ES2834318T3 - Formulaciones de agonistas de receptores de la somatostatina - Google Patents
Formulaciones de agonistas de receptores de la somatostatina Download PDFInfo
- Publication number
- ES2834318T3 ES2834318T3 ES13724299T ES13724299T ES2834318T3 ES 2834318 T3 ES2834318 T3 ES 2834318T3 ES 13724299 T ES13724299 T ES 13724299T ES 13724299 T ES13724299 T ES 13724299T ES 2834318 T3 ES2834318 T3 ES 2834318T3
- Authority
- ES
- Spain
- Prior art keywords
- formulation
- pasireotide
- weight
- formulations
- formulation according
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/403—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with carbocyclic rings, e.g. carbazole
- A61K31/404—Indoles, e.g. pindolol
- A61K31/4045—Indole-alkylamines; Amides thereof, e.g. serotonin, melatonin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/04—Peptides having up to 20 amino acids in a fully defined sequence; Derivatives thereof
- A61K38/08—Peptides having 5 to 11 amino acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/04—Peptides having up to 20 amino acids in a fully defined sequence; Derivatives thereof
- A61K38/12—Cyclic peptides, e.g. bacitracins; Polymyxins; Gramicidins S, C; Tyrocidins A, B or C
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- A61K38/22—Hormones
- A61K38/31—Somatostatins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/08—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing oxygen, e.g. ethers, acetals, ketones, quinones, aldehydes, peroxides
- A61K47/10—Alcohols; Phenols; Salts thereof, e.g. glycerol; Polyethylene glycols [PEG]; Poloxamers; PEG/POE alkyl ethers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/08—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing oxygen, e.g. ethers, acetals, ketones, quinones, aldehydes, peroxides
- A61K47/14—Esters of carboxylic acids, e.g. fatty acid monoglycerides, medium-chain triglycerides, parabens or PEG fatty acid esters
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/24—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing atoms other than carbon, hydrogen, oxygen, halogen, nitrogen or sulfur, e.g. cyclomethicone or phospholipids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
- A61K9/0024—Solid, semi-solid or solidifying implants, which are implanted or injected in body tissue
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/10—Dispersions; Emulsions
- A61K9/127—Synthetic bilayered vehicles, e.g. liposomes or liposomes with cholesterol as the only non-phosphatidyl surfactant
- A61K9/1274—Non-vesicle bilayer structures, e.g. liquid crystals, tubules, cubic phases or cochleates; Sponge phases
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/12—Antidiarrhoeals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/18—Drugs for disorders of the alimentary tract or the digestive system for pancreatic disorders, e.g. pancreatic enzymes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/12—Drugs for disorders of the metabolism for electrolyte homeostasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/02—Drugs for disorders of the endocrine system of the hypothalamic hormones, e.g. TRH, GnRH, CRH, GRH, somatostatin
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K7/00—Peptides having 5 to 20 amino acids in a fully defined sequence; Derivatives thereof
- C07K7/64—Cyclic peptides containing only normal peptide links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/16—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing nitrogen, e.g. nitro-, nitroso-, azo-compounds, nitriles, cyanates
- A61K47/18—Amines; Amides; Ureas; Quaternary ammonium compounds; Amino acids; Oligopeptides having up to five amino acids
- A61K47/183—Amino acids, e.g. glycine, EDTA or aspartame
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Engineering & Computer Science (AREA)
- Epidemiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Organic Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Diabetes (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Immunology (AREA)
- Dermatology (AREA)
- Endocrinology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Gastroenterology & Hepatology (AREA)
- Molecular Biology (AREA)
- Biophysics (AREA)
- Neurosurgery (AREA)
- Zoology (AREA)
- Dispersion Chemistry (AREA)
- Biomedical Technology (AREA)
- Rheumatology (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Emergency Medicine (AREA)
- Urology & Nephrology (AREA)
- Pain & Pain Management (AREA)
- Physical Education & Sports Medicine (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Biochemistry (AREA)
- Genetics & Genomics (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Una pre-formulación que comprende una mezcla de baja viscosidad de: a) 20-50% en peso de al menos un diacilglicerol; b) 20-54% en peso de al menos una fosfatidilcolina (PC); c) 0,1-35% en peso de al menos un disolvente monoalcohólico orgánico biocompatible; d) 1 a 20% en peso de disolvente polar; e) 5 a 150 mg/ml de al menos un agonista peptídico del receptor de somatostatina que comprende pasireotida, calculado como base libre; y f) opcionalmente al menos un antioxidante; en la que la relación de los componentes a:b está en el intervalo 40:60 a 54:46; en la que la pre-formulación tiene una viscosidad de 1-1000 mPas a 20ºC; en la que la pre-formulación forma, o es capaz de formar, al menos una estructura de fase cristalina líquida al entrar en contacto con un exceso de fluido acuoso.
Description
DESCRIPCIÓN
Formulaciones de agonistas de receptores de la somatostatina
CAMPO DE LA INVENCIÓN
La presente invención se refiere a precursores de formulación (pre-formulaciones) para la generación in situ de composiciones para la liberación controlada de agentes activos peptídicos, y a métodos de tratamiento con tales formulaciones. En particular, la invención se refiere a pre-formulaciones de alta carga de componentes anfifílicos y al menos un agente activo peptídico que comprende pasireotida para aplicación parenteral, que experimentan una transición de fase tras la exposición a fluidos acuosos, tales como fluidos corporales, formando así una composición de liberación controlada.
ANTECEDENTES DE LA INVENCION
Muchos agentes bioactivos, incluyendo sustancias farmacéuticas, nutrientes, vitaminas, etc., tienen una “ventana funcional”. Es decir, existe un intervalo de concentraciones en las que se puede observar que estos agentes proporcionan algún efecto biológico. Cuando la concentración en la parte apropiada del cuerpo (por ejemplo, localmente o como se demuestra mediante la concentración sérica) cae por debajo de un cierto nivel, no se puede atribuir ningún efecto beneficioso al agente. De manera similar, generalmente existe un nivel de concentración superior por encima del cual no se obtiene ningún beneficio adicional al aumentar la concentración. En algunos casos, el aumento de la concentración por encima de un nivel particular da como resultado efectos indeseables o incluso peligrosos.
Algunos agentes bioactivos tienen una semivida biológica prolongada y/o una ventana funcional amplia, y de este modo pueden administrarse ocasionalmente, manteniendo una concentración biológica funcional durante un período de tiempo sustancial (por ejemplo, 6 horas a varios días). En otros casos, la velocidad de aclaramiento es alta y/o la ventana funcional es estrecha, y de este modo, para mantener una concentración biológica dentro de esta ventana, se requieren dosis regulares (o incluso continuas) de una pequeña cantidad. Esto puede ser particularmente difícil cuando las vías de administración no orales (por ejemplo, administración parenteral) son deseables o necesarias, ya que la autoadministración puede ser difícil y provocar de este modo inconvenientes y/o cumplimiento deficiente. En tales casos, sería ventajoso que una sola administración proporcionara un agente activo a un nivel terapéutico durante todo el período durante el cual se necesita actividad.
Existe un enorme potencial en el uso de péptidos (incluyendo las proteínas) para tratar diversas enfermedades, así como en la profilaxis y en la mejora de la salud y el bienestar general de los sujetos. Sin embargo, el rendimiento de los agentes peptídicos administrados generalmente está limitado debido a la escasa biodisponibilidad, que a su vez es causada por la rápida degradación de péptidos y proteínas en fluidos biológicos. Esto aumenta la dosis que debe administrarse, y en muchos casos restringe las vías de administración eficaces. Estos efectos se exageran aún más por la permeabilidad a menudo limitada de péptidos y proteínas a través de las membranas biológicas.
Los péptidos y proteínas que se administran al cuerpo de los mamíferos (por ejemplo, por vía oral, intramuscular, etc.) están sujetos a degradación por diversas enzimas y sistemas proteolíticos presentes en todo el cuerpo. Los sitios bien conocidos de actividad de peptidasas incluyen el estómago (por ejemplo, pepsina) y el tubo digestivo (por ejemplo, tripsina, quimotripsina, y otros), pero otras peptidasas (por ejemplo, aminopeptidasas, carboxipeptidasas, etc.) se encuentran en todo el cuerpo. Tras la administración oral, la degradación gástrica e intestinal reduce la cantidad de péptido o proteína que potencialmente podría absorberse a través del revestimiento de la superficie intestinal, y de ese modo disminuye su biodisponibilidad. De manera similar, los péptidos y proteínas libres en el torrente sanguíneo de mamíferos también están sujetos a degradación enzimática (por ejemplo, por proteasas plasmáticas, etc.).
Algunos pacientes sometidos a tratamiento requerirán típicamente que se mantenga una dosis terapéutica durante un período considerable y/o un tratamiento continuo durante muchos meses o años. De este modo, un sistema de depósito que permita la carga y liberación controlada de una dosis mayor durante un período más largo ofrecería una ventaja considerable con respecto a sistemas de administración convencionales.
Los péptidos pueden administrarse mediante sistemas tales como el sistema de administración Alkermes Medisorb® que consiste en microesferas de polímeros biodegradables. Tales formulaciones de microesferas poliméricas deben administrarse generalmente por medio de una aguja de tamaño considerable, típicamente de calibre 20 o más ancha. Esto es necesario como resultado de la naturaleza de los sistemas de dosificación poliméricos utilizados, que son típicamente suspensiones poliméricas.
Evidentemente, sería ventajoso proporcionar un sistema de baja viscosidad, tal como una disolución homogénea, dispersión de partículas finas, o fase L2, que podría administrarse fácilmente a través de una aguja estrecha, disminuyendo así las molestias del paciente durante el procedimiento. Esta facilidad de administración es particularmente significativa cuando los pacientes estén en un régimen de autoadministración y ya puedan autoadministrarse varias veces al día. Proporcionar una formulación sostenida con una duración de unos pocos días, pero que sea lo suficientemente compleja de administrar como para requerir tratamiento por parte de un profesional sanitario no será una ventaja para todos los pacientes en comparación con la autoadministración dos veces al día o
diaria, y es probable que sea más costoso. Proporcionar una formulación que tenga una duración suficientemente larga para justificar una visita a un profesional sanitario para su administración, y/o una preparación que pueda ser autoadministrada, y reducir el tiempo de preparación de los profesionales sanitarios o de los pacientes antes de la administración real, son cuestiones importantes.
Los polímeros de polilactato, poliglicolato y polilactato-co-glicolato típicamente utilizados para degradar formulaciones de liberación lenta también son la causa de cierta irritación en al menos algunos pacientes. En particular, estos polímeros contienen típicamente una cierta proporción de impurezas ácidas tales como ácido láctico y glicólico, que irritarán el lugar de la inyección en la administración. Cuando el polímero se descompone, el ácido láctico y el ácido glicólico son los productos de degradación, por lo que se produce una mayor irritación. Como resultado de los efectos combinados de la administración con aguja ancha y el contenido irritante, la incomodidad en el sitio de administración y la formación de tejido cicatricial conjuntivo son mayores de lo deseable.
Desde el punto de vista de la administración de fármacos, las composiciones de depósito de polímero generalmente tienen la desventaja de aceptar solo cargas de fármaco relativamente bajas y de tener un perfil de liberación de “estallido/retraso”. La naturaleza de la matriz polimérica, especialmente cuando se aplica como disolución o prepolímero, provoca un estallido inicial de liberación de fármaco cuando se administra la composición por primera vez. A esto le sigue un período de baja liberación, mientras comienza la degradación de la matriz, seguido finalmente de un aumento en la tasa de liberación hasta el perfil sostenido deseado. Este perfil de liberación de estallido/retraso puede causar que la concentración in vivo del agente activo estalle por encima de la ventana funcional inmediatamente después de la administración, y después caiga nuevamente por la parte inferior de la ventana funcional durante el período de retraso antes de alcanzar una concentración funcional sostenida durante un período de tiempo. Evidentemente, desde un punto de vista funcional y toxicológico, este perfil de liberación de estallido/retraso es indeseable, y podría ser peligroso. También puede limitar la concentración de equilibrio que puede proporcionarse debido al peligro de efectos adversos en el punto “pico”. La presencia de una fase de retraso puede requerir además una dosificación suplementaria con inyecciones repetidas durante el período de inicio del tratamiento de depósito a fin mantener una dosis terapéutica, mientras que las concentraciones de activo proporcionadas desde el depósito son subfuncionales. Para ciertos polipéptidos en particular, sería ventajoso minimizar el efecto de “estallido” inmediato tras la administración de una composición, a fin de evitar efectos secundarios tales como hipoglucemia.
Una clase de hormonas peptídicas que se beneficia particularmente de una concentración in vivo muy estable, de “estallido bajo”, son los análogos de somatostatina tal como Pasireotida (SOM230). Los ensayos in vivo sugieren que estos péptidos son particularmente beneficiosos cuando se mantienen a una concentración plasmática estacionaria, y, como hormona reguladora, es particularmente probable que Pasireotida se beneficie de un nivel plasmático estable. Esto no solo sugiere que una composición de depósito sería una ventaja para evitar “picos” en la concentración tras la administración y/o la dosificación diaria repetida, sino que además dicha composición de depósito debería tener un perfil de liberación lo más plano posible durante el período terapéutico.
Las formulaciones de liberación controlada se generan típicamente a partir de polímeros biocompatibles en forma de, por ejemplo, implantes o perlas inyectables. La formulación líder actual de Pasireotida, por ejemplo (Pasireotida LAR), comprende micropartículas de poli(D,L-lactida-co-glicolida). Las formulaciones de microesferas poliméricas deben administrarse generalmente por medio de una aguja de tamaño considerable, típicamente de calibre 20 o más ancha. Esto es necesario como resultado de la naturaleza de los sistemas de dosificación poliméricos utilizados, que son típicamente suspensiones poliméricas. Sería una ventaja proporcionar un sistema de baja viscosidad, tal como una disolución homogénea, dispersión de partículas finas, o fase L2, que pudiera administrarse fácilmente a través de una aguja estrecha, disminuyendo así las molestias del paciente durante el procedimiento. La facilidad de administración es particularmente importante cuando los pacientes se autoadministren, pero también reduce la carga para los profesionales sanitarios cuando lleven a cabo la administración.
La fabricación de microperlas y suspensiones de PLGA es, además, una dificultad considerable con ciertos sistemas de depósito existentes. En particular, dado que las perlas son particuladas, generalmente no pueden filtrarse de forma estéril, y además, dado que el copolímero de PLGA se funde a temperatura elevada, no pueden tratarse térmicamente para esterilidad. Como resultado, el complejo procedimiento de fabricación debe realizarse de forma aséptica.
Otros problemas con las microesferas de polímero biodegradables incluyen la reconstitución compleja antes de la inyección y la estabilidad de almacenamiento limitada, debido tanto a la agregación como a la degradación del sistema de suministro y/o del agente activo.
Se ha descrito una composición de liberación lenta a base de lípidos para ciertos péptidos. Por ejemplo, el documento WO2006/131730 describe un sistema de depósito de lípidos para GLP-1 y análogos del mismo. Ésta es una formulación muy eficaz, pero la concentración de agente activo que se puede incluir en la formulación está limitada por su solubilidad. Evidentemente, una concentración más alta de agente activo permite la posibilidad de productos de depósito de mayor duración, productos que mantienen una concentración sistémica más alta, y productos que tienen un volumen de inyección más pequeño, todos los cuales son factores de considerable ventaja en circunstancias apropiadas. De este modo, sería de considerable valor establecer una forma por la cual podrían incluirse concentraciones más altas de agentes activos en una formulación de depósito a base de lípidos, e identificar
combinaciones de agente activo y sistema de administración que sean particularmente eficaces desde el punto de vista de la carga, estabilidad, fabricación y/o liberación controlada.
Los presentes inventores han establecido ahora que al proporcionar una pre-formulación que comprende al menos un diacilglicerol neutro, al menos una fosfatidilcolina, al menos un disolvente monoalcohólico orgánico biocompatible, al menos un disolvente polar, al menos un agente activo peptídico que comprende pasireotida (SOM230), y opcionalmente al menos un antioxidante en una fase de baja viscosidad, tal como disolución molecular o fase L2 (micelar inversa), se puede generar una pre-formulación que aborde muchos de los defectos de las formulaciones de depósito conocidas, y que se puede aplicar para proporcionar una liberación controlada del agente activo pasireotida. Mediante el uso de componentes específicos en relaciones cuidadosamente seleccionadas, y en particular con una mezcla de pasireotida, un alcohol y un disolvente polar, se puede generar una formulación de depósito que tiene una combinación de propiedades que supera el rendimiento incluso de las composiciones previas de liberación controlada lipídicas, y que proporciona una ventaja con respecto a las composiciones conocidas, tal como pasireotida LAR.
En particular, la pre-formulación muestra un perfil de liberación muy ventajoso, es fácil de fabricar, puede filtrarse de forma estéril, tiene baja viscosidad (lo que permite una administración fácil y menos dolorosa, típicamente a través de una aguja estrecha), permite que se incorpore un nivel elevado de agente bioactivo (permitiendo así potencialmente que se use una cantidad más pequeña de composición y/o agente activo), requiere una inyección poco profunda, y/o forma una composición de depósito no laminar deseada in vivo que tiene un perfil de liberación “sin estallido”. Las composiciones también se forman a partir de materiales que son no tóxicos, biotolerables y biodegradables, que pueden administrarse por vía intramuscular o inyección s.c., y son adecuados para la autoadministración. Además, la pre-formulación puede tener un nivel muy bajo de irritación en la inyección, y en casos preferidos, no causa irritación en el lugar de la inyección (incluyendo la irritación transitoria).
Algunas de las formulaciones de la presente invención generan una fase cristalina líquida no laminar después de la administración. El uso de estructuras de fase no laminar (tales como fases cristalinas líquidas no laminares) en la administración de agentes bioactivos está ahora relativamente bien establecido. Un sistema de depósito de lípidos más eficaz se describe en el documento WO2005/117830, y en ese documento se describe un depósito de lípidos muy preferido. Sin embargo, queda margen para lograr formulaciones de depósito que tengan un rendimiento mejorado en varios aspectos, y en particular, se pueden lograr mejoras sorprendentes mediante una selección y optimización cuidadosas de la gama de componentes y proporciones descritas en trabajos anteriores.
Las ventajas de las composiciones de la presente invención con respecto a las formulaciones poliméricas, tales como microesferas de PLGA, incluyen la facilidad de fabricación (incluyendo la esterilización), las propiedades de manipulación y uso combinadas con una baja liberación inicial (“perfil sin estallido”) del agente activo. Esto puede definirse de manera que el área bajo la curva de concentración plasmática frente al tiempo, durante las primeras 24 horas de un período de dosificación de un mes, es menos del 20% del área bajo la curva para toda la curva (medida o extrapolada del tiempo 0 hasta infinito, o desde el tiempo 0 hasta el último punto de tiempo de muestreo), más preferiblemente menos del 15%, y lo más preferible menos del 10%. Además, se puede definir de manera que la concentración plasmática máxima de agente activo in vivo después de la inyección de la pre-formulación (Cmax) no es más de 10 veces, preferiblemente no más de 8 veces, y lo más preferible no más de 5 veces la concentración plasmática promedio durante el período terapéutico (Cave) (es decir, Cmax/Cave <10, preferiblemente <8, más preferiblemente <5).
SUMARIO DE LA INVENCIÓN
La presente invención proporciona una formulación farmacéutica que comprende una combinación apropiada de excipientes lipídicos, disolvente alcohólico orgánico, disolvente polar, agente activo peptídico que comprende pasireotida, y ciertos componentes opcionales, que se puede usar como una formulación precursora de depósito (denominada aquí de forma breve como pre-formulación) para abordar una o más de las necesidades descritas anteriormente. Los inventores han establecido que, optimizando estos componentes, se pueden generar composiciones de depósito de pasireotida y formulaciones precursoras correspondientes con una combinación de propiedades muy ventajosa.
En un primer aspecto, la invención proporciona por tanto una pre-formulación que comprende una mezcla de baja viscosidad de:
a. 20-50% en peso de al menos un diacilglicerol;
b. 20-54% en peso de al menos una fosfatidilcolina (PC);
c. 0,1-35% en peso de al menos un disolvente monoalcohólico orgánico biocompatible;
d. 1 a 20% en peso de disolvente polar
e. 5 a 150 mg/ml de al menos un agonista peptídico del receptor de somatostatina que comprende pasireotida (calculado como la base libre);
f. opcionalmente al menos un antioxidante;
en la que la relación de los componentes a:b está en el intervalo de 40:60 a 54:46;
en la que la pre-formulación tiene una viscosidad de 1-1000 mPas a 20°C;
en la que la pre-formulación forma, o es capaz de formar, al menos una estructura de fase cristalina líquida al entrar en contacto con un exceso de fluido acuoso.
Dichas composiciones comprenderán preferiblemente dioleato de glicerol (GDO), PC de soja y/o PC de alta pureza (tal como PC con al menos 95% de grupos de cabeza de PC y al menos 95% de grupos acilo de C16 a C20 que tienen 0 a 3 insaturaciones), etanol, agua/propilenglicol y/o EDTA como componentes a), b), c), d) y f) respectivamente. El componente e) comprende o consiste en pasireotida o una sal de la misma, preferiblemente en la que dicha sal es una sal biotolerable, tal como la seleccionada de las sales de cloruro, acetato, pamoato y tartrato, lo más preferible la sal de pamoato, como se describe aquí.
Las preformulaciones son muy útiles para la liberación controlada y sostenida del agente activo peptídico, especialmente aquellas que requieren o se benefician de un perfil de liberación muy plano y/o de “estallido” mínimo tras la administración.
Los agonistas peptídicos del receptor de somatostatina en las formulaciones de la presente invención son de forma preferible farmacéuticamente activos. Es decir, proporcionan un efecto terapéutico, paliativo y/o profiláctico cuando se administran a un sujeto adecuado (que típicamente necesita un efecto de este tipo). En una realización adicional, la invención proporciona por lo tanto una pre-formulación como se describe aquí para uso en el tratamiento de la enfermedad de Cushing, acromegalia, diabetes mellitus tipo I o tipo II, especialmente sus complicaciones, por ejemplo angiopatía, retinopatía proliferativa diabética, edema macular diabético, nefropatía, neuropatía y fenómeno del amanecer, y otros trastornos metabólicos relacionados con la liberación de insulina o glucagón, por ejemplo obesidad, por ejemplo obesidad mórbida u obesidad hipotalámica o hiperinsulinémica, fístula enterocutánea y pancreaticocutánea, síndrome del intestino irritable, enfermedades inflamatorias, por ejemplo enfermedad de Grave, enfermedad inflamatoria intestinal, psoriasis o artritis reumatoide, enfermedad renal poliquística, síndrome de dumping, síndrome de diarrea acuosa, diarrea relacionada con el SIDA, diarrea inducida por quimioterapia, pancreatitis aguda o crónica y tumores secretores de hormonas gastrointestinales (por ejemplo, tumores GEP, por ejemplo vipomas, glucagonomas, insulinomas, carcinoides, y similares), neoplasias de linfocitos, por ejemplo linfomas o leucemias, carcinoma hepatocelular, así como hemorragia gastrointestinal, por ejemplo hemorragia esofágica por varices.
En todos los aspectos apropiados de la invención, las afecciones preferidas son la enfermedad de Cushing y la acromegalia.
Una de las ventajas de las formulaciones de la presente invención con respecto a muchas otras composiciones de liberación controlada es que son estables al almacenamiento en su forma final, y de este modo, se requiere poca o ninguna preparación en el momento de la administración. Esto permite que las pre-formulaciones estén listas para administrar y también se suministren en forma conveniente, lista para administrar. En un aspecto adicional, la invención proporciona por tanto un dispositivo de administración precargado que contiene una pre-formulación como se describe aquí, siendo dicho dispositivo una jeringa o cilindro de jeringa, un inyector sin aguja, un inyector multiusos o de un solo uso, un cartucho o un vial. Un dispositivo de este tipo proporcionará generalmente una sola administración o múltiples administraciones de una composición que suministrará, por ejemplo, una dosis de agonista del receptor de somatostatina en el intervalo de 0,2 a 3 mg/día de pasireotida.
BREVE SUMARIO DE LAS FIGURAS ADJUNTAS
Figura 1. Diagrama de flujo que describe la preparación de muestras de lípidos/SOM230 (pasireotida) para el cribado de solubilidad.
Figura 2. Inyectabilidad (segundos/ml) medida a una fuerza constante de 20 N en función de la composición de la formulación y que usa una jeringa de vidrio Luer-Lock de 1 ml de largo con una aguja de 23 G 5/8” (16 mm). Los números de las formulaciones se refieren al ID de la muestra (los dos últimos dígitos) en la Tabla 3. Los puntos de datos representan la media de medidas duplicadas.
Figura 3. Inyectabilidad (segundos por ml), medida a una fuerza constante de 20 N, en función de la viscosidad de la formulación y utilizando la configuración de jeringa y aguja indicada.
Figura 4. Comparación de los datos de estabilidad en formulaciones de lípidos/pasireotida diferenciadas por su respectiva composición de disolvente (relación en peso SPC/GDO constante a 50/50), con tres formas de sal de SOM230 (pasireotida) diferentes (pamoato (Pm), acetato (Ac) e hidrocloruro (Cl)) después del almacenamiento durante 2 semanas a 60°C. La concentración de base libre de pasireotida al inicio fue en todos los casos aproximadamente 30 mg/ml.
Figura 5. Concentraciones plasmáticas medias de SOM230 (pamoato de pasireotida) después de inyección s.c. en ratas. Las barras de error denotan la desviación estándar (n = 6). Datos obtenidos en PK-12-437.
Figura 6. Concentraciones plasmáticas medias de SOM230 (pamoato de pasireotida) después de inyección s.c. en ratas. Las barras de error denotan la desviación estándar (n = 6). Datos obtenidos en PK-12-438.
Figura 7. Linealidad de la dosis con respecto a la exposición (AUC) en el estudio PK-12-438. Las barras de error representan la desviación estándar.
Figura 8. Linealidad de la dosis con respecto a la Cmax en el estudio PK-12-438. Las barras de error representan la desviación estándar.
Figura 9. Concentraciones plasmáticas medias de SOM230 (pamoato de pasireotida) después de inyección s.c. en ratas. Las barras de error denotan la desviación estándar (n = 6). Datos obtenidos en PK-12-451.
Figura 10. La pureza de SOM230 después del almacenamiento a 5°C. La leyenda de la figura se refiere al número de lote respectivo como se indica en la Tabla 19.
Figura 11. La pureza de SOM230 después del almacenamiento a 25°C/60% HR. La leyenda de la figura se refiere al número de lote respectivo como se indica en la Tabla 19.
DESCRIPCIÓN DETALLADA DE LA INVENCIÓN
Las formulaciones de la presente invención generan una fase cristalina líquida no laminar después de la administración. El uso de estructuras de fase no laminar (tales como fases cristalinas líquidas) en el suministro de agentes bioactivos está ahora relativamente bien establecido. Un sistema de depósito de lípidos más eficaz para uso general se describe en el documento WO2005/117830, y una matriz lipídica adecuada para uso en la presente invención se describe en términos generales en ese documento, cuya descripción completa se incorpora aquí como referencia. Para una descripción de las estructuras de fase más favorables de tales formulaciones, se llama la atención sobre la discusión en el documento WO2005/117830, y particularmente en su página 29.
Todos los % se especifican en peso aquí, excepto que se indique de otro modo. Además, el % en peso indicado es el % de la pre-formulación total que incluye todos los componentes indicados aquí cuando el contexto lo permita. Los porcentajes en peso de pasireotida se calcularán sobre la base del peso del ácido libre, independientemente de si se usa el ácido o una sal del mismo. Las pre-formulaciones pueden consistir de forma opcional esencialmente solo en los componentes indicados aquí (incluyendo, cuando sea apropiado, componentes opcionales adicionales indicados aquí a continuación y en las reivindicaciones adjuntas), y en un aspecto, consisten completamente en tales componentes. Una formulación se indica como “que consiste esencialmente en” ciertos componentes aquí, cuando los componentes especificados proporcionan la naturaleza esencial de esa formulación, tal como cuando los componentes especificados constituyen al menos el 95%, preferiblemente al menos el 98%, de la formulación.
Los sistemas basados en lípidos descritos aquí comprenden componentes lipídicos a) y b), más disolvente orgánico monoalcohólico (c), disolvente polar (d), agonista peptídico del receptor de somatostatina que comprende pasireotida (e), y componentes antioxidantes opcionales (f).
Preferiblemente, la pre-formulación según la invención es una disolución molecular o tiene una estructura de fase L2 (antes de la administración). Preferiblemente, la pre-formulación forma una fase no laminar (por ejemplo, cristalina líquida) después de la administración. Este cambio de fase se produce típicamente mediante la absorción de fluido acuoso del entorno fisiológico, como se indica aquí.
Los presentes inventores han establecido ahora sorprendentemente que mediante la elección apropiada de tipos, cantidades absolutas y relaciones de componentes lipídicos junto con un agonista peptídico del receptor de somatostatina que comprende pasireotida y al menos dos disolventes que incluyen un alcohol y al menos un disolvente polar, las propiedades de liberación de las composiciones de depósito formadas a partir de las pre-formulaciones de la invención pueden resultar muy ventajosas. En particular, mediante el uso de una mezcla de un alcohol y un disolvente polar (especialmente en las relaciones en peso cercanas a 1:1 descritas aquí (por ejemplo, entre 10:1-1:3, preferiblemente 5:1-1:2, y más preferiblemente 2:1 -2:3)) se pueden mantener las ventajas del disolvente alcohólico en el perfil de liberación, mientras que se pueden mejorar otras propiedades como la comodidad en la administración y/o la viscosidad de la formulación. Alternativamente o además de esto, el perfil de liberación del agonista del receptor de somatostatina se puede nivelar notablemente, siendo la concentración plasmática máxima in vivo solo un pequeño múltiplo de la concentración promedio o incluso mínima durante el período de dosificación. Tales ventajas se aplican incluso en comparación con otras composiciones de depósito de lípidos, que en sí mismas ofrecen patrones previamente inalcanzables en liberación controlada.
Es importante, particularmente con ciertos agentes activos peptídicos, tales como los análogos de la somatostatina (por ejemplo, pasireotida), controlar la concentración máxima (Cmax) del fármaco en el plasma a un nivel igual o menor que el tolerable para el sujeto, por ejemplo para evitar efectos secundarios tales como rubor o náuseas intensas, mientras proporciona o alcanza un nivel terapéuticamente eficaz durante el período de liberación deseado. Generalmente, la concentración promedio durante el período de liberación antes de que se administre la siguiente dosis, Cave, se encuentra dentro del intervalo terapéutico. El control sobre las concentraciones máxima (Cmax) y mínima (Cmin) también es importante para lograr el tratamiento deseado a lo largo del tiempo. En una realización, el
estallido inicial (por ejemplo, durante las primeras 12 horas después de la administración) no es la Cmax del perfil de liberación.
Tanto si el estallido inicial es también la Cmax o no, preferiblemente la relación Cmax/Cave es menor que 50, preferiblemente menor o igual a 15, más preferiblemente menor o igual a 10, incluso más preferiblemente menor o igual a 5. Además, se prefiere que la relación Cmax/Cmin no sea mayor de 50, preferiblemente menor o igual a 15, más preferiblemente menor o igual a 10, incluso más preferiblemente menor o igual a 5. Cmax se define como se conoce en la técnica, como la concentración plasmática máxima o pico observada durante el período de liberación antes de que se administre la siguiente dosis, y Cave se define como la concentración plasmática promedio durante ese período de liberación. Cmin es correspondientemente la concentración mínima durante ese período. Cave se puede calcular calculando el fármaco presente en el plasma como área bajo la curva (AUC) durante el período de tiempo seleccionado, generalmente el período completo de liberación antes de la administración de la siguiente dosis, y dividiendo entre ese período de tiempo.
Componente a) - Diacil glicerol
Los intervalos para el componente a) son 20-50%, tal como 33-60% (por ejemplo 43-60%, 30 a 43% o 30-40%), particularmente 38 a 43%, alrededor de 32% (por ejemplo ±2) y/o alrededor de 40% (por ejemplo, ±2). Los intervalos del componente b) son 20-54% en peso, preferiblemente 30-45%, más preferiblemente 38 a 43%.
Las relaciones de a:b son 40:60 a 54:46, o 42:58 a 48:52. Las relaciones de alrededor de 50:50 (por ejemplo, ±2) y alrededor de 45:55 (por ejemplo, ±3 a:b) son muy efectivas.
El componente “a”, como se indica aquí, es al menos un diacil glicerol (DAG), y de este modo tiene dos grupos de “cola” no polares. Los dos grupos no polares pueden tener el mismo o diferente número de átomos de carbono, y cada uno puede estar independientemente saturado o insaturado. Ejemplos de grupos no polares incluyen grupos alquilo y alquenilo de C6-C32, que normalmente están presentes como ésteres de ácidos carboxílicos de cadena larga. Estos se describen a menudo con referencia al número de átomos de carbono y al número de insaturaciones en la cadena de carbono. De este modo, CX:Z indica una cadena hidrocarbonada que tiene X átomos de carbono y Z insaturaciones. Los ejemplos incluyen particularmente grupos lauroílo (C12:0), miristoílo (C14:0), palmitoílo (C16:0), fitanoílo (C16:0), palmitoleoílo (C16:1), estearoílo (C18:0), oleoílo (C18:1), elaidoílo (C18:1), linoleoílo (C18:2), linolenoílo (C18:3), araquidonoílo (C20:4), behenoílo (C22:0) y lignoceroílo (C24:9). De este modo, las cadenas no polares típicas se basan en los ácidos grasos de lípidos de ésteres naturales, incluyendo ácidos caproico, caprílico, cáprico, láurico, mirístico, palmítico, fitánico, palmitólico, esteárico, oleico, elaídico, linoleico, linolénico, araquidónico, behénico o lignocérico, o los correspondientes alcoholes. Las cadenas no polares preferidas son los ácidos palmítico, esteárico, oleico y linoleico, particularmente ácido oleico.
Se pueden usar mezclas de cualquier número de diacil lípidos como componente a). Preferiblemente, este componente incluirá al menos una porción de lípidos de C18 (por ejemplo, DAG que tiene uno o más (es decir, uno o dos) grupos no polares C18:0, C18:1, C18:2 o C18:3), tales como dioleato de glicerol (GDO) y/o dilinoleato de glicerol (GDL). Un ejemplo muy preferido es DAG que comprende al menos 50%, preferiblemente al menos 80%, e incluso comprende sustancialmente 100% de GDO.
Dado que GDO y otros diacilgliceroles son productos derivados de fuentes naturales, generalmente existe una cierta proporción de lípidos “contaminantes” que tienen otras longitudes de cadena, etc. En un aspecto, GDO, como se usa aquí, se usa así para indicar cualquier grado comercial de GDO con impurezas concomitantes (es decir, GDO de pureza comercial). Estas impurezas pueden separarse y eliminarse mediante purificación, pero siempre que el grado sea consistente, esto rara vez es necesario. Sin embargo, si es necesario, “GDO” puede ser GDO esencialmente puro químicamente, tal como GDO al menos 80% puro, preferiblemente al menos 85% puro, y más preferiblemente al menos 90% puro.
Componente b) - Fosfatidilcolina
El componente “b” en las matrices lipídicas de la presente invención es al menos una fosfatidilcolina (PC). Al igual que con el componente a), este componente comprende un grupo de cabeza polar y al menos un grupo de cola no polar. La diferencia entre los componentes a) y b) radica principalmente en el grupo polar. De este modo, las porciones no polares pueden derivar de manera adecuada de los ácidos grasos o los alcoholes correspondientes considerados anteriormente para el componente a. Al igual que con el componente a), la PC contendrá dos grupos no polares. Nuevamente, se prefieren los grupos de C18, y pueden combinarse con cualquier otro grupo no polar adecuado, particularmente grupos de C16.
La porción de fosfatidilcolina, incluso más adecuadamente que cualquier porción de diacil glicerol, puede derivar de una fuente natural. Las fuentes adecuadas de fosfolípidos incluyen huevo, corazón (por ejemplo, bovino), cerebro, hígado (por ejemplo, bovino), y fuentes vegetales, incluyendo la soja. Dichas fuentes pueden proporcionar uno o más constituyentes del componente b, que pueden comprender cualquier mezcla de fosfolípidos. Se puede usar cualquier PC individual o mezcla de PC de estas u otras fuentes, pero las mezclas que comprenden PC de soja o PC de huevo son muy adecuadas. El componente de PC contiene preferiblemente al menos 50% de PC de soja o PC de huevo,
más preferiblemente al menos 75% de PC de soja o PC de huevo, y lo más preferible PC de soja o PC de huevo esencialmente pura.
En todos los aspectos de la invención, el componente b) comprende PC. Preferiblemente, la PC deriva de la soja. Preferiblemente, la PC comprende ácidos grasos 18:2 como componente de ácido graso primario con 16:0 y/o 18:1 como componentes de ácido graso secundario. Estos están presentes preferiblemente en la PC en una relación de entre 1,5:1 y 6:1. Se prefiere PC que tiene aproximadamente 60-65% de 18:2, 10 a 20% de 16:0, 5-15% de 18:1, siendo el resto predominantemente otros ácidos grasos de 16 carbonos y 18 carbonos, y es típicamente PC de soja.
En una realización alternativa pero igualmente preferida, también aplicable a todos los aspectos de la invención, el componente de PC puede comprender dioleoil PC sintética. Se cree que esto proporciona una mayor estabilidad, y así será particularmente preferible para las composiciones que necesitan ser estables al almacenamiento a largo plazo y/o que tienen un período de liberación prolongado in vivo. En esta realización, el componente de PC contiene preferiblemente al menos 50% de dioleoil PC sintética, más preferiblemente al menos 75% de dioleoil PC sintética, y lo más preferible dioleoil PC sintética esencialmente pura. Cualquier PC restante es preferiblemente PC de soja o huevo como anteriormente.
En una realización, las formulaciones precursoras de la presente invención están compuestas al menos parcialmente de DOPC sintética (es decir, PC que tiene al menos 95% de grupos de cabeza de PC y al menos 90% de grupos oleoil acilo) y tiene una estabilidad de almacenamiento a 15-25°C, definida como menos del 5% de degradación del péptido según lo evaluado por la pureza del péptido, de al menos 6 meses, más preferiblemente al menos 12 meses, y lo más preferible al menos 24 meses.
Dado que las pre-formulaciones de la invención se van a administrar a un sujeto para la liberación controlada de un agente activo peptídico, es importante que los componentes sean biocompatibles. A este respecto, las matrices lipídicas preferidas para uso en las pre-formulaciones de la presente invención son muy ventajosas ya que tanto PC como DAGs se toleran bien y se descomponen in vivo en componentes que están presentes naturalmente en el cuerpo de los mamíferos.
Las PC sintéticas o altamente purificadas, tales como dioleoil fosfatidilcolina (DOPC) y palmitoil oleoil fosfatidilcolina (POPC), así como las otras diversas PC de alta pureza descritas aquí, son muy apropiadas como todo o parte del componente b).
En una realización muy preferida, el componente b) es una PC de “alta pureza” como sigue:
b. al menos un componente fosfolipídico que comprende fosfolípidos que tienen
i. grupos de cabeza polares que comprenden al menos 95% de fosfatidilcolina, y
ii. dos cadenas de acilo que tienen cada una independientemente 16 a 20 carbonos, en las que al menos una cadena de acilo tiene al menos una insaturación en la cadena de carbono, y no hay más de cuatro insaturaciones en dos cadenas de carbono;
Normalmente, esto puede ser PC con al menos 95% de grupos de cabeza de PC y al menos 95% de cadenas de acilo de C16 a C20 que tienen 0 a 3 insaturaciones.
La dioleoil PC sintética es muy preferible 1,2-dioleoil-sn-glicero-3-fosfocolina, y otros componentes de PC sintética incluyen DDPC (1,2-didecanoil-sn-glicero-3-fosfocolina); DEPC (1,2-dierucoil-sn-glicero-3-fosfocolina); DLOPC (1,2-dilinoleoil-sn-glicero-3-fosfocolina); DLPC (1,2-dilauroil-sn-glicero-3-fosfocolina); DMPC (1,2-dimiristoil-sn-glicero-3-fosfocolina); DOPC (1,2-dioleoil-sn-glicero-3-fosfocolina); DPPC (1,2-dipalmitoil-sn-glicero-3-fosfocolina); DSPC (1,2-distearoil-sn-glicero-3-fosfocolina); MPPC (1-miristoil-2-palmitoil-sn-glicero-3-fosfocolina); MSPC (1 -miristoil-2-estearoil-sn-glicero-3-fosfocolina); PMPC (1-palmitoil-2-miristoil-sn-glicero-3-fosfocolina); POPC (1 -palmitoil-2-oleoilsn-glicero-3-fosfocolina); PSPC (1-palmitoil-2-estearoil-sn-glicero-3-fosfocolina); SMPC (1-estearoil-2-miristoil-snglicero-3-fosfocolina); SOPC (1-estearoil-2-oleoil-sn-glicero-3-fosfocolina); y SPPC (1-estearoil-2-palmitoil-sn-glicero-3-fosfocolina), o cualquier combinación de los mismos.
En algunas circunstancias, tales como la ausencia de agentes conservantes tal como EDTA, el uso de PC sintéticas o altamente purificadas (por ejemplo, DOPC) puede proporcionar una mayor estabilidad al agonista del receptor de somatostatina en las formulaciones. De este modo, en una realización, el componente b) puede comprender (por ejemplo, puede comprender al menos 75%) PC sintéticas o altamente purificadas (por ejemplo, pureza >90%) (por ejemplo, DOPC). Esto puede ocurrir particularmente en ausencia de agentes quelantes tal como EDTA. En una realización alternativa, el componente b) puede comprender (por ejemplo, comprender al menos 75%) PC de origen natural, tal como PC de soja o PC de huevo. Esto será particularmente cuando se incluye al menos un componente estabilizante (tal como un antioxidante, quelante, etc.) en la formulación precursora.
Una combinación particularmente favorecida de componentes a) y b) son GDO con PC, especialmente GDO con PC de soja y/o PC de “alta pureza”. Cantidades apropiadas de cada componente adecuadas para la combinación son las
cantidades indicadas aquí para los componentes individuales en cualquier combinación. Esto se aplica también a cualquier combinación de componentes indicados aquí, cuando el contexto lo permita.
La relación de los componentes a:b está en el intervalo 40:60 a 54:46. Preferiblemente, la relación a:b está en el intervalo 45:55 a 54:46, más preferiblemente 47:53 a 53:47. Lo más preferible, la relación a:b es aproximadamente 50:50.
En una realización aplicable a todos los aspectos de la invención, se prefiere si la relación a:b está en el intervalo 40:60 a 49:51. En una realización alternativa, la relación puede estar en el intervalo 42:58 a 52:48.
Componente c) - disolvente monoalcohólico orgánico
El componente c) de las pre-formulaciones de la invención es un disolvente monoalcohólico orgánico. Dado que la pre-formulación debe generar una composición de depósito después de la administración (por ejemplo in vivo), típicamente al entrar en contacto con un exceso de fluido acuoso, es deseable que este disolvente sea tolerable para el sujeto y sea capaz de mezclarse con el fluido acuoso y/o difundirse o disolverse fuera de la pre-formulación en el fluido acuoso. De este modo, se prefieren los disolventes que tienen al menos una moderada solubilidad en agua.
Lo más preferible, el componente c) comprende o consiste en etanol, propanol, ispropanol, alcohol bencílico, o mezclas de los mismos. Lo más preferible, el componente c) comprende o consiste en etanol.
En una realización preferida, el disolvente es tal que una adición relativamente pequeña a una mezcla que comprende a) y b) (es decir, preferiblemente por debajo del 15%) produce grandes reducciones de viscosidad, de un orden de magnitud o más. Como se describe aquí, la adición de disolvente monoalcohólico orgánico al 10% puede dar una reducción de dos o más órdenes de magnitud en la viscosidad con respecto a la composición sin disolvente, o con respecto a un depósito que contiene solo un disolvente polar tal como agua, o glicerol.
La cantidad de componente c) en la pre-formulación tendrá un efecto considerable sobre varias características. En particular, la viscosidad y la velocidad (y duración) de liberación se alterarán significativamente con el nivel de disolvente. De este modo, la cantidad de disolvente será al menos suficiente para proporcionar una mezcla de baja viscosidad, pero además se determinará para proporcionar la velocidad de liberación deseada. Esto puede determinarse mediante métodos de rutina a la vista de los Ejemplos más abajo. Normalmente, un nivel de 0,1 a 35%, particularmente 5 a 25% de disolvente, proporcionará propiedades de liberación y viscosidad adecuadas. Este será preferiblemente 5 a 16% (por ejemplo, 6 a 14%), y una cantidad de alrededor de 8% (por ejemplo, 8±2%) es muy eficaz.
Como se indicó anteriormente, la cantidad de componente c) en las pre-formulaciones de la invención será al menos suficiente para proporcionar una mezcla de baja viscosidad (por ejemplo, una disolución molecular, véase anteriormente) de los componentes a), b), c) y d) y opcionalmente f), y se determinará fácilmente para cualquier combinación particular de componentes mediante métodos estándar.
El comportamiento de fase puede analizarse mediante técnicas como la observación visual en combinación con microscopía de luz polarizada, técnicas de dispersión y difracción de rayos X, resonancia magnética nuclear, y microscopía electrónica de crio-transmisión (crio-TEM) para buscar disoluciones, fases L2 o L3 , o fases cristalinas líquidas o, como en el caso de crioTEM, fragmentos dispersos de dichas fases. La viscosidad puede medirse directamente por medios estándar. Como se describió anteriormente, una viscosidad práctica apropiada es la que puede inyectarse eficazmente con jeringa y filtrarse particularmente de forma estéril. Esto se evaluará fácilmente como se indica aquí.
Los disolventes orgánicos monoalcohólicos típicos adecuados para uso en la invención incluyen al menos un disolvente seleccionado de etanol, propanol, isopropanol, y alcohol bencílico, particularmente etanol.
Una combinación muy preferida para los componentes a), b) y c) es PC de soja y/o “PC de alta pureza”, GDO y etanol. Como se indicó anteriormente, las cantidades apropiadas de cada componente adecuadas para la combinación son las cantidades indicadas aquí para los componentes individuales, en cualquier combinación.
Es preferible que poco o nada del componente c) contenga hidrocarburos sustituidos con halógeno, ya que estos tienden a tener una menor biocompatibilidad. Por ejemplo, el contenido de disolventes orgánicos halogenados puede ser menor que 0,5%, preferiblemente menor que 0,1%.
El componente c), como se usa aquí, puede ser un solo disolvente o una mezcla de disolventes adecuados, pero generalmente será de baja viscosidad. Esto es importante debido a que uno de los aspectos clave de la presente invención es que proporciona pre-formulaciones que son de baja viscosidad, y una función principal de un disolvente adecuado es reducir esta viscosidad. Esta reducción será una combinación del efecto de la menor viscosidad del disolvente y el efecto de las interacciones moleculares entre el disolvente y la composición lipídica. Una observación de los presentes inventores es que los disolventes de baja viscosidad que contienen oxígeno descritos aquí tienen interacciones moleculares muy ventajosas e inesperadas con las partes lipídicas de la composición, proporcionando así una reducción no lineal de la viscosidad con la adición de un pequeño volumen de disolvente.
La viscosidad del componente c) de disolvente de “baja viscosidad” (disolvente único o mezcla) no debería ser normalmente superior a 18 mPas a 20°C. Esta es preferiblemente no mayor que 15 mPas, más preferiblemente no mayor que 10 mPas, y lo más preferible no mayor que 7 mPas a 20°C.
Componente d) - Disolvente polar
Algunos de los beneficios particulares de las composiciones de la presente invención provienen del hallazgo inesperado de que el uso de un disolvente alcohólico en combinación con un disolvente polar, tal como un diol o agua, permite una mejora significativa en el rendimiento de ciertas composiciones de liberación controlada a base de lípidos. En particular, se ha observado que la adición de un diol (tal como propilenglicol) o agua permite aumentar la proporción de alcohol sin afectar negativamente el perfil de liberación, y/o permite una mejora en el perfil de liberación, y/o permite una carga más alta del agonista del receptor de somatostatina. Por “afectar negativamente al perfil de liberación” se pretende indicar que la relación de Cmax/Cave aumenta y/o la relación de Cmax/Cmin aumenta (por ejemplo, aumenta en un factor de al menos 1,2). De manera similar, una mejora en el perfil de liberación indica que la relación de Cmax/Cave y/o Cmax/Cmin disminuye (por ejemplo, disminuye en un factor de al menos 1,2).
Los disolventes polares típicos tendrán una constante dieléctrica comparativamente alta correspondiente a su alta polaridad. De este modo, los disolventes polares adecuados generalmente tendrán una constante dieléctrica de al menos 28 a 25°C, más preferiblemente al menos 30 a 25°C. Los ejemplos muy adecuados incluyen agua (~80), propilenglicol (~32) y N-metil-2-pirrolidona (NMP, ~32).
Aunque se ha sugerido anteriormente que las composiciones de liberación controlada de lípidos deberían formularse sustancialmente en ausencia de agua, para evitar la conversión en fases cristalinas líquidas de alta viscosidad, ahora se ha establecido además que una cantidad pequeña y cuidadosamente controlada de un disolvente polar tal como el agua puede proporcionar beneficios considerables. En particular, la inclusión de este disolvente polar (preferiblemente que comprende agua) permite mejoras adicionales en el control de la liberación inicial del agonista del receptor de somatostatina, permite una carga estable más alta de algunos agonistas peptídicos del receptor de somatostatina, proporciona una formación de depósitos más rápida, y/o proporciona una incomodidad más reducida con la inyección. Cualquiera de estos factores proporciona potencialmente una mejora significativa en el contexto de la administración de fármacos terapéuticos, la salud del paciente y/o el cumplimiento del paciente.
De este modo, las pre-formulaciones de la presente invención también deben contener un disolvente polar, componente d). Una cantidad adecuada será típicamente 1 a 20% en peso de la pre-formulación, particularmente 1,2-20% en peso, especialmente 2-18% en peso. Más preferiblemente, el componente d) está presente en el intervalo 5-15% en peso, especialmente 6-12% en peso. El componente d) es preferiblemente agua, propilenglicol, o mezclas de los mismos. En un aspecto preferido, las pre-formulaciones de la invención contienen etanol como componente c), con agua y/o propilenglicol como componente d).
En una realización, la pre-formulación comprende al menos 1,5% (por ejemplo, al menos 4,5%) de agua como parte del componente d) (en peso de la composición total), siendo el resto propilenglicol. Se prefiere al menos 5% de agua, siendo el resto del componente d) PG. El componente d) puede comprender o consistir en agua.
En una realización alternativa, el componente d) puede comprender o consistir en propilenglicol.
Preferiblemente, el nivel total de componentes c) y d) no es más de 35% en peso, preferiblemente no más de 30% en peso, más preferiblemente no más de 25% en peso, lo más preferible no más de 20% en peso. Por ejemplo, el nivel total de los componentes c) y d) puede estar en el intervalo 10-30% en peso, preferiblemente 12-25% en peso, lo más preferible 15-20% en peso.
La relación de componentes c) y d) también tendrá ventajas potenciales en las composiciones de la invención. En particular, mediante la inclusión de algún disolvente polar que sea miscible con el componente monoalcohólico (especialmente agua), se puede eliminar sustancialmente la ligera sensación que puede producirse en el lugar de la inyección por el contenido de alcohol. De este modo, en una realización, la relación de componentes c):d) puede estar en el intervalo 30:70 a 70:30, más preferiblemente 40:60 a 60:40. En una realización, la cantidad de componente alcohólico c) en peso no es mayor que la cantidad de disolvente polar d). Las relaciones de c):d) que oscilan de 30:70 a 50:50 son así apropiadas en tal realización. Cantidades aproximadamente iguales de componentes c) y d) son muy apropiadas.
Una combinación muy preferida para el aspecto de matriz lipídica es PC de soja y/o PC de C16 a C20 de al menos 95% de pureza, tal como DOPC (como se describe aquí), con GDO, etanol y agua/propilenglicol, o mezclas de los mismos. El disolvente puede ser, por ejemplo, etanol y agua en ausencia de PG, etanol y PG en ausencia de agua, o una mezcla de los tres. Como se indicó anteriormente, las cantidades apropiadas de cada componente adecuadas para la combinación son las cantidades indicadas aquí para los componentes individuales, en cualquier combinación.
Componente e) - Agente activo peptídico (agonista del receptor de somatostatina)
Las pre-formulaciones de la presente invención contienen al menos un agonista peptídico del receptor de somatostatina que comprende pasireotida. Los péptidos adecuados para uso en los agonistas peptídicos del receptor
de somatostatina necesarios pueden ser de origen natural o derivados de péptidos naturales, o pueden ser moléculas peptídicas modificadas químicamente o totalmente sintéticas. Cualquier aminoácido puede estar comprendido en los péptidos, incluyendo los descritos aquí, y los péptidos pueden modificarse química o enzimáticamente. De manera similar, los agonistas peptídicos del receptor de somatostatina pueden ser lineales o ciclados por medio de una o más interacciones covalentes o no covalentes.
La pasireotida, por ejemplo, comprende una porción cíclica de seis restos peptídicos (véase más abajo).
Los agentes activos peptídicos típicos estarán en el intervalo de 500 a 100.000 uma de peso molecular, y evidentemente pueden incluir agonistas proteicos del receptor de somatostatina. En una realización, los polipéptidos pueden tener al menos una carga catiónica a pH neutro y/o fisiológico, y lo más preferible, requerirán al menos un contraión aniónico a pH 6,5 o superior, preferiblemente a pH 7,5 o superior. Este contraión será fisiológicamente aceptable, y de este modo puede ser un haluro o el ión de un ácido fisiológicamente aceptable. Los contraiones de acetato, pamoato y tartrato y/o iones cloruro son particularmente preferidos, y por lo tanto, en una realización de la invención, el agonista del receptor de somatostatina es pamoato de pasireotida.
En particular, los presentes inventores han establecido sorprendentemente que el pamoato de pasireotida es sorprendentemente más estable al almacenamiento cuando se formula con los excipientes lipídicos descritos aquí que la correspondiente sal de acetato o de cloruro. Esto es particularmente sorprendente, ya que el acetato es la forma más comúnmente utilizada de muchos agentes activos de péptidos pequeños, tales como el análogo de somatostatina octreotida. Además, en trabajos anteriores se ha demostrado que la sal de cloruro es sorprendentemente eficaz en formulaciones de ciertos principios activos, tal como la octreotida. Sin embargo, en el presente caso, el almacenamiento a 60°C en las formulaciones de la presente invención ilustró una estabilidad marcadamente superior para el pamoato con respecto al acetato e incluso al cloruro de pasireotida (véanse los ejemplos y las Figuras a continuación). De este modo, la sal de pamoato es la forma preferida del agonista del receptor de somatostatina que comprende pasireotida.
En los agentes activos peptídicos de la presente invención, los péptidos pueden contener solo aminoácidos seleccionados de los 20 a-aminoácidos indicados en el código genético, o más preferiblemente pueden contener sus isómeros y otros aminoácidos naturales y no naturales (generalmente a, b o g aminoácidos) y sus análogos y derivados.
Los derivados de aminoácidos son especialmente útiles en los extremos de los péptidos, en los que el grupo amino o carboxilato terminal puede estar sustituido por o con cualquier otro grupo funcional, tal como hidroxi, alcoxi, carboxi (en el extremo N-terminal), éster, amida, tio, amido, amino (en el extremo C-terminal), alquilamino, di- o tri-alquilamino, alquilo (mediante lo cual se quiere decir aquí alquilo de C1-C20, preferiblemente alquilo de C1-C18, por ejemplo metilo, etilo, n-propilo, isopropilo, n-butilo, iso-, sec- o t-butilo, etc.), arilo (por ejemplo, fenilo, bencilo, naftilo, etc.), heteroarilo, u otros grupos funcionales, preferiblemente con al menos un heteroátomo y que preferiblemente que tienen no más de 20 átomos en total, más preferiblemente no más de 10, y lo más preferible no más de 6 átomos (opcionalmente excluyendo los hidrógenos).
En la presente invención, el agonista peptídico del receptor de somatostatina comprende pasireotida, que es un análogo de somatostatina. El agonista del receptor de somatostatina también puede comprender otros péptidos tales como otros análogos peptídicos de somatostatina, y puede incluir octreotida, somatostatina 14 y/o somatostatina 28.
La somatostatina tiene dos formas activas producidas por la escisión alternativa de una única preproteína: una de 14 aminoácidos y la otra de 28 aminoácidos. La somatostatina 1-14 es una hormona peptídica cíclica que tiene la secuencia Ala-Gly-Cys-Lys-Asn-Phe-Phe-Trp-Lys-Thr-Phe-Thr-Ser-Cys, en la que los dos restos de cistina están conectados por un puente de disulfuro para generar una hélice b tipo II en la secuencia de unión clave de Phe-Trp-Lys-Thr. La somatostatina es una hormona peptídica natural también conocida como factor inhibidor de la liberación de la hormona del crecimiento, y tiene un papel como antagonista de la insulina, el glucógeno y algunas otras hormonas en la liberación de somatotropina (hormona del crecimiento humano). La semivida biológica de la somatostatina natural es muy corta (1-3 minutos), y de este modo, en sí misma es difícil de formular como una sustancia terapéutica viable. Sin embargo, las composiciones de depósito de lípidos de la presente invención son altamente efectivas para agentes activos de vida corta, y un número creciente de análogos de somatostatina están disponibles con actividades más altas y/o tiempos de aclaramiento más largos in vivo. La pasireotida es uno de tales análogos, y forma el agente activo peptídico esencial de las composiciones de la presente invención.
Los análogos de somatostatina, que incluyen pasireotida, pero también tal como octreotida, lanreotida, vapreotida y péptidos relacionados, se usan o están indicados en el tratamiento de una variedad de afecciones en las que se administran típicamente durante un período prolongado. En una realización de la invención, el agente activo peptídico comprende o consiste en pasireotida así como otro análogo de somatostatina seleccionado del grupo que consiste en octreotida, lanreotida y vapreotida. En una realización, el agente activo peptídico de la invención comprende o consiste en pasireotida y octreotida. En una realización adicional, la pasireotida puede formar el único agente activo.
La octreotida, por ejemplo, es el octapéptido sintético con secuencia D-Phe-Cys-Phe-D-Trp-Lys-Thr-Cys-Thr-ol (puente de disulfuro 2-7), y se administra típicamente como la sal de acetato. Varios estudios clínicos también
presentan el pamoato de octreotida. Este derivado retiene la hélice b clave Phe-(D)Trp-Lys-Thr, pero, a diferencia de la hormona natural, tiene una semivida terminal de alrededor de 1,7 horas. La octreotida se usa en el tratamiento de afecciones que incluyen tumores carcinoides y acromegalia, y después de una dosis inicial, generalmente se administra durante un período sostenido de semanas, o más comúnmente muchos meses o años. Además, los análogos de somatostatina están indicados en el tratamiento de muchos cánceres, ya que se encuentra que una amplia variedad de tumores expresa receptores de somatostatina. De particular interés son los que expresan el receptor “sst(2)” y/o “sst(5)”.
Como se usa aquí, la expresión “agonista del receptor de somatostatina” se usa para indicar un compuesto que tiene una función agonista en uno o más receptores de somatostatina (SSTR). Hay cinco tipos conocidos de SSTR (SSTR1 -SSTR5), que muestran una afinidad igualmente alta por SST-14. Los agonistas del receptor de somatostatina más investigados, incluyendo la octreotida, muestran una alta selectividad por SSTR2 y SSTR5. De este modo, en una realización preferida, los agonistas del receptor de somatostatina como se indica aquí tienen una función agonista en los receptores de somatostatina, que incluyen SSTR2 y/o SSTR5.
La formulación “simple” más común de octreotida es “Sandostatina” (RTM) de Novartis. Esta es una disolución para inyección subcutánea (s.c), y una dosis de 100 mg alcanza una concentración pico de 5,2 ng/ml a las 0,4 horas después de la inyección. La duración de la acción puede ser de hasta 12 horas, pero la dosificación s.c. se lleva a cabo generalmente cada 8 horas. Evidentemente, la inyección s.c. 3 veces al día durante períodos de meses o años no es un régimen de dosificación ideal.
Para evitar la necesidad de múltiples inyecciones diarias de octreotida, se encuentra disponible una formulación adicional; “Sandostatina LAR”(RTM), nuevamente de Novartis. Esta es una formulación de octreotida en microesferas de ácido poli(láctico-co-glicólico) que, después de la reconstitución en un diluyente acuoso, puede administrarse mediante inyección intramuscular (i.m.).
La pasireotida es un análogo de somatostatina dirigido a múltiples receptores con alta afinidad por los subtipos de receptores de somatostatina sstr1,2,3 y sstr5, que se ha desarrollado para el tratamiento de enfermedades neuroendocrinas. Actualmente se han desarrollado dos formulaciones de pasireotida: una formulación de liberación inmediata para inyección subcutánea (sc) y una formulación de liberación de acción prolongada (LAR). La estructura de la pasireotida es la siguiente:

La pasireotida fue desarrollada inicialmente por Novartis Pharma como tratamiento para la enfermedad/síndrome de Cushing y la acromegalia, pero tiene aplicabilidad potencial en el tratamiento de varias afecciones para las que están indicados los análogos de la somatostatina tal como la octreotida, incluyendo los tumores carcinoides.
Después de una sola dosis subcutánea de pasireotida, los niveles plasmáticos humanos alcanzan un pico rápido, a alrededor de 15 minutos a 1 hora después de la dosificación, con una semivida inicial de 2-3 horas después de ese pico. Aunque la semivida de aclaramiento es mayor para las fases posteriores del declive, está claro que la Cmax/Cave para tal liberación será bastante alta.
Pasireotida LAR es una formulación de pasireotida de acción prolongada que aborda algunos de los problemas anteriores. Sin embargo, este es un sistema basado en micropartículas de polímero con las limitaciones inherentes de dicho sistema, como se conocen en la técnica y se describen aquí anteriormente.
Los tumores carcinoides son tumores intestinales que surgen de células especializadas con funciones paracrinas (células APUD). El tumor primario suele estar en el apéndice, en el que es clínicamente benigno. Los tumores carcinoides intestinales metastásicos secundarios segregan cantidades excesivas de sustancias vasoactivas, incluyendo serotonina, bradicinina, histamina, prostaglandinas, y hormonas polipeptídicas. El resultado clínico es el síndrome carcinoide (un síndrome de rubor cutáneo episódico, cianosis, calambres abdominales, y diarrea en un paciente con valvulopatía cardiaca y, con menor frecuencia, asma y artropatía). Estos tumores pueden crecer en cualquier parte del tubo digestivo (y en los pulmones), con aproximadamente 90% en el apéndice. El resto ocurre en el íleo, el estómago, el colon o el recto. Actualmente, el tratamiento del síndrome carcinoide comienza con la inyección de bolo i.v. seguida de infusión i.v. Cuando se ha establecido un efecto suficiente sobre los síntomas, se inicia el tratamiento con una formulación de depósito de octreotida, formulada en microesferas de ácido poli(láctico-co-glicólico) (PLGA). Sin embargo, durante las dos primeras semanas o más después de la inyección del depósito, se recomiendan inyecciones s.c. diarias con octreotida para compensar la lenta liberación de las esferas de PLGA.
La acromegalia es un trastorno hormonal crónico e insidioso poco común que ocurre cuando la glándula pituitaria produce un exceso de hormona del crecimiento (GH). Afecta con mayor frecuencia a adultos de mediana edad, y puede provocar una muerte prematura.
La diabetes mellitus, la hipertensión y el aumento del riesgo de enfermedad cardiovascular son las consecuencias para la salud más graves de la acromegalia. Además, los pacientes con acromegalia tienen un mayor riesgo de desarrollar pólipos en el colon, que pueden volverse cancerosos. La prevalencia de acromegalia es de aproximadamente 60 casos por millón de habitantes, y la incidencia es de 3,3 casos nuevos por millón por año. La palabra acromegalia proviene de las palabras griegas para “extremidades” (acro) y “grande” (megalia), debido a que uno de los síntomas más comunes de esta afección es el crecimiento anormal de las manos y los pies.
La acromegalia es causada por la sobreproducción prolongada de hormona del crecimiento (GH) y la producción excesiva de factor de crecimiento similar a la insulina-I (IGF-I). En el 98 por ciento de los casos, la sobreproducción de GH es causada por un adenoma hipofisario. La tasa de producción de GH y la agresividad del tumor varían de un paciente a otro. Generalmente, los tumores más agresivos se observan en pacientes más jóvenes.
La acromegalia es una enfermedad grave que a menudo se diagnostica tarde. Las tasas de morbimortalidad son elevadas, en particular, debido a los trastornos cardiovasculares, cerebrovasculares y respiratorios y neoplasias asociados.
El tratamiento actual de la acromegalia generalmente se inicia mediante un período de inyecciones s.c. tres veces al día (dosis diaria óptima = 300 mg de octreotida). Después de la última dosis s.c. y de que se observa que se proporciona un efecto adecuado, entonces se comienza el tratamiento con una formulación de depósito de octreotida formulada en microesferas de ácido poli(láctico-co-glicólico) (PLGA). Los ajustes de dosis se realizan después de la medida de los biomarcadores (HG e IGF-1), normalmente después de unos 3 meses.
La fórmula de liberación lenta de octreotida existente se basa en un tipo de formulación de depósito de polímero que se degrada bien establecido. Típicamente, tales formulaciones se basan en un polímero biodegradable tal como poli(ácido láctico) (PLA) y/o poli(ácido láctico-co-glicólico) (PLGA), y pueden estar en forma de disolución en un disolvente orgánico, un prepolímero mezclado con un iniciador, partículas de polímero encapsuladas, o (como en el caso de octreotida) microesferas de polímero.
En una realización típica, el agonista peptídico del receptor de somatostatina que comprende pasireotida se formulará generalmente como 0,02 a 12% en peso de la formulación total. Los valores típicos serán 0,1 a 10%, preferiblemente 0,2 a 8%, más preferiblemente 0,5 a 6% (por ejemplo, 1 a 3%). Estos niveles se pueden aplicar a todos los aspectos de la invención, cuando el contexto lo permita. Otro intervalo preferido está entre 0,5 y 4% en peso, más preferiblemente 1 -3% en peso, y lo más preferible 1,5-2,5% en peso.
En una realización de la presente invención, la concentración de pasireotida en las formulaciones precursoras de la presente invención es mayor que 4% y la relación de componentes a)/b) es menor que 1. Es decir, el porcentaje en peso del componente a) es menor que el del componente b). En particular, en esta realización de alto contenido de pasireotida, la relación de a) a b) puede estar entre 49:51 y 40:60, preferiblemente 48:52 a 42:58.
En una realización relacionada, el agonista peptídico del receptor de somatostatina se puede formular a un nivel que no se puede alcanzar fácilmente en ausencia del componente de disolvente polar de la mezcla. En tal realización, el contenido de pasireotida es típicamente al menos 0,7%, preferiblemente al menos 1%, más preferiblemente al menos 1,8%, o al menos 2% en peso de formulación. Con la presente invención se pueden conseguir niveles de al menos 3% y al menos 4%, así como niveles de carga de hasta 8, 10 o 12%. Tales composiciones de la presente invención típicamente no solo contienen un nivel muy alto de agonista peptídico del receptor de somatostatina como se indica, sino que además son estables al almacenamiento con baja o nula degradación del agonista del receptor de somatostatina (por ejemplo, menos de 5%) durante períodos considerables, como se indica aquí. Dichos períodos serán generalmente al menos un mes a 25°C, o al menos un mes a 5°C, preferiblemente al menos 3 meses, más preferiblemente al menos 6 meses, lo más preferible 12 a 24 meses a 5°C, o alternativamente a 25°C. Estos grados
de estabilidad son aplicables a todos los aspectos de la invención, cuando el contexto lo permita, y se refieren a la estabilidad tanto del agonista del receptor de somatostatina como del comportamiento de fase de la pre-formulación.
En una realización relacionada, en la situación en la que un agonista peptídico del receptor de somatostatina es altamente soluble en el componente de alcohol, puede ser una ventaja limitar esta solubilidad de este agente. Sin pretender imponer ninguna teoría, se cree que la solubilidad excesiva en este componente de alcohol puede provocar que el alcohol transporte una cantidad significativa de agonista del receptor de somatostatina fuera de la composición de depósito a medida que se forma in vivo. Por lo tanto, en una realización de la presente invención, el disolvente polar se usa para controlar la solubilidad del agonista del receptor de somatostatina en la pre-formulación para ayudar a controlar el perfil de liberación.
El agonista peptídico del receptor de somatostatina comprende pasireotida; por tanto, las dosis adecuadas para su inclusión en la formulación, y de este modo el volumen de formulación utilizado, dependerán de la velocidad de liberación (controlada, por ejemplo, por el tipo de disolvente y la cantidad utilizada, el contenido de antioxidantes, etc.) y de la duración de la liberación, así como del nivel terapéutico deseado, la actividad del agente específico, y la velocidad de aclaramiento del agente activo particular escogido. Normalmente, una cantidad de alrededor de 0,05 a 40 mg por semana de duración del depósito, preferiblemente 0,1 a 20 mg por semana de duración (por ejemplo, 1 a 5 mg por semana) durante una duración de 1 a 24 semanas, preferiblemente de 2 a 16 (por ejemplo, 3, 4, 8, 10 o 12) semanas. En una realización alternativa, la pre-formulación puede formularse para dosificación semanal (por ejemplo, cada 7±1 días). Una dosis total de 0,05 a 250 mg por dosis sería adecuada para proporcionar un nivel terapéutico entre 7 y 168 días. Esta será preferiblemente 0,1 a 200 mg, por ejemplo 0,2 a 150 mg, 0,1 a 100 mg, 20 a 160 mg, etc. Evidentemente, la estabilidad del agente activo y los efectos sobre la velocidad de liberación significarán que la carga a duración puede no ser una relación lineal. Un depósito administrado cada 30 días podría tener, por ejemplo, 0,2 a 20 mg, o un depósito de 90 días podría tener 30 a 60 mg de agonista del receptor de somatostatina.
Evidentemente también, la semivida biológica del agente activo específico será particularmente importante. La semivida de la somatostatina es inferior a 5 minutos, por lo que para una liberación sostenida se necesitará una cantidad relativamente grande (por ejemplo, hacia el extremo superior del intervalo). Para un análogo tal como la pasireotida, con una semivida mucho más larga (2-3 horas como mínimo), la cantidad necesaria será evidentemente menor. Los expertos en la técnica establecerán fácilmente los niveles apropiados para los principios activos específicos con referencia al nivel terapéutico conocido, la duración deseada de la acción, y el volumen que se va a inyectar. Un buen cálculo básico sería multiplicar una dosis diaria típica del agente activo por el número de días de duración del depósito. Después, la formulación puede ensayarse para determinar la linealidad de liberación, y puede ajustarse según sea apropiado.
En una realización muy preferida, el aspecto de la matriz lipídica es PC de soja o PC de “alta pureza” (tal como DOPC), GDO, etanol, y agua/propilenglicol o mezclas de los mismos, y el agonista peptídico del receptor de somatostatina comprende pasireotida. Como se indicó anteriormente, las cantidades apropiadas de cada componente adecuadas para la combinación son las cantidades indicadas aquí para los componentes individuales, en cualquier combinación.
En una realización preferida, GLP-1, análogos de GLP-1 y agonistas del receptor de GLP-1 y/o antagonistas del receptor de GLP-1 no están presentes en las formulaciones precursoras de la invención.
Componente opcional f) - Antioxidante
El componente f) es un antioxidante. Lo más preferible, se selecciona de ácido ascórbico, palmitato de ascorbilo, ácido etilendiaminotetraacético (EDTA) y sales del mismo, y ácido cítrico.
En todos los aspectos de la invención, el componente f) está típicamente presente en una relación en peso de antioxidante a agonista peptídico del receptor de somatostatina de 1:50 a 1:6000, preferiblemente 1:100 a 1:1300, y lo más preferible de 1:150 a 1:1250. Dado que los antioxidantes típicos tienen un peso molecular más bajo que los agonistas peptídicos del receptor de somatostatina, la proporción en peso de antioxidante puede ser relativamente pequeña. Por ejemplo, con un antioxidante de pequeño peso molecular (por ejemplo, menos de 500 uma), 0,0001 a 0,5% de la composición puede ser antioxidante, preferiblemente 0,0005 a 0,2%, más preferiblemente 0,0008 a 0,1%, por ejemplo 0,001 a 0,02%.
Desafortunadamente, muchos antioxidantes comunes no son muy compatibles con los sistemas lipídicos. De hecho, los presentes inventores han establecido previamente que algunos antioxidantes comúnmente usados en sistemas previos pueden causar una mayor degradación de los agentes activos en un sistema lipídico. Esto se aplica particularmente a los agentes activos peptídicos. Por lo tanto, los presentes inventores han analizado una variedad de compuestos y clases de antioxidantes potenciales para uso con sistemas de matriz basados en lípidos, y han descubierto sorprendentemente que una clase particular de antioxidantes es excepcionalmente adecuada para uso en estos sistemas.
El componente antioxidante se incluye generalmente en el intervalo 0,0001 a 0,5% en peso de la pre-formulación total. Se prefiere particularmente alrededor de 0,0005 a 0,015% de antioxidante (particularmente EDTA), especialmente en combinación con los otros componentes e intervalos preferidos indicados aquí anteriormente y más abajo.
Los datos de estabilidad que utilizan varios antioxidantes diferentes demuestran que los antioxidantes de EDTA son sorprendentemente más eficientes que otros antioxidantes suprimiendo la degradación oxidativa de los agentes bioactivos. El EDTA como antioxidante también puede mostrar un efecto sinérgico en combinación con los antioxidantes de la presente invención, para mantener la estabilidad química y física del agente activo peptídico y la pre-formulación completa. EDTA tiene un efecto estabilizador sobre el agente activo.
Por “estabilizador” se indica un aumento en la estabilidad física y química del agonista del receptor de somatostatina disuelto o disperso. Un aumento de la estabilidad puede demostrarse mediante la estabilidad química y/o física de un agonista peptídico del receptor de somatostatina en una formulación lipídica durante un período mayor del que se observaría en ausencia de un antioxidante. Esto se evaluaría preferiblemente en condiciones de almacenamiento típico, tales como 2-82C, 25°C y/o temperatura ambiente. Esto se describe con más detalle aquí a continuación.
En una realización preferida de la invención, los antioxidantes se excluyen de las pre-formulaciones.
Componentes adicionales opcionales
En una realización particularmente preferida de la presente invención, las composiciones (preformulaciones y depósitos resultantes) no incluyen agentes de fragmentación, tales como agente de fragmentación de poli(óxido de etileno) o poli(etilenglicol) (PEG), por ejemplo un lípido y/o tensioactivo injertado con PEG.
Por ejemplo, las composiciones preferiblemente no incluyen agentes de fragmentación tales como Polisorbato 80 (P80, monooleato de polioxietileno (20) sorbitán), u otros Polisorbatos (por ejemplo, Polisorbato 20), fosfolípidos PEGilados (PEG-lípidos tales como DSPE-PEG(2000), DSPE-PEG(5000), DOPE-PEG(2000) y DOPE-PEG(5000)), Solutol HS 15, ácidos grasos PEGilados (por ejemplo, PEG-oleato), copolímeros de bloques tales como Pluronic® F127 y Pluronic® F68, derivados etoxilados de aceite de ricino (por ejemplo, Chremophores), ésteres de ácido graso de glicerilo PEGilado (tal como TMGO-15 de Nikko Chemicals) y tocoferoles PEGilados (tal como succinato de d-alfa tocoferil poli(etilenglicol) 1000, conocido como vitamina E TPGS de Eastman.
Los formatos de dosis única deben permanecer estables y potentes durante el almacenamiento antes del uso, pero son desechables después de un solo uso. En una realización, un formato de dosis única es estable en condiciones de refrigeración (por ejemplo, 0-5 o 2-8°C) durante al menos 12 meses. Además, tal pre-formulación puede ser estable a temperatura ambiente (por ejemplo, 25°C) durante al menos 12 meses. Los formatos de dosis múltiples no solo deben permanecer estables y potentes en almacenamiento antes del uso, sino que también deben permanecer estables, potentes y relativamente libres de bacterias (y en particular esencialmente libres de crecimiento bacteriano) durante el período de administración del régimen de uso de dosis múltiples después del primer uso en el que se ha comprometido un sello. Por esta razón, los formatos de dosis múltiple a menudo requieren un agente antimicrobiano o microbiano-estático, por ejemplo agente bacteriostático, conservante.
Sin embargo, la producción de preparaciones farmacéuticas conservadas que contienen proteínas o agentes activos peptídicos a menudo ha resultado difícil, ya que cuando se utilizan conservantes, estos dan lugar a problemas de estabilidad. A menudo, las proteínas se inactivan y se forman agregados, lo que a veces puede dar lugar a intolerancia en el lugar de la inyección o inmunogenicidad al agente activo. Esto puede agravarse aún más con excipientes o componentes de formulación adicionales.
En un aspecto, cada una de las realizaciones aquí puede contener opcionalmente un agente antimicrobiano o microbiano-estático, que incluye agentes bacteriostáticos y conservantes. Dichos agentes incluyen cloruro de benzalconio, m-cresol, alcohol bencílico u otros conservantes fenólicos. Se pueden usar concentraciones típicas como las conocidas en la técnica.
Los componentes adicionales aparte de los mencionados como componentes a) a f), cuando estén presentes, estarán presentes preferiblemente en una cantidad de 0 a 5% (por ejemplo, 0,01% a 5%) en peso, preferiblemente no más de 2% en peso, y más preferiblemente no más de 1% en peso.
En una realización, los componentes a) y b) (que tienen en cuenta cualquier impureza inherente a la naturaleza de estos componentes) constituyen al menos el 95% de los componentes lipídicos de la composición. Preferiblemente, al menos el 99% del contenido total de lípidos de la pre-formulación consiste en los componentes a) y b). Preferiblemente, el componente lipídico de la pre-formulación consiste esencialmente en los componentes a) y b).
Administración
Las pre-formulaciones de la presente invención se formulan generalmente para administrarse por vía parenteral. Esta administración generalmente no será un método intravascular, pero preferiblemente será subcutánea (s.c.), intracavitaria o intramuscular (i.m.). Típicamente, la administración será por inyección, término que se usa aquí para indicar cualquier método en el que la formulación se hace pasar a través de la piel, tal como con aguja, catéter o inyector sin aguja (libre aguja).
La administración parenteral preferida es por vía intramuscular o inyección s.c., más preferiblemente por inyección s.c. Una característica importante de la composición de la invención es que puede administrarse tanto por vía i.m. como
por s.c. y otras vías sin toxicidad o efectos locales significativos. También es adecuada para administración intracavital. La inyección s.c. tiene la ventaja de ser menos profunda y menos dolorosa para el sujeto que la inyección i.m. (profunda) utilizada para algunos depósitos actuales, y es técnicamente más adecuada en el presente caso, ya que combina la facilidad de inyección con un bajo riesgo de efectos secundarios locales. Es una observación sorprendente de los presentes inventores que las formulaciones proporcionan una liberación sostenida del agente activo durante un período de tiempo predecible mediante inyección tanto subcutánea como intramuscular. Por tanto, esto permite variar ampliamente el sitio de inyección, y permite administrar la dosis sin una consideración detallada de la profundidad del tejido en el sitio de inyección.
Las pre-formulaciones de lípidos preferidas de la presente invención proporcionan composiciones de depósito cristalinas líquidas no laminares tras la exposición a fluidos acuosos, especialmente in vivo. Como se usa aquí, la expresión “no laminar” se usa para indicar una fase cristalina líquida normal o más preferiblemente invertida (tal como una fase cúbica o hexagonal invertida), o la fase L3, o cualquier combinación de las mismas. La expresión cristalina líquida indica todas las fases cristalinas líquidas hexagonales, todas las cúbicas, y/o todas sus mezclas. Hexagonal, como se usa aquí, indica hexagonal “normal” o “invertido” (preferiblemente invertido), y “cúbico” indica cualquier fase cristalina líquida cúbica a menos que se especifique lo contrario. El lector experto no tendrá dificultad en identificar aquellas composiciones que tengan un comportamiento de fase apropiado por referencia a la descripción y los ejemplos proporcionados aquí, y al documento WO2005/117830, pero el área de composición más favorecida para el comportamiento de fase es cuando la relación de componentes a:b está en la región de 40:60 a 54:46. Las relaciones de alrededor de 50:50 (por ejemplo, ±2) son muy preferidas para la mayoría de las formulaciones (aunque se indican aquí ciertas excepciones), lo más preferible alrededor de 50:50.
Es importante apreciar que las pre-formulaciones de la presente invención son de baja viscosidad. Como resultado, estas pre-formulaciones no deben estar en ninguna fase cristalina líquida a granel, ya que todas las fases cristalinas líquidas tienen una viscosidad significativamente más alta que la que podría administrarse con una jeringa o un dispensador inyectable similar. De este modo, las pre-formulaciones de la presente invención estarán en un estado cristalino no líquido, tal como una disolución, fase L2 o L3, particularmente disolución o L2. La fase L2, tal como se usa aquí, es preferiblemente una fase L2 “hinchada” que contiene más de 5% en peso, preferiblemente más de 7%, y lo más preferible más de 9% de disolvente orgánico monoalcohólico (componente c) que tiene un efecto reductor de la viscosidad. Las pre-formulaciones de la invención que están en fase L2 forman un conjunto preferido de pre formulaciones, y éstas generalmente contendrán al menos 2% de agua como disolvente polar.
Como se usa aquí, la expresión “mezcla de baja viscosidad” se usa para indicar una mezcla que puede administrarse fácilmente a un sujeto, y en particular puede administrarse fácilmente por medio de un montaje estándar de jeringa y aguja. Esto puede estar indicado, por ejemplo, por la capacidad de dispensarse desde una jeringa desechable de 1 ml a través de una aguja de pequeño calibre. Preferiblemente, las mezclas de baja viscosidad se pueden dispensar a través de una aguja de calibre 19, preferiblemente menor que calibre 19, más preferiblemente una aguja de calibre 23 (o lo más preferible incluso, de calibre 27) mediante presión manual. En una realización particularmente preferida, la mezcla de baja viscosidad debería ser una mezcla capaz de pasar a través de una membrana de filtración estéril estándar, tal como un filtro de jeringa de 0,22 pm. Un intervalo típico de viscosidades adecuadas sería 1 a 1000 mPas, más preferiblemente 10 a 750 mPas, y lo más preferible 25 a 500 mPas a 20°C.
Se ha observado que mediante la adición de pequeñas cantidades de disolvente monoalcohólico orgánico de baja viscosidad, como se indica aquí, se puede proporcionar un cambio de viscosidad muy significativo. Por ejemplo, la adición de solo 5% de disolvente a una mezcla de lípidos puede reducir la viscosidad 100 veces, y la adición de 10% puede reducir la viscosidad hasta 10.000 veces. Para conseguir este efecto sinérgico no lineal en la reducción de la viscosidad, es importante que se emplee un disolvente de viscosidad apropiadamente baja y polaridad adecuada. Dichos disolventes incluyen los descritos a continuación aquí. Las mezclas de baja viscosidad preferidas incluyen disoluciones moleculares, que incluyen dispersiones del agonista peptídico del receptor de somatostatina en una disolución molecular de los otros componentes.
Tras la administración, las pre-formulaciones a base de lípidos preferidas de la presente invención experimentan una transición de estructura de fase desde una mezcla de baja viscosidad a una composición de depósito de alta viscosidad (generalmente adherente al tejido). Generalmente esto será una transición de una mezcla molecular, fase hinchada L2 y/o L3, a una o más fases cristalinas líquidas (de alta viscosidad) tales como fases cristalinas líquidas hexagonales o cúbicas invertidas, o mezclas de las mismas. También pueden tener lugar otras transiciones de fase después de la administración. Obviamente, la transición de fase completa no es necesaria para el funcionamiento de la invención, pero al menos una capa superficial de la mezcla administrada formará una estructura cristalina líquida. Generalmente, esta transición será rápida para al menos la región de la superficie de la formulación administrada (esa parte en contacto directo con el aire, superficies corporales y/o fluidos corporales). Esto será más preferiblemente de unos pocos segundos o minutos (por ejemplo, desde 1 segundo hasta 30 minutos, preferiblemente hasta 10 minutos, más preferiblemente 5 minutos o menos). El resto de la composición puede cambiar de fase a una fase cristalina líquida más lentamente por difusión y/o a medida que se dispersa la región superficial.
Sin pretender imponer ninguna teoría, se cree que tras la exposición a un exceso de fluido acuoso, las pre formulaciones de la invención pierden parte o todo el disolvente orgánico incluido en ellas (por ejemplo, por difusión) y absorben el fluido acuoso del entorno corporal (por ejemplo, el entorno in vivo). Para las pre-formulaciones de lípidos,
al menos una parte de la formulación genera preferiblemente una estructura de fase no laminar, particularmente cristalina líquida. En la mayoría de los casos, estas estructuras no laminares son muy viscosas y no se disuelven o dispersan fácilmente en el entorno in vivo. El resultado es un “depósito” monolítico generado in vivo con solo un área limitada de exposición a los fluidos corporales. Además, debido a que la estructura no laminar tiene grandes regiones polares, apolares y de frontera, el depósito de lípidos es muy eficaz para solubilizar y estabilizar agentes activos tales como péptidos, y protegerlos de los mecanismos de degradación. A medida que la composición de depósito formada a partir de la pre-formulación se degrada gradualmente durante un período de días, semanas o meses, el agente activo se libera gradualmente y/o se difunde fuera de la composición. Dado que el entorno dentro de la composición de depósito está relativamente protegido, las pre-formulaciones de la invención son muy adecuadas para agentes activos con una semivida biológica relativamente corta (véase más arriba).
Mediante la incorporación de al menos 5% (por ejemplo, al menos 10%) de un disolvente polar (especialmente al menos 5% de agua y/o PG) en las pre-formulaciones, se cree que la velocidad de transición de fase a una fase no laminar (por ejemplo, cristalina líquida) en la superficie de la pre-formulación inyectada se puede mejorar en comparación con las composiciones que contienen disolventes orgánicos en ausencia sustancial de agua. De este modo, se mejora el rendimiento del depósito resultante, y se logra un control adicional sobre la liberación del agente activo.
Los sistemas de depósito formados por las formulaciones de la presente invención son muy eficaces para proteger al agente activo de la degradación, y de este modo permiten un período de liberación prolongado. De este modo, las formulaciones de la invención pueden proporcionar depósitos in vivo de agonistas peptídicos del receptor de somatostatina que requieren la administración solo una vez cada 5 a 90 días, preferiblemente 5 a 60 días, más preferiblemente 6 a 32. Evidentemente, es deseable un período de liberación estable más largo para la comodidad y el cumplimiento del paciente, así como para exigir menos tiempo de profesionales sanitarios si la composición no se va a autoadministrar. Cuando la composición se va a autoadministrar, el cumplimiento del paciente puede ser ayudado por una administración semanal (por ejemplo, cada 7 días, opcionalmente ±1 día) o mensual (por ejemplo, cada 28 o 30 días (opcionalmente ±7 días) de modo que la necesidad de administrar no se olvida.
Una ventaja considerable de los precursores de depósito de la presente invención es que son fases homogéneas estables. Es decir, pueden almacenarse durante períodos considerables (preferiblemente al menos 6 meses) a temperatura ambiente o del frigorífico, sin separación de fases. Además de proporcionar un almacenamiento ventajoso y una administración fácil, esto permite seleccionar la dosis de agonista peptídico del receptor de somatostatina (por ejemplo, pasireotida) en referencia a la especie, edad, sexo, peso, y/o condición física del sujeto individual, por medio de la inyección de un volumen seleccionado.
De este modo, la presente invención proporciona métodos que comprenden la selección de una cantidad de dosificación específica para un individuo, particularmente por peso del sujeto. El medio para esta selección de dosis es la elección del volumen de administración.
En un aspecto preferido, la presente invención proporciona una pre-formulación que comprende los componentes a), b), c), d), f), y al menos un agonista peptídico del receptor de somatostatina que comprende pasireotida como se indica aquí. Las cantidades de estos componentes estarán típicamente en el intervalo 36-44% de a), 36-44% de b), 3-18% de c) y 5-18% de d) (preferiblemente incluyendo al menos 2% de agua), estando presente el agonista peptídico del receptor de somatostatina que comprende pasireotida en una cantidad de 1% a 8%, en el que la relación de a:b está en el intervalo de 40:60 a 54:46.
Típicamente, el componente f) está presente en una relación molar de antioxidante a agonista peptídico del receptor de somatostatina de 1:50 a 1:6000, preferiblemente 1:100 a 1:1300, y lo más preferible 1:150 a 1:1250. Dado que los antioxidantes típicos tienen un peso molecular más bajo que el agonista peptídico del receptor de somatostatina (por ejemplo, un análogo de somatostatina, por ejemplo octreotida), la proporción en peso de antioxidante puede ser relativamente pequeña. Por ejemplo, con un antioxidante de pequeño peso molecular (por ejemplo, menos de 500 uma), el 0,001 a 5% de la composición puede ser antioxidante, preferiblemente 0,002 a 2%, más preferiblemente 0,002 a 0,15%, por ejemplo 0,002 a 0,015%.
Las pre-formulaciones de la presente invención son muy ventajosas por cuanto son estables al almacenamiento prolongado en su forma final “lista para la administración”. Como resultado, pueden ser fácilmente suministradas para su administración por profesionales sanitarios o por pacientes o sus cuidadores, quienes no necesitan ser profesionales sanitarios completamente capacitados y pueden no tener la experiencia o las habilidades para realizar preparaciones complejas. Esto es particularmente importante en enfermedades de larga duración y efectos lentos tal como la diabetes.
La pre-formulación de la invención preferiblemente excluirá cualquier GLP-1, análogos de GLP-1, y agonistas y/o antagonistas del receptor de GLP-1. Las pre-formulaciones de la invención preferiblemente excluirán las siguientes pre-formulaciones:

en la que EtOH es etanol, PC es fosfatidilcolina de soja LIPOID S100 o fosfatidilcolina de huevo LIPOID E 80 (marcada con *), y GDO es dioleato de glicerol que tiene la calidad (según AC) de la siguiente manera:
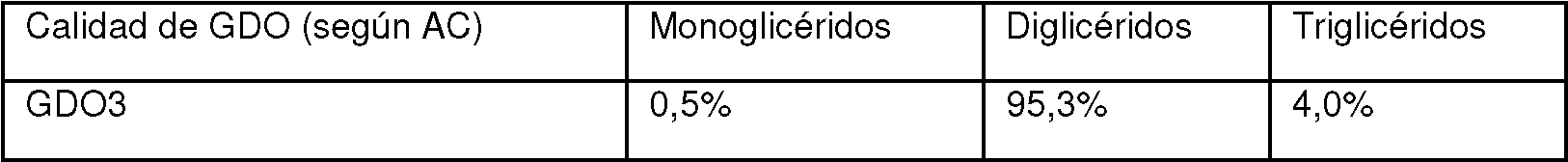
Dispositivos
En un aspecto aún adicional, la presente invención proporciona un dispositivo de administración desechable (que también debe incluir un componente del dispositivo) precargado con una dosis medida de una pre-formulación de la presente invención. Un dispositivo de este tipo contendrá típicamente una dosis única lista para la administración, y generalmente estará envasada de forma estéril de manera que la composición se almacene dentro del dispositivo hasta la administración. Los dispositivos adecuados incluyen cartuchos, y jeringas y cilindros de jeringa, ya sea con agujas integradas o con accesorios estándar (por ejemplo, luer) adaptados para aceptar una aguja desechable adecuada. Los dispositivos igualmente apropiados incluyen un inyector sin aguja, un autoinyector multiusos o de un solo uso combinado con una jeringa precargada, un cartucho, opcionalmente combinado con un dispositivo de pluma multiusos, o un vial. Evidentemente, tales jeringas y cartuchos precargados pueden ser para cualquier dispositivo de inyección apropiado, tal como un inyector de uso múltiple o de un solo uso o una unidad de inyección sin aguja.
Los dispositivos de la invención pueden contener preferiblemente la pre-formulación de la invención que administra una dosis en el intervalo de 5 a 150 mg/ml, preferiblemente 10 a 100 mg/ml, lo más preferible 10 a 70 o 10 a 90 mg/ml, por ejemplo 20 a 60 o 20 a 80 mg/ml, tal como 20 a 60 o 30 a 60 mg/ml.
En una realización aplicable a todos los aspectos de la invención, los dispositivos de la invención pueden contener una dosis única de 1 a 200 mg, por ejemplo 1 a 150 mg (por ejemplo, 1 a 120 mg) de agonista peptídico del receptor de somatostatina que comprende pasireotida, preferiblemente pamoato de pasireotida.
Los dispositivos de la invención pueden contener un agonista peptídico del receptor de somatostatina que comprende pasireotida, preferiblemente pamoato de pasireotida, a alrededor de 0,1 a 6 mg (por ejemplo, 0,2 a 4 mg) por día entre administraciones programadas, por ejemplo alrededor de 0,6 (por ejemplo, 0,6 a 3) mg por día, particularmente 1 a 2 mg/día.
Los dispositivos de la invención pueden contener un volumen total para la administración de no más de 5 ml, por ejemplo no más de 2 ml, tal como aproximadamente 1,5 ml.
Características y combinaciones preferidas
En combinación con las características y características preferidas aquí indicadas, las pre-formulaciones de la invención pueden tener una o más de las siguientes características preferidas, independientemente o en combinación:
todas las proporciones indicadas aquí se pueden variar opcionalmente en hasta 10% de la cantidad especificada, opcional y preferiblemente en hasta 5%;
el componente a) comprende, consiste esencialmente en, o consiste preferiblemente en GDO;
el componente b) comprende, consiste esencialmente en, o consiste preferiblemente en PC de soja y/o “PC de alta pureza”, tal como DOPC;
el componente c) comprende, consiste esencialmente en, o consiste preferiblemente en un alcohol de 1, 2, 3 o 4 carbonos, preferiblemente isopropanol, o más preferiblemente etanol;
el componente d) comprende, consiste esencialmente en, o consiste preferiblemente en un disolvente polar tal como agua, propilenglicol, o mezclas de los mismos;
el componente f) comprende, consiste esencialmente en, o consiste preferiblemente en ácido ascórbico, palmitato de ascorbilo, ácido etilendiaminotetraacético (EDTA), y/o ácido cítrico;
la pre-formulación contiene al menos un agonista peptídico del receptor de somatostatina que comprende pasireotida, preferiblemente pamoato de pasireotida;
la pre-formulación tiene una viscosidad baja como se indica aquí.
La pre-formulación comprende formas de una fase cristalina líquida como se indica aquí tras la administración in vivo. La pre-formulación genera un depósito tras la administración in vivo, depósito el cual libera al menos un agonista del receptor de somatostatina a un nivel terapéutico durante un período de al menos 7 días, preferiblemente al menos 28 días, más preferiblemente al menos 60 días.
La pre-formulación tiene una carga más alta de agonista peptídico del receptor de somatostatina que comprende pasireotida que la que es estable en la misma formulación en ausencia del componente d).
La pre-formulación tiene una carga más alta de agonista peptídico del receptor de somatostatina que comprende pasireotida que la que se puede obtener mediante el equilibrio a 25°C de la misma formulación en ausencia del componente d).
En combinación con las características y características preferidas indicadas aquí, la pre-formulación para uso en el método o métodos de tratamiento de la presente invención puede tener una o más de las siguientes características preferidas, independientemente o en combinación:
el método comprende la administración de al menos una formulación con una o más características preferidas como se indicó anteriormente;
el método comprende la administración de al menos una formulación como se indica aquí mediante inyección i.m., s.c., o preferiblemente s.c. profunda;
el método comprende la administración por medio de un dispositivo de administración precargado como se indica aquí;
el método comprende la administración a través de una aguja de calibre no mayor de 20, preferiblemente calibre menor de 20, y lo más preferible calibre 23 o menor;
el método comprende una sola administración cada 20 a 100 días, preferiblemente 28 a 60 días (por ejemplo 30 45 días).
En combinación con las características y características preferidas indicadas aquí, los dispositivos precargados de la invención pueden tener una o más de las siguientes características preferidas, independientemente o en combinación: contienen una formulación preferida como se indica aquí;
comprenden una aguja de calibre menor de 20, preferiblemente calibre no mayor de 23;
contienen una dosis única de 1 a 300 mg de agonista peptídico del receptor de somatostatina que comprende pasireotida, preferiblemente 1 a 200 mg, más preferiblemente 5-150 mg, por ejemplo 10-100 mg, lo más preferible 20-70 mg, y especialmente de forma preferible 30-60 mg.
Contienen una mezcla homogénea de una composición de la invención en forma lista para inyectar.
Contienen una formulación de los componentes a) a c) para combinación con un agonista peptídico del receptor de somatostatina que comprende pasireotida, con lo que se forma una pre-formulación de la invención.
Contienen un agonista peptídico del receptor de somatostatina que comprende pasireotida para la combinación con una formulación de los componentes a) a d) y opcionalmente f), con lo que se forma una pre-formulación de la invención.
Contienen un volumen total para la administración de no más de 5 ml, preferiblemente no más de 3 ml, por ejemplo no más de 2 ml, más preferiblemente no más de 1,5 ml.
La invención se ilustrará ahora adicionalmente con referencia a los siguientes Ejemplos no limitativos y las Figuras adjuntas.
EJEMPLOS
Materiales
SPC Fosfatidilcolina de soja (Lipid S100) - Lipoid
GDO Dioleato de glicerol (Rylo DG19 Pharma) - Danisco
DOPC Dioleoil fosfatidilcolina - NOF
EtOH Etanol - Solveco
PG Propilenglicol - Fischer
WFI Agua para preparaciones inyectables - Apoteket
EDTA Ácido etilendiaminotetraacético, sal disódica - Sigma Aldrich
SOM230(pm) SOM230 pamoato (pamoato de pasireotida) - Novartis Pharma
SOM230(Ac) SOM230 acetato (acetato de pasireotida) - Novartis Pharma
Ejemplo 1: Cribado de solubilidad
Las formulaciones de mezcla de lípidos de placebo así como las formulaciones del lípido respectivo individual solo se prepararon en viales 10R según la Tabla 1. El tamaño de las muestras fue 6 g. Se prepararon muestras de SOM230 (pamoato de pasireotida) del tipo de formulación respectivo con una carga de fármaco del 3% en peso (corregido por pureza y contenido de péptido) en viales 2R, excepto para el tipo de formulación 15, en la que solo se evaluó el 1% en peso de S230. Se dejó que las muestras se equilibraran a temperatura ambiente; las muestras con los tipos de formulación 1-8 en rotación de un extremo a otro, y las muestras con los tipos de formulación 9-12 en agitación magnética. El diagrama de flujo de la Figura 1 describe el procedimiento de preparación de las muestras. El cribado cubrió las siguientes variables de formulación:
• Relación ponderal de lípidos (relación ponderal SPC/GDO)
• Naturaleza y concentración del codisolvente
• Formulaciones de un solo lípido
• Disolvente único (PG solo)
Tabla 1. Composiciones de formulaciones de lípidos de placebo (% en peso)
utilizadas para el cribado de solubilidad.


Las muestras se estudiaron mediante inspección visual, y se anotó el aspecto. Se añadió polvo de fármaco SOM230 adicional en etapas de 1% en peso (corregido por pureza y contenido de péptido según CoA) a cualquier muestra homogénea y transparente con la excepción del tipo de formulación 15. El mezclamiento a temperatura ambiente y la observación visual continuaron después de la adición de más polvo de fármaco SOM230.
Los resultados del cribado de solubilidad se resumen en la Tabla 2. Se concluye que, para algunos tipos de formulación, se puede alcanzar una concentración de dosis de hasta al menos 10% en peso o aproximadamente 100 mg de base libre de SOM230/ml. Además, PG parece ser un disolvente relativamente bueno para SOM230 (> 1% en peso) en comparación con la escasa solubilidad en EtOH (<< 1% en peso).
Se lograron mayores cargas de fármaco de SOM230, hasta 10% en peso (o aproximadamente 100 mg/ml (corregido)), utilizando una combinación de codisolvente de EtOH/PG a concentraciones de 10% en peso cada una. Las formulaciones que comprenden EtOH solo como codisolvente no fueron tan eficaces como la combinación de EtOH/PG o EtOH/WFI para la carga de SOM230. Las formulaciones que comprenden solo el excipiente de lípido respectivo individual (tipos de formulación 13 y 14) mostraron una menor capacidad de carga de fármaco (< 3% en peso) en comparación con las mezclas de lípidos con una composición de disolvente equivalente, lo que indica fuertes efectos sinérgicos de mejora de la solubilidad al combinar los componentes de la formulación de la invención.
Tabla 2. Resultados del cribado de solubilidad. Para conocer las composiciones de la formulación, véase la Tabla 1.


Los resultados del análisis de solubilidad se resumen a continuación:
• Se encontró que múltiples tipos de formulación permiten niveles de carga de fármaco de 30-60 mg/ml
• El aumento de los niveles de codisolvente y la combinación de EtOH y PG o EtOH y agua aumentaron la solubilidad de SOM230
• Se verificó una carga de fármaco de (al menos) 10% en peso (aproximadamente 100 mg/ml) para al menos una variante de formulación
• La combinación de SPC y GDO aumentó la solubilidad del SOM230 de forma sinérgica en comparación con las mezclas de lípidos individuales
• PG pareció ser un disolvente relativamente bueno para SOM230
Ejemplo 2 - Inyectabilidad, densidad y viscosidad
Se prepararon y usaron formulaciones según la Tabla 3 para evaluar la inyectabilidad, densidad y viscosidad. Las formulaciones de lípidos/SOM230 con cargas de fármaco de 3 y 6% en peso, respectivamente, se prepararon en viales de vidrio para inyección de 15R.
Tabla 3. Composiciones de formulaciones de lípidos/SOM230 (% en peso).

El procedimiento de preparación de las muestras fue como se describe en la Figura 1. Se añadió una barra de agitación magnética a las muestras (tamaño de muestra 6 g), seguido de agitación magnética a temperatura ambiente. Las muestras se estudiaron mediante inspección visual, y se anotó el tiempo aproximado para completar la disolución.
Las formulaciones con 30 mg/g fueron transparentes y homogéneas en 48 h. Las muestras con 60 mg/g fueron homogéneas y transparentes en 3 días. No se hicieron esfuerzos especiales (como aumentar la velocidad de agitación) para acelerar los tiempos de disolución para estas preparaciones.
Inyectabilidad (caudales)
La inyectabilidad o el caudal de cada formulación se evaluó según el siguiente método: se aplica una fuerza constante a la configuración de jeringa seleccionada llena con la formulación respectiva. A continuación, se realiza la inyección a temperatura ambiente en un vial vacío, y se anota el peso de la formulación inyectada, y se mide el tiempo para completar la inyección. El volumen de inyección por ensayo de inyección fue aprox. 0,4-0,6 ml, y se realizaron ensayos por duplicado. La fuerza constante aplicada fue 20 N. El formato de jeringa y aguja utilizado para los ensayos de inyectabilidad se da en la Tabla 4.
Tabla 4. Configuración de jeringa y aguja.
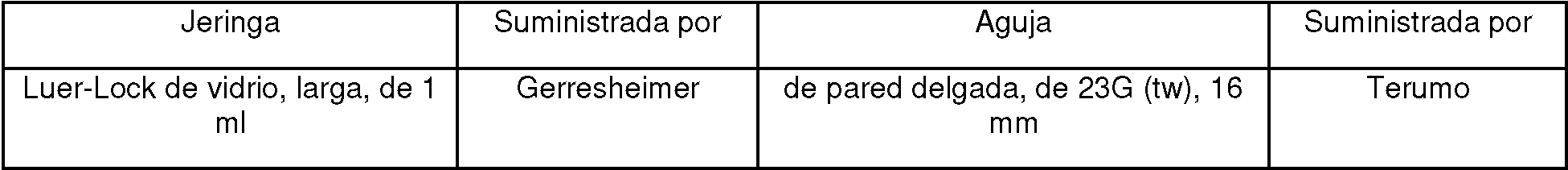
Los tapones utilizados fueron de West (4432/50 grey B240 Westar®) con vástago de émbolo 55103 suministrado por Fresenius Kabi (FKA).
Los resultados de la inyectabilidad se resumen en la Figura 2. La inyectabilidad se da como segundos por ml (inverso del caudal). Los valores se convirtieron de segundos por gramo a segundos por ml mediante el uso de valores de densidad de la formulación (véase más abajo). El tiempo para la inyección, usando la fuerza constante de 20 N, varió entre aproximadamente 8-30 segundos dependiendo de la formulación.
Densidad
La densidad de cada formulación se determinó usando un densímetro Anton Paar DMA 4500 M (1360) a 20°C. Se realizaron ensayos individuales en cada formulación. Los resultados se presentan en la Tabla 5.
Tabla 5. Medidas de densidad en formulaciones de lípidos/SOM230. Las composiciones de formulación se proporcionan en la Tabla 3.

Viscosidad
La viscosidad se midió usando un instrumento Bohlin Visco 88 BV (cilindro interior giratorio, cilindro exterior estacionario) a tres ajustes de velocidad (tres velocidades de cizallamiento). Se realizaron ensayos individuales para cada formulación.
Las medidas se realizaron a temperatura ambiente (ambiente). Los resultados se muestran en la Tabla 6 como viscosidad media para los diferentes ajustes de velocidad. No se pudo observar ninguna diferencia en los valores de viscosidad (dentro de la variación experimental) entre las diferentes velocidades de cizallamiento que indican el comportamiento newtoniano.
Tabla 6. Medidas de viscosidad en formulaciones de lípidos/SOM230.
Las composiciones de formulación se proporcionan en la Tabla 3.
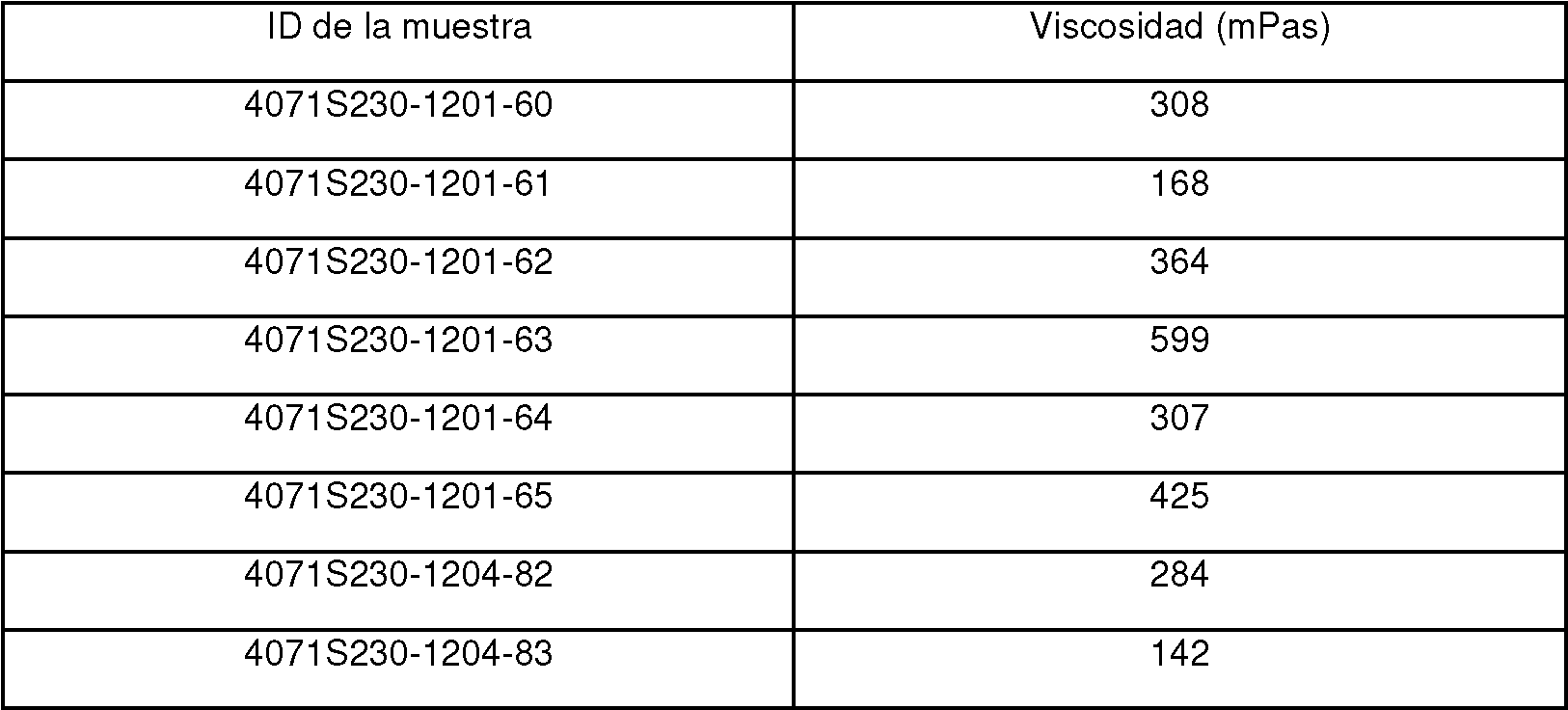
Cabe señalar que al duplicar la carga de fármaco de SOM230 de aproximadamente 30 a 60 mg/ml, la viscosidad casi se duplicó para las formulaciones con EtOH/PG, mientras que el aumento de viscosidad para las correspondientes formulaciones de EtOH/WFI fue solo alrededor de 20%. El vínculo entre la inyectabilidad y la viscosidad se ilustra en la Figura 3. Como se esperaba para este tipo de formulaciones, la inyectabilidad (calculada como tiempo para una inyección de 1 ml) es casi linealmente proporcional a la viscosidad.
Ejemplo 3 - Estudios comparativos de formulación con acetato de SOM230 e hidrocloruro de SOM230
Preparación de hidrocloruro de SOM230
La sal de hidrocloruro de SOM230 se preparó a partir de acetato de SOM230 usando un procedimiento de intercambio iónico. La columna de intercambio iónico se preparó poniendo lana de vidrio (grado HPLC) en el fondo de una columna de cromatografía de vidrio de 200 ml de Sigma-Aldrich. Se añadió una mezcla de aproximadamente 20 ml de forma de cloruro Dowex 1x2 (Sigma-Aldrich) y agua destilada (relación en volumen 1:1), seguido de un trozo de lana de vidrio en la parte superior de la columna. La columna se aclaró con agua destilada, y se midió la conductividad. Cuando la conductividad estaba por debajo de 35 mS/cm, se añadió a la columna un volumen de 40 ml de WFI.
Para el intercambio iónico, se preparó una muestra de acetato de SOM230 (SOM230(Ac)) en un matraz Pyrex de 100 ml. La muestra se diluyó con WFI hasta un volumen final de 65,55 ml y una concentración de 3,8 mg de SOM230(Ac)/ml. La disolución de SOM230(Ac) se transfirió a la columna de intercambio iónico usando una pipeta de plástico desechable. El matraz Pyrex se enjuagó con 20 ml adicionales de WFI, que también se transfirieron a la columna. Se recogieron fracciones de muestra de 20 ml a medida que la columna se enjuagaba con WFI. Se midió la conductividad en cada fracción, y se recogieron las fracciones hasta que la conductividad estuvo por debajo de 75 mS/cm. Todas las fracciones se reunieron en un matraz de fondo redondo de 1000 ml (volumen reunido, aproximadamente 180 ml).
El matraz de fondo redondo de la etapa anterior se mantuvo a 2-8°C hasta uso posterior. El matraz se montó en un Rotavapor, y se puso aproximadamente al 80% de la velocidad máxima de rotación. La disolución se congeló superficialmente bajando el matraz giratorio en un baño de EtOH que contenía hielo seco y una mezcla de EtOH al 99,5% y al 96% (relación en volumen 1:1), durante 10 minutos. Después de la congelación superficial, el matraz de fondo redondo se mantuvo a -80°C durante 30 minutos antes de que comenzara la liofilización.
El material se liofilizó durante casi 30 h. El polvo de hidrocloruro de SOM230 (SOM230(Cl)) obtenido se transfirió posteriormente y se pesó en un matraz Pyrex de 250 ml. La cantidad total de SOM230(Cl) recuperada fue 0,215 g, lo que dio como resultado un rendimiento del 86% del procedimiento de intercambio iónico. El polvo peptídico se almacenó en un congelador (< -15°C) hasta uso posterior.
Análisis farmacéutico de hidrocloruro de SOM230
Una comparación de los datos de pureza (HPLC) para el SOM230(Ac) y el SOM230(Cl) indicó que la integridad del material de SOM230 permaneció intacta a través del proceso de intercambio iónico. El contenido de cloruro se determinó por HPLC al 5,15% en peso, que correspondía bien al contenido de acetato determinado de 8,90% en peso en el polvo de fármaco SOM230(Ac) original cuando se tiene en cuenta la relación en peso de acetato/cloruro. El análisis del polvo de fármaco de SOM230(Cl) no detectó ninguna presencia de iones acetato, lo que demuestra un procedimiento de intercambio iónico completo y exitoso.
Solubilidad
Se prepararon formulaciones lipídicas con SOM230(Ac) y SOM230(Cl), respectivamente, como se describe en el Ejemplo 1 según las Tablas 8 y 9. La concentración diana fue una concentración de SOM230 (base libre) de aproximadamente 30 mg/ml.
Tabla 8. Composiciones (% en peso) de formulaciones de lípidos/SOM230(Ac).

Tabla 9. Composiciones (% en peso) de formulaciones de lípidos/SOM230(Cl).

Se dejó que las muestras se equilibraran a temperatura ambiente en un agitador magnético (500 rpm) después de un breve mezclamiento en vórtice. La inspección visual se realizó después de 1,5 y 24 horas. Todas las formulaciones indicadas en las Tablas 8 y 9 fueron transparentes y homogéneas después de 24 h de mezclamiento, lo que indica una buena solubilidad tanto de las formas de sal de acetato como de hidrocloruro. Según el mismo procedimiento, se prepararon formulaciones de pamoato de SOM230 (SOM230(Pm)) con composición de lípidos y codisolventes idéntica a las descritas en las Tablas 8 y 9.
Comparación de estabilidad
Las formulaciones de lípidos/SOM230(Pm), lípidos/SOM230(Cl) y lípidos/SOM230(Ac) se dividieron en dos viales 2R que se colocaron a 60°C, y la cantidad restante de la preparación se analizó por HPLC (punto de tiempo cero).
Las muestras almacenadas a 60°C se extrajeron después de 2 semanas de almacenamiento para inspección visual y análisis por HPLC.
Los resultados del análisis de HPLC se proporcionan en la Figura 4, y muestran la diferencia en el perfil de estabilidad entre las formas de sal para las variantes de formulación investigadas (debido a que la relación en peso de SPC/GDO era equivalente para todas las formas de sal, las variantes de formulación se diferencian por su composición de disolvente en la Figura 4). Los resultados después del almacenamiento durante 2 semanas a 60°C indican claramente que el pamoato de SOM230 es la forma de sal más estable en las formulaciones lipídicas. La sal de hidrocloruro de SOM230 también fue más estable que la sal de acetato, pero no tan estable como la sal de pamoato.
Ejemplo 4 - Estudios PK in vivo I y II
Formulaciones
Las formulaciones utilizadas para el estudio farmacocinético (PK) I in vivo en rata (estudio n° PK-12-437) se describen en la Tabla 10. Para todas las formulaciones, se seleccionó una carga constante de SOM230 (sal de pamoato) correspondiente a 30 mg de base libre de SOM230/ml. Se investigaron combinaciones de diferentes disolventes, EtOH, EtOH/PG y EtOH/WFI. El objetivo principal fue caracterizar los perfiles PK de las diferentes variantes de formulación del pamoato de SOM230.
Tabla 10. Formulaciones de lípidos/SOM230 seleccionadas para el estudio PK I (PK-12-437). Las composiciones se proporcionan en % en peso.

Las formulaciones utilizadas para el estudio PK II in vivo en rata (estudio n° PK-12-438) se describe en la Tabla 11. El objetivo principal era caracterizar los efectos del aumento de la carga de SOM230 en el perfil PK. Para este estudio, se usó una combinación de EtOH y agua (WFI) como disolvente para las formulaciones a una concentración fija del 10% en peso de cada componente como se indica en la Tabla 11. La formulación 4071S230-C se investigó en ambos estudios PK, proporcionando un puente entre los estudios.
Tabla 11. Formulaciones de lípidos/SOM230 seleccionadas para el estudio PK II (PK-12-438).
Las composiciones se proporcionan en % en peso.

La fabricación de las formulaciones en las Tablas 10 y 11 se realizó esencialmente como se describe en el Ejemplo 1, con la adición de una etapa de filtración estéril después del mezclamiento completo en formulaciones líquidas homogéneas. Las formulaciones se filtraron de forma estéril bajo una presión de nitrógeno de 2,5 bares utilizando un filtro de membrana PVDF de 0,2 micrómetros de Millipore.
Rendimiento del estudio in vivo
Las formulaciones en la Tabla 10 (PK-12-437) se inyectaron por vía subcutánea a ratas macho Sprague-Dawley (peso corporal aprox. 330 g) a un volumen de dosis de 0,2 ml por animal (6 mg de SOM230/animal), mientras que las formulaciones en la Tabla 11 (PK-12-438) se inyectaron a un volumen de dosis de 0,1 ml/animal correspondiente a 3, 4, 5 y 6 mg de SOM230/animal para 4071S230-C, 4071S230-E, 4071S230-F y 4071S230-G, respectivamente. Se recogió sangre para la farmacocinética antes de la dosis, y 1 hora, 6 horas, 1 día, 7 días, 14 días, 21 días, 28 días y
35 días después de la dosis. Se recogieron muestras de sangre de 0,5 ml mediante sangrado sublingual en tubos de ensayo tratados con EDTA (Capiject 3T-MQK, Terumo Medical Corporation). La sangre se colocó en hielo inmediatamente después de la recolección, y se centrifugó (aproximadamente 1500xg, a 5°C durante 10 min) dentro de 30 a 60 minutos. El plasma se transfirió a tubos de ensayo de propileno translúcidos de 1,5 ml debidamente etiquetados (tubos de microcentrífuga, Plastibrand, Buch & Holm), y se almacenó por debajo de -70°C hasta el bioanálisis mediante ensayo ELISA.
Farmacocinética
Los perfiles PK de las respectivas formulaciones en las Tablas 10 y 11 se proporcionan en las Figuras 5 y 6.
Como se desprende de los datos en la Figura 5, los perfiles farmacocinéticos son muy planos, con relaciones de concentraciones plasmáticas Cmax/C28d entre alrededor de 3,1-4,9, en las que Cmax es la concentración plasmática máxima observada y C28d es la concentración plasmática observada 28 días después de la inyección. En términos de relaciones CmaX/Cpromedio, en las que Cpromedio es la concentración plasmática promedio a lo largo de la duración diana de 28 días, esta relación es incluso menor que la relación Cmax/C28d respectiva. De este modo, la liberación inicial (o estallido) es baja, seguida de niveles plasmáticos consistentes, cumpliendo con los requisitos PK para formulaciones de depósito eficaces. Los principales parámetros PK obtenidos en PK-12-437 se tabulan en la Tabla 12.
Tabla 12. Parámetros PK obtenidos en el estudio PK n° PK-12-437. Las composiciones de formulación se proporcionan en la Tabla 10.

Los datos en la Figura 6 muestran nuevamente que la velocidad de liberación es constante a lo largo del tiempo, lo que da como resultado perfiles PK muy planos. Las relaciones de concentraciones plasmáticas Cmax/C28d estaban entre alrededor de 3,4-9,2 para las formulaciones indicadas en la Tabla 11. En términos de relaciones de Cmax/Cpromedio, esta relación es incluso más baja que la relación respectiva de Cmax/C28d. Los principales parámetros PK obtenidos en PK-12-438 se tabulan en la Tabla 13.
Tabla 13. Parámetros PK obtenidos en el estudio PK n° PK-12-438. Las composiciones de formulación se proporcionan en la Tabla 11.

La linealidad de la dosis con respecto a la exposición (AUC) se indicó claramente como se muestra en la Figura 7 (R2 = 0,977). La linealidad de la dosis con respecto a Cmax también se mostró como se indica en la Figura 8 (R2 = 0,975).
Ejemplo 5 - Ensayo de estabilidad exploratorio
Las formulaciones evaluadas en los estudios PK (Tablas 10 y 11) también se sometieron a ensayos de estabilidad exploratorios. Además de estas formulaciones, se fabricó una formulación adicional que comprende el antioxidante ácido etilendiaminotetraacético disódico (EDTA). La composición de formulación de la formulación adicional se proporciona en la Tabla 14 (véanse las Tablas 10 y 11 para las composiciones de las otras formulaciones).
Tabla 14. Formulación de lípidos/SOM230 que comprende antioxidante incluido en el ensayo exploratorio de estabilidad. Composición en % en peso.

Sumario del montaje del estudio exploratorio de estabilidad
Cada formulación se llenó en viales 2R con 0,8 g por vial, seguido de lavado con nitrógeno durante 5 segundos y cierre con tapones de caucho recubiertos de teflón y tapas alabeadas de aluminio. Las condiciones de almacenamiento para las formulaciones proporcionadas en las Tablas 10 y 14 fueron 5°C, 252C/60% HR, 40°C/75% HR y 60°C, mientras que las formulaciones proporcionadas en la Tabla 11 solo se evaluaron a 5°C y 25°C/60% HR. Las muestras siempre se dejaron equilibrar durante 60 min a temperatura ambiente antes de comenzar el análisis de HPLC UV-DAD (detección de matriz de diodos).
Resultados
Los contenidos de péptidos analizados (HPLC UV-DAD) después de hasta 8 semanas de almacenamiento se resumen en las Tablas 15 y 16, mientras que los resultados de pureza del péptido, calculados como el área del pico de SOM230 dividida entre el área total del pico de SOM230 y sustancias relacionadas, se resumen en las Tablas 17 y 18.

Tabla 16. Análisis del contenido de SOM230 (por HPLC UV-DAD) de formulaciones (véase la Tabla 11) almacenadas a 5 y 25°C hasta 8 semanas.

Tabla 17. Análisis de pureza de SOM230 (por HPLC UV-DAD) de formulaciones (véanse las Tablas 10 y 14 almacenadas a 5, 25, 40 y 60°C hasta 8 semanas.


Tabla 18. Análisis de pureza de SOM230 (por HPLC UV-DAD) de formulaciones (véase la Tabla 11) almacenadas a 5 y 25°C hasta 8 semanas.
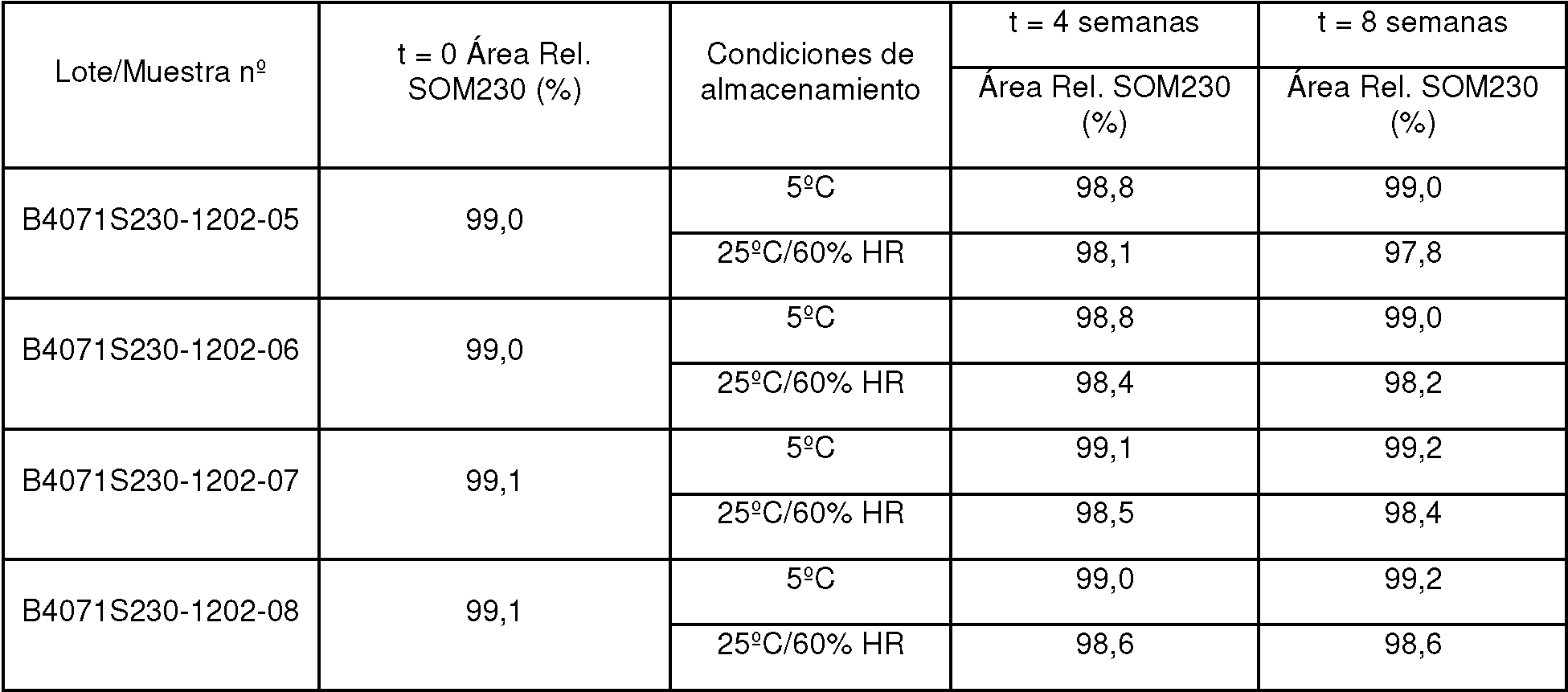
Las siguientes conclusiones se extrajeron basándose en los datos de contenido y pureza del péptido: No se detectó ningún cambio en el contenido o pureza de SOM230 a 5°C (dentro de la variabilidad experimental).
Solo se detectaron pequeños cambios en el contenido y la pureza del péptido a 25°C. Dependiendo del tipo de formulación, la pureza del péptido disminuyó en un 2,6-4,2% después de 8 semanas a 40°C, con una tendencia a disminuir la velocidad de degradación con el tiempo de almacenamiento.
Se detectó un efecto positivo de la inclusión de EDTA, como se muestra comparando B4071S230-1202-03 (sin EDTA) y 4071S230-1202-109 (con EDTA).
Ejemplo 6 - Estudio PK III in vivo (PK-12-451)
Formulaciones
Las formulaciones utilizadas para el estudio farmacocinético (PK) III in vivo en rata (estudio n° PK-12-451) se describe en la Tabla 19. Las concentraciones de SOM230 investigadas corresponden a 40 mg y 50 mg de base libre de SOM230/ml para las respectivas formulaciones. Se utilizó una combinación de etanol (EtOH) y propilenglicol (PG) para todas las formulaciones. El objetivo principal fue caracterizar los perfiles PK de las diferentes variantes de formulación del pamoato de SOM230.
Tabla 19. Formulaciones de lípidos/SOM230 seleccionadas para el estudio PK III (PK-12-451). Las composiciones se proporcionan en % en peso.


La fabricación de las formulaciones en la Tabla 19 se realizó esencialmente como se describe en el Ejemplo 1 con la adición de una etapa de filtración estéril después del mezclamiento completo en formulaciones líquidas homogéneas. Las formulaciones se filtraron de forma estéril bajo una presión de nitrógeno de 2,5 bares utilizando un filtro de membrana PVDF de 0,2 micrómetros de Millipore.
Rendimiento del estudio in vivo
Las formulaciones de la Tabla 19 (PK-12-451) se inyectaron por vía subcutánea a ratas macho Sprague-Dawley (peso corporal aprox. 330 g) a un volumen de dosis de 0,1 ml por animal (6 mg de SOM230/animal) correspondiente a 4 y 5 mg de SOM230/animal para 4071S230-H, 4071S230-J, 4071S230-K y 4071S230-I, respectivamente. La sangre para el análisis farmacocinético se recogió 1 hora, 6 horas, 1 día, 3 días, 7 días, 14 días, 21 días, 28 días y 35 días después de la dosificación. Se recogieron muestras de sangre de 0,5 ml mediante sangrado sublingual en tubos de ensayo tratados con EDTA (Capiject 3T-MQK, Terumo Medical Corporation). La sangre se colocó en hielo inmediatamente después de la recolección y se centrifugó (aproximadamente 1500xg, a 5°C durante 10 min) dentro de 30 a 60 minutos. El plasma se transfirió a tubos de ensayo de propileno translúcidos de 1,5 ml debidamente etiquetados (tubos de microcentrífuga, Plastibrand, Buch & Holm), y se almacenó por debajo de -70°C hasta el bioanálisis mediante ensayo de ELISA.
Los perfiles PK de las respectivas formulaciones en la Tabla 19 se proporcionan en la Figura 10.
Como se desprende de los datos en la Figura 9, los perfiles PK son generalmente planos con niveles plasmáticos algo más altos durante los primeros 14 días para 4071S230-I. Las relaciones de concentraciones plasmáticas Cmax/C28días variaron en el intervalo 2,6-8,4 dependiendo de la variante de formulación.
Un resultado digno de mención es el hecho de que se obtuvo la relación más baja de niveles plasmáticos de Cmax/C28días, y por tanto, desde esta perspectiva, se obtuvo el perfil PK más atractivo para 4071S230-J que comprende DOPC en lugar de SPC (véase la Tabla 19 para las composiciones).
Los principales parámetros PK obtenidos en PK-12-451 se tabulan en la Tabla 20.
Tabla 20. Parámetros PK obtenidos en el estudio farmacocinético PK-12-451. Las composiciones de formulación se proporcionan en la Tabla 19.

Ejemplo 7 - Ensayos exploratorios de estabilidad adicionales
Sumario de los ensayos de estabilidad exploratorios
Las composiciones de las formulaciones investigadas en el estudio de estabilidad se proporcionan en la Tabla 19. Cada formulación se llenó en viales 2R con 1,0 g por vial, seguido de lavado con nitrógeno durante 5 segundos y cierre con tapones de caucho recubiertos de teflón y tapas alabeadas de aluminio. Las condiciones de almacenamiento fueron 5°C y 25°C/60% HR (compatible con ICH). Las muestras siempre se dejaron equilibrar durante 60 min a temperatura ambiente antes de comenzar el análisis de HPLC UV-DAD (detección de matriz de diodos).
Análisis de pureza de SOM230 (por HPLC UV-DAD) hasta 12 semanas
Los resultados del análisis de pureza de SOM230 después del almacenamiento hasta 12 semanas se presentan en las Figuras 10 y 11. No se detectó ningún cambio en la pureza de SOM230 a 5°C. Las sustancias relacionadas totales (RS = 100% - pureza del péptido encontrada) observadas después de 12 semanas a 25°C estaban en el intervalo de 1,4-1,9%, estando los valores de partida en la liberación (tiempo cero) en el intervalo de 0,9-1,1%. Se encontró que el polvo del fármaco de SOM230 (pamoato) contiene alrededor de 0,7% de sustancias relacionadas, y por tanto este nivel debe tomarse como nivel de referencia. Ajustado para el nivel de referencia de polvo de fármaco de SOM230, las RS relacionadas totales, o los productos de degradación totales, encontrados después de 12 semanas a 25°C estaban en el intervalo de 0,7-1,3%, mientras que el aumento de las RS totales hasta 12 semanas, con tiempo cero como referencia, estaba en el intervalo 0,3-0,9%, mostrando la formulación basada en DOPC (B4071S30-1206-11, véase la Tabla 19) las RS totales más bajas.
Ejemplo 8 - Otras composiciones de SOM230 que comprenden DOPC
Las formulaciones lipídicas de SOM230 que comprenden DOPC se prepararon como se describe en el Ejemplo 1 dando como resultado, líquidos homogéneos después del procedimiento de mezclamiento. Las composiciones de formulación se proporcionan en la Tabla 21.
Tabla 21. Formulaciones de lípidos/SOM230 que comprenden DOPC. Las composiciones se proporcionan en % en peso.

Las formulaciones (0,2 g) se inyectaron en 5 ml de disolución salina amortiguada con fosfato (PBS pH 7,4) usando una jeringa Luer-Lock desechable de 1 ml equipada con una aguja de 16 mm de pared delgada 23G. Todas las formulaciones formaron geles de cristal líquido coherentes en contacto con PBS.
Ejemplo 9 - Composiciones de SOM230 que comprenden DOPC y diferente contenido de disolvente
Las formulaciones lipídicas de SOM230 que comprenden DOPC y un contenido de disolvente diferente se prepararon esencialmente como se describe en el Ejemplo 1 con la adición de una etapa de filtración estéril después del mezclamiento completo en formulaciones líquidas homogéneas. Las formulaciones se filtraron de forma estéril bajo una presión de nitrógeno de 2,5 bares utilizando un filtro de membrana PVDF de 0,2 micrómetros de Millipore. Las composiciones de formulación se proporcionan en la Tabla 22.
Tabla 22. Formulaciones de lípidos/SOM230 que comprenden DOPC y diferente contenido de disolvente. Las composiciones se proporcionan en % en peso.

Las formulaciones (0,2 g) se inyectaron en 5 ml de disolución salina amortiguada con fosfato (PBS pH 7,4) usando una jeringa Luer-Lock desechable de 1 ml equipada con una aguja de 16 mm de pared delgada 23G. Todas las formulaciones formaron geles de cristal líquido coherentes en contacto con PBS.
Ejemplo 10 - Composiciones de SOM230 de alta carga de fármaco
Las formulaciones lipídicas de alta carga de SOM230 que comprenden DOPC y diferente contenido de disolvente se preparan esencialmente como se describe en el Ejemplo 1 con la adición de una etapa de filtración estéril después del mezclamiento completo en formulaciones líquidas homogéneas. Las formulaciones se filtraron de forma estéril bajo una presión de nitrógeno de 2,5 bares utilizando un filtro de membrana PVDF de 0,2 micrómetros de Millipore. Las composiciones de formulación se proporcionan en la Tabla 23.
Tabla 23. Composiciones lipídicas de alta carga del fármaco de SOM230

Las formulaciones (0,2 g) se inyectan en 5 ml de disolución salina amortiguada con fosfato (PBS pH 7,4) usando una jeringa Luer-Lock desechable de 1 ml equipada con una aguja de 16 mm de pared delgada 23G. Todas las formulaciones forman geles de cristal líquido coherentes en contacto con PBS.
Claims (19)
1. Una pre-formulación que comprende una mezcla de baja viscosidad de:
a) 20-50% en peso de al menos un diacilglicerol;
b) 20-54% en peso de al menos una fosfatidilcolina (PC);
c) 0,1-35% en peso de al menos un disolvente monoalcohólico orgánico biocompatible;
d) 1 a 20% en peso de disolvente polar;
e) 5 a 150 mg/ml de al menos un agonista peptídico del receptor de somatostatina que comprende pasireotida, calculado como base libre; y
f) opcionalmente al menos un antioxidante;
en la que la relación de los componentes a:b está en el intervalo 40:60 a 54:46;
en la que la pre-formulación tiene una viscosidad de 1 -1000 mPas a 20°C;
en la que la pre-formulación forma, o es capaz de formar, al menos una estructura de fase cristalina líquida al entrar en contacto con un exceso de fluido acuoso.
2. Una pre-formulación según la reivindicación 1, en la que el agonista peptídico del receptor de somatostatina comprende o consiste en pasireotida o una sal de la misma, preferiblemente en la que dicha sal de pasireotida se selecciona del grupo que consiste en cloruro de pasireotida, acetato de pasireotida, pamoato de pasireotida y tartrato de pasireotida.
3. Una pre-formulación según la reivindicación 1, en la que el agonista peptídico del receptor de somatostatina consiste en pamoato de pasireotida.
4. La pre-formulación según la reivindicación 1 o la reivindicación 2, en la que el agonista peptídico del receptor de la somatostatina consiste en pasireotida y octreotida.
5. Una pre-formulación según cualquiera de las reivindicaciones 1 a 4, en la que dicha pre-formulación comprende una dosis de agonista peptídico del receptor de somatostatina en el intervalo de 10 a 100 mg/ml.
6. Una pre-formulación según cualquiera de las reivindicaciones 1 a 5, en la que el componente a) comprende o consiste en dioleato de glicerol (GDO).
7. Una pre-formulación según cualquiera de las reivindicaciones 1 a 6, en la que el componente b) comprende o consiste en PC de soja, DOPC, o PC con al menos 95% de grupos de cabeza de PC y al menos 95% de cadenas de acilo de C16 a C20 que tienen de 0 a 3 insaturaciones.
8. Una pre-formulación según cualquiera de las reivindicaciones 1 a 7, en la que el componente c) comprende o consiste en etanol, propanol, isopropanol, o mezclas de los mismos, preferiblemente etanol.
9. Una pre-formulación según cualquiera de las reivindicaciones 1 a 8, en la que el componente d) tiene una constante dieléctrica de al menos 28 a 25°C.
10. Una pre-formulación según cualquiera de las reivindicaciones 1 a 9, en la que el componente d) comprende agua o propilenglicol, o mezclas de los mismos.
11. Una pre-formulación según las reivindicaciones 1 a 10, que comprende al menos uno de:
a) a un nivel de 30-43% en peso;
b) a un nivel de 30-45% en peso; y
c) a un nivel de 0,1-12% en peso.
12. Una pre-formulación según las reivindicaciones 1 a 11, en la que el componente d) está presente a un nivel de 5 a 20% en peso, más preferiblemente 8 a 15% en peso.
13. Una pre-formulación según cualquiera de las reivindicaciones 1 a 12, en la que los componentes c) y d) combinados están presentes a un nivel total en el intervalo de 10-30% en peso, preferiblemente en el intervalo de 12-25% en peso.
14. Una pre-formulación según cualquiera de las reivindicaciones 1 a 13, en la que la relación de componentes a:b está en el intervalo 45:55 a 54:46.
15. Una pre-formulación según cualquiera de las reivindicaciones 1 a 14, en la que d) es PG, y la relación de componentes c:d está en el intervalo 90:10 a 25:75.
16. Un dispositivo de administración precargado que contiene una pre-formulación como se reivindica en cualquiera de las reivindicaciones 1 a 15, siendo dicho dispositivo una jeringa o cilindro de jeringa, un inyector sin aguja, un inyector multiusos o de un solo uso, un cartucho o un vial.
17. El dispositivo de la reivindicación 16, que contiene una dosis única de 10 a 100 mg de agonista peptídico del receptor de somatostatina, preferiblemente pamoato de pasireotida.
18. El dispositivo de cualquiera de las reivindicaciones 16 o 17, que contiene un agonista peptídico del receptor de somatostatina, preferiblemente pamoato de pasireotida, a alrededor de 0,2 a 4 mg por día entre administraciones programadas.
19. Una pre-formulación según cualquiera de las reivindicaciones 1 a 15, para uso en el tratamiento de la enfermedad de Cushing, acromegalia, diabetes mellitus tipo I o tipo II y/o sus complicaciones, síndrome del intestino irritable, enfermedades inflamatorias, enfermedad inflamatoria intestinal, psoriasis o artritis reumatoide, enfermedad renal poliquística, síndrome de dumping, síndrome de diarrea acuosa, diarrea relacionada con el SIDA, diarrea inducida por quimioterapia, pancreatitis aguda o crónica, y tumores secretores de hormonas gastrointestinales, neoplasias malignas de linfocitos, o hemorragia gastrointestinal.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/EP2012/059917 WO2012160213A1 (en) | 2011-05-25 | 2012-05-25 | Controlled release peptide formulations |
| US201261730613P | 2012-11-28 | 2012-11-28 | |
| PCT/EP2013/060739 WO2013174978A1 (en) | 2012-05-25 | 2013-05-24 | Somatostatin receptor agonist formulations |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2834318T3 true ES2834318T3 (es) | 2021-06-17 |
Family
ID=49623188
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES13724299T Active ES2834318T3 (es) | 2012-05-25 | 2013-05-24 | Formulaciones de agonistas de receptores de la somatostatina |
Country Status (23)
| Country | Link |
|---|---|
| US (3) | US11672843B2 (es) |
| JP (1) | JP6374380B2 (es) |
| KR (1) | KR102139080B1 (es) |
| CN (1) | CN104487050B (es) |
| AU (1) | AU2013265210B2 (es) |
| BR (1) | BR112014029425A2 (es) |
| CA (1) | CA2874367C (es) |
| CL (1) | CL2014003185A1 (es) |
| CO (1) | CO7160067A2 (es) |
| DK (1) | DK2861209T3 (es) |
| EA (1) | EA035495B1 (es) |
| ES (1) | ES2834318T3 (es) |
| HK (1) | HK1207985A1 (es) |
| IL (1) | IL235740A0 (es) |
| IN (1) | IN2014DN09831A (es) |
| MA (1) | MA37672A1 (es) |
| MX (1) | MX350964B (es) |
| NZ (1) | NZ702028A (es) |
| PE (1) | PE20150195A1 (es) |
| PH (1) | PH12014502614A1 (es) |
| SG (1) | SG11201407678YA (es) |
| TN (1) | TN2014000483A1 (es) |
| WO (1) | WO2013174978A1 (es) |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005117830A1 (en) * | 2004-06-04 | 2005-12-15 | Camurus Ab | Liquid depot formulations |
| EP4427814A3 (en) * | 2011-05-25 | 2024-11-20 | Camurus AB | Controlled release peptide formulations |
| WO2018060213A1 (en) * | 2016-09-27 | 2018-04-05 | Camurus Ab | Formulations containing a somatostatin receptor agonist |
| CN113018248B (zh) * | 2019-12-23 | 2022-07-22 | 南京清普生物科技有限公司 | 一种缓释递药系统 |
| CN114240934B (zh) * | 2022-02-21 | 2022-05-10 | 深圳大学 | 一种基于肢端肥大症的图像数据分析方法及系统 |
Family Cites Families (58)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB2209937B (en) | 1987-09-21 | 1991-07-03 | Depiopharm S A | Water insoluble polypeptides |
| US4938763B1 (en) | 1988-10-03 | 1995-07-04 | Atrix Lab Inc | Biodegradable in-situ forming implants and method of producing the same |
| US5340802A (en) | 1989-06-30 | 1994-08-23 | Abbott Laboratories | Peptide analog type-B CCK receptor ligands |
| CA2535463A1 (en) | 1989-07-07 | 1991-01-08 | Novartis Ag | Octreotide-pamoate and its use in sustained release formulations of water soluble peptides |
| PH30995A (en) | 1989-07-07 | 1997-12-23 | Novartis Inc | Sustained release formulations of water soluble peptides. |
| MY107937A (en) | 1990-02-13 | 1996-06-29 | Takeda Chemical Industries Ltd | Prolonged release microcapsules. |
| US5531925A (en) | 1991-10-04 | 1996-07-02 | Gs Biochem Ab | Particles, method of preparing said particles and uses thereof |
| WO1993006921A1 (en) | 1991-10-04 | 1993-04-15 | Gs Biochem Ab | Particles, method of preparing said particles and uses thereof |
| US6228383B1 (en) | 1994-03-03 | 2001-05-08 | Gs Development Ab | Use of fatty acid esters as bioadhesive substances |
| JP4137179B2 (ja) | 1994-03-30 | 2008-08-20 | ジーエス ディベロップメント エービー | 生物付着性物質としての脂肪酸エステルの使用 |
| SE518578C2 (sv) | 1994-06-15 | 2002-10-29 | Gs Dev Ab | Lipidbaserad komposition |
| FR2725369B1 (fr) | 1994-10-07 | 1997-01-03 | Oreal | Composition cosmetique ou dermatologique constituee d'une emulsion huile dans eau a base de globules huileux pourvus d'un enrobage cristal liquide lamellaire |
| WO1997013528A1 (en) | 1995-10-12 | 1997-04-17 | Gs Development Ab | A pharmaceutical composition for administration of an active substance to or through a skin or mucosal surface |
| US6458924B2 (en) | 1996-08-30 | 2002-10-01 | Novo Nordisk A/S | Derivatives of GLP-1 analogs |
| DE69730093T2 (de) | 1996-10-31 | 2006-07-20 | Takeda Pharmaceutical Co. Ltd. | Zubereitung mit verzögerter Freisetzung |
| AU6919598A (en) | 1997-04-17 | 1998-11-13 | Dumex-Alpharma A/S | A novel bioadhesive drug delivery system based on liquid crystals |
| US6638621B2 (en) | 2000-08-16 | 2003-10-28 | Lyotropic Therapeutics, Inc. | Coated particles, methods of making and using |
| US20040018241A1 (en) | 1997-09-26 | 2004-01-29 | Noven Pharmaceuticals, Inc. | Bioadhesive compositions and methods for topical administration of active agents |
| US6320017B1 (en) | 1997-12-23 | 2001-11-20 | Inex Pharmaceuticals Corp. | Polyamide oligomers |
| BE1011899A6 (fr) | 1998-04-30 | 2000-02-01 | Ucb Sa | Compositions pharmaceutiques gelifiables utilisables. |
| SI1140148T1 (sl) | 1998-12-22 | 2006-04-30 | Lilly Co Eli | Stabilna formulacija raztopine glukagonu podobnega peptida-1 |
| US6011067A (en) | 1999-06-11 | 2000-01-04 | Thione International, Inc. | Antioxidant composition for the treatment of psoriasis and related diseases |
| GB0018891D0 (en) * | 2000-08-01 | 2000-09-20 | Novartis Ag | Organic compounds |
| US20020153508A1 (en) | 2000-06-29 | 2002-10-24 | Lynch Matthew Lawrence | Cubic liquid crystalline compositions and methods for their preparation |
| US7008646B2 (en) | 2001-02-20 | 2006-03-07 | Patrick Thomas Spicer | Cubic liquid crystalline compositions and methods for their preparation |
| US6936187B2 (en) | 2001-02-21 | 2005-08-30 | Matthew Lawrence Lynch | Functionalized cubic liquid crystalline phase materials and methods for their preparation and use |
| US6656385B2 (en) | 2001-02-21 | 2003-12-02 | The Procter & Gamble Company | Functionalized cubic liquid crystalline phase materials and methods for their preparation and use |
| US6991809B2 (en) | 2001-06-23 | 2006-01-31 | Lyotropic Therapeutics, Inc. | Particles with improved solubilization capacity |
| AU2002316811A1 (en) | 2001-06-28 | 2003-03-03 | Novo Nordisk A/S | Stable formulation of modified glp-1 |
| EP1474163A2 (en) | 2002-01-10 | 2004-11-10 | Imperial College Innovations Limited | Modification of feeding behavior |
| US7622118B2 (en) | 2002-07-15 | 2009-11-24 | Board Of Regents, The University Of Texas System | Cancer treatment methods using selected antibodies to aminophospholipids |
| US7731947B2 (en) | 2003-11-17 | 2010-06-08 | Intarcia Therapeutics, Inc. | Composition and dosage form comprising an interferon particle formulation and suspending vehicle |
| GB0305941D0 (en) | 2003-03-14 | 2003-04-23 | Camurus Ab | Composition |
| GB0307777D0 (en) | 2003-04-04 | 2003-05-07 | Medical Res Council | Conjugate compounds |
| WO2005014162A1 (en) | 2003-08-04 | 2005-02-17 | Camurus Ab | Method for improving the properties of amphiphile particles |
| JP5518282B2 (ja) | 2003-09-01 | 2014-06-11 | ノヴォ ノルディスク アー/エス | 安定なペプチドの製剤 |
| GB0322033D0 (en) | 2003-09-19 | 2003-10-22 | Camurus Ab | Composition |
| WO2006075124A1 (en) * | 2005-01-14 | 2006-07-20 | Camurus Ab | Somatostatin analogue formulations |
| PL1682091T3 (pl) | 2003-11-07 | 2017-09-29 | Camurus Ab | Kompozycje lipidów i kationowych peptydów |
| WO2005117830A1 (en) * | 2004-06-04 | 2005-12-15 | Camurus Ab | Liquid depot formulations |
| WO2005063213A1 (en) | 2003-12-19 | 2005-07-14 | Biodelivery Sciences International, Inc. | Rigid liposomal cochleate and methods of use and manufacture |
| GB0401513D0 (en) | 2004-01-23 | 2004-02-25 | Camurus Ab | Compositions |
| EP1713446B1 (en) | 2004-01-23 | 2010-12-01 | Camurus Ab | Ternary non-lamellar lipid compositions |
| CA2575906C (en) | 2004-08-04 | 2014-04-15 | Camurus Ab | Compositions forming non-lamellar dispersions |
| JP5144277B2 (ja) * | 2005-01-14 | 2013-02-13 | カムルス エービー | 局所用生体付着性製剤 |
| ATE501710T1 (de) | 2005-01-14 | 2011-04-15 | Camurus Ab | Somatostatin-analog-formulierungen |
| US9757461B2 (en) | 2005-01-14 | 2017-09-12 | Camurus Ab | GnRH analogue formulations |
| KR101245022B1 (ko) * | 2005-01-21 | 2013-03-19 | 카무러스 에이비 | 약제학적 지질 조성물 |
| CA2609810C (en) | 2005-06-06 | 2012-05-22 | Camurus Ab | Glp-1 analogue formulations |
| WO2007061896A1 (en) | 2005-11-17 | 2007-05-31 | Zogenix, Inc. | Delivery of viscous formulations by needle-free injection |
| GB0602639D0 (en) * | 2006-02-09 | 2006-03-22 | Novartis Ag | Organic compounds |
| AR066677A1 (es) * | 2007-05-24 | 2009-09-02 | Novartis Ag | Formulacion de pasireotida. composicion farmaceutica para liberacion prolongada. microparticulas. |
| GB0711656D0 (en) | 2007-06-15 | 2007-07-25 | Camurus Ab | Formulations |
| GB0716385D0 (en) | 2007-08-22 | 2007-10-03 | Camurus Ab | Formulations |
| PL2310042T3 (pl) * | 2008-07-08 | 2013-05-31 | Novartis Ag | Zastosowanie pasyreotydu do leczenia endogennej hipoglikemii hiperinsulinemicznej |
| GB0815435D0 (en) | 2008-08-22 | 2008-10-01 | Camurus Ab | Formulations |
| EP4427814A3 (en) * | 2011-05-25 | 2024-11-20 | Camurus AB | Controlled release peptide formulations |
| US9585959B2 (en) * | 2011-12-05 | 2017-03-07 | Camurus Ab | Robust controlled-release formulations |
-
2013
- 2013-05-24 ES ES13724299T patent/ES2834318T3/es active Active
- 2013-05-24 JP JP2015513209A patent/JP6374380B2/ja active Active
- 2013-05-24 CA CA2874367A patent/CA2874367C/en active Active
- 2013-05-24 KR KR1020147036398A patent/KR102139080B1/ko active Active
- 2013-05-24 MX MX2014014379A patent/MX350964B/es active IP Right Grant
- 2013-05-24 MA MA37672A patent/MA37672A1/fr unknown
- 2013-05-24 WO PCT/EP2013/060739 patent/WO2013174978A1/en not_active Ceased
- 2013-05-24 EA EA201491928A patent/EA035495B1/ru not_active IP Right Cessation
- 2013-05-24 BR BR112014029425A patent/BR112014029425A2/pt not_active Application Discontinuation
- 2013-05-24 PE PE2014002215A patent/PE20150195A1/es not_active Application Discontinuation
- 2013-05-24 HK HK15108712.3A patent/HK1207985A1/xx unknown
- 2013-05-24 AU AU2013265210A patent/AU2013265210B2/en active Active
- 2013-05-24 DK DK13724299.6T patent/DK2861209T3/da active
- 2013-05-24 NZ NZ702028A patent/NZ702028A/en not_active IP Right Cessation
- 2013-05-24 CN CN201380034030.8A patent/CN104487050B/zh active Active
- 2013-05-24 SG SG11201407678YA patent/SG11201407678YA/en unknown
- 2013-05-24 IN IN9831DEN2014 patent/IN2014DN09831A/en unknown
- 2013-05-24 US US14/401,559 patent/US11672843B2/en active Active
-
2014
- 2014-11-17 IL IL235740A patent/IL235740A0/en unknown
- 2014-11-21 TN TN2014000483A patent/TN2014000483A1/fr unknown
- 2014-11-24 PH PH12014502614A patent/PH12014502614A1/en unknown
- 2014-11-24 CL CL2014003185A patent/CL2014003185A1/es unknown
- 2014-12-18 CO CO14278535A patent/CO7160067A2/es unknown
-
2023
- 2023-04-26 US US18/307,124 patent/US20230372436A1/en not_active Abandoned
-
2025
- 2025-01-17 US US19/028,330 patent/US20260000730A1/en active Pending
Also Published As
| Publication number | Publication date |
|---|---|
| EA035495B1 (ru) | 2020-06-25 |
| JP6374380B2 (ja) | 2018-08-15 |
| CA2874367A1 (en) | 2013-11-28 |
| AU2013265210B2 (en) | 2016-09-15 |
| BR112014029425A2 (pt) | 2017-06-27 |
| CN104487050A (zh) | 2015-04-01 |
| DK2861209T3 (da) | 2020-12-21 |
| CN104487050B (zh) | 2019-08-20 |
| SG11201407678YA (en) | 2014-12-30 |
| JP2015520762A (ja) | 2015-07-23 |
| CO7160067A2 (es) | 2015-01-15 |
| CL2014003185A1 (es) | 2015-04-24 |
| AU2013265210A1 (en) | 2014-12-04 |
| MA37672A1 (fr) | 2016-07-29 |
| IN2014DN09831A (es) | 2015-08-07 |
| MX350964B (es) | 2017-09-27 |
| MX2014014379A (es) | 2015-02-05 |
| TN2014000483A1 (en) | 2016-03-30 |
| IL235740A0 (en) | 2015-01-29 |
| KR20150016977A (ko) | 2015-02-13 |
| US20230372436A1 (en) | 2023-11-23 |
| WO2013174978A1 (en) | 2013-11-28 |
| HK1207985A1 (en) | 2016-02-19 |
| US20260000730A1 (en) | 2026-01-01 |
| PH12014502614A1 (en) | 2015-01-21 |
| PE20150195A1 (es) | 2015-02-27 |
| KR102139080B1 (ko) | 2020-07-29 |
| US11672843B2 (en) | 2023-06-13 |
| EA201491928A1 (ru) | 2015-09-30 |
| CA2874367C (en) | 2020-08-18 |
| US20150105332A1 (en) | 2015-04-16 |
| NZ702028A (en) | 2016-07-29 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20250121035A1 (en) | Controlled release peptide formulations | |
| ES2604205T3 (es) | Formulaciones de liberación lenta que contienen somatostatina cíclica | |
| US20260000730A1 (en) | Somatostatin receptor agonist formulations | |
| AU2012260821A1 (en) | Controlled release peptide formulations | |
| EP2861209B1 (en) | Somatostatin receptor agonist formulations | |
| NZ617828B2 (en) | Controlled release peptide formulations | |
| HK1195253B (en) | Controlled release peptide formulations | |
| HK1195253A (en) | Controlled release peptide formulations |