ES2933807T3 - Proceso para la preparación de un inhibidor de quinasa estructurado de sulfonamida - Google Patents
Proceso para la preparación de un inhibidor de quinasa estructurado de sulfonamida Download PDFInfo
- Publication number
- ES2933807T3 ES2933807T3 ES18715893T ES18715893T ES2933807T3 ES 2933807 T3 ES2933807 T3 ES 2933807T3 ES 18715893 T ES18715893 T ES 18715893T ES 18715893 T ES18715893 T ES 18715893T ES 2933807 T3 ES2933807 T3 ES 2933807T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- formula
- process according
- acid
- catalyst
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 238000000034 method Methods 0.000 title claims abstract description 34
- 238000002360 preparation method Methods 0.000 title claims abstract description 23
- 229940043355 kinase inhibitor Drugs 0.000 title abstract description 4
- 239000003757 phosphotransferase inhibitor Substances 0.000 title abstract description 4
- 229940124530 sulfonamide Drugs 0.000 title abstract description 4
- 150000003456 sulfonamides Chemical class 0.000 title abstract description 4
- 150000001875 compounds Chemical class 0.000 claims abstract description 114
- 150000003839 salts Chemical class 0.000 claims abstract description 25
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 claims description 31
- 239000003054 catalyst Substances 0.000 claims description 25
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 claims description 24
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 claims description 16
- PFWWSGFPICCWGU-UHFFFAOYSA-N cyclopropanesulfonyl chloride Chemical compound ClS(=O)(=O)C1CC1 PFWWSGFPICCWGU-UHFFFAOYSA-N 0.000 claims description 16
- 235000019253 formic acid Nutrition 0.000 claims description 16
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 claims description 15
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 claims description 14
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 claims description 12
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 claims description 10
- 150000007530 organic bases Chemical class 0.000 claims description 8
- 229910052763 palladium Inorganic materials 0.000 claims description 8
- 229940098779 methanesulfonic acid Drugs 0.000 claims description 7
- LXNAVEXFUKBNMK-UHFFFAOYSA-N palladium(II) acetate Substances [Pd].CC(O)=O.CC(O)=O LXNAVEXFUKBNMK-UHFFFAOYSA-N 0.000 claims description 7
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 claims description 7
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 claims description 7
- VLDIOICWDGMAJP-UHFFFAOYSA-N 5-(2,4-difluorophenyl)benzene-1,3-diamine Chemical compound FC1=CC=C(C2=CC(N)=CC(N)=C2)C(F)=C1 VLDIOICWDGMAJP-UHFFFAOYSA-N 0.000 claims description 6
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 claims description 6
- 238000010438 heat treatment Methods 0.000 claims description 6
- 150000007524 organic acids Chemical class 0.000 claims description 6
- 229910000027 potassium carbonate Inorganic materials 0.000 claims description 6
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 claims description 6
- VQCWSOYHHXXWSP-UHFFFAOYSA-N 4-bromo-2-fluoro-1-nitrobenzene Chemical compound [O-][N+](=O)C1=CC=C(Br)C=C1F VQCWSOYHHXXWSP-UHFFFAOYSA-N 0.000 claims description 5
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 claims description 4
- 239000003960 organic solvent Substances 0.000 claims description 4
- 150000001282 organosilanes Chemical class 0.000 claims description 4
- UCNGGGYMLHAMJG-UHFFFAOYSA-N 1-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrazole Chemical compound C1=NN(C)C=C1B1OC(C)(C)C(C)(C)O1 UCNGGGYMLHAMJG-UHFFFAOYSA-N 0.000 claims description 3
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 claims description 3
- 229910000024 caesium carbonate Inorganic materials 0.000 claims description 3
- RSIHJDGMBDPTIM-UHFFFAOYSA-N ethoxy(trimethyl)silane Chemical compound CCO[Si](C)(C)C RSIHJDGMBDPTIM-UHFFFAOYSA-N 0.000 claims description 3
- 229910000029 sodium carbonate Inorganic materials 0.000 claims description 3
- 239000002253 acid Substances 0.000 claims description 2
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 claims description 2
- 229940092714 benzenesulfonic acid Drugs 0.000 claims description 2
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 claims description 2
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 claims 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 claims 1
- WMSPXQIQBQAWLL-UHFFFAOYSA-N cyclopropanesulfonamide Chemical compound NS(=O)(=O)C1CC1 WMSPXQIQBQAWLL-UHFFFAOYSA-N 0.000 abstract description 5
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical group C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 abstract description 5
- 108091008794 FGF receptors Proteins 0.000 abstract description 2
- 206010028980 Neoplasm Diseases 0.000 abstract description 2
- 108091000080 Phosphotransferase Proteins 0.000 abstract description 2
- 229940124639 Selective inhibitor Drugs 0.000 abstract description 2
- 108091008605 VEGF receptors Proteins 0.000 abstract description 2
- 102100033177 Vascular endothelial growth factor receptor 2 Human genes 0.000 abstract description 2
- 235000010290 biphenyl Nutrition 0.000 abstract description 2
- 201000011510 cancer Diseases 0.000 abstract description 2
- 102000052178 fibroblast growth factor receptor activity proteins Human genes 0.000 abstract description 2
- 102000020233 phosphotransferase Human genes 0.000 abstract description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 39
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 38
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 36
- 239000000203 mixture Substances 0.000 description 30
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 27
- 239000000047 product Substances 0.000 description 26
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 24
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 20
- 238000001914 filtration Methods 0.000 description 20
- 238000006243 chemical reaction Methods 0.000 description 17
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 15
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 14
- YIIIBLKLGXCLTP-UHFFFAOYSA-N N-[3-(2,4-difluorophenyl)-5-[4-(1-methylpyrazol-4-yl)-2-nitroanilino]phenyl]cyclopropanesulfonamide Chemical compound FC1=C(C=CC(=C1)F)C1=CC(=CC(=C1)NC1=C(C=C(C=C1)C=1C=NN(C=1)C)[N+](=O)[O-])NS(=O)(=O)C1CC1 YIIIBLKLGXCLTP-UHFFFAOYSA-N 0.000 description 12
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 11
- 229910052757 nitrogen Inorganic materials 0.000 description 10
- -1 1-methyl-1H-pyrazol-4-yl ring Chemical group 0.000 description 9
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 9
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 9
- 238000004128 high performance liquid chromatography Methods 0.000 description 9
- ZJFCBQXPTQSTCZ-UHFFFAOYSA-N n-[3-(2,4-difluorophenyl)-5-[5-(1-methylpyrazol-4-yl)benzimidazol-1-yl]phenyl]cyclopropanesulfonamide Chemical compound C1=NN(C)C=C1C1=CC=C(N(C=N2)C=3C=C(C=C(NS(=O)(=O)C4CC4)C=3)C=3C(=CC(F)=CC=3)F)C2=C1 ZJFCBQXPTQSTCZ-UHFFFAOYSA-N 0.000 description 9
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 8
- 238000001035 drying Methods 0.000 description 7
- 239000007787 solid Substances 0.000 description 7
- 239000002904 solvent Substances 0.000 description 7
- OLDMYNWXIGPOCI-UHFFFAOYSA-N 1-bromo-3,5-dinitrobenzene Chemical compound [O-][N+](=O)C1=CC(Br)=CC([N+]([O-])=O)=C1 OLDMYNWXIGPOCI-UHFFFAOYSA-N 0.000 description 6
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 6
- 239000000706 filtrate Substances 0.000 description 6
- 238000003756 stirring Methods 0.000 description 6
- 238000005406 washing Methods 0.000 description 6
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 5
- 238000001816 cooling Methods 0.000 description 5
- 239000000543 intermediate Substances 0.000 description 5
- 239000011541 reaction mixture Substances 0.000 description 5
- 239000012453 solvate Substances 0.000 description 5
- 239000000725 suspension Substances 0.000 description 5
- BQLBGVYCDBBWPP-UHFFFAOYSA-N 3-N-(4-bromo-2-nitrophenyl)-5-(2,4-difluorophenyl)benzene-1,3-diamine Chemical compound BrC1=CC(=C(C=C1)NC=1C=C(C=C(C=1)N)C1=C(C=C(C=C1)F)F)[N+](=O)[O-] BQLBGVYCDBBWPP-UHFFFAOYSA-N 0.000 description 4
- 238000004587 chromatography analysis Methods 0.000 description 4
- 238000004821 distillation Methods 0.000 description 4
- 238000000746 purification Methods 0.000 description 4
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 3
- 239000002178 crystalline material Substances 0.000 description 3
- 238000000605 extraction Methods 0.000 description 3
- 239000001257 hydrogen Substances 0.000 description 3
- 229910052739 hydrogen Inorganic materials 0.000 description 3
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 3
- QQLRSCZSKQTFGY-UHFFFAOYSA-N (2,4-difluorophenyl)boronic acid Chemical compound OB(O)C1=CC=C(F)C=C1F QQLRSCZSKQTFGY-UHFFFAOYSA-N 0.000 description 2
- NMSKVMZQOZYMNA-UHFFFAOYSA-N 1-(3,5-dinitrophenyl)-2,4-difluorobenzene Chemical group FC1=C(C=CC(=C1)F)C1=CC(=CC(=C1)[N+](=O)[O-])[N+](=O)[O-] NMSKVMZQOZYMNA-UHFFFAOYSA-N 0.000 description 2
- XZKOSLLFCXVPNE-UHFFFAOYSA-N 3-N-(4-bromo-2-nitrophenyl)-5-(2,4-difluorophenyl)benzene-1,3-diamine methanesulfonic acid Chemical compound S(C)(=O)(=O)O.BrC1=CC(=C(C=C1)NC=1C=C(C=C(C1)N)C1=C(C=C(C=C1)F)F)[N+](=O)[O-] XZKOSLLFCXVPNE-UHFFFAOYSA-N 0.000 description 2
- CYTROQTYTIQTQK-UHFFFAOYSA-N 5-(2,4-difluorophenyl)-3-N-[4-(1-methylpyrazol-4-yl)-2-nitrophenyl]benzene-1,3-diamine Chemical compound FC1=C(C=CC(=C1)F)C1=CC(=CC(=C1)N)NC1=C(C=C(C=C1)C=1C=NN(C=1)C)[N+](=O)[O-] CYTROQTYTIQTQK-UHFFFAOYSA-N 0.000 description 2
- KRHYYFGTRYWZRS-UHFFFAOYSA-N Fluorane Chemical compound F KRHYYFGTRYWZRS-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- DXRFFDWUBXLCHV-UHFFFAOYSA-N N-[3-(4-bromo-2-nitroanilino)-5-(2,4-difluorophenyl)phenyl]cyclopropanesulfonamide Chemical compound BrC1=CC(=C(C=C1)NC=1C=C(C=C(C=1)C1=C(C=C(C=C1)F)F)NS(=O)(=O)C1CC1)[N+](=O)[O-] DXRFFDWUBXLCHV-UHFFFAOYSA-N 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- 239000012043 crude product Substances 0.000 description 2
- 238000004090 dissolution Methods 0.000 description 2
- 229910000040 hydrogen fluoride Inorganic materials 0.000 description 2
- 238000005984 hydrogenation reaction Methods 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 2
- 238000010992 reflux Methods 0.000 description 2
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 2
- WDCYWAQPCXBPJA-UHFFFAOYSA-N 1,3-dinitrobenzene Chemical compound [O-][N+](=O)C1=CC=CC([N+]([O-])=O)=C1 WDCYWAQPCXBPJA-UHFFFAOYSA-N 0.000 description 1
- 238000005160 1H NMR spectroscopy Methods 0.000 description 1
- LBLYYCQCTBFVLH-UHFFFAOYSA-N 2-Methylbenzenesulfonic acid Chemical compound CC1=CC=CC=C1S(O)(=O)=O LBLYYCQCTBFVLH-UHFFFAOYSA-N 0.000 description 1
- MTJGVAJYTOXFJH-UHFFFAOYSA-N 3-aminonaphthalene-1,5-disulfonic acid Chemical compound C1=CC=C(S(O)(=O)=O)C2=CC(N)=CC(S(O)(=O)=O)=C21 MTJGVAJYTOXFJH-UHFFFAOYSA-N 0.000 description 1
- MCSXGCZMEPXKIW-UHFFFAOYSA-N 3-hydroxy-4-[(4-methyl-2-nitrophenyl)diazenyl]-N-(3-nitrophenyl)naphthalene-2-carboxamide Chemical compound Cc1ccc(N=Nc2c(O)c(cc3ccccc23)C(=O)Nc2cccc(c2)[N+]([O-])=O)c(c1)[N+]([O-])=O MCSXGCZMEPXKIW-UHFFFAOYSA-N 0.000 description 1
- UQEANKGXXSENNF-UHFFFAOYSA-N 4-bromo-1-fluoro-2-nitrobenzene Chemical compound [O-][N+](=O)C1=CC(Br)=CC=C1F UQEANKGXXSENNF-UHFFFAOYSA-N 0.000 description 1
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- UQFQONCQIQEYPJ-UHFFFAOYSA-N N-methylpyrazole Chemical group CN1C=CC=N1 UQFQONCQIQEYPJ-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- PBCJIPOGFJYBJE-UHFFFAOYSA-N acetonitrile;hydrate Chemical compound O.CC#N PBCJIPOGFJYBJE-UHFFFAOYSA-N 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 235000019270 ammonium chloride Nutrition 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 239000004305 biphenyl Substances 0.000 description 1
- 239000011449 brick Substances 0.000 description 1
- 150000004649 carbonic acid derivatives Chemical class 0.000 description 1
- 239000003610 charcoal Substances 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- VRLDVERQJMEPIF-UHFFFAOYSA-N dbdmh Chemical compound CC1(C)N(Br)C(=O)N(Br)C1=O VRLDVERQJMEPIF-UHFFFAOYSA-N 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- 238000011010 flushing procedure Methods 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 235000011087 fumaric acid Nutrition 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 230000001473 noxious effect Effects 0.000 description 1
- 238000010899 nucleation Methods 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 238000006798 ring closing metathesis reaction Methods 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 150000003460 sulfonic acids Chemical class 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-N sulfuric acid Substances OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/02—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings
- C07D231/10—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D231/12—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C211/00—Compounds containing amino groups bound to a carbon skeleton
- C07C211/43—Compounds containing amino groups bound to a carbon skeleton having amino groups bound to carbon atoms of six-membered aromatic rings of the carbon skeleton
- C07C211/44—Compounds containing amino groups bound to a carbon skeleton having amino groups bound to carbon atoms of six-membered aromatic rings of the carbon skeleton having amino groups bound to only one six-membered aromatic ring
- C07C211/52—Compounds containing amino groups bound to a carbon skeleton having amino groups bound to carbon atoms of six-membered aromatic rings of the carbon skeleton having amino groups bound to only one six-membered aromatic ring the carbon skeleton being further substituted by halogen atoms or by nitro or nitroso groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C311/00—Amides of sulfonic acids, i.e. compounds having singly-bound oxygen atoms of sulfo groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/14—Sulfonamides having sulfur atoms of sulfonamide groups bound to carbon atoms of rings other than six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/02—Systems containing only non-condensed rings with a three-membered ring
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Enzymes And Modification Thereof (AREA)
Abstract
La presente invención se refiere a un proceso mejorado para la preparación de un inhibidor de quinasa estructurado de sulfonamida, a saber, N-(2',4'-difluoro-5-(5-(1-metil-1H-pirazol-4-il)-1H -benzo[d]imidazol-1-il)-[1,1'-bifenil]-3-il)ciclopropanosulfonamida (1A) y sus sales farmacéuticamente aceptables. El compuesto de fórmula (1A) es un inhibidor selectivo de las familias de cinasas FGFR/VEGFR y es útil en el tratamiento del cáncer. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Proceso para la preparación de un inhibidor de quinasa estructurado de sulfonamida
Campo técnico
La presente invención se refiere a un proceso mejorado para la preparación de un inhibidor de quinasa estructurado de sulfonamida, a saber, N-(2',4'-difluoro-5-(5-( 1-metil-1H-pirazol-4-il)-1H-benzo[d]imidazol-1-il)-[1,1'-bifenil]-3-il)ciclopropanosulfon-amida (1A) y sales farmacéuticamente aceptables de la misma.
Antecedentes de la invención
El compuesto N-(2',4'-difluoro-5-(5-( 1-metil-1 H-pirazol-4-il)-1 H-benzo[d]imidazol-1-il)-[1,1'-bifenil]-3-il)ciclopropanosulfonamida de fórmula (1A) y derivados de la misma se han descrito en el documento WO 2013/053983. El compuesto de fórmula (1A) y sus sales farmacéuticamente aceptables son inhibidores selectivos de las familias de quinasas FGFR/VEGFR y son útiles en el tratamiento del cáncer.

El documento WO 2013/053983 desvela un proceso para la preparación del compuesto de fórmula (1A) de acuerdo con el Esquema 1:
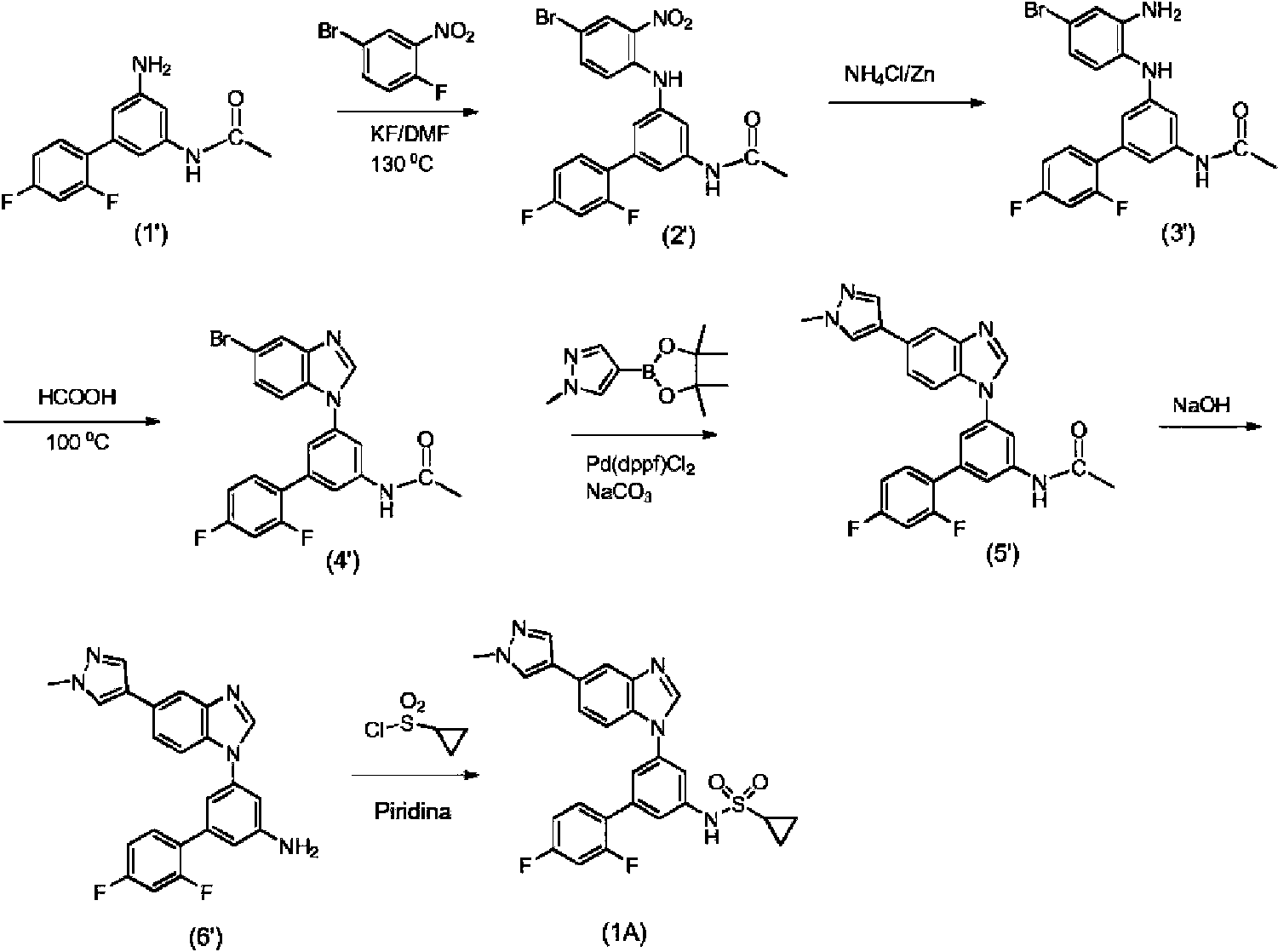
ESQUEMA 1
El proceso del Esquema 1 tiene varios inconvenientes. La primera etapa de reacción con 4-bromo-1-fluoro-2-nitrobenceno implica condiciones duras, liberación de fluoruro de hidrógeno nocivo, necesidad de purificar el producto por cromatografía y bajo rendimiento (45 %). La posterior conversión del compuesto (2') en el compuesto (4') por reducción del grupo nitro con NH4Cl/Zn seguido del cierre del anillo con HCOOH requiere el aislamiento del compuesto
intermedio (3'). La adición del anillo 1-metil-1 H-pirazol-4-ilo al compuesto (4') para obtener el compuesto (5') requiere una gran cantidad de costoso catalizador de paladio mientras que el rendimiento sigue siendo bajo. También la reacción de hidrólisis al compuesto (6') y la última etapa de crear un enlace de sulfonamina sufren bajos rendimientos. Por lo tanto, existe una necesidad de un proceso más económico que sea adecuado para la fabricación del compuesto de fórmula (1A) a gran escala.
Sumario de la invención
Ahora se ha descubierto que el compuesto de fórmula (1A) puede prepararse usando un proceso que es más práctico y económico y adecuado para su uso a gran escala. Los inconvenientes del método descrito en el documento WO 2013/053983 pueden evitarse en gran medida.
Por lo tanto la presente invención proporciona un proceso para la preparación de un compuesto de fórmula (1A) o una sal farmacéuticamente aceptable del mismo

que comprende las etapas de
a) hacer reaccionar 2',4'-difluoro-[1,1'-bifenil]-3,5-diamina de fórmula (V)

b) tratar el producto obtenido de la etapa (a) con un ácido orgánico y aislar la sal orgánica formada;
c) liberar el compuesto de fórmula (IV) de su forma de sal;
d) ya sea (i) haciendo reaccionar el compuesto de fórmula (IV) con cloruro de ciclopropanosulfonilo para obtener un compuesto de fórmula (III)

y posteriormente hacer reaccionar el compuesto de fórmula (III) con 1-metil-4-(4,4,5,5-tetrametiM,3,2-dioxaborlan-2-il)-1 H-pirazol;
o
(ii) hacer reaccionar el compuesto de fórmula (IV) con 1-metil-4-(4,4,5,5-tetrametiM,3,2-dioxaborlan-2-N)-1H-pirazol para obtener un compuesto de fórmula (IIIb)

y posteriormente hacer reaccionar el compuesto de fórmula (IIIb) con cloruro de ciclopropanosulfonilo; para obtener un compuesto de fórmula (II);
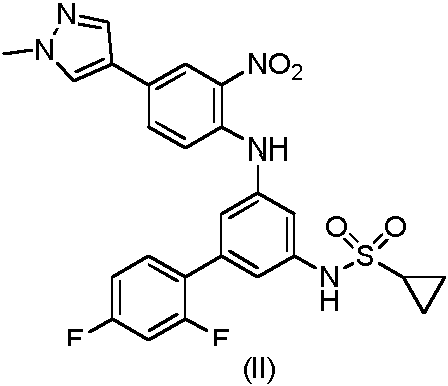
e) hidrogenar el compuesto de fórmula (II) en ácido fórmico en presencia de un catalizador para obtener un compuesto de fórmula (1A), que opcionalmente se convierte en su sal farmacéuticamente aceptable.
En otro aspecto, la presente divulgación proporciona un proceso para la preparación de un compuesto de fórmula (IV)

que comprende las etapas de
a) hacer reaccionar 2',4'-difluoro-[1,1'-bifenil]-3,5-diamina de fórmula (V)

con 4-bromo-2-fluoronitrobenceno en presencia de una base orgánica y un organosilano para obtener un compuesto de fórmula (IV)

b) tratar el producto obtenido de la etapa (a) con un ácido orgánico y aislar la sal orgánica formada;
c) liberar el compuesto de fórmula (IV) de su forma de sal.
En otro aspecto, la presente divulgación proporciona el uso del compuesto de fórmula (IV) en la preparación del compuesto de fórmula (1A), en donde el compuesto de fórmula (IV) se prepara de acuerdo con el método desvelado anteriormente.
En otro aspecto, la presente divulgación proporciona un proceso para la preparación de un compuesto de fórmula (1A) o una sal farmacéuticamente aceptable del mismo
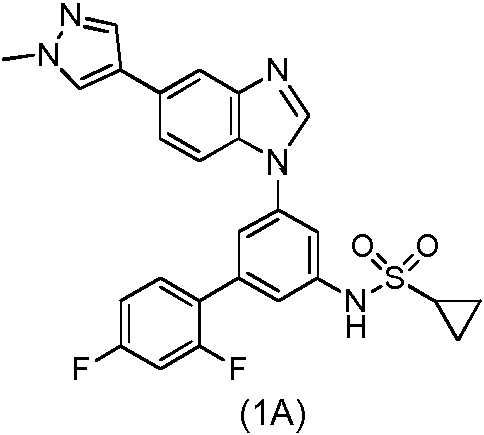
que comprende la etapa de hidrogenar el compuesto de fórmula (II)

en ácido fórmico en presencia de un catalizador para obtener un compuesto de fórmula (1A), que opcionalmente se convierte en su sal farmacéuticamente aceptable.
En otro aspecto, la presente invención proporciona nuevos intermedios de fórmula (V), (IV), (III), (IIIb) y (II).
En otro aspecto, la presente divulgación proporciona el uso de cualquiera de los intermedios de fórmula (V), (IV), (III), (IIIb) o (II) en la preparación del compuesto de fórmula (1A) o una sal farmacéuticamente aceptable del mismo. Descripción detallada de la invención
De acuerdo con la presente invención, 2',4'-difluoro-[1,1'-bifenil]-3,5-diamina de fórmula (V)

se hace reaccionar primero con 4-bromo-2-fluoronitrobenceno en presencia de una base orgánica y un organosilano para obtener un compuesto de fórmula (IV)

en alto rendimiento sin etapas de extracción y destilación y sin necesidad de purificación por cromatografía.
La reacción se lleva a cabo a temperatura elevada en un disolvente adecuado tal como dimetil sulfóxido. La base orgánica puede ser cualquier base orgánica adecuada conocida en la técnica, por ejemplo, N,N-diisopropiletilamina (DIPEA). Se ha descubierto que la reacción se lleva a cabo convenientemente en presencia de un compuesto de organosilano que elimina el fluoruro de hidrógeno nocivo y altamente corrosivo que se libera en la reacción. Puede usarse cualquier compuesto de organosilano adecuado, prefiriéndose el etoxitrimetilsilano. La reacción se lleva a cabo a temperatura elevada tal como 80-110 °C, por ejemplo 90-100 °C, hasta que se completa la reacción, normalmente durante menos de 8 h, por ejemplo, durante 5 h. La mezcla de reacción puede enfriarse después, se añade adecuadamente agua y metanol y se deja precipitar el producto, por ejemplo, a 20 °C. El compuesto de fórmula (IV) obtenido puede aislarse después, por ejemplo, por filtración, lavado con agua y metanol y secado a presión reducida a aproximadamente 60 °C.
El compuesto de fórmula (IV) obtenido normalmente contiene un producto de sobrerreacción que se forma cuando el compuesto de fórmula (V) reacciona dos veces con 4-bromo-2-fluoronitrobenceno. Este producto de sobrerreacción es difícil de aislar del compuesto (IV) y es problemático en las siguientes etapas del proceso.
Se ha descubierto que el producto de sobrereacción puede retirarse convenientemente tratando el compuesto de fórmula (IV) obtenido en la etapa anterior con un ácido orgánico y aislando la sal orgánica formada. Las sales orgánicas adecuadas incluyen, por ejemplo, ácidos sulfónicos tales como ácido metanosulfónico, ácido p-toluenosulfónico, ácido o-toluenosulfónico y ácido etanosulfónico, o ácido bencenosulfónico, ácido benzoico, ácido tartárico, ácido fumárico y similares. Se prefieren ácido metanosulfónico o ácido p-toluenosulfónico, siendo el ácido metanosulfónico el más preferido. La formación de la sal se lleva a cabo disolviendo el compuesto (IV) en un disolvente orgánico adecuado, por ejemplo, en la mezcla de tolueno y 2-butanona, mediante calentamiento, por ejemplo, a aproximadamente 70 -80 °C. El ácido orgánico, por ejemplo, ácido metanosulfónico, puede añadirse después en la cantidad de aproximadamente 1 equivalente con respecto al compuesto (IV) seguido de agitación, por ejemplo, durante aproximadamente 30 a 60 min. A continuación, la mezcla se enfría a una temperatura donde la sal orgánica del compuesto (IV) empieza a precipitar. Cuando se usa ácido metanosulfónico, la mezcla se enfría adecuadamente a aproximadamente 55 - 65 °C. La sal precipitada puede recogerse después, por ejemplo, mediante filtración.
A continuación, el compuesto (IV) puede liberarse de su forma de sal calentando la sal en un disolvente orgánico adecuado, por ejemplo, en la mezcla de tolueno y 2-butanona, por ejemplo, a aproximadamente 70 - 80 °C y añadiendo una base orgánica, tal como trietilamina, a la mezcla. El compuesto (IV) puede precipitarse después enfriando la mezcla, por ejemplo, a aproximadamente 15 - 25 °C. El compuesto precipitado (IV) que ahora está sustancialmente libre del producto de reacción excesiva puede aislarse, por ejemplo, mediante filtración, lavado con 2-propanol y secado al vacío, por ejemplo, a aproximadamente 60 °C.
La conversión del compuesto (IV) en el compuesto (II) incluye la adición de un grupo 1-metil-1H-pirazol y un grupo ciclopropanosulfonilo a la estructura del compuesto (IV). Estas dos etapas pueden llevarse a cabo secuencialmente en cualquier orden. En consecuencia, la conversión del compuesto de fórmula (IV) en el compuesto (II) puede llevarse a cabo ya sea (i) tratando el compuesto de fórmula (IV) con cloruro de ciclopropanosulfonilo para obtener un compuesto de fórmula (III)

y posteriormente hacer reaccionar el compuesto de fórmula (III) con 1-metil-4-(4,4,5,5-tetrametil-1,3,2-dioxaborlan-2-il)-1 H-pirazol;
o
(ii) tratar el compuesto de fórmula (IV) con 1-metil-4-(4,4,5,5-tetrametil-1,3,2-dioxaborlan-2-il)-1H-pirazol para
obtener un compuesto de fórmula (IIIb)

y posteriormente hacer reaccionar el compuesto de fórmula (IIIb) con cloruro de ciclopropanosulfonilo.
El compuesto (III) puede obtenerse con alto rendimiento sin etapas de extracción y destilación y sin necesidad de purificación por cromatografía tratando el compuesto (IV) con cloruro de ciclopropanosulfonilo en presencia de una base, convenientemente piridina que opcionalmente también puede funcionar como disolvente, opcionalmente en un disolvente adecuado tal como, por ejemplo, acetato de etilo a temperaturas que varían de aproximadamente 10 °C a aproximadamente 65 °C, para proporcionar el compuesto (III). El compuesto (III) puede precipitarse adecuadamente de la mezcla de reacción mediante la adición de un ácido, por ejemplo, ácido acético, agua y etanol o 2-propanol, calentar la mezcla, por ejemplo, a aproximadamente 40 - 75 °C y después enfriar, por ejemplo, a aproximadamente 0 - 25 °C. El compuesto (III) puede aislarse, por ejemplo, mediante filtración, lavado con agua y etanol o 2-propanol y secado al vacío a temperatura elevada, por ejemplo, a 60 °C.
El compuesto (II) puede obtenerse con alto rendimiento sin etapas de extracción y destilación y sin necesidad de purificación por cromatografía tratando el compuesto (III) con 1-metil-4-(4,4,5,5-tetrametil-1,3,2-dioxaborlan-2-il)-1H-pirazol en un disolvente adecuado tal como, por ejemplo, una mezcla de agua y DMSO, en presencia de una base, un catalizador y carbón activado. Las bases adecuadas incluyen carbonatos tales como carbonato potásico, carbonato de cesio y carbonato de sodio, prefiriéndose el carbonato potásico. Los catalizadores adecuados incluyen catalizadores de paladio tales como acetato de paladio (II) que pueden formar complejos con trifenilfosfina. Normalmente, se ha descubierto que 0,0050 - 0,01 equivalentes de acetato de paladio (II) son suficientes. La mezcla de reacción se calienta preferentemente a aproximadamente 90 - 110 °C durante varias horas, por ejemplo, 4 horas, después de lo cual la mezcla se enfría, por ejemplo, a aproximadamente 75 - 85 °C, seguido de la adición de etanol. La mezcla de reacción puede filtrarse después. El filtrado se enfría, por ejemplo, a aproximadamente 55 - 65 °C, seguido de la adición de agua. La mezcla puede después enfriarse adicionalmente, por ejemplo, a aproximadamente 0 - 10 °C y el compuesto precipitado (II) puede aislarse, por ejemplo, mediante filtración, lavado con agua y etanol y secado al vacío a temperatura elevada, por ejemplo, a 60 °C.
El compuesto (IIIb) puede obtenerse tratando el compuesto (IV) con 1-metil-4-(4,4,5,5-tetrametil-1,3,2-dioxaborlan-2-il)-1H-pirazol usando el mismo procedimiento como se ha descrito anteriormente para la preparación del compuesto (II).
El compuesto (II) puede obtenerse a partir del compuesto (IIIb) tratando el compuesto (IIIb) con cloruro de ciclopropanosulfonilo usando el mismo procedimiento que se describió anteriormente para la preparación del compuesto (III).
Finalmente, el compuesto (II) puede convertirse directamente en un compuesto de fórmula (1A) hidrogenando el compuesto (II) en ácido fórmico en presencia de un catalizador sin necesidad de aislar el compuesto intermedio que tiene un grupo nitro reducido a un grupo amina. El catalizador adecuado incluye catalizadores de paladio tales como paladio al 5 % sobre carbón vegetal. El Compuesto (II), el ácido fórmico y el catalizador se cargan en el reactor inertizado con nitrógeno seguido de la introducción de hidrógeno gaseoso. El reactor se calienta a aproximadamente 30 °C y la hidrogenación continúa hasta que se completa la reacción. El catalizador se retira por filtración y el filtrado se calienta, por ejemplo, a aproximadamente 100 °C. Parte del ácido fórmico puede eliminarse por destilación a presión reducida. En lo sucesivo, se añade adecuadamente 2-propanol y agua a la mezcla de reacción mientras se mantiene la temperatura por encima de aproximadamente 70 °C. El compuesto (1A) puede precipitarse, opcionalmente con siembra, enfriando la mezcla lentamente, por ejemplo, durante aproximadamente 8-10 horas a aproximadamente 0 °C. El compuesto (IA) puede aislarse, por ejemplo, mediante filtración, lavado con 2-propanol y secado al vacío a temperatura elevada, por ejemplo, a 60 °C.
El compuesto obtenido (1A) está normalmente en forma de solvato 1:1 con ácido fórmico. El ácido fórmico puede retirarse del compuesto (1A), por ejemplo, calentando el solvato en un disolvente orgánico adecuado tal como, por ejemplo, 2-butanona, metanol, acetato de etilo, tolueno, metil ferc-butil éter y diclorometano. Por ejemplo, calentar el solvato en 2-butanona a unos 75 °C durante aproximadamente 2 h seguido de enfriar la mezcla, aislar el precipitado por filtración, lavar con 2-butanona y secar al vacío a aproximadamente 60 °C produjo el compuesto (1A) sin ácido fórmico.
Si se desea, el compuesto (1A) puede convertirse en una sal farmacéuticamente aceptable del mismo mediante métodos conocidos en la técnica.
El compuesto de partida (V)

puede prepararse, por ejemplo
a) haciendo reaccionar 1-bromo-3,5-dinitrobenceno de fórmula (VII)

con ácido 2,4-difluorofenilborónico a temperatura elevada en presencia de catalizador, por ejemplo, un catalizador de paladio, y una base, por ejemplo, trimetilamina, en un disolvente adecuado, por ejemplo, disolvente de acetonitrilo-agua, para obtener 2,4-difluoro-3',5'-dinitro-1,1'-bifenilo de fórmula (VI)

; y
b) reducir el compuesto de fórmula (VI) por hidrogenación con gas hidrógeno en presencia de catalizador, por ejemplo, catalizador de paladio, en un disolvente adecuado, por ejemplo, acetato de etilo, para obtener el compuesto de fórmula (V).
La invención se ilustra además mediante los siguientes ejemplos no limitantes.
Ejemplo 1. Preparación de 1-Bromo-3,5-dinitrobenceno (VII)
A un matraz inertizado (N2) se añadió ácido sulfúrico concentrado (500 ml) seguido de 1,3-dinitrobenceno (100 g, 1 equivalente). La masa se agitó hasta su completa disolución. La masa se enfrió a 15 ± 5 °C y se añadió ácido acético (200 ml). La masa se enfrió aún más a 0±5 °C. Se añadió 1,3-dibromo-5,5-dimetilhidantoína (130,9 g, 0,77 equivalentes) en cinco partes iguales a intervalos de 15 min. La masa se agitó durante 1 h y después se calentó a 25 ± 5 °C durante varias horas, seguido de agitación durante 24 h.
A otro matraz se añadió agua (1 l) que se enfrió a 5±5 °C. La masa de reacción se añadió al agua fría durante 1-2 h mientras se mantenía la temperatura < 20 °C. La suspensión resultante se agitó durante 1 h a 25±5 °C. El producto se recogió por filtración y se lavó con agua (500 ml).
Se cargó un matraz a 25±5 °C con agua (1 l) seguido de bicarbonato de sodio (100 g) y se agitó hasta la disolución completa. La torta húmeda obtenida anteriormente se cargó en la solución de bicarbonato. La masa se agitó durante 30-40 min. El producto se recogió por filtración y se lavó con agua (500 ml).
La torta húmeda se volvió a cargar en un matraz junto con agua (1 l). La masa se calentó a 50±5 °C y se agitó durante 1 h. El material se filtró, se lavó con agua (500 ml) y se secó en un horno de vacío a 45±5 °C para dar 135 g (91,9 %) de material cristalino amarillo con una pureza del 99,4 % en HPLC.
Ejemplo 2. Preparación de 2,4-Difluoro-3',5'-dinitro-1,1'-bifenilo (VI)
A un matraz inertizado (N2) se cargó acetonitrilo (600 ml), agua (10 ml) y trietilamina (169,5 ml, 3 equivalentes). Se añadió 1-bromo-3,5-dinitrobenceno (VII) (100 g, 1 equivalente) y la masa se calentó a 70±5 °C en atmósfera de
nitrógeno. La solución se agitó durante 30 min antes de enfriarla a 15±5 °C. Se añadió ácido 2,4-difluorofenilborónico (76,7 g, 1,2 equivalentes) seguido de acetato de paladio (II) (1,28 g, 0,0047 equivalentes). La mezcla se calentó a reflujo (80-85 °C) durante 2 h y se mantuvo durante 5-6 h. La masa se enfrió a 25±5 °C seguido de la adición de dietanolamina (68,1 g, 1,6 equivalentes) y agua (550 ml). La masa se agitó durante 1 hora, después de lo cual los sólidos se recogieron por filtración y se lavaron con agua (210 ml). El producto bruto se secó en una estufa de vacío durante 6 h a 45±5 °C.
Se cargó un matraz inertizado con acetonitrilo (210 ml) y el producto bruto. La masa se calentó a reflujo (85±5 °C) y se agitó durante 30 min. La masa se enfrió durante 3 h a 25±5 °C y se agitó durante una hora adicional. La masa se filtró y se lavó con hexano (100 ml). El material se secó en un horno de vacío a 45±5 °C para dar 86,0 g (75,8 %) de material cristalino de color amarillo pálido a marrón con una pureza del 98,5 % por HPLC.
Ejemplo 3. Preparación de 2',4'-Difluoro-[1,1'-bifenil]-3,5-diamina (V)
Acetato de etilo (1000 ml), 2,4-Difluoro-3',5'-dinitro-1,1'-bifenilo (VI) (100 g, 1 equivalente) y Pd/C (5 g, 10 % Pd sobre carbón, 50 % húmedo con agua) se cargaron en un autoclave inerte (N2). El sistema se lavó con nitrógeno varias veces antes de introducir hidrógeno (0,5 MPa [5 bar]). La masa de reacción se calentó a 40-45 °C y se agitó durante 5-7 h. Una vez completada la reacción, el sistema se lavó a fondo con nitrógeno y el sistema se enfrió a 25±5 °C. El catalizador se filtró y se lavó con acetato de etilo (250 ml). El filtrado se lavó con agua (2 x 700 ml). Se añadió carbón activado (5 g, 5 % en peso) y la mezcla se agitó durante 1 h. El carbón vegetal se filtró y se lavó con acetato de etilo (250 ml). El acetato de etilo se eliminó por destilación al vacío (T < 45 °C). Se añadió tolueno (200 ml) y se eliminó por destilación al vacío (T < 45 °C). Se añadió tolueno (200 ml) y la mezcla se calentó a 40±5 °C. La masa se agitó durante 10 min antes de enfriarla a 25±5 °C durante 2 h. El producto se recogió por filtración y se lavó con tolueno (100 ml). El producto se secó en un horno de vacío a 45±5 °C para dar 70,0 g (89,0 %) de material cristalino amarillo.
Ejemplo 4. Preparación de N3-(4-bromo-2-nitrofenil)-2',4'-difluoro-[1,1'-bifenil]-3,5-diamina (IV)

Un matraz inertizado con N2 se cargó con dimetil sulfóxido (250 ml) seguido de 2',4'-difluoro-[1,1'-bifenil]-3,5-diamina (V) (50 g, 1,0 equivalente). Después se añadieron posteriormente N,N-diisopropiletilamina (9,79 ml, 0,3 equivalentes), etoxitrimetilsilano (38,7 ml, 1,1 equivalentes) y finalmente 4-bromo-2-fluoronitrobenceno (1,0 equivalente). La temperatura del baño se ajustó a 100 °C (temperatura del lote 96-99 °C) y la solución de color rojo oscuro se agitó durante 5 h. El contenido se enfrió a 60±5 °C, después de lo cual se añadió metanol (250 ml). Se añadió agua (250 ml) durante 40 min mientras se mantenía la temperatura del lote a 60±5 °C. Después la masa se enfrió a 20±5 °C durante 1 h y después se agitó aún más a esa temperatura durante 2-3 h. El producto se recogió por filtración. La torta se lavó con agua (200 ml) seguido de metanol (200 ml). El producto se secó en un horno de vacío a 60 °C para dar 86,2 g (92,5 %) de un sólido de color rojo ladrillo a un % de pureza por HPLC del 93,9.
Ejemplo 5. Purificación de N3-(4-bromo-2-nitrofenil)-2',4'-difluoro-[1,1'-bifenil]-3,5-diamina (IV) formando un intermedio de sal mesilato (IVb)

Un matraz inertizado con N2 se cargó con tolueno (860 ml) y 2-butanona (430 ml) seguido de N3-(4-bromo-2-nitrofenil)-2',4'-difluoro-[1,1'-bifenil]-3,5-diamina (IV) (87,4 g, 1,0 equivalente). La mezcla se calentó a 75±5 °C y se agitó hasta su completa disolución. Se añadió ácido metanosulfónico (14,86 ml, 1,1 equivalentes) durante 17 min. La suspensión espesa de color naranja resultante se agitó durante 40 min y después se enfrió a 60±5 °C. Después de agitar durante 60 min, la sal mesilato de N3-(4-bromo-2-nitrofenil)-2',4'-difluoro-[1,1'-bifenil]-3,5-diamina (IVb) se recogió por filtración y se lavó con tolueno (160 ml).
La torta húmeda de sal de mesilato (IVb) se cargó en un matraz inertizado junto con 2-propanol (570 ml) y tolueno (285 ml). La suspensión espesa se calentó a 75±5 °C. Después se añadió trietilamina (31,9 ml, 1,1 equivalentes) durante 30 min. La masa ahora de color rojo brillante se dejó enfriar a 20±5 °C durante la noche. El producto se recogió por filtración y la torta se lavó dos veces con 2-propanol enfriado con hielo (85 ml). El producto se secó en un horno de vacío a 60 °C para dar 75,4 g (86,3 %) de N3-(4-bromo-2-nitrofenil)-2',4-difluoro-[1,1'-bifenil]-3,5-diamina (IV) de 99,95 % de pureza por HPLC.
Ejemplo 6. Preparación de N-(5-((4-bromo-2-nitrofenil)amino)-2',4'-difluoro-[1,1'-bifenil]-3-il)ciclopropanosulfonamida (III)

A un matraz inertizado con N2 se añadió piridina seca (110 ml) seguida de N3-(4-bromo-2-nitrofenil)-2',4'-difluoro-[1,1-bifenil]-3,5-diamina (IV) (36,4 g). La suspensión roja se enfrió a 10±2 °C, después de lo cual se añadió cloruro de ciclopropanosulfonilo (9,71 ml, 1,1 equivalentes) durante 10 min. Después de la adición, la mezcla se agitó durante 1 h seguido de 1 h más de agitación a 20±5 °C. A la solución ahora de color rojo oscuro se añadió agua (7,80 ml, 5 equivalentes) y la mezcla resultante se agitó durante 30 min.
En otro matraz se preparó una solución compuesta por AcOH (127 ml), 2-propanol (110 ml) y agua (145 ml). La solución se calentó a 45±5 °C y se sembraron cristales de N-(5-((4-bromo-2-nitrofenil)amino)-2',4'-difluoro-[1,1-bifenil]-3-il)ciclopropanosulfonamida (III). La masa de reacción se añadió lentamente a esta mezcla mientras se mantenía una temperatura de 45±5 °C. La masa se calentó después a 70±5 °C y se agitó durante 1 hora antes de enfriarla a 20±5 °C. El producto se recogió por filtración y se lavó con 2 x 100 ml de agua y 45 ml de 2-propanol enfriado con hielo. El producto se secó en un horno de vacío a 60 °C para dar 42,9 g (94,5 %) de un sólido cristalino naranja a un % de pureza por HPLC del 98,4.
Ejemplo 7. Preparación de N-(5-((4-bromo-2-nitrofenil)amino)-2',4'-difluoro-[1,1'-bifenil]-3-il)ciclopropanosulfonamida (III) (método alternativo)
A un reactor de 1 l inertizado con nitrógeno se cargó acetato de etilo (210 ml), piridina (67,1 ml, 5 equivalentes) y N3-(4-bromo-2-nitrofenil)-2',4'-difluoro-[1,1'-bifenil]-3,5-diamina (IV) (70 g, 1 equivalente). A esta masa espesa se añadió cloruro de ciclopropanosulfonilo (20,37 ml, 1,2 equivalentes) durante 10 min. La mezcla resultante se calentó a 60±2,5 °C. Después de la finalización de la reacción (aproximadamente 4 h) ácido acético (glacial, 66,8 ml, 7 equivalentes) se añadió seguido de etanol (420 ml). Se añadió lentamente agua (175 ml) mientras se mantenía la temperatura a 60±5 °C. La solución se sembró y después se enfrió a 40 °C durante 2 h. La masa resultante se enfrió a 0 °C durante 3 h y se agitó durante 30 min antes de la filtración. La torta se lavó con agua (210 ml) y etanol (210 ml). Después de secar al vacío (60 °C) se obtuvieron 78,2 g de un sólido cristalino de color naranja brillante con una pureza del 99,8 % en HPLC.
Ejemplo 8. Preparación de 2',4'-difluoro-N3-(4-(1-metil-1H-pirazol-4-il)-2-nitrofenil)-[1,1'-bifenil]-3,5-diamina (II-Ib)
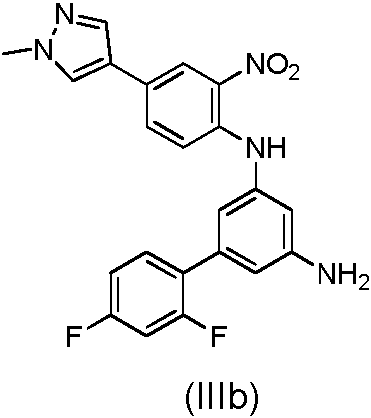
A un matraz inertizado con N2 se añadió dimetilsulfóxido (140 ml) y agua (40 ml). A esta solución se añadió N3-(4-bromo-2-nitrofenil)-2',4'-difluoro-[1,1'-bifenil]-3,5-diamina (IV) (20,0 g, 1 equivalente) seguido de carbonato potásico (8,55 g, 1,3 equivalentes), 1-metil-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-1H-pirazol (12,38 g, 1,25 equivalentes) y carbón activado (2 g, CN1). La mezcla resultante se desgasificó ajustando la presión a 100 mbar durante unos minutos y volviendo a llenar el matraz con nitrógeno. El procedimiento se repitió dos veces. Finalmente se añadieron acetato de paladio (II) (0,08 g, 0,0075 equivalentes) y trifenilfosfina (0,281 g, 0,0225 equivalentes) y la mezcla se calentó a 100±5 °C durante 4 h. Después de alcanzar los 100±5 °C, la mezcla se enfrió de nuevo a 80±5 °C y se
añadió etanol (60 ml). La masa se filtró y la torta se lavó con etanol (20 ml).
La temperatura del filtrado se ajustó a 80±5 °C y se añadió agua (80 ml) durante 30 min. La masa se dejó en agitación durante 30 min y después se dejó enfriar a 20±5 °C. El producto se aisló por filtración y la torta se lavó con agua (40 ml) y etanol (40 ml). El producto se secó en un horno de vacío a 60 °C para dar 20,55 g de un sólido rojo a un % de pureza por HPLC del 99,9.
Ejemplo 9. Preparación de N-(2',4-difluoro-5-((4-(1-metiMH-pirazol-4-il)-2-nitrofenil)aminoH1,1-bifenil]-3-il)ciclopropanosulfonamida (II) del compuesto (III)

A un matraz inertizado con N2 se añadió dimetilsulfóxido (600 ml) y agua (170 ml). A esta solución se añadió N-(5-((4-bromo-2-nitrofenil)-2',4'-difluoro-[1,1'-bifenil]-3-il)ciclopropanosulfonamida) (III) (85,0 g, 1 equivalente) seguido de carbonato potásico (29,1 g, 1,3 equivalentes), 1-metil-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-1H-pirazol (42,2 g, 1,25 equivalentes) y carbón activado (8,5 g, Norit CN1 o SX Ultra). La mezcla resultante se desgasificó ajustando la presión a 100 mbar durante unos minutos y volviendo a llenar el matraz con nitrógeno. El procedimiento se repitió dos veces. Finalmente se añadieron acetato de paladio (II) (0,27 g, 0,0075 equivalentes) y trifenilfosfina (0,96 g, 0,0225 equivalentes) y la mezcla se calentó a 100±5 °C durante 4 h. Después de alcanzar los 100±5 °C, la mezcla se enfrió de nuevo a 80±5 °C y se añadió etanol (255 ml). La masa se filtró y la torta se lavó con etanol (85 ml). La temperatura del filtrado se ajustó a 60±5 °C y se añadió agua (467 ml) durante 1 h. La masa se dejó en agitación durante 1 h y después se dejó enfriar a 10 °C/min a 5±5 °C. El producto se aisló por filtración y la torta se lavó con agua (2 x 60 ml) y etanol (2 x 85 ml). El producto se secó en un horno de vacío a 60 °C para dar 76,9 g (90,2 %) de un sólido rojo a un % de pureza por HPLC del 98,3.
Ejemplo 10. Preparación de N-(2',4'-difluoro-5-((4-(1-metil-1H-pirazol-4-il)-2-nitrofenil)amino)-[1,1'-bifenil]-3-il)ciclopropanosulfonamida (II) del compuesto (IIIb)
Se cargó un matraz con piridina (20 ml) y 2',4'-difluoro-N3-(4-(1-metil-1H-pirazol-4-il)-2-nitrofenil)-[1,1'-bifenil]-3,5-diamina (llb) (5 g, 1 equivalente). La masa gelatinosa se diluyó con 20 ml de piridina. Se añadió cloruro de ciclopropanosulfonilo (1,33 ml, 1,1 equivalentes) a la mezcla durante 1 min. Después de 1 h, se añadieron otros 1,33 ml de cloruro de ciclopropanosulfonilo. Después de 1 h de agitación adicional, se añadió agua (1,07 ml, 5 equivalentes) y la masa se agitó durante 15 min. Se añadió ácido acético (20 ml) y la mezcla se calentó a 45±5 °C. Se añadió 2-propanol (20 ml) seguido de agua (20 ml). Se dejó enfriar la masa a 20±5 °C. El producto se recogió por filtración y se lavó con agua (2 x 20 ml) y 2-propanol (20 ml). El producto se secó en un horno de vacío a 60 °C para dar N-(2',4'-difluoro-5-((4-(1-metil-1H-pirazol-4-il)-2-nitrofenil)amino)-[1,1'-bifenil]-3-il)ciclopropanosulfonamida (ll) (5,65 g, 90,6 %) con una pureza del 99,2 %.
Ejemplo 11. Preparación de N-(2',4'-difluoro-5-(5-(1-metil-1H-pirazol-4-il)-1H-benzo[d]imidazol-1-il)-[1,1'-bifenil]-3-il)ciclopropanosulfonamida (1A)

Se inertizó un reactor con nitrógeno y se cargó con ácido fórmico (280 ml), N-(2',4'-difluoro-5-((4-(1-metil-1H-pirazol-4-il)-2-nitrofenil)amino)-[1,1'-bifenil]-3-il)ciclopropanosulfonamida (40 g) y paladio al 5 % sobre carbón vegetal (4,86 g, 50 % humedecido con agua, 0,015 equivalentes de Pd). Se introdujo hidrógeno (0,2 MPa [2 bares]) después de un lavado minucioso con nitrógeno y el contenido del reactor se calentó a 30±2 °C. La suspensión rojo oscuro se disolvió completamente a medida que avanzaba la reacción. La reacción se detuvo reemplazando el hidrógeno por nitrógeno cuando el líquido cambió de rojo a incoloro (4 h). El catalizador se filtró y el filtrado se calentó a 100±5 °C durante 1 h.
Se destilaron 120 ml de ácido fórmico a presión reducida (10 kPa [100 mbar], 46 °C). Se añadió 2-propanol (106 ml) y la solución se calentó a 80±5 °C. Se añadió agua (240 ml) mientras se mantenía la temperatura > 70 °C. La solución se sembró y la mezcla se enfrió primero de 5 °C/h a 50 °C y después de 10 °C/h a 20 °C. La masa se enfrió aún más a 0±5 °C durante 2 h y después el producto se recogió por filtración. La torta se lavó con 2-propanol (50 ml) y se secó a 60 °C en un horno de vacío. El rendimiento fue de 37,4 g (89,0 %) de un sólido cristalino de color beige claro con una pureza del 99,51 % por HPLC. El compuesto obtenido (1A) estaba en forma de solvato 1:1 con ácido fórmico.
El ácido fórmico se retiró del solvato de ácido fórmico 1:1 (12,3 g) añadiéndolo a un matraz junto con 2-butanona (98,4 ml). La mezcla se calentó a 75 °C y se agitó durante 2 h. La masa se dejó enfriar a temperatura ambiente, después de lo cual se filtró y se lavó con 2-butanona (12,3 ml). El producto se secó en un horno de vacío a 60 °C para dar 10,62 g (94,2 %) del compuesto (1A) como un polvo blanquecino. El compuesto estaba libre de ácido fórmico por 1H-RMN.
Claims (15)
1. Un proceso para la preparación de un compuesto de fórmula (1A) o una sal farmacéuticamente aceptable del mismo

que comprende las etapas de
a) hacer reaccionar 2',4'-difluoro-[1,1'-bifenil]-3,5-diamina de fórmula (V)

con 4-bromo-2-fluoronitrobenceno en presencia de una base orgánica y un organosilano, para obtener un compuesto de fórmula (IV)

b) tratar el producto obtenido de la etapa (a) con un ácido orgánico y aislar la sal orgánica formada;
c) liberar el compuesto de fórmula (IV) de su forma de sal;
d) ya sea (i) haciendo reaccionar el compuesto de fórmula (IV) con cloruro de ciclopropanosulfonilo para obtener un compuesto de fórmula (III)

y posteriormente hacer reaccionar el compuesto de fórmula (III) con 1-metil-4-(4,4,5,5-tetrametil-1,3,2-dioxaborlan-2-il)-1H-pirazol;
o
(ii) hacer reaccionar el compuesto de fórmula (IV) con 1-metil-4-(4,4,5,5-tetrametil-1,3,2-dioxaborlan-2-il)-1H-pirazol para obtener un compuesto de fórmula (IIIb)

y posteriormente hacer reaccionar el compuesto de fórmula (IIIb) con cloruro de ciclopropanosulfonilo; para obtener un compuesto de fórmula (II);

e) hidrogenar el compuesto de fórmula (II) en ácido fórmico en presencia de un catalizador para obtener un compuesto de fórmula (1A), que opcionalmente se convierte en su sal farmacéuticamente aceptable.
2. Un proceso de acuerdo con la reivindicación 1 en donde la base orgánica usada en la etapa a) es N,N-diisopropiletilamina (DIPEA).
3. Un proceso de acuerdo con la reivindicación 1 o 2, en donde el organosilano usado en la etapa a) es etoxitrimetilsilano.
4. Un proceso de acuerdo con cualquiera de las reivindicaciones anteriores, en donde el ácido orgánico usado en la etapa b) es un ácido sulfónico, preferentemente ácido metanosulfónico, ácido p-toluenosulfónico, ácido otoluenosulfónico, ácido etanosulfónico o ácido bencenosulfónico.
5. Un proceso de acuerdo con cualquiera de las reivindicaciones anteriores, en donde la etapa c) se lleva a cabo calentando la sal en un disolvente orgánico en presencia de una base orgánica.
6. Un proceso de acuerdo con cualquiera de las reivindicaciones anteriores, en donde la etapa d) se lleva a cabo haciendo reaccionar el compuesto de fórmula (IV) con cloruro de ciclopropanosulfonilo para obtener un compuesto de fórmula (III) y posteriormente haciendo reaccionar el compuesto de fórmula (III) con 1-metil-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-1H-pirazol para obtener un compuesto de fórmula (II).
7. Un proceso de acuerdo con la reivindicación 6, en donde el compuesto de fórmula (IV) se hace reaccionar con cloruro de ciclopropanosulfonilo en presencia de una base, preferentemente piridina.
8. Un proceso de acuerdo con la reivindicación 6 o 7, en donde el compuesto de fórmula (III) se hace reaccionar con 1-metil-4-(4,4,5,5-tetrametil-1,3,2-dioxaborlan-2-il)-1H-pirazol en presencia de una base, preferentemente carbonato potásico, carbonato de cesio o carbonato sódico, y un catalizador, preferentemente un catalizador de paladio o acetato de paladio (II) complejado con trifenilfosfina.
9. Un proceso de acuerdo con cualquiera de las reivindicaciones anteriores, en donde la etapa d) se lleva a cabo haciendo reaccionar el compuesto de fórmula (IV) con 1-metil-4-(4,4,5,5-tetrametil-1,3,2-dioxaborlan-2-il)-1H-pirazol para obtener un compuesto de fórmula (I II b) y posteriormente hacer reaccionar el compuesto de fórmula (IIIb) con cloruro de ciclopropanosulfonilo para obtener un compuesto de fórmula (II).
10. Un proceso de acuerdo con la reivindicación 9, en donde el compuesto de fórmula (IV) se hace reaccionar con 1-metil-4-(4,4,5,5-tetrametil-1,3,2-dioxaborlan-2-il)-1H-pirazol en presencia de una base y un catalizador.
11. Un proceso de acuerdo con la reivindicación 10, en donde la base es un carbonato, preferentemente carbonato potásico, carbonato de cesio o carbonato de sodio.
12. Un proceso de acuerdo con la reivindicación 10 u 11, en donde el catalizador es un catalizador de paladio, preferentemente acetato de paladio (II) complejado con trifenilfosfina.
13. Un proceso de acuerdo con cualquiera de las reivindicaciones 9-12, en donde el compuesto de fórmula (IIIb) se hace reaccionar con cloruro de ciclopropanosulfonilo en presencia de una base, preferentemente piridina.
14. Un proceso de cualquiera de las reivindicaciones anteriores, en donde el catalizador usado en la etapa e) es un catalizador de paladio, preferentemente paladio sobre carbón.
15. Un compuesto que es de fórmula (V), (IV), (III), (IIIb) o (II)

Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FI20175272 | 2017-03-23 | ||
| PCT/FI2018/050214 WO2018172616A1 (en) | 2017-03-23 | 2018-03-22 | Process for the preparation of a sulfonamide structured kinase inhibitor |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2933807T3 true ES2933807T3 (es) | 2023-02-14 |
Family
ID=61906763
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES18715893T Active ES2933807T3 (es) | 2017-03-23 | 2018-03-22 | Proceso para la preparación de un inhibidor de quinasa estructurado de sulfonamida |
Country Status (8)
| Country | Link |
|---|---|
| US (2) | US10870637B2 (es) |
| EP (2) | EP3601262B1 (es) |
| JP (2) | JP7205018B2 (es) |
| CN (2) | CN115124470B (es) |
| BR (1) | BR112019019589A2 (es) |
| ES (1) | ES2933807T3 (es) |
| FI (1) | FI3601262T3 (es) |
| WO (1) | WO2018172616A1 (es) |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| MY203097A (en) * | 2018-09-06 | 2024-06-09 | Aurigene Oncology Ltd | Novel hydrochloride salt forms of a sulfonamide structured kinase inhibitor |
| EP4006027B1 (en) | 2019-07-26 | 2025-10-01 | CGeneTech (Suzhou, China) Co., Ltd. | Pyridine derivative as fgfr and vegfr dual inhibitors for the treatment of cancer |
| BR112022017758A2 (pt) * | 2020-03-05 | 2022-11-29 | Aurigene Discovery Tech Ltd | Composições farmacêuticas de um inibidor de quinase |
| CA3206502A1 (en) * | 2021-01-26 | 2022-08-04 | Zhengxia CHEN | Crystal form of methylpyrazole-substituted pyridoimidazole compound and preparation method therefor |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8513276B2 (en) * | 2006-12-22 | 2013-08-20 | Astex Therapeutics Limited | Imidazo[1,2-a]pyridine compounds for use in treating cancer |
| PE20120008A1 (es) * | 2009-01-12 | 2012-01-24 | Icagen Inc | Derivados de fenoxi bencenosulfonamida |
| UA111382C2 (uk) * | 2011-10-10 | 2016-04-25 | Оріон Корпорейшн | Інгібітори протеїнкінази |
| US20150152083A1 (en) * | 2012-06-06 | 2015-06-04 | Irm Llc, A Delaware Limited Liability Company | Compounds and Compositions for Modulating EGFR Activity |
| MY203097A (en) * | 2018-09-06 | 2024-06-09 | Aurigene Oncology Ltd | Novel hydrochloride salt forms of a sulfonamide structured kinase inhibitor |
| WO2020135878A1 (zh) * | 2018-12-29 | 2020-07-02 | 南京明德新药研发有限公司 | 作为fgfr和vegfr双重抑制剂的咪唑并吡啶衍生物 |
| EP3985005A4 (en) * | 2019-06-14 | 2023-07-19 | CGeneTech (Suzhou, China) Co., Ltd. | FUSED RING COMPOUND AS DUAL FGFR AND VEGFR INHIBITOR |
-
2018
- 2018-03-22 JP JP2019551696A patent/JP7205018B2/ja active Active
- 2018-03-22 EP EP18715893.6A patent/EP3601262B1/en active Active
- 2018-03-22 US US16/495,865 patent/US10870637B2/en not_active Expired - Fee Related
- 2018-03-22 BR BR112019019589-0A patent/BR112019019589A2/pt active Search and Examination
- 2018-03-22 WO PCT/FI2018/050214 patent/WO2018172616A1/en not_active Ceased
- 2018-03-22 FI FIEP18715893.6T patent/FI3601262T3/fi active
- 2018-03-22 EP EP22199077.3A patent/EP4137487A1/en not_active Withdrawn
- 2018-03-22 CN CN202210807243.0A patent/CN115124470B/zh active Active
- 2018-03-22 CN CN201880018317.4A patent/CN110446704B/zh active Active
- 2018-03-22 ES ES18715893T patent/ES2933807T3/es active Active
-
2020
- 2020-11-20 US US16/953,468 patent/US11332460B2/en active Active
-
2022
- 2022-10-17 JP JP2022166520A patent/JP7377330B2/ja active Active
Also Published As
| Publication number | Publication date |
|---|---|
| WO2018172616A1 (en) | 2018-09-27 |
| US11332460B2 (en) | 2022-05-17 |
| EP3601262A1 (en) | 2020-02-05 |
| JP2023011667A (ja) | 2023-01-24 |
| CN110446704B (zh) | 2022-07-05 |
| CN115124470B (zh) | 2024-08-27 |
| US20210139462A1 (en) | 2021-05-13 |
| CN115124470A (zh) | 2022-09-30 |
| US10870637B2 (en) | 2020-12-22 |
| JP2020512323A (ja) | 2020-04-23 |
| JP7205018B2 (ja) | 2023-01-17 |
| BR112019019589A2 (pt) | 2020-04-22 |
| FI3601262T3 (fi) | 2023-01-13 |
| US20200102291A1 (en) | 2020-04-02 |
| JP7377330B2 (ja) | 2023-11-09 |
| CN110446704A (zh) | 2019-11-12 |
| EP4137487A1 (en) | 2023-02-22 |
| EP3601262B1 (en) | 2022-11-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2933807T3 (es) | Proceso para la preparación de un inhibidor de quinasa estructurado de sulfonamida | |
| RU2554081C2 (ru) | Способ синтеза соединений диарилтиогидантоина и диарилгидантоина | |
| ES2881373T3 (es) | Un procedimiento mejorado para la preparación de apixabán e intermedios del mismo | |
| ES2923278T3 (es) | Intermedios útiles para la síntesis de derivados de aminopirimidinas, proceso de preparación del mismo y proceso de preparación de aminopirimidina derivados utilizando los mismos | |
| CN112047888B (zh) | 一种合成恩杂鲁胺的方法 | |
| WO2012028925A2 (en) | An improved process for the preparation of telmisartan | |
| JP7731005B2 (ja) | ベンズイミダゾール誘導体の調製方法 | |
| CN102351778A (zh) | 一种盐酸阿比朵尔的制备方法 | |
| ES2451350T3 (es) | Procedimientos para la preparación de compuestos de 2-(1-feniletil)isoindolin-1-ona | |
| CN102001979A (zh) | 2-(2’,2’-二氟乙氧基)-6-三氟甲基苯基丙基硫醚的制备方法 | |
| HK40081621A (en) | Process for the preparation of a sulfonamide structured kinase inhibitor | |
| ES2844100T3 (es) | Proceso para la preparación de anagrelida y análogos de la misma | |
| CA2882138C (en) | Process for the synthesis of substituted gamma lactams | |
| HK40021800B (en) | Process for the preparation of a sulfonamide structured kinase inhibitor | |
| HK40021800A (en) | Process for the preparation of a sulfonamide structured kinase inhibitor | |
| BR122025011693A2 (pt) | Processo para a preparação de um inibidor de quinase estruturado por sulfonamida | |
| CN116813553A (zh) | 一种多氮杂芳环化合物的选择性氘代n-甲基化方法 | |
| KR100334462B1 (ko) | 에틸 2-(2,3,4,5-테트라플루오로벤조일)-3(s)-(1-히드록시프로프-2-일아미노)아크릴레이트의 제조방법 | |
| KR20190039087A (ko) | 정제된 세니크리비록 및 세니크리비록을 제조하기 위한 정제된 중간체 | |
| HK40051429B (zh) | 四环化合物的制备方法 |