ES2829253T3 - Resolución de compuestos de beta-aminosulfona racémicos - Google Patents
Resolución de compuestos de beta-aminosulfona racémicos Download PDFInfo
- Publication number
- ES2829253T3 ES2829253T3 ES18716164T ES18716164T ES2829253T3 ES 2829253 T3 ES2829253 T3 ES 2829253T3 ES 18716164 T ES18716164 T ES 18716164T ES 18716164 T ES18716164 T ES 18716164T ES 2829253 T3 ES2829253 T3 ES 2829253T3
- Authority
- ES
- Spain
- Prior art keywords
- ethoxy
- methoxyphenyl
- amine
- methylsulfonyl
- formula
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 COc1c(*)cc(C(CS(C)(=O)=O)N)cc1 Chemical compound COc1c(*)cc(C(CS(C)(=O)=O)N)cc1 0.000 description 3
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/44—Iso-indoles; Hydrogenated iso-indoles
- C07D209/48—Iso-indoles; Hydrogenated iso-indoles with oxygen atoms in positions 1 and 3, e.g. phthalimide
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C233/00—Carboxylic acid amides
- C07C233/01—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C233/45—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by carboxyl groups
- C07C233/46—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by carboxyl groups with the substituted hydrocarbon radical bound to the nitrogen atom of the carboxamide group by an acyclic carbon atom
- C07C233/47—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by carboxyl groups with the substituted hydrocarbon radical bound to the nitrogen atom of the carboxamide group by an acyclic carbon atom having the carbon atom of the carboxamide group bound to a hydrogen atom or to a carbon atom of an acyclic saturated carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C317/00—Sulfones; Sulfoxides
- C07C317/26—Sulfones; Sulfoxides having sulfone or sulfoxide groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton
- C07C317/28—Sulfones; Sulfoxides having sulfone or sulfoxide groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton with sulfone or sulfoxide groups bound to acyclic carbon atoms of the carbon skeleton
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Procedimiento para la preparación de una sal de N-formil leucina enantioméricamente pura de 1-(3-etoxi-4- metoxifenil)-2-(metilsulfonil)etan-1-amina (4) **(Ver fórmula)** comprendiendo dicho procedimiento las etapas de: a) proporcionar 1-(3-etoxi-4-20 metoxifenil)-2-(metilsulfonil)etan-1-amina en forma de una mezcla racémica (1') o en forma de una mezcla enantioméricamente enriquecida de la misma (1"); **(Ver fórmula)** b) resolver ópticamente dicha mezcla racémica (1') o enantioméricamente enriquecida (1") mediante tratamiento con una N-formil leucina enantioméricamente pura en presencia de un disolvente para producir las correspondientes sales de N-formil-leucina enantioméricamente puras (4).
Description
DESCRIPCIÓN
Resolución de compuestos de beta-aminosulfona racémicos
Campo de la invención
[0001] La presente invención proporciona un procedimiento industrialmente viable y ventajoso para la preparación de la (S)-beta-aminosulfona que tiene el nombre IUPAC (S)-2-(3-etoxi-4-metoxifenil)-1-(metilsulfonil)-et-2-ilamina y la fórmula (1) representada a continuación, partiendo de la correspondiente mezcla racémica. Dicha (S)-betaaminosulfona es un intermedio útil para la preparación de W-(2-((1S)-1-(3-etoxi-4-metoxifenil)-2-(metilsulfonil)etil)-2,3-dihidro-1,3-dioxo-1H-isoindol-4-il)acetamida, también conocida como Apremilast (compuesto que tiene la fórmula que se muestra a continuación), siendo ésta última adecuada para tratar, prevenir y/o controlar la psoriasis o la artritis psoriásica.

Antecedentes de la invención
[0002] Apremilast es un inhibidor de molécula pequeña oral novedoso de la fosfodiesterasa 4 (PDE4) que funciona intracelularmente para modular una red de mediadores proinflamatorios y antiinflamatorios. La fosfodiesterasa 4 es una PDE específica del monofosfato de adenosina cíclico (AMPc) y es la PDE dominante en las células inflamatorias. La inhibición de PDE4 eleva los niveles intracelulares de AMPc, que a su vez regula a la baja la respuesta inflamatoria al modular la expresión del factor de necrosis tumoral alfa (TNF-a), interleucina (IL)-23, IL-17 y otras citocinas proinflamatorias. La elevación de AMPc también aumenta las citocinas antiinflamatorias. Estos mediadores proinflamatorios y antiinflamatorios se han relacionado con la psoriasis y la artritis psoriásica.
[0003] En base a estos efectos, se está desarrollando Apremilast para su uso en el tratamiento de diversas afecciones inflamatorias mediadas por el sistema inmunitario, tales como psoriasis, artritis reumatoide, enfermedad de Behget y espondilitis anquilosante.
[0004] Como es el caso de la gran mayoría de fármacos, Apremilast es una molécula quiral y sólo el enantiómero [5] de beta-aminosulfona (1) es útil para la preparación de la misma. En general, una mezcla de enantiómeros (R, S) puede contener los dos enantiómeros en cualquier proporción entre sí. La pureza enantiomérica se expresa generalmente como "exceso enantiomérico" o ee y se define, por ejemplo, para el enantiómero (S), como [(S-R)/(R+S)] x 100, donde S y R son respectivamente las cantidades de los enantiómeros (S) y (R) (tal como se determina, por ejemplo, mediante CG o HPLC en una fase estacionaria quiral o polarimetría).
[0005] La beta-aminosulfona (1) podría obtenerse en la forma enantiomérica deseada (S) mediante síntesis enantioselectiva directa o mediante síntesis no enantioselectiva, seguida de separación de la mezcla de enantiómeros y aislamiento del de interés. Las rutas sintéticas no enantioselectivas conducen a una mezcla racémica, es decir, una mezcla en la que dos posibles enantiómeros de un compuesto están presentes en cantidades iguales; en el presente documento, (1) designará un enantiómero enantioméricamente puro, mientras que con (1') se indicará la mezcla racémica (racemato) del compuesto (1), y con (1") una mezcla enantioméricamente enriquecida del mismo.
[0006] Los procedimientos enantioselectivos se basan en catálisis asimétrica, tal como, por ejemplo, hidrogenación o transferencia catalítica asimétrica, aminación reductora asimétrica, reducción asimétrica a través de oxazaborolidina quiral. Sin embargo, estas rutas sintéticas requieren el uso de materiales costosos, por ejemplo, metales de transición, tales como Rh o Ru, y también pueden ser ineficientes e inconvenientes debido a las condiciones extremas que a menudo se requieren (temperaturas muy bajas, condiciones estrictamente anhidras, altas presiones), lo que limita la utilidad de estas rutas en procedimientos industriales viables. Además, en algunos casos, la catálisis asimétrica puede carecer de enantioselectividad de modo que se requieren otras etapas, por ejemplo, cristalización, de todos modos para obtener la pureza óptica deseada.
[0007] Por consiguiente, en la industria, generalmente, se prefiere adoptar rutas sintéticas no enantioselectivas y aislar el enantiómero de interés al final de la síntesis con procedimientos denominados de resolución de la mezcla de enantiómeros.
[0008] El apremilast y otros compuestos similares se describieron por primera vez en la solicitud de patente internacional WO 00/25777 A1. El procedimiento allí descrito implica, como etapas clave, una reacción aldólica entre 3-etoxi-4-metoxibenzaldehído y dimetilsulfona en presencia de n-butil litio, bis (trimetilsilil) amida de litio y trifluoruro de boro dietileterato; la reacción de W-(1-(3-etoxi-4-metoxifenil)-2-(metilsulfonil)etil)-1,1,1-trimetilsilanamina con ácido clorhídrico; la reacción entre 1-(3-etoxi-4-metoxifenil)-2-(metilsulfonil)etan-1-amina y W-(1,3-dioxo-1,3-dihidroisobenzofuran-4-il)acetamida, y la separación final de isómeros mediante cromatografía en fase estacionaria quiral.

[0009] A escala industrial, el uso de esta vía de síntesis tiene varios inconvenientes causados por el pobre rendimiento de la reacción aldólica o la etapa tardía en la que se realiza la separación de enantiómeros de Apremilast. En este último aspecto, vale la pena señalar que, al convertir un compuesto racémico en un producto final enantioméricamente puro (como en el caso de Apremilast), se puede alcanzar un rendimiento máximo del 50%, lo que conduce a una pérdida de al menos el 50% del producto final.
[0010] En la solicitud internacional WO 03/080049 A1 de Celgene se ha descrito un procedimiento alternativo para la preparación de Apremilast que implica una etapa de resolución óptica. Según esta solicitud de patente, una mezcla racémica (1') de 1-(3-etoxi-4-metoxifenil)-2-(metilsulfonil)etan-1-amina puede resolverse ópticamente mediante tratamiento con una sal de aminoácido quiral seleccionada del grupo que consiste en los isómeros L de alanina, arginina, asparagina, ácido aspártico, cisteína, glutamina, ácido glutámico, glicina, histidina, isoleucina, leucina, lisina, metionina, fenilalanina, prolina, serina, treonina, triptófano, tirosina, valina, ornitina, ácido 4-aminobutírico, ácido 2-amino isobutírico, ácido 3-amino propiónico, norleucina, norvalina, hidroxiprolina, sarcosina, citrulina, ácido cisteico, terc-butilglicina, terc-butilalanina, fenilglicina,ciclohexilalanina o preferiblemente W-acetil-leucina.
[0011] A pesar de la gran variedad de sales que caen dentro de esta descripción, en un informe experimental presentado posteriormente el Solicitante del documento WO 03/080049 A1 comparó el uso de A/-acetil-L-leucina y N-acetil-L-fenilalanina como agentes de resolución y mostró que la N-acetil-L-leucina proporcionó la sal de aminoácido con un 77% de ee. después de una primera cristalización, mientras que la N-acetil-L-fenilalalnina solo proporcionó un 14% de ee.
[0012] Un inconveniente asociado con este procedimiento deriva del uso de N-acetil-L-leucina para separar los dos enantiómeros de la 1-(3-etoxi-4-metoxifenil)-2-(metilsulfonil)etan-1-amina, que conduce al aislamiento, después de dos etapas de cristalización, de un producto con un exceso enantiomérico del 98,5 % y un rendimiento global del 25% (con respecto a la 1-(3-etoxi-4-metoxifenil)-2-(metilsulfonil)etan-1-amina racémica).
[0013] Basándose en el hecho de que, a fin de cumplir con la especificación de ICH, todos los compuestos farmacéuticamente aceptables deben tener un grado de pureza de al menos el 99,8 %, un producto que tiene un exceso enantiomérico del 98,5% debe ser sometido a al menos una etapa de cristalización adicional destinada a aumentar su perfil de pureza, lo que inevitablemente da como resultado una disminución adicional del rendimiento general y, como consecuencia, un mayor impacto económico del procedimiento.
[0014] La solicitud de patente EP 2431371 A1 describe la resolución de 1-(3-etoxi-4-metoxifenil)-2-(metilsulfonil) etan-1-amina (1') a través de la formación de sal diastereomérica con N-acetil-L-Valina (N-Ac-L-Val), tal como se muestra a continuación:

[0015] Aunque no se hace referencia al enriquecimiento diastereomérico, el procedimiento se caracteriza por rendimientos bajos, lo que limita su escalado industrial.
[0016] El objetivo de la presente invención es dar a conocer un nuevo procedimiento químico para preparar Apremilast o intermedios útiles en la síntesis del mismo, caracterizado por altos rendimientos y que proporciona los compuestos deseados con una pureza apropiada para su uso en productos farmacéuticos.
Características de la invención
[0017] Estos objetivos se consiguen con la presente invención que, en un primer aspecto de la misma, se refiere a un procedimiento para la preparación de una sal de N-formil leucina enantioméricamente pura de 1-(3-etoxi-4-metoxifenil)-2-(metilsulfonil)etan-1-amina (4):
comprendiendo dicho procedimiento las siguientes operaciones:
a) proporcionar 1-(3-etoxi-4-metoxifenil)-2-(metilsulfonil)etan-1-amina en forma de una mezcla racémica (1') o en forma de una mezcla enantioméricamente enriquecida de la misma
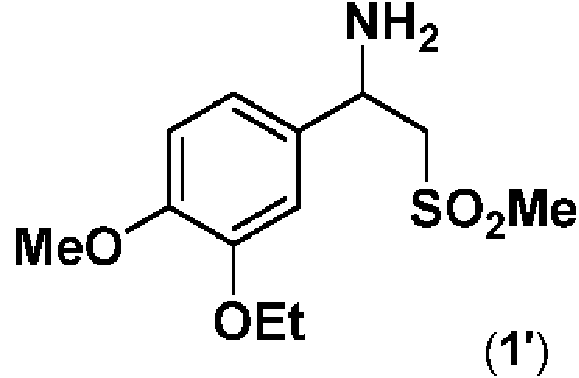
b) resolver ópticamente dicha mezcla racémica (1') o enantioméricamente enriquecida (1") mediante tratamiento con una W-formil leucina enantioméricamente pura en presencia de un disolvente, para producir las correspondientes sales de W-formil-leucina enantioméricamente puras (4).
[0018] En una posible variante de este aspecto de la invención, se lleva a cabo una etapa adicional b') después de la etapa b), que comprende desbloquear la sal enantioméricamente pura (4) preparada en la etapa b) para producir una 1-(3-etoxi-4-metoxifenil)-2-(metilsulfonil)etan-1-amina (1) enantioméricamente pura.
Descripción detallada de la invención
[0019] Todos los términos utilizados en la presente solicitud, a menos que se indique lo contrario, deben interpretarse en su significado ordinario, tal como se conocen en el campo técnico. Otras definiciones más específicas para algunos términos usados en la presente solicitud se proporcionan a continuación y pretenden aplicarse uniformemente a la descripción completa y las reivindicaciones, a menos que se indique lo contrario.
[0020] En general, la nomenclatura utilizada en esta solicitud se basa en AUTONOM™ v.4.0, un sistema computerizado del Beilstein Institute para la generación de nomenclatura sistemática IUPAC. Si hay una discrepancia entre una estructura representada y el nombre que se le da a esa estructura, la estructura representada debe considerarse correcta. Además, si la estereoquímica de una estructura o una parte de una estructura no se indica, por ejemplo, con líneas en negrita o discontinuas, la estructura o parte de la estructura debe interpretarse como que abarca todos los estereoisómeros existentes de la misma.
[0021] Los compuestos preparados mediante los procedimientos de la presente invención pueden tener uno o más centros estereogénicos y pueden existir y pueden ser utilizados o aislados en formas enantioméricamente puras, como mezclas enantioméricas enriquecidas, así como formas diastereoméricamente puras o como mezclas diastereoméricas enriquecidas. Debe entenderse que los procedimientos de la presente invención pueden dar lugar a cualquiera de las formas anteriores o una combinación de las mismas. Debe entenderse además que los productos de los procedimientos descritos en el presente documento se pueden aislar como formas enantiomérica y diastereoméricamente puras o como mezclas enantiomérica y diastereoméricamente enriquecidas.
[0022] El término "arilo" se refiere a cualquier sustituyente derivado de un hidrocarburo aromático monocíclico o policíclico mediante eliminación de un átomo de hidrógeno de un átomo de carbono del anillo (por ejemplo fenilo, tolilo, 1-naftilo o 2-naftilo).
[0023] El signo "*" (asterisco) presente en algunas fórmulas de esta descripción indica un centro estereogénico (asimétrico), aunque la ausencia de asteriscos no implica necesariamente que el compuesto carece de un estereocentro. Tales fórmulas pueden referirse al racemato o a enantiómeros o diastereómeros individuales, que pueden ser o no sustancialmente puros.
[0024] El término "racémico" se refiere a una muestra de un compuesto quiral que contiene los isómeros (+) y (-) en la misma cantidad.
[0025] El término "enantioméricamente enriquecido", tal como se usa en este documento, significa que uno de los enantiómeros de un compuesto está presente en exceso comparado con el otro enantiómero.
[0026] El término "enantioméricamente puro", tal como se usa en este documento, significa que la pureza enantiomérica es habitualmente al menos aproximadamente el 96%, preferiblemente al menos el 99%, más preferiblemente al menos el 99,5%.
[0027] El símbolo ..... (enlace discontinuo) presente en algunas de las fórmulas de la descripción y de las reivindicaciones indica que el sustituyente esta dirigido por debajo del plano de la hoja.
[0028] El símbolo — (enlace en cuña) presente en algunas de las fórmulas de la descripción y de las reivindicaciones indica que el sustituyente está dirigido por encima del plano de la hoja.
[0029] Los compuestos obtenidos mediante las transformaciones químicas de la presente invención se pueden utilizar en las siguientes etapas sin purificación adicional u, opcionalmente, pueden separarse y purificarse empleando procedimientos convencionales bien conocidos por los expertos en la técnica, tales como recristalización, cromatografía en columna, o transformándolos en una sal o en un cocristal con un coformador apropiado, o lavando con un disolvente orgánico o con una solución acuosa, opcionalmente ajustando el pH.
[0030] Se entenderá que cualquier compuesto descrito en este documento también puede describir cualquier sal o cocristal del mismo.
[0031] El término "semilla" se refiere a una sustancia cristalina que se añade a una solución de la misma sustancia para inducir su cristalización. La siembra con un isómero óptico específico a menudo tiene el efecto útil de promover la cristalización de la sustancia en la misma forma de la semilla.
[0032] Cabe señalar que, durante la conversión de la 1-(3-etoxi-4-metoxifenil)-2-(metilsulfonil)etan-1-amina (1) enantioméricamente pura o la sal W-formil-leucina de la misma (4) en una 1,3-dioxoisoindolin-4-ilacetamida enantioméricamente pura (6), no se observa racemización (conversión del enantiómero principal en el menor que conduce a un ee. inferior). En consecuencia, el ee detectado en la 1-(3-etoxi-4-metoxifenil)-2-(metilsulfonil)etan-1-amina pura (1) o en la sal de W-formil-leucina de la misma (4), se retiene sin cambios en la 1,3-dioxoisoindolin-4-ilacetamida pura de fórmula (6).
[0033] De acuerdo con su aspecto más general, la presente invención se refiere a un procedimiento para la preparación de una sal de W-formil-leucina enantioméricamente pura de 1-(3-etoxi-4-metoxifenil)-2-(metilsulfonil) etan-1-amina (4).
[0034] La primera operación del procedimiento de la invención, a), consiste en la provisión de una 1-(3-etoxi-4-metoxifenil)-2-(metilsulfonil)etan-1-amina (1') o una mezcla enriquecida enantioméricamente de la misma (1"). Esta operación se puede realizar según dos rutas alternativas de síntesis a.i) y a.ii).
[0035] La ruta sintética a.i) conduce a la preparación de 1-(3-etoxi-4-metoxifenil)-2-(metilsulfonil)etan-1-amina en forma de una mezcla racémica (1'). Este esquema se puede desarrollar, por ejemplo, de acuerdo con los procedimientos detallados en el documento WO 2010/030345 A2 (a través de 3-etoxi-4-metoxibenzonitrilo) o, preferiblemente, implica las siguientes etapas:
d) hacer reaccionar 3-etoxi-4-metoxibenzaldehído (9) con un compuesto de fórmula R1-NH2 y un compuesto de fórmula R2-S(O)OH, o una sal del mismo para obtener una aminosulfona de fórmula (2):

en la que:
R1 es un grupo protector de amino; y
R2 es un alquilo C1-C6, un alquilo C1-C6 sustituido, un arilo C6-C10 o un arilo C6-C10 sustituido;
e) convertir la aminosulfona de fórmula (2) en una sulfona de fórmula (3) mediante tratamiento con un carbanión derivado de dimetilsulfona:
f) desproteger la sulfona (3) para obtener 1-(3-etoxi-4-metoxifenil)-2-(metilsulfonil)etan-1-amina en forma de una mezcla racémica (1').

Los grupos protectores de amina útiles para la presente invención son, por ejemplo, carbamatos (en los que el átomo de nitrógeno está unido a un grupo de fórmula -C(O)OR, en la que R es, por ejemplo, carbamato de metilo, etilo, terc-butoxi, alilo, bencilo, p-metoxibencilo, p-nitrobencilo, p-clorobencilo o 2,4-diclorobencilo), amidas (en las que el átomo de nitrógeno está unido a un grupo de fórmula -C(O)-R', en la que R' es, por ejemplo, hidrógeno, metilo, clorometilo, triclorometilo, trifluorometilo, fenilo o bencilo) o fosfinoamidas (en las que el átomo de nitrógeno está unido a un grupo de fórmula -P(O)-(R")2, en la que R' es, por ejemplo, arilo, preferiblemente fenilo).
[0036] La etapa d) incluye la conversión de 3-etoxi-4-metoxibenzaldehído (9) en una aminosulfona de fórmula (2). Esta operación se puede realizar convenientemente haciendo reaccionar 3-etoxi-4-metoxibenzaldehído (9) con un compuesto de fórmula R1-NH2 y un compuesto de fórmula R2-S(O)OH o una sal del compuesto de fórmula R2-S(O)OH, en el que los sustituyentes adoptan los significados indicados anteriormente. La etapa d) se lleva a cabo convenientemente en presencia de un ácido, preferiblemente seleccionado del grupo que consiste en un ácido de Lewis (más preferiblemente, clorotrimetilsilano) y un ácido carboxílico (más preferiblemente, ácido fórmico).
[0037] Preferiblemente, la etapa d) se lleva a cabo en presencia de una sal del compuesto de fórmula R2-S(O)OH (generalmente referido como sulfinato). Más preferiblemente, dicho sulfinato es un compuesto de fórmula R2-S(O)OM, en la que los sustituyentes adoptan los significados indicados anteriormente y M es un ión, tal como sodio, potasio, litio, calcio, magnesio, cobre o cesio. Incluso más preferiblemente, dicha sal del compuesto de fórmula R2-S(O)OH es p-toluenosulfinato de sodio.
[0038] El compuesto de fórmula R1-NH2 se selecciona preferiblemente del grupo que consiste en carbamatos (preferiblemente, carbamato de terc-butilo o bencilo), amidas (por ejemplo, formamida o acetamida), fosfinoamidas (preferiblemente, difenilfosfinoamida) o mezclas de los mismos.
[0039] Preferiblemente, la etapa d) se lleva a cabo en al menos un disolvente, más preferiblemente, en una mezcla que comprende agua y al menos un disolvente seleccionado del grupo que consiste en disolventes alcohólicos (por ejemplo, metanol, etanol, 2-propanol, 1-butanol, 2-butanol, terc-butanol y mezclas de los mismos) y éteres (por ejemplo, tetrahidrofurano). Según una realización preferida de este aspecto de la invención, la etapa d) se lleva a cabo en una mezcla que comprende un alcohol (preferiblemente, 1-butanol) y agua, teniendo dicha mezcla, en una realización aún más preferida, una relación del disolvente alcohólico con respecto al agua de aproximadamente 1:0,5 vol/vol a aproximadamente 1:10 vol/vol. Preferiblemente, la relación de disolvente alcohólico con respecto a agua es de aproximadamente 1:2 vol/vol a aproximadamente 1:4 vol/vol.
[0040] En la etapa d) la temperatura de reacción puede estar entre de 0 °C a 40 °C, preferiblemente de aproximadamente 10 °C a aproximadamente 20 °C. En general, cuanto mayor es la temperatura de reacción, menor es el tiempo de reacción.
[0041] La cantidad de ácido usado opcionalmente en la etapa d) (preferiblemente un ácido de Lewis o un ácido carboxílico) es convenientemente de aproximadamente 1 a aproximadamente 2 equivalentes, preferiblemente de 1,05 a 1,50 equivalentes, más preferiblemente, de 1,10 a 1,40, incluso más preferiblemente, de 1,15 a 1,35, con respecto a la cantidad molar del aldehído de partida (9).
El compuesto de fórmula R2-S(O)OH o la sal del mismo se utilizan en la etapa d) en cantidades convenientemente de 1 a 2 equivalentes, preferiblemente de 1,01 a 1,50 equivalentes, más preferiblemente de 1,05 a 1,40, incluso más preferiblemente de 1,08 a 1,35, con respecto a la cantidad molar del aldehído de partida (9).
La cantidad del compuesto de fórmula R1-NH2 usado en la etapa d) es convenientemente de 1 a 2 equivalentes, preferiblemente de 1,01 a 1,50 equivalentes, más preferiblemente de 1,05 a 1,40, incluso más preferiblemente de 1,08 a 1,35, con respecto a la cantidad molar del aldehído de partida (9).
[0042] Los inventores han observado que, en la mezcla de reacción, la aminosulfona (2) está en equilibrio con el aldehído de partida (9), y que el equilibrio se puede desplazar hacia la aminosulfona de reacción (2) al permitir que este compuesto cristalice fuera de la mezcla de reacción, llevando así la reacción a una finalización sustancial.
[0043] Dichas condiciones de cristalización se pueden determinar fácilmente por una persona experta en la técnica por medio de una investigación preliminar que incluya la dispersión de la aminosulfona (2) en el disolvente o mezcla de disolventes utilizados en la etapa d seleccionado) a fin de determinar su equilibrio de solubilidad.
[0044] En una realización de este aspecto de la invención, la aminosulfona precipitada (2) se tritura y, finalmente, se lava con acetato de iso-propilo y posteriormente se convierte en una sulfona de fórmula (3).
[0045] La aminosulfona de fórmula (2) así obtenida, opcionalmente aislada, se convierte posteriormente en una sulfona de fórmula (3), según la etapa e).
[0046] La etapa e) se puede realizar tratando la aminosulfona de fórmula (2), opcionalmente convertida en la imina correspondiente (10) como se detalla a continuación, con un carbanión de dimetilsulfona. Esta etapa se puede llevar a cabo en cualquier disolvente adecuado, tales como éteres (preferiblemente éter dietílico, tetrahidrofurano, 2-metiltetrahidrofurano, terc-butil metil éter), N-metil pirrolidona, N, N-dimetilacetamida, N, N-dimetilformamida, glima, diglima, tolueno, xileno, hexano o mezclas de los mismos; a una temperatura de -80 a -30 °C, preferiblemente de -60 a -50 °C, por ejemplo a -55 °C.
[0047] El carbanión de dimetilsulfona puede prepararse de acuerdo con técnicas convencionales en síntesis orgánica, por ejemplo, mediante el tratamiento de dimetilsulfona con una base fuerte. Las bases fuertes adecuadas se seleccionan preferiblemente del grupo que consiste en reactivos de organolitio, reactivos de organomagnesio o amidas de sodio, litio, potasio o magnesio. La preparación del carbanión de dimetilsulfona normalmente tiene lugar a una temperatura de -80 a -30 °C, por ejemplo de -60 a -50 °C, en un éter (preferiblemente tetrahidrofurano, éter dietílico, 2-metiltetrahidrofurano, terc-butil metil éter, bis(2-metoxietil) éter) opcionalmente en mezcla con un hidrocarburo, alifático o aromático (preferiblemente hexano, tolueno o xileno). Preferiblemente, el disolvente utilizado es una mezcla de tetrahidrofurano y n-hexano.
[0048] Los reactivos de organolitio útiles para este propósito son, por ejemplo n-butil litio, sec-butil litio, fenil litio, neopentil litio, propil litio, terc-butil litio o preferiblemente hexil litio; los reactivos de organomagnesio son, por ejemplo, un haluro de terc-butilmagnesio o isopropilmagnesio (preferiblemente cloruro de terc-butilmagnesio o isopropilmagnesio); las amidas de litio, sodio, potasio o magnesio se seleccionan, por ejemplo, del grupo que comprende bis(trimetilsilil) amidas de sodio, litio o potasio (NaHMDS, LiHMDS o KHMDS), diisopropilamida de litio (LDA) o bis(diisopropilamida) de magnesio (MDA).
[0049] En el caso cuando un reactivo de organomagnesio se utiliza para preparar el carbanión de dimetilsulfona, las condiciones de reacción convenientes incluyen la presencia de al menos un haluro alcalino (preferiblemente un cloruro), por ejemplo un cloruro de litio, sodio, potasio o cesio, o de un haluro de zinc o cobre. Más preferiblemente, el carbanión de dimetilsulfona se prepara mediante una mezcla de cloruro de isopropilmagnesio y cloruro de litio.
[0050] Alternativamente, dicho carbanión se puede preparar mediante el tratamiento de dimetilsulfona con un hidruro (preferiblemente hidruro de sodio, litio o potasio) o un terc-butóxido de metal alcalino (tal como terc-butóxido de sodio o potasio) a una temperatura de -40 a 50 °C, por ejemplo de -30 a 40 °C, en un éter (preferiblemente tetrahidrofurano), en N, N-dimetilacetamida o en N, N-dimetilformamida.
[0051] La base fuerte se utiliza preferiblemente en cantidad estequiométrica con respecto a la cantidad molar de dimetilsulfona utilizada.
[0052] La proporción molar del carbanión de dimetilsulfona con respecto a la aminosulfona de fórmula (2) es normalmente de 1 a 6, preferiblemente de 3 a 4.
[0053] En una posible variante de esta operación, la aminosulfona de fórmula (2) se convierte en la imina correspondiente (10):

en la que R1 puede adoptar cualquiera de los significados indicados anteriormente;
que posteriormente se trata con el carbanión de dimetilsulfona.
[0054] Dicha imina (10) puede preparse, por ejemplo, mediante tratamiento de la aminosulfona (2) con una de las bases mencionadas anteriormente para operar la etapa e). Alternativamente, dicha etapa se puede realizar tratando la aminosulfona (2) con un hidróxido o un carbonato de un metal alcalino (tal como K2CO3, Na2CO3, Li2CO3, Cs2CO3, KOH, NaOH, LiOH) en un disolvente miscible en agua (por ejemplo, metanol, etanol, tetrahidrofurano, dimetoxietano, dioxano o una mezcla de los mismos) opcionalmente en mezcla con agua. La cantidad del hidróxido o carbonato del metal alcalino utilizada es normalmente de 1 a 5 equivalentes, preferiblemente de 1,5 a 2 equivalentes, en comparación con la cantidad molar de la aminosulfona (2) utilizada.
[0055] Preferiblemente, el carbanión de dimetilsulfona utilizado en la etapa e) es LiCH2SO2Me, más preferiblemente, LiCH2SO2Me preparado in situ por reacción de Me2SO2 con nBuLi o, incluso más preferiblemente, con nHexLi.
[0056] Preferiblemente, en el caso cuando nHexLi se utiliza para preparar el carbanión de dimetilsulfona, la relación de compuesto intermedio (2) con respecto a dimetilsulfona y nHexLi es de aproximadamente 1:2:2 a aproximadamente 1:6:6 en base molar, preferiblemente aproximadamente 1:4:4 en base molar.
[0057] La siguiente etapa f), implica la conversión de sulfona (3), opcionalmente aislada, en una mezcla racémica de 1-(3-etoxi-4-metoxifenil)-2-(metilsulfonil)etan-1-amina (1'). Esta operación se puede realizar usando cualquiera de los procedimientos generalmente conocidos en el campo para eliminar un grupo protector de amino, por ejemplo, uno de los descritos en Theodora W. Green, Protective Groups in Organic Synthesis, John Wiley & Sons (1999), páginas 503-598, que se incorporan en el presente documento como referencia. Preferiblemente, en el caso en el que R1 forma, junto con el átomo de nitrógeno al que está unido, un grupo carbamato, dicha etapa de desprotección se puede realizar de acuerdo con los procedimientos detallados en las páginas 504-540 del texto referido anteriormente, mientras que, en el caso donde R1 forme, junto con el átomo de nitrógeno al que está unido, una amida o un grupo fosfinoamida, dicha etapa de desprotección se puede realizar según los procedimientos descritos respectivamente en las páginas 551-561 y 598 del mismo texto. De acuerdo con una realización aún más preferida de este aspecto de la invención, en el caso en el que R1 es un benciloxicarbonilo, la etapa f) se puede llevar a cabo mediante tratamiento con hidrógeno en presencia de un catalizador (por ejemplo, paladio, platino o níquel) opcionalmente soportado sobre un portador apropiado, tal como carbonato de carbono o bario en un alcohol (preferiblemente, metanol o etanol) o una mezcla de los mismos con agua. Por el contrario, cuando R1 es un grupo terc-butoxicarbonilo, puede llevarse a cabo de acuerdo con uno de los procedimientos descritos en Theodora W. Green, Protective Groups in Organic Synthesis, John Wiley & Sons (1999), páginas 520-522. Preferiblemente, la etapa f) se realiza tratando la sulfona (3) con una solución de cloruro de hidrógeno en agua o en un disolvente orgánico.
[0058] La ruta sintética alternativa a.ii) conduce a la preparación de una mezcla enantioméricamente enriquecida de 1-(3-etoxi-4-metoxifenil)-2-(metilsulfonil)etan-1-amina (1"). Esta etapa puede llevarse a cabo de acuerdo con procedimientos conocidos en la técnica, por ejemplo los detallados en WO 2013/126360 A2 o en WO 2013/126495 A2.
[0059] La 1-(3-etoxi-4-metoxifenil)-2-(metilsulfonil)etan-1-amina (1') o (1") opcionalmente aislada proporcionada en la etapa a), se trata posteriormente, según la etapa b), con una W-formil leucina enantioméricamente pura, preferiblemente W-formil-L-leucina, en presencia de un disolvente para obtener las correspondientes sales diastereoméricas de W-formil leucina que finalmente se purifican ópticamente por resolución de la sal diastereomérica a la sal de W-formil-leucina enantiomérica pura de 1-(3-etoxi-4-metoxifenil)-2-(metilsulfonil)etan-1-amina (4).
[0060] La formación y el fraccionamiento de las sales diastereoméricas se realizan generalmente por calentamiento hasta una temperatura próxima al punto de ebullición del disolvente utilizado, seguido de enfriamiento hasta una temperatura entre 0 y 40 °C, preferiblemente entre 10 y 30 °C. La formación de la sal se completa en unos minutos, pero el tiempo de reacción puede extenderse a varias horas sin causar ninguna alteración. La relación molar de W-formil leucina con respecto a 1-(3-etoxi-4-metoxifenil)-2-(metilsulfonil)etan-1-amina (1') o (1") es normalmente de 0,5 a 1,5, preferiblemente 1. Ejemplos de disolventes adecuados para la formación y fraccionamiento de las sales son alcoholes (por ejemplo, etanol, 2-propanol, 1-butanol, 2-butanol o preferiblemente metanol), opcionalmente en mezcla con agua.
[0061] El volumen del disolvente es normalmente de 5 ml a 20 ml por gramo de 1-(3-etoxi-4-metoxifenil)-2-(metilsulfonil)etan-1-amina (1') o (1") utilizada; preferiblemente, dicho volumen es de 6 a 12 ml por gramo de 1-(3-etoxi-4-metoxifenil)-2-(metilsulfonil)etan-1-amina (1') o (1"), incluso más preferiblemente de 8 a 11 ml.
[0062] En una posible variante de esta operación, una semilla del isómero ópticamente activo deseado se añade a la solución de las sales diastereoméricas antes de enfriarla.
[0063] Si el exceso enantiomérico de la sal de W-formil leucina (4) todavía no es adecuado para el uso previsto, se lleva a cabo trituración en caliente, según la etapa g), a temperatura de reflujo en los mismos disolventes alcohólicos usados para realizar la etapa b), preferiblemente en metanol. Durante dicha trituración en caliente, la relación de
disolvente alcohólico con respecto a la sal de A/-form¡l leucina (4) es de 6:1 v/p a 4:1 v/p, preferiblemente de 5:1 v/p a 4,5:1 v/p.
[0064] Una variante del procedimiento objeto de este aspecto de la invención incluye una etapa adicional b'), llevada a cabo después de la etapa b), que comprende desbloquear la sal (4) enantioméricamente pura recuperada en la etapa b) para producir la correspondiente 1-(3-etox¡-4-metox¡fen¡l)-2-(met¡lsulfon¡l)etan-1-am¡na (1) enantioméricamente pura. Preferiblemente, dicho desbloqueo se lleva a cabo poniendo en contacto la sal pura preparada en la etapa b) con un hidróxido o un carbonato de un metal alcalino (tal como K2CO3, Na2CO3, LhCO3, Cs2CO3, KOH, NaOH, LiOH) en agua o en una mezcla que comprende agua y al menos un disolvente, preferiblemente seleccionado del grupo que consiste en disolventes miscibles en agua (por ejemplo, metanol, etanol, tetrahidrofurano, dioxano y una mezcla de los mismos); éteres (por ejemplo, tetrahidrofurano, 2-metiltetrahidrofurano); ésteres (por ejemplo, acetato de etilo, acetato de isopropilo, acetato de n-propilo, acetato de n-butilo); hidrocarburos alifáticos o aromáticos (por ejemplo, hexano, heptano o tolueno) y disolventes halogenados (preferiblemente, diclorometano). La cantidad del hidróxido o carbonato del metal alcalino utilizada es normalmente de 1 a 5 equivalentes, preferiblemente de 1,05 a 4 equivalentes, más preferiblemente de 1,1 a 3 equivalentes, en comparación con la cantidad molar de la sal (4) enantioméricamente pura.
[0065] De acuerdo con una realización alternativa y preferida de este aspecto de la invención, el desbloqueo de la etapa b') se lleva a cabo en agua a una temperatura de 20 a 100 °C. Incluso más preferiblemente, el desbloqueo de la etapa b') se lleva a cabo en agua a una temperatura de 40 a 100 °C, seguido de enfriamiento hasta una temperatura de 0 a 35 °C para promover la cristalización de la 1-(3-etox¡-4-metox¡fen¡l)-2-(met¡lsulfon¡l)etan-1-am¡na (1) enantioméricamente pura.
[0066] En otra realización preferida de la invención, la sal W-formil-leucina (4) enantioméricamente pura preparada en la etapa b), o la 1-(3-etox¡-4-metox¡fen¡l)-2-(met¡lsulfon¡l)etan-1-am¡na (1) enantioméricamente pura producida en la etapa b') se hacen reaccionar adicionalmente, en una etapa c) opcional, con W-(1,3-dioxo-1,3-dihidroisobenzofuran^-i^acetamida (5) para producir una 1,3-d¡oxo¡so¡ndol¡n-4-¡lacetam¡da enantioméricamente pura de fórmula (6), un cocristal, un solvato o un hidrato de la misma:

[0067] La W-(1,3-d¡oxo-1,3-d¡h¡dro¡sobenzofuran-4-¡l)acetam¡da (5) usada como reactivo en esta etapa está disponible comercialmente; alternativamente, se puede preparar de acuerdo con técnicas estándar en síntesis orgánica, por ejemplo, de acuerdo con el procedimiento descrito en el documento US 4.456.595 A.
[0068] El acoplamiento entre la 1-(3-etox¡-4-metox¡fen¡l)-2-(met¡lsulfon¡l)etan-1-am¡na (1) pura o la sal de W-formil leucina pura (4) y W-(1,3-d¡oxo-1,3-d¡h¡dro¡sobenzofuran-4-¡l)acetam¡da (5) se lleva a cabo, en la etapa c) opcional, a una temperatura de 30 °C hasta el punto de ebullición del disolvente utilizado, preferiblemente de 40 a 90 °C, por ejemplo a 75°C, en un disolvente adecuado, preferiblemente una mezcla de W, W-dimetilacetamida y ácido acético.
[0069] La cantidad de W-(1,3-d¡oxo-1,3-d¡h¡dro¡sobenzofuran-4-¡l)acetam¡da (5) es normalmente de 1 a 2 equivalentes, preferiblemente de 1,03 a 1,05 equivalentes, con respecto a la cantidad molar de la 1-(3-etoxi-4-metox¡fen¡l)-2-(met¡lsulfon¡l)etan-1-am¡na (1) enantioméricamente pura o de la sal pura (4).
[0070] En una realización más preferida del mismo, el procedimiento objeto de este aspecto de la invención puede comprender una etapa opcional adicional h) que incluye la cristalización de la 1,3-d¡oxo¡so¡ndol¡n-4-¡lacetam¡da enantioméricamente pura de fórmula (6) preparada de acuerdo con la etapa c).
[0071] Dicha cristalización se realiza, generalmente, calentando una solución de la 1,3-d¡oxo¡so¡ndol¡n-4-¡lacetam¡da de fórmula (6) hasta una temperatura próxima al punto de ebullición del disolvente utilizado, seguido de enfriamiento hasta una temperatura de 0 a 30 °C. Los ejemplos de disolventes adecuados para el objetivo son agua; alcoholes (por ejemplo, metanol, etanol); hidrocarburos, alifáticos o aromáticos (preferiblemente hexano, heptano o tolueno); acetatos (preferiblemente acetato de etilo o acetato de isopropilo); éteres (preferiblemente, terc-butil metil éter o tetrahidrofurano); cetonas (preferiblemente, acetona o 2-butanona); disolventes clorados (preferiblemente, diclorometano) y mezclas de los mismos.
[0072] Preferiblemente, la sal de A-formil-leucina enantioméricamente pura de 1-(3-etoxi-4-metoxifenil)-2-(metilsulfonil)etan-l-amina (4), preparada en la operación b) del procedimiento, es una sal de A-formil-L-leucina de fórmula (4A):

[0073] En este caso, el procedimiento de la invención conduce, después de la etapa opcional c), a la formación de Apremilast, un cocristal, un solvato o un hidrato del mismo.
[0074] En otra realización de la misma, este aspecto de la invención se refiere a un procedimiento para la preparación de una 1,3-dioxoisoindolin-4-ilacetamida enantioméricamente pura de fórmula (6) (preferiblemente Apremilast), un cocristal, un solvato o una hidrato de la misma, que comprende hacer reaccionar 1-(3-etoxi-4-metoxifenil)-2-(metilsulfonil)etan-1-amina (1) o su sal de A-formil leucina (4) con A-(1,3-dioxo-1,3-dihidroisobenzofuran-4-il)acetamida (5).
[0075] El procedimiento de la presente invención alcanza el objeto propuesto ya que, sorprendentemente, sólo se requiere una cristalización y una trituración en caliente de la sal de A-formil-leucina de 1-(3-etoxi-4-metoxifenil)-2-(metilsulfonil)etan-1-amina (4) para lograr una beta-aminosulfona (1) con una pureza óptica adecuada para usos farmacéuticos. En comparación, el procedimiento de WO 03/080049 A1, además de mayores volúmenes de disolvente, requiere más de dos etapas de cristalización de la sal diastereomérica correspondiente para lograr el mismo objetivo. Por tanto, es sorprendente que pueda obtenerse una mejora notable del rendimiento de la resolución de la mezcla racémica simplemente cambiando el radical unido al átomo de nitrógeno de la leucina, de acetilo a formilo.
[0076] El uso de la 1-(3-etoxi-4-metoxifenil)-2-(metilsulfonil)etan-1-amina (1) enantioméricamente pura para preparar directamente Apremilast es ventajoso, ya que el Apremilast puede aislarse mediante precipitación inducida por dilución con un antidisolvente cuando la reacción alcanza su finalización sustancial.
[0077] La invención se ilustra adicionalmente mediante los siguientes Ejemplos.
Ejemplo 1
Resolución de la mezcla racémica (1') con A-Formil-L-Leucina (A-CHO-L-Leu).
[0078] En un reactor de vidrio con camisa de 3 litros, equipado con agitador mecánico, termómetro, condensador y mantenido bajo gas inerte, se cargaron a 20-25 °C beta-aminosulfona racémica (1') (200 g, 0731 moles), A-Formil-L-leucina (115 g, 0,722 moles) y metanol (2200 ml). Con agitación, la temperatura se llevó a reflujo del sistema (aproximadamente 65 °C) y se mantuvo en estas condiciones hasta que se obtuvo una solución. A continuación, la solución se dejó enfriar hasta 40° C y, a continuación, se mantuvo con agitación a 40° C hasta que se observó la formación de una suspensión espesa. La suspensión se calentó hasta 45 - 47 ° C para homogeneizar la masa durante 30 minutos adicionales y, a continuación, se enfrió hasta 20 - 25 °C durante un período de tres a cuatro horas. El sólido se filtró mediante un embudo Buckner y se lavó con metanol frío (3 x 100 ml). La torta húmeda se secó al horno a 45-50 °C a presión reducida hasta peso constante, produciendo así 110 g (0,256 moles) del producto en bruto (rendimiento molar de la beta-aminosulfona racémica 35,0%, 97,65 de ee, S:R 98,8:1,2).
[0079] El sólido en bruto (110 g) se mezcló con 500 ml de metanol fresco y la mezcla se llevó hasta temperatura de reflujo (65 °C) y se mantuvo en estas condiciones durante dos horas. A continuación, la suspensión se enfrió hasta 20-25 °C durante un período de tres a cuatro horas y se mantuvo en estas condiciones durante dos horas más. Finalmente, la suspensión se filtró mediante un embudo Buckner y se lavó con metanol (3 x 100 ml). La torta húmeda se secó en horno a 45 - 50 °C a presión reducida hasta peso constante, produciendo así 95 g (0,220 moles) de la sal diastereomérica (4') (99,8% de ee, S:R 99,9:0,1, rendimiento molar total de beta-aminosulfona racémica: 30%).
Ejemplo 2
[0080] Este ejemplo es representativo de la etapa d) de la presente invención.

[0081] En un reactor de vidrio con camisa de 3 litros, equipado con agitador mecánico, termómetro, condensador y mantenido bajo gas inerte, se cargaron a 20-25 °C 3-etoxi-4-metoxibenzaldehído (200 g), carbamato de terc-butilo (144 g), p-toluenosulfinato de sodio (216 g), 1-butanol (130 ml) y agua (1080 ml). La adición de los disolventes sobre los sólidos dio como resultado una endotermia latente del sistema (disolución endotérmica). Con agitación, se ajustó la temperatura a 20-25°C, se añadieron 68 g de ácido fórmico y se mantuvo el sistema en estas condiciones durante 48 horas adicionales, para que el producto intermedio (11) precipitara abundantemente. A continuación, el sistema se diluyó con 800 ml de agua y se mantuvo en agitación durante 24 horas adicionales.
Después de enfriar a 0-5°C, se añadió lentamente a la suspensión una solución acuosa de hidrogenocarbonato de sodio al 15% (p/p) hasta un pH estable de 7,0 - 7,3. La mezcla resultante se filtró finalmente a presión reducida y la torta húmeda se lavó con 3 x 100 ml de agua. El sólido húmedo se cargó nuevamente en el mismo reactor y se agregaron 1200 ml de acetato de isopropilo. La suspensión resultante se agitó durante una hora a 20-25°C, a continuación se filtró y la torta húmeda se lavó con 3 x 60 ml de acetato de isopropilo. El producto final se secó al vacío a 40°C, proporcionando así 300 g de intermedio (11).
Ejemplo 3
[0082] Este ejemplo es representativo de las etapas e) y f) de la presente invención.

[0083] Se cargaron dimetilsulfona (310 g) y tetrahidrofurano (1800 ml) a 20-25 °C en un reactor de vidrio con camisa de 10 litros equipado con agitador mecánico, termómetro, condensador y se mantuvo bajo gas inerte. La suspensión resultante se enfrió hasta -60 °C. Con agitación, se añadió n-hexil litio (solución 2,3 M en hexano, 1440 ml) a una velocidad tal que la temperatura de reacción no excediera de -40 °C (reacción exotérmica). A continuación, se añadió al reactor una solución preparada previamente de intermedio activado (11) (360 g) en tetrahidrofurano (3600 ml) a una velocidad tal que la temperatura de reacción no excediera de -55°C. Al final de la adición, el sistema se mantuvo a -55 °C durante una hora adicional. A continuación, la masa se vertió rápidamente en otro reactor que contenía 1260 mL de agua a 20-25 °C. El reactor se enjuagó con 360 ml de tetrahidrofurano y el enjuague se combinó con la mezcla inactivada. La temperatura de la masa se ajustó a 20-25 °C y se dejó que las capas se separaran. La fase acuosa inferior se transfirió a otro reactor adecuado y se contraextrajo con 540 ml de tolueno a 45-50 °C. Las capas orgánicas combinadas se concentraron a presión reducida (25-40 mbar; T = 50-55 °C) hasta un residuo viscoso.
Después de haber añadido tolueno (1080 ml), agua (270 ml) y ácido clorhídrico (12 N, 170 g), la solución resultante se calentó hasta 65-70 °C y se mantuvo bajo agitación en estas condiciones durante una hora adicional. La mezcla se enfrió hasta 25-30 °C y se dejó que las capas se separaran. La capa acuosa inferior se transfirió a otro reactor, donde se añadieron 1440 ml de 1-butanol y 240 g de solución acuosa de hidróxido de sodio al 30% (p/p), manteniendo la temperatura interna por debajo de 50 °C. La temperatura de la masa se ajustó a 45-50 °C y se dejó que las capas se separaran. La fase acuosa inferior se transfirió a otro reactor adecuado y se contraextrajo con 180 ml de 1-butanol. Las capas orgánicas combinadas se concentraron a presión reducida (25-40 mbar; T = 55-60 °C) hasta un residuo viscoso. La masa se diluyó con 540 ml de 1-butanol, se calentó hasta 75-80 °C y se filtró a través de una almohadilla de celita para eliminar las partículas insolubles. El reactor y los conductos se enjuagaron con 180 ml de 1-butanol y las fases alcohólicas combinadas se dejaron enfriar hasta 0-5 °C para que cristalizara el compuesto (1'). El sólido obtenido se filtró, la torta húmeda se lavó con 2 x 180 ml de 1-butanol y el producto final se secó al vacío a 45-50 °C, proporcionando así 145 g del compuesto (1').
1H RMN (CDCI3): 51,43-1,46 (t, 3H), 2,88 (s, 3H), 3,18-3,32 (m, 2H), 3,83 (s, 3H), 4,07-4,11 (q, 2H), 4,55-4,58 (d, 1H), 6,81-6,89 (m, 3H), 8,71 (2H).
Ejemplo 4
[0084] Este ejemplo es representativo de la etapa adicional b') de la presente invención.
[0085] A una solución de carbonato de sodio (17,5 g, 165 mmol) en agua (220 ml) a 20-25 °C, se añadió la sal de N-formil-L-Leucina (4') (65,0 g, 150 mmol) bajo agitación. La mezcla se calentó hasta 65-70 °C hasta que se obtuvo una solución, a continuación se enfrió hasta 50-55°C y se mantuvo en las mismas condiciones hasta que se produjo la cristalización (aproximadamente 30 minutos). La suspensión se enfrió hasta 25 °C durante 3-4 horas y el sólido se filtró y se lavó con agua. El sólido se secó a presión reducida a 55 °C para producir 40 g de 1-(3-etoxi-4-metoxifenil)-2-(metilsulfonil)etan-1-amina (1) enantioméricamente pura (rendimiento del 97% de la sal 4').
Claims (15)
1. Procedimiento para la preparación de una sal de W-formil leucina enantioméricamente pura de 1-(3-etoxi-4-metoxifenil)-2-(metilsulfonil)etan-1-amina (4)

comprendiendo dicho procedimiento las etapas de:
a) proporcionar 1-(3-etoxi-4-metoxifenil)-2-(metilsulfonil)etan-1-amina en forma de una mezcla racémica (1') o en forma de una mezcla enantioméricamente enriquecida de la misma

b) resolver ópticamente dicha mezcla racémica (1') o enantioméricamente enriquecida (1") mediante tratamiento con una W-formil leucina enantioméricamente pura en presencia de un disolvente para producir las correspondientes sales de W-formil-leucina enantioméricamente puras (4).
2. Procedimiento, según la reivindicación 1, en el que la 1-(3-etoxi-4-metoxifenil)-2-(metilsulfonil)etan-1-amina (1') racémica se prepara según las siguientes etapas:
d) hacer reaccionar 3-etoxi-4-metoxibenzaldehído (9) con un compuesto de fórmula R1-NH2 y un compuesto de fórmula R2-S(O)OH, o una sal del mismo para obtener una aminosulfona de fórmula (2):

en la que:
R1 es un grupo protector de amino; y
R2 es un alquilo C1-C6, un alquilo C1-C6 sustituido, un arilo C6-C10 o un arilo C6-C10 sustituido;
e) convertir la aminosulfona de fórmula (2) en una sulfona de fórmula (3) mediante tratamiento con un carbanión derivado de dimetilsulfona:

f) desproteger la sulfona (3) para obtener 1-(3-etoxi-4-metoxifenil)-2-(metilsulfonil)etan-1-amina (1') en forma de una mezcla racémica.

3. Procedimiento, según cualquiera de las reivindicaciones 1 y 2, en el que, en la etapa b), se añade una semilla del isómero ópticamente activo deseado durante la resolución de la sal diastereomérica.
4. Procedimiento, según cualquiera de las reivindicaciones 1 a 3, en el que el volumen de disolvente utilizado en la etapa b) está entre 5 ml y 20 ml por gramo de 1-(3-etoxi-4-metoxifenil)-2-(metilsulfonilo)etan-1-amina (1') o (1"). 5. Procedimiento según cualquiera de las reivindicaciones 1 a 4, en el que la relación molar de W-formil leucina con respecto a 1-(3-etoxi-4-metoxifenil)-2-(metilsulfonil)etan-1-amina (1') o (1") está entre 0,5 y 1,
5.
6. Procedimiento, según cualquiera de las reivindicaciones 1 a 5, que comprende, además, una etapa g) llevada a cabo después de la etapa b), que comprende triturar en caliente la sal de W-formil leucina de 1-(3-etoxi-4-metoxifenil)-2-(metilsulfonil)etan-1-amina (4) en un disolvente.
7. Procedimiento, según cualquiera de las reivindicaciones 1 a 6, en el que el disolvente utilizado en la etapa b) o en la etapa g) es un alcohol.
8. Procedimiento, según la reivindicación 7, en el que dicho alcohol se selecciona del grupo que consiste en metanol, etanol, 2-propanol, 1-butanol, 2-butanol, terc-butanol o mezclas de los mismos.
9. Procedimiento, según cualquiera de las reivindicaciones 1 a 8, que comprende, además, una etapa b') que incluye desbloquear la sal enantioméricamente pura (4) preparada en la etapa b) para producir una 1-(3-etoxi-4-metoxifenil). -2-(metilsulfonil)etan-1-amina (1) enantioméricamente pura.
10. Procedimiento, según cualquiera de las reivindicaciones 1 a 9, que comprende, además, una etapa c) que incluye hacer reaccionar 1-(3-etoxi-4-metoxifenil)-2-(metilsulfonil)etan-1-amina (1) o su sal de W-formil leucina (4) con W-(1,3-dioxo-1,3-dihidroisobenzofuran-4-il)acetamida (5) para producir una 1,3-dioxoisoindolin-4-ilacetamida enantioméricamente pura de fórmula (6), un cocristal, un solvato o un hidrato de la misma:

11. Procedimiento, según la reivindicación 10, que comprende, además, una etapa h), que incluye cristalizar la 1,3-dioxoisoindolin-4-ilacetamida enantioméricamente pura de fórmula (6) producida en la etapa c).
12. Sal de W-formil leucina enantioméricamente pura de 1-(3-etoxi-4-metoxifenil)-2-(metilsulfonil)etan-1-amina (4):

13. Sal de W-formil leucina, según la reivindicación 13, que tiene la fórmula (4A):

14. Uso de la sal de A/-form¡l leucina, según la reivindicación 13, para la preparación de Apremilast, un cocristal, un solvato o un hidrato del mismo.
15. Uso de la sal de W-formil leucina, según la reivindicación 12, para la preparación de una 1,3-d¡oxo¡so¡ndol¡n-4-ilacetamida enantioméricamente pura de fórmula (6), tal como se muestra en la reivindicación 10, un cocristal, un solvato o un hidrato de la misma.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP17382175 | 2017-04-04 | ||
| PCT/EP2018/057898 WO2018184936A1 (en) | 2017-04-04 | 2018-03-28 | Resolution of racemic beta-aminosulfone compounds |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2829253T3 true ES2829253T3 (es) | 2021-05-31 |
Family
ID=58501429
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES18716164T Active ES2829253T3 (es) | 2017-04-04 | 2018-03-28 | Resolución de compuestos de beta-aminosulfona racémicos |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US11149003B2 (es) |
| EP (1) | EP3606908B1 (es) |
| ES (1) | ES2829253T3 (es) |
| HR (1) | HRP20201376T1 (es) |
| HU (1) | HUE051684T2 (es) |
| SI (1) | SI3606908T1 (es) |
| WO (1) | WO2018184936A1 (es) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| HRP20201376T1 (hr) | 2017-04-04 | 2020-12-11 | Quimica Sintetica, S.A. | Rezolucija racemičnih beta-aminosulfonskih spojeva |
Family Cites Families (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4456595A (en) | 1982-12-06 | 1984-06-26 | E. R. Squibb & Sons, Inc. | Carboxy and substituted carboxy aroly peptides |
| US6020358A (en) | 1998-10-30 | 2000-02-01 | Celgene Corporation | Substituted phenethylsulfones and method of reducing TNFα levels |
| US6962940B2 (en) | 2002-03-20 | 2005-11-08 | Celgene Corporation | (+)-2-[1-(3-Ethoxy-4-methoxyphenyl)-2-methylsulfonylethyl]-4-acetylaminoisoindoline-1,3-dione: methods of using and compositions thereof |
| HRP20130356T1 (en) | 2008-09-10 | 2013-05-31 | Celgene Corporation | Processes for the preparation of aminosulfone compounds |
| BRPI1012770B8 (pt) | 2009-05-14 | 2021-05-25 | Tianjin Hemay Bio Tech Co Ltd | derivados de tiofeno |
| AR090100A1 (es) | 2012-02-21 | 2014-10-22 | Celgene Corp | Procesos para la preparacion de la (s)-1-(3-etoxi-4-metoxifenil)-2-metanosulfoniletilamina |
| US9126906B2 (en) | 2012-02-21 | 2015-09-08 | Celgene Corporation | Asymmetric synthetic processes for the preparation of aminosulfone compounds |
| WO2016146990A1 (en) * | 2015-03-19 | 2016-09-22 | Cipla Limited | Improved process for the preparation of apremilast |
| EP3659997A1 (en) * | 2015-11-13 | 2020-06-03 | API Corporation | Method for producing lacosamide and intermediate thereof |
| CN105348172B (zh) * | 2015-12-04 | 2017-11-14 | 新发药业有限公司 | (s)‑1‑(4‑甲氧基‑3‑乙氧基)苯基‑2‑甲磺酰基乙胺的制备及阿普斯特的制备方法 |
| HRP20201376T1 (hr) | 2017-04-04 | 2020-12-11 | Quimica Sintetica, S.A. | Rezolucija racemičnih beta-aminosulfonskih spojeva |
-
2018
- 2018-03-28 HR HRP20201376TT patent/HRP20201376T1/hr unknown
- 2018-03-28 ES ES18716164T patent/ES2829253T3/es active Active
- 2018-03-28 EP EP18716164.1A patent/EP3606908B1/en active Active
- 2018-03-28 WO PCT/EP2018/057898 patent/WO2018184936A1/en not_active Ceased
- 2018-03-28 SI SI201830111T patent/SI3606908T1/sl unknown
- 2018-03-28 HU HUE18716164A patent/HUE051684T2/hu unknown
- 2018-03-28 US US16/500,671 patent/US11149003B2/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| SI3606908T1 (sl) | 2020-10-30 |
| EP3606908B1 (en) | 2020-06-17 |
| EP3606908A1 (en) | 2020-02-12 |
| HUE051684T2 (hu) | 2021-03-29 |
| US20200199072A1 (en) | 2020-06-25 |
| US11149003B2 (en) | 2021-10-19 |
| WO2018184936A1 (en) | 2018-10-11 |
| HRP20201376T1 (hr) | 2020-12-11 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR100572687B1 (ko) | 광학적으로 순수한 4-하이드록시-2-옥소-1-피롤리딘아세트아미드의 제조방법 | |
| JP4170753B2 (ja) | オキサゾリジノン化合物の製造方法 | |
| ES2398579T3 (es) | Una síntesis asimétrica de ácido (S)-(+)-3-(aminometil)-5-metilhexanoico | |
| ES2607089T3 (es) | Método de producción de un compuesto intermedio para sintetizar un medicamento | |
| ES2909825T3 (es) | Procedimientos para la preparación de oxazolinas diastereomérica y enantioméricamente enriquecidas | |
| US10858324B2 (en) | Processes for the preparation of pyrimidinylcyclopentane compounds | |
| WO2015037460A1 (ja) | 光学活性な3-(ビフェニル-4-イル)-2-[(t-ブトキシカルボニル)アミノ]プロパン-1-オールの製造方法 | |
| TWI397380B (zh) | 含磷之α胺基酸之製造方法及其製造中間體 | |
| ES2829253T3 (es) | Resolución de compuestos de beta-aminosulfona racémicos | |
| KR101430116B1 (ko) | 스트레커 반응용 촉매를 사용하는 키랄성 α-아미노나이트릴의 제조방법 | |
| Sibi et al. | Synthesis of silicon containing alanines | |
| TW202229229A (zh) | 4-硼-l-苯基丙胺酸及其中間體之製造方法 | |
| ES2866476T3 (es) | Compuestos de beta-aminosulfona racémicos | |
| JP2013010732A (ja) | プロリン化合物の製造方法 | |
| JP5569938B2 (ja) | ピロリジン誘導体及びその製造方法 | |
| JP2017014109A (ja) | 光学活性2,6−ジメチルチロシン誘導体の製造法 | |
| WO2000053571A1 (fr) | PROCÉDÉS DE PRÉPARATION D'α-AMINOCÉTONES | |
| EP1078919B1 (en) | Synthesis of alpha-amino-alpha',alpha'-dihaloketones and process for the preparation of beta)-amino acid derivatives by the use of the same | |
| JP2021512162A (ja) | (6s、15s)−3,8,13,18−テトラアザイコサン−6,15−ジオールの製造方法 | |
| JP3855323B2 (ja) | 3−アミノ−2−オキソ−1−ハロゲノプロパン誘導体の製造方法 | |
| JP4187822B2 (ja) | 光学活性4−ヒドロキシ−2−ピロリドンの製造方法 | |
| JP3778843B2 (ja) | 光学活性アミン誘導体および合成法 | |
| CN115996730A (zh) | 用于合成阳离子脂质的方法 | |
| WO2017098975A1 (ja) | ビフェニル化合物の製造方法 | |
| CN103435534A (zh) | 具有环丙烷结构s-脯氨酸的制备方法 |