 en el modelo de Ramos subcutáneo. A ratones desnudos (N = 10 por grupo) con tumores que promediaban aproximadamente 0,35 cm3 (0,20-0,55 cm3) se les administró 0,25 mg de Vmab-SN-38 dos veces por semana durante 4 semanas.
FIG. 12C. Eficacia comparativa de los conjugados epratuzumab (Emab)-SN-38 y veltuzumab (Vmab)-SN-38 en el modelo de Ramos subcutáneo. A ratones desnudos (N = 10 por grupo) con tumores que promediaban aproximadamente 0,35 cm3 (0,20-0,55 cm3) se les administró 0,5 mg de Emab-SN-38 dos veces por semana durante 4 semanas.
FIG. 12D. Eficacia comparativa de los conjugados epratuzumab (Emab)-SN-38 y veltuzumab (Vmab)-SN-38 en el modelo de Ramos subcutáneo. A ratones desnudos (N = 10 por grupo) con tumores que promediaban aproximadamente 0,35 cm3 (0,20-0,55 cm3) se les administró 0,5 mg de Vmab-SN-38 dos veces por semana durante 4 semanas.
FIG. 13A. Especificidad del conjugado anti-CD22-SN-38 de Emab (línea continua) frente a un conjugado irrelevante de labetuzumab (Lmab)-SN-38 (línea discontinua) en ratones desnudos con tumores de Ramos subcutáneos. A los animales se les administraron dosis dos veces por semana de 75 μg de cada conjugado por dosis (54,5 mg/kg de SN-38, basado en un peso promedio de 22 g) por vía intraperitoneal durante 4 semanas. Supervivencia basada en el tiempo hasta la progresión (TTP) a 3,0 cm3, con tumores que comienzan en un tamaño promedio de 0,4 cm3. En cada panel se muestran los valores de P que comparan la mediana de supervivencia (mostrada) para el conjugado Emab-SN-38 con Lmab-SN-38. C, curvas de supervivencia (gris sólido) para otro grupo de animales que recibieron inyecciones intraperitoneales semanales de irinotecán (6,5 μg/dosis; equivalentes de SN-38 aproximadamente iguales a la dosis de 250 μg del conjugado Emab-SN-38).
FIG. 13b . Especificidad del conjugado anti-CD22-SN-38 de Emab (línea continua) frente a un conjugado irrelevante de labetuzumab (Lmab)-SN-38 (línea discontinua) en ratones desnudos con tumores de Ramos subcutáneos. A los animales se les administraron dosis dos veces por semana de 125 μg de cada conjugado por dosis (91 mg/kg de SN-38, basado en un peso promedio de 22 g) por vía intraperitoneal durante 4 semanas. Supervivencia basada en el tiempo hasta la progresión (TTP) a 3,0 cm3, con tumores que comienzan en un tamaño promedio de 0,4 cm3. En cada panel se muestran los valores de p que comparan la mediana de supervivencia (mostrada) para el conjugado Emab-SN-38 con Lmab-SN-38. C, curvas de supervivencia (gris sólido) para otro grupo de animales que recibieron inyecciones intraperitoneales
semanales de irinotecán (6,5 μg/dosis; equivalentes de SN-38 aproximadamente iguales a la dosis de 250 μg del conjugado Emab-SN-38).
FIG. 13C. Especificidad del conjugado anti-CD22-SN-38 de Emab (línea continua) frente a un conjugado irrelevante de labetuzumab (Lmab)-SN-38 (línea discontinua) en ratones desnudos con tumores de Ramos subcutáneos. A los animales se les administraron dosis dos veces por semana de 250 mg de cada conjugado por dosis (182 mg/kg de SN-38, basado en un peso promedio de 22 g) por vía intraperitoneal durante 4 semanas. Supervivencia basada en el tiempo hasta la progresión (TTP) a 3,0 cm3, con tumores que comienzan en un tamaño promedio de 0,4 cm3. En cada panel se muestran los valores de p que comparan la mediana de supervivencia (mostrada) para el conjugado Emab-SN-38 con Lmab-SN-38. También se muestran las curvas de supervivencia (gris sólido) para otro grupo de animales que recibieron inyecciones intraperitoneales semanales de irinotecán (6,5 μg/dosis; equivalentes de SN-38 aproximadamente igual a la dosis de 250 μg del conjugado Emab-SN-38).
FIG. 14. Antecedentes de tratamiento previo del paciente, antes de administrar IMMU-130 (labetuzumab-SN-38). El tratamiento previo incluyó colectomía/hepatectomía (lóbulo parcial) por CCR en estadio IV, terapia de ablación por radiofrecuencia de metástasis hepáticas, resección en cuña de metástasis pulmonares y quimioterapia con irinotecán/oxaliplatino, Folfirinox, Folfirinox bevacizumab, bevacizumab 5-FU/leucovorina, FolFiri, Folfiri cetuximab y cetuximab solo. El paciente recibió dosis de 16 mg/kg de IMMU-130 por infusión intravenosa lenta cada dos semanas para un total de 17 dosis de tratamiento.
DESCRIPCIÓN DETALLADA DE LA INVENCIÓN
Definiciones
[0041] En la descripción que sigue, se utilizan una serie de términos y se proporcionan las siguientes definiciones para facilitar la comprensión del objeto reivindicado. Los términos que no están expresamente definidos en este documento se utilizan de acuerdo con sus significados simples y ordinarios.
[0042] A menos que se especifique lo contrario, un o una significa "uno o más".
[0043] El término aproximadamente se usa aquí para significar más o menos el diez por ciento (10%) de un valor. Por ejemplo, "alrededor de 100" se refiere a cualquier número entre 90 y 110.
[0044] Un anticuerpo, como se usa en este documento, se refiere a una molécula de inmunoglobulina de longitud completa (es decir, de origen natural o formada por procesos de recombinación de fragmentos de genes de inmunoglobulina normales) (por ejemplo, un anticuerpo IgG) o una porción de unión a antígeno de una molécula de inmunoglobulina, tal como un fragmento de anticuerpo. Un anticuerpo o fragmento de anticuerpo puede conjugarse o derivatizarse de otro modo dentro del alcance del objeto reivindicado. Dichos anticuerpos incluyen, entre otros, IgG1, IgG2, IgG3, IgG4 (y subformas de IgG4), así como isotipos de IgA. Como se usa a continuación, la abreviatura "MAb" se puede usar indistintamente para referirse a un anticuerpo, fragmento de anticuerpo, anticuerpo monoclonal o anticuerpo multiespecífico.
[0045] Un fragmento de anticuerpo es una porción de un anticuerpo como F(ab')2 , F(ab)2 , Fab', Fab, Fv, scFv (Fv de cadena sencilla), anticuerpos de dominio único (DAB o VHH) y similares. incluyendo las medias moléculas de IgG4 citadas anteriormente (van der Neut Kolfschoten et al. (Science 2007; 317 (14 de septiembre): 1554-1557). Independientemente de la estructura, un fragmento de anticuerpo de uso se une con el mismo antígeno que es reconocido por el anticuerpo intacto. El término "fragmento de anticuerpo" también incluye proteínas sintéticas o modificadas genéticamente que actúan como un anticuerpo uniéndose a un antígeno específico para formar un complejo. Por ejemplo, los fragmentos de anticuerpos incluyen fragmentos aislados que consisten en regiones variables, como el Fragmentos "Fv" que consisten en las regiones variables de las cadenas pesada y ligera y moléculas polipeptídicas de cadena sencilla recombinantes en las que las regiones variables ligeras y pesadas están conectadas por un conector peptídico ("proteínas scFv"). Los fragmentos pueden construirse de diferentes maneras para producen formas de unión multivalentes y/o multiespecíficas.
[0046] Un anticuerpo desnudo es generalmente un anticuerpo completo que no está conjugado con un agente terapéutico. Un anticuerpo desnudo puede mostrar efectos terapéuticos y/o citotóxicos, por ejemplo, mediante funciones dependientes de Fc, como la fijación del complemento (CDC) y ADCc (citotoxicidad celular dependiente de anticuerpos). Sin embargo, otros mecanismos, como la apoptosis, la antiangiogénesis, la actividad antimetastásica, la actividad antiadhesiva, la inhibición de la adhesión heterotípica u homotípica y la interferencia en las vías de señalización, también pueden proporcionar un efecto terapéutico. Los anticuerpos desnudos incluyen anticuerpos policlonales y monoclonales, anticuerpos naturales o recombinantes, tales como anticuerpos quiméricos, humanizados o humanos y fragmentos de los mismos. En algunos casos, un "anticuerpo desnudo" también puede referirse a un fragmento de anticuerpo "desnudo". Tal como se define en el presente documento, "desnudo" es sinónimo de "no conjugado" y significa no unido o conjugado con un agente terapéutico.
[0047] Un anticuerpo quimérico es una proteína recombinante que contiene los dominios variables de las cadenas pesada y ligera del anticuerpo, incluidas las regiones determinantes de complementariedad (CDR) de un anticuerpo derivado de una especie, preferiblemente un anticuerpo de roedor, más preferiblemente un anticuerpo murino, mientras que los dominios constantes de la molécula de anticuerpo se derivan de los de un anticuerpo humano. Para aplicaciones veterinarias, los dominios constantes del anticuerpo quimérico pueden derivarse del de otras especies, como primates, gatos o perros.
[0048] Un anticuerpo humanizado es una proteína recombinante en la que las CDR de un anticuerpo de una especie; por ejemplo, un anticuerpo murino, se transfieren desde las cadenas variables pesadas y ligeras del anticuerpo murino a los dominios variables pesados y ligeros humanos (regiones marco). Los dominios constantes de la molécula de anticuerpo se derivan de los de un anticuerpo humano. En algunos casos, los residuos específicos de la región estructural del anticuerpo humanizado, en particular aquellos que están en contacto o cerca de las secuencias CDR, pueden modificarse, por ejemplo, reemplazarse con los residuos correspondientes del murino, roedor, primate subhumano u otro original. anticuerpo.
[0049] Un anticuerpo humano es un anticuerpo obtenido, por ejemplo, de ratones transgénicos que han sido "diseñados" para producir anticuerpos humanos en respuesta a una exposición antigénica. En esta técnica, los elementos de los loci de la cadena pesada y ligera humana se introducen en cepas de ratones derivadas de líneas de células madre embrionarias que contienen interrupciones específicas de los loci endógenos de la cadena pesada y la cadena ligera. Los ratones transgénicos pueden sintetizar anticuerpos humanos específicos para varios antígenos, y los ratones pueden usarse para producir hibridomas secretores de anticuerpos humanos. Los métodos para obtener anticuerpos humanos a partir de ratones transgénicos se describen en Green et al., Nature Genet. 7:13 (1994), Lonberg y col., Nature 368:856 (1994), y Taylor y col., Int. inmune 6:579 (1994). También se puede construir un anticuerpo totalmente humano mediante métodos de transfección genética o cromosómica, así como tecnología de presentación en fagos, todos los cuales son conocidos en la técnica. Véase, por ejemplo, McCafferty et al., Nature 348:552-553 (1990) para la producción de anticuerpos humanos y fragmentos de los mismos in vitro, a partir de repertorios de genes de dominio variable de inmunoglobulina de donantes no inmunizados. En esta técnica, los genes de dominio variable de anticuerpos humanos se clonan en marco en un gen de proteína de cubierta mayor o menor de un bacteriófago filamentoso y se muestran como fragmentos de anticuerpos funcionales en la superficie de la partícula de fago. Debido a que la partícula filamentosa contiene una copia de ADN monocatenario del genoma del fago, las selecciones basadas en las propiedades funcionales del anticuerpo también dan como resultado la selección del gen que codifica el anticuerpo que exhibe esas propiedades. De esta forma, el fago imita algunas de las propiedades de la célula B. La presentación de fagos se puede realizar en una variedad de formatos, para su revisión, véase, por ejemplo, Johnson y Chiswell, Current Opinion in Structural Biology 3:5564-571 (1993). Los anticuerpos humanos también pueden ser generados por células B activadas in vitro. Véanse las patentes EE. UU. números 5.567.610 y 5.229.275.
[0050] Un agente terapéutico es un átomo, molécula o compuesto que es útil en el tratamiento de una enfermedad. Los ejemplos de agentes terapéuticos incluyen, entre otros, anticuerpos, fragmentos de anticuerpos, inmunoconjugados, fármacos, agentes citotóxicos, agentes proapopoptóticos, toxinas, nucleasas (incluidas ADNasas y ARNasas), hormonas, inmunomoduladores, quelantes, compuestos de boro, agentes fotoactivos. o colorantes, radionúclidos, oligonucleótidos, ARN de interferencia, siARN, ARNi, agentes antiangiogénicos, agentes quimioterapéuticos, citocinas, quimiocinas, profármacos, enzimas, proteínas de unión o péptidos o combinaciones de los mismos.
[0051] Un inmunoconjugado es un anticuerpo, fragmento de anticuerpo de unión a antígeno, complejo de anticuerpo o proteína de fusión de anticuerpo que se conjuga con al menos un agente terapéutico. La conjugación puede ser covalente o no covalente. Preferiblemente, la conjugación es covalente.
[0052] Tal como se usa en el presente documento, el término proteína de fusión de anticuerpos es una molécula de unión a antígeno producida de forma recombinante en la que uno o más anticuerpos naturales, anticuerpos de cadena sencilla o fragmentos de anticuerpos se unen a otro resto, como una proteína o un péptido, una toxina, una citoquina, una hormona, etc. En ciertas formas de realización preferidas, la proteína de fusión puede comprender dos o más anticuerpos iguales o diferentes, fragmentos de anticuerpos o anticuerpos monocatenarios fusionados, que pueden unirse al mismo epítopo, epítopos diferentes en el mismo antígeno, o diferentes antígenos.
[0053] Un inmunomodulador es un agente terapéutico que, cuando está presente, altera, suprime o estimula el sistema inmunitario del organismo. Normalmente, un inmunomodulador de uso estimula las células inmunitarias para que proliferen o se activen en una cascada de respuesta inmunitaria, como macrófagos, células dendríticas, células B y/o células T. Sin embargo, en algunos casos, un inmunomodulador puede suprimir la proliferación o activación de células inmunitarias. Un ejemplo de un inmunomodulador como se describe en este documento es una citocina, que es una pequeña proteína soluble de aproximadamente 5-20 kDa que es liberada por una población celular (p. ej., linfocitos T cebados) al entrar en contacto con antígenos específicos, y que actúa como un mediador intercelular entre las células. Como comprenderá el experto en la materia, los ejemplos de citocinas incluyen linfocinas, monocinas, interleucinas y varias moléculas de señalización relacionadas, como el factor de necrosis tumoral (TNF) y los
interferones. Las quimiocinas son un subconjunto de las citocinas. Ciertas interleucinas e interferones son ejemplos de citocinas que estimulan la proliferación de células T u otras células inmunitarias. Los interferones ejemplares incluyen interferón-a, interferón-p, interferón-y e interferón-A.
[0054] CPT es una abreviatura de camptotecina, y como se usa en la presente solicitud, CPT representa la propia camptotecina o un análogo o derivado de la camptotecina, tal como SN-38. Las estructuras de la camptotecina y algunos de sus análogos, con la numeración indicada y los anillos etiquetados con las letras AE, se dan en la fórmula 1 en el Gráfico 1 a continuación.
Gráfico 1
[0055]

CPT: R1 = R2 = R3 = H
10-Hidroxi-CPT: R1 = OH; R2 = R3 = H
CPT-11: R1 =

R2 = etilo; R3 = H
SN-38: R1 = OH; R2 = etilo; R3 = H
Topotecan: R1 = OH; R2 = H; R3 = CH2-N(CH3)2
Técnicas generales de anticuerpos
[0056] Las técnicas para preparar anticuerpos monoclonales contra prácticamente cualquier antígeno diana son bien conocidas en la técnica. Véase, por ejemplo, Kohler y Milstein, Nature 256: 495 (1975), y Coligan et al. (eds.), CUTTENT PROTOCOLS IN IMMUNOLOGY, VOL. 1, páginas 2.5.1-2.6.7 (John Wiley & Sons 1991). El experto en la materia se dará cuenta de que, cuando se van a tratar sujetos humanos, los anticuerpos se unen preferentemente a antígenos humanos. Brevemente, los anticuerpos monoclonales se pueden obtener inyectando ratones con una composición que comprende un antígeno, extirpando el bazo para obtener linfocitos B, fusionando los linfocitos B con células de mieloma para producir hibridomas, clonando los hibridomas, seleccionando clones positivos que producen anticuerpos contra el antígeno, cultivando los clones que producen anticuerpos contra el antígeno y aislando los anticuerpos de los cultivos de hibridomas.
[0057] Los MAbs se pueden aislar y purificar a partir de cultivos de hibridomas mediante una variedad de técnicas bien establecidas. Dichas técnicas de aislamiento incluyen cromatografía de afinidad con Proteína A o Proteína G Sepharose, cromatografía de exclusión por tamaño y cromatografía de intercambio iónico. Ver, por ejemplo, Coligan en las páginas 2.7.1-2.7.12 y páginas 2.9.1-2.9.3. También, véase Baines et al., “Purification of Immunoglobulin G (IgG)”, en METHODS IN MOLECULAR BIOLOGY, VOL. 10, páginas 79-104 (The Humana Press, Inc. 1992).
[0058] Después de la generación inicial de anticuerpos contra el inmunógeno, los anticuerpos pueden secuenciarse y prepararse posteriormente mediante técnicas recombinantes. La humanización y quimerización de anticuerpos murinos y fragmentos de anticuerpos son bien conocidas por los expertos en la técnica, como se analiza a continuación.
[0059] El experto en la materia se dará cuenta de que, según la invención, tal como se define en las reivindicaciones, se puede utilizar una amplia variedad de anticuerpos conocidos en la técnica. Los anticuerpos de uso pueden obtenerse comercialmente de una amplia variedad de fuentes conocidas. Por ejemplo, una variedad de líneas de hibridomas secretores de anticuerpos están disponibles en la Colección Americana de Cultivos Tipo (ATCC, Manassas, VA). Se ha depositado en la ATCC una gran cantidad de anticuerpos contra varios objetivos de enfermedades, incluidos, entre otros, antígenos asociados a tumores, y/o se han publicado secuencias de región variable y están disponibles para su uso en los métodos y composiciones reivindicados. Véanse, por ejemplo, las
patentes EE. UU. números 7.312.318; 7.282.567; 7.151.164; 7.074.403; 7.060.802; 7.056.509; 7.049.060; 7.045.132 7.041.803 7.041.802 7.041.293 7.038.018 7.037.498 7.012.133 7.001.598 6.998.468; 6.994.976 6.994.852 6.989.241 6.974.863 6.965.018 6.964.854 6.962.981 6.962.813 6.956.107 6.951.924; 6.949.244 6.946.129 6.943.020; 6.939.547 6.921.645 6.921.645 6.921.533 6.919.433 6.919.078 6.916.475; 6.905.681 6.899.879 6.893.625; 6.887.468 6.887.466 6.884.594 6.881.405 6.878.812 6.875.580 6.872.568; 6.867.006 6.864.062 6.861.511 6.861.227 6.861.226 6.838.282 6.835.549 6.835.370 6.824.780 6.824.778; 6.812.206 6.793.924 6.783.758; 6.770.450 6.767.711 6.764.688 6.764.681 6.764.679 6.743.898 6.733.981; 6.730.307 6.720.155 6.716.966 6.709.653 6.693.176 6.692.908 6.689.607 6.689.362 6.689.355 6.682.737; 6.682.736 6.682.734 6.673.344; 6.653.104 6.652.852 6.635.482 6.630.144 6.610.833 6.610.294 6.605.441; 6.605.279 6.596.852 6.592.868 6.576.745 6.572; 856; 6.566.076 6.562.618; 6.545.130; 6.544.749; 6.534.058; 6.528.625 6.528.269 6.521.227 6.518.404 6.511.665 6.491.915 6.488.930 6.482.598 6.482.408 6.479.247; 6.468.531 6.468.529 6.465.173 6.461.823 6.458.356 6.455.044 6.455.040 6.451.310 6.444.206 6.441.143; 6.432.404 6.432.402 6.419.928 6.413.726 6.406.694 6.403.770 6.403.091 6.395.276 6.395.274 6.387.350; 6.383.759 6.383.484 6.376.654 6.372.215 6.359.126 6.355.481 6.355.444 6.355.245 6.355.244 6.346.246; 6.344.198 6.340.571 6.340.459 6.331.175 6.306.393 6.254.868 6.187.287 6.183.744 6.129.914 6.120.767; 6.096.289 6.077.499 5.922.302 5.874.540 5.814.440 5.798.229 5.789.554 5.776.456 5.736.119 5.716.595; 5.677.136 5.587.459 5.443.953 5.525.338. Estos son solo ejemplos y se conocen en la técnica una amplia variedad de otros anticuerpos y sus hibridomas. El experto en la materia se dará cuenta de que las secuencias de anticuerpos o los hibridomas secretores de anticuerpos contra casi cualquier antígeno asociado a una enfermedad pueden obtenerse mediante una simple búsqueda en las bases de datos de ATCC, NCBI y/o USPTO para anticuerpos contra un objetivo de interés asociado a una enfermedad seleccionado. Los dominios de unión a antígeno de los anticuerpos clonados pueden amplificarse, escindirse, ligarse en un vector de expresión, transfectarse en una célula huésped adaptada y usarse para la producción de proteínas, usando técnicas estándar bien conocidas en la técnica. Los anticuerpos aislados pueden conjugarse con agentes terapéuticos que inducen roturas de cadenas de ADN, como camptotecinas o antraciclinas, utilizando las técnicas descritas en el presente documento.
Anticuerpos quiméricos y humanizados
[0060] Un anticuerpo quimérico es una proteína recombinante en la que las regiones variables de un anticuerpo humano han sido reemplazadas por las regiones variables de, por ejemplo, un anticuerpo de ratón, incluidas las regiones determinantes de complementariedad (CDR) del anticuerpo de ratón. Los anticuerpos quiméricos muestran una inmunogenicidad disminuida y una mayor estabilidad cuando se administran a un sujeto. Los métodos para construir anticuerpos quiméricos son bien conocidos en la técnica (p. ej., Leung et al., 1994, Hybridoma 13:469).
[0061] Un anticuerpo monoclonal quimérico puede humanizarse transfiriendo las CDR de ratón de las cadenas variables pesadas y ligeras de la inmunoglobulina de ratón a los dominios variables correspondientes de un anticuerpo humano. Las regiones marco de ratón (FR) en el anticuerpo monoclonal quimérico también se reemplazan con secuencias FR humanas. Para preservar la estabilidad y la especificidad antigénica del monoclonal humanizado, uno o más residuos FR humanos pueden ser reemplazados por los residuos homólogos de ratón. Pueden usarse anticuerpos monoclonales humanizados para el tratamiento terapéutico de sujetos. Las técnicas para la producción de anticuerpos monoclonales humanizados son bien conocidas en la técnica. (Ver, por ejemplo, Jones et al., 1986, Nature, 321:522; Riechmann et al., Nature, 1988, 332:323; Verhoeyen et al., 1988, Science, 239:1534; Carter et al., 1992, Proc. Nat'l Acad. Sci. EE. UU., 89:4285; Sandhu, Crit. Rev. Biotech., 1992, 12:437; Tempest et al., 1991, Biotechnology 9:266; Singer et al., J. Immun., 1993, 150:2844.)
[0062] Otras formas de realización de la presente invención como se define en las reivindicaciones pueden referirse a anticuerpos de primates no humanos. Pueden encontrarse técnicas generales para generar anticuerpos terapéuticamente útiles en babuinos, por ejemplo, en Goldenberg et al., WO 91/11465 (1991), y en Losman et al., Int. J. Cancer 46: 310 (1990). En otra forma de realización, un anticuerpo puede ser un anticuerpo monoclonal humano. Dichos anticuerpos pueden obtenerse de ratones transgénicos que han sido diseñados para producir anticuerpos humanos específicos en respuesta a la exposición antigénica, como se analiza a continuación.
Anticuerpos humanos
[0063] Los métodos para producir anticuerpos completamente humanos utilizando enfoques combinatorios o animales transgénicos transformados con loci de inmunoglobulina humana son conocidos en la técnica (p. ej., Mancini et al., 2004, New Microbiol. 27:315-28; Conrad and Scheller, 2005, Comb. Chem. High Throughput Screen. 8:117-26; Brekke and Loset, 2003, Curr. Opin. Phamacol. 3:544-50). Se espera que dichos anticuerpos totalmente humanos muestren incluso menos efectos secundarios que los anticuerpos quiméricos o humanizados y que funcionen in vivo como anticuerpos humanos esencialmente endógenos. En ciertas formas de realización de la presente invención, como se define en las reivindicaciones, se pueden utilizar anticuerpos humanos producidos por dichas técnicas.
[0064] En una alternativa, la técnica de presentación en fagos puede usarse para generar anticuerpos humanos (p. ej., Dantas-Barbosa et al., 2005, Genet. Mol. Res. 4:126-40). Los anticuerpos humanos pueden generarse a partir de seres humanos normales o de seres humanos que presenten un estado de enfermedad particular, como el cáncer
(Dantas-Barbosa et al., 2005). La ventaja de construir anticuerpos humanos a partir de un individuo enfermo es que el repertorio de anticuerpos circulantes puede estar sesgado hacia anticuerpos contra antígenos asociados a enfermedades.
[0065] En un ejemplo no limitativo de esta metodología, Dantas-Barbosa et al. (2005) construyeron una biblioteca de presentación en fagos de fragmentos de anticuerpos Fab humanos de pacientes con osteosarcoma. En general, el ARN total se obtuvo de los linfocitos sanguíneos circulantes (Id.) Los Fab recombinantes se clonaron de los repertorios de anticuerpos de cadena p, y y k y se insertaron en una biblioteca de presentación de fagos (Id.) Los ARN se convirtieron en ADNc y se usaron para producir ADNc de Fab bibliotecas que usan cebadores específicos contra las secuencias de inmunoglobulina de cadena pesada y ligera (Marks et al., 1991, J. Mol. Biol. 222:581-97). La construcción de la biblioteca se realizó de acuerdo con Andris-Widhopf et al. (2000, en: Phage Display Laboratory Manual, Barbas et al. (eds), 1a edición, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY págs. 9.1 a 9.22). Los fragmentos Fab finales se digirieron con endonucleasas de restricción y se insertaron en el genoma del bacteriófago para crear la biblioteca de presentación de fagos. Dichas bibliotecas pueden examinarse mediante métodos estándar de presentación en fagos. El experto en la materia se dará cuenta de que esta técnica es solo un ejemplo y que puede utilizarse cualquier método conocido para producir y examinar anticuerpos humanos o fragmentos de anticuerpos mediante expresión en fagos.
[0066] En otra alternativa, los animales transgénicos que han sido manipulados genéticamente para producir anticuerpos humanos pueden usarse para generar anticuerpos contra esencialmente cualquier objetivo inmunogénico, usando protocolos de inmunización estándar como se discutió anteriormente. Los métodos para obtener anticuerpos humanos a partir de ratones transgénicos se describen en Green et al., Nature Genet. 7:13 (1994), Lonberg y col., Nature 368:856 (1994), y Taylor y col., Int. inmune 6:579 (1994). Un ejemplo no limitativo de dicho sistema es el XENOMOUSE® (p. ej., Green et al., 1999, J. Immunol. Methods 231:11-23) de Abgenix (Fremont, CA). En XENOMOUSE® y animales similares, los genes de anticuerpos de ratón han sido desactivados y reemplazados por genes de anticuerpos humanos funcionales, mientras que el resto del sistema inmunitario del ratón permanece intacto.
[0067] El XENOMOUSE® se transformó con YAC (cromosomas artificiales de levadura) configurados en la línea germinal que contenían porciones de los loci kappa de IgH e Ig humanos, incluida la mayoría de las secuencias de la región variable, junto con genes accesorios y secuencias reguladoras. El repertorio de regiones variables humanas puede usarse para generar células B productoras de anticuerpos, que pueden procesarse en hibridomas mediante técnicas conocidas. Un XENOMOUSE® inmunizado con un antígeno diana producirá anticuerpos humanos mediante la respuesta inmunitaria normal, que pueden ser recolectados y/o producidos mediante técnicas estándar discutidas anteriormente. Hay disponible una variedad de cepas de XENOMo Us E®, cada una de las cuales es capaz de producir una clase diferente de anticuerpo. Se ha demostrado que los anticuerpos humanos producidos transgénicamente tienen potencial terapéutico, al tiempo que conservan las propiedades farmacocinéticas de los anticuerpos humanos normales (Green et al., 1999). El experto en la materia se dará cuenta de que las composiciones y los métodos reivindicados no se limitan al uso del sistema XENOMOUSE® sino que pueden utilizar cualquier animal transgénico que haya sido manipulado genéticamente para producir anticuerpos humanos.
Producción de fragmentos de anticuerpos
[0068] Algunas formas de realización de la invención, como se define en las reivindicaciones, pueden referirse a fragmentos de anticuerpos. Dichos fragmentos de anticuerpos pueden obtenerse, por ejemplo, mediante digestión con pepsina o papaína de anticuerpos completos mediante métodos convencionales. Por ejemplo, se pueden producir fragmentos de anticuerpos mediante la escisión enzimática de anticuerpos con pepsina para proporcionar un fragmento 5S denominado F(ab')2. Este fragmento puede escindirse adicionalmente utilizando un agente reductor de tiol y, opcionalmente, un grupo de bloqueo para los grupos sulfhidrilo resultantes de la escisión de los enlaces disulfuro, para producir fragmentos monovalentes 3,5s Fab'. Alternativamente, una escisión enzimática usando pepsina produce dos fragmentos Fab monovalentes y un fragmento Fc. Los métodos ejemplares para producir fragmentos de anticuerpos se describen en las patentes de EE. UU. n.° 4.036.945; Patente de EE. UU. n.° 4.331.647; Nisonoff et al., 1960, Arch. Biochem. Biophys., 89:230; Porter, 1959, Biochem. J., 73:119; Edelman y col., 1967, METHODS IN ENZYMOLOGY, página 422 (Academic Press), y Coligan y col. (eds.), 1991, CURRENT PROTOCOLS IN IMMUNOLOGY, (John Wiley & Sons).
[0069] También se pueden usar otros métodos de escisión de anticuerpos, como la separación de cadenas pesadas para formar fragmentos monovalentes de cadena ligera-pesada, escisión adicional de fragmentos u otras técnicas enzimáticas, químicas o genéticas, siempre que los fragmentos se unan al antígeno que se reconoce. por el anticuerpo intacto. Por ejemplo, los fragmentos Fv comprenden una asociación de cadenas Vh y Vl. Esta asociación puede ser no covalente, como se describe en Inbar et al., 1972, Proc. Nat'l Acad. Sci. EE. UU., 69:2659. Alternativamente, las cadenas variables pueden estar unidas por un enlace disulfuro intermolecular o entrecruzadas por productos químicos como el glutaraldehído. Véase Sandhu, 1992, Crit. Rev. Biotech., 12:437.
[0070] Preferiblemente, los fragmentos Fv comprenden cadenas Vh y Vl conectadas por un conector peptídico. Estas proteínas de unión a antígeno de cadena sencilla (scFv) se preparan mediante la construcción de un gen estructural
que comprende secuencias de ADN que codifican los dominios Vh y Vl, conectados por una secuencia conectora de oligonucleótidos. El gen estructural se inserta en un vector de expresión que posteriormente se introduce en una célula huésped, como E. coli. Las células huésped recombinantes sintetizan una sola cadena polipeptídica con un péptido enlazador que une los dos dominios V. Los métodos para producir scFv son bien conocidos en la técnica. Véase Whitlow et al., 1991, Methods: A Companion to Methods in Enzymology 2:97; Bird y col., 1988, Science, 242:423; Patente de EE. UU. n.° 4.946.778; Pack et al., 1993, Bio/Technology, 11:1271, y Sandhu, 1992, Crit. Rev. Biotech., 12:437.
[0071] Otra forma de fragmento de anticuerpo es un anticuerpo de dominio único (dAb), a veces denominado anticuerpo de cadena única. Las técnicas para producir anticuerpos de dominio único son bien conocidas en la técnica (ver, por ejemplo, Cossins et al., Protein Expression and Purification, 2007, 51:253-59; Shuntao et al., Molec Immunol 2006, 43:1912-19 Tanha y col., J. Biol. Chem. 2001,276:24774-780). Otros tipos de fragmentos de anticuerpos pueden comprender una o más regiones determinantes de complementariedad (CDR). Los péptidos CDR ("unidades mínimas de reconocimiento") pueden obtenerse mediante la construcción de genes que codifican la CDR de un anticuerpo de interés. Dichos genes se preparan, por ejemplo, utilizando la reacción en cadena de la polimerasa para sintetizar la región variable a partir del ARN de las células productoras de anticuerpos. Véase Larrick et al., 1991, Methods: A Companion to Methods in Enzymology 2:106; Ritter et al. (eds.), 1995, MONOCLONAL ANTIBODIES: PRODUCTION, ENGINEERING AND CLINICAL APPLICATION, páginas 166-179 (Cambridge University Press); Birch et al., (eds.), 1995, MONOCLONAL ANTIBODIES: PRINCIPLES AND APPLICATIONS, páginas 137-185 (Wiley-Liss, Inc.).
Variaciones de anticuerpos
[0072] En ciertas formas de realización, las secuencias de anticuerpos, como las porciones Fc de los anticuerpos, se pueden variar para optimizar las características fisiológicas de los conjugados, como la vida media en suero. Los métodos de sustitución de secuencias de aminoácidos en proteínas son ampliamente conocidos en la técnica, como la mutagénesis dirigida al sitio (p. ej., Sambrook et al., Molecular Cloning, A laboratory manual, 2a ed., 1989). En formas de realización preferidas, la variación puede implicar la adición o eliminación de uno o más sitios de glicosilación en la secuencia Fc (p. ej., Patente de EE. UU. n.° 6.254.868). En otras formas de realización preferidas, se pueden realizar sustituciones de aminoácidos específicos en la secuencia Fc (p. ej., Hornick et al., 2000, J Nucl Med 41:355-62; Hinton et al., 2006, J Immunol 176:346-56; Petkova et al., 2006, Int Immunol 18:1759-69, patente de EE. UU. n.° 7.217.797).
Antígenos diana y anticuerpos ejemplares
[0073] En una forma de realización preferida de la invención como se define en las reivindicaciones, se usan anticuerpos que reconocen y/o se unen a antígenos humanos que se expresan en niveles elevados en las células diana y que se expresan predominantemente o exclusivamente en células enfermas frente a tejidos normales. Más preferiblemente, los anticuerpos se internalizan rápidamente después de la unión. Un ejemplo de anticuerpo de internalización rápida es el anticuerpo LL1 (anti-CD74), con una tasa de internalización de aproximadamente 8 x 106 moléculas de anticuerpo por célula por día (p. ej., Hansen et al., 1996, Biochem J. 320:293-300). Por lo tanto, un anticuerpo de "internalización rápida" puede ser uno con una tasa de internalización de aproximadamente 1 x 106 a aproximadamente 1 x 107 moléculas de anticuerpo por célula por día. Los anticuerpos de uso en las composiciones y métodos reivindicados pueden incluir MAb con propiedades como las indicadas anteriormente. Ejemplos de anticuerpos de uso para la terapia de, por ejemplo, cáncer incluyen pero no se limitan a LL1 (anti-CD74), LL2 o RFB4 (anti-CD22), veltuzumab (hA20, anti-CD20), rituxumab (anti-CD20), obinutuzumab (GA101, anti-CD20), lambrolizumab (anti-receptor PD-1), nivolumab (anti-receptor PD-1), ipilimumab (anti-CTLA-4), RS7 (glicoproteína-1 anti-epitelial (EGP-1, también conocido como Trop-2)), PAM4 o KC4 (ambos anti-mucina), MN-14 (anti-antígeno carcinoembrionario (CEA, también conocido como CD66e o CEACAM5), MN-15 o MN-3 (anti-CEACAM6), Mu-9 (antiantígeno-p específico de colon), Immu 31 (una anti-alfa-fetoproteína), R1 (anti-IGF-1R), A19 (anti-CD19), TAG-72 (p. ej., CC49), Tn, J591 o HuJ591 (antiPSMA (antígeno de membrana específico de la próstata)), AB-PG1-XG1-026 (dímero anti-PSMA), D2/B (anti-PSMA), G250 (un MAb IX antianhidrasa carbónica), L243 (anti-HLA-DR) alemtuzumab (anti-CD52), bevacizumab (anti-VEGF), cetuximab (anti-EGFR), gemtuzumab (anti-CD33), ibritumomab tiuxetan (anti-CD20); panitumumab (anti-EGFR) tositumomab (anti-CD20); PAM4 (también conocido como clivatuzumab, antimucina) y trastuzumab (anti-ErbB2). Dichos anticuerpos son conocidos en la técnica (p. ej., patentes de EE. UU. n.° 2; 7.074.403; 7.230.084; 7.238.785; 7.238.786; 7.256.004; 7.282.567; 7.300.655; 7.312.318; 7.585.491, 7.612.180, 7.642.239 y publicación de solicitud de patente de EE. UU. n.° 20050271671, 20060193865, 20060210475, 20070087001.) Los anticuerpos específicos conocidos de uso incluyen hPAM4 (patente de EE. UU. n.° 7.282.567), hA20 (Patente de EE. UU. n.° 7,151,164), hA19 (Patente de EE. UU. n.° 7.109.304), hIMMU-31 (patente de EE. UU. n.° 7.300.655), hLL1 (patente de EE. UU. n.° 7.312.318), hLL2 (patente de EE. UU. n.° 5.789.554), hMu-9 (patente de EE. n.° de patente 7.612.180), hMN-14 (patente de EE. UU. n.° 6.676.924), hMN-15 (patente de EE. UU. n.° 8.287.865), hR1 (solicitud de patente de EE. UU. 14/061,1767), hRS7 (patente de EE. UU. n.° 7.238.785), hMN-3 (patente de EE. UU. n.° 7.541.440), AB-PG1-XG1-026 (solicitud de patente de EE. UU. 11/983.372, depositada como ATCC PTA-4405 y PTA-4406) y D2/B (WO 2009/130575).
[0074] Otros antígenos útiles a los que se pueden dirigir usando los conjugados descritos incluyen anhidrasa carbónica IX, B7, CCL19, CCL21, CSAp, HER-2/neu, BrE3, CD1, CD1a, CD2, CD3, CD4, CD5, CD8, CD11A, CD14,
CD15, CD16, CD18, CD19, CD20 (p. ej., C2B8, hA20, 1F5 MAb), CD21, CD22, CD23, CD25, CD29, CD30, CD32b, CD33, CD37, CD38, CD40, CD40L, CD44, CD45, CD46, CD52, CD54, CD55, CD59, CD64, CD67, CD70, CD74, CD79a, CD80, CD83, CD95, CD126, CD133, CD138, CD147, CD154, CEACAM5, CEACAM6, CTLA-4, alfafetoproteína (AFP), VEGF (p. ej., AVASTIN®, variante de empalme de fibronectina), fibronectina ED-B (p. ej., L19), EGP-1 (Trop-2), EGP-2 (p. ej., 17-1A), receptor EGF (ErbB1) (p. ej., ERBITUX®), ErbB2, ErbB3, Factor H, FHL-1, Flt-3, receptor de folato, Ga 733,GRO-p, HMGB-1, factor inducible por hipoxia (HIF), HM1.24, HER-2/neu, insulina factor de crecimiento similar (ILGF), IFN-y, IFN-α, IFN-β, IFN-λ, IL-2R, IL-4R, IL-6R, IL-13R, IL-15R, IL-17R, IL-18R, IL-2, IL-6, IL-8, IL-12, IL-15, IL-17, IL-18, IL-25, IP-10, IGF-1R, Ia, HM1.24, gangliósidos, HCG, el antígeno HLA-DR al que se une L243, antígenos CD66, es decir, CD66a-d o una combinación de los mismos, MAGE, mCRP, MCP-1, MIP-1A, MIP-1B, factor inhibidor de la migración de macrófagos (MIF), MUC1, MUC2, MUC3, MUC4, MUC5ac, factor de crecimiento placentario (PIGF), PSA (antígeno prostático específico), PSMA, antígeno PAM4, receptor PD-1, NCA-95, NCA-90, A3, A33, Ep-CAM, KS-1, Le(y), mesotelina, S100, tenascina, TAC, antígeno Tn, antígenos de Thomas-Friedenreich, antígenos de necrosis tumoral, antígenos de angiogénesis tumoral, TNF-α, receptor TRAIL (R1 y R2), Trop- 2, VEGFR, RANTES, T101, así como antígenos de células madre cancerosas, factores del complemento C3, C3a, C3b, C5a, C5 y un producto oncogénico.
[0075] Un análisis exhaustivo de objetivos de antígenos adecuados (designación de grupo o CD) en tumores hematopoyéticos malignos células, como se muestra por citometría de flujo y que puede ser una guía para seleccionar anticuerpos adecuados para la inmunoterapia conjugada con fármacos, es Craig and Foon, Blood prepublicado en línea 15 de enero de 2008; DOL 10.1182/blood-2007-11 -120535.
[0076] Los antígenos CD66 consisten en cinco glicoproteínas diferentes con estructuras similares, CD66a-e, codificadas por los miembros de la familia de genes del antígeno carcinoembrionario (CEA), BCG, CGM6, NCA, CGM1 y CEA, respectivamente. Estos antígenos CD66 (por ejemplo, CEACAM6) se expresan principalmente en granulocitos, células epiteliales normales del tracto digestivo y células tumorales de diversos tejidos. También se incluyen como dianas adecuadas para los cánceres los antígenos testiculares del cáncer, como NY-ESO-1 (Theurillat et al., Int. J. Cancer 2007; 120(11 ):2411-7), así como CD79a en la leucemia mieloide (Kozlov et al., Cancer Genet. Cytogenet.
2005; 163(1):62-7) y también enfermedades de células B, y CD79b para linfoma no Hodgkin (Poison et al., Blood 110(2):616-623). Varios de los antígenos antes mencionados se describen en la solicitud provisional de los EE. UU. con número de serie 60/426,379, titulada "Use of Multi-specific, Non-covalent Complexes for Targeted Delivery of Therapeutics", presentada el 15 de noviembre de 2002. Las células madre cancerosas, que son atribuidas a ser poblaciones de células malignas precursoras más resistentes a la terapia (Hill and Perris, J. Natl. Cancer Inst. 2007; 99:1435-40), tienen antígenos que pueden ser objetivo en ciertos tipos de cáncer, como CD 133 en cáncer de próstata (Maitland et al., Ernst Schering Found. Sympos. Proc. 2006; 5:155-79), cáncer de pulmón de células no pequeñas (Donnenberg et al., J. Control Release 2007; 122(3):385-91), y glioblastoma (Beier et al., Cancer Res. 2007; 67(9):4010-5), y CD44 en cáncer colorrectal (Dalerba er al., Proc. Natl. Acad. Sci. EE. UU. 2007; 104(24)10158-63), cáncer de páncreas (Li et al., Cancer Res. 2007; 67(3):1030-7), y en carcinoma de células escamosas de cabeza y cuello (Prince et al., Proc. Natl. Acad. Sci. EE. UU. 2007; 104(3)973-8).
[0077] Para la terapia del mieloma múltiple, se han descrito anticuerpos dirigidos adecuados contra, por ejemplo, CD38 y CD138 (Stevenson, Mol Med 2006; 12(11-12):345-346; Tassone et al., Blood 2004; 104(12): 3688-96), CD74 (Stein et al., ibid.), CS1 (Tai et al., Blood 2008; 112(4):1329-37) y CD40 (Tai et al., 2005; Cancer Res. 65(13):5898-5906).
[0078] El factor inhibidor de la migración de macrófagos (MIF) es un importante regulador de la inmunidad innata y adaptativa y de la apoptosis. Se ha informado que CD74 es el receptor endógeno para MIF (Leng et al., 2003, J Exp Med 197:1467-76). El efecto terapéutico de los anticuerpos antagonistas anti-CD74 en las vías intracelulares mediadas por MIF puede ser útil para el tratamiento de una amplia variedad de estados patológicos, como cánceres de vejiga, próstata, mama, pulmón, colon y leucemia linfocítica crónica (p. ej., Meyer-Siegler et al., 2004, BMC Cancer 12:34, Shachar & Haran, 2011, Leuk Lymphoma 52:1446-54); enfermedades autoinmunes como la artritis reumatoide y el lupus eritematoso sistémico (Morand & Leech, 2005, Front Biosci 10:12-22; Shachar & Haran, 2011, Leuk Lymphoma 52:1446-54); enfermedades renales tales como rechazo de aloinjertos renales (Lan, 2008, Nephron Exp Nephrol.
109:e79-83); y numerosas enfermedades inflamatorias (Meyer-Siegler et al., 2009, Mediators Inflamm epub 22 de marzo de 2009; Takahashi et al., 2009, Respir Res 10:33; Milatuzumab (hLL1) es un anticuerpo anti-CD74 ejemplar de uso terapéutico para tratamiento de enfermedades mediadas por MIF.
[0079] Los anticuerpos anti-TNF-α son conocidos en la técnica y pueden ser útiles para tratar enfermedades inmunitarias, tales como enfermedades autoinmunes, disfunción inmunitaria (p. ej., enfermedad de injerto contra huésped, rechazo de trasplante de órganos) o diabetes. Los anticuerpos conocidos contra TNF-α incluyen el anticuerpo humano CDP571 (Ofei et al., 2011, Diabetes 45:881-85); anticuerpos murinos MTNFAI, M2TNFAI, M3TNFAI, M3TNFABI, M302B y M303 (Thermo Scientific, Rockford, IL); infliximab (Centocor, Malvern, PA); certolizumab pegol (UCB, Bruselas, Bélgica); y adalimumab (Abbott, Abbott Park, IL). Estos y muchos otros anticuerpos anti-TNF-α conocidos pueden usarse en los métodos y composiciones reivindicados. Otros anticuerpos de uso para la terapia de enfermedades autoinmunes o de desregulación inmunitaria incluyen, entre otros, anticuerpos de células anti-B tales como veltuzumab, epratuzumab, milatuzumab o hL243; tocilizumab (receptor anti-IL-6);
basiliximab (anti-CD25); daclizumab (anti-CD25); efalizumab (anti-CD11a); muromonab-CD3 (receptor anti-CD3); anti-CD40L (UCB, Bruselas, Bélgica); natalizumab (integrina anti-a4) y omalizumab (anti-IgE).
[0080] La diabetes tipo 1 y tipo 2 se puede tratar con anticuerpos conocidos contra antígenos de células B, como Cd 22 (epratuzumab y hRFB4), CD74 (milatuzumab), CD19 (hA19), CD20 (veltuzumab) o HLA-DR (hL243). (ver, por ejemplo, Winer et al., 2011, Nature Med 17:610-18). También se han propuesto anticuerpos anti-CD3 para la terapia de la diabetes tipo 1 (Cernea et al., 2010, Diabetes Metab Rev 26:602-05).
[0081] En otra forma de realización preferida de la invención como se define en las reivindicaciones, se usan anticuerpos que se internalizan rápidamente y luego se vuelven a expresar, procesar y presentar en las superficies celulares, lo que permite la captación y acumulación continua del conjugado circulante por parte de la célula. Un ejemplo de un par anticuerpo/antígeno más preferido es LL1, un MAb anti-CD74 (cadena invariable, chaperona específica de clase II, Ii) (véanse, por ejemplo, las patentes de EE. UU. números 6.653.104; 7.312.318). El antígeno CD74 está altamente expresado en linfomas de células B (incluido el mieloma múltiple) y leucemias, ciertos linfomas de células T, melanomas, cánceres de colon, pulmón y riñón, glioblastomas y otros tipos de cáncer (Ong et al., Immunology 98: 296-302 (1999)). Una revisión del uso de anticuerpos CD74 en el cáncer está contenida en Stein et al., Clin Cancer Res. 15 de septiembre de 2007; 13 (18 Pt 2): 5556s-5563s.
[0082] Las enfermedades que se tratan preferentemente con anticuerpos anti-CD74 incluyen, pero no se limitan a, linfoma no Hodgkin, enfermedad de Hodgkin, melanoma, cáncer de pulmón, renal, colónico, glioblastoma multiforme, histiocitomas, leucemias mieloides y mieloma múltiple. La expresión continua del antígeno CD74 durante cortos períodos de tiempo en la superficie de las células diana, seguida de la internalización del antígeno y la reexpresión del antígeno, permite que el anticuerpo dirigido contra LL1 se internalice junto con cualquier resto de fármaco que lleve. Esto permite que se acumule dentro de tales células una concentración alta y terapéutica de conjugado de LL1 -fármaco. Los conjugados de LL1-fármaco internalizados pasan por los lisosomas y los endosomas, y la fracción del fármaco se libera en una forma activa dentro de las células diana.
[0083] Los anticuerpos de uso para tratar enfermedades autoinmunes o disfunciones del sistema inmunológico (p. ej., enfermedad de injerto contra huésped, rechazo de trasplante de órganos) son conocidas en la técnica y pueden conjugarse con SN-38 usando los métodos y composiciones descritos. Los anticuerpos de uso para tratar la enfermedad de disfunción autoinmune/inmune pueden unirse a antígenos ejemplares que incluyen, entre otros, BCL-1, BCL-2, BCL-6, CD1a, CD2, CD3, CD4, CD5, CD7, CD8, CD10, CD11b, CD11c, CD13, CD14, CD15, CD16, CD19, CD20, CD21, CD22, CD23, CD25, CD33, CD34, CD38, CD40, CD40L, CD41a, CD43, CD45, CD55, CD74, TNF-αlfa, interferón y HLA-DR. Los anticuerpos que se unen a estos y otros antígenos diana, discutidos anteriormente, pueden usarse para tratar enfermedades autoinmunes o disfunciones inmunes. Las enfermedades autoinmunes que pueden tratarse con anticuerpos o inmunoconjugados pueden incluir púrpura trombocitopénica idiopática aguda, púrpura trombocitopénica idiopática crónica, dermatomiositis, corea de Sydenham, miastenia grave, lupus eritematoso sistémico, nefritis lúpica, fiebre reumática, síndromes poliglandulares, penfigoide ampolloso, diabetes mellitus, Henoch-Púrpura de Schonlein, nefritis posestreptocócica, eritema nodoso, arteritis de Takayasu, vasculitis asociadas a ANCA, enfermedad de Addison, artritis reumatoide, esclerosis múltiple, sarcoidosis, colitis ulcerosa, eritema multiforme, nefropatía IgA, poliarteritis nodosa, espondilitis anquilosante, síndrome de Goodpasture, tromboangitis obliterante, síndrome de Sjogren, cirrosis biliar primaria, tiroiditis de Hashimoto, tirotoxicosis, esclerodermia, hepatitis crónica activa, polimiositis/dermatomiositis, policondritis, penfigoide ampolloso, pénfigo vulgar, granulomatosis de Wegener, nefropatía membranosa, esclerosis lateral amiotrófica, tabes dorsal, arteritis de células gigantes/polimialgia, anemia perniciosa, glomerulonefritis rápidamente progresiva, psoriasis o alveolitis fibrosante.
[0084] Los anticuerpos discutidos anteriormente y otros anticuerpos conocidos contra antígenos asociados a enfermedades pueden usarse como conjugados de CPT, más preferiblemente conjugados de SN-38, en la práctica de la invención como se define en las reivindicaciones.
Anticuerpos biespecíficos y multiespecíficos
[0085] Los anticuerpos biespecíficos son útiles en varias aplicaciones biomédicas. Por ejemplo, un anticuerpo biespecífico con sitios de unión para un antígeno de superficie de células tumorales y para un receptor de superficie de células T puede dirigir la lisis de células tumorales específicas por parte de las células T. Se han usado con éxito anticuerpos biespecíficos que reconocen gliomas y el epítopo CD3 en células T en el tratamiento de tumores cerebrales en pacientes humanos (Nitta, et al. Lancet. 1990; 355:368-371). Un anticuerpo biespecífico preferido es un anticuerpo anti-CD3 x anti-CD19. Alternativamente, un anticuerpo anti-CD3 o un fragmento del mismo puede unirse a un anticuerpo o fragmento contra otro antígeno asociado a células B, como anti-CD3 x anti-CD20, anti-CD3 x anti-CD22, anti-CD3 x anti-HLA-DR o anti-CD3 x anti-CD74. En ciertas formas de realización de la invención como se define en las reivindicaciones, las técnicas y composiciones para la conjugación de agentes terapéuticos descritas en el presente documento pueden usarse con anticuerpos biespecíficos o multiespecíficos como restos de direccionamiento.
[0086] Se conocen numerosos métodos para producir anticuerpos biespecíficos o multiespecíficos, como se describe, por ejemplo, en la Patente de Estados Unidos n.° 7.405.320. Los anticuerpos biespecíficos se pueden producir mediante el método del cuadroma, que implica la fusión de dos hibridomas diferentes, cada uno de los cuales produce un anticuerpo monoclonal que reconoce un sitio antigénico diferente (Milstein y Cuello, Nature, 1983; 305:537-540).
[0087] Otro método para producir anticuerpos biespecíficos usa reticuladores heterobifuncionales para unir químicamente dos anticuerpos monoclonales diferentes (Staerz, et al. Nature, 1985; 314:628-631; Perez, et al. Nature, 1985; 316:354-356). Los anticuerpos biespecíficos también se pueden producir mediante la reducción de cada uno de los dos anticuerpos monoclonales parentales a las respectivas medias moléculas, que luego se mezclan y se dejan reoxidar para obtener la estructura híbrida (Staerz y Bevan. Proc Natl Acad Sci EE. UU. 1986; 83: 1453-1457). Otra alternativa implica el entrecruzamiento químico de dos o tres fragmentos Fab' purificados por separado usando enlazadores apropiados. (Véase, por ejemplo, la solicitud de patente europea 0453082).
[0088] Otros métodos incluyen mejorar la eficiencia de generar hibridomas híbridos mediante la transferencia génica de distintos marcadores seleccionables a través de vectores lanzadera derivados de retrovirus en los respectivos hibridomas parentales, que se fusionan posteriormente (DeMonte, et al. Proc Natl Acad Sci EE. UU. 1990, 87:2941-2945); o transfección de una línea celular de hibridoma con plásmidos de expresión que contienen los genes de cadena ligera y pesada de un anticuerpo diferente.
[0089] Vh y Vl afines se pueden unir con un conector peptídico de composición y longitud apropiadas (que generalmente consta de más de 12 residuos de aminoácidos) para formar un Fv monocatenario (scFv) con actividad de unión. Los métodos de fabricación de scFv se describen en las patentes de EE. UU. n.° 4.946.778 y Patente de EE. UU. n.° 5.132.405. La reducción de la longitud del conector peptídico a menos de 12 residuos de aminoácidos evita el emparejamiento de los dominios Vh y Vl en la misma cadena y fuerza el emparejamiento de los dominios Vh y Vl con dominios complementarios en otras cadenas, lo que da como resultado la formación de multímeros funcionales. Las cadenas polipeptídicas de los dominios Vh y Vl que se unen con conectores entre 3 y 12 residuos de aminoácidos forman predominantemente dímeros (denominados diacuerpos). Con enlazadores entre 0 y 2 residuos de aminoácidos, se favorecen los trímeros (denominados triacuerpo) y tetrámeros (denominados tetracuerpo), pero los patrones exactos de oligomerización parecen depender de la composición, así como de la orientación de los dominios V (VH-enlazador-VL o VL-enlazador-VH), además de la longitud del enlazador.
[0090] Estas técnicas para producir anticuerpos multiespecíficos o biespecíficos presentan diversas dificultades en términos de bajo rendimiento, necesidad de purificación, baja estabilidad o la mano de obra intensiva de la técnica. Más recientemente, se ha utilizado una técnica conocida como "dock and lock" (DNL) para producir combinaciones de prácticamente todos los anticuerpos deseados, fragmentos de anticuerpos y otras moléculas efectoras (véanse, por ejemplo, las patentes de EE. UU. Nos.7.521.056; 7.527.787; 7.534.866; 7.550.143; 7.666.400; 7.858.070; 7.871.622; 7.906.121; 7.906.118; 8.163.291; 7.901.680; 7.981.398; 8.003.111 y 8.034.352). La técnica utiliza dominios de unión a proteínas complementarios, denominados dominios de anclaje (AD) y dominios de dimerización y acoplamiento (DDD), que se unen entre sí y permiten el ensamblaje de estructuras complejas, que van desde dímeros, trímeros, tetrámeros, quintameros y hexámeros. Estos forman complejos estables con alto rendimiento sin necesidad de una purificación extensa. La técnica DNL permite el ensamblaje de anticuerpos monoespecíficos, biespecíficos o multiespecíficos. Cualquiera de las técnicas conocidas en la técnica para producir anticuerpos biespecíficos o multiespecíficos puede utilizarse en la práctica de la invención como se define en las reivindicaciones.
[0091] En varias formas de realización de la invención como se define en las reivindicaciones, un conjugado como se describe en el presente documento puede ser parte de un anticuerpo multiespecífico compuesto. Dichos anticuerpos pueden contener dos o más sitios de unión a antígeno diferentes, con diferentes especificidades. El compuesto multiespecífico puede unirse a diferentes epítopos del mismo antígeno o, alternativamente, puede unirse a dos antígenos diferentes. Algunas de las combinaciones de objetivos más preferidas incluyen las enumeradas en la Tabla 1. Esta es una lista de ejemplos de combinaciones preferidas, pero no pretende ser exhaustiva.
Tabla 1. Algunos ejemplos de anticuerpos multiespecíficos

[0092] Otras combinaciones más, como las que se prefieren para las terapias contra el cáncer, incluyen anticuerpos CD20 CD22, anticuerpos CD74 CD20, anticuerpos CD74 CD22, anticuerpos CEACAM5 (CeA) CEACAM6 (NCA), factor de crecimiento similar a la insulina (ILGf ) anticuerpos CEACAM5, EGP-1 (p. ej., RS-7) anticuerpos ILGF, CEACAM5 anticuerpos EGFR, IL6 anticuerpos CEACAM6, anticuerpos CD74 y HLA-DR, y anticuerpos CD22 y HLA-DR. Dichos anticuerpos no solo necesitan usarse en combinación, sino que pueden combinarse como proteínas de fusión de varias formas, tales como IgG, Fab, scFv y similares, como se describe en las Patentes de EE. UU. N.os 6.083.477; 6.183.744 y 6.962.702 y números de publicación de solicitud de patente de EE. UU.20030124058; 20030219433; 20040001825; 20040202666; 20040219156; 20040219203; 20040235065; 20050002945; 20050014207; 20050025709; 20050079184; 20050169926; 20050175582; 20050249738; 20060014245 y 20060034759.
DOCK-AND-LOCKO (DNLO)
[0093] En formas de realización preferidas, un anticuerpo bivalente o multivalente está formado como un complejo DOCK-AND-LOCK® (DNL®) (véanse, por ejemplo, las patentes de EE. UU. N.os 7.521.056; 7.527.787; 7.534.866; 7.550.143; 7.666.400; 7.858.070; 7.871.622; 7.906.121; 7.906.118; 8.163.291; 7.901.680; 7.981.398; 8.003.111 y 8.034.352). En general, la técnica aprovecha las interacciones de unión específicas y de alta afinidad que se producen entre una secuencia de dimerización y dominio de acoplamiento (DDD) de las subunidades reguladoras (R) de cAMP dependiente proteína quinasa (PKA) y una secuencia de dominio de anclaje (AD) derivada de cualquiera de una variedad de proteínas AkAP (Baillie et al., FEBS Letters. 2005; 579: 3264. Wong and Scott, Nat. Rev. Mol. Cell Biol.
2004; 5: 959). Los péptidos DDD y AD pueden unirse a cualquier proteína, péptido u otra molécula. Debido a que las secuencias de DDD se dimerizan espontáneamente y se unen a la secuencia de AD, la técnica permite la formación de complejos entre cualquier molécula seleccionada que pueda estar unida a las secuencias de DDD o AD.
[0094] Aunque el complejo DNL® estándar comprende un trímero con dos moléculas unidas a DDD unidas a una molécula unida a AD, las variaciones en la estructura del complejo permiten la formación de dímeros, trímeros, tetrámeros, pentámeros, hexámeros y otros multímeros. En algunas formas de realización, el complejo DNL® puede comprender dos o más anticuerpos, fragmentos de anticuerpos o proteínas de fusión que se unen al mismo determinante antigénico o a dos o más antígenos diferentes. El complejo DNL® también puede comprender uno o más efectores, como proteínas, péptidos, inmunomoduladores, citocinas, interleucinas, interferones, proteínas de unión, ligandos peptídicos, proteínas transportadoras, toxinas, ribonucleasas como onconasa, oligonucleótidos inhibidores como siARN, antígenos o xenoantígenos, polímeros como PEG, enzimas, agentes terapéuticos, hormonas, agentes citotóxicos, agentes antiangiogénicos, agentes proapoptóticos o cualquier otra molécula o agregado.
[0095] La PKA, que juega un papel central en una de las vías de transducción de señales mejor estudiadas desencadenadas por la unión del segundo mensajero AMPc a las subunidades R, se aisló por primera vez del músculo esquelético de conejo en 1968 (Walsh et al., J. Biol. Chem. 1968; 243:3763). La estructura de la holoenzima consta de dos subunidades catalíticas mantenidas en forma inactiva por las subunidades R (Taylor, J. Biol. Chem. 1989; 264:8443). Las isoenzimas de PKA se encuentran con dos tipos de subunidades R (RI y RII), y cada tipo tiene isoformas a y p (Scott, Pharmacol. Ther. 1991; 50: 123). Así, las cuatro isoformas de las subunidades reguladoras de PKA son RIa, RIp, RIIa y RIIp. Las subunidades R se han aislado sólo como dímeros estables y se ha demostrado que el dominio de dimerización consta de los primeros 44 residuos amino-terminales de RIIa (Newlon et al., Nat. Struct. Biol. 1999; 6:222). Como se analiza a continuación, porciones similares de las secuencias de aminoácidos de otras subunidades reguladoras están involucradas en la dimerización y el acoplamiento, cada una ubicada cerca del extremo N-terminal de la subunidad reguladora. La unión de cAMP a las subunidades R conduce a la liberación de subunidades catalíticas activas para un amplio espectro de actividades de serina/treonina quinasa, que se orientan hacia sustratos seleccionados a través de la compartimentación de PKA a través de su acoplamiento con AKAP (Scott et al., J. Biol. Chem. 1990;265;21561)
[0096] Desde que se caracterizó la primera AKAP, la proteína 2 asociada a microtúbulos, en 1984 (Lohmann et al., Proc. Natl. Acad. Sci EE. UU. 1984; 81:6723), más de 50 AKAP que se localizan en varios sitios subcelulares, incluida la membrana plasmática, el citoesqueleto de actina, el núcleo, las mitocondrias y el retículo endoplásmico, se han identificado con diversas estructuras en especies que van desde la levadura hasta los seres humanos (Wong y Scott, Nat. Rev. Mol. Cell Biol. 2004;5:959). La AD de AKAP para PKA es una hélice anfipática de 14-18 residuos (Carr et al., J. Biol. Chem. 1991; 266:14188). Las secuencias de aminoácidos de AD varían bastante entre los AKAP individuales, y las afinidades de unión reportadas para los dímeros RII varían de 2 a 90 nM (Alto et al., Proc. Natl. Acad. Sci. EE. UU. 2003; 100: 4445). Los AKAP solo se unirán a las subunidades R diméricas. Para RIIa humana, la AD se une a una superficie hidrofóbica formada por los 23 residuos amino-terminales (Colledge y Scott, Trends Cell Biol. 1999; 6:216). Por lo tanto, el dominio de dimerización y el dominio de unión a AKAP del RIIa humano están ubicados dentro de la misma secuencia de 44 aminoácidos N-terminal (Newlon et al., Nat. Struct. Biol. 1999; 6:222; Newlon et al., EMBO J. 2001;20:1651), que se denomina DDD en este documento.
[0097] Hemos desarrollado una tecnología de plataforma para utilizar la DDD de las subunidades reguladoras de la PKA humana y la AD de AKAP como un excelente par de módulos enlazadores para acoplar dos entidades cualesquiera, denominadas A y B en lo sucesivo, en un complejo no covalente, que podría ser más bloqueado en un complejo DNL® a través de la introducción de residuos de cisteína tanto en el DDD como en el AD en posiciones estratégicas para facilitar la formación de enlaces disulfuro. La metodología general del enfoque es la siguiente. La entidad A se construye uniendo una secuencia DDD a un precursor de A, lo que da como resultado un primer componente que en lo sucesivo se denominará a. Debido a que la secuencia DDD efectuaría la formación espontánea de un dímero, A estaría compuesto por a 2 . La entidad B se construye uniendo una secuencia de AD a un precursor de B, lo que da como resultado un segundo componente que en lo sucesivo se denominará b. El motivo dimérico de DDD contenido en a 2 creará un sitio de acoplamiento para unirse a la secuencia de AD contenida en b, facilitando así una fácil asociación de a 2 y b para formar un complejo trimérico binario compuesto por a 2 b. Este evento de unión se hace irreversible con una reacción posterior para asegurar covalentemente las dos entidades a través de puentes disulfuro, lo que ocurre de manera muy eficiente según el principio de concentración local efectiva porque las interacciones de unión iniciales deberían llevar los grupos tiol reactivos colocados tanto en DDD como en AD en proximidad (Chmura et al., Proc. Natl. Acad. Sci. EE. UU. 2001; 98:8480) para ligar sitio específico. Usando varias combinaciones de enlazadores, módulos adaptadores y precursores, se puede producir y usar una amplia variedad de construcciones DNL® de diferente estequiometría (ver, por ejemplo, US N.os 7.550.143; 7.521.056; 7.534.866; 7.527.787 y 7.666.400).
[0098] Al unir el DDD y el AD lejos de los grupos funcionales de los dos precursores, también se espera que dichas ligaciones específicas del sitio conserven las actividades originales de los dos precursores. Este enfoque es de naturaleza modular y potencialmente se puede aplicar para unir, de forma covalente y específica de sitio, una amplia gama de sustancias, incluidos péptidos, proteínas, anticuerpos, fragmentos de anticuerpos y otros restos efectores con una amplia gama de actividades. Utilizando el método de proteína de fusión para construir efectores conjugados de AD y DDD descritos en los Ejemplos a continuación, prácticamente cualquier proteína o péptido puede incorporarse en una construcción DNL®. Sin embargo, la técnica no es limitante y pueden utilizarse otros métodos de conjugación.
[0099] Se conocen una variedad de métodos para hacer proteínas de fusión, incluyendo la síntesis, hibridación y/o amplificación de ácidos nucleicos para producir un ácido nucleico de doble cadena sintético que codifica una proteína de fusión de interés. Dichos ácidos nucleicos de doble cadena pueden insertarse en vectores de expresión para la producción de proteínas de fusión mediante técnicas estándar de biología molecular (véase, por ejemplo, Sambrook et al., Molecular Cloning, A lab. manual, 2a ed., 1989). En dichas formas de realización preferidas, el resto AD y/o DDD puede unirse al extremo N-terminal o C-terminal de una proteína o péptido efector. Sin embargo, el experto en la materia se dará cuenta de que el sitio de unión de un resto AD o DDD a un resto efector puede variar, dependiendo de la naturaleza química del resto efector y la(s) parte(s) del resto efector involucradas en su actividad fisiológica. La unión específica del sitio de una variedad de restos efectores se puede realizar utilizando técnicas conocidas en la técnica, como el uso de reactivos de reticulación bivalentes y/u otras técnicas de conjugación química.
[0100] En varias formas de realización de la invención como se define en las reivindicaciones, un anticuerpo o fragmento de anticuerpo puede incorporarse en un complejo DNL®, por ejemplo, uniendo un resto DDD o AD al extremo C-terminal de la cadena pesada del anticuerpo, como se describe en detalle a continuación. En formas de realización más preferidas, el resto DDD o AD, más preferentemente el resto AD, puede unirse al extremo C-terminal de la cadena ligera del anticuerpo (véase, por ejemplo, la solicitud de patente de EE. 24/13).
Relaciones estructura-función en fracciones AD y DDD
[0101] Para diferentes tipos de construcciones de DNL®, se pueden utilizar diferentes secuencias de AD o DDD. A continuación, se proporcionan secuencias ejemplares de DDD y AD.
DDD1
SHIQIPPGLTELLQGYTVEVLRQQPPDLVEFAVEYFTRLREARA (SEQ ID NO: 1)
DDD2
CGHIQIPPGLTELLQGYTVEVLRQQPPDLVEFAVEYFTRLREARA (SEQ ID NO: 2)
AD1
QIEYLAKQIVDNAIQQA (SEQ ID NO: 3)
AD2
CGQIEYLAKQIVDNAIQQAGC (SEQ ID NO: 4)
[0102] El experto en la materia se dará cuenta de que DDD1 y DDD2 se basan en la secuencia DDD de la isoforma RIla humana de la proteína quinasa A. Sin embargo, en formas de realización alternativas, los restos DDD y AD pueden basarse en la secuencia DDD de la forma Rla humana. de proteína quinasa A y una secuencia AKAP correspondiente, como se ejemplifica en DDD3, DDD3C y AD3 a continuación.
DDD3
SLRECELYVQKHNIQALLKDSIVQLCTARPERPMAFLREYFERLEKEEAK (SEQ ID NO:5)
DDD3C
MSCGGSLRECELYVQKHNIQALLKDSIVQLCTARPERPMAFLREYFERLEKEEAK (SEQ ID NO:6)
AD3
CGFEELAWKIAKMIWSDVFQQGC (SEQ ID NO:7)
[0103] En otras formas de realización alternativas de la invención como se define en las reivindicaciones, se pueden utilizar otras variantes de secuencia de restos AD y/o DDD en la construcción de los complejos DNL®. Por ejemplo, solo hay cuatro variantes de secuencias DDD de PKA humana, que corresponden a los restos DDD de PKA Rla, RIla, Rlp y RIlp. La secuencia RIla DDD es la base de DDD1 y DDD2 descrita anteriormente. Las cuatro secuencias DDD de PKA humana se muestran a continuación. La secuencia DDD representa los residuos 1-44 de RIla, 1-44 de RIlp, 12-61 de Rla y 13-66 de Rlp. (Tenga en cuenta que la secuencia de DDD1 se modifica ligeramente de la fracción humana PKA RIla DDD).
PKA Rla
SLRECELYVQKHNIQALLKDVSIVQLCTARPERPMAFLREYFEKLEKEEAK (SEQ ID NO:8)
PKA Rlp
SLKGCELYVQLHGIQQVLKDCIVHLCISKPERPMKFLREHF EKLEKEENRQILA (SEQ ID NO: 9)
PKA RIla
SHIQIPPGLTELLQGYTVEVGQQPPDLVDFAVEYFTRLREARRQ (SEQ ID NO: 10)
PKA RIlp
SIEIPAGLTELLQGFTVEVLRHQPADLLEFALQHFTRLQQENER (SEQ ID NO: 11)
[0104] Las relaciones estructura-función de los dominios AD y DDD han sido objeto de investigación. (Véase, por ejemplo, Burns-Hamuro et al., 2005, Protein Sci 14:2982-92; Carr et al., 2001, J Biol Chem 276:17332-38; Alto et al., 2003, Proc Natl Acad Sci EE. UU 100:4445-50; Hundsrucker et al., 2006, Biochem Y 396:297-306; Stokka et al., 2006, Biochem J 400:493-99; Gold et al., 2006, Mol Cell 24:383-95 Kinderman et al., 2006, Mol Cell 24:397-408.)
[0105] Por ejemplo, Kinderman et al. (2006, Mol Cell 24:397-408) examinaron la estructura cristalina de la interacción de unión de a D-DDD y concluyeron que la secuencia de DDD humana contenía una serie de residuos de aminoácidos conservados que eran importantes en la formación de dímeros o en la unión de AKAP, subrayados en SEQ ID NO: 1 a continuación. (Consulte la Figura 1 de Kinderman et al., 2006). El artesano experto se dará cuenta de que al diseñar variantes de secuencia de la secuencia DDD, sería deseable evitar cambiar cualquiera de los residuos subrayados, mientras que se podrían realizar sustituciones de aminoácidos conservadoras para residuos que son menos críticos para la dimerización y la unión de AKAP.
SHIQIPPGLTELLQGYTVEVLRQQPPDLVEFAVEYFTRLREARA (SEQ ID NO: 1)
[0106] Como se analiza con más detalle a continuación, se han caracterizado sustituciones conservativas de aminoácidos para cada uno de los veinte L-aminoácidos comunes. Por lo tanto, basándose en los datos de Kinderman (2006) y las sustituciones de aminoácidos conservadoras, en la Tabla 2 se muestran secuencias DDD alternativas potenciales basadas en SEQ ID NO: 1. Al diseñar la Tabla 2, solo se consideraron sustituciones de aminoácidos altamente conservadoras. Por ejemplo, los residuos cargados solo se sustituyeron por residuos de la misma carga, los residuos con cadenas laterales pequeñas se sustituyeron por residuos de tamaño similar, las cadenas laterales de hidroxilo solo se sustituyeron por otros hidroxilos, etc. Debido al efecto único de la prolina sobre los aminoácidos estructura secundaria, ningún otro residuo fue sustituido por prolina. Un número limitado de secuencias de restos DDD alternativas potenciales se muestra en SEQ ID NO: 12 a s Eq ID NO: 31 a continuación. El experto en la materia se dará cuenta de que se puede construir un número casi ilimitado de especies alternativas dentro del género de fracciones DDD mediante técnicas estándar, por ejemplo, utilizando un sintetizador de péptidos comercial o técnicas de mutagénesis dirigida al sitio bien conocidas. El efecto de las sustituciones de aminoácidos sobre la unión del resto de AD también puede determinarse fácilmente mediante ensayos de unión estándar, por ejemplo, como se describe en Alto et al. (2003, Proc Natl Acad Sci EE. UU 100:4445-50).
Tabla 2. Sustituciones de aminoácidos conservadores en DDD1 (SEQ ID NO:1). Secuencia de consenso divulgada como SEQ ID NO:87.


THIQIPPGLTELLQGYTVEVLRQQPPDLVEFAVEYFTRLREARA (SEQ ID NO: 12) SKIQIPPGLTELLQGYTVEVLRQQPPDLVEFAVEYFTRLREARA (SEQ ID NO: 13) SRIQIPPGLTELLQGYTVEVLRQQPPDLVEFAVEYFTRLREARA (SEQ ID NO: 14) SHINIPPGLTELLQGYTVEVLRQQPPDLV EFAVEYFTRLREARA (SEQ ID NO: 15) SHIQIPPALTELLQGYTVEVLRQQPPDLVEFAVEYFTRLREARA (SEQ ID NO: 16) SHIQIPPGLSELLQGYTVEVLRQQPPDLVEFAVEYFTRLREARA (SEQ ID NO: 17) SHIQIPPGLTDLLQGYTVEVLRQQPPDLVEFAVEYFTRLREARA (SEQ ID NO: 18) SHIQIPPGLTELLNGYTVEVLRQQPPDLVEFAVEYFTRLREARA (SEQ ID NO: 19) SHIQIPPGLTELLQAYTVEVLRQQPPDLVEFAVEYFTRLREARA (SEQ ID NO: 20) SHIQIPPGLTELLQGYSVEVLRQQPPDLVEFAVEYFTRLREARA (SEQ ID NO: 21) SHIQIPPGLTELLQGYTVDVLRQQPPDLVEFAVEYFTRLREARA (SEQ ID NO: 22) SHIQIPPGLTELLQGYTVEVLKQQPPDLVEFAVEYFTRLREARA (SEQ ID NO: 23) SHIQIPPGLTELLQGYTVEVLRNQPPDLVEFAVEYFTRLREARA (SEQ ID NO: 24) SHIQIPPGLTELLQGYTVEVLRQNPPDLVEFAVEYFTRLREARA (SEQ ID NO: 25) SHIQIPPGLTELLQGYTVEVLRQQPPELVEFAVEYFTRLREARA (SEQ ID NO: 26) SHIQIPPGLTELLQGYTVEVLRQQPP DLVDFAVEYFTRLREARA (SEQ ID NO:27) SHIQIPPGLTELLQGYTVEVLRQQPPDLVEFLVEYFTRLREARA (SEQ ID NO:28) SHIQIPPGLTELLQGYTVEVLRQQPPDLVEFIVEYFTRLREARA (SEQ ID NO: 29) SHIQIPPGLTELLQGYTVEVLRQQPPDLVEFVVEYFTRLREARA (SEQ ID NO: 30) SHIQIPPGLTELLQGYTVEVLRQQPPDLVEFAVDYFTRLREARA (SEQ ID NO: 31)
[0107] Alto et al. (2003, Proc Natl Acad Sci EE. UU. 100:4445-50) realizaron un análisis bioinformático de la secuencia AD de varias proteínas AKAP para diseñar una secuencia AD selectiva de RII denominada AKAP-IS (SEQ ID NO:3), con una constante de unión para DDD de 0,4 nM. La secuencia de AKAP-IS se diseñó como un péptido antagonista de la unión de AKAP a PKA. Los residuos en la secuencia de AKAP-IS en los que las sustituciones tendían a disminuir la unión a DDD están subrayados en SEQ ID NO:3 a continuación. El experto en la materia se dará cuenta de que al diseñar variantes de secuencia de la secuencia de AD, se evitaría deseablemente cambiar cualquiera de los residuos subrayados, mientras que se podrían realizar sustituciones de aminoácidos conservadoras para residuos que son menos críticos para la unión de DDD. La Tabla 3 muestra sustituciones de aminoácidos conservadoras potenciales
en la secuencia de AKAP-IS (AD1, SEQ ID NO: 3), similar a la que se muestra para DDD1 (SEQ ID NO: 1) en la Tabla 2 anterior.
[0108] Un número limitado de secuencias de restos de AD alternativas potenciales se muestra en SEQ ID NO: 32 a SEQ ID NO: 49 a continuación. Nuevamente, un gran número de especies dentro del género de posibles secuencias de restos de AD podrían ser preparadas, probadas y utilizadas por el experto en la materia, basándose en los datos de Alto et al. (2003). Se observa que la figura 2 de Alto (2003) muestra un número incluso grande de posibles sustituciones de aminoácidos que se pueden realizar, mientras se retiene la actividad de unión a los restos DDD, en base a experimentos de unión reales.
AKAP-IS
QIEYLAKQIVDNAIQQA (SEQ ID NO: 3)
Tabla 3. Sustituciones de aminoácidos conservadores en AD1 (SEQ ID NO: 3). Secuencia de consenso divulgada como SEQ ID NO:88.

NIEYLAKQIVDNAIQQA (SEQ ID NO: 32)
QLEYLAKQIVDNAIQQA (SEQ ID NO: 33)
QVEYLAKQIVDNAIQQA (SEQ ID NO: 34)
QIDYLAKQIVDNAIQQA (SEQ ID NO: 35)
QIEFLAKQIVDNAIQQA (SEQ ID NO: 36)
QIETLAKQIVDNAIQQA (SEQ ID NO: 37)
QIESLAKQIVDNAIQQA (SEQ ID NO: 38)
QIEYIAKQIVDNAIQQA (SEQ ID NO: 39)
QIEYVAKQIVDNAIQQA (SEQ ID NO: 40)
QIEYLARQIVDNAIQQA (SEQ ID NO: 41)
QIEYLAKNIVDNAIQQA (SEQ ID NO: 42)
QIEYLAKQIVENAIQQA (SEQ ID NO: 43)
QIEYLAKQIVDQAIQQA (SEQ ID NO: 44)
QIEYLAKQIVDNAINQA (SEQ ID NO: 45)
QIEYLAKQIVDNAIQNA (SEQ ID NO: 46)
QIEYLAKQIVDNAIQQL (SEQ ID NO: 47)
QIEYLAKQIVDNAIQQI (SEQ ID NO: 48)
QIEYLAKQIVDNA IQQV (SEQ ID NO: 49)
[0109] Gold et al. (2006, Mol Cell 24:383-95) utilizaron cristalografía y detección de péptidos para desarrollar una secuencia SuperAKAPIS (SEQ ID NO:50), que presenta una selectividad cinco veces mayor para la isoforma RII de PKA en comparación con la isoforma RI. Los residuos subrayados indican las posiciones de las sustituciones de aminoácidos, en relación con la secuencia AKAP-IS, que aumentaron la unión al resto DDD de RI la. En esta secuencia, el residuo Q N-terminal se numera como el residuo número 4 y el residuo A C-terminal es el residuo número 20. Los residuos en los que se podían hacer sustituciones para afectar la afinidad por RI la eran los residuos 8, 11, 15, 16, 18, 19 y 20 (Gold et al., 2006). Se contempla que en ciertas formas de realización alternativas de la invención como se define en las reivindicaciones, la secuencia SuperAKAP-IS puede sustituirse por la secuencia del resto AKAP-IS AD para preparar construcciones DNL®. Otras secuencias alternativas que podrían sustituirse por la secuencia AKAP-IS AD se muestran en SEQ ID NO: 51-53. Las sustituciones relativas a la secuencia AKAP-IS están subrayadas. Se anticipa que, como con la secuencia de AD2 que se muestra en SEQ ID NO: 4, el resto de AD también puede incluir los residuos N-terminales adicionales de cisteína y glicina y los residuos C-terminales de glicina y cisteína.
SuperAKAP-IS
QIEYVAKQIVDYAIHQA (SEQ ID NO:50)
Secuencias AKAP alternativas
QIEYKAKQIVDHAIHQA (SEQ ID NO:51)
QIEYHAKQIVDHAIHQA (SEQ ID NO:52)
QIEYVAKQIVDHAIHQA (SEQ ID NO:53)
[0110] Figura 2 de Gold et al. dio a conocer secuencias de unión a DDD adicionales de una variedad de proteínas AKAP, que se muestran a continuación.
AKAP específicos de RII
[0111]
AKAP-KL
PLEYQAGLLVQNAIQQAI (SEQ ID NO:54)
AKAP79
LLIETASSLVKNAIQLSI (SEQ ID NO:55)
AKAP-Lbc
LIEEAASRIVDAVIEQVK (SEQ ID NO:56)
AKAP específicos de RI
[0112]
AKAPce
ALYQFADRFSELVISEAL (SEQ ID NO:57)
RIAD LEQVANQLADQIIKEAT (SEQ ID NO:58)
PV38
FEELAWKIAKMIWSDVF (SEQ ID NO:59)
AKAP de doble especificidad
[0113]
AKAP 7
ELVRLSKRLVENAVLKAV (SEQ ID NO: 60)
MAP2D
TAEEVSARIVQVVTAEAV (SEQ ID NO: 61)
DAKAPI QIKQAAFQLISQVILEAT (SEQ ID NO: 62)
DAKAP2
LAWKIAKMIVSDVMQQ (SEQ ID NO: 63)
[0114] Stokka et al. (2006, Biochem J 400:493-99) también desarrollaron competidores peptídicos de la unión de AKAP a PKA, que se muestran en SEQ ID NO:64-66. Los antagonistas peptídicos se designaron como Ht31 (SEQ ID NO: 64), RIAD (SEQ ID NO: 65) y PV-38 (SEQ ID NO: 66). El péptido Ht-31 mostró mayor afinidad por la isoforma RII de PKA, mientras que RIAD y PV-38 mostraron mayor afinidad por RI.
Ht31
DLIEAASRIVDAVIEQVKAAGAY (SEQ ID NO: 64)
RIAD LEQYANQLADQIIKEATE (SEQ ID NO: 65)
PV-38
FEELAWKIAKMIWSDVFQQC (SEQ ID NO: 66)
[0115] Hundsrucker et al. (2006, Biochem J 396:297-306) desarrollaron aún otros competidores peptídicos para la unión de AKAP a PKA, con una constante de unión tan baja como 0,4 nM a la DDD de la forma RII de PKA. Las secuencias de varios péptidos antagonistas de AKAP se proporcionan en la Tabla 1 de Hundsrucker et al., reproducidas en la Tabla 4 a continuación. AKAPIS representa un péptido de unión a la subunidad RII sintético. Todos los demás péptidos se derivan de los dominios de unión a RII de los AKAP indicados.
Tabla 4. Secuencias peptídicas de AKAP
Secuencia peptídica

[0116] Los residuos que estaban altamente conservados entre los dominios AD de diferentes proteínas AKAP se indican a continuación subrayando con referencia a la secuencia IS de AKAP (SEQ ID NO: 3). Los residuos son los mismos que los observados por Alto et al. (2003), con la adición del residuo de alanina C-terminal. (Ver FIG. 4 de Hundsrucker et al. (2006).) Las secuencias de antagonistas peptídicos con afinidades particularmente altas por la secuencia RII DDD fueron las de AKAP-IS, AKAP76-wtpep, AKAP7 6-L304T-pep y AKAP76-L308D-pep.
AKAP-IS
QIEYLAKQIVDNAIQQA (SEQ ID NO: 3)
[0117] Carr et al. (2001, J Biol Chem 276:17332-38) examinaron el grado de homología de secuencia entre diferentes secuencias DDD de unión a AKAP de proteínas humanas y no humanas e identificaron residuos en las secuencias DDD que parecían ser los más conservados entre diferentes DDD. fracciones Estos se indican a continuación subrayando con referencia a la secuencia humana PKA RIla DDD de SEQ ID NO:1. Los residuos que se conservaron particularmente se indican en cursiva. Los residuos se superponen, pero no son idénticos a los sugeridos por Kinderman et al. (2006) como importante para la unión a proteínas AKAP. El experto en la materia se dará cuenta de que al diseñar variantes de secuencia de DDD, lo más preferible sería evitar cambiar los residuos más conservados (en cursiva), y también sería preferible evitar cambiar los residuos conservados (subrayados), mientras que las sustituciones conservativas de aminoácidos pueden considerarse para residuos que no están subrayados ni en cursiva.
S HIQ JFP GLTELLQGV TVE VLRQ QPFDLV EEAVE YFTRL REARA (SEO ID NO: 1)
[0118] Un conjunto modificado de sustituciones de aminoácidos conservadoras para la secuencia DDD1 (SEQ ID NO: 1), basado en los datos de Carr et al. (2001) se muestra en la Tabla 5. Incluso con este conjunto reducido de secuencias sustituidas, existen numerosas secuencias de restos DDD alternativas posibles que pueden ser producidas, probadas y utilizadas por el experto en la materia sin experimentación indebida. El experto en la materia podría derivar fácilmente tales secuencias de aminoácidos de DDD alternativas como las descritas anteriormente para la Tabla 2 y la Tabla 3.
Tabla 5. Sustituciones de aminoácidos conservadores en DDD1 (SEQ ID NO:1). Secuencia de consenso divulgada como SEQ ID NO:89.


[0119] El experto en la materia se dará cuenta de que estas y otras sustituciones de aminoácidos en las secuencias de aminoácidos de DDD o AD pueden utilizarse para producir especies alternativas dentro del género de fracciones de AD o DDD, usando técnicas que son estándar en el campo y solo experimentación de rutina.
Alotipos de anticuerpos
[0120] La inmunogenicidad de los anticuerpos terapéuticos está asociada con un mayor riesgo de reacciones a la infusión y una menor duración de la respuesta terapéutica (Baert et al., 2003, N Engl J Med 348:602-08). El grado en que los anticuerpos terapéuticos inducen una respuesta inmunitaria en el huésped puede estar determinado en parte por el alotipo del anticuerpo (Stickler et al., 2011, Genes and Immunity 12:213-21). El alotipo del anticuerpo está relacionado con las variaciones de la secuencia de aminoácidos en ubicaciones específicas en las secuencias de la región constante del anticuerpo. Los alotipos de anticuerpos IgG que contienen una región constante de tipo y de cadena pesada se denominan alotipos Gm (1976, J Immunol 117:1056-59).
[0121] Para los anticuerpos humanos IgG1 comunes, el alotipo más prevalente es G1m1 (Stickler et al., 2011, Genes and Immunity 12:213-21). Sin embargo, el alotipo G1m3 también ocurre con frecuencia en caucásicos (Id.). Se ha informado que los anticuerpos G1 m1 contienen secuencias alotípicas que tienden a inducir una respuesta inmunitaria cuando se administran a receptores que no son G1m1 (nG1m1), como los pacientes G1m3 (Id.). Los anticuerpos de alotipo distintos de G1m1 no son tan inmunogénicos cuando se administran a pacientes G1m1 (Id.).
[0122] El alotipo humano G1m1 comprende los aminoácidos ácido aspártico en la posición Kabat 356 y leucina en la posición Kabat 358 en la secuencia CH3 de la cadena pesada IgG1. El alotipo nG1m1 comprende los aminoácidos ácido glutámico en la posición de Kabat 356 y metionina en la posición de Kabat 358. Tanto los alotipos G1m1 como nG1m1 comprenden un residuo de ácido glutámico en la posición de Kabat 357 y los alotipos a veces se denominan alotipos DEL y EEM. Se muestra un ejemplo no limitante de las secuencias de la región constante de la cadena pesada para los anticuerpos alotipo G 1m 1 y nG 1m 1 para los anticuerpos ejemplares rituximab (SEQ ID NO: 85) y veltuzumab (SEQ ID NO: 86).
Secuencia de región variable de cadena pesada de rituximab (SEQ ID NO: 85)
ASTKGPS VTPL A PSSKSTSGGT A ALGCL YK D YFPCPVT VSWNSGALTSG VHTFPA VL
QSSGLYSLS SV VTVP S S SLGTQT YICN VNHKPSNTK VDKKAEPK SCDKTHTCPPCP AP
ELLGGPS VFJ .FPPKPKDTLMISRTPEVTC V W D V SHEDPE VKFNW YVDGVEVI1N AK
TKPREEQ YN S T Y R W S VLTVLHQDWLNGKE YK CKVSNKAL P APIEKTISKAKGQPRE
PQVYTLPPSRDELTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDG
SFFL Y SKLT VDKSRWQQGN VFSC S VMHE ALHNH YTQKSLSLSPGK
Región variable de cadena pesada de veltuzumab (SEQ ID NO: 86
ASTKGfPSVFPLAPSSKSTSGGTAALOCLVKDYFPEPVTVSWNSGALTSGVHTFPAVL
QS SGLYSLSSWTVP S S SLGTQTY1CNVNHKPSNTK VDKRVEPK SCDKTHTCPPCP AP
ELLGGPSVFLFPE'KPKDTLMISRTPEVTCWVDVSI ICDPEVKFNWYVDGVEVHNAK
TKPREEQYNS T Y R W S VL TVLHQDWLNGKE YK CKVSNKALPAP IE KTIS KAKGQ PRE
PQVYTLPP SREE MTKNQV SLTCLVKGFYP S DIA VEWE S NGQPENNYKT TPPVLD SDG
SF F L Y SKLTVDKSRW QQGN VFSC S VMHE ALHNHYTQKSL SLSPGK
[0123] Jefferis y Lefranc (2009, mAb 1:1-7) revisaron las variaciones de secuencia características de los alotipos de IgG y su efecto sobre la inmunogenicidad. Informaron que el alotipo G1m3 se caracteriza por un residuo de arginina en la posición 214 de Kabat, en comparación con un residuo de lisina en Kabat 214 en el alotipo G1m17. El alotipo nG 1m 1,2 se caracterizó por ácido glutámico en la posición de Kabat 356, metionina en la posición de Kabat 358 y alanina en la posición de Kabat 431. El alotipo G1 m1,2 se caracterizó por ácido aspártico en la posición de Kabat 356, leucina en la posición de Kabat 358 y glicina en la posición 431 de Kabat. Además de las variantes de secuencia de la región constante de la cadena pesada, Jefferis y Lefranc (2009) informaron variantes alotípicas en la región constante de la cadena ligera kappa, con el alotipo Km1 caracterizado por valina en la posición 153 de Kabat y leucina en la posición 191 de Kabat, el alotipo Km1,2 por alanina en la posición 153 de Kabat y leucina en la posición 191 de Kabat, y el alotipo Km3 caracterizado por alanina en la posición 153 de Kabat y valina en la posición 191 de Kabat.
[0124] Con respecto a los anticuerpos terapéuticos, veltuzumab y rituximab son, respectivamente, anticuerpos IgG1 humanizados y quiméricos contra CD20, de uso para la terapia de una amplia variedad de neoplasias malignas hematológicas. La Tabla 6 compara las secuencias alotípicas de rituximab frente a veltuzumab. Como se muestra en la Tabla 6 , rituximab (G1m17,1) es un alotipo IgG1 de DEL, con una variación de secuencia adicional en la posición 214 de Kabat (CH1 de cadena pesada) de lisina en rituximab frente a arginina en veltuzumab. Se ha informado que veltuzumab es menos inmunogénico en sujetos que rituximab (ver, por ejemplo, Morchhauser et al., 2009, J Clin Oncol 27:3346-53; Goldenberg et al., 2009, Blood 113:1062-70; Robak & Robak, 2011, BioDrugs 25:13-25), efecto que se ha atribuido a la diferencia entre anticuerpos humanizados y quiméricos. Sin embargo, la diferencia en los alotipos entre los alotipos EEM y DEL probablemente también explica la menor inmunogenicidad de veltuzumab.

[0125] Para reducir la inmunogenicidad de los anticuerpos terapéuticos en individuos de genotipo nG 1m 1, es deseable seleccionar el alotipo del anticuerpo para que corresponda al alotipo G1m3, caracterizado por arginina en Kabat 214, y el alotipo nulo nG1m1,2, caracterizado por ácido glutámico en la posición 356 de Kabat, metionina en la posición 358 de Kabat y alanina en la posición 431 de Kabat. Sorprendentemente, se encontró que la administración subcutánea repetida de anticuerpos G1m3 durante un largo período de tiempo no dio como resultado una respuesta inmunitaria significativa. En formas de realización alternativas, la cadena pesada de IgG4 humana en común con el alotipo G1m3 tiene arginina en Kabat 214, ácido glutámico en Kabat 356, metionina en Kabat 359 y alanina en Kabat 431. Dado que la inmunogenicidad parece relacionarse al menos en parte con los residuos en esas ubicaciones, el uso de la secuencia de la región constante de la cadena pesada de IgG4 humana para anticuerpos terapéuticos también es una forma de realización preferida. Las combinaciones de anticuerpos IgG1 G1m3 con anticuerpos IgG4 también pueden ser útiles para la administración terapéutica.
Sustituciones de aminoácidos
[0126] En formas de realización alternativas, los métodos y composiciones descritos pueden implicar la producción y el uso de proteínas o péptidos con uno o más residuos de aminoácidos sustituidos. Por ejemplo, las secuencias de DDD y/o AD usadas para hacer construcciones de DNL® pueden modificarse como se discutió anteriormente.
[0127] El experto en la materia sabrá que, en general, las sustituciones de aminoácidos normalmente implican la sustitución de un aminoácido por otro aminoácido de propiedades relativamente similares (es decir, sustituciones de aminoácidos conservativas). Las propiedades de los diversos aminoácidos y el efecto de la sustitución de aminoácidos sobre la estructura y función de la proteína han sido objeto de extensos estudios y conocimientos en la técnica.
[0128] Por ejemplo, se puede considerar el índice hidropático de los aminoácidos (Kyte & Doolittle, 1982, J. Mol. Biol., 157:105-132). El carácter hidropático relativo del aminoácido contribuye a la estructura secundaria de la proteína resultante, que a su vez define la interacción de la proteína con otras moléculas. A cada aminoácido se le ha asignado un índice hidropático en base a sus características de hidrofobicidad y carga (Kyte & Doolittle, 1982), estos son: isoleucina (+4.5); valina (+4,2); leucina (+3,8); fenilalanina (+2,8); cisteína/cistina (+2,5); metionina (+1,9); alanina (+1,8); glicina (-0,4); treonina (-0,7); serina (-0,8); triptófano (-0,9); tirosina (-1,3); prolina (-1,6); histidina (-3,2); glutamato (-3,5); glutamina (-3,5); aspartato (-3,5); asparagina (-3,5); lisina (-3,9); y arginina (-4,5). Al hacer sustituciones conservativas, se prefiere el uso de aminoácidos cuyos índices hidropáticos están dentro de ± 2, se prefieren más dentro de ± 1 y se prefieren aún más dentro de ± 0,5.
[0129] La sustitución de aminoácidos también puede tener en cuenta la hidrofilia del residuo de aminoácido (p. ej., patente de EE. UU. n.° 4.554.101). Se han asignado valores de hidrofilia a los residuos de aminoácidos: arginina (+3,0); lisina (+3,0); aspartato (+3,0); glutamato (+3,0); serina (+0,3); asparagina (+0,2); glutamina (+0,2); glicina (0); treonina (-0,4); prolina (-0,5 -,1); alanina (-0,5); histidina (-0,5); cisteína (-1,0); metionina (-1,3); valina (-1,5); leucina (-1,8); isoleucina (-1,8); tirosina (-2,3); fenilalanina (-2,5); triptófano (-3,4). Se prefiere la sustitución de aminoácidos por otros de hidrofilicidad similar.
[0130] Otras consideraciones incluyen el tamaño de la cadena lateral del aminoácido. Por ejemplo, generalmente no sería preferible reemplazar un aminoácido con una cadena lateral compacta, como glicina o serina, con un aminoácido con una cadena lateral voluminosa, por ejemplo, triptófano o tirosina. También se considera el efecto de varios residuos de aminoácidos en la estructura secundaria de la proteína. Mediante estudios empíricos, se ha determinado y se conoce en la técnica el efecto de diferentes residuos de aminoácidos sobre la tendencia de los dominios proteicos a adoptar una estructura secundaria de hélice alfa, hoja beta o giro inverso (véase, por ejemplo, Chou & Fasman, 1974, Biochemistry, 13:222-245, 1978, Ann. Rev. Biochem., 47: 251-276, 1979, Biophys. J., 26:367-384).
[0131] Sobre la base de tales consideraciones y extensos estudios empíricos, se han construido tablas de sustituciones de aminoácidos conservativas y son conocidas en la técnica. Por ejemplo: arginina y lisina; glutamato y aspartato; serina y treonina; glutamina y asparagina; y valina, leucina e isoleucina. Alternativamente: Ala (A) leu, ile, val; Arg (R) gln, asn, lys; Asn (N) his, asp, lys, arg, gln; Asp (D) asn, glu; Cys (C) ala, ser; Gln (Q) glu, asn; Glu (E) gln, asp; Gly (G) ala; Su (H) asn, gln, lys, arg; Ile (I) val, met, ala, phe, leu; Leu (L) val, met, ala, phe, ile; Lys (K) gln, asn, arg; Met (M) phe, ile, leu; Phe (F) leu, val, ile, ala, tyr; Pro (P) ala; Ser (S), thr; Thr (T) ser; Trp (W) phe, tyr; Tyr (Y) trp, phe, thr, ser; Val (V) ile, leu, met, phe, ala.
[0132] Otras consideraciones para las sustituciones de aminoácidos incluyen si el residuo está ubicado o no en el interior de una proteína o si está expuesto al solvente. Para residuos interiores, las sustituciones conservadoras incluirían: Asp y Asn; Ser y Thr; ser y ala; Thr y Ala; Ala y Gly; ile y Val; Val y Leu; Leu e Ilé; Leu y Met; Phé y Tyr; Tyr y Trp. (Consulte, por ejemplo, el sitio web de PROWL en rockefeller.edu) Para residuos expuestos a solventes, las sustituciones conservadoras incluirían: Asp y Asn; Asp y Glú; Glu y Gln; Glu y Ala; Gly y Asn; Ala y Pro; Ala y Gly; Ala y Ser; Ala y Lys; Ser y Thr; Lys y Arg; Val y Leu; Leu e Ilé; ile y Val; Phé y Tyr. (Id.) Se han construido varias matrices para ayudar en la selección de sustituciones de aminoácidos, como la matriz de puntuación PAM250, la matriz de Dayhoff, la matriz de Grantham, la matriz de McLachlan, la matriz de Doolittle, la matriz de Henikoff, la matriz de Miyata, la matriz de Fitch, la matriz de Jones, Rao matriz, matriz de Levin y matriz de Risler (Idem.)
[0133] Al determinar las sustituciones de aminoácidos, también se puede considerar la existencia de enlaces intermoleculares o intramoleculares, como la formación de enlaces iónicos (puentes salinos) entre residuos con carga positiva (p. ej., His, Arg, Lys) y residuos con carga negativa (p. ej., Asp, Glu) o enlaces disulfuro entre residuos de cisteína cercanos.
[0134] Los métodos para sustituir cualquier aminoácido por cualquier otro aminoácido en una secuencia de proteína codificada están bien conocidos y objeto de experimentación rutinaria para el experto en la materia, por ejemplo mediante la técnica de mutagénesis dirigida al sitio o mediante síntesis y ensamblaje de oligonucleótidos que codifican un aminoácido sustitución y empalme en una construcción de vector de expresión.
Avímeros
[0135] En ciertas formas de realización de la invención, como se define en las reivindicaciones, los restos de unión descritos en el presente documento pueden comprender una o más secuencias de avimer. Los avimeros son una clase de proteínas de unión algo similares a los anticuerpos en sus afinidades y especificidades por varias moléculas diana. Fueron desarrollados a partir de dominios de receptores extracelulares humanos mediante reproducción aleatoria de exones in vitro y presentación en fagos. (Silverman et al., 2005, Nat. Biotechnol. 23:1493-94; Silverman et al., 2006, Nat. Biotechnol. 24:220). Las proteínas de múltiples dominios resultantes pueden comprender múltiples dominios de unión independientes, que pueden mostrar una afinidad (en algunos casos subnanomolar) y una especificidad mejoradas en comparación con las proteínas de unión de un solo epítopo. (Id.) En varias formas de realización, los avimeros se pueden unir a, por ejemplo, secuencias DDD y/o AD para usar en los métodos y
composiciones reivindicados. Se describen detalles adicionales sobre los métodos de construcción y uso de avimeros, por ejemplo, en las publicaciones de solicitud de patente de EE. UU. n.° 20040175756, 20050048512, 20050053973, 20050089932 y 20050221384.
Exhibición de fagos
[0136] Ciertas formas de realización de la invención como se define en las reivindicaciones pueden abarcar péptidos de unión y/o miméticos de péptidos de diversas moléculas, células o tejidos diana. Los péptidos de unión pueden identificarse mediante cualquier método conocido en la técnica, que incluye, entre otros, la técnica de expresión en fagos. Se conocen bien en la técnica diversos métodos de presentación en fagos y técnicas para producir diversas poblaciones de péptidos. Por ejemplo, la patente de EE. u U. números 5.223.409; 5.622.699 y 6.068.829 describen métodos para preparar una biblioteca de fagos. La técnica de presentación en fagos implica la manipulación genética de bacteriófagos para que se puedan expresar pequeños péptidos en su superficie (Smith y Scott, 1985, Science 228:1315-1317; Smith y Scott, 1993, Meth. Enzymol. 21:228-257). Además de los péptidos, en la superficie de las partículas de fago también pueden mostrarse dominios de proteínas más grandes, como anticuerpos monocatenarios (Arap et al., 1998, Science 279:377-380).
[0137] Las secuencias de aminoácidos dirigidas selectivas para un órgano, tejido, tipo de célula o molécula diana determinados pueden aislarse mediante paneo (Pasqualini y Ruoslahti, 1996, Nature 380:364-366; Pasqualini, 1999, The Quart. J. Nucl. Med. 43:159-162). En resumen, se administra una biblioteca de fagos que contienen péptidos dirigidos putativos a un organismo intacto o a órganos, tejidos, tipos de células o moléculas diana aisladas y se recogen muestras que contienen fagos unidos. El fago que se une a una diana puede eluirse de un órgano, tejido, tipo de célula o molécula diana diana y luego amplificarse cultivándolos en la bacteria huésped.
[0138] El fago se puede propagar en la bacteria huésped entre rondas de cribado. En lugar de ser lisadas por el fago, la bacteria puede secretar múltiples copias del fago que muestran un inserto particular. Si se desea, el fago amplificado puede exponerse de nuevo a los órganos, tejidos, tipos de células o moléculas diana diana y recogerse para rondas adicionales de cribado. Se pueden realizar múltiples rondas de cribado hasta que se obtenga una población de aglutinantes selectivos o específicos. La secuencia de aminoácidos de los péptidos puede determinarse mediante la secuenciación del ADN correspondiente al inserto del péptido dirigido en el genoma del fago. El péptido dirigido identificado puede entonces producirse como un péptido sintético mediante técnicas estándar de química de proteínas (Arap et al., 1998, Smith et al., 1985).
[0139] Se puede usar un protocolo de sustracción para reducir aún más la unión de fagos de fondo. El propósito de la sustracción es eliminar fagos de la biblioteca que se unen a objetivos distintos del objetivo de interés. Alternativamente, la biblioteca de fagos se puede examinar previamente frente a una célula, tejido u órgano de control. Por ejemplo, los péptidos de unión a tumores pueden identificarse después de preseleccionar una biblioteca frente a una línea celular normal de control. Después de la sustracción, la biblioteca puede examinarse frente a la molécula, célula, tejido u órgano de interés. Se conocen y se pueden usar otros métodos de protocolos de sustracción, por ejemplo, como se describe en las patentes EE. UU. números 5.840.841, 5.705.610, 5.670.312 y 5.492.807.
Aptámeros
[0140] Un resto de direccionamiento de uso puede ser un aptámero. Los métodos para construir y determinar las características de unión de los aptámeros son bien conocidos en la técnica. Por ejemplo, dichas técnicas se describen en las patentes EE. UU. números 5.582.981, 5.595.877 y 5.637.459. Los métodos para la preparación y selección de aptámeros que se unen a dianas particulares de interés son bien conocidos, por ejemplo, las Patentes de EE. UU. n.° 5.475.096 y Patente de EE. UU. n.° 5.270.163.
[0141] Los aptámeros se pueden preparar mediante cualquier método conocido, incluidos los métodos sintéticos, recombinantes y de purificación, y se pueden usar solos o en combinación con otros ligandos específicos para el mismo objetivo. En general, se necesita un mínimo de aproximadamente 3 nucleótidos, preferiblemente al menos 5 nucleótidos, para efectuar la unión específica. Pueden ser factibles aptámeros de secuencias más cortas que 10 bases, aunque pueden preferirse aptámeros de 10, 20, 30 ó 40 nucleótidos.
[0142] Los aptámeros pueden aislarse, secuenciarse y/o amplificarse o sintetizarse como moléculas de ADN o ARN convencionales. Alternativamente, los aptámeros de interés pueden comprender oligómeros modificados. Cualquiera de los grupos hidroxilo normalmente presentes en los aptámeros puede reemplazarse por grupos fosfonato, grupos fosfato, protegerse con un grupo protector estándar o activarse para preparar enlaces adicionales con otros nucleótidos, o puede conjugarse con soportes sólidos. Uno o más enlaces fosfodiéster pueden reemplazarse por grupos de enlace alternativos, como P(O)O reemplazado por P(O)S, P(O)NR 2, P(O)R, P(O)OR', CO, o Cn R2, donde R es H o alquilo (1-20C) y R' es alquilo (1-20C); además, este grupo puede unirse a nucleótidos adyacentes a través de O o S. No todos los enlaces en un oligómero necesitan ser idénticos.
Aficuerpos y Fynómeros
[0143] Ciertos ejemplos pueden utilizar aficuerpos en lugar de anticuerpos. Los aficuerpos están disponibles comercialmente en Affibody AB (Solna, Suecia). Los aficuerpos son proteínas pequeñas que funcionan como miméticos de anticuerpos y son útiles para unir moléculas diana. Los aficuerpos se desarrollaron mediante ingeniería combinatoria en un andamio de proteína alfa helicoidal (Nord et al., 1995, Protein Eng 8:601-8; Nord et al., 1997, Nat Biotechnol 15:772-77). El diseño del aficuerpo se basa en una estructura de haz de tres hélices que comprende el dominio de unión a IgG de la proteína A (Nord et al., 1995; 1997). Se pueden producir aficuerpos con una amplia gama de afinidades de unión mediante la aleatorización de trece aminoácidos implicados en la actividad de unión a Fc de la proteína A bacteriana (Nord et al., 1995; 1997). Después de la aleatorización, la biblioteca amplificada por PCR se clonó en un vector de fagémido para el cribado mediante la presentación en fago de las proteínas mutantes. La biblioteca de presentación en fagos puede examinarse frente a cualquier antígeno conocido, usando técnicas de selección de presentación en fagos estándar (p. ej., Pasqualini y Ruoslahti, 1996, Nature 380:364-366; Pasqualini, 1999, Quart. J. Nucl. Med. 43:159- 162), con el fin de identificar uno o más aficuerpos contra el antígeno diana.
[0144] Se ha demostrado que un aficuerpo marcado con 177Lu específico para HER2/neu se dirige a xenoinjertos que expresan HER2 in vivo (Tolmachev et al., 2007, Cancer Res 67:2773-82). Aunque la toxicidad renal debida a la acumulación del compuesto radiomarcado de bajo peso molecular fue inicialmente un problema, la unión reversible a la albúmina redujo la acumulación renal, lo que permitió la terapia basada en radionucleidos con aficuerpo marcado (Id.).
[0145] Recientemente se ha demostrado la viabilidad de utilizar aficuerpos radiomarcados para la obtención de imágenes de tumores in vivo (Tolmachev et al., 2011, Bioconjugate Chem 22:894-902). Un NOTA derivado de maleimida se conjugó con el aficuerpo anti-HER2 y se marcó radiactivamente con 111In (Id.). La administración a ratones que portaban el xenoinjerto de DU-145 que expresaba HER2, seguida de imágenes de cámara gamma, permitió la visualización del xenoinjerto (Id.).
[0146] Los fenómeros también pueden unirse a antígenos diana con una afinidad y especificidad similares a las de los anticuerpos. Los fenómeros se basan en el dominio Fyn SH3 humano como andamio para el ensamblaje de moléculas de unión. El dominio Fyn SH3 es una proteína de 63 aminoácidos completamente humana que se puede producir en bacterias con altos rendimientos. Los fonómeros se pueden unir para producir una proteína de unión multiespecífica con afinidades por dos o más dianas antigénicas diferentes. Los fenómeros están disponibles comercialmente en COVAGEN AG (Zurich, Suiza).
[0147] El experto en la materia se dará cuenta de que se pueden usar aficuerpos o fenómeros como moléculas de direccionamiento en la práctica de los métodos y composiciones reivindicados.
Protocolos de conjugación
[0148] El protocolo de conjugación preferido se basa en una reacción de tiol-maleimida, tiol-vinilsulfona, tiolbromoacetamida o tiol-yodoacetamida que es fácil a pH neutro o ácido. Esto evita la necesidad de condiciones de pH más altas para las conjugaciones como, por ejemplo, serían necesarias cuando se usan ésteres activos. Más detalles de los protocolos de conjugación ejemplares se describen a continuación en la sección de Ejemplos.
Tratamiento terapéutico
[0149] En otro aspecto, la presente descripción se refiere a un método para tratar a un sujeto, que comprende administrar una cantidad terapéuticamente eficaz de un anticuerpo o inmunoconjugado como se describe en el presente documento a un sujeto, preferiblemente en combinación con un inhibidor de PARP, un inhibidor de microtúbulos, un inhibidor de quinasa de Bruton y/o o inhibidor de PI3K. Las enfermedades que se pueden tratar con los anticuerpos o inmunoconjugados descritos en este documento incluyen, entre otras, tumores malignos de células B (p. ej., linfoma no Hodgkin, linfoma de células del manto, mieloma múltiple, linfoma de Hodgkin, linfoma difuso de células B grandes, linfoma de Burkitt, linfoma folicular, leucemia linfocítica aguda, leucemia linfocítica crónica, leucemia de células pilosas) usando, por ejemplo, un anticuerpo anti-CD22 como el MAb hLL2 (epratuzumab, consulte la Patente de EE. UU. N° 6.183.744), contra otro epítopo CD22 (hRFB4) o anticuerpos contra otros antígenos de células B, como CD19, CD20, CD21, CD22, CD23, CD37, CD40, CD40L, CD52, CD74, CD80 o HLA-DR. Otras enfermedades incluyen, pero no se limitan a, adenocarcinomas del epitelio del sistema digestivo derivado del endodermo, cánceres como el cáncer de mama y el cáncer de pulmón de células no pequeñas y otros carcinomas, sarcomas, tumores gliales, leucemias mieloides, etc. En particular, los anticuerpos contra un antígeno, por ejemplo, un antígeno oncofetal, producido por o asociado con un tumor sólido maligno o neoplasia hematopoyética, por ejemplo, una neoplasia gastrointestinal, de estómago, de colon, de esófago, de hígado, de pulmón, de mama, de páncreas, de hígado, de próstata, de ovario, de testículo, de cerebro, de hueso o se utilizan ventajosamente un tumor linfático, un sarcoma o un melanoma. Dichas terapias pueden administrarse una o varias veces, según el estado de la enfermedad y la tolerabilidad del conjugado, y también pueden usarse opcionalmente en combinación con otras modalidades terapéuticas, como cirugía, radiación externa, radioinmunoterapia, inmunoterapia, quimioterapia, terapia antisentido,
interferencia Terapia de ARN, terapia génica y similares. Cada combinación se adaptará al tipo de tumor, estadio, condición del paciente y terapia previa, y otros factores considerados por el médico tratante.
[0150] Como se usa en el presente documento, el término "sujeto" se refiere a cualquier animal (es decir, vertebrados e invertebrados) incluidos, entre otros, mamíferos, incluidos los seres humanos. No se pretende que el término se limite a una edad o sexo en particular. Por lo tanto, el término abarca sujetos adultos y recién nacidos, así como fetos, ya sean hombres o mujeres. Las dosis proporcionadas en este documento son para humanos, pero se pueden ajustar al tamaño de otros mamíferos, así como a niños, de acuerdo con el peso o el tamaño del metro cuadrado. Cuando se administra un anticuerpo o inmunoconjugado a un sujeto humano, el experto en la materia se dará cuenta de que el antígeno diana al que se une el anticuerpo o el inmunoconjugado será un antígeno humano.
[0151] En una forma de realización preferida de la invención como se define en las reivindicaciones, el inmunoconjugado que comprende un anticuerpo antiEGP-1 (anti-Trop-2) es el Mab hRS7 que puede usarse para tratar carcinomas tales como carcinomas de esófago, páncreas, pulmón, estómago, colon y recto, vejiga urinaria, mama, ovario, útero, riñón y próstata, como se describe en la Patente de Estados Unidos n.° 7.238.785; 7.517.964 y 8.084.583. Un anticuerpo hRS7 es un anticuerpo humanizado que comprende secuencias de la región determinante de la complementariedad (CDR) de cadena ligera CDR1 (KASQDVSIAVA, SEQ ID NO: 90); CDR2 (SASYRYT, SEQ ID NO: 91); y CDR3 (QQHYITPLT, SEQ ID NO: 92) y secuencias CDR de cadena pesada CDR1 (NYGMN, SEQ ID NO: 93); CDR2 (WINTYTGEPTYTDDFKG, SEQ ID NO: 94) y CDR3 (GGFGSSYWYFDV, SEQ ID NO: 95).
[0152] Los anticuerpos o inmunoconjugados que comprenden un anticuerpo anti-CEACAM5 (p. ej., hMN-14, labretuzumab) y/o un anticuerpo anti-CEACAM6 (p. ej., hMN-3 o hMN-15) pueden usarse para tratar cualquiera de una variedad de cánceres que expresan CEACAM5 y/o CEACAM6, como se describe en las Patentes de EE. UU. Nos. 7.541.440; 7.951.369; 5.874.540; 6.676.924 y 8.267.865. Los tumores sólidos que se pueden tratar con anti-CEACAM5, anti-CEACAM6 o una combinación de ambos incluyen, entre otros, mama, pulmón, páncreas, esófago, tiroides medular, ovario, colon, recto, vejiga urinaria, boca y cánceres de estómago. La mayoría de los carcinomas, incluidos los cánceres gastrointestinales, respiratorios, genitourinarios y de mama, expresan CEACAM5 y pueden tratarse con los anticuerpos o inmunoconjugados del sujeto. Un anticuerpo hMN-14 es un anticuerpo humanizado que comprende secuencias CDR de región variable de cadena ligera CDr 1 (KASQDVGTSVA; SEQ ID NO: 96), CDR2 (WTSTRHT; SEQ ID NO: 97) y CDR3 (QQYSLYRS; SEQ ID NO: 98), y las secuencias CDR de región variable de cadena pesada CDR1 (TYWMS; SEQ ID NO: 99), CDR2 (EIHPDSSTINYAPSLKD; SEQ ID NO: 100) y CDR3 (LYFGFpWFAY; SEQ ID NO: 101). Un anticuerpo hMN-3 es un anticuerpo humanizado que comprende secuencias CDR de región variable de cadena ligera CDR1 (RSSQSIVHSNGNTYLE, SEQ ID NO: 102), CDR2 (KVSNRFS, SEQ ID NO: 103) y CDR3 (FQGSHVPPT, SEQ ID NO: 104) y las secuencias CDR de cadena pesada CDR1 (NYGMN, SEQ ID NO: 105), CDR2 (WINTYTGEPTYADDFKG, SEQ ID NO: 106) y CDR3 (KGWMDFNSSLDY, SEQ ID NO: 107). Un anticuerpo hMN-15 es un anticuerpo humanizado que comprende secuencias CDR de región variable de cadena ligera SASSRVSYIH (SEQ ID NO: 108); GTSTLAS (SEQ ID NO: 109); y QQWSYNPPT (SEQ ID NO: 110); y secuencias CDR de región variable de cadena pesada DYYMS (SEQ ID NO: 111); FIANKANGHTTDYSPSVKG (SEQ ID NO: 112); y DMGIRWNFDV (SEQ ID NO: 113).
[0153] Los anticuerpos o inmunoconjugados que comprenden un anticuerpo anti-CD74 (p. ej., hLL1, milatuzumab, descritos en las patentes de EE. limitado a cáncer renal, de pulmón, intestinal, de estómago, de mama, de próstata o de ovario, así como varios cánceres hematológicos, tales como mieloma múltiple, leucemia linfocítica crónica, leucemia linfoblástica aguda, linfoma no Hodgkin y linfoma de Hodgkin. Un anticuerpo hLL1 es un anticuerpo humanizado que comprende las secuencias CDR de cadena ligera CDR1 (RSSQSLVHRNGNTYLH; SEQ ID NO: 114), CDR2 (TVSNRFS; SEQ ID NO: 115) y CDR3 (SQSSHVPPT; SEQ ID NO: 116) y la cadena pesada secuencias CDR de región variable CDR1 (NYGVN; SEQ ID NO: 117), CDR2 (WINPNTGEPTFDDDFKG; SEQ ID NO: 118) y CDR3 (SRGKNEAWFAY; SEQ ID NO: 119). Los anticuerpos o inmunoconjugados que comprenden un anticuerpo anti-CD22 (p. ej., hLL2, epratuzumab, descritos en las patentes de EE. variedad de cánceres que expresan CD22, incluidos, entre otros, formas indolentes de linfomas de células B, formas agresivas de linfomas de células B, leucemias linfáticas crónicas, leucemias linfáticas agudas, linfoma no Hodgkin, linfoma de Hodgkin, linfoma de Burkitt, linfoma folicular o Linfoma difuso de células B. Un anticuerpo hLL2 es un anticuerpo humanizado que comprende secuencias CDR de cadena ligera CDR1 (KSSQSVLYSANHKYLA, SEQ ID NO: 120), CDR2 (WASTRES, SEQ ID NO: 121) y CDR3 (HQYLSSWTF, SEQ ID NO: 122) y la cadena pesada CDR secuencias CDR1 (SYWLH, SEQ ID NO: 123), CDR2 (YINPRNDYTEYNQNFKD, SEQ ID NO: 124) y CDR3 (RDITTFY, SEQ ID NO: 125).
[0154] Los anticuerpos o inmunoconjugados que comprenden anticuerpos anti-CSAp, tales como el MAb hMu-9, pueden usarse para tratar cánceres colorrectales, pancreáticos y de ovario como se describe en las Patentes de EE. UU. N.os 6.962.702; 7.387.772; 7.414.121; 7.553.953; 7.641.891 y 7.670.804. Además, los anticuerpos o inmunoconjugados que comprenden el mAb hPAM4 se pueden usar para tratar el cáncer de páncreas u otros tumores sólidos, como se describe en las patentes EE. UU. números 7.238.786 y 7.282.567. Un anticuerpo hMu-9 es un anticuerpo humanizado que comprende secuencias CDR de cadena ligera CDR1 (RSSQSIVHSNGNTYLE, SEQ ID NO: 126), CDR2 (KVSNRFS, SEQ ID NO: 127) y CDR3 (FQGSRVPYT, SEQ ID NO: 128), y secuencias CDR de cadena variable CDR1 (EYVIT, SEQ ID NO: 129), CDR2 (EIYPGSGSTSYNEKFK, SEQ ID NO: 130) y CDR3 (EDL, SEQ ID NO: 131). Un anticuerpo hPAM4 es un anticuerpo humanizado que comprende secuencias CDR de región
variable de cadena ligera CDR1 (SASSSVSSSYLY, SEQ ID NO: 132); CDR2 (STSNLAS, SEQ ID NO: 133); y CDR3 (HQWNRYPYT, SEQ ID NO: 134); y secuencias CDR de cadena pesada CDR1 (SYVLH, SEQ ID NO: 135); CDR2 (YINPYNDGTQYNEKFKG, SEQ ID NO: 136) y CDR3 (GFGGSYGFAY, SEQ ID NO: 137).
[0155] Los anticuerpos o inmunoconjugados que comprenden un MAb anti-alfa fetoproteína (AFP), como IMMU31, pueden usarse para tratar carcinoma hepatocelular, tumores de células germinales y otros tumores productores de AFP usando formas de anticuerpos humanizados, quiméricos y humanos, como se describe en la patente de EE. UU. n.° 7.300.655. Un anticuerpo IMMU31 es un anticuerpo humanizado que comprende las secuencias CDR de cadena pesada CDR1 (SYVIH, SEQ ID NO: 138), CDR2 (YIHPYNGGTKYNEKFKG, SEQ ID NO: 139) y CDR3 (SGGGDPFAY, SEQ ID NO: 140) y la cadena ligera CDR1 (KASQDINKYIG, SEQ ID NO: 141), CDR2 (YTSALLP, SEQ ID NO: 142) y CDR3 (LQYDDLWT, SEQ ID NO: 143).
[0156] Los anticuerpos o inmunoconjugados que comprenden un MAb anti-HLA-DR, como hL243 (IMMU-114), pueden usarse para tratar linfoma, leucemia, mieloma múltiple, cánceres de piel, esófago, estómago, colon, recto, páncreas, pulmón, mama, ovario, vejiga, endometrio, cuello uterino, testículos, riñón, hígado, melanoma u otros tumores productores de HLA-DR, como se describe en la Patente de EE. UU. n.° 7.612.180. Un anticuerpo hL243 es un anticuerpo humanizado que comprende las secuencias CDR de cadena pesada CDR1 (NYGMN, SEQ Id NO: 144), CDR2 (WINTYTREPTYADDFKG, SEQ ID NO: 145) y CDR3 (DITAVVPTGFDY, SEQ ID NO: 146) y CDR de cadena ligera secuencias CDR1 (RASENIYSNLA, SEQ ID NO: 147), CDR2 (AASNLAD, SEQ ID NO: 148) y CDR3 (QHFWTTPWA, SEQ ID NO: 149).
[0157] Los anticuerpos o inmunoconjugados que comprenden un MAb anti-CD20, como veltuzumab (hA20), 1F5, obinutuzumab (GA101) o rituximab, pueden usarse para tratar linfoma, leucemia, púrpura trombocitopénica inmune, lupus eritematoso sistémico, síndrome de Sjogren, síndrome de Evans, artritis, arteritis, pénfigo vulgar, rechazo de injerto renal, rechazo de injerto cardíaco, artritis reumatoide, linfoma de Burkitt, linfoma no Hodgkin, linfoma folicular, linfoma linfocítico pequeño, linfoma difuso de células B, linfoma de la zona marginal, leucemia linfocítica crónica, leucemia linfocítica aguda, diabetes mellitus tipo I, GVHD o esclerosis múltiple, como se describe en las patentes EE. UU. números 7.435.803 o 8.287.864. Un anticuerpo hA20 (veltuzumab) es un anticuerpo humanizado que comprende las secuencias CDR de cadena ligera CDRL1 (RASSSVSYIH, SEQ ID NO: 150), CDRL2 (ATSNLAS, SEQ ID NO: 151) y CDRL3 (QQWTSNPPT, SEQ ID NO: 152) y pesado secuencias CDR de cadena CDRH1 (SYNMH, SEQ ID NO: 153), CDRH2 (AIYPGNGDTSYNQKFKG, SEQ ID NO: 154) y CDRH3 (STYYGGDWYFDV, SEQ ID NO: 155). Los anticuerpos o inmunoconjugados que comprenden un MAb anti-CD19, tal como hA19, pueden usarse para tratar linfomas y leucemias relacionados con células B, tales como linfoma no Hodgkin, leucemia linfocítica crónica o leucemia linfoblástica aguda. Otros estados de enfermedad que pueden tratarse incluyen enfermedades autoinmunes, como trombocitopenia inmune aguda o crónica, dermatomiositis, corea de Sydenham, miastenia grave, lupus eritematoso sistémico, nefritis lúpica, fiebre reumática, síndromes poliglandulares, penfigoide ampolloso, diabetes mellitus, púrpura de Henoch-Schonlein, nefritis posestreptocócica, eritema nudoso, arteritis de Takayasu, enfermedad de Addison, artritis reumatoide, esclerosis múltiple, sarcoidosis, colitis ulcerosa, eritema multiforme, nefropatía IgA, poliarteritis nodosa, espondilitis anquilosante, síndrome de Goodpasture, tromboangitis ubiterante, síndrome de Sjogren, primario cirrosis biliar, tiroiditis de Hashimoto, tirotoxicosis, esclerodermia, hepatitis crónica activa, polimiositis/dermatomiositis, policondritis, pénfigo vulgar, granulomatosis de Wegener, nefropatía membranosa, esclerosis lateral amiotrófica, tabes dorsal, arteritis de células gigantes/polimialgia, anemia perniciosa, glomerulonefritis rápidamente progresiva, psoriasis y alveolitis fibrosante, como se describe en las patentes EE. UU. números 7.109.304, 7.462.352, 7.902.338, 8.147.831 y 8.337.840. Un anticuerpo hA19 es un anticuerpo humanizado que comprende las secuencias CDR de cadena ligera CDR1 KASQSVDy Dg DSYLN (SEQ ID n O: 156); CDR2 DASNLVS (SEQ ID NO: 157); y CDR3 QQSTEDPWT (SEQ ID NO: 158) y las secuencias CDR de cadena pesada CDR1 SYWMN (SEQ ID NO: 159); CDR2 QIWPGDGDTNYNGKFKG (SEQ ID NO: 160) y CDR3 RETTTVGRYYYAMDY (SEQ ID NO: 161). Los anticuerpos o inmunoconjugados que comprenden anticuerpos antitenascina pueden usarse para tratar tumores hematopoyéticos y sólidos, y los conjugados que comprenden anticuerpos contra tenascina pueden usarse para tratar tumores sólidos, preferiblemente cánceres cerebrales como glioblastomas.
[0158] En una forma de realización preferida de la invención como se define en las reivindicaciones, los anticuerpos que se usan en el tratamiento de enfermedades humanas son versiones de anticuerpos humanos o humanizados (injertados con CDR), aunque se pueden usar versiones murinas y quiméricas de anticuerpos. Se prefieren principalmente moléculas de IgG de la misma especie como agentes de suministro para minimizar las respuestas inmunitarias. Esto es particularmente importante cuando se consideran tratamientos repetidos. Para los seres humanos, es menos probable que un anticuerpo IgG humano o humanizado genere una respuesta inmunitaria anti-IgG en los pacientes. Los anticuerpos como hLL1 y hLL2 se internalizan rápidamente después de unirse al antígeno de internalización en las células diana, lo que significa que el fármaco transportado también se internaliza rápidamente en las células. Sin embargo, los anticuerpos que tienen velocidades de internalización más lentas también pueden usarse para efectuar una terapia selectiva.
[0159] Los anticuerpos o inmunoconjugados pueden usarse para tratar enfermedades autoinmunes o disfunciones del sistema inmunitario (p. ej., enfermedad de injerto contra huésped, rechazo de trasplante de órganos). Los
anticuerpos de uso para tratar la enfermedad de disfunción autoinmune/inmune pueden unirse a antígenos ejemplares que incluyen, entre otros, BCL-1, BCL-2, BCL-6, CD1a, CD2, CD3, CD4, CD5, CD7, CD8, CD10, CD11b, CD11c, CD13, CD14, CD15, CD16, CD19, CD20, CD21, CD22, CD23, CD25, CD33, CD34, CD38, CD40, CD40L, CD41a, CD43, CD45, CD55, CD56, CCD57, CD59, CD64, CD71, CD74, CD79a, CD79b, CD117, CD138, FMC-7 y HLA-DR. Los anticuerpos que se unen a estos y otros antígenos diana, discutidos anteriormente, pueden usarse para tratar enfermedades autoinmunes o disfunciones inmunes. Las enfermedades autoinmunes que pueden tratarse con anticuerpos o inmunoconjugados pueden incluir púrpura trombocitopénica idiopática aguda, púrpura trombocitopénica idiopática crónica, dermatomiositis, corea de Sydenham, miastenia grave, lupus eritematoso sistémico, nefritis lúpica, fiebre reumática, síndromes poliglandulares, penfigoide ampolloso, diabetes mellitus, Henoch-Púrpura de Schonlein, nefritis posestreptocócica, eritema nodoso, arteritis de Takayasu, vasculitis asociadas a ANCA, enfermedad de Addison, artritis reumatoide, esclerosis múltiple, sarcoidosis, colitis ulcerosa, eritema multiforme, nefropatía IgA, poliarteritis nodosa, espondilitis anquilosante, síndrome de Goodpasture, tromboangitis obliterante, síndrome de Sjogren, cirrosis biliar primaria, tiroiditis de Hashimoto, tirotoxicosis, esclerodermia, hepatitis crónica activa, polimiositis/dermatomiositis, policondritis, penfigoide ampolloso, pénfigo vulgar, granulomatosis de Wegener, nefropatía membranosa, esclerosis lateral amiotrófica, tabes dorsal, arteritis de células gigantes/polimialgia, anemia perniciosa, glomerulonefritis rápidamente progresiva, psoriasis o alveolitis fibrosante.
[0160] En otra modalidad preferida de la invención como se define en las reivindicaciones, un agente terapéutico usado en combinación con los inmunoconjugados puede comprender uno o más isótopos. Los isótopos radiactivos útiles para tratar tejidos enfermos incluyen, entre otros: 111In, 177Lu, 212Bi, 213Bi, 211At, 62Cu, 67Cu, 90Y, 1251,131I, 32P, 33P, 47Sc, 111Ag 67Ga, 142Pr, 153Sm, 161Tb, 166Dy, 166Ho, 186Re, 188Re, 189Re, 212Pb, 223Ra, 225Ac, 59Fe, 75Se, 77As, 89Sr, 9 Mo, 105Rh, 109Pd, 143Pr, 149Pm, 169Er, 194Ir, 198Au, 199Au, 227Th y 211Pb. El radionucleido terapéutico tiene preferiblemente una energía de desintegración en el rango de 20 a 6000 keV, preferiblemente en el rango de 60 a 200 keV para un emisor Auger, 100-2500 keV para un emisor beta y 4000-6000 keV para un emisor alfa. Las energías máximas de desintegración de los nucleidos emisores de partículas beta útiles son preferiblemente de 20 a 5000 keV, más preferiblemente de 100 a 4000 keV y lo más preferiblemente de 500 a 2500 keV. También se prefieren los radionúclidos que se desintegran sustancialmente con partículas emisoras Auger. Por ejemplo, Co-58, Ga-67, Br-80m, Tc-99m, Rh-103m, Pt-109, In-111, Sb-119, I-125, Ho-161, Os-189m e Ir-192. Las energías de desintegración de los nucleidos emisores de partículas beta útiles son preferentemente <1.000 keV, más preferentemente <100 keV y lo más preferentemente <70 keV. También se prefieren los radionúclidos que se desintegran sustancialmente con la generación de partículas alfa. Dichos radionucleidos incluyen, entre otros: Dy-152, At-211, Bi-212, Ra-223, Rn-219, Po-215, Bi-211, Ac-225, Fr-221, At-217, Bi-213, Th-227 y Fm-255. Las energías de desintegración de los radionucleidos emisores de partículas alfa útiles son preferentemente de 2000 a 10000 keV, más preferentemente de 3000 a 8000 keV y lo más preferentemente de 4000 a 7000 keV. Los radioisótopos potenciales adicionales de uso incluyen 11C, 13N, 15O, 75Br, 198Au, 224Ac, 126I, 133I, 77Br, 113mIn, 95Ru, 97Ru, 103Ru, 105Ru, 107Hg, 203Hg, 121mTe, 122mTe, 125mTe, 165Tm, 167Tm, 168Tm, 197Pt, 109Pd, 105Rh, 142Pr, 143Pr, 161Tb, 166Ho, 199Au, 57Co, 58Co, 51Cr, 59Fe, 75Se, 201Tl, 225Ac, 76Br, 169Yb y similares.
[0161] Los radionúclidos y otros metales pueden administrarse, por ejemplo, utilizando grupos quelantes unidos a un anticuerpo o inmunoconjugado. Los quelatos macrocíclicos como NOTA, DOTA y TETA son útiles con una variedad de metales y radiometales, más particularmente con radionúclidos de galio, itrio y cobre, respectivamente. Dichos complejos de quelato de metal se pueden hacer muy estables adaptando el tamaño del anillo al metal de interés. Se pueden usar otros quelatos de tipo anillo, tales como poliéteres macrocíclicos para complejar 223Ra.
[0162] Los agentes terapéuticos de uso en combinación con los anticuerpos o inmunoconjugados descritos en este documento también incluyen, por ejemplo, fármacos quimioterapéuticos tales como alcaloides de la vinca, antraciclinas, epidofilotoxinas, taxanos, antimetabolitos, inhibidores de tirosina quinasa, inhibidores de tirosina quinasa de Bruton, inhibidores de microtúbulos, inhibidores de PARP, PI3K inhibidores, agentes alquilantes, antibióticos, inhibidores de Cox-2, agentes antimitóticos, antiangiogénicos y proapoptóticos, particularmente doxorrubicina, metotrexato, taxol, otras camptotecinas y otras de estas y otras clases de agentes anticancerígenos, y similares. Otros fármacos quimioterapéuticos contra el cáncer incluyen mostazas nitrogenadas, sulfonatos de alquilo, nitrosoureas, triacenos, análogos de ácido fólico, análogos de pirimidina, análogos de purina, complejos de coordinación de platino, hormonas y similares. Los agentes quimioterapéuticos adecuados se describen en REMINGTON'S PHARMACEUTICAL SCIENCES, 19a ed. (Mack Publishing Co. 1995), y en THE PARMACOLOGICAL BASIS OF THERAPEUTICS DE GOODMAN Y GILMAN, 7 a ed. (MacMillan Publishing Co. 1985), así como ediciones revisadas de estas publicaciones. Otros agentes quimioterapéuticos adecuados, tales como fármacos experimentales, son conocidos por los expertos en la técnica.
[0163] Ejemplos de fármacos de uso incluyen, entre otros, 5-fluorouracilo, afatinib, aplidina, azaribina, anastrozol, antraciclinas, axitinib, AVL-101, AVL-291, bendamustina, bleomicina, bortezomib, bosutinib, briostatina-1, busulfán, caliqueamicina, camptotecina, carboplatino, 10-hidroxicamptotecina, carmustina, celebrex, clorambucilo, cisplatino (CDDP), inhibidores de la Cox-2, irinotecán (CPT-11), SN-38, carboplatino, cladribina, camptoteca, crizotinib, ciclofosfamida, citarabina, dacarbazina, dasatinib, dinaciclib, docetaxel, dactinomicina, daunorrubicina, doxorrubicina, 2-pirrolinodoxorrubicina (2PDOX), pro-2PDOX, ciano-morfolino doxorrubicina, doxorrubicina glucurónido, epirrubicina glucurónido, erlotinib, estramustina, epidofilotoxina, erlotinib, entinostat, agentes de unión al receptor de estrógeno,
etopósido (VP16), glucurónido de etopósido, fosfato de etopósido, exemestano, fingolimod, floxuridina (FUdR), 3',5'-O-dioleoil-FudR (FUdR-dO), fludarabina, flutamida, inhibidores de la farnesil-proteína transferasa, flavopiridol, fostamatinib, ganetespib, GDC-0834, GS- 1101, gefitinib, gemcitabina, hidroxiurea, ibrutinib, idarubicin, idelalisib, ifosfamida, imatinib, L-asparaginasa, lapatinib, lenolidamida, leucovorin, LFM-A13, lomustina, mecloretamina, melfalano, mercaptopurina, 6 -mercaptopurina, metotrexato, mitoxantrona, mitramicina, mitomicina, mitotano, navelbina, neratinib, nilotinib, nitrosurea, olaparib, plicomicina, procarbazina, paclitaxel, PCI-32765, pentostatina, PSI-341, raloxifeno, semustina, sorafenib, estreptozocina, SU11248, sunitinib, tamoxifeno, temazolomida (una forma acuosa de DTIC), transplatino, talidomida, tioguanina, tiotepa, tenipósido, topotecan, uracilo mostaza, vatalanib, vinorelbina, vinblastina, vincristina, vinca alcaloides y ZD1839. Dichos agentes pueden ser parte de los conjugados descritos en el presente documento o, alternativamente, pueden administrarse en combinación con los conjugados descritos, ya sea antes, simultáneamente o después del conjugado. Alternativamente, se pueden usar uno o más anticuerpos desnudos terapéuticos como se conocen en la técnica en combinación con los conjugados descritos. Anteriormente se describen ejemplos de anticuerpos desnudos terapéuticos.
[0164] En formas de realización preferidas de la invención, como se define en las reivindicaciones, un agente terapéutico para usar en combinación con un conjugado de anticuerpo que rompe el ADN (p. ej., un SN-38-ADC) es un inhibidor de microtúbulos, como un alcaloide de la vinca, un taxano, un maitansinoide o una auristatina. Los ejemplos de inhibidores de microtúbulos conocidos incluyen paclitaxel, vincristina, vinblastina, mertansina, epotilona, docetaxel, discodermolida, combrestatina, podofilotoxina, CI-980, fenilahistinas, esteganacinas, curacinas, 2-metoxiestradiol, E7010, metoxibencenosuflonamidas, vinorelbina, vinflunina, vindesina, dolastatinas, espongistatina, rizoxina, tasidotina, halicondrinas, hemiasterlinas, criptoficina 52, MMAE y mesilato de eribulina.
[0165] En una forma de realización preferida alternativa de la invención como se define en las reivindicaciones, un agente terapéutico que se utilizará en combinación con un ADC que rompe el ADN, como un conjugado de anticuerpo SN-38, es un inhibidor de PARP, como olaparib, talazoparib (BMN-673), rucaparib, veliparib, Ce P 9722, MK 4827, BGB-290, ABT-8 8 8 , AG014699, BSI-201, CEP-8983 o 3-aminobenzamida.
[0166] Alternativamente, un agente terapéutico usado en combinación con un anticuerpo o inmunoconjugado es un inhibidor de la quinasa de Bruton, como ibrutinib (PCI-32765), PCI-45292, CC-292 (AVL-292), ONO-4059, GDC-0834, LFM-A13 o RN486.
[0167] En otra alternativa más, un agente terapéutico utilizado en combinación con un anticuerpo o inmunoconjugado es un inhibidor de PI3K, como idelalisib, Wortmannin, demethoxyviridin, perifosine, PX-8 6 6 , IPI-145 (duvelisib), BAY 80-6946, BEZ235, RP6530, TGR1202, SF1126, INK1117, GDC-0941, BKM120, XL147, XL765, Palomid 529, GSK1059615, ZSTK474, PWT33597, IC87114, TG100-115, CAL263, PI-103, GNE477, CUDC-907, A EZS-136 o LY294002.
[0168] Los agentes terapéuticos que pueden usarse en la práctica de la invención como se define en las reivindicaciones junto con los anticuerpos o inmunoconjugados también pueden comprender toxinas conjugadas con restos de direccionamiento. Las toxinas que pueden usarse a este respecto incluyen ricina, abrina, ribonucleasa (RNasa), DNasa I, enterotoxina A estafilocócica, proteína antiviral de hierba carmín, gelonina, toxina diftérica, exotoxina de Pseudomonas y endotoxina de Pseudomonas. (Véase, por ejemplo, Pastan. et al., Cell (1986), 47:641, y Sharkey y Goldenberg, CA Cancer J Clin. 2006 Jul-Ago;56(4):226-43.) Toxinas adicionales adecuadas para su uso en este documento son conocidos por los expertos en la técnica y se describen en el documento US 6.077.499.
[0169] Otra clase más de agente terapéutico puede comprender uno o más inmunomoduladores. Los inmunomoduladores de uso pueden seleccionarse de una citocina, un factor de crecimiento de células madre, una linfotoxina, un factor hematopoyético, un factor estimulante de colonias (CSF), un interferón (IFN), eritropoyetina, trombopoyetina y una combinación de los mismos. Específicamente útiles son las linfotoxinas como el factor de necrosis tumoral (TNF), los factores hematopoyéticos, como la interleucina (IL), el factor estimulante de colonias, como el factor estimulante de colonias de granulocitos (G-CSF) o el factor estimulante de colonias de granulocitos y macrófagos (GMCSF), interferón, como los interferones-a, -p, -y o -A, y factor de crecimiento de células madre, como el denominado “factor S1”. Entre las citoquinas se incluyen hormonas de crecimiento tales como hormona de crecimiento humana, hormona de crecimiento humana N-metionil y hormona de crecimiento bovina; hormona paratiroidea; tiroxina; insulina; proinsulina; relaxina; prorelaxina; hormonas de glicoproteína tales como hormona estimulante del folículo (FSH), hormona estimulante de la tiroides (TSH) y hormona luteinizante (LH); factor de crecimiento hepático; prostaglandina, factor de crecimiento de fibroblastos; prolactina; lactógeno placentario, proteína OB; factor de necrosis tumoral-a y -p; sustancia inhibidora de Müller; péptido asociado a gonadotropina de ratón; inhibina; activina; factor de crecimiento vascular endotelial; integrina; trombopoyetina (TPO); factores de crecimiento nervioso tales como NGF-p; factor de crecimiento plaquetario; factores de crecimiento transformante (TGF) tales como TGF-a y TGF-p; factor de crecimiento tipo insulina-I y -II; eritropoyetina (EPO); factores osteoinductivos; interferones tales como interferón-a, -p y -y; factores estimulantes de colonias (CSF) tales como macrófagos CSF (M-CSF); interleucinas (IL) como IL-1, IL,-1a, IL-2, IL-3, IL-4, IL-5, IL-6 , IL-7, IL-8 , IL-9, IL-10, IL-11, IL-12; IL-13, IL-14, IL-15, IL-16, IL-17, IL-18 , IL-21, IL-25, LIF, kit-ligando o FLT-3, angiostatina, trombospondina, endostatina, necrosis tumoral factor y linfotoxina (LT). Como se usa en el presente documento, el término citocina incluye proteínas de fuentes
naturales o de cultivos celulares recombinantes y equivalentes biológicamente activos de las citocinas de secuencia nativa.
[0170] Las quimioquinas de uso incluyen RANTES, MCAF, MIP1-alfa, MIP1-Beta e IP-10.
[0171] El experto en la materia se dará cuenta de que el uso de los inmunoconjugados y fármacos objeto de acuerdo con la presente invención, tal como se define en las reivindicaciones, puede abarcar además el uso de uno o más agentes terapéuticos, como un segundo anticuerpo, un segundo fragmento de anticuerpo, un segundo inmunoconjugado, radionúclido, toxina, fármaco, agente quimioterapéutico, radioterapia, quimiocina, citocina, inmunomodulador, enzima, hormona, oligonucleótido, RNAi o siARN. Preferiblemente, el agente terapéutico es un inhibidor de PARP, un inhibidor de microtúbulos, un inhibidor de quinasa de Bruton o un inhibidor de PI3K. Los ejemplos de inhibidores de PARP conocidos incluyen olaparib, talazoparib (BMN-673), rucaparib, veliparib, CEP 9722, MK 4827, BGB-290, ABT-8 8 8 , AG014699, BSI-201, Ce P-8983 o 3-aminobenzamida. Los ejemplos de inhibidores de microtúbulos conocidos incluyen, pero no se limitan a, alcaloide de la vinca, taxanos, maitansinoides, auristatinas, paclitaxel, vincristina, vinblastina, mertansina, epotilona, docetaxel, discodermolida, combrestatina, podofilotoxina, CI-980, fenilahistinas, esteganacinas, curacinas, 2-metoxiestradiol, E7010, metoxibencenosuflonamidas, vinorelbina, vinflunina, vindesina, dolastatinas, espongistatina, rizoxina, tasidotina, halicondrinas, hemiasterlinas, criptoficina 52, MMAE y mesilato de eribulina. Los ejemplos de inhibidores de la quinasa Bruton incluyen ibrutinib (PCI-32765), PCI-45292, CC-292 (AVL-292), ONO-4059, GDC-0834, LFM-A13 o RN486. Los inhibidores ejemplares de PI3K incluyen idelalisib, Wortmannin, demetoxiviridina, perifosina, PX-866 , IPI-145 (duvelisib), BAY 80-6946, BEZ235, RP6530, TGR1202, SF1126, INK1117, GDC-0941, BKM120, XL147, XL765, Palomid 529, GSK1059615, ZSTK474, PWT33597, IC87114, TG100-115, CAL263, PI-103, GNE477, CUDC-907, AEZS-136 o LY294002. Dichos agentes terapéuticos adicionales pueden administrarse por separado, en combinación o unidos a los anticuerpos o inmunoconjugados en cuestión.
Formulación y administración
[0172] Las rutas adecuadas de administración de los anticuerpos, inmunoconjugados y/o fármacos incluyen, sin limitación, inyecciones parenterales, subcutáneas, rectales, transmucosas, intestinales, intramusculares, intramedulares, intratecales, intraventriculares directas, intravenosas, intravítreas, intraperitoneales, intranasales o intraoculares. Las vías de administración preferidas son las parenterales. Alternativamente, se puede administrar el compuesto de manera local en lugar de sistémica, por ejemplo, mediante inyección del compuesto directamente en un tumor sólido. Ciertos fármacos, como los inhibidores de microtúbulos, los inhibidores de PARP, los inhibidores de la quinasa de Bruton o los inhibidores de PI3K pueden diseñarse para administrarse por vía oral.
[0173] Los anticuerpos o inmunoconjugados se pueden formular según métodos conocidos para preparar composiciones farmacéuticamente útiles, en las que el anticuerpo o inmunoconjugado se combina en una mezcla con un excipiente farmacéuticamente adecuado. La solución salina tamponada con fosfato estéril es un ejemplo de un excipiente farmacéuticamente adecuado. Otros excipientes adecuados son bien conocidos por los expertos en la técnica. Ver, por ejemplo, Ansel et al., PHARMACEUTICAL DOSAGE FORMS AND DRUG DELIVERY SYSTEMS, 5a Edición (Lea & Febiger 1990), y Gennaro (ed.), REMINGTON'S PHARMACEUTICAL SCIENCES, 18a Edición (Mack Publishing Company 1990), y ediciones revisadas del mismo.
[0174] En una forma de realización preferida, el anticuerpo o inmunoconjugado se formula en tampón biológico de Good (pH 6-7), usando un tampón seleccionado del grupo que consiste en ácido N-(2-acetamido)-2-aminoetanosulfónico (ACES); ácido N-(2-acetamido)iminodiacético (ADA); ácido N,N-bis(2-hidroxietil)-2-aminoetanosulfónico (BES); ácido 4-(2-hidroxietil)piperazina-1-etanosulfónico (HEPES); ácido 2-(N-morfolino)etanosulfónico (MES); ácido 3-(N-morfolino)propanosulfónico (MOPS); ácido 3-(N-morfolinil)-2-hidroxipropanosulfónico (MOPSO); y ácido piperazina-N,N'-bis(2-etanosulfónico) [Pipas]. Los tampones más preferidos son MES o MOPS, preferiblemente en el rango de concentración de 20 a 100 mM, más preferiblemente alrededor de 25 mM. El más preferido es MES 25 mM, pH 6,5. La formulación puede comprender además trehalosa 25 mM y polisorbato 80 al 0,01% v/v como excipientes, con la concentración final de tampón modificada a 22,25 mM como resultado de los excipientes añadidos. El método preferido de almacenamiento es como una formulación liofilizada de los conjugados, almacenados en el rango de temperatura de -20 °C a 2 °C, siendo el almacenamiento más preferido de 2 °C a 8 °C.
[0175] El anticuerpo o inmunoconjugado se puede formular para administración intravenosa mediante, por ejemplo, inyección en bolo, infusión lenta o infusión continua. Preferiblemente, el anticuerpo de la presente invención se infunde durante un período de menos de aproximadamente 4 horas, y más preferiblemente, durante un período de menos de aproximadamente 3 horas. Por ejemplo, los primeros 25 a 50 mg podrían infundirse en 30 minutos, preferiblemente incluso 15 minutos, y el resto infundirse durante las siguientes 2 a 3 horas. Las formulaciones para inyección se pueden presentar en forma de dosificación unitaria, por ejemplo, en ampollas o en envases multidosis, con un conservante añadido. Las composiciones pueden adoptar formas tales como suspensiones, soluciones o emulsiones en vehículos oleosos o acuosos, y pueden contener agentes de formulación tales como agentes de suspensión, estabilización y/o
dispersión. Alternativamente, el ingrediente activo puede estar en forma de polvo para la reconstitución con un vehículo adecuado, por ejemplo, agua estéril libre de pirógenos, antes de su uso.
[0176] Pueden emplearse métodos farmacéuticos adicionales para controlar la duración de la acción del conjugado terapéutico. Las preparaciones de liberación controlada se pueden preparar mediante el uso de polímeros para complejar o adsorber el inmunoconjugado. Por ejemplo, los polímeros biocompatibles incluyen matrices de poli(etilenoco-acetato de vinilo) y matrices de un copolímero de polianhídrido de un dímero de ácido esteárico y ácido sebácico. Sherwood y col., Bio/Technology 10: 1446 (1992). La tasa de liberación de un anticuerpo o inmunoconjugado de dicha matriz depende del peso molecular, la cantidad de anticuerpo o inmunoconjugado dentro de la matriz y el tamaño de las partículas dispersas. Saltzman y col., Biophys. J. 55: 163 (1989); Sherwood y col., supra. Otras formas de dosificación sólidas se describen en Ansel et al., PHARMACEUTICAL DOSAGE FORMS AND DRUG DELIVERY SYSTEMS, 5a Edición (Lea & Febiger 1990), y Gennaro (ed.), REMINGTON'S PHARMACEUTICAL SCIENCES, 18a Edición (Mack Publishing Company 1990), y ediciones revisadas de los mismos.
[0177] Generalmente, la dosificación de un anticuerpo o inmunoconjugado administrado para humanos variará dependiendo de factores tales como la edad, el peso, la altura, el sexo, el estado médico general y el historial médico previo del paciente. Puede ser deseable proporcionar al receptor una dosis de inmunoconjugado que esté en el rango de aproximadamente 1 mg/kg a 24 mg/kg como una infusión intravenosa única, aunque también se puede administrar una dosis más baja o más alta según lo dicten las circunstancias. Una dosis de 1 a 20 mg/kg para un paciente de 70 kg, por ejemplo, es de 70 a 1400 mg o de 41 a 824 mg/m2 para un paciente de 1,7 m. La dosis puede repetirse según sea necesario, por ejemplo, una vez por semana durante 4 a 10 semanas, una vez por semana durante 8 semanas o una vez por semana durante 4 semanas. También se puede administrar con menos frecuencia, como cada dos semanas durante varios meses, o mensualmente o trimestralmente durante muchos meses, según sea necesario en una terapia de mantenimiento. Las dosis preferidas pueden incluir, entre otras, 1 mg/kg, 2 mg/kg, 3 mg/kg, 4 mg/kg, 5 mg/kg, 6 mg/kg, 7 mg/kg, 8 mg/kg, 9 mg/kg, 10 mg/kg, 11 mg/kg, 12 mg/kg, 13 mg/kg, 14 mg/kg, 15 mg/kg, 16 mg/kg, 17 mg/kg y 18 mg/ kg. La dosificación se administra preferiblemente múltiples veces, una o dos veces por semana, o con una frecuencia tan baja como una vez cada 3 o 4 semanas. Se puede usar un programa de dosificación mínimo de 4 semanas, más preferiblemente de 8 semanas, más preferiblemente de 16 semanas o más. El programa de administración puede comprender la administración una o dos veces por semana, en un ciclo seleccionado del grupo que consiste en: (i) semanal; (ii) cada dos semanas; (iii) una semana de terapia seguida de dos, tres o cuatro semanas de descanso; (iv) dos semanas de terapia seguidas de una, dos, tres o cuatro semanas de descanso; (v) tres semanas de terapia seguidas de una, dos, tres, cuatro o cinco semanas de descanso; (vi) cuatro semanas de terapia seguidas de una, dos, tres, cuatro o cinco semanas de descanso; (vii) cinco semanas de terapia seguidas de una, dos, tres, cuatro o cinco semanas de descanso; (viii) mensual y (ix) cada 3 semanas. El ciclo puede repetirse 2, 4, 6 , 8 , 10, 12, 16 o 2 0 veces o más.
[0178] Alternativamente, un anticuerpo o inmunoconjugado puede administrarse como una dosis cada 2 o 3 semanas, repetida para un total de al menos 3 dosis. O bien, dos veces por semana durante 4-6 semanas. Si la dosis se reduce a aproximadamente 200-300 mg/m2 (340 mg por dosis para un paciente de 1,7 m o 4,9 mg/kg para un paciente de 70 kg), se puede administrar una o incluso dos veces por semana durante 4 a 10 semanas. Alternativamente, el programa de dosificación puede reducirse, es decir, cada 2 o 3 semanas durante 2-3 meses. Sin embargo, se ha determinado que incluso dosis más altas, como 12 mg/kg una vez a la semana o una vez cada 2-3 semanas, pueden administrarse mediante infusión intravenosa lenta, para ciclos de dosificación repetidos. El programa de dosificación puede repetirse opcionalmente en otros intervalos y la dosificación puede administrarse a través de varias vías parenterales, con el ajuste apropiado de la dosis y el programa.
[0179] Los ejemplos de cánceres que pueden tratarse de acuerdo con la invención como se define en las reivindicaciones incluyen, pero no se limitan a, carcinoma, linfoma, glioblastoma, melanoma, sarcoma y leucemia, mieloma o neoplasias malignas linfoides. Más ejemplos particulares de tales cánceres se indican a continuación e incluyen: cáncer de células escamosas (p. ej., cáncer epitelial de células escamosas), sarcoma de Ewing, tumor de Wilms, astrocitomas, glioblastomas, cáncer de pulmón, incluido el cáncer de pulmón de células pequeñas, cáncer de pulmón de células no pequeñas, adenocarcinoma de pulmón y carcinoma escamoso de pulmón, cáncer de peritoneo, cáncer gástrico o de estómago, incluido el cáncer gastrointestinal, cáncer de páncreas, glioblastoma multiforme, cáncer de cuello uterino, cáncer de ovario, cáncer de hígado, cáncer de vejiga, hepatoma, carcinoma hepatocelular, tumores neuroendocrinos, cáncer medular de tiroides, carcinoma diferenciado de tiroides, cáncer de mama, cáncer de ovario, cáncer de colon, cáncer de recto, cáncer de endometrio o carcinoma uterino, carcinoma de glándulas salivales, cáncer de riñón o renal, cáncer de próstata, cáncer de vulva, carcinoma anal, carcinoma de pene, así como cáncer de cabeza y cuello. El término "cáncer' incluye células o tumores malignos primarios (p. ej., aquellos cuyas células no han migrado a sitios en el cuerpo del sujeto que no sean el sitio del tumor o malignidad original) y células o tumores malignos secundarios (p. ej., aquellos que surgen de la metástasis, la migración de células malignas o células tumorales a sitios secundarios que son diferentes del sitio del tumor original).
[0180] Otros ejemplos de cánceres o neoplasias malignas incluyen, entre otros: leucemia linfoblástica aguda infantil, leucemia linfoblástica aguda, leucemia linfocítica aguda, leucemia mieloide aguda, carcinoma adrenocortical, cáncer hepatocelular (primario) en adultos, cáncer de hígado (primario) en adultos, Leucemia linfocítica, leucemia mieloide
aguda en adultos, linfoma de Hodgkin en adultos, leucemia linfocítica en adultos, linfoma no Hodgkin en adultos, cáncer primario de hígado en adultos, sarcoma de tejido blando en adultos, linfoma relacionado con el SIDA, neoplasias malignas relacionadas con el SIDA, cáncer anal, astrocitoma, cáncer de las vías biliares, cáncer de vejiga, cáncer de huesos, glioma de tronco encefálico, tumores cerebrales, cáncer de mama, cáncer de pelvis renal y uréter, linfoma del sistema nervioso central (primario), linfoma del sistema nervioso central, astrocitoma cerebeloso, astrocitoma cerebral, cáncer de cuello uterino, cáncer de cuello uterino infantil (primario). Cáncer hepatocelular, cáncer de hígado (primario) infantil, leucemia linfoblástica aguda infantil, leucemia mieloide aguda infantil, glioma de tronco encefálico infantil, astrocitoma cerebeloso infantil, astrocitoma cerebral infantil, tumores extracraneales de células germinales infantiles, enfermedad de Hodgkin infantil, linfoma de Hodgkin infantil, hipotalámico infantil y glioma de la vía visual, leucemia linfoblástica infantil, meduloblastoma infantil, linfoma no Hodgkin infantil, tumores neuroectodérmicos primitivos pineales y supratentoriales infantiles, cáncer de hígado primario infantil, rabdomiosarcoma infantil, sarcoma de tejido blando infantil, glioma hipotalámico y de la vía visual infantil, leucemia linfocítica crónica, Leucemia mielógena crónica, cáncer de colon, linfoma cutáneo de células T, carcinoma de células de los islotes pancreáticos endocrinos, cáncer de endometrio, ependimoma, cáncer epitelial, cáncer de esófago, sarcoma de Ewing y tumores relacionados, cáncer de páncreas exocrino, tumor extracraneal de células germinales, tumor extragonadal de células germinales, Cáncer de vías biliares extrahepáticas, cáncer de ojo, cáncer de mama femenino, enfermedad de Gaucher, cáncer de vesícula biliar, cáncer gástrico, tumor carcinoide gastrointestinal, tumores gastrointestinales, tumores de células germinales, tumor trofoblástico gestacional, leucemia de células pilosas, cáncer de cabeza y cuello, cáncer hepatocelular, linfoma de Hodgkin, hipergammaglobulinemia, cáncer de hipofaringe, cánceres intestinales, melanoma intraocular, carcinoma de células de los islotes, cáncer de páncreas de células de los islotes, sarcoma de Kaposi, cáncer de riñón, cáncer de laringe, cáncer de labio y cavidad oral, cáncer de hígado, cáncer de pulmón, trastornos linfoproliferativos, macroglobulinemia, cáncer de mama masculino, mesotelioma maligno, timoma maligno, meduloblastoma, melanoma, mesotelioma, cáncer de cuello escamoso primario oculto metastásico, cáncer de cuello escamoso primario metastásico, cáncer de cuello escamoso metastásico, mieloma múltiple, mieloma múltiple/neoplasia de células plasmáticas, síndrome mielodisplásico, leucemia mielógena, leucemia mieloide, trastornos mieloproliferativos, cáncer de cavidad nasal y senos paranasales, cáncer nasofaríngeo, neuroblastoma, linfoma no Hodgkin, cáncer de piel no melanoma, cáncer de pulmón de células no pequeñas, metástasis primaria oculta Cáncer de cuello escamoso, cáncer de orofaringe, osteosarcoma/sarcoma fibroso maligno, osteosarcoma/histiocitoma fibroso maligno, osteosarcoma/histiocitoma fibroso maligno del hueso, cáncer epitelial de ovario, tumor de células germinales de ovario, tumor de ovario de bajo potencial maligno, cáncer de páncreas, paraproteinemias, policitemia vera, cáncer de paratiroides, cáncer de pene, feocromocitoma, tumor hipofisario, linfoma primario del sistema nervioso central, cáncer primario de hígado, cáncer de próstata, cáncer rectal, cáncer de células renales, cáncer de pelvis renal y uréter, retinoblastoma, rabdomiosarcoma, cáncer de glándulas salivales, sarcoidosis sarcomas, Sezary cáncer de piel, cáncer de pulmón de células pequeñas, cáncer de intestino delgado, sarcoma de tejido blando, cáncer de cuello escamoso, cáncer de estómago, tumores neuroectodérmicos y pineales supratentoriales primitivos, linfoma de células T, cáncer testicular, timoma, cáncer de tiroides, cáncer de células de transición del Pelvis renal y uréter, transición de pelvis renal y cáncer de uréter, tumores trofoblásticos, cáncer de células de uréter y pelvis renal, cáncer de uretra, cáncer de útero, sarcoma de útero, cáncer de vagina, glioma hipotalámico y de la vía visual, cáncer de vulva, macroglobulinemia de Waldenstrom, tumor de Wilms, y cualquier otra enfermedad hiperproliferativa, además de la neoplasia, localizada en un sistema de órganos mencionado anteriormente.
[0181] La invención, tal como se define en las reivindicaciones, puede usarse para tratar afecciones malignas o premalignas y para prevenir la progresión a un estado neoplásico o maligno, incluidos, entre otros, los trastornos descritos anteriormente. Dichos usos están indicados en condiciones conocidas o sospechosas de progresión previa a neoplasia o cáncer, en particular, donde se ha producido un crecimiento celular no neoplásico consistente en hiperplasia, metaplasia o, más particularmente, displasia (para una revisión de tales condiciones de crecimiento anormal, ver Robbins y Angell, Basic Pathology, 2.a edición, WB Saunders Co., Filadelfia, págs. 68-79 (1976)).
[0182] La displasia es frecuentemente un precursor del cáncer y se encuentra principalmente en el epitelio. Es la forma más desordenada de crecimiento celular no neoplásico, que implica una pérdida en la uniformidad de las células individuales y en la orientación arquitectónica de las células. La displasia ocurre característicamente cuando existe irritación o inflamación crónica. Los trastornos displásicos que se pueden tratar incluyen, entre otros, displasia ectodérmica anhidrótica, displasia anterofacial, displasia torácica asfixiante, displasia atriodigital, displasia broncopulmonar, displasia cerebral, displasia cervical, displasia condroectodérmica, displasia cleidocraneal, displasia ectodérmica congénita, displasia craneodiafisaria, displasia craneocarpotarsiana, displasia craneometafisaria, displasia de dentina, displasia diafisaria, displasia ectodérmica, displasia del esmalte, displasia encefalooftálmica, displasia epifisaria hemimelia, displasia epifisaria múltiple, displasia epifisaria punctata, displasia epitelial, displasia faciodigitogenital, displasia fibrosa familiar de los maxilares, displasia plegada blanca familiar, displasia fibromuscular, displasia fibrosa del hueso, displasia ósea florida, displasia renal-retiniana hereditaria, displasia ectodérmica hidrótica, displasia ectodérmica hipohidrótica, displasia tímica linfopénica, displasia mamaria, displasia mandibulofacial, displasia metafisaria, displasia de Mondini, displasia fibrosa monostótica, displasia mucoepitelial, displasia epifisaria múltiple, displasia oculoauriculovertebral, displasia oculodentodigital, displasia oculovertebral, displasia odontogénica, displasia oftalmomandibulomélica, displasia cementosa periapical, displasia fibrosa poliostótica, displasia
espondiloepifisaria pseudoacondroplásica, displasia retiniana, displasia septoóptica, displasia espondiloepifisaria y displasia ventriculoradial.
[0183] Los trastornos preneoplásicos adicionales que se pueden tratar incluyen, entre otros, trastornos disproliferativos benignos (p. ej., tumores benignos, afecciones fibroquísticas, hipertrofia tisular, pólipos o adenomas intestinales y displasia esofágica), leucoplasia, queratosis, enfermedad de Bowen, piel de Farmer, queilitis solar y queratosis solar.
[0184] En formas de realización preferidas, la invención tal como se define en las reivindicaciones se usa para inhibir el crecimiento, la progresión y/o la metástasis de cánceres, en particular los enumerados anteriormente.
[0185] Otras enfermedades, trastornos y/o afecciones hiperproliferativas incluyen, entre otros, progresión y/o metástasis de tumores malignos y trastornos relacionados, como la leucemia (incluidas las leucemias agudas; p. ej., leucemia linfocítica aguda, leucemia mielocítica aguda [incluidas las mieloblásticas, promielocítica, mielomonocítica, monocítica y eritroleucemia]) y leucemias crónicas (p. ej., leucemia mielocítica [granulocítica] crónica y leucemia linfocítica crónica), policitemia vera, linfomas (p. ej., enfermedad de Hodgkin y enfermedad no Hodgkin), mieloma múltiple, macroglobulinemia de Waldenstrom, enfermedad de cadena pesada y tumores sólidos que incluyen, entre otros, sarcomas y carcinomas como fibrosarcoma, mixosarcoma, liposarcoma, condrosarcoma, sarcoma osteogénico, cordoma, angiosarcoma, endoteliosarcoma, linfangiosarcoma, linfangioendoteliosarcoma, sinovioma, mesotelioma, tumor de Ewing, leiomiosarcoma, rabdom yosarcoma, carcinoma de colon, cáncer de páncreas, cáncer de mama, cáncer de ovario, cáncer de próstata, carcinoma de células escamosas, carcinoma de células basales, adenocarcinoma, carcinoma de glándulas sudoríparas, carcinoma de glándulas sebáceas, carcinoma papilar, adenocarcinomas papilares, cistadenocarcinoma, carcinoma medular, carcinoma broncogénico, células renales carcinoma, hepatoma, carcinoma de las vías biliares, coriocarcinoma, seminoma, carcinoma embrionario, tumor de Wilm, cáncer de cuello uterino, tumor testicular, carcinoma de pulmón, carcinoma de pulmón de células pequeñas, carcinoma de vejiga, carcinoma epitelial, glioma, astrocitoma, meduloblastoma, craneofaringioma, ependimoma, pinealoma, emangioblastoma, neuroma acústico, oligodendroglioma, meningioma, melanoma, neuroblastoma y retinoblastoma.
[0186] Las enfermedades autoinmunes que pueden tratarse con anticuerpos o inmunoconjugados pueden incluir trombocitopenias inmunitarias agudas y crónicas, dermatomiositis, corea de Sydenham, miastenia grave, lupus eritematoso sistémico, nefritis lúpica, fiebre reumática, síndromes poliglandulares, penfigoide ampolloso, diabetes mellitus, púrpura de Henoch-Schonlein, nefritis posestreptocócica, eritema nodoso, arteritis de Takayasu, vasculitis asociada con ANCA, enfermedad de Addison, artritis reumatoide, esclerosis múltiple, sarcoidosis, colitis ulcerosa, eritema multiforme, nefropatía IgA, poliarteritis nodosa, espondilitis anquilosante, síndrome de Goodpasture, tromboangitis obliterante, síndrome de Sjogren, cirrosis biliar primaria, tiroiditis de Hashimoto, tirotoxicosis, esclerodermia crónica hepatitis activa, polimiositis/dermatomiositis, policondritis, penfigoide ampolloso, pénfigo vulgar, granulomatosis de Wegener, nefropatía membranosa, esclerosis lateral amiotrófica, tabes dorsal, arteritis de células gigantes/polimialgia, anemia perniciosa, glomerulonefritis rápidamente progresiva, psoriasis o alveolitis fibrosante.
Kits
[0187] La presente divulgación también se refiere a kits que contienen componentes adecuados para tratar tejido enfermo en un paciente. Los kits de ejemplo pueden contener al menos un anticuerpo o inmunoconjugado como se describe en este documento. Un kit también puede incluir un fármaco seleccionado entre inhibidores de microtúbulos, inhibidores de PARP, inhibidores de quinasa de Bruton y/o inhibidores de PI3K. Si la composición que contiene los componentes para la administración no está formulada para la administración a través del canal alimentario, como la administración oral, se puede incluir un dispositivo capaz de administrar los componentes del kit a través de alguna otra ruta. Un tipo de dispositivo, para aplicaciones como la administración parenteral, es una jeringa que se usa para inyectar la composición en el cuerpo de un sujeto. También se pueden usar dispositivos de inhalación.
[0188] Los componentes del kit pueden empaquetarse juntos o separarse en dos o más contenedores. En algunas formas de realización, los recipientes pueden ser viales que contienen formulaciones liofilizadas estériles de una composición que es adecuada para la reconstitución. Un kit también puede contener uno o más tampones adecuados para la reconstitución y/o dilución de otros reactivos. Otros recipientes que pueden usarse incluyen, pero no se limitan a, una bolsa, bandeja, caja, tubo o similar. Los componentes del kit pueden empaquetarse y mantenerse estériles dentro de los contenedores. Otro componente que se puede incluir son las instrucciones para una persona que utilice un kit para su uso.
EJEMPLOS
Ejemplo 1. Terapia combinada con ADC IMMU-132 e inhibidores de microtúbulos o inhibidores de PARP
[0189] En los ensayos clínicos actuales (ClinicalTrials.gov, NCT01631552), los pacientes con cáncer de mama triple negativo (TNBC) tratados con IMMU-132, que está compuesto por el metabolito activo del irinotecán, SN-38,
conjugado con un anticuerpo anti-Trop-2, muestra una toxicidad manejable y respuestas muy alentadoras en casos recidivantes/refractarios.
[0190] La letalidad sintética es un concepto en el que una célula que alberga uno de dos posibles defectos genéticos o proteicos es viable, mientras que una célula que contiene ambos defectos es inviable. Las mutaciones BRCA1/2 están vinculadas a deficiencias en la reparación del ADN y están asociadas con TNBC. Otros mecanismos de reparación implican la poli(adenosina difosforribosa) polimerasa (PARP), que las células cancerosas pueden utilizar para superar la pérdida de BRACA1/2. El tratamiento de las células TNBC con IMMU-132 o paclitaxel da como resultado la escisión y desactivación de PARP, mientras que la molécula pequeña de olaparib inhibe directamente la PARP. Por lo tanto, se investigó el fundamento de la combinación de IMMU-132 con paclitaxel u olaparib para eliminar de forma efectiva la actividad de PARP en xenoinjertos de TNBC para determinar si estas combinaciones resultarán en letalidad sintética.
[0191] El propósito de este estudio fue determinar si la combinación de un conjugado de anticuerpo y fármaco que induce roturas de cadenas de ADN, como sacituzumab govitecan (también conocido como IMMU-132, un anti-Trop-2 hRS7-CL2A-SN-38), con microtúbulos inhibidores (p. ej., paclitaxel o mesilato de eribulina) o inhibidores de la poli(adenosina difosforribosa) polimerasa (PARP) (p. ej., olaparib) en el cáncer (p. ej., ratones desnudos portadores de xenoinjertos de TNBC) mejora los efectos antitumorales. El experto en la materia se dará cuenta de que los efectos superiores inesperados de la combinación de conjugados de anticuerpo-SN-38 con PARP o inhibidores de microtúbulos no se limitan al anticuerpo, fármaco, inhibidor de PARP o inhibidor de microtúbulos ejemplares específicos, sino que son característicos de las clases de anticuerpos contra antígenos asociados a tumores (TAA), fármacos que inducen roturas de cadenas de ADN, inhibidores de PARP e inhibidores de microtúbulos.
Procedimientos experimentales
[0192] En un ejemplo no limitativo, ratones portadores de xenoinjertos humanos de TNBC (cáncer de mama triple negativo) (MDA-MB-468 o HCC1806; ~0,3 cm3) se trataron con la dosis máxima tolerada de paclitaxel (15 mg/kg semanales x 5 semanas). e IMMU-132 a 10 mg/kg o 12,5 mg/kg los días 1, 8 , 22 y 29. Se trataron ratones con tumores HCC1806 (~0,28 cm3) durante 2 ciclos con IMMU-132 (12,5 mg/kg) y 0,5 mg/kg de mesilato de eribulina (equivalente a la dosis humana de 1,4 mg/m2) semanalmente durante 2 semanas en un ciclo de 21 días. Los estudios que examinaron la inhibición de PARP utilizaron ratones con tumores MDA-MB-468 (~0,32 cm3) tratados con olaparib (50 mg/kg, qdx5d, x 4 semanas; 33 % de la dosis humana equivalente a 800 mg diarios) e IMMU-132 (10 mg/kg, dos veces por semana x 4 semanas). Olaparib se administró como inyecciones i.p. diariamente durante 5 días seguidos con dos días de descanso antes de repetir (qdx5). Esto se hizo durante cuatro semanas. IMMU-132 se administró i.p. dos veces por semana durante cuatro semanas. Los animales de control recibieron el anti-CD20 ADC hA20-CL2A-SN-38 no dirigido a tumores, ya sea solo o en combinación con olaparib. El criterio principal de valoración fue la mediana del tiempo de supervivencia (MST), definido como el tiempo que tardan los tumores en progresar hasta 1,0 cm3.
[0193] En formas de realización alternativas, el ensayo de efectos sinérgicos puede determinarse mediante ensayo in vitro. Puede usarse un ensayo clonogénico para determinar la fracción de supervivencia de las células (Ibrahim et al., 2012, Cancer Discovery 2:1036-47). Brevemente, se sembraron 350-800 células en placas de cultivo celular de fondo plano de 6 pocillos por duplicado. Veinticuatro horas después de la siembra, las células se lavan y se agrega medio fresco en presencia o ausencia de dosis crecientes de ADC y/o PARP o inhibidor de microtúbulos (p. ej., olaparib) solos o en combinación. Los medios que contienen el fármaco y/o se actualizan el día 4. Las colonias se fijan y tiñen después de 7 días de tratamiento con 1,5 ml de glutaraldehído al 6,0 % y cristal violeta al 0,5 % y las colonias se cuentan mediante procedimientos estándar. La fracción sobreviviente (SF) de las células se calcula de la siguiente manera:

donde
[0194] La interacción entre ADC y PARP o inhibidor de microtúbulos se evalúa utilizando el método de análisis de efectos de fármacos múltiples de Chou y Talalay (1984, Adv Enzyme Regul 22:27-55). Este método describe cuantitativamente la interacción entre dos o más fármacos, donde los valores inferiores a 1 indican interacciones sinérgicas, los valores superiores a 1 indican interacciones antagónicas y los valores iguales a 1 indican interacciones aditivas.
Resultados
[0195] Los ratones con tumores MDA-MB-468 que recibieron la combinación de IMMU-132 y paclitaxel exhibieron efectos antitumorales superiores (FIG. 1A-1C), con una reducción del tumor de >11 veces, en comparación con una reducción de 1,4 veces en la IMMU- 132 solo (P=0,0003; área bajo la curva, AUC) o aumento de 11,4 veces en el tamaño del tumor en ratones tratados con paclitaxel solo (P<0,0001; AUC).
[0196] En MDA-MB-468, la combinación de 200 μg de IMMU-132 más paclitaxel tiene efectos antitumorales superiores en términos de área bajo la curva (AUC) en comparación con todos los demás grupos (Tabla 7, P<0,0013). Reducir la cantidad de IMMU-132 administrada con paclitaxel a 100 mg también da como resultado efectos antitumorales significativos en comparación con ratones tratados con paclitaxel solo, IMMU-132 solo (100 mg) o animales no tratados (Tabla 8, P< 0,0328). No se pueden realizar más comparaciones entre las curvas de crecimiento con paclitaxel o grupos de control no tratados, ya que cada uno comenzó a perder ratones debido a la progresión de la enfermedad (es decir, TV>1,0 cm3) a partir del día 49 de la terapia.
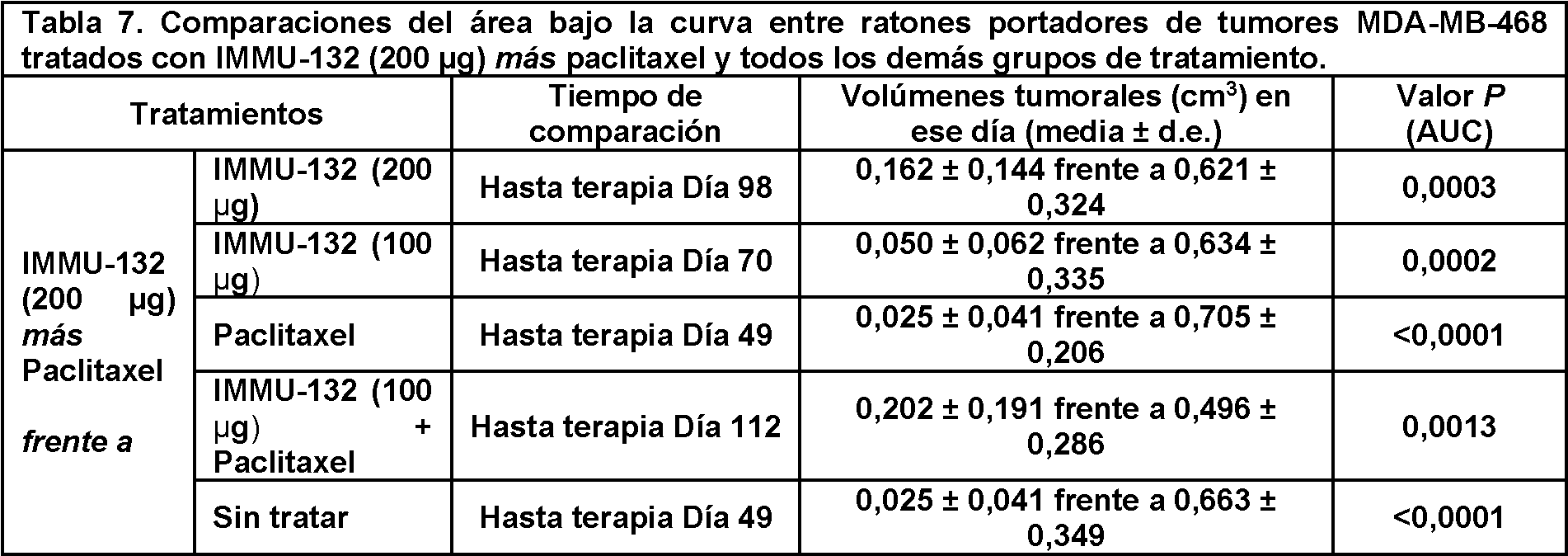

[0197] En los xenoinjertos de HCC1806 de rápido progreso (FIG. 2A-2B), la combinación de IMMU-132 más paclitaxel demostró tener un efecto antitumoral superior en comparación con la monoterapia con IMMU-132 (P = 0,0195, AUC17 días). Este es un tumor muy agresivo con un tiempo medio de supervivencia (MST) de solo 10 días después del inicio de la terapia para los animales de control no tratados (18 días después de la inoculación de células tumorales). En términos de supervivencia, la combinación, que alcanzó su MST de 38 días, proporcionó un beneficio de supervivencia significativo en comparación con todas las demás terapias (P <0,017; rango logarítmico). Cabe señalar que esto se logró con una dosis baja de solo 0,25 mg, que sería la dosis equivalente en humanos de solo 1 mg/kg.
[0198] Los ratones tratados con la combinación de IMMU-132 más mesilato de eribulina (FIG. 3A-3E) mostraron una respuesta antitumoral significativamente mayor que todos los demás grupos de monoterapia (P < 0,0432; prueba t pareada). Esto resultó en un beneficio de supervivencia significativo para la combinación (MST = 23 días) en comparación con la monoterapia con eribulina o IMMU-132 (MST = 18 y 14 días, respectivamente; P <0,0044; rango logarítmico).
[0199] Asimismo, la combinación de la terapia IMMU-132 con olaparib fue superior a la terapia con un solo agente en ratones que tenían tumores MDA-MB-468 (P<0,0032; AUC) (FIG. 4). Los resultados se resumen en la Tabla 9.
Todos los tratamientos combinados con IMMU-132 fueron bien tolerados.

[0200] Determinación de la relación fármaco-anticuerpo (DAR). Se evaluaron cinco lotes clínicos de IMMU-132 mediante HPLC de interacción hidrofóbica (HIC-HPLC), que resolvió tres picos que representan especies con DAR de 6 , 7 y 8 , con la mayor fracción que comprende un DAR = 8 (no se muestra). IMMU-132 se produjo consistentemente mediante este proceso de fabricación, con un DAR (PAR AVE) general de 7,60 ± 0,03 entre los cinco lotes clínicos. Los resultados de HIC-HPLC se confirmaron mediante cromatografía líquida-espectrometría de masas (LC-MS). El análisis mostró que >99 % de los 8 grupos sulfhidrilo disponibles se acoplaron con el enlazador CL2A, con o sin SN-38 (no se muestra). No se detectaron cadenas pesadas o ligeras no sustituidas (o cubiertas con N-etilmaleimida). Por lo tanto, la diferencia en DAR entre las especies resulta de la liberación de SN-38 del enlazador durante la fabricación y no de una relación de sustitución inicial más baja. Una vez preparado y liofilizado, IMMU-132 se ha mantenido estable durante varios años.
[0201] Efecto de DAR sobre la farmacocinética y la eficacia antitumoral en ratones. Los ratones que portaban xenoinjertos de carcinoma gástrico humano Trop-2+ (NCI-N87) recibieron 2 tratamientos con 7 días de diferencia, cada uno con dosis iguales de proteína (0,5 mg) de IMMU-132 con DAR de 6,89, 3,28 o 1,64. Los animales tratados con ADC que tenían un DAR de 6,89 tuvieron una mediana de tiempo de supervivencia (MST) significativamente mejorada en comparación con los ratones que recibieron ADC con DAR de 3,38 o 1,64 (MST = 39 días frente a 25 y 21 días, respectivamente; P <0,0014) (no mostrada). No hubo diferencia entre los grupos tratados con los conjugados 3.28 o 1.64 DAR y el grupo de control con solución salina.
[0202] Para dilucidar aún más la importancia de una DAR más alta, a los ratones que tenían tumores gástricos NCI-N87 se les administró 0,5 mg de IMMU-132 con una DAR de 6,89 dos veces por semana durante dos semanas (no se muestra). Otro grupo recibió el doble de la dosis de proteína (1 mg) de un conjugado IMMU-132 con un DAR de 3,28. Aunque ambos grupos recibieron la misma cantidad total de SN-38 (36 mg) con cada esquema de dosificación, los tratados con el conjugado DAR 6,89 inhibieron el crecimiento tumoral significativamente más que los animales con tumores tratados con el conjugado DAR 3,28 (P = 0,0227; AUC) (no se muestra). Además, el tratamiento con el DAR más bajo no fue significativamente diferente de los controles sin tratar. En conjunto, estos estudios indican que una DAR más baja reduce la eficacia.
[0203] Se realizó un examen del comportamiento farmacocinético de los conjugados preparados en estas diferentes proporciones en ratones sin tumor que recibieron 0,2 mg de cada conjugado, hRS7 IgG no conjugado o hRS7 IgG que se redujo y luego se tapó con N-etilmaleimida. Se tomó suero a 5 intervalos de 0,5 a 168 h y se analizó por ELISA para hRS7 IgG. No hubo diferencia significativa en la eliminación de estos conjugados en comparación con la IgG no conjugada (no se muestra). Por lo tanto, el nivel de sustitución no afectó a la farmacocinética de los conjugados e, igualmente importante, la reducción de los enlaces disulfuro entre cadenas no pareció desestabilizar el anticuerpo.
[0204] Mecanismo de acción de IMMU-132 en TNBC. La vía apoptótica utilizada por IMMU-132 se examinó en la línea celular TNBC, MDA-MB-468, y en la línea celular HER2+ SK-BR-3, para confirmar que el ADC funciona sobre la base de su SN-38 incorporado. Las células se expusieron a SN-38 1 μM, el equivalente de SN-38 de IMMU-132 o el equivalente proteico de hRS7. Se recogieron las células y se realizaron transferencias de Western. SN-38 solo y mediada por IMMU-132 >2 veces la regulación positiva de p2 1 WAF1/Cip1 dentro de las 24 h en MDA-MB-468, y a las 48 h, la cantidad de p2 1 WAF1/Cip1 en estas células comenzó a disminuir (31 % y 43 % con SN-38 o iMm U-132, respectivamente) (no se muestra). Curiosamente, en la línea tumoral HER2+ SκΒR-3, ni SN-38 ni IMMU-132 mediaron la regulación positiva de p2 1 WAF1/Cip1 por encima de los niveles constitutivos en las primeras 24 h, pero como se observa en las células MDA-MB-468 después Exposición de 48 h a SN-38 o IMMU-132, la cantidad de p2 1 WAF1/Cip1 disminuyó >57 % (no se muestra). Tanto SN-38 como IMMU-132 dieron como resultado la escisión de pro-caspasa-3 en sus fragmentos activos dentro de las 24 h, pero se observó el mayor grado de fragmentos activos después de la exposición durante 48 h. Es de destacar que en ambas líneas celulares, IMMU-132 medió un mayor grado de escisión de procaspasa-3, con el nivel más alto observado después de 48 h en comparación con las células expuestas a SN-38 (no
se muestra). Finalmente, tanto SN-38 como IMMU-132 mediaron la escisión de la poli ADP ribosa polimerasa (PARP), comenzando a las 24 h, con una escisión casi completa después de 48 h (no se muestra). En conjunto, estos resultados confirman que IMMU-132 tiene un mecanismo de acción similar al de SN-38 libre cuando se administra in vitro.
[0205] Entrega de SN-38 por IMMU-132 frente a irinotecán en un modelo de xenoinjerto de tumor humano. Los productos constitutivos derivados del irinotecán o IMMU-132 se determinaron en el suero y los tumores de ratones implantados s.c. con un xenoinjerto de cáncer de páncreas humano (Capan-1) al que se administró irinotecán (773 mg; equivalentes de SN-38 = 448 mg) e IMMU- 132 (1,0 mg; s N-38 equivalentes = 16 mg). Después de la administración, en 5 intervalos, 3 animales de cada grupo fueron sacrificados con suero extraído para los productos de interés.
[0206] El irinotecán se eliminó muy rápidamente del suero y se observó conversión a SN-38 y SN-38G en 5 minutos (no se muestra). Ninguno de los productos fue detectado a las 24 h. Las AUC durante un período de 6 horas fueron 21,0, 2,5 y 2,8 μg/mlh para irinotecán, SN-38 y SN-38G, respectivamente (conversión de SN-38 en ratones = [2,5 2,8)/21 = 25,2%]). Los animales que recibieron IMMU-132 tenían concentraciones mucho más bajas de SN-38 libre en el suero, pero se detectó a las 48 h (no se muestra). El SN-38G libre se detectó solo a las 1 y 6 horas, y fue de 3 a 7 veces más bajo que el SN-38 libre (no mostrado).
[0207] En los tumores Capan-1 extirpados de animales tratados con irinotecán, los niveles de irinotecán fueron altos durante 6 h, pero indetectables a las 24 h (AUC 5min-6 h = 48,4 μg/gh). SN-38 fue mucho menor y se detectó solo durante 2 h (es decir, AUC 5min-2 h = 0,4 μg/gh), con valores de SN-38G casi 3 veces más altos (AUC = 1,1 μg/gh) (no mostrada). Los tumores tomados de animales que recibieron IMMU-132 no tenían ningún SN-38 o SN-38G libre detectable, sino que todo el SN-38 en el tumor estaba unido a IMMU-132. Es importante destacar que, dado que no se detectó SN-38G en los tumores, esto sugiere que SN-38 unido a IMMU-132 no estaba glucuronizado. El AUC de SN-38 unido a IMMU-132 en estos tumores fue de 54,3 μg/gh, que es 135 veces mayor que la cantidad de SN-38 en los tumores de animales tratados con irinotecán durante el período de 2 h. que se pudo detectar SN-38, aunque los ratones que recibieron irinotecán recibieron 28 veces más equivalentes de SN-38 que los que recibieron IMMU-132 (es decir, 448 frente a 16 μg de equivalentes de SN-38, respectivamente).
Conclusiones
[0208] IMMU-132 es un anticuerpo anti-Trop-2 humanizado conjugado con 7,6 moléculas de SN-38, el metabolito activo del irinotecán, un inhibidor de la topoisomerasa I. Clínicamente, IMMU-132 ha mostrado una toxicidad manejable y respuestas alentadoras en pacientes con TNBC recidivante/refractario (ClinicalTrials.gov, NCT01631552). La terapia IMMU-132 sola demostró efectos antitumorales significativos en xenoinjertos de TNBC humanos a una dosis equivalente en humanos que es 5 veces menor que la que se usa clínicamente (es decir, 10 mg/kg). Dado que los estudios preclínicos indican que IMMU-132 se puede combinar con dos inhibidores de microtúbulos diferentes o un inhibidor de PARP con una actividad antitumoral significativamente mejorada, estos datos respaldan el uso de IMMU-132 y otros conjugados de anticuerpo-fármaco (ADC) que causan el ADN. se rompe, en combinación con inhibidores de microtúbulos y/o inhibidores de PARP en general, así como otros agentes quimioterapéuticos que se dirigen a la división celular a través de la inhibición de microtúbulos o mecanismos de reparación de ADN. Una clase de ADC preferida está representada por conjugados de anticuerpos anti-Trop-2 en pacientes con cánceres positivos para Trop-2, incluidos, entre otros, TNBC, cáncer de colon metastásico, SCLC y NSCLC, ya que este es un objetivo que se expresa en grandes cantidades en una gran cantidad de cánceres, y se localiza en la superficie celular y en el citoplasma de las células cancerosas. Sin embargo, también se pueden usar otros objetivos de antígenos del cáncer para el ADC en esta aplicación si contienen un fármaco que también provoca roturas en el ADN, por ejemplo, al dirigirse a la topoisomerasa I, como SN-38.
[0209] La sinergia se logró cuando IMMU-132 se combinó con inhibidores de PARP (p. ej., olaparib) en líneas tumorales de TNBC que tenían defectos de BRCA1/2, así como expresión de tipo salvaje, incluido uno con solo un defecto de PTEN. Esto sugiere que IMMU-132 puede generar sinergia con cualquier tumor que tenga algún tipo de interrupción en las vías de recombinación homóloga de ADN. En combinación con olaparib, la terapia IMMU-132 logró efectos antitumorales significativos superiores a los observados con la monoterapia con cada uno, lo que resultó en un beneficio de supervivencia significativo. IMMU-132 combinado con inhibidores de microtúbulos (p. ej., paclitaxel o mesilato de eribulina) también mejoró significativamente la eficacia en comparación con la monoterapia con cada agente.
[0210] En general, estos datos evidencian la ventaja significativa inesperada de la terapia de combinación con un conjugado de anticuerpo y fármaco (ADC) que se dirige a las células cancerosas e induce roturas de la cadena de ADN, como IMMU-132, e inhibidores de microtúbulos o inhibidores de PARP. Apuntar a la vía de reparación del ADN PARP en tumores TNBC mutantes BRCA1/2 al combinar la terapia IMMU-132 con paclitaxel u olaparib logró letalidad sintética en este modelo de enfermedad sin toxicidad observable. En una forma de realización ejemplar, la combinación de IMMU-132 y un inhibidor de microtúbulos o PARP es útil para tratar cánceres positivos para Trop-2, como TNBC. Estos datos proporcionan la justificación para el uso de IMMU-132 en combinación con otros
quimioterapéuticos que también se dirigen a los mecanismos de reparación del ADN en pacientes con TNBC o tumores similares.
Ejemplo 2. Terapia combinada con IMMU-114 (anti-HLA-DR) más inhibidor de quinasa de Bruton en leucemia linfocítica crónica (LLC)
Diseño experimental.
[0211] Se sembraron células de leucemia de células B crónicas humanas (JVM-3) en una placa de 96 pocilios. A continuación, las células se incubaron con varias dosis del inhibidor de quinasa Bruton ibrutinib (1 x 10-5 a 3,9 x 10-9 M) más una cantidad constante de IMMU-114 (0,25, 0,5, 0,75 o 1 nM) durante 96 h. La viabilidad celular se evaluó mediante ensayo MTS y se generaron curvas de dosis/respuesta basadas en el porcentaje de inhibición del crecimiento en relación con las células de control no tratadas. Los valores de Cl50 se determinaron usando Prism Graph-Pad. Los datos se normalizaron y se generaron isobologramas para demostrar el efecto de IMMU-114 sobre ibrutinib en células JVM-3.
Resultados
[0212] Un ejemplo de como se ha mostrado en la FIG. 5A como IMMU-114 desplazó el Cl50 de ibrutinib. Las células incubadas con ibrutinib solo dieron como resultado una Cl50 de 2,2 x 10-6M. Sin embargo, cuando se combina 0,5 nM de IMMU-114 con ibrutinib, el valor de Cl50 se reduce al doble a 1,08 x 10-6M. cabe señalar que IMMU-114 a 0,5 nM solo da como resultado una inhibición del crecimiento de aproximadamente un 22 %. Asimismo, IMMU-114 0,75 nM desplazó el valor de Cl50 de ibrutinib hacia abajo 5,7 veces.
[0213] Estos datos, junto con los resultados de otros dos ensayos, se normalizaron para producir un isobolograma con el fin de determinar si esta interacción era sinérgica, aditiva o antagónica (FIG. 6A). Como se muestra, cuando las células JVM-3 se incubaron con ibrutinib combinado con IMMU-114, es evidente un efecto aditivo. Estos datos demuestran que, si bien cada agente por sí solo es capaz de inhibir el crecimiento de la LLC in vitro, cuando se combina se logra un efecto aditivo que respalda la combinación de terapias con IMMU-114 y quinasa Bruton en pacientes con LLC.
Ejemplo 3. Terapia combinada con IMMU-114 (anti-HLA-DR) más inhibidor de fosfatidilinositida 3-quinasa (PI3K) en leucemia linfocítica crónica (LLC)
Diseño experimental.
[0214] Se sembraron células B de leucemia crónica humana (JVM-3) en una placa de 96 pocillos. Luego, las células se incubaron con varias dosis de un inhibidor de PI3K idelalisib (1 x 10'5 a 3,9 x 10'9 M) más una cantidad constante de IMMU-114 (0,25, 0,5, 0,75 o 1 nM) durante 96 h. La viabilidad celular se evaluó mediante ensayo MTS y se generaron curvas de dosis/respuesta basadas en el porcentaje de inhibición del crecimiento en relación con las células de control no tratadas. Los valores de Cl50 se determinaron usando Prism Graph-Pad. Los datos se normalizaron y se generaron isobologramas para demostrar el efecto de IMMU-114 sobre ibrutinib en células JVM-3.
Resultados
[0215] Al igual que con ibrutinib, IMMU-114 cambió la Cl50 de idelalisib en células JVM-3 (FIG. 5B). Las células incubadas con idelalisib solo dieron como resultado una Cl50 de 1,51 x 10-6M. Sin embargo, cuando se combinan 0,5 nM de IMMU-114 con idelalisib, el valor de Cl50 cae 2 veces a 0,77 x 10-6M (IMMU -114 a 0,5 nM solo da como resultado una inhibición del crecimiento aproximada del 26 %. Asimismo, IMMU-114 1 nM desplazó el valor de Cl50 de ibrutinib hacia abajo >18 veces.
[0216] Estos datos, junto con los resultados de otro ensayo, se normalizaron para producir un isobolograma (FIG.
6B). Como se muestra, se logró un efecto aditivo cuando las células JVM-3 se incubaron con idelalisib combinado con Im Mu -114. Estos datos demuestran la utilidad de combinar IMMU-114 con un inhibidor de PI3K para tratar clínicamente la LLC.
Ejemplo 4. Tratamiento de leucemia linfocítica crónica recidivante con IMMU-114 e ibrutinib
[0217] Una mujer de 65 años con antecedentes de LLC, según la definición del Taller Internacional sobre Leucemia Linfocítica Crónica y las clasificaciones de la Organización Mundial de la Salud, presenta recidiva de la enfermedad después de tratamientos previos con fludarabina, dexametasona y rituximab, así como un régimen de CVP. Ahora tiene fiebre y sudores nocturnos asociados con un agrandamiento generalizado de los ganglios linfáticos, una producción reducida de hemoglobina y plaquetas, así como un recuento de leucocitos que aumenta rápidamente. Su LDH está elevada y la beta-2-microglobulina es casi el doble de lo normal. El paciente recibe terapia con anti-HLA-DR IMMU-114 (IgG4 hL243) a 200 mg administrados por vía subcutánea dos veces por semana. El anticuerpo se
administra en combinación con el inhibidor de la quinasa Bruton ibrutinib, administrado por vía oral en una dosis de 420 mg al día. Después de 6 semanas, la evaluación de los valores hematológicos y de laboratorio de la paciente indica que ha mostrado una respuesta parcial a la terapia combinada.
Ejemplo 5. Tratamiento de linfoma no Hodgkin (LNH) en recaída/refractario con IMMU-114 e idelalisib
[0218] Diecisiete pacientes con LNH folicular que no han respondido a rituximab y a un agente alquilante, o que han tenido una recaída dentro de los 6 meses posteriores a la recepción de esas terapias, reciben 200 mg de IMMU-114, inyectados s.c. dos veces por semana, en combinación con el inhibidor de PI3K idelalisib, administrado por vía oral a razón de 150 mg dos veces al día, hasta que la enfermedad progrese o el paciente se retire del estudio. Las respuestas se evalúan mediante tomografías computarizadas, con otras evaluaciones que incluyen eventos adversos, niveles sanguíneos de células B, niveles séricos de IMMU-114 y títulos humanos anti-IMMU-114 (HAHA).
[0219] Solo se observan reacciones de inyección transitorias ocasionales, de leves a moderadas, y no se observan otros problemas de seguridad, excepto neutropenia hasta el Grado 3 (pero reversible después de interrumpir el tratamiento hasta que se reduce al Grado 1). Se observa un agotamiento transitorio de células B (hasta aproximadamente un 25%). La tasa de respuesta objetiva (respuestas parciales más respuestas completas más respuestas completas no confirmadas) es del 47 % (8/17) con una tasa de respuesta completa/respuesta completa no confirmada del 24 % (4/17). Cuatro de las ocho respuestas objetivas continúan durante 30 semanas o más. Todas las muestras de suero evaluadas para anticuerpos humanos anti-IMMU-114 (HAHA) son negativas.
Ejemplo 6. Conjugación de productos SN-38 bifuncionales a anticuerpos levemente reducidos
[0220] El MAb humanizado anti-CEACAM5, hMN-14 (también conocido como labetuzumab), el MAb humanizado anti-CD22, hLL2 (también conocido como epratuzumab), el MAb humanizado anti-CD20, hA20 (también conocido como veltuzumab), el En estos estudios se utilizaron MAb humanizado EGP-1, hRS7, y MAb humanizado anti-mucina, hPAM4 (también conocido como clivatuzumab). Cada anticuerpo se redujo con ditiotreitol (DTT), utilizado en un exceso molar de 50 a 70 veces, en PBS 40 mM, pH 7,4, que contenía EDTA 5,4 mM, a 37 °C (baño) durante 45 min. El producto reducido se purificó mediante cromatografía de exclusión por tamaño y/o diafiltración, y se intercambió el tampón por un tampón adecuado a pH 6,5. El contenido de tiol se determinó mediante el ensayo de Ellman y estuvo en el rango de 6,5 a 8,5 SH/IgG. Alternativamente, los anticuerpos se redujeron con Tris (2-carboxietil) fosfina (TCEP) en tampón fosfato a un pH en el rango de 5-7, seguido de conjugación in situ. El MAb reducido se hizo reaccionar con un exceso molar de ~ 10 a 15 veces de CL2A-SN-38 (preparado como se describe en la Patente de EE. UU. n.° 7.999.083) utilizando DMSO a 7-15 % v/v como cosolvente, e incubando durante 20 min a temperatura ambiente. El conjugado se purificó por SEC centrifugada, paso a través de una columna hidrofóbica y finalmente por ultrafiltracióndiafiltración. El producto se ensayó para SN-38 mediante la absorbancia a 366 nm y la correlación con los valores estándar, mientras que la concentración de proteína se dedujo de la absorbancia a 280 nm, corregida por el exceso de absorbancia de SN-38 a esta longitud de onda. De esta forma se determinaron las relaciones de sustitución SN-38/MAb. Los conjugados purificados se almacenaron como formulaciones liofilizadas en viales de vidrio, se taparon al vacío y se almacenaron en un congelador a -20 °C. En la Tabla 10 se muestran las relaciones de sustitución molar (MSR) de SN-38 obtenidas para algunos de estos conjugados, que típicamente estaban en el rango de 5 a 7. El experto en la materia se dará cuenta de que el método de conjugación se puede aplicar a cualquier anticuerpo de uso en los métodos descritos.
Tabla 10: Relaciones de sustitución molar (MSR) SN-38/MAb en algunos conjugados

Ejemplo 7. Eficacias terapéuticas in vivo en modelos preclínicos de carcinoma pancreático o de colon humano
[0221] Se trataron ratones desnudos atímicos inmunocomprometidos (hembra), portadores de xenoinjertos subcutáneos de tumores pancreáticos o de colon humanos con conjugado CL2A-SN-38 específico o conjugado de control, o se dejaron sin tratar. Se observaron las eficacias terapéuticas de los conjugados específicos. FIG. 7 muestra un modelo de tumor pancreático Capan 1, en el que los conjugados CL2A-SN-38 específicos de los anticuerpos hRS7 (anti-EGP-1), hPAM4 (anti-mucina) y hMN-14 (anti-CEACAM5) mostraron mejores eficacias que el control hA20. -Conjugado CL2A-SN-38 (anti-CD20) y control sin tratar. De manera similar, en un modelo BXPC3 de cáncer de páncreas humano, el hRS7-CL2A-SN-38 específico mostró una mejor eficacia terapéutica que los tratamientos de control (FIG. 8). Asimismo, en un modelo LS174T agresivo de carcinoma de colon humano, el tratamiento con hMN-14-CL2A-SN-38 específico fue más eficaz que la ausencia de tratamiento (FIG. 9).
Ejemplo 8. Terapia in vivo de metástasis pulmonares de tumores colónicos humanos GW-39 en ratones desnudos usando hMN-14-[CL1-SN-38] y hMN-14-[CL2-SN-38]
[0222] Se estableció un modelo metastásico de pulmón de carcinoma de colon en ratones desnudos mediante inyección iv de suspensión de tumor de colon humano GW-39, y la terapia se inició 14 días después. Conjugados de anticuerpos anti-CEACAM5 específicos, hMN14-CL1-SN-38 y hMN14-CL2-SN-38, así como conjugados de control de MAb anti-CD22 no dirigidos, hLL2-CL1-SN-38 y hLL2-CL2-SN-38 y Se inyectaron mezclas equivalentes de hMN14 y SN-38 en un programa de dosis de q4d x 8, usando diferentes dosis. FIG. 10 (MSR = relación de sustitución molar de SN-38/anticuerpo) muestra efectos terapéuticos selectivos debido a los conjugados de hMN-14. A dosis equivalentes de 250 mg, los ratones tratados con hMN14-CL1-SN-38 o hMN14-CL2-SN-38 mostraron una mediana de supervivencia de más de 107 días. Los ratones tratados con los anticuerpos conjugados de control hLL2-CL1-SN-38 y hLL2-CL2-SN-38, que no se dirigen específicamente a las células de cáncer de pulmón, mostraron una mediana de supervivencia de 56 y 77 días, mientras que los ratones tratados con hMN14 IgG no conjugado y libres SN-38 mostró una mediana de supervivencia de 45 días, comparable con el control de solución salina sin tratar de 43,5 días. Se observó claramente un aumento significativo y sorprendente en la eficacia del conjugado de anticuerpo-SN-38 dirigido a células cancerosas conjugado, que fue sustancialmente más eficaz que el anticuerpo no conjugado y el agente quimioterapéutico libre solos. También se observó la sensibilidad a la dosis del efecto terapéutico del anticuerpo conjugado. Estos resultados demuestran la clara superioridad de los conjugados de anticuerpo SN-38 en comparación con el efecto combinado del anticuerpo no conjugado y el SN-38 libre en el mismo sistema de cáncer de pulmón humano in vivo.
Ejemplo 9. Uso del conjugado anti-Trop-2 IgG-SN-38 humanizado para el tratamiento eficaz de diversos cánceres epiteliales
Resumen
[0223] El propósito de este estudio fue evaluar la eficacia de un conjugado de anticuerpo-fármaco (ADC) SN-38-anti-Trop-2 contra varios tipos de tumores sólidos humanos, y evaluar su tolerabilidad en ratones y monos, este último con reactividad cruzada tisular. a hRS7 similar a los humanos. Dos derivados de SN-38, CL2-SN-38 y CL2A-SN-38, se conjugaron con el anticuerpo humanizado antiTrop-2, hRS7. Los inmunoconjugados se caracterizaron in vitro en cuanto a estabilidad, unión y citotoxicidad. La eficacia se probó en cinco modelos diferentes de xenoinjerto de tumor sólido humano que expresaban el antígeno Trop-2. La toxicidad se evaluó en ratones y en monos Cynomolgus.
[0224] Los conjugados de hRS7 de los dos derivados de SN-38 fueron equivalentes en sustitución de fármacos (~6), unión celular (Kd ~ 1,2 nmol/l), citotoxicidad (Cl50 ~ 2,2 nmol/L) y estabilidad sérica in vitro (t/k ~ 20 horas). La exposición de las células al ADC demostró vías de señalización que conducen a la escisión de PARP, pero se observaron diferencias frente a SN-38 libre en la regulación positiva de p53 y p21. Efectos antitumorales significativos fueron producidos por hRS7-SN-38 a dosis no tóxicas en ratones portadores de tumores Calu-3 (P ≤ 0.05), Capan-1 (P < 0,018), BxPC-3 (P < 0,005) y COLO 205 (P < 0,033) en comparación con los ADC de control no dirigidos. Los ratones toleraron una dosis de 2 x 12 mg/kg (equivalentes de SN-38) con solo elevaciones de corta duración en niveles de enzimas hepáticas ALT y AST. Los monos Cynomolgus infundidos con 2 x 0,96 mg/kg exhibieron solo disminuciones transitorias en los recuentos sanguíneos, aunque, lo que es más importante, los valores no cayeron por debajo de los rangos normales.
[0225] Concluimos que el ADC anti-Trop-2 hRS7-CL2A-SN-38 proporcionó efectos antitumorales significativos y específicos contra una variedad de tipos de tumores sólidos humanos. Fue bien tolerado en monos, con una expresión tisular de Trop-2 similar a la de los humanos. (Cardillo et al., 2011, Clin Cancer Res 17:3157-69).
[0226] El tratamiento exitoso con irinotecán de pacientes con tumores sólidos ha sido limitado debido en gran parte a la baja tasa de conversión del profármaco CPT-11 en el metabolito activo SN-38. Otros han examinado formas no dirigidas de SN-38 como un medio para eludir la necesidad de esta conversión y administrar SN-38 de forma pasiva a los tumores. Conjugamos SN-38 covalentemente con un anticuerpo anti-Trop-2 humanizado, hRS7. Este conjugado de anticuerpo y fármaco tiene efectos antitumorales específicos en una variedad de modelos de xenoinjerto de cáncer humano sc, incluido el carcinoma de pulmón de células no pequeñas, carcinomas de pulmón de células escamosas, colorrectal y pancreático, todos en dosis no tóxicas (p. ej., ≤3,2 mg/ kg de dosis equivalente de SN-38).
[0227] Trop-2 se expresa ampliamente en muchos cánceres epiteliales, pero también en algunos tejidos normales y, por lo tanto, se realizó un estudio de aumento de dosis en monos Cynomolgus para evaluar la seguridad clínica de este conjugado. Los monos toleraron 24 mg de equivalentes de SN-38/kg con solo toxicidades menores y reversibles. Dada su orientación al tumor y su perfil de seguridad, hRS7-SN-38 puede proporcionar una mejora en el tratamiento de los tumores sólidos que responden al irinotecán.
Introducción
[0228] El antígeno de superficie celular del trofoblasto humano (Trop-2), también conocido como GA733-1 (antígeno gástrico 733-1), EGP-1 (glucoproteína epitelial-1) y TACSTD2 (transductor de señales de calcio asociado a tumores), se expresa en una variedad de carcinomas humanos y tiene significado pronóstico en algunos, estando asociado con una enfermedad más agresiva (ver, por ejemplo, Alberti et al., 1992, Hybridoma 11:539-45; Stein et al., 1993, Int J Cancer 55:938-46;Stein et al., 1994, Int J Cancer Suppl. 8:98-102). Los estudios del papel funcional de Trop-2 en una línea celular de cáncer de páncreas de ratón transfectada con Trop-2 murino revelaron una mayor proliferación en condiciones de suero bajo, migración y crecimiento independiente del anclaje in vitro, y una tasa de crecimiento mejorada con evidencia de aumento de Ki-67 expresión in vivo y una mayor probabilidad de metástasis (Cubas et al., 2010, Mol Cancer 9:253).
[0229] La distribución del antígeno Trop-2 en muchos cánceres epiteliales lo convierte en un objetivo terapéutico atractivo. Stein y colegas (1993, Int J Cancer 55:938-46) caracterizaron un anticuerpo, denominado RS7-3G11 (RS7), que se unía a EGP-1, que estaba presente en varios tumores sólidos, pero el antígeno también se expresaba. en algunos tejidos normales, generalmente en menor intensidad, o en regiones restringidas. Se documentaron eficacias terapéuticas y dirigidas en una serie de xenoinjertos de tumores humanos usando RS7 radiomarcado (Shih et al., 1995, Cancer Res 55:5857s-63s; Stein et al., 1997, Cancer 80:2636-41; Govindan et al., 2004, Breast Cancer Res Treat 84: 173-82), pero este anticuerpo de internalización no mostró actividad terapéutica en forma no conjugada (Shih et al., 1995, Cancer Res 55: 5857s-63s). Sin embargo, in vitro ha demostrado actividad de citotoxicidad celular dependiente de anticuerpos (ADCC) contra carcinomas positivos para Trop-2.
[0230] Reportamos la preparación de conjugados anticuerpo-fármaco (ADC) utilizando una IgG anti-CEACAM5 (CD66e) acoplada a varios derivados de SN-38, un inhibidor de la topoisomerasa-I que es el componente activo del irinotecán, o CPT-11 (Moon et al. al., 2008, J Med Chem 51:6916-26; Govindan et al., 2009, Clin Cancer Res 15:6052-61). Los derivados variaron en sus propiedades de estabilidad sérica in vitro, y los estudios in vivo encontraron que una forma (designada como CL2) era más efectiva para prevenir o detener el crecimiento de xenoinjertos de cáncer de páncreas y colón humanos que otros enlaces con mayor o menor estabilidad.
[0231] Es importante destacar que estos efectos ocurrieron en dosis no tóxicas, y las pruebas iniciales no lograron determinar una toxicidad limitante de la dosis (Govindan et al., 2009, Clin Cancer Res 15:6052-61). Estos resultados fueron alentadores, pero también sorprendentes, porque el anticuerpo CEACAM5 no se internaliza, una propiedad que se considera crítica para el éxito de un ADC. Especulamos que la actividad terapéutica del conjugado anti-CEACAM5-SN-38 podría estar relacionada con la liberación lenta de SN-38 dentro del tumor después de la localización del anticuerpo. Debido a que el irinotecán funciona mejor cuando las células están expuestas durante la fase S de su ciclo de crecimiento, se espera que una liberación sostenida mejore las respuestas. De hecho, el SN-38 acoplado a agentes extensores del plasma no dirigidos, como el polietilenglicol (PEG) o las micelas, ha demostrado una mayor eficacia que el irinotecán o el SN-38 solo (p. ej., Koizumi et al., 2006, Cancer Res 66:10048- 56), dando un apoyo adicional a este mecanismo.
[0232] Dada la amplia reactividad del anticuerpo RS7 con los cánceres epiteliales y su capacidad de internalización, planteamos la hipótesis de que un conjugado RS7-SN-38 podría beneficiarse no solo de la liberación sostenida del fármaco, sino también de la administración intracelular directa. Por lo tanto, preparamos y probamos la eficacia de los conjugados SN-38 utilizando una versión humanizada del anticuerpo murino RS7 (hRS7), en el que el SN-38 se unió al anticuerpo utilizando un enlazador CL2A mejorado, lo que mejoró la calidad del conjugado y su eficacia in vivo sin alterar su estabilidad in vitro. Este nuevo derivado (designado CL2A) es un agente preferido para el acoplamiento de SN-38 a anticuerpos.
[0233] En este documento, mostramos la eficacia del conjugado hRS7-SN-38 en varias líneas celulares de cáncer epitelial implantadas en ratones desnudos en dosis no tóxicas, y otros estudios revelan que se podrían tolerar dosis sustancialmente más altas. Más importante aún, los estudios de toxicidad en monos que también expresan Trop-2 en tejidos similares a los humanos mostraron que hRS7-SN-38 fue tolerado en cantidades apreciablemente mayores que la dosis terapéuticamente eficaz en ratones.
Materiales y métodos
[0234] Líneas celulares, anticuerpos y quimioterapéuticos. Todas las líneas celulares de cáncer humano utilizadas en este estudio se adquirieron de la American Type Culture Collection. Estos incluyen Calu-3 (carcinoma de pulmón de células no pequeñas), SK-MES-1 (carcinoma de pulmón de células escamosas), COLO 205 (adenocarcinoma de colon), Capan-1 y BxPC-3 (adenocarcinomas de páncreas) y PC-3 (adenocarcinomas prostáticos). IgG RS7 humanizado y anticuerpos anti-CD20 (hA20 IgG, veltuzumab) y anti-CD22 (hLL2 IgG, epratuzumab) humanizados de control se prepararon en Immunomedics, Inc. El irinotecán (20 mg/ml) se obtuvo de Hospira, Inc.
[0235] Inmunoconjugados SN-38 y aspectos in vitro. La síntesis de CL2-SN-38 se ha descrito previamente (Moon et al., 2008, J Med Chem 51:6916-26). Su conjugación con hRS7 IgG y la estabilidad en suero se realizaron como se describe (Moon et al., 2008, J Med Chem 51:6916-26; Govindan et al., 2009, Clin Cancer Res 15:6052-61). Las preparaciones de CL2A-SN-38 (MW 1480) y su conjugado hRS7 y los estudios de estabilidad, unión y citotoxicidad se
realizaron como se describió anteriormente (Moon et al., 2008, J Med Chem 51:6916-26). Se prepararon lisados celulares y se realizó inmunotransferencia para p21Waf1/Cip, p53 y PARP (poli-ADP-ribosa polimerasa).
[0236] Estudios terapéuticos in vivo. Para todos los estudios en animales, las dosis de inmunoconjugados de SN-38 e irinotecán se muestran en equivalentes de SN-38. Sobre la base de una proporción media de sustitución de SN-38/IgG de 6, una dosis de 500 μg de ADC para un ratón de 20 g (25 mg/kg) contiene 0,4 mg/kg de SN-38. Las dosis de irinotecán también se muestran como equivalentes de SN-38 (es decir, 40 mg de irinotecán/kg equivalen a 24 mg/kg de SN-38). Se compraron ratones desnudos atímicos (nu/nu) hembra NCr, de 4 a 8 semanas de edad, y ratones macho SwissWebster, de 10 semanas de edad, de Taconic Farms. Los estudios de tolerabilidad se realizaron en monos Cynomolgus (Macaca fascicularis; 2,5-4 kg macho y hembra) por SNBL USA, Ltd. A los animales se les implantó por vía subcutánea diferentes líneas celulares de cáncer humano. El volumen del tumor (TV) se determinó mediante mediciones en 2 dimensiones utilizando calibres, con volúmenes definidos como: L x w2/2, donde L es la dimensión más larga del tumor y w es la más corta. Los tumores variaban en tamaño entre 0,10 y 0,47 cm3 cuando comenzó la terapia. Los regímenes de tratamiento, las dosis y el número de animales en cada experimento se describen en los Resultados. El hRS7-CL2A-SN-38 liofilizado y el ADC de control se reconstituyeron y diluyeron según se requería en solución salina estéril. Todos los reactivos se administraron por vía intraperitoneal (0,1 ml), excepto el irinotecán, que se administró por vía intravenosa. El régimen de dosificación estuvo influenciado por nuestras investigaciones previas, donde el ADC se administró cada 4 días o dos veces por semana durante períodos de tiempo variables (Moon et al., 2008, J Med Chem 51: 6916-26; Govindan et al., 2009, Clin Cancer Res 15:6052-61). Esta frecuencia de dosificación reflejó una consideración de la vida media en suero del conjugado in vitro, para permitir una exposición más continua al ADC.
[0237] Estadísticas. Las curvas de crecimiento se determinaron como cambio porcentual en el TV inicial a lo largo del tiempo. El análisis estadístico del crecimiento tumoral se basó en el área bajo la curva (AUC). Los perfiles de crecimiento tumoral individual se obtuvieron a través de modelos de curvas lineales. Se empleó una prueba f para determinar la igualdad de varianza entre grupos antes del análisis estadístico de las curvas de crecimiento. Se usó una prueba t de 2 colas para evaluar la significación estadística entre los diversos grupos de tratamiento y controles, excepto para el control de solución salina, donde se usó una prueba t de 1 cola (significación en P < 0,05). Las comparaciones estadísticas de AUC se realizaron solo hasta el momento en que el primer animal dentro de un grupo fue sacrificado debido a la progresión.
[0238] Farmacocinética y biodistribución. Se inyectaron hRS7-CL2A-SN-38 y hRS7 IgG radiomarcados con 111In en ratones desnudos que tenían tumores SK-MES-1 s.c. (~0,3 cm3). A un grupo se le inyectó por vía intravenosa 20 μCi (250 μg de proteína) de 111In-hRS7-CL2A-SN-38, mientras que otro grupo recibió 20 μCi (250 μg de proteína) de 111InhRS7 IgG. En varios puntos de tiempo, los ratones (5 por punto de tiempo) fueron anestesiados, sangrados mediante punción intracardíaca y luego sacrificados. Los tumores y diversos tejidos se extrajeron, pesaron y contaron mediante centelleo y para determinar el porcentaje de dosis inyectada por gramo de tejido (% DI/g). A un tercer grupo se le inyectó 250 μg de hRS7-CL2A-SN-38 sin marcar 3 días antes de la administración de 111In-hRS7-CL2A-SN-38 y se le realizó la necropsia. Se usó una prueba t de 2 colas para comparar la captación de hRS7-CL2A-SN-38 y hRS7 IgG después de determinar la igualdad de varianza usando la prueba f. El análisis farmacocinético del aclaramiento sanguíneo se realizó utilizando el software WinNonLin (Parsight Corp.).
[0239] Tolerabilidad en ratones Swiss-Webster y monos Cynomolgus. Brevemente, los ratones se clasificaron en 4 grupos cada uno para recibir inyecciones i.p. de 2 ml de un control de tampón de acetato de sodio o 3 dosis diferentes de hRS7-CL2A-SN-38 (4, 8 o 12 mg/kg de SN-38) en los días 0 y 3 seguido de la recolección de sangre y suero, como se describe en Resultados. Monos Cynomolgus (3 machos y 3 hembras; 2,5-4,0 kg) se les administraron 2 dosis diferentes de hRS7-CL2A-SN-38. Las dosis, tiempos y número de monos desangrados para la evaluación de posibles toxicidades hematológicas y químicas séricas se describen en los Resultados.
Resultados
[0240] Estabilidad y potencia de hRS7-CL2A-SN-38. Se usaron dos enlaces diferentes para conjugar SN-38 con hRS7 IgG. El primero se denomina CL2-SN-38 y se ha descrito previamente (Moon et al., 2008, J Med Chem 51:6916-26; Govindan et al., 2009, Clin Cancer Res 15:6052-61). Se realizó un cambio en la síntesis del enlazador CL2 en el sentido de que se eliminó el resto de fenilalanina. Este cambio simplificó la síntesis, pero no afectó el resultado de la conjugación (p. ej., tanto CL2-SN-38 como CL2A-SN-38 incorporaron ~6 SN-38 por molécula de IgG). Las comparaciones lado a lado no encontraron diferencias significativas en la estabilidad del suero, la unión al antígeno o la citotoxicidad in vitro (no se muestra).
[0241] Para confirmar que el cambio en el conector SN-38 de CL2 a CL2A no afectó la potencia in vivo, se compararon hRS7-CL2A y hRS7-CL2-SN-38 en ratones con tumores COLO 205 o Capan-1 (no se muestra), utilizando 0,4 mg o 0,2 mg/kg de s N-38 dos veces por semana durante x 4 semanas, respectivamente, y con tumores iniciales de 0,25 cm3 de tamaño en ambos estudios. Tanto los conjugados hRS7-CL2A como CL2-SN-38 inhibieron significativamente el crecimiento tumoral en comparación con los no tratados (AUC14 días P < 0,002 frente a solución salina en el modelo COLO 205; AUC21 días P < 0,001 frente a solución salina en el modelo Capan-1), y una ADC de
control anti-CD20 no dirigido, hA20-CL2A-SN-38 (AUC14 días P < 0,003 en el modelo COLO-205; AUC35 días: P < 0,002 en el modelo Capan-1). Al final del estudio (día 140) en el modelo Capan-1, el 50 % de los ratones tratados con hRS7-CL2A-SN-38 y el 40 % de los ratones hRS7-CL2-SN-38 estaban libres de tumor, mientras que sólo el 20 % de los animales tratados con hA20-ADC no tenían signos visibles de enfermedad.
[0242] Mecanismo de acción. Los estudios de citotoxicidad in vitro demostraron que hRS7-CL2A-SN-38 tenía valores de Cl50 en el rango de nmol/L contra varias líneas de tumores sólidos diferentes (Tabla 11). El Cl50 con SN-38 libre fue menor que el conjugado en todas las líneas celulares. Aunque no hubo correlación entre la expresión de Trop-2 y la sensibilidad a hRS7-CL2A-SN-38, la relación Cl50 del ADC versus el SN-38 libre fue menor en las células con mayor expresión de Trop-2, lo que probablemente refleja la mayor capacidad de internalizar el fármaco cuando hay más antígeno presente.

[0243] Se sabe que SN-38 activa varias vías de señalización en las células, lo que lleva a la apoptosis. Nuestros estudios iniciales examinaron la expresión de 2 proteínas involucradas en eventos de señalización temprana (p21WAF1/Cip1 y p53) y 1 evento apoptótico tardío [escisión de poli-ADP-ribosa polimerasa (PARP)] in vitro (no se muestra). En BxPC-3, SN-38 condujo a un aumento de 20 veces en la expresión de p2 1 WAF1/Cip1, mientras que hRS7-CL2A-SN-38 resultó en solo un aumento de 10 veces, un hallazgo consistente con la mayor actividad con SN-38 libre en esta línea celular (Tabla 11). Sin embargo, hRS7-CL2A-SN-38 aumentó la expresión de p2 1 WAF1/Cip1 en Calu-3 más de 2 veces sobre SN-38 libre (no mostrado).
[0244] Se observó una mayor disparidad entre los eventos de señalización mediados por hRS7-CL2A-SN-38 y SN-38 libre en la expresión de p53. Tanto en BxPC-3 como en Calu-3, la regulación al alza de p53 con SN-38 libre no fue evidente hasta las 48 horas, mientras que hRS7-CL2A-SN-38 reguló al alza a p53 en 24 horas (no se muestra). Además, la expresión de p53 en células expuestas al ADC fue mayor en ambas líneas celulares en comparación con SN-38 (no mostrado). Curiosamente, aunque hRS7 IgG no tuvo un efecto apreciable en la expresión de p2 1 WAF1/Cip1, indujo la regulación positiva de p53 tanto en BxPC-3 como en Calu-3, pero solo después de una exposición de 48 horas. En términos de eventos apoptóticos posteriores, la escisión de PARP fue evidente en ambas líneas celulares cuando se incubaron con SN-38 o el conjugado (no mostrado). La presencia de la PARP escindida fue mayor a las 24 horas en BxPC-3, lo que se correlaciona con una alta expresión de p21 y su menor Cl50. El mayor grado de escisión con SN-38 libre sobre el ADC fue consistente con los hallazgos de citotoxicidad.
[0245] Eficacia de hRS7-SN-38. Debido a que Trop-2 se expresa ampliamente en varios carcinomas humanos, se realizaron estudios en varios modelos de cáncer humano diferentes, que comenzaron con una evaluación del enlace hRS7-CL2-SN-38, pero luego se usaron conjugados con el enlace CL2A. Los ratones desnudos portadores de Calu-3 que recibieron 0,04 mg de SN-38/kg de hRS7-CL2-SN-38 cada 4 días x 4 tuvieron una respuesta significativamente mejorada en comparación con los animales a los que se les administró la cantidad equivalente de hLL2-CL2-SN-38 (TV = 0,14 ± 0,22 cm3 frente a 0,80 ± 0,91 cm3, respectivamente, AUC42 días P < 0,026; FIG. 11A). Se observó una respuesta a la dosis cuando se aumentó la dosis a 0,4 mg/kg de SN-38. Con este nivel de dosis más alto, todos los ratones que recibieron el conjugado específico de hRS7 se "curaron" en 28 días y permanecieron libres de tumores hasta el final del estudio el día 147, mientras que los tumores volvieron a crecer en los animales tratados con el ADC irrelevante (específico vs. irrelevante AUC98 días: P = 0,05). En ratones que recibieron la mezcla de hRS7 IgG y SN-38, los tumores progresaron >4,5 veces el día 56 (TV = 1,10 ± 0,88 cm3; AUC56 días P < 0,006 frente a hRS7-CL2-SN-38).
[0246] También se examinó la eficacia en xenoinjertos de tumores colónicos humanos (COLO 205) y pancreáticos (Capan-1). En animales portadores de tumores COLO 205, (FIG. 11B), hRS7-CL2-SN-38 (0,4 mg/kg, q4dx8) impidió el crecimiento tumoral durante el período de tratamiento de 28 días con tumores significativamente más pequeños en
comparación con ADC anti-CD20 de control (hA20-CL2-SN-38), o hRS7 IgG (TV = 0,16 ± 0,09 cm3, 1,19 ± 0,59 cm3 y 1,77 ± 0,93 cm3, respectivamente; AUC28 días P < 0,016). La MTD de irinotecan (24 mg SN-38/kg, q2dx5) fue tan efectiva como hRS7-CL2-SN-38, porque el suero de ratón puede convertir el irinotecan en SN-38 de manera más eficiente que el suero humano, pero la dosis de SN-38 en el irinotecán (2.400 μg acumulativos) fue 37,5 veces mayor que con el conjugado (64 μg en total).
[0247] Los animales que portaban Capan-1 no mostraron una respuesta significativa al irinotecán solo cuando se les administró una dosis de SN-38 equivalente al conjugado hRS7-CL2-SN-38 (p. ej., en el día 35, el tamaño tumoral promedio fue de 0,04 x 0,05 cm3 en animales 0,4 mg de SN-38/kg hRS7-SN-38 frente a 1,78 x 0,62 cm3 en animales tratados con irinotecán que recibieron 0,4 mg/kg de SN-38; AUCdía 35 P < 0,001; FIG. 11C). Cuando la dosis de irinotecán se aumentó 10 veces a 4 mg/kg de SN-38, la respuesta mejoró, pero aún no fue tan significativa como el conjugado al nivel de dosis de 0,4 mg/kg de SN-38 (TV = 0,17 ± 0,18 cm3 vs. 1,69 ± 0,47 cm3, AUCdía49 P < 0,001). Una dosis igual de hA20-CL2-SN-38 no dirigido también tuvo un efecto antitumoral significativo en comparación con los animales tratados con irinotecán, pero el conjugado específico de hRS7 fue significativamente mejor que el ADC irrelevante (TV = 0,17 ± 0,18 cm3 frente a 0,80 ± 0,68 cm3, AUCdía49 P < 0,018).
[0248] Los estudios con el ADC hRS7-CL2A-SN-38 se ampliaron luego a otros 2 modelos de cánceres epiteliales humanos. En ratones portadores de tumores pancreáticos humanos BxPC-3 (FIG. 11D), hRS7-CL2A-SN-38 nuevamente inhibió significativamente el crecimiento tumoral en comparación con ratones de control tratados con solución salina o una cantidad equivalente de hA20-CL2A-SN-38 no dirigido (TV = 0,24 ± 0,11 cm3 frente a 1,17 ± 0,45 cm3 y 1,05 ± 0,73 cm3, respectivamente; AUCdía21 P < 0,001), o irinotecán administrado a una dosis equivalente de SN-38 10 veces superior (TV = 0,27 ± 0,18 cm3 frente a 0,90 ± 0,62 cm3, respectivamente, AUCdía25 P < 0,004). Curiosamente, en ratones portadores de tumores de pulmón de células escamosas humanas SK-MES-1 tratados con 0,4 mg/kg de ADC (FIG. 11E), la inhibición del crecimiento tumoral fue superior a la solución salina o hRS7 IgG no conjugada (TV = 0,36 x 0,25 cm3 frente a 1,02 ± 0,70 cm3 y 1,30 ± 1,08 cm3, respectivamente; AUC28 días, P < 0,043), pero no apuntar a hA20-CL2A-SN-38 o la MTD de irinotecan proporcionó los mismos efectos antitumorales que el hRS7-SN-38 específico conjugado. En todos los estudios murinos, el ADC hRS7-SN-38 fue bien tolerado en términos de pérdida de peso corporal (no mostrado).
[0249] Biodistribución de hRS7-CL2A-SN-38. Las biodistribuciones de hRS7-CL2A-SN-38 o hRS7 IgG no conjugada se compararon en ratones que portaban xenoinjertos de carcinoma de pulmón de células escamosas humanas SK-MES-1 (no mostrado), utilizando los respectivos sustratos marcados con 111In. Se realizó un análisis farmacocinético para determinar el aclaramiento de hRS7-CL2A-SN-38 en relación con hRS7 no conjugado (no mostrado). El ADC se eliminó más rápido que la cantidad equivalente de hRS7 no conjugado, y el ADC exhibió una vida media y un tiempo de residencia medio aproximadamente un 40 % más cortos. No obstante, esto tuvo un impacto mínimo en la captación del tumor (no mostrado). Aunque hubo diferencias significativas en los puntos temporales de 24 y 48 horas, a las 72 horas (captación máxima) las cantidades de ambos agentes en el tumor eran similares. Entre los tejidos normales, las diferencias hepáticas y esplénicas fueron las más llamativas (no se muestra). A las 24 horas posteriores a la inyección, había >2 veces más hRS7-CL2ASN-38 en el hígado que hRS7 IgG. Por el contrario, en el bazo había 3 veces más hRS7 IgG parental presente en la captación máxima (punto de tiempo de 48 horas) que hRS7-CL2A-SN-38. La captación y aclaramiento en el resto de los tejidos generalmente reflejaron diferencias en la concentración en sangre.
[0250] Debido a que se administraron dosis dos veces por semana para la terapia, la captación tumoral en un grupo de animales que recibieron por primera vez una dosis previa de 0,2 mg/kg (250 μg de proteína) del ADC hRS73 días antes de la inyección del anticuerpo marcado con 111In fue menor. examinado. La captación tumoral de 111In-hRS7-CL2A-SN-38 en ratones predosificados se redujo sustancialmente en cada punto de tiempo en comparación con los animales que no recibieron la predosis (p. ej., a las 72 horas, la captación tumoral predosificada fue del 12,5 % ± 3,8 % DI/g frente a 25,4 % ± 8,1 % DI/g en animales que no recibieron la predosis; P = 0,0123). La dosificación previa no tuvo un impacto apreciable en el aclaramiento sanguíneo o la absorción tisular (no mostrado). Estos estudios sugieren que en algunos modelos tumorales, la acumulación tumoral del anticuerpo específico puede reducirse con la(s) dosis(es) anterior(es), lo que probablemente explica por qué la especificidad de una respuesta terapéutica podría disminuir al aumentar las dosis de ADC y por qué no es necesario aumentar más la dosis indicada.
[0251] Tolerabilidad de hRS7-CL2A-SN-38 en ratones Swiss-Webster y monos Cynomolgus. Los ratones Swiss-Webster toleraron 2 dosis durante 3 días, cada una de 4, 8 y 12 mg de SN-38/kg de hRS7-CL2A-SN-38, con una pérdida de peso transitoria mínima (no se muestra). No se produjo toxicidad hematopoyética y las químicas séricas solo revelaron niveles elevados de transaminasa de aspartato (AST) y transaminasa de alanina (no mostrada). Siete días después del tratamiento, la AST aumentó por encima de los niveles normales (>298 U/l) en los 3 grupos de tratamiento (no se muestra), con la mayor proporción de ratones en el grupo de 2 x 8 mg/kg. Sin embargo, a los 15 días posteriores al tratamiento, la mayoría de los animales estaban dentro del rango normal. Los niveles de ALT también estuvieron por encima del rango normal (>77 U/l) dentro de los 7 días de tratamiento (no se muestra) y con evidencia de normalización el día 15. Los hígados de todos estos ratones no mostraron evidencia histológica de daño tisular (no se muestra). En cuanto a la función renal, solo los niveles de glucosa y cloruro estaban algo elevados en los grupos tratados. A 2 x 8 mg/kg, 5 de 7 ratones tenían niveles de glucosa ligeramente elevados (rango de 273-320 mg/dl, extremo superior de lo normal 263 mg/dl) que volvieron a la normalidad 15 días después de la inyección. De
manera similar, los niveles de cloruro estaban levemente elevados, con un rango de 116 a 127 mmol/l (extremo superior del rango normal 115 mmol/l) en los 2 grupos de dosis más altas (57 % en el grupo de 2 x 8 mg/kg y 100 % del grupo de 2 x 12 mg/kg), y permaneció elevada hasta 15 días después de la inyección. Esto también podría ser indicativo de toxicidad gastrointestinal, porque la mayor parte del cloruro se obtiene a través de la absorción en el intestino; sin embargo, al terminar, no hubo evidencia histológica de daño tisular en ningún sistema de órganos examinado (no mostrado).
[0252] Debido a que los ratones no expresan Trop-2 unida por hRS7, se requirió un modelo más adecuado para determinar el potencial del conjugado hRS7 para uso clínico. Los estudios de inmunohistología revelaron la unión en múltiples tejidos tanto en humanos como en monos Cynomolgus (mama, ojo, tracto gastrointestinal, riñón, pulmón, ovario, trompa de Falopio, páncreas, paratiroides, próstata, glándula salival, piel, timo, tiroides, amígdala, uréter, orina vejiga y útero; no se muestra). En base a esta reactividad cruzada, se realizó un estudio de tolerabilidad en monos.
[0253] El grupo que recibió 2 x 0,96 mg SN-38/kg de hRS7-CL2A-SN-38 no tuvo eventos clínicos significativos después de la infusión y hasta la finalización del estudio. La pérdida de peso no superó el 7,3 % y volvió a los pesos de aclimatación el día 15. Se observaron disminuciones transitorias en la mayoría de los datos del hemograma (no se muestran), pero los valores no cayeron por debajo de los rangos normales. No se encontraron valores anormales en las químicas del suero. La histopatología de los animales a los que se les realizó la necropsia el día 11 ( 8 días después de la última inyección) mostró cambios microscópicos en los órganos hematopoyéticos (timo, ganglios linfáticos mandibulares y mesentéricos, bazo y médula ósea), órganos gastrointestinales (estómago, duodeno, yeyuno, íleon, ciego, colon y recto), los órganos reproductivos femeninos (ovario, útero y vagina) y en el lugar de la inyección. Estos cambios variaron de mínimos a moderados y se revirtieron por completo al final del período de recuperación (día 32) en todos los tejidos, excepto en el timo y el tracto gastrointestinal, que tendían a una recuperación total en este punto de tiempo posterior.
[0254] Con el nivel de dosis de 2 x 1,92 mg de SN-38/kg del conjugado, hubo 1 muerte por complicaciones gastrointestinales y supresión de la médula ósea, y otros animales dentro de este grupo mostraron eventos adversos similares, pero más graves que los 2 x 0,96 mg/kg grupo. Estos datos indican que las toxicidades limitantes de la dosis fueron idénticas a las del irinotecán; a saber, intestinal y hematológico. Por lo tanto, la MTD para hRS7-CL2A-SN-38 se encuentra entre 2 x 0,96 y 1,92 mg SN-38/kg, lo que representa una dosis equivalente en humanos de 2 x 0,3 a 0,6 mg/kg SN-38.
Discusión
[0255] Trop-2 es una proteína expresada en muchos tumores epiteliales, incluidos los cánceres de pulmón, mama, colorrectal, páncreas, próstata y ovario, lo que la convierte en un objetivo potencialmente importante para la administración de agentes citotóxicos. El anticuerpo RS7 se internaliza cuando se une a Trop-2 (Shih et al., 1995, Cancer Res 55:5857s-63s), lo que permite la administración intracelular directa de citotóxicos.
[0256] La conjugación de fármacos quimioterapéuticos con anticuerpos se ha explorado durante más de 30 años. Debido a que una parte sustancial de un ADC no es procesada por el tumor, sino por los tejidos normales, existe el riesgo de que estos agentes sean demasiado tóxicos para los sistemas de órganos normales antes de alcanzar el nivel terapéutico en los tumores. Al igual que con cualquier tratamiento, la ventana terapéutica es un factor clave que determina el potencial de un ADC y, por lo tanto, en lugar de examinar fármacos "ultratóxicos", elegimos SN-38 como el componente farmacológico del ADC dirigido a Trop-2.
[0257] SN-38 es un potente inhibidor de la topoisomerasa-I, con valores Cl50 en el rango nanomolar en varias líneas celulares. Es la forma activa del profármaco, irinotecan, que se usa para el tratamiento del cáncer colorrectal y que también tiene actividad en los cánceres de pulmón, mama y cerebro. Razonamos que un SN-38 dirigido directamente, en forma de ADC, sería una terapia significativamente mejorada sobre CPT-11, al superar la bioconversión baja y variable del paciente de este último a SN-38 activo.
[0258] El péptido Phe-Lys insertado en el derivado CL2 original permitió una posible escisión a través de la catepsina B. En un esfuerzo por simplificar el proceso de síntesis, en CL2A, se eliminó la fenilalanina y, por lo tanto, se eliminó el sitio de escisión de la catepsina B. Curiosamente, este producto tenía un perfil cromatográfico mejor definido en comparación con el perfil amplio obtenido con CL2 (no se muestra), pero lo que es más importante, este cambio no tuvo un impacto negativo en la unión, la estabilidad o la potencia del conjugado en las pruebas de lado a lado. Estos datos sugieren que SN-38 en CL2 se liberó del conjugado principalmente por la escisión en el enlace de carbonato de bencilo sensible al pH con el anillo de lactona de SN-38 y no por el sitio de escisión de la catepsina B.
[0259] La citoxicidad in vitro de hRS7 ADC frente a una variedad de líneas de células tumorales sólidas tuvo consistentemente valores Cl50 en el rango de nmol/L. Sin embargo, las células expuestas a SN-38 libre demostraron un valor Cl50 más bajo en comparación con el ADC. Esta disparidad entre SN-38 libre y conjugado también se informó para ENZ-2208 (Sapra et al., 2008, Clin Cancer Res 14:1888-96) y NK012 (Koizumi et al., 2006, Cancer Res 66:10048-56). ENZ-2208 utiliza un PEG ramificado para unir entre 3,5 y 4 moléculas de SN-38 por PEG, mientras que NK012
es una nanopartícula de micela que contiene un 20 % de SN-38 en peso. Con nuestro ADC, esta disparidad (es decir, la proporción de potencia con SN-38 libre frente a conjugado) disminuyó a medida que aumentaron los niveles de expresión de Trop-2 en las células tumorales, lo que sugiere una ventaja para la administración dirigida del fármaco. En términos de estabilidad sérica in vitro, las formas CL2- y CL2A-SN-38 de hRS7-SN-38 produjeron una t/y de ~20 horas, lo que contrasta con la breve t/y de 12,3 minutos informada para ENZ-2208 (Zhao et al., 2008, Bioconjug Chem 19:849-59), pero similar al 57 % de liberación de SN-38 de NK012 en condiciones fisiológicas después de 24 horas (Koizumi et al., 2006, Cancer Res 66:10048-56).
[0260] El tratamiento de ratones portadores de tumores con hRS7-SN-38 (ya sea con CL2-SN-38 o CL2A-SN-38) inhibió significativamente el crecimiento del tumor en 5 modelos de tumores diferentes. En 4 de ellos, se observaron regresiones tumorales y, en el caso de Calu-3, todos los ratones que recibieron la dosis más alta de hRS7-SN-38 estaban libres de tumores al finalizar el estudio. A diferencia de los humanos, el irinotecán se convierte de manera muy eficiente en SN-38 mediante una esterasa plasmática en ratones, con una tasa de conversión superior al 50 % y con una mayor eficacia en ratones que en humanos. Cuando se administró irinotecán a niveles de SN-38 10 veces más altos o equivalentes, hRS7-SN-38 fue significativamente mejor en el control del crecimiento tumoral. Solo cuando se administró irinotecán en su MTD de 24 mg/kg q2dx5 (37,5 veces más SN-38) igualó la eficacia de hRS7-SN-38. En pacientes, esperaríamos que esta ventaja favoreciera aún más a hRS7-CL2A-SN-38, porque la bioconversión de irinotecán sería sustancialmente menor.
[0261] También demostramos en algunas líneas celulares que expresan antígenos, como SK-MES-1, que el uso de un ADC de unión a antígeno no garantiza mejores respuestas terapéuticas que un conjugado irrelevante que no se une. Este no es un hallazgo inusual o inesperado. De hecho, los conjugados de SN-38 que no se unen mencionados anteriormente mejoran la actividad terapéutica en comparación con el irinotecán, por lo que se espera que un conjugado de IgG-SN-38 irrelevante tenga cierta actividad. Esto está relacionado con el hecho de que los tumores tienen vasos inmaduros con fugas que permiten el paso de macromoléculas mejor que los tejidos normales. Con nuestro conjugado, el 50 % del SN-38 se liberará en ~ 13 horas cuando el pH se reduzca a un nivel que imite los niveles lisosomales (p. ej., pH 5,3 a 37 °C; datos no mostrados), mientras que al pH neutro de suero, la tasa de liberación se reduce casi 2 veces. Si un conjugado irrelevante entra en un microambiente tumoral ácido, se espera que libere algo de SN-38 localmente. Otros factores, como la fisiología del tumor y las sensibilidades innatas al fármaco, también desempeñarán un papel en la definición de esta actividad "básica". Sin embargo, un conjugado específico con un tiempo de residencia más largo debería tener una potencia mejorada sobre esta respuesta de referencia siempre que haya suficiente antígeno para capturar el anticuerpo específico. Los estudios de biodistribución en el modelo SKMES-1 también mostraron que si el antígeno tumoral se satura como consecuencia de dosis sucesivas, se reduce la captación tumoral del conjugado específico, lo que produce resultados terapéuticos similares a los encontrados con un conjugado irrelevante.
[0262] Aunque es difícil hacer comparaciones directas entre nuestro ADC y los informes publicados de otros agentes de entrega SN-38, se pueden hacer algunas observaciones generales. En nuestros estudios de terapia, la dosis individual más alta fue de 0,4 mg/kg de SN-38. En el modelo Calu-3, solo se administraron 4 inyecciones para una dosis total acumulada de 1,6 mg/kg de SN-38 o 32 μg de SN-38 en un ratón de 20 g. Se realizaron múltiples estudios con ENZ-2208 usando su MTD de 10 mg/kg x 5, y los estudios preclínicos con NK012 involucraron su MTD de 30 mg/kg x 3. Por lo tanto, se obtuvieron efectos antitumorales significativos con hRS7-SN-38 a 30 veces y 55 veces menos equivalentes de SN-38 que las dosis notificadas en ENZ-2208 y NK012, respectivamente. Incluso con 10 veces menos ADC de hRS7 (0,04 mg/kg), se observaron efectos antitumorales significativos, mientras que no se presentaron dosis más bajas de ENZ-2208, y cuando la dosis de NK012 se redujo 4 veces a 7,5 mg/kg, la eficacia se perdió (Koizumi et al., 2006, Cancer Res 66:10048-56). Los ratones normales no mostraron toxicidad aguda con una dosis acumulada durante 1 semana de 24 mg/kg de SN-38 (1500 mg/kg del conjugado), lo que indica que la MTD era mayor. Por lo tanto, los animales portadores de tumores fueron tratados eficazmente con cantidades de equivalentes de SN-38 de 7,5 a 15 veces menores.
[0263] Como inhibidor de la topoisomerasa-I, SN-38 induce un daño significativo en el ADN de una célula, con una regulación al alza de p53 y p2 1 WAF1/Cip1 que da como resultado la activación de la caspasa y la escisión de PARP. Cuando expusimos las células BxPC-3 y Calu-3 a nuestro ADC, tanto p53 como p2 1 WAF1/Cip1 aumentaron por encima de los niveles basales. Además, la escisión de PARP también fue evidente en ambas líneas celulares, lo que confirma un evento apoptótico en estas células. De interés fue la mayor regulación positiva de p2 1 WAF1/Cip1 en BxPC-3 y Calu-3 en relación con p53 tanto por SN-38 libre como por nuestro hRS7-SN-38. Esto puede ser indicativo del estado mutacional de p53 en estas 2 líneas celulares y el uso de una vía independiente de p53 para la apoptosis mediada por p2 1 WAF1/Cip1.
[0264] Una observación interesante fue la regulación ascendente temprana de p53 tanto en BxPC-3 como en Calu-3 a las 24 horas mediada por el hRS7-ADC en relación con el SN-38 libre. Incluso la IgG hRS7 desnuda podría aumentar p53 en estas líneas celulares, aunque solo después de una exposición de 48 horas. La sobreexpresión de Trop-2 y el entrecruzamiento por parte de los anticuerpos se ha relacionado con varios eventos de señalización relacionados con MAPK, así como con la liberación de calcio intracelular. Si bien la unión de hRS7 no fue suficiente para inducir la apoptosis en BxPC-3 y Calu-3, como lo demuestra la falta de escisión de PARP, puede ser suficiente
para cebar una célula, de modo que la inclusión de SN-38 conjugado con hRS7 puede conducir a un mayor efecto sobre la inhibición del crecimiento tumoral. Actualmente se están realizando estudios para comprender qué vías están involucradas con la entrega de hRS7 de SN-38 y cómo pueden diferir del SN-38 libre, y qué efecto puede tener el estado de p53 en esta señalización.
[0265] Los estudios de biodistribución revelaron que hRS7-CL2A-SN-38 tenía una absorción tumoral similar a la hRS7 IgG parental, pero se eliminaba sustancialmente más rápido con una absorción hepática dos veces mayor, lo que puede deberse a la hidrofobicidad de SN-38. Eliminándose el ADC a través del hígado, se esperaba que las toxicidades hepáticas y gastrointestinales fueran limitantes de la dosis. Aunque los ratones tenían evidencia de aumento de las transaminasas hepáticas, la toxicidad gastrointestinal fue leve en el mejor de los casos, con solo una pérdida transitoria de peso y no se observaron anomalías en el examen histopatológico. Curiosamente, no se observó toxicidad hematológica. Sin embargo, los monos mostraron un perfil de toxicidad idéntico al esperado para el irinotecán, siendo la toxicidad gastrointestinal y hematológica la dosis limitante.
[0266] Debido a que Trop-2 reconocido por hRS7 no se expresa en ratones, era de vital importancia realizar estudios de toxicidad en monos que tienen una expresión tisular de Trop-2 similar a la de los humanos. Los monos toleraron 0,96 mg/kg/dosis (~12 mg/m2) con toxicidad leve y reversible, que se extrapola a una dosis humana de ~0,3 mg/kg/dosis (~11 mg/m2). En un ensayo clínico de fase I de NK012, los pacientes con tumores sólidos toleraron 28 mg/m2 de SN-38 cada 3 semanas con neutropenia de grado 4 como toxicidad limitante de la dosis (Hamaguchi et al., 2010, Clin Cancer Res 16:5058-66). Del mismo modo, los ensayos clínicos de fase I con ENZ-2208 revelaron una neutropenia febril limitante de la dosis, recomendándose administrar 10 mg/m2 cada 3 semanas o 16 mg/m2 si se administraba G-CSF a los pacientes. Debido a que los monos toleraron una dosis equivalente humana acumulada de 22 mg/m2, es posible que, aunque hRS7 se una a varios tejidos normales, la MTD para un solo tratamiento del ADC de hRS7 podría ser similar a la de otros agentes SN-38 no dirigidos. De hecho, la especificidad del anticuerpo anti-Trop-2 no pareció desempeñar un papel en la definición de la DLT, porque el perfil de toxicidad fue similar al del irinotecán. Más importante aún, si la actividad antitumoral se puede lograr en humanos como en ratones que respondieron con una dosis equivalente humana de solo 0,03 mg de equivalentes/kg/dosis de SN-38, entonces se podrían lograr respuestas antitumorales clínicamente significativas.
[0267] En conclusión, los estudios de toxicología en monos, combinados con modelos de xenoinjerto de cáncer humano in vivo en ratones, han indicado que este ADC dirigido a Trop-2 es un tratamiento eficaz en varios tumores de diferente origen epitelial.
Ejemplo 10. Eficacia del anticuerpo anti-Trop-2 conjugado con una forma de profármaco de 2-pirrolinodoxorrubicina (2-PDox)
[0268] Se preparó una forma de profármaco de 2-PDox (denominada pro-2-PDox) y se conjugó con anticuerpos como se describe en la solicitud de patente de EE. UU. 14/175.089. A menos que se indique lo contrario a continuación, el número de fracciones de fármaco por molécula de anticuerpo estaba en el intervalo de aproximadamente 6,5 a aproximadamente 7,5.
[0269] Estudios de unión celular in vitro: La retención de la unión del anticuerpo se confirmó mediante ensayos de unión celular que compararon la unión del anticuerpo conjugado con el no conjugado (Chari, 2008, Acc Chem Res 41:98-107). La potencia del conjugado se probó en un ensayo MTS de 4 días usando células diana apropiadas. El ADC anti-Trop-2 (hRS7-pro-2-PDox) exhibió valores Cl50 de 0,35-1,09 nM en líneas celulares de cáncer gástrico (NCI-N87), pancreático (Capan-1) y de mama (MDA-MB-468) humanas, con fármaco libre que exhibe una potencia de 0,02 0,07 nM en las mismas líneas celulares. En estudios adicionales, se observó que hRS7-pro-2-PDox era citotóxico para MDA-MB-468, AG S, NCI-N87 y líneas celulares de tumor sólido Capan-1 (no se muestra).
[0270] No se observaron diferencias significativas en la unión del resto de anticuerpo a células de carcinoma gástrico NCI-N87 entre hRS7 no conjugado y pro-2-PDox-hRS7 conjugado con 6 moléculas de pro-2-PDox por anticuerpo (no mostrado). Se concluye que la conjugación de pro-2-PDox a anticuerpos no afecta la actividad de unión antígenoanticuerpo.
[0271] Estabilidad del suero: La estabilidad del suero del ADC anti-Trop-2 (hRS7-pro-2-PDox) se determinó mediante incubación en suero humano a una concentración de 0,2 mg/mL a 37 °C. El incubado se analizó por HPLC usando cromatografía de interacción hidrofóbica de butilo (HIC). El análisis mostró que no hubo liberación de fármaco libre del conjugado, lo que sugiere una alta estabilidad sérica del conjugado. Cuando se repitió el mismo experimento con el conjugado de hRS7-doxorrubicina, que contenía el mismo enlazador escindible que hRS7-pro-2-PDox, y donde se verificó de forma independiente que el fármaco libre se liberaba con una vida media de 96 h, se observó una clara formación de un se observó el pico correspondiente a la doxorrubicina libre en HIC HPLC.
[0272] Sorprendentemente, se determinó que el conjugado de pro-2-PDox se sujetaba firmemente al anticuerpo porque entrecruzaba las cadenas peptídicas del anticuerpo. El entrecruzamiento estabiliza la unión del fármaco al anticuerpo para que el fármaco solo se libere intracelularmente después de que se haya metabolizado el anticuerpo.
El entrecruzamiento ayuda a minimizar la toxicidad, por ejemplo, la cardiotoxicidad, que resultaría de la liberación del fármaco libre en circulación. El uso anterior de conjugados peptídicos 2-PDox fracasó porque el fármaco entrecruzaba el péptido con otras proteínas o péptidos in vivo. Con el presente anti-Trop-2 ADC, el pro-2-PDox se une a grupos disulfuro tiol intercatenarios mientras está en forma de profármaco. La protección del profármaco se elimina rápidamente in vivo poco después de la inyección y la porción 2-PDox resultante del conjugado entrecruza las cadenas peptídicas del anticuerpo, formando un entrecruzamiento intramolecular dentro de la molécula del anticuerpo. Esto estabiliza el ADC y evita el entrecruzamiento con otras moléculas en circulación.
[0273] Estudios preclínicos in vivo: El tamaño del tumor se determinó mediante mediciones con calibre de longitud (L) y ancho (W) con el volumen del tumor calculado como (L x W2)/2. Se midieron los tumores y se pesaron los ratones dos veces por semana. Los ratones se sacrificaron si sus tumores alcanzaban un tamaño > 1 cm3, perdían más del 15 % de su peso corporal inicial o se volvían moribundos de otro modo. El análisis estadístico de los datos de crecimiento tumoral se basó en el área bajo la curva (AUC) y el tiempo de supervivencia. Los perfiles de crecimiento tumoral individual se obtuvieron a través de modelos de curvas lineales. Se empleó una prueba f para determinar la igualdad de varianza entre grupos antes del análisis estadístico de las curvas de crecimiento. Se utilizó una prueba t de dos colas para evaluar la significación estadística entre todos los diversos grupos de tratamiento y los controles no específicos. Para el análisis de control de solución salina, se usó una prueba t de una cola para evaluar la significación. Los estudios de supervivencia se analizaron mediante diagramas de Kaplan-Meier (log-rank), utilizando el paquete de software Prism GraphPad Software (v4.03) (Advanced Graphics Software, Inc.; Encinitas, CA). Todas las dosis en los experimentos preclínicos se expresan en cantidades de anticuerpos. En términos de fármaco, 100 μg de anticuerpo (5 mg/kg) en un ratón de 20 g, por ejemplo, lleva 1,4 μg-2,8 μg (0,14-0,17 mg/kg) de dosis equivalente de pro-2-PDox cuando se utiliza un ADC con 3-6 fármacos/IgG.
[0274] Una sola dosis i.v. de > 300 μg [~ 10 μg de pro-2-PDox] del ADC anti-Trop-2 fue letal, pero todos los animales toleraron 4 dosis de 45 μg administradas en 2 semanas. Usando este régimen de dosificación, examinamos el efecto terapéutico de anti-Trop-2 hRS7-pro-2-PDox en 2 modelos de xenoinjerto de tumor humano, Capan-1 (cáncer de páncreas) y NCI-N87 (cáncer gástrico). La terapia comenzó 7 días después del trasplante del tumor en ratones desnudos. En el modelo Capan-1 establecido de 7 días de edad, el 100 % de los tumores establecidos retrocedieron rápidamente, sin evidencia de nuevo crecimiento (no mostrado). Este resultado se reprodujo en un experimento repetido (no mostrado). El conjugado anti-Trop-2 de pro-2-PDox fue mucho más efectivo que el mismo fármaco conjugado con un anticuerpo (hMN-14) contra CEACAM5, que también se expresa en el cáncer de páncreas, o un anticuerpo contra CD20 (hA20), lo cual no es. Todos los tratamientos fueron superiores al control de solución salina.
[0275] Se observaron resultados similares en el modelo NCI-N87 establecido (no mostrado), donde un segundo curso de terapia, administrado después del día 70, se toleró con seguridad y condujo a regresiones adicionales del tumor residual (no mostrado). El conjugado de hRS7-SN-38 internalizado, dirigido a Trop-2, proporcionó mejores respuestas terapéuticas que un conjugado de un anticuerpo anti-CEACAM5 de internalización deficiente, hMN-14 (no mostrado). Un ADC anti-CD20 no dirigido, hA20-pro-2-PDox, fue ineficaz, lo que indica una eficacia terapéutica selectiva (no se muestra). Los datos de un xenoinjerto de cáncer de mama (MDA-MB-468) y un segundo xenoinjerto de cáncer de páncreas (no mostrado) mostraron el mismo patrón, con el ADC anti-Trop-2 significativamente más eficaz en comparación con el ADC no dirigido o el control de solución salina. En ambos casos, la administración de ADC anti-Trop-2 produjo una clara inhibición del crecimiento tumoral hasta el final del estudio.
[0276] PK y toxicidad de hRS7-pro-2-PDox con sustituciones de 6 , 8 o 3,7 fármaco/IgG - Se sabe que los conjugados de fármaco-anticuerpo (ADC) que contienen hasta 8 fármacos ultratóxicos/MAb se eliminan más rápido que los MAb no modificados y aumentan la eliminación. toxicidad diana, hallazgo que ha llevado a las tendencias actuales de uso de sustituciones de fármacos de ≤ 4 (Hamblett et al., 2004, Clin Cáncer Res 10:7063-70). Los ADC se prepararon y evaluaron con proporciones medias de sustitución de fármaco/MAb (MSR) de ~6:1 y ~3:1. Se administraron grupos de ratones normales (n = 5), i.v., dosis únicas de hRS7 o hRS7-pro-2-PDox sin modificar con sustitución de fármaco de 6 , 8 o 3,7 (misma dosis de proteína), y se recogieron muestras de suero a los 30 min, 4 h, 24 h, 72 h y 168 h después de la inyección. Estos fueron analizados por ELISA para determinar la concentración de anticuerpos. No hubo diferencias significativas en las concentraciones séricas en varios momentos, lo que indica que estos mostraron una eliminación similar de la sangre. Los parámetros farmacocinéticos (Cmax, AUC, etc.) también fueron similares. Los ADC con mayor o menor sustitución de fármaco tuvieron una tolerabilidad similar en ratones desnudos, cuando se administraron a la misma dosis de fármaco conjugado.
[0277] Eficacia terapéutica a la dosis mínima efectiva (MED) - Anti-Trop-2 ADC (hRS7-pro-2-PDox), se evaluó en ratones desnudos portadores de xenoinjertos de cáncer gástrico humano NCI-N87 mediante la administración de una dosis única de proteína en bolo de 9 mg/ kg, 6,75 mg/kg, 4,5 mg/kg, 2,25 mg/kg o 1 mg/kg. La terapia se inició cuando el volumen tumoral medio (mTV) fue de 0,256 cm3. El día 21, el mTV en el grupo de control de solución salina (grupo sin tratamiento) fue de 0,801 ± 0,181 cm3, que fue significativamente mayor que el de los ratones tratados con una dosis de 9, 6,75, 4,5 o 2,25 mg/kg con un mTV de 0,211 ± 0,042 cm3, 0,239 ± 0,0.054 cm3, 0,264 ± 0,087 cm3, y 0,567 ± 0,179 cm3, respectivamente (P<0.0047, prueba t de una cola). A partir de estos, la dosis efectiva mínima se estimó en 2,25 mg/kg, mientras que 9 mg/kg representaban MTD.
Ejemplo 11. ADC anti-Trop-2 que comprende hRS7 y Paclitaxel
[0278] Se preparó un nuevo conjugado de anticuerpo y fármaco (ADC) conjugando paclitaxel (TAXOL®) con el anticuerpo antitrop-2 humano hRS7 (hRS7-paclitaxel). El producto final tenía una proporción media de sustitución de fármaco a anticuerpo de 2,2. Este ADC se probó in vitro usando dos líneas celulares positivas para Trop-2 diferentes como dianas: BxPC-3 (adenocarcinoma pancreático humano) y MDA-MB-468 (carcinoma de mama triple negativo humano). Un día antes de añadir el ADC, se recogieron células del cultivo de tejidos y se sembraron en placas de 96 pocillos a razón de 2000 células por pocillo. Al día siguiente, las células se expusieron a paclitaxel libre (6,1 x 10 -11 a 4 x 10-6M) o el fármaco equivalente de hRS7-paclitaxel. A modo de comparación, hRS7-SN-38 y SN-38 libre también se probaron en un rango de 3,84 x 10-12 a 2,5 x 10-7 M. Las placas se incubaron a 37 °C durante 96 h. Después de este período de incubación, se añadió un sustrato MTS a todas las placas y se leyó el desarrollo del color a intervalos de media hora hasta que los pocillos de control no tratados tuvieran una lectura de DO492nm de aproximadamente 1 ,0. La inhibición del crecimiento se midió como un porcentaje de crecimiento en relación con las células no tratadas utilizando el software Microsoft Excel y Prism (regresión no lineal para generar curvas de respuesta a la dosis sigmoideas que producen valores de Cl50).
[0279] El ADC hRS7-paclitaxel exhibió actividad citotóxica en la línea celular de mama MDA-MB-468 (no mostrada), con un valor Cl50 aproximadamente 4,5 veces mayor que hRS7-SN-38. El paclitaxel libre era mucho más potente que el SN-38 libre (no mostrado). Mientras que la Cl50 para SN-38 libre fue de 1,54 x 10'9 M, la Cl50 para paclitaxel libre fue inferior a 6,1 x 10'11 M. Se obtuvieron resultados similares para la línea celular pancreática BxPC-3 (no mostrada) en en el que el ADC hRS7-paclitaxel tenía un valor Cl50 aproximadamente 2,8 veces mayor que el ADC hRS7-SN-38. Estos resultados muestran la eficacia del paclitaxel conjugado anti-Trop-2 in vitro, con valores de Cl50 en el rango nanomolar, similares al ADC hRS7-SN-38.
Ejemplo 12. Citotoxicidad de Anti-Trop-2 ADC (MAB650-SN-38)
[0280] Se fabricó un ADC anti-Trop-2 novedoso con SN-38 y MAB650, lo que arrojó una proporción media de sustitución de fármaco por anticuerpo de 6,89. Se realizaron ensayos de citotoxicidad para comparar los ADC MAB650-SN-38 y hRS7-SN-38 usando dos líneas celulares de adenocarcinoma pancreático humano diferentes (BxPC-3 y Capan-1) y una línea celular de carcinoma de mama triple negativo humano (MDA-MB-468) como objetivos.
[0281] Un día antes de añadir los ADC, las células se recogieron del cultivo de tejidos y se sembraron en placas de 96 pocillos. Al día siguiente, las células se expusieron a hRS7-SN-38, MAB650-SN-38 y SN-38 libre en un rango de fármaco de 3.84 x 10'12 a 2.5 x 10'7 M. Se usó MAB650 no conjugado como control en dosis equivalentes de proteína como el MAB650-SN-38. Las placas se incubaron a 37°C durante 96 h. Después de este período de incubación, se añadió un sustrato MTS a todas las placas y se leyó el desarrollo del color a intervalos de media hora hasta que se alcanzó una DO492nm de aproximadamente 1,0 para las células no tratadas. La inhibición del crecimiento se midió como un porcentaje de crecimiento en relación con las células no tratadas utilizando el software Microsoft Excel y Prism (regresión no lineal para generar curvas de respuesta a la dosis sigmoidea que producen valores de Cl50).
[0282] hRS7-SN-38 y MAB650-SN-38 tuvieron efectos inhibidores del crecimiento similares (no mostrados) con valores de Cl50 en el rango bajo de nM que es típico para los SN-38-ADC en estas líneas celulares. En la línea celular de adenocarcinoma de páncreas humano Capan-1 (no mostrada), el ADC hRS7-SN-38 mostró un Cl50 de 3,5 nM, en comparación con 4,1 nM para el ADC m Ab650-SN-38 y 1,0 nM para el SN-38 libre. En la línea celular de adenocarcinoma pancreático BxPC-3 humana (no mostrada), el ADC hRS7-SN-38 mostró un Cl50 de 2,6 nM, en comparación con 3,0 nM para el ADC MAB650-SN-38 y 1,0 nM para SN-38 libre. En la línea celular de adenocarcinoma gástrico humano NCI-N87 (no mostrada), el ADC hRS7-SN-38 mostró un Cl50 de 3,6 nM, en comparación con 4,1 nM para el ADC MAB650-SN-38 y 4,3 nM para el SN-38 libre.
[0283] En resumen, en estos ensayos in vitro, los conjugados SN-38 de dos anticuerpos anti-Trop-2, hRS7 y MAB650, mostraron eficacias iguales contra varias líneas de células tumorales, similar a la de SN-38 libre. Debido a que la función de direccionamiento de los anticuerpos anti-Trop-2 sería un factor mucho más significativo in vivo que in vitro, los datos respaldan que los ADC anti-Trop-2-SN-38 como clase serían altamente eficaces in vivo, como demostrado en los Ejemplos anteriores para hRS7-SN-38.
Ejemplo 13. Citotoxicidad de Anti-Trop-2 ADC (162-46.2-SN-38)
[0284] Se fabricó un nuevo ADC anti-Trop-2 con SN-38 y 162-46.2, lo que produjo una proporción de sustitución de fármaco por anticuerpo de 6,14. Se realizaron ensayos de citotoxicidad para comparar los ADC 162-46.2-SN-38 y hRS7-SN-38 usando dos líneas celulares positivas para Trop-2 diferentes como objetivos, el adenocarcinoma pancreático humano BxPC-3 y el carcinoma de mama negativo triple humano MDA-MB-468.
[0285] Un día antes de añadir el ADC, se recogieron células del cultivo de tejidos y se sembraron en placas de 96 pocillos a razón de 2000 células por pocillo. Al día siguiente, las células se expusieron a hRS7-SN-38, 162-46.2-SN-38 o SN-38 libre en un rango de fármaco de 3.84 x 10'12 a 2.5 x 10'7 M. 162-46.2 y hRS7 no conjugados se usaron
como controles a las mismas dosis equivalentes de proteína que 162-46.2-SN-38 y hRS7-SN-38, respectivamente. Las placas se incubaron a 37°C durante 96 h. Después de este período de incubación, se añadió un sustrato MTS a todas las placas y se leyó el desarrollo del color a intervalos de media hora hasta que los pocillos de control no tratados tuvieran una lectura de DO492nm de aproximadamente 1,0. La inhibición del crecimiento se midió como un porcentaje de crecimiento en relación con las células no tratadas utilizando el software Microsoft Excel y Prism (regresión no lineal para generar curvas de respuesta a la dosis sigmoideas que producen valores de Cl50).
[0286] El 162-46.2-SN-38 ADC tuvo valores Cl50 similares en comparación con hRS7-SN-38 (no mostrado). Cuando se ensayó contra la línea celular de adenocarcinoma de páncreas humano BxPC-3 (no se muestra), hRS7-SN-38 tenía un Cl50 de 5,8 nM, en comparación con 10,6 nM para 162-46.2-SN-38 y 1,6 nM para SN-38 libre. Cuando se ensayó contra la línea celular de adenocarcinoma de mama humano MDA-MB-468 (no se muestra), hRS7-SN-38 tenía un Cl50 de 3,9 nM, en comparación con 6,1 nM para 162-46.2-SN-38 y 0,8 nM para SN-38 libre. Los anticuerpos libres solos mostraron poca citotoxicidad para cualquiera de las líneas celulares de cáncer positivas para Trop-2.
[0287] En resumen, al comparar las eficacias in vitro de tres anticuerpos anti-Trop-2 diferentes conjugados con el mismo fármaco citotóxico, los tres ADC exhibieron efectos citotóxicos equivalentes contra una variedad de líneas celulares de cáncer positivas para Trop-2. Estos datos respaldan que la clase de anticuerpos anti-Trop-2, incorporados en ADC conjugados con fármacos, son agentes terapéuticos anticancerígenos eficaces para tumores sólidos que expresan Trop-2.
Ejemplo 14. Ensayos clínicos con ADC anti-Trop-2 IMMU-132 que comprende anticuerpo hRS7 conjugado con SN-38
Resumen
[0288] El presente ejemplo informa los resultados de un ensayo clínico de fase I y una extensión de fase II en curso con IMMU-132, un ADC del anticuerpo anti-Trop-2 hRS7 humanizado que se internaliza conjugado con un enlazador sensible al pH a SN-38 (relación media de fármaco-anticuerpo = 7,6). Trop-2 es una proteína transmembrana de tipo I, transductora de calcio, expresada a alta densidad ( ~ 1 x 105), frecuencia y especificidad por muchos carcinomas humanos, con una expresión tisular normal limitada. Los estudios preclínicos en ratones desnudos portadores de xenoinjertos de tumor pancreático humano Capan-1 han revelado que IMMU-132 es capaz de administrar hasta 120 veces más SN-38 a tumor que el derivado de una terapia con irinotecán tolerado al máximo.
[0289] El presente ejemplo informa sobre el ensayo de fase I inicial de 25 pacientes (pts) que habían fracasado en múltiples terapias previas (algunos incluidos los fármacos inhibidores de la topoisomerasa-I/II), y la extensión de fase II en curso que ahora informa sobre 69 pts, incluso en cáncer colorrectal (CRC), pulmón de células pequeñas y no pequeñas (SCLC, NSCLC, respectivamente), cáncer de mama triple negativo (TNBc ), pancreático (PDC), esofágico y otros cánceres.
[0290] se detectó en suero, pero se expresó fuertemente (> 2+) en la mayoría de los tumores archivados. En un diseño de ensayo 3+3, IMMU-132 se administró los días 1 y 8 en ciclos repetidos de 21 días, comenzando con 8 mg/kg/dosis, luego 12 y 18 mg/kg antes de la neutropenia limitante de la dosis. Para optimizar el tratamiento acumulativo con retrasos mínimos, la fase II se centra en 8 y 10 mg/kg (n=30 y 14, respectivamente). En 49 pts que informaron EA relacionados en este momento, se produjo neutropenia > G3 en el 28 % (4 % G4). Las toxicidades no hematológicas más comunes inicialmente en estos pacientes han sido fatiga (55 %; > G3 = 9 %), náuseas (53 %; > G3 = 0 %), diarrea (47 %; > G3 = 9 %), alopecia (40%) y vómitos (32%; > G3 = 2%). Se encontró homocigosis UGT1A1 * 28/ * 28 en 6 pacientes, 2 de los cuales tenían toxicidades hematológicas y gastrointestinales más graves. En la Fase I y las fases de expansión, ahora hay 48 pts (excluyendo PDC) que son evaluables por RECIST/CT para la mejor respuesta. Siete (15 %) de los pacientes tuvieron una respuesta parcial (RP), incluidos pacientes con CCR (N = 1), TNBC (N = 2), SCLC (N = 2), NSCLC (N = 1) y cánceres de esófago (N = 1), y otros 27 pts (56%) tenían enfermedad estable (DE), para un total de 38 pts (79%) con respuesta a la enfermedad; 8 de 13 pacientes con PDC evaluables por TC (62 %) tenían SD, con una mediana de tiempo hasta la progresión (TTP) de 12,7 semanas en comparación con 8,0 semanas en su última terapia anterior. El TTP para los 48 pts restantes es de más de 12,6 semanas (rango de 6,0 a 51,4 semanas). Plasma CEA y CA19-9 se correlacionaron con las respuestas. No se detectaron anticuerpos anti-hRS7 o antiSN-38 a pesar de la dosificación durante meses. El conjugado se eliminó del suero dentro de los 3 días, de acuerdo con los estudios en animales in vivo donde el 50 % del SN-38 se liberó diariamente, con >95 % del SN-38 en el suero unido a la IgG en una forma no glucuronizada. y en concentraciones hasta 100 veces más altas que el SN-38 notificado en pacientes que recibieron irinotecán. Estos resultados muestran que el ADC que contiene hRS7-SN-38 es terapéuticamente activo en cánceres sólidos metastásicos, con diarrea y neutropenia manejables.
Farmacocinética
[0291] Se usaron dos métodos ELISA para medir el aclaramiento de IgG (captura con anticuerpo anti-idiotipo hRS7) y el conjugado intacto (captura con anti-SN-38 IgG/sonda con anticuerpo anti-idiotipo anti-hRS7). SN-38 se midió por HPLC. La fracción total de IMMU-132 (conjugado intacto) se eliminó más rápidamente que la IgG (no se muestra), lo
que refleja la liberación gradual conocida de SN-38 del conjugado. La determinación por HPLC de SN-38 (sin unir y TOTAL) mostró que >95 % de SN-38 en el suero estaba unido a la IgG. Las bajas concentraciones de SN-38G sugieren que el SN-38 unido a la IgG está protegido de la glucuronidación. La comparación de ELISA para conjugado y SN-38 HPLC reveló que ambos se superponen, lo que sugiere que ELISA es un sustituto para monitorear la eliminación de SN-38.
[0292] En la Tabla 12 se proporciona un resumen del régimen de dosificación y la encuesta del paciente.
Tabla 12. Parámetros del ensayo clínico

Estado del ensayo clínico
[0293] Se informó un total de 69 pacientes (incluidos 25 pacientes en Fase I) con diversos cánceres metastásicos que tenían una mediana de 3 terapias previas. Ocho pacientes tuvieron progresión clínica y se retiraron antes de la evaluación por TC. Trece pacientes con cáncer de páncreas evaluable por CT se informaron por separado. La mediana de TTP (tiempo hasta la progresión) en pacientes con PDC fue de 11,9 semanas (rango de 2 a 21,4 semanas) en comparación con la mediana de 8 semanas de TTP para la última terapia anterior.
[0294] Un total de 48 pacientes con diversos tipos de cáncer tuvieron al menos 1 evaluación por TC a partir de la cual se determinaron la mejor respuesta (no se muestra) y el tiempo hasta la progresión (TTP; no se muestra). Para resumir los datos de Best Response, de 8 pacientes evaluables con TNBC (cáncer de mama triple negativo), hubo 2 PR (respuesta parcial), 4 SD (enfermedad estable) y 2 PD (enfermedad progresiva) para una respuesta total [PR DE] de 6 / 8 (75%). Para SCLC (cáncer de pulmón de células pequeñas), de 4 pacientes evaluables hubo 2 PR, 0 SD y 2 PD para una respuesta total de 2/4 (50%). Para CCR (cáncer colorrectal), de 18 pacientes evaluables hubo 1 PR, 11 SD y 6 PD para una respuesta total de 12/18 (67%). Para el cáncer de esófago, de 4 pacientes evaluables hubo 1 PR, 2 Sd y 1 Pd para una respuesta total de 3/4 (75%). Para NSCLC (cáncer de pulmón de células no pequeñas), de 5 pacientes evaluables hubo 1 PR, 3 SD y 1 PD para una respuesta total de 4/5 (80%). Sobre todos los pacientes tratados, de 48 pacientes evaluables hubo 7 PR, 27 SD y 14 PD para una respuesta total de 34/48 (71%). Estos resultados demuestran que el ADC anti-TROP-2 (hRS7-SN-38) mostró una eficacia clínica significativa contra una amplia gama de tumores sólidos en pacientes humanos.
[0295] Los efectos secundarios informados de la terapia (eventos adversos) se resumen en la Tabla 13. Como se desprende de los datos de la Tabla 13, la eficacia terapéutica de hRS7-SN-38 se logró a dosis de ADC que mostraron un nivel aceptablemente bajo de efectos secundarios adversos.
Tabla 13.

[0296] Las respuestas parciales ejemplares al ADC anti-Trop-2 se confirmaron mediante datos de TC (no mostrados). Como RP ejemplar en CCR, una mujer de 62 años diagnosticada por primera vez con CCR se sometió a una hemicolectomía primaria. Cuatro meses después, se sometió a una resección hepática por metástasis hepáticas y recibió 7 meses de tratamiento con FOLFOX y 1 mes de 5FU. Presentó múltiples lesiones principalmente en el hígado (3+ Trop-2 por inmunohistología), ingresando al ensayo hRS7-SN-38 con una dosis inicial de 8 mg/kg aproximadamente 1 año después del diagnóstico inicial. En su primera evaluación por TC, se logró una RP, con una reducción del 37 % en las lesiones diana (no se muestra). El paciente continuó el tratamiento, logrando una reducción máxima de 65% de disminución después de 10 meses de tratamiento (no se muestra) con disminución de CEA de 781 ng/ml a 26,5 ng/ml), antes de progresar 3 meses más tarde.
[0297] Como PR ejemplar en NSCLC, un hombre de 65 años fue diagnosticado con NSCLC en estadio IIIB (células cuadradas). El tratamiento inicial de caboplatino/etopósido (3 meses) junto con 7000 cGy XRT resultó en una respuesta que duró 10 meses. A continuación, comenzó con la terapia de mantenimiento Tarceva, que continuó hasta que se consideró para el ensayo IMMU-132, además de someterse a una laminectomía lumbar. Recibió primera dosis de IMMU-132 después de 5 meses de Tarceva, presentando en ese momento lesión de 5,6 cm en pulmón derecho con abundante derrame pleural. Acababa de completar su sexta dosis dos meses después cuando la primera TC mostró que la lesión diana primaria se había reducido a 3,2 cm (no se muestra).
[0298] Como PR ejemplar en SCLC, una mujer de 65 años fue diagnosticada con SCLC pobremente diferenciado. Después de recibir carboplatino/etopósido (inhibidor de Topo-II) que finalizó a los 2 meses sin respuesta, seguido de topotecán (inhibidor de Topo-I) que finalizó a los 2 meses, también sin respuesta, recibió XRT local (3000 cGy) que finalizó 1 mes después. Sin embargo, al mes siguiente la progresión había continuado. El paciente comenzó con IMMU-132 al mes siguiente (12 mg/kg; reducido a 6,8 mg/kg; expresión de Trop-2 3+), y después de dos meses de IMMU-132, una reducción del 38 % en las lesiones diana, incluida una se produjo una reducción sustancial de la lesión pulmonar principal (no mostrada). El paciente progresó 3 meses después de recibir 12 dosis.
[0299] Estos resultados son significativos porque demuestran que el ADC anti-Trop-2 fue eficaz, incluso en pacientes que habían fracasado o progresado después de múltiples terapias previas.
[0300] En conclusión, a las dosis utilizadas, la toxicidad principal fue una neutropenia manejable, con pocas toxicidades de grado 3. IMMU-132 mostró evidencia de actividad (PR y SD duradero) en pacientes recidivantes/refractarios con cáncer de mama triple negativo, cáncer de pulmón de células pequeñas, cáncer de pulmón de células no pequeñas, cáncer colorrectal y cáncer de esófago, incluidos pacientes con antecedentes de recaída en terapia con inhibidores de la topoisomerasa-I. Estos resultados muestran la eficacia del ADC anti-Trop-2 en una amplia gama de cánceres que son resistentes a las terapias existentes.
Ejemplo 15. Anti-CD22 (epratuzumab) SN-38 conjugado para la terapia de malignidades hematológicas
Resumen
[0301] Anteriormente descubrimos que los anticuerpos de internalización lenta conjugados con SN-38 podrían usarse con éxito cuando se preparan con un enlazador que permite que aproximadamente el 50 % del SN-38 unido a IgG se disocie en el suero cada 24 horas. En este estudio, se examinó la eficacia de los conjugados SN-38 preparados con epratuzumab (de internalización rápida) y veltuzumab (de internalización lenta), IgG anti-CD22 y anti-CD20 humanizados, respectivamente, para el tratamiento de tumores malignos de células B. Ambos conjugados de anticuerpo y fármaco tenían una actividad nanomolar similar contra una variedad de líneas celulares de linfoma/leucemia humana, pero la liberación lenta de SN-38 comprometía la discriminación de potencia in vitro incluso contra un conjugado irrelevante. Cuando SN-38 se unió de manera estable al conjugado anti-CD22, su potencia se redujo de 40 a 55 veces. Por lo tanto, se realizaron estudios adicionales solo con el conector menos estable que se disocia lentamente. In vivo, se encontró una actividad antitumoral similar entre CD22 y el conjugado de anticuerpofármaco CD20 en ratones con xenoinjertos de Ramos, aunque Ramos expresó 15 veces más CD20 que CD22, lo que sugiere que la internalización del conjugado epratuzumab-SN-38 (Emab- SN-38) potenció su actividad. Emab-SN-38 fue más eficaz que un conjugado IgG-SN-38 irrelevante que no se une in vivo, eliminando la mayoría de los xenoinjertos de Ramos bien establecidos en dosis no tóxicas. Los estudios in vitro e in vivo mostraron que Emab-SN-38 podría combinarse con veltuzumab no conjugado para un tratamiento más eficaz. Por lo tanto, Emab-SN-38 es activo en el linfoma y la leucemia en dosis muy por debajo de los niveles tóxicos y, por lo tanto, representa un nuevo agente prometedor con potencial terapéutico solo o combinado con la terapia con anticuerpos anti-CD20. (Sharkey et al., 2011, Mol Cancer Ther 11:224-34).
Introducción
[0302] Un esfuerzo significativo se ha centrado en la terapia biológica de la leucemia y el linfoma, donde se recibieron anticuerpos no conjugados (p. ej., rituximab, alemtuzumab, ofatumumab), radioinmunoconjugados (90Y-ibritumomab tiuxetan, 131I-tositumomab) y un fármaco conjugado (gemtuzumab ozogamicina). Aprobación de la Administración de Drogas y Alimentos de los Estados Unidos (FDA). Otro conjugado de anticuerpo y fármaco (ADC), brentuximab vedotin (SGN-35; anti-CD30-auristatin E), recibió recientemente la aprobación acelerada de la FDA para el linfoma de Hodgkin y los linfomas anaplásicos de células grandes. También hay una serie de otros ADC en desarrollo preclínico y clínico que se dirigen a CD19, CD22, CD37, CD74 y CD79b.
[0303] Los anticuerpos contra todos estos objetivos son opciones lógicas para los portadores de fármacos, porque se internalizan. La internalización y la especificidad de CD22 lo han convertido en un objetivo particularmente importante para la leucemia y los linfomas, con al menos 3 conjugados anti-CD22 diferentes en investigación clínica, incluido CMC-544 (calicheamicina conjugada lábil en ácido), un conjugado anti-CD22-maitansina (MCC-DM1 unido de forma estable) y CAT-3888 (formalmente BL22; una proteína de fusión de cadena sencilla de exotoxina de Pseudomonas). El agente activo en todos estos conjugados tiene una potencia subnanomolar (es decir, los llamados ultratóxicos).
[0304] Recientemente desarrollamos métodos para conjugar anticuerpos con SN-38, un inhibidor de la topoisomerasa I con potencia nanomolar baja que se deriva del profármaco irinotecán (Govindan et al., 2009, Clin Cancer Res 15:6052-62; Moon et al., 2008, J Med Chem 51:6916-26). Inicialmente se examinaron cuatro químicas de enlace SN-38 utilizando conjugados preparados con un anticuerpo anti-CEACAM5 de internalización lenta (Govindan et al., 2009, Clin Cancer Res 15:6052-62; Moon et al., 2008, J Med Chem 51:6916-26). Los conjugados retuvieron la unión a CEACAM5 pero diferían en la tasa de disociación de SN-38 en suero humano, con vidas medias que variaban de aproximadamente 10 a 67 horas (Govindan et al., 2009, Clin Cancer Res 15:6052-62). Finalmente, se seleccionó el enlazador denominado CL2, con estabilidad intermedia (~ 50 % disociado en 24-35 horas), para un mayor desarrollo. CL2 se modificó recientemente, eliminando la fenilalanina en el dipéptido escindible de catepsina B para simplificar y mejorar los rendimientos de fabricación. El nuevo derivado, denominado CL2A, retiene el enlace de carbonato sensible al pH con el SN-38, pero ya no es escindido selectivamente por la catepsina B. Sin embargo, tiene una estabilidad sérica idéntica y una actividad in vivo mejorada en comparación con el enlazador CL2 original (Cardillo et al. al., 2011, Clin Cancer Res 17:3157-69). Debido a que se encontró una eficacia significativa sin toxicidad con el antiCEACAM5-SN-38 de internalización lenta, postulamos que su actividad se vio favorecida por la liberación lenta de SN-38 del anticuerpo después de que se localizó en un tumor. Por lo tanto, el objetivo principal de este informe fue evaluar las perspectivas terapéuticas de los conjugados preparados con el enlazador CL2A con dos anticuerpos que son altamente específicos para los cánceres de células B pero difieren en sus propiedades de internalización y expresión de antígenos.
[0305] Epratuzumab (Emab) es un anti-CD22 IgG1 humanizado de internalización rápida (p. ej., > 50 % en 1 hora) que se ha evaluado ampliamente en linfoma y leucemia en forma no conjugada o conjugada. Veltuzumab (Vmab) es un anticuerpo anti-CD20 humanizado que también se está estudiando clínicamente pero se internaliza lentamente (p. ej., ~10 % en 1 hora). El CD20 generalmente se expresa a niveles mucho más altos que el CD22 en el linfoma no Hodgkin, mientras que el CD22 se expresa preferentemente en la leucemia linfoblástica aguda (LLA), pero no en el mieloma múltiple. Ambos anticuerpos son efectivos en pacientes como agentes no conjugados, pero solo veltuzumab es activo en modelos de xenoinjerto murino (Stein et al., 2004, Clin Cancer Res 10:2868-76). Sobre la base de estudios previos que mostraron que 90Y-Emab combinado con veltuzumab no conjugado tenía mayor eficacia en modelos de NHL (Mattes et al., 2008, Clin Cancer Res 14:6154-60), también examinamos el Emab-SN-38 combinación de Vmab,
ya que esto podría proporcionar un beneficio adicional sin competir por el mismo antígeno diana o tener toxicidad adicional.
Materiales y métodos
[0306] Líneas celulares. Ramos, Raji, Daudi (linfomas de Burkitt) y JeKo-1 (linfoma de células del manto) se adquirieron de la American Type Culture Collection. REH, RS4;11, m N-60 y 697 (ALL) se adquirieron de Deutsche Sammlung von Mikroorganismen und Zellkulturen. WSU-FSCCL (NHL folicular) fue un regalo del Dr. Mitchell R. Smith (Fox Chase Cancer Center, Filadelfia, PA). Todas las líneas celulares se cultivaron en una incubadora con CO2 humidificado (5 %) a 37 °C en medios suplementados recomendados que contenían entre un 10 y un 20 % de suero de ternera fetal y se controlaron periódicamente para Mycoplasma.
[0307] Anticuerpos y métodos de conjugación. Epratuzumab y veltuzumab son anticuerpos monoclonales IgG1 anti-CD22 y anti-CD20 humanizados, respectivamente. Labetuzumab (Lmab), un IgG1 anti-CEACAM5 humanizado, y RS7, un anticuerpo anti-Trop-2 humanizado (ambos de Immunomedics, Inc.), se usaron como controles irrelevantes no vinculantes. En este documento, Emab-SN-38, Vmab-SN-38 y Lmab-SN-38 se refieren a conjugados preparados usando el enlazador CL2A que se describió anteriormente. Los estudios in vitro en suero humano mostraron que aproximadamente el 50 % de la fracción SN-38 activa se libera de la IgG cada día (Cardillo et al., 2011, Clin Cancer Res 17:3157-69). Otro enlazador, denominado CL2E, es estable en suero humano durante 14 días, pero contiene un sitio de escisión de catepsina B para facilitar la liberación de SN-38 cuando se procesa en lisosomas. El método para preparar CL2E y las estructuras de los enlazadores CL2A y CL2E se dan en los ejemplos anteriores. Los conjugados contenían aproximadamente 6 unidades de SN-38 por IgG (p. ej., 1,0 mg del conjugado de IgG-SN-38 contiene ~16 μg de SN-38).
[0308] Unión celular y citotoxicidad in vitro. La citometría de flujo se llevó a cabo usando los anticuerpos irrelevantes y específicos no conjugados incubados durante 1 hora a 4 °C, y la unión se reveló usando IgG anti-humana de cabra específico al fragmento (FITC)-Fcy de isotiocianato de fluoresceína (Jackson ImmunoResearch), también incubados durante 1 hora a 4°C. La mediana de la fluorescencia se determinó en un citómetro de flujo FACSCALIBUR® (Becton Dickinson) usando un paquete de software CellQuest.
[0309] La citotoxicidad se determinó utilizando el ensayo de reducción de colorante MTS (Promega). Curvas de dosis-respuesta [con/sin Fcy F(ab')2 antihumano de cabra; Jackson ImmunoResearch] se generaron a partir de la media de determinaciones por triplicado, y los valores de Cl50 se calcularon utilizando el software PRISM® GraphPad (v5), con comparaciones estadísticas utilizando una prueba F en las curvas de mejor ajuste para los datos. La significación se fijó en P < 0,05.
[0310] Inmunotransferencia. Después de 24 o 48 horas de exposición a los agentes de prueba, los marcadores de apoptosis temprana (expresión de p21) y tardía (escisión de PARP) se revelaron mediante transferencia de Western.
[0311] Estudios in vivo. El modelo Ramos subcutáneo se inició implantando 1 x 107 células (0,2 ml) de cultivo (>95 % de viabilidad) en ratones desnudos hembra de 4 a 6 semanas de edad (Taconic). Tres semanas después de la implantación, los animales con tumores que oscilaban entre 0,4 y 0,8 cm3 (medidos con calibre, L x W x D) se segregaron en grupos de animales, cada uno con el mismo rango de tamaños de tumor. El tamaño del tumor y los pesos corporales se midieron al menos una vez por semana, y los animales se retiraron del estudio cuando los tumores crecieron hasta 3,0 cm3 o si experimentaron una pérdida de peso corporal del 20% o más. el intravenoso Los modelos WSU-FSCCL y 697 se iniciaron mediante inyección intravenosa de 2,5 x 106 y 1 x 107 células, respectivamente, en mujeres con inmunodeficiencia combinada grave (SCID) ratones (Taconic). El tratamiento comenzó 5 días después de la administración de las células WSU-FSCCL y 7 días después de la inoculación 697. Los animales se observaron diariamente, usando parálisis de las patas traseras u otros signos de morbilidad como puntos finales de supervivencia sustitutos. Todos los tratamientos se administraron por vía intraperitoneal en ≤ 0,2 ml. Las dosis específicas y la frecuencia se dan en la sección de Resultados. Debido a que los ratones convierten el irinotecán en SN-38 de manera eficiente, la dosis de irinotecán se ajustó sobre la base de los equivalentes de SN-38; Los equivalentes molares de SN-38 se basan en el 1,6 % de la masa de ADC y el 60 % de la masa de irinotecán.
[0312] La eficacia se expresó en una curva de Kaplan-Meier, utilizando el tiempo hasta la progresión (TTP) como criterios de valoración de supervivencia alternativos, como se indicó anteriormente. El análisis estadístico se realizó mediante una prueba de rango logarítmico utilizando el software PRISM® GraphPad (significación, P < 0,05).
Resultados
[0313] Expresión de antígenos y citotoxicidad in vitro. Todas las líneas celulares fueron altamente susceptibles a SN-38, con valores de CE50 que oscilaron entre 0,13 nmol/L para Daudi y 2,28 nmol/l para RS4;11 (Tabla 14). Con la excepción de 697 y RS4;11, el conjugado anti-CD22 de Emab-SN-38 fue de 2 a 7 veces menos efectivo que SN-38. Este es un hallazgo común con nuestros conjugados SN-38 dirigidos, así como con otros no dirigidos. A pesar de las diferencias en la expresión del antígeno, Emab-SN-38 y Vmab-SN-38 tenían potencias similares a las del conjugado
Lmab-SN-38 anti-CEACAM5 que no se une, lo que probablemente se debió a la disociación de aproximadamente el 90% de SN-38 durante el ensayo MTS de 4 días. Otros procedimientos in vitro que utilizaron tiempos de exposición más cortos también fueron ineficaces para discriminar las diferencias en las potencias de los conjugados. Por ejemplo, la tinción con anexina V después de una exposición de 1 día no logró encontrar diferencias entre las células tratadas y no tratadas (no se muestra). También se examinó la regulación al alza de la escisión de p21 y PARP como marcadores tempranos y tardíos de apoptosis, respectivamente. Ramos no expresó p21. Sin embargo, se detectó la escisión de PARP, pero solo después de una exposición de 48 horas, y se expresó más fuertemente en las células tratadas con SN-38 (no se muestra). La línea celular WSU-FSCCL expresó p21, pero ni la regulación positiva de p21 ni la escisión de PARP fueron evidentes hasta 48 horas después de la exposición a Emab-SN-38. Sin embargo, ambos se observaron después de una exposición de 24 horas con SN-38 libre (no se muestra). Si bien la mayor intensidad y la activación más temprana de los eventos apoptóticos con SN-38 libre son consistentes con su menor CE50 sobre la forma conjugada con IgG, los resultados indicaron que se requeriría un período de exposición de al menos 48 horas, pero en este momento, aproximadamente el 75% del SN-38 se liberaría del conjugado.

[0314] Nuevamente examinamos la escisión de PARP y la expresión de p21, esta vez en células tratadas con Emab-SN-38 Vmab. Confirmando el estudio anterior en Ramos, la escisión de PARP ocurre primero solo después de una exposición de 48 horas al conjugado, sin cambios en la expresión en presencia de un anticuerpo de entrecruzamiento (no se muestra). La exposición a veltuzumab durante más de 48 horas no tuvo efecto sobre la escisión de PARP, pero la escisión fue fuerte dentro de las 24 horas cuando se agregó un anticuerpo de entrecruzamiento (no se muestra). Sin embargo, cuando se combinó veltuzumab solo (sin reticulante) con Emab-SN-38, se produjo la escisión de PARP después de una exposición de 24 horas (no se muestra), lo que indica que veltuzumab podría inducir un inicio más rápido de la apoptosis, incluso en ausencia de entrecruzamiento La única diferencia notable en la línea celular WSU-FSCCL fue que la combinación mejoró mucho la expresión de p21 a las 48 horas (no se muestra), lo que nuevamente sugiere una aceleración de la inducción de apoptosis cuando veltuzumab se combina con el conjugado Emab-SN-38. El retraso en la inducción de apoptosis en WSU-FSCCL en comparación con Ramos probablemente se explica por la menor expresión de CD22 y CD20.
[0315] Los agentes ultratóxicos suelen utilizar conectores que son muy estables en el suero, ya que su liberación prematura aumentaría la toxicidad, pero estos conjugados deben internalizarse para que el fármaco se administre de manera óptima. Debido a que epratuzumab se internaliza rápidamente, examinamos si podría beneficiarse de un SN-38 unido de manera más estable, comparando la citotoxicidad in vitro del conjugado Emab-SN-38 ligado a CL2A con el conjugado CL2E-SN-38 estable en suero. Ambos conjugados tenían una afinidad de unión similar (no se muestra), pero el Emab-CL2E-SN-38 más estable era aproximadamente 40 a 55 veces menos potente que el conjugado CL2A en 3 líneas celulares (no se muestra). Si bien faltaba especificidad con los conjugados CL2A, el Emab-CL2ESN-38 fue consistentemente aproximadamente dos veces más potente que el conjugado Lmab-anti-CEACAM5-CL2E-SN-38 que no se une (no se muestra). Llegamos a la conclusión de que era poco probable que el conjugado ligado de forma más estable fuera apropiado para un conjugado de veltuzumab de internalización lenta y, por lo tanto, continuamos nuestra investigación solo con conjugados de SN-38 ligados a CL2A.
[0316] Debido a las limitaciones de los ensayos in vitro, la eficacia se evaluó en modelos de xenoinjertos. Como se indica en la Tabla 14, todas las líneas celulares de linfoma tienen una expresión mucho mayor de CD20 que de CD22. Daudi tuvo la expresión más alta de CD22 y CD20, pero es muy sensible in vivo a veltuzumab no conjugado y las pruebas in vitro revelaron la mayor sensibilidad a SN-38 (Tabla 14). Es probable que estas propiedades dificulten la evaluación de las diferencias en la actividad atribuida al conjugado s N-38 frente al anticuerpo no conjugado, en particular cuando el epratuzumab no conjugado no es un tratamiento eficaz en animales. Debido a que Ramos se había utilizado previamente para mostrar una ventaja al combinar 90Y-Emab con veltuzumab (Mattes et al., 2008, Clin Cancer Res 14:6154-60), elegimos comenzar con una comparación de Emab-SN-38 y conjugados Vmab-SN-38 en la línea celular Burkitt humana Ramos. A pesar de que la citometría de flujo mostró una expresión 15 veces mayor de CD20 que de CD22, la inmunohistología de los xenoinjertos de Ramos mostró abundantes CD22 y CD20, con CD22 aparentemente expresado de manera más uniforme que CD20 (no se muestra).
[0317] Los xenoinjertos de Ramos en animales no tratados progresaron rápidamente, alcanzando el tamaño de terminación de 3,0 cm3 desde su tamaño inicial de 0,4 cm3 en 6 días (no se muestra), y como se informó anteriormente, ni veltuzumab ni epratuzumab afectaron apreciablemente la progresión de Ramos bien establecido. xenoinjertos (Sharkey et al., 2009, J Nucl Med 50:444-53). En consonancia con los hallazgos previos utilizando otros conjugados de SN-38, ninguno de los animales tratados con un régimen de tratamiento de 0,5 mg/dosis de 4 semanas, dos veces por semana, tuvo una pérdida de peso apreciable. Ambos conjugados fueron altamente efectivos para controlar el crecimiento tumoral, con un 80% o más de los animales sin evidencia de tumor al final del tratamiento de 4 semanas (FIG. 12A-12D). La dosis de 0,25 mg de Vmab-SN-38 controló mejor el crecimiento durante las primeras 4 semanas, pero a 0,5 mg se observó un control temprano del crecimiento similar para ambos conjugados. Por lo tanto, a pesar de una expresión 15 veces mayor de CD20 que de CD22, EmabSN-38 se comparó favorablemente con Vmab-SN-38. Por lo tanto, los estudios restantes se centraron en Emab-SN-38 solo o en combinación con veltuzumab no conjugado.
[0318] Emab-SN-38 dosis-respuesta y especificidad. Se observó una relación dosis-respuesta para los conjugados específicos EmabSN-38 y Lmab-SN-38 irrelevantes, pero Emab-SN-38 tuvo un control de crecimiento significativamente mejor en 2 de los 3 niveles probados, y con una fuerte tendencia a favor del conjugado específico. a la dosis intermedia (FIG. 13A-13C). De nuevo, 0,25 mg de Emab-SN-38 extirparon la mayoría de los tumores; en este documento, 7 de 10 animales estaban libres de tumores al final del período de seguimiento de 12 semanas, sin cambios en el peso corporal. Los animales que recibieron irinotecán solo (6,5 μg/dosis; aproximadamente los mismos equivalentes de SN-38 que 0,25 mg de conjugado) tuvieron una mediana de supervivencia de 1,9 semanas, con 3 de 11 animales libres de tumores al final del estudio, lo que no fue significativamente diferente de la mediana de supervivencia de 3,45 semanas para el conjugado Lmab-SN-38 irrelevante (P = 0,452; FIG. 13C).
[0319] En el modelo de leucemia diseminada 697, la mediana de supervivencia de los animales tratados con solución salina fue de solo 17 días desde la inoculación del tumor. Los animales que recibieron epratuzumab no conjugado más irinotecán (los mismos equivalentes molares de SN-38 que 0,5 mg del conjugado) tuvieron la misma mediana de supervivencia, mientras que los animales que recibieron 0,5 mg de Emab-SN-38 dos veces por semana a partir de los 7 días de la inoculación del tumor sobrevivieron hasta los 24,5 días., significativamente más largo que los animales no tratados (P < 0,0001) o para epratuzumab no conjugado administrado con irinotecan (P = 0,016). Sin embargo, Emab
SN-38 no fue significativamente mejor que el conjugado irrelevante (mediana de supervivencia = 22 días; P = 0,304), lo que probablemente refleja la baja expresión de CD22 en esta línea celular.
[0320] Emab-SN-38 combinado con Vmab anti-CD20 no conjugado. Anteriormente informamos mejores respuestas cuando 90Y-Emab se combinó con veltuzumab no conjugado en el modelo de Ramos subcutáneo (Mattes et al., 2008, Clin Cancer Res 14: 6154-60) y, por lo tanto, esta posibilidad se examinó con Emab-SN-38. En un estudio piloto, a 5 animales portadores de veltuzumab (0,1 mg), se les administró 0,1 mg de Emab-SN-38 o Emab-SN-38 Vmab a tumores de Ramos subcutáneos con un promedio aproximado de 0,3 cm3 (todos los agentes administrados dos veces por semana durante 4 semanas). La mediana de Tt P a 2,0 cm3 fue de 22, 14 y más de 77 días, respectivamente (veltuzumab frente a Emab-SN-38 solo, P = 0,59; Emab-SN-38 Vmab frente a Emab-SN-38, P = 0,0145), proporcionando una indicación inicial de que la combinación de veltuzumab con Emab-SN-38 mejoró la respuesta terapéutica global. En un estudio de seguimiento que también usó un régimen de tratamiento de 4 semanas dos veces por semana, 6 de 11 animales que recibieron 0,1 mg de Emab-SN-38 más 0,1 mg de veltuzumab no tuvieron evidencia de tumores 16 semanas después del inicio del tratamiento, mientras que la mediana de supervivencia para los animales que recibieron veltuzumab solo o con 0,1 mg del Lmab-SN-38 de control fue de 1,9 y 3,3 semanas, respectivamente, con 3 de 11 animales sin tumor a las 16 semanas en cada uno de estos grupos (no se muestra). A pesar de la mediana de TTP más larga y más sobrevivientes, no se encontraron diferencias significativas entre los grupos. Por lo tanto, en el modelo de Ramos, que tiene abundante CD20 y niveles moderados de CD22, el conjugado Emab-SN-38 administrado a niveles de dosis no tóxicos no fue significativamente mejor que la terapia anti-CD20 no conjugada, pero la adición de Emab-SN-38 a La terapia anti-CD20 no conjugada pareció mejorar la respuesta sin toxicidad. Es importante recalcar que los conjugados SN-38 se administran a niveles muy inferiores a su dosis máxima tolerada, por lo que estos resultados no deben interpretarse como que la terapia anti-CD20 no conjugada es igual a la del conjugado Emab-SN-38.
[0321] Se realizaron dos estudios adicionales en un modelo implantado por vía intravenosa utilizando la línea celular de NHL folicular WSU-FSCCL que tiene una baja expresión de CD20 y CD22 (no se muestra). El tiempo medio de supervivencia para los animales tratados con solución salina fue de 40 a 42 días desde la implantación del tumor. El irinotecán solo (no se muestra), administrado en una dosis que contenía los mismos equivalentes de SN-38 que 0,3 mg de ADC, aumentó la mediana de supervivencia (49 frente a 40 días, respectivamente; P = 0,042), pero 14 de 15 animales sucumbieron a progresión de la enfermedad el día 49, el mismo día que se eliminaron los últimos 4 de 15 animales en el grupo de solución salina (no mostrado). A pesar de su expresión de CD20 relativamente baja, veltuzumab solo (35 μg dos veces por semana x 4 semanas) fue efectivo en este modelo. La mediana de supervivencia aumentó a 91 días en el primer estudio, con 2 curaciones (día 161), y a 77 días en el segundo, pero sin supervivientes después de 89 días (veltuzumab solo frente a tratamiento con solución salina, P < 0,001 en ambos estudios). El epratuzumab no conjugado (0,3 mg/dosis) combinado con irinotecán y veltuzumab tuvo la misma mediana de supervivencia que el veltuzumab solo, lo que sugiere que ni el epratuzumab ni el irinotecán contribuyeron a la respuesta neta.
[0322] Como era de esperar debido a la baja expresión de CD22 por WSU-FSCCL, Emab-SN-38 solo no fue tan efectivo como en Ramos. Con la dosis de 0,15 mg, no se observaron beneficios significativos sobre el grupo de solución salina, pero con 0,3 mg, la mediana de supervivencia aumentó a 63 días, proporcionando una mejora significativa en comparación con los animales tratados con solución salina (P = 0,006). El segundo estudio, que utilizó 0,3 mg de Emab-SN-38, confirmó una mayor supervivencia en comparación con el grupo de solución salina (75 frente a 40 días; P < 0,0001). La especificidad de esta respuesta no fue evidente en el primer estudio, donde la mediana de supervivencia del conjugado irrelevante Lmab-SN-38 y Emab-SN-38 no fue diferente a niveles de dosis de 0,15 o 0,3 mg (42 vs. 49 días y 63 frente a 63 días para los conjugados Emab-SN-38 frente a anti-CEACAM5-SN-38 en los 2 niveles de dosis, respectivamente). Sin embargo, en el segundo estudio, la dosis de 0,3 mg de Emab-SN-38 proporcionó una supervivencia significativamente mejor que el conjugado irrelevante (75 frente a 49 días; P < 0,0001). Una vez más, la dificultad para mostrar especificidad en este modelo probablemente esté relacionada con la baja expresión de CD22.
[0323] La combinación del Emab-SN-38 específico con veltuzumab aumenta sustancialmente la supervivencia, con evidencia de respuestas más sólidas que el control Lmab-SN-38. Por ejemplo, en el primer estudio, los animales tratados con veltuzumab más 0,15 o 0,3 mg del conjugado de control tuvieron una mediana de supervivencia de 98 y 91 días, respectivamente, que fue similar a la de veltuzumab solo (91 días; no se muestra). Sin embargo, veltuzumab más 0,15 mg del conjugado específico Emab-SN-38 aumentó la mediana de supervivencia a 140 días. Si bien esta mejora no fue significativamente mayor que la de veltuzumab solo (P = 0,257), cuando la dosis de Emab-SN-38 se aumentó a 0,3 mg con veltuzumab, 6 de 10 animales permanecieron vivos al final del estudio, lo que proporcionó una ventaja de supervivencia significativa. sobre el conjugado de control más veltuzumab (P = 0,0002). En un segundo estudio, la mediana de supervivencia de veltuzumab solo fue más corta que en el primero (77 frente a 91 días); sin embargo, la mediana de supervivencia del conjugado de control con veltuzumab fue nuevamente de 91 días, lo que ahora produjo una ventaja de supervivencia significativa sobre veltuzumab solo. (P < 0,0001). La combinación del conjugado específico Emab-SN-38 con veltuzumab extendió la mediana de supervivencia a 126 días, que fue significativamente más larga que la mediana de supervivencia de 75 y 77 días para Emab-SN-38 y veltuzumab solo,
respectivamente (P < 0,0001 para cada uno). Sin embargo, en este estudio, no cumplió del todo los requisitos para una mejora estadística sobre la combinación con el conjugado anti-CEACAM5-SN-38 de control (P = 0,078).
Discusión
[0324] En los últimos 10 años, los ADC han logrado avances sustanciales en la terapia del cáncer, pero también ha habido algunos contratiempos. Las ganancias ocurrieron en gran medida cuando los investigadores eligieron examinar agentes que eran demasiado tóxicos para usarse solos, pero cuando se combinaron con un anticuerpo, estos llamados ultratóxicos produjeron respuestas sustancialmente mejoradas en las pruebas preclínicas. La reciente aprobación de brentuximab vedotina, un conjugado de auristatina, en el linfoma de Hodgkin y el éxito clínico con el conjugado de trastuzumab-DM1 anti-HER2-maitansina como agente único en el cáncer de mama refractario al trastuzumab no conjugado sugieren que estos ADC que contienen agentes ultratóxicos se están convirtiendo en un tratamiento aceptado modalidades. Sin embargo, los conjugados preparados con agentes que son potentes en el rango picomolar pueden tener un mayor riesgo de toxicidad, ya que la reciente decisión de retirar gemtuzumab ozogamicina, el conjugado anti-CD33-calicheamicina, del mercado sugiere (Ravandi, 2011, J Clin Oncol 29:349-51). Por lo tanto, el éxito de un ADC puede depender de la identificación de las químicas apropiadas para unir el fármaco y el anticuerpo, así como de la definición de un objetivo adecuado que se exprese lo suficiente para permitir una administración adecuada y selectiva del agente citotóxico.
[0325] Desarrollamos un enlazador para acoplar SN-38 a IgG que permite que SN-38 se libere lentamente del conjugado en suero (alrededor del 50 % por día). Con este enlazador, un anticuerpo que se interioriza lentamente podría ser una terapéutica eficaz, quizás porque el conjugado localizado en un tumor libera localmente una cantidad suficiente de fármaco, incluso sin interiorizarse. El conector CL2A también se usó recientemente con un anticuerpo contra Trop-2 que se informó que se internalizaba rápidamente (Cardillo et al., 2011, Clin Cancer Res 17: 3157-69). Por tanto, parece que el mecanismo de liberación lenta es beneficioso para los anticuerpos internalizantes y no internalizantes.
[0326] En este informe, ampliamos nuestra evaluación del enlazador CL2A al comparar conjugados SN-38 preparados con epratuzumab, una IgG anti-CD22 de internalización rápida, y veltuzumab, una IgG anti-CD20 de internalización lenta, para el tratamiento de tumores malignos de células B, y ahora hemos encontrado combinaciones con agentes que rompen el ADN, como inhibidores de microtúbulos e inhibidores de PARP que muestran efectos antitumorales sinérgicos. Estudios previos con el progenitor murino de epratuzumab habían indicado que la mayor parte del anticuerpo se internaliza en 1 hora y el 50 % de CD22 se vuelve a expresar en la superficie celular en 5 horas (Shih et al., 1994, Int J Cancer 56:538-45). Este proceso de internalización y reexpresión permitiría el suministro intracelular que podría compensar la menor expresión superficial de CD22. Debido a que muchas de las neoplasias malignas de células B expresan mucho más CD20 que CD22, un conjugado dirigido a CD20 podría administrar más moles de fármaco al liberar su carga tóxica después de localizarse en el tumor.
[0327] Estudios de citotoxicidad in vitro no pudieron discriminar la potencia de los conjugados específicos o incluso de un conjugado irrelevante debido a la liberación de SN-38 del conjugado al medio. De hecho, el SN-38 solo fue un poco más potente que los conjugados, lo que puede reflejar su capacidad acelerada para ingresar a la célula y activar la topoisomerasa I. Porque otros estudios revelaron que los conjugados requerían una exposición de 48 horas antes de que pudieran verse los primeros signos de apoptosis., llegamos a la conclusión de que las pruebas in vitro no podrían discriminar la potencia de estos 2 conjugados y, por lo tanto, recurrimos a estudios in vivo.
[0328] En modelos de xenoinjerto, ambos conjugados tenían una actividad antitumoral similar contra los tumores de Ramos, que la citometría de flujo había indicado que expresaba casi 15 veces más CD20 que CD22. Esto apoyó la selección del conjugado Emab anti-CD22-SN-38 especialmente porque podría combinarse con la terapia Vmab anti-CD20 sin conjugar sin preocuparse de que cualquiera de los agentes interfiriera con la unión del otro agente. De hecho, si se usara un conjugado anti-CD20-SN-38, la dosis total de proteína IgG dada probablemente estaría por debajo del nivel típicamente necesario para tratamientos efectivos con anticuerpos anti-CD20 no conjugados, ya que la toxicidad limitante de la dosis estaría impulsada por el contenido SN-38. Agregar más antiCD20 sin marcar a un conjugado anti-CD20-SN-38 podría reducir la captación del conjugado y potencialmente disminuir su eficacia. Sin embargo, como mostramos anteriormente en estudios combinados que utilizaron epratuzumab radiomarcado con veltuzumab no conjugado, se puede obtener un beneficio de ambos agentes administrados en sus dosis máximas efectivas y seguras. Los estudios in vitro mostraron que veltuzumab, incluso en ausencia del entrecruzamiento que se usa para mejorar la señalización, aceleró los eventos apoptóticos iniciados con Emab-SN-38. Por lo tanto, siempre que el conjugado Emab-SN-38 fuera tan efectivo como el conjugado anti-CD20, seleccionar el conjugado Emab-SN-38 es una opción lógica porque permite una terapia de combinación más efectiva, incluso en tumores donde uno o ambos antígenos tienen baja expresión.
[0329] Debido a que la mayoría de los ADC que usan fármacos ultratóxicos están unidos de manera estable, también probamos un conjugado anti-CD22-SN-38 estable en suero, pero escindible intracelularmente, pero determinamos que era de 40 a 55 veces menos potente que el enlazador CL2A. Otros han examinado una variedad de fármacos ultratóxicos conjugados con anticuerpos anti-CD20 o anti-CD22, encontrando que los conjugados de internalización
son generalmente más activos, pero también observaron que incluso los anticuerpos de internalización lenta podrían ser efectivos si el fármaco liberado penetrara la membrana celular. Si bien el enlazador de tipo CL2A puede ser apropiado para SN-38, puede no ser óptimo para un agente más tóxico, en el que incluso una liberación pequeña y sostenida en el suero aumentaría la toxicidad y comprometería la ventana terapéutica.
[0330] Emab-SN-38 fue activo a una dosis acumulada de 0,6 mg en ratones portadores de Ramos (75 mg dos veces por semana durante 4 semanas), lo que se extrapola a una dosis humana de solo 2,5 mg/kg. Por lo tanto, Emab-SN-38 debería tener una amplia ventana terapéutica en los pacientes. Además, se combinó una dosis eficaz y segura del conjugado anti-Trop-2-SN-38 con una dosis máxima tolerada de un anticuerpo marcado con 90Y sin un aumento apreciable de la toxicidad pero con una eficacia mejorada (Sharkey et al., 2011)., Mol Cancer Ther 10:1072-81). Por lo tanto, el perfil de seguridad y eficacia de estos conjugados de anticuerpos SN-38 es muy favorable para otras terapias combinadas.
[0331] Aunque el irinotecán no se usa de forma rutinaria para el tratamiento de cánceres hematopoyéticos, SN-38 fue tan potente en líneas celulares de linfoma y leucemia como en tumores sólidos (Cardillo et al., 2011, Clin Cancer Res 17:3157-69). En la línea celular WSUFSCCL, los conjugados de IgG específicos e irrelevantes fueron significativamente mejores que el irinotecán, mientras que en Ramos, la mediana de Tt P con el conjugado irrelevante fue más larga pero no significativamente mejor que el irinotecán. Estos resultados son consistentes con otros estudios que han demostrado que una IgG no específica es un excelente portador de fármacos y más potente in vivo que el fármaco libre o los conjugados preparados con albúmina o polietilenglicol (PEG)-Fc. Si bien el conjugado de PEG-SN-38 tuvo efectos antitumorales significativos, se administró en sus cantidades máximas toleradas, que oscilaron entre 10 y 30 mg/kg de equivalentes de SN-38 (Sapra et al., 2009, Haematologica 94:1456-9). Por el contrario, la dosis acumulada máxima de SN-3863 administrado durante 4 semanas a animales portadores de Ramos fue de solo 1,6 mg/kg (es decir, la dosificación de 0,25 mg de Emab-SN-38 administrada dos veces por semana durante 4 semanas) y esto no era tóxico.
[0332] La actividad terapéutica específica de Emab-SN-38 pareció mejorar en las líneas celulares con mayor expresión de CD22. Por ejemplo, en Ramos, se registraron efectos terapéuticos específicos de Emab-SN-38 solo en 2 de los 3 niveles de dosis diferentes examinados, y se extirpó por completo una cantidad considerable de tumores. Por el contrario, en WSU-FSCCL que tenía una expresión de CD22 unas 2,5 veces menor, Emab-SN-38 mejoró significativamente la supervivencia en comparación con el conjugado antiCEACAM5-SN-38 irrelevante en 1 de 2 estudios. Sin embargo, es importante enfatizar que cuando se usa en combinación con la terapia anti-CD20 no conjugada, Emab-SN-38 amplifica la respuesta terapéutica. Por lo tanto, la combinación de estos dos tratamientos podría aumentar la respuesta incluso en situaciones en las que CD22 no se expresa en gran medida.
[0333] En conclusión, usando el ligador CL2A-SN-28 menos estable, el conjugado anti-CD22-SN-38 de Emab fue igualmente activo en dosis no tóxicas in vivo como un conjugado similar anti-CD20-SN-38, a pesar del hecho de que la expresión de CD20 fue más de un múltiplo log mayor que CD22. Las respuestas terapéuticas se beneficiaron de la combinación de Emab-SN-38 con la terapia anti-CD20 de Vmab no conjugado, incluso cuando la expresión de CD22 era baja, lo que sugiere que la terapia de combinación podría mejorar las respuestas en varias neoplasias malignas de células B cuando ambos antígenos están presentes. los estudios sugieren que esta combinación es muy potente en diversos modelos preclínicos de linfoma y leucemia, pero parece tener menos toxicidad para el huésped. Además, las combinaciones de este ADC con microtúbulos e inhibidores de PARP pueden ser sinérgicas para inhibir el crecimiento tumoral y prolongar la supervivencia del huésped portador del tumor. Este resultado es sorprendente, ya que se informó anteriormente que el inhibidor de PARP iniparib no logró sensibilizar a las células cancerosas. a la terapia combinada con agentes anticancerígenos estándar, como cisplatino, gemcitabina o paclitaxel (Bao et al., 2015, Oncotarget [Epub antes de la impresión, 22 de septiembre de 2015])
Ejemplo 16. Conjugados Anti-CD74 (Milatuzumab) SN-38 para el tratamiento de cánceres humanos CD74+
Resumen
[0334] CD74 es un objetivo atractivo para los conjugados de anticuerpo y fármaco (ADC), porque se internaliza y se recicla después de la unión del anticuerpo. CD74 se asocia principalmente con cánceres hematológicos, pero también se expresa en cánceres sólidos. Por lo tanto, se examinó la utilidad de los ADC preparados con el anticuerpo anti-CD74 humanizado, milatuzumab, para la terapia de tumores sólidos que expresan Cd 74. Se prepararon milatuzumabdoxorrubicina y dos conjugados de milatuzumab-SN-38 con enlazadores escindibles (CL2A y CL2E), que difieren en su estabilidad en suero y en cómo liberan SN-38 en el lisosoma. La expresión de CD74 se determinó mediante citometría de flujo e inmunohistología. Se realizaron estudios de citotoxicidad in vitro y terapéuticos in vivo en las líneas celulares de cáncer humano A-375 (melanoma), HuH-7 y Hep-G2 (hepatoma), Capan-1 (pancreático) y NCI-N87 (gástrico), y Linfoma de Raji Burkitt. El ADC de milatuzumab-SN-38 se comparó con los ADC de SN-38 preparados con anticuerpos anti-Trop-2 y anti-CEACAM6 en xenoinjertos que expresaban sus antígenos diana.
[0335] Milatuzumab-doxorrubicina fue más efectivo en el modelo de linfoma, mientras que en A-375 y Capan-1, solo el milatuzumab-CL2A-SN-38 mostró un beneficio terapéutico. A pesar de una expresión de superficie mucho más baja
de CD74 que Trop-2 o CEACAM6, milatuzumab-CL2A-SN-38 tuvo una eficacia similar en Capan-1 como anti-Trop-2 CL2A-SN-38, pero en NCI-N87, el anti-CEACAM6 y los conjugados anti-Trop-2 fueron superiores. Los estudios en 2 líneas celulares de hepatoma a un nivel de dosis única mostraron un beneficio significativo sobre los animales tratados con solución salina, pero no contra un conjugado de IgG irrelevante. CD74 es un objetivo adecuado para los ADC en algunos xenoinjertos de tumores sólidos, con eficacia influenciada en gran medida por la uniformidad de la expresión de CD74, y con conjugados de SN-38 ligados a CL2A que proporcionan las mejores respuestas terapéuticas.
Introducción
[0336] CD74, denominada cadena invariable o Ii, es una glicoproteína transmembrana de tipo II que se asocia con HLADR e inhibe la unión de péptidos antigénicos a la estructura de presentación de antígenos de clase II. Sirve como molécula chaperona, dirigiendo los complejos de cadena invariante a endosomas y lisosomas, molécula accesoria en la maduración de células B, utilizando una vía mediada por NF-kB, y en respuestas de células T a través de interacciones con CD44 (Naujokas et al., 1993, Cell 74:257-68), y es un receptor de la citocina proinflamatoria, factor inhibidor de la migración de macrófagos (Leng et al., 2003, J Exp Med 197:1467-76), que está implicado en activando la proliferación celular y las vías de supervivencia.
[0337] En tejidos humanos normales, CD74 se expresa principalmente en células B, monocitos, macrófagos, células dendríticas, células de Langerhans, subconjuntos de células T activadas y epitelio tímico (no se muestra), y se expresa en más del 90 % de los tumores de células B (Burton y col., 2004, Clin Cancer Res 10:6606-11; Stein y col., 2004, Blood 104:3705-11). Los primeros estudios tenían datos contradictorios sobre la presencia de CD74 en la membrana, en parte porque los anticuerpos contra la cadena invariable eran específicos para la porción citoplásmica de la molécula, pero también porque hay relativamente pocas copias en la superficie y su vida media en la superficie celular es muy corto. Aproximadamente el 80% del CD74 en la superficie celular está asociado con el antígeno HLA-DR del MHC II (Roche et al., 1993, PNAS USA 90:8581-85). Usando el anticuerpo murino anti-CD74, LL1, se estimó que la línea celular de linfoma Raji Burkitt tenía 4.8 x 104 copias/célula, pero debido al rápido tránsito intracelular, se internalizaron y catabolizaron ~ 8 x 106 moléculas de anticuerpo por día (Hansen et al., 1996, Biochem J320:293-300). Por lo tanto, la internalización de CD74 es muy dinámica, con el anticuerpo siendo movido rápidamente desde la superficie y descargado dentro de la célula, seguido por la reexpresión de CD74 en la superficie. La internalización de Fab' se produce tan rápidamente como la unión de IgG, lo que indica que no se requiere la unión bivalente. Estudios posteriores con una versión injertada de CDR de LL1 murino, milatuzumab (hLL1), encontraron que el anticuerpo podría alterar la proliferación de células B, la migración y la expresión de moléculas de adhesión (Stein et al., 2004, Blood 104: 3705-11; Qu et al., 2002, Proc Am Assoc Cancer Res 43:255; Frolich et al., 2012, Arthritis Res Ther 14:R54), pero las excepcionales propiedades de internalización del anticuerpo anti-CD74 lo convirtieron en un vehículo eficaz para la administración intracelular de la terapéutica del cáncer (p. ej., Griffiths et al., 2003, Clin Cancer Res 9:6567-71). Con base en los resultados preclínicos de eficacia y toxicología, los ensayos clínicos de Fase I con milatuzumab-doxorrubicina en mieloma múltiple (Kaufman et al., 2008, ASH Annual Meeting Abstracts, 112:3697), así como en linfoma no Hodgkin y leucemia linfocítica crónica, han sido iniciados.
[0338] Curiosamente, CD74 también se expresa en cánceres no hematopoyéticos, como gástrico, renal, de vejiga urinaria, cánceres de pulmón de células no pequeñas, ciertos sarcomas y glioblastoma (p. ej., Gold et al., 2010, Int J Clin Exp Pathol 4: 1-12), por lo que puede ser una diana terapéutica para tumores sólidos que expresen este antígeno. Dado que un conjugado de milatuzumab-doxorrubicina fue muy activo en modelos de cánceres hematológicos, era una elección lógica para esta evaluación. Sin embargo, recientemente desarrollamos procedimientos para acoplar el inhibidor de topoisomerasa I altamente potente, SN-38, a anticuerpos. SN-38 es la forma activa del irinotecán, cuya farmacología y metabolismo son bien conocidos. Estos conjugados tienen una potencia nanomolar en líneas de células tumorales sólidas y se encontró que eran activos con anticuerpos que no estaban internalizados activamente. Estudios anteriores indicaron una preferencia por un enlazador (CL2A) que permitía que SN-38 se disociara del conjugado en el suero con una vida media de ~1 día, en lugar de otros enlazadores que eran más o menos estables en el suero. Sin embargo, dada la excepcional capacidad de internalización de milatuzumab, se desarrolló un nuevo enlazador que es altamente estable en el suero, pero que puede liberar SN-38 cuando se introduce en el lisosoma.
[0339] La investigación actual examina las perspectivas de uso de estos tres conjugados anti-CD74 de milatuzumab, uno con doxorrubicina y dos conjugados de SN-38, para una terapia eficaz principalmente contra tumores sólidos.
Materiales y métodos
[0340] Líneas de células tumorales humanas. Las líneas celulares de linfoma Raji Burkitt, A-375 (melanoma), Capan-1 (adenocarcinoma pancreático), NCI-N87 (carcinoma gástrico), hepatoma Hep-G2 y mieloma MC/CAR se adquirieron de American Tissue Culture Collection (Manassas, VA). La línea celular de hepatoma HuH-7 se adquirió del Banco de Recursos de Investigación de Ciencias de la Salud de Japón (Osaka, Japón). Todas las líneas celulares se cultivaron en una incubadora con CO2 humidificado (5 %) a 37 °C en los medios recomendados que contenían entre un 10 % y un 20 % de suero de ternera fetal y suplementos. Las células se pasaron <50 veces y se revisaron periódicamente para detectar micoplasma.
[0341] Anticuerpos y métodos de conjugación. Milatuzumab (anti-CD74 MAb), epratuzumab (anti-CD22), veltuzumab (anti-CD20), labetuzumab (anti-CEACAM5), hMN15 (anti-CEACAM6) y hRS7 (anti-Trop-2) son monoclonales IgG 1 humanizados. anticuerpos Los conectores CL2A y CL2E y sus derivados SN-38 se prepararon y conjugaron con anticuerpos como se describe en los Ejemplos anteriores. Los conjugados de milatuzumab-doxorrubicina se prepararon como se describió previamente (Griffiths et al., 2003, Clin Cancer Res 9:6567-71). Todos los conjugados se prepararon mediante reducción con disulfuro de la IgG, seguido de reacción con los correspondientes derivados de maleimida de estos enlazadores. Los análisis espectrofotométricos estimaron que la relación de sustitución molar de fármaco:IgG era de 5-7 (1,0 mg de la proteína contiene ~16 μg de SN-38 o 25 μg de doxorrubicina equivalente).
[0342] Unión celular y citotoxicidad in vitro. Los ensayos para comparar la unión celular de milatuzumab no conjugado y conjugado con células positivas para antígeno y las pruebas de citotoxicidad utilizaron el método de reducción de colorante MTS (Promega, Madison, WI).
[0343] Citometría de flujo e inmunohistología. La citometría de flujo se realizó de una manera que proporcionó una evaluación del antígeno unido a membrana o de membrana y citoplasmático solamente. La inmunohistología se realizó en secciones fijadas en formalina e incluidas en parafina de xenoinjertos de tumores subcutáneos, tinción sin métodos de recuperación de antígenos, utilizando anticuerpos a 10 μg/mL que se revelaron con un conjugado de IgG antihumana.
[0344] Estudios in vivo. Se compraron ratones desnudos hembra (4-8 semanas de edad) o ratones SCID hembra (7 semanas de edad) de Taconic (Germantown, NY) y se usaron después de una cuarentena de 1 semana. Todos los agentes, incluidos los controles de solución salina, se administraron por vía intraperitoneal dos veces por semana durante 4 semanas. Las dosis específicas se dan en los Resultados. La toxicidad se evaluó mediante mediciones de peso semanales. Para el modelo de linfoma de Raji Burkitt, se inyectaron por vía intravenosa ratones SCID con 2,5 x 106 células Raji en 0,1 ml de medio. Cinco días después, los animales recibieron una única inyección intravenosa (0,1 ml) del conjugado o solución salina (N = 10/grupo). Los ratones se observaron diariamente en busca de signos de angustia y parálisis, y se les practicó la eutanasia cuando se desarrolló parálisis en las patas traseras, >15 % de pérdida del peso inicial o si estaban moribundos (criterios de valoración sustitutos de la supervivencia).
[0345] Los tumores subcutáneos se midieron con un calibrador en dos dimensiones, y el volumen del tumor (TV) se calculó como L x W^/2, donde L es el diámetro más largo y w es el más corto. Las mediciones se realizaron al menos una vez a la semana, y los animales se sacrificaron cuando los tumores crecieron hasta 1,0 cm3 (es decir, punto final de supervivencia sustituto). La línea celular de melanoma A-375 (6 x 106 células en 0,2 ml) se implantó en ratones desnudos y la terapia se inició cuando los tumores promediaron 0,23 ± 0,06 cm3 (N = 8/grupo). Capan-1 se implantó por vía subcutánea en ratones desnudos usando una combinación de suspensión tumoral de tumores sometidos a pases en serie (0,3 ml de una suspensión tumoral al 15 % p/v) combinados con 8 x 106 células de cultivo tisular. Los tratamientos se iniciaron cuando TV promedió 0,27 ± 0,05 cm3 (N = 10/grupo). Los xenoinjertos de tumor gástrico NCI-N87 se iniciaron inyectando subcutáneamente 0,2 ml de una mezcla 1:1 (v/v) de matrigel y 1 x 107 células del cultivo terminal. La terapia se inició cuando la TV promedió 0,249 ± 0,045 cm3 (N = 7/grupo). Se siguió el mismo procedimiento para desarrollar los xenoinjertos de hepatoma Hep-G2 y HuH-7 en ratones desnudos. La terapia se inició cuando Hep-G2 promedió 0,364 ± 0,062 cm3 (N = 5/grupo) y HuH-7 promedió 0,298 ± 0,055 cm3 (N = 5/grupo).
[0346] La eficacia se expresa en las curvas de supervivencia de Kaplan-Meier, utilizando los puntos finales sustitutos mencionados anteriormente para determinar los tiempos medios de supervivencia. El análisis se realizó mediante una prueba de rangos logarítmicos (Mantel-Cox) utilizando el software Prism GraphPad (LaJolla, CA), con una significancia de P <0,05.
Resultados
[0347] Expresión de CD74 en líneas de células tumorales humanas y xenoinjertos. Seis líneas celulares derivadas de 4 tipos de tumores sólidos diferentes se identificaron como positivas para CD74 basándose principalmente en el análisis de células permeabilizadas (Tabla 15), ya que la MFI de CD74 solo de membrana en las líneas celulares de tumores sólidos muy a menudo era <2 veces mayor que el MFI de fondo (excepto la línea celular de melanoma A-375). La expresión de CD74 superficial en Raji fue >5 veces mayor que en las líneas celulares de tumor sólido, pero el CD74 total en las células Raji permeabilizadas fue similar a la mayoría de las líneas celulares de tumor sólido.

[0348] La inmunohistología mostró que los xenoinjertos subcutáneos de Raji tenían una tinción intensa y en gran parte uniforme, con un marcaje prominente en la superficie celular (no mostrado). La línea celular de hepatoma Hep-G2 tuvo la captación más uniforme de los tumores sólidos, con tinción moderadamente fuerte, pero predominantemente citoplásmica (no se muestra), seguida por la línea celular de melanoma A-375 que tuvo una tinción algo menos uniforme con tinción más intensa, aunque principalmente expresión citoplasmática (no mostrada). Las líneas celulares de carcinoma gástrico de páncreas Capan-1 (no mostrada) y NCI-N87 (no mostrada) tenían una tinción CD74 de moderada (Capan-1) a intensa (NCIN87), pero no estaba uniformemente distribuida. La línea celular de hepatoma HuH-7 (no mostrada) tenía la tinción menos uniforme y más débil.
[0349] Inmunorreactividad de los conjugados. Los valores de Kd para los conjugados de milatuzumab no conjugado, milatuzumab-CL2A- y CL2ESN-38 no fueron significativamente diferentes, con un promedio de 0,77 nM, 0,59 nM y 0,80 nM, respectivamente. Los valores de Kd para el milatuzumab no conjugado y conjugado con doxorrubicina medidos en la línea celular de mieloma múltiple Mc /CAR fueron 0,5 ± 0,02 nM y 0,8 ± 0,2 nM, respectivamente (Sapra et al., 2008, Clin Cancer Res 14:1888-96).
[0350] Liberación de fármacos in vitro y estabilidades séricas de los conjugados. Los mecanismos de liberación de SN-38 de los enlazadores CL2A y CL2E rematados con mercaptoetanol se determinaron en un entorno que simulaba parcialmente las condiciones lisosomales, a saber, pH bajo (pH 5,0), y en presencia o ausencia de catepsina B. El CL2E-SN- El sustrato 38 era inerte a pH 5 en ausencia de la enzima (no se muestra), pero en presencia de catepsina B, la escisión en el sitio Phe-Lys avanzó rápidamente, con una vida media de 34 min (no se muestra). La formación de SN-38 activo requiere la ciclación intramolecular del enlace carbamato en la décima posición de SN-38, que ocurrió más lentamente, con una vida media de 10,7 h (no se muestra).
[0351] Como se esperaba, la catepsina B no tuvo ningún efecto sobre la liberación de SN-38 activo en el enlazador CL2A. Sin embargo, CL2A tiene un enlace de carbonato de bencilo escindible, liberando SN-38 activo a una velocidad similar a la del enlazador CL2E a pH 5,0, con una vida media de ~ 10,2 h (no mostrado). El conjugado de milatuzumabdoxorrubicina, que tiene un enlace de acilhidrazona sensible al pH, tuvo una vida media de 7 a 8 h a pH 5,0 (no se muestra).
[0352] Si bien todos estos enlazadores liberan el fármaco a velocidades relativamente similares en condiciones relevantes para los lisosomas, tienen estabilidades muy diferentes en el suero. Milatuzumab-CL2A-SN-38 liberó el 50 % de SN-38 libre en 21,55 ± 0,17 h (no se muestra), en consonancia con otros conjugados de CL2A-SN-38. Sin embargo, el conjugado CL2E-SN-38 era muy inerte, con una vida media extrapolada a -2100 h. El conjugado de milatuzumab-doxorrubicina liberó el 50 % de la doxorrubicina en 98 h, lo que fue similar a otros 2 conjugados de anticuerpo-doxorrubicina (no mostrados).
[0353] Citotoxicidad. Una cuestión importante relacionada con la evaluación de estos conjugados fue la potencia relativa de la doxorrubicina libre y el s N-38 en las líneas de células tumorales sólidas y hematopoyéticas. Nuestro grupo informó previamente que SN-38 era activo en varias líneas celulares de linfoma de células B y leucemia aguda, con potencias que oscilaban entre 0,13 y 2,28 nM (Sharkey et al., 2011, Mol Cancer Ther 11: 224-34). La potencia de SN-38 en 4 de las líneas de células tumorales sólidas que se usaron posteriormente para estudios de terapia in vivo osciló entre 2,0 y 6 nM (no se muestra). La doxorrubicina tuvo una respuesta mixta, con una potencia de 3-4 nM en las líneas celulares de linfoma Raji y melanoma A-375, pero fue casi 10 veces menos potente contra las líneas celulares Capan-1, NCI-N87 y Hep G2. Otros estudios que compararon la potencia de SN-38 con la doxorrubicina encontraron: cáncer de colon LS174T, 18 frente a 18 (potencia nM de SN-38 frente a doxorrubicina, respectivamente); cáncer de mama MDA-MB-231, 2 frente a 2 nM; cáncer de ovario SK-OV-4, 18 frente a 90 nM; adenocarcinoma de pulmón Calu-3, 32 frente a 582 nM; cáncer de páncreas Capan-2, 37 frente a 221 nM; y cáncer de pulmón de células pequeñas NCI-H466, 0,1 frente a 2 nM. Por lo tanto, SN-38 fue de 5 a 20 veces más potente que la doxorrubicina en
4 de estas 6 líneas celulares, con una potencia similar en LS174T y MDA-MB-231. En conjunto, estos datos indican que la doxorrubicina es menos eficaz contra los tumores sólidos que el SN-38, mientras que el SN-38 parece ser igualmente eficaz en los tumores sólidos y hematopoyéticos.
[0354] Como era de esperar, las 3 formas conjugadas fueron a menudo un orden de magnitud menos potentes que el fármaco libre in vitro, ya que se espera que ambos fármacos se transporten fácilmente a las células, mientras que los conjugados de fármacos requieren la unión de anticuerpos para transportar el fármaco dentro de la célula (no se muestra). El conjugado de SN-38 ligado a CL2A es una excepción, ya que más del 90 % del SN-38 se libera del conjugado al medio durante el período de ensayo de 4 días (Cardillo et al., 2011, Clin Cancer Res 17: 3157-69; Sharkey et al., 2011, Mol Cancer Ther 11:224-34). Por tanto, incluso si el conjugado se internalizara rápidamente, sería difícil discernir las diferencias entre el fármaco libre y el fármaco unido a CL2A.
[0355] El SN-38 estable vinculado a CL2E se desempeñó comparativamente bien en la línea celular Raji, en comparación con el SN-38 libre, pero tuvo una potencia sustancialmente menor (7 a 16 veces) en las 4 líneas celulares de tumor sólido, lo que sugiere la relativamente baja la expresión superficial de CD74 puede desempeñar un papel en la minimización del transporte de fármacos en estos tumores sólidos. El conjugado de milatuzumab-doxorrubicina tuvo diferencias sustanciales en su potencia en comparación con la doxorrubicina libre en todas las líneas celulares, que fue de una magnitud similar a la de los conjugados CL2E-SN-38 con SN-38 libre en las líneas celulares de tumor sólido.
[0356] En las 6 líneas celulares adicionales mencionadas anteriormente, el conjugado de milatuzumab-CL2A-SN-38 fue de 9 a 60 veces más potente que el conjugado de milatuzumab-doxorrubicina (no se muestra), pero nuevamente, este resultado estuvo influenciado en gran medida por el hecho de que el conjugado ligado a CL2a libera la mayor parte de su SN-38 en el medio durante el período de incubación de 4 días, mientras que el conjugado de doxorrubicina liberaría como máximo el 50 % de su fármaco durante este mismo tiempo. El milatuzumab ligado a CL2E no se examinó en estas otras líneas celulares.
[0357] Terapia in vivo de xenoinjertos de tumores humanos. Estudios previos in vivo con milatuzumab-doxorrubicina o conjugados SN-38 preparados con varios anticuerpos habían indicado que eran eficaces a dosis mucho más bajas que su dosis máxima tolerada (Griffiths et al., 2003, Clin Cancer Res 9:6567-71; Sapra et al., 2005, Clin Cancer Res 11:5257-64; Govindan et al., 2009, Clin Cancer Res 15:6052-61; Cardillo et al., 2011, Clin Cancer Res 17:3157-69; Sharkey et al., 2011, Mol Cancer Ther 11:224-34) y, por lo tanto, las pruebas in vivo se centraron en comparar cantidades similares, pero fijas, de cada conjugado a niveles bien tolerados.
[0358] Los estudios iniciales primero examinaron los conjugados de doxorrubicina y SN-38 en un modelo de linfoma Raji diseminado para evaluar cómo se compara el conjugado de milatuzumab-doxorrubicina con los 2 conjugados de SN-38 (no se muestra). Todos los conjugados específicos fueron significativamente mejores que el conjugado de labetuzumab-SN-38 no dirigido o los animales tratados con solución salina, que tuvieron una mediana de supervivencia de solo 20 días (P <0,0001). A pesar de que los estudios in vitro indicaron una ventaja de hasta 8 veces para los conjugados de SN-38 en Raji, la mejor supervivencia se observó con los conjugados de milatuzumab-doxorrubicina, donde todos los animales recibieron una dosis única de 17,5 mg/kg (350 mg). y H10 animales que recibieron 2,0 mg/kg (40 mg) estaban vivos al finalizar el estudio (día 112) (p. ej., dosis de 17,5 mg/kg de milatuzumab-doxorrubicina frente a milatuzumab-CL2A-SN-38, P = 0,0012). La supervivencia fue significativamente menor para los conjugados CL2E-SN-38 más estables (P< 0,0001 y P = 0,0197, dosis de 17,5 y 2,0 mg/kg para CL2A frente a CL2E, respectivamente), aunque los estudios in vitro sugirieron que ambos conjugados libera SN-38 activo a tasas similares cuando se internaliza.
[0359] Se examinaron cinco líneas celulares de tumor sólido, comenzando con la línea celular de melanoma A-375, ya que tenía la mejor respuesta in vitro tanto a la doxorrubicina como a la SN-38. Los xenoinjertos A-375 crecieron rápidamente, teniendo los animales de control tratados con solución salina una supervivencia media de sólo 10,5 días (no se muestra). Una dosis dos veces por semana de 12,5 mg/kg (0,25 mg por animal) del conjugado milatuzumabCL2A-SN-38 prolongó la supervivencia a 28 días (P = 0,0006), que fue significativamente mejor que el conjugado epratuzumab-CL2A-SN-38 de control que tenía una mediana de supervivencia de 17,5 días (P = 0,0089), siendo esta última no significativamente diferente de los animales tratados con solución salina (P = 0,1967). El conjugado milatuzumab-CL2A proporcionó una supervivencia significativamente mayor que el conjugado milatuzumab-CL2E-SN-38 (P = 0,0014), que tuvo la misma mediana de supervivencia de 14 días que su control conjugado epratuzumab-CL2E-SN-38. A pesar de administrar una dosis dos veces mayor de milatuzumabdoxorrubicina que los conjugados SN-38, la mediana de supervivencia no fue mejor que la de los animales tratados con solución salina (10,5 días).
[0360] Al igual que con el modelo de melanoma A-375, en Capan-1, solo el conjugado SN-38 ligado a CL2A fue efectivo, con una mediana de supervivencia de 35 días, significativamente diferente de los animales no tratados (P <0.036) (no mostrado), incluso a una dosis más baja (5 mg/kg; 100 mg por animal) (P <0,02). Ni el milatuzumab-CL2E ni los conjugados de epratuzumab-CL2A-SN-38 no dirigidos, o una dosis dos veces mayor del conjugado de milatuzumab-doxorrubicina, proporcionaron ninguna ventaja de supervivencia (p = 0,44 frente a solución salina). Es
de destacar que en el mismo estudio con animales que recibieron la misma dosis del conjugado internalizante anti-Trop-2 CL2A-SN-38 (hRS7-SN-38; IMMU-132), la mediana de supervivencia fue igual a milatuzumab-CL2A- SN-38 (no se muestra). El conjugado hRS7-CL2A-SN-38 se había identificado previamente como un ADC de interés para tratar una variedad de tumores sólidos (Cardillo et al., 2011, Clin Cancer Res 17:3157-69). El MFI para hRS7 de unión a superficie en Capan-1 fue de 237 (no se muestra), en comparación con 22 para milatuzumab (consulte la Tabla 15). Por lo tanto, a pesar de tener una expresión de antígeno de superficie sustancialmente más baja, el conjugado milatuzumab-CL2A-SN-38 funcionó tan bien como el conjugado hRS7-CL2A-SN-38 en este modelo.
[0361] Dado que el conjugado de milatuzumab-doxorrubicina tuvo resultados terapéuticos inferiores en 2 de los xenoinjertos de tumor sólido, el enfoque cambió para comparar los conjugados de milatuzumab-SN-38 con los conjugados de SN-38 preparados con otros anticuerpos humanizados contra Trop-2 (hRS7) o CEACAM6 (hMN-15), que se expresan más en la superficie de muchos tumores sólidos (Blumenthal et al., 2007, BMC Cancer 7:2; Stein et al., 1993, Int J Cancer 55:938-46). Se examinaron tres modelos de xenoinjerto adicionales.
[0362] En el modelo de tumor gástrico, NCI-N87, los animales que recibieron 17,5 mg/kg/dosis (350 mg) de milatuzumab-CL2A-SN-38 proporcionaron cierta mejora en la supervivencia, pero no alcanzaron la significación estadística en comparación con los tratados con solución salina. animales (31 frente a 14 días; P = 0,0760) o al conjugado anti-CD20-CL2A-SN39 de veltuzumab que no se une (21 días; P = 0,3128) (no se muestra). Sin embargo, los conjugados hRS7- y hMN-15-CL2A mejoraron significativamente la mediana de supervivencia a 66 y 63 días, respectivamente (P = 0,0001). El MFI para Trop-2 y CEACAM6 expresados en superficie fueron 795 y 1123, respectivamente, mucho más altos que c D74 que fue solo 5 (ver Tabla 15). La inmunohistología mostró una expresión citoplásmica relativamente intensa de CD74 en el xenoinjerto de esta línea celular, pero lo que es más importante, estaba dispersa, apareciendo solo en bolsas definidas dentro del tumor (no se muestra). CEACAM6 y Trop-2 se expresaron más uniformemente que CD74 (no se muestra), estando CEACAM6 más intensamente presente tanto en el citoplasma como en la membrana, y Trop-2 principalmente en la membrana. Por lo tanto, la supervivencia mejorada con los conjugados anti-CEACAM6 y anti-Trop-2 muy probablemente refleja tanto una mayor densidad de antígenos como una expresión más uniforme en NCI-N87.
[0363] En la línea celular de hepatoma Hep-G2 (no mostrada), la inmunohistología mostró una expresión muy uniforme con una tinción citoplasmática moderada de CD74, y la citometría de flujo indicó una expresión superficial relativamente baja (MFI = 9). El MFI con hMN-15 fue 175 y la inmunohistología mostró una expresión citoplásmica y de membrana bastante uniforme de CEACAM6, con bolsas aisladas de tinción de membrana muy intensa (no mostrada). Un estudio en animales con xenoinjertos de Hep-G2 encontró que el milatuzumab-CL2A-SN-38 extendió la supervivencia a 45 días en comparación con los 21 días en el grupo tratado con solución salina (P = 0,0048), mientras que el hMN-15-CL2A-SN-38 conjugado mejoró la supervivencia a 35 días. Hubo una tendencia a favor del conjugado de milatuzumab sobre hMN-15-CL2A-SN-38, pero no alcanzó significación estadística (46 frente a 35 días; P = 0,0802). Sin embargo, el conjugado de veltuzumab-CL2A-SN-38 que no se une proporcionó una ventaja de supervivencia similar a la del conjugado de milatuzumab. Previamente, observamos que los resultados terapéuticos con conjugados que no se unen podrían ser similares a los del conjugado específico ligado a CL2A, particularmente a dosis más altas de proteínas, pero la titulación de los conjugados específicos y de control generalmente se revela de manera selectiva. Por lo tanto, ninguno de los conjugados específicos proporcionó una ventaja terapéutica selectiva a estas dosis en esta línea celular.
[0364] Otro estudio que usó la línea celular de hepatoma HuH-7 (no se muestra), que tenía una expresión de superficie similar, pero niveles citoplasmáticos ligeramente más bajos que Hep-G2 (consulte la Tabla 15), encontró que el conjugado hMN-15-SN-38 proporcionaba una mayor (35 frente a 18 días), aunque no significativamente diferente, ventaja de supervivencia que el conjugado milatuzumab-CL2A (P = 0,2944). Si bien los conjugados de hMN-15 y milatuzumab fueron significativamente mejores que los animales tratados con solución salina (P = 0,008 y 0,009, respectivamente), nuevamente, ninguno de los conjugados fue significativamente diferente del conjugado de veltuzumab-SN-38 no dirigido a este nivel de dosis (P = 0,4602 y 0,9033, respectivamente). La expresión superficial de CEACAM6 fue relativamente baja en esta línea celular (MFI = 81), y la inmunohistología mostró que tanto CD74 (no mostrado) como CEACAM6 (no mostrado) eran muy débiles y muy dispersos.
Discusión
[0365] El enfoque del conjugado anticuerpo-fármaco (ADC) para la quimioterapia selectiva de tumores es un área de considerable interés actual (p. ej., Govindan et al., 2012, Expert Opin Biol Ther 12:873-90; Sapra et al., 2011, Expert Opin Biol Ther 20:1131-49 Los éxitos clínicos recientes (Pro et al., 2012, Expert Opin Biol Ther 12:1415-21; LoRusso et al., 2011, Clin Cancer Res 17:437-47) se han producido en un en gran parte con la adopción de fármacos supertóxicos en lugar de los agentes quimioterapéuticos convencionales que se habían utilizado anteriormente. Sin embargo, la selección del objetivo, el anticuerpo y el enlazador del fármaco son factores que influyen en el rendimiento óptimo de un ADC. Por ejemplo, en el caso de trastuzumab-DM1, HER2 es abundante en tumores que expresan este antígeno, el anticuerpo se internaliza y el propio anticuerpo tiene actividad antitumoral, todo lo cual podría combinarse para mejorar el resultado terapéutico. En marcado contraste, CD74 se expresa a un nivel mucho más bajo en la superficie de las células, pero sus propiedades únicas de internalización y reexpresión en la superficie han permitido
que un ADC anti-CD74 de milatuzumab sea eficaz en modelos de xenoinjerto de cáncer hematopoyético incluso con un fármaco moderadamente tóxico., como la doxorrubicina (Griffiths et al., 2003, Clin Cancer Res 9:6567-71; Sapra et al., 2005, Clin Cancer Res 11:5257-64). Aunque la doxorrubicina se usa con más frecuencia en los cánceres hematopoyéticos, mientras que el SN-38 y otras camptotecinas se administran a pacientes con tumores sólidos, decidimos evaluar la utilidad de la doxorrubicina y los conjugados de SN-38 de milatuzumab en tumores sólidos. El conjugado de milatuzumab-doxorrubicina fue efectivo en modelos de xenoinjerto de varios cánceres hematológicos, lo que llevó a su prueba clínica (NCT01101594 y NCT01585688), mientras que varios conjugados de SN-38 fueron efectivos en modelos de tumores sólidos y hematológicos, lo que llevó a la creación de 2 nuevos conjugados de SN-38. buscado en ensayos clínicos de Fase I de cáncer colorrectal y epitelial diverso (NCT01270698 y NCT01631552).
[0366] La doxorrubicina y SN-38 no conjugadas in vitro tuvieron una potencia similar a la de la doxorrubicina contra la línea celular de linfoma Raji, pero SN-38 fue más potente en varias líneas celulares de tumores sólidos diferentes. Curiosamente, in vivo, el conjugado de milatuzumabdoxorrubicina proporcionó la mejor respuesta en Raji en comparación con los conjugados de milatuzumab-SN-38. Sin embargo, en Capan-1 y A-375, milatuzumab doxorrubicina fue menos eficaz que el conjugado de milatuzumab SN-38 ligado a CL2A, aunque las pruebas in vitro indicaron que A-375 era igualmente sensible a la doxorrubicina libre que a la SN-38 libre. Otras dos líneas celulares, cáncer de mama MDA-MB-231 y cáncer de colon LS174T, también tenían una potencia similar con doxorrubicina libre como SN-38 in vitro, pero dado que las pruebas in vitro indicaron que SN-38 era igualmente eficaz en cánceres sólidos y hematológicos, y dado que SN-38 tiene una potencia de 5 a 20 veces mayor que la doxorrubicina en la mayoría de las líneas celulares de tumores sólidos evaluadas, decidimos centrarnos en los 2 conjugados de milatuzumab-SN-38 para la terapia de tumores sólidos. Sin embargo, para evaluar mejor la utilidad de los conjugados de milatuzumab-SN-38, incluimos una evaluación comparativa con los ADC de SN-38 preparados con anticuerpos contra otros antígenos que están presentes en una variedad de tumores sólidos.
[0367] Anteriormente habíamos investigado las respuestas terapéuticas con el conjugado SN-38 ligado a CL2A anti-Trop-2 hRS7 internalizado en la línea celular Capan-1 (Cardillo et al., 2011, Clin Cancer Res 17:3157-69), y por lo tanto el Se comparó la eficacia de los conjugados milatuzumab y hRS7 SN-38. En este estudio, ambos conjugados mejoraron significativamente la supervivencia en comparación con los anticuerpos de control, y los conjugados de SN-38 ligados a CL2A de cada uno fueron superiores a los conjugados ligados a CL2E. Dado que la citometría de flujo había indicado que la expresión de Trop-2 era mayor que la de CD74 en Capan-1, este resultado sugirió que las capacidades de transporte de CD74, que se sabía que eran excepcionales, eran más eficientes que las de Trop-2. Sin embargo, es bien sabido que la accesibilidad del antígeno (es decir, barreras de membrana frente a citoplasma, fisiológicas y de "sitio de unión") y la distribución entre las células dentro de un tumor son factores críticos que influyen en todas las formas de terapia dirigida, en particular aquellas que dependen de una concentración intracelular adecuada. suministro de un producto a células individuales (Thurber et al., 2008, Adv Drug Del Rev 60: 1421-34). En situaciones en las que el antígeno no se expresa uniformemente en todas las células dentro del tumor, tener un agente dirigido que libere lentamente su carga útil después de localizarse en el tumor, como los conjugados ligados a CL2A, permitiría que el fármaco se difunda a las células testigo no dirigidas. mejorando así su rango de eficacia. De hecho, la alta expresión de antígeno podría impedir potencialmente la penetración del tumor según el efecto de barrera del sitio de unión, pero el mecanismo de liberación extracelular podría proporcionar un mecanismo para que el fármaco se difunda dentro del tumor. También se cree que este mecanismo ayuda a la eficacia de otros conjugados que hemos examinado utilizando anticuerpos de internalización deficiente, como anti-CEACAM5 y anti-CEACAM6 utilizados en este documento. Los conjugados basados en milatuzumab dependen más de la interacción directa del anticuerpo con la célula tumoral, aprovechando la rápida internalización y reexpresión de CD74 que puede compensar su menor abundancia en la superficie de las células. Sin embargo, esta ventaja se vería mitigada cuando el CD74 esté muy disperso dentro del tumor, y sin un mecanismo para retener el conjugado dentro del tumor, se perdería el beneficio de la liberación lenta del fármaco del conjugado. Una revisión previa de tumores gastrointestinales humanos realizada por nuestro grupo sugiere que a menudo tienen un alto nivel de expresión con buena uniformidad (Gold et al., 2010, Int J Clin Exp Pathol 4:1-12).
Ejemplo 17. Uso de hRS7-SN-38 (IMMU-132) para tratar el cáncer de mama metastásico refractario a la terapia
[0368] La paciente era una mujer de 57 años con cáncer de mama en estadio IV, triple negativo (ER/PR negativo, HER-neu negativo), diagnosticado originalmente en 2005. Se sometió a una lumpectomía de la mama izquierda en 2005, seguida de ACT de dosis densa en un entorno adyuvante en septiembre de 2005. A continuación, recibió radioterapia, que finalizó en noviembre. La recurrencia local de la enfermedad se identificó cuando la paciente palpó un bulto en el seno contralateral (derecho) a principios de 2012 y luego se trató con quimioterapia CMF (ciclofosfamida, metotrexato, 5-fluorouracilo). Su enfermedad recurrió en el mismo año, con lesiones metastásicas en la piel de la pared torácica. A continuación, recibió un régimen de quimioterapia con carboplatino TAXOL®, durante el cual se produjo una trombocitopenia. Su enfermedad progresó y comenzó con doxorrubicina semanal, que continuó durante 6 dosis. La enfermedad de la piel también estaba progresando. Una exploración FDG-PET el 26/09/12 mostró progresión de la enfermedad en la pared torácica y ganglios axilares sólidos y agrandados. El paciente recibió oxicodona para el control del dolor.
[0369] Recibió IXEMPRA® desde octubre de 2012 hasta febrero de 2013 (cada 2 semanas durante 4 meses), cuando la lesión de la pared torácica se abrió y sangró. A continuación, le recetaron XELODA®, que no fue bien tolerado debido a la neuropatía en sus manos y pies, así como estreñimiento. Las lesiones cutáneas fueron progresivas y luego se inscribió en el ensayo IMMU-132 después de dar su consentimiento informado. El paciente también tenía antecedentes médicos de hipertiroidismo y trastornos visuales, con alto riesgo de enfermedad del SNC (sin embargo, la resonancia magnética cerebral fue negativa para enfermedad del SNC). En el momento de la inscripción en este ensayo, sus lesiones cutáneas (objetivo) en la mama derecha medían 4,4 cm y 2,0 cm en el diámetro más grande. Tenía otra lesión no diana en la mama derecha y un ganglio linfático agrandado en la axila derecha e izquierda.
[0370] La primera infusión de IMMU-132 (12 mg/kg) se inició el 12 de marzo de 2013, la cual fue bien tolerada. Su segunda infusión se retrasó debido a una reducción del recuento absoluto de neutrófilos (RAN) de Grado 3 (0,9) en el día programado de la infusión, una semana después. Tras una semana de retraso y tras recibir NEULASTA®, se le administró su segunda IMMU-132, con una reducción del 25% de la dosis a 9 mg/kg. A partir de entonces, ha estado recibiendo IMMU-132 según lo programado según el protocolo, una vez por semana durante 2 semanas, luego una semana de descanso. Su primera evaluación de respuesta el 17 de mayo de 2013, después de 3 ciclos de terapia, mostró una disminución del 43 % en la suma del diámetro largo de las lesiones diana, lo que constituye una respuesta parcial según los criterios RECIST. Continúa el tratamiento al nivel de dosis de 9 mg/kg. Su salud general y sus síntomas clínicos mejoraron considerablemente desde que comenzó el tratamiento con IMMU-132.
Ejemplo 18. Uso de hRS7-SN-38 (IMMU-132) para tratar cáncer de pulmón de células no pequeñas metastásico refractario
[0371] Se trata de un hombre de 60 años diagnosticado de cáncer de pulmón de células no pequeñas. El paciente recibe regímenes de quimioterapia de carboplatino, bevacizumab durante 6 meses y muestra una respuesta, y luego, después de progresar, recibe ciclos adicionales de quimioterapia con carboplatino, etopósido, TAXOTERE®, gemcitabina durante los próximos 2 años, con respuestas ocasionales que no duran más de 2 meses. El paciente presenta entonces una masa mediastínica izquierda de 6,5 x 4 cm y derrame pleural.
[0372] Después de firmar el consentimiento informado, el paciente recibe IMMU-132 a una dosis de 18 mg/kg cada dos semanas. Durante las dos primeras inyecciones, se experimentan períodos breves de neutropenia y diarrea, con 4 deposiciones en 4 horas, pero se resuelven o responden a medicamentos sintomáticos en 2 días. Después de un total de 6 infusiones de IMMU-132, la evaluación por TC de la lesión índice muestra una reducción del 22 %, justo por debajo de una respuesta parcial pero una reducción definitiva del tumor. El paciente continúa con esta terapia durante otros dos meses, cuando se observa por TC una respuesta parcial de reducción tumoral del 45% de la suma de los diámetros de la lesión índice, constituyendo así una respuesta parcial según los criterios RECIST.
Ejemplo 19. Uso de hRS7-SN-38 (IMMU-132) más olaparib para tratar cáncer de pulmón de células pequeñas metastásico refractario
[0373] Esta es una mujer de 65 años con un diagnóstico de cáncer de pulmón de células pequeñas, que afecta el pulmón izquierdo, los ganglios linfáticos mediastínicos y evidencia de metástasis en el lóbulo parietal izquierdo del cerebro en la resonancia magnética. La quimioterapia previa incluye carboplatino, etopósido y topotecán, pero no se observó respuesta. La radioterapia tampoco logra controlar su enfermedad. A continuación, recibe una terapia combinada con IMMU-132 más olaparib en un ciclo de 21 días. Olaparib se administra a 200 mg dos veces al día en los días 1-10 del ciclo. IMMU-132 se administra a 8 mg/kg los días 1 y 8 del ciclo. Después de 3 ciclos, hay una reducción del 31 % de la suma de los diámetros más largos de los tumores de pulmón y ganglios linfáticos por TC, mientras que la supuesta metástasis cerebral ya no se detecta. Esto constituye un PR por RECIST 1. 1, porque la reducción se confirma 4 semanas más tarde (35 % de reducción de la suma de todas las lesiones diana. Continúa su dosificación de IMMU-132 cada 3 semanas durante otros 3 meses y continúa mostrando y mejora subjetiva de su condición.
Ejemplo 20. Terapia de un paciente con cáncer gástrico con enfermedad metastásica en estadio IV con hRS7-SN-38 (IMMU-132) más paclitaxel
[0374] Este paciente es un varón de 60 años con antecedentes de tabaquismo y períodos de consumo excesivo de alcohol durante un período de 40 años. Experimenta pérdida de peso, incomodidad para comer y dolor que no se alivia con los antiácidos, dolor abdominal frecuente, dolor lumbar y, más recientemente, ganglios palpables en ambas axilas. Busca consejo médico y, después de un estudio, se muestra que tiene un adenocarcinoma, incluidas algunas características escamosas, en la unión gastroesofágica, según una biopsia a través de un gastroscopio. Los estudios radiológicos (TC y FDG-PET) también revelan enfermedad metastásica en axila derecha e izquierda, región mediastínica, columna lumbar e hígado (2 tumores en el lóbulo derecho y 1 en el izquierdo, todos de entre 2 y 4 cm de diámetro). Se reseca su tumor gástrico y luego se lo somete a un ciclo de quimioterapia con epirrubicina, cisplatino y 5-fluorouracilo. Después de 4 meses y un período de descanso de 6 semanas, se cambia a quimioterapia con docetaxel, que tampoco logra controlar su enfermedad, según la progresión confirmada por mediciones de TC de los tumores metastásicos y cierto deterioro general.
[0375] Luego, el paciente recibe una terapia combinada con IMMU-132 (hRS7-SN-38) y paclitaxel en un ciclo de 21 días. Paclitaxel se administra a dosis de 175 mg/m2 los días 1, 7 y 14 del ciclo. IMMU-132 se administra a 10 mg/kg los días 1 y 8 del ciclo. Después de 3 ciclos, se realizan estudios de TC para evaluar el estado de su enfermedad. Las infusiones son bien toleradas, con algunas náuseas y diarreas leves, y también con neutropenia Grado 3, controlada con medicamentos sintomáticos y con G-CSF (Neulasta®) para la neutropenia. Los estudios de TC revelan que la suma de sus lesiones metastásicas de índice ha disminuido en un 28 %, por lo que continúa con esta terapia durante otros 5 ciclos. Los estudios de TC de seguimiento muestran que la enfermedad permanece reducida en aproximadamente un 35 % según los criterios RECIST con respecto a sus mediciones iniciales antes de la terapia combinada, y su estado general también parece haber mejorado, con el paciente recuperando una actitud optimista hacia el control de su enfermedad.
Ejemplo 21. Terapia de paciente con cáncer de colon avanzado refractario a quimioinmunoterapia previa, utilizando únicamente IMMU-130 (labetuzumab-SN-38)
[0376] El paciente es un hombre de 50 años con antecedentes de cáncer de colon metastásico en estadio IV, diagnosticado por primera vez en 2008 y sometido a una colectomía y hepatectomía parcial para los cánceres de colon primario y metastásico, respectivamente. A continuación, recibió quimioterapia, como se indica en la FIG. 14, que incluía irinotecán, oxaliplatino, FOLFIRINOX (5-fluorouracilo, leucovorina, irinotecán, oxaliplatino) y bevacizumab, así como bevacizumab combinado con 5-fluorouracilo/leucovorina, durante casi 2 años. A partir de entonces, recibió cursos de cetuximab, ya sea solo o combinado con quimioterapia FOLFIRI (leucovorina, 5-fluorouracilo, irinotecán) durante el año siguiente o más. En 2009, recibió terapia de ablación por radiofrecuencia de sus metástasis hepáticas mientras estaba bajo quimioinmunoterapia, y a finales de 2010 se sometió a una resección en cuña de sus metástasis pulmonares, que se repitió unos meses después, a principios de 2011. A pesar de haber recibido quimioinmunoterapia en 2011, aparecieron nuevas metástasis pulmonares a finales de 2011, y en 2012 se visualizaron metástasis tanto pulmonares como hepáticas. Su título inicial de antígeno carcinoembrionario (CEA) en plasma fue de 12,5 ng/ml justo antes de someterse a la terapia de anticuerpos y fármacos con IMMU-130. Las lesiones índice elegidas por el radiólogo para medir el cambio de tamaño del tumor mediante tomografía computarizada fueron el lóbulo medio del pulmón derecho y las metástasis hepáticas, ambas con un total de 91 mm como la suma de sus diámetros más largos en la línea de base antes de la terapia de IMMU-130 (anti-CEACAM5-SN-38).
[0377] Este paciente recibió dosis de 16 mg/kg de IMMU-130 por infusión IV lenta cada dos semanas para un total de 17 dosis de tratamiento. El paciente toleró bien la terapia, teniendo solo náuseas, diarrea y fatiga de grado 1 después del primer tratamiento, lo que ocurrió después de los tratamientos 4 y 5, pero no después, porque recibió medicación para estos efectos secundarios. Después del tratamiento 3, mostró alopecia (grado 2), que estuvo presente durante la terapia posterior. Las náuseas, la diarrea y los vómitos ocasionales duraron solo 2 o 3 días, y su fatiga después de la primera infusión duró 2 semanas. Por lo demás, el paciente toleró bien la terapia. Debido a la larga duración de la recepción de este anticuerpo humanizado (injertado con CDR) conjugado con SN-38, se midió su sangre en busca de anticuerpos anti-labetuzumab y no se detectó ninguno, incluso después de 16 dosis.
[0378] Las primeras mediciones de tomografía computarizada (TC) se realizaron después de 4 tratamientos y mostraron un cambio del 28,6 % con respecto a la suma de las mediciones realizadas al inicio, antes de esta terapia, en las lesiones índice. Después de 8 tratamientos, esta reducción pasó a ser del 40,6%, constituyendo así una remisión parcial según los criterios RECIST. Esta respuesta se mantuvo durante otros 2 meses, cuando sus mediciones de TC indicaron que las lesiones índice eran un 31,9 % menores que las mediciones iniciales, pero algo más altas que la disminución anterior del 40,6 % medido. Por lo tanto, con base en mediciones cuidadosas de TC de las lesiones índice en el pulmón y el hígado, este paciente, que había fracasado con la quimioterapia y la inmunoterapia previas, incluido el irinotecán (molécula principal de SN-38), mostró una respuesta objetiva al metabolito activo del irintotecán (o camptotequina), SN-38, cuando se dirige a través del anticuerpo humanizado anti-CEACAM5, labetuzumab (hMN-14). Fue sorprendente que aunque el irinotecán (CPT-11) actúa liberando SN-38 in vivo, el anticuerpo antiCEACAM5 conjugado con SN-38 demostró ser eficaz en un paciente con cáncer colorrectal al inducir una respuesta parcial después de que el paciente no respondiera previamente a su última terapia que contiene irinotecán. La reducción del título de CEA en plasma del paciente también corroboró los hallazgos de la TC: cayó del nivel inicial de 12,6 ng/ml a 2,1 ng/ml después de la tercera dosis de terapia, y estuvo entre 1,7 y 3,6 ng/ml entre las dosis 8 y 12. Normalmente se considera que el título plasmático normal de CEA está entre 2,5 y 5,0 ng/ml, por lo que esta terapia efectuó una normalización de su título CEA en la sangre.
Ejemplo 22. Terapia de un paciente con cáncer de colon avanzado con IMMU-130 más mesilato de eribulina
[0379] Esta paciente es una mujer de 75 años inicialmente diagnosticada con cáncer de colon metastásico (Estadio IV). Tiene una hemicolectomía parcial derecha y resección de su intestino delgado y luego recibe las terapias FOLFOX, FOLFOX bevacizumab, FOLFIRI ramucirumab y FOLFIRI cetuximab durante un año y medio, cuando muestra progresión de la enfermedad, con diseminación de la enfermedad al fondo de saco posterior, omento, con ascitis en la pelvis y derrame pleural en el lado derecho de la cavidad torácica. Su título basal de CEA en plasma justo antes de esta terapia es de 15 ng/ml. Recibe terapia combinada con mesilato de eribulina (1,4 mg/m2) e IMMU-130 (anti
CEACAM5-SN-38) 10 mg/kg, ambos administrados los días 1 y 8 de un ciclo de 21 días, que se tolera muy bien, sin ninguna toxicidad hematológica o no hematológica importante. Dentro de los 2 meses de terapia, su título de CEA en plasma se reduce modestamente a 1,3 ng/ml, pero en la evaluación de 8 semanas muestra una reducción del 21 % de las lesiones tumorales índice, que aumenta a una reducción del 27 % a las 13 semanas. Sorprendentemente, la ascitis y el derrame pleural del paciente disminuyen (desapareciendo este último) en este momento, mejorando así notablemente el estado general del paciente. La paciente continúa con su terapia de investigación.
Ejemplo 23. Paciente con cáncer gástrico con enfermedad metastásica en estadio IV tratada con IMMU-130 más rucaparib
[0380] El paciente es un varón de 52 años que consultó por malestar gástrico y dolor relacionado con la alimentación desde hacía unos 6 años, y con pérdida de peso en los últimos 12 meses. La palpación del área del estómago revela un bulto firme que luego se gastroscopia, revelando una masa ulcerosa en la parte inferior del estómago. Se realiza una biopsia y se diagnostica como un adenocarcinoma gástrico. Las pruebas de laboratorio no revelan cambios anormales específicos, excepto que las pruebas de función hepática, LDH y CEA plasmático están elevados, siendo este último de 10,2 ng/ml. A continuación, la paciente se somete a una PET de cuerpo entero, que revela, además del tumor gástrico, enfermedad metastásica en la axila izquierda y en el lóbulo hepático derecho (2 metástasis pequeñas). Al paciente se le reseca el tumor gástrico y luego se le toman medidas basales de TC de sus tumores metastásicos. Cuatro semanas después de la cirugía, recibe 3 cursos de quimioterapia combinada que consisten en un régimen de cisplatino y 5-fluorouracilo (CF), pero no lo tolera bien, por lo que se cambia al tratamiento con docetaxel. Parece que la enfermedad se estabiliza durante aproximadamente 4 meses, según las tomografías computarizadas, pero luego las quejas del paciente de mayor pérdida de peso, dolor abdominal, pérdida de apetito y fatiga extrema hacen que se repitan los estudios de tomografía computarizada, que muestran un aumento en el tamaño de las metástasis. por una suma del 20% y una lesión sospechosa en el sitio de la resección gástrica original.
[0381] A continuación, el paciente recibe una terapia combinada con IMMU-130 (anti-CEACAM5-SN-38, 8 mg/kg) y el inhibidor de PARP rucaparib (12 mg/m2) en un programa semanal. Lo tolera bien, pero después de 3 semanas muestra neutropenia de grado 3 y diarrea de grado 1. Su cuarta infusión se pospone una semana y luego se reinstituyen las infusiones semanales, sin evidencia de diarrea o neutropenia para las siguientes 4 inyecciones. A continuación, el paciente se somete a un estudio de TC para medir el tamaño de su tumor metastásico y para ver el área original de la resección gástrica. El radiólogo mida, según los criterios RECIST, una disminución de la suma de las lesiones metastásicas, respecto al valor basal previo a la terapia combinada, del 23%. No parece haber ninguna lesión clara en el área de la resección gástrica original. El título de CEA en plasma del paciente en este momento es de 7,2 ng/ml, que está muy por debajo del valor inicial de 14,5 ng/ml. El paciente continúa con la terapia combinada semanal a las mismas dosis, y después de un total de 13 infusiones, sus estudios de TC muestran que una metástasis hepática ha desaparecido y la suma de todas las lesiones metastásicas se reduce en un 41 %, lo que constituye una respuesta parcial de RECIST. El estado general del paciente mejora y reanuda sus actividades habituales mientras continúa recibiendo una terapia de mantenimiento de 8 mg/kg IMMU-130 cada tres semanas durante otras 4 inyecciones. En la última medición de CEA en sangre, el valor es de 4,8 ng/ml, que está dentro del rango normal para un fumador, como es el caso de este paciente. Las mediciones séricas de HAHA no revelan un anticuerpo o anticuerpos anti-SN-38, por lo que la terapia parece no ser inmunogénica.
Ejemplo 24. Terapia de cáncer de mama metastásico trip le negativo recidivante con IMMU-130 más veliparib
[0382] Una mujer de 58 años con cáncer de mama metastásico triple negativo tratado previamente con bevacizumab más paclitaxel, sin respuesta, presenta metástasis en varias costillas, vértebras lumbares, lesión solitaria de 3 cm de diámetro en pulmón izquierdo, con considerable hueso dolor y fatiga. Recibe terapia combinada con anti-CEACAM5 IMMU-130 (hMN-14-SN-48) más veliparib. IMMU-130 se administra a 12 mg/kg los días 1 y 8 de un ciclo de 21 días, mientras que veliparib se administra una vez al día a 60 mg. Excepto por neutropenia transitoria de grado 2 y algo de diarrea inicial, tolera bien la terapia, que luego se repite, después de un descanso de 2 meses, para otro curso. El examen radiológico indica que tiene respuesta parcial por criterios RECIST, porque la suma de los diámetros de las lesiones índice disminuye en un 39%. Su estado general, incluido el dolor óseo, también mejora y vuelve casi al mismo nivel de actividad que antes de su enfermedad.
Ejemplo 25. Terapia de carcinoma de colon metastásico, generalmente refractivo, recidivante con hMN-15-SN-38 más paclitaxel
[0383] Mujer de 46 años con cáncer de colon metastásico estadio IV, con antecedente de resección de la lesión primaria que además presentaba metástasis hepáticas sincrónicas a ambos lóbulos hepáticos, así como un foco único de diseminación al pulmón derecho; estas metástasis miden, por TC, entre 2 y 5 cm de diámetro. Se somete a varios ciclos de quimioterapia durante un período de 3 años, incluidos 5-fluorouracilo, leucovorina, irinotecán, oxaliplatino, cetuximab y bevacizumab. En dos ocasiones, hay evidencia de estabilización de la enfermedad o una respuesta a corto plazo, pero no hay reducción del 30% o más de sus lesiones medidas. Su título de CEA en plasma al inicio del estudio antes de la terapia combinada es de 46 ng/mL, y sus lesiones índice totales miden una suma de 92 mm.
[0384] La terapia de combinación con hMN-15-SN-38 y paclitaxel se instituye en un ciclo de 21 días. Se administra hMN-l5-SN-38 a 12 mg/kg los días 1 y 8 y paclitaxel a dosis de 135 mg/m2 los días 1, 7 y 14 del ciclo. Este ciclo se repite 3 veces, con solo neutropenia transitoria y efectos secundarios gastrointestinales (náuseas, vómitos, diarrea). Sorprendentemente, a pesar de no responder a la terapia con FOLFIRI (que incluye irinotecán o CPT-11), la paciente muestra una respuesta parcial según los criterios RECIST después de completar su terapia. A continuación, se la coloca en un programa de mantenimiento de esta terapia una vez al mes durante los próximos 6 meses. Las exploraciones de seguimiento muestran que su enfermedad permanece bajo control como una respuesta parcial (RP) y, en general, la paciente se encuentra en buenas condiciones con un estado funcional de Karnofsky del 90 %.
Ejemplo 26. Terapia de cáncer de ovario metastásico recidivante con IMMU-130 más olaparib
[0385] Una mujer de 66 años con cáncer de ovario en estadio IV de FIGO positivo para mutación BRCA1 se somete a cirugía primaria y posoperatorio de paclitaxel y carboplatino (TC). Después de un intervalo de 20 meses sin platino, la TC confirma un nivel elevado de CA125 y la recurrencia en el peritoneo. Después de un nuevo tratamiento con TC, se produce una reacción de hipersensibilidad al carboplatino, que se cambia a nedaplatino. Una respuesta completa se confirma por TC. Después de una PFI de 8 meses, se confirma un nivel sérico elevado de CA125 y recurrencia en el peritoneo y el hígado.
[0386] A continuación, ella recibe una terapia combinada con anti-CEACAM5 IMMU-130 (hMN-14-SN-38) más oliparib. IMMU-130 se administra a 10 mg/kg los días 1 y 8 de un ciclo de 21 días, mientras que oliparib se administra a 200 mg dos veces al día los días 1 a 10 del ciclo. Excepto por neutropenia transitoria de grado 2 y algo de diarrea inicial, tolera bien la terapia, que luego se repite, después de un descanso de 2 meses, para otro curso. El examen radiológico indica que tiene respuesta parcial por criterios RECIST, porque la suma de los diámetros de las lesiones índice disminuyen en un 45%. Su estado general también mejora y vuelve casi al mismo nivel de actividad que antes de su enfermedad.
Ejemplo 27. Paciente con cáncer de colon con enfermedad metastásica en estadio IV tratado con conjugado anti-CSAp-SN-38 más paclitaxel
[0387] Esta paciente presenta metástasis de cáncer de colon en el lóbulo izquierdo del hígado y en ambos pulmones, luego de una resección de un adenocarinoma de colon sigmoide de 9 cm, seguido de quimioinmunoterapia con FOLIFIRI y cetuximab durante 6 meses, y luego FOLFOX seguido de bevacizumab por un período adicional de aproximadamente 9 meses. Diez meses después de la resección inicial y luego del comienzo de la terapia, la enfermedad estable que se creía presente muestra progresión por el crecimiento de las lesiones y la aparición de una nueva metástasis en la glándula suprarrenal izquierda. Su c Ea en plasma en este momento es de 52 ng/ml y su estado general parece haberse deteriorado, con dolores abdominales, fatiga y anemia limítrofe, lo que sugiere una posible hemorragia interna.
[0388] A continuación ella recibe una terapia combinada con un conjugado SN-38 de anticuerpo hMu-9 (anti-CSAp) y paclitaxel en un ciclo de 21 días. El anticuerpo o inmunoconjugado se administra los días 1 y 8 del ciclo a dosis de 12 mg/kg, y el paclitaxel a dosis de 175 mg/m2 los días 1, 7 y 14 del ciclo, que se repite para ciclos de tratamiento adicionales, midiendo sus hemogramas cada semana y recibiendo medicamentos con atropina para controlar las reacciones gastrointestinales. Se observa alopecia de grado 2 después del primer ciclo de tratamiento, pero solo neutropenia de grado 1. Después de 3 ciclos de tratamiento, su título de CEA en plasma se reduce a 19 ng/ml, y en este momento sus mediciones de TC muestran una disminución de las lesiones índice en el hígado y los pulmones en un 24,1 %. Después de 3 ciclos adicionales de terapia, muestra una reducción en la TC de las lesiones índice del 31,4 % y una reducción del tamaño de la masa suprarrenal en aproximadamente un 40 %. Se considera que este paciente responde a la terapia combinada y continúa con esta terapia. Su estado general parece haber mejorado, con menos fatiga, sin dolor o malestar abdominal y, en general, con más energía.
Ejemplo 28. Tratamiento de cáncer de páncreas metastásico con inmunoconjugado anti-MUC5ac-SN-38 más rucaparib
[0389] Este paciente de 44 años tiene antecedentes de carcinoma de páncreas metastásico, con adenocarcinoma ductal de páncreas inoperable en la cabeza del páncreas, y que muestra metástasis en los lóbulos izquierdo y derecho del hígado, el primero mide 3 x 4 cm y el segundo mide 2 x 3 cm El paciente recibe un curso de gemcitabina pero no muestra una respuesta objetiva. Cuatro semanas después, recibe terapia combinada con hPAM4-SN-38 iv a una dosis de 8 mg/kg dos veces por semana durante 2 semanas, con una semana de descanso, más rucaparib (12 mg/m2) una vez por semana. Esto se repite durante otros 2 ciclos. Los estudios de TC se realizan una semana después y muestran una reducción total en la masa tumoral (todos los sitios) del 32 % (respuesta parcial), junto con una caída en el título de CA19-9 en sangre de 220 al inicio a 75 en el momento de la evaluación radiológica. El paciente muestra solo náuseas y vómitos de grado 1 después de cada tratamiento y una neutropenia de grado 2 al final del último ciclo de tratamiento, que se resuelve 4 semanas después. No se administra premedicación para prevenir las reacciones a la infusión.
Ejemplo 29. Uso de anti-CD74 hLL1-SN-38 más oliparib para tratar el cáncer de colon metastásico refractario a la terapia (mCRC)
[0390] El paciente es un hombre de 67 años que presenta cáncer de colon metastásico. A continuación, de la colectomía transversa poco después del diagnóstico, el paciente recibe 4 ciclos de quimioterapia FOLFOX en un entorno neoadyuvante antes de la hepatectomía parcial para extirpar las lesiones metastásicas en el lóbulo izquierdo del hígado. A esto le sigue un régimen adyuvante de FOLFOX para un total de 10 ciclos de FOLFOX.
[0391] La TC muestra metástasis en el hígado. Su lesión diana es un tumor de 3,0 cm en el lóbulo izquierdo del hígado. Las lesiones no diana incluyeron varias masas hipoatenuadas en el hígado. El CEA inicial es de 685 ng/mL.
[0392] A continuación se trata al paciente con milatuzumab anti-CD74 conjugado con SN-38 (hLL1-SN38) en combinación con oliparib. hLL1-SN-38 (10 mg/kg) se administra los días 1 y 8 de un ciclo de 21 días, mientras que oliparib se administra a 200 mg dos veces al día los días 1 a 10 del ciclo. El paciente experimenta náuseas (Grado 2) y fatiga (Grado 2) durante el primer ciclo y continúa el tratamiento sin mayores eventos adversos. La primera evaluación de respuesta realizada (después de 3 ciclos) muestra una reducción de la lesión objetivo en un 26 % mediante tomografía computarizada (TC) y su nivel de CEA disminuye a 245 ng/mL. En la segunda evaluación de respuesta (después de 8 ciclos), la lesión diana se ha reducido en un 35 %. Su salud general y sus síntomas clínicos han mejorado considerablemente.
Ejemplo 30. Tratamiento de leucemia linfocítica crónica recidivante con IMMU-114-SN-38 y paclitaxel
[0393] Un hombre de 67 años con antecedentes de LLC, según la definición del Taller Internacional sobre Leucemia Linfocítica Crónica y las clasificaciones de la Organización Mundial de la Salud, presenta recaída de la enfermedad después de terapias previas con fludarabina, dexametasona y rituximab, así como un régimen de CVP. Ahora tiene fiebre y sudores nocturnos asociados con un agrandamiento generalizado de los ganglios linfáticos, una producción reducida de hemoglobina y plaquetas, así como un recuento de leucocitos que aumenta rápidamente. Su LDH está elevado y la beta-2-microglobulina es casi el doble de lo normal. El paciente recibe terapia con conjugado anti-HLA-DR IMMU-114-SN-38 (IgG4 hL243-SN-38) en combinación con paclitaxel en un ciclo de 21 días. Se administra IMMU-114-SN-38 a 8 mg/kg los días 1 y 8 y se administra paclitaxel a dosis de 175 mg/m2 los días 1, 7 y 14 del ciclo, luego se repite el ciclo. Después de 4 ciclos, la evaluación muestra que los parámetros hematológicos del paciente mejoran y sus células de LLC circulantes parecen estar disminuyendo en número. La terapia se reanuda por otros 3 ciclos, después de lo cual sus valores hematológicos y de laboratorio indican que tiene una respuesta parcial.
Ejemplo 31. Tratamiento de paciente con linfoma fo licu lar con hA19-SN-38, rucaparib y paclitaxel
[0394] Varón de 60 años que consulta por dolor abdominal y presencia de masa palpable. El paciente tiene estudios de TC y FDG-PET que confirman la presencia de la masa con adenopatías patológicas en el mediastino, axilas y ganglios del cuello. Las pruebas de laboratorio son normales excepto por LDH elevado y beta-2-microglobulina. La biopsia de médula ósea revela varios agregados linfoides paratrabeculares y perivasculares. Estos son linfocíticos con expresión de CD20, CD19 y CD10 por inmunotinción. El diagnóstico final es linfoma folicular de grado 2, estadio IVA, con una puntuación FLIPI de 4. El diámetro más largo del ganglio afectado más grande es de 7 cm. El paciente recibe una terapia combinada con un anticuerpo monoclonal IgG (hA19) anti-CD19 humanizado conjugado con SN-38, más rucaparib y paclitaxel, en un ciclo de 21 días. El ADC se administra a 6 mg/kg los días 7 y 14, rucaparib a 10 mg/m2 los días 1, 8 y 15 y paclitaxel a 125 mg/m2 los días 1, 7 y 14 del ciclo. Después de 5 ciclos, las evaluaciones de la médula ósea y las imágenes (TC) muestran una respuesta parcial, donde las lesiones medibles disminuyen en aproximadamente un 60 % y la médula ósea está mucho menos infiltrada. Además, los títulos de LDH y beta-2-microglobulina también disminuyen.
Ejemplo 32. Tratamiento de LLA de células B precursoras en recaída con hA20-SN-38 más olaparib
[0395] Esta mujer de 51 años ha estado en tratamiento para LLA de células B precursoras con cromosoma Filadelfia negativo, que muestra la tinción de células de LLA para CD19, CD20, CD 10, CD38 y CD45. Más del 20% de los linfoblastos de la médula y la sangre expresan CD19 y CD20. El paciente había recibido tratamiento previo con clofarabina y citarabina, lo que resultó en una toxicidad hematológica considerable, pero sin respuesta. También se inició un ciclo de citarabina en dosis altas (ara-C), pero el paciente no pudo tolerarlo. Se le administra hA20-SN-38 (veltuzumab-SN-38) y oliparib en un ciclo de 21 días, con dosis de hA20-SN-38 por infusión de 6 mg/kg los días 7 y 14 y oliparib a 200 mg dos veces al día en los días 1-10 del ciclo.
[0396] Después de 3 ciclos, sorprendentemente, muestra una mejoría en los recuentos de sangre y médula, suficientes para determinar una respuesta parcial. Después de un descanso de 2 meses debido a la neutropenia (Grado 3), la terapia se reanuda durante otros 4 cursos. En este momento, ha mejorado mucho y se está considerando una terapia de mantenimiento para tratar de llevarla a una etapa en la que pueda ser candidata para un trasplante de células madre.
Ejemplo 33. Tratamiento de linfoma con Anti-CD22-SN-38 más paclitaxel
[0397] El paciente es un hombre de 62 años con linfoma difuso de células B grandes (DLBCL) recidivante. Después de 6 cursos de quimioinmunoterapia RCHOP, ahora presenta una diseminación extensa de los ganglios linfáticos en los ganglios linfáticos mediastino, axilar e inguinal. Se le administra anti-CD22 epratuzumab-SN-38 más paclitaxel en un ciclo de 21 días. El ADC se administra a dosis de 12 mg/kg los días 1 y 7 y el paclitaxel se administra a 175 mg/m2 los días 1, 7 y 14 del ciclo. Después de 3 ciclos, se evalúa al paciente mediante tomografía computarizada y se mide la masa tumoral total y muestra una disminución del 35 % (respuesta parcial), que parece mantenerse durante los siguientes 3 meses. Los efectos secundarios son solo trombocitopenia y náuseas y vómitos de grado 1 después del tratamiento, que se resuelven en 2 semanas. No se administra preterapia para reducir las reacciones a la infusión.
Ejemplo 34. Terapia de primera línea de linfoma fo licu lar usando veltuzumab-SN-38 y rucaparib
[0398] La paciente es una mujer de 41 años que presenta linfoma folicular de bajo grado, con ganglios linfáticos cervicales y axilares bilaterales medibles (2-3 cm cada uno), masa mediastínica de 4 cm de diámetro y agrandamiento del bazo. Se le administra veltuzumab-SN-38 (anti-CD20-SN-38) en combinación con rucaparib, administrándose ADC a 10 mg/kg el día 1 y rucaparib a 12 mg/m2 los días 1, 8 y 15. Después 4 ciclos, sus medidas tumorales por TC muestran una reducción del 80%. A continuación, recibe 2 cursos adicionales de terapia y las mediciones de TC indican que se logró una respuesta completa. Esto se confirma mediante imágenes FDG-PET.
Ejemplo 35. Producción y uso de pro-2-pirrolinodoxorrubicina (P2PDox)
[0399] Se sintetizó pro-2-pirrolinodoxorrubicina (P2PDox) y se conjugó con anticuerpos como se describe en la Patente de Estados Unidos n.° 8.877.202. Los ADC P2PDox ejemplares se describen en la Tabla 16 a continuación.
Tabla 16. Conjugados de anticuerpo P2PDox de ejemplo

[0400] También se prepararon conjugados para hPAM4-P2PDox, hLL2-P2PDox y RFB4-P2PDox, con una recuperación y pureza de proteínas similares (no se muestra).
[0401] Estudios de unión celular in vitro: La retención de la unión del anticuerpo se confirmó mediante ensayos de unión celular que compararon la unión del conjugado con el anticuerpo no conjugado (Chari, 2008, Acc Chem Res 41:98-107). La potencia del conjugado se probó en un ensayo MTS de 4 días usando células diana apropiadas. El conjugado hRS7-P2PDox exhibió valores Cl50 de 0,35-1,09 nM en líneas celulares de cáncer humano gástrico (NCI-N87), pancreático (Capan-1) y de mama (MDA-MB-468), con el fármaco libre exhibiendo 0,02-0,07 potencia nM en las mismas líneas celulares.
[0402] Estabilidad del suero: La estabilidad del suero del conjugado prototípico de P2PDox, hRS7-P2PDox, se determinó incubando en suero humano a una concentración de 0,2 mg/mL a 37 °C. El incubado se analizó mediante HPLC usando una columna de cromatografía de interacción hidrófoba de butilo (HIC) en en el que hubo una buena separación del tiempo de retención entre el pico debido al fármaco libre y el debido al conjugado o a las especies de mayor peso molecular. Este análisis mostró que no hubo liberación de fármaco libre del conjugado, lo que sugiere una alta estabilidad sérica del conjugado. Cuando se repitió el mismo experimento con el conjugado hRS7-doxorrubicina, que contenía el mismo enlazador escindible que hRS7-P2PDox, y donde se verificó de forma independiente que el fármaco libre se liberaba con una vida media de 96 h, se observó una formación clara del pico del fármaco libre, a saber pico de doxorrubicina, se observó en HIC HPLC.
[0403] Sorprendentemente, se determinó que el conjugado de P2PDox se sujetaba firmemente al anticuerpo porque entrecruzaba las cadenas peptídicas del anticuerpo. El entrecruzamiento estabiliza la unión del fármaco al anticuerpo para que el fármaco solo se libere intracelularmente después de que se haya metabolizado el anticuerpo. El entrecruzamiento ayuda a minimizar la toxicidad, por ejemplo, la cardiotoxicidad, que resultaría de la liberación del fármaco libre en circulación. El uso anterior de conjugados peptídicos 2-PDox fracasó porque el fármaco entrecruzaba el péptido con otras proteínas o péptidos in vivo. Con los presentes conjugados, el P2PDox se une a grupos disulfuro tiol intercatenarios mientras está en forma de profármaco. La protección del profármaco se elimina rápidamente in vivo
poco después de la inyección y la porción 2-PDox resultante del conjugado entrecruza las cadenas peptídicas del anticuerpo, formando un entrecruzamiento intramolecular dentro de la molécula del anticuerpo. Esto estabiliza el ADC y evita el entrecruzamiento con otras moléculas en circulación.
EJEMPLO 34. Estudios in vivo
[0404] General - El tamaño del tumor se determinó mediante mediciones con calibre de longitud (L) y ancho (W) con el volumen del tumor calculado como (L x W2)/2. Se midieron los tumores y se pesaron los ratones dos veces por semana. Los ratones se sacrificaron si sus tumores alcanzaban un tamaño > 1 cm3, perdían más del 15 % de su peso corporal inicial o se volvían moribundos de otro modo. El análisis estadístico para los datos del crecimiento del tumor se basó en el área bajo la curva (AUC) y el tiempo de supervivencia. Los perfiles de crecimiento tumoral individual se obtuvieron a través de modelos de curvas lineales. Se empleó una prueba f para determinar la igualdad de varianza entre grupos antes del análisis estadístico de las curvas de crecimiento. Se utilizó una prueba t de dos colas para evaluar la significación estadística entre todos los diversos grupos de tratamiento y los controles no específicos. Para el análisis de control de solución salina, se usó una prueba t de una cola para evaluar la significación. Los estudios de supervivencia se analizaron utilizando diagramas de Kaplan-Meier (análisis de rango logarítmico), utilizando el paquete de software Prism GraphPad Software (v4.03) (Advanced Graphics Software, Inc.; Encinitas, CA). Todas las dosis en los experimentos preclínicos se expresan en cantidades de anticuerpos. En términos de fármaco, 100 μg de anticuerpo (5 mg/kg) en un ratón de 20 g, por ejemplo, transporta 1,4 μg-2,8 μg (0,14-0,17 mg/kg) de dosis equivalente de P2PDox cuando se usa un ADC con 3-6 fármacos/IgG.
[0405] Una sola dosis i.v. de > 300 mg [~ 10 μg de P2PDox] del conjugado fue letal, pero todos los animales toleraron 4 dosis de 45 μg administradas en 2 semanas. Usando este régimen de dosificación, examinamos el efecto terapéutico de hRS7-P2PDox en 2 modelos de xenoinjerto de tumor humano, Capan-1 (cáncer de páncreas) y NCI-N87 (cáncer gástrico). La terapia comenzó 7 días después del trasplante del tumor en ratones desnudos. En el modelo Capan-1 establecido de 7 días de edad, el 100 % de los tumores establecidos retrocedieron rápidamente, sin evidencia de nuevo crecimiento (no mostrado). Este resultado se reprodujo en un experimento repetido (no mostrado). Se hicieron hallazgos similares en el modelo NCI-N87 establecido (no mostrado), donde un segundo curso de terapia, administrado después del día 70, se toleró con seguridad y condujo a regresiones adicionales del tumor residual (no mostrado). El conjugado de hRS7-SN-38 internalizado, dirigido a Trop-2, proporcionó mejores respuestas terapéuticas que un conjugado de un anticuerpo anti-CEACAM5 de internalización deficiente, hMN-14 (no mostrado). Un ADC anti-CD20 no dirigido, hA20-P2PDox, fue ineficaz, lo que indica una eficacia terapéutica selectiva (no se muestra). Los datos de un xenoinjerto de cáncer de mama (MDA-MB-468) y un segundo xenoinjerto de cáncer de páncreas (no mostrado) reiteran la misma tendencia de los efectos antitumorales significativos y específicos del conjugado.
[0406] PK y toxicidad de hRS7-P2PDox con sustituciones de 6,8 o 3,7 fármaco/IgG: Se sabe que los conjugados de fármaco-anticuerpo (ADC) que contienen hasta 8 fármacos ultratóxicos/MAb se eliminan más rápido que los MAb no modificados y aumentan la toxicidad fuera del objetivo, un hallazgo que ha llevado a las tendencias actuales de utilizar sustituciones de fármacos de ≤ 4 (Hamblett et al., 2004, Clin Cáncer Res 10:7063-70). Los conjugados se prepararon y evaluaron con proporciones medias de sustitución de fármaco/MAb (MSR) de ~6:1 y ~3:1. Se administraron grupos de ratones normales (n = 5), i.v., dosis únicas de hRS7 o hRS7-P2PDox sin modificar con sustitución de fármaco de 6,8 o 3,7 (misma dosis de proteína), y se recogieron muestras de suero a los 30 min, 4 h, 24 h, 72 h y 168 h después de la inyección. Estos fueron analizados por ELISA para determinar la concentración de anticuerpos (no mostrado). No hubo diferencias significativas en las concentraciones séricas en varios momentos, lo que indica que estos se eliminaron de manera similar. Los parámetros farmacocinéticos (Cmax, AUC, etc.) fueron similares. Los conjugados con mayor o menor sustitución de fármaco tuvieron una tolerabilidad similar en ratones desnudos, cuando se administraron a la misma dosis de fármaco conjugado.
[0407] Eficacia terapéutica a la dosis mínima eficaz (MED): El conjugado de anticuerpo anti-TROP-2, hRS7-P2PDox, se evaluó en ratones desnudos que portaban xenoinjertos de cáncer gástrico humano NCI-N87 mediante la administración de una dosis única de proteína en bolo de 9 mg/kg, 6,75 mg/kg, 4,5 mg/kg, 2,25 mg/kg o 1 mg/kg. La terapia se inició cuando el volumen tumoral medio (mTV) fue de 0,256 cm3. El día 21, el mTV en el grupo de control de solución salina (grupo sin tratamiento) fue de 0,801 ± 0,181 cm3, que fue significativamente mayor que el de los ratones tratados con una dosis de 9, 6,75, 4,5 o 2,25 mg/kg con un mTv de 0,211 ± 0,042 cm3, 0,239 ± 0,0.054 cm3, 0,264 ± 0,087 cm3, y 0,567 ± 0,179 cm3, respectivamente (P<0,0047, prueba t de una cola). A partir de estos, se consideró que la dosis efectiva mínima era de 2,25 mg/kg, mientras que 9 mg/kg representaban m Td .
[0408] MTD del anticuerpo-P2PDox: un estudio de MTD que comparó los conjugados 2-PDox y P2PDox del anticuerpo prototipo, hLL1, en ratones demostró que el conjugado P2PDox era mucho más potente (no se muestra). La MTD de una única inyección iv estuvo entre 100 y 300 mg. A continuación, se determinó la MTD de inyecciones múltiples, en un programa de cada cuatro días para un total de cuatro inyecciones (q4dx4), usando dosis de proteína entre 25 μg y 150 μg por inyección. A estas dosis, se administró a los animales una dosis acumulativa de entre 100 y 600 μg. Tabla 17 a continuación resume los diversos grupos.
Tabla 17. Dosis y horario para MTD de anticuerpo-P2PDox

[0409] Sólo aquellos ratones tratados con 25 μg de P2PDox-ADC continúan sin mostrar signos de toxicidad (no mostrado). Esta es una dosis acumulativa de 100 μg que también fue la dosis tolerada cuando se administró como una inyección única (no se muestra). Por lo tanto, la MTD para inyecciones múltiples de un P2PDox-ADC en ratones es de 25 μg q4dx4 de este experimento. Un análisis más detallado de los datos y la repetición del experimento establecieron que la MTD para la dosificación fraccionada era de 45 μg de dosis de proteína del conjugado, administrada cada 4 días durante 2 semanas (programa de 45 μg, q4dx4).
[0410] Estudios de unión: no se observaron diferencias significativas en la unión del resto del anticuerpo a las células de carcinoma gástrico NCI-N87 entre hRS7 no conjugado y P2PDox-hRS7 conjugado con 6 moléculas de P2PDox por anticuerpo (no se muestra). La falta de efecto de la conjugación sobre la unión del anticuerpo al antígeno diana se confirmó para los conjugados P2PDox-hMN-15 (antiCEACAM6), P2PDox-hLL2 (anti-CD22) y P2PDox-hMN-24 (anti-CEACAM5). Se concluye que la conjugación de P2PDox a anticuerpos no afecta la actividad de unión antígenoanticuerpo.
[0411] Estudios de citotoxicidad: se examinó la citotoxicidad de los conjugados P2PDox-mAb para las células diana. hRS7-P2PDox y hMN-15-P2PDox fueron citotóxicos para las líneas celulares de tumor sólido MDA-MB-468, AG S, NCI-N87 y Capan-1 (no se muestra). hMN-14-P2PDox fue citotóxico para las líneas tumorales pancreáticas humanas Capan-1, BxPC-3 y AsPC-1 y las líneas tumorales gástricas y colónicas humanas AGS, NCI-N87 y LS147T (no mostrado). hLL2-P2PDOx fue citotóxico para las líneas tumorales hematopoyéticas Daudi, Raji, Ramos y JVM-3 (no mostrado). Los valores Cl50 para los conjugados estaban en el rango de concentración nanomolar (no mostrado).
EJEMPLO 35. Estudios in vivo adicionales
[0412] Se realizaron más estudios de eficacia in vivo en ratones desnudos implantados con xenoinjertos de cáncer gástrico humano NCI-N87 (no mostrado). Un ciclo de tratamiento con 4 x 45 μg de hRS7-P2PDox hizo retroceder rápidamente todos los tumores (no mostrado). Se inició un segundo ciclo de tratamiento aproximadamente 2 meses después del final del primer ciclo, lo que resultó en una regresión completa de todos menos uno de los animales tratados con hRS7-P2PDox. Los conjugados hA20, hLL1 y hMN-14 tuvieron poco efecto sobre la progresión tumoral (no mostrado). La administración de P2PDox-hMN-15 dio como resultado una regresión retardada del cáncer gástrico, que fue menos eficaz que el conjugado hRS7.
[0413] Se examinó el efecto de variar el programa de dosificación sobre la eficacia antitumoral (no mostrado). El experimento comenzó 9 días después de la implantación del tumor cuando el volumen tumoral medio para todos los grupos fue de 0,383 cm3 y finalizó el día 93 (84 días después del inicio de la terapia). En este estudio, una dosis única de 180 μg, dos dosis semanales de 90 μg y q4dx4 de 45 μg dieron como resultado una supervivencia significativamente mejorada (no se muestra). Para el control de solución salina, sobrevivieron 0 de 9 ratones (no se muestra). Para los ratones que recibieron 45 μg q4dx4 de hRS7-P2PDox, 8 de 9 ratones estaban vivos el día 94 (no se muestra). Para los ratones que recibieron 90 μg semanales x 2 de hRS7-P2PDox, 9 de 9 ratones estaban vivos el día 94 (no se muestra). Para los ratones que recibieron una dosis única de 180 μg de hRS7-P2PDox, 8 de 9 ratones estaban vivos el día 94 (no se muestra). Con el mismo programa de dosificación, el conjugado de hA20 de control no tuvo efecto sobre la supervivencia (no se muestra). Un estudio de toxicidad mostró que los tres programas de dosificación de hRS7-P2PDox dieron como resultado niveles de toxicidad igualmente bajos (no se muestra).
[0414] El conjugado hRS7-P2PDox también fue efectivo en el cáncer de páncreas Capan-1 (no mostrado) y fue más efectivo para inhibir el crecimiento tumoral que un conjugado hRS7-SN-38 (no mostrado). El conjugado hPAM4-P2PDox también fue más eficaz para inhibir el crecimiento del cáncer de páncreas humano Capan-1 que un conjugado hPAM4-SN-38 (no mostrado). A los 63 días después de la inyección del tumor Capan-1 (con la terapia comenzando 1 día después de la inoculación), 0 de 10 ratones estaban vivos en el control de solución salina, 10 de 10 ratones estaban vivos en los ratones tratados dos veces por semana x 2 semanas con 45 μg de hPAM4-P2PDox, 2 de 10 ratones estaban vivos en ratones tratados dos veces por semana x 2 semanas con 45 μg de hA20-P2PDox, 0 de 10 ratones estaban vivos en ratones tratados dos veces por semana x 4 semanas con 250 μg de hPAM4-SN-38, y 0 de 10 ratones estaban vivos en ratones tratados dos veces por semana x 4 semanas con 250 μg de h20-SN-38.
[0415] hRS7-P2PDox fue sustancialmente más eficaz que hRS7-SN-38 para inhibir el crecimiento del cáncer de páncreas PxPC-3 (no se muestra) y fue ligeramente más eficaz que hRS7-SN-38 para inhibir el crecimiento del cáncer de mama MDA-MB-468 (no se muestra).
[0416] Se examinó el efecto de diferentes dosis únicas de hRS7-P2PDox sobre el crecimiento de xenoinjertos de carcinoma gástrico NCI-N87. Usando una dosis única, el efecto máximo sobre el crecimiento tumoral se observó a 90 mg o más (no se muestra). Una dosis única de 45 mg fue el mínimo requerido para ver un beneficio de supervivencia significativo en comparación con el control de solución salina (no se muestra).
[0417] La actividad ADCC de varios conjugados hRS7-ADC se determinó en comparación con hRS7 IgG (no se muestra). Las PBMC se purificaron a partir de sangre adquirida en el Centro de Sangre de Nueva Jersey. Se usó una línea celular de adenocarcinoma de páncreas humano Trop-2-positiva (BxPC-3) como línea celular diana con una proporción de efector a diana de 100:1. La ADCC mediada por hRS7 IgG se comparó con hRS7-Pro-2-PDox, hRS7-CL2A-SN-38 y hRS7-NEM reducida y protegida. Todos se usaron a 33,3 nM. La actividad general fue baja, pero significativa (no se muestra). Hubo una lisis específica del 8,5% para hRS7 IgG que no fue significativamente diferente de hRS7-Pro-2-PDox. Ambos fueron significativamente mejores que el control hLL2 y hRS7-NEM y hRS7-SN-38 (P <0,02, prueba t de dos colas). No hubo diferencia entre hRS7-NEM y hRS7-SN-38.
Ejemplo 36. Tratamiento de linfoma no Hodgkin (LNH) con anti-CD22 P2PDox-epratuzumab más oliparib
[0418] Se prepara un ADC de P2PDox-epratuzumab como se describe anteriormente. Diecisiete pacientes con antecedentes no tratados o LNH recidivante reciben 4 dosis de 70, 100 o 150 mg de P2PDox-epratuzumab inyectados i.v. en los días 1 y 14 de un ciclo de 28 días. Oliparib a 200 mg al día se administra los días 1-7 y 14-21 del ciclo. Las respuestas se evalúan mediante tomografías computarizadas, con otras evaluaciones que incluyen eventos adversos, niveles sanguíneos de células B, niveles séricos de epratuzumab y títulos de antiepratuzumab humano (HAHA).
[0419] Solo se observan reacciones de inyección transitorias ocasionales, de leves a moderadas, y no se observan otros problemas de seguridad, excepto neutropenia hasta el Grado 3 (pero reversible después de interrumpir el tratamiento hasta que se reduce al Grado 1). Se observa un agotamiento transitorio de células B (hasta aproximadamente un 25 %) en todos los niveles de dosificación de P2PDox-epratuzumab en combinación con oliparib. La tasa de respuesta objetiva (respuestas parciales más respuestas completas más respuestas completas no confirmadas) es del 47 % (8/17) con una tasa de respuesta completa/respuesta completa no confirmada del 24 % (4/17). Cuatro de las ocho respuestas objetivas continúan durante 30 semanas o más. Se observan respuestas objetivas en todos los niveles de dosis de P2PDox-epratuzumab más oliparib. Todas las muestras de suero evaluadas para anticuerpos humanos anti-epratuzumab (HAHA) son negativas.
Ejemplo 37. Tratamiento de cáncer de mama trip le negativo con P2PDox-hRS7 más paclitaxel
[0420] El ADC anti-Trop-2 P2PDox-hRS7 se prepara como se describe anteriormente. Los pacientes con cáncer de mama triple negativo que han fracasado en al menos dos terapias estándar reciben 70 mg de P2PDox-hRS7 inyectados por vía i.v. el día 1 de un ciclo de 21 días, con paclitaxel administrado a 175 mg/m2 los días 1, 7 y 14 del ciclo. ciclo. Después de 3 ciclos, se observan respuestas objetivas, con una disminución promedio del volumen tumoral del 35%. Todas las muestras de suero evaluadas para anticuerpos humanos anti-hRS7 (HAHA) son negativas.
Ejemplo 38. Tratamiento de cáncer de colon metastásico con P2PDox-hMN-14 más olaparib
[0421] Un hombre de 52 años con cáncer de colon metastásico (3-5 cm de diámetro) en los lóbulos hepáticos izquierdo y derecho, así como una metástasis de 5 cm en el pulmón derecho y un valor elevado de CEA en sangre de 130 ng/mL, es tratados con la combinación de anti-CEACAM5 hMN-14-P2PDox más olaparib. Se administra una dosis de 150 mg de hMN-14-P2PDox los días 1 y 14 de un ciclo de 28 días. Oliparib se administra a 200 mg dos veces al día en los días 1-10 del ciclo. Tras la evaluación por TC después de 2 ciclos, se mide una reducción del 25 % de los diámetros medios totales de las 3 lesiones diana, lo que constituye una buena respuesta estable de la enfermedad según los criterios RECIST1.1. Al mismo tiempo, su título de CEA en sangre se reduce a 30 ng/mL. Los cursos repetidos de terapia continúan a medida que su neutropenia se normaliza.
Ejemplo 39. Almacenamiento de inmunoconjugados
[0422] Los conjugados de ADC se purificaron y se intercambió el tampón con ácido 2-(N-morfolino)etanosulfónico (MES), pH 6,5, y se formularon posteriormente con trehalosa (concentración final de 25 mM) y polisorbato 80 (concentración final de 0,01 % v/v), siendo la concentración de tampón final de 22,25 mM como resultado de la adición del excipiente. Los conjugados formulados se liofilizan y almacenan en viales sellados, con almacenamiento a 2 °C-8 °C. Los inmunoconjugados liofilizados son estables en las condiciones de almacenamiento y mantienen sus actividades fisiológicas.
Claims (13)
1. Un inmunoconjugado que se une a T rop-2 para su uso en un método para tratar el cáncer que expresa T rop-2, en el que el método comprende:
a) administrar el inmunoconjugado a un sujeto humano con cáncer; y
b) administrar al sujeto al menos un agente terapéutico seleccionado del grupo que consiste en un inhibidor de PARP y un inhibidor de microtúbulos
en el que el inmunoconjugado comprende un anticuerpo anti-Trop-2 conjugado con SN-38.
2. Al menos un agente terapéutico seleccionado del grupo que consiste en un inhibidor de PARP y un inhibidor de microtúbulos, para usar en un método para tratar el cáncer que expresa Trop-2, en el que el método comprende: a) administrar a un sujeto humano con el cáncer un inmunoconjugado que se une a Trop-2; y
b) administrar al sujeto al menos un agente terapéutico
en el que el inmunoconjugado comprende un anticuerpo anti-Trop-2 conjugado con SN-38.
3. El inmunoconjugado para uso de la reivindicación 1 y el al menos un agente terapéutico para uso de la reivindicación 2, donde la combinación de anticuerpo o inmunoconjugado y agente terapéutico tiene un efecto sinérgico en la inhibición del crecimiento tumoral.
4. El inmunoconjugado para el uso de la reivindicación 1 y el al menos un agente terapéutico para el uso de la reivindicación 2, en el que el anticuerpo anti-Trop-2 es hRS7.
5. El inmunoconjugado para uso de la reivindicación 1 y el al menos un agente terapéutico para uso de la reivindicación 2, donde el inhibidor de PARP se selecciona del grupo que consiste en olaparib, talazoparib (BMN-673), rucaparib, veliparib, CEP 9722, MK 4827, BGB-290, ABT-888, AG014699, BSI-201, CEP-8983 y 3-aminobenzamida; o el inhibidor de microtúbulos se selecciona del grupo que consiste en un alcaloide de la vinca, un taxano, un maitansinoide, una auristatina, vincristina, vinblastina, paclitaxel, mertansina, demecolcina, nocodazol, epotilona, docetaxel, disodermolida, colchicina, combrestatina, podofilotoxina, CI-980, fenilahistinas, esteganacinas, curacinas, 2-metoxiestradiol, E7010, metoxibencenosuflonamidas, vinorelbina, vinflunina, vindesina, dolastatinas, espongistatina, rizoxina, tasidotina, halicondrinas, hemiasterlinas, criptoficina 52, MMAE y mesilato de eribulina.
6. El inmunoconjugado para uso de la reivindicación 5 y el al menos un agente terapéutico para uso de la reivindicación 5, en el que el inhibidor de PARP es olaparib; o el inhibidor de microtúbulos es paclitaxel o mesilato de eribulina.
7. El inmunoconjugado para el uso de la reivindicación 1 y el al menos un agente terapéutico para el uso de la reivindicación 2, en el que el cáncer es un tumor sólido y el tratamiento da como resultado una reducción del tamaño del tumor de al menos un 15 %, al menos un 20 %, al menos el 30%, o al menos el 40%.
8. El inmunoconjugado para el uso de la reivindicación 1 y el al menos un agente terapéutico para el uso de la reivindicación 2, en el que el cáncer es metastásico y el método comprende además reducir el tamaño o eliminar las metástasis.
9. El inmunoconjugado para uso de la reivindicación 1 y el al menos un agente terapéutico para uso de la reivindicación 2, en el que el cáncer es refractario a otras terapias pero responde a la combinación de conjugado de anticuerpofármaco y agente terapéutico.
10. El inmunoconjugado para uso de la reivindicación 9 y el al menos un agente terapéutico para uso de la reivindicación 9, en el que el paciente no ha respondido a la terapia con irinotecan, antes del tratamiento con el conjugado de anticuerpo-fármaco.
11. El inmunoconjugado para el uso de la reivindicación 1 y el al menos un agente terapéutico para el uso de la reivindicación 2, en el que el método comprende además administrar al sujeto una o más modalidades terapéuticas adicionales seleccionadas del grupo que consiste en anticuerpos no conjugados, anticuerpos radiomarcados, anticuerpos conjugados por fármaco, anticuerpos conjugados con toxina, terapia génica, quimioterapia, péptidos terapéuticos, terapia con citocinas, oligonucleótidos, radioterapia localizada, cirugía y terapia con ARN de interferencia.
12. El inmunoconjugado para el uso de la reivindicación 11 y el al menos un agente terapéutico para el uso de la reivindicación 11, donde la modalidad terapéutica comprende el tratamiento con un agente seleccionado del grupo que consiste en 5-fluorouracilo, afatinib, aplidina, azaribina, anastrozol, antraciclinas, axitinib, AVL-101, AVL-291, bendamustina, bleomicina, bortezomib, bosutinib, briostatina-1, busulfán, caliqueamicina, camptotecina, carboplatino,
10-hidroxicamptotecina, carmustina, celebrex, clorambucilo, cisplatino (CDDP), inhibidores de la Cox-2, irinotecán (CPT-11), SN-38, carboplatino, cladribina, camptotecas, ciclofosfamida, crizotinib, citarabina, dacarbazina, dasatinib, dinaciclib, docetaxel, dactinomicina, daunorrubicina, doxorrubicina, 2-pirrolinodoxorrubicina (2P-DOX), cianomorfolino doxorrubicina, glucurónido de doxorrubicina, glucurónido de epirrubicina, erlotinib, estramustina, epidofilotoxina, erlotinib, entinostat, agentes de unión al receptor de estrógeno, etopósido (VP16), glucurónido de etopósido, fosfato de etopósido, exemestano, fingolimod, flavopiridol, floxuridina (FUdR), 3',5' -O-dioleoil-FudR (FUdR-dO), fludarabina, flutamida, inhibidores de la farnesil-proteína transferasa, fostamatinib, ganetespib, GDC-0834, GS-1101, gefitinib, gemcitabina, hidroxiurea, ibrutinib, idarrubicina, idelalisib, ifosfamida, imatinib, L-asparaginasa, lapatinib, lenolidamida, leucovorina, LFM-A13, lomustina, mecloretamina, melfalán, mercaptopurina, 6-mercaptopurina, metotrexato, mitoxantrona, mitramicina, mitomicina, mitotano, navelbina, neratinib, nilotinib, nitrosurea, olaparib, plicomicina, procarbazina, paclitaxel, PCI-32765, pentostatina, PSI-341, raloxifeno, semustina, sorafenib, estreptozocina, SU11248, sunitinib, tamoxifeno, temazolomida (una forma acuosa de DTIC), transplatino, talidomida, tioguanina, tiotepa, tenipósido, topotecán, uracilo mostaza, vatalanib, vinorelbina, vinblastina, vincristina, alcaloides de la vinca y ZD1839.
13. El inmunoconjugado para el uso de la reivindicación 1 y el al menos un agente terapéutico para el uso de la reivindicación 2, en el que el cáncer se selecciona del grupo que consiste en cáncer de mama triple negativo, cáncer de mama, cáncer de ovario, cáncer de endometrio cáncer, cáncer urotelial, cáncer uterino, cáncer de pulmón no microcítico, cáncer de pulmón microcítico, cáncer de estómago, cáncer de vejiga urinaria, cáncer renal, próstata cáncer y cáncer colorrectal.
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201562184331P | 2015-06-25 | 2015-06-25 | |
| US201562201361P | 2015-08-05 | 2015-08-05 | |
| US201562250715P | 2015-11-04 | 2015-11-04 | |
| US201562263134P | 2015-12-04 | 2015-12-04 | |
| US15/069,208 US10137196B2 (en) | 2012-12-13 | 2016-03-14 | Dosages of immunoconjugates of antibodies and SN-38 for improved efficacy and decreased toxicity |
| PCT/US2016/038986 WO2016210108A1 (en) | 2015-06-25 | 2016-06-23 | Combining anti-hla-dr or anti-trop-2 antibodies with microtubule inhibitors, parp inhibitors, bruton kinase inhibitors or phosphoinositide 3-kinase inhibitors significantly improves therapeutic outcome in cancer |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2953441T3 true ES2953441T3 (es) | 2023-11-13 |
Family
ID=57586368
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES16815285T Active ES2953441T3 (es) | 2015-06-25 | 2016-06-23 | Combinación de anticuerpos anti-hla-dr o anti-Trop-2 con inhibidores de microtúbulos, inhibidores de parp, inhibidores de la cinasa de bruton o inhibidores de la fosfoinositida 3-cinasa mejora significativamente el resultado terapéutico en el cáncer |
Country Status (11)
| Country | Link |
|---|---|
| EP (2) | EP4257134A3 (es) |
| JP (2) | JP6980980B2 (es) |
| CN (3) | CN114796501A (es) |
| AU (1) | AU2016281622C1 (es) |
| CA (1) | CA2983597A1 (es) |
| ES (1) | ES2953441T3 (es) |
| IL (1) | IL256150B (es) |
| PL (1) | PL3313443T3 (es) |
| PT (1) | PT3313443T (es) |
| SI (1) | SI3313443T1 (es) |
| WO (1) | WO2016210108A1 (es) |
Families Citing this family (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10058621B2 (en) | 2015-06-25 | 2018-08-28 | Immunomedics, Inc. | Combination therapy with anti-HLA-DR antibodies and kinase inhibitors in hematopoietic cancers |
| DK2694075T3 (en) | 2011-04-01 | 2016-08-01 | Curis Inc | Phosphoinositide 3-kinase inhibitor with zinc-binding moiety |
| US10478494B2 (en) | 2015-04-03 | 2019-11-19 | Astex Therapeutics Ltd | FGFR/PD-1 combination therapy for the treatment of cancer |
| CN114796501A (zh) * | 2015-06-25 | 2022-07-29 | 免疫医疗公司 | 抗体与治疗剂的组合治疗癌症的方法 |
| WO2017180565A1 (en) * | 2016-04-14 | 2017-10-19 | Immunomedics, Inc. | Combination therapy with anti-hla-dr antibodies and kinase inhibitors in hematopoietic cancers |
| PL3585442T3 (pl) * | 2017-02-24 | 2024-03-11 | Immunomedics, Inc. | Terapia drobnokomórkowego raka płuca (sclc) koniugatem przeciwciało-lek (adc) hamującym topoizomerazę i celującym w trop-2 |
| CN110392570A (zh) * | 2017-03-27 | 2019-10-29 | 免疫医疗公司 | 用沙妥珠单抗格维替康和rad51抑制剂治疗表达trop-2的三阴性乳腺癌 |
| WO2018187074A1 (en) * | 2017-04-03 | 2018-10-11 | Immunomedics, Inc. | Subcutaneous administration of antibody-drug conjugates for cancer therapy |
| BR112019023591A2 (pt) * | 2017-05-09 | 2020-05-26 | Tesaro, Inc. | Terapias de combinação para tratar câncer |
| JP2020520921A (ja) | 2017-05-18 | 2020-07-16 | テサロ, インコーポレイテッド | 癌を処置する併用療法 |
| EP3641782A4 (en) | 2017-06-20 | 2021-03-24 | Madrigal Pharmaceuticals, Inc. | COMBINED THERAPIES INCLUDING TARGETED THERAPEUTICS |
| CN107446050A (zh) * | 2017-08-11 | 2017-12-08 | 百奥泰生物科技(广州)有限公司 | Trop2阳性疾病治疗的化合物及方法 |
| MX2020003770A (es) | 2017-09-30 | 2020-07-29 | Tesaro Inc | Terapias de combinacion para tratar cancer. |
| MX2020003799A (es) | 2017-10-06 | 2020-11-06 | Tesaro Inc | Terapias de combinacion y usos de las mismas. |
| KR20200117992A (ko) * | 2017-12-11 | 2020-10-14 | 트리페이스 리서치 앤드 디벨로프먼트 쓰리 코포레이션 | 항-cd22 항체-메이탄신 컨쥬게이트, 그것의 조합, 및 사용 방법 |
| AU2019279206A1 (en) | 2018-05-31 | 2020-12-03 | Daiichi Sankyo Company, Limited | Anti-human TLR7 antibody |
| CN108743947B (zh) * | 2018-07-04 | 2020-12-15 | 复旦大学附属肿瘤医院 | 一种治疗b细胞淋巴瘤的药物组合物 |
| JP7458981B2 (ja) | 2018-08-06 | 2024-04-01 | 第一三共株式会社 | 抗体-薬物コンジュゲートとチューブリン阻害剤の組み合わせ |
| AU2019340376A1 (en) * | 2018-09-11 | 2021-04-08 | Curis Inc. | Combination therapy with a phosphoinositide 3-kinase inhibitor with a zinc binding moiety |
| WO2020094670A1 (en) * | 2018-11-05 | 2020-05-14 | Synaffix B.V. | Antibody-conjugates for targeting of tumours expressing trop-2 |
| CN109985238A (zh) * | 2019-04-16 | 2019-07-09 | 中山大学 | 一种基于免疫检查点阻断的抗肿瘤药物组合物及其应用 |
| GB202011993D0 (en) | 2020-07-31 | 2020-09-16 | Adc Therapeutics Sa | ANTI-IL 13Ra2 antibodies |
| EP4291238A1 (en) * | 2021-02-10 | 2023-12-20 | Great Novel Therapeutics Biotech & Medicals Corporation | A pharmaceutical combination and method for overcoming immune suppression or stimulating immune response against cancer |
| WO2023175483A1 (en) * | 2022-03-16 | 2023-09-21 | Astrazeneca Uk Limited | A scoring method for an anti-trop2 antibody‑drug conjugate therapy |
| CN117122680B (zh) * | 2023-06-12 | 2024-04-16 | 上海市肿瘤研究所 | 一种可有效抑制bap1失活突变型葡萄膜黑色素瘤脏器转移的靶向抑制剂 |
Family Cites Families (222)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4036945A (en) | 1976-05-03 | 1977-07-19 | The Massachusetts General Hospital | Composition and method for determining the size and location of myocardial infarcts |
| US4331647A (en) | 1980-03-03 | 1982-05-25 | Goldenberg Milton David | Tumor localization and therapy with labeled antibody fragments specific to tumor-associated markers |
| US4554101A (en) | 1981-01-09 | 1985-11-19 | New York Blood Center, Inc. | Identification and preparation of epitopes on antigens and allergens on the basis of hydrophilicity |
| US5776093A (en) | 1985-07-05 | 1998-07-07 | Immunomedics, Inc. | Method for imaging and treating organs and tissues |
| US5525338A (en) | 1992-08-21 | 1996-06-11 | Immunomedics, Inc. | Detection and therapy of lesions with biotin/avidin conjugates |
| US6861511B1 (en) | 1986-06-04 | 2005-03-01 | Bayer Corporation | Detection and quantification of neu related proteins in the biological fluids of humans |
| US4946778A (en) | 1987-09-21 | 1990-08-07 | Genex Corporation | Single polypeptide chain binding molecules |
| US5567610A (en) | 1986-09-04 | 1996-10-22 | Bioinvent International Ab | Method of producing human monoclonal antibodies and kit therefor |
| US6893625B1 (en) | 1986-10-27 | 2005-05-17 | Royalty Pharma Finance Trust | Chimeric antibody with specificity to human B cell surface antigen |
| IL85035A0 (en) | 1987-01-08 | 1988-06-30 | Int Genetic Eng | Polynucleotide molecule,a chimeric antibody with specificity for human b cell surface antigen,a process for the preparation and methods utilizing the same |
| US5132405A (en) | 1987-05-21 | 1992-07-21 | Creative Biomolecules, Inc. | Biosynthetic antibody binding sites |
| US5831034A (en) | 1987-11-13 | 1998-11-03 | Hermann Katinger | Human monoclonal anti-HIV-I-antibodies |
| US6511665B1 (en) | 1987-11-25 | 2003-01-28 | Immunex Corporation | Antibodies to interleukin-1 receptors |
| US5223409A (en) | 1988-09-02 | 1993-06-29 | Protein Engineering Corp. | Directed evolution of novel binding proteins |
| US6362325B1 (en) | 1988-11-07 | 2002-03-26 | Advanced Research And Technology Institute, Inc. | Murine 4-1BB gene |
| US6352694B1 (en) | 1994-06-03 | 2002-03-05 | Genetics Institute, Inc. | Methods for inducing a population of T cells to proliferate using agents which recognize TCR/CD3 and ligands which stimulate an accessory molecule on the surface of the T cells |
| US5571714A (en) | 1988-12-22 | 1996-11-05 | Celtrix Pharmaceuticals, Inc. | Monoclonal antibodies which bind both transforming growth factors β1 and β2 and methods of use |
| US6432402B1 (en) | 1989-05-25 | 2002-08-13 | Sloan-Kettering Institute For Cancer Research | Anti-idiotypic antibody which induces an immune response against a glycosphingolipid and use thereof |
| DK0432249T3 (da) | 1989-06-02 | 1997-02-17 | Univ Johns Hopkins Med | Monoklonale antistoffer over for beta-kæden i leukocytadhæsionsreceptoren, fremgangsmåder til fremstilling af disse antistoffer og anvendelse heraf |
| EP0438803B1 (en) | 1990-01-26 | 1997-03-12 | Immunomedics, Inc. | Vaccines against cancer and infectious diseases |
| IL97459A0 (en) | 1990-03-09 | 1992-06-21 | Hybritech Inc | Trifunctional antibody-like compounds as a combined diagnostic and therapeutic agent |
| US7041293B1 (en) | 1990-04-03 | 2006-05-09 | Genentech, Inc. | HIV env antibodies |
| US5229275A (en) | 1990-04-26 | 1993-07-20 | Akzo N.V. | In-vitro method for producing antigen-specific human monoclonal antibodies |
| US5252296A (en) | 1990-05-15 | 1993-10-12 | Chiron Corporation | Method and apparatus for biopolymer synthesis |
| US5496938A (en) | 1990-06-11 | 1996-03-05 | Nexstar Pharmaceuticals, Inc. | Nucleic acid ligands to HIV-RT and HIV-1 rev |
| KR970002255B1 (ko) | 1990-06-11 | 1997-02-26 | 넥스스타 파아마슈티컬드, 인크. | 핵산 리간드 |
| US5637459A (en) | 1990-06-11 | 1997-06-10 | Nexstar Pharmaceuticals, Inc. | Systematic evolution of ligands by exponential enrichment: chimeric selex |
| US5270163A (en) | 1990-06-11 | 1993-12-14 | University Research Corporation | Methods for identifying nucleic acid ligands |
| US5582981A (en) | 1991-08-14 | 1996-12-10 | Gilead Sciences, Inc. | Method for identifying an oligonucleotide aptamer specific for a target |
| US6764681B2 (en) | 1991-10-07 | 2004-07-20 | Biogen, Inc. | Method of prophylaxis or treatment of antigen presenting cell driven skin conditions using inhibitors of the CD2/LFA-3 interaction |
| US5854416A (en) | 1991-11-14 | 1998-12-29 | The United States Of America As Represented By The Department Of Health And Human Services | Streptococcus pneumoniae 37-KDA surface adhesin a protein and nucleic acids coding therefor |
| US5474771A (en) | 1991-11-15 | 1995-12-12 | The Trustees Of Columbia University In The City Of New York | Murine monoclonal antibody (5c8) recognizes a human glycoprotein on the surface of T-lymphocytes, compositions containing same |
| AU3657693A (en) | 1992-01-29 | 1993-09-01 | Regents Of The University Of California, The | Carcinoma associated antigen (SK1) monoclonal antibodies against SK1, methods of producing these antibodies and use therfor |
| CA2131692A1 (en) | 1992-03-09 | 1993-09-16 | Sybille Muller | An anti-idiotypic antibody and its use in diagnosis and therapy in hiv-related disease |
| US6129914A (en) | 1992-03-27 | 2000-10-10 | Protein Design Labs, Inc. | Bispecific antibody effective to treat B-cell lymphoma and cell line |
| JP3353209B2 (ja) | 1992-04-03 | 2002-12-03 | ジェネンテク,インコーポレイテッド | αvβ3インテグリンに対する抗体 |
| US6096289A (en) | 1992-05-06 | 2000-08-01 | Immunomedics, Inc. | Intraoperative, intravascular, and endoscopic tumor and lesion detection, biopsy and therapy |
| EP0643583B1 (en) | 1992-05-06 | 2000-07-26 | Immunomedics, Inc. | Intraoperative, intravascular and endoscopic tumor and lesion detection and therapy |
| WO1993023556A1 (en) | 1992-05-08 | 1993-11-25 | Genentech, Inc. | Antibodies to leukemia inhibitory factor |
| US5686072A (en) | 1992-06-17 | 1997-11-11 | Board Of Regents, The University Of Texas | Epitope-specific monoclonal antibodies and immunotoxins and uses thereof |
| US5397703A (en) | 1992-07-09 | 1995-03-14 | Cetus Oncology Corporation | Method for generation of antibodies to cell surface molecules |
| EP1005870B1 (en) | 1992-11-13 | 2009-01-21 | Biogen Idec Inc. | Therapeutic application of chimeric antibodies to human B lymphocyte restricted differentiation antigen for treatment of B cell lymphoma |
| IL109666A0 (en) | 1993-05-17 | 1994-08-26 | Immunomedics Inc | A targeting composition containing a biotin- or avidinprotein conjugate and methods for the use thereof |
| AU695124B2 (en) | 1993-05-28 | 1998-08-06 | Scripps Research Institute, The | Methods and compositions for inhibiting CD14 mediated cell activation |
| US6084067A (en) | 1993-07-26 | 2000-07-04 | Dana-Farber Cancer Institute | CTLA4/CD28 ligands and uses therefor |
| UA40577C2 (uk) | 1993-08-02 | 2001-08-15 | Мерк Патент Гмбх | Біспецифічна молекула, що використовується для лізису пухлинних клітин, спосіб її одержання, моноклональне антитіло (варіанти), фармацевтичний препарат, фармацевтичний набір (варіанти), спосіб видалення пухлинних клітин |
| AU7831594A (en) | 1993-09-09 | 1995-03-27 | Duke University | A method of promoting cellular function |
| WO1995014040A1 (en) | 1993-11-19 | 1995-05-26 | Baylor College Of Medicine | Monoclonal antibodies specific for human interleukin-5 |
| US5492807A (en) | 1993-11-19 | 1996-02-20 | Santi; Daniel V. | Method of obtaining diagnostic reagents, assays and therapeutics based on clinical manifestations of a disease |
| US5443953A (en) | 1993-12-08 | 1995-08-22 | Immunomedics, Inc. | Preparation and use of immunoconjugates |
| US6432404B1 (en) | 1993-12-23 | 2002-08-13 | Icos Corporation | Methods of inhibiting locomotor damage following spinal cord injury with α D-specific antibodies |
| US5922572A (en) | 1994-01-25 | 1999-07-13 | Human Genome Sciences, Inc. | Polynucleotides encoding haemopoietic maturation factor |
| US6074642A (en) | 1994-05-02 | 2000-06-13 | Alexion Pharmaceuticals, Inc. | Use of antibodies specific to human complement component C5 for the treatment of glomerulonephritis |
| US6100389A (en) | 1995-04-21 | 2000-08-08 | Human Genome Sciences, Inc. | Polynucleotides encoding a human chemotactic protein |
| US5798100A (en) | 1994-07-06 | 1998-08-25 | Immunomedics, Inc. | Multi-stage cascade boosting vaccine |
| US6303769B1 (en) | 1994-07-08 | 2001-10-16 | Immunex Corporation | Lerk-5 dna |
| ES2251723T3 (es) | 1994-08-12 | 2006-05-01 | Immunomedics, Inc. | Inmunoconjugados y anticuerpos humanizados especificos para linfoma de celulas b y celulas de leucemia. |
| US5587459A (en) | 1994-08-19 | 1996-12-24 | Regents Of The University Of Minnesota | Immunoconjugates comprising tyrosine kinase inhibitors |
| US6458349B1 (en) | 1995-06-02 | 2002-10-01 | Human Genome Sciences, Inc. | Chemokine β-4 polypeptides |
| US6174995B1 (en) | 1994-08-23 | 2001-01-16 | Haodong Li | Human chemokines, CKβ4 and CKβ10/MCP-4 |
| US6709653B1 (en) | 1994-09-16 | 2004-03-23 | Human Genome Sciences, Inc. | Antibodies specific for human inositol monophosphatase H1 |
| US5874540A (en) | 1994-10-05 | 1999-02-23 | Immunomedics, Inc. | CDR-grafted type III anti-CEA humanized mouse monoclonal antibodies |
| US5750370A (en) | 1995-06-06 | 1998-05-12 | Human Genome Sciences, Inc. | Nucleic acid encoding human endothlein-bombesin receptor and method of producing the receptor |
| US6538121B1 (en) | 1994-11-01 | 2003-03-25 | Human Genome Sciences, Inc. | Interleukin-1 β converting enzyme like apoptosis protease-3 and 4 |
| US5677136A (en) | 1994-11-14 | 1997-10-14 | Systemix, Inc. | Methods of obtaining compositions enriched for hematopoietic stem cells, compositions derived therefrom and methods of use thereof |
| US6521227B1 (en) | 1999-11-18 | 2003-02-18 | Peter L. Hudson | Polynucleotides encoding prostatic growth factor and process for producing prostatic growth factor polypeptides |
| US6537764B1 (en) | 1995-01-19 | 2003-03-25 | Children's Medical Center Corporation | Method of identifying inhibitors of C—C chemokine receptor 3 |
| US5786204A (en) | 1995-01-20 | 1998-07-28 | Human Genome Sciences, Inc. | Human prostatic specific reductase |
| US6312691B1 (en) | 1996-01-26 | 2001-11-06 | Jeffrey L. Browning | Lymphotoxin-α/β complexes and anti-lympotoxin-β receptor antibodies as anti-tumor agents |
| US6949244B1 (en) | 1995-12-20 | 2005-09-27 | The Board Of Trustees Of The University Of Kentucky | Murine monoclonal anti-idiotype antibody 11D10 and methods of use thereof |
| US5783404A (en) | 1995-04-13 | 1998-07-21 | Amgen Inc. | Methods and compositions for determining HER-2/neu expression using monoclonal antibodies |
| DE69527089T2 (de) | 1995-04-19 | 2003-01-02 | Polymun Scient Immunbio Forsch | Monoklonale antikörper gegen hiv-1 und davon hergestellte impfstoffe |
| US5686292A (en) | 1995-06-02 | 1997-11-11 | Genentech, Inc. | Hepatocyte growth factor receptor antagonist antibodies and uses thereof |
| US5773292A (en) | 1995-06-05 | 1998-06-30 | Cornell University | Antibodies binding portions, and probes recognizing an antigen of prostate epithelial cells but not antigens circulating in the blood |
| US6605441B1 (en) | 1995-06-05 | 2003-08-12 | Human Genome Sciences, Inc. | Antibodies against fibroblast growth factor 11 |
| US5773252A (en) | 1995-06-05 | 1998-06-30 | Human Genome Sciences, Inc. | Fibroblast growth factor 15 |
| WO1996040875A1 (en) | 1995-06-07 | 1996-12-19 | Novartis Ag | Methods for obtaining compositions enriched for hematopoietic stem cells and antibodies for use therein |
| US5622699A (en) | 1995-09-11 | 1997-04-22 | La Jolla Cancer Research Foundation | Method of identifying molecules that home to a selected organ in vivo |
| US6068829A (en) | 1995-09-11 | 2000-05-30 | The Burnham Institute | Method of identifying molecules that home to a selected organ in vivo |
| US6340459B1 (en) | 1995-12-01 | 2002-01-22 | The Trustees Of Columbia University In The City Of New York | Therapeutic applications for the anti-T-BAM (CD40-L) monoclonal antibody 5C8 in the treatment of reperfusion injury in non-transplant recipients |
| US5783186A (en) | 1995-12-05 | 1998-07-21 | Amgen Inc. | Antibody-induced apoptosis |
| US5945309A (en) | 1996-03-19 | 1999-08-31 | Human Genome Sciences, Inc. | Cytostatin III nucleic acids encoding |
| EP0939653B1 (en) | 1996-03-20 | 2005-08-31 | Immunomedics, Inc. | Humanization of an anti-carcinoembryonic antigen anti-idiotype antibody and use as a tumor vaccine and for targeting applications |
| EP0888125B1 (en) | 1996-03-20 | 2004-05-26 | Immunomedics, Inc. | GLYCOSYLATED IgG ANTIBODIES |
| FR2746398B1 (fr) | 1996-03-21 | 1998-04-30 | Bio Merieux | Anticorps specifique de staphylococcus aureaus, et utilisations |
| US6174992B1 (en) | 1997-03-21 | 2001-01-16 | Human Genome Sciences, Inc. | Human endometrial specific steroid-binding factor I, II and III |
| US6962981B1 (en) | 1996-03-25 | 2005-11-08 | Medarex, Inc. | Monoclonal antibodies specific for the extracellular domain of prostate-specific membrane antigen |
| US6004780A (en) | 1996-03-26 | 1999-12-21 | Human Genome Sciences, Inc. | Growth factor HTTER36 |
| US6110463A (en) | 1996-03-29 | 2000-08-29 | North Carolina State University | Anti-Cryptosporidium parvum preparations |
| US6066617A (en) | 1996-04-03 | 2000-05-23 | Human Genome Sciences, Inc. | Human cystatin F |
| JP2000510119A (ja) | 1996-05-03 | 2000-08-08 | イムノメディクス,インコーポレイテッド | ガンに対する標的コンビネーション免疫療法 |
| US6107090A (en) | 1996-05-06 | 2000-08-22 | Cornell Research Foundation, Inc. | Treatment and diagnosis of prostate cancer with antibodies to extracellur PSMA domains |
| US6136311A (en) | 1996-05-06 | 2000-10-24 | Cornell Research Foundation, Inc. | Treatment and diagnosis of cancer |
| US6764688B2 (en) | 1996-09-03 | 2004-07-20 | Kaneka Corporation | Method for inducing immunosuppressive cells and a culture device to be used therefor |
| DE69731977T2 (de) | 1996-10-09 | 2005-12-08 | Canterbury Health Ltd. | Spezifische antikörper für dendritische zellen |
| US6653104B2 (en) | 1996-10-17 | 2003-11-25 | Immunomedics, Inc. | Immunotoxins, comprising an internalizing antibody, directed against malignant and normal cells |
| ES2191827T3 (es) | 1996-10-17 | 2003-09-16 | Immunomedics Inc | Conjugado de toxina no antigenica y proteina de fusion de un sistema receptor de penetracion intracelular. |
| US6812327B1 (en) | 1996-10-25 | 2004-11-02 | Human Genome Sciences, Inc. | Neutrokine-alpha polypeptides |
| US6528625B1 (en) | 1996-10-28 | 2003-03-04 | Millennium Pharmaceuticals, Inc. | Anti-CCR5 antibodies and kits comprising same |
| WO1998021334A2 (en) | 1996-11-13 | 1998-05-22 | Morphogenesis, Inc. | Antibody mg1 recognizing a small subset of human hematopoietic cells |
| US6455040B1 (en) | 1997-01-14 | 2002-09-24 | Human Genome Sciences, Inc. | Tumor necrosis factor receptor 5 |
| US6605699B1 (en) | 1997-01-21 | 2003-08-12 | Human Genome Sciences, Inc. | Galectin-11 polypeptides |
| US6046031A (en) | 1997-01-21 | 2000-04-04 | Human Genome Sciences, Inc. | Metalloproteinases |
| JP4450870B2 (ja) | 1997-01-28 | 2010-04-14 | ヒューマン ジノーム サイエンシーズ, インコーポレイテッド | 死ドメイン含有レセプター4(dr4:死レセプター4)、tnf−レセプタースーパーファミリーのメンバーおよびtrailへの結合 |
| WO1998043089A1 (en) | 1997-03-03 | 1998-10-01 | Bristol-Myers Squibb Company | Monoclonal antibodies to human cd6 |
| US6951924B2 (en) | 1997-03-14 | 2005-10-04 | Human Genome Sciences, Inc. | Antibodies against secreted protein HTEBYII |
| US6919433B2 (en) | 1997-03-14 | 2005-07-19 | Human Genome Sciences, Inc. | Antibodies to protein HPMBQ91 |
| US6872568B1 (en) | 1997-03-17 | 2005-03-29 | Human Genome Sciences, Inc. | Death domain containing receptor 5 antibodies |
| US6306393B1 (en) | 1997-03-24 | 2001-10-23 | Immunomedics, Inc. | Immunotherapy of B-cell malignancies using anti-CD22 antibodies |
| US6183744B1 (en) | 1997-03-24 | 2001-02-06 | Immunomedics, Inc. | Immunotherapy of B-cell malignancies using anti-CD22 antibodies |
| EP0975674B1 (en) | 1997-05-02 | 2005-08-17 | THE GOVERNMENT OF THE UNITED STATES OF AMERICA, as represented by the Secretary, Department of Health and Human Services | Immunotoxins, comprising an onc protein, directed against malignant cells |
| US6284474B1 (en) | 1998-04-23 | 2001-09-04 | The Regents Of The University Of California | Detection and diagnosis of conditions associated with lung injury |
| US6372217B1 (en) | 1997-06-03 | 2002-04-16 | Regents Of The University Of Minnesota | Methods for the treatment of CD7+ viral infection with TXU-7-PAP |
| US6545130B2 (en) | 1997-06-04 | 2003-04-08 | Albert Einstein College Of Medicine Of Yeshiva University | Monoclonal antibodies to mycobacterium tuberculosis and a modified ELISA assay |
| US6358508B1 (en) | 1997-06-11 | 2002-03-19 | Human Genome Sciences, Inc. | Antibodies to human tumor necrosis factor receptor TR9 |
| JP4303883B2 (ja) | 1997-09-18 | 2009-07-29 | ジェネンテック・インコーポレーテッド | DcR3ポリペプチドというTNFR相同体 |
| US6689607B2 (en) | 1997-10-21 | 2004-02-10 | Human Genome Sciences, Inc. | Human tumor, necrosis factor receptor-like proteins TR11, TR11SV1 and TR11SV2 |
| US6355244B1 (en) | 1997-11-17 | 2002-03-12 | University Of Kentucky Research Foundation | Methods and compositions for the treatment of psoriasis |
| US6610833B1 (en) | 1997-11-24 | 2003-08-26 | The Institute For Human Genetics And Biochemistry | Monoclonal human natural antibodies |
| AU1682699A (en) | 1997-12-25 | 1999-07-19 | Japan Tobacco Inc. | Monoclonal antibody against connective tissue growth factor and medicinal uses thereof |
| US6956107B2 (en) | 1998-02-20 | 2005-10-18 | Tanox, Inc. | Inhibitors of complement activation |
| US6861227B2 (en) | 1998-03-19 | 2005-03-01 | Human Genome Sciences, Inc. | Antibodies to cytokine receptor common gamma chain like |
| US6200765B1 (en) | 1998-05-04 | 2001-03-13 | Pacific Northwest Cancer Foundation | Non-invasive methods to detect prostate cancer |
| AU742626B2 (en) | 1998-05-20 | 2002-01-10 | Immunomedics Inc. | Therapeutics using a bispecific anti-HLA class II invariant chain X anti-pathogen antibody |
| US6881405B2 (en) | 1998-06-15 | 2005-04-19 | Altarex Medical Corp. | Reagents and methods for inducing an immune response to prostate specific antigen |
| US7387772B1 (en) | 1999-06-22 | 2008-06-17 | Immunimedics, Inc. | Chimeric, human and humanized anti-CSAP monoclonal antibodies |
| US7138103B2 (en) | 1998-06-22 | 2006-11-21 | Immunomedics, Inc. | Use of bi-specific antibodies for pre-targeting diagnosis and therapy |
| US7405320B2 (en) | 1998-06-22 | 2008-07-29 | Immunomedics, Inc. | Therapeutic and diagnostic conjugates for use with multispecific antibodies |
| US6528269B1 (en) | 1998-06-22 | 2003-03-04 | Case Western Reserve University | Immunological agents specific for prion protein (PRP) |
| US6962702B2 (en) | 1998-06-22 | 2005-11-08 | Immunomedics Inc. | Production and use of novel peptide-based agents for use with bi-specific antibodies |
| US6312689B1 (en) | 1998-07-23 | 2001-11-06 | Millennium Pharmaceuticals, Inc. | Anti-CCR2 antibodies and methods of use therefor |
| AUPP525198A0 (en) | 1998-08-13 | 1998-09-03 | Medvet Science Pty. Ltd. | Monoclonal antibody inhibitor of GM-CSF, IL-3 and IL-5 and other cytokines and uses thereof |
| US6572856B1 (en) | 1998-09-10 | 2003-06-03 | The University Of Virginia Patent Foundation | Methods for the prevention and treatment of cancer using anti-C3b(i) antibodies |
| US6818749B1 (en) | 1998-10-31 | 2004-11-16 | The United States Of America As Represented By The Department Of Health And Human Services | Variants of humanized anti carcinoma monoclonal antibody cc49 |
| EP1127170A4 (en) | 1998-11-05 | 2005-04-27 | Univ Leland Stanford Junior | HUMAN MONOCLONAL ANTIBODIES TO HUMAN PAN-HEPATITIS C VIRUS |
| DK1140175T3 (da) | 1998-12-21 | 2006-08-14 | Ludwig Inst Cancer Res | Antistoffer mod trunkeret VEGF-D og anvendelser deraf |
| EE05627B1 (et) | 1998-12-23 | 2013-02-15 | Pfizer Inc. | CTLA-4 vastased inimese monoklonaalsed antikehad |
| WO2000039327A1 (en) | 1998-12-23 | 2000-07-06 | Human Genome Sciences, Inc. | Peptidoglycan recognition proteins |
| US6488930B1 (en) | 1999-01-15 | 2002-12-03 | Millennium Pharmaceuticals, Inc. | Anti-CCR4 antibodies and methods of use therefor |
| ATE269357T1 (de) | 1999-04-28 | 2004-07-15 | Univ Texas | Zusammensetzungen und verfahren zur krebsbehandlung durch die selektive hemmung von vegf |
| US7666400B2 (en) | 2005-04-06 | 2010-02-23 | Ibc Pharmaceuticals, Inc. | PEGylation by the dock and lock (DNL) technique |
| US7527787B2 (en) | 2005-10-19 | 2009-05-05 | Ibc Pharmaceuticals, Inc. | Multivalent immunoglobulin-based bioactive assemblies |
| US7829064B2 (en) | 1999-05-10 | 2010-11-09 | Immunomedics, Inc. | Anti-CD74 immunoconjugates and methods |
| US7550143B2 (en) | 2005-04-06 | 2009-06-23 | Ibc Pharmaceuticals, Inc. | Methods for generating stably linked complexes composed of homodimers, homotetramers or dimers of dimers and uses |
| US7534866B2 (en) | 2005-10-19 | 2009-05-19 | Ibc Pharmaceuticals, Inc. | Methods and compositions for generating bioactive assemblies of increased complexity and uses |
| US6946129B1 (en) | 1999-06-08 | 2005-09-20 | Seattle Genetics, Inc. | Recombinant anti-CD40 antibody and uses thereof |
| EP1194167B1 (en) | 1999-06-09 | 2009-08-19 | Immunomedics, Inc. | Immunotherapy of autoimmune disorders using antibodies which target b-cells |
| US20020002270A1 (en) | 1999-06-16 | 2002-01-03 | Raymond P. Zinkowski | Purified antigen for alzheimer's disease, and methods of obtaining and using same |
| US6355481B1 (en) | 1999-06-18 | 2002-03-12 | Emory University | Hybridoma cell line and monoclonal antibody for huntingtin protein |
| US6964854B1 (en) | 1999-07-13 | 2005-11-15 | Science & Technology Corporation | Compositions and methods useful for the diagnosis and treatment of heparin induced thrombocytopenia/thrombosis |
| US6693176B1 (en) | 1999-07-23 | 2004-02-17 | University Of Massachusetts | Antitumor antibodies, proteins, and uses thereof |
| US6376654B1 (en) | 1999-08-13 | 2002-04-23 | Molecular Discoveries, Llc | Myeloma cell and ovarian cancer cell surface glycoproteins, antibodies thereto, and uses thereof |
| US6716966B1 (en) | 1999-08-18 | 2004-04-06 | Altarex Corp. | Therapeutic binding agents against MUC-1 antigen and methods for their use |
| WO2001016183A1 (en) | 1999-08-30 | 2001-03-08 | U.S. Army Medical Research Institute Of Infectious Diseases | Monoclonal antibodies and vaccines against epitopes on the ebola virus glycoprotein |
| US6824780B1 (en) | 1999-10-29 | 2004-11-30 | Genentech, Inc. | Anti-tumor antibody compositions and methods of use |
| US20030031670A1 (en) | 1999-11-08 | 2003-02-13 | Jack R. Wands | Diagnosis and treatment of malignant neoplasms |
| US6835370B2 (en) | 1999-11-08 | 2004-12-28 | Rhode Island Hospital | Diagnosis and treatment of malignant neoplasms |
| US6994852B1 (en) | 1999-11-12 | 2006-02-07 | Temple University-Of The Commonwealth System Of Higher Education | Inhibition of angiogenesis by antibodies against high molecular weight kininogen domain 5 |
| US6994976B1 (en) | 1999-11-19 | 2006-02-07 | Tittle Thomas V | Tr3-specific binding agents and methods for their use |
| US6319675B1 (en) | 1999-11-24 | 2001-11-20 | Millennium Pharmaceuticals, Inc. | Methods for detecting and/or identifying agents which bind and/or modulate function of “bonzo” chemokine receptor |
| US6835549B2 (en) | 2000-02-24 | 2004-12-28 | University Of Medicine & Dentistry Of New Jersey | Immunoassay method for the diagnosis of gastric intestinal metaplasia associated with gastric carcinoma |
| DK1265929T3 (da) | 2000-03-23 | 2009-11-16 | Genentech Inc | Anti-C2/C2a-inhibitorer til komplement aktivering |
| CA2408549A1 (en) | 2000-05-11 | 2001-11-15 | Altarex Corp. | Therapeutic method and composition utilizing antigen-antibody complexation and presentation by dendritic cells |
| AU2001275285A1 (en) | 2000-06-06 | 2001-12-17 | Bristol-Myers Squibb Company | B7-related nucleic acids and polypeptides and their uses for immunomodulation |
| WO2002010764A2 (en) | 2000-07-31 | 2002-02-07 | The Government Of The United States Of America, As Represented By The Secretary Of The Department Of Health And Human Services | SPECIFIC BINDING AGENTS FOR KSHV vIL-6 THAT NEUTRALIZE A BIOLOGICAL ACTIVITY |
| US7060802B1 (en) | 2000-09-18 | 2006-06-13 | The Trustees Of Columbia University In The City Of New York | Tumor-associated marker |
| ATE464374T1 (de) | 2000-10-02 | 2010-04-15 | Oklahoma Med Res Found | Assay zum schnellen nachweis vom humanen aktivierten protein c und hochspezifischer monoklonaler antikörper dagegen |
| US6534058B2 (en) | 2000-10-10 | 2003-03-18 | Tanox, Inc. | Anti-C5 monoclonal antibodies |
| US7138496B2 (en) | 2002-02-08 | 2006-11-21 | Genetastix Corporation | Human monoclonal antibodies against human CXCR4 |
| NZ569856A (en) | 2001-01-05 | 2010-03-26 | Pfizer | Antibodies to insulin-like growth factor 1 receptor |
| US6743898B2 (en) | 2001-03-15 | 2004-06-01 | Ochsner Clinic Foundation | Monoclonal antibodies that suppress B cell growth and/or differentiation |
| WO2002078638A2 (en) | 2001-03-30 | 2002-10-10 | University Of Massachusetts | Morpholino imaging and therapy |
| US6824778B2 (en) | 2001-04-23 | 2004-11-30 | The United States Of America As Represented By The Secretary Of The Army | Prophylactic and therapeutic monoclonal antibodies |
| US20050053973A1 (en) | 2001-04-26 | 2005-03-10 | Avidia Research Institute | Novel proteins with targeted binding |
| US20050089932A1 (en) | 2001-04-26 | 2005-04-28 | Avidia Research Institute | Novel proteins with targeted binding |
| US20040175756A1 (en) | 2001-04-26 | 2004-09-09 | Avidia Research Institute | Methods for using combinatorial libraries of monomer domains |
| US20050048512A1 (en) | 2001-04-26 | 2005-03-03 | Avidia Research Institute | Combinatorial libraries of monomer domains |
| DE60236861D1 (de) | 2001-04-26 | 2010-08-12 | Amgen Mountain View Inc | Kombinatorische bibliotheken von monomerdomänen |
| US6962813B2 (en) | 2001-05-21 | 2005-11-08 | The Brigham And Women's Hospital, Inc. | P. aeruginosa mucoid exopolysaccharide specific binding peptides |
| US7049060B2 (en) | 2001-11-05 | 2006-05-23 | Ortho-Clinical Diagnostics, Inc. | HCV anti-core monoclonal antibodies |
| AU2003208415B2 (en) | 2002-02-14 | 2009-05-28 | Immunomedics, Inc. | Anti-CD20 antibodies and fusion proteins thereof and methods of use |
| US8287864B2 (en) | 2002-02-14 | 2012-10-16 | Immunomedics, Inc. | Structural variants of antibodies for improved therapeutic characteristics |
| US7282567B2 (en) | 2002-06-14 | 2007-10-16 | Immunomedics, Inc. | Monoclonal antibody hPAM4 |
| US8877901B2 (en) | 2002-12-13 | 2014-11-04 | Immunomedics, Inc. | Camptothecin-binding moiety conjugates |
| AU2003209447B8 (en) | 2002-03-01 | 2008-10-23 | Immunomedics, Inc. | RS7 antibodies |
| AU2003215732B2 (en) | 2002-03-01 | 2009-12-17 | Immunomedics, Inc. | Internalizing anti-CD74 antibodies and methods of use |
| US7238786B2 (en) | 2002-06-14 | 2007-07-03 | Immunomedics, Inc. | Monoclonal antibody cPAM4 |
| US7906118B2 (en) | 2005-04-06 | 2011-03-15 | Ibc Pharmaceuticals, Inc. | Modular method to prepare tetrameric cytokines with improved pharmacokinetics by the dock-and-lock (DNL) technology |
| US7300655B2 (en) | 2002-08-01 | 2007-11-27 | Immunomedics, Inc. | Alpha-fetoprotein Immu31 antibodies and fusion proteins and methods of use thereof |
| US7541440B2 (en) | 2002-09-30 | 2009-06-02 | Immunomedics, Inc. | Chimeric, human and humanized anti-granulocyte antibodies and methods of use |
| US7217797B2 (en) | 2002-10-15 | 2007-05-15 | Pdl Biopharma, Inc. | Alteration of FcRn binding affinities or serum half-lives of antibodies by mutagenesis |
| EP1572242B1 (en) | 2002-12-13 | 2014-04-16 | Immunomedics, Inc. | Immunoconjugates with an intracellularly-cleavable linkage |
| US7534427B2 (en) | 2002-12-31 | 2009-05-19 | Immunomedics, Inc. | Immunotherapy of B cell malignancies and autoimmune diseases using unconjugated antibodies and conjugated antibodies and antibody combinations and fusion proteins |
| US20040202666A1 (en) | 2003-01-24 | 2004-10-14 | Immunomedics, Inc. | Anti-cancer anthracycline drug-antibody conjugates |
| MXPA05007973A (es) | 2003-01-28 | 2005-09-20 | Schering Corp | Anticuerpos especificos para celulas dendriticas plasmacitoides. |
| CA2529027C (en) | 2003-06-13 | 2013-09-10 | Immunomedics, Inc. | D-amino acid peptides |
| US7902338B2 (en) | 2003-07-31 | 2011-03-08 | Immunomedics, Inc. | Anti-CD19 antibodies |
| US7109304B2 (en) | 2003-07-31 | 2006-09-19 | Immunomedics, Inc. | Humanized anti-CD19 antibodies |
| US20050079184A1 (en) | 2003-08-08 | 2005-04-14 | Immunomedics, Inc. | Bispecific antibodies for inducing apoptosis of tumor and diseased cells |
| JP5150103B2 (ja) | 2004-02-13 | 2013-02-20 | イミューノメディクス、インコーポレイテッド | 組み換え細胞毒性RNAseを含む融合タンパク質 |
| US8003111B2 (en) | 2005-04-06 | 2011-08-23 | Ibc Pharmaceuticals, Inc. | Dimeric alpha interferon pegylated site-specifically shows enhanced and prolonged efficacy in vivo |
| US8034352B2 (en) | 2005-04-06 | 2011-10-11 | Ibc Pharmaceuticals, Inc. | Tetrameric cytokines with improved biological activity |
| CN101171034B (zh) | 2005-03-03 | 2014-06-18 | 免疫医疗公司 | 人源化l243抗体 |
| AU2006232920B2 (en) | 2005-04-06 | 2011-09-29 | Ibc Pharmaceuticals, Inc. | Methods for generating stably linked complexes composed of homodimers, homotetramers or dimers of dimers and uses |
| CN101534865A (zh) | 2005-10-19 | 2009-09-16 | Ibc药品公司 | 生物活性装配体的制备方法及其用途 |
| CA2651285C (en) | 2006-05-15 | 2014-08-19 | Immunomedics, Inc. | Methods and compositions for treatment of human immunodeficiency virus infection with conjugated antibodies or antibody fragments |
| US8267865B2 (en) | 2007-02-16 | 2012-09-18 | University Of Rochester | Sonoelastographic shear velocity imaging using crawling wave excitation |
| ITTO20080313A1 (it) | 2008-04-22 | 2009-10-23 | Marco Colombatti | Anticorpo monoclonale isolato o suo frammento legante l'antigene specifico di membrana della prostata, suoi coniugati e suoi usi |
| CN102186499B (zh) | 2008-08-20 | 2015-05-20 | Ibc医药公司 | 用于癌症治疗的对接和锁定(dnl)疫苗 |
| EP3903829B1 (en) | 2009-02-13 | 2023-05-03 | Immunomedics, Inc. | Immunoconjugates with an intracellularly-cleavable linkage |
| EP2478110B1 (en) | 2009-09-16 | 2016-01-06 | Immunomedics, Inc. | Class i anti-cea antibodies and uses thereof |
| CA2787054A1 (en) * | 2010-01-11 | 2011-07-14 | Center For Molecular Medicine And Immunology | Enhanced cytotoxicity of anti-cd74 and anti-hla-dr antibodies with interferon-gamma |
| US9382329B2 (en) * | 2012-08-14 | 2016-07-05 | Ibc Pharmaceuticals, Inc. | Disease therapy by inducing immune response to Trop-2 expressing cells |
| EP2918292B1 (en) * | 2012-11-08 | 2019-12-11 | Nippon Kayaku Kabushiki Kaisha | Polymeric compound having camptothecin compound and anti-cancer effect enhancer bound thereto, and use of same |
| HUE057977T2 (hu) * | 2012-12-13 | 2022-06-28 | Immunomedics Inc | Ellenanyagok és SN-38 immunkonjugátumainak dózisai javított hatásossággal és csökkentett toxicitással |
| US9107960B2 (en) * | 2012-12-13 | 2015-08-18 | Immunimedics, Inc. | Antibody-SN-38 immunoconjugates with a CL2A linker |
| US9492566B2 (en) * | 2012-12-13 | 2016-11-15 | Immunomedics, Inc. | Antibody-drug conjugates and uses thereof |
| US8877202B2 (en) | 2013-02-07 | 2014-11-04 | Immunomedics, Inc. | Pro-drug form (P2PDOX) of the highly potent 2-pyrrolinodoxorubicin conjugated to antibodies for targeted therapy of cancer |
| EP3038600B1 (en) * | 2013-08-27 | 2020-06-03 | Northeastern University | Nanoparticle drug delivery system and method of treating cancer and neurotrauma |
| EP3049443A4 (en) | 2013-09-27 | 2017-04-05 | Immunomedics, Inc. | Anti-trop-2 antibody-drug conjugates and uses thereof |
| EP3066470A4 (en) * | 2013-11-05 | 2017-05-31 | Immunomedics, Inc. | Humanized anti-ceacam5 antibody and uses thereof |
| CN114796501A (zh) * | 2015-06-25 | 2022-07-29 | 免疫医疗公司 | 抗体与治疗剂的组合治疗癌症的方法 |
-
2016
- 2016-06-23 CN CN202210425682.5A patent/CN114796501A/zh active Pending
- 2016-06-23 AU AU2016281622A patent/AU2016281622C1/en active Active
- 2016-06-23 PT PT168152858T patent/PT3313443T/pt unknown
- 2016-06-23 JP JP2017566640A patent/JP6980980B2/ja active Active
- 2016-06-23 ES ES16815285T patent/ES2953441T3/es active Active
- 2016-06-23 WO PCT/US2016/038986 patent/WO2016210108A1/en active Application Filing
- 2016-06-23 CA CA2983597A patent/CA2983597A1/en active Pending
- 2016-06-23 SI SI201631735T patent/SI3313443T1/sl unknown
- 2016-06-23 CN CN202210414951.8A patent/CN114796500A/zh active Pending
- 2016-06-23 EP EP23175990.3A patent/EP4257134A3/en active Pending
- 2016-06-23 EP EP16815285.8A patent/EP3313443B9/en active Active
- 2016-06-23 CN CN201680036918.9A patent/CN107735104B/zh active Active
- 2016-06-23 PL PL16815285.8T patent/PL3313443T3/pl unknown
-
2017
- 2017-12-06 IL IL256150A patent/IL256150B/en unknown
-
2021
- 2021-08-31 JP JP2021141615A patent/JP2021185187A/ja active Pending
Also Published As
| Publication number | Publication date |
|---|---|
| JP2021185187A (ja) | 2021-12-09 |
| JP6980980B2 (ja) | 2021-12-15 |
| AU2016281622C1 (en) | 2021-11-18 |
| EP4257134A3 (en) | 2024-01-24 |
| PT3313443T (pt) | 2023-08-30 |
| AU2016281622B2 (en) | 2021-07-22 |
| SI3313443T1 (sl) | 2023-11-30 |
| EP3313443B1 (en) | 2023-06-07 |
| AU2016281622A1 (en) | 2017-11-09 |
| JP2018520140A (ja) | 2018-07-26 |
| CA2983597A1 (en) | 2016-12-29 |
| CN107735104A (zh) | 2018-02-23 |
| EP4257134A2 (en) | 2023-10-11 |
| IL256150B (en) | 2021-10-31 |
| EP3313443A1 (en) | 2018-05-02 |
| IL256150A (en) | 2018-02-28 |
| EP3313443A4 (en) | 2019-01-23 |
| CN107735104B (zh) | 2022-05-03 |
| CN114796500A (zh) | 2022-07-29 |
| PL3313443T3 (pl) | 2023-11-06 |
| EP3313443B9 (en) | 2023-10-04 |
| CN114796501A (zh) | 2022-07-29 |
| WO2016210108A1 (en) | 2016-12-29 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2953441T3 (es) | Combinación de anticuerpos anti-hla-dr o anti-Trop-2 con inhibidores de microtúbulos, inhibidores de parp, inhibidores de la cinasa de bruton o inhibidores de la fosfoinositida 3-cinasa mejora significativamente el resultado terapéutico en el cáncer | |
| US10954305B2 (en) | Combination of ABCG2 inhibitors with sacituzumab govitecan (IMMU-132) overcomes resistance to SN-38 in Trop-2 expressing cancers | |
| US11192955B2 (en) | Efficacy of anti-Trop-2-SN-38 antibody drug conjugates for therapy of tumors relapsed/refractory to checkpoint inhibitors | |
| US9707302B2 (en) | Combining anti-HLA-DR or anti-Trop-2 antibodies with microtubule inhibitors, PARP inhibitors, bruton kinase inhibitors or phosphoinositide 3-kinase inhibitors significantly improves therapeutic outcome in cancer | |
| CA2884313C (en) | Dosages of immunoconjugates of antibodies and sn-38 for improved efficacy and decreased toxicity | |
| US20210046185A1 (en) | Dosages of immunoconjugates of antibodies and sn-38 for improved efficacy and decreased toxicity | |
| ES2819573T3 (es) | Método para producir inmunoconjugados de anticuerpo-SN-38 con un enlazador CL2A | |
| AU2014293670B2 (en) | Antibody-SN-38 immunoconjugates with a CL2A linker | |
| US11253606B2 (en) | Combining anti-HLA-DR or anti-Trop-2 antibodies with microtubule inhibitors, PARP inhibitors, Bruton kinase inhibitors or phosphoinositide 3-kinase inhibitors significantly improves therapeutic outcome in cancer | |
| US20240139324A1 (en) | Dosages of immunoconjugates of antibodies and sn-38 for improved efficacy and decreased toxicity | |
| ES2886927T3 (es) | Inmunoconjugados de anticuerpo-SN-38 con un enlazador CL2A |