ES2929200T3 - Tienopirroles sustituidos como inhibidores de PAD4 - Google Patents
Tienopirroles sustituidos como inhibidores de PAD4 Download PDFInfo
- Publication number
- ES2929200T3 ES2929200T3 ES19759134T ES19759134T ES2929200T3 ES 2929200 T3 ES2929200 T3 ES 2929200T3 ES 19759134 T ES19759134 T ES 19759134T ES 19759134 T ES19759134 T ES 19759134T ES 2929200 T3 ES2929200 T3 ES 2929200T3
- Authority
- ES
- Spain
- Prior art keywords
- substituted
- alkyl
- independently selected
- occurrence
- ring
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D495/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms
- C07D495/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D495/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/439—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom the ring forming part of a bridged ring system, e.g. quinuclidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D519/00—Heterocyclic compounds containing more than one system of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring system not provided for in groups C07D453/00 or C07D455/00
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Epidemiology (AREA)
- Rheumatology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Immunology (AREA)
- Pain & Pain Management (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Physical Education & Sports Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
Abstract
La presente invención proporciona compuestos de fórmula (I) útiles como inhibidores de PAD4, composiciones de los mismos y métodos para tratar trastornos relacionados con PAD4. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Tienopirroles sustituidos como inhibidores de PAD4
Antecedentes de la invención
La PAD4 es un miembro de la familia de enzimas peptidilarginina deiminasa (PAD) capaces de catalizar la citrulinación de arginina en citrulina en secuencias peptídicas. La PAD4 es responsable de la deiminación o citrulinación de diversas proteínas in vitro e in vivo, con las consecuencias de diversas respuestas funcionales en diversas enfermedades (Jones J. E. et al., Curr. Opin. Drug Discov. Devel., 12(5), (2009), 616-627). Los ejemplos de enfermedades ilustrativas incluyen artritis reumatoide, enfermedades con contribuciones neutrófilas a la patogenia (por ejemplo, vasculitis, lupus eritematoso sistémico, colitis ulcerosa) además de indicaciones oncológicas. Los inhibidores de la PAD4 también tienen una aplicabilidad más amplia como herramientas y terapias para enfermedades humanas a través de mecanismos epigenéticos.
Los inhibidores de PAD4 tienen utilidad contra la artritis reumatoide (AR). La AR es una enfermedad autoinmunitaria que afecta aproximadamente al 1 % de la población (Wegner N. et al., Immunol. Rev., 233(1) (2010), 34-54). Se caracteriza por inflamación de las articulaciones que conduce a la destrucción debilitante de hueso y cartílago. Se ha sugerido una asociación genética débil entre los polimorfismos de PAD4 y la susceptibilidad a la AR, aunque de manera inconsistente, en varios estudios de población (Kochi Y. et al., Ann. Rheum. Dis., 70, (2011),512-515). Se ha detectado PAD4 (junto con un miembro de la familia PAD2) en el tejido sinovial, donde es responsable de la deiminación de diversas proteínas articulares. Se presume que este proceso conduce a una ruptura de la tolerancia y al inicio de respuestas inmunitarias a sustratos citrulinados tales como fibrinógeno, vimentina y colágeno en las articulaciones de la AR. Estos anticuerpos anti-proteína citrulinada (ACPA, Anti-Citrullinated Protein Antibodies) contribuyen a la patogenia de la enfermedad y también pueden usarse como una prueba de diagnóstico para la AR (por ejemplo, la prueba CCP2 disponible comercialmente o la prueba de proteína cíclica citrulinada 2). Además, el aumento de la citrulinación también puede ofrecer contribuciones directas adicionales a la patogenia de la enfermedad a través de su capacidad para afectar directamente a la función de varios mediadores inflamatorios y articulares (por ejemplo, fibrinógeno, antitrombina, múltiples quimiocinas). En un subconjunto más pequeño de pacientes con AR, se pueden medir los anticuerpos anti-PAD4 y pueden correlacionarse con una forma más erosiva de la enfermedad.
Los inhibidores de PAD4 también son útiles para la reducción de la actividad patológica de los neutrófilos en diversas enfermedades. Los estudios sugieren que el proceso de formación de la trampa extracelular de neutrófilos (NET, Neutrophil Extracellular Trap), un mecanismo de defensa innato por el cual los neutrófilos pueden inmovilizar y destruir patógenos, está asociado con la citrulinación de histonas y es deficiente en ratones nuligénicos para PAD4 (Neeli I. et al., J. Immunol., 180, (2008), 1895-1902 y Li P. et al, J. Exp. Med., 207(9), (2010), 1853-1862). Por lo tanto, los inhibidores de PAD4 pueden tener aplicabilidad para enfermedades en las que la formación de NET en los tejidos contribuye a lesiones locales y patologías de la enfermedad. Dichas enfermedades incluyen, aunque sin limitación, vasculitis de vasos pequeños (Kessenbrock K. et al., Nat. Med., 15(6), (2009), 623-625), lupus eritematoso sistémico (Hakkim A. et al., Proc. Natl. Acad. Sci. EE.UU., 107(21), (2010), 9813-9818 y Villanueva E. et al, J. Immunol., 187(1), (2011), 538-52), colitis ulcerosa (Savchenko A. et al., Pathol. Int., 61(5), (2011), 290-7), fibrosis quística, asma (Dworski R. et al., J. Allergy Clin. Immunol., 127(5), (2011), 1260-6), trombosis venosa profunda (Fuchs T. et al., Proc. Natl. Acad. Sci. EE.UU., 107(36), (2010), 15880-5), periodontitis (Vitkov L. et al., Ultrastructural Pathol., 34(1), (2010), 25-30), septicemia (Clark S. R. et al., Nat. Med., 13(4), (2007), 463-9), apendicitis (Brinkmann V. et al., Science, 303, (2004), 1532-5) e ictus. Además, hay evidencia de que las NET pueden contribuir a la patología en enfermedades que afectan a la piel, por ejemplo, en el lupus eritematoso cutáneo (Villanueva E. et al., J. Immunol., 187(1), (2011), 538-52) y la psoriasis (Lin A. M. et al., J. Immunol., 187(1), (2011), 490-500), por lo que un inhibidor de PAD4 puede mostrar beneficios para combatir las enfermedades cutáneas NET, cuando se administra por vía sistémica o cutánea. Los inhibidores de la PAD4 pueden afectar a funciones adicionales dentro de los neutrófilos y tienen una aplicabilidad más amplia a las enfermedades neutrofílicas.
Los estudios han demostrado la eficacia de los inhibidores de PAD como herramienta (por ejemplo, cloroamidina) en varios modelos animales de enfermedad, incluida la artritis inducida por colágeno (Willis V. C. et al., J. Immunol., 186(7), (2011), 4396-4404), colitis experimental inducida por dextrano sulfato sódico (DSS) (Chumanevich A. A. et al., Am. J. Physiol. Gastrointest. Liver Physiol., 300(6), (2011), G929-G938), reparación de la médula espinal (Lange S. et al., Dev. Biol., 355(2), (2011), 205-14) y encefalomielitis autoinmune experimental (EAE). El informe de colitis DSS también demuestra que la cloroamidina impulsa la apoptosis de las células inflamatorias tanto in vitro como in vivo, lo que sugiere que los inhibidores de PAD4 pueden ser eficaces de manera más general en enfermedades inflamatorias generalizadas.
Los inhibidores de PAD4 también son útiles en el tratamiento de cánceres (Slack.J. L. et al., Cell. Mol. Life Sci., 68(4), (2011), 709-720). Se ha demostrado la sobreexpresión de PAD4 en numerosos cánceres (Chang X. et al., BMC Cancer, 9, (2009), 40). Se ha sugerido un papel antiproliferativo para los inhibidores de PAD4 a partir de la observación de que PAD4 citrulina residuos de arginina en histonas en los promotores de genes diana de p53 tales como p21, que están implicados en la detención del ciclo celular y la inducción de apoptosis (Li P. et al., Mol. Cell
Biol., 28(15), (2008), 4745-4758).
Se ha informado sobre la síntesis de inhibidores reversibles de PAD4 a través de la arilación C-H de bencimidazol catalizada por cobre (Guo. Z et al., Sci, China Chem, 62(5), (2019), 592-596). Los documentos WO 2017/100594 y WO 2017/147102 desvelan compuestos útiles como inhibidores de la PAD4, composiciones de los mismos y métodos para tratar trastornos relacionados con PAD4. El documento WO 2019/161803 describe compuestos inhibidores de PAD4 que tienen un efecto inhibidor sobre PAD4 y pueden usarse para tratar diversas afecciones.
El papel mencionado anteriormente de PAD4 en la deiminación de residuos de arginina en histonas puede ser indicativo de un papel de PAD4 en la regulación epigenética de la expresión génica. PAD4 es el miembro primario de la familia PAD que se observa que reside en el núcleo, así como y en el citoplasma. La evidencia preliminar de que PAD4 puede actuar como una histona desmetiliminasa, así como una deiminasa es inconsistente y no está probada. Sin embargo, puede reducir la metilación de la histona-arginina (y, por tanto, la regulación epigenética asociada con esta marca) indirectamente a través del agotamiento de los residuos de arginina disponibles mediante la conversión en citrulina. Los inhibidores de PAD4 son útiles como herramientas epigenéticas o terapéuticas para afectar a la expresión de diversos genes diana en entornos de enfermedad adicionales. A través de dichos mecanismos, los inhibidores de PAD4 también pueden ser eficaces para controlar los niveles de citrulinación en las células madre y, por lo tanto, pueden afectar terapéuticamente al estado de pluripotencia y el potencial de diferenciación de diversas células madre incluyendo, pero sin limitación, células madre embrionarias, células madre neuronales, células madre hematopoyéticas y células madre cancerosas. Por consiguiente, sigue existiendo la necesidad insatisfecha de identificar y desarrollar inhibidores de PAD4 para el tratamiento de trastornos mediados por PAD4.
Sumario de la invención
Se ha descubierto ahora que los compuestos de Fórmula (I) son útiles como inhibidores de PAD4:

en donde cada uno del anillo A, R1, R2, R3, L, R4, R7 y otras variables es como se define en el presente documento.
En algunas realizaciones, un compuesto proporcionado demuestra selectividad por PAD4 con respecto a PAD2. La presente invención también proporciona composiciones farmacéuticamente aceptables que comprenden un compuesto proporcionado. Los compuestos proporcionados son útiles en el tratamiento de diversos trastornos asociados con PAD4. Dichos trastornos se describen con detalle en el presente documento e incluyen, por ejemplo, artritis reumatoide, vasculitis, lupus eritematoso sistémico, colitis ulcerosa, cáncer, fibrosis quística, asma, lupus eritematoso cutáneo y psoriasis.
Descripción detallada de la invención
1. Descripción general de ciertos aspectos de la invención
En algunas realizaciones, dichos compuestos incluyen los de las fórmulas descritas en el presente documento, o una sal farmacéuticamente aceptable de los mismos, en donde cada variable es como se define en el presente documento y se describe en las realizaciones. Dichos compuestos tienen la estructura de Fórmula (I):

o una de sus sales farmacéuticamente aceptables, en donde:
El anillo A es heterociclilo de 4 a 15 miembros sustituido con 1-4 R7;
Ri se selecciona entre -CH3 y CD3;
R2 se selecciona entre H, alquilo C1-3 sustituido con 0-5 Re, -(CH2)r-cicloalquilo C3-6 con 0-5 Re;
L se selecciona entre -(CRdRd)0-3-, -NRa-, -S(O)p- y -C(=O)-;
R3 se selecciona entre H, F, Cl, Br y -ORb;
R4 se selecciona entre H, F, Cl, Br, -CN, -ORb, alquilo C1-6 sustituido con 1-5 R5, arilo sustituido con 1-5 R5, cicloalquilo C3-12 sustituido con 1-5 R5 y heterociclilo que comprende átomos de carbono y 1-3 heteroátomos seleccionados entre N, NR6, O y S y sustituido con 1-5 R5;
R5, en cada caso, se selecciona independientemente entre H, F, Cl, Br, alquilo C1-4 sustituido con 0-4 Re, alquenilo C2-4 sustituido con 0-4 Re, alquinilo C2-4 sustituido con 0-4 Re, nitro, -(CH2)rS(O)pRc, -(CH2)rS(O)pNRaRa, -(CH2)rNRaS(O)pRc, -(CH2)rORb, -(CH2)rCN, -(CH2)rNRaRa, -(CH2)rNRaC(=O)Rb, -(CH2)rNRaC(=O)NRaRa, -(CH2)rC(=O)ORb, -(CH2)rC(=O)Rb, -(CH2)rOC(=O)Rb, -(CH2)rC(=O)NRaRa, P(=O)(Oalquilo ^ .4)2, -P(=O)(alquilo C1-4)2, cicloalquilo C3-6 sustituido con 0-4 Re, arilo sustituido con 0-4 Re, heterociclilo sustituido con 0-4 Re y heteroarilo sustituido con 0-4 Re;
R6, en cada caso, se selecciona independientemente entre H, alquilo C1-3 sustituido con 0-5 Re, -S(O)pRc, -C(=O)Rb, -C(=O)(CH2)rNRaRa, -C(=O)(CH2)rNRaC(=O)Rb, -C(=O)ORb, -S(O)pNRaRa, -(CH2)r-cicloalquilo C3-6 sustituido con 0-4 Re, -(CH2)r-arilo sustituido con 0-4 Re, -(CH2)r-heterociclilo sustituido con 0-4 Re y -(CH2)rheteroarilo sustituido con 0-4 Re;
R7 se selecciona entre H, F, Cl, CN, alquilo C1-3, =N-ORb, -(CH2)rORb, -(CH2)rNRaRa, -NRaC(=NH)alquilo C1-3, -NRaC(=O)ORb, carbociclilo y un heterociclilo; como alternativa, dos grupos R7 se toman juntos para formar carbociclilo o heterociclilo;
Ra, en cada caso, se selecciona independientemente entre H, alquilo C1-6 sustituido con 0-5 Re, alquenilo C2-6 sustituido con 0-5 Re, alquinilo C2-6 sustituido con 0-5 Re, -(CH2)r-carbociclilo C3-10 sustituido con 0-5 Re y -(CH2)rheterociclilo sustituido con 0-5 Re; o Ra y Ra junto con el átomo de nitrógeno al que ambos están unidos forman un anillo heterocíclico sustituido con 0-5 Re;
Rb, en cada caso, se selecciona independientemente entre H, alquilo C1-6 sustituido con 0-5 Re, alquenilo C2-6 sustituido con 0-5 Re, alquinilo C2-6 sustituido con 0-5 Re, -(CH2)r-carbociclilo C3-10 sustituido con 0-5 Re y -(CH2)rheterociclilo sustituido con 0-5 Re;
Rc, en cada caso, se selecciona independientemente entre alquilo C1-6 sustituido con 0-5 Re, alquenilo C2-6 sustituido con 0-5 Re, alquinilo C2-6 sustituido con 0-5 Re, -(CH2)r-carbociclilo C3--6 sustituido con 0-5 Re y -(CH2)rheterociclilo sustituido con 0--5 Re;
Rd, en cada caso, se selecciona independientemente entre H y alquilo C1-6 sustituido con 0-5 Re;
Re, en cada caso, se selecciona independientemente entre alquilo C1-6 sustituido con 0-5 Rf, alquenilo C2-6, alquinilo C2-6, -(CH2)r-cicloalquilo C3-6, -(CH2)r-arilo, F, Cl, Br, CN, NO2, =O, -C(=O)OH, -C(=O)Oalquilo C1-4, -(CH2)rOH y -(CH2)rOalquilo C1-4;
Rf, en cada caso, se selecciona independientemente entre H, F, Cl, Br, CN, OH, alquilo C1-5 opcionalmente sustituido con OH, cicloalquilo C3-6 y fenilo;
p, en cada caso, se selecciona independientemente entre cero, 1 y 2;
r, en cada caso, se selecciona independientemente entre cero, 1, 2, 3 y 4; y
proporcionado cuando L se selecciona entre -NRa-, -S(O)p- y -C(=O)-, R4 se selecciona entre arilo sustituido con 1-5 R5, cicloalquilo C3-12 sustituido con 1-5 R5 y heterociclilo que comprende átomos de carbono y 1-3 heteroátomos seleccionados entre N, NR6, O y S y sustituido con 1-5 R5.
2. Definiciones
A lo largo de la memoria descriptiva y las reivindicaciones adjuntas, una fórmula o nombre químico dado abarcará todos los estereoisómeros e isómeros ópticos y racematos del mismo cuando existan dichos isómeros. A menos que se indique de otra manera, todas las formas quirales (enantioméricas y diastereoméricas) y racémicas están dentro del alcance de la presente invención. Muchos isómeros geométricos de dobles enlaces C=C, dobles enlaces C=N, sistemas anulares y similares pueden estar presentes también en los compuestos y todos estos isómeros estables están contemplados en la presente invención. Se describen los isómeros geométricos cis y trans (o E y Z) de los compuestos de la presente invención y pueden aislarse en forma de una mezcla de isómeros o como formas isoméricas separadas. Los presentes compuestos pueden aislarse en formas ópticamente activas o racémicas. Las formas ópticamente activas se pueden preparar por resolución de las formas racémicas o mediante síntesis a partir de materiales de partida ópticamente activos. Se considera que todos los procesos usados para preparar los compuestos de la presente invención y los intermedios elaborados con los mismos forman parte de la presente invención. Cuando se preparan productos enantioméricos o diastereoméricos, estos se pueden separar por métodos convencionales, por ejemplo, por cromatografía o cristalización fraccionada. Dependiendo de las condiciones de proceso los productos finales de la presente invención se obtienen tanto en forma libre (neutral) como de sal. Tanto la forma libre como las sales de estos productos finales están dentro del alcance de la invención. Si se desea, una forma de un compuesto se puede convertir en otra forma. Una base libre o ácida se puede convertir en una sal; una sal se puede convertir en el compuesto libre o en otra sal; una mezcla de compuestos isoméricos de la presente invención se puede separar en los isómeros individuales. Los compuestos de la presente invención, la forma libre y las sales de los mismos, pueden existir en múltiples formas tautoméricas, en las que los átomos de hidrógeno se transponen a otras partes de las moléculas y los enlaces químicos entre los átomos de las moléculas se reordenan en consecuencia. Debe entenderse que todas las formas tautoméricas, en la medida en que puedan existir, se incluyen dentro de la invención.
Como se usa en el presente documento, el término "alquilo" o "alquileno" pretende incluir grupos hidrocarburo alifáticos saturados, tanto de cadena ramificada como lineal, que tienen el número especificado de átomos de carbono. Como ejemplos, se pretende que "alquilo C1 a C12" o "alquilo C1-12" (o alquileno) incluyan los grupos alquilo C1, C2, C3, C4, C5, Ce, C7, C8, C9, C10, C11 y C12; se pretende que "alquilo C4 a C18" o "alquilo C4-18" (o alquileno) incluya los grupos alquilo C4, C5, Ce, C7, C8, C9, C10, C11, C12, C13, C14, C15, C16, C17 y C18. Además, por ejemplo, "alquilo C1 a Ce" o "alquilo C1-6" representa alquilo que tiene de 1 a 6 átomos de carbono. El grupo alquilo puede estar sin sustituir o sustituido con al menos un hidrógeno que está reemplazado por otro grupo químico. Los ejemplos de grupos alquilo incluyen, aunque sin limitación, metilo (Me), etilo (Et), propilo (por ejemplo, n-propilo e isopropilo), butilo (por ejemplo, n-butilo, isobutilo, t-butilo) y pentilo (por ejemplo, n-pentilo, isopentilo, neopentilo). Cuando se usa "alquilo Co" o "alquileno Co", se pretende indicar un enlace directo.
"Alquenilo" o "alquenileno" pretende incluir cadenas de hidrocarburo tanto de configuración lineal como ramificada que tienen el número especificado de átomos de carbono y uno o más, preferentemente uno o dos, dobles enlaces carbono-carbono que pueden aparecer en cualquier punto estable a lo largo de la cadena. Por ejemplo, se pretende que "alquenilo C2 a C6" o "alquenilo C2-6" (o alquenileno), incluya los grupos alquenilo C2, C3, C4, C5 y C6. Los ejemplos de alquenilo incluyen, aunque sin limitación, etenilo, 1-propenilo, 2-propenilo, 2-butenilo, 3-butenilo, 2-pentenilo, 3, pentenilo, 4-pentenilo, 2-hexenilo, 3-hexenilo, 4-hexenilo, 5-hexenilo, 2-metil-2-propenilo y 4-metil-3-pentenilo.
"Alquinilo" o "alquinileno" pretende incluir cadenas hidrocarburo de configuración tanto lineal como ramificada que tienen uno o más, preferentemente de uno a tres, triples enlaces carbono-carbono que pueden aparecer en cualquier punto estable a lo largo de la cadena. Por ejemplo, se pretende que "alquinilo C2 a Ce" o "alquinilo C2-6" (o alquinileno) incluya los grupos alquinilo C2, C3, C4, C5 y C6; tales como etinilo, propinilo, butinilo, pentinilo y hexinilo.
El término "alcoxi" o "alquiloxi" se refiere a un grupo -O-alquilo. Por ejemplo, se pretende que "alcoxi C1 a C6" o "alcoxi C1-6" (o alquiloxi) incluya los grupos alcoxi C1, C2, C3, C4, C5 y C6. Los ejemplos de grupos alcoxi incluyen, aunque sin limitación, metoxi, etoxi, propoxi (por ejemplo, n-propoxi e isopropoxi) y t-butoxi. De forma análoga, "alquiltio" o "tioalcoxi" representa un grupo alquilo tal como se ha definido anteriormente con el número indicado de átomos de carbono unidos a través de un puente de azufre; por ejemplo, metil-S- y etil-S-.
"Halo" o "halógeno" incluye flúor, cloro, bromo y yodo. "Haloalquilo" pretende incluir grupos hidrocarburo alifáticos saturados tanto de cadena ramificada como lineal que tienen el número especificado de átomos de carbono, sustituidos con 1 o más halógenos. Los ejemplos de haloalquilo incluyen, aunque sin limitación, fluorometilo, difluorometilo, trifluorometilo, triclorometilo, pentafluoroetilo, pentacloroetilo, 2,2,2-trifluoroetilo, heptafluoropropilo y heptacloropropilo. Los ejemplos de haloalquilo también incluyen "fluoroalquilo", lo que pretende incluir grupos hidrocarburo alifáticos saturados, tanto de cadena ramificada como lineal, que tienen el número especificado de átomos de carbono, sustituidos por 1 o más átomos de flúor.
El término "cicloalquilo" se refiere a grupos alquilo ciclados, incluyendo sistemas anulares monocíclicos, bicíclicos o policíclicos. Por ejemplo, se pretende que "cicloalquilo C3 a C6" o "cicloalquilo C3-6" incluya los grupos cicloalquilo C3, C4, C5 y C6. Los ejemplos de grupos cicloalquilo incluyen, aunque sin limitación, ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo y norbornilo. Se incluyen en la definición de "cicloalquilo" los grupos cicloalquilo ramificados tales como 1-metilciclopropilo y 2-metilciclopropilo. El término "cicloalquenilo" se refiere a grupos alquenilo ciclados. Se pretende que cicloalquenilo C4-6 incluya grupos cicloalquenilo C4, C5 y C6. Los ejemplos de grupos cicloalquenilo incluyen, aunque sin limitación, ciclobutenilo, ciclopentenilo y ciclohexenilo.
Como se usa en el presente documento, se pretende que "carbociclo", "carbociclilo" o "residuo carbocíclico" signifique cualquier anillo hidrocarbonado monoclíclico o bicíclico de 3, 4, 5, 6, 7 u 8 miembros o bicíclico o tricíclico de 7, 8-, 9-, 10-, 11, 12 o 13 miembros, cualquiera de los cuales puede estar saturado, parcialmente insaturado, insaturado o aromático. Los ejemplos de dichos carbociclos incluyen, aunque sin limitación, ciclopropilo, ciclobutilo, ciclobutenilo, ciclopentilo, ciclopentenilo, ciclohexilo, cicloheptenilo, cicloheptilo, cicloheptenilo, adamantilo, ciclooctilo, ciclooctenilo, ciclooctadienilo, [3.3.0]biciclooctano, [4.3.0]biciclononano, [4.4.0]biciclodecano (decalina), [2.2.2]biciclooctano, fluorenilo, fenilo, naftilo, indanilo, adamantilo, antracenilo y tetrahidronaftilo (tetralina). Como se ha mostrado anteriormente, los anillos puenteados también están incluidos en la definición de carbociclo (por ejemplo, [2.2.2]biciclooctano). Los carbociclos preferidos, a menos que se indique otra cosa, son ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, fenilo, indanilo y tetrahidronaftilo. Cuando se usa el término "carbociclo", se pretende incluir "arilo". Se produce un anillo puenteado cuando uno o más, preferentemente de uno a tres, átomos de carbono enlazan dos átomos de carbono no adyacentes. Los puentes preferidos son uno o dos átomos de carbono. Cabe apreciar que un puente siempre convierte un anillo monocíclico en un anillo tricíclico. Cuando un anillo está puenteado, los sustituyentes citados para el anillo también pueden estar presentes en el puente.
Como se usa en el presente documento, la expresión "carbociclo bicíclico" o "grupo carbocíclico bicíclico" pretende indicar un sistema de anillo carbocíclico estable de 9 o 10 miembros que contiene dos anillos condensados y consiste en átomos de carbono. De los dos anillos condensados, un anillo es un anillo benzo condensado a un segundo anillo; y el segundo anillo es un anillo de carbono de 5 o 6 miembros que está saturado, parcialmente
insaturado o insaturado. El grupo carbocíclico bicíclico puede estar unido a su grupo colgante en cualquier átomo de carbono que dé como resultado una estructura estable. El grupo carbocíclico bicíclico descrito en el presente documento puede estar sustituido en cualquier carbono si el compuesto resultante es estable. Son ejemplos de un grupo carbocíclico bicíclico, pero sin limitación, naftilo, 1,2-dihidronaftilo, 1,2,3,4-tetrahidronaftilo e indanilo.
Grupos "arilo" se refiere a hidrocarburos aromáticos monocíclicos o bicíclicos, lo que incluye, por ejemplo, fenilo y naftilo. Los restos arilo se conocen bien y se describen, por ejemplo, en Lewis, R. J., ed., Hawley's Condensed Chemical Dictionary, 15a edición, John Wiley & Sons, Inc., Nueva York (2007). "Arilo C6-10" se refiere a fenilo y naftilo.
Como se usa en el presente documento, se pretende que el término "heterociclo", "heterociclilo" o "grupo heterocíclico" signifique un anillo monocíclico o bicíclico de 3, 4, 5, 6 o 7 miembros o policíclico heterocíclico de 7, 8, 9, 10, 11, 12, 13 o 14 miembros que está saturado, parcialmente insaturado o totalmente insaturado y que contiene átomos de carbono y 1, 2, 3 o 4 heteroátomos seleccionados independientemente entre el grupo que consiste en N, O y S; y que incluye cualquier grupo policíclico en el que cualquiera de los anillos heterocíclicos definidos anteriormente está condensado con un anillo de benceno. Los heteroátomos de nitrógeno y azufre pueden estar opcionalmente oxidados (es decir, N ^O y S(O)p, en donde p es 0, 1 o 2). El átomo de nitrógeno puede estar sustituido o sin sustituir (es decir, N o NR en el que R es H u otro sustituyente, si se define). El anillo heterocíclico puede estar unido a su grupo colgante en cualquier heteroátomo o átomo de carbono que dé como resultado una estructura estable. Los anillos heterocíclicos descritos en el presente documento pueden estar sustituidos en un átomo de carbono o en uno de nitrógeno en caso de que el compuesto resultante sea estable. Un nitrógeno del heterociclo puede estar opcionalmente cuaternizado. Se prefiere que cuando el número total de átomos S y O en el heterociclo exceda de 1, entonces estos heteroátomos no sean adyacentes entre sí. Se prefiere que el número total de átomos de S y O en el heterociclo no sea mayor de 1. Cuando se usa el término "heterociclo", se pretende incluir heteroarilo.
Los ejemplos de heterociclos incluyen, aunque sin limitación, acridinilo, azetidinilo, azocinilo, benzoimidazolilo, benzofuranilo, benzotiofuranilo, benzotiofenilo, benzoxazolilo, benzoxazolinilo, benzotiazolilo, benzotriazolilo, benzotetrazolilo, benzoisoxazolilo, benzoisotiazolilo, benzoimidazolinilo, carbazolilo, 4aH-carbazolilo, carbolinilo, cromanilo, cromenilo, cinnolinilo, decahidroquinolinilo, 2H,6H-1,5,2-ditiazinilo, dihidrofuro[2,3-b]tetrahidrofurano, furanilo, furazanilo, imidazolidinilo, imidazolinilo, imidazolilo, 1H-indazolilo, imidazolopiridinilo, indolenilo, indolinilo, indolizinilo, indolilo, 3H-indolilo, isatinoílo, isobenzofuranilo, isocromanilo, isoindazolilo, isoindolinilo, isoindolilo, isoquinolinilo, isotiazolilo, isotiazolopiridinilo, isoxazolilo, isoxazolopiridinilo, metilenodioxifenilo, morfolinilo, naftiridinilo, octahidroisoquinolinilo, oxadiazolilo, 1,2,3-oxadiazolilo, 1,2,4-oxadiazolilo, 1,2,5-oxadiazolilo, 1,3,4-oxadiazolilo, oxazolidinilo, oxazolilo, oxazolopiridinilo, oxazolidinilperimidinilo, oxindolilo, pirimidinilo, fenantridinilo, fenantrolinilo, fenazinilo, fenotiazinilo, fenoxatiinilo, fenoxazinilo, ftalazinilo, piperazinilo, piperidinilo, piperidonilo, 4-piperidonilo, piperonilo, pteridinilo, purinilo, piranilo, pirazinilo, pirazolidinilo, pirazolinilo, pirazolopiridinilo, pirazolilo, piridazinilo, piridooxazolilo, piridoimidazolilo, piridotiazolilo, piridinilo, pirimidinilo, pirrolidinilo, pirrolinilo, 2-pirrolidonilo, 2H-pirrolilo, pirrolilo, quinazolinilo, quinolinilo, 4H-quinolizinilo, quinoxalinilo, quinuclidinilo, tetrazolilo, tetrahidrofuranilo, tetrahidroisoquinolinilo, tetrahidroquinolinilo, 6H-1,2,5-tiadiazinilo, 1,2,3-tiadiazolilo, 1,2,4-tiadiazolilo, 1,2,5-tiadiazolilo, 1,3,4-tiadiazolilo, tiantrenilo, tiazolilo, tienilo, tiazolopiridinilo, tienotiazolilo, tienooxazolilo, tienoimidazolilo, tiofenilo, triazinilo, 1,2,3-triazolilo, 1,2,4-triazolilo, 1,2,5-triazolilo, 1,3,4-triazolilo y xantenilo. También se incluyen anillos condensados y compuestos espiro que contienen, por ejemplo, los heterociclos anteriores.
Los ejemplos de heterociclos de 5 a 10 miembros incluyen, aunque sin limitación, piridinilo, furanilo, tienilo, pirrolilo, pirazolilo, pirazinilo, piperazinilo, piperidinilo, imidazolilo, imidazolidinilo, indolilo, tetrazolilo, isoxazolilo, morfolinilo, oxazolilo, oxadiazolilo, oxazolidinilo, tetrahidrofuranilo, tiadiazinilo, tiadiazolilo, tiazolilo, triazinilo, triazolilo, benzoimidazolilo, 1H-indazolilo, benzofuranilo, benzotiofuranilo, benzotetrazolilo, benzotriazolilo, benzoisoxazolilo, benzoxazolilo, oxindolilo, benzoxazolinilo, benzotiazolilo, benzoisotiazolilo, isatinoílo, isoquinolinilo, octahidroisoquinolinilo, tetrahidroisoquinolinilo, tetrahidroquinolinilo, isoxazolopiridinilo, quinazolinilo, quinolinilo, isotiazolopiridinilo, tiazolopiridinilo, oxazolopiridinilo, imidazolopiridinilo y pirazolopiridinilo.
Los ejemplos de heterociclos de 5 a 6 miembros incluyen, aunque sin limitación, piridinilo, furanilo, tienilo, pirrolilo, pirazolilo, pirazinilo, piperazinilo, piperidinilo, imidazolilo, imidazolidinilo, indolilo, tetrazolilo, isoxazolilo, morfolinilo, oxazolilo, oxadiazolilo, oxazolidinilo, tetrahidrofuranilo, tiadiazinilo, tiadiazolilo, tiazolilo, triazinilo y triazolilo. También se incluyen anillos condensados y compuestos espiro que contienen, por ejemplo, los heterociclos anteriores.
Como se usa en el presente documento, la expresión "heterociclo bicíclico" o "grupo heterocíclico bicíclico" pretende indicar un sistema anular heterocíclico estable de 9 o 10 miembros que contiene dos anillos condensados y que consiste en átomos de carbono y 1, 2, 3 o 4 heteroátomos seleccionados independientemente del grupo que consiste en N, O y S. De los dos anillos condensados, un anillo es un anillo aromático monocíclico de 5 o 6 miembros que comprende un anillo heteroarilo de 5 miembros, un anillo heteroarilo de 6 miembros o un anillo benzo, cada uno condensado a un segundo anillo. El segundo anillo es un anillo monocíclico de 5 o 6 miembros el cual está saturado, parcialmente insaturado o insaturado y comprende un heterociclo de 5 miembros, un heterociclo de 6 miembros o un carbociclo (con la condición de que el primer anillo no sea benzo cuando el segundo anillo sea un
carbociclo).
El grupo heterocíclico bicíclico puede estar unido a su grupo colgante en cualquier heteroátomo o átomo de carbono que dé como resultado una estructura estable. El grupo heterocíclico bicíclico descrito en el presente documento puede estar sustituido en un átomo de carbono o en uno de nitrógeno si el compuesto resultante es estable. Se prefiere que cuando el número total de átomos S y O en el heterociclo exceda de 1, entonces estos heteroátomos no sean adyacentes entre sí. Se prefiere que el número total de átomos de S y O en el heterociclo no sea superior a 1. Los ejemplos de grupo heterocíclico bicíclico son, pero sin limitación, quinolinilo, isoquinolinilo, ftalazinilo, quinazolinilo, indolilo, isoindolilo, indolinilo, 1H-indazolilo, benzoimidazolilo, 1,2,3,4-tetrahidroquinolinilo, 1,2,3,4-tetrahidroisoquinolinilo, 5,6,7,8-tetrahidro-quinolinilo, 2,3-dihidro-benzofuranilo, cromanilo, 1,2,3,4-tetrahidroquinoxalinilo y 1,2,3,4-tetrahidro-quinazolinilo.
Como se usa en el presente documento, se pretende que la expresión "grupo heterocíclico aromático" o "heteroarilo" signifique hidrocarburos aromáticos monocíclicos y policíclicos estables que incluyen al menos un miembro de anillo de heteroátomos tal como azufre, oxígeno o nitrógeno. Los grupos heteroarilo incluyen, sin limitación, piridilo, pirimidinilo, pirazinilo, piridazinilo, triazinilo, furilo, quinolilo, isoquinolilo, tienilo, imidazolilo, tiazolilo, indolilo, pirroílo, oxazolilo, benzofurilo, benzotienilo, benzotiazolilo, isoxazolilo, pirazolilo, triazolilo, tetrazolilo, indazolilo, 1,2,4-tiadiazolilo, isotiazolilo, purinilo, carbazolilo, benzoimidazolilo, indolinilo, benzodioxolanilo y benzodioxano. Los grupos heteroarilo están sustituidos o sin sustituir. El átomo de nitrógeno está sustituido o sin sustituir (es decir, N o NR en el que R es H u otro sustituyente, si se define). Los heteroátomos de nitrógeno y azufre pueden estar opcionalmente oxidados (es decir, N ^O y S(O)p, en donde p es 0, 1 o 2).
Los ejemplos de heteroarilos de 5 a 6 miembros incluyen, aunque sin limitación, piridinilo, furanilo, tienilo, pirrolilo, pirazolilo, pirazinilo, imidazolilo, imidazolidinilo, tetrazolilo, isoxazolilo, oxazolilo, oxadiazolilo, oxazolidinilo, tiadiazinilo, tiadiazolilo, tiazolilo, triazinilo y triazolilo.
Los anillos con puentes también están incluidos en la definición de heterociclo. Se produce un anillo puenteado cuando uno o más, preferentemente de uno a tres, átomos (es decir, C, O, N o S) unen dos átomos de carbono o nitrógeno no adyacentes. Los ejemplos de anillos puenteados incluyen, aunque sin limitación, un átomo de carbono, dos átomos de carbono, un átomo de nitrógeno, dos átomos de nitrógeno y un grupo de carbono-nitrógeno. Cabe apreciar que un puente siempre convierte un anillo monocíclico en un anillo tricíclico. Cuando un anillo está puenteado, los sustituyentes citados para el anillo también pueden estar presentes en el puente.
El término "contraión" se usa para representar una especie cargada negativamente tal como cloruro, bromuro, hidróxido, acetato y sulfato o una especie cargada positivamente, tal como sodio (Na+), potasio (K+), amonio (RnNHm+ donde n = 0-4 y m = 0-4) y similares.
Cuando se usa un anillo punteado dentro de una estructura de anillo, esto indica que la estructura de anillo puede estar saturada, parcialmente saturada o insaturada.
Como se usa en el presente documento, la expresión "grupo protector de amina" significa cualquier grupo conocido en la técnica de síntesis orgánica para la protección de grupos amina que sea estable a un agente reductor de éster, una hidrazina disustituida, R4-M y R7-M, un nucleófilo, un agente reductor de hidrazina, un activador, una base fuerte, una base de amina impedida y un agente de ciclación. Tales grupos protectores de amina que encajan en estos criterios incluyen los enumerados en Wuts, P.G.M. et al., Protecting Groups in Organic Synthesis, 4a Edición, Wiley (2007) y The Peptides: Analysis, Synthesis, Biology, Vol. 3, Academic Press, Nueva York (1981). Los ejemplos de grupos protectores de amina incluyen, aunque sin limitación, los siguientes: (1) los de tipo acilo, tales como formilo, trifluoroacetilo, ftalilo y p-toluenosulfonilo; (2) los de tipo carbamato aromático, tales como benciloxicarbonilo (Cbz) y benciloxicarbonilos sustituidos, 1-(p-bifenil)-1-metiletoxicarbonilo y 9-fluorenilmetiloxicarbonilo (Fmoc); (3) los de tipo carbamato alifático, tales como terc-butiloxicarbonilo (Boc), etoxicarbonilo, diisopropilmetoxicarbonilo y aliloxicarbonilo; (4) los tipo alquil carbamato cíclicos, tales como ciclopentiloxicarbonilo y adamantiloxicarbonilo; (5) los de tipo alquilo, tales como trifenilmetilo y bencilo; (6) trialquilsilano tal como trimetilsilano; (7) los tipos que contienen tiol, tales como feniltiocarbonilo y ditiasuccinoílo; y (8) los de tipo alquilo, tales como trifenilmetilo, metilo y bencilo; y los de tipo alquilo sustituidos, tales como 2,2,2-tricloroetilo, 2-feniletilo, y f-butilo; y los de tipo trialquilsilano, tales como trimetilsilano.
Como se cita en el presente documento, el término "sustituido" significa que al menos un átomo de hidrógeno se reemplaza con un grupo distinto de hidrógeno, con la condición de que las valencias normales se mantengan y que la sustitución dé como resultado un compuesto estable. Los dobles enlaces de anillo, tal como se usan en el presente documento, son dobles enlaces que se forman entre dos átomos adyacentes del anillo (por ejemplo, C=C, C=N o N=N).
En los casos donde hay átomos de nitrógeno (por ejemplo, aminas) en los compuestos de la presente invención, estos se pueden convertir en N-óxidos mediante tratamiento con un agente oxidante (por ejemplo, mCPBA y/o peróxido de hidrógenos) para proporcionar otros compuestos de esta invención. De esta manera, se considera que
los átomos de nitrógeno mostrados y reivindicados incluyen tanto el nitrógeno mostrado como su derivado de N-óxido (N^O).
Cuando cualquier variable aparece más de una vez en cualquier constituyente o fórmula para un compuesto, su definición cada vez que aparece es independiente de su definición en cualquier otro caso. De esta manera, por ejemplo, si se muestra que un grupo está sustituido con 0-3 R, entonces dicho grupo puede sustituirse opcionalmente con hasta tres grupos R y cada vez que aparece R se selecciona independientemente a partir de la definición de R.
Cuando se muestra un enlace a un sustituyente que cruza un enlace que conecta dos átomos en un anillo, entonces dicho sustituyente puede unirse a cualquier átomo del anillo. Cuando se enumera un sustituyente sin indicar el átomo en el que dicho sustituyente está unido al resto del compuesto de una fórmula dada, entonces dicho sustituyente puede estar unido a través de cualquier átomo de dicho sustituyente.
Solo se permiten las combinaciones de sustituyentes y/o variables en caso de que dichas combinaciones den como resultado compuestos estables.
En el presente documento la expresión "farmacéuticamente aceptable" se emplea para referirse a aquellos compuestos, materiales, composiciones y/o formas de dosificación que son, dentro del alcance del buen criterio médico, adecuados para su uso en contacto con los tejidos de seres humanos y animales sin excesiva toxicidad, irritación, respuesta alérgica y/u otro problema o complicación, acorde con una relación beneficio/riesgo razonable.
Como se usa en el presente documento, "sales farmacéuticamente aceptables" se refiere a derivados de los compuestos divulgados en los que el compuesto precursor se modifica fabricando sales ácidas o básicas del mismo. Los ejemplos de sales farmacéuticamente aceptables incluyen, aunque sin limitación, sales de ácidos minerales u orgánicos de grupos básicos tales como aminas; y sales alcalinas u orgánicas de grupos ácidos tales como ácidos carboxílicos. Las sales farmacéuticamente aceptables incluyen las sales no tóxicas convencionales o las sales de amonio cuaternario del compuesto precursor formadas, por ejemplo, de ácidos orgánicos o inorgánicos no tóxicos. Son ejemplos de sales de adición de ácidos no tóxicas farmacéuticamente aceptables las sales de un grupo amino formadas con ácidos inorgánicos, tales como ácido clorhídrico, ácido bromhídrico, ácido fosfórico, ácido sulfúrico y ácido perclórico o con ácidos orgánicos, tales como ácido acético, ácido oxálico, ácido maleico, ácido tartárico, ácido cítrico, ácido succínico o ácido malónico o usando otros métodos usados en la técnica, tales como intercambio iónico. Otras sales farmacéuticamente aceptables incluyen las sales adipato, alginato, ascorbato, aspartato, bencenosulfonato, benzoato, bisulfato, borato, butirato, alcanforato, alcanforsulfonato, citrato, ciclopentanopropionato, digluconato, dodecilsulfato, etanosulfonato, formiato, fumarato, glucoheptonato, glicerofosfato, gluconato, hemisulfato, heptanoato, hexanoato, yodhidrato, 2-hidroxi-etanosulfonato, lactobionato, lactato, laurato, lauril sulfato, malato, maleato, malonato, metanosulfonato, 2-naftalenosulfonato, nicotinato, nitrato, oleato, oxalato, palmitato, pamoato, pectinato, persulfato, 3-fenilpropionato, fosfato, pivalato, propionato, estearato, succinato, sulfato, tartrato, tiocianato, p-toluenosulfonato, undecanoato, valerato y similares.
Las sales derivadas de bases adecuadas incluyen sales de metales alcalinos, metales alcalinotérreos, de amonio y N+(alquilo C-m )4. Las sales de metales alcalinos o alcalinotérreos representativas incluyen sodio litio, potasio, calcio, magnesio y similares. Otras sales farmacéuticamente aceptables incluyen, cuando sea adecuado, amonio no tóxico, amonio cuaternario y cationes de amina formados usando contraiones, tales como haluro, hidróxido, carboxilato, sulfato, fosfato, nitrato, sulfonato de alquilo inferior y sulfonato de arilo.
Las sales farmacéuticamente aceptables de la presente invención se pueden sintetizar a partir del compuesto precursor que contiene un resto básico o ácido mediante métodos convencionales. En general, dichas sales se pueden preparar haciendo reaccionar las formas de ácido o de base libre de estos compuestos con una cantidad estequiométrica de la base o el ácido apropiado en agua o en un disolvente orgánico o en una mezcla de los dos; en general, se prefieren los medios no acuosos como éter, acetato de etilo, etanol, isopropanol o acetonitrilo. Se encuentran listas de sales adecuadas en Allen, Jr., L.V., ed., Remington: The Science and Practice of Pharmacy, 22a edición, Pharmaceutical Press, Londres, RU (2012).
La presente invención pretende incluir todos los isótopos de los átomos que aparecen en los presentes compuestos. Los isótopos incluyen aquellos átomos que tienen el mismo número atómico pero diferentes números másicos. A modo de ejemplo general y sin limitación, los isótopos de hidrógeno incluyen deuterio y tritio. Los isótopos de carbono incluyen 13C y 14C. Los compuestos marcados con isótopos de la invención pueden prepararse generalmente por técnicas convencionales conocidas por los expertos en la materia o por procesos análogos a los descritos en el presente documento, usando un reactivo marcado con isótopos apropiado en lugar del reactivo no marcado empleado de otro modo.
El término "solvato" significa una asociación física de un compuesto de esta invención con una o más moléculas de disolvente, ya sea orgánico o inorgánico. Esta asociación física incluye enlaces de hidrógeno. En ciertos casos el solvato podrá aislarse, por ejemplo, cuando se incorporan una o más moléculas de disolvente a la red cristalina del sólido cristalino. Las moléculas de disolvente en el solvato pueden estar presentes en una disposición regular y/o
una disposición no ordenada. El solvato puede comprender una cantidad tanto estequiométrica como no estequiométrica de las moléculas de disolvente. "Solvato" abarca solvatos tanto en fase de solución como aislables. Los solvatos a modo de ejemplo incluyen, aunque sin limitación, hidrato, etanolatos, metanolatos e isopropanolatos. Los métodos de solvatación se conocen habitualmente en la técnica.
Las expresiones "afinidad medible" e "inhibir de forma medible", tal como se usan en el presente documento, significan un cambio medible en la actividad de PAD4 entre una muestra que comprende un compuesto de la presente invención, o composición del mismo, y PAD4, y una muestra equivalente que comprende PAD4 en ausencia de dicho compuesto o composición del mismo.
Las abreviaturas tal como se usan en el presente documento, se definen como sigue: "1 x" para una vez, "2 x" para dos veces, "3 x" para tres veces, "°C" para grados Celsius, "equiv." para equivalente o equivalentes, "g" para gramo o gramos, "mg" para miligramo o miligramos, "l" para litro o litros, " ml" para mililitro o mililitros, "jl" para microlitro o microlitros, "N" para normal, "M" para molar, "mmol" para milimol o milimoles, "min" para minuto o min, "h" para hora o h, "ta" para temperatura ambiente, "TR" para tiempo de retención, "atm" para atmósfera, "kPa, psi" para kilopascal (libras por pulgada cuadrada), "conc." para concentrado, "ac." para "acuoso", "sat." para saturado, "PM" para peso molecular, "pf' para punto de fusión, "MS" o "Espec. de Masas" para espectrometría de masas, "ESI" para espectroscopia de masas con ionización por electronebulización, "HR" para alto rendimiento, "HRMS" para espectrometría de masas de alto rendimiento, "LCMS" para cromatografía liquida-espectrometría de masas, "HpLC" para cromatografía líquida de alto rendimiento, "RP HPLC" para HPLC de fase inversa, "TLC" o "tlc" para cromatografía de capa fina, "RMN" para espectroscopía de resonancia magnética nuclear, "nOe" para espectroscopía nuclear de efecto Overhauser, "1H" para protón, "8" para delta, "s" para singlete, "d" para doblete, "t" para triplete, "c" para cuartete, "m" para multiplete, "a" para ancho, "Hz" para hercio y "a", "p", "R", "S", "E", "Z" y "ee" son designaciones estereoquímicas familiares para un experto en la técnica. Como se usa en el presente documento, la expresión "sal farmacéuticamente aceptable" se refiere a las sales que son, dentro del alcance del buen criterio médico, adecuadas para su uso en contacto con los tejidos de seres humanos o animales inferiores sin excesiva toxicidad, irritación, respuesta alérgica y similares, y son proporcionadas con una relación beneficio/riesgo razonable.
AcOH u HOAc ácido acético
ACN acetonitrilo
Alq Alquilo
AlMea Trimetilaluminio
BBr3 tribromuro de boro
Bn bencilo
Boc ferc-butilo oxicarbonilo
reactivo BOP hexafluorofosfato de benzotriazol-1-iloxitris(dimetilamino)fosfonio Bu butilo
/-Bu isobutilo
t-Bu ferc-butilo
t-BuOH ferc-butanol
Cbz carbobenciloxi
CDCla deutero-cloroformo
CDaOD deutero-metanol
CH2Cl2 diclorometano
CHaCN acetonitrilo
CHCla cloroformo
DCM diclorometano
DIEA, DIPEA o diisopropiletilamina
base de Hunig, DMF dimetilformamida
DMSO dimetilsulfóxido
Et etilo
EtaN o TEA trietilamina
Et2O éter dietílico
EtOAc acetato de etilo
EtOH etanol
HCl ácido clorhídrico
HPLC cromatografía líquida de alto rendimiento
K2CO3 carbonato potásico
K2HPO4 hidrogenofosfato potásico
LCMS cromatografía líquida-espectrometría de masas
LiHMDS bis(trimetilsilil)amida de litio
LG grupo saliente
Me metilo
MeOH metanol
MgSO4 sulfato de magnesio
MsOH o MSA ácido metilsulfónico
NaCl cloruro sódico
Na2CO3 carbonato sódico
NaHCO3 bicarbonato sódico
NaOH hidróxido sódico
Na2SO4 sulfato de sodio
NH3 amoniaco
NH4Cl cloruro de amonio
NH4OAc acetato amónico
Pd(OAc)2 acetato de paladio (II)
Pd(dppf)Cl2 Dicloruro de [1,1'-bis(difenilfosfino)ferroceno]paladio
Pd(PPh3)4 tetraquis(trifenilfosfina)paladio (0)
PG grupo protector
Ph fenilo
Pr propilo
i-Pr isopropilo
i-PrOH o IPA isopropanol
Tr tiempo de retención
SiO2 óxido de sílice
SFC (Supercritica l F lu id Chrom atography) cromatografía de fluidos supercríticos
TBAI yoduro de tetrabutilamonio
TEA trietilamina
TFA ácido trifluoroacético
TFAA anhídrido trifluoroacético
THF tetrahidrofurano
TiCl4 tetracloruro de titanio
T3P anhídrido cíclico del ácido 1-propanofosfónico
3. Descripción de compuestos ilustrativos
En un primer aspecto, la presente invención proporciona un compuesto de Fórmula (I):

o una de sus sales farmacéuticamente aceptables, en donde:
El anillo A es heterociclilo de 4 a 15 miembros sustituido con 1-4 R7;
R1 se selecciona entre -CH3 y CD3;
R2 se selecciona entre H, alquilo C1-3 sustituido con 0-5 Re y -(CH2)r-cicloalquilo C3-6 con 0-5 Re;
R3 se selecciona entre H, F, Cl, Br y -ORb;
L se selecciona entre -(CRdRd)0-3-, -NRa-, -S(O)p- y -C(=O)-;
R4 se selecciona entre H, F, Cl, Br, -CN, -ORb, alquilo C1-6 sustituido con 1-5 R5, arilo sustituido con 1-5 R5, cicloalquilo C3-12 sustituido con 1-5 R5 y heterociclilo que comprende átomos de carbono y 1-3 heteroátomos seleccionados entre N, NR6, O y S y sustituido con 1-5 R5;
R5, en cada caso, se selecciona independientemente entre H, F, Cl, Br, alquilo C1-4 sustituido con 0-4 Re, alquenilo C2-4 sustituido con 0-4 Re, alquinilo C2-4 sustituido con 0-4 Re, nitro, -(CH2)rS(O)pRc, -(CH2)rS(O)pNRaRa, -(CH2)rNRaS(O)pRc, -(CH2)rORb, -(CH2)rCN, -(CH2)rNRaRa, -(CH2)rNRaC(=O)Rb, -(CH2)rNRaC(=O)NRaRa, -(CH2)rC(=O)ORb, -(CH2)rC(=O)Rb, -(CH2)rOC(=O)Rb, -(CH2)rC(=O)NRaRa, -P(=O)(Oalquilo ^.4)2, -P(=O)(alquilo C1-4)2, cicloalquilo C3-6 sustituido con 0-4 Re, arilo sustituido con 0-4 Re, heterociclilo sustituido con 0-4 Re y heteroarilo sustituido con 0-4 Re;
R6, en cada caso, se selecciona independientemente entre H, alquilo C1-3 sustituido con 0-5 Re, -S(O)pRc, -C(=O)Rb, -C(=O)(CH2)rNRaRa, -C(=O)(CH2)rNRaC(=O)Rb, -C(=O)ORb, -S(O)pNRaRa, -(CH2)r-cicloalquilo C3-6 sustituido con 0-4 Re, -(CH2)r-arilo sustituido con 0-4 Re, -(CH2)r-heterociclilo sustituido con 0-4 Re y -(CH2)rheteroarilo sustituido con 0-4 Re;
R7 se selecciona entre H, F, Cl, CN, alquilo C1-3, =N-ORb, -(CH2)rORb, -(CH2)rNRaRa, -NRaC(=NH)alquilo C1.3, -NRaC(=O)ORb, carbociclilo y heterociclilo; como alternativa, dos grupos R7 se toman juntos para formar carbociclilo o heterociclilo;
Ra, en cada caso, se selecciona independientemente entre H, alquilo C1-6 sustituido con 0-5 Re, alquenilo C2-6
sustituido con 0-5 Re, alquinilo C2-6 sustituido con 0-5 Re, -(CH2)r-carbociclilo C3-10 sustituido con 0-5 Re y -(CH2)rheterocidilo sustituido con 0-5 Re; o Ra y Ra junto con el átomo de nitrógeno al que ambos están unidos forman un anillo heterocíclico sustituido con 0-5 Re;
Rb, en cada caso, se selecciona independientemente entre H, alquilo C1-6 sustituido con 0-5 Re, alquenilo C2-6 sustituido con 0-5 Re, alquinilo C2-6 sustituido con 0-5 Re, -(CH2)r-carbociclilo C3-10 sustituido con 0-5 Re y -(CH2)rheterociclilo sustituido con 0-5 Re;
Rc, en cada caso, se selecciona independientemente entre alquilo C1-6 sustituido con 0-5 Re, alquenilo C2-6 sustituido con 0-5 Re, alquinilo C2-6 sustituido con 0-5 Re, -(CH2)r-carbociclilo C3--6 sustituido con 0-5 Re y -(CH2)rheterociclilo sustituido con 0--5 Re;
Rd, en cada caso, se selecciona independientemente entre H y alquilo C1-6 sustituido con 0-5 Re;
Re, en cada caso, se selecciona independientemente entre alquilo C1-6 sustituido con 0-5 Rf, alquenilo C2-6, alquinilo C2-6, -(CH2)r-cicloalquilo C3-6, -(CH2)r-arilo, F, Cl, Br, CN, NO2, =O, -C(=O)OH, -C(=O)Oalquilo C1.4, -(CH2)rOH y -(CH2)rOalquilo C1.4;
Rf, en cada caso, se selecciona independientemente entre H, F, Cl, Br, CN, OH, alquilo C1-5 opcionalmente sustituido con OH, cicloalquilo C3-6 y fenilo;
p, en cada caso, se selecciona independientemente entre cero, 1 y 2;
r, en cada caso, se selecciona independientemente entre cero, 1, 2, 3 y 4; y
proporcionado cuando L se selecciona entre -NRa-, -S(O)p- y -C(=O)-, R4 se selecciona entre arilo sustituido con 1-5 R5, cicloalquilo C3-12 sustituido con 1-5 R5 y heterociclilo que comprende átomos de carbono y 1-3 heteroátomos seleccionados entre N, NR6, O y S y sustituido con 1-5 R5.
En un segundo aspecto, la presente invención proporciona un compuesto de Fórmula (II):

o una de sus sales farmacéuticamente aceptables, dentro del alcance del primer aspecto, en donde:
el Anillo A se selecciona entre

R2 se selecciona entre -CH3, -CH2CH3 -y CH2-ciclopropilo;
R3 se selecciona entre H, F, Cl, Br y -Oalquilo C1-4;
R4 se selecciona entre F, Cl, Br, alquilo C1-5 sustituido con 1-4 R5


R5, en cada caso, se selecciona independientemente entre H, F, Cl, Br, alquilo C1-4, alquenilo C2-4, alquinilo C2-4, nitro, -S(O)pRc, -S(O)pNRaRa, -NRaS(O)pRc, - ORb, -CN, -NRaRa, -NRaC(=O)Rb, -NRaC(=O)NRaRa, -C(=O)ORb, -C(=O)Rb, -OC(=O)Rb, -C(=O)NRaRa, -P(=O)(alquilo C1-4)2, cicloalquilo C3.6 sustituido con 0-4 Re, arilo sustituido con 0-4 Re, heterociclilo sustituido con 0-4 Re y heteroarilo sustituido con 0-4 Re;
R6, en cada caso, se selecciona independientemente entre H, alquilo C1-3 sustituido con 0-5 Re, -S(O)pRc, -C(=O)Rb, -C(=O)NRaRa, -C(=O)ORb, -S(O)pNRaRa, -(CH2)r-cicloalquilo C3-6 sustituido con 0-4 Re, -(CH2)r-arilo sustituido con 0-4 Re, -(CH2)r-heterociclilo sustituido con 0-4 Re y -(CH2)r-heteroarilo sustituido con 0-4 Re;
Ra, en cada caso, se selecciona independientemente entre H, alquilo C1-6 sustituido con 0-5 Re, alquenilo C2-6 sustituido con 0-5 Re, alquinilo C2-6 sustituido con 0-5 Re, -(CH2)r-carbociclilo C3-10 sustituido con 0-5 Re y -(CH2)rheterociclilo sustituido con 0-5 Re; o Ra y Ra junto con el átomo de nitrógeno al que ambos están unidos forman un anillo heterocíclico sustituido con 0-5 Re;
Rb, en cada caso, se selecciona independientemente entre H, alquilo C1-6 sustituido con 0-5 Re, alquenilo C2-6 sustituido con 0-5 Re, alquinilo C2-6 sustituido con 0-5 Re, -(CH2)r-carbociclilo C3-10 sustituido con 0-5 Re y -(CH2)rheterociclilo sustituido con 0-5 Re;
Rc, en cada caso, se selecciona independientemente entre alquilo C1-6 sustituido con 0-5 Re, alquenilo C2-6 sustituido con 0-5 Re, alquinilo C2-6 sustituido con 0-5 Re, -(CH2)r-carbociclilo C3--6 sustituido con 0-5 Re y -(CH2)rheterociclilo sustituido con 0--5 Re;
Re, en cada caso, se selecciona independientemente entre alquilo C1-6 sustituido con 0-5 Rf, alquenilo C2-6, alquinilo C2-6, -(CH2)r-cicloalquilo C3-6, -(CH2)r-arilo, F, Cl, Br, CN, NO2, =O, C(=O)OH, -C(=O)Oalquilo C1.4, -(CH2)rOH y -(CH2)rOalquilo C1.4;
Rf, en cada caso, se selecciona independientemente entre H, F, Cl, Br, CN, OH, alquilo C1-5 opcionalmente sustituido con OH, cicloalquilo C3-6 y fenilo;
p, en cada caso, se selecciona independientemente entre cero, 1 y 2; y
r, en cada caso, se selecciona independientemente entre cero, 1, 2, 3 y 4.
En un tercer aspecto, la presente invención proporciona un compuesto de Fórmula (III):

o una de sus sales farmacéuticamente aceptables, en donde:
el Anillo A se selecciona entre

R2 se selecciona entre -CH3 y CH2-ciclopropilo;
R3 es -Oalquilo C1-4;
R4 se selecciona entre

R5, en cada caso, se selecciona independientemente entre H, F, Cl, Br, alquilo C1-4, -ORb, -CN, -S(O)PRc, S(O)pNRaRa, -NRaS(O)pRc, -NRaC(=O)NRaRa, -C(=O)ORb, -C(=O)Rb, -OC(=O)Rb, -C(=O)NRaRa y -NHC(=O)Rb; R6, en cada caso, se selecciona independientemente entre H, alquilo C1-3 sustituido con 0-5 Re, -C(=O)Rb, C(=O)ORb,

Ra, en cada caso, se selecciona independientemente entre H, alquilo Ci -6 sustituido con 0-5 Re, -(CH2)rcarbociclilo C3-10 sustituido con 0-5 Re y -(CH2)r-heterociclilo sustituido con 0-5 Re; o Ra y Ra junto con el átomo de nitrógeno al que ambos están unidos forman un anillo heterocíclico sustituido con 0-5 Re;
Rb, en cada caso, se selecciona independientemente entre H, alquilo C1-6 sustituido con 0-5 Re, alquenilo C2-6 sustituido con 0-5 Re, alquinilo C2-6 sustituido con 0-5 Re, -(CH2)r-carbociclilo C3-10 sustituido con 0-5 Re y -(CH2)rheterociclilo sustituido con 0-5 Re;
Re, en cada caso, se selecciona independientemente entre alquilo C1-6 sustituido con 0-5 Rf, alquenilo C2-6, alquinilo C2-6, -(CH2)r-cicloalquilo C3-6, -(CH2)r-arilo, F, Cl, Br;
Rf, en cada caso, se selecciona independientemente entre H, F, Cl, Br, CN, OH, alquilo C1-5 opcionalmente sustituido con OH, cicloalquilo C3-6 y fenilo;
r, en cada caso, se selecciona independientemente entre cero, 1,2, 3 y 4.
En un cuarto aspecto, la presente invención proporciona un compuesto o una sal farmacéuticamente aceptable del mismo, en donde:
el Anillo A es

R2 se selecciona entre -CH3 y CH2-ciclopropilo;
R3 es -OCH3;
R4 se selecciona entre

R5, en cada caso, se selecciona independientemente entre H, alquilo C1-4, CN, OH, -C(=O)NRaRa y NHC(=O)alquilo C1-4; y
R6, en cada caso, se selecciona independientemente entre H, metilo, etilo y -C(=O)-alquilo C1-4.
En un quinto aspecto, la presente invención proporciona un compuesto de Fórmula (IV):

o una de sus sales farmacéuticamente aceptables, dentro del alcance del primer aspecto, en donde:
el Anillo A se selecciona entre

R2 se selecciona entre -CH3, -CH2CH3 -y CH2-ciclopropilo;
R3 se selecciona entre H, F, Cl, Br y -Oalquilo C1-4;
R4 se selecciona entre H, F, Cl, Br, alquilo C1-3 sustituido con 0-4 Re,

R5, en cada caso, se selecciona independientemente entre H, F, Cl, Br, alquilo C1-4;
R6, en cada caso, se selecciona independientemente entre H, alquilo C1.6 sustituido con 0-5 Re, -(CH2)r arilo sustituido con 0-4 Re y -(CH2)r-heterociclilo sustituido con 0-4 Re;
Re, en cada caso, se selecciona independientemente entre alquilo C1-6 sustituido con 0-5 Rf, alquenilo C2-6, alquinilo C2-6, -(CH2)r-cicloalquilo C3-6, -(CH2)r-arilo, F, Cl, Br, CN, NO2, =O, C(=O)OH, -C(=O)Oalquilo C1.4, -(CH2)rOH y -(CH2)rOalquilo C1.4;
Rf, en cada caso, se selecciona independientemente entre H, F, Cl, Br, CN, OH, alquilo C1-5
opcionalmente sustituido con OH, cicloalquilo C3-6 y fenilo; y
r, en cada caso, se selecciona independientemente entre cero, 1, 2, 3 y 4.
En un sexto aspecto, la presente invención proporciona un compuesto o una sal farmacéuticamente aceptable del mismo, dentro del alcance del quinto aspecto, en donde:
el Anillo A se selecciona entre

R2 es -CH2-ciclopropilo;
R3 es -Oalquilo C1-4;
R4 se selecciona entre H, F, Cl, Br, alquilo C1-3,

y
R6 se selecciona entre H y alquilo C1-3.
En un séptimo aspecto, la presente invención proporciona un compuesto o una sal farmacéuticamente aceptable del mismo, dentro del alcance del segundo aspecto, en donde:
L es -(CHRd)0-;
R4 es alquilo C1-5 sustituido por 1-3 R5;
R5, en cada caso, se selecciona independientemente entre H, F, Cl, Br, alquilo C1-4, - ORb, -CN, -NRaRa y -C(=O)NRaRa,
Ra, en cada caso, se selecciona independientemente entre H y alquilo C1-6; y
Rb, en cada caso, se selecciona independientemente entre H y alquilo C1-6.
En un octavo aspecto, la presente invención proporciona un compuesto de Fórmula (V):

en donde:
el Anillo A se selecciona entre
y
R2 se selecciona entre -CH3 y CH2-ciclopropilo;
R3 es -Oalquilo C1-4;
R4 se selecciona entre

R5, en cada caso, se selecciona independientemente entre H, F, Cl, Br, alquilo C1-4, -ORb, -CN, -C(=O)NRaRa y NHC(=O)Rb;
R6, en cada caso, se selecciona independientemente entre H, alquilo C1-3, -C(=O)Rb y -C(=O)ORb;
Ra, en cada caso, se selecciona independientemente entre H y alquilo C1-6; y
Rb, en cada caso, se selecciona independientemente entre H y alquilo C1-6.
En un noveno aspecto, la presente invención proporciona un compuesto de fórmula (I) o una sal farmacéuticamente aceptable del mismo, en donde:
El anillo A es heterociclilo de 4 a 15 miembros sustituido con 1-4 R7;
R1 se selecciona entre -CH3 y CD3;
R2 se selecciona entre H, alquilo C1-3 sustituido con 0-5 Re y -(CH2)r-cicloalquilo C3-6 con 0-5 Re;
R3 se selecciona entre F, Cl, Br y -ORb;
L se selecciona entre -(CRdRd)o-3-, -NRa-, -S(O)p- y -C(=O)-;
R4 se selecciona entre H, F, Cl, Br, -CN, -ORb, alquilo C1.6 sustituido con 1-5 R5, arilo sustituido con 1-5 R5, cicloalquilo C3-12 sustituido con 1-5 R5 y heterociclilo que comprende átomos de carbono y 1-3 heteroátomos seleccionados entre N, NR6, O y S y sustituido con 1-5 R5;
R5, en cada caso, se selecciona independientemente entre H, F, Cl, Br, nitro, =O, -alquilo C1-4 sustituido con 0-4 Re, alquenilo C2-4 sustituido con 0-4 Re, alquinilo C2-4 sustituido con 0-4 Re, -(CH2)rCN, -(CH2)rORb, (CH2)rS(O)pRc, -(CH2)rS(O)pNRaRa, -(CH2)rNRaS(O)pRc, -(CH2)rNRaRa, -(CH2)rNRaC(=O)Rb, -(CH2)rNRaC(=O)NRaRa, -(CH2)rC(=O)ORb, -(CH2)rC(=O)Rb, -(CH2)rOC(=O)Rb, -(CH2)rC(=O)NRaRa, P(=O)(Oalquilo ^ .4)2, -P(=O)(alquilo C1-4)2, cicloalquilo C3-6 sustituido con 0-4 Re, arilo sustituido con 0-4 Re, heterociclilo sustituido con 0-4 Re y heteroarilo sustituido con 0-4 Re;
R6, en cada caso, se selecciona independientemente entre H, alquilo C1-3 sustituido con 0-5 Re, -C(=O)Rb, -C(=O)(CH2)rNRaRa, -C(=O)(CH2)rNRaC(=O)Rb, -C(=O)ORb, -S(O)pRc, -S(O)pNRaRa, -(CH2)r-cicloalquilo C3-6 sustituido con 0-4 Re, -(CH2)r-arilo sustituido con 0-4 Re, -(CH2)r-heterociclilo sustituido con 0-4 Re y -(CH2)rheteroarilo sustituido con 0-4 Re;
R7 se selecciona entre H, F, Cl, CN, alquilo C1-3, =N-ORb, -(CH2)rORb, -(CH2)rNRaRa, -NRaC(=NH)alquilo C1-3, -NRaC(=O)ORb, carbociclilo y un heterociclilo; como alternativa, dos grupos R7 se toman juntos para formar carbociclilo o heterociclilo;
Ra, en cada caso, se selecciona independientemente entre H, alquilo C1-6 sustituido con 0-5 Re, alquenilo C2-6 sustituido con 0-5 Re, alquinilo C2-6 sustituido con 0-5 Re, -(CH2)r-carbociclilo C3-10 sustituido con 0-5 Re y -(CH2)rheterociclilo sustituido con 0-5 Re; o Ra y Ra junto con el átomo de nitrógeno al que ambos están unidos forman un anillo heterocíclico sustituido con 0-5 Re;
Rb, en cada caso, se selecciona independientemente entre H, alquilo C1-6 sustituido con 0-5 Re, alquenilo C2-6 sustituido con 0-5 Re, alquinilo C2-6 sustituido con 0-5 Re, -(CH2)r-carbociclilo C3-10 sustituido con 0-5 Re y -(CH2)rheterociclilo sustituido con 0-5 Re;
Rc, en cada caso, se selecciona independientemente entre alquilo C1-6 sustituido con 0-5 Re, alquenilo C2-6 sustituido con 0-5 Re, alquinilo C2-6 sustituido con 0-5 Re, -(CH2)r-carbociclilo C3--6 sustituido con 0-5 Re y -(CH2)rheterociclilo sustituido con 0--5 Re;
Rd, en cada caso, se selecciona independientemente entre H y alquilo C1-6 sustituido con 0-5 Re;
Re, en cada caso, se selecciona independientemente entre alquilo C1-6 sustituido con 0-5 Rf, alquenilo C2-6, alquinilo C2-6, -(CH2)r-cicloalquilo C3-6, -(CH2)r-arilo, F, Cl, Br, CN, NO2, =O, -C(=O)OH, -C(=O)Oalquilo C1-4, -(CH2)rOH y -(CH2)rOalquilo C ^;
Rf, en cada caso, se selecciona independientemente entre H, F, Cl, Br, CN, OH, alquilo C1-5 opcionalmente sustituido con OH, cicloalquilo C3-6 y fenilo;
p, en cada caso, se selecciona independientemente entre cero, 1 y 2;
r, en cada caso, se selecciona independientemente entre cero, 1,2, 3 y 4; y
proporcionado cuando L se selecciona entre -NRa-, -S(O)p- y -C(=O)-, R4 se selecciona entre arilo sustituido con 1-5 R5, cicloalquilo C3-12 sustituido con 1-5 R5 y heterociclilo que comprende átomos de carbono y 1-3 heteroátomos seleccionados entre N, NR6, O y S y sustituido con 1-5 R5.
En un décimo aspecto, la presente invención proporciona un compuesto de Fórmula (II): o una de sus sales farmacéuticamente aceptables, en donde:
el Anillo A se selecciona entre

R2 se selecciona entre -CH3, -CH2CH3 -y CH2-ciclopropilo;
R3 se selecciona entre H, F, Cl, Br y -Oalquilo C1-4;
R4 se selecciona entre F, Cl, Br, alquilo C1-5 sustituido con 1-4 R5,


R5, en cada caso, se selecciona independientemente entre H, F, Cl, Br, alquilo C1-4, alquenilo C2-4, alquinilo C2-4, nitro, -CN, ORb, -S(O)pRc, -S(O)pNRaRa, -NRaS(O)pRc, -NRaRa, -NRaC(=O)Rb, -NRaC(=O)NRaRa, -C(=O)ORb, -C(=O)Rb, -OC(=O)Rb, -C(=O)NRaRa, -P(=O)(alquilo C1-4)2, cicloalquilo C3-6 sustituido con 0-4 con 0-4 Re, heterociclilo sustituido con 0-4 Re y heteroarilo sustituido con 0-4 Re;
Re, en cada caso, se selecciona independientemente entre H, alquilo C1-6 sustituido con 0-5 Re, -C(=O)Rb, -C(=O)NRaRa, -C(=O)ORb, -S(O)pRc, -S(O)pNRaRa, -(CH2)r-cicloalquilo C3-6 sustituido con 0-4 Re, -(CH2)r-arilo sustituido con 0-4 Re, -(CH2)r-heterociclilo sustituido con 0-4 Re y -(CH2)r-heteroarilo sustituido con 0-4 Re;
Ra, en cada caso, se selecciona independientemente entre H, alquilo C1-6 sustituido con 0-5 Re, alquenilo C2-6 sustituido con 0-5 Re, alquinilo C2-6 sustituido con 0-5 Re, -(CH2)r-carbociclilo C3-10 sustituido con 0-5 Re y -(CH2)rheterociclilo sustituido con 0-5 Re; o Ra y Ra junto con el átomo de nitrógeno al que ambos están unidos forman un anillo heterocíclico sustituido con 0-5 Re;
Rb, en cada caso, se selecciona independientemente entre H, alquilo C1-6 sustituido con 0-5 Re, alquenilo C2-6 sustituido con 0-5 Re, alquinilo C2-6 sustituido con 0-5 Re, -(CH2)r-carbociclilo C3-10 sustituido con 0-5 Re y -(CH2)rheterociclilo sustituido con 0-5 Re;
Rc, en cada caso, se selecciona independientemente entre alquilo C1-6 sustituido con 0-5 Re, alquenilo C2-6 sustituido con 0-5 Re, alquinilo C2-6 sustituido con 0-5 Re, -(CH2)r-carbociclilo C3--6 sustituido con 0-5 Re y -(CH2)rheterociclilo sustituido con 0--5 Re;
Re, en cada caso, se selecciona independientemente entre alquilo C1-6 sustituido con 0-5 Rf, alquenilo C2-6, alquinilo C2-6, -(CH2)r-cicloalquilo C3-6, -(CH2)r-arilo, F, Cl, Br, CN, NO2, =O, C(=O)OH, -C(=O)Oalquilo C1.4, -(CH2)rOH y -(CH2)rOalquilo C1.4;
Rf, en cada caso, se selecciona independientemente entre H, F, Cl, Br, CN, OH, alquilo C1-5 opcionalmente sustituido con OH, cicloalquilo C3-6 y fenilo;
p, en cada caso, se selecciona independientemente entre cero, 1 y 2; y
r, en cada caso, se selecciona independientemente entre cero, 1, 2, 3 y 4.
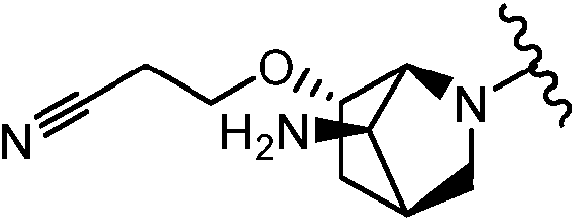
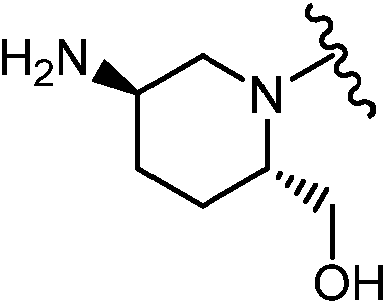
Como se ha definido anteriormente y se describe en el presente documento, L es -(CRdRd)0-3-, -NRa-, -S(O)p- o -C(=O)-. En algunas realizaciones, L está ausente cuando L es -(CRdRd)ü-. En algunas realizaciones, L es -CH2-. En algunas realizaciones, L es -CH2CH2-. En algunas realizaciones, L es -CH2CH2CH2-. En algunas realizaciones, L es -NRa-, Ra es H o alquilo C1-3. En algunas realizaciones, Lis -NH-. En determinadas realizaciones, L se selecciona entre los grupos funcionales representados en los ejemplos siguientes.
Como se ha definido anteriormente y se describe en el presente documento, R1 se selecciona entre -CH3 y CD3. En algunas realizaciones, R1 es CH3. En algunas realizaciones, R1 es CD3.
Como se ha definido anteriormente y se describe en el presente documento, R2 es hidrógeno, alquilo C1-3 sustituido con 0-5 Re o cicloalquilo C3-6 sustituido con 0-5 Re. En algunas realizaciones, R2 es hidrógeno. En algunas realizaciones, R2 es alquilo C1-2 sustituido con cicloalquilo C3-6. En algunas realizaciones, R2 es cicloalquilo C3-6. En algunas realizaciones, R2 es metilo. En algunas realizaciones, R2 es etilo. En algunas realizaciones, R2 es ciclopropilo. En algunas realizaciones, R2 es ciclobutilo. En algunas realizaciones, R2 es ciclopentilo. En algunas realizaciones, R2 es ciclohexilo. En algunas realizaciones, R2 es ciclopropilmetilo. En algunas realizaciones, R2 es ciclobutilmetilo. En algunas realizaciones, R2 es ciclopentilmetilo. En algunas realizaciones, R2 es ciclohexilmetilo. En algunas realizaciones, R2 es ciclopropiletilo. En algunas realizaciones, R2 es ciclobutiletilo. En algunas realizaciones, R2 es ciclopentiletilo. En algunas realizaciones, R2 es ciclohexiletilo. En algunas realizaciones, R2 es -CH2-ciclopropilo o -CH2-ciclobutilo. En algunas realizaciones, R2 es -CH2-ciclobutilo opcionalmente sustituido con metilo y -OH. En determinadas realizaciones, R2 se selecciona entre los grupos funcionales representados en los ejemplos
siguientes.
Como se ha definido anteriormente y se describe en el presente documento, R3 se selecciona entre F, Cl, Br, -ORb o alquilo C1-3 sustituido con 0-5 Re. En algunas realizaciones, R3 es F, Cl, Br. En algunas realizaciones, R3 es F, En algunas realizaciones, R3 es alquilo C1-3. En algunas realizaciones, R3 es metilo. En algunas realizaciones, R3 es etilo. En algunas realizaciones, R3 es propilo. En algunas realizaciones, R3 es ORb. En algunas realizaciones, R3 es -OCH3. En algunas realizaciones, R3 es -OCH2CH3. En algunas realizaciones, R3 es -OCH2CH2CH3. En determinadas realizaciones, R3 es -OCH(F)2. En determinadas realizaciones, R3 se selecciona entre los grupos funcionales representados en los ejemplos siguientes.
Como se ha definido anteriormente y se describe en el presente documento, cada R4 es


En algunas realizaciones, R4 es

En algunas realizaciones, R4 es

En algunas realizaciones, R4 es

En algunas realizaciones, R4 es

En algunas realizaciones, R4 es

En algunas realizaciones, R4 es

En algunas realizaciones, R4 es

En algunas realizaciones, R4 es

En algunas realizaciones, R4 es

En algunas realizaciones, R4 es

En algunas realizaciones, R4

En algunas realizaciones, R4 es

En algunas realizaciones, R4 es

En determinadas realizaciones, R4 se selecciona entre los grupos funcionales representados en los ejemplos siguientes.
Como se ha definido anteriormente y se describe en el presente documento, R5 es H, F, Cl, Br, CN, alquilo C1-4 sustituido con 0-5 Re, alquenilo C2-4, alquinilo C2-4, nitro, -S(O)pRc, -S(O)pNRaRa,-NRaS(O)pRc, -(CHRd)rORb, -(CH2)rNRaRa, -NRaC(=O)Rb, NRaC(=O)ORb-NRaC(=O)NRaRa, -C(=O)Rb, -C(=O)ORb, C(=O)NRaRa, -OC(=O)Rb, cicloalquilo C3.6 sustituido con 0-4 Re, arilo sustituido con 0-4 Re y heterociclilo sustituido con 0-4 Re
En algunas realizaciones, R5 es F. En algunas realizaciones, R5 es alquilo C1-4. En algunas realizaciones, R5 es -OH u O-alquilo C1-3. En algunas realizaciones, R5 es -NHS(O)2 alquenilo C2-4. En determinadas realizaciones, R5 se selecciona entre los grupos funcionales representados en los ejemplos siguientes.
Como se ha definido anteriormente y se describe en el presente documento, R6 es H, alquilo C1-3 sustituido con 0-4 Re, -S(O)pRc, -C(=O)Rb, -(CH2)r-C(=O)NRaRa, -C(=O)(CH2)rNRaC(=O)Rb, -C(=O)ORb,-S(O)pNRaRa, arilo sustituido con 0-4 Re y heterociclilo sustituido con 0-4 Re.
En algunas realizaciones, R6 es H. En algunas realizaciones, R6 es metilo o isopropilo. En algunas realizaciones, R6 es -(CH2)2C(=O)NH2. En algunas realizaciones, R6 es-(CH2)2OH. En algunas realizaciones, R6 es -C(=O)alquilo C1-4. En determinadas realizaciones, R6 se selecciona entre los grupos funcionales representados en los ejemplos siguientes.
Como se ha definido anteriormente y se describe en el presente documento, R7 es H, F, Cl, alquilo C1-3, -NRaRa o-NRaC(=O)ORb. En algunas realizaciones, R7 es NH2. En algunas realizaciones, R7 es F.
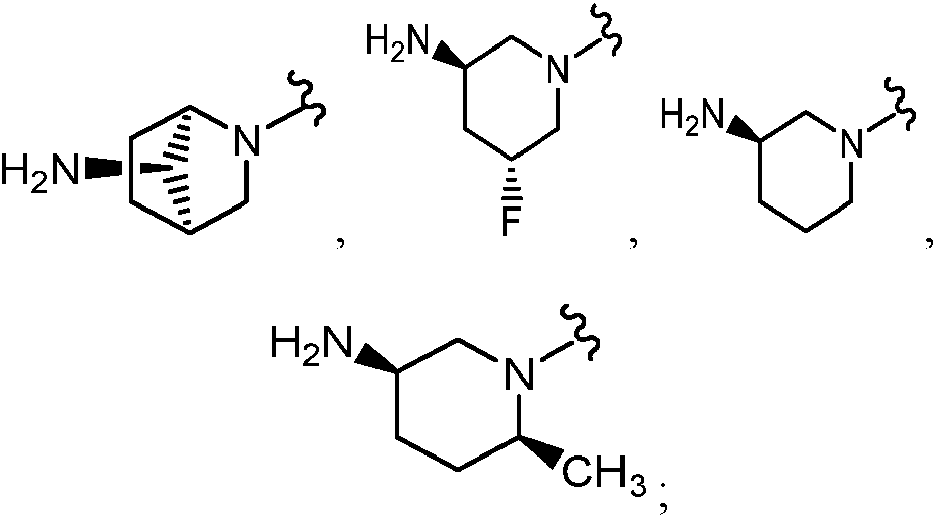
Como se ha definido anteriormente, el Anillo A es

En algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es


En algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es


En algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es
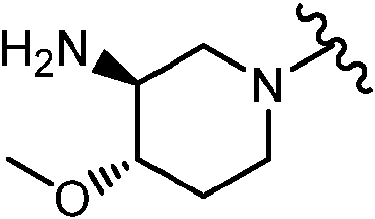
En algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es

. En algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es
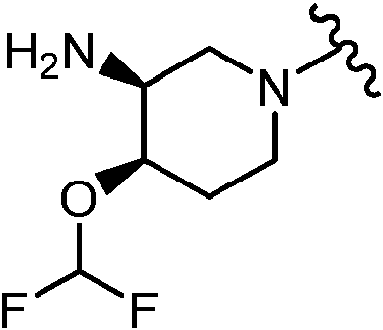
En algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es
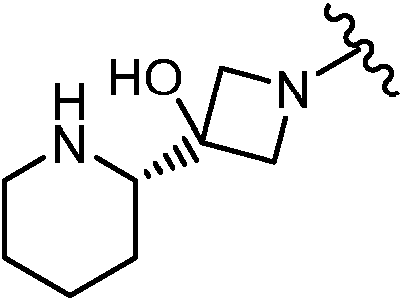
En algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es
En algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es
En algunas realizaciones, el Anillo A es

En
algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es

En algunas realizaciones, el Anillo A es

En determinadas realizaciones, el anillo A se selecciona entre los grupos funcionales representados en los ejemplos siguientes.
Como se ha definido anteriormente y se describe en el presente documento, r es 0-4. En algunas realizaciones, r es 0. En algunas realizaciones, r es 1. En algunas realizaciones, r es 2. En algunas realizaciones, r es 3. En algunas realizaciones, r es 4.
En algunas realizaciones, el Anillo A es

L está ausente; R2 es ciclopropilmetilo; R3 es H, F o -OCH3; R4 es

y R5 es H, F, Cl, CN, alquilo C 1-4, alquilo C 1-4 sustituido con OH, NH2 y COOH, S alquilo C 1-4, S(O)2alquilo C 1-4, S(O)2NH-ciclopropilo, -(CH2)0.1 NHS(O)2alquilo C 1-4 , N(Rd)S(O)2alquenilo C2-4,-(CH2)ü.1Oh , Oalquilo C 1-4, -(CH2)ü-1 NH2 , -(CH2)0 -1NHC(=O)alquilo C 1.4, -NRdC(=O)alquenilo C 2-4, -NHC(=O)alquinilo C2-4, -(CH2)0-1C(=O)OH, -C(=O)Oalquilo C 1.4, -NHC(=O)Oalquilo C 1.4, -NHC(=O)O(CH2)2Oalquilo C 1.4 , -NHC(=O)OCH2-ciclopropilo, -NHC(=O)NH2 , -C(=O)NHalquilo C 1.4, CONH(CH2)1-2C(=O)OH, -(CH2)0-1C(=O)NH2 , -(CH2)0 -1C(=O)NHalquilo C 1.4, C(=O)NH-piridina, -C(=O)NH(CH2)2N(alquilo C 1-4)2 ,-C(=O)NH(CH2)2OH, -C(=O)NH(CH2)2S(O)2alquilo C 1-4 y -OC(=O)alquilo C 1-4,

En algunas realizaciones, el Anillo A es

L es -CH2-; R2 es ciclopropilmetilo; R3 es H, F o -OCH3; R4 es
o 
y R5 es H, F, Cl, CN, alquilo C1-4, alquilo C1-4 sustituido con OH, NH2 y COOH, S alquilo C1-4, S(O)2alquilo C1-4, S(O)2NH-ciclopropilo, -(CH2)0 -1NHS(O)2alquilo C1-4, N(Rd)S(O)2alquenilo C2-4, -(CH2)0-1OH, Oalquilo C1-4, -(CH2)0-1 NH2 ,-(CH2)0 -1NHC(=O)alquilo C1.4, -NRdC(=O)alquenilo C2-4, -NHC(=O)alquinilo C2-4, -(CH2)0-1C(=O)OH, -C(=O)Oalquilo C1-4, -NHC(=O)Oalquilo C1-4, -NHC(=O)O(CH2)2Oalquilo C1-4,-NHC(=O)OCH2-ciclopropilo, -NHC(=O)NH2 , -C(=O)NHalquilo C1.4, CONH(CH2)1-2C(=O)OH, -(CH2)0-1C(=O)NH2 , -(CH2)0-1C(=O)NHalquilo C1.4, C(=O)NH-piridina,-C(=O)NH(CH2)2N(alquilo C1-4)2, -C(=O)NH(CH2)2OH, -C(=O)NH(CH2)2S(O)2alquilo C1-4 y -OC(=O)alquilo C1-4,

En algunas realizaciones, el Anillo A es

L está ausente; R2 es ciclopropilmetilo; R3 es H, F o -OCH3; R4 es
o 
y R5 es H, F, Cl, CN, alquilo C1-4, alquilo C1.4 sustituido con OH, NH2 y COOH, S alquilo C1-4, S(O)2alquilo C1.4, S(O)2NH-ciclopropilo, -(CH1 )0 -1NHS(O)2alquilo C1.4, N(Rd)S(O)2alquenilo C2-4, -(CH2)0-1OH, Oalquilo C1.4, -(CH2)0
iNH2 ,-(CH2)o-iNHC(=O)alquilo C-m , -NRdC(=O)alquenilo C2-4, -NHC(=O)alquinilo C2-4, -(CH2)o-iC(=O)OH, -C(=O)Oalquilo C 1.4, -NHC(=O)Oalquilo C 1.4, -NHC(=O)O(CH2 )2Oalquilo C i -4,-NHC(=O)OCH2-ciclopropilo, -NHC(=O)NH2 , -C(=O)NHalquilo C 1.4, CONH(CH2)i-2C(=O)OH, -(CH2)o-iC(=O)NH2 , -(CH2)o-iC(=O)NHalquilo C 1.4, C(=O)NH-piridina,-C(=O)NH(CH2)2N(alquilo ^ .4)2, -C(=O)NH(CH2)2OH, -C(=O)NH(CH2)2S(O)2alquilo C 1.4 y -OC(=O)alquilo C-i_4,

En algunas realizaciones, el Anillo A es

L es -CH2-; R2 es ciclopropilmetilo; R3 es H, F o -OCH3; R4 es

y R5 es H, F, Cl, CN, alquilo C 1-4, alquilo C 1.4 sustituido con OH, NH2 y COOH, S alquilo C 1-4, S(O)2alquilo C 1.4, S(O)2NH-ciclopropilo, -(CH2)0-iNHS(O)2alquilo C 1.4, N(Rd)S(O)2alquenilo C2-4, -(CH2 )0-iOH, Oalquilo C 1-4, -(CH2)0-iNH2 ,-(CH2)0-iNHC(=O)alquilo C 1.4, -NRdC(=O)alquenilo C2-4, -NHC(=O)alquinilo C2-4, -(CH2)0-iC(=O)OH, -C(=O)Oalquilo C 1-4, -NHC(=O)Oalquilo C 1-4, -NHC(=O)O(CH2 )2Oalquilo C 1-4,-NHC(=O)OCH2-ciclopropilo, -NHC(=O)NH2 , -C(=O)NHalquilo C 1.4, CONH(CH2)i-2C(=O)OH, -(CH2)0-iC(=O)NH2 , -(CH2)0-iC(=O)NHalquilo C 1.4, C(=O)NH-piridina,-C(=O)NH(CH2)2N(alquilo C 1-4)2, -C(=O)NH(CH2)2OH, -C(=O)NH(CH2)2S(O)2alquilo C 1-4 y -OC(=O)alquilo C 1-4,


En algunas realizaciones, el Anillo A es

L está ausente o es -CH2-; R2 es ciclopropilmetilo; R3 es H, F o -OCH3; R4 es

En algunas realizaciones, el Anillo A es

L está ausente o es -CH2-; R2 es ciclopropilmetilo; R3 es H, F o -OCH3; R4 es

En algunas realizaciones, el Anillo A es

L está ausente o es -CH2-; R2 es ciclopropilmetilo; R3 es H, F o -OCH3; R4 es

En algunas realizaciones, el Anillo A es

L está ausente o es -CH2-; R2 es ciclopropilmetilo; R3 es H, F o -OCH3; R4 es

En algunas realizaciones, el Anillo A es

L está ausente o es -CH2-; R2 es ciclopropilmetilo; R3 es H, F o -OCH3; R4 es

En algunas realizaciones, el Anillo A es

L está ausente o es -CH2-; R2 es ciclopropilmetilo; R3 es H, F o -OCH3; R4 es

R6 es H, alquilo C1-3 y -C(=O)Rb.
En algunas realizaciones, el Anillo A es

L está ausente o es -CH2-; R2 es ciclopropilmetilo; R3 es H, F o -OCH3; R4 es
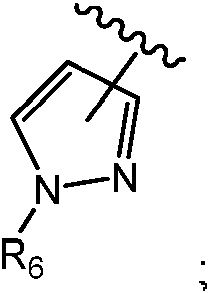
R6 es H, alquilo C1-3 y -C(=O)Rb.
En algunas realizaciones, el compuesto de Fórmula (I) se selecciona entre los ejemplos representados a continuación. En determinadas realizaciones, la presente invención proporciona cualquier compuesto descrito anteriormente y en el presente documento, o una sal farmacéuticamente aceptable del mismo. En algunas realizaciones, la presente invención proporciona cualquier compuesto descrito anteriormente y en el presente documento en forma aislada.
4. Usos, Formulación y administración
Composiciones farmacéuticamente aceptables
De acuerdo con otra realización, la invención proporciona una composición que comprende un compuesto de la presente invención o un derivado farmacéuticamente aceptable del mismo y un excipiente, adyuvante o vehículo farmacéuticamente aceptable. La cantidad de compuesto en las composiciones de la presente invención es tal que es eficaz para inhibir de forma medible la PAD4, en una muestra biológica o en un paciente. En determinadas realizaciones, la cantidad de compuesto en las composiciones de la presente invención es tal que es eficaz para inhibir de forma medible la PAD4, en una muestra biológica o en un paciente. En determinadas realizaciones, una composición de la presente invención se formula para su administración a un paciente que necesita dicha composición. En algunas realizaciones, una composición de la presente invención se formula para su administración oral a un paciente.
El término "sujeto", tal como se usan en el presente documento, se usa indistintamente con el término "paciente" y significa un animal, preferentemente, un mamífero. En algunas realizaciones, un sujeto o paciente es un ser humano. En otras realizaciones, un sujeto (o paciente) es un sujeto (o paciente) veterinario. En algunas realizaciones, un sujeto (o paciente) veterinario es un sujeto canino, un sujeto felino o equino.
La expresión "excipiente, adyuvante o vehículo farmacéuticamente aceptable" se refiere a un excipiente, adyuvante, o vehículo no tóxico que no destruye la actividad farmacológica del compuesto con el que se formula. Los excipientes, adyuvantes o vehículos farmacéuticamente aceptables que pueden usarse en las composiciones de la presente invención incluyen, aunque sin limitación, intercambiadores iónicos, alúmina, estearato de aluminio, lecitina, proteínas séricas, tales como seroalbúmina humana, sustancias tamponantes tales como fosfatos, glicina, ácido sórbico, sorbato potásico, mezclas de glicéridos parciales de ácidos grasos vegetales saturados, agua, sales o electrolitos, tales como sulfato de protamina, hidrogenofosfato disódico, hidrogenofosfato potásico, cloruro de sodio, sales de cinc, sílice coloidal, trisilicato de magnesio, polivinilpirrolidona, sustancias a base de celulosa, polietilenglicol, carboximetilcelulosa sódica, poliacrilatos, ceras, polímeros de bloques de polietileno-polioxipropileno, polietilenglicol y lanolina.
Las composiciones de la presente invención pueden administrarse por vía oral, vía parenteral, mediante pulverizador por inhalación, por vía tópica, por vía rectal, por vía nasal, por vía bucal, por vía vaginal o a través de un depósito implantado. El término "parenteral", como se usa en el presente documento, incluye inyección subcutánea, intravenosa, intramuscular, intraarticular, intrasinovial, intraesternal, intratecal, intrahepática, intralesional e intracraneal o técnicas de infusión. Preferentemente, las composiciones se administran por vía oral, por vía intraperitoneal o por vía intravenosa. Las formas inyectables estériles de las composiciones de la presente invención pueden ser una suspensión acuosa u oleaginosa. Estas suspensiones pueden formularse de acuerdo con técnicas conocidas en la técnica usando agentes de dispersión o humectación y agentes de suspensión adecuados. La preparación inyectable estéril también puede ser una solución o suspensión inyectable estéril en un diluyente o disolvente no tóxico parenteralmente aceptable, por ejemplo en forma de una solución en 1,3-butanodiol. Entre los vehículos y disolventes aceptables que pueden emplearse se encuentran agua, solución de Ringer y solución isotónica de cloruro sódico. Además, convencionalmente se usan aceites no volátiles estériles en forma de un disolvente o un medio de suspensión.
Con este propósito, puede emplearse cualquier aceite suave no volátil incluyendo mono- o diglicéridos sintéticos. Los ácidos grasos, tales como ácido oleico y sus derivados de glicéridos, son útiles en la preparación de inyectables, como lo son los aceites naturales farmacéuticamente aceptables, tales como aceite de oliva o aceite de ricino, especialmente en sus versiones polioxietiladas. Estas soluciones o suspensiones oleaginosas también pueden contener un diluyente o dispersante alcohólico de cadena larga, tales como carboximetilcelulosa o agentes dispersantes similares que se usan comúnmente en la formulación de formas farmacéuticas farmacéuticamente aceptables incluyendo emulsiones y suspensiones. Otros tensioactivos comúnmente usados, tales como los Tween, Span y otros agentes emulsionantes o potenciadores de la biodisponibilidad que se usan habitualmente en la fabricación de formas farmacéuticas sólidas, líquidas u otras farmacéuticamente aceptables también pueden usarse con fines de formulación.
Las composiciones farmacéuticamente aceptables de la presente invención pueden administrarse por vía oral en cualquier forma farmacéutica oralmente aceptable, incluyendo, pero sin limitación, cápsulas, comprimidos,
suspensiones o soluciones acuosas. En el caso de los comprimidos para su uso oral, los vehículos comúnmente usados incluyen lactosa y almidón de maíz. Los agentes lubricantes, tales como estearato de magnesio, también se añaden normalmente. Para la administración oral en forma de una cápsula, los diluyentes útiles incluyen lactosa y almidón de maíz deshidratado. Cuando se necesitan suspensiones acuosas para uso oral, el principio activo se combina con agentes emulsionantes y de suspensión. Si se desea, también pueden añadirse determinados agentes edulcorantes, aromatizantes o colorantes.
Como alternativa, las composiciones farmacéuticamente aceptables de la presente invención pueden administrarse en forma de supositorios para administración rectal. Estos pueden prepararse mezclando el agente con un excipiente no irritante adecuado que es sólido a temperatura ambiente, pero líquido a temperatura rectal y, por lo tanto, se derretirá en el recto para liberar el fármaco. Dichos materiales incluyen manteca de cacao, cera de abeja y polietilenglicoles.
Las composiciones farmacéuticamente aceptables de la presente invención también pueden administrarse por vía tópica, especialmente cuando la diana de tratamiento incluye áreas u órganos fácilmente accesibles mediante aplicación tópica, incluyendo enfermedades de los ojos, la piel o el tracto intestinal inferior. Las formulaciones tópicas adecuadas se preparan fácilmente para cada una de estas áreas u órganos.
La aplicación tópica para el tracto intestinal inferior puede lograrse en una formulación de supositorio rectal (véase anteriormente) o en una formulación de enema adecuada. También pueden usarse parches transdérmicos por vía tópica.
Para aplicaciones tópicas, las composiciones farmacéuticamente aceptables proporcionadas pueden formularse en una pomada adecuada que contiene el componente activo suspendido o disuelto en uno o más vehículos. Los vehículos para administración tópica de los compuestos de la presente invención incluyen, aunque sin limitación, aceite mineral, vaselina líquida, vaselina blanca, propilenglicol, polioxietileno, compuesto de polioxipropileno, cera emulsionante y agua. Como alternativa, las composiciones farmacéuticamente aceptables proporcionadas pueden formularse en una loción o crema adecuados que contiene los componentes activos suspendidos o disueltos en uno o más vehículos farmacéuticamente aceptables. Los vehículos adecuados incluyen, aunque sin limitación, aceite mineral, monoestearato de sorbitán, polisorbato 60, cera de ésteres cetílicos, alcohol cetearílico, 2-octildodecanol, alcohol bencílico y agua.
Para su uso oftálmico, siempre que las composiciones farmacéuticamente aceptables puedan formularse como suspensiones micronizadas en isotónico, de pH ajustado, preferentemente, como soluciones en solución salina estéril isotónica, de pH ajustado, tanto con un conservante como sin él, tal como cloruro de benzalconio. Como alternativa, para usos oftálmicos, las composiciones farmacéuticamente aceptables pueden formularse en una pomada tal como vaselina.
Las composiciones farmacéuticamente aceptables de la presente invención también pueden administrarse por aerosol nasal o inhalación. Dichas composiciones se preparan de acuerdo con técnicas bien conocidas en la técnica de la formulación farmacéutica y pueden prepararse como soluciones en solución salina, empleando alcohol bencílico u otros conservantes adecuados, promotores de la absorción para potenciar la biodisponibilidad, fluorocarbonos y/u otros agentes solubilizantes o dispersantes convencionales.
Lo más preferentemente, las composiciones farmacéuticamente aceptables de la presente invención se formulan para administración oral. Dichas formulaciones pueden administrarse con o sin alimento. En algunas realizaciones, las composiciones farmacéuticamente aceptables de la presente invención se administran sin alimento. En otras realizaciones, las composiciones farmacéuticamente aceptables de la presente invención se administran con alimento.
Las composiciones farmacéuticamente aceptables de la presente invención pueden administrarse a seres humanos y otros animales por vía oral, por vía rectal, vía parenteral, por vía intracisternal, por vía intravaginal, por vía intraperitoneal, por vía tópica (en forma de polvos, pomadas o gotas), por vía bucal, como un pulverizador oral o nasal o similares, dependiendo de la gravedad de la infección que se va a tratar. En determinadas realizaciones, los compuestos de la invención pueden administrarse por vía oral o parenteral a niveles de dosificación de aproximadamente 0,01 mg/kg a aproximadamente 50 mg/kg y, preferentemente, de aproximadamente 1 mg/kg a aproximadamente 25 mg/kg, de peso corporal del sujeto por día, una o más veces al día, para obtener el efecto terapéutico deseado.
Las formas farmacéuticas líquidas para administración oral incluyen, aunque sin limitación, emulsiones, microemulsiones, soluciones, suspensiones, jarabes y elixires farmacéuticamente aceptables. Además de los compuestos activos, las formas farmacéuticas líquidas pueden contener diluyentes inertes usados habitualmente en la técnica, tales como, por ejemplo, agua u otros disolventes, agentes solubilizantes y emulsionantes tales como alcohol etílico, alcohol isopropílico, carbonato de etilo, acetato de etilo, alcohol bencílico, benzoato de bencilo, propilenglicol, 1,3-butilenglicol, dimetilformamida, aceites (en particular, aceites de semillas de algodón, cacahuete, maíz, germen, oliva, ricino y sésamo), glicerol, alcohol tetrahidrofurfurílico, polietilenglicoles y ésteres de ácidos
grasos de sorbitán y mezclas de los mismos. Además de diluyentes inertes, las composiciones orales también pueden incluir adyuvantes tales como agentes humectantes, agentes emulsionantes y de suspensión, edulcorantes, saborizantes y agentes perfumantes.
Las preparaciones inyectables, por ejemplo, las suspensiones acuosas u oleosas inyectables estériles pueden formularse de acuerdo con la técnica conocida utilizando agentes dispersantes o humectantes y agentes de suspensión adecuados. La preparación inyectable estéril también puede ser una solución, suspensión o emulsión inyectable estéril en un diluyente o disolvente no tóxico por vía parenteralmente aceptable, por ejemplo, como una solución en 1,3-butanodiol. Entre los vehículos y disolventes aceptables que pueden emplearse se encuentran agua, solución de Ringer, U.S.P. y solución de cloruro de sodio isotónica. Además, convencionalmente se usan aceites no volátiles estériles en forma de un disolvente o un medio de suspensión. Para este fin, puede emplearse cualquier aceite no volátil suave, incluyendo mono- o diglicéridos sintéticos. Además, se usan ácidos grasos tales como ácido oleico en la preparación de inyectables.
Las formulaciones inyectables pueden esterilizarse, por ejemplo, mediante filtración a través de un filtro de retención de bacterias o mediante incorporación de agentes esterilizantes en forma de composiciones sólidas estériles que pueden disolverse o dispersarse en agua estéril u otro medio inyectable estéril antes de su uso.
Para prolongar el efecto de un compuesto de la presente invención, es deseable con frecuencia ralentizar la absorción del compuesto de inyección subcutánea o intramuscular. Esto puede lograrse mediante el uso de una suspensión líquida de material cristalino o amorfo con mala solubilidad en agua. La velocidad de absorción del compuesto depende entonces de su velocidad de disolución que, a su vez, puede depender del tamaño del cristal y de la forma cristalina. Como alternativa, se realiza absorción retardada de una forma de compuesto administrado por vía parenteral disolviendo o suspendiendo el compuesto en un vehículo de aceite. Las formas de depósito inyectables se preparan formando matrices microencapsuladas del compuesto en polímeros biodegradables tales como polilactida-poliglicólido. Dependiendo de la proporción de compuesto con respecto a polímero y de la naturaleza del polímero concreto empleado, puede controlarse la velocidad de liberación del compuesto. Los ejemplos de otros polímeros biodegradables incluyen poli(ortoésteres) y poli(anhídridos). También se preparan formulaciones de depósito inyectables atrapado el compuesto en liposomas o microemulsiones que son compatibles con los tejidos corporales.
Las composiciones para administración rectal o vaginal son, preferentemente, supositorios que pueden prepararse mezclando los compuestos de la presente invención con excipientes o vehículos adecuados no irritantes tales como manteca de cacao, polietilenglicol o una cera para supositorios que son sólidos a temperatura ambiente pero líquidos a temperatura corporal y, por lo tanto, se derriten en el recto o en la cavidad vaginal y liberan el compuesto activo.
Las formas farmacéuticas sólidas para administración oral incluyen cápsulas, comprimidos, píldoras, polvos y gránulos. En dichas formas farmacéuticas sólidas, el compuesto activo se mezcla con al menos un excipiente o transportador inerte, farmacéuticamente aceptable tal como citrato de sodio o fosfato de dicalcio y/o a) cargas o diluyentes tales como almidones, lactosa, sacarosa, glucosa, manitol y ácido silícico, b) aglutinantes tales como, por ejemplo, carboximetilcelulosa, alginatos, gelatina, polvinilpirrolidona, sacarosa y acacia, c) humectantes tales como glicerol, d) agentes disgregantes tales como agar-agar, carbonato cálcico, almidón de patata o tapioca, ácido algínico, determinados silicatos y carbonato de sodio, e) agentes retardantes de la solución tales como parafina, f) aceleradores de la absorción tales como compuestos de amonio cuaternario, g) agentes humectantes tales como, por ejemplo, alcohol cetílico y monoestearato de glicerol, h) absorbentes tales como caolín y arcilla bentonítica e i) lubricantes tales como talco, estearato cálcico, estearato de magnesio, polietilenglicoles sólidos, lauril sulfato sódico y mezclas de los mismos. En el caso de cápsulas, comprimidos y píldoras, la forma farmacéutica también puede comprender agentes tamponantes.
También pueden emplearse composiciones sólidas de tipo similar como cargas en cápsulas de gelatina de relleno blando y duro usando excipientes tales como lactosa o azúcar de la leche, así como polietilenglicoles de alto peso molecular y similares. Las formas farmacéuticas sólidas de comprimidos, grageas, cápsulas, píldoras y gránulos pueden prepararse con recubrimientos y cubiertas tales como recubrimientos entéricos y otros recubrimientos bien conocidos en la técnica de la formulación farmacéutica. Opcionalmente, pueden contener agentes opacificantes y también pueden tener una composición que libere el principio o principios activos solo o con preferencia, en una parte determinada del tracto intestinal, opcionalmente, de una manera retardada. Los ejemplos de composiciones de inclusión que pueden usarse incluyen sustancias poliméricas y ceras. También pueden emplearse composiciones sólidas de tipo similar como cargas en cápsulas de gelatina de relleno blando y duro usando excipientes tales como lactosa o azúcar de la leche, así como polietilenglicoles de alto peso molecular y similares.
Los compuestos activos también pueden estar en forma microencapsulada con uno o más excipientes como se ha indicado anteriormente. Las formas farmacéuticas sólidas de comprimidos, grageas, cápsulas, píldoras y gránulos pueden prepararse con recubrimientos y cubiertas tales como recubrimientos entéricos, recubrimientos de control de la liberación y otros recubrimientos bien conocidos en la técnica de la formulación farmacéutica. En dichas formas farmacéuticas sólidas, el compuesto activo puede mezclarse con al menos un diluyente inerte tales como sacarosa, lactosa o almidón. Dichas formas farmacéuticas también pueden comprender, como es práctica normal, sustancias
adicionales distintas de diluyentes inertes, por ejemplo, lubricantes para la formación de comprimidos y otros adyuvantes para la formación de comprimidos tales como estearato de magnesio y celulosa microcristalina. En el caso de cápsulas, comprimidos y píldoras, las formas de dosificación también pueden comprender agentes tamponantes. Opcionalmente, pueden contener agentes opacificantes y también pueden tener una composición que libere el principio o principios activos solo o con preferencia, en una parte determinada del tracto intestinal, opcionalmente, de una manera retardada. Los ejemplos de composiciones de inclusión que pueden usarse incluyen sustancias poliméricas y ceras.
Las formas farmacéuticas para administración tópica o transdérmica de un compuesto de la presente invención incluyen pomadas, pastas, cremas, lociones, geles, polvos, soluciones, pulverizadores, inhalantes o parches. El componente activo se mezcla en condiciones estériles con un transportador farmacéuticamente aceptable y cualquier conservante o tampón necesario, según sea necesario. Formulación oftálmica, gotas óticas y/o colirios también se contemplan en el alcance de la presente invención. Además, la presente invención contempla el uso de parches transdérmicos, que tienen la ventaja añadida de proporcionar un suministro controlado de un compuesto al organismo. Dichas formas farmacéuticas pueden prepararse disolviendo o dispensando el compuesto en el medio apropiado. También pueden usarse potenciadores de la absorción para aumentar el flujo del compuesto a través de la piel. La velocidad puede controlarse proporcionando una membrana de control de la velocidad o dispersando el compuesto en una matriz polimérica o gel.
La cantidad de los compuestos de la presente invención que puede combinarse con los materiales transportadores para producir una composición en una forma farmacéutica unitaria variará dependiendo del hospedador tratado, del modo particular de administración. Preferentemente, las composiciones proporcionadas deben formularse de tal manera que se pueda administrar una dosificación de entre 0,01 -100 mg/kg de peso corporal/día del inhibidor a un paciente que recibe estas composiciones.
Un compuesto de la presente invención se puede administrar solo o en combinación con uno o más compuestos terapéuticos diferentes, adoptando la posible terapia de combinación la forma de combinaciones fijas o la administración de un compuesto de la invención y uno o más compuestos terapéuticos diferentes que se escalonan o se administran independientemente entre sí, o la administración combinada de combinaciones fijas y uno o más compuestos terapéuticos diferentes. Algunos ejemplos de dichos otros agentes terapéuticos incluyen corticoesteroides, rolipram, calfostina, medicamentos antiinflamatorios supresores de citocinas (CSAID), interleucina-10, glucocorticoides, salicilatos, óxido nítrico y otros inmunosupresores; inhibidores de la translocación nuclear, tales como desoxipergualina (DSG); fármacos antiinflamatorios no esteroideos (AINE) tales como ibuprofeno, celecoxib y rofecoxib; corticoides, tales como prednisona o dexametasona; agentes antivíricos tales como abacavir; agentes antiproliferativos tales como metotrexato, leflunomida, FK506 (tacrolimus, Prograf); fármacos citotóxicos tales como azatioprina y ciclofosfamida; inhibidores de TNF-h tales como tenidap, anticuerpos anti-TNF o el receptor de TNF soluble, y rapamicina (sirólimus o Rapamune), o derivados de los mismos. Un compuesto de la presente invención puede administrarse además especialmente para terapia tumoral en combinación con quimioterapia, radioterapia, inmunoterapia, fototerapia, intervención quirúrgica o una combinación de estas. La terapia a largo plazo es igualmente posible como lo es la terapia adyuvante en el contexto de otras estrategias de tratamiento, como se ha descrito anteriormente. Otros tratamientos posibles son la terapia para mantener el estado del paciente después de la regresión tumoral o incluso quimioterapia preventiva, por ejemplo, en pacientes en riesgo.
Esos agentes adicionales pueden administrarse por separado de una composición que contiene el compuesto de la invención, como parte de un régimen de dosificación múltiple. Como alternativa, esos agentes pueden ser parte de una forma de dosificación individual, mezclados junto con un compuesto de la presente invención en una única composición. Si se administran como parte de un régimen de dosificación múltiple, los dos principios activos pueden suministrarse de forma simultánea, secuencialmente o dentro de un periodo de tiempo entre sí, normalmente dentro de cinco horas entre sí.
Como se usa en el presente documento, el término "combinación", "combinado", y términos relacionados se refieren a la administración simultánea o secuencial de agentes terapéuticos de acuerdo con la presente invención. Por ejemplo, un compuesto de la presente invención puede administrarse con otro agente terapéutico de forma simultánea o secuencial en formas de dosificación unitarias separadas o conjuntamente en una forma de dosificación unitaria individual. Por consiguiente, la presente invención proporciona una forma de dosificación unitaria individual que comprende un compuesto de la presente invención, un agente terapéutico adicional y un excipiente, adyuvante o vehículo farmacéuticamente aceptable.
La cantidad tanto de un compuesto de la invención como de agente terapéutico adicional (en las composiciones que comprenden un agente terapéutico adicional como se describe anteriormente) que puede combinarse con los materiales vehículo para producir una forma farmacéutica unitaria variará dependiendo del hospedador tratado y del modo particular de administración. Preferentemente, las composiciones de la presente invención deben formularse de tal manera que se pueda administrar una dosis de entre 0,01 - 100 mg/kg de peso corporal/día de un compuesto de la invención.
En las composiciones que comprenden un agente terapéutico adicional, ese agente terapéutico adicional y el
compuesto de la presente invención pueden actuar de manera sinérgica. Por lo tanto, la cantidad de agente terapéutico adicional en dichas composiciones será inferior al necesario en una monoterapia que utilice solamente dicho agente terapéutico.
La cantidad de agente terapéutico adicional presente en las composiciones de la presente invención no será mayor que la cantidad que normalmente se administraría en una composición que comprende ese agente terapéutico como el único agente activo. Preferentemente, la cantidad de agente terapéutico adicional en las composiciones divulgadas en el presente documento variará de aproximadamente el 50% al 100% de la cantidad normalmente presente en una composición que comprende ese agente como el único agente terapéuticamente activo.
Debe entenderse también que un régimen de dosificación y tratamiento específico para cualquier paciente particular dependerán de diversos factores, incluyendo la actividad del compuesto específico empleado, la edad, el peso corporal, el estado de salud general, el sexo, la dieta, el tiempo de administración, la tasa de excreción, la combinación farmacológica y el criterio del médico a cargo del tratamiento y la gravedad de la enfermedad particular que se esté tratando. La cantidad de un compuesto de la presente invención en la composición también variará dependiendo del compuesto concreto en composición.
5. Usos de compuestos y composiciones farmacéuticamente aceptables
Los compuestos y composiciones que se describen en el presente documento son generalmente útiles para la inhibición de PAD4.
La actividad de un compuesto utilizado en la presente invención como inhibidor de PAD4, puede ensayarse in vitro, in vivo o en una línea celular. Los ensayos in vitro incluyen ensayos que determinan la inhibición de PAD4. Las condiciones detalladas para ensayar un compuesto utilizado en la presente invención como inhibidor de PAD4 se exponen en los Ejemplos a continuación. En algunas realizaciones, un compuesto proporcionado inhibe PAD4 selectivamente en comparación con PAD2.
Como se usa en el presente documento, los términos "tratamiento", "tratar", y "tratando" se refieren a revertir, aliviar, retrasar el inicio de o inhibir el progreso de una enfermedad o trastorno o de uno o más síntomas de los mismos, como se describe en el presente documento. El tratamiento puede administrarse después de que se hayan desarrollado uno o más síntomas. Como alternativa, el tratamiento puede administrarse en ausencia de síntomas. Por ejemplo, el tratamiento puede administrarse a un individuo susceptible antes de la aparición de los síntomas (por ejemplo, a la vista de antecedentes de síntomas y/o a la vista de factores genéticos u otros factores de susceptibilidad). El tratamiento también puede continuar después de que los síntomas se hayan resuelto, por ejemplo, para prevenir o retrasar la recaída.
Los compuestos proporcionados son inhibidores de PAD4 y, por lo tanto, son útiles para tratar uno o más trastornos asociados con la actividad de PAD4. De esta manera, en determinadas realizaciones, la presente invención proporciona un compuesto de la presente invención o una composición farmacéuticamente aceptable del mismo, para su uso en un método para tratar un trastorno mediado por PAD4 que comprende la etapa de administrar a un paciente que lo necesita un compuesto de la presente invención, o una composición farmacéuticamente aceptable del mismo.
Un trastorno mediado por PAD4 es una enfermedad, afección o trastorno mediado por una actividad inapropiada de PAD4. Un trastorno mediado por PAD4 se puede seleccionar entre el grupo que consiste en artritis reumatoide, vasculitis, lupus eritematoso sistémico, colitis ulcerosa, cáncer, fibrosis quística, asma, lupus eritematoso cutáneo y psoriasis. El trastorno mediado por una actividad inapropiada de PAD4 es artritis reumatoide. El trastorno mediado por una actividad inapropiada de PAD4 es lupus sistémico. El trastorno mediado por una actividad inapropiada de PAD4 es vasculitis. El trastorno mediado por una actividad inapropiada de PAD4 es lupus eritematoso cutáneo. El trastorno mediado por una actividad inapropiada de PAD4 es psoriasis.
En una realización, se proporciona un compuesto de la presente invención o una composición farmacéuticamente aceptable del mismo, para su uso en un método de tratamiento de artritis reumatoide, vasculitis, lupus eritematoso sistémico, colitis ulcerosa, cáncer, fibrosis quística, asma, lupus eritematoso cutáneo o psoriasis, método que comprende administrar a un sujeto humano que lo necesite, una cantidad terapéuticamente eficaz de un compuesto proporcionado o una sal farmacéuticamente aceptable del mismo.
En una realización, se proporciona un compuesto de la presente invención o una composición farmacéuticamente aceptable del mismo, para su uso en un método de tratamiento de artritis reumatoide, método que comprende administrar a un sujeto humano que lo necesite, una cantidad terapéuticamente eficaz de un compuesto proporcionado o una sal farmacéuticamente aceptable del mismo. En una realización, se proporciona un compuesto de la presente invención o una composición farmacéuticamente aceptable del mismo, para su uso en un método de tratamiento de lupus sistémico, método que comprende administrar a un sujeto humano que lo necesite, una cantidad terapéuticamente eficaz de un compuesto proporcionado o una sal farmacéuticamente aceptable del mismo. En una realización, se proporciona un compuesto de la presente invención o una composición
farmacéuticamente aceptable del mismo, para su uso en un método de tratamiento de la vasculitis, método que comprende administrar a un sujeto humano que lo necesite, una cantidad terapéuticamente eficaz de un compuesto proporcionado o una sal farmacéuticamente aceptable del mismo. En una realización, se proporciona un compuesto de la presente invención o una composición farmacéuticamente aceptable del mismo, para su uso en un método de tratamiento de lupus eritematoso cutáneo, método que comprende administrar a un sujeto humano que lo necesite, una cantidad terapéuticamente eficaz de un compuesto proporcionado o una sal farmacéuticamente aceptable del mismo. En una realización, se proporciona un compuesto de la presente invención o una composición farmacéuticamente aceptable del mismo, para su uso en un método de tratamiento de la psoriasis, método que comprende administrar a un sujeto humano que lo necesite, una cantidad terapéuticamente eficaz de un compuesto proporcionado o una sal farmacéuticamente aceptable del mismo.
Un trastorno mediado por PAD4 se selecciona entre el grupo que consiste en lesión pulmonar inducida por ácido, acné (PAPA), leucemia linfocítica aguda, síndrome de dificultad respiratoria aguda, enfermedad de Addison, hiperplasia suprarrenal, insuficiencia adrenocortical, envejecimiento, SIDA, hepatitis alcohólica, hepatitis alcohólica, enfermedad de hígado alcohólico, asma inducida por alérgenos, broncopulmonar alérgico, aspergilosis, conjuntivitis alérgica, alopecia, enfermedad de Alzheimer, amiloidosis, esclerosis lateral amiotrófica, y pérdida de peso, angina de pecho, angioedema, displasia ectodérmica anhidrótica-ID, espondilitis anquilosante, segmento anterior, inflamación, síndrome antifosfolípido, estomatitis aftosa, apendicitis, artritis, asma, ateroesclerosis, dermatitis atópica, enfermedades autoinmunitarias, hepatitis autoinmunitaria, inflamación inducida por picadura de abeja, enfermedad de Behcet, síndrome de Behcet, parálisis de campanas, beriliosis, síndrome de Blau, dolor de huesos, bronquiolitis, quemaduras, bursitis, cáncer, hipertrofia cardíaca, síndrome del túnel carpiano, trastornos catabólicos, cataratas, aneurisma cerebral, inflamación inducida por irritantes químicos, corioretinitis, insuficiencia cardíaca crónica, enfermedad pulmonar crónica del prematuro, leucemia linfocítica crónica, enfermedad pulmonar obstructiva crónica, colitis, síndrome de dolor regional complejo, enfermedad del tejido conjuntivo, úlcera corneal, enfermedad de Crohn, síndromes periódicos asociados a la criopirina, criptococosis, fibrosis quística, deficiencia del antagonista del receptor de interleucina-1 (DIRA), dermatitis, endotoxemia de dermatitis, dermatomiositis, glioma pontino intrínseco difuso, endometriosis, endotoxemia, epicondilitis, eritroblastopenia, polineuropatía amiloidótica familiar, urticaria familiar por frío, fiebre mediterránea familiar, retraso del crecimiento fetal, glaucoma, enfermedad glomerular, nefritis glomerular, gota, artritis gotosa, enfermedad de injerto contra huésped, enfermedades intestinales, lesión craneal, cefalea, pérdida de audición, cardiopatía, anemia hemolítica, púrpura de Henoch-Scholein, hepatitis, síndrome de fiebre periódica hereditaria, herpes zóster y simple, VIH-1, enfermedad de Hodgkin, enfermedad de Huntington, enfermedad de la membrana hialina, hiperamonemia, hipercalcemia, hipercolesterolemia, hiperinmunoglobulinemia D con fiebre recurrente (HIDS), anemias hipoplásicas y otras, anemia hipoplásica, púrpura trombocitopénica idiopática, incontinencia pigmentaria, mononucleosis infecciosa, enfermedad inflamatoria intestinal, enfermedad pulmonar inflamatoria, neuropatía inflamatoria, dolor inflamatorio, inflamación inducida por picadura de insecto, iritis, inflamación inducida por irritantes, isquemia/reperfusión, artritis reumatoide juvenil, queratitis, nefropatía, lesión renal causada por infecciones parasitarias, lesión renal causada por infecciones parasitarias, profilaxis del rechazo del trasplante de riñón, leptospiriosis, leucemia, síndrome de Loeffler, lesión pulmonar, lesión pulmonar, lupus, lupus, nefritis lúpica, linfoma, meningitis, mesotelioma, enfermedad mixta del tejido conjuntivo, síndrome de Muckle-Wells (urticaria, sordera, amiloidosis), esclerosis múltiple, pérdida muscular, distrofia muscular, miastenia grave, miocarditis, micosis fungoide, micosis fungoide, síndrome mielodisplásico, miositis, sinusitis nasal, enterocolitis necrosante, enfermedad inflamatoria multisistémica de inicio neonatal (NOMID), síndrome nefrótico, neuritis, enfermedades neuropatológicas, asma no inducida por alérgenos, obesidad, alergia ocular, neuritis óptica, trasplante de órgano, artrosis, otitis media, enfermedad de Paget, dolor, pancreatitis, enfermedad de Parkinson, pénfigo, pericarditis, fiebre periódica, periodontitis, endometriosis peritoneal, tos ferina, faringitis y adenitis (síndrome PFAPA), inflamación inducida por irritantes vegetales, neumonía, neumonitis, infección por Pneumocystis, inflamación inducida por hiedra venenosa/aceite de urushiol, poliarteritis nodosa, policondritis, enfermedad renal poliquística, polimiositis, psoriasis, psoriasis, psoriasis, psoriasis, enfermedades de estrés psicosocial, enfermedad pulmonar, hipertensión pulmonar, fibrosis pulmonar, pioderma gangrenoso, artritis estéril piógena, enfermedad renal, enfermedad retinal, carditis reumática, enfermedad reumática, artritis reumatoide, sarcoidosis, seborrea, sepsis, dolor grave, células falciformes, anemia de células falciformes, enfermedad inducida por sílice, síndrome de Sjogren, dermatopatías, apnea del sueño, tumores sólidos, lesión de la médula espinal, síndrome de Stevens-Johnson, accidente cerebrovascular, hemorragia subaracnoidea, quemadura solar, arteritis temporal, tenosinovitis, trombocitopenia, tiroiditis, trasplante de tejido, síndrome periódico asociado al receptor del TNF (TRAPS), toxoplasmosis, trasplante, lesión cerebral traumática, tuberculosis, diabetes de tipo 1, diabetes de tipo 2, colitis ulcerosa, urticaria, uveítis, granulomatosis de Wegener, enfermedad pulmonar intersticial, artritis psoriásica, artritis idiopática juvenil, síndrome de Sjogren, vasculitis asociada a anticuerpos anti-neutrófilos (ANCA), síndrome de anticuerpos antifosfolipídicos, sepsis, trombosis venosa profunda, fibrosis, enfermedad de Alzheimer, esclerodermia y síndrome CREST.
En una realización, la invención proporciona un compuesto proporcionado, o una sal farmacéuticamente aceptable del mismo, para su uso en terapia. En otra realización, la invención proporciona un compuesto proporcionado, o una sal farmacéuticamente aceptable del mismo, para su uso en el tratamiento de un trastorno mediado por la actividad inapropiada de PAD4. En otra realización, la invención proporciona un compuesto proporcionado, o una sal farmacéuticamente aceptable del mismo, para su uso en el tratamiento de la artritis reumatoide, vasculitis, lupus eritematoso sistémico, colitis ulcerosa, cáncer, fibrosis quística, asma, lupus eritematoso cutáneo o psoriasis. En otra
realización, la invención proporciona un compuesto proporcionado, o una sal farmacéuticamente aceptable del mismo, para su uso en el tratamiento de la artritis reumatoide. En otra realización, la invención proporciona un compuesto proporcionado, o una sal farmacéuticamente aceptable del mismo, para su uso en el tratamiento del lupus sistémico. En otra realización, la invención proporciona un compuesto proporcionado, o una sal farmacéuticamente aceptable del mismo, para su uso en el tratamiento de la vasculitis. En otra realización, la invención proporciona un compuesto proporcionado, o una sal farmacéuticamente aceptable del mismo, para su uso en el tratamiento del lupus eritematoso cutáneo. En otra realización, la invención proporciona un compuesto proporcionado, o una sal farmacéuticamente aceptable del mismo, para su uso en el tratamiento de la psoriasis. En una realización adicional, la invención proporciona una composición farmacéutica para el tratamiento o profilaxis de un trastorno mediado por la actividad inapropiada de PAD4 que comprende un compuesto proporcionado, o una sal farmacéuticamente aceptable del mismo. En una realización adicional, la invención proporciona una composición farmacéutica para el tratamiento o la profilaxis de la artritis reumatoide, vasculitis, lupus eritematoso sistémico, colitis ulcerosa, cáncer, fibrosis quística, asma, lupus eritematoso cutáneo o psoriasis, que comprende un compuesto proporcionado, o una sal farmacéuticamente aceptable del mismo. En una realización adicional, la invención proporciona una composición farmacéutica para el tratamiento o la profilaxis de la artritis reumatoide que comprende un compuesto proporcionado, o una sal farmacéuticamente aceptable del mismo. En una realización adicional, la invención proporciona una composición farmacéutica para el tratamiento o la profilaxis del lupus sistémico que comprende un compuesto proporcionado, o una sal farmacéuticamente aceptable del mismo. En una realización adicional, la invención proporciona una composición farmacéutica para el tratamiento o la profilaxis de la vasculitis que comprende un compuesto proporcionado, o una sal farmacéuticamente aceptable del mismo. En una realización adicional, la invención proporciona una composición farmacéutica para el tratamiento o la profilaxis del lupus eritematoso cutáneo que comprende un compuesto proporcionado, o una sal farmacéuticamente aceptable del mismo. En una realización adicional, la invención proporciona una composición farmacéutica para el tratamiento o la profilaxis de la psoriasis que comprende un compuesto proporcionado, o una sal farmacéuticamente aceptable del mismo.
Todas las características de cada uno de los aspectos de la invención se aplican a todos los demás aspectos mutatis mutandis.
Con el fin de que la invención descrita en el presente documento se pueda entender de forma más completa, se exponen los siguientes ejemplos. Debe entenderse que estos ejemplos son únicamente para fines ilustrativos y no deben interpretarse como limitantes de la presente invención de ningún modo.
Como se presenta en los siguientes Ejemplos, en ciertas realizaciones ilustrativas, los compuestos se preparan de acuerdo con los siguientes procedimientos generales. Se apreciará que, aunque los métodos generales representan la síntesis de ciertos compuestos de la presente invención, los siguientes métodos generales y otros métodos conocidos por un experto en la materia, pueden aplicarse a todos los compuestos y subclases y especies de cada uno de estos compuestos, como se describe en el presente documento.
Ciertos compuestos de la presente invención se prepararon de acuerdo con los Esquemas descritos más adelante. Para la síntesis de compuestos que implican química fotorredox, véase el Esquema 1

Para la síntesis de compuestos que implican química fotorredox de aminas protegidas cíclicas, bicíclicas, tricíclicas seguidas de desprotección y posterior acoplamiento véase el Esquema 2. Obsérvese que la amina cíclica utilizada en la etapa fotorredox también puede estar en una forma completamente elaborada que no requiere etapas posteriores de desprotección y acoplamiento.
Esquema 2

a
Lámpara Kessil donde “B” es un anillo saturado con o sin donde p.| es un grupo heteroátomos embebidos; E es RbCO-, RbOCO-protector ortogonal tal como 2) Desproteger P RcS02-, RaRaNCO-, etc.
CBZ o BOC 3) Enfriar con y Rr Rs son sustituyentes.
Donde X es un halógeno RbCOOH,
RbOCOCI, R0SO2CI,
RaRaNCOCI, etc.
4) Des proteger Pi
Para la síntesis de compuestos que implican química fotorredox de ácidos carboxílicos clínicos, bicíclicos o tricíclicos protegidos (por ejemplo, ésteres), seguidos de desprotección y posterior acoplamiento véase el Esquema 3. Obsérvese que el ácido carboxílico cíclica utilizada en la etapa fotorredox también puede estar en una forma completamente elaborada que no requiere etapas posteriores de desprotección y acoplamiento.
Esquema 3

donde P-i es un grupo Na^CO^
Lampara Kessil
donde‘ B es un amNo
protector ortogonal tal como saturado con o sin
CBZ o BOC heteroatomos embebidos
Donde X es un halógeno 2) Desproteger P y Rr R son sustituyentes
3) Acoplar con HNRaR
4) Desproteger P
Para la síntesis de compuestos que implican las reacciones de Suzuki, Stille u otras de acoplamiento cruzado aromático, véase el Esquema 4.
Esquema 4
donde P-i es un grupo protector
ortogononal adecuado, tal como donde B es un anillo aromático o CBZ o BOC heteroaromatico o un anillo parcialmente donde X es un halógeno y insaturado que contiene cero o un donde Y es un ester o acido boromco, heteroatomos y R R5 son sustituyentes. sal trifluoroacetato, haluro de cinc, haluro
de magnesio, trialquilestano u otra pareja
de acoplamiento fammarpara el experto en la técnica
Para la síntesis de productos acoplados del tipo de reacción de Buchwald, véase el Esquema 5.
Scheme 5

donde P-i es un grupo donde R6 es alquilo o 
protector ortogononal aromatico/heteroaromatico Rr R6 son sustituyentes adecuado, tal como y cíclico o aciclico o heteroatomo o
CBZ o BOC halógeno;
donde X es un halógeno E es un anillo monociclico o biciclico o
aromático o heteroaromatico o parcialmente
heteroaromatico y
W = -NHR6 o -OH o -SH
Descripción de los métodos de LCMS analíticos:
Método 1: Columna: Waters XBridge C18, 2,1 mm x 50 mm, partículas de 1,7 pm; Fase móvil A: 5:95 de acetonitrilo:agua con acetato amónico 10 mM; Fase móvil B: 95:5 de acetonitrilo:agua con acetato amónico 10 mM; Temperatura: 50 °C; Gradiente: de B al 0 % a B al 100 % durante 3 min, a continuación, una retención de 0,75 min en B al 100 %; Flujo: 1 ml/min; Detección: MS y UV (220 nm).
Método 2: Columna: Waters XBridge C18, 2,1 mm x 50 mm, partículas de 1,7 pm; Fase móvil A: 5:95 acetonitrilo:agua con ácido trifluoroacético al 0,1 % ; Fase móvil B: 95:5 acetonitrilo:agua con ácido trifluoroacético al 0,1 % ; Temperatura: 50 °C; Gradiente: de B al 0 % a B al 100 % durante 3 min, a continuación, una retención de 0,75 min en B al 100 % ; Flujo: 1 ml/min; Detección: MS y UV (220 nm).
Método A: Columna: Waters Acquity UPLC BEH C18, 2,1 * 50 mm, partículas de 1,7 pm; Fase móvil A: ACN:agua 5:95 con NH4OAc 10 mM; Fase móvil B: ACN:agua 95:5 con NH4OAc 10 mM; Temperatura: 50 °C; Gradiente: 0 -100% de B durante 3 minutos, después una retención de 0,75 minutos a 100 % de B; Flujo: 1,11 ml/min; Detección: UV a 220 nm.
Método B: Columna: Waters Acquity UPLC BEH C18, 2,1 * 50 mm, partículas de 1,7 pm; Fase móvil A: 5:95 de ACN:agua con TFA al 0,1 % ; Fase móvil B: 95:5 de ACN:agua con t Fa al 0,1 % ; Temperatura: 50 °C; Gradiente: 0 -100 % de B durante 3 minutos, después una retención de 0,75 minutos a 100 % de B; Flujo: 1,11 ml/min; Detección: UV a 220 nm.
Método C: Columna: PHENOMENEX® Luna 3 pm C18 (2,0 x 30 mm); Fase móvil A: 10:90 de MeOH:agua con TFA al 0,1 % ; Fase móvil B: 90:10 de MeOH:agua con t Fa al 0,1 % ; Gradiente: 0-100 % de B durante 2 minutos, después una retención de 1 minutos a 100 % de B; Flujo: 1 ml/min; Detección: UV a 220 nm.
Método D: Waters Acquity UPLC BEH C18, 2,1 * 50 mm, partículas de 1,7 pm; Fase móvil A: agua con TFA al 0,05 % ; Fase móvil B: ACN con TFA al 0,05 % ; Gradiente: 2-98 % de B durante 1 minuto, después una retención de 0,5 minutos a 98 % de B; Flujo: 0,8 ml/min; Detección: UV a 220 o 254 nm.
Ejemplo 1
((1R,4R,7R)-7-amino-2-azabiciclo[2.2.11heptan-2-il)(2-(3-bromo-4-(ciclopropilmetil)-4H-tieno[3,2-b1pirrol-5-il)-7-metox¡-1-met¡l-1H-benzo[d1¡m¡dazol-5-¡l)metanona

Intermedio 1A: 3-bromo-4-(c¡cloprop¡lmet¡l)-4H-t¡enof3,2-b1p¡rrol-5-carbox¡lato de ciclopropilmetilo

En un vial de centelleo de 40 ml, se trató una mezcla en agitación de ácido 3-bromo-4H-tieno[3,2-b]p¡rrol-5-carboxílico (1 g, 4,06 mmol) y carbonato de potasio (2,247 g, 16,26 mmol) en DMF (5 ml) con (bromometil)ciclopropano (1,577 ml, 16,26 mmol). El vial se cerró herméticamente y la reacción se agitó a 70 °C durante 3 horas, punto en el que se juzgó que se había completado mediante LCMS. La mezcla se diluyó con acetato de etilo (75 ml) y la solución turbia se lavó una vez con agua, tres veces con cloruro de litio al 10 % y una vez con salmuera, después se secó sobre sulfato sódico y se concentró al vacío. El residuo se adsorbió en Celite y se cromatografió mediante MPLC en una columna de gel de sílice de 40 g, eluyendo a 40 ml/min con un gradiente del 0 % al 30 % de acetato de etilo/hexanos sobre 14 volúmenes de columna. Las fracciones que contenían el producto deseado se combinaron y concentraron al vacío para producir el compuesto del título (1,33 g, 3,75 mmol, 92 % de rendimiento) como un sólido ámbar. RMN 1H (499 MHz, cloroformo-d) 87,25 (s, 1H), 7,24 (s, 1H), 4,79 (d, J = 7,2 Hz, 2H), 4,13 (d, J = 7,2 Hz, 2H), 1,46 - 1,37 (m, 1H), 1,31 - 1,21 (m, 1H), 0,66-0,60 (m, 2H), 0,52 - 0,46 (m, 4H), 0,41 - 0,36 (m, 2h ). MS ESI m/z = 353,9 (M+H). Tiempo de retención de HPLC 1,25 minutos, Método D.
Intermedio 1B: Ácido 3-bromo-4-(c¡cloprop¡lmet¡l)-4H-t¡eno[3,2-b1p¡rrol-5-carboxíl¡co

Una solución en agitación de 3-bromo-4-(cidopropilmetil)-4H-tieno[3,2-b]pirrol-5-carboxilato de ciclopropilmetilo (1,27 g, 3,58 mmol) en metanol/THF (3:1) (20 ml) se trató con hidróxido de sodio 1 M (11,11 ml, 11,11 mmol). La reacción se agitó a 70 °C durante 3 horas, punto en el que se juzgó que se había completado mediante LCMS. Los disolventes orgánicos se evaporaron y la solución acuosa restante se lavó dos veces con éter dietílico, luego se trató con HCl 1 M (14 ml). La solución turbia se extrajo 4X con acetato de etilo, luego, las fases orgánicas combinadas se lavaron una vez con salmuera, se secaron sobre sulfato de sodio y se concentraron al vacío para producir el compuesto del título (0,99 g, 3,30 mmol, 92 % de rendimiento) como un sólido incoloro. RMN 1H (500 mHz , DMSO-da) 812,76 (s a, 1H), 7,71 (s, 1H), 7,23 (s, 1H), 4,70 (d, J = 7,0 Hz, 2H), 1,37 - 1,26 (m, 1H), 0,48 - 0,37 (m, 4H).
Intermedio 1C: 2-(3-bromo-4-(c¡cloprop¡lmet¡l)-4H-t¡eno[3.2-b1p¡rrol-5-il)-7-metox¡-1-met¡l-1H-benzo[d1¡m¡dazol-5-carboxilato de metilo

Una solución en agitación de 3-amino-5-metoxi-4-(metilamino)benzoato de metilo (0,832 g, 3,96 mmol), ácido 3-bromo-4-(cicloprop¡lmet¡l)-4H-t¡eno[3,2-b]p¡rrol-5-carboxíl¡co (0,99 g, 3,30 mmol) y base de Hunig (1,440 ml, 8,25 mmol) en DMF (10 ml) se trató con HATU (1,505 g, 3,96 mmol). La reacción se agitó en una atmósfera de nitrógeno a temperatura ambiente durante 60 horas, en ese punto, la LCMS juzgó que se había completado basándose en la desaparición del material inicial. La mezcla de reacción se concentró al vacío y el residuo se recogió en acetato de etilo (250 ml) y cloruro de litio al 10 % (100 ml). La mezcla turbia se filtró, las capas se separaron y la fase de acetato de etilo se lavó dos veces con cloruro de litio al 10 % y una vez con salmuera. La fase orgánica se secó sobre sulfato de sodio, se concentró al vacío y el residuo se recogió en ácido acético (15 ml). La reacción se calentó a 75 °C y se agitó durante 90 minutos, punto en el que se juzgó que se había completado mediante LCMS. La mezcla se concentró al vacío y el residuo se recogió en acetato de etilo (100 ml). Se añadió una barra de agitación magnética al matraz y la mezcla agitada se trató cuidadosamente con carbonato de sodio medio saturado hasta que cesó el desprendimiento de gas. La mezcla se agitó durante 10 minutos más, tiempo durante el cual precipitó un sólido incoloro. Los sólidos se recogieron mediante filtración, se aclaró abundantemente con agua y acetato de etilo, seguido de una pequeña cantidad de metanol y se secaron al vacío para producir 700 mg de un sólido amarillo pálido. La NMR y la LCMS son consistentes con el producto esperado. El filtrado y los lavados combinados se transfirieron a un embudo de decantación y se separaron las capas. La fase orgánica se lavó dos veces con carbonato de sodio saturado y las fases acuosas combinadas se extrajeron dos veces con acetato de etilo (50 ml). Las fases orgánicas combinadas se lavaron una vez con salmuera, se secaron sobre sulfato sódico y se concentraron al vacío para producir 0,85 g de un sólido pegajoso de color ámbar. El material se agitó en metanol hirviendo (15 ml) durante 5 minutos, después la solución se dejó enfriar a temperatura ambiente. Los sólidos resultantes se recogieron mediante filtración, se enjuagaron 3 veces con metanol y se secaron al vacío para producir 525 mg de un sólido ámbar. La RMN es consistente con el producto esperado. Las dos cosechas de sólidos se combinaron para producir el compuesto del título (1,25 g, 2,64 mmol, 80 % de rendimiento). RMN 1H (499 MHz, DMSO-d6) 87,94 (d, J = 1,3 Hz, 1H), 7,61 (s, 1H), 7,39 (d, J = 1,2 Hz, 1H), 7,08 (s, 1H), 4,59 (d, J = 7,0 Hz, 2H), 4,09 (s, 3H), 4,02 (s, 3H), 3,89 (s, 3H), 1,13 - 1,02 (m, 1H), 0,34 - 0,28 (m, 2H), 0,00 - -0,05 (m, 2H). MS ESI m/z = 476,1 (m+H). Tiempo de retención de Hp LC 1,10 minutos, Método D.
Intermedio 1D: Ácido 2-(3-bromo-4-(ciclopropilmetil)-4H-tienol[3,2-b1pirrol-5-il)-7-metoxi-1-metil-1H-benzo[d1imidazol-5-carboxílico
Una solución en agitación de 2-(7-bromo-1-(ciclopropilmetil)-1H-indol-2-il)-7-metoxi-1-metil-1H-benzo[d]imidazol-5-carboxilato de metilo (1,25 g, 2,67 mmol) en metanol/THF (2:1) (15 ml) se trató con hidróxido de sodio 1 M (8,01 ml, 8,01 mmol). La reacción se agitó a 50 °C durante 18 horas, punto en el que se juzgó que se había completado mediante LCMS. Los disolventes orgánicos se eliminaron en el evaporador rotatorio y la mezcla acuosa heterogénea restante se ajustó a pH 5 con HCl 1 M. La mezcla se agitó enérgicamente durante 30 minutos, luego se extrajo 4 veces con acetato de etilo (75 ml). Las fases orgánicas combinadas se lavaron una vez con salmuera, se secaron sobre sulfato de sodio y se concentraron al vacío para producir el compuesto del título (1,12 g, 2,433 mmol, 91 % de rendimiento) como un sólido incoloro. RMN 1H (499 MHz, DMSO-da) 812,85 (s a, 1H), 7,92 (d, J = 1,2 Hz, 1H), 7,61 (s, 1H), 7,39 (d, J = 1,2 Hz, 1H), 7,07 (s, 1H), 4,59 (d, J = 7,0 Hz, 2H), 4,09 (s, 3H), 4,01 (s, 3H), 1,12 - 1,02 (m, 1H), 0,35 - 0,27 (m, 2H), 0,00 - -0,07 (m, 2H). MS ESI m/z = 460,1 (M+H). Tiempo de retención de HPLC 0,93 minutos, Método D.
Intermedio 1E: ((1R.4R.7R)-2-(2-(3-bromo-4-(c¡cloprop¡lmet¡l)-4H-t¡enol[3,2-b1p¡rrol-5-¡l)-7-metox¡-1-met¡l-1H-benzo[d1¡m¡dazol-5-carbonil)-2-azab¡c¡clo[2.2.11heptan-7-¡l)carbamato de tere-butilo
Una solución en agitación de ((1R,4R,7R)-2-azabiciclo[2.2.1]heptan-7-¡l)carbamato de tere-butilo (0,568 g, 2,68 mmol), ácido 2-(3-bromo-4-(c¡cloprop¡lmet¡l)-4H-t¡eno[3,2-b]p¡rrol-5-il)-7-metox¡-1-met¡l-1H-benzo[d]¡m¡dazol-5-carboxílico (1,12 g, 2,433 mmol) y trietilamina (1,017 ml, 7,30 mmol) en diclorometano (20 ml) se trató con BOP (1,130 g, 2,55 mmol). La reacción se agitó a temperatura ambiente durante 18 horas, punto en el que se juzgó que se había completado mediante LCMS. La mezcla se concentró al vacío y el residuo se recogió en acetato de etilo (300 ml). La solución turbia se lavó 3X con agua, 3 veces con hidróxido de sodio 1 M y una vez con salmuera, después se secó sobre sulfato sódico y se concentró al vacío. El residuo se cromatografió mediante MPLC sobre una columna de gel de sílice de 80 g, eluyendo a 60 ml/min con un gradiente del 0 % al 10 % de acetona/hexanos sobre 15 volúmenes de columna. Las fracciones que contenían el producto deseado se combinaron y concentraron al vacío para producir el compuesto del título (1,65 g, 2,52 mmol, 104 % de rendimiento) como un sólido blanco. RMN 1H (499 MHz, CLOROFORMO-d) 87,48 (d, J = 1,1 Hz, 1H), 7,14 (s, 1H), 7,05 - 6,97 (m, 1H), 6,72-6,67 (m, 1H), 4,69 - 4,47 (m, 3H), 4,35 (s a, 1H), 4,12 (s, 3H), 4,03 (s, 3H), 3,90 - 3,71 (m, 2H), 3,31 - 3,18 (m, 1H), 2,54 (s a, 1H), 2,11 - 1,81 (m, 3H), 1,72 (s a, 1H), 1,53-1,36 (m, 9H), 1,15 - 1,05 (m, 1H), 0,40 - 0,27 (m, 2H), 0,01 --0 ,16 (m, 2H). MS ESI m/z = 654,2 (M+H). Tiempo de retención de HPLC 0,97 minutos, Método D.
Ejemplo 1: ((1R.4R.7R)-7-am¡no-2-azab¡c¡clo[2.2.11heptan-2-¡l)(2-(3-bromo-4-(c¡cloprop¡lmet¡l)-4H-t¡eno[3.2-b1p¡rrol-5-¡l)-7-metoxi-1-met¡l-1H-benzo[d1¡m¡dazol-5-¡l)metanona
Una solución en agitación de ((1R,4R,7R)-2-(2-(3-bromo-4-(ciclopropilmetil)-4H-tieno[3,2-b]pirrol-5-il)-7-metoxi-1-met¡l-1H-benzo[d]im¡dazol-5-carbon¡l)-2-azab¡c¡clo[2.2.1]heptan-7-¡l)carbamato de tere-butilo (20 mg, 0,031 mmol) en diclorometano (1 ml) se trató con HCl 4 M en dioxano (1 ml, 4,00 mmol). La reacción se agitó a temperatura ambiente durante 1 hora, punto en el que se juzgó que se había completado mediante LCMS. La mezcla se concentró al vacío. El material en bruto se purificó a través de LC/MS preparativa con las siguientes condiciones: Columna: XBridge C18, 200 mm x 19 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: un mantenimiento de 0 minutos al 31 % de B, 31-71 % de B durante 20 minutos, después, una retención de 4 minutos a 100 % de B; Caudal: 20 ml/min; Temperatura de la columna: 25 °C. La recogida de fracciones se activó por señales de MS y UV. Las fracciones que contenían el producto deseado se combinaron y se secaron por evaporación por centrifugación. A continuación, el material purificado se diluyó con DMF, se trató con Si-piridina y se agitó durante un mínimo de 2 horas. La mezcla resultante se filtró y se secó mediante evaporación centrífuga para producir el compuesto del título, (17,1 mg, 0,031 mmol, 101 % de rendimiento). RMN 1H (500 MHz, DMSO-d6) 87,58 (d, J =
2.3 Hz, 1H), 7,52 - 7,36 (m, 1H), 7,04 (d, J = 1,7 Hz, 1H), 7,02 - 6,94 (m, 1H), 4,61 (d a, J = 5,1 Hz, 2H), 4,11 (s, 3H), 4.04 (s, 3H), 3,77 - 3,59 (m, 3H), 3,20 - 3,06 (m, 1H), 2,35 - 2,20 (m, 1H), 2,13 - 1,70 (m, 4H), 1,56 - 1,40 (m, 1H), 1,09 (d a, J = 3,2 Hz, 1H), 0,37 (d a, J = 7,9 Hz, 2H), 0,02 (s a, 2H). MS ESI m/z = 554,0.2 (M+H). Tiempo de retención de HPLC 1,97 minutos, Método 1.
Ejemplo 2
((1R,4R,7R)-7-amino-2-azabiciclo[2.2.1lheptan-2-il)(2-(4-(ciclopropilmetil)-3-(oxetan-3-il)-4H-tieno[3,2-blpirrol-5-il)-7-metoxi-1-metil-1H-benzo[dlimidazol-5-il)metanona

En un vial de aprox. 7,4 ml (2 dracmas líquidas), una mezcla en agitación de 3-yodooxetano (28,1 mg, 0,153 mmol), ((1R,4R,7R)-2-(2-(3-bromo-4-(ciclopropilmetil)-4H-tienol[3,2-b]pirrol-5-il)-7-metoxi-1-metil-1H-benzo[d]imidazol-5-carbonil)-2-azabiciclo[2.2.1]heptan-7-il)carbamato de tere-butilo (50 mg, 0,076 mmol), tris(trimetilsilil)silano (0,035 ml, 0,115 mmol), Ir(dF(CF3)ppy)2(dtbbpy)PF6 (2,57 mg, 2,291 ^mol) y carbonato de sodio (32,4 mg, 0,306 mmol) en 1,4-dioxano (2 ml) se desgasificó con nitrógeno burbujeante durante 10 minutos. En un vial separado, una mezcla en agitación de complejo de éter dimetílico de etilenglicol de cloruro de níquel (II) (2,52 mg, 0,011 mmol) y 4,4'-di-ferebutil-2,2'-bipiridina (3,49 mg, 0,013 mmol) en 1,4-dioxano (1 ml) se desgasificó con nitrógeno durante 20 minutos. El complejo de níquel se transfirió al que contenía la otra mezcla, el vial se selló y la reacción se agitó a temperatura ambiente bajo una lámpara Kessil azul durante 18 horas, punto en el que se juzgó que se había completado mediante LCMS. La mezcla se diluyó con diclorometano (2 ml) y se filtró y el filtrado se concentró al vacío. El residuo se recogió en diclorometano (2 ml) y la solución se trató con TFA (200 l^, 2,60 mmol). La reacción se agitó a temperatura ambiente durante 2 horas, punto en el que se juzgó que se había completado mediante LCMS. La mezcla se concentró al vacío y el residuo se concentró tres veces a partir de diclorometano para eliminar el TFA residual. El material en bruto se purificó a través de LC/MS preparativa con las siguientes condiciones: Columna: XBridge C18, 200 mm x 19 mm, partículas de 5 ^m; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: un mantenimiento de 0 minutos al 16 % de B, 16-56 % de B durante 30 minutos, después, una retención de 4 minutos a 100 % de B; Caudal: 20 ml/min; Temperatura de la columna: 25 °C. La recogida de fracciones se activó por señales de MS y UV. Las fracciones que contenían el producto deseado se combinaron y se secaron por evaporación por centrifugación. A continuación, el material purificado se diluyó con DMF, se trató con Si-piridina y se agitó durante un mínimo de 2 horas. La mezcla resultante se filtró y se secó mediante evaporación centrífuga para producir el compuesto del título, (14,9 mg, 0,028 mmol, 36,5 % de rendimiento). RMN 1H (500 MHz, DMSO-da) 57,24 (s, 1H), 7,19 - 7,06 (m, 1H), 6.75 - 6,64 (m, 2H), 4,80 - 4,73 (m, 2H), 4,59 (t a, J = 6,0 Hz, 2H), 4,51 - 4,41 (m, 1H), 4,04 (s a, 2H), 3,84 (s, 3H), 3.75 (s, 3H), 3,57 -3,26 (m, 3H), 3,14 -2,92 (m, 1H), 2,87 -2,74 (m, 1H), 2,05 - 1,91 (m, 1H), 1,82-1,62 (m, 2H), 1,59 - 1,43 (m, 1H), 1,27 - 1,03 (m, 1H), 0,64 - 0,53 (m, 1H), 0,01 (d a, J = 7,6 Hz, 2H), -0,47 - -0,62 (m, 2H). MS ESI m/z = 532,2 (M+H). Tiempo de retención de Hp Lc 1,42 minutos, Método 1.
Los siguientes compuestos de la Tabla 1 se pueden preparar mediante los procedimientos descritos en el Ejemplo 2, sustituyendo el haluro de alquilo apropiado por 3-bromo-1-propanol. Para ejemplos en los que el sustituyente en la posición 7 del indol contiene una amina básica, se usó el haluro de aminoalquilo protegido con Boc apropiado, y el grupo Boc se escindió durante el paso final de desprotección.
Tabla 1

continuación

continuación

Ejemplos 18 y 19
((1R.4R.7R)-7-am¡no-2-azab¡c¡clor2.2.11heptan-2-¡l)(2-(3-(azet¡d¡n-3-ilmet¡l)-4-(c¡cloprop¡lmet¡l)-4H-t¡eno [3.2-bjp¡rrol-5-¡l)-7-metox¡-1-met¡l-1H-benzo[d1¡m¡dazol-5-¡l)metanona y (2-(3-(3-am¡no-2-(cloromet¡l)prop¡l)-4-(c¡cloprop¡lmet¡l)-4H
t¡eno[3.2-blp¡rrol-5-¡l)-7-metox¡-1-met¡l-1H-benzo[dl¡m¡dazol-5-¡l)((1R.4R.7R)-7-am¡no-2-azab¡c¡clo[2.2.1lheptan-2-¡l)metanona

Intermed¡o 18/19A: 3-((5-(5-((1R.4R.7R)-7-((terc-butox¡carbon¡l)am¡no)-2-azab¡c¡clo[2.2.1lheptano-2-carbon¡l)-7-metox¡-1-met¡l-1H-benzo[dl¡m¡dazol-2-¡l)-4-(c¡cloprop¡lmet¡l)-4H-t¡eno[3.2-blp¡rrol-3-¡l)met¡l)azet¡d¡na-1-carbox¡lato de terc-but¡lo

En un v¡al de aprox. 7.4 ml (2 dracmas líqu¡das). una mezcla ag¡tada de 3-(yodomet¡l)azet¡d¡na-1-carbox¡lato de terc-but¡lo (137 mg. 0.463 mmol). ((1R.4R.7R)-2-(2-(7-bromo-1-(c¡cloprop¡lmet¡l)-1H-¡ndol-2-¡l)-7-metox¡-1-met¡l-1H-benzo[dl ¡m¡dazol-5-carbon¡l)-2-azab¡c¡clo[2.2.1lheptan-7-¡l)carbamato de terc-but¡lo (150 mg. 0.231 mmol). tr¡s(tr¡met¡ls¡l¡l)s¡lano (0.107 ml. 0.347 mmol). Ir(dF(CF3)ppy)2(dtbbpy)PF6 (7.78 mg. 6.94 pmol) y carbonato de sod¡o (98 mg. 0.925 mmol) en 1.4-d¡oxano (2 ml) se desgas¡f¡có con n¡trógeno burbujeante durante 10 m¡nutos. En un v¡al separado. una mezcla en ag¡tac¡ón de complejo de éter d¡metíl¡co de et¡lengl¡col de cloruro de níquel (II) (7.62 mg.
0.035 mmol) y 4.4'-d¡-terc—but¡l-2.2'-b¡p¡r¡d¡na (10.55 mg. 0.039 mmol) en 1.4-d¡oxano (1 ml) se desgas¡f¡có con n¡trógeno durante 20 m¡nutos. El complejo de níquel se transf¡r¡ó al que contenía la otra mezcla. el v¡al se selló y la reacc¡ón se ag¡tó a temperatura amb¡ente bajo una lámpara Kess¡l azul durante 60 horas. punto en el que se juzgó que se había completado med¡ante LCMS. La mezcla se d¡luyó con d¡clorometano (2 ml) y se f¡ltró. y el f¡ltrado se usó tal cual en la s¡gu¡ente etapa. MS ESI m/z = 745.5 (M+H). T¡empo de retenc¡ón de HPLC 0.97 m¡nutos. Método D.
Ejemplo 18 y 19: ((1R.4R.7R)-7-am¡no-2-azab¡c¡clo[2.2.1lheptan-2-¡l)(2-(3-(azet¡d¡n-3-¡lmet¡l)-4-(c¡cloprop¡lmet¡l)-4H-t¡eno [3.2-blp¡rrol-5-¡l)-7-metox¡-1-met¡l-1H-benzo[dl¡m¡dazol-5-¡l)metanona y (2-(3-(3-am¡no-2-(cloromet¡l)prop¡l)-4-(c¡cloprop¡lmet¡l)-4H-t¡eno[3.2-blp¡rrol-5-¡l)-7-metox¡-1-met¡l-1H-benzo[dl¡m¡dazol-5-¡l)((1R.4R.7R)-7-am¡no-2-azab¡c¡clo[2.2.1lheptan-2-¡l)metanona
Una soluc¡ón en ag¡tac¡ón de 3-((5-(5-((1 R.4R.7R)-7-((terc-butox¡carbon¡l)am¡no)-2-azab¡c¡clo[2.2.1lheptano-2-carbon¡l)-7-metox¡-1-met¡l-1H-benzo[dl¡m¡dazol-2-¡l)-4-(c¡cloprop¡lmet¡l)-4H-t¡eno[3.2-blp¡rrol-3-¡l)met¡l)azet¡d¡na-1-carbox¡lato de terc-but¡lo (56.6 mg. 0.076 mmol) en d¡clorometano/d¡oxano 1:1 (4 ml) con HCl 4 M en d¡oxano (2 ml.
8.00 mmol). La reacc¡ón se ag¡tó a temperatura amb¡ente durante 2 horas. punto en el que se juzgó que se había completado med¡ante LCMS. Se detectaron dos productos: la azet¡d¡na deseada (m/z = 545) y el producto de ad¡c¡ón de HCl/apertura del an¡llo (m/z = 581). La mezcla se concentró al vacío. El mater¡al en bruto se pur¡f¡có a través de LC/MS preparat¡va con las s¡gu¡entes cond¡c¡ones: Columna: XBr¡dge C18. 200 mm x 19 mm. partículas de 5 pm; Fase móv¡l A: 5:95 de aceton¡tr¡lo: agua con acetato de amon¡o 10 mM; Fase móv¡l B: 95:5 de aceton¡tr¡lo: agua con acetato de amon¡o 10 mM; Grad¡ente: un manten¡m¡ento de 0 m¡nutos al 10% de B. 10-50% de B durante 20 m¡nutos. después. una retenc¡ón de 4 m¡nutos a 100 % de B; Caudal: 20 ml/m¡n; Temperatura de la columna: 25 °C. La recog¡da de fracc¡ones se act¡vó por señales de UV. Las fracc¡ones que contenían el producto deseado se comb¡naron y se secaron por evaporac¡ón por centr¡fugac¡ón. A cont¡nuac¡ón. el mater¡al pur¡f¡cado se d¡luyó con
DMF, se trató con Si-piridina y se agitó durante un mínimo de 2 horas. La mezcla resultante se filtró y se secó mediante evaporación centrífuga para producir:
Ejemplo 18: (3,3 mg, 5,69 pmol, 7,49 % de rendimiento). RMN 1H (500 MHz, DMSO-da) 87,42 -7,30 (m, 1H), 7,02 -6,88 (m, 3H), 4,45 (d a, J = 6,1 Hz, 2H), 4,16 (s a, 1H), 4,07 (s a, 3H), 3,98 (s a, 3H), 3,78 (d a, J = 6,4 Hz, 1H), 3,49 - 3,16 (m, 4H), 3,06 (d a, J = 11,0 Hz, 1H), 2,99 (s, 1H), 2,28 -2 ,14 (m, 1H), 2,05 - 1,83 (m, 2H), 1,82 - 1,71 (m, 1H), 1,45 (s a, 1H), 1,38 (s a, 1H), 1,23 (s a, 2H), 1,02 - 0,82 (m, 3H), 0,26 (s a, 2H), -0,24 (s a, 2H). MS ESI m/z = 545,5 (M+H). Tiempo de retención de Hp Lc 0,94 minutos, Método 2.
Ejemplo 19: (11,8 mg, 0,020 mmol, 26,5 % de rendimiento). RMN 1H (500 MHz, DMSO-d6) 87,19-7,07 (m, 1H), 6,99 - 6,85 (m, 1H), 6,76 - 6,61 (m, 2H), 4,18 (t a, J = 7,2 Hz, 2H), 3,95 (t a, J = 9,6 Hz, 1H), 3,83 (d a, J = 4,1 Hz, 3H), 3,74 (s a, 3H), 3,60 (s a, 3H), 3,40 (d a, J = 10,4 Hz, 1H), 3,02 (d a, J = 7,1 Hz, 1H), 2,93 - 2,83 (m, 1H), 2,74 (d a, J = 5,6 Hz, 1H), 2,60 -2,47 (m, 1H), 2,19 -2,07 (m, 1H), 1,78 - 1,52 (m, 3H), 1,38 - 1,20 (m, 1H), 1,00 (s a, 1H), 0,79 -0,56 (m, 2H), 0,03 (d a, J = 4,4 Hz, 2H), -0,49 (s a, 2H). (El recuento de protones es bajo debido al algoritmo de supresión de agua utilizado durante el procesamiento de datos). MS ESI m/z = 581,4 (M+H). Tiempo de retención de HPLC 1,31 minutos, Método 1.
Ejemplo 20
((1R,4R,7R)-7-amino-2-azabiciclo[2.2.11heptan-2-il)(2-(4-(ciclopropilmetil)-3-(2-azaspiro[3.31heptan-6-il)-4H-tieno[3,2-b1pirrol-5-il)-7-metoxi-1-metil-1H-benzo[d1imidazol-5-il)metanona

Intermedio 20A: 6-(tosiloxi)-2-azaespiro[3,31heptano-2-carboxilato de tere-butilo

En un vial de centelleo de 20 ml, una solución agitada de 6-hidroxi-2-azaspiro[3,31heptano-2-carboxilato de terebutilo (291 mg, 1,364 mmol) y trietilamina (0,285 ml, 2,047 mmol) en diclorometano (5 ml) se trató con tosil-Cl (286 mg, 1,501 mmol) seguido de DMAP (16,67 mg, 0,136 mmol). El vial se cerró herméticamente y la de reacción se agitó a temperatura ambiente durante 18 horas. La TLC (50 % de EtOAc/hexano, KMnO4) indicó que la reacción no se había completado. La mezcla se trató con trietilamina (0,076 ml, 0,544 mmol) y tosil-Cl (50 mg, 0,262 mmol) y la reacción se agitó a temperatura ambiente durante 18 horas, punto en el que se juzgó que se había completado mediante TLC. La mezcla se diluyó con diclorometano (5 ml) y se lavó dos veces con agua, dos veces con NaOH 1 M, dos veces con HCl 1 M y una vez con salmuera, después se secó sobre sulfato sódico y se concentró al vacío. El residuo se cromatografió mediante MPLC sobre una columna de gel de sílice de 24 g, eluyendo a 40 ml/min con un gradiente del 5 % al 50 % de acetona/hexanos sobre 13 volúmenes de columna. Las fracciones que contenían el producto deseado se combinaron y concentraron al vacío para producir el compuesto del título (425 mg, 1,157 mmol, 85 % de rendimiento) como un sólido incoloro. RMN 1H (499 MHz, CLOROFORMO-d) 87,81-7,75 (m, 2H), 7,37 (d, J = 8,0 Hz, 2H), 4,70 (quint., J = 7,2 Hz, 1H), 3,86 (d, J = 2,1 Hz, 4H), 2,54 -2,46 (m, 5H), 2,36 -2,28 (m, 2H), 1,42 (s, 9H).
Intermedio 20B: 6-yodo -2-azaespiro[3.31heptano-2-carboxilato de tere-butilo

En un vial de aprox. 7,4 ml (2 dracmas líquidas), una solución de 6-(tos¡lox¡)-2-azasp¡ro[3,3]heptano-2-carbox¡lato de fere-butilo (420 mg, 1,143 mmol) en metiletilcetona (5 ml) se trató con yoduro de sodio (685 mg, 4,57 mmol). El vial se cerró herméticamente y la reacción se agitó a 100 °C durante 3 horas, luego se almacenó en el congelador durante la noche. La reacción se agitó a 100 °C durante 2 horas, punto en el que se juzgó que se había completado mediante LCMS. La mezcla se diluyó con diclorometano (10 ml) y se filtró y el filtrado se concentró al vacío. El residuo se cromatografió mediante MPLC sobre una columna de gel de sílice de 12 g, eluyendo a 30 ml/min con un gradiente del 50% al 100% de acetona/hexanos sobre 12 volúmenes de columna, luego con acetato de etilo al 100%. Las fracciones que contenían el producto deseado se agruparon y concentraron al vacío para producir el compuesto del título (267 mg, 0,826 mmol, 72,3% de rendimiento). RMN 1H (499 MHz, cloroformo-d) 8 4,31 (d, J=7,8 Hz, 1H), 3,96 (d, J = 15,1 Hz, 4H), 2,99 -2,89 (m, 2H), 2,77 -2,68 (m, 2H), 1,45 (s, 9H).
Ejemplo 20: ((1R.4R.7R)-7-am¡no-2-azab¡c¡clo[2.2.11heptan-2-¡l)(2-(4-(c¡cloprop¡lmet¡l)-3-(2-azasp¡ro[3.31heptan-6-¡l)-4H-t¡eno[3.2-b1p¡rrol-5-¡l)-7-metox¡-1-met¡l-1H-benzo[d1¡m¡dazol-5-¡l)metanona
El compuesto del título se preparó a partir de ((1R,4R,7R)-2-(2-(3-bromo-4-(c¡cloprop¡lmet¡l)-4H-t¡eno[3,2-b]p¡rrol-5-il)-7-metoxi-1- met¡l-1H-benzo[d]¡m¡dazol-5-carbon¡l)-2-azab¡c¡clo[2.2.1]heptan-7-¡l)carbamato de fere-butilo y 6-yodo-2-azaespiro[3.3]heptano-2-carboxilato de fere-butilo usando el procedimiento descrito en el Ejemplo 2. RMN 1H (500 MHz, DMSO-d6) 87,28-7,04 (m, 1H), 6,95 (s, 1H), 6,78 - 6,64 (m, 2H), 4,14 (d a, J = 6,6 Hz, 2H), 3,92 (s, 2H), 3,84 (s, 3H), 3,76 (s, 3H), 3,74 (s, 2H), 3,51 - 3,33 (m, 2H), 3,32 - 3,04 (m, 1H), 2,99 - 2,91 (m, 1H), 2,60 - 2,49 (m, 2H), 2,27 (s a, 9H), 1,71 (s a, 3H), 0,62 (s a, 1H), 0,01 (d a, J = 7,8 Hz, 2H), -0,48 (s a, 2H). MS ESI m/z = 571,2 (M+H). Tiempo de retención de HpLC 1,14 minutos, Método 2.
Ejemplos 21 y 22 ((1R,4R,7R)-7-am¡no-2-azab¡c¡clo[2.2.1]heptan-2-¡l)(2-(4-(c¡cloprop¡lmet¡l)-3-(3-h¡drox¡c¡clobu¡l)-4H-t¡eno[3.2-b1p¡rrol-5-¡l)-7-metox¡-1-met¡l-1H-benzo[d1¡m¡dazol-5-¡l)metanona. isómero 1 e isómero 2

La mezcla isomérica de e/s/frans-((1R,4R,7R)-7-am¡no-2-azab¡c¡clo[2.2.1]heptan-2-¡l)(2-(4-(c¡cloprop¡lmet¡l)-3-(3-h¡drox¡c¡clobut¡l)-4H-t¡eno[3,2-b]p¡rrol-5-¡l)-7-metox¡-1-met¡l-1H-benzo[d]¡m¡dazol-5-¡l)metanona del Ejemplo 7 se separó mediante LC/MS preparativa con las siguientes condiciones: Columna: XBridge C18, 200 mm x 19 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: un mantenimiento de 0 minutos al 16 % de B, 16-56 % de B durante 20 minutos, después, una retención de 4 minutos a 100 % de B; Caudal: 20 ml/min; Temperatura de la columna: 25 °C. La recogida de fracciones se activó por señales de MS y UV. Las fracciones que contenían el producto deseado se combinaron y se secaron por evaporación por centrifugación. El material se purificó de nuevo mediante LC/MS preparativa con las condiciones siguientes: Columna: XBridge C18, 200 mm x 30 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con ácido trifluoroacético al 0,1 %; Fase móvil B: 95:5 de acetonitrilo: agua con ácido trifluoroacético al 0,1 %; Gradiente: un mantenimiento de 0 minutos al 3% de B, 3-43% de B durante 20 minutos, después, una retención de 2 minutos a 100% de B; Caudal: 45 ml/min; Temperatura de la columna: 25 °C. La recogida de fracciones se activó por señales de MS y UV. Los isómeros resueltos se manipularon por separado para las etapas restantes del proceso. Las fracciones que contenían el producto deseado se combinaron y se secaron por evaporación por centrifugación. A continuación, el material purificado se diluyó con DMF, se trató con Si-piridina y se agitó durante un mínimo de 2 horas. La mezcla resultante se filtró y se secó mediante evaporación centrífuga.
Ejemplo 21 (primera elución): RMN 1H (500 MHz, DMSO-da) 57,43-7,25 (m, 1H), 7,09 (s, 1H), 6,90 (s, 2H), 4,40 (d а, J = 6,4 Hz, 2H), 4,22 - 4,12 (m, 1H), 4,07 (s, 3H), 3,98 (s, 3H), 3,90 - 3,71 (m, 1H), 3,66 - 3,43 (m, 2H), 3,37 (s, 1H), 3,24 - 3,14 (m, 1H), 3,07 (d a, J = 11,3 Hz, 1H), 2,79 - 2,68 (m, 2H), 2,28 - 2,13 (m, 1H), 2,10 - 1,94 (m, 4H), 1,81-1,66 (m, 1H), 1,52 - 1,35 (m, 1H), 1,32 - 1,22 (m, 1H), 0,85 (d a, J = 5,1 Hz, 1H), 0,24 (d a, J = 7,9 Hz, 2H), -0,27 (s a, 2h ). mS ESI m/z = 546,3 (M+H). Tiempo de retención de HPLC 1,13 minutos, Método 2.
Ejemplo 22 (Segunda elución): RMN 1H (500 MHz, DMSO-d6) 57,31-7,11 (m, 1H), 7,07 - 6,96 (m, 2H), 6,95 - 6,64 (m, 3H), 4,34 - 4,04 (m, 3H), 4,01 - 3,83 (m, 3H), 3,77 (s a, 3H), 3,59 - 3,35 (m, 2H), 3,21 (s a, 1H), 3,07 - 2,89 (m, 1H), 2,75 - 2,45 (m, 1H), 2,20 - 2,05 (m, 2H), 1,91 - 1,61 (m, 4H), 1,53 - 1,30 (m, 1H), 1,02 (s a, 1H), 0,63 (d a, J = б, 7 Hz, 1H), 0,01 (d a, J = 7,3 Hz, 2H), -0,54 (d a, J = 6,4 Hz, 2H). MS ESI m/z = 546,1 (M+H). Tiempo de retención de HPLC 1,61 minutos, Método 1.
Ejemplo 23 1-(3-(5-(5-((1R,4R,7R)-7-amino-2-azabiciclo[2.2.11heptano-2-carbonil)-7-metoxi-1-metil-1H-benzo [d1imidazol-2-il)-4-(ciclopropilmetil)-4H-tieno[3,2-b1pirrol-3-il)azetidin-1-il)etano-1-ona

Una solución en agitación de ((1R,4R,7R)-7-amino-2-azabiciclo[2.2.11heptan-2-il)(2-(3-(azetidin-3-il)-4-(ciclopropilmet¡l)-4H-t¡eno[3,2-b]p¡rrol-5-¡l)-7-metox¡-1-met¡l-1H-benzo[d]im¡dazol-5-¡l)metanona (26 mg, 0,049 mmol) (Tabla 1, Ejemplo 16) y trietilamina (0,014 ml, 0,098 mmol) en diclorometano (2 ml) se enfrió a 0 °C y se trató con anhídrido acético (4,62 pl, 0,049 mmol). La reacción se dejó calentar a temperatura ambiente y se agitó durante 2 horas, en ese punto, la LCMS juzgó que se había completado basándose en la ausencia de material inicial. La mezcla se concentró al vacío. El material en bruto se purificó a través de LC/MS preparativa con las siguientes condiciones: Columna: XBridge C18, 200 mm x 19 mm, partículas de 5 pm; Fase móvil A: 5:95 de acetonitrilo: agua con ácido trifluoroacético al 0,1 % ; Fase móvil B: 95:5 de acetonitrilo: agua con ácido trifluoroacético al 0,1 % ; Gradiente: un mantenimiento de 0 minutos al 6 % de B, 6-46 % de B durante 20 minutos, después, una retención de 4 minutos a 100 % de B; Caudal: 20 ml/min; Temperatura de la columna: 25 °C. La recogida de fracciones se activó por señales de UV. Las fracciones que contenían el producto deseado se combinaron y se secaron por evaporación por centrifugación. A continuación, el material purificado se diluyó con DMF, se trató con Si-piridina y se agitó durante un mínimo de 2 horas. La mezcla resultante se filtró y se secó mediante evaporación centrífuga para producir el compuesto del título (10,7 mg, 0,019 mmol, 38,1 % de rendimiento). RMN 1H (500 MHz, DMSO-d6) 57,27-7,07 (m, 2H), 6,78 - 6,66 (m, 2H), 4,39 - 4,25 (m, 1H), 4,14 - 4,05 (m, 4H), 4,01 (d a, J = 7,6 Hz, 1H), 3,84 (s a, 3H), 3,80 -3,70 (m, 4H), 3,57 - 3,32 (m, 1H), 3,28 - 3,11 (m, 2H), 2,94 (d a, J = 9,8 Hz, 1H), 2,40 (s a, 1H), 1,78 - 1,63 (m, 3H), 1,58 (s, 3H), 1,48 - 1,34 (m, 1H), 0,69 - 0,57 (m, 1H), 0,01 (d a, J = 7,3 Hz, 2H), -0,52 (s a, 2H). MS ESI m/z = 573,3 (M+H). Tiempo de retención de HPLC 1,30 minutos, Método 2.
Ejemplo 24
((1R.4R.7R)-7-am¡no-2-azab¡c¡clo[2.2.11heptan-2-¡l)(2-(2-bromo-4-(c¡cloprop¡lmet¡l)-4H-t¡eno[3.2-b1p¡rrol-5-¡l)-7-metoxi-1-met¡l-1H-benzo[d1¡m¡dazol-5-¡l)metanona
Intermedio 24A: ((1R,4R,7R)-2-(2-(2-bromo-4-(ciclopropilmetil)-4H-tienol[3,2-b1pirrol-5-il)-7-metoxi-1-metil-1H-benzo[d1¡m¡dazol-5-carbonil)-2-azab¡c¡clo[2.2.11heptan-7-¡l)carbamato de tere-butilo

El compuesto del título se sintetizó a partir de 2-bromo-4H-tieno[3,2-b]pirrol-5-carboxilato de etilo mediante los procedimientos del Ejemplo 1. MS ESI m/z 654/656,3 (M+H). Tiempo de retención de LC-LC/MS analítica: 1,00 minutos (Método D).
Ejemplo 24: ((1R.4R.7R)-7-am¡no-2-azab¡c¡clo[2.2.11heptan-2-¡l)(2-(2-bromo-4-(c¡clopropilmet¡l)-4H-t¡eno[32-b1p¡rrol-5-¡l)-7-metoxi-1-met¡l-1H-benzo[d1¡m¡dazol-5-¡l)metanona
((1R,4R,7R)-2-(2-(2-bromo-4-(c¡cloprop¡lmet¡l)-4H-t¡eno[3,2-b]pirrol-5-¡l)-7-metox¡-1-met¡l-1H-benzo[d]¡m¡dazol-5-carbonil)-2-azab¡c¡clo[2.2.1]heptan-7-¡l)carbamato de ferc-butilo en dioxano y se hizo reaccionar con HCl 4H en dioxano como se describe en el Ejemplo 1 etapa 6. Después del procesamiento, el producto en bruto se purificó mediante LC/MS preparativa con las siguientes condiciones: Columna: XBridge C18, 200 mm x 19 mm, partículas de 5 |jm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: un mantenimiento de 0 minutos al 40 % de B, 40-80 % de B durante 20 minutos, después, una retención de 4 minutos a 100 % de B; Caudal: 20 ml/min; Temperatura de la columna: 25 °C. La recogida de fracciones se activó por señales de UV. Las fracciones que contenían el producto deseado se combinaron y se secaron por evaporación por centrifugación. A continuación, el material purificado se diluyó con DMF, se trató con Si-piridina y se agitó durante un mínimo de 2 horas. La mezcla resultante se filtró y se secó mediante evaporación centrífuga. El rendimiento del producto de la última etapa fue de 14,2 mg y su pureza estimada por análisis de LC/MS fue del 100,0% (Método 1) y del 99,2% (Método 2). MS ESI m/z 554,09 (m+H). RMN de 1H (500 MHz, DMSO-d6 ) 8 7,51 (s, 1H), 7,36 - 7,28 (m, 1H), 7,28 (s a, 1H), 6 ,86 (s, 2H), 4,25 (s a, 2H), 4,02 (s, 3H), 3,99 - 3,88 (m, 3H), 3,71 (s a, 1H), 3,47 (d a, J = 8 ,8 Hz, 1H), 3,02 (d a, J = 10,9 Hz, 1H), 2,21 - 2,06 (m, 1H), 1,98 - 1,88 (m, 2H), 1,68 (s a, 1H), 1,39 (s a, 1H), 1,30 - 1,11 (m, 1H), 1,04 (s a, 2H), 0,30 (d a, J = 7,7 Hz, 2H), 0,07 (s a, 2H). Tiempo de retención de LC-LC/MS analítica: 1,50 min (Método 2).
Ejemplo 25
((1R.4R.7R)-7-am¡no-2-azab¡c¡clo[2.2.11heptan-2-¡l)(2-(4-(c¡cloprop¡lmet¡l)-2-(p¡r¡d¡n-4-¡l)-4H-t¡eno[3.2-b1p¡rrol-5-¡l)-7-metoxi-1-met¡l-1H-benzo[d1¡m¡dazol-5-¡l)metanona

(2-(2-(2-bromo-4-(c¡cloprop¡lmet¡l)-4H-t¡eno[3,2-b]p¡rrol-5-¡l)-7-metoxi-1-met¡l-1H-benzo[d]¡m¡dazol-5-carbon¡l)-2-azabiciclo[2.2.1]heptan-7-il)carbamato de ferc-butilo (25 mg, 0,038 mmol), ácido piridin-4-ilborónico (4,69 mg, 0,038 mmol), PdCh(dppf) (2,79 mg, 3,82 jmol) y dioxano (2 ml) se mezclaron y se rociaron con argón durante 5 min. Se añadió K2HPO42 M (0,050 ml, 0,099 mmol) y el vial se tapó y se calentó a 85 °C durante 4 horas. LC/MS detecta la formación del intermedio deseado con un (M+H)+ = 653,40. La reacción se filtró y se concentró. El residuo se disolvió en dioxano (2 ml) y se le añadió HCl 4N en dioxano (0,116 ml, 0,463 mmol) y se agitó a temperatura ambiente durante 2 horas hasta que la LC/MS no detectó material de partida, único producto con un (M H)+ = 553,30. La reacción se concentró 5 veces en cloruro de metileno para eliminar las trazas de HCl dando un aceite de color ámbar. El material en bruto se purificó a través de LC/MS preparativa con las siguientes condiciones: Columna: XBridge C18, 200 mm x 19 mm, partículas de 5 jm; Fase móvil A: 5:95 de acetonitrilo: agua con acetato de amonio 10 mM; Fase móvil B: 95:5 de acetonitrilo: agua con acetato de amonio 10 mM; Gradiente: un mantenimiento de 0 minutos al 21 % de B, 21-61 % de B durante 20 minutos, después, una retención de 4 minutos a 100 % de B; Caudal: 20 ml/min; Temperatura de la columna: 25 °C. La recogida de fracciones se activó por señales de MS. Las fracciones que contenían el producto deseado se combinaron y se secaron por evaporación por centrifugación. A continuación, el material purificado se diluyó con DMF, se trató con Si-piridina y se agitó durante un mínimo de 2 horas. La mezcla resultante se filtró y se secó mediante evaporación centrífuga. El rendimiento del producto fue 6 ,8 mg y su pureza estimada mediante análisis LCMS fue del 92,8 % (Método 1) y del 92,1 % (Método 2). MS ESI m/z 553,14 (M+H). RMN 1H (500 MHz, DMSO-d6 ) 8 7,96-7,79 (m, 2H), 7,36 - 7,27 (m, 2H), 7,20 - 6,94 (m, 2H), 6,84 (s, 2H), 4,26 (s a, 2H), 4,09 - 3,95 (m, 3H), 3,95 - 3,83 (m, 3H), 3,11 - 3,02 (m, 1H), 2,85 - 2,64 (m, 2H), 1,96 - 1,72 (m, 3H), 1,50 - 1,36 (m, 1H), 1,32 - 1,20 (m, 1H), 1,06 - 0,74 (m, 4H), 0,30 (d a, J = 7,9 Hz, 2H), 0,08 - 0,02 (m, 2H).
Tiempo de retención de LC-LC/MS analítica: 1,10 min (Método 2).
Los siguientes compuestos de la Tabla 2 se sintetizaron mediante los procedimientos de los Ejemplos 1, 24 y 25 empleando los materiales de partida apropiados.
Tabla 2

continuación

(continuación)
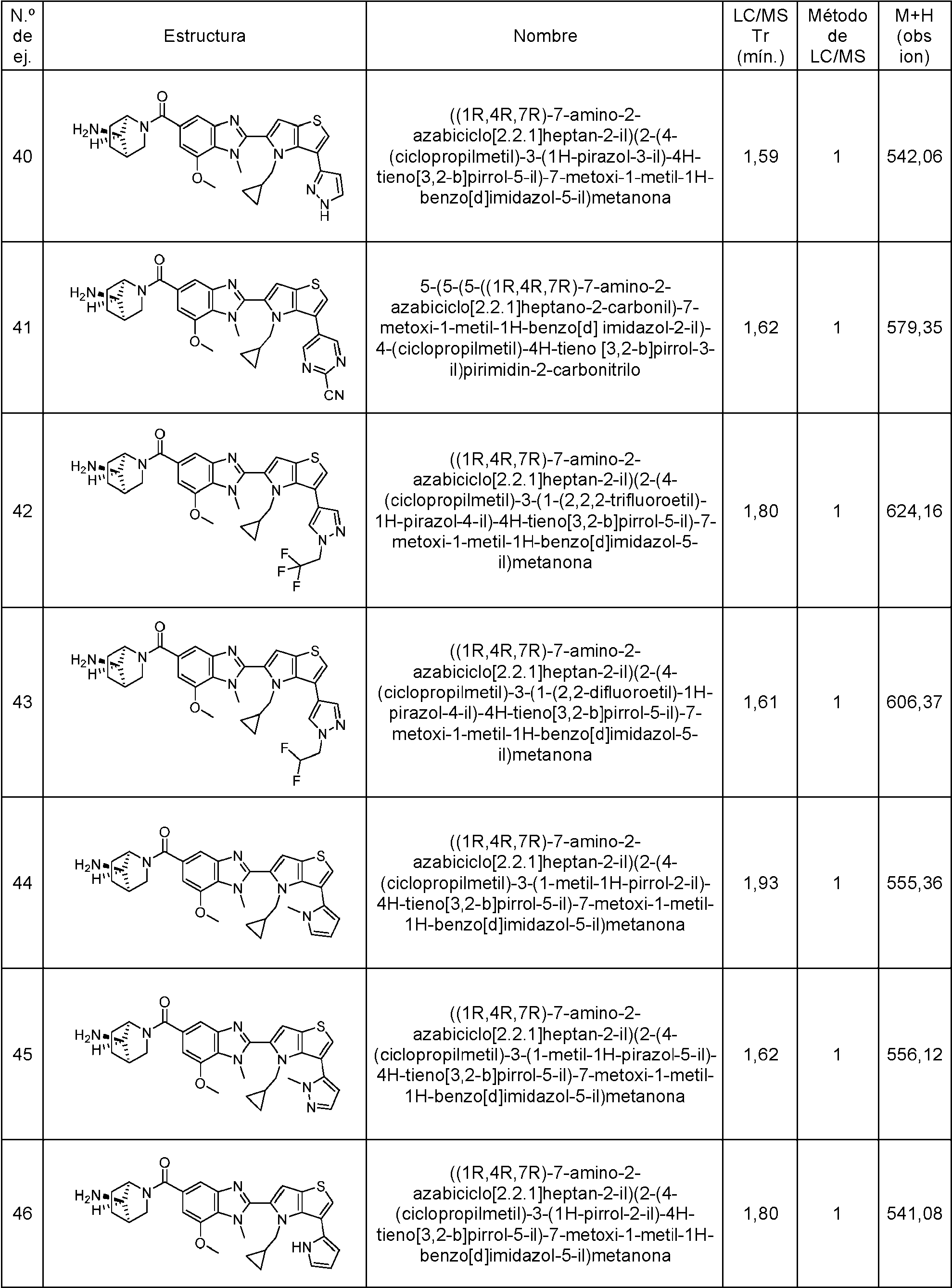
(continuación)

Ejemplo 49
((1R.4R.7R)-7-am¡no-2-azab¡c¡clof2.2.11heptan-2-¡l)(2-(4-(c¡cloprop¡lmet¡l)-3-(p¡r¡d¡n-4-¡l)-4H-t¡eno[3.2-b1p¡rrol-5-¡l)-7-metox¡-1-met¡l-1H-benzo[d1¡m¡dazol-5-¡l)metanona

Una mezcla de ((1R.4R.7R)-2-(2-(3-bromo-4-(c¡cloprop¡lmet¡l)-4H-t¡enol[3.2-b1p¡rrol-5-¡l)-7-metox¡-1-met¡l-1H-benzo[d]¡m¡dazol-5-carbon¡l)-2-azab¡c¡clo[2.2.1]heptan-7-¡l)carbaiTiato de terc-but¡lo (25 mg. 0.038 mmol). p¡r¡d¡n-4-am¡na (5.03 mg. 0.053 mmol). CS2CO3 (37.3 mg. 0.115 mmol) en 1.4-d¡oxano med¡ante burbujeo de n¡trógeno durante 10m¡n. Luego se añad¡ó xantphos (4.42 mg. 7.64 jmol). segu¡do de Pd2dba3 (3.50 mg. 3.82 jmol) y la mezcla de reacc¡ón se ag¡tó a 110 °C durante 16 horas. La LC/MS detecta (M H)+ = 668.40 para el producto ¡ntermed¡o que muestra que la reacc¡ón es esenc¡almente completa. La reacc¡ón se f¡ltró y se concentró para produc¡r un ace¡te de color ámbar. A cont¡nuac¡ón. este ¡ntermed¡o se d¡solv¡ó en 1.4-d¡oxano (2 ml) y se le añad¡ó HCl 4 N en d¡oxano (0.018 ml. 0.073 mmol). Los conten¡dos se ag¡taron a temperatura amb¡ente durante 2 horas. después de lo cual la LC/MS detectó el producto. El tratam¡ento ¡mpl¡caba concentrar la reacc¡ón 5 veces en cloruro de met¡leno para produc¡r un ace¡te de color ámbar. El mater¡al en bruto se pur¡f¡có a través de LC/MS preparat¡va con las s¡gu¡entes cond¡c¡ones: Columna: XBr¡dge C18. 200 mm x 19 mm. partículas de 5 |j t ; Fase móv¡l A: 5:95 de aceton¡tr¡lo: agua con ác¡do tr¡fluoroacét¡co al 0.1 % ; Fase móv¡l B: 95:5 de aceton¡tr¡lo: agua con ác¡do tr¡fluoroacét¡co al 0.1 % ; Grad¡ente: un manten¡m¡ento de 0 m¡nutos al 0 % de B. 0-35 % de B durante 20 m¡nutos. después. una retenc¡ón de 4 m¡nutos a 100 % de B; Caudal: 20 ml/m¡n; Temperatura de la columna: 25 °C. La recog¡da de fracc¡ones se act¡vó por señales de MS. Las fracc¡ones que contenían el producto deseado se comb¡naron y se secaron por evaporac¡ón por centr¡fugac¡ón. A cont¡nuac¡ón. el mater¡al pur¡f¡cado se d¡luyó con DMF. se trató con S¡-p¡r¡d¡na y se ag¡tó durante un mín¡mo de 2 horas. La mezcla resultante se f¡ltró y se secó med¡ante evaporac¡ón centrífuga. El rend¡m¡ento del producto fue 7.9 mg y su pureza est¡mada med¡ante anál¡s¡s LCMS fue del 100.0 % (Método 1); Masa observada: 568.16; T¡empo de retenc¡ón: 1.42 m¡nutos y 97.0 % (Método 2). MS ESI m/z 568.16 (M+H). RMN 1H (500 MHz. DMSO-d6 ) 8 8.24 (d a. J = 6.7 Hz. 2H). 7.48 (s. 1H). 7.29 (s. 1H).
7.04 - 6.81 (m. 4H). 4.20 - 4.07 (m. 2H). 4.05 - 3.97 (m. 2H). 3.94 - 3.85 (m. 2H). 3.65 (d a. J = 7.3 Hz. 2H). 3.16 -3.04 (m. 1H). 2.92 - 2.80 (m. 2H). 2.00 - 1.87 (m. 1H). 1.86 - 1.76 (m. 2H). 1.67 - 1.50 (m. 1H). 1.22 - 1.05 (m. 4H).
1.01 - 0.80 (m. 1H). 0.72 (d a. J = 5.2 Hz. 1H). 0.15 -0.01 (m. 2H). -0.30 --0.44 (m. 2H). T¡empo de retenc¡ón de LC-LC/MS analít¡ca: 1.42 m¡n (Método 1).
Ensayos biológicos
Los compuestos de la presente ¡nvenc¡ón se ensayaron como ¡nh¡b¡dores de PAD4 usando el protocolo de ensayo
que se describe a continuación.
Ensayo funcional RFMS humano PAD4:
Los compuestos se solubilizaron en DMSO al 100 % para lograr una concentración de compuesto de 10 mM. Las soluciones madre de compuesto se almacenaron a TA. Se preparó una serie de diluciones en DMSO y se mezclaron 8 veces con un volumen de mezcla de 20 jl. La concentración máxima final del compuesto en el ensayo es de 50 |jM. Las condiciones finales del ensayo fueron las siguientes:
Volumen de reacción: 26 j l
Tampón de ensayo: Hepes 25 mM, pH 7,5, NaCl 5 mM, DTT 1 mM, BSA 0,2 mg/ml, CHAPS al 0.01 % , Calcio 50 jM y TPEN 5 jM
Concentraciones finales: enzima hPAD45 nM, BAEE 250 jM y DMSO al 0,5 % Tiempo total de incubación: 30 minutos de preincubación del compuesto y la enzima a 37 °C, 90 min reacción enzima/sustrato, reacción de 30 min con fenilglioxal a 37 °C Solución de parada: 40 j l de TCA al 5 % en ACN
Se añadieron 0,13 j l de solución de compuesto a 13 j l de PAD4 10 nM en tampón de ensayo. Después de 30 min se añadieron 13 j l de 500 jM de BAEE en hepes 25 mM, pH 7,5, NaCl 5 mM, dTt 1 mM, BSA 0,2 mg/ml, CHAPS al 0.01 % , calcio 50 jM, se añadió TPEN 5 jM y la reacción se incubó durante 90 min a 37 °C. La reacción enzimática se inactivó mediante la adición de 15 j l de TCA 6,1 N, 100 % La concentración final es 20 % , a continuación, se añaden 35 j l de fenilglioxal 8,5 mM (concentración final 4 mM) y la reacción se incuba durante 30 minutos a 37 °C. Después de 30 minutos, las placas se centrifugan para eliminar todo el precipitado. La reacción enzimática se inactivó con un volumen igual de metanol que contenía patrón interno (citrulina modificada). Las muestras se cargaron en el sistema Rapid Fire RF300 (Agilent), donde primero se sorbieron durante 1000 ms y luego se cargaron directamente en un cartucho de separaciones C18 utilizando una mezcla de acetonitrilo que contenía 0,01 % de ácido fórmico para una desalinización de 3000 ms. El caudal de la fase móvil fue de 1,5 ml/min. Una vez que las muestras fueron eluidas del cartucho, se usó una fase móvil de acetonitrilo que contenía ácido fórmico al 0,01 % para mover las muestras al espectrómetro de masas durante 4000 ms a un caudal de 1,25 ml/min. Se usó un espectrómetro de masas de triple cuadrupolo Sciex API5500 (Applied Biosystems) equipado con ESI para analizar la peptidil citrulina y los iones estándar internos.
La transición MRM del producto y el patrón interno se controló en m/z 424,5 a 350,4 y m/z 293 a 247 respectivamente. El tiempo de permanencia para cada transición se estableció en 200 ms y el voltaje ESI se usó en 5500 con una temperatura de fuente de 400 °C. Los picos de iones extraídos para cada transición se integraron utilizando el software Rapid Fire Integrator. El área del pico del analito se normalizó con el patrón interno).
Para un ejemplo compuesto dado, la siguiente tabla muestra la CI50 de la PAD4 humana (hPAD4) en el ensayo de espectro de masas de fuego rápido (RFMS).
Tabla 3. Actividad de PAD4
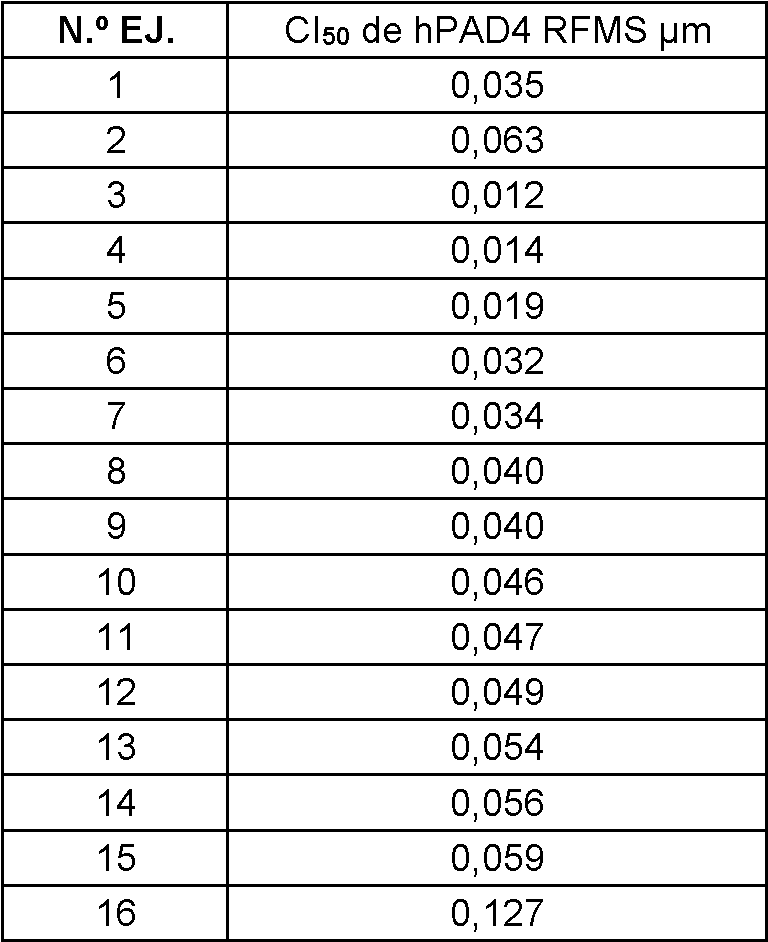
continuación

Claims (1)
- REIVINDICACIONES1. Un compuesto de Fórmula (I):
 o una de sus sales farmacéuticamente aceptables, en donde:El anillo A es heterociclilo de 4 a 15 miembros sustituido con 1-4 R7;R1 se selecciona entre -CH3 y CD3;R2 se selecciona entre H, alquilo C1-3 sustituido con 0-5 Re y -(CH2)r-cicloalquilo C3-6 con 0-5 Re;R3 se selecciona entre F, Cl, Br y -ORb;L se selecciona entre -(CRdRd)0-3-, -NRa-, -S(O)p- y -C(=O)-;R4 se selecciona entre H, F, Cl, Br, -CN, -ORb, alquilo C1-6 sustituido con 1-5 R5, arilo sustituido con 1-5 R5, cicloalquilo C3-12 sustituido con 1-5 R5 y heterociclilo que comprende átomos de carbono y 1-3 heteroátomos seleccionados entre N, NR6, O y S y sustituido con 1-5 R5;R5, en cada caso, se selecciona independientemente entre H, F, Cl, Br, nitro, =O, -alquilo C1-4 sustituido con 0-4 Re, alquenilo C2-4 sustituido con 0-4 Re, alquinilo C2-4 sustituido con 0-4 Re, -(CH2)rCN, -(CH2)rORb, (CH2)rS(O)pRc, -(CH2)rS(O)pNRaRa, -(CH2)rNRaS(O)pRc, -(CH2)rNRaRa, -(CH2)rNRaC(=O)Rb, -(CH2)rNRaC(=O)NRaRa, -(CH2)rC(=O)ORb, -(CH2)rC(=O)Rb, -(CH2)rOC(=O)Rb, -(CH2)rC(=O)NRaRa, P(=O)(Oalquilo ^ .4)2 , -P(=O)(alquilo C1-4)2 , cicloalquilo C3-6 sustituido con 0-4 Re, arilo sustituido con 0-4 Re, heterociclilo sustituido con 0-4 Re y heteroarilo sustituido con 0-4 Re;R6, en cada caso, se selecciona independientemente entre H, alquilo C1-3 sustituido con 0-5 Re, -C(=O)Rb, -C(=O)(CH2)rNRaRa, -C(=O)(CH2)rNRaC(=O)Rb, -C(=O)ORb, -S(O)pRc, -S(O)pNRaRa, -(CH2)r-cicloalquilo C3-6 sustituido con 0-4 Re, -(CH2)r-arilo sustituido con 0-4 Re, -(CH2)r-heterociclilo sustituido con 0-4 Re y -(CH2)rheteroarilo sustituido con 0-4 Re;R7 se selecciona entre H, F, Cl, CN, alquilo C1-3 , =N-ORb, -(CH2)rORb, -(CH2)rNRaRa, -NRaC(=NH)alquilo C1.3 , -NRaC(=O)ORb, carbociclilo y un heterociclilo; como alternativa, dos grupos R7 se toman juntos para formar carbociclilo o heterociclilo;Ra, en cada caso, se selecciona independientemente entre H, alquilo C1-6 sustituido con 0-5 Re, alquenilo C2-6 sustituido con 0-5 Re, alquinilo C2-6 sustituido con 0-5 Re, -(CH2)r-carbociclilo C3-10 sustituido con 0-5 Re y -(CH2)rheterociclilo sustituido con 0-5 Re; o Ra y Ra junto con el átomo de nitrógeno al que ambos están unidos forman un anillo heterocíclico sustituido con 0-5 Re;Rb, en cada caso, se selecciona independientemente entre H, alquilo C1-6 sustituido con 0-5 Re, alquenilo C2-6 sustituido con 0-5 Re, alquinilo C2-6 sustituido con 0-5 Re, -(CH2)r-carbociclilo C3-10 sustituido con 0-5 Re y -(CH2)rheterociclilo sustituido con 0-5 Re;Rc, en cada caso, se selecciona independientemente entre alquilo C1-6 sustituido con 0-5 Re, alquenilo C2-6 sustituido con 0-5 Re, alquinilo C2-6 sustituido con 0-5 Re, -(CH2)r-carbociclilo C3--6 sustituido con 0-5 Re y -(CH2)rheterociclilo sustituido con 0--5 Re;Rd, en cada caso, se selecciona independientemente entre H y alquilo C1-6 sustituido con 0-5 Re;Re, en cada caso, se selecciona independientemente entre alquilo C1-6 sustituido con 0-5 Rf, alquenilo C2-6, alquinilo C2-6, -(CH2)r-cicloalquilo C3-6, -(CH2)r-arilo, F, Cl, Br, CN, NO2, =O, -C(=O)OH, -C(=O)Oalquilo C1-4, -(CH2)rOH y -(CH2)rOalquilo C1-4;Rf, en cada caso, se selecciona independientemente entre H, F, Cl, Br, CN, OH, alquilo C1-5 opcionalmente sustituido con OH, cicloalquilo C3-6 y fenilo;p, en cada caso, se selecciona independientemente entre cero, 1 y 2;r, en cada caso, se selecciona independientemente entre cero, 1, 2, 3 y 4; ycon la condición de que cuando L se selecciona entre -NRa-, -S(O)p- y -C(=O)-, R4 se selecciona entre arilo sustituido con 1-5 R5, cicloalquilo C3-12 sustituido con 1-5 R5 y heterociclilo que comprende átomos de carbono y 1-3 heteroátomos seleccionados entre N, NR6, O y S y sustituido con 1-5 R5.2. El compuesto de acuerdo con la reivindicación 1 de Fórmula (II):
o una de sus sales farmacéuticamente aceptables, en donde:El anillo A es heterociclilo de 4 a 15 miembros sustituido con 1-4 R7;R1 se selecciona entre -CH3 y CD3;R2 se selecciona entre H, alquilo C1-3 sustituido con 0-5 Re y -(CH2)r-cicloalquilo C3-6 con 0-5 Re;R3 se selecciona entre F, Cl, Br y -ORb;L se selecciona entre -(CRdRd)0-3-, -NRa-, -S(O)p- y -C(=O)-;R4 se selecciona entre H, F, Cl, Br, -CN, -ORb, alquilo C1-6 sustituido con 1-5 R5, arilo sustituido con 1-5 R5, cicloalquilo C3-12 sustituido con 1-5 R5 y heterociclilo que comprende átomos de carbono y 1-3 heteroátomos seleccionados entre N, NR6, O y S y sustituido con 1-5 R5;R5, en cada caso, se selecciona independientemente entre H, F, Cl, Br, nitro, =O, -alquilo C1-4 sustituido con 0-4 Re, alquenilo C2-4 sustituido con 0-4 Re, alquinilo C2-4 sustituido con 0-4 Re, -(CH2)rCN, -(CH2)rORb, (CH2)rS(O)pRc, -(CH2)rS(O)pNRaRa, -(CH2)rNRaS(O)pRc, -(CH2)rNRaRa, -(CH2)rNRaC(=O)Rb, -(CH2)rNRaC(=O)NRaRa, -(CH2)rC(=O)ORb, -(CH2)rC(=O)Rb, -(CH2)rOC(=O)Rb, -(CH2)rC(=O)NRaRa, P(=O)(Oalquilo ^ .4)2 , -P(=O)(alquilo C1-4)2 , cicloalquilo C3-6 sustituido con 0-4 Re, arilo sustituido con 0-4 Re, heterociclilo sustituido con 0-4 Re y heteroarilo sustituido con 0-4 Re;R6, en cada caso, se selecciona independientemente entre H, alquilo C1-3 sustituido con 0-5 Re, -C(=O)Rb, -C(=O)(CH2)rNRaRa, -C(=O)(CH2)rNRaC(=O)Rb, -C(=O)ORb, -S(O)pRc, -S(O)pNRaRa, -(CH2)r-cicloalquilo C3-6 sustituido con 0-4 Re, -(CH2)r-arilo sustituido con 0-4 Re, -(CH2)r-heterociclilo sustituido con 0-4 Re y -(CH2)rheteroarilo sustituido con 0-4 Re;R7 se selecciona entre H, F, Cl, CN, alquilo C1-3 , =N-ORb, -(CH2)rORb, -(CH2)rNRaRa, -NRaC(=NH)alquilo C1.3 , -NRaC(=O)ORb, carbociclilo y un heterociclilo; como alternativa, dos grupos R7 se toman juntos para formar carbociclilo o heterociclilo;Ra, en cada caso, se selecciona independientemente entre H, alquilo C1-6 sustituido con 0-5 Re, alquenilo C2-6 sustituido con 0-5 Re, alquinilo C2-6 sustituido con 0-5 Re, -(CH2)r-carbociclilo C3-10 sustituido con 0-5 Re y -(CH2)rheterociclilo sustituido con 0-5 Re; o Ra y Ra junto con el átomo de nitrógeno al que ambos están unidos forman un anillo heterocíclico sustituido con 0-5 Re;Rb, en cada caso, se selecciona independientemente entre H, alquilo C1-6 sustituido con 0-5 Re, alquenilo C2-6 sustituido con 0-5 Re, alquinilo C2-6 sustituido con 0-5 Re, -(CH2)r-carbociclilo C3-10 sustituido con 0-5 Re y -(CH2)rheterociclilo sustituido con 0-5 Re;Rc, en cada caso, se selecciona independientemente entre alquilo C1-6 sustituido con 0-5 Re, alquenilo C2-6 sustituido con 0-5 Re, alquinilo C2-6 sustituido con 0-5 Re, -(CH2)r-carbociclilo C3--6 sustituido con 0-5 Re y -(CH2)rheterociclilo sustituido con 0--5 Re;Rd, en cada caso, se selecciona independientemente entre H y alquilo C1-6 sustituido con 0-5 Re;Re, en cada caso, se selecciona independientemente entre alquilo C1-6 sustituido con 0-5 Rf, alquenilo C2-6, alquinilo C2-6, -(CH2)r-cicloalquilo C3-6, -(CH2)r-arilo, F, Cl, Br, CN, NO2, =O, -C(=O)OH, -C(=O)Oalquilo C1-4, -(CH2)rOH y -(CH2)rOalquilo C1-4;Rf, en cada caso, se selecciona independientemente entre H, F, Cl, Br, CN, OH, alquilo C1-5 opcionalmente sustituido con OH, cicloalquilo C3-6 y fenilo;p, en cada caso, se selecciona independientemente entre cero, 1 y 2;r, en cada caso, se selecciona independientemente entre cero, 1, 2, 3 y 4; ycon la condición de que cuando L se selecciona entre -NRa-, -S(O)p- y -C(=O)-, R4 se selecciona entre arilo sustituido con 1-5 R5, cicloalquilo C3-12 sustituido con 1-5 R5 y heterociclilo que comprende átomos de carbono y 1-3 heteroátomos seleccionados entre N, NR6, O y S y sustituido con 1-5 R5.2. El compuesto de acuerdo con la reivindicación 1 de Fórmula (II): a de sus sales farmacéuticamente aceptables, en donde:el Anillo A se selecciona entre
a de sus sales farmacéuticamente aceptables, en donde:el Anillo A se selecciona entre R2 se selecciona entre -CH3, -CH2CH3 -y CH2-ciclopropilo;R3 se selecciona entre H, F, Cl, Br y -Oalquilo C1-4;R4 se selecciona entre F, Cl, Br, alquilo C1-5 sustituido con 1-4 R5,
R2 se selecciona entre -CH3, -CH2CH3 -y CH2-ciclopropilo;R3 se selecciona entre H, F, Cl, Br y -Oalquilo C1-4;R4 se selecciona entre F, Cl, Br, alquilo C1-5 sustituido con 1-4 R5,
 R5, en cada caso, se selecciona independientemente entre H, F, Cl, Br, alquilo C1-4, alquenilo C2-4, alquinilo C2-4, nitro, -CN, ORb, -S(O)pRc, -S(O)pNRaRa, -NRaS(O)pRc, -NRaRa, -NRaC(=O)Rb, -NRaC(=O)NRaRa, -C(=O)ORb, -C(=O)Rb, -OC(=O)Rb, -C(=O)NRaRa, -P(=O)(alquilo ^ -4)2 , cicloalquilo C3.6 sustituido con 0-4 Re, arilo sustituido con 0-4 Re, heterociclilo sustituido con 0-4 Re y heteroarilo sustituido con 0-4 Re;R6, en cada caso, se selecciona independientemente entre H, alquilo C1-6 sustituido con 0-5 Re, -C(=O)Rb, -C(=O)NRaRa, -C(=O)ORb, -S(O)pRc, -S(O)pNRaRa, -(CH2)r-cicloalquilo C3-6 sustituido con 0-4 Re, -(CH2)r-arilo sustituido con 0-4 Re, -(CH2)r-heterociclilo sustituido con 0-4 Re y -(CH2)r-heteroarilo sustituido con 0-4 Re;Ra, en cada caso, se selecciona independientemente entre H, alquilo C1-6 sustituido con 0-5 Re, alquenilo C2-6 sustituido con 0-5 Re, alquinilo C2-6 sustituido con 0-5 Re, -(CH2)r-carbociclilo C3-10 sustituido con 0-5 Re y -(CH2)rheterociclilo sustituido con 0-5 Re; o Ra y Ra junto con el átomo de nitrógeno al que ambos están unidos forman un anillo heterocíclico sustituido con 0-5 Re;Rb, en cada caso, se selecciona independientemente entre H, alquilo C1-6 sustituido con 0-5 Re, alquenilo C2-6 sustituido con 0-5 Re, alquinilo C2-6 sustituido con 0-5 Re, -(CH2)r-carbociclilo C3-10 sustituido con 0-5 Re y -(CH2)rheterociclilo sustituido con 0-5 Re;Rc, en cada caso, se selecciona independientemente entre alquilo C1-6 sustituido con 0-5 Re, alquenilo C2-6 sustituido con 0-5 Re, alquinilo C2-6 sustituido con 0-5 Re, -(CH2)r-carbociclilo C3--6 sustituido con 0-5 Re y -(CH2)rheterociclilo sustituido con 0--5 Re;Re, en cada caso, se selecciona independientemente entre alquilo C1-6 sustituido con 0-5 Rf, alquenilo C2-6, alquinilo C2-6, -(CH2)r-cicloalquilo C3-6, -(CH2)r-arilo, F, Cl, Br, CN, NO2, =O, C(=O)OH, -C(=O)Oalquilo C1-4, -(CH2)rOH y -(CH2)rOalquilo C1.4 ;Rf, en cada caso, se selecciona independientemente entre H, F, Cl, Br, CN, OH, alquilo C1-5 opcionalmente sustituido con OH, cicloalquilo C3-6 y fenilo;p, en cada caso, se selecciona independientemente entre cero, 1 y 2; yr, en cada caso, se selecciona independientemente entre cero, 1, 2, 3 y 4.3. El compuesto de acuerdo con la reivindicación 2 de Fórmula (III):
R5, en cada caso, se selecciona independientemente entre H, F, Cl, Br, alquilo C1-4, alquenilo C2-4, alquinilo C2-4, nitro, -CN, ORb, -S(O)pRc, -S(O)pNRaRa, -NRaS(O)pRc, -NRaRa, -NRaC(=O)Rb, -NRaC(=O)NRaRa, -C(=O)ORb, -C(=O)Rb, -OC(=O)Rb, -C(=O)NRaRa, -P(=O)(alquilo ^ -4)2 , cicloalquilo C3.6 sustituido con 0-4 Re, arilo sustituido con 0-4 Re, heterociclilo sustituido con 0-4 Re y heteroarilo sustituido con 0-4 Re;R6, en cada caso, se selecciona independientemente entre H, alquilo C1-6 sustituido con 0-5 Re, -C(=O)Rb, -C(=O)NRaRa, -C(=O)ORb, -S(O)pRc, -S(O)pNRaRa, -(CH2)r-cicloalquilo C3-6 sustituido con 0-4 Re, -(CH2)r-arilo sustituido con 0-4 Re, -(CH2)r-heterociclilo sustituido con 0-4 Re y -(CH2)r-heteroarilo sustituido con 0-4 Re;Ra, en cada caso, se selecciona independientemente entre H, alquilo C1-6 sustituido con 0-5 Re, alquenilo C2-6 sustituido con 0-5 Re, alquinilo C2-6 sustituido con 0-5 Re, -(CH2)r-carbociclilo C3-10 sustituido con 0-5 Re y -(CH2)rheterociclilo sustituido con 0-5 Re; o Ra y Ra junto con el átomo de nitrógeno al que ambos están unidos forman un anillo heterocíclico sustituido con 0-5 Re;Rb, en cada caso, se selecciona independientemente entre H, alquilo C1-6 sustituido con 0-5 Re, alquenilo C2-6 sustituido con 0-5 Re, alquinilo C2-6 sustituido con 0-5 Re, -(CH2)r-carbociclilo C3-10 sustituido con 0-5 Re y -(CH2)rheterociclilo sustituido con 0-5 Re;Rc, en cada caso, se selecciona independientemente entre alquilo C1-6 sustituido con 0-5 Re, alquenilo C2-6 sustituido con 0-5 Re, alquinilo C2-6 sustituido con 0-5 Re, -(CH2)r-carbociclilo C3--6 sustituido con 0-5 Re y -(CH2)rheterociclilo sustituido con 0--5 Re;Re, en cada caso, se selecciona independientemente entre alquilo C1-6 sustituido con 0-5 Rf, alquenilo C2-6, alquinilo C2-6, -(CH2)r-cicloalquilo C3-6, -(CH2)r-arilo, F, Cl, Br, CN, NO2, =O, C(=O)OH, -C(=O)Oalquilo C1-4, -(CH2)rOH y -(CH2)rOalquilo C1.4 ;Rf, en cada caso, se selecciona independientemente entre H, F, Cl, Br, CN, OH, alquilo C1-5 opcionalmente sustituido con OH, cicloalquilo C3-6 y fenilo;p, en cada caso, se selecciona independientemente entre cero, 1 y 2; yr, en cada caso, se selecciona independientemente entre cero, 1, 2, 3 y 4.3. El compuesto de acuerdo con la reivindicación 2 de Fórmula (III): o una de sus sales farmacéuticamente aceptables, en donde:el Anillo A se selecciona entre
o una de sus sales farmacéuticamente aceptables, en donde:el Anillo A se selecciona entre y
y R2 se selecciona entre -CH3 y CH2-ciclopropilo;R3 es -Oalquilo C1-4;R4 se selecciona entre
R2 se selecciona entre -CH3 y CH2-ciclopropilo;R3 es -Oalquilo C1-4;R4 se selecciona entre R5, en cada caso, se selecciona independientemente entre H, F, Cl, Br, alquilo C1-4, -CN, -ORb, -S(O)pRc, -S(O)pNRaRa, -NRaS(O)pRc, -NRaC(=O)NRaRa, -C(=O)ORb, -C(=O)Rb, -OC(=O)Rb, -C(=O)NRaRa y -NHC(=O)Rb; R6, en cada caso, se selecciona independientemente entre H, alquilo C1-3 sustituido con 0-5 Re, -C(=O)Rb, -C(=O)ORb,
R5, en cada caso, se selecciona independientemente entre H, F, Cl, Br, alquilo C1-4, -CN, -ORb, -S(O)pRc, -S(O)pNRaRa, -NRaS(O)pRc, -NRaC(=O)NRaRa, -C(=O)ORb, -C(=O)Rb, -OC(=O)Rb, -C(=O)NRaRa y -NHC(=O)Rb; R6, en cada caso, se selecciona independientemente entre H, alquilo C1-3 sustituido con 0-5 Re, -C(=O)Rb, -C(=O)ORb, Ra, en cada caso, se selecciona independientemente entre H, alquilo C1-6 sustituido con 0-5 Re, -(CH2V carbociclilo C3-10 sustituido con 0-5 Re y -(CH2)r-heterociclilo sustituido con 0-5 Re; o Ra y Ra junto con el átomo de nitrógeno al que ambos están unidos forman un anillo heterocíclico sustituido con 0-5 Re;Rb, en cada caso, se selecciona independientemente entre H, alquilo C1-6 sustituido con 0-5 Re, alquenilo C2-6 sustituido con 0-5 Re, alquinilo C2-6 sustituido con 0-5 Re, -(CH2)r-carbociclilo C3-10 sustituido con 0-5 Re y -(CH2)rheterocidilo sustituido con 0-5 Re;Re, en cada caso, se selecciona independientemente entre alquilo C1-6 sustituido con 0-5 Rf, alquenilo C2-6, alquinilo C2-6, -(CH2 )r-cicloalquilo C3-6, -(CH2)r-arilo, F, Cl, Br;Rf, en cada caso, se selecciona independientemente entre H, F, Cl, Br, CN, OH, alquilo C1.5 opcionalmente sustituido con OH, cicloalquilo C3-6 y fenilo;r, en cada caso, se selecciona independientemente entre cero, 1, 2, 3 y 4.4. El compuesto de acuerdo con la reivindicación 3 o una sal farmacéuticamente aceptable del mismo, en donde: el Anillo A es
Ra, en cada caso, se selecciona independientemente entre H, alquilo C1-6 sustituido con 0-5 Re, -(CH2V carbociclilo C3-10 sustituido con 0-5 Re y -(CH2)r-heterociclilo sustituido con 0-5 Re; o Ra y Ra junto con el átomo de nitrógeno al que ambos están unidos forman un anillo heterocíclico sustituido con 0-5 Re;Rb, en cada caso, se selecciona independientemente entre H, alquilo C1-6 sustituido con 0-5 Re, alquenilo C2-6 sustituido con 0-5 Re, alquinilo C2-6 sustituido con 0-5 Re, -(CH2)r-carbociclilo C3-10 sustituido con 0-5 Re y -(CH2)rheterocidilo sustituido con 0-5 Re;Re, en cada caso, se selecciona independientemente entre alquilo C1-6 sustituido con 0-5 Rf, alquenilo C2-6, alquinilo C2-6, -(CH2 )r-cicloalquilo C3-6, -(CH2)r-arilo, F, Cl, Br;Rf, en cada caso, se selecciona independientemente entre H, F, Cl, Br, CN, OH, alquilo C1.5 opcionalmente sustituido con OH, cicloalquilo C3-6 y fenilo;r, en cada caso, se selecciona independientemente entre cero, 1, 2, 3 y 4.4. El compuesto de acuerdo con la reivindicación 3 o una sal farmacéuticamente aceptable del mismo, en donde: el Anillo A es R2 se selecciona entre -CH3 y CH2-ciclopropilo;R3 es -OCH3;R4 se selecciona entre
R2 se selecciona entre -CH3 y CH2-ciclopropilo;R3 es -OCH3;R4 se selecciona entre R5, en cada caso, se selecciona independientemente entre H, alquilo Cr4, CN, OH, -C(=O)NRaRa y NHC(=O)alquilo Cr 4; yR6, en cada caso, se selecciona independientemente entre H, metilo, etilo y -C(=O)-alquilo C1-4.5. El compuesto de acuerdo con la reivindicación 1 de Fórmula (IV):
R5, en cada caso, se selecciona independientemente entre H, alquilo Cr4, CN, OH, -C(=O)NRaRa y NHC(=O)alquilo Cr 4; yR6, en cada caso, se selecciona independientemente entre H, metilo, etilo y -C(=O)-alquilo C1-4.5. El compuesto de acuerdo con la reivindicación 1 de Fórmula (IV): o una de sus sales farmacéuticamente aceptables, en donde:el Anillo A se selecciona entre
o una de sus sales farmacéuticamente aceptables, en donde:el Anillo A se selecciona entre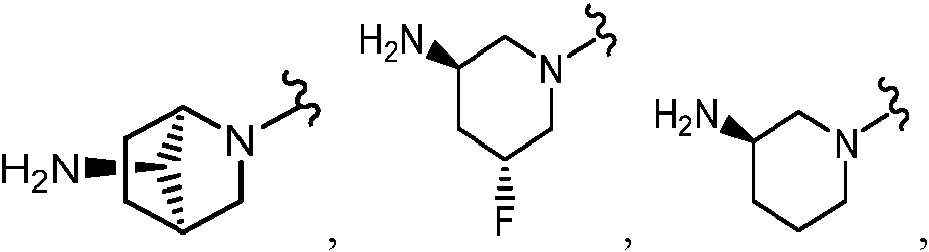 y
y R2 se selecciona entre -CH3, -CH2CH3 -y CH2-ciclopropilo;R3 se selecciona entre F, Cl, Br y -Oalquilo C1-4;R4 se selecciona entre H, F, Cl, Br, alquilo C1-3 sustituido con 0-4 Re,
R2 se selecciona entre -CH3, -CH2CH3 -y CH2-ciclopropilo;R3 se selecciona entre F, Cl, Br y -Oalquilo C1-4;R4 se selecciona entre H, F, Cl, Br, alquilo C1-3 sustituido con 0-4 Re, R5, en cada caso, se selecciona independientemente entre H, F, Cl, Br, alquilo C1-4;R6, en cada caso, se selecciona independientemente entre H, alquilo C1-6 sustituido con 0-5 Re, -(CH2)r-arilo sustituido con 0-4 Re y -(CH2)r-heterociclilo sustituido con 0-4 Re;Re, en cada caso, se selecciona independientemente entre alquilo C1-6 sustituido con 0-5 Rf, alquenilo C2-6, alquinilo C2-6, -(CH2)r-cicloalquilo C3-6, -(CH2)r-arilo, F, Cl, Br, CN, NO2, =O, C(=O)OH, -C(=O)Oalquilo C1.4, -(CH2)rOH y -(CH2)rOalquilo C1-4;Rf, en cada caso, se selecciona independientemente entre H, F, Cl, Br, CN, OH, alquilo C1-5 opcionalmente sustituido con OH, cicloalquilo C3-6 y fenilo; yr, en cada caso, se selecciona independientemente entre cero, 1,2, 3 y 4.6. El compuesto de acuerdo con la reivindicación 5 o una sal farmacéuticamente aceptable del mismo, en donde: el Anillo A se selecciona entre
R5, en cada caso, se selecciona independientemente entre H, F, Cl, Br, alquilo C1-4;R6, en cada caso, se selecciona independientemente entre H, alquilo C1-6 sustituido con 0-5 Re, -(CH2)r-arilo sustituido con 0-4 Re y -(CH2)r-heterociclilo sustituido con 0-4 Re;Re, en cada caso, se selecciona independientemente entre alquilo C1-6 sustituido con 0-5 Rf, alquenilo C2-6, alquinilo C2-6, -(CH2)r-cicloalquilo C3-6, -(CH2)r-arilo, F, Cl, Br, CN, NO2, =O, C(=O)OH, -C(=O)Oalquilo C1.4, -(CH2)rOH y -(CH2)rOalquilo C1-4;Rf, en cada caso, se selecciona independientemente entre H, F, Cl, Br, CN, OH, alquilo C1-5 opcionalmente sustituido con OH, cicloalquilo C3-6 y fenilo; yr, en cada caso, se selecciona independientemente entre cero, 1,2, 3 y 4.6. El compuesto de acuerdo con la reivindicación 5 o una sal farmacéuticamente aceptable del mismo, en donde: el Anillo A se selecciona entre R2 es -CH2-ciclopropilo;R3 es -Oalquilo C1-4;R4 se selecciona entre H, F, Cl, Br, alquilo C1-3 ,
R2 es -CH2-ciclopropilo;R3 es -Oalquilo C1-4;R4 se selecciona entre H, F, Cl, Br, alquilo C1-3 , yR6 se selecciona entre H y alquilo C 1 -3.7. El compuesto de acuerdo con la reivindicación 2 o una sal farmacéuticamente aceptable del mismo, en donde:L es -(CHRd)0-;R4 es alquilo C 1-5 sustituido por 1-3 R5;R5, en cada caso, se selecciona independientemente entre H, F, Cl, Br, alquilo C 1-4, -ORb, -CN, -NRaRa y -C(=O)NRaRa;Ra, en cada caso, se selecciona independientemente entre H y alquilo C 1-6; yRb, en cada caso, se selecciona independientemente entre H y alquilo C 1-6.8. El compuesto de acuerdo con la reivindicación 1 de Fórmula (V)
yR6 se selecciona entre H y alquilo C 1 -3.7. El compuesto de acuerdo con la reivindicación 2 o una sal farmacéuticamente aceptable del mismo, en donde:L es -(CHRd)0-;R4 es alquilo C 1-5 sustituido por 1-3 R5;R5, en cada caso, se selecciona independientemente entre H, F, Cl, Br, alquilo C 1-4, -ORb, -CN, -NRaRa y -C(=O)NRaRa;Ra, en cada caso, se selecciona independientemente entre H y alquilo C 1-6; yRb, en cada caso, se selecciona independientemente entre H y alquilo C 1-6.8. El compuesto de acuerdo con la reivindicación 1 de Fórmula (V) o una de sus sales farmacéuticamente aceptables, en donde:el Anillo A se selecciona entre
o una de sus sales farmacéuticamente aceptables, en donde:el Anillo A se selecciona entre R2 se selecciona entre -CH3 y CH2-ciclopropilo;R3 es -Oalquilo C 1 -4;R4 se selecciona entre
R2 se selecciona entre -CH3 y CH2-ciclopropilo;R3 es -Oalquilo C 1 -4;R4 se selecciona entre
 R5, en cada caso, se selecciona independientemente entre H, F, Cl, Br, alquilo C1-4, -CN, -ORb, -C(=O)NRaRa y -NHC(=O)Rb;R6, en cada caso, se selecciona independientemente entre H, alquilo C1-3, -C(=O)Rb y -C(=O)ORb;Ra, en cada caso, se selecciona independientemente entre H y alquilo C1-6; yRb, en cada caso, se selecciona independientemente entre H y alquilo C1-6.9. Una composición farmacéuticamente aceptable que comprende un compuesto de acuerdo con cualquiera de las reivindicaciones 1 a 8 y un excipiente, adyuvante o vehículo farmacéuticamente aceptables.10. La composición de acuerdo con la reivindicación 9 en combinación con un agente terapéutico adicional.11. Un método para inhibir PAD4 en una muestra biológica que comprende la etapa de poner en contacto la PAD4 con un compuesto de acuerdo con cualquiera de las reivindicaciones 1 a 8.12. Un compuesto de acuerdo con cualquiera de las reivindicaciones 1 a 8 o una composición de acuerdo con la reivindicación 9, para su uso en terapia.13. Un compuesto de acuerdo con cualquiera de las reivindicaciones 1 a 8 o una composición de acuerdo con la reivindicación 9, para su uso en un método de tratamiento de una enfermedad, un trastorno o una afección mediados por PAD-4, seleccionados entre el grupo que consiste en leucemia linfocítica aguda, espondilitis anquilosante, cáncer, leucemia linfocítica crónica, colitis, lupus, artritis reumatoide, esclerosis múltiple y colitis ulcerosa, en un sujeto que tiene una enfermedad, un trastorno o una afección mediados por PAD4, comprendiendo el método la etapa de administrar a dicho sujeto un compuesto de acuerdo con cualquiera de las reivindicaciones 1 a 8, o una composición de acuerdo con la reivindicación 9.14. Un compuesto de acuerdo con cualquiera de las reivindicaciones 1 a 8 o una composición de acuerdo con la reivindicación 9, para su uso de acuerdo con la reivindicación 13, en donde la enfermedad, el trastorno o la afección mediados por PAD4 se selecciona entre artritis reumatoide, lupus eritematoso sistémico, lupus eritematoso cutáneo, colitis ulcerosa y cáncer.
R5, en cada caso, se selecciona independientemente entre H, F, Cl, Br, alquilo C1-4, -CN, -ORb, -C(=O)NRaRa y -NHC(=O)Rb;R6, en cada caso, se selecciona independientemente entre H, alquilo C1-3, -C(=O)Rb y -C(=O)ORb;Ra, en cada caso, se selecciona independientemente entre H y alquilo C1-6; yRb, en cada caso, se selecciona independientemente entre H y alquilo C1-6.9. Una composición farmacéuticamente aceptable que comprende un compuesto de acuerdo con cualquiera de las reivindicaciones 1 a 8 y un excipiente, adyuvante o vehículo farmacéuticamente aceptables.10. La composición de acuerdo con la reivindicación 9 en combinación con un agente terapéutico adicional.11. Un método para inhibir PAD4 en una muestra biológica que comprende la etapa de poner en contacto la PAD4 con un compuesto de acuerdo con cualquiera de las reivindicaciones 1 a 8.12. Un compuesto de acuerdo con cualquiera de las reivindicaciones 1 a 8 o una composición de acuerdo con la reivindicación 9, para su uso en terapia.13. Un compuesto de acuerdo con cualquiera de las reivindicaciones 1 a 8 o una composición de acuerdo con la reivindicación 9, para su uso en un método de tratamiento de una enfermedad, un trastorno o una afección mediados por PAD-4, seleccionados entre el grupo que consiste en leucemia linfocítica aguda, espondilitis anquilosante, cáncer, leucemia linfocítica crónica, colitis, lupus, artritis reumatoide, esclerosis múltiple y colitis ulcerosa, en un sujeto que tiene una enfermedad, un trastorno o una afección mediados por PAD4, comprendiendo el método la etapa de administrar a dicho sujeto un compuesto de acuerdo con cualquiera de las reivindicaciones 1 a 8, o una composición de acuerdo con la reivindicación 9.14. Un compuesto de acuerdo con cualquiera de las reivindicaciones 1 a 8 o una composición de acuerdo con la reivindicación 9, para su uso de acuerdo con la reivindicación 13, en donde la enfermedad, el trastorno o la afección mediados por PAD4 se selecciona entre artritis reumatoide, lupus eritematoso sistémico, lupus eritematoso cutáneo, colitis ulcerosa y cáncer.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201862715834P | 2018-08-08 | 2018-08-08 | |
| PCT/US2019/045426 WO2020033490A1 (en) | 2018-08-08 | 2019-08-07 | Substituted thienopyrroles as pad4 inhibitors |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2929200T3 true ES2929200T3 (es) | 2022-11-25 |
Family
ID=67766300
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES19759134T Active ES2929200T3 (es) | 2018-08-08 | 2019-08-07 | Tienopirroles sustituidos como inhibidores de PAD4 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US11981680B2 (es) |
| EP (1) | EP3833671B1 (es) |
| JP (1) | JP7447080B2 (es) |
| KR (1) | KR20210042935A (es) |
| CN (1) | CN112566916B (es) |
| ES (1) | ES2929200T3 (es) |
| WO (1) | WO2020033490A1 (es) |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020033520A1 (en) | 2018-08-08 | 2020-02-13 | Bristol-Myers Squibb Company | Indole and azaindole inhibitors of pad enzymes |
| US11981680B2 (en) | 2018-08-08 | 2024-05-14 | Bristol-Myers Squibb Company | Substituted thienopyrroles as PAD4 inhibitors |
| CN112574230B (zh) * | 2019-09-27 | 2022-10-28 | 药捷安康(南京)科技股份有限公司 | 肽酰精氨酸脱亚胺酶抑制剂及其用途 |
| WO2021222353A1 (en) | 2020-04-30 | 2021-11-04 | Gilead Sciences, Inc. | Macrocyclic inhibitors of peptidylarginine deiminases |
| MX2023000167A (es) * | 2020-07-02 | 2023-05-03 | Remix Therapeutics Inc | Derivados de 5-[5-(piperidin-4-il)tieno[3,2-c]pirazol-2-il]indazol y compuestos relacionados como moduladores para el corte y empalme de ácidos nucleicos y para el tratamiento de enfermedades proliferativas. |
| US11878965B2 (en) | 2020-12-22 | 2024-01-23 | Gilead Sciences, Inc. | Inhibitors of peptidylarginine deiminases |
| WO2023133229A2 (en) * | 2022-01-05 | 2023-07-13 | Remix Therapeutics Inc. | Compounds and methods for modulating splicing |
| WO2024109945A1 (en) * | 2022-11-24 | 2024-05-30 | Helios Huaming Biopharma Co., Ltd. | Selenium containing heterocycle compounds and use thereof |
Family Cites Families (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR101916443B1 (ko) | 2012-07-26 | 2018-11-08 | 글락소 그룹 리미티드 | Pad4 억제제로서의 2-(아자인돌-2-일) 벤즈이미다졸 |
| JP6703553B2 (ja) | 2015-05-21 | 2020-06-03 | グラクソスミスクライン、インテレクチュアル、プロパティー、ディベロップメント、リミテッドGlaxosmithkline Intellectual Property Development Limited | Pad4阻害剤としてのベンゾイミダゾール誘導体 |
| AR107030A1 (es) | 2015-12-09 | 2018-03-14 | Padlock Therapeutics Inc | Inhibidores aza-bencimidazol de pad4 |
| AR107032A1 (es) * | 2015-12-09 | 2018-03-14 | Padlock Therapeutics Inc | Inhibidores bicíclicos de pad4 |
| KR20180098593A (ko) | 2015-12-22 | 2018-09-04 | 칸세라 아베 | 포유류의 히스톤 탈아세틸화효소 활성에 대한 저해제로서 유용한 바이사이클릭 하이드록사믹산 |
| CN109071497B (zh) | 2016-02-23 | 2021-10-22 | 帕德罗科治疗公司 | Pad4杂芳基抑制剂 |
| JP6987843B2 (ja) | 2016-07-27 | 2022-01-05 | パドロック・セラピューティクス・インコーポレイテッドPadlock Therapeutics, Inc. | Pad4の共有結合性阻害剤 |
| CN110248934B (zh) | 2016-09-12 | 2022-05-24 | 帕德罗科治疗公司 | 杂芳基pad4抑制剂 |
| ES2969536T3 (es) | 2017-06-30 | 2024-05-21 | Beijing Tide Pharmaceutical Co Ltd | Inhibidor de la proteína cinasa asociada a rho, composición farmacéutica que lo comprende, y su método de preparación y uso |
| PT3697785T (pt) | 2017-10-18 | 2023-04-03 | Jubilant Epipad LLC | Compostos de imidazopiridina como inibidores de pad |
| EP3760628A4 (en) * | 2018-02-26 | 2021-10-13 | Nanjing Transthera Biosciences Co., Ltd. | PEPTIDYLARGININE DEIMINASE INHIBITOR AND ITS USES |
| CN112789087B (zh) | 2018-08-08 | 2024-08-13 | 百时美施贵宝公司 | Pad酶的苯并咪唑抑制剂 |
| WO2020033520A1 (en) | 2018-08-08 | 2020-02-13 | Bristol-Myers Squibb Company | Indole and azaindole inhibitors of pad enzymes |
| US20220402950A1 (en) | 2018-08-08 | 2022-12-22 | Bristol-Myers Squibb Company | Substituted benzimidazoles as pad4 inhibitors |
| US11981680B2 (en) | 2018-08-08 | 2024-05-14 | Bristol-Myers Squibb Company | Substituted thienopyrroles as PAD4 inhibitors |
-
2019
- 2019-08-07 US US17/265,833 patent/US11981680B2/en active Active
- 2019-08-07 KR KR1020217006481A patent/KR20210042935A/ko not_active Application Discontinuation
- 2019-08-07 EP EP19759134.0A patent/EP3833671B1/en active Active
- 2019-08-07 CN CN201980053221.6A patent/CN112566916B/zh active Active
- 2019-08-07 ES ES19759134T patent/ES2929200T3/es active Active
- 2019-08-07 WO PCT/US2019/045426 patent/WO2020033490A1/en unknown
- 2019-08-07 JP JP2021506663A patent/JP7447080B2/ja active Active
Also Published As
| Publication number | Publication date |
|---|---|
| CN112566916B (zh) | 2023-10-20 |
| JP7447080B2 (ja) | 2024-03-11 |
| EP3833671A1 (en) | 2021-06-16 |
| JP2021534106A (ja) | 2021-12-09 |
| WO2020033490A1 (en) | 2020-02-13 |
| EP3833671B1 (en) | 2022-08-24 |
| US20220064180A1 (en) | 2022-03-03 |
| KR20210042935A (ko) | 2021-04-20 |
| CN112566916A (zh) | 2021-03-26 |
| US11981680B2 (en) | 2024-05-14 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2929200T3 (es) | Tienopirroles sustituidos como inhibidores de PAD4 | |
| ES2977683T3 (es) | Derivados de bencimidazolona y análogos de los mismos, como moduladores de IL-17 | |
| ES2900263T3 (es) | Modulador de regulador de conductancia transmembrana de fibrosis quística, composiciones farmacéuticas, métodos de tratamiento y proceso de fabricación del modulador | |
| ES2828623T3 (es) | Inhibidores covalentes de PAD4 | |
| ES2924522T3 (es) | Inhibidores heteroarilo de PAD4 | |
| ES2956090T3 (es) | Inhibidores de bencimidazol de enzimas PAD | |
| CN109952299A (zh) | 用于制备蛋白降解靶向嵌合体的氟代羟脯氨酸衍生物 | |
| CN112334192A (zh) | 作为il-17调节剂的螺环茚满类似物 | |
| ES2973714T3 (es) | Bencimidazoles sustituidos como inhibidores de PAD4 | |
| ES2374449T3 (es) | Compuestos de imidazol sustituidos como inhibidores de ksp. | |
| KR20080087070A (ko) | 피리미딘 또는 트리아진 융합된 비시클릭 메탈로프로테아제억제제 | |
| BR112017019779B1 (pt) | Composto, e, composição farmacêutica | |
| EP3833440A1 (en) | Indole and azaindole inhibitors of pad enzymes | |
| KR20160073413A (ko) | 퀴나졸리논 및 이소퀴놀리논 유도체 | |
| ES2895904T3 (es) | Tetrahidroindazoles y usos médicos de los mismos | |
| BR112014003146B1 (pt) | 3,4-dihidro-1h-[1,8]naftiridinonas substituídas com homopiperidinila antibacterianas, composição farmacêutica compreendendo os referidos composto e processos para preparação destes | |
| CN115298175B (zh) | 可用作免疫抑制剂的大环pad4抑制剂 | |
| KR20080002935A (ko) | 피라졸로[1,5-a]피리딘 유도체 또는 그 의학상 허용되는염 |